UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 8-K
CURRENT REPORT
Pursuant to Section 13 or 15(d) of
The Securities Exchange Act of 1934
Date of Report (Date of earliest event reported): January 13, 2025 (January 13, 2025)
REGENERON PHARMACEUTICALS, INC.
(Exact name of registrant as specified in its charter)
New York
(State or other jurisdiction of incorporation)
| 000-19034 | 13-3444607 | |
|
(Commission File Number) |
(I.R.S. Employer Identification No.) |
|
| 777 Old Saw Mill River Road, Tarrytown, New York | 10591-6707 | |
| (Address of principal executive offices) | (Zip Code) | |
Registrant’s telephone number, including area code: (914) 847-7000
Check the appropriate box below if the Form 8-K filing is intended to simultaneously satisfy the filing obligation of the registrant under any of the following provisions (see General Instruction A.2. below):
¨ Written communications pursuant to Rule 425 under the Securities Act (17 CFR 230.425)
¨ Soliciting material pursuant to Rule 14a-12 under the Exchange Act (17 CFR 240.14a-12)
¨ Pre-commencement communications pursuant to Rule 14d-2(b) under the Exchange Act (17 CFR 240.14d-2(b))
¨ Pre-commencement communications pursuant to Rule 13e-4(c) under the Exchange Act (17 CFR 240.13e-4(c))
Securities registered pursuant to Section 12(b) of the Act:
| Title of each class | Trading Symbol(s) | Name of each exchange on which registered |
| Common Stock – par value $0.001 per share | REGN | NASDAQ Global Select Market |
Indicate by check mark whether the registrant is an emerging growth company as defined in Rule 405 of the Securities Act of 1933 (§ 230.405 of this chapter) or Rule 12b-2 of the Securities Exchange Act of 1934 (§ 240.12b-2 of this chapter).
Emerging growth company ¨
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ¨
| Item 2.02. | Results of Operations and Financial Condition. |
On January 13, 2025, Regeneron Pharmaceuticals, Inc. (“Regeneron” or the “Company”) issued a press release titled “Regeneron Provides Business Updates and Highlights from Broad Clinical Pipeline at the 43rd Annual J.P. Morgan Healthcare Conference.” The press release and the Company’s January 13, 2025 conference presentation contain certain preliminary (unaudited) financial information for the fourth quarter and full year 2024 and are being furnished to the Securities and Exchange Commission as Exhibits 99.1 and 99.2 to this Current Report on Form 8-K, respectively, and incorporated by reference in this Item 2.02.
Additionally, the Company currently expects that its financial results calculated in accordance with U.S. generally accepted accounting principles (“GAAP”) and its non-GAAP financial results for the fourth quarter 2024 will include an acquired in-process research and development (“IPR&D”) charge of approximately $14 million on a pre-tax basis. This charge primarily relates to asset acquisitions. This acquired IPR&D charge is expected to negatively impact each of GAAP and non-GAAP net income per diluted share for the fourth quarter 2024 by approximately $0.11.
Acquired IPR&D charges may include IPR&D acquired in connection with asset acquisitions as well as premiums paid on equity securities and up-front, opt-in, and certain development milestone payments related to collaboration and licensing agreements. Regeneron does not forecast such acquired IPR&D charges due to the uncertainty of the future occurrence, magnitude, and timing of these transactions in any given period.
Regeneron’s results for the fourth quarter and full year 2024 have not been finalized and are subject to Regeneron’s financial statement closing procedures. There can be no assurance that actual results will not differ from the preliminary (unaudited) estimates described or incorporated by reference herein.
| Item 7.01. | Regulation FD Disclosure. |
The information set forth under Item 2.02 of this Current Report on Form 8-K is incorporated by reference herein. Copies of the press release and the presentation referenced in Item 2.02 are furnished as Exhibits 99.1 and 99.2 to this Current Report on Form 8-K and are incorporated by reference in this Item 7.01.
The information included or incorporated in Item 2.02 and the information included or incorporated in Item 7.01 of this Current Report on Form 8-K, including Exhibits 99.1 and 99.2, shall not be deemed “filed” for purposes of Section 18 of the Securities Exchange Act of 1934, as amended, nor shall such information and exhibit be deemed incorporated by reference in any filing under the Securities Act of 1933, as amended, except as shall be expressly set forth by specific reference in such a filing.
| Item 9.01. | Financial Statements and Exhibits. |
(d) Exhibits.
Note Regarding Forward-Looking Statements
This Current Report on Form 8-K (this “Report”) includes forward-looking statements that involve risks and uncertainties relating to future events and the future performance of Regeneron Pharmaceuticals, Inc. (“Regeneron” or the “Company”), and actual events or results may differ materially from these forward-looking statements. Words such as “anticipate,” “expect,” “intend,” “plan,” “believe,” “seek,” “estimate,” variations of such words, and similar expressions are intended to identify such forward-looking statements, although not all forward-looking statements contain these identifying words. These statements concern, and these risks and uncertainties include, among others, Regeneron’s expectations with respect to commercialization of its marketed products, competitive and other relevant developments affecting the market share of Regeneron’s marketed products, and other relevant factors (whether within or without Regeneron’s control) impacting the degree to which commercialization of Regeneron’s marketed products is successful, as well as the impact of any of the foregoing on Regeneron’s results of operations; and Regeneron’s expected acquired in-process research and development charge for the quarterly period ended December 31, 2024 and its expected impact on GAAP and non-GAAP net income per diluted share for this period as discussed in this Report. A more complete description of these and other material risks can be found in Regeneron’s filings with the U.S. Securities and Exchange Commission. Any forward-looking statements are made based on management’s current beliefs and judgment, and the reader is cautioned not to rely on any forward-looking statements made by Regeneron. Regeneron does not undertake any obligation to update (publicly or otherwise) any forward-looking statement, including without limitation any financial projection or guidance, whether as a result of new information, future events, or otherwise.
Note Regarding Non-GAAP Financial Measures
This Report references non-GAAP net income per diluted share, which is a financial measure that is not calculated in accordance with U.S. Generally Accepted Accounting Principles (“GAAP”). This non-GAAP financial measure is computed by excluding certain non-cash and/or other items from the related GAAP financial measure. The Company also includes a non-GAAP adjustment for the estimated income tax effect of reconciling items. The Company makes such adjustments for items the Company does not view as useful in evaluating its operating performance. Management uses this and other non-GAAP measures for planning, budgeting, forecasting, assessing historical performance, and making financial and operational decisions, and also provides forecasts to investors on this basis. Additionally, such non-GAAP measures provide investors with an enhanced understanding of the financial performance of the Company's core business operations. However, there are limitations in the use of such non-GAAP financial measures as they exclude certain expenses that are recurring in nature. Furthermore, the Company's non-GAAP financial measures may not be comparable with non-GAAP information provided by other companies. Any non-GAAP financial measure presented by Regeneron should be considered supplemental to, and not a substitute for, measures of financial performance prepared in accordance with GAAP.
SIGNATURES
Pursuant to the requirements of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned hereunto duly authorized.
| REGENERON PHARMACEUTICALS, INC. | |
| /s/ Joseph J. LaRosa | |
| Joseph J. LaRosa | |
| Executive Vice President, General Counsel and Secretary |
Date: January 13, 2025
Exhibit 99.1

Press Release
Regeneron Provides Business Updates and Highlights from Broad Clinical Pipeline at the 43rd Annual J.P. Morgan Healthcare Conference
Dupixent® is now used to treat over a million patients globally, with continued growth and expansion in multiple indications for diseases in which type 2 inflammation plays a role
EYLEA HD® and EYLEA® remained the U.S. anti-VEGF category leader in 2024; aggregate U.S. net product sales were $6 billion for full-year 2024, up 1% based on preliminary (unaudited) results
EYLEA HD pre-filled syringe (PFS) submission completed; launch expected by mid-2025
Libtayo® exceeded $1 billion dollars in 2024 annual net sales, and becomes the first and only immunotherapy to show a statistically significant clinical benefit as adjuvant therapy in high-risk cutaneous squamous cell carcinoma (CSCC)
Linvoseltamab Biologics License Application (BLA) resubmitted following resolution of third-party manufacturing issues; launch anticipated mid-2025
Approximately 40 investigational candidates in industry-leading pipeline cover dozens of disease states with expansive market potential
Regeneron collaborates with Truveta and leading American health systems to massively extend its DNA-linked healthcare database to further advance scientific innovation and healthcare delivery
Tarrytown, N.Y., January 13, 2025 – Regeneron Pharmaceuticals, Inc. (NASDAQ: REGN) today will share corporate progress and highlights from the Company’s broad and diverse investigational pipeline while presenting at the annual J.P. Morgan Healthcare Conference. The presentation is scheduled for 2:15 p.m. Pacific Time (5:15 p.m. Eastern Time) and may be accessed from the "Investors & Media" page of Regeneron's website.
“The Regeneron name is synonymous with innovation, brought to life through proprietary technologies and world-class science that produce medicines that make a meaningful impact on patients’ lives,” said Leonard S. Schleifer, M.D., Ph.D., Board co-Chair, President and Chief Executive Officer of Regeneron. “Thanks to our long-term and consistent R&D investment, we have – in addition to our four blockbuster medicines – one of the industry’s largest, most promising and most diverse clinical pipelines. Our therapeutic candidates tackle a myriad of diseases, with the most advanced programs addressing an aggregate commercial market opportunity expected to exceed $220 billion by 2030. We are well positioned for future growth and more confident than ever in the power of Regeneron’s science.”
Marketed Products
Dupixent Updates
| · | Dupixent® (dupilumab) is now used to treat over a million patients globally. The recent approval and launch in chronic obstructive pulmonary disease (COPD) has had a successful start, with coverage secured from the top commercial and Medicare payers and Dupixent now well positioned to address approximately 300,000 patients in the U.S. |
| · | There is continued growth potential in existing and additional indications for diseases in which type 2 inflammation may play a role, including chronic spontaneous urticaria (CSU) with an expected U.S. Food and Drug Administration (FDA) decision by April 18, 2025, and bullous pemphigoid, for which a supplemental Biologics License Application (sBLA) was submitted in the fourth quarter of 2024. |
EYLEA HD and EYLEA Updates
| · | On a combined basis, EYLEA HD® (aflibercept) Injection 8 mg and EYLEA® (aflibercept) Injection 2 mg remained the U.S. anti-VEGF category leader in 2024. Based on preliminary (unaudited) results, the products achieved 1% year-over-year growth by reaching $6 billion in aggregate U.S. net product sales for the year and $1.5 billion in aggregate U.S. net product sales for the fourth quarter of 2024, despite increasing competition. EYLEA HD U.S. net product sales were $305 million in the fourth quarter of 2024. EYLEA U.S. net product sales were $1.19 billion in the fourth quarter of 2024. |
| · | Combined EYLEA HD and EYLEA U.S. net product sales for the fourth quarter of 2024 were favorably impacted by approximately $85 million as a result of higher wholesaler inventory levels for EYLEA, partially offset by lower wholesaler inventory levels for EYLEA HD. |
| · | The Company filed an application with the FDA for use of the EYLEA HD pre-filled syringe (PFS) with U.S. approval and launch expected by mid-2025. |
| · | Longer term data in wet age-related macular degeneration (wAMD) and diabetic macular edema (DME) are under FDA review with a PDUFA date of April 20, 2025 to potentially extend dosing intervals for EYLEA HD up to every-24 weeks. |
| · | The Company plans to submit a sBLA for EYLEA HD for every four-week dosing and for retinal vein occlusion (RVO) in the first quarter of 2025 to potentially maximize dosing flexibility and address more retinal diseases. |
Libtayo Updates
| · | Libtayo® (cemiplimab) exceeded $1 billion in sales for 2024 and remains foundational to Regeneron’s oncology portfolio. |
| · | As announced this morning, a Phase 3 study demonstrated that Libtayo is the only immunotherapy to show a statistically significant and clinically meaningful benefit in high-risk cutaneous squamous cell carcinoma (CSCC) in the adjuvant setting; a recent Phase 3 trial with Keytruda® failed in the same setting.1 Specifically, adjuvant Libtayo demonstrated a 68% reduction in the risk of disease recurrence or death, compared to placebo (hazard ratio: 0.32; 95% confidence interval: 0.20-0.51; p<0.0001). Grade ≥3 adverse events occurred in 24% (n = 49 of 205) and 14% (n = 29 of 204) of patients in the Libtayo arm and the placebo arm, respectively. Detailed results will be presented at an upcoming medical meeting and will be shared with regulatory authorities with a plan for FDA submission in the first half of 2025. |
Phase 3 and Other Major Pipeline Opportunities
Regeneron is progressing numerous promising drug candidates across diverse disease states, with advanced programs that together have a total addressable commercial market expected to exceed $220 billion by 2030. Some near-term highlights include:
| · | Itepekimab (IL-33) for COPD: Based on genetic data linking IL-33 with increased risk of COPD and Phase 2 results, Regeneron’s next innovation in COPD offers potential for benefit in a broader population, including former smokers, non-cystic fibrosis bronchiectasis and other indications. Results are expected from the Phase 3 AERIFY study in the second half of 2025, with a potential BLA submission to follow. |
| · | Fianlimab (LAG3) for melanoma: Combining fianlimab and Libtayo, two potentially best-in-class checkpoint inhibitors, has the potential for differentiated efficacy and safety versus the current standard-of-care. Results from the first Phase 3 study in first-line metastatic melanoma are expected in the second half of 2025, with a potential BLA submission to follow. |
| · | Linvoseltamab (BCMAxCD3) for multiple myeloma: Linvoseltamab has potential to be the best-in-class BCMAxCD3 bispecific with its differentiated clinical profile, dosing regimen and administration method. The linvoseltamab BLA has been resubmitted following resolution of third-party manufacturing issues, with launch anticipated in mid-2025. Phase 3 programs in earlier lines of therapy using linvoseltamab monotherapy and novel combinations are also underway. |
| · | Odronextamab (CD20xCD3) for lymphoma: Ordspono™ (odronextamab) has been approved in the European Union for relapsed/refractory follicular lymphoma (FL) and diffuse large B-cell lymphoma (DLBCL) after two or more lines of systemic therapy, and enrollment is underway for a confirmatory study to support resubmission of the BLA for FL to the FDA in the first quarter of 2025. A broad and differentiated Phase 3 program is also underway to investigate odronextamab in earlier lines of FL and DLBCL. As reported at the American Society of Hematology annual meeting, odronextamab monotherapy showed complete responses in 12 out of 12 evaluable patients with first-line FL in the safety lead-in portion of the Phase 3 program. |
| · | Factor XI for anticoagulation: Regeneron’s two-pronged approach to anticoagulation is being evaluated for its potential to control thombosis while minimizing bleeding risk in a variety of patient populations and clinical settings. Two Factor XI antibodies, REGN7508 (catalytic domain) and REGN9933 (A2 domain), will advance to pivotal trials in 2025 on the basis of positive proof-of-concept data announced in December 2024. Current standards of care for thrombosis disorders have challenges including elevated risk of bleeding resulting in underutilization, presenting an unmet need for more specific inhibition of the intrinsic coagulation pathway. |
| · | Multiple approaches to obesity: Regeneron is studying various combinations with GLP-based therapies to potentially improve quality of weight loss by preserving lean muscle, as well as improve maintenance of weight loss following GLP-1/GIP discontinuations. A Phase 2 study of trevogrumab and semaglutide with and without garetosmab is now fully enrolled and a Phase 2 study testing combinations of tirzepatide and mibavademab is ongoing, with initial data expected from both in the second half of 2025. |
| · | BCMAxCD3/Dupixent in severe allergy: Combining linvoseltamab and Dupixent has the potential to eliminate immunoglobulin E (IgE), the key driver of allergic reactions, and thus potentially reverse severe allergies. A trial in patients with severe food allergies is ongoing, with initial clinical data shared in today’s presentation showing profound reduction of IgE in the first patient treated with this two-drug approach. |
| · | C5 Combo (pozelimab and cemdisiran) in complement-mediated diseases: Regeneron’s differentiated siRNA and antibody combination approach has the potential to address multiple complement-mediated diseases, such as generalized myasthenia gravis (Phase 3 results expected in the second half of 2025), paroyxsmal noctural hemoglobinuria (Phase 3 registrational data expected in 2026+) and geographic atrophy, an advanced form of dry AMD (Phase 3 pivotal program underway). |
DNA Sequence-Linked Healthcare Database
Regeneron continues to grow its leadership in genetics-driven drug discovery and is building the world’s largest DNA sequence-linked healthcare database, designed to unlock profound insights into how genetics impact health and aid in the development new genetic-based therapies and optimized healthcare services.
| · | The Regeneron Genetics Center® has sequenced nearly three million people to date, all with deidentified linked healthcare records. |
| · | A newly announced strategic collaboration with Truveta, Inc. is expected to dramatically expand the size of this database, with sequencing and linked Electronic Health Records for up to 10 million additional individuals from Truveta’s network of leading U.S. health systems. |
| · | On the basis of its industry-leading capabilities, Regeneron Genetics Center was selected by UK BioBank consortium members to complete proteomic assay data generation for the recently announced UK Biobank Pharma Proteomics Project. |
“Regeneron continues to diversify our commercial, clinical and research portfolios by relentlessly pushing the boundaries of innovation and technology,” said George D. Yancopoulos, M.D., Ph.D., Board co-Chair, President and Chief Scientific Officer of Regeneron. “In 2025, we will progress dozens of promising new assets and expand the reach of our important established medicines to help even more patients in need. We remain at the forefront of biotechnology’s most remarkable era of drug discovery, striving to change the practice of medicine with approaches spanning antibodies, bispecifics, gene editing, gene silencing, gene therapy and cell therapy supported by DNA sequence- and proteomics-linked healthcare database.”
The unapproved uses of EYLEA, EYLEA HD, Dupixent, Libtayo and pozelimab noted here are investigational and have not been fully evaluated by any regulatory authority. Cemdisiran, itepekimab, fianlimab, linvoseltamab, REGN7508, REGN9933, trevogrumab and garetosmab are investigational and have also not been fully evaluated by any regulatory authority. Odronextamab is approved in the European Union as Ordspono™ to treat R/R FL or DLBCL after two or more lines of systemic therapy, but the safety and efficacy of odronextamab has not been fully evaluated by any other regulatory authority.
About Regeneron
Regeneron (NASDAQ: REGN) is a leading biotechnology company that invents, develops and commercializes life-transforming medicines for people with serious diseases. Founded and led by physician-scientists, our unique ability to repeatedly and consistently translate science into medicine has led to numerous approved treatments and product candidates in development, most of which were homegrown in our laboratories. Our medicines and pipeline are designed to help patients with eye diseases, allergic and inflammatory diseases, cancer, cardiovascular and metabolic diseases, neurological diseases, hematologic conditions, infectious diseases, and rare diseases.
Regeneron pushes the boundaries of scientific discovery and accelerates drug development using our proprietary technologies, such as VelociSuite®, which produces optimized fully human antibodies and new classes of bispecific antibodies. We are shaping the next frontier of medicine with data-powered insights from the Regeneron Genetics Center® and pioneering genetic medicine platforms, enabling us to identify innovative targets and complementary approaches to potentially treat or cure diseases.
For more information, please visit www.Regeneron.com or follow Regeneron on LinkedIn, Instagram, Facebook or X.
#
DUPIXENT IMPORTANT SAFETY INFORMATION AND U.S. INDICATIONS
DUPIXENT® (dupilumab) is a prescription medicine used:
| · | to treat adults and children 6 months of age and older with moderate-to-severe eczema (atopic dermatitis or AD) that is not well controlled with prescription therapies used on the skin (topical), or who cannot use topical therapies. DUPIXENT can be used with or without topical corticosteroids. It is not known if DUPIXENT is safe and effective in children with atopic dermatitis under 6 months of age. |
| · | with other asthma medicines for the maintenance treatment of moderate-to-severe eosinophilic or oral steroid dependent asthma in adults and children 6 years of age and older whose asthma is not controlled with their current asthma medicines. DUPIXENT helps prevent severe asthma attacks (exacerbations) and can improve your breathing. DUPIXENT may also help reduce the amount of oral corticosteroids you need while preventing severe asthma attacks and improving your breathing. It is not known if DUPIXENT is safe and effective in children with asthma under 6 years of age. |
| · | with other medicines for the maintenance treatment of chronic rhinosinusitis with nasal polyps (CRSwNP) in adults and children 12 years of age and older whose disease is not controlled. It is not known if DUPIXENT is safe and effective in children with chronic rhinosinusitis with nasal polyps under 12 years of age. |
| · | to treat adults and children 1 year of age and older with eosinophilic esophagitis (EoE), who weigh at least 33 pounds (15 kg). It is not known if DUPIXENT is safe and effective in children with eosinophilic esophagitis under 1 year of age, or who weigh less than 33 pounds (15 kg). |
| · | to treat adults with prurigo nodularis (PN). It is not known if DUPIXENT is safe and effective in children with prurigo nodularis under 18 years of age. |
| · | with other medicines for the maintenance treatment of adults with inadequately controlled chronic obstructive pulmonary disease (COPD) and a high number of blood eosinophils (a type of white blood cell that may contribute to your COPD). DUPIXENT is used to reduce the number of flare-ups (the worsening of your COPD symptoms for several days) and can improve your breathing. It is not known if DUPIXENT is safe and effective in children with chronic obstructive pulmonary disease under 18 years of age. |
Do not use if you are allergic to dupilumab or to any of the ingredients in DUPIXENT®.
Before using DUPIXENT, tell your healthcare provider about all your medical conditions, including if you:
| · | have eye problems. |
| · | have a parasitic (helminth) infection. |
| · | are scheduled to receive any vaccinations. You should not receive a “live vaccine” right before and during treatment with DUPIXENT. |
| · | are pregnant or plan to become pregnant. It is not known whether DUPIXENT will harm your unborn baby. |
| o | A pregnancy registry for women who take DUPIXENT during pregnancy collects information about the health of you and your baby. To enroll or get more information call 1-877-311-8972 or go to https://mothertobaby.org/ongoing-study/dupixent/. |
| · | are breastfeeding or plan to breastfeed. It is not known whether DUPIXENT passes into your breast milk. |
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements.
Especially tell your healthcare provider if you are taking oral, topical, or inhaled corticosteroid medicines; have asthma and use an asthma medicine; or have atopic dermatitis, chronic rhinosinusitis with nasal polyps, eosinophilic esophagitis, prurigo nodularis, or chronic obstructive pulmonary disease and also have asthma. Do not change or stop your other medicines, including corticosteroid medicine or other asthma medicine, without talking to your healthcare provider. This may cause other symptoms that were controlled by those medicines to come back.
DUPIXENT can cause serious side effects, including:
| · | Allergic reactions. DUPIXENT can cause allergic reactions that can sometimes be severe. Stop using DUPIXENT and tell your healthcare provider or get emergency help right away if you get any of the following signs or symptoms: breathing problems or wheezing, swelling of the face, lips, mouth, tongue or throat, fainting, dizziness, feeling lightheaded, fast pulse, fever, hives, joint pain, general ill feeling, itching, skin rash, swollen lymph nodes, nausea or vomiting, or cramps in your stomach-area. |
| · | Eye problems. Tell your healthcare provider if you have any new or worsening eye problems, including eye pain or changes in vision, such as blurred vision. Your healthcare provider may send you to an ophthalmologist for an exam if needed. |
| · | Inflammation of your blood vessels. Rarely, this can happen in people with asthma who receive DUPIXENT. This may happen in people who also take a steroid medicine by mouth that is being stopped or the dose is being lowered. It is not known whether this is caused by DUPIXENT. Tell your healthcare provider right away if you have: rash, chest pain, worsening shortness of breath, a feeling of pins and needles or numbness of your arms or legs, or persistent fever. |
| · | Joint aches and pain. Some people who use DUPIXENT have had trouble walking or moving due to their joint symptoms, and in some cases needed to be hospitalized. Tell your healthcare provider about any new or worsening joint symptoms. Your healthcare provider may stop DUPIXENT if you develop joint symptoms. |
The most common side effects include:
| · | Eczema: injection site reactions, eye and eyelid inflammation, including redness, swelling, and itching, sometimes with blurred vision, dry eye, cold sores in your mouth or on your lips, and high count of a certain white blood cell (eosinophilia). |
| · | Asthma: injection site reactions, high count of a certain white blood cell (eosinophilia), pain in the throat (oropharyngeal pain), and parasitic (helminth) infections. |
| · | Chronic Rhinosinusitis with Nasal Polyps: injection site reactions, eye and eyelid inflammation, including redness, swelling, and itching, sometimes with blurred vision, high count of a certain white blood cell (eosinophilia), gastritis, joint pain (arthralgia), trouble sleeping (insomnia), and toothache. |
| · | Eosinophilic Esophagitis: injection site reactions, upper respiratory tract infections, cold sores in your mouth or on your lips, and joint pain (arthralgia). |
| · | Prurigo Nodularis: eye and eyelid inflammation, including redness, swelling, and itching, sometimes with blurred vision, herpes virus infections, common cold symptoms (nasopharyngitis), dizziness, muscle pain, and diarrhea. |
| · | Chronic Obstructive Pulmonary Disease: injection sites reactions, common cold symptoms (nasopharyngitis), high count of a certain white blood cell (eosinophilia), viral infection, back pain, inflammation inside the nose (rhinitis), diarrhea, gastritis, joint pain (arthralgia), toothache, headache, and urinary tract infection. |
Tell your healthcare provider if you have any side effect that bothers you or that does not go away. These are not all the possible side effects of DUPIXENT. Call your doctor for medical advice about side effects. You are encouraged to report negative side effects of prescription drugs to the FDA. Visit www.fda.gov/medwatch, or call 1-800-FDA-1088.
Use DUPIXENT exactly as prescribed by your healthcare provider. It’s an injection given under the skin (subcutaneous injection). Your healthcare provider will decide if you or your caregiver can inject DUPIXENT. Do not try to prepare and inject DUPIXENT until you or your caregiver have been trained by your healthcare provider. In children 12 years of age and older, it’s recommended DUPIXENT be administered by or under supervision of an adult. In children 6 months to less than 12 years of age, DUPIXENT should be given by a caregiver.
Please see accompanying full Prescribing Information including Patient Information.
#
EYLEA AND EYLEA HD IMPORTANT SAFETY INFORMATION AND U.S. INDICATIONS
INDICATIONS
EYLEA HD® (aflibercept) Injection 8 mg is a prescription medicine approved for the treatment of patients with Wet Age-Related Macular Degeneration (AMD), Diabetic Macular Edema (DME), and Diabetic Retinopathy (DR).
EYLEA® (aflibercept) Injection 2 mg is a prescription medicine approved for the treatment of patients with Wet Age-Related Macular Degeneration (AMD), Macular Edema following Retinal Vein Occlusion (RVO), Diabetic Macular Edema (DME), Diabetic Retinopathy (DR), and Retinopathy of Prematurity (ROP) (0.4 mg).
IMPORTANT SAFETY INFORMATION
| · | EYLEA HD and EYLEA are administered by injection into the eye. You should not use EYLEA HD or EYLEA if you have an infection in or around the eye, eye pain or redness, or known allergies to any of the ingredients in EYLEA HD or EYLEA, including aflibercept. |
| · | Injections into the eye with EYLEA HD or EYLEA can result in an infection in the eye, retinal detachment (separation of retina from back of the eye) and, more rarely, serious inflammation of blood vessels in the retina that may include blockage. Call your doctor right away if you or your baby (if being treated with EYLEA for Retinopathy of Prematurity) experience eye pain or redness, light sensitivity, or a change in vision after an injection. |
| · | In some patients, injections with EYLEA HD or EYLEA may cause a temporary increase in eye pressure within 1 hour of the injection. Sustained increases in eye pressure have been reported with repeated injections, and your doctor may monitor this after each injection. |
| · | In infants with Retinopathy of Prematurity (ROP), treatment with EYLEA will need extended periods of ROP monitoring. |
| · | There is a potential but rare risk of serious and sometimes fatal side effects, related to blood clots, leading to heart attack or stroke in patients receiving EYLEA HD or EYLEA. |
| · | The most common side effects reported in patients receiving EYLEA HD were cataract, increased redness in the eye, increased pressure in the eye, eye discomfort, pain, or irritation, blurred vision, vitreous (gel-like substance) floaters, vitreous detachment, injury to the outer layer of the eye, and bleeding in the back of the eye. |
| · | The most common side effects reported in patients receiving EYLEA were increased redness in the eye, eye pain, cataract, vitreous detachment, vitreous floaters, moving spots in the field of vision, and increased pressure in the eye. |
| · | The most common side effects reported in pre-term infants with ROP receiving EYLEA were separation of the retina from the back of the eye, increased redness in the eye, and increased pressure in the eye. Side effects that occurred in adults |
are considered applicable to pre-term infants with ROP, though not all were seen in clinical studies.
| · | You may experience temporary visual changes after an EYLEA HD or EYLEA injection and associated eye exams; do not drive or use machinery until your vision recovers sufficiently. |
| · | For additional safety information, please talk to your doctor and see the full Prescribing Information for EYLEA HD and EYLEA. |
You are encouraged to report negative side effects of prescription drugs to the FDA. Visit www.fda.gov/medwatch or call 1-800-FDA-1088.
Please click here for full Prescribing Information for EYLEA HD and EYLEA.
#
LIBTAYO IMPORTANT SAFETY INFORMATION AND U.S. INDICATIONS
LIBTAYO® (cemiplimab-rwlc) is a prescription medicine used to treat:
| · | People with a type of skin cancer called cutaneous squamous cell carcinoma (CSCC) that has spread or cannot be cured by surgery or radiation. |
| · | People with a type of skin cancer called basal cell carcinoma (BCC) when your BCC cannot be removed by surgery (locally advanced BCC) or when it has spread (metastatic BCC) and have received treatment with a hedgehog pathway inhibitor (HHI), or cannot receive treatment with a HHI. |
| · | Adults with a type of lung cancer called non-small cell lung cancer (NSCLC). |
| o | LIBTAYO may be used in combination with chemotherapy that contains a platinum medicine as your first treatment when your lung cancer has not spread outside your chest (locally advanced lung cancer) and you cannot have surgery or chemotherapy with radiation, or your lung cancer has spread to other areas of your body (metastatic lung cancer), and your tumor does not have an abnormal “EGFR,” “ALK,” or “ROS1” gene. |
| o | LIBTAYO may be used alone as your first treatment when your lung cancer has not spread outside your chest (locally advanced lung cancer) and you cannot have surgery or chemotherapy with radiation, or your lung cancer has spread to other areas of your body (metastatic lung cancer), and your tumor tests positive for high “PD-L1,” and your tumor does not have an abnormal “EGFR,” “ALK,” or “ROS1” gene. |
It is not known if Libtayo is safe and effective in children.
IMPORTANT SAFETY INFORMATION FOR U.S. PATIENTS
What is the most important information I should know about LIBTAYO?
LIBTAYO is a medicine that may treat certain cancers by working with your immune system. LIBTAYO can cause your immune system to attack normal organs and tissues in any area of your body and can affect the way they work. These problems can sometimes become severe or life-threatening and can lead to death. You can have more than one of these problems at the same time. These problems may happen anytime during treatment or even after your treatment has ended.
Call or see your healthcare provider right away if you develop any new or worsening signs or symptoms, including:
| · | Lung problems: cough, shortness of breath, or chest pain |
| · | Intestinal problems: diarrhea (loose stools) or more frequent bowel movements than usual, stools that are black, tarry, sticky or have blood or mucus, or severe stomach-area (abdomen) pain or tenderness |
| · | Liver problems: yellowing of your skin or the whites of your eyes, severe nausea or vomiting, pain on the right side of your stomach-area (abdomen), dark urine (tea colored), or bleeding or bruising more easily than normal |
| · | Hormone gland problems: headache that will not go away or unusual headaches, eye sensitivity to light, eye problems, rapid heartbeat, increased sweating, extreme tiredness, weight gain or weight loss, feeling more hungry or thirsty than usual, urinating more often than usual, hair loss, feeling cold, constipation, your voice gets deeper, dizziness or fainting, or changes in mood or behavior, such as decreased sex drive, irritability, or forgetfulness |
| · | Kidney problems: decrease in your amount of urine, blood in your urine, swelling of your ankles, or loss of appetite |
| · | Skin problems: rash, itching, skin blistering or peeling, painful sores or ulcers in mouth or nose, throat, or genital area, fever or flu-like symptoms, or swollen lymph nodes |
| · | Problems can also happen in other organs and tissues. These are not all of the signs and symptoms of immune system problems that can happen with LIBTAYO. Call or see your healthcare provider right away for any new or worsening signs or symptoms, which may include: chest pain, irregular heartbeat, shortness of breath or swelling of ankles, confusion, sleepiness, memory problems, changes in mood or behavior, stiff neck, balance problems, tingling or numbness of the arms or legs, double vision, blurry vision, sensitivity to light, eye pain, changes in eyesight, persistent or severe muscle pain or weakness, muscle cramps, low red blood cells, or bruising |
| · | Infusion reactions that can sometimes be severe or life-threatening. Signs and symptoms of infusion reactions may include: nausea, vomiting, chills or shaking, itching or rash, flushing, shortness of breath or wheezing, dizziness, feel like passing out, fever, back or neck pain, or facial swelling |
| · | Rejection of a transplanted organ. Your healthcare provider should tell you what signs and symptoms you should report and monitor you, depending on the type of organ transplant that you have had |
| · | Complications, including graft-versus-host disease (GVHD), in people who have received a bone marrow (stem cell) transplant that uses donor stem cells (allogeneic). These complications can be serious and can lead to death. |
These complications may happen if you underwent transplantation either before or after being treated with LIBTAYO. Your healthcare provider will monitor you for these complications
Getting medical treatment right away may help keep these problems from becoming more serious. Your healthcare provider will check you for these problems during your treatment with LIBTAYO. Your healthcare provider may treat you with corticosteroid or hormone replacement medicines. Your healthcare provider may also need to delay or completely stop treatment with LIBTAYO if you have severe side effects.
Before you receive LIBTAYO, tell your healthcare provider about all your medical conditions, including if you:
| · | have immune system problems such as Crohn’s disease, ulcerative colitis, or lupus |
| · | have received an organ transplant |
| · | have received or plan to receive a stem cell transplant that uses donor stem cells (allogeneic) |
| · | have received radiation treatment to your chest area |
| · | have a condition that affects your nervous system, such as myasthenia gravis or Guillain-Barré syndrome |
| · | are pregnant or plan to become pregnant. LIBTAYO can harm your unborn baby |
Females who are able to become pregnant:
| o | Your healthcare provider will give you a pregnancy test before you start treatment |
| o | You should use an effective method of birth control during your treatment and for at least 4 months after your last dose of LIBTAYO. Talk to your healthcare provider about birth control methods that you can use during this time |
| o | Tell your healthcare provider right away if you become pregnant or think you may be pregnant during treatment with LIBTAYO |
| · | are breastfeeding or plan to breastfeed. It is not known if LIBTAYO passes into your breast milk. Do not breastfeed during treatment and for at least 4 months after the last dose of LIBTAYO |
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements.
The most common side effects of LIBTAYO when used alone include tiredness, muscle or bone pain, rash, diarrhea, and low levels of red blood cells (anemia). The most common side effects of LIBTAYO when used in combination with platinum-containing chemotherapy include hair loss, muscle or bone pain, nausea, tiredness, numbness, pain, tingling, or burning in your hands or feet, and decreased appetite. These are not all the possible side effects of LIBTAYO. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. You may also report side effects to Regeneron Pharmaceuticals at 1-877-542-8296.
Please see full Prescribing Information, including Medication Guide.
#
Forward-Looking Statements and Use of Digital Media
This press release includes forward-looking statements that involve risks and uncertainties relating to future events and the future performance of Regeneron Pharmaceuticals, Inc. (“Regeneron” or the “Company”), and actual events or results may differ materially from these forward-looking statements. Words such as “anticipate,” “expect,” “intend,” “plan,” “believe,” “seek,” “estimate,” variations of such words, and similar expressions are intended to identify such forward-looking statements, although not all forward-looking statements contain these identifying words. These statements concern, and these risks and uncertainties include, among others, the nature, timing, and possible success and therapeutic applications of products marketed or otherwise commercialized by Regeneron and/or its collaborators or licensees (collectively, "Regeneron's Products") and product candidates being developed by Regeneron and/or its collaborators or licensees (collectively, "Regeneron's Product Candidates") and research and clinical programs now underway or planned, including without limitation Dupixent® (dupilumab), EYLEA HD® (aflibercept) Injection 8 mg, EYLEA® (aflibercept) Injection, Libtayo® (cemiplimab), Ordspono™ (odronextamab), itepekimab, linvoseltamab, fianlimab, pozelimab in combination with cemdisiran, REGN7508, REGN9933, other of Regeneron's Product Candidates discussed or referenced in this press release, and the use of human genetics in Regeneron's research programs; the likelihood and timing of achieving any of the anticipated milestones discussed or referenced in this press release; the likelihood, timing, and scope of possible regulatory approval and commercial launch of Regeneron's Product Candidates and new indications for Regeneron's Products, including those listed above and/or otherwise discussed in this press release; uncertainty of the utilization, market acceptance, and commercial success of Regeneron’s Products and Regeneron’s Product Candidates and the impact of studies (whether conducted by Regeneron or others and whether mandated or voluntary), including the studies discussed or referenced in this press release, on any of the foregoing or any potential regulatory approval of Regeneron’s Products and Regeneron’s Product Candidates; Regeneron’s expectations with respect to commercialization of Regeneron’s Products (including Dupixent, EYLEA HD, EYLEA, and Libtayo), competitive and other relevant developments affecting the market share of Regeneron’s Products, and other relevant factors (whether within or without Regeneron’s control) impacting the degree to which commercialization of Regeneron’s marketed products is successful, as well as the impact of any of the foregoing on Regeneron’s results of operations; the ability of Regeneron’s collaborators, licensees, suppliers, or other third parties (as applicable) to perform manufacturing, filling, finishing, packaging, labeling, distribution, and other steps related to Regeneron’s Products and Regeneron’s Product Candidates; the ability of Regeneron to manage supply chains for multiple products and product candidates; safety issues resulting from the administration of Regeneron’s Products and Regeneron’s Product Candidates in patients, including serious complications or side effects in connection with the use of Regeneron’s Products and Regeneron’s Product Candidates in clinical trials; determinations by regulatory and administrative governmental authorities which may delay or restrict Regeneron’s ability to continue to develop or commercialize Regeneron’s Products and Regeneron’s Product Candidates; ongoing regulatory obligations and oversight impacting Regeneron’s Products, research and clinical programs, and business, including those relating to patient privacy; the availability and extent of reimbursement of Regeneron’s Products from third-party payers, including private payer healthcare and insurance programs, health maintenance organizations, pharmacy benefit management companies, and government programs such as Medicare and Medicaid; coverage and reimbursement determinations by such payers and new policies and procedures adopted by such payers; competing drugs and product candidates that may be superior to, or more cost effective than, Regeneron’s Products and Regeneron’s Product Candidates (including biosimilar versions of Regeneron’s Products); the extent to which the results from the research and development programs conducted by Regeneron and/or its collaborators or licensees (such as those that may result from the strategic collaboration with Truveta, Inc. discussed in this press release) may be replicated in other studies and/or lead to advancement of product candidates to clinical trials, therapeutic applications, or regulatory approval; unanticipated expenses; the costs of developing, producing, and selling products; the ability of Regeneron to meet any of its financial projections or guidance and changes to the assumptions underlying those projections or guidance; the potential for any license, collaboration, or supply agreement, including Regeneron’s agreements with Sanofi and Bayer (or their respective affiliated companies, as applicable), as well as the collaboration with Truveta, Inc. discussed in this press release, to be cancelled or terminated; the impact of public health outbreaks, epidemics, or pandemics (such as the COVID-19 pandemic) on Regeneron's business; and risks associated with intellectual property of other parties and pending or future litigation relating thereto (including without limitation the patent litigation and other related proceedings relating to EYLEA), other litigation and other proceedings and government investigations relating to the Company and/or its operations (including the pending civil proceedings initiated or joined by the U.S. Department of Justice and the U.S. Attorney's Office for the District of Massachusetts), the ultimate outcome of any such proceedings and investigations, and the impact any of the foregoing may have on Regeneron’s business, prospects, operating results, and financial condition. A more complete description of these and other material risks can be found in Regeneron’s filings with the U.S. Securities and Exchange Commission, including its Form 10-K for the year ended December 31, 2023 and its Form 10-Q for the quarterly period ended September 30, 2024. Any forward-looking statements are made based on management’s current beliefs and judgment, and the reader is cautioned not to rely on any forward-looking statements made by Regeneron. Regeneron does not undertake any obligation to update (publicly or otherwise) any forward-looking statement, including without limitation any financial projection or guidance, whether as a result of new information, future events, or otherwise.
Regeneron uses its media and investor relations website and social media outlets to publish important information about the Company, including information that may be deemed material to investors. Financial and other information about Regeneron is routinely posted and is accessible on Regeneron's media and investor relations website (https://investor.regeneron.com) and its LinkedIn page (https://www.linkedin.com/company/regeneron-pharmaceuticals).
|
Contacts:
Media Relations Christina Chan Tel: +1 914-847-8827 Christina.chan@regeneron.com |
Investor Relations Ryan Crowe Tel: +1 914-847-8790 Ryan.crowe@regeneron.com |
[1] Data not yet published. https://www.merck.com/news/merck-provides-update-on-phase-3-keynote-867-and-keynote-630-trials/. All trademarks used are the property of their respective owners. The studies had differences in trial design specifics and no head-to-head comparisons have been conducted.
Exhibit 99.2

J.P. Morgan Healthcare Conference JANUARY 13, 2025 This non - promotional presentation contains investigational data as well as forward - looking statements; actual results may vary m aterially.

2 Strategy & Business Update J.P. Morgan Healthcare Conference 2025 Leonard S.

Schleifer, MD, PhD Co - Founder, Board Co - Chair, President & Chief Executive Officer 3 Note regarding forward - looking statements This presentation includes forward - looking statements that involve risks and uncertainties relating to future events and the future performance of Regeneron Pharmaceuticals, Inc . ("Regeneron" or the "Company"), and actual events or results may differ materially from these forward - looking statements . Words such as "anticipate," "expect," "intend," "plan," "believe," "seek," "estimate," variations of such words, and similar expressions are intended to identify such forward - looking statements, although not all forward - looking statements contain these identifying words . These statements concern, and these risks and uncertainties include, among others, the nature, timing, and possible success and therapeutic applications of products marketed or otherwise commercialized by Regeneron and/or its collaborators or licensees (collectively, "Regeneron's Products") and product candidates being developed by Regeneron and/or its collaborators or licensees (collectively, "Regeneron's Product Candidates") and research and clinical programs now underway or planned, including without limitation EYLEA HD ® (aflibercept) Injection 8 mg, EYLEA ® (aflibercept) Injection, Dupixent ® (dupilumab) Injection, Libtayo ® (cemiplimab) Injection, Praluent ® (alirocumab) Injection, Kevzara ® (sarilumab) Injection, Evkeeza ® (evinacumab) Injection, Veopoz (pozelimab) Injection, Ordspono (odronextamab), itepekimab, fianlimab, garetosmab, linvoseltamab, Regeneron's other oncology programs (including its costimulatory bispecific portfolio), REGN 5713 - 5715 , nexiguran ziclumeran (Nex - z, NTLA - 2001 ), REGN 1908 - 1909 , mibavademab, DB - OTO, Regeneron's and its collaborators' earlier - stage programs, and the use of human genetics in Regeneron's research programs ; the likelihood and timing of achieving any of the anticipated milestones discussed or referenced in this presentation ; safety issues resulting from the administration of Regeneron's Products and Regeneron's Product Candidates in patients, including serious complications or side effects in connection with the use of Regeneron's Products and Regeneron's Product Candidates in clinical trials ; the likelihood, timing, and scope of possible regulatory approval and commercial launch of Regeneron's Product Candidates and new indications for Regeneron's Products, such as those listed above ; the extent to which the results from the research and development programs conducted by Regeneron and/or its collaborators may be replicated in other studies and/or lead to advancement of product candidates to clinical trials, therapeutic applications, or regulatory approval ; ongoing regulatory obligations and oversight impacting Regeneron's Products, research and clinical programs, and business, including those relating to patient privacy ; determinations by regulatory and administrative governmental authorities which may delay or restrict Regeneron's ability to continue to develop or commercialize Regeneron's Products and Regeneron's Product Candidates ; competing drugs and product candidates that may be superior to, or more cost effective than, Regeneron's Products and Regeneron's Product Candidates (including biosimilar versions of Regeneron's Products) ; uncertainty of the utilization, market acceptance, and commercial success of Regeneron's Products and Regeneron's Product Candidates and the impact of studies (whether conducted by Regeneron or others and whether mandated or voluntary) or recommendations and guidelines from governmental authorities and other third parties on the commercial success of Regeneron's Products and Regeneron's Product Candidates ; Regeneron's ability to manufacture and manage supply chains for multiple products and product candidates ; the ability of Regeneron's collaborators, suppliers, or other third parties (as applicable) to perform manufacturing, filling, finishing, packaging, labeling, distribution, and other steps related to Regeneron's Products and Regeneron's Product Candidates ; the availability and extent of reimbursement of Regeneron's Products from third - party payors, including private payor healthcare and insurance programs, health maintenance organizations, pharmacy benefit management companies, and government programs such as Medicare and Medicaid ; coverage and reimbursement determinations by such payors and new policies and procedures adopted by such payors ; unanticipated expenses ; the costs of developing, producing, and selling products ; Regeneron's ability to meet any of its financial projections or guidance and changes to the assumptions underlying those projections or guidance ; the potential for any license or collaboration agreement, including Regeneron's agreements with Sanofi and Bayer (or their respective affiliated companies, as applicable), to be cancelled or terminated ; the impact of public health outbreaks, epidemics, or pandemics (such as the COVID - 19 pandemic) on Regeneron's business ; and risks associated with intellectual property of other parties and pending or future litigation relating thereto (including without limitation the patent litigation and other related proceedings relating to EYLEA), other litigation and other proceedings and government investigations relating to the Company and/or its operations (including the pending civil proceedings initiated or joined by the U . S . Department of Justice and the U . S . Attorney's Office for the District of Massachusetts), the ultimate outcome of any such proceedings and investigations, and the impact any of the foregoing may have on Regeneron's business, prospects, operating results, and financial condition . A more complete description of these and other material risks can be found in Regeneron's filings with the U . S . Securities and Exchange Commission . Any forward - looking statements are made based on management's current beliefs and judgment, and the reader is cautioned not to rely on any forward - looking statements made by Regeneron . Regeneron does not undertake any obligation to update (publicly or otherwise) any forward - looking statement, including without limitation any financial projection or guidance, whether as a result of new information, future events, or otherwise .

Differentiated technology platforms have delivered 4 ‘blockbuster’ products discovered by Regeneron Unprecedented research and discovery capabilities drive best - in - class pipeline of ~40 product candidates — Includes many near - term opportunities with potential to address therapeutic categories expected to exceed an aggregate of $220 billion in 2030 Regeneron Genetics Center ® has created the world’s largest DNA sequence - linked healthcare database. Large - scale proteomics - linked database underway — For drug discovery and development as well as healthcare analytics and management 4 Driven by science and innovation Note: Definitions for all acronyms and abbreviations in this presentation can be found on slide 41.

Driving long - term shareholder value creation 5 * Based on preliminary, unaudited results. Continued strong execution across our in - line brands Dupixent now treating over 1 million patients worldwide across 7 approved indications, with new indications expected in 2025 • COPD launch underway; potential U.S. launches for CSU and BP in 2025 EYLEA HD + EYLEA U.S. net product sales grew 1% * in 2024 • EYLEA HD pre - filled syringe submission completed; mid - 2025 launch planned • 2 nd year of PHOTON and PULSAR data under FDA review (April 20 PDUFA) • EYLEA HD FDA submissions for RVO and Q4W dosing planned for Q1 2025 LIBTAYO to be Regeneron’s fourth ‘blockbuster’ product • First immunotherapy to demonstrate statistically significant disease free survival benefit in high - risk adjuvant CSCC Potential best - in - class opportunities across large and growing therapeutic categories Advancing our differentiated pipeline 1 2 3 Positioning for the future with genetics, proteomics, & targeted medicine Expanding the world’s largest DNA sequence - linked healthcare database and empowering it with large - scale proteomics to prepare for the future of healthcare analytics & management 6 Continued growth and expansion in multiple Type 2 indications Q3 2024 Dupixent global net sales of $3.8B (+23% YoY), annualizing at over $15 billion This slide contains investigational indications for dupilumab that have not been approved by any regulatory authority.
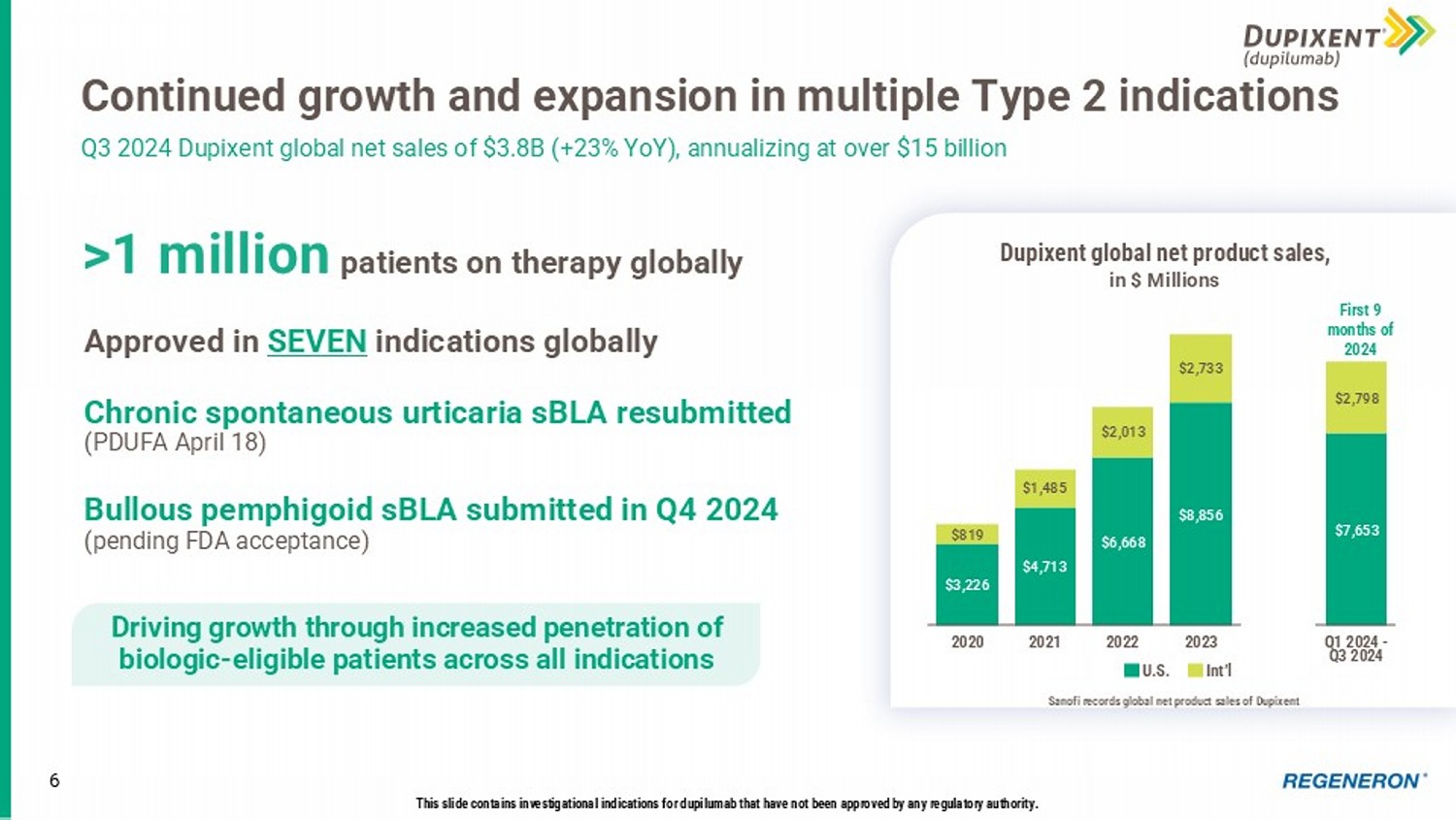
Approved in SEVEN indications globally Chronic spontaneous urticaria sBLA resubmitted (PDUFA April 18) Bullous pemphigoid sBLA submitted in Q4 2024 (pending FDA acceptance) >1 million patients on therapy globally Driving growth through increased penetration of biologic - eligible patients across all indications $3,226 $4,713 $6,668 $8,856 $7,653 $819 $1,485 $2,013 $2,733 $2,798 2020 2021 2022 2023 Q1 2024 - Q3 2024 First 9 months of 2024 Dupixent global net product sales , in $ Millions U.S.
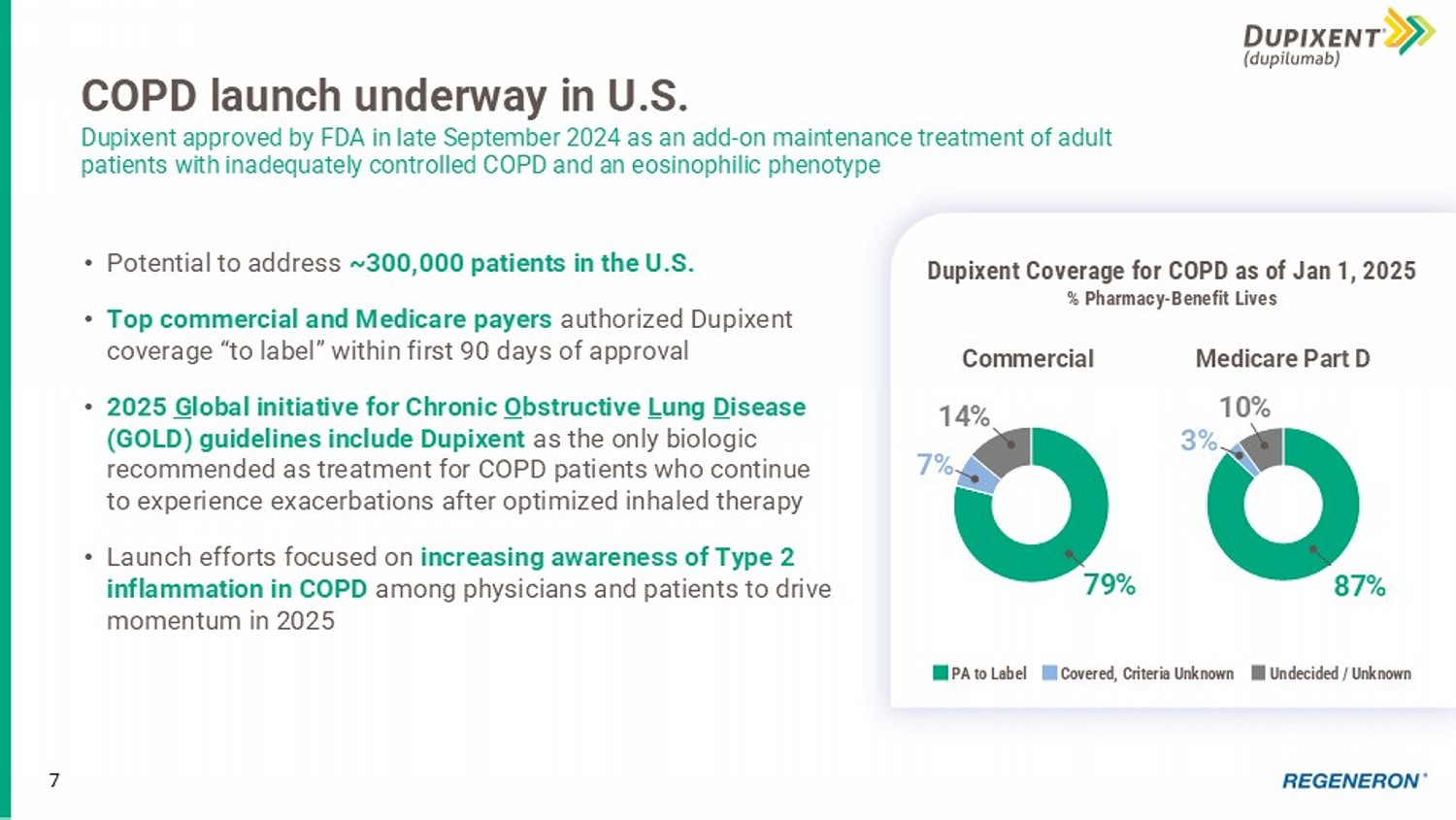
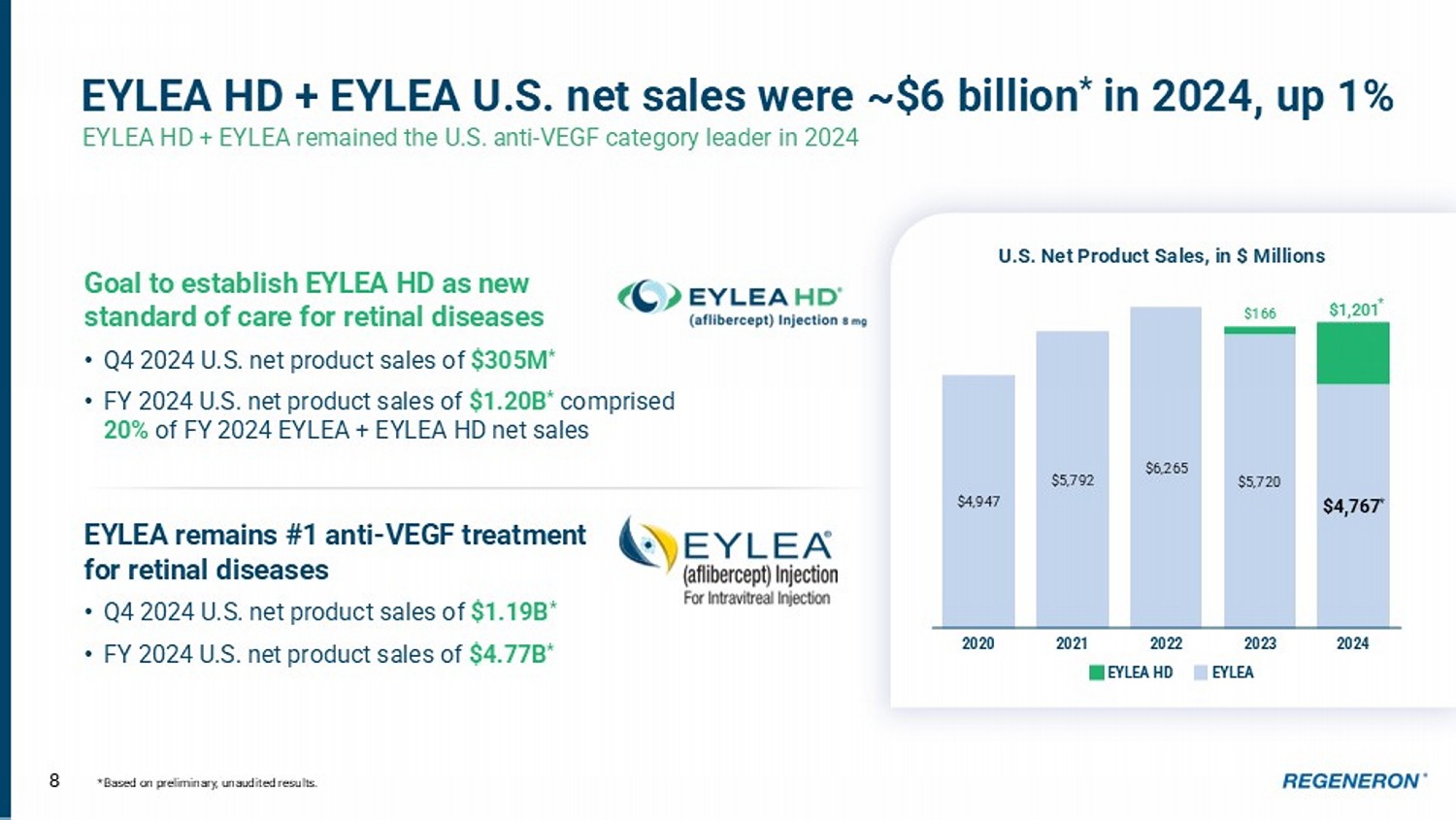
Int’l Sanofi records global net product sales of Dupixent 7 COPD launch underway in U.S. Dupixent approved by FDA in late September 2024 as an add - on maintenance treatment of adult patients with inadequately controlled COPD and an eosinophilic phenotype • Potential to address ~300,000 patients in the U.S. • Top commercial and Medicare payers authorized Dupixent coverage “to label” within first 90 days of approval • 2025 G lobal initiative for Chronic O bstructive L ung D isease (GOLD) guidelines include Dupixent as the only biologic recommended as treatment for COPD patients who continue to experience exacerbations after optimized inhaled therapy • Launch efforts focused on increasing awareness of Type 2 inflammation in COPD among physicians and patients to drive momentum in 2025 79% 7% 14% Commercial Dupixent Coverage for COPD as of Jan 1, 2025 % Pharmacy - Benefit Lives PA to Label Undecided / Unknown Covered, Criteria Unknown 87% 3% 10% Medicare Part D $4,947 $5,792 $6,265 $5,720 $4,767 * $166 $1,201 2020 2021 2022 2023 2024 8 EYLEA HD + EYLEA U.S. net sales were ~$6 billion * in 2024, up 1% EYLEA HD + EYLEA remained the U.S. anti - VEGF category leader in 2024 *Based on preliminary, unaudited results. U.S. Net Product Sales, in $ Millions Goal to establish EYLEA HD as new standard of care for retinal diseases • Q4 2024 U.S. net product sales of $305M * • FY 2024 U.S. net product sales of $1.20B * comprised 20% of FY 2024 EYLEA + EYLEA HD net sales EYLEA remains #1 anti - VEGF treatment for retinal diseases • Q4 2024 U.S. net product sales of $1.19B * • FY 2024 U.S. net product sales of $4.77B * EYLEA HD EYLEA *

9 Strengthening EYLEA HD product profile in 2025 Delivering key enhancements to EYLEA HD product offering to further unlock ongoing launch Opportunity for EYLEA HD to have broadest indication set with greatest dosing flexibility in anti - VEGF category Convenient Administration • Pre - Filled Syringe (PFS) submission completed; U.S. launch anticipated by mid - 2025 • Same PFS device approved in Europe in 2024 • Strong physician preference; 95% of EYLEA administered via PFS Addressing More Retinal Diseases • Positive Phase 3 data in retinal vein occlusion (RVO) announced in December 2024 • RVO was ~17% of EYLEA sales in 2024 • sBLA submission in Q1 2025 Maximizing Dosing Flexibility • sBLA submission in Q1 2025 for every - 4 - week dosing (Q4W) for wAMD, DME, and DR indications Extended Dosing Intervals • 2 nd year of PHOTON and PULSAR data under FDA review ( April 20 PDUFA ) • Potential to offer wAMD and DME patients the longest dosing interval (up to every - 24 - week dosing) of any approved anti - VEGF therapy • Best - in - class efficacy and durability profile provide potential to become the new standard - of - care for retinal diseases • Safety profile consistent with the established safety profile of EYLEA • Long - term data from PHOTON and PULSAR extension studies and real - world experience continue to support differentiated profile Planned for 2025 Strong and consistent growth • WW net sales $850M through 9 months of 2024 (+36% YoY) • Expanding global commercia l footprint 2020 2021 2022 2023 Q1 2024 - Q3 2024 $ 850 M First 9 months of 2024 10 Key growth driver and foundational to oncology portfolio LIBTAYO to become Regeneron’s next internally - discovered drug to reach >$1B in annual net sales Prior to July 1, 2022, Sanofi recorded net product sales of Libtayo outside the United States.
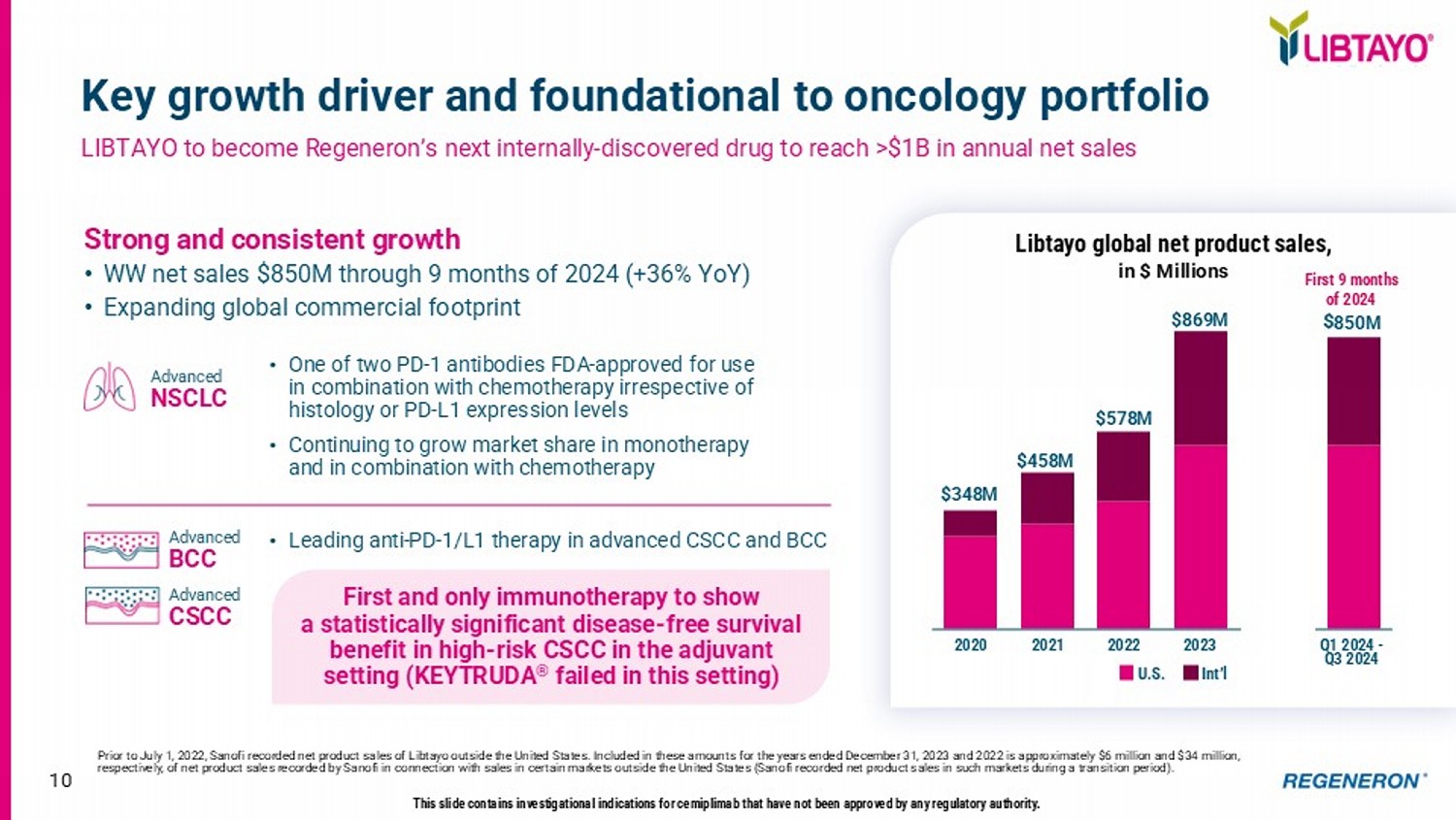
Included in these amounts for the years ended December 31, 2023 and 2022 is approximately $6 million and $34 million, respectively, of net product sales recorded by Sanofi in connection with sales in certain markets outside the United States ( San ofi recorded net product sales in such markets during a transition period). $458M $869M $348M $578M U.S. Int’l • One of two PD - 1 antibodies FDA - approved for use in combination with chemotherapy irrespective of histology or PD - L1 expression levels • Continuing to grow market share in monotherapy and in combination with chemotherapy • Leading anti - PD - 1/L1 therapy in advanced CSCC and BCC Advanced NSCLC Advanced BCC Advanced CSCC This slide contains investigational indications for cemiplimab that have not been approved by any regulatory authority.

First and only immunotherapy to show a statistically significant disease - free survival benefit in high - risk CSCC in the adjuvant setting (KEYTRUDA ® failed in this setting) Libtayo global net product sales , in $ Millions 11 Value Proposition Indication(s) Anticipated Launch Year Product Category First and only biologic approved for eosinophilic COPD COPD with Type 2 inflammation 2024 Dupixent Eosinophilic COPD Potential first - in class opportunity to address up to 1 million former smokers with COPD globally COPD in former smokers 2026 itepekimab COPD in former smokers First and only immunotherapy to show a statistically significant DFS benefit in high - risk adjuvant CSCC Adjuvant CSCC 2025 - 2026 Libtayo Non - melanoma skin cancers Emerging as a potentially differentiated treatment option in multiple solid tumors Melanoma, NSCLC, HNSCC 2026 (Melanoma) fianlimab + Libtayo Solid tumors Potentially best - in - class BCMA bispecific to disrupt current treatment paradigm in earlier lines Multiple myeloma & pre - cursor conditions 2025 (3L+ MM only) linvoseltamab Myeloma Potentially best - in - class CD20 bispecific (in FL) to disrupt current treatment paradigm in earlier lines FL, DLBCL 2025 (3L+ FL only) odronextamab Lymphoma Complete inhibition of C5 has potential to improve efficacy and convenience gMG, PNH, GA 2027 (gMG) pozelimab + cemdisiran Complement - mediated diseases Potential to improve efficacy and safety relative to current standards of care Coagulation disorders 2028 REGN7508 & REGN9933 Anticoagulants Potential to improve quality of weight loss when combined with GLP - 1 therapy Obesity, T2DM 2028 trevogrumab ± garetosmab Obesity Groundbreaking approach to potentially reverse severe food allergy IgE - mediated food allergies TBD Dupixent + linvoseltamab Food allergy treatment Differentiated pipeline opportunities to potentially address categories expected to exceed $220 billion annually in 2030 12 Research & Pipeline Update J.P. Morgan Healthcare Conference 2025 George D. Yancopoulos, MD, PhD Co - Founder, Board Co - Chair, President & Chief Scientific Officer

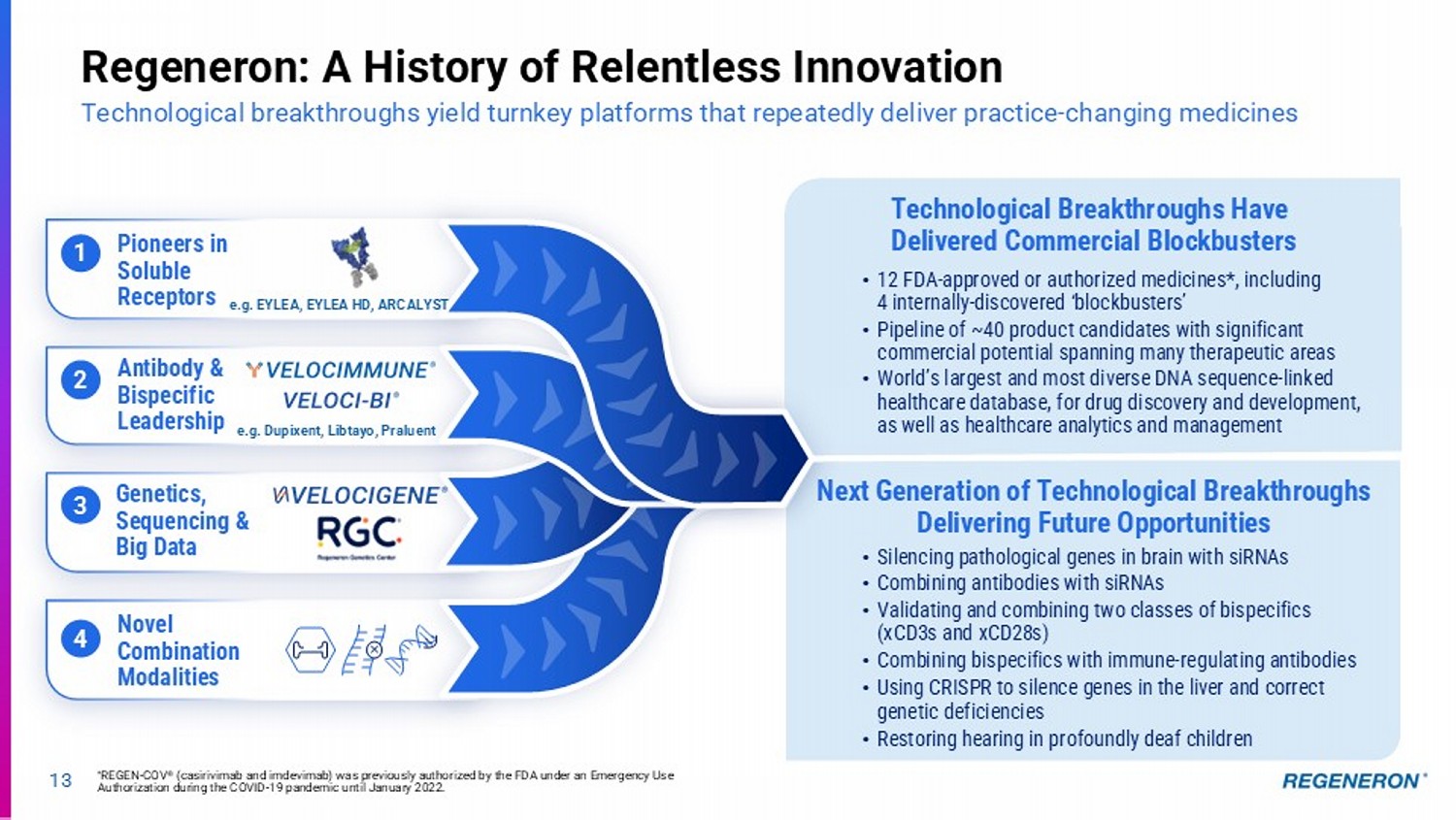
3 4 2 1 13 Regeneron: A History of Relentless Innovation Technological breakthroughs yield turnkey platforms that repeatedly deliver practice - changing medicines Genetics, Sequencing & Big Data Novel Combination Modalities Antibody & Bispecific Leadership Technological Breakthroughs Have Delivered Commercial Blockbusters Pioneers in Soluble Receptors • Silencing pathological genes in brain with siRNAs • Combining antibodies with siRNAs • Validating and combining two classes of bispecifics (xCD3s and xCD28s) • Combining bispecifics with immune - regulating antibodies • Using CRISPR to silence genes in the liver and correct genetic deficiencies • Restoring hearing in profoundly deaf children e.g. EYLEA, EYLEA HD, ARCALYST e.g. Dupixent, Libtayo, Praluent Next Generation of Technological Breakthroughs Delivering Future Opportunities • 12 FDA - approved or authorized medicines*, including 4 internally - discovered ‘blockbusters’ • Pipeline of ~40 product candidates with significant commercial potential spanning many therapeutic areas • World’s largest and most diverse DNA sequence - linked healthcare database, for drug discovery and development, as well as healthcare analytics and management * REGEN - COV ® (casirivimab and imdevimab) was previously authorized by the FDA under an Emergency Use Authorization during the COVID - 19 pandemic until January 2022.
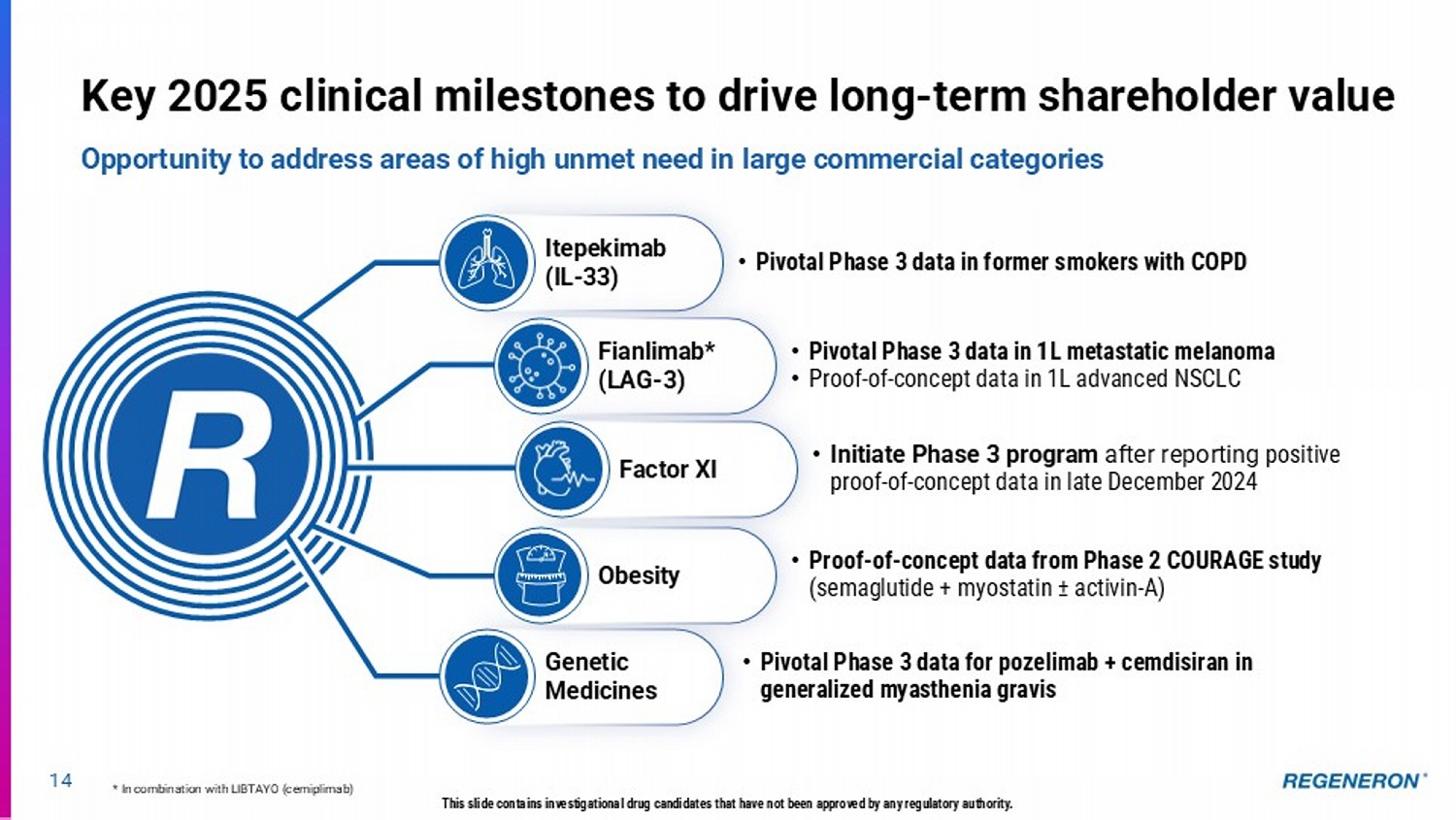
14 This slide contains investigational drug candidates that have not been approved by any regulatory authority. Key 2025 clinical milestones to drive long - term shareholder value Opportunity to address areas of high unmet need in large commercial categories * In combination with LIBTAYO (cemiplimab) Itepekimab (IL - 33) • Pivotal Phase 3 data in former smokers with COPD Genetic Medicines • Pivotal Phase 3 data for pozelimab + cemdisiran in generalized myasthenia gravis • Proof - of - concept data from Phase 2 COURAGE study (semaglutide + myostatin ± activin - A) Obesity Fianlimab* (LAG - 3) • Pivotal Phase 3 data in 1L metastatic melanoma • Proof - of - concept data in 1L advanced NSCLC Factor XI • Initiate Phase 3 program after reporting positive proof - of - concept data in late December 2024 P - value 0.091 0.001 0.002 0.005 15 Itepekimab (IL - 33): Regeneron’s next innovation in COPD with pivotal results anticipated in 2H 2025 Building upon Dupixent’s clinical success, potential for benefit in broader COPD population LOF: loss - of - function GOF: gain - of - function This slide contains investigational drug candidates that have not been approved by any regulatory authority.
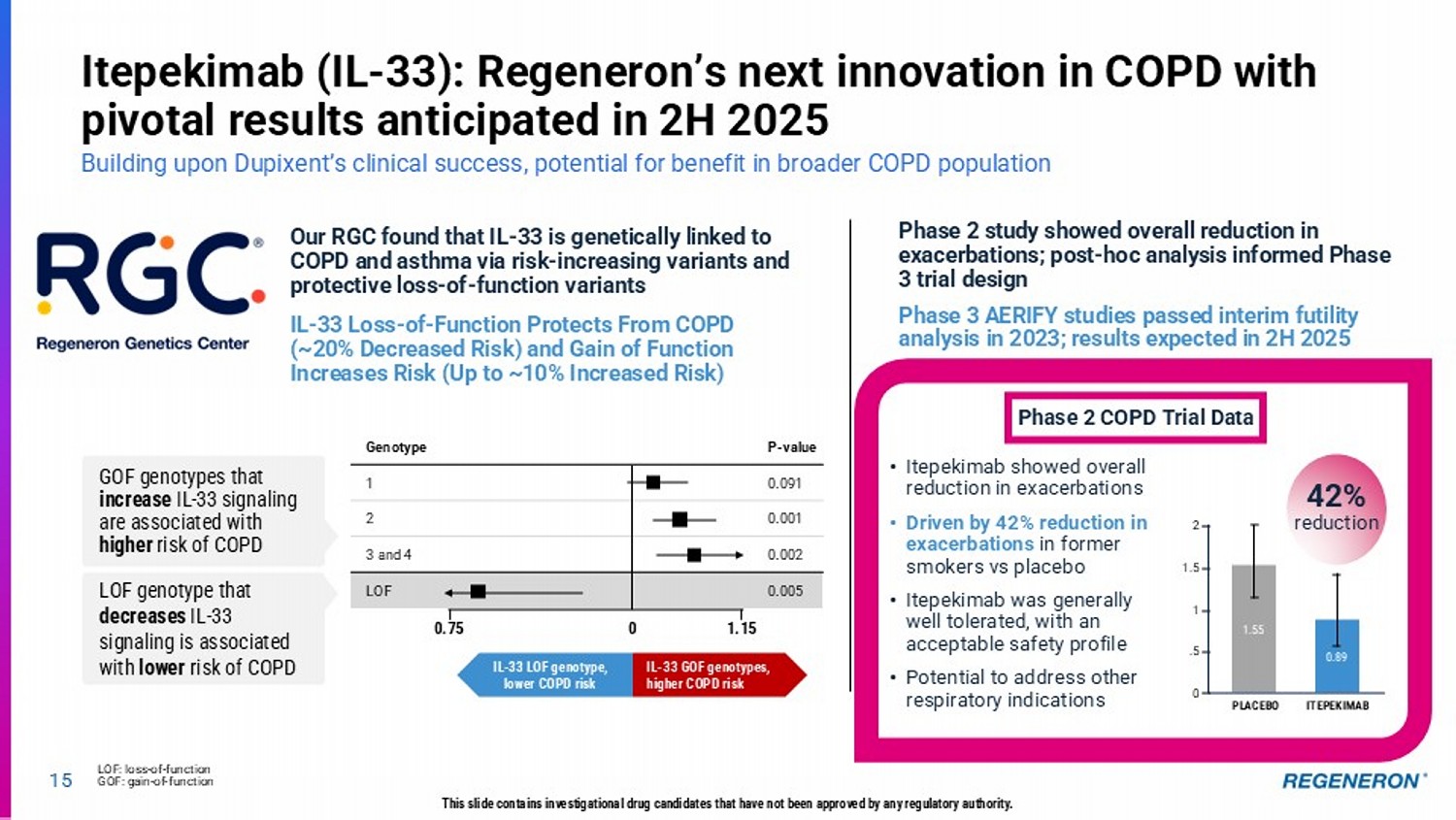
• Itepekimab showed overall reduction in exacerbations • Driven by 42% reduction in exacerbations in former smokers vs placebo • Itepekimab was generally well tolerated, with an acceptable safety profile • Potential to address other respiratory indications Our RGC found that IL - 33 is genetically linked to COPD and asthma via risk - increasing variants and protective loss - of - function variants IL - 33 Loss - of - Function Protects From COPD (~20% Decreased Risk) and Gain of Function Increases Risk (Up to ~10% Increased Risk) Phase 2 study showed overall reduction in exacerbations; post - hoc analysis informed Phase 3 trial design Phase 3 AERIFY studies passed interim futility analysis in 2023; results expected in 2H 2025 Phase 2 COPD Trial Data 42% reduction 1.55 0.89 ITEPEKIMAB PLACEBO 2 1.5 1 .5 0 Genotype 1 2 3 and 4 LOF IL - 33 GOF genotypes, higher COPD risk IL - 33 LOF genotype, lower COPD risk 0.75 0 1.15 LOF genotype that decreases IL - 33 signaling is associated with lower risk of COPD GOF genotypes that increase IL - 33 signaling are associated with higher risk of COPD Regeneron’s oncology strategy: Using the immune system to defeat cancer with 5 classes of immunomodulatory agents Signal 1 CD3 Bispecifics Designed to link killer T cells with cancer cells Signal 2 Costimulatory Bispecifics Activating killer T cells via costimulation Signal 3 (e.g., Targeted Cytokines) Designed to selectively recruit immune cells to the tumor microenvironment T Cell checkpoint inhibitors LIBTAYO: anti - PD1 Fianlimab: anti - LAG3 Designed to overcome T cell suppression Antibody Drug Conjugates Designed to directly and selectively kill tumor cells Lung Cancer NSCLC; potential for SCLC Indication areas of focus Dermato - Oncology CSCC; BCC; Melanoma Genitourinary Prostate; RCC; potential for bladder Gyn - Onc Ovarian; endometrial; cervical GI CRC; esophageal / gastric; HCC HNSCC Hematological Lymphomas, Myelomas, Myeloid malignancy 16 □ REGN has clinically validated the first 3 classes □ Can be used across multiple tumor types and in combination Regeneron has clinically validated these first 3 classes, several with potentially best - in - class clinical efficacy Earlier - stage Programs

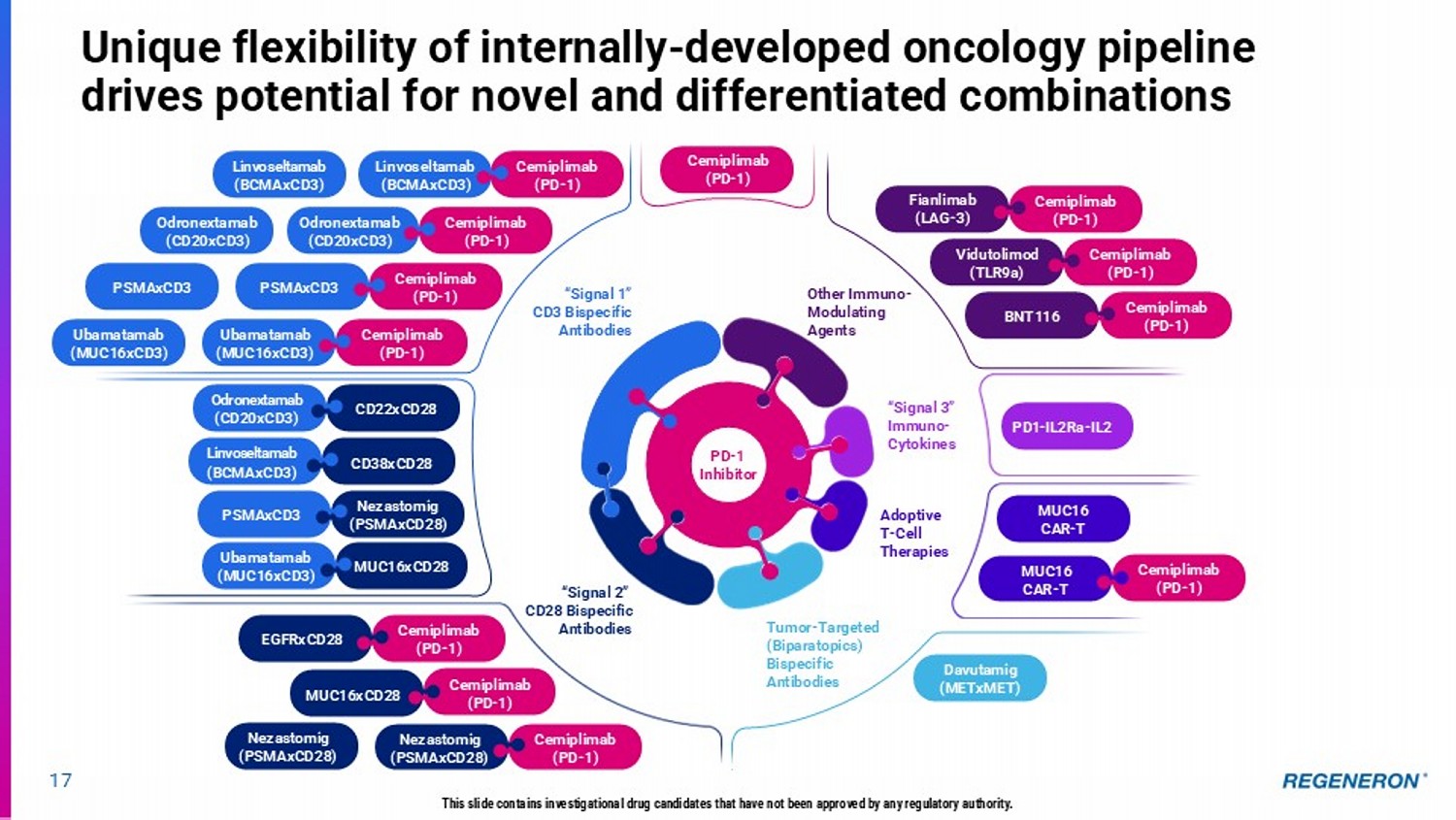
Unique flexibility of internally - developed oncology pipeline drives potential for novel and differentiated combinations Cemiplimab (PD - 1) PD1 - IL2Ra - IL2 MUC16 CAR - T Davutamig (METxMET) MUC16 CAR - T Cemiplimab (PD - 1) PD - 1 Inhibitor Odronextamab (CD20xCD3) Linvoseltamab (BCMAxCD3) PSMAxCD3 Ubamatamab (MUC16xCD3) Cemiplimab (PD - 1) Linvoseltamab (BCMAxCD3) “Signal 1” CD3 Bispecific Antibodies “Signal 2” CD28 Bispecific Antibodies Other Immuno - Modulating Agents “Signal 3” Immuno - Cytokines Tumor - Targeted (Biparatopics) Bispecific Antibodies Adoptive T - Cell Therapies Odronextamab (CD20xCD3) CD22xCD28 Cemiplimab (PD - 1) Odronextamab (CD20xCD3) Cemiplimab (PD - 1) PSMAxCD3 Cemiplimab (PD - 1) Ubamatamab (MUC16xCD3) Linvoseltamab (BCMAxCD3) CD38xCD28 PSMAxCD3 Nezastomig (PSMAxCD28) Fianlimab (LAG - 3) Cemiplimab (PD - 1) Vidutolimod (TLR9a) Cemiplimab (PD - 1) BNT116 Cemiplimab (PD - 1) Ubamatamab (MUC16xCD3) MUC16xCD28 Cemiplimab (PD - 1) EGFRxCD28 Nezastomig (PSMAxCD28) Cemiplimab (PD - 1) Nezastomig (PSMAxCD28) Cemiplimab (PD - 1) MUC16xCD28 17 This slide contains investigational drug candidates that have not been approved by any regulatory authority.
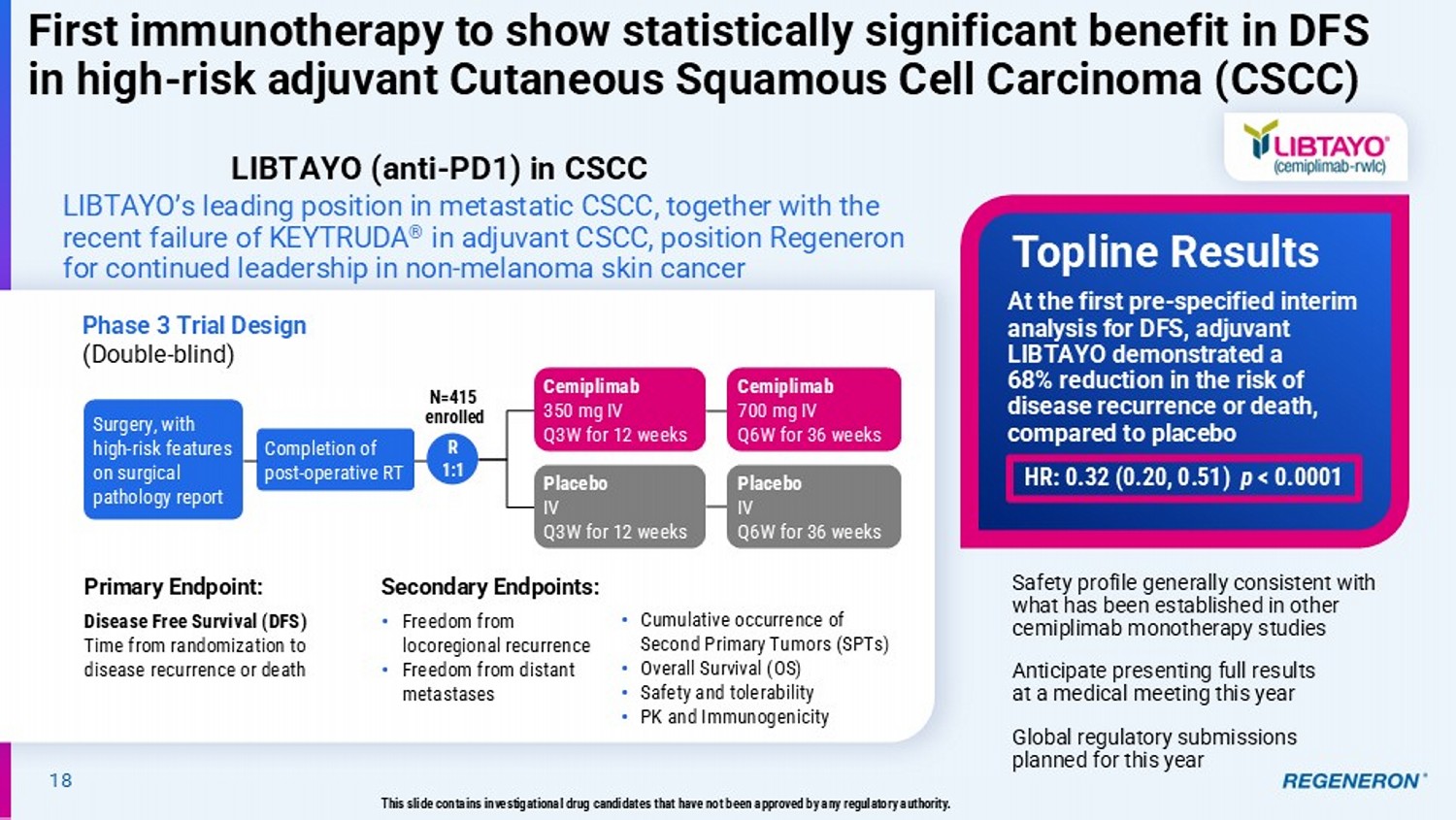
18 First immunotherapy to show statistically significant benefit in DFS in high - risk adjuvant Cutaneous Squamous Cell Carcinoma (CSCC) LIBTAYO’s leading position in metastatic CSCC, together with the recent failure of KEYTRUDA ® in adjuvant CSCC, position Regeneron for continued leadership in non - melanoma skin cancer Phase 3 Trial Design (Double - blind) Surgery, with high - risk features on surgical pathology report N=415 enrolled Primary Endpoint: Disease Free Survival (DFS) Time from randomization to disease recurrence or death Secondary Endpoints: • Freedom from locoregional recurrence • Freedom from distant metastases Safety profile generally consistent with what has been established in other cemiplimab monotherapy studies Anticipate presenting full results at a medical meeting this year Global regulatory submissions planned for this year Topline Results R 1:1 Completion of post - operative RT Cemiplimab 350 mg IV Q3W for 12 weeks Cemiplimab 700 mg IV Q6W for 36 weeks Placebo IV Q3W for 12 weeks Placebo IV Q6W for 36 weeks • Cumulative occurrence of Second Primary Tumors (SPTs) • Overall Survival (OS) • Safety and tolerability • PK and Immunogenicity This slide contains investigational drug candidates that have not been approved by any regulatory authority.
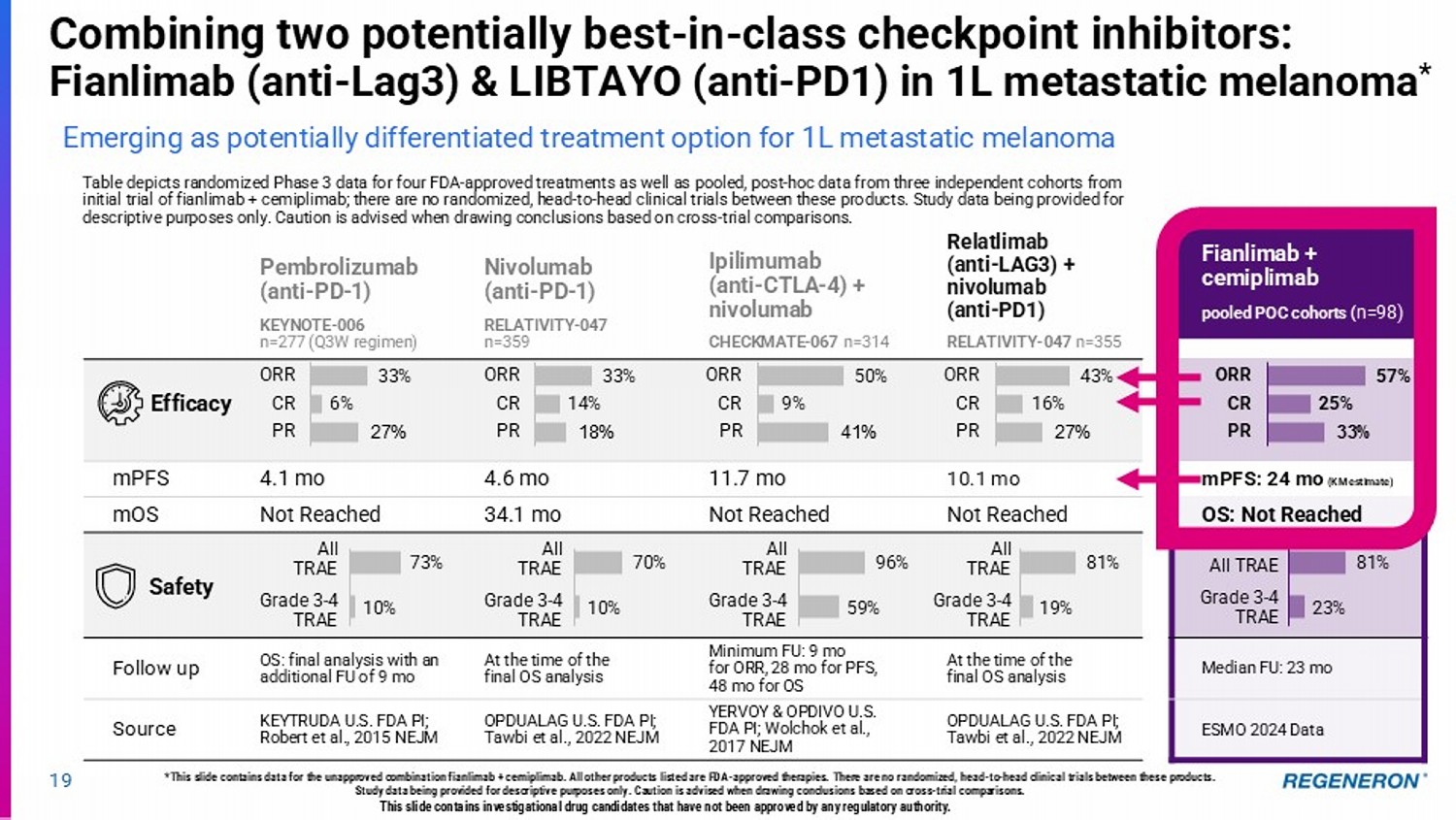
At the first pre - specified interim analysis for DFS, adjuvant LIBTAYO demonstrated a 68% reduction in the risk of disease recurrence or death, compared to placebo HR: 0.32 (0.20, 0.51) p < 0.0001 LIBTAYO (anti - PD1) in CSCC Fianlimab + cemiplimab pooled POC cohorts ( n=98) Relatlimab (anti - LAG3) + nivolumab (anti - PD1) RELATIVITY - 047 n=355 Ipilimumab (anti - CTLA - 4) + nivolumab CHECKMATE - 067 n=314 Nivolumab (anti - PD - 1) RELATIVITY - 047 n=359 Pembrolizumab (anti - PD - 1) KEYNOTE - 006 n=277 (Q3W regimen) mPFS: 24 mo (KM estimate) 10.1 mo 11.7 mo 4.6 mo 4.1 mo mPFS OS: Not Reached Not Reached Not Reached 34.1 mo Not Reached mOS All TRAE Grade 3 - 4 TRAE All TRAE Grade 3 - 4 TRAE All TRAE Grade 3 - 4 TRAE All TRAE Grade 3 - 4 TRAE All TRAE Grade 3 - 4 TRAE Median FU: 23 mo At the time of the final OS analysis Minimum FU: 9 mo for ORR, 28 mo for PFS, 48 mo for OS At the time of the final OS analysis OS: final analysis with an additional FU of 9 mo Follow up ESMO 2024 Data OPDUALAG U.S. FDA PI; Tawbi et al., 2022 NEJM YERVOY & OPDIVO U.S. FDA PI; Wolchok et al., 2017 NEJM OPDUALAG U.S. FDA PI; Tawbi et al., 2022 NEJM KEYTRUDA U.S. FDA PI; Robert et al., 2015 NEJM Source 19 Table depicts randomized Phase 3 data for four FDA - approved treatments as well as pooled, post - hoc data from three independent c ohorts from initial trial of fianlimab + cemiplimab; there are no randomized, head - to - head clinical trials between these products. Study dat a being provided for descriptive purposes only. Caution is advised when drawing conclusions based on cross - trial comparisons. *This slide contains data for the unapproved combination fianlimab + cemiplimab. All other products listed are FDA - approved ther apies. There are no randomized, head - to - head clinical trials between these products. Study data being provided for descriptive purposes only. Caution is advised when drawing conclusions based on cross - trial compar isons. Safety Efficacy 33% 6% 27% ORR CR PR 33% 14% 18% ORR CR PR 50% 9% 41% ORR CR PR 43% 16% 27% ORR CR PR 57% 25% 33% ORR CR PR 73% 10% 70% 10% 96% 59% 81% 19% 81% 23% Combining two potentially best - in - class checkpoint inhibitors: Fianlimab (anti - Lag3) & LIBTAYO (anti - PD1) in 1L metastatic melanoma * Emerging as potentially differentiated treatment option for 1L metastatic melanoma This slide contains investigational drug candidates that have not been approved by any regulatory authority.
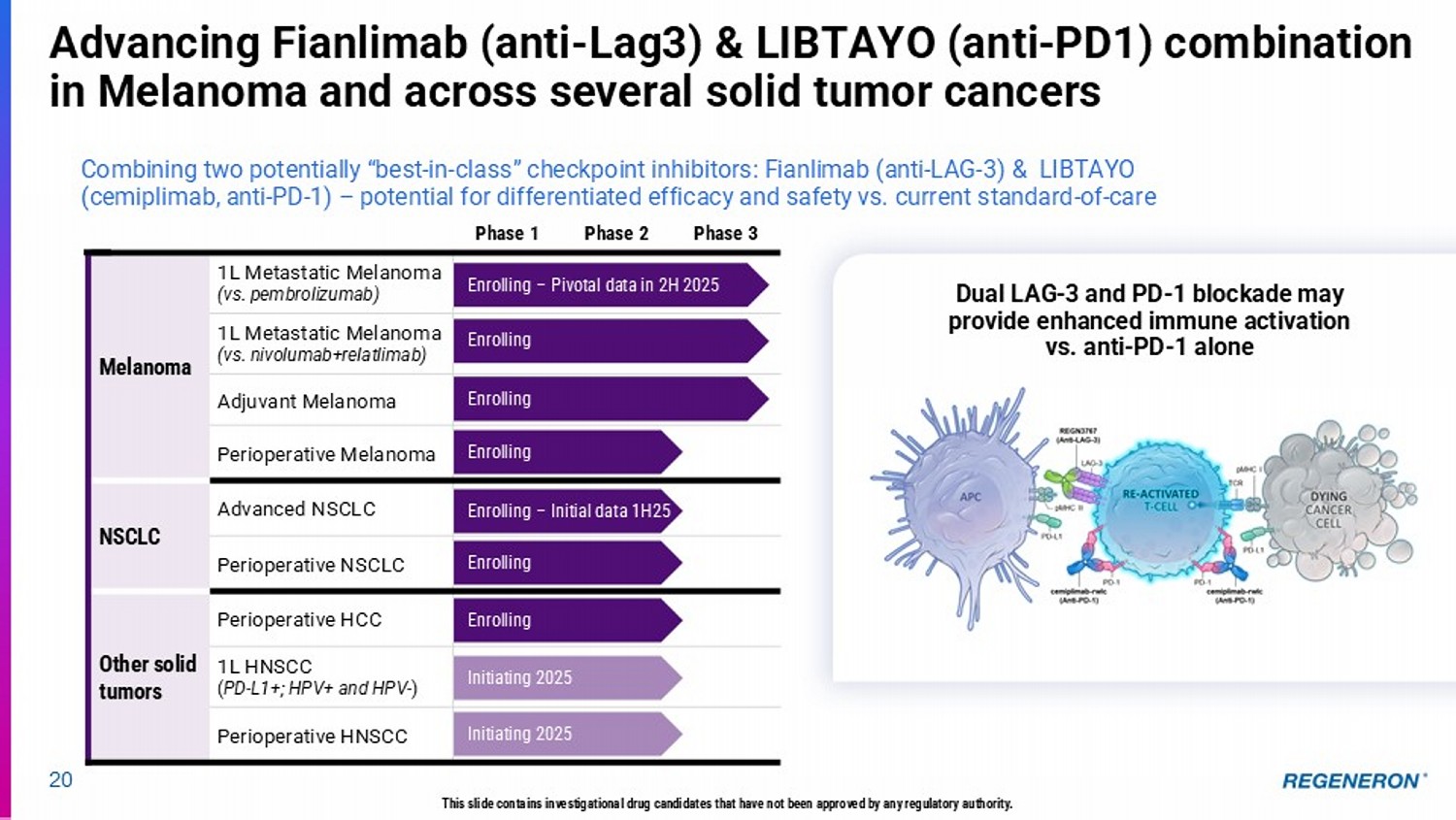
Phase 3 Phase 2 Phase 1 1L Metastatic Melanoma (vs. pembrolizumab) Melanoma 1L Metastatic Melanoma (vs. nivolumab+relatlimab) Adjuvant Melanoma Perioperative Melanoma Advanced NSCLC NSCLC Perioperative NSCLC Perioperative HCC Other solid tumors 1L HNSCC ( PD - L1+; HPV+ and HPV - ) Perioperative HNSCC 20 Advancing Fianlimab (anti - Lag3) & LIBTAYO (anti - PD1) combination in Melanoma and across several solid tumor cancers Combining two potentially “best - in - class” checkpoint inhibitors: Fianlimab (anti - LAG - 3) & LIBTAYO (cemiplimab, anti - PD - 1) – potential for differentiated efficacy and safety vs. current standard - of - care Enrolling – Pivotal data in 2H 2025 Enrolling Enrolling Enrolling – Initial data 1H25 Enrolling Initiating 2025 Enrolling Initiating 2025 Enrolling Dual LAG - 3 and PD - 1 blockade may provide enhanced immune activation vs. anti - PD - 1 alone This slide contains investigational drug candidates that have not been approved by any regulatory authority.
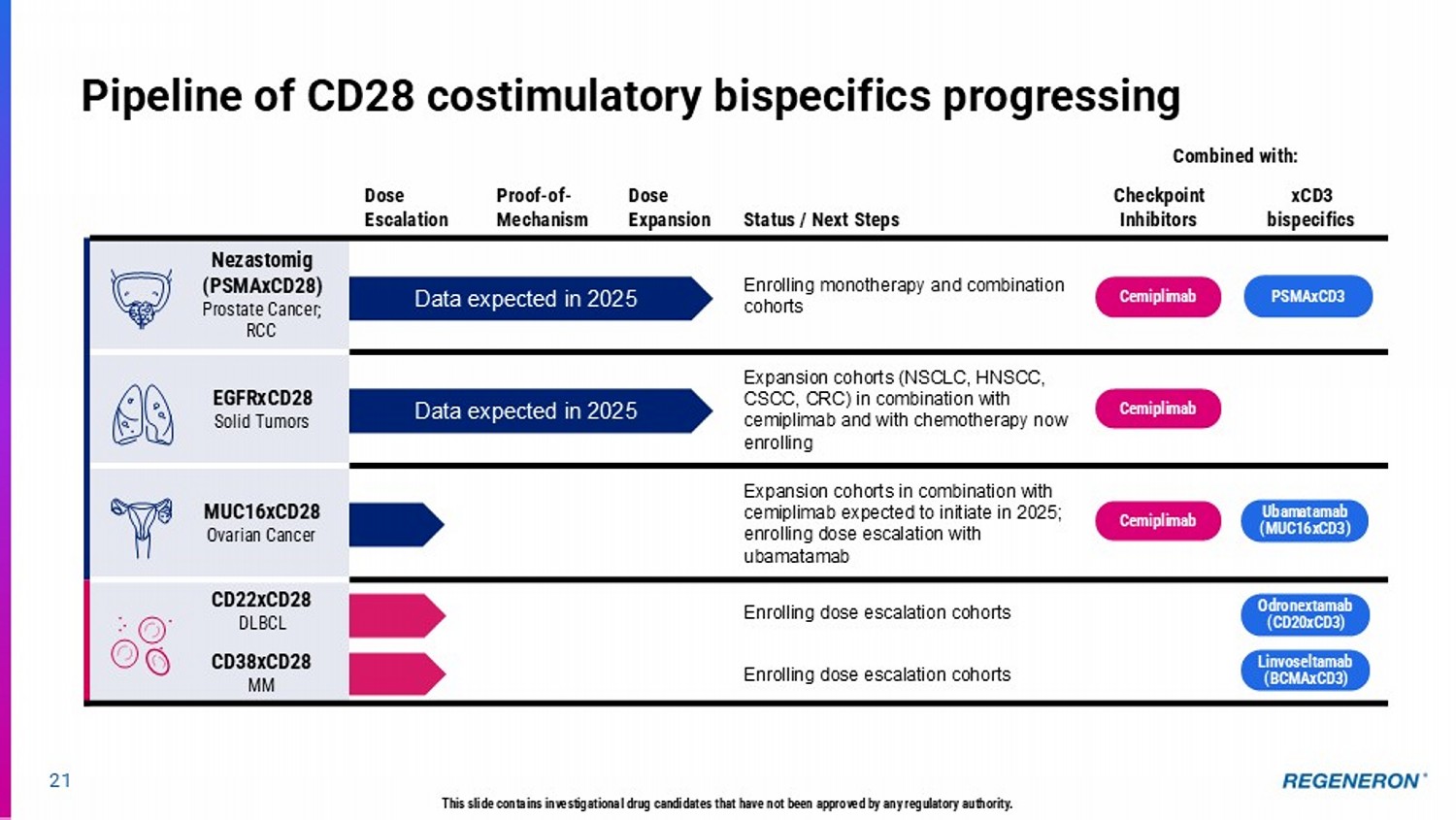
Combined with: Status / Next Steps Dose Expansion Proof - of - Mechanism Dose Escalation xCD3 bispecifics Checkpoint Inhibitors Enrolling monotherapy and combination cohorts Nezastomig (PSMAxCD28) Prostate Cancer; RCC Expansion cohorts (NSCLC, HNSCC, CSCC, CRC) in combination with cemiplimab and with chemotherapy now enrolling EGFRxCD28 Solid Tumors Expansion cohorts in combination with cemiplimab expected to initiate in 2025; enrolling dose escalation with ubamatamab MUC16xCD28 Ovarian Cancer Enrolling dose escalation cohorts CD22xCD28 DLBCL Enrolling dose escalation cohorts CD38xCD28 MM 21 Pipeline of CD28 costimulatory bispecifics progressing D ata expected in 2025 Data expected in 2025 Cemiplimab PSMAxCD3 Cemiplimab Cemiplimab Ubamatamab (MUC16xCD3) Odronextamab (CD20xCD3) Linvoseltamab (BCMAxCD3) This slide contains investigational drug candidates that have not been approved by any regulatory authority.

Regeneron’s differentiated CD3 bispecifics 22 LINVOSELTAMAB (BCMAxCD3) Multiple myeloma (MM) Linvoseltamab has the potential to be the best - in - class BCMAxCD3 bispecific with its differentiated clinical profile, dosing, and administration ORDSPONO (odronextamab, CD20xCD3) Non - Hodgkin lymphoma (NHL) Regeneron’s first approved bispecific antibody (in EU) in relapsed/refractory (R/R) follicular lymphoma (FL) and diffuse large B - cell lymphoma (DLBCL) T Cell Receptor/ CD3 Tumor - specific target This slide contains investigational drug candidates that have not been approved by any regulatory authority.
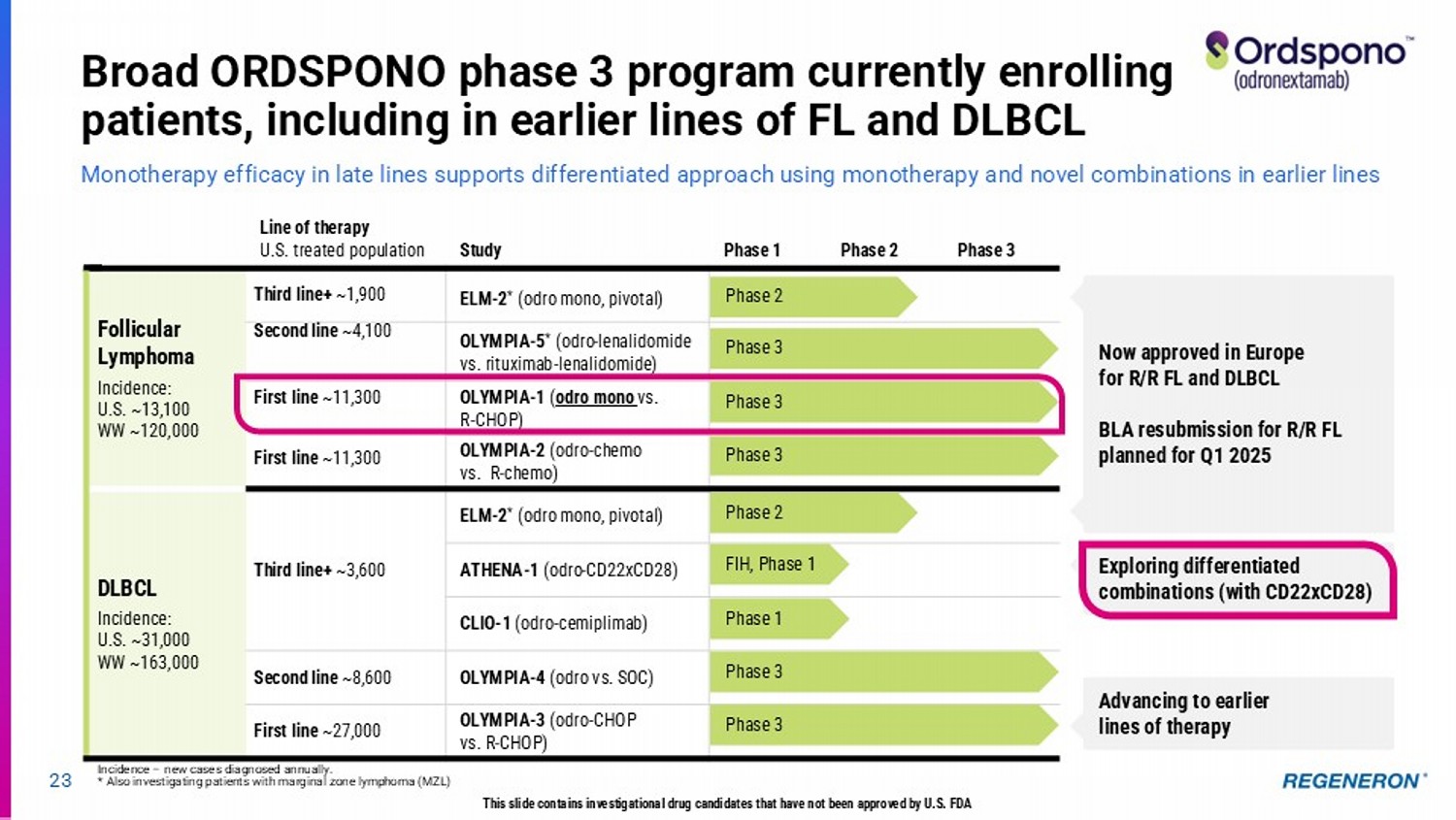
Approved in Europe in 2024 Enrollment underway for confirmatory study to support BLA resubmission for FL BLA resubmission planned for Q1 2025 71% ORR / 50% CR in r/r MM † Nearly double the CR rate of other bispecifics at similar follow - up 80% ORR / 73% CR in r/r FL Highest response rate observed in the class in this late - line setting Third - party fill/finish manufacturer currently in compliance BLA recently resubmitted: FDA approval anticipated by mid - 2025 Differentiated Phase 3 programs in earlier lines of therapy using monotherapy and novel combinations underway for both ORDSPONO and linvoseltamab † Median follow up of 14 months Exploring differentiated combinations (with CD22xCD28) 23 Broad ORDSPONO phase 3 program currently enrolling patients, including in earlier lines of FL and DLBCL Monotherapy efficacy in late lines supports differentiated approach using monotherapy and novel combinations in earlier lines Phase 3 Phase 2 Phase 1 Study Line of therapy U.S. treated population ELM - 2 * (odro mono, pivotal) Third line+ ~1,900 Follicular Lymphoma Incidence: U.S. ~13,100 WW ~120,000 OLYMPIA - 5 * (odro - lenalidomide vs. rituximab - lenalidomide) Second line ~4,100 OLYMPIA - 1 ( odro mono vs. R - CHOP) First line ~11,300 First line ~11,300 OLYMPIA - 2 (odro - chemo vs. R - chemo) ELM - 2 * (odro mono, pivotal) Third line+ ~3,600 DLBCL Incidence: U.S. ~31,000 WW ~163,000 ATHENA - 1 (odro - CD22xCD28) CLIO - 1 (odro - cemiplimab) OLYMPIA - 4 (odro vs. SOC) Second line ~8,600 OLYMPIA - 3 (odro - CHOP vs. R - CHOP) First line ~27,000 Now approved in Europe for R/R FL and DLBCL BLA resubmission for R/R FL planned for Q1 2025 Incidence – new cases diagnosed annually. * Also investigating patients with marginal zone lymphoma (MZL) Advancing to earlier lines of therapy Phase 2 Phase 3 FIH, Phase 1 Phase 3 Phase 3 Phase 1 Phase 3 Phase 3 Phase 2 This slide contains investigational drug candidates that have not been approved by U.S.
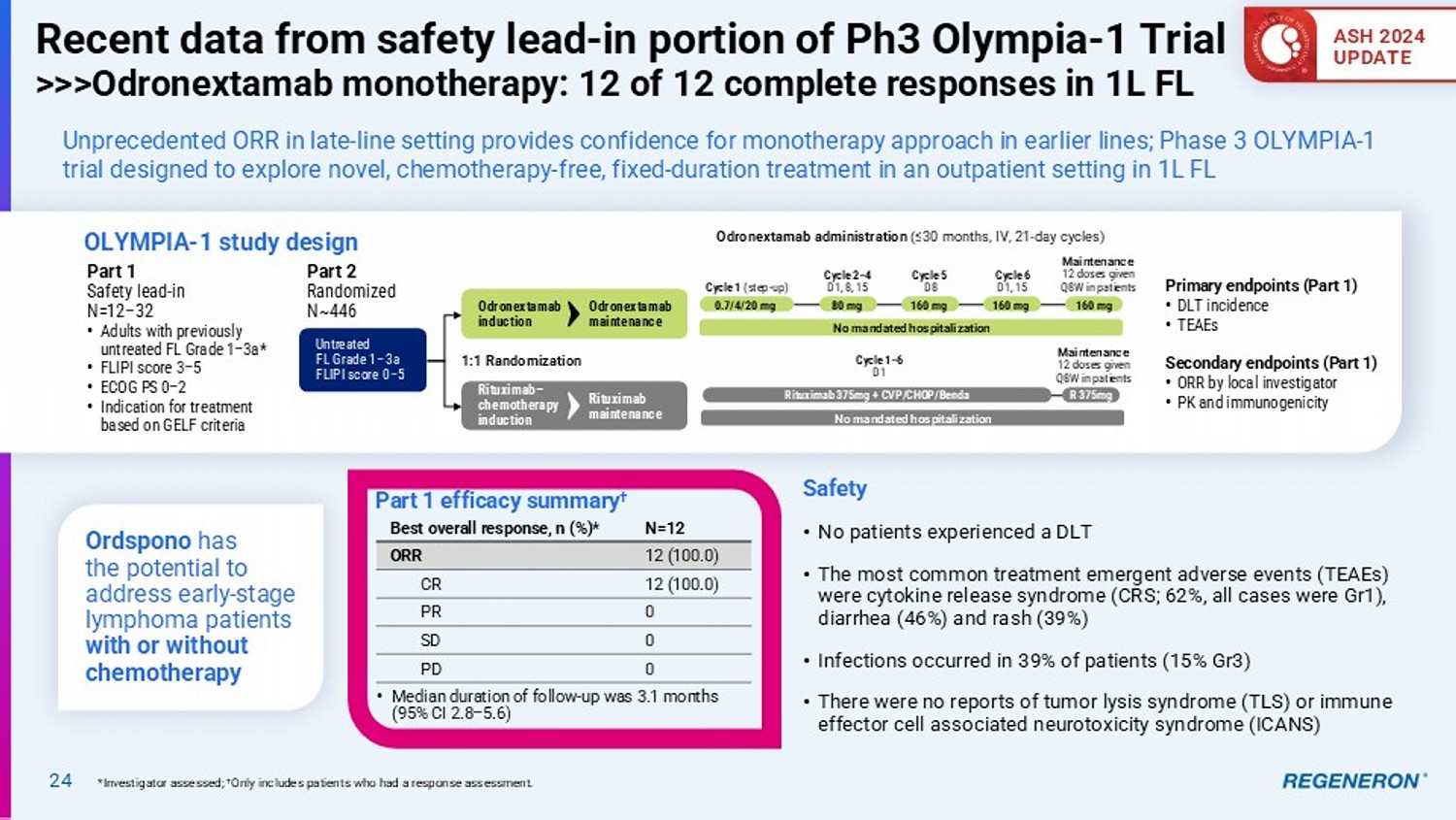
FDA Unprecedented ORR in late - line setting provides confidence for monotherapy approach in earlier lines; Phase 3 OLYMPIA - 1 trial designed to explore novel, chemotherapy - free, fixed - duration treatment in an outpatient setting in 1L FL 24 Recent data from safety lead - in portion of Ph3 Olympia - 1 Trial >>>Odronextamab monotherapy: 12 of 12 complete responses in 1L FL *Investigator assessed; † Only includes patients who had a response assessment.
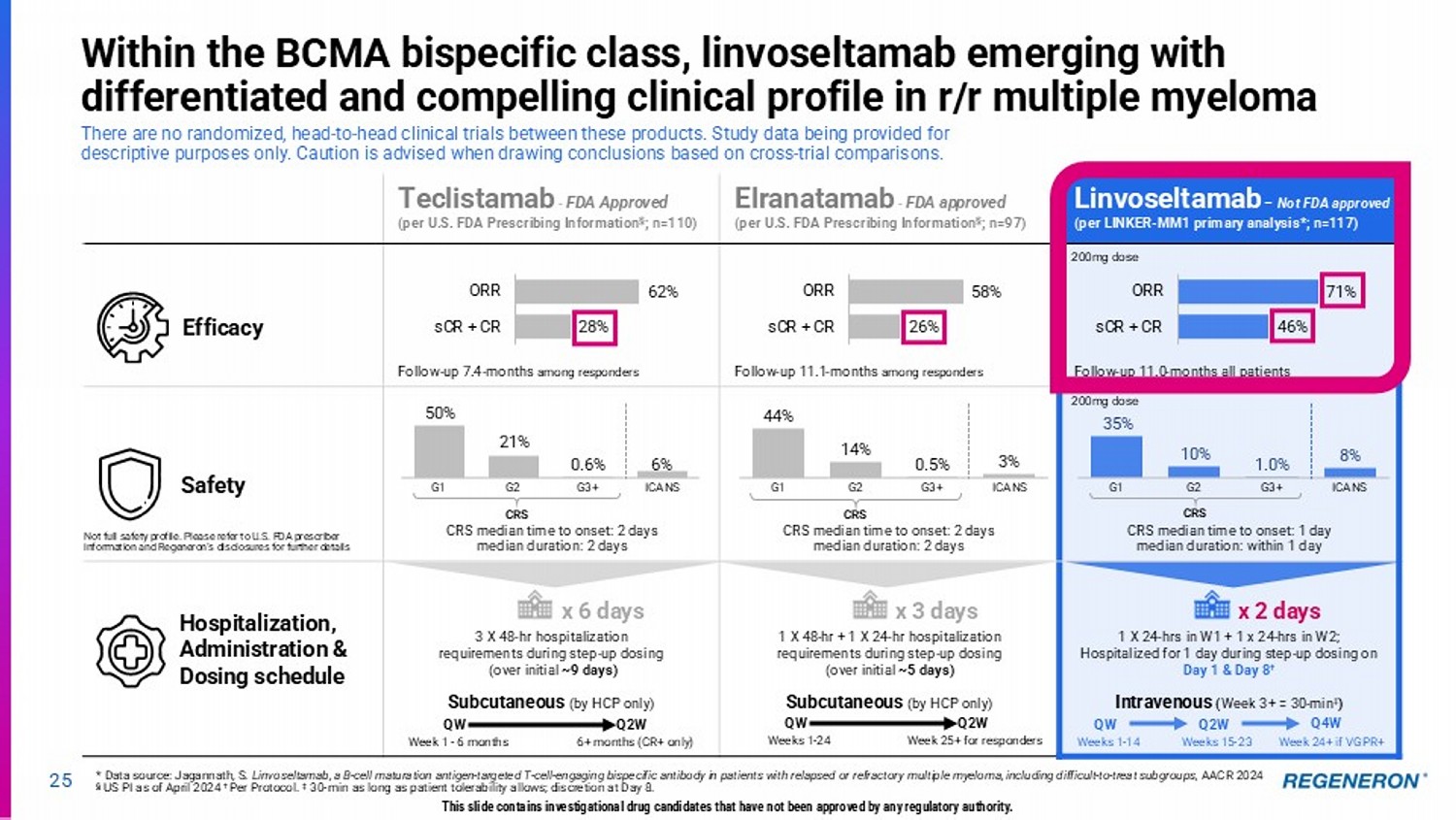
Safety • No patients experienced a DLT • The most common treatment emergent adverse events (TEAEs) were cytokine release syndrome (CRS; 62%, all cases were Gr1), diarrhea (46%) and rash (39%) • Infections occurred in 39% of patients (15% Gr3) • There were no reports of tumor lysis syndrome (TLS) or immune effector cell associated neurotoxicity syndrome (ICANS) N=12 Best overall response, n (%)* 12 (100.0) ORR 12 (100.0) CR 0 PR 0 SD 0 PD • Median duration of follow - up was 3.1 months (95% CI 2.8 – 5.6) Part 1 efficacy summary † OLYMPIA - 1 study design Part 1 Safety lead - in N=12 – 32 • Adults with previously untreated FL Grade 1 – 3a* • FLIPI score 3 – 5 • ECOG PS 0 – 2 • Indication for treatment based on GELF criteria Part 2 Randomized N~446 Ordspono has the potential to address early - stage lymphoma patients with or without chemotherapy ASH 2024 UPDATE Primary endpoints (Part 1) • DLT incidence • TEAEs Secondary endpoints (Part 1) • ORR by local investigator • PK and immunogenicity Untreated FL Grade 1 – 3a FLIPI score 0 – 5 Odronextamab administration (≤30 months, IV, 21 - day cycles) Cycle 1 (step - up) 0.7/4/20 mg Cycle 2 – 4 D1, 8, 15 80 mg Cycle 5 D8 160 mg Cycle 6 D1, 15 160 mg Maintenance 12 doses given Q8W in patients 160 mg No mandated hospitalization Odronextamab induction Odronextamab maintenance Rituximab – chemotherapy induction Rituximab maintenance 1:1 Randomization No mandated hospitalization Cycle 1 – 6 D1 Rituximab 375mg + CVP/CHOP/Benda Maintenance 12 doses given Q8W in patients R 375mg * Data source: Jagannath, S. Linvoseltamab, a B - cell maturation antigen - targeted T - cell - engaging bispecific antibody in patients with relapsed or refractory multiple myeloma, including difficult - to - treat subgroups, AACR 2024 § US PI as of April 2024 † Per Protocol. ‡ 30 - min as long as patient tolerability allows; discretion at Day 8. Linvoseltamab – Not FDA approved (per LINKER - MM1 primary analysis*; n=117) Elranatamab - FDA approved (per U.S. FDA Prescribing Information § ; n=97) Teclistamab - FDA Approved (per U.S. FDA Prescribing Information § ; n=110) Follow - up 11.0 - months all patients Follow - up 11.1 - months among responders Follow - up 7.4 - months among responders CRS median time to onset: 1 day median duration: within 1 day CRS median time to onset: 2 days median duration: 2 days CRS median time to onset: 2 days median duration: 2 days 1 X 24 - hrs in W1 + 1 x 24 - hrs in W2; Hospitalized for 1 day during step - up dosing on Day 1 & Day 8 † Intravenous ( Week 3+ = 30 - min ‡ ) 1 X 48 - hr + 1 X 24 - hr hospitalization requirements during step - up dosing (over initial ~5 days) Subcutaneous (by HCP only) 3 X 48 - hr hospitalization requirements during step - up dosing (over initial ~9 days) Subcutaneous (by HCP only) 44% 14% 0.5% 3% G1 G2 G3+ ICANS 50% 21% 0.6% 6% G1 G2 G3+ ICANS 25 Within the BCMA bispecific class, linvoseltamab emerging with differentiated and compelling clinical profile in r/r multiple myeloma CRS Safety Hospitalization, Administration & Dosing schedule Efficacy Week 25+ for responders Weeks 1 - 24 Q2W QW Q4W Week 24+ if VGPR+ Weeks 1 - 14 Weeks 15 - 23 QW Q2W CRS 62% 28% ORR sCR + CR 200mg dose 35% 10% 1.0% 8% G1 G2 G3+ ICANS CRS 200mg dose 58% 26% ORR sCR + CR 71% 46% ORR sCR + CR x 6 days x 3 days x 2 days Not full safety profile. Please refer to U.S. FDA prescriber information and Regeneron’s disclosures for further details This slide contains investigational drug candidates that have not been approved by any regulatory authority. 6+ months (CR+ only) Week 1 - 6 months Q2W QW There are no randomized, head - to - head clinical trials between these products. Study data being provided for descriptive purposes only. Caution is advised when drawing conclusions based on cross - trial comparisons.

26 Broad linvoseltamab development program to evaluate monotherapy and simplified combinations in earlier stages of disease Unprecedented late - line responses rates provide confidence to explore monotherapy and novel combinations in earlier disease settings to simplify treatment approaches † Linvoseltamab mono vs. EPd (Elotuzumab + Pomalidomide + dexamethasone); 3L+ in the U.S.; earlier line of therapy eligible in som e geographies based on regional SOC Incidence – new cases diagnosed annually. *Prevalence provided instead of incidence as MGUS is a slow progressing disease. Phase 3 Phase 2 Phase 1 Study Line of therapy U.S. treated population LINKER - MM3 § (Linvo vs. EPd) Third line+ ~4,000 in 4L+/ ~8,000 in 3L Multiple Myeloma Incidence: U.S. ~35,000 WW >176,000 LINKER - MM1 (Linvo mono) (Linvo + CD38xCD28) LINKER - MM2 (cohorts of Linvo + SOC / novel therapies) Second line ~16,000 LINKER - MM4 (Linvo mono) First line ~30,000 Studies in maintenance, transplant ineligible, transplant eligible Study 2256 (Linvo mono) High Risk (HR) Smoldering MM Multiple Myeloma Precursor Conditions LINKER - MGUS1 (Linvo mono) HR MGUS / non - HR Smoldering MM LINKER - AL2 (Linvo mono) Second line+ AL Amyloidosis Incidence: U.S. ~4,500 Phase 3 Phase 1 Phase 3s planned Phase 1/2 FIH/Phase 1/2 Phase 2 Phase 2 FIH/Phase 1/2 Phase 1/2 BLA resubmitted, approval anticipated by mid - 2025 U.S. Epidemiology MM Precursor Conditions (clinically detected cases only, actual population may be higher; estimates not as well - characterized as MM) 1,200 – 1,600 HR SMM , incidence: 3,000 – 3,500 Non - HR SMM , incidence: 11,000 – 19,000 HR MGUS , prevalence * : This slide contains investigational drug candidates that have not been approved by any regulatory authority.
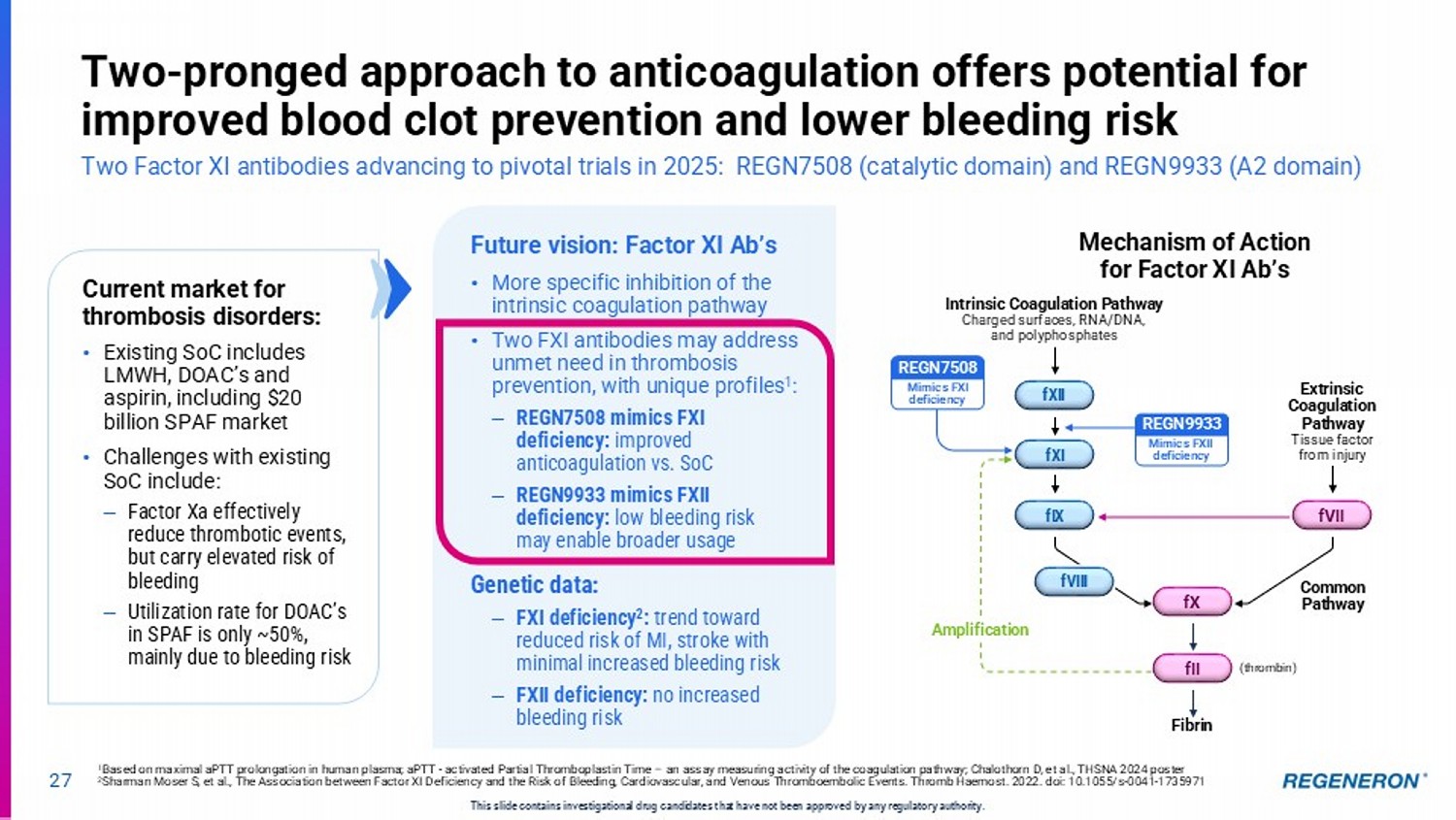
Exploring differentiated combinations (with CD38xCD28) Advancing to earlier lines of therapy 27 Two - pronged approach to anticoagulation offers potential for improved blood clot prevention and lower bleeding risk Two Factor XI antibodies advancing to pivotal trials in 2025: REGN7508 (catalytic domain) and REGN9933 (A2 domain) 1 Based on maximal aPTT prolongation in human plasma; aPTT - activated Partial Thromboplastin Time – an assay measuring activity o f the coagulation pathway; Chalothorn D, et al., THSNA 2024 poster 2 Sharman Moser S, et al., The Association between Factor XI Deficiency and the Risk of Bleeding, Cardiovascular, and Venous Th rom boembolic Events. Thromb Haemost. 2022. doi: 10.1055/s - 0041 - 1735971 Future vision: Factor XI Ab’s • More specific inhibition of the intrinsic coagulation pathway • T wo FXI antibodies may address unmet need in thrombosis prevention, with unique profiles 1 : – REGN7508 mimics FXI deficiency: improved anticoagulation vs. SoC – REGN9933 mimics FXII deficiency: low bleeding risk may enable broader usage Genetic data: – FXI deficiency 2 : trend toward reduced risk of MI, stroke with minimal increased bleeding risk – FXII deficiency: no increased bleeding risk This slide contains investigational drug candidates that have not been approved by any regulatory authority.
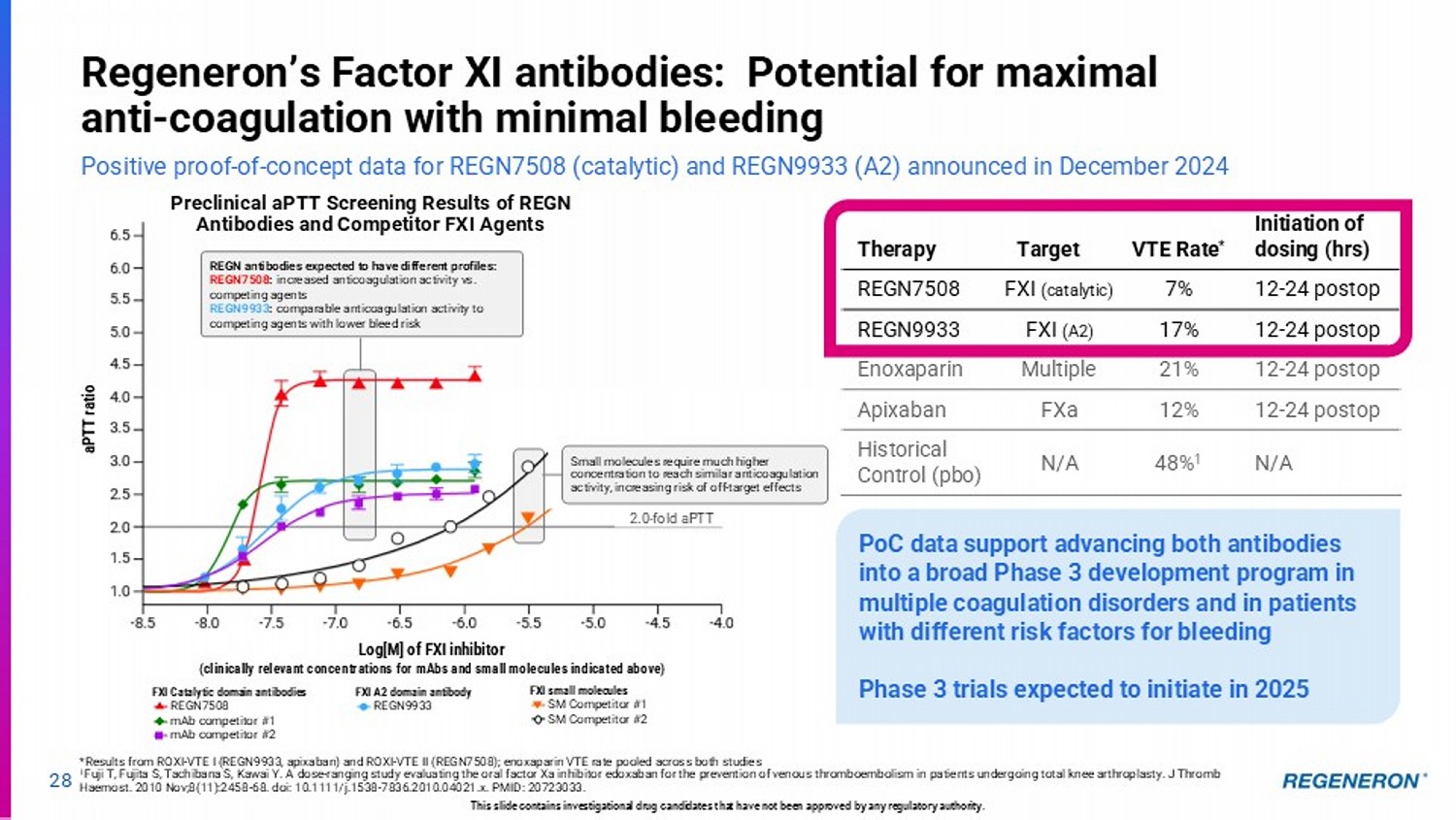
fXII fXI fIX fII fX Intrinsic Coagulation Pathway Charged surfaces, RNA/DNA, and polyphosphates Common Pathway Fibrin fVII fVIII (thrombin) Extrinsic Coagulation Pathway Tissue factor from injury Amplification Mechanism of Action for Factor XI Ab’s Current market for thrombosis disorders: • Existing SoC includes LMWH, DOAC’s and aspirin, including $20 billion SPAF market • Challenges with existing SoC include: – Factor Xa effectively reduce thrombotic events, but carry elevated risk of bleeding – Utilization rate for DOAC’s in SPAF is only ~50%, mainly due to bleeding risk Mimics FXI deficiency REGN7508 Mimics FXII deficiency REGN9933 28 Regeneron’s Factor XI antibodies: Potential for maximal anti - coagulation with minimal bleeding Positive proof - of - concept data for REGN7508 (catalytic) and REGN9933 (A2) announced in December 2024 *Results from ROXI - VTE I (REGN9933, apixaban) and ROXI - VTE II (REGN7508); enoxaparin VTE rate pooled across both studies 1 Fuji T, Fujita S, Tachibana S, Kawai Y. A dose - ranging study evaluating the oral factor Xa inhibitor edoxaban for the prevention of venous thromboembolism in patients undergoing total knee arthroplasty. J Thromb Haemost. 2010 Nov;8(11):2458 - 68. doi: 10.1111/j.1538 - 7836.2010.04021.x. PMID: 20723033. This slide contains investigational drug candidates that have not been approved by any regulatory authority. PoC data support advancing both antibodies into a broad Phase 3 development program in multiple coagulation disorders and in patients with different risk factors for bleeding Phase 3 trials expected to initiate in 2025 Initiation of dosing (hrs) VTE Rate * Target Therapy 12 - 24 postop 7% FXI (catalytic) REGN7508 12 - 24 postop 17% FXI (A2) REGN9933 12 - 24 postop 21% Multiple Enoxaparin 12 - 24 postop 12% FXa Apixaban N/A 48% 1 N/A Historical Control (pbo) )TZGQNSL 9NRJ1NSJ aPTT ratio REGN antibodies expected to have different profiles: REGN7508 : increased anticoagulation activity vs. competing agents REGN9933 : comparable anticoagulation activity to competing agents with lower bleed risk Log[M] of FXI inhibitor (clinically relevant concentrations for mAbs and small molecules indicated above) FXI Catalytic domain antibodies REGN7508 mAb competitor #1 mAb competitor #2 FXI A2 domain antibody REGN9933 FXI small molecules SM Competitor #1 SM Competitor #2 Preclinical aPTT Screening Results of REGN Antibodies and Competitor FXI Agents 2.0 - fold aPTT Small molecules require much higher concentration to reach similar anticoagulation activity, increasing risk of off - target effects 29 Regeneron’s approach to obesity: novel combinations with leading medicines aim to improve quality of weight loss GLP - 1 based therapies, such as semaglutide (sema) and tirzepatide, are emerging as standards of care for weight loss; however, up to 40% of weight loss from these agents is due to decreases in muscle mass 1 1 Wilding, Diabetes Obes Metab, 2022; PMID: 35441470, 2 Mastaitis J, et al.
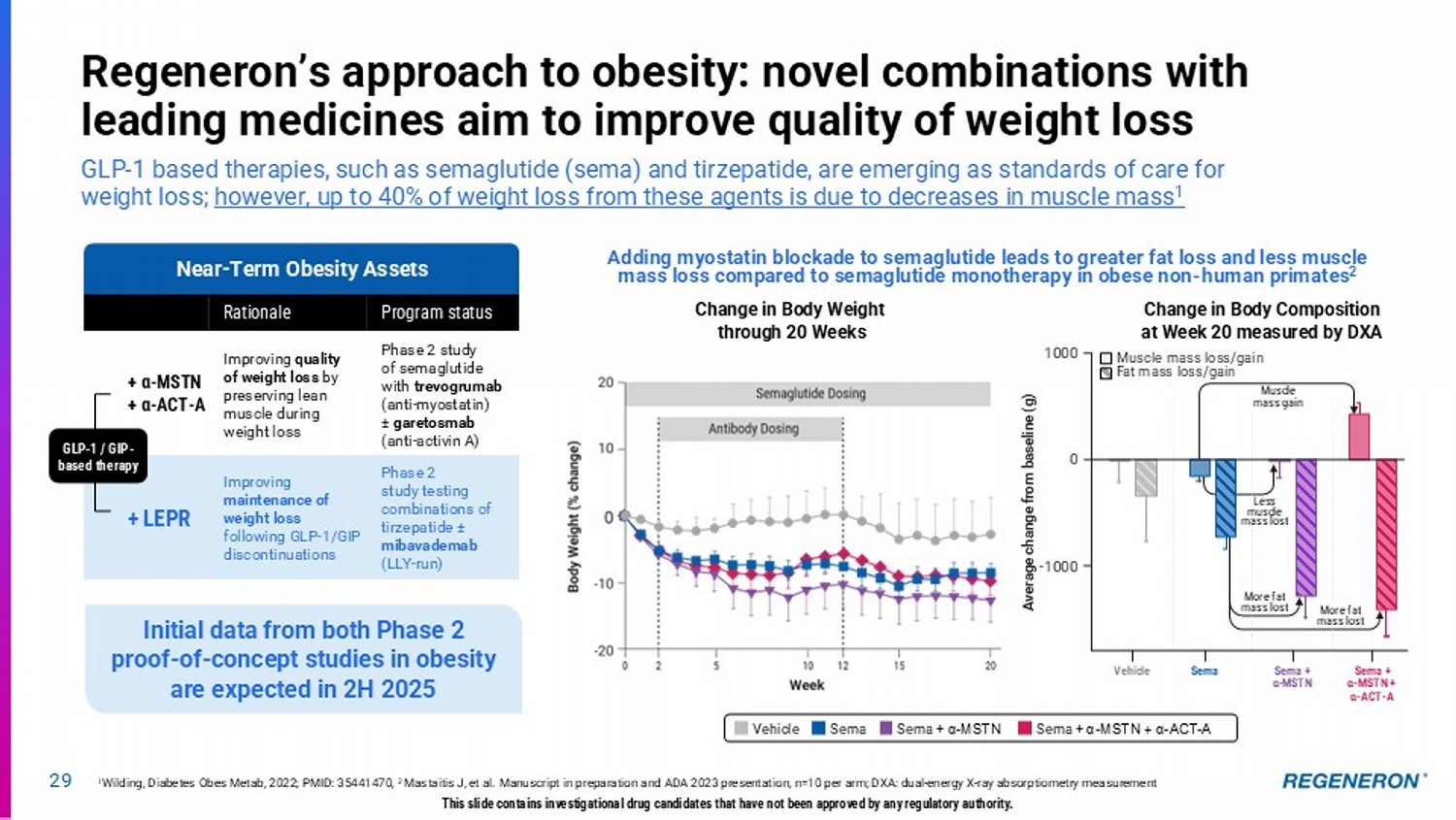
Manuscript in preparation and ADA 2023 presentation, n=10 per arm; DXA: dual - energy X - ray absorptiometry mea surement Near - Term Obesity Assets Program status Rationale Phase 2 study of semaglutide with trevogrumab (anti - myostatin) ± garetosmab (anti - activin A) Improving quality of weight loss by preserving lean muscle during weight loss + α - MSTN + α - ACT - A Phase 2 study testing combinations of tirzepatide ± mibavademab (LLY - run) Improving maintenance of weight loss following GLP - 1/GIP discontinuations + LEPR Adding myostatin blockade to semaglutide leads to greater fat loss and less muscle mass loss compared to semaglutide monotherapy in obese non - human primates 2 This slide contains investigational drug candidates that have not been approved by any regulatory authority.
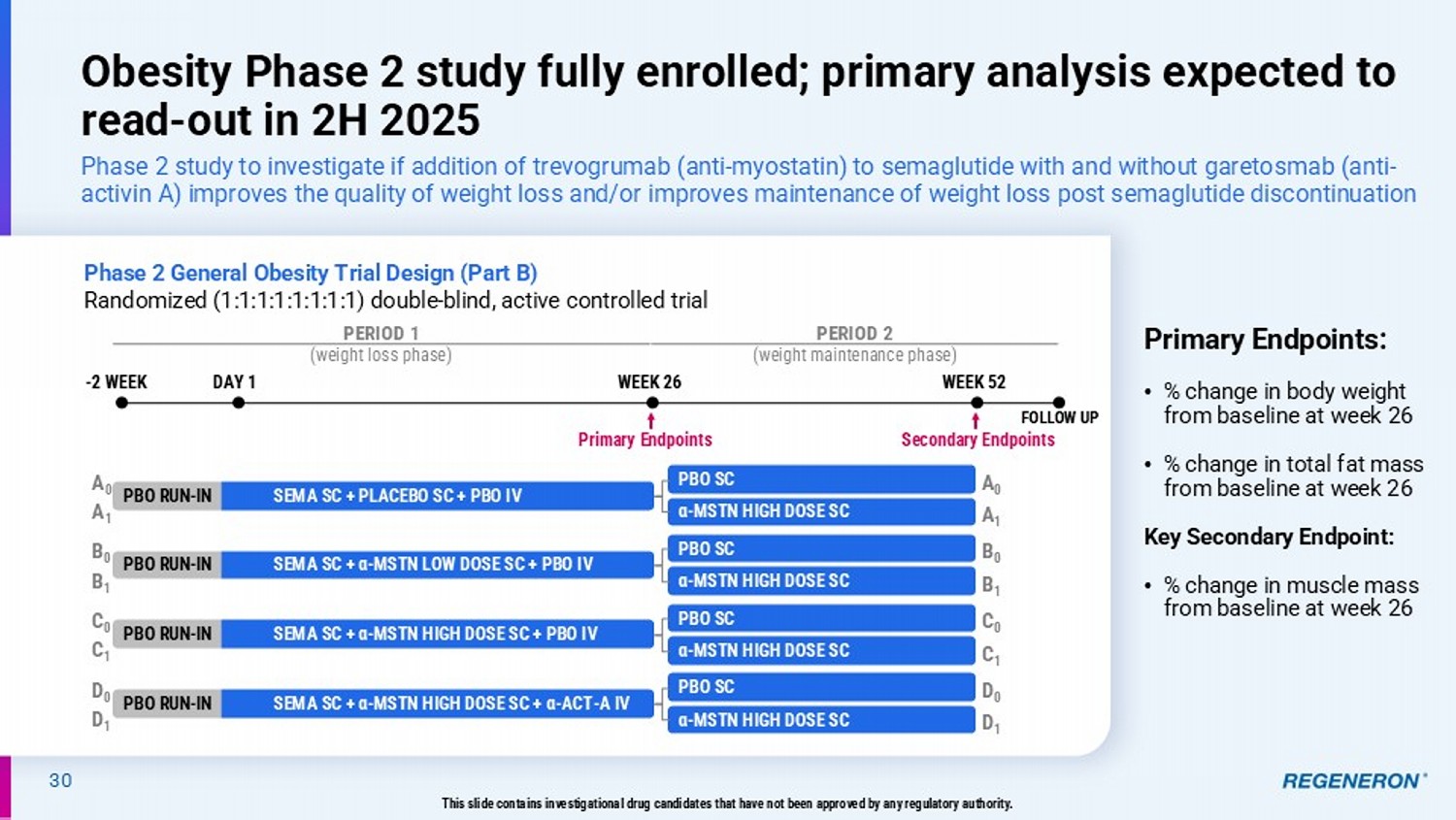
GLP - 1 / GIP - based therapy Change in Body Weight through 20 Weeks Average change from baseline (g) Vehicle Sema Sema + α - MSTN Sema + α - MSTN + α - ACT - A Change in Body Composition at Week 20 measured by DXA Sema + α - MSTN + α - ACT - A Sema + α - MSTN Sema Vehicle Muscle mass loss/gain F at mass loss/gain 1000 - 1000 0 More fat mass lost Less muscle mass lost Initial data from both Phase 2 proof - of - concept studies in obesity are expected in 2H 2025 Muscle mass gain More fat mass lost 30 Obesity Phase 2 study fully enrolled; primary analysis expected to read - out in 2H 2025 Phase 2 study to investigate if addition of trevogrumab (anti - myostatin) to semaglutide with and without garetosmab (anti - activin A) improves the quality of weight loss and/or improves maintenance of weight loss post semaglutide discontinuation Phase 2 General Obesity Trial Design (Part B) Randomized (1:1:1:1:1:1:1:1) double - blind, active controlled trial PERIOD 1 (weight loss phase) PERIOD 2 (weight maintenance phase) FOLLOW UP - 2 WEEK DAY 1 WEEK 26 WEEK 52 Primary Endpoints Secondary Endpoints A 0 A 1 B 0 B 1 C 0 C 1 D 0 D 1 PBO RUN - IN SEMA SC + PLACEBO SC + PBO IV PBO SC α - MSTN HIGH DOSE SC PBO RUN - IN SEMA SC + α - MSTN LOW DOSE SC + PBO IV PBO SC α - MSTN HIGH DOSE SC PBO RUN - IN SEMA SC + α - MSTN HIGH DOSE SC + PBO IV PBO SC α - MSTN HIGH DOSE SC PBO RUN - IN SEMA SC + α - MSTN HIGH DOSE SC + α - ACT - A IV PBO SC α - MSTN HIGH DOSE SC A 0 A 1 B 0 B 1 C 0 C 1 D 0 D 1 This slide contains investigational drug candidates that have not been approved by any regulatory authority.

Primary Endpoints: • % change in body weight from baseline at week 26 • % change in total fat mass from baseline at week 26 Key Secondary Endpoint: • % change in muscle mass from baseline at week 26 Leveraging decades of expertise to develop a robust pre - clinical obesity and cardiometabolic pipeline 31 Our next wave of therapeutics focuses on GLP1 - independent mechanisms and targeting muscle growth and improved metabolism Goal: To bring next - generation muscle and/or neuro - targeted therapies (androgens, siRNAs, gene therapies) to patients as the next cornerstone of healthy weight management therapy Opportunity to combine novel, first - in - class muscle and/or neuro - targeting agents with appropriate weight loss interventions to provide benefit to distinct patient populations Goal: To provide the best suite of antibody + GLP1 combination therapies – either as co - formulations or ‘unimolecular’ solutions – to improve quality of weight loss and long - term health outcomes Our first wave of therapeutics focuses on improving GLP1 - based weight loss by preserving muscle 32 Novel treatment approach for potentially reversing severe allergy: Linvoseltamab (BCMAxCD3) plus Dupixent (anti - IL4R α ) This slide contains investigational drug candidates that have not been approved by any regulatory authority.
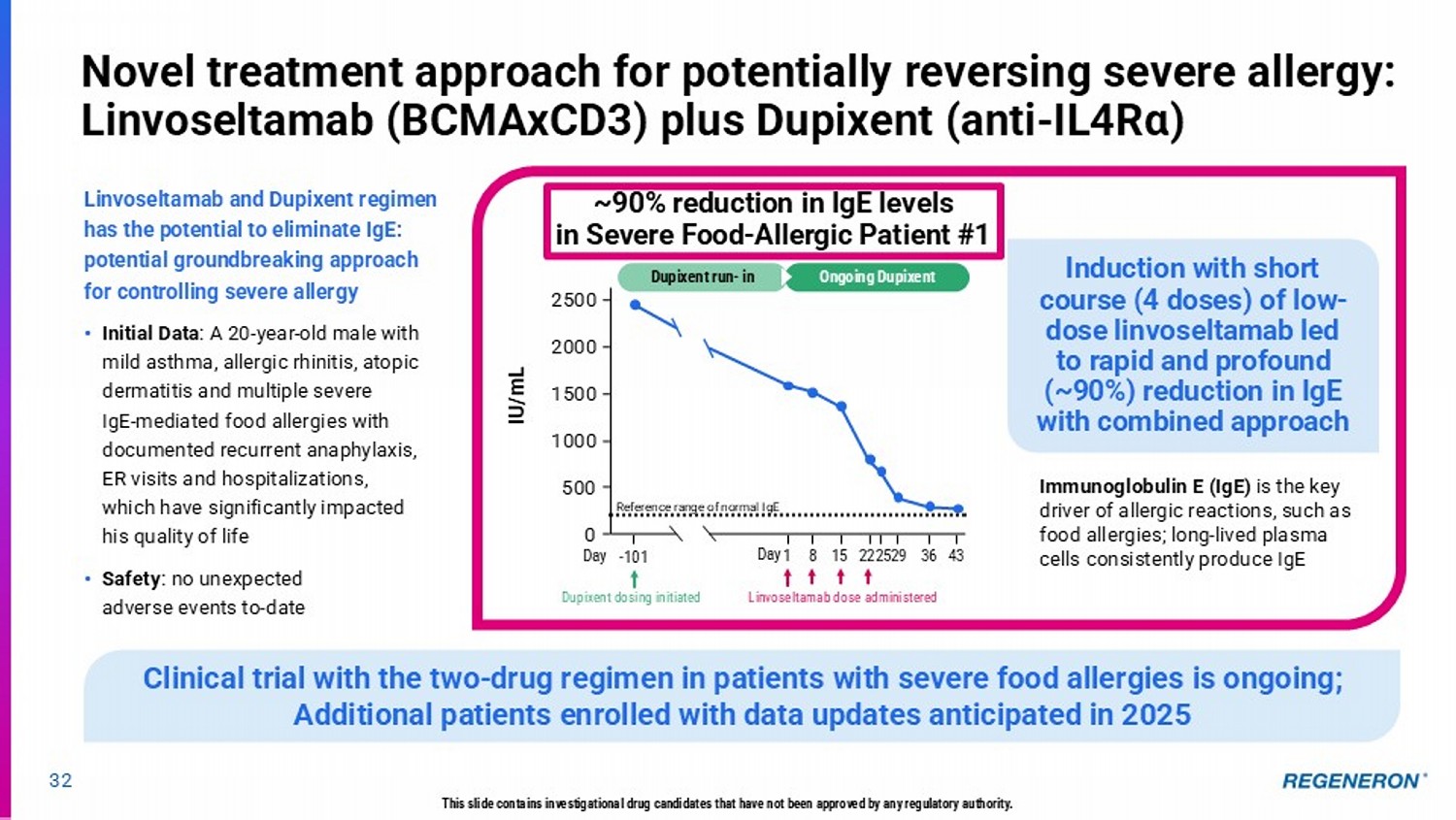
Linvoseltamab and Dupixent regimen has the potential to eliminate IgE: potential groundbreaking approach for controlling severe allergy • Initial Data : A 20 - year - old male with mild asthma, allergic rhinitis, atopic dermatitis and multiple severe IgE - mediated food allergies with documented recurrent anaphylaxis, ER visits and hospitalizations, which have significantly impacted his quality of life • Safety : no unexpected adverse events to - date Clinical trial with the two - drug regimen in patients with severe food allergies is ongoing; Additional patients enrolled with data updates anticipated in 2025 Induction with short course (4 doses) of low - dose linvoseltamab led to rapid and profound (~90%) reduction in IgE with combined approach Immunoglobulin E (IgE) is the key driver of allergic reactions, such as food allergies; long - lived plasma cells consistently produce IgE ~90% reduction in IgE levels in Severe Food - Allergic Patient #1 Dupixent run - in Ongoing Dupixent 2500 IU/mL - 101 2000 1500 1000 500 0 1 8 15 22 25 29 36 43 Linvoseltamab dose administered Day Day Reference range of normal IgE Dupixent dosing initiated Genetic Medicines Viral vector (e.g.
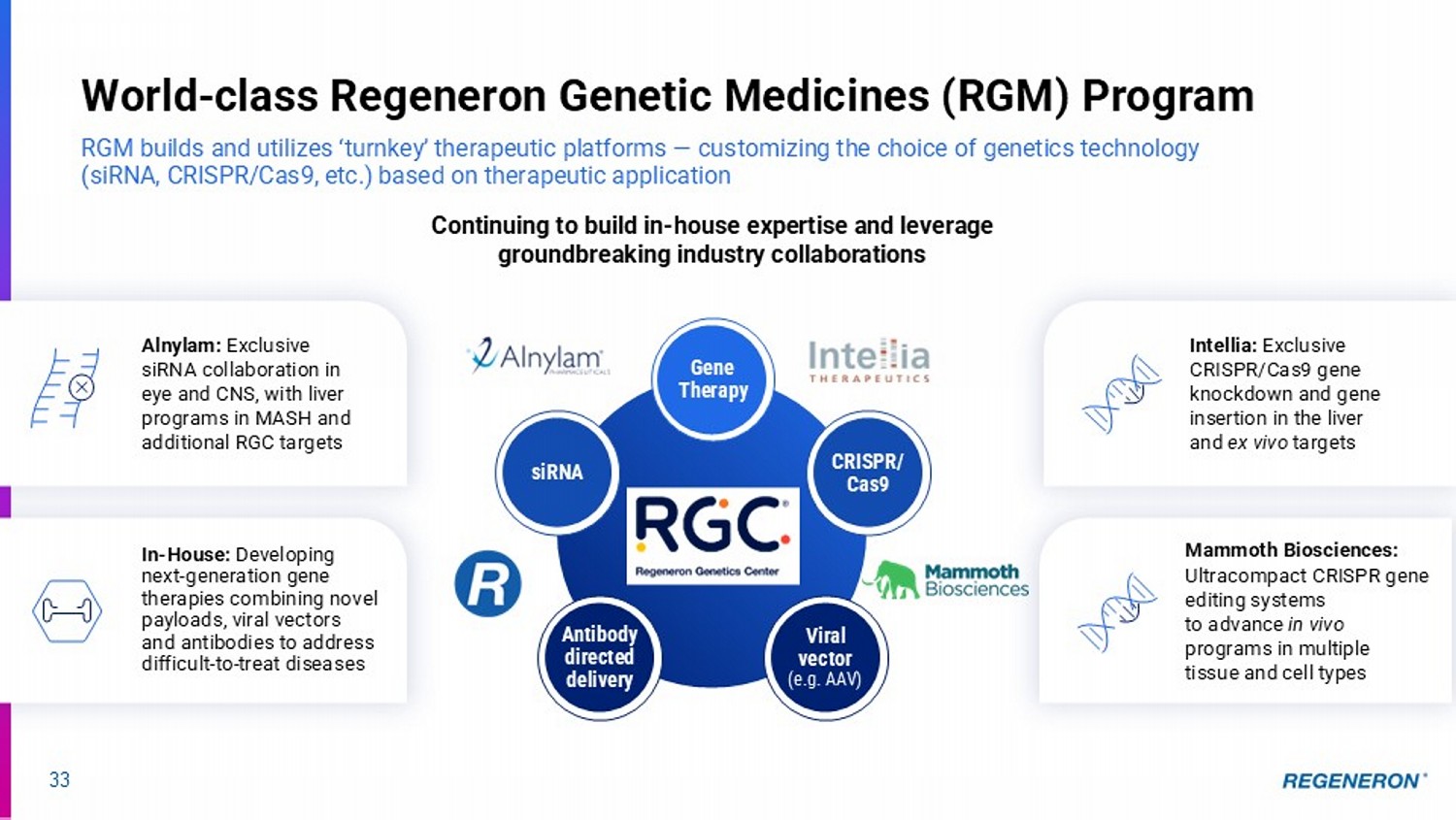
AAV) A ntibody directed delivery siRNA CRISPR/ Cas9 Gene Therapy 33 World - class Regeneron Genetic Medicines (RGM) Program RGM builds and utilizes ‘turnkey’ therapeutic platforms — customizing the choice of genetics technology (siRNA, CRISPR/Cas9, etc.) based on therapeutic application Continuing to build in - house expertise and leverage groundbreaking industry collaborations In - House: Developing next - generation gene therapies combining novel payloads, viral vectors and antibodies to address difficult - to - treat diseases Alnylam: Exclusive siRNA collaboration in eye and CNS, with liver programs in MASH and additional RGC targets Intellia: Exclusive CRISPR/Cas9 gene knockdown and gene insertion in the liver and ex vivo targets Mammoth Biosciences: Ultracompact CRISPR gene editing systems to advance in vivo programs in multiple tissue and cell types 34 DB - OTO demonstrates the potential to provide hearing to deaf children (from infancy to adolescence) DB - OTO is an AAV - based dual - vector gene therapy delivered to the inner ear to enable hearing in children Behavioral pure tone audiogram – a plot of softest sounds a patient can hear in an individual ear *Arrows indicate no response at maximum level tested This slide contains investigational drug candidates that have not been approved by any regulatory authority.
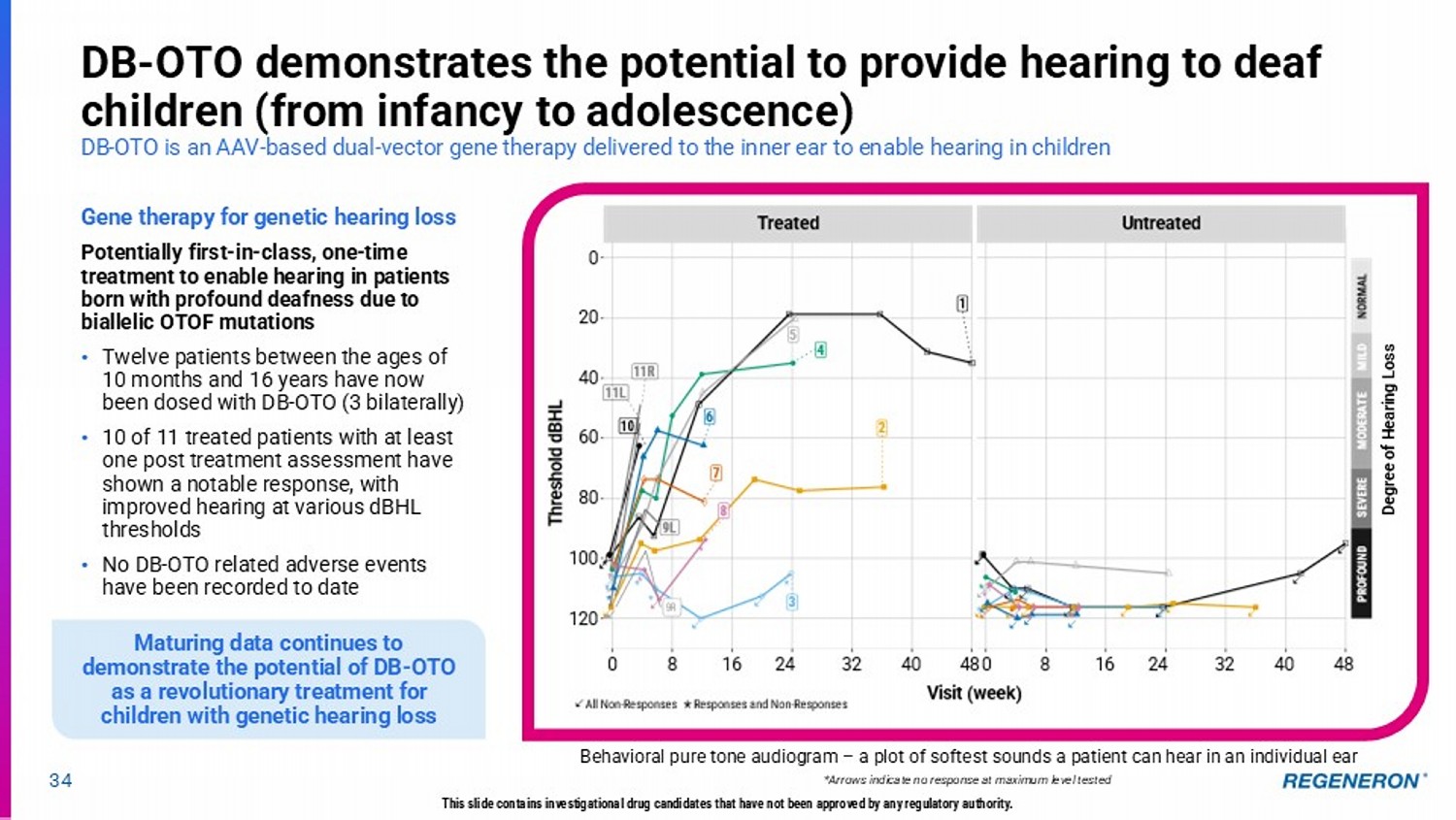

Gene therapy for genetic hearing loss Potentially first - in - class, one - time treatment to enable hearing in patients born with profound deafness due to biallelic OTOF mutations • Twelve patients between the ages of 10 months and 16 years have now been dosed with DB - OTO (3 bilaterally) • 10 of 11 treated patients with at least one post treatment assessment have shown a notable response, with improved hearing at various dBHL thresholds • No DB - OTO related adverse events have been recorded to date 9MWJXMTQII'-1 ;NXNY \JJP &QQ3TS 7JXUTSXJX 7JXUTSXJXFSI3TS 7JXUTSXJX 7 1 1 574+4:3) 8*;*7* 24)*7 & 9* 2.1) 3472&1 9 WJFYJI :SYWJFYJI Maturing data continues to demonstrate the potential of DB - OTO as a revolutionary treatment for children with genetic hearing loss Degree of Hearing Loss Program Status 35 2025 U.S. Prevalence (patients): ~1.1M Worldwide market sales * (2025e): ~$1.0B Estimated market sales CAGR* (2025 - 2030): ~34% Geographic Atrophy 2025 U.S. Prevalence (patients): ~6k Worldwide market sales * (2025e): ~$2.0B Estimated market sales CAGR* (2025 - 2030): ~12% Paroxysmal Nocturnal Hemoglobinuria 2025 U.S. Prevalence (patients): ~90k Worldwide market sales * (2025e): ~$5.0B Estimated market sales CAGR* (2025 - 2030): ~17% Myasthenia Gravis This slide contains investigational drug candidates that have not been approved by any regulatory authority. • Phase 3 pivotal program initiated in 2H 2024 • Study fully enrolled • Phase 3 results expected in 2H 2025 • Cohort A (exploratory): Updated Phase 3 data recently reported • Cohort B (registrational): Study enrolling, data expected in 2026+ * Evaluate Pharma Despite competitive markets, there is opportunity to improve upon the current standard of care with prolonged and complete inhibition of complement protein C5 (for multiple diseases) siRNA (cemdisiran) lowers C5 target burden, allowing antibody (pozelimab) to more effectively block C5 function Our differentiated siRNA + antibody approach has the potential to address multiple complement - mediated diseases Primary Endpoint: % change in lactate dehydrogenase (LDH) from baseline to week 26 in PNH patients 36 Pozelimab + C emdisiran (Poze - Cemdi) enables complete, rapid, uninterrupted and durable inhibition of terminal complement Results from an exploratory cohort in the pivotal PNH trial; safety profile of poze - cemdi was generally consistent with approved C5 inhibitors LDH is a well - accepted biomarker of hemolysis – with adequate control and normalization defined as ≤1.5 and ≤1.0 times ULN, resp ectively This slide contains investigational drug candidates that have not been approved by any regulatory authority.
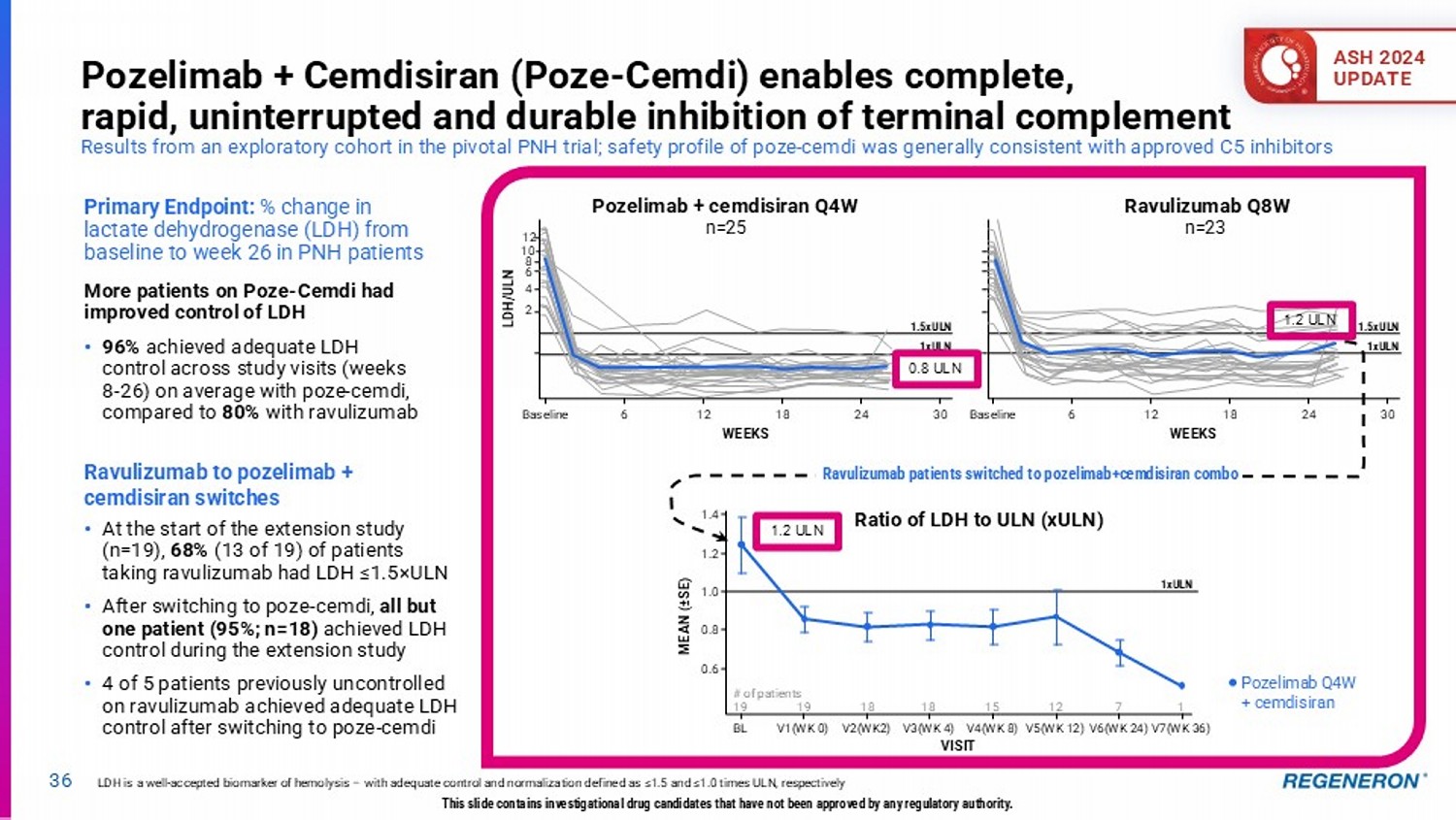
More patients on Poze - Cemdi had improved control of LDH • 96% achieved adequate LDH control across study visits (weeks 8 - 26) on average with poze - cemdi, compared to 80% with ravulizumab ASH 2024 UPDATE • At the start of the extension study (n=19), 68% (13 of 19) of patients taking ravulizumab had LDH ≤1.5 × ULN • After switching to poze - cemdi, all but one patient (95%; n=18) achieved LDH control during the extension study • 4 of 5 patients previously uncontrolled on ravulizumab achieved adequate LDH control after switching to poze - cemdi Ravulizumab to pozelimab + cemdisiran switches Ravulizumab Q8W n=23 Pozelimab + cemdisiran Q4W n=25 Baseline 6 12 18 24 30 12 Baseline LDH/ULN 10 8 6 4 6 12 18 24 30 2 WEEKS 1xULN 1.5xULN 1xULN 1.5xULN WEEKS 1.4 1.2 1.0 0.8 0.6 MEAN ( ± SE) BL V1(WK 0) V2(WK2) V3(WK 4) V4(WK 8) V5(WK 12) V6(WK 24) V7(WK 36) 19 19 18 18 15 12 7 1 # of patients 1xULN Ratio of LDH to ULN (xULN) VISIT Pozelimab Q4W + cemdisiran Ravulizumab patients switched to pozelimab+cemdisiran combo 0.8 ULN 1.2 ULN 1.2 ULN 37 Regeneron genetic medicines pipeline Phase 3 Phase 2 Phase 1 Select Pre - IND Candidates CIDEB * CIDEB siRNA MASH ALN - HTT02 * HTT siRNA Huntington’s Disease Factor 9 † F9 CRISPR + AAV Hemophilia B GAA † GAA CRISPR + AAV Pompe Disease GJB2/DB - 103 GJB2 AAV GJB2 - related Hearing Loss DB - OTO | OTOF AAV Dual Vector Gene Therapy OTOF - related Hearing Loss ALN - PNP * PNPLA3 siRNA NAFLD Rapirosiran * HSD17B13 siRNA MASH Pozelimab + Cemdisiran * C5 Antibody + C5 siRNA Myasthenia Gravis; Paroxysmal Nocturnal Hemoglobinuria; Geographic Atrophy Nexiguran ziclumeran (Nex - z, NTLA - 2001) † CRISPR/Cas9 Transthyretin Amyloidosis with cardiomyopathy (ATTR - CM); H ereditary transthyretin amyloidosis with polyneuropathy (ATTRv - PN) This slide contains investigational drug candidates that have not been approved by any regulatory authority.
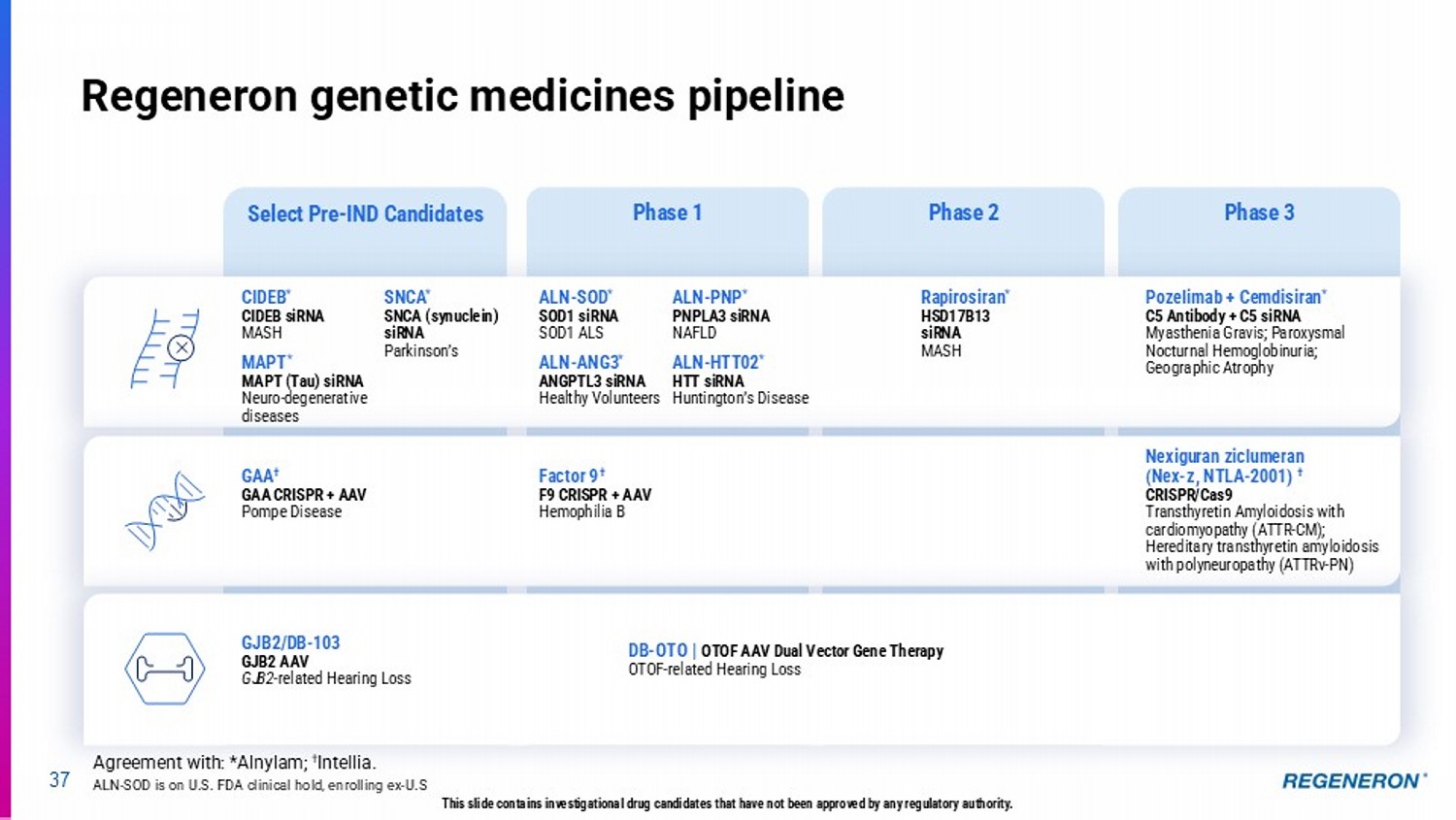
ALN - SOD * SOD1 siRNA SOD1 ALS Agreement with: *Alnylam; † Intellia. ALN - SOD is on U.S.

FDA clinical hold, enrolling ex - U.S ALN - ANG3 * ANGPTL3 siRNA Healthy Volunteers MAPT * MAPT (Tau) siRNA Neuro - degenerative diseases SNCA * SNCA (synuclein) siRNA Parkinson’s 38 2025 key upcoming milestones EYLEA HD • RVO sBLA acceptance (1H) and FDA decision (2H) • Pre - filled s yringe FDA decision and launch ( mid ) • Addition of 2 - year data in wAMD and DME to FDA label (PDUFA April 20) • Addition of Q4W dosing to FDA label for all indications (2H) Dupixent / I&I • Report pivotal data for itepekimab in COPD (2H); submit BLA (2H) • Dupixent - CSU FDA decision ( PDUFA April 18 ) • Dupixent - BP sBLA acceptance (1H) and FDA decision (2H); EU submission (1H) • Initiate additional Phase 3 studies for itepekimab (1H) • Report additional data for Dupixent + BCMA in severe food allergies Internal Medicine • Report proof - of - concept data of combination of semaglutide and trevogrumab with and without garetosmab in obesity (2H) • Report proof - of - concept data for mibavademab with tirzepatide in obesity (2H) • Report Phase 3 data for garetosmab in FOP (2H) Solid Organ Oncology • Submit sBLA for Libtayo in adjuvant CSCC (1H) • Report results from Phase 3 study of fianlimab + cemiplimab vs. pembrolizumab monotherapy in 1L metastatic melanoma (2H); submit BLA pending results (2H) • Report initial Phase 2 data for fian limab + cemiplimab in 1L advanced NSCLC (1H) • Report additional data for ubamatamab (MUC16xCD3) in ovarian cancer • Report additional data across solid tumor costimulatory bispecific programs: • Nezastomig (PSMAxCD28) + cemiplimab in mCRPC • EGFRxCD28 + cemiplimab -- dose expansion cohorts • MUC16xCD28 + ubamatamab in ovarian cancer Hematology • Resubmit BLA for odronextamab in R/R follicular lymphoma (Q1); FDA decision (2H) • Resubmit BLA for linvoseltamab in R/R multiple myeloma ; FDA decision (mid) • Initiate Phase 3 program for Factor XI antibodies across multiple indications Genetic Medicines • Report additional data for DB - OTO (mid) • Report pivotal Phase 3 data for pozelimab +cemdisiran in gMG (2H) This slide contains investigational drug candidates that have not been approved by any regulatory authority. x Q&A 39 Leonard S. Schleifer, MD, PhD Co - Founder, Board Co - Chair, President & Chief Executive Officer George D. Yancopoulos, MD, PhD Co - Founder, Board Co - Chair, President & Chief Scientific Officer



40 Three responsibility focus areas reflect our “doing well by doing good” ethos OUR MISSION Use the power of science to repeatedly bring new medicines to people with serious diseases 2 Foster a culture of integrity and excellence 1 Improve the lives of people with serious diseases 3 Build sustainable communities • Pipeline innovation • Access to medicine and fair pricing • Patient advocacy • Product quality and safety • Diverse, healthy and engaged workforce • Ethics and integrity • Responsible supply chain • STEM education – sponsorship of top science competitions: – Regeneron Science Talent Search – Regeneron International Science and Engineering Fair • Environmental sustainability All trademarks included are the property of their respective owners 41 Abbreviations and Definitions Definition Abbreviation First line 1L Adeno - associated virus AAV Amyotrophic lateral sclerosis ALS Activated Partial Thromboplastin Time aPTT Basal cell carcinoma BCC B - cell maturation antigen BCMA Biologics license application BLA Bullous pemphigoid BP Chimeric antigen receptor T - cell CAR - T Confidence Interval CI Central nervous system CNS Chronic obstructive pulmonary disease COPD Complete response CR Colorectal Cancer CRC Cytokine release syndrome CRS Chronic sinusitis with nasal polyposis CRSwNP Cutaneous squamous cell carcinoma CSCC Chronic spontaneous urticaria CSU Decibel hearing loss dB HL Disease - Free Survival DFS Diffuse large B - cell lymphoma DLBCL Dose - limiting toxicity DLT Diabetic macular edema DME Direct oral anticoagulants DOAC Definition Abbreviation Diabetic retinopathy DR Dual - energy X - ray absorptiometry DXA Eastern Cooperative Oncology Group ECOG Epidermal growth factor receptor EGFR First in human FIH Follicular lymphoma FL Follicular Lymphoma International Prognostic Index FLIPI Geographic atrophy GA Alpha glucosidase GAA Groupe d'Etude des Lymphomes Folliculaires GELF Gastrointestinal GI Gastric inhibitory polypeptide GIP Glucagon - like peptide 1 GLP - 1 Generalized myasthenia gravis gMG Gain of function GOF Hepatocellular carcinoma HCC Healthcare Provider HCP Head and neck squamous cell carcinoma HNSCC Human Papillomavirus HPV Hazard Ratio HR Huntingtin HTT Immune effector cell - associated neurotoxicity syndrome ICANS Immunoglobulin - E IgE Initial new drug application IND Definition Abbreviation Kaplan - Meier curve KM Lymphocyte - activation gene 3 LAG - 3 Lactate dehydrogenase LDH Leptin receptor LEPR Low molecular weight heparin LMWH Loss of function LOF Microtubule - associated protein tau MAPT Metabolic Dysfunction - Associated Steatohepatitis MASH Metastatic castration - resistant prostate cancer mCRPC Monoclonal gammopathy of unknown significance MGUS Multiple myeloma MM Median overall survival mOS Median progression - free survival mPFS Mucin 16 MUC16 Non - alcoholic fatty liver disease NAFLD Not Estimable NE Non - human primate NHP Not Reached NR (Non - )small cell lung cancer (N)SCLC Overall Response Rate ORR Placebo PBO Progressive disease PD Programmed cell death protein/(ligand) 1 PD - 1/PD - (L)1 Prescription Drug User Fee Act PDUFA l 7JLJSJWTS5MFWRFHJZYNHFQX.SH&QQ7NLMYX7JXJW[JI Definition Abbreviation Pharmacokinetic PK Paroxysmal nocturnal hemoglobinuria PNH Proof - of - concept POC Partial response PR Prostate - specific membrane antigen PSMA Relapsed/Refractory R/R Renal cell carcinoma RCC Regeneron Genetics Center RGC Radiotherapy RT Retinal vein occlusion RVO Supplemental biologics license application sBLA Subcutaneous SC Stringent complete response sCR Stable disease SD Small interfering RNA siRNA Standard of care SOC Stroke Prevention in Atrial Fibrillation SPAF Type 2 diabetes mellitus T2DM Treatment - emergent adverse events TEAE Treatment - related adverse events TRAE Upper limit of normal ULN Vascular endothelial growth factor VEGF Venous thromboembolism VTE Wet age - related macular degeneration wAMD