UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, DC 20549
FORM 10-K
(Mark One)
☒ ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the fiscal year ended December 31, 2024
or
☐ TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the transition period from ___________ to ___________
Commission file number: 001-41575
Lipella Pharmaceuticals Inc.
(Exact name of registrant as specified in its charter)
| Delaware | 20-2388040 | |
| (State or other jurisdiction of | (I.R.S. Employer | |
| incorporation or organization) | Identification No.) | |
7800 Susquehanna St., Suite 505
Pittsburgh, PA 15208
(Address of principal executive offices) (Zip Code)
Registrant’s telephone number, including area code: (412) 894-1853
Securities registered pursuant to Section 12(b) of the Act:
| Title of each class: | Trading Symbol |
Name of each exchange on which registered: |
| Common Stock, par value $0.0001 per share | LIPO | The Nasdaq Stock Market LLC |
Securities registered pursuant to Section 12(g) of the Act:
None
(Title of class)
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☐ No ☒
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ☐ No ☒
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ☒ No ☐
Indicate by check mark whether the registrant has submitted electronically, every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files). Yes ☒ No ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer | ☐ | Accelerated filer | ☐ |
| Non-accelerated filer | ☒ | Smaller reporting company | ☒ |
| Emerging growth company | ☒ |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report. ☐
If securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant included in the filing reflect the correction of an error to previously issued financial statements. ☐
Indicate by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive-based compensation received by any of the registrant’s executive officers during the relevant recovery period pursuant to §240.10D-1(b). ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes ☐ No ☒
The aggregate market value of common stock held by non-affiliates of the registrant, based on the closing price for the registrant’s common stock on June 30, 2024 (the last business day of the second quarter of the registrant’s current fiscal year), was $2,623,125.60.
The registrant had 2,548,811 shares of its common stock outstanding as of March 26, 2025.
References in this Annual Report on Form 10-K to the “Company,” “Lipella,” “we,” “us,” or “our” mean Lipella Pharmaceuticals Inc. unless otherwise expressly stated or the context indicates otherwise.
Documents Incorporated By Reference: None.
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
This Annual Report on Form 10-K (this “Report”) contains “forward-looking statements” within the meaning of the Private Securities Litigation Reform Act of 1995, Section 27A of the Securities Act of 1933, as amended (the “Securities Act”), and Section 21E of the Securities Exchange Act of 1934, as amended (the “Exchange Act”). Forward-looking statements include information concerning our strategy, future operations, future financial position, future revenue, projected expenses, prospects and plans and objectives of management. Forward-looking statements include all statements that are not historical facts and can be identified by terms such as “anticipate,” “believe,” “continue,” “could,” “estimate,” “expect,” “intend,” “may,” “plan,” “potential,” “predict,” “project,” “seek,” “should,” “target,” “will,” “would” or similar expressions and the negatives of those terms.
Forward-looking statements contained in this Report include, but are not limited to, statements about the following:
| ● | the initiation, timing, progress preclinical and clinical trials for other product candidates, including statements regarding the timing of initiation and completion of studies or trials and related preparatory work, the period during which the results of the trials will become available and our research and development programs; |
| ● | the timing, scope or results of regulatory filings and approvals for our product candidates, including timing of final U.S. Food and Drug Administration (“FDA”) marketing and other regulatory approvals of our lead product candidates, LP-10 and LP-310; and our other product candidates, including, but not limited to, LP-410 and LP-50; |
| ● | our ability to achieve certain accelerated or “orphan drug” designations from the FDA; |
| ● | our estimates regarding the potential market opportunity for LP-10, LP310 or any of our other product candidates; |
| ● | our research and development programs for our product candidates; |
| ● | our plans and ability to successfully develop and commercialize LP-10, LP-310, or any of our other product candidates; |
| ● | our ability to identify and develop new product candidates; |
| ● | our ability to identify, recruit and retain key personnel; |
| ● | our commercialization, marketing and manufacturing capabilities and strategy; |
| ● | the implementation of our business model, strategic plans for our business, product candidates and technology; |
| ● | the scalability and commercial viability of our proprietary manufacturing methods and processes; |
| ● | the rate and degree of market acceptance and clinical utility of our product candidates and gene therapy, in general; |
| ● | our competitive position; |
| ● | our intellectual property position and our ability to protect and enforce our intellectual property; |
| ● | our financial performance; |
| ● | developments and projections relating to our competitors and our industry; |
| ● | our ability to establish and maintain collaborations or obtain additional funding; |
| ● | our estimates regarding expenses, future revenue, capital requirements and needs for or ability to obtain additional financing; |
| ● | the impact of laws and regulations; |
| ● | our expectations regarding the time during which an emerging growth company under the JOBS Act; and |
| ● | the impact of global economic and political developments on our business, including capital market disruptions, the Ukraine-Russia and Israel-Hamas wars, economic sanctions and economic slowdowns or recessions, including any that may result from such developments and public health concerns, which could harm our commercialization efforts as well as the value of our common stock and our ability to access capital markets. |
Forward-looking statements are subject to a number of risks, uncertainties and assumptions, including those described in “Risk Factors” and elsewhere in this Report. Moreover, we operate in a very competitive and rapidly changing environment, and new risks emerge from time to time. It is not possible for our management to predict all risks, nor can we assess the impact of all factors on our business or the extent to which any factor, or combination of factors, may cause actual results to differ materially from those contained in any forward-looking statements we may make. In light of these risks, uncertainties and assumptions, the forward-looking events and circumstances discussed in this Report may not occur and actual results could differ materially and adversely from those anticipated or implied in the forward-looking statements. Given these uncertainties, you should not place undue reliance on these forward-looking statements. Also, forward-looking statements represent our management’s beliefs and assumptions only as of the date of this Report. You should read this Report and the documents that we have filed as exhibits hereto completely and with the understanding that our actual future results may be materially different from what we expect.
Except as required by law, we assume no obligation to update these forward-looking statements publicly, or to update the reasons actual results could differ materially from those anticipated in these forward-looking statements, even if new information becomes available in the future.
For discussion of factors that we believe could cause our actual results to differ materially from expected and historical results, see “Item 1A - Risk Factors” below. These and other factors could cause results to differ materially from those expressed in the estimates made by the independent parties and by us.
TABLE OF CONTENTS
PART I
Item 1. Business
Overview
We are a clinical-stage biotechnology company that was incorporated under the laws of the State of Delaware in February 2005. We are focused on developing new drugs by reformulating the active agents in existing generic drugs and optimizing these reformulations for new applications.
We believe that our strategy combines many of the cost efficiencies and risk abatements derived from using existing generic drugs with potential patent protections for our proprietary formulations; this strategy allows us to expedite, protect, and monetize our product candidates. Additionally, we maintain a therapeutic focus on diseases with significant, unaddressed morbidity and mortality where no approved drug therapy currently exists. We believe that this focus can potentially help reduce the cost, time and risk associated with obtaining marketing approval. We have not yet commercialized any products, and we do not expect to generate revenue from sales of any product candidates for several years.
Our Product Candidates: LP-10, LP-310, LP-410, and LP-50
Consistent with our strategy, we are currently addressing two indications via development of our product candidates, which we have designated as LP-10 for the indication of hemorrhagic cystitis (“HC”) and LP-310 for the indication of oral lichen planus (“OLP”), which is chronic, uncontrolled urinary blood loss that results from certain chemotherapies (such as alkylating agents) or pelvic radiation therapy (also called “radiation cystitis”). Many radiation cystitis patients experience severe morbidity (and in some cases, mortality), and currently, there is no therapy for their condition approved by the FDA, or, to our knowledge, any other regulatory body. LP-310 employs a formulation similar to LP-10, for the treatment of OLP. OLP is a chronic, T-cell-mediated, autoimmune oral mucosal disease, and LP-310 contains tacrolimus which inhibits T-lymphocyte activation. Symptoms of OLP include painful burning sensations, bleeding and irritation with tooth brushing, painful, thickened patches on the tongue, and discomfort when speaking, chewing or swallowing. These symptoms frequently cause weight loss, nutritional deficiency, anxiety, depression, and scarring from erosive lesions. OLP can also be a precursor to cancer, predominately squamous cell carcinoma, with a malignant transformation rate of approximately one percent.
LP-10 is the development name of our reformulation of tacrolimus (an approved generic active agent) specifically optimized for topical deposition to the internal surface of the urinary bladder lumen using a proprietary drug delivery platform that we have developed and that we refer to as our metastable liposome drug delivery platform (our “Platform”). We are developing LP-10 and our Platform to be, to our knowledge, the first drug candidate and drug delivery technology that could be successful in treating cancer survivors who acquire HC. Our first issued U.S. patent covering LP-10 expires July 11, 2035, unless extended for regulatory delay (up to 14 years), our second issued U.S. patent covering the method of making LP-10 expires November 9, 2034, unless extended for regulatory delay, and our third U.S. patent covering particular LP-10 tacrolimus formulations expires November 9, 2034, unless extended for regulatory delay. Our issued Australian patent covering LP-10 expires October 22, 2034. The Canadian patent, issued on August 23, 2022, expires October 22, 2034. The European patent, issued on June 7, 2023, expires October 22, 2034. We also have a corresponding patent application pending in the U.S. (U.S.S.N. 18/924,830). We also have a pending U.S. patent application on an improvement to the technology. We have received FDA “orphan drug” designation covering LP-10 and plan to apply for additional regulatory designations in the event we achieve qualifying results in the current phase 2a clinical trial for LP-10. Market data exclusivity may be available in the US and other jurisdictions in which regulatory approval is obtained for the Company’s product, regardless of patent status.
The safety and efficacy of LP-10 was evaluated in a 13-subject, open-label, multi-center, dose-escalation, phase 2a clinical trial in patients experiencing complications associated with a rare but highly morbid disease called “radiation-induced hemorrhagic cystitis” or “radiation cystitis.” This phase 2a clinical trial commenced on February 15, 2021, and we reported the trial’s summary results in the first quarter of 2023. There is currently no FDA approved drug therapy available for radiation cystitis patients, who are all cancer survivors who received pelvic radiation therapy to treat solid pelvic tumors, including prostate and ovarian cancers and who are now dealing with therapy-associated complications, including urinary bleeding (a radiation cystitis symptom). LP-10’s active ingredient, tacrolimus, which has a well-known pharmacology and toxicology, addresses a reduction (or cessation) of uncontrolled urinary bleeding.
LP-310 is the development name of our oral, liposomal formulation of tacrolimus (the same approved generic active agent in LP-10) specifically optimized for local delivery to oral mucosa. We believe that our approach of using metastable liposomal tacrolimus as a treatment for OLP is novel. To date, upon review of relevant FDA public data resources on approved drugs and biologics, we are not aware of any other liposomal products developed to treat such disease. We received investigational new drug (“IND”) approval from the FDA regarding LP-310 in the third quarter of 2023. We initiated a Phase 2a multicenter dose escalation of using LP-310 for the treatment of OLP in 2024. In this first cohort, eight participants received a dose of 0.25 mg LP-310, with promising initial results. No product-related serious adverse events were reported. Pharmacokinetic data demonstrated that whole blood tacrolimus levels in all patients were either undetectable or minimal, highlighting LP-310’s potential to deliver localized therapeutic effects while minimizing systemic exposure. Additionally, all patients tolerated LP-310 without significant adverse reactions. The trial is expected to be completed in the second quarter of 2025. The top line data from the first two dose cohorts of this trial has been selected for podium presentation at the 2025 American Association of Oral Medicine and European Association of Oral Medicine Joint Meeting that will be held in Las Vegas, NV on May 15, 2025.
Our issued U.S. and Australian patents covering LP-310 expire July 11, 2035, November 9, 2034 and October 22, 2034, respectively. The Canadian patent, issued on August 23, 2022, expires October 22, 2034. The European patent, issued on June 7, 2023, expires October 22, 2034. We also have a corresponding patent application pending in the U.S. (U.S.S.N. 18/924,830). We also have a pending U.S. patent application on an improvement to the technology. As noted above, patent term extensions may be available in Europe, Canada and the US for regulatory delay. Market data exclusivity is also applicable in many jurisdictions, regardless of patent status. Approval of a 505(b)(1) or 505(b)(2) application can result in five or three years of such exclusivity, respectively.
In a third program, Lipella is also developing an oral, liposomal formulation of tacrolimus, LP-410, for the treatment of oral graft-versus-host disease (“GVHD”). LP-410 is an oral rinse, similar to LP-310, but will have a different containment system. Hematopoietic cell transplantation (“HCT”) is used to treat a wide range of malignancies, hematologic and immune deficiency states, and autoimmune diseases. GVHD is a clinical syndrome where donor-derived immunocompetent T-cells react against patient tissues directly or through exaggerated inflammatory responses following HCT. Oral GVHD is a rare and serious disease, with a prevalence of approximately 30,000 patients in the US annually in 2023 (Bachier et al., 2019; Bachier et al., 2021, Orphanet 2023). GVHD remains a major cause of morbidity and mortality with patients who undergo HCT treatment, with chronic GVHD being the leading cause of non-malignant fatality for such patients who receive such HCT treatments.
Topical and local management of symptomatic oral GVHD can reduce oral symptoms that can interfere with oral function and quality of life and can reduce the need for more intensive immunosuppressive systemic therapies. However, there is currently no FDA approved local drug treatment of oral GVHD (Martini et al., 2022).
Lipella has developed LP-410 for the topical delivery directly to the mouth surface. LP-410 targets the underlying mechanisms of oral GVHD, potentially providing a safe and effective treatment option for affected individuals. Lipella received orphan designation approval, on November 11, 2023, for tacrolimus for the treatment of oral GVHD. We received IND approval from the FDA for LP-410’s treatment of oral GVHD on March 5, 2024.The issued and pending patents regarding LP-310 are relevant to LP-410 as well. We plan to expand and specify patent coverage of LP-410 in the patent application currently pending in the U.S. (U.S.S.N. 18/924,830).
In a fourth program, Lipella is also developing an intravesical formulation of immunoglobulins including checkpoint inhibitors, referred to as LP-50. LP-50 is an intravesical formulation of immunoglobulins including local, intravesical PD-1 (i.e. checkpoint) inhibition, intended for the treatment of non-muscle invasive bladder cancer (“NMIBC”), offering the potential for increasing efficacy while minimizing systemic toxicity. Additional information regarding this preclinical program is included in the International Journal of Molecular Sciences 2024, 25(9), 4945, titled “Enhancing Therapeutic Efficacy and Safety of Immune Checkpoint Inhibition for Bladder Cancer: A Comparative Analysis of Injectable vs. Intravesical Administration,” as well as in US patent publication number 2024/0115503 titled “Intravesical Delivery of Hydrophilic Therapeutic Agents Using Liposomes.” We have a corresponding patent application pending in the U.S. (U.S.S.N 18/011,635).
Our Metastable Liposome Drug Delivery Platform
We have developed a proprietary technology, referred to as our Platform, which is optimized for local hydrophobic drug delivery to body cavities having endothelial surfaces. Our process employs liposomal technology protected by issued patents in the United States, Australia, and Canada. We also have a corresponding patent application pending in the U.S. (U.S.S.N. 18/924,830) and a corresponding European Patent, issued on June 7, 2023. This technology involves direct drug delivery to the urinary bladder mucosa, and, we believe, has the potential to improve efficacy (by increasing drug concentration at the site of injury) and to reduce the possibility of side effects (by reducing the drug’s exposure to unrelated organs). The first body-cavity application for which we intend to utilize our Platform is the urinary bladder, which has been designed to deliver LP-10. We are also developing an oral cavity product for the treatment of OLP and oral GVHD using our Platform (liposomal-tacrolimus). We are continuing to research and develop products for additional body cavities, including the anal-rectal cavity (radiation proctitis) and the esophagus (eosinophilic esophagitis). We have a pending U.S. patent application on a new embodiment of this technology.
We predict that our Platform will provide a superior approach for treating inflammatory urinary bladder conditions compared to other delivery mechanisms and that certain inherent features of the metastable liposomes, combined with our intravesical formulations, provides our Platform with several advantages over existing bladder drug delivery methodologies in current clinical practice for inflammatory bladder applications. These advantageous characteristics include the following:
| ● | non-inflammatory (without the use of ethanol or other alcohols for solubility); |
| ● | large payload capacity of hydrophobic agents (10% by mass); |
| ● | urothelial affinity, which results in efficient drug transfer; |
| ● | low systemic distribution (large particle size); |
| ● | reproducible manufacturing and scalability; and |
| ● | prior clinical experience utilizing the liposomal delivery vehicle. |
The following table summarizes our therapeutic candidate pipeline and discovery research programs:

Figure 1
Our Strengths
We believe we are uniquely positioned to employ liposome technology in the development of intravesical treatments for urinary bladder and oral indications due, in part, to our particular strengths, including:
| ● | our proprietary Platform, which we believe will allow us to develop a pipeline of products to treat urinary bladder diseases as well as diseases of other body cavities; |
| ● | our clinical development strategy intended to maximize efficiencies by repurposing existing therapeutics for new proprietary indications and formulations; |
| ● | our clinical programs, which are designed to qualify, and take advantage of, accelerated regulatory approval pathways and designations that provide marketing exclusivity; |
| ● | take advantage of product exclusivity through patent protection of our novel formulations and indications for use; |
| ● | our product candidates, LP-10, LP-310, LP-410 and LP-50, which are being developed to address HC, OLP, oral GVHD and NMIBC, in accordance with our capital-efficient strategy via: |
| ○ | the “505(b)(2) regulatory pathway” strategy, which refers to requests for marketing approval from the FDA upon submission of an abbreviated new drug application (“aNDA”) pursuant to Section 505 of the Federal Food, Drug, and Cosmetic Act, as amended (the “FDCA”) and permits us to rely on existing data pertaining to the generic active ingredient; we anticipate referencing relevant publicly available data, including the publicly disclosed FDA drug approval package for tacrolimus in the preparation and submission of our aNDA for LP-10 and LP-310; |
| ○ | a known mechanism of action being combined with a new drug delivery method (our Platform) and a new site of delivery; and |
| ○ | our receipt of FDA “orphan drug” designations covering LP-10 and LP-410; |
| ● | our in-house manufacturing pilot plant (our “Facility”), which positions us to maximize scalability, quality and reliability, and permits us to better develop and maintain our trade secrets; |
| ● | our experienced scientific team, which has expertise in urology and liposomal drug development; and |
| ● | our management team, which has a track record in clinical development of local therapeutics for urinary bladder indications. |
Our Strategy
We are, to our knowledge, currently developing the first drug candidate and proprietary drug delivery platform that could be successful in treating cancer survivors who acquire HC and we intend to apply our proprietary drug delivery technology to the oral mucosa for the treatment of OLP and oral GVHD. Our development programs are designed to address opportunities for capital efficient drug discovery and development, especially research programs that reposition existing therapeutics for new indications that exploit new formulations. The key elements of the strategy that we are employing to achieve our goals are:
| ● | Advance the development of our lead product candidates, LP-10, to treat HC patients, and LP-310, to treat OLP. We designed LP-10 as a differentiated therapy for the treatment of cancer survivors with HC risks. We believe that LP-10 could be approved by the FDA as an effective therapy against HC due to its ability to exploit the known irreversible local vasoconstriction of tacrolimus (the active ingredient of LP-10) and take advantage of tacrolimus’ well-known anti-inflammatory properties. Our Platform permits a relatively high local drug concentration while also avoiding potential systemic toxicity. LP-10 has completed a phase 2a open-label, dose-escalation clinical trial for patients experiencing moderate to severe HC. Based on these recent results and the preclinical profile, we believe LP-10 has the potential to deliver meaningful clinical benefits over the currently available standard of care. A phase 2a multi-center dose-escalation study of LP-310 for the treatment of OLP is expected to be completed in the second quarter of 2025 and the top line data from the first two dose cohorts of this trial has been selected for podium presentation at the 2025 American Association of Oral Medicine and European Association of Oral Medicine Joint Meeting that will be held in Las Vegas, NV on May 15, 2025. |
| ● | Leverage our differentiated research and discovery approach to expand our product candidate pipeline. We expect to maintain a pipeline of additional product candidates, including LP-410 and LP-50, consistent with our strategy of developing proprietary 505(b)(2) regulatory pathway assets to address highly morbid indications where no adequate treatment(s) exists. We believe that our drug design approach, which involves proprietary repositioning of existing therapeutics (i.e., development of new applications using existing, approved active agents), integrated with our Platform, will allow us to efficiently design and validate novel product candidates that target inflammatory conditions of mucosal membranes. |
| ● | Maximize the clinical impact and value of our pipeline by relying on the 505(b)(2) regulatory pathway and, accordingly, deliver value to the stockholders. We believe the targeted nature of our research and discovery approach fosters efficient and focused clinical development. We intend to continue to build a lean, experienced team to develop product candidates in a capital-efficient manner. We intend to retain the commercialization rights to product candidates; however, we may opportunistically enter into strategic collaborations in certain geographic or clinical settings to maximize the value of our product pipeline. |
| ● | Continue to seek new therapies for rare diseases that can be evaluated with relatively small clinical trials, with an intent to minimize clinical development costs. Rare diseases that present severe morbidity and mortality are potentially eligible for accelerated regulatory approval pathways, such as the FDA’s “orphan drug” designation and designations under one or more of the FDA’s expedited development and review programs, which are associated with significantly lower development costs to obtain marketing approval for promising drug candidates. |
Our product development strategy involves combining intellectual property protection for novel formulations and indications for approved active pharmaceutical ingredients (“APIs”) with regulatory efficiencies provided by obtaining FDA designations that make our product candidates eligible for certain incentives that expedite development and review. We believe that this product development strategy is more capital efficient compared to traditional discovery of a new chemical entity because the safety and mechanisms of the approved APIs for the novel formulations of our product candidates are better understood and established. In the United States, approval of API products follows the “505(b)(2) regulatory pathway”; which permits us to rely on existing research and development (“R&D”) data pertaining to the generic active ingredient. The 505(b)(2) regulatory pathway often provides an alternate path to FDA approval for new or improved formulations or new uses of previously approved products. Using a 505(b)(2) new drug application (“NDA”), we expect to reduce the cost, time and risk that would otherwise be associated with bringing these programs to market. See “Government Regulation Applicable To Our Business – The 505(b)(2) NDA Regulatory Pathway” below for more information.
LP-10 and the Intended Treatment of HC
We completed our phase 2a clinical trial of LP-10 and reported top-line results in January 2023. LP-10 relies on intravesical vasoconstrictive and anti-inflammatory drug therapy for our intended treatment of HC, a rare and severe consequence of cancer therapy for which there is currently no approved treatment. HC affects the bladder lining and is caused by the protein-cross-linking effects of chemotherapy as well as longer-term effects from radiation-induced damage to urothelial tissue. In HC patients, the urothelial damage results in significant urinary bleeding, inducing the need for blood transfusions. Those cancer patients who acquire HC suffer from pain and discomfort that accompanies their bleeding. Based on information from the American Cancer Society as well as published reports on the incidence of HC resulting from either chemo or radiation therapy, we believe there are approximately 60,000 patients annually in the United States who suffer from a severe form of radiation-induced HC and an estimated 60,000 patients annually with systemic chemotherapy-induced HC. We received “orphan drug” designation from the FDA for the use of tacrolimus (including LP-10) for the treatment of HC.
We believe that our approach of using metastable liposomal tacrolimus as a treatment for HC, which has not yet been approved by the FDA, is novel. To date, we are not aware of any other liposomal products developed for clinical urinary bladder instillation. The current standard of care for HC patients is limited to measures such as irrigation and cauterization, which seek to reduce or halt the urinary bleeding of HC but often do not work effectively. There is no approved treatment for HC, and there are currently no other drug treatments for HC in clinical development of which we are aware. LP-10 is designed to be an acute treatment for HC to be administered via urinary catheter either at a hospital or doctor’s office within 30 minutes, which would be repeated daily for a total of four instillations in the same number of days. LP-10 seeks to treat HC via two mechanisms: high local vasoconstriction and longer-term anti-inflammation.
On December 23, 2019, we received IND approval from the FDA for LP-10, including approval for LP-10’s proposed clinical protocol, and central investigational review board (“IRB”) approval of our IND-approved clinical protocol, as well as approval for the investigator’s brochure and patient’s informed consent associated with LP-10. From 2020 to 2022, we signed clinical trial agreements in connection with eight clinical sites to conduct the dose-escalation, phase 2a clinical trial of LP-10. We completed the phase 2a dose-escalation trial (reporting results in January 2023) and intend to apply for FDA accelerated approval pathways, and the design of a pivotal phase 2b well-controlled clinical trial. If successful, a pivotal phase 3 trial can be requested, and we believe the results of an LP-10 phase 3 trial could support the submission of an NDA for LP-10 to the FDA through the 505(b)(2) regulatory pathway and a Marketing Authorization Application to the EMA in Europe. However, there can be no assurance that we will obtain such designation from, or be permitted to use such pathway by, the FDA, who is ultimately responsible for making such determinations.
Background on HC
HC is characterized by the presence of sustained hematuria and lower urinary tract symptoms in the absence of active tumor and other conditions or infections that cause excessive bleeding, (Gorzynska et al. 2005). Urologic adverse events caused by HC include frequency, dysuria, urgency, nocturia, suprapubic pain, bladder infection, fatigue and both microscopic and gross hematuria.
Bleeding from HC ranges from non-visible (or microscopic) hematuria to gross (visible) hematuria with clots (Decker et al. 2009). Moderately severe cases of HC involve massive bleeding and clot formation. Severe HC is a challenging condition to treat and may give rise to serious complications, leading to prolonged hospitalization and/or mortality (Decker et al. 2009; Mukhtar and Woodhouse 2010) and HC cases resulting from chemotherapy are reported to have a mortality rate approaching 4% (Rastinehad et al. 2007). Even mild cases of HC can cause disabling symptoms (e.g., frequency, urgency and pelvic pain, often localized to the bladder or urethra) (Payne et al. 2013). A standardized grading system (Droller et al. 1982) to classify the severity of HC has been proposed, which is shown in Figure 2 below:

Figure 2
HC can be classified as early- or late-onset (Zwaans et al. 2016). HC can also develop weeks to months after treatment in 20%–25% of patients who receive high doses of cyclophosphamide. The effects of radiation-induced HC may be acute or delayed, occurring long after radiation treatment has ended, from two months to 15 years later (Zwaans et al. 2018; Manikandan et al. 2010).
Prevalence
At the suggestion of the FDA’s Office of Orphan Products Development, we have measured annual cyclophosphamide and ifosphamide use in a large commercial database for private health plans between 2008 and 2010 and, based on guidance from the FDA, applied a 40% rate of HC in such patient database. The information from the database, combined with the FDA’s recommended guidance, results in a prevalence of consequential HC to potentially reach 60,000 new cases per year in the United States. This methodology implicitly assumes that the prevalence of use observed in private health plans (including Medicare beneficiaries enrolled in private plans) is generalizable to the nation as a whole, and such figure represents our conservative estimate of the number of new cases per year after applying the FDA’s recommended 40% rate to the figures in such patient database. HC resulting from pelvic radiation therapy (occurring in the prostate, rectum and uterine corpus) is less common than HC resulting from chemotherapy and is believed to be proportional to the incidence of the “primary neoplasia” (the original malignancy). Such incidence of HC is based on a combined estimate of the incidence of both chemotherapy-induced HC and radiation-induced HC from (i) peer-reviewed literature estimating the proportion of cyclophosphamide and ifosphamide recipients that acquire chemotherapy-induced HC after undergoing chemotherapy, as applied to a national chemotherapy incidence measurement study, and (ii) peer-reviewed literature containing estimates of the proportion of cancers treated with pelvic radiation therapy and the number of years patients survive post-radiation therapy, in addition to pelvic cancer incidence estimates publicly available from sources such as the American Cancer Society. According to the American Cancer Society publication Cancer Facts & Figures 2023 (available at: https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2023/2023-cancer-facts-and-figures.pdf), there are an estimated 288,300 new cases of prostate cancer in the U.S. each year, 153,020 new cases of rectum and colon cancer in the U.S. each year, and 66,200 cases new cases of uterine corpus cancer in the U.S. each year. Based, in part, on this data, we estimate the U.S. incidence of HC to be in the range of approximately 100,000 to 200,000 cases per year.
Existing Treatment Options
There is currently no standard therapy available for patients with HC, and there are no guidelines available on how HC should be optimally managed. Current HC treatments are regarded as ineffective, risky, or both. Such treatments include general medical management (e.g., estrogens, pentosan-polysulfate, and hyperbaric oxygen (“HBO”)), instillation therapy (e.g., aminocaproic-acid, alum, silver-nitrate, formalin, and fibrin glue), embolization and surgery (e.g., coagulation and cystectomy). The moderately severe cases of HC involve massive bleeding as well as clot formations that require evacuation. The most severe cases require surgical intervention (e.g., urinary diversion or cystectomy) (Sant 2002; Perez-Brayfield and Kirsch 2009). In addition, we believe current treatments pose significant patient risk: interventional fulguration of bleeding sites rarely works and exposes sick, frail patients to surgical risks; treatment with aminocaprotic acid often leads to dangerous clots; treatment with silver nitrate can cause bladder perforation or kidney failure; and treatment with formalin significantly reduces bladder functionality and causes excruciating pain (Vicente, Rios et al. 1990).
HBO treatments for HC may decrease and prevent the risk of bleeding but cannot treat ongoing bleeding, in part because therapy takes up to 40 sessions over a period of two to three months. Cystectomy causes significant morbidity and is generally an option of last resort; in some cases where cystectomy is conducted, old and/or frail patients can bleed to death. There are no other products in development of which we are aware that are indicated for the treatment of HC. Should LP-10 ultimately receive FDA market approval, we believe it will address this unmet medical need and provide a benefit over existing products while fitting into the existing treatment algorithm as a treatment for refractory HC.
LP-10’s Mechanisms of Action – Tacrolimus
LP-10’s API tacrolimus has been approved by the FDA for systemic use for inhibiting transplant rejection and as topical ointment for moderate to severe atopic dermatitis. Tacrolimus acts by inhibition of IL-2-dependent T-cell activation and has a direct inhibitory effect on cell-mediated immunity (Kino et al., 1987; Tamura et al., 2002). Tacrolimus prolongs the survival of the host and transplanted graft in animal transplant models of liver, kidney, heart, bone marrow, small bowel and pancreas, lung and trachea, skin, cornea and limb. In animals, tacrolimus has been demonstrated to suppress some humoral immunity and, to a greater extent, cell-mediated reactions such as allograft rejection, delayed type hypersensitivity, collagen-induced arthritis, experimental allergic encephalomyelitis and graft versus host disease. Tacrolimus inhibits T-lymphocyte activation, though the exact mechanism is not known. Experimental evidence suggests that tacrolimus binds to an intracellular protein named FKBP-12. A complex molecule comprising tacrolimus-FKBP-12, calcium, calmodulin and calcineurin is formed and the phosphatase activity of calcineurin is inhibited. This effect may prevent the dephosphorylation and translocation of the nuclear factor of activated T-cells, a nuclear component thought to initiate gene transcription for the formation of lymphokines (such as interleukin-2, gamma interferon). The net result is the inhibition of T-lymphocyte activation (i.e., immunosuppression).
The urothelium is the primary site of tissue damage in the general pathophysiology of cystitis (Erdogan et al. 2002). Recent studies have highlighted the overexpression of genes related to immune and inflammatory responses, including activation of CD4+ T-helper type-1-related chemokines in general cystitis (Trompeter et al. 2002; Almawi and Melemedjian 2000). Expression of chemokines precedes infiltration of immune cells and elevation of chemokines is an established signature of the inflammatory phenotype in bladder pain. Many of the symptoms of HC are related to inflammation of urothelial tissues. We believe that our application of the liposomal tacrolimus could potentially have a two-fold effect of (i) inhibiting calcineurin and the related response, and (ii) causing acute arteriole vasoconstriction to suppress HC (see Figure 2 above). Calcineurin inhibition is the well-known tacrolimus intracellular signal transduction mechanism that impairs the ability of certain immune cells to activate, and tacrolimus’ vasoconstrictive properties are referenced, for example, in section “5.7 Nephrotoxicity” of the Label (prescribing information) associated with the “PROGRAF® (tacrolimus) injection (for intravenous use) Initial U.S. Approval: 1994.”
Non-Clinical Study Results Involving Intravesical Tacrolimus
The following is a summary of non-clinical studies conducted with rats and dogs that were sponsored by the Company or conducted in collaboration with Company scientists. Results from animal studies are not always predictive of results of subsequent human clinical trials:
Effect of intravesical-tacrolimus on chemotherapy-induced HC
In September 2010, the effect of intravesical-tacrolimus was examined in a rat model for chemotherapy-induced, intraperitoneal injection of cyclophosphamide (200 mg/kg; i.p.) HC. This study demonstrated that cyclophosphamide-induced hyperactivity (i.e., decrease in inter-contraction interval) was suppressed in rats with intravesical LP-10 treatment but not in the rat groups left untreated (sham) or treated with empty liposomes (vehicle control) (Chuang et al. 2010). This result indicates that liposomal tacrolimus may mitigate cyclophosphamide injury in an animal model (Neurology and Urodynamics 30:421-427 (2011)).
Effect of intravesical-tacrolimus on radiation-induced HC
In October 2012, the efficacy of intravesical-tacrolimus was also examined in a rat model for radiation-induced HC. A 40 Gy radiation dose induced statistically significant reductions in the intermicturition interval recorded during metabolic urination patterns. Irradiated rats were randomly assigned to receive a single instillation of saline or intravesical-tacrolimus. Intravesical-tacrolimus increased the post-irradiation intermicturition intervals (p <0.001). Rat bladders that were harvested six weeks after the 40 Gy irradiation doses and two weeks after saline instillation showed edematous changes accompanying infiltration of inflammatory cells and hyperplastic urothelial changes. In contrast, bladder from group treated with intravesical-tacrolimus shows minimal edematous change, consistent with the hypothesis that the intravesical-tacrolimus had an anti-inflammatory effect (J. of Urology 194, 578-584 (2015)).
Pharmacokinetics of sphingomyelin formulated tacrolimus
A 2013 study examined levels of tacrolimus in blood, urine and bladder tissue following a single dose of liposome formulated tacrolimus instilled in the bladder of rats under anesthesia as compared to intravesical instillation of tacrolimus or intraperitoneal injection of tacrolimus in other rat groups. The tacrolimus dose was constant in all formulations at 200g/ml. At different times, blood, urine and bladder samples were collected. Tacrolimus levels in samples were analyzed using microparticle enzyme immunoassay. The area under curve (“AUC”) of liposome tacrolimus in serum at 0 to 24 hours was significantly lower than that of tacrolimus instillation or injection. Non-compartmental pharmacokinetic data analysis revealed maximum concentration of liposomal tacrolimus and tacrolimus in blood and urine at one and at two hours, respectively. Urine AUC (0–24 hours) after intravesical administration was significantly higher than in the intraperitoneal group (p < 0.05). Bladder tacrolimus AUC (0–24 hours) did not differ significantly between the groups. Single dose pharmacokinetics revealed that bladder instillation of liposome tacrolimus significantly decreased systemic exposure to instilled tacrolimus. This appears to indicate that a reduction in systemic exposure helps to limit the potential side effects of the tacrolimus by concentrating the dose to only one organ (J. of Urology Vol. 189, 1553-1558 (2013)).
LP-10 Toxicology Studies
In 2018, we completed chronic toxicology studies in rats and dogs, which were the two species of animals that we agreed to study in the course of our pre-IND communications with the FDA. The completion of such studies is normally required prior to requesting IND approval. The in-life phase of the toxicology rat study was performed between February and March 2018 and the in-life phase of the dog toxicology study was performed in March 2018. Such studies were company-sponsored and conducted by qualified vendors specializing in good laboratory practice in-vivo toxicology studies. The animals in such 2018 studies were assessed for morbidity, mortality, clinical observations and weekly body weight. Full sets of standard tissues, including urinary tract tissues, were collected and weighed and histopathology evaluations were conducted from all such animals. The studies concluded that no significant local and systemic toxicity resulted from the administration of LP-10 by intravesical instillation in either rats or dogs.
LP-10’s Addressable Market
LP-10 has been designed for the approximately one million cancer survivors in the United States today who have had pelvic radiation therapy and are at risk for HC. Based on the managed care database study that we sponsored in 2012 as part of our approved request for FDA “orphan drug” designation of tacrolimus for HC, approximately 60,000 of these patients annually experience severe chronic bladder bleeding that is often fatal. LP-10 has been developed to address this form of bleeding, as well as bladder bleeding associated with breast cancer patients who are taking systemic cyclophosphamide or ifosfamide, leading to chemotherapy-related cystitis experienced by an estimated 60,000 patients annually in the United States, inferring an addressable market in excess of 120,000 patients annually.
Figure 3

(1) American Cancer Society Cancer Facts and Figures 2023, (2) derived from a Company-sponsored study, (3) based on the Company’s 40% estimate, (4) American Cancer Society Cancer Treatment and Survivorship Fact and Figures 2022-2024, (5) based on the Company’s 30% estimate (6) 8% estimate, (7) based on the Company’s estimate, (8) $20,000 average revenue per each of an estimated 60,000 patients treated per year.
Figure 3 above illustrates the potential sources of revenue for LP-10. LP-10 is not currently approved for any indication; however, if clinical development is successful and we receive marketing approval for LP-10, we estimate the average LP-10 price to exceed $20,000 per patient-year domestically. This estimate is based on costs of HBO therapy, which is an option for patients with mild cases. HBO therapy can cost approximately $15,000 for a course of 30 sessions. Our price estimate also includes the potential for associated reductions in direct medical expenditures, especially for severe cases. We estimate the peak demand, at this price, to be, approximately 60,000 patients annually, which represents an approximate 50% market penetration in the U.S. Based on such price and demand estimates, we believe there is potential to receive up to $1.2 billion in annual gross revenue.
Our Lead Drug Candidates, LP-10, LP-310, and Our Product Pipeline
Five fundamental aspects of our LP-10 drug candidate make it an excellent fit for our strategy (see Figure 4 below). First, our API has a well-known mechanism of action. Second, published non-clinical studies involving animals, which are described above, demonstrate the potential for significant efficacy in our intended indication and route of administration. Third, we are fortunate to have had a successful human experience with intravesical tacrolimus (Dave et. al. Int Urol Nephrol 2015). Fourth, we believe we can take advantage of accelerated regulatory approval pathways for LP-10; we have already received “orphan drug” designation from the FDA that grants us product exclusivity, and we plan to apply for designations under one or more of the FDA’s expedited development and review programs. Fifth, we believe that the revenue potential for LP-10 could be significant. We believe our focus on capital-efficient drug development provides us with additional opportunities as we evaluate potential drug candidates for other rare diseases, especially those associated with locally delivering drugs to body cavities. When evaluating opportunities, we ensure that both the indication as well as the regulatory pathway are conducive to capital-efficient drug development. Our product candidate pipeline includes product candidates that could treat OLP (LP-310) and oral GVHD (LP-410). On November 10, 2023, we received the FDA’s IND approval for a Phase 2a dose escalation clinical trial regarding LP-310, and we were granted “orphan drug” designation for LP-410 the treatment of oral GVHD in November 2023. We submitted a Phase 2a IND for LP-310 treatment of OLP in the first quarter of 2024 and proceeded into clinical trial that is expected to be completed in the second quarter of 2025. We believe that our current product candidate pipeline could enable us to apply our drug delivery technology (our Platform) for multiple types of severe, rare diseases, and in the future, could enable us to address additional broader indications associated with endothelial inflammation. Local delivery often allows us to avert known risk factors by only locally applying the effective dose.
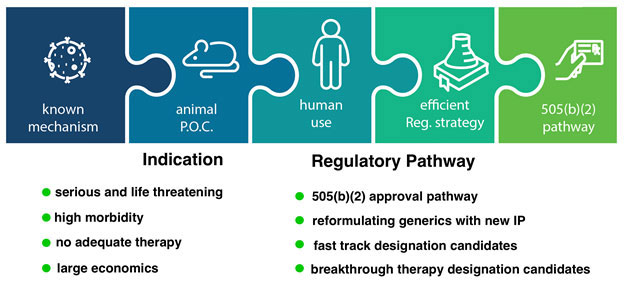
Figure 4
We are currently evaluating several potential product candidates for additional indications (including radiation proctitis and eosinophilic esophagitis).
LP-10’s Regulatory Status
In 2019, we completed the required manufacturing and toxicology program to submit an IND request to the FDA to begin testing LP-10 in human subjects. We submitted the IND request in September 2019 and received approval from the FDA within 30 days of submission to begin a clinical study involving LP-10. In December 2019, we received an advice letter from the FDA recommending several modifications to our proposed clinical protocol for LP-10, which we accommodated. We also submitted and received approval from the FDA for the trial’s associated investigator brochure and the proposed documentation of patient consent. Both of these documents, in addition to the clinical protocol, were submitted to Adverra, our central IRB, and we subsequently received IRB approval to conduct our clinical trial. In February 2020, the first patient was dosed in LP-10’s FDA phase 2a open-label, dose-escalation clinical trial for patients experiencing moderate to severe HC, which is intended to demonstrate proof-of-concept in humans. We reported summary results from LP-10’s phase 2a clinical trial in January 2023. We had a type-C meeting with the FDA on November 7, 2023, during which we agreed on a submission of a well-controlled phase 2b double-blind placebo-controlled trial with gross hematuria as assessed by patient report outcome as primary endpoint. This submission has been completed, and we are proceeding toward the initiation of a Phase2b clinical trial.
LP-10’s FDA “Orphan Drug” Designation Status
In 2010, we submitted a request to the FDA for “orphan drug” designation covering LP-10 and subsequently received approval for such designation in July 2012. This provides us with marketing exclusivity and permits us to benefit from shorter FDA review periods and reduced regulatory fees for LP-10. We intend to apply for similar “orphan drug” designations in additional jurisdictions, including Europe and Japan, as well as additional regulatory classifications, such as the FDA’s Breakthrough Therapy and Fast Track designations, in the United States. We expect that any designations that we have received, or may in the future receive, will confer additional advantages during LP-10’s development. However, there can be no assurance that we will obtain such designations from the FDA, who is ultimately responsible for making such determinations.
LP-10’s Clinical Status
Our multi-center, open-label dose-escalation phase 2a LP-10 clinical trial involved a total of thirteen subjects who received tacrolimus doses in one or two instillations of 2, 4 or 8mg via a pre-liposomal lyophilate reconstituted in 40 milliliters of sterile water. Subjects were cancer survivors with a history of pelvic radiotherapy who developed moderate to severe HC refractory to conventional therapy. The study was IRB-approved at nine clinical sites within the FDA’s jurisdiction.
Four subjects were enrolled in the 2mg group, four subjects were enrolled in the 4mg group, and five subjects were enrolled in the 8mg group. All subjects were male, with a median age of 67 years. Nine of the thirteen subjects had a history of prostate cancer and had been previously treated with external beam radiation. Two of the thirteen subjects had a history of lymphoma previously treated with radiation, and two had a history of bladder cancer previously treated with radiation.
The last subject of the LP-10 phase 2a study completed the last visit in October 2022. We reported top-line data from this trial in January 2023. All twenty-three LP-10 instillations in the 2mg, 4 mg and 8mg groups were well-tolerated by all thirteen subjects without related adverse events or elevated blood tacrolimus levels. For multiple subjects, hematuria and urinary symptoms improved, and cystoscopic bleeding and ulceration sites decreased. There was a complete response in three of the subjects, a partial response in seven of the subjects and no response in three of the subjects. We believe that such data and instillation safety findings indicate LP-10’s tolerability in HC patients and evidence LP-10’s potential use for the treatment of HC.
The results of the LP-10 phase-2a clinical trial have been published in the journal of International Urology and Nephrology, on September 19, 2023 (Hafron J. et al. Int. Urol. Nephrol. September 2023, Springer).
LP-310 and the Intended Treatment of OLP
LP-310 is currently in clinical development. LP-310 uses immunosuppressive and anti-inflammatory drug therapy to treat OLP, which is a chronic immune-mediated mucosal disease characterized by ulcerative lesions in the oral cavity. To date, upon review of relevant FDA public data resources on approved drugs and biologics, we are not aware of any other liposomal products developed to treat OLP. Patients are currently treated with off-label steroids for managing painful, erosive or ulcerative lesions. Yet, there are virtually no steroids formulated for topical drug delivery to lesions in the mouth. For severe and difficult-to-treat lesions, systemic steroids, and other immunosuppressive agents (e.g., hydroxychloroquin) are often needed, even though OLP is localized. Creams, gels and ointments do not adhere to oral mucosa and are easily swallowed, while mouthwashes and steroid inhalers have extremely short contact time with lesions. For severe and difficult-to-treat lesions, systemic steroids are often needed, even though OLP is localized. As a result, we believe there is great unmet medical need for this disease.
We believe that our approach of using metastable liposomal tacrolimus as a treatment for OLP, which has not yet been approved by the FDA, is novel. Tacrolimus has been used as an off-label oral treatment of OLP, and it has been shown to be effective based on systematic review and meta-analysis (Sun et al., 2019), which we believe is indicative of a rationale for using tacrolimus to treat OLP. Twenty-one trials involving 965 patients were included in this meta-analysis that concluded, in part, that treatment with tacrolimus may be an alternative approach when OLP does not respond to the standard protocols.
Background on OLP
OLP is a chronic T-cell-mediated mucosal disease that affects more than 1% of the global population, or more than 6 million people in the U.S. and Europe, according to González-Moles et. al., Oral Diseases 27(4):813-828 May 2021, “Worldwide prevalence of oral lichen planus: A systematic review and meta-analysis.” OLP is generally divided into three clinical subtypes: reticular, atrophic or erythematous, and erosive and/or ulcerative. Although lichen planus can be found on other areas of the body, such as with cutaneous lichen planus (“LP”), OLP has a chronic course, with little chance for spontaneous resolution, and most therapies that are currently available are palliative rather than curative. Based on peer-reviewed medical literature, OLP has a prevalence ranging from 1-2%, and females twice as likely as men to have the disease. The age on onset is generally between 30-60 years. Although cutaneous LP is associated with approximately 15% of OLP cases, OLP is associated with approximately 75% of patients with cutaneous LP.
Symptoms vary, but the disease is typically characterized by white reticular changes, erythema and painful ulcerative lesions in the oral cavity, accompanied by inflammation and severe pain. The precise cause is unknown, although an autoreactive immune process is suspected by most experts in the field. OLP is most frequently located bilaterally on the buccal mucosa (the inside lining of the cheeks and floor of the mouth), but can also appear on the tongue, palatal mucosa, gingiva and lips. Because of the long-lasting nature of the disease and painful symptoms, which can be spontaneous or triggered by acidic, crunchy and spicy food, patients require ongoing care and monitoring. Patients with OLP also have an approximately 1% likelihood of being diagnosed with oral cancer as a result of OLP (between 0.4% to 5% over a 20-year period, with an annual rate between 0.2% to 0.5%), making early detection and treatment imperative.
Some cases of OLP are caused by a hypersensitivity reaction to mercury and formaldehyde or medications such as ACE inhibitors, thiazide diuretics, beta blockers, gold salts, sulfasalazine, sulfonylureas and penicillamine. The new biologic agents such as TNF alpha inhibitors may also cause lichen planus-like eruptions. Patients with hypothyroidism, including Hashimoto thyroiditis, also develop OLP and it is unclear whether it is thyroid disease that predisposes an individual to OLP, or whether the drugs used to treat such disease also cause OLP. Hepatitis C virus infection has also been associated with the development of OLP in southern European countries. As mentioned above, we are not aware of any approved treatments for OLP, and we do not believe that current treatments are sufficiently effective.
LP-310’s Mechanisms of Action
LP-310 contains the API tacrolimus, like LP-10. Recent studies have highlighted that OLP pathophysiology is initiated by cellular-mediated immunity, most importantly, the increased production of T-helper 1(Th1) cytokines (Chamani et al., 2015). The oral mucosa is the primary site of tissue damage in the pathophysiology of OLP (Alrashdan et al., 2016). For a discussion of the tacrolimus API, on which LP-310 relies for its delivery to the oral cavity, and the effect tacrolimus has on T-lymphocyte activation, see “– LP-10’s Mechanisms of Action – Tacrolimus” above. We believe that our application of the liposomal tacrolimus to the oral cavity to address OLP could exploit this mechanism with a high local and low systemic distribution.
LP-310’s Addressable Market
LP-310 is not currently approved for any indication; however, if clinical development is successful and we receive marketing approval for LP-310, based upon the economics of existing oral cavity drug products, we project that the treatment of OLP will cost approximately $4,000 annually per patient. Most OLP patients are treated by dentists, who are relatively accessible compared to other medical specialists (in the United States there are approximately 200,000 dentists and ear, nose and throat physicians). Currently, dentists routinely recommend and prescribe instill agents as oral rinses and the procedure is simple and easy to teach. Given the absence of FDA approved treatment of OLP, we estimate revenue of approximately $4,000 per course of therapy, resulting in a total addressable market that exceeds $980 million. These estimates are based on the prices of other brand intravesical products as well as our preliminary estimates of the potential for reduction in medical expenditures associated with intractable cases.
LP-310’s Regulatory Status
On April 8, 2021, we successfully completed a pre-IND meeting to confirm the specific IND manufacturing, analytical, toxicology requirements for LP-310 as an oral rinse for the treatment of OLP. On October 17, 2023, the FDA approved an IND application for a multi-center, phase-2a, dose-escalation clinical trial to assess the safety and efficacy of LP-310 in patients with symptomatic OLP.
We began the clinical trial process for LP-310 and initiated the first clinical site in the second quarter of 2024. In the fourth quarter of 2024, we announced the completion of dosing for the first cohort in its multi-center Phase 2a clinical trial of LP-310. In this first cohort, eight participants received a dose of 0.25 mg LP-310, with promising initial results. No product-related serious adverse events were reported. Pharmacokinetic data demonstrated that whole blood tacrolimus levels in all patients were either undetectable or minimal, highlighting LP-310’s potential to deliver localized therapeutic effects while minimizing systemic exposure. Additionally, all patients tolerated LP-310 without significant adverse reactions.
The trial is expected to be completed in the second quarter of 2025. The top line data from the first two dose cohorts of this trial has been selected for podium presentation at the 2025 American Association of Oral Medicine and European Association of Oral Medicine Joint Meeting that will be held in Las Vegas, NV on May 15, 2025. Lipella anticipates the submission to the FDA of a request for Breakthrough Designation for LP-310 in the second half of 2025 and plans to commence a Phase 2b clinical trial for such product.
LP-410 and the Intended Treatment of oral GVHD
Lipella Pharmaceuticals Inc. is also developing an oral, liposomal formulation of tacrolimus, LP-410, for the treatment of oral GVHD. LP-410 is an oral rinse, similar to LP-310, but will have a different containment system. LP-410 targets the underlying mechanisms of oral GVHD, potentially providing a safe and effective treatment option for affected individuals. Lipella received “orphan drug” designation approval, on November 11, 2023, for tacrolimus for the treatment of oral GVHD. On March 5, 2024, the FDA approved an IND application for LP-410’s treatment of oral GVHD.
Background on oral GVHD
HCT is used to treat a wide range of malignancies, hematologic and immune deficiency states, and autoimmune diseases. GVHD is a clinical syndrome where donor-derived immunocompetent T cells react against patient tissues directly or through exaggerated inflammatory responses following HCT. Oral GVHD is a rare and serious disease, with a prevalence of approximately 30,000 patients in the US annually (Bachier et al., 2019; Bachier et al., 2021, Orphanet 2023). GVHD remains a major cause of morbidity and mortality with patients who undergo HCT treatment, with chronic GVHD being the leading cause of non-malignant fatality for such patients who receive HCT treatment.
Topical and local management of symptomatic oral GVHD can reduce oral symptoms that can interfere with oral function and quality of life and can reduce the need for more intensive immunosuppressive systemic therapies. However, there is currently no FDA approved local drug treatment of oral GVHD.
LP-50 and the Intended Treatment of NMIBC
Lipella is also developing an intravesical formulation of immunoglobulins including checkpoint inhibitors, LP-50, for the treatment of NMIBC. LP-50 offers the potential for increasing efficacy while minimizing systemic toxicity. Additional information regarding this preclinical program is included in the International Journal of Molecular Sciences 2024, 25(9), 4945, titled “Enhancing Therapeutic Efficacy and Safety of Immune Checkpoint Inhibition for Bladder Cancer: A Comparative Analysis of Injectable vs. Intravesical Administration,” as well as in US patent publication number 2024/0115503 titled “Intravesical Delivery of Hydrophilic Therapeutic Agents Using Liposomes.”
Facility
We have approximately 2,000 square feet of combined laboratory, office and warehouse space at our principal executive offices that we use in our research and development efforts. The lease for our principal executive offices has a five-year term that ends on May 31, 2024, and the lease provides us with an option to extend the term for an additional five years.
We believe our Facility contains all of the various components necessary to support our research, and it includes a current good manufacturing practices (“cGMP”)-capable manufacturing capability with a dedicated pilot-scale manufacturing. The space is divided into a production area and office space, with the production area subdivided into a clean space (Class 10,000) and sterile space (Class 100 (ISO class 5) clean room). Our Facility includes a pre-fabricated soft-wall, 6’x10’ class-100 clean room for aseptic formulation.
We maintain an internal LP-10 pilot manufacturing facility. We plan to file for an NDA utilizing the 505(b)(2) regulatory pathway for LP-10, which, if approved, may rapidly increase our manufacturing compliance needs. Even if we are able to pursue the 505(b)(2) regulatory pathway strategy, however, there is no assurance that we will be successful developing and/or commercializing LP-10 in a rapid or accelerated manner.
We are in a continuous process of complying with increasing regulatory requirements as the development of LP-10 and our other product candidates progresses. Currently, our manufacturing process primarily involves facility-dependent sterility protocols surrounding a five-step batch process. The simplicity of our process provides a strong incentive to continue investing internally in manufacturing compliance.
Compliance with stage-appropriate cGMPs is a prerequisite for FDA approval of a drug product for use in a clinical trial. cGMP regulations increase as a product candidate enters each subsequent clinical trial phase and as the scope of a proposed trial increases. Compliance with all cGMP regulations is a requirement for NDA approval and commercialization of LP-10. We expect to increase the cGMP manufacturing capabilities at our Facility to ensure full-scale compliant production of LP-10.
We believe that our manufacturing program will be able to support any future clinical trials involving LP-10. We currently lease industrial space used for cGMP manufacturing and analytical support. The space includes a non-porous epoxy floor, ideally suited for sterile environments, such as those used in hospital surgical rooms and sterile processing facilities. We have completed initial characterization and quality control release testing to confirm consistency of production of LP-10. Any applicable revised information and data will be provided to the FDA as part of an amended Chemistry, Manufacturing and Controls (“CMC”) section of the IND application prior to and in conjunction with use in any subsequent clinical trial.
Our Analytical Laboratory, Equipment & Supplies
Our current preparatory and biochemical/biophysical analysis capabilities include: ultra-centrifugation, high performance liquid chromatography; differential scanning calorimetry; gas chromatography; cross-polarization microscopy; fluorescent microscopy; near-infra-red imaging; and particle size analysis. In addition to the analytical equipment and sterile cleanroom, our Facility it contains a laminar flow hood for sterile procedures outside of the cleanroom, two Labconco lyophylizers (each with a 50-vile capacity), multiple incubators, a laboratory oven, an autoclave, various mass balances, vortexes, a heat stage for our optical microscope, various freezers and refrigerators, chemical and flammable storage cabinets, sterile disposables (including clothing, materials and vials), and raw materials, including APIs and lipids.
Suppliers
We obtain our raw material supply of LP-10 from multiple vendors who have a drug master file with the FDA. It is supplied as a white, lyophilized powder (a pre-liposomal lyophilate) formulated from sphingomyelin phospholipids, and tacrolimus. One vial of LP-10 drug product contains 80mg of tacrolimus and sphingomyelin (10% tacrolimus by weight) and is supplied to a clinic as a powder to be reconstituted with sterile water for injection prior to instillation. Quality control samples from each batch would be submitted for release testing according to established product specifications for identity and purity, residual solvent quantification, sterility assurance, and bacterial endotoxins.
Intellectual Property
Protection of our intellectual property is an important part of our business. On May 5, 2020, we were issued U.S. patent number 10,639,278 (the “278 Patent”) from the United States Patent and Trademark Office (“USPTO”), which does not expire until July 11, 2035. On June 14, 2022, we were issued U.S. patent number 11,357,725 (the “725 Patent”), which does not expire until November 9, 2034. Further, on May 28, 2020, we were issued one patent in Australia (No. 2014340137) (the “Australia Patent”), which does not expire until October 22, 2034. On August 23, 2022, we were issued a patent in Canada (No. 2,927,356) (the “Canadian Patent”), which does not expire until October 22, 2034. On June 7, 2023, we were issued a European patent (No. 3060197) (the “European Patent”), which does not expire until October 22, 2034. The European Patent was nationally validated in Switzerland, Spain, and Great Britain and a request for unitary effect was granted covering Austria, Belgium, Bulgaria, Denmark, Estonia, Finland, France, Germany, Italy, Latvia, Lithuania, Luxembourg, Malta, The Netherlands, Portugal, Slovenia, and Sweden. Each of the aforementioned patents cover aspects of our Platform technology relating to uses for delivering hydrophobic therapeutic, prophylactic or diagnostic agents to the body cavities, including LP-10 and LP-310, as well as methods of making formulations for delivering such hydrophobic agents. We are also actively prosecuting corresponding utility patent applications in the U.S. We intend to seek additional patent applications in the U.S. as well as in other jurisdictions, such as Europe, for our other proprietary technologies relating to intravesical immunoglobulin delivery and any future discoveries that we deem appropriate to protect. A U.S. patent application on the intravesical immunoglobulin delivery formulation is pending. Regulatory exclusivity should also be available in those countries in which regulatory approval is required, including Europe, Canada and others.
In addition to patents, we rely on trade secrets and know-how relating to our Platform technology and the product candidates we are developing using our Platform to develop and maintain our competitive position. However, trade secrets can be difficult to protect. We intend to protect our proprietary technology and processes, and maintain ownership of certain technologies, in part, through licenses as well as confidentiality agreements and invention assignment agreements with our employees, consultants and commercial partners.
Government Regulation Applicable to Our Business
In the United States, the FDA regulates drug products, including liposomally delivered products, under the FDCA, the Public Health Service Act (the “PHSA”), and regulations and guidance implementing these laws. The FDCA, PHSA and their corresponding regulations govern, among other things, the testing, manufacturing, safety, efficacy, labeling, packaging, storage, record keeping, distribution, reporting, advertising and other promotional practices involving drug products. Applications to the FDA are required before conducting human clinical testing of drug products. FDA approval also must be obtained before marketing of drug products. The process of obtaining regulatory approvals and the subsequent compliance with appropriate federal, state, local and foreign statutes and regulations require the expenditure of substantial time and financial resources and we may not be able to obtain the required regulatory approvals to successfully develop and commercialize our product candidates, including LP-10, LP-310, LP-410 and LP-50.
U.S. Drug Development Process
The FDA must approve a product candidate before it may be legally marketed in the United States. The process required by the FDA before a drug product candidate may be marketed in the United States generally involves the following:
| ● | completion of preclinical laboratory tests and in vivo studies in accordance with the FDA’s current good laboratory practice (“GLP”) regulations and applicable requirements for the humane use of laboratory animals or other applicable regulations; |
| ● | submission to the FDA of an application for an IND exemption, which allows human clinical trials to begin unless FDA objects within 30 days; |
| ● | approval by an independent IRB reviewing each clinical site before each clinical trial may be initiated; |
| ● | performance of adequate and well-controlled human clinical trials according to the FDA’s current GCP regulations, and any additional requirements for the protection of human research subjects and their health information, to establish the safety and efficacy of the proposed drug product candidate for its intended use; |
| ● | preparation and submission to the FDA of an NDA for marketing approval that includes substantial evidence of safety, purity and potency from results of nonclinical testing and clinical trials; |
| ● | review of the product by an FDA advisory committee, if applicable; |
| ● | satisfactory completion of an FDA inspection of the manufacturing facility or facilities where the drug product candidate is produced to assess compliance with cGMP requirements and to assure that the facilities, methods and controls are adequate to preserve the drug product candidate’s identity, safety, strength, quality, potency and purity; |
| ● | potential FDA audit of the nonclinical and clinical trial sites that generated the data in support of the NDA; and |
| ● | payment of user fees and FDA review and approval, or licensure, of the NDA. |
Before testing any drug product candidate in humans, including a liposomal intravesical product candidate, the product candidate must undergo preclinical testing. Preclinical tests, also referred to as nonclinical studies, include laboratory evaluations of product chemistry, toxicity and formulation, as well as in vivo studies to assess the potential safety and activity of the product candidate and to establish a rationale for therapeutic use. The conduct of the preclinical tests must comply with federal regulations and requirements including GLPs.
Concurrent with clinical trials, companies usually must complete some long-term preclinical testing, such as animal studies of reproductive adverse events and carcinogenicity, and must also develop additional information about the chemistry and physical characteristics of the drug and finalize a process for manufacturing the drug in commercial quantities in accordance with cGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the drug candidate and, among other things, the manufacturer must develop methods for testing the identity, strength, quality and purity of the final drug product. Additionally, appropriate packaging must be selected and tested and stability studies must be conducted to demonstrate that the drug candidate does not undergo unacceptable deterioration over its shelf life.
The clinical trial sponsor must submit the results of the preclinical tests, together with manufacturing information, analytical data, any available clinical data or literature and a proposed clinical protocol, to the FDA as part of an IND. Some preclinical testing may continue even after the IND is submitted. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA places the clinical trial on a clinical hold. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. The FDA also may impose clinical holds on a drug product candidate at any time before or during clinical trials due to safety concerns or non-compliance. If the FDA imposes a clinical hold, trials may not recommence without FDA authorization and then only under terms authorized by the FDA.
Human Clinical Trials Under an IND
Clinical trials involve the administration of the drug product candidate to healthy volunteers or patients under the supervision of qualified investigators which generally are physicians not employed by, or under, the control of the trial sponsor. Clinical trials are conducted under written study protocols detailing, among other things, the objectives of the clinical trial, dosing procedures, subject selection and exclusion criteria and the parameters to be used to monitor subject safety, including stopping rules that assure a clinical trial will be stopped if certain adverse events should occur. Each protocol and any amendments to the protocol must be submitted to the FDA as part of the IND. An IND automatically becomes effective 30 days after receipt by the FDA, unless before that time the FDA raises concerns or questions related to a proposed clinical trial and places the trial on clinical hold, including concerns that human research subjects will be exposed to unreasonable health risks. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. Accordingly, submission of an IND may or may not result in the FDA allowing clinical trials to commence. Clinical trials must be conducted and monitored in accordance with the FDA’s regulations comprising the current GCP requirements, including the requirement that all research subjects provide informed consent. Further, each clinical trial must be reviewed and approved by an IRB at or servicing each institution at which the clinical trial will be conducted. An IRB is charged with protecting the welfare and rights of trial participants and considers items such as whether the risks to individuals participating in the clinical trials are minimized and are reasonable in relation to anticipated benefits. The IRB also approves the form and content of the informed consent that must be signed by each clinical trial subject, or their legal representative, reviews and approves the study protocol, and must monitor the clinical trial until completed.
U.S. Review and Approval Processes
The results of the preclinical tests and clinical trials, together with detailed information relating to the product’s CMC and proposed labeling, among other things, are submitted to the FDA as part of an NDA requesting approval to market the product for one or more indications. Under the Prescription Drug User Fee Act, as amended (“PDUFA”), each NDA must be accompanied by a significant user fee. The FDA adjusts the PDUFA user fees on an annual basis. Fee waivers or reductions are available in certain circumstances, including a waiver of the application fee for the first application filed by a small business. Additionally, no user fees are assessed on NDAs for product candidates designated as orphan drugs unless the product candidate also includes a non-orphan indication.
“Orphan Drug” Designation
Under the Orphan Drug Act of 1983, the FDA may designate a drug product as an “orphan drug” if it is intended to treat a rare disease or condition (generally meaning that it affects fewer than 200,000 individuals in the United States, or more than 200,000 individuals in the United States in cases in which there is no reasonable expectation that the cost of developing and making a drug product available in the United States for treatment of the disease or condition will be recovered from sales of the product). “Orphan drug” designation must be requested before submitting an NDA. After the FDA grants “orphan drug” designation, the identity of the therapeutic agent and its potential orphan use are disclosed publicly by the FDA. “Orphan drug” designation does not convey any advantage in the regulatory review and approval process, nor does it shorten the duration of such process.
If a product with “orphan drug” status receives FDA approval for the disease or condition for which it has such designation, the product is entitled to “orphan drug” exclusivity, meaning that the FDA may not approve any other applications to market the same drug product for the same indication for seven years, except in limited circumstances, such as a showing of clinical superiority to the product with “orphan drug” exclusivity or if the party holding the exclusivity fails to assure the availability of sufficient quantities of the drug to meet the needs of patients with the disease or condition for which the drug was designated. Competitors, however, may receive approval of different drug products for the same indication for which the orphan drug has exclusivity or obtain approval for the same drug product but for a different indication for which the orphan drug has exclusivity. Orphan medicinal product status in the European Union has similar, but not identical, benefits.
The 505(b)(2) NDA Regulatory Pathway
Most drug products obtain FDA marketing approval pursuant to a full Section 505(b)(1) NDA or an aNDA. A third alternative is a Section 505(b)(2) NDA, which enables the applicant to rely, in part, on studies not conducted by, or for, the applicant and for which the applicant has not obtained a right of reference or use, such as the FDA’s findings of safety and/or effectiveness for a similar previously approved product, or published literature, in support of its application. For example, we anticipate referencing relevant publicly available data, including the publicly disclosed FDA drug approval package for tacrolimus, in the preparation and submission of our aNDA for LP-10 and LP-310. However, the FDA is responsible for ultimately determining if the Company may utilize this pathway for LP-10, LP-310, or any of our other product candidates and has presently not provided the Company with any indication that it may use such pathway. There is no guarantee that the FDA will make such a determination with respect to LP-10 or any of our other product candidates.
505(b)(2) NDAs often provide a path to FDA approval for new or improved formulations or new uses of previously approved products. Section 505(b)(2) permits the filing of an NDA where at least some of the information required for approval comes from studies not conducted by, or for, the applicant and for which the applicant has not obtained a right of reference. If the 505(b)(2) applicant can establish that reliance on the FDA’s previous approval is scientifically appropriate, it may eliminate the need to conduct certain preclinical or clinical studies of the new product. The FDA may also require companies to perform additional studies or measurements to support the change from the approved product. The FDA may then approve the new product candidate for all, or some, of the label indications for which the referenced product has been approved, as well as for any new indication sought by the Section 505(b)(2) applicant.
The FDCA also provides three years of marketing exclusivity for an NDA, 505(b)(2) NDA or supplement to an existing NDA if new clinical investigations, other than bioavailability studies, that were conducted or sponsored by the applicant are deemed by the FDA to be essential to the approval of the application, for example, new indications, dosages or strengths of an existing drug. This three-year exclusivity covers only the conditions of use associated with the new clinical investigations and does not prohibit the FDA from approving aNDAs for drugs containing the original active agent. Five-year and three-year exclusivity will not delay the submission or approval of a full NDA. However, an applicant submitting a full NDA would be required to conduct or obtain a right of reference to all of the preclinical studies and adequate and well-controlled clinical trials necessary to demonstrate safety and effectiveness.
To the extent that the Section 505(b)(2) applicant is relying on studies conducted for an already approved product, the applicant is required to certify to the FDA concerning any patents listed for the approved product in the Orange Book to the same extent that an aNDA applicant would. Thus approval of a 505(b)(2) NDA can be stalled until all the listed patents claiming the referenced product have expired, until any non-patent exclusivity, such as exclusivity for obtaining approval of a new chemical entity, listed in the Orange Book for the referenced product has expired, and, in the case of a Paragraph IV certification and subsequent patent infringement suit, until the earlier of 30 months, settlement of the lawsuit or a decision in the infringement case that is favorable to the Section 505(b)(2) applicant.
Expedited Development and Review Programs
In addition, the FDA is authorized to expedite the review of NDAs in several ways, including:
| ● | Fast Track Designation |
To obtain Fast Track designation for a drug product candidate, the sponsor of such drug may request the FDA to designate such drug for a specific indication concurrent with or after the filing of the related IND. Drug products are eligible for Fast Track designation if they are intended to treat a serious or life-threatening condition and demonstrate the potential to address unmet medical needs for the condition. We believe that LP-10, and the specific indication for which it is being studied, meets the qualifications for Fast Track designation; however, the FDA is responsible for ultimately determining if LP-10 meets such qualifications, and there is no guarantee that the FDA will make such a determination with respect to LP-10. In addition to other benefits, such as the ability to have greater interactions with the FDA, the FDA may initiate review of certain sections of a Fast Track NDA before the application is complete, a process known as a “rolling review.” Any drug product candidate submitted to the FDA for marketing, including for Fast Track designation, may be eligible for other types of FDA designations intended to expedite drug development and review, such as Breakthrough Therapy designation, priority review and accelerated approval. Our initial request to obtain Fast Track designation covering LP-10 in July 2021 was denied by the FDA in September 2021. Lipella anticipates repeating its submission for Fast Track designation at the conclusion of the Phase 2b study for LP-10.
| ● | Breakthrough Therapy Designation |
To qualify for the FDA’s Breakthrough Therapy designation, product candidates must be intended to treat a serious or life-threatening disease or condition and preliminary clinical evidence must indicate that such product candidates may demonstrate substantial improvement on one or more clinically significant endpoints compared to existing therapies.
The FDA will seek to ensure that the sponsor of a breakthrough therapy product candidate receives intensive guidance on an efficient drug development program, intensive involvement of senior managers and experienced staff on a proactive, collaborative and cross-disciplinary review, and rolling review. We believe that LP-10, and the specific indication for which it is being studied, meets the qualifications for Breakthrough Therapy designation, and we intend to apply for such status. Our initial Breakthrough Therapy designation was denied by the FDA on November 16, 2023. The FDA determined that refractory HC meets the criteria for a serious or life-threatening disease or condition, however, the clinical evidence does not yet demonstrate substantial improvement and the designation as a Breakthrough Therapy cannot be granted at this time. Lipella anticipates repeating its submission for Breakthrough Therapy Designation at the conclusion of the Phase 2b study for LP-10. In addition, Lipella anticipates the submission for Breakthrough Therapy Designation for LP-310 treatment of OLP in the second half of 2025, after the conclusion of the Phase2a clinical trial.
| ● | Accelerated Approval |
Drug or drug products studied for their safety and effectiveness in treating serious or life-threatening illnesses and that provide meaningful therapeutic benefits over existing treatments may receive accelerated approval from the FDA. Accelerated approval means that a product candidate may be approved on the basis of adequate and well-controlled clinical trials establishing that the product candidate has an effect on a surrogate endpoint that is reasonably likely to predict a clinical benefit, or on the basis of an effect on a clinical endpoint other than survival or irreversible morbidity or mortality or other clinical benefit, taking into account the severity, rarity and prevalence of the condition and the availability or lack of alternative treatments. As a condition of receipt of accelerated approval, the FDA may require that a sponsor of a drug product candidate requesting such approval perform adequate and well-controlled post-marketing clinical trials. In addition, the FDA currently requires, as a condition for accelerated approval, pre-approval of promotional materials relating to such drug candidate. Given the availability of direct efficacy measures in the case of LP-10 for HC, it is unlikely that a surrogate measurement would accelerate approval of LP-10. However, we remain open to the possibility of discovering relevant surrogate measurements during the clinical development of LP-10 that would accelerate the approval process. Lipella is also exploring inflammatory saliva cytokines measurement as a potential surrogate measure to accelerate approval of LP-310 treatment of OLP.
Fast Track designation, Breakthrough Therapy designation and accelerated approval do not change the standards for approval but may expedite the development or approval process.
Post-Approval Requirements
Rigorous and extensive FDA regulation of drug products continues after approval, particularly with respect to cGMP requirements. Manufacturers are required to comply with applicable requirements in the cGMP regulations, including quality control, quality assurance and maintenance of records and documentation. Other post-approval requirements applicable to drug products include reporting of cGMP deviations that may affect the identity, potency, purity and overall safety of a distributed product; recordkeeping requirements; reporting of adverse effects; reporting updated safety and efficacy information; and complying with electronic record and signature requirements. After an NDA is approved, the product also may be subject to official lot release, which is a potential marketing requirement related to manufacturing quality. If the product is subject to official release by the FDA, the manufacturer submits samples of each lot of product to the FDA, together with a release protocol, showing a summary of the manufacturing history of the lot and the results of all tests performed on the lot. The FDA also may perform certain confirmatory tests on lots of some products before releasing the lots for distribution. In addition, the FDA conducts laboratory research related to the regulatory standards on the safety, purity, potency and effectiveness of drug products. A sponsor also must comply with the FDA’s advertising and promotion requirements, such as the prohibition on promoting products for uses or in patient populations that are not described in the product’s approved labeling (known as “off-label use”).
Discovery of previously unknown problems or the failure to comply with the applicable regulatory requirements may result in restrictions on the marketing of a product or withdrawal of the product from the market, as well as possible civil or criminal sanctions. In addition, changes to the manufacturing process or facility generally require prior FDA approval before being implemented and other types of changes to the approved product, such as adding new indications and additional labeling claims, are also subject to further FDA review and approval.
Although our primary market is in the United States, we plan to commercialize LP-10 and our other product candidates in additional jurisdictions, including Europe, Canada, Mexico and Australia. Each of these jurisdictions may currently have, and may in the future adopt, laws, directives and regulations, that could affect our plan to test, obtain approval of and commercialize LP-10 and such other product candidates. We plan to develop an international regulatory strategy regarding such additional jurisdictions in the United States.
Our Team and History
Dr. Jonathan Kaufman, our Chief Executive Officer, and Dr. Michael Chancellor, our Chief Medical Officer, co-founded the Company in 2005. Prior to founding the Company, Dr. Kaufman was employed in the manufacturing division of Merck & Co. Inc (NYSE: MRK), and, subsequently, helped co-found Knopp Biosciences LLC, a privately held drug discovery and development company, and served as chief financial officer of Semprus Biosciences Corp. (a biomedical company acquired by Teleflex Incorporated (NYSE: TFX)). Dr. Chancellor has conducted more than 75 clinical trials and has consulted with numerous biotech companies developing urology products. We believe that Drs. Kaufman and Chancellor have a complimentary skillset combining basic and clinical research experience with entrepreneurial finance experience in the biotech sector.
Our Competition
The biotechnology and pharmaceutical industries are highly competitive. We are not aware of other existing clinical programs addressing new products for HC or OLP or GVHD. However, there are several pharmaceutical companies that are developing intravesical technologies for other indications, including transitional cell carcinoma (which is a superficial, non-muscle invasive form of bladder cancer). These companies and/or new entrants may potentially compete with LP-10, LP-310 and any other products that we develop in the future that use novel delivery technologies. We intend to rely on the market data exclusivity associated with obtaining FDA “orphan drug” designation covering LP-10, as well as our issued U.S. patents, the 278 Patent and the 725 Patent, our issued Australian Patent, our issued Canadian Patent, our issued European Patent, which may be extended for regulatory delay, and pending and future patent applications in order to maintain our competitive advantages in this space.
Employees and Human Capital Resources
As of March 26, 2025, we had five full-time employees and two part-time employees. None of our employees is subject to a collective bargaining agreement or represented by a trade or labor union. We consider our relationship with our employees to be good.
Our human capital resources objectives include, as applicable, identifying, recruiting, retaining, incentivizing and integrating our existing and additional employees. The principal purposes of our equity incentive plans are to attract, retain and motivate selected employees, advisors, consultants and directors through stock-based compensation awards. Also, we rely on cash-based performance bonus awards to incentivize such individuals.
Corporate Information
Our principal executive offices are located at 7800 Susquehanna Street, Suite 505, Pittsburgh, PA 15208 and include our Facility. Our telephone number is (412) 894-1853. We maintain an Internet website at www.lipella.com. The information contained on our website is not incorporated by reference into this Report.
We make available free of charge under the “Investors” section of our website all of our filings with the Securities and Exchange Commission (the “SEC”), including our annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K, proxy statements and amendments to such documents, each of which is provided on our website as soon as reasonably practicable after we electronically file or furnish, as applicable, the information with the SEC.
Implications of Being an Emerging Growth and Smaller Reporting Company
We qualify as an “emerging growth company” as defined in the Jumpstart Our Business Startups Act of 2012 (the “JOBS Act”). An emerging growth company may take advantage of relief from certain reporting requirements and other burdens that are otherwise applicable generally to public companies. These provisions include:
| ● | reduced obligations with respect to financial data; |
| ● | an exception from compliance with the auditor attestation requirements of Section 404(b) of the Sarbanes-Oxley Act of 2002 (the “Sarbanes-Oxley Act”); |
| ● | reduced disclosure about our executive compensation arrangements in our periodic reports, proxy statements and registration statements; and |
| ● | exemptions from the requirements of holding non-binding advisory votes on executive compensation or golden parachute arrangements. |
We may take advantage of these provisions for up to five years or such earlier time that we no longer qualify as an emerging growth company. We would cease to be an emerging growth company upon the earliest of:
| ● | the last day of the fiscal year on which we have $1.235 billion or more in annual revenue, |
| ● | the date on which we become a “large accelerated filer” (i.e., as of our fiscal year end, the total market value of our common equity securities held by non-affiliates is $700 million or more as of June 30), |
| ● | the date on which we issue more than $1.0 billion of non-convertible debt over a three-year period, or |
| ● | the last day of our fiscal year following the fifth anniversary of the date of the completion of our initial public offering (“IPO”). |
We may choose to take advantage of some but not all of these reduced reporting burdens.
In addition, under the JOBS Act, emerging growth companies can take advantage of an extended transition period and delay adopting new or revised accounting standards until such time as those standards apply to private companies. We have elected to use this extended transition period and, as a result, we will adopt new or revised accounting standards on the relevant dates on which adoption of such standards is required for private companies. If we were to subsequently elect instead to comply with public company effective dates, such election would be irrevocable pursuant to the JOBS Act.
Also, we are a “smaller reporting company” (and may continue to qualify as such even after we no longer qualify as an emerging growth company). For as long as we qualify as a “smaller reporting company,” we may provide reduced disclosure in the public filings that we make with the SEC than larger public companies, such as the inclusion of only two years of audited financial statements and only two years of management’s discussion and analysis of financial condition and results of operations disclosure.
As a result of qualifying as an emerging growth company and a smaller reporting company, to the extent we take advantage of the allowable reduced reporting burdens, the information that we provide to our stockholders may be different than what you might receive from other public reporting companies in which you hold equity interests.
Item 1A. Risk Factors
Our business, financial condition and operating results are subject to a number of risks and uncertainties, including those that are known to us and identified below and others that may arise from time to time. The following is a summary of the principal risk factors described in this section:
| ● | we may be unable to continue as a going concern and have incurred net losses since inception, with expected losses for the foreseeable future; |
| ● | we will need to raise additional funding in order to receive approval for our product candidates; |
| ● | our ability to obtain reimbursement or funding for our programs from the federal government may be impacted by reductions in federal spending; |
| ● | we are substantially dependent on the success of our lead product candidates, LP-10 and LP-310, and any delays or failure to commercialize them may harm our business; |
| ● | our product candidates are in the early stages of development, and we cannot guarantee regulatory approval, timely or successful progression through clinical trials, commercial potential, or the time and cost of development; |
| ● | even if we obtain regulatory approval for a product candidate, it will remain subject to regulatory oversight; |
| ● | we may never obtain approval for our product candidates outside of the United States; |
| ● | “orphan drug” designations may not effectively provide us with exclusive marketing rights, and our competitors may be able to obtain “orphan drug” exclusivity before us; |
| ● | we face significant competition in a rapidly changing industry, and our competitors may achieve regulatory approval before us or develop therapies that are more advanced or effective than ours; |
| ● | delays or disruptions in our manufacturing process may hinder product development and commercialization efforts, and third parties that we may need to utilize to conduct our product manufacturing may not perform satisfactorily; |
| ● | our ability to expand our market development or enter into agreements with third parties to market and sell our product candidates is uncertain; |
| ● | changes in pricing regulations could restrict product pricing; |
| ● | failure to obtain or maintain adequate insurance coverage and reimbursement for our products could limit our ability to market those products and generate product revenue; |
| ● | failure to comply with federal and state healthcare fraud and abuse laws, false claims laws and health information privacy and security laws could result in substantial penalties; |
| ● | our future depends on retaining, attracting, and motivating qualified personnel, and any personnel misconduct or non-compliance with regulatory standards could harm operations; |
| ● | we may face difficulties in protecting our intellectual property rights due to our inability to enforce our rights in certain parts of the world or changes in U.S. patent law, and third parties may initial legal proceedings alleging infringement of their intellectual property rights; |
| ● | we rely on third parties, which requires us to share our trade secrets; |
| ● | we have been notified by The Nasdaq Stock Market LLC (“Nasdaq”) of our failure to comply with certain continued listing requirements and if we are unable to regain compliance, we could be delisted from the Nasdaq Capital Market; |
| ● | certain stockholders may have effective control over actions requiring stockholder approval; |
| ● | provisions in our certificate of incorporation, bylaws and other agreements, as well as certain provisions of Delaware law, may deter, delay, or prevent a change in our control or unsolicited acquisition proposals, limit our rights and rights of our shareholders to take action against our management or limit our shareholder’s ability to obtain a favorable judicial forum for certain disputes; |
| ● | raising additional capital may cause dilution to our existing stockholders, restrict our operations or require us to relinquish rights to our technologies; |
| ● | we do not anticipate paying any cash dividends on our capital stock in the foreseeable future; |
| ● | as a smaller reporting public company, we face increased costs and new compliance initiatives. |
These risk factors could cause our actual results to differ materially from those suggested by forward-looking statements in this Report and elsewhere, and may adversely affect our business, financial condition or operating results. If any of these risk factors should occur, moreover, the trading price of our securities could decline, and investors in our securities could lose all or part of their investment in our securities. These risk factors should be carefully considered in evaluating our prospects.
Risks Related to Our Business
The report of the independent registered public accounting firm on our 2024 and 2023 financial statements contains a going concern qualification.
The report of the independent registered public accounting firm covering our financial statements for the years ended December 31, 2024 and December 31, 2023 stated that certain factors, including that we have suffered recurring losses from operations and have an accumulated deficit at December 31, 2024, raised substantial doubt as to our ability to continue as a going concern. Because we are not yet producing sufficient revenue to sustain our operating costs, we are dependent upon raising capital to continue our business. If we are unable to raise capital, we may be unable to continue as a going concern.
We have incurred net losses since inception. We expect to incur losses for the foreseeable future and may never achieve or maintain profitability.
There are numerous risks and uncertainties associated with pharmaceutical product development, and we are unable to accurately predict the timing or amount of increased expenses or when, or if, we will be able to achieve profitability.
As of December 31, 2024 we had an accumulated deficit of approximately $15,340,107, which includes a net loss of approximately $5,016,264 for the year ended December 31, 2024, as compared to an accumulated deficit of approximately $10,323,843, which includes a net loss of approximately $4,618,965 for the year ended December 31, 2023. Historically, we have financed our operations through a combination of grant revenue and equity financing, however our goals for the foreseeable future will likely require significant equity financing. Our ability to achieve significant profitability depends on our ability to successfully complete the development of, and obtain the regulatory approvals necessary to commercialize, LP-10 and/or our other product candidates, which may not occur for several years, if ever. The net losses we incur may fluctuate significantly from quarter to quarter.
If we are required by the FDA, the European Medicines Agency (“EMA”) or other regulatory authorities to perform studies in addition to those currently expected, or if there are any delays in completing our clinical trials or the development of LP-10 and/or our other product candidates, our expenses could increase and revenue could be further delayed. We anticipate that our expenses will increase substantially if, and as, we:
| ● | continue our research and the clinical development of LP-10 and our other product candidates; |
| ● | initiate additional clinical trials and preclinical studies for any additional product candidates that we may pursue in the future; |
| ● | prepare an NDA for filing with the FDA, a marketing authorization application, and approvals in certain other countries; |
| ● | ramp-up our in-house commercial-scale cGMP manufacturing facility; |
| ● | manufacture material for clinical trials or potential commercial sales; |
| ● | further develop our product candidate portfolio; |
| ● | establish a sales, marketing and distribution infrastructure to commercialize any product candidate for which we may obtain marketing approval; |
| ● | develop, maintain, expand and protect our intellectual property portfolio; and/or |
| ● | acquire or in-license other product candidates and technologies. |
To become and remain profitable, we must develop and eventually commercialize one or more product candidates with significant market potential. This will require us to be successful in a range of challenging activities, including completing the clinical trials, developing and validating commercial scale manufacturing processes, obtaining marketing approval for this product candidate, manufacturing, marketing and selling any future product candidates for which we may obtain marketing approval and satisfying any post-marketing requirements. If we were required to discontinue development of LP-10 or LP-310, if LP-10 or LP-310 do not receive regulatory approval, if we do not obtain our targeted indication(s) for LP-10 or LP-310, or if LP-10 or LP-310 fails to achieve sufficient market acceptance for any indication, we could be delayed by many years in our ability to achieve profitability for such assets. Lipella has additional pipeline assets, including but not limited to LP-410 and LP-50. As with LP-10 and LP-310, if LP-410 or LP-50 do not receive regulatory approval, if we do not obtain our targeted indication(s) for such assets, or if such assets fail to achieve sufficient market acceptance for any indication, we could be delayed by many years in our ability to achieve profitability for such assets. Our failure to become and remain profitable would decrease the value of our company and could impair our ability to raise capital, maintain our research and development efforts, expand our business or continue our operations. A decline in the value of our company also could cause you to lose all or part of your investment.
We will need to raise additional funding in order to receive approval for LP-10 or any other product candidate. Such funding may not be available on acceptable terms, or at all. Failure to obtain this necessary capital when needed may force us to delay, limit or terminate certain of our product development efforts or other operations.
To complete the process of obtaining regulatory approval for LP-10 and our other product candidates and to build the sales, marketing and distribution infrastructure that we believe will be necessary to commercialize such product candidates, if approved, we will require substantial additional funding. In addition, if we obtain marketing approval for LP-10 and any of our other product candidates, we expect to incur significant expenses related to product sales, medical affairs, marketing, manufacturing and distribution. We also anticipate that we will require substantial additional funding for LP-310, LP-410, LP-50 and product candidates that we decide to develop in the future.
Our future capital requirements will depend on many factors, including:
| ● | the progress, timing, results and costs of any future trials for LP-10, LP-310 and our other product candidates; |
| ● | the progress, timing and costs of manufacturing for our planned pivotal clinical trials for our product candidates; |
| ● | the continued development and the filing of an IND application for our product candidates; |
| ● | the initiation, scope, progress, timing, costs and results of drug discovery, laboratory testing, manufacturing, preclinical studies and clinical trials for any other product candidates that we may pursue in the future, if any; |
| ● | the costs of building and maintaining our own commercial-scale cGMP manufacturing facilities, including costs of maintaining our Facility; |
| ● | the outcome, timing and costs of seeking regulatory approvals; |
| ● | the costs associated with the manufacturing process development and evaluation of third-party manufacturers; |
| ● | the costs of future activities, including product sales, medical affairs, marketing, manufacturing and distribution, in the event we receive marketing approval for LP-10 or any other product candidates that we may develop; |
| ● | the extent to which the costs of our product candidates, if approved, will be paid by health maintenance, managed care, pharmacy benefit and similar healthcare management organizations, or will be reimbursed by government authorities, private health coverage insurers and other third-party payors; |
| ● | the costs of commercialization activities for LP-10 and our other product candidates if we receive marketing approval for LP-10 or such other product candidates and those that we may develop, including the costs and timing of establishing product sales, medical affairs, marketing, distribution and manufacturing capabilities; |
| ● | subject to receipt of marketing approval, if any, revenue received from commercial sale of LP-10 or any of our other product candidates; |
| ● | the terms and timing of any future collaborations, licensing, consulting or other arrangements that we may establish; |
| ● | the amount and timing of any payments we may be required to make, or that we may receive, in connection with the licensing, filing, prosecution, maintenance, defense and enforcement of any patents or other intellectual property rights, including milestone and royalty payments and patent prosecution fees that we are obligated to pay pursuant to our license agreements, if any; |
| ● | our ability to establish and maintain collaborations and licenses on favorable terms, if at all; and |
| ● | the extent to which we acquire or in-license other product candidates and technologies. |
Identifying potential product candidates and conducting preclinical testing and clinical trials is a time-consuming, expensive and uncertain process that takes years to complete, and we may never generate the necessary data or results required to obtain marketing approval and achieve product sales. Our product candidates, if approved, may not achieve commercial success. Our product revenues, if any, will be derived from or based on sales of product candidates that may not be commercially available for many years, if at all. Accordingly, we will need to continue to rely on additional financing to achieve our business objectives. Any additional fundraising efforts may divert our management from their day-to-day activities, which may adversely affect our ability to develop and commercialize our product candidates. Moreover, the terms of any financing may adversely affect the holdings or the rights of our stockholders and the issuance of additional securities, whether equity or debt, by us, or the possibility of such issuance, may cause the market price of our shares to decline. The sale of additional equity or convertible securities would dilute all our stockholders. The incurrence of indebtedness would result in increased fixed payment obligations and a portion of our operating cash flows, if any, being dedicated to the payment of principal and interest on such indebtedness, and we may be required to agree to certain restrictive covenants, such as limitations on our ability to incur additional debt, limitations on our ability to acquire, sell or license intellectual property rights and other operating restrictions that could adversely impact our ability to conduct our business. Furthermore, existing stockholders may not agree with our financing plans or the terms of such financings. Adequate additional financing may not be available to us on acceptable terms, or at all. The terms of additional financing may be impacted by, among other things, general market conditions, and the market’s perception of our product candidates.
We are currently supported partially by government grant awards, which may not be available to us in the future, and such grant awards are subject to guidelines regulating our research.
We have received and intend to continue to seek funding under grant award programs, including a program funded by the National Institutes of Health (“NIH”). To continue to fund a portion of our future research and development programs, we may also require grant funding from similar governmental agencies in the future. However, funding by the NIH or other governmental agencies may be significantly reduced or eliminated in the future for a number of reasons. For example, some programs are subject to a yearly appropriations process overseen by the U.S. Congress. In addition, we may not receive full funding under current or future grants because of budgeting constraints of the agency administering the program or unsatisfactory progress on the study being funded. Therefore, we cannot provide any assurance that we will receive any future grant funding from any government agencies, or, that if received, we will receive the full amount of the particular grant award. Any such reductions could delay the development of our product candidates and the introduction of new products.
Any research conducted under such federal grants will subject us to federal regulation regarding how we conduct our research and we will be obligated to abide by the agreement terms relating to those grants. There are also ethical guidelines promulgated by various governments and research institutions that we are required to follow in respect of our research. These guidelines are orientated towards research and experimentation involving humans and animals. Failure to follow the regulations, agreement terms and accepted scientific practices would jeopardize our grants and our results and the use of the results in further research and approval circumstances, which could have a material adverse effect on our results of operations and financial condition. In addition, any failure to comply with applicable laws or regulations affecting such grant awards could harm our business and divert our management’s attention.
Our ability to obtain reimbursement or funding for our programs from the federal government may be impacted by possible reductions in federal spending.
U.S. federal government agencies currently face potentially significant spending reductions. The U.S. federal budget remains in flux, however, which could, among other things, result in a cut to Medicare payments to providers and otherwise affect federal spending on clinical and pre-clinical research and development. The Medicare program is frequently mentioned as a target for spending cuts. The full impact on our business of any future cuts in Medicare or other programs is uncertain. In addition, we cannot predict any impact which the actions of the current Presidential administration and the U.S. Congress may have on the federal budget. If federal spending is reduced, anticipated budgetary shortfalls may also impact the ability of relevant agencies, such as the FDA or the NIH, to continue to function at current levels. Amounts allocated to federal grants and contracts may be reduced or eliminated. These reductions may also impact the ability of relevant agencies to timely review and approve drug research and development, manufacturing, and marketing activities, which may delay our ability to develop, market and sell any products we may develop.
We are substantially dependent on the success of our lead product candidates, LP-10 and LP-310. If we are unable to commercialize LP-10 or LP-310, or experience significant delays in doing so, our business will be materially harmed.
Our ability to generate product revenues, which may not occur for several years, if ever, currently depends heavily on the successful development and commercialization of LP-10 and LP-310. The success of LP-10 and LP-310 will depend on a number of factors, including the following:
| ● | successful completion of clinical development; |
| ● | receipt of marketing approvals from applicable regulatory authorities; |
| ● | establishing commercial manufacturing arrangements with third-party manufacturers; |
| ● | Obtaining, maintaining, and extending patent and trade secret protection and regulatory exclusivity; |
| ● | protecting our rights in our intellectual property portfolio; |
| ● | establishing sales, marketing and distribution capabilities; |
| ● | launching commercial sales of LP-10 and LP-310, if and when approved, whether alone or in collaboration with others; |
| ● | acceptance of LP-10 and LP-310, if and when approved, by patients, the medical community and third-party payors; |
| ● | effectively competing with other therapies; and |
| ● | maintaining a continued acceptable safety profile of LP-10 and LP-310, following approval. |
If we do not achieve one or more of these factors in a timely manner or at all, we could experience significant delays or an inability to successfully commercialize LP-10 and LP-310, which would materially harm our business. We have not yet demonstrated our ability to successfully complete development of any product candidates, obtain marketing approvals, manufacture a commercial scale product, or arrange for a third party to do so on our behalf, or conduct sales and marketing activities necessary for successful product commercialization.
Assuming we obtain marketing approval for any of our product candidates, we will need to transition our focus from research and development to supporting commercial activities. We may encounter unforeseen expenses, difficulties, complications and delays and may not be successful in such a transition.
We are early in our efforts to develop LP-10, LP-310, and our other product candidates. If we are unable to advance LP-10, LP-310, and such other candidates through clinical trials, obtain regulatory approval and ultimately commercialize such product candidates, or if we experience significant delays in doing so, our business will be materially harmed.
We are early in our development of LP-10, which will begin its phase 2b clinical trial, LP-310, which is expected to complete its multi-center Phase 2a clinical trial in the second quarter of 2025, and LP-410, which recently received FDA clearance for a Phase 2a clinical trial. The development and commercialization of LP-10, LP-310 and LP-410 (or any other product candidate that we may develop) is subject to many uncertainties, including the following:
| ● | successful enrollment and completion of clinical trials; |
| ● | positive results from our current and planned future clinical trials; |
| ● | receipt of regulatory approvals from applicable regulatory authorities; |
| ● | successful development of our internal manufacturing processes on an ongoing basis and maintenance of our potential future arrangements with third-party manufacturers for clinical supply; |
| ● | commercial launch of such product candidate, if and when approved, whether alone or in collaboration with others; and |
| ● | acceptance of such product candidate, if and when approved, by patients, the medical community and third-party payors. |
We must conduct extensive clinical trials to demonstrate the safety and efficacy of each drug candidate for its intended indications. Clinical trials are expensive, time consuming and uncertain as to outcome. We cannot guarantee that any clinical trials will be conducted as planned or completed on schedule, if at all. A failure of one or more clinical trials can occur at any stage of testing. Events that may prevent successful or timely completion of clinical development include:
| ● | delays in reaching a consensus with regulatory authorities on trial design; |
| ● | delays in opening sites and recruiting suitable patients to participate in our clinical trials; |
| ● | imposition of a clinical hold by regulatory authorities as a result of a serious adverse event or concerns with a class of drug candidates, or after an inspection of our clinical trial operations or trial sites; |
| ● | suspension of our clinical trials if it is determined that we or our collaborators are failing to conduct a trial in accordance with regulatory requirements, including the FDA’s cGCP regulations; |
| ● | delays in having patients complete participation in a trial or return for post-treatment follow-up; |
| ● | occurrence of serious adverse events associated with the drug candidate that are viewed to outweigh its potential benefits; or |
| ● | changes in regulatory requirements and guidance that require amending or submitting new clinical protocols. |
Additionally, if the results of our clinical trials are inconclusive or if there are safety concerns or serious adverse events associated with our drug candidates, we may:
| ● | be delayed in obtaining marketing approval, if at all, or be required to conduct additional confirmatory safety and/or efficacy studies; |
| ● | obtain approval for indications or patient populations that are not as broad as intended or desired; |
| ● | obtain approval with labeling that includes significant use or distribution restrictions or safety warnings; |
| ● | be subject to additional post-marketing testing requirements; |
| ● | be required to perform additional clinical trials to support approval or be subject to additional post-marketing testing requirements; |
| ● | have regulatory authorities withdraw, or suspend, their approval of the drug or impose restrictions on its distribution; |
| ● | be subject to the addition of labeling statements, such as warnings or contraindications; |
| ● | be sued; or |
| ● | experience damage to our reputation. |
In addition, if we make manufacturing or formulation changes to any of our other product candidates, we may need to conduct additional studies to bridge our modified product candidate to earlier versions. If we elect, or are required, to delay, suspend, or terminate any clinical trial of any of our product candidates at any stage, it could shorten any periods during which we may have the exclusive right to commercialize such product candidates or allow our competitors to bring products to market before we do, which could limit our potential revenue or impair our ability to successfully commercialize our current product candidates now, or such other product candidate in the future, and may harm our business, financial condition, results of operations and prospects. Any such significant changes, delays, setbacks or failures we experience, including our inability to obtain regulatory approval for or successfully commercialize our product candidates, particularly LP-10 and LP-310, would materially harm our business, financial condition, results of operations and prospects.
We are heavily dependent on the success of our product candidates, which are in the early stages of clinical development. Although we have reported positive results from our phase 2a clinical trials for LP-10 and LP-310, we cannot give any assurance that our trial results are indicative of success for future trials or commercialization.
We have reported positive top-line results from our completed phase 2a clinical trial evaluating the safety and efficacy of LP-10 and our recently completed dosing of the first cohort of our phase 2a clinical trial evaluating the safety and efficacy of LP-310. The top-line results from such clinical trials does not indicate or guarantee the future success for future clinical trials or for commercialization for commercialization of LP-10 or any of our other products. There can be no assurance that the data from such trial for LP-10, LP-310 or any future trial for LP-10, LP-310 or any of our other product candidates in our planned indications will be sufficiently supportive to rely on Fast Track designation or to obtain regulatory approval for such products. If our data is not supportive of, or the FDA will not allow us to apply for, Fast Track designation of LP-10, LP-310 or such other products, we cannot predict when, if ever, we will be able to seek the FDA approval for LP-10, LP-310 or such other products.
In addition, none of our product candidates have advanced into a pivotal clinical trial for our proposed indications, and it may be years before any such clinical trial is initiated and completed, if at all. We are not permitted to market or promote any of our product candidates before we receive regulatory approval from the FDA or comparable foreign regulatory authorities, and we may never receive such regulatory approval for any of our product candidates. If we do not receive regulatory approvals for our product candidates, we may not be able to continue our operations.
Even if we complete the necessary clinical trials for LP-10, LP-310 or for any of our other product candidates in the future, such as LP-410 or LP-50, we cannot predict when, or if, we will obtain regulatory approval to commercialize such other product candidates, and the approval may be for a narrower indication than we seek.
We cannot commercialize a product candidate until the appropriate regulatory authorities have reviewed and approved such product candidate. Even if LP-10 and LP-310 meet the applicable safety and efficacy standards in clinical trials, the regulatory authorities may not complete their review processes in a timely manner, or we may not be able to obtain regulatory approval for LP-10 and LP-310. Additional delays may result if an FDA Advisory Committee or other regulatory authority recommends non-approval or restrictions on approval for LP-10 and LP-310. In addition, we may experience delays or rejections based upon additional government regulation from future legislation or administrative action, or changes in regulatory authority policy during the period of LP-10’s or LP-310’s product development, clinical trials and review process. Similar issues could arise with respect to LP-410 and LP-50 in the event such products enter their planned clinical trial phases, as well as any of our other product candidates we develop in the future.
Regulatory authorities also may approve a product candidate for more limited indications than requested or they may impose significant limitations in the form of narrow indications, warnings or a post-approval safety monitoring program. These regulatory authorities may require precautions or contra-indications with respect to conditions of use or they may grant approval subject to the performance of costly post-marketing clinical trials. In addition, regulatory authorities may not approve the labeling claims that are necessary or desirable for the successful commercialization of LP-10, LP-310 or another product candidate. Any of the foregoing scenarios could materially harm the commercial prospects for LP-10, LP-310 or our other product candidates and materially and adversely affect our business, financial condition, results of operations and prospects.
LP-10 or LP-310 may cause undesirable side effects or have other properties that could delay or prevent its regulatory approval, limit its commercial potential, or result in significant negative consequences following any potential marketing approval.
In addition to side effects caused by LP-10 and LP-310, the administration process or related procedures also can cause adverse side effects. If in the future we are unable to demonstrate that such adverse events were caused by the administration process or related procedures, the FDA, the EMA or other regulatory authorities could order us to cease further development of, or deny approval of, LP-10 and LP-310, for any or all targeted indications. Even if we can demonstrate that any serious adverse events are not product-related, such occurrences could affect patient recruitment or the ability of enrolled patients to complete our clinical trials. Any of these occurrences may harm our ability to develop other product candidates, and may harm our business, financial condition and prospects significantly.
Additionally, if LP-10 or LP-310 receives marketing approval, the FDA could require us to adopt a post-approval safety monitoring program to ensure that the benefits outweigh its risks, which may include, among other things, a medication guide outlining the risks of the product for distribution to patients and a communication plan to health care practitioners. Furthermore, if we or others later identify undesirable side effects caused by LP-10 and LP-310, several potentially significant negative consequences could result, including:
| ● | regulatory authorities may suspend or withdraw approvals of such product candidate; |
| ● | regulatory authorities may require additional warnings on the label; |
| ● | we may be required to change the way a product candidate is administered or conduct additional clinical trials; |
| ● | we could be sued and held liable for harm caused to patients; and |
| ● | our reputation may suffer. |
Any of these events could prevent us from achieving or maintaining market acceptance of LP-10 and LP-310 and could significantly harm our business, financial condition, results of operations and prospects.
Our pipeline of products, including LP-10, LP-310, LP-410 and LP-50, are each based on novel technology, which makes it difficult to predict the time and cost of development and of subsequently obtaining regulatory approval.
The regulatory approval process and clinical trial requirements of the FDA, EMA and other regulatory authorities for novel product candidates such as ours can be more expensive and take longer than for other, better known or more extensively studied product candidates. It is difficult to determine how long it will take or how much it will cost to obtain regulatory approvals for our product candidates in either the United States or the European Union or how long it will take to commercialize our product candidates. Approvals by the European Commission may not be indicative of what the FDA may require for approval.
Regulatory requirements governing drug and biologic products have changed frequently and may continue to change in the future. In addition, adverse developments in clinical trials of similar drug and biologic products conducted by others may cause the FDA or other oversight bodies to change the requirements for approval of our product candidates. Similarly, the EMA may issue new guidelines concerning the development and marketing authorization for gene therapy medicinal products and require that we comply with these new guidelines.
These regulatory review committees and advisory groups and the new guidelines they promulgate may lengthen the regulatory review process, require us to perform additional studies, increase our development costs, lead to changes in regulatory positions and interpretations, delay or prevent approval and commercialization of any product candidate or lead to significant post-approval limitations or restrictions. As we advance our product candidates, we will be required to consult with these regulatory and advisory groups and comply with applicable guidelines. If we fail to do so, we may be required to delay or discontinue development of any product candidate. These additional processes may result in a review and approval process that is longer than we otherwise would have expected. Delay or failure to obtain, or unexpected costs in obtaining, the regulatory approval necessary to bring a potential product to market could decrease our ability to generate sufficient product revenue, and our business, financial condition, results of operations and prospects would be materially and adversely affected.
Even if we obtain regulatory approval for a product candidate, each approved product candidate will remain subject to regulatory oversight.
Even if we obtain regulatory approval for a product candidate, each approved product candidate will be subject to ongoing regulatory requirements for manufacturing, labeling, packaging, storage, advertising, promotion, sampling, record-keeping and submission of safety and other post-market information. Any regulatory approvals that we receive for any product candidate may also be subject to a post-approval safety monitoring program or limitations on the approved indicated uses for which the product may be marketed or to the conditions of approval or contain requirements for potentially costly post-marketing testing, including phase 4 clinical trials, and surveillance to monitor the quality, safety and efficacy of the product.
In addition, product manufacturers and their facilities are subject to payment of user fees and continual review and periodic inspections by the FDA and other regulatory authorities for compliance with cGMP requirements and adherence to commitments made in the NDA or foreign marketing application. If we, or a regulatory authority, discover previously unknown problems with a product, such as adverse events of unanticipated severity or frequency, or problems with the facility where the product is manufactured or disagrees with the promotion, marketing or labeling of that product, a regulatory authority may impose restrictions relative to that product, the manufacturing facility or us, including requiring recall or withdrawal of the product from the market or suspension of manufacturing.
If we fail to comply with applicable regulatory requirements following approval of any of our product candidates, a regulatory authority may:
| ● | issue a warning letter asserting that we are in violation of the law; |
| ● | seek an injunction or impose administrative, civil or criminal penalties or monetary fines; |
| ● | suspend or withdraw regulatory approval; |
| ● | suspend any ongoing clinical trials; |
| ● | refuse to approve a pending NDA or comparable foreign marketing application (or any supplements thereto) submitted by us or our strategic partners; |
| ● | restrict the marketing or manufacturing of the product; |
| ● | seize or detain the product or otherwise require the withdrawal of the product from the market; |
| ● | refuse to permit the import or export of product candidates; or |
| ● | refuse to allow us to enter into supply contracts, including government contracts. |
Any government investigation of alleged violations of law could require us to expend significant time and resources in response and could generate negative publicity. The occurrence of any event or penalty described above may inhibit our ability to commercialize any of our product candidates and adversely affect our business, financial condition, results of operations and prospects.
The FDA’s policies, and those of equivalent foreign regulatory agencies, may change and additional government regulations may be enacted that could prevent, limit or delay regulatory approval of any of our product candidates. We cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative action, either in the United States or abroad. If we are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we are not able to maintain regulatory compliance, we may lose any marketing approval that we may have obtained and we may not achieve or sustain profitability, which would materially and adversely affect our business, financial condition, results of operations and prospects.
Even if we obtain and maintain approval for our product candidates from the FDA, we may never obtain approval for them outside of the United States, which could limit our market opportunities and adversely affect our business.
Approval of a product candidate in the United States by the FDA does not ensure approval of such product candidate by regulatory authorities in other countries or jurisdictions, and approval by one foreign regulatory authority does not ensure approval by regulatory authorities in other foreign countries or by the FDA. Approval, marketing and sales of any of our product candidates outside of the United States will be subject to the regulatory requirements governing clinical trials and marketing approval in those countries. Approval procedures vary among jurisdictions and can involve requirements and administrative review periods different from, and more onerous than, those in the United States, including additional preclinical studies or clinical trials, as clinical trials in one country may not be accepted by regulatory authorities in other countries. Regulatory approval for any of our product candidates may be withdrawn. In many countries outside the United States, a product candidate must be approved for reimbursement before it can be approved for sale in that country. In some cases, the price that we intend to charge for our product candidates, if approved, is also subject to approval.
For example, we intend to submit a marketing authorization application to the EMA for approval of LP-10 and LP-310 in the European Union, but obtaining such approval from the European Commission following the opinion of the EMA is a lengthy and expensive process. Even if LP-10 and LP-310 are approved, the FDA or the European Commission, as the case may be, may limit the indications for which the product may be marketed, require extensive warnings on the product labeling or require expensive and time-consuming additional clinical trials or reporting as conditions of approval. Obtaining foreign regulatory approvals and compliance with foreign regulatory requirements could result in significant delays, difficulties and costs for us and could delay or prevent the introduction of our product candidates in certain countries.
If we fail to comply with the regulatory requirements, our target market will be reduced and our ability to realize the full market potential of any of our product candidates will be harmed and our business, financial condition, results of operations and prospects will be adversely affected.
While we have obtained “orphan drug” designations covering LP-10, and LP-410 from the FDA, such designations may not effectively provide us with exclusive marketing rights for LP-10 or LP-410, and we may be unable to obtain “orphan drug” designation covering any of our other product candidates. If our competitors are able to obtain “orphan drug” exclusivity before us covering products that constitute the same drug and treat the same indications as our product candidates, we may not be able to have competing products approved by the applicable regulatory authority for a significant period of time.
Under the Orphan Drug Act of 1983, the FDA may designate a product candidate as an “orphan drug” if it is intended to treat a rare disease or condition, which is generally defined as having a patient population of fewer than 200,000 individuals in the United States, or a patient population greater than 200,000 in the United States where there is no reasonable expectation that the cost of developing the drug will be recovered from sales in the United States.
Generally, if a product candidate with an “orphan drug” designation receives the first marketing approval for the indication for which it has such designation, the product is entitled to a period of marketing exclusivity, which precludes the FDA from approving another marketing application for a product that constitutes the same drug treating the same indication for that marketing exclusivity period, except in limited circumstances. If another sponsor receives such approval before we do (regardless of our “orphan drug” designation), we will be precluded from receiving marketing approval for our product for the applicable exclusivity period. The applicable period is seven years in the United States, however even after an orphan drug is approved, the FDA may subsequently approve another drug for the same condition if the FDA concludes that the latter drug is not the same drug or is clinically superior in that it is shown to be safer, more effective or makes a major contribution to patient care. In the EU, marketing authorization may be granted to a similar medicinal product for the same orphan indication if:
| ● | the second applicant can establish in its application that its medicinal product, although like the orphan medicinal product already authorized, is safer, more effective or otherwise clinically superior; |
| ● | the holder of the marketing authorization for the original orphan medicinal product consents to a second orphan medicinal product application; or |
| ● | the holder of the marketing authorization for the original orphan medicinal product cannot supply enough quantities of orphan medicinal product. |
On July 6, 2012, the FDA granted “orphan drug” designation covering LP-10 (or any other formulation of tacrolimus) for the treatment of HC and we may seek “orphan drug” designation from the FDA covering our future product candidates. On November 11, 2023 the FDA granted “orphan drug” designation covering LP-410 for treatment of oral GVHD.
Even though we have obtained such “orphan drug” designations, providing us with exclusivity for LP-10 and LP-410 for certain indications, such exclusivity may not effectively protect a product candidate from competition because different drugs can be approved for the same condition.
If the FDA does not conclude that LP-10, LP-310 or any of our other product candidates satisfy the requirements for the 505(b)(2) regulatory approval pathway, or if the requirements for approval of such product candidates under Section 505(b)(2) are not as we expect, the approval pathway for such product candidates will likely take significantly longer, cost significantly more and encounter significantly greater complications and risks than anticipated, and in any case may not be successful.
We intend to seek FDA approval through the 505(b)(2) regulatory pathway for LP-10, LP-310 and certain of our other product candidates, although we have not received any indication from the FDA that the 505(b)(2) regulatory pathway will be available for LP-10, LP-310, or any of our other product candidates. The Drug Price Competition and Patent Term Restoration Act of 1984, also known as the Hatch-Waxman Act, added Section 505(b)(2) to the FDCA. Section 505(b)(2) permits the filing of an NDA where at least some of the information required for approval comes from studies that were not conducted by or for the applicant. We anticipate referencing relevant publicly available data, including the publicly disclosed FDA drug approval package for tacrolimus, in the preparation and submission of our aNDA for LP-10 and LP-310.
If the FDA does not allow us to pursue the 505(b)(2) regulatory pathway for our product candidates as anticipated, we may need to conduct additional clinical trials, provide additional data and information and meet additional standards for regulatory approval. If this were to occur, the time and financial resources required to obtain FDA approval for our product candidates would likely substantially increase. Moreover, the inability to pursue the 505(b)(2) regulatory pathway could result in new competitive products reaching the market faster than our product candidates, which could materially adversely impact our competitive position and prospects. Even if we are permitted to pursue the 505(b)(2) regulatory pathway for a product candidate, we cannot assure you that we will receive the requisite or timely approvals for commercialization of such product candidate.
In addition, notwithstanding the approval of a number of products by the FDA under Section 505(b)(2) over the last few years, certain competitors and others have objected to the FDA’s interpretation of Section 505(b)(2). We expect that our competitors could file citizens’ petitions with the FDA in an attempt to persuade the FDA that our product candidates, or the clinical studies that support their approval, contain deficiencies. If the FDA’s interpretation of Section 505(b)(2) is successfully challenged, the FDA may be required to change its Section 505(b)(2) policies and practices, which could delay or even prevent the FDA from approving any NDA that we submit under Section 505(b)(2).
FDA designations to expedite drug development and review, including “orphan drug” designation, Breakthrough Therapy designation, and/or Fast Track designation, even if granted for any of our product candidates, may not lead to a faster development, regulatory review or approval process and do not increase the likelihood that any of our product candidates will receive marketing approval in the United States.
We have received “orphan drug” designation covering LP-10 and LP-410 from the FDA, but there is no assurance that any of our other product candidates will receive a similar designation from the FDA or that we will receive Breakthrough Therapy or Fast Track designations covering any of our product candidates (including LP-10 and LP-410) from the FDA. Our initial request to obtain Fast Track designation covering LP-10 in July 2021 was denied by the FDA in September 2021; however, we are still seeking to obtain Fast Track designation covering LP-10. In addition, we anticipate the submission for Breakthrough Designation Request for LP-310 for the treatment of OLP in the second half of 2025. Further, even if we do receive favorable designations from the FDA, the receipt of any of these designations covering any of our product candidates may not result in a faster development process, review or approval of such product candidates compared to products considered for approval under conventional FDA procedures and does not assure ultimate approval by the FDA.
If we are not successful in discovering, developing and commercializing additional product candidates, our ability to expand our business and achieve our strategic objectives would be impaired.
Although we focus a substantial amount of our efforts on the potential approval of LP-10 and LP-310, a key component of our strategy is to discover, develop and potentially commercialize a portfolio of other product candidates, including LP-410 and LP-50, to treat orphan diseases and ultimately, non-orphan diseases. Identifying new product candidates requires substantial technical, financial and human resources, whether any product candidates are ultimately identified. Even if we identify product candidates that initially show promise, we may fail to successfully develop and commercialize such product candidates for many reasons, including the following:
| ● | the research methodology used may not be successful in identifying potential product candidates; |
| ● | competitors may develop alternatives that render our product candidates obsolete; |
| ● | product candidates we develop may nevertheless be covered by third parties’ patents or other exclusive rights; |
| ● | a product candidate may, on further study, be shown to have harmful side effects or other characteristics that indicate it is unlikely to be effective or otherwise does not meet applicable regulatory criteria; |
| ● | a product candidate may not be capable of being produced in commercial quantities at an acceptable cost, or at all; and |
| ● | a product candidate may not be accepted as safe and effective by patients, the medical community or third-party payors. |
As we have limited resources, we may forego or delay pursuit of opportunities with certain programs or product candidates or for indications that later prove to have greater commercial potential. Our spending on current and future research and development programs may not yield any commercially viable products. If we do not accurately evaluate the commercial potential for a particular product candidate, we may relinquish valuable rights to that product candidate through strategic collaboration, licensing or other arrangements in cases in which it would have been more advantageous for us to retain sole development and commercialization rights to such product candidate. Alternatively, we may allocate internal resources to a product candidate in a therapeutic area in which it would have been more advantageous to enter into a partnering arrangement.
If any of these events occur, we may be forced to abandon our development efforts with respect to a particular product candidate or fail to develop a potentially successful product candidate, which could have a material adverse effect on our business, financial condition, results of operations and prospects.
We face significant competition in an environment of rapid technological change and the possibility that our competitors may achieve regulatory approval before us or develop therapies that are more advanced or effective than ours, which may adversely affect our financial condition and our ability to successfully market or commercialize our product candidates.
Many of our potential competitors, alone or with their strategic partners, have substantially greater financial, technical and other resources, such as larger research and development, clinical, marketing and manufacturing organizations. Mergers and acquisitions in the biotechnology and pharmaceutical industries may result in even more resources being concentrated among a smaller number of competitors. Our commercial opportunity could be reduced or eliminated if competitors develop and commercialize products that are safer, more effective, have fewer or less severe side effects, are more convenient or are less expensive than any product candidate that we may develop. Also, competitors may obtain FDA or other regulatory approval for their products more rapidly or earlier than we may obtain approval for ours, which could result in our competitors establishing a strong market position before we are able to enter the market. Additionally, technologies developed by our competitors may render our product candidates, including, in particular, LP-10 and LP-310, uneconomical or obsolete, and we may not be successful in marketing these and our other product candidates against competitors.
In addition, as a result of the expiration or successful challenge of our patent rights, we could face more competition from our competitors’ products. The availability of our competitors’ products could limit the demand, and the price we are able to charge, for any product candidate that we may develop and commercialize.
Risks Related to Manufacturing
Delays in obtaining regulatory approvals of the process and facilities needed to manufacture any of our product candidates, including LP-10 and LP-310, or disruptions in our manufacturing process may delay or disrupt our product development and commercialization efforts.
Before we can begin to commercially manufacture any of our product candidates, including LP-10 or LP-310, in a manufacturing facility, whether in a third-party facility or in a facility that we maintain and operate, the facility must pass a pre-approval inspection by the FDA, and a manufacturing authorization must be obtained from the appropriate regulatory authorities. The timeframe required to obtain such approvals is uncertain. In order to obtain approval, we will need to ensure that all our processes, methods and equipment are compliant with cGMP and perform extensive audits of vendors, contract laboratories and suppliers. If any of our vendors, contract laboratories or suppliers is found to be out of compliance with cGMP, we may experience delays or disruptions in manufacturing while we work with these third parties to remedy the violation or while we work to identify suitable replacement vendors. The cGMP requirements govern quality control of the manufacturing process and documentation policies and procedures. In complying with cGMP, we will be obligated to expend time, money and effort in production, record keeping and quality control to assure that the product meets applicable specifications and other requirements. If we fail to comply with these requirements, we would be subject to possible regulatory action and may not be permitted to sell any product candidate that we may develop.
In addition, the manufacturing process used to produce our product candidates is complex, novel and has not been validated for commercial use. In order to produce enough quantities of our product candidates for future clinical trials and initial U.S. commercial demand, we will need to increase the scale of our manufacturing process. The production of our product candidates requires processing steps that are more complex than those required for most chemical pharmaceuticals. We employ multiple steps to control our manufacturing process to assure that the process works and that each of our products candidates will be made strictly and consistently in compliance with the process. Problems with the manufacturing process, even minor deviations from the normal process, could result in product defects or manufacturing failures that result in lot failures, product recalls, product liability claims or insufficient inventory. We may encounter problems achieving adequate quantities and quality of clinical-grade materials that meet FDA, EMA or other applicable standards or specifications with consistent and acceptable production yields and costs.
Although we have established our Facility, we may need to utilize third parties to conduct our product manufacturing for the near future. Therefore, we are subject to the risk that these third parties may not perform satisfactorily.
Even if we obtain the validation from the FDA of our Facility, we intend to maintain third-party manufacturing capabilities in order to provide multiple sources of supply. In the event that these third-party manufacturers do not successfully carry out their contractual duties, meet expected deadlines or manufacture LP-10 or LP-310 in accordance with regulatory requirements, or if there are disagreements between us and these third-party manufacturers, we will not be able to complete, or may be delayed in completing, the preclinical studies required to support future IND submissions of other product candidates or the clinical trials required for approval of LP-10 or LP-310. In such instances, we may need to locate an appropriate replacement third-party relationship, which may not be readily available or on the same economic terms, which would cause additional delay or increased expense prior to the approval of LP-10 or LP-310 and would thereby have a material adverse effect on our business, financial condition, results of operations and prospects.
If we or our third-party manufacturer fails to comply with applicable cGMP regulations, the FDA and foreign regulatory authorities can impose regulatory sanctions including, among other things, refusal to approve a pending application for a new product candidate or suspension or revocation of a pre-existing approval. Such an occurrence may cause our business, financial condition, results of operations and prospects to be materially harmed.
Any contamination in our manufacturing process, shortages of raw materials or failure of any of our key suppliers to deliver necessary components could result in delays in our clinical development or marketing schedules.
Given the nature of sterile product manufacturing, there is a risk of contamination. Any contamination could materially adversely affect our ability to produce any of our product candidates, including LP-10, on schedule and could, therefore, harm our results of operations and cause reputational damage.
Some of the raw materials required in our manufacturing process are derived from biologic sources. Such raw materials are difficult to procure and may be subject to contamination or recall. A material shortage, contamination, recall or restriction on the use of biologically derived substances in the manufacture of any of our product candidates, including LP-10 and LP-310, could adversely impact or disrupt the commercial manufacturing or the production of clinical material, which could materially and adversely affect our development timelines and our business, financial condition, results of operations and prospects.
Risks Related to Commercialization of Our Product Candidates
If we are unable to expand our market development capabilities or enter into agreements with third parties to market and sell our product candidates, we may be unable to generate any product revenue.
We currently have a small market development organization. To successfully commercialize LP-10, LP-310, LP-410 or LP-50, if approved, and any other products that may result from our development programs, we plan to expand our capabilities to promote market access and build awareness, either on our own or with one or more third parties. The development of our own market development team will be expensive and time-consuming and could delay any product launch. Moreover, we cannot be certain that we will be able to successfully develop this capability. We may enter into collaboration agreements regarding any of our product candidates with third parties to utilize their established marketing and distribution capabilities, but we may be unable to enter into such agreements on favorable terms, if at all. If any future collaborators do not commit sufficient resources to commercialize our products, or we are unable to develop the necessary capabilities on our own, we will be unable to generate sufficient product revenue to sustain our business. We compete with many companies that currently have extensive, experienced and well-funded medical affairs, marketing and sales operations to recruit, hire, train and retain marketing and sales personnel. We also face competition in our search for third parties to assist us with the sales and marketing efforts of our product candidates. Without an internal team or the support of a third party to perform marketing and sales functions, we may be unable to compete successfully against these more established companies.
Our efforts to educate the medical community and third-party payors on the benefits of our product candidates may require significant resources and may never be successful. Such efforts may require more resources than are typically required due to the complexity and uniqueness of our potential products. If any of our product candidates is approved but fails to achieve market acceptance among physicians, patients or third-party payors, we will not be able to generate significant revenues from such product, which could have a material adverse effect on our business, financial condition, results of operations and prospects.
If the market opportunities for LP-10 or LP-310 are smaller than we believe they are, our product revenues may be adversely impacted, and our business may suffer.
We are currently primarily focusing our research and product development efforts on LP-10 for HC and LP-310 for OLP. Our understanding of both the number of people who have these diseases, as well as the subset of people with these diseases who have the potential to benefit from treatment with LP-10 or LP-310, are based on estimates in published literature. These estimates may prove to be incorrect, and new studies may reduce the estimated incidence or prevalence of this disease. The number of patients in the United States, the EU and elsewhere may turn out to be lower than expected or these patients may not be otherwise amenable to treatment with LP-10 or LP-310, or may become increasingly difficult to identify and access, all of which would adversely affect our business, financial condition, results of operations and prospects.
Further, there are several factors that could contribute to making the actual number of patients who receive LP-10 or LP-310 less than the potentially addressable market. These include the lack of widespread availability of, and limited reimbursement for, new therapies in many underdeveloped markets. These risks could similarly apply to LP-410 and LP-50, each of which we are simultaneously developing.
Government price controls or other changes in pricing regulation could restrict the amount that we are able to charge for any of our product candidates that may be approved in the future, including LP-10 and LP-310, which would adversely affect our revenue and results of operations.
We expect that coverage and reimbursement of pharmaceutical costs may be increasingly restricted both in the United States and abroad. The escalating cost of health care has led to increased pressure on the health care industry to reduce costs. Drug pricing by pharmaceutical companies recently has come under increased scrutiny and continues to be subject to intense political and public debate in the United States and abroad. Government and private third-party payors have proposed health care reforms and cost reductions. A number of federal and state proposals to control the cost of health care, including the cost of drug treatments, have been made in the United States. Specifically, there have been several recent U.S. Congressional inquiries and proposed bills designed to, among other things, bring more transparency to drug pricing, review the relationship between pricing and manufacturer patient programs and reform government program reimbursement methodologies for drugs. In some international markets, the government controls the pricing, which can affect the profitability of drugs. Current government regulations and possible future legislation regarding health care may affect coverage and reimbursement for medical treatment by third-party payors, which may render our product candidates, if approved, not commercially viable or may adversely affect our anticipated future revenues and gross margins.
We cannot predict the extent to which our business may be affected by these or other potential future legislative or regulatory developments. However, future price controls or other changes in pricing regulation or negative publicity related to the pricing of pharmaceutical drugs generally could restrict the amount that we are able to charge for our future products, which would adversely affect our anticipated revenue and results of operations.
The insurance coverage and reimbursement status of newly approved products is uncertain. Failure to obtain or maintain adequate coverage and reimbursement for our products, if approved, could limit our ability to market those products and decrease our ability to generate product revenue.
We expect that coverage and reimbursement by government and private payors will be essential for most patients to be able to afford any of our product candidates that receive approval. Accordingly, sales of our product candidates will depend substantially, both domestically and abroad, on the extent to which the costs of our product candidates will be paid by health maintenance, managed care, pharmacy benefit and similar healthcare management organizations, or will be reimbursed by government authorities, private health coverage insurers and other third-party payors. Coverage and reimbursement by a third-party payor may depend upon several factors, including the third-party payor’s determination that use of a product is:
| ● | a covered benefit under its health plan; |
| ● | safe, effective and medically necessary; |
| ● | appropriate for the specific patient; |
| ● | cost-effective; and |
| ● | neither experimental nor investigational. |
Obtaining coverage and reimbursement for a product from third-party payors is a time-consuming and costly process that could require us to provide to the payor supporting scientific, clinical and cost-effectiveness data. We may not be able to provide data sufficient to gain acceptance with respect to coverage and reimbursement. If coverage and reimbursement are not available, or are available only at limited levels, we may not be able to successfully commercialize our product candidates. Even if coverage is provided, the approved reimbursement amount may not be adequate to realize a sufficient return on our investment.
There is significant uncertainty related to third-party coverage and reimbursement of newly approved products. In the United States, third-party payors, including government payors such as the Medicare and Medicaid programs, play an important role in determining the extent to which new drugs and biologics will be covered and reimbursed. The Medicare and Medicaid programs increasingly are used as models for how private payors and government payors develop their coverage and reimbursement policies.
Outside the United States, international operations generally are subject to extensive government price controls and other market regulations and increasing emphasis on cost-containment initiatives in the European Union, Canada and other countries may put pricing pressure on us. In many countries, the prices of medical products are subject to varying price control mechanisms as part of national health systems. It also can take a significant amount of time after approval of a product to secure pricing and reimbursement for such product in many counties outside the United States. In general, the prices of medicines under such systems are substantially lower than in the United States. Other countries allow companies to fix their own prices for medical products but monitor and control company profits. Additional foreign price controls or other changes in pricing regulation could restrict the amount that we are able to charge for our product candidates. Accordingly, in markets outside the United States, the reimbursement for our products may be reduced compared with the United States and may be insufficient to generate commercially reasonable product revenues.
Moreover, increasing efforts by government and third-party payors in the United States and abroad to cap or reduce healthcare costs may cause such organizations to limit both coverage and the level of reimbursement for new products approved and, as a result, they may not cover or provide adequate payment for our product candidates. Payors increasingly are considering new metrics as the basis for reimbursement rates, such as average sales price, average manufacturer price, and “actual acquisition cost.” Therefore, it may be difficult to project the impact of these evolving reimbursement metrics on the willingness of payors to cover candidate products that we or our partners are able to commercialize. We expect to experience pricing pressures in connection with the sale of any of our product candidates due to the trend toward managed healthcare, the increasing influence of health maintenance organizations and additional legislative changes. The downward pressure on healthcare costs in general, particularly prescription drugs and surgical procedures and other treatments, has become intense. As a result, increasingly high barriers are being erected to the entry of new products such as ours.
Healthcare legislative reform measures may have a material adverse effect on our business and results of operations.
In the United States and some foreign jurisdictions, there have been, and continue to be, several legislative and regulatory changes and proposed changes regarding the healthcare system that could prevent or delay marketing approval of our product candidates, restrict or regulate post-approval activities, and affect our ability to profitably sell any product candidates for which we obtain marketing approval.
For example, in March 2010, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act (collectively, the “PPACA”), was passed, which substantially changed the way healthcare is financed by both the government and private insurers, and significantly impacted the U.S. pharmaceutical industry.
Further, there has been heightened governmental scrutiny over the manner in which manufacturers set prices for their marketed products, which have resulted in several recent Congressional inquiries and proposed and enacted bills designed to, among other things, bring more transparency to product pricing, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for products. In addition, the United States government, state legislatures, and foreign governments have shown significant interest in implementing cost containment programs, including price-controls, restrictions on reimbursement and requirements for substitution of generic products for branded prescription drugs to limit the growth of government paid health care costs. For example, the United States government has passed legislation allowing the federal government to negotiate prices for some drugs covered under Medicare and requiring pharmaceutical manufacturers to provide rebates and discounts to certain entities and governmental payors to participate in federal healthcare programs. Further, Congress and the current administration have each indicated that it will continue to seek new legislative and/or administrative measures to control drug costs. Individual states in the United States have also been increasingly passing legislation and implementing regulations designed to control pharmaceutical product pricing.
Any reduction in reimbursement from Medicare or other government programs may result in a similar reduction in payments from private payors. The implementation of cost containment measures or other healthcare reforms, including those governing enrollment in federal healthcare programs, reimbursement changes, fraud and abuse enforcement, and expansion of new programs could result in reduced demand for our product candidates, including LP-10, and may prevent us from being able to generate revenue, attain profitability, or commercialize our products.
We may be subject, directly or indirectly, to federal and state healthcare fraud and abuse laws, false claims laws and health information privacy and security laws. If we are unable to comply, or have not fully complied, with such laws, we could face substantial penalties.
If we obtain FDA approval for any of our product candidates, including LP-10 and LP-310, and begin the process of commercialization in the United States, our operations will be directly, or indirectly through our prescribers, customers and purchasers, subject to various federal and state fraud and abuse laws and regulations, including, without limitation, the federal Anti-Kickback Statute, federal civil and criminal false claims laws and the Physician Payments Sunshine Act and regulations. These laws will impact, among other things, our proposed sales, marketing and educational programs. In addition, we may be subject to patient privacy laws by both the federal government and the states in which we conduct our business as well as other jurisdictions. The laws that will affect our operations include, but are not limited to:
| ● | the federal Anti-Kickback Statute, which prohibits, among other things, persons or entities from knowingly and willfully soliciting, receiving, offering or paying any remuneration (including any kickback, bribe or rebate), directly or indirectly, overtly or covertly, in cash or in kind, in return for the purchase, recommendation, leasing or furnishing of an item or service reimbursable under a federal healthcare program, such as the Medicare and Medicaid programs. This statute has been interpreted to apply to arrangements between pharmaceutical manufacturers on the one hand, and prescribers, purchasers and formulary managers on the other. The PPACA amended the intent requirement of the federal Anti-Kickback Statute to clarify that a person or entity does not have to have actual knowledge of this statute or specific intent to violate it; |
| ● | federal civil and criminal false claims laws and civil monetary penalty laws which prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment or approval from Medicare, Medicaid or other government payors that are false or fraudulent. The PPACA provides that a claim for items or services resulting from an Anti-Kickback Statute violation is a false claim under the federal False Claims Act. Cases against pharmaceutical manufacturers support the view that certain marketing practices, including off-label promotion, may implicate the False Claims Act; |
| ● | the federal Health Insurance Portability and Accountability Act of 1996 (“HIPAA”), which created new federal criminal statutes that prohibit a person from knowingly and willfully executing a scheme or from making false or fraudulent statements to defraud any healthcare benefit program, regardless of the payor (e.g., public or private); |
| ● | HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act (“HITECH”), and its implementing regulations, and as amended again by the final HIPAA omnibus rule, Modifications to the HIPAA Privacy, Security, Enforcement, and Breach Notification Rules under HITECH and the Genetic Information Nondiscrimination Act; |
| ● | other modifications to HIPAA, which impose certain requirements relating to the privacy, security and transmission of individually identifiable health information without appropriate authorization by entities subject to the rule, such as health plans, health care clearinghouses and health care providers; |
| ● | federal transparency laws, including the federal Physician Payment Sunshine Act, that require certain manufacturers of drugs, devices, biologics and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program, with specific exceptions, to report annually to the Centers for Medicare & Medicaid Services information related to: (i) payments or other “transfers of value” made to physicians and teaching hospitals and (ii) ownership and investment interests held by physicians and their immediate family members; |
| ● | state and foreign law equivalents of each of the above federal laws, state laws that require drug manufacturers to report information related to payments and other transfers of value to physicians and other healthcare providers or marketing expenditures and state laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and may not have the same effect, thus complicating compliance efforts in certain circumstances, such as specific disease states; and |
| ● | state and foreign laws that govern the privacy and security of health information in some circumstances, many of which differ from each other in significant ways and often are not preempted by HIPAA, thus complicating compliance efforts. |
Because of the breadth of these laws and the narrowness of the statutory exceptions and safe harbors available, it is possible that some of our business activities could be subject to challenge under one or more of such laws. If our operations are found to be in violation of any of the laws described above or any other government regulations that apply to us, we may be subject to penalties, including civil and criminal penalties, damages, fines, exclusion from participation in government health care programs, such as Medicare and Medicaid, imprisonment and the curtailment or restructuring of our operations, any of which could adversely affect our ability to operate our business and our results of operations.
Efforts to ensure that our business arrangements with third parties will comply with applicable healthcare laws and regulations will involve substantial costs. It is possible that governmental authorities will conclude that our business practices may not comply with current or future statutes, regulations or case law involving applicable fraud and abuse or other healthcare laws and regulations. If our operations are found to be in violation of any of these laws or any other governmental regulations that may apply to us, we may be subject to significant civil, criminal and administrative penalties, damages, fines, imprisonment, exclusion of products from government funded healthcare programs, such as Medicare and Medicaid, and the curtailment or restructuring of our operations.
The risk of our being found in violation of these laws is increased by the fact that many of them have not been fully interpreted by the regulatory authorities or the courts, and their provisions are open to a variety of interpretations. Any action against us for violation of these laws, even if we successfully defend against it, could cause us to incur significant legal expenses and divert our management’s attention from the operation of our business. The shifting compliance environment and the need to build and maintain a robust and expandable systems to comply with multiple jurisdictions with different compliance and/or reporting requirements increases the possibility that a healthcare company may run afoul of one or more of the requirements.
Risks Related to Our Operations
If we are unable to manage expected growth in the scale and complexity of our operations, our performance may suffer.
If we are successful in executing our business strategy, we will need to expand our managerial, operational, financial and other systems and resources to manage our operations, continue our research and development activities and, in the longer term, build a commercial infrastructure to support commercialization of any of our product candidates that are approved for sale. Future growth would impose significant added responsibilities on members of management. It is likely that our management, finance, development personnel, systems and facilities currently in place may not be adequate to support this future growth. Our need to effectively manage our operations, growth and product candidates requires that we continue to develop more robust business processes and improve our systems and procedures in each of these areas. We may be unable to successfully implement these tasks on a larger scale and, accordingly, may not achieve our research, development and growth goals.
Our future success depends on our ability to retain key employees and scientific advisors and to attract, retain and motivate qualified personnel.
The success of the Company is highly dependent upon certain key management and technical personnel, the loss of whose services may a material adverse impact on the Company’s business, financial condition, results of operations and the achievement of our objectives. Dr. Chancellor, our Chief Medical Officer and a director, and Dr. Kaufman, our Chief Executive Officer and a director, have played key roles in the founding, management, technology development and/or promotion of the Company. There can be no assurance that either of these persons will remain with the Company in the future due to circumstances either within or outside of their control. Additionally, our employees and scientific advisors are at-will employees and consultants, and the loss of one or more of them might impede the achievement of our research, development and commercialization objectives.
Recruiting and retaining other qualified employees and scientific advisors for our business, including scientific and technical personnel, also will be critical to our success. The Company currently does not hold key man insurance on the lives of its executives; even if the Company does seek to obtain such insurance, there is no assurance as to the availability of such insurance or the cost thereof if such insurance is available, or the ability to find a qualified replacement for its executives. In addition, failure to succeed in preclinical or clinical trials or applications for marketing approval may make it more challenging to recruit and retain qualified personnel. Further development of the Company’s products will require additional personnel, particularly qualified scientific and technical personnel. The Company currently has limited personnel and other resources. The success of the Company will be dependent on attracting and retaining key employees, including management. The inability to recruit, or loss of services of certain executives, key employees or advisors may impede the progress of our research, development and commercialization objectives and have a material adverse effect on our business, financial condition, results of operations and prospects.
Our employees, principal investigators and advisors may engage in misconduct or other improper activities, including non-compliance with regulatory standards and requirements.
We are exposed to the risk of fraud or other misconduct by our employees, principal investigators and advisors. Principal investigators are physicians who we utilize to lead the conduct of our clinical trials and assist us with the development of our drug product candidates, including LP-10 and LP-310. Misconduct by these parties could include intentional failures to comply with FDA regulations or the regulations applicable in the EU and other jurisdictions, provide accurate information to the FDA, the EMA and other regulatory authorities, comply with healthcare fraud and abuse laws and regulations in the United States and abroad, report financial information or data accurately or disclose unauthorized activities to us. Sales, marketing and business arrangements in the healthcare industry are subject to extensive laws and regulations intended to prevent fraud, misconduct, kickbacks, self-dealing and other abusive practices. These laws and regulations restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, customer incentive programs and other business arrangements. Such misconduct also could involve the improper use of information obtained in the course of clinical trials or interactions with the FDA or other regulatory authorities, which could result in criminal and civil penalties or sanctions and cause serious harm to our reputation. It is not always possible to identify and deter employee misconduct, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from government investigations or other actions or lawsuits stemming from a failure to comply with these laws or regulations. If any such actions are instituted against us and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, financial condition, results of operations and prospects, including the imposition of significant fines, criminal penalties, or other sanctions.
In addition, principal investigators for our clinical trials may serve as scientific advisors or consultants to us from time to time and receive compensation in connection with such services. Under certain circumstances, we may be required to report some of these relationships to the FDA. The FDA may conclude that a financial relationship between us and a principal investigator has created a conflict of interest or otherwise affected interpretation of the trial. The FDA may therefore question the integrity of the data generated at the applicable clinical trial site and the utility of the clinical trial itself may be jeopardized. This could result in a delay in approval, or rejection, of our marketing applications by the FDA and may ultimately lead to the denial of marketing approval of our current and future drug candidates.
If we fail to comply with environmental, health and safety laws and regulations, we could become subject to fines or penalties or incur costs that could have a material adverse effect on the success of our business.
We are subject to numerous environmental, health and safety laws and regulations, including those governing laboratory procedures and the generation, handling, use, storage, treatment, manufacture, transportation and disposal of, and exposure to, hazardous materials and wastes, as well as laws and regulations relating to occupational health and safety. Our operations involve the use of hazardous and flammable materials, including chemicals and biologic materials. Our operations also produce hazardous waste products. We generally contract with third parties for the disposal of these materials and wastes. We cannot eliminate the risk of contamination or injury from these materials. In the event of contamination or injury resulting from our use of hazardous materials, we could be held liable for any resulting damages, and any liability could exceed our resources. Also, we could incur significant costs associated with civil or criminal fines and penalties. We do not carry specific biological or hazardous waste insurance coverage, and our property, casualty and general liability insurance policies specifically exclude coverage for damages and fines arising from biological or hazardous waste exposure or contamination. Accordingly, in the event of contamination or injury, we could be held liable for damages or be penalized with fines in an amount exceeding our resources, and our clinical trials or regulatory approvals could be suspended.
Although we maintain workers’ compensation insurance for certain costs and expenses, we may incur due to injuries to our employees resulting from the use of hazardous materials or other work-related injuries, this insurance may not provide adequate coverage against potential liabilities. We do not maintain insurance for toxic tort claims that may be asserted against us in connection with our storage or disposal of biologic, hazardous or radioactive materials.
Also, we may incur substantial costs to comply with current or future environmental, health and safety laws and regulations, which have tended to become more stringent over time. These current or future laws and regulations may impair our research, development or production efforts. Failure to comply with these laws and regulations also may result in substantial fines, penalties or other sanctions or liabilities, which could materially adversely affect our business, financial condition, results of operations and prospects.
A pandemic, epidemic or outbreak of an infectious disease, may materially and adversely affect our business and our financial results and could cause a disruption to the development of our product candidates.
Public health crises such as pandemics, epidemics or similar outbreaks could adversely impact our business, preclinical or clinical trial operations, including our ability to recruit and retain patients and principal investigators and site staff. Public health crises may also affect employees of third-party contract research organizations (“CROs”) located in geographies where we carry out our clinical trials.
In addition, the patient populations that our core product candidates target may be particularly susceptible to pandemics, epidemics or adverse public health developments, which may make it more difficult for us to identify patients able to enroll in our future clinical trials and may impact the ability of enrolled patients to complete any such trials. Any negative impact that adverse public health development may have on patient enrollment or the treatment or execution of our product candidates could cause costly delays to clinical trial activities, which could adversely affect our ability to obtain regulatory approval for and to commercialize our product candidates, could materially increase our operating expenses and could have a material adverse effect on our financial results.
Unfavorable global economic conditions could adversely affect our business, financial condition or results of operations.
Our results of operations could be adversely affected by general conditions in the global economy and in the global financial markets, including conditions that are outside of our control, such as conflict, loss of life and disaster connected to ongoing armed conflicts between Ukraine and Russia in Europe and Israel and Hamas in the Middle East, and the foreign and domestic government sanctions imposed on Russia as a result of its invasion of Ukraine. There continues to be volatility and disruptions in the capital and credit markets, and a severe or prolonged economic downturn, including, but not limited to as a result of such events, could result in a variety of risks to our business, such as weakened demand for our product candidates and our ability to raise additional capital when needed on acceptable terms, if at all. A weak or declining economy could strain our suppliers, possibly resulting in supply disruption, or cause delays in payments for our services by third-party payors or our collaborators. Any of the foregoing could harm our business and we cannot anticipate all the ways in which the current economic climate and financial market conditions could adversely impact our business.
Our internal computer systems, or those of our collaborators or other contractors or consultants, may fail or suffer security breaches, which could result in a material disruption of our product development programs.
We receive, process, store, and transmit, often electronically, confidential data of others, which increase cybersecurity risks. Our internal computer systems and those of our current and any future collaborators and other contractors or consultants are vulnerable to damage from computer viruses, unauthorized access, natural disasters, terrorism, war and telecommunication and electrical failures, and we are also subject to occurrences of theft, improper disclosure of confidential information and the deletion or modification of records. Data security breaches - whether by employees or others - which may expose sensitive data to unauthorized persons, could lead to the loss of trade secrets or other intellectual property, result in demands for ransom or other forms of blackmail, or lead to the public exposure of personal information (including sensitive personal information) of our employees, clinical trial patients, customers, and others. Such attacks are of ever-increasing levels of sophistication and are made by groups and individuals with a wide range of motives (including industrial espionage or extortion) and expertise, including by organized criminal groups, “hacktivists”, nation states, and others.
While we have not experienced any such material system failure, accident or security breach to date, if such an event were to occur and cause interruptions in our operations, it could result in a material disruption of our development programs and our business operations, whether due to a loss of our trade secrets or other proprietary information or other similar disruptions. For example, the loss of clinical trial data from completed or future clinical trials could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data. To the extent that any disruption or security breach were to result in a loss of, or damage to, our data or applications, or inappropriate disclosure of confidential or proprietary information, we could incur civil or criminal penalties, be exposed to liabilities, our reputation or competitive position could be harmed and the further development and commercialization of our product candidates could be delayed.
Our business continuity and disaster recovery plans may not adequately protect us from a serious disaster.
Natural disasters could severely disrupt our operations or the operations of manufacturing facilities and have a material adverse effect on our business, financial condition, results of operations and prospects. If a natural disaster, power outage or other event occurred that prevented us from using all or a significant portion of our headquarters, that damaged critical infrastructure, such as manufacturing facilities, or that otherwise disrupted operations, it may be difficult or, in certain cases, impossible for us to continue our business for a substantial period of time. The disaster recovery and business continuity plans that we have in place currently are limited and may not prove adequate in the event of a serious disaster or similar event. Substantially all of our current supply of LP-10 and LP-310 are located at our Facility. We are in the early stages of investigating the construction of an additional manufacturing facility and establishing a relationship with a third-party contract manufacturer as a back-up supplier for the commercial supply of our products, if necessary, but there is no assurance that we will establish such a relationship in a timely manner, on acceptable terms, or at all. We may incur substantial expenses as a result of the limited nature of our disaster recovery and business continuity plans, which could have a material adverse effect on our business, financial condition, results of operations and prospects.
Product liability lawsuits against us could cause us to incur substantial liabilities and to limit commercialization of any products that we may develop.
Our business exposes us to significant potential product liability risks that are inherent in the development, manufacturing, marketing and sale of human device and drug products. Product liability claims could delay or prevent completion of its development programs, clinical or otherwise. If we succeed in marketing and selling products, such claims could result in a recall of any products or a limitation or other change in the indications for which they may be used. If we cannot successfully defend ourselves against claims that our product candidates or drugs caused injuries, we will incur substantial liabilities. Regardless of merit or eventual outcome, liability claims may result in:
| ● | decreased demand for any product candidates or drugs that we may develop; |
| ● | injury to our reputation and significant negative media attention; |
| ● | withdrawal of clinical trial participants; |
| ● | significant costs to defend the related litigation; |
| ● | substantial monetary awards paid to trial participants or patients; |
| ● | loss of revenue; |
| ● | reduced resources of our management to pursue our business strategy; and |
| ● | the inability to commercialize any products that we may develop. |
Risks Related to Our Intellectual Property
If we are unable to obtain and maintain adequate U.S. and foreign patent protection for our product candidates, including LP-10, LP-310, LP-410, LP-50 and any future product candidates that we may develop, and/or our Platform, or if the scope of the patent protection obtained is not sufficiently broad, our competitors could develop and commercialize products and technologies similar or identical to ours, and our ability to successfully commercialize such products and technologies may be adversely affected.
Our success depends, in large part, on our ability to obtain and maintain patent protection in the United States and other countries with respect to LP-10, additional product candidates in our product pipeline, current and future innovations related to our Platform, and our institutional knowledge. The patent prosecution process is expensive, time-consuming and complex; we may not be able to file, prosecute, maintain, and/or enforce all necessary or desirable patent applications and issued patents at a reasonable cost or in a timely manner. We currently have the 278 Patent, the 725 Patent, the Australia Patent, the Canadian Patent, and the European Patent covering aspects of our Platform technology and its uses in delivering hydrophobic therapeutic, prophylactic or diagnostic agents to body cavities, as well as methods of making formulations for delivering such hydrophobic agents. We also have a corresponding patent application pending in the U.S. (U.S.S.N. 18/924,830). The Australian Patent, the Canadian Patent, and the European Patent each expire on October 22, 2034. We also have a pending U.S. patent application on an improvement to the technology. Such patents may be extendable for regulatory delay, but there is no guarantee that they will be extended. The Company may also be able to rely on market data exclusivity for the Company’s products, but there is no guarantee the Company will be able to do so.
It is possible that none of our pending patent applications will result in issued patents in a timely fashion or at all, and even if we are granted the patents that we are currently pursuing in foreign jurisdictions, the patents may not be issued in a form that will provide us with the full scope of protection that we desire, they may not prevent competitors or other third parties from competing with us, and/or they may not otherwise provide us with a competitive advantage. Our competitors, or other third parties, may be able to circumvent our patents by developing similar or alternative technologies or products in a non-infringing manner, or through the use of post-issuance legal or administrative proceedings challenging the validity or scope of our patents. For example, there is no assurance that the 278 Patent or the 725 Patent, or any other patent that we are granted, will prevent third parties from developing competing technologies. Moreover, our patent estate, including the 278 Patent and the 725 Patent, may not preclude third parties from having or obtaining intellectual property rights that could interfere with our freedom to use our Platform. Even assuming patents issue from our pending and future patent applications, changes in either the patent laws or interpretation of the patent laws in the United States and foreign jurisdictions may diminish the value of our patents or narrow their scope of protection.
We will not be able to protect our intellectual property rights in certain parts of the world.
Filing, prosecuting and defending patents on each and every one of our product candidates, current and future innovations related to our Platform, and our institutional knowledge in all countries throughout the world would be prohibitively expensive, and intellectual property rights in some countries outside the United States may differ in scope from those eventually granted in the United States. Thus, in some cases, we will not have the opportunity to obtain patent protection for certain technologies in some jurisdictions outside the United States. In addition, the laws of some foreign countries do not protect intellectual property rights to the same extent as federal and state laws in the United States. Consequently, we will not be able to prevent third parties from utilizing our inventions in certain countries outside the United States, even in jurisdictions where we do pursue patent protection. Competitors may use our technologies in jurisdictions where we have not pursued and obtained patent protection to develop their own products and, further, may export otherwise infringing products to territories where we have patent protection, but enforcement is not as strong as that in the United States. These products may compete with our product candidates, and our patents or other intellectual property rights will not be effective or sufficient to prevent them from competing.
Many companies have encountered significant problems in protecting and defending intellectual property rights in foreign jurisdictions. The legal systems of certain countries, particularly certain developing countries, do not favor the enforcement of patents, trade secrets and other intellectual property protection, particularly those relating to biotechnology products. Such challenges in enforcing rights in these countries could make it difficult for us to stop the infringement of our patents, if pursued and obtained, or marketing of competing products in violation of our proprietary rights generally. Proceedings to enforce our current and future patent rights in foreign jurisdictions could result in substantial costs and may divert our efforts and attention from other aspects of our business; could put our patents at risk of being invalidated or interpreted narrowly; could put any future patent applications, including continuation and divisional applications, at risk of not issuing; and could provoke third parties to assert claims against us. We may not prevail in any lawsuits that we initiate, and the damages or other remedies awarded, if any, may not be commercially meaningful. Accordingly, our efforts to enforce any intellectual property rights in certain parts of the world stemming from intellectual property that we develop will be inadequate to obtain a significant commercial advantage in these foreign jurisdictions.
Third parties may initiate legal proceedings alleging that we are infringing their intellectual property rights, the outcome of which would be uncertain and could have a material adverse effect on the success of our business.
Our commercial success depends upon our ability (and the ability of any potential future collaborators) to develop, manufacture, market and sell our product candidates, and to freely use our proprietary technologies (e.g., without infringing the rights and intellectual property of others). Many companies and institutions have filed, and continue to file, patent applications related to various aspects of drug delivery therapy. In some instances, patent applications or patents may be abandoned or allowed to lapse, resulting in partial or complete loss of patent rights in a relevant jurisdiction. The biotechnology and pharmaceutical industries are characterized by extensive and complex litigation regarding patents and other intellectual property rights. We may in the future become party to, or be threatened with, adversarial proceedings or litigation regarding intellectual property rights with respect to LP-10, LP-310 or any other product candidate, or related technologies, including, for example, derivation proceedings, post grant review challenges, and inter partes review before the USPTO. For example, a third party may bring an inter partes review challenging our patents and any future patent that may be granted to us. Our competitors or other third parties may assert infringement claims against us, alleging that our therapeutics, manufacturing methods, formulations or administration methods are covered by their patents. Moreover, we may face patent infringement claims from non-practicing entities that have no relevant product revenue, and against whom our patent portfolio may therefore have no deterrent effect. Further, while the existing patents may be extendable for regulatory delay, there is no guarantee that they will be extended. The Company may also be able to rely on market data exclusivity for the Company’s products, but there is no guarantee the Company will be able to do so.
Patent and other types of intellectual property litigation can involve complex factual and legal questions, and their outcomes are uncertain. If we are found, or believe there is a risk that we may be found, to infringe a third party’s valid and enforceable intellectual property rights, we could be required (or may choose) to obtain a license from such a third party to continue developing, manufacturing and marketing our technologies. However, we may not be able to obtain any required license on commercially reasonable terms, if at all. Even if we were able to obtain a license, it could be non-exclusive, thereby giving our competitors and other third parties access to the same technologies licensed to us, and further, it could require us to make substantial licensing and royalty payments. We could be forced, including by court order, to cease developing, manufacturing and commercializing the infringing technologies, including LP-10 or LP-310. We also could be found liable for monetary damages, including treble damages and attorneys’ fees, if we are found to have willfully infringed a patent or other intellectual property right. A finding of infringement could prevent us from manufacturing and commercializing our technologies, including LP-10 or LP-310, or force us to cease some or all our business operations. Claims that we have misappropriated the confidential information or trade secrets of third parties could have a similar negative impact on our business, financial condition, results of operations and prospects.
Intellectual property litigation could cause us to spend substantial resources and distract our personnel from their normal responsibilities.
Litigation or other legal proceedings relating to intellectual property claims, with or without merit, is unpredictable and generally expensive and time consuming. Competitors may infringe our current or future patents, should such patents issue, or we may be required to defend against claims of infringement or other unauthorized use of intellectual property. Even if resolved in our favor, litigation or other legal proceedings relating to intellectual property claims may cause us to incur significant expenses and could distract our scientific and management personnel from their normal responsibilities. Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation. In addition, there could be public announcements of the results of hearings, motions, or other interim proceedings or developments, and if securities analysts or investors perceive these results to be negative, it could have a substantial adverse effect on the price of our shares of common stock, par value $0.0001 per share (“Common Stock”). Such litigation or proceedings could substantially increase our operating losses and reduce the resources available for development activities or any future sales, marketing, or distribution activities.
We may not have sufficient financial or other resources to adequately conduct such litigation or proceedings. Some of our competitors may be able to sustain the costs of such litigation or proceedings more effectively than we can because of their greater financial resources. Accordingly, despite our efforts, we may not be able to prevent third parties from infringing, misappropriating, or successfully challenging our intellectual property rights. Uncertainties resulting from the initiation and continuation of patent litigation or other proceedings could have a material adverse effect on our ability to compete in the marketplace.
Changes in U.S. patent law could diminish the value of patents in general, thereby impairing our ability to protect our product candidates.
Patent reform legislation could increase the uncertainties and costs surrounding the prosecution of patent applications and the enforcement or defense of issued patents. As patent reform legislation can inject serious uncertainty into the patent prosecution and litigation processes, it is not clear what impact future patent reform legislation will have on the operation of our business. However, such future legislation, and its implementation, could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of any issued patents, all of which could have a material adverse effect on our business, financial condition, results of operations and prospects.
Moreover, the patent positions of companies engaged in the development and commercialization of biologics and pharmaceuticals are particularly uncertain. We cannot assure you that our efforts to seek patent protection for our technology and product candidates will not be negatively impacted by the future court decisions or changes in guidance or procedures issued by the USPTO. These decisions, and any guidance issued by the USPTO (or changes thereto), could have a material adverse effect on our existing patent portfolio and our ability to protect and enforce our intellectual property rights in the future.
Intellectual property rights and regulatory exclusivity rights do not necessarily address all potential threats.
The degree of future protection afforded by our intellectual property rights is uncertain because intellectual property rights have limitations and may not adequately protect our business or permit us to maintain our competitive advantage. For example:
| ● | others may be able to make products that are similar to, or competitive with, our product candidates but that are not covered by the claims of our current patents or of patents that we may own or license in the future; |
| ● | we, or any future license partners or collaborators, might not have been the first to file patent applications covering certain aspects of the concerned technologies; |
| ● | others may independently develop similar or alternative technologies, or duplicate any of our technologies, potentially without falling within the scope of our current or future issued claims, thus not infringing our intellectual property rights; |
| ● | it is possible that our filed or future patent applications will not lead to issued patents; |
| ● | issued patents to which we currently hold rights or to which we may hold rights in the future may be held invalid or unenforceable, including as a result of legal or administrative challenges by our competitors; |
| ● | others may have access to any future intellectual property rights licensed to us on a non-exclusive basis; |
| ● | our competitors might conduct research and development activities in countries where we do not have or pursue patent rights, and then use the information learned from such activities to develop competitive products for sale in our major commercial markets; |
| ● | we may not develop additional proprietary technologies that are patentable; |
| ● | the patents or other intellectual property rights of others may have an adverse effect on our business; and |
| ● | we may choose not to file a patent application covering certain of our trade secrets or know-how, and a third party may subsequently file a patent covering such intellectual property. |
Should any of these events occur, they could significantly harm our business, financial condition, results of operations and prospects.
If we are unable to protect the confidentiality of our trade secrets, our business and competitive position would be harmed. Our reliance on third parties requires us to share our trade secrets, which increases the possibility that a competitor will discover them or that our trade secrets will be misappropriated or disclosed.
In addition to patent protection, we also rely on the protection of trade secrets, know-how, confidential and proprietary information and regulatory exclusion. However, trade secrets are difficult to protect. To maintain the confidentiality of trade secrets and proprietary information, we rely in part on confidentiality agreements with our employees, consultants, outside scientific collaborators, sponsored researchers, and/or other advisors, and inventions agreements with employees, consultants, and advisors, to protect our trade secrets and other proprietary information. These agreements require that all confidential information developed by the individual or made known to the individual by us during the course of the individual’s relationship with us be kept confidential and not disclosed to third parties.
Our agreements with employees and consultants also provide that inventions conceived by the individual in the course of rendering services to us will be our exclusive property. Despite these efforts, we cannot provide any assurances that all such agreements have been duly executed, and these agreements may not effectively prevent disclosure of confidential information and may not provide an adequate remedy in the event of unauthorized disclosure of confidential information. In addition, others may independently discover our trade secrets and proprietary information.
In the event of unauthorized use or disclosure of trade secrets or proprietary information, these agreements, even if obtained, may not provide sufficient protection for our trade secrets or other confidential information. Further, to the extent that our employees, consultants or contractors use technology or know-how owned by others in their work for the Company, disputes may arise as to the rights in related inventions. This can be of particular concern with respect to university collaborators with us, who typically have preexisting obligations to their universities to assign intellectual property rights, which university rights generally are superior to assignment rights that we might receive from such individuals. The disclosure of our trade secrets would impair our competitive position and could harm our business.
Enforcing a claim that a party illegally disclosed or misappropriated a trade secret is difficult, expensive and time-consuming, and the outcome is unpredictable. In addition, some courts inside and outside the United States are less willing or unwilling to protect trade secrets. Moreover, third parties may still obtain this information or may come upon this or similar information independently, and we would have no right to prevent them from using that technology or information to compete with us. Trade secrets will over time be disseminated within the industry through independent development, the publication of journal articles, and the movement of personnel skilled in the art from company to company or academic to industry scientific positions. Though our agreements with third parties typically restrict the ability of our advisors, employees, collaborators, licensors, suppliers, third-party contractors, and/or consultants to publish data potentially relating to our trade secrets, our agreements may contain certain limited publication rights. If any of our trade secrets were to be lawfully obtained or independently developed by a competitor, we would have no right to prevent such competitor from using that technology or information to compete with us, which could harm our competitive position. Because from time to time we expect to rely on third parties in the development, manufacture, and distribution of our products and provision of our services, we must, at times, share trade secrets with them. Despite employing the contractual and other security precautions described above, the need to share trade secrets increases the risk that such trade secrets become known by our competitors, are inadvertently incorporated into the technology of others, or are disclosed or used in violation of these agreements. If any of these events occurs or if we otherwise lose protection for our trade secrets, the value of this information may be greatly reduced and our competitive position would be harmed. If we do not apply for patent protection prior to such publication or if we cannot otherwise maintain the confidentiality of our proprietary technology and other confidential information, then our ability to obtain patent protection or to protect our trade secret information may be jeopardized.
Risks Related to our Common Stock
The market price and trading volume of our Common Stock may experience rapid and substantial volatility, which could cause purchasers of our Common Stock to incur substantial losses.
The market price of our Common Stock may fluctuate dramatically, and may decline rapidly, regardless of any developments in our business. Overall, there are various factors, many of which are beyond our control, that could negatively affect the market price of our Common Stock or result in fluctuations in the price or trading volume of our Common Stock, including:
| ● | actual or anticipated variations in our annual or quarterly results of operations, including our earnings estimates and whether we meet market expectations with regard to our earnings; |
| ● | our current inability to pay dividends or other distributions; |
| ● | publication of research reports by analysts or others about us or the industry in which we operate, including the pharmaceutical or biotechnology industry which may be unfavorable, inaccurate, inconsistent or not disseminated on a regular basis; |
| ● | changes in market valuations of similar companies; |
| ● | market reaction to any additional equity, debt or other securities that we may issue in the future, and which may or may not dilute the holdings of our existing stockholders; |
| ● | additions or departures of key personnel; |
| ● | actions by institutional or significant stockholders; |
| ● | short interest in our Common Stock or our other securities and the market response to such short interest; |
| ● | the dramatic increase in the number of individual holders of our Common Stock and their participation in social media platforms targeted at speculative investing; |
| ● | speculation in the press or investment community about our company or industries in which we operate; |
| ● | strategic actions by us or our competitors, such as acquisitions or other investments; |
| ● | legislative, administrative, regulatory or other actions affecting our business, our industry, including positions taken by the FDA; |
| ● | investigations, proceedings, or litigation that involve or affect us; and |
| ● | the occurrence of any of the other risk factors included in this Report; and |
| ● | general market and economic conditions. |
Our Common Stock is currently listed on the Nasdaq Capital Market. We have been notified by Nasdaq of our failure to comply with certain continued listing requirements and, if we are unable to regain compliance with all applicable continued listing requirements and standards of Nasdaq, our Common Stock could be delisted from the Nasdaq Capital Market.
Our Common Stock is currently listed on the Nasdaq Capital Market. In order to maintain that listing, we must satisfy minimum financial and other continued listing requirements and standards, including those regarding director independence and independent committee requirements, minimum stockholders’ equity, minimum share price, and certain corporate governance requirements.
As disclosed in our Current Report on Form 8-K filed with the SEC on April 19, 2024, we received a written notification from Nasdaq’s Listing Qualifications Department (the “Staff”) on April 17, 2024 notifying us that we were not in compliance with the minimum bid price requirement for continued listing on the Nasdaq Capital Market, as set forth under Nasdaq Listing Rule 5550(a)(2) (the “Minimum Bid Price Requirement”), because the closing bid price of our Common Stock was below $1.00 per share for the previous thirty (30) consecutive business days. As disclosed in our Current Report on Form 8-K filed with the SEC on August 23, 2024, we received a letter from the Staff on August 21, 2024 stating that we were not in compliance with Nasdaq Listing Rule 5550(b)(1), which requires companies listed on the Nasdaq Capital Market to maintain a minimum of $2,500,000 in stockholders’ equity for continued listing (the “Stockholders’ Equity Requirement”). We reported stockholders’ equity of $1,703,798 in our Quarterly Report on Form 10-Q for the quarter ended June 30, 2024, and, as a result, we were not in compliance with such requirement.
As disclosed in our Current Report on Form 8-K filed with the SEC on October 18, 2024, the Staff notified us on October 16, 2024 that it would delist the Common Stock from the Nasdaq Capital Market, and in response, we timely requested an appeal of such notice to the Nasdaq Hearings Panel (the “Panel”). We received a written communication from the Staff advising us that we had regained compliance with the Minimum Bid Price Requirement as of November 26, 2024, and we presented in front of the Panel at a hearing on December 12, 2024. As disclosed in the Current Report on Form 8-K filed by the Company with the SEC on January 13, 2025, the Panel notified us via letter on January 10, 2025 (the “January Letter”) that it had granted the Company’s request for continued listing on the Nasdaq Capital Market, subject to the Company demonstrating compliance with the Stockholders’ Equity Requirement, including the achievement of interim milestones as follows: (i) on or before April 14, 2025, the Company must file a public disclosure describing any transactions undertaken by the Company to increase its equity and providing an indication of its equity following those transactions; and (ii) on or before April 14, 2025, the Company must provide the Staff with an update on its fundraising plans, updated income projections for the next 12 months, with all underlying assumptions clearly stated, and a description of how the Company intends to achieve, if necessary, and maintain compliance with the Minimum Bid Price Requirement.
There can be no assurances that we will be able to comply with all of the obligations placed on us by the Panel pursuant to the January Letter, and, assuming that we are able to comply with such obligations, that we will be able to continue to comply with all applicable Nasdaq listing requirements now or in the future, including the Stockholders’ Equity Requirement or the Minimum Bid Price Requirement. If we fail to meet all of the conditions listed in the January Letter, our Common Stock may be delisted from the Nasdaq Capital Market. Additionally, assuming that we are able to comply with all such obligations, if we fail to comply with all applicable Nasdaq continued listing standards now or in the future, our Common Stock may be subject to delisting from the Nasdaq Capital Market.
In the event that our Common Stock is delisted from the Nasdaq Capital Market, as a result of our failure to comply with the obligations in the January Letter, our failure to comply with the Stockholders’ Equity Requirement or due to our failure to continue to comply with any other requirement for continued listing on the Nasdaq Capital Market, and the Common Stock is not eligible for listing on another exchange, trading in the shares of our Common Stock could be conducted in the over-the-counter market or on an electronic bulletin board established for unlisted securities such as the Pink Market or another over-the-counter market operated by the OTC Markets Group Inc. In such event, it could become more difficult to dispose of, or obtain accurate price quotations for, our Common Stock, and it would likely be more difficult to obtain coverage by securities analysts and the news media, which could cause the price of our Common Stock to decline further. Also, it may be difficult for us to raise additional capital if we are not listed on a national exchange.
If there are substantial sales of shares of our Common Stock, the price of our Common Stock could decline.
As of March 26, 2025, our directors, executive officers and principal stockholders, and their respective affiliates, beneficially own 466,808 shares of Common Stock, or approximately 17% of our outstanding shares of Common Stock. If these stockholders sell, or indicate an intent to sell, substantial amounts of our Common Stock in the public market after the expiration of such lock-up period, the trading price of our Common Stock could decline significantly.
Because certain of our stockholders control a significant number of shares of our Common Stock, they may have effective control over actions requiring stockholder approval.
As of March 26, 2025, our directors, executive officers and principal stockholders, and their respective affiliates, beneficially own 466,808 shares of Common Stock, or approximately 17% of our outstanding shares of Common Stock. As a result, these stockholders, acting together, would have the ability to control the outcome of matters submitted to our stockholders for approval, including the election of directors and any merger, consolidation or sale of all or substantially all of our assets. In addition, these stockholders, acting together, would have the ability to control the management and affairs of our Company. Accordingly, this concentration of ownership might harm the market price of our Common Stock by:
| ● | delaying, deferring or preventing a change in corporate control; |
| ● | impeding a merger, consolidation, takeover or other business combination involving us; or |
| ● | discouraging a potential acquirer from making a tender offer or otherwise attempting to obtain control of us. |
Provisions in our Second Amended and Restated Certificate of Incorporation, as amended (our “Certificate of Incorporation”), our amended and restated by-laws (our “Bylaws”) and under Delaware law could make an acquisition of us, which may be beneficial to our stockholders, more difficult and may prevent attempts by our stockholders to replace or remove our current management.
Provisions in our Certificate of Incorporation and our Bylaws, as currently in effect, may discourage, delay or prevent a merger, acquisition or other change in control of us that stockholders may consider favorable, including transactions in which you might otherwise receive a premium for your shares of Common Stock. These provisions also could limit the price that investors might be willing to pay in the future for shares of our Common Stock, thereby depressing the market price of our Common Stock. In addition, because our board of directors is responsible for appointing the members of our management team, these provisions may frustrate or prevent any attempts by our stockholders to replace or remove our current management by making it more difficult for stockholders to replace members of our board of directors. Among other things, these provisions:
| ● | allow the authorized number of our directors to be changed only by resolution of our board of directors; |
| ● | establish advance notice requirements for stockholder proposals that can be acted on at stockholder meetings and nominations to our board of directors; |
| ● | require that stockholder actions must be effected at a duly called stockholder meeting and prohibit actions by our stockholders by written consent; |
| ● | limit who may call special stockholder meetings; |
| ● | authorize our board of directors to issue capital stock without stockholder approval; and |
| ● | require the approval of a majority of the directors to amend or repeal certain provisions of our Bylaws. |
Moreover, because we are incorporated in Delaware, we are governed by the provisions of Section 203 of the General Corporation Law of the State of Delaware (the “DGCL”), which prohibits a person who owns in excess of 15% of our outstanding voting stock from merging or combining with us for a period of three years after the date of the transaction in which the person acquired in excess of 15% of our outstanding voting stock, unless the merger or combination is approved in a prescribed manner.
Our Certificate of Incorporation and our Bylaws provide that the Court of Chancery of the State of Delaware will be the exclusive forum for substantially all disputes between us and our stockholders, which could limit our stockholders’ ability to obtain a favorable judicial forum for disputes with us or our directors, officers or employees.
Our Certificate of Incorporation and our Bylaws provide that the Court of Chancery of the State of Delaware is the exclusive forum for any derivative action or proceeding brought on our behalf; any action asserting a breach of fiduciary duty; any action asserting a claim against us arising pursuant to the DGCL, our Certificate of Incorporation or Bylaws; or any action asserting a claim against us that is governed by the internal affairs doctrine. Notwithstanding the foregoing, the exclusive forum provision does not apply to suits brought to enforce any liability or duty created by the Securities Act, the Exchange Act or any other claim for which the federal courts have exclusive or concurrent jurisdiction. Section 27 of the Exchange Act creates exclusive federal jurisdiction over all suits brought to enforce any duty or liability created by the Exchange Act of the rules and regulations thereunder, and Section 22 of the Securities Act creates concurrent jurisdiction for federal and state courts over all suits brought to enforce any duty or liability created by the Securities Act or the rules and regulations thereunder, and notwithstanding the provisions of our Certificate of Incorporation and our Bylaws, compliance with the federal securities laws and the rules and regulations thereunder may not be waived by our investors. The choice of forum provision may limit a stockholder’s ability to bring a claim in a judicial forum that it finds favorable for disputes with us or our directors, officers or other employees, which may discourage such lawsuits against us and our directors, officers and other employees. Alternatively, if a court were to find the choice of forum provision contained in our Certificate of Incorporation and our Bylaws to be inapplicable or unenforceable in an action, we may incur additional costs associated with resolving such action in other jurisdictions, which could materially and adversely affect our business, financial condition, and results of operation.
Raising additional capital may cause dilution to our existing stockholders, restrict our operations or require us to relinquish rights to our technologies, including LP-10 and LP-310.
We may seek additional capital through a combination of public and private equity offerings, debt financings, collaborations and licensing arrangements. To the extent that we raise additional capital through the sale of equity or debt securities, the ownership interest of our stockholders will be diluted, and the terms may include liquidation or other preferences that adversely affect your rights as a stockholder. The incurrence of indebtedness would result in increased fixed payment obligations and could involve restrictive covenants, such as limitations on our ability to incur additional debt, limitations on our ability to acquire or license intellectual property rights and other operating restrictions that could adversely impact our ability to conduct our business. If we raise additional funds through strategic partnerships and alliances and licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies or any of our product candidates or grant licenses on terms unfavorable to us.
Because we do not anticipate paying any cash dividends on our capital stock in the foreseeable future, capital appreciation, if any, will be your sole source of gain.
We have never declared or paid cash dividends on our capital stock. We currently intend to retain all of our future earnings, if any, to finance the growth and development of our business. In addition, the terms of any future debt agreements may preclude us from paying dividends. As a result, capital appreciation, if any, of our Common Stock will be your sole source of gain for the foreseeable future.
In the event that our Common Stock is delisted from the Nasdaq Capital Market, U.S. broker-dealers may be discouraged from effecting transactions in shares of our Common Stock because they may be considered penny stocks and thus be subject to the penny stock rules.
The SEC has adopted a number of rules to regulate “penny stock” that restricts transactions involving stock which is deemed to be penny stock. Such rules include Rules 3a51-1, 15g-1, 15g-2, 15g-3, 15g-4, 15g-5, 15g-6, 15g-7, and 15g-9 under the Exchange Act. These rules may have the effect of reducing the liquidity of penny stocks. “Penny stocks” generally are equity securities with a price of less than $5.00 per share (other than securities registered on certain national securities exchanges or quoted on the Nasdaq Capital Market if current price and volume information with respect to transactions in such securities is provided by the exchange or system). Our shares of Common Stock may in the future constitute, “penny stock” within the meaning of the rules. The additional sales practice and disclosure requirements imposed upon U.S. broker-dealers for sales of penny stocks may discourage such broker-dealers from effecting transactions in shares of our Common Stock, which could severely limit the market liquidity of such shares and impede their sale in the secondary market.
A U.S. broker-dealer selling penny stock to anyone other than an established customer or “accredited investor” (generally, an individual with a net worth in excess of $1,000,000 or an annual income exceeding $200,000, or $300,000 together with his or her spouse) must make a special suitability determination for the purchaser and must receive the purchaser’s written consent to the transaction prior to sale, unless the broker-dealer or the transaction is otherwise exempt. In addition, the “penny stock” regulations require the U.S. broker-dealer to deliver, prior to any transaction involving a “penny stock”, a disclosure schedule prepared in accordance with SEC standards relating to the “penny stock” market, unless the broker-dealer or the transaction is otherwise exempt. A U.S. broker-dealer is also required to disclose commissions payable to the U.S. broker-dealer and the registered representative and current quotations for the securities. Finally, a U.S. broker-dealer is required to submit monthly statements disclosing recent price information with respect to the “penny stock” held in a customer’s account and information with respect to the limited market in “penny stocks”.
Stockholders should be aware that, according to the SEC, the market for “penny stocks” has suffered in recent years from patterns of fraud and abuse. Such patterns include: (i) control of the market for the security by one or a few broker-dealers that are often related to the promoter or issuer; (ii) manipulation of prices through prearranged matching of purchases and sales and false and misleading press releases; (iii) “boiler room” practices involving high-pressure sales tactics and unrealistic price projections by inexperienced sales persons; (iv) excessive and undisclosed bid-ask differentials and markups by selling broker-dealers; and (v) the wholesale dumping of the same securities by promoters and broker-dealers after prices have been manipulated to a desired level, resulting in investor losses. Our management is aware of the abuses that have occurred historically in the penny stock market. Although we do not expect to be in a position to dictate the behavior of the market or of broker-dealers who participate in the market, management will strive within the confines of practical limitations to prevent the described patterns from being established with respect to our securities.
General Risk Factors
Market and economic conditions may negatively impact our business, financial condition and share price.
Concerns over medical epidemics, energy costs, geopolitical issues, the U.S. mortgage market and a deteriorating real estate market, unstable global credit markets and financial conditions, and volatile oil prices have led to periods of significant economic instability, diminished liquidity and credit availability, declines in consumer confidence and discretionary spending, diminished expectations for the global economy and expectations of slower global economic growth, increased unemployment rates, and increased credit defaults in recent years. Our general business strategy may be adversely affected by any such economic downturns volatile business environments and continued unstable or unpredictable economic and market conditions. If these conditions continue to deteriorate or do not improve, it may make any necessary debt or equity financing more difficult to complete, more costly, and more dilutive. Failure to secure any necessary financing in a timely manner and on favorable terms could have a material adverse effect on our growth strategy, financial performance, and share price and could require us to delay or abandon development or commercialization plans.
If securities analysts do not publish research or reports about our business or if they publish negative evaluations of our Common Stock, the price of our Common Stock could decline.
The trading market for our Common Stock may rely, in part, on the research and reports that industry or financial analysts publish about us or our business. If securities analysts do not commence coverage of us, the trading price of our Common Stock could decrease. Additionally, if one or more of the analysts covering our business downgrade their evaluations of our Common Stock, the price of our Common Stock could decline. If one or more of these analysts cease to cover our Common Stock, we could lose visibility in the market for our Common Stock, which in turn could cause our Common Stock price to decline.
We are an “emerging growth company” and the reduced disclosure requirements applicable to emerging growth companies may make our securities less attractive to investors.
We are an “emerging growth company,” as defined in the JOBS Act, and we may take advantage of certain exemptions and relief from various reporting requirements that are applicable to other public companies that are not “emerging growth companies.” In particular, while we are an “emerging growth company”: (i) we will not be required to comply with the auditor attestation requirements of Section 404(b) of the Sarbanes-Oxley Act; (ii) we will be exempt from any rules that may be adopted by the Public Company Accounting Oversight Board requiring mandatory audit firm rotations or a supplement to the auditor’s report on financial statements; (iii) we will be subject to reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements; and (iv) we will not be required to hold nonbinding advisory votes on executive compensation or stockholder approval of any golden parachute payments not previously approved. We have taken advantage of reduced reporting burdens in this Report.
In addition, under the JOBS Act, emerging growth companies can take advantage of an extended transition period and delay adopting new or revised accounting standards until such time as those standards apply to private companies. We have elected to use this extended transition period and, as a result, we will adopt new or revised accounting standards on the relevant dates on which adoption of such standards is required for private companies. If we were to subsequently elect instead to comply with public company effective dates, such election would be irrevocable pursuant to the JOBS Act. Investors may find our securities less attractive if we rely on the exemptions and relief granted by the JOBS Act. If some investors find our securities less attractive as a result, there may be a less active trading market for our Common Stock and the per share price of our Common Stock price may decline or become more volatile.
We will incur increased costs as a result of operating as a smaller reporting public company, and our management will be required to devote substantial time to new compliance initiatives.
As a smaller reporting public company, and particularly after we are no longer an emerging growth company, we will incur significant legal, accounting and other expenses that we did not incur as a private company. In addition, the Sarbanes-Oxley Act and rules subsequently implemented by the SEC and Nasdaq have imposed various requirements on public companies, including establishment and maintenance of effective disclosure and financial controls and corporate governance practices. Our management and other personnel will need to devote a substantial amount of time to these compliance initiatives. Moreover, these rules and regulations will increase our legal and financial compliance costs and will make some activities more time consuming and costly. For example, we expect that these rules and regulations may make it more difficult and more expensive for us to obtain director and officer liability insurance.
Pursuant to Section 404 of the Sarbanes-Oxley Act (“Section 404”), we will be required to furnish a report by our management on our internal control over financial reporting, including an attestation report on internal control over financial reporting issued by our independent registered public accounting firm. However, while we remain an emerging growth company, we will not be required to include an attestation report on internal control over financial reporting issued by our independent registered public accounting firm. To achieve compliance with Section 404 within the prescribed period, we will be engaged in a process to document and evaluate our internal control over financial reporting, which is both costly and challenging. In this regard, we will need to continue to dedicate internal resources, potentially engage outside consultants and adopt a detailed work plan to assess and document the adequacy of internal control over financial reporting, continue steps to improve control processes as appropriate, validate through testing that controls are functioning as documented and implement a continuous reporting and improvement process for internal control over financial reporting. Despite our efforts, there is a risk that neither we nor our independent registered public accounting firm will be able to conclude within the prescribed timeframe that our internal control over financial reporting is effective as required by Section 404. This could result in an adverse reaction in the financial markets due to a loss of confidence in the reliability of our financial statements.
Our business could be adversely impacted if there are deficiencies in our disclosure controls and procedures or our internal control over financial reporting.
The design and effectiveness of our disclosure controls and procedures and our internal control over financial reporting may not prevent all errors, misstatements or misrepresentations. There can be no guarantee that our disclosure controls and procedures and internal control over financial reporting will be effective in accomplishing all control objectives all of the time. Deficiencies, including any material weaknesses, in our disclosure controls and procedures or internal control over financial reporting could result in misstatements of our results of operations or our financial statements or could otherwise materially and adversely affect our business, reputation, results of operations, financial condition or liquidity.
Item 1B. Unresolved Staff Comments.
None.
Item 1C. Cybersecurity
Risk management and strategy
We are an R&D stage pharmaceutical company, with no commercial operations or revenue streams. Since our IPO, our sole business activity has been ongoing research into our drug therapies. Therefore, we do not consider that we face significant cybersecurity risk and have not adopted a formal cybersecurity risk management program or process for assessing cybersecurity risk currently. We assess material risks from cybersecurity threats on an ongoing basis, including any potential unauthorized occurrence on or conducted through our information systems that may result in adverse effects on the confidentiality, integrity, or availability of our information systems or any information residing therein. As our Company grows, we plan to develop a more robust and detailed strategy for cybersecurity in alignment with nationally accepted standards. We have not encountered cybersecurity challenges that have materially impaired our operations or financial standing. For additional information regarding risks from cybersecurity threats, please refer to Item 1A, “Risk Factors,” in this annual report on Form 10-K.
Governance
Our management and board of directors recognize the critical importance of maintaining the trust and confidence of our business partners and employees, including the importance of managing cybersecurity risks as part of our larger risk management program. While all of our personnel play a part in managing cybersecurity risks, one of the key functions of our board of directors is informed oversight of our risk management process, including risks from cybersecurity threats. Our board of directors is responsible for monitoring and assessing strategic risk exposure, and our executive officers are responsible for the day-to-day management of the material risks that we face. In general, we seek to address cybersecurity risks through a cross-functional approach that is focused on preserving the confidentiality, integrity, and availability of the information that we collect and store by identifying, preventing, and mitigating cybersecurity threats and effectively responding to cybersecurity incidents when they occur.
Item 2. Properties.
Our principal executive offices are located at 7800 Susquehanna Street, Suite 505, Pittsburgh, PA 15208, containing approximately 2,000 square feet of combined laboratory, office and warehouse space that we use in our research and development efforts. The lease for our principal executive offices has a five-year term that ends on June 30, 2025, and the lease provides us with an option to extend the term for an additional five years. We believe our executive offices, including our Facility, are adequate to meet our current needs, although we may seek to negotiate new leases or evaluate additional or alternate space for our operations. We believe appropriate alternative space will be readily available on commercially reasonable terms.
Item 3. Legal Proceedings
From time to time, we may be involved in legal proceedings arising in the ordinary course of our business. We are not presently a party to any legal proceedings that, in the opinion of our management, would have a material adverse effect on our business. Regardless of outcome, litigation can have an adverse impact on us due to defense and settlement costs, diversion of management resources, negative publicity and reputation harm, and other factors.
Item 4. Mine Safety Disclosures Item 5.
Not applicable.
PART II
Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities.
Market Information
Our Common Stock trades on the Nasdaq Capital Market under the symbol “LIPO.”
Holders of our Common Stock
As of March 26, 2025, there were 15 holders of record of our Common Stock. This number does not include shares of Common Stock held by brokerage clearing houses, depositories or others in unregistered form.
Dividends
We have never declared or paid cash dividends on our capital stock. We currently intend to retain all available funds and any future earnings for use in the operation of our business and do not anticipate paying any dividends on our Common Stock in the foreseeable future. Any future determination to declare dividends will be made at the discretion of our board of directors and will depend on our financial condition, operating results, capital requirements, general business conditions and other factors that our board of directors may deem relevant.
Securities Authorized for Issuance under Equity Compensation Plans
Reference is made to “Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters—Securities Authorized for Issuance under Equity Compensation Plans” for the information required by this item.
Recent Sales of Unregistered Securities
On February 5, 2024, we issued 24,509 shares of Common Stock in exchange for services rendered by a third party and on March 4, 2024, we issued 62,500 shares of Common Stock for the exercise of the same number of pre-funded warrants. Such shares of Common Stock were issued pursuant to the exemption from the registration requirements of the Securities Act provided in Section 4(a)(2) of the Securities Act and/or Regulation D promulgated thereunder.
Purchases of Equity Securities by the Issuer and Affiliated Purchasers
None.
Item 6. [Reserved]
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations.
You should read the following discussion and analysis of our financial condition and results of operations together with our financial statements and related notes appearing in this Report. Some of the information contained in this discussion and analysis or set forth elsewhere in this Report, including information with respect to our plans and strategy for our business and related financing, includes forward-looking statements that involve risks and uncertainties. As a result of many factors, including those factors set forth in the “Risk Factors” section of this Report, our actual results could differ materially from the results described in or implied by the forward-looking statements contained in the following discussion and analysis.
Overview
We are a clinical-stage biotechnology company focused on developing new drugs by reformulating the active agents in existing generic drugs and optimizing these reformulations for new applications. We believe that this strategy combines many of the cost efficiencies and risk abatements derived from using existing generic drugs with potential patent protections for our proprietary formulations; this strategy allows us to expedite, protect, and monetize our product candidates. Additionally, we maintain a therapeutic focus on diseases with significant, unaddressed morbidity and mortality where no approved drug therapy currently exists. We believe that this focus can potentially help reduce the cost, time and risk associated with obtaining marketing approval.
Consistent with our strategy, we are currently addressing two indications via development of our product candidates, LP-10 for HC, and LP-310 for OLP. HC is chronic, uncontrolled urinary blood loss that results from certain chemotherapies (such as alkylating agents) or pelvic radiation therapy (also called “radiation cystitis”). Many radiation cystitis patients experience severe morbidity (and in some cases, mortality), and currently, there is no therapy for their condition approved by the FDA, or, to our knowledge, any other regulatory body.
LP-310 employs a formulation similar to LP-10, for the treatment of OLP. OLP is a chronic, T-cell-mediated, autoimmune oral mucosal disease, and LP-310 contains tacrolimus which inhibits T-lymphocyte activation. Symptoms of OLP include painful burning sensations, bleeding and irritation with tooth brushing, painful, thickened patches on the tongue, and discomfort when speaking, chewing or swallowing. These symptoms frequently cause weight loss, nutritional deficiency, anxiety, depression, and scarring from erosive lesions. OLP can also be a precursor to cancer, predominately squamous cell carcinoma, with a malignant transformation rate of approximately one percent.
LP-10 is the development name of our reformulation of tacrolimus (an approved generic active agent) specifically optimized for topical deposition to the internal surface of the urinary bladder lumen using a proprietary drug delivery platform that we have developed and that we refer to as our metastable liposome drug delivery platform (our “Platform”). We are developing LP-10 and our Platform to be, to our knowledge, the first drug candidate and drug delivery technology that could be successful in treating cancer survivors who acquire HC.
LP-310 is the development name of our oral, liposomal formulation of tacrolimus (the same approved generic active agent in LP-10) specifically optimized for local delivery to oral mucosa. We believe that our approach of using metastable liposomal tacrolimus as a treatment for OLP is novel. To date, upon review of relevant FDA public data resources on approved drugs and biologics, we are not aware of any other liposomal products developed to treat such disease. In the fourth quarter of 2024, we announced the completion of dosing for the first cohort in its multi-center Phase 2a clinical trial of LP-310. No product-related serious adverse events were reported. Pharmacokinetic data demonstrated that whole blood tacrolimus levels in all patients were either undetectable or minimal, highlighting LP-310’s potential to deliver localized therapeutic effects while minimizing systemic exposure. Additionally, all patients tolerated LP-310 without significant adverse reactions. The trial is expected to be completed in the second quarter of 2025. The top line data from the first two dose cohorts of this trial has been selected for podium presentation at the 2025 American Association of Oral Medicine and European Association of Oral Medicine Joint Meeting that will be held in Las Vegas, NV on May 15, 2025.
Additionally, the Company is developing an oral, liposomal formulation of tacrolimus, LP-410, for the treatment of oral GVHD. GVHD is a clinical syndrome where donor-derived immunocompetent T cells react against patient tissues directly or through exaggerated inflammatory responses following HCT. GVHD remains a major cause of morbidity and mortality for patients who undergo HCT treatment, with chronic GVHD being the leading cause of nonmalignant fatality for such patients who receive such HCT treatment. Topical and local management of symptomatic oral GVHD can reduce oral symptoms that can interfere with oral function and quality of life and can reduce the need for more intensive immunosuppressive systemic therapies. However, there is currently no FDA approved local drug treatment of oral GVHD. On November 11, 2023, we received “orphan drug” designation from the FDA for LP-410 for oral GVHD. We received IND approval from the FDA for LP-410’s treatment of oral GVHD on March 5, 2024.
Lipella is also developing an intravesical formulation of immunoglobulins including checkpoint inhibitors, LP-50, for the treatment of NMIBC, offering the potential for increasing efficacy while minimizing systemic toxicity. Additional information regarding this preclinical program is included in the International Journal of Molecular Sciences 2024, 25(9), 4945, titled “Enhancing Therapeutic Efficacy and Safety of Immune Checkpoint Inhibition for Bladder Cancer: A Comparative Analysis of Injectable vs. Intravesical Administration,” as well as in US patent publication number 2024/0115503 titled “Intravesical Delivery of Hydrophilic Therapeutic Agents Using Liposomes.”
Our Platform includes proprietary drug delivery technologies optimized for use with epithelial tissues that coat lumenal surfaces, such as the colon, the various tissues lining the mouth and esophagus and the tissues lining the bladder and urethra. The Company has two issued patents in the United States that should exclude competitors from making, selling or using our LP-10 and LP-310 formulations in the United States until July 11, 2035. We also have issued patents in Australia, Canada, and Europe that do not expire until October 22, 2034. Corresponding patent applications are pending in the United States Patent Offices. We also have a pending United States patent application on an improvement to the technology. In some jurisdictions, such as the US, Europe, Canada, and some Asian countries, such patents may be extendable for regulatory delay. Market data exclusivity may also be available for the approved products.
Since our inception in 2005, we have focused primarily on business planning, progressing our lead product candidates, including progressing LP-10 through clinical development, raising capital, organizing and staffing our company.
On December 22, 2022, we completed our IPO and issued and sold 1,217,391 shares of Common Stock at a price to the public of $5.75 per share, which when accounting for the reverse split stock split that occurred on November 7, 2024, converts to 152,173 shares at a price of $46.00 per share. The aggregate net proceeds from the IPO were approximately $5.0 million after deducting underwriting discounts and commissions of approximately $630,000 and offering expenses of approximately $1.16 million.
Recent Developments of the Company
Offering of Series B Preferred Stock
On March 14, 2025, the Company completed a best efforts private placement offering of an aggregate of 72,000 shares of Series B non-voting convertible preferred stock, par value $0.0001 per share, of the Company (the “Series B Preferred Stock”), inclusive of 12,000 shares of Series B Preferred Stock pursuant to exercise of the placement agent’s over-allotment option (the “Over-allotment Option”) (the “Offering”), with Spartan Capital Securities, LLC (“Spartan”) providing placement agent and consulting services in connection therewith. The Series B Preferred Stock were sold to the investors for a purchase price of $100 per share, with such shares of Series B Preferred Stock convertible into an aggregate of 2,560,315 shares of Common Stock. The Company received net proceeds of $5,906,000 in connection with the Offering, after deducting placement agent fees and expenses.
In connection with the Offering, the Company and Spartan entered into that certain (i) placement agent agreement, dated December 5, 2024 (the “Placement Agent Agreement”) as amended by that certain amendment to consulting agreement and placement agent agreement, made as of December 10, 2024, between the Company and Spartan (the “Amendment”), as further amended by that certain second amendment to placement agent agreement, dated February 23, 2025, by and between the Company and Spartan (the “Placement Agent Agreement Amendment”) and (ii) consulting agreement and advisory agreement, made as of December 5, 2024 (the “Consulting Agreement”), as amended by the Amendment, as further amended by that certain second amendment to consulting agreement and advisory agreement, dated February 28, 2025, by and between the Company and Spartan (the “Consulting Agreement Amendment”), pursuant to which the Company paid Spartan an aggregate of $1,224,000 in placement agent and consulting fees and issued to Spartan and its designee (i) an aggregate of 840,000 shares of the Company’s Series C voting convertible preferred stock, par value $0.0001 per share (“Series C Preferred Stock”), and (ii) placement agent warrants to purchase up to 256,031 shares of Common Stock in connection with the Offering. Pursuant to the Placement Agent Agreement Amendment, Spartan and the Company agreed to, among other things, (i) remove restrictions on the Company’s ability to conduct at-the-market transactions and modify certain variable rate transaction provisions, (ii) modify the Company’s tail fee obligation to apply only to investors who were participants in the Offering and (iii) clarify the Company’s obligations with respect to filing a registration statement in connection with a potential subsequent “best efforts” offering of up to an additional $6,000,000 of shares of Series B Preferred Stock at the same terms offered to investors in the Offering (the “Mirror Offering”). Pursuant to the Consulting Agreement Amendment, Spartan and the Company agreed to modify certain terms of the Consulting Agreement, as amended by the Amendment, to require the Company to compensate Spartan with additional cash fees and stock compensation on a pro rata basis in the event that shares of Series B Preferred Stock are sold pursuant to Spartan’s exercise of the Over-allotment Option and the $1,200,000 over-allotment option for the Mirror Offering.
Offering of Warrants to Purchase Series B Preferred Stock
On March 17, 2025, the Company entered into an additional placement agent agreement with Spartan (the “Subsequent Placement Agent Agreement”), pursuant to which Spartan agreed to serve as exclusive placement agent for the private offer and sale (the “Subsequent Offering”) of warrants (the “Warrants”) to purchase up to 72,000 shares of Series B Preferred Stock at a price per Warrant equal to $0.125 in order to comply with applicable Nasdaq rules, with each Warrant exercisable for shares of Series B Preferred Stock at $100 per share, and such share of Series B Preferred Stock convertible into shares of Common Stock at a conversion price equal to $2.16 per share, which was the Minimum Price of the Common Stock immediately prior to the execution of the Subsequent Subscription Agreements (as defined below). On March 17, 2025 and in connection with the Subsequent Offering, the Company entered into subscription agreements (each, a “Subsequent Subscription Agreement”) and registration rights agreements with investors in the Subsequent Offering, who were also participants in the Offering, for the purchase and sale of an aggregate of 72,000 Warrants. In connection with the Subsequent Offering, the Company and Spartan entered into (i) the Subsequent Placement Agent Agreement and (ii) an additional consulting agreement and advisory agreement, dated March 17, 2025, pursuant to which Spartan agreed to provide placement agent and consulting services in connection with the Subsequent Offering and will receive certain compensation in consideration for such services, including, but not limited to, purchase warrants to purchase a number of shares of Common Stock equal to ten percent (10%) of the shares of Common Stock issuable upon conversion of the shares of Series B Preferred Stock that may be issued upon the Warrant exercises, (ii) a 2% non-accountable fee based on the aggregate number of proceeds received from Warrant exercises and (iii) shares of Series C Preferred Stock to be issued on a pro rata basis at the time of and based upon the amount of such proceeds received from Warrant exercises. Additionally, in connection with the Subsequent Offering, Dr. Kaufman and Spartan entered into another irrevocable proxy and power of attorney, effective as of March 17, 2025 (the “Subsequent Irrevocable Proxy”), pursuant to which, among other things, Spartan agreed to grant to Dr. Kaufman all voting power over and power of attorney with respect to all shares of Series C Preferred Stock, and all shares of Common Stock issuable upon conversion of shares of Series C Preferred Stock and upon exercise of Common Stock purchase warrants issued or issuable to Spartan or its Attribution Parties (as defined in the Subsequent Irrevocable Proxy) in connection with the Warrant exercises.
Nasdaq Notifications
On April 17, 2024 and on August 21, 2024, the Company received letters from the Staff stating that it was not in compliance with the Minimum Bid Price Requirement and the Stockholders’ Equity Requirement, respectively. On October 16, 2024, the Company received a letter from the Staff stating that although the Company submitted a plan to regain compliance with the Stockholders’ Equity Requirement on October 4, 2024, the Common Stock would be delisted from the Nasdaq Capital Market unless such determination is appealed to the Panel by October 23, 2024. On October 17, 2024, the Company requested a hearing before the Panel to appeal such determination and the hearing occurred on December 12, 2024.
On January 10, 2025, the Company received the January Letter from the Panel granting the Company’s request for continued listing on Nasdaq, subject to the Company demonstrating compliance with the Stockholders’ Equity Requirement, including the achievement of interim milestones as follows: (i) on or before April 14, 2025, the Company must file a public disclosure describing any transactions undertaken by the Company to increase its equity and providing an indication of its equity following those transactions; and (ii) on or before April 14, 2025, the Company must provide the Staff with an update on its fundraising plans, updated income projections for the next 12 months, with all underlying assumptions clearly stated, and a description of how the Company intends to achieve, if necessary, and maintain compliance with the Minimum Bid Price Requirement.
Results of Operations
Comparison of the Fiscal Years Ended December 31, 2024 and 2023
The following table summarizes our results of operations for the fiscal years ended December 31, 2024 and 2023 (in thousands):
| 2024 | 2023 | Increase (Decrease) |
||||||||||
| (in thousands) | ||||||||||||
| Revenue | $ | 536 | 450 | 86 | ||||||||
| Operating expenses: | ||||||||||||
| R&D | 3,613 | 3,039 | 574 | |||||||||
| General and administrative | 2,004 | 2,157 | (153) | |||||||||
| Total operating expenses | 5,617 | 5,196 | 421 | |||||||||
| Loss from operations | (5,081 | ) | (4,746 | ) | (335 | ) | ||||||
| Other income (expense) | 64 | 127 | (63 | ) | ||||||||
| Net loss | $ | (5,016 | ) | (4,619 | ) | (397 | ) | |||||
Grants and Other Revenue
We have not yet commercialized any products, and we do not expect to generate revenue from sales of any product candidates for several years. We recognize revenue from grants when the related costs are incurred and the right to payment is realized. For the year ended December 31, 2023, we derived revenue from a grant awarded by the NIH on September 15, 2022 of approximately $673,000 (the “NIH Grant”). The NIH approved an additional year of funding under the NIH Grant in June 2023, increasing the total funding provided under the NIH Grant to $1,353,000.
For the year ended December 31, 2024, we received approximately $536,000 in connection with the NIH Grant, recognized as revenue, compared to a total of $450,000 in connection with the NIH Grant recognized as revenue in the year ended December 31, 2023. The increase in annual grant revenue from 2024 to 2023 is related to the time devoted to the grant work and subcontractor costs.
Operating Expenses
Our operating expenses consist of (i) R&D expenses and (ii) general and administrative expenses.
Research and Development Expenses
R&D costs primarily consist of direct costs associated with consultants and materials, biologic storage, third party CRO costs and contract development and manufacturing expenses, salaries and other personnel-related expenses. R&D costs are expensed as incurred. More specifically, these costs include:
| ● | costs of funding research performed by third parties that conduct R&D and nonclinical and clinical activities on our behalf; |
| ● | costs of manufacturing drug supply and drug product; |
| ● | costs of conducting nonclinical studies and clinical trials of our product candidates; |
| ● | consulting and professional fees related to R&D activities, including equity-based compensation to non-employees; |
| ● | costs related to compliance with clinical regulatory requirements; and |
| ● | employee-related expenses, including salaries, benefits and stock-based compensation expense for our R&D personnel. |
Costs for certain activities are recognized based on an evaluation of the progress to completion of specific tasks using data, such as information provided to us by our vendors, and analyzing the progress of our nonclinical and clinical studies or other services performed. Significant judgments and estimates are made in determining the accrued expense balances at the end of any reporting period. Advance payments that we make for goods or services to be received in the future for use in R&D activities are recorded as prepaid expenses. Such amounts are recognized as an expense as the goods are delivered or the related services are performed, or until it is no longer expected that the goods will be delivered or the services rendered.
We expect that our R&D expenses will increase substantially in connection with our clinical development activities for our LP-10 program. At this time, we cannot accurately estimate or know the nature, timing and costs of the efforts that will be necessary to complete the clinical development of, or obtain regulatory approval for, any of our current or future product candidates. This is due to the numerous risks and uncertainties associated with product development and commercialization, including the specific factors set forth in the section titled “Risk Factors.” If any events described in the applicable risk factors included in the section titled “Risk Factors” occur, then the costs and timing associated with the development of any of our product candidates could significantly change. We may never succeed in obtaining regulatory approval for, of commercialization of, LP-10 or any of our other product candidates.
The following table summarizes our R&D expenses for the years ended December 31, 2024 and 2023 (in thousands):
| Years Ended | Increase | |||||||||||
| December 31, | (Decrease) | |||||||||||
| 2024 | 2023 | (in thousands) | ||||||||||
| Direct R&D expenses for the LP-10 and LP-310 product candidates program: | ||||||||||||
| Employee-related costs | $ | 504 | $ | 488 | $ | 16 | ||||||
| Employee stock option expense | 432 | 616 | (184 | ) | ||||||||
| Outsourced R&D | 1,276 | 477 | 799 | |||||||||
| Facility-related costs | 289 | 262 | 27 | |||||||||
| Platform development, early-stage research and unallocated expenses: | ||||||||||||
| Employee-related costs | 396 | 317 | 79 | |||||||||
| Employee stock option expense | 192 | 400 | (208 | ) | ||||||||
| Outsourced R&D | 312 | 288 | 24 | |||||||||
| Facility-related costs | 212 | 191 | 21 | |||||||||
| Total research and development expenses | $ | 3,613 | $ | 3,039 | $ | 574 | ||||||
R&D expenses increased by approximately $574,000, to approximately $3,613,000 for the year ended December 31, 2024, from approximately $3,039,000 for year ended December 31, 2023. The increase in R&D expenses was primarily attributable to an increase in outside services of $823,000, the majority of which was for our current clinical trial for LP-310, offset by reduction in employee stock option expense of $392,000. There was also an increase in personnel and facility costs of $143,000.
General and Administrative Expenses
General and administrative expenses consist primarily of management, overhead costs, outside services fees and other related costs, including stock-based compensation. General and administrative expenses also include board of directors’ expenses and professional fees for legal, patent, consulting, accounting, auditing, tax services and insurance costs.
General and administrative expenses were $2,004,000 for the year ended December 31, 2024, compared to $2,157,000 for the year ended December 31, 2023, a decrease of approximately $153,000. Stock option expense decreased by $215,000 for the year ended December 31, 2024 compared to the year ended December 31, 2023, correlating with changes to the share price. Insurance expense also decreased by $17,000 year over year, and personnel costs for G&A decreased by $69,000. There was a net increase in outside and professional services, including investor relations, accounting and tax, and legal services, of $111,000. Additionally, for the year ended December 31, 2024, rent increased approximately $17,000, related to additional space added to our lease.
We expect that our general and administrative expenses will increase as our organization and personnel needs grow to support continued R&D activities and the potential commercialization of our product candidates, including, but not limited to LP-10. We believe that these increases will likely include increased costs related to the hiring of additional personnel and fees to consultants, attorneys and accountants, among other expenses. Additionally, we expect to incur increased expenses associated with being a public company, including costs of additional personnel, accounting, audit, legal, regulatory and tax-related services associated with maintaining compliance with exchange listing and SEC requirements, director and officer insurance costs, and investor and public relations costs.
Other Income (Expense)
Other Income for the year ended December 31, 2024 was $64,000, compared to an other income of $127,000 in the year ended December 31, 2023. The decrease was due primarily to a $75,000 decrease in interest income on our liquid investments. This was offset by a savings of $11,000 in interest expense on notes payable in the year ended December 31, 2024 versus the prior year.
Liquidity and Capital Resources
Sources of Liquidity
We have not yet commercialized any products, and we do not expect to generate revenue from sales of any product candidates for several years, if at all. Cash and cash equivalents totaled $2.2 million as of December 31, 2024. We consider all highly liquid investments that mature in 90 days or less when purchased to be cash equivalents.
We have incurred operating losses and experienced negative operating cash flows for the years ended December 31, 2024 and 2023, and we anticipate that we will continue to incur losses for the foreseeable future. Our net loss totaled $5,016,264 for the year ended December 31, 2024 and $4,618,965 for the year ended December 31, 2023.
From inception through December 31, 2024, we have funded our operations primarily through (i) private equity financings (from which we have raised an aggregate of approximately $11 million), (ii) grants received from the U.S. government (from which we have received an aggregate of approximately $10.5 million since inception), (iii) the IPO (from which we raised net proceeds of approximately $5.0 million), (iv) a private placement transaction in October 2023 in which we raised net proceeds of approximately $1.0 million, (v) a registered direct offering in July 2024 (from which we raised net proceeds of approximately $1.3 million, and (vi) additional issuances of equity through a private placement in 2024 netting approximately $1.8 million. Until such time, if ever, as we can generate substantial revenue, we expect to finance our cash needs through a combination of public or private equity offerings and debt financings or other sources, such as potential collaboration agreements, strategic alliances and licensing arrangements.
Cash Flows
The following table provides information regarding our cash flows for each of the periods presented (in thousands):
| Fiscal Years Ended | ||||||||
| December 31, | ||||||||
| Dollars in thousands | 2024 | 2023 | ||||||
| Net cash (used) provided in operating activities | $ | (3,951 | ) | (3,150 | ) | |||
| Net cash (used) provided in investing activities | — | (14 | ) | |||||
| Net cash provided in financing activities | 2,842 | 1,336 | ||||||
| Net increase(decrease) in cash and cash equivalents | $ | (1,109 | ) | (1,828 | ) | |||
Net Cash (Used) Provided in Operating Activities
Net Cash used in operating activities for the year ended December 31, 2024 was approximately $3,951,000. This comprised a net loss of $5,016,000 for the year, offset by non-cash stock option expense of $749,000 and shares issued for services expense of $200,000. Changes in operating assets and liabilities provided in net cash inflows of $113,000.
Net cash used in operating activities for the year ended December 31, 2023 was approximately $3,150,000. This was composed of a net loss of $4,619,000 for the year, offset by non-cash stock option expense of $1,355,000 and shares issued for services of $122,000 expenses. There were changes in operating assets and liabilities that increased cash by $30,000.
Net Cash (Used) Provided in Investing Activities
There were no cash flows from investing activities in the year ended December 31, 2024. Net cash used in investing activities was $14,000 for the year ended December 31, 2023, related to the purchase of laboratory equipment.
Net Cash Provided by Financing Activities
For the year ended December 31, 2024, net cash provided by financing activities was $2,842,000, representing proceeds from issuance of preferred stock, common stock, and pre-funded warrants.
Net cash provided by financing activities for the year ended December 31, 2023 was $1,336,000, comprising primarily $1,612,000 in proceeds received from the issuance warrants through the October 2023 private placement, net of issuance costs. These were offset by $275,000 in debt repayments.
Funding Requirements
We expect our expenses to increase substantially in connection with our ongoing R&D activities, particularly as we continue R&D, advance clinical trials of LP-10 and LP-310, and advance the preclinical development of our other programs. In addition, we expect to incur additional costs associated with operating as a public company. As a result, we expect to incur substantial operating losses and negative operating cash flows for the foreseeable future.
Based on our current operating plan, we believe that our existing cash and cash equivalents will be sufficient to fund our operations and capital expenses into 2024. However, we have based this estimate on assumptions that may prove to be wrong, and we could exhaust our capital resources sooner than we expect.
Because of the numerous risks and uncertainties associated with research, development and commercialization of LP-10, LP-310, and our other and future product candidates, we are unable to estimate the exact amount of our working capital requirements. Our future funding requirements will depend on, and could increase significantly as a result of, many factors, including, but not limited to, those referenced above in “— Results of Operations — Operating Expenses — Research and Development Expenses”.
Going Concern
The financial statements of the Company have been prepared on a going concern basis, which contemplates the realization of assets and the discharge of liabilities in the normal course of business. We have generated losses from operations since inception. The Company expects operating losses to continue in the foreseeable future because of additional costs and expenses related to research and development activities, plans to expand its product portfolio, and increasing its market share. The Company’s ability to transition to attaining profitable operations is dependent upon achieving a level of revenues adequate to support its cost structure. The timing and amount of our actual expenditures will be based on many factors, including cash flows from operations and the anticipated growth of our business.
Management of the Company may raise additional funds through the issuance of equity securities or debt. There can be no assurance that such financing will be available at terms acceptable to the Company, if at all. Failure to generate sufficient cash flows from operations and raise additional capital could have a material adverse effect on the Company’s ability to achieve its intended business objectives. These factors raise substantial doubt about the Company’s ability to continue as a going concern. The accompanying financial statements do not include any adjustments that might be necessary if the Company is unable to continue as a going concern.
Off-Balance Sheet Arrangements
We did not have for the years ended December 31, 2024 or 2023, and we do not currently have, any off-balance sheet arrangements, as defined under applicable SEC rules.
Contractual Obligations
We did not have during the years ended December 31, 2024 and 2023, and we do not currently have, any material contractual obligations, such as license agreements or similar arrangements, other than as described below and in the financial notes.
Employment Agreements
We are party to employment agreements with each of Drs. Kaufman and Chancellor and Mr. Johnston, executive officers of the Company, the material terms of each of which are described in the section entitled “Executive Compensation – Employment Agreements”.
Lease Agreement
We are party to a lease agreement, dated June 1, 2019, with Bridgeway Development Corporation, as amended, for the lease of 2,690 square feet of office and lab and manufacturing space in Pittsburgh, Pennsylvania commencing on July 1, 2020 (the “Lease”). The current lease term expires on June 30, 2025 and we have the right to exercise a one-time option to extend the term of the lease for an additional five-year term. The annual base rent under the lease is approximately $67,000. On July 26, 2023, the Company entered into a second lease for additional space in the same building (the “Additional Lease”), commencing August 1, 2023 and co-terminating with the existing Lease on June 30, 2025. Annual rent under the Additional Lease was approximately $28,000. As space became available in the immediate proximity to our existing offices at the beginning of 2024, we terminated the Fourth Floor Lease upon mutual agreement with the landlord and replaced it with a lease for Suite 504 (“the Suite 504 Lease”). The Suite 504 Lease became effective January 1, 2024, and the term co-terminates with the Lease. The annual base rent for the current year for the Suite 504 Lease is approximately $29,000. See Note 13 of the notes to our audited financial statements included in this Report.
Service Agreements
We enter into service agreements in the normal course of business with CROs and for clinical trials, preclinical research studies and testing, manufacturing, and other services and products for operating purposes. These contracts do not contain any minimum purchase commitments. Certain agreements provide for termination rights subject to termination fees or wind down costs. Under such agreements, we are contractually obligated to make certain payments to vendors, mainly to reimburse them for their unrecoverable outlays incurred prior to cancellation. The exact amounts of such obligations are dependent on the timing of termination, and the exact terms of the relevant agreement and cannot be reasonably estimated. The expense we incurred pursuant to these agreements for the year ended December 31, 2024 was approximately $1,359,000, compared to approximately $675,000 for the year ended December 31, 2023. The increase was due to our current phase 2a clinical trial for LP-310 that started in 2024.
Critical Accounting Policies and Significant Judgments and Estimates
This management’s discussion and analysis is based on our financial statements, which have been prepared in accordance with U.S. generally accepted accounting principles. The preparation of these financial statements requires us to make judgments and estimates that affect the reported amounts of assets and liabilities, the disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of expenses during the reported periods. We base our estimates on historical experience, known trends and events, and various other factors that we believe to be reasonable under the circumstances. Actual results may differ from these estimates under different assumptions or conditions. On an ongoing basis, we evaluate our judgments and estimates in light of changes in circumstances, facts and experience. The effects of material revisions in estimates, if any, will be reflected in the financial statements prospectively from the date of change in estimates.
While our accounting policies are described in more detail in the notes to our financial statements included in this Report, we believe the following accounting policies used in the preparation of our financial statements require the most significant judgments and estimates. See Note 3 to our audited financial statements included elsewhere in this Report for a description of our other significant accounting policies.
Accrued Expenses
As part of the process of preparing our financial statements, we are required to estimate our accrued third-party R&D expenses as of each balance sheet date. This process involves reviewing open contracts and purchase orders, communicating with our personnel to identify services that have been performed on our behalf, and estimating the level of service performed and the associated cost incurred for the service when we have not yet been invoiced or otherwise notified of the actual cost. The majority of our service providers invoice us monthly in arrears for services performed or when contractual milestones are met. We make estimates of our accrued expenses as of each balance sheet date based on facts and circumstances known to us at that time. We periodically confirm the accuracy of our estimates with the service providers and make adjustments if necessary. The significant estimates in our accrued R&D expenses include the costs incurred for services performed by our vendors in connection with R&D activities for which we have not yet been invoiced.
We base our expenses related to R&D activities on our estimates of the services received and efforts expended pursuant to quotes and contracts with vendors that conduct R&D activities on our behalf. The financial terms of these agreements are subject to negotiation, vary from contract to contract and may result in uneven payment flows. There may be instances in which payments made to our vendors will exceed the level of services provided and result in a prepayment of the R&D expense. In accruing service fees, we estimate the time period over which services will be performed and the level of effort to be expended in each period. If the actual timing of the performance of services or the level of effort varies from our estimate, we adjust the accrual or prepaid balance accordingly. Non-refundable advance payments for goods and services that will be used in future R&D activities are expensed when the activity has been performed or when the goods have been received rather than when the payment is made.
Although we do not expect our estimates to be materially different from amounts incurred, if our estimates of the status and timing of services performed differ from the actual status and timing of services performed, it could result in us reporting amounts that are too high or too low in any particular period. To date, there have been no material differences between our estimates of such expenses and the amounts incurred.
Stock-Based Compensation
We measure stock-based compensation based on the grant date fair value of the stock-based awards and recognize stock-based compensation expense on a straight-line basis over the requisite service period of the awards, which is generally the vesting period of the respective award. For non-employee awards, compensation expense is recognized as the services are provided, which is generally ratably over the vesting period. We account for forfeitures as they occur. On January 1, 2018, we adopted, using the modified retroactive approach, the guidance of Accounting Standard Update 2018-07, Compensation — Stock Compensation (Topic 718) — Improvements to Nonemployee Share-Based Payment Accounting (“ASU 2018-07”), and account for awards to non-employees using the grant date fair value without subsequent periodic remeasurement. The adoption of ASU 2018-07 did not have a material effect on our financial statements.
We classify stock-based compensation expense in our statements of operations in the same manner in which the award recipient’s salary and related costs are classified or in which the award recipient’s service payments are classified. In future periods, we expect stock-based compensation expense to increase, due in part to our existing unrecognized stock-based compensation expense and as we grant additional stock-based awards to continue to attract and retain our employees.
We determine the fair value of restricted common stock awards granted based on the fair value of our Common Stock. We have historically determined the fair value of the underlying Common Stock based on input from management and the board of directors and the Company’s enterprise value determined utilizing various methods, including the “back-solve” method. The total enterprise value, determined from the back-solve method, is historically then allocated to the various outstanding equity instruments, including the underlying Common Stock, utilizing the option pricing method (“OPM”) or a hybrid of the probability-weighted expected return method (“PWERM”) and the OPM.
The fair value of each stock option grant is estimated on the date of grant using the Black-Scholes option-pricing model, which requires inputs based on certain subjective assumptions, including the expected stock price volatility, the expected term of the option, the risk-free interest rate for a period that approximates the expected term of the option, and our expected dividend yield. As the public market for our Common Stock has been limited and prior to the IPO there was no such public market, we have historically determined the volatility for awards granted based on an analysis of reported data for a group of guideline companies that issued options with substantially similar terms. The expected volatility has been determined using a weighted-average of the historical volatility measures of this group of guideline companies. We expect to continue estimating expected volatility based on the group of guideline companies until we have adequate historical data regarding the volatility of our own traded stock price. The expected term of our stock options granted to employees and non-employees has been determined utilizing the “simplified” method for awards that qualify as “plain-vanilla” options. The risk-free interest rate is determined by reference to the U.S. Treasury yield curve in effect at the time of grant of the award for time periods approximately equal to the expected term of the award. We have not paid, and do not anticipate paying, dividends on our Common Stock; therefore, the expected dividend yield is assumed to be zero.
As there was no public market for our Common Stock prior to the IPO, the estimated fair value of our Common Stock prior to our IPO had been approved by our board of directors, with input from management, as of the date of each award grant, considering our most recently available independent third-party valuations of our Common Stock and any additional objective and subjective factors that we believed were relevant and which may have changed from the date of the most recent valuation through the date of each award grant. We estimated the value of our equity using the market approach and a precedent transaction method which “back-solves” the equity value that yields a specific value for our Series A Preferred Stock. We allocated the equity value to our Common Stock and shares of our Series A Preferred Stock using either an OPM or a hybrid method, which is a hybrid between the OPM and the PWERM. The hybrid method we have historically utilized estimates the probability-weighted value across multiple scenarios but uses the OPM to estimate the allocation of value within at least one of the scenarios. In addition to the OPM, the hybrid method considered the IPO scenario in which the shares of our Series A Preferred Stock converted to Common Stock. The future value of the Common Stock in the IPO scenario is discounted back to the valuation date at an appropriate risk adjusted discount rate. In the hybrid method, the present value indicated for each scenario is probability weighted to arrive at an indication of value for our Common Stock.
In addition to considering the results of the valuations, management considered various objective and subjective factors to determine the fair value of our Common Stock as of each grant date, which may be a date later than the most recent third-party valuation date, including:
| ● | the prices of our Series A Preferred Stock sold to or exchanged between outside investors in arm’s length transactions, if any, and the rights, preferences and privileges of our Series A Preferred Stock as compared to those of our Common Stock, including the liquidation preferences of our Series A Preferred Stock; |
| ● | the progress of our R&D efforts, including the status of preclinical studies; |
| ● | the lack of liquidity of our equity as a private company; |
| ● | our stage of development and business strategy and the material risks related to our business and industry; |
| ● | the achievement of enterprise milestones; |
| ● | the valuation of publicly traded companies in the life sciences and biotechnology sectors, as well as recently completed mergers and acquisitions of peer companies; |
| ● | any external market conditions affecting the biotechnology industry, and trends within the biotechnology industry; |
| ● | the likelihood of achieving a liquidity event for the holders of our Series A Preferred Stock and Common Stock, such as an IPO, or a sale of the Company, given prevailing market conditions; and |
| ● | the analysis of IPOs and the market performance of similar companies in the biopharmaceutical industry. |
There are significant judgments and estimates inherent in these valuations. These judgments and estimates include assumptions regarding our future operating performance, the stage of development of our programs, the timing of a potential offering, or other liquidity event, and the determination of the appropriate valuation methodology at each valuation date. The assumptions underlying these valuations represent management’s best estimates, which involve inherent uncertainties and the application of management judgment. As a result, if factors or expected outcomes change and we use significantly different assumptions or estimates, our stock-based compensation expense could be materially different. Subsequent to the completion of the IPO, the fair value of our Common Stock will be determined based on the market price of our Common Stock on Nasdaq.
The following table sets forth by grant date, after giving effect to the reverse stock splits of the Company’s outstanding shares of Common Stock, the stock options granted during the years ended December 31, 2024 and December 31, 2023, including the (i) number of shares of our Common Stock issuable upon exercise of such stock options, (ii) per share exercise price of such options and (iii) estimated fair value per share of our Common Stock on each such date. We did not grant any shares of restricted Common Stock during this period.
| Grant date |
Number of shares of Stock issuable upon stock options granted |
Exercise price per share of Common Stock |
Estimated fair value share of Common at grant date |
|||||||||
| 03/15/2024 | 55,000 | $ | 6.16 | $ | 4.40 | |||||||
| 06/16/2023 | 53,000 | $ | 17.52 | $ | 12.00 | |||||||
The per share values at each such grant date, which we applied to determine the per share estimated fair value of the respective awards for accounting purposes, were based upon (a) the calculations described above used to determine the fair value of our Common Stock as of each grant date if pre-IPO, or (b) the closing trading price of the Common Stock on the date of issuance.
Emerging Growth Company Status
In April 2012, the JOBS Act was enacted. Section 107 of the JOBS Act provides that an “emerging growth company” may take advantage of the extended transition period provided in Section 7(a)(2)(B) of the Securities Act for complying with new or revised accounting standards. Therefore, an emerging growth company can delay the adoption of certain accounting standards until those standards would otherwise apply to private companies. We have irrevocably elected to avail ourselves of this extended transition period and, as a result, we will not be required to adopt new or revised accounting standards on the relevant dates on which adoption of such standards is required for other public companies.
In addition, as an emerging growth company, we may take advantage of specified reduced disclosure and other requirements that are otherwise applicable generally to public companies. These provisions include, among other things:
| ● | reduced disclosure about the compensation paid to our executive officers; |
| ● | not being required to submit to our stockholders’ advisory votes on executive compensation or golden parachute arrangements; |
| ● | an exemption from the auditor attestation requirement in the pursuant to the Sarbanes-Oxley Act of 2002; and |
| ● | an exemption from compliance with any new requirements adopted by the Public Company Accounting Oversight Board requiring mandatory audit firm rotation. |
We may take advantage of these exemptions until such time that we are no longer an emerging growth company.
We would cease to be an emerging growth company upon the earliest of:
| ● | the last day of the fiscal year on which we have $1.235 billion or more in annual revenue, |
| ● | the date on which we become a “large accelerated filer” (i.e., as of our fiscal year end, the total market value of our common equity securities held by non-affiliates is $700 million or more as of June 30), |
| ● | the date on which we issue more than $1.0 billion of non-convertible debt over a three-year period, or |
| ● | the last day of our fiscal year following the fifth anniversary of the date of the completion of the IPO. |
We may choose to take advantage of some but not all of these exemptions.
Recent Accounting Pronouncements
We have reviewed all recently issued accounting pronouncements and have determined that, other than as disclosed in Note 3 to our audited financial statements included in this Report, such standards will not have a material impact on our financial statements or do not otherwise apply to our operations.
Item 7A. Quantitative and Qualitative Disclosures about Market Risk.
We are not required to provide the information required by this Item 7A as we are a smaller reporting company.
Item 8. Financial Statements and Supplementary Data.
The Company’s financial statements, notes to the financial statements, and the reports of the Company’s independent registered accountants required to be filed in response to this Item 8 begin on page F-1 of this Report.
Item 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure.
None.
Item 9A. Controls and Procedures
Evaluation of Disclosure Controls and Procedures
As required by Rule 13a-15 under the Exchange Act, we have carried out an evaluation of the effectiveness of our disclosure controls and procedures as of the end of the period covered by this Report. This evaluation was carried out under the supervision and with the participation of our management, including our Chief Executive Officer and Chief Financial Officer.
Disclosure controls and procedures are controls and other procedures that are designed to ensure that information required to be disclosed in our reports filed or submitted under the Exchange Act is recorded, processed, summarized and reported, within the time periods specified in the SEC’s rules and forms. Disclosure controls and procedures include controls and procedures designed to ensure that information required to be disclosed in our company’s reports filed under the Exchange Act is accumulated and communicated to management, including our Chief Executive Officer and Chief Accounting Officer, to allow timely decisions regarding required disclosure. In designing and evaluating the disclosure controls and procedures, management recognizes that any controls and procedures, no matter how well designed and operated, cannot provide absolute assurance that the objectives of the controls system are met, and no evaluation of controls can provide absolute assurance that all control issues and instances of fraud, if any, within a company have been detected. Based on the evaluation of our disclosure controls and procedures as of December 31, 2024, our Chief Executive Officer and our Chief Financial Officer concluded that, as of such date, our disclosure controls and procedures were effective.
Management’s Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Exchange Act Rule 13a-15(f). Under the supervision and with the participation of our management, including our Chief Executive Officer and Chief Financial Officer, we are required to conduct an evaluation of the effectiveness of our internal control over financial reporting as of December 31, 2024. Management has completed an evaluation and concluded that, based on the criteria set forth in the report entitled Internal Control-Integrated Framework published by the Committee of Sponsoring Organizations of the Treadway Commission (2013), our internal control over financial reporting was effective as of December 31, 2024. Our auditors are not required to formally opine on the effectiveness of our internal control over financial reporting pursuant to Section 404 until we are no longer an “emerging growth company” as defined in the JOBS Act.
Limitations of the Effectiveness of Internal Control
Our management, including our Chief Executive Officer and Chief Financial Officer, does not expect that our disclosure controls and procedures will prevent all errors and all fraud. A control system, no matter how well conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met. Further, the design of a control system must reflect the fact that there are resource constraints, and the benefits of controls must be considered relative to their costs. Because of the inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that all control issues and instances of fraud, if any, have been detected. These inherent limitations include, but are not limited to, the realities that judgments in decision making can be faulty and that breakdowns can occur because of simple errors. Additionally, controls can be circumvented by the individual acts of a person, by collusion of two or more people, or by management override of the control. The design of any system of controls is also based in part upon certain assumptions about the likelihood of future events, and there can be no assurance that any design will succeed in achieving its stated goals under all potential future conditions. Because of the inherent limitations in a cost-effective control system, misstatements due to error or fraud may occur and not be detected.
Changes in Internal Control over Financial Reporting
There were no changes in the Company’s internal control over financial reporting during the year ended December 31, 2024, that have materially affected, or are reasonably likely to materially affect, the Company’s internal control over financial reporting.
Item 9B. Other Information
None.
Item 9C. Disclosure Regarding Foreign Jurisdictions That Prevent Inspections
Not applicable.
PART III
Item 10. Directors, Executive Officers and Corporate Governance
Our executive officers and directors and their ages and positions are as follows:
| Name | Age | Position |
| Jonathan Kaufman, PhD, MBA | 58 | President, Chief Executive Officer, Secretary and Treasurer and Chairman of the Board of Directors |
| Michael Chancellor, MD | 66 | Chief Medical Officer and Director |
| Douglas Johnston, CPA | 40 | Chief Financial Officer |
| Lori Birder, PhD | 64 | Director |
| Daniel Cohen | 60 | Director |
| Byong (Christopher) Kim, PhD | 54 | Director |
| Ryan Pruchnic, MBA | 50 | Director |
| Naoki Yoshimura, MD, PhD | 67 | Director |
The following is a biographical summary of the experience of each of our executive officers, directors and significant employees:
Executive Officers
Jonathan Kaufman, PhD, MBA, has served as a director and as our Chief Executive Officer, President, Secretary and Treasurer since the Company’s inception in 2005. From 2016 to 2019, Dr. Kaufman was a managing director and registered representative of Pickwick Capital Partners, LLC, a growth equity firm, and from 2015 to 2018 he consulted for multiple biotechnology companies, including Menogenix Inc., and Frequency Therapeutics Inc. (Nasdaq: FREQ). Previously, Dr. Kaufman served as chief financial officer of Semprus Biosciences Corp. (acquired by Teleflex Incorporated (NYSE: TFX)), chief science officer at LaunchCyte LLC, a company that creates, seeds and harvests life science innovations from top U.S. academic institutions, fellow in the department of radiation at the Hospital of the University of Pennsylvania, consultant to GlaxoSmithKline plc. (NYSE: GSK). Also, Dr. Kaufman served on the new technology committee during his employment at Merck & Co., Inc. (NYSE: MRK). Dr. Kaufman is the co-founder of Knopp Biosciences LLC, a privately held drug discovery and development company, and until March 2022 had served on the board of directors of Reaction Biology Corporation, a pre-clinical contract research organization that provides a full suite of preclinical drug discovery services. Dr. Kaufman received an MBA from the Wharton School, a PhD from the University of Pennsylvania School of Medicine, an MS from Brown University, and a BS from Carnegie Mellon University. We believe that Dr. Kaufman is qualified to serve on our board of directors due to his extensive business experience and knowledge in the life science industry.
Michael Chancellor, MD, has served as a director and as our Chief Medical Officer since 2008 and has been a consultant to the Company since 2005. Since 2008, Dr. Chancellor has served as a professor and research director at the William Beaumont School of Medicine. He is co-founder of Cook Myosite Incorporated, company that develops and commercializes technology related to the collection, selection, and expansion of human skeletal muscle cells for the treatment of various disorders and a wholly owned subsidiary of Cook Medical Incorporated, a medical device company. Dr. Chancellor has been principal investigator in more than 75 clinical trials has authored hundreds of publications regarding the treatment of urinary bladder dysfunction, has received more than 90 awards in connection with his work with urinary bladder dysfunction, and is generally considered an international key opinion leader in the industry. Dr. Chancellor is a board-certified urologist, previously holding the positions of instructor at the College of Physicians and Surgeons Columbia University, associate professor at Jefferson Medical College, and professor at University of Pittsburgh School of Medicine. He received an MD from the Medical College of Wisconsin and completed his urology residency at the University of Michigan and his neurourology and female urology fellowship at Columbia University. We believe that Dr. Chancellor is qualified to serve on our board of directors due to his extensive business experience as an executive in the pharmaceutical industry and his depth of knowledge and substantial experience as a research scientist.
Douglas Johnston, CPA, has served as the Company’s Chief Financial Officer since November 9, 2022, and previously served in a similar capacity as the Company’s Vice President of Finance since October 2021. Mr. Johnston has more than 15 years of experience, including working with global pharmaceutical companies and early-stage pharmaceutical and technology companies. Most recently, Mr. Johnston served as the chief financial officer of Apogee IT Services (“Apogee”) from 2017 to 2021. Prior to Apogee, Mr. Johnston served from 2015 to 2017 as senior manager of finance for Mylan N.V. (specialty division), a global generic and specialty pharmaceuticals company, director of finance from 2013 to 2015 at Forever, Inc., a digital archive and internet storage company, assistant controller from 2011 to 2013 at Kadmon Corporation, a biopharmaceutical company that discovers, develops and markets transformative therapies for unmet medical needs and is a subsidiary of Sanofi S.A. (Nasdaq: SNY), and prior to that, he served as an audit manager at Deloitte Touche Tohmatsu Limited. Also, Mr. Johnston is the co-founder of Stonewall Finance, LLC. Mr. Johnston received a bachelor’s degree in accounting from Washington and Jefferson College, is a certified public accountant licensed in Pennsylvania (inactive), and is an active member of the American Institute of Certified Public Accountants.
Independent Directors
Lori A. Birder, PhD, has served as a director of the Company since June 2023. Since 2001, Dr. Birder has been a tenured Professor of Medicine and Pharmacology and Chemical Biology at the University of Pittsburgh School of Medicine. Dr. Birder’s research has been durably funded by the NIH, including an NIH MERIT award, and focusses on understanding mechanisms underlying lower urinary tract dysfunction with chronic stress, pain and aging. Dr. Birder has published more than 200 peer-reviewed articles, book chapters and reviews. She has organized and chaired a number of symposia and workshops involving chronic visceral pain and aging, is a member of several scientific and editorial boards and scientific societies (e.g., International Continence Society-ICS, International Neurourology Society-INUS, International Society for the Study of Bladder Pain Syndrome-ESSIC and the Society of Urodynamics, Female Pelvic Medicine & Urogenital Reconstruction-SUFU) and serves as an ICS Board of Trustee member and the Founding Editor-in-Chief for the open access International Continence Society (ICS) journal ‘Continence’. We believe that Dr. Birder is qualified to serve on our board of directors due to her experience in the field of genitourinary research and her deep knowledge of the pharmaceutical industry.
Daniel Cohen, MBA, has served as a director of the Company since March 21, 2023. Since April 2024, Mr. Cohen has served in the role of Commercialization Director of Alphabet Inc. (formerly Google, NASDAQ: GOOG), and since 2018, Mr. Cohen has served as managing member and founder at Brightdrum LLC, a management consulting firm that works to accelerate growth of technology ventures. From 2021 to 2023, Mr. Cohen also served as an executive at Mojo Vision, a technology company, where he led healthcare product strategy and medical device product management. As a serial entrepreneur, Mr. Cohen founded and served as CEO of five startups with exits, including Personity, a mobile infrastructure software company, acquired by Openwave Systems (now Enea, STO: ENEA), and USConnect, an enterprise software company, acquired by IKON (now Canon, NYSE: CAJ). Mr. Cohen has also held executive leadership roles in product management, strategic partnerships, and business operations in companies including Alphabet, Inc., (NASDAQ: GOOG) and Yahoo. Mr. Cohen has worked across multiple technology sectors including health tech, IoT, consumer web, and enterprise SaaS. Mr. Cohen is coauthor of eight patents including innovations in ophthalmic medical devices, mobile communications, user interfaces, security, presence, messaging, and peer-to-peer networks. Mr. Cohen holds a dual BS degree in electrical engineering and computer engineering from Carnegie Mellon University, and an MBA from the Wharton School of the University of Pennsylvania. We believe that Mr. Cohen is qualified to serve on our board of directors because of his extensive experience in operating and advising diverse technology companies and commercializing innovation.
Byong (Christopher) Kim, PhD, has served as a director of the Company since March 2022 and is a venture capitalist with a focus on drug discovery. Since 2015, Dr. Kim has served as managing director at Novatio Ventures, which invests in seed- to early-stage life sciences companies originating from the U.S., Canada and Korea. He has also served as a member of the selection committee for BaseLaunch since July 2020, an accelerator firm located in Basel, Switzerland which has supported ventures that have since raised over $390M since its founding in 2018. Additionally, since 2016, Dr. Kim has served as an executive vice president and board member of Bridge Biotherapeutics, Inc., a clinical stage biotech company that went public on the Korean stock exchange KOSDAQ in December 2019. Mr. Kim holds a B.S. in Biology from the University of California at Irvine, a PhD in Developmental Biology from the University of Texas at MD Anderson, and an MBA from Carnegie Mellon University. We believe that Dr. Kim is qualified to serve on our board of directors because of his experience evaluating and financing early-stage biotechnology companies.
Ryan Pruchnic, MBA, has served as a director of the Company since September 2021. Mr. Pruchnic has been employed by Cook Myosite since 2001, and currently serves as managing vice president at Cook MyoSite. Mr. Pruchnic received a bachelor’s degree in biology and a master’s degree in exercise physiology from the University of Pittsburgh and an MBA from the Joseph M. Katz Graduate School of Business at the University of Pittsburgh. While working as a research scientist investigating the experimental uses of skeletal muscle-derived cells for urinary tract tissue augmentation, Mr. Pruchnic was part of the original team that custom-built the cell isolation and manufacturing technology for use in human clinical trials. Mr. Pruchnic has authored and coauthored numerous peer-reviewed scientific journal articles and book chapters relating to gene and cell therapy research for musculoskeletal disorders. Currently, Mr. Pruchnic oversees the day-to-day operations and the manufacturing, quality testing and releasing of the cellular product for human use in clinical investigations at Cook MyoSite, including leading the regulatory and clinical initiatives. We believe that Mr. Pruchnic is qualified to serve on our board of directors due to his experience building a substantial global research initiative in applied regenerative medicine.
Naoki Yoshimura, MD, PhD, has served as a director of the Company since September 2021. Dr. Yoshimura is a professor and the endowed chair of neurological research in the department of urology at the University of Pittsburgh School of Medicine, where he has been employed since 1996. Dr. Yoshimura also serves on the appeals committee of the University of Pittsburgh School of Medicine. Dr. Yoshimura’s research interests include understanding the mechanism inducing hyperexcitability of visceral afferent pathways innevating the lower urinary tract in relation with pathophysiological conditions such as spinal cord injury, peripheral nerve injury, inflammation, and diabetes mellitus, and identifying the role of neurotrophic factors in controlling the activity of visceral afferent neurons. Since 2006, Dr. Yoshimura has served on the board of directors of the Comfortable Urology Network, a non-profit organization. In addition, Dr. Yoshimura has served as a research officer for and on the board of directors of the International Neuro-Urology Society since its establishment in 2016. Dr. Yoshimura is a published author of more than 300 articles, abstracts, and book chapters, teaches several courses at the University of Pittsburgh School of Medicine, and mentors a number of students, residents, and research fellows each year. Dr. Yoshimura is also principal investigator on several sponsored research projects and holds a number of patents in his field. Dr. Yoshimura serves as a member of the editorial board for the Journal of the Japanese Continence Society and the International Journal of Urology. We believe that Dr. Yoshimura is qualified to serve on our board of directors due to his extensive experience in urinary bladder research and related consulting experience with the pharmaceutical industry.
Significant Employees
Michele Gruber has served in various roles for the Company since 2009 and currently serves as the Company’s Director of Operations, a position she has held since March 2010. She has participated in the development of multiple Company product candidates for the treatment of urologic diseases, as well as the design and conduct of urologic clinical trials. Mrs. Gruber’s early work in the chemistry field included development of calibration standards for a Macromizer MALDI-TOF mass spectrometry as well as analytical work in the biofuels industry. Mrs. Gruber was responsible for the development of GMP manufacturing and validation and stability testing of LP-10 and has been similarly responsible for LP-310 in this regard, including preparation of the related IND package. Mrs. Gruber holds a Bachelor’s degree in Chemistry from Carnegie Mellon University.
Janet Okonski has served as the Company’s Director of Clinical Operations since August 2021, where she is responsible for the Company’s clinical trial data management as well as communications with the Company’s clinical trial sites and clinical research vendors, including medical monitoring, clinical laboratories for body-fluid analysis, safety monitoring and overall data management. For more than twenty years prior, she was employed as a clinical research director at the University of Pittsburgh’s Department of Urology. Mrs. Okonski has experience managing over 40 clinical trials in all phases of research, including translational clinical research, and her experience also includes participating in clinical trial design and budgeting, preparing FDA regulatory submissions (including IND applications) and clinical trial subject recruitment, retention and data collection. Mrs. Okonski holds a Bachelor’s degree from Indiana University of Pennsylvania.
Family Relationships
There are no family relationships among any of our directors or executive officers.
Involvement in Certain Legal Proceedings
None.
Code of Ethics
We have a written code of conduct and ethics that applies to our directors, officers, employees and contractors, including our principal executive officer, principal financial officer, principal accounting officer or controller, and persons performing similar functions. The code of conduct and ethics is available on our website at www.lipella.com. We intend to disclose future amendments to such code, or any waivers of its requirements, applicable to any principal executive officer, principal financial officer, principal accounting officer or controller, or persons performing similar functions or our directors on our website identified above or in a current report on Form 8-K that we would file with the SEC.
Delinquent Section 16(a) Reports
Under the securities laws of the United States, our directors, executive (and certain other) officers, and any persons holding ten percent or more of our Common Stock must report on their ownership of the Common Stock and any changes in that ownership to the SEC. Specific due dates for these reports have been established. During the fiscal year ended December 31, 2024, all reports required to be filed by such persons pursuant to Section 16(a) were filed on a timely basis.
Corporate Governance
Board of Directors
Our board of directors (the “Board”) consists of seven members. The number of directors is fixed from time to time by our Board, subject to the terms of our Certificate of Incorporation and our Bylaws. Each of our current directors will continue to serve as a director until the election and qualification of his or her successor, or until his or her earlier death, disqualification, resignation, or removal.
Director Independence
As our Common Stock is listed on Nasdaq, our determination of the independence of directors is made using the definition of “independent director” contained in Nasdaq Rule 5605(a)(2). As of March 26, 2025, our Board has affirmatively determined that Drs. Birder, Kim and Yoshimura and Messrs. Cohen and Pruchnic are “independent directors,” as that term is defined in the Nasdaq Rules. Under the Nasdaq Rules, our Board must be composed of a majority of “independent directors.” Additionally, subject to certain limited exceptions, our Board’s audit, compensation, and nominating and corporate governance committees also must be composed of all independent directors.
Audit committee members must also satisfy the independence criteria set forth in Rule 10A-3 under the Exchange Act. Under the rules of Nasdaq, a director will only qualify as an “independent director” if, in the opinion of that company’s board of directors, that person does not have a relationship that would interfere with the exercise of independent judgment in carrying out the responsibilities of a director.
To be considered to be independent for purposes of Rule 10A-3 of the Exchange Act, a member of an audit committee of a listed company may not, other than in his capacity as a member of our audit committee, our board of directors, or any other committee of our board of directors: (1) accept, directly or indirectly, any consulting, advisory, or other compensatory fee from the listed company or any of its subsidiaries; or (2) be an affiliated person of the listed company or any of its subsidiaries.
Our Board has undertaken a review of its composition, the composition of its committees and the independence of each director. Based upon information requested from and provided by each director concerning his or her background, employment and affiliations, including family relationships, our Board has determined that the following members of our Board have a relationship that would interfere with the exercise of independent judgment in carrying out the responsibilities of a director: Dr. Kaufman and Dr. Chancellor, and that other than such directors, each of our directors is “independent” as that term is defined under the listing requirements and rules of Nasdaq. In making this determination, our Board considered the current and prior relationships that each non-employee director has with the Company and all other facts and circumstances our Board deemed relevant in determining their independence, including the beneficial ownership of our Common Stock by each non-employee director.
Board Meetings and Attendance
During the 2024 fiscal year, the Board conducted its annual meeting virtually in September 2024. Ongoing and ad-hoc business throughout 2024 was conducted remotely on an as needed basis, and by means of unanimous written consent.
Annual Meeting Attendance
Although we do not have a formal policy regarding attendance by members of the Board at our annual meeting of shareholders, the Board encourages all of its members to attend the annual meeting of shareholders. In September 2024, three directors attended our 2024 annual meeting of shareholders virtually.
Role of our Board of Directors in Risk Oversight
One of the key functions of the Board is informed oversight of our risk management process. The Board does not anticipate having a standing risk management committee, but rather anticipates administering this oversight function directly through our Board as a whole, as well as through various standing committees of our Board that address risks inherent in their respective areas of oversight. In particular, our Board is responsible for monitoring and assessing strategic risk exposure and our audit committee has the responsibility to consider and discuss our major financial risk exposures and the steps our management has to take to monitor and control such exposures, including guidelines and policies to govern the process by which risk assessment and management is undertaken. The audit committee also monitors compliance with legal and regulatory requirements. Our compensation committee also assesses and monitors whether our compensation plans, policies and programs comply with applicable legal and regulatory requirements.
Committees of Our Board of Directors
Our board of directors has established an audit committee, a compensation committee and a nominating and corporate governance committee, each of which has the composition and the responsibilities described below. Members serve on these committees until their resignation or until otherwise determined by our board of directors. Each committee operates under a charter approved by our board of directors. Copies of each committee’s charter are available on the investor relations section of our website at www.lipella.com, under Corporate Governance.
Nominating and Corporate Governance Committee
The members of our nominating and corporate governance committee are Drs. Yoshimura and Kim and Mr. Pruchnic. Dr. Yoshimura serves as the chairperson of our nominating and corporate governance committee. The composition of our nominating and corporate governance committee meets the requirements for independence under the current Nasdaq listing standards and SEC rules and regulations. Our nominating and corporate governance committee oversees and assists our board of directors in reviewing and recommending nominees for election as directors and is responsible for, among other things:
| ● | identifying, considering and recommending candidates for membership on our board of directors; |
| ● | developing and maintaining corporate governance policies applicable to us; |
| ● | overseeing the process of evaluating the performance of our board of directors; and |
| ● | advising our board of directors on other corporate governance matters. |
Our nominating and corporate governance committee operates under a written charter which satisfies the applicable rules of the SEC and the listing standards of Nasdaq.
Audit Committee
The members of our audit committee are Mr. Pruchnic and Drs. Kim and Birder. Mr. Pruchnic serves as the chairperson of our audit committee. Drs. Birder and Kim and Mr. Pruchnic each meet the requirements for independence under the current Nasdaq listing standards and SEC rules and regulations. Each member of our audit committee is financially literate. In addition, our board of directors has determined that Mr. Pruchnic is an “audit committee financial expert” as defined in Item 407(d)(5)(ii) of Regulation S-K promulgated under the Securities Act. This designation does not impose any duties, obligations or liabilities that are greater than are generally imposed on members of our audit committee and our board of directors. Our audit committee oversees our corporate accounting and financial reporting process, assists our board of directors in monitoring our financial systems and is responsible for, among other things:
| ● | our accounting and financial reporting processes and internal controls, including our financial statement audits and the integrity of our financial statements; |
| ● | our compliance with applicable laws (including U.S. federal securities laws and other legal and regulatory requirements); |
| ● | our design, implementation and performance of the Company’s internal control function; |
| ● | our policies with respect to risk assessment and risk management pertaining to the financial, accounting and tax matters of the Company; |
| ● | reviewing and approving related person transactions; |
| ● | selecting and hiring our registered independent public accounting firm; |
| ● | the qualifications, independence and performance of our independent auditors; and |
| ● | the preparation of the audit committee report to be included in our annual proxy statements. |
Our audit committee operates under a written charter that satisfies the applicable rules of the SEC and the listing standards of Nasdaq.
Compensation Committee
The members of our compensation committee are Drs. Yoshimura, Birder and Kim. Dr. Kim serves as the chairperson of our compensation committee. The composition of our compensation committee meets the requirements for independence under the current Nasdaq listing standards and SEC rules and regulations. Each member of such committee is: (i) an outside director, as defined pursuant to Section 162(m) of the Internal Revenue Code of 1986, as amended (the “Code”); and (ii) a non-employee director, as defined in Rule 16b-3 promulgated under the Exchange Act. Our compensation committee oversees our compensation policies, plans and benefits programs and is responsible for, among other things:
| ● | evaluating, recommending, approving and reviewing executive officer and director compensation arrangements, plans, policies and programs; |
| ● | overseeing the Company’s compensation policies, plans and benefit programs, and being responsible for the Company’s overall compensation philosophy; |
| ● | administering our cash-based and equity-based compensation plans; and |
| ● | making recommendations to our board of directors regarding any other board of director responsibilities relating to executive compensation. |
Our compensation committee operates under a written charter which satisfies the applicable rules of the SEC and the listing standards of Nasdaq.
Director Nomination Procedures
There have been no material changes to the procedures by which security holders may recommend nominees to our Board.
Insider Trading Arrangements and Policies
We have a written insider trading policy that applies to our directors, officers, employees and contractors, including our principal executive officer, principal financial officer, principal accounting officer or controller, and persons performing similar functions. We intend to disclose future amendments to such policy, or any waivers of its requirements, applicable to any principal executive officer, principal financial officer, principal accounting officer or controller, or persons performing similar functions or our directors on our website identified above or in a current report on Form 8-K that we would file with the SEC.
Our directors and executive officers may adopt written plans, known as Rule 10b5-1 plans, in which they will contract with a broker to buy or sell shares of our Common Stock on a periodic basis. Under a Rule 10b5-1 plan, a broker executes trades pursuant to parameters established by the director or officer when entering into the plan, without further direction from them. The director or officer may amend a Rule 10b5-1 plan in some circumstances and may terminate a plan at any time. Our directors and executive officers also may buy or sell additional shares outside of a Rule 10b5-1 plan when they are not in possession of material non-public information subject to compliance with the terms of our insider trading policy. Prior to 180 days after the date of the pricing of our initial public offering of the Company, subject to early termination, the sale of any shares under such plans would be prohibited by the lock-up agreement that the director or officer has entered into with the underwriters.
Item 11. Executive Compensation.
Our named executive officers for the fiscal year ended December 31, 2024, which consist of our principal executive officer and the next two most highly compensated executive officers, are:
| ● | Jonathan Kaufman, our Chief Executive Officer; |
| ● | Michael Chancellor, our Chief Medical Officer; and |
| ● | Douglas Johnston, our Chief Financial Officer |
Summary Compensation Table for Fiscal Years 2024 and 2023
The following table sets forth all plan and non-plan compensation for the last two fiscal years paid to individuals who served as the Company’s principal executive officers and the Company’s two other most highly compensated executive officers serving as executive officers at the end of the last completed fiscal year, as required by Item 402(m)(2) of Regulation S-K of the Securities Act. We refer to these individuals collectively as our “named executive officers.”
| Name and Principal Position |
Year | Salary | Bonus | Stock Awards | Option Awards (1) |
Non-Equity Incentive Plan Compensation |
Nonqualified deferred compensation earnings |
All Other Compensation |
Total | |||||||||||||||||||||||||||
| Jonathan Kaufman, | 2024 | $ | 233,300 | — | — | $ | 44,000 | (2) | — | — | — | $ | 277,300 | |||||||||||||||||||||||
| Chief Executive Officer | 2023 | $ | 204,133 | — | — | $ | 119,250 | (2) | — | — | — | $ | 323,383 | |||||||||||||||||||||||
| Michael Chancellor, | 2024 | $ | 225,000 | — | — | $ | 44,000 | (3) | — | — | — | $ | 269,000 | |||||||||||||||||||||||
| Chief Medical Officer | 2023 | $ | 195,868 | — | — | $ | 119,250 | (3) | — | — | — | $ | 315,118 | |||||||||||||||||||||||
| Douglas Johnston, | 2024 | $ | 165,000 | — | — | $ | 27,500 | (4) | — | — | — | $ | 192,500 | |||||||||||||||||||||||
| Chief Financial Officer | 2023 | $ | 165,000 | $ | 20,000 | — | $ | 75,000 | (4) | — | — | — | $ | 260,000 | ||||||||||||||||||||||
| (1) | Amounts reflect the aggregate grant date fair value of the stock options granted to each named executive officer during the fiscal year ended December 31, 2023 and 2024, as computed in accordance with Financial Accounting Standards Board ASC 718. |
| (2) | During the fiscal year ended December 31, 2024, the Company granted Dr. Kaufman stock options exercisable for up to an aggregate of 10,000 shares of Common Stock, which had a grant date fair value of $4.40 per share. During the fiscal year ended December 31, 2023, the Company granted Dr. Kaufman stock options exercisable for up to an aggregate of 9,938 shares of Common Stock, which had a grant date fair value of $12.00 per share. |
| (3) |
During the fiscal year ended December 31, 2024, the Company granted Dr. Chancellor stock options exercisable for up to an aggregate of 10,000 shares of Common Stock, which had a grant date fair value of $4.40 per share. During the fiscal year ended December 31, 2023, the Company granted Dr. Chancellor stock options exercisable for up to an aggregate of 9,938 shares of Common Stock, which had a grant date fair value of $12.00 per share.
|
| (4) | During the fiscal year ended December 31, 2024, the Company granted Mr. Johnston stock options exercisable for up to an aggregate of 6,250 shares of Common Stock, which had a grant date fair value of $4.40 per share. During the fiscal year ended December 31, 2023, the Company granted Mr. Johnston stock options exercisable for up to an aggregate of 6,250 shares of Common Stock, which had a grant date fair value of $12.00 per share. |
Employment Agreements
Jonathan Kaufman
On July 17, 2020, Dr. Kaufman and the Company entered into an employment agreement appointing Dr. Kaufman as Chief Executive Officer of the Company (the “Kaufman Agreement”). The Kaufman Agreement establishes an employment term of two years beginning on July 17, 2020, which term will be automatically extended for successive one-year periods unless either party notifies the other party of its intention not to renew upon at least 90 days’ written notice prior to the applicable renewal date. The Kaufman Agreement provides Dr. Kaufman with an annual base salary of $183,300. In addition, Dr. Kaufman may be entitled to receive equity awards under the company’s stock incentive plans, as well as reimbursement of business expenses and bonus compensation, at the discretion of the board of directors, depending upon relevant factors, including but not limited to fundraising success, continued ongoing grant revenue, successful progress in the clinic and the Company’s financial position. The Kaufman Agreement also provides that Dr. Kaufman will participate in employee benefits plans, practices and programs maintained by the Company. On August 4, 2023, the Company and Dr. Kaufman entered into an amendment agreement, dated August 4, 2023 (the “Kaufman Amendment”), to the Kaufman Agreement. The Kaufman Amendment amended the Kaufman Agreement solely to increase Dr. Kaufman’s annual base salary by $50,000, to $233,300.
Pursuant to the Kaufman Agreement, either party may terminate such agreement for any reason upon 90 days’ advance written notice. In the event that Dr. Kaufman is terminated by the Company for Cause or by Dr. Kaufman Without Good Reason (as such terms are defined in the Kaufman Agreement), Dr. Kaufman is entitled to any accrued but unpaid base salary, employee benefits and reimbursement of unreimbursed expenses incurred until the date of termination. In the event that Dr. Kaufman is terminated by the Company for Without Cause or by Dr. Kaufman for Good Reason (as such terms are defined in the Kaufman Agreement), Dr. Kaufman is entitled to his base salary for six months following such date of termination and all unvested stock options held by Dr. Kaufman under the Company’s stock incentive plans will immediately vest. Additionally, in the event of termination without Cause by the Company or for Good Reason by Dr. Kaufman within 12 months of a Change in Control (as defined in the Kaufman Agreement), Dr. Kaufman is entitled to a receive a lump sum payment of two times the annual base salary within 60 days following such termination and reimbursement for certain health insurance premium payments.
Michael Chancellor
On July 17, 2020, Dr. Chancellor and the Company entered into an employment agreement appointing Dr. Chancellor as Chief Medical Officer of the Company (the “Chancellor Agreement”). The Chancellor Agreement establishes an employment term of two years beginning on July 17, 2020, which term will be automatically extended for successive one-year periods unless either party notifies the other party of its intention not to renew upon at least 90 days’ written notice prior to the applicable renewal date. The Chancellor Agreement provides that Dr. Chancellor was initially to be paid an annual base salary of $45,650, provided that if the Company achieves adequate financial liquidity and net working capital in connection with a subsequent private offering, such salary may be increased up to a maximum of $175,000. This amount is currently covered by federal grant revenue. In addition, Dr. Chancellor may be entitled to receive equity awards under the Company’s stock incentive plans, as well as reimbursement of business expenses and bonus compensation at the discretion of the board of directors. The Chancellor Agreement also provides that Dr. Chancellor will participate in employee benefits plans, practices and programs maintained by the Company. On August 4, 2023, the Company and Dr. Chancellor entered into an amendment agreement, dated August 4, 2023 (the “Chancellor Amendment”), to Chancellor Agreement. The Chancellor Amendment amended the Chancellor Agreement solely to increase Dr. Chancellor’s annual base salary by $50,000, to $225,000.
Pursuant to the Chancellor Agreement, either party may terminate such agreement for any reason upon 90 days’ advance written notice. In the event that Dr. Chancellor is terminated by the Company for Cause or by Dr. Chancellor Without Good Reason (as such terms are defined in the Chancellor Agreement), Dr. Chancellor is entitled to any accrued but unpaid base salary, employee benefits and reimbursement of unreimbursed expenses incurred until the date of termination. In the event that Dr. Chancellor is terminated by the Company for Without Cause or by Dr. Chancellor for Good Reason (as such terms are defined in the Chancellor Agreement), Dr. Chancellor is entitled to his base salary for six months following such date of termination and all unvested stock options held by Dr. Chancellor under the Company’s stock incentive plans will immediately vest. Additionally, in the event of termination without Cause by the Company or for Good Reason by Dr. Chancellor within 12 months of a Change in Control (as defined in the Chancellor Agreement), Dr. Chancellor is entitled to a receive a lump sum payment of two times the annual base salary within 60 days following such termination and reimbursement for certain health insurance premium payments.
Douglas Johnston
Effective November 1, 2022, Mr. Johnston and the Company entered into an employment agreement appointing Mr. Johnston as Chief Financial Officer of the Company (the “Johnston Agreement”). The Johnston Agreement establishes an employment term of two years beginning on November 1, 2022, which term will be automatically extended for successive one-year periods unless either party notifies the other party of its intention not to renew upon at least 90 days’ written notice prior to the applicable renewal date. The Johnston Agreement provides that Mr. Johnston will be paid an annual base salary of $165,000. In addition, Mr. Johnston may be entitled to receive equity awards under the Company’s stock incentive plans, as well as reimbursement of business expenses and bonus compensation at the discretion of the board of directors. The Johnston Agreement also provides that Mr. Johnston will participate in employee benefits plans, practices and programs maintained by the Company.
Pursuant to the Johnston Agreement, either party may terminate such agreement for any reason upon 90 days’ advance written notice. In the event that Mr. Johnston is terminated by the Company for Cause or by Mr. Johnston Without Good Reason (as such terms are defined in the Johnston Agreement), Mr. Johnston is entitled to any accrued but unpaid base salary, employee benefits and reimbursement of unreimbursed expenses incurred until the date of termination. In the event that Mr. Johnston is terminated by the Company for Without Cause or by Mr. Johnston for Good Reason (as such terms are defined in the Johnston Agreement), Mr. Johnston is entitled to his base salary for six months following such date of termination and all unvested stock options held by Mr. Johnston under the Company’s stock incentive plans will immediately vest. Additionally, in the event of termination without Cause by the Company or for Good Reason by Mr. Johnston within 12 months of a Change in Control (as defined in the Johnston Agreement), Mr. Johnston is entitled to a receive a lump sum payment of two times the annual base salary within 60 days following such termination and reimbursement for certain health insurance premium payments.
Stonewall Finance, LLC, of which Mr. Johnston is a partner and co-founder, and the Company previously were party to an agreement, dated October 14, 2021 and which terminated on October 22, 2022, pursuant to which Mr. Johnston had served and performed financial and accounting services for the Company and pursuant to which Mr. Johnston received cash payments from the Company of $4,000 per month.
Outstanding Equity Awards at 2024 Fiscal Year End
The following table provides information relating to the vested and unvested option and stock awards held by our named executive officers as of December 31, 2024. Each award to each named executive officer is shown separately. All such option awards were fully vested at December 31, 2024.
| Option Awards | Stock Awards | |||||||||||||||||||||||||||||||||
| Name and Principal Position |
Number
of Securities Underlying Unexercised Options Exercisable |
Number of Securities Underlying Unexercised Options Unexercisable |
Equity Incentive Plan Awards: Number of Securities Underlying Unexercised Unearned Options |
Option Exercise Price |
Option Expiration Date |
Number of Shares or Units of Stock Unvested |
Market Value of Shares of Units of Stock Unvested |
Equity Incentive Plan Awards: Number of Unearned Unvested Shares |
Equity Incentive Plan Awards: Market or Payout Value of Unearned Unvested Shares |
|||||||||||||||||||||||||
| Jonathan Kaufman, | — | — | 10,000 | 10.00 | 10/14/2015 | — | — | — | — | |||||||||||||||||||||||||
| Chief Executive Officer | — | — | 25,000 | 10.00 | 10/15/2025 | — | — | — | — | |||||||||||||||||||||||||
| — | — | 12,500 | 10.00 | 10/12/2027 | — | — | — | — | ||||||||||||||||||||||||||
| — | — | 12,500 | 40.00 | 03/31/2031 | — | — | — | — | ||||||||||||||||||||||||||
| — | — | 20,000 | 40.00 | 09/03/2031 | — | — | — | — | ||||||||||||||||||||||||||
| — | — | 9,937 | 17.52 | 06/16/2033 | — | — | — | — | ||||||||||||||||||||||||||
| — | — | 10,000 | 6.16 | 03/15/2034 | — | — | — | — | ||||||||||||||||||||||||||
| Michael Chancellor, | — | — | 10,000 | 10.00 | 10/14/2015 | — | — | — | — | |||||||||||||||||||||||||
| Chief Medical Officer | — | — | 25,000 | 10.00 | 10/15/2025 | — | — | — | — | |||||||||||||||||||||||||
| — | — | 12,500 | 10.00 | 10/12/2027 | — | — | — | — | ||||||||||||||||||||||||||
| — | — | 12,500 | 40.00 | 03/31/2031 | — | — | — | — | ||||||||||||||||||||||||||
| — | — | 20,000 | 40.00 | 09/03/2031 | — | — | — | — | ||||||||||||||||||||||||||
| — | — | 9,937 | 17.52 | 06/16/2033 | — | — | — | — | ||||||||||||||||||||||||||
| 10,000 | 6.16 | 03/15/2034 | ||||||||||||||||||||||||||||||||
| Douglas Johnston, | — | — | 6,250 | 17.52 | 06/16/2033 | — | — | — | — | |||||||||||||||||||||||||
| Chief Financial Officer | — | — | 6,250 | 6.16 | 03/15/2034 | — | — | — | — | |||||||||||||||||||||||||
Director Compensation for Fiscal Year 2024
We have not implemented a formal policy with respect to compensation payable to our non-employee directors. From time to time, we have granted equity awards to attract individuals to join our board of directors and for their continued service thereon. During the fiscal year ended 2024, our independent directors received no cash compensation, but our independent directors were granted options to purchase an aggregate of 15,625 shares of Common Stock at fair market value as of the date of issuance, expiring ten years from such issuance. In addition, we reimburse our directors for expenses associated with attending meetings of our board of directors and its committees. Our board of directors is still in the process of considering the non-employee director compensation policy.
| Name | Fees Earned or Paid in Cash ($) |
Stock Awards ($) |
Stock Option Awards ($)(1) |
Non-Equity Incentive Plan Compensation ($) |
Nonqualified Deferred Compensation Earnings ($) |
All Other Compensation ($)(2) |
Total ($) |
|||||||||||||||||||||
| Byong (Christopher) Kim | — | — | $ | 13,750 | — | — | — | $ | 13,750 | |||||||||||||||||||
| Ryan Pruchnic | — | — | 13,750 | — | — | — | 13,750 | |||||||||||||||||||||
| Naoki Yoshimura | — | — | 13,750 | — | — | — | 13,750 | |||||||||||||||||||||
| Lori Birder | — | — | 13,750 | — | — | — | 13,750 | |||||||||||||||||||||
| Daniel Cohen | — | — | 13,750 | — | — | — | 13,750 | |||||||||||||||||||||
| (1) | Each of our independent directors received stock options to purchase up to 3,125 shares of Common Stock at an aggregate grant date fair value of $4.40 per share. Such options were fully vested as of December 31, 2024, at an exercise price of $6.16 per share. |
(2) |
The Company reimbursed our directors for travel-related expenses. |
Policies on the Timing of Option Grants in Relation to the Release of Material Non-public Information
We do not have any formal policy that requires the Company to grant, or avoid granting, equity-based compensation at certain times. We do not grant equity awards in anticipation of the release of material non-public information that is likely to result in changes to the price of our Common Stock, and do not time the public release of such information based on award grant dates. The timing of any equity grants to executive officers or directors in connection with new hires, promotions, or other non-routine grants is tied to the event giving rise to the award (such as an executive officer’s commencement of employment or promotion effective date).
During the year ended December 31, 2024, there were no equity grants made to our executive officers during any period beginning four business days before the filing of a periodic report or current report disclosing material non-public information and ending one business day after the filing or furnishing of such report with the SEC.
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters
The following table sets forth certain information regarding the beneficial ownership of our capital stock as of March 26, 2025 by (a) each person, or group of affiliated persons, who is known to us to own beneficially 5% or more of our outstanding equity securities; (b) each of our directors; (c) each of our named executive officers; and (d) all of our named executive officers and directors as a group. Except as otherwise indicated in the footnotes below, we believe, based on the information provided to us, that all persons listed below have sole voting power and investment power with respect to their shares of Common Stock or other equity securities that they beneficially own, subject to community property laws where applicable.
For purposes of this table, a person or group of persons is deemed to have “beneficial ownership” of any shares of Common Stock or other equity securities of the Company that such person has the right to acquire within sixty (60) days of March 26, 2025. For purposes of computing the percentage of outstanding shares of our Common Stock or other equity securities of the Company held by each person or group of persons named above, any shares that such person or persons has the right to acquire within sixty (60) days of March 26, 2025 is deemed to be outstanding, but is not deemed to be outstanding for the purpose of computing the percentage ownership of any other person. The inclusion herein of any shares of Common Stock or other equity securities of the Company listed as beneficially owned does not constitute an admission of beneficial ownership. Unless otherwise identified, the address of our directors and executive officers is 7800 Susquehanna Street, Suite 505, Pittsburgh, PA 15208.
| Name of and Address of Beneficial Owner(1): | Shares of Common Stock Beneficially Owned (2) |
Percentage of Common Stock Beneficially Owned (2) |
||||||
| Directors and executive officers | ||||||||
| Jonathan Kaufman (3) | 212,293 | 8.01 | % | |||||
| Michael Chancellor (4) | 209,640 | 7.91 | % | |||||
| Douglas Johnston (5) | 12,500 | * | % | |||||
| Lori Birder (6) | 3,125 | * | ||||||
| Byong (Christopher) Kim (7) | 7,500 | * | ||||||
| Ryan Pruchnic (8) | 7,500 | * | ||||||
| Naoki Yoshimura (9) | 8,000 | * | ||||||
| Daniel Cohen (10) | 6,250 | * | ||||||
| All executive officers and directors as a group (8 persons) | 466,808 | 16.71 | % | |||||
| * | Less than 1% |
| (1) | Except as otherwise indicated, the persons named in the table above have sole voting and investment power with respect to all shares of Common Stock shown as beneficially owned by them. |
| (2) | Based on 2,548,811 shares of common stock issued and outstanding as of March 26, 2025 and excluding all Advisory Shares issuable to Spartan in connection with this Offering and assuming no exercise of the Placement Agent Warrants. |
| (3) | Number of shares of common stock beneficially owned consists of (i) 112,356 shares of common stock and (ii) 99,937 shares of common stock issuable upon the exercise of stock options held by Dr. Kaufman. Such stock options held by Dr. Kaufman are exercisable for shares of common stock at prices ranging from $6.16 to $40.00 per share. |
| (4) | Number of shares of common stock beneficially owned consists of (i) 109,703 shares of common stock and (ii) 99,937 shares of common stock issuable upon the exercise of stock options held by Dr. Chancellor. Such stock options held by Dr. Chancellor are exercisable for shares of common stock at prices ranging from $6.16 to $40.00 per share. |
| (5) | Number of shares of common stock beneficially owned consists of 12,500 shares of common stock issuable upon the exercise of stock options held by Mr. Johnston. Such stock options held by Mr. Johnston are exercisable for shares of common stock at prices ranging from $6.16 to $17.52 per share. |
| (6) | Number of shares of common stock beneficially owned consists of 3,125 shares of common stock issuable upon the exercise of stock options held by Dr. Birder. Such stock options held by Dr. Birder are exercisable for shares of common stock at $6.16 per share. |
| (7) | Number of shares of common stock beneficially owned consists of 7,500 shares of common stock issuable upon the exercise of stock options held by Dr. Kim. Such stock options held by Dr. Kim are exercisable for shares of common stock at prices ranging from $6.16 to $40.00 per share. |
| (8) | Number of shares of common stock beneficially owned consists of 7,500 shares of common stock issuable upon the exercise of stock options held by Mr. Pruchnic. Such stock options held by Mr. Pruchnic are exercisable for shares of common stock at prices ranging from $6.16 to $40.00 per share. |
| (9) | Number of shares of Common Stock beneficially owned consists of 8,000 shares of common stock issuable upon the exercise of stock options held by Dr. Yoshimura. Such stock options held by Dr. Yoshimura are exercisable for shares of common stock at prices ranging from $6.16 to $40.00 per share. |
| (10) | Number of shares of common stock beneficially owned consists of 6,250 shares of common stock issuable upon the exercise of stock options held by Mr. Cohen. Such stock options held by Mr. Cohen are exercisable for shares of common stock at prices ranging from $6.16 to $17.52 per share. |
Securities Authorized for Issuance under Equity Compensation Plans
| Plan Category | Number of Securities to Be Issued upon Exercise of Outstanding Options, Warrants and Rights (3) |
Weighted Average Exercise Price of Outstanding Options, Warrants and Rights (3) |
Number of Securities
Remaining Available for Future Issuance under the Plan (excluding securities reflected in column (a) (3)) |
|||||||||
| (a) | (b) | (c) | ||||||||||
| Equity compensation plans approved by security holders (1) | 134,750 | $ | 10.00 | — | ||||||||
| Equity compensation plans approved by security holders (2) | 226,875 | $ | 25.53 | 73,125 | ||||||||
| Equity compensation plans not approved by security holders | — | — | — | |||||||||
| Total | 361,625 | 73,125 | ||||||||||
| (1) | Represents the shares of Common Stock authorized for issuance under the 2008 Stock Incentive Plan (as amended and restated from time to time, the “2008 Plan”), which was approved by the Company’s stockholders on March 6, 2009. The 2008 Plan terminated in accordance with its terms in 2018. An aggregate of 300,000 shares were authorized for issuance under the 2008 Plan. As of December 31, 2024, options to purchase an aggregate of 134,750 shares of our Common Stock at an exercise price of $10.00 per share were outstanding under the 2008 Plan. |
| (2) | Represents the shares of Common Stock authorized for issuance under the 2020 Stock Incentive Plan (as amended and restated from time to time, the “2020 Plan”) in July 2020. The Board approved an amended and restated version of the 2020 Plan on November 10, 2022, subject to stockholder approval which was subsequently obtained. An aggregate of 125,000 shares were originally authorized for issuance under the 2020 Plan, and after the amendment in 2022 and our subsequent reverse stock splits, an aggregate of 175,000 shares were authorized under the 2020 Plan. On November 21, 2023, our stockholders approved an amendment to the 2020 Plan to increase the number of shares of Common Stock authorized for issuance thereunder from 175,000 shares to 300,000 shares. As of December 31, 2024, options to purchase an aggregate of 226,874 shares of our Common Stock at exercise prices ranging from $6.16 to $40.00 per share, with a weighted-average exercise price of $25.53 per share, were outstanding under the 2020 Plan, with 73,126 shares of our Common Stock remaining available for future issuance. Unissued shares subject to awards that expire, are forfeited, or are cancelled will again become available for issuance under the 2020 Plan. |
| (3) | As of December 31, 2024. |
Equity Incentive Plans
2008 Stock Incentive Plan
Our board of directors adopted and our stockholders approved the 2008 Plan in March 2008. The 2008 Plan terminated in accordance with its terms in 2018; however, awards outstanding under the 2008 Plan continue to be governed by their existing terms.
Share Reserve. An aggregate of 300,000 shares were authorized for issuance under the 2008 Plan. As of December 31, 2024, options to purchase an aggregate of 134,750 shares of our Common Stock at an exercise price of $10.00 per share were outstanding under the 2008 Plan.
Administration. Our board of directors, or a committee thereof, has administered the 2008 Plan since its adoption; however, we intend for the compensation committee of our board of directors to administer the 2008 Plan to the extent such administration is necessary in light of the termination of the 2008 Plan.
Eligibility. Employees, officers, members of our board of directors, consultants and advisors were eligible to participate in the 2008 Plan. However, only employees were eligible to receive incentive stock options.
Types of Awards. The 2008 Plan provides for the following types of awards granted with respect to shares of our Common Stock:
| ● | incentive and non-statutory stock options to purchase shares of our Common Stock; and |
| ● | direct award or sale of shares of our Common Stock, including restricted shares. |
Options. The exercise price for options granted under the 2008 Plan is determined by our board of directors but may not be less than 100% of the fair market value of our Common Stock on the grant date. Optionees may pay the exercise price in cash or cash equivalents or by one, or any combination of, the following forms of payment, as permitted by:
| ● | surrender of shares of Common Stock that the optionee already owns; |
| ● | delivery of a full-recourse promissory note, with the option shares pledged as security against the principal and accrued interest on the note; |
| ● | an immediate sale of the option shares through a company-approved broker, if the shares of our Common Stock are publicly traded; |
| ● | a number of vested shares subject to the option having an aggregate fair market value no greater than the aggregate exercise price, or the sum of such exercise price plus all or a portion of the minimum amount required to be withheld under applicable law; or |
| ● | other methods permitted by the DGCL. |
In general, we have granted options that vest over a three-year period or less. Options expire at the time determined by the Board, but in no event more than ten years after they are granted, and generally expire earlier if the optionee’s service terminates.
Restricted Shares. Restricted shares may be awarded or sold under the 2008 Plan in return for cash or cash equivalents or, as permitted by the Board in its sole discretion, in exchange for services rendered to us, by delivery of a full-recourse promissory note or through any other means permitted by applicable law. Restricted shares vest as determined by the Board.
Corporate Transactions. In the event that we are a party to a merger or consolidation or in the event of a sale of all or substantially all of our stock or assets, awards granted under the 2008 Plan will be subject to the agreement governing such transaction or, in the absence of such agreement, in the manner determined by the Board. Such treatment may include, without limitation, one or more of the following with respect to outstanding awards:
| ● | the continuation, assumption or substitution of an award by the surviving entity or its parent; |
| ● | cancellation of the vested portion of the award in exchange for a payment equal to the excess, if any, of the value of the shares subject to the award over any exercise price per share applicable to the award; |
| ● | cancellation of the award without payment of any consideration; |
| ● | suspension of the optionee’s right to exercise the option during a limited period of time preceding the completion of the transaction; or |
| ● | termination of any right the optionee has to exercise the option prior to vesting in the shares subject to the option. |
The Board is not obligated to treat all awards in the same manner. The Board has the discretion, at any time, to provide that an award under the 2008 Plan will vest on an accelerated basis in connection with a corporate transaction or to amend or modify an award so long as such amendment or modification is not inconsistent with the terms of the 2008 Plan or would not result in the impairment of a participant’s rights without the participant’s consent.
Changes in Capitalization. In the event of certain specified changes in the capital structure of our Common Stock, such as a stock split, reverse stock split, stock dividend, reclassification or any other increase or decrease in the number of issued shares of stock effective without receipt of consideration by us, proportionate adjustments will automatically be made in (i) each of the number and kind of shares available for future grants under the 2008 Plan, (ii) the number and kind of shares covered by each outstanding award, (iii) the exercise price per share subject to each outstanding option and (iv) any repurchase price applicable to shares granted under the 2008 Plan. In the event of an extraordinary cash divided that has a material effect on the fair market value of our Common Stock, a recapitalization, spin-off or other similar occurrence, the Board at its sole discretion may make appropriate adjustments to one or more of the items described above.
Amendments or Termination. Pursuant to the 2008 Plan, the administrator could amend, suspect or terminate the 2008 Plan, subject to stockholder approval in the case of an amendment requiring stockholder approval under the Internal Revenue Code, any rule of a stock exchange on which the Company’s securities are listed or any other applicable law. The 2008 Plan was terminated pursuant to its terms in 2018, but as noted above, awards outstanding under the 2008 Plan will remain outstanding and will continue to be governed by the 2008 Plan and any outstanding related award agreements.
2020 Stock Incentive Plan
Our board of directors adopted and our stockholder approved the 2020 Plan in July 2020, and the Board approved an amended and restated version of the 2020 Plan on November 10, 2022, subject to stockholder approval which was subsequently obtained. The material terms of the 2020 Plan are summarized below and reflects such amended and restated version.
Share Reserve. An aggregate of 125,000 shares were originally authorized for issuance under the 2020 Plan, and after the amendment in 2022 and our subsequent reverse stock splits, an aggregate of 175,000 shares were authorized under the 2020 Plan. On November 21, 2023, our stockholders approved an amendment to the 2020 Plan to increase the number of shares of Common Stock authorized for issuance thereunder from 175,000 shares to 300,000 shares. As of December 31, 2024, options to purchase an aggregate of 226,874 shares of our Common Stock at exercise prices ranging from $6.16 to $40.00 per share, with a weighted-average exercise price of $25.53 per share, were outstanding under the 2020 Plan, with 73,126 shares of our Common Stock remaining available for future issuance. Unissued shares subject to awards that expire, are forfeited, or are cancelled will again become available for issuance under the 2020 Plan.
Administration. Our Board, or a committee thereof, has administered the 2020 Plan since its adoption; however, following the IPO, the compensation committee of our Board has administered the 2020 Plan. Our compensation committee has complete discretion to make all decisions relating to the 2020 Plan and outstanding awards.
Eligibility. Key employees, directors and consultants and other persons who render services of special importance to our management, operation or development are eligible to participate in the 2020 Plan. However, only employees are eligible to receive incentive stock options.
Types of Awards. The 2020 Plan provides for the following types of awards granted with respect to shares of our Common Stock:
| ● | incentive and non-statutory stock options to purchase shares of our Common Stock; |
| ● | stock appreciation rights to purchase shares of our Common Stock; |
| ● | restricted stock units to acquire our Common Stock; |
| ● | direct award or sale of shares of our Common Stock, including restricted shares; and |
| ● | Other Common Stock based awards. |
Options. The exercise price for options granted under the 2020 Plan is determined by our board of directors, but may not be less than 100% of the fair market value of our Common Stock on the grant date. Optionees may pay the exercise price in cash or cash equivalents or by one, or any combination of, the following forms of payment, as permitted by the administrator in its sole discretion:
| ● | surrender of shares of Common Stock that the optionee already owns; |
| ● | delivery of a recourse promissory note, with the option shares pledged as security (along with other security as required by the Board) against the principal and accrued interest on the note; |
| ● | an immediate sale of the option shares through a company-approved broker, if the shares of our Common Stock are publicly traded; |
| ● | surrendering a number of vested shares subject to the option having an aggregate fair market value no greater than the aggregate exercise price, or the sum of such exercise price plus all or a portion of the amount required to be withheld under applicable law; or |
| ● | other methods permitted by the DGCL. |
Options vest as determined by the administrator. In general, we have granted options that vest over a three-year period. Options expire at the time determined by the Board, but in no event more than ten years after they are granted, and generally expire earlier if the optionee’s service terminates.
Stock Appreciation Rights.
The exercise price for stock appreciation rights granted under the 2020 Plan is determined by our Board, but may not be less than 100% of the fair market value of our Common Stock on the grant date. Stock appreciation rights may be settled in cash or our shares of Common Stock, as determined by the Board at the time of grant. A recipient may pay the exercise price in cash or cash equivalents or by one, or any combination of, the following forms of payment, as permitted by the administrator in its sole discretion:
| ● | surrender of shares of Common Stock that the recipient already owns; |
| ● | delivery of a recourse promissory note, with the stock appreciation right shares pledged as security (along with other security as required by the Board) against the principal and accrued interest on the note; |
| ● | an immediate sale of the stock appreciation right shares through a company-approved broker, if the shares of our Common Stock are publicly traded; |
| ● | surrendering a number of vested shares subject to the stock appreciation right having an aggregate fair market value no greater than the aggregate exercise price, or the sum of such exercise price plus all or a portion of the amount required to be withheld under applicable law; or |
| ● | other methods permitted by the DGCL. |
Stock appreciation rights vest as determined by the administrator. We have not granted any stock appreciation rights under the 2020 Plan.
Restricted Stock Units. Restricted stock units (a promise to deliver a number of our shares of Common Stock at a future date) may be awarded or sold under the 2020 Plan in return for cash or cash equivalents or, as permitted by the Board in its sole discretion, in exchange for services rendered to us. Restricted stock units may vest based on time or performance, as determined by the Board. We have not granted any restricted stock units under the 2020 Plan.
Restricted Shares. Restricted shares may be awarded or sold under the 2020 Plan in return for cash or cash equivalents or, as permitted by the Board in its sole discretion, in exchange for services rendered to us, by delivery of a full-recourse promissory note or through any other means permitted by applicable law. Restricted shares may vest based on time or performance, as determined by the Board.
Corporate Transactions. If we (i) merge or consolidate with another entity and our Common Stock is converted into or exchanged for the right to receive cash, securities or other property, or is cancelled, (ii) transfer or dispose of all of our Common Stock for cash, securities or other property pursuant to a share exchange or other transaction, (iii) sell or otherwise dispose of all or substantially all of our assets, or (iv) liquidate or dissolve (each, a “Reorganization Event”), then the Board may provide for any combination of the following:
| ● | the continuation, assumption or substitution of an award by the surviving entity or its parent (with or without similar vesting restrictions); |
| ● | after notice, provide for the accelerated vesting, exercisability and/or delivery, as applicable, immediately prior to the Reorganization Event and a lapse of the award upon occurrence of the Reorganization event; |
| ● | provide for a cash payment equivalent to what a holder of Common Stock would receive as a result of the Reorganization Event with respect to the vested portion of the award (less any exercise price or other amount paid, and any applicable withholdings) in exchange for a cancellation of the award; and/or; or |
| ● | in the event there is a liquidation or dissolution of the Company, provide that the vested portion of the award be converted into a right to receive liquidation proceeds (less any exercise price or other amount paid, and any applicable withholdings). |
The Board is not obligated to treat all awards in the same manner. The Board has the discretion, at any time, to provide that an award under the 2020 Plan will vest on an accelerated basis in connection with a corporate transaction or to amend or modify an award so long as such amendment or modification is not inconsistent with the terms of the 2020 Plan or would not result in the impairment of a participant’s rights without the participant’s consent.
Amendments or Termination. The Board may at any time amend, suspect or terminate the 2020 Plan, subject to stockholder approval in the case of an amendment requiring stockholder approval under the Internal Revenue Code, any rule of a stock exchange on which the Company’s securities are listed or any other applicable law. The 2020 Plan will terminate automatically ten years after the date on which the most recent amendment and restatement of such plan became effective, unless terminated earlier pursuant to its terms.
Item 13. Certain Relationships and Related Transactions, and Director Independence
Transactions with Related Parties
Other than as described below, except compensation arrangements, since the past two fiscal years, there have been no transactions, whether directly or indirectly, between us and any of the Company’s officers, directors, beneficial owners of more than 5% of outstanding shares of Common Stock or outstanding shares of a class of voting preferred stock, or their family members, that exceeded the lesser of (i) $120,000 or (ii) one percent (1%) of the average of the Company’s total assets at year-end for the last two fiscal years, and in which any of our directors, executive officers or beneficial holders of more than 5% of any class of our capital stock, or any immediate family member of, or person sharing the household with, any of these individuals, had or will have a direct or indirect material interest.
In August 2009 and January 2015, we issued an aggregate of $100,000 in promissory notes to our co-founder, Dr. Chancellor, of which an aggregate face value of approximately $75,000 had been outstanding immediately prior to our IPO (the “Chancellor Notes”). In connection with the IPO, the Company and Dr. Chancellor entered into a note cancellation and stock purchase agreement, pursuant to which the Chancellor Notes were cancelled and in connection therewith, Dr. Chancellor was issued an aggregate of 2,868 shares of Common Stock.
As of December 31, 2022, Jonathan Kaufman, the Company’s Chief Executive Officer, had contributed an aggregate of $250,000 in cash to the Company to support its continued operations, in the form of a note payable. This note was paid in full at maturity, and had a balance of $0 at December 31, 2023.
On March 13, 2024, the Company entered into Affiliate Stock Purchase Agreements with each of Drs. Kaufman and Chancellor, pursuant to which each purchased $100,000 of shares of Common Stock in cash from the Company at the then market price of $5.52 per share, resulting in the issuance of 18,113 shares of Common Stock to each of them. The shares were issued pursuant to an exemption from registration under Section 4(a)(2) of the Securities Act.
Review, Approval or Ratification of Transactions with Related Parties
We have adopted a written related-person transactions policy that provides that our executive officers, directors, nominees for election as a director, beneficial owners of more than 5% of our Common Stock and any members of the immediate family of the foregoing persons, are not permitted to enter into a material related-person transaction with us without the review and approval of our audit committee, or a committee composed solely of independent directors in the event it is inappropriate for our audit committee to review such transaction due to a conflict of interest. Such policy provides that any request for us to enter into a transaction with an executive officer, director, nominee for election as a director, beneficial owner of more than 5% of our Common Stock or with any of their immediate family members or affiliates, in which the amount involved exceeds the lesser of (i) $120,000 or (ii) one percent of the average of the Company’s total assets at year-end for the last two fiscal years, will be presented to our audit committee for review, consideration and approval. In approving or rejecting any such proposal, we expect that our audit committee will consider the relevant facts and circumstances available and deemed relevant to the audit committee, including, but not limited to, whether the transaction is on terms no less favorable than terms generally available to an unaffiliated third party under the same or similar circumstances and the extent of the related person’s interest in the transaction.
Item 14. Principal Accountant Fees and Services.
Urish Popeck & Co., LLC is our independent registered public accounting firm and performed the audits of our financial statements for the years ended December 31, 2024 and 2023. The following table sets forth all fees billed or to be billed for such periods:
| 2024 | 2023 | |||||||
| Audit fees (1) | $ | 127,319 | $ | 76,617 | ||||
| Audit-related fees (2) | — | — | ||||||
| Tax fees (3) | 16,805 | 14,108 | ||||||
| All other fees | — | — | ||||||
| Total | $ | 144,124 | $ | 90,725 | ||||
(1) “Audit fees” include fees for professional services rendered in connection with the audit of our annual financial statements, review of our quarterly condensed financial statements and advisory services on accounting matters that were addressed during the annual audit and quarterly review. This category also includes fees for services that were incurred in connection with statutory and regulatory filings or engagements, such as consents and review of documents filed with the SEC.
(2) “Audit-related fees” include fees billed for professional services rendered that are reasonably related to the performance of the audit or review of our financial statements including subscription for the online library of accounting research literature and are not reported under “Audit Fees”.
(3) “Tax fees” include fees for tax compliance. Tax compliance fees encompass a variety of permissible services, including technical tax advice related to federal and state income tax matters, and assistance with tax audits.
Policy on Audit Committee Pre-Approval of Audit and Permissible Non-Audit Services of Independent Auditors
Our Audit Committee pre-approves all audit and non-audit services provided by the independent auditors prior to the engagement of the independent auditors with respect to such services. The chairman of our Audit Committee has been delegated the authority by such committee to pre-approve interim services by the independent auditors other than the annual audit. The chairman of our Audit Committee must report all such pre-approvals to the entire Audit Committee at the next committee meeting.
PART IV
Item 15. Exhibits and Financial Statement Schedules.
| (a) | The following documents are filed as part of this Report: |
| (1) | Financial Statements: |
The audited balance sheets of the Company as of December 31, 2024 and December 31, 2023, the related statements of operations, changes in stockholders’ equity and cash flows for the years then ended, the footnotes thereto, and the report of Urish Popeck & Co., LLC, an independent registered public accounting firm, are filed herewith.
| (2) | Financial Schedules: |
None. Financial statement schedules have been omitted because they are either not applicable or the required information is included in the financial statements or notes thereto.
| (3) | Exhibits: |
The exhibits listed in the accompanying index to exhibits are filed with this Report or incorporated by reference into this Item 15(a)(3) as part of this Report.
(b) The following are exhibits to this Report and, if incorporated by reference, we have indicated the document previously filed with the SEC in which the exhibit was included.
| ● | Certain of the agreements filed as exhibits to this Report contain representations and warranties by the parties to the agreements that have been made solely for the benefit of such parties. These representations and warranties: |
| ● | may have been qualified by disclosures that were made to the other parties in connection with the negotiation of the agreements, which disclosures are not necessarily reflected in the agreements; |
| ● | may apply standards of materiality that differ from those of a reasonable investor; and |
| ● | were made only as of specified dates contained in the agreements and are subject to subsequent developments and changed circumstances. |
Accordingly, these representations and warranties may not describe the actual state of affairs as of the date that these representations and warranties were made or at any other time. Investors should not rely on them as statements of fact.
* Filed or furnished herewith, as applicable.
+ Indicates management contract or compensatory plan.
# The certifications furnished in Exhibits 32.1 and 32.2 hereto are deemed to be furnished with this Report and will not be deemed to be “filed” for purposes of Section 18 of the Exchange Act, except to the extent that the registrant specifically incorporates it by reference.
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this Report to be signed on its behalf by the undersigned, thereunto duly authorized.
| Lipella Pharmaceuticals Inc. | |||
| Date: March 27, 2025 | By: | /s/ Jonathan Kaufman | |
| Name: Jonathan Kaufman | |||
| Title: President and Chief Executive Officer | |||
| Date: March 27, 2025 | By: | /s/ Douglas Johnston | |
| Name: Douglas Johnston | |||
| Title: Chief Financial Officer | |||
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| Date: | March 27, 2025 | By: | /s/ Jonathan Kaufman |
| Jonathan Kaufman | |||
| Director | |||
| Date: | March 27, 2025 | By: | /s/ Michael Chancellor |
| Michael Chancellor | |||
| Director | |||
| Date: | March 27, 2025 | By: | /s/ Lori Birder |
| Lori Birder | |||
| Director | |||
| Date: | March 27, 2025 | By: | /s/ Byong (Christopher) Kim |
| Byong (Christopher) Kim | |||
| Director | |||
| Date: | March 27, 2025 | By: | /s/ Ryan Pruchnic |
| Ryan Pruchnic | |||
| Director | |||
| Date: | March 27, 2025 | By: | /s/ Naoki Yoshimura |
| Naoki Yoshimura | |||
| Director | |||
| Date: | March 27, 2025 | By: | /s/ Daniel Cohen |
|
Daniel Cohen Director |
LIPELLA PHARMACEUTICALS INC.
INDEX TO FINANCIAL STATEMENTS
| Page | |
| Report of Independent Registered Public Accounting Firm (PCAOB ID 1013) | F-2 |
| Audited Financial Statements | |
| Balance Sheets | F-3 |
| Statements of Operations | F-4 |
| Statements of Stockholders’ Equity (Deficit) | F-5 |
| Statements of Cash Flows | F-6 |
| Notes to the Financial Statements | F-7 |
F-
Report of Independent Registered Public Accounting Firm
Urish Popeck & Co., LLC Pittsburgh, PA
Stockholders and Board of Directors
Lipella Pharmaceuticals Inc.
Pittsburgh, PA
Opinion on the Financial Statements
We have audited the accompanying balance sheets of Lipella Pharmaceuticals Inc. (the “Company”) as of December 31, 2024 and 2023, the related statements of operations, stockholders’ equity (deficit), and cash flows for the years then ended, and the related notes (collectively referred to as the “financial statements”). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company at December 31, 2024 and 2023, and the results of its operations and its cash flows for the years then ended, in conformity with accounting principles generally accepted in the United States of America.
Going Concern Uncertainty
The accompanying financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 2 to the financial statements, the Company has suffered recurring losses from operations and has an accumulated deficit at December 31, 2024. These conditions raise substantial doubt about its ability to continue as a going concern. Management’s plans in regard to these matters are also described in Note 2. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Basis for Opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
/s/ Urish Popeck & Co., LLC
We have served as the Company's auditor since 2020.
Pittsburgh, PA
March 27, 2025
F-
Lipella Pharmaceuticals Inc.
Balance Sheets
| December 31, 2024 | December 31, 2023 | |||||||
| Assets | ||||||||
| Current Assets | ||||||||
| Cash and cash equivalents | $ | 2,184,863 | $ | 3,293,738 | ||||
| Grants receivable | 84,713 | 32,286 | ||||||
| Prepaid expenses | 347,676 | 103,256 | ||||||
| Total Current Assets | 2,617,252 | 3,429,280 | ||||||
| Property and Equipment | ||||||||
| Furniture, fixtures and equipment | 140,294 | 140,294 | ||||||
| Furniture, fixtures and equipment (Accumulated Depreciation) | (130,430 | ) | (127,544 | ) | ||||
| Furniture and fixtures, net | 9,864 | 12,750 | ||||||
| Other Assets | ||||||||
| Operating lease right of use asset | 46,754 | 135,144 | ||||||
| Total Assets | 2,673,870 | 3,577,174 | ||||||
| Liabilities and Stockholders’ Equity | ||||||||
| Current liabilities | ||||||||
| Accounts payable | 388,191 | 138,016 | ||||||
| Accrued expenses | 237,886 | 77,280 | ||||||
| Operating lease liability | 47,605 | 89,223 | ||||||
| Payroll liability | 80,735 | 80,836 | ||||||
| Total Current Liabilities | 754,417 | 385,355 | ||||||
| Operating lease liability, net of current portion | — | 47,371 | ||||||
| Total Liabilities | 754,417 | 432,726 | ||||||
| Stockholders’ Equity: | ||||||||
| Preferred stock, $0.0001 par value; 20,000,000 shares authorized; 25,975 shares Series B and 303,041 shares Series C issued and outstanding at December 31, 2024, -0- shares issued and outstanding at December 31, 2023 | $ | 33 | $ | — | ||||
| Common stock, $0.0001 par value; 200,000,000 shares authorized, 1,208,919 shares issued and outstanding at December 31, 2024 and 756,743 shares issued and outstanding at December 31, 2023 | 121 | 75 | ||||||
| Additional paid-in capital | 17,259,406 | 13,468,216 | ||||||
| Accumulated deficit | (15,340,107 | ) | (10,323,843 | ) | ||||
| Total stockholders' equity | 1,919,453 | 3,144,448 | ||||||
| Total liabilities and stockholders' equity | $ | 2,673,870 | $ | 3,577,174 | ||||
The accompanying notes are an integral part of these financial statements.
F-
Lipella Pharmaceuticals Inc.
Statements of Operations
| For the year ended December 31, | ||||||||
| 2024 | 2023 | |||||||
| Grant revenues | $ | 536,357 | $ | 449,617 | ||||
| Total revenues | 536,357 | 449,617 | ||||||
| Cost and expenses | ||||||||
| Research and development | 3,613,187 | 3,038,836 | ||||||
| General and administrative | 2,003,712 | 2,156,734 | ||||||
| Total costs and expenses | 5,616,899 | 5,195,570 | ||||||
| Loss from operations | (5,080,542 | ) | (4,745,953 | ) | ||||
| Other income (expense) | ||||||||
| Interest income, net | 64,278 | 137,836 | ||||||
| Interest expense related party | — | (10,848 | ) | |||||
| Total other income/(expense) | 64,278 | 126,988 | ||||||
| Loss before income taxes | (5,016,264 | ) | (4,618,965 | ) | ||||
| Provision for income taxes | — | — | ||||||
| Net Loss | $ | (5,016,264 | ) | $ | (4,618,965 | ) | ||
| Loss per share of Common Stock | ||||||||
| Basic | (4.79 | ) | (6.16 | ) | ||||
| Dilutive | (4.79 | ) | (6.16 | ) | ||||
| Weighted-average of shares of Common Stock outstanding: | ||||||||
| Basic | 1,048,185 | 750,383 | ||||||
| Dilutive | 1,048,185 | 750,383 | ||||||
The accompanying notes are an integral part of these financial statements.
F-
Lipella Pharmaceuticals Inc.
Statements of Stockholder’s Equity (Deficit)
| Series B
Convertible Preferred Stock |
Series C
Convertible Preferred Stock |
Common Stock | Additional | Accumulated | ||||||||||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | Shares | Amount | paid-in capital | Deficit | Total | ||||||||||||||||||||||||||||
| Balances, December 31, 2022 | — | $ | — | — | $ | — | 717,993 | $ | 72 | $ | 10,380,403 | $ | (5,704,878 | ) | $ | 4,675,596 | ||||||||||||||||||||
| Net loss | — | — | — | — | — | — | — | (4,618,965 | ) | (4,618,965 | ) | |||||||||||||||||||||||||
| Stock-based compensation | — | — | — | — | — | — | 1,355,016 | — | 1,355,016 | |||||||||||||||||||||||||||
| Issuance of warrants, net of issuance costs | — | — | 1,611,601 | 1,611,601 | ||||||||||||||||||||||||||||||||
| Warrants converted to Common Stock | 31,250 | 3 | (3 | ) | — | |||||||||||||||||||||||||||||||
| Shares issued for services | — | — | — | — | 7,500 | 1 | 121,199 | — | 121,200 | |||||||||||||||||||||||||||
| Balances, December 31, 2023 | — | — | — | — | 756,743 | 75 | 13,468,216 | (10,323,843 | ) | 3,144,448 | ||||||||||||||||||||||||||
| BS Check | ||||||||||||||||||||||||||||||||||||
| Net loss | — | — | — | — | — | — | — | (5,016,264 | ) | (5,016,264 | ) | |||||||||||||||||||||||||
| Share based compensation | — | — | — | — | — | — | 749,396 | — | 749,396 | |||||||||||||||||||||||||||
| Pre-funded warrants converted to Common Stock | — | — | — | — | 341,566 | 34 | (34 | ) | — | — | ||||||||||||||||||||||||||
| Issuance of Common Stock | — | — | — | — | 36,226 | 4 | 199,996 | — | 200,000 | |||||||||||||||||||||||||||
| Shares issued for services | — | — | — | — | 24,509 | 3 | 199,998 | — | 200,000 | |||||||||||||||||||||||||||
| Issuance of Common Stock and warrants, net of issuance costs of $468,588 | — | — | — | — | 49,875 | 5 | 820,327 | — | 820,332 | |||||||||||||||||||||||||||
| Issuance of Preferred Stock and warrants, net of issuance costs of $775,960 | 25,975 | 3 | 303,041 | 30 | — | — | 1,821,507 | — | 1,821,540 | |||||||||||||||||||||||||||
| Balances, December 31, 2024 | 25,975 | $ | 3 | 303,041 | $ | 30 | 1,208,919 | $ | 121 | $ | 17,259,406 | $ | (15,340,107 | ) | $ | 1,919,453 | ||||||||||||||||||||
The accompanying notes are an integral part of these financial statements.
F-
Lipella Pharmaceuticals Inc.
Statements of Cash Flows
| For the year ended December 31, |
||||||||
| 2024 | 2023 | |||||||
| Cash flow from operating activities: | ||||||||
| Net loss | $ | (5,016,264 | ) | $ | (4,618,965 | ) | ||
| Adjustments to reconcile net loss to net cash provided by (used in) operating activities: | ||||||||
| Depreciation and amortization | 2,886 | 1,685 | ||||||
| Shares issued for services | 200,000 | 121,200 | ||||||
| Non-cash stock option expense | 749,396 | 1,355,016 | ||||||
| Changes in operating assets and liabilities: | ||||||||
| Operating right of use asset | (599 | ) | (2,249 | ) | ||||
| Grants receivable | (52,427 | ) | 81,369 | |||||
| Prepaid expense | (244,420 | ) | 460,634 | |||||
| Accounts payable | 250,175 | (246,341 | ) | |||||
| Accrued expenses | 160,606 | (315,525 | ) | |||||
| Payroll liability | (100 | ) | 13,004 | |||||
| Net cash used in operating activities | (3,950,747 | ) | (3,150,172 | ) | ||||
| Cash flow from investing activities: | ||||||||
| Purchase of Property, plant and equipment | — | (14,434 | ) | |||||
| Net cash used in investing activities | — | (14,434 | ) | |||||
| Cash flow from financing activities: | ||||||||
| Proceeds from issuance of Preferred Stock, Common Stock and pre-funded warrants, net of issuance costs | 2,841,872 | 1,611,601 | ||||||
| Repayment of notes payable, related party | — | (250,000 | ) | |||||
| Repayment of notes payable | — | (25,000 | ) | |||||
| Net cash provided by financing activities | 2,841,872 | 1,336,601 | ||||||
| Net decrease in cash, cash equivalents | (1,108,875 | ) | (1,828,005 | ) | ||||
| Cash, and cash equivalents at beginning of period | 3,293,738 | 5,121,743 | ||||||
| Cash, and cash equivalents at end of period | $ | 2,184,863 | $ | 3,293,738 | ||||
| Supplemental disclosure of cash flow information: | ||||||||
| Interest paid | $ | (11,908 | ) | $ | (11,358 | ) | ||
| Income taxes paid | — | — | ||||||
| Supplemental disclosure of cash flow information: | ||||||||
| Issuance Common Stock options for consulting services | — | — | ||||||
The accompanying notes are an integral part of these financial statements.
F-
Lipella Pharmaceuticals Inc.
NOTES TO FINANCIAL STATEMENTS
1. Background
Lipella Pharmaceuticals Inc. (the “Company”, “we”, “us” or “our”) is a clinical-stage biotechnology company focused on developing new drugs by reformulating the active agents in existing generic drugs and optimizing these reformulations for new applications. Our operations consist of research, preclinical development and clinical development activities, and our most advanced program is in Phase 2a clinical development. Since our inception in 2005, we have historically financed our operations through a combination of federal grant revenue, licensing revenue, manufacturing revenue, as well as equity and debt financing. On December 19, 2022, a reverse stock split (hereafter, the “Stock Split”) was effected, with a 2.5 to 1 share conversion ratio for all shares of common stock outstanding. Subsequently, a reverse stock split was effected on November 8, 2024 (the “2024 Stock Split”, and together with the Stock Split, the “Stock Splits”) with an 8 to 1 share conversion ratio for all shares of common stock outstanding. The Company’s outstanding share and per share amounts in these financial statements have been adjusted to give effect to the Stock Splits, for all periods presented. For more information, see Note 11 - Common Stock.
2. Going Concern
The accompanying financial statements have been prepared in conformity with accounting principles generally accepted in the United States of America (“GAAP”), which contemplate continuation of the Company as a going concern. The Company has not established a source of revenues sufficient to cover its operating costs and will require significant additional capital to continue its research and development programs, including progressing our clinical product candidates to commercialization and preparing for commercial-scale manufacturing and sales.
The Company’s net loss for the years ended December 31, 2024 and 2023 was $5,016,264 and $4,618,965, respectively. Since inception, the Company has incurred losses and has an accumulated deficit of $15,340,107 at December 31, 2024. At December 31, 2024, the Company had available cash and cash equivalents of $2,184,863 and a net working capital of $1,862,835. The Company anticipates operating losses to continue for the foreseeable future due to, among other things, costs related to research, development of our product candidates, conducting preclinical studies and clinical trials, and our administrative organization. Management’s plans with respect to operations include the sustained and aggressive development and marketing of pharmaceutical products both domestically and abroad, and raising additional capital through sales of equity securities as may be necessary to pursue our business plans and sustain operations until such time as the Company can achieve profitability. These funds, and our funds available under existing government contracts, should be sufficient to enable us to meet our obligations as they come due for at least the next twelve months from the issuance date of these financial statements.
If we are unable to obtain additional capital (which is not assured at this time), our long-term business plan may not be accomplished, and we may be forced to curtail or cease operations. These factors individually and collectively raise substantial doubt about our ability to continue as a going concern. The accompanying financial statements do not include any adjustments that may result from this uncertainty.
3. Accounting Policies
Use of Estimates
The preparation of financial statements in conformity with GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements. Actual results could differ from those estimates.
Cash and Cash Equivalents
Cash and cash equivalents are comprised of deposits at major financial banking institutions, commercial paper, and highly liquid investments with an original maturity of three months or less at the date of purchase. Cash equivalents are primarily invested in money market funds. The carrying amount of our cash equivalents approximates fair value due to the short maturity of the investments. The Company regularly monitors the financial condition of the institutions in which it has depository accounts and believes the risk of loss is minimal.
F-
Marketable Debt Securities
Marketable debt securities consist of debt investments with original maturities greater than three months. The Company classifies its marketable debt securities as available-for-sale. Accordingly, these investments are recorded at fair value, which is based on quoted market prices. When the fair value is below the amortized cost the amount of the expected credit loss is estimated. The credit-related impairment amount is recognized in net income; the remaining impairment amount and unrealized gains are reported as a component of accumulated other comprehensive income in stockholders’ equity. Credit losses are recognized through the use of an allowance for credit losses account and subsequent improvements in expected credit losses are recognized as a reversal of the allowance account. If the Company has the intent to sell the security or if it is more likely than not that the Company will be required to sell the security prior to recovery of its amortized cost basis, the allowance for credit loss is written off and the excess of the amortized cost basis of the asset over its fair value is recorded in net income.
Grants Receivable
Grants receivable result from drawdown requests to various federal agencies for reimbursement of costs incurred during the operation of its research and development programs. Grants receivable are reported at net realizable value.
Accounts Receivable
We report accounts receivable at net realizable value. When required we estimate losses on uncollectible accounts receivable based upon historical data. Such allowance for doubtful accounts is estimated based upon management’s assessment of individual accounts. The Company concluded that an allowance for doubtful accounts is not considered necessary at December 31, 2024 and 2023.
Prepaid Expenses
Our insurance policies have a 12-month term and annually renew each June and December. Premiums are paid in advance either, annually, quarterly, or monthly. The collective value of any prepaid portions of policy terms is record at cost. Contracts involving pre-payment are capitalized and amortized in accordance with performance. In addition, costs directly associated with equity issuances for which the proceeds have not been received are deferred and will be recognized as an offset to proceeds received.
Long-lived Assets (equipment)
Fixed assets are recorded at cost and depreciated over their estimated useful lives.
Laboratory and office equipment, not covered by federal grant financing, are depreciated on the straight-line basis over the estimated useful lives (three to ten years). Leasehold improvements are amortized over the shorter of the lease term or estimated useful life. Long-lived assets are evaluated for impairment when events or changes in circumstances indicate that the carrying amount of the asset or related group of assets may not be recoverable. If the expected future undiscounted cash flows are less than the carrying amount of the asset, an impairment loss is recognized at that time. Measurement of impairment may be based upon appraisal, market value of similar assets or discounted cash flows.
Equipment expenditures that are covered by federal grant financing are depreciated using the activity method (or variable charge approach) employing an intended purpose that expires during or by the conclusion of the funded project. Such equipment acquisition expenditures are therefore effectively expensed if the timing of the intended purpose is within the same reporting period as delivery.
F-
At December 31, 2024, the Company had long-lived assets of $140,294, with accumulated depreciation of $130,430, for a net value $9,864. At December 31, 2023, the Company had long-lived assets of $140,294, with accumulated depreciation of $127,544, for a net value of $12,750. Depreciation expense was $2,886 for the year ended December 31, 2024 and $1,685 for December 31, 2023.
Accounts Payable
Accounts payable are short term liabilities with product/service vendors including any credit-card liability.
Accrued Expenses
Accrued expenses are recorded when incurred but have not been paid by year-end. See Note 6 – Accrued Expenses, related to the balance at December 31, 2024 and 2023.
Unearned Grant Revenue
Unearned grant revenue results from drawdown requests to various federal agencies for reimbursement of costs prior to being incurred during the operation of research and development programs. At December 31, 2024 and 2023, there were no unearned grant revenues.
Revenue Recognition
On January 1, 2018, the Company adopted Accounting Standards Update (“ASU”) No. 2014-09, Revenue from Contracts with Customers (“Topic 606”), to account for revenue. The deliverables under our arrangements are evaluated under Topic 606 which requires an entity to recognize revenue in a manner that depicts the transfer of promised goods or services to customers in an amount that reflects the consideration to which the entity expects to be entitled in exchange for those goods or services.
The Company recognizes revenue when persuasive evidence of an arrangement exists, delivery occurs, the fee is fixed or determinable, and collectability is reasonably assured.
The Company’s contract revenue consists primarily of amounts earned under contracts with third-party customers and reimbursed expenses under such contracts. The Company analyzes its agreements to determine whether the elements can be separated and accounted for individually or as a single unit of accounting. Allocation of revenue to individual elements that qualify for separate accounting is based on the separate selling prices determined for each component, and total contract consideration is then allocated pro rata across the components of the arrangement.
In general, the Company applies the following steps when recognizing revenue from contracts with customers: (i) identify the contract, (ii) identify the performance obligations, (iii) determine the transaction price, (iv) allocate the transaction price to the performance obligations and (v) recognize revenue when a performance obligation is satisfied.
Recognition of revenue is driven by satisfaction of the performance obligations using one of two methods: revenue is either recognized over time or at a point in time. Revenue is generally recognized as the performance obligations are satisfied, which generally occurs when control of the goods or services has been transferred to the customer or once the customer is able to use those goods and/or services as well as obtains substantially all of its benefits.
F-
The Company primarily generates contract revenue under the following types of contracts:
Fixed-fee
Under a fixed-fee contract, the Company charges a fixed agreed upon amount for a deliverable. Fixed-fee contracts have fixed deliverables upon completion of the project. Typically, the Company recognizes revenue for fixed-fee contracts as delivery is made and title transfers to the customer, and collection is reasonably assured.
Time and materials
Under a time and materials contract, the Company charges customers an hourly rate plus reimbursement for other project specific costs. The Company recognizes revenue for time and material contracts based on the number of hours devoted to the project multiplied by the customer’s billing rate plus other project specific costs incurred.
Payment terms vary, but are generally due within 60 days.
Contract Assets
A contract asset is our right to payment for goods and services already transferred to a customer if that right to payment is conditional on something other than the passage of time. Generally, we will recognize a contract asset when it has fulfilled a contract obligation but must perform other obligations before being entitled to payment. At December 31, 2024 and 2023, there were no contract assets related to contract revenues.
Contract Liabilities
A contract liability is our obligation to transfer goods or services to a customer when the customer prepays consideration.
Contract liabilities consist primarily of consideration received on project work to be performed whereby the Company expects to recognize related revenue at a later date, upon satisfaction of the contract obligations. Contract liabilities may also be described as deferred revenue. At December 31, 2024 and 2023, there were no contract liabilities related to contract revenues.
Disaggregation of Revenues
For the Company’s time and material projects, the Company recognizes revenue over time. This is generally due to the customer simultaneously receiving the benefit, while the Company is owed for its services to date and has an enforceable right and/or the Company would incur significant re-work of the specified item. The Company uses a method to best depict the transfer of control which is generally hours incurred (input method) or units produced (output method). For each of the years ended December 31, 2024 and 2023, the Company recognized $0 in revenues over time.
Grant Revenues
The Company has concluded its government grants are not within the scope of Topic 606, as government entities do not meet the definition of a “customer” as defined by Topic 606, because there is not considered to be a transfer of control of goods or services to the government entity funding the grant. Grant revenue, which is not within the scope of Topic 606, consists of funding under cost reimbursement programs primarily from federal and non-profit foundation sources for qualified research and development activities performed by us, and as such, are not based on estimates that are susceptible to change. Such amounts are invoiced and recorded as revenue as grant-funded activities are performed, with any advance funding recorded as deferred revenue until the activities are performed. The Company believes the recognition of revenue as costs are incurred and amounts become earned/realizable is analogous to the concept of transfer of control of a service over time under Topic 606. The Company’s grant revenues are primarily with the National Institutes of Health (“NIH”).
F-
Recently Adopted Accounting Pronouncements
In January 2019, we adopted ASU 2016-02, Leases (“Topic 842”), which requires lessees to put most leases with a term greater than 12 months on their balance sheets, but recognize expenses on their statement of operations in a manner similar to current accounting practice. Under the guidance, lessees initially recognize a lease liability for the obligation to make lease payments and a right-of-use (“ROU”) asset for the right to use the underlying asset for the lease term. The lease liability is measured at the present value of the lease payments over the lease term. The ROU asset is measured at the lease liability amount, adjusted for lease prepayments, lease incentives received and the lessee’s initial direct costs. The Company used the package of practical expedients permitted under the transition guidance that allowed us to not reassess: (1) whether any expired or existing contracts are or contain leases, (2) lease classification for any expired or existing leases and (3) initial direct costs for any expired or existing leases. The Company elected the practical expedient that allows lessees to treat the lease and non-lease components of leases as a single lease component. Additionally, the Company elected the hindsight practical expedient to determine the reasonably certain lease terms for existing leases. The adoption of this standard had no impact to the Company during 2019, as the Company’s lease was less than 12 months.
In September 2016, the FASB issued ASU 2016-13, Financial Instruments - Credit Losses, with additional updates and amendments being issued in 2018, 2019, 2020 and 2022 (collectively, “ASU 326”). The new standard updates the impairment model for financial assets measured at amortized cost, known as the Current Expected Credit Loss model. For trade and other receivables, held-to-maturity debt securities, loans, and other instruments, entities are required to use a new forward-looking “expected loss” model that generally results in the earlier recognition of an allowance for credit losses. The Company adopted ASU 326 on a modified retrospective basis as of January 1, 2024 with no impact to the Company.
In November 2023, the Financial Accounting Standards Board (“FASB”) issued ASU 2023-07, Segment Reporting (Topic 280): Improvements to Reportable Segment Disclosures (“ASU 2023-07”). The main provisions of ASU 2023-07 require (i) enhanced disclosures about significant segment expenses, (ii) extension of certain annual disclosures to interim periods and (iii) certain qualitative information on the chief operating decision maker. We adopted the annual reporting requirement under ASU 2023-07 for the year ended December 31, 2024. The adoption did not have a material impact on our consolidated financial statements. See Note 15 - Segment Information for additional information.
Recently Issued Accounting Pronouncements
In December 2023, the FASB issued ASU 2023-09, Income Taxes (Topic 740): Improvements to Income Tax Disclosures, to enhance the transparency and decision-usefulness of income tax disclosures, particularly in the rate reconciliation table and disclosures about income taxes paid. The ASU’s amendments are effective for annual periods beginning after December 15, 2024 on a prospective basis. Early adoption is permitted. We are currently evaluating the impact of adopting this ASU on our consolidated financial statements and related disclosures.
Lease Obligations
The Company determines if an agreement is a lease at inception. The Company evaluates the lease terms to determine whether the lease will be accounted for as an operating or finance lease. Operating leases are included in operating lease ROU assets, operating lease liabilities, current portion, and operating lease liabilities, net of current portion in our balance sheets.
ROU assets represent our right to use an underlying asset for the lease term and lease liabilities represent our obligation to make lease payments arising from the lease. Operating lease ROU assets and liabilities are recognized at a commencement date based on the present value of lease payments over the lease term. As most of our leases do not provide an implicit rate, we use our incremental borrowing rate based on the information available at commencement date in determining the present value of lease payments. The Company uses the implicit rate when readily determinable. The operating lease ROU asset also includes any lease payments made and excludes lease incentives. Our lease terms may include options to extend or terminate the lease when it is reasonably certain that we will exercise that option. Lease expense for lease payments is recognized on a straight-line basis over the lease term.
A lease that transfers substantially all of the benefits and risks incidental to ownership of property are accounted for as finance leases. At the inception of a finance lease, an asset and finance lease obligation is recorded at an amount equal to the lesser of the present value of the minimum lease payments and the property’s fair market value. Finance lease obligations are classified as either current or long-term based on the due dates of future minimum lease payments, net of interest. At December 31, 2024 and 2023, there were no finance leases.
Research and Development
The Company accounts for research and development costs in accordance with Accounting Standard Codification (“ASC”) 730-10, Research and Development (“ASC 730-10”). Under ASC 730-10, all research and development costs must be charged to expense as incurred. Accordingly, internal research and development costs are expensed as incurred. Third-party research and development costs are expensed when the contracted work has been performed. Research and development expenses include salaries and benefits, facilities and other overhead expenses, external clinical trial expenses, research related manufacturing services, contract services and other outside expenses.
F-
Patent Costs and Rights
Costs of applying for, prosecuting and maintaining patents and patent rights are expensed as incurred due to uncertainty of future economic benefit.
Clinical Trial Costs
Clinical trial costs are charged to us and recognized as the tasks are completed by the contractor(s) or, alternatively, may be invoiced in accordance with agreed-upon payment schedules and recognized based on estimates of work completed to date. These costs are included in research and development expenses in the accompanying statements of operations.
The Company recognizes the fair value of stock option award expenses consistent with vesting periods, using the Black-Scholes options pricing model to estimate fair value of option awards. Such expenses are categorized as research and development or general and administrative depending on the role of each option recipient. Service condition forfeitures are recognized when they occur.
The Company recognizes income tax expense or benefit in the statement of operations and the tax effects of exercised or vested awards are treated as discrete items in the reporting period in which they occur. The Company also recognizes excess tax benefits regardless of whether the benefit reduces taxes payable in the current period. Excess tax benefits are classified along with other income tax cash flows as an operating activity in the statement of cash flows. Regarding forfeitures, the Company accounts for them when they occur.
Warrants
The Company accounts for common stock warrants as either liabilities or as equity instruments depending on the specific terms of the warrant agreements. Generally, warrants are classified as liabilities, as opposed to equity, if the agreement includes the potential for a cash settlement or an adjustment to the exercise price, and warrant liabilities are recorded at their fair values at each balance sheet date. For the Company, our warrants at December 31, 2024 and 2023 do not contain the cash settlement or adjustment to exercise price. See Note 12 related to issued warrants.
Income Taxes
The Company accounts for income taxes in accordance with the asset and liability method of accounting for income taxes prescribed by FASB ASC Topic 740, “Accounting for Income Taxes” (“ASC 740”). Under the asset and liability method of ASC 740, deferred tax assets and liabilities are recognized for the future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax basis and operating loss and tax credit carry-forwards. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to the taxable income in the years in which those temporary differences are expected to be recovered or settled. Under ASC 740, the effect on deferred tax assets and liabilities of a change in tax rates is recognized in income in the period that includes the enactment dates. The Company evaluates its deferred income taxes to determine if a valuation allowance should be established against the deferred tax assets or if the valuation allowance should be reduced based on consideration of all available evidence, both positive and negative, using a “more likely than not” standard.
F-
The Company follows FASB ASC Topic 740-10, “Accounting for Uncertainty in Income Taxes”, which prescribes a recognition threshold and measurement attribute for financial statement recognition and measurement of a tax position taken, or expected to be taken, in a tax return. For those benefits to be recognized, a tax position must be more likely than not to be sustained upon examination by taxing authorities. At December 31, 2024 and 2023, the Company had no material uncertain tax positions to be accounted for in the financial statements. The Company recognizes interest and penalties, if any, related to unrecognized tax benefits in interest expense.
Concentration Of Credit Risk
The Company’s grant revenues and receivables were with the NIH. The NIH is an agency of the United States Department of Health & Human Services and the Company believes amounts are fully collectible from this agency. There were no contract revenues for the years ended December 31, 2024 and 2023.
Basic net loss per common share of common stock is computed by dividing the net loss for the period by the weighted-average number of shares of common stock outstanding during the period. Shares of the Company’s common stock underlying pre-funded warrants are included in the calculation of basic and diluted earnings per share. Diluted net loss per share of common stock is computed giving effect to all dilutive common stock equivalents, consisting of common stock options and warrants. Diluted net loss per share of common stock for the year ended December 31, 2024 and 2023 is the same as basic net loss per share of common stock as the common stock equivalents were anti-dilutive due to the net loss.
| December 31, | ||||||||
| 2024 | 2023 | |||||||
| Shares of common stock issuable under equity incentive plans outstanding | 361,624 | 306,625 | ||||||
| Shares of common stock issuable upon exercise of warrants | 311,389 | 328,029 | ||||||
| Shares of Common Stock issuable upon conversion of Series B Preferred stock | 972,162 | — | ||||||
| Shares of Common Stock issuable upon conversion of Series C Preferred stock | 97,216 | — | ||||||
| Common stock equivalents excluded from diluted net loss per share | 1,742,391 | 634,654 | ||||||
4. Fair Value Measurements and Marketable Debt Securities
In accordance with ASC 820, Fair Value Measurements and Disclosures, the Company measure its assets and liabilities at fair value. We apply the three-level valuation hierarchy as described in the ASC, which is based upon the transparency of input as of the measurement date. The three levels of inputs as defined are:
Level 1 - Inputs to the valuation methodology are quoted prices (unadjusted) for identical assets or liabilities in active markets.
Level 2 - Inputs to the valuation methodology include quoted prices for similar assets and liabilities in active markets, and inputs that are observable for the asset or liability, either directly or indirectly, for substantially the full term of the financial instruments.
Level 3 - Inputs to the valuation methodology are unobservable and significant to the fair value measurement.
At December 31, 2024 and 2023, the Company’s financial instruments consist primarily of: cash and cash equivalents, marketable securities, accounts payable and accrued liabilities. For cash equivalents, accounts payable and accrued liabilities, the carrying amounts of these financial instruments as of December 31, 2024 and 2023 were considered representative of their fair values due to their short term to maturity.
F-
At December 31, 2023 and 2024, the fair value input levels for cash equivalents are summarized below:
| December 31, 2024 | Level 1 | Level 2 | Level 3 | Total | ||||||||||||
| Cash Equivalents (maturity less than 90 days) | ||||||||||||||||
| Commercial Paper | $ | — | $ | — | $ | — | $ | — | ||||||||
| Corporate bonds | — | — | — | — | ||||||||||||
| Money market funds | 1,692,141 | — | — | 1,692,141 | ||||||||||||
| Total Cash equivalents | 1,692,141 | — | — | 1,692,141 | ||||||||||||
| Marketable Securities | — | — | — | — | ||||||||||||
| Total Cash equivalents and marketable securities | $ | 1,692,141 | $ | — | $ | — | $ | 1,692,141 | ||||||||
| December 31, 2023 | Level 1 | Level 2 | Level 3 | Total | ||||||||||||
| Cash Equivalents (maturity less than 90 days) | ||||||||||||||||
| Commercial Paper | $ | — | $ | — | $ | — | $ | — | ||||||||
| Corporate bonds | — | — | — | — | ||||||||||||
| Money market funds | 3,052,648 | — | — | 3,052,648 | ||||||||||||
| Total Cash equivalents | 3,052,648 | — | — | 3,052,648 | ||||||||||||
| Marketable Securities | — | — | — | — | ||||||||||||
| Total Cash equivalents and marketable securities | $ | 3,052,648 | $ | — | $ | — | $ | 3,052,648 | ||||||||
There were no marketable debt securities as of December 31, 2024 and December 31, 2023.
5. Prepaid Expenses
At December 31, 2024, prepaid expenses consisted of $66,128 of prepaid operating expenses, $13,864 in pre-paid testing services related to our clinical trial, and $267,684 of deposits with a third party CRO for our current clinical trial. At December 31, 2023, prepaid expenses consisted of $88,554 of prepaid insurance, and $14,702 of prepaid expenses.
On January 12, 2024, the Company financed certain insurance policies for an aggregate of $362,500 to be paid in nine equal monthly instalments of $31,531 with an initial downpayment of $90,625. The agreement bears interest at 10.392% per annum. As of December 31, 2024 the financing agreement was repaid in full.
6. Accrued Expenses
At December 31, 2024 accrued expenses consisted of $237,886 in accrued professional service expenses. At December 31, 2023 accrued expenses consisted of $19,575 in accrued clinical expenses, $5,655 in professional service expenses, and $52,050 in franchise tax expense.
7. Notes Payable – Related Party
In October and November 2022, the Company received cash contributions of $150,000 and $100,000, respectively, from Dr. Jonathan Kaufman, the Company’s Chief Executive Officer, to support its continued operations. In consideration for such contributions, the Company issued Mr. Kaufman a note payable due within one year with an aggregate face value of $250,000. The note accrued interest at a rate of 8.75% annually. The note and accrued interest were paid in full in June 2023. The balance of the note payable and accrued interest was $0 at December 31, 2023 and 2024.
8. Letter of Credit
The Company had a letter of credit with a bank permitting it to borrow an aggregate of $50,000 which was due upon demand. The letter of credit was collateralized by substantially all of the Company’s assets and personally guaranteed by Dr. Jonathan Kaufman, our co-founder and chief executive officer. The outstanding advances under the line of credit bear interest at the lending bank’s prime rate plus 3.10%. The outstanding balance at December 31, 2024 and 2023 was $0, respectively. The letter of credit has been paid in full since February 2023.
F-
The Company has two stock incentive plans (each, a “Stock Option Plan”), each of which provides for the grant of both incentive stock options and nonqualified stock options. Under the terms of the Stock Option Plans, the maximum number of shares of common stock for which incentive and/or nonqualified options may be issued is 434,750 shares. This number is comprised of 134,750 options already issued and outstanding (non-expired) from the 2008 stock option plan, and 300,000 options as the maximum issuable under the 2020 stock option plan. Incentive stock options are granted with an exercise price determined by the Board of Directors (the “Board”). Unless otherwise provided for in an associated board consent, vesting terminates once the optionee is no longer affiliated with the Company. These options generally expire 10 years from the date of the grant. Stock options are granted with an exercise price not less than the fair market value of the underlying common stock on the date of the grant. Unless otherwise specified by the Board, all grants vest fully over a three-year period, provided that the employee continues to be employed. Vesting terminates once the optionee is no longer an employee. If an employee leaves the Company prior to fully vesting their option awards, the remaining unvested portion is considered forfeited, and the earlier recognition of the unvested shares is reversed during the period of forfeiture. As of December 31, 2024, there were no unrecognized compensation costs related to non-vested share-based compensation arrangements granted to be recognized over the remaining vesting period of less than one year.
The Company recognized $749,396 of compensation costs for the year ended December 31, 2024 and $1,355,016 of compensation costs for the year ended December 31, 2023.
| Shares |
Weighted- Average Exercise Price Per Share ($) |
Weighted- Remaining Contractual
|
Aggregate intrinsic value ($) |
||||||||||||||
| Outstanding as of December 31, 2022 | 256,750 | $ | 22.72 | 5.51 | $ | 605.687 | |||||||||||
| Granted | 52,999 | $ | 17.52 | — | |||||||||||||
| Expired | — | — | |||||||||||||||
| Cancelled | (3,125 | ) | $ | 17.52 | |||||||||||||
| Exercised | — | — | |||||||||||||||
| Outstanding as of December 31, 2023 | 306,624 | $ | 21.84 | 5.19 | $ | — | |||||||||||
| Granted | 55,000 | $ | 6.16 | ||||||||||||||
| Expired | — | — | |||||||||||||||
| Cancelled | — | — | |||||||||||||||
| Exercised | — | — | |||||||||||||||
| Outstanding as of December 31, 2024 | 361,624 | $ | 19.74 | 5.07 | $ | — | |||||||||||
| Vested as of December 31, 2024 | 361,624 | ||||||||||||||||
| Exercisable as of December 31, 2024 | 361,624 | ||||||||||||||||
| Exercisable as of December 31, 2023 | 284,042 | ||||||||||||||||
F-
|
Number of Shares underlying stock options |
Weighted- Average Fair Value Grant Date |
||||||||
| Nonvested at December 31, 2022 | 54,332 | $ | 22.56 | ||||||
| Granted | 53,000 | 17.52 | |||||||
| Vested | (84.750 | ) | 14.32 | ||||||
| Expired | — | — | |||||||
| Nonvested at December 31, 2023 | 22,582 | $ | 22.48 | ||||||
| Granted | 55,000 | 4.40 | |||||||
| Vested | (77,582 | ) | 5.74 | ||||||
| Expired | — | — | |||||||
| Nonvested at December 31, 2024 | — | $ | — | ||||||
Stock Option Grants - During the years ended December 31, 2024 and 2023, the Company issued stock options on the following dates:
On June 16, 2023, the Company issued 53,000 stock options at a $17.52 strike price, vesting immediately upon issuance.
On March 15, 2024, the Company issued 55,000 stock options at a $6.16 strike price, vesting immediately upon issuance.
| 2024 | 2023 | |||||||
| Weighted-average exercise price of options granted | $ | 4.40 | 12.00 | |||||
| Expected volatility | 86.175 | % | 83.5 | % | ||||
| Expected life (in years) | 5.17 | 5.04 | ||||||
| Risk-free interest rate (range) | 4.33 | % | 3.99 | % | ||||
| Expected dividend yield | $ | — | — | |||||
10. Preferred Stock
The Company’s Amended and Restated Certificate of Incorporation authorizes the issuance of 20,000,000 shares of Preferred Stock, par value $0.0001 per share. Below is a summary of the various Series of preferred stock to date:
Series A: There were no shares of Series A Stock outstanding at December 31, 2024 or December 31, 2023. The Series A Preferred Stock was cancelled and eliminated by the Company on April 11, 2024.
F-
Series B: On December 20, 2024, the Certificate of Designation of Preferences, Rights and Limitations of Series B Preferred Stock (the “Series B Certificate of Designation”), authorizing 144,000 shares of Series B non-voting convertible preferred stock, par value $0.0001 per share, of the Company (the “Series B Preferred Stock”) and establishing the rights, preferences, privileges, qualifications, restrictions, and limitations of the Series B Preferred Stock became effective upon the Company’s filing with the Secretary of State of the State of Delaware (the “Delaware Secretary of State”) of the Series B Certificate of Designation and a Certificate of Correction to the Series B Certificate of Designation on such date (the “Certificate of Correction”), which corrected the effective date of the Series B Certificate of Designation to December 20, 2024.
Pursuant to the Series B Certificate of Designation, each share of Series B Preferred Stock is convertible at any time into such number of shares of Common Stock obtained by the quotient of (i) the product of (x) such number of shares of Preferred Stock being converted by such holder and (y) a stated value of $100 and (ii) the Minimum Price (as defined in Rule 5635(d)(1)(A) of The Nasdaq Stock Market LLC). Holders of shares of Series B Preferred Stock do not have any voting rights other than certain limited voting rights with respect to actions which may adversely affect the Series B Preferred Stock. A holder of shares of Series B Preferred Stock cannot convert such shares to the extent that such holder would own more than 4.99% (or at the election of such holder, 9.99%) of outstanding Common Stock immediately after such conversion. The Series B Preferred Stock ranks senior to the Common Stock and any class or series of capital stock created after the Series B Preferred Stock with respect to the preferences as to dividends, distributions and payments upon the liquidation, dissolution and winding up of the Company. The Series B Certificate of Designation also contains certain standard adjustment provisions as are customarily included in similar derivative securities.
On December 23, 2024, the Company issued an aggregate of 22,295 shares of Series B Preferred Stock at a purchase price of $100 per share, for an aggregate of $2,229,500, in connection with a best efforts private offering in one or more closings of up to $6,000,000 of shares of Series B Preferred Stock (the “Offering”). The shares of Series B Preferred Stock were convertible into 854,214 shares of common stock, par value $0.0001 per share, of the Company (“Common Stock”) at a conversion price of $2.61 per share, subject to customary adjustments.
On December 31, 2024, in connection with the Offering, the Company issued and sold to certain investors an aggregate of 3,680 shares of Series B Preferred Stock and received gross proceeds of $368,000. Such shares of Series B Preferred Stock were convertible into 117,948 shares of Common Stock, at a conversion price of $3.12 per share, subject to customary adjustments.
Series C: On December 23, 2024, the Company filed the Certificate of Designation of Preferences, Rights and Limitations of Series C Convertible Preferred Stock (the “Series C Certificate of Designation”), with the Delaware Secretary of State, authorizing 1,050,000 shares of the Company’s Series C voting convertible preferred stock, par value $0.0001 per share (the “Series C Preferred Stock”), establishing the rights, preferences, privileges, qualifications, restrictions, and limitations relating to the Series C Preferred Stock. Pursuant to the Series C Certificate of Designation, each share of Series C Preferred Stock is convertible into one share of Common Stock at any time only on or after the date on which the applicable Registration Statement (as defined in the Series C Certificate of Designation) has been declared effective by the SEC. A holder of shares of Series C Preferred Stock cannot convert such shares to the extent that such holder would own more than 4.99% (or at the election of such holder, 9.99%) of outstanding Common Stock immediately after such conversion. The Series C Preferred Stock ranks pari passu to all shares of Common Stock and junior to all other shares of capital stock of the Company authorized or designated before or after the date of designation of the Series C Preferred Stock with respect to the preferences as to dividends, distributions and payments upon the liquidation, dissolution and winding up of the Company. The Series C Certificate of Designation also contains certain standard adjustment provisions as are customarily included in similar derivative securities.
The Company and Spartan Capital Securities, LLC (“Spartan”) entered into that certain (i) placement agent agreement, dated December 5, 2024, as amended by that certain amendment to consulting agreement and placement agent agreement, made as of December 10, 2024, between the Company and Spartan (the “Amendment”), as further amended by that certain second amendment to placement agent agreement, dated February 23, 2025, by and between the Company and Spartan and (ii) consulting agreement and advisory agreement, made as of December 5, 2024, as amended by the Amendment, as further amended by that certain second amendment to consulting agreement and advisory agreement, dated February 28, 2025, by and between the Company and Spartan (collectively, the “Spartan Agreements”), pursuant to which Spartan agreed to provide placement agent and consulting services in connection with the Offering. Also in connection with the Spartan Agreements, pursuant to that certain irrevocable proxy and power of attorney between Spartan and Dr. Kaufman (the “Proxy”), Spartan agreed to grant to Dr. Kaufman all voting power over and power of attorney with respect to all such shares of Series C Preferred Stock, and all shares of Common Stock issuable upon conversion of such shares or exercise of common stock purchase warrants (each, a “Placement Agent Warrant”), issued or issuable to Spartan or its designee in connection with the Offering.
On December 23, 2024, in accordance with the Spartan Agreements, the Company issued to Spartan and its designee an aggregate of 260,108 shares of Series C Preferred Stock at a conversion price of $1.00 per share, subject to customary adjustments, and a Placement Agent Warrant to purchase up to 85,421 shares of Common Stock at an exercise price of $1.00 per share, subject to customary adjustments thereunder. See Note 12 - Warrants for further discussion of the Placement Agent Warrants.
F-
On December 31, 2024, in connection with an additional closing of the Offering, the Company issued Spartan and its designee an aggregate of 42,933 shares of Series C Preferred Stock, and a Placement Agent Warrant to purchase up to 11,795 shares of Common Stock.
The Company’s Second Amended and Restated Certificate of Incorporation authorizes the issuance of 200,000,000 shares of Common Stock. On December 19, 2022, a reverse stock split was effected, with a 2.5 to 1 share conversion ratio for all shares of common stock outstanding, and on November 7, 2024, a reverse stock split was effected with an 8 to 1 shares conversion ratio for all shares outstanding. The Company’s outstanding share and per share amounts in these financial statements have been adjusted to give effect to the Stock Splits, for all periods presented. There were 1,208,919 shares of common stock outstanding as of December 31, 2024, and there were 756,743 shares of common stock outstanding as of December 31, 2023.
During the year ended December 31, 2022, 2,868 shares of Common Stock were issued in forgiveness of two related party notes, along with accrued interest on the notes, with a value of $138,810. See Note 7 – Notes Payable – Related Party, for more information. On December 22, 2022, we completed an initial public offering (“IPO”) and listing on the Nasdaq Capital Market (“Nasdaq”) of our Common Stock at a price to the public of $46 per share, which resulted in issuance of an additional 152,174 shares. The aggregate net proceeds from the IPO were approximately $5.0 million after deducting underwriting discounts and commissions of $630,000 and offering expenses of approximately $1,160,000.
On November 28, 2023, we issued 31,250 shares of Common Stock for the exercise of the same number of pre-funded warrants. See Note 12 – Warrants, for details of the pre-funded warrants. During the year ended December 31, 2023, the Company also issued 7,500 shares of Common Stock in exchange for services rendered by a third party.
On February 5, 2024, we issued 24,509 shares of Common Stock in exchange for services rendered by a third party.
On March 4, 2024, we issued 62,500 shares of Common Stock for the exercise of the same number of pre-funded warrants.
On March 13, 2024, the Company entered into Affiliate Stock Purchase Agreements with each of Jonathan H. Kaufman, the Company’s Chief Executive Officer and Chairman of its board of directors, and Michael B. Chancellor, the Company’s Chief Medical Officer and a member of its board of directors, pursuant to which each of Drs. Kaufman and Chancellor purchased $100,000 of shares of common stock of the Company, in cash from the Company at $5.52 per share, based on the official closing price of The Nasdaq Stock Market LLC for the Common Stock on March 13, 2024, resulting in the issuance of 18,113 shares of Common Stock to each of Drs. Kaufman and Chancellor.
On May 3 2024, we issued 70,691 shares of Common Stock for the exercise of the same number of pre-funded warrants.
On August 1, 2024, we issued 49,875 shares of our Common Stock to an institutional investor at an offering price of $4.96 per share, along with 208,375 pre-funded warrants to purchase the same number of shares of Common Stock (the “August 2024 Offering”). See Note 12- Warrants, for details of the pre-funded warrants.
On August 16, 2024, 12,500 shares of Common Stock were issued for the exercise of the same number of pre-funded warrants. On September 17, 2024, 28,125 shares of Common Stock were issued for the exercise of the same number of pre-funded warrants. On September 24, 2024, 25,000 shares of Common Stock were issued for the exercise of the same number of pre-funded warrants. On September 26, 2024, an additional 25,000 shares of Common Stock were issued for the exercise of the same number of pre-funded warrants. On October 15, 2024, 117,750 shares of Common Stock were issued for the exercise of the remaining pre-funded warrants.
F-
The Common Stock is subject to and qualified by the rights of the Preferred Stock. Upon the dissolution or liquidation of the Company, the holders of Common Stock will be entitled to receive all assets of the Company available for distribution to its stockholders, subject to any preferential rights of any then outstanding Preferred Stock.
Notice of Failure to Satisfy Nasdaq Minimum Bid Price Requirement
As disclosed in the Company’s Current Report on Form 8-K filed with the SEC on April 19, 2024, on April 17, 2024, the Company received a written notification (the “April Nasdaq Letter”) from the Nasdaq Listing Qualifications staff (the “Staff”) of The Nasdaq Stock Market LLC notifying the Company that, based upon the closing bid price of the Common Stock for the last 30 consecutive business days, the Company was not in compliance with the requirement to maintain a minimum bid price of $1.00 per share of its Common Stock, as set forth in Nasdaq Listing Rule 5550(a)(2) (the “Minimum Bid Price Requirement”). The April Nasdaq Letter had no immediate effect on the listing of the Common Stock, which continues to trade on Nasdaq.
Nasdaq provided the Company with 180 calendar days, or until October 14, 2024, to regain compliance with the Minimum Bid Price Requirement.
As disclosed in the Company’s Current Report on Form 8-K filed with the SEC on August 23, 2024, the Company received a letter from the Staff on August 21, 2024 stating that it was not in compliance with Nasdaq Listing Rule 5550(b)(1), which requires companies listed on Nasdaq to maintain a minimum of $2,500,000 in stockholders’ equity for continued listing (the “Stockholders’ Equity Requirement”). The Company reported stockholders’ equity of $1,703,798 in its Quarterly Report on Form 10-Q for the quarter ended June 30, 2024, and, as a result, it was not in compliance with the Stockholders’ Equity Requirement.
As disclosed in our Current Report on Form 8-K filed with the SEC on October 18, 2024, the Staff notified the Company on October 16, 2024 that it would delist the Common Stock from Nasdaq, and in response, the Company timely requested an appeal of such notice to a Nasdaq hearing panel (the “Panel”). The Nasdaq hearing was held on December 12, 2024.
12. Warrants
On October 23, 2023, the Company entered into a securities purchase agreement (the “Purchase Agreement”) with an institutional investor for the issuance and sale in a private placement (the “2023 Private Placement”) of pre-funded common stock purchase warrants (“Pre-Funded Warrants”) to purchase up to 164,473 shares of Common Stock, with an exercise price of $0.001 per share, and common stock purchase warrants (the “2023 Warrants”) to purchase up to 164,473 shares of Common Stock, with an exercise price of $11.20 per share. The gross proceeds to the Company from the 2023 Private Placement were approximately $2.0 million, before deducting placement agent fees and expenses and offering expenses payable by the Company. The 2023 Warrants and the Pre-Funded Warrants are immediately exercisable for three years from issuance and are subject to 4.99% and 9.99% beneficial ownership limitations (as applicable). The combined purchase price for one Pre-Funded Warrant and one accompanying 2023 Warrant was $12.152. The closing of the 2023 Private Placement contemplated by the Purchase Agreement occurred on October 25, 2023. All of the Pre-Funded Warrants from the 2023 Private Placement were converted to Common Stock on a one-for-one basis as follows: 31,250 pre-funded warrants converted on November 2, 2023, 62,500 pre-funded warrants were converted on March 4, 2024, and 70,691 pre-funded warrants were converted on May 3, 2024.
F-
On August 1, 2024, the Company conducted another private placement offering (the “August 2024 Offering”). In connection therewith, the Company issued pre-funded warrants to purchase up to 208,375 shares of Common Stock at a price of $4.952, with an exercise price of $0.008 per share. The pre-funded warrants were immediately exercisable, subject to 4.99% and 9.99% beneficial ownership limitations (as applicable) and were exercisable at any time until exercised in full. The securities issued in the August 2024 Offering were issued pursuant to a prospectus supplement, dated July 31, 2024, to the registration statement on Form S-3 (File No. 333-276815), declared effective by the SEC on February 8, 2024. In connection with the August 2024 Offering and pursuant to an engagement letter with H.C. Wainwright & Co. LLC (the “August 2024 Placement Agent”), who served as the placement agent for the August 2024 Offering, the Company agreed to issue to the August 2024 Placement Agent, as partial compensation for its services as the August 2024 Placement Agent, warrants to purchase up to 19,368 shares of Common Stock, which were exercisable immediately, subject to 4.99% and 9.99% beneficial ownership limitations (as applicable), expire on July 31, 2029, and have an exercise price of $6.20 per share. The gross proceeds to the Company from the August 2024 Offering were approximately $1,280,920, before deducting placement agent fees and expenses and offering expenses payable by the Company. All of the pre-funded warrants from the August 2024 Offering were converted to Common Stock on a one-for-one basis as follows: 12,500 pre-funded warrants were converted on August 16, 2024, 28,125 pre-funded warrants were converted on September 17, 2024, 25,000 pre-funded warrants were converted on September 24, 2024, 25,000 pre-funded warrants were converted on September 26, 2024, and 117,750 pre-funded warrants were converted on October 15, 2024.
In connection with the sale of the Series B Preferred Stock on December 23, 2024, and in accordance with the Spartan Agreements, the Company issued to the Placement Agent and its designee a Placement Agent Warrant to purchase up to 85,421 shares of Common Stock. On December 31, 2024, pursuant to the Spartan Agreements, the Company issued Spartan an additional Placement Agent Warrant to purchase up to 11,795 shares of Common Stock. The Placement Agent Warrants are immediately exercisable at a price of $1.00 per share of Common Stock, or through a cashless exercise according to a formula where the number of shares received is reduced by the difference in the fair market value per share and the exercise price per share. The Placement Agent Warrants expire five years from issuance. In addition, pursuant to the Proxy, Spartan agreed to grant to Dr. Kaufman all voting power over and power of attorney with respect to all shares of Common Stock issuable upon exercise of the Placement Agent Warrants. The Placement Agent Warrants qualified for equity accounting treatment, as they do not have any continuing or performance obligations, they are not linked to debt instruments, and do not require net cash settlement. Subsequently, all 97,216 Placement Agent Warrants were converted to 64,702 shares of Common Stock on January 23, 2025 via the cashless exercise option.
The Company has warrants outstanding to private investors convertible to 17,995 shares of Common Stock. The warrants were issued at various dates prior to December 31, 2021 with an exercise share price of $40, and expiring at various dates between October 2025 and June 2026.
The Company had no warrant liabilities at December 31, 2024 and 2023.
13. Commitments and contingencies
Operating Leases
On January 1, 2019, the Company adopted ASC Topic 842, Leases, which requires operating leases to be recorded as ROU assets and lease liabilities on the balance sheet. ROU assets represent our right to use the leased asset for the lease term and lease liabilities represent our obligation to make lease payments. Operating lease ROU assets and liabilities are recognized at commencement date based on the present value of lease payments over the lease term. As most of the Company’s leases do not provide an implicit rate, the Company uses its estimated incremental borrowing rate at the commencement date to determine the present value of lease payments. The operating lease ROU assets also include any lease payments made and exclude lease incentives.
F-
The Company entered into a lease agreement beginning July 1, 2020, for the Company’s principal headquarters on the fifth floor of 7800 Susquehanna Street, Pittsburgh, Pennsylvania, which includes office space and sterile manufacturing operations (the “Lease”). The Lease has a five-year term and includes an option for renewal, which is not reasonably certain and is excluded from the right of use calculation. On July 26, 2023, the Company entered a second lease for additional space on the fourth floor of the same building (the “Fourth Floor Lease”), commencing August 1, 2023 and co-terminating with the existing Lease on September 30, 2025. Subsequently effective January 1, 2024, the Company terminated the Fourth Floor Lease early at no penalty upon mutual agreement with the landlord and replaced it with a lease of additional space that had become available immediately adjacent to our existing offices (the “Suite 504 Lease”, and together with the “Lease”, “the Leases”). The Suite 504 Lease term co-terminates with the Lease. Future minimum rent payments under the Leases as December 31, 2024 are as follows:
| Total minimum lease payments 2025 | $ | 48,518 | ||
| Less: amount representing interest | $ | (913 | ) | |
| Present value of minimum lease payments | $ | 47,605 |
As of December 31, 2024, the Company had an ROU asset of $46,754 and a current operating lease liability of $47,605. As of December 31, 2023, the Company had an ROU asset of $135,144, and current and non-current operating lease liabilities of $89,223 and $47,371, respectively, recorded on the balance sheets. The lease expense for the year ended December 31, 2024 and 2023 was $96,930 and $77,763, respectively. The weighted average remaining lease term and discount rate was 0.5 year and 6.5%, respectively.
Contract Commitments
The Company enters into contracts in the normal course of business with CROs, CMOs, universities, and other third parties for preclinical research studies, clinical trials and testing and manufacturing services. These contracts generally do not contain minimum purchase commitments and are cancelable by us upon prior written notice although, purchase orders for clinical materials are generally non-cancelable. Payments due upon cancellation consist only of payments for services provided or expenses incurred, including non-cancelable obligations of our service providers, up to the date of cancellation or upon the completion of a manufacturing run.
14. Income Taxes
As of December 31, 2024, the Company had a U.S. federal net operating loss of approximately $6,474,046. Such operating losses may be used to reduce future taxable income and tax liabilities. The net operating loss expires at various dates between 2029 and 2042. Additionally, the Company has federal net operating loss carryforwards generated after 2017 of approximately $5,544,802 that have an indefinite life but with a limited usage of 80% of the taxable income in any given tax year. In addition, any sustained ownership changes defined by Section 382 of the Internal Revenue Code may subject the Company's net operating losses and other carryforward to an annual limitation. State net operating loss carryforwards may be used to reduce future taxable income and liabilities and will expire at various dates between 2029 and 2042. The Company’s state net operating loss usage is limited to 40% of the taxable income in any given tax year.
The primary components of the deferred tax assets are as follows:
| December 31, | 2024 | 2023 | ||||||
| Deferred tax assets: | ||||||||
| Net Operating loss carryforwards | $ | 1,792,928 | $ | 1,159,128 | ||||
| Interest on related party note | — | — | ||||||
| Stock Option Expense | 1,008,073 | 811,849 | ||||||
| R&D Expense | 1,391,484 | 872,178 | ||||||
| Other | 25,609 | 12,842 | ||||||
| Subtotal | 4,218,094 | 2,855,997 | ||||||
| Valuation allowance | (4,218,094 | ) | (2,855,997 | ) | ||||
| Net deferred income tax assets (liabilities) | — | — | ||||||
F-
Because of our cumulative losses, substantially all the deferred tax assets have been fully offset by a valuation allowance. We have not paid income taxes for the years ended December 31, 2024 and 2023.
The income tax provision attributable to loss before income tax benefit for the years ended December 31, 2024 and 2023 differed from the amounts computed by applying the U.S. federal statutory tax rate of 21.0% as a result of the following:
| For the years ended December 31, |
||||||||
| 2024 | 2023 | |||||||
| Statutory federal income tax rate | 21.0 | % | 21.0 | % | ||||
| State taxes, net of federal tax benefit | 6.71 | % | 7.11 | % | ||||
| Non-deductible parking expenses | 0.0 | % | 0.0 | % | ||||
| Change in valuation allowance | (27.71 | )% | (28.11 | )% | ||||
| Effective tax rate | 0.0 | % | 0.0 | % | ||||
The change in valuation allowance for the year ending December 31, 2024 was an increase of $1,362,097.
The Company’s 2020 through 2024 tax years remain subject to examination by the Internal Revenue Services for federal tax purposes and the Pennsylvania Department of Taxation for state purposes.
15. Segment Information
We report segment information based on ASC 280 Segment Reporting (“ASC 280”), which defines operating segments as components of a company that engage in activities from which it may recognize revenues and incur expenses, and for which operating results are regularly reviewed by the entity’s chief operating decision maker (“CODM”) to make decisions regarding resource allocation and assess performance, and for which discrete financial information is available. We determined that the Company is a single operating and reportable segment (“Products”) focused on treating the indications previously described through discovery, development, and commercialization of our novel therapeutics. Our chief executive officer acts as the CODM and allocates resources and assesses performance at a consolidated level.
Our Products segment measurement of profit is the consolidated loss from operations, and its measure of total assets is consolidated total assets. There was no difference in our consolidated balance sheet and our segment balance sheet. Our CODM uses loss from operations predominantly in the annual budget and forecasting process to monitor variances in budget versus actual results along with consolidated total assets to determine resource allocation. The following is a summary of the significant revenue and expense categories, and net (loss) provided to the CODM during the years ended December 31, 2024 and 2023:
Schedule of segment results
| For the year ended December 31, | 2024 | 2023 | ||||||
| Segment Results | ||||||||
| Grant Revenue | $ | 536,357 | 449,617 | |||||
| Operating expenses: | ||||||||
| Research and development | ||||||||
| Salaries and benefits, insurance | 899,519 | 804,925 | ||||||
| Outsourced R&D | 1,587,620 | 764,854 | ||||||
| Stock option expense | 624,711 | 1,015,287 | ||||||
| Facility and other overhead | 501,337 | 453,770 | ||||||
| Total Research and development | 3,613,187 | 3,038,836 | ||||||
| General and administrative | ||||||||
| Salaries and benefits, insurance | 639,921 | 709,223 | ||||||
| Outside services | 1,023,316 | 912,531 | ||||||
| Stock option expense | 124,685 | 339,729 | ||||||
| Facility and other overhead | 215,791 | 195,251 | ||||||
| Total General and Administrative | 2,003,712 | 2,156,734 | ||||||
| Total operating expenses | 5,616,899 | 5,195,570 | ||||||
| Loss from operations | (5,080,542 | ) | (4,745,953 | ) | ||||
| Other income (expense) | 64,278 | 126,988 | ||||||
| Segment loss from operations | $ | (5,016,264 | ) | (4,618,965 | ) | |||
| Adjusting and Reconciling items | — | — | ||||||
| Loss from operations | $ | (5,016,264 | ) | $ | (4,618,965 | ) | ||
F-
16. Subsequent Events
Subsequent events have been evaluated through the date of the independent auditors’ report, which is the date the financial statements were available to be issued.
On January 9, 2025, the Company financed certain insurance policies for an aggregate of $331,420 to be paid in nine equal monthly instalments of $28,870 with an initial downpayment of $82,855. The agreement bears interest at 10.75% per annum.
In January and February 2025, substantially all of the Series B and all of the Series C Preferred Stock, and Placement Agent Warrants issued and outstanding as of December 31, 2024 were converted to Common Stock as follows:
| ● | 97,216 Placement Agent Warrants were converted to 64,702 shares of Common Stock on January 23, 2025 via the cashless exercise option. |
| ● | 22,295 shares of Series B Preferred Stock were converted to 972,152 shares of Common Stock |
| ● | 303,041 shares of Series C Preferred Stock were converted to 303,041 shares of Common Stock |
Subsequent to December 31, 2024, additional investments were sold to private investors under the Spartan Agreements, in the following dates and amounts (collectively, the “2025 Closings”):
| ● | On February 25, 2025, we sold and issued 34,750 shares of Series B Preferred Stock for gross proceeds of $3,475,000. After closing costs, net proceeds to the Company were $2,884,250. Such Series B shares are convertible to 1,158,327 shares of Common Stock upon registration of the underlying shares. |
| ● | On February 26, 2025, we sold and issued 3,130 shares of Series B Preferred Stock for gross proceeds of $313,000. After closing costs, net proceeds to the Company were $239,790. Such Series B shares are convertible to 100,000 shares of Common Stock upon registration of the underlying shares. |
| ● | On March 3, 2025, we sold and issued 7,260 shares of Series B Preferred Stock for gross proceeds of $726,000. After closing costs, net proceeds to the Company were $602,580. Such Series B shares are convertible to 289,241 shares of Common Stock upon registration of the underlying shares. |
| ● | On March 12, 2025, we sold and issued 885 shares of Series B Preferred Stock for gross proceeds of $88,500. After closing costs, net proceeds to the Company were $73,455. Such Series B shares are convertible to 40,596 shares of Common Stock upon registration of the underlying shares. |
In connection with the 2025 Closings, the Company issued 536,959 shares of Series C Preferred Stock to Spartan and its designee, and such shares of Series C Preferred Stock are convertible into 536,959 shares of Common Stock upon registration of the underlying shares to the Placement Agent and its designee. We also issued Placement Agent Warrants exercisable for 158,817 shares of Common Stock to Spartan and its designee.
The issuance of these shares of Series B Preferred Stock in connection with the 2025 Closings completed the capital raise under the Spartan Agreements, resulting in $7,200,000 of gross proceeds raised.
F-
Offering of Warrants to Purchase Series B Preferred Stock
On March 17, 2025, the Company entered into an additional placement agent agreement with Spartan (the “Subsequent Placement Agent Agreement”), pursuant to which Spartan agreed to serve as exclusive placement agent for the private offer and sale (the “Subsequent Offering”) of warrants (the “Warrants”) to purchase up to 72,000 shares of Series B Preferred Stock at a price per Warrant equal to $0.125 in order to comply with applicable Nasdaq rules, with each Warrant exercisable for shares of Series B Preferred Stock at $100 per share, and such share of Series B Preferred Stock convertible into shares of Common Stock at a conversion price equal to $2.16 per share, which was the Minimum Price of the Common Stock immediately prior to the execution of the Subsequent Offering Subscription Agreements (as defined below).
On March 17, 2025 and in connection with the Subsequent Offering, the Company formally entered into subscription agreements (each, a "Subsequent Subscription Agreement") and registration rights agreements with investors in the Subsequent Offering, who were also participants in the 2025 Closings, for the purchase and sale of an aggregate of 72,000 of Warrants. In connection with the Subsequent Offering, the Company and Spartan entered into (i) the Subsequent Placement Agent Agreement and (ii) an additional consulting agreement and advisory agreement, dated March 17, 2025, pursuant to which Spartan agreed to provide placement agent and consulting services in connection with the Subsequent Offering and will receive certain compensation in consideration for such services, including, but not limited to, purchase warrants to purchase a number of shares of Common Stock equal to ten percent (10%) of the shares of Common Stock issuable upon conversion of the shares of Series B Preferred Stock that may be issued upon the Warrant exercises, (ii) a 2% non-accountable fee based on the aggregate number of proceeds received from Warrant exercises and (iii) shares of the Company's Series C voting convertible preferred stock, $0.0001 par value per share, to be issued on a pro rata basis at the time of and based upon the amount of such proceeds received from Warrant exercises.
F-
Additionally, in connection with the Subsequent Offering, Dr. Kaufman and Spartan entered into another irrevocable proxy and power of attorney, effective as of March 17, 2025 (the “Subsequent Offering Irrevocable Proxy”), pursuant to which, among other things, Spartan agreed to grant to Dr. Kaufman all voting power over and power of attorney with respect to all shares of Series C Preferred Stock, and all shares of Common Stock issuable upon conversion of shares of Series C Preferred Stock and upon exercise of Common Stock purchase warrants issued or issuable to Spartan or its Attribution Parties (as defined in the Subsequent Irrevocable Proxy) in connection with Warrant exercises.
Updates to Nasdaq Notifications
On January 10, 2025, the Company received a decision from the Panel granting the Company’s request for continued listing on Nasdaq, subject to the Company demonstrating compliance with the following: (i) on or before April 14, 2025, the Company must file a public disclosure describing any transactions undertaken by the Company to increase its equity and providing an indication of its equity following those transactions; and (ii) on or before April 14, 2025, the Company must provide the Panel with an update on its fundraising plans, updated income projections for the next 12 months, with all underlying assumptions clearly stated, and a description of how the Company intends to achieve, if necessary, and maintain compliance with the Minimum Bid Price Requirement.
F-
The Warrants were offered to the March Investors, and the March Offering Placement Agent Warrants and shares of Series C Preferred Stock will be issued to Spartan and its designee, as applicable, pursuant to the exemption from the registration requirements of the Securities Act provided in Section 4(a)(2) of the Securities Act and/or Regulation D promulgated thereunder.
Lease extension
On March 20, 2025, we notified Bridgeway Capital of our intent to renew our Leases for our headquarters at the end of their existing term, on June 30, 2025. The renewal will be for five years beginning July 1, 2025, as per the terms of our existing lease agreements. Rent under the Leases will be as follows:
Schedule of lease extension
| For the year ended | Total | |||
| December 31, 2025 (6 months only) | $ | 30,087 | ||
| December 31, 2026 | 61,435 | |||
| December 31, 2027 | 63,957 | |||
| December 31, 2028 | 66,478 | |||
| December 31, 2029 | 68,999 | |||
| December 31, 2030 (expires June 30, 2030) | 35,130 | |||
| Total rents under the Leases' renewal | $ | 326,087 | ||
F-
Exhibit 4.2
DESCRIPTION OF SECURITIES
REGISTERED PURSUANT TO SECTION 12 OF THE
SECURITIES EXCHANGE ACT OF 1934
As of December 31, 2024, Lipella Pharmaceuticals Inc. (the “Company,” “we,” “us” or “our”) has one class of securities registered under Section 12 of the Securities Exchange Act of 1934, as amended (the “Exchange Act”): our common stock, par value $0.0001 per share (our “Common Stock”).
General
The following description of our Common Stock is a summary and does not purport to be complete. It is subject to and qualified in its entirety by reference to our Second Amended and Restated Certificate of Incorporation, as amended (our “Charter”) and our Second Amended and Restated Bylaws (our “Bylaws”), each of which are incorporated by reference as an exhibit to the Annual Report on Form 10-K of which this Exhibit 4.2 is a part. Copies of these documents can be accessed through hyperlinks to those documents in the list of exhibits in our Annual Report on Form 10-K for the fiscal year ending December 31, 2024. We encourage you to read our Charter, our Bylaws, and the applicable provisions of the Delaware General Corporation Law (the “DGCL”) for additional information.
Authorized Capital Shares
Our authorized capital shares consist of (a) 200,000,000 shares of Common Stock and (b) 20,000,000 shares of “blank check” preferred stock, par value $0.0001 per share (our “Preferred Stock”), of which 144,000 shares of Preferred Stock were designated as Series B non-voting convertible preferred stock, par value $0.0001 per share, and 1,050,000 shares of Preferred Stock were designated as Series C voting convertible preferred stock, par value $0.0001 per share. The outstanding shares of our Common Stock are fully paid and nonassessable.
Voting Rights
The holders of shares of Common Stock vote together as one class on all matters as to which holders of Common Stock are entitled to vote. Except as otherwise required by applicable law, all voting rights are vested in and exercised by the holders of Common Stock with each share of Common Stock being entitled to one vote, including in all elections of directors. The vote of the holders of a majority of the issued and outstanding shares of Common Stock entitled to vote thereon is sufficient to authorize, affirm, ratify or consent to such act or action, except as otherwise provided by law.
Dividend Rights
Subject to the rights of holders of outstanding shares of preferred stock, if any, holders of Common Stock are entitled to receive such dividends and distributions and other distributions in cash, stock or property of the Company when, as and if declared thereon by the board of directors from time to time out of assets or funds of the Company legally available therefor.
Liquidation Rights
Subject to the rights of holders of outstanding shares of preferred stock, if any, upon our liquidation, dissolution or winding up, the holders of our Common Stock will be entitled to share ratably in the net assets and funds legally available for distribution to stockholders after the payment of all of our debts and other liabilities.
Other Rights and Preferences
Holders of our Common Stock have no pre-emptive rights or other subscription rights, conversion rights, registration rights, redemption or sinking fund provisions by virtue of only holding such shares.
Anti-Takeover Provisions
Provisions of the DGCL, our Charter, and our Bylaws could make it more difficult to acquire us by means of a tender offer, a proxy contest or otherwise, or to remove incumbent officers and directors. These provisions, summarized below, are expected to discourage certain types of coercive takeover practices and takeover bids that our board of directors may consider inadequate and to encourage persons seeking to acquire control of us to first negotiate with our board of directors. We believe that the benefits of increased protection of our ability to negotiate with the proponent of an unfriendly or unsolicited proposal to acquire or restructure us outweigh the disadvantages of discouraging takeover or acquisition proposals because, among other things, negotiation of these proposals could result in improved terms for our stockholders.
Section 203 of the DGCL. We are subject to Section 203 of the DGCL, which generally prohibits a publicly held Delaware corporation from engaging in any “business combination” with any interested stockholder for a period of three (3) years after the date that such stockholder became an interested stockholder, with the following exceptions:
● before such date, the board of directors of the corporation approved either the business combination or the transaction that resulted in the stockholder becoming an interested stockholder;
● upon completion of the transaction that resulted in the stockholder becoming an interested stockholder, the interested stockholder owned at least 85% of the voting stock of the corporation outstanding at the time the transaction began, excluding for purposes of determining the voting stock outstanding (but not the outstanding voting stock owned by the interested stockholder) those shares owned (i) by persons who are directors and also officers and (ii) employee stock plans in which employee participants do not have the right to determine confidentially whether shares held subject to the plan will be tendered in a tender or exchange offer; or
● on or after such date, the business combination is approved by the board of directors and authorized at an annual or special meeting of the stockholders, and not by written consent, by the affirmative vote of at least 66 2/3% of the outstanding voting stock that is not owned by the interested stockholder.
In general, Section 203 of the DGCL defines a “business combination” to include the following:
● any merger or consolidation involving the corporation and the interested stockholder;
● any sale, transfer, pledge or other disposition of 10% or more of the assets of the corporation involving the interested stockholder;
● subject to certain exceptions, any transaction that results in the issuance or transfer by the corporation of any stock of the corporation to the interested stockholder;
● any transaction involving the corporation that has the effect of increasing the proportionate share of the stock of any class or series of the corporation beneficially owned by the interested stockholder; or
● the receipt by the interested stockholder of the benefit of any loans, advances, guarantees, pledges or other financial benefits provided by or through the corporation.
In general, Section 203 of the DGCL defines an “interested stockholder” as an entity or person who, together with the person’s affiliates and associates, beneficially owns, or within three (3) years prior to the time of determination of interested stockholder status did own, 15% or more of the outstanding voting stock of the corporation.
Anti-Takeover Effects of Certain Provisions of our Bylaws
Our Bylaws provide that directors may be removed by the stockholders with or without cause upon the vote of a majority of the holders of Common Stock then entitled to vote. Except as otherwise provided in our Bylaws and our Charter, any vacancies or newly created directorships on our board of directors resulting from any increase in the authorized number of directors elected by all of the stockholders having the right to vote as a single class may be filled by a majority of the directors then in office, although less than a quorum, or by a sole remaining director.
Our Bylaws also provide that only our chairman of the board of directors, chief executive officer, president or one or more stockholders holding shares in the aggregate entitled to cast not less than ten percent of the votes at that meeting may call a special meeting of stockholders.
The combination of these provisions makes it more difficult for our existing stockholders to replace our board of directors as well as for another party to obtain control of us by replacing our board of directors. Since our board of directors has the power to retain and discharge our officers, these provisions could also make it more difficult for existing stockholders or another party to effect a change in management. In addition, the authorization of undesignated preferred stock makes it possible for our board of directors to issue preferred stock with voting or other rights or preferences that could impede the success of any attempt to change our control.
These provisions are intended to enhance the likelihood of continued stability in the composition of our board of directors and its policies and to discourage coercive takeover practices and inadequate takeover bids. These provisions are also designed to reduce our vulnerability to hostile takeovers and to discourage certain tactics that may be used in proxy fights. However, such provisions could have the effect of discouraging others from making tender offers for our shares and may have the effect of delaying changes in our control or management. As a consequence, these provisions may also inhibit fluctuations in the market price of our Common Stock that could result from actual or rumored takeover attempts. We believe that the benefits of these provisions, including increased protection of our potential ability to negotiate with the proponent of an unfriendly or unsolicited proposal to acquire or restructure the Company, outweigh the disadvantages of discouraging takeover proposals, because negotiation of takeover proposals could result in an improvement of their terms.
Listing
Our Common Stock is listed on The Nasdaq Capital Market under the symbol “LIPO”.
Transfer Agent and Registrar
The transfer agent and registrar for our Common Stock is Nevada Agency and Transfer Company. The transfer agent’s address is 50 West Liberty Street, Suite 880, Reno NV 89501 and its telephone number is (775) 322-0626.
Exhibit 23.1
Consent of Independent Registered Public Accounting Firm
We hereby consent to the incorporation by reference in the Registration Statements on Form S-3 No.’s 333-284172 and 333-276815, From S-1 No.’s 333-275245 and 333-266397, and Form S-8 No.’s 333-277961 and 333-272387 of Lipella Pharmaceuticals Inc. (the Company) of our report dated March 27, 2025, relating to the financial statements, which appear in this Annual Report on Form 10-K for the year ended December 31, 2024. Our report contains an explanatory paragraph regarding the Company’s ability to continue as a going concern.
/s/ Urish Popeck & Co., LLC
Pittsburgh, PA
March 27, 2025
Exhibit 31.1
CERTIFICATION
OF PRINCIPAL EXECUTIVE OFFICER
PURSUANT TO 18 U.S.C. SECTION 1350,
AS ADOPTED PURSUANT TO SECTION 302 OF
THE SARBANES-OXLEY ACT OF 2002
I, Jonathan Kaufman, certify that:
| 1. | I have reviewed this annual report on Form 10-K of Lipella Pharmaceuticals Inc.; |
| 2. | Based on my knowledge, this report does not contain any untrue statement of a material fact or omit to state a material fact necessary to make the statements made, in light of the circumstances under which such statements were made, not misleading with respect to the period covered by this report; |
| 3. | Based on my knowledge, the financial statements, and other financial information included in this report, fairly present in all material respects the financial condition, results of operations and cash flows of the registrant as of, and for, the periods presented in this report; |
| 4. | The registrant’s other certifying officer and I are responsible for establishing and maintaining disclosure controls and procedures (as defined in Exchange Act Rules 13a-15(e) and 15d-15(e)) and internal control over financial reporting (as defined in Exchange Act Rules 13a-15(f) and 15d-15(f)) for the registrant and have: |
| (a) | Designed such disclosure controls and procedures, or caused such disclosure controls and procedures to be designed under our supervision, to ensure that material information relating to the registrant, is made known to us by others within those entities, particularly during the period in which this report is being prepared; |
| (b) | Designed such internal control over financial reporting, or caused such internal control over financial reporting to be designed under our supervision, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles; |
| (c) | Evaluated the effectiveness of the registrant’s disclosure controls and procedures and presented in this report our conclusions about the effectiveness of the disclosure controls and procedures, as of the end of the period covered by this report based on such evaluation; and |
| (d) | Disclosed in this report any change in the registrant’s internal control over financial reporting that occurred during the registrant’s most recent fiscal quarter (the registrant’s fourth fiscal quarter in the case of an annual report) that has materially affected, or is reasonably likely to materially affect, the registrant’s internal control over financial reporting; and |
| 5. | The registrant’s other certifying officer and I have disclosed, based on our most recent evaluation of internal control over financial reporting, to the registrant’s auditors and the audit committee of the registrant’s board of directors (or persons performing the equivalent functions): |
| (a) | All significant deficiencies and material weaknesses in the design or operation of internal control over financial reporting which are reasonably likely to adversely affect the registrant’s ability to record, process, summarize and report financial information; and |
| (b) | Any fraud, whether or not material, that involves management or other employees who have a significant role in the registrant’s internal control over financial reporting. |
| Date: March 27, 2025 | By: | /s/ Jonathan Kaufman |
| Jonathan Kaufman |
Exhibit 31.2
CERTIFICATION
OF PRINCIPAL FINANCIAL OFFICER
PURSUANT TO 18 U.S.C. SECTION 1350,
AS ADOPTED PURSUANT TO SECTION 302 OF
THE SARBANES-OXLEY ACT OF 2002
I, Douglas Johnston, certify that:
| 1. | I have reviewed this annual report on Form 10-K of Lipella Pharmaceuticals Inc.; |
| 2. | Based on my knowledge, this report does not contain any untrue statement of a material fact or omit to state a material fact necessary to make the statements made, in light of the circumstances under which such statements were made, not misleading with respect to the period covered by this report; |
| 3. | Based on my knowledge, the financial statements, and other financial information included in this report, fairly present in all material respects the financial condition, results of operations and cash flows of the registrant as of, and for, the periods presented in this report; |
| 4. | The registrant’s other certifying officer and I are responsible for establishing and maintaining disclosure controls and procedures (as defined in Exchange Act Rules 13a-15(e) and 15d-15(e)) and internal control over financial reporting (as defined in Exchange Act Rules 13a-15(f) and 15d-15(f)) for the registrant and have: |
| (a) | Designed such disclosure controls and procedures, or caused such disclosure controls and procedures to be designed under our supervision, to ensure that material information relating to the registrant, is made known to us by others within those entities, particularly during the period in which this report is being prepared; |
| (b) | Designed such internal control over financial reporting, or caused such internal control over financial reporting to be designed under our supervision, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles; |
| (c) | Evaluated the effectiveness of the registrant’s disclosure controls and procedures and presented in this report our conclusions about the effectiveness of the disclosure controls and procedures, as of the end of the period covered by this report based on such evaluation; and |
| (d) | Disclosed in this report any change in the registrant’s internal control over financial reporting that occurred during the registrant’s most recent fiscal quarter (the registrant’s fourth fiscal quarter in the case of an annual report) that has materially affected, or is reasonably likely to materially affect, the registrant’s internal control over financial reporting; and |
| 5. | The registrant’s other certifying officer and I have disclosed, based on our most recent evaluation of internal control over financial reporting, to the registrant’s auditors and the audit committee of the registrant’s board of directors (or persons performing the equivalent functions): |
| (a) | All significant deficiencies and material weaknesses in the design or operation of internal control over financial reporting which are reasonably likely to adversely affect the registrant’s ability to record, process, summarize and report financial information; and |
| (b) | Any fraud, whether or not material, that involves management or other employees who have a significant role in the registrant’s internal control over financial reporting. |
| Date: March 27, 2025 | By: | /s/ Douglas Johnston |
| Douglas Johnston | ||
| Chief Financial Officer | ||
| (Principal Financial and Accounting Officer) |
Exhibit 32.1
CERTIFICATION
OF PRINCIPAL EXECUTIVE OFFICER
PURSUANT TO 18 U.S.C. SECTION 1350,
AS ADOPTED PURSUANT TO SECTION 906 OF
THE SARBANES-OXLEY ACT OF 2002
In connection with the Annual Report of Lipella Pharmaceuticals Inc. (the “Company”) on Form 10-K for the period ended December 31, 2024, as filed with the Securities and Exchange Commission on the date hereof (the “Report”), I, Jonathan Kaufman, Chief Executive Officer of Lipella Pharmaceuticals Inc., certify, pursuant to 18 U.S.C. §1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002, that:
| (1) | The Report fully complies with the requirements of Section 13(a) or 15(d), as applicable, of the Securities Exchange Act of 1934; and |
| (2) | The information contained in the Report fairly presents, in all material respects, the financial condition and results of operations of the Company. |
| Date: March 27, 2025 | By: | /s/ Jonathan Kaufman |
| Jonathan Kaufman | ||
| Chief Executive Officer | ||
| (Principal Executive Officer) |
Exhibit 32.2
CERTIFICATION
OF PRINCIPAL FINANCIAL OFFICER
PURSUANT TO 18 U.S.C. SECTION 1350,
AS ADOPTED PURSUANT TO SECTION 906 OF
THE SARBANES-OXLEY ACT OF 2002
In connection with the Annual Report of Lipella Pharmaceuticals Inc. (the “Company”) on Form 10-K for the period ended December 31, 2024, as filed with the Securities and Exchange Commission on the date hereof (the “Report”), I, Douglas Johnston, Chief Financial Officer of Lipella Pharmaceuticals Inc., certify, pursuant to 18 U.S.C. §1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002, that:
| (1) | The Report fully complies with the requirements of Section 13(a) or 15(d), as applicable, of the Securities Exchange Act of 1934; and |
| (2) | The information contained in the Report fairly presents, in all material respects, the financial condition and results of operations of the Company. |
| Date: March 27, 2025 | By: | /s/ Douglas Johnston |
| Douglas Johnston | ||
| Chief Financial Officer (Principal Financial and Accounting Officer) |
Exhibit 97.1
LIPELLA PHARMACEUTICALS INC. (the “Company”)
CLAWBACK POLICY
Effective as of December 1, 2023
Background
The Board of Directors of the Company (the “Board”) believes that it is in the best interests of the Company and its shareholders to create and maintain a culture that emphasizes integrity and accountability and that reinforces the Company’s pay-for-performance compensation philosophy. The Compensation Committee of the Board (the “Compensation Committee”) and the Board have therefore adopted this policy, which provides for the recoupment (or clawback) of certain executive compensation in the event of an accounting restatement resulting from material noncompliance with financial reporting requirements under the federal securities laws of the United States (the “Policy”). This Policy is designed to comply with Section 10D of the Securities Exchange Act of 1934, as amended (the “Exchange Act”), Rule 10D-1 promulgated under the Exchange Act (“Rule 10D -1”) and the listing standards of The Nasdaq Stock Market LLC (“Nasdaq”) under Nasdaq Listing Rule 5608.
Administration
This Policy shall be administered by the Compensation Committee. Any determinations made by the Compensation Committee shall be final and binding on all affected individuals. Subject to any limitation under applicable law, the Compensation Committee may authorize and empower any officer or employee of the Company to take any and all actions necessary or appropriate to carry out the purpose and intent of this Policy (the “Authorized Officers”) (other than with respect to any recovery under this Policy involving such officer or employee).
Covered Executives
This Policy applies to the Company’s current and former executive officers, as determined by the Board in accordance with Section 10D of the Exchange Act and the listing standards of the Nasdaq (“Covered Executives”).
Recoupment; Accounting Restatement
In the event the Company is required to prepare an accounting restatement of its financial statements due to the Company’s material noncompliance with any financial reporting requirement under the securities laws, the Compensation Committee will require prompt reimbursement or forfeiture of any excess Incentive Compensation (as defined below) received by any Covered Executive during the three completed fiscal years immediately preceding the date on which the Company is required to prepare an accounting restatement. For the sake of clarity, recoupment is required in the event of any restatement that either: (a) corrects an error in previously issued financial statements that is material to the previously issued financial statements; or (b) corrects an error not material to previously issued financial statements, but that would result in a material misstatement if (i) the error was left uncorrected in the then current period; or (ii) the error correction was recognized in the then current period. The Company’s obligation to recover erroneously awarded compensation is not dependent on if or when the restated financial statements are filed. For purposes of determining the relevant recovery period, the date that the Company is required to prepare an accounting restatement as described above is the earlier to occur of: (A) the date the Board, a committee of the Board, the Authorized Officers, or officers of the Company authorized to take such action if Board action is not required, concludes, or reasonably should have concluded, that the Company is required to prepare an accounting restatement as described above; or (B) the date a court, regulator, or other legally authorized body directs the Company to prepare an accounting restatement as described above. In accordance with Nasdaq Rule 5608(e), this Policy is applicable to Incentive Compensation (as described below) received on or after October 2, 2023.
Incentive Compensation
For purposes of this Policy, “Incentive Compensation” means any of the following, provided that such compensation is granted, earned or vested based wholly or in part on the attainment of a financial reporting measure affected by the restated financial statements:
| ● | Annual bonuses and other short- and long-term cash incentives. |
| ● | Stock options. |
| ● | Stock appreciation rights. |
| ● | Restricted stock. |
| ● | Restricted stock units. |
| ● | Performance shares. |
| ● | Performance share units. |
Financial reporting measures are measures that are determined and presented in accordance with the accounting principles used in preparing the Company’s financial statements, and any measures that are derived wholly or in part from such measures. Stock price and total stockholder return are also financial reporting measures. A financial reporting measure need not be presented within the financial statements or included in a filing with the Securities and Exchange Commission. The Company’s financial reporting measures may include, but are not limited to, the following:
| ● | Revenues. |
| ● | Net Income. |
| ● | Assets & Liabilities. |
| ● | Shareholder Equity. |
| ● | Operating, Inventing, and Financing Cash flow |
| ● | Liquidity measures such as Current Assets & Current Liabilities. |
This Policy applies to all Incentive Compensation received by a Covered Executive:
| ● | After beginning service as an executive officer; |
| ● | Who served as an executive officer at any time during the performance period for that Incentive Compensation; |
| ● | While the Company has a class of securities listed on a national securities exchange or a national securities association; and |
| ● | During the three completed fiscal years immediately preceding the date that the Company is required to prepare an accounting restatement as described in this Policy. In addition to these last three completed fiscal years, this Policy applies to any transition period (that results from a change in the Company’s fiscal year) within or immediately following those three completed fiscal years. However, a transition period between the last day of the Company’s previous fiscal year end and the first day of its new fiscal year that comprises a period of nine to 12 months would be deemed a completed fiscal year. |
Incentive Compensation is deemed received in the Company’s fiscal period during which the financial reporting measure specified in the Incentive Compensation award is attained, even if the payment or grant of the Incentive Compensation occurs after the end of that period.
Excess Incentive Compensation: Amount Subject to Recovery
The amount to be recovered will be the excess of the Incentive Compensation paid to the Covered Executive based on the erroneous data over the Incentive Compensation that would have been paid to the Covered Executive had it been based on the restated results, as determined by the Compensation Committee, and without regard to any taxes paid by or withheld from the Covered Executive. If the Compensation Committee cannot determine the amount of excess Incentive Compensation received by the Covered Executive directly from the information in the accounting restatement, then it will make its determination based on a reasonable estimate of the effect of the accounting restatement. For Incentive Compensation based on stock price or total stockholder return, where the amount of erroneously awarded compensation is not subject to mathematical recalculation directly from the information in an accounting restatement, the amount will be based on a reasonable estimate of the effect of the accounting restatement on the stock price or total stockholder return upon which the Incentive Compensation was received. In such case, the Company shall maintain documentation of the determination of that reasonable estimate and provide such documentation to Nasdaq.
Method of Recoupment
The Compensation Committee will determine, in its sole discretion, the method for recouping Incentive Compensation hereunder which may include, without limitation:
| ● | Requiring reimbursement of cash Incentive Compensation previously paid; |
| ● | Seeking recovery of any gain realized on the vesting, exercise, settlement, sale, transfer, or other disposition of any equity-based awards; |
| ● | Offsetting the recouped amount from any compensation otherwise owed by the Company to the Covered Executive in accordance with applicable law; |
| ● | Cancelling outstanding vested or unvested equity awards; and/or |
| ● | Taking any other remedial and recovery action permitted by law, as determined by the Compensation Committee. |
No Indemnification
The Company shall not indemnify any Covered Executives against the loss of any Incentive Compensation recovered under this Policy or from any consequence arising therefrom.
Interpretation
The Compensation Committee is authorized to interpret and construe this Policy and to make all determinations necessary, appropriate or advisable for the administration of this Policy. It is intended that this Policy be interpreted in a manner that is consistent with the requirements of Section 10D of the Exchange Act, Rule 10D-1 and any applicable rules or standards adopted by the Securities and Exchange Commission or Nasdaq.
Effective Date
This Policy shall be effective as of the date it is adopted by the Board (the “Effective Date”) and, in accordance with Nasdaq Rule 5608(e), shall apply to Incentive Compensation that is received by Covered Executives on or after October 2, 2023.
Amendment; Termination
The Board may amend this Policy from time to time in its discretion and shall amend this Policy as it deems necessary to reflect regulations adopted by the Securities and Exchange Commission under Section 10D of the Exchange Act and to comply with any rules or standards adopted by Nasdaq. The Board may terminate this Policy at any time.
Other Recoupment Rights
The Board intends that this Policy will be applied to the fullest extent of applicable law. The Board and/or Compensation Committee may require that any employment agreement, equity award agreement, or similar agreement entered into or amended on or after the Effective Date shall, as a condition to the grant of any benefit thereunder, require a Covered Executive to agree to abide by the terms of this Policy. Any right of recoupment under this Policy is in addition to, and not in lieu of: (a) any other remedies or rights of recoupment that may be available to the Company pursuant to the terms of any similar policy in any employment agreement, equity award agreement or similar agreement and any other legal remedies available to the Company, including termination of employment or institution of legal proceedings; and (b) any statutory recoupment requirement, including Section 304 of the Sarbanes-Oxley Act of 2022. For the avoidance of doubt, any amounts paid to the Company pursuant to Section 304 of the Sarbanes-Oxley Act of 2022 shall be considered (and may be credited) in determining any amounts recovered under this Policy.
Impracticability
The Compensation Committee shall recover any excess Incentive Compensation in accordance with this Policy unless such recovery would be impracticable, as determined in accordance with Rule 10D-1(b)(1)(iv) under the Exchange Act and the listing standards of Nasdaq. In order for the Company to determine that recovery would be impracticable, the Company’s Compensation Committee must conclude the following:
| a) | The direct expense paid to a third party to assist in enforcing this Policy would exceed the amount to be recovered after making a reasonable attempt to recover such Incentive Compensation. Note that the attempt(s) to recover must be documented by the Company and such documentation provided to Nasdaq; or |
| b) | Recovery would likely cause an otherwise tax-qualified retirement plan under which benefits are broadly available to Company employees to fail to meet the requirements for qualified pension, profit-sharing and stock bonus plans under Section 401(a)(13) of the U.S. Internal Revenue Code or the minimum vesting standards under Section 411(a) of the U.S. Internal Revenue Code. |
Successors
This Policy shall be binding and enforceable against all Covered Executives and their beneficiaries, heirs, executors, administrators or other legal representatives.
Exhibit Filing
A copy of this Policy shall be filed as an exhibit to the Company’s annual report on Form 10-K.