false2023FY0001745114trueP3YP1YP3YP3Y00017451142023-01-012023-12-310001745114dei:BusinessContactMember2023-01-012023-12-310001745114moln:AmericanDepositorySharesTheNasdaqStockMarketLLCMember2023-01-012023-12-3100017451142023-12-31xbrli:sharesiso4217:CHF00017451142022-12-3100017451142022-01-012022-12-3100017451142021-01-012021-12-31iso4217:CHFxbrli:shares00017451142021-12-3100017451142020-12-310001745114ifrs-full:IssuedCapitalMember2020-12-310001745114ifrs-full:AdditionalPaidinCapitalMember2020-12-310001745114ifrs-full:RetainedEarningsMember2020-12-310001745114ifrs-full:RetainedEarningsMember2021-01-012021-12-310001745114ifrs-full:AdditionalPaidinCapitalMember2021-01-012021-12-310001745114ifrs-full:IssuedCapitalMember2021-01-012021-12-310001745114ifrs-full:IssuedCapitalMember2021-12-310001745114ifrs-full:AdditionalPaidinCapitalMember2021-12-310001745114ifrs-full:RetainedEarningsMember2021-12-310001745114ifrs-full:RetainedEarningsMember2022-01-012022-12-310001745114ifrs-full:AdditionalPaidinCapitalMember2022-01-012022-12-310001745114ifrs-full:IssuedCapitalMember2022-01-012022-12-310001745114ifrs-full:TreasurySharesMember2022-01-012022-12-310001745114ifrs-full:IssuedCapitalMember2022-12-310001745114ifrs-full:AdditionalPaidinCapitalMember2022-12-310001745114ifrs-full:TreasurySharesMember2022-12-310001745114ifrs-full:RetainedEarningsMember2022-12-310001745114ifrs-full:RetainedEarningsMember2023-01-012023-12-310001745114ifrs-full:AdditionalPaidinCapitalMember2023-01-012023-12-310001745114ifrs-full:IssuedCapitalMember2023-01-012023-12-310001745114ifrs-full:IssuedCapitalMember2023-12-310001745114ifrs-full:AdditionalPaidinCapitalMember2023-12-310001745114ifrs-full:TreasurySharesMember2023-12-310001745114ifrs-full:RetainedEarningsMember2023-12-31moln:segment0001745114moln:LaboratoryEquipmentMember2023-01-012023-12-310001745114ifrs-full:OfficeEquipmentMember2023-01-012023-12-310001745114ifrs-full:ComputerEquipmentMember2023-01-012023-12-310001745114ifrs-full:ComputerSoftwareMember2023-01-012023-12-310001745114moln:VSAOMember2023-01-012023-12-31xbrli:pure0001745114moln:VoluntaryComplementaryPlanMember2023-01-012023-12-31moln:employee0001745114moln:VoluntaryComplementaryPlanMember2022-01-012022-12-310001745114moln:A2021LicenseAndCollaborationAgreementWithNovartisDARPINConjugatedRadioligandTherapiesMember2023-12-31iso4217:USD0001745114moln:A2021LicenseAndCollaborationAgreementWithNovartisDARPINConjugatedRadioligandTherapiesMembersrt:MaximumMember2021-12-142021-12-140001745114moln:NovartisMembermoln:A2021LicenseAndCollaborationAgreementWithNovartisDARPINConjugatedRadioligandTherapiesMember2023-01-012023-12-310001745114moln:NovartisMember2023-01-012023-12-310001745114moln:NovartisMembermoln:A2021LicenseAndCollaborationAgreementWithNovartisDARPINConjugatedRadioligandTherapiesMember2022-01-012022-12-310001745114moln:NovartisMembermoln:A2021LicenseAndCollaborationAgreementWithNovartisDARPINConjugatedRadioligandTherapiesMember2021-01-012021-12-310001745114moln:NovartisOptionAndEquityRightsAgreementMember2020-01-012020-12-310001745114moln:NovartisOptionAndEquityRightsAgreementMember2022-01-012022-12-310001745114moln:NovartisOptionAndEquityRightsAgreementMembermoln:CommercialSupplyOfProductsMember2022-01-012022-12-310001745114moln:FederalOfficeOfPublicHealthMember2021-01-012021-12-310001745114moln:FederalOfficeOfPublicHealthMember2021-12-310001745114moln:LicenseAndCollaborationAgreementWithFOPHMember2022-01-012022-12-310001745114moln:LicenseAndCollaborationAgreementWithAmgenIncMember2018-01-012018-12-310001745114moln:LicenseAndCollaborationAgreementWithAmgenIncMember2022-01-012022-12-310001745114country:CH2023-01-012023-12-310001745114country:CH2022-01-012022-12-310001745114country:CH2021-01-012021-12-310001745114country:US2023-01-012023-12-310001745114country:US2022-01-012022-12-310001745114country:US2021-01-012021-12-310001745114moln:LicenseAndCollaborationAgreementWithNovartisAGMember2023-01-012023-12-310001745114moln:LicenseAndCollaborationAgreementWithNovartisAGMember2022-01-012022-12-310001745114moln:LicenseAndCollaborationAgreementWithNovartisAGMember2021-01-012021-12-310001745114moln:LicenseAndCollaborationAgreementWithFOPHMember2023-01-012023-12-310001745114moln:LicenseAndCollaborationAgreementWithFOPHMember2021-01-012021-12-310001745114moln:LicenseAndCollaborationAgreementWithAmgenIncMember2023-01-012023-12-310001745114moln:LicenseAndCollaborationAgreementWithAmgenIncMember2021-01-012021-12-310001745114ifrs-full:GrossCarryingAmountMembermoln:LabEquipmentMember2022-12-310001745114ifrs-full:GrossCarryingAmountMemberifrs-full:OfficeEquipmentMember2022-12-310001745114ifrs-full:GrossCarryingAmountMemberifrs-full:ComputerEquipmentMember2022-12-310001745114ifrs-full:RightofuseAssetsMemberifrs-full:GrossCarryingAmountMember2022-12-310001745114ifrs-full:LeaseholdImprovementsMemberifrs-full:GrossCarryingAmountMember2022-12-310001745114ifrs-full:GrossCarryingAmountMember2022-12-310001745114ifrs-full:GrossCarryingAmountMembermoln:LabEquipmentMember2023-01-012023-12-310001745114ifrs-full:GrossCarryingAmountMemberifrs-full:OfficeEquipmentMember2023-01-012023-12-310001745114ifrs-full:GrossCarryingAmountMemberifrs-full:ComputerEquipmentMember2023-01-012023-12-310001745114ifrs-full:RightofuseAssetsMemberifrs-full:GrossCarryingAmountMember2023-01-012023-12-310001745114ifrs-full:LeaseholdImprovementsMemberifrs-full:GrossCarryingAmountMember2023-01-012023-12-310001745114ifrs-full:GrossCarryingAmountMember2023-01-012023-12-310001745114ifrs-full:GrossCarryingAmountMembermoln:LabEquipmentMember2023-12-310001745114ifrs-full:GrossCarryingAmountMemberifrs-full:OfficeEquipmentMember2023-12-310001745114ifrs-full:GrossCarryingAmountMemberifrs-full:ComputerEquipmentMember2023-12-310001745114ifrs-full:RightofuseAssetsMemberifrs-full:GrossCarryingAmountMember2023-12-310001745114ifrs-full:LeaseholdImprovementsMemberifrs-full:GrossCarryingAmountMember2023-12-310001745114ifrs-full:GrossCarryingAmountMember2023-12-310001745114ifrs-full:AccumulatedDepreciationAndAmortisationMembermoln:LabEquipmentMember2022-12-310001745114ifrs-full:AccumulatedDepreciationAndAmortisationMemberifrs-full:OfficeEquipmentMember2022-12-310001745114ifrs-full:AccumulatedDepreciationAndAmortisationMemberifrs-full:ComputerEquipmentMember2022-12-310001745114ifrs-full:AccumulatedDepreciationAndAmortisationMemberifrs-full:RightofuseAssetsMember2022-12-310001745114ifrs-full:AccumulatedDepreciationAndAmortisationMemberifrs-full:LeaseholdImprovementsMember2022-12-310001745114ifrs-full:AccumulatedDepreciationAndAmortisationMember2022-12-310001745114ifrs-full:AccumulatedDepreciationAndAmortisationMembermoln:LabEquipmentMember2023-01-012023-12-310001745114ifrs-full:AccumulatedDepreciationAndAmortisationMemberifrs-full:OfficeEquipmentMember2023-01-012023-12-310001745114ifrs-full:AccumulatedDepreciationAndAmortisationMemberifrs-full:ComputerEquipmentMember2023-01-012023-12-310001745114ifrs-full:AccumulatedDepreciationAndAmortisationMemberifrs-full:RightofuseAssetsMember2023-01-012023-12-310001745114ifrs-full:AccumulatedDepreciationAndAmortisationMemberifrs-full:LeaseholdImprovementsMember2023-01-012023-12-310001745114ifrs-full:AccumulatedDepreciationAndAmortisationMember2023-01-012023-12-310001745114ifrs-full:AccumulatedDepreciationAndAmortisationMembermoln:LabEquipmentMember2023-12-310001745114ifrs-full:AccumulatedDepreciationAndAmortisationMemberifrs-full:OfficeEquipmentMember2023-12-310001745114ifrs-full:AccumulatedDepreciationAndAmortisationMemberifrs-full:ComputerEquipmentMember2023-12-310001745114ifrs-full:AccumulatedDepreciationAndAmortisationMemberifrs-full:RightofuseAssetsMember2023-12-310001745114ifrs-full:AccumulatedDepreciationAndAmortisationMemberifrs-full:LeaseholdImprovementsMember2023-12-310001745114ifrs-full:AccumulatedDepreciationAndAmortisationMember2023-12-310001745114moln:LabEquipmentMember2023-12-310001745114ifrs-full:OfficeEquipmentMember2023-12-310001745114ifrs-full:ComputerEquipmentMember2023-12-310001745114ifrs-full:RightofuseAssetsMember2023-12-310001745114ifrs-full:LeaseholdImprovementsMember2023-12-310001745114ifrs-full:GrossCarryingAmountMembermoln:LabEquipmentMember2021-12-310001745114ifrs-full:GrossCarryingAmountMemberifrs-full:OfficeEquipmentMember2021-12-310001745114ifrs-full:GrossCarryingAmountMemberifrs-full:ComputerEquipmentMember2021-12-310001745114ifrs-full:RightofuseAssetsMemberifrs-full:GrossCarryingAmountMember2021-12-310001745114ifrs-full:LeaseholdImprovementsMemberifrs-full:GrossCarryingAmountMember2021-12-310001745114ifrs-full:GrossCarryingAmountMember2021-12-310001745114ifrs-full:GrossCarryingAmountMembermoln:LabEquipmentMember2022-01-012022-12-310001745114ifrs-full:GrossCarryingAmountMemberifrs-full:OfficeEquipmentMember2022-01-012022-12-310001745114ifrs-full:GrossCarryingAmountMemberifrs-full:ComputerEquipmentMember2022-01-012022-12-310001745114ifrs-full:RightofuseAssetsMemberifrs-full:GrossCarryingAmountMember2021-01-012021-12-310001745114ifrs-full:LeaseholdImprovementsMemberifrs-full:GrossCarryingAmountMember2022-01-012022-12-310001745114ifrs-full:GrossCarryingAmountMember2022-01-012022-12-310001745114ifrs-full:RightofuseAssetsMemberifrs-full:GrossCarryingAmountMember2022-01-012022-12-310001745114ifrs-full:AccumulatedDepreciationAndAmortisationMembermoln:LabEquipmentMember2021-12-310001745114ifrs-full:AccumulatedDepreciationAndAmortisationMemberifrs-full:OfficeEquipmentMember2021-12-310001745114ifrs-full:AccumulatedDepreciationAndAmortisationMemberifrs-full:ComputerEquipmentMember2021-12-310001745114ifrs-full:AccumulatedDepreciationAndAmortisationMemberifrs-full:RightofuseAssetsMember2021-12-310001745114ifrs-full:AccumulatedDepreciationAndAmortisationMemberifrs-full:LeaseholdImprovementsMember2021-12-310001745114ifrs-full:AccumulatedDepreciationAndAmortisationMember2021-12-310001745114ifrs-full:AccumulatedDepreciationAndAmortisationMembermoln:LabEquipmentMember2022-01-012022-12-310001745114ifrs-full:AccumulatedDepreciationAndAmortisationMemberifrs-full:OfficeEquipmentMember2022-01-012022-12-310001745114ifrs-full:AccumulatedDepreciationAndAmortisationMemberifrs-full:ComputerEquipmentMember2022-01-012022-12-310001745114ifrs-full:AccumulatedDepreciationAndAmortisationMemberifrs-full:RightofuseAssetsMember2022-01-012022-12-310001745114ifrs-full:AccumulatedDepreciationAndAmortisationMemberifrs-full:LeaseholdImprovementsMember2022-01-012022-12-310001745114ifrs-full:AccumulatedDepreciationAndAmortisationMember2022-01-012022-12-310001745114moln:LabEquipmentMember2022-12-310001745114ifrs-full:OfficeEquipmentMember2022-12-310001745114ifrs-full:ComputerEquipmentMember2022-12-310001745114ifrs-full:RightofuseAssetsMember2022-12-310001745114ifrs-full:LeaseholdImprovementsMember2022-12-310001745114ifrs-full:GrossCarryingAmountMemberifrs-full:ComputerSoftwareMember2022-12-310001745114ifrs-full:GrossCarryingAmountMemberifrs-full:ComputerSoftwareMember2023-01-012023-12-310001745114ifrs-full:GrossCarryingAmountMemberifrs-full:ComputerSoftwareMember2023-12-310001745114ifrs-full:AccumulatedDepreciationAndAmortisationMemberifrs-full:ComputerSoftwareMember2022-12-310001745114ifrs-full:AccumulatedDepreciationAndAmortisationMemberifrs-full:ComputerSoftwareMember2023-01-012023-12-310001745114ifrs-full:AccumulatedDepreciationAndAmortisationMemberifrs-full:ComputerSoftwareMember2023-12-310001745114ifrs-full:ComputerSoftwareMember2023-12-310001745114ifrs-full:GrossCarryingAmountMemberifrs-full:ComputerSoftwareMember2021-12-310001745114ifrs-full:GrossCarryingAmountMemberifrs-full:ComputerSoftwareMember2022-01-012022-12-310001745114ifrs-full:AccumulatedDepreciationAndAmortisationMemberifrs-full:ComputerSoftwareMember2021-12-310001745114ifrs-full:AccumulatedDepreciationAndAmortisationMemberifrs-full:ComputerSoftwareMember2022-01-012022-12-310001745114ifrs-full:ComputerSoftwareMember2022-12-310001745114moln:CashAndCashEquivalentsMemberifrs-full:FinancialAssetsAtAmortisedCostCategoryMember2023-12-310001745114ifrs-full:TradeReceivablesMemberifrs-full:FinancialAssetsAtAmortisedCostCategoryMember2023-12-310001745114moln:AccruedIncomeMemberifrs-full:FinancialAssetsAtAmortisedCostCategoryMember2023-12-310001745114moln:ShortTermTimeDepositsMemberifrs-full:FinancialAssetsAtAmortisedCostCategoryMember2023-12-310001745114ifrs-full:FinancialAssetsAtAmortisedCostCategoryMember2023-12-310001745114moln:CashAndCashEquivalentsMemberifrs-full:FinancialAssetsAtAmortisedCostCategoryMember2022-12-310001745114ifrs-full:TradeReceivablesMemberifrs-full:FinancialAssetsAtAmortisedCostCategoryMember2022-12-310001745114moln:AccruedIncomeMemberifrs-full:FinancialAssetsAtAmortisedCostCategoryMember2022-12-310001745114moln:ShortTermTimeDepositsMemberifrs-full:FinancialAssetsAtAmortisedCostCategoryMember2022-12-310001745114ifrs-full:FinancialAssetsAtAmortisedCostCategoryMember2022-12-310001745114ifrs-full:FinancialLiabilitiesAtAmortisedCostCategoryMembermoln:TradePayablesMember2023-12-310001745114ifrs-full:FinancialLiabilitiesAtAmortisedCostCategoryMembermoln:AccruedProjectCostsAndRoyaltiesMember2023-12-310001745114ifrs-full:FinancialLiabilitiesAtAmortisedCostCategoryMemberifrs-full:LeaseLiabilitiesMember2023-12-310001745114ifrs-full:FinancialLiabilitiesAtAmortisedCostCategoryMembermoln:OtherNonEmployeeRelatedAccruedExpensesMember2023-12-310001745114ifrs-full:FinancialLiabilitiesAtAmortisedCostCategoryMember2023-12-310001745114ifrs-full:FinancialLiabilitiesAtAmortisedCostCategoryMembermoln:TradePayablesMember2022-12-310001745114ifrs-full:FinancialLiabilitiesAtAmortisedCostCategoryMembermoln:AccruedProjectCostsAndRoyaltiesMember2022-12-310001745114ifrs-full:FinancialLiabilitiesAtAmortisedCostCategoryMemberifrs-full:LeaseLiabilitiesMember2022-12-310001745114ifrs-full:FinancialLiabilitiesAtAmortisedCostCategoryMembermoln:OtherNonEmployeeRelatedAccruedExpensesMember2022-12-310001745114ifrs-full:FinancialLiabilitiesAtAmortisedCostCategoryMember2022-12-310001745114currency:CHF2023-12-310001745114currency:CHF2022-12-310001745114currency:EUR2023-12-310001745114currency:EUR2022-12-310001745114currency:USD2023-12-310001745114currency:USD2022-12-310001745114currency:GBP2023-12-310001745114currency:GBP2022-12-31moln:positionmoln:bank00017451142022-08-310001745114ifrs-full:OrdinarySharesMember2023-01-012023-12-31moln:vote0001745114ifrs-full:OrdinarySharesMember2023-12-3100017451142022-04-132022-04-1300017451142022-04-1200017451142022-04-1300017451142022-08-012022-08-310001745114moln:ConditionalShareCapitalMember2023-12-310001745114moln:ConditionalShareCapitalMember2023-01-012023-12-310001745114moln:ConditionalShareCapitalMember2022-12-310001745114moln:ShareOptionsPSUsAndRSUsMember2023-01-012023-12-310001745114moln:ShareOptionsPSUsAndRSUsMember2022-01-012022-12-310001745114moln:ShareOptionsPSUsAndRSUsMember2021-01-012021-12-310001745114ifrs-full:NotLaterThanOneYearMember2023-12-310001745114ifrs-full:NotLaterThanOneYearMember2022-12-310001745114ifrs-full:LaterThanOneYearAndNotLaterThanTwoYearsMember2022-12-310001745114moln:NovartisMember2022-12-310001745114moln:NovartisMember2023-12-310001745114moln:AmgenIncMember2021-12-310001745114moln:AmgenIncMember2022-01-012022-12-310001745114moln:AmgenIncMember2022-12-310001745114moln:NovartisMember2021-12-310001745114moln:NovartisMember2022-01-012022-12-310001745114moln:FederalOfficeOfPublicHealthMember2022-01-012022-12-310001745114moln:FederalOfficeOfPublicHealthMember2022-12-310001745114moln:ResearchAndDevelopmentExpenseMember2023-01-012023-12-310001745114moln:ResearchAndDevelopmentExpenseMember2022-01-012022-12-310001745114moln:ResearchAndDevelopmentExpenseMember2021-01-012021-12-310001745114ifrs-full:SellingGeneralAndAdministrativeExpenseMember2023-01-012023-12-310001745114ifrs-full:SellingGeneralAndAdministrativeExpenseMember2022-01-012022-12-310001745114ifrs-full:SellingGeneralAndAdministrativeExpenseMember2021-01-012021-12-310001745114moln:UniversityOfZurichMember2023-01-012023-12-310001745114ifrs-full:PresentValueOfDefinedBenefitObligationMember2022-12-310001745114ifrs-full:PresentValueOfDefinedBenefitObligationMember2021-12-310001745114ifrs-full:PresentValueOfDefinedBenefitObligationMember2023-01-012023-12-310001745114ifrs-full:PresentValueOfDefinedBenefitObligationMember2022-01-012022-12-310001745114ifrs-full:PresentValueOfDefinedBenefitObligationMember2023-12-310001745114ifrs-full:PlanAssetsMember2022-12-310001745114ifrs-full:PlanAssetsMember2021-12-310001745114ifrs-full:PlanAssetsMember2023-01-012023-12-310001745114ifrs-full:PlanAssetsMember2022-01-012022-12-310001745114ifrs-full:PlanAssetsMember2023-12-310001745114ifrs-full:Level1OfFairValueHierarchyMember2023-12-310001745114ifrs-full:Level1OfFairValueHierarchyMember2022-12-310001745114ifrs-full:Level2OfFairValueHierarchyMember2023-12-310001745114ifrs-full:Level2OfFairValueHierarchyMember2022-12-310001745114ifrs-full:ActuarialAssumptionOfDiscountRatesMember2023-12-310001745114ifrs-full:ActuarialAssumptionOfDiscountRatesMember2022-12-310001745114moln:ActuarialAssumptionOfInterestRateOnRetirementSavingsCapitalMember2022-12-310001745114moln:ActuarialAssumptionOfInterestRateOnRetirementSavingsCapitalMember2023-12-310001745114ifrs-full:ActuarialAssumptionOfExpectedRatesOfSalaryIncreasesMember2022-12-310001745114ifrs-full:ActuarialAssumptionOfExpectedRatesOfSalaryIncreasesMember2023-12-310001745114ifrs-full:ActuarialAssumptionOfLifeExpectancyAfterRetirementMember2022-12-310001745114ifrs-full:ActuarialAssumptionOfLifeExpectancyAfterRetirementMember2023-12-310001745114moln:ActiveMembersMember2023-01-012023-12-310001745114moln:ActiveMembersMember2022-01-012022-12-310001745114moln:PensionersMember2023-01-012023-12-310001745114moln:PensionersMember2022-01-012022-12-310001745114moln:ESOP2009AndESOP2014Member2023-01-012023-12-310001745114moln:ESOP2009AndESOP2014Member2023-12-310001745114moln:ESOP2009AndESOP2014Member2022-12-31moln:plan0001745114moln:RestrictedShareUnitsRSUMember2023-01-012023-12-310001745114moln:PerformanceShareUnitsPSUEmployeesExcludingManagementBoardMember2023-01-012023-12-31moln:tranche0001745114moln:PerformanceShareUnitsPSUManagementBoardMember2023-01-012023-12-310001745114moln:PerformanceShareUnitsPSUMember2023-01-012023-12-310001745114moln:PerformanceShareUnitsPSUEmployeesExcludingManagementBoardMembersrt:MinimumMember2023-01-012023-12-310001745114moln:PerformanceShareUnitsPSUEmployeesExcludingManagementBoardMembersrt:MaximumMember2023-01-012023-12-310001745114moln:PerformanceShareUnitsPSUMember2023-12-310001745114moln:RestrictedShareUnitsRSUMember2023-12-310001745114moln:PerformanceShareUnitsPSUMember2022-12-310001745114moln:RestrictedShareUnitsRSUMember2022-12-310001745114moln:RestrictedShareUnitsRSUMember2022-01-012022-12-310001745114moln:PerformanceShareUnitsPSUMember2022-01-012022-12-310001745114srt:MinimumMember2023-01-012023-12-310001745114srt:MaximumMember2023-01-012023-12-310001745114srt:MinimumMember2022-01-012022-12-310001745114srt:MaximumMember2022-01-012022-12-310001745114moln:RestrictedShareUnitsRSUMembersrt:MinimumMember2023-01-012023-12-310001745114moln:RestrictedShareUnitsRSUMembersrt:MinimumMember2022-01-012022-12-310001745114moln:PerformanceShareUnitsPSUManagementBoardMember2022-01-012022-12-310001745114moln:PerformanceShareUnitsPSUEmployeesExcludingManagementBoardMembersrt:MinimumMember2022-01-012022-12-310001745114moln:ExercisePriceRangeOneMember2023-12-310001745114moln:ExercisePriceRangeOneMember2023-01-012023-12-310001745114moln:ExercisePriceRangeTwoMember2023-12-310001745114moln:ExercisePriceRangeTwoMember2023-01-012023-12-310001745114moln:ExercisePriceRangeThreeMember2023-12-310001745114moln:ExercisePriceRangeOneMember2022-12-310001745114moln:ExercisePriceRangeOneMember2022-01-012022-12-310001745114moln:ExercisePriceRangeTwoMember2022-12-310001745114moln:ExercisePriceRangeTwoMember2022-01-012022-12-310001745114moln:ExercisePriceRangeThreeMember2022-12-310001745114moln:MolecularPartnersIncMember2023-01-012023-12-310001745114moln:MolecularPartnersIncMember2022-01-012022-12-310001745114moln:MolecularPartnersIncMember2021-01-012021-12-310001745114moln:ExpiringIn2027Member2023-12-310001745114moln:ExpiringIn2027Member2022-12-310001745114moln:ExpiringIn2028Member2023-12-310001745114moln:ExpiringIn2028Member2022-12-310001745114moln:ExpiringIn2030Member2023-12-310001745114moln:ExpiringIn2030Member2022-12-310001745114srt:MinimumMember2023-01-012023-12-310001745114srt:MaximumMember2023-01-012023-12-310001745114moln:LeaseFacilitiesSchlierenMember2020-01-012020-12-310001745114ifrs-full:LaterThanOneYearAndNotLaterThanTwoYearsMember2023-12-310001745114ifrs-full:LaterThanTwoYearsAndNotLaterThanFiveYearsMember2023-12-310001745114ifrs-full:LaterThanFiveYearsMember2023-12-310001745114ifrs-full:LaterThanTwoYearsAndNotLaterThanFiveYearsMember2022-12-310001745114ifrs-full:LaterThanFiveYearsMember2022-12-310001745114ifrs-full:CurrencyRiskMembercurrency:USD2023-01-012023-12-310001745114ifrs-full:CurrencyRiskMembercurrency:USD2022-01-012022-12-310001745114ifrs-full:CurrencyRiskMembercurrency:USD2021-01-012021-12-310001745114ifrs-full:CurrencyRiskMembercurrency:EUR2023-01-012023-12-310001745114ifrs-full:CurrencyRiskMembercurrency:EUR2022-01-012022-12-310001745114ifrs-full:CurrencyRiskMembercurrency:EUR2021-01-012021-12-310001745114currency:CHFifrs-full:InterestRateRiskMember2023-01-012023-12-310001745114currency:CHFifrs-full:InterestRateRiskMember2022-01-012022-12-310001745114currency:CHFifrs-full:InterestRateRiskMember2021-01-012021-12-310001745114currency:USDifrs-full:InterestRateRiskMember2023-01-012023-12-310001745114currency:USDifrs-full:InterestRateRiskMember2022-01-012022-12-310001745114currency:USDifrs-full:InterestRateRiskMember2021-01-012021-12-310001745114currency:EURifrs-full:InterestRateRiskMember2023-01-012023-12-310001745114currency:EURifrs-full:InterestRateRiskMember2022-01-012022-12-310001745114currency:EURifrs-full:InterestRateRiskMember2021-01-012021-12-310001745114moln:CashAndCashEquivalentsMemberifrs-full:CreditRiskMember2023-12-310001745114moln:CashAndCashEquivalentsMemberifrs-full:CreditRiskMember2022-12-310001745114ifrs-full:TradeReceivablesMemberifrs-full:CreditRiskMember2023-12-310001745114ifrs-full:TradeReceivablesMemberifrs-full:CreditRiskMember2022-12-310001745114moln:AccruedIncomeMemberifrs-full:CreditRiskMember2023-12-310001745114moln:AccruedIncomeMemberifrs-full:CreditRiskMember2022-12-310001745114moln:ShortTermTimeDepositsMemberifrs-full:CreditRiskMember2023-12-310001745114moln:ShortTermTimeDepositsMemberifrs-full:CreditRiskMember2022-12-310001745114ifrs-full:CreditRiskMember2023-12-310001745114ifrs-full:CreditRiskMember2022-12-31
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM 20-F
(Mark One)
|
|
|
|
|
|
| ☐ |
REGISTRATION STATEMENT PURSUANT TO SECTION 12(b) OR (g) OF THE SECURITIES EXCHANGE ACT OF 1934 |
OR
|
|
|
|
|
|
| ☒ |
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2023
OR
|
|
|
|
|
|
| ☐ |
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to
OR
|
|
|
|
|
|
| ☐ |
SHELL COMPANY REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
Date of event requiring this shell company report
Commission File Number 001-40488
MOLECULAR PARTNERS AG
(Exact name of registrant as specified in its charter and translation of registrant’s name into English)
Switzerland
(Jurisdiction of incorporation or organization)
Wagistrasse 14
8952 Zurich-Schlieren
Switzerland
(Address of principal executive offices)
Patrick Amstutz
Chief Executive Officer
Molecular Partners AG
Wagistrasse 14
8952 Zurich-Schlieren
Switzerland
Tel: +41 44 755 77 00
(Name, Telephone, E-mail and/or Facsimile number and Address of Company Contact Person)
Securities registered or to be registered pursuant to Section 12(b) of the Act.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Title of each class |
|
Trading Symbol |
|
Name of each exchange on which registered |
|
American depositary shares (each representing
one ordinary share, CHF 0.10 nominal value per share)
|
|
MOLN |
|
The Nasdaq Stock Market LLC |
Ordinary shares, CHF 0.10 nominal value per shares |
|
* |
|
The Nasdaq Stock Market LLC |
* Not for trading, but only in connection with the listing on the Nasdaq Global Select Market of the American depositary shares.
Securities registered or to be registered pursuant to Section 12(g) of the Act. None
Securities for which there is a reporting obligation pursuant to Section 15(d) of the Act. None
Indicate the number of outstanding shares of each of the issuer’s classes of capital or ordinary shares as of the close of the period covered by the annual report.
Ordinary shares, CHF 0.10 nominal value per share: 36,354,297 ordinary shares outstanding as of December 31, 2023
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. ☐ Yes ☒ No
If this report is an annual or transition report, indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934. ☐ Yes ☒ No
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. ☒ Yes ☐ No
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files).
☒ Yes ☐ No
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Large accelerated filer |
|
☐ |
|
Accelerated filer |
|
☐ |
| Non-accelerated filer |
|
☒ |
|
Emerging growth company |
|
☒ |
If an emerging growth company that prepares its financial statements in accordance with U.S. GAAP, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
† The term “new or revised financial accounting standard” refers to any update issued by the Financial Accounting Standards Board to its Accounting Standards Codification after April 5, 2012.
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report. ☐
If the securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant in the filing reflect the correction of an error to previously filed financial statements. ☐
Indicate by checkmark whether any of those error corrections are restatements that required a recovery analysis of incentive-based compensation received by any of the registrant’s executive officers during the relevant recovery period pursuant to §240.10D-1(b). ☐
Indicate by check mark which basis of accounting the registrant has used to prepare the financial statements included in this filing:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
U.S. GAAP ☐ |
|
International Financial Reporting Standards as issued
by the International Accounting Standards Board ☒
|
|
Other ☐ |
If “Other” has been checked in response to the previous question, indicate by check mark which financial statement item the registrant has elected to follow. ☐ Item 17 ☐ Item 18
If this is an annual report, indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). ☐ Yes ☒ No
TABLE OF CONTENTS
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
PAGE |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Item 1. |
|
|
|
|
Item 2. |
|
Offer Statistics and Expected Timetable |
|
|
Item 3. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Item 4. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Item 4A. |
|
|
|
|
Item 5. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Item 6. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
F. Disclosure of a Registrant's Action to Recover Erroneously Awarded Compensation |
|
155 |
Item 7. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Item 8. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Item 9. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Item 10. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Item 11. |
|
|
|
|
Item 12. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Item 13. |
|
|
|
|
Item 14. |
|
|
|
Item 15. |
|
|
|
|
Item 16A. |
|
|
|
|
Item 16B. |
|
|
|
|
Item 16C. |
|
|
|
|
Item 16D. |
|
|
|
|
Item 16E. |
|
|
|
|
Item 16F. |
|
|
|
|
Item 16G. |
|
|
|
|
Item 16H. |
|
|
|
|
| Item 16I. |
|
|
|
|
Item 16J. |
|
|
|
|
Item 16K. |
|
Cybersecurity |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Item 17. |
|
|
|
|
Item 18. |
|
|
|
|
Item 19. |
|
|
|
|
INTRODUCTION
Unless the context requires otherwise, references in this Annual Report on Form 20-F to the “Company,” “Molecular Partners,” “we,” “us” and “our” refer to Molecular Partners AG and its wholly-owned subsidiary.
We own trademark registrations for “Molecular Partners®” and “DARPin®” in Switzerland, the European Union, the United States and Japan. All other trade names, trademarks and service marks of other companies appearing in this Annual Report on Form 20-F are the property of their respective holders. Solely for convenience, the trademarks and trade names in this Annual Report on Form 20-F may be referred to without the ® and ™ symbols, but such references should not be construed as any indicator that their respective owners will not assert, to the fullest extent under applicable law, their rights thereto. We do not intend to use or display other companies’ trademarks and trade names to imply a relationship with, or endorsement or sponsorship of us by, any other companies.
We present our consolidated financial statements in CHF and in accordance with IFRS® Accounting Standards ("IFRS") as issued by the International Accounting and Standards Board. None of the financial statements were prepared in accordance with generally accepted accounting principles in the United States.
The terms “dollar,” “USD” or “$” refer to U.S. dollars and the terms “Swiss Francs” or “CHF” refer to the legal currency of Switzerland. Unless otherwise indicated, all references to currency amounts in this Annual Report on Form 20-F are in U.S. dollars.
We have made rounding adjustments to some of the figures included in this Annual Report on Form 20-F. Accordingly, numerical figures shown as totals in some tables may not be an arithmetic aggregation of the figures that preceded them.
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
This Annual Report on Form 20-F contains forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended, that are based on our management’s beliefs and assumptions and on information currently available to our management. All statements other than present and historical facts and conditions contained in this Annual Report on Form 20-F, including statements regarding our future results of operations and financial positions, business strategy, plans and our objectives for future operations, are forward-looking statements. When used in this Annual Report on Form 20-F, the words “anticipate,” “believe,” “can,” “could,” “estimate,” “expect,” “intend,” “designed,” “may,” “might,” “plan,” “potential,” “predict,” “objective,” “should,” or the negative of these and similar expressions identify forward-looking statements. Forward-looking statements include, but are not limited to, statements about:
•the initiation, timing, progress and results of our clinical trials and preclinical studies, and our research and development programs;
•our ability to advance product candidates into, and successfully complete, clinical trials;
•the timing of regulatory filings and the likelihood of favorable regulatory outcomes and approvals;
•the regulatory treatment of our product candidates;
•regulatory developments in the European Union, United States and other countries;
•the commercialization of our product candidates, if and once approved;
•the pricing and reimbursement of our product candidates, if and once approved;
•our ability to contract on commercially reasonable terms with third-party suppliers and manufacturers;
•the implementation of our business model and strategy and the development of our product candidates and platforms;
•the scope of protection we are able to establish, obtain and maintain for intellectual property rights covering our product candidates and technology and our ability to protect and enforce such rights;
•our ability to operate our business without infringing on, misappropriating or otherwise violating the intellectual property rights of others;
•the ability of third parties with whom we contract to successfully conduct, supervise and monitor clinical trials for our product candidates;
•estimates of our expenses, future revenues, earnings, capital requirements and our needs for additional financing;
•the timing and amount of milestone and royalty payments that we may receive under our strategic collaboration agreements;
•our ability to obtain additional funding for our operations;
•the potential benefits of our strategic collaboration agreements and our ability to enter into future strategic arrangements;
•our ability to maintain and establish collaborations or obtain additional funding;
•the rate and degree of market acceptance of, and pricing for, our product candidates;
•our financial performance;
•the impact of macro-economic factors, including a health pandemic, rising inflation, the U.S Federal Reserve and other financial regulatory agencies raising interest rates, the Russia-Ukraine war and the Israel-Hamas war, on our business, operations and prospects and on our clinical trials;
•our ability to attract and retain key scientific and management personnel;
•developments relating to our competitors and our industry, including competing therapies;
•the future trading price of the ADSs and impact of securities analysts reports on these prices; and
•other risks and uncertainties, including those listed under the caption “Risk Factors.”
You should refer to the section of this Annual Report on Form 20-F titled “Item 3.D-Risk Factors” for a discussion of important factors that may cause our actual results to differ materially from those expressed or implied by our forward-looking statements. As a result of these factors, we cannot assure you that the forward-looking statements in this Annual Report on Form 20-F will prove to be accurate. Furthermore, if our forward-looking statements prove to be inaccurate, the inaccuracy may be material.
In light of the significant uncertainties in these forward-looking statements, you should not regard these statements as a representation or warranty by us or any other person that we will achieve our objectives and plans in any specified time frame or at all. We undertake no obligation to publicly update any forward-looking statements, whether as a result of new information, future events or otherwise, except as required by law.
You should read this Annual Report on Form 20-F and the documents that we reference in this Annual Report on Form 20-F and have filed as exhibits to this Annual Report on Form 20-F completely and with the understanding that our actual future results may be materially different from what we expect. We qualify all of our forward-looking statements by these cautionary statements.
In addition, statements that “we believe” and similar statements reflect our beliefs and opinions on the relevant subject. These statements are based upon information available to us as of the date of this Annual Report on Form 20-F, and while we believe such information forms a reasonable basis for such statements, such information may be limited or incomplete. Our statements should not be read to indicate that we have conducted an exhaustive inquiry into, or review of, all potentially available relevant information. These statements are inherently uncertain and investors are cautioned not to unduly rely upon these statements.
This Annual Report on Form 20-F contains market data and industry forecasts that were obtained from industry publications. These data involve a number of assumptions and limitations, and you are cautioned not to give undue weight to such estimates. We have not independently verified any third-party information. While we believe the market position, market opportunity and market size information included in this Annual Report on Form 20-F is generally reliable, such information is inherently imprecise.
SUMMARY OF RISK FACTORS
Our business faces significant risks. If any of the following risks are realized, our business, financial condition and results of operations could be materially and adversely affected. You should carefully review and consider the full discussion of our risk factors set forth under the caption “Risk Factors” in Item 3.D. in Part I of this Annual Report on Form 20-F. An investment in our ADSs involves a high degree of risk. Any of the factors set forth under “Risk Factors” may limit our ability to successfully execute our business strategy. You should carefully consider all of the information set forth in this Annual Report on Form 20-F and, in particular, should evaluate the specific factors set forth under “Risk Factors” in deciding whether to invest in our securities. Among these risks are the following:
•We have incurred significant losses since our inception, we expect to incur losses in future periods and may not achieve profitability in the upcoming years. We may need substantial additional funding in order to complete the development and commercialization of our product candidates. Failure to obtain this necessary capital when needed may force us to delay, limit or terminate certain of our product development or research operations.
•We may need substantial additional funding in order to complete the development and commercialization of our product candidates. Failure to obtain this necessary capital when needed may force us to delay, limit or terminate certain of our product development or research operations.
•Raising additional capital may cause dilution to holders of our ordinary shares or ADSs, and an inability to raise capital may restrict our operations or require us to relinquish rights to our technologies or product candidates.
•We are heavily dependent on the success of our DARPin platform to identify and develop product candidates. If we or our collaborators are unable to successfully develop and commercialize product candidates based on our platform or experience significant delays in doing so, our business may be harmed.
•All of our product candidates are in preclinical or various stages of clinical development. Clinical drug development is a lengthy and expensive process with uncertain timelines and uncertain outcomes. If clinical trials of our product candidates, particularly MP0317, MP0533 and product candidates that we have licensed to our partners are prolonged, delayed or not commercially viable, we or our collaborators may be unable to obtain required regulatory approvals, and therefore may be unable to commercialize our product candidates on a timely basis or at all, which will adversely affect our business.
•Preclinical drug development is uncertain. Some or all of our preclinical programs may experience delays or may never advance to clinical trials, which would adversely affect our ability to obtain regulatory approvals or commercialize these product candidates on a timely basis or at all, which would have an adverse effect on our business.
•Positive results from early preclinical studies of our product candidates would not necessarily be predictive of the results of later preclinical studies and any ongoing or future clinical trials of our product candidates. If we were to achieve positive results from preclinical studies, but were unable to then replicate those positive results in our later preclinical studies and ongoing or future clinical trials, we might be unable to successfully develop, obtain regulatory or marketing approval and commercialize our product candidates.
•Interim, topline and preliminary data from our clinical trials that we announce or publish may change as more patient data become available and are subject to audit and verification procedures that could result in material changes in the final data.
•Because the number of patients in certain of our clinical trials may be small, the results from such trials may be less reliable than results achieve in larger clinical trials.
•If any of our product candidates has negative side effects, public perception of our DARPin platform and commercial opportunities for all of our current and future product candidates could be adversely affected.
•We face significant competition for our drug discovery and development efforts, and if we do not compete effectively, our commercial opportunities will be reduced or eliminated.
•Our financial prospects are dependent upon the research, manufacture, development and marketing efforts of our licensees. Our licensees may act in their best interest rather than in our best interest, which could materially adversely affect our business, financial condition and results of operations.
•We rely on patents and other intellectual property rights to protect our product candidates and the DARPin technology, the prosecution, grant, enforcement, defense and maintenance of which may be challenging and costly. Failure to obtain, maintain, enforce or protect these rights adequately could harm our ability to compete and impair our business.
•Third parties may initiate legal proceedings alleging that we are infringing, misappropriating, or otherwise violating their intellectual property rights, the outcome of which would be uncertain and could have a material adverse effect on the success of our business. Intellectual property litigation could cause us to spend substantial resources and distract our personnel from their normal responsibilities and negative outcomes could result in adverse effects on our business.
•The base patents relating to the DARPin base technology we use to generate our DARPin product candidates has expired, and our competitors may use the technology claimed in such patents, which may materially adversely affect our business and competitive position.
•Certain significant shareholders own a substantial number of our securities and as a result, may be able to exercise significant influence over the outcome of shareholder votes. These shareholders may have different interests from us or your interests.
•We depend on our information technology systems, and any failure of these systems could harm our business. Security incidents and other disruptions could compromise sensitive information related to our business or prevent us from accessing critical information and expose us to liability, which could adversely affect our business, results of operations and financial condition.
PART I
Item 1. Identity of Directors, Senior Management and Advisers.
Not applicable.
Item 2. Offer Statistics and Expected Timetable.
Not applicable.
Item 3. Key Information.
A. [Reserved]
B. Capitalization and Indebtedness
Not applicable.
C. Reasons for the Offer and Use of Proceeds
Not applicable.
D. Risk Factors
Investing in the ADSs involves a high degree of risk. You should carefully consider the risks and uncertainties described below and the other information in this Annual Report on Form 20-F before making an investment decision. Our business, financial condition or results of operations could be adversely affected if any of these risks occurs, and as a result, the market price of the ADSs could decline and you could lose all or part of your investment. This report also contains forward-looking statements that involve risks and uncertainties. See "Special Note Regarding Forward-Looking Statements." Our actual results could differ materially and adversely from those anticipated in these forward-looking statements as a result of certain factors.
Risks Related to Our Financial Position and Need for Additional Capital
We have incurred significant losses since our inception, we expect to incur losses in future periods and may not maintain profitability in the upcoming years. We may need substantial additional funding in order to complete the development and commercialization of our product candidates. Failure to obtain this necessary capital when needed may force us to delay, limit or terminate certain of our product development or research operations.
Since our inception, we have incurred significant operating losses, including negative net results, attributable to shareholders. As of December 31, 2023, we had cumulative losses of CHF 191.8 million. For the year ended December 31, 2023 we recorded negative net result, attributable to shareholders of CHF 62.0 million and for the year ended December 31, 2022, we incurred positive net result, attributable to shareholders of CHF 117.9 million. The positive result in 2022 was largely driven by the January 2022 payment of CHF 150 million by Novartis Pharma AG, or Novartis, following exercise of its option under a collaboration agreement with us.
Our historical losses resulted principally from costs incurred in research and development, preclinical testing, clinical development of our product candidates as well as costs incurred for research programs and from selling, general and administrative costs associated with our operations. In the future, we intend to continue to conduct research and development, preclinical testing, clinical trials and regulatory compliance activities that, together with anticipated selling, general and administrative expenses, may result in incurring losses in future periods. Our losses, among other things, will continue to cause our working capital and shareholders' equity to decrease. We anticipate that our expenses will increase substantially if and as we:
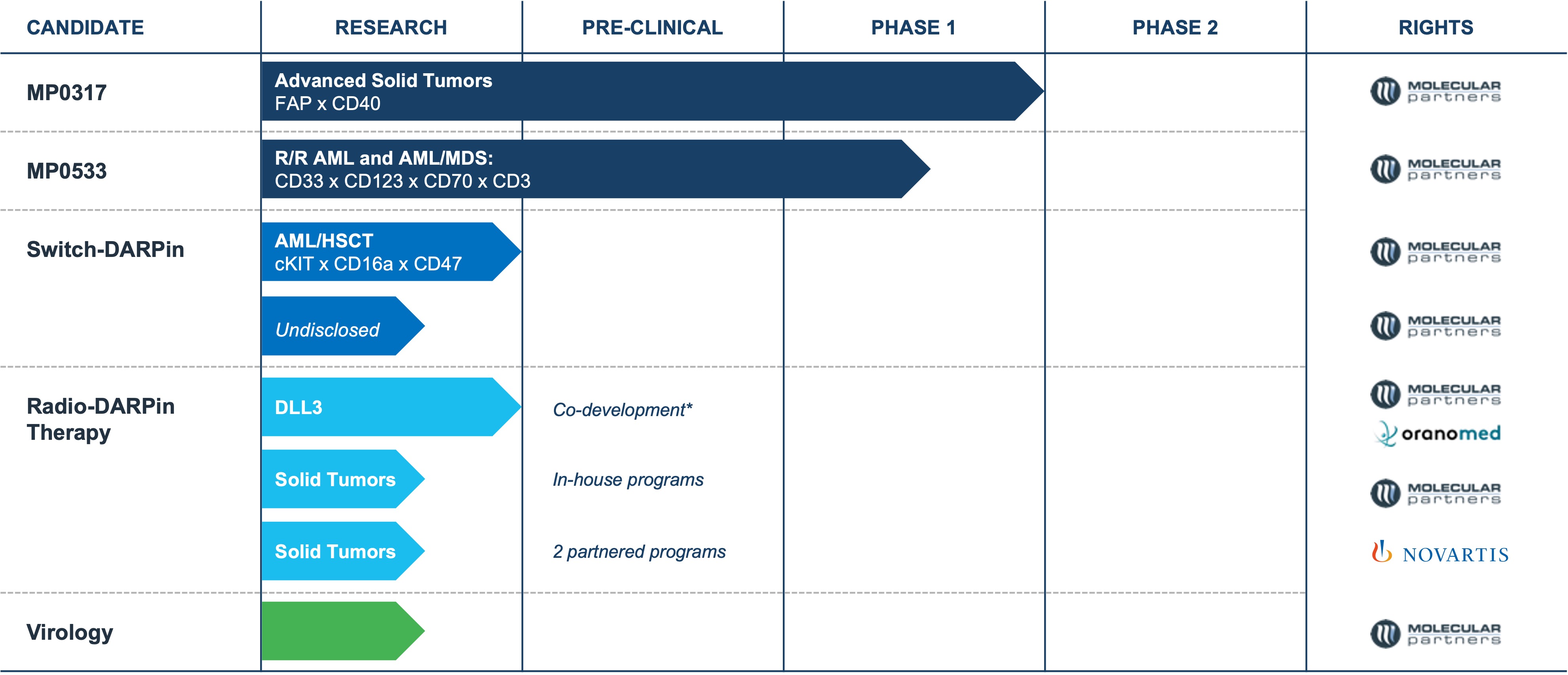
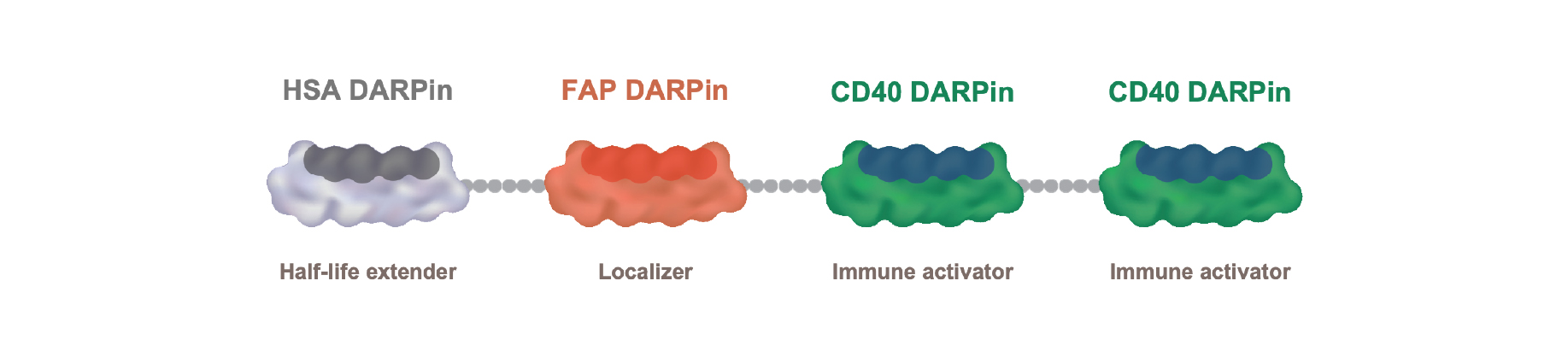
•complete the Phase 1 clinical trial and potentially initiate a new clinical trial of MP0317 (in combination with other therapies), one of our product candidates in our oncology program;
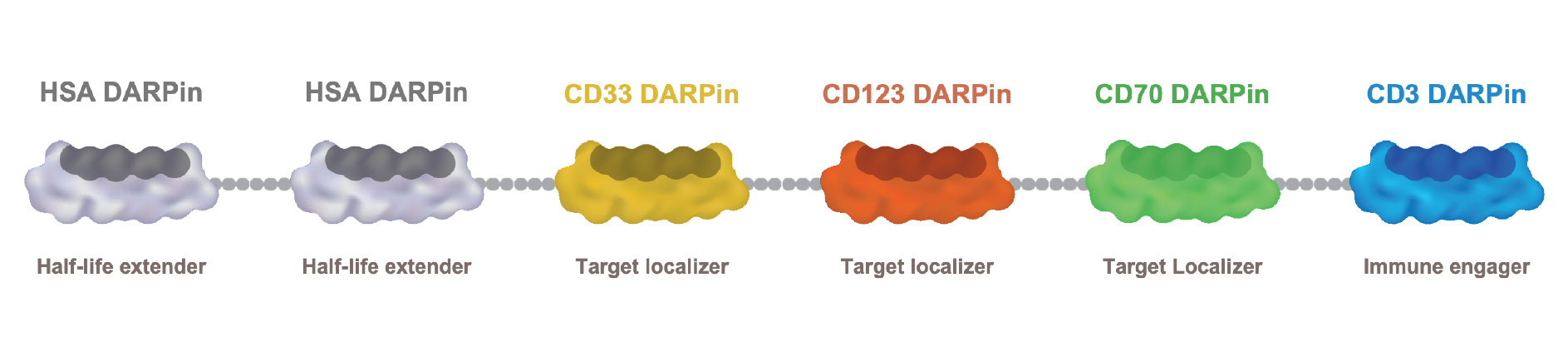
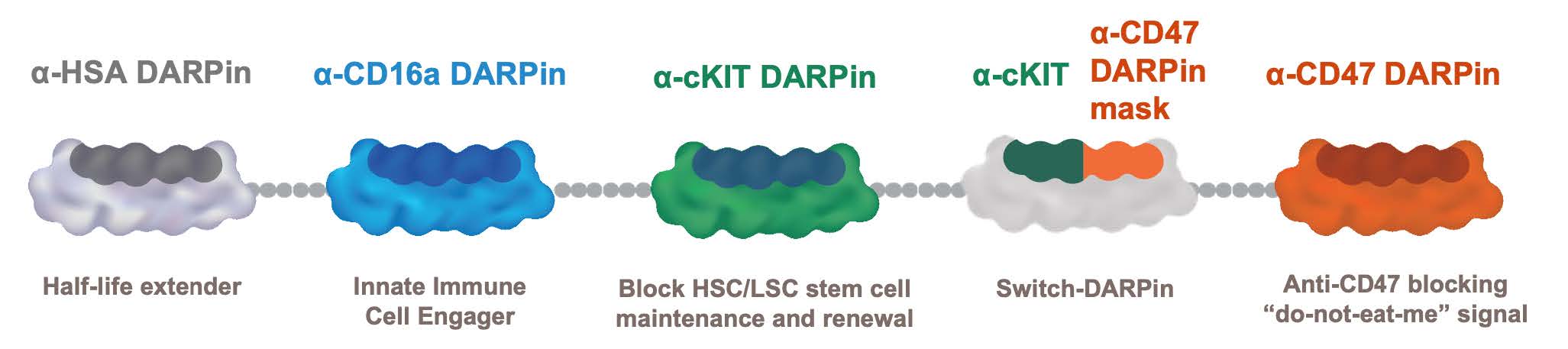
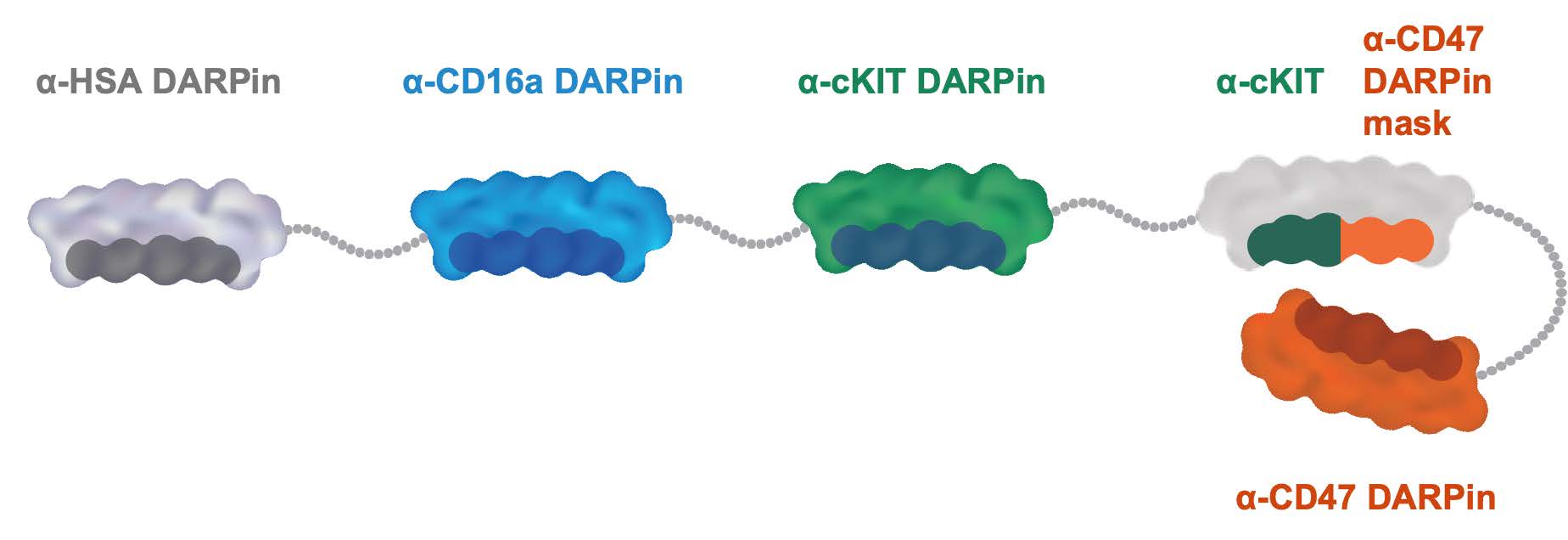
•continue to prepare for and complete and potentially expand the Phase 1 clinical trial of MP0533, our CD3 T-cell engaging candidate against acute myeloid leukemia or AML;
•seek to enhance our Designed Ankyrin Repeat Protein, or DARPin, technology and build on our proprietary product pipeline;
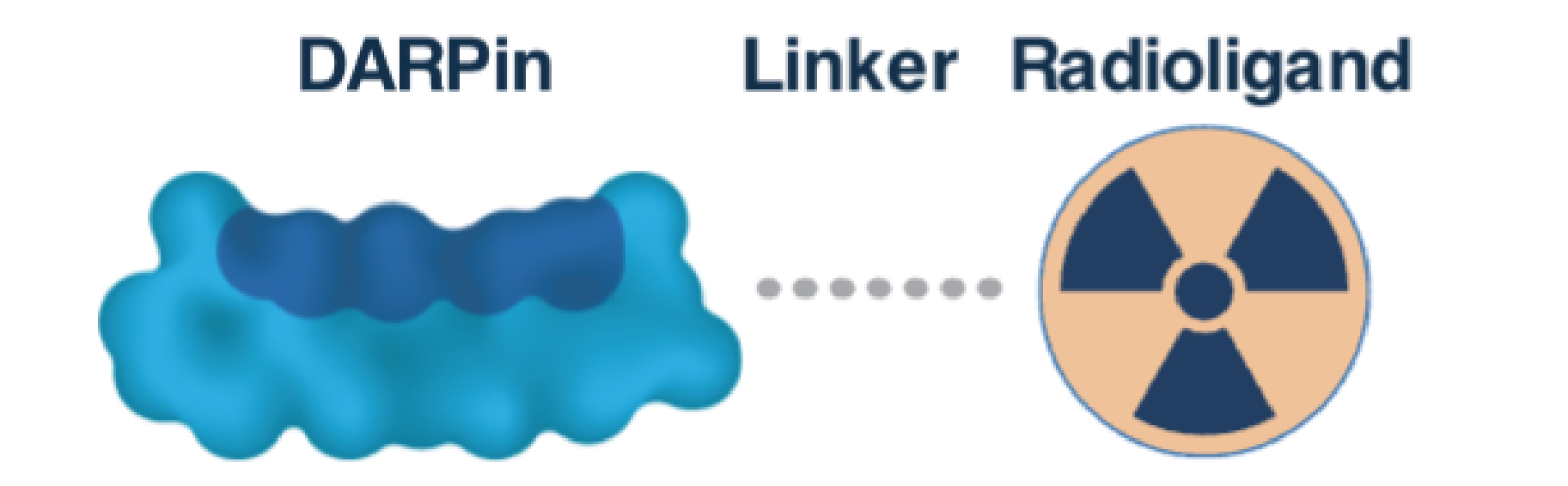
•continue our research activities for developing suitable candidates within the Radio DARPin Therapy, or RDT, space, such as the DLL3 program in collaboration with Orano Med SAS, a subsidiary of Orano SA, or Orano Med;
•continue the research and development of our other clinical- and preclinical-stage product candidates and discovery stage programs including within the radioligand therapeutic space;
•continue the research and development of our other product candidates;
•seek regulatory approvals for any product candidates that successfully complete clinical trials;
•establish a sales, marketing and distribution infrastructure and scale-up manufacturing capabilities to commercialize any product candidates for which we may obtain regulatory approval;
•obtain, maintain, expand, protect and enforce our intellectual property and other proprietary rights and obtain licenses to third-party intellectual property;
•add clinical, regulatory, scientific, operational, financial, legal, intellectual property, compliance and management information systems and personnel, including personnel to support our product development and potential future commercialization efforts; and
•experience any delays or encounter any issues relating to any of the above, including failed studies, ambiguous trial results, safety issues, other regulatory challenges or third party supply or manufacturing issues.
Since our inception in 2004, we have invested most of our resources in developing our product candidates, building our intellectual property portfolio, developing our supply chain, conducting business planning, raising capital and providing general and administrative support for these operations. We do not currently have any approved products and have never generated any revenue from product sales.
To become profitable, we must succeed in developing and eventually commercializing products that generate significant revenue. This will require us or our licensees to be successful in a range of challenging activities, including completing preclinical testing and clinical trials of our product candidates, discovering and developing additional product candidates, obtaining regulatory approval for any product candidates that successfully complete clinical trials, establishing manufacturing and marketing capabilities and ultimately selling any products for which we may obtain regulatory approval. We are only in the preliminary stages of most of these activities. We may never succeed in these activities and, even if we do, may never generate revenue that is significant enough to sustain or increase profitability on a quarterly or annual basis. Our failure to become profitable would decrease the value of our company and could impair our ability to raise capital, maintain our research and development efforts, expand our business or continue our operations.
If we are required by the U.S. Food and Drug Administration, or FDA, the European Medicines Agency, or EMA, or other comparable foreign authorities to perform studies in addition to those we currently anticipate, or if there are any delays in completing our clinical trials or the development of any of our product candidates, our expenses could increase and revenue could be further delayed.
Even if we do generate product royalties or product sales, we may never sustain profitability on a quarterly or annual basis. Our failure to sustain profitability would depress the market price of the ADSs and could impair our ability to raise capital, expand our business, diversify our product offerings or continue our operations. A decline in the market price of the ADSs also could cause you to lose all or a part of your investment.
We may need substantial additional funding in order to complete the development and commercialization of our product candidates. Failure to obtain this necessary capital when needed may force us to delay, limit or terminate certain of our product development or research operations.
To date, we have funded our operations through public and private placements of equity securities, upfront, milestone, option exercise, reservation fee, expense reimbursement, sponsored research payments received from our collaborators, recharging of third party costs and interest income from the investment of our cash, cash equivalents and financial assets. We expect to require additional funding in the future to sufficiently finance our operations and advance development of our product candidates. On July 1, 2022, we entered into a sales agreement with Leerink Partners LLC (previously known as SVB Securities LLC), or the Sales Agreement, to sell ordinary shares from time to time at our discretion under an “at the market” program, with aggregate gross sales proceeds of up to $100.0 million.
We expect that our existing cash, cash equivalents, together with anticipated funding through collaborations, will enable us to fund our operating expenses and capital expenditure requirements well into 2026. We have based this estimate on assumptions that may prove to be wrong, and we could use our capital resources sooner than we currently expect. Our future capital requirements for MP0317 and MP0533 or our preclinical programs will depend on many factors, including:
•the progress, timing and completion of preclinical testing and clinical trials for our current or any future product candidates;
•the number of potential new product candidates we identify and decide to develop;
•the costs involved in growing our organization to the size needed to allow for the research, development and potential commercialization of our current or any future product candidates;
•the costs involved in filing patent applications, maintaining and enforcing patents or defending against infringement, misappropriation or other claims raised by third parties;
•the maintenance of our existing license and collaboration agreements and the entry into new license and collaboration agreements;
•the time and costs involved in obtaining regulatory approval for our product candidates and any delays we may encounter as a result of evolving regulatory requirements or adverse results with respect to any of our product candidates;
•selling and marketing activities undertaken in connection with the potential commercialization of our current or any future product candidates, if approved, and costs involved in the creation of an effective sales and marketing organization; and
•the amount of revenues, if any, we may derive either directly or in the form of milestone and royalty payments from future sales of our product candidates, if approved.
Our ability to raise additional funds will depend on financial, economic and market conditions and other factors, over which we may have no or limited control. Further, as a Swiss corporation, we have less flexibility to raise capital than U.S. companies, particularly in a quick and efficient manner. As a result, we may not be able to access the capital markets as frequently as comparable U.S. companies. See the Risk Factor entitled “Our status as a Swiss corporation means that our shareholders enjoy certain rights that may limit our flexibility to raise capital, issue dividends and otherwise manage ongoing capital needs” for additional information related to our ability to timely raise capital. If adequate funds are not available on commercially acceptable terms or at all when needed, we may be forced to delay, reduce or terminate the development or commercialization of all or part of our research programs or product candidates or we may be unable to take advantage of future business opportunities.
The effects of health epidemics in regions where we, or the third parties on which we rely, have business operations could adversely impact our business, including our preclinical studies and clinical trials, as well as the business or operations of third parties with whom we conduct business.
Our business could be adversely affected by health epidemics in regions where we have concentrations of clinical trial sites or other business operations, and could cause significant disruption in the operations of third party manufacturers and contract research organizations, or CROs, upon whom we rely.
As a result of a pandemic, we may experience disruptions that could impact our business, preclinical studies and clinical trials, including:
•delay of submissions to, and approvals of, regulatory authorities;
•interruption or delays in the operations of regulatory authorities, which may impact review and approval timelines, including delays in receiving approval from local regulatory authorities to initiate our planned clinical trials;
•interruption of, or delays in receiving, supplies of our product candidates or target material from our contract manufacturing organizations, or CMOs, and other suppliers due to staffing shortages, shortages in supply of production materials, production slowdowns or stoppages and disruptions in delivery systems;
•interruptions in preclinical studies due to restricted or limited operations at our facilities; limitations on employee resources that would otherwise be focused on the conduct of our preclinical studies and clinical trials, including because of sickness of employees or their families or the desire of employees to avoid contact with large groups of people; and
•interruption or delays to our sourced discovery and clinical activities.
The extent to which any future health epidemic outbreaks of contagious disease ultimately impacts our business, preclinical studies and clinical trials will depend on future developments, which are highly uncertain and cannot be predicted with confidence, such as the ultimate geographic spread of the disease, the duration of the pandemic, the emergence of variants, the transition to endemic status, travel restrictions and social distancing in Switzerland, the United States and other countries, business closures or business disruptions and the effectiveness of actions taken in countries around the world to contain and treat the disease.
Raising additional capital may cause dilution to holders of our ordinary shares or ADSs, and an inability to raise capital may restrict our operations or require us to relinquish rights to our technologies or product candidates.
Until such time, if ever, as we can generate substantial product revenues, we expect to finance our operations with our existing cash, cash equivalents and current financial assets, proceeds from debt or equity offerings, revenue from our collaborations and interest income from the investment of our cash, cash equivalents and financial assets. In order to further advance the development of our product candidates, discover additional product candidates and pursue our other business objectives, however, we will need to seek additional funds.
We cannot guarantee that future financing will be available in sufficient amounts or on commercially reasonable terms, if at all. Moreover, the terms of any financing may adversely affect the holdings or the rights of holders of our ordinary shares or ADSs and the issuance of additional securities, whether equity or debt, by us, or the possibility of such issuance, may cause the market price of our ADSs and our ordinary shares to decline. The sale of additional equity or convertible securities would dilute all of our existing shareholders and the terms of these securities may include liquidation or other preferences that adversely affect the rights of our shareholders. The incurrence of indebtedness could result in increased fixed payment obligations and we may be required to agree to certain restrictive covenants, such as limitations on our ability to incur additional debt, limitations on our ability to acquire, sell or license intellectual property rights and other operating restrictions that could adversely impact our ability to conduct our business. We could also be required to seek funds through arrangements with collaborators or others at an earlier stage than otherwise would be desirable and we may be required to relinquish rights to some of our technologies or product candidates or otherwise agree to terms unfavorable to us, any of which may have a material adverse effect on our business, operating results and prospects. Further, any additional fundraising efforts may divert our management from its day-to-day activities, which may adversely affect our ability to develop and commercialize our product candidates.
If we are unable to obtain funding on a timely basis, we may be required to significantly curtail, delay or discontinue one or more of our research or development programs or the commercialization of any of our product candidates, or be unable to expand our operations or otherwise capitalize on our business opportunities, as desired, which could materially affect our business, financial condition and results of operations.
Risks Related to the Development and Clinical Testing of Our Product Candidates
We are heavily dependent on the success of our DARPin platform to identify and develop product candidates. If we or our collaborators are unable to successfully develop and commercialize product candidates based on our platforms or experience significant delays in doing so, our business may be harmed.
We are heavily dependent on the success of our DARPin platform technology and the product candidates currently in our core programs. Our commercial prospects will be heavily dependent on product candidates identified and developed using our DARPin platform. To date, we have invested substantially all of our efforts and financial resources to identify, acquire intellectual property for, and develop our DARPin platform technology and our programs, including conducting preclinical studies and early-stage clinical trials, and providing general and administrative support for these operations.
We may not be successful in our efforts to further develop our DARPin platform technology and current product candidates. We are not permitted to market or promote any of our product candidates before we receive regulatory approval from the FDA, European Commission (granted on the basis of a positive opinion from the Committee for Medicinal Products for Human Use of the European Medicines Agency, or EMA and commonly referred to as EMA approval) or comparable foreign regulatory authorities, and we may never receive such regulatory approval for any of our product candidates. Each of our product candidates will require significant additional clinical development, management of preclinical, clinical, and manufacturing activities, regulatory approval, adequate manufacturing supply, a commercial organization, and significant marketing efforts before we generate any revenue from product sales, if at all.
All of our product candidates are in preclinical or various stages of clinical development. Clinical drug development is a lengthy and expensive process with uncertain timelines and uncertain outcomes. If clinical trials of our product candidates, particularly MP0317, MP0533 and product candidates that we have licensed to our partners are prolonged, delayed or not commercially viable, we or our collaborators may be unable to obtain required regulatory approvals, and therefore may be unable to commercialize our product candidates on a timely basis or at all, which will adversely affect our business.
To obtain the requisite regulatory approvals to market and sell any of our product candidates, we or our collaborators for such candidates must demonstrate through extensive preclinical studies and clinical trials that our products are safe, pure and potent or effective in humans. Further, the process of obtaining regulatory approval is expensive, often takes many years following the commencement of clinical trials and can vary substantially based upon the type, complexity and novelty of the product candidates involved, as well as the target indications and patient population. Prior to obtaining approval to commercialize a product candidate in the United States or in other countries, we or our potential future collaborators must demonstrate with substantial evidence from adequate and well-controlled clinical trials, and to the satisfaction of the FDA or comparable foreign regulatory authorities, that such product candidates are safe and effective for their intended uses. Additionally, clinical testing is expensive and can take many years to complete, and its outcome is inherently uncertain. Failure can occur at any time during the clinical trial process and our future clinical trial results may not be successful.
We may experience delays in our ongoing clinical trials and we do not know whether planned clinical trials will begin on time, need to be redesigned, enroll patients on time or be completed on schedule, if at all.
Clinical trials can be delayed, suspended, or terminated for a variety of reasons, including the following:
•delays in or failure to obtain regulatory approval to commence a trial;
•delays in or failure to reach agreement on acceptable terms with prospective CROs and clinical trial sites, the terms of which can be subject to extensive negotiation and may vary significantly among different CROs and trial sites;
•delays in or failure to obtain institutional review board, or IRB, or ethics committee approval at each site;
•delays in or failure to recruit suitable patients to participate in a trial;
•failure to have patients complete a trial or return for post-treatment follow-up;
•clinical sites deviating from trial protocol or dropping out of a trial;
•adding new clinical trial sites;
•manufacturing sufficient quantities of product candidate for use in clinical trials;
•third-party actions claiming infringement by our product candidates in clinical trials and obtaining injunctions interfering with our progress;
•safety or tolerability concerns could cause us or our collaborators, as applicable, to suspend or terminate a trial if we or our collaborators find that the participants are being exposed to unacceptable health risks;
•changes in regulatory requirements, policies and guidelines;
•lower than anticipated retention rates of patients and volunteers in clinical trials;
•our third-party research contractors failing to comply with regulatory requirements or meet their contractual obligations to us in a timely manner, or at all;
•delays in establishing the appropriate dosage levels in clinical trials;
•the difficulty in certain countries in identifying the sub-populations that we are trying to treat in a particular trial, which may delay enrollment and reduce the power of a clinical trial to detect statistically significant results; and
•the quality or stability of the product candidate falling below acceptable standards.
We could encounter delays if a clinical trial is suspended or terminated by us, by the IRBs of the institutions in which such trials are being conducted or ethics committees, by the Data Review Committee, or DRC, or Data Safety Monitoring Board, or DSMB, for such trial or by the EMA, the FDA or other regulatory authorities. Such authorities may impose such a suspension or termination due to a number of factors, including failure to conduct the clinical trial in accordance with regulatory requirements or our clinical protocols, inspection of the clinical trial operations or trial site by the EMA, the FDA or other regulatory authorities resulting in the imposition of a clinical hold, unforeseen safety issues or adverse side effects, including those relating to the class to which our product candidates belong, failure to demonstrate a benefit from using a drug, changes in governmental regulations or administrative actions or lack of adequate funding to continue the clinical trial. For example, we have faced and may face in the future bioburden during drug substance production campaigns or particles in drug product preparations at our CMOs which led or may lead to regulatory actions, including from the FDA. While we and our partners endeavor to maintain appropriate backup supply with respect to our product candidates, and not all such bioburden or particles result in regulatory action or delays, we cannot assure that any such issues would not result in delays in our clinical trials or product development or other adverse impacts on our business.
If we experience delays in the completion of, or termination of, any clinical trial of our product candidates, the commercial prospects of our product candidates will be harmed. In addition, any delays in completing our clinical trials will increase our costs, slow down our product candidate development and approval process and jeopardize our ability to commence product sales and generate revenues. Significant clinical trial delays could also allow our competitors to bring products to market before we do or shorten any periods during which we have the exclusive right to commercialize our product candidates and impair our ability to commercialize our product candidates and may harm our business and results of operations. Any of these occurrences may harm our business, financial condition and prospects significantly. In addition, many of the factors that cause, or lead to, a delay in the commencement or completion of clinical trials may also ultimately lead to the denial of regulatory approval of our product candidates or result in the development of our product candidates being stopped early.
Clinical trials must be conducted in accordance with the FDA, the EMA and other applicable regulatory authorities' legal requirements and regulations, and are subject to oversight by these governmental agencies, IRBs at the medical institutions where the clinical trials are conducted or ethics committees. In addition, clinical trials must be conducted with supplies of our product candidates produced under cGMP requirements and other regulations. Furthermore, we rely on CROs and clinical trial sites to ensure the proper and timely conduct of our clinical trials and while we have agreements governing their committed activities, we have limited influence over their actual performance. We depend on our collaborators and on medical institutions and CROs to conduct our clinical trials in compliance with GCP requirements. To the extent our collaborators or the CROs or investigators fail to enroll participants for our clinical trials, fail to conduct the study to GCP standards or are delayed for a significant time in the execution of trials, including achieving full enrollment, we may be affected by increased costs, program delays or both, which may harm our business.
Further, conducting clinical trials in multiple countries presents additional risks that may delay completion of our clinical trials. These risks include the failure of enrolled patients in foreign countries to adhere to clinical protocol as a result of differences in healthcare services or cultural customs, managing additional administrative burdens associated with adhering to GCP, regulations and other foreign regulatory schemes, as well as political and economic risks relevant to such foreign countries.
In addition, future clinical trials that could be conducted in countries outside Switzerland, the European Union and the United States may subject us to further delays and expenses as a result of increased shipment costs, additional regulatory requirements and the engagement of non-European Union and non-U.S. CROs, as well as expose us to risks associated with clinical investigators who are unknown to the FDA or the EMA, and different standards of diagnosis, screening and medical care.
We may not be successful in our efforts to use and expand our platform to build a pipeline of product candidates with commercial value.
A key element of our strategy is to use and expand our platform to build a pipeline of product candidates and progress these product candidates through clinical development. So far none of the product candidates originating from our platform has received marketing approval from the FDA or other regulatory authorities. The scientific discoveries that form the basis for our efforts to discover and develop targeted oncology therapeutic candidates for cancer patients are relatively new. The scientific evidence to support the feasibility of developing product candidates based on these discoveries is both preliminary and limited. There can be no assurance that any development problems we may experience in the future related to our platform will not cause significant delays or unanticipated costs or that such development problems can be solved. Even if we are successful in building our pipeline of product candidates, the potential product candidates that we identify may not be suitable for clinical development or generate acceptable clinical data, including as a result of being shown to have characteristics that indicate that they are unlikely to be products that will receive marketing approval from the FDA or other regulatory authorities or achieve market acceptance.
Preclinical drug development is uncertain. Some or all of our preclinical programs may experience delays or may never advance to clinical trials, which would adversely affect our ability to obtain regulatory approvals or commercialize these product candidates on a timely basis or at all, which would have an adverse effect on our business.
In order to obtain FDA or EMA approval to market a new pharmaceutical or biological product we must demonstrate proof of safety, purity and potency or efficacy in humans. To meet these requirements we will have to conduct adequate and well-controlled clinical trials. Before we can commence clinical trials for a product candidate, we must complete extensive preclinical testing and studies that support our planned Investigational New Drug application, or IND, in the United States, or a Clinical Trial Authorization Application, or CTA, in Europe. We cannot be certain of the timely completion or outcome of our preclinical testing and studies and cannot predict if the FDA or EMA will accept our proposed clinical programs or if the outcome of our preclinical testing and studies will ultimately support the further development of these product candidates. Thus, we cannot be sure that we will be able to submit INDs or CTAs for our preclinical programs on the timelines we expect, if at all, and we cannot be sure that submission of INDs or CTAs will result in the FDA or EMA allowing clinical trials to begin.
Conducting preclinical testing is a lengthy, time-consuming and expensive process. The length of time may vary substantially according to the type, complexity, novelty and intended use of the product candidate, and often can be several years or more per product candidate. Delays associated with product candidates for which we are directly conducting preclinical testing and studies may cause us to incur additional operating expenses. We may encounter similar or different safety issues in this trial or our other clinical trials in the future. Moreover, we may continue to be affected by delays associated with the preclinical testing and studies of certain product candidates conducted by our potential partners over which we have no control. The commencement and rate of completion of preclinical studies and studies for a product candidate may be delayed by many factors, including, for example:
•the inability to generate sufficient preclinical or other in vivo or in vitro data to support the initiation of clinical studies;
•delays in reaching a consensus with regulatory agencies on study design; and
•the FDA or EMA not allowing us to rely on previous findings of safety and efficacy for other similar but approved products and published scientific literature.
Moreover, even if clinical trials do begin for our preclinical programs, our development efforts may not be successful, and clinical trials that we conduct or that third parties conduct on our behalf may not demonstrate sufficient safety, purity and potency or efficacy to obtain the requisite regulatory approvals for any of our product candidates or product candidates employing our technology.
Positive results from early preclinical studies of our product candidates would not necessarily be predictive of the results of later preclinical studies and any ongoing or future clinical trials of our product candidates. If we were to achieve positive results from preclinical studies, but were unable to then replicate those positive results in our later preclinical studies and ongoing future clinical trials, we might be unable to successfully develop, obtain regulatory or marketing approval for and commercialize our product candidates.
Any positive results from our preclinical studies of our product candidates may not necessarily be predictive of the results from required later preclinical studies and clinical trials, and there can be no assurance that any of our clinical trials will ultimately be successful or support further clinical development of any of our product candidates.
For example, even if biological activity in patient samples following administration of initial doses is observed in our clinical trials, there can be no assurance that such biological activity will be similarly observed and maintained following administration of additional doses or any drop in biological activity could be overcome with additional development regarding more frequent dosing regimens. Similarly, even if we are able to complete our planned preclinical studies or any future clinical trials of our product candidates according to our current development timeline, the positive results from such preclinical studies and clinical trials of our product candidates may not be replicated in subsequent preclinical studies or clinical trial results. In addition, positive results in later stage clinical trials of one of our product candidates in an indication may not be predictive of the safety or efficacy of our other product candidates in other indications, even if they employ a similar mechanism of action.
Many companies in the pharmaceutical and biotechnology industries have suffered significant setbacks in late-stage clinical trials after achieving positive results in early-stage development and we cannot be certain that we will not face similar setbacks. For example, our therapeutics in oncology, ophthalmology and virology has in the past and may in the future result in the creation of anti-drug antibodies that can neutralize the effects of the therapeutic, require that higher doses be used to obtain a therapeutic effect or cause adverse events. Whether anti-drug antibodies will be created and how they react can often not be predicted from nonclinical or even clinical studies, and their detection or appearance can be delayed. These setbacks have been caused by, among other things, preclinical and other nonclinical findings made while clinical trials were underway, or safety or efficacy observations made in preclinical studies and clinical trials, including previously unreported adverse events. Moreover, preclinical, nonclinical and clinical data are often susceptible to varying interpretations and analyses and many companies that believed their product candidates performed satisfactorily in preclinical studies and clinical trials nonetheless failed to obtain FDA or EMA approval.
Some of our product candidates utilize a novel mechanism of action which may result in greater research and development expenses, regulatory issues that could delay or prevent approval, or discovery of unknown or unanticipated adverse effects.
Some of our product candidates, such as MP0317 and MP0533, the lead product candidates from our oncology program, and also the potential future RDT product candidates, utilize novel mechanisms of action which may result in greater research and development expenses, regulatory and development or CMC and supply chain issues that could delay or prevent approval, or discovery of unknown or unanticipated adverse effects. Regulatory approval of novel product candidates such as ours can be more expensive, riskier and take longer than for other, more well-known or extensively studied pharmaceutical or biopharmaceutical product candidates due to our and regulatory agencies’ lack of experience with the novel mechanisms of action. The novelty of our mechanism of action may lengthen the regulatory review process, require us to conduct additional studies or clinical trials, increase our development costs, lead to changes in regulatory positions and interpretations, delay or prevent approval and commercialization of our product candidates or lead to significant post-approval limitations or restrictions. The novel mechanisms of action also means that fewer people are trained in or experienced with product candidates of such type, which may make it more difficult to find, hire and retain personnel for research, development and manufacturing positions. Any such events could adversely impact our business prospects, financial condition and results of operations.
Failure to successfully validate, develop and obtain regulatory approval for companion diagnostics, if needed, could harm our product development strategy.
As one of the key elements of our clinical development strategy, we seek to identify patient subsets within a disease category who may derive selective and meaningful benefit from the product candidates we are developing.
In collaboration with partners, we may develop companion diagnostics to help us to more accurately identify patients within a particular subset, both during our clinical trials and in connection with the commercialization of our product candidates.
Companion diagnostics are subject to regulation by the FDA and comparable foreign regulatory authorities as medical devices and require separate regulatory approval prior to commercialization. The FDA generally expects contemporaneous regulatory approvals of the companion diagnostic and the therapeutic product. We do not develop companion diagnostics internally and thus we are dependent on the sustained cooperation and effort of third-party collaborators in developing and obtaining regulatory approval for these companion diagnostics. We and our collaborators may encounter difficulties in developing and obtaining approval for the companion diagnostics, including issues relating to selectivity/specificity, analytical validation, reproducibility or clinical validation. Any delay or failure by our collaborators to develop or obtain regulatory approval of the companion diagnostics could delay or prevent approval of our product candidates.
In addition, our collaborators may encounter production difficulties that could constrain the supply of the companion diagnostics, and both they and we may have difficulties gaining acceptance of the use of the companion diagnostics in the clinical community. If such companion diagnostics fail to gain market acceptance, it would have an adverse effect on our ability to derive revenues from sales of our products. In addition, the diagnostic company with whom we contract may decide to discontinue selling or manufacturing the companion diagnostic that we anticipate using in connection with development and commercialization of our product candidates or our relationship with such diagnostic company may otherwise terminate.
We may not be able to enter into arrangements with another diagnostic company to obtain supplies of an alternative diagnostic test for use in connection with the development and commercialization of our product candidates or do so on commercially reasonable terms, which could adversely affect and/or delay the development or commercialization of our product candidates.
Interim, topline and preliminary data from our clinical trials that we announce or publish may change as more patient data become available and are subject to audit and verification procedures that could result in material changes in the final data.
From time to time, we may publish interim, topline or preliminary data from our clinical trials. Preliminary and interim data from our clinical trials may change as more patient data become available. Preliminary or interim data from our clinical trials are not necessarily predictive of final results. Preliminary and interim data are subject to the risk that one or more of the clinical outcomes may materially change as patient enrollment continues, more patient data become available and we issue our final clinical trial report. Interim, topline and preliminary data also remain subject to audit and verification procedures that may result in the final data being materially different from the preliminary data we previously published. As a result, preliminary and interim data should be viewed with caution until the final data are available. Adverse changes in the final data compared to the interim data could significantly harm our business prospects.
Further, others, including regulatory agencies, may not accept or agree with our assumptions, estimates, calculations, conclusions or analyses or may interpret or weigh the importance of data differently, which could impact the value of the particular program, the approvability or commercialization of the particular product candidate or product and our company in general. In addition, the information we choose to publicly disclose regarding a particular preclinical study or clinical trial is based on what is typically extensive information, and you or others may not agree with what we determine is the material or otherwise appropriate information to include in our disclosure, and any information we determine not to disclose may ultimately be deemed significant with respect to future decisions, conclusions, views, activities or otherwise regarding a particular product, product candidate or our business.
If the preliminary and interim data that we report differ from actual results, or if others, including regulatory authorities, disagree with the conclusions reached, our ability to obtain approval for, and commercialize, our product candidates may be harmed, which could harm our business, operating results, prospects or financial condition.
Because the number of patients in certain of our clinical trials may be small, the results from such trials may be less reliable than results achieved in larger clinical trials.
A study design that is considered appropriate includes a sufficiently large sample size with appropriate statistical power to allow a meaningful interpretation of the results. The preliminary results of studies with smaller sample sizes can be disproportionately influenced by the impact the treatment had on a few individuals, which limits the ability to generalize the results across a broader community, thus making the study results less reliable than studies with a larger number of subjects.
Our product candidates may have serious adverse, undesirable or unacceptable side effects which may delay or prevent marketing approval. If side effects are identified during the development of our product candidates or following approval, if any, we may need to abandon our development of such product candidates, the commercial profile of any approved label may be limited, or we may be subject to other significant negative consequences following marketing approval, if any.
Undesirable side effects that may be caused by our product candidates could cause us or regulatory authorities to interrupt, delay or halt clinical trials and could result in a more restrictive label or the delay or denial of regulatory approval by the FDA, the EMA or other comparable foreign authorities. While our preclinical studies and clinical trials for our product candidates to date have generally been well tolerated from a risk-benefit perspective, the results from ongoing and future trials may not support this conclusion.
The results of future clinical studies may show that our product candidates cause undesirable or unacceptable side effects or even death. In such an event, our trials could be suspended or terminated and the FDA, the EMA or comparable foreign regulatory authorities could order us to cease further development of or deny approval of our product candidates for any or all targeted indications. The drug-related side effects could affect patient recruitment or the ability of enrolled patients to complete the trial or result in potential product liability claims. Any of these occurrences may harm our prospects significantly. Further, because all of our product candidates and preclinical programs are based on our DARPin technology, any adverse safety or efficacy findings related to any product candidate or preclinical program may adversely impact the viability of our other product candidates or preclinical programs.
Additionally, if any of our product candidates receives marketing approval and we or others later identify undesirable or unacceptable side effects caused by such products, a number of potentially significant negative consequences could result, including:
•regulatory authorities may withdraw approvals of such products and require us to take our approved product off the market;
•regulatory authorities may require the addition of labeling statements, specific warnings, a contraindication or field alerts to physicians and pharmacies;
•regulatory authorities may require a medication guide outlining the risks of such side effects for distribution to patients, or that we implement a risk evaluation and mitigation strategy, or REMS, plan to ensure that the benefits of the product outweigh its risks;
•regulatory authorities may require additional clinical trials;
•we may be required to change the way the product is administered, conduct additional clinical trials or change the labeling of the product;
•sales of the product may decrease significantly;
•we may be subject to litigation or product liability claims; and
•our reputation may suffer.
Any of these events could prevent us, our collaborators or our potential future partners from achieving or maintaining market acceptance of the affected product or could substantially increase commercialization costs and expenses, which in turn could delay or prevent us from generating significant revenue from the sale of our products.
If any of our product candidates has negative side effects, public perception of our DARPin platform and commercial opportunities for all of our current and future product candidates could be adversely affected.
Adverse side effects that may be caused by any of our product candidates could negatively impact the public perception of and commercial opportunities for all of our product candidates. The clinical and commercial success of our product candidates will depend in part on the absence of negative side effects caused by our product candidates. Even if an adverse side effect that results from one of our product candidates is unlikely to occur in our other product candidates, all of our product candidates could be adversely affected because the negative side effect may be perceived to be a likely side effect of all of our product candidates. Infusion-related reactions have been seen in MP0310, MP0317 and MP0533, and, while manageable, such reactions may limit the tolerability profile of a product candidate or its potential for combination with other medication. These adverse events may negatively affect the perception of the DARPin platform, the commercial opportunity for our product candidates or cause us to suspend clinical trials. In addition, if public perception is influenced by claims that radioligands or specific therapies within radioligands are unsafe or less safe than available alternatives, our product candidates may not be accepted by the general public or the medical community.
We face significant competition for our drug discovery and development efforts, and if we do not compete effectively, our commercial opportunities will be reduced or eliminated.
The market for pharmaceutical products is highly competitive. Our competitors include many established pharmaceutical companies, biotechnology companies, universities and other research or commercial institutions, many of which have substantially greater financial, research and development resources than us. Large pharmaceutical companies, in particular, have extensive experience in clinical testing, obtaining regulatory approvals, recruiting patients and manufacturing pharmaceutical products. Smaller and early stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. These third parties compete with us in recruiting and retaining qualified scientific and management personnel, establishing clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies complementary to, or necessary for, the development of our products. The fields in which we operate are characterized by rapid technological change and innovation. There can be no assurance that our competitors are not currently developing, or will not in the future develop, technologies and products that are equally or more effective or are more economically attractive as any of our current or future technology or product. Competing products or platforms may gain faster or greater market acceptance than our products or platform and medical advances or rapid technological development by competitors may result in our product candidates or platforms becoming non-competitive or obsolete before we are able to recover our research and development and commercialization expenses.
Additionally, certain of our product candidates may be administered in combination with approved pharmaceutical products. Our ability to develop and ultimately commercialize our product candidates used in combination with other therapies will depend on our ability to access these drugs on commercially reasonable terms for the clinical trials and their availability for use with the commercialized product, if approved. We cannot be certain that current or potential future commercial relationships will provide us with a sufficient supply of these drugs on commercially reasonable terms or at all. If we, our product candidates or our platforms do not compete effectively, it may have an adverse effect on our business and results of operation.
We depend on enrollment of patients in our clinical trials for our product candidates. If we are unable to enroll patients in our clinical trials, our research and development efforts and business could be adversely affected.
Identifying and qualifying patients to participate in our clinical trials is critical to our success. Patient enrollment depends on many factors, including the size and nature of the patient population, eligibility criteria for the trial, the proximity of patients to clinical sites, the design of the clinical protocol, the availability of competing clinical trials, the availability of new drugs approved for the indication the clinical trial is investigating, and clinicians' and patients' perceptions as to the potential advantages of the drug being studied in relation to other available therapies. Since some of our product candidates could be focused on addressing sub-groups of cancer patients, there are limited patient pools from which to draw in order to complete our clinical trials in a timely and cost-effective manner. Furthermore, if the actual number of patients with these pathologies is smaller than we anticipate, we may encounter difficulties in enrolling patients in our clinical trials, thereby delaying or preventing development and approval of our drug candidates. Even once enrolled we may be unable to retain a sufficient number of patients to complete any of our trials.
Furthermore, our efforts to build relationships with patient communities may not succeed, which could result in delays in patient enrollment in our clinical trials. In addition, any negative results we may report in clinical trials of one of our product candidates may make it difficult or impossible to recruit and retain patients in other clinical trials of that same product candidate. Delays in the completion of any clinical trial of our product candidates will increase our costs, slow down our product candidate development and approval process and delay or potentially jeopardize our ability to commence product sales and generate revenue. In addition, some of the factors that cause, or lead to, a delay in the commencement or completion of clinical trials may also ultimately lead to the denial of regulatory approval of our product candidates.
Additionally, our ability to successfully initiate, enroll and complete clinical trials in foreign countries is subject to numerous risks unique to conducting business in foreign countries, including:
•different standards for the conduct of clinical trials;
•difficulty in identifying and partnering with qualified local consultants, physicians and partners; and
•the potential burden of complying with a variety of foreign laws, medical standards and regulatory requirements, including the regulation of pharmaceutical and biotechnology research and products.
We may become exposed to costly and damaging liability claims, either when testing our product candidates in the clinic or at the commercial stage, and our product liability insurance may not cover all damages from such claims.
We are exposed to potential product liability and professional indemnity risks that are inherent in the research, development, manufacturing, marketing and use of pharmaceutical products.
Currently, we have no products that have been approved for commercial sale; however, the current and future use of product candidates by us and our corporate collaborators in clinical trials, and the potential sale of any approved products in the future, may expose us to liability claims. These claims might be made by patients who use the product, healthcare providers, pharmaceutical companies, our corporate collaborators or others selling such products. Any claims against us, regardless of their merit, could be difficult and costly to defend and could adversely affect the market for our product candidates or any prospects for commercialization of our product candidates. Although the clinical trial process is designed to identify and assess potential side effects, it is always possible that a drug, even after regulatory approval, may exhibit unforeseen side effects. If any of our product candidates were to cause adverse side effects during clinical trials or after approval of the product candidate, we may be exposed to substantial liabilities. Physicians and patients may not comply with any warnings that identify known potential adverse effects and patients who should not use our product candidates. Regardless of the merits or eventual outcome, liability claims may result in:
•decreased demand for our products due to negative public perception and injury to our reputation;
•withdrawal of clinical trial participants or difficulties in recruiting new trial participants;
•initiation of investigations by regulators;
•costs to defend or settle the related litigation;
•a diversion of management's time and our resources;
•substantial monetary awards to trial participants or patients;
•product recalls, withdrawals or labeling, marketing or promotional restrictions;
•loss of revenues from product sales; and
•the inability to commercialize any of our product candidates, if approved.
Although we maintain adequate clinical trial insurance for our product candidates, it is possible that our liabilities could exceed our insurance coverage. We intend to expand our insurance coverage to include the sale of commercial products if we obtain marketing approval for any of our product candidates. However, we may not be able to maintain insurance coverage at a reasonable cost or obtain insurance coverage that will be adequate to satisfy any liability that may arise. If a successful product liability claim or series of claims is brought against us for uninsured liabilities or in excess of insured liabilities, our assets may not be sufficient to cover such claims and our business operations could be impaired. Should any of the events described above occur, this could have an adverse effect on our business and results of operations.
We conduct clinical trials for our product candidates outside the United States, and the FDA and similar foreign regulatory authorities may not accept data from such trials.
We also conduct clinical trials outside the United States, including in Europe and are likely to continue to do so in these or other foreign jurisdictions. The acceptance of trial data from clinical trials conducted outside the United States by the FDA may be subject to certain conditions. In cases where data from clinical trials conducted outside the United States are intended to serve as the sole basis for marketing approval in the United States, the FDA will generally not approve the application on the basis of foreign data alone unless (i) the data are applicable to the U.S. population and medical practice; (ii) the trials were performed by clinical investigators of recognized competence and (iii) the data may be considered valid without the need for an on-site inspection by the FDA or, if the FDA considers such an inspection to be necessary, the FDA is able to validate the data through an on-site inspection or other appropriate means.
Additionally, the FDA’s clinical trial requirements, including sufficient size of patient populations and statistical powering, must be met. Many foreign regulatory bodies have similar approval requirements. In addition, such foreign trials would be subject to the applicable local laws of the foreign jurisdictions where the trials are conducted. There can be no assurance that the FDA or any similar foreign regulatory authority will accept data from trials conducted outside of the United States or the applicable jurisdiction. If the FDA or any similar foreign regulatory authority does not accept such data, it would result in the need for additional trials, which would be costly and time-consuming and delay aspects of our business plan, and which may result in our product candidates not receiving approval or clearance for commercialization in the applicable jurisdiction.
The regulatory approval processes of the FDA, the EMA and comparable foreign authorities are lengthy, time consuming and inherently unpredictable, and if we are ultimately unable to obtain regulatory approval for our product candidates, our business will be substantially harmed.
The time required to obtain approval by the FDA, the EMA and comparable foreign authorities is unpredictable but typically takes many years, if obtained at all, following the commencement of clinical trials and depends upon numerous factors, including the substantial discretion of the regulatory authorities. In addition, approval policies, regulations, or the type and amount of clinical data necessary to gain approval may change during the course of a product candidate’s clinical development and may vary among jurisdictions. We have not obtained regulatory approval for any product candidate or product candidates licensed to our partners and it is possible that none of such existing product candidates or any product candidates we may seek to develop in the future will ever obtain regulatory approval.
Our product candidates could fail to receive regulatory approval for many reasons, including the following:
•the FDA, the EMA or comparable foreign regulatory authorities may disagree with the design or implementation of our clinical trials, including the size of our clinical trials or the doses tested;
•we may be unable to demonstrate to the satisfaction of the FDA, the EMA or comparable foreign regulatory authorities that a product candidate is safe, pure and potent or effective for its proposed indication;
•the results of clinical trials may not meet the level of statistical significance required by the FDA, the EMA or comparable foreign regulatory authorities for approval;
•we may be unable to demonstrate that a product candidate's clinical and other benefits outweigh its safety risks;
•the FDA, the EMA or comparable foreign regulatory authorities may disagree with our interpretation of data from preclinical studies or clinical trials or may require us to test additional dose regimens of our product candidates;
•the data collected from clinical trials of our product candidates may not be sufficient to support the submission of a Biologics License Application, or BLA, to the FDA or other submission or to obtain regulatory approval in the United States, the European Union or elsewhere;
•the FDA, the EMA or comparable foreign regulatory authorities may find deficiencies with or fail to approve the manufacturing processes or facilities of third-party manufacturers with which we contract for clinical and commercial supplies; and
•the approval policies or regulations of the FDA, the EMA or comparable foreign regulatory authorities may significantly change in a manner rendering our clinical data insufficient for approval.
This lengthy approval process as well as the unpredictability of future clinical trial results may result in our failing to obtain regulatory approval to market any of our product candidates, which would significantly harm our business. The FDA, the EMA and other comparable foreign authorities have substantial discretion in the approval process, and determining when or whether regulatory approval will be obtained for any of our product candidates. Even if we believe the data collected from clinical trials of our product candidates are promising, such data may not be sufficient to support approval by the FDA, the EMA or any other regulatory authority.
We or our partners may seek fast-track designation for some or all of our product candidates, but we may not receive such designation, and even if we do, it may not lead to a faster development or regulatory review or approval process, and will not increase the likelihood that such product candidates will receive marketing approval.
We or our partners may seek fast-track designation and review for some or all of our product candidates. If a drug is intended for the treatment of a serious or life-threatening condition or disease, and nonclinical or clinical data demonstrate the potential to address an unmet medical need, the product may qualify for FDA fast track designation, for which sponsors must apply. The FDA has broad discretion whether or not to grant this designation. Thus, even if we or our collaborators believe a particular product candidate is eligible for this designation, such as we received for ensovibep in June 2021, the FDA may decide not to grant it. Moreover, even if we do receive fast track designation, we or our collaborators may not experience a faster development process, review or approval compared to conventional FDA procedures. In addition, the FDA may withdraw fast track designation if it believes that the designation is no longer supported by data from the clinical development program.
Even if our product candidates obtain regulatory approval, we will be subject to ongoing obligations and continued regulatory review, which may result in significant additional expense. Additionally, our product candidates, if approved, could be subject to labeling and other restrictions and market withdrawal and we may be subject to penalties if we fail to comply with regulatory requirements or experience unanticipated problems with our products.
If the FDA, the EMA or a comparable foreign regulatory authority approves any of our product candidates, the manufacturing processes, labeling, packaging, distribution, adverse event reporting, storage, advertising, promotion, recordkeeping, exporting and importing for the product will be subject to extensive and ongoing regulatory requirements. These requirements include submissions of safety and other post-marketing information and reports, registration, as well as continued compliance with cGMPs and GCPs for any clinical trials that we conduct post-approval, all of which may result in significant expense and limit our ability to commercialize such products. In addition, any regulatory approvals that we receive for our product candidates may also be subject to limitations on the approved indicated uses for which the product may be marketed or to the conditions of approval, or contain requirements for potentially costly post marketing testing, including Phase 4 clinical trials, and surveillance to monitor the safety and efficacy of the product candidate.
Later discovery of previously unknown problems with our product candidates, including adverse events of unanticipated severity or frequency, or with our third-party manufacturers or manufacturing processes, or failure to comply with regulatory requirements, may result in, among other things:
•restrictions on the marketing or manufacturing of our product candidates, withdrawal of the product from the market or voluntary or mandatory product recalls;
•restrictions on product distribution or use, or requirements to conduct post-marketing studies or clinical trials;
•fines, restitutions, disgorgement of profits or revenues, warning letters, untitled letters or holds on clinical trials;
•refusal by the FDA, the EMA or comparable foreign regulatory authorities to approve pending applications or supplements to approved applications filed by us or suspension or revocation of approvals;
•product seizure or detention, or refusal to permit the import or export of our product candidates;
•negative impact to our reputation; and
•injunctions or the imposition of civil or criminal penalties.
The occurrence of any event or penalty described above may inhibit our ability to commercialize our product candidates and generate revenue and could require us to expend significant time and resources in response and could generate negative publicity.
In addition, if any of our product candidates is approved, our product labeling, advertising and promotion will be subject to regulatory requirements and continuing regulatory review. The FDA strictly regulates the promotional claims that may be made about drug products. In particular, a product may not be promoted for uses that are not approved by the FDA as reflected in the product’s approved labeling. If we receive marketing approval for a product candidate, physicians may nevertheless prescribe it to their patients in a manner that is inconsistent with the approved label based on the physician’s independent medical judgement. If we are found to have promoted such off-label uses, we may become subject to significant liability. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses, and a company that is found to have improperly promoted off-label uses may be subject to significant sanctions. The federal government has levied large civil and criminal fines against companies for alleged improper promotion and has enjoined several companies from engaging in off-label promotion. The FDA has also requested that companies enter into consent decrees or permanent injunctions under which specified promotional conduct is changed or curtailed.
Our product candidates are classified as biologics in the United States and, therefore, can only be sold if we obtain a BLA from the FDA. The holder of a BLA is obligated to monitor and report adverse events and any failure of a product to meet the specifications in the BLA. The holder of a BLA must also submit new or supplemental applications and obtain FDA approval for certain changes to the approved product, product labeling or manufacturing process. Failure to comply with a BLA or any other ongoing regulatory obligation may result in suspension of approval to manufacture or distribute the relevant product, as well as fines or imprisonment for violations.
The FDA’s and other regulatory authorities’ policies may change and additional government regulations may be enacted that could prevent, limit or delay regulatory approval of our product candidates. If we or one of our distributors, licensees or co-marketers are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we are not able to maintain regulatory compliance, we may lose any marketing approval that we may have obtained and we may not achieve or sustain profitability. We also cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative or executive action, either in the United States or in other countries. For example, the policies and executive actions of the Biden administration may impact our business and industry. It is difficult to predict how these policies and executive actions will be implemented, and the extent to which they will impact the FDA’s ability to exercise its regulatory authority. If these policies or executive actions impose constraints on FDA’s ability to engage in oversight and implementation activities in the normal course, our business may be negatively impacted.
Due to our limited resources and access to capital, we must, and have in the past decided to, prioritize development of certain product candidates over other potential candidates. These decisions may prove to have been wrong and may adversely affect our revenues.
Because we have limited resources and access to capital to fund our operations, we must decide which product candidates to pursue and the amount of resources to allocate to each. Our decisions concerning the allocation of research, collaboration, management and financial resources toward particular compounds, product candidates or therapeutic areas may not lead to the development of viable commercial products and may divert resources away from better opportunities. Similarly, our decisions to delay, terminate or collaborate with third parties in respect of certain product development programs may also prove not to be optimal and could cause us to miss valuable opportunities. If we make incorrect determinations regarding the market potential of our product candidates or misread trends in the biopharmaceutical industry, in particular for our lead product candidates, our business, financial condition and results of operations could be adversely affected.
Risks Related to Commercialization of Our Product Candidates
Enacted and future legislation may increase the difficulty and cost for us to obtain marketing approval of and commercialize our product candidates and may affect the prices we may set.
In the United States, the European Union and other foreign jurisdictions, there have been a number of legislative and regulatory changes to the healthcare system that could affect our future results of operations. In particular, there have been and continue to be a number of initiatives at the United States federal and state levels that seek to reduce healthcare costs and improve the quality of healthcare. For example, in March 2010, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act of 2010, or collectively the ACA, became law. The ACA is a sweeping law intended to broaden access to health insurance, reduce or constrain the growth of healthcare spending, enhance remedies against fraud and abuse, add new transparency requirements for the healthcare and health insurance industries, impose new taxes and fees on the health industry and impose additional health policy reforms.
The ACA, among other things, increased the minimum level of Medicaid rebates payable by manufacturers of brand name drugs; required collection of rebates for drugs paid by Medicaid managed care organizations; required manufacturers to participate in a coverage gap discount program, under which they must agree to offer point-of-sale discounts (increased to 70 percent, effective as of January 1, 2019) off negotiated prices of applicable brand drugs to eligible beneficiaries during their coverage gap period, as a condition for the manufacturer’s outpatient drugs to be covered under Medicare Part D; imposed a non-deductible annual fee on pharmaceutical manufacturers or importers who sell certain “branded prescription drugs” to specified federal government programs, implemented a new methodology by which rebates owed by manufacturers under the Medicaid Drug Rebate Program are calculated for drugs that are inhaled, infused, instilled, implanted, or injected expanded the types of entities eligible for the 340B drug discount program; expanded eligibility criteria for Medicaid programs; created a new Patient-Centered Outcomes Research Institute to oversee, identify priorities in, and conduct comparative clinical effectiveness research, along with funding for such research; and established a Center for Medicare Innovation at CMS to test innovative payment and service delivery models to lower Medicare and Medicaid spending, potentially including prescription drug spending.
There have been executive, judicial and Congressional challenges to certain aspects of the ACA. While Congress has not passed comprehensive repeal legislation, several bills affecting the implementation of certain taxes under the ACA have been signed into law.
The Tax Cuts and Jobs Act of 2017, or the Tax Act, includes a provision that repealed, effective January 1, 2019, the tax-based shared responsibility payment imposed by the ACA on certain individuals who fail to maintain qualifying health coverage for all or part of a year that is commonly referred to as the “individual mandate.” In addition, the 2020 federal spending package permanently eliminated, effective January 1, 2020, the ACA-mandated “Cadillac” tax on high-cost employer-sponsored health coverage and medical device tax and, effective January 1, 2021, also eliminated the health insurer tax. On June 17, 2021 the U.S. Supreme Court dismissed a challenge on procedural grounds that argued the ACA is unconstitutional in its entirety because the “individual mandate” was repealed by Congress. Further, on August 16, 2022, President Biden signed the Inflation Reduction Act of 2022, or IRA, into law, which among other things, extends enhanced subsidies for individuals purchasing health insurance coverage in ACA marketplaces through plan year 2025. The IRA also eliminates the “donut hole” under the Medicare Part D program beginning in 2025 by significantly lowering the beneficiary maximum out-of-pocket cost and creating a new manufacturer discount program. It is possible that the ACA will be subject to judicial or Congressional challenges in the future. It is unclear how such challenges, and the healthcare reform measures of the Biden administration will impact the ACA and our business.
In addition, other legislative changes have been proposed and adopted since the ACA was enacted. For example, on August 2, 2011, the Budget Control Act of 2011, among other things, includes aggregate reductions of Medicare payments to providers of 2% per fiscal year. These reductions went into effect on April 1, 2013 and, due to subsequent legislative amendments to the statute will remain in effect until 2032 unless additional U.S. Congressional action is taken. Additionally, on March 11, 2021, President Biden signed the American Rescue Plan Act of 2021 into law, which eliminates the statutory Medicaid drug rebate cap, currently set at 100% of a drug’s average manufacturer price, for single source and innovator multiple source drugs, beginning January 1, 2024. On January 2, 2013, the American Taxpayer Relief Act of 2012 was signed into law, which, among other things, further reduced Medicare payments to several types of providers, including hospitals, imaging centers and cancer treatment centers, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years.
Recently there has also been heightened governmental scrutiny over the manner in which manufacturers set prices for their marketed products. Specifically, there have been several recent U.S. Congressional inquiries and proposed and enacted federal and state legislation designed to, among other things, bring more transparency to drug pricing, reduce the cost of prescription drugs under Medicare, review the relationship between pricing and manufacturer patient programs and reform government program reimbursement methodologies for drugs. At the federal level, in July 2021, the Biden administration released an executive order, “Promoting Competition in the American Economy,” with multiple provisions aimed at prescription drugs. In response to Biden’s executive order, on September 9, 2021, HHS released a Comprehensive Plan for Addressing High Drug Prices that outlines principles for drug pricing reform and sets out a variety of potential legislative policies that Congress could pursue as well as potential administrative actions HHS can take to advance these principles. In addition, the IRA, among other things (i) directs HHS to negotiate the price of certain high-expenditure, single-source drugs and biologics covered under Medicare and (ii) imposes rebates under Medicare Part B and Medicare Part D to penalize price increases that outpace inflation. These provisions will take effect progressively starting in fiscal year 2023. On August 29, 2023, HHS announced the list of the first ten drugs that will be subject to price negotiations, although the Medicare drug price negotiation program is currently subject to legal challenges. In response to the Biden administration’s October 2022 executive order, on February 14, 2023, HHS released a report outlining three new models for testing by the CMS Innovation Center which will be evaluated on their ability to lower the cost of drugs, promote accessibility, and improve quality of care. It is unclear whether the models will be utilized in any health reform measures in the future. Further, on December 7, 2023, the Biden administration announced an initiative to control the price of prescription drugs through the use of march-in rights under the Bayh-Dole Act. On December 8, 2023, the National Institute of Standards and Technology published for comment a Draft Interagency Guidance Framework for Considering the Exercise of March-In Rights which for the first time includes the price of a product as one factor an agency can use when deciding to exercise march-in rights.
While march-in rights have not previously been exercised, it is uncertain if that will continue under the new framework. It is unclear whether these or similar policy initiatives will be implemented in the future. In addition, Congress is considering additional health reform measures.
At the state level, legislatures are increasingly passing legislation and implementing regulations designed to control pharmaceutical and biological product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing. Legally mandated price controls on payment amounts by third-party payors or other restrictions could harm our business, results of operations, financial condition and prospects. In addition, regional healthcare authorities and individual hospitals are increasingly using bidding procedures to determine what pharmaceutical products and which suppliers will be included in their prescription drug and other healthcare programs. This could reduce the ultimate demand for our products or put pressure on our product pricing, which could negatively affect our business, results of operations, financial condition and prospects.
We expect that additional U.S. federal healthcare reform measures will be adopted in the future, any of which could limit the amounts that the U.S. federal government will pay for healthcare products and services, which could result in reduced demand for our product candidates or additional pricing pressures.
In the European Union, similar political, economic and regulatory developments may affect our ability to profitably commercialize our current or any future products. In addition to continuing pressure on prices and cost containment measures, legislative developments at the European Union or member state level may result in significant additional requirements or obstacles that may increase our operating costs. The delivery of healthcare in the European Union, including the establishment and operation of health services and the pricing and reimbursement of medicines, is almost exclusively a matter for national, rather than European Union, law and policy. National governments and health service providers have different priorities and approaches to the delivery of health care and the pricing and reimbursement of products in that context. In general, however, the healthcare budgetary constraints in most European Union member states have resulted in restrictions on the pricing and reimbursement of medicines by relevant health service providers. Coupled with ever-increasing European Union and national regulatory burdens on those wishing to develop and market products, this could prevent or delay marketing approval of our product candidates, restrict or regulate post-approval activities and affect our ability to commercialize any products for which we obtain marketing approval. In international markets, reimbursement and healthcare payment systems vary significantly by country, and many countries have instituted price ceilings on specific products and therapies.
We cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative action, either in the United States or in other countries. If we or our collaborators are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we or our collaborators are not able to maintain regulatory compliance, our product candidates may lose any regulatory approval that may have been obtained and we may not achieve or sustain profitability, which would adversely affect our business.
We may be subject to healthcare laws, regulation and enforcement. Our failure to comply with these laws could harm our results of operations and financial conditions.
Although we do not currently have any products on the market, our current and future operations may be directly, or indirectly through our relationships with healthcare providers, healthcare institutions, patients, customers and third-party payors, subject to various U.S. federal and state healthcare laws and regulations, including, without limitation, the U.S. federal Anti-Kickback Statute. Healthcare providers, physicians and others play a primary role in the recommendation and prescription of any products for which we obtain marketing approval.
These laws impact, among other things, our proposed sales, marketing and education programs and constrain our business and financial arrangements and relationships with third-party payors, healthcare professionals and healthcare institutions who participate in our clinical research programs, healthcare professionals and others who recommend, purchase, or provide our approved products, and other parties through which we market, sell and distribute our products for which we obtain marketing approval. In addition, we may be subject to patient data privacy and security regulation by both the U.S. federal government and the states in which we conduct our business. Finally, our current and future operations are subject to additional healthcare-related statutory and regulatory requirements and enforcement by regulatory authorities in jurisdictions in which we conduct our business. The laws that may affect our ability to operate include:
•the U.S. federal Anti-Kickback Statute, which prohibits, among other things, individuals or entities from knowingly and willfully soliciting, offering, receiving or paying any remuneration (including any kickback, bribe, or rebate), directly or indirectly, overtly or covertly, in cash or in kind, to induce or reward either the referral of an individual for, or the purchase, lease, order or recommendation of, any good, facility, item or service, for which payment may be made, in whole or in part, under U.S. federal and state healthcare programs such as Medicare and Medicaid;
•the U.S. federal civil and criminal false claims and civil monetary penalties laws, including, without limitation, the civil False Claims Act (which can be enforced through "qui tam," or whistleblower actions, by private citizens on behalf of the federal government), which prohibits, among other things, individuals or entities from knowingly presenting, or causing to be presented, to the U.S. federal government, claims for payment or approval that are false or fraudulent or for knowingly making a false statement to avoid, decrease or conceal an obligation to pay money to the U.S. federal government;
•the U.S. federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, which prohibits, among other things, knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program, or knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false statement, in connection with the delivery of, or payment for, healthcare benefits, items or services;
•HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act of 2009, or HITECH, and its implementing regulations, and as amended again by the Final HIPAA Omnibus Rule, published in January 2013, which imposes certain obligations, including mandatory contractual terms, with respect to safeguarding the privacy, security and transmission of individually identifiable health information without appropriate authorization by certain health plans, healthcare clearinghouses and healthcare providers, known as covered entities, as well as their business associates that perform certain services involving the use, disclosure or transmission of individually identifiable health information for or on behalf of a covered entity, and their covered subcontractors;
•the U.S. Federal Food, Drug, and Cosmetic Act, or FDCA, which prohibits, among other things, the adulteration or misbranding of drugs, biologics and medical devices;
•the U.S. federal physician payment transparency legislation, commonly referred to as Physician Payments Sunshine Act, and its implementing regulations, which requires certain manufacturers of drugs, devices, biologics and medical supplies that are reimbursable under Medicare, Medicaid, or the Children's Health Insurance Program to report annually to CMS information related to certain payments and other transfers of value to physicians (defined to include doctors, dentists, optometrists, podiatrists and chiropractors), other healthcare professionals (such as physician assistants and nurse practitioners), and teaching hospitals, as well as ownership and investment interests held by physicians and their immediate family members;
•analogous state laws and regulations, including: state anti-kickback and false claims laws, which may apply to our business practices, including but not limited to, research, distribution, sales and marketing arrangements and claims involving healthcare items or services reimbursed by any third-party payor, including private insurers; state laws that require pharmaceutical companies to comply with the pharmaceutical industry's voluntary compliance guidelines and the relevant compliance guidance promulgated by the U.S. federal government, or otherwise restrict payments that may be made to healthcare providers and other potential referral sources; and state laws and regulations that require drug manufacturers to file reports relating to pricing and marketing information, which requires tracking gifts and other remuneration and items of value provided to healthcare professionals and entities, state and local laws that require the registration of pharmaceutical sales representatives; and state laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and often are not preempted by HIPAA, thus complicating compliance efforts; and
•European and other foreign law equivalents of each of the above laws, including reporting requirements detailing interactions with and payments to healthcare providers.
It is possible that governmental authorities will conclude that our business practices may not comply with current or future statutes, regulations or case law involving applicable fraud and abuse or other healthcare laws and regulations. If our operations are found to be in violation of any of these laws or any other governmental regulations that may apply to us, we may be subject to significant civil, criminal and administrative penalties, damages, fines, disgorgement, imprisonment, exclusion from participation in government funded healthcare programs, such as Medicare and Medicaid or comparable foreign programs, additional reporting requirements and oversight if we become subject to a corporate integrity agreement or similar agreement to resolve allegations of non-compliance with these laws, reputational harm and the curtailment or restructuring of our operations.
The risk of us being found in violation of these laws is increased by the fact that many of them have not been fully interpreted by the regulatory authorities or the courts, and their provisions are open to a variety of interpretations. For example, the definition of the “remuneration” under the U.S. federal Anti-Kickback Statute has been interpreted to include anything of value. Further, courts have found that if “one purpose” of remuneration is to induce referrals, the U.S. federal Anti-Kickback Statute is violated.
Additionally, recent healthcare reform legislation has strengthened federal and state healthcare fraud and abuse laws. For example, the ACA amends the intent requirement of the U.S. federal Anti-Kickback Statute and criminal healthcare fraud statutes to clarify that liability under these statutes does not require a person or entity to have actual knowledge of the statutes or a specific intent to violate them in order to have committed a violation. Moreover, the ACA provides that the government may assert that a claim that includes items or services resulting from a violation of the U.S. federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the civil False Claims Act. Because of the breadth of these laws and the narrowness of the statutory exceptions and regulatory safe harbors available, it is possible that some of our business activities could be subject to challenge under one or more of such laws.
Efforts to ensure that our business arrangements with third parties will comply with applicable healthcare laws and regulations will involve substantial costs. Any action against us for violation of these laws, even if we successfully defend against it, could cause us to incur significant legal expenses and divert our management's attention from the operation of our business. The shifting compliance environment and the need to build and maintain robust and expandable systems to comply with multiple jurisdictions with different compliance or reporting requirements increases the possibility that a healthcare company may run afoul of one or more of the requirements.
The successful commercialization of our product candidates will depend in part on the extent to which governmental authorities and health insurers establish adequate reimbursement levels and pricing policies. Failure to obtain or maintain coverage and adequate reimbursement for our product candidates, if approved, could limit our ability to market those products and decrease our ability to generate revenue.
The availability of coverage and adequate reimbursement by third-party payors, including governmental healthcare programs such as Medicare and Medicaid, private health insurers and managed care organizations, is essential for most patients to be able to afford products such as our product candidates, assuming approval. Our ability to achieve acceptable levels of coverage and reimbursement for products by third-party payors will have an effect on our ability to successfully commercialize, and attract additional collaboration partners to invest in the development of our product candidates. Assuming we obtain coverage for a given product by a third-party payor, the resulting reimbursement payment rates may not be adequate or may require co-payments that patients find unacceptably high. We cannot be sure that coverage and reimbursement in the United States, the European Union or elsewhere will be available for any product that we may develop, and any reimbursement that may become available may be decreased or eliminated in the future.
Third-party payors increasingly are challenging prices charged for pharmaceutical products and services, and many third-party payors may refuse to provide coverage and reimbursement for particular drugs when an equivalent generic drug or a less expensive therapy is available. It is possible that a third-party payor may consider our product candidate and other therapies as substitutable and only offer to reimburse patients for the less expensive product. Even if we show improved efficacy or improved convenience of administration with our product candidate, pricing of existing drugs may limit the amount we will be able to charge for our product candidate. Third-party payors may deny or revoke the reimbursement status of a given drug product or establish prices for new or existing marketed products at levels that are too low to enable us to realize an appropriate return on our investment in product development. If coverage and reimbursement is not available or is available only at limited levels, we may not be able to successfully commercialize our product candidates, and may not be able to obtain a satisfactory financial return on products that we may develop.
There is significant uncertainty related to the third-party payor coverage and reimbursement of newly approved products. In the United States, third-party payors play an important role in determining the extent to which new drugs and biologics will be covered. The Medicare and Medicaid programs increasingly are used as models for how private payors and other governmental payors develop their coverage and reimbursement policies for drugs and biologics. Some third-party payors may require pre-approval of coverage for new or innovative devices or drug therapies before they will reimburse health care providers who use such therapies. It is difficult to predict at this time what third-party payors will decide with respect to the coverage and reimbursement for our product candidates.
Obtaining and maintaining reimbursement status is time-consuming and costly. No uniform policy for coverage and reimbursement for drug products exists among third-party payors in the United States. Therefore, coverage and reimbursement for drug products can differ significantly from payor to payor. As a result, the coverage determination process is often a time-consuming and costly process that will require us to provide scientific and clinical support for the use of our products to each payor separately, with no assurance that coverage and adequate reimbursement will be applied consistently or obtained in the first instance. Furthermore, rules and regulations regarding reimbursement change frequently, in some cases at short notice, and we believe that changes in these rules and regulations are likely.
In addition, companion diagnostic tests require coverage and reimbursement separate and apart from the coverage and reimbursement for their companion pharmaceutical or biological products. Similar challenges to obtaining coverage and reimbursement, applicable to pharmaceutical or biological products, will apply to companion diagnostics. Additionally, if any companion diagnostic provider is unable to obtain reimbursement or is inadequately reimbursed, that may limit the availability of such companion diagnostic, which would negatively impact prescriptions for our product candidates, if approved.
Outside the United States, international operations are generally subject to extensive governmental price controls and other market regulations, and we believe the increasing emphasis on cost-containment initiatives in Europe, Canada and other countries has and will continue to put pressure on the pricing and usage of our product candidates. In many countries, the prices of medical products are subject to varying price control mechanisms as part of national health systems. Other countries allow companies to fix their own prices for medical products, but monitor and control company profits. Additional foreign price controls or other changes in pricing regulation could restrict the amount that we are able to charge for our product candidates. Accordingly, in markets outside the United States, the reimbursement for our products may be reduced compared with the United States and may be insufficient to generate commercially reasonable revenue and profits.
The delivery of healthcare in the European Union, including the establishment and operation of health services and the pricing and reimbursement of medicines, is almost exclusively a matter for national, rather than European Union, law and policy. National governments and health service providers have different priorities and approaches to the delivery of healthcare and the pricing and reimbursement of products in that context. In general, however, the healthcare budgetary constraints in most European Union member states have resulted in restrictions on the pricing and reimbursement of medicines by relevant health service providers. Coupled with ever-increasing European Union and national regulatory burdens on those wishing to develop and market products, this could prevent or delay marketing approval of our product candidates, restrict or regulate post-approval activities and affect our ability to commercialize any products for which we obtain marketing approval.
Moreover, increasing efforts by governmental and third-party payors in the European Union, the United States and abroad to cap or reduce healthcare costs may cause such organizations to limit both coverage and the level of reimbursement for newly approved products and, as a result, they may not cover or provide adequate payment for our product candidates. We expect to experience pricing pressures in connection with the sale of any of our product candidates due to the trend toward managed healthcare, the increasing influence of health maintenance organizations and additional legislative changes. The downward pressure on healthcare costs in general, particularly prescription drugs and surgical procedures and other treatments, has become very intense. As a result, increasingly high barriers are being erected to the entry of new products.
The future commercial success of our product candidates will depend on the degree of market acceptance of our potential products among physicians, patients, healthcare payors and the medical community.
Our product candidates are at varying stages of development and we may never have a product that is commercially successful. To date, we have no product authorized for marketing. Our lead product candidates are in the relatively early stages of clinical development. Our lead product candidates will require further clinical investigation, regulatory review, significant marketing efforts and substantial investment before they can provide us with any revenues. Furthermore, if and when available on the market, our products may not achieve an adequate level of acceptance by physicians, patients and the medical community, and we may not become profitable. If our products are not accepted, we may need to increase our efforts to educate the medical community and third-party payors on the benefits of our products, which may require significant resources and may never be successful.
Market acceptance of our future products by physicians, patients and healthcare payors will depend on a number of factors, many of which are beyond our control, including:
•the wording of the product label;
•changes in the standard of care as well as recommendations from relevant national and/or international associations for the targeted indications for any product candidate;
•sales, marketing and distribution support;
•potential product liability claims;
•acceptance by physicians, patients and healthcare payors of each product as safe and effective;
•relative convenience, ease of use, ease of administration and other perceived advantages over alternative products;
•availability of coverage and adequate reimbursement from third-party payors and the willingness of patients to pay out-of-pocket in the absence of adequate reimbursement;
•prevalence and severity of adverse events or publicity;
•limitations, precautions or warnings listed in the summary of product characteristics, patient information leaflet, package labeling or instructions for use;
•the cost of treatment with our products in relation to alternative treatments;
•the extent to which products are approved for inclusion and reimbursed on formularies of hospitals and managed care organizations; and
•whether our products are designated in the label, under physician treatment guidelines or under reimbursement guidelines as a first-line, second-line, third-line or last-line therapy.
If our product candidates fail to gain market acceptance, this will have a material adverse impact on our ability to generate revenues to provide a satisfactory, or any, return on our investments. Even if a potential product displays a favorable efficacy and safety profile in preclinical studies and clinical trials, market acceptance of the product will not be fully known until after it is launched. Furthermore, even if some products achieve market acceptance, the market may prove not to be large enough to allow us to generate significant revenues.
We have never commercialized a product candidate before and may lack the necessary expertise, personnel and resources to successfully commercialize our products on our own or together with suitable collaboration partners.
We do not have a sales or marketing infrastructure and have no experience in the sale or marketing of pharmaceutical products. To achieve commercial success for any approved product, we must develop or acquire a sales and marketing organization, outsource these functions to third parties or enter into collaboration or license arrangements with third parties.
To the extent possible, we may establish our own sales and marketing capabilities and promote our product candidates if and when regulatory approval has been obtained in the major European Union countries and the United States for certain of our product candidates. There are risks involved should we decide to establish our own sales and marketing capabilities or enter into arrangements with third parties to perform these services. Even if we establish sales and marketing capabilities, we may fail to launch or market our products effectively since we have no experience in the sales and marketing of pharmaceutical products.
In addition, recruiting and training a sales force is expensive and time consuming and could delay any product launch. In the event that any such launch is delayed or does not occur for any reason, we would have prematurely or unnecessarily incurred these commercialization expenses, and our investment would be lost if we cannot retain or reposition our sales and marketing personnel. Factors that may inhibit our efforts to commercialize our products on our own include:
•our inability to build a supply chain with sufficient coverage and capacity to fully support the sales and marketing efforts of any future products;
•our inability to recruit, train and retain adequate numbers of effective sales and marketing personnel;
•the inability of sales personnel to obtain access to or our failure to educate adequate numbers of physicians on the benefits of any future products;
•the lack of complementary products to be offered by sales personnel, which may put us at a competitive disadvantage relative to companies with more extensive product lines;
•unforeseen costs and expenses associated with creating an independent sales and marketing organization; and
•costs of marketing and promotion above those anticipated by us.
If we enter into arrangements with third parties to perform sales and marketing services, our product revenues or profitability could be lower than if we were to market and sell any products that we develop ourselves. Such collaborative arrangements may place the commercialization of our products outside of our control and would make us subject to a number of risks including that we may not be able to control the amount or timing of resources that our collaborative partner devotes to our products or that our collaborator's willingness or ability to complete its obligations, and our obligations under our arrangements, may be adversely affected by business combinations or significant changes in our collaborator's business strategy. In addition, we may not be successful in entering into arrangements with third parties to sell and market our products or may be unable to do so on terms that are favorable to us. Acceptable third parties may fail to devote the necessary resources and attention to sell and market our products effectively.
If we do not establish sales and marketing capabilities successfully, either on our own or in collaboration with third parties, we may not be successful in commercializing our products, which in turn would have a material adverse effect on our business, prospects, financial condition and results of operations.
Our product candidates for which we intend to seek approval as biologic products may face competition sooner than anticipated.
The Biologics Price Competition and Innovation Act of 2009, or BPCIA, created an abbreviated approval pathway for biological products that are biosimilar to or interchangeable with an FDA-licensed reference biological product. Under the BPCIA, an application for a biosimilar product may not be submitted to the FDA until four years following the date that the reference product was first licensed by the FDA. In addition, the approval of a biosimilar product may not be made effective by the FDA until 12 years from the date on which the reference product was first licensed. During this 12-year period of exclusivity, another company may still market a competing version of the reference product if the FDA approves a full BLA for the competing product containing the sponsor's own preclinical data and data from adequate and well-controlled clinical trials to demonstrate the safety, purity and potency of their product. The law is complex and is still being interpreted and implemented by the FDA. As a result, its ultimate impact, implementation and meaning are subject to uncertainty.
We believe that any of our product candidates approved as a biological product under a BLA should qualify for the 12-year period of exclusivity. However, there is a risk that this exclusivity could be shortened due to U.S. Congressional action or otherwise, or that the FDA will not consider our product candidates to be reference products for competing products, potentially creating the opportunity for generic competition sooner than anticipated. Other aspects of the BPCIA, some of which may impact the BPCIA exclusivity provisions, have also been the subject of recent litigation. Moreover, the extent to which a biosimilar, once approved, will be substituted for any one of our reference products in a way that is similar to traditional generic substitution for non-biological products is not yet clear, and will depend on a number of marketplace and regulatory factors that are still developing.
The European Union provides opportunities for data and market exclusivity related to marketing authorizations. Upon receiving a marketing authorization, innovative medicinal products are generally entitled to receive eight years of data exclusivity and 10 years of market exclusivity. Data exclusivity, if granted, prevents regulatory authorities in the European Union from referencing the innovator’s data to assess a generic application or biosimilar application for eight years from the date of authorization of the innovative product, after which a generic or biosimilar marketing authorization application can be submitted, and the innovator’s data may be referenced. The market exclusivity period prevents a successful generic or biosimilar applicant from commercializing its product in the European Union until 10 years have elapsed from the initial marketing authorization of the reference product in the European Union. The overall ten-year period may, occasionally, be extended for a further year to a maximum of 11 years if, during the first eight years of those ten years, the marketing authorization holder obtains an authorization for one or more new therapeutic indications which, during the scientific evaluation prior to their authorization, are held to bring a significant clinical benefit in comparison with existing therapies. However, there is no guarantee that a product will be considered by the European Union’s regulatory authorities to be a new chemical/biological entity, and products may not qualify for data exclusivity.
In the EU, there is a special regime for biosimilars, or biological medicinal products that are similar to a reference medicinal product but that do not meet the definition of a generic medicinal product. For such products, the results of appropriate preclinical or clinical trials must be provided in support of an application for MA. Guidelines from the EMA detail the type of quantity of supplementary data to be provided for different types of biological product.
We also believe that our product candidates in the EEA should benefit from this data and market exclusivity. As with the U.S., however, if competitors obtain marketing authorization for their biosimilar products, our products may become subject to competition from these biosimilars, with the attendant competitive pressure and consequences.
If we do not achieve our projected development and commercialization goals in the timeframes we announce and expect, the commercialization of our product candidates or any future product candidates may be delayed, and our business will be harmed.
For planning purposes, we estimate the timing of achieving various scientific, clinical, regulatory, and other product development objectives. These milestones may include our expectations regarding the commencement or completion of scientific studies and clinical trials, regulatory submissions or commercialization objectives. From time to time, we may publicly announce the expected timing of some of these milestones, such as the completion of an ongoing clinical trial, the initiation of clinical trials, receipt of regulatory approval, or the commercial launch of a product. The achievement of many of these milestones may be outside of our control. All of these milestones are based on a variety of assumptions, which may cause the timing of achieving the milestones to vary considerably from our estimates, including:
•our available capital resources or capital constraints we experience;
•the rate of progress, costs, and results of our clinical trials and research and development activities, including the extent of scheduling conflicts with participating clinicians and collaborators;
•our ability to identify and enroll patients who meet clinical trial eligibility criteria;
•our receipt of approvals by the FDA, EMA and comparable foreign regulatory authorities, and the timing thereof;
•other actions, decisions, or rules issued by regulators;
•our ability to access sufficient, reliable, and affordable supplies of materials used in the manufacture of our product candidates;
•our ability to manufacture and supply clinical trial materials to our clinical sites on a timely basis;
•the efforts of our collaborators with respect to the commercialization of our approved products, if any; and
•the securing of, costs related to, and timing issues associated with, commercial product manufacturing, as well as sales and marketing activities.
If we fail to achieve announced milestones in the timeframes we expect, the commercialization of our current or any future product candidates may be delayed, and our business, results of operations, financial condition, and prospects may be adversely affected.
Risks Related to Our Business and Industry
Nearly all aspects of our activities are subject to substantial regulation. No assurance can be given that any of our product candidates will fulfill regulatory compliance. Failure to comply with such regulations could result in delays, suspension, refusals and withdrawal of approvals, as well as fines.
The international biopharmaceutical and medical technology industry is highly regulated by the FDA, the EMA and other comparable foreign authorities and by other national or supra-national regulatory authorities that impose substantial requirements covering nearly all aspects of our activities notably on research and development, manufacturing, preclinical tests, clinical trials, labeling, marketing, sales, storage, record keeping, promotion and pricing of our product candidates. Such regulation is further subject to regular review by the FDA, the EMA and other comparable foreign authorities which may result in changes in applicable regulation. If we do not comply with one or more of these requirements in a timely manner, or at all, our product development could experience significant delays as a result of the FDA, the EMA or other comparable regulatory authorities recommending non-approval or restrictions on approval of a product candidate, leading to an inability to successfully commercialize any of our product candidates, which would materially harm our business. Any failure of any of our product candidates in clinical studies or to receive regulatory approval could have a material adverse effect on our business, results of operations and financial condition. If any of our product candidates fails to obtain approval on the basis of any applicable condensed regulatory approval process, this will prevent such product candidate from obtaining approval in a shortened time frame, or at all, resulting in increased expenses which would materially harm our business.
Compliance with requirements laid down by local regulatory authorities is necessary in each country where we, or any of our partners or licensees, conduct said activities in whole or in part. Local regulatory authorities notably include the EMA and the FDA. In order to market our future products in regions such as the European Economic Area, United States of America, Asia Pacific and many other foreign jurisdictions, we must obtain separate regulatory approvals. The approval procedures vary among countries and can require additional clinical testing, and the time required to obtain approval may differ from that required to obtain for example FDA or EMA approval.
Moreover, clinical studies conducted in one country may not be accepted by regulatory authorities in other countries. Approval by the FDA or EMA does not ensure approval by the comparable foreign authorities in other countries, and approval by one or more foreign regulatory authorities does not ensure approval by regulatory authorities in other foreign countries or by the FDA or EMA.
There can be no assurance that our product candidates will fulfil the criteria required to obtain necessary regulatory approval to access the market. Also, at this time, we cannot guarantee or know the exact nature, precise timing and detailed costs of the efforts that will be necessary to complete the remainder of the development of our research programs and products candidates. Each of the FDA, the EMA and other comparable foreign authorities may impose its own requirements, may discontinue an approval or revoke a license, may refuse to grant approval, or may require additional data before granting approval, notwithstanding that approval may have been granted by the FDA, the EMA or one or more other comparable foreign authority. The FDA, the EMA or other comparable foreign authorities may also approve a product candidate for fewer or more limited indications or patient sub-segments than requested or may grant approval subject to the performance of post-marketing studies. The EMA's, the FDA's or other regulatory authority's approval may be delayed, limited or denied for a number of reasons, most of which are beyond our control. Such reasons could include, among others, the production process or site not meeting the applicable requirements for the manufacture of regulated products, or the products not meeting applicable requirements for safety, purity or potency, or efficacy, during the clinical development stage or after marketing. No assurance can be given that clinical trials will be approved the FDA, the EMA or other comparable foreign authorities or that products will be approved for marketing by such regulatory authorities in any pre-determined indication or intended use. Any of the FDA, the EMA and other comparable foreign authorities may disagree with our interpretation of data submitted for their review.
We and our collaborative partners are, or may become subject to, numerous ongoing other regulatory obligations, such as data protection, environmental, health and safety laws and restrictions on the experimental use of animals. The costs of compliance with such applicable regulations, requirements or guidelines could be substantial, and failure to comply could result in sanctions, including fines, injunctions, civil penalties, denial of applications for marketing authorization of our products, delays, suspension or withdrawal of approvals, license revocation, seizures or recalls of products, operating restrictions and criminal prosecutions, any of which could significantly increase our or our collaborative partners' costs or delay the development and commercialization of our product candidates.
Changes in funding for the FDA and other government agencies could hinder their ability to hire and retain key leadership and other personnel, prevent new or existing product candidates from being developed or commercialized in a timely manner, or otherwise prevent those agencies from performing normal functions on which the operation of our business may rely, which could negatively impact our business.
The ability of the FDA to review and approve new products can be affected by a variety of factors, including government budget and funding levels, ability to hire and retain key personnel, and accept payment of user fees, and statutory, regulatory, and policy changes. Average review times at the agency have fluctuated in recent years as a result. In addition, government funding of other government agencies on which our operations may rely, including those that fund research and development activities is subject to the political process, which is inherently fluid and unpredictable.
Disruptions at the FDA and other agencies may also slow the time necessary for new drugs to be reviewed and/or approved by necessary government agencies, which would adversely affect our business. For example, over the last several years, the U.S. government has shut down several times, and certain regulatory agencies, such as the FDA, have had to furlough critical FDA and other government employees and stop critical activities.
If a prolonged government shutdown occurs, it could significantly impact the ability of the FDA to timely review and process our regulatory submissions, which could have a material adverse effect on our business. Further, in our operations as a public company, future government shutdowns could impact our ability to access the public markets and obtain necessary capital in order to properly capitalize and continue our operations.
Our research and development activities could be affected or delayed as a result of possible restrictions on animal testing.
Certain laws and regulations require us to test our product candidates on animals before initiating clinical trials involving humans. Animal testing activities have been the subject of controversy and adverse publicity. Animal rights groups and other organizations and individuals have attempted to stop animal testing activities by pressing for legislation and regulation in these areas and by disrupting these activities through protests and other means. To the extent the activities of these groups are successful, our research and development activities may be interrupted, delayed or become more expensive.
Because we are subject to environmental, health and safety laws and regulations, we may become exposed to liability and substantial expenses in connection with environmental compliance or remediation activities which may adversely affect our business.
Our operations, including our research, development, testing and manufacturing activities, are subject to numerous environmental, health and safety laws and regulations. These laws and regulations govern, among other things, the controlled use, handling, release and disposal of, and the maintenance of a registry for, hazardous materials and biological materials, such as radioactive compounds and byproducts, chemical solvents, human cells, carcinogenic compounds, mutagenic compounds and compounds that have a toxic effect on reproduction, laboratory procedures and exposure to blood-borne pathogens. We cannot eliminate, and we cannot guarantee that our third-party partners would eliminate the risk of contamination or injury from these materials. In the event of contamination or injury resulting from our use of hazardous materials, we could be held liable for any resulting damages, and any liability could exceed our resources. We also incur significant costs associated with civil or criminal fines and penalties for failure to comply with such laws and regulations.
Our professional liability insurance and our accident insurance, which cover for costs and expenses we may incur due to environmental liability that may be asserted against us or due to injuries to our employees resulting from the use of hazardous materials, may not provide adequate coverage against potential liabilities.
As with other companies engaged in activities similar to ours, we face a risk of environmental liability inherent in our current and historical activities, including liability relating to releases of or exposure to hazardous or biological materials. Environmental, health and safety laws and regulations are becoming more stringent. We may be required to incur substantial expenses in connection with future environmental compliance or remediation activities, in which case, our production and development efforts may be interrupted or delayed and our financial condition and results of operations may be adversely affected.
Further with respect to the operations of our third-party contract manufacturers, it is possible that if they fail to operate in compliance with applicable environmental, health and safety laws and regulations or properly dispose of wastes associated with our product candidates or products, we could be held liable for any resulting damages, suffer reputational harm or experience a disruption in the manufacture and supply of our product candidates or products, if approved.
The use of hazardous materials, including radioactive and biological materials, in our research and development efforts imposes certain compliance costs on us and may subject us to liability for claims arising from the use or misuse of these materials.
Our research, development and manufacturing activities involve the controlled use of hazardous materials, including chemicals, radioactive and biological materials, such as radioisotopes. We are subject to federal, state, local and foreign environmental laws and regulations governing, among other matters, the handling, storage, use and disposal of these materials and some waste products. In addition, we are required to obtain and maintain a hazardous materials license, pursuant to which we are required to perform annual self-audits, and that may result in random inspections by regulators. If such audit or inspection were to result in adverse findings, it may impact our ability to maintain our license, which would in turn adversely affect our ability to conduct our business. Additionally, we cannot completely eliminate the risk of contamination or injury from these materials and we could be held liable for any damages that result, which could exceed our financial resources. We currently maintain insurance coverage for injuries resulting from the hazardous materials we use; however, future claims may exceed the amount of our coverage. Also, we do not have insurance coverage for pollution cleanup and removal. Currently the costs of complying with such federal, state, local and foreign environmental regulations are not significant, and consist primarily of waste disposal expenses. However, they could become expensive, and current or future environmental laws or regulations may impair our research, development, production and commercialization efforts.
Our employees, independent contractors, principal investigators, CROs, consultants, vendors and collaboration partners may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements, which could have a material adverse effect on our business.
We are exposed to the risk that our employees, independent contractors, principal investigators, CROs, consultants, vendors and collaboration partners may engage in fraudulent conduct or other illegal activities. Misconduct by these parties could include intentional, reckless and negligent conduct or unauthorized activities that violate: the regulations of the FDA, the EMA and other comparable foreign authorities, including those laws that require the reporting of true, complete and accurate information to such authorities; manufacturing standards; federal and state data privacy, security, fraud and abuse and other healthcare laws and regulations in the United States and abroad; or laws that require the reporting of true, complete and accurate financial information and data. Specifically, sales, marketing and business arrangements in the healthcare industry are subject to extensive laws and regulations intended to prevent fraud, misconduct, kickbacks, self-dealing and other abusive practices. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, customer incentive programs and other business arrangements. Activities subject to these laws could also involve the improper use or misrepresentation of information obtained in the course of clinical trials or creating fraudulent data in our preclinical studies or clinical trials, which could result in regulatory sanctions and cause serious harm to our reputation. It is not always possible to identify and deter misconduct by employees and other third parties, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to comply with such laws or regulations. Additionally, we are subject to the risk that a person could allege such fraud or other misconduct, even if none occurred. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business and results of operations, including the imposition of significant civil, criminal and administrative penalties, damages, monetary fines, disgorgements, possible exclusion from participation in Medicare, Medicaid and other U.S.
federal healthcare programs, individual imprisonment, additional reporting requirements and oversight if we become subject to a corporate integrity agreement or similar agreement to resolve allegations of non-compliance with these laws, other sanctions, contractual damages, reputational harm, diminished profits and future earnings and curtailment of our operations.
Our high dependency on public perception of our products may negatively influence the success of these products.
If any of our product candidates are approved for commercial sale, we will be highly dependent upon consumer perceptions of the safety and quality of our products. We could be adversely affected if we were subject to negative publicity or if any of our products or any similar products distributed by other companies prove to be, or are asserted to be, harmful to patients. Because of our dependence upon consumer perception, any adverse publicity associated with illness or other adverse effects resulting from patients' use or misuse of our products or any similar products distributed by other companies could have a material adverse impact on our business and results of operations.
Future adverse events in research into the oncology and virology fields that we focus our research efforts on, or the biopharmaceutical industry more generally, could also result in greater governmental regulation, stricter labeling requirements and potential regulatory delays in the testing or approvals of our products. Any increased scrutiny could delay or increase the costs of obtaining regulatory approval for our product candidates.
Failure to successfully identify, develop and commercialize additional products or product candidates could impair our ability to grow.
Although a substantial amount of our efforts will focus on the continued preclinical and clinical testing and potential approval of our product candidates in our current pipeline, a key element of our long-term growth strategy is to develop and market additional product candidates. Because we have limited managerial resources, research programs to identify product candidates will require substantial additional technical, financial and human resources, whether or not any product candidates are ultimately identified. The success of this strategy depends partly upon our ability to identify, select and develop promising product candidates and products. Our DARPin platform and future platforms may fail to discover and to generate additional product candidates that are suitable for further development. All product candidates are prone to risks of failure typical of pharmaceutical product development, including the possibility that a product candidate may not be suitable for clinical development as a result of its harmful side effects, limited efficacy or other characteristics that indicate that it is unlikely to be a product that will receive approval by the FDA, the EMA and other comparable foreign regulatory authorities and achieve market acceptance. If we do not successfully develop and commercialize product candidates based upon our DARPin technology approach, we may not be able to obtain product or collaboration revenues in future periods, which would adversely affect our business and results of operations.
We may expend our limited resources to pursue a particular DARPin product candidate or indication and fail to capitalize on DARPin product candidates or indications that may be more profitable or for which there is a greater likelihood of success.
Because we have limited financial and managerial resources, we focus on research programs and DARPin product candidates for specific indications, mode of actions or targets. As a result, we may forego or delay pursuit of opportunities with other DARPin product candidates or other indications that later prove to have greater commercial potential. Our resource allocation decisions may cause us to fail to capitalize on viable commercial products or profitable market opportunities. Our spending on current and future research and development programs and DARPin product candidates for specific indications may not yield any commercially viable products.
If we do not accurately evaluate the commercial potential or target market for a particular DARPin product candidate, we may relinquish valuable rights to that product candidate through collaboration, licensing or other royalty arrangements in cases in which it would have been advantageous for us to retain sole development and commercialization rights.
Service or supply failures, or other failures, business interruptions or other disasters affecting the manufacturing facilities of any party participating in the supply chain would adversely affect our ability to supply our products, if approved.
Our product candidates are biologics and require processing steps that are more difficult than those required for most chemical pharmaceuticals. Accordingly, multiple steps are needed to control the manufacturing processes. Problems with these manufacturing processes, even minor deviations from the normal process or from the materials used in the manufacturing process, which may not be detectable by us in a timely manner, could lead to product defects or manufacturing failures, resulting in lot failures, product recalls, product liability claims and insufficient inventory.
Also, certain raw materials or other products necessary for the manufacture and formulation of our product candidates, some of which are difficult to source, are provided by single-source unaffiliated third-party suppliers. In addition, we rely on certain third parties to perform filling, finishing, distribution, laboratory testing and other services related to the manufacture of our product candidates, and to supply various raw materials and other products, including for the RDT where we will rely on isotope providers and third-parties capabilities in radio-labelling and product supply. We would be unable to obtain these raw materials, other products, or services for an indeterminate period of time if any of these third parties were to cease or interrupt production or otherwise fail to supply these materials, products, or services to us for any reason, including due to regulatory requirements or actions (including recalls), adverse financial developments at or affecting the supplier, failure by the supplier to comply with cGMPs, contamination, business interruptions, or labor shortages or disputes. In any such circumstances, we may not be able to engage a backup or alternative supplier or service provider in a timely manner or at all. This, in turn, could materially and adversely affect our ability to supply product candidates, which could materially and adversely affect our business and future prospects.
We may develop our DARPin platform and other current or future product candidates, in combination with other therapies, which exposes us to additional risks.
We may develop our DARPin platform and other current or future product candidates in combination with one or more currently approved therapies. Even if any product candidates we develop were to receive marketing approval or be commercialized for use in combination with other existing therapies, we would continue to be subject to the risks that the FDA or comparable foreign regulatory authorities could revoke approval of the therapy used in combination with our DARPin platform or any other current or future product candidates or that safety, efficacy, manufacturing, or supply issues could arise with these existing therapies. This could result in our own product candidates being removed from the market or being less successful commercially.
We may also evaluate our DARPin platform or any other current or future product candidates in combination with one or more other therapies that have not yet been approved for marketing by the FDA or comparable foreign regulatory authorities. We will not be able to market and sell our DARPin product candidates or any product candidate we develop in combination with any such unapproved therapies that do not ultimately obtain marketing approval. These unapproved therapies may face the same risks described with respect to our product candidates, including the emergence of adverse events and delays in their clinical trials. If the FDA or comparable foreign regulatory authorities do not approve these other therapies or revoke their approval of, or if safety, efficacy, manufacturing, or supply issues arise with, the therapies we choose to evaluate in combination with our DARPin product candidates or any other product candidate we develop, we may be unable to obtain approval of or market our DARPin product candidates or any other product candidate we develop.
We are subject to stringent and changing obligations related to data privacy and security. Our actual or perceived failure to comply with such obligations could lead to government enforcement actions (which could include civil or criminal penalties), private litigation, and/or adverse publicity and could negatively affect our operating results and business.
In the ordinary course of business, we collect, receive, store, process, generate, use, transfer, disclose, make accessible, protect, secure, dispose of, transmit, and share (commonly known as processing) personal data and other sensitive information, including proprietary and confidential business data, trade secrets, intellectual property, data we collect about trial participants in connection with clinical trials, and sensitive third-party data. Our data processing activities subject us to numerous data privacy and security obligations, such as various laws, regulations, guidance, industry standards, external and internal privacy and security policies, contracts, and other obligations that govern the processing of personal data by us and on our behalf. In the United States, numerous federal and state laws, rules and regulations, including federal health information privacy laws, state data breach notification laws, state health information privacy laws, and federal and state consumer protection laws that govern the collection, processing of personal information, including health-related personal information, could apply to our operations or the operations of our collaborators. For example, HIPAA, as amended by HITECH, imposes specific requirements relating to the privacy, security, and transmission of individually identifiable health information. We may obtain health information from third parties (including research institutions from which we obtain clinical trial data) that are subject to privacy and security requirements under HIPAA. Depending on the facts and circumstances, we may be subject to civil, criminal, and administrative penalties if we knowingly obtain, use, or disclose individually identifiable health information in a manner that is not authorized or permitted by HIPAA.
Additionally, the California Consumer Privacy Act, or CCPA, imposes obligations on covered businesses. These obligations include, but are not limited to, providing specific disclosures in privacy notices and affording California residents certain rights related to their personal data. The CCPA allows for statutory fines for noncompliance (up to $7,500 per violation) and includes a private right of action for certain data breaches. Although the CCPA exempts some data processed in the context of clinical trials, the CCPA may increase compliance costs and potential liability with respect to other personal data we maintain about California residents. In addition the California Privacy Rights Act of 2020, or CPRA, effective January 1, 2023, expands the CCPA, including by adding a new right for individuals to correct their personal information and establishing a new regulatory agency to implement and enforce the law. Other states have enacted data privacy laws as well. For example, Virginia passed the Consumer Data Protection Act, and Colorado passed the Colorado Privacy Act, both of which become effective in 2023. While these states, like the CCPA, also exempt some data processed in the context of clinical trials, these developments further complicate compliance efforts, and increase legal risk and compliance costs for us and the third parties upon whom we rely. Data privacy and security laws have been proposed at the federal, state, and local levels in recent years, which could further complicate compliance efforts.
Outside the United States, an increasing number of laws, regulations, and industry standards apply to data privacy and security. For example, the European Union’s General Data Protection Regulation, or EU GDPR, and the United Kingdom’s GDPR, or UK GDPR, impose strict requirements for processing personal data. For example, under the EU GDPR, government regulators may impose temporary or definitive bans on data processing, as well as fines of up to 20 million euros or 4% of annual global revenue, whichever is greater. Further, companies may face private litigation related to processing of personal data brought not only by individuals but also by classes of data subjects or consumer protection organizations authorized at law to represent their interests. Additionally, EU member states are also able to legislate separately on health and genetic data, and we must comply with these local laws where we operate.
The revised Swiss Federal Act on Data Protection, or DPA, entered into force on September 1, 2023 and also applies to the collection and processing of personal data, including health-related information, by companies located in Switzerland, or in certain circumstances, by companies located outside of Switzerland.
Certain jurisdictions have enacted data localization laws and cross-border personal data transfer laws, which could make it more difficult to transfer information across jurisdictions (such as transferring or receiving personal data that originates in the EU or in other foreign jurisdictions). Existing mechanisms that facilitate cross-border personal data transfers may change or be invalidated. For example, absent appropriate safeguards or other circumstances, the EU GDPR generally restricts the transfer of personal data to countries outside of the European Economic Area, or EEA, such as the United States, which are not considered by the European Commission to provide an adequate level of data protection. The European Commission released a set of “Standard Contractual Clauses,” or SCCs, that are designed to be a valid mechanism to facilitate personal data transfers out of the EEA to these jurisdictions. Currently, these SCCs are a valid mechanism to transfer personal data outside of the EEA, but this mechanism is subject to legal challenge. Additionally, the SCCs impose additional compliance burdens, such as conducting transfer impact assessments to determine whether additional security measures are necessary to protect the at-issue personal data. In addition, Switzerland and the UK similarly restrict personal data transfers outside of those jurisdictions to countries such as the United States that do not provide an adequate level of personal data protection. If we cannot implement a valid compliance mechanism for cross-border data transfers, we may face increased exposure to regulatory actions, substantial fines, and injunctions against processing or transferring personal data from Europe or other foreign jurisdictions. The inability to import personal data to the United States could significantly and negatively impact our business operations, including by limiting our ability to conduct clinical trial activities in Europe and elsewhere; limiting our ability to collaborate with parties that are subject to such cross-border data transfer or localization laws; or requiring us to increase our personal data processing capabilities and infrastructure in foreign jurisdictions at significant expense.
We are also bound by contractual obligations related to data privacy and security, and our efforts to comply with such obligations may not be successful. For example, certain privacy laws, such as the GDPR and the CCPA, require our collaborators to impose specific contractual restrictions on their service providers. We publish privacy policies, and other statements, such as compliance with certain certifications or self-regulatory principles, regarding data privacy and security. If these policies, materials or statements are found to be deficient, lacking in transparency, deceptive, unfair, or misrepresentative of our practices, we may be subject to investigation, enforcement actions by regulators or other adverse consequences.
Our obligations related to data privacy and security are quickly changing in an increasingly stringent fashion, creating some uncertainty as to the effective future legal framework. Additionally, these obligations may be subject to differing applications and interpretations, which may be inconsistent or conflict among jurisdictions. Preparing for and complying with these obligations requires significant resources and may necessitate changes to our information technologies, systems, and practices and to those of any third parties that process personal data on our behalf. Although we endeavor to comply with all applicable data privacy and security obligations, we may at times fail (or be perceived to have failed) to do so. Moreover, despite our efforts, our personnel or third parties upon whom we rely may fail to comply with such obligations, which could negatively impact our business operations and compliance posture. For example, any failure by a third-party processor to comply with applicable law, regulations, or contractual obligations could result in adverse effects, including inability to or interruption in our ability to operate our business and proceedings against us by governmental entities or others.
If we fail, or are perceived to have failed, to address or comply with data privacy and security obligations, we could face significant consequences. These consequences may include, but are not limited to government enforcement actions (which could include civil, criminal and administrative penalties), private litigation, and/or adverse publicity, additional reporting requirements and/or oversight, bans on processing personal data, orders to destroy or not use personal data, imprisonment of company officials.
Moreover, clinical trial subjects, employees and other individuals about whom we or our potential collaborators obtain personal information, as well as the providers who share this information with us, may limit our ability to collect, use and disclose the information. Any of these events could have a material adverse effect on our reputation, business, or financial condition, including but not limited to interruptions or stoppages in our business operations (including, as relevant, clinical trials), inability to process personal data or to operate in certain jurisdictions, limited ability to develop or commercialize our products, expenditure of time and resources to defend any claim or inquiry, adverse publicity, or revision or restructuring of our operations.
We are subject to anti-corruption laws, as well as export control laws, customs laws, sanctions laws and other laws governing our operations. If we fail to comply with these laws, we could be subject to civil or criminal penalties, other remedial measures and legal expenses, which could adversely affect our business, results of operations and financial condition.
We are subject to other laws and regulations governing our international operations, including regulations administered by the governments of the United States, and authorities in the European Union, including applicable export control regulations, economic sanctions on countries and persons, customs requirements and currency exchange regulations, collectively referred to as the trade control laws.
The Foreign Corrupt Practices Act, or FCPA, prohibits any U.S. individual or business from paying, offering, authorizing payment or offering of anything of value, directly or indirectly, to any foreign official, political party or candidate for the purpose of influencing any act or decision of the foreign entity in order to assist the individual or business in obtaining or retaining business. The FCPA also obligates companies whose securities are listed in the United States to comply with certain accounting provisions requiring the company to maintain books and records that accurately and fairly reflect all transactions of the corporation, including international subsidiaries, and to devise and maintain an adequate system of internal accounting controls for international operations.
Compliance with the FCPA is expensive and difficult, particularly in countries in which corruption is a recognized problem. In addition, the FCPA presents particular challenges in the biopharmaceutical industry, because, in many countries, hospitals are operated by the government, and doctors and other hospital employees are considered foreign officials. Certain payments to hospitals in connection with clinical trials and other work have been deemed to be improper payments to government officials and have led to FCPA enforcement actions.
There is no assurance that we will be effective in ensuring our compliance with all applicable anti-corruption laws, including the FCPA, the Swiss anti-corruption laws or other legal requirements, including trade control laws. If we are not in compliance with the FCPA, the Swiss anti-corruption laws and other anti-corruption laws or trade control laws, we may be subject to criminal and civil penalties, disgorgement and other sanctions and remedial measures, and legal expenses, which could have an adverse impact on our business, financial condition, results of operations and liquidity. Likewise, any investigation of any potential violations of the FCPA, the Swiss anti-corruption laws, other anti-corruption laws or trade control laws by U.S. or other authorities could also have an adverse impact on our reputation, our business, results of operations and financial condition.
In addition, changes in our products and product candidates or changes in applicable export or import laws and regulations may create delays in the introduction or provision of our products and product candidates in other jurisdictions, prevent others from using our products and product candidates or, in some cases, prevent the export or import of our products and product candidates to certain countries, governments or persons altogether. Any limitation on our ability to export or provide our products and product candidates could adversely affect our business, financial condition and results of operations.
Moreover, a growing number of investors, regulators, self-regulatory organizations and other stakeholders have expressed an interest in Environmental, Social and Corporate Governance, or ESG, matters, and are requiring more robust ESG disclosures. The related legislative landscape in Switzerland and in the EU has been evolving accordingly. According to new provisions in the Swiss Code of Obligations that became effective on 1 January 2022, publicly listed companies exceeding, together with the Swiss and foreign undertakings they control, certain thresholds must each year, as of the financial year beginning on 1 January 2023, prepare a report on non-financial matters. The report must cover environmental matters, in particular the CO2 goals, social issues, employee-related issues, respect for human rights and combating corruption. The report must contain the information required to understand the state of the company, the performance and results of its business, and the effects of its activity on these non-financial matters. The relevant thresholds are: At least 500 full-time equivalent positions on annual average in two successive financial years and exceeding one of the following thresholds in two successive financial years (ii) balance sheet total of CHF 20 million or sales revenue of CHF 40 million. In addition, under Switzerland’s legislation regarding the due diligence and transparency in relation to conflict minerals and metals and child labor in the supply chain, we may in the future be required to establish (1) a supply chain policy on conflict minerals and metals and child labor, (2) a supply chain traceability system, (3) as an early warning mechanism for risk identification, a reporting procedure that allows interested parties to raise reasonable concerns about the existence of a potential or actual adverse impact related to conflict minerals and metals and child labor, and (4) a risk management plan which identifies and assesses the risks in the supply chain according to the likelihood of occurrence and severity of adverse impacts and eliminates, prevents or minimizes such risks on that basis. If the new due diligence obligations will apply to us in the future, we will be required to publish an annual report regarding compliance with our conflict minerals and metals and child labor due diligence obligations.
In the EU, for example, EU Directive No 2464/2022 on Corporate Sustainability Reporting, or CSRD, was adopted and entered into force on 5 January 2023, amending the current EU Accounting Directive No 2013/34. The CSRD introduces new mandatory reporting obligations that will require in-scope companies to publish audited sustainability information. The CSRD is supplemented by EU Delegated Regulation No 2023/2772 which establishes the first set of European Sustainability Reporting Standards, or ESRS, which are applicable to in-scope EU entities. Further reporting standards are due to be adopted by June 2026, including for in-scope non-EU entities.
The CSRD and ESRS require certain mandatory disclosures, as well as disclosures of certain "material" sustainability matters in the company's own operations, those of their subsidiaries and those of their value chain. Like the Swiss legislation the identification of material sustainability matters in the CSRD also requires a "double materiality" assessment. This means that in-scope entities will have to assess both financial materiality (i.e., sustainability matters which generate risks or opportunities that affect, or could reasonably be expected to affect, the company's financial position, financial performance, cash flows, access to finance or cost of capital over the short-, medium- or long-term) and impact materiality (i.e., the company's material actual or potential, positive or negative impacts on people or the environment over the short-medium and long-term). Sustainability matters are material if they satisfy one or both of these materiality tests.
The CSRD applies to entities with securities admitted to trading on an EU-regulated market, as well as large EU companies, EU parents of a "large group", and to listed EU small or medium-sized enterprises, amongst others. It will also apply to non-EU companies that have a certain threshold of EU-generated turnover and an in-scope EU subsidiary or EU branch meeting the turnover thresholds. Companies subject to the CSRD are required to fulfil their reporting obligations in accordance with a staggered timeline depending on the category of company. The first reports are expected in 2025 for the 2024 financial year, predominantly for entities with securities admitted to trading on an EU-regulated market, and in 2026 for the 2025 financial year for many other EU companies (including EU subsidiaries of non-EU parents) that are not listed on an EU-regulated market but meet the relevant threshold tests.
In response to new ESG initiatives and regulations we may voluntarily elect, or be required, to adopt strategies, policies, or procedures related to ESG matters and report on these. Reporting on ESG goals and objectives may cause us to expend significant capital and human resources, and could divert management’s attention from central operational matters. Reports could also lead to the disclosure of information that which may have a negative impact on our operations and reputation which may lead to additional exposure. Failure to accurately comply with any ESG reporting obligations may result in enforcement actions, sanctions, reputational harm or private litigation.
Risks Related to Our Dependence on Third Parties
We rely, and expect to continue to rely, on third parties, including independent clinical investigators and CROs, to conduct our preclinical studies and clinical trials. If these third parties do not successfully carry out their contractual duties or meet expected deadlines, we may not be able to obtain regulatory approval for or commercialize our product candidates and our business could be harmed.
We have relied upon and plan to continue to rely upon third parties, including independent clinical investigators and third-party CROs mandated by us or by our partners, to conduct our preclinical studies and clinical trials and to monitor and manage data for our ongoing preclinical and clinical programs. We rely on these parties for execution of our preclinical studies and clinical trials, and control only certain aspects of their activities. Nevertheless, we are responsible for ensuring that each of our studies and trials is conducted in accordance with the applicable protocol, legal and regulatory requirements and scientific standards, and our reliance on these third parties does not relieve us of our regulatory responsibilities. We and our third party contractors and CROs are required to comply with GCP requirements, which are regulations and guidelines enforced by the FDA, the EMA and comparable foreign regulatory authorities for all of our products in clinical development. Regulatory authorities enforce these GCPs through periodic inspections of trial sponsors, principal investigators and trial sites. If we, our investigators or any of our CROs fail to comply with applicable GCPs, the clinical data generated in our clinical trials may be deemed unreliable and the FDA, the EMA or comparable foreign regulatory authorities may require us to perform additional clinical trials before approving our marketing applications. We cannot assure you that upon inspection by a given regulatory authority, such regulatory authority will determine that any of our clinical trials comply with GCP regulations. In addition, our clinical trials must be conducted with product produced under cGMP regulations. Our failure to comply with these regulations may require us to repeat clinical trials, which would delay the regulatory approval process.
Further, these investigators and CROs are not our employees and we will not be able to control, other than by contract, the amount of resources, including time, which they devote to our product candidates and clinical trials. If independent investigators or CROs fail to devote sufficient resources to the development of our product candidates, or if their performance is substandard, it may delay or compromise the prospects for approval and commercialization of any product candidates that we develop. In addition, the use of third-party service providers requires us to disclose our proprietary information to these parties, which could increase the risk that this information will be misappropriated.
Our CROs have the right to terminate their agreements with us in the event of an uncured material breach. In addition, some of our CROs have an ability to terminate their respective agreements with us if it can be reasonably demonstrated that the safety of the subjects participating in our clinical trials warrants such termination, if we make a general assignment for the benefit of our creditors or if we are liquidated.
We also face the risk of potential infringement, unauthorized disclosure, misappropriation or other violation of our intellectual property by our third party contractors or CROs, which may reduce our trade secret protection and allow our potential competitors or other third parties to access and exploit our proprietary technology. Our third party contractors or CROs also may use our proprietary information and intellectual property in such a way as to invite litigation or other intellectual property-related proceedings that could jeopardize or invalidate our proprietary information and intellectual property.
For more information regarding our intellectual property, see “Risk Factors—Risks Related to Intellectual Property.”
There are a limited number of third-party service providers that specialize or have the expertise required to achieve our business objectives. If any of our relationships with these third-party CROs or clinical investigators terminate, we may not be able to enter into arrangements with alternative CROs or investigators or to do so on commercially reasonable terms. Switching or adding additional CROs (or investigators) involves additional cost and requires management time and focus. In addition, there is a natural transition period when a new CRO commences work. As a result, delays occur, which can materially impact our ability to meet our desired clinical development timelines. Though we carefully manage our relationships with our CROs, there can be no assurance that we will not encounter similar challenges or delays in the future or that these delays or challenges will not have a material adverse impact on our business, financial condition and prospects.
We rely and will continue to rely on collaborative partners regarding the development of our research programs and product candidates. If we are not able to maintain our current relationships or enter into new strategic relationships our business, financial condition, commercialization prospects and results of operations may be adversely affected.
We are, and expect to continue to be, dependent on partnerships with partners relating to the development and commercialization of our existing and future research programs and product candidates.
We currently have collaborative research relationships with the University of Bern for MP0533, Orano Med for Radio-DARPin therapies, University of Freiburg for Radio-DARPin therapies and with Novartis, primarily for Radio-DARPin therapies. We have had and will continue to have discussions on potential partnering opportunities with various pharmaceutical companies. If we fail to enter into or maintain collaborations on reasonable terms or at all, our ability to develop our existing or future research programs and product candidates could be delayed, the commercial potential of our products could change and our costs of development and commercialization could increase. Furthermore, we may find that our programs require the use of intellectual property rights and other proprietary rights held by third parties, and the growth of our business may depend in part on our ability to acquire, in-license or use these intellectual property and other proprietary rights.
Our dependence on collaborative partners subjects us to a number of risks, including, but not limited to, the following:
•we may not be able to control the amount and timing of resources that the collaboration partner devotes to our research programs and product candidates;
•for collaboration agreements where we are solely or partially responsible for funding development expenses through a defined milestone event, the payments we receive from the collaboration partner may not be sufficient to cover the expenses we have or would need to incur in order to achieve that milestone event;
•we may be required to relinquish significant rights, including intellectual property or other proprietary rights, marketing and distribution rights;
•our anticipated payments under any partnership agreement (e.g., royalty payments for licensed products) may not materialize;
•we rely on the information and data received from third parties regarding their research programs and product candidates and will not have control of the process conducted by the third party in gathering and composing such data and information.
•if rights to develop and commercialize our product candidates subject to collaborations revert to us for any reason (for example MP0310 and abicipar), we may not have sufficient financial resources to develop such product candidates, which may result in us failing to recognize any value from our investments in developing such product candidates and/or requiring us to divert our resources elsewhere;
•a collaborative partner may decide not to pursue, or discontinue the collaborative development of, our product candidates;
•a collaborative partner may develop a competing product either by itself or in collaboration with others, including one or more of our competitors;
•our collaborative partners' willingness or ability to complete their obligations under our partnership arrangements may be adversely affected by business combinations or significant changes in a collaborative partner's business strategy;
•we may experience delays in, or increases in the costs of, the development of our research programs and product candidates due to the termination or expiration of collaborative research and development arrangements;
•we may have disagreements with collaborative partners, including disagreements over proprietary rights, contract interpretation or the preferred course of development, that might cause delays or termination of the research, development or commercialization of product candidates, might lead to additional responsibilities for us with respect to product candidates, or might result in litigation or arbitration, any of which would be time-consuming and expensive;
•collaborative partners may not properly maintain, enforce or defend our intellectual property rights or other proprietary information or may such use proprietary information in such a way as to invite litigation or other intellectual property-related proceedings that could jeopardize or invalidate our intellectual property or proprietary information or expose us to potential litigation; or
•collaborative partners may infringe, misappropriate or otherwise violate the intellectual property or other proprietary rights of third parties, which may expose us to litigation and potential liability, and collaborators may also allege that we are liable for potential infringement, misappropriation or other violations of third-party intellectual property or proprietary rights during the research and development work for the collaboration.
We face significant competition in seeking appropriate collaborative partners. Our ability to reach a definitive agreement for a partnership will depend, among other things, upon an assessment of the collaborator's resources and expertise, the terms and conditions of the proposed partnership and the proposed collaborator's evaluation of a number of factors. These factors may include the design or results of clinical trials, the likelihood of regulatory approval, the potential market for the subject product candidate, the costs and complexities of manufacturing and delivering such product candidate to patients, the potential of competing products, the existence of uncertainty with respect to our ownership of technology, which can exist if there is a challenge to such ownership regardless of the merits of the challenge and industry and market conditions generally. The collaborator may also consider alternative product candidates or technologies for similar indications that may be available to collaborate on and whether such a partnership could be more attractive than the one with us.
We may not be able to negotiate collaborations on a timely basis, on acceptable terms, or at all. If we are unable to do so, we may have to curtail the development of the product candidate for which we are seeking to collaborate, reduce or delay its development program or one or more of our other development programs, delay its potential commercialization or reduce the scope of any sales or marketing activities or increase our expenditures and undertake development or commercialization activities at our own expense. If we elect to increase our expenditures to fund development or commercialization activities on our own, we may need to obtain additional capital, which may not be available to us on acceptable terms or at all. If we do not have sufficient funds, we may not be able to further develop product candidates or bring them to market and generate product revenue.
We rely completely on third parties to manufacture our preclinical and clinical drug supplies and we intend to rely on third parties to provide us with required target material for developing and selecting product candidates as well as to produce commercial supplies of any approved product candidate.
We do not currently have the infrastructure or capability internally to manufacture our product candidates for use in the conduct of our clinical studies or for commercial supply, if our products are approved. Instead, we rely on, and expect to continue to rely on CMOs. We currently rely mainly on a few CMOs for the manufacturing of our product candidate materials. Any replacement of our CMOs could require significant effort and expertise because there may be a limited number of qualified CMOs. Reliance on third-party providers may expose us to more risk than if we were to manufacture our product candidates ourselves. We are dependent on our CMOs for the production of our product candidates in accordance with relevant regulations (such as cGMP), which includes, among other things, quality control, quality assurance and the maintenance of records and documentation. Moreover, many of the third parties with whom we contract may also have relationships with other commercial entities, including our competitors, for whom they may also be conducting product development activities that could harm our competitive position.
If we were to experience an unexpected delay in receiving required target material or loss of supply of or if any supplier were unable to meet our demand for any of our product candidates, we could experience delays in our research or planned clinical studies or commercialization. We could be unable to find alternative suppliers of acceptable quality, in the appropriate volumes and at an acceptable cost. Moreover, our suppliers are subject to strict manufacturing requirements and rigorous testing requirements, which could limit or delay production. The long transition periods necessary to switch manufacturers and suppliers, if necessary, would significantly delay our clinical studies and the commercialization of our products, if approved, which would adversely affect our business and results of operation.
In complying with the manufacturing regulations of the FDA, the EMA and other comparable foreign authorities, we and our third-party suppliers must spend significant time, money and effort in the areas of design and development, testing, production, record-keeping and quality control to assure that the products meet applicable specifications and other regulatory requirements. The failure to comply with these requirements could result in an enforcement action against our CMOs and subsequently against us, including the seizure of products and shutting down of production. We and any of these third-party suppliers may also be subject to audits by the FDA, the EMA or other comparable foreign authorities. If any of our third-party suppliers fails to comply with cGMP or other applicable manufacturing regulations, our ability to develop and commercialize the products could suffer significant interruptions. For example, we have faced and may face in the future bioburden during drug substance production campaigns or particles in drug product preparations at our CMOs which led or may lead to regulatory actions, including from the FDA. While we and our partners endeavor to maintain appropriate backup supply with respect to our product candidates, and not all such bioburden or particles result in regulatory action or delays, we cannot assure that any such issues would not result in delays in our clinical trials or product development or other adverse impacts on our business.
We face risks inherent in relying on our CMOs, as any disruption, such as a fire, natural hazards or vandalism at any such CMO could significantly interrupt our manufacturing capability. Our CMOs currently do not have alternative production plans in place or disaster-recovery facilities available. In case of a disruption, we will have to establish alternative manufacturing sources. This would require substantial capital on our part, which we may not be able to obtain on commercially acceptable terms or at all. Additionally, we would likely experience months of manufacturing delays as the CMO builds or locates replacement facilities and seeks and obtains necessary regulatory approvals. If this occurs, we will be unable to satisfy manufacturing needs on a timely basis, if at all.
The manufacturing of all of our product candidates requires using cells which are stored in a cell bank. We have one master cell bank for each product manufactured in accordance with cGMP. Working cell banks have not yet been manufactured. Half of each master cell bank is stored at a separate site so that in case of a catastrophic event at one site we believe sufficient vials of the master cell banks are left at the alternative storage site to continue manufacturing. We believe sufficient working cell banks could be produced from the vials of the master cell bank stored at a given site to assure product supply for the future. However, it is possible that we could lose multiple cell banks and have our manufacturing significantly impacted by the need to replace these cell banks, which could materially adversely affect our business, prospects, financial condition and results of operations.
We do not and will not have access to all information regarding the product candidates we license to our collaboration partners. Consequently, our ability to inform our shareholders about the status of such product candidates, and to make informed operational and investment decisions about the product candidates to which we have retained development and commercialization rights, may be limited.
We do not and will not have access to all information regarding the product candidates being developed and potentially commercialized by Novartis, including potentially material information about clinical trial design and execution, safety reports from clinical trials, spontaneous safety reports if the product is later approved and marketed, regulatory affairs, process development, manufacturing, marketing and other areas known by Novartis. In addition, we have confidentiality obligations under our agreement with Novartis. Thus, our ability to keep our shareholders informed about the status of product candidates under our collaboration will be limited by the degree to which Novartis keeps us informed and allows us to disclose such information to the public.
Our financial prospects are dependent upon the manufacture, development and marketing efforts of our licensees. Our licensees may act in their best interest rather than in our best interest, which could materially adversely affect our business, financial condition and results of operations.
We rely on our licensees to manufacture, fund and conduct the clinical development and commercialization of product candidates, and our licensees have complete control over such activities. Our ability to generate revenue in the near term will depend primarily on the successful development, regulatory approval, marketing and commercialization of product candidates by our licensees. Such success is subject to significant uncertainty, and we have limited control over the manufacturing processes of such product candidates as well as the resources, time and effort that licensees may devote to such product candidates. Any of several events or factors could have a material adverse effect on our ability to generate revenue from our licensee’s potential commercialization of product candidates.
In addition, our licensees have the right to make decisions regarding the development and commercialization of product candidates under the collaborations without consulting us and may make decisions with which we do not agree.
For example, in April 2022, Amgen, our collaboration partner for MP0310 (AMG 506), informed us of its decision to return global rights of MP0310 to us following a strategic pipeline review and in January 2024, Novartis, our collaboration partner for ensovibep, informed us of its decision to return the global rights for ensovibep to us following the end of COVID-19 pandemic. Conflicts between our licensees and us may arise if there is a dispute about the progress of the clinical development of a product candidate, the achievement and payment of a milestone amount or the ownership of intellectual property developed during the course of our collaboration agreements. If any of our licenses terminate with our licensees, it may be necessary for us to assume responsibility at our own expense for the development of the applicable product candidates. In that event, we would likely be required to limit the size and scope of one or more of our programs or increase our expenditures and seek additional funding, which may not be available on acceptable terms or at all, which would materially adversely affect our business, financial condition and results of operations.
Risks Related to Intellectual Property
We rely on patents and other intellectual property rights to protect our product candidates and the DARPin technology, the prosecution, grant, enforcement, defense and maintenance of which may be challenging and costly. Failure to obtain, maintain, enforce or protect these rights adequately could harm our ability to compete and impair our business.
Our commercial success depends in part on obtaining and maintaining patents and other forms of intellectual property rights for our product candidates, methods used to manufacture those products and methods for treating patients using those products, or on licensing in such rights. Failure to obtain, maintain, enforce, protect or extend adequate patent and other intellectual property rights could adversely affect our ability to develop and market our products and product candidates or pursue collaborations with partners for our product candidates.
We cannot be certain that patents will be issued or granted with respect to applications that are currently pending, or that issued or granted patents will not later be found to be invalid or unenforceable. The patent position of biopharmaceutical companies is generally uncertain because it involves complex legal and factual considerations, and has been the subject of much litigation in recent years. The standards applied by the United States Patent and Trademark Office, or USPTO, the European Patent Office, or EPO, and other foreign patent offices in granting patents are not always identical or applied uniformly or predictably. For example, there is no uniform worldwide policy regarding patentable subject matter or the scope of claims allowable in biopharmaceutical patents. Consequently, patents may not issue from our pending patent applications or, if issued, patents may vary in scope depending on the jurisdiction. As such, we do not know the degree of future protection that we will have on our proprietary products and technology in the various jurisdictions. The scope of patent protection that the USPTO, the EPO and other foreign patent offices will grant with respect to the DARPin product candidates in our product pipeline is uncertain. It is possible that the USPTO, the EPO and other foreign patent offices will not allow broad claims that cover DARPin product candidates closely related to our product candidates or to the specific protein building blocks. As a result, upon receipt of EMA or FDA approval, competitors may be free to market other products almost identical to ours, thereby decreasing our market share.
The patent prosecution process is expensive, time-consuming and complex, and we and our current or future licensors, licensees or collaboration partners may not be able to prepare, file and prosecute all necessary or desirable patent applications at a reasonable cost or in a timely manner. It is also possible that we or our licensors, licensees or collaboration partners will fail to identify patentable aspects of inventions made in the course of development and commercialization activities before it is too late to obtain patent protection for them. Although we enter into non-disclosure and confidentiality agreements with parties who have access to confidential or patentable aspects of our research and development output, such as our employees, collaborators, CROs, contract manufacturers, consultants, advisors and other third parties, any of these parties may breach the agreements and disclose such output before a patent application is filed, thereby jeopardizing our ability to seek patent protection.
Further, the issuance, scope, validity, enforceability and commercial value of our and our current or future licensors’, licensees' or collaboration partners’ patent rights are highly uncertain. Our and our licensors’ pending and future patent applications may not result in patents being issued that protect our technology or products, in whole or in part, or that effectively prevent others from commercializing competitive technologies and products. Moreover, in some circumstances, we may not have the right to control the preparation, filing, prosecution and maintenance of the licensed patent applications or other intellectual property, or to maintain the patents, or may not have the first right to enforce the intellectual property. We may need to enter into new license or royalty agreements, covering technology that we license from or license to third parties or have developed in collaboration with our collaboration partners and are reliant on patent procurement activities of our licensors, licensees or collaboration partners. Therefore, we may not be able to adequately influence the patent prosecution or enforcement of these patents and patent applications, or prevent inadvertent lapses of coverage due to failure to pay maintenance fees and we cannot be certain that these patents and patent applications will be prepared, filed, prosecuted, maintained, enforced and defended in a manner consistent with the best interests of our business and that does not compromise the patent rights. If our current or future licensors, licensees or collaboration partners fail to obtain, maintain, protect or enforce such patents and other intellectual property rights, such rights may be reduced or lost. If our licensors, licensees or collaboration partners are not fully cooperative or disagree with us as to the preparation, filing, prosecution, maintenance, defense or enforcement of any licensed patent rights, such patent rights could be compromised. The patent examination process may require us or our licensors, licensees or collaboration partners to narrow the scope of the claims of our or our licensors’, licensees’ or collaboration partners’ pending and future patent applications, which may limit the scope of patent protection that may be obtained. We cannot assure you that all of the potentially relevant prior art relating to our patents and patent applications has been found. If such prior art exists, it may invalidate patents in whole or in part or prevent patents from issuing from pending patent applications. Even if patents do successfully issue and even if such patents cover our product candidates, third parties may initiate an opposition, interference, re-examination, post-grant review, inter partes review, nullification, revocation, derivation, or other actions in court or before patent offices challenging the validity, enforceability or scope of such patents, which may result in the patent claims being narrowed, invalidated, or held unenforceable. Such proceedings have a higher impact in the biopharmaceutical industry than in other industries, given that biopharmaceutical products are often protected by only one or few patents. Our and our licensors’, licensees’ or collaboration partners’ patent applications cannot be enforced against third parties practicing the technology claimed in such applications unless and until a patent issues from such applications, and then only to the extent the issued claims cover the practiced technology. An adverse determination in any such proceeding could reduce the scope of, invalidate, or render unenforceable our patent rights, and allow third parties to commercialize our technology or products and compete directly with us, without payment to us. Such proceedings also may result in substantial cost and require significant time from our scientists and management, even if the eventual outcome is favorable to us.
Moreover, the coverage claimed in a patent application can be significantly reduced before the patent is issued, and the issuance of a patent is not conclusive as to its inventorship, scope, validity or enforceability. Even if our patent applications or those of our licensors, licensees or collaboration partners issue as patents, they may not issue in a form that will provide us with any meaningful protection, prevent competitors or other third parties from competing with us, or otherwise provide us with any competitive advantage. Any patents may be challenged, narrowed, circumvented or invalidated by third parties. Consequently, we do not know whether any of our DARPin platform advances or product candidates will be protectable or remain protected by valid and enforceable patents. In addition, our competitors or other third parties may be able to circumvent our patents by developing similar or alternative technologies or products in a non-infringing manner.
In addition, publications of discoveries in the scientific literature often lag behind the actual discoveries, and patent applications are confidential for a period of time after filing, and some remain so until issued. Therefore, we cannot be certain that we or our licensors, licensees or collaborators were the first to make the inventions claimed in any patent application, or were the first to file any patent application related to a product candidate. Furthermore, as to the United States, if third parties have filed such patent applications on or before March 15, 2013, an interference proceeding can be initiated by such third parties to determine who was the first to invent any of the subject matter covered by the patent claims of our applications. If third parties have filed such applications after March 15, 2013, a derivation proceeding can be initiated by such third parties to determine whether our invention was derived from theirs. Even where we have a valid and enforceable patent, we may not be able to exclude others from practicing our invention where the other party can show that they used the invention in commerce before our filing date, or if the other party is able to obtain a compulsory license.
Given the amount of time required for the development, testing and regulatory review of new product candidates, patents protecting such candidates might expire before or shortly after such product candidates are commercialized. As a result, our intellectual property may not provide us with sufficient rights to exclude others from commercializing products similar or identical to ours, including generic versions of such products. Moreover, it is possible that some future patents and patent applications owned or in-licensed by us may be co-owned with third parties, including our collaboration partners and other third parties with whom we conduct research and development. If we are unable to obtain an exclusive license to any such third party co-owners’ interest in such patents or patent applications, such co-owners may be able to license their rights to other third parties, including our competitors, and our competitors could market competing products and technology. In addition, we may need the cooperation of any such co-owners of our patents in order to enforce such patents against third parties, and such cooperation may not be provided to us.
Furthermore, it is possible that some future patents and patent applications owned or in-licensed by us may be subject to a reservation of rights by one or more third parties. For example, this may happen if the research resulting in certain of our owned or in-licensed patent rights and technology was funded in part by the U.S. government. As a result, the government may have certain rights, or march-in rights, to such patent rights and technology. When new technologies are developed with government funding, the government generally obtains certain rights in any resulting patents, including a non-exclusive license authorizing the government to use the invention for noncommercial purposes. These rights may permit the government to disclose our confidential information to third parties and to exercise march-in rights to use or allow third parties to use our licensed technology. The government can exercise its march-in rights if it determines that action is necessary because we fail to achieve practical application of the government-funded technology, because action is necessary to alleviate health or safety needs, to meet requirements of federal regulations, or to give preference to U.S. industry. In addition, our rights in such inventions may be subject to certain requirements to manufacture products embodying such inventions in the United States. Any of the foregoing could have a material adverse effect on our competitive position, business, financial conditions, results of operations, and prospects.
We may fail in enforcing our intellectual property rights and issued patents covering one or more of our product candidates or DARPin technology or our intellectual property rights and issued patents could be found invalid or unenforceable if challenged in court.
To protect our competitive position, we or our licensors or collaboration partners may from time to time need to resort to litigation in order to enforce or defend any patents or other intellectual property rights owned by or licensed to us, or to determine or challenge the scope or validity of patents or other intellectual property rights of third parties.
Enforcement of intellectual property rights is difficult, unpredictable and expensive, and many of our or our licensors’ or collaboration partners’ adversaries in these proceedings may have the ability to dedicate substantially greater resources to prosecuting these legal actions than we or our licensors or collaboration partners can. Accordingly, despite our or our licensors' or collaboration partners’ efforts, we or our licensors or collaboration partners may not be able to prevent third parties from infringing upon, misappropriating or otherwise violating intellectual property rights we own or control, particularly in countries where the laws may not protect those rights as fully as in the European Union and the United States. We may fail in enforcing our rights, in which case our competitors may be permitted to use our technology without being required to pay us any license fees. In addition, litigation involving our patents carries the risk that one or more of our patents will be held invalid (in whole or in part, on a claim-by-claim basis) or unenforceable. Such an adverse court ruling could allow third parties to commercialize our products or use our DARPin technology, and then compete directly with us, without payment to us.
If we or one of our licensors or collaboration partners were to initiate legal proceedings against a third party to enforce a patent covering one of our product candidates or our technology, including our DARPin technology, the defendant could counterclaim that our patent is invalid or unenforceable. In patent litigation in the United States and Europe, defendant counterclaims alleging invalidity or unenforceability are commonplace. Third parties could also raise challenges to the validity of patent claims before administrative bodies in the United States, Europe or other foreign jurisdictions, even outside the context of litigation. Such mechanisms include re-examination, post-grant review, inter partes review, interference proceedings, derivation proceedings and equivalent proceedings in foreign jurisdictions (e.g., opposition proceedings). Such proceedings could result in the revocation of, cancellation of or amendment to our patent claims in such a way that they no longer cover our technology or DARPin platform, or any product candidates that we may develop. A claim for a validity challenge may be based on failure to meet any of several statutory requirements, including lack of novelty, obviousness or non-enablement. A claim for unenforceability could involve an allegation that someone connected with prosecution of the patent withheld relevant information from or made a misleading statement to the USPTO, the EPO or other patent offices during prosecution. The outcome following legal assertions of invalidity and unenforceability is unpredictable. With respect to the validity of our issued patents, for example, we cannot be certain that there is no invalidating prior art, of which we, our licensors or collaboration partners and the patent examiner were unaware during prosecution. If a third party were to prevail on a legal assertion of invalidity or unenforceability, we would lose part or all of the patent protection afforded by the affected patent. Such a loss of patent protection could have a material adverse impact on our business. Further, litigation could result in substantial costs and diversion of management resources, and reputational harm, regardless of the outcome, which could harm our business and financial results.
Third parties may initiate legal proceedings alleging that we are infringing, misappropriating, or otherwise violating their intellectual property rights, the outcome of which would be uncertain and could have a material adverse effect on the success of our business. Intellectual property litigation could cause us to spend substantial resources and distract our personnel from their normal responsibilities and negative outcomes could result in adverse effects on our business.
Our success depends, in part, on our ability to operate without infringing the patents and other proprietary intellectual property rights of third parties. This is generally referred to as having the “freedom to operate.” The biotechnology and pharmaceutical industries in which we plan to operate are subject to frequent and extensive litigation regarding patents and other intellectual property rights. In addition, companies producing therapeutics in the oncology and virology fields have employed intellectual property litigation as a means to gain an advantage over their competitors.
As a result, we may be required to defend against claims of intellectual property infringement, misappropriation or other violation that may be asserted by third parties against us and, if the outcome of any such litigation is adverse to us, it may affect our ability to compete effectively.
Our competitive position may suffer if patents issued to third parties or other third-party intellectual property rights cover our products or elements thereof, our manufacture or uses relevant to our products or development plans, the targets of our product candidates, or other attributes of our product candidates or our technology. In such cases, we may not be in a position to develop or commercialize the applicable products or product candidates unless we successfully pursue litigation to nullify or invalidate the third-party intellectual property right concerned, or enter into a license agreement with the intellectual property right holder, which may not be available on commercially reasonable terms, or at all. In the event that a relevant patent has not expired at the time of approval of such product candidate and the patent owner were to bring an infringement action against us, we may have to argue that our product, its manufacture, importation or use does not infringe, misappropriate or otherwise violate a valid claim of the patent in question. Alternatively, if we were to challenge the validity of any issued U.S. patent in court, we would need to overcome a statutory presumption of validity that attaches to every U.S. patent. This means that in order to prevail, we would need to present clear and convincing evidence as to the invalidity of the patent's claims. There is no assurance that a court would find in our favor on questions of infringement or validity. In the event that a patent is successfully asserted against us such that the patent is found to be valid and enforceable and infringed by our product, unless we obtain a license to such a patent, which may not be available on commercially reasonable terms or at all, we could be prevented from continuing to develop or commercialize our product. Similarly, the targets for certain of our product candidates have also been the subject of research by other companies, which have filed patent applications or own issued patents on aspects related to the targets or their uses. There can be no assurance that any such patents will not be asserted against us or that we will not need to seek licenses from such third parties. We may not be able to secure such licenses on acceptable terms, if at all, and any such litigation would be costly and time-consuming.
It is also possible that we failed to identify relevant patents or applications. For example, certain U.S. applications filed after November 29, 2000 that will not be filed outside the United States may remain confidential until patents issue. In general, patent applications in the United States and elsewhere are published approximately 18 months after the earliest filing from which priority is claimed, with such earliest filing date being commonly referred to as the priority date. Therefore, patent applications covering our products or platform technology could have been filed by others without our knowledge. Furthermore, we operate in a highly competitive field, and given our limited resources, it is unreasonable to monitor all patent applications purporting to gain broad coverage in the areas in which we are active. Additionally, claims in pending patent applications which have been published can, subject to certain limitations, be later amended in a manner that could cover our platform technologies, our products or the use of our products.
Third-party intellectual property right holders, including our competitors, may actively bring infringement, misappropriation or other claims against us. We may not be able to successfully settle or otherwise resolve such claims. If we are unable to successfully settle future claims on terms acceptable to us, we may be required to engage in or continue costly, unpredictable and time-consuming litigation and may be prevented from or experience substantial delays in marketing our products.
If we fail in any such dispute, in addition to being forced to pay damages, we or our licensees may be temporarily or permanently prohibited from commercializing any of our product candidates that are held to be infringing, misappropriating or violating any third-party intellectual property rights.
We might also be forced to redesign product candidates so that we no longer infringe, misappropriate or otherwise violate third-party intellectual property rights, which may result in significant cost or delay to us or be technically infeasible, or to seek a license to any such third-party intellectual property rights that we are found to infringe, misappropriate or otherwise violate, which license may not be available on commercially reasonable terms, or at all. Even if we or our licensors or collaboration partners obtain a license, it may be non-exclusive, thereby giving our competitors access to the same technologies licensed to us or our licensors or collaboration partners, and it could require us or our licensors or collaboration partners to make substantial royalty and other payments. In addition, we could be found liable for monetary damages, including treble damages and attorneys' fees, if we are found to have willfully infringed a patent. Any of these events, even if we were to ultimately prevail, could require us to divert substantial financial and management resources that we would otherwise be able to devote to our business.
Our involvement in litigation, and in any interference, derivation, reexamination, inter partes review, post grant review, opposition or other post-grant proceedings or other intellectual property proceedings inside and outside of the European Union or the United States, even if resolved in our favor, may cause us to incur significant expenses, distract our technical and management personnel from their normal responsibilities and cause substantial delays in marketing our products. In addition, there could be public announcements of the results of hearings, motions, other interim proceedings or developments, or of final verdicts and if securities analysts or investors perceive these results to be negative, this could have a substantial adverse effect on our share price. Such litigation or proceedings could substantially increase our operating losses and reduce our resources available for development activities. We may not have sufficient financial or other resources to adequately conduct such litigation or proceedings. Some of our competitors may be able to sustain the costs of such litigation or proceedings more effectively than we can because of their substantially greater financial resources. Uncertainties resulting from the initiation and continuation of patent litigation or other proceedings could have an adverse effect on our ability to compete in the marketplace.
In addition, if the breadth or strength of protection provided by our or our licensors’ or collaboration partners’ patents and patent applications is challenged or threatened, it could dissuade companies from collaborating with us to license, develop or commercialize current or future product candidates. Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation. Any of the foregoing could have a material adverse effect on our competitive position, business, financial conditions, results of operations and prospects.
Our rights to develop and commercialize our technology and product candidates are subject, in part, to the terms and conditions of licenses granted to us by others, and we may not be successful in obtaining or maintaining additional necessary rights related to our product candidates through acquisitions and in-licenses.
We rely upon licenses to certain patent rights and other intellectual property from third parties that are important or necessary to the development of our product candidates. We may also need to obtain additional licenses to advance the development and commercialization of any product candidates we may develop. Additionally, we have in the past collaborated and may in the future collaborate with U.S. and/or European academic institutions to accelerate our preclinical research or development under written agreements with these institutions. In some instances, these institutions provide us with an option to negotiate a license to any of the institution’s rights in technology resulting from the collaboration. Regardless of such option, we may be unable to negotiate a license within the specified timeframe or under terms that are acceptable to us. If we are unable to do so, the institution may offer the intellectual property rights to other parties, potentially blocking our ability to pursue our applicable product candidate or program.
Our current license and collaborations agreements also impose, and we expect that future agreements will likely impose various reporting, prosecution, diligence, fee payment, royalty and other obligations on us. If there is any conflict, dispute, disagreement or issue of non-performance between us and our licensing or collaboration partners regarding our rights or obligations under the agreements, including any such conflict, dispute or disagreement arising from our alleged failure to satisfy payment obligations under any such agreement, we may owe damages, the counterparty may have a right to terminate the affected agreement, and our and our licensees' ability to utilize the affected intellectual property in drug discovery and development efforts, and our ability to enter into collaboration or marketing agreements for an affected product candidate, may be adversely affected. Our business could also suffer if a licensor or collaborator fails to abide by the terms of the agreement, if any licensed patents or other rights are found to be invalid or unenforceable, or if we are unable to enter into necessary licenses on acceptable terms or at all. Any of these events could have a material adverse effect on our competitive position, business, financial conditions, results of operations, and prospects. For more information regarding our license and collaboration agreements, see “Item 4.B — Business—License and Collaboration Agreements.”
In addition, we may not have the right to control the preparation, filing, prosecution, maintenance, enforcement and defense of patents and patent applications covering the technology that we license from third parties. Therefore, we cannot be certain that these patents and patent applications will be prepared, filed, prosecuted, maintained, enforced and defended in a manner consistent with the best interests of our business. If our current or future licensors fail to prosecute, maintain, enforce and defend such patents, or lose rights to those patents or patent applications, the rights we have licensed may be reduced or eliminated, and our right to develop and commercialize any of our products that are subject of such licensed rights could be adversely affected.
Our current or future licensors may have relied on third party consultants or collaborators or on funds from third parties such that our licensors are not the sole and exclusive owners of the patents we in-licensed. This could have a material adverse effect on our competitive position, business, financial conditions, results of operations and prospects.
Because our programs may require the use of proprietary rights held by third parties, the growth of our business may depend in part on our ability to acquire, in-license, maintain or use these proprietary rights. We may be unable to acquire or in-license, on reasonable terms or at all, any compositions, methods of use, processes, or other third-party intellectual property rights from third parties that we identify as necessary for our product candidates. The licensing and acquisition of third-party intellectual property rights is a competitive area, and a number of more established companies may pursue strategies to license or acquire third-party intellectual property rights that we may consider attractive. These established companies may have a competitive advantage over us due to their size, cash resources and greater clinical development and commercialization capabilities. In addition, companies that perceive us to be a competitor may be unwilling to assign or license rights to us. We also may be unable to license or acquire third-party intellectual property rights on terms that would allow us to make an appropriate return on our investment. If we are unable to successfully obtain a license to third-party intellectual property rights necessary for the development of a product candidate or program, we may have to abandon development of that product candidate or program and our business and financial condition could suffer.
If in the future we do undertake any acquisitions, the process of integrating an acquired business, technology, service, products or product candidates into our business may result in unforeseen operating difficulties and expenditures, including diversions of resources and management’s attention from our core business, or any acquired intellectual property may be subject to claims of invalidity or unenforceability or held to be invalid.
In addition, we may fail to retain key executives and employees of the companies we acquire, which may reduce the value of the acquisition or give rise to additional integration costs. Future acquisitions could result in additional issuances of equity securities that would dilute the ownership of existing shareholders. Future acquisitions could also result in the incurrence of debt, actual or contingent liabilities or the amortization of expenses related to other intangible assets, any of which could adversely affect our operating results. In addition, we may fail to realize the anticipated benefits of any acquisition. Any of the foregoing could have a material adverse effect on our competitive position, business, financial conditions, results of operations and prospects.
If our trademarks and trade names are not adequately protected, then we may not be able to build name recognition in our markets of interest and our business may be adversely affected.
Our registered or unregistered trademarks or trade names may be challenged, infringed, circumvented, declared generic or determined to be infringing on other marks or names. We may not be able to protect or enforce our rights to these trademarks and trade names, which we need to build name recognition among potential partners or customers in our markets of interest. If we are unable to establish name recognition based on our trademarks and trade names, then we may not be able to compete effectively and our business may be adversely affected. If other entities use trademarks similar to ours in different jurisdictions, or have rights senior to ours, it could interfere with our use of our current trademarks throughout the world.
If we do not obtain protection under the Hatch-Waxman Act Amendments and similar non-U.S. legislation for extending the term of patents covering each of our product candidates, our business may be materially harmed.
Patents have a limited duration. In the United States, if all maintenance fees are timely paid, the natural expiration of a patent is generally 20 years from its earliest effective U.S. non-provisional filing date. Various extensions may be available, but the life of a patent, and the protection it affords, is limited. Even if patents covering our product candidates, their manufacture, or use are obtained, once the patent life has expired, we may be open to competition from competitive medications, including biosimilar medications or generic versions of such products. Given the amount of time required for the development, testing and regulatory review of new product candidates, patents protecting such candidates might expire before or shortly after such candidates are commercialized. As a result, our owned and licensed patent portfolio may not provide us with sufficient rights to exclude others from commercializing products similar or identical to ours, at least not long enough to recoup the costs incurred in developing our products.
Depending upon the timing, duration and conditions of FDA marketing approval of our product candidates, one or more of our U.S. patents may be eligible for limited patent term extension under the Drug Price Competition and Patent Term Restoration Act of 1984, referred to as the Hatch-Waxman Act and similar legislation in the European Union and several other relevant countries around the world. The Hatch-Waxman Act permits a patent term extension of up to five years for a patent covering an approved product as compensation for effective patent term lost during product development and the FDA regulatory review process. The patent term extension cannot extend the remaining term of a patent beyond a total of 14 years from the date of product approval, only one patent applicable to an approved drug may be extended, and only those claims covering the approved drug, a method for using it, or a method for manufacturing it may be extended. However, we may not receive an extension if we fail to exercise due diligence during the testing phase or regulatory review process, fail to apply within applicable deadlines, fail to apply prior to expiration of relevant patents or otherwise fail to satisfy applicable requirements. Moreover, the length of the extension could be less than we request. If we are unable to obtain patent term extension or the term of any such extension is less than we request, the period during which we can enforce our patent rights for the applicable product will be shortened and our competitors may be able to enter the market with competing products sooner than we expect, and our business, financial condition, results of operations, and prospects could be materially harmed.
The base patents relating to the DARPin base technology we use to generate our DARPin product candidates has expired, and our competitors may use the technology claimed in such patents, which may materially adversely affect our business and competitive position.
The base patents that we had licensed from the University of Zurich in 2004 expired in September 2021 (except for one patent in the United States) and we terminated the license agreement effective October 2021 and the remaining U.S. patent expired in August 2023. Our competitors may be able to utilize the technology claimed in such patents to develop product candidates that compete with ours. This could harm our reputation as being the leader in the DARPin technology, and could have an adverse effect on our competitive position, business, financial conditions, results of operations and prospects.
We enjoy only limited geographical protection with respect to certain patents and may face difficulties in certain jurisdictions, which may diminish the value of our intellectual property rights.
Filing, prosecuting and defending patents on product candidates in all countries throughout the world would be prohibitively expensive, and the laws of foreign countries may not protect our rights to the same extent as the laws of the United States and the European Union. Consequently, we may not be able to prevent third parties from practicing our inventions in all countries outside the United States and the European Union, or from selling or importing products made using our inventions in and into all countries outside the United States and the European Union.
We often file our first patent application, or our priority filing, at the EPO or the USPTO. International applications under the Patent Cooperation Treaty, or PCT, are usually filed within twelve months after the priority filing. Based on the PCT filing, national and regional patent applications may be filed in additional jurisdictions where we believe our product candidates may be marketed. We have so far not filed for patent protection in all national and regional jurisdictions where such protection may be available. In addition, we may decide to abandon national and regional patent applications before grant. Finally, the grant proceeding of each national/regional patent is an independent proceeding which may lead to situations in which applications might in some jurisdictions be refused by the relevant patent offices, while granted by others. It is also quite common that depending on the country, the scope of patent protection may vary for the same product candidate or technology.
Competitors may use our or our licensors’ or collaboration partners’ technologies in jurisdictions where we have not obtained patent protection to develop their own products and, further, may export otherwise infringing products to territories where we and our licensors or collaboration partners have patent protection, but enforcement is not as strong as that in the United States and the European Union. These products may compete with our product candidates, and our and our licensors' or collaboration partners' patents or other intellectual property rights may not be effective or sufficient to prevent them from competing.
The laws of some jurisdictions do not protect intellectual property rights to the same extent as the laws in the United States and the European Union, and companies have encountered significant difficulties in protecting and defending such rights in such jurisdictions. If we or our licensors encounter difficulties in protecting, or are otherwise precluded from effectively protecting, the intellectual property rights important for our business in such jurisdictions, the value of these rights may be diminished and we may face additional competition from others in those jurisdictions.
Some countries have compulsory licensing laws under which a patent owner may be compelled to grant licenses to third parties. In addition, some countries limit the enforceability of patents against government agencies or government contractors.
In these countries, the patent owner may have limited remedies, which could materially diminish the value of such patent. If we or any of our licensors is forced to grant a license to third parties with respect to any patents relevant to our business, our competitive position may be impaired and our business and results of operations may be adversely affected.
Proceedings to defend or enforce our and our licensors' or collaboration partners' patent rights in foreign jurisdictions could result in substantial costs and divert our and our licensors' or collaboration partners' efforts and attention from other aspects of our business, could put our and our licensors' or collaboration partners' patents at risk of being invalidated or interpreted narrowly, could put our and our licensors' or collaboration partners' patent applications at risk of not issuing and could provoke third parties to assert claims against us or our licensors or collaboration partners. We or our licensors or collaboration partners may not prevail in any lawsuits that we or our licensors or collaboration partners initiate and the damages or other remedies awarded, if any, may not be commercially meaningful. Accordingly, our efforts to enforce our intellectual property and proprietary rights around the world may be inadequate to obtain a significant commercial advantage from the intellectual property that we develop or license. If we or our licensors or collaborators encounter difficulties in protecting, or are otherwise precluded from effectively protecting, the intellectual property rights important for our business in such jurisdictions, the value of these rights may be diminished and we may face additional competition from others in those jurisdictions.
For example, the complexity and uncertainty of European patent laws have also increased in recent years. In Europe, a new unitary patent system has become effective and a new European patent court started operations in 2023, which may significantly impact European patents, including those granted before the introduction of such a system. Under the unitary patent system, European patent applications have the option, upon grant of a European patent, of becoming a Unitary Patent which will be subject to the jurisdiction of the new Unified Patent Court (UPC). As the UPC is a new court system, there is no precedent for the court, increasing the uncertainty of any litigation. Patents granted before the implementation of the UPC will have the option of opting out of the jurisdiction of the UPC and remaining as national patents in the UPC countries. Patents that remain under the jurisdiction of the UPC will be potentially vulnerable to a single UPC-based revocation challenge that, if successful, could invalidate the patent in all countries who are signatories to the UPC. We cannot predict with certainty the long-term effects of any potential changes.
In addition, a decree was adopted by the Russian government in March 2022 as a response to economic sanctions imposed by various other governments, allowing Russian companies and individuals to exploit inventions owned by patentees that have citizenship or nationality in, are registered in, or have a primary place of business or profit-making activities in the U.S. and other countries that Russia has deemed unfriendly without consent or compensation. Consequently, we would not be able to prevent third parties from practicing our inventions protected by Russian patents in Russia or from manufacturing, selling, using or importing products made using our inventions in and into Russia. Accordingly, our competitive position may be impaired, and our business, financial condition, results of operations and prospects may be adversely affected.
If we fail to comply with our obligations under the agreements pursuant to which we license intellectual property rights from third parties, or otherwise experience disruptions to our business relationships with our licensors, we could lose the rights to intellectual property that are important to our business.
We are a party to agreements under which we are granted rights to intellectual property that are important to our business and we expect that we may need to enter into additional license agreements in the future. Under certain license agreements, we may not control the preparation, filing, prosecution or maintenance of the licensed intellectual property, or may not have the first right to enforce or defend the intellectual property.
In those cases, we may not be able to adequately influence patent prosecution, enforcement or defense, or prevent inadvertent lapses of coverage due to failure to pay maintenance fees and we cannot be certain that these patents and patent applications will be prepared, filed, prosecuted, maintained, enforced, and defended in a manner consistent with the best interests of our business and that does not compromise the patent rights. Existing license agreements impose, and we expect that future license agreements will impose, various development obligations as well as other obligations, such as payment of royalties. If we fail to comply with our obligations under these agreements, the licensor may have the right to terminate the license. The termination of any license agreements or failure to adequately protect such license agreements could prevent us from commercializing product candidates covered by the licensed intellectual property. For more information regarding our license and collaboration agreements, see “Business—License and Collaboration Agreements.”
Disputes may arise regarding intellectual property subject to a licensing agreement, including:
•the scope of rights granted under the license agreement and other interpretation-related issues;
•the extent to which our technology and processes infringe, misappropriate or otherwise violate intellectual property of the licensor that is not subject to the licensing agreement;
•the sublicensing of patent and other rights under any current or future collaboration relationships;
•our diligence obligations under the license agreement and what activities satisfy those diligence obligations; and
•the ownership of inventions and know-how resulting from the joint creation or use of intellectual property by our licensors and us and our partners.
In addition, the agreements under which we currently license intellectual property or technology from third parties are complex, and certain provisions in such agreements may be susceptible to multiple interpretations. The resolution of any contract interpretation disagreement that may arise could narrow what we believe to be the scope of our rights to the relevant intellectual property or technology, or increase what we believe to be our financial or other obligations under the relevant agreement, either of which could have a material adverse effect on our business, financial condition, results of operations, and prospects. Moreover, if disputes over intellectual property that we have licensed prevent or impair our ability to maintain our current licensing arrangements on commercially acceptable terms, we may be unable to successfully develop and commercialize the affected product candidates, which could have a material adverse effect on our business, financial conditions, results of operations, and prospects.
It is possible that we may be unable to obtain any necessary additional licenses at a reasonable cost or on reasonable terms, if at all. In that event, we may be required to expend significant time and resources to redesign our technology, product candidates, or the methods for manufacturing them or to develop or license replacement technology, all of which may not be feasible on a technical or commercial basis. If we are unable to do so, we may be unable to develop or commercialize the affected product candidates, which could harm our business, financial condition, results of operations, and prospects significantly. We cannot provide any assurances that third party patents do not exist which might be enforced against our current technology, including our DARPin product candidates, manufacturing methods or future methods or products resulting in either an injunction prohibiting our manufacture or sales, or, with respect to our sales, an obligation on our part to pay royalties or other forms of compensation to third parties, which could be significant.
Intellectual property rights do not necessarily address all potential threats to our competitive advantage.
The degree of future protection afforded by our intellectual property rights is uncertain because intellectual property rights have limitations, and may not adequately protect our business, or permit us to maintain our competitive advantage. For example:
•others may be able to make compounds that are similar or substantially equivalent to our product candidates but that are not covered by the claims of the patents that we own or have exclusively licensed.
•the patents of third parties, including patents related to repeat protein technology, may have an adverse effect on our business.
•we or our current or future licensors or strategic partners might not have been the first to conceive or reduce to practice the inventions covered by the issued patent or pending patent application that we own or have licensed.
•we or our current or future licensors or strategic partners might not have been the first to file patent applications covering certain of our or their inventions.
•others may independently develop similar or alternative technologies or duplicate any of our technologies without infringing, misappropriating or otherwise violating our intellectual property rights;
•it is possible that our current or future patent applications will not lead to issued patents.
•issued patents that we own or license may not provide us with any competitive advantage, or may be held invalid or unenforceable, including as a result of legal challenges by our competitors.
•our competitors might conduct research and development activities in countries where we do not have patent rights and then use the information learned from such activities to develop competitive products for sale in our major commercial markets;
•third parties performing manufacturing or testing for us using our products or technologies could use the intellectual property of third parties without obtaining a proper license, rendering us susceptible to claims of infringement, misappropriation or other violation of such third parties’ intellectual property rights;
•we may not develop additional technologies that are patentable; and
•the patents of others may have an adverse effect on our business; in particular, our product candidates may in the future be tested for new indications, and if one proves to be effective against a specific new indication, we may be confronted with existing patents covering such indication.
Should any of these events occur, they could have a material adverse effect on our business, financial condition, results of operations, and prospects.
Changes in patent laws or patent jurisprudence could diminish the value of patents in general, thereby impairing our ability to protect our products.
Our success is heavily dependent on the extent of our intellectual property rights, particularly patents. Obtaining, defending and enforcing patents in the biopharmaceutical industry involves both technological and legal complexity and is costly, time-consuming and inherently uncertain. Changes in either the patent laws or interpretation of the patent laws in the European Union, United States or other jurisdictions could increase the uncertainties and costs surrounding the prosecution of patent applications and the enforcement or defense of issued patents.
The Leahy-Smith America Invents Act, or the AIA, was enacted in the United States in September 2011, resulting in significant changes to the U.S. patent system.
For example, assuming that other requirements for patentability are met, prior to March 2013, in the United States, the first to invent the claimed invention was entitled to the patent, while outside the United States, the first to file a patent application was entitled to the patent. As of March 16, 2013, under the AIA, the United States transitioned to a "first-to-file" system in which, assuming that other requirements for patentability are met, the first inventor to file a patent application will be entitled to the patent on an invention regardless of whether a third party was the first to invent the claimed invention. Therefore, a third party that files a patent application in the USPTO before us could therefore be awarded a patent covering an invention even if we had made the invention before it was made by the third party. This will require us to be cognizant of the time from invention to filing of a patent application, and circumstances could prevent or dissuade us from promptly filing patent applications on our inventions.
The AIA also includes a number of significant changes that affect the way patent applications will be prosecuted and also may affect patent litigation. These include changes that limit where a patentee may file a patent infringement suit and that allow third party submissions of prior art to the USPTO during patent prosecution and additional procedures to attack the validity of a patent by USPTO-administered post-grant proceedings, including post-grant review, inter partes review, and derivation proceedings. Because of a lower evidentiary standard in USPTO proceedings compared to the evidentiary standard in U.S. federal courts necessary to invalidate a patent claim, a third party could potentially provide evidence in a USPTO proceeding sufficient for the USPTO to hold a claim invalid even though the same evidence would be insufficient to invalidate the claim if first presented in a district court action. Accordingly, a third party may attempt to use USPTO procedures to invalidate our patent claims that would not have been invalidated if first challenged by the third party as a defendant in a district court action. The AIA and its implementation could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents, all of which could have a material adverse effect on our business, financial condition, results of operations, and prospects.
In addition, the patent positions of companies in the development and commercialization of biologics and pharmaceuticals are particularly uncertain. The U.S. Supreme Court has ruled on several patent cases in recent years, either narrowing the scope of patent protection available in certain circumstances or weakening the rights of patent owners in certain situations. In addition to increasing uncertainty with regard to our ability to obtain patents in the future, this combination of events has created uncertainty with respect to the value of patents, once obtained. Depending on decisions by the U.S. Congress, the federal courts and the USPTO, and other applicable bodies in the European Union and other foreign jurisdictions, the laws and regulations governing patents could change in unpredictable ways that could weaken our ability to obtain new patents or to defend and enforce our existing patents and patents that we might obtain in the future.
If we are unable to protect the confidentiality of our trade secrets, our business and competitive position would be harmed.
We consider proprietary trade secrets, confidential know-how and unpatented know-how to be important to our business and competitive position. We may rely on trade secrets or confidential know-how to protect our technology, especially where patent protection is believed to be of limited value. However, trade secrets and confidential know-how are difficult to protect.
To protect this type of information against disclosure or appropriation by competitors, our policy is to require our employees, consultants, contractors, CROs and advisors to enter into confidentiality agreements with us.
We cannot guarantee that we have entered into such agreements with each party that may have or have had access to our trade secrets or proprietary technology and processes and, despite these efforts, any of these parties may unintentionally or willfully breach the agreements and use or disclose our confidential information to competitors, and such agreements may not provide an adequate remedy in the event of unauthorized disclosure or use of confidential information. Enforcing a claim that a third party illegally disclosed or misappropriated trade secrets or confidential know-how is expensive, time-consuming and unpredictable. In addition, the enforceability of confidentiality agreements and trade secrets may vary from jurisdiction to jurisdiction. Furthermore, if a third party lawfully obtained or independently developed any of our trade secrets, we would have no right to prevent such third party from using that technology or information to compete with us or from disclosing it to others, which could harm our competitive position. Additionally, if the steps taken to maintain our trade secrets are deemed inadequate, we may have insufficient recourse against third parties for misappropriating our trade secrets.
Failure to effectively maintain and protect trade secrets or confidential know-how could adversely affect our competitive position. Moreover, our competitors may independently develop substantially equivalent proprietary information and may even apply for patent protection in respect of the same. If successful in obtaining such patent protection, our competitors may be able to limit our use of our trade secrets or confidential know-how.
We may be subject to claims by third parties asserting that we or our employees have infringed, misappropriated or otherwise violated their intellectual property, or claiming ownership of what we regard as our own intellectual property.
Many of our consultants and employees, including our senior management, were previously employed at other biotechnology or pharmaceutical companies, including our competitors or potential competitors. Some of these consultants and employees executed proprietary rights, non-disclosure and non-competition agreements in connection with such previous employment. Although we try to ensure that our consultants and employees do not use the proprietary information or know-how of others in their work for us, we may be subject to claims that we or these consultants and employees have used or disclosed confidential information or intellectual property, including trade secrets or other proprietary information, of any such consultant's or employee's current or former employer, or have breached their non-competition agreement. Litigation may be necessary to defend against such claims.
In addition, we or our licensors may be subject to claims that former employees, consultants, collaborators or other third parties have an interest in our owned or in-licensed patents or other intellectual property as an inventor or co-inventor. While it is our policy to require our consultants and employees who may be involved in the development of intellectual property to execute agreements assigning such intellectual property to us, we may be unsuccessful in executing such an agreement with each party who in fact develops intellectual property that we regard as our own. Our and their assignment agreements may not be self-executing or may be breached, and we may be forced to bring claims against third parties, or defend claims they may bring against us, to determine the ownership of what we regard as our intellectual property.
If we fail in prosecuting or defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel or sustain damages. Such intellectual property rights could be awarded to a third party, and we could be required to obtain a license from such third party to commercialize our technology or products. Such a license may not be available on commercially reasonable terms or at all. Even if we successfully prosecute or defend against such claims, litigation could result in substantial costs and distract management.
Obtaining and maintaining our patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements.
Periodic maintenance and annuity fees on any of our owned or licensed patents and patent applications are due to be paid to the USPTO, the EPO and other foreign patent agencies in several stages over the lifetime of the patents and patent applications. The USPTO, the EPO and various foreign governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other similar provisions during the patent application process. While an inadvertent lapse can in many cases be cured by payment of a late fee or by other means in accordance with the applicable rules, there are situations in which noncompliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. Non-compliance events that could result in abandonment or lapse of a patent or patent application include failure to respond to official actions within prescribed time limits, non-payment of fees and failure to properly legalize and submit formal documents. If we or our licensors or collaboration partners fail to maintain the patents and patent applications covering our product candidates, our competitors might be able to enter the market earlier with similar products or technology, which would have an adverse effect on our business.
Risks Related to Our Organization and Operations
Our future growth and ability to compete depends on retaining our key personnel and recruiting additional qualified personnel.
Our success depends upon the continued contributions of our key management, scientific and technical personnel, many of whom have been instrumental for us and have substantial experience with our therapies and related technologies. These key management individuals include the members of our board of directors and executive management, including Dr. Patrick Amstutz, our Chief Executive Officer, Dr. Alexander Zürcher, our Chief Operating Officer, Dr. Michael Tobias Stumpp, our EVP Projects, Dr. Philippe Legenne, our acting Chief Medical Officer and Renate Gloggner, our EVP People and Culture.
The loss of key managers and senior scientists could delay our research and development activities. In addition, our ability to compete in the highly competitive biotechnology and pharmaceutical industries, and particularly, in the oncology field, depends upon our ability to attract and retain highly qualified management, scientific and medical personnel. Many other biotechnology and pharmaceutical companies and academic institutions that we compete against for qualified personnel have greater financial and other resources, different risk profiles and a longer history in the industry than we do. Therefore, we might not be able to attract or retain these key persons on conditions that are economically acceptable. Furthermore, we will need to recruit new managers and qualified scientific personnel to develop our business if we expand into fields that will require additional skills. Additionally, there is a larger pool of qualified scientific and medical personnel in the United States than in Switzerland, and we may need to increase our presence in the United States in order to attract and retain the necessary human resources. Our inability to attract and retain these key persons could prevent us from achieving our objectives and implementing our business strategy, which could have an adverse effect on our business and prospects.
We expect to expand our development, regulatory and sales and marketing capabilities and, as a result, we may encounter difficulties in managing our growth, which could disrupt our operations.
We expect to experience growth in the number of our employees and the scope of our operations, particularly in the areas of drug development, regulatory affairs, sales and marketing and support functions such as finance, human resources, legal, intellectual property, information technology and administration. To manage our anticipated future growth, we must continue to implement and improve our managerial, operational and financial systems, expand our facilities and continue to recruit and train additional qualified personnel.
Due to our limited resources and the limited experience of our management team in managing a company with such anticipated growth, we may not be able to effectively manage the expansion of our operations or recruit and train additional qualified personnel. The expansion of our operations may lead to significant costs and may divert our management and business development resources. Any inability to manage growth could delay the execution of our business plans or disrupt our operations.
We may not be able to integrate efficiently or achieve the expected benefits of any acquisitions of complementary businesses, product candidates or technologies.
Since our inception in 2004, we have grown organically without any acquisitions. Should we in the future contemplate to acquire any complementary business, product candidates or technologies, our ability to integrate and manage acquired businesses, product candidates or technologies effectively will depend upon a number of factors including the size of the acquired business, the complexity of any product candidate or technology and the resulting difficulty of integrating the acquired business's operations, if any. Our relationship with current employees or employees of any acquired business may become impaired. We may also be subject to unexpected claims and liabilities arising from such acquisitions. These claims and liabilities could be costly to defend, could be material to our financial position and might exceed either the limitations of any applicable indemnification provisions or the financial resources of the indemnifying parties.
Our business is subject to economic, political, regulatory and other risks associated with international operations.
Our business is subject to risks associated with conducting business internationally. Accordingly, our future results could be harmed by a variety of factors, including:
•economic weakness, including inflation, increase of interest rates, or political instability in particular economies and markets;
•differing regulatory requirements for drug approvals;
•differing jurisdictions could present different issues for securing, maintaining or obtaining freedom to operate in such jurisdictions;
•potentially reduced ability to obtain, maintain, protect and enforce intellectual property rights and other proprietary rights;
•difficulties in compliance with different, complex and changing laws, regulations and court systems of multiple jurisdictions and compliance with a wide variety of foreign laws, treaties and regulations;
•changes in regulations and customs, tariffs and trade barriers;
•changes in currency exchange rates of the euro, U.S. dollar and Swiss franc and currency controls;
•changes in a specific country's or region's political or economic environment;
•trade protection measures, import or export licensing requirements or other restrictive actions by governments;
•differing reimbursement regimes and price controls in certain international markets;
•negative consequences from changes in tax laws;
•compliance with tax, employment, immigration and labor laws for employees living or traveling abroad, including, for example, the variable tax treatment in different jurisdictions of stock options granted under our employee stock plan;
•workforce uncertainty in countries where labor unrest is more common than in the United States;
•litigation or administrative actions resulting from claims against us by current or former employees or consultants individually or as part of class actions, including claims of wrongful terminations, discrimination, misclassification or other violations of labor law or other alleged conduct;
•litigation resulting from claims against us by third parties, including claims of breach of noncompete and confidentiality provisions of our employees' former employment agreements with such third parties;
•difficulties associated with staffing and managing international operations, including differing labor relations;
•production shortages resulting from any events affecting raw material supply or manufacturing capabilities abroad; and
•business interruptions resulting from cyber-attacks, geo-political actions, including war (such as the Russia-Ukraine war and the Israel-Hamas war) and terrorism, or natural disasters including earthquakes, typhoons, floods and fires.
Additionally, in connection with the ongoing war between Russia and Ukraine, the U.S. government and other governments have imposed enhanced export controls on certain products and sanctions on certain industry sectors and parties in Russia, and have indicated they will consider imposing additional sanctions and other similar measures in the near future. Although we do not have any operations in Russia or Ukraine, further escalation of geopolitical tensions could have a broader impact that expands into other markets where we do business, which could adversely affect our business, our supply chain or our collaborators.
Exchange rate fluctuations or abandonment of the euro currency may materially affect our results of operations and financial condition.
Due to the international scope of our operations, our assets, earnings and cash flows are influenced by movements in exchange rates of several currencies, particularly regarding U.S. dollars, euros, and Swiss francs. Our functional currency is the Swiss franc and the majority of our operating expenses are paid in Swiss francs, but we also may receive payments from our business partners in U.S. dollars or euros and we regularly acquire services, consumables and materials in U.S. dollars, euros and Swiss francs. Further, potential future revenue may be derived from abroad, particularly from the United States and the European Union. As a result, our business and share price may be affected by fluctuations in foreign exchange rates between the Swiss franc, the euro, the U.S. dollar and these other currencies, which may also have a significant impact on our reported results of operations and cash flows from period to period. Besides our natural hedging, currently, we do not have any exchange rate hedging arrangements in place.
In addition, the possible abandonment of the euro by one or more members of the European Union could materially affect our business in the future. Despite measures taken by the European Union to provide funding to certain European Union member states in financial difficulties and by a number of European countries to stabilize their economies and reduce their debt burdens, it is possible that the euro could be abandoned in the future as a currency by countries that have adopted its use. This could lead to the re-introduction of individual currencies in one or more European Union member states, or in more extreme circumstances, the abandonment of the euro or the dissolution of the European Union. The effects on our business of a potential dissolution of the European Union, the exit of one or more European Union member states from the European Union or the abandonment of the euro as a currency, are impossible to predict with certainty, and any such events could have a material adverse effect on our business, financial condition and results of operations.
Unfavorable global economic conditions, including as a result of the ongoing war between Russia and Ukraine as well as Israel and Hamas, could have a negative impact on our operations, which could materially and adversely affect our business, financial condition, results of operations, prospects and market price of our ordinary shares.
Global economic instability and unfavorable conditions could materially and adversely affect our business. The war between Russia and Ukraine is ongoing. The impact to Ukraine as well as actions taken by other countries, including new and stricter sanctions imposed by Canada, the United Kingdom, the European Union, the United States. and other countries against officials, individuals, regions, and industries in Russia and Ukraine, and actions taken by Russia in response to such sanctions, and responses of countries and political bodies to such sanctions, tensions, and military actions and the potential for more widespread conflict, have resulted in supply chain disruptions, increases in inflation, financial market volatility and capital markets disruption. In addition, the war between Israel and Hamas is ongoing. Any resulting instability and unfavorable economic conditions from the wars could disrupt our and our collaborators’ supply chains and adversely affect our and our collaborators’ ability to conduct ongoing and future clinical trials of our product candidates. The extent and duration of the wars, sanctions and resulting economic, market and other disruptions are impossible to predict, but could be substantial. Any such disruptions may heighten the impact of the other risks described in this report on Form 20-F.
The United Kingdom's referendum vote in favor of withdrawal from the European Union could adversely affect our ability to develop, manufacture and commercialize our product candidates in the United Kingdom.
Following the result of a referendum in 2016, the United Kingdom left the European Union on January 31, 2020, commonly referred to as Brexit. Pursuant to the formal withdrawal arrangements agreed between the United Kingdom and the European Union, the United Kingdom was subject to a transition period until December 31, 2020, or the Transition Period, during which European Union rules continued to apply. The Trade and Cooperation Agreement, or the Trade and Cooperation Agreement, which outlines the future trading relationship between the United Kingdom and the European Union was agreed in December 2020 and formally entered into force on May 1, 2021.
Since a significant proportion of the regulatory framework in the United Kingdom applicable to our business and our product candidates is derived from European Union directives and regulations, Brexit has had, and will continue to have, a material impact on the regulatory regime with respect to the development, manufacture, importation, approval and commercialization of our product candidates in the United Kingdom. For example, Great Britain is no longer covered by the centralized procedures for obtaining European Union-wide marketing authorizations from the EMA, and a separate marketing authorization will be required to market our product candidates in Great Britain. It is currently unclear whether the Medicines & Healthcare products Regulatory Agency in the United Kingdom is sufficiently prepared to handle the increased volume of marketing authorization applications that it is likely to receive. Any delay in obtaining, or an inability to obtain, any marketing approvals, as a result of Brexit or otherwise, would delay or prevent us from commercializing our product candidates in the United Kingdom and limit our ability to generate revenue and achieve and sustain profitability. We could face significant additional expenses to obtain regulatory approval for our products in the United Kingdom.
We are exposed to unanticipated changes in tax laws and regulations, adjustments to our tax provisions, exposure to additional tax liabilities, or forfeiture of our tax assets.
The determination of our provision for income taxes and other tax liabilities requires significant judgment, including the adoption of certain accounting policies and our determination of whether we will be able to obtain a future tax benefit from our deferred tax assets. We cannot guarantee that our interpretations will not be challenged by the relevant tax authorities, or that the relevant tax laws and regulations, or the interpretation thereof, including through tax rulings, by the relevant tax authorities, will not be subject to change. Any adverse outcome of such a challenge may lead to adjustments in the amounts recorded in our financial statements, and could have a material adverse effect on our operating results and financial condition.
We are subject to laws and regulations on tax levies and other charges or contributions in different countries, including transfer pricing and tax regulations applicable to the compensation of personnel and third parties. Transactions between current group companies, as well as additional companies that may form part of our group in the future, are subject to transfer pricing regulations, which may be subject to change or our existing transfer pricing system could be challenged by the relevant tax authority, and any such changes or challenges could adversely affect us.
Our effective tax rate could be adversely affected by changes in tax laws, treaties and regulations, both internationally and domestically, or the interpretation thereof by the relevant tax authorities, including changes to the U.K. research and development tax credit regime or the "patent box" regime, possible changes to the corporate income tax base, changes to the additional deduction for expenditure on research and development personnel in Switzerland and other tax incentives. An increase in our effective tax rate could have an adverse effect on our business, financial position, results of operations and cash flows.
In addition, we may not be able to use, or changes in tax regulations may affect the use of, certain tax loss carryforwards that we have generated in prior years. For instance, as of December 31, 2023, we had substantial tax loss carry forwards. In general, some of these tax loss carry forwards may be forfeited in whole, or in part, as a result of various transactions, or their utilization may be restricted by statutory law in the relevant jurisdiction. Any corporate reorganization by us or any transaction relating to our shareholding structure may result in partial or complete forfeiture of tax loss carry forwards. If not used, tax loss carryforwards for Swiss corporate income tax purposes expire seven years after the tax year in which they were incurred. Due to our limited future / expected income, there is a high risk that our tax loss carryforwards will expire in part or in their entirety and therefore will not be able to be used to offset future taxable income for Swiss corporate income tax purposes. Furthermore, any tax loss carry forwards that we report on our Swiss tax returns are subject to review and confirmation by the competent Swiss tax authorities in their tax assessment of the tax year for which the tax loss carryforwards are used to offset taxable income. Consequently, we are exposed to the risk that the competent Swiss tax authorities may not accept the reported tax loss carryforwards in part or in their entirety.
Changes in our effective tax rate or tax liability may have an adverse effect on our results of operations.
Our effective tax rate could increase due to several factors, including:
•changes in the relative amounts of income before taxes in the jurisdictions in which we operate that have differing statutory tax rates;
•changes in tax laws, tax treaties, and regulations or the interpretation of them;
•changes to our assessment about our ability to realize any deferred tax assets that are based on estimates of our future results, the prudence and feasibility of possible tax planning strategies, and the economic and political environments in which we do business;
•the outcome of any current or future tax audits, examinations, or administrative appeals; and
•any limitations or adverse findings regarding our ability to do business in some jurisdictions.
Any of these developments could adversely affect our business, results of operations and financial condition.
As a result of changes in, or in the interpretation of, tax laws, treaties, rulings, regulations or agreements of Switzerland or any other country in which we currently operate or may in the future operate, the loss of a major tax dispute or a challenge to our operating structure, intercompany pricing policies or the taxable presence of our existing or any future subsidiaries in certain countries, or other factors, our effective income tax rates may increase in the future, which could adversely affect our net income and cash flows.
We operate in multiple jurisdictions and our profits are taxed pursuant to the tax laws of these jurisdictions. The tax laws applicable to our business activities, however, are subject to changes in interpretation. Our tax position could be adversely impacted by changes in tax rates, tax laws, tax practice, tax treaties or tax regulations or changes in the interpretation thereof by the tax authorities in jurisdictions in which we currently do or may in the future elect to do business. Our effective income tax rate may be affected by changes in or interpretations of tax laws, treaties, rulings, regulations or agreements in any such jurisdiction, the resolution of issues arising from any future tax audits with various tax authorities, utilization of net operating loss and tax credit carryforwards, changes in geographical allocation of income and expense, and changes in management’s assessment of matters such as the realizability of deferred tax assets. In the past, we have experienced fluctuations in our effective income tax rate. Our actual tax rate may vary from our expectation and that variance may be material. Our effective income tax rate in a given fiscal year reflects a variety of factors that may not be present in the succeeding fiscal year or years. There is no assurance that our effective income tax rate will not change in future periods.
The standard effective corporate tax rate in Schlieren, Canton of Zurich, can change from time to time. The standard combined (federal, cantonal, municipal) effective corporate income tax rate, except for dividend income for which we could claim a participation exemption, for 2023 in Schlieren, Canton of Zurich, is approximately 19.3%.
We urge our shareholders to consult with their legal and tax advisors with respect to the potential tax consequences of investing in or holding our ADSs and ordinary shares.
The price of our ADSs may be volatile and may fluctuate due to factors beyond our control.
The market price of our ADSs and our ordinary shares may fluctuate significantly due to a variety of factors, many of which are beyond our control, including:
•positive or negative results of testing and clinical trials reported or conducted by us, strategic partners or competitors;
•delays in entering into strategic relationships with respect to development or commercialization of our product candidates or entering into strategic relationships on terms that are not deemed to be favorable to us;
•technological innovations or commercial product introductions by us or competitors;
•changes in government regulations;
•developments concerning proprietary rights, including patents and litigation matters;
•public concern relating to the commercial value or safety of any of our product candidates;
•financing or other corporate transactions;
•publication of research reports or comments by securities or industry analysts;
•general market conditions in the pharmaceutical industry or in the economy as a whole;
•impact of macroeconomic factors, including a health pandemic, rising inflation, the U.S. Federal Reserve raising interest rates, the Russia-Ukraine war and the Israel-Hamas war, on the global economy or financial markets; or
•price and volume fluctuations attributable to inconsistent trading volume levels of our ADSs and/or ordinary shares.
These and other market and industry factors may cause the market price and demand for our ADSs and ordinary shares to fluctuate substantially, regardless of our actual operating performance, which may limit or prevent investors from readily selling their ADSs or ordinary shares and may otherwise negatively affect the liquidity of our ADSs and ordinary shares. In addition, the stock market in general, and biopharmaceutical companies in particular, have experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of these companies.
We have and will continue to incur increased costs as a result of operating as a U.S.-listed public company, and our board of directors will be required to devote substantial time to compliance initiatives and corporate governance practices.
As a public company in the United States, and particularly after we no longer qualify as an emerging growth company, we have and will continue to incur significant legal, accounting and other expenses that we did not incur as a public company listed only on the SIX Swiss Exchange. We are a corporation (Aktiengesellschaft), organized under the laws of Switzerland in accordance with articles 620 et seqq. CO and subject to the listing rules and the applicable regulations for companies listed on the SIX Swiss Exchange, the Sarbanes-Oxley Act of 2002, the Dodd-Frank Wall Street Reform and Consumer Protection Act, the listing requirements of the Nasdaq Stock Market, or Nasdaq, and other applicable securities rules and regulations that impose various requirements on non-U.S. reporting public companies, including the establishment and maintenance of effective disclosure and financial controls and certain additional corporate governance practices. Our board of directors and other personnel are required to devote a substantial amount of time to these compliance initiatives. Moreover, these rules and regulations will increase our legal and financial compliance costs and will make some activities more time-consuming and costly.
Pursuant to Section 404 of the Sarbanes-Oxley Act of 2002, or Section 404, we are required to furnish a report on our internal control over financial reporting. However, while we remain an emerging growth company, we will not be required to include an attestation report on internal control over financial reporting issued by our independent registered public accounting firm. To achieve compliance with Section 404 within the prescribed period, we are documenting and evaluating our internal control over financial reporting, which is both costly and challenging. In this regard, we continue to dedicate internal resources, continue to engage outside consultants and adopt a detailed work plan to assess and document the adequacy of internal control over financial reporting, continue steps to improve control processes as appropriate, validate through testing that controls are functioning as documented and implement a continuous reporting and improvement process for internal control over financial reporting.
Despite our efforts, there is a risk that we will not be able to conclude, within the prescribed timeframe or at all, that our internal control over financial reporting is effective as required by Section 404. If we identify one or more material weaknesses, it could result in an adverse reaction in the financial markets due to a loss of confidence in the reliability of our financial statements.
Certain significant shareholders may own a substantial number of our securities and as a result, may be able to exercise significant influence over the outcome of shareholder votes. These shareholders may have different interests from us or your interests.
We have a number of significant shareholders. For an overview of our current significant shareholders, please see "Principal Shareholders."
Currently, we are not aware that any of our existing shareholders have entered or will enter into a shareholders' agreement with respect to the exercise of their voting rights. Nevertheless, depending on the level of attendance at our general meetings of shareholders, or the General Meeting, these significant shareholders could have the ability to significantly influence the outcome of decisions taken at any such General Meeting. Any such voting by these shareholders may not be in accordance with our interests or those of our other shareholders. Among other consequences, this concentration of ownership may have the effect of delaying or preventing a change in control and might therefore negatively affect the market price of our ADSs.
Future sales, or the possibility of future sales, of a substantial number of our securities could adversely affect the price of the shares and dilute shareholders.
If our existing shareholders sell, or indicate an intent to sell, substantial amounts of our securities in the public market, the trading price of our ADSs and our ordinary shares could decline significantly. As of December 31, 2023, we had 36,354,297 ordinary shares outstanding including 3,500,000 treasury shares held through our wholly-owned subsidiary Molecular Partners Inc. and 3,000,000 ADS representing our ordinary shares issued and outstanding. In addition, ordinary shares subject to outstanding options under our equity incentive plans and the ordinary shares reserved for future issuance under our equity incentive plan will become eligible for sale in the public market in the future, subject to certain legal and contractual limitations.
We intend to register all ordinary shares that we may issue under our equity compensation plans. Once we register these ordinary shares, they can be freely sold in the public market upon issuance, subject to volume limitations applicable to affiliates and any applicable lock-up agreements.
Provisions of our articles of association or Swiss corporate law might deter acquisition bids for us that might be considered favorable and prevent or frustrate any attempt to replace or remove the then board of directors.
Provisions of our articles of association may make it more difficult for a third party to acquire control of us. For example, our board of directors is authorized to deny the preemptive rights of shareholders and allocate them to third parties as a defense of an actual, threatened or potential takeover bid, in relation to which our board of directors, upon consultation with an independent financial adviser retained by it, has not recommended to the shareholders acceptance on the basis that the board of directors has not found the takeover bid to be financially fair to the shareholders.
In addition, several provisions of Swiss corporate law and certain other provisions of Swiss law, such as obligations to disclose significant shareholdings and merger control regulations, that apply to us may make an unsolicited tender offer, merger, change in management or other change in control of our company more difficult.
These provisions could discourage potential takeover attempts that other shareholders may consider to be in their best interest and could adversely affect the market price of our securities. These provisions may also have the effect of depriving ADS holders of the opportunity to sell their ADSs at a premium. In addition, the board of directors of Swiss companies may in certain instances, and subject to prior authorization by the shareholders, deter or frustrate public takeover bids through dilutive issuances of equity securities (pursuant to the authorized capital) or through share buy-backs.
Fluctuations in exchange rates may increase the risk of holding ADSs and ordinary shares.
Due to the international scope of our operations, our assets, earnings and cash flows are influenced by movements in exchange rates of several currencies, particularly the euro, U.S. dollar and Swiss franc. Our functional currency is the Swiss franc, and the majority of our operating expenses are paid in Swiss franc, but we also receive or may receive payments from business partners in U.S. dollars, and we regularly acquire services, consumables and materials in U.S. dollars and euros. Further, potential future revenue may be derived from abroad, particularly from the United States or the European Union. As a result, our business and the price of our ADSs and ordinary shares may be affected by fluctuations in foreign exchange rates between the Swiss franc and these other currencies, which may also have a significant impact on our reported results of operations and cash flows from period to period. Besides natural hedging, currently, we do not have any exchange rate hedging arrangements in place.
Moreover, because our ordinary shares currently trade on the SIX Swiss Exchange in Swiss francs, and our ADSs trade on the Nasdaq Global Select Market in U.S. dollars, fluctuations in the exchange rate between the U.S. dollar and the Swiss franc may result in temporary differences between the value of our ADSs and the value of our ordinary shares, which may result in heavy trading by investors seeking to exploit such differences.
Our ordinary shares and ADSs are traded on more than one market and this may result in price variations and adversely affect the liquidity and value of the ADSs; in addition, investors may not be able to easily move ordinary shares for trading between such markets. Furthermore, because of this dual listing, securities and stock exchange laws, regulations and rules apply to us that may be irreconcilable or otherwise difficult to comply with contemporaneously.
Our ordinary shares have traded on the SIX Swiss Exchange since 2014 and our ADSs have traded on the Nasdaq Global Select Market since June 2021. Trading in our ADSs or ordinary shares on these markets takes place in different currencies (U.S. dollars on the Nasdaq Global Select Market and Swiss Francs on the SIX Swiss Exchange), at different times (resulting from different time zones, different trading days and different public holidays in the United States and Switzerland) and among a different investor base. The trading prices of our ordinary shares and our ADSs on these two markets may differ due to these and other factors. Any decrease in the price of our ordinary shares on the SIX Swiss Exchange could cause a decrease in the trading price of our ADSs on the Nasdaq Global Select Market. Investors could seek to sell or buy our ordinary shares to take advantage of any price differences between the markets through a practice referred to as arbitrage. Any arbitrage activity could create unexpected volatility in both our share prices on one exchange and the ordinary shares available for trading on the other exchange. In addition, holders of ADSs cannot immediately surrender their ADSs and withdraw the underlying ordinary shares for trading on the other market without effecting necessary procedures with the depositary. This could result in time delays and additional cost for holders of ADSs.
Because different types of our equity securities are admitted to trading and listed on two different stock exchanges in two different jurisdictions, two sets of securities laws and regulations and stock exchange rules apply to us contemporaneously. It cannot be excluded that the laws, regulations and/or rules of one jurisdiction or trading venue may require us to effect disclosures or filings or grant shareholders and/or holders of our ADSs certain rights that would be unlawful under the laws, regulations and/or rules of the respective other jurisdiction or trading venue.
For this or other reasons, it may prove difficult or impossible for us to at all times comply with the laws, regulations and/or rules of both jurisdictions and trading venues at the same time.
Holders of ADSs are not treated as holders of our ordinary shares.
Holders of our ADSs are not treated as holders of our ordinary shares, unless they withdraw the ordinary shares underlying their ADSs in accordance with the deposit agreement and applicable laws and regulations. The depositary is the holder of the ordinary shares underlying our ADSs. Holders of our ADSs therefore do not have any rights as holders of our ordinary shares, other than the rights that they have pursuant to the deposit agreement.
Holders of ADSs may be subject to limitations on the transfer of their ADSs and the withdrawal of the underlying ordinary shares.
ADSs are transferable on the books of the depositary. However, the depositary may close its books at any time or from time to time when it deems expedient in connection with the performance of its duties. The depositary may refuse to deliver, transfer or register transfers of ADSs generally when our books or the books of the depositary are closed, or at any time if we or the depositary think it is advisable to do so because of any requirement of law, government or governmental body, or under any provision of the deposit agreement, or for any other reason, subject to the right of ADS holders to cancel their ADSs and withdraw the underlying ordinary shares. Temporary delays in the cancellation of your ADSs and withdrawal of the underlying ordinary shares may arise because the depositary has closed its transfer books or we have closed our transfer books, the transfer of ordinary shares is blocked to permit voting at a shareholders' meeting or we are paying a dividend on our ordinary shares. In addition, ADS holders may not be able to cancel their ADSs and withdraw the underlying ordinary shares when they owe money for fees, taxes and similar charges and when it is necessary to prohibit withdrawals in order to comply with any laws or governmental regulations that apply to ADSs or to the withdrawal of ordinary shares or other deposited securities.
We are entitled to amend the deposit agreement and to change the rights of ADS holders under the terms of such agreement, or to terminate the deposit agreement, without the prior consent of the ADS holders.
We are entitled to amend the deposit agreement and to change the rights of the ADS holders under the terms of such agreement, without the prior consent of the ADS holders. We and the depositary may agree to amend the deposit agreement in any way we decide is necessary or advantageous to us or to the depositary. Amendments may reflect, among other things, operational changes in the ADS program, legal developments affecting ADSs or changes in the terms of our business relationship with the depositary. In the event that the terms of an amendment are materially disadvantageous to ADS holders, ADS holders will only receive 30 days’ advance notice of the amendment, and no prior consent of the ADS holders is required under the deposit agreement. Furthermore, we may decide to direct the depositary to terminate the ADS facility at any time for any reason. For example, terminations may occur if we become the subject of a takeover or a going-private transaction. If the ADS facility will terminate, ADS holders will receive at least 30 days’ prior notice, but no prior consent is required from them. Under the circumstances that we decide to make an amendment to the deposit agreement that is disadvantageous to ADS holders or terminate the deposit agreement, the ADS holders may choose to sell their ADSs or surrender their ADSs and become direct holders of the underlying ordinary shares, but will have no right to any compensation whatsoever.
ADSs holders may not be entitled to a jury trial with respect to claims arising under the deposit agreement, which could result in less favorable outcomes to the plaintiff(s) in any such action.
The deposit agreement governing the ADSs representing our ordinary shares provides that, to the fullest extent permitted by law, holders and beneficial owners of ADSs irrevocably waive the right to a jury trial of any claim they may have against us or the depositary arising out of or relating to the ADSs or the deposit agreement.
If this jury trial waiver provision is not permitted by applicable law, an action could proceed under the terms of the deposit agreement with a jury trial. If we or the depositary opposed a jury trial demand based on the waiver, the court would determine whether the waiver was enforceable based on the facts and circumstances of that case in accordance with the applicable state and federal law. To our knowledge, the enforceability of a contractual pre-dispute jury trial waiver in connection with claims arising under the federal securities laws has not been finally adjudicated by the United States Supreme Court. However, we believe that a contractual pre-dispute jury trial waiver provision is generally enforceable, including under the laws of the State of New York, which govern the deposit agreement, by a federal or state court in the City of New York, which has non-exclusive jurisdiction over matters arising under the deposit agreement. In determining whether to enforce a contractual pre-dispute jury trial waiver provision, courts will generally consider whether a party knowingly, intelligently and voluntarily waived the right to a jury trial. We believe that this is the case with respect to the deposit agreement and the ADSs. It is advisable that you consult legal counsel regarding the jury waiver provision before entering into the deposit agreement.
If you or any other holders or beneficial owners of ADSs bring a claim against us or the depositary in connection with matters arising under the deposit agreement or the ADSs, including claims under federal securities laws, you or such other holder or beneficial owner may not be entitled to a jury trial with respect to such claims, which may have the effect of limiting and discouraging lawsuits against us and/or the depositary. If a lawsuit is brought against us and/or the depositary under the deposit agreement, it may be heard only by a judge or justice of the applicable trial court, which would be conducted according to different civil procedures and may result in different outcomes than a trial by jury would have had, including results that could be less favorable to the plaintiff(s) in any such action, depending on, among other things, the nature of the claims, the judge or justice hearing such claims, and the venue of the hearing.
No condition, stipulation or provision of the deposit agreement or ADSs serves as a waiver by any holder or beneficial owner of ADSs or by us or the depositary of compliance with any substantive provision of the U.S. federal securities laws and the rules and regulations promulgated thereunder.
Moreover, as the jury trial waiver relates to claims arising out of or relating to the ADSs or the deposit agreement, we believe that, as a matter of construction of the clause, the waiver would likely to continue to apply to ADS holders who withdraw the ordinary shares from the ADS facility with respect to claims arising before the cancellation of the ADSs and the withdrawal of the ordinary shares, and the waiver would most likely not apply to ADS holders who subsequently withdraw the ordinary shares represented by ADSs from the ADS facility with respect to claims arising after the withdrawal. However, to our knowledge, there has been no case law on the applicability of the jury trial waiver to ADS holders who withdraw the ordinary shares represented by the ADSs from the ADS facility.
You will not have the same voting rights as the holders of our ordinary shares and may not receive voting materials in time to be able to exercise your right to vote.
Except as described in this Annual Report on Form 20-F and the deposit agreement, holders of the ADSs are not able to exercise voting rights attached to the ordinary shares represented by the ADSs. Under the terms of the deposit agreement, holders of the ADSs may instruct the depositary to vote the ordinary shares underlying their ADSs.
Otherwise, holders of ADSs are not able to exercise their right to vote unless they withdraw the ordinary shares underlying their ADSs in accordance with the deposit agreement and applicable laws and regulations to vote them in person or by proxy in accordance with applicable Swiss laws and regulations and our articles of association. Even so, ADS holders may not know about a meeting far enough in advance to withdraw those ordinary shares. If we ask for the instructions of holders of the ADSs, the depositary, upon timely notice from us, will notify ADS holders of the upcoming vote and arrange to deliver our voting materials to them. Upon our request, the depositary will mail to holders a shareholder meeting notice that contains, among other things, a statement as to the manner in which voting instructions may be given. We cannot guarantee that ADS holders will receive the voting materials in time to ensure that they can instruct the depositary to vote the ordinary shares underlying their ADSs. In addition, regardless of whether timely voting instructions are provided to the depositary, at our request, the depositary will represent all ordinary shares underlying the ADSs for the purpose of establishing a quorum at a meeting of our shareholders. A shareholder is only entitled to participate in, and vote at, the meeting of shareholders, provided that its shares are recorded in its name at midnight (Central European Time) at the end of the 28th day preceding the date of the meeting of shareholders. In addition, the depositary's liability to ADS holders for failing to execute voting instructions or for the manner of executing voting instructions is limited by the deposit agreement. As a result, holders of ADSs may not be able to exercise their right to give voting instructions or to vote in person or by proxy and they may not have any recourse against the depositary or us if their ordinary shares are not voted as they have requested or if their shares cannot be voted.
A beneficial owner of our ordinary shares that is not registered in our shareholders register may not be able to exercise certain rights attached to the ordinary shares.
The financial rights attached to our ordinary shares transfer to a holder of those shares upon purchasing such shares in a stock market transaction. Any voting rights or rights related to voting rights only transfer once the acquirer has been registered in the shareholders' register as shareholder of such ordinary shares. A beneficial owner that is not directly registered in the shareholders' register can enjoy the financial rights, voting rights and rights related to voting rights only through the entity that acts as nominee or depositary for those ordinary shares and is recorded in the shareholders' register as the shareholder of record of those shares. This is also the case if you hold ADSs. It is possible that a nominee or a depositary will be unwilling to exercise certain rights attached to the ordinary shares, such as rights that require litigation. Therefore, failing to register in the shareholders' register may result in your inability to exercise certain rights as a shareholder.
We do not expect to pay dividends in the foreseeable future.
We have not paid any dividends since our incorporation. Even if future operations lead to significant levels of distributable profits, we currently intend that any earnings will be reinvested in our business and that dividends will not be paid until we have an established revenue stream to support continuing dividends. In addition, payment of any future dividends to shareholders would be subject to shareholder approval at our General Meeting, upon proposal of the board of directors, which proposal would be subject to the approval of the majority of the non-executive directors after taking into account various factors including our business prospects, cash requirements, financial performance and new product development. In addition, certain limitations apply to the payment of future dividends pursuant to Swiss law and our articles of association. In addition, payment of future cash dividends may be made only if our shareholders' equity exceeds the sum of our paid-in and called-up share capital plus the reserves required to be maintained by Swiss law or by our articles of association. Accordingly, investors cannot rely on cash dividend income from ADSs and any returns on an investment in the ADSs will likely depend entirely upon any future appreciation in the price of the ADSs.
You may not receive distributions on our ordinary shares represented by our ADS or any value for them if it is illegal or impractical to make them available to holders of ADSs.
The depositary for our ADSs will pay to you or distribute the cash dividends or other distributions it or the custodian receives on our ordinary shares or other deposited securities after deducting its fees and expenses. You will receive these distributions in proportion to the number of our ordinary shares your ADSs represent. However, in accordance with the limitations set forth in the deposit agreement, it may be unlawful or impractical to make a distribution available to holders of ADSs. We have no obligation to take any other action to permit the distribution of our ADSs, ordinary shares, rights or anything else to holders of our ADSs. This means that you may not receive the distributions we make on our ordinary shares or any value from them if it is unlawful or impracticable to make them available to you. These restrictions may have a material adverse effect on the value of your ADSs.
Holders of our ordinary shares outside Switzerland and ADS holders may not be able to exercise pre-emptive rights.
Under Swiss law, shareholders may receive certain pre-emptive rights to subscribe on a pro rata basis for issuance of equity or other securities that are convertible into equity. Due to laws and regulations in their respective jurisdictions, however, non-Swiss shareholders may not be able to exercise such rights unless we take action to register or otherwise qualify the rights offering under the laws of that jurisdiction. There can be no assurance that we would take any such action and we reserve the right to determine whether we should take such action in any jurisdiction. If shareholders in such jurisdictions were unable to exercise their subscription rights, their ownership interest in the Company would be diluted.
ADS holders have no pre-emptive rights to subscribe to newly issued shares unless we grant such rights to the foreign depositary. The right to exercise such pre-emptive rights is set out in the agreement between the ADS holder and the depositary.
We are a Swiss corporation. The rights of our shareholders may be different from the rights of shareholders in companies governed by the laws of U.S. jurisdictions.
We are a Swiss corporation. Our corporate affairs are governed by our articles of association and organizational rules and by the laws governing companies, including listed companies, incorporated in Switzerland. The rights of our shareholders and the responsibilities of members of our board of directors may be different from the rights and obligations of shareholders and directors of companies governed by the laws of U.S. jurisdictions. In the performance of its duties, our board of directors is required by Swiss law to consider the interests of our company, and may also have regard to the interests of our shareholders, our employees and other stakeholders, in all cases with due observation of the principles of reasonableness and fairness. It is possible that some of these parties will have interests that are different from, or in addition to, your interests as a holder of ADSs. Swiss corporate law limits the ability of our shareholders to challenge resolutions made or other actions taken by our board of directors in court. Our shareholders generally are not permitted to file a suit to reverse a decision or an action taken by our board of directors but are instead only permitted to seek damages for breaches of fiduciary duty. As a matter of Swiss law, shareholder claims against a member of our board of directors for breach of fiduciary duty would have to be brought to the competent courts in Schlieren, Canton of Zurich, Switzerland, or where the relevant member of our board of directors is domiciled. In addition, under Swiss law, any claims by our shareholders against us must be brought exclusively to the competent courts in Schlieren, Canton of Zurich, Switzerland.
On January 1, 2023, legislation that modernized certain aspects of Swiss corporate law (the Swiss corporate law reform (Aktienrechtsrevision)) entered into force. The new legislation altered the rights of shareholders under Swiss law, and as a consequence the rights of holders of our ADSs. The Swiss corporate law reform is subject to certain transitional periods as provided for therein.
In particular, Swiss stock corporations registered with the Commercial Register on January 1, 2023, are required to amend their articles of incorporation and organizational regulations in line with the new legislation within a transitional period of two years (i.e., until January 1, 2025). See "Item 10. - Memorandum and Articles of Association - Swiss Corporate Law Reform." There can be no assurance that Swiss law will not once again change in the future, which could adversely affect the rights of our shareholders or holders of our ADSs. Furthermore, there can be no guarantee that Swiss law does or will protect our shareholders or the holders of our ADSs in a similar fashion as the laws of U.S. jurisdictions would, in particular as regards corporate law principles, if we were a U.S.-incorporated company.
Our ordinary shares are issued under the laws of Switzerland, which may not provide investors with the same protections provided by incorporation in Delaware.
We are organized under the laws of Switzerland. A further summary of applicable Swiss law is contained in this Annual Report on Form 20-F. There can be no assurance that Swiss law will not change in the future or that it will provide investors with the same protections afforded to investors of a Delaware corporation, which could adversely affect the rights of investors.
Claims of U.S. civil liabilities may not be enforceable against us.
We are incorporated under the laws of Switzerland and our registered office and domicile is located in Schlieren, Switzerland. Substantially all of our assets are located outside the United States. A number of our directors and executive officers are not residents of the United States. As a result, it may not be possible for investors to effect service of process within the United States upon such persons or to enforce judgments obtained against them or us in U.S. courts, including judgments predicated upon the civil liability provisions of the U.S. federal securities laws. We have been advised by our Swiss counsel that there is doubt as to the enforceability in Switzerland of original actions, or in actions for enforcement of judgments of U.S. courts, of civil liabilities to the extent solely predicated upon the federal and state securities laws of the United States. Original actions against persons in Switzerland based solely upon the U.S. federal or state securities laws are governed, among other things, by the principles set forth in the Swiss Federal Act on Private International Law.
The United States currently does not have a treaty with Switzerland providing for the reciprocal recognition and enforcement of judgments, other than arbitration awards, in civil and commercial matters. Consequently, a final judgment for payment given by a court in the United States, whether or not predicated solely upon U.S. securities laws, would not automatically be recognized or enforceable in Switzerland. In order to obtain a judgment which is enforceable in Switzerland, the party in whose favor a final and conclusive judgment of the U.S. court has been rendered will be required to file its claim with a court of competent jurisdiction in Switzerland. Such party may submit to the Swiss court the final judgment rendered by the U.S. court. If and to the extent that the Swiss court finds that the jurisdiction of the U.S. court has been based on grounds which are internationally acceptable and that proper legal procedures have been observed, the court of Switzerland will, in principle, give binding effect to the judgment of the U.S. court, unless such judgment contravenes principles of public policy of Switzerland. Also, mandatory provisions of Swiss law may be applicable regardless of any other law that would otherwise apply. Swiss courts may deny the recognition and enforcement of punitive damages or other awards. Moreover, a Swiss court may reduce the amount of damages granted by a U.S. court and recognize damages only to the extent that they are necessary to compensate actual losses or damages. Enforcement and recognition of judgments of U.S. courts in Switzerland are solely governed by the provisions of the Swiss Federal Private International Law Act. This statute provides in principle that a judgment rendered by a non-Swiss court may be enforced in Switzerland only if:
•the non-Swiss court had jurisdiction pursuant to the Swiss Federal Act on Private International Law;
•the judgment of such non-Swiss court has become final and non-appealable;
•the judgment does not contravene Swiss public policy;
•the court procedures and the service of documents leading to the judgment were in accordance with the due process of law; and
•no proceeding involving the same position and the same subject matter was first brought in Switzerland, or adjudicated in Switzerland, or was earlier adjudicated in a third state and this decision is recognizable in Switzerland.
Based on the lack of a treaty as described above, U.S. investors may not be able to enforce against us or members of our board of directors or certain experts named herein who are residents of Switzerland or countries other than the United States any judgments obtained in U.S. courts in civil and commercial matters, including judgments under the U.S. federal securities laws.
Our status as a Swiss corporation means that our shareholders enjoy certain rights that may limit our flexibility to raise capital, issue dividends and otherwise manage ongoing capital needs.
Swiss law reserves for approval by shareholders certain corporate actions over which a board of directors would have authority in some other jurisdictions. For example, the payment of dividends and cancellation of treasury shares must be approved by shareholders. Swiss law also requires that our shareholders themselves resolve to, or authorize our board of directors to, increase our share capital. While our shareholders may authorize share capital that can be issued by our board of directors without additional shareholder approval, Swiss law limits this authorization to 50% of the share capital registered in the commercial register at the time of the authorization. The authorization, furthermore, has a limited duration of up to five years and must be renewed by the shareholders from time to time thereafter in order to be available for raising capital. For an overview of the changes in Swiss corporate law due to the Swiss corporate law reform that came into effect on January 1, 2023, see "Item 10. - Memorandum and Articles of Association - Swiss Corporate Law Reform." Additionally, subject to specified exceptions, including exceptions explicitly described in our articles of association, Swiss law grants preemptive rights to existing shareholders to subscribe for new issuances of shares, which may be limited or withdrawn only under certain limited conditions. Swiss law also does not provide as much flexibility in the various rights and regulations that can attach to different categories of shares as do the laws of some other jurisdictions. These Swiss law requirements relating to our capital management may limit our flexibility, and situations may arise where greater flexibility would have provided benefits to our shareholders. For changes to Swiss corporate law potentially affecting the rights of the holders of our ADSs, see also "Item 10. - Memorandum and Articles of Association - Swiss Corporate Law Reform"
We are a foreign private issuer and, as a result, we are not subject to U.S. proxy rules and are subject to Exchange Act reporting obligations that, to some extent, are more lenient and less frequent than those of a U.S. domestic public company.
We report under the Securities Exchange Act of 1934, as amended, or the Exchange Act, as a non-U.S. company with foreign private issuer status. Because we qualify as a foreign private issuer under the Exchange Act, we are exempt from certain provisions of the Exchange Act that are applicable to U.S. domestic public companies, including (i) the sections of the Exchange Act regulating the solicitation of proxies, consents or authorizations in respect of a security registered under the Exchange Act; (ii) the sections of the Exchange Act requiring insiders to file public reports of their stock ownership and trading activities and liability for insiders who profit from trades made in a short period of time; and (iii) the rules under the Exchange Act requiring the filing with the SEC of quarterly reports on Form 10-Q containing unaudited financial and other specified information, or current reports on Form 8-K, upon the occurrence of specified significant events.
In addition, foreign private issuers are not required to file their annual report on Form 20-F until 120 days after the end of each fiscal year, while U.S. domestic issuers that are accelerated filers are required to file their annual report on Form 10-K within 75 days after the end of each fiscal year. Foreign private issuers are also exempt from the Regulation Fair Disclosure, aimed at preventing issuers from making selective disclosures of material information. As a result of the above, you may not have the same protections afforded to shareholders of companies that are not foreign private issuers.
As a foreign private issuer and as permitted by the listing requirements of Nasdaq, we rely on certain home country governance practices rather than the corporate governance requirements of Nasdaq.
We are a foreign private issuer. As a result, in accordance with Nasdaq Listing Rule 5615(a)(3), we comply with home country governance requirements and certain exemptions thereunder rather than complying with certain of the corporate governance requirements of Nasdaq.
Swiss law does not require that a majority of our board of directors consist of independent directors. Our board of directors therefore may include fewer independent directors than would be required if we were subject to Nasdaq Listing Rule 5605(b)(1). In addition, we are not subject to Nasdaq Listing Rule 5605(b)(2), which requires that independent directors regularly have scheduled meetings at which only independent directors are present.
Although Swiss law also requires that we adopt a compensation committee, we follow home country requirements with respect to such committee. As a result, our practice varies from the requirements of Nasdaq Listing Rule 5605(d), which sets forth certain requirements as to the responsibilities, composition and independence of compensation committees. We have opted out of shareholder approval requirements for the issuance of securities in connection with certain events such as the acquisition of stock or assets of another company, the establishment of or amendments to equity-based compensation plans for employees, a change of control of us and certain private placements. To this extent, our practice varies from the independent director oversight of director nominations requirements of Nasdaq Listing Rule 5605(e).
Furthermore, in accordance with Swiss law and generally accepted business practices, our articles of association do not provide quorum requirements generally applicable to general meetings of shareholders. Our practice thus varies from the requirement of Nasdaq Listing Rule 5620(c), which requires an issuer to provide in its bylaws for a generally applicable quorum, and that such quorum may not be less than one-third of the outstanding voting stock. To this extent, our practice varies from the requirements of Nasdaq Listing Rule 5635, which generally requires an issuer to obtain shareholder approval for the issuance of securities in connection with such events.
As a result of the above, you may not have the same protections afforded to shareholders of companies that are not foreign private issuers.
We may lose our foreign private issuer status which would then require us to comply with the Exchange Act's domestic reporting regime and cause us to incur significant legal, accounting and other expenses.
We may no longer be a foreign private issuer as of June 30 for a given fiscal year (the end of our second fiscal quarter for a given fiscal year), which would require us to comply with all of the periodic disclosure and current reporting requirements of the Exchange Act applicable to U.S. domestic issuers as of January 1 of such year. In order to maintain our current status as a foreign private issuer, either (a) a majority of our ordinary shares must be either directly or indirectly owned of record by non-residents of the United States or (b)(i) a majority of our executive officers or directors may not be U.S. citizens or residents, (ii) more than 50% of our assets cannot be located in the United States and (iii) our business must be administered principally outside the United States.
If we lost foreign private issuer status, we would be required to comply with the Exchange Act reporting and other requirements applicable to U.S. domestic issuers, which are more detailed and extensive than the requirements for foreign private issuers. We would be required to change our accounting from reporting under IFRS to reporting under U.S. generally accepted accounting principles. We would also be required to make changes in our corporate governance practices in accordance with various SEC and Nasdaq rules. The regulatory and compliance costs to us under U.S. securities laws if we are required to comply with the reporting requirements applicable to a U.S. domestic issuer may be significantly higher than the cost we would incur as a foreign private issuer. As a result, we expect that a loss of foreign private issuer status would increase our legal and financial compliance costs and would make some activities highly time consuming and costly. We also expect that if we were required to comply with the rules and regulations applicable to U.S. domestic issuers, it would make it more difficult and expensive for us to obtain director and officer liability insurance, and we may be required to accept reduced coverage or incur substantially higher costs to obtain coverage. These rules and regulations could also make it more difficult for us to attract and retain qualified members of our board of directors.
We are an "emerging growth company" and we cannot be certain if the reduced reporting requirements applicable to "emerging growth companies" will make our ADSs less attractive to investors.
We are an "emerging growth company," as defined in the U.S. Jumpstart Our Business Startups Act of 2012, or the JOBS Act. For as long as we continue to be an "emerging growth company," we may take advantage of exemptions from various reporting requirements that are applicable to other public companies that are not "emerging growth companies," including not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act, exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and shareholder approval of any golden parachute payments not previously approved. We may take advantage of these exemptions until we are no longer an "emerging growth company." We could be an "emerging growth company" for up to the last day of the fiscal year ending after the fifth anniversary of our initial U.S. public offering (June 2021), although circumstances could cause us to lose that status earlier, including if the aggregate market value of our ordinary shares held by non-affiliates exceeds $700 million as of any June 30 (the end of our second fiscal quarter) before that time, in which case we would no longer be an "emerging growth company" as of the following December 31 (our fiscal year-end).
We cannot predict if investors will find our ordinary shares less attractive because we may rely on the exemptions and reduced disclosure obligations applicable to emerging growth companies. If some investors find our ordinary shares less attractive as a result, there may be a less active trading market for our ordinary shares and our share price may be more volatile.
If we fail to maintain an effective system of internal control over financial reporting, we may not be able to accurately report our financial results or prevent fraud. As a result, shareholders could lose confidence in our financial and other public reporting, which would harm our business and the trading price of our shares or ADSs.
Effective internal controls over financial reporting are necessary for us to provide reliable financial reports and, together with adequate disclosure controls and procedures, are designed to prevent fraud. Any failure to implement required new or improved controls, or difficulties encountered in their implementation could cause us to fail to meet our reporting obligations. In addition, any testing by us conducted in connection with Section 404 of the Sarbanes-Oxley Act, or any subsequent testing by our independent registered public accounting firm, may reveal deficiencies in our internal controls over financial reporting that are deemed to be material weaknesses or that may require prospective or retroactive changes to our financial statements or identify other areas for further attention or improvement.
Inadequate internal controls could also cause investors to lose confidence in our reported financial information, which could have a negative effect on the trading price of ADSs representing our shares or our shares.
Management is required to assess the effectiveness of our internal controls annually beginning with this annual report on Form 20-F to be filed with the SEC. However, for as long as we are an “emerging growth company” under the JOBS Act, our independent registered public accounting firm will not be required to attest to the effectiveness of our internal controls over financial reporting pursuant to Section 404 of the Sarbanes-Oxley Act. An independent assessment of the effectiveness of our internal controls could detect problems that our management’s assessment might not. Undetected material weaknesses in our internal controls could lead to financial statement restatements requiring us to incur the expense of remediation and could also result in an adverse reaction in the financial markets due to a loss of confidence in the reliability of our financial statements.
If we are classified as a passive foreign investment company, U.S. Holders of our ADSs may be subject to adverse U.S. federal income tax consequences.
For U.S. federal income tax purposes, we generally will be classified as a passive foreign investment company, or PFIC, for any taxable year, in which, after the application of certain look-through rules with respect to our subsidiaries, at least 75% of our gross income is passive income, or at least 50% of the average value (determined on the basis of a weighted quarterly average) of our assets for the taxable year is attributable to assets that produce passive income or are held for the production of passive income, including cash. For purposes of these tests, passive income includes, among other things, dividends, interest, gains from commodities and securities transactions, the excess of gains over losses from the disposition of assets which produce passive income (including amounts derived by reason of the temporary investment of funds raised in offerings of our ADSs) and rents and royalties other than rents and royalties which are received from unrelated parties in connection with the active conduct of a trade or business. Generally, in determining whether a non-U.S. corporation is a PFIC, a non-U.S. corporation that directly or indirectly owns at least 25% by value of the shares of another corporation is treated as holding and receiving directly its proportionate share of assets and income of such corporation.
Based upon our analysis of the value of our assets and the nature and composition of our income and assets, we believe that we were a PFIC for the taxable year ended December 31, 2023. However, the determination of whether or not we are a PFIC for any taxable year is a factual determination made annually after the end of each taxable year, and because the applicable law is subject to varying interpretations, we cannot provide any assurance regarding our PFIC status and our U.S. counsel expresses no opinion with respect to our PFIC status for any taxable year. If we are characterized as a PFIC for any taxable year during which U.S. Holders (as defined in “Item 10. E Taxation – Material U.S. Federal Income Tax Consequences for U.S. Holders” below) hold our ADSs, U.S. Holders of our ADSs may suffer adverse tax consequences regardless of whether we continue to qualify as a PFIC, including having gains realized on the sale of our ADSs treated as ordinary income rather than capital gain, the loss of the preferential rate applicable to dividends received on our ADSs by individuals who are U.S. Holders, and having interest charges apply to certain distributions by us and gains from the sales of our ADSs.
The tax consequences that would apply if we are classified as a PFIC would be different from those described above if a U.S. Holder of our ADSs were able to make a valid qualified electing fund, or QEF, election, or, in some circumstances, a “mark-to-market” election. We currently expect to provide U.S. Holders of our ADSs with the information necessary to make a QEF election if we were treated as a PFIC for any taxable year, although there is no assurance that we will do so.
For further discussion of the PFIC rules and the adverse U.S. federal income tax consequences in the event we are classified as a PFIC, as well as certain elections that may be available to U.S. shareholders, see “Item 10. E Taxation – Material U.S. Federal Income Tax Consequences for U.S. Holders.”
General Risks
We depend on our information technology systems, and any failure of these systems could harm our business. Security incidents, and other disruptions could compromise sensitive information related to our business or prevent us from accessing critical information and expose us to liability, which could adversely affect our business, results of operations and financial condition.
We collect and maintain information in digital and other forms that is necessary to conduct our business, and we are increasingly dependent on information technology systems and infrastructure to operate our business. In the ordinary course of our business, we may process large amounts of confidential and sensitive data, including personal data (such as health-related data), intellectual property, and proprietary business information (collectively, sensitive information). We have also outsourced elements of our information technology infrastructure, and as a result a number of third-party vendors may have access to sensitive information. Our ability to monitor these third parties’ information security practices is limited, and these third parties may not have adequate information security measures in place. We may share or receive sensitive information with or from third parties.
Cyberattacks, malicious internet-based activity, and online and offline fraud are prevalent and have generally increased as the number, intensity and sophistication of attempted attacks and intrusions from around the world have increased. These threats come from a variety of sources, including traditional computer “hackers,” threat actors, personnel (such as through theft or misuse), sophisticated nation states, and nation-state-supported actors. Some actors now engage and are expected to continue to engage in cyber-attacks, including without limitation nation-state actors for geopolitical reasons and in conjunction with military conflicts and defense activities. During times of war and other major conflicts, we and the third parties upon which we rely may be vulnerable to a heightened risk of these attacks, including cyber-attacks that could materially disrupt our systems and operations, supply chain, and ability to produce, sell and distribute our products. Further, cyberattacks include AI-driven attacks, bypassing multi-factor authentication, supply chain and critical infrastructure vulnerabilities, targeted ransomware campaigns, identity attacks, machine learning and generative AI usage, deepfake and synthetic media attacks, and increased focus on small and mid-market businesses.
We and the third parties upon which we rely may also be subject to a variety of evolving threats, including but not limited to social-engineering attacks (including through phishing attacks), malicious code (such as viruses and worms), malware (including as a result of advanced persistent threat intrusions), denial-of-service attacks (such as credential stuffing), ransomware attacks, supply-chain attacks, software bugs, server malfunctions, software or hardware failures, loss of data or other information technology assets, adware, telecommunications failures, and other similar threats. Ransomware attacks, including by organized criminal threat actors, nation-states, and nation-state-supported actors, are becoming increasingly prevalent and severe and can lead to significant interruptions in our operations, loss of data and income, reputational harm, and diversion of funds. Extortion payments may alleviate the negative impact of a ransomware attack, but we may be unwilling or unable to make such payments due to, for example, applicable laws or regulations prohibiting such payments. Similarly, supply-chain attacks have increased in frequency and severity, and we cannot guarantee that third parties and infrastructure in our supply chain or our third-party partners’ supply chains have not been compromised or that they do not contain exploitable defects or bugs that could result in a breach of or disruption to our information technology systems or the third-party information technology systems that support us and our business operations.
In addition, the prevalent use of mobile devices that access confidential information increases the risk of lost or stolen devices and security incidents, which could lead to the loss of sensitive information. Remote work has become more common and has increased risks to our information technology systems and data, as more of our employees utilize network connections, computers and devices outside our premises or network, including working at home, while in transit and in public locations. Additionally, future or past business transactions (such as acquisitions or integrations) could expose us to additional cybersecurity risks and vulnerabilities, as our systems could be negatively affected by vulnerabilities present in acquired or integrated entities’ systems and technologies. Furthermore, we may discover security issues that were not found during due diligence of such acquired or integrated entities, and it may be difficult to integrate companies into our information technology environment and security program.
Any of the previously identified or similar threats could cause a security incident or other interruption. A security incident or other interruption could result in unauthorized, unlawful, or accidental acquisition, modification, destruction, loss, alteration, encryption, disclosure of, or access to our sensitive information. A security incident or other interruption could disrupt our ability (and that of third parties upon whom we rely) to conduct our business. For example, the loss of clinical trial data from completed or ongoing or planned clinical trials could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data.
We may expend significant resources or modify our business activities (including our clinical trial activities) to try to protect against security incidents. Certain data privacy and security obligations may require us to implement and maintain specific security measures, industry-standard or reasonable security measures to protect our information technology systems and sensitive information.
While we have implemented security measures designed to protect against security incidents, there can be no assurance that these measures will be effective. We experience such security incidents of varying degrees from time to time, and we incur costs in protecting against or remediating such security incidents.
We take steps to detect and remediate vulnerabilities, but we may be unable in the future to detect vulnerabilities in our information technology systems because such threats and techniques change frequently, are often sophisticated in nature, and may not be detected until after a security incident has occurred. Despite our efforts to identify and remediate vulnerabilities, if any, in our information technology systems, our efforts may not be successful. These vulnerabilities pose material risks to our business. Further, we may experience delays in developing and deploying remedial measures designed to address any such identified vulnerabilities.
Applicable data privacy and security obligations may require us to notify relevant stakeholders of security incidents. Such disclosures are costly, and the disclosure or the failure to comply with such requirements could lead to adverse consequences. If we (or a third party upon whom we rely) experience a security incident or are perceived to have experienced a security incident, we may experience adverse consequences. These consequences may include governmental enforcement actions (for example, investigations, fines, penalties, audits, and inspections), additional reporting requirements and/or oversight, restrictions on processing sensitive information (including personal data), litigation (including class claims), indemnification obligations, negative publicity, reputational harm, monetary fund diversions, interruptions in our operations (including availability of data); financial loss; and other similar harms.
Our contracts may not contain limitations of liability, and even where they do, there can be no assurance that limitations of liability in our contracts are sufficient to protect us from liabilities, damages, or claims related to our data privacy and security obligations. We cannot be sure that our insurance coverage will be adequate or sufficient to protect us from or to mitigate liabilities arising out of our privacy and security practices, that such coverage will continue to be available on commercially reasonable terms or at all, or that such coverage will pay future claims.
If securities or industry analysts cease coverage of us, or publish inaccurate or unfavorable research about our business, the price of the ADSs and our trading volume could decline.
The trading market for the ADSs and our ordinary shares depends in part on the research and reports that securities or industry analysts publish about us or our business. If no or too few securities or industry analysts cover us, the trading price for the ADSs and our ordinary shares would likely be negatively affected. If one or more of the analysts who cover us downgrade the ADSs or our ordinary shares or publish inaccurate or unfavorable research about our business, the price of the ADSs and our ordinary shares would likely decline. If one or more of these analysts cease coverage of us or fail to publish reports on us regularly, demand for the ADSs and our ordinary shares could decrease, which might cause the price of the ADSs and our ordinary shares and trading volume to decline.
We may be subject to securities litigation, which is expensive and could divert management attention and adversely impact our business.
The market price of our ordinary shares has been and may continue to be volatile. Companies that have experienced volatility in the market price of their ordinary shares are often subject to securities class action litigation. For example, in July 2022, a securities class action complaint was filed in the U.S. District Court for the Southern District of New York against us, our directors and certain of our current and former executive officers. After several proceedings, on February 29, 2024, the court ordered the case closed. However, any future securities litigation could result in substantial costs and diversion of management’s attention and resources, which could adversely impact our business. Any adverse determination in litigation could also subject us to significant liabilities. See Part I, Item 8.A “Consolidated Statements and Other Financial Information – Legal Proceedings” for more information.
Item 4. Information on the Company.
A. History and Development of the Company
Our corporate name is Molecular Partners AG. We were incorporated in Switzerland as an Aktiengesellschaft, or AG, on November 22, 2004 and are subject to article 620 et seq. of the Swiss Code of Obligations. Our principal executive offices are located at Wagistrasse 14, 8952 Schlieren, Switzerland. We are registered with the commercial register of the Canton of Zurich under number CHE-112.115.136. In November 2014, we completed the initial public offering of our ordinary shares on the SIX Swiss Exchange. In June 2021, we completed the initial public offering of our ADSs on the Nasdaq Global Select Market. Our telephone number at our principal executive offices in Switzerland is +41 44 755 77 00.
Molecular Partners AG is the sole shareholder of Molecular Partners Inc., a Delaware corporation, with a registered office at 245 Main Street, Cambridge, Massachusetts 02142, with a secondary address at 33, Bradford Street, Concord, Massachusetts 01742. Molecular Partners Inc. is our agent for service of process in the United States.
Our website address is www.molecularpartners.com. The reference to our website is an inactive textual reference only and information contained in, or that can be accessed through, our website or any other website cited in this registration statement is not part of this Annual Report on Form 20-F. The SEC maintains a website at www.sec.gov that contains reports, proxy and information statements, and other information regarding issuers that file electronically with the SEC.
B. Business Overview
We are a clinical-stage biotechnology company pioneering the design and development of DARPin therapeutics for medical challenges that other drug modalities cannot readily address. We have programs in various stages of preclinical and clinical development, currently with main focus on oncology. DARPin (Designed Ankyrin Repeat Protein) therapeutics are a new class of custom-built protein drugs based on natural binding proteins that have the potential to unlock new dimensions of multi-functionality and multi-target specificity in drug design. Our DARPin platform allows us to generate candidates with multiple mechanisms of action, or MoAs, - such as immune cell engagers and radiotherapy - to address complex biological problems. We believe the DARPins' flexible architecture, intrinsic potential for high affinity and specificity, small size and high stability offer benefits to drug design over other currently available protein-based therapeutics.
Our DARPin candidates have been extensively tested in preclinical studies and clinical trials, including in more than 2,500 patients, and have been observed to be highly active and generally well-tolerated.
Leveraging our DARPin platform, we have designed product candidates with multiple MoAs that we believe have the potential to offer patients therapeutic options with higher efficacy and fewer adverse events as compared to the current standard of care. Among these multiple MoAs, DARPin product candidates have been designed to block growth factors, localize activity, conditionally activate immune cells, deliver cytotoxic payloads and radionuclides, neutralize viruses, adjust half-life as needed, and initiate cell death. We apply these features across our portfolio to elicit a specific therapeutic response.
We believe that our DARPin platform has the potential to yield novel product candidates with broad therapeutic applications given their ability to overcome many of the limitations of antibody and other conventional protein-based therapeutics. By harnessing DARPins’ intrinsic advantages and leveraging our two decades of experience and leadership with DARPins, we believe our DARPin platform can close the gap between small molecule and antibody medicines as a new therapeutic modality poised to offer clinical breakthroughs.
Our Pipeline
We believe our DARPin therapies have the potential to address defined medical problems that are not addressable by other drug classes. We currently remain focused on oncology through our robust pipeline of clinical and preclinical programs, with particular attention to MP0533 for the treatment of acute myeloid leukemia, or AML, the Radio-DARPin Therapy, or RDT, platform and pipeline, and the multispecific cKIT x CD16a x CD47 Switch-DARPin program and other next-generation immune cell engagers leveraging the Switch-DARPin platform.
Our pipeline chart as of March 2024 is illustrated below:
*The co-development agreement with Orano Med includes up to three potential oncology targets including DLL3.
While our DARPin candidates have distinct therapeutic features and particular targets, each DARPin therapeutic modality can be utilized across multiple programs. Our pipeline programs benefit from the learnings of earlier discoveries, such as:
•The use of fibroblast activation protein, or FAP, as a localized activator for MP0317, and previously MP0310;
•Multi-specificity and avidity-driven selectivity to boost tumor specificity for our candidate MP0533, a tetra-specific T cell engager for AML;
•"Stealth DARPins" whereby the DARPin backbone surface is engineering and which are used in our Radio-DARPin programs, allowing for reduced kidney accumulation; and
•Switch-DARPins, that combine two distinct target specificities on a single DARPin domain, resulting in targeted and conditional effector functions and safer systemic delivery, as seen in our cKIT x CD16a x CD47 multispecific program as next generation conditioning regiment for HSCT (hematopoietic stem cell transplantation, or HSCT).
We have strategic collaboration agreements with Novartis, Orano Med, and other third party collaborators.
Our Team
We were founded in 2004 by the inventors of our DARPin platform. Our senior management, which includes two of our company’s co-founders, have significant prior experience in oncology, research, drug development and finance. Members of our team have served as senior executives at other well-established companies including Amgen, Bavarian Nordic, GSK, Genentech, J&J, Novartis, Roche and Tesaro. Additionally, our board of directors includes current and former senior executives of AbbVie, Biogen, Novartis, Roche and Takeda (Millennium Pharmaceuticals, Shire).
As our name indicates, partnership and collaboration are at the core of our company, our research activities and our therapeutic designs. Molecular Partners embodies an international working environment comprised of over 165 individuals from numerous disciplines who contribute to our shared values of scientific excellence, respectful teamwork and personal aspiration. Stemming from our long-standing goal of improving the lives of patients with cancer, we are a group dedicated to moving the needle of medicine. We foster true innovation and creative thinking to advance our therapeutic product candidates, and we continue to be inspired by the difference we can make for our patients. Our team members possess a curiosity and a passion to advance our shared goal of providing better treatment options for patients with serious diseases.
Our Strategy
We are committed to leveraging our proprietary platform to design DARPin-unique solutions for challenges other therapies cannot readily address. DARPins have several intrinsic properties that differentiate them from other modalities. We combine these unique properties with insights from our deep clinical experience and understanding of underlying disease biology to create molecules that offer new solutions to patients with high medical need and that can be tested in preclinical models with translatable value. Key aspects of our strategy include the following:
a.Rapidly advance our tetra-specific T-cell engaging DARPin, MP0533, for the treatment of patients with AML and high-risk MDS. Presently in a Phase 1/2a clinical trial, we plan to establish the safety and tolerability profile of the program, and aim to identify a therapeutic dose level for additional clinical evaluation. Once a dose level is identified, we intend to quickly expand our clinical trials to evaluate which patients may derive the most benefit from MP0533.
b.Unlock novel biological solutions and expand therapeutic applications of clinically validated DARPin approaches. As the inventors of the DARPin class of drugs, we believe we are the world leaders in DARPin engineering and research. With this expertise, we have developed a strategy of unlocking various technical hurdles which may limit other discovery platforms, and then expanding our clinical product candidates based upon each technological solution. Examples of this include the tumor-localized activation of our oncology program MP0317, the avidity-driven mechanism of MP0533, the "Stealth DARPin" which we invented to reduce kidney accumulation of our radio-pharmaceutical programs, and the Switch-DARPin which enables a logic-gated, conditional activation of immunity.
c.Continue to optimize and expand our pipeline of Radio-DARPin Therapeutics (RDTs). Significant efforts have been put into demonstrating both reduced kidney accumulation, and increased tumor accumulation of our RDTs. These platform learnings will allow us to explore multiple tumor targets not readily addressable by other therapeutic platforms. This is intended to be demonstrated by our DLL3 program, as well as other targets which we plan to introduce in 2024 and the coming years.
d.Continue a strategic approach to in-house versus partnered development. To unlock and expand the full potential of our DARPin platform, we intend to independently develop and commercialize product candidates in our core focus areas, where we believe we have an established clinical and regulatory approval pathway and the resources to commercialize successfully. To complement this approach, we also plan to collaborate with biopharmaceutical companies on product candidates that have promising utility in target areas or patient populations requiring greater global development capabilities or those outside of our strategic focus.
i.This strategy has allowed us to pursue major therapeutic innovations for the DARPin platform, often in parallel, across our oncology and virology focus areas. To this end, we continue to support our partners across our portfolio as we advance our Radio DARPin-Therapy Platform, both independently and in collaboration with Novartis and Orano Med; and our research collaboration with the University of Bern for MP0533.
ii.We will also seek to collaborate with companies developing complementary technology to our platform when we see the strategic rationale to combine our industry-leading DARPin capabilities with other modalities. This is shown in our partnership with Orano Med. Both MP and Orano Med have agreed to co-develop multiple 212Pb-based Radio-DARPin Therapies using Orano Med's capabilities in Targeted Alpha Therapy as the cell killing isotope to attack solid tumors. The DLL3 program is the first target in the Orano Med collaboration.
Our DARPin Platform
Our DARPin platform was invented over twenty years ago by the co-founders of our company, who were then researchers at the University of Zurich. DARPin molecules were discovered as a result of our co-founders’ quest to find a versatile protein-based therapeutic class that was highly differentiated from antibodies. The ability to design multi-specific molecules, along with the ease of use and manufacturing, made the DARPin technology an ideal platform for us from which to develop treatments beyond traditional protein therapeutics. The foundational technology we use in our DARPin platform to generate our product candidates was initially licensed to us by the University of Zurich. Leveraging our DARPin platform, we have designed product candidates with MoAs that we believe have the potential to offer therapeutic options to patients with higher efficacy and fewer adverse events compared to the current standard of care. Among these multiple MoAs, DARPin product candidates have been designed to block growth factors, localize activity, conditionally activate, neutralize viruses, adjust half-life as needed and initiate cell death. We apply these features across our portfolio to elicit a specific therapeutic response.
For more than two decades, we have pioneered DARPins as a new class of therapeutics, evolving our capabilities and mastery of DARPin design with an increasing focus on novel platforms and MoAs that are highly differentiated from other drug classes. The intrinsic advantages of DARPins include:
•Derivation from natural binding proteins:
•DARPins are based on natural protein binders that mediate protein interactions in most living cells: ankyrin repeat domains. Evolved by nature and engineered by us, we believe ankyrin repeat domains are the ideal foundation for an efficient, versatile and innovative approach to biologic drug design. An individual DARPin is a radically simple unit consisting of a robust backbone, or scaffold, supporting a binding surface that is shaped to bind its target with exquisite precision and strength. Unlike larger, more complex binding proteins, the basic repeating unit can be engineered against a vast array of different targets with very low risk of off-target effects or interactions outside the binding surface.
•High affinity and specificity:
◦DARPin’s intrinsic potential for high affinity and high specificity mean DARPin candidates can tightly bind to their targets. This binding strength is matched by the specificity of DARPins to bind only to the intended target, limiting the potential of off-target effects.
• Small size:
◦Even when linked together, multispecific DARPins are smaller than large proteins such as antibodies, which allows a potentially greater tissue penetration. Additionally, every dose given to a patient contains more molecules per gram than larger molecules like antibodies.
•Multispecificity:
◦DARPins can be used in a radically simple format with single-target specificity or can be easily be linked together to enable multispecific drug candidates. DARPin candidates comprised of up to six DARPins and five target specificities have been tested in the clinic without impacting affinity, potency, stability, or production yields compared to the single DARPin units.
•“Either-or” specificity:
◦The repeat structure of DARPins allows to fuse two different DARPins with different target specificities into one DARPin domain thereby enabling mutually exclusive, “Either-Or” binding properties for either of the targets. We believe this opens the possibility of creating “smart drugs” that are conditionally activated only where activity is desired.
•High stability:
◦The very high stability intrinsic to DARPins allows novel engineering approaches, such as those applied to the DARPin backbone surface enabling the Stealth-DARPin design developed for RDT, without impact on the structure and binding characteristics of the engineered DARPins.
We believe that our DARPin platform has the potential to yield novel product candidates with broad therapeutic application given their ability to overcome many of the limitations of antibody and other conventional protein-based therapeutics.
Benefits and Advantages of our DARPin Platform over Traditional Approaches
We believe the benefits of our DARPin platform include:
•Ability of DARPin product candidates to target multiple escape pathways in parallel When cancer cells or a virus are targeted by conventional therapies, they often develop resistance by loss of target, mutational escape or activating multiple escape pathways at once. To create effective products, we believe that we must understand the dynamics of these escape pathways and then target their key components in parallel. We believe our DARPin product candidates are ideally suited for this approach because of their ability to bind to multiple targets and inhibit multiple escape pathways at once. Our approach allows us to efficiently test product candidates to determine the affinity and target binding of our DARPin proteins in the relevant setting. The most effective combination of DARPin proteins is assembled into one DARPin product candidate for further product development. These DARPin product candidates can demonstrate cooperative binding, leading to high potency and preventing escape.
•Capacity to find and address new biology on known targets
Using our DARPin approach, we are able to select DARPin proteins that bind to known targets in novel ways, thereby unlocking additional therapeutic solutions. For example, we can achieve conditional activation where the molecule will activate only in the presence of a particular tumor-associated antigen, or TAA. One of our product candidates, MP0533 utilizes the power of multi-specific targeting to potentially enhance efficacy and minimize tumor resistance through simultaneously targeting three known antigens associated with AML, which, to our knowledge, had not previously been addressed in one molecule. In addition, the RDT platform highlights the potential to use DARPins specifically tailored for use with radio-isotopes, can potentially expand the universe of targets for the treatment of cancer with radiotherapy.
•Flexible architecture to engage and locally activate immune cells
Immune-mediated therapies rely on the activation of a patient’s immune response to fight tumors. In some cases, blocking negative checkpoint signals can produce a deep and durable effect in stopping the growth of, and regressing, tumors. We believe that our DARPin platform is well suited for the combined approaches of blocking negative checkpoint signals and engaging and activating immune cells. We have unlocked approaches that utilize DARPins to direct tumor-localized activation of immune cells, resulting in the selective activation of immune cells within a tumor, which may potentially avoid systemic adverse events. Previously, we have designed two of our DARPin product candidates, MP0310 and MP0317, to cluster, thereby locally activating immune cells more effectively. MP0310 is a tumor-restricted 4-1BB immune-cell activator for the potential treatment of FAP-positive cancers, and MP0317 activates CD40, also in an FAP-dependent manner. Our current Phase 1 program for the treatment of AML and MDS, MP0533, has shown initial clinical activity with our first T-cell engaging CD3 DARPin. Since demonstrating this initial activity, the use of CD3 DARPins in future programs could prove highly useful in treating cancers. Additionally, our Switch-DARPin platform uses a dual-binding logic-gated DARPin (i.e. the Switch) to provide an "on/off" function to the multispecific DARPin candidate. The Switch function is modulated according to the presence of defined targets as well as their relative proximity and affinity to the Switch, thereby allowing conditional activation of targets. The goal of the Switch-DARPin platform is the activation of a highly specific targeted immune response.
•High stability and microbial manufacturing to enable high yields at scale
All or our DARPin product candidates are constructed to benefit from high-yield microbial manufacturing. Unlike manufacturing using mammalian cell lines, productions of DARPin molecules via microbial manufacturing allow for several key competitive advantages, including the ability to manufacture clinical batches every seven to ten days, versus a thirty-day mammalian campaign. This advantage is critical to allow drug supply on a global scale. Additional benefits include high production yield of raw drug substance, 12-15g/L for example, as well as high thermal stability, with certain programs demonstrating shelf stability at 4 degrees centigrade for several years.
Background of Our DARPin Platform: A Source of Virtually Unlimited Binding Proteins
The fundamental building block for all of our DARPin product candidates is the single DARPin protein. A DARPin protein consists of an engineered protein base structure, which we refer to as the scaffold. The DARPin scaffold is formed from consecutive copies of ankyrin repeat proteins, which are chains of 33 amino acids stacked together. The scaffold can be generated to provide a binding site to specifically recognize, or permit binding to, a desired target protein or other molecule, similar to how monoclonal antibodies can be generated to recognize a single target antigen.
We have developed and upgraded our libraries to include over one trillion DARPin proteins, each of which can potentially bind to a specific target structure. From this library we can screen and select DARPins within weeks that are highly specific and have high affinity for any given target structure. We use these selected DARPins to build our product candidates.
DARPins are small, with a molecular weight of approximately 14–18 kilodaltons, or approximately the tenth of the size of a monoclonal antibody. We believe this smaller size enables increased tissue penetration and a higher potency at lower doses. The natural biophysical properties of DARPins, including high affinity due to the rigidity of the scaffold and high solubility of the base structure, enable more distinct specificity for a particular target, or a specific site on a particular target, such as an epitope. These benefits have the potential to increase activity and efficacy of our product candidates for their targets.
How We Use DARPins
We can select DARPins to bind to a given target and form the basis of a product candidate, or we can genetically assemble DARPins into DARPin product candidates using different linkers. This allows us to screen our libraries that contain over one trillion DARPins to select those with the optimal properties. We believe this process is more difficult with multi-specific antibodies or other complex proteins. Further, we can add additional elements either to increase the half-life of our product candidates to match the therapeutic need or to add functionality. While antibodies generally have a long systemic half-life, most repeat proteins have a short half-life. The half-life of a single DARPin is usually a few hours when injected into the blood stream. To increase the half-life of DARPin product candidates, we have created multiple proprietary, patent protected, specific DARPins that bind to human serum albumin, or HSA. HSA is the most abundant protein in human blood and has a half-life of approximately three weeks. When administered intravenously, the HSA-DARPin binds to its target to extend its half-life to the same period as HSA. This approach allows us to tailor the half-life of our individual product candidates.
Our accumulated preclinical and clinical experience of developing and testing DARPin candidates has allowed us to establish an intellectual property portfolio that, as of December 31, 2023, included over 200 granted patents and over 200 additional pending U.S. and foreign patent applications across more than 30 patent families, covering both core and derivative aspects of our DARPin platform.
Our Oncology Programs
Cancer Background and Treatment
The rapid development of immune-mediated therapies for multiple types of cancer has transformed the oncology treatment landscape and improved the long-term outlook for many cancer patients. Rather than targeting treatments directly at the tumor, these therapies generally engage the immune system to promote its recognition and eradication of tumor cells. Key features of immune-mediated therapy include specificity, breadth of response, and memory. These features can contribute to complete tumor regressions, often providing more durable clinical outcomes and improved quality of life relative to other therapies. However, despite the early success observed with immune mediated therapies, it has become clear that these treatments can currently help only a minority of patients and are more effective in some tumor types than others. This limit arises from various factors, including differential target expression patterns by cancer cells (and expression thereof on healthy cells), variable immune responses to the treatment, and cancer immune-escape via mutagenesis and proliferation of non-targeted cellular populations and an immune-suppressive tumor microenvironment.
We believe that, through years of building our DARPin expertise, we have developed DARPin candidates that can unlock and expand these immune-mediated capabilities through several mechanisms, which include targeting immuno-stimulatory proteins through multi-specific DARPin candidates and using delayed and/or conditional activation of our immune engagers. These attributes allow us to optimize the potency, localization and/or exposure of our product candidates and reduce the risk of off-target toxicity in order to improve their therapeutic index and overall profile.
Tumor-Localized Immune Activation: MP0317 and MP0533 Product Candidates
We are currently developing and assessing two oncology product candidates which activate immune cells at the tumor site:
•MP0533, which targets CD3, CD70, CD123 and CD33 for the treatment of AML and MDS;
•MP0317, which allows for tumor-restricted immune-cell CD40 activation for the treatment of FAP positive cancers;
Development of our MP0317 and MP0533 product candidates has leveraged the learnings from our first-generation product candidates in our oncology program, MP0250, MP0274 and MP0310. Those candidates have shown efficacy and tolerability in preclinical studies and clinical trials in patient populations who were resistant and /or refractory to previous standard of care treatments.
However, MP0317 and MP0533 both utilize novel mechanisms of action, which may result in greater research and development expenses, regulatory issues that could delay or prevent approval, or the discovery of unknown or unanticipated adverse effects. See “Risk Factors—Risks Related to the Development and Clinical Testing of Our Product Candidates—Some of our product candidates utilize a novel mechanism of action which may result in greater research and development expenses, regulatory issues that could delay or prevent approval, or discovery of unknown or unanticipated adverse effects.”
MP0533: DARPin Product Candidate Targeting CD3, CD70, CD123 and CD33 for the Treatment of AML (r/r AML and AML/MDS)
MP0533 is our novel tetra-specific T cell-engaging DARPin, which simultaneously targets the antigens CD33, CD123 and CD70 on AML cells as well as the immune activator CD3 on T cells.
The unmet medical need in AML remains high. Despite the achievement of remission for a majority of patients, up to 70% of adults and 30% of children will not survive beyond five years after initial clinical response due to relapsing disease. Further, the treatment of relapsed/refractory AML, or r/r AML, is therapeutically challenging due to high relapse rates with current standard of care treatments and the aggressive nature of the disease. Currently, a variety of highly potent mono-targeting TCE (T cell engager) and CAR-T therapies have entered clinical development, but those therapies are often accompanied by dose limiting toxicities, such as cytokine release syndrome, or CRS, and myelotoxicities, preventing dose escalation to induce robust anti-tumor efficacy. More selective therapies addressing the growing number of subclasses and rationally designed target combinations are needed to allow for extended dose escalation with a more acceptable safety profile and to achieve more durable responses.
In AML, leukemic stem cells, or LSCs, produce all the leukemic cells in the patient and therefore a lasting cure for this disease is dependent on eradication of these cells. However, LSCs are relatively resistant to standard therapies. For example, these cells are less sensitive to killing by daunorubicin and cytarabine, two commonly used chemotherapeutic agents. This is partially due to increased expression by LSCs of multidrug resistance genes, and also to their quiescent state, which reduces the effects of cytotoxic agents that target rapidly replicating cells. It is therefore essential to target LSCs, in addition to AML blasts, to achieve durable disease control.
Some cancer antigens are also present on many healthy cells, but at a lower density, and as such it is difficult to select any single target to sufficiently differentiate between cancer cells and healthy tissue. To overcome this limitation and increase specificity, we leveraged our unique DARPin platform to generate a multi-specific T-cell engager (TCE) DARPin molecule, targeting CD33, CD70 and CD123, by a fine-tuned and tailored avidity-driven affinity to these TAAs, in conjunction with our CD3-binding DARPin molecule.
In avidity-driven selectivity, the presence of two or more binding targets on the cell, and the molecular interaction with these targets increases the effective concentration of the binder and the resulting binding strength. This dependency of binding strength on the presence of more than one cancer antigen conveys a far superior selectivity to these multi-specific binders. This approach is a concept that is well known in the scientific community but has so far been limited by the availability of an optimal therapeutic platform to address the associated technical challenges. In order to find the right target combination, the optimal affinity to increase tumor specificity via avidity, as well as the best molecular architecture, we took advantage of our unique modular DARPin platform and screened thousands of combinations of multi-specific DARPin molecules, binding simultaneously to the three different TAAs — CD33, CD70 and CD123. We combined our three TAA-binding DARPins with our CD3-binding T-cell engaging DARPin into our candidate, MP0533.
Our approach allows for the design of multi-specific TCEs which are designed to simultaneously target CD33, CD70 and CD123, three well-known AML antigens that are co-expressed on approximately 50% of AML cells and of which at least two are expressed on approximately 70% of AML cells. Healthy cells generally express only one or none of these TAAs. To further optimize our molecules, we have devised a concentration dependent MoA utilizing moderate affinity binders rather than high affinity ones. When such a DARPin encounters a cell expressing only one antigen, we believe there should only be a transient interaction and the DARPin should quickly disengage the target with limited cytotoxic effect. However, when there are two or three targets, the mechanism of avidity driven selectivity is activated.
In preclinical tests against AML cells, we observed MP0533 delivered highly potent and specific activity and the potential for a reduced effect on healthy normal cells. As well as its increased selectivity, MP0533's ability to target three TAAs simultaneously gives it additional potential to counteract target escape mechanisms expected due to tumor heterogeneity. In addition, this mechanism is designed to capture a larger population of AML patients due to its ability to engage with any two of these targets simultaneously, while maintaining specificity.
In December 2021, we announced a research collaboration with the University of Bern, to advance the development of MP0533, into the clinic. The collaboration aims to leverage our DARPin technology and the University of Bern group’s expertise in AML, and specifically in LSCs.
In an oral presentation at the 64th American Society of Hematology (ASH) Annual Meeting in December 2022, we presented preclinical results showing MP0533 can induce preferential killing of cells expressing two or three tumor-associated antigens, or TAAs, compared to cells expressing a single TAA.
In January 2023, the first patient was dosed in our Phase 1/2a clinical trial of MP0533 for patients with r/r AML and AML/MDS. In December 2023, we presented positive initial data from the first four dosing cohorts at the 65th American Society of Hematology (ASH Annual Meeting and Exposition. The results from the first 11 patients treated with MP0533 indicated an acceptable safety profile as of the data cut-off across all four dosing regimens, or DRs, with no dose-limiting toxicities observed. The most frequently reported MP0533-related adverse events were infusion-related reactions and CRS (all Grade 1-2). Two responders were observed at the time of presentation, including a patient achieving complete response (CR) in DR 4 and another patient with morphological leukemia-free state (MLFS) in DR 3. These responses are particularly notable for having occurred at dose levels below those predicted as therapeutically active.
The Phase 1/2a clinical trial is currently on track with dosing in DR 6 currently ongoing. We expect to present data from further cohorts receiving MP0533 in the first half of 2024. Based upon current safety and tolerability data from the ongoing trial as well as discussions with treating investigators and key opinion leaders, we intend to file a protocol amendment to expand enrollment to additional higher dose cohorts of MP0533 beyond the initially planned highest cohort (DR 7). The goal of the additional higher doses will be to explore the full potential efficacy of MP0533. We expect to enroll patients in the added higher cohorts in the second half of 2024.
MP0317: DARPin Product Candidate Targeting FAP x CD40
•Designed to activate CD40 only in FAP-high tumor tissue.
•Localized activation by FAP targeting underpins the therapeutic benefits while expanding the range of immune cell activation.
•Designed to reinforce the effect of other immune stimulating therapies.
•Positive interim Phase 1 clinical trial data presented at ASCO in the second quarter of 2023.
•Additional positive dose-escalation data from the Phase 1 clinical trial in patients with advanced solid tumors presented at the Society for Immunotherapy of Cancer, or SITC, in the fourth quarter of 2023.
The tumor-localized immune agonist MP0317 is one of our product candidates currently being developed in our oncology pipeline. MP0317 comprises a localizer to FAP and immune stimulator binding to CD40. FAP is found in the tumor stroma in high density and its binding is intended to create a cluster of CD40 on immune cells enabling immune activation.
MP0317 is designed to simultaneously engage FAP and CD40 to create tightly bound clusters around tumors, which are necessary to induce CD40-mediated local immune activation. CD40 plays a critical role in antigen presentation and the monocyte maturation process, and therefore, indirectly, T-cell activation. One of the main functions of CD40 signaling is to enhance antigen-presentation to T-cells by activating dendritic cells, or DCs. CD40 engagement on the surface of DCs promotes cytokine and chemokine production, induces expression of costimulatory molecules, and facilitates the cross-presentation of antigens. This step increases the interaction of DCs with T-cells by upregulating surface proteins such as CD54 and CD86, thereby activating the surface proteins.
Agonist anti-CD40 antibody treatments have been associated with mild to moderate toxicity in the clinic, which we believe is related to on-target but off tumor effects causing CRS and liver toxicity.
Aiming to avoid CD40-related toxicity, we developed MP0317 to work as a locally activated CD40 engager, designed to only activate the immune system when both FAP and CD40 are simultaneously engaged. We expect this localizing mechanism to reduce the likelihood of extra-tumoral systemic side effects and allow an increase of the therapeutic index.
In November 2021, we announced the first patient had been dosed in our Phase 1 clinical trial evaluating the safety and tolerability of MP0317. The open-label dose escalation study is designed to assess the safety and tolerability as well as pharmacokinetics and pharmacodynamics of MP0317 as a monotherapy in patients with solid tumors known to express fibroblast activation protein (FAP) and CD40.
In November 2023, we presented additional positive dose-escalation data from the Phase 1 clinical trial of MP0317 in patients with advanced solid tumors at the 38th Annual Meeting of SITC. These data from 46 patients supported our earlier reported findings at the 2023 ASCO Annual Meeting of MP0317-induced CD40 activation and related remodeling of the tumor microenvironment. At the time of the presentation, MP0317, as a monotherapy, continued to display a favorable safety profile across all dosing cohorts up to the highest planned dose (0.03-10 mg/kg in every 3-weeks or weekly schedules).
Enrollment in the Phase 1 clinical trial of MP0317 has concluded. We expect to report the full dataset from the Phase 1 dose-escalation trial in the first half of 2024.
Our Radio DARPin Therapy (RDT) Platform
Radiation therapy, particularly external beam radiation, is a frequently used approach to treating cancer. Due to its limited selectivity, this treatment can only be considered for localized or oligometastatic disease: radiation therapy often affects healthy tissues resulting in harmful side effects, which limits the amount of radioactivity that can be given to a patient. As a consequence, hard-to-reach tumor lesions or micrometastases are left untreated resulting in progression or relapse of the disease. Targeted radiotherapies delivering radioisotope selectively to the tumor while sparing healthy tissues have continue to make significant progress with positive clinical results. A key limiting factor in expanding this treatment modality to a broad range of relevant cancer types is the lack of vectors matching targeted radiotherapy requirements and spanning a broad tumor target space.
Our Radio-DARPin Therapy (RDT) platform represents a unique targeting approach for highly effective and selective delivery of radioactive payloads to a broad range of tumors while sparing healthy tissues. The unique nature of DARPins as an engineered protein drug class may allow us to overcome the limitations of other radioligand therapies. DARPins have great intrinsic properties as vector – such as small size, high affinity and specificity – to enable robust, tumor-specific delivery of therapeutic radionuclides.
We have built on these innate advantages by making further engineering advancements across our RDT portfolio. We have designed our candidates to minimize kidney retention, one of the key challenges of the broader radiotherapy class, through our use of Stealth-DARPins— DARPins whose backbone is surface engineered to be excreted by kidneys in urine instead of being re-absorbed. These results were presented at several conferences in 2023 including AACR and at SNMMI. Building on these results, we established a half-life engineering (HLE) toolbox which led to increased tumor uptake across multiple tumor targets, which we presented at EANM 2023 and at the J.P. Morgan Healthcare Conference in January 2024.
Taken together, our half-life engineering (HLE) toolbox combined with Stealth-DARPin technology has enabled us to reach improved tumor uptake and reduced kidney reabsorption, which supports expansion of the RDT pipeline and the development of a first generation of RDT pipeline candidates, including for Delta-like ligand 3 (DLL3). We presented relevant data on our DLL3 RDT program in January 2024.
In December 2021, we announced a new collaboration with Novartis in the form of a license and collaboration agreement to develop, manufacture and commercialize DARPin-based radiotherapeutics. By harnessing the power of radioactive atoms, or radionuclides, and applying it to cancers through targeted radioligand therapy, DARPin-based radiotherapeutics have the potential to selectively deliver molecularly targeted radiation to tumor cells anywhere in the body, while sparing healthy tissue. DARPins have significant potential to enable robust, tumor-specific delivery of radionuclides owing to their small size in combination with high specificity and affinity.
The collaboration combines our industry-leading ability to rapidly generate high-affinity DARPins and the radioligand therapies, or RLT, capabilities and expertise of Novartis. Under the terms of the agreement, we will collaborate with Novartis to discover DARPin-based radiotherapeutic candidates that target specific tumor associated antigens.
Novartis is responsible for all clinical development and commercialization activities. Under the terms of the agreement, we received a $20 million upfront payment from Novartis in January 2022, and are entitled to total potential development, regulatory and commercialization milestone payments of up to $560 million based on future achievements, and up to low double-digit percent of royalties to the extent that sales occur.
Furthermore, in January 2024, we entered a strategic collaboration with Orano Med to co-develop 212Pb-based RDTs for patients with solid tumors. The deal combines the power of DARPins, as a highly differentiated modality for tumor-targeted delivery of radioisotopes, with Orano Med’s leading capabilities in Targeted Alpha Therapy and supply to further advance our RDT platform and expand our RDT portfolio. 212-Pb has ideal properties for radiotherapeutic applications: a very clean decay chain, which releases one high-energy alpha particle, and relatively short decay half-life of 10.6 h, which ensures that the majority of the radioactivity is deposited at the tumor site resulting in efficient cancer cell killing. The short half-life is also beneficial for waste management.
The tumor-associated protein Delta-like ligand 3 (DLL3) was selected as the target of our lead RDT program to be advanced into IND-enabling studies in the first half of 2024. Expression of DLL3 is low in healthy tissues but significantly increased in certain tumor types, providing an opportunity for selective targeting through the high affinity and specificity offered by DARPins.
The initiation of clinical studies and first-in-human data are expected in 2025 through our co-development agreement with Orano Med. We also expect to nominate additional targets and RDT candidates in 2024.
Switch-DARPin Platform
Our Switch-DARPin platform represents a further evolution of our capabilities to deliver multispecific candidates to address different disease needs. It uses a dual-binding logic-gated DARPin (the Switch) to provides an "on/off" function to the multispecific DARPin candidate. The Switch’s function is modulated according to the presence of defined antigens as well as their relative proximity and affinity to the Switch. The goal of the Switch-DARPin platform is the activation of a targeted immune response.
In 2023, we debuted our Switch-DARPin platform and presented data supporting its mechanism of action at PEGS 2023. In January 2024, we introduced the first multi-specific program from the platform targeting the proteins cKIT, CD16a and CD47.
The multispecific cKIT x CD16a x CD47 Switch-DARPin program is designed to induce exhaustive killing of stem cells that express cKIT to optimize the outcomes of current high- or reduced-intensity conditioning and HSCT for AML patients. The program thereby strives to further increase long-term disease control post HSCT for AML patients eligible for high-intensity induction therapy, including those with a poor cytogenetic risk profile. It may also provide an alternative approach with a better safety profile for patients currently not eligible for standard high-intensity conditioning.
The intent is to extend the access to potentially curative HSCT for more patients with AML and beyond, including the potential to treat patients with severe autoimmune disease and those receiving certain cell-based therapies.
The target-by-target rationale for this program’s design is:
•cKIT is critical for stem cell maintenance and renewal and thus expressed on both hematopoietic and leukemic stem cells.
•The CD16a DARPin engages NK cells and macrophages to selectively kill cKIT-positive cells.
•The Switch-DARPin conditionally blocks the CD47 “don’t eat me” signal in the presence of cKIT, leveraging the power of CD47 inhibition without its associated toxicity to healthy cells.
We expect to present initial preclinical data from the first Switch-DARPin program in the first half of 2024 and to run preclinical proof-of-concept studies in the second half of 2024, which should provide strong translational efficacy data.
Other Programs
Our continued expansion of our capabilities and those of our DARPin candidates is due in part to our deep clinical experience with DARPin programs, across development stages through to the registrational phase. Our work today is informed by the past development of abicipar, a product candidate for the treatment of neovascular age-related macular degeneration and Diabetic Macular Edema; ensovibep, our trispecific candidate for COVID-19; and MP0310, which we designed to target both localizing target fibroblast activation protein (FAP) on tumor cells and 4-1BB, an immune modulatory protein on T cells. All programs showed activity and an acceptable safety profile in the clinic. These programs are no longer in active development.
In January 2024, Novartis returned the rights to the ensovibep program, previously under investigation for the treatment of COVID-19, to Molecular Partners. Clinical work on the ensovibep program ended in 2022 and the program remains terminated.
Intellectual Property
Our success depends in part on our ability to obtain, maintain, enforce and defend patents and other intellectual property and proprietary protection for our product candidates and technology, to preserve the confidentiality of our trade secrets, to operate without infringing, misappropriating or otherwise violating patents and other proprietary rights of others, and to prevent others from infringing, misappropriating or otherwise violating our patent and other proprietary rights. We seek to protect our proprietary position by, among other methods, filing patent applications covering our proprietary technology, improvements thereof, product candidates, and other inventions in Europe, the United States, and Japan, as well as in other relevant jurisdictions that are important to the development of our business, including Australia, Canada, South Korea and China. To protect aspects of our business that are not amenable to, or that we do not consider appropriate for, patent protection, we rely on trade secrets, know-how, confidential information and continuing technological innovation. We also rely on in-licensing opportunities to develop and maintain our proprietary position. We may further rely on statutory market exclusivity and patent term extensions that may be available for our product candidates once they achieve regulatory approval.
We maintain three categories of patent protection for, respectively, our DARPin platform, key single-binding domain DARPin proteins binding to specific targets and our DARPin product candidates. The first category of protection covers our DARPin platform:
•In an effort to stay a leader in the field of repeat protein technology, we have continued to work on improving the basic ankyrin repeat protein technology and have filed patent applications covering these improvements. Furthermore, we have enhanced our efforts to innovate in the ankyrin repeat protein field, including by developing new molecular designs, by generating ankyrin repeat proteins with novel modes of action, and by applying the ankyrin repeat protein technology to new disease areas and new target classes. We have translated this enhanced innovation into the generation of new intellectual property and have expanded our patent portfolio in the last couple of years. Taken together, we have made progress in protecting improvements of the DARPin base technology and innovative new aspects and applications of the DARPin technology in newly filed patent applications. However, we can provide no assurances that any such patent applications will be issued as patents.
•One example of a patent family that we own in this category is based on international patent application WO 2012/069655, relating to DARPin binding proteins comprising certain improved N-terminal capping modules. As of December 31, 2023, we owned three issued U.S. patents, nine issued foreign patents (i.e. patents granted in jurisdictions other than the U.S.) and one pending foreign patent application in this family. Any issued patents in this family are expected to expire in 2031. The disclosed improvement of the DARPin platform is included, for example, in our DARPin product candidates MP0310, MP0317 and MP0533.
•Another example of a patent family that we own in this category is based on international patent application WO 2023/110983, relating to DARPin domains which have binding specificity for two different targets, wherein binding of such a DARPin domain to its two targets is mutually exclusive. Such novel DARPin domains (also called “Switch DARPins”) may be used as molecular switches, such as, e.g., switches to control activation or deactivation of a therapeutic molecule. As of December 31, 2023, we owned one pending PCT international patent application in this family. Any patents that may issue in this family are expected to expire in 2042. The disclosed Switch DARPin platform is applied, for example, in our multispecific cKIT x CD16a x CD47 Switch DARPin program.
•Other patent applications falling in this category have been filed and are currently being prosecuted.
A second category of protection covers our key single-binding domain DARPin proteins binding to specific targets. These single domain DARPin binding proteins can be used in multiple DARPin product candidates. Our patent applications and corresponding patents directed to key single domain DARPin binding proteins currently include:
•One example of a patent family that we own in this category is based on international patent application WO 2010/060748, relating to single domain DARPin binding proteins with specificity for vascular endothelial growth factor A, or VEGF-A. As of December 31, 2023, we owned one issued U.S. patent and 31 issued foreign patents in this family. Any issued patents in this family are expected to expire in 2029, with the exception of one U.S. patent that received patent term adjustment and is expected to expire in 2031. VEGF-specific DARPin binding proteins are used, for example, in our DARPin product candidate abicipar.
•Another example of a patent family in this category is based on international patent application WO 2012/069654, relating to single domain DARPin binding proteins with specificity for human serum albumin, or HSA. As of December 31, 2023, we owned two issued U.S. patents, 22 issued foreign patents and three pending foreign patent applications in this family. Any issued patents in this family are expected to expire in 2031. HSA-specific DARPin binding proteins are used, for example, in our DARPin product candidates MP0310, MP0317 and MP0533.
•Another example of a patent family in this category is based on international patent application WO 2020/245173, relating to single domain DARPin binding proteins with specificity for fibroblast activation protein, or FAP. As of December 31, 2023, we owned one pending U.S. patent application and thirteen pending foreign patent applications in this family. Any patents that may be granted in this family are expected to expire in 2040. FAP-specific DARPin binding proteins are used in our DARPin product candidates MP0310 and MP0317.
•Another example of a patent family in this category is based on international patent application WO 2020/245175, relating to single domain DARPin binding proteins with specificity for 4-1BB. As of December 31, 2023, we owned one pending U.S. patent application and six pending foreign patent applications in this family. Any patents that may be granted in this family are expected to expire in 2040. 4-1BB-specific DARPin binding proteins are used in our DARPin product candidate MP0310.
•Another example of a patent family in this category is based on international patent application WO 2020/245171, relating to improved single domain DARPin binding proteins with specificity for HSA. As of December 31, 2023, we owned one pending U.S. patent application and 13 pending foreign patent applications in this family. Any patents that may be granted in this family are expected to expire in 2040. Disclosed HSA-specific DARPin binding proteins are used in our DARPin product candidates MP0310, MP0317 and MP0533.
•Another example of a patent family in this category is based on international patent application WO 2021/229076, relating to single domain DARPin binding proteins with specificity for CD40. As of December 31, 2023, we owned one pending U.S. patent application and five pending foreign patent applications in this family. Any patents that may be granted in this family are expected to expire in 2041. CD40-specific DARPin binding proteins are used in our DARPin product candidate MP0317.
•Another example of a patent family in this category is based on international patent application WO 2022/129428, relating to single domain DARPin binding proteins with specificity for CD3. As of December 31, 2023, we owned one pending U.S. patent application and thirteen pending foreign patent applications in this family. Any patents that may be granted in this family are expected to expire in 2041. CD3-specific DARPin binding proteins are used in our DARPin product candidate MP0533.
•Another example of a patent family in this category is based on international patent application WO 2022/190010, relating to single domain DARPin binding proteins with specificity for CD33. As of December 31, 2023, we owned one pending U.S. patent application and five pending foreign patent applications in this family. Any patents that may be granted in this family are expected to expire in 2042. CD33-specific DARPin binding proteins are used in our DARPin product candidate MP0533.
•Other patent applications falling in this category have been filed and are currently being prosecuted.
A third category of protection covers the composition of matter of certain of our DARPin product candidates (e.g., the specific combination and structure of DARPin binding proteins and additional elements that constitute the DARPin product candidate) as well as other product-specific inventions (e.g.
formulation, manufacturing process or dosing schedule). Our patent applications and corresponding patents related to our DARPin product candidates currently include:
•One example of a patent family that we own in this category is based on international patent application WO 2011/135067, relating to abicipar. As of December 31, 2023, we owned four issued U.S. patents, one pending U.S. patent application, 64 issued foreign patents and two pending foreign patent applications in this family. Any issued patents in this family are expected to expire in 2031, not considering any patent term extensions that may be available in various jurisdictions if abicipar obtains regulatory approval there.
•Another example of a patent family in this category is based on international patent application WO 2020/245746, relating to MP0310. As of December 31, 2023, we owned one pending U.S. patent application and 22 pending foreign patent applications in this family. Any patents that may be granted in this patent family are expected to expire in 2040, not considering any patent term extensions that may be available in various jurisdictions if MP0310 obtains regulatory approval there.
•Another example of a patent family in this category is based on international patent application WO 2021/229067, relating to MP0317. As of December 31, 2023, we owned two pending U.S. patent applications and 20 pending foreign patent applications in this family. Any patents that may be granted in this patent family are expected to expire in 2041, not considering any patent term extensions that may be available in various jurisdictions if MP0317 obtains regulatory approval there.
•Another example of a patent family in this category is based on international patent application WO 2022/190016, relating to MP0533. As of December 31, 2023, we owned one issued U.S. patent, one pending U.S. patent application and 17 pending foreign patent applications in this family. Any issued patents in this patent family are expected to expire in 2042, not considering any patent term extensions that may be available in various jurisdictions if MP0533 obtains regulatory approval there.
•Other patent applications falling in this category have been filed.
The actual protection afforded by a patent may vary on a product-by-product basis and from country to country and can depend upon many factors, including the type of patent, the scope of its coverage, the availability of regulatory-related extensions, the availability of legal remedies in a particular country and the validity and enforceability of the patent.
The term of an individual patent depends upon the legal term for patents in the countries in which they are granted. In most jurisdictions, including the United States and countries that are members of the European Patent Convention, the patent term is generally 20 years from the earliest effective filing date of a non-provisional patent application in the applicable country. In the United States, a patent’s term may, in certain cases, be lengthened by patent term adjustment, which compensates a patentee for administrative delays by the USPTO in granting a patent, or may be shortened if a patent is terminally disclaimed over a co-owned patent or patent application having an earlier expiration date or over a non-commonly owned patent or patent application having an earlier expiration date that was filed as a result of activities undertaken within the scope of a joint research agreement. In addition, patent term provisions are available in the United States, the member states of the European Union and certain other jurisdictions to extend the term of a patent that covers an approved drug to recapture a portion of the term effectively lost as a result of the regulatory review period. However, in the United States, the restoration period cannot be longer than five years, the total patent term including the restoration period must not exceed 14 years following FDA approval, only one patent applicable to each regulatory review period may be extended and only those claims covering the approved drug, a method for using it or a method of manufacturing it may be extended.
In the future, if and when our product candidates, including abicipar, MP0310, MP0317 and MP0533, receive approval by the FDA, EMA or any other relevant jurisdiction’s regulatory authorities, we expect, where possible, to apply for patent term extensions on issued patents covering those products, depending upon the length of the clinical trials for each product candidate and other factors. The expiration dates referred to above are without regard to potential patent term extensions that may be available to us and without regard to potential patent term adjustments or terminal disclaimers that may become applicable.
Notwithstanding our efforts, we cannot be sure that patents will be granted with respect to any patent applications we have licensed or filed or may license or file in the future, and we cannot be sure that any patents we have licensed or that have been granted to us, or any patents that may be licensed or granted to us in the future, will not be challenged, invalidated, rendered unenforceable or circumvented or that such patents will be commercially useful in protecting our technologies or product candidates.
We may rely, in some circumstances, on trade secrets and know-how to protect our technology. However, trade secrets and know-how can be difficult to protect. We seek to protect our proprietary technology and processes, in part, through confidentiality agreements and invention assignment agreements with our employees, consultants, scientific advisors, contractors and commercial partners. We also seek to preserve the integrity and confidentiality of our data and trade secrets by maintaining physical security of our premises and physical and electronic security of our information technology systems.
We own registrations for certain trademarks, including “Molecular Partners”, in Switzerland, the European Union, the United States and Japan. Further, we intend to build up a trademark portfolio for our technologies and product candidates as potential branding and commercialization approaches.
For more information regarding the risks related to our intellectual property, please see “Risk Factors—Risks Related to Intellectual Property.”
License and Collaboration Agreements
Research and Development Collaboration and Option Agreement with Orano Med in the area of Radio-DARPin Therapies, or the Orano Med Agreement
On January 5, 2024, we announced we entered into a co-development agreement with Orano Med to co-develop 212Pb-based Radio-DARPin Therapies (RDT). Under the terms of the co-development agreement, our previously disclosed RDT target DLL3 (delta-like ligand 3) will be included in the collaboration with Orano Med. Both companies are developing additional radioligand therapy candidates in partnership with other companies, with us having announced our first collaboration with Novartis in December 2021.
Expression of DLL3 is low in healthy tissue but significantly increased in certain tumor types, such as small-cell lung cancer, providing an opportunity for selective tumor-targeting. DLL3 will be exclusively developed by us and Orano Med as a RDT target.
We maintain the option to explore DLL3 for targeted therapy outside of the radiotherapy space. Both companies have committed to sharing the cost of preclinical and clinical development, including for supply of their respective materials.
License and Collaboration Agreement with Novartis in the Area of DARPin Conjugated Radioligand Therapies
On December 14, 2021, we entered into a license and collaboration agreement with Novartis to develop DARPin-conjugated radioligand therapeutic candidates for oncology, or the Novartis Radioligand Agreement. Under the agreement, both parties will collaborate on the discovery and optimization of the therapeutic candidates.
We are primarily responsible for the generation of DARPins for tumor-specific delivery of radioligands. We are eligible to invoice Novartis for our employee-related expenses associated with the research activities. Novartis is responsible for all clinical development and commercialization activities. In January 2022, Novartis paid us an upfront fee of $20 million (CHF 18.6 million). Additionally, we are eligible to receive milestone payments of up to $560 million, relating to development, regulatory and commercialization activities plus tiered royalties based on commercial sale levels from mid-single digit to low double-digit percentages of royalties on net sales of products commercialized by Novartis.
Option and Equity Rights Agreement with Novartis for Ensovibep
In October 2020, we entered into an agreement with Novartis, granting Novartis the exclusive option to in-license global rights in relation to MP0420 (ensovibep), or the Option and Equity Rights Agreement. Under the terms of the agreement, we in 2020 received an upfront, non-refundable fee of CHF 20 million for the technology transfer and manufacturing of MP0420.
Ensovibep License Agreement
In January 2022, following positive Phase 2 clinical trial results, Novartis exercised its option for ensovibep, triggering a milestone payment of CHF 150 million due to us, which was received in 2022. Relatedly, we were eligible to invoice Novartis CHF 13.1 million for other items related to ensovibep.
On January 2023 Novartis informed us that it has submitted a request to withdraw, with an effective date of January 25, 2023 the Emergency Use Authorization (EUA) application from the U.S. Food and Drug Administration (FDA) for ensovibep.
On January 5 2024, the License Agreement for ensovibep has been terminated and Novartis has returned the rights to the ensovibep program to us. Clinical work on the ensovibep program ended in 2022 and the program remains terminated.
Reservation agreement with the Swiss Federal Office of Public Health / Bundesamt für Gesundheit
On August 11, 2020, we announced the reservation by the Federal Office of Public Health, or FOPH, of a defined number of initial doses of our anti-COVID-19 candidate, MP0420. Under the terms of the agreement, we received a reservation fee of CHF 7.0 million. With the exercise of the option by Novartis in January 2022 and the subsequent assignment of the agreement to Novartis, we recognized the CHF 7.0 million as revenue in 2022.
License and Collaboration Agreement with Amgen
In December 2018, we entered into a license and collaboration agreement with Amgen for the clinical development and commercialization of MP0310 / AMG 506, or the Amgen Collaboration Agreement. Under the agreement we received a non-refundable upfront payment of USD 50 million.
On April 26, 2022 we announced that Amgen, had informed us of its decision to return the global rights of MP0310 following a strategic pipeline review. With no remaining performance obligations under the Amgen Collaboration Agreement, we recognized the remaining balance of the Amgen contract liability of CHF 9.7 million as revenue in 2022.
Government Regulation and Product Approval
As a biopharmaceutical company that operates in the United States, we are subject to extensive regulation. Government authorities in the United States (at the federal, state and local level) and in other countries extensively regulate, among other things, the research, development, testing, manufacturing, quality control, approval, labeling, packaging, storage, record-keeping, promotion, advertising, distribution, post-approval monitoring and reporting, marketing and export and import of biopharmaceutical products such as those we are developing.
Our product candidates must be approved by the FDA before they may be legally marketed in the United States and by the appropriate foreign regulatory agency before they may be legally marketed in foreign countries. Generally, our activities in other countries will be subject to regulation that is similar in nature and scope as that imposed in the United States, although there can be important differences. Additionally, some significant aspects of regulation in Europe are addressed in a centralized way, but country-specific regulation remains essential in many respects. The process for obtaining regulatory marketing approvals and the subsequent compliance with appropriate federal, state, local and foreign statutes and regulations require the expenditure of substantial time and financial resources.
U.S. Product Development Process
In the United States, the FDA regulates pharmaceutical and biological products under the Federal Food, Drug and Cosmetic Act, or the FDCA, the Public Health Service Act, or the PHSA, and their implementing regulations. The process of obtaining regulatory approvals and the subsequent compliance with appropriate federal, state, local and foreign statutes and regulations require the expenditure of substantial time and financial resources. Failure to comply with the applicable U.S. requirements at any time during the product development process, approval process or after approval, may subject an applicant to administrative or judicial sanctions. FDA sanctions could include, among other actions, refusal to approve pending applications, withdrawal of an approval, a clinical hold, warning letters, product recalls or withdrawals from the market, product seizures, total or partial suspension of production or distribution injunctions, fines, refusals of government contracts, restitution, disgorgement or civil or criminal penalties. Any agency or judicial enforcement action could have a material adverse effect on us. The process required by the FDA before a biological product may be marketed in the United States generally involves the following:
•completion of nonclinical laboratory tests and animal studies according to good laboratory practices, or GLPs, and applicable requirements for the humane use of laboratory animals or other applicable regulations;
•submission to the FDA of an IND, which must become effective before human clinical trials may begin;
•approval by an independent Institutional Review Board, or IRB, or ethics committee at each clinical site before the trial is commenced;
•performance of adequate and well-controlled human clinical trials according to the FDA’s regulations commonly referred to as good clinical practices, or GCPs, and any additional requirements for the protection of human research patients and their health information, to establish the safety and efficacy of the proposed biological product for its intended use;
•submission to the FDA of a Biologics Licensing Application, or BLA, for marketing approval that includes substantial evidence of safety, purity, and potency from results of nonclinical testing and clinical trials;
•satisfactory completion of an FDA Advisory Committee review, if applicable;
•satisfactory completion of an FDA inspection of the manufacturing facility or facilities where the biological product is produced to assess compliance with cGMP, to assure that the facilities, methods and controls are adequate to preserve the biological product’s identity, strength, quality and purity;
•potential FDA audit of the nonclinical study and clinical trial sites that generated the data in support of the BLA; and
•FDA review and approval or licensure of the BLA.
Before testing any biological product candidate, including our product candidates, in humans, the product candidate enters the preclinical testing stage. Preclinical tests, also referred to as nonclinical studies, include laboratory evaluations of product chemistry, toxicity and formulation, as well as animal studies to assess the potential safety and activity of the product candidate. The conduct of the preclinical tests must comply with federal regulations and requirements including GLPs. The clinical trial sponsor must submit the results of the preclinical tests, together with manufacturing information, analytical data, any available clinical data or literature and a proposed clinical protocol, to the FDA as part of the IND. Some preclinical testing may continue even after the IND is submitted. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA raises concerns or questions regarding the proposed clinical trials and places the trial on a clinical hold within that 30-day time period. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. The FDA may also impose clinical holds on a biological product candidate at any time before or during clinical trials due to safety concerns or non-compliance. If the FDA imposes a clinical hold, trials may not recommence without FDA authorization and then only under terms authorized by the FDA. Accordingly, we cannot be sure that submission of an IND will result in the FDA allowing clinical trials to begin, or that, once begun, issues will not arise that suspend or terminate such trials.
Clinical trials involve the administration of the biological product candidate to patients under the supervision of qualified investigators, generally physicians not employed by or under the trial sponsor’s control. Clinical trials are conducted under protocols detailing, among other things, the objectives of the clinical trial, dosing procedures, subject selection and exclusion criteria, and the parameters to be used to monitor subject safety, including stopping rules that assure a clinical trial will be stopped if certain adverse events should occur. Each protocol and any amendments to the protocol must be submitted to the FDA as part of the IND. Clinical trials must be conducted and monitored in accordance with the FDA’s regulations comprising the GCP requirements, including the requirement that all research patients provide informed consent. Further, each clinical trial must be reviewed and approved by an independent IRB at or servicing each institution at which the clinical trial will be conducted. An IRB is charged with protecting the welfare and rights of trial participants and considers such items as whether the risks to individuals participating in the clinical trials are minimized and are reasonable in relation to anticipated benefits. The IRB also approves the form and content of the informed consent that must be signed by each clinical trial subject or his or her legal representative and must monitor the clinical trial until completed. Some studies also include oversight by an independent group of qualified experts organized by the clinical study sponsor, known as a data safety monitoring board, which provides authorization for whether or not a study may move forward at designated check points based on access to certain data from the study and may halt the clinical trial if it determines that there is an unacceptable safety risk for subjects or other grounds, such as no demonstration of efficacy. There are also requirements governing the reporting of ongoing clinical studies and clinical study results to public registries.
Human clinical trials are typically conducted in three sequential phases that may overlap or be combined:
•Phase 1. The biological product is initially introduced into healthy human subjects and tested for safety. In the case of some products for severe or life-threatening diseases, especially when the product may be too inherently toxic to ethically administer to healthy volunteers, the initial human testing is often conducted in patients.
•Phase 2. The biological product is evaluated in a limited patient population to identify possible adverse effects and safety risks, to preliminarily evaluate the efficacy of the product for specific targeted diseases and to determine dosage tolerance, optimal dosage and dosing schedule.
•Phase 3. Clinical trials are undertaken to further evaluate dosage, clinical efficacy, potency, and safety in an expanded patient population at geographically dispersed clinical trial sites. These clinical trials are intended to establish the overall risk to benefit ratio of the product and provide an adequate basis for product labeling.
Post-approval clinical trials, sometimes referred to as Phase 4 clinical trials, may be conducted after initial marketing approval. These clinical trials are used to gain additional experience from the treatment of patients in the intended therapeutic indication, particularly for long-term safety follow-up. During all phases of clinical development, regulatory agencies require extensive monitoring and auditing of all clinical activities, clinical data, and clinical trial investigators. Annual progress reports detailing the results of the clinical trials must be submitted to the FDA. Written IND safety reports must be promptly submitted to the FDA and the investigators for serious and unexpected adverse events, any findings from other studies, tests in laboratory animals or in vitro testing that suggest a significant risk for human patients, or any clinically important increase in the rate of a serious suspected adverse reaction over that listed in the protocol or investigator brochure. Phase 1, Phase 2 and Phase 3 clinical trials may not be completed successfully within any specified period, if at all. The FDA or the sponsor or its data safety monitoring board may suspend or terminate a clinical trial at any time on various grounds, including a finding that the research patients are being exposed to an unacceptable health risk, including risks inferred from other unrelated immunotherapy trials. Similarly, an IRB can suspend or terminate approval of a clinical trial at its institution if the clinical trial is not being conducted in accordance with the IRB’s requirements or if the biological product has been associated with unexpected serious harm to patients.
Concurrently with clinical trials, companies usually complete additional studies and must also develop additional information about the physical characteristics of the biological product as well as finalize a process for manufacturing the product in commercial quantities in accordance with cGMP requirements. To help reduce the risk of the introduction of adventitious agents with use of biological products, the PHSA emphasizes the importance of manufacturing control for products whose attributes cannot be precisely defined. The manufacturing process must be capable of consistently producing quality batches of the product candidate and, among other things, the sponsor must develop methods for testing the identity, strength, quality, potency and purity of the final biological product. Additionally, appropriate packaging must be selected and tested and stability studies must be conducted to demonstrate that the biological product candidate does not undergo unacceptable deterioration over its shelf life.
U.S. Review and Approval Processes
After the completion of clinical trials of a biological product, FDA approval of a BLA must be obtained before commercial marketing of the biological product. The BLA submission must include results of product development, laboratory and animal studies, human trials, information on the manufacture and composition of the product, proposed labeling and other relevant information. The testing and approval processes require substantial time and effort and there can be no assurance that the FDA will accept the BLA for filing and, even if filed, that any approval will be granted on a timely basis, if at all.
Under the Prescription Drug User Fee Act, or PDUFA, as amended, each BLA must be accompanied by a significant user fee. The FDA adjusts the PDUFA user fees on an annual basis. PDUFA also imposes an annual program fee for biological products. Fee waivers or reductions are available in certain circumstances, including a waiver of the application fee for the first application filed by a small business.
Additionally, no user fees are assessed on BLAs for products designated as orphan drugs, unless the product also includes a non-orphan indication.
Within 60 days following submission of the application, the FDA reviews a BLA submitted to determine if it is substantially complete before the agency accepts it for filing. The FDA may refuse to file any BLA that it deems incomplete or not properly reviewable at the time of submission and may request additional information. In this event, the BLA must be resubmitted with the additional information. The resubmitted application also is subject to review before the FDA accepts it for filing. Once the submission is accepted for filing, the FDA begins an in-depth substantive review of the BLA. The FDA reviews the BLA to determine, among other things, whether the proposed product is safe, potent, and/or effective for its intended use, and has an acceptable purity profile, and whether the product is being manufactured in accordance with cGMP to assure and preserve the product’s identity, safety, strength, quality, potency and purity. The FDA may refer applications for novel biological products or biological products that present difficult questions of safety or efficacy to an advisory committee, typically a panel that includes clinicians and other experts, for review, evaluation and a recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions. During the biological product approval process, the FDA also will determine whether a Risk Evaluation and Mitigation Strategy, or REMS, is necessary to assure the safe use of the biological product. A REMS is a safety strategy to manage a known or potential serious risk associated with a medicine and to enable patients to have continued access to such medicines by managing their safe use, and could include medication guides, physician communication plans, or elements to assure safe use, such as restricted distribution methods, patient registries and other risk minimization tools. If the FDA concludes a REMS is needed, the sponsor of the BLA must submit a proposed REMS. The FDA will not approve a BLA without a REMS, if required.
Before approving a BLA, the FDA will inspect the facilities at which the product is manufactured. The FDA will not approve the product unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications. Additionally, before approving a BLA, the FDA will typically inspect one or more clinical sites to assure that the clinical trials were conducted in compliance with IND trial requirements and GCP requirements. To assure cGMP and GCP compliance, an applicant must incur significant expenditure of time, money and effort in the areas of training, record keeping, production, and quality control.
Notwithstanding the submission of relevant data and information, the FDA may ultimately decide that the BLA does not satisfy its regulatory criteria for approval and deny approval. Data obtained from clinical trials are not always conclusive and the FDA may interpret data differently than we interpret the same data. If the agency decides not to approve the BLA in its present form, the FDA will issue a complete response letter that describes all of the specific deficiencies in the BLA identified by the FDA. The deficiencies identified may be minor, for example, requiring labeling changes, or major, for example, requiring additional clinical trials. Additionally, the complete response letter may include recommended actions that the applicant might take to place the application in a condition for approval. If a complete response letter is issued, the applicant may either resubmit the BLA, addressing all of the deficiencies identified in the letter, or withdraw the application.
If a product receives regulatory approval, the approval may be limited to specific diseases and dosages or the indications for use may otherwise be limited, which could restrict the commercial value of the product. Further, the FDA may require that certain contraindications, warnings or precautions be included in the product labeling. The FDA may impose restrictions and conditions on product distribution, prescribing, or dispensing in the form of a risk management plan, or otherwise limit the scope of any approval.
In addition, the FDA may require post marketing clinical trials, sometimes referred to as Phase 4 clinical trials, designed to further assess a biological product’s safety and effectiveness, and testing and surveillance programs to monitor the safety of approved products that have been commercialized.
In addition, under the Pediatric Research Equity Act, or PREA, a BLA or supplement to a BLA must contain data to assess the safety and effectiveness of the product for the claimed indications in all relevant pediatric subpopulations and to support dosing and administration for each pediatric subpopulation for which the product is safe and effective. The FDA may grant deferrals for submission of data or full or partial waivers. Unless otherwise required by regulation, PREA does not apply to any product for an indication for which orphan designation has been granted. However, if only one indication for a product has orphan designation, a pediatric assessment may still be required for any applications to market that same product for the non-orphan indication or indications.
Post-Approval Requirements
Any products for which we receive FDA approvals are subject to continuing regulation by the FDA, including, among other things, record-keeping requirements, reporting of adverse experiences with the product, providing the FDA with updated safety and efficacy information, product sampling and distribution requirements, and complying with FDA promotion and advertising requirements, which include, among others, standards for direct-to-consumer advertising, restrictions on promoting products for uses or in patient populations that are not described in the product’s approved uses (known as “off-label use”), limitations on industry-sponsored scientific and educational activities, and requirements for promotional activities involving the internet. Although a physician may prescribe a legally available product for an off-label use, if the physician deems such product to be appropriate in his/her professional medical judgment, a manufacturer may not market or promote off-label uses. Violations, including actual or alleged promotion of products for unapproved or off-label uses, are subject to enforcement letters, inquiries and investigations, and civil and criminal sanctions by the FDA or comparable foreign bodies. Any actual or alleged failure to comply with labeling and promotion requirements may result in fines, warning letters, mandates to corrective information to healthcare practitioners, injunctions, or civil or criminal penalties.
In addition, quality control and manufacturing procedures must continue to conform to applicable manufacturing requirements after approval to ensure the long-term stability of the product. cGMP regulations require among other things, quality control and quality assurance as well as the corresponding maintenance of records and documentation and the obligation to investigate and correct any deviations from cGMP. Manufacturers and other entities involved in the manufacture and distribution of approved products are required to register their establishments with the FDA and certain state agencies, and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with cGMP and other laws. Accordingly, manufacturers must continue to expend time, money, and effort in the area of production and quality control to maintain cGMP compliance. In addition, changes to the manufacturing process are strictly regulated, and depending on the significance of the change, may require prior FDA approval before being implemented. Other types of changes to the approved product, such as adding new indications and claims, are also subject to further FDA review and approval. Discovery of problems with a product after approval may result in restrictions on a product, manufacturer, or holder of an approved BLA, including, among other things, recall or withdrawal of the product from the market. Other potential consequences include, among other things:
•restrictions on the marketing or manufacturing of the product;
•fines, warning letters, untitled letters, or clinical holds;
•refusal of the FDA to approve pending applications or supplements to approved applications, or suspension or revocation of product approvals;
•adverse publicity, FDA mandated corrective advertising or communications with doctors;
•product seizure or detention, or refusal to permit the import or export of products; or
•injunctions or the imposition of civil or criminal penalties.
The FDA also may require post-marketing testing, known as Phase 4 testing, and surveillance to monitor the effects of an approved product. Newly discovered or developed safety or effectiveness data may require changes to a product’s approved labeling, including the addition of new warnings and contraindications, and also may require the implementation of other risk management measures. Also, new government requirements, including those resulting from new legislation, may be established, or the FDA’s policies may change, which could delay or prevent regulatory approval of our products under development.
U.S. Marketing Exclusivity
The Biologics Price Competition and Innovation Act, or BPCIA, amended the PHSA to authorize the FDA to approve similar versions of innovative biologics, commonly known as biosimilars. A competitor seeking approval of a biosimilar must file an application to establish its molecule as highly similar to an approved innovator biologic, among other requirements. The BPCIA, however, bars the FDA from approving biosimilar applications referencing that biologic for 12 years after an innovator biological product receives initial marketing approval. During this 12-year period of exclusivity, another company may still market a competing version of the reference product if the FDA approves a full BLA for the competing product containing that applicant’s own preclinical data and data from adequate and well-controlled clinical trials to demonstrate the safety, purity and potency of its product. Pediatric exclusivity is another type of regulatory market exclusivity in the United States. Pediatric exclusivity, if granted, adds six months to existing exclusivity periods and patent terms. This six-month exclusivity, which runs from the end of other exclusivity protection or patent term, may be granted based on the voluntary completion of a pediatric trial in accordance with an FDA-issued “Written Request” for such a trial.
U.S. Healthcare Laws
A biopharmaceutical company’s operations may be directly, or indirectly through relationships with healthcare providers, healthcare institutions, patients, customers and third-party payors, subject to various federal and state healthcare laws and regulations. These laws impact, among other things, sales, marketing and education programs and may constrain business and financial arrangements and relationships with third-party payors, healthcare professionals and healthcare institutions who participate in a biopharmaceutical company’s clinical research programs, healthcare professionals and others who recommend, purchase, or provide a biopharmaceutical company’s approved drug products, and other parties through which it markets, sells and distributes its approved drug products. In addition, a biopharmaceutical company may be subject to patient data privacy and security regulation by both the federal government and the states in which it conducts its business. The laws that may affect a biopharmaceutical company’s ability to operate include:
•the federal Anti-Kickback Statute, which prohibits, among other things, individuals or entities from knowingly and willfully soliciting, offering, receiving or paying any remuneration (including any kickback, bribe, or rebate), directly or indirectly, overtly or covertly, in cash or in kind, to induce or reward either the referral of an individual for, or the purchase, lease, order or recommendation of, any good, facility, item or service, for which payment may be made, in whole or in part, under federal and state healthcare programs such as Medicare and Medicaid;
•the federal civil and criminal false claims laws, including, without limitation, the civil False Claims Act (which can be enforced through "qui tam," or whistleblower actions, by private citizens on behalf of the federal government), and the federal civil monetary penalties law, which prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, to the federal government, claims for payment or approval that are false or fraudulent or for knowingly making a false statement to avoid, decrease or conceal an obligation to pay money to the federal government;
•the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, which prohibits, among other things, knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program, or knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false statement, in connection with the delivery of, or payment for, healthcare benefits, items or services;
•HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act of 2009, or HITECH, and its implementing regulations, and as amended again by the Final HIPAA Omnibus Rule, published in January 2013, which imposes certain obligations, including mandatory contractual terms, with respect to safeguarding the privacy, security and transmission of individually identifiable health information without appropriate authorization on certain health plans, healthcare clearinghouses and healthcare providers, known as covered entities, as well as their business associates that perform certain services involving the use, disclosure or transmission of individually identifiable health information for or on behalf of a covered entity, and their covered subcontractors;
•the Federal Food, Drug, and Cosmetic Act which prohibits, among other things, the adulteration or misbranding of drugs, biologics and medical devices;
•the federal physician payment transparency legislation commonly referred to as the Physician Payments Sunshine Act, and its implementing regulations, which requires certain manufacturers of drugs, devices, biologics and medical supplies (with certain exceptions) that are reimbursable under Medicare, Medicaid, or the Children's Health Insurance Program to report annually to the Centers for Medicare & Medicaid, or CMS, information related to certain payments and other transfers of value to physicians (defined to include doctors, dentists, optometrists, podiatrists and chiropractors), certain other health care professionals (such as physicians assistants and nurse practitioners), and teaching hospitals, as well as ownership and investment interests held by physicians and their immediate family members; and
•analogous state laws and regulations, including: state anti-kickback and false claims laws, which may apply to our business practices, including but not limited to, research, distribution, sales and marketing arrangements and claims involving healthcare items or services reimbursed by any third-party payor, including private insurers; state laws that require pharmaceutical companies to comply with the pharmaceutical industry's voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government, or otherwise restrict payments that may be made to healthcare providers and other potential referral sources; state laws and regulations that require drug manufacturers to file reports relating to pricing and marketing information, which requires tracking gifts and other remuneration and items of value provided to healthcare professionals and entities; state and local laws that require the registration of pharmaceutical sales representatives; and state laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and often are not preempted by HIPAA, thus complicating compliance efforts.
It is possible that governmental authorities will conclude that a biopharmaceutical company’s business practices do not comply with current or future statutes, regulations or case law involving applicable fraud and abuse or other healthcare laws and regulations. If a biopharmaceutical company’s operations are found to be in violation of any of these laws or any other governmental regulations that may apply to it, it may be subject to significant civil, criminal and administrative penalties, damages, fines, disgorgement, imprisonment, exclusion from participation in government funded healthcare programs, such as Medicare and Medicaid, additional reporting requirements and oversight if subject to a corporate integrity agreement or similar agreement to resolve allegations of non-compliance with these laws, reputational harm and the curtailment or restructuring of operations.
The risk of being found in violation of these laws is increased by the fact that many of them have not been fully interpreted by the regulatory authorities or the courts, and their provisions are open to a variety of interpretations. For example, the definition of "remuneration" under the federal Anti-Kickback Statute has been interpreted to include anything of value. Further, courts have found that if "one purpose" of remuneration is to induce referrals, the federal Anti-Kickback Statute is violated.
Additionally, recent healthcare reform legislation has strengthened federal and state healthcare fraud and abuse laws. For example, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act of 2010, or collectively the ACA, amends the intent requirement of the federal Anti-Kickback Statute and criminal healthcare fraud statutes to clarify that liability under these statutes does not require a person or entity to have actual knowledge of the statutes or a specific intent to violate them in order to have committed a violation. Moreover, the ACA provides that the government may assert that a claim that includes items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the civil False Claims Act. Because of the breadth of these laws and the narrowness of the statutory exceptions and regulatory safe harbors available, it is possible that some of a biopharmaceutical company’s business activities could be subject to challenge under one or more of such laws.
U.S. Healthcare Reform
In the United States, there have been a number of legislative and regulatory changes at the federal and state levels which seek to reduce healthcare costs and improve the quality of healthcare. For example, in March 2010, the ACA became law. The ACA is a sweeping law intended to broaden access to health insurance, reduce or constrain the growth of healthcare spending, enhance remedies against fraud and abuse, add new transparency requirements for the healthcare and health insurance industries, impose new taxes and fees on the health industry and impose additional health policy reforms.
The ACA, among other things, increased the minimum level of Medicaid rebates payable by manufacturers of brand name drugs; required collection of rebates for drugs paid by Medicaid managed care organizations; required manufacturers to participate in a coverage gap discount program, under which they must agree to offer point-of-sale discounts (increased to 70 percent, effective as of January 1, 2019) off negotiated prices of applicable brand drugs to eligible beneficiaries during their coverage gap period, as a condition for the manufacturer’s outpatient drugs to be covered under Medicare Part D; imposed a non-deductible annual fee on pharmaceutical manufacturers or importers who sell certain “branded prescription drugs” to specified federal government programs, implemented a new methodology by which rebates owed by manufacturers under the Medicaid Drug Rebate Program are calculated for drugs that are inhaled, infused, instilled, implanted, or injected expanded the types of entities eligible for the 340B drug discount program; expanded eligibility criteria for Medicaid programs; created a new Patient-Centered Outcomes Research Institute to oversee, identify priorities in, and conduct comparative clinical effectiveness research, along with funding for such research; and established a Center for Medicare Innovation at CMS to test innovative payment and service delivery models to lower Medicare and Medicaid spending, potentially including prescription drug spending.
There have been executive, judicial and Congressional challenges to certain aspects of the ACA. While Congress has not passed comprehensive repeal legislation, several bills affecting the implementation of certain taxes under the ACA have been signed into law. The Tax Cuts and Jobs Act of 2017, or the Tax Act, included a provision, which repealed, effective January 1, 2019, the tax-based shared responsibility payment imposed by the ACA on certain individuals who fail to maintain qualifying health coverage for all or part of a year that is commonly referred to as the “individual mandate.” In addition, the 2020 federal spending package permanently eliminated, effective January 1, 2020, the ACA-mandated “Cadillac” tax on high-cost employer-sponsored health coverage and medical device tax and, effective January 1, 2021, also eliminated the health insurer tax. On June 17, 2021, the U.S. Supreme Court dismissed a challenge on procedural grounds that argue the Affordable Care Act is unconstitutional in its entirety because the “individual mandate” was repealed by Congress. Further, on August 16, 2022, President Biden signed the Inflation Reduction Act of 2022, or IRA, into law, which among other things, extends enhanced subsidies for individuals purchasing health insurance coverage in ACA marketplaces through plan year 2025. The IRA also eliminates the “donut hole” under the Medicare Part D program beginning in 2025 by significantly lowering the beneficiary maximum out-of-pocket cost and creating a new manufacturer discount program. It is possible that the ACA will be subject to judicial or Congressional challenges in the future. It is unclear how any such challenges and the healthcare reform measures of the Biden administration will impact the ACA.
In addition, other legislative changes have been proposed and adopted since the ACA was enacted. For example, on August 2, 2011, the Budget Control Act of 2011, among other things, includes aggregate reductions of Medicare payments to providers of 2% per fiscal year. These reductions went into effect on April 1, 2013 and, due to subsequent legislative amendments to the statute will remain in effect through 2032 unless additional Congressional action is taken. On January 2, 2013, the American Taxpayer Relief Act of 2012 was signed into law, which, among other things, further reduced Medicare payments to several types of providers, including hospitals, imaging centers and cancer treatment centers, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years. Additionally, on March 11, 2021, President Biden signed the American Rescue Plan Act of 2021 into law, which eliminates the statutory Medicaid drug rebate cap, currently set at 100% of a drug’s average manufacturer price, for single source and innovator multiple source drugs, beginning on January 1, 2024.
Further, there has also been heightened governmental scrutiny over the manner in which manufacturers set prices for their marketed products. Specifically, there have been several recent Congressional inquiries and proposed and enacted federal and state legislation designed to, among other things, bring more transparency to drug pricing, reduce the cost of prescription drugs under Medicare, review the relationship between pricing and manufacturer patient programs and reform government program reimbursement methodologies for drugs. At the federal level in July 2021, the Biden administration released an executive order, “Promoting Competition in the American Economy,” with multiple provisions aimed at prescription drugs. In response to Biden’s executive order, on September 9, 2021, the U.S. Department of Health and Human Services, or HHS, released a Comprehensive Plan for Addressing High Drug Prices that outlines principles for drug pricing reform and sets out a variety of potential legislative policies that Congress could pursue as well as potential administrative actions HHS can take to advance these principles. In addition, the IRA, among other things (i) directs HHS to negotiate the price of certain high-expenditure, single-source drugs and biologics covered under Medicare and (ii) imposes rebates under Medicare Part B and Medicare Part D to penalize price increases that outpace inflation. These provisions take effect progressively starting in fiscal year 2023. On August 29, 2023, HHS announced the list of the first ten drugs that will be subject to price negotiations, although the Medicare drug price negotiation program is currently subject to legal challenges. In response to the Biden administration’s October 2022 executive order, on February 14, 2023, HHS released a report outlining three new models for testing by the CMS Innovation Center which will be evaluated on their ability to lower the cost of drugs, promote accessibility, and improve quality of care.
It is unclear whether the models will be utilized in any health reform measures in the future. Further, on December 7, 2023, the Biden administration announced an initiative to control the price of prescription drugs through the use of march-in rights under the Bayh-Dole Act. On December 8, 2023, the National Institute of Standards and Technology published for comment a Draft Interagency Guidance Framework for Considering the Exercise of March-In Rights which for the first time includes the price of a product as one factor an agency can use when deciding to exercise march-in rights. While march-in rights have not previously been exercised, it is uncertain if that will continue under the new framework.
At the state level, legislatures are increasingly passing legislation and implementing regulations designed to control pharmaceutical and biological product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing. In addition, regional healthcare authorities and individual hospitals are increasingly using bidding procedures to determine what pharmaceutical products and which suppliers will be included in their prescription drug and other healthcare programs.
Clinical Trials in the European Union
Clinical trials of medicinal products in the European Union must be conducted in accordance with European Union and national regulations and the international council for harmonization, or ICH, guidelines on GCP. Additional GCP guidelines from the EC, focusing in particular on traceability, apply to clinical trials of advanced therapy medicinal products. The sponsor must take out a clinical trial insurance policy, and in most European Union countries, the sponsor is liable to provide “no fault” compensation to any study subject injured in the clinical trial.
Prior to commencing a clinical trial, the sponsor must obtain a clinical trial authorization from the competent authority, and a positive opinion from an independent ethics committee. The application for a clinical trial authorization must include, among other things, a copy of the trial protocol and an investigational medicinal product dossier containing information about the manufacture and quality of the medicinal product under investigation. Previously, in the European Union, pursuant to the EU Clinical Trials Directive 2001/20/EC, a CTA had to be submitted to each country’s national regulatory authority in which the clinical trial was to take place, together with an independent ethics committee, much like the FDA and IRB, respectively. Although the Directive had sought to harmonize the EU clinical trials regulatory framework, EU Member States transposed and applied the provisions of the Directive differently, leading to significant variation in the regulatory regimes of the member states. In 2014, a new Clinical Trials Regulation 536/2014, replacing the current Directive, was adopted. The new Regulation is directly applicable in all EU Member States (without national implementation) and entered into application on 31 January 2022. The new Regulation seeks to simplify and streamline the approval of clinical trials in the European Union. Pursuant to the Regulation, the sponsor shall submit a single CTA via the EMA's Clinical Trials Information System, or CTIS, which will cover all regulatory and ethics assessments from the member states concerned.
Any submissions made from January 31, 2023 onwards must be made through CTIS and all trials authorized pursuant to the Directive that are still ongoing on January 31, 2025 must be made through CTIS. Once the CTA is approved in accordance with a member state's requirements, clinical trial development may proceed. Approval and monitoring of clinical trials in the European Union is, as it was under the Directive, the responsibility of individual member states, but compared to the position prior to the applicability of the Clinical Trials Regulation there is likely to be more collaboration, information-sharing, and decision-making between member states.
The new Regulation also aims to streamline and simplify the rules on safety reporting and introduces enhanced transparency requirements, such as mandatory submission of a summary of the clinical trial results to a new E.U. Database. The requirements and process governing the conduct of clinical trials, product licensing, pricing and reimbursement vary from country to country. In all cases, the clinical trials are conducted in accordance with GCP and the applicable regulatory requirements and the ethical principles that have their origin in the Declaration of Helsinki. Medicines used in clinical trials must be manufactured in accordance with cGMP.
During the development of a medicinal product the EMA and national medicines regulators within the European Union provide the opportunity for dialogue and guidance on the development program. At the EMA level, this is usually done in the form of scientific advice, which is given by the Scientific Advice Working Party of the Committee for Medicinal Products for Human Use. A fee is incurred with each scientific advice procedure. Advice from the EMA is typically provided based on questions concerning, for example, quality (chemistry, manufacturing and controls testing), nonclinical testing and clinical studies, and pharmacovigilance plans and risk-management programs.
Marketing Authorizations in the European Union
In order to market a new medicinal product in the European Union, a company must submit a marketing authorization application, or MAA, to either the EMA using the centralized procedure, or competent authorities in European Union Member States using the other procedures (decentralized procedure, national procedure, or mutual recognition procedure). A marketing authorization, or MA may only be granted to an applicant established in the European Union, or Norway, Iceland, and Liechtenstein, who are members of the European Economic Area, or European Economic Area. Medicinal products can only be commercialized after obtaining an MA pursuant to one of the three processes outlined below:
•the Centralized MA, which is issued by the European Commission through the Centralized Procedure, based on the scientific opinion of the Committee for Medicinal Products for Human Use of the EMA, and which is valid throughout the entire territory of the European Union/European Economic Area. The Centralized Procedure is mandatory for certain types of products, such as (i) biotechnology medicinal products such as genetic engineering, (ii) orphan medicinal products, (iii) medicinal products containing a new active substance indicated for the treatment of AIDS, cancer, neurodegenerative disorders, diabetes, autoimmune and viral diseases and (iv) advanced-therapy medicines, such as gene therapy, somatic cell therapy or tissue-engineered medicines. The Centralized Procedure is optional for products containing a new active substance not yet authorized in the European Union/European Economic Area, or for products that constitute a significant therapeutic, scientific or technical innovation or which are in the interest of public health in the European Union.
•Decentralized Procedure MAs are available for products not falling within the mandatory scope of the Centralized Procedure. An identical dossier is submitted to the competent authorities of each of the Member States in which the MA is sought, one of which is selected by the applicant as the Reference Member State, or RMS, to lead the evaluation of the regulatory submission. The competent authority of the RMS prepares a draft assessment report, a draft summary of the product characteristics, or SmPC, and a draft of the labeling and package leaflet as distilled from the preliminary evaluation, which are sent to the other Member States (referred to as the Concerned Member States) for their approval. If the Concerned Member States raise no objections, based on a potential serious risk to public health, to the assessment, SmPC, labeling, or packaging proposed by the RMS, the RMS records the agreement, closes the procedure and informs the applicant accordingly. Each Member State concerned by the procedure is required to adopt a national decision to grant a national MA in conformity with the approved assessment report, SmPC and the labeling and package leaflet as approved. Where a product has already been authorized for marketing in a Member State of the European Economic Area, the granted national MA can be used for mutual recognition in other Member States through the Mutual Recognition Procedure, or MRP, resulting in progressive national approval of the product in the European Union/European Economic Area.
•National MAs, which are issued by a single competent authority of the Member States of the European Economic Area and only covers their respective territory, are also available for products not falling within the mandatory scope of the Centralized Procedure. Once a product has been authorized for marketing in a Member State of the European Economic Area through the National Procedure, this National MA can also be recognized in other Member States through the Mutual Recognition Procedure.
Under the procedures described above, before granting the MA, the EMA or the competent authority(ies) of the member state(s) of the European Economic Area prepare an assessment of the risk-benefit balance of the product against the scientific criteria concerning its quality, safety and efficacy.
Data Exclusivity in the European Union
Under Regulation (EC) No 726/2004/EC and Directive 2001/83/EC (each as amended), the European Union has adopted a harmonized approach to data and market protection or exclusivity (known as the 8 + 2 + 1 formula). The data exclusivity period begins to run on the date when the first MA is granted in the European Union. It confers on the MA holder of the reference medicinal product eight years of data exclusivity and ten years of market exclusivity. A reference medicinal product is defined to mean a medicinal product authorized based on a full dossier consisting of pharmaceutical and preclinical testing results and clinical trial data, such as a medicinal product containing a new active substance. The ten-year market protection can be extended cumulatively to a maximum period of eleven years if during the first eight years of those ten years of protection period, the MA holder obtains an authorization for one or more new therapeutic indications that are deemed to bring a significant clinical benefit compared to existing therapies.
The exclusivity period means that an applicant for a generic medicinal product is not permitted to rely on preclinical pharmacological, toxicological, and clinical data contained in the file of the reference medicinal product of the originator until the first eight years of data exclusivity have expired. Thereafter, a generic product application may be submitted and generic companies may rely on the preclinical and clinical data relating to the reference medicinal product to support approval of the generic product. However, a generic product cannot market until ten years have elapsed from the initial authorization of the reference medicinal product or eleven years if the protection period is extended, based on the formula of 8+2+1.
In addition to the above, where an application is made for a new indication for a well-established substance, a non-cumulative period of one year of data exclusivity may be granted, provided that significant preclinical or clinical studies were carried out in relation to the new indication. Finally, where a change of classification of a medicinal product has been authorized on the basis of significant preclinical tests or clinical trials, the competent authority shall not refer to the results of those tests or trials when examining an application by another applicant for or holder of marketing authorization for a change of classification of the same substance for one year after the initial change was authorized.
The 8 + 2 + 1 exclusivity scheme applies to products that have been authorized in the European Union by the European Commission through the Centralized Procedure or the competent authorities of the Member States of the European Economic Area nationally, including through the Decentralized and Mutual Recognition procedures.
For a medicinal product which has received orphan designation under Regulation 141/2000, it will, benefit from a period of ten years of orphan market exclusivity which essentially constitutes a period of market monopoly. During this period of orphan market exclusivity, no European Union regulatory authority is permitted to accept or approve an application for marketing authorization for a similar medicinal product or an extension application for the same therapeutic indication. This period can be extended cumulatively to a total of twelve years if the marketing authorization holder or applicant complies with the requirements for an agreed pediatric investigation plan pursuant to Regulation 1901/2006.
Post Authorization Obligations in the European Union
The holder of a Centralized MA or National MA is subject to various obligations under the applicable European Union laws, such as pharmacovigilance obligations, requiring it to, among other things, report and maintain detailed records of adverse reactions, and to submit periodic safety update reports, or PSURs, to the competent authorities. All new marketing authorization applications must include a risk management plan, or RMP, describing the risk management system that the company will put in place and documenting measures to prevent or minimize the risks associated with the product. The regulatory authorities may also impose specific obligations as a condition of the marketing authorization. Such risk-minimization measures or post-authorization obligations may include additional safety monitoring, more frequent submission of PSURs, or the conduct of additional clinical trials or post-authorization safety studies. RMPs and PSURs are routinely available to third parties requesting access, subject to limited redactions. All advertising and promotional activities for the product must be consistent with the approved summary of product characteristics, and therefore all off-label promotion is prohibited. Direct-to-consumer advertising of prescription medicines is also prohibited in the European Union. The holder must also ensure that the manufacturing and batch release of its product is in compliance with the applicable requirements. The MA holder is further obligated to ensure that the advertising and promotion of its products complies with applicable European Union laws and industry code of practice as implemented in the domestic laws of the Member States of the European Union/European Economic Area. The advertising and promotional rules are enforced nationally by the European Union/European Economic Area Member States.
Pediatric Development in the European Union
In the European Union, companies developing a new medicinal product must agree to a Pediatric Investigation Plan, or PIP, with the EMA and must conduct pediatric clinical trials in accordance with that PIP, unless a deferral or waiver applies, (e.g., because the relevant disease or condition occurs only in adults). The marketing authorization application for the product must include the results of pediatric clinical trials conducted in accordance with the PIP, unless a waiver applies, or a deferral has been granted, in which case the pediatric clinical trials must be completed at a later date. Products that are granted a marketing authorization on the basis of the pediatric clinical trials conducted in accordance with the PIP are eligible for a six month extension of the protection under a supplementary protection certificate (if any is in effect at the time of approval) or, in the case of orphan medicinal products, a two year extension of the orphan market exclusivity. This pediatric reward is subject to specific conditions and is not automatically available when data in compliance with the PIP are developed and submitted.
Pricing and Reimbursement in the European Union
Governments influence the price of medicinal products in the European Union through their pricing and reimbursement rules and control of national healthcare systems that fund a large part of the cost of those products to consumers. Some jurisdictions operate positive and negative list systems under which products may only be marketed once a reimbursement price has been agreed. To obtain reimbursement or pricing approval, some of these countries may require the completion of clinical trials that compare the cost-effectiveness of a particular product candidate to currently available therapies.
Other European Union Member States allow companies to fix their own prices for medicines, but monitor and control company profits. The downward pressure on healthcare costs in general, particularly prescription medicines, has become very intense. As a result, increasingly high barriers are being erected to the entry of new products.
Brexit and the Regulatory Framework in the United Kingdom
Following the result of a referendum in 2016, the United Kingdom officially withdrew from the European Union on January 31, 2020, commonly referred to as Brexit. Pursuant to the formal withdrawal arrangements agreed between the United Kingdom and the European Union, the United Kingdom was subject to a transition period until December 31, 2020, during which European Union rules continued to apply. A trade and cooperation agreement, or the Trade and Cooperation Agreement, which outlines the future trading relationship between the United Kingdom and the European Union was agreed in December 2020 and formally entered into force on May 1, 2021.
Great Britain is no longer covered by the European Union’s procedures for the grant of marketing authorizations (Northern Ireland is covered by the centralized authorization procedure and can be covered under the decentralized or mutual recognition procedures). Various national procedures are now available to place a drug on the market in the United Kingdom, Great Britain, or Northern Ireland, with the main national procedure having a maximum timeframe of 150 days (excluding time taken to provide any further information or data required). The data exclusivity periods in the United Kingdom are currently in line with those in the European Union, but the Trade and Cooperation Agreement provides that the periods for both data and market exclusivity are to be determined by domestic law, and so there could be divergence in the future.
Orphan designation in Great Britain following Brexit is, unlike in the European Union, not available pre-marketing authorization. Applications for orphan designation are made at the same time as an application for a marketing authorization. The criteria to be granted an orphan drug designation are essentially identical to those in the European Union, but based on the prevalence of the condition in Great Britain. It is therefore possible that conditions that were or would have been designated as orphan conditions in Great Britain prior to the end of the Transition Period are or would no longer be and that conditions that were not or would not have been designated as orphan conditions in the European Union will be designated as such in Great Britain.
The European Union’s regulatory environment for clinical trials has been harmonized as part of the Clinical Trials Regulation, which entered into application on January 31, 2022. The MHRA has opened a consultation on proposed revisions to United Kingdom clinical trials legislation, but it is currently unclear as to what extent the United Kingdom will seek to align its regulations with the European Union.
Coverage and Reimbursement
The availability of coverage and adequate reimbursement by third-party payors, including governmental healthcare programs such as Medicare and Medicaid, private health insurers and managed care organizations, is essential for most patients to be able to afford drug products. Achieving acceptable levels of coverage and reimbursement for drug products by third-party payors affects a biopharmaceutical company’s ability to successfully commercialize, and attract collaboration partners to invest in, the development its drug products. Even if coverage is obtained from a third-party payor for a given drug product, the resulting reimbursement rates may not be adequate or may require co-payments that patients find unacceptably high. There is no guarantee that coverage and reimbursement will be provided for a given drug product, and any reimbursement that may become available can be decreased or eliminated in the future.
Third-party payors are increasingly challenging prices charged for drug products and services, and many third-party payors may refuse to provide coverage and reimbursement for particular drug products when an equivalent generic drug product or a less expensive therapy is available. It is possible that a third-party payor may consider a drug product and other therapies as substitutable and only offer to reimburse patients for the less expensive drug product or therapy. Even if a drug product shows improved efficacy or improved convenience of administration, pricing of existing drug products may limit the amount that can be charged for a new drug product. Third-party payors may deny or revoke the reimbursement status of a given drug product or establish prices for new or existing marketed drug products at levels that are too low to enable a biopharmaceutical company to realize an appropriate return on its investment in drug product development.
There is significant uncertainty related to third-party payor coverage and reimbursement of newly approved drug products. In the United States, third-party payors play an important role in determining the extent to which new drugs products will be covered. The Medicare and Medicaid programs increasingly are used as models for how private payors and other governmental payors develop their coverage and reimbursement policies for drug products. Some third-party payors may require pre-approval of coverage for new or innovative devices or drug products before they will reimburse health care providers who use such therapies.
Obtaining and maintaining reimbursement status is time-consuming and costly. No uniform policy for coverage and reimbursement for drug products exists among third-party payors in the United States. Therefore, coverage and reimbursement for drug products can differ significantly from payor to payor. As a result, the coverage determination process is often a time-consuming and costly process that requires the provision of scientific and clinical support for the use of a drug product to each payor separately. Furthermore, rules and regulations regarding reimbursement change frequently and, in some cases, upon short notice.
In addition, companion diagnostic tests require coverage and reimbursement separate and apart from the coverage and reimbursement for their companion pharmaceutical or biological products. Similar challenges to obtaining coverage and reimbursement, applicable to pharmaceutical or biological products, will apply to companion diagnostics. Additionally, if any companion diagnostic provider is unable to obtain reimbursement or is inadequately reimbursed, that may limit the availability of such companion diagnostic, which would negatively impact prescriptions for our product candidates, if approved.
Outside the United States, international operations are generally subject to extensive governmental price controls and other market regulations. Increasing emphasis on cost-containment initiatives in Europe, Canada and other countries puts pressure on the pricing and usage of drug products. In many countries, the prices of drug products are subject to varying price control mechanisms as part of national health systems. Other countries allow companies to fix their own prices for drug products, but monitor and control company profits.
The delivery of healthcare in the European Union, including the establishment and operation of health services and the pricing and reimbursement of drug products, is almost exclusively a matter for national, rather than European Union, law and policy. National governments and health service providers have different priorities and approaches to the delivery of healthcare and the pricing and reimbursement of drug products in that context. In general, however, the healthcare budgetary constraints in most European Union member states have resulted in restrictions on the pricing and reimbursement of drug products by relevant health service providers.
Increasing efforts by governmental and third-party payors in the European Union, the United States and abroad to cap or reduce healthcare costs can cause such organizations to limit coverage and reimbursement for drug products. Additionally, a trend toward managed healthcare, and the influence of health maintenance organizations, have increased pricing pressure on the sale of drug products. The downward pressure on healthcare costs in general, particularly prescription drugs and surgical procedures and other treatments, has become very intense.
Competition
We compete in a highly innovative industry characterized by a rapidly growing understanding of disease biology, evolving technologies and strong intellectual property barriers to entry. While we believe that our DARPin platform and product candidates, strategic collaborations and scientific expertise may provide us with competitive advantages, our business may be impacted competitively from many different sources. We compete with a wide range of pharmaceutical companies, biotechnology companies, academic institutions and other research organizations for novel therapeutic biological targets, new technologies for optimizing antibodies, new scaffolds such as antibody fragments and other small protein-based therapeutics, talent, financial resources, intellectual property rights and collaboration opportunities. Many of our competitors have substantially greater scientific, research and product development capabilities, and greater financial, manufacturing, marketing and sales, and human resources than we do. In addition, there is intense competition for securing clinical trial sites as well as recruiting and registering patients for clinical trials. Many specialized biotechnology companies have formed collaborations with large, established companies to support the research, development and commercialization of products that may be competing with ours. Accordingly, our competitors may be more successful than we may be in developing, commercializing and achieving widespread market acceptance. Any product candidates that we successfully develop and commercialize from our platforms may compete with existing products and new products that may become available in the future.Finally, we also face competition on the DARPin technology, with other companies filing intellectual property on DARPins and striking collaborations with partners to develop DARPin therapeutics; however only we have gathered 20 years of experience with DARPin therapeutics in clinical development.
Competition in the oncology space is intense: common methods of treatment for cancer patients with cancer include surgery, radiation and drug therapy, and approved drugs and methods are widely accepted by physicians, patients and third-party payors. Yet, and in spite of recent progress, and intense competition, unmet medical needs remain high in oncology and call for new treatment options for patients. Competition is especially fierce in hematologic cancers, where several novel approaches including immune engagers, antibody-drug conjugates and cell therapies are becoming part of clinical practice, with next generation molecules and novel targeted approaches under development. AML is a particularly aggressive hematologic cancer and despite recent advancements in the field, most patients relapse after initial treatment. Less than a third of patients are still alive after five years, likely due to the inability of current approaches to effectively eliminate all AML cells (blasts and leukemic stem cells). MP0533, if approved, will enter a competitive market populated by other biologics and small molecules. However, we believe that MP0533 by virtue of its unique multi-targeting molecular design, has the potential to become a more effective and tolerable treatment option for AML and MDS patients, where significant unmet need remains.
Our multispecific cKIT x CD16a x CD47 Switch-DARPin is designed as a next-generation conditioning regimen for hematopoietic stem cell transplantation (HSCT) in AML and beyond. HSCT is an effective treatment against hematological conditions, and it is often performed after administration of intensive chemotherapy regimens and immunosuppressants to maximize its efficacy. However, many older and frailer patients are often ineligible for such conditioning regimens, and instead undergo more tolerable treatments that are associated with relatively shorter durability and poorer outcomes.
Additionally, novel therapies recently approved for non-malignant hematologic conditions such as sickle cell disease also require a conditioning step. Many patients affected by such non-lethal conditions need a safer option to avoid treatment-related toxicities and other complications such as infertility. The field of HSCT conditioning continues to see multiple treatment modalities and new approaches emerging from different competitors that our Switch-DARPin program will likely face. By acting only on hematopoietic stem cells, our Switch-DARPin candidate has the potential to offer a more tolerable, highly effective option to prepare patients with malignant and non-malignant hematologic conditions for HSCT and maximize their chances to increase long-term disease control or even be cured in some cases.
Radioligand therapy (RLT) is a rapidly emerging field in oncology, with over 100 companies developing therapeutic candidates across indications. In addition to a few already approved drugs, several alpha and beta emitting isotopes are being tested, as well as a variety of vectors, including monoclonal antibodies, small molecules and other low molecular weight protein-based scaffolds. Our DARPin radioconjugates, beginning with our frontrunner DLL3 program, will face competition from several other RLT developers. However, we believe DARPin features make them especially well suited as vectors due to their tunability and manufacturing properties.
If approved for solid tumors, MP0317 would compete with agents that are currently in development such as monoclonal antibodies, or mAbs, including bispecifics and fusion proteins, and small molecule approaches.
Our commercial opportunity could be reduced or eliminated if our competitors’ products prove to be safer and more tolerable, more effective, more convenient to dose, less expensive, faster to approve, or more effectively marketed and reimbursed than any of our product candidates that may gain regulatory approval. In addition, the level of generic competition and the availability of reimbursement from government and other third-party payors will impact the commercial viability of our programs.
Manufacturing
We do not currently own or operate manufacturing facilities for the production of clinical or commercial quantities of our DARPin product candidates. We utilize third-party contract manufacturers for the manufacture of drug substances and product candidates for human use. Since we rely on third-party contract manufacturers to produce our proprietary product candidates, we have recruited personnel with experience to manage the third-party contract manufacturers that will produce our proprietary product candidates in clinical or commercial quantities.
We design and develop the manufacturing process for the mono-DARPin proteins and multi-DARPin product candidates that are included in our DARPin product candidates, whether or not they are partnered. For purposes of our and our partner’s DARPin preclinical studies, we supply high quality gram scale DARPin material that we produce in our own facilities. We have fermenter volumes up to ten liters, which provides us with sufficient capacity to produce the quantities needed for DARPin preclinical studies.
Employees
As of December 31, 2023, we had 167.5 full-time equivalent employees (December 31, 2022: 175.3 full-time equivalents). None of our employees are represented by collective bargaining agreements. We believe that we maintain good relations with our employees. At each date shown, we had the following number of full-time equivalent employees, broken out by department.
The majority of our employees are based in Zurich, Switzerland. Three of our employees are based in the United States of America (two as of December 31, 2022)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Full-time equivalent employees |
At December 31, |
|
At December 31, |
|
2023 |
|
2022 |
| Function |
|
|
|
| Research and development |
138.5 |
|
|
142.7 |
|
| Selling, general and administrative |
29.0 |
|
|
32.6 |
|
Total |
167.5 |
|
|
175.3 |
|
C. Organizational Structure.
The following diagram illustrates our corporate structure:
D. Property, Plants and Equipment.
We lease our principal executive office and laboratory space, animal facility and other facilities, consisting of an aggregate of 3,200 square meters, in Zurich-Schlieren, Switzerland. The leases for our principal executive office and laboratory space expire on December 31, 2026. We also have an office in Massachusetts for our U.S. subsidiary, Molecular Partners Inc. consisting of 19 square meters.
We believe our current facilities are sufficient to meet our short-term needs. If we need to add new facilities or expand existing facilities as we add employees, we believe that suitable additional space will be available to accommodate any such expansion of our operations.
Item 4A. Unresolved Staff Comments.
Not applicable.
Item 5. Operating and Financial Review and Prospects.
Overview
We are a clinical-stage biotechnology company pioneering the design and development of DARPin therapeutics for medical challenges that other drug modalities cannot readily address. We have programs in various stages of preclinical and clinical development, currently with main focus on oncology.
DARPin therapeutics are a new class of custom-built protein drug candidates based on natural binding proteins that have the potential to unlock new dimensions of multi-functionality and multi-target specificity in drug design. Our DARPin candidates have been extensively tested in preclinical studies and clinical trials, including in more than 2,500 patients, and have been observed to be highly active and generally well-tolerated. By harnessing DARPins’ intrinsic advantages and leveraging our two decades of experience and leadership with DARPins, we believe our DARPin platform can close the gap between small molecule and antibody medicines as a new therapeutic modality poised to offer clinical breakthroughs.
We were founded in 2004 by the inventors of DARPins. Our senior management, which includes two of our group’s co-founders, has significant prior experience in oncology, research, drug development and finance. Members of our leadership team have served as senior executives at other well-established companies including Amgen, Bavarian Nordic, Genentech, GSK, J&J, Novartis, Roche, and Tesaro. Additionally, our board of directors includes current and former senior executives of AbbVie, Biogen, Novartis, Roche and Takeda (Millennium Pharmaceuticals, Shire). We have collaboration agreements with Novartis and Orano Med, as well as other third party collaborators.
Our operations to date have focused upon organizing and staffing our company, business planning, raising capital, developing our DARPin platform and conducting research, preclinical studies and clinical trials with DARPin therapeutics. We do not have any products approved for sale.
For the year ended December 31, 2023, we incurred a negative net result, attributable to shareholders of CHF 62.0 million; for the year ended December 31, 2022, we incurred a positive net result, attributable to shareholders of CHF 117.9 million; and for the year ended December 31, 2021, we incurred a negative net result of CHF 63.8 million. As of December 31, 2023, we had cumulative losses of CHF 191.8 million.
From inception through December 31, 2023, we have received a total of CHF 446.2 million in funding from our major partnership agreements and we have obtained a total of CHF 322.5 million in eight equity financing rounds, net of cost of capital increases. Since November 2014, we have been listed on the SIX Swiss Exchange, or SIX, under the symbol “MOLN.” Since June 16, 2021, we have also been listed on the Nasdaq Global Select Market, under the symbol “MOLN.” As of December 31, 2023, we had cash and cash equivalents plus short-term time deposits of CHF 186.9 million.
Macroeconomic Considerations
Unfavorable conditions in the economy both in the United States and abroad may negatively affect the growth of our business and our results of operations. For example, macroeconomic events, including a health pandemic, rising inflation, the U.S. Federal Reserve and other financial regulatory agencies raising interest rates, the Russia-Ukraine war and the Israel-Hamas war, have led to economic uncertainty globally. The effect of macroeconomic conditions may not be fully reflected in our results of operations until future periods. If, however, economic uncertainty increases or the global economy worsens, our business, financial condition and results of operations may be harmed. For further discussion of the potential impacts of macroeconomic events on our business, financial condition, and operating results, see the section titled “Risk Factors.”
Licensing and Collaboration Agreements
Research and Development Collaboration and Option Agreement with Orano Med in the area of Radio DARPin Therapies, or the Orano Med Agreement
On January 5, 2024 we announced we entered into a co-development agreement with Orano Med to co-develop 212Pb-based Radio Darpin Therapies (RDT). Under the terms of the co-development agreement, our previously disclosed RDT target DLL3 (delta-like ligand 3) will be included in the collaboration with Orano Med.
Both companies are developing additional radioligand therapy candidates in partnership with other companies, with us having announced our first collaboration with Novartis in December 2021. DLL3 will be exclusively developed by Molecular Partners and Orano Med as a RDT target.
We maintain the option to explore DLL3 for targeted therapy outside of the radiotherapy space. Both companies have committed to sharing the cost of preclinical and clinical development, including for supply of their respective materials.
License and Collaboration Agreement with Novartis in the Area of DARPIN-Conjugated Radioligand Therapies, or the Novartis Radioligand Agreement
On December 14, 2021, we entered into the Novartis Radioligand Agreement with Novartis to develop DARPin-conjugated radioligand therapeutic candidates for oncology.
In January 2022, we received a non-refundable upfront payment of $20 million (CHF 18.6 million) from Novartis. In addition, we are eligible to receive milestone payments of up to $560 million relating to development, regulatory and commercialization activities plus tiered royalties based on commercial sale levels from mid-single digit to low double-digit percentages of net sales of licensed products for a specified period beginning with the first commercial sale of a licensed product in a given country of sale and expiring on the latest of (a) the expiration of the last valid claim covering such product, (b) ten years after such sale, and (c) expiration of regulatory exclusivity.
Novartis Option and Equity Rights Agreement
In October 2020, we entered into an agreement with Novartis, granting Novartis the exclusive option to in-license global rights in relation to MP0420 (ensovibep). Under the terms of the Option and Equity Rights Agreement, in 2020 we received an upfront, non-refundable fee of CHF 20 million for the technology transfer and manufacturing of MP0420.
Ensovibep License Agreement
In January 2022, following positive Phase 2 clinical trial results, Novartis exercised its option for ensovibep, triggering a milestone payment of CHF 150 million due to us, which was received in 2022. Relatedly, we were eligible to invoice Novartis CHF 13.1 million for other items related to ensovibep.
In January 2023, Novartis informed us that it has submitted a request to withdraw, with an effective date of January 25, 2023, the Emergency Use Authorization (EUA) application from the FDA for ensovibep.
On January 5 2024, the Ensovibep License Agreement for the treatment of COVID-19 was terminated and Novartis has returned the rights to the ensovibep program to us. Clinical work on the ensovibep program ended in 2022 and the program remains terminated.
Reservation Agreement with the Swiss Federal Office of Public Health / Bundesamt für Gesundheit, or the FOPH Agreement
On August 11, 2020, we announced the reservation by the FOPH of a defined number of initial doses of our anti-COVID-19 candidate, MP0420. Under the terms of the agreement, we received a reservation fee of CHF 7.0 million which resulted in a contract liability of CHF 7.0 million. With the exercise of the option by Novartis in January 2022 and the subsequent assignment of the agreement to Novartis, we recognized the CHF 7.0 million contract liability as revenue in 2022.
License and Collaboration Agreement with Amgen, or the Amgen Collaboration Agreement
In December 2018, we entered into the Amgen Collaboration Agreement for the clinical development and commercialization of MP0310 / AMG 506.
Under the agreement, we granted to Amgen an exclusive worldwide, royalty-bearing, sublicensable license under our patents and know-how relating to MP0310 / AMG 506 to develop and commercialize MP0310 / AMG 506.
In April 2022, Amgen informed us of its decision to return the global rights of MP0310 to us following a strategic pipeline review.
Royalties and License Fees
We currently hold a non-exclusive perpetual license from the University of Zurich on patent applications and patents relating to Phage Display technology. The amount we are required to pay is CHF 10,000 per annum.
Components of Results of Operations
Revenues
As described above, we have entered into licensing and collaboration agreements pursuant to which we generally have been and will be entitled to upfront fees and milestone payments upon the achievement of predetermined development, regulatory and sales events. Our revenue to date has primarily consisted of amounts received under our collaboration agreements, including upfront fees, option exercise fees, milestone payments and sponsored research payments. In addition, under the collaboration agreements, we will generally be entitled to royalty payments on the net sales of products ultimately developed and commercialized under our partnerships. For any of our proprietary product candidates that we have not yet licensed, we may decide to retain all or a portion of the commercialization rights. To date, we have not generated any revenue from commercial product sales.
Our revenue may vary substantially from quarter to quarter and year to year, depending on the structure and timing of milestone events, as well as the development and marketing strategies of commercialization partners from whom we will be entitled to receive royalty and other payments. We believe that period-to-period comparisons of our results of operations are therefore not meaningful and should not be relied on or to be indicative of our future performance.
Operating expenses
Our operating expenses consist primarily of costs associated with research, preclinical studies and clinical testing, personnel-related costs and, to a lesser extent, royalty and license fees, facility expenses, professional fees for legal, tax, audit and strategic purposes, administrative expenses and depreciation of property, plant and equipment.
We expect our operating expenses to increase as compared to prior periods in connection with our ongoing activities, particularly as we continue the development of our proprietary product candidates, expand our proprietary product pipeline and invest in our DARPin platform. Our operating expenses may vary substantially from period to period mainly driven by the timing of enrollment of patients in clinical trials and other research and development activities. Following our listing on the Nasdaq Global Select Market in June 2021, we have and expect to continue to incur additional costs associated with operating as a public company in the United States.
Research and development expenses
Research and development expenses consist primarily of compensation and other expenses related to:
•Research and development personnel;
•Preclinical studies and clinical trials of our product candidates, including the costs of manufacturing the product candidates;
•Research and services under our partnership agreements; and
•Attributable facility expenses, including depreciation and amortization of equipment and any intangible research and development assets.
From inception through December 31, 2023, we cumulatively have spent approximately CHF 480 million on research and development activities which we classify as research and development expense for financial reporting purposes.
At this time, we cannot reasonably estimate the nature, timing and estimated costs of the efforts that will be necessary to complete the development of, or the period, if any, in which material net cash inflows may commence from, any of our product candidates. This is due to numerous risks and uncertainties associated with developing product candidates, including the uncertainty of:
•the scope, rate of progress, results and cost of our clinical trials, preclinical studies and other related activities;
•the cost of manufacturing clinical supplies and establishing commercial supplies of our product candidates and any products that we may develop;
•the number and characteristics of product candidates that we pursue;
•the cost, timing, and outcomes of regulatory approvals;
•the cost and timing of establishing sales, marketing and distribution capabilities; and
•the terms and timing of any collaborative, licensing and other arrangements that we may establish, including any required milestone and royalty payments thereunder.
A change in the outcome of any of these variables with respect to the development of any of our current or future product candidates could mean a significant change in the costs and timing associated with the development of such product candidates.
At this time, due to the inherently unpredictable nature of preclinical and clinical development and given the early stage of many of our programs and/or product candidates, we generally do not track our internal research and development expenses on a program-by-program basis as they primarily relate to personnel, research and consumable costs, which are deployed across multiple projects under development. A portion of our research and development costs are external costs, which we do track on a program-by-program basis following the program's nomination to the development candidate stage. Included in table below are our external research and development expenses as well as external clinical and regulatory costs, presented by our most significant programs:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
For the year ended |
|
|
For the year ended |
|
|
For the year ended |
|
|
December 31, 2023 |
|
|
December 31, 2022 |
|
|
December 31, 2021 |
|
|
in CHF thousands |
|
in CHF thousands |
|
in CHF thousands |
| External research and development expense |
|
|
|
|
|
| MP0250 |
112 |
|
|
|
144 |
|
|
|
856 |
|
|
| MP0274 |
(29) |
|
|
|
402 |
|
|
|
946 |
|
|
| MP0310 |
628 |
|
|
|
2,085 |
|
|
|
2,353 |
|
|
| MP0317 |
3,739 |
|
|
|
2,967 |
|
|
|
2,863 |
|
|
| MP0533 |
5,609 |
|
|
|
6,647 |
|
|
|
2,211 |
|
|
| MP0420 / MP0423 |
6 |
|
|
|
1,258 |
|
|
|
13,224 |
|
|
| Other research and development expense |
6,333 |
|
|
|
5,037 |
|
|
|
4,248 |
|
|
| Total |
16,398 |
|
|
|
18,540 |
|
|
|
26,701 |
|
|
We charge all research and development expenses, including internal patent filing and patent maintenance costs, to research and development expenses when incurred, as the criteria for capitalization are currently not met.
Selling, general and administrative expenses
Our selling, general and administrative costs principally consist of salaries and related benefits, including share-based compensation, for personnel in our executive, finance and other administrative functions. Other selling, general and administrative costs include facility-related costs and professional services fees for auditing, tax and general legal services, as well as expenses associated with the requirements of being a listed public company listed both in Switzerland on the SIX and in the United States on Nasdaq.
Financial income and financial expenses
Financial income consists primarily of interest earned on our cash and cash equivalents and short-term time deposits as well as realized and unrealized gains of foreign exchange. The financial expenses are driven by realized and unrealized foreign exchange losses and negative interest on certain cash balances.
Income taxes and taxation
Income taxes
We have operating entities in two jurisdictions. In Switzerland, due to losses incurred to-date, we have not paid any income taxes since inception. For our U.S. based activities, we have paid the required tax amounts of both federal and state taxes, which are not material to our financial results.
Deferred taxes
We are entitled under Swiss laws to carry forward any losses incurred for a period of seven years and can offset our losses carried forward against future taxes. As of December 31, 2023, in Switzerland, we had tax loss carry-forwards totaling CHF 144.5 million. No deferred tax assets have been recognized for these tax loss carry-forwards because as of December 31, 2023, it was not probable that such loss carry-forwards can be utilized in the foreseeable future. In addition, no deferred tax assets were recognized on other deductible temporary differences (e.g.
pension liabilities) due to the significant tax losses carried forward.
A. Operating Results
Analysis of Results of Operations
Comparison of Operations for the Years Ended December 31, 2023, 2022 and 2021
The following table sets forth summaries of our statements of income for the years ended December 31, 2023, 2022 and 2021 (in thousands CHF):
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
For the year ended
December 31, 2023
|
|
For the year ended
December 31, 2022
|
|
For the year ended
December 31, 2021
|
|
in CHF thousands |
|
in CHF thousands |
|
in CHF thousands |
| Revenues and other income |
|
|
|
|
|
| Revenues from research and development collaborations |
7,038 |
|
|
|
189,556 |
|
|
|
9,330 |
|
|
| Other income |
— |
|
|
|
44 |
|
|
|
424 |
|
|
| Total revenues and other income |
7,038 |
|
|
|
189,600 |
|
|
|
9,754 |
|
|
| Operating expenses |
|
|
|
|
|
|
| Research and development expenses |
(48,784) |
|
|
|
(50,749) |
|
|
|
(55,718) |
|
|
| Selling, general and administrative expenses |
(19,362) |
|
|
|
(22,238) |
|
|
|
(17,454) |
|
|
| Total operating expenses |
(68,146) |
|
|
|
(72,987) |
|
|
|
(73,172) |
|
|
| Operating result |
(61,108) |
|
|
|
116,613 |
|
|
|
(63,418) |
|
|
| Financial income |
4,279 |
|
|
|
1,859 |
|
|
|
191 |
|
|
| Financial expenses |
(5,155) |
|
|
|
(619) |
|
|
|
(556) |
|
|
| Result before income taxes |
(61,984) |
|
|
|
117,853 |
|
|
|
(63,783) |
|
|
| Income taxes |
— |
|
|
|
— |
|
|
|
(2) |
|
|
| Net result, attributable to shareholders |
(61,984) |
|
|
|
117,853 |
|
|
|
(63,785) |
|
|
Revenues and other income
In the year ended December 31, 2023, we recognized total revenues and other income of CHF 7.0 million, a decrease of CHF 182.6 million compared to the previous year. Revenues in the year ended December 31, 2023, were exclusively driven by the Novartis Radioligand Agreement. The revenue in 2022 largely related to the Novartis Option and Equity Rights Agreement.
The revenue in the year ended December 31, 2021, related largely to the Amgen Collaboration Agreement]. In 2023 and 2022 we also recorded other income related to agency fees that were invoiced to Novartis.
Operating expenses (including depreciation and amortization)
In the year ended December 31, 2023, total operating expenses decreased by CHF 4.9 million to CHF 68.1 million (2022: CHF 73.0 million, 2021: CHF 73.2 million). These costs included CHF 5.7 million in non-cash effective share-based compensation and pension costs as well as CHF 2.4 million in depreciation.
The two major expense categories were consistently personnel expenses of CHF 40.0 million (59% of total operating expenses) and research consumables and project costs totaling CHF 15.9 million (23% of total operating expenses).
Research and development expenses
Total research and development expenses in the year ended December 31, 2023, were CHF 48.7 million (2022: CHF 50.7 million, 2021: CHF 55.7 million). We charge all research and development expenses, including internal patent filing and patent maintenance costs, to the statement of operations when incurred.
Selling, general and administrative expenses
Total selling, general and administrative expenses in the year ended December 31, 2023, decreased by CHF 2.9 million (13%) to CHF 19.4 million (2022: CHF 22.3 million, 2021: CHF 17.5 million), mainly reflecting the first full 12 month period of directors and officers insurance cost and professional services costs associated with our June 2021 listing on the Nasdaq Global Select Market as well as growth in personnel costs for the administrative functions.
Financial income / financial expense
In the year ended December 31, 2023, we recorded a net financial gain of CHF 0.9 million, mainly driven by interest income and exchange losses on the cash positions held in foreign currencies, whereas in the year ended December 31, 2022, there was a net financial gain of CHF 1.2 million also mainly driven by interest income and foreign exchange gains (2021: net financial loss of CHF 0.4 million). Financial income / expense in all three years are primarily driven by foreign exchange results on the cash positions held in foreign currencies.
Income taxes
The Swiss legal entity of the Company did not have to pay or accrue any income taxes during the reporting period, as the Company incurred a taxable loss in 2023. Including the net operating loss for the year ended December 31, 2023, the remaining tax losses of CHF 144.5 million may be used as tax loss carryforwards to offset future taxable income over a period of seven years. No deferred tax assets have been recognized for these tax loss carryforwards, because at December 31, 2023, we determined it was unlikely that such loss carryforwards could be utilized in the foreseeable future.
Future taxable income in Switzerland will be subject to federal, cantonal and communal income taxes. The applicable income tax rate in Switzerland for the year ended December 31, 2023 was 19.3%.
B. Liquidity and Capital Resources
From inception through December 31, 2023, we have raised an aggregate of CHF 322.5 million of net proceeds from the sale of our ordinary shares to founders and investors and collected cash under our partnership agreements in an aggregate amount of CHF 446.2 million. Our primary uses of cash is to fund our ongoing research and development activities and other operating expenses. We currently have no ongoing material financing commitments, such as lines of credit or guarantees.
As of December 31, 2023, we had CHF 186.9 million in cash and cash equivalents and short-term time deposits. We are investing our cash in risk-free money market instruments in line with our treasury guidelines to accommodate our financial needs over time.
We expect our expenses to increase in connection with our ongoing activities, particularly as we continue the research and development of, continue or initiate clinical trials of, and seek marketing approval for, our product candidates. In addition, if we obtain marketing approval for any of our product candidates, we expect to incur significant commercialization expenses related to program sales, marketing, manufacturing and distribution to the extent that such sales, marketing and distribution are not the responsibility of current or future collaborators. Furthermore, following our June 2021 listing on the Nasdaq Global Select Market, we have and will continue to incur additional costs associated with operating as a public company in the United States. If we are unable to raise capital when needed or on attractive terms, we would be forced to delay, reduce or eliminate our research and development programs or future commercialization efforts.
Comparison of cash and cash equivalents and short-term time deposits as of December 31, 2023, 2022 and 2021 and cash flows for the years ended December 31, 2023, 2022 and 2021.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
As of and for the year ended |
|
As of and for the year ended |
|
|
As of and for the year ended |
|
|
December 31, 2023 |
|
December 31, 2022 |
|
|
December 31, 2021 |
|
|
in CHF thousands |
|
in CHF thousands |
|
in CHF thousands |
| Cash and cash equivalents |
67,309 |
|
|
|
87,946 |
|
|
|
71,813 |
|
|
| Short-term time deposits |
119,580 |
|
|
|
161,198 |
|
|
|
61,000 |
|
|
Total |
186,889 |
|
|
|
249,144 |
|
|
|
132,813 |
|
|
|
|
|
|
|
|
| Net cash (used in) from operating activities |
(59,005) |
|
|
|
118,566 |
|
|
|
(90,953) |
|
|
| Net cash from (used in) investing activities |
44,637 |
|
|
|
(101,121) |
|
|
|
(22,237) |
|
|
| Net cash (used in) from financing activities |
(1,167) |
|
|
|
(1,570) |
|
|
|
50,581 |
|
|
| Exchange (loss) gain on cash positions |
(5,102) |
|
|
|
258 |
|
|
|
701 |
|
|
|
|
|
|
|
|
|
| Net (decrease) increase in cash and cash equivalents |
(20,637) |
|
|
|
16,133 |
|
|
|
(61,907) |
|
|
All short-term time deposits at December 31, 2023 and 2022 were held with Swiss banks. Please refer to note 25 to our consolidated financial statements as of and for the year ended December 31, 2023 included elsewhere in this Annual Report on Form 20-F.
Net cash from (used in) operating activities
During the year ended December 31, 2023, operating activities used CHF 59.0 million of cash, primarily as a result of the negative net result attributable to shareholders of CHF 62.0 million.
During the year ended December 31, 2022, operating activities generated CHF 118.6 million of cash, primarily as a result of the positive net result attributable to shareholders of CHF 117.8 million driven by the revenue generated by the option exercise by Novartis.
During the year ended December 31, 2021, operating activities used CHF 91.0 million of cash, primarily as a result of the negative net result attributable to shareholders of CHF 63.8 million together with a reduction in contract liability and an increase in trade and other receivables following the Novartis license and collaboration agreement signed in December 2021.
Net cash used in investing activities
During the years ended December 31, 2023, 2022 and 2021, cash used in, or from investing activities primarily related to movements in investments in short-term time deposits of CHF 41.6 million, and cash used in investing activities of CHF 100.2 million and CHF 21.0 million, respectively.
During the years ended December 31, 2023, 2022 and 2021, we recorded a cash outflow for the acquisition of property, plant and equipment and intangible assets of CHF 0.8 million, CHF 1.4 million and CHF 1.3 million, respectively.
During the years ended December 31, 2023, 2022 and 2021, we recorded a cash inflow on interest received of CHF 3.8 million, CHF 0.5 million and CHF 0.1 million, respectively.
Net cash from (used in) financing activities
During the year ended December 31, 2023, net cash used in financing activities was CHF 1.2 million, driven primarily by payments of our lease liabilities. During the year ended December 31, 2022, net cash used in financing activities was CHF 1.6 million, driven primarily by payments of our lease liabilities and the investment related to the creation of treasury shares. During the year ended December 31, 2021, net cash from financing activities was CHF 50.6 million, primarily related to the proceeds from issuance of new shares, net of transaction costs, following our initial public offering of ADSs in the U.S. in June 2021.
Funding requirements
We believe that our existing cash and cash equivalents and short-term time deposits as of December 31, 2023, will be sufficient to fund our operating expenses and capital expenditure requirements well into 2026. However, our present and future funding requirements may change and will depend on many factors, including, among other things:
•timelines for preclinical and clinical development programs;
•change in product development plans needed to address any set-backs in our research and development activities;
•scope, prioritization and number of clinical trials and research and development activities;
•rate of progress and cost of the clinical trials, and other research and development activities;
•terms and timing of any collaborative, licensing and other arrangements that may be established;
•costs and timing of preparing, filing, prosecuting, maintaining, defending and enforcing patent claims and other intellectual property rights;
•the need or decision to acquire or license complementary compounds, technologies or complementary businesses or companies;
•regulatory approval, manufacturing or commercialization of our product candidates for which we receive marketing approval through partners;
•costs, timing and outcome of regulatory review of our product candidates;
•costs and timing of future commercialization activities, including drug manufacturing, marketing, sales and distribution, for any of our product candidates for which we receive marketing approval;
•changes in regulatory policies or laws that affect our operations; and
•competing medical treatment and market developments.
We expect our operating expenses to increase over the next several years as we expand our research and development activities. Until such time as we can generate significant revenue from product sales or royalties, if ever, we expect to finance our operations through a combination of public or private equity and debt financings or other sources, including payments upon achievement of certain development, regulatory and sales milestone events and royalty payments under our existing partnership agreements, and future collaborations with other third parties. To the extent that we raise additional capital through the sale of equity or convertible debt securities, our shareholders' ownership interest may be diluted, and the terms of any additional securities may include liquidation or other preferences that adversely affect the rights of our shareholders. Debt financing, if available, may involve agreements that include covenants limiting or restricting our ability to take specific actions, such as incurring additional debt, making capital expenditures or declaring dividends. If we raise funds through additional collaborations, strategic alliances or licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies, future revenue streams, research programs or product candidates or to grant licenses on terms that may not be favorable to us. If we are unable to raise additional funds through equity or debt financings when needed, we may be required to delay, limit, reduce or terminate our product development or future commercialization efforts or grant rights to develop and market product candidates that we would otherwise prefer to develop and market ourselves.
C. Research and Development
For a discussion of our research and development activities, see “Item 4.B-Business Overview” and “Item 5.A-Operating Results.”
D. Trend Information
For a discussion of trends, see “Item 5.A-Operating Results” and “Item 5.B-Liquidity and Capital Resources.”
E. Critical Accounting Estimates
Not applicable.
Item 6. Directors, Senior Management and Employees.
A. Directors and Senior Management.
The following table sets forth information regarding our executive officers, also referred to as members of the Management Board, and directors as of December 31, 2023. Unless otherwise stated, the business address for our directors and executive officers is c/o Molecular Partners AG, Wagistrasse 14, 8952 Schlieren, Switzerland.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Name |
|
Age |
|
Position |
| Executive Officers |
|
|
|
|
| Dr. Patrick Amstutz |
|
48 |
|
Chief Executive Officer and Director |
| Renate Gloggner |
|
53 |
|
EVP People and Community |
| Dr. Nicolas Leupin |
|
50 |
|
Former Chief Medical Officer |
| Dr. Michael Tobias Stumpp |
|
51 |
|
EVP Projects |
| Alexander Zürcher |
|
48 |
|
Chief Operating Officer |
| Non-Employee Directors |
|
|
|
|
| William M. Burns |
|
76 |
|
Chairman of the Board |
| Dr. Agnete Fredriksen |
|
46 |
|
Director |
| Dr. Dominik Höchli |
|
56 |
|
Director |
| Steven H. Holtzman |
|
69 |
|
Director |
| Sandip Kapadia |
|
53 |
|
Director |
| Dr. Vito J. Palombella |
|
61 |
|
Director |
| Dr. Michael Vasconcelles |
|
60 |
|
Director |
Executive Officers
Dr. Patrick Amstutz, Ph.D., one of our founders, has served as our Chief Executive Officer since November 2016, as an executive director since 2017 and as a member of the Company’s management team since its inception in 2004. Previously, he served as our Chief Operating Officer from 2014 to 2016 and as our Chief Business Officer from 2006 to 2014. Since 2017, Dr. Amstutz has served as Vice-President of the Board of the Swiss Biotech Association. Dr. Amstutz holds a Master of Science from the ETH Zurich and a Ph.D. in molecular biology from the University of Zurich. Our board of directors believes that Dr. Amstutz's leadership of our company since its inception as well as his scientific background provide him with the qualifications and skills to serve as a director.
Renate Gloggner has served as EVP People and Community and a Member of the Management Board of Molecular Partners since July 2022. She joined the Company in October 2021. Prior to joining Molecular Partners, Renate held European and International Human Resource leadership positions at two US companies, Global Blood Therapeutics and Tesaro Bio. In both companies, she built strong teams with an engaging culture in the European headquarter as well as in several European countries, allowing these teams to successfully gain market access and launch products. Renate began her career in biotech at Biogen and Amgen working in a variety of HR roles in the international headquarter as well as in country roles. She holds an MBA from the University of Bern, Switzerland and an executive coaching degree from the University of the West of England, Bristol.
On August 24, 2023, the Company announced the departure of the Chief Medical Officer Dr. Nicolas Leupin, M.D., PhD by end of 2023 for personal reasons.
Dr. Nicolas Leupin, M.D., Ph.D., served as our Chief Medical Officer since September 2019 until December 31, 2023. Dr. Leupin is a medical oncologist with a successful track record in drug development, most recently as Chief Medical Officer of argenx from 2016 to 2019, a clinical-stage biotechnology company developing antibody-based therapies for treatment of severe autoimmune diseases and cancer. In that role he led the company’s global clinical strategy and execution, successfully supporting the company’s transformation into a late-stage clinical company, and was responsible for translating preclinical hypotheses into innovative proof-of concept clinical trials. Prior to argenx, Dr. Leupin held roles of increasing responsibility at Celgene, where he supported the clinical development of several drug candidates in lymphoma and multiple myeloma, resulting in regulatory filings in Europe and the United States.
Dr. Michael Tobias Stumpp, Ph.D., one of our founders, has served as EVP Projects and a Member of the Management Board of Molecular Partners. Michael is a co-founder of Molecular Partners and was part of the team that invented the DARPin technology. Michael previously served as Chief Scientific Officer of Molecular Partners, in which capacity he oversaw development of the DARPin pipeline. He started his scientific career at the ETH Zurich and then progressed to the Imperial College London and the Tokyo Institute of Technology. Michael has published his research in many international, peer-reviewed scientific journals and presented his findings at numerous congresses.
Alexander Zürcher has served as Chief Operating Officer and a Member of the Management Board of Molecular Partners since 2022. Prior to this role, he served as SVP of Development, where he oversaw project and portfolio management, manufacturing, pharmacology, and quality assurance activities. Alexander has also previously been VP Operations and Director of CMC. He has more than 20 years of industry experience, with prior work in drug development as Director of Drug Product Development at Cytos Biotechnology and Head of R&D Operations at Spirig Pharma. Alexander holds a M.Sc. degree in biology from the University of Basel, as well as a Certificate of Advanced Studies in Business Management from the University of Zurich.
Non-Employee Directors
William M. Burns has served as Chairman of our board of directors since April 2018 and a director since October 2017. His professional career has been spent in the Life Sciences sector. His career in Roche took him to CEO of the Pharma Division and to the Boards of Genentech and Chugai. From 2010 to 2014 he also served as a Non-Executive Director of F Hoffmann La Roche. He is currently chair of Vestergaard sarl, vice chair of Mesoblast in Australia and is a Trustee of the Institute of Cancer Research in London. He also serves on a Cancer Advisory board to the Universities of Aachen/Bonn/Cologne and Dusseldorf. Mr. Burns holds an honors degree in economics from the University of Strathclyde, Glasgow, Scotland. Our board of directors believes that Mr. Burns' experience with the healthcare and pharmaceutical industries and his broad management experience provide him with the qualifications and skills to serve as a director.
Dr. Agnete Fredriksen has served as a director since April 2021. Dr. Fredriksen has served as a co-founder and chief business officer of Nykode Therapeutics AS (formerly Vaccibody AS) since August 2022, as chief innovation and strategy officer from July 2021 to July 2022 and as president and chief scientific officer from 2017 to June 2021. Nykode Therapeutics is a clinical-stage biopharmaceutical company dedicated to the discovery and development of novel immunotherapies for cancer and infectious diseases. Prior to founding Vaccibody Dr. Fredriksen previously held researcher roles at Affitech AS, a private technology transfer company, and Medinnova AS, a technology transfer company. Dr. Fredriksen is the author of numerous scientific papers in the field of immunology, immunotherapy and vaccines, and has been awarded several patents in the field of immunotherapy. She holds an MSc and a Ph.D. from the Institute of Immunology, Oslo University Hospital, Rikshospitalet in Oslo, Norway. Our board of directors believes that Dr. Fredriksen’s experience in immunotherapy and vaccine development, as well as her medical and scientific background, provide her with the qualifications and skills to serve as a director.
Dr. Dominik Höchli has served as a director since April 2021. He has more than 20 years of experience as a marketing and medical affairs executive. Since spring 2021 he is the CEO of Catapult Therapeutics, a clinical stage biotech company in the Netherlands. Previously he worked at AbbVie as Vice President, Head of Global Medical Affairs and member of the R&D and the Commercial leadership team. He led global product launches for major blockbuster products, including HUMIRA, Maviret, Venetoclax and Skyrizi, and his leadership experience ranges from smaller country organizations to large global functions.
He began his corporate career at McKinsey & Co. Dr. Höchli is a Swiss national and obtained his medical degree (M.D.) from the University of Bern in Switzerland. Our board of directors believes that Dr. Höchli’s over 20 years of experience as a marketing and medical affairs executive, as well as his broad business experience provide him with the qualifications and skills to serve as a director.
Steven H. Holtzman has served as a director since May 2014. He has served as chair of the board of directors of, and strategic business advisor to, CAMP4 Therapeutics Corporation, a private biopharmaceutical company, since October 2019, executive chair of the board of directors of, and a strategic business advisor to, Qihan Biotech, a private biopharmaceutical company, since April 2019, and as a founder, a strategic business advisor, and a member and the lead independent director of the board of directors of Shoreline Bio, a private biopharmaceutical company, since June 2020. From July 2016 to January 2020, Mr. Holtzman was the first President and Chief Executive Officer and a member of the board of directors of Decibel Therapeutics, Inc., a public biopharmaceutical company. From January 2011 to March 2016, he served as the Executive Vice President of Corporate Development at Biogen, Inc., a public biopharmaceutical company. From 2001 to 2010, he served as a founder, chair of the board of directors, and Chief Executive Officer of Infinity Pharmaceuticals, Inc., a public biopharmaceutical company. Additionally, Mr. Holtzman was Chief Business Officer of Millennium Pharmaceuticals, Inc., a public biopharmaceutical company, from May 1994 to June 2001, and a founder, member of the board of directors, and Executive Vice President of DNX Corporation, a public biopharmaceutical company, from August 1986 to March 1994. He is a trustee of The Berklee College of Music and a Senior Fellow at the Belfer Center for Science and International Affairs at the Harvard Kennedy School. He received his B.A. in Philosophy from Michigan State University and his B.Phil. in Philosophy from Corpus Christi College, Oxford University, which he attended as a Rhodes Scholar. Our board of directors believes that Mr. Holtzman's experience in the biotechnology industry and his broad management experience provide him with the qualifications and skills to serve as a director.
Sandip Kapadia has served as a director since April 2020. Mr. Kapadia brings over 25 years of science industry experience and has served as the Chief Financial Officer (CFO) for Harmony Biosciences since March 2021. Previously Mr. Kapadia was CFO for Intercept Pharmaceuticals. Before Intercept, Mr. Kapadia served in various leadership capacities within finance for more than 19 years at Novartis International AG and Novartis affiliates in the United Kingdom, Netherlands, Switzerland and the US. Mr. Kapadia received a B.S. in Accounting from Montclair State University and an M.B.A. from Rutgers University, and is also a US Certified Public Accountant. Mr. Kapadia currently serves on the board of directors of Passage Bio, and previously on the board of directors of VectivBio Holding AG and Therachon AG. We believe that Mr. Kapadia is qualified to serve on our board of directors due to his leadership experience in the biopharmaceutical industry and finance expertise. Our board of directors believes that Mr. Kapadia’s over 25 years of experience in the life science industry and his broad finance and management experience provide him with the qualifications and skills to serve as a director.
Dr. Vito J. Palombella, Ph.D., has served as a director since April 2020. Vito J. Palombella, Ph.D., has over 30 years of scientific leadership and experience advancing first-in-class therapeutic programs, as well as a successful track record of building drug discovery and development organizations. Currently, Dr. Palombella is the chief scientific officer of TRIANA Biomedicines. Prior to joining TRIANA, Dr. Palombella was the Chief Scientific Officer of Surface Oncology from January 2016 to August 2023, where he was responsible for drug discovery and preclinical development. Prior to Surface Oncology, Dr. Palombella was executive vice president and chief scientific officer from May 2010 to January 2016, and vice president, biology/research, from 2004 to 2010, at Infinity Pharmaceuticals, Inc., where he was responsible for drug discovery and preclinical development. Prior to that, he was director of molecular biology and protein chemistry at Syntonix Pharmaceuticals, and senior director of cell and molecular biology at Millennium Pharmaceuticals and held a number of positions at LeukoSite and ProScript.
Dr. Palombella was involved in the discovery and development of bortezomib (Velcade®), a proteasome inhibitor, and duvelisib (Copiktra®), a PI3K-d/g inhibitor, both for cancer therapy. Dr. Palombella earned his bachelor’s degree in microbiology from Rutgers University and a master’s degree and doctorate degree in viral oncology and immunology from the New York University Medical Center and completed his post-doctoral training at Harvard University. Our board of directors believes that Dr. Palombella’s over 30 years of scientific leadership and experience, as well as his medical and scientific background, provide him with the qualifications and skills to serve as a director.
Dr. Michael Vasconcelles, M.D., is currently Executive Vice President, Research, Development, and Medical Affairs at Immunogen. He was most recently the chief medical officer and Head of the Medical and Scientific Organization at Flatiron Health, a healthcare technology and services company focused on creating digital solutions to accelerate cancer research and improving patient care. Prior to joining Flatiron Health in 2019, Dr. Vasconcelles served as the Chief Medical Officer of Unum Therapeutics Inc. (Unum) from 2015-2019. A Cambridge, MA-based cell and gene therapy company, Prior to Unum, Dr. Vasconcelles spent several years at Takeda/Millennium, where he was Senior Vice President, Head of the Oncology Therapy Area Unit and member of the R&D Executive Team, accountable for strategic and operational oversight of the oncology research and development portfolio globally. Prior to Takeda/Millennium, Dr. Vasconcelles was Group Vice President and the Global Therapeutic Area Head, Transplant and Oncology, at Genzyme Corporation, where he was responsible for clinical development of the transplant and oncology portfolio and a member of the Transplant and Oncology Business Unit Management Team. Following Sanofi’s acquisition of Genzyme, Dr. Vasconcelles joined Sanofi Oncology as Head, Personalized Medicine and Companion Diagnostics. From 1996 -2021, Dr. Vasconcelles was a faculty member of the Harvard Medical School and an associate physician at Brigham and Women’s Hospital and Dana-Farber Cancer Institute. He received both his B.A. and M.D. from Northwestern University. Our board of directors believes that Dr.Vasconcelles’ extensive experience in the life sciences industry and clinical development programs, as well as his medical and scientific background, provide him with the qualifications and skills to serve as a director.
The table below provides certain highlights of the composition of our board members and nominees. Each of the categories listed in the below table has the meaning as it is used in Nasdaq Rule 5605(f).
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Board Diversity Matrix |
| Country of Principal Executive Offices: |
Switzerland |
| Foreign Private Issuer |
Yes |
| Disclosure Prohibited under Home Country Law |
No |
| Total Number of Directors |
8 |
|
Female |
Male |
Non- Binary |
Did Not Disclose Gender |
| Part I: Gender Identity |
|
| Directors |
1 |
7 |
0 |
0 |
| Part II: Demographic Background |
|
| Underrepresented Individual in Home Country Jurisdiction |
2 |
| LGBTQ+ |
1 |
| Did Not Disclose Demographic Background |
0 |
Family Relationships
There are no family relationships among any of our executive officers or directors.
B. Compensation.
Compensation of Executive Officers and Directors
The aggregate compensation paid by us to our executive officers and directors, including share-based compensation, for the year ended December 31, 2023, was CHF 5,288,000. Out of this amount a total of CHF 417,000 related to pension benefits and social security contributions.
Director Compensation
As required by the "Say on Pay" rules, our articles of association set out the principles for the elements of the compensation of the members of our board of directors. The compensation of the members of our board of directors may consist of fixed and variable compensation. The total compensation takes into account the position and level of responsibility of the respective member of the board of directors, including board and committee chairmanship and membership and a travel fee. Members of our board of directors are paid for their service over one year starting with their election at the ordinary shareholders’ meeting and ending with the subsequent ordinary shareholders’ meeting. Our shareholders at the 2022 annual general meeting held on April 13, 2022, set the maximum aggregate amount of compensation for the board of directors for their term of office until the 2023 general meeting at CHF 1,091,400.
Our shareholders at the 2023 annual general meeting held on April 4, 2023, set the maximum aggregate amount of compensation for the board of directors for their term of office until the 2024 general meeting at CHF 1,091,400.
For the year ended December 31, 2023, the compensation of the members of our board of directors consisted of fixed compensation only. Compensation of the members of our board of directors for the year ended December 31, 2023 consisted of a fixed cash fee and restricted share units, or RSUs. The following table sets out information regarding the compensation earned by our directors for service on our board of directors during the year ended December 31, 2023. Dr. Amstutz, our Chief Executive Officer and a member of our board of directors, does not receive any additional compensation for his service as a director.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Name |
|
Fees Earned |
|
RSUs |
|
Total(1) |
|
|
in CHF thousands |
| William M. Burns |
|
125 |
|
170 |
|
|
295 |
|
| Steven H. Holtzman |
|
50 |
|
85 |
|
|
135 |
|
Sandip Kapadia |
|
45 |
|
85 |
|
|
130 |
|
Vito J. Palombella |
|
40 |
|
85 |
|
|
125 |
|
Michael Vasconcelles |
|
50 |
|
85 |
|
|
135 |
|
Dr. Agnete Fredriksen |
|
40 |
|
85 |
|
|
125 |
|
Dr. Dominik Höchli |
|
48 |
|
85 |
|
|
133 |
|
Dr. Patrick Amstutz(2) |
|
— |
|
— |
|
|
— |
|
(1)The total compensation awarded to the members of the board of directors shown in this table does not include the payments of TCHF 17 we made in 2023 to cover the mandatory employer social security contribution on the base fees and the vesting of the RSUs 2020. In addition, upon vesting of the RSUs 2023 in 2026, we will be obliged to make employer contributions to social security pursuant to applicable mandatory law. As an estimate based on currently applicable contribution rates, the employer contributions on the RSUs 2023 expected to vest in 2026 will amount to TCHF 26.
(2)For our Chief Executive Officer’s compensation other than in connection with his service on our board of directors, please refer to “— Executive Compensation.”
As of December 31, 2023, all members of our board of directors were non-executives, except for Dr. Amstutz. None of the members of our board of directors has any significant business connections with the Company or was a member of the Management Board of the Company, except for Dr. Amstutz, who has been a member of the Management Board since the Company's inception in 2004.
Except as described in the section of this Annual Report on Form 20-F entitled “Related Party Transactions—Agreements with Our Directors and Executive Officers”, there are no arrangements or understandings between us and any of our directors providing for benefits upon termination of their service as our directors.
Executive Compensation
The compensation of the Management Board, also referred to herein as our executive officers, may consist of fixed and variable compensation. Fixed compensation comprises the base salary and the corresponding pension contributions.
Variable compensation comprises short-term and long-term variable compensation elements:
•the short-term variable compensation, paid as a cash bonus, is determined exclusively by the achievement of predefined annual corporate goals; and
•the long-term variable compensation, granted as Performance Share Units, or PSUs, is determined based on (i) the achievement of annual corporate goals, (ii) the achievement of long-term value-driving milestones outside of such corporate goals and (iii) the development of the share price of the Company.
The following table sets out information regarding compensation earned by members of the Management Board during the year ended December 31, 2023.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Name and principal position |
|
Salary |
|
Bonus(1) |
|
Equity Awards |
|
Non-Equity
Incentive Plan
Compensation
|
|
All Other
Compensation
(2)(3)
|
|
Total(4) |
|
|
in CHF thousands |
| Dr. Patrick Amstutz |
|
385 |
|
|
182 |
|
|
385 |
|
|
0 |
|
|
95 |
|
1,047 |
|
| Chief Executive Officer, Director and Co-Founder |
|
|
|
|
|
|
|
|
|
|
|
|
| Total Management Board |
|
1,690 |
|
|
674 |
|
|
1,429 |
|
|
0 |
|
|
400 |
|
4,193 |
|
(1)Represents amounts earned in 2023.
(2)Represents pension and social security contributions during 2023 on base salary, bonus and the vesting of the PSUs 2020.
(3)In addition, upon vesting of the PSUs 2023 in 2026, we will be obliged to make employer contributions to social security pursuant to applicable mandatory law. As an estimate based on currently applicable contribution rates, the employer contributions on the PSUs 2023 expected to vest in 2026 will amount to approximately TCHF 76 (assuming 100% target achievement and full vesting of the PSUs).
(4)The total compensation awarded to members of the Management Board shown in this table does not include the items mentioned in the foregoing note (3).
Executive Compensation Arrangements
For a discussion of our employment arrangements with our executive officers, see the section of this Annual Report on Form 20-F entitled “Related Party Transactions—Agreements with Our Directors and Executive Officers—Employment Arrangements.” Except for the arrangements described in the section of this Annual Report on Form 20-F entitled “Related Party Transactions—Agreements with Our Directors and Executive Officers—Employment Arrangements,” there are no arrangements or understanding between us and any of our other executive officers providing for benefits upon termination of their employment, other than as required by applicable law.
Adoption of Clawback Policy
On November 14, 2023, in accordance with Rule 10D-1 promulgated under the Exchange Act and Nasdaq Listing Rule 5608, we adopted an incentive compensation recoupment policy which is filed herewith as Exhibit 97.1.
Limitations on Liability and Indemnification Matters
Under Swiss corporate law, an indemnification of a director or member of the executive management in relation to potential personal liability is not effective to the extent the director or member of the executive management intentionally or negligently violated his or her corporate duties towards the company (certain views advocate that at least a grossly negligent violation is required to exclude the indemnification). Most violations of corporate law are regarded as violations of duties towards the company rather than towards the shareholders. In addition, indemnification of controlling persons is not permitted under Swiss corporate law, including shareholders of the company.
Nevertheless, the articles of association of a Swiss corporation may set forth that the company shall indemnify and hold harmless to the extent permitted by the law, the directors and executive managers out of assets of the company against threatened, pending or completed actions. However, our articles of association do not provide for such an indemnification provision.
Within the same limitations, articles of association of a Swiss corporation may also provide that the directors shall be entitled to the reimbursement of all expenses incurred in the interests of the corporation. Our articles of association contain such a provision.
In addition, a corporation may enter into and pay for directors' and officers' liability insurance which typically covers negligent acts as well.
We extended liability insurance for our directors and officers, including insurance coverage for liability under the Securities Act. We believe that this insurance is necessary to attract qualified directors and executive officers.
Equity Incentives
We believe that our ability to grant incentive awards is a valuable and necessary compensation tool that allows us to attract and retain the best available personnel for positions of substantial responsibility, provides additional incentives to directors, executive officers, and employees and promotes the success of our business. Historically, we have granted several different equity incentive instruments to our directors, employees and other service providers, including:
•Restricted Share Units, or RSUs, granted to our directors;
•Performance Share Units, or PSUs, granted to our executive officers and employees; and
•share options granted to employees, directors and selected advisors.
Our articles of association authorize the board of directors to issue one or more participation plans and/or policies. An amendment or renewal of the relevant provision in our articles of association must be approved by an absolute majority of the votes represented at the general meeting of shareholders. Once our board of directors' authority is approved by our shareholders, the maximum aggregate amounts of the variable compensation elements actually granted to the directors and executive officers must be approved by an absolute majority of the votes represented at the general meeting of shareholders and shall continue for the duration of the current financial year. Compensation may be paid out prior to approval by the general meeting of shareholders subject to subsequent approval. If the general meeting of shareholders does not approve a proposal of the board of directors, the board of directors must newly determine the maximum aggregate amount or maximum partial amounts taking into account all relevant factors and submit such amounts for approval to the same general meeting of shareholders, to an extraordinary general meeting of shareholders or to the next ordinary general meeting of shareholders.
Share Options
Prior to our initial public offering on SIX Swiss Exchange on November 5, 2014, which we refer to as our Swiss IPO, our board of directors established three share option plans: (i) the Employee Share Option Plan 2007, or ESOP 2007, (ii) the Employee Share Option Plan 2009, or ESOP 2009, and (iii) the Employee Share Option Plan 2014, or ESOP 2014, with similar features as the ESOP 2009, but no longer providing for accelerated vesting of options in the event of our Swiss IPO. Each option entitles its holder to purchase one of our shares at the pre-defined exercise price. The number of options granted to each participant was determined by the board of directors based on a participant’s position and level of responsibility. As a rule, the options vested quarterly over a four-year period. At the end of the option term, the unexercised options expire without value.
As of December 31, 2023, no options were outstanding under the ESOP 2007, and an aggregate of 282,105 options were outstanding under the ESOP 2009 and ESOP 2014, together. As of December 31, 2023, all of the outstanding options were fully vested.
Following our Swiss IPO in 2014, no further grants were made under any of the ESOP 2007, ESOP 2009 or ESOP 2014, and we do not intend to make any further grants under any of these plans in the future. For additional information, see Note 18 to our consolidated financial statements as of and for the year ended December 31, 2023 included elsewhere in this Annual Report on Form 20-F.
Restricted Share Units (RSUs)
Under the LTI Plans, described in “—Long-Term Incentive Plans” below, members of our board of directors are eligible to be granted RSUs. RSUs are contingent rights to receive a certain number of our shares at the end of a three-year blocking period. RSUs vest over a one-year period from their date of grant, following the lapse of which they are no longer subject to forfeiture if a member of our board resigns. The number of shares to be received is not variable, i.e., the number of shares does not depend on the achievement of certain pre-defined performance metrics. In certain circumstances, including a change of control, a full or partial early vesting of the RSUs may occur.
As of December 31, 2023, 182,678 RSUs were outstanding.
Performance Share Units (PSUs)
Under the LTI Plans, described in “—Long-Term Incentive Plans” below, executive officers and employees are eligible to be granted PSUs. PSUs are contingent rights to receive a variable number of our shares either in aggregate at the end of a three-year cliff-vesting period or in annual installments over a three-year vesting period. The number of PSUs granted to a plan participant is calculated by dividing the CHF amount approved for the respective individual by the fair value of each PSU at the grant date based on the average share price in the two months preceding the grant date. While the PSUs are designed to allow the beneficiaries to participate in the long-term share price development, the number of shares to be earned in relation to a PSU depends on (i) the achievement of annual corporate goals for the respective year, (ii) the achievement of long-term value-driving milestones outside of such corporate goals during such year and (iii) the development of the share price of the Company. In accordance with these parameters, the number of shares to be issued based on the PSUs can be between zero and 150% of the number of PSUs granted. Even after the determination of goal achievement, participants may lose their entitlements in full or in part depending on certain conditions relating to their employment.
In certain circumstances, including a change of control, a full or partial accelerated vesting of the PSUs may occur.
As of December 31, 2023, 1,347,983 PSUs were outstanding.
Long-Term Incentive Plans
Our long-term incentive plans established in March of 2015, March of 2016, March of 2017, March of 2018, March of 2019, March of 2020, March of 2021, March of 2022 and March of 2023, respectively, which we collectively refer to as the LTI Plans, are rolled out annually. This allows our board of directors to review and adjust the terms and targets of the LTI Plans on an annual basis. Employees generally receive the grants on April 1 of each calendar year. With respect to members of the Management Board, the annual grants are usually made on April 1 subject to approval of the ordinary shareholders’ meeting at which the necessary amounts for variable compensation are approved by the shareholders. With respect to members of our board of directors, the annual grants are made following the ordinary shareholders’ meeting, at which the necessary amounts for variable compensation are approved by the shareholders.
C. Board Practices.
We currently have eight directors, four of whom are citizens or residents of the United States.
Our articles of association provide that our board of directors shall consist of a minimum of three members and maximum of eleven members. All directors (including the chairperson of the board of directors) are appointed to and removed from the board of directors exclusively by shareholders’ resolution for a maximum term of office of one year, extending until completion of the next annual shareholders' meeting. Directors may be re-elected at any time. In the event the office of the chairperson is vacant, the board of directors shall appoint a new chairperson from its members for the remaining term of office. The board of directors may elect a vice-chairperson from its members each year immediately following the annual shareholders' meeting for a term ending at the closing of the following annual shareholders' meeting. The board of directors shall further appoint the secretary, who need not be a member of the board of directors. The secretary shall be entitled to participate in the deliberations and discussions of the board of directors, but shall not vote, unless he or she is a member of the board of directors.
The following table sets forth the names of our directors, the year of their initial appointment as directors and the expiration dates of their current term:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Name |
|
Current Position |
|
Year of Initial Appointment |
|
Term Expiration Year(1) |
| William M. Burns |
|
Chairman of the Board |
|
2017 |
|
2024 |
| Dr. Patrick Amstutz |
|
Chief Executive Officer, Director and Co-Founder |
|
2017 |
|
2024 |
| Steven H. Holtzman |
|
Director |
|
2014 |
|
2024 |
| Sandip Kapadia |
|
Director |
|
2020 |
|
2024 |
| Dr. Vito J. Palombella |
|
Director |
|
2020 |
|
2024 |
| Dr. Michael Vasconcelles |
|
Director |
|
2020 |
|
2024 |
| Dr. Agnete Fredriksen |
|
Director |
|
2021 |
|
2024 |
| Dr. Dominik Höchli |
|
Director |
|
2021 |
|
2024 |
(1)At the end of the general meeting of shareholders during the year in which their term office expires, in each case as indicated.
Director Independence
As a foreign private issuer, under Nasdaq rule 560(a)(2), we are not required to have independent directors on our board of directors, except to the extent that our audit committee is required to consist of independent directors, subject to certain phase-in schedules.
Nevertheless, our board of directors has undertaken a review of the independence of the directors and considered whether any director has a material relationship with us that could compromise his or her ability to exercise independent judgment in carrying out his or her responsibilities. Based upon information requested from, and provided by, each director concerning such director’s background, employment and affiliations, including family relationships, our board of directors determined that William M. Burns, Agnete Fredriksen, Dominik Höchli, Steven H. Holtzman, Sandip Kapadia, Vito J. Palombella and Michael Vasconcelles are “independent directors” as defined under applicable Nasdaq rules and the independence requirements contemplated by Rule 10A-3 under the Securities Exchange Act of 1933, as amended, or the Exchange Act. In making these determinations, our board of directors considered the current and prior relationships that each non-employee director has with us and all other facts and circumstances that our board of directors deemed relevant in determining the director’s independence, including the number of ordinary shares beneficially owned by the director and his or her affiliated entities (if any).
Role of the Board in Risk Oversight
Our board of directors is primarily responsible for the oversight of our risk management activities and has delegated to the Audit and Finance Committee the responsibility to assist our board of directors in this task. The Audit and Finance Committee also monitors the issues relating to the preparation and supervision of accounting and financial information. The Audit and Finance Committee, among other things, monitors the effectiveness of the internal control and risk management systems with regard to the procedures relating to the preparation and processing of accounting and financial information, without undermining the independence of the board of directors. While our board of directors oversees our risk management, our management is responsible for day-to-day risk management processes. Our board of directors expects our management to consider risk and risk management in each business decision, to proactively develop and monitor risk management strategies and processes for day-to-day activities and to effectively implement risk management strategies adopted by the board of directors. We believe this division of responsibilities is the most effective approach for addressing the risks we face.
Corporate Governance Practices
The Sarbanes-Oxley Act of 2002, as well as related rules subsequently implemented by the SEC, requires foreign private issuers, including our company, to comply with various corporate governance practices. In addition, Nasdaq rules provide that foreign private issuers may follow home country practice in lieu of the Nasdaq corporate governance standards, subject to certain exceptions and except to the extent that such exemptions would be contrary to U.S. federal securities laws. However, if the laws of a foreign private issuer’s home country require that any such matter be approved by the board of directors or the shareholders, the audit committee’s responsibilities or powers with respect to such matter may instead be advisory. Under Swiss law, the audit committee may only have an advisory role and appointment of our statutory auditors, in particular, must be decided by the shareholders at our annual meeting.
Because we are a foreign private issuer, our members of our board of directors, executive board members and senior management are not subject to short-swing profit and insider trading reporting obligations under Section 16 of the Exchange Act. They are, however, subject to the obligations to report changes in share ownership under Section 13 of the Exchange Act and related SEC rules.
Board Committees
On January 1, 2023, Swiss ordinance against excessive compensation in listed stock corporations, known as the “Say on Pay” rule, was incorporated in the Swiss Code of Obligations. Pursuant to Swiss corporate law, companies listed on the SIX Swiss Exchange are required to establish a compensation committee. Our board of directors has established an Audit and Finance Committee, a Nomination and Compensation Committee and a Research and Development Committee, which operate pursuant to our articles of association, the charter of the Audit and Finance Committee, the charter of the Nomination and Compensation Committee and the charter of the Research and Development Committee. The composition and functioning of all of our committees is designed to comply with all applicable requirements of Swiss law, the Exchange Act, Nasdaq and SEC rules and regulations.
Audit and Finance Committee
Our Audit and Finance Committee assists our board of directors in its oversight of our corporate accounting and financial reporting by making an independent assessment of the quality of the external auditors, our financial statements and our internal controls. Sandip Kapadia, Dr. Dominik Höchli and Steven Holtzman currently serve on our Audit and Finance Committee. Mr. Kapadia is chairperson of our Audit and Finance Committee. Our board of directors has determined that each of Mr. Kapadia, Dr. Höchli and Mr. Holtzman is independent within the meaning of the applicable Nasdaq listing rules and the independence requirements contemplated by Rule 10A-3 under the Exchange Act. Our board of directors has further determined that Mr. Kapadia is an “audit committee financial expert” as defined by SEC rules and regulations and that each of the members of the Audit and Finance Committee qualifies as financially sophisticated under the applicable exchange listing rules. The principal duties and responsibilities of our Audit and Finance Committee include (1) analyzing economic and financial information and (2) ensuring the accuracy and honesty of our company’s financial statements, as well as the quality of the information provided.
Our board of directors has specifically assigned the following duties to the Audit and Finance Committee:
•assessing the quality and effectiveness of the external audit;
•assessing the quality of the internal control system, including risk management and the efficiency and state of compliance and monitoring with applicable norms within the Company;
•reviewing the stand-alone Swiss statutory and consolidated financial statements as well as all reporting prepared by the external auditor;
•deciding whether the year-end stand-alone Swiss statutory and consolidated financial statements be recommended to the board of directors for presentation to the general shareholders’ meeting;
•assessing the performance and the fees charged by the external auditors and ascertain their independence;
•annually review written disclosures from the external auditors delineating all relationships between the external auditors and the Company and take appropriate action to oversee the independence of the external auditors;
•reviewing the scope of the prospective external audit, the estimated fees thereof and any other matters pertaining to such audit;
•approve the annual engagement letter of external auditor, including the scope of the audit and the fees and terms for the planned audit
•pre-approve all audit review or attest services and permitted non-audit services by the external auditors
•taking notice of all comments from the external auditors on accounting procedures and systems of control;
•reviewing with the external auditors and/or the SVP Finance as principal financial officer / CEO any questions, comments or suggestions they may have regarding the internal control, risk management, accounting practices and procedures of the Company and its subsidiaries;
•discussing with the management any legal matters that may have a material impact on the Company's financial statements and any material reports or inquiries from regulatory or governmental agencies which could materially impact the Company's contingent liabilities and risks;
•reviewing with management and the external auditors, as appropriate, the Company’s MD&A disclosures;
•annually reviewing and discussing with management the management’s report in relation to internal controls over financial reporting pursuant to the Sarbanes-Oxley Act of 2002;
•reviewing and approving in advance any transaction that could be within the scope of a related party transaction;
•establishing procedures for the receipt, retention and treatment of complaints received by the Company regarding accounting, internal accounting controls or auditing matters and the confidential and anonymous submission by employees of concerns regarding questionable accounting or auditing matters;
•supporting the board of directors with regard to the financial planning as well as the principles of accounting and financial control;
•evaluating management’s principles and proposals for, and formulate recommendations to the board of directors in regard to financial planning (capital structure, management of resources, inter-company financing), dividend policy and capital market relations;
•reviewing proposed concepts of financial objectives such as costs of capital, enhancement of shareholders’ value, Company objectives, project objectives (capital expenditures and M&A); and
•reviewing finance policy and operations in treasury, controlling, insurance, taxes and investment and acquisitions.
Nomination and Compensation Committee
Our Nomination and Compensation Committee assists our board of directors in establishing and reviewing the compensation strategy and guidelines as well as in preparing the compensation plans and proposals to the general meeting of shareholders regarding the compensation of the board of directors and executive officers. William M. Burns, Steven H. Holtzman and Dr. Michael Vasconcelles currently serve on the Nomination and Compensation Committee. Mr. Burns is the chairperson of our Nomination and Compensation Committee. We are subject to the Swiss Code of Obligations rules against excessive compensation in listed stock corporations, known as the “Say on Pay” rule. As a result of the Say on Pay rule, the members of the Nomination and Compensation Committee must be elected by our shareholders and the aggregate compensation of our board of directors and executive officers must also be approved by our shareholders.
The principal duties and responsibilities of our Nomination and Compensation Committee include:
•reviewing and making recommendations regarding the compensation strategy and guidelines of the Company;
•reviewing and making recommendations regarding the compensation of the members of the board of directors and the executive management;
•reviewing and making recommendations regarding compensation plans (cash and/or equity-based plans), and where appropriate or required, make recommendations to adopt, amend and terminate such plans;
•administering the compensation plans;
•reviewing and making recommendations regarding any employment agreements (including any benefits) for members of the executive management;
•reviewing and making recommendations regarding the proposals of the board of directors for the aggregate amount of the compensation of the board of directors and of the executive management to be submitted to the annual general shareholders' meeting for approval;
•ensuring that any reporting obligation with respect to compensation matters, specifically any necessary disclosures in the annual report and/or compensation report, are met;
•reviewing considerations relating to the composition of the board of directors, including the size and the criteria for membership on the board of directors;
•evaluating candidates to the board of directors and making recommendations to the board of directors in this respect; and
•evaluating candidates to the Management Board and making recommendations to the board of directors in this respect.
Research and Development Committee
The Research and Development Committee (i) provides strategic advice and brings recommendations to the Management Board and the board of directors regarding current and planned research and development programs, (ii) provides strategic advice to the board of directors regarding emerging science and technology issues and trends, and (iii) conducts a review of the effectiveness and competitiveness of our research and development function. Dr. Michael Vasconcelles, Dr. Agnete Fredriksen, Dr. Vito J. Palombella and Dr. Dominik Höchli currently serve on the Research and Development Committee. Dr. Vasconcelles is the chairperson of the Research and Development Committee.
Code of Conduct
We have adopted a Code of Conduct which is applicable to all of our employees, executive officers and directors. The Code of Conduct is available on our website at www.molecularpartners.com. The Audit and Finance Committee of our board of directors is responsible for overseeing the Code of Conduct and is required to approve any waivers of the Code of Conduct for employees, executive officers and directors. We expect that any amendments to the Code of Conduct will be disclosed on our website.
D. Employees.
As of December 31, 2023, we had 167.5 full-time equivalent employees (December 31, 2022: 175.3 full-time equivalents). None of our employees are represented by collective bargaining agreements. We believe that we maintain good relations with our employees. At each date shown, we had the following number of full time employees, broken out by department. The majority of our employees are based in Zurich, Switzerland. Three of our employees are based in the United States of America.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Full-time equivalent employees |
At December 31, |
|
At December 31, |
|
2023 |
|
2022 |
| Function |
|
|
|
| Research and development |
138.5 |
|
|
142.7 |
|
| Selling, general and administrative |
29.0 |
|
|
32.6 |
|
Total |
167.5 |
|
|
175.3 |
|
E. Share Ownership.
The following table shows the number of ordinary shares, options, RSUs and PSUs held by the individual members of the board of directors and the Management Board, as of December 31, 2023. For more information regarding the share ownership of our directors and executive officers, see "Item 6.B - Compensation" and "Item 7.A - Major shareholders."
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Name |
|
Shares |
|
Options |
|
RSUs |
|
PSUs |
| William M. Burns |
|
26,951 |
|
|
— |
|
|
45,668 |
|
|
— |
| Steven H. Holtzman |
|
14,717 |
|
|
20,000 |
|
|
22,835 |
|
|
— |
| Sandip Kapadia |
|
3,507 |
|
|
— |
|
|
22,835 |
|
|
— |
| Vito J. Palombella |
|
3,505 |
|
|
— |
|
|
22,835 |
|
|
— |
| Michael Vasconcelles |
|
3,507 |
|
|
— |
|
|
22,835 |
|
|
— |
| Dr. Agnete Fredriksen |
|
— |
|
|
— |
|
|
22,835 |
|
|
— |
| Dr. Dominik Höchli |
|
— |
|
|
— |
|
|
22,835 |
|
|
— |
| Dr. Patrick Amstutz |
|
735,095 |
|
|
70,080 |
|
|
— |
|
97,306 |
|
| Dr. Michael Tobias Stumpp |
|
767,524 |
|
|
36,070 |
|
|
— |
|
63,293 |
|
| Dr. Nicolas Leupin |
|
29,168 |
|
|
— |
|
|
— |
|
73,793 |
|
| Renate Gloggner |
|
23,006 |
|
|
— |
|
|
— |
|
58,339 |
|
| Alexander Zürcher |
|
25,706 |
|
|
13,040 |
|
|
— |
|
57,369 |
|
F. Disclosure of a Registrant's Action to Recover Erroneously Awarded Compensation.
Not applicable.
Item 7. Major Shareholders and Related Party Transactions
A. Major shareholders.
The following table and accompanying footnotes set forth, as of December 31, 2023, information regarding beneficial ownership of our ordinary shares by:
•each person, or group of affiliated persons, known by us to beneficially own more than 3% of our ordinary shares;
•each of our executive officers;
•each of our directors; and
•all of our executive officers and directors as a group.
Beneficial ownership is determined according to the rules of the SEC and generally means that a person has beneficial ownership of a security if he, she or it possesses sole or shared voting power or investment power with respect to that security, including ordinary shares that vest within 60 days of December 31, 2023 and options and warrants that are currently exercisable or exercisable within 60 days of December 31, 2023. Shares issuable under PSUs or RSUs that vest within 60 days of December 31, 2023 and shares subject to options currently exercisable or exercisable within 60 days of December 31, 2023 are deemed to be outstanding for computing the percentage ownership of the person holding these free shares, options and the percentage ownership of any group of which the holder is a member, but are not deemed outstanding for computing the percentage of any other person.
Except as indicated in the footnotes below, we believe, based on the information furnished or otherwise known to us, that the persons named in the table below have sole voting and investment power with respect to all shares shown that they beneficially own, subject to community property laws where applicable. The information does not necessarily indicate beneficial ownership for any other purpose, including for purposes of Sections 13(d) and 13(g) of the Securities Act or applicable Swiss law.
Our calculation of the percentage of beneficial ownership is based on 36,044,706 of our ordinary shares registered with the commercial register of the canton of Zurich as of December 31, 2023 and includes 3,500,000 treasury shares indirectly held through our wholly-owned subsidiary Molecular Partners Inc.
Except as otherwise indicated in the following table, addresses of the directors, executive officers and named beneficial owners are in care of Molecular Partners AG, Wagistrasse 14, 8952 Schlieren, Switzerland.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Shares Beneficially Owned |
| Name of Beneficial Owner |
|
|
|
Number |
|
Percentage |
Principal Shareholders |
|
|
|
|
|
|
Entities affiliated with Biotechnology Value Fund, L.P.(2)(3) |
|
|
|
8,696,205 |
|
|
|
24.13 |
% |
Entities affiliated with Suvretta Capital Management, LLC(1)(2)(4) |
|
|
|
1,750,000 |
|
|
|
4.86 |
% |
Novartis Pharma AG(1)(5) |
|
|
|
1,739,130 |
|
|
|
4.82 |
% |
UBS Fund Management (Switzerland) AG(1)(2)(6) |
|
|
|
995,989 |
|
|
|
2.76 |
% |
| Directors and Executive Officers |
Dr. Patrick Amstutz(7) |
|
|
|
805,175 |
|
|
|
2.23 |
% |
Renate Gloggner (11) |
|
|
|
23,006 |
|
|
|
0.06 |
% |
Dr. Nicolas Leupin(12) |
|
|
|
29,168 |
|
|
|
0.08 |
% |
Dr. Michael Tobias Stumpp(8) |
|
|
|
803,594 |
|
|
|
2.23 |
% |
William M. Burns(9) |
|
|
|
26,951 |
|
|
|
0.07 |
% |
Dr. Agnete Fredriksen |
|
|
|
— |
|
|
|
— |
% |
Dr. Dominik Höchli |
|
|
|
— |
|
|
|
— |
% |
Steven H. Holtzman(10) |
|
|
|
34,717 |
|
|
|
0.10 |
% |
Sandip Kapadia |
|
|
|
3,507 |
|
|
|
0.01 |
% |
Dr. Vito Palombella |
|
|
|
3,505 |
|
|
|
0.01 |
% |
Dr. Michael Vasconcelles |
|
|
|
3,507 |
|
|
|
0.01 |
% |
Alexander Zürcher(13) |
|
|
|
38,746 |
|
|
|
0.11 |
% |
All current directors and executive officers as a group (12 individuals)(14) |
|
|
|
1,771,876 |
|
|
|
4.92 |
% |
(1)Number of voting rights carried by shares as reported by our shareholders in notifications filed with SIX Swiss Exchange.
(2)The information reported is in part derived from reports filed with the SEC pursuant to the Exchange Act.
(3)Based on a Schedule 13D filed with the SEC on November 20, 2023, the shares provided in the table above consist of 443,221 ADSs and 8,696,205 ordinary shares of the Company held by Biotechnology Value Fund, L.P. ("BVF"), Biotechnology Value Fund II, L.P. ("BVF2"), Biotechnology Value Trading Fund OS, L.P. ("Trading Fund OS") and a managed account for BVF Partners L.P. (the "Partners Managed Account," and collectively, the "BVF Funds"). BVF GP LLC is the general partner of BVF and may be deemed to beneficially own the shares beneficially owned by BVF. BVF II GP LLC is the general partner of BVF2 and may be deemed to beneficially own the shares beneficially owned by BVF2. BVF Partners OS Ltd. is the general partner of Trading Fund OS and may be deemed to beneficially own the shares beneficially owned by Trading Fund OS. BVF GP Holdings LLC, as the sole member of BVF GP LLC and BVF II GP LLC, may be deemed to beneficially own the shares beneficially owned by BVF and BVF2. BVF Partners L.P. ("Partners"), as the investment manager of BVF, BVF2 and Trading Fund OS, and the sole member of BVF Partners OS Ltd, may be deemed to beneficially own the shares held by BVF, BVF2, Trading Fund OS and the Partners Managed Account. BVF Inc., as the general partner of Partners, may be deemed to beneficially own the shares beneficially owned by Partners. Mark Lampert, as a director and officer of BVF Inc., may be deemed to beneficially own the shares beneficially owned by BVF Inc. Each of Partners, BVF Inc. and Mark Lampert disclaims beneficial ownership of the shares beneficially owned by BVF, BVF2, Trading Fund OS, and the Partners Managed Account. The address for the BVF Funds is 44 Montgomery St., 40th Floor, San Francisco, California 94104. According to a Schedule 13D filed with the SEC on November 20, 2023, Mark N. Lampert (Biotechnology Value Funds) held 8,696,205 shares (corresponding to 23.9% of voting rights), consisting of 443,221 ADSs and 8,696,205 ordinary shares.
(4)Includes 400,000 ADSs purchased in the Company's initial public offering of ADSs in the U.S. Shares of the Company are held by Suvretta Master Fund, Ltd., Averill Master Fund, Ltd., Vitruvius US Equity and Suvretta Long Master Fund, Ltd. Suvretta Capital Management, LLC is the beneficial owner and may exercise voting power over the shares. Aaron Cowen may also be deemed to have shared dispositive power and voting power with respect to the shares by virtue of his role at Suvretta Capital Management, LLC. The address for Suvretta Capital Management, LLC is 540 Madison Avenue, 7th Floor, New York, New York 10022.
(5)Shares of the Company are held by Novartis Pharma AG. Novartis Pharma AG is a direct wholly-owned subsidiary of Novartis AG, which is the beneficial owner and may exercise voting power over the shares. The address of Novartis AG is Lichtstrasse 35, 4056 Basel, Switzerland.
(6)Shares of the Company are held by UBS Fund Management (Switzerland) AG. UBS Fund Management (Switzerland) AG is a direct wholly-owned subsidiary of UBS Group AG, which is the beneficial owner and may exercise voting power over the shares.
(7)Consists of 735,095 ordinary shares and 70,080 ordinary shares issuable upon exercise of options that are exercisable within 60 days of December 31, 2023.
(8)Consists of 767,524 ordinary shares and 36,070 ordinary shares issuable upon exercise of options that are exercisable within 60 days of December 31, 2023.
(9)Consists of 26,951 ordinary shares.
(10)Consists of 14,717 ordinary shares and 20,000 ordinary shares issuable upon exercise of options that are exercisable within 60 days of December 31, 2023.
(11)Consists of 23,006 ordinary shares.
(12)Consists of 29,168 ordinary shares.
(13)Consists of 25,706 ordinary shares and 13,040 ordinary shares issuable upon exercise of options that are exercisable within 60 days of December 31, 2023.
(14)Consists of 1,632,686 ordinary shares and 139,190 ordinary shares issuable upon exercise of options that are exercisable within 60 days of December 31, 2023.
In June 2021, we completed our initial public offering in the U.S. and listed our ADSs on the Nasdaq Global Select Market. In the initial public offering, we issued and sold 3,000,000 ADSs representing 3,000,000 ordinary shares.
To our knowledge, other than as provided in the table above, our other filings with the SEC and this Annual Report on Form 20-F, the significant changes in the percentage ownership held by our principal shareholders since January 1, 2019 are as a result of the transactions described in the final prospectus related to our initial public offering dated June 15, 2021, filed with the SEC on June 16, 2021 pursuant to Rule 424(b), under the heading “Related Party Transactions-Agreements with Shareholders” and the dilution resulting from our initial public offering of ADSs in the U.S.
As of December 31, 2023, our issued share capital as recorded in the commercial register of the Canton of Zurich was CHF 3,604,470.60, consisting of 36,044,706 ordinary shares with a nominal value of CHF 0.10 each. All shares rank pari passu with each other and no preferred shares exist.
As of December 31, 2023, to the best of our knowledge and assuming that all of our ordinary shares represented by ADSs are held by residents of the United States, we estimate that 30% of our issued ordinary shares (including ordinary shares underlying ADSs) as identified in publicly available filings were held in the United States by approximately 5 holders of record. The actual number of holders is potentially greater than these numbers of record holders, and includes beneficial owners whose ordinary shares or ADSs are held in street name by brokers and other nominees. This number of holders of record also does not include holders whose shares may be held in trust by other entities.
B. Related Party Transactions.
Since January 1, 2023, we have engaged in the following transactions with our directors, executive officers and holders of more than 3% of our outstanding voting securities and their affiliates, which we refer to as our related parties.
Agreements with Our Directors and Executive Officers
Employment Arrangements
We have entered into customary employment agreements with all of our executive officers. These agreements provide for a base salary and annual incentive bonus opportunity, as well as participation in our equity incentive plans. These agreements generally require advance notice of termination of six months.
Indemnification Agreements
We have entered into indemnification agreements with each of our directors and executive officers. See the section of this Annual Report on Form 20-F entitled “Item 6.B - Compensation—Compensation of Executive Officers—Limitations on Liability and Indemnification Matters.”
Insofar as indemnification for liabilities arising under the Securities Act may be permitted to directors, officers or persons controlling us pursuant to the foregoing provisions, we have been informed that in the opinion of the SEC such indemnification is against public policy as expressed in the Securities Act and is therefore unenforceable.
Agreements with Shareholders
See “Item 4.B - Business Overview—License and Collaboration Agreements" for information regarding the Novartis Option Agreement, the Ensovibep License Agreement and the Novartis Radioligand Agreement with Novartis.
Related Party Transactions Policy
We have adopted a related person transaction policy that sets forth our procedures for the identification, review, consideration and approval or ratification of related person transactions. For purposes of our policy only, a related person transaction is a transaction, arrangement or relationship, or any series of similar transactions, arrangements or relationships, in which we and any related person are, were or will be participants in which the amount involved exceeds $120,000 or which is unusual in its nature or conditions. Transactions involving compensation for services provided to us as an employee, consultant or director are not covered by this policy. A related person is any enterprise that controls, is controlled by or is under common control with the Company, or in which the Company has significant influence or which has significant influence over the Company; an individual owning, directly or indirectly, an interest in the voting power of the Company that gives them significant influence over the Company, and close members of any such individual’s family; key management personnel, including directors and senior management and close members of such individuals' families; and any enterprise in which a substantial interest in the voting power of the Company is owned, directly or indirectly, by any person described in the foregoing list or over which such a person is able to exercise significant influence, including enterprises owned by directors or major shareholders of the Company and enterprises that have a member of key management in common with the Company.
Under the policy, any proposed transaction that has been identified as a related person transaction may be consummated or materially amended only following approved by our Audit and Finance Committee, or, if Audit and Finance Committee approval would be inappropriate, another independent body of our board of directors. Any transaction that was not a related person transaction when originally consummated or any transaction that was not initially identified as a related person transaction prior to consummation shall be submitted for review and ratification by our Audit and Finance Committee, The presentation of such related party transactions shall include a description of, among other things, the parties thereto, the interests, direct and indirect, of the related persons, the purpose and material facts of the transaction,, the benefits to the Company of the transaction and whether the transaction is on terms that are comparable to the terms available to or from, as the case may be, an unrelated third- party or to or from employees generally, and management's recommendation with respect to the transaction. The Audit and Finance Committee shall approve only those related person transactions that, in light of known circumstances, are in, or are not inconsistent with, the best interests of the Company and its shareholders, as the Audit and Finance Committee determines in the good faith.
In addition, while related party transaction policies are generally not required by statutory Swiss law, our articles of association provide for the following rules in connection with transactions with members of the board of directors and the executive management:
•We may enter into mandate or other agreements with the members of our board of directors regarding their compensation as directors for a fixed term or for an indefinite term. The duration and termination are subject to term of office and the law.
•We may enter into employment agreements with the members of the executive management for a fixed term or for an indefinite term. The duration of fixed term agreements may not exceed one year. A renewal of a fixed term agreement is permissible. Agreements for an indefinite term may have a termination notice period of a maximum of one year.
•We may enter into non-competition agreements with members of the executive management for the period after the termination of the employment agreement. The duration of any such non-competition undertaking by a member of the executive management shall not exceed two years, and the consideration paid for a non-competition undertaking shall not exceed the sum of the total annual compensation of the respective member of the executive management last paid.
•Loans to members of the board of directors and the executive management may be granted, provided they are at standard market rates and the aggregate amount of the loan extended to the member of the board of directors or executive management does not exceed 200% of the total annual compensation of the respective member of the executive management last paid or payable for the first time.
•Subject to the approval by the meeting of shareholders, we may grant to members of our board of directors or the executive management post-retirement benefits beyond the occupational benefit scheme, if such post-retirement benefits do not exceed 100% of the total annual compensation of the respective member last paid. In case of capital settlements, the value is determined by recognized actuarial methods.
C. Interests of Experts and Counsel.
Not applicable.
Item 8. Financial Information
A. Consolidated Statements and Other Financial Information.
Consolidated Financial Statements
Our consolidated financial statements are appended at the end of this Annual Report, starting at page F-1, and incorporated herein by reference.
Dividend Distribution Policy
We have never declared or paid any dividends on our ordinary shares and we do not anticipate paying dividends on our equity securities in the foreseeable future. Instead, we intend to retain any earnings for use in the operation and expansion of our business, including for continued advancement of our proprietary DARPin product candidates, investment in research and development, building up our late-stage clinical development and, eventually, commercialization abilities. As a result, investors in our ordinary shares or ADSs will benefit in the foreseeable future only if the ordinary shares or ADSs appreciate in value.
In order for us to declare and pay dividends, the distribution must be approved by shareholders holding an absolute majority of the ordinary shares represented at the general meeting of shareholders. Our board of directors may propose distributions in the form of a common dividend or in the form of a distribution of cash or property that is based upon a reduction of our share capital recorded in the commercial register.
Common dividends may be paid only if we have sufficient distributable profits from previous years (Gewinnvortrag) or freely distributable reserves to allow the distribution of a dividend, in each case, as presented on our annual statutory standalone balance sheet prepared in accordance with Swiss company law after deduction of allocated statutory reserves and reserves required by our articles of association (Statuten). Our auditor must confirm that a proposal made by the board of directors to shareholders regarding the appropriation of our available earnings conforms to the requirements of the Swiss Code of Obligations of March 30, 1911, as amended, the CO, and our articles of association. In order for us to pay dividends to our shareholders out of reserves from capital contributions (Reserven aus Kapitaleinlagen), a shareholders’ meeting must approve by the absolute majority of votes represented the reclassification of such reserves from capital contributions to freely distributable reserves (frei verfügbare Reserven) (to the extent permissible by the CO). Furthermore, dividends can be paid out of reserves from capital reserves only if the same amount is paid out of the annual profit or ordinary reserves. Dividends and distributions against reserves from capital contributions are usually due and payable after the shareholders’ resolution relating to the allocation of profit and distribution against reserves from capital contributions (if applicable) has been passed at the shareholders’ meeting or at a later date as determined by the shareholders’ dividend resolution. Under Swiss law, the statute of limitations with respect to dividend payments is five years. Dividends not collected within five years after their due date accrue to us and will be allocated to our general reserves. Dividends paid on ordinary shares are subject to Swiss federal withholding tax, except if paid out of reserves from capital contributions. See “Swiss Tax Implications for U.S. Holders—Swiss Tax Considerations—Swiss Federal Withholding Tax” for a summary of certain Swiss tax consequences regarding dividends and other distributions distributed to holders of our ordinary shares. As of December 31, 2023, we had reserves from capital contributions in an aggregate amount of CHF 327,226,672, of which CHF 179,226,672 were legal capital reserves and CHF 148,000,000 were free reserves.
A distribution of cash or property that is based on a reduction of our share capital requires a special audit report confirming that the claims of our creditors remain fully covered by our assets despite the reduction in the share capital recorded in the commercial register. Upon approval by the general meeting of the shareholders of the capital reduction, our board of directors must give public notice of the capital reduction in the Swiss Official Gazette of Commerce three times and notify our creditors that they may request, within two months of the third publication, satisfaction of or security for their claims. Distributions of cash or property that are based upon a capital reduction are not subject to Swiss federal withholding tax. See “Swiss Tax Implications for U.S. Holders—Swiss Tax Considerations—Swiss Federal Withholding Tax” for a summary of certain Swiss tax consequences regarding distributions paid on the ordinary shares that are based upon a capital reduction. For a description of share capital reductions under the revised Swiss corporate law that entered into force in 2023, see "Item 10.B. Memorandum and Articles of Association Swiss Corporate Law Reform."
Dividend distributions, if any in the future, will be declared and paid in Swiss francs and converted into U.S. dollars with respect to our ADSs.
Our board of directors determines the date on which the dividend entitlement starts. Dividends are usually due and payable shortly after the shareholders have passed the resolution approving the payment, but shareholders may also resolve at the ordinary general meeting of shareholders to pay dividends in quarterly or other installments.
Legal Proceedings
From time to time we may become involved in legal proceedings or be subject to claims arising in the ordinary course of our business.Regardless of the outcome, litigation can have an adverse impact on us because of defense and settlement costs, diversion of management resources and other factors.
On July 12, 2022, a putative class action complaint was filed in the U.S. District Court for the Southern District of New York against the Company, its directors, and certain of its executive officers. On May 23, 2023, an amended complaint was filed. The amended complaint alleged that the defendants violated federal securities laws by, among other things, making misrepresentations and omissions regarding its product candidate MP0310 and an associated licensing agreement. The amended complaint sought unspecified compensatory damages, as well as an award of reasonable attorneys’ fees and other costs, on behalf of persons and/or entities which purchased the Company's American Depositary Shares (ADSs) pursuant to certain offering documents issued in connection with the Company's initial public offering of ADSs. The Company and named individual defendants moved to dismiss the amended complaint on July 24, 2023. Plaintiffs filed their opposition on September 7, 2023 and the Company and named individual defendants filed their reply brief on October 5, 2023. On February 5, 2024, the court dismissed the amended complaint without prejudice and gave plaintiff the opportunity to amend the complaint by February 26, 2024. On February 23, 2024, plaintiff filed a stipulation of dismissal with prejudice. On February 29, 2024, the court ordered the case closed.
B. Significant Changes.
A discussion of significant changes in our business can be found under "Item 4. Information on the Company - 4.B. Business Overview." Our ADSs have been listed on the Nasdaq Global Select Market under the symbol “MOLN” since June 16, 2021.
Item 9. The Offer and Listing.
A. Offer and Listing Details.
Prior to June 16, 2021, there was no public trading market for ADSs. Our ordinary shares have been listed on the SIX Swiss Exchange, or SIX, under the symbol “MOLN” since November 5, 2014. Prior to November 5, 2014, there was no public trading market for ordinary shares.
B. Plan of Distribution.
Not applicable.
C. Markets.
Our ADSs have been listed on the Nasdaq Global Select Market under the symbol “MOLN” since June 16, 2021, and our ordinary shares have been listed on SIX under the symbol “MOLN” since November 5, 2014.
D. Selling Shareholders.
Not applicable.
E. Dilution.
Not applicable.
F. Expenses of the Issue.
Not applicable.
Item 10. Additional Information.
A. Share Capital.
Not applicable.
B. Memorandum and Articles of Association.
Please see the information set forth in Exhibit 2.3 “Description of Securities” and the copy of our Articles of Association filed as Exhibit 1.1, which are each incorporated herein by reference.
C. Material Contracts.
In addition to the contracts described elsewhere in this Annual Report, the following are summaries of each material contract to which we are a party for the two years preceding the date of this Annual Report.
Sales Agreement
On July 1, 2022, we entered into the Sales Agreement with Leerink Partners LLC (previously known as SVB Securities LLC to sell ordinary shares from time to time at our discretion under an “at the market”, or ATM, program, with aggregate gross sales proceeds of up to $100.0 million. The Sales Agreement provides that the commission payable to Leerink Partners LLC (previously known as SVB Securities LLC, as sales agent, for sales of ordinary shares under the ATM program shall be up to 3.0% of the aggregate gross proceeds of any ordinary shares sold under the Sales Agreement. The Sales Agreement contains customary representations and warranties of the parties and indemnification and contribution provisions, including our agreement to indemnify Leerink Partners LLC (previously known as SVB Securities LLC against certain liabilities, including liabilities under the Securities Act. We and Leerink Partners LLC (previously known as SVB Securities LLC have the right, by giving written notice as specified in the Sales Agreement, to terminate the Sales Agreement. As of March [14], 2024, we have not made any sales under the Sales Agreement.
For additional information on our material contracts, please see “Item 4. - Information on the Company,” “Item 6. - Directors, Senior Management and Employees,” and “Item 7.B. - Related Party Transactions” of this Annual Report.
D. Exchange Controls.
There are no Swiss governmental laws, decrees or regulations that restrict, in a manner material to us, the export or import of capital, including any foreign exchange controls, or that generally affect the remittance of dividends or other payments to non-residents or non-citizens of Switzerland who hold our ordinary shares.
E. Taxation.
Swiss Federal, Cantonal and Communal Individual Income Tax and Corporate Income Tax
Non-Resident Shareholders
Holders of ADSs representing our shares who are not resident in Switzerland for tax purposes, and who, during the relevant taxation year, have not engaged in a trade or business carried on through a permanent establishment or fixed place of business situated in Switzerland for tax purposes (all such shareholders are hereinafter referred to as the Non-Resident Shareholders), will not be subject to any Swiss federal, cantonal and communal income tax on dividends and similar cash or in-kind distributions on ADSs representing our shares (including dividends on liquidation proceeds and stock dividends) (hereinafter referred to as the Dividends), distributions based upon a capital reduction (Nennwertrückzahlungen) or paid out of reserves from capital contributions (Reserven aus Kapitaleinlagen) on shares underlying the ADSs, or capital gains realized on the sale or other disposition of ADSs (see, however, "Swiss Federal Withholding Tax" for a summary of Swiss federal withholding tax on Dividends).
Resident Private Shareholders
Swiss resident individuals who hold their ADSs as private assets (all such shareholders are hereinafter referred to as the Resident Private Shareholders) are required to include Dividends, but not distributions based upon a capital reduction (Nennwertrückzahlungen) or paid out of reserves from capital contributions (Reserven aus Kapitaleinlagen) of the shares underlying the ADSs, in their personal income tax return and are subject to Swiss federal, cantonal and communal income tax on any net taxable income for the relevant taxation period, including the Dividends, but not the distributions based upon a capital reduction (Nennwertrückzahlungen) or paid out of reserves from capital contributions (Reserven aus Kapitaleinlagen).
Capital gains resulting from the sale or other dispositions of ADSs are not subject to Swiss federal, cantonal and communal income tax, and conversely, capital losses are not tax-deductible for Resident Private Shareholders. See "Domestic Commercial Shareholders" for a summary of the taxation treatment applicable to Swiss resident individuals, who, for income tax purposes, are classified as "professional securities dealers."
Domestic Commercial Shareholders
Corporate and individual shareholders who are resident in Switzerland for tax purposes and corporate and individual shareholder who are not resident in Switzerland, and who, in each case, hold their ADSs as part of a trade or business carried on in Switzerland, in the case of corporate and individual shareholders not resident in Switzerland, through a permanent establishment or fixed place of business situated, for tax purposes, in Switzerland, are required to recognize Dividends, distributions based upon a capital reduction (Nennwertrückzahlungen) or paid out of reserves from capital contributions (Reserven aus Kapitaleinlagen) received on shares underlying the ADSs and capital gains or losses realized on the sale or other disposition of ADSs in their income statement for the relevant taxation period and are subject to Swiss federal, cantonal and communal individual or corporate income tax, as the case may be, on any net taxable earnings for such taxation period. The same taxation treatment also applies to Swiss-resident private individuals who, for income tax purposes, are classified as "professional securities dealers" for reasons of, inter alia, frequent dealing, or leveraged investments in ADSs and other securities (the shareholders referred to in this section, the Domestic Commercial Shareholders). Domestic Commercial Shareholders who are corporate taxpayers may be eligible for dividend relief (Beteiligungsabzug) in respect of Dividends and distributions based upon a capital reduction (Nennwertrückzahlungen) or paid out of reserves from capital contributions (Reserven aus Kapitaleinlagen) if the shares underlying the ADSs held by them as part of a Swiss business have an aggregate market value of at least CHF 1 million.
Swiss Cantonal and Communal Private Wealth Tax and Capital Tax
Non-Resident Shareholders
Non-Resident Shareholders are not subject to Swiss cantonal and communal private wealth tax or capital tax.
Resident Private Shareholders and Domestic Commercial Shareholders
Resident Private Shareholders and Domestic Commercial Shareholders who are individuals are required to report their ADSs as part of private wealth or their Swiss business assets, as the case may be, and will be subject to Swiss cantonal and communal private wealth tax on any net taxable wealth (including the ADSs), in the case of Domestic Commercial Shareholders to the extent the aggregate taxable wealth is allocated in Switzerland. Domestic Commercial Shareholders who are corporate taxpayers are subject to Swiss cantonal and communal capital tax on taxable capital to the extent the aggregate taxable capital is allocated to Switzerland.
Swiss Federal Withholding Tax
Dividends that the Company pays on the shares underlying the ADSs are subject to Swiss Federal withholding tax (Verrechnungssteuer) at a rate of 35% on the gross amount of the Dividend. The Company is required to withhold the Swiss federal withholding tax from the Dividend and remit it to the Swiss Federal Tax Administration. Distributions based upon a capital reduction (Nennwertrückzahlungen) or paid out of reserves from capital contributions (Reserven aus Kapitaleinlagen) are not subject to Swiss federal withholding tax.
The Swiss federal withholding tax on a Dividend will be refundable in full to a Resident Private Shareholder and to a Domestic Commercial Shareholder, who, in each case, inter alia, as a condition to refund, duly reports the Dividend in his or her individual income tax return as income or recognizes the Dividends in its income statement as earnings, as applicable.
A Non-Resident Shareholder may be entitled to a partial refund of the Swiss federal withholding tax on Dividend if the country of his or her residence for tax purposes has entered into a bilateral treaty for the avoidance of double taxation with Switzerland and the conditions of such treaty are met. Such shareholders should be aware that the procedures for claiming tax treaty benefits (and the time required for obtaining a refund) might be different from country to country. For example, a shareholder who is resident of the U.S. for the purposes of the bilateral treaty between the U.S. and Switzerland is eligible for a refund of the amount of the withholding tax in excess of the 15% treaty rate, provided such shareholder: (i) qualifies for benefits under this treaty and qualifies as beneficial owner of the Dividends; (ii) hold, directly or indirectly, less than 10% of the voting stock of the Company; (iii) does not qualify as a pension scheme or retirement arrangement for the purpose of the bilateral treaty; and (iv) does not conduct business through a permanent establishment or fixed base in Switzerland to which the ADSs are attributable. Such an eligible U.S. shareholder may apply for a refund of the amount of the withholding tax in excess of the 15% treaty rate. The applicable refund request form may be filed with the Swiss Federal Tax Administration following receipt of the dividend and the relevant deduction certificate, however no later than December 31 of the third year following the calendar year in which the dividend was payable.
Swiss Federal Stamp Taxes
Any dealings in the ADSs, where a bank or another securities dealer in Switzerland, as defined in the Swiss Federal Stamp Tax Act, acts as intermediary or is a party to the transaction, are, subject to certain exemptions provided for in the Swiss Federal Stamp Tax Act, subject to Swiss securities turnover tax at an aggregate tax rate of up to 0.15% of the consideration paid for such ADSs.
International Automatic Exchange of Information in Tax Matters
On November 19, 2014, Switzerland signed the Multilateral Competent Authority Agreement, which is based on article 6 of the OECD/Council of Europe administrative assistance convention and is intended to ensure the uniform implementation of automatic exchange of information, or the AEOI. The Federal Act on the International Automatic Exchange of Information in Tax Matters, or the AEOI Act, entered into force on January 1, 2017. The AEOI Act is the legal basis for the implementation of the AEOI standard in Switzerland.
The AEOI is being introduced in Switzerland through bilateral agreements or multilateral agreements. The agreements have, and will be, concluded on the basis of guaranteed reciprocity, compliance with the principle of specialty (i.e., the information exchanged may only be used to assess and levy taxes (and for criminal tax proceedings) and adequate data protection.
Based on such multilateral agreements and bilateral agreements and the implementing laws of Switzerland, Switzerland exchanges data in respect of financial assets, including the Shares, held in, and income derived thereon and credited to, accounts or deposits with a paying agent in Switzerland for the benefit of individuals resident in an EU member state or in a treaty state.
Swiss Facilitation of the Implementation of the U.S. Foreign Account Tax Compliance Act
Switzerland has concluded an intergovernmental agreement with the U.S. to facilitate the implementation of FATCA. The agreement ensures that the accounts held by U.S.
persons with Swiss financial institutions are disclosed to the U.S. tax authorities either with the consent of the account holder or by means of group requests within the scope of administrative assistance. Information will not be transferred automatically in the absence of consent, and instead will be exchanged only within the scope of administrative assistance on the basis of the double taxation agreement between the U.S. and Switzerland. On October 8, 2014, the Swiss Federal Council approved a mandate for negotiations with the U.S. on changing the current direct-notification-based regime to a regime where the relevant information is sent to the Swiss Federal Tax Administration, which in turn provides the information to the U.S. tax authorities.
Material U.S. Federal Income Tax Consequences for U.S. Holders
The following discussion describes the material U.S. federal income tax consequences relating to the ownership and disposition of our ADSs by U.S. Holders (as defined below). This discussion applies to U.S. Holders that hold our ADSs as capital assets (generally, property held for investment) within the meaning of Section 1221 of the U.S. Internal Revenue Code of 1986, as amended, or the Code. This discussion is based on the Code, U.S. Treasury Regulations promulgated thereunder, the income tax treaty between the United States and Switzerland, or the Treaty, and administrative and judicial interpretations thereof, all as in effect on the date hereof and all of which are subject to change, possibly with retroactive effect. This discussion does not address all of the U.S. federal income tax consequences that may be relevant to specific U.S. Holders in light of their particular circumstances or to U.S. Holders subject to special treatment under U.S. federal income tax law (such as certain financial institutions, insurance companies, broker‑dealers and traders in securities or other persons that generally mark their securities to market for U.S. federal income tax purposes, tax‑exempt entities, retirement plans, regulated investment companies, real estate investment trusts, certain former citizens or residents of the United States, persons who hold ADSs as part of a “straddle,” “conversion transaction,” “synthetic security” or integrated investment, persons who received their ADSs as compensatory payments, persons that have a “functional currency” other than the U.S. dollar, persons that own directly, indirectly or through attribution 10% or more of the voting power or value of our ADSs, corporations that accumulate earnings to avoid U.S. federal income tax, S-corporations, partnerships (including entities or arrangements treated as partnerships for U.S. federal income tax purposes) and other pass‑through entities, and investors in such pass‑through entities, or persons who hold our ADSs in connection with a trade or business, permanent establishment or fixed place of business outside the United States, including a permanent establishment in Switzerland). This discussion relates only to U.S. federal income taxes and does not address any other taxes, including but not limited to, U.S. state or local or non‑U.S. tax consequences or any U.S. federal estate, gift or alternative minimum tax consequences, or the special tax accounting rules under Section 451(b) of the Code.
As used in this discussion, the term “U.S. Holder” means a beneficial owner of our ADSs that is, for U.S. federal income tax purposes, (1) an individual who is a citizen or resident of the United States, (2) a corporation (or entity treated as a corporation for U.S. federal income tax purposes) created or organized in or under the laws of the United States, any state thereof, or the District of Columbia, (3) an estate the income of which is subject to U.S. federal income tax regardless of its source or (4) a trust (x) with respect to which a court within the United States is able to exercise primary supervision over its administration and one or more United States persons have the authority to control all of its substantial decisions or (y) that has elected under applicable U.S. Treasury Regulations to be treated as a domestic trust for U.S. federal income tax purposes.
If a partnership (including an entity or arrangement treated as a partnership for U.S. federal income tax purposes) holds our ADSs, the U.S. federal income tax consequences relating to an investment in the ADSs will depend in part upon the status and activities of such entity or arrangement and the particular partner. Any such entity or arrangement should consult its own tax advisor regarding the U.S. federal income tax consequences applicable to it and its partners of the purchase, ownership and disposition of our ADSs.
The discussion below assumes that the representations contained in the deposit agreement are true and that the obligations in the deposit agreement and any related agreement will be complied with in accordance with their terms. A U.S. Holder of our ADSs will generally be treated for U.S. federal income tax purposes as holding the ordinary shares represented by the ADSs, and, accordingly, no gain or loss will be recognized upon an exchange of the ADSs for the ordinary shares.
Passive Foreign Investment Company Consequences
In general, a corporation organized outside the United States will be treated as a passive foreign investment company, or PFIC, for U.S. federal income tax purposes for any taxable year in which, after the application of certain look-through rules with respect to income and assets of its subsidiaries, either (1) at least 75% of its gross income is “passive income,” or (2) on average at least 50% of its assets, determined on a quarterly basis, for the taxable year are assets that produce passive income or are held for the production of passive income. Passive income for this purpose generally includes, among other things, dividends, interest, royalties, rents, and gains from the sale or exchange of property that gives rise to passive income. Assets that produce or are held for the production of passive income generally include cash (unless held in a non-interest bearing account for short-term working capital needs), marketable securities, and other assets that may produce passive income. Generally, in determining whether a non‑U.S. corporation is a PFIC, a proportionate share of the income and assets of each corporation in which it owns, directly or indirectly, at least a 25% interest (by value) is taken into account.
Based upon our analysis of the value of our assets and the nature and composition of our income and assets, we believe that we were a PFIC for the taxable year ended December 31, 2023. However, the determination of whether or not we are a PFIC is a fact-intensive determination made annually after the end of the taxable year and the applicable law is subject to varying interpretations. For instance, the value of our assets may be determined in large part by reference to the market price of our ADSs, which is likely to continue to fluctuate. Accordingly, we cannot provide any assurance regarding, and our U.S. counsel expresses no opinion with respect to, our PFIC status for any taxable year. Furthermore, there can be no assurance that the U.S. Internal Revenue Service, or the IRS, will agree with our conclusion or that the IRS will not successfully challenge our position.
If we are a PFIC in any taxable year during which a U.S. Holder owns our ADSs, the U.S. Holder could be liable for additional taxes and interest charges under the “PFIC excess distribution regime” upon (1) a distribution made during a taxable year that is greater than 125% of the average annual distributions made in the three preceding taxable years, or, if shorter, the U.S. Holder’s holding period for the ADSs, and (2) any gain recognized on a sale, exchange or other disposition, including a pledge, of the ADSs, whether or not we continue to be a PFIC. Under the PFIC excess distribution regime, the tax on such distribution or gain would be determined by allocating the distribution or gain ratably over the U.S. Holder’s holding period for the ADSs. The amount allocated to the current taxable year (i.e., the year in which the distribution occurs or the gain is recognized) and any year prior to the first taxable year in which we are a PFIC will be taxed as ordinary income earned in the current taxable year. The amount allocated to other taxable years will be taxed at the highest marginal rates in effect for individuals or corporations, as applicable, for ordinary income for each such taxable year, and an interest charge, generally applicable to underpayments of tax, will be added to the tax.
If we are a PFIC for any taxable year during which a U.S. Holder holds our ADSs, we must generally continue to be treated as a PFIC by that holder for all succeeding years during which the U.S. Holder holds the ADSs, unless we cease to meet the requirements for PFIC status and the U.S. Holder makes a “deemed sale” election with respect to the ADSs.
If this election is made, the U.S. Holder will be deemed to sell the ADSs it holds at their fair market value on the last day of the last taxable year in which we qualified as a PFIC. Any gain recognized from such deemed sale will be taxed under the PFIC excess distribution regime, and any loss will not be recognized. The U.S. Holder’s tax basis in its ADSs will be increased by the amount of gain recognized, and the U.S. Holder’s holding period for its ADSs will start on the day after the last day of the last taxable year in which we qualified as a PFIC. After the deemed sale election, the U.S. Holder’s ADSs will not be treated as shares of a PFIC unless we subsequently become a PFIC.
If we are a PFIC for any taxable year during which a U.S. Holder holds our ADSs and at any time have a non‑U.S. corporate subsidiary that is also a PFIC (i.e., a lower‑tier PFIC), such U.S. Holder generally will be treated as owning a proportionate amount (by value) of the shares of the lower‑tier PFIC and will be taxed under the PFIC excess distribution regime on distributions by the lower‑tier PFIC and on gain from the disposition of shares of the lower‑tier PFIC even though such U.S. Holder would not receive the proceeds of those distributions or dispositions. Each U.S. Holder is advised to consult its tax advisors regarding the application of the PFIC rules to our non‑U.S. subsidiaries, if any.
If we are a PFIC for any taxable year during which a U.S. Holder holds our ADSs, such U.S. Holder will not be subject to tax under the PFIC excess distribution regime on distributions or gain recognized on the ADSs if such U.S. Holder makes a valid “mark‑to‑market” election for our ADSs. A mark‑to‑market election is available to a U.S. Holder only for “marketable stock.” Our ADSs will be marketable stock as long as they remain listed on the Nasdaq Global Select Market and are regularly traded, other than in de minimis quantities, on at least 15 days during each calendar quarter. If a mark‑to‑market election is in effect, a U.S. Holder generally will take into account, as ordinary income for each taxable year of the U.S. Holder, any excess of the fair market value of the U.S. Holder’s ADSs held at the end of such taxable year over the U.S. Holder's adjusted tax basis in such ADSs. The U.S. Holder will also take into account, as an ordinary loss for each taxable year, any excess of its adjusted tax basis in such ADSs over their fair market value at the end of the taxable year, but only to the extent of the excess of amounts previously included in income over ordinary losses deducted as a result of the mark‑to‑market election. The U.S. Holder’s tax basis in its ADSs will be adjusted to reflect any income or loss recognized as a result of the mark‑to‑market election. Any gain from a sale, exchange or other disposition of the ADSs in any taxable year in which we are a PFIC will be treated as ordinary income and any loss from such sale, exchange or other disposition would be treated first as ordinary loss (to the extent of any net mark‑to‑market gains previously included in income) and thereafter as capital loss.
A mark‑to‑market election will not apply to our ADSs for any taxable year during which we are not a PFIC but will remain in effect with respect to any subsequent taxable year in which we become a PFIC. Such election generally will not apply to any lower-tier PFICs that we may organize or acquire in the future, unless shares of such lower-tier PFICs are themselves marketable stock. Accordingly, a U.S. Holder may continue to be subject to tax under the PFIC excess distribution regime with respect to any lower‑tier PFICs that we may organize or acquire in the future notwithstanding the U.S. Holder’s mark‑to‑market election for our ADSs.
The tax consequences that would apply if we are a PFIC would also be different from those described above if a U.S. Holder were able to make a valid qualified electing fund, or QEF, election for taxable years during which the U.S. Holder holds our ADSs and in which we are a PFIC. Instead, a U.S. Holder that makes a QEF election is required for each taxable year to include in income (i) the U.S. Holder’s pro rata share of the PFIC’s ordinary earnings as ordinary income or (ii) the U.S. Holder’s pro rata share of the PFIC’s net capital gains as capital gain, regardless of whether such earnings or gain have in fact been distributed, for each taxable year that the entity is classified as a PFIC. If a U.S.
Holder makes a QEF election with respect to us, any distributions paid by us out of our earnings and profits that were previously included in the U.S. Holder’s income under the QEF election would not be taxable to the U.S. Holder. A U.S. Holder will increase its tax basis in its ADSs by an amount equal to any income included under the QEF election and will decrease its tax basis by any amount distributed on the ADSs that is not included in the U.S. Holder’s income. If a U.S. Holder has made a QEF election with respect to its ADSs, any gain or loss recognized by the U.S. Holder on a sale or other disposition of such ADSs will constitute capital gain or loss. In addition, if a U.S. Holder makes a timely QEF election, our ADSs will not be considered shares in a PFIC in years in which we are not a PFIC, even if the U.S. Holder had held ADSs in prior years in which we were a PFIC.
U.S. Holders should consult their tax advisors regarding making QEF elections in their particular circumstances. If a U.S. Holder does not make and maintain a QEF election for the U.S. Holder’s entire holding period for our ADSs by making the election for the first year in which the U.S. Holder owns our ADSs, the U.S. Holder will be subject to the adverse PFIC rules discussed above unless the U.S. Holder can properly make a “purging election” with respect to our ADSs in connection with the U.S. Holder’s QEF election. A purging election may require the U.S. Holder to recognize taxable gain on the U.S. Holder’s ADSs.
In order to comply with the requirements of a QEF election, a U.S. Holder must receive certain information from us. The QEF election is made on a shareholder-by-shareholder basis and can be revoked only with the consent of the IRS. A shareholder makes a QEF election by attaching a completed IRS Form 8621, including the information provided in a PFIC annual information statement, to a timely filed U.S. federal income tax return and by filing a copy of the form with the IRS. We expect to provide the information necessary for a U.S. Holder to make a QEF election if we were treated as a PFIC for any taxable year, although there is no assurance that we will do so. There is no assurance that we have not been previously classified as a PFIC during a U.S. Holder’s holding period for our ADSs. Accordingly, U.S. holders may be unable to make a timely QEF election with respect to our ADSs.
U.S. Holders should consult their tax advisors to determine whether any of these above elections would be available and if so, what the consequences of the alternative treatments would be in their particular circumstances.
Each U.S. person (as defined in the Code) that is an investor of a PFIC is generally required to file an annual information return on IRS Form 8621 containing such information as the U.S. Treasury Department may require. The failure to file IRS Form 8621 could result in the imposition of penalties and the extension of the statute of limitations with respect to U.S. federal income tax.
The U.S. federal income tax rules relating to PFICs are very complex. U.S. Holders are strongly urged to consult their own tax advisors with respect to the impact of PFIC status on the ownership and disposition of our ADSs, the consequences to them of an investment in a PFIC, any elections available with respect to the ADSs and the IRS information reporting obligations with respect to the ownership and disposition of ADSs of a PFIC.
Distributions
We do not anticipate declaring or paying dividends to holders of our ADSs in the foreseeable future. However, if we make a distribution contrary to this expectation, subject to the discussion above under “- Passive Foreign Investment Company Consequences,” a U.S. Holder that receives a distribution with respect to our ADSs generally will be required to include the gross amount of such distribution in gross income as a dividend when actually or constructively received to the extent of the U.S. Holder’s pro rata share of our current or accumulated earnings and profits (as determined under U.S. federal income tax principles). To the extent a distribution received by a U.S.
Holder is not a dividend because it exceeds the U.S. Holder’s pro rata share of our current and accumulated earnings and profits, it will be treated first as a taxfree return of capital and reduce (but not below zero) the adjusted tax basis of the U.S. Holder’s ADSs. To the extent the distribution exceeds the adjusted tax basis of the U.S. Holder’s ADSs, the excess will be taxed as capital gain. Because we may not account for our earnings and profits in accordance with U.S. federal income tax principles, U.S. Holders should expect all distributions to them to be treated as dividends. The amount of any dividend income paid in a currency other than the U.S. dollar will be the U.S. dollar amount calculated by reference to the exchange rate in effect on the date of actual or constructive receipt, regardless of whether the payment is in fact converted into U.S. dollars at that time. If the dividend is converted into U.S. dollars on the date of receipt, a U.S. Holder should not be required to recognize foreign currency gain or loss in respect of the dividend income. A U.S. Holder may have foreign currency gain or loss if the dividend is converted into U.S. dollars after the date of receipt.
Distributions on our ADSs that are treated as dividends generally will constitute income from sources outside the United States for U.S. foreign tax credit purposes and generally will constitute passive category income. Subject to certain complex conditions and limitations, Swiss taxes withheld on any distributions on our ADSs at a rate not exceeding the rate provided by the Treaty may be eligible for credit against a U.S. Holder’s U.S. federal income tax liability. The rules relating to the determination of the U.S. foreign tax credit are complex, and U.S. Holders should consult their tax advisors regarding the availability of a foreign tax credit in their particular circumstances and the possibility of claiming a deduction (in lieu of the foreign tax credit) for any foreign taxes paid or withheld.
Dividends paid by a “qualified foreign corporation” are eligible for taxation to non-corporate U.S. Holders at a reduced capital gains rate rather than the marginal tax rates generally applicable to ordinary income provided that certain requirements are met, including holding period and the absence of certain risk reduction transaction requirements. Each U.S. Holder is advised to consult its tax advisors regarding the availability of the reduced tax rate on dividends with regard to its particular circumstances. Prospective investors should be aware, however, that dividends paid by a company that is a PFIC in the taxable year in which the distribution is paid or in the preceding taxable year are not eligible to be taxed at such reduced rate. Distributions on our ADSs that are treated as dividends generally will not be eligible for the “dividends received” deduction generally allowed to corporate shareholders with respect to dividends received from U.S. corporations.
A non-U.S. corporation (other than a corporation that is classified as a PFIC for the taxable year in which the dividend is paid or the preceding taxable year) generally will be considered to be a qualified foreign corporation (a) if it is eligible for the benefits of a comprehensive tax treaty with the United States which the Secretary of Treasury of the United States determines is satisfactory for purposes of this provision and which includes an exchange of information provision, or (b) with respect to any dividend it pays on ADSs that are readily tradable on an established securities market in the United States. We believe that we qualify as a resident of Switzerland for purposes of, and are eligible for the benefits of the Treaty, although there can be no assurance in this regard. Further, the IRS has determined that the Treaty is satisfactory for purposes of the qualified dividend rules and that it includes an exchange of information provision. Our ADSs will generally be considered to be readily tradable on an established securities market in the United States if they are listed on Nasdaq Global Select Market, as we intend our ADSs to be. U.S. Holders should consult their own tax advisors regarding the availability of the lower rate for dividends paid with respect to our ADSs.
Sale, Exchange or Other Disposition of Our ADSs
Subject to the discussion above under “- Passive Foreign Investment Company Consequences,” a U.S. Holder generally will recognize capital gain or loss for U.S.
federal income tax purposes upon the sale, exchange or other disposition of our ADSs in an amount equal to the difference, if any, between the amount realized (i.e., the amount of cash plus the fair market value of any property received) on the sale, exchange or other disposition and such U.S. Holder’s adjusted tax basis in the ADSs. Such capital gain or loss generally will be long-term capital gain taxable at a reduced rate for non-corporate U.S. Holders or long-term capital loss if, on the date of sale, exchange or other disposition, the ADSs were held by the U.S. Holder for more than one year. Any capital gain of a non-corporate U.S. Holder that is not long-term capital gain will be taxed at ordinary income rates. The deductibility of capital losses is subject to limitations. Any gain or loss recognized from the sale or other disposition of the ADSs will generally be gain or loss from sources within the United States for U.S. foreign tax credit purposes.
Medicare Tax
Certain U.S. Holders that are individuals, estates or trusts and whose income exceeds certain thresholds generally are subject to a 3.8% tax on all or a portion of their net investment income, which may include their gross dividend income and net gains from the disposition of our ADSs. If you are a United States person that is an individual, estate or trust, you are encouraged to consult your tax advisors regarding the applicability of this Medicare tax to your income and gains in respect of your investment in our ADSs.
Information Reporting and Backup Withholding
U.S. Holders may be required to file certain U.S. information reporting returns with the IRS with respect to their investment in our ADSs, including, among others, IRS Form 8938 (Statement of Specified Foreign Financial Assets). As described above under “- Passive Foreign Investment Company Consequences,” each U.S. Holder who is a shareholder of a PFIC must file an annual report containing certain information.
Dividends on and proceeds from the sale or other disposition of our ADSs may be subject to U.S. backup withholding unless the U.S. Holder establishes a basis for exemption. Backup withholding generally would apply if the holder fails to (1) provide an accurate United States taxpayer identification number (usually on IRS Form W-9), or (2) otherwise establish an exemption from information reporting and backup withholding. Backup withholding is not an additional tax. Any amounts withheld under the backup withholding rules generally will be allowed as a refund or a credit against a U.S. Holder’s U.S. federal income tax liability if the required information is furnished by the U.S. Holder on a timely basis to the IRS.
U.S. Holders paying more than US$100,000 for our ADSs may be required to file IRS Form 926 (Return by a U.S. Transferor of Property to a Foreign Corporation) reporting this payment. Substantial penalties may be imposed upon a U.S. Holder that fails to comply with the required information reporting.
U.S. Holders should consult their own tax advisors regarding the backup withholding and information reporting rules.
The discussion above is for general informational purposes only and is not tax advice. Prospective investors in our ADSs should consult their tax advisors regarding the U.S. federal, state, and local and non-U.S. income and non-income tax consequences of the ownership and disposition of our ADSs in their particular circumstances, including information reporting requirements and the impact of any potential change in law.
F. Dividends and Paying Agents.
Not applicable.
G. Statement by Experts.
Not applicable.
H. Documents on Display.
We are subject to the information reporting requirements of the Exchange Act applicable to foreign private issuers and under those requirements will file reports with the SEC. Those reports may be inspected without charge at the locations described below. As a foreign private issuer, we are exempt from the rules under the Exchange Act related to the furnishing and content of proxy statements, and our officers, directors and principal shareholders are exempt from the reporting and short-swing profit recovery provisions contained in Section 16 of the Exchange Act. In addition, we are not required under the Exchange Act to file periodic reports and financial statements with the SEC as frequently or as promptly as United States companies whose securities are registered under the Exchange Act. Nevertheless, we will file with the SEC an Annual Report on Form 20-F containing financial statements that have been examined and reported on, with and opinion expressed by an independent registered public accounting firm.
We maintain a corporate website at www.molecularpartners.com. We intend to post our Annual Report on Form 20-F on our website promptly following it being filed with the SEC. Information contained on, or that can be accessed through, our website does not constitute a part of this Annual Report on Form 20-F. We have included our website address in this Annual Report on Form 20-F solely as an inactive textual reference.
The Securities and Exchange Commission maintains a website (www.sec.gov) that contains reports, proxy and information statements and other information regarding registrants, such as Molecular Partners, that file electronically with the SEC.
With respect to references made in this Annual Report on Form 20-F to any contract or other document of our company, such references are not necessarily complete and you should refer to the exhibits attached or incorporated by reference to this Annual Report on Form 20-F for copies of the actual contract or document.
I. Subsidiary Information.
Not required.
J. Annual Report to Security Holders
The Company intends to submit any annual report provided to security holders in electronic format as an exhibit to a current report on Form 6-K.
Item 11. Quantitative and Qualitative Disclosures About Market Risk.
We operate primarily in Switzerland, Europe and in the United States and are therefore exposed to market risk, which represents the risk of loss that may impact our financial position due to adverse changes in financial market prices and rates.
As of December 31, 2023, we had cash and cash equivalents plus short-term time deposits of CHF 186.9 million.
Foreign exchange risk
We operate primarily in Switzerland, Europe and in the United States and our functional currency is the Swiss franc, and as a result, we are exposed to (1) transactional foreign exchange risk when we enter into a transaction in a currency other than our functional currency and (2) translational foreign exchange risk when we translate our financial statements from USD into our functional currency.
In order to reduce our foreign exchange exposure, we may enter into currency contracts with selected high-quality financial institutions to hedge against foreign currency exchange rate risks. The Group’s primary exposure to financial risk is due to fluctuation of exchange rates between CHF, USD and EUR. Our hedging policy is (1) to maximize natural hedging by matching expected future cash flows in the different currencies and (2), if market conditions allow and as the need arises, to consider hedging some of the remaining expected net currency exposure. However, due to market volatilities and uncertainties in the cash flows, a 100% hedging of the currency exposure is impossible.
Credit risk
The maximum credit risk on financial instruments corresponds to the carrying amounts of our cash and cash equivalents, short-term time deposits and receivables. We have not entered into any guarantees or similar obligations that would increase the risk over and above the carrying amounts. As of December 31, 2023, substantially all of our cash and cash equivalents, and short-term time deposits were held at major financial institutions located in Switzerland. We believe that these financial institutions are of high credit quality and continually monitor the credit worthiness of these financial institutions. We enter into partnerships with partners that have the appropriate credit history and a commitment to ethical business practices. Other receivables with credit risk mainly include interest receivables.
Item 12. Description of Securities Other than Equity Securities.
A. Debt Securities.
Not applicable.
B. Warrants and Rights.
Not applicable.
C. Other Securities.
Not applicable.
D. American Depositary Shares.
Fees and Charges
Holders of our ADSs are required to pay the following service fees to the depositary under the terms of our deposit agreement:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Service |
|
Fees |
|
|
•Issuance of ADSs (e.g., an issuance of ADS upon a deposit of ordinary shares, upon a change in the ADS(s)-to- common share(s) ratio, or for any other reason), excluding ADS issuances as a result of distributions of ordinary shares) |
|
Up to U.S. $0.05 per ADS issued |
|
|
•Cancellation of ADSs (e.g., a cancellation of ADSs for delivery of deposited property, upon a change in the ADS(s)-to- common share(s) ratio, or for any other reason) |
|
Up to U.S. $0.05 per ADS canceled |
|
|
•Distribution of cash dividends or other cash distributions (e.g., upon a sale of rights and other entitlements) |
|
Up to U.S. $0.05 per ADS held |
|
|
•Distribution of ADSs pursuant to stock dividends, other free stock distributions or exercise of rights to purchase additional ADSs. |
|
Up to U.S. $0.05 per ADS held |
|
|
•Distribution of securities other than ADSs or rights to purchase additional ADSs (e.g., upon a spin-off) |
|
Up to U.S. $0.05 per ADS held |
|
|
•ADS Services |
|
Up to U.S. $0.05 per ADS held on the applicable record date(s) established by the depositary |
|
|
|
•Registration of ADS Transfers (e.g., upon a registration of the transfer of registered ownership of ADSs, upon a transfer of ADSs into DTC and vice versa, or for any other reason). |
|
Up to U.S. $0.05 per ADS transferred |
|
|
|
•Conversion of ADSs of one series for ADSs of another series (e.g., upon conversion of Partial Entitlement ADSs for Full Entitlement ADSs, or upon conversion of Restricted ADSs into freely transferable ADSs, and vice versa). |
|
Up to U.S. $0.05 per ADS converted |
Holders of ADSs are also responsible to pay certain fees, expenses, taxes and governmental charges such as:
•taxes (including applicable interest and penalties) and other governmental charges; •such registration fees as may from time to time be in effect for the registration of ordinary shares or other deposited securities on the share register and applicable to transfers of ordinary shares or other deposited securities to or from the name of the custodian, the depositary or any nominees upon the making of deposits and withdrawals, respectively;
•such cable, telex and facsimile transmission and delivery expenses as are expressly provided in the deposit agreement to be at the expense of the person depositing ordinary shares or withdrawing deposited property or of the holders and beneficial owners of ADSs;
•in connection with the conversion of foreign currency, the fees, expenses, spreads, taxes and other charges of the depositary and/or conversion service providers (which may be a division, branch or affiliate of the depositary). Such fees, expenses, spreads, taxes, and other charges shall be deducted from the foreign currency;
•any reasonable and customary out-of-pocket expenses incurred in such conversion and/or on behalf of the holders and beneficial owners in complying with currency exchange control or other governmental requirements; and
•the fees, charges, costs and expenses incurred by the depositary, the custodian, or any nominee in connection with the ADR program.
ADS fees and charges for (i) the issuance of ADSs, and (ii) the cancellation of ADSs are charged to the person for whom the ADSs are issued (in the case of ADS issuances) and to the person for whom ADSs are cancelled (in the case of ADS cancellations). In the case of ADSs issued by the depositary into DTC, the ADS issuance and cancellation fees and charges may be deducted from distributions made through DTC, and may be charged to the DTC participant(s) receiving the ADSs being issued or the DTC participant(s) holding the ADSs being cancelled, as the case may be, on behalf of the beneficial owner(s) and will be charged by the DTC participant(s) to the account of the applicable beneficial owner(s) in accordance with the procedures and practices of the DTC participants as in effect at the time.
ADS fees and charges in respect of distributions and the ADS service fee are charged to the holders as of the applicable ADS record date. In the case of distributions of cash, the amount of the applicable ADS fees and charges is deducted from the funds being distributed. In the case of (i) distributions other than cash and (ii) the ADS service fee, holders as of the ADS record date will be invoiced for the amount of the ADS fees and charges and such ADS fees and charges may be deducted from distributions made to holders of ADSs. For ADSs held through DTC, the ADS fees and charges for distributions other than cash and the ADS service fee may be deducted from distributions made through DTC, and may be charged to the DTC participants in accordance with the procedures and practices prescribed by DTC and the DTC participants in turn charge the amount of such ADS fees and charges to the beneficial owners for whom they hold ADSs.
In the case of (i) registration of ADS transfers, the ADS transfer fee will be payable by the ADS holder whose ADSs are being transferred or by the person to whom the ADSs are transferred, and (ii) conversion of ADSs of one series for ADSs of another series, the ADS conversion fee will be payable by the holder whose ADSs are converted or by the person to whom the converted ADSs are delivered.
In the event of refusal to pay the depositary fees or other charges, the depositary may, under the terms of the deposit agreement, refuse the requested service until payment is received or may set off the amount of the depositary fees or other charges from any distribution to be made to the ADS holder.
The fees and charges holders of ADSs may be required to pay may vary over time and may be changed by us and by the depositary. Holders of ADSs will receive prior notice of such changes.
The depositary may reimburse us for certain expenses incurred by us in respect of the ADR program, by making available a portion of the depositary fees charged in respect of the ADR program or otherwise, upon such terms and conditions as we and the depositary may agree from time to time.
PART II
Item 13. Defaults, Dividend Arrearages and Delinquencies.
Not applicable.
Item 14. Material Modifications to the Rights of Security Holders and Use of Proceeds.
A. Not applicable.
B. Not applicable.
C. Not applicable.
D. Not applicable.
E. Use of Proceeds.
Not applicable
Item 15. Controls and Procedures.
A. Disclosure Controls and Procedures
We maintain “disclosure controls and procedures,” as defined in Rules 13a-15(e) and 15d-15(e) under the Securities Exchange Act of 1934, as amended, or the Exchange Act, that are designed to ensure that information required to be disclosed by us in the reports that we file or submit under the Exchange Act is recorded, processed, summarized and reported, within the time periods specified in the SEC’s rules and forms, and is accumulated and communicated to our management, including our Chief Executive Officer and SVP Finance, as appropriate to allow timely decisions regarding required disclosure. Our management, with the participation of our Chief Executive Officer and SVP Finance, has evaluated the effectiveness of our disclosure controls and procedures (as defined in Exchange Act Rules 13a-15(e) and 15d-15(e)) as of December 31, 2023. Based on such evaluation, our Chief Executive Officer and SVP Finance as principal financial officer, have concluded that, as of December 31, 2023, our disclosure controls and procedures were effective.
B. Management’s Annual Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal controls over financial reporting (as defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act) and for the assessment of the effectiveness of our internal control over financial reporting. Under the supervision and with the participation of our Chief Executive Officer (principal executive officer) and SVP Finance (principal financial officer), management assessed our internal control over financial reporting based upon the framework in Internal Control - Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO).
Based on this assessment, our management has concluded that our internal control over financial reporting was effective at the reasonable assurance level as of December 31, 2023.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements, and can only provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
C. Attestation Report of the Registered Public Accounting Firm
This Annual Report does not include an attestation report of our registered public accounting firm regarding the effectiveness of our internal control over financial reporting due to an exemption provided by the JOBS Act for emerging growth companies.
D. Changes in Internal Control Over Financial Reporting
There has been no change in our internal control over financial reporting (as defined in Rule 13a-15(f) under the Exchange Act) that occurred during the period covered by this Annual Report that has materially affected, or is reasonably likely to materially affect, internal control over financial reporting.
Item 16. Reserved.
Not applicable.
Item 16A. Audit Committees Financial Expert.
Our board of directors has further determined that Sandip Kapadia is an “audit committee financial expert” as defined by SEC rules and regulations and that each of the members of the Audit and Finance Committee qualifies as financially sophisticated under the applicable exchange listing rules. Mr. Kapadia is independent as such term is defined in Rule 10A-3 under the Exchange Act and under the listing standards of Nasdaq.
Item 16B. Code of Business Conduct and Ethics.
We have adopted a Code of Conduct which is applicable to all of our employees, executive officers and directors. The Code of Conduct is available on our website at www.molecularpartners.com. The Audit and Finance Committee of our board of directors is responsible for overseeing the Code of Conduct and is required to approve any waivers of the Code of Conduct for employees, executive officers and directors. We expect that any amendments to the Code of Conduct will be disclosed on our website.
Item 16C. Principal Accountant Fees and Services.
The aggregate fees for services rendered by KPMG AG, Zurich, Switzerland (PCAOB ID 3240), for professional services were as follows:
|
|
|
|
|
|
|
|
|
| in CHF thousands |
2023 |
2022 |
| Audit fees |
571 |
|
643 |
|
| Audit related fees |
— |
|
— |
|
| Tax fees |
— |
|
— |
|
| All other fees |
— |
|
— |
|
|
|
|
| Balance at December 31 |
571 |
|
643 |
|
Audit Fees
Audit fees include the standard audit work performed each fiscal year necessary to allow the auditor to issue an opinion on our Consolidated Financial Statements and to issue an opinion on the local statutory financial statements of the Company and its subsidiaries. Audit fees also include services that can be provided only by the auditor such as reviews of quarterly financial results, review of the registration statement filed with the SEC and comfort letters delivered to underwriters in connection with equity offerings.
Audit related fees
These services consisting primarily of agreed-upon procedure reports, accounting consultations and other attest services related to financial reporting that are not required by statute or regulation.
Tax fees
Fees for tax services represent income tax and indirect tax compliance services as well as tax advisory services.
All other fees
Fees for other services not included in the above three categories.
Pre-Approval Procedures and Policies
In accordance with the requirements of the U.S. Sarbanes-Oxley Act of 2002 and rules issued by the SEC, we utilize a procedure for the review and pre-approval of any services performed by KPMG. The procedure requires that all proposed engagements of KPMG for audit and permitted non-audit services are submitted to the Audit and Finance Committee for approval prior to the beginning of any such services. In accordance with this policy, all services performed by and fees paid to KPMG in 2023 and 2022 were approved by the Audit and Finance Committee.
Item 16D. Exemptions from the Listing Standards for Audit Committees.
Not applicable.
Item 16E. Purchases of Equity Securities by the Issuer and Affiliated Purchasers.
Not applicable.
Item 16F. Change in Registrant’s Certifying Accountant.
Not applicable.
Item 16G. Corporate Governance.
Summary of Significant Corporate Governance Differences From Nasdaq Listing Standards
We are a "foreign private issuer" as defined by the SEC. As a result, in accordance with Nasdaq Listing Rule 5615(a)(3), we comply with home country governance requirements and certain exemptions thereunder rather than complying with certain of the corporate governance requirements of Nasdaq.
Swiss law does not require that a majority of our board of directors consist of independent directors. Our board of directors therefore may include fewer independent directors than would be required if we were subject to Nasdaq Listing Rule 5605(b)(1). In addition, we are not subject to Nasdaq Listing Rule 5605(b)(2), which requires that independent directors regularly have scheduled meetings at which only independent directors are present. We believe seven of the Board members satisfy the “independence” requirements of the Nasdaq rules.
Although Swiss law also requires that we adopt a compensation committee, we follow home country requirements with respect to such committee. As a result, our practice varies from the requirements of Nasdaq Listing Rule 5605(d), which sets forth certain requirements as to the responsibilities, composition and independence of compensation committees. To this extent, our practice varies from the independent director oversight of director nominations requirements of Nasdaq Listing Rule 5605(e).
We have opted out of shareholder approval requirements for the issuance of securities in connection with certain events such as the acquisition of stock or assets of another company, the establishment of or amendments to equity-based compensation plans for employees, a change of control of us and certain private placements. To this extent, our practice varies from the requirements of Nasdaq Listing Rule 5635, which generally requires an issuer to obtain shareholder approval for the issuance of securities in connection with such events.
In accordance with Swiss law and generally accepted business practices, our articles of association do not provide quorum requirements generally applicable to general meetings of shareholders. Our practice thus varies from the requirement of Nasdaq Listing Rule 5620(c), which requires an issuer to provide in its bylaws for a generally applicable quorum, and that such quorum may not be less than one-third of the outstanding voting stock.
Item 16H. Mine Safety Disclosure.
Not applicable.
Item 16I. Disclosure Regarding Foreign Jurisdictions that Prevent Inspection.
Not applicable.
Item 16J. Insider Trading Policies.
Pursuant to applicable SEC transition guidance, the disclosure required by Item 16J will be applicable to us starting the fiscal year ending December 31, 2024.
Item 16K. Cybersecurity.
Risk Management and Strategy
We have implemented and maintain various information security processes designed to identify, assess and manage material risks from cybersecurity threats to our critical computer networks, third party hosted services, communications systems, hardware and software, and our critical data, including intellectual property, confidential information that is proprietary, strategic or competitive in nature, and clinical trial data (“Information Systems and Data”).
Our information security function, legal team, and management board help identify, assess and manage the Company’s cybersecurity threats and risks. We identify and assess risks from cybersecurity threats by monitoring and evaluating our threat environment using various methods including, for example manual and automated tools, evaluating our and our industry’s risk profile, evaluating threats reported to us, audits, conducting threat assessments, and conducting tabletop incident response exercises.
Depending on the environment, we implement and maintain various technical, physical, and organizational measures, processes, and policies designed to manage and mitigate material risks from cybersecurity threats to our Information Systems and Data, including, for example: an incident response plan, disaster recovery/business continuity plans, risk assessments, network security controls, access controls, vendor risk management processes, employee training, and written IT policies.
Our assessment and management of material risks from cybersecurity threats are integrated into our overall risk management processes. For example, we prioritize and mitigate cybersecurity threats that are more likely to lead to a material impact to our business.
We use third-party service providers to assist us from time to time to identify, assess, and manage material risks from cybersecurity threats, including for example professional services firms, cybersecurity consultants, penetrating testing firms, and forensic investigators.
We use third-party service providers to perform a variety of functions throughout our business, such as data hosting, contract research, and contract manufacturing. We have a vendor management program to manage cybersecurity risks associated with our use of these providers. Depending on the nature of the services provided, the sensitivity of the Information Systems and Data at issue, and the identity of the provider, our vendor management process may involve different assessments designed to help identify cybersecurity risks associated with a provider and imposing contractual obligations related to cybersecurity on the provider.
For a description of the risks from cybersecurity threats that may materially affect the Company and how they may do so. See “Risk Factors—General Risks—We depend on our information technology systems, and any failure of these systems could harm our business. Security incidents, and other disruptions could compromise sensitive information related to our business or prevent us from accessing critical information and expose us to liability, which could adversely affect our business, results of operations and financial condition.”
Governance
Our board of directors addresses the Company’s cybersecurity risk management as part of its general oversight function. The board of directors’ audit and finance committee is responsible for overseeing Company’s cybersecurity risk management processes, including oversight and mitigation of risks from cybersecurity threats.
Our cybersecurity risk assessment and management processes are implemented and maintained by certain Company management, including:
•Baris Arican, Vice President of Information Technology. Mr. Arican has over 20 years experience in information technology/security at life sciences companies, including as the Chief Information Officer.
•Michael Pitzner, General Counsel and Compliance Officer and Senior Vice President of Legal and Business Development. Mr. Pitzner worked in various senior legal roles at other life sciences companies prior to joining our company.
•Robert Hendriks, Senior Vice President Finance and Principal Financial Officer. Mr. Hendriks worked in various senior finance roles at other life sciences companies prior to joining our company.
Company management is responsible for hiring appropriate personnel, communicating key priorities to relevant personnel, helping prepare for cybersecurity incidents, approving cybersecurity processes.
Our cybersecurity incident response processes are designed to escalate certain cybersecurity incidents to members of management depending on the circumstances, including to Messers Arican, Pitzner and Hendriks, who work with the Company’s incident response team to help the Company mitigate and remediate cybersecurity incidents of which they are notified. In addition, the Company’s incident response process includes reporting to the audit of the board of directors for certain cybersecurity incidents.
The board receives periodic reports from Mr. Arican concerning the Company’s significant cybersecurity threats and risk and the processes the Company has implemented to address them. The board also receives various reports, summaries or presentations related to cybersecurity threats, risk and mitigation.
PART III
Item 17. Financial Statements.
See pages F-1 through page F-
41 of this Annual Report on Form 20-F
Item 18. Financial Statements.
Not applicable.
Item 19. Exhibits.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Incorporated by Reference |
Exhibit |
|
Description |
|
Schedule/
Form
|
|
File
Number
|
|
Exhibit |
|
File
Date
|
|
|
|
|
|
|
|
|
|
|
|
| 1.1* |
|
|
|
|
|
|
|
|
|
|
1.2 |
|
|
|
20-F |
|
001-40488 |
|
1.2 |
|
3/09/2023 |
2.1 |
|
|
|
20-F |
|
001-40488 |
|
2.1 |
|
3/09/2023 |
2.2 |
|
|
|
20-F |
|
001-40488 |
|
2.2 |
|
3/09/2023 |
2.3 |
|
|
|
20-F |
|
001-40488 |
|
2.3 |
|
3/09/2023 |
| 4.1# |
|
|
|
F-1 |
|
333-255447 |
|
10.1 |
|
4/22/2021 |
| 4.2† |
|
|
|
F-1 |
|
333-255447 |
|
10.3 |
|
4/22/2021 |
| 4.3† |
|
|
|
F-1 |
|
333-255447 |
|
10.6 |
|
4/22/2021 |
4.4† |
|
|
|
20-F |
|
001-40488 |
|
4.4 |
|
3/09/2023 |
4.5† |
|
|
|
20-F |
|
001-40488 |
|
4.5 |
|
3/09/2023 |
4.6*† |
|
|
|
|
|
|
|
|
|
|
4.7# |
|
|
|
F-1 |
|
333-255447 |
|
10.8 |
|
4/22/2021 |
4.8# |
|
|
|
F-1 |
|
333-255447 |
|
10.9 |
|
4/22/2021 |
4.9# |
|
|
|
F-1 |
|
333-255447 |
|
10.10 |
|
4/22/2021 |
4.10# |
|
|
|
F-1 |
|
333-255447 |
|
10.11 |
|
4/22/2021 |
4.11# |
|
|
|
20-F |
|
001-40488 |
|
4.10 |
|
3/09/2023 |
4.12# |
|
|
|
20-F |
|
001-40488 |
|
4.11 |
|
3/09/2023 |
4.13# |
|
|
|
20-F |
|
001-40488 |
|
4.12 |
|
3/09/2023 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
4.14# |
|
|
|
20-F |
|
001-40488 |
|
4.13 |
|
3/09/2023 |
4.15# |
|
|
|
20-F |
|
001-40488 |
|
4.14 |
|
3/09/2023 |
4.16*# |
|
|
|
|
|
|
|
|
|
|
4.17*# |
|
|
|
|
|
|
|
|
|
|
4.18# |
|
|
|
F-1 |
|
333-255447 |
|
10.14 |
|
4/22/2021 |
4.19# |
|
|
|
F-1 |
|
333-255447 |
|
10.15 |
|
4/22/2021 |
4.20# |
|
|
|
F-1 |
|
333-255447 |
|
10.16 |
|
4/22/2021 |
4.21# |
|
|
|
F-1 |
|
333-255447 |
|
10.17 |
|
4/22/2021 |
4.22# |
|
|
|
20-F |
|
001-40488 |
|
4.19 |
|
3/09/2023 |
4.23# |
|
|
|
20-F |
|
001-40488 |
|
4.20 |
|
3/09/2023 |
4.24*# |
|
|
|
|
|
|
|
|
|
|
| 8.1 |
|
|
|
F-1 |
|
333-255447 |
|
21.1 |
|
4/22/2021 |
| 12.1* |
|
|
|
|
|
|
|
|
|
|
| 12.2* |
|
|
|
|
|
|
|
|
|
|
| 13.1** |
|
|
|
|
|
|
|
|
|
|
| 15.1* |
|
|
|
|
|
|
|
|
|
|
| 97.1* |
|
|
|
|
|
|
|
|
|
|
| 101.INS* |
|
Inline XBRL Instance Document |
|
|
|
|
|
|
|
|
| 101.SCH* |
|
Inline XBRL Taxonomy Extension Schema Document |
|
|
|
|
|
|
|
|
| 101.CAL* |
|
Inline XBRL Taxonomy Extension Calculation Linkbase Document |
|
|
|
|
|
|
|
|
| 101.DEF* |
|
Inline XBRL Taxonomy Extension Definition Linkbase Document |
|
|
|
|
|
|
|
|
| 101.LAB* |
|
Inline XBRL Taxonomy Extension Label Linkbase Document |
|
|
|
|
|
|
|
|
| 101.PRE* |
|
Inline XBRL Taxonomy Extension Presentation Linkbase Document |
|
|
|
|
|
|
|
|
| 104 |
|
Cover Page Interactive Data File (formatted as inline XBRL and contained in Exhibit 101) |
|
|
|
|
|
|
|
|
* Filed herewith.
** Furnished herewith.
† Certain portions of this exhibit (indicated by asterisks) have been redacted because they are both not material and are the type that the Registrant treats as private or confidential.
# Indicates a management contract or any compensatory plan, contract or arrangement.
The registrant hereby certifies that it meets all of the requirements for filing on Form 20-F and that it has duly caused and authorized the undersigned to sign this annual report on its behalf.
|
|
|
|
|
|
|
|
| Molecular Partners AG |
|
| /s/ Patrick Amstutz |
| By: |
Patrick Amstutz |
| Title: |
Chief Executive Officer
(Principal Executive Officer)
|
Date: March 14, 2024
Index to Financial Statements
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Consolidated Financial Statements as of and for the years ended December 31, 2023 and December 31, 2022 and December 31, 2021 |
|
Page |
|
|
|
|
|
|
|
|
|
|
|
|
Report of Independent Registered Public Accounting Firm
To the Shareholders and Board of Directors
Molecular Partners AG
Opinion on the Consolidated Financial Statements
We have audited the accompanying consolidated statements of financial position of Molecular Partners AG and subsidiary (the Group) as of December 31, 2023 and 2022, the related consolidated statements of comprehensive income, cash flows, and changes in equity for each of the years in the three-year period ended December 31, 2023, and the related notes to the consolidated financial statements (collectively, the consolidated financial statements). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Group as of December 31, 2023 and 2022, and the results of its operations and its cash flows for each of the years in the three‑year period ended December 31, 2023, in conformity with IFRS® Accounting Standards (IFRS) as issued by the International Accounting Standards Board.
Basis for Opinion
These consolidated financial statements are the responsibility of the Group’s management. Our responsibility is to express an opinion on these consolidated financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Group in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud. The Group is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits, we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Group’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. We believe that our audits provide a reasonable basis for our opinion.
Critical Audit Matters
Critical audit matters are matters arising from the current period audit of the consolidated financial statements that were communicated or required to be communicated to the audit committee and that: (1) relate to accounts or disclosures that are material to the consolidated financial statements and (2) involved our especially challenging, subjective, or complex judgments. We determined that there are no critical audit matters.
/s/ KPMG AG
We have served as the Group’s auditor since 2009.
Zurich, Switzerland
March 12, 2024
Consolidated Statement of Financial Position
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| as of December 31, |
Note |
|
2023 |
|
2022 |
| in CHF thousands |
|
|
| Assets |
|
|
|
|
|
| Property, plant and equipment |
6 |
|
|
5,681 |
|
|
|
7,235 |
|
|
| Intangible assets |
7 |
|
|
212 |
|
|
|
271 |
|
|
| Total non-current assets |
|
|
5,893 |
|
|
|
7,506 |
|
|
|
|
|
|
|
|
| Short-term time deposits |
11 |
|
|
119,580 |
|
|
|
161,198 |
|
|
| Other current assets |
9 |
|
|
3,617 |
|
|
|
4,589 |
|
|
| Trade and other receivables |
10 |
|
|
1,953 |
|
|
|
1,019 |
|
|
| Cash and cash equivalents |
11 |
|
|
67,309 |
|
|
|
87,946 |
|
|
| Total current assets |
|
|
192,459 |
|
|
|
254,752 |
|
|
|
|
|
|
|
|
| Total assets |
|
|
198,352 |
|
|
|
262,258 |
|
|
|
|
|
|
|
|
| Shareholders' equity and liabilities |
|
|
|
|
|
| Share capital |
12 |
|
|
3,635 |
|
|
|
3,604 |
|
|
| Additional paid-in capital |
|
|
365,530 |
|
|
|
360,323 |
|
|
| Treasury share reserve |
|
|
|
(981) |
|
|
|
(981) |
|
|
| Cumulative losses |
|
|
(191,755) |
|
|
|
(127,780) |
|
|
| Total shareholders' equity |
|
|
176,429 |
|
|
|
235,166 |
|
|
|
|
|
|
|
|
| Contract liability |
15 |
|
|
— |
|
|
|
3,637 |
|
|
| Lease liability |
22 |
|
|
2,444 |
|
|
|
3,652 |
|
|
| Employee benefits |
18.1 |
|
|
5,063 |
|
|
|
2,552 |
|
|
| Total non-current liabilities |
|
|
7,507 |
|
|
|
9,841 |
|
|
|
|
|
|
|
|
| Trade and other payables |
13 |
|
|
1,328 |
|
|
|
2,143 |
|
|
| Accrued expenses |
14 |
|
|
7,547 |
|
|
|
7,501 |
|
|
| Contract liability |
15 |
|
|
4,333 |
|
|
|
6,409 |
|
|
| Lease liability |
22 |
|
|
1,208 |
|
|
|
1,198 |
|
|
| Total current liabilities |
|
|
14,416 |
|
|
|
17,251 |
|
|
| Total liabilities |
|
|
21,923 |
|
|
|
27,092 |
|
|
|
|
|
|
|
|
| Total shareholders' equity and liabilities |
|
|
198,352 |
|
|
|
262,258 |
|
|
See accompanying notes, which form an integral part of these consolidated financial statements.
Consolidated Statement of Comprehensive Income
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| for the year ended December 31, |
Note |
|
2023 |
|
2022 |
|
2021 |
| in CHF thousands |
|
|
|
|
|
|
|
|
|
|
|
| Revenues and other income |
|
|
|
|
|
|
|
| Revenues from research and development collaborations |
|
|
7,038 |
|
|
|
189,556 |
|
|
|
9,330 |
|
|
| Other income |
|
|
|
— |
|
|
|
44 |
|
|
|
424 |
|
|
| Total revenues and other income |
5 |
|
|
7,038 |
|
|
|
189,600 |
|
|
|
9,754 |
|
|
|
|
|
|
|
|
|
|
| Operating expenses |
|
|
|
|
|
|
|
| Research and development expenses |
16 |
|
|
(48,784) |
|
|
|
(50,749) |
|
|
|
(55,718) |
|
|
| Selling, general and administrative expenses |
16 |
|
|
(19,362) |
|
|
|
(22,238) |
|
|
|
(17,454) |
|
|
| Total operating expenses |
|
|
(68,146) |
|
|
|
(72,987) |
|
|
|
(73,172) |
|
|
|
|
|
|
|
|
|
|
| Operating result |
|
|
(61,108) |
|
|
|
116,613 |
|
|
|
(63,418) |
|
|
|
|
|
|
|
|
|
|
| Financial income |
19 |
|
|
4,279 |
|
|
|
1,859 |
|
|
|
191 |
|
|
| Financial expenses |
19 |
|
|
(5,155) |
|
|
|
(619) |
|
|
|
(556) |
|
|
| Net finance result |
|
|
(876) |
|
|
|
1,240 |
|
|
|
(365) |
|
|
|
|
|
|
|
|
|
|
| Result before income taxes |
|
|
(61,984) |
|
|
|
117,853 |
|
|
|
(63,783) |
|
|
|
|
|
|
|
|
|
|
| Income taxes |
20 |
|
|
— |
|
|
|
— |
|
|
|
(2) |
|
|
| Net result, attributable to shareholders |
|
|
(61,984) |
|
|
|
117,853 |
|
|
|
(63,785) |
|
|
|
|
|
|
|
|
|
|
| Other comprehensive result |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Items that will not be reclassified to profit or loss |
|
|
|
|
|
|
|
| Remeasurement of net pension liabilities, net of tax |
18.1 |
|
|
(1,975) |
|
|
|
5,334 |
|
|
|
8,012 |
|
|
|
|
|
|
|
|
|
|
| Items that are or may be reclassified subsequently to profit or loss |
|
|
|
|
|
|
|
| Exchange differences on translating foreign operations |
|
|
(16) |
|
|
|
(17) |
|
|
|
(3) |
|
|
|
|
|
|
|
|
|
|
| Other comprehensive result, net of tax |
|
|
(1,991) |
|
|
|
5,317 |
|
|
|
8,009 |
|
|
|
|
|
|
|
|
|
|
| Total comprehensive result, attributable to shareholders |
|
|
(63,975) |
|
|
|
123,170 |
|
|
|
(55,776) |
|
|
|
|
|
|
|
|
|
|
| Basic net result per share (in CHF) |
21 |
|
|
(1.89) |
|
|
|
3.63 |
|
|
|
(2.06) |
|
|
| Diluted net result per share (in CHF) |
21 |
|
|
(1.89) |
|
|
|
3.54 |
|
|
|
(2.06) |
|
|
See accompanying notes, which form an integral part of these consolidated financial statements.
Consolidated Statement of Cash Flows
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| for the year ended December 31, |
Note |
|
2023 |
|
2022 |
|
2021 |
| in CHF thousands |
|
|
|
|
|
|
|
|
|
|
|
|
| Net result attributable to shareholders |
|
|
|
(61,984) |
|
|
|
117,853 |
|
|
|
(63,785) |
|
|
| Adjustments for: |
|
|
|
|
|
|
|
|
|
| Depreciation and amortization |
6/7 |
|
|
2,420 |
|
|
|
2,388 |
|
|
|
2,565 |
|
|
| Share-based compensation costs |
18 |
|
|
5,207 |
|
|
|
5,088 |
|
|
|
4,085 |
|
|
| Change in employee benefits |
|
|
|
535 |
|
|
|
1,147 |
|
|
|
1,073 |
|
|
| Income tax |
20 |
|
|
— |
|
|
|
— |
|
|
|
2 |
|
|
| Financial income |
19 |
|
|
(4,279) |
|
|
|
(1,859) |
|
|
|
(191) |
|
|
| Financial expenses |
19 |
|
|
5,155 |
|
|
|
619 |
|
|
|
556 |
|
|
| Changes in working capital: |
|
|
|
|
|
|
|
|
|
| Change in other current assets |
|
|
|
1,424 |
|
|
|
1,787 |
|
|
|
(4,445) |
|
|
| Change in trade and other receivables |
|
|
|
(933) |
|
|
|
25,264 |
|
|
|
(23,374) |
|
|
| Change in trade and other payables |
|
|
|
(812) |
|
|
|
(5,339) |
|
|
|
1,656 |
|
|
| Change in contract liability |
15 |
|
|
(5,713) |
|
|
|
(25,190) |
|
|
|
(10,651) |
|
|
| Change in accrued expenses |
|
|
|
45 |
|
|
|
(2,434) |
|
|
|
2,290 |
|
|
| Exchange (loss) gain on working capital positions |
|
|
|
(21) |
|
|
|
(98) |
|
|
|
(144) |
|
|
| Interest paid |
|
|
|
(34) |
|
|
|
(646) |
|
|
|
(583) |
|
|
| Income taxes paid |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
| Other financial expense |
|
|
|
(15) |
|
|
|
(14) |
|
|
|
(8) |
|
|
| Net cash (used in) from operating activities |
|
|
|
(59,005) |
|
|
|
118,566 |
|
|
|
(90,953) |
|
|
|
|
|
|
|
|
|
|
|
|
| Proceeds from investments in short-term time deposits |
|
|
|
319,443 |
|
|
|
199,219 |
|
|
|
67,876 |
|
|
| Investments in short-term time deposits |
|
|
|
(277,825) |
|
|
|
(299,417) |
|
|
|
(88,876) |
|
|
| Acquisition of property, plant and equipment |
6 |
|
|
(575) |
|
|
|
(1,177) |
|
|
|
(933) |
|
|
| Acquisition of intangible assets |
7 |
|
|
(233) |
|
|
|
(240) |
|
|
|
(374) |
|
|
| Interest received |
|
|
|
3,827 |
|
|
|
494 |
|
|
|
70 |
|
|
| Net cash from (used in) investing activities |
|
|
|
44,637 |
|
|
|
(101,121) |
|
|
|
(22,237) |
|
|
|
|
|
|
|
|
|
|
|
|
| Proceeds from issuance of new shares, net of transaction costs |
12 |
|
|
— |
|
|
|
— |
|
|
|
51,493 |
|
|
| Investments in treasury shares |
12 |
|
|
— |
|
|
|
(631) |
|
|
|
— |
|
|
| Proceeds from exercise of stock options, net of transaction costs |
12 |
|
|
31 |
|
|
|
250 |
|
|
|
267 |
|
|
| Payment of lease liabilities |
|
|
|
(1,198) |
|
|
|
(1,189) |
|
|
|
(1,179) |
|
|
| Net cash (used in) from financing activities |
|
|
|
(1,167) |
|
|
|
(1,570) |
|
|
|
50,581 |
|
|
|
|
|
|
|
|
|
|
|
|
| Exchange (loss) gain on cash positions |
|
|
|
(5,102) |
|
|
|
258 |
|
|
|
701 |
|
|
| Net (decrease) increase in cash and cash equivalents |
|
|
|
(20,637) |
|
|
|
16,133 |
|
|
|
(61,907) |
|
|
|
|
|
|
|
|
|
|
|
|
| Cash and cash equivalents at January 1 |
|
|
|
87,946 |
|
|
|
71,813 |
|
|
|
133,721 |
|
|
|
|
|
|
|
|
|
|
|
|
| Cash and cash equivalents at December 31 |
11 |
|
|
67,309 |
|
|
|
87,946 |
|
|
|
71,813 |
|
|
See accompanying notes, which form an integral part of these consolidated financial statements.
Consolidated Statement of Changes in Equity
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Share capital |
|
Additional paid-in capital |
|
Treasury share reserve |
|
Cumulative losses |
|
Total shareholders' equity |
| in CHF thousands |
|
|
|
|
| At January 1, 2021 |
2,915 |
|
|
|
299,479 |
|
|
|
— |
|
|
|
(195,174) |
|
|
|
107,220 |
|
|
| Net result |
— |
|
|
|
— |
|
|
|
— |
|
|
|
(63,785) |
|
|
|
(63,785) |
|
|
Remeasurement of net pension liabilities (1) |
— |
|
|
|
— |
|
|
|
— |
|
|
|
8,012 |
|
|
|
8,012 |
|
|
| Exchange differences on translating foreign operations |
— |
|
|
|
— |
|
|
|
— |
|
|
|
(3) |
|
|
|
(3) |
|
|
Total comprehensive loss |
— |
|
|
|
— |
|
|
|
— |
|
|
|
(55,776) |
|
|
|
(55,776) |
|
|
Share-based compensation costs (1) |
— |
|
|
|
4,085 |
|
|
|
— |
|
|
|
— |
|
|
|
4,085 |
|
|
Issuance of new shares, net of transaction costs(2) |
300 |
|
|
|
51,193 |
|
|
|
— |
|
|
|
— |
|
|
|
51,493 |
|
|
Exercise of stock options, net of transaction costs (2) |
14 |
|
|
|
253 |
|
|
|
— |
|
|
|
— |
|
|
|
267 |
|
|
| At December 31, 2021 |
3,229 |
|
|
|
355,010 |
|
|
|
— |
|
|
|
(250,950) |
|
|
|
107,289 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| At January 1, 2022 |
3,229 |
|
|
|
355,010 |
|
|
|
— |
|
|
|
(250,950) |
|
|
|
107,289 |
|
|
| Net result |
— |
|
|
|
— |
|
|
|
— |
|
|
|
117,853 |
|
|
|
117,853 |
|
|
Remeasurement of net pension liabilities (1) |
— |
|
|
|
— |
|
|
|
— |
|
|
|
5,334 |
|
|
|
5,334 |
|
|
| Exchange differences on translating foreign operations |
— |
|
|
|
— |
|
|
|
— |
|
|
|
(17) |
|
|
|
(17) |
|
|
Total comprehensive income |
— |
|
|
|
— |
|
|
|
— |
|
|
|
123,170 |
|
|
|
123,170 |
|
|
Share-based compensation costs (1) |
— |
|
|
|
5,088 |
|
|
|
— |
|
|
|
— |
|
|
|
5,088 |
|
|
Issuance of new shares, net of transaction costs(2) |
350 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
350 |
|
|
Issuance of treasury shares incl. transaction costs (2) |
— |
|
|
|
— |
|
|
|
(981) |
|
|
|
— |
|
|
|
(981) |
|
|
Exercise of stock options, net of transaction costs (2) |
25 |
|
|
|
225 |
|
|
|
— |
|
|
|
— |
|
|
|
250 |
|
|
| At December 31, 2022 |
3,604 |
|
|
|
360,323 |
|
|
|
(981) |
|
|
|
(127,780) |
|
|
|
235,166 |
|
|
|
|
|
|
|
|
|
|
|
|
| At January 1, 2023 |
3,604 |
|
|
|
360,323 |
|
|
|
(981) |
|
|
|
(127,780) |
|
|
|
235,166 |
|
|
| Net result |
— |
|
|
|
— |
|
|
|
— |
|
|
|
(61,984) |
|
|
|
(61,984) |
|
|
Remeasurement of net pension liabilities (1) |
— |
|
|
|
— |
|
|
|
— |
|
|
|
(1,975) |
|
|
|
(1,975) |
|
|
Exchange differences on translating foreign operations |
— |
|
|
|
— |
|
|
|
— |
|
|
|
(16) |
|
|
|
(16) |
|
|
Total comprehensive loss |
— |
|
|
|
— |
|
|
|
— |
|
|
|
(63,975) |
|
|
|
(63,975) |
|
|
Share-based compensation costs (1) |
— |
|
|
|
5,207 |
|
|
|
— |
|
|
|
— |
|
|
|
5,207 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Exercise of stock options, net of transaction costs (2) |
31 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
31 |
|
|
| At December 31, 2023 |
3,635 |
|
|
|
365,530 |
|
|
|
(981) |
|
|
|
(191,755) |
|
|
|
176,429 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(1) See note 18
(2) See note 12
See accompanying notes, which form an integral part of these consolidated financial statements.
Notes to the IFRS Consolidated Financial Statements
1. General information
Molecular Partners AG ("Company") and its subsidiary (collectively "Molecular Partners" or, "Group") is a clinical-stage biotech company pioneering the design and development of DARPin therapeutics for medical challenges other drug modalities cannot readily address. The Company has programs in various stages of pre-clinical and clinical development, with oncology as its main focus. Molecular Partners leverages the advantages of DARPins to provide unique solutions to patients through its proprietary programs as well as through partnerships with leading pharmaceutical companies.
The Company was founded on November 22, 2004, and is domiciled at Wagistrasse 14, 8952 Schlieren, Canton of Zurich, Switzerland. It is subject to the provisions of the articles of association and to article 620 et seq. of the Swiss Code of Obligations, which describe the legal requirements for limited companies (“Aktiengesellschaften”).
Molecular Partners Inc. is a wholly owned subsidiary of Molecular Partners AG. Molecular Partners Inc. was incorporated in the United States in the State of Delaware on October 8, 2018. Molecular Partners Inc. is based in Cambridge, Massachusetts.
These audited consolidated financial statements as of and for the year ended December 31, 2023 comprise Molecular Partners AG and Molecular Partners Inc.
The Company’s shares are listed on the SIX Swiss Exchange (Ticker: MOLN) since November 5, 2014 and on the Nasdaq Global Select Market (Ticker: MOLN) since June 16, 2021.
2. Summary of material accounting policies
Basis of preparation
These consolidated financial statements have been prepared in accordance with the IFRS® Accounting Standards ("IFRS") as issued by the IASB. The accounting policies set forth below have been consistently applied to all years presented. Unless stated otherwise, all financial statements are presented in thousands of Swiss Francs (“TCHF”).
The consolidated financial statements have been prepared under the historical cost convention. The preparation of financial statements in conformity with IFRS requires the use of certain critical accounting estimates. It also requires management to exercise its judgment in the process of applying the Group's accounting policies. The areas involving a higher degree of judgment or complexity, or areas where assumptions and estimates are significant to the financial statements are disclosed in note 4 “Critical accounting estimates and judgments”.
Based on the Group's cash and short-term time deposits positions at December 31, 2023, the Group deemed there to be no material uncertainties that would cast doubt on the Group's ability to operate on a going concern basis.
The consolidated financial statements as of and for the year ended December 31, 2023 were approved for issuance by the Company's Board of Directors on March 12, 2024.
Due to rounding, the numbers presented in the financial statements might not precisely equal those included in the accompanying notes.
Basis of consolidation
(i) Subsidiaries
Subsidiaries are entities controlled by the Company. The Company controls an entity when it is exposed to, or has rights to, variable returns from its involvement with the entity and has the ability to affect those returns through its power over the entity. The financial statements of subsidiaries are included in the consolidated financial statements from the date on which control commences until the date on which control ceases.
(ii) Transactions eliminated on consolidation
Intra-group balances and transactions, and any unrealized income and expenses arising from intra-group transactions, are eliminated.
New or revised IFRS standards and interpretations
The following new or revised standards that became effective during 2023 did not have a material effect on these consolidated financial statements:
•Disclosure of Accounting Policies - Amendments to IAS 1
•Definition of Accounting Estimates / Amendments to IAS 8
Several new or revised standards have been published that are not yet effective and that have not been early adopted. No significant impacts on the Group's consolidated financial statements are expected.
Segment reporting
The Group operates in one segment, focusing on the discovery, development and prospective commercialization of a new class of biopharmaceutical products. The executive management, acting together as the chief operating decision makers, assess the financial performance and allocate resources on an aggregated level, and monitor the Group's operating expenses. Accounting policies applied are the same for both internal and external reporting purposes. The Group derives its research and collaboration revenues from research and development collaborations with third parties.
Foreign currency translation / transactions
The consolidated financial statements are presented in thousands of CHF. The presentation currency of the Group is the functional currency of the Company. Foreign currency transactions are translated into the functional currency using the exchange rates prevailing at the dates of the transactions. Foreign exchange gains and losses resulting from the settlement of such transactions and from the translation at year-end exchange rates of monetary assets and liabilities denominated in foreign currencies are recognized in profit or loss.
The results and financial position of foreign operations that have a functional currency different from the presentation currency are translated into the presentation currency as follows:
•assets and liabilities are translated at the closing rate at the date of the respective balance sheet;
•income and expenses for each consolidated statement of comprehensive income are translated at average exchange rates (unless this is not a reasonable approximation of the cumulative effect of the rates prevailing on the transaction dates, in which case income and expenses are translated at the exchange rates at the dates of the transactions); and
•all resulting exchange differences are recognized in other comprehensive result.
Property, plant and equipment
Laboratory equipment, Office equipment, IT hardware and Leasehold improvements are stated at historical cost less accumulated depreciation and any impairment. Historical cost includes expenditures that are directly attributable to the acquisition of the items. Depreciation is calculated on a straight-line basis over the expected useful lives of the individual assets or asset categories. The applicable estimated useful lives are as follows:
|
|
|
|
|
|
| Laboratory equipment: |
5 years |
| Office equipment: |
3 years |
| IT hardware: |
2 years |
Leasehold improvements and right-of-use assets are depreciated using the straight line method over the shorter of their estimated useful life and the lease term.
The assets’ residual values and useful lives are reviewed, and adjusted if appropriate, at each reporting date. An asset’s carrying amount is written down to its recoverable amount, if the asset's carrying amount exceeds its estimated recoverable amount.
Intangible assets
Intangible assets are solely comprised of software. They are stated at historical cost less accumulated amortization and any impairment. Historical cost includes expenditures that are directly attributable to the acquisition of the items. Amortization is calculated on a straight-line basis over the expected useful lives of the individual assets or asset categories. The applicable estimated useful life of intangible assets is determined to be two years.
Leases
At inception of a contract, the Group assesses whether a contract is, or contains a lease. This is the case if the contract conveys the right to control the use of an identified asset for a period of time in exchange for consideration. The Group has elected not to recognize right-of-use assets and lease liabilities for leases of low-value assets (threshold of CHF 5,000) and short-term leases. Short-term leases are leases with a lease term of twelve months or less that do not contain a purchase option. For all other leases the Group recognizes a right-of-use asset and a lease liability at the lease commencement date.
The Group does not provide residual value guarantees and does not have any leases not yet commenced to which it is committed. The Group is presenting right-of-use assets in Property, Plant and Equipment, whereas lease liabilities are presented separately within current and non-current liabilities in the consolidated statement of financial position.
Financial assets at amortized costs
Classification
Cash and cash equivalents / short-term deposits / trade and other receivables (except for VAT and withholding taxes) (and when applicable accrued interest income) are all considered held-to-collect items and are labeled under financial assets measured at amortized costs, with the following definition / accounting policy:
Financial assets measured at amortized cost are assets that meet both of the following conditions: (1) the asset is held within a business model whose objective is to hold assets in order to collect contractual cash flows; and (2) the contractual terms of the financial asset give rise on specified dates to cash flows that are solely payments of principal and interest on the principal amount outstanding.
They arise when the Group provides money, goods or services directly to a debtor with no intention of trading the receivable. They are included in current assets, except for maturities longer than 12 months after the balance sheet date which are classified as non-current assets. Interest income on the short-term deposit is accounted for on the statement of comprehensive income as financial income.
Measurement
Initially, financial assets, except for trade receivables, are measured at their fair value plus, in the case of financial assets not at fair value through profit or loss, transaction costs that are directly attributable to the acquisition or issue of the financial asset; for the Group these are considered to be immaterial. Trade receivables are initially measured at their transaction price.
Subsequent measurement for the financial assets mentioned above which are classified as measured at amortized cost, is based on the effective interest method, reduced by any impairment loss.
For trade receivables, the Group applies a simplified approach which requires expected credit losses to be recognized from initial recognition (measuring the loss allowance at an amount equal to lifetime expected credit losses). This takes into consideration past history, combined with predictive information which accounts for the specific circumstances of the customer (e.g., credit rating etc.), and other relevant factors such as the economic environment.
Other financial assets at amortized costs
Other receivables generally arise from transactions outside the usual operating activities of the Group.
Financial liabilities at amortized costs
Trade payables and non-employee related accrued expense are measured at amortized costs and classified as financial liabilities.
Cash and cash equivalents
Cash includes cash at banks. The Group considers all short-term, highly liquid investments convertible into known amounts of cash with maturities of three months or less from the date of acquisition to be cash equivalents, provided that they are subject to an insignificant risk of changes in value. The cash flow statement is based on cash and cash equivalents.
Share capital / Additional paid-in capital
Common shares are classified as equity. Incremental costs directly attributable to the issue of new shares are shown in equity as a deduction from the proceeds. The Group has not paid any dividends since its inception and does not anticipate paying dividends in the foreseeable future.
Treasury shares
The amount of the consideration paid for the acquisition of treasury shares, which includes directly attributable costs, is recognized as a deduction from equity. When treasury shares are sold subsequently, the amount received is recognized as an increase in equity, and the resulting surplus or deficit on the transaction is presented in additional paid-in capital.
Income taxes
Income taxes include current and deferred taxes. Current income taxes are recognized on taxable profits at applicable tax rates.
Deferred taxes are calculated using the balance sheet liability method. Deferred income taxes reflect the net tax effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. Deferred tax assets and liabilities are measured using the tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled based on tax rates enacted or substantially enacted at the balance sheet date.
Deferred tax assets are recognized if it is probable that sufficient taxable profits will be available against which the deferred tax assets can be utilized. At each balance sheet date, the Group reassesses unrecognized deferred tax assets and the carrying amount of recognized deferred tax assets. The Group recognizes a previously unrecognized deferred tax asset to the extent that it has become probable that future taxable profit will allow the deferred tax asset to be recovered. The Group conversely reduces the carrying amount of a deferred tax asset to the extent that it is no longer probable that sufficient taxable profit will be available to allow the benefit of part or the entire deferred tax asset to be utilized.
The amount of deferred tax liabilities and deferred tax assets reflects the tax consequences on the balance sheet date of the Group's expectation of recovery or settlement of the carrying amounts of its assets and liabilities. Deferred tax assets and liabilities are not discounted and are classified as non-current assets and liabilities in the statement of financial position. They are offset against each other if they relate to the same taxable entity and tax authority.
Molecular Partners Inc, the Group's U.S. subsidiary, is subject to U.S. federal and Massachusetts and New York state minimal tax.
Employee benefits
Postretirement benefits (pension plans)
The Company provides retirement, death and disability benefits to its Swiss employees in line with local customs and requirements through two separate plans, which are both accounted for as defined benefit plans.
The first plan is the compulsory defined benefit plan which is funded through employer (60%) and employee (40%) contributions to VSAO, a Switzerland based plan. This Company-wide plan has been in place since inception of the Company and all employees of the Company are eligible to its benefits. On retirement, the plan participant will receive his or her accumulated savings, which consist of all contributions paid in by the employer and the employee (net of any withdrawals) and the interest granted on those savings at the discretion of the pension foundation.
At that time, the plan participant has the right to choose between a lump-sum payment and an annuity, or a combination thereof. The annuity is calculated using a fixed conversion rate determined by the pension foundation. The VSAO’s plan assets are pooled and the Company’s share is calculated based on its share of retirement savings. Additional funding requirements may be determined by the pension foundation in case of a severe underfunding. Should the Company withdraw from the plan, the withdrawal may qualify as a partial liquidation under Swiss law.
The second plan is a voluntary complementary defined management benefit scheme established as of January 1, 2014, in which only employees with a certain management level and / or above a certain salary level are eligible to participate. 29 of the 29 eligible employees participated in this plan as of December 31, 2023 (2022: 33 out of 33).
This plan is set up as a collective foundation with Swiss Life, a Switzerland-based insurance company, for which contributions are 30% funded by the employee and 70% funded by the Company. The purpose of this voluntary plan is to allow higher savings opportunity in a tax effective manner and risk benefits for senior management. In addition, plan participants are entitled to a lump sum payment of five times their annual base salary in case of death. This is a fully insured Swiss pension plan that covers all investment and actuarial risks, including invalidity and death.
The VSAO pension plan accounts for over 90% of both the Company’s defined benefit obligation and plan assets. The liability recognized in the statement of financial position in respect of defined benefit pension plans is the present value of the defined benefit obligations at the balance sheet date less the fair value of plan assets.
The defined benefit obligation is calculated annually by independent actuaries using the projected unit credit method. The present value of the defined benefit obligation is determined by discounting the estimated future cash outflows. Pension liabilities are determined on an actuarial basis using a number of assumptions, such as the discount rate and expected salary increases applied to determine the defined benefit obligation and an estimate of the fair value of plan assets attributable to the Company. In determining the appropriate discount rate, for example, the Company considers the interest rates of high-quality corporate bonds that are denominated in the currency in which the benefits will be paid, and that have terms to maturity approximating the terms of the related pension liability. In determining the fair value of plan assets, the Company adds to the participants’ savings a share of the pension plan’s technical and fluctuation reserves. Additional information is disclosed in note 18.1.
Current and past service costs as well as the net interest on the defined benefit obligation are recognized in profit or loss in the period in which they are incurred, and are presented as part of personnel expenses. Remeasurements of the defined benefit pension plans are recognized in other comprehensive result.
The Group has set up a 401k plan for its U.S. based employees. Under the plan the U.S. entity matches the employee's contribution and provides a true-up in matched contributions at year end. The 401k plan qualifies as a defined contribution plan and the associated expenses, that are deemed immaterial, are presented under operating expenses in the statement of comprehensive income.
The Group has set up a defined contribution plan for its UK based employees. Under the plan the Company and the employee both contribute into the plan. The associated expenses, that are deemed immaterial, are presented under operating expenses in the statement of comprehensive income.
Share-based compensation
The Group operates share-based compensation plans that qualify as equity-settled plans. The fair value of the employee services received in exchange for the grant of equity instruments is recognized as an expense. The total amount to be expensed over the vesting period is determined by reference to the fair value of the equity instruments granted, which is determined at grant date. The fair values are determined by management with the assistance of an independent valuation expert. At each reporting date, estimates of the number of equity instruments that are expected to vest are revised. The impact of the revision of the previous estimates, if any, is recognized as part of share-based compensation (non-cash effective) with a corresponding adjustment to equity. When the vested equity instruments are exercised, any proceeds received net of any directly attributable transaction costs are credited to share capital (nominal value) and additional paid-in capital.
Bonus plan
The Group recognizes an accrual where contractually obliged or where there is a past practice that has created a constructive obligation. Bonuses are based on a formula that takes into consideration the achievement of the Group’s goals.
Revenue recognition
As a guiding principle of IFRS 15, revenues from research and development collaboration agreements are recognized when earned based upon the performance requirements of the respective agreements. For revenue arrangements with separately identifiable components (separate performance obligations), the revenue recognition criteria are applied to each component. The transaction price is determined as the consideration expected to be received from the arrangement and is allocated amongst the separate components based on their relative stand-alone selling prices. The corresponding amount of transaction price allocated to each component is recognized as revenue when (or as) the Group satisfies the performance obligation by transferring the good or service to the customer, which generally is over time for upfront payments or at a point in time for milestone payments and development option payments. Payments received in excess of revenue recognized are recorded as contract liabilities.
Revenues may include fees such as upfront payments received in connection with out-licensing of products and/or access the knowledge without transfer of a license as well as R&D support and services, participation in Joint Steering Committees and other involvement in collaboration agreements. In exchange for these non-refundable upfront fees, the Group does not immediately transfer a good or a service to the customer, rather the upfront fee consists of an advance payment for future services and the right to access the underlying intellectual property of the Group. For such arrangements, the Group has determined that the promised goods and services are not distinct and are accounted for as one performance obligation. The Group recognizes revenue for this performance obligation over time using an input-based method to measure its progress towards complete satisfaction of the performance obligation. Accordingly, revenue is recognized over time based on the percentage of actual costs incurred to date relative to the Group's estimate of total costs expected to satisfy the performance obligation. Estimated costs are reviewed and updated routinely for contracts in progress to reflect any changes of which the Group becomes aware. The cumulative effect of any change in estimate is recorded in the period when the change in estimate is determined.
Revenues could include fees such as milestone and development option payments received in connection with out-licensing of products and in connection with discovery alliances. Upon meeting the set milestone or upon a development option being exercised, the Group obtains a right to a non-refundable payment and the customer has typically acquired the right to use the underlying intellectual property, without any remaining performance obligations for the Group. Consequently, the related revenues are typically recognized at a point in time, either when the milestone is met or the option is exercised by the customer.
Revenue could also include reservation fees that will be recognized into revenue in case of successful development of a final drug and exercise or lapse of the related reservation right or, alternatively, in case the results from the research will not justify further development of the drug.
Consideration payable to a customer is recorded as a reduction of the arrangement's transaction price, if it relates to the same arrangement, thereby reducing the amount of revenue recognized, unless the payment is for a distinct good or service received from the customer consistent with IFRS 15.
The details of the accounting policy, based on the type of payments received, are set out below. Under IFRS 15, revenue is recognized as or when a customer obtains control of the services. Determining the timing of the transfer of control - at a point in time or over time - requires judgment.
|
|
|
|
|
|
|
|
|
| Type of payments received |
|
Timing of revenue recognition |
| Revenue recognition of upfront payments |
|
Upfront payments received in connection with out-licensing arrangements are typically non-refundable fees for which the Group does not transfer a good or a service to the customer, rather the upfront payments consists of an advance payment for future services and/or an acquisition of the right to the current or future access to the underlying intellectual property of the Group. For such arrangements, the Group has determined that the promised goods and services are not distinct and are accounted for as one performance obligation. The Group recognizes revenue for this performance obligation over time using an input based method to measure its progress towards complete satisfaction of the performance obligation. |
|
|
|
| Revenue recognition of milestone payments |
|
Milestone payments received in connection with out-licensing or other arrangements are typically non-refundable fees entitling the Group to a right to payment upon such milestone being met. At that time, the customer has typically acquired the right to use the underlying intellectual property or additional knowledge about drug candidate(s), without any remaining performance obligation of the Group. Considering the uncertainty surrounding the outcome of such development activities, the revenue is consequently recognized at a point in time, when the milestone is reached. At this stage it is highly probable that a reversal of the cumulative revenue will not occur. |
|
|
|
| Revenue recognition of payments received for development options exercises |
|
Development option payments received in connection with out-licensing arrangements are typically non-refundable fees entitling the Group to a right to payment upon such option being exercised. At that time, the customer has typically acquired the right to use the underlying intellectual property, without any remaining performance obligations of the Group. Considering the fact that the exercise of any option is outside the control of the Group, revenue for options that provide the right to use is recognized at a point in time at the effective exercise of the option. At this stage it is highly probable that a reversal of the cumulative revenue will not occur. |
|
|
|
| Revenue recognition for reservation fees |
|
Reservation fees received are typically non-refundable fees. The timing of revenue recognition depends on whether development of the final drug is successful. If development is successful, revenue will be recognized when the related reservation right is exercised or lapses (as the exercise of any reservation right is outside the control of the Group). Alternatively, revenue will be recognized at the point in time when the results from the research will not justify further development of the drug. At this stage it is highly probable that a reversal of the cumulative revenue will not occur. |
Research and development expenses
Research and development expenses as disclosed in note 16 consist primarily of compensation and other expenses related to:
•research and development personnel;
•preclinical studies and clinical trials of the Group's product candidates, including the costs of manufacturing the product candidates;
•research and services performed under collaboration agreements;
•research and development services outsourced to research institutions; and
•attributable facility expenses, including depreciation of equipment and amortization.
Internal development costs are capitalized as intangible assets only when there is an identifiable asset that can be completed that will generate probable future economic benefits, and when the cost of such an asset can be measured reliably. The Group does not currently have any such internal development costs that qualify for capitalization as intangible assets.
The Group charges all research and development expenses, including internal patent filing and patent maintenance costs, to profit or loss when incurred, as the criteria for recognition as an asset are not currently met.
3. Financial risk management
Financial risk factors
The Group is subject to risks common to companies in the biopharmaceutical industry, including, but not limited to, uncertainties regarding the effectiveness and safety of new drugs, new and unproven technologies, development process and outcome of clinical trials, rigorous governmental regulation and uncertainty regarding regulatory approvals, long product development cycles, continuing capital requirements to fund research and development, history of operating losses and uncertainty of future profitability, uncertainty regarding commercial success and acceptance, third party reimbursements, uncertainties regarding patents and legally protected products or technologies, uncertainty regarding third party intellectual property rights, dependence on third parties, dependence on publicly available scientific findings and research data, lack of experience with production facilities, dependence on third party manufacturers and service providers, competition, concentration of operations, product liability, dependence on important employees, environment, health, data protection and safety, lack of experience in marketing and sales, litigation, currency fluctuation risks and other financial risks, volatility of market value, as well as limited liquidity and shares eligible for future sale.
The Group is developing several products currently not generating constant revenue streams which results in volatile cash flow from operating activities. Currently and in the periods presented, the Group’s revenues stem mainly from irregular and difficult to predict income from product out-licensing, milestone payments and fees from R&D collaboration agreements. This will likely remain the same at least until the first product reaches the market on the Group’s own or through a partner. This results in a lack of regular positive operating cash flow, which may expose the Group to financing risks in the medium-term. Furthermore, management has taken actions to manage financial risks, such as foreign exchange risk and liquidity risk.
Molecular Partners conducts research and development activities primarily in Switzerland, the European Union and the United States. As a result, the Group is exposed to a variety of financial risks, such as foreign exchange rate risk, credit risk, liquidity risk, cash-flow and interest rate risk. The Group’s overall financial risk management program focuses on the unpredictability of financial markets and seeks to minimize potential adverse effects on the financial performance of the Group. Further details are disclosed under note 25.
Capital management
The Group is not regulated and not subject to specific capital requirements. The amount of equity depends on the Group’s funding needs and statutory capital requirements. The Group monitors capital periodically on an interim and annual basis. From time to time, the Group may take appropriate measures or propose capital increases to its shareholders to ensure the necessary capital remains intact. The Group did not have any short-term or long-term debt outstanding as of December 31, 2023 and 2022.
4. Accounting estimates and judgments
The Group’s accounts are prepared on a going concern basis. The preparation of the consolidated financial statements in conformity with IFRS requires that management and the Board of Directors make estimates and assumptions which affect the amounts of the assets and liabilities, contingent liabilities, as well as the income and expenses reported in the consolidated financial statements. These estimates take into consideration historic experience as well as developments in the economic circumstances and are further based on management’s best knowledge of current events and actions that the Group may undertake in the future. These estimates are subject to risks and uncertainties. The actual results can deviate from these estimates.
5. Revenue, other income and entity-wide disclosures
The Group assesses and estimates the progress of its projects with alliance partners at each reporting date.
License and Collaboration Agreement with Novartis in the Area of DARPIN-Conjugated Radioligand Therapies, or the Novartis Radioligand Agreement
On December 14, 2021, the Group entered into a License and Collaboration Agreement with Novartis to develop DARPin-conjugated radioligand therapeutic candidates for oncology. Under the agreement, both parties will collaborate on the discovery and optimization of the therapeutic candidates. The Group will be primarily responsible for the generation of DARPins for tumor-specific delivery of radioligands. The Group is eligible to invoice Novartis for its employee-related expenses associated with the research activities. Novartis is responsible for all clinical development and commercialization activities. As of December 31, 2021 the Group recognized a receivable for the upfront fee of USD 20 million (CHF 18.6 million) payable from Novartis in trade and other receivables and a corresponding contract liability in the consolidated statement of financial position. In January 2022, Novartis paid Molecular Partners the upfront fee. The Group will be eligible to receive milestone payments of up to USD 560 million, relating to development, regulatory and commercialization activities, plus tiered royalties based on commercial sale levels from mid-single digit to low double-digit percentages of royalties on net sales of products commercialized by Novartis.
The Group identified one combined performance obligation consisting of the license and the research activities to be provided. Revenue related to the upfront payment of USD 20 million (CHF 18.6 million) is being recognized over time in line with the progress made over the duration of the contractually agreed research plan. Progress towards completion of the research plan is based on the cost-based method and is measured by employee costs on the related research activities as specified in the agreement relative to the total employee costs estimated to be incurred. During 2023, the Group recognized total revenue of CHF 7.0 million of which CHF 5.7 million related to the recognition of the upfront fee and CHF 1.3 million related to the recharge of employee-related expenses (2022 total revenue of CHF 9.8 million, 2021: CHF nil).
In June and December 2023, the Group increased its estimate of the total future costs required to satisfy the performance obligation under this Novartis collaboration. This change in estimate affects the allocation of revenue over time and has no impact on the total amount recognized or to be recognized into revenue under the agreement with Novartis.
The increase in total estimated future costs is primarily related to the continued development of various DARPin-conjugated radioligand therapeutic candidates.
Future milestone payments and royalties under the agreement will be recognized as revenue at a point in time, when a milestone is achieved or any subsequent sales by Novartis occur.
Novartis Option and Equity Rights Agreement
In October 2020, the Group entered into the Option and Equity Rights Agreement with Novartis, granting Novartis the exclusive option to in-license global rights in relation to MP0420 (ensovibep). Under the terms of the agreement, in 2020, the Group received an upfront, non-refundable fee of CHF 20 million for the technology transfer and manufacturing of MP0420. As of December 31, 2021, the entire CHF 20 million has been utilized for the manufacturing of commercial supply for MP0420.
Ensovibep License Agreement
In January 2022, following positive Phase 2 clinical trial results, Novartis exercised its option for ensovibep, triggering a milestone payment of CHF 150 million to the Group, which was received in 2022. Relatedly, the Group was eligible to invoice Novartis CHF 13.1 million for other items related to ensovibep.
In January 2023 Novartis informed the Group that it has submitted a request to withdraw, with an effective date of January 25, 2023 the Emergency Use Authorization (EUA) application from the U.S. Food and Drug Administration (FDA) for ensovibep. Ensovibep is not presently in clinical development.
On January 5, 2024, Novartis has agreed the termination of the License Agreement for ensovibep, previously under investigation for the treatment of SARS Cov-2, and Novartis has returned the rights to the ensovibep program to the Company. Clinical work on the ensovibep program ended in 2022, and the program remains terminated.
Reservation Agreement with the Swiss Federal Office of Public Health / Bundesamt für Gesundheit, or the FOPH Agreement
On August 11, 2020, the Group announced the reservation by the FOPH of a defined number of initial doses of the Group's anti-COVID-19 candidate, MP0420. Under the terms of the agreement, the Group received a reservation fee of CHF 7.0 million which resulted in a contract liability of CHF 7.0 million. With the exercise of the option by Novartis in January 2022 and the subsequent assignment of the agreement to Novartis, the Group recognized the CHF 7.0 million as revenue in 2022.
License and Collaboration Agreement with Amgen, or the Amgen Collaboration Agreement
In December 2018, the Group entered into a license and collaboration agreement with Amgen for the clinical development and commercialization of MP0310 / AMG 506. Under the agreement the Group received a non-refundable upfront payment of USD 50 million. The Group recognized the related revenue using the cost -based method to measure it progress by reference to actual costs incurred in relation to the Group's best estimate of total expected costs to satisfy the performance obligation.
On April 26, 2022 the Group announced that Amgen, had informed the Group of its decision to return the global rights of MP0310 following a strategic pipeline review. With no remaining performance obligations under the Amgen Collaboration Agreement, the Group recognized the remaining balance of the Amgen contract liability of TCHF 9,653 as revenue in 2022.
During the years ended December 31, 2023, 2022 and 2021, the Group recognized revenues as disclosed in the table below. Revenues in the table below are attributable to individual countries and are based on the location of the Group's alliance partner.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Revenues by country |
|
|
|
|
|
| in CHF thousands, for the years ended December 31 |
2023 |
|
2022 |
|
2021 |
| Revenues Switzerland |
7,038 |
|
|
|
179,903 |
|
|
|
— |
|
|
| Revenues USA |
— |
|
|
|
9,653 |
|
|
|
9,330 |
|
|
| Total revenues |
7,038 |
|
|
|
189,556 |
|
|
|
9,330 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Analysis of revenue by major alliance partner |
|
|
|
|
|
| in CHF thousands, for the years ended December 31 |
2023 |
|
2022 |
|
2021 |
| Novartis AG, Switzerland |
7,038 |
|
|
|
172,903 |
|
|
|
— |
|
|
| FOPH, Switzerland |
— |
|
|
|
7,000 |
|
|
|
— |
|
|
| Amgen Inc., USA |
— |
|
|
|
9,653 |
|
|
|
9,330 |
|
|
| Total revenues |
7,038 |
|
|
|
189,556 |
|
|
|
9,330 |
|
|
Other income
In the first quarter of 2021 the Group entered into an agreement with Novartis to facilitate manufacturing of MP0420 drug supply at a third party supplier. The related agency services earned during 2022 amounted to TCHF 44 (2021: TCHF 424) and are presented as Other income in the consolidated statement of comprehensive income. No such services were performed during 2023.
6. Property, plant and equipment
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| in CHF thousands |
Lab equipment |
|
Office equipment |
|
IT hardware |
|
Right-of-use assets |
|
Leasehold improvements |
|
Total |
| 2023 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Cost |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| At January 1, 2023 |
9,646 |
|
|
|
731 |
|
|
|
1,315 |
|
|
|
9,616 |
|
|
|
624 |
|
|
|
21,932 |
|
|
| Additions |
397 |
|
|
|
6 |
|
|
|
163 |
|
|
|
— |
|
|
|
9 |
|
|
|
575 |
|
|
| Disposals |
(303) |
|
|
|
(14) |
|
|
|
(167) |
|
|
|
— |
|
|
|
— |
|
|
|
(484) |
|
|
| At December 31, 2023 |
9,740 |
|
|
|
723 |
|
|
|
1,311 |
|
|
|
9,616 |
|
|
|
633 |
|
|
|
22,023 |
|
|
| Accumulated depreciation |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| At January 1, 2023 |
(7,660) |
|
|
|
(687) |
|
|
|
(1,172) |
|
|
|
(4,815) |
|
|
|
(364) |
|
|
|
(14,697) |
|
|
| Depreciation charge for the year |
(711) |
|
|
|
(27) |
|
|
|
(120) |
|
|
|
(1,200) |
|
|
|
(70) |
|
|
|
(2,128) |
|
|
| Disposals |
303 |
|
|
|
14 |
|
|
|
167 |
|
|
|
— |
|
|
|
— |
|
|
|
484 |
|
|
| At December 31, 2023 |
(8,068) |
|
|
|
(700) |
|
|
|
(1,125) |
|
|
|
(6,015) |
|
|
|
(434) |
|
|
|
(16,342) |
|
|
| Carrying amount at December 31, 2023 |
1,672 |
|
|
|
23 |
|
|
|
186 |
|
|
|
3,601 |
|
|
|
199 |
|
|
|
5,681 |
|
|
The right-of-use assets relate to the facilities the Group is leasing in Schlieren, Switzerland.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| in CHF thousands |
Lab equipment |
|
Office equipment |
|
IT hardware |
|
Right-of-use assets |
|
Leasehold improvements |
|
Total |
| 2022 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Cost |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| At January 1, 2022 |
8,754 |
|
|
|
711 |
|
|
|
1,199 |
|
|
|
9,616 |
|
|
|
607 |
|
|
|
20,887 |
|
|
| Additions |
1,019 |
|
|
|
20 |
|
|
|
121 |
|
|
|
— |
|
|
|
17 |
|
|
|
1,177 |
|
|
| Disposals |
(127) |
|
|
|
— |
|
|
|
(5) |
|
|
|
— |
|
|
|
— |
|
|
|
(132) |
|
|
| At December 31, 2022 |
9,646 |
|
|
|
731 |
|
|
|
1,315 |
|
|
|
9,616 |
|
|
|
624 |
|
|
|
21,932 |
|
|
| Accumulated depreciation |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| At January 1, 2022 |
(7,164) |
|
|
|
(653) |
|
|
|
(1,012) |
|
|
|
(3,615) |
|
|
|
(298) |
|
|
|
(12,741) |
|
|
| Depreciation charge for the year |
(623) |
|
|
|
(34) |
|
|
|
(165) |
|
|
|
(1,200) |
|
|
|
(66) |
|
|
|
(2,088) |
|
|
| Disposals |
127 |
|
|
|
— |
|
|
|
5 |
|
|
|
— |
|
|
|
— |
|
|
|
132 |
|
|
| At December 31, 2022 |
(7,660) |
|
|
|
(687) |
|
|
|
(1,172) |
|
|
|
(4,815) |
|
|
|
(364) |
|
|
|
(14,697) |
|
|
| Carrying amount at December 31, 2022 |
1,986 |
|
|
|
44 |
|
|
|
143 |
|
|
|
4,802 |
|
|
|
260 |
|
|
|
7,235 |
|
|
7. Intangible assets
|
|
|
|
|
|
|
|
|
| in CHF thousands |
Software |
| 2023 |
| Cost |
|
|
| At January 1, 2023 |
2,122 |
|
|
| Additions |
233 |
|
|
| Disposals |
(59) |
|
|
| At December 31, 2023 |
2,296 |
|
|
| Accumulated amortization |
|
|
| At January 1, 2023 |
(1,851) |
|
|
| Amortization charge for the year |
(292) |
|
|
| Disposals |
59 |
|
|
| At December 31, 2023 |
(2,084) |
|
|
| Carrying amount at December 31, 2023 |
212 |
|
|
|
|
|
|
|
|
|
|
|
| in CHF thousands |
Software |
| 2022 |
| Cost |
|
|
| At January 1, 2022 |
1,904 |
|
|
| Additions |
240 |
|
|
| Disposals |
(22) |
|
|
| At December 31, 2022 |
2,122 |
|
|
| Accumulated amortization |
|
|
| At January 1, 2022 |
(1,574) |
|
|
| Amortization charge for the year |
(299) |
|
|
| Disposals |
22 |
|
|
| At December 31, 2022 |
(1,851) |
|
|
| Carrying amount at December 31, 2022 |
271 |
|
|
8. Financial instruments
|
|
|
|
|
|
|
|
|
| in CHF thousands |
Financial assets at amortized costs |
| 2023 |
| Cash and cash equivalents |
67,309 |
|
|
| Trade receivables |
295 |
|
|
| Accrued income |
1,131 |
|
|
| Short-term time deposits |
119,580 |
|
|
| Balance at December 31 |
188,315 |
|
|
| 2022 |
|
|
| Cash and cash equivalents |
87,946 |
|
|
| Trade receivables |
521 |
|
|
| Accrued income |
679 |
|
|
| Short-term time deposits |
161,198 |
|
|
| Balance at December 31 |
250,344 |
|
|
The above mentioned amounts were neither past due nor impaired at the end of the respective reporting period. Please also see note 25.
|
|
|
|
|
|
|
|
|
| in CHF thousands |
Financial liabilities at amortized cost |
| 2023 |
| Trade payables |
410 |
|
|
| Accrued project costs and royalties |
1,827 |
|
|
| Lease liabilities |
3,652 |
|
|
| Other non-employee related accrued expenses |
704 |
|
|
| Balance at December 31 |
6,593 |
|
|
| 2022 |
|
|
| Trade payables |
997 |
|
|
| Accrued project costs and royalties |
2,167 |
|
|
| Lease liabilities |
4,850 |
|
|
| Other non-employee related accrued expenses |
556 |
|
|
| Balance at December 31 |
8,570 |
|
|
The carrying amount of financial assets and financial liabilities not measured at fair value (except for lease liabilities) is a reasonable approximation of fair value.
9. Other current assets
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| in CHF thousands |
2023 |
|
2022 |
| Prepayments |
2,486 |
|
|
|
3,910 |
|
|
| Accrued income |
1,131 |
|
|
|
679 |
|
|
| Balance at December 31 |
3,617 |
|
|
|
4,589 |
|
|
Accrued income relates to interest income accrued on the Group's balances of cash and cash equivalents and short-term time deposits.
10. Trade and other receivables
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| in CHF thousands |
2023 |
|
2022 |
| Trade receivables |
295 |
|
|
|
521 |
|
|
| Value added tax |
253 |
|
|
|
250 |
|
|
| Withholding tax |
1,339 |
|
|
|
173 |
|
|
| Other receivables |
66 |
|
|
|
75 |
|
|
| Balance at December 31 |
1,953 |
|
|
|
1,019 |
|
|
Trade receivables are denominated in the following currencies:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| in CHF thousands |
2023 |
|
2022 |
| CHF |
— |
|
|
|
160 |
|
|
| EUR |
— |
|
|
|
— |
|
|
| USD |
295 |
|
|
|
361 |
|
|
| Balance at December 31 |
295 |
|
|
|
521 |
|
|
11. Cash and cash equivalents and short-term time deposits
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| in CHF thousands |
2023 |
|
2022 |
| Cash at bank in CHF |
57,379 |
|
|
|
67,611 |
|
|
| Cash at bank in EUR |
4,948 |
|
|
|
7,685 |
|
|
| Cash at bank in USD |
4,829 |
|
|
|
12,520 |
|
|
| Cash at bank in GBP |
153 |
|
|
|
130 |
|
|
| Total cash at bank at December 31 |
67,309 |
|
|
|
87,946 |
|
|
|
|
|
|
|
|
| Short-term time deposits in CHF |
77,500 |
|
|
|
110,000 |
|
|
| Short-term time deposits in EUR |
— |
|
|
|
4,938 |
|
|
| Short-term time deposits in USD |
42,080 |
|
|
|
46,260 |
|
|
| Total short-term deposits at December 31 |
119,580 |
|
|
|
161,198 |
|
|
All short-term time deposits at December 31, 2023 and 2022 were held with Swiss banks. As of December 31, 2023, the deposits denominated in CHF contained six positions with three banks, the deposits denominated in USD contained five positions with two banks. As of December 31, 2022, there were six deposits denominated in CHF with three banks, where the short-term time deposits denominated in USD contained three positions with three banks and the short-term time deposits in EUR contained one position with one bank. Please refer to note 25.
12. Shareholders’ equity
In August 2022, the Company issued 3,500,000 common shares at par value CHF 0.10 per share. The shares were fully subscribed for by Molecular Partners Inc., a fully owned subsidiary of the Company. As of December 31, 2023 and 2022, all 3,500,000 common shares were held as treasury shares of the Company. The purpose of the share issuance was to replenish the Company’s pool of treasury shares that the Company can use in the future to raise funds, including in connection with the Company’s at-the-market sales program for American Depositary Shares established in July 2022.
The total amount presented as Treasury shares reserve in the consolidated statement of financial position, is comprised of CHF 350,000 of the nominal value of the treasury shares and CHF 631,336 of transaction costs incurred directly related to the issuance.
The amount of CHF 350,000 was a non-cash transaction for the Group.
Classes of share capital
Ordinary share capital
On December 31, 2023, the Company’s issued share capital amounted to CHF 3,635,430 divided into 36,354,297 fully paid registered shares with a par value of CHF 0.10 each. Ordinary shares are entitled to one vote per share and rank equally with regard to the Company's residual assets and dividends (if any should be declared in the future).
|
|
|
|
|
|
|
|
|
|
Ordinary shares |
| Shares in issue at December 31, 2020 |
29,146,992 |
|
|
| Issued in relation to June 2021 IPO |
3,000,000 |
|
|
| Issued in relation to vesting of PSU, RSU and options |
145,656 |
|
|
| Shares in issue at December 31, 2021 |
32,292,648 |
|
|
| Issued in relation to creation of treasury shares in August 2022 |
3,500,000 |
|
|
| Issued in relation to vesting of PSU, RSU and options |
252,058 |
|
|
| Shares in issue at December 31, 2022 |
36,044,706 |
|
|
| Issued in relation to vesting of PSU, RSU and options |
309,591 |
|
|
| Shares in issue at December 31, 2023 |
36,354,297 |
|
|
The Company’s share capital registered with the Swiss Commercial Register on December 31, 2023 amounted to CHF 3,604,471 divided into 36,044,706 fully paid up registered shares with a par value of CHF 0.10 per share.
The capital increases in 2023 triggered by the option exercises and the vesting of Performance Share Units ("PSU") and Restricted Share Units ("RSU"), from the RSU Plan 2020 and PSU Plans 2020, 2021 and 2022 was registered with the Commercial Register on January 31, 2024.
Authorized share capital
On December 31, 2023, the Company had an authorized share capital in the amount of up to CHF 457,316 which allows for the issuance of up to 4,573,162 fully paid up registered shares with a par value of CHF 0.10 per share, which is valid until April 13, 2024. This authorized capital of up to CHF 457,316 equates to approximately 13% of the existing share capital. As approved by the annual general meeting on April 13, 2022, the authorized share capital was increased by CHF 350,000 from CHF 457,316 to CHF 807,316. In August 2022, the authorized share capital was subsequently reduced by CHF 350,000 from CHF 807,316 to CHF 457,316 due to the creation of treasury shares.
The Board of Directors is authorized to determine the issue price, type of payment, time of the issuance, conditions for the exercise of the preemptive rights and the date from which the shares carry the right to dividends. The Board of Directors can issue new shares by means of an underwriting arrangement by a bank or another third party with a subsequent offer of these shares to the existing shareholders or third parties (if the preemptive rights of the existing shareholders have been denied or not been duly exercised). The Board of Directors is authorized to permit, to restrict or to deny the trade of preemptive rights. The Board of Directors may permit preemptive rights that have been granted but not exercised to expire or it may place these rights respectively the shares as to which preemptive rights have been granted but not exercised, at market conditions or use them for other purposes in the interest of the Group.
The Board of Directors is further authorized to restrict or deny the preemptive rights of shareholders and to allocate them to third parties: (a) for the acquisition of companies, parts of companies or participations, for the acquisition of products, intellectual property or licenses, for investment projects or for the financing or refinancing of such transactions through a placement of shares, (b) for the purpose of broadening the shareholder constituency or in connection with a listing of shares on domestic or foreign stock exchanges, (c) if the issue price of the new shares is determined by reference to the market price, (d) for purposes of granting an over-allotment option (greenshoe) of up to 20% of the total number of shares in a placement or sale of shares to the respective initial purchasers or underwriters, (e) following a shareholder or a group of shareholders acting in concert having accumulated shareholdings in excess of 15% of the share capital registered with the commercial register of the Canton of Zurich, without having submitted to the other shareholders a take-over offer recommended by the Board of Directors, or (f) for the defense of an actual, threatened or potential takeover bid, in relation to which the Board of Directors has not recommended to the shareholders acceptance on the basis that the Board of Directors has not found the takeover bid to be financially fair to the shareholders.
Conditional share capital
As of December 31, 2023 the Company’s share capital was allowed to be increased by an amount not to exceed CHF 105,337 through the issuance of up to 1,053,372 fully paid up shares with a par value of CHF 0.10 per share through the direct or indirect issuance of shares, options or preemptive rights granted to employees, members of the Board of Directors or members of any advisory boards. During 2023, the share capital was increased out of this conditional capital for employee participation (Article 3b of the Articles of Association). As a result, the available conditional capital for employee participation was reduced by CHF 30,959 from CHF 136,296 to CHF 105,337.
In addition, the share capital may be increased by an amount not to exceed CHF 226,087 through the issuance of up to 2,260,870 fully paid up shares with a par value of CHF 0.10 per share through the exercise or mandatory exercise of conversion, exchange, option, warrant or similar rights for the subscription of shares granted to shareholders or third parties alone or in connection with bonds, notes, options, warrants or other securities or contractual obligations by or of the Company. During 2023, this conditional capital for financing transactions and other purposes (Article 3c of the Articles of Association) remained unchanged.
In 2023, 2022 and 2021 the cash proceeds from the exercise of share options and the vesting of Performance Share Units (“PSU”) and Restricted Share Units ("RSU"), amounted to CHF 30,959, CHF 251,957 and CHF 269,552 respectively and all resulted from the issuance of new shares (conditional share capital).
13. Trade and other payables
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| in CHF thousands |
2023 |
|
2022 |
| Trade payables |
410 |
|
|
|
997 |
|
|
| Social security |
918 |
|
|
|
1,146 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance at December 31 |
1,328 |
|
|
|
2,143 |
|
|
Trade payables are denominated in the following currencies:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| in CHF thousands |
2023 |
|
2022 |
| CHF |
227 |
|
|
|
790 |
|
|
| EUR |
161 |
|
|
|
104 |
|
|
| USD |
22 |
|
|
|
103 |
|
|
|
|
|
|
|
|
| Balance at December 31 |
410 |
|
|
|
997 |
|
|
14. Accrued expenses
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| in CHF thousands |
2023 |
|
2022 |
| Accrued project costs and royalties |
1,827 |
|
|
|
2,167 |
|
|
| Accrued payroll and bonuses |
5,012 |
|
|
|
4,763 |
|
|
| Other |
708 |
|
|
|
571 |
|
|
| Balance at December 31 |
7,547 |
|
|
|
7,501 |
|
|
|
|
|
|
|
|
15. Contract liability
The Group expects the contract liability to be recognized as revenue as follows:
|
|
|
|
|
|
|
|
|
| in CHF thousands |
Contract liability |
| Expected revenue recognition in year one after balance sheet date |
4,333 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Balance at December 31, 2023 |
4,333 |
|
|
|
|
|
|
|
|
|
|
|
| in CHF thousands |
Contract liability |
| Expected revenue recognition in year one after balance sheet date |
6,409 |
|
|
| Expected revenue recognition in year two after balance sheet date |
3,637 |
|
|
|
|
|
|
|
|
|
|
|
| Balance at December 31, 2022 |
10,046 |
|
|
The table below presents the movement on the contract liability:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Contract liability at January 1, |
|
Additions |
|
Recognized as revenue |
|
|
|
|
Contract liability at December 31, |
| in CHF thousands |
2023 |
|
|
|
|
|
|
2023 |
| Novartis |
10,046 |
|
|
|
— |
|
|
|
(5,713) |
|
|
|
|
|
|
4,333 |
|
|
| Balance |
10,046 |
|
|
|
— |
|
|
|
(5,713) |
|
|
|
|
|
|
4,333 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Contract liability at January 1, |
|
Additions |
|
Recognized as revenue |
|
|
|
|
Contract liability at December 31, |
| in CHF thousands |
2022 |
|
|
|
|
|
|
2022 |
| Amgen |
9,653 |
|
|
|
— |
|
|
|
(9,653) |
|
|
|
|
|
|
— |
|
|
| Novartis |
18,584 |
|
|
|
— |
|
|
|
(8,538) |
|
|
|
|
|
|
10,046 |
|
|
| FOPH |
7,000 |
|
|
|
— |
|
|
|
(7,000) |
|
|
|
|
|
|
— |
|
|
| Balance |
35,237 |
|
|
|
— |
|
|
|
(25,191) |
|
|
|
|
|
|
10,046 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| in CHF thousands |
Current |
|
Non-current |
|
Contract liability |
| Novartis |
4,333 |
|
|
|
— |
|
|
|
4,333 |
|
|
| Balance at December 31, 2023 |
4,333 |
|
|
|
— |
|
|
|
4,333 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| in CHF thousands |
Current |
|
Non-current |
|
Contract liability |
| Novartis |
6,409 |
|
|
|
3,637 |
|
|
|
10,046 |
|
|
| Balance at December 31, 2022 |
6,409 |
|
|
|
3,637 |
|
|
|
10,046 |
|
|
16. Additional information on the nature of expenses
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Research and development expenses |
|
|
|
|
|
| in CHF thousands |
2023 |
|
|
2022 |
|
|
2021 |
|
| Research consumables and external research and development expenses |
(15,892) |
|
|
|
(17,154) |
|
|
|
(26,342) |
|
|
Personnel expenses (1), see also note 18 |
(28,376) |
|
|
|
(28,101) |
|
|
|
(25,647) |
|
|
| Depreciation and amortization |
(2,053) |
|
|
|
(1,971) |
|
|
|
(2,016) |
|
|
| Intellectual property |
(853) |
|
|
|
(957) |
|
|
|
(636) |
|
|
| Facility expenses |
(940) |
|
|
|
(854) |
|
|
|
(758) |
|
|
| Other research and development expenses |
(660) |
|
|
|
(703) |
|
|
|
(259) |
|
|
| Royalties and license fees, see also note 17 |
(10) |
|
|
|
(1,010) |
|
|
|
(60) |
|
|
| Total year ended December 31 |
(48,784) |
|
|
|
(50,749) |
|
|
|
(55,718) |
|
|
|
|
|
|
|
|
| Selling, general and administrative expenses |
|
|
|
|
|
| in CHF thousands |
2023 |
|
|
2022 |
|
|
2021 |
|
Personnel expenses (2), see also note 18 |
(11,640) |
|
|
|
(11,788) |
|
|
|
(10,604) |
|
|
| Other administrative expenses |
(7,283) |
|
|
|
(9,965) |
|
|
|
(6,242) |
|
|
| Depreciation and amortization |
(367) |
|
|
|
(416) |
|
|
|
(549) |
|
|
| Facility expenses |
(72) |
|
|
|
(69) |
|
|
|
(60) |
|
|
| Total year ended December 31 |
(19,362) |
|
|
|
(22,238) |
|
|
|
(17,454) |
|
|
|
|
|
|
|
|
|
|
|
| Total operating expenses |
(68,146) |
|
|
|
(72,987) |
|
|
|
(73,172) |
|
|
|
|
|
|
|
|
|
|
|
(1) Research and development non-cash effective pension and share-based compensation costs were TCHF 3,447 in 2023, TCHF 3,856 in 2022 and TCHF 3,045 in 2021.
(2) Selling, general and administrative non-cash effective pension and share based compensation costs were TCHF 2,260 in 2023, TCHF 2,329 in 2022 and TCHF 2,113 in 2021.
17. Royalties and license fees
The Group holds a non-exclusive perpetual license from the University of Zurich on patent applications and patents relating to Phage Display technology. The amount payable by the Group is CHF 10,000 per annum.
18. Personnel expenses
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| in CHF thousands |
2023 |
|
2022 |
|
2021 |
| Salaries |
(27,022) |
|
|
|
(27,737) |
|
|
|
(25,909) |
|
|
| Share-based compensation (non-cash effective) |
(5,207) |
|
|
|
(5,088) |
|
|
|
(4,085) |
|
|
| Pension costs |
(2,632) |
|
|
|
(3,192) |
|
|
|
(3,059) |
|
|
| Social security costs |
(2,201) |
|
|
|
(2,399) |
|
|
|
(2,535) |
|
|
| Other personnel expenses |
(2,954) |
|
|
|
(1,473) |
|
|
|
(663) |
|
|
| Total year ended December 31 |
(40,016) |
|
|
|
(39,889) |
|
|
|
(36,251) |
|
|
|
|
|
|
|
|
| Full-time equivalents and head count |
2023 |
|
2022 |
|
2021 |
| Average number of full-time equivalents |
167.8 |
|
167.4 |
|
158.3 |
| Full-time equivalents at year end |
167.5 |
|
175.3 |
|
163.2 |
| Headcount at year end |
182 |
|
|
191 |
|
|
177 |
|
18.1 Pension costs and liabilities
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| in CHF thousands |
2023 |
|
2022 |
| Defined benefit pension plans |
|
|
|
| Actuarial assumptions |
|
|
|
| Discount rate at January 1 |
2.25 |
% |
|
0.40 |
% |
Discount rate at December 31 (1) |
1.50 |
% |
|
2.25 |
% |
| Future salary increases at December 31 |
2.00 |
% |
|
2.00 |
% |
| Mortality tables |
BVG2020 GT |
|
BVG2020 GT |
| Date of last actuarial valuation |
31.12.2023 |
|
31.12.2022 |
| Reconciliation of the amount recognized in the statement of financial position |
|
|
|
|
|
| Defined benefit obligation at December 31 |
56,347 |
|
|
52,529 |
|
| Fair value of plan assets at December 31 |
51,627 |
|
|
50,284 |
|
Net defined benefit liability at December 31 (2) |
4,720 |
|
|
2,245 |
|
| Components of defined benefit cost in profit or loss |
|
|
|
|
|
| Current service cost (employer) |
2,507 |
|
|
3,137 |
|
| Past service cost |
43 |
|
|
— |
|
| Interest expense on defined benefit obligation |
1,182 |
|
|
231 |
|
| Interest income on plan assets |
(1,126) |
|
|
(203) |
|
| Administrative cost excl. cost for managing plan assets |
26 |
|
|
27 |
|
| Defined benefit cost recognized in profit or loss |
2,632 |
|
|
3,192 |
|
| thereof service cost and administrative cost |
2,576 |
|
|
3,164 |
|
| thereof net interest expense on the net defined benefit liability |
56 |
|
|
28 |
|
| Reconciliation of net defined benefit liability |
|
|
|
|
|
| Net defined benefit liability at January 1 |
2,245 |
|
|
6,483 |
|
Defined benefit cost recognized in profit or loss (3) |
2,632 |
|
|
3,192 |
|
| Remeasurement of net pension liabilities |
1,975 |
|
|
(5,334) |
|
Contributions by the employer (3) |
(2,132) |
|
|
(2,096) |
|
Net defined benefit liability at December 31 (2) |
4,720 |
|
|
2,245 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Reconciliation of defined benefit obligation |
|
|
|
|
|
| Defined benefit obligation at January 1 |
52,529 |
|
|
54,461 |
|
| Interest expenses on defined benefit obligation |
1,182 |
|
|
231 |
|
| Current service cost (employer) |
2,507 |
|
|
3,137 |
|
| Contributions by plan participants |
1,344 |
|
|
1,317 |
|
| Benefits (paid)/deposited |
(3,918) |
|
|
2,032 |
|
| Past service cost |
43 |
|
|
— |
|
| Administrative cost (excl. cost for managing plan assets) |
26 |
|
|
27 |
|
| Actuarial (gain)/loss on defined benefit obligation |
2,634 |
|
|
(8,676) |
|
| Defined benefit obligation at December 31 |
56,347 |
|
|
52,529 |
|
| Reconciliation of amount recognized in OCI |
|
|
|
|
|
| Actuarial (gain) / loss on changes in financial assumptions |
3,644 |
|
|
(12,222) |
|
| Actuarial (gain) / loss on changes in demographic assumptions |
(10) |
|
|
— |
|
| Actuarial (gain) / loss arising from experience adjustments |
(1,000) |
|
|
3,546 |
|
| Actuarial (gain)/loss on defined benefit obligation |
2,634 |
|
|
(8,676) |
|
| Return on plan assets excluding interest income |
(659) |
|
|
3,342 |
|
| Remeasurement of net pension liabilities |
1,975 |
|
|
(5,334) |
|
| Reconciliation of fair value of plan assets |
|
|
|
|
|
| Fair value of plan assets at January 1 |
50,284 |
|
|
47,979 |
|
| Interest income on plan assets |
1,126 |
|
|
203 |
|
| Contributions by the employer |
2,132 |
|
|
2,096 |
|
| Contributions by plan participants |
1,344 |
|
|
1,317 |
|
| Benefits (paid)/deposited |
(3,918) |
|
|
2,032 |
|
| Return on plan assets excl. interest income |
659 |
|
|
(3,342) |
|
| Fair value of plan assets at December 31 |
51,627 |
|
|
50,284 |
|
| Best estimate of contributions of next year |
|
|
|
|
|
| Contributions by the employer |
2,156 |
|
|
2,231 |
|
| Plan asset classes |
|
|
|
|
|
| Cash and cash equivalents |
7,684 |
|
|
7,896 |
|
| Equity instruments |
21,810 |
|
|
20,754 |
|
| Debt instruments (e.g. bonds) |
9,047 |
|
|
8,200 |
|
| Real estate funds |
1,821 |
|
|
1,793 |
|
| Others |
1,792 |
|
|
1,748 |
|
| Total plan assets at fair value (quoted market price) |
42,154 |
|
|
40,391 |
|
| Others |
9,473 |
|
|
9,892 |
|
| Total plan assets at fair value (non-quoted market price) |
9,473 |
|
|
9,892 |
|
| Total plan assets at fair value at December 31 |
51,627 |
|
|
50,284 |
|
| thereof entity's own transferable financial instruments |
— |
|
|
— |
|
| thereof property occupied or other assets used by the entity |
— |
|
|
— |
|
Sensitivity (4) |
|
|
|
|
|
Defined benefit obligation at December 31 with discount rate -0.25% |
58,683 |
|
|
54,524 |
|
Defined benefit obligation at December 31 with discount rate +0.25% |
54,179 |
|
|
50,672 |
|
Defined benefit obligation at December 31 with interest rate on retirement savings capital -0.25% |
55,437 |
|
|
51,699 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Defined benefit obligation at December 31 with interest rate on retirement savings capital +0.25% |
57,283 |
|
|
53,383 |
|
Defined benefit obligation at December 31 with salary increases -0.25% |
55,974 |
|
|
52,253 |
|
Defined benefit obligation at December 31 with salary increases +0.25% |
56,707 |
|
|
52,768 |
|
Defined benefit obligation at December 31 with life expectancy +1 year |
57,071 |
|
|
53,090 |
|
Defined benefit obligation at December 31 with life expectancy -1 year |
55,619 |
|
|
51,961 |
|
| Maturity profile of defined benefit obligation |
|
|
|
|
|
| Weighted average duration of defined obligation in years at December 31 |
16.2 |
|
|
15.0 |
|
| Weighted average duration of defined obligation in years at December 31 for active members |
16.1 |
|
|
14.8 |
|
| Weighted average duration of defined obligation in years at December 31 for pensioners |
17.3 |
|
|
16.3 |
|
|
|
(1) Discount rates are based on industry benchmarks related to benefits with a 20 year duration.
(2) In liabilities for employee benefits, as presented in the consolidated statement of financial position included are also TCHF 343 (2022: TCHF 307; 2021: TCHF 257) for accrued sabbatical cost.
(3) The sum of these two positions represent the non-cash effective pension costs recognized in the profit and loss section of the consolidated statement of comprehensive income of which TCHF 390 are research and development costs (2022: TCHF 846; 2021: TCHF 837) and TCHF 110 are selling, general and administrative costs (2022: TCHF 250; 2021: TCHF 235).
(4) For the most important parameters which influence the pension obligation of the Company a sensitivity analysis was performed. The discount rate and the assumption for salary increases were modified by a certain percentage value. Sensitivity on mortality was calculated by changing the mortality with a constant factor for all age groups. With this procedure the Company could change the longevity for most of the age categories by one year longer or shorter than the baseline value.
The table below presents the amounts that are reflected in the statement of comprehensive income for the periods indicated:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| in CHF thousands |
2023 |
|
2022 |
|
2021 |
|
|
|
|
|
|
|
|
|
| Components of defined benefit cost in profit or loss |
|
|
|
|
|
|
|
|
| Current service cost (employer) |
2,507 |
|
|
|
3,137 |
|
|
|
3,097 |
|
|
| Past service cost |
43 |
|
|
|
— |
|
|
|
(94) |
|
|
| Interest expense on defined benefit obligation |
1,182 |
|
|
|
231 |
|
|
|
114 |
|
|
| Interest income on plan assets |
(1,126) |
|
|
|
(203) |
|
|
|
(86) |
|
|
| Administrative cost excl. cost for managing plan assets |
26 |
|
|
|
27 |
|
|
|
27 |
|
|
| Defined benefit cost recognized in profit or loss |
2,632 |
|
|
|
3,192 |
|
|
|
3,059 |
|
|
| thereof service cost and administrative cost |
2,576 |
|
|
|
3,164 |
|
|
|
3,031 |
|
|
| thereof net interest expense on the net defined benefit liability |
56 |
|
|
|
28 |
|
|
|
28 |
|
|
|
|
|
|
|
|
|
|
|
| Reconciliation of amount recognized in OCI |
|
|
|
|
|
|
|
|
| Actuarial (gain) / loss on changes in financial assumptions |
3,644 |
|
|
|
(12,222) |
|
|
|
(2,303) |
|
|
| Actuarial (gain) / loss on changes in demographic assumptions |
(10) |
|
|
|
— |
|
|
|
(2,432) |
|
|
| Actuarial (gain) / loss arising from experience adjustments |
(1,000) |
|
|
|
3,546 |
|
|
|
(773) |
|
|
| Actuarial (gain)/loss on defined benefit obligation |
2,634 |
|
|
|
(8,676) |
|
|
|
(5,508) |
|
|
| Return on plan assets excluding interest income |
(659) |
|
|
|
3,342 |
|
|
|
(2,504) |
|
|
| Remeasurement of net pension liabilities |
1,975 |
|
|
|
(5,334) |
|
|
|
(8,012) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
18.2 Share-based compensation
18.2.1 Employee Share Option Plans (“ESOP”)
1. ESOP 2009 established in December 2009
2. ESOP 2014 established in July 2014
An ESOP is an incentive tool that fosters the entrepreneurial spirit and performance by way of financial participation in the Group's long term success. It gives employees, members of the Board of Directors and selected advisors a beneficial opportunity to purchase shares of the Company. Each option entitles its holder to purchase one share of the Company at a pre-defined exercise price. The number of options granted to each participant was determined by the Board of Directors based on a participant’s position and level of responsibility. The options generally vested quarterly over four years, with vesting of 25% after one year. At the end of the 10 year option term, unexercised options expire without value.
As of December 31, 2023 and December 31, 2022, an aggregate of 282,105 options were outstanding under the ESOP 2009 and ESOP 2014. All these options are fully vested at the reporting date.
Since the initial public offering of the Company on the SIX Swiss Exchange on November 5, 2014, no further option grants have been made under any of these two share option plans.
18.2.2 Long Term Incentive (“LTI”) Plans: Restricted Share Units ("RSU'') and Performance Share Units ("PSU")
•LTI plans 2019 established in March 2019
•LTI plans 2020 established in March 2020
•LTI plans 2021 established in March 2021
•LTI plans 2022 established in March 2022
•LTI plans 2023 established in March 2023
Under the LTI plans, members of the Board of Directors are eligible to be granted RSUs, whereas members of the Management Board and other employees are eligible to be granted PSUs.
RSUs are contingent rights to receive a certain number of shares of the Company at the end of a three-year blocking period. The number of RSUs per plan participant is a function of the approved CHF amount per position divided by the fair value of each RSU as at the grant date. In certain circumstances, including a change of control, a full or partial accelerated vesting of the RSUs may occur. RSUs vest over a one-year period from date of grant.
PSUs are contingent rights to receive a variable number of shares of the Company. Since 2021, PSUs granted to employees (except for members of the Management Board) will vest in three tranches of one third each. The first tranche of the PSUs shall vest on the first anniversary of the grant date, the second tranche on the second anniversary of the grant date and the third tranche on the third anniversary of the grant date. For the members of the Management Board PSUs will vest at the end of a three year cliff-vesting period. PSUs granted to all employees under PSU plans prior to 2021 will continue to vest at the end of a three-year cliff-vesting period.
The number of PSUs per plan participant is a function of the approved CHF amount per position divided by the fair value of each PSU as of the grant date. While the PSUs are designed to let the beneficiaries participate in the long-term share price development, the number of shares to be earned in relation to a PSU also depends on the achievement of pre-defined corporate goals for the respective year. Accordingly, the number of shares to be issued based on the PSUs can be between zero and 150% of the number of PSUs granted. Even after the determination of goal achievement, participants may lose their entitlements in full or in part depending on certain conditions relating to their employment. In certain circumstances, including a change of control, a full or partial accelerated vesting of the PSUs may occur.
The LTI plans are issued annually, which allows the Board of Directors to review the terms and determine the targets on an annual basis. Employees generally receive the grants on April 1 of each calendar year, or for new employees on the first day of the calendar quarter after the start of their employment. Members of the Management Board and the Board of Directors receive the annual grants after the approval of the ordinary shareholders’ meeting.
As of December 31, 2023, 1,347,983 PSUs and 182,678 RSUs were outstanding. As of December 31, 2022, 604,800 PSUs and 96,001 RSUs were outstanding.
18.2.3 Conditions attached to and measurement of fair values of equity-settled share-based payment arrangements
The following table provides the conditions as well as the inputs used in the measurement of the values at grant dates:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| RSU/PSU, conditions and assumptions |
2023 |
2022 |
| Nature of arrangement |
Grant of PSU/RSU |
Grant of PSU/RSU |
| Grant date RSU |
April 4, 2023 |
April 13, 2022 |
| Grant dates PSU |
Jan 1 - Oct 1 |
Jan 1 - Oct 1 |
| Number of RSU granted |
120,144 |
|
33,015 |
|
| Number of PSU granted |
1,162,228 |
|
307,137 |
|
| Weighted average exercise price (CHF) |
0.10 |
|
0.10 |
|
| Share price (CHF) |
3.86 - 6.16 |
6.55 - 18.88 |
| Vesting period for RSU (years) |
1.00 |
1.00 |
| Full contractual life for RSU (years) |
3.00 |
3.00 |
| Vesting period for PSU (years), Management Board |
3.00 |
3.00 |
| Vesting period for PSU (years), employees excluding Management Board |
3.00 (pro-rata annual vesting) |
3.00 (pro-rata annual vesting) |
| Full contractual life for PSU (years) |
3.00 |
3.00 |
| Settlement |
Common Shares |
Common Shares |
| Expected volatility on Common shares |
67.08 - 77.51 |
64.69 - 76.84 |
| Risk-free interest rate p. a. (%) / CHF LIBOR / Common shares |
(0.24) - 1.17 |
(0.54) - (0.71) |
| Expected volatility on NBI |
23.36 - 28.66 |
25.89 - 28.16 |
| Risk-free interest rate p. a. (%) / USD LIBOR / NBI |
5.30 - 6.04 |
0.58 - 4.78 |
| Expected volatility on SPI |
13.20 - 17.27 |
15.57 - 17.02 |
| Risk-free interest rate p. a. (%) / CHF LIBOR / SPI |
(0.24) - 1.17 |
(0.54) - (0.71) |
| Expected dividend (CHF) |
— |
|
|
— |
|
|
| Weighted average fair value of rights granted (CHF) |
5.20 |
|
17.08 |
|
| Latest expiry date |
Sep 30, 2026 |
Sep 30, 2025 |
| Valuation model |
Monte Carlo |
Monte Carlo |
Additional comments:
•Expected volatility: Historical share prices of the Company have been used.
•The indices, Nasdaq Biotechnology Index ('NBI") and Swiss performance Index ("SPI") are used as inputs in determining the fair values for the 2022 and 2023 PSU Plans.
The movements in the number of all issued RSUs, PSUs and share options are as follows:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Share option / PSU / RSU movements |
Total (numbers) |
|
Weighted average exercise price (CHF) |
|
Options (numbers) |
|
Weighted average exercise price (CHF) |
|
PSU/RSU (numbers) |
|
Weighted average exercise price (CHF) |
| Balance outstanding at December 31, 2021 |
962,022 |
|
|
|
2.35 |
|
|
|
318,902 |
|
|
|
6.87 |
|
|
|
643,120 |
|
|
|
0.10 |
|
|
| Granted |
340,152 |
|
|
|
0.10 |
|
|
|
— |
|
|
|
— |
|
|
|
340,152 |
|
|
|
0.10 |
|
|
(Performance adjustment) (1) |
— |
|
|
|
0.10 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
0.10 |
|
|
(Forfeited) (2) |
(63,990) |
|
|
|
0.10 |
|
|
|
— |
|
|
|
— |
|
|
|
(63,990) |
|
|
|
0.10 |
|
|
| (Expired) |
(3,220) |
|
|
|
5.40 |
|
|
|
(3,220) |
|
|
|
5.40 |
|
|
|
— |
|
|
|
— |
|
|
(Exercised options, vested PSU / RSU) (3) |
(252,058) |
|
|
|
1.00 |
|
|
|
(33,577) |
|
|
|
6.85 |
|
|
|
(218,481) |
|
|
|
0.10 |
|
|
| Balance outstanding at December 31, 2022 |
982,906 |
|
|
|
2.05 |
|
|
|
282,105 |
|
|
|
6.89 |
|
|
|
700,801 |
|
|
|
0.10 |
|
|
| Granted |
1,282,372 |
|
|
|
0.10 |
|
|
|
— |
|
|
|
— |
|
|
|
1,282,372 |
|
|
|
0.10 |
|
|
(Performance adjustment) (1) |
(79,703) |
|
|
|
0.10 |
|
|
|
— |
|
|
|
— |
|
|
|
(79,703) |
|
|
|
0.10 |
|
|
(Forfeited) (2) |
(63,218) |
|
|
|
0.10 |
|
|
|
— |
|
|
|
— |
|
|
|
(63,218) |
|
|
|
0.10 |
|
|
| (Expired) |
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
|
— |
|
|
(Exercised options, vested PSU / RSU) (3) |
(309,591) |
|
|
|
0.10 |
|
|
|
— |
|
|
|
|
|
|
(309,591) |
|
|
|
0.10 |
|
|
| Balance outstanding at December 31, 2023 |
1,812,766 |
|
|
|
1.16 |
|
|
|
282,105 |
|
|
|
6.89 |
|
|
|
1,530,661 |
|
|
|
0.10 |
|
|
|
|
(1) Performance adjustments indicate forfeitures due to non-market performance conditions not achieved
(2) Forfeited due to service conditions not fulfilled
(3) The weighted average share prices at the dates of exercising options during the year 2022 amounted to CHF 22.35. There were no options exercised in 2023.
The following table applies to all share options, PSUs and RSUs outstanding at December 31, 2023:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Exercise price
CHF |
Options /
PSU/RSU
(number) |
|
Remaining life
(years) |
|
Thereof exercisable options |
| Options |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 6.06 |
15,450 |
|
|
|
0.4 |
|
15,450 |
|
|
| 6.94 |
266,655 |
|
|
|
0.7 |
|
266,655 |
|
|
| PSU/RSU |
|
|
|
|
|
|
|
| 0.10 |
1,530,661 |
|
|
|
1.4 |
|
|
|
| Total |
1,812,766 |
|
|
|
|
|
282,105 |
|
|
The following table applies to all share options, PSUs and RSUs outstanding at December 31, 2022:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Exercise price
CHF |
Options /
PSU/RSU
(number) |
|
Remaining life
(years) |
|
Thereof exercisable options |
| Options |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 6.06 |
15,450 |
|
|
|
1.4 |
|
|
15,450 |
|
|
| 6.94 |
266,655 |
|
|
|
1.7 |
|
|
266,655 |
|
|
| PSU/RSU |
|
|
|
|
|
|
|
|
| 0.10 |
700,801 |
|
|
|
1.1 |
|
|
|
|
| Total |
982,906 |
|
|
|
|
|
|
282,105 |
|
|
The non-cash costs for share-based payments recognized in the statement of comprehensive income can be attributed to the Group’s two functions as follows:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| in CHF thousands |
2023 |
|
2022 |
|
2021 |
| Research and development |
3,057 |
|
|
|
3,010 |
|
|
|
2,208 |
|
|
| Selling, general and administrative |
2,150 |
|
|
|
2,078 |
|
|
|
1,877 |
|
|
| Total year ended December 31 |
5,207 |
|
|
|
5,088 |
|
|
|
4,085 |
|
|
19. Financial income and financial expense
Financial income
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| in CHF thousands |
2023 |
|
2022 |
|
2021 |
| Interest income on financial assets held at amortized costs |
4,279 |
|
|
|
1,142 |
|
|
|
99 |
|
|
| Net foreign exchange gain |
— |
|
|
|
717 |
|
|
|
92 |
|
|
| Total year ended December 31 |
4,279 |
|
|
|
1,859 |
|
|
|
191 |
|
|
Financial expense
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| in CHF thousands |
2023 |
|
2022 |
|
2021 |
| Net foreign exchange loss |
(5,106) |
|
|
|
— |
|
|
|
— |
|
|
| Negative interest on financial assets held at amortized costs |
— |
|
|
|
(562) |
|
|
|
(495) |
|
|
| Interest expense on leases |
(34) |
|
|
|
(43) |
|
|
|
(53) |
|
|
| Other financial expenses |
(15) |
|
|
|
(14) |
|
|
|
(8) |
|
|
| Total year ended December 31 |
(5,155) |
|
|
|
(619) |
|
|
|
(556) |
|
|
20. Income Taxes
Current taxes
The Company generated taxable losses in 2023 and 2021 whereas in 2022 the Company generated a taxable profit in Switzerland. In 2023, the Company did not have to pay or accrue any income taxes during the reporting period, as the Company incurred a taxable loss in 2023. Any potential future taxable income will be subject to Swiss federal, cantonal and communal income taxes. The Company’s applicable income tax rate (after tax) for the year 2023 is 19.3% (2022: 19.4%; 2021: 19.4%).
Molecular Partners Inc., which is incorporated in the United States in the State of Delaware, is subject to statutory U.S. federal corporate income taxes and minimal state taxes for Massachusetts and New York.
For the year ended December 31, 2023, current income tax expense of TCHF 0.4 (or in thousands of US Dollars ("TUSD") 0.5) was recognized by the Group's U.S. based subsidiary for estimated U.S. tax obligations of the subsidiary based on intra-Group activity (for the year ended December 31, 2022: tax expense of TCHF 0.3 (TUSD 0.3) and for the year ended December 31, 2021: tax credit of TCHF 2 (TUSD 2)). The tax expense amount comprises of the sum of the minimal taxes payable for federal taxes and for the various states in which Molecular Partners Inc. is liable for taxes. The applicable income tax rates are 21% U.S. federal tax plus 8.00% state tax (Massachusetts) and 6.50% (New York).
Deferred taxes
The Company's net loss before income taxes amounted to TCHF 56,285 in 2023 whereas the prior years generated a net income before income taxes of TCHF 124,020 in 2022 and a net loss before income taxes of TCHF 58,632 in 2021. The cumulative tax losses as of December 31, 2023 of TCHF 144,483 may be used as tax loss carry forwards to offset future taxable income over a period of seven years.
No deferred tax assets have been recognized for these tax loss carry forwards, because as of December 31, 2023, it was not considered probable that such loss carry forwards can be utilized in the foreseeable future. In addition, no deferred tax positions were recognized on other deductible temporary differences (e.g., pension liabilities under IAS 19 for a total of TCHF 4,720, see also note 18.1) due to the tax losses carried forward. Income tax expense has been calculated for the year ending December 31, 2023, based on the effective income tax rate expected for the full financial year, being 0% on December 31, 2023.
Given the facts above, as well as the fact that the Company incurred no significant tax expense in the reporting periods presented, a numerical reconciliation of the effective tax rate is not provided. The primary reconciling item is the effect of unrecognized deferred tax assets for tax losses and deductible temporary differences.
The following table shows the expiry of tax loss carry forwards for the Company, for which no deferred tax asset was recognized:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| in CHF thousands |
2023 |
|
2022 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 2027 |
(29,566) |
|
|
|
(29,566) |
|
|
| 2028 |
(58,632) |
|
|
|
(58,632) |
|
|
| 2030 |
(56,285) |
|
|
|
— |
|
|
|
|
|
|
|
|
| Total tax loss carry forwards as at December 31 |
(144,483) |
|
|
|
(88,198) |
|
|
21. Earnings per share
Basic earnings per share is calculated by dividing the net result attributable to the shareholders of the Company by the weighted average number of shares issued and outstanding during the reporting period, excluding any shares held as treasury shares. Diluted earnings per share additionally takes into account the potential conversion of all dilutive potential ordinary shares.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2023 |
|
2022 |
|
2021 |
| Weighted average number of shares used in computing basic earnings per share |
32,770,665 |
|
|
|
32,469,957 |
|
|
|
31,005,171 |
|
|
| Weighted average number of shares used in computing diluted earnings per share |
32,770,665 |
|
|
|
33,265,567 |
|
|
|
31,005,171 |
|
|
At December 31, 2023 and December 31, 2021, all potential ordinary shares were anti-dilutive (1,526,976 and 835,422). At December 31, 2022, the number of shares that were dilutive, is 795,610.
22. Leases
The Group leases office and laboratory facilities in Schlieren, Switzerland. These leases generally have terms between 2 and 10 years and contain extension or terminations options exercisable by the Group up to one year before the end of the non‑cancellable contract period. These terms are used to maximize operational flexibility in terms of managing contracts. The options to extend are held by the Company and the termination options are held both by the Company and the lessor. As of December 31, 2020, the Group exercised the option to extend the lease on its facilities in Schlieren by five years with a new lease term ending on December 31, 2026. The earliest contractual termination date for both the lessor and the Group on the major real estate lease is December 31, 2025. For information about the right-of-use assets please also see note 6.
Set out below are the carrying amounts of the lease liabilities and the movements during the period:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| in CHF thousands |
2023 |
|
2022 |
| as at January 1, |
4,850 |
|
|
|
6,039 |
|
|
| Additions / new leases |
— |
|
|
|
— |
|
|
| Remeasurements |
— |
|
|
|
— |
|
|
| Recognition of interest on lease liabilities |
34 |
|
|
|
43 |
|
|
| Payments |
(1,232) |
|
|
|
(1,232) |
|
|
| Balance as at December 31, |
3,652 |
|
|
|
4,850 |
|
|
|
|
|
|
|
|
| Current |
1,208 |
|
|
|
1,198 |
|
|
| Non-current |
2,444 |
|
|
|
3,652 |
|
|
| Balance as at December 31, |
3,652 |
|
|
|
4,850 |
|
|
|
|
The following are the expense amounts recognized in the consolidated statement of comprehensive income.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| in CHF thousands |
2023 |
|
2022 |
|
2021 |
| Depreciation on right-of-use assets |
1,200 |
|
|
|
1,200 |
|
|
|
1,200 |
|
|
| Interest expense on lease liabilities |
34 |
|
|
|
43 |
|
|
|
53 |
|
|
| Short term leases |
— |
|
|
|
— |
|
|
|
— |
|
|
| Total amount recognized in profit or loss |
1,234 |
|
|
|
1,243 |
|
|
|
1,253 |
|
|
The total cash outflow for leases for the year ended December 31, 2023 amounted to TCHF 1,232 (year ended December 31, 2022 TCHF 1,232; year ended December 31, 2021 TCHF 1,232).
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Contractual maturities of financial liabilities at December 31, 2023 |
| in CHF thousands |
|
Less than 1 year |
Between 1 and 2 years |
Between 2 and 5 years |
More than 5 years |
Total contractual cash-flows |
Carrying Amount lease liabilities |
| Lease liabilities |
1,232 |
|
|
1,232 |
|
|
1,232 |
|
|
— |
|
|
3,696 |
|
|
3,652 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Contractual maturities of financial liabilities at December 31, 2022 |
| in CHF thousands |
|
Less than 1 year |
Between 1 and 2 years |
Between 2 and 5 years |
More than 5 years |
Total contractual cash-flows |
Carrying Amount lease liabilities |
| Lease liabilities |
1,232 |
|
|
1,232 |
|
|
2,464 |
|
|
— |
|
|
4,928 |
|
|
4,850 |
|
|
23. Related party disclosures
Compensation costs of key management, which includes executive management and the Board of Directors, are as follows:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| in CHF thousands |
2023 |
|
2022 |
|
2021 |
| Short-term employee benefits |
2,761 |
|
|
|
3,159 |
|
|
|
2,423 |
|
|
| Post-employment benefits |
253 |
|
|
|
297 |
|
|
|
203 |
|
|
| Share-based compensation |
1,914 |
|
|
|
2,111 |
|
|
|
1,784 |
|
|
| Total year ended December 31 |
4,928 |
|
|
|
5,567 |
|
|
|
4,410 |
|
|
24. Capital commitments
As of December 31, 2023 and December 31, 2022, the Group did not have any capital commitments.
25. Financial risk management
Foreign exchange risk
In order to reduce its foreign exchange exposure, Molecular Partners may enter into currency contracts with selected high-quality financial institutions to hedge against foreign currency exchange rate risks. The Group’s primary exposure to financial risk is due to fluctuation of exchange rates between CHF, USD and EUR.
During 2023 and 2022, the Group did not enter into any forward currency transactions. No forward currency transactions were outstanding as of December 31, 2023 and 2022.
The following table demonstrates the sensitivity to a reasonably possible change in exchange rates for the Group's main foreign currencies, USD and EUR, with all other variables held constant, of the Group’s result before taxes. There is no direct impact on the Group’s equity.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| in % and CHF thousands |
Incr./Decr. exchange rate |
|
Effect on result before tax (in TCHF) |
| USD Positions |
|
|
|
|
|
| 2023 |
+10 |
% |
|
|
4,718 |
|
|
|
-10 |
% |
|
|
(4,718) |
|
|
| 2022 |
+10 |
% |
|
|
5,904 |
|
|
|
-10 |
% |
|
|
(5,904) |
|
|
| 2021 |
+10 |
% |
|
|
6,633 |
|
|
|
-10 |
% |
|
|
(6,633) |
|
|
| EUR Positions |
|
|
|
|
|
| 2023 |
+10 |
% |
|
|
479 |
|
|
|
-10 |
% |
|
|
(479) |
|
|
| 2022 |
+10 |
% |
|
|
1,252 |
|
|
|
-10 |
% |
|
|
(1,252) |
|
|
| 2021 |
+10 |
% |
|
|
2,019 |
|
|
|
-10 |
% |
|
|
(2,019) |
|
|
Interest rate risk
Molecular Partners earns interest on cash and cash equivalents, and its profit and loss may be influenced by changes in market interest rates. The Group does invest its cash balances into a variety of current and deposit accounts in four different Swiss banks to optimize interest. In addition, the Group does invest a portion of its cash into risk free money market investments in line with its treasury guidelines.
The Group strives to optimize the net balance of interest paid and interest received by monitoring the interest rates applicable over the major currencies the Group holds as well as the offered holding periods.
The following table demonstrates the sensitivity of the main currencies used in the Group, to reasonably possible changes in interest rates, with all other variables held constant, of the Group’s results before tax. There is no direct impact on the Group’s equity.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| in % and CHF thousands |
Incr./Decr. interest rate |
|
|
Effect on result before tax (in TCHF) |
| CHF Positions |
|
|
|
|
|
| 2023 |
+0.5 |
% |
|
|
674 |
|
|
|
-0.5 |
% |
|
|
(674) |
|
|
| 2022 |
+0.5 |
% |
|
|
888 |
|
|
|
-0.5 |
% |
|
|
(888) |
|
|
| 2021 |
+0.5 |
% |
|
|
323 |
|
|
|
-0.5 |
% |
|
|
(323) |
|
|
| USD Positions |
|
|
|
|
|
| 2023 |
+0.5 |
% |
|
|
235 |
|
|
|
-0.5 |
% |
|
|
(235) |
|
|
| 2022 |
+0.5 |
% |
|
|
294 |
|
|
|
-0.5 |
% |
|
|
(294) |
|
|
| 2021 |
+0.5 |
% |
|
|
234 |
|
|
|
-0.5 |
% |
|
|
(234) |
|
|
| EUR Positions |
|
|
|
|
|
| 2023 |
+0.5 |
% |
|
|
25 |
|
|
|
-0.5 |
% |
|
|
(25) |
|
|
| 2022 |
+0.5 |
% |
|
|
63 |
|
|
|
-0.5 |
% |
|
|
(63) |
|
|
| 2021 |
+0.5 |
% |
|
|
102 |
|
|
|
-0.5 |
% |
|
|
(102) |
|
|
Credit risk
The maximum credit risk on financial assets corresponds to the carrying amounts of the Group’s cash and cash equivalents, short-term time deposits and receivables. The Group has not entered into any guarantees or similar obligations that would increase the risk over and above the carrying amounts.
The cash and cash equivalents and short-term deposits are considered low risk and were held at Swiss banks with Standard & Poor long-term credit ratings as of December 31, 2023 of AAA (Zürcher Kantonalbank), AA (Luzerner Kantonalbank) and A+ (UBS and Credit Suisse) and therefore any impact resulting from the expected credit loss model is considered immaterial. Analysis performed included assessing the cumulative default rates by credit rating category and applying these rates to the cash and short-term deposit balances at reporting dates. The calculated loss allowance based on the ECL is considered immaterial.
The Group enters into agreements with partners that have appropriate credit history and a commitment to ethical business practices.
The maximum credit risk as of the balance sheet date was as follows:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Credit risk |
|
|
|
| in CHF thousands |
2023 |
|
2022 |
| Cash and cash equivalents |
67,309 |
|
|
|
87,946 |
|
|
| Trade receivables |
295 |
|
|
|
521 |
|
|
| Accrued income |
1,131 |
|
|
|
679 |
|
|
| Short-term time deposits |
119,580 |
|
|
|
161,198 |
|
|
| Total credit risk as at December 31 |
188,315 |
|
|
|
250,344 |
|
|
Liquidity risk
Liquidity risk is the risk that the Group will encounter difficulties in meeting the obligations associated with its financial liabilities that are settled by delivering cash or another financial asset. The Group’s liquidity risk is considered low by management due to the financial assets at the reporting date, giving the Group a secure source of funding for its research and development activities.
26. Putative Class Action
On July 12, 2022, a putative class action complaint was filed in the U.S. District Court for the Southern District of New York against the Company, its directors, and certain of its executive officers. On May 23, 2023, an amended complaint was filed. The amended complaint alleged that the defendants violated federal securities laws by, among other things, making misrepresentations and omissions regarding its product candidate MP0310 and an associated licensing agreement. The amended complaint sought unspecified compensatory damages, as well as an award of reasonable attorneys’ fees and other costs, on behalf of persons and/or entities which purchased the Company's American Depositary Shares (ADSs) pursuant to certain offering documents issued in connection with the Company's initial public offering of ADSs. The Company and named individual defendants moved to dismiss the amended complaint on July 24, 2023. Plaintiffs filed their opposition on September 7, 2023 and the Company and named individual defendants filed their reply brief on October 5, 2023. On February 5, 2024, the court dismissed the amended complaint without prejudice and gave plaintiff the opportunity to amend the complaint by February 26, 2024. On February 23, 2024, plaintiff filed a stipulation of dismissal with prejudice On February 29, 2024 the court ordered the case closed.
27. Events after the balance sheet date
On January 5, 2024 the Group announced it entered into a co-development agreement with Orano Med to co-develop 212Pb-based Radio Darpin Therapies (RDT). Under the terms of the co-development agreement, Molecular Partner’s previously disclosed RDT target DLL3 (delta-like ligand 3) will be included in the collaboration with Orano Med. Both companies are developing additional radioligand therapy candidates in partnership with other companies, with Molecular Partners having announced its first collaboration with Novartis in December 2021.
Expression of DLL3 is low in healthy tissue but significantly increased in certain tumor types, such as small-cell lung cancer, providing an opportunity for selective tumor-targeting. DLL3 will be exclusively developed by Molecular Partners and Orano Med as a RDT target.
Molecular Partners maintains the option to explore DLL3 for targeted therapy outside of the radiotherapy space. Both companies commit to sharing the cost of preclinical and clinical development with additional commitments to supply of their respective materials.
Additional agreements are being put in place for future development and commercialization of any potential programs that proceed into the clinical stage of development.
On January 5, 2024, Novartis has agreed the termination of the License Agreement for ensovibep, previously under investigation for the treatment of SARS Cov-2, and Novartis has returned the rights to the ensovibep program to the Company. Clinical work on the ensovibep program ended in 2022, and the program remains terminated.
No other events occurred between the balance sheet date and the date on which these consolidated financial statements were approved by the Board of Directors that would require adjustment to the consolidated financial statements or disclosure under this heading.
EX-1.1
2
a11articlesofincorporation.htm
EX-1.1
Document
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Statuten
von Molecular Partners AG
vom 16. Januar 2024
|
|
|
Articles of Incorporation of
Molecular Partners Ltd
as of January 16, 20241
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
1 This is a translation of the original German version. In case of any discrepancy, the German version shall prevail. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Abschnitt 1:
Firma, Sitz, Zweck und Dauer der Gesellschaft |
|
|
Section 1:
Name, Place of Incorporation, Purpose and Duration of the Company |
|
|
|
|
|
|
|
|
Artikel 1 |
|
|
Article 1 |
| Firma, Sitz |
1 |
Unter der Firma
Molecular Partners AG (Molecular Partners SA) (Molecular Partners Ltd) (die Gesellschaft)
besteht eine Aktiengesellschaft, die den vorliegenden Statuten und den Vorschriften des 26. Titels des Schweizerischen Obligationenrechts (das OR) untersteht.
|
Name, Place of Incorporation |
1 |
Under the name
Molecular Partners AG (Molecular Partners SA) (Molecular Partners Ltd) (the Company)
there exists a corporation as defined in title 26 of the Swiss Code of Obligations (CO) and in these Articles of Incorporation.
|
|
2 |
Der Sitz der Gesellschaft ist in Schlieren, Kanton Zürich. Die Dauer der Gesellschaft ist unbeschränkt. |
|
2 |
The registered office of the Company is in Schlieren, Canton of Zurich. The duration of the Company is unlimited. |
|
|
|
|
|
|
| Zweck |
|
Artikel 2 |
Purpose |
|
Article 2 |
|
1 |
Zweck der Gesellschaft ist die Forschung, Entwicklung, Herstellung und der Verkauf von Produkten in den Gebieten der Biotechnologie, der Pharmazie, Medizintechnologie, Diagnose und Therapie sowie der Kauf, Verkauf und die Verwendung von Patenten und Lizenzen auf diesem Gebiet. Die Gesellschaft kann alle Geschäfte tätigen, die geeignet erscheinen, den Zweck der Gesellschaft zu fördern, oder die mit diesem zusammenhängen. |
|
1 |
The Company's purpose is to research, develop, produce and sell products in the fields of biotechnology, pharamceutica, medical technology, diagnosis and therapy as well as to purchase, sell and use patents and licences in this field. The Company may engage in all types of transactions that appear appropriate to promote the purpose of the Company or that are related thereto. |
|
2 |
Die Gesellschaft kann Grundstücke im In- und Ausland erwerben, verwalten, belasten, verwerten und verkaufen sowie andere Gesellschaften finanzieren. |
|
2 |
The Company may acquire, administer, encumber, exploit or sell real estate in Switzerland and aborad and may also finance other companies. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
3 |
Die Gesellschaft kann Zweigniederlassungen und Tochtergesellschaften im In- und Ausland errichten und sich an anderen Unternehmen beteiligen oder mit diesen fusionieren. |
|
3 |
The Company may establish branches and subsidiaries within Switzerland or abroad and may acquire participations in other companies. |
|
4 |
Bei der Verfolgung ihres Gesellschaftszwecks strebt die Gesellschaft die Schaffung von langfristigem, nachhaltigem Wert an. |
|
4 |
In pursuing its purpose, the Company strives to create long-term, sustainable value. |
|
|
|
|
|
|
|
|
Abschnitt 2:
Aktienkapital |
|
|
Section 2:
Share Capital |
|
|
|
|
|
|
|
|
Artikel 3 |
|
|
Article 3 |
| Aktienkapital |
1 |
Das Aktienkapital der Gesellschaft beträgt CHF 3‘635‘429.70 und ist eingeteilt in 36‘354‘297 Namenaktien mit einem Nennwert von je CHF 0.10. |
Share Capital |
1 |
The share capital of the Company is CHF 3,635,429.70 and is divided into 36,354,297 registered shares. Each registered share has a par value of CHF 0.10. |
|
2 |
Die Aktien sind voll liberiert. |
|
2 |
The shares are fully paid up. |
|
|
|
|
|
|
|
|
Artikel 3a |
|
|
Article 3a |
| Genehmigtes Aktienkapital |
1 |
Der Verwaltungsrat ist ermächtigt, jederzeit bis zum 13. April 2024 das Aktienkapital im Maximalbetrag von CHF 457'316.20 durch Ausgabe von höchstens 4'573'162 vollständig zu liberierenden Namenaktien mit einem Nennwert von je CHF 0.10 zu erhöhen. Erhöhungen in Teilbeträgen sind gestattet. |
Authorized Share Capital |
1 |
The board of directors is authorized to increase the share capital, at any time until April 13, 2024, by a maximum amount of CHF 457,316.20 by issuing a maximum of 4,573,162 fully paid up registered shares with a par value of CHF 0.10 each. An increase of the share capital in partial amounts shall be permissible. |
|
2 |
Zeichnung und Erwerb der neuen Aktien sowie jede nachfolgende Übertragung der Aktien unterliegen den Beschränkungen von Artikel 5 dieser Statuten. |
|
2 |
The acquisition of shares and each subsequent transfer of the shares shall be subject to the restrictions of Article 5 of these Articles of Incorporation. |
|
3 |
Der Verwaltungsrat legt den Ausgabebetrag, die Art der Einlagen, den Zeitpunkt der Ausgabe, die Bedingungen der Bezugsrechtsausübung und den Beginn der Dividendenberechtigung fest. Dabei kann der Verwaltungsrat neue Aktien mittels Festübernahme durch eine Bank oder einen anderen Dritten und anschliessendem Angebot an die bisherigen Aktionäre oder an Dritte (sofern die Bezugsrechte der bisherigen Aktionäre aufgehoben sind oder nicht gültig ausgeübt werden) ausgeben. Der Verwaltungsrat ist ermächtigt, den Handel mit Bezugsrechten zu ermöglichen, zu beschränken oder auszuschliessen. Nicht ausgeübte Bezugsrechte kann der Verwaltungsrat verfallen lassen, oder er kann diese bzw. Aktien, für welche Bezugsrechte eingeräumt, aber nicht ausgeübt werden, zu Marktkonditionen platzieren oder anderweitig im Interesse der Gesellschaft verwenden. |
|
3 |
The board of directors shall determine the issue price, the type of payment, the time of the issuance, the conditions for the exercise of the preemptive rights and the date from which the shares carry the right to dividends. The board of directors can issue new shares by means of an underwriting by a bank or another third party with a subsequent offer of these shares to the existing shareholders or third parties (if the preemptive rights of the existing shareholders have been denied or not been duly exercised). The board of directors is authorized to permit, to restrict or to deny the trade of preemptive rights. The board of directors may permit preemptive rights that have been granted but not exercised to expire or it may place these rights respectively the shares as to which preemptive rights have been granted but not exercised, at market conditions or use them for other purposes in the interest of the Company. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
4 |
Der Verwaltungsrat ist ferner ermächtigt, das Bezugsrecht der Aktionäre zu beschränken oder aufzuheben und Dritten zuzuweisen:
(a)für die Übernahme von Unternehmen, Unternehmensteilen oder Beteiligungen, den Erwerb von Produkten, Immaterialgütern oder Lizenzen oder für Investitionsvorhaben oder für die Finanzierung oder Refinanzierung solcher Transaktionen durch eine Aktienplatzierung; oder
(b)zum Zwecke der Erweiterung des Aktionärskreises oder im Zusammenhang mit der Kotierung der Aktien an inländischen oder an ausländischen Börsen; oder
(c)wenn der Ausgabebetrag der neuen Aktien unter Berücksichtigung des Marktpreises festgesetzt wird; oder
(d)für die Einräumung einer Mehrzuteilungsoption (Greenshoe) von bis zu 20% der zu platzierenden oder zu verkaufenden Aktien an die betreffenden Erstkäufer oder Festübernehmer im Rahmen einer Aktienplatzierung oder eines Aktienverkaufs; oder
(e)wenn ein Aktionär oder eine Gruppe von in gemeinsamer Absprache handelnden Aktionären mehr als 15% des im Handelsregister eingetragenen Aktienkapitals der Gesellschaft auf sich vereinigt hat, ohne den übrigen Aktionären ein vom Verwaltungsrat empfohlenes Übernahmeangebot zu unterbreiten; oder
(f)zur Abwehr eines unterbreiteten, angedrohten oder potentiellen Übernahmeangebotes, welches der Verwaltungsrat, nach Konsultation mit einem von ihm beigezogenen unabhängigen Finanzberater, den Aktionären nicht zur Annahme empfohlen hat, weil der Verwaltungsrat das Übernahmeangebot in finanzieller Hinsicht gegenüber den Aktionären nicht als fair beurteilt hat.
|
|
4 |
The board of directors is further authorized to restrict or deny the preemptive rights of shareholders and to allocate them to third parties:
(a)for the acquisition of companies, parts of companies or participations, for the acquisition of products, intellectual property or licenses, or for investment projects or for the financing or refinancing of such transactions through a placement of shares; or
(b)for the purpose of broadening the shareholder constituency or in connection with a listing of shares on domestic or foreign stock exchanges; or
(c)if the issue price of the new Shares is determined by reference to the market price; or
(d)for purposes of granting an over-allotment option (Greenshoe) of up to 20% of the total number of Shares in a placement or sale of Shares to the respective initial purchasers or underwriters; or
(e)following a shareholder or a group of shareholders acting in concert having accumulated shareholdings in excess of 15% of the share capital registered in the commercial register without having submitted to the other shareholders a takeover offer recommended by the board of directors; or
(f)for the defense of an actual, threatened or potential takeover bid, in relation to which the board of directors, upon consultation with an independent financial adviser retained by it, has not recommended to the shareholders acceptance on the basis that the board of directors has not found the takeover bid to be financially fair to the shareholders.
|
|
|
|
|
|
|
|
|
Artikel 3b |
|
|
Article 3b |
| Bedingtes Aktienkapital für Mitarbeiterbeteiligung |
1 |
Das Aktienkapital kann sich durch Ausgabe von höchstens 1'053’372 voll zu liberierenden Namenaktien im Nennwert von je CHF 0.10 um höchstens CHF 105'337.20 erhöhen durch direkte oder indirekte Ausgabe von Aktien, Optionen oder diesbezüglichen Bezugsrechten an Mitarbeiter und Mitglieder des Verwaltungsrats der Gesellschaft und ihrer Konzerngesellschaften sowie an Mitglieder von Beiräten. |
Conditional Share Capital for Employee Participation |
1 |
The share capital may be increased in an amount not to exceed CHF 105,337.20 through the issuance of up to 1,053,372 fully paid up registered shares with a par value of CHF 0.10 per share through the direct or indirect issuance of shares, options or preemptive rights thereof granted to employees and members of the board of directors of the Company or its subsidiaries as well as to members of any advisory boards. |
|
2
|
Bei der Ausgabe von Aktien, Optionen oder diesbezüglichen Bezugsrechten sind das Bezugsrecht wie auch das Vorwegzeichnungsrecht der Aktionäre der Gesellschaft ausgeschlossen. Die Ausgabe von Aktien, Optionen oder diesbezüglichen Bezugsrechten erfolgt gemäss einem oder mehreren vom Verwaltungsrat zu erlassenden Beteiligungsplänen und/oder Reglementen und unter Beachtung von Abschnitt 4 dieser Statuten. |
|
2 |
The preemptive rights and advance subscription rights of the shareholders of the Company shall be excluded in connection with the issuance of any shares, options or preemptive rights thereof. Shares, options or preemptive rights thereof shall be issued in accordance with one or more participation plans and/or policies to be issued by the board of directors and in accordance with Section 4 of these Articles of Incorporation. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
3
|
Die neuen Aktien, welche durch Mitarbeiter, Mitglieder des Verwaltungsrats der Gesellschaft und ihrer Konzerngesellschaften oder Mitglieder von Beiräten im Rahmen eines Mitarbeiterbeteiligungsprogramms direkt oder indirekt erworben werden, sowie jede nachfolgende Übertragung der Aktien unterliegen den Beschränkungen von Artikel 5 dieser Statuten. |
|
3 |
The new shares directly or indirectly acquired by employees, members of the board of directors of the Company or its subsidiaries or members of any advisory boards in connection with an employee participation program and any subsequent transfer of such shares shall be restricted by Article 5 of these Articles of Incorporation. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Artikel 3c |
|
|
Article 3c |
| Bedingtes Aktienkapital für Finanzierungen, Akquisitionen und andere Zwecke |
1 |
Das Aktienkapital kann sich durch Ausgabe von höchstens 2'260'870 voll zu liberierenden Namenaktien im Nennwert von je CHF 0.10 um höchstens CHF 226'087 erhöhen durch die Ausübung oder Zwangsausübung von Wandel-, Tausch-, Options-, Bezugs- oder ähnlichen Rechten auf den Bezug von Aktien, welche Aktionären oder Dritten allein oder in Verbindung mit Anleihensobligationen, Darlehen, Optionen, Warrants oder anderen Finanzmarktinstrumenten oder vertraglichen Verpflichtungen der Gesellschaft oder einer ihrer Gruppengesellschaften eingeräumt werden (nachfolgend zusammen die Finanzinstrumente). |
Conditional Share Capital for Financing, Acquisitions and other Purposes |
1 |
The share capital may be increased in an amount not to exceed CHF 226,087 through the issuance of up to 2,260,870 fully paid up registered shares with a par value of CHF 0.10 per share through the exercise or mandatory exercise of conversion, exchange, option, warrant or similar rights for the subscription of shares granted to shareholders or third parties alone or in connection with bonds, notes, options, warrants or other securities or contractual obligations by or of the Company or any of its group companies (hereinafter collectively, the Financial Instruments). |
|
2 |
Bei der Ausgabe von Aktien bei Ausübung der Finanzinstrumente ist das Bezugsrecht der Aktionäre ausgeschlossen. Zum Bezug der neuen Aktien, die bei Ausübung von Finanzinstrumenten ausgegeben werden, sind die jeweiligen Inhaber der Finanzinstrumente berechtigt. Die Bedingungen der Finanzinstrumente sind durch den Verwaltungsrat festzulegen. |
|
2 |
The preemptive rights of the shareholders shall be excluded in connection with the issuance of shares upon the exercise of any Financial Instruments. The then-current owners of such Financial Instruments shall be entitled to acquire the new shares issued upon conversion, exchange or exercise of any Financial Instruments. The conditions of the Financial Instruments shall be determined by the board of directors. |
|
3 |
Der Verwaltungsrat ist ermächtigt, die Vorwegzeichnungsrechte der Aktionäre im Zusammenhang mit der Ausgabe von Finanzinstrumenten durch die Gesellschaft oder eine ihrer Gruppengesellschaften zu beschränken oder aufzuheben, falls (1) die Ausgabe zum Zwecke der Finanzierung oder Refinanzierung der Übernahme von Unternehmen, Unternehmensteilen, Beteiligungen oder Investitionen, oder (2) die Ausgabe auf nationalen oder internationalen Finanzmärkten oder im Rahmen einer Privatplatzierung erfolgt.
Wird das Vorwegzeichnungsrecht weder direkt noch indirekt durch den Verwaltungsrat gewährt, gilt Folgendes:
(a)Die Finanzinstrumente sind zu marktüblichen Bedingungen auszugeben oder einzugehen; und
(b)der Umwandlungs-, Tausch- oder sonstige Ausübungspreis der Finanzinstrumente ist unter Berücksichtigung des Marktpreises im Zeitpunkt der Ausgabe der Finanzinstrumente festzusetzen; und
(c)die Finanzinstrumente sind höchstens während 10 Jahren ab dem jeweiligen Zeitpunkt der betreffenden Ausgabe oder des betreffenden Abschlusses wandel-, tausch- oder ausübbar.
|
|
3 |
The board of directors shall be authorized to withdraw or limit the advance subscription rights of the shareholders in connection with the issuance by the Company or one of its group companies of Financial Instruments if (1) the issuance is for purposes of financing or refinancing the acquisition of an enterprise, parts of an enterprise, participations or investments or (2) the issuance occurs in national or international capital markets or through a private placement.
If the advance subscription rights are neither granted directly nor indirectly by the board of directors, the following shall apply:
(a)the Financial Instruments shall be issued or entered into at market conditions; and
(b)the conversion, exchange or exercise price of the Financial Instruments shall be set with reference to the market conditions prevailing at the date on which the Financial Instruments are issued; and
(c)the Financial Instruments may be converted, exchanged or exercised during a maximum period of 10 years from the date of the relevant issuance or entry.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
4 |
Die neuen Aktien, welche über die Ausübung von Finanzinstrumenten direkt oder indirekt erworben werden, sowie jede nachfolgende Übertragung der Aktien unterliegen den Beschränkungen von Artikel 5 dieser Statuten. |
|
4 |
The new shares directly or indirectly acquired through the exercise of Financial Instruments and any subsequent transfer of such shares shall be restricted by Article 5 of these Articles of Incorporation. |
|
|
|
|
|
|
|
|
Artikel 4 |
|
|
Article 4 |
| Aktienzertifikate und Bucheffekten |
1 |
Die Gesellschaft kann ihre Namenaktien als Wertrechte nach Artikel 973c oder 973d OR, als Bucheffekten im Sinne des Bucheffektengesetzes oder als Einzel- oder Globalurkunden ausgeben. Der Gesellschaft steht es im Rahmen der gesetzlichen Vorgaben frei, ihre in einer dieser Formen ausgegebenen Namenaktien jederzeit und ohne Zustimmung der Aktionäre in eine andere Form umzuwandeln. Die Gesellschaft trägt dafür die Kosten. |
Share Certificates and Intermediated Securities |
1 |
The Company may issue its registered shares as uncertificated securities pursuant to article 973c or 973d CO, as intermediated securities in the sense of the Federal Act on Intermediated Securities, or in the form of single or global certificates. Subject to applicable law, the Company may convert its registered shares from one form into another form at any time and without the approval of the shareholders. The Company shall bear the cost associated with any such conversion. |
|
2 |
Der Aktionär hat keinen Anspruch auf Umwandlung von in bestimmter Form ausgegebenen Namenaktien in eine andere Form. Insbesondere hat der Aktionär keinen Anspruch auf die Verbriefung der Mitgliedschaft in einem Wertpapier. Jeder Aktionär kann jedoch von der Gesellschaft jederzeit die Ausstellung einer Bescheinigung über die von ihm gemäss Aktienbuch gehaltenen Namenaktien verlangen. |
|
2 |
The shareholder has no right to demand a conversion of the form of the registered shares. In particular, the shareholder has no claim to the certification of the membership in a security. Each shareholder may, however, at any time request a written confirmation from the Company of the registered shares held by such shareholder, as reflected in the share register. |
|
3 |
Bucheffekten, denen Namenaktien der Gesellschaft zugrunde liegen, können nicht durch Zession übertragen werden. An diesen Bucheffekten können auch keine Sicherheiten durch Zession bestellt werden. |
|
3 |
Intermediated securities based on registered shares of the Company cannot be transferred by way of assignment. Further, a security interest in any such intermediated securities cannot be granted by way of assignment. |
|
|
Artikel 5 |
|
|
Article 5 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Aktienbuch, Übertragungsbeschränkungen, Nominees |
1 |
Die Gesellschaft führt für die Namenaktien ein Aktienbuch, in welches die Eigentümer und Nutzniesser mit Namen und Vornamen (bei juristischen Personen die Firma), Kontaktdaten und Staatsangehörigkeit (bei juristischen Personen der Sitz) eingetragen werden. Wechselt eine im Aktienbuch eingetragene Person ihre Kontaktdaten, so hat sie dies der Gesellschaft mitzuteilen. Mitteilungen der Gesellschaft gelten als rechtsgültig erfolgt, wenn sie an die im Aktienbuch zuletzt eingetragenen Kontaktdaten des Aktionärs bzw. Zustellungsbevollmächtigten gesendet werden. |
Share Register, Transfer Restrictions, Nominees |
1 |
The Company shall maintain a share register that lists the surname, first name, contact information and citizenship (in the case of legal entities, the company name and company seat) of the holders and usufructuaries of the registered shares. A person recorded in the share register shall notify the Company of any change in contact information. Communications from the Company shall be deemed to have been validly made if sent to the shareholder's or authorized delivery agent's last registered contact information in the share register. |
|
2 |
Erwerber von Namenaktien werden auf Gesuch als Aktionäre mit Stimmrecht im Aktienbuch eingetragen, falls sie ausdrücklich erklären, diese Namenaktien im eigenen Namen und für eigene Rechnung erworben zu haben, keine Vereinbarung über die Rücknahme oder die Rückgabe entsprechender Aktien besteht und sie das mit den Aktien verbundene wirtschaftliche Risiko tragen. |
|
2 |
An acquirer of registered shares shall be recorded upon request in the share register as a shareholder with voting rights, if such acquirer expressly declares to have acquired the registered shares in his own name and for his own account, that there is no agreement on the redemption of the relevant shares and that they bear the economic risk associated with the shares. |
|
3 |
Der Verwaltungsrat trägt einzelne Personen, die im Eintragungsgesuch nicht ausdrücklich erklären, die Namenaktien auf eigene Rechnung zu halten (Nominees), mit Stimmrecht im Aktienbuch ein, wenn der Nominee mit dem Verwaltungsrat eine Vereinbarung über seine Stellung abgeschlossen hat und einer anerkannten Bank- oder Finanzaufsicht untersteht. |
|
3 |
The board of directors records persons who do not declare to hold the registered shares for their own account (Nominees) as shareholders with voting rights in the share register, if such Nominee has entered into an agreement regarding its position with the board of directors and is subject to a recognized banking or finance supervision. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
4 |
Der Verwaltungsrat kann nach Anhörung des eingetragenen Aktionärs oder Nominees Eintragungen im Aktienbuch mit Rückwirkung auf das Datum der Eintragung streichen, wenn diese durch falsche Angaben zustande gekommen sind. Der Betroffene muss über die Streichung informiert werden. |
|
4 |
After hearing the registered shareholder concerned, the board of directors may cancel the registration of such shareholder as a shareholder with voting rights in the share register with retroactive effect as of the date of registration, if such registration was made based on false or misleading information. The relevant shareholder shall be informed of the cancellation. |
|
5 |
Der Verwaltungsrat regelt die Einzelheiten und trifft die zur Einhaltung der vorstehenden Bestimmungen notwendigen Anordnungen. Er kann in besonderen Fällen Ausnahmen von der Nomineeregelung bewilligen. Er kann seine Aufgaben delegieren. |
|
5 |
The board of directors shall regulate the details and issue the instructions necessary for compliance with the preceding provisions. In special cases, it may grant exemptions from the rule concerning Nominees. The board of directors may delegate its duties. |
|
|
|
|
|
|
|
|
Artikel 6 |
|
|
Article 6 |
| Rechtsausübung |
1 |
Die Gesellschaft anerkennt nur einen Vertreter pro Aktie. |
Exercise of Rights |
1 |
The Company shall only accept one representative per share. |
|
2 |
Das Stimmrecht und die damit zusammenhängenden Rechte aus einer Namenaktie können der Gesellschaft gegenüber nur von einem Aktionär, Nutzniesser oder Nominee, der mit Stimmrecht im Aktienbuch eingetragen ist, ausgeübt werden. |
|
2 |
Voting rights and appurtenant rights associated therewith may be exercised in relation to the Company by a shareholder, usufructuary of shares or nominee only to the extent that such person is recorded in the share register as a shareholder with voting rights. |
|
|
|
|
|
|
|
|
Abschnitt 3:
Organe |
|
|
Section 3:
Corporate Bodies |
|
|
|
|
|
|
|
|
Artikel 7 |
|
|
Article 7 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Organe |
|
Die Organe der Gesellschaft sind:
(a) die Generalversammlung;
(b) der Verwaltungsrat;
(c) die Revisionsstelle.
|
Corporate Bodies |
|
The Company's bodies are:
(a) the general meeting of shareholders;
(b) the board of directors;
(c) the auditors.
|
|
|
|
|
|
|
|
|
A. Generalversammlung |
|
|
A. General Meeting of Shareholders |
|
|
|
|
|
|
|
|
Artikel 8 |
|
|
Article 8 |
| Befugnisse |
|
Oberstes Organ der Gesellschaft ist die Generalversammlung der Aktionäre. Ihr stehen folgende unübertragbare Befugnisse zu:
(a)die Festsetzung und Änderung der Statuten;
(b)die Wahl der Mitglieder des Verwaltungsrats, des Präsidenten des Verwaltungsrats, der Mitglieder des Vergütungsausschusses, des unabhängigen Stimmrechtsvertreters und der Revisionsstelle;
(c)die Genehmigung des Lageberichts und der Konzernrechnung;
(d)die Genehmigung der Jahresrechnung sowie die Beschlussfassung über die Verwendung des Bilanzgewinnes, insbesondere die Festsetzung der Dividende;
(e)die Festsetzung der Zwischendividende und die Genehmigung des dafür erforderlichen Zwischenabschlusses;
(f)die Beschlussfassung über die Rückzahlung der gesetzlichen Kapitalreserve;
(g)die Genehmigung der Vergütung des Verwaltungsrats und der Geschäftsleitung gemäss Art. 28 dieser Statuten;
(h)die Entlastung der Mitglieder des Verwaltungsrats und der mit der Geschäftsleitung betrauten Personen;
(i)die Dekotierung der Beteiligungspapiere der Gesellschaft; und
(j)die Beschlussfassung über die Gegenstände, die der Generalversammlung durch das Gesetz oder die Statuten vorbehalten sind.
|
Powers |
|
The general meeting of shareholders is the supreme corporate body of the Company. It has the following non-delegable powers:
(a)adoption and amendment of the Articles of Incorporation;
(b)election of the members of the board of directors, the chairman of the board of directors, the members of the compensation committee, the independent voting rights representative and the auditors;
(c)approval of the annual management report and the consolidated financial statements;
(d)approval of the annual financial statements and decision on the allocation of profits shown on the balance sheet, in particular with regard to dividends;
(e)the determination of interim dividends and the approval of the interim financial statements required for this purpose;
(f)the resolution on the repayment of the statutory capital reserve;
(g)approval of the compensation of the board of directors and of the executive management pursuant to Article 28 of these Articles of Incorporation;
(h)granting discharge to the members of the board of directors and the persons entrusted with the executive management;
(i)the delisting of the Company's equity securities; and
(j)passing of resolutions as to all matters reserved by law or under these Articles of Incorporation to the authority of the general meeting of shareholders.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Artikel 9 |
|
|
Article 9 |
| Ordentliche und ausserordentliche Generalversammlungen |
1 |
Die ordentliche Generalversammlung findet alljährlich innerhalb von sechs Monaten nach Schluss des Geschäftsjahres statt. |
Ordinary and Extraordinary General Meeting of Shareholders |
1 |
The ordinary general meeting of shareholders shall be held each year within six months after the close of the fiscal year of the Company. |
| 2 |
Ausserordentliche Generalversammlungen finden statt, wenn der Verwaltungsrat oder die Revisionsstelle es für angezeigt erachten oder wenn es eine Generalversammlung beschliesst. Darüber hinaus können Aktionäre, die zusammen mindestens 5 Prozent des Aktienkapitals oder der Stimmen vertreten, gemeinsam schriftlich unter Angabe des Verhandlungsgegenstandes und des Antrages, bei Wahlen der Namen der vorgeschlagenen Kandidaten, die Einberufung einer ausserordentlichen Generalversammlung verlangen. |
2 |
Extraordinary general meetings of shareholders shall be held when deemed necessary by the board of directors or the auditors. Furthermore, extraordinary general meetings of shareholders shall be convened upon resolution of a general meeting of shareholders or if this is requested by one or more shareholders who represent an aggregate of at least 5 percent of the share capital or votes and who submit a written request specifying the agenda items and the proposals, in case of elections the name of the proposed candidates. |
|
|
|
|
|
|
|
|
Artikel 10 |
|
|
Article 10 |
| Einberufung |
1 |
Die Generalversammlung wird durch den Verwaltungsrat, nötigenfalls die Revisionsstelle, spätestens 20 Tage vor der Versammlung einberufen. Das Einberufungsrecht steht auch den Liquidatoren zu. |
Notice |
1 |
Notice of a general meeting of shareholders shall be given by the board of directors or, if necessary, by the auditors, no later than twenty calendar days prior to the date of the general meeting of shareholders. The liquidators may also call the general meeting of shareholders. |
|
2 |
Die Einberufung erfolgt durch einmalige Bekanntmachung im Publikationsorgan der Gesellschaft. Namenaktionäre können überdies schriftlich orientiert werden. |
|
2 |
Notice of the general meeting of shareholders shall be given by way of a one-time announcement in the official means of publication of the Company. In addtion, shareholders of record may be informed by ordinary mail. |
|
3 |
Spätestens 20 Tage vor der ordentlichen Generalversammlung sind der Geschäftsbericht, der Vergütungsbericht, und die Revisionsberichte zugänglich zu machen. |
|
3 |
The annual report, the compensation report and the auditors' reports shall be made available to the shareholders no later than twenty calendar days prior to the annual general meeting of shareholders. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
4 |
In der Einberufung sind bekanntzugeben:
1. Datum, Beginn, Art und Ort der Generalversammlung;
2. die Verhandlungsgegenstände;
3. die Anträge des Verwaltungsrates samt kurzer Begründung;
4. gegebenenfalls die Anträge der Aktionäre samt kurzer Begründung; und
5. der Name und die Adresse des unabhängigen Stimmrechtsvertreters.
|
|
4 |
The notice shall include:
1. date, beginning, mode and venue of the general meeting of shareholders;
2. the agenda;
3. the proposals of the board of directors together with a brief statement of the reasons;
4. proposals of the shareholders, if any, together with a brief statement of the reasons; and
5. name and address of the independent voting rights representative.
|
|
|
|
|
|
|
|
|
Artikel 10a |
|
|
Article 10a |
| Tagungsort |
1 |
Der Verwaltungsrat bestimmt den Tagungsort der Generalversammlung, welche in der Schweiz oder im Ausland durchgeführt werden kann. |
Venue |
1 |
The board of directors shall determine the venue of the general meeting of shareholders, which may be held in Switzerland or abroad. |
|
2 |
Der Verwaltungsrat kann bestimmen, dass die Generalversammlung an verschiedenen Orten gleichzeitig durchgeführt wird, sofern die Voten der Teilnehmer unmittelbar in Bild und Ton an sämtliche Tagungsorte übertragen werden. |
|
2 |
The board of directors can determine that the general meeting of shareholders be held simultaneously at different locations, provided that the contributions of the participants are transmitted directly in video and audio to all venues. |
|
3 |
Der Verwaltungsrat kann vorsehen, dass die Generalversammlung auf elektronischem Weg ohne Tagungsort durchgeführt wird. |
|
3 |
The board of directors may also provide that the general meeting of shareholders will be held by electronic means without a venue. |
|
|
|
|
|
|
|
|
Artikel 11 |
|
|
Article 11 |
| Traktandierung |
1 |
Aktionäre, die alleine oder zusammen über mindestens 0.5 Prozent des Aktienkapitals oder der Stimmen verfügen, können die Traktandierung eines Verhandlungsgegenstandes oder die Aufnahme eines Antrages zu einem Verhandlungsgegenstand in die Einberufung der Generalversammlung verlangen. Ein solches Gesuch muss der Gesellschaft mindestens 45 Kalendertage vor der Versammlung schriftlich unter Angabe des Verhandlungsgegenstandes und des Antrags oder der Anträge zugehen. |
Agenda |
1 |
One or more shareholders whose combined shareholdings represent at least 0.5 percent of the share capital or votes may request that an item be included on the agenda of a general meeting of shareholders or that a proposal relating to an agenda item be included in the notice convening the general meeting of shareholders. Such a request must be received by the Company in writing at least 45 calendar days prior to the general meeting of shareholders, specifying the agenda item and the proposal or proposals. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2 |
Über Anträge zu nicht gehörig angekündigten Verhandlungsgegenständen kann die Generalversammlung keine Beschlüsse fassen; ausgenommen sind Anträge auf Einberufung einer ausserordentlichen Generalversammlung und auf Durchführung einer Sonderuntersuchung. |
|
2 |
No resolutions may be passed at a general meeting of shareholders concerning agenda items for which proper notice was not given. This provision shall not apply, however, to proposals made during a general meeting of shareholders to convene an extraordinary general meeting of shareholders or to initiate a special investigation. |
|
3 |
Zur Stellung von Anträgen im Rahmen der Verhandlungsgegenstände und zu Verhandlungen ohne Beschlussfassung bedarf es keiner vorgängigen Ankündigung. |
|
3 |
No previous notification shall be required for proposals concerning items included on the agenda and for debates as to which no vote is taken. |
|
|
|
|
|
|
|
|
Artikel 12 |
|
|
Article 12 |
| Vorsitz der Generalversammlung, Stimmenzähler, Protokoll |
1 |
Der Präsident des Verwaltungsrats führt den Vorsitz in der Generalversammlung. Bei seiner Abwesenheit führt der Vizepräsident des Verwaltungsrats den Vorsitz. Ist auch dieser abwesend, so wird der Vorsitzende durch den Verwaltungsrat gewählt. |
Acting Chair, Vote Counters, Minutes |
1 |
At the general meeting of shareholders, the Chairman of the board of directors or, in his absence, the Vice-Chairman or, in his absence, any other person designated by the board of directors shall take the chair. |
|
2 |
Der Vorsitzende bezeichnet einen Protokollführer und die Stimmenzähler, die nicht Aktionäre sein müssen. Das Protokoll ist vom Vorsitzenden und vom Protokollführer zu unterzeichnen. |
|
2 |
The acting chair of the general meeting of shareholders shall appoint the secretary and the vote counters, none of whom need be shareholders. The minutes of the general meeting of shareholders shall be signed by the acting chair and the secretary. |
|
3 |
Die Beschlüsse und Wahlergebnisse sind unter Angabe der genauen Stimmenverhältnisse innerhalb von 15 Kalendertagen nach der Generalversammlung auf elektronischem Weg zugänglich zu machen; jeder Aktionär kann verlangen, dass ihm das Protokoll innerhalb von 30 Kalendertagen nach der Generalversammlung zugänglich gemacht wird. |
|
3 |
The resolutions and election results shall be made available electronically within 15 calendar days after the general meeting of shareholders, stating the exact proportion of votes; each shareholder may request that the minutes be made available to him within 30 calendar days after the general meeting of shareholders. |
|
|
|
|
|
|
|
|
Artikel 13 |
|
|
Article 13 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Stimmrecht, Vertretung |
1 |
Jede mit Stimmrecht im Aktienbuch eingetragene Aktie berechtigt zu einer Stimme. |
Voting Rights, Representation |
1 |
Each share registered in the share register grants one vote. |
|
2 |
Der Verwaltungsrat erlässt die Verfahrensvorschriften über die Teilnahme und Vertretung an der Generalversammlung. Ein Aktionär kann sich an der Generalversammlung nur durch den unabhängigen Stimmrechtsvertreter (mittels schriftlicher oder elektronischer Vollmacht), seinen gesetzlichen Vertreter oder (mittels schriftlicher Vollmacht) durch einen anderen Bevollmächtigten, der nicht Aktionär zu sein braucht, vertreten lassen. Alle von einem Aktionär gehaltenen Aktien können nur von einer Person vertreten werden. |
|
2 |
The board of directors shall issue procedural rules regarding participation in and representation at the general meeting of shareholders. A shareholder may be represented only by the independent voting rights representative (unabhängiger Stimmrechtsvertreter) (by way of a written or electronic proxy), his legal representative or, by means of a written proxy, by any other proxy who need not be a shareholder. All shares held by one shareholder must be represented by only one representative. |
|
3 |
Die Generalversammlung wählt den unabhängigen Stimmrechtsvertreter für eine Amtsdauer bis zum Abschluss der nächsten ordentlichen Generalversammlung. Wiederwahl ist möglich. Hat die Gesellschaft aus irgendwelchen Gründen keinen unabhängigen Stimmrechtsvertreter, bezeichnet der Verwaltungsrat für die nächste stattfindende Generalversammlung einen unabhängigen Stimmrechtsvertreter. |
|
3 |
The general meeting of shareholders shall elect the independent voting rights representative at a general meeting of shareholders for a term of office extending until completion of the next ordinary general meeting of shareholders. Re-election is possible. If the company does not have an independent voting rights representative for whatever reason, the board of directors shall appoint the independent voting rights representative for the next meeting of shareholders. |
|
4 |
Der Verwaltungsrat regelt die Anforderungen an die Vollmachten und Weisungen an den unabhängigen Stimmrechtsvertreter. |
|
4 |
The board of directors shall issue the particulars for the proxy of and for providing instructions to the independent voting rights representative. |
|
|
|
|
|
|
|
|
Artikel 14 |
|
|
Article 14 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Beschlüsse, Wahlen |
1 |
Die Generalversammlung beschliesst und wählt, soweit das Gesetz und die Statuten es nicht anders bestimmen, mit der Mehrheit der vertretenen Aktienstimmen. |
Resolutions and Elections |
1 |
Unless otherwise required by law or these Articles of Incorporation, the general meeting of shareholders shall take resolutions and decide elections upon a majority of the votes represented at the general meeting of shareholders. |
|
2 |
Ein Beschluss der Generalversammlung, der mindestens zwei Drittel der vertretenen Stimmen und die Mehrheit der vertretenen Aktiennennwerte auf sich vereinigt, ist erforderlich für:
(a)die Änderung des Gesellschaftszweckes;
(b)die Zusammenlegung von Aktien;
(c)die Einführung von Stimmrechtsaktien;
(d)die Beschränkung der Übertragbarkeit von Namenaktien und die Aufhebung einer solchen Beschränkung;
(e)die Einführung eines bedingten Kapitals oder die Einführung eines Kapitalbands;
(f)die Kapitalerhöhung aus Eigenkapital, gegen Sacheinlage oder durch Verrechnung mit einer Forderung und die Gewährung von besonderen Vorteilen;
(g)die Einschränkung oder Aufhebung des Bezugsrechtes;
(h)die Einführung des Stichentscheids des Vorsitzenden in der Generalversammlung;
(i)die Dekotierung der Beteiligungspapiere der Gesellschaft
(j)die Verlegung des Sitzes der Gesellschaft;
(k)den Wechsel der Währung des Aktienkapitals;
(l)die Einführung einer statutarischen Schiedsklausel; und
(m)die Auflösung der Gesellschaft.
|
|
2 |
The approval of at least two-thirds of the votes and the majority of the par value of shares, each as represented at a general meeting of shareholders, shall be required for resolutions with respect to:
(a)The amendment or modification of the purpose of the Company;
(b)the combination of shares;
(c)the creation of shares with privileged voting rights;
(d)the restriction on the transferability of registered shares and the cancelation of such restriction;
(e)the introduction of conditional share capital or the introduction of a capital range;
(f)an increase of the share capital through the conversion of capital surplus, through contribution in kind, by set-off against a claim, or the granting of special privileges;
(g)the limitation or withdrawal of preemptive rights;
(h)the introduction of the casting vote of the acting chair in the general meeting of shareholders;
(i)the delisting of the Company's equity securities;
(j)the relocation of the registered office of the Company;
(k)the change of currency of the share capital;
(l)the introduction of an arbitration clause in the Articles of Incorporation; and
(m)the dissolution of the Company.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
3 |
Die Abstimmungen und Wahlen erfolgen offen, es sei denn, dass die Generalversammlung schriftliche Abstimmung respektive Wahl (einschliesslich elektronische Abstimmungsverfahren) beschliesst oder der Vorsitzende dies anordnet. |
|
3 |
Resolutions and elections shall be decided by a show of hands, unless a written ballot (including electronic voting systems) is resolved by the general meeting of shareholders or is ordered by the acting chair of the general meeting of shareholders. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
B. Verwaltungsrat |
|
|
B. Board of Directors |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Artikel 15 |
|
|
Article 15 |
| Anzahl der Verwaltungsräte |
|
Der Verwaltungsrat besteht aus mindestens 3 und höchstens 11 Mitgliedern. |
Number of Directors |
|
The board of directors shall consist of no less than 3 and no more than 11 members. |
|
|
|
|
|
|
|
|
Artikel 16 |
|
|
Article 16 |
| Wahl, Amtsdauer |
1 |
Die Mitglieder des Verwaltungsrats und der Präsident des Verwaltungsrats werden von der Generalversammlung einzeln für eine Amtsdauer bis zum Abschluss der nächsten ordentlichen Generalversammlung gewählt. Findet die ordentliche Generalversammlung später als sechs Monate nach Abschluss des Geschäftsjahres statt, so dauert die Amtsdauer dennoch bis zum Abschluss der ordentlichen Generalversammlung. |
Election, Term of Office |
1 |
The shareholders shall elect the members of the board of directors and the chair of the board of directors individually at a general meeting of shareholders for a term of office extending until completion of the next ordinary general meeting of shareholders. If the ordinary general meeting of shareholders is held more than six months after the end of the financial year, the term of office shall nevertheless continue until the end of the ordinary general meeting of shareholders. |
|
2 |
Die Mitglieder des Verwaltungsrats sind jederzeit wieder wählbar. |
|
2 |
Members of the board of directors may be re-elected at any time. |
|
3 |
Ist das Präsidium vakant, bezeichnet der Verwaltungsrat aus seiner Mitte einen neuen Präsidenten für eine Amtsdauer bis zum Abschluss der nächsten ordentlichen Generalsversammlung. |
|
3 |
If the office of the chair of the board of directors is vacant, the board of directors shall appoint the chair from among its members for a term of office extending until completion of the next ordinary general meeting of shareholders. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Artikel 17 |
|
|
Article 17 |
| Organisation des Verwaltungsrats, Ersatz der Auslagen |
1 |
Vorbehältlich der Wahl des Präsidenten des Verwaltungsrats und der Mitglieder des Vergütungsausschusses durch die Generalversammlung konstituiert sich der Verwaltungsrat selbst. Er kann aus seiner Mitte einen oder mehrere Vize-Präsidenten wählen sowie einen Sekretär bezeichnen, der nicht Mitglied des Verwaltungsrats zu sein braucht. |
Organization of the Board of Directors, Reimbursement of Expenses |
1 |
Except for the election of the chairman of the board of directors and the members of the compensation committee by the general meeting of shareholders, the board of directors shall constitute itself. It may elect from among its members one or several vice-chairmen and appoint a secretary who need not be a member of the board of directors. |
|
2 |
Der Verwaltungsrat ordnet im Übrigen im Rahmen von Gesetz und Statuten seine Organisation und Beschlussfassung durch ein Organisationsreglement. |
|
2 |
Subject to applicable law and these Articles of Incorporation, the board of directors shall establish the particulars of its organization in organizational regulations. |
|
3 |
Die Mitglieder des Verwaltungsrats haben Anspruch auf Ersatz ihrer im Interesse der Gesellschaft aufgewendeten Auslagen. |
|
3 |
The members of the board of directors shall be entitled to the reimbursement of all expenses incurred in the interests of the Company. |
|
|
|
|
|
|
|
|
Artikel 18 |
|
|
Article 18 |
| Einberufung, Beschlussfassung, Protokoll |
1 |
Sitzungen des Verwaltungsrats werden vom Präsidenten oder im Falle seiner Verhinderung vom Vize-Präsidenten oder einem anderen Mitglied des Verwaltungsrats einberufen, so oft dies als notwendig erscheint oder wenn ein Mitglied es schriftlich oder per E-Mail oder einer anderen Art der elektronischen Übermittlung unter Angabe der Gründe verlangt. Sitzungen können auch per Telefon- oder Videokonferenz durchgeführt werden. |
Invitation, Resolutions, Minutes |
1 |
The chairman or, should he be unable to do so, the vice-chairman or any other member of the board of directors shall convene meetings of the board of directors if and when the need arises or whenever a member indicating the reasons so requests in writing or via e-mail or another form of electronic communication. Meetings may also be held by telephone or video conference. |
|
2 |
Der Verwaltungsrat fasst seine Beschlüsse mit der Mehrheit der abgegebenen Stimmen. Der Vorsitzende hat den Stichentscheid. |
|
2 |
Resolutions of the board of directors shall be adopted upon a majority of the votes cast. In the event of a tie, the chairman shall have the casting vote. |
|
3 |
Zur Beschlussfähigkeit des Verwaltungsrats ist die Anwesenheit der Mehrheit seiner Mitglieder erforderlich. Kein Präsenzquorum ist erforderlich für die Anpassungs- und Feststellungsbeschlüsse des Verwaltungsrats im Zusammenhang mit Kapitalerhöhungen oder einem Wechsel der Währung des Aktienkapitals. |
|
3 |
In order to pass resolutions, at least a majority of the members of the board of directors must be present. No attendance quorum shall be required for confirmation or amendment resolutions of the board of directors in connection with capital increase or a change in the currency of the share capital. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
4 |
Beschlüsse können auch auf schriftlichem Weg oder in elektronischer Form gefasst werden, sofern nicht ein Mitglied mündliche Beratung verlangt. |
|
4 |
Resolutions may be passed by way of written consent or electronically, provided that no member requests oral deliberation. |
|
5 |
Die Beschlüsse sind in einem Protokoll festzuhalten, das vom Sitzungspräsidenten und dem Sekretär zu unterzeichnen ist. |
|
5 |
The resolutions shall be confirmed in the minutes, which are to be signed by the acting chair and the secretary. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Artikel 19 |
|
|
Article 19 |
| Befugnisse des Verwaltungsrates |
1 |
Der Verwaltungsrat kann in allen Angelegenheiten Beschluss fassen, die nicht nach Gesetz, Statuten oder Reglement einem anderen Organ der Gesellschaft übertragen sind. |
Powers of the Board of Directors |
1 |
The board of directors may pass resolutions with respect to all matters that are not reserved to the general meeting of shareholders or any other corporate body by law or under these Articles of Incorporation. |
|
2 |
Er hat folgende unübertragbare und unentziehbare Aufgaben:
(a)die Oberleitung der Gesellschaft und die Erteilung der nötigen Weisungen;
(b)die Festlegung der Organisation;
(c)die Ausgestaltung des Rechnungswesens, der Finanzkontrolle sowie der Finanzplanung;
(d)die Ernennung und Abberufung der mit der Geschäftsführung und der Vertretung betrauten Personen und die Regelung von deren Zeichnungsberechtigung;
(e)die Oberaufsicht über die mit der Geschäftsführung betrauten Personen, namentlich im Hinblick auf die Befolgung der Gesetze, Statuten, Reglemente und Weisungen;
(f)die Erstellung des Geschäftsberichts und des Vergütungsberichts sowie gegebenenfalls andere gesetzlich vorgeschriebene Berichte;
(g)die Vorbereitung der Generalversammlung und die Ausführung ihrer Beschlüsse;
(h)die Beschlussfassung über nachträgliche Leistung von Einlagen auf nicht vollständig liberierten Aktien und daraus folgende Statutenänderungen;
(i)die Beschlussfassung über die Veränderung des Aktienkapitals, soweit dies in der Kompetenz des Verwaltungsrates liegt, die Feststellung von Kapitalveränderungen, die Erstellung des Kapitalerhöhungsberichts und die Vornahme der entsprechenden Statutenänderungen (einschliesslich Löschungen);
(j)die gemäss Fusionsgesetz unübertragbaren und unentziehbaren Aufgaben und Befugnisse des Verwaltungsrats;
(k)die Einreichung eines Gesuchs um Nachlassstundung und die Benachrichtigung des Richters im Falle der Überschuldung;
(l)andere durch Gesetz oder Statuten dem Verwaltungsrat vorbehaltene Aufgaben und Befugnisse.
|
|
2 |
The board of directors has the following non-delegable and inalienable duties:
(a)the ultimate direction of the business of the Company and the issuance of the necessary instructions;
(b)the determination of the organization of the Company;
(c)the administration of accounting, financial control and financial planning;
(d)the appointment and removal of the persons entrusted with executive management and their representation of the Company;
(e)the ultimate supervision of the persons entrusted with management of the Company, specifically in view of their compliance with the law, these Articles of Incorporation, the regulations and directives;
(f)the preparation of the business report, the compensation report and other reports as required by law, if any;
(g)the preparation of the general meetings of shareholders as well as the implementation of the resolutions adopted by the general meetings of shareholders;
(h)the adoption of resolutions regarding the subsequent payment of capital with respect to non-fully paid up shares and the amendments to the Articles of Incorporation related thereto;
(i)the adoption of resolutions on the change of the share capital to the extent that such power is vested in the board of directors, the ascertainment of capital changes, the preparation of the report on the capital increase, and the respective amendments of the Articles of Incorporation (including deletions);
(j)the non-delegable and inalienable duties and powers of the board of directors pursuant to the Merger Act;
(k)the submission of a petition for debt-restructuring moratorium and the notification of the court if liabilities exceed assets;
(l)any other matter reserved to the board of directors by the law or the Articles of Incorporation.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
3 |
Im Übrigen kann der Verwaltungsrat die Geschäftsführung sowie die Vertretung der Gesellschaft im Rahmen der gesetzlichen Bestimmungen durch Erlass eines Organisationsreglements ganz oder teilweise an einzelne oder mehrere seiner Mitglieder oder an andere natürliche Personen übertragen. |
|
3 |
The board of directors may delegate the executive management of the Company in whole or in part to one or several individual directors or to individuals other than directors pursuant to organizational regulations. |
|
|
|
|
|
|
|
|
C. Der Vergütungsausschuss |
|
|
C. The Compensation Committee |
|
|
|
|
|
|
|
|
Artikel 20 |
|
|
Article 20 |
| Anzahl der Mitglieder |
|
Der Vergütungsausschuss besteht aus mindestens zwei Mitgliedern. |
Number of Members |
|
The compensation committee shall consist of no less than two members. |
|
|
|
|
|
|
|
|
Artikel 21 |
|
|
Article 21 |
| Wahl und Amtsdauer |
1 |
Die Mitglieder des Vergütungsausschusses werden von der Generalversammlung einzeln für eine Amtsdauer bis zum Abschluss der nächsten ordentlichen Generalversammlung gewählt. Wählbar sind nur Mitglieder des Verwaltungsrates. |
Election and Term of Office |
1 |
The general meeting of shareholders shall elect the members of the compensation committee individually for a term of office extending until completion of the next ordinary general meeting of shareholders. Only members of the board of directors may be elected. |
| 2 |
Die Mitglieder des Vergütungsausschusses sind jederzeit wieder wählbar. |
2 |
Members of the compensation committee may be re-elected at any time. |
|
3 |
Bei Vakanzen im Vergütungsausschuss kann der Verwaltungsrat aus seiner Mitte Ersatzmitglieder für eine Amtsdauer bis zum Abschluss der nächsten ordentlichen Generalversammlung bezeichnen. |
|
3 |
If there are vacancies on the compensation committee, the board of directors shall appoint from among its members substitutes for a term of office extending until completion of the next ordinary general meeting of shareholders. |
|
|
|
|
|
|
|
|
Artikel 22 |
|
|
Article 22 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Organisation des Vergütungsausschusses |
1 |
Der Vergütungsausschuss konstituiert sich unter Vorbehalt der Kompetenzen der Generalversammlung und des Verwaltungsrats selbst. Der Verwaltungsrat bezeichnet den Vorsitzenden des Vergütungsausschusses. |
Organization of the Compensation Committee |
1 |
The compensation committee constitutes itself subject to the powers of the general meeting of shareholders and the board of directors. The board of directors shall elect the chair of the compensation committee. |
|
2 |
Im Übrigen erlässt der Verwaltungsrat ein Reglement über die Organisation und Beschlussfassung des Vergütungsausschusses. |
|
2 |
The board of directors shall establish the particulars of the organization and adoption of resolutions of the compensation committee in regulations. |
|
|
|
|
|
|
|
|
Artikel 23 |
|
|
Article 23 |
| Befugnisse des Vergütungsausschusses |
1 |
Der Vergütungsausschuss unterstützt den Verwaltungsrat bei der Festsetzung und Überprüfung der Vergütungsstrategie und -richtlinien sowie bei der Vorbereitung der Anträge zuhanden der Generalversammlung betreffend die Vergütung des Verwaltungsrats und der Geschäftsleitung und kann dem Verwaltungsrat Anträge zu weiteren Vergütungsfragen unterbreiten. |
Powers of the Compensation Committee |
1 |
The compensation committee shall support the board of directors in establishing and reviewing the compensation strategy and guidelines as well as in preparing the proposals to the general meeting of shareholders regarding the compensation of the board of directors and of the executive management, and may submit proposals to the board of directors in other compensation-related issues. |
|
2 |
Der Verwaltungsrat legt in einem Reglement fest, für welche Funktionen des Verwaltungsrats und der Geschäftsleitung der Vergütungsausschuss dem Verwaltungsrat Vorschläge für die Leistungswerte, Zielwerte und die Vergütung unterbreitet und für welche Funktionen er selbst im Rahmen der Statuten und der vom Verwaltungsrat erlassenen Vergütungsrichtlinien die Leistungswerte, Zielwerte und die Vergütung festsetzt. |
|
2 |
The board of directors shall determine in regulations for which positions of the board of directors and of the executive management, the compensation committee shall submit proposals for the performance metrics, target values and the compensation to the board of directors, and for which positions it shall itself determine, in accordance with the Articles of Incorporation and the compensation guidelines established by the board of directors, the performance metrics, target values and the compensation. |
|
3 |
Der Verwaltungsrat kann dem Vergütungsausschuss weitere Aufgaben zuweisen, die in einem Reglement festgehalten werden. |
|
3 |
The board of directors may determine in regulations to delegate further authorities and duties to the compensation committee. |
|
|
|
|
|
|
|
|
D. Die Revisionsstelle |
|
|
D. Auditors |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Artikel 24 |
|
|
Article 24 |
| Wahl, Amtsdauer |
1 |
Die Generalversammlung wählt die Revisionsstelle. |
Election, Term of Office |
1 |
The auditors shall be elected by the general meeting of shareholders. |
|
2 |
Die Revisionsstelle wird von der Generalversammlung für eine Amtsdauer eines Geschäftsjahres gewählt. Ihre Amtszeit endet mit der Genehmigung der Jahresrechnung für das betreffende Geschäftsjahr durch die Generalversammlung. Wiederwahl ist möglich. |
|
2 |
The shareholders shall elect the auditors at a general meeting of shareholders for a term of office extending one financial year. Their term of office ends with the approval of the annual financial statements of the respective financial year by the general meeting of shareholders. Re-election is possible. |
|
|
|
|
|
|
|
|
Artikel 25 |
|
|
Article 25 |
| Prüfungs-, Berichterstattungspflicht |
|
Die Revisionsstelle nimmt ihre Prüfungs- und Berichterstattungspflichten in Übereinstimmung mit dem Gesetz wahr. |
Duty of Auditing and Reporting |
|
The auditors shall perform their duties to audit and report in accordance with the law. |
|
|
|
|
|
|
|
|
Artikel 26 |
|
|
Article 26 |
| Besondere Abklärungen, Zwischenrevisionen |
|
Der Verwaltungsrat kann die Revisionsstelle jederzeit beauftragen, besondere Abklärungen, insbesondere Zwischenrevisionen, durchzuführen und darüber Bericht zu erstatten. |
Special Audits, Interim Audits |
|
The board of directors may at any time request the auditors to conduct special audits, including interim audits, and to submit a respective report. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Abschnitt 4:
Vergütung der Mitglieder des Verwaltungsrates und der Geschäftsleitung |
|
|
Section 4:
Compensation of the Board of Directors and the Executive Management |
|
|
Artikel 27 |
|
|
Article 27 |
| Grundsätze der Vergütungen |
1 |
Die Vergütung der Mitglieder des Verwaltungsrats kann fixe und variable Vergütungselemente umfassen. Die Gesamtvergütung berücksichtigt Funktion und Verantwortungsstufe des Empfängers. |
General Compensation Principles |
1 |
Compensation of the members of the board of directors may consist of fixed and variable compensation. Total compensation shall take into account the position and level of responsibility of the recipient. |
|
2 |
Die Vergütung der Mitglieder der Geschäftsleitung besteht aus fixen und variablen Vergütungselementen. Die fixe Vergütung umfasst das Basissalär und weitere nicht-variable Vergütungselemente. Die variable Vergütung kann kurzfristige und langfristige variable Vergütungselemente umfassen. |
|
2 |
Compensation of the members of the executive management consists of fixed and variable compensation elements. Fixed compensation comprises the base salary and other non-variable compensation elements. Variable compensation may comprise short-term and long-term variable compensation elements. |
|
3 |
Die kurzfristigen variablen Vergütungselemente orientieren sich an Leistungswerten, die das Erreichen von operativen, strategischen, finanziellen oder anderen Zielen, das Ergebnis der Gesellschaft, des Konzerns oder Teilen davon und/oder individuelle Ziele berücksichtigen, und deren Erreichung sich in der Regel während eines einjährigen Zeitraums bemisst. Je nach erreichter Leistung kann sich die Vergütung auf einen vordefinierten Multiplikator des Zielwerts belaufen. |
|
3 |
Short-term variable compensation elements shall be governed by performance metrics that take into account the achievement of operational, strategic, financial or other objectives, the results of the Company, the group or parts thereof and/or individual targets, and achievement of which is generally measured during a one-year period. Depending on achieved performance, the compensation may amount to a multiplier of target level. |
|
4 |
Die langfristigen variablen Vergütungselemente orientieren sich an Leistungswerten, welche die Entwicklung des Aktienkurses oder Aktienergebnisses absolut oder im Verhältnis zu Vergleichsgruppen oder Indices und/oder das Ergebnis der Gesellschaft, des Konzerns oder Teilen davon und/oder das Erreichen von operativen, strategischen, finanziellen oder anderen Zielen absolut oder im Vergleich zum Markt, anderen Unternehmen oder vergleichbaren Richtgrössen und/oder Elemente zwecks Mitarbeiterbindung berücksichtigen. Die Zielerreichung bemisst sich in der Regel während eines mehrjährigen Zeitraums, sowie an Elementen zwecks Mitarbeiterbindung. Je nach erreichter Leistung kann sich die Vergütung auf einen vordefinierten Multiplikator des Zielwerts belaufen. |
|
4 |
Long-term variable compensation elements shall be governed by performance metrics that take into account the development of the share price or share performance in absolute terms or in relation to peer groups or indices and/or the results of the Company, the group or parts thereof and/or the achievement of operational, strategic, financial or other objectives in absolute terms or in relation to the market, other companies or comparable benchmarks and/or retention elements. An achievement of the objectives is generally measured over a period of several years. Depending on achieved performance, the compensation may amount to a multiplier of target level. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
5 |
Der Verwaltungsrat oder, soweit an ihn delegiert, der Vergütungsausschuss legen Leistungs- und Zielwerte sowie deren Gewichtung und Erreichung fest. |
|
5 |
The board of directors or, to the extent delegated to it, the compensation committee shall determine the performance metrics and target levels of the short- and long-term variable compensation elements, as well as their achievement. |
|
6 |
Die Vergütung kann in der Form von Geld, Aktien oder Sach- oder Dienstleistungen ausgerichtet werden werden; Vergütung der Mitglieder der Geschäftsleitung kann zusätzlich in der Form von aktienbasierten Instrumenten oder Einheiten ausgerichtet werden. Der Verwaltungsrat oder, soweit an ihn delegiert, der Vergütungsausschuss legen Zuteilungs-, Vesting-, Ausübungs- und Verfallsbedingungen fest. Sie können insbesondere vorsehen, dass aufgrund des Eintritts im Voraus bestimmter Ereignisse, wie eines Kontrollwechsels oder der Beendigung des Arbeits- oder Mandatsverhältnisses, Vesting-, Ausübungs- und Verfallsbedingungen weitergelten, verkürzt oder aufgehoben werden, Vergütungen unter der Annahme der Erreichung von Zielwerten ausgerichtet werden oder Vergütungen verfallen. Die Gesellschaft kann die erforderlichen Aktien auf dem Markt erwerben, aus Beständen eigener Aktien entnehmen oder unter Verwendung von bedingtem oder genehmigtem Kapital bereitstellen. |
|
6 |
Compensation may be paid in the form of cash, shares, or in the form of other types of benefits benefits; for the executive management, compensation may in addition be paid in the form of share-based instruments or units. The board of directors or, to the extent delegated to it, the compensation committee shall determine grant, vesting, exercise and forfeiture conditions. In particular, they may provide for continuation, acceleration or removal of vesting, exercise and forfeiture conditions, for payment or grant of compensation based upon assumed target achievement, or for forfeiture, in each case in the event of pre-determined events such as a change-of-control or termination of an employment or mandate agreement. The Company may procure the required shares through purchases in the market, from treasury shares or by using contingent or authorized share capital. |
|
7 |
Die Vergütung kann durch die Gesellschaft oder durch von ihr kontrollierte Unternehmen ausgerichtet werden. |
|
7 |
Compensation may be paid by the Company or companies controlled by it. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Artikel 28 |
|
|
Article 28 |
| Genehmigung der Vergütungen |
1 |
Die Generalversammlung genehmigt die Anträge des Verwaltungsrats in Bezug auf die maximalen Gesamtbeträge der
(a)Vergütung des Verwaltungsrats für die kommende Amtsdauer; und
(b)der fixen Vergütung der Geschäftsleitung für die Periode vom 1. Juli des laufenden bis zum 30. Juni des folgenden Jahres; und
(c)der variablen Vergütungselemente der Geschäftsleitung für das laufende Geschäftsjahr.
|
Approval of Compensation |
1 |
The general meeting of shareholders shall approve the proposals of the board of directors in relation to the maximum aggregate amounts of:
(a)the compensation of the board of directors for the next term of office; and
(b)of the fixed compensation of the executive management for the period of July 1 of the current year until June 30 of the following year; and
(c)of the variable compensation elements of the executive management for the current financial year.
|
|
2 |
Der Verwaltungsrat kann der Generalversammlung abweichende, zusätzliche oder bedingte Anträge in Bezug auf die maximalen Gesamtbeträge, mehrere maximale Teilbeträge für die gleichen oder andere Zeitperioden und/oder einzelne Vergütungselemente und/oder in Bezug auf Zusatzbeträge für besondere Vergütungselemente zur Genehmigung vorlegen. |
|
2 |
The board of directors may submit for approval by the general meeting of shareholders deviating, additional or conditional proposals relating to the maximum aggregate amount or maximum partial amounts for the same or different periods and/or specific compensation components and/or in relation to additional amounts for specific compensation components. |
|
3 |
Die Vergütung kann vor der Genehmigung durch die Generalversammlung unter Vorbehalt der nachträglichen Genehmigung ausgerichtet werden. |
|
3 |
Compensation may be paid out prior to approval by the general meeting of shareholders subject to subsequent approval. |
|
4 |
Genehmigt die Generalversammlung einen Antrag des Verwaltungsrats nicht, setzt der Verwaltungsrat den entsprechenden (maximalen) Gesamtbetrag oder (maximale) Teilbeträge unter Berücksichtigung aller relevanten Faktoren neu fest und unterbreitet den oder die so festgesetzten Beträge der gleichen Generalversammlung, einer ausserordentlichen Generalversammlung oder der nächsten ordentlichen Generalversammlung zur Genehmigung. |
|
4 |
If the general meeting of shareholders does not approve a proposal of the board of directors, the board of directors newly determines the maximum aggregate amount or maximum partial amounts taking into account all relevant factors and submits such amounts for approval to the same general meeting of shareholders, to an extraordinary general meeting of shareholders or to the next ordinary general meeting of shareholders. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
5 |
Werden variable Vergütungen prospektiv genehmigt, legt der Verwaltungsrat der Generalversammlung den Vergütungsbericht zur Konsultativabstimmung vor. |
|
5 |
If variable compensation is approved prospectively, the board of directors shall submit the compensation report to the general meeting of shareholders for a consultative vote. |
|
|
|
|
|
|
|
|
Artikel 29 |
|
|
Article 29 |
| Zusatzbetrag |
1 |
Die Gesellschaft oder von ihr kontrollierte Gesellschaften sind ermächtigt, Mitgliedern der Geschäftsleitung, die während einer Periode, für welche die Vergütung der Geschäftsleitung bereits genehmigt ist, in die Geschäftsleitung eintreten oder befördert werden, einen Zusatzbetrag auszurichten, sofern der für die betreffende Periode bereits genehmigte Gesamtbetrag für deren Vergütung nicht ausreicht. |
Supplementary Amount |
1 |
The Company or companies under its control shall be authorized to pay a supplementary amount of compensation ratified by the shareholders at a general meeting of shareholders to members of the executive management who joined or were promoted during a compensation period for which the maximum aggregate amount of compensation has already been approved, but is insufficient to cover compensation of such members of the executive management. |
|
2 |
Der Zusatzbetrag darf je Vergütungsperiode je Mitglied 50% des letzten genehmigten maximalen Gesamtbetrags der Vergütung der Geschäftsleitung nicht übersteigen. |
|
2 |
The supplementary amount per compensation period per member shall not exceed 50% of the maximum aggregate amount of compensation of the executive management last approved. |
|
|
|
|
|
|
|
|
Abschnitt 5:
Verträge mit Mitgliedern des Verwaltungsrats und der Geschäftsleitung |
|
|
Section 5:
Agreements regarding Compensation with Members of the Board of Directors and the Executive Management |
|
|
|
|
|
|
|
|
Artikel 30 |
|
|
Article 30 |
| Verträge mit Mitgliedern des Verwaltungsrats und der Geschäftsleitung |
1 |
Die Gesellschaft oder von ihr kontrollierte Gesellschaften können mit Mitgliedern des Verwaltungsrats befristete oder unbefristete Verträge über deren Mandat und Vergütung abschliessen. Dauer und Beendigung richten sich nach Amtsdauer und Gesetz. |
Agreements with Members of the Board of Directors and the Executive Management |
1 |
The Company or companies under its control may enter into mandate or other agreements with the members of the board of directors regarding their compensation as directors for a fixed term or for an indefinite term. The duration and termination are subject to term of office and the law. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2 |
Die Gesellschaft oder von ihr kontrollierte Gesellschaften können mit Mitgliedern der Geschäftsleitung befristete oder unbefristete Arbeitsverträge abschliessen. Befristete Arbeitsverträge haben eine Höchstdauer von einem Jahr. Eine Erneuerung ist zulässig. Unbefristete Verträge haben eine Kündigungsfrist von maximal einem Jahr. |
|
2 |
The Company or companies under its control may enter into employment agreements with the members of the executive management for a fixed term or for an indefinite term. The duration of fixed term agreements may not exceed one year. A renewal of a fixed term agreement is permissible. Agreements for an indefinite term may have a termination notice period of a maximum of one year. |
|
3 |
Die Gesellschaft oder von ihr kontrollierte Gesellschaften können mit Mitgliedern der Geschäftsleitung Konkurrenzverbote für die Zeit nach Beendigung des Arbeitsverhältnisses vereinbaren. Deren Dauer soll zwei Jahre nicht übersteigen. Zur Abgeltung eines solchen Konkurrenzverbots darf eine Entschädigung pro Jahr ausgerichtet werden, deren Höhe die letzte Gesamtjahresvergütung des betreffenden Mitglieds der Geschäftsleitung und in keinem Fall den Durchschnitt der Vergütungen der letzten drei Geschäftsjahre übersteigen. |
|
3 |
The Company or companies under its control may enter into non-competition agreements with members of the executive management for the period after the termination of the employment agreement. The duration of any such non-competition undertaking by a member of the executive management shall not exceed two years, and the consideration paid for a non-competition undertaking shall not exceed the sum of the total annual compensation of the respective member of the executive management last paid and in no event exceed the average of the compensation of the last three financial years. |
|
|
|
|
|
|
|
|
Abschnitt 6:
Darlehen, Kredite und Vorsorgeleistungen an die Mitglieder des Verwaltungsrats und der Geschäftsleitung |
|
|
Section 6:
Loans, Credits, Post-Retirement Benefits to members of the Board of Directors and the Executive Management |
|
|
|
|
|
|
|
|
Artikel 31 |
|
|
Article 31 |
| Darlehen und Kredite |
|
Kredite an Mitglieder des Verwaltungsrats und der Geschäftsleitung dürfen von der Gesellschaft oder von ihr kontrollierten Gesellschaften nur zu Marktbedingungen und nur solange ausgerichtet werden, als die Gesamtsumme der insgesamt ausstehenden Kredite an dieses Mitglied des Verwaltungsrats oder der Geschäftsleitung einschliesslich der zu gewährenden Kredite das Zweifache der letztmalig an dieses Mitglied bezahlten oder erstmaligen Jahresvergütung nicht übersteigt. |
Loans and Credits |
|
Credits to members of the board of directors and the executive management can solely be granted at standard market rates and the aggregate amount of credit to the member of the board of directors or executive management may not exceed double the total annual compensation of the respective member of the executive management last paid or payable for the first time. |
|
|
|
|
|
|
|
|
Artikel 32 |
|
|
Article 32 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Vorsorgeleistungen ausserhalb der beruflichen Vorsorge |
|
Vorbehältlich der Genehmigung durch die Generalversammlung gemäss Artikel 28 dieser Statuten können die Gesellschaft oder von ihr kontrollierte Gesellschaften an Mitglieder des Verwaltungsrates und der Geschäftsleitung Vorsorgeleistungen ausserhalb der beruflichen Vorsorge ausrichten, soweit solche Vorsorgeleistungen 100% der letztmalig an dieses Mitglied bezahlten Jahresvergütung nicht übersteigen. Im Fall von Kapitalabfindungen wird der Wert aufgrund anerkannter versicherungsmathematischer Methoden ermittelt. |
Post-Retirement Benefits beyond Occupational Benefit Scheme |
|
Subject to the approval by the meeting of shareholders pursuant to Article 28 of these Articles of Incorporation, the Company or companies under its control may grant to members of the board of directors or the executive management post-retirement benefits beyond the occupational benefit scheme, if such post-retirement benefits do not exceed 100% of the total annual compensation of the respective member last paid. In case of capital settlements, the value is determined by recognized actuary methods. |
|
|
|
|
|
|
|
|
Abschnitt 7:
Mandate ausserhalb des Konzerns |
|
|
Section 7:
Mandates Outside the Group |
|
|
|
|
|
|
|
|
Artikel 33 |
|
|
Article 33 |
| Mandate ausserhalb des Konzerns |
1 |
Kein Mitglied des Verwaltungsrates kann mehr als 15 zusätzliche Mandate wahrnehmen, wovon nicht mehr als 4 in börsenkotierten Unternehmen. |
Mandates Outside the Group |
1 |
No member of the board of directors may hold more than 15 additional mandates of which no more than 4 may be in listed companies. |
|
2 |
Kein Mitglied der Geschäftsleitung kann mehr als 5 zusätzliche Mandate wahrnehmen, wovon nicht mehr als 1 in einem börsenkotierten Unternehmen. Jedes dieser Mandate bedarf der Genehmigung durch den Präsidenten des Verwaltungsrates. Die Mitglieder der Geschäftsleitung dürfen keine Verwaltungsratsmandate in anderen börsenkotierten Unternehmen wahrnehmen. |
|
2 |
No member of the executive management may hold more than 5 additional mandates of which no more than 1 may be in a listed company. Each of these mandates is subject to the approval by the Chairperson of the board of directors. Members of the executive management are not allowed to hold chairs of the board of directors of other listed companies. |
|
3 |
Die folgenden Mandate fallen nicht unter die Beschränkungen gemäss Absatz 1 und 2 dieses Artikels: |
|
3 |
The following mandates shall not be subject to the limitations set forth in paragraphs 1 and 2 of this Article: |
|
|
(a) Mandate in Unternehmen, die durch die Gesellschaft kontrolliert werden oder die Gesellschaft kontrollieren; |
|
|
(a) mandates in companies which are controlled by the Company or which control the Company; |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(b) Mandate, die auf Anordnung der Gesellschaft oder von ihr kontrollierten Gesellschaften wahrgenommen werden. Kein Mitglied des Verwaltungsrates oder der Geschäftsleitung kann mehr als 10 solche Mandate wahrnehmen; und |
|
|
(b) mandates held at the request of the Company or companies controlled by it. No member of the board of directors or of the executive management shall hold more than 10 such mandates; and |
|
|
(c) Mandate in Vereinen, Verbänden, Stiftungen, Trusts, Personalfürsorgestiftungen, Bildungseinrichtungen und ähnlichen Organisationen. Kein Mitglied des Verwaltungsrates oder der Geschäftsleitung kann mehr als 10 solche Mandate wahrnehmen. |
|
|
(c) mandates in associations, professional or trade associations, foundations, trusts, employee welfare foundations, educational institutions, and similar organizations. No member of the board of directors or of the executive management shall hold more than 10 such mandates. |
|
4 |
Als Mandate gelten Mandate in vergleichbaren Funktionen bei anderen Unternehmen mit wirtschaftlichem Zweck. Mandate in verschiedenen Rechtseinheiten, die unter einheitlicher Kontrolle oder gleicher wirtschaftlicher Berechtigung stehen, gelten als 1 Mandat. |
|
4 |
Mandates shall mean mandates in comparable functions at other enterprises with an economic purpose. Mandates in different legal entities that are under joint control or same beneficial ownership are deemed 1 mandate. |
|
|
|
|
|
|
|
|
Abschnitt 8:
Geschäftsjahr, Gewinnverteilung |
|
|
Section 8:
Fiscal Year, Profit Allocation |
|
|
|
|
|
|
|
|
Artikel 34 |
|
|
Article 34 |
| Geschäftsjahr |
|
Das Geschäftsjahr der Gesellschaft wird vom Verwaltungsrat festgesetzt. |
Fiscal Year |
|
The board of directors determines the fiscal year. |
|
|
|
|
|
|
|
|
Artikel 35 |
|
|
Article 35 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Verteilung des Bilanzgewinnes, Reserven |
1 |
Über den Bilanzgewinn verfügt die Generalversammlung im Rahmen der gesetzlichen Vorschriften. Der Verwaltungsrat unterbreitet ihr seine Anträge. |
Allocation of Profits,
Reserves |
1 |
The profit shown on the annual statutory balance sheet shall be allocated by the general meeting of shareholders in accordance with applicable law. The board of directors shall submit its proposals to the general meeting of shareholders. |
|
2 |
Neben der gesetzlichen Reserve kann die Generalversammlung weitere Reserven schaffen. |
|
2 |
Further reserves may be taken in addition to the reserves required by law by the general meeting of shareholders. |
|
3 |
Dividenden, die während fünf Jahren von ihrem Verfalltag an nicht bezogen worden sind, fallen der Gesellschaft zu und werden der allgemeinen Reserve zugeteilt. |
|
3 |
Dividends that have not been collected within five years after their payment date shall enure to the Company and be allocated to the general statutory reserves. |
|
|
|
|
|
|
|
|
Abschnitt 9:
Auflösung, Liquidation |
|
|
Section 9:
Winding-Up and Liquidation |
|
|
|
|
|
|
|
|
Artikel 36 |
|
|
Article 36 |
| Auflösung, Liquidation |
1 |
Die Generalversammlung kann jederzeit die Auflösung und Liquidation der Gesellschaft nach Massgabe der gesetzlichen und statutarischen Vorschriften beschliessen. |
Winding-Up, Liquidation |
1 |
The general meeting of shareholders may at any time resolve on the winding-up and liquidation of the Company pursuant to applicable law and the provisions set forth in these Articles of Incorporation. |
|
2 |
Die Liquidation wird durch den Verwaltungsrat durchgeführt, sofern sie nicht durch die Generalversammlung anderen Personen übertragen wird. |
|
2 |
The liquidation shall be effected by the board of directors, unless the general meeting of shareholders shall appoint other persons as liquidators. |
|
3 |
Die Liquidation der Gesellschaft erfolgt nach Massgabe der Art. 742 ff. OR. Die Liquidatoren sind ermächtigt, Aktiven (Grundstücke eingeschlossen) auch freihändig zu verkaufen. |
|
3 |
The liquidation of the Company shall be effectuated pursuant to art. 742 et seq. CO. The liquidators are authorized to sell assets (including real estate) in the open market. |
|
4 |
Nach erfolgter Tilgung der Schulden wird das Vermögen unter den Aktionären nach Massgabe der einbezahlten Beträge verteilt. |
|
4 |
Upon discharge of all liabilities, the assets of the Company shall be distributed to the shareholders pursuant to the amounts paid in. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Abschnitt 10:
Mitteilungen, Bekanntmachungen |
|
|
Section 10:
Communications, Announcements |
|
|
|
|
|
|
|
|
Artikel 37 |
|
|
Article 37 |
| Mitteilungen, Publikationsorgan |
1 |
Publikationsorgan der Gesellschaft ist das Schweizerische Handelsamtsblatt. Der Verwaltungsrat kann weitere Publikationsorgane bezeichnen. |
Communications, Official Means of Publication |
1 |
The official means of publication of the Company shall be the Swiss Official Gazette of Commerce. The board of directors may designate additional means of publication. |
|
2 |
Mitteilungen der Gesellschaft an die Aktionäre können nach Wahl des Verwaltungsrates gültig durch Publikation im Schweizerischen Handelsamtsblatt oder in einer Form, die den Nachweis durch Text ermöglicht, erfolgen. |
|
2 |
Notices by the Company to the shareholders may, at the election of the board of directors, be validly given by publication in the Swiss Official Gazette of Commerce or in a form that allows proof by text. |
EX-4.6
3
a46molecularpartners-resea.htm
EX-4.6
Document
Exhibit 4.6
[***] = CERTAIN CONFIDENTIAL INFORMATION CONTAINED IN THIS DOCUMENT, MARKED BY [***], HAS BEEN OMITTED BECAUSE IT IS BOTH (I) NOT MATERIAL AND (II) IS THE TYPE THAT THE REGISTRANT TREATS AS PRIVATE OR CONFIDENTIAL.
RESEARCH AND DEVELOPMENT COLLABORATION
AND OPTION AGREEMENT
This Research and Development Collaboration and Option Agreement (this “Agreement”) is made and entered into effective as of 5 January 2024 (the “Effective Date”) by and between Orano Med, a French simplified joint-stock company, with registered office at 125 avenue de Paris, 92320 Chatillon, France (“Orano Med”), and Molecular Partners, a Swiss corporation, with registered office at Wagistrasse 14, 8952 Schlieren, Switzerland (“Molecular Partners”). Molecular Partners and Orano Med may be referred to herein individually as a “Party” or collectively as the “Parties”.
RECITALS
WHEREAS, Orano Med has developed experience and know-how in the field of targeted alpha-emitter therapies using a lead-212 isotope (“[***]”), production of high-purity [***], design and production of chelating agents as well as transportation of such radionuclide, and wishes to extend the range of therapies using [***].
WHEREAS, Molecular Partners has developed, owns or otherwise controls know-how, experience and proprietary information pertaining to designed repeat proteins, including DARPin proteins, and their research, development, clinical use, and commercialization.
WHEREAS, the Parties have entered into a research and development collaboration dated 20 July 2022 (“R&D Agreement”) to conduct certain pre-clinical research on the combination of Molecular Partners’ DARPin proteins and Orano Med’s [***] aimed at a first target – DLL3 – and potentially additional or substitute targets, and the Parties’ collaboration under such R&D Agreement is on-going.
WHEREAS, Molecular Partners and Orano Med wish to build on and extend their collaboration with the (a) short-term aim to bring a DLL3-DARPin-[***] candidate to clinical proof of concept to validate the approach and on-going collaboration, (b) the mid-term aim to both reach late-stage development with the target DLL3, and build a pipeline of DARPin-[***] candidates, and (c) the long-term aim to achieve successful commercialization of DARPin-[***] based radiopharmaceutical products.
NOW, THEREFORE, in consideration of the foregoing premises and the mutual promises and covenants contained herein, the Parties agree as follows:
Section 1
DEFINITIONS
The terms in this Agreement with initial letters capitalized, whether used in the singular or the plural, shall have the meaning set forth below or, if not listed below, the meaning designated in places throughout this Agreement.
1.1“[***]” shall have the meaning set forth in the Recitals.
1.2“Acquiror Group” has meaning set forth in Section 2.5(a) of this Agreement.
1.3“Additional Candidates” means [***], which may be included to this Agreement as Collaboration Products pursuant to Section 4.2, in such delivery form and for such indications as the Parties may agree during the Term.
1.4“Affiliates” means, with respect to any Person, another Person which controls, is controlled by, or is under common control with such Person, but only when such control exists. With respect to this Section, a Person shall be deemed to control another Person if such Person possesses, directly or indirectly, the power to direct or cause the direction of the management and policies of such other Person, whether through the ownership of voting securities, by contract, or otherwise. Without limiting the generality of the foregoing, a Person shall be deemed to control another Person if any of the following conditions is met: (a) in the case of corporate entities, direct or indirect ownership of fifty percent (50%) or more of the stock or shares having the right to vote for the election of directors, and (b) in the case of non-corporate entities, direct or indirect ownership of fifty percent (50%) or more of the equity interest with the power to direct the management and policies of such non-corporate entities.
1.5“Agreement” shall have the meaning set forth in the preamble to this Agreement, as it may be amended by the Parties from time to time.
1.6“Alternative Research Partners” has meaning set forth in Section 2.6(a) of this Agreement.
1.7“Alternative Research Program(s)” has meaning set forth in Section 2.6(a) of this Agreement.
1.8“Alternative Target(s)” has meaning set forth in Section 2.6(a) of this Agreement.
1.9“Applicable Law(s)” means to the extent applicable to a Party and its activities under this Agreement or any R&D Program, any and all applicable laws, binding rules and regulations (whether international, federal, national or local) that may be in effect from time to time and applicable to a Party and/or its activities under this Agreement. For the avoidance of doubt, Applicable Laws shall include, without limitation, Applicable Standards and Nuclear Activity Laws. For the sake of clarity, data protection and privacy Applicable Laws related to privacy and data protections issues, including without limitation the EU General Data Protection Regulation 2016/679 (“GDPR”) of the European Parliament and of the Council of 27 April 2016 “on the protection of natural persons with regard to the processing of Personal Data and on the free movement of such data”, the Data Protection Act 2018 and any successor UK legislation and the retained EU law version of General Data Protection Regulation ((EU) 2016/679) (UK GDPR), and Health Insurance Portability and Accountability Act of 1996 (“HIPAA”), shall be hereinafter referred to as “Data Protection Applicable Laws”.
1.10“Applicable Standards” means all good practice requirements and guidelines of any geographical area applicable to a Party’s performance of its obligations hereunder, notably in the United States, the European Union, the United Kingdom and Switzerland, including the list below, and only to the extent applicable to that Party and/or its activities and obligations under this Agreement:
|
|
|
|
|
|
| Standard |
Document(s) (or current equivalent) |
|
|
|
|
|
|
cGDP or GDP |
All applicable current good distribution practice standards that promote the safety and integrity of the pharmaceutical supply chain, including requirements with respect to data integrity and compliance set forth in 21 CFR Parts 210 and 211 and 21 CFR Part 11 regarding electronic record-keeping requirements, European Commission Directive 2001/83/EC, and Directive 2011/62/EU concerning falsified medicines and any implementing acts, delegated acts and guidance thereunder, including the European Commission Guideline 2013/C 343/01 on Good Distribution Practice of medicinal products for human use and the European Commission Guideline 2015/C 95/01 on principles of Good Distribution Practice of active substances for medicinal products for human use, or in any similar set of laws, regulations, rules, or practices that are applicable in countries where activities are or will be carried out under this Agreement, including WHO Guidelines; |
cGMP or GMP |
All applicable current principles and guidelines of good manufacturing practice for medicinal products for human use as set forth in the current Good Manufacturing Practice Regulations of the U.S. Code of Federal Regulations, including 21 C.F.R. Sections 210 and 211 and applicable FDA guidance documents, compliance policy guides and guidelines; EU GMP Directive 2003/94/EC on good manufacturing practice in respect of medicinal products for human use and investigations medical products for human use, Volume 4 of the European Commission’s Rules governing medicinal products in the European Union, the current version of the ICH Q7 guideline “Good Manufacturing Practice Guide for Active Pharmaceutical Ingredients” and the Guide to Good Manufacturing Practice for Medicinal Products (PE 009-15), or in any similar set of laws, regulations, rules, or practices that are applicable in countries where the Collaboration Product is or will be manufactured, released or supplied to give effect to this Agreement, including WHO Guidelines; |
cGVP or GVP |
All applicable current principles and guidelines of good pharmacovigilance practice for medicinal products for human use, as set forth in FDA regulations for investigational and post-marketing safety reporting requirements, 21 CFR Parts 312, 314 and 600; EU Directive 2001/83/EC, Commission Implementing Regulation 520/2012 on the performance of pharmacovigilance activities and the EMA’s Guideline on Good Pharmacovigilance Practice, or in any similar set of laws, regulations, rules, or practices that are applicable in countries where development activities are or will be carried out under this Agreement, including WHO Guidelines; |
|
|
|
|
|
|
| GLP |
All applicable current principles and guidelines of good laboratory practice for medicinal products for human use including those set forth in U.S. Code of Federal Regulations 21 Part 58, the principles set out in Annex I to Directive 2004/10/EC and all national legislation implementing that Directive (as applicable), Good Laboratory Regulations 1999 (SI 1999/3106), or in any similar set of laws, regulations, rules, or practices that are applicable in countries where development activities are or will be carried out under this Agreement; |
cGxP or GxP |
GMP, GVP, GLP and GDP; |
| Other Standards |
•All general biologics products standards applicable to the activities of a Party under this Agreement, including the EMA’s Biological Guidelines for Active Substances and EMA’s Biological Guidance for Finished Product, including ICH guideline Q11 on development and manufacture of drug substances (chemical entities and biotechnological/ biological entities), the FDA’s Guidance for Industry Q7A Good Manufacturing Practice Guide For Active Pharmaceutical Ingredients; and
•All good practice standards applicable to the transport, handling, storage and processing of radio-active materials or hazards in the any geographical area applicable to a Party’s performance of its obligations hereunder.
|
In the event of any conflict between the aforementioned practices, national laws and regulations, the stricter standard shall apply.
1.11“Assuming Party” has meaning set forth in Section 16.6.
1.12“Background IPR” means Molecular Partners Background and/or Orano Med Background, as the context requires.
1.13“Baseball Arbitration” shall have the meaning set forth in Section 6.5(a).
1.14“BLA” means a Biologics License Application as described in U.S. 21 C.F.R. §601.2, including any amendments thereto, or any corresponding foreign application in any applicable foreign jurisdiction in the Territory.
1.15“[***]” shall have the meaning set forth in Section 5.5 of this Agreement.
1.16“Breaching Party” shall have the meaning set forth in Section 16.2(a) of this Agreement.
1.17“Brief” shall have the meaning set forth in Section 6.5(a)i of this Agreement.
1.18“Bundle” shall have the meaning set forth in Section 1.121 of this Agreement.
1.19“Business Day” means a day other than Saturday, Sunday or any day on which banking institutions in Paris, France or Zurich, Switzerland are authorized or obligated by Applicable Law to close.
1.20“Calendar Quarter” means each three (3) months period commencing January 1, April 1, July 1 or October 1, provided, however, that (a) the first Calendar Quarter of the Term shall extend from the Effective Date to the end of the first full Calendar Quarter thereafter, and (b) the last Calendar Quarter of the Term shall end on the date of termination or expiration of this Agreement.
1.21“Calendar Year” means the period beginning on January 1 and ending on December 31 of the same year, provided, however, that (a) the first Calendar Year of the Term shall commence on the Effective Date and end on December 31 of the same year and (b) the last Calendar Year of the Term shall commence on January 1 of the Calendar Year in which this Agreement terminates or expires and end on the date of termination or expiration of this Agreement.
1.22“Causing Party” has meaning set forth in the Section 16.6.
1.23“CD(s)” means companion diagnostics using [***] (or any other radio-imaging agent) for the First Product or any Additional Candidate.
1.24“Change of Control” means, with respect to a Party, the (a) acquisition of beneficial ownership, directly or indirectly, by a Third Party of a majority or more of the then-outstanding securities or other voting interest of such Party; (b) any merger, reorganization, consolidation or business combination involving such Party and a Third Party that results in the holders of beneficial ownership (other than by virtue of obtaining irrevocable proxies) of the voting securities or other voting interests of such Party immediately prior to such merger, reorganization, consolidation, or business combination ceasing to hold beneficial ownership of at least fifty percent (50%) of the combined voting power of the surviving entity immediately after such merger, reorganization, consolidation or business combination; or (c) any sale, lease, exchange, contribution, or other transfer (in one (1) transaction or a series of related transactions) to a Third Party of all or substantially all of such Party’s assets to the extent, with respect to Molecular Partners, the same relates to the DARPin Technology or, with respect to Orano Med, as relates to the TAT Technology.
1.25“Clinical Supply Agreement” shall have the meaning set forth in Section 8.1 of this Agreement.
1.26“Clinical Transfer Price” means, with respect to each Collaboration Product the price agreed by the Parties and set forth in the Clinical Supply Agreement. For the avoidance of doubt, the Clinical Transfer Price shall include [***] as well as [***] and/or [***], and the [***]; and shall exclude [***] and [***].
1.27“Clinical Trial” means a clinical trial conducted on sufficient numbers of human subjects that is designed to (a) establish that a pharmaceutical product is reasonably safe for continued testing, (b) investigate the safety and efficacy of the pharmaceutical product for its intended use, and to define warnings, precautions and adverse reactions that may be associated with the pharmaceutical product in the dosage range to be prescribed or (c) support Regulatory Approval of such pharmaceutical product or label expansion or extension of such pharmaceutical product. Clinical Trials shall include Phase 0, Phase 1, Phase 2, Phase 2b, Phase 3 Clinical Trials and Registration Enabling Trials.
1.28“Clinical Trial Application” or “CTA” means an application to a Regulatory Authority for purposes of requesting the ability to start or continue a clinical trial, which CTA may consist of, or include, an IND or IMPD, as applicable.
1.29“CMO” means any Third-Party contract manufacturing organizations involved in a Collaboration Product’s manufacture and supply.
1.30“Cost of Goods Sold” or “COGS” means the sum of any amounts that a Party actually pays to a Third Party (or to Molecular Partners with respect to the DARPin Technology supply) to procure supply of Collaboration Products or any components thereof, including the cost of any transfer, shipping, or delivery, customs duties, excise taxes, warehousing costs, or similar charges applied thereto, in each case as determined in accordance with IFRS.
1.31“Collaboration” shall have the meaning set forth in Section 2.1 of this Agreement.
1.32“Collaboration Coordinator” shall have the meaning set forth in Section 3.1(a) of this Agreement.
1.33“Collaboration Costs” shall have the meaning set forth in Section 5.1 of this Agreement.
1.34“Collaboration Product” means (a) the First Product, (b) any Additional Candidate and/or (c) any CD.
1.35“Commercial Supply Agreement” shall have the meaning set forth in Section 8.2 of this Agreement.
1.36“Commercialization Plan” shall have the meaning set forth in Exhibit E of this Agreement.
1.37“Commercialization Requirements” shall have the meaning set forth in Section 6.2(b) of this Agreement.
1.38“Commercial Transfer Price” means, unless otherwise agreed (i) with respect to each Collaboration Product manufactured by Orano Med, the price agreed by the Parties for that Collaboration Product in the Commercial Supply Agreement, which shall include, [***], as well as the [***], provided that such [***] and/or (ii) with respect to each Collaboration Product manufactured by a Third Party CMO following a technology transfer, [***], and the applicable [***].
1.39“Commercially Reasonable Efforts” means on a Collaboration Product-by-Collaboration Product basis, with respect to the efforts to be expended by any Party with respect to any objective, such reasonable, diligent, and good faith efforts as such Party would normally use to accomplish a similar objective under similar circumstances, applying such level of efforts as required to carry out such obligation in a sustained manner and consistent with such level of efforts as those which biotechnology companies at similar stage of development, manufacturing and/or commercialization would devote to a product of similar stage of development, manufacture and/or commercialization, market potential, profit potential or strategic value, based on conditions then prevailing.
1.40“Commercialization Trademarks” shall have the meaning set forth in Section 12.13 of this Agreement.
1.41“Commercialization” or “Commercialize” means all activities, before or after Regulatory Approval, that are directed to the commercial exploitation of a medicinal product, including pre-launch, launch, and post-launch marketing, promotion (including advertising and detailing), medical affairs activities, medical science liaison activities, sponsored product or continuing medical education activities, post-Regulatory Approval clinical studies (that are not required to obtain or maintain such Regulatory Approval), obtaining pricing and reimbursement approvals (whether or not required to obtain or maintain such Regulatory Approval), in each case with respect to such product, any importing, exporting, offering for sale, distribution, marketing and sale of such product, identifying, screening, treating or diagnosing patients as potential users of such product, as well as customer support and invoicing, and interacting with Regulatory Authorities regarding any of the foregoing.
1.42“Competing CoC” shall have the meaning set forth in Section 2.5(a) of this Agreement.
1.43“Competing Program” means the research, development, manufacture or commercialization of any medicinal product in the Exclusive Field.
1.44“Confidential Information” means any information provided orally, visually, in writing or other form by or on behalf of one Party (or an Affiliate or representative of such Party) to the other Party (or to an Affiliate or representative of such other Party) in connection with the R&D Agreement, the Letter Agreement, or this Agreement, whether prior to, on, or after the Effective Date, including information relating to (a) the terms of this Agreement, (b) the identities of targets targeted by a Collaboration Product, (c) any information relating to the Manufacture or exploitation of any Collaboration Product, (d) any Know-How with respect thereto developed by or on behalf of the disclosing Party or its Affiliates, and (e) the scientific, regulatory, or business affairs or other activities of either Party. For purposes of this Agreement, regardless of which Party discloses such Confidential Information to the other, (i) each Party’s [***] Background Technology, Background IPR and Sole Ownership shall be Confidential Information owned by such Party (collectively with any other Confidential Information disclosed by a Party to the other, the “Solely Owned Confidential Information”), and (ii) all the Joint [***] Technology, all the Joint IPR pursuant to Section 12.4 and any other information obtained through the Collaboration, including without limitation the Data but excluding each Party’s Solely Owned Confidential Information, shall be Confidential Information jointly owned by both Parties (the “Jointly Owned Confidential Information”).
1.45“Continuing Party” shall have the meaning set forth in Section 9.1(c) of this Agreement.
1.46“Control” or “Controlled” means, with respect to particular information, data, result or IPR, that the applicable Party owns or has a license to such information or Intellectual Property Right and has the ability to grant a right, license or sublicense to the other Party and any Subcontractor or Third Party as provided for herein without violating the terms of any agreement or other financial or scientific arrangement with any Third Party.
1.47“CRO” means any Third-Party contract research organizations used to implement Clinical Trials, including but not limited to laboratories and Third Parties involved in a Collaboration Product’s supply and Clinical Trial’s coordination, but, for clarity, excluding clinical trial sites and any Third Parties who are individuals.
1.48“Cure Period” shall have the meaning set forth in Section 16.2(a) of this Agreement.
1.49“DARPIN Margin” means a margin of [***] of the DARPin COGS from time to time.
1.50“DARPin Technology” means any [***], but excluding [***].
1.51“Data” means any and all pre-clinical and clinical data generated and Controlled by either Party as a result of the Development under an R&D Plan, including without limitation Patient characteristics, raw data, analyses of Patients biological samples, clinical outcome data, recruitment rates, etc.
1.52“Deficit Party” shall have the meaning set forth in Section 5.3(d) of this Agreement.
1.53“Demanding Party” shall have the meaning set forth in Section 11.4(a) of this Agreement.
1.54“Development” or “Develop” means, with respect to a medicinal product, any research activities, the design, protocol development and performance of all pre-clinical and clinical development, including translational studies, safety, toxicology and tolerability studies, pharmacology/efficacy, test method development and stability testing, statistical analysis and report writing, process development, method development, GMP manufacturing process development, formulation, formulation development and optimization, quality control development, statistical analysis, Clinical Trials (yet excluding post-Regulatory Approval clinical studies that are not required to obtain or maintain Regulatory Approval), regulatory affairs (including preparation for a Regulatory Approval application submission and other submission-related activities), product approval and registration activities, manufacturing of clinical supplies and regulatory activities that are required to obtain Regulatory Approval of an MAA for such medicinal product in the Territory, interacting with Regulatory Authorities regarding the foregoing, and all other activities necessary to conduct Clinical Trials or seek, obtain and maintain Regulatory Approval. “Development” shall not include Commercialization.
1.55“Dispute” shall have the meaning set forth in Section 17.3 of this Agreement.
1.56“Drug Master File” means a drug master file compiled for submission to a Regulatory Authority to support Regulatory Approval of a medicinal product in a given country or jurisdiction and providing information on the manufacturing facilities and manufacturing processes for making of such product or the active pharmaceutical ingredient of such product, including information on activities relating to manufacturing, processing, formulating, packaging and storage thereof.
1.57“Effective Date” shall have the meaning set forth in the preamble to this Agreement.
1.58“Engaging Party” shall have the meaning set forth in Section 2.6(b) of this Agreement.
1.59“Excedent Party” shall have the meaning set forth in Section 5.3(d) of this Agreement.
1.60“Exclusive Field” shall have the meaning set forth in Section 2.4(a) of this Agreement.
1.61“Exclusive First Option Period” means, with respect of each Collaboration Product, the period [***], and ending [***].
1.62“Exclusive Second Option Period” means, with respect of each Collaboration Product, the period [***], and ending [***].
1.63“Executive Officers” means the [***] of Molecular Partners and the [***] of Orano Med (or their respective designees).
1.64“Existing Contracts” shall have the meaning set forth in Section 2.4(b) of this Agreement.
1.65“Exit Decision” shall have the meaning set forth in Section 9.1(a) of this Agreement.
1.66“Exit License” means the licenses granted in Section 9.4 and the right of reference granted in Section 9.5.
1.67“Exiting Party” shall have the meaning set forth in Section 9.1(a) of this Agreement.
1.68“Expert” means a Third Party individual who is a qualified professional with expertise in the matter in dispute as demonstrated by at least fifteen (fifteen) years of experience in (as applicable to the grounds for the dispute) the development, manufacture and/or commercialization and/or licensing of medicinal products (e.g. having occupied at least one senior position within a large pharmaceutical company relating to product development, manufacture, commercialization and/or licensing), but excluding any current or former employee or consultant of either Party. Such person shall be fluent in the English language.
1.69“Expert Mediation” shall have the meaning set forth in Section 3.3(e) of this Agreement.
1.70“Exploitation” or “Exploit” means to make, have made, use, have used, offer to sell, sell, have sold, import, export and otherwise practice or exploit, including to research, Develop, have Developed, Manufacture, have Manufactured, Commercialize and/or have Commercialized.
1.71“Exploiting Party” means (a) in case of a successful and completed Option exercise pursuant to Section 6, the Party who has exercised the Option for a given Collaboration Product; or (b) in case of an Exit Decision, the Continuing Party with respect to the Collaboration Product for which the Exit Decision was exercised.
1.72“Facility(ies)” has the meaning set out in Schedule D.
1.73“[***]” has the meaning set out in Section 2.6(f) of this Agreement.
1.74“FDA” means the United States Food and Drug Administration, or any successor agency having the same or similar authority.
1.75“Field” means therapeutic and diagnostic uses in oncologic indications in humans.
1.76“Firewall” means certain steps to establish the robust and workable tangible and intangible segregation of the Development and Commercialization activities relating to any Competing Program from the Development and Commercialization activities with respect to the concerned Collaboration Product under this Agreement and applicable security protocols, as follows: (a) no personnel in the core project team involved in performing the Development or Commercialization of the Competing Program have access to non-public plans or non-public information relating to the Development or Commercialization of the concerned Collaboration Product or any other Confidential Information of the Remaining Party; (b) no personnel in the core project team of the concerned Collaboration Product program have access to non-public plans or information relating to the Development or Commercialization activities under such Competing Program in the Field, and (c) the Party subject to a Competing CoC does not use in the conduct of any Competing Program any Confidential Information or IPR of the Remaining Party, provided that for clarity, these restrictions shall not apply to (i) individuals involved at a senior management or executive level who are involved in decision-making regarding programs and products generally, and who are not involved in day-to-day activities or decision-making in connection with the concerned Collaboration Product or Competing Program, nor to (ii) individuals involved in support function activities (including quality oversight and management) in connection with the concerned Collaboration Product or Competing Program as applicable, as long as, in the case of (i), such individuals have no access to material Confidential Information that may be taken into account to conduct Development or Commercialization activities for a Competing Program and in each case of (i) and (ii) such individuals do not use the Confidential Information of the Remaining Party for the benefit of the Competing Program except for information related to safety and quality concerns related to the Collaboration Product to the extent required by Applicable Law or under existing quality agreements provided always on a reciprocal basis. For the purposes of the foregoing, the core project team includes all individuals having access to material Confidential Information that may be taken into account to conduct Development or Commercialization activities for a Competing Program.
1.77“First Commercial Sale” means the first sale (or other commercial transfer or disposition for value considered as Net Sales under this Agreement) of a Collaboration Product to a Third Party (other than to an Affiliate or sublicensee) by the Exploiting Party in any country of the Territory. Sales for test marketing, clinical-trial purposes or compassionate use shall not constitute a First Commercial Sale of such Collaboration Product.
1.78“First Product” means the first radiopharmaceutical product combining DARPin Technology and [***] and potentially Joint [***] Technology and targeting DLL3 or a different target substituted and agreed upon by Parties in accordance with Section 4.2(a), in such delivery form and for such indications as the Parties may agree during the Term.
1.79“FTE” means the equivalent of the work of one (1) qualified full-time person specialized in the clinical field and suited for the work to be performed by a Party under an R&D Plan, where “full time” is based upon a total of [***] working hours per Calendar Year of work.
1.80“FTE Costs” means, for a given period, the FTE Rate multiplied by the applicable number of FTEs performing relevant activities in accordance with an R&D Plan during such period but for clarity, excluding any FTEs performing administrative, supportive and managerial activities, including but not limited to coordination, management, Legal, IP or BD.
1.81“FTE Rate” means [***] representing the fully burdened rate for an FTE, whereby the FTE Rate shall be adjusted annually, beginning with January 1, 2025 on the basis of the average inflation rate in the Eurozone (in comparison to the base value such rate on January 1, 2024). FTE Rate includes [***].
1.82“[***]” means [***] that can be [***]. For the avoidance of doubt the [***].
1.83“[***] Background Technology” means all right, title and interest in any and all Patent Rights, Know-How and other IPR concerning the [***].
1.84“[***] Project Plan” means the separate project plan agreed upon by the Parties in the Letter Agreement.
1.85“ICF” shall have the meaning set forth in Section 4.6 of this Agreement.
1.86“IFRS” means International Financial Reporting Standards, as generally and consistently applied by the applicable Party.
1.87“IMPD” means an Investigational Medicinal Product Dossier which includes all data required by Regulatory Authorities in the European Union and the UK for the performance of Clinical Trials in one or more European Union member states and/or the UK.
1.88“IND” means an Investigational New Drug Application as defined in the United States Food, Drug and Cosmetic Act, as amended, and regulations promulgated thereunder, or any successor application or procedure required to initiate clinical testing of a drug in humans in the United States, or the equivalent in any other jurisdiction such as an application for a Clinical Trial approval in the European Union.
1.89“Indemnify” shall have the meaning set forth in Section 15.1 of this Agreement.
1.90“Intellectual Property Right(s)” or “IPR” means any and all Patent Rights, Know-How and all other Intellectual Property Rights worldwide, including without limitation, enhancements, design rights, copyrights, trademarks, trade names, trade secrets, all rights of whatsoever nature in computer programs, codes, databases, algorithms, software, data, and all other intangible rights and privileges of a nature similar to any of the foregoing (whether or not protectable under Applicable Laws, whether or not registered), including all granted registrations and applications of, and the right to apply for, the same.
1.91“Joint Exit Decision” shall have the meaning set forth in Section 9.1(b) of this Agreement.
1.92“Joint IPR” shall have the meaning set forth in Section 12.4(a) of this Agreement.
1.93“Joint [***] Technology” shall mean any and all right, title and interest in and to any results that (i) are discovered, developed or invented in course of performance of the [***] Project Plan pursuant to the Letter Agreement or the R&D Plan pursuant to this Agreement (x) solely by, for or on behalf of a Party or (y) jointly by, for or on behalf of both Parties, and (ii) are related to (x) the development of the [***], or to (y) any modification or improvement of a Party’s [***] Background Technology to the extent such a modification or improvement directly relates to [***] and (iii) do not fall within the scope of Molecular Partners [***] Background Technology or Orano Med [***] Background Technology.
1.94“Joint Patent Application” shall have the meaning set forth in Section 12.9(b) of this Agreement.
1.95“Joint Steering Committee” or “JSC” shall have the meaning set forth in Section 3.2(a) of this Agreement.
1.96“Jointly Owned Confidential Information” shall have the meaning set forth in Section 1.44 of this Agreement.
1.97“JSC Chair” shall have the meaning set forth in Section 3.2(b) of this Agreement.
1.98“Know-How” means any scientific or technical information, results and data of any type, in any tangible or intangible form, that is not in the public domain or otherwise publicly known, including discoveries, inventions, trade secrets, devices, databases, practices, protocols, methods, processes, specifications, techniques, concepts, ideas, formulations, formulae, data (including clinical and non-clinical, pharmacological, biological, chemical, toxicological, clinical test, safety, clinical and analytical information, quality control, trial, stability and manufacturing processes and techniques data), case reports forms, medical records, data analyses, reports, studies and procedures, designs for experiments and tests and results of experimentation and testing (including results of research or development), summaries and information contained in submissions to and information from ethical committees, or Regulatory Authorities, and manufacturing process and development information, in each case to the extent not claimed or disclosed in a patent or patent application. The fact that an item is known to the public shall not be taken to exclude the possibility that a compilation including the item, or a development relating to the item, is (and remains) not known to the public. “Know-How” excludes Patent Rights.
1.99“Launch Preparedness Plan” shall have the meaning set forth in Section 6.2(b) of this Agreement.
1.100“Lead Prosecuting Party” shall have the meaning set forth in Section 12.9(a) of this Agreement.
1.101“Letter Agreement” means that letter agreement entered into by the Parties and dated [***] to collaborate on the development of an optimized [***].
1.102“License Agreement” shall have the meaning set forth in Section 6.5 of this Agreement.
1.103“License and Supply Agreements” shall have the meaning set forth in Section 6.5 of this Agreement.
1.104“Logistics Costs” shall mean with respect to a Collaboration Product, the costs of [***], and such other elements as may be specified in the Clinical Supply Agreement or the Commercial Supply Agreement and may include costs incurred by [***]. For the avoidance of doubt, unless otherwise agreed, [***].
1.105“Losses” shall have the meaning set forth in Section 15.1 of this Agreement.
1.106“MAA” means a marketing authorization application or equivalent application, and all amendments and supplements thereto, filed with an applicable Regulatory Authority in the Territory (including a New Drug Application filed pursuant to the requirements of the FDA).
1.107“Manufacture” or “Manufacturing” with respect to the manufacture and supply of the Collaboration Product has the meaning in Section 8.3 and Schedule D.
1.108“Manufacturing and Logistics Requirements” has the meaning set forth in Section 6.2(b).
1.109“Mediation Expert(s)” shall have the meaning set forth in Section 3.3(e).
1.110“Molecular Partners” shall have the meaning set forth in the preamble to this Agreement.
1.111“Molecular Partners Background” means all right, title and interest in any and all Patent Rights, Know-How and other IPR are that (i) Controlled by Molecular Partners as of the Effective Date or during the Term other than in the course of performance of the R&D Program pursuant to this Agreement and (ii) necessary or reasonably useful for the research, Development, Manufacture or Commercialization of Collaboration Products. For the sake of clarity, Molecular Partners Background includes without limitation Molecular Partners’ IPR pertaining to designed repeat proteins, including DARPin proteins, and their research, development, manufacture and clinical use.
1.112“Molecular Partners [***] Background Technology” means [***] Background Technology Controlled by Molecular Parties before the Effective Date of the Letter Agreement, or acquired or generated by Molecular Partners independently from the Collaboration according to the R&D Agreement or this Agreement and without the use of the Confidential Information of Orano Med.
1.113“Molecular Partners Indemnitees” shall have the meaning set forth in Section 15.1 of this Agreement.
1.114“Molecular Partners Owned IP” shall have the meaning set forth in Section 12.2(b) of this Agreement.
1.115“Molecular Partners Owned Patents” shall have the meaning set forth in Section 12.2(b) of this Agreement.
1.116“MTA” shall have the meaning set forth in Section 4.2(b) of this Agreement.
1.117“Necessary Support” shall have the meaning set forth in Section 9.3 of this Agreement.
1.118“Negotiation Period” shall have the meaning set forth in Section 6.5 of this Agreement.
1.119“Net Loss” shall have the meaning set forth in Section 1.120 of this Agreement.
1.120“Net Profit” means with respect to a Collaboration Product, on a Calendar Quarter-by-Calendar Quarter basis during the Term (except where an alternative time period is expressly stated, e.g. monthly), an amount equal to Net Sales minus the Operating Expenses. [***].
1.121“Net Sales” means the gross amounts billed, invoiced or otherwise recognized as revenue in accordance with IFRS with respect to sales of Collaboration Products by the Exploiting Party, its Affiliates or its permitted subcontractors to independent or unaffiliated Third Party customers in bona fide, arm’s-length transactions, less the following permitted deductions: (a) amounts repaid, credits and allowances granted for defective or damaged Collaboration Products, returns, recalls or rejections of Collaboration Products or for billing errors or adjustments (including write-offs); (b) customary and reasonable trade, cash and quantity discounts, allowances and credits (including chargebacks); (c) excise taxes, sales taxes, duties, VAT, clawbacks and other similar taxes directly related to the sale; (d) customary rebates and chargebacks made with respect to sales paid for by competent Regulatory Authorities, their
agencies and purchasers and reimbursers, public or private hospitals, managed health care organizations, group purchase organizations or trade customers; (e) price protection adjustments; (f) amounts provided or credited to customers through coupons and other discount programs; (g) wholesaler fees and other expenses actually incurred for distribution and logistics (including freight, postage, shipping, customs, duties and shipping insurance and other transportation charges directly related to the distribution of Collaboration Products); (h) delayed ship order credits, discounts or payments related to the impact of price increases between purchase and shipping dates or retroactive price reductions; (i) fee for service payments to customers for any non-separable services (including compensation for maintaining agreed inventory levels and providing information); and (j) other reductions or specifically identifiable amounts deducted for reasons similar to those listed above in accordance with IFRS,
provided that:
if any Collaboration Product(s) is sold by the Exploiting Party or any of its Affiliates to customers together with any other product(s) (including CDs) (“Other Products”) as a package or bundle (“Bundle”), that (i) benefits such Other Products at the expense of the Collaboration Product(s) or (ii) benefits the Collaboration Product(s) at the expenses of the Other Products, such Bundle shall be disaggregated for the purposes of revenue recognition such that each product included in such Bundle is attributed the fair and appropriate level of revenue.
Collaboration Products are considered “sold” when billed out or invoiced in connection with an arms’ length sale. If a Collaboration Product is delivered to a Third Party before being invoiced (or is not invoiced), Net Sales will be calculated at the time all of the revenue recognition criteria under IFRS are met. Net Sales shall not include transfers or dispositions for no profit or for charitable purposes to the extent such transfer and dispositions are in accordance with Applicable Laws and with prevailing industry standards in the relevant country.
1.122“Non-Breaching Party” shall have the meaning set forth in Section 16.2(a) of this Agreement.
1.123“Non-Engaging Party” shall have the meaning set forth in Section 2.6(b) of this Agreement.
1.124“Nuclear Activity” or “Nuclear Activities” means the storing, handling, processing, transporting, use, packaging and disposal of radio-active materials supplied by Orano Med and in relation with [***] the Collaboration Products.
1.125“Nuclear Activity Laws” means to the extent applicable to a Party and any of its activities under this Agreement, all laws, regulations, binding rules and binding good practice standards and guidelines relating to the Nuclear Activities.
1.126“Officials” shall have the meaning set forth in Section 14.6(a) of this Agreement.
1.127“Operating Expenses” means with respect to a Collaboration Product during commercial supply on a Calendar Quarter-by-Calendar Quarter basis during the Term, the sum of (a) (i) whilst Collaboration Product is manufactured by Orano Med, [***] and (ii) whilst Collaboration Product is manufactured by a Third Party CMO following a technology transfer [***], and (b) the [***] and [***], including (i) [***]; (ii) [***]; (iii) [***]; and (iv) [***], in each case during the relevant Calendar Quarter and to the extent not already deducted from Net Sales or previously charged through the Clinical Transfer Price.
1.128“Option” shall have the meaning set forth in Section 6.1 of this Agreement.
1.129“Opt-Out Point” means any of the following GO/NO GO decision points upon completion of a given activity/milestone under an R&D Plan or, as applicable, decision points for commitment on cost items in an R&D Budget: (a) upon [***]; (b) upon [***]; (c) upon the [***]; as such points (a) – (c) may be more specifically described in R&D Plan, and (d) [***], provided that [***], provided that [***].
1.130“Orano Med” shall have the meaning set forth in the preamble of this Agreement.
1.131“Orano Med Background” means all right, title and interest in any and all Patent Rights, Know-How and other IPR that are (i) Controlled by Orano Med as of the Effective Date or during the Term other than in the course of performance of the R&D Program pursuant to this Agreement and (ii) necessary or reasonably useful for the research, Development, Manufacture or Commercialization of Collaboration Products. For the sake of clarity, Orano Med Background includes without limitation the TAT Technology.
1.132“Orano Med [***] Background Technology” means [***] Background Technology Controlled by Orano Med before the Effective Date of the Letter Agreement, or acquired or generated by Orano Med independently from the collaboration according to the R&D Agreement or this Agreement and without the use of Confidential Information of Molecular Partners.
1.133“Orano Med Indemnitees” shall have the meaning set forth in Section 15.2 of this Agreement.
1.134“Orano Med Owned IP” shall have the meaning set forth in Section 12.3(a) of this Agreement.
1.135“Orano Med Owned Patents” shall have the meaning set forth in Section 12.3(b) of this Agreement.
1.136“Orano Med Reserved Technology” shall mean [***] and all IPR related thereto.
1.137“Other Products” shall have the meaning set forth in Section 1.121 of this Agreement.
1.138“Out-Licensing” means licensing out divesting, disposing of, or otherwise transferring of rights of IPR, including Background IPR and Joint IPR, necessary or reasonably useful for Development, Manufacture and Commercialization of a Collaboration Product to a Third Party, whether by a Party solely or the Parties jointly in accordance with Section 7.
1.139“Out-Licensing Revenues” means all compensation received by a Party from any Third Party in consideration for an Out-Licensing of a Collaboration Product, including [***], yet excluding [***].
1.140“Out-Licensing Revenues Report” shall have the meaning set forth in Section 10.3(b) of this Agreement.
1.141“Out-of-Pocket Costs” means costs and expenses paid to Third Parties (or payable to Third Parties and accrued in accordance with IFRS) by a Party or any of its Affiliates and directly incurred in the conduct of any applicable activities under an R&D Plan. Out-of-Pocket Costs include [***], but exclude costs which are [***].
1.142“Owed Party” shall have the meaning set forth in Section 11.1(a) of this Agreement.
1.143“Owing Party” shall have the meaning set forth in Section 11.1(a) of this Agreement.
1.144“Party” or “Parties” shall have the meaning set forth in the preamble to this Agreement.
1.145“Patent Rights” means (a) all national, regional and international patents and patent applications, including provisional patent applications, and all rights to apply for the same, (b) all patent applications filed either from such patents, patent applications or provisional applications or from an application claiming priority from either of these, including divisionals, continuations, continuations-in-part, provisionals, converted provisionals and continued prosecution applications, (c) any and all patents that have issued or in the future issue from the foregoing patent applications ((a) and (b)), including utility models, petty patents and design patents and certificates of invention, (d) any and all extensions or restorations by existing or future extension or restoration mechanisms, including revalidations, renewals, reissues, re-examinations (including without limitation, ex partes reexaminations, inter partes reviews, inter partes reexaminations, post grant reviews and supplemental examinations) and extensions (including any supplementary protection certificates and the like) of the foregoing patents or patent applications ((a), (b), and (c)) and (e) any other form of government-issued right substantially similar to of any of the foregoing described in (a), (b), (c), or (d)).
1.146“Patient(s)” means an individual selected to participate in each Clinical Trial of a Collaboration Product and who has signed an informed consent form in accordance with the terms of the relevant protocol.
1.147“Permitted Recipient” shall have the meaning set forth in Section 13.3(a) of this Agreement.
1.148“Person” means an individual, sole proprietorship, partnership, limited partnership, limited liability partnership, corporation, limited liability company, business trust, joint stock company, trust, unincorporated association, joint venture or other similar entity or organization, including a government or political subdivision, department or agency of a government.
1.149“Publishing Party” shall have the meaning set forth in Section 13.5(a).
1.150“Profit Sharing Report” shall have the meaning set forth in Section 10.3(a) of this Agreement.
1.151“Profit Share” means (i) [***]; or (ii) [***]:
|
|
|
|
|
|
|
|
|
| Profit Share Trigger |
[***] |
[***] |
| [***]: |
[***] |
[***] |
| [***]: |
[***] |
[***] |
| [***]: |
[***] |
[***] |
| [***]: |
[***] |
[***] |
1.152“Quarterly Excess” shall have the meaning set forth in Section 5.3(d) of this Agreement.
1.153“Quarterly Report” shall have the meaning set forth in Section 5.3(a) of this Agreement.
1.154“R&D Agreement” shall have the meaning set forth in the Recitals.
1.155“R&D Budget” shall have the meaning set forth in Section 4.3(a) of this Agreement.
1.156“R&D Plan” shall have the meaning set forth in Section 4.3(a) of this Agreement.
1.157“R&D Program” shall have the meaning set forth in Section 4.1 of this Agreement.
1.158“R&D Term” means the period starting upon the Effective Date, or with respect to any subsequent Collaboration Product Developed hereunder, commencing on the initiation of an R&D Plan for such Collaboration Product and ending on the earlier of (a) completion of all activities set forth in the corresponding R&D Plan, or (b) exercise of an Exit Decision by either Party, or (c) the effective date of a termination of this Agreement.
1.159“Registration Enabling Trial” means a Clinical Trial of a Collaboration Product that is designed to, and for which a competent Regulatory Authority in the Territory has provided guidance that the design of such Clinical Trial is sufficient to, ascertain efficacy and safety of such Collaboration Product in support of the preparation and submission of Regulator Approval for Commercialization of such Collaboration Product to such competent Regulatory Authority, regardless of whether such Clinical Trial is referred to as a Phase 2, Phase 2b or Phase 3 Clinical Trial or otherwise, provided that for countries where Regulatory Authorities will not make this determination or provide this guidance ahead of a Clinical Trial, the Parties shall jointly agree in the R&D Plan when / which Clinical Trial shall for that country constitute a Registration Enabling Trial and such Clinical Trial shall comprise a Registration Enabling Trial for the purposes of this Agreement. If a Clinical Trial of a Collaboration Product is not initially designed as a Registration Enabling Trial but is later re-designed, converted or expanded into such a trial, then it shall be deemed to be a Registration Enabling Trial as of the date of such re-design, conversion or expansion. If the Sponsor or, if different, the Party responsible for regulatory filing and communication publicly describes (including in public announcements or information on its website) a Clinical Trial of a Collaboration Product that has not otherwise been classified as a Registration Enabling Trial pursuant to this Section as such a trial or a Phase 3 Trial, then such Clinical Trial shall be deemed to be a Registration Enabling Trial as of the first date that such description is available to the public.
1.160“Regulatory Approval” means, with respect to a country, region or regulatory jurisdiction, any and all approvals, licenses, registrations or authorizations of any competent Regulatory Authority necessary for the Development (including the conduct of Clinical Trials), manufacture, Commercialization (including distribution, marketing, promotion, offer for sale, use, import, reimbursement, export or sale) or other exploitation of medicinal products in such country, region or regulatory jurisdiction, and shall include any licenses, approvals, registrations, consents or authorisations required under the Nuclear Activity Laws to commence or carry on any of the activities contemplated by this Agreement, including any activities under an R&D Program (“Nuclear Activity Licences”).
1.161“Regulatory Authority” means (on a Collaboration Product-by-Collaboration Product basis and country-by-country basis) any competent regulatory, governmental authority in the United States, in the European Union, the United Kingdom, Switzerland or in other applicable jurisdictions (whether national, multinational, federal, provincial and/or local) that are involved in the review and approval of Clinical Trials and in the review and approval of applications related to the manufacture, development and/or commercialization of medicinal products, as well as, all governmental, regulatory and other authorities with competence to oversee and enforce the Nuclear Activity Laws.
1.162“Regulatory Documentation” means all regulatory applications, submissions, filings, dossiers, notifications, registrations, or other documentation provided to Regulatory Authorities in connection with the development, manufacturing and commercialization of a radiopharmaceutical medicinal product alone or in combination, including but not limited to all CTAs, INDs and amendments thereto, MAAs, NDAs or BLAs, and amendments thereto, correspondence as well as minutes of material communication with Regulatory Authorities, periodic safety update reports, adverse event files, complaint files, inspection reports and manufacturing process records, in each case together with all supporting documents (including documents with respect to Data) and shall, include all like documents submitted to Regulatory Authorities in respect of or in relation to [***] and required for any Nuclear Activities under this Agreement. Regulatory Documentation excludes Drug Master Files.
1.163“Regulatory Hold” means that a Regulatory Authority has issued an order (a) to suspend or withdraw an authorization required for a facility to manufacture medicinal products for GMP release, (b) to delay a proposed clinical trial or (c) to suspend an ongoing Clinical Trial of a medicinal product in any country or group of countries.
1.164“Remaining Party” shall have the meaning set forth in Section 2.5(b)i of this Agreement.
1.165“Requesting Party” shall have the meaning set forth in the Section 5.5(b) of this Agreement.
1.166“Reversion Terms” has meaning set forth in the Section 16.7.
1.167“ROFR 1” shall have the meaning set forth in the Section 2.6(b) of this Agreement.
1.168“ROFR 1 Notice” shall have the meaning set forth in the Section 2.6(b) of this Agreement.
1.169“ROFR 2” shall have the meaning set forth in the Section 2.6(c) of this Agreement.
1.170“ROFR 2 Notice” shall have the meaning set forth in the Section 2.6(c) of this Agreement.
1.171“Sole Ownership” refers generally to Orano Med Owned IP and/or Molecular Partners Owned IP as the context may require.
1.172“Sole Patent Rights” refers generally to Orano Med Owned Patents and/or Molecular Partners Owned Patents.
1.173“Solely Owned Confidential Information” shall have the meaning set forth in Section 1.44 of this Agreement.
1.174“Sponsor” shall have the meaning of Article 2.2 (14) of Regulation (EU) 536/2014 of 16 April 2014 of the European Parliament and of the Council ‘on clinical trials on medicinal products for human use’ (repealing Directive 2001/20/EC) and of Section 1.60 of ICH E6(R2) GCP Guidance.
1.175“Subcontractors” shall have the meaning set forth in Section 2.3 of this Agreement.
1.176“Substitute Target” shall have the meaning set forth in Section 4.2(a) of this Agreement.
1.177“Supply Exit” shall have the meaning set forth in Section 8.3(b) of this Agreement.
1.178“TAT Technology” means Orano Med’s IPR pertaining to the [***], including its use for targeted alpha-emitter therapy, its production, and chelating agents’ design and production for targeted alpha-emitter therapy using [***], but excluding [***].
1.179“Term” shall have the meaning set forth in Section 16.1 hereunder.
1.180“Term Sheet” shall have the meaning set forth in the Recitals.
1.181“Territory” means worldwide.
1.182“Third Party” means any Person or entity other than Molecular Partners and Orano Med and their respective Affiliates.
1.183“Third Party Claim” shall have the meaning set forth in Section 15.1 of this Agreement.
1.184“Third Party Agreement” shall have the meaning set forth in Section 5.5(b) of this Agreement.
1.185“Third Party Agreement Payments” means any payments (e.g., upfront payments, milestones, royalties, and maintenance payments) due by a Party to any Third Party under license agreements or other written agreements granting rights to Intellectual Property Right owned or otherwise controlled by such Third Party.
1.186“Third Party IPR” shall have the meaning set forth in Section 5.5(a) of this Agreement.
1.187“Unitary Patent” shall have the meaning set forth in Section 12.9(b)ii of this Agreement.
1.188“UPC Notice” shall have the meaning set forth in Section 12.9(b) of this Agreement.
1.189“UPC State” shall have the meaning set forth in Section 12.9(b)iii of this Agreement.
1.190“UPC System” shall have the meaning set forth in Section 12.9(b)i of this Agreement.
1.191“[***]” shall have the meaning set forth in Section 5.5(c) of this Agreement.
1.192“Withholding Taxes” shall have the meaning set forth in Section 11.2(d) of this Agreement.
Section 2
COLLABORATION SCOPE
2.1Collaboration. The main objectives of the Parties’ collaboration under this Agreement (“Collaboration”) are, on a Collaboration Product-by-Collaboration Product basis,
(a)to conduct each R&D Program pursuant to the terms and conditions set forth in Section 4 and Section 5, whereby the Parties agree that (i) they shall be jointly and equally responsible for the cost and expenses incurred in the conduct of each R&D Program pursuant to Section 5; (ii) each Party shall be entitled to exit the joint Development of a Collaboration Product at certain Opt-Out Points by way of an Exit Decision pursuant to Section 9; and (iii) to share in accordance with the Profit Share the Net Profits achieved through Commercialization of a Collaboration Product or Out-Licensing Revenues achieved through an Out-Licensing;
(b)to grant to each other, and potentially to exercise, the mutual option rights for further Development and Commercialization of the First Product (or subsequent Collaboration Products) pursuant to the terms and conditions set forth in Section 6 and, upon successful Option exercise by a Party, the post-Option licenses pursuant to Section 6.6;
(c)to achieve an Out-Licensing of the First Product (and/or subsequent Collaboration Products) pursuant to Section 7 in the event that (i) neither Party exercises its Option or (ii) both Parties, despite their intention to exercise the Option, fail to establish the concerned Party’s financial and commercial abilities to further Develop and Commercialise the First Product (or subsequent Collaboration Products) to the other Party’s reasonable satisfaction as determined pursuant to Section 6.4; and
(d)to establish the Manufacture and supply of the First Product (and subsequent Collaboration Products) by Orano Med, using Molecular Partners’ supplied DARPin proteins pursuant to the terms and conditions set forth in Section 8.
2.2Performance.
(a)Each Party agrees to act in good faith in performing its obligations under this Agreement. Both Parties shall perform their respective activities under an R&D Plan, and more generally pursuant to this Agreement, and shall notify the other Party as promptly as possible in the event of any material delay or event that is likely to adversely affect the advance and progress of the Collaboration as contemplated by this Agreement, in particular progress under an R&D Plan. Each Party shall (a) perform its obligations under an R&D Plan with reasonable due care and in conformity with Applicable Laws, (b) use Commercially Reasonable Efforts; and (c) procure appropriate quality assurance, quality controls and review procedures to secure good standard performance of its obligations hereunder. To the extent required by Applicable Laws, each Party agrees to provide the other Party any reasonable information and assistance in order to enable the other Party to comply with its obligations under Applicable Laws.
(b)Each Party agrees that with respect to the Collaboration Product(s) it shall not deprioritise the performance of its respective activities under this Agreement in favour of its other then current business activities or collaborations that relate to the development, commercialisation or manufacture of a product that is not a Collaboration Product and shall use Commercially Reasonable Efforts to invest the level of resource and of care sufficient to perform its activities thereunder in light of the stage of development, market potential, profit potential and strategic value of the Collaboration Product, based on conditions then prevailing.
2.3Subcontracting. Each Party shall have the right to subcontract any portion of its activities or obligations hereunder to subcontractors including Affiliates and any Third Parties (“Subcontractors”), subject to consulting with the JSC and obtaining its approval with respect to Development or Manufacturing activities where a Party wishes to sub-contract significant and/or material parts of its obligations to a Third Party (i,e., other than its Affiliates), including, in the case of Orano Med, any subcontracting or sublicensing of the Manufacture of [***] or finished Collaboration Product. Each Party shall remain solely and fully liable for the performance of its Subcontractors. Each Party shall ensure that each of its Subcontractors, in such Party’s reasonable and good faith determination, is reasonably capable and qualified to duly and diligently exercise the rights subcontracted to it hereunder and performs its obligations pursuant to the terms of this Agreement, including its Exhibits attached hereto. For clarity, to the extent that a Party has an obligation under this Agreement to perform an action or to meet the agreed criteria, and such Party subcontracts such obligation, such Party shall be responsible for any failure by such Party’s Subcontractor to perform the action or meet the agreed criteria. Each Party shall obtain and maintain copies of documents relating to the obligations performed by such Subcontractors that are held by or under the Control of such Subcontractors and that are required to be provided to the other Party under this Agreement. To the extent the results obtained through the activities performed by Subcontractors in connection with the Collaboration may be within a Party’s Control, such Party shall provide the other Party with such results.
2.4Exclusivity.
(a)The Parties and/or their Affiliates shall exclusively (both on a direct and indirect basis) collaborate on, and thereby undertake not to directly or indirectly conduct (including through the grant of a license), participate in, fund, on its own or in collaboration with any Third Party, any programs directed to (i) [***]; and/or (ii) [***].
(b)Notwithstanding the foregoing, Orano Med acknowledges that [***] and agrees that, [***] and that such [***].
(c)In the event of an Exit Decision or if this Agreement should be terminated pursuant to Section 16.2, the exclusivity obligations in the Exclusive Field as set forth in this Section 2.4 shall [***], while the [***] provided always that the [***], or is [***], and that [***].
2.5Exclusivity Obligations in case of Change of Control.
(a)In the event that (i) either Party becomes subject to a Change of Control after the Effective Date, and (ii), as of the closing date of such transaction or the date the concerned Party enters into such transaction, the Third Party involved in the Change of Control or its Affiliates at such time (the “Acquiror Group”) is engaged in the Development manufacture, or Commercialization of a Competing Program (such Change of Control, a “Competing CoC”), then the Party being subject to the Competing CoC shall notify the other Party as soon as reasonably possible after the first public disclosure or signing of the transaction (provided this may be disclosed without breaching regulatory requirements).
(b)Upon the Competing CoC becoming effective:
i.the Party being subject to the Competing CoC shall [***]; and
ii.[***]; or
iii.[***].
2.6External Validation of New Targets, Inclusion to Collaboration.
(a)Notwithstanding the foregoing, and notwithstanding the exclusive licences granted elsewhere in this Agreement or the attribution of Intellectual Property rights pursuant to Section 12 below, either Party shall be entitled to conduct, alone or in partnership with Third Party partners (“Alternative Research Partners”), research programs prior to initiation of a Phase 1 Clinical Trial for the identification and validation of new targets (other than for the avoidance of doubt, DLL3 or if substituted, the target of the First Product and if selected, the target of any Additional Candidate) (“Alternative Target(s)” and/or “Alternative Research Program(s)”).
(b)The Party engaging in an Alternative Research Program that could potentially subsequently be covered by Section 2.4 of this Agreement (the “Engaging Party”) shall seek to inform the other Party (the “Non-Engaging Party”) by notice in writing (“ROFR 1 Notice”) [***] of the [***] and of the [***] such [***]. If the Non-Engaging Party should [***], the Parties shall [***] (“ROFR 1”). [***].
(c)Where the Engaging Party fails to inform the Non-Engaging Party of the Existence of such Alternative Research Program before engaging with an Alternative Research Partner, it shall (in each case to the extent it is legally permitted to do so) inform the Non-Engaging Party by notice in writing (“ROFR 2 Notice”) [***], and as to the [***]. If the Non-Engaging Party [***] (collectively referred to as “ROFR 2”). For the avoidance of doubt, this Section shall not apply where [***].
(d)If the Engaging Party is not, through its arrangements or agreement(s) with the Alternative Research Partner, contractually permitted to comply with the disclosures required under Section 2.6(c) and/or Section 2.6(f), it shall confirm the same to the Non-Engaging Party within the same timeframe as the ROFR 2 Notice and the terms of Section 2.6(f) shall then apply without the need for the Parties to adhere to Section 2.6(e) and the Parties shall either (at the Engaging Party’s election) [***].
(e)In the event that the Parties wish to [***], both Parties shall [***]. [***],
i.the Parties agree and acknowledge that such [***]. However, unless otherwise agreed, [***]; and
ii.the Parties shall [***], whereby (x) [***]; and (y) [***].
(f)In the event that the Parties and, as the case may be, the Alternative Research Partner cannot reach an agreement on ROFR 1 or ROFR 2 within the time periods referenced above, then [***] provided that [***]. For purposes of the foregoing, “[***]” shall mean [***].
(g)[***] shall not [***] with respect to the [***] if [***].
(h)Save with respect to a determination under Section 2.6(d), if the Parties [***].
Section 3
GOVERNANCE
3.1Collaboration Coordinators.
(a)To support the conduct of the Collaboration, management of the Parties’ joint interest in a Collaboration Product and coordination under this Agreement, immediately after the Effective Date hereof, each Party shall notify to the other Party the name and contact information of its respective coordinator for this Agreement (each a “Collaboration Coordinator”). Each Party may in its sole discretion replace its Collaboration Coordinator at any time by notice in writing to the other Party.
(b)Each Collaboration Coordinator shall possess a general understanding of this Agreement and of key matters relating to the research and Development of a Collaboration Product set forth in an R&D Plan, including matters relating to the conduct of Clinical Trials.
(c)The role of the Collaboration Coordinator is to act as a key point of contact between the Parties to facilitate a successful Collaboration hereunder and resolution of deadlocks or disputes that may arise hereunder. Collaboration Coordinators shall oversee interactions between the Parties through meetings of the JSC. The Collaboration Coordinators shall attend, or cause its nominee with his proxy to attend, all JSC meetings and may bring to the attention of the JSC any financial, scientific or technical matters or issues either of them reasonably believes should be discussed.
3.2Joint Steering Committee.
(a)Establishment, Responsibilities. As soon as possible after the Effective Date, the Parties shall form a joint committee (the “Joint Steering Committee” or “JSC”), consisting of [***] representatives of each of Orano Med and Molecular Partners, which shall have the following responsibility for coordinating all activities under, and pursuant to, this Agreement:
i.The JSC shall coordinate and facilitate the exchange of information between the Parties in compliance with this Agreement in order to ensure that significant regulatory, technical scientific or safety issues concerning the Collaboration are addressed consistently and in a timely manner.
ii.The JSC shall discuss and agree upon an R&D Plan and a R&D Budget for each Collaboration Product and any updates thereto pursuant to Section 4.3(c).
iii.The JSC shall discuss and decide on the substitution of DLL3 as the target for the First Product and any Additional Candidates pursuant to Section 4.2.
iv.The JSC shall (without prejudicing any pre-agreed timelines and subject to Section 4.3(d)) review, comment on and approve all regulatory filings and communications for the Collaboration Products and shall discuss and decide on matters concerning Clinical Trials performed under a R&D Plan.
v.The JSC shall discuss and agree upon estimated Collaboration Costs, any updates thereto and, if applicable, cost overruns pursuant to Section 5.4.
vi.The JSC shall facilitate the Parties’ joint efforts to achieve an Out-Licensing on and subject to Section 7.2.
vii.The JSC may discuss issues relating to any proposed publication, communication or other disclosure regarding a Collaboration Product or this Collaboration.
viii.The JSC will establish the Launch Preparedness Plan and, at the request of either Party upon exercise of the Option by the other, it will review and discuss evidence relating to the achievement of the requirements thereunder.
ix.The JSC shall discuss and, as applicable, try to resolve any disputed financial aspects arising from Collaboration Costs overruns, Quarterly Reports or related supporting documentation and more generally, the JSC shall try to resolve any dispute or claim arising out of, relating to or in connection with any provision of this Agreement.
x.The JSC may agree to establish one or more subcommittees operating under its authority to address technical, financial, intellectual property and operational issues arising from the Collaboration and establish the operating rules and delegated competencies thereof.
(b)Meetings, JSC Chair. The JSC shall meet as soon as practicable after the Effective Date and then no less than on a quarterly basis or more often as reasonably considered necessary at the request of either Party, acting through its Collaboration Coordinator. The JSC may meet in person or by means of teleconference, Internet conference, videoconference or other similar communications equipment. Each Party shall be responsible for its expenses, including travel costs incurred for attending the JSC. The Parties’ Collaboration Coordinators will alternately be responsible for hosting the JSC meetings as chair (the “JSC Chair”). The JSC Chair shall be responsible for calling meetings of the JSC with at least one (1) month prior notice, for leading the meetings and for distributing an agenda for the JSC. The JSC Chair will include on the agenda any item within the scope of the responsibility of the JSC that is requested to be included by a Party, and will distribute the agenda to the Parties no less than one (1) week before any meeting of the JSC, together with a written update report which contains information about overall progress under the Collaboration, in particular under an R&D Plan.
(c)Minutes. All JSC meeting minutes shall be drafted by the JSC Chair, reviewed by the other Collaboration Coordinator and ultimately be approved by both Parties prior to formal adoption.
(d)Quorum, Consensus. [***] of the JSC will constitute a quorum for any meeting, provided that at least [***] representative from each Party is present. Decisions of the JSC shall be based on the consensus of representatives of both Parties. In the event that the JSC cannot agree on a matter at a JSC meeting or within a period of [***] hereafter, such matter or dispute shall be escalated to the Parties’ respective Executive Officers, who shall confer in good faith on the resolution of the issue and attempt to decide on such matter or settle such dispute within [***], or such longer period as may be mutually agreed. Any final decision mutually agreed to by the Executive Officers shall be conclusive and binding on the Parties.
3.3Resolution of Disputes. In the event that the Parties’ respective Executive Officers cannot decide on the matter or settle such dispute within the designated period set forth in Section 3.2(d), the following shall apply during the R&D Term with respect to any given Collaboration Product:
(a)[***] shall have final decision-making authority for [***], provided that such decision is [***]. Such final decision-making authority will [***] with regard to the [***]. The final decision-making authority for [***] will [***].
(b)[***] shall have final decision-making authority with respect to [***] provided that [***] shall have the final decision-making authority regarding [***].
(c)If the decision submitted to the JSC relates to actual or potential R&D Budget overrun of external Collaboration Costs resulting from (i) [***], (ii) [***], or (iii) [***], the Parties shall include such overrun in the R&D Budget.
(d)Any other matters shall be subject to mutual agreement of the Parties and, if not resolved by the Executive Officers within the designated period set forth in Section 3.2(d) [***], shall be resolved pursuant to Section 3.3(e). Such other matters include, but are not limited to:
i.[***];
ii.[***];
iii.[***];
iv.[***];
v.[***]; and
vi.[***].
(e)If the Parties are unable to resolve the matter of dispute through their Executive officers as set forth in Section 3.2(d), the Parties shall submit the matter to [***]:
i.[***].
ii.[***].
iii.[***].
iv.[***].
(f)After the relevant R&D Term, the decision making with respect to any subsequent Development activity, registration and Commercialization of a Collaboration Product will vest in [***].
(g)The inclusion of Additional Candidates in the R&D Program (including the agreement on a corresponding R&D Plan, R&D Budget and which Party would have final decision-making authority for the execution of any envisaged R&D activities) shall be [***]. [***], the candidate proposed for inclusion as an Additional Candidate [***].
3.4Scope of the JSC Governance. Notwithstanding the creation of the JSC, each Party shall retain the rights, powers and discretion granted to it hereunder, and no committee of any kind (including the JSC) shall be delegated or vested with rights, powers, or discretion unless such delegation or vesting is expressly provided herein, or the Parties expressly so agree in writing. Apart from the updates of an R&D Plan, R&D Budget and related Collaboration Costs, the JSC shall have no power to amend or modify this Agreement, and no decision of the JSC or any other committee created by the Parties shall be in contravention of any terms and conditions of this Agreement.
3.5Dissolution of the JSC. Subject to neither Party having exercised an Exit Decision, the JSC shall remain intact and active upon termination of the R&D Term and shall extend to any subsequent collaboration of the Parties pursuant to this Agreement, including the subsequent Development (including conduct of Clinical Trials), registration and Commercialization of subsequent Collaboration Products (beyond the First Product).
3.6Communication between the Parties. Any and all the communication, including without limitation, those at the JSC and between Executive Officers, all the reports and documents provided to the other Party, shall be in English unless otherwise agreed in writing between the Parties.
Section 4
R&D PROGRAM
4.1R&D Program; Mutual Licenses for R&D Program.
(a)The Parties undertake to (i) jointly research and Develop the First Product through to successful completion of a Registration Enabling Trial in such countries as the Parties shall agree in the R&D Plan, unless either Party exercises an Exit Decision; and (ii) potentially research and Develop Additional Candidates; and (iii) potentially research and Develop CDs (each of (i)-(iii), a “R&D Program”). In the event that a Registration Enabling Trial is completed in one country in the Territory to which an R&D Program relates, or any country in respect of which a multi-jurisdictional Clinical Trial is being conducted, this shall not impact on either Party’s obligations to supply the finished Collaboration Product or the components integrated in said Collaboration Product for clinical purposes to the other Party in respect of that R&D Program in accordance with the Clinical Supply Agreement, which obligations shall endure for the longer of: (i) the term of those obligations as set out in the Clinical Supply Agreement; and (ii) until a Registration Enabling Trial has successfully completed in each country to which an R&D Program applies, or as otherwise agreed by the Parties in writing.
(b)Molecular Partners shall grant and herewith grants to Orano Med an exclusive, worldwide license under Molecular Partners’ interest in the Joint IPR (i) to Manufacture the Collaboration Products as required under this Agreement and (ii) to carry out all of Orano Med’s other responsibilities under the R&D Program, including any activities assigned to it under an R&D Plan, save that Molecular Partners shall retain the rights to (1) use such Joint IPR to carry out all of Molecular Partners’ responsibilities under the R&D Program, including any activities assigned to it under an R&D Plan (e.g., conduct of a Clinical Trial), (2) use or sub-license such Joint IPR to manufacture and conjugate DARPins pursuant to this Agreement, and (3) grant a licence to a CMO as envisaged pursuant to Section 8.3, and to that extent the license granted under this subclause 4.1(b) shall be a sole rather than an exclusive licence.
(c)Molecular Partners shall grant and herewith grants to Orano Med a non¬exclusive, worldwide license under Molecular Partners Background and Sole Ownership (i) to Manufacture the Collaboration Products as required under this Agreement, and (ii) to carry out all of Orano Med’s other responsibilities under the R&D Program, including any activities assigned to it under an R&D Plan.
(d)Orano Med shall grant and herewith grants to Molecular Partners an exclusive, worldwide license under Orano Med’s interest in the Joint IPR to carry out all of Molecular Partners’ responsibilities under the R&D Program, including any activities assigned to it under an R&D Plan (e.g., conduct of a Clinical Trial), save that Orano Med shall retain the rights to use such Joint IPR (1)(i) to Manufacture the Collaboration Products as required under this Agreement and (ii) to carry out all of Orano Med’s other responsibilities under the R&D Program, including any activities assigned to it under an R&D Plan, and (2) to grant a licence to a CMO as envisaged pursuant to Section 8.3, and to that extent the license granted under this subclause 4.1(d) shall be a sole rather than an exclusive licence.
(e)Orano Med shall grant and herewith grants to Molecular Partners a non-exclusive, worldwide license under Orano Med Background and Sole Ownership to carry out all of Molecular Partners’ responsibilities under the R&D Program, including any activities assigned to it under an R&D Plan (e.g., conduct of a Clinical Trial).
(f)It is understood that the licenses under this Agreement that are stated “exclusive” shall be exclusive (including vis a vis the grantor) with respect to the Collaboration Product, and shall not extend to the use of different components therein outside of a Collaboration Product and the licenses which are stated “non-exclusive” shall mean that the grantor shall be entitled to grant non-exclusive licenses to others but shall not grant any such license with respect to the Collaboration Product and shall carve out the Collaboration Product from any such other licenses. Further, each Party shall retain the rights to use its respective IPR to perform its responsibilities hereunder, as may be subcontracted when permitted pursuant to the Agreement.
4.2Substitution of target.
(a)The Parties may decide to substitute the target DLL3 in the First Product by a different target (such target, the “Substitute Target”). Either Party may propose to the other Party to introduce a Substitute Target in to the R&D Program and Plan in place of the target DLL3 provided that both Parties agree on the Substitute Target and that such substitution occurs no later than the earlier of (i) [***] and (ii) [***]. A Party wishing to introduce a Substitute Candidate shall notify the other Party in writing and shall provide all relevant technical, scientific, safety and therapeutic information about the proposed target, including how it will impact the R&D Plan, Program and Budget and the benefits and risks of substitution over the target DLL3.
(b)Should the Parties wish to jointly engage in additional pre-clinical activities for Additional Candidates, they will enter into a Materials Transfer Agreement (“MTA”) substantially in the form of the MTA entered into by the Parties in relation to DLL3. They shall ensure that such MTA comprises [***]. Upon finalization of such pre-clinical studies for Additional Candidates, the Parties will decide, under the supervision of the JSC, whether or not to include such Additional Candidates under this Agreement. Should the Parties decide to include such Additional Candidates under this Agreement, such Additional Candidates shall be considered as a Collaboration Product, and all rights and obligations in relation thereto shall apply unless otherwise agreed. As of the date of entering into this Agreement, it is not Parties’ initial intention to engage in more than [***] additional pre-clinical programs for new targets, [***] of which would be taken to the clinical stage.
4.3R&D Plans and Budgets.
(a)The R&D Program shall be performed, on a Collaboration Product-by-Collaboration Product basis and, where relevant, a country-by-country basis, in accordance with a general work plan which shall describe, in reasonable detail, the research and Development activities and phases to be performed under the R&D Program for the relevant Collaboration Product, including pre-clinical study designs, clinical trial descriptions for all proposed targets, specific activities and deliverables to be performed and provided by the Parties, estimated timelines, and key milestones (“R&D Plan”) as well as the associated estimated budgets integrated thereto by phase (“R&D Budget”). The R&D Budget shall specify the estimated costs and expenses of Development (including external and internal, and notably CMC development costs) for the relevant Collaboration Product in each Calendar Year of the corresponding R&D Term, including the Clinical Transfer Price and all Logistics Costs. The initial R&D Budget will cover [***].
(b)As of the Effective Date, the Parties have agreed on an R&D Plan for the First Product as the first Collaboration Product attached at Exhibit B hereto and on a corresponding R&D Budget, attached hereto as Exhibit C.
(c)The JSC shall review and, as required, update the then current R&D Plans and R&D Budgets at least once per Calendar Quarter; this is with the exception of the [***] of the Term, during which period the R&D Budget and R&D Plans should only be updated as deemed strictly necessary by the JSC. However, each Party must diligently inform the other Party of any foreseeable significant change of costs of a Clinical Trial at the earliest anticipation and shall request a meeting of the JSC to discuss the necessity of an update to the relevant then current R&D Budget.
(d)Under the R&D Program for the First Product and under the oversight of the JSC,
i.Molecular Partners shall be responsible for certain pre-clinical/clinical and other Development activities with respect to the First Product subject to its reliance on input from Orano Med with respect to development and integration of the TAT Technology as set out in the corresponding R&D Plan;
ii.Orano Med shall be responsible for certain pre-clinical/clinical and other Development activities with respect to the First Product, subject to its reliance on input from Molecular Partners with respect to Development and integration of the DARPin technology as set out in the corresponding R&D Plan; and
iii.the roles and responsibilities of the Parties for subsequent targets (Collaboration Product targeting proteins other than DLL3) shall be separately agreed by the JSC upon program initiation.
4.4Clinical Trials.
(a)Subject to the Opt-Out provisions set out under Section 6 below, Molecular Partners shall be the Sponsor for all Clinical Trials of the First Product, and Orano Med shall be the Sponsor for the first Additional Candidate. For Additional Candidates which the Parties agree to pursue, the Parties shall agree in the JSC who will be the Sponsor of the respective Clinical Trials and the same shall apply for any testing to be performed in the course of Development of any CDs which the Parties agree to pursue. If the Parties are unable to agree on the clinical trial design or protocol development for any Clinical Trial or the Clinical Trial footprint (site mapping), the matter shall be referred to the JSC for resolution.
(b)The Party determined as Sponsor of a Clinical Trial pursuant to this Agreement shall:
i.subject to Section 4.9, provide regulatory lead for the Clinical Trials set forth in an R&D Plan and shall accordingly ensure that all directions and formalities vis-a-vis any Regulatory Authority and/or ethics committee with jurisdiction over such Clinical Trials are complied with, whereby the other Party shall fully cooperate with the Sponsor to comply with such directions and formalities, including but not limited to, with respect to CTA/IND/BLA submissions. For the avoidance of doubt, whilst both Parties are responsible for compliance with applicable Nuclear Activity Laws for their respective activities pursuant to the R&D Plan, Orano Med shall be responsible for setting up the supply chain for the supply of the Collaboration Products, to the exclusion of those components supplied by Molecular Partners or its subcontractors, including as to [***] (as the case may be), [***], [***] and/or [***] (and associated logistics arrangements) in compliance with Nuclear Activity Laws. If and to the extent any such activities were to be carried out by Molecular Partners, it shall endeavour to develop a suitable compliance strategy (at Molecular Partners’ cost) that Molecular Partners can adopt to ensure its compliance with Nuclear Activity Laws (as applicable);
ii.conduct the Clinical Trials under an R&D Plan as Sponsor, prepare and submit Regulatory Documentation and perform related activities (including obtaining IEC/IRB approvals, obtaining signed informed consents, etc.);
iii.list the Clinical Trials in an R&D Plan on www.clinicaltrials.gov, EudraCT or other public registry in any country in which any such Clinical Trial is being conducted in accordance with Applicable Law and in accordance with its internal policies relating to clinical trial registration; iv.select suppliers, and manage contract negotiation with suppliers, with regard to all contracts relating to the Clinical Trials of an R&D Plan as may be needed for Clinical Trial activities under the R&D Program and Collaboration (including as to supply of IMP, pharmacovigilance, site, CRO or other agreements) and provide relevant contracts, including without limitation with CRO, to the other Party for review comments and approval, as per an approval process and timeline agreed by the Parties in the R&D Plan;
v.negotiate and, following review and commenting by the other Party, enter into contracts relating to the Clinical Trials of an R&D Plan as may be needed for Clinical Trial activities under the R&D Program and Collaboration (including as to supply of IMP, pharmacovigilance, site, CRO or other agreements) and perform related activities thereunder (including approving contract deliverables; monitoring and approving performance etc.);
vi.notify the other Party in the event of an inspection by a Regulatory Authority(ies) relating to the Clinical Trials set forth in an R&D Plan and the results of any such inspection;
vii.notify the other Party in the event of any serious adverse events related to a Collaboration Product and provide the other Party with the copies of relevant safety reports submitted to the Regulatory Authority(ies);
viii.report to the other Party immediately if any Regulatory Authority(ies) places a Regulatory Hold or any restriction on any aspect of one of the Clinical Trials set forth in an R&D Plan;
ix.ensure compliance with all Applicable Laws pertaining to safety reporting for the Clinical Trials set forth in an R&D Plan and related safety activities in relation to a Collaboration Product;
x.provide the other Party with (i) email updates on a monthly basis regarding the progress of patient recruitment versus the forecasting and (ii) a report on at least a quarterly basis or upon the other Party’s reasonable request, on the status of the Clinical Trials set forth in an R&D Plan, including but not limited to information data regarding the number and status of sites, the number of screened subjects, the ongoing, discontinued and completed subjects, first patient first visit (“FPFV”), last patient first visit, (“LPFV”), last patient last visit (“LPLV”), a Collaboration Product relevant design information and available immunogenicity results and safety updates as contemplated by the protocol and/or routinely performed by the Sponsor in its normal course of management of such Clinical Trials;
xi.provide the other Party with the reports for the Clinical Trials set forth in an R&D Plan, which includes not only the final version of such report but also the progress of such Clinical Trials and interim data of the Clinical Trials as reasonably requested by the other Party; and xii.assume any other responsibilities as may be agreed upon by the Parties.
(c)The Party not acting as Sponsor of a given Clinical Trial set forth in an R&D Plan shall:
i.in accordance with guidance issued by the JSC review and comment on a timely basis on all main contracts relating to the Clinical Trials of an R&D Plan as provided to it by the Sponsor (including contracts as to supply of IMP, pharmacovigilance, or other agreements);
ii.provide the Sponsor with technical and consulting support together with relevant documentation for CTA/IND/BLA regulatory submissions for the Clinical Trials set forth in an R&D Plan;
iii.provide the Sponsor with any reasonable assistance as required by any interactions of the Sponsor with Regulatory Authorities in relation to the Collaboration;
iv.be available for the auditing or qualification activities of the other Party; and
v.assume any other responsibilities as may be agreed upon by the Parties.
(d)Modifications of Protocols. The Sponsor shall not modify any a protocol of a given Clinical Trial set forth in an R&D Plan without the approval of the JSC, provided that such approval shall not be withheld to the extent such modification is required by any competent Regulatory Authority with jurisdiction or authority over such Clinical Trial or the GMP manufacturing facility of a Collaboration Product. In such case, the Sponsor shall promptly inform the other Party thereof and the Parties shall negotiate in good faith any adjustments to an R&D Budget which may become necessary due to the modification.
4.5Regulatory Filings, Ownership.
(a)Responsibility. For the First Product and subject to Section 4.9, Molecular Partners (or if Molecular Partners is not the Exploiting Party, Orano Med) will be responsible for the preparation and filing of all Regulatory Documentation under the relevant R&D Program, including (as applicable) filing the CTA/IND/BLA, and other regulatory filings, and for all regulatory communications concerning the First Product (subject to Orano Med’s right and obligation, as long as it is the Manufacturer of the Collaboration Product, to submit filings comprising Regulatory Documents relating to the Manufacturing of the Collaboration Product or as relates to [***]), subject to review and approval at the JSC and with reasonable support from Orano Med. For the second Collaboration Product, Orano Med (or if Orano Med is not the Exploiting Party, Molecular Partners) will be responsible for the preparation and filing of all Regulatory Documentation under the relevant R&D Program and the first sentence shall apply mutatis mutandis (except that the requirement on Orano Med to have the right and obligation, as long as it is the Manufacturer of the Collaboration Product, to submit filings comprising Regulatory Documents relating to the Manufacturing of the Collaboration Product or as relates to 212 Pb shall remain). For any subsequent Additional Candidates on which the Parties agree to pursue, the Parties shall agree through the JSC which Party will be appointed as Sponsor and be responsible for the preparation and filing of all Regulatory Documentation.
(b)Submission of Regulatory Documentation; Participation Rights.
i.The Party determined as responsible for preparation and filing of Regulatory Documentation for a given Collaboration Product pursuant to subclause 4.5(a) shall submit the Regulatory Documentation to Regulatory Authorities under its name and responsibility, subject (y) to review, commenting and approval by the JSC pursuant to Section 4.4(b)ii and (x) to review and commenting of the other Party pursuant to Section 4.4(b)iii. Each Party shall notify the other Party of any material Regulatory Documentation submitted to or received from the Regulatory Authorities as soon as possible and no later than within [***] of receipt or awareness and shall provide the other Party with copies thereof.
ii.During the R&D Program, the JSC shall have the right to review, comment on and approve all regulatory filings and communications for the Collaboration Products. The responsible Party shall send to the JSC drafts of all material Regulatory Documentation intended to be submitted to a Regulatory Authority at least [***] prior to submission, or solely in the event of urgent submissions or as mutually agreed by the Parties, any shorter reasonable period of time. The Collaboration Coordinators shall promptly call a meeting of the JSC to discuss such draft of Regulatory Documentation with the aim to provide comments to the responsible Party by no later than [***] (or in urgent cases a shorter period of time) following receipt of the draft by the JSC. The responsible Party shall consider any comments received by it in due time in good faith, shall reflect such comments in the Regulatory Documentation and shall re-submit the updated drafts to the JSC for approval as soon as reasonably feasible. Upon receipt of the updated drafts, the Collaboration Coordinators shall promptly call a second meeting of the JSC to review and approve the updated drafts and the JSC shall provide its approval or objection to the responsible Party immediately following such meeting. In the event that the JSC does not approve the updated draft, the dispute escalation mechanism pursuant to Section 3.3 shall apply. In no event shall the responsible Party submit to a Regulatory Authority a Regulatory Documentation without prior approval of the JSC, unless it is mandated to do so by a Regulatory Authority.
iii.The Party not responsible for preparation and filing of Regulatory Documentation for a given Collaboration Product pursuant to this Agreement shall have the right, until such Party exercises an Exit Decision, to review and comment on all regulatory filings and communications for the relevant Collaboration Product, not forming part of the closed part of the Drug Master File. The responsible Party shall send to the other Party drafts of all material Regulatory Documentation intended to be submitted to a Regulatory Authority at least [***] prior to submission, or solely in the event of urgent submissions or as mutually agreed by the Parties, any shorter reasonable period of time. The other Party shall provide its comments, if any, to the responsible Party by no later than
[***] (or in urgent cases a shorter period of time) following receipt of the draft. The responsible Party shall consider any comments received by it in due time in good faith and may implement the comments received at its sole discretion. Notwithstanding the foregoing, after its Exit Decision, Orano Med shall remain entitled to review and comment on all CMC Regulatory Documentation prior to submission to the extent related to TAT Technology, and Molecular Partners shall have an equivalent right to review and comment on all Regulatory Documentation prior to submission, to the extent related to the DARPin Technology.
iv.To the extent this may be accommodated, the Party not responsible for preparation and filing of Regulatory Documentation for a given Collaboration Product under this Agreement shall be entitled to attend all scheduled meetings of the responsible Party with Regulatory Authorities. The responsible Party shall inform the other Party about any envisaged meeting with a Regulatory Authority in the Territory as soon as reasonably practicable upon it becoming aware of any such meeting and shall allow the other Party, at the other Party’s request and sole cost and expense, to participate in any meeting with Regulatory Authorities in the Territory, physically or, if possible, via electronic media. Independently of whether or not the other Party participates in any meeting of the responsible Party with a Regulatory Authority, the responsible Party shall prepare and send to the other Party written minutes of any meeting with such Regulatory Authority in sufficient detail to reasonably enable a non-present recipient to assess the agenda, content and outcome of such meeting within [***] following such meeting.
(c)Drug Master Files; Rights of Reference.
i.Subject to the licenses and rights granted hereunder, Orano Med shall own and maintain all right, title and interest in and to all Drug Master Files for [***] and [***], including in the Field in the Territory. Subject to the licenses and rights granted to hereunder, Molecular Partners shall own and maintain all right, title and interest in and to all Drug Master Files for DARPin proteins, including in the Field in the Territory.
ii.The Party responsible for submission of Regulatory Documentation for a given Collaboration Product shall be entitled to reference the Regulatory Documentation of the other Party, including to use the open part of any Drug Master File owned or otherwise Controlled by the other Party for Development and Commercialization of the relevant Collaboration Product in the Field in the Territory and accordingly (i) Molecular Partners shall be entitled to access and reference the open part of any Drug Master File owned or otherwise Controlled by Orano Med with respect to Pb212 and [***] and with respect to any linkers or chelators supplied by Orano Med and (ii) Orano Med shall be entitled to access and reference the open part of any Drug Master File owned or otherwise Controlled by Molecular Partners with respect to the relevant DARpin proteins and with respect to any linkers or chelators supplied by Molecular Partners. On such responsible Party’s request, the Party owning the Drug Master File, if any, will provide the closed part of such Drug Master Files directly to the competent Regulatory Authority in the Territory by itself or through a Third Party agent in the Territory, provided that if there are local requirements that a Drug Master File be submitted by a single party in the relevant country, the Party owning the Drug Master File shall permit the other Party (or its appointee) to submit the Drug Master File, including the closed part, to the Regulatory Authority under strict obligations of confidentiality and non-use for other purposes. The Party owning the Drug Master File will be solely responsible to meet all local requirements of the respective competent Regulatory Authority in the Territory regarding the substance of the Drug Master File in its own name and behalf. If a Party has not filed a Drug Master File, it shall provide access to the responsible Party, upon the latter request, to all necessary CMC Data and information to be submitted to the Regulatory Authority.
iii.Where necessary, the Party granting rights of reference hereunder shall provide appropriate notification of the other Party’s access and reference rights to the applicable Regulatory Authorities.
4.6Data. Notwithstanding Section 12, the Parties agree that any and all Data shall be jointly owned in equal shares by the Parties to the extent such Data are related to joint activities and not either Party’s Background IPR. The Parties shall grant to each other the right to use the Data, in such manner as set forth in Section 4.1. The Parties acknowledge and agree that any use of the Data which requires the consent of Patient must comply with the terms of the corresponding Patient’s Informed Consent Form (“ICF”).
4.7Records.
(a)In connection with the Collaboration, each Party shall prepare, maintain, and retain or shall cause to be prepared, maintained, and retained complete and accurate written records, accounts, notes, reports and data with respect to its activities conducted pursuant to an R&D Plan in conformity with Applicable Laws and standard pharmaceutical industry practices, provided that in no case shall written documentation be maintained for less than [***] following the Calendar Year to which such records pertain or such longer period as required by Applicable Law. Such records shall fully and properly reflect all work done and results achieved in the performance of the development activities in good scientific manner appropriate for regulatory and patent purposes. Upon a Party’s written request, solely with respect to information, records or data to which such Party is entitled to receive pursuant to other provisions of this Agreement, the other Party shall send legible copies of the aforesaid information to the requesting Party during the Term and for a minimum of [***] following the Term.
(b)The Parties acknowledge the importance of ensuring that the performance of activities under an R&D Plan is undertaken in accordance with the following good Data management practices: (i) Data shall be generated using sound scientific techniques and processes; (ii) Data shall be accurately and reasonably contemporaneously recorded in accordance with good scientific practices by Persons conducting research hereunder; (iii) Data shall be analyzed appropriately without bias in accordance with good scientific practices; and (iv) all Data and results shall be stored securely and shall be easily retrievable.
4.8Governmental Inspection. If any Regulatory Authority conducts or gives notice to either Party of its intent to conduct an inspection or audit of such Party or its facilities (including any facilities of any Third Party performing activities for or on behalf of such Party) that specifically relates to such Party’s performance hereunder, or that could materially affect such Party’s ability to perform hereunder and in accordance herewith, such Party shall promptly provide notice of such inspection or audit to the other Party and shall provide updates from time-to-time, including upon such other Party’s reasonable request, regarding the results of such audit or inspection, including any corrective steps to be taken. If such Party receives sufficient prior notice of such inspection or audit, it shall use reasonable efforts to permit the other Party to be present at such inspection or audit to the extent applicable to any Collaboration Product or such other Party’s performance, and shall permit such other Party to provide input (and shall consider and incorporate relevant comments in good faith) in connection with responses provided to any such Regulatory Authority that relate specifically to any Collaboration Product or to such other Party’s performance hereunder, provided that for clarity, the Party being inspected or audited shall have the right to make the final decision in connection with any such responses (following escalation to Executive Officers where reasonably practicable given time constraints in connection with any such inspection or audit). The rights in this Section 4.8 shall no longer apply if a Party exercises its Exit Decision except with respect to any activities conducted by such Party prior to the exercise of the Exit Decision, or activities under a Clinical Supply Agreement or Commercial Supply Agreement.
4.9Compliance with Nuclear Activity Laws.
(a)Each Party shall conduct its activities under the R&D Plan in compliance with Nuclear Activity Laws including with respect to its respective responsibilities in the R&D Plan (including those of a sponsor of a Clinical Trial) and shall respectively obtain and maintain all Nuclear Activity Licenses for the duration of such activities under the relevant R&D Program as designated in the R&D Plan (and Orano Med shall be responsible for identifying where those licenses are required), provided that the Parties will collaborate to develop a compliance strategy and if requested by Molecular Partners, Orano Med will provide reasonable support and training to Molecular Partners and all Orano Med’s reasonable costs related to the requested support or training of Molecular Partners shall be included in the R&D Budget.
(b)Upon Option exercise, the Exploiting Party shall be responsible at all times for compliance with Nuclear Activity Laws of its respective activities pursuant to the Agreement and shall obtain and to the extent it is required to do so with respect to its activities under this Agreement maintain all Nuclear Activity Licenses for the duration of such activities under this Agreement. For the avoidance of doubt Orano Med has primary responsibility for establishing and maintain the supply chain for each Collaboration Product (and associated compliance with Nuclear Activity Laws) unless the Parties agree otherwise under this Agreement. To the extent requested by Molecular Partners, and as long as it supplies the Collaboration Products, Orano Med shall provide all reasonable assistance to Molecular Partners to support its compliance with any obligations it may have under Nuclear Activity Laws as the Exploiting Party, to the extent within Orano Med’s expertise. Subject to the foregoing, this assistance may include: (i) training of personnel; (ii) advising on applicable Nuclear Activity Laws and consulting on compliance; (iii) assisting with applications to Regulatory Authorities and with reporting obligations, if any; and (iv) conducting risk or impact assessments and supporting Molecular Partners to develop procedures and solutions to comply with Nuclear Activity Laws. Orano Med’s reasonable costs related to such assistance shall be borne by Molecular Partners and treated as Operating Expenses, to the extent strictly related to the Collaboration Products.
(c)Without limiting the foregoing, each Party shall ensure that with respect to its activities:
i.all personnel responsible for Nuclear Activities under this Agreement are appropriately qualified, skilled and trained for the relevant Nuclear Activities and that all sites at which Nuclear Activities are carried out accord with Nuclear Activity Laws and are appropriately registered or licensed, as required; and
ii.traceability systems are put in place in relation to the Collaboration Products, as required under Nuclear Activity Laws.
(d)Further, and to the extent applicable, Orano Med shall be responsible for ensuring, as long as it supplies the Collaboration Product, that:
i.the [***] is produced in compliance with all Nuclear Activity Laws and that the Collaboration Products are Manufactured, transported, stored, packaged and handled in accordance with Nuclear Activity Laws; and
ii.secure and compliant logistics solutions are implemented for the delivery of Collaboration Product to applicable delivery sites, and that these solutions are designed and implemented in accordance with Nuclear Activity Laws.
Section 5
R&D PROGRAM COST SHARING
5.1General Principle. During the Development of a Collaboration Product jointly conducted by both Parties, and until either Party exercises an Exit Decision, the internal costs and external costs and expenses of the Parties (in each case as identified in the R&D Budget) which are incurred in the course of the Parties’ performance of their activities under an R&D Plan and R&D Budget, including Manufacturing, regulatory, pre-clinical and clinical and other activities described in the R&D Plan (“Collaboration Costs”) shall be equally borne and shared by the Parties.
5.2Scope of Collaboration Costs, Excluded Costs.
(a)Collaboration Costs may include, as applicable, for the supply of a Collaboration Product during a Calendar Quarter, and to the extent included in the R&D Budget,
i.[***];
ii.[***];
iii.[***]; and
iv.[***].
(b)Each Party shall be solely responsible at its own cost for filing, registering, prosecuting, maintaining and defending its own Intellectual Property Rights, except otherwise agreed by the Parties for specific IPR. Costs for filing, registering, prosecuting, maintaining and defending Joint IPR, and any other IPR the Parties may agree, shall be equally borne and shared by the Parties.
(c)Third Party License Payments will be [***].
5.3Reconciliation. Invoicing.
(a)Each Party shall use standard industry systems and processes to record the number of hours and FTEs working on an R&D Plan, as consistently applied by such Party to all (internal or external) programs.
(b)The specific detail regarding the reporting, reconciliation and invoicing will be agreed by the JSC, however unless otherwise agreed within [***] following the end of a given Calendar Quarter, on a Collaboration Product-by-Collaboration Product basis, each Party shall send a report to the other Party outlining in reasonable detail all internal and external costs and expenses actually invoiced to or incurred by it during the prior three calendar months in the course of such Party’s performance of an R&D Plan, whereby such report shall be sent by electronic means with reasonable supporting documentation and calculations (such as invoices for Out-of-Pocket Costs) and such other information as reasonably required by the other Party to review and assess such costs and expenses (a “Quarterly Report”). At either Party’s reasonable request, an appointee from the finance team of the other Party will be reasonably available to discuss and answer questions regarding such Quarterly Report.
(c)The Parties shall accept all internal and external costs and expenses as Collaboration Costs equitable for cost sharing, if they fulfil the following criteria: (i) actually incurred by or invoiced to a Party in the performance of such Party’s activities under an R&D Plan, (ii) reasonably documented, (iii) provided for as Collaboration Costs under an R&D Budget or accepted pursuant to Section 5.4, and (iv) not yet captured in a prior Calendar Quarter’s reconciliation calculation. The Parties’ Collaboration Coordinators (possibly through the JSC) shall use good faith efforts to discuss and resolve any disputed aspects of the Quarterly Reports or related supporting documentation within [***] following receipt by each Party’s report hereunder.
(d)In application of the principles set forth in Section 5.1, both Collaboration Coordinators (or, if applicable, a competent subcommittee established for this purpose by the JSC) will cooperate to calculate the relative portion of the quarterly Collaboration Costs invoiced to or incurred by each Party and determine the amount payable by one Party (the Party whose relative portion is under [***] of the relevant quarterly Collaboration Costs, referred to herein as the “Deficit Party”) to the other Party (the Party whose relative portion exceeds [***] of the relevant quarterly actually realized part of the budgeted Collaboration Costs, referred to herein as the “Excedent Party”) to achieve a [***] sharing for the actual Collaboration Costs incurred each quarter to the extent in accordance with the R&D Budget, such amount being referred to herein as the “Quarterly Excess”. For the avoidance of doubt, the Quarterly Excess shall be determined with respect to the time period relevant to each Quarterly Report.
(e)The Excedent Party shall issue an invoice to the Deficit Party in the amount of the Quarterly Excess. The payment terms set forth in Section 11 shall apply.
5.4Cost Overruns.
(a)In the event that external costs and expenses actually incurred by both Parties in the course of their performance of an R&D Plan in a given Calendar Year in the aggregate exceed the amount budgeted for external costs in the corresponding R&D Budget for such Calendar Year the Party first becoming aware of a potential overrun in external costs shall immediately notify the other Party to discuss such overruns at the JSC, including an analysis of costs and expenses, the allocation of such cost overruns between the Parties or, as applicable, a potential amendment of an R&D Budget and each Party has the right to call for an audit of the other Parties’ external costs pursuant to Section 11.4. The Parties undertake to use Commercially Reasonable Efforts to ensure that potential costs overruns are discussed and agreed in, and as relevant budgeted, by the JSC before they are actually incurred.
(b)Unless otherwise approved by the JSC, in the event that costs actually incurred by either Party in the course of its performance of an R&D Plan in a given Calendar Year in the aggregate exceed the amount budgeted for such Party’s costs in the corresponding R&D Budget for such Calendar Year, such excess amounts shall be borne [***].
5.5Third Party Agreement Payments.
(a)On a Collaboration Product-by-Collaboration Product basis, if at the Effective Date a Party Controls any IPR of a Third Party (“Third Party IPR”) that it determines, in its reasonable judgment, is necessary in order to Exploit any Collaboration Product(s), then such Party shall bear the cost of any payments to such Third Parties for the use of such Third Party IPR.
(b)If after the Effective Date a Party determines, in its reasonable judgment, that any IPR of a Third Party is necessary or reasonably useful to Exploit any Collaboration Product(s) pursuant to this Agreement, then such Party shall notify the JSC of such Third Party IPR, along with (where reasonably practical) a proposal of the terms upon which such Third Party IPR are available to license or acquire for use in connection with the applicable Collaboration Products, including any amounts payable to such Third Party in consideration of such license or other rights, as applicable, including royalties, milestones or other amounts arising from either Party’s practice of the applicable Third Party IPR under the Collaboration (such payments, the “Third Party Agreement Payments”). The JSC shall determine whether or not to seek a license under, or to acquire such Third Party IPR for the purposes of Exploiting such Collaboration Product(s). If the JSC decides to seek such a license or acquisition, then the Party which determined the need for the Third Party IPR (the “Requesting Party”) [***] the agreement pursuant to which a license under or acquisition of such Third Party IPR would be obtained (such agreement, the “Third Party Agreement”), provided that [***].
(c)If the Parties cannot agree through the JSC on whether to enter into any such Third Party Agreement, or upon the terms of such Third Party Agreement, then [***]. The Exploiting Party (or prior to Option exercise or an Exit Decision, the Requesting Party), shall have the final decision on whether to enter into such Third Party Agreement as long as [***].
i.With respect to [***] (or Requesting Party where applicable), the Exploiting Party shall have the right to treat [***] Third Party Agreement Payments as Operating Expenses for purposes of profit sharing. [***].
ii.With respect to [***], if the Exploiting Party (or Requesting Party where applicable) decides to enter into a Third Party Agreement, then the Exploiting Party (or Requesting Party where applicable) shall have the right to treat [***] of the payment under such Third Party Agreement as Operating Expenses for purposes of profit sharing.
Section 6
OPTION AND CONSEQUENCES OF OPTION EXERCISE
6.1Option.
(a)Each Party hereby grants to the other Party, on a Collaboration Product-by-Collaboration Product basis and subject to the terms and conditions in this Section 6, an exclusive option (“Option”) to obtain from the granting Party:
i.an exclusive, worldwide license, with the right to grant sublicenses to a sublicensee, with written consent from the other Party, under the granting Party’s interest in the Joint IPR to Exploit a Collaboration Product, save that the granting Party shall retain the rights to use such Joint IPR to carry out any ongoing obligations under subclauses 4.1(b) to 4.1(e) (as applicable to the granting Party), and to that extent that licence granted shall be a sole rather than an exclusive licence;
ii.a non-exclusive worldwide license, with the right to grant sublicenses to a sublicensee with the prior written consent of the other Party, under the granting Party’s Background IPR or Sole Ownership to Exploit a Collaboration Product; and
iii.for purposes of the above, subject to Section 6.7, such consent to a sublicense shall not be unreasonably withheld conditioned or delayed provided always that the proposed sublicense remains subject to and is consistent with the terms and conditions of the present Agreement (including with regard to IPR, confidentiality and exclusivity obligations). As between the Parties, the sublicensing Party shall remain liable for the performance of the sublicensed activities.
6.2Option for the First Product, Rights of Exercise.
(a)At the request of either Party [***], the Parties shall appoint a mutually agreed consultant to perform within a reasonable timeframe a market study determining the potential of the Collaboration Product broken down per key markets and countries, and more particularly in the [***], and [***], and shall share the costs of this study, which may be updated from time to time as reasonably requested by either Party. If the Parties cannot agree on a consultant within [***], each Party shall appoint one Expert, who shall appoint the required consultant by mutual agreement by the two Experts, within [***], or such longer period as may be mutually agreed by the Parties.
(b)Thereafter, the Parties shall meet through the JSC to discuss and agree a launch preparedness plan (the “Launch Preparedness Plan”) detailing (i) the Commercialization investments expected from either Party as a Commercialization party [***] (the “Commercialization Requirements”) and (ii) the Facilities and logistics solutions presented by Orano Med [***] (“Manufacturing and Logistics Requirements”). [***]. For the avoidance of doubt, the Launch Preparedness Plan shall serve to determine whether the Option can be exercised or not as set forth in subclause (c) below, but shall not be binding on the Parties as the extent of the obligations of the Parties with respect to the Manufacture, supply and Commercialization of the Collaboration Product which shall be those set forth in the License and Supply Agreements. The Launch Preparedness Plan may only be updated or modified by agreement of the Parties at the JSC.
(c)Molecular Partners shall have the first right to exercise an Option for the First Product by providing to Orano Med during the Exclusive First Option Period, (i) a written notice of Option exercise for the First Product, which notice shall be accompanied by reasonable evidence that Molecular Partners satisfies the Commercialization Requirements criteria with respect to the First Product or is reasonably expected to satisfy them as at the time of launch of the First Product upon Orano Med confirming that it has met the Manufacturing and Logistics Requirements (or is reasonably expected to meet them at the time of the launch of the First Product), provided that such Commercialization Requirements shall be adjusted to the extent that (i) Orano Med cannot reasonably demonstrate at that time that it has met (or is reasonably expected to meet) the Manufacturing and Logistics Requirements set out in the applicable Launch Preparedness Plan or (ii) the Parties have agreed to adjust the Commercialization Requirements or the Manufacturing and Logistics Requirements.
(d)Promptly upon receipt of the documentation referred to in Section 6.2(c)from Molecular Partners, Orano Med – in its capacity as receiving Party – shall perform the evaluation and approval process set forth in Section 6.4.
(e)In the event that (i) the first Option right of Molecular Partners for the First Product has lapsed due to non-service of a notice to exercise the first Option right for the First Product pursuant to Section 6.2(c) or (ii) the Option exercise by Molecular Partners is not determined as successful and completed pursuant to Section 6.4, then Orano Med shall have the second right to exercise an Option for the First Product by providing to Molecular Partners at any time during the Exclusive Second Option Period, (x) a written notice of Option exercise for the First Product and (y) reasonable evidence that Orano Med satisfies the Commercialization Requirements and the Manufacturing and Logistics Requirements.
(f)Promptly upon receipt of the documentation referred to in Section 6.2(f), Molecular Partners – in its capacity as receiving Party – shall perform the evaluation and approval process set forth in Section 6.4.
(g)In the event the first Option right exercise by Molecular Partners pursuant to Section 6.2(c) is determined as successful and completed pursuant to Section 6.4, Molecular Partners shall (subject to Section 6.2(j) and Sections 4.5(a) and 4.9) be responsible for the preparation and filing of all Regulatory Documentation to the extent required.
(h)In the event the second Option exercise by Orano Med pursuant to Section 6.2(e) is determined as successful and completed pursuant to Section 6.4, Orano Med shall (subject to Section 6.2(j) and Sections 4.5(a) and 4.9) be responsible for the preparation and filing of all Regulatory Documentation to the extent required.
(i)In the event that (i) the second Option right of Orano Med for the First Product has lapsed due to non-service of a notice to exercise the second Option right for the First Product pursuant to Section 6.2(e) or (ii) the Option exercise by Orano Med is not determined as successful and completed pursuant to Section 6.4, the Parties shall undertake Commercially Reasonable Efforts to achieve an Out-Licensing of the First Product on and subject to Section 7.
(j)As soon as the Option exercise by Molecular Partners or, as applicable, by Orano Med, is successful and completed pursuant to Section 6.4, the Parties will negotiate in good faith the terms of and enter into License and Supply Agreements under which they would collaborate in connection with Developing, seeking Regulatory Approvals of and Commercializing the Collaboration Products, as provided under Section 6.5 below.
6.3Option for Additional Candidates; Rights of Exercise.
(a)With regard to the first Additional Candidate (i.e.: the first Collaboration Product included into this Collaboration in addition to the First Product), Orano Med will have the first right to exercise the Option for such first Additional Candidate during the Exclusive First Option Period, and Molecular Partners shall have the second right to exercise the Option for such first Additional Candidate. The exercise of the Option for the first Additional Candidate shall be performed pursuant to the process set forth in Section 6.2 and Section 6.2 shall apply mutatis mutandis, whereby Orano Med shall have the first right to exercise the Option, Molecular Partners shall have the second right to exercise the Option and the concerned Collaboration Product shall be the first Additional Product (in lieu of the First Product), provided that Orano Med shall remain responsible for the Manufacture of the Collaboration Product and submission of the Manufacturing and Logistics Requirements unless otherwise agreed by the Parties in writing or provided in the Clinical Supply Agreement or Commercial Supply Agreement.
(b)Regarding any subsequent Additional Candidate (i.e.: any Collaboration Product included into this Collaboration in addition to the First Product and the first Additional Candidate), the Parties shall discuss no later than [***] for such subsequent Additional Candidate who should have the first right to exercise the Option in relation to such subsequent Additional Candidate. Failing an agreement, the Parties shall undertake Commercially Reasonable Efforts to achieve an Out-Licensing of the applicable Collaboration Product on and subject to Section 7, whereby, in such case both Molecular Partners and Orano Med shall be entitled to participate in such competitive process and to match the offer of any Third Party bidding for an Out-Licensing. The Out-Licensing of a subsequent Additional Candidate shall be without prejudice to the possibility for the Parties to jointly Develop the concerned Additional Candidate until its Commercialization.
6.4Evaluation and Approval Process.
(a)From time to time through the JSC and promptly upon receipt of a written notice of Option exercise and accompanying documentation issued by the other Party within its applicable Exclusive First/Second Option Period, the receiving Party shall review the evidence submitted by (i) the issuing Party to confirm whether it satisfies the Commercialization Requirements and (ii) the submission by Orano Med as to whether Orano Med meets the Manufacturing and Logistics Requirements and taking account of the interdependency of the requirements in (i) and (ii). Any and all questions in relation to this assessment shall be handled in the JSC. The receiving Party shall not unreasonably withhold or delay its approval with respect to an Option exercise. When Orano Med is responsible for fulfilment of both the Commercialization Requirements and the Manufacture and Logistics Requirements then Molecular Partners’ written confirmation is required that Orano Med has met the Manufacture and Logistics Requirements for the applicable Collaboration Product.
(b)If a Party wishes to challenge the other Party’s notice setting forth its determination as to the achievement or not of the Commercialization Requirements, it shall send a notice of objection to the issuing Party no later than [***] after receipt of such notice, outlining in reasonable detail its reasoning for challenging such determination. In this event, the Parties shall discuss the notice of objection with a view to settling any challenges raised by the reviewing Party and notably if the failure to meet the Commercialisation Requirements results from a failure to meet Manufacturing and Logistics Requirements. If the Parties fail to settle the matter (notably by adjusting the Commercial Requirements if failure to meet such requirements results from a failure to meet Manufacturing and Logistics Requirements) within [***], the matter shall be resolved in accordance with Section 17.3 hereunder, which shall apply mutatis mutandis. In the event that upon Option exercise, the receiving Party (or where Molecular Partners is approving the Manufacturing and Logistics Requirements, Molecular Partners) (i) fails to send the notice of objection in due form and within the timeline set forth above or (ii) approves the exercise of the Option from the issuing Party, the Option exercise by the issuing Party shall be successful and effective.
6.5License and Supply Agreements.
Upon exercise and successful completion and effectiveness of the Option for a given Collaboration Product by either Party, the Parties shall negotiate in good faith the terms and conditions of a license under which they would collaborate in Developing, seeking Regulatory Approvals of and the Commercialization of such Collaboration Product (the “License Agreement”) and a Commercial Supply Agreement (as specified and defined in Section 8.2(a) below) (together with the License Agreement, the “License and Supply Agreements”) for up to [***] (the “Negotiation Period”), which period may be extended by mutual agreement of the Parties, provided that the financial terms of such License and Supply Agreements shall be those as set forth in this Agreement including the Profit Share of Net Profit and Out-Licensing Revenues and associated terms. The License and Supply Agreements shall further (i) spell out the consequences of failure by the Parties of meeting their respective obligations under the Commercialization Requirements and the Manufacture and Logistics Requirements, (ii) reflect the license grants pursuant to Section 6.6, and (iii) shall contain such applicable terms and conditions as are customary for agreements of its type.
(a)If the Parties are unable to agree on terms for the License and Supply Agreements within the Negotiation Period, then at the request of either Party, either Party may give notice to the other of its intention to submit the matter to “baseball” style arbitration (“Baseball Arbitration”) in accordance with the procedure set out below.
i.Within [***] of the notice referred to above, each Party will provide the other Party with a proposal and written memorandum in support of its position regarding the dispute, as well as any documentary evidence it wishes to provide in support thereof, including under this Section 6.5 its preferred terms for the License and Supply Agreements (each a “Brief”).
ii.If the Parties cannot resolve the dispute within [***] days following exchange of each Party’s respective Brief, either Party may refer the dispute to Baseball Arbitration.
iii.Any matter submitted to Baseball Arbitration shall be finally settled [***].
iv.The arbitrator will be a professional in collaborations and licensing experienced in the valuation of pharmaceutical products with [***] of experience in the pharmaceutical and life sciences industries, including the conduct of development, manufacture and commercialization collaborations.
v.The cost of the arbitration will be borne [***].
vi.Except in a proceeding in a court of competent jurisdiction to enforce the results of the arbitration or as otherwise required by Applicable Law, neither Orano Med nor Molecular Partners nor any arbitrator may disclose [***] without the prior written agreement of Orano Med and Molecular Partners.
vii.Within [***] after such matter is referred to arbitration, each Party will provide the arbitrator with its Brief, which may be revised from the form provided to the other Party pursuant to paragraph (a) above, and the arbitrator will provide each Party’s Brief to the other Party after it receives it from both Parties.
viii.Within [***] after a Party submits its Brief, the other Party will have the right to respond thereto. The response and any material in support thereof will be provided to the arbitrator and the other Party.
ix.The arbitrator will have the right to meet with the Parties to clarify the record and determine the schedule, as necessary. Within [***] of the receipt by the arbitrator of both Parties’ responses (or expiration of the [***] period if any Party fails to submit a response), the arbitrator will deliver his/her decision regarding the expedited dispute in writing; provided, that the arbitrator will select only one of the resolutions proposed by either Party, which may not be altered or modified in any way.
x.[***], the arbitrator shall select the resolution that s/he considers to be the resolution that is more consistent [***].
6.6License Grant Post Option Exercise. Subject to the conditions of exercise and successful completion and effectiveness of the Option for a given Collaboration Product by a Party, the other Party shall grant and hereby grants to the exercising Party, with regard to the Collaboration Product concerned by the exercised Option,
(a)an exclusive, worldwide license, with the right to grant sublicenses, subject to Section 6.7 below, to a sublicensee reasonably acceptable to the granting Party, under the granting Party’s interest in the Joint IPR to Exploit such Collaboration Product, save that the granting Party shall retain the rights to use such Joint IPR to carry out any ongoing obligations under subclauses 4.1(b) to 4.1(e) (as applicable to the granting Party), and to that extent that licence granted shall be a sole rather than an exclusive licence; and
(b)a non-exclusive, worldwide license, with the right to grant sublicenses to a sublicensee reasonably acceptable to the granting Party, under the granting Party’s Background IPR or Sole Ownership to Exploit such Collaboration Product.
6.7Unless agreed otherwise in the Launch Preparedness Plan, the Exploiting Party shall have no right to sublicence within [***] following the Option exercise in the countries covered by the Launch Preparedness Plan, without the approval of the other Parties which cannot be unreasonably withheld, conditioned or delayed. [***].
Section 7
OUT-LICENSING
7.1Conditions for Out-Licensing Efforts. If, with regard to the First Product or any subsequent Collaboration Product, neither Party exercises its Option, or neither Option exercised by a Party is deemed as successful and completed pursuant to Section 6, or one Party exercises an Exit Decision pursuant to Section 9, and the other Party does not wish to assume the development of a Collaboration Product post Opt-Out, the Parties shall undertake Commercially Reasonable Efforts to achieve an Out-Licensing to a Third Party, on terms agreeable for both Parties, for Development and Commercialization of such Collaboration Product (for which no Option was exercised, successful and completed).
7.2Joint Efforts Towards Out-Licensing.
(a)Through the JSC, the Parties shall agree on adequate activities to be performed by the leading Party and, if applicable, the other Party in preparation of an Out-Licensing and on a respective budget and cost sharing principle. The Parties shall discuss and determine in the JSC whether, when and to what extent external advisors shall be involved to support or enable the Parties’ efforts to achieve an Out-Licensing (e.g., M&A advisors, investment banks, etc.) and how the costs shall be shared.
(b)The JSC shall establish, itself or with support of external advisors, a list of potential (sub)licensees and shall update the list quarterly. In preparation of the transaction process, the JSC shall agree upon a set of non-confidential data to be shared with potential bidders (e.g., a non-confidential teaser slide deck) as well as a Confidential Information memorandum or presentation to be shared with potential Third Party (sub)licensees solely under confidentiality obligations and restrictions on use no less restrictive than the ones hereunder.
(c)The Parties shall agree in the JSC which Party shall lead the negotiations with respect to a particular potential (sub)licensee. Prior to sharing a term sheet or any substantive mark-up thereof with a Third Party (sub)licensee, the leading Party shall [***].
Section 8
MANUFACTURING AND SUPPLY
8.1Clinical Supply. Through the rights granted to Orano Med under Section 4.1, Orano Med shall have the exclusive right to Manufacture the First Product (and other subsequent Collaboration Products) using Molecular Partners’ supplied DARPin proteins during the Term. With regard to the Manufacture and supply of pre-clinical supply and of clinical batches of a Collaboration Product for the performance of Clinical Trials until a Registration Enabling Trial has successfully completed in each country to which an R&D Program applies, or as otherwise agreed by the Parties in writing, the Parties shall negotiate in good faith and enter into a pre-clinical and clinical supply agreement (“Clinical Supply Agreement”) no later than [***] following the Effective Date in accordance with the terms set forth in Exhibit D.
(a)Orano Med shall make Commercially Reasonable Efforts to Manufacture (as defined in Schedule D) and supply the Collaboration Product (in finished product form) in the countries and in quantities sufficient for the conduct of the Clinical Trials set forth in an R&D Plan(s). Orano Med shall make Commercially Reasonable Efforts to develop a reliable, safe and secure logistics solution for supply and delivery of the finished Collaboration Product to customers.
(b)Molecular Partners shall make Commercially Reasonable Efforts to supply DARPin proteins to Orano Med for the Manufacturing of Collaboration Products. For clarity, the Parties may agree in the Clinical Supply Agreement that [***].
(c)Each Party shall respectively comply with Applicable Laws and Applicable Standards throughout the pre-clinical/clinical manufacturing phase and scale up to commercial supply, and shall share the required Regulatory Documentation with the other Party. Orano Med will be responsible for ensuring that Collaboration Product complies with all local requirements under local Nuclear Activity Laws, including with respect to labelling, packaging, transport, handling and other related activities and will use Commercially Reasonable Efforts to ensure all facilities in the Collaboration Product’s supply chain (other than those managed by Molecular Partners) are GMP compliant and appropriately licensed, authorised or approved (as applicable) for any Nuclear Activities.
8.2Commercial Supply.
(a)In addition to the License Agreement provided for under Section 6.5, the Parties shall negotiate in good faith and enter into a commercial supply agreement for the Manufacture and commercial supply of Collaboration Product by Orano Med from the Orano Med Facilities and any other sites agreed in the Launch Preparedness Plan (with continued associated supply of DARPin proteins by Molecular Partners) (the “Commercial Supply Agreement”). The Parties will enter into a quality agreement to allocate responsibility for quality activities under the Commercial Supply Agreement and will enter into a pharmacovigilance agreement to allocate responsibility for pharmacovigilance activities related to the commercial supply of the Collaboration Product, either together with entering into the Commercial Supply Agreement or otherwise within the time periods specified in the Commercial Supply Agreement.
(b)The Commercial Supply Agreement shall be based on the principles agreed in the Clinical Supply Agreement and, inter alia, include a detailed program of Manufacture and details on facilities, applicable liabilities, process for making changes to Manufacturing process, safety stock requirements, key performance indicators and a definition of and terms to address serious supply failures.
8.3Continuity of supply - Technology Transfer.
(a)Manufacturing and supply continuity arrangements shall be addressed in the R&D Plan. For [***] supply Orano Med undertakes as part of its business continuity planning to ensure that [***] as required under the R&D Plan, [***]. The details will be set forth in the Clinical Supply Agreement and Commercial Supply Agreement.
(b)The Clinical Supply Agreement and the Commercial Supply Agreement shall provide that following [***], in the event of repeated and significant failures to Manufacture and supply a Collaboration Product by a Party or its designated CMO (such failure to be respectively defined in the Clinical Supply Agreement and the Commercial Supply Agreement), the Party responsible for such Manufacture and supply will [***].
(c)The Commercial Supply Agreement shall also provide the right for Orano Med terminate the Commercial Supply Agreement, thereby triggering a Supply Exit (i) after the [***], or (ii) [***].
(d)In case of a Supply Exit:
i.Orano Med shall [***] reasonably acceptable to both Parties; and
ii.the Exploiting Party (acting reasonably) shall [***]; and
(e)Orano Med shall [***]:
i.[***];
ii.[***]; and
iii.[***].
(f)In case of a Supply Exit the Profit Share of the Party initially responsible for the Manufacturing and supply which requests the Supply Exit shall [***] with respect to those Collaboration Products concerned by the Supply Exit and [***]. For the avoidance of doubt, in case of a Supply Exit concerning certain Collaboration Products only, Orano Med shall no longer be the exclusive supplier for such Collaboration Products concerned by the Supply Exit.
(g)If a Supply Exit has occurred in both the EU and the United States with respect to a Collaboration Product, then [***].
Section 9
EXIT BY A PARTY
9.1Exit Decision, Continuing Party.
(a)Either Party (in such capacity, the “Exiting Party”) is entitled to stop its participation and involvement in the Development in respect of a Collaboration Product or in respect of a Collaboration Product for a particular country in the Territory as agreed in the relevant R&D Plan without cause (“Exit Decision”) following a given Opt-Out Point, provided that for purposes of the foregoing, [***]. To exercise such Exit Decision, the Exiting Party shall notify the other Party of its Exit Decision in writing within [***] following a given Opt-Out Point, whereby the effective date of the Exit Decision shall be subject to a [***] notice period commencing upon the delivery of the written notice regarding the Exit Decision from the Exiting Party to the other Party. Any Exit Decision shall become effective upon expiry of the last day of the [***] notice period.
(b)Within [***] of its receipt of a notice of Exit from the Exiting Party, which period shall be extended for a further [***] upon written request of the other Party, the other Party is entitled to refuse to assume the sole continued Development of a Collaboration Product beyond the Opt-Out Point to which the Exit Decision of the Exiting Party relates, in which case it shall provide written notice to the Exiting Party (“Joint Exit Decision”). In such case, the Parties will seek an Out-Licensing of the Collaboration Product pursuant to Section 7 above. Alternatively, Parties may jointly decide to put an end to the Development of the Collaboration Product and the Parties will share equally in the wind down costs.
(c)If the other Party (i) has not requested a prolongation of the initial [***] period in writing before expiry of such period or (ii) has not exercised its Joint Exit Decision towards the Exiting Party within the first or, if applicable, second [***] period, then the other Party shall continue the Development of a Collaboration Product alone after the effective date of the Exit Decision pursuant to Section 9.1(a) (the other Party in such capacity, the “Continuing Party”). In such case, the Exiting Party shall provide to the Continuing Party the Necessary Support pursuant to Section 9.3 and the Exit License pursuant to Sections 9.4 and 9.5.
(d)Upon the effective date of the Exit Decision the Exiting Party shall be dismissed and relieved from any active involvement in the performance of an R&D Plan to which the Exit Decision pertains as well as from any corresponding cost sharing obligations pursuant to Section 5, provided the Exiting Party shall not thereafter be able to recover any amounts paid in respect of its cost share prior to the effective date of the Exit Decision.
9.2Rights and Obligations of the Continuing Party.
(a)The Continuing Party shall
i.pursue the Development of a Collaboration Product to which the Exit Decision pertains at its own cost with no more referral to the JSC,
ii.use Commercially Reasonable Efforts to Develop and Commercialize a Collaboration Product to which the Exit Decision pertains (including to make reasonable effort to find Out-Licensing opportunities);
iii.update the Exiting Party, subject to any restrictions that may be imposed by Applicable Law regarding data sharing, on an annual basis within [***], of the Development and Commercialization of a Collaboration Product to which the Exit Decision pertains under clinical and commercial aspects and of the related budgets for Development and Commercialization,
iv.comply with the profit-sharing principles set forth in Section 10.3,
v.assume all liabilities relating to the continued Development of a Collaboration Product to which the Exit Decision pertains and hold the Exiting Party harmless against any Third Party claims arising out of activities conducted by or on behalf of the Continuing Party after the effective date of the Exit Decision, except to the extent such Third Party claims arise out of the negligence or intentional misconduct of the Exiting Party at any time or accrued prior to the effective date of the Exit Decision.
(b)The Continuing Party shall be released from its exclusivity obligations under Section 2.4, yet solely with regard to the target which is targeted by the concerned Collaboration Product to which the Exit Decision pertains (e.g., DLL3 with respect to the first Collaboration Product).
9.3Necessary Support. In the event the Exit Decision becomes effective after the filing of IND for the concerned Collaboration Product, each Party, in its capacity as Exiting Party, shall provide to the other Party, in its capacity as Continuing Party, the necessary support set forth in below (“Necessary Support”).
(a)In case of Orano Med being the Exiting Party: Orano Med shall provide to Molecular Partners such assistance and support, using Commercially Reasonable Efforts, as necessary or reasonably useful to enabling Molecular Partners to continue further Development of such Collaboration Product. Such reasonable support and assistance shall not exceed [***] and shall be reimbursed by Molecular Partners to Orano Med at an hourly rate of [***] and subject to reimbursement of reasonable, documented and authorized expenses associated with the provision of such support and assistance by Orano Med.
(b)In case of Molecular Partners being the Exiting Party: Molecular Partners shall provide to Orano Med such assistance and support, using Commercially Reasonable Efforts, as necessary or reasonably useful to enabling Orano Med to continue further Development of such Collaboration Product. Such reasonable support and assistance shall not exceed [***] and shall be reimbursed by Orano Med to Molecular Partners at an hourly rate of [***] and subject to reimbursement of reasonable, documented and authorized expenses associated with the provision of such support and assistance by Molecular Partners.
(c)For the avoidance of doubt, the Necessary Support shall include the following:
i.if the Exiting Party is Orano Med, Orano Med’s supply obligations shall continue notwithstanding its Exit Decision and any license grants by Orano Med hereunder shall continue, under reasonable conditions to be determined under the Clinical Supply Agreement or, as applicable, Commercial Supply Agreement;
ii.if the Exiting Party is Molecular Partners, the supplyobligations of Molecular Partners for the DARPin proteins (directly or through CMO) shall continue notwithstanding its Exit Decision, and any license grants by Molecular Partners hereunder shall in such case also continue, under reasonable conditions to be determined under the Clinical Supply Agreement or, as applicable, Commercial Supply Agreement; and
iii.cost related to Joint IPR shall remain equally shared by the Parties notwithstanding an Exit Decision (except the Exiting Party decides not to participate to such Joint IPR costs in which case Section 12.9(d) shall apply and its rights to the Joint IPR shall be wholly assigned to the Continuing Party).
9.4Grant of Exit License. In consideration for the benefit of continued profit sharing described in Section 10 (as adjusted pursuant to Section 10.3) from the continued Development of a Collaboration Product to which the Exit Decision pertains by the Continuing Party, the Exiting Party shall grant, and hereby grants, to the Continuing Party the following licenses with effective as of the effective date of the Exit Decision:
(a)an exclusive, worldwide license or, if the Exit Decision pertains to a particular country in the Territory, for such countries to which the Exit Decision pertains, with the right to grant sublicenses to a sublicensee reasonably acceptable to the granting Party, under the granting Party’s interest in the Joint IPR to Exploit such Collaboration Product; and
(b)a non-exclusive worldwide license or, if the Exit Decision pertains to a particular country in the Territory, for such countries to which the Exit Decision pertains, with the right to grant sublicenses to a sublicensee reasonably acceptable to the granting Party, under the granting Party’s Background IPR or Sole Ownership to Exploit such Collaboration Product.
(c)It is understood between the Parties that the licenses under this Section 9.4 shall be exclusive with respect to the Collaboration Product, to which the Exit Decision pertains, but that these licences shall not extend to any Exploitation of the respective IPR outside of such Collaboration Product (e.g., they shall not extend to the use of components of such Collaboration Product outside of the Collaboration Products or on their own).
(d)For clarity, the Exit License shall (i) enable the Continuing Party to search for, evaluate and negotiate Out-Licensing opportunities and (ii) include the right for the Continuing Party to use all Data related to a Collaboration Product to which the Exit Decision pertains generated by both Parties before the effective exit date of the Exit Decision.
9.5Right of Reference. In consideration for the benefit of continued profit sharing described in Section 10 (as adjusted pursuant to Section 10.3) from the continued Development of a Collaboration Product to which the Exit Decision pertains by the Continuing Party, the Exiting Party shall grant and hereby grants to the Continuing Party a right of reference and access to all Regulatory Documentation submitted to any Regulatory Authority in the Territory for Collaboration Products to which the Exit Decision pertains that are in the Exiting Party’s or any of its Affiliates’ name or are otherwise Controlled by the Exiting Party or any of its Affiliates. Where the Continuing Party wishes to submit or file Regulatory Documentation to a given Regulatory Authority in the Territory or to obtain or maintain a Regulatory Approval for a Collaboration Product in the Territory to which the Exit Decision pertains, the Exiting Party shall provide appropriate notification of the Continuing Party’s access and reference rights to the applicable Regulatory Authorities, where necessary.
9.6Transfer of Regulatory Approvals and Third Party agreements. If, at the effective date of the Exit Decision, the Exiting Party or any of its Affiliates (i) holds any Regulatory Approvals for the Collaboration Product to which the Exit Decision pertains, which are capable of being transferred to the Continuing Party; and/or (ii) has entered into any Third Party Agreements which relate solely to the Development of a Collaboration Product to which the Exit Decision pertains, in each case the Exiting Party shall disclose the same to the Continuing Party and shall promptly transfer such Regulatory Approvals and/or Third Party agreements to the Continuing Party on written request and at the cost of the Continuing Party.
Section 10
PROFIT SHARING
10.1General Principle.
(a)The Exploiting Party shall book the sales of the Collaboration Product and shall pay to the other Party the Profit Share of the Net Profit generated by it from Commercialization of each Collaboration Product on worldwide basis.
(b)For the avoidance of doubt, in the event that there is a Net Loss, in a given Calendar Quarter, [***].
(c)For the avoidance of doubt, this Profit Share of Net Profit is distinct from and without prejudice to any additional revenue a Party may derive from the supply of the Collaboration Products.
10.2Shared Out-Licensing Revenues in Case of Out-Licensing.
(a)For Collaboration Products which are (sub)licensed to a Third Party in an Out-Licensing in the absence of an Exit Decision by a Party hereto, Out-Licensing Revenues shall be shared on a [***].
(b)For Products which are (sub)licensed to a Third Party in an Out-Licensing subsequent to an Exit Decision, the Continuing Party shall pay to the other Party a portion equal to the Profit Share of the Net Profits generated by the Continuing Party with respect to such Product.
10.3Reporting on Sales and Net Profit and Out Licensing Revenues.
(a)The Exploiting Party shall provide to the other Party as promptly as practicable, but in any event within [***], a written report setting forth, on a country-by-country basis, so far as is permitted under Applicable Law, (i) the amount of Net Sales generated with a Collaboration Product during the previous calendar month in such country, (ii) the Operating Expenses incurred at or invoiced to the Exploiting Party for Commercialization of such Collaboration Product during such calendar month, stating the relevant cost items per individual item as reasonably required to assess the calculation of such Operating Expenses, and its alignment with Commercialization Plan, and (iii) the amount of the monthly Net Profit payable by or to the Exploiting Party by or to the other Party with regard to a Collaboration Product and applicable calendar month, including the method applied by the Exploiting Party to calculate such monthly Net Profit (“Profit Sharing Report”).
(b)The Exploiting Party shall provide to the other Party as promptly as practicable, but in any event within [***], a written report setting forth, on a country-by-country basis, the amount of Out-Licensing Revenues during such calendar month in such country, and the amount of the Out-Licensing Revenues payable by or to the Exploiting Party by or to the other Party with regard to a Collaboration Product and calendar month, (“Out-Licensing Revenues Report(s)”).
(c)If the other Party has objections against any report provided to it by the Exploiting Party, the other Party shall send a written objection notification to the Exploiting Party in which the other Party shall explain its reasoning for the objection. Upon the Exploiting Party’s receipt of the objection notification, the Parties shall promptly schedule a meeting of the Executive Officers to discuss and prepare a decision of the Parties to resolve or settle the dispute. If the Executive Officers cannot agree on a proposal for a respective decision or if the Parties cannot mutually agree to resolve or settle the dispute, the dispute shall be escalated to arbitration pursuant to Section 17.3.
Section 11
PAYMENT TERMS, TAXES, AUDIT RIGHTS
11.1Payment Terms.
(a)For all payments or costs for which a Party (the “Owing Party”) is obligated to pay to or to reimburse the other Party (the “Owed Party”) pursuant to this Agreement (and for which no specific provision is made hereunder for such payment), the Owed Party shall send to the Owing Party an invoice for such amount within [***] of (i) receipt of all relevant Profit Sharing Reports and Out-Licensing Revenues Reports pertaining to that Calendar Quarter (i.e. the third of each report relating to that Calendar Quarter) from the Exploiting Party by the other Party or (ii) any other point in time on which it is determined under this Agreement that an amount is payable by the Owing Party to the Owed Party, which invoice shall include a reference to the section of this Agreement under which the Owed Party is requesting reimbursement or payment.
(b)Unless otherwise agreed between the Parties payment with respect to each such invoice shall be due within [***] after receipt by the Owing Party thereof. Any undisputed invoice which is not paid by its due date shall incur a late payment fee at the rate of [***] per month, compounded monthly, or at the highest rate permitted under the governing law as set forth in Section 17.2, if less.
(c)All amounts due hereunder shall be paid by the paying Party in Euros by electronic funds transmission to the receiving Party’s designated bank account as indicated in the respective invoice or as the receiving Party may from time to time otherwise designate by written notice to the paying Party. Any costs of bank wire transfer shall be borne by the paying Party.
11.2Taxes and Withholding.
(a)All payments to be paid by the paying Party to the receiving Party under this Agreement shall be understood as net amounts for value added tax (VAT) or equivalent tax purposes. The payment of such VAT (or equivalent tax), if due, is within the sole responsibility of the paying Party. In this case, the receiving Party shall provide the paying Party with an appropriate VAT invoice.
(b)Each Party will pay all taxes imposed on its share of income arising directly or indirectly from the efforts of, or the receipt of any payment by, such Party under this Agreement in its relevant tax jurisdiction.
(c)All payments under this Agreement shall be made without any deduction or withholding for or on account of any tax, except as set forth in this Section 11.2.
(d)The Parties agree to cooperate with each other and use Commercially Reasonable Efforts to minimize under Applicable Law obligations for any and all income or other taxes required by Applicable Laws to be withheld or deducted from any of the royalty and other payments made by or on behalf of a Party hereunder (“Withholding Taxes”).
(e)The Deficit Party shall, if so required by Applicable Law, deduct from any amounts that it is required to pay to the Excedent Party an amount equal to such Withholding Taxes. Such Withholding Taxes shall be paid to the proper taxing authority for the Deficit Party’s account and, if available, evidence of such payment shall be secured and sent to Excedent Party within [***] of such payment. The Deficit Party shall, at the Excedent Party’s sole cost and expense, as mutually agreed by the Parties, do all such lawful acts and things and sign all such lawful deeds and documents as the Excedent Party may reasonably request to enable the Deficit Party to avail itself of any Applicable Laws (including notably any double taxation treaties) with the goal of paying the sums due to the Excedent Party hereunder without deducting any Withholding Taxes.
11.3Record Keeping. During the Term of this Agreement, each Party shall maintain complete and accurate books and records of account, in accordance with its applicable generally accepted accounting standards, of all transactions and other activities under this Agreement, sufficient to confirm (a) the accuracy of all reports furnished by such Party to the other Party under this Agreement; (b) the accuracy of the determination of Net Sales and Operating Expenses and respective Profit Sharing Report; and (c) all other payments paid or payable by such Party to the other Party under this Agreement.
11.4Audit.
(a)At the request and expense of one Party (the “Demanding Party”), the other Party shall permit an independent accounting expert appointed by the Demanding Party and reasonably acceptable to the other Party, at reasonable times and upon reasonable notice, to examine only those records as may be reasonably necessary to determine, with respect to any Calendar Year ending not more than three (3) years prior to the Demanding Party’s request or with respect to any period referenced in a dispute under Section 5.4, the correctness or completeness of any Out-Licensing Revenues Report, Profit Sharing Report and other payments as well as supporting documents submitted to the Demanding Party hereunder. The foregoing right of review may be exercised only once per year and shall not exceed [***] in duration. Results of any such examination shall be (i) made available to the audited Party and (ii) subject to the confidentiality obligations hereunder.
(b)Any examination conducted under this Section 11.4 shall be at the expense of the Demanding Party, unless such inspection reveals any underpayment of the payments due for the audited period by at least [***], in which case the full costs of such inspection for such period shall be borne by the other Party.
(c)In the event of a dispute regarding the findings of the inspection of the applicable records of the audited Party, the Parties shall work in good faith to resolve the dispute in accordance with the provisions of Section 17.3. If the Parties are unable to resolve the dispute within the time period stipulated in Section 17.3, the dispute shall be submitted for decision to a certified public accounting firm mutually selected by each Party’s certified public accountants or to such other Third Party as the Parties shall mutually agree. The decision of such expert shall be final and the costs of such decision as well as the initial audit shall be borne between the Parties in such manner as such expert shall determine. Not later than [***] after such decision and in accordance with such decision, the Owing Party shall make any additional payments to the Owed Party.
Section 12
INTELLECTUAL PROPERTY
12.1Background IPR. Subject to Section 12.2 and 12.3, each Party is and shall remain the sole and exclusive owner of all its Background IPR and this Agreement shall not affect such ownership. Except as expressly stated in this Agreement, no licenses nor any other rights, implied or otherwise, are granted by a Party to the other Party under its Background IPR. For clarity, each Party shall remain the sole and exclusive owner of all Patent Rights, Know-How and other IPR related to a Collaboration Product which the Party owns or otherwise Controls on or before the Effective Date or which is generated by a Party outside of the R&D Program.
12.2Molecular Partners Owned Foreground IP.
(a)All right, title and interest in and to any and all Patent Rights, Know-How and other IPR that [***] (“Molecular Partners Owned IP”) shall be allocated to and solely owned by Molecular Partners.
(b)Molecular Partners shall solely own any Patent Rights that claim or that are directed to Molecular Partners Owned IP (“Molecular Partners Owned Patents”). Molecular Partners shall have the [***] to prepare, file, prosecute (including any proceedings relating to reissues, reexaminations, protests, interferences, oppositions, post-grant reviews or similar proceedings and requests for patent extensions) and maintain any Molecular Partners Owned Patents at its own expense. If Molecular Partners elects not to prosecute any Molecular Partners Owned Patents, it shall notify Orano Med, [***].
(c)Orano Med shall fully disclose in writing to Molecular Partners any and all Molecular Partners Owned IP, which is discovered, developed, or invented by or on behalf of Orano Med, independent of whether or not such is protectable under any Applicable Laws within [***] from Orano Med’s awareness of such discovery, development or invention. Orano Med agrees to transfer and assign, and to cause its Affiliates or subcontractors to transfer and assign, and herewith transfers and assigns to Molecular Partners all of Orano Med’s right, title and interest in and to all Molecular Partners Owned IP. Molecular Partners herewith accepts such transfer and assignment.
12.3Orano Med Owned Foreground IP.
(a)All right, title and interest in and to any and all Patent Rights, Know-How and other IPR that [***] (“Orano Med Owned IP”) shall be allocated to and solely owned by Orano Med.
(b)Orano Med shall solely own any Patent Rights that claim or that are directed to Orano Med Owned IP (“Orano Med Owned Patents”). Orano Med shall have the [***] to prepare, file, prosecute (including any proceedings relating to reissues, reexaminations, protests, interferences, oppositions, post-grant reviews or similar proceedings and requests for patent extensions) and maintain any Orano Med Owned Patents at its own expense. If Orano Med elects not to prosecute any Orano Med Owned Patents, it shall notify Molecular Partners, [***].
(c)Molecular Partners shall fully disclose in writing to Orano Med any and all Orano Med Owned IP, which is discovered, developed, or invented by or on behalf of Molecular Partners, independent of whether or not such is protectable under any Applicable Laws within [***] from Molecular Partners’ awareness of such discovery, development or invention. Molecular Partners agrees to transfer and assign, and to cause its Affiliates or subcontractors to transfer and assign, and herewith transfers and assigns to Orano Med all of Molecular Partners’ right, title and interest in and to all Orano Med Owned IP. Orano Med herewith accepts such transfer and assignment.
12.4Jointly Owned Foreground IP.
(a)Any and all right, title and interest in and to any Patent Rights, Know-How or other IPR that [***] (“Joint IPR”) shall be allocated to both Parties in equal shares and shall be jointly owned by the Parties. For clarity, Joint IPR includes [***]. Recourse to any Third Party’s [***].
(b)Both Parties shall jointly own in equal shares any Patent Rights that claim or that are directed to Joint IPR.
(c)Each Party hereby agrees to transfer and assign, and to cause its Affiliates and Subcontractors to transfer and assign, and hereby transfers and assigns to the other Party, who accepts such transfer and assignment, such portion of its right, title, and interest in, to and under the Joint IPR as required for both Parties to jointly own an equal and undivided part of the Joint IPR.
12.5[***] Technology Collaboration, ownership and exploitation.
(a)Collaboration. The Parties will collaborate to Develop an optimized [***], as agreed in the [***] Project Plan.
(b)[***] Background Technology.
i.Each Party is and shall remain the sole and exclusive owner of all its [***] Background Technology and this Agreement shall not affect such ownership. Except as expressly stated in this Agreement, no licenses nor any other rights, implied or otherwise, are granted by a Party to the other Party under its [***] Background Technology.
ii.Molecular Partners shall grant and herewith grants to Orano Med (i) a non-exclusive, worldwide license under Molecular Partners [***] Background Technology to carry out Orano Med’s responsibilities under the [***] Project Plan and (ii) a non-exclusive, worldwide, perpetual, fully paid-up, royalty free, sub-licensable license to the Molecular Partners [***] Background Technology to the extent such license is required and necessary for Orano Med to Exploit and license any of its rights in and to the Joint [***] Technology.
iii.Orano Med shall grant and herewith grants to Molecular Partners (i) a non-exclusive, worldwide license under Orano Med [***] Background Technology to carry out Molecular Partners’ responsibilities under the [***] Project Plan and (ii) a non-exclusive, worldwide, perpetual, fully paid-up, royalty free, sub-licensable license to the Orano Med [***] Background Technology to the extent such license is necessary for Molecular Partners to Exploit and license any of its rights in and to the Joint [***] Technology.
(c)Jointly Owned Foreground [***] Technology.
i.All Joint [***] Technology shall be allocated to both Parties in equal shares and shall be jointly owned by the Parties.
ii.Both Parties shall jointly own in equal shares any Patent Rights that claim or that are directed to Joint [***] Technology. Parties agree in this respect that Sections 6.2.3.1 (Joint Patents) and 6.3 (Lawsuits) of the R&D Agreement shall apply mutatis mutandis.
iii.Each Party hereby agrees to transfer and assign, and to cause its Affiliates to transfer and assign, and hereby transfers and assigns to the other Party, who accepts such transfer and assignment, such portion of its right, title, and interest in, to and under the Joint [***] Technology as required for both Parties to jointly own an equal and undivided part of the Joint [***] Technology.
(d)Use of Joint [***] Technology.
i.Neither Party may sell, transfer, or assign any Joint [***] Technology without prior written consent of the other Party, except in the case of a full or partial sale, transfer or assignment of the business to which the [***] technology relates or to an Affiliate.
ii.Each Party will have an equal and undivided interest in the Joint [***] Technology, and each Party will be entitled to Exploit or license the Joint [***] Technology, without the consent of or duty to account to the other Party subject to the exclusive rights and licences granted herein, provided that such Exploitation or license does not incur conflicts that may adversely impact the collaboration between the Parties, and provided that (i) Orano Med may not Exploit or license the Joint [***] Technology in conjunction or combination with any DARPin Technology save to the extent permitted under the R&D Agreement or this Agreement, and (ii) Molecular Partners may not Exploit or license the Joint [***] Technology in conjunction or combination with [***] save to the extent permitted under the R&D Agreement or this Agreement.
iii.Molecular Partners shall grant and herewith grants to Orano Med a non-exclusive, worldwide, perpetual, fully paid-up, royalty free, sub-licensable license to the Molecular Partners [***] Background Technology (but excluding all DARPin Technology comprised therein) to the extent such license is required and necessary for Orano Med to Exploit or license any of its rights in and to the Joint [***] Technology.
(e)Orano Med shall grant and herewith grants to Molecular Partners a non-exclusive, worldwide, perpetual, fully paid-up, royalty free, sub-licensable license to the Orano Med [***] Background Technology (but excluding any rights to [***] comprised therein) to the extent such license is necessary for Molecular Partners to Exploit or license any of its rights in and to the Joint [***] Technology.
12.6Other Foreground IP.
(a)Any and all right, title and interest in and to any Patent Rights, Know-How or other IPR that [***] shall be owned by either or both Parties based on inventorship.
12.7Inventorship; Employee Inventions.
(a)Determination of inventorship will be made in accordance with US patent laws. The Parties will name only the true and real inventors according to their contributions to each invention.
(b)To the extent in line with Applicable Laws and unless equivalent measures are already provided on the basis of Applicable Laws, each Party shall ensure that employees and subcontractors of such Party or its respective Affiliates performing activities under this Agreement shall, prior to commencing such work, be bound by written invention assignment obligations requiring: (i) prompt reporting of any Patent Rights, Know-How, or other Intellectual Property Rights arising from such work; (ii) assignment to the applicable Party or Affiliate of his or her rights, title, and interests in, to and under any Patent Rights, Know-How, or other Intellectual Property Rights arising from such work; and (iii) performance of all acts and signing, executing, acknowledging, and delivering any and all documents required for effecting the obligations and purposes of this Agreement. Each Party shall comply with and shall be solely responsible for all applicable country-specific inventor remuneration laws and regulations, if inventor remuneration obligations are triggered by an employee of such Party or its Affiliates.
12.8Use of Joint IPR.
(a)Unless otherwise authorized under this Agreement, neither Party may sell, transfer or assign any Joint IPR (including but not limited to in case of Out-Licensing) without prior written consent of the other Party and provided that such consent shall not be required for an assignment by a Party (a) to an Affiliate of the assigning Party, or (b) subject always to Section 2.5 above, in a Change of Control. Any such assignment of Joint IPR to an Affiliate shall be made only on the condition (to be recorded in such assignment) that, if such Affiliate ceases to become an Affiliate of the assigning Party, then the Affiliate shall be obliged, at that time, to assign back to the assigning Party all of its rights to and in such Joint IPR at its own cost, and shall execute all necessary documents and take all necessary steps to record and perfect such assignment back.
(b)Subject to the mutual licenses granted by the Parties to each other under this Agreement under the Joint IPR, and subject to the exclusivity obligations of the Parties, each Party will have an equal and undivided interest in the Joint IPR, [***].
12.9Maintenance and Prosecution of Joint IPR.
(a)Whenever required or appropriate, both Parties shall discuss and agree the patenting strategy for any Joint IPR which may arise, including deciding on the timing for the filing of any patent application, the countries in which patent applications should be filed and the Party that will take the lead in drafting, filing, prosecuting, maintaining and/or defending particular Joint IPR (the “Lead Prosecuting Party”). The level of generalization of any jointly owned inventions shall be discussed if and when the filing of a patent application is envisaged so as to afford the Parties the broadest possible Patent Right. Unless agreed otherwise, or if the Parties cannot agree, then (i) [***] shall have the first right (but no obligation) to be the Lead Prosecuting Party for any patent applications regarding Joint IPR relating to the First Product, and (ii) [***] shall have the first right (but no obligation) to be the Lead Prosecuting Party for any patent applications regarding Joint IPR relating to the first Additional Candidate. If the Party with such first prosecution right chooses not to lead the prosecution, then it shall notify the other Party, which shall have the right (but no obligation) to lead such prosecution.
(b)The Parties acknowledge and agree that unless otherwise agreed, the Lead Prosecuting Party shall have the first right (but not the obligation) to file a patent application (including any provisional, substitution, divisional, continuation, continuation in part, reissue, renewal, reexamination, extension, supplementary protection certificate and the like) in respect of any Joint IPR (each, a “Joint Patent Application”), using patent counsel selected by the Lead Prosecuting Party and reasonably acceptable to the other Party. Notwithstanding the foregoing, when deciding whether to (i) [***], or (ii) [***], following which the Parties shall [***]. For the purposes of this subclause 12.9(b):
i.“UPC System” means the system for a new Unified Patent Court established pursuant to the Regulation (EU) No 1257/2012 on the Unitary Patent, the Agreement on a Unified Patent Court dated 19 February 2013, the Protocol to the UPC Agreement dated October 2015, and the Rules of Procedure of the Unified Patent Court dated 8 July 2022, each as amended from time to time.
ii.“Unitary Patent” means the new European patent with unitary effect covering multiple UPC States and administered by the European Patent Office as established pursuant to Regulation (EU) No 1257/2012.
iii.“UPC State” means any EU Member State that at the relevant date has signed and ratified the Agreement on a Unified Patent Court dated 19 February 2013.
(c)In any event, the Parties shall consult and reasonably cooperate with one another to jointly agree on (i) the preparation, filing, prosecution (including prosecution strategy) and maintenance of such Joint Patent Application and Orano Med and Molecular Partners shall equally share the expenses associated therewith, and (ii) the maintenance and defense of any joint Patent Rights and shall equally share the expenses associated therewith. The Lead Prosecuting Party shall provide to the other Party copies of any papers related to such Joint Patent Applications reasonably in advance of any submission to the relevant patent office or other registry so that the other Party can comment and provide input. The Lead Prosecuting Party shall not finally abandon or disclaim any claims that it has been informed are of interest to the other Party without prior consultation with the other Party. For the avoidance of doubt, both the Lead Prosecuting Party and the other Party shall be both fully and equally considered as the beneficial owners of the rights derived from any Joint IPR.
(d)If a Party does not want to file a patent application for a Joint IPR (either generally or with respect to a particular country) or at any point after the initial filing wishes to discontinue the prosecution, maintenance or defense of a Joint Patent Application or joint Patent, the other Party, at its sole option, may file such a patent application and/or may continue such prosecution, maintenance or defense at its sole expense. In such event, the opting-out Party shall execute such documents and perform such acts, at the other Party’s expense, as may be reasonably necessary in a timely manner to assign such Joint IPR to the other Party (in such country or all countries, as applicable) to allow the other Party to continue such Joint IPR. Any Joint Patent Application or Joint IPR so assigned shall thereafter be owned solely by the other Party and treated as Sole Patent Rights hereunder.
12.10Separation of Patent Rights.
(a)The Parties will use good faith efforts to separate Molecular Partners Owned Patents, Orano Med Owned Patents and jointly owned Patent Rights into separate filings to the extent possible and without adversely impacting prosecution and maintenance of Molecular Partners Owned Patents, Orano Med Owned Patents and jointly-owned Patent Rights with a view to minimizing the inclusion of one Party’s Sole Ownership in the other Party’s Sole Patent Rights and/or in any jointly-owned Patent Right.
(b)In the interest of isolating where possible each Party’s Sole Patent Rights and/or any jointly-owned Patent Rights in distinct instruments, each Party shall make good faith efforts to supply to the other Party information regarding contemplated applications of its Sole Patent Rights within reasonable timeframe before the relevant filing and in sufficient details to allow the other Party to advise of its views concerning the separation of Patent Rights as per this Section 12.10. Each Party shall, upon receipt of such views from the other Party, take necessary reasonable actions, including amendments to the patent application in preparation, to ensure that its patent application complies with the terms and conditions hereunder.
(c)Any dispute arising out of, relating to or in connection with this Section 12.10 shall be escalated to the Parties’ respective Executive Officers, who shall attempt to settle such dispute within [***], or such longer period as may be mutually agreed. In the event that the Parties’ respective Executive Officers cannot settle such issue within the designated period, the issue shall be subject to a dispute resolution procedure pursuant to Section 17.3 hereof.
12.11Infringement of Patent Rights by Third Parties.
(a)Infringement of Molecular Partners Owned Patents. For all infringement or misappropriation by a Third Party of Molecular Partners Owned Patents, of which Molecular Partners or its in-house patent counsel becomes aware anywhere in the world, [***] shall have [***] to prosecute such infringement as it may determine in its sole and absolute discretion, and [***] shall bear all related expenses and retain all related recoveries. [***] shall reasonably cooperate with [***] or its designee, at [***]’ request and expense, in any such action.
(b)Infringement of Orano Med Owned Patents. For all infringement or misappropriation by a Third Party of Orano Med Owned Patents, of which Orano Med or its in-house patent counsel becomes aware anywhere in the world, [***] shall have [***] to prosecute such infringement as it may determine in its sole and absolute discretion, and [***] shall bear all related expenses and retain all related recoveries. [***] shall reasonably cooperate with [***] or its designee, at [***]’s request and expense, in any such action.
(c)Infringement of jointly owned Patent Rights. For all infringement or misappropriation by a Third Party of jointly owned Patent Rights or other Joint IPR, the Parties will discuss in good faith and determine the strategy and their respective roles to deal with the situation on a case-by-case basis. Unless agreed otherwise, or if the Parties cannot agree, then (i) [***] shall have the first right (but no obligation) to lead the enforcement of jointly owned Patent Rights or other Joint IPR relating to First Product, and (ii) [***] shall have the first right (but no obligation) to lead the enforcement of jointly owned Patent Rights or other Joint IPR relating to the first Additional Candidate. If the Party with such first enforcement right chooses not to lead the enforcement, then it shall notify the other Party, which shall have the right (but no obligation) to lead such enforcement.
12.12Infringement of Third Party IPR.
(a)Notice. If a Party is or otherwise becomes aware that the activities relating to the Collaboration (i) may infringe a patent, copyright or other proprietary right by a Third Party anywhere in the world, or (ii) are the subject of a claim of infringement of a patent, copyright or other proprietary right by a Third Party anywhere in the world, such Party shall promptly notify the other Party and, without regard to which Party first became aware, or is charged with said infringement and the venue of such claim, the Parties shall promptly confer to discuss the potential infringement and/or claim.
(b)Defence. If both Parties are charged with infringement pursuant to a claim described in Section 12.12(a), the Parties shall defend such claim jointly, unless they agree otherwise. If only one Party is charged with infringement, such Party will have the sole right but not the obligation to defend such claim.
(c)If the Parties jointly defend the claim, Molecular Partners and Orano Med shall bear any costs and expenses of the defense of any such Third Party infringement claim in accordance with such share ratio as agreed by both Parties taking into account how much the claim relates to each Party’s Background IPR or Sole Ownership, with the understanding that if the claim relates solely to one Party’s Background IPR or Sole Ownership, such Party will bear one hundred percent (100%) of the costs and expenses of the defense of such claim and shall have the sole right, but not the obligation, to defend, settle and otherwise handle the disposition of such claim
(d)Neither Party shall enter into any settlement concerning activities under this Agreement or the Collaboration that affects the other Party’s rights under this Agreement, the Commercial Supply Agreement or Clinical Supply Agreement or imposes any obligations on the other Party, including any admissions of wrongdoing on behalf of the other Party, without such other Party’s prior written consent, not to be unreasonably withheld or delayed, except that a Party may settle any claim that solely relates to its Background IPR or Sole Ownership without the consent of the other Party as long as such other Party’s rights under this Agreement and the Collaboration are not adversely impacted (in which case, it will obtain such other Party’s prior written consent, not to be unreasonably withheld or delayed).
12.13Trademarks. The Exploiting Party shall own and Control any and all trademarks developed during the Term for the purposes of Commercialization of the First Product or other Collaboration Product, (the “Commercialization Trademarks”), for which the Exploiting Party exercised an Option in the Territory. If and to the extent that the other Party has any rights to such Commercialization Trademarks, following the exercise of the Option such Party shall promptly assign its rights to such Commercialization Trademarks to the Exploiting Party. Where the Parties agree to an Out-Licensing to a Third Party with respect to the First Product or other Collaboration Product, the relevant Commercialization and Trademarks, if any, shall be jointly owned by the Parties unless otherwise agreed with such Third Party.
Section 13
CONFIDENTIALITY
13.1Confidential Information. Except to the extent expressly authorized by this Agreement or otherwise agreed in writing by the Parties, during the Term and for [***] thereafter (save with respect to trade secrets, for which, subject to Section 13.2, there shall be no end date to the confidentiality provided hereunder), the receiving Party shall keep confidential and shall not publish or otherwise disclose, and shall not use for any purpose other than as expressly provided for in this Agreement, any Confidential Information furnished or otherwise made known to it, directly or indirectly, by the other Party, and each Party shall keep confidential and shall not publish or otherwise disclose the terms of this Agreement except as permitted herein. Each Party may use the other Party’s Confidential Information only to the extent required to accomplish the purposes of this Agreement, including exercising its rights and performing its obligations under this Agreement. Each Party will use at least the same standard of care as it uses to protect its own proprietary or confidential information (but no less than reasonable care) to ensure that its employees, agents, consultants, contractors, and other representatives do not disclose or make any unauthorized use of the other Party’s Confidential Information. Each Party will promptly notify the other upon discovery of any loss or unauthorized use or disclosure of the other Party’s Confidential Information.
13.2Exceptions. The obligations in Section 13.1 shall not apply with respect to any portion of Confidential Information that a Party can demonstrate contemporaneously by tangible records or other competent proof that such information:
(a)was already known to the receiving Party or its Permitted Recipient, other than under an obligation of confidentiality at the time that such Party obtains such information;
(b)was generally available to the public or otherwise part of the public domain at the time that such Party obtains such information;
(c)became generally available to the public or otherwise part of the public domain after its obtaining and other than through any act or omission of such Party in breach of this Agreement;
(d)was disclosed to such Party or its Permitted Recipient, other than under an obligation of confidentiality, by a Third Party who had no obligation to such Party not to disclose such information to others; or
(e)was independently discovered or developed by a Party or its Permitted Recipient without the use of or reference to the Solely Owned Confidential Information or Jointly Owned Confidential Information.
13.3Authorized Disclosure.
(a)The receiving Party may disclose the Confidential Information of the other Party to its Affiliates and its or their respective employees, consultants, or Subcontractors solely on a need-to-know basis for the purpose of conducting the receiving Party’s activities under this Agreement (collectively “Permitted Recipient”) that (1) any such Permitted Recipient must be bound by obligations of confidentiality at least as restrictive as those set forth in this Agreement, and (2) the receiving Party remains liable for the compliance of its Permitted Recipient with such obligations.
(b)Further, the receiving Party may, without the prior written consent of the other Party but on not less than [***]’ prior written notice of such disclosure; or (where it is not possible to give such prior notice) with notification to the other Party in writing within [***] of such disclosure, disclose the Solely Owned Confidential Information of the other Party or the Jointly Owned Confidential Information to the extent such disclosure is reasonably necessary in the following instances:
i.prosecuting or defending litigation;
ii.complying with Applicable Law or the rules or regulations of any securities exchange on which such Party’s stock is listed, whereby each Party shall provide the other Party with advance notice of legally required disclosures to the extent practicable, and to the extent possible, at least [***] prior to such disclosure;
iii.disclosure, in connection with the performance of this Agreement, to ethics committees and institutional review boards (collectively,
“IRBs”), institutional biosafety committees (“IBCs”), CROs, and investigators involved with the Clinical Trials set forth in an R&D Plan, each of whom prior to disclosure must be bound by similar terms of confidentiality and non-use at least equivalent in scope to those set forth in this Section 13 under the responsibility of the receiving Party;
iv.disclosure that is deemed necessary by either Party to be disclosed to its respective Affiliates, agents, consultants or actual or prospective licensees (or other bona fide collaborators) in furtherance of the development, manufacture, licensing and/or commercialization of such Party’s proprietary products or technologies, on the condition that such Third Parties are authorised undertake such activities and to receive such a disclosure under this Agreement and agree to be bound by confidentiality and non-use obligations that are substantially consistent with (and no less onerous than) the confidentiality and non-use provisions contained in this Agreement;
v.disclosure to its attorneys, accountants, auditors and other advisors on a need to know basis provided such individuals or entities are bound to confidentiality and nondisclosure requirements by professional rules of conduct or nondisclosure agreements under the responsibility of the receiving Party, and to actual or prospective acquirers, lenders, financers, or investors as may be necessary to comply with the terms, or in connection with their evaluation, of such potential or actual acquisition, loan, financing, or investment; on the condition that such acquires, lenders, financers, or investors agree to be bound by confidentiality and non-use obligations that are substantially consistent with the confidentiality and non-use provisions contained in this Agreement under the responsibility of the receiving Party;
vi.disclosure entailed by filing and prosecuting Patent Rights as contemplated by Section 12 hereof;
vii.disclosure to Regulatory Authorities in connection with the Development of a Collaboration Product, in particular conduct of Clinical Trials, including disclosure of relevant safety information related to the Clinical Trials set forth in an R&D Plan to investigators, institutional review boards and/or ethics committees and Regulatory Authorities; and
viii.disclosure on www.clinicaltrials.gov or other comparable European or national websites.
13.4Press Releases. The Parties agree to issue an initial joint press release at or shortly after the Effective Date within the time-period as required by relevant securities laws. It is understood that each Party may desire or be required to issue subsequent press releases relating to this Agreement or activities hereunder. The Parties agree to consult with each other reasonably and in good faith with respect to the text and timing of such press releases prior to the issuance thereof. Following the initial joint press release announcing this Agreement, either Party shall be free to disclose, without the other Party’s prior written consent, the existence of this Agreement, the identity of the other Party, and those terms of the Agreement which have already been publicly disclosed in accordance with this Section.
13.5Publications.
(a)During the Term, the disclosure by either Party relating to any aspect of the Collaboration, including a Collaboration Product in any publication or presentation shall be in accordance with the procedure set forth in this Section. Prior to either Party’s exercise of its Exit Decision, a Party (“Publishing Party”) shall provide the JSC with a copy of any proposed publication or presentation at least [***] prior to submission for publication or presentation to provide the JSC with an opportunity to review and approve the Publishing Party’s proposed publication or presentation. The Publishing Party may not publish any publication or presentation without the JSC’s review and approval and the approval of both Parties.
(b)If the JSC notifies the Publishing Party in writing, within [***] after the Publishing Party’s submission of the copy of the proposed publication or presentation to the JSC, that such publication or presentation, (i) contains an invention, solely or jointly conceived or reduced to practice by the non-Publishing Party, for which the non-Publishing Party reasonably desires to obtain patent protection or (ii) could be expected to have a material adverse effect on the commercial value of any Confidential Information disclosed by the non-Publishing Party to the Publishing Party, the Publishing Party shall prevent or delay such publication or presentation for a period of time specified by the JSC. In the case of inventions, a delay shall be for a period reasonably sufficient to permit the timely preparation and filing of a patent application(s) on such invention, and in no event less than [***] from the date of the Publishing Notice.
(c)After either Party’s exercise of an Exit Decision only the Continuing Party shall have the right to publish in connection with a Collaboration Product or the wider Collaboration as it relates to that Collaboration Product, including with respect to any Joint IPR, and the Exiting Party shall not make any publication or presentation relating to any Collaboration Product or the wider Collaboration as it relates to that Collaboration Product, including with respect to any Joint IPR.
(d)In the event that a Party desires to use the other Party’s trademark for any publication, communication or other public disclosure, such Party shall obtain the prior written consent of the other Party for such use.
13.6The Parties agree that all Confidential Information containing personal data shall be handled in accordance with any and all Applicable Laws, including without limitation, Data Protection Applicable Laws and any other laws related to privacy.
Section 14
REPRESENTATIONS AND WARRANTIES
14.1Authority and Binding Agreement. Molecular Partners and Orano Med each represents and warrants to the other that (a) it has the corporate power and authority and the legal right to enter into this Agreement and perform its obligations hereunder; (b) it has taken all necessary corporate action on its part required to authorize the execution and delivery of the Agreement and the performance of its obligations hereunder; and (c) the Agreement has been duly executed and delivered on behalf of each Party and constitutes a legal, valid and binding obligation of such Party that is enforceable against it in accordance with its terms subject to bankruptcy, insolvency, reorganization, arrangement, winding-up, moratorium, and similar laws of general application affecting the enforcement of creditors’ rights generally, and subject to general equitable principles, including the fact that the availability of equitable remedies, such as injunctive relief or specific performance, is in the discretion of the applicable court.
14.2No Conflicts. Molecular Partners and Orano Med each represents and warrants that, it has not entered, and shall not enter, into any agreement with any Third Party that is in conflict with the rights granted to the other Party under this Agreement, and has not taken any action that would in any way prevent it from granting the rights granted to the other Party under this Agreement, or that would otherwise materially conflict with or adversely affect the rights granted to the other Party under this Agreement. Without limiting the foregoing, Molecular Partners represents and warrants that the Existing Contract does not conflict with this Agreement.
14.3No Adverse Proceedings. Except as otherwise notified in writing to the other Party on and before the execution of this Agreement, to the best of its knowledge, there is not pending or, to the knowledge of such Party, threatened, against such Party, any claim, suit, action or governmental proceeding that would, if adversely determined, materially impair the ability of such Party to perform its obligations under this Agreement, including without limitation such claims for its Background IPR infringing the Third Party’s IPR.
14.4No Debarment. Each Party hereby certifies to the other that it has not used, and will not use the services of any person disqualified, debarred, banned, subject to debarment or convicted of a crime for which a person could be debarred by the FDA under 21 U.S.C. 335a, as amended (or subject to a similar sanction of any other Regulatory Authority), in any capacity in connection with any of the services or work provided under any element of the Collaboration and that this certification may be relied upon in any applications to the FDA or any other Regulatory Authority. It is understood and agreed that this certification imposes a continuing obligation upon each Party to notify the other promptly of any change in the truth of this certification.
14.5Personal Data. Molecular Partners and Orano Med each represents and warrants that it shall comply with any Data Protection Applicable Laws of the related countries or jurisdiction to use, disclose, process, transfer or administrate any personal data for the purpose of the Collaboration, and, if necessary, separately enter into the agreement(s) of the detailed terms and conditions for such personal data to satisfy and comply with all the requirements under the respective Data Protection Applicable Laws, including without limitation GDPR. Each Party shall cooperate with each other in good faith in ensuring compliance with the Data Protection Applicable Laws.
14.6Ethical Business Practices.
(a)Molecular Partners and Orano Med each represents and warrants that neither it nor its Affiliates will promise, offer or make any payment, either directly or indirectly, of money or other assets, including the compensation such Party derives from this Agreement, to government or political party officials, officials of International Public Organizations, candidates for public office, or representatives of other businesses or persons acting on behalf of any of the foregoing (collectively “Officials”) where such payment would constitute a violation of any Applicable Law. In addition, regardless of legality, neither it nor its Affiliates will promise, offer or make any payment either directly or indirectly to Officials if such payment is for the purpose of improperly influencing decisions or actions with respect to the subject matter of this Agreement.
(b)Each Party represents and warrants that it shall comply at all times with all applicable laws and regulations, including, but not limited to, those pertaining to export control and those preventing and fighting against corruption and influence peddling (French Sapin II law, US FCPA, UK Bribery Act), bribery, money laundering, extortion, embezzlement, rebate payoff, influence payment, kickback, or other unlawful payment, facilitation payments, and shall take reasonable measures to ensure their respective shareholders, directors, officers, employees, agents, representatives and subcontractors, so comply. From the Effective Date, each Party undertakes to inform the other Party without any delay, of any event which would contradict the declarations and guarantees defined in this Article 14.6. In the event that a Party is determined by final judicial decision to have violated the requirements of this Article and fails to cure same within [***] after written notice from the other Party, and to the reasonable satisfaction of such other Party, such other Party may, by prior written notice, and in addition to any other rights or remedies, terminate this Agreement, for cause.
(c)Without limiting the foregoing, each Party undertakes and warrants to comply with the principles set forth in their respective codes of ethics and business conduct which for Orano Med is available on the website www.orano.group and for Molecular Partners is available at Ethics & Compliance | Molecular Partners, in each case to the extent applicable to the activities covered under this Agreement.
14.7Compliance with Applicable Law. Molecular Partners and Orano Med each represents and warrants that it and its Affiliates will perform their obligations under this Agreement and each R&D Program and R&D Plan in accordance with Applicable Laws and that each Party has and will maintain all applicable Regulatory Approvals required to conduct its activities under this Agreement.
14.8No Knowledge of Infringement. Orano Med represents and warrants that as of the Effective Date and to its actual knowledge the practice of the Orano Med Background Technology to the extent required for the Exploitation of the Collaboration Product does not infringe any issued Patent Rights of any Third Party or, if and when issued, any claim within any patent application of any Third Party. Molecular Partners represents and warrants that as of the Effective Date and to its actual knowledge the practice of the Molecular Partners Background Technology to the extent required for the Exploitation of the Collaboration Product does not infringe any issued Patent Rights of any Third Party or, if and when issued, any claim within any patent application of any Third Party.
14.9NO WARRANTY. EXCEPT AS EXPRESSLY PROVIDED HEREIN, NEITHER PARTY MAKES WARRANTIES, EXPRESS OR IMPLIED, INCLUDING ANY WARRANTY OF MERCHANTABILITY OR FITNESS FOR A PARTICULAR PURPOSE OR USE, AND NON-INFRINGEMENT OF THIRD PARTY INTELLECTUAL PROPERTY RIGHTS, WITH RESPECT TO THE COLLABORATION PRODUCT.
Section 15
INDEMNIFICATION
15.1Orano Med Indemnification. Orano Med hereby agrees to defend, hold harmless and indemnify (collectively, “Indemnify”) Molecular Partners, its Affiliates, and its and their agents, directors, officers, and employees (the “Molecular Partners Indemnitees”) from and against any and all documented liabilities, expenses and/or losses, including without limitation reasonable legal expenses and attorneys’ fees (collectively “Losses”) resulting from any suits, claims, actions and demands from a Third Party (each, a “Third Party Claim”) to the extent that they arise or result from (a) the gross negligence or intentional misconduct of Orano Med, any Orano Med Indemnitee conducting activities on behalf of Orano Med under this Agreement; or (b) any material breach by Orano Med of any representation, warranty or covenant under this Agreement; but excluding, in each case, any such Losses to the extent arising from Molecular Partners’ breach of its obligations under this Agreement.
15.2Molecular Partners Indemnification. Molecular Partners hereby agrees to Indemnify Orano Med, its Affiliates, and its and their agents, directors, officers, and employees (the “Orano Med Indemnitees”) from and against any and all Losses resulting from Third Party Claims to the extent that they arise or result from (a) the gross negligence or intentional misconduct of Molecular Partners, any Molecular Partners Indemnitee conducting activities on behalf of Molecular Partners under this Agreement; or (b) any material breach, by Molecular Partners of any representation, warranty or covenant under this Agreement; but excluding, in each case, any such Losses to the extent arising from Orano Med’s breach of its obligations under this Agreement.
15.3Indemnification Procedure. Each Party’s agreement to Indemnify the other Party is conditioned on the performance of the following by the Party seeking indemnification: (a) promptly notifying the Indemnifying Party in writing that the Party seeking indemnification has received the Third Party Claim; provided that, any delay in complying with the requirements of this Section 15.3(a) will only limit the Indemnifying Party’s obligation to the extent of the prejudice caused to the Indemnifying Party by such delay; (b) permitting the Indemnifying Party to assume, if it requests so, full control and responsibility to investigate, prepare for and defend against any such Third Party Claim; (c) providing necessary information and reasonable assistance to the Indemnifying Party, at the Indemnifying Party’s expense, in the investigation of, preparation for and defense of any Third Party Claim; and (d) not compromising or settling such Third Party Claim without the Indemnifying Party’s written consent, provided that such consent shall not be unreasonably withheld nor delayed. The Party seeking indemnification shall, in order to receive the Indemnification for the Loss, provide the Indemnifying Party with the written evidence for any Loss incurred to such Party.
15.4Separate Defense of Claims. In the event that the Parties cannot agree as to the application of Sections 15.1, 15.2 and/or 15.3 to any particular Loss, the Parties may conduct separate defences of such Loss. Each Party further reserves the right to claim indemnity from the other in accordance with Sections 15.1, 15.2 and/or 15.3 upon resolution of the underlying claim.
15.5Insurance. Each Party shall maintain commercially reasonable levels of insurance or other adequate and commercially reasonable forms of protection or self-insurance to satisfy its indemnification obligations under this Agreement. Each Party shall provide the other Party with written notice at least [***] prior to the cancellation, non-renewal or material change in such insurance or self-insurance which would materially adversely affect the rights of the other Party hereunder. The maintenance of any insurance shall not constitute any limit or restriction on damages available to a Party under this Agreement.
15.6LIMITATION OF LIABILITY.
NEITHER PARTY SHALL BE LIABLE TO THE OTHER PARTY FOR INDIRECT, INCIDENTAL, CONSEQUENTIAL OR SPECIAL DAMAGES, INCLUDING BUT NOT LIMITED TO LOST PROFITS, ARISING FROM OR RELATING TO THIS AGREEMENT AND/OR SUCH PARTY’S PERFORMANCE HEREUNDER, REGARDLESS OF ANY NOTICE OF THE POSSIBILITY OF SUCH DAMAGES AND REGARDLESS OF THE CAUSE OF ACTION (WHETHER IN CONTRACT, TORT, BREACH OF WARRANTY OR OTHERWISE).
EACH PARTY’S MAXIMUM, CUMULATIVE LIABILITY ARISING OUT OF OR RELATING TO THE COLLABORATION PURSUANT TO THIS AGREEMENT AND/OR SUCH PARTY’S PERFORMANCE RELATING THERETO, REGARDLESS OF THE CAUSE OF ACTION (WHETHER IN CONTRACT, TORT, BREACH OF WARRANTY, INDEMNITY OR OTHERWISE), WILL NOT EXCEED FOR ALL CLAIMS UNDER THE COLLABORATION [***], PROVIDED, HOWEVER, SUCH LIMITATION SHALL NOT APPLY TO EITHER PARTY’S INDEMNIFICATION OBLIGATION ARISING OUT OF, RELATING TO OR RESULTING FROM (1) ANY BREACH OF SUCH PARTY’S EXCLUSIVITY OBLIGATIONS, REPRESENTATIONS AND WARRANTIES AND CONFIDENTIALITY OBLIGATIONS, OR (2) THIRD PARTY CLAIMS OR (3) GROSS NEGLIGENCE OR INTENTIONAL MISCONDUCT.
NOTHING IN THIS SECTION 15.6 SHALL LIMIT OR RESTRICT A PARTY’S LIABILITY TO THE EXTENT THAT IT MAY NOT BE SO EXCLUDED UNDER APPLICABLE LAWS, IN PARTICULAR UNDER BELGIAN LAW, INCLUDING (I) ANY STATUTORY LIABILITY UNDER MANDATORY LAWS, SUCH AS PRODUCT LIABILITY, PHARMACEUTICAL LIABILITY ETC. AND (II) ANY LIABILITY IN TORT FOR DEATH OR PERSONAL INJURY CAUSED BY THAT PARTY’S NEGLIGENCE, OR LIABILITY FOR FRAUD.
FOR THE SAKE OF CLARITY, NOTHING CONSTRUED HEREIN SHALL PREVENT EITHER PARTY’S RIGHT TO SEEK PRELIMINARY AND PERMANENT INJUNCTIVE RELIEF, WITHOUT THE NECESSITY OF PROVIDING ACTUAL DAMAGES. SUCH RIGHT SHALL BE CUMULATIVE AND IN ADDITION TO ANY OTHER RIGHTS OR REMEDIES TO WHICH EITHER PARTY MAY BE ENTITLED.
Section 16
TERM AND TERMINATION
16.1Term. This Agreement shall be effective as of the Effective Date and, unless terminated earlier pursuant to Section 16.2, 16.3, or 16.4 or any other termination right expressly stated in this Agreement by the Parties in writing, shall continue in effect in respect of each Collaboration Product from the Effective Date until completion of all Commercialization activities for such Collaboration Product (the “Term”).
16.2Termination for Material Breach.
(a)Notice and Cure Period. If a Party (the “Breaching Party”) is in material breach, the other Party (the “Non-Breaching Party”) shall have the right to give the Breaching Party written notice specifying the nature of such material breach. The Breaching Party shall have a period of [***] after receipt of such notice to cure such material breach (the “Cure Period”) in a manner reasonably acceptable to the Non-Breaching Party. For the avoidance of doubt, this provision is not intended to restrict in any way either Party’s right to notify the other Party of any other breach or to demand the cure of any other breach.
(b)Termination Right. The Non-Breaching Party shall have the right to terminate this Agreement in respect of any Collaboration Product or country or countries in the Territory directly affected by the material breach (provided that with respect to any country, the materiality of the breach will be assessed at the level of the country), upon written notice, in the event that the Breaching Party has not cured such material breach within the Cure Period, provided, however, that if such breach is capable of cure but cannot reasonably be cured within the Cure Period, and the Breaching Party notifies the Non-Breaching Party of its intent to cure such material breach, commences actions to cure such material breach within the Cure Period and thereafter diligently continue such actions, the Breaching Party shall have an additional [***] to cure such breach. If a dispute over such termination occurs between the Parties and such dispute is submitted to the arbitration pursuant to Section 17.3, such termination shall not be effective until a conclusion of the dispute resolution procedures in Section 17.4, as applicable, resulting in a determination that there has been a material breach that was not cured within the Cure Period.
16.3Termination for Bankruptcy. Either Party may terminate this Agreement by providing a written notice to the other Party at any time if, (i) the other Party files in any court or agency pursuant to any statute or regulation of any state, country or jurisdiction, a petition in bankruptcy or insolvency or for reorganization or for an arrangement or for the appointment of a receiver or trustee of such other Party or of such other Party’s assets, (ii) the other Party proposes a written agreement of composition or extension of its debts, (iii) the other Party shall be served with an involuntary petition against it, filed in any insolvency proceeding, and such petition shall not be dismissed or stayed within [***] after the filing thereof, (iv) the other Party proposes or becomes a party to any dissolution or liquidation, or (v) the other Party makes an assignment for the benefit of its creditors.
16.4Other Termination Rights.
(a)The Agreement may, without any cause, be terminated by the mutual written consent of the Parties on a Collaboration Product-by-Product basis.
(b)At any time after the earlier of (i) [***] or (ii) [***].
16.5General Effects of Termination.
(a)Accrued Rights and Obligations. Termination of this Agreement shall not release either Party from its obligations accrued prior to the effective date of termination nor deprive either Party from any rights that this Agreement has conferred on such Party. Such obligations and rights shall survive termination of this Agreement. Termination of this Agreement by either Party shall be in addition to and not in lieu of any other remedies available to such Party, at law and in equity.
(b)Survival. The following Sections of this Agreement and all definitions relating thereto shall survive any expiration or termination of this Agreement for any reason: Sections 2.4 (to the extent provided in Section 2.4(c)) and 2.6, Section 5 (in relation to Collaboration Costs validly incurred prior to expiration or termination of this Agreement), Section 7, Section 9, Section 10, Section 11, Section 12, Section 13, Section 15, Section 16 and Section 17, as well as any other Sections of this Agreement which expressly or by implication are intended to survive expiration or termination of the Agreement and each Section necessary for a Party to enforce the foregoing provisions.
16.6Consequences in case of termination pursuant to Section 16.2 or 16.4(b). Upon any termination of this Agreement pursuant to Section 16.2 by the non-breaching Party or pursuant to Section 16.4(b) by the Continuing Party, the non-breaching Party or, as applicable, the Exiting Party (“Assuming Party”) shall be entitled to take over the exploitation of the respective Collaboration Product(s) from the other Party (“Causing Party”) subject to the following provisions (on a country-by-country or Collaboration Product-by-Collaboration Product basis (as applicable)), unless otherwise provided in the applicable License and Supply Agreements:
(a)all licenses granted by one Party to the other Party under this Agreement shall terminate and revert to the granting Party;
(b)to the extent permitted by Applicable Law, and upon written request by the Assuming Party, the Causing Party shall transfer to the Assuming Party all Regulatory Documentation and Regulatory Approvals prepared or obtained by or on behalf of the Causing Party prior to the date of such termination, to the extent transferable;
(c)upon written request by the Assuming Party, the Causing Party shall transfer to the Assuming Party [***];
(d)upon written request by the Assuming Party and subject to the payment by the Assuming Party to the Causing Party of compensation for such license under the Reversion Terms, the Causing Party shall grant to the Assuming Party [***]. This licence shall not extend to any Exploitation of the respective IPR outside of such Collaboration Product (e.g., they shall not extend to the use of components of such Collaboration Product outside of the respective Collaboration Product or on their own);
(e)upon written request by the Assuming Party within [***] after termination, the Causing Party shall provide [***];
(f)the Causing Party shall return to the Assuming Party (or at the Assuming Party’s request, destroy) all relevant records and materials in its possession or Control containing or comprising the Know-How or other Confidential Information of the Assuming Party; and
(g)the Causing Party, its Affiliates or its sublicensees shall cease all Commercialization of Collaboration Products in the Territory (or applicable country in the Territory) in a prompt manner and in accordance with Applicable Laws
16.7Consideration Payable by the Assuming Party. With regard to the consideration payable by the Assuming Party for the Causing Party’s obligations set forth in Section 16.6, the Parties shall use good faith efforts to negotiate and agree upon [***] (the “Reversion Terms”), within [***] (or such longer period of time agreed by the Parties) following the effective date of termination. If the Parties are unable to agree on the Reversion Terms [***].
Section 17
MISCELLANEOUS
17.1Entire Agreement. The Parties acknowledge that this Agreement shall govern all activities of the Parties with respect to the Collaboration from the Effective Date forward. This Agreement, including the Exhibits hereto, sets forth the complete, final and exclusive agreement between the Parties concerning the subject matter hereof and supersedes all prior agreements and understandings between the Parties with respect to such subject matter, save to the extent the same may be modified by the Clinical Supply Agreement, Commercial Supply Agreement and License Agreement contemplated by the Parties once the same are agreed by the Parties and in force. Save as aforesaid, there are no covenants, promises, agreements, warranties, representations, conditions or understandings, either oral or written, between the Parties with respect to such subject matter other than as are set forth in this Agreement. All Exhibits attached hereto are incorporated herein as part of this Agreement.
17.2Governing Law. Injunctive relief. This Agreement shall be governed and construed in accordance with the internal laws of Belgium, excluding any choice of law rules that may direct the application of the laws of another jurisdiction. Nothing contained in this Agreement shall deny either Party the right to seek injunctive or other equitable relief from a court of competent jurisdiction in the context of a bona fide emergency or prospective irreparable harm, and such an action may be filed or maintained notwithstanding any ongoing discussions between the Parties.
17.3Dispute Resolution. Unless otherwise provided in this Agreement, in the event of any dispute, controversy or claim arising out of, relating to or in connection with any provision of this Agreement (each a “Dispute”), the Parties shall refer the Dispute to the Executive Officers (or designees with similar authority to resolve such Dispute), who shall attempt in good faith to resolve such Dispute. If the Executive Officers cannot resolve such Dispute within [***] of the matter being referred to them in writing, then such Dispute is to be settled by binding arbitration in Brussels, Belgium in accordance with the Rules of Arbitration of the International Chamber of Commerce as in effect on the date of signing of this Agreement, except as modified herein. The arbitration tribunal shall consist of three (3) arbitrators with each Party appointing one (1) arbitrator and the two (2) arbitrators thus appointed choosing the third arbitrator who will act the presiding arbitrator of the arbitration tribunal. The language of the arbitration shall be English. The arbitration award shall be final and binding on the parties, and the parties agree to be bound thereby and to act accordingly. The arbitrators shall have the power to grant any remedy or relief that they deem just and equitable and that is in accordance with the terms of this Agreement, including specific performance, and including injunctive relief, whether interim or final, and any such relief and any interim, provisional or conservatory measure ordered by the arbitrators may be specifically enforced by any court of competent jurisdiction. Each Party retains the right to seek interim, provisional or conservatory measures from judicial authorities, and any such request shall not be deemed incompatible with the agreement to arbitrate or a waiver of the right to arbitrate.
17.4Force Majeure. The Parties shall be excused from the performance of their obligations under this Agreement to the extent that such performance is prevented by force majeure and the non-performing Party promptly provides written notice of the prevention to the other Party. For purposes of this Agreement, force majeure shall mean acts of God, civil disturbances, fires, earthquakes, acts of terrorism, floods, explosions, riots, war, rebellion, or failure or default of public utilities or common carriers or similar conditions beyond the control of the Parties. In the event of force majeure, either Party, as the case may be, will immediately notify the other Party of such inability and of the period for which such inability is expected to continue. The Party giving such notice will thereupon be excused from such of its obligations under this Agreement as it is thereby disabled from performing for so long as such Party is so disabled, up to a maximum of [***], after which time the Party not affected by the force majeure may terminate this Agreement. To the extent possible, each Party will use Commercially Reasonable Efforts to minimize the duration of any force majeure.
17.5Notices. Any notice required or permitted to be given under this Agreement shall be in writing, shall specifically refer to this Agreement and shall be deemed to have been sufficiently given for all purposes if such notice is timely and is: (a) mailed by first class certified or registered mail, postage prepaid, return receipt requested, (b) sent by express delivery service, or (c) personally delivered, except in the event this Agreement specifies, or the Parties mutually agree in writing that, the notice may be delivered by email. Unless otherwise specified by one Party to the other in writing, the respective mailing addresses of the Parties shall be as described below.
For Molecular Partners: [***]
With a copy to: [***]
For Orano Med: [***]
With a copy to: [***]
Any such communication shall be deemed to have been received when delivered or when transmitted in the case of permitted email. It is understood and agreed that this Section 17.5 is not intended to govern the day-to-day business communications necessary between the Parties in performing their duties, in due course, under the terms of this Agreement.
17.6No Waiver; Modifications. It is agreed that no waiver by a Party hereto of any breach or default of any of the covenants or agreements herein set forth shall be deemed a waiver as to any subsequent and/or similar breach or default. No amendment, modification, release or discharge shall be binding upon the Parties unless in writing and duly executed by authorized representatives of both Parties.
17.7No Strict Construction. This Agreement has been prepared jointly and shall not be strictly construed against either Party. No presumption as to construction of this Agreement shall apply against either Party with respect to any ambiguity in the wording of any provision(s) of this Agreement irrespective of which Party may be deemed to have authored the ambiguous provision(s).
17.8Independent Contractor. The Parties are independent contractors of each other, and the relationship between the Parties shall not constitute a partnership, joint venture or agency under this Agreement. Neither Party shall be the agent of the other or have any authority to act for, or on behalf of, the other Party in any matter.
17.9Assignment. Except as otherwise expressly provided herein, neither this Agreement nor any of the rights or obligations hereunder (including any licensed IPR) may be assigned by either Party without the prior written consent of the other Party (such consent not to be unreasonably withheld, conditioned or delayed); provided, that such consent shall not be required for an assignment by a Party (a) to an Affiliate of the assigning Party (provided, however, that a Party assigning to an Affiliate shall remain fully and unconditionally liable and responsible to the non-assigning Party hereto for the performance and observance of all such duties and obligations by such Affiliate), or (b) subject always to Section 2.5 above, to any Third Party in a Change of Control, so long as such Affiliate or Third Party agrees in writing to be bound by the terms of this Agreement. An assignment to an Affiliate shall terminate, and all rights so assigned shall revert to the assigning Party, if and when such Affiliate ceases to be an Affiliate of the assigning Party. Any attempted assignment in violation hereof shall be void ab initio and of no effect.
17.10Headings. The captions to the several Sections and Sections hereof are not a part of this Agreement but are included merely for convenience of reference only and shall not affect its meaning or interpretation.
17.11Counterparts. This Agreement may be executed in two (2) counterparts, each of which shall be deemed an original, but all of which together shall constitute one (1) and the same instrument. This Agreement may be executed by facsimile or electronic (e.g., pdf) signatures and such signatures shall be deemed to bind each Party hereto as if they were original signatures.
17.12Severability. If any provision of this Agreement is held to be illegal, invalid or unenforceable under any present or future law, and if the rights or obligations of a Party under this Agreement will not be materially and adversely affected thereby, (a) such provision shall be fully severable, (b) this Agreement shall be construed and enforced as if such illegal, invalid or unenforceable provision had never comprised a part hereof, (c) the remaining provisions of this Agreement shall remain in full force and effect and shall not be affected by the illegal, invalid or unenforceable provision or by its severance here from and (d) in lieu of such illegal, invalid or unenforceable provision, there shall be added automatically as a part of this Agreement a legal, valid and enforceable provision as similar in terms to such illegal, invalid or unenforceable provision as may be possible and reasonably acceptable to the Parties.
17.13No Benefit to Third Parties. The representations, warranties and agreements set forth in this Agreement are for the sole benefit of the Parties, and they shall not be construed as conferring any rights on any other parties.
17.14Construction.
(a)General. Except as otherwise explicitly specified to the contrary, (a) references to a Section, Section or Exhibit means a Section or Section of, or Exhibit to, this Agreement and all subsections thereof, unless another agreement is specified; (b) references to a particular statute or regulation include all rules and regulations promulgated thereunder and any successor statute, rules or regulations then in effect, in each case including the then-current amendments thereto; (c) words in the singular or plural form include the plural and singular form, respectively; (d) the terms “including,” “include(s),” “such as,” and “for example” used in this Agreement mean including the generality of any description preceding such term and will be deemed to be followed by “without limitation”; and (e) the words “hereof,” “herein,” “hereunder,” “hereby” and derivative or similar words refer to this Agreement. No presumption as to construction of this Agreement shall apply against either Party with respect to any ambiguity in the wording of any provision(s) of this Agreement irrespective of which Party may be deemed to have authored the ambiguous provision(s).
(b)No Response. Where a provision of this Agreement provides for a Party to respond within a designated period following written notice from the other Party, and if such Party fails to respond, then the failure to respond shall not be deemed to create or imply (except where expressly stated otherwise in this Agreement): (i) that the non-responding Party agrees or disagrees with the proposed action to be taken by the other Party, (ii) any amendment, change or waiver of the terms of this Agreement, or (iii) any consent that an action proposed to be taken may be taken if it conflicts with the terms of this Agreement and/or waiver of any rights it may have to seek remedies at law or in equity for breach of this Agreement as a result of the action taken.
IN WITNESS WHEREOF, the Parties hereto, intending to be legally bound hereby, have caused this Agreement to be executed by their duly authorized representatives as of the Effective Date.
|
|
|
|
|
|
|
Molecular Partners
/s/ Patrick Amstutz
Name: Patrick Amstutz
Title: CEO, Molecular Partners
Date: 5/1/2024 | 17:34:57 CET
|
Orano Med
/s/ Julien Dodet
Name: Julien Dodet
Title: CEO, Orano Med
Date: 5/1/2024 | 17:55:15 CET
|
|
/s/ Michael Pitzner
Name: Michael Pitzner
Title: General Counsel,
Molecular Partners
Date: 5/1/2024 | 17:35:40 CET
|
|
Attached:
i) Exhibit A – Existing Contracts;
ii) Exhibit B – R&D Plan;
iii) Exhibit C – R&D Budget;
iv) Exhibit D - Manufacturing Key Terms;
v) Exhibit E - Certain Commercialization Key Terms; and
vi) Exhibit F – Launch Preparedness Plan.
EXHIBIT A – EXISTING CONTRACTS
[***]
EXHIBIT B – R&D PLAN
[***]
EXHIBIT D - MANUFACTURING KEY TERMS
[***]
EXHIBIT E - CERTAIN COMMERCIALIZATION KEY TERMS
[***]
EXHIBIT F – LAUNCH PREPAREDNESS REQUIREMENTS
[***]
EX-4.16
4
a416psuplan2024_employeeso.htm
EX-4.16
Document
|
|
|
|
|
|
| Molecular Partners AG: Performance Share Plan 2024 - Employees |
|
Exhibit 4.16
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Molecular Partners AG
Performance Share Plan 2024
Employees
|
|
|
|
|
|
|
|
| Molecular Partners AG: Performance Share Plan 2024 - Employees |
|
Table of Contents
Annexes
Annex 1: Definitions
Annex 2: Forms of PSU Award Agreement
|
|
|
|
|
|
| Molecular Partners AG: Performance Share Plan 2024 - Employees |
|
Performance Share Plan 2024
1.Purpose
The purpose of this performance share plan (Plan) is to establish a framework that enables the Company to provide certain eligible persons with a variable long-term incentive to contribute to the future success and prosperity of the Company and to better align their interests with those of the Company and its shareholders by granting Performance Share Units (each a PSU) to them.
2.Definitions and Interpretation
(a)Capitalized terms used in this Plan shall have the meaning set forth in Annex 1.
(b)Where this Plan refers to employer, employee or employment, such terms shall apply by analogy if the relevant eligible person or Participant is not engaged as an employee, but under a different type of contract or in a different capacity, e.g. as a consultant under a mandate agreement or as a member of a corporate body (e.g. member of a board of directors or advisory board).
3.Responsibilities and Administration
(a)This Plan has been approved and issued by the Board of Directors and any amendments or new editions of this Plan or new or other plans shall require the approval by the Board of Directors. The Board of Directors shall be in charge of approving, upon recommendation of the nomination and compensation committee of the Board of Directors (Nomination and Compensation Committee), the maximum number of PSUs that may be granted under this Plan and of shares to be allocated from the Company's conditional capital or otherwise in connection with such PSUs.
(b)The Nomination and Compensation Committee shall be responsible for the implementation and administration of the Plan and shall make recommendations to amend or renew or terminate the Plan or to replace it with new editions or other plans. It may delegate, under its supervision, the implementation and administration, as well as grants of PSUs to one or several administrators (Administrator).
(c)All resolutions, decisions, determinations and interpretations made by the Nomination and Compensation Committee or, upon delegation by the Nomination and Compensation Committee, an Administrator pursuant to this Plan, including any amendments or withdrawals of grants, are final and binding, unless approval by the Board of Directors or the shareholders' meeting is required.
(d)Any technical or administrative task in connection with the Plan may be outsourced by the Nomination and Compensation Committee or the relevant Administrator to a third party service
|
|
|
|
|
|
| Molecular Partners AG: Performance Share Plan 2024 - Employees |
|
provider, e.g. the bank in charge with the creation of the Shares (each a Plan Service Provider).
4.Eligibility and Participation
(a)As a rule, employees (other than members of the Management Board) as well as selected consultants may become eligible to participate in this Plan. The decision on eligibility is reserved to the Nomination and Compensation Committee or the relevant Administrator.
(b)Nothing in this Plan shall provide any rights to eligible persons or any other person nor create any obligation of the Company to grant PSUs based on this Plan or otherwise. This Plan is only applicable in connection with a mutually signed PSU award agreement (PSU Award Agreement) among the Company (or the employer Group Company) and the eligible person, substantially in the form attached hereto as Annex 2. The right to receive PSUs shall accrue exclusively to those eligible persons who have, in accordance with this Plan, been duly and validly offered, and have signed and returned, their individual PSU Award Agreement by the relevant due date (each a Participant).
(c)Grants of PSUs based on this Plan are discretionary and shall not create any entitlement to participate in future grants or in future participation, incentive or benefit plans, including future performance share plans, regardless of the length of time a person has previously been allocated PSUs or other entitlements under this Plan or other plans.
(d)Neither the grant of PSUs, nor the transfer of Shares in connection with this Plan shall confer upon any Participant any right to continue to be employed by any Group Company.
5.Grant of PSUs
(a)Grants of PSUs shall be exclusively made by way of PSU Award Agreements. The PSU Award Agreement shall set forth the number of PSUs and certain other terms and conditions of such grant. Except as otherwise determined in a PSU Award Agreement, PSUs shall be granted to the Participants free of charge.
(b)One PSU represents a conditional entitlement to purchase a number of Shares at the nominal value of a Share. The number of Shares which shall be allocated to a Participant upon vesting shall be determined pursuant to a vesting multiple as described in Section 10 hereof (the Vesting Multiple), subject to, and in accordance with, the terms and conditions of this Plan and the PSU Award Agreement.
(c)The date of grants shall be determined by the Nomination and Compensation Committee or the relevant Administrator and set out in the PSU Award Agreement (the Grant Date).
(d)A change of the regular working quota (Arbeitspensum) during the Vesting Period shall not lead to an adjustment of PSUs already granted. New Participants admitted to the Plan after the Grant Date may, if any, be granted a pro rata number of PSUs for that year, i.e. for the period between
|
|
|
|
|
|
| Molecular Partners AG: Performance Share Plan 2024 - Employees |
|
the beginning of their employment and the next regular Grant Date, as an interim grant or as additional PSUs on the next regular Grant Date.
6.No Securities
PSUs are neither Shares nor securities of any kind and no shareholder rights or similar rights are attached to the PSUs. The Participants will only obtain shareholder rights (including voting and dividend rights) upon actual transfer of Shares, if any, according to the terms and conditions of the Plan and upon entry into the share register, subject to, and in accordance with, the restrictions and procedures set out in article 5 of the Company's articles of incorporation.
7.No Transfer
PSUs granted under this Plan and the PSU Award Agreement are personal and non-transferable. Participants shall not be permitted to sell, donate, pledge, assign or otherwise dispose of the PSUs to third parties other than as provided for in the Plan. In case of death of a Participant, Section 14 hereof shall apply.
8.Vesting and Delivery of Shares
(a)Unless otherwise set out in this Plan (in particular in Section 14) or in the PSU Award Agreement, the PSUs granted by a PSU Award Agreement shall vest in three tranches of one third each. If the number of PSUs granted by a PSU Award Agreement cannot be divided by three, the two tranches vesting first shall be rounded up to the next integer and the tranche vesting last shall be rounded down and, if necessary, reduced in order to get to integers that add up to the total number of PSUs granted by the PSU Award Agreement.
(b)The first tranche of the PSUs shall vest on the first anniversary of the Grant Date, the second tranche on the second anniversary of the Grant Date and the third tranche on the third anniversary of the Grant Date (each, with respect to the relevant tranche, the Vesting Date) The period between the Grant Date and the Vesting Date shall, with respect to the relevant tranche, be deemed the Vesting Period.
(c)Subject to Section 14(b) and (c) below, no PSU shall vest if, during the relevant Vesting Period, the relevant Participant's employment is terminated (i.e. effective date of termination) or another reason for termination occurs other than through termination by the Participant for cause (wichtige Gründe) within the meaning of Article 337 CO.
(d)The relevant number of Shares shall be delivered by or on behalf of the Company to the Participant upon and subject to signing an acquisition declaration and payment of the nominal value of the Shares by the Participant. Alternatively, the Company may provide for cash-less acquisition or vesting-sale arrangements through a Plan Service Provider or otherwise. Delivery of Shares or other consideration shall, subject to the further conditions of delivery being met, occur no later than three months following the Vesting Date of the relevant PSUs.
|
|
|
|
|
|
| Molecular Partners AG: Performance Share Plan 2024 - Employees |
|
9.Underlying Shares
Shares to be delivered to Participants shall, subject to adjustment, if any, pursuant to Section 16, be registered shares of the Company with a nominal value of CHF 0.10 each (each a Share). Such Shares shall, at the discretion of the Company, be sourced from conditional share capital, from treasury shares or from other sources. Unless otherwise determined by the Board of Directors or the Nomination and Compensation Committee, Shares shall be sourced from the Company's conditional share capital and a respective maximum number of Shares out of conditional capital shall be deemed reserved, accordingly.
10.Vesting Multiple
(a)The Vesting Multiple shall not be lower than 0 nor higher than 1.5 (one point five). Within such range, the Vesting Multiple shall be determined by the Board of Directors upon proposal by the Nomination and Compensation Committee based on its assessment of the achievement of the goals set out in the score card (LTI Score Card) attached to the PSU Award Agreement or otherwise communicated by the Company to the Participant in connection with the grant (Goals). The Goals may include any corporate goals, i.e. strategic, operating or financial goals of the Company or the Group, any personal goals and performance of the relevant Participant and/or any goals relating to the total shareholder return or share price development. The LTI Score Card may allocate a percentage weighting to each Goal for purposes of deriving the Vesting Multiple or require a global assessment of the achievement of goals.
(b)The Vesting Multiple shall, unless the nature of the Goals demands otherwise, be determined for all tranches of the PSUs granted by a PSU Award Agreement in the year following the year of grant. Notwithstanding such determination, Vesting shall occur only at the time and subject to the conditions otherwise set out in this Plan or in the PSU Award Agreement.
(c)Depending on the corporate goals set out in the LTI Score Card, the Vesting Multiple may be fixed (if all elements of Goal achievement are known at the time of determination) or variable (e.g. depending on further stock price development throughout the remainder of the Vesting Period).
11.Taxes and Social Security Contributions
(a)Any Participant shall be responsible for reporting the receipt of any income under the Plan, however made, to the appropriate tax and social security authorities. Income, capital gain or other taxes due on the granting of PSUs, on the allocation of Shares and the subsequent sale of Shares or on a respective cash equivalent are in the sole responsibility of the Participant.
(b)The grant, vesting, delivery or sale of Shares or other relevant event in connection with the PSUs may be subject to the withholding of tax and social security contributions by the Company or, if different, the employer Group Company. The Company and the relevant employer Group Company shall be entitled to deduct or withhold a sufficient portion of the value otherwise due to be released under this Plan or of any other payment to the relevant Participant to satisfy any withholding requirement in connection therewith. Without limitation, withholding arrangements may include the sale of Shares to be delivered for PSU awards on behalf of a Participant and withholding of proceeds or deductions from salary or bonus payments, or require a payment from the Participant to the Company or the employer Group Company before settlement of the PSU awards.
|
|
|
|
|
|
| Molecular Partners AG: Performance Share Plan 2024 - Employees |
|
(c)The Company shall have the right (but no obligation, unless required by applicable law) to notify the tax and social security authorities of the grant of PSU awards, Shares or related events.
12.Disclosure Requirements
Any Participant shall be responsible to promptly comply with any applicable disclosure requirements under securities law and stock exchange regulations in connection with the receipt of grants of PSUs or Shares or upon the sale of Shares, including any disclosure requirements triggered by the thresholds for the ownership of shares and/or rights to obtain shares under Article 120 Financial Market Infrastructure Act and any management transaction notifications under Article 56 of the SIX Swiss Exchange listing rules. See also the Company's public disclosure, reporting and securities trading policy.
13.Other Obligations of the Participant
The Company is entitled to block or prohibit the issuance or release of Shares otherwise due to be issued or released if the Participant has any outstanding obligations (whether in connection with the Plan or otherwise arising in connection with the Participant's employment) to any Group Company, until the Participant has satisfied such outstanding obligations.
14.Termination of Employment and Forfeiture
(a)If (i) a Participant's employment is terminated or (ii) another reason for termination occurs during the Vesting Period, other than through termination by the Participant for cause (wichtige Gründe) within the meaning of Article 337 CO and except as set out in Section 14(b) and (c) below, all remaining unvested PSUs shall cease and be forfeited on the effective date of termination
(b)If a Participant’s employment agreement is terminated by the Company or a Group Company for reasons not pertaining to the Participant, the next unvested tranche of PSUs granted would vest pro rata (based on the effective date of termination) at the end of the relevant Vesting Period with the remaining PSUs lapsing without further effect. For clarity, the pro rata calculation under this Section 14 shall be determined on a monthly basis (tranche 1: month 1 to 12; tranche 2: month 13 to 24; tranche 3: month 25 to 36; respectively), based on the complete months of employment worked during the relevant Vesting Period.
(c)If the employment agreement terminates by reason of (i) death, permanent illness or disability of the Participant or (ii) retirement, a pro rata number of PSUs granted to the Participant shall vest immediately with the remaining PSUs lapsing without further effect. In the case of (i) here before, the pro rata calculation will be made assuming that employment lasted one year longer (but in any case not longer than the end of the regular three-year Vesting Period).
|
|
|
|
|
|
| Molecular Partners AG: Performance Share Plan 2024 - Employees |
|
(d)The Management Board may, taking into consideration the objectives of the Plan, at its sole discretion and with final and binding effect grant further exceptions from the forfeiture as per Section 14(a) above and assess whether a Participant’s employment agreement was terminated for reasons not pertaining to the Participant pursuant to Section 14(b) above. Two Management Board members need to approve and sign such specific PSU agreement pursuant to this Section 14(d).
15.Change of Control
(a)For purposes of this Plan, a change of control shall mean the occurrence of any of the following events (each a Change of Control):
(i)the acquisition in one or more transaction by any person or group of persons acting in concert, directly or indirectly, of the beneficial ownership of Shares and | or rights to acquire Shares representing 50% or more of the voting rights pertaining to the total number of Shares issued and registered in the commercial register;
(ii)any facts or circumstances that require any person or group of persons acting in concert to launch a mandatory offer within the meaning of applicable takeover regulations;
(iii)a public offer for Shares by any person or persons (other than in order to implement a new parent company held by the same owners of Shares), for such number of Shares that, by itself or together with Shares already held, triggers the duty to extend the offer to all Shares outstanding, if and when such offer becomes unconditional (subject only to conditions, if any, that survive following the regular offer period);
(iv)the reorganization, merger, scheme of arrangement, consolidation, liquidation or similar transaction of the Company otherwise than through a transaction by which the persons who beneficially held Shares representing 100% of the voting rights pertaining to the total number of Shares issued and registered in the commercial register prior to such transaction receive or continue to hold shares representing more than 50% of the voting rights pertaining to the total number of outstanding shares of the new or continuing entity.
(b)In the event of a Change of Control of the Company, the following shall apply for all PSUs in respect of which the Vesting Date has not occurred by the date of the Change of Control:
(i)all PSUs will vest immediately;
(ii)the Vesting Multiple with respect to each PSU allocation will be determined by the Nomination and Compensation Committee at the time of the Change of Control unless the Vesting Multiple has already been determined prior to the Change of Control; and
|
|
|
|
|
|
| Molecular Partners AG: Performance Share Plan 2024 - Employees |
|
(iii)the PSUs will be paid out in Shares, unless the Nomination and Compensation Committee resolves to repurchase or exchange PSUs or decides upon another solution to provide the Participants with the vesting value of the PSUs.
(c)If based on a good faith assessment of the particular circumstances and effects of a Change of Control event, such event does not fall into the category and nature of cases, circumstances and consequences addressed by the definition of Change of Control, which are deemed to justify an early vesting, the Board of Directors may, based on a recommendation by the Nomination and Compensation Committee, decide to replace the consequences set forth above, by other terms that more appropriately and fairly address the situation.
(d)If an event does not fall under the definition of Change of Control, but has substantially comparable effects as a Change of Control event, the Board of Directors may, based on a recommendation by the Nomination and Compensation Committee, decide to treat such event like a Change of Control event, providing, however, such adjustments to the consequences set forth above, that adequately and fairly address the differences to an actual Change of Control.
16.Corporate Events
In the event of a stock dividend, extraordinary cash dividend, recapitalization, reorganization, merger, consolidation, split-up, spin-off, combination, exchange of shares, issuance of options or other rights to purchase Shares at a price substantially below fair market value, or other extraordinary corporate events, which significantly dilute the value of the Shares underlying the PSUs such that an adjustment is required in order to preserve the benefits intended to be made available under this Plan, then the PSUs and related terms shall be adjusted and/or, if deemed appropriate, a cash payment to Participants or persons having outstanding PSUs shall be made to compensate such dilution. Such adjustment shall be resolved by the Board of Directors at its sole discretion with final and binding effect, based on a recommendation by the Nomination and Compensation Committee and taking into consideration the acquired rights of the Participants and the objectives of the Plan.
17.Data Protection
(a)By accepting a grant of PSUs, each Participant consents to the collection and processing of personal data relating to the Participant in connection with such grant and the performance of this Plan and the PSU Award Agreement by the Company, the Board of Directors, the Administrator and any other person or entity the Company may find appropriate for the administration of the Plan. The data may be used by the aforementioned parties to perform their rights and obligations in connection with this Plan, issue certificates (if any), issue statements, disclosure and communications relating to the Plan and the PSUs, to provide for cash-less grants or sale mechanics and to generally administer and manage the Plan or keep records of participation levels.
|
|
|
|
|
|
| Molecular Partners AG: Performance Share Plan 2024 - Employees |
|
(b)Each Participant consents to the disclosure of such personal data by any Group Company to the Board of Directors, any Administrator or any Plan Service Provider and any other person or entity (including, without limitation, to third parties for due diligence purposes, or to tax authorities) as the Company may find appropriate. Such disclosure may include the transfer or processing of such personal data in jurisdictions other than Switzerland or the jurisdiction of the employer Group Company.
18.Legal and regulatory restrictions
(a)Neither the Shares nor the PSUs have been or will be registered or listed in any jurisdiction other than, if and to the extent required, Switzerland.
(b)Nothing in this Plan is intended to be deemed a public offering of, or solicitation of investments in, securities of the Company nor a private offering of securities into any jurisdiction or to any person in circumstances that would require compliance with licensing, filing, prospectus, registration or similar requirements in connection therewith. If and to the extent that the grant of PSUs or the delivery of Shares pursuant to this Plan or the extension of eligibility under this Plan into any jurisdiction or to any person conflicts with any securities, stock exchange or other laws and regulations or would trigger any licensing, filing, prospectus, registration or similar requirement (other than the regular listing of the Shares at SIX Swiss Exchange and their registration in the commercial register and in the book entry system to create intermediated securities (Bucheffekten)), such grant, delivery or extension shall be deemed null and void. In such case, the Company may (without obligation) decide in its own discretion whether and how to compensate the relevant persons in lieu of such grant, delivery or extension.
(c)Any Participant shall be required to observe trading or other bans as well as the prohibitions of insider trading and market manipulation in connection with the PSUs and any shares granted thereunder.
19.Amendment and Termination
In exceptional cases, the Board of Directors may terminate, suspend or amend this Plan at its sole discretion with regard to all or some future or past PSU grants. Any adverse economic effects of such termination, suspension or amendment on grants already made pursuant to a PSU Award Agreement shall be fairly compensated in cash, by adjustment of other terms of the grant, by replacement by other grants or benefits, or otherwise.
20.Severability
The invalidity or non-enforceability of any one or more provisions of this Plan shall not affect the validity or enforceability of any other provisions of this Plan, which shall remain in full force and effect. The invalid provisions shall be replaced by valid provisions that economically come as close as possible to the original (invalid) provisions.
|
|
|
|
|
|
| Molecular Partners AG: Performance Share Plan 2024 - Employees |
|
21.Governing Law and Jurisdiction
(a)This Plan and any PSU Award Agreement shall be governed by, and construed in accordance with, the substantive laws of Switzerland.
(b)Any disputes arising under or in connection with this Plan, including any disputes under or in connection with the PSU Award Agreement shall be submitted to the exclusive jurisdiction of the courts at the domicile of the Company (currently Schlieren, Canton of Zurich, Switzerland).
22.Entry into Force
As per approval of the Board of Directors, this Plan shall enter into force as of March 12, 2024.
|
|
|
|
|
|
| Molecular Partners AG: Performance Share Plan 2024 - Employees |
|
Annex 1
Definitions
As used in this Plan in capitalized form, the following terms shall have the following meaning:
Administrator shall have the meaning set forth in Section 3 above.
Board of Directors shall mean the board of directors of the Company.
Change of Control shall have the meaning set forth in Section 15 above.
CO shall mean the Swiss Code of Obligations as amended.
Company shall mean Molecular Partners AG or any successor or replacement company or a new parent company, all as may be designated by the Board of Directors in the future.
Nomination and Compensation Committee shall have the meaning set forth in Section 3 above.
Compensation Ordinance shall mean the Federal Ordinance against Excessive Compensation in Listed Companies of November 20, 2013, as may be amended or replaced
Goal shall have the meaning set forth in Section 10 above.
Grant Date shall have the meaning set forth in Section 5 above.
Group shall mean all Group Companies.
Group Company shall mean the Company and any company or entity of which at least 50% of the ownership or voting rights are directly or indirectly owned or otherwise controlled by the Company.
LTI Score Card shall have the meaning set forth in Section 10 above.
Management Board shall mean the members of the top level executive management, i.e. those managers whose compensation is subject to the Compensation Ordinance.
Participant shall mean any eligible person to whom the Company has granted PSUs through a PSU Award Agreement based on this Plan.
Plan shall have the meaning set forth in Section 1 above.
Plan Service Provider shall have the meaning set forth in Section 3 (e) above.
PSU shall have the meaning set forth in Section 1 above.
PSU Award Agreement shall have the meaning set forth in Section 4 above.
Shares shall have the meaning set forth in Section 9 above.
Vesting Date shall have the meaning set forth in Section 8 above.
|
|
|
|
|
|
| Molecular Partners AG: Performance Share Plan 2024 - Employees |
|
Vesting Multiple shall be the multiple determined in accordance with Section 10 above.
Vesting Period shall have the meaning set forth in Section 8 above.
|
|
|
|
|
|
| Molecular Partners AG: Performance Share Plan 2024 - Employees |
|
Annex 2
PSU Award Agreement 2024
This agreement (Agreement) is made as of the Grant Date set forth below by and between Molecular Partners AG (the Company) and
«first_name» «last_name», «Address1», «Zip_Code» «City» (the Participant)
in connection with the Performance Share Plan 2024 for employees (the PSU Plan), issued by the Company.
Capitalized terms used, but not defined herein, shall have the meaning assigned to them in the PSU Plan. Subject to the terms and conditions of the PSU Plan, the Company hereby grants to you the following PSUs:
|
|
|
|
|
|
Number of PSUs |
«total_final_no_of_PSUs» |
| Grant Date |
«Grant_Date» |
| Start Vesting Date |
«Start_Vesting» |
| Vesting Date |
«End_Vesting_1», «End_Vesting_2», «End_Vesting_3» |
| Agreement number |
«Agreement_no» |
The grants and any rights associated therewith are personal and not transferable. The number of Shares that may be allocated according to the PSU Plan shall be determined by the Nomination and Compensation Committee in accordance with the PSU Plan and the LTI Score Card setting out the goals relevant for this award. The PSU Plan and the LTI Score Card have separately been brought to the Participant's attention prior to entering into this Agreement and the Participant hereby acknowledges to have taken notice of the PSU Plan and the LTI Score Card (available on MPConnect, Our People, LTI_PSU). Please note that the PSU Plan includes a number of restrictions and conditions, which may lead to a complete loss of any entitlements hereunder.
By entering this Agreement, you accept the grant of the PSUs in accordance with this Agreement and the PSU Plan. In order to do so, please tick the Accept button in your Credit Suisse e-banking login within the next 10 days.
This grant of PSUs is being made, without obligation, at the sole and unrestricted discretion of the Company. The PSU Plan, your eligibility thereunder, the grant of PSUs or the allocation of Shares in connection therewith shall not confer upon you any right to participate in the PSU Plan or to receive grants of PSUs or Shares in the future.
This Agreement shall be governed by, and construed in accordance with, the substantive laws of Switzerland. Any disputes arising under or in connection with this Agreement shall be submitted to the exclusive jurisdiction of the courts at the domicile of the Company (currently Schlieren, Canton of Zurich, Switzerland).
Molecular Partners AG
Patrick Amstutz, CEO Robert Hendriks, VP Finance
|
|
|
|
|
|
| Molecular Partners AG: Performance Share Plan 2024 - Employees |
|
PSU Award Agreement 2024
This agreement (Agreement) is made as of the Grant Date set forth below by and between Molecular Partners AG (the Company) and
«first_name» «last_name», «Address1», «Zip_Code» «City» (the Participant)
in connection with the Performance Share Plan 2024 for employees (the PSU Plan), issued by the Company.
Capitalized terms used, but not defined herein, shall have the meaning assigned to them in the PSU Plan. Subject to the terms and conditions of the PSU Plan, the Company hereby grants to you the following PSUs:
|
|
|
|
|
|
Number of PSUs |
«total_final_no_of_PSUs» |
| Grant Date |
«Grant_Date» |
| Start Vesting Date |
«Start_Vesting» |
| Vesting Date |
«End_Vesting_1», «End_Vesting_2», «End_Vesting_3» |
| Agreement number |
«Agreement_no» |
The grants and any rights associated therewith are personal and not transferable. The number of Shares that may be allocated according to the PSU Plan shall be determined by the Nomination and Compensation Committee in accordance with the PSU Plan and the LTI Score Card setting out the goals relevant for this award. The PSU Plan and the LTI Score Card have separately been brought to the Participant's attention prior to entering into this Agreement and the Participant hereby acknowledges to have taken notice of the PSU Plan and the LTI Score Card (available on MPConnect, Our People, LTI_PSU). Please note that the PSU Plan includes a number of restrictions and conditions, which may lead to a complete loss of any entitlements hereunder.
By entering this Agreement, you accept the grant of the PSUs in accordance with this Agreement and the PSU Plan. In order to do so, please tick the Accept button in your Credit Suisse e-banking login within the next 10 days.
This grant of PSUs is being made, without obligation, at the sole and unrestricted discretion of the Company. The PSU Plan, your eligibility thereunder, the grant of PSUs or the allocation of Shares in connection therewith shall not confer upon you any right to participate in the PSU Plan or to receive grants of PSUs or Shares in the future.
This Agreement shall be governed by, and construed in accordance with, the substantive laws of Switzerland. Any disputes arising under or in connection with this Agreement shall be submitted to the exclusive jurisdiction of the courts at the domicile of the Company (currently Schlieren, Canton of Zurich, Switzerland).
Notwithstanding any provison of the PSU Plan or this Agreement, the following terms and conditions will apply in the case of any Participant who is or becomes a United States taxpayer (including a Participant who is a United States citizen or resident, or a person who is otherwise subject to the income tax laws of the United States):
|
|
|
|
|
|
| Molecular Partners AG: Performance Share Plan 2024 - Employees |
|
1. The Company is required to issue the Shares underlying a PSU at the time the PSU vests (as set forth in the Plan and this Agreement), provided that the Company may require the signing of an acquisition declaration and payment of nominal value for the Shares as provided in Section 8(c) of the Plan (and subject to the satisfaction of any tax withholding obligations under Section 11 of the Plan). Notwithstanding the above, in no event will the Shares be delivered to the Participant later than thirty (30) days after the date the PSU is considered to vest under the terms of the Plan and this Agreement (unless any such delay or non-delivery of the Shares is required under the provisions below in order to comply with Section 409A of the Code).
2. To the greatest extent possible the PSUs are intended to be exempt from the application of Section 409A of the Internal Revenue Code of 1986, as amended (Code), including but not limited to by reason of complying with the “short-term deferral” rule set forth in Treasury Regulation Section 1.409A-1(b)(4) and any ambiguities herein shall be interpreted accordingly. Notwithstanding the foregoing, if it is determined that a PSU fails to satisfy the requirements of the short-term deferral rule and is otherwise not exempt from, and determined to be deferred compensation subject to Section 409A of the Code, then such PSU shall comply with Section 409A to the extent necessary to avoid adverse personal tax consequences and any ambiguities herein shall be interpreted accordingly.
3. If a PSU is treated as deferred compensation under Section 409A of the Code, and the delivery or issuance of the underlying Shares will otherwise occur upon a termination of employment with, or service to, the Group Company, then (to the extent necessary to avoid the imposition of taxes or adverse consequences under Section 409A) the Shares will not be delivered or issued to the Participant unless and until such termination of employment or service also qualifies as a “separation from service” (Separation from Service) under Treasury Regulation Section 1.409A-1(h).
4. If a PSU is treated as deferred compensation under Section 409A of the Code, and the delivery or issuance of the underlying Shares will otherwise occur upon a Change of Control, then (to the extent necessary to avoid the imposition of taxes or adverse consequences under Section 409A) the Shares will not be delivered or issued unless and until such Change of Control also qualifies as a “change in control event” under Treasury Regulation Section 1.409A-3(i)(5).
5. If it is determined that the PSUs are deferred compensation subject to Section 409A and the Participant is a “Specified Employee” (within the meaning set forth in Section 409A(a)(2)(B)(i) of the Code) as of the date of the Participant’s Separation from Service, then the issuance of any Shares that would otherwise be made upon the date of the Participant’s Separation from Service or within the first six (6) months thereafter will not be made on the originally scheduled date(s) and will instead be issued in a lump sum on the date that is six (6) months and one day after the date of the Separation from Service, with the balance of the Shares issued thereafter in accordance with the original vesting and issuance schedule set forth above, but if and only if such delay in the issuance of the Shares is necessary to avoid the imposition of adverse taxation on the Participant in respect of the Shares under Section 409A of the Code. Each installment of shares that vests is intended to constitute a “separate payment” for purposes of Treasury Regulation Section 1.409A-2(b)(2).
Molecular Partners AG
Patrick Amstutz, CEO Robert Hendriks, VP Finance
EX-4.24
6
a424rsuplan2024.htm
EX-4.24
Document
|
|
|
|
|
|
| Molecular Partners AG: Restricted Share Plan 2024 |
|
Exhibit 4.24
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Molecular Partners AG
Restricted Share Plan 2024
|
|
|
|
|
|
|
|
| Molecular Partners AG: Restricted Share Plan 2024 |
|
Table of Contents
Annexes
Annex 1: Definitions
|
|
|
|
|
|
| Molecular Partners AG: Restricted Share Plan 2024 |
|
Restricted Share Plan 2024
1.Purpose
Annex 2: Form of RSU Award Agreement The purpose of this restricted share plan (Plan) is to establish a framework that enables the Company to provide certain eligible persons with a variable long-term incentive to contribute to the future success and prosperity of the Company and to better align their interests with those of the Company and its shareholders by granting Restricted Share Units (each a RSU) to them.
2.Definitions and Interpretation
Capitalized terms used in this Plan shall have the meaning set forth in Annex 1.
3.Responsibilities and Administration
(a)This Plan has been approved and issued by the Board of Directors and any amendments or new editions of this Plan or new or other plans shall require the approval by the Board of Directors. The Board of Directors shall be in charge of approving, upon recommendation of the nomination and compensation committee of the Board of Directors (Nomination and Compensation Committee), the maximum number of RSUs that may be granted under this Plan.
(b)The Nomination and Compensation Committee shall be responsible for the implementation and administration of the Plan and shall make recommendations to amend, renew or terminate the Plan or to replace it with new editions or other plans. It may delegate, under its supervision, the implementation and administration, as well as grants of RSUs other than to members of the Board of Directors to one or several administrators (Administrator).
(c)Any grants of RSUs to members of the Board of Directors shall be approved by the Board of Directors based on individual recommendations of the Nomination and Compensation Committee and will, as long as shareholder approval for the aggregate amount of compensation is outstanding, be conditional upon such shareholder approval. In case that the amount approved by the shareholders does not cover the full amount of contemplated aggregate compensation for the year of grant, the entitlements to long-term compensation may be reduced by the Nomination and Compensation Committee in its sole discretion.
(d)All resolutions, decisions, determinations and interpretations made by the Nomination and Compensation Committee or, upon delegation by the Nomination and Compensation Committee, an Administrator pursuant to this Plan, including any amendments or withdrawals of grants, are final and binding, unless approval by the Board of Directors or the shareholders' meeting is required.
(e)Any technical or administrative task in connection with the Plan may be outsourced by the Nomination and Compensation Committee or the relevant Administrator to a third party service
|
|
|
|
|
|
| Molecular Partners AG: Restricted Share Plan 2024 |
|
provider, e.g. the bank in charge with the creation of the Shares (each a Plan Service Provider).
4.Eligibility and Participation
(a)As a rule, members of the Board of Directors and selected consultants may become eligible to participate in this Plan. The decision on eligibility is reserved to the Nomination and Compensation Committee or, other than to members of the Board of Directors, the relevant Administrator.
(b)Nothing in this Plan shall provide any rights to eligible persons or any other person nor create any obligation of the Company to grant RSUs based on this Plan or otherwise. This Plan is only applicable in connection with a mutually signed RSU award agreement (RSU Award Agreement) among the Company (or the relevant Group Company) and the eligible person, substantially in the form attached hereto as Annex 2. The right to receive RSUs shall accrue exclusively to those eligible persons who have, in accordance with this Plan, been duly and validly offered and have signed and returned their individual RSU Award Agreement by the relevant due date (each a Participant).
(c)Grants of RSUs based on this Plan are discretionary and shall not create any entitlement to participate in future grants or in future participation, incentive or benefit plans, including future restricted share plans, regardless of the length of time a person has previously been allocated RSUs or other entitlements under this Plan or other plans.
(d)Neither the grant of RSUs, nor the transfer of Shares in connection with this Plan shall confer upon any Participant any right to a continued Relationship with any Group Company.
5.Grant of RSUs
(a)Grants of RSUs shall be exclusively made by way of RSU Award Agreements. The RSU Award Agreement shall set forth the number of RSUs and certain other terms and conditions of such grant. Except as otherwise determined in a RSU Award Agreement, RSUs shall be granted to the Participants free of charge.
(b)One RSU represents a conditional entitlement to purchase one Share at the nominal value of the Share. The number of Shares corresponding to the number vested RSUs shall be allocated to a Participant upon vesting, subject to, and in accordance with, the terms and conditions of this Plan and the RSU Award Agreement.
(c)The date of grant shall be determined by the Nomination and Compensation Committee or the relevant Administrator and set out in the RSU Award Agreement (the Grant Date).
|
|
|
|
|
|
| Molecular Partners AG: Restricted Share Plan 2024 |
|
6.No Securities
RSUs are neither Shares nor securities of any kind and no shareholder rights or similar rights are attached to the RSUs. The Participants will only obtain shareholder rights (including voting and dividend rights) upon actual transfer of Shares, if any, according to the terms and conditions of the Plan and upon entry into the share register, subject to, and in accordance with, article 5 of the Company's articles of incorporation.
7.No Transfer
RSUs granted under this Plan and the RSU Award Agreement are personal and non-transferable. Participants shall not be permitted to sell, donate, pledge, assign or otherwise dispose of the RSUs to third parties other than as provided for in the Plan. In case of death of a Participant, Section 13 hereof shall apply.
8.Vesting and Delivery of Shares
(a)Unless otherwise set out in this Plan (in particular in Section 13) or in the RSU Award Agreement, RSUs shall vest in the third calendar year following the year of grant. In such case, vesting shall occur on the third anniversary of the Grant Date (the Vesting Date). The period between the Grant Date and the Vesting Date shall be deemed the Vesting Period.
(b)Subject to section 13(b) below, no RSU shall vest if, during the first year of the Vesting Period (or, in case of a member of the Board of Directors, prior to the end of a full term of office), the relevant Participant's Relationship is terminated or another reason for termination of such Relationship occurs other than through termination by the Participant for cause (wichtige Gründe), set by a Group Company.
(c)The Shares shall be delivered by or on behalf of the Company to the Participant upon signing an acquisition declaration and payment of the nominal value of the Shares by the Participant. Instead, the Company may provide for cash-less acquisition or vesting-sale arrangements through a Plan Service Provider or otherwise.
9.Underlying Shares
Shares to be delivered to Participants shall, subject to adjustment, if any, pursuant to Section 15, be registered shares of the Company with a nominal value of CHF 0.10 each (each a Share). Such Shares shall, at the discretion of the Nomination and Compensation Committee and the Board of Directors, be sourced from conditional share capital, from treasury shares or from other sources. Unless otherwise determined by the Board of Directors or the Nomination and Compensation Committee, Shares shall be sourced from the Company's conditional share capital and a respective number of Shares out of conditional capital shall be deemed reserved, accordingly.
|
|
|
|
|
|
| Molecular Partners AG: Restricted Share Plan 2024 |
|
10.Taxes and Social Security Contributions
(a)Any Participant shall be responsible for reporting the receipt of any income under the Plan, however made, to the appropriate tax and social security authorities. Income, capital gain or other taxes due on the granting of RSUs, on the allocation of Shares and the subsequent sale of Shares or a respective cash equivalent are in the sole responsibility of the Participant.
(b)The grant, vesting, delivery or sale of Shares or other relevant event in connection with the RSUs may be subject to the withholding of tax and social security contributions by the Company or, if different, the relevant Group Company. The Company and the relevant Group Company, shall be entitled to deduct or withhold a sufficient portion of the value otherwise due to be released under this Plan or of any other payment to the relevant Participant to satisfy any withholding requirement in connection therewith. Without limitation, withholding arrangements may include the sale of Shares to be delivered for RSU awards on behalf of a Participant and withholding of proceeds or deductions from salary or bonus payments, or require a payment from the Participant to the Company or the relevant Group Company before settlement of the RSU awards.
(c)The Company shall have the right (but no obligation, unless required by applicable law) to notify the tax and social security authorities of the grant of RSU awards, Shares or related events.
11.Disclosure Requirements
Any Participant shall be responsible to promptly comply with any applicable disclosure requirements under securities law and stock exchange regulations in connection with the receipt of grants of RSUs or Shares or upon the sale of Shares, including any disclosure requirements triggered by the thresholds for the ownership of shares and|or rights to obtain shares under Article 120 Financial Market Infrastructure Act and any management transaction notifications under Article 56 of the SIX Swiss Exchange listing rules. See also the Company's public disclosure, reporting and securities trading policy.
12.Other Obligations of the Participant
The Company is entitled to block or prohibit the issuance or release of Shares otherwise due to be issued or released if the Participant has any outstanding obligations (whether in connection with the Plan or otherwise arising in connection with the Participant's Relationship with the Company) to any Group Company, until the Participant has satisfied such outstanding obligations.
13.Termination of Relationship
(a)If a Participant's Relationship is terminated or another reason for termination occurs during the first year of the Vesting Period (or, in case of a member of the Board of Directors, prior to the end of a full term of office), other than through termination by the Participant for cause (wichtige Gründe) and except as set out in subsection 13 (b) below, all RSUs shall cease and be forfeited on the effective date of termination.
|
|
|
|
|
|
| Molecular Partners AG: Restricted Share Plan 2024 |
|
(b)If the Relationship terminates during the first year of the Vesting Period (or, in case of a member of the Board of Directors, prior to the end of a full term of office) by reason of death, permanent illness or disability of the Participant, a pro rata number of RSUs granted to the Participant shall vest immediately with the remaining RSUs lapsing without further effect. The Board of Directors may, at its sole discretion and with final and binding effect, based on a recommendation by the Nomination and Compensation Committee and taking into consideration the objectives of the Plan, grant further exceptions from the forfeiture clause as per section 13 (a) above.
14.Change of Control
(a)For purposes of this Plan, a change of control shall mean the occurrence of any of the following events (each a Change of Control):
(i)the acquisition in one or more transaction by any person or group of persons acting in concert, directly or indirectly, of the beneficial ownership of Shares and | or rights to acquire Shares representing 50% or more of the voting rights pertaining to the total number of Shares issued and registered in the commercial register;
(ii)any facts or circumstances that require any person or group of persons acting in concert to launch a mandatory offer within the meaning of applicable takeover regulations;
(iii)a public offer for Shares by any person or persons (other than in order to implement a new parent company held by the same owners of Shares), for such number of Shares that, by itself or together with Shares already held, triggers the duty to extend the offer to all Shares outstanding, if and when such offer becomes unconditional (subject only to conditions, if any, that survive following the regular offer period);
(iv)the reorganization, merger, scheme of arrangement, consolidation, liquidation or similar transaction of the Company otherwise than through a transaction by which the persons who beneficially held Shares representing 100% of the voting rights pertaining to the total number of Shares issued and registered in the commercial register prior to such transaction receive or continue to hold shares representing more than 50% of the voting rights pertaining to the total number of outstanding shares of the new or continuing entity.
(b)In the event of a Change of Control of the Company, the following shall apply for all RSUs in respect of which the Vesting Date has not occurred by the date of the Change of Control:
(i)all RSUs will vest immediately; and
(ii)the RSUs will be paid out in Shares, unless the Nomination and Compensation Committee resolves to repurchase or exchange RSUs or decides upon another solution to provide the Participants with the vesting value of the RSUs.
|
|
|
|
|
|
| Molecular Partners AG: Restricted Share Plan 2024 |
|
(c)If based on a good faith assessment of the particular circumstances and effects of a Change of Control event, such event does not fall into the category and nature of cases, circumstances and consequences addressed by the definition of Change of Control, which are deemed to justify an early vesting, the Board of Directors may, based on a recommendation by the Nomination and Compensation Committee, decide to replace the consequences set forth above, by other terms that more appropriately and fairly address the situation.
(d)If an event does not fall under the definition of Change of Control, but has substantially comparable effects as a Change of Control event, the Board of Directors may, based on a recommendation by the Nomination and Compensation Committee, decide to treat such event like a Change of Control event, providing, however, such adjustments to the consequences set forth above, that adequately and fairly address the differences to an actual Change of Control.
15.Corporate Events
In the event of a stock dividend, extraordinary cash dividend, recapitalization, reorganization, merger, consolidation, split-up, spin-off, combination, exchange of shares, issuance of options or other rights to purchase Shares at a price substantially below fair market value, or other extraordinary corporate events, which significantly dilute the value of the Shares underlying the RSUs such that an adjustment is required in order to preserve the benefits intended to be made available under this Plan, then the RSUs and related terms shall be adjusted and/or, if deemed appropriate, a cash payment to Participants or persons having outstanding RSUs shall be made to compensate such dilution. Such adjustment shall be resolved by the Board of Directors at its sole discretion with final and binding effect, based on a recommendation by the Nomination and Compensation Committee and taking into consideration the acquired rights of the Participants and the objectives of the Plan.
16.Data Protection
(a)By accepting a grant of RSUs, each Participant consents to the collection and processing of personal data relating to the Participant in connection with such grant and the performance of this Plan and the RSU Award Agreement by the Company, the Board of Directors, the Administrator and any other person or entity the Company may find appropriate for the administration of the Plan. The data may be used for the aforementioned parties to perform their rights and obligations in connection with this Plan, issue certificates (if any), issue statements, disclosure and communications relating to the Plan and the RSUs, to provide for cash-less grants or sale mechanics and to generally administer and manage the Plan or keep records of participation levels.
(b)Each Participant consents to the disclosure of such personal data by any Group Company to the Board of Directors, any Administrator or any Plan Service Provider and any other person or entity (including, without limitation, to third parties for due diligence purposes, or to tax authorities) as the Company may find appropriate. Such disclosure may include the transfer or processing of such personal data in jurisdictions other than Switzerland or the jurisdiction of the relevant Group Company.
|
|
|
|
|
|
| Molecular Partners AG: Restricted Share Plan 2024 |
|
17.Legal and regulatory restrictions
(a)Neither the Shares nor the RSU have been or will be registered or listed in any jurisdiction other than, if and to the extent required, Switzerland.
(b)Nothing in this Plan is intended to be deemed a public offering of, or solicitation of investments in, securities of the Company nor a private offering of securities into any jurisdiction or to any person in circumstances that would require compliance with licensing, filing, prospectus, registration or similar requirements in connection therewith. If and to the extent that the grant of RSUs or the delivery of Shares pursuant to this Plan or the extension of eligibility under this Plan into any jurisdiction or to any person conflicts with any securities, stock exchange or other laws and regulations or would trigger any licensing, filing, prospectus, registration or similar requirement (other than the regular listing of the Shares at SIX Swiss Exchange and their registration in the commercial register and in the book entry system to create intermediated securities (Bucheffekten)), such grant, delivery or extension shall be deemed null and void. In such case, the Company may (without obligation) decide in its own discretion whether and how to compensate the relevant persons in lieu of such grant, delivery or extension.
(c)Any Participant shall be required to observe trading or other bans as well as the prohibitions of insider trading and market manipulation in connection with the RSUs and any shares granted thereunder.
(d)Any grant of RSUs to members of the Board of Directors that qualifies as prohibited payment under the Compensation Ordinance or otherwise, shall be null and void.
(e)If and to the extent that any term of this Plan, such as terms providing for early vesting, in particular in case of termination of Relationship or Change of Control, should, at the time of the relevant event, qualify as providing additional value to a member of the Board of Directors in a manner that would violate the Compensation Ordinance or other legal provisions, such additional value shall be otherwise compensated, e.g. by a relevant deduction from cash compensation or other proceeds.
18.Amendment and Termination
In exceptional cases, the Board of Directors may terminate, suspend or amend this Plan at its sole discretion with regard to all or some future or past RSU grants. Any adverse economic effects of such termination, suspension or amendment on grants already made pursuant to a RSU Agreement shall be fairly compensated in cash, by adjustment of other terms of the grant, by replacement by other grants or benefits, or otherwise.
|
|
|
|
|
|
| Molecular Partners AG: Restricted Share Plan 2024 |
|
19.Severability
The invalidity or non-enforceability of any one or more provisions of this Plan shall not affect the validity or enforceability of any other provisions of this Plan, which shall remain in full force and effect. The invalid provisions shall be replaced by valid provisions that economically come as close as possible to the original (invalid) provisions.
20.Governing Law and Jurisdiction
(a)This Plan and any RSU Award Agreement shall be governed by, and construed in accordance with, the substantive laws of Switzerland.
(b)Any disputes arising under or in connection with this Plan, including any disputes under or in connection with the RSU Award Agreement shall be submitted to the exclusive jurisdiction of the courts at the domicile of the Company (currently Schlieren, Canton of Zurich, Switzerland).
21.Entry into Force
As per the approval of the Board of Directors, this Plan shall enter into force as of March 12, 2024.
|
|
|
|
|
|
| Molecular Partners AG: Restricted Share Plan 2024 |
|
Annex 1
Definitions
As used in this Plan in capitalized form, the following terms shall have the following meaning:
Administrator shall have the meaning set forth in Section 3 above.
Board of Directors shall mean the board of directors of the Company.
Change of Control shall have the meaning set forth in Section 14 above.
CO shall mean the Swiss Code of Obligations as amended.
Company shall mean Molecular Partners AG or any successor or replacement company or a new parent company, all as may be designated by the Board of Directors in the future.
Nomination and Compensation Committee shall have the meaning set forth in Section 3 above.
Compensation Ordinance shall mean the Federal Ordinance against Excessive Compensation in Listed Companies of November 20, 2013, as may be amended or replaced
Grant Date shall have the meaning set forth in Section 5 above.
Group Company shall mean the Company and any company or entity of which at least 50% of the ownership or voting rights are directly or indirectly owned or otherwise controlled by the Company.
Participant shall mean any eligible person to whom the Company has granted RSUs through a RSU Award Agreement based on this Plan.
Plan shall have the meaning set forth in Section 1 above.
Plan Service Provider shall have the meaning set forth in Section 3 (e) above.
Relationship shall mean the board relationship and any relating contractual relationship of a member of the Board of Directors and the consultancy or other legal or contractual relationship of any other Participant.
RSU shall have the meaning set forth in Section 1 above.
RSU Award Agreement shall have the meaning set forth in Section 4 above.
Shares shall have the meaning set forth in Section 9 above.
Vesting Date shall have the meaning set forth in Section 8 above.
Vesting Period shall have the meaning set forth in Section 8 above.
|
|
|
|
|
|
| Molecular Partners AG: Restricted Share Plan 2024 |
|
|
|
|
|
|
|
| Molecular Partners AG: Restricted Share Plan 2024 |
|
Annex 2
RSU Award Agreement 2024
This agreement (Agreement) is made as of the Grant Date set forth below by and between Molecular Partners AG (the Company) and
«Vorname» «Nachname», «Adresse», «PLZ» «Ort» (the Participant)
in connection with the Restricted Share Plan 2024 (the RSU Plan), issued by the Company.
Capitalized terms used, but not defined herein, shall have the meaning assigned to them in the RSU Plan.
Subject to the terms and conditions of the RSU Plan, the Company hereby grants to you the following RSUs:
|
|
|
|
|
|
Number of RSUs |
«Final_no_of_RSUs_rounded» |
| Grant Date |
«Grant_Date» |
| Start Vesting Date |
«Start_Vesting» |
| Vesting Date |
«End_Vesting» |
| Agreement number |
«Agreement_no» |
The grants and any rights associated therewith are personal and not transferable. Please note that the RSU Plan includes a number of restrictions and conditions, which may lead to a complete loss of any entitlements hereunder.
By entering this Agreement, you accept the grant of the RSUs in accordance with this Agreement and the RSU Plan attached hereto. In order to do so, please sign and return this Agreement no later than by April 30, 2024 to the Secretary of the Board of Directors.
This grant of RSUs is being made, without obligation, at the sole and unrestricted discretion of the Company. The RSU Plan, your eligibility thereunder, the grant of RSUs or the allocation of Shares in connection therewith shall not confer upon you any right to participate in the RSU Plan or to receive grants of RSUs or Shares in the future.
This Agreement shall be governed by, and construed in accordance with, the substantive laws of Switzerland. Any disputes arising under or in connection with this Agreement shall be submitted to the exclusive jurisdiction of the courts at the domicile of the Company (currently Schlieren, Canton of Zurich, Switzerland).
Molecular Partners AG
__________________________ __________________________
By: Patrick Amstutz, CEO By: Robert Hendriks, VP Finance
Accepted and agreed by the Participant on (Date, Signature):
|
|
|
|
|
|
| Molecular Partners AG: Restricted Share Plan 2024 |
|
__________________________ __________________________
Attachments: RSU Plan 2024
EX-12.1
7
a121section302certification.htm
EX-12.1
Document
Exhibit 12.1
CERTIFICATION*
I, Patrick Amstutz, certify that:
1. I have reviewed this annual report on Form 20-F of Molecular Partners AG (the "company");
2. Based on my knowledge, this report does not contain any untrue statement of a material fact or omit to state a material fact necessary to make the statements made, in light of the circumstances under which such statements were made, not misleading with respect to the period covered by this report;
3. Based on my knowledge, the financial statements, and other financial information included in this report, fairly present in all material respects the financial condition, results of operations and cash flows of the company as of, and for, the periods presented in this report;
4. The company’s other certifying officer(s) and I are responsible for establishing and maintaining disclosure controls and procedures (as defined in Exchange Act Rules 13a-15(e) and 15d-15(e)) and internal control over financial reporting (as defined in Exchange Act Rules 13a-15(f) and 15d-15(f)) for the company and have:
(a) Designed such disclosure controls and procedures, or caused such disclosure controls and procedures to be designed under our supervision, to ensure that material information relating to the company, including its consolidated subsidiaries, is made known to us by others within those entities, particularly during the period in which this report is being prepared;
(b) Designed such internal control over financial reporting, or caused such internal control over financial reporting to be designed under our supervision, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles;
(c) Evaluated the effectiveness of the company’s disclosure controls and procedures and presented in this report our conclusions about the effectiveness of the disclosure controls and procedures, as of the end of the period covered by this report based on such evaluation; and
(d) Disclosed in this report any change in the company’s internal control over financial reporting that occurred during the period covered by the annual report that has materially affected, or is reasonably likely to materially affect, the company’s internal control over financial reporting; and
5. The company’s other certifying officer(s) and I have disclosed, based on our most recent evaluation of internal control over financial reporting, to the company’s auditors and the audit committee of the company’s board of directors (or persons performing the equivalent functions):
(a) All significant deficiencies and material weaknesses in the design or operation of internal control over financial reporting which are reasonably likely to adversely affect the company’s ability to record, process, summarize and report financial information; and
(b) Any fraud, whether or not material, that involves management or other employees who have a significant role in the company’s internal control over financial reporting.
Date: March 14, 2024
|
|
|
|
|
|
|
/s/ Patrick Amstutz |
|
Chief Executive Officer
(Principal Executive Officer)
|
EX-12.2
8
a122section302certification.htm
EX-12.2
Document
Exhibit 12.2
CERTIFICATION*
I, Robert Hendriks, certify that:
1. I have reviewed this annual report on Form 20-F of Molecular Partners AG (the "company");
2. Based on my knowledge, this report does not contain any untrue statement of a material fact or omit to state a material fact necessary to make the statements made, in light of the circumstances under which such statements were made, not misleading with respect to the period covered by this report;
3. Based on my knowledge, the financial statements, and other financial information included in this report, fairly present in all material respects the financial condition, results of operations and cash flows of the company as of, and for, the periods presented in this report;
4. The company’s other certifying officer(s) and I are responsible for establishing and maintaining disclosure controls and procedures (as defined in Exchange Act Rules 13a-15(e) and 15d-15(e)) and internal control over financial reporting (as defined in Exchange Act Rules 13a-15(f) and 15d-15(f)) for the company and have:
(a) Designed such disclosure controls and procedures, or caused such disclosure controls and procedures to be designed under our supervision, to ensure that material information relating to the company, including its consolidated subsidiaries, is made known to us by others within those entities, particularly during the period in which this report is being prepared;
(b) Designed such internal control over financial reporting, or caused such internal control over financial reporting to be designed under our supervision, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles;
(c) Evaluated the effectiveness of the company’s disclosure controls and procedures and presented in this report our conclusions about the effectiveness of the disclosure controls and procedures, as of the end of the period covered by this report based on such evaluation; and
(d) Disclosed in this report any change in the company’s internal control over financial reporting that occurred during the period covered by the annual report that has materially affected, or is reasonably likely to materially affect, the company’s internal control over financial reporting; and
5. The company’s other certifying officer(s) and I have disclosed, based on our most recent evaluation of internal control over financial reporting, to the company’s auditors and the audit committee of the company’s board of directors (or persons performing the equivalent functions):
(a) All significant deficiencies and material weaknesses in the design or operation of internal control over financial reporting which are reasonably likely to adversely affect the company’s ability to record, process, summarize and report financial information; and
(b) Any fraud, whether or not material, that involves management or other employees who have a significant role in the company’s internal control over financial reporting.
Date: March 14, 2024
|
|
|
| /s/ Robert Hendriks |
|
SVP Finance
(Principal Financial Officer)
|
EX-13.1
9
a131section906certification.htm
EX-13.1
Document
Exhibit 13.1
CERTIFICATION
Pursuant to the requirement set forth in Rule 13a-14(b) of the Securities Exchange Act of 1934, as amended, (the “Exchange Act”) and Section 1350 of Chapter 63 of Title 18 of the United States Code (18 U.S.C. §1350), Patrick Amstutz, Chief Executive Officer of Molecular Partners AG (the “Company”), and Robert Hendriks, SVP Finance of the Company, each hereby certifies that, to the best of his or her knowledge:
1.The Company’s Annual Report on Form 20-F for the fiscal year ended December 31, 2023, to which this Certification is attached as Exhibit 13.1 (the “Annual Report”), fully complies with the requirements of Section 13(a) or Section 15(d) of the Exchange Act; and
2.The information contained in the Annual Report fairly presents, in all material respects, the financial condition and results of operations of the Company.
Dated: March 14, 2024
In Witness Whereof, the undersigned have set their hands hereto as of the 14th day of March, 2024
|
|
|
|
|
|
|
|
|
| /s/ Patrick Amstutz |
|
/s/ Robert Hendriks |
|
|
|
| Patrick Amstutz |
|
Robert Hendriks |
| Chief Executive Officer |
|
SVP Finance |
“This certification accompanies the Form 20-F to which it relates, is not deemed filed with the Securities and Exchange Commission and is not to be incorporated by reference into any filing of Molecular Partners AG, under the Securities Act of 1933, as amended, or the Exchange Act, as amended (whether made before or after the date of the Form 20-F), irrespective of any general incorporation language contained in such filing.”
EX-15.1
10
a151auditorconsent.htm
EX-15.1
Document
Consent of Independent Registered Public Accounting Firm
We consent to the incorporation by reference in the registration statement (No. 333-265960) on Form F-3 of our report dated March 14, 2024, with respect to the consolidated financial statements of Molecular Partners AG.
/s/ KPMG
Zurich, Switzerland
March 14, 2024
EX-97.1
11
a971molecularpartners-dodd.htm
EX-97.1
Document
Exhibit 97.1
Molecular Partners AG
Incentive Compensation Recoupment Policy
1.Introduction
The Nomination and Compensation Committee (the “Compensation Committee”) of the Board of Directors (the “Board”) and the Board of Molecular Partners AG, a stock corporation (Aktiengesellschaft) organized under the laws of Switzerland (the “Company”), have determined that it is in the best interests of the Company and its shareholders, to adopt this Incentive Compensation Recoupment Policy (this “Policy”) providing for the Company’s recoupment of Recoverable Incentive Compensation that is received by Covered Officers of the Company under certain circumstances. Certain capitalized terms used in this Policy have the meanings given to such terms in Section 3 below.
This Policy is designed to comply with, and shall be interpreted to be consistent with, Section 10D of the Exchange Act, Rule 10D-1 promulgated thereunder (“Rule 10D-1”) and Nasdaq Listing Rule 5608 (the “Listing Standards”).
2.Effective Date
This Policy shall apply to all Incentive Compensation that is received by a Covered Officer on or after October 2, 2023 (the “Effective Date”). Incentive Compensation is deemed “received” in the Company’s fiscal period in which the Financial Reporting Measure specified in the Incentive Compensation award is attained, even if the payment or grant of such Incentive Compensation occurs after the end of that period.
3.Definitions
“Accounting Restatement” means an accounting restatement that the Company is required to prepare due to the material noncompliance of the Company with any financial reporting requirement under the securities laws, including any required accounting restatement to correct an error in previously issued financial statements that is material to the previously issued financial statements, or that would result in a material misstatement if the error were corrected in the current period or left uncorrected in the current period.
“Accounting Restatement Date” means the earlier to occur of (a) the date that the Board, a committee of the Board authorized to take such action, or the officer or officers of the Company authorized to take such action if Board action is not required, concludes, or reasonably should have concluded, that the Company is required to prepare an Accounting Restatement, or (b) the date that a court, regulator or other legally authorized body directs the Company to prepare an Accounting Restatement.
“Administrator” means the Compensation Committee.
“Code” means the U.S. Internal Revenue Code of 1986, as amended, and the regulations promulgated thereunder.
“Covered Officer” means each current and former Executive Officer.
“Exchange” means the Nasdaq Stock Market.
“Exchange Act” means the U.S. Securities Exchange Act of 1934, as amended.
“Executive Officer” means the Company’s chief executive officer, principal financial officer, principal accounting officer (or if there is no such accounting officer, the controller), any officers of the Company in charge of a principal business unit, division, or function (such as sales, administration, or finance), any other officer who performs a policy-making function, or any other person who performs similar policy-making functions for the Company. Executive officers of the Company’s parent(s) or subsidiaries are deemed executive officers of the Company if they perform such policy-making functions for the Company.
Policy-making function is not intended to include policy-making functions that are not significant. Identification of an executive officer for purposes of this Policy would include at a minimum executive officers identified pursuant to Item 401(b) of Regulation S-K promulgated under the Exchange Act.
“Financial Reporting Measures” means measures that are determined and presented in accordance with the accounting principles used in preparing the Company’s financial statements, and any measures derived wholly or in part from such measures, including Company share price and total shareholder return (“TSR”). A measure need not be presented in the Company’s financial statements or included in a filing with the SEC in order to be a Financial Reporting Measure.
“Incentive Compensation” means any compensation that is granted, earned or vested based wholly or in part upon the attainment of a Financial Reporting Measure.
“Lookback Period” means the three completed fiscal years immediately preceding the Accounting Restatement Date, as well as any transition period (resulting from a change in the Company’s fiscal year) within or immediately following those three completed fiscal years (except that a transition period of at least nine months shall count as a completed fiscal year). Notwithstanding the foregoing, the Lookback Period shall not include fiscal years completed prior to the Effective Date.
“Recoverable Incentive Compensation” means Incentive Compensation received by a Covered Officer during the Lookback Period that exceeds the amount of Incentive Compensation that would have been received had such amount been determined based on the Accounting Restatement, computed without regard to any taxes paid (i.e., on a gross basis without regarding to tax withholdings and other deductions). For any compensation plans or programs that take into account Incentive Compensation, the amount of Recoverable Incentive Compensation for purposes of this Policy shall include, without limitation, the amount contributed to any notional account based on Recoverable Incentive Compensation and any earnings to date on that notional amount. For any Incentive Compensation that is based on share price or TSR, where the Recoverable Incentive Compensation is not subject to mathematical recalculation directly from the information in an Accounting Restatement, the Administrator will determine the amount of Recoverable Incentive Compensation based on a reasonable estimate of the effect of the Accounting Restatement on the share price or TSR upon which the Incentive Compensation was received. The Company shall maintain documentation of the determination of that reasonable estimate and provide such documentation to the Exchange in accordance with the Listing Standards.
“SEC” means the U.S. Securities and Exchange Commission.
4.Recoupment
(a)Applicability of Policy. This Policy applies to Incentive Compensation received by a Covered Officer (i) after beginning services as an Executive Officer, (ii) who served as an Executive Officer at any time during the performance period for such Incentive Compensation, (iii) while the Company had a class of securities listed on a U.S. national securities exchange or a national securities association, and (iv) during the Lookback Period.
(b)Recoupment Generally. Pursuant to the provisions of this Policy, if there is an Accounting Restatement, the Company must reasonably promptly recoup the full amount of the Recoverable Incentive Compensation, unless the conditions of one or more subsections of Section 4(c) of this Policy are met and the Compensation Committee, or, if such committee does not consist solely of independent directors, a majority of the independent directors serving on the Board, has made a determination that recoupment would be impracticable. Recoupment is required regardless of whether the Covered Officer engaged in any misconduct and regardless of fault, and the Company’s obligation to recoup Recoverable Incentive Compensation is not dependent on whether or when any restated financial statements are filed.
(c)Impracticability of Recovery. Recoupment may be determined to be impracticable if, and only if:
(i)the direct expense paid to a third party to assist in enforcing this Policy would exceed the amount of the applicable Recoverable Incentive Compensation; provided that, before concluding that it would be impracticable to recover any amount of Recoverable Incentive Compensation based on expense of enforcement, the Company shall make a reasonable attempt to recover such Recoverable Incentive Compensation, document such reasonable attempt(s) to recover, and provide that documentation to the Exchange in accordance with the Listing Standards;
(ii)recoupment of the applicable Recoverable Incentive Compensation would violate home country law where that law was adopted prior to November 28, 2022; provided that, before concluding that it would be impracticable to recover any amount of Recoverable Incentive Compensation based on violation of home country law, the Company shall obtain an opinion of home country counsel, acceptable to the Exchange, that recoupment would result in such a violation, and shall provide such opinion to the Exchange in accordance with the Listing Standards; or
(iii)recoupment of the applicable Recoverable Incentive Compensation would likely cause an otherwise tax-qualified retirement plan, under which benefits are broadly available to employees of the Company, to fail to meet the requirements of Code Section 401(a)(13) or Code Section 411(a) and regulations thereunder.
(d)Sources of Recoupment. To the extent permitted by applicable law, the Administrator shall, in its sole discretion, determine the timing and method for recouping Recoverable Incentive Compensation hereunder, provided that such recoupment is undertaken reasonably promptly. The Administrator may, in its discretion, seek recoupment from a Covered Officer from any of the following sources or a combination thereof, whether the applicable compensation was approved, awarded, granted, payable or paid to the Covered Officer prior to, on or after the Effective Date: (i) direct repayment of Recoverable Incentive Compensation previously paid to the Covered Officer; (ii) cancelling prior cash or equity-based awards (whether vested or unvested and whether paid or unpaid); (iii) cancelling or offsetting against any planned future cash or equity-based awards; (iv) forfeiture of deferred compensation, subject to compliance with Code Section 409A; and (v) any other method authorized by applicable law or contract. Subject to compliance with any applicable law, the Administrator may effectuate recoupment under this Policy from any amount otherwise payable to the Covered Officer, including amounts payable to such individual under any otherwise applicable Company plan or program, e.g., base salary, bonuses or commissions and compensation previously deferred by the Covered Officer. The Administrator need not utilize the same method of recovery for all Covered Officers or with respect to all types of Recoverable Incentive Compensation.
(e)No Indemnification of Covered Officers. Notwithstanding any indemnification agreement, applicable insurance policy or any other agreement or provision of the Company’s certificate of incorporation or bylaws to the contrary, no Covered Officer shall be entitled to indemnification or advancement of expenses in connection with any enforcement of this Policy by the Company, including paying or reimbursing such Covered Officer for insurance premiums to cover potential obligations to the Company under this Policy.
(f)Indemnification of Administrator. Any members of the Administrator, and any other members of the Board who assist in the administration of this Policy, shall not be personally liable for any action, determination or interpretation made with respect to this Policy and shall be indemnified by the Company to the fullest extent under applicable law and Company policy with respect to any such action, determination or interpretation. The foregoing sentence shall not limit any other rights to indemnification of the members of the Board under applicable law or Company policy.
(g)No “Good Reason” for Covered Officers. Any action by the Company to recoup or any recoupment of Recoverable Incentive Compensation under this Policy from a Covered Officer shall not be deemed (i) “good reason” for resignation or to serve as a basis for a claim of constructive termination under any benefits or compensation arrangement applicable to such Covered Officer, or(ii) to constitute a breach of a contract or other arrangement to which such Covered Officer is party.
5.Administration
Except as specifically set forth herein, this Policy shall be administered by the Administrator. The Administrator shall have full and final authority to make any and all determinations required under this Policy. Any determination by the Administrator with respect to this Policy shall be final, conclusive and binding on all interested parties and need not be uniform with respect to each individual covered by this Policy. In carrying out the administration of this Policy, the Administrator is authorized and directed to consult with the full Board or such other committees of the Board as may be necessary or appropriate as to matters within the scope of such other committee’s responsibility and authority. Subject to applicable law, the Administrator may authorize and empower any employee of the Company to take any and all actions that the Administrator, in its sole discretion, deems necessary or appropriate to carry out the purpose and intent of this Policy (other than with respect to any recovery under this Policy involving such employee).
6.Severability
If any provision of this Policy or the application of any such provision to a Covered Officer shall be adjudicated to be invalid, illegal or unenforceable in any respect, such invalidity, illegality or unenforceability shall not affect any other provisions of this Policy, and the invalid, illegal or unenforceable provisions shall be deemed amended to the minimum extent necessary to render any such provision or application enforceable.
7.No Impairment of Other Remedies
Nothing contained in this Policy, and no recoupment or recovery as contemplated herein, shall limit any claims, damages or other legal remedies the Company or any of its affiliates may have against a Covered Officer arising out of or resulting from any actions or omissions by the Covered Officer. This Policy does not preclude the Company from taking any other action to enforce a Covered Officer’s obligations to the Company, including, without limitation, termination of employment and/or institution of civil proceedings. This Policy is in addition to the requirements of Section 304 of the Sarbanes-Oxley Act of 2002 (“SOX 304”) that are applicable to the Company’s Chief Executive Officer and Chief Financial Officer and to any other compensation recoupment policy and/or similar provisions in any employment, equity plan, equity award, or other individual agreement, to which the Company is a party or which the Company has adopted or may adopt and maintain from time to time; provided, however, that compensation recouped pursuant to this policy shall not be duplicative of compensation recouped pursuant to SOX 304 or any such compensation recoupment policy and/or similar provisions in any such employment, equity plan, equity award, or other individual agreement except as may be required by law.
8.Amendment; Termination
The Administrator may amend, terminate or replace this Policy or any portion of this Policy at any time and from time to time in its sole discretion. The Administrator shall amend this Policy as it deems necessary to comply with applicable law or any Listing Standard.
9.Successors
This Policy shall be binding and enforceable against all Covered Officers and, to the extent required by Rule 10D-1 and/or the applicable Listing Standards, their beneficiaries, heirs, executors, administrators or other legal representatives.
10. Required Filings
The Company shall make any disclosures and filings with respect to this Policy that are required by law, including as required by the SEC.
* * * * *