UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-Q
☒ QUARTERLY REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the quarterly period ended June 30, 2025
☐ TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
Commission file number: 001-39717
LIXTE BIOTECHNOLOGY HOLDINGS, INC.
(Exact name of registrant as specified in its charter)
| Delaware | 20-2903526 | |
| (State or other jurisdiction of | (I.R.S. Employer | |
| incorporation or organization) | Identification Number) |
680 East Colorado Boulevard, Suite 180
Pasadena, California 91101
(Address of principal executive offices, including Zip Code)
(631) 830-7092
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
| Title of each class | Trading Symbol(s) | Name of each exchange on which registered | ||
| Common Stock, par value $0.0001 per share | LIXT | The Nasdaq Stock Market LLC | ||
| Warrants to Purchase Common Stock, par value $0.0001 per share | LIXTW | The Nasdaq Stock Market LLC |
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days.
Yes ☒ No ☐
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files).
Yes ☒ No ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer”, “accelerated filer”, “smaller reporting company”, and “emerging growth company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer ☐ | Accelerated filer ☐ |
| Non-accelerated filer ☒ | Smaller reporting company ☒ |
| Emerging growth company ☐ |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act).
Yes ☐ No ☒
As of August 5, 2025, the Company had 4,561,363 shares of common stock issued and outstanding.
LIXTE BIOTECHNOLOGY HOLDINGS, INC.
AND SUBSIDIARY
TABLE OF CONTENTS
|
|
PART I - FINANCIAL INFORMATION
ITEM 1. CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
LIXTE BIOTECHNOLOGY HOLDINGS, INC.
AND SUBSIDIARY
CONDENSED CONSOLIDATED BALANCE SHEETS
|
June 30, 2025 |
December 31, 2024 |
|||||||
| (Unaudited) | ||||||||
| ASSETS | ||||||||
| Current assets: | ||||||||
| Cash | $ | 887,212 | $ | 1,038,952 | ||||
| Prepaid insurance | 14,597 | 20,898 | ||||||
| Other prepaid expenses | 87,943 | 85,653 | ||||||
| Total current assets | 989,752 | 1,145,503 | ||||||
| Deferred offering costs | 198,826 | — | ||||||
| Total assets | $ | 1,188,578 | $ | 1,145,503 | ||||
| LIABILITIES AND STOCKHOLDERS’ EQUITY | ||||||||
| Current liabilities: | ||||||||
| Accounts payable and accrued expenses, including $10,000 and $27,500 to related parties at June 30, 2025 and December 31, 2024, respectively | $ | 92,149 | $ | 83,206 | ||||
| Research and development contract liabilities | 256,147 | 235,078 | ||||||
| Accrued offering costs | 188,826 | — | ||||||
| Total current liabilities | 537,122 | 318,284 | ||||||
| Commitments and contingencies | - | - | ||||||
| Stockholders’ equity: | ||||||||
| Preferred Stock, $0.0001 par value; authorized – 10,000,000 shares; issued and outstanding –0 350,000 shares of Series A Convertible Preferred Stock, $10.00 per share stated value, convertible into0 72,917 shares of common stock | — | 3,500,000 | ||||||
| Common stock, $0.0001 par value; authorized – 100,000,000 shares; issued and outstanding – 2,756,991 shares and 2,249,290 shares at June 30, 2025 and December 31, 2024, respectively | 276 | 225 | ||||||
| Additional paid-in capital | 54,204,101 | 49,394,687 | ||||||
| Accumulated deficit | (53,552,921 | ) | (52,067,693 | ) | ||||
| Total stockholders’ equity | 651,456 | 827,219 | ||||||
| Total liabilities and stockholders’ equity | $ | 1,188,578 | $ | 1,145,503 | ||||
See accompanying notes to condensed consolidated financial statements.
|
|
LIXTE BIOTECHNOLOGY HOLDINGS, INC.
AND SUBSIDIARY
CONDENSED CONSOLIDATED STATEMENTS OF OPERATIONS
(Unaudited)
| Three Months Ended | Six Months Ended | |||||||||||||||
| June 30, | June 30, | |||||||||||||||
| 2025 | 2024 | 2025 | 2024 | |||||||||||||
| Revenues | $ | — | $ | — | $ | — | $ | — | ||||||||
| Costs and expenses: | ||||||||||||||||
| Research and development costs | 60,648 | 210,708 | 152,105 | 329,772 | ||||||||||||
| General and administrative costs | 714,161 | 798,448 | 1,329,644 | 1,646,263 | ||||||||||||
| Total costs and expenses | 774,809 | 1,009,156 | 1,481,749 | 1,976,035 | ||||||||||||
| Loss from operations | (774,809 | ) | (1,009,156 | ) | (1,481,749 | ) | (1,976,035 | ) | ||||||||
| Interest income | 365 | 2,233 | 806 | 5,092 | ||||||||||||
| Interest expense | (1,810 | ) | (4,154 | ) | (4,945 | ) | (11,340 | ) | ||||||||
| Foreign currency gain | 581 | 158 | 660 | 42 | ||||||||||||
| Net loss | $ | (775,673 | ) | $ | (1,010,919 | ) | $ | (1,485,228 | ) | $ | (1,982,241 | ) | ||||
| Net loss per common share – basic and diluted | $ | (0.29 | ) | $ | (0.45 | ) | $ | (0.57 | ) | $ | (0.88 | ) | ||||
| Weighted average common shares outstanding – basic and diluted | 2,720,533 | 2,249,290 | 2,596,509 | 2,249,290 | ||||||||||||
See accompanying notes to condensed consolidated financial statements.
|
|
LIXTE BIOTECHNOLOGY HOLDINGS, INC.
AND SUBSIDIARY
CONDENSED CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ EQUITY
(Unaudited)
Three Months and Six Months Ended June 30, 2025 and 2024
|
Series A Convertible Preferred Stock |
Common Stock | Additional | Total | |||||||||||||||||||||||||
| Shares | Amount | Shares | Par Value |
Paid-in Capital |
Accumulated Deficit |
Stockholders’ Equity |
||||||||||||||||||||||
| Three months ended June 30, 2025: | ||||||||||||||||||||||||||||
| Balance, March 31, 2025 | 350,000 | $ | 3,500,000 | 2,684,074 | $ | 268 | $ | 50,436,110 | $ | (52,777,248 | ) | $ | 1,159,130 | |||||||||||||||
| Conversion of Series A convertible preferred stock | (350,000 | ) | (3,500,000 | ) | 72,917 | 8 | 3,499,992 | — | — | |||||||||||||||||||
| Stock-based compensation expense | — | — | — | — | 267,999 | — | 267,999 | |||||||||||||||||||||
| Net loss | — | — | — | — | — | (775,673 | ) | (775,673 | ) | |||||||||||||||||||
| Balance, June 30, 2025 | — | $ | — | 2,756,991 | $ | 276 | $ | 54,204,101 | $ | (53,552,921 | ) | $ | 651,456 | |||||||||||||||
| Six months ended June 30, 2025: | ||||||||||||||||||||||||||||
| Balance, December 31, 2024 | 350,000 | $ | 3,500,000 | 2,249,290 | $ | 225 | $ | 49,394,687 | $ | (52,067,693 | ) | $ | 827,219 | |||||||||||||||
| Proceeds from sale of securities in registered direct offering, net of offering costs | — | — | 434,784 | 43 | 914,185 | — | 914,228 | |||||||||||||||||||||
| Stock options issued to settle accrued payable | — | — | — | — | 27,500 | — | 27,500 | |||||||||||||||||||||
| Conversion of Series A convertible preferred stock | (350,000 | ) | (3,500,000 | ) | 72,917 | 8 | 3,499,992 | — | — | |||||||||||||||||||
| Stock-based compensation expense | — | — | — | — | 367,737 | — | 367,737 | |||||||||||||||||||||
| Net loss | — | — | — | — | — | (1,485,228 | ) | (1,485,228 | ) | |||||||||||||||||||
| Balance, June 30, 2025 | — | $ | — | 2,756,991 | $ | 276 | $ | 54,204,101 | $ | (53,552,921 | ) | $ | 651,456 | |||||||||||||||
(continued)
|
|
LIXTE BIOTECHNOLOGY HOLDINGS, INC.
AND SUBSIDIARY
CONDENSED CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ EQUITY
(Unaudited)
(Continued)
Three Months and Six Months Ended June 30, 2025 and 2024
|
Series A Convertible Preferred Stock |
Common Stock | Additional | Total | |||||||||||||||||||||||||
| Shares | Amount | Shares | Par Value |
Paid-in Capital |
Accumulated Deficit |
Stockholders’ Equity |
||||||||||||||||||||||
| Three months ended June 30, 2024: | ||||||||||||||||||||||||||||
| Balance, March 31, 2024 | 350,000 | $ | 3,500,000 | 2,249,290 | $ | 225 | $ | 49,079,192 | $ | (49,453,050 | ) | $ | 3,126,367 | |||||||||||||||
| Stock-based compensation expense | — | — | — | — | 130,691 | — | 130,691 | |||||||||||||||||||||
| Net loss | — | — | — | — | — | (1,010,919 | ) | (1,010,919 | ) | |||||||||||||||||||
| Balance, June 30, 2024 | 350,000 | $ | 3,500,000 | 2,249,290 | $ | 225 | $ | 49,209,883 | $ | (50,463,969 | ) | $ | 2,246,139 | |||||||||||||||
| Six months ended June 30, 2024: | ||||||||||||||||||||||||||||
| Balance, December 31, 2023 | 350,000 | $ | 3,500,000 | 2,249,290 | $ | 225 | $ | 48,976,265 | $ | (48,481,728 | ) | $ | 3,994,762 | |||||||||||||||
| Stock-based compensation expense | — | — | — | — | 233,618 | — | 233,618 | |||||||||||||||||||||
| Net loss | — | — | — | — | — | (1,982,241 | ) | (1,982,241 | ) | |||||||||||||||||||
| Balance, June 30, 2024 | 350,000 | $ | 3,500,000 | 2,249,290 | $ | 225 | $ | 49,209,883 | $ | (50,463,969 | ) | $ | 2,246,139 | |||||||||||||||
See accompanying notes to condensed consolidated financial statements.
|
|
LIXTE BIOTECHNOLOGY HOLDINGS, INC.
AND SUBSIDIARY
CONDENSED CONSOLIDATED STATEMENTS OF CASH FLOWS
(Unaudited)
| Six Months Ended June 30, | ||||||||
| 2025 | 2024 | |||||||
| Cash flows from operating activities: | ||||||||
| Net loss | $ | (1,485,228 | ) | $ | (1,982,241 | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||
| Stock-based compensation expense included in - | ||||||||
| Research and development costs | — | — | ||||||
| General and administrative costs | 367,737 | 233,618 | ||||||
| Changes in operating assets and liabilities: | ||||||||
| (Increase) decrease in - | ||||||||
| Advances on research and development contract services | — | 78,016 | ||||||
| Prepaid insurance | 6,301 | (5,442 | ) | |||||
| Other prepaid expenses | (2,290 | ) | (39,824 | ) | ||||
| Increase (decrease) in - | ||||||||
| Accounts payable and accrued expenses | 36,443 | (25,354 | ) | |||||
| Research and development contract liabilities | 21,069 | 132,961 | ||||||
| Net cash used in operating activities | (1,055,968 | ) | (1,608,266 | ) | ||||
| Cash flows from financing activities: | ||||||||
| Proceeds from sale of securities in registered direct offering, net of offering costs |
914,228 | — | ||||||
| Payment of deferred offering costs | (10,000 | ) | — | |||||
| Net cash provided by financing activities | 904,228 | — | ||||||
| Cash: | ||||||||
| Net decrease | (151,740 | ) | (1,608,266 | ) | ||||
| Balance at beginning of period | 1,038,952 | 4,203,488 | ||||||
| Balance at end of period | $ | 887,212 | $ | 2,595,222 | ||||
| Supplemental disclosures of cash flow information: | ||||||||
| Cash paid for - | ||||||||
| Interest | $ | 4,945 | $ | 11,340 | ||||
| Income taxes | $ | — | $ | — | ||||
| Non-cash investing and financing activities: | ||||||||
| Settlement of accrued compensation to Board of Directors by issuance of stock options | $ | 27,500 | $ | — | ||||
| Conversion of Series A Convertible Preferred Stock into common stock | $ | 3,500,000 | $ | — | ||||
| Accrual of deferred offering costs | $ | 188,826 | $ | — | ||||
See accompanying notes to condensed consolidated financial statements.
|
|
LIXTE BIOTECHNOLOGY HOLDINGS, INC.
AND SUBSIDIARY
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
Three Months and Six Months Ended June 30, 2025 and 2024
1. Organization and Basis of Presentation
The condensed consolidated financial statements of Lixte Biotechnology Holdings, Inc., a Delaware corporation), including its wholly-owned Delaware subsidiary, Lixte Biotechnology, Inc. (collectively, the “Company”), at June 30, 2025, and for the three months and six months ended June 30, 2025 and 2024, are unaudited. In the opinion of management of the Company, all adjustments, including normal recurring accruals, have been made that are necessary to present fairly the financial position of the Company as of June 30, 2025, and the results of its operations for the three months and six months ended June 30, 2025 and 2024, and its cash flows for the six months ended June 30, 2025 and 2024. Operating results for the interim periods presented are not necessarily indicative of the results to be expected for a full fiscal year. The condensed consolidated balance sheet at December 31, 2024 has been derived from the Company’s audited consolidated financial statements at such date.
The condensed consolidated financial statements and related notes have been prepared pursuant to the rules and regulations of the Securities and Exchange Commission (“SEC”). Accordingly, certain information and footnote disclosures normally included in financial statements prepared in accordance with generally accepted accounting principles have been omitted pursuant to such rules and regulations. These condensed consolidated financial statements should be read in conjunction with the financial statements and other information included in the Company’s Annual Report on Form 10-K for the fiscal year ended December 31, 2024, as filed with the SEC.
Business
The Company is a clinical-stage biopharmaceutical company focused on identifying new targets for cancer drug development and developing and commercializing cancer therapies. The Company’s corporate office is located in Pasadena, California.
The Company’s product pipeline is primarily focused on inhibitors of Protein Phosphatase 2A, which is used to enhance cytotoxic agents, radiation, immune checkpoint blockers and other cancer therapies. The Company believes that inhibitors of protein phosphatases have significant therapeutic potential for a broad range of cancers. The Company is focusing on the clinical development of a specific protein phosphatase inhibitor, referred to as LB-100.
The Company’s activities are subject to significant risks and uncertainties, including the need for additional capital. The Company has not yet commenced any revenue-generating operations, does not have positive cash flows from operations, relies on stock-based compensation for a substantial portion of employee and consultant compensation, and is dependent on periodic infusions of equity capital to fund its operating requirements.
Nasdaq Compliance
The Company’s common stock and public warrants are traded on the Nasdaq Capital Market under the symbols “LIXT” and “LIXTW”, respectively.
On June 2, 2023, the Company effected a 1-for-10 reverse split of its outstanding shares of common stock in order to remain in compliance with the $1.00 minimum closing bid price requirement of the Nasdaq Stock Market LLC (“Nasdaq”).
On August 19, 2024, the Company received a letter from the Listing Qualifications Department (the “Staff”) of Nasdaq indicating that the Company was not in compliance with the minimum stockholders’ equity requirement of $2,500,000 for continued listing on the Nasdaq Capital Market under Listing Rule 5550(b)(1) (the “Stockholders’ Equity Requirement”).
|
|
On October 3, 2024, the Company submitted a plan to the Staff to regain compliance with the Stockholders’ Equity Requirement, which outlined the Company’s proposed initiatives to regain compliance by raising equity capital through various registered equity offerings.
On October 21, 2024, the Staff provided notice (the “Notice”) to the Company that it had granted an extension through February 18, 2025 to regain compliance with the Stockholders’ Equity Requirement, which required that the Company complete its capital raising initiatives and evidence compliance with the Stockholders’ Equity Requirement through filing a Current Report on Form 8-K with the SEC providing certain required information.
As of February 18, 2025, the Company had not regained compliance with the Stockholders’ Equity Requirement. On February 19, 2025, the Company received a Staff determination letter stating that the Company did not meet the terms of the extension because it did not complete its proposed financing initiatives to regain compliance. The Company timely requested a Hearing before a Nasdaq Hearings Panel (the “Panel”), which automatically stayed Nasdaq’s suspension or delisting of the Company’s common stock and public warrants pending the Panel’s decision.
On April 17, 2025, the Company received notice that the Panel had granted the Company an extension in which to regain compliance with all continued listing rules of the Nasdaq Capital Market. The Panel’s determination followed a hearing on April 3, 2025, at which the Panel considered the Company’s plan to regain compliance with the Stockholders’ Equity Requirement. As a result of the extension, the Panel granted the Company’s request for continued listing on the Nasdaq Capital Market, provided that the Company demonstrates compliance with the Stockholders’ Equity Requirement and all other continued listing requirements for the Nasdaq Capital Market by July 3, 2025.
On July 2, 2025, the Company closed a private placement for gross proceeds of $5,050,000, consisting of shares of common stock, pre-funded warrants to purchase shares of common stock, warrants to purchase shares of common stock, and shares of Series B Convertible Preferred Stock, and on July 8, 2025, the Company closed a registered direct offering for gross proceeds of $1,500,000, consisting of shares of common stock and pre-funded warrants to purchase shares of common stock.
On July 15, 2025, the Company received notice from Nasdaq that the Panel found that the Company was in compliance with the Stockholders’ Equity Requirement. The Company was also notified that it will remain subject to a “Panel Monitor”, as that term is defined in Nasdaq Listing Rule 5815(d)(4)(B), for a period of one year from the date of the Nasdaq notice, through July 15, 2026. If, during the term of the Panel Monitor, the Company does not continue to remain in compliance with the Stockholders’ Equity Requirement, the Company will not be provided with the opportunity to submit a compliance plan for review by the Listing Qualifications Staff and must instead request a hearing before the Panel to address the deficiency, with such request staying any further action with respect to the Company’s listing on Nasdaq pending completion of the hearing process.
The Company is undertaking measures to maintain compliance under Nasdaq’s continued listing requirements and to remain listed on the Nasdaq Capital Market. However, there can be no assurances that the Company will ultimately be able to maintain compliance with the Stockholders’ Equity Requirement, or be able to maintain compliance with all other applicable requirements for continued listing on the Nasdaq Capital Market. The Company’s failure to meet these requirements would result in the Company’s securities being delisted from the Nasdaq Capital Market.
Going Concern
For the six months ended June 30, 2025, the Company recorded a net loss of $1,485,228 and used cash in operations of $1,055,968. At June 30, 2025, the Company had cash of $887,212 available to fund its operations.
Because the Company is currently engaged in various early-stage clinical trials, it is expected that it will take a significant amount of time and resources to develop any product or intellectual property capable of generating sustainable revenues. Accordingly, the Company’s business is unlikely to generate any sustainable operating revenues in the next several years and may never do so. Even if the Company is able to generate revenues through licensing its technology, product sales or other commercial activities, there can be no assurance that the Company will be able to achieve and maintain positive earnings and operating cash flows. At June 30, 2025, the Company’s remaining financial contractual commitments pursuant to clinical trial agreements and clinical trial monitoring agreements not yet incurred aggregated approximately $524,000, which are currently scheduled to be incurred through approximately December 31, 2027.
|
|
The Company’s consolidated financial statements have been presented on the basis that it will continue as a going concern, which contemplates the realization of assets and satisfaction of liabilities in the normal course of business. The Company has no recurring source of revenues and has experienced negative operating cash flows since inception. The Company has financed its working capital requirements through the recurring sale of its equity securities. These factors raise substantial doubt about the Company’s ability to continue as a going concern within one year after the date the consolidated financial statements are issued. The consolidated financial statements also do not reflect any adjustments relating to the recoverability of assets and liabilities that might be necessary if the Company is unable to continue as a going concern.
The Company’s ability to continue as a going concern is dependent upon its ability to raise additional equity capital to fund its research and development activities, including its ongoing clinical trials. The amount and timing of future cash requirements depends in substantial part on the pace, design and results of the Company’s clinical trial program, which, in turn, depends on the availability of operating capital to fund such activities.
Based on current operating plans, the Company estimates that its existing cash resources at June 30, 2025, together with the net proceeds from the July 2, 2025 private placement, and the July 8, 2025 registered direct offering, will provide sufficient working capital to fund the Company’s operations as currently configured, including its ongoing clinical trial program with respect to the development of the Company’s lead anti-cancer clinical compound LB-100, for at least the next 12 months. However, existing cash resources will not be sufficient to complete the development of and to obtain regulatory approval for the Company’s product candidate, which would require significant additional operating capital.
In addition, as a result of the appointment of a new Chairman and Chief Executive Officer in June 2025, the completion of the July 2025 equity financings, and other changes in senior management and the Board of Directors in July 2025, the Company’s operating strategies and business plans may change, including the incurrence of additional personnel and operating costs, which may require that the Company raise additional capital to fund operations. However, as market conditions present uncertainty as to the Company’s ability to secure additional funds, there can be no assurances that the Company will be able to secure additional financing on acceptable terms, as and when necessary, to continue to fund its operations.
The Company’s independent registered public accounting firm included an explanatory paragraph in their report with respect to this uncertainty that accompanied the Company’s audited consolidated financial statements as of and for the year ended December 31, 2024, in which they expressed substantial doubt about the Company’s ability to continue as a going concern. The Company’s consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
If cash resources are insufficient to satisfy the Company’s ongoing cash requirements, the Company would be required to scale back or discontinue its clinical trial program, as well as its licensing and patent prosecution efforts and its technology and product development efforts, or obtain funds, if available, through strategic alliances, joint ventures or other transaction structures that could require the Company to relinquish rights to and/or control of LB-100, or to curtail or discontinue operations entirely.
2. Summary of Significant Accounting Policies
Principles of Consolidation
The accompanying consolidated financial statements of the Company have been prepared in accordance with United States generally accepted accounting principles (“GAAP”) and include the financial statements of Lixte Biotechnology Holdings, Inc. and its wholly-owned subsidiary, Lixte Biotechnology, Inc. Intercompany balances and transactions have been eliminated in consolidation.
|
|
Segment Information
The Company’s Chief Executive Officer is the Company’s Chief Operating Decision Maker (“CODM”) and evaluates performance and makes operating decisions about allocating resources based on internal financial data presented on a consolidated basis. Because the CODM evaluates financial performance on a consolidated basis, the Company has determined that it currently operates in a single reportable segment, which consists of the development of a drug class called Protein Phosphatase 2A inhibitors, and is comprised of the consolidated financial results of the Company. The CODM uses consolidated net income (loss) as the sole measure of segment profit or loss. The required segment information, including significant segment expenses, is presented at Note 3.
Use of Estimates
The preparation of financial statements in conformity with GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities at the date of the financial statements and the reported amounts of expenses during the reporting period. Some of those judgments can be subjective and complex, and therefore, actual results could differ materially from those estimates under different assumptions or conditions. Management bases its estimates on historical experience and on various assumptions that are believed to be reasonable in relation to the financial statements taken, as a whole, under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Management regularly evaluates the key factors and assumptions used to develop the estimates utilizing currently available information, changes in facts and circumstances, historical experience, and reasonable assumptions. After such evaluations, if deemed appropriate, those estimates are adjusted accordingly. Actual results could differ from those estimates. Significant estimates include those related to assumptions used in the calculation of accruals for clinical trial costs and other potential liabilities, and valuing equity instruments issued for services.
Cash
Cash is held in a cash bank deposit program maintained by Morgan Stanley Wealth Management, a division of Morgan Stanley Smith Barney LLC (“Morgan Stanley”). Morgan Stanley is a FINRA-regulated broker-dealer. The Company’s policy is to maintain its cash balances with financial institutions in the United States with high credit ratings and in accounts insured by the Federal Deposit Insurance Corporation (the “FDIC”) and/or by the Securities Investor Protection Corporation (the “SIPC”). The Company periodically has cash balances in financial institutions in excess of the FDIC and SIPC insurance limits of $250,000 and $500,000, respectively. Morgan Stanley Wealth Management also maintains supplemental insurance coverage for the cash balances of its customers. The Company has not experienced any losses to date resulting from this policy.
Research and Development
Research and development costs consist primarily of fees paid to consultants and contractors, and other expenses relating to the negotiation, design, development, conduct and management of clinical trials with respect to the Company’s clinical compound and product candidate. Research and development costs also include the costs to manufacture compounds used in research and clinical trials, which are charged to operations as incurred. The Company’s inventory of LB-100 for clinical use has been manufactured separately in the United States and in the European Union in accordance with the laws and regulations of such jurisdictions.
Research and development costs are generally charged to operations ratably over the life of the underlying contracts, unless the achievement of milestones, the completion of contracted work, the termination of an agreement, or other information indicates that a different expensing schedule is more appropriate. However, payments for research and development costs that are contractually defined as non-refundable are charged to operations as incurred.
Obligations incurred with respect to mandatory scheduled payments under agreements with milestone provisions are recognized as charges to research and development costs in the Company’s consolidated statement of operations based on the achievement of such milestones, as specified in the respective agreement. Obligations incurred with respect to mandatory scheduled payments under agreements without milestone provisions are accounted for when due, are recognized ratably over the appropriate period, as specified in the respective agreement, and are recorded as liabilities in the Company’s consolidated balance sheet, with a corresponding charge to research and development costs in the Company’s consolidated statement of operations.
|
|
Payments made pursuant to contracts are initially recorded as advances on research and development contract services in the Company’s consolidated balance sheet and are then charged to research and development costs in the Company’s consolidated statement of operations as those contract services are performed. Expenses incurred under contracts in excess of amounts advanced are recorded as research and development contract liabilities in the Company’s consolidated balance sheet, with a corresponding charge to research and development costs in the Company’s consolidated statement of operations. The Company reviews the status of its various clinical trial and research and development contracts on a quarterly basis.
Prepaid Insurance
Prepaid insurance represents the premiums paid for directors and officers insurance coverage and for general liability insurance coverage in excess of the amortization of the total policy premium charged to operations at each balance sheet date. Such amount is determined by amortizing the total policy premium charged on a straight-line basis over the respective policy period. As the policy premiums incurred are generally amortizable over the ensuing twelve-month period, they are recorded as a current asset in the Company’s consolidated balance sheet at each reporting date and appropriately amortized to the Company’s consolidated statement of operations for each reporting period.
Offering Costs
Offering costs consist of costs incurred with respect to equity financing transactions, including legal fees. Such costs are deferred and charged to additional paid-in capital upon the successful completion of such financings, or are charged to operations if and when such financings are abandoned or terminated.
Patent and Licensing Legal and Filing Fees and Costs
Due to the significant uncertainty associated with the successful development of commercially viable products based on the Company’s research efforts and related patent applications, all patent and licensing legal and filing fees and costs related to the development and protection of the Company’s intellectual property are charged to operations as incurred. Patent and licensing legal and filing fees and costs were $17,303 and $63,612 for the three months ended June 30, 2025 and 2024, respectively, and $73,386 and $146,823 for the six months ended June 30, 2025 and 2024, respectively. Patent and licensing legal and filing fees and costs are included in general and administrative costs in the Company’s consolidated statement of operations.
Concentration of Risk
The Company periodically contracts with vendors and consultants to provide services related to the Company’s operations. Charges incurred for these services can be for a specific period (typically one year) or for a specific project or task. Costs and expenses incurred that represented 10% or more of general and administrative costs or research and development costs for the three months ended June 30, 2025 and 2024 are described below.
Research and development costs for the three months ended June 30, 2025 include charges from five vendors and consultants representing 20.7%, 19.5%, 17.9%, 17.8% and 11.2%, respectively, of total research and development costs. Research and development costs for the three months ended June 30, 2024 include charges from three vendors and consultants representing 37.0%, 31.9% and 10.4%. respectively, of total research and development costs.
General and administrative costs for the three months ended June 30, 2025 and 2024 include charges from legal firms and other vendors for general licensing and patent prosecution costs relating to the Company’s intellectual properties representing 3.3% and 8.0%, respectively of total general and administrative costs. General and administrative costs for the three months ended June 30, 2025 also includes a charge from a vendor representing 11.9% of total general and administrative costs. General and administrative costs for the three months ended June 30, 2024 include charges from two vendors and consultants representing 16.5% and 15.6%, respectively, of total general and administrative costs. General and administrative costs for the three months ended June 30, 2025 and 2024 also included charges for the fair value of stock options granted to directors and corporate officers representing 33.7% and 12.9%, respectively, of total general and administrative costs.
|
|
Research and development costs for the six months ended June 30, 2025 include charges from five vendors and consultants representing 22.9%, 18.5%, 16.3%, 16.2% and 12.7%, respectively, of total research and development costs. Research and development costs for the six months ended June 30, 2024 include charges from three vendors and consultants representing 40.7%, 23.7% and 10.8%, respectively, of total research and development costs.
General and administrative costs for the six months ended June 30, 2025 and 2024 include charges from legal firms and other vendors for general licensing and patent prosecution costs relating to the Company’s intellectual properties representing 5.5% and 8.9%. respectively, of total general and administrative costs. General and administrative costs for the six months ended June 30, 2025 also include charges from a vendor/consultant representing 12.5% of total general and administrative costs. General and administrative costs for the six months ended June 30, 2024 also include charges from two vendors and consultants representing 15.1% and 12.2%, respectively, of total general and administrative costs. General and administrative costs for the six months ended June 30, 2025 and 2024 also included charges for the fair value of stock options granted to directors and corporate officers representing 23.5% and 12.5%, respectively, of total general and administrative costs.
Income Taxes
The Company accounts for income taxes under an asset and liability approach for financial accounting and reporting for income taxes. Accordingly, the Company recognizes deferred tax assets and liabilities for the expected impact of differences between the financial statements and the tax basis of assets and liabilities.
The Company records a valuation allowance to reduce its deferred tax assets to the amount that is more likely than not to be realized. Due to the uncertainty of the Company’s ability to realize the benefit of the deferred tax assets, the net deferred tax assets are fully offset by a valuation allowance at June 30, 2025 and December 31, 2024. In the event the Company was to determine that it would be able to realize its deferred tax assets in the future in excess of its recorded amount, an adjustment to the deferred tax assets would be credited to operations in the period such determination was made. Should the Company determine that it would not be able to realize all or part of its deferred tax assets in the future, an adjustment to the deferred tax assets would be charged to operations in the period such determination was made.
The Company is subject to U.S. federal income taxes and income taxes of various state tax jurisdictions. As the Company’s net operating losses have yet to be utilized, all previous tax years remain open to examination by Federal authorities and other jurisdictions in which the Company currently operates or has operated in the past. The Company did not have any unrecognized tax benefits as of June 30, 2025 or December 31, 2024, and does not anticipate any material amount of unrecognized tax benefits through December 31, 2025.
The Company accounts for uncertainties in income tax law under a comprehensive model for the financial statement recognition, measurement, presentation, and disclosure of uncertain tax positions taken or expected to be taken in income tax returns as prescribed by GAAP. The tax effects of a position are recognized only if it is “more-likely-than-not” to be sustained by the taxing authority as of the reporting date. If the tax position is not considered “more-likely-than-not” to be sustained, then no benefits of the position are recognized. The Company had not recorded any liability for uncertain tax positions as of June 30, 2025 or December 31, 2024. Subsequent to June 30, 2025, any interest and penalties related to uncertain tax positions will be recognized as a component of income tax expense.
The Company periodically issues common stock and stock options to officers, directors, employees, contractors and consultants for services rendered. Options vest and expire according to terms established at the issuance date of each grant. Stock grants, which are generally time vested, are measured at the grant date fair value and charged to operations ratably over the vesting period.
|
|
The Company accounts for stock-based payments to officers, directors, employees, contractors, and consultants by measuring the cost of services received in exchange for equity awards utilizing the grant date fair value of the awards, with the cost recognized as compensation expense on the straight-line basis in the Company’s financial statements over the vesting period of the awards. Recognition of compensation expense for non-employees is in the same period and manner as if the Company had paid cash for the services.
The fair value of stock options granted as stock-based compensation is determined utilizing the Black-Scholes option-pricing model, and is affected by several variables, the most significant of which are the expected life of the stock option, the exercise price of the stock option as compared to the fair market value of the common stock on the grant date, and the estimated volatility of the common stock. Unless sufficient historical exercise data is available, the expected life of the stock option is calculated as the mid-point between the vesting period and the contractual term (the “simplified method”). The estimated volatility is based on the historical volatility of the Company’s common stock, calculated utilizing a look-back period approximately equal to the contractual life of the stock option being granted. The risk-free interest rate is based on the U.S. Treasury yield curve in effect at the time of grant. The fair market value of the common stock is determined by reference to the quoted market price of the Company’s common stock on the grant date. The expected dividend yield is based on the Company’s expectation of dividend payouts and is assumed to be zero.
The Company recognizes the fair value of stock-based compensation awards in general and administrative costs and in research and development costs, as appropriate, in the Company’s consolidated statements of operations. The Company issues new shares of common stock to satisfy stock option exercises.
Warrants
The Company accounts for warrants as either equity-classified or liability-classified instruments based on an assessment of the warrant’s specific terms and applicable authoritative guidance in Accounting Standards Codification (“ASC”) 480, Distinguishing Liabilities from Equity (“ASC 480”), and ASC 815, Derivatives and Hedging (“ASC 815”). The assessment considers whether the warrants are freestanding financial instruments pursuant to ASC 480, meet the definition of a liability pursuant to ASC 480, and whether the warrants meet all of the requirements for equity classification under ASC 815, including whether the warrants are indexed to the Company’s own common stock and whether the warrant holders could potentially require “net cash settlement” in a circumstance outside of the Company’s control, among other conditions for equity classification. The Company has determined that the warrants issued in the July 2023 equity financing, the February 2025 equity financing, and the July 2025 equity financings (see Note 4) meet the requirements for equity classification. This assessment, which requires the use of professional judgment, is conducted when the warrants are issued and at the end each subsequent quarterly period while the warrants are outstanding. For issued or modified warrants that meet all of the criteria for equity classification, the warrants are required to be recorded as a component of additional paid-in capital at the time of issuance. For issued or modified warrants that do not meet all of the criteria for equity classification, the warrants are required to be liability-classified and recorded at their initial fair value on the date of issuance and remeasured at fair value at each balance sheet date thereafter. Changes in the estimated fair value of the warrants that are liability-classified are recognized as a non-cash gain or loss in the statement of operations at each balance sheet date. At June 30, 2025 and December 31, 2024, the Company did not have any liability-classified warrants.
The Company’s computation of earnings (loss) per share (“EPS”) includes basic and diluted EPS. Basic EPS is measured as the income (loss) attributable to common stockholders divided by the weighted average common shares outstanding for the period. Diluted EPS is similar to basic EPS but presents the dilutive effect on a per share basis of potential common shares (e.g., preferred shares, warrants and stock options) as if they had been converted at the beginning of the respective periods presented, or issuance date, if later. Potential common shares that have an anti-dilutive effect (i.e., those that increase income per share or decrease loss per share) are excluded from the calculation of diluted EPS.
Loss per common share is computed by dividing net loss by the weighted average number of common shares outstanding during the respective periods. Basic and diluted loss per common share was the same for all periods presented because all preferred shares, warrants and stock options outstanding were anti-dilutive.
|
|
Schedule of Anti-dilutive Securities Excluded from Computation of Earnings Per Share
| June 30, | ||||||||
| 2025 | 2024 | |||||||
| Series A Convertible Preferred Stock | — | 72,917 | ||||||
| Common stock warrants | 1,275,758 | 808,365 | ||||||
| Common stock options | 744,726 | 605,348 | ||||||
| Total | 2,020,484 | 1,486,630 | ||||||
Foreign Currency Translation
The consolidated financial statements are presented in the United States dollar, which is the functional and reporting currency of the Company.
The Company periodically incurs a cost or expense in a foreign jurisdiction denominated in a local currency. The Company purchases the required foreign currency to pay such cost or expense on an as-needed basis. Such cost or expense is converted into United States dollars for financial statement purposes based on the foreign currency conversion rate in effect on the transaction date. The Company purchases the requisite foreign currency to pay such cost or expense on an as-needed basis. Any gain or loss resulting from the purchase of the foreign currency is included as foreign currency gain (loss) in the consolidated statement of operations.
During the three months ended June 30, 2025 and 2024, the Company incurred various costs and expenses denominated in Euros, which were converted into United States dollars at the average rate of 1.1338 and 1.0766 Euros per United States dollar, respectively. During the six months ended June 30, 2025 and 2024, the Company incurred various costs and expenses denominated in Euros, which were converted into United States dollars at the average rate of 1.0927 and 1.0813 Euros per United States dollar, respectively. As of June 30, 2025 and December 31, 2024, the Company did not hold any currencies other than the United States dollar in its bank accounts, and was not a party to any foreign currency forward or exchange contracts.
Fair Value of Financial Instruments
The authoritative guidance with respect to fair value established a fair value hierarchy that prioritizes the inputs to valuation techniques used to measure fair value into three levels and requires that assets and liabilities carried at fair value be classified and disclosed in one of three categories, as presented below. Disclosure as to transfers in and out of Levels 1 and 2, and activity in Level 3 fair value measurements, is also required.
Level 1. Observable inputs such as quoted prices in active markets for an identical asset or liability that the Company has the ability to access as of the measurement date. Financial assets and liabilities utilizing Level 1 inputs include active-exchange traded securities and exchange-based derivatives.
Level 2. Inputs, other than quoted prices included within Level 1, which are directly observable for the asset or liability or indirectly observable through corroboration with observable market data. Financial assets and liabilities utilizing Level 2 inputs include fixed income securities, non-exchange-based derivatives, mutual funds, and fair-value hedges.
Level 3. Unobservable inputs in which there is little or no market data for the asset or liability which requires the reporting entity to develop its own assumptions. Financial assets and liabilities utilizing Level 3 inputs include infrequently traded non-exchange-based derivatives and commingled investment funds and are measured using present value pricing models.
|
|
The Company determines the level in the fair value hierarchy within which each fair value measurement falls in its entirety, based on the lowest level input that is significant to the fair value measurement in its entirety. In determining the appropriate levels, the Company performs an analysis of the assets and liabilities at each reporting period end.
The carrying value of financial instruments, which consists of accounts payable and accrued expenses is considered to be representative of their respective fair values due to the short-term nature of those instruments.
Recent Accounting Pronouncements
In November 2024, the Financial Accounting Standards Board (“FASB”) issued Accounting Standards Update (“ASU”) 2024-03, Income Statement – Reporting Comprehensive Income – Expense Disaggregation Disclosures (Subtopic 220-40). ASU 2024-03 amends the FASB Accounting Standards Codification to require specified information about certain costs and expenses in the notes to the financial statements at each interim and annual reporting period, including disclosure of the amounts of purchases of inventory; employee compensation; depreciation; intangible asset amortization; and depreciation, depletion, and amortization included in each relevant expense caption on the face of the income statement within continuing operations that contains any of the expense categories previously listed. Disclosure will also be required of the total amount of selling expenses and an entity’s definition of selling expenses in annual reporting periods. ASU 2024-03 does not change or remove current expense disclosure requirements, but does affect where and how this information is presented in the notes to the financial statements. ASU 2024-03 is effective for the Company for annual reporting periods beginning January 1, 2027, and interim periods within annual reporting periods beginning January 1, 2028. Early adoption is permitted. The Company is in the process of evaluating ASU 2024-03 to determine its impact on the Company’s consolidated financial statement presentation and related disclosures.
In January 2025, the FASB issued ASU 2025-01, Income Statement – Reporting Comprehensive Income – Expense Disaggregation Disclosures (Subtopic 220-40), Clarifying the Effective Date. ASU 2025-01 clarifies the effective date of ASU 2024-03 for all public business entities that do not have an annual reporting period that ends on December 31 (referred to as non-calendar year-end entities). All public business entities are required to adopt the disclosure requirements in the first annual reporting period beginning after December 15, 2026, and interim reporting periods within annual reporting periods beginning after December 15, 2027. As the Company’s annual reporting period ends on December 31, ASU-2025-01 did not have any impact on the Company’s process of evaluating ASU-2024-03 to determine its impact on the Company’s consolidated financial statement presentation and related disclosures.
Management does not believe that any other recently issued, but not yet effective, authoritative guidance, if currently adopted, would have a material impact on the Company’s financial statements, including their presentation and related disclosures.
Reclassifications
As a result of the adoption of ASU 2023-07 effective January 1, 2024, certain reclassifications have been made to the prior year statement of operations to conform it to the current year presentation. In presenting general and administrative costs on the Company’s consolidated statement of operations for the three months ended June 30, 2024, $306,354 of compensation to related parties, $63,612 of patent and licensing legal and filing fees and costs, and $428,482 of other costs and expenses were shown separately. In presenting the Company’s consolidated statement of operations for the three months ended June 30, 2024, the Company has combined these categories into general and administrative costs in the accompanying consolidated statement of operations for the three months ended June 30, 2024. In presenting general and administrative costs on the Company’s consolidated statement of operations for the six months ended June 30, 2024, $624,016 of compensation to related parties, $146,823 of patent and licensing legal and filing fees and costs, and $875,424 of other costs and expenses were shown separately. In presenting the Company’s consolidated statement of operations for the six months ended June 30, 2024, the Company has combined these categories into general and administrative costs in the accompanying consolidated statement of operations for the six months ended June 30, 2024. These reclassifications had no effect on the reported results of operations, including loss from operations and net loss.
|
|
3. Segment Information
The Company’s chief operating decision maker (“CODM”) has been identified as the Company’s Chief Executive Officer (“CEO”). The Company’s CODM evaluates performance and makes operating decisions about allocating resources based on financial data presented on a consolidated basis. Because the CODM evaluates financial performance on a consolidated basis, the Company has determined that it currently has a single operating segment which is comprised of the consolidated financial results of the Company.
The following table presents the significant segment expenses (10% or greater) and other segment items regularly reviewed by the Company’s CODM and included in research and development costs for the three months and six months ended June 30, 2025 and 2024.
Schedule of Information by Segment
| Three Months Ended | Six Months Ended | |||||||||||||||
| June 30, | June 30, | |||||||||||||||
| 2025 | 2024 | 2025 | 2024 | |||||||||||||
| Clinical and related oversight costs | $ | 11,601 | $ | 97,947 | $ | 27,470 | $ | 107,977 | ||||||||
| Preclinical research focused on development of additional novel anti-cancer compounds | 27,592 | 104,829 | 70,362 | 209,309 | ||||||||||||
| Compound maintenance | 20,265 | 5,976 | 53,083 | 9,870 | ||||||||||||
| Regulatory service costs | 1,190 | 1,956 | 1,190 | 2,616 | ||||||||||||
| Total research and development costs | $ | 60,648 | $ | 210,708 | $ | 152,105 | $ | 329,772 | ||||||||
The following table presents a summary of research and development costs for the three months and six months ended June 30, 2025 and 2024 based on the respective geographical regions where such costs were incurred.
| Three Months Ended | Six Months Ended | |||||||||||||||
| June 30, | June 30, | |||||||||||||||
| 2025 | 2024 | 2025 | 2024 | |||||||||||||
| United States | $ | 41,928 | $ | 114,345 | $ | 100,499 | $ | 148,928 | ||||||||
| Spain | 18,720 | 29,244 | 51,606 | 44,478 | ||||||||||||
| China | — | — | — | 2,282 | ||||||||||||
| Netherlands | — | 67,119 | — | 134,084 | ||||||||||||
| Total research and development costs | $ | 60,648 | $ | 210,708 | $ | 152,105 | $ | 329,772 | ||||||||
The following table presents the significant segment expenses (10% or greater) and other segment items regularly reviewed by the Company’s CODM and included in general and administrative costs for the three months and six months ended June 30, 2025 and 2024.
| Three Months Ended | Six Months Ended | |||||||||||||||
| June 30, | June 30, | |||||||||||||||
| 2025 | 2024 | 2025 | 2024 | |||||||||||||
| Compensation to related parties: | ||||||||||||||||
| Cash-based | $ | 123,285 | $ | 175,663 | $ | 232,016 | $ | 390,398 | ||||||||
| Stock-based | 267,999 | 130,691 | 367,737 | 233,618 | ||||||||||||
| Patent and licensing legal and filing fees and costs | 17,303 | 63,612 | 73,386 | 146,823 | ||||||||||||
| Other consulting and professional fees | 176,551 | 191,529 | 381,867 | 363,972 | ||||||||||||
| Insurance expense | 64,277 | 126,873 | 128,553 | 253,727 | ||||||||||||
| Other costs and expenses, net | 64,746 | 110,080 | 146,085 | 257,725 | ||||||||||||
| Total general and administrative costs | $ | 714,161 | $ | 798,448 | $ | 1,329,644 | $ | 1,646,263 | ||||||||
The following table presents the Company’s total assets by segment at June 30, 2025 and December 31, 2024.
|
June 30, 2025 |
December 31, 2024 |
|||||||
| Research and development assets | $ | 20,041 | $ | 39,298 | ||||
| Corporate assets (primarily cash) | 1,168,537 | 1,106,205 | ||||||
| Total assets | $ | 1,188,578 | $ | 1,145,503 | ||||
|
|
4. Stockholders’ Equity
Preferred Stock
The Company is authorized to issue a total of 10,000,000 shares of preferred stock, par value $0.0001 per share.
On March 17, 2015, the Company filed a Certificate of Designations, Preferences, Rights and Limitations of its Series A Convertible Preferred Stock with the Delaware Secretary of State to amend the Company’s certificate of incorporation. The Company has designated a total of 350,000 shares as Series A Convertible Preferred Stock, which are non-voting and are not subject to increase without the written consent of a majority of the holders of the Series A Convertible Preferred Stock or as otherwise set forth in the Preferences, Rights and Limitations. The holders of each tranche of 175,000 shares of the Series A Convertible Preferred Stock are entitled to receive a per share dividend equal to 1% of the annual net revenue of the Company divided by 175,000, until converted or redeemed. Each share of Series A Convertible Preferred Stock was convertible, at the option of the holder, into 0.20833 shares of common stock (subject to customary anti-dilution provisions) and the Series A Convertible Preferred Stock is subject to mandatory conversion at the conversion rate in the event of a merger or sale transaction resulting in gross proceeds to the Company of at least $21,875,000. The Series A Convertible Preferred Stock had a liquidation preference based on its assumed conversion into shares of common stock. The Series A Convertible Preferred Stock did not have any cash liquidation preference rights or any registration rights. Based on the attributes of the Series A Convertible Preferred Stock as previously described, the Company accounted for the Series A Convertible Preferred Stock as a permanent component of stockholders’ equity. The 350,000 outstanding shares of Series A Convertible Preferred Stock were converted into a total of 72,917 shares of common stock pursuant to a notice of conversion dated May 16, 2025.
As of June 30, 2025 and December 31, 2024, the Company had 9,650,000 shares of undesignated preferred stock, which may be issued with such rights and powers as the Board of Directors may designate. The Company expects to amend its certificate of incorporation to eliminate the Series A Convertible Preferred Stock classification.
On July 2, 2025, the Company closed a private placement for gross proceeds of $5,050,000, consisting, in part, of shares of Series B Convertible Preferred Stock (see Note 9).
Common Stock
The Company is authorized to issue a total of 100,000,000 shares of common stock, par value $0.0001 per share. As of June 30, 2025 and December 31, 2024, the Company had 2,756,991 shares and 2,249,290 shares, respectively, of common stock issued and outstanding.
July 20, 2023 equity offering
Effective July 20, 2023, the Company sold 180,000 shares of common stock at a price of $6.00 per share and pre-funded warrants to purchase 403,334 shares of common stock at a price of $5.9999 per pre-funded warrant to an institutional investor in a registered direct offering. The pre-funded warrants had an exercise price of $0.0001 per share, were immediately exercisable upon issuance, and were valid and exercisable until all pre-funded warrants were exercised in full. During the period from July 24, 2023 through August 7, 2023, the 403,334 pre-funded warrants, exercisable at $0.0001 per common share, were exercised for total cash proceeds of $41, resulting in the issuance of 403,334 shares of common stock. The pre-funded warrants were determined to be common stock equivalents.
|
|
In a concurrent private placement to the institutional investor, the Company also sold warrants to purchase 583,334 shares of common stock. Each common warrant had an initial exercise price of $6.00 per share, was immediately exercisable upon issuance, and expires five years thereafter on July 20, 2028. The common warrants and the shares of common stock issuable upon exercise of the common warrants were not registered under the Securities Act of 1933, as amended (the “Securities Act”) and were offered pursuant to the exemption provided in Section 4(a)(2) under the Securities Act and Rule 506(b) promulgated thereunder. The shares of common stock issuable upon exercise of the warrants were registered for resale in a registration statement on Form S-3 declared effective by the SEC on May 2, 2024.
The registered direct offering and the concurrent private placement generated gross proceeds of $3,499,964. The total cash costs of the registered direct offering and the private placement were $362,925, resulting in net proceeds of $3,137,039. Pursuant to the placement agent agreement, the Company granted the placement agent warrants to purchase 35,000 shares of common stock at an exercise price of $6.60 per share and expiring on July 20, 2028. The net proceeds from the registered direct offering and the concurrent private placement were used for general working capital purposes.
The exercise prices of the warrants issued to the institutional investor (exercisable at $6.00 per share) and to the placement agent (exercisable at $6.60 per share) are subject to customary adjustments for stock splits, stock dividends, stock combinations, reclassifications, reorganizations, or similar events affecting the Company’s common stock. In addition, the warrants issued to the institutional investor contain a “fundamental transaction” provision which provides that if any defined fundamental transactions are within the Company’s control and are consummated, the holder of the unexercised common stock warrants would be entitled to receive, at its option, in exchange for extinguishment of such warrants, cash consideration equal to a Black-Scholes valuation amount, as defined in the warrant agreement.
The fundamental transaction provision includes (i) a sale, lease, assignment, transfer, conveyance or other disposition of all or substantially all of the assets of the Company in one or a series of related transactions, or (ii) a change in control of the Company by which it, directly or indirectly, in one or more related transactions, consummates a stock or share purchase agreement or other business combination with another person or group, whereby such other person or group acquires more than 50% of the voting power of the common equity of the Company.
If such fundamental transaction is not within the Company’s control, including not being approved by the Company’s Board of Directors, the warrant holder would only be entitled to receive the same type or form of consideration (and in the same proportion) equal to the Black-Scholes valuation amount of the remaining unexercised portion of the warrant on the date of consummation of such fundamental transaction as the holders of the Company’s common stock receive. Accordingly, these warrants are classified as a component of permanent stockholders’ equity. The Company will account for any cash payment for a warrant redemption as a distribution from stockholders’ equity, as and when a fundamental transaction is consummated and such cash payment is required to be made.
February 13, 2025 equity offering
Effective February 13, 2025, the Company sold, in a registered direct offering, an aggregate of 434,784 shares of the Company’s common stock at an offering price of $2.415 per share, and in a concurrent private placement, warrants to purchase an aggregate of 434,784 shares of common stock. The common stock warrants were immediately exercisable for a term of five years from issuance at an exercise price of $2.29 per share.
The common stock warrants and the shares of common stock underlying the common stock warrants were not registered under the Securities Act, and were issued in reliance on an exemption from the registration requirements of the Securities Act afforded by Section 4(a)(2) thereof. The shares of common stock issuable upon exercise of the common stock warrants were registered for resale in a registration statement on Form S-1 declared effective by the SEC on April 10, 2025.
The registered direct offering and the concurrent private placement generated gross proceeds of $1,050,003 before deducing the placement agent’s fee and related offering costs of $135,775, resulting in net proceeds of $914,228. Pursuant to the placement agent agreement, the Company granted the placement agent warrants to purchase 32,609 shares of common stock at an exercise price of $3.0188 per share and expiring on February 11, 2030. The net proceeds from the registered direct offering and the concurrent private placement will be used for general working capital purposes.
|
|
The exercise prices of the warrants issued to the institutional investors (exercisable at $2.29 per share) and to the placement agent (exercisable at $3.0188 per share) are subject to customary adjustments for stock splits, stock dividends, stock combinations, reclassifications, reorganizations, or similar events affecting the Company’s common stock. In addition, the warrants issued to the institutional investor and to the placement agent contain a “fundamental transaction” provision which provides that if any defined fundamental transactions are within the Company’s control and are consummated, the holder of the unexercised common stock warrants would be entitled to receive, at its option, in exchange for extinguishment of such warrants, cash consideration equal to a Black-Scholes valuation amount, as defined in the warrant agreement.
The fundamental transaction provision includes (i) a sale, lease, assignment, transfer, conveyance or other disposition of all or substantially all of the assets of the Company in one or a series of related transactions, or (ii) a change in control of the Company by which it, directly or indirectly, in one or more related transactions, consummates a stock or share purchase agreement or other business combination with another person or group, whereby such other person or group acquires more than 50% of the voting power of the common equity of the Company.
If such fundamental transaction is not within the Company’s control, including not being approved by the Company’s Board of Directors, the warrant holder would only be entitled to receive the same type or form of consideration (and in the same proportion) equal to the Black-Scholes valuation amount of the remaining unexercised portion of the warrant on the date of consummation of such fundamental transaction as the holders of the Company’s common stock receive. Accordingly, these warrants are classified as a component of permanent stockholders’ equity. The Company will account for any cash payment for a warrant redemption as a distribution from stockholders’ equity, as and when a fundamental transaction is consummated and such cash payment is required to be made.
July 2, 2025 equity offering
On July 2, 2025, the Company closed a private placement for gross proceeds of $5,050,000, consisting, in part, of shares of common stock, pre-funded warrants to purchase shares of common stock, and warrants to purchase shares of common stock (see Note 9). The pre-funded warrants were determined to be common stock equivalents.
The exercise prices of the warrants issued to the purchasers and to the placement agent are subject to customary adjustments for stock splits, stock dividends, stock combinations, reclassifications, reorganizations, or similar events affecting the Company’s common stock. In addition, the warrants issued contain a “fundamental transaction” provision whereby in the event of a fundamental transaction (including a sale or transfer of assets or ownership of the Company as defined in the warrant agreement) within the Company’s control, the holders of the unexercised common stock warrants would be entitled to receive, in exchange for extinguishment of the warrants, cash consideration equal to a Black-Scholes valuation, as defined in the warrant agreement. If such fundamental transaction is not within the Company’s control, the warrant holders would only be entitled to receive the same form of consideration (and in the same proportion) as the holders of the Company’s common stock.
Accordingly, in the event of a change in control of the Company or a sale or transfer of all or substantially all of the Company’s assets, as defined in the July 2, 2025 warrants, to the extent that the warrants are outstanding at the effective date that such a transaction is closed, this “fundamental transaction” provision would entitle the holders to substantial cash consideration, thus reducing the amounts to be retained by the Company or potentially distributable to the Company’s stockholders.
July 8, 2025 equity offering
On July 8, 2025, the Company closed a registered direct offering for gross proceeds of $1,500,000, consisting of shares of common stock and pre-funded warrants to purchase shares of common stock (see Note 9). The pre-funded warrants were determined to be common stock equivalents.
|
|
Common Stock Warrants
A summary of common stock warrant activity, including warrants to purchase common stock that were issued in conjunction with the Company’s public offerings, is presented below.
Schedule of Warrants Outstanding
| Number of Shares |
Weighted Average Exercise Price |
Weighted Average Remaining Contractual Life (in Years) |
|||||||||||
| Warrants outstanding at December 31, 2024 | 808,365 | $ | 16.407 | ||||||||||
| Issued | 467,393 | 2,341 | |||||||||||
| Exercised | — | — | |||||||||||
| Expired | — | — | |||||||||||
| Warrants outstanding at June 30, 2025 | 1,275,758 | $ | 11.254 | 3.27 | |||||||||
| Warrants exercisable at December 31, 2024 | 808,365 | $ | 16.407 | ||||||||||
| Warrants exercisable at June 30, 2025 | 1,275,758 | $ | 11.254 | 3.27 | |||||||||
At June 30, 2025, the outstanding warrants are exercisable at the following prices per common share:
Schedule of Warrants Outstanding and Exercisable
| Exercise Prices | Warrants Outstanding (Shares) |
|||||
| $ | 2.2900 | 434,784 | ||||
| $ | 3.0188 | 32,609 | ||||
| $ | 6.0000 | 583,334 | ||||
| $ | 6.6000 | 35,000 | ||||
| $ | 20.0000 | 29,000 | ||||
| $ | 37.0000 | 11,331 | ||||
| $ | 57.0000 | 149,700 | ||||
| 1,275,758 | ||||||
The warrants exercisable at $57.00 per share at June 30, 2025 consist of 1,497,000 publicly-traded warrants, described herein on a pre-split 1-for-10 basis, that were issued as part of the Company’s November 2020 public offering of units, and are exercisable for a period of five years thereafter. As a result of the 1-for-10 reverse split of the Company’s common stock effective June 2, 2023, each such publicly-traded warrant currently now represents the right to purchase 1/10th of a share of common stock at the original exercise price of $5.70 per share. Accordingly, the exercise of 10 warrants, each exercisable at $5.70, are required to acquire one share of post-split common stock, which is equivalent to a purchase price of $57.00 per share.
Based on the closing fair market value of $0.905 per common share on June 30, 2025, there was no intrinsic value attributed to exercisable but unexercised common stock warrants at June 30, 2025.
Information with respect to the issuance of common stock in connection with various stock-based compensation arrangements is provided at Note 6.
5. Related Party Transactions
Related party transactions include transactions with the Company’s officers, directors and affiliates.
|
|
Employment Agreements with Officers
During July and August 2020, the Company entered into one-year employment agreements with each of its executive officers at that time, consisting of Dr. John S. Kovach, Eric J. Forman, Dr. James S. Miser, and Robert N. Weingarten, payable monthly, as described below. These employment agreements were automatically renewable for additional one-year periods unless terminated by either party upon 60 days written notice prior to the end of the applicable one-year period, or by death, or by termination for cause. Except as noted below, these employment agreements were automatically renewed for additional one-year periods in July and August 2021, 2022, 2023 and 2024.
The Company entered into an employment agreement with Dr. Kovach dated July 15, 2020, effective October 1, 2020, to provide for Dr. Kovach to continue to act as the Company’s President, Chief Executive Officer and Chief Scientific Officer, with an annual salary of $250,000. The employment agreement with Dr. Kovach terminated upon his death on October 5, 2023.
The Company entered into an employment agreement with Dr. James S. Miser, M.D., effective August 1, 2020, to act as the Company’s Chief Medical Officer, with an annual salary of $150,000. Effective May 1, 2021, Dr. Miser’s annual salary was increased to $175,000. Dr. Miser was required to devote at least 50% of his business time to the Company’s activities. On May 29, 2024, the Company elected not to renew its employment agreement with Dr. Miser, as a result of which such employment agreement expired on July 31, 2024. During the three months and six months ended June 30, 2024, the Company paid $43,750 and $87,500, respectively, to Dr. Miser under this employment agreement, which costs are included in general and administrative costs in the Company’s consolidated statements of operations for such periods.
The Company entered into an employment agreement with Eric J. Forman effective July 15, 2020, as amended on August 12, 2020, to act as the Company’s Chief Administrative Officer, with an annual salary of $120,000. Mr. Forman is the son-in-law of Gil Schwartzberg (deceased), a former member of the Company’s Board of Directors who died on October 30, 2022 and was a significant stockholder of and consultant to the Company, and is the son of Dr. Stephen Forman, a member of the Company’s Board of Directors. Julie Forman, the wife of Mr. Forman and the daughter of Gil Schwartzberg, is Vice President of Morgan Stanley Wealth Management, at which firm the Company’s cash is on deposit and with which the Company maintains a continuing banking relationship. Effective May 1, 2021, Mr. Forman’s annual salary was increased to $175,000. Additionally, effective November 6, 2022, Mr. Forman was promoted to Vice President and Chief Operating Officer with an annual salary of $200,000. The employment agreement with Mr. Forman terminated upon his resignation as an officer of the Company effective December 31, 2024. During the three months and six months ended June 30, 2024, the Company paid $50,000 and $100,000, respectively, to Mr. Forman under this employment agreement, which costs are included in general and administrative costs in the Company’s consolidated statements of operations for such periods. Additionally, Mr. Forman was provided a monthly office rent allowance, pursuant to which the Company paid $3,780 and $9,098 during the three months and six months ended June 30, 2024.
The Company entered into an employment agreement with Robert N. Weingarten effective August 12, 2020 to act as the Company’s Vice President and Chief Financial Officer, with an annual salary of $120,000. Effective May 1, 2021, Mr. Weingarten’s annual salary was increased to $175,000. During the three months ended June 30, 2025 and 2024, the Company paid $43,750 and $43,750, respectively, to Mr. Weingarten under this employment agreement, which costs are included in general and administrative costs in the Company’s consolidated statements of operations for such periods. During the six months ended June 30, 2025 and 2024, the Company paid $87,500 and $87,500, respectively, to Mr. Weingarten under this employment agreement, which costs are included in general and administrative costs in the Company’s consolidated statements of operations for such periods.
The Company entered into an employment agreement with Bastiaan van der Baan effective September 26, 2023 to act as the Company’s President and Chief Executive Officer and as Vice Chairman of the Board of Directors, with an annual salary of $150,000. Effective October 6, 2023, Mr. van der Baan was appointed as Chairman of the Board of Directors upon the death of Dr. Kovach on October 5, 2023. Effective June 16, 2025, the employment agreement was amended to provide that Mr. van der Baan will serve as President and Chief Scientific Officer of the Company. Mr. van der Baan’s annual salary may be increased from time to time at the sole discretion of the Board of Directors. In addition, Mr. van der Baan is eligible to receive an annual bonus as determined at the sole discretion of the Board of Directors. The term of the employment agreement is for three years and is automatically renewable for additional one-year periods unless terminated by either party, subject to early termination provisions as described in the employment agreement. During the three months ended June 30, 2025 and 2024, the Company paid $39,724 and $38,163, respectively, to Mr. van der Baan under this employment agreement, which costs are included in general and administrative costs in the Company’s consolidated statements of operations for such periods. During the six months ended June 30, 2025 and 2024, the Company paid $77,201 and $76,579, respectively, to Mr. van der Baan under this employment agreement, which costs are included in general and administrative costs in the Company’s consolidated statements of operations for such periods.
|
|
On May 31, 2024, the Company entered into a consulting agreement with Dr. Jan H.M. Schellens, M.D., Ph.D. Pursuant to the agreement, effective July 1, 2024, the Company engaged Dr. Schellens as a consultant, and, effective August 1, 2024, as the Company’s Chief Medical Officer. The term of the agreement is in effect from July 1, 2024 until the earliest of (i) termination by either party upon sixty days’ notice, (ii) Dr. Schellens’ death or disability, or (iii) termination by the Company for breach as provided in the agreement. Under the agreement, Dr. Schellens provides his services for two days per week with the specific days in each week based on arrangements agreed to from time to time between Dr. Schellens and the Company’s Chief Executive Officer. The Company pays Dr. Schellens an annual compensation of 104,000 Euros (approximately $122,000 as of June 30, 2025), payable on a monthly basis. During the three months ended June 30, 2025 and 2024, the Company paid $29,811 and $0, respectively, to Dr. Schellens under this consulting agreement, which costs are included in general and administrative costs in the Company’s consolidated statements of operations for such periods. During the six months ended June 30, 2025 and 2024, the Company paid $57,315 and $0, respectively, to Dr. Schellens under this consulting agreement, which costs are included in general and administrative costs in the Company’s consolidated statements of operations for such periods. Effective as of July 31, 2025, the Company agreed to accept the resignation of Dr. Schellens and to terminate his consulting agreement, to allow Dr. Schellens to pursue other employment opportunities.
Effective as of June 15, 2022, Dr. René Bernards was appointed to the Company’s Board of Directors as an independent director. Dr. Bernards is a leader in the field of molecular carcinogenesis and is employed by the Netherlands Cancer Institute in Amsterdam. Upon his appointment, it was agreed that Dr. Bernards would receive annual compensation for his services on the Board of Directors only in the form of cash, in lieu of the annual June 30 grant of stock options as provided to the Company’s other non-officer directors. During the three months ended June 30, 2025 and 2024, the Company recorded charges to general and administrative costs in the consolidated statements of operations of $0 and $0, respectively, with respect to his annual cash board compensation. During the six months ended June 30, 2025 and 2024, the Company recorded charges to general and administrative costs in the consolidated statements of operations of $0 and $10,000, respectively, with respect to his annual cash board compensation.
In conjunction with the Company’s efforts to preserve cash during 2024, effective with the quarter ended June 30, 2024, Dr. Bernards agreed to receive equity-based compensation for his services on the Board of Directors, for the quarters ended June 30, 2024 through December 31, 2024. In order to reconcile his Board of Directors compensation with that of the other non-officer directors, Dr. Bernards has agreed to receive the same Board of Directors compensation, both in form and amount, as the other non-officer directors for the year ending December 31, 2025.
Previously, on October 8, 2021, the Company had entered into a Development Collaboration Agreement (subsequently amended and extended) with the Netherlands Cancer Institute, Amsterdam, one of the world’s leading comprehensive cancer centers, and Oncode Institute, Utrecht, a major independent cancer research center, to identify the most promising drugs to be combined with LB-100, and potentially LB-100 analogues, to be used to treat a range of cancers, as well as to identify the specific molecular mechanisms underlying the identified combinations (see Note 8).
Effective June 16, 2025, the Company entered into an employment agreement with Geordan Pursglove pursuant to which Mr. Pursglove was appointed as the Company’s Chief Executive Officer and Chairman of the Board of Directors for a term of three years, subject to automatic termination if the Company did not complete a successful financing that would enable it to maintain its listing on the Nasdaq Capital Market by July 3, 2025, which was accomplished on July 2, 2025. Under the employment agreement, Mr. Pursglove will receive an annual salary of $240,000, which may be increased from time to time in the sole discretion of the Board of Directors. At his election, Mr. Pursglove’s compensation will be payable in cash and/or restricted shares of common stock, or a combination thereof. He will also be eligible to receive an annual bonus as determined in the sole discretion of the Board of Directors in the form of cash or equity, or a combination thereof. Mr. Pursglove will not receive any additional compensation for serving as Chairman of the Board of Directors. During the three months and six months ended June 30, 2025, the Company paid $10,000 to Mr. Pursglove under this employment agreement, which cost is included in general and administrative costs in the Company’s consolidated statements of operations for such periods.
|
|
Effective as of July 3, 2025, the end of the first trading day of the Company’s common stock immediately following the successful completion of the above referenced financing, as an inducement to Mr. Pursglove to join the Company, as a signing bonus, Mr. Pursglove was granted a stock option to purchase 350,000 shares of the Company’s common stock at an exercise price of $2.83 per share (the closing market price on the grant date), for a term of five years, exercisable on a cashless basis and vesting 50% on the grant date, 25% on September 30, 2025, and 25% on December 31, 2025, subject to continued service. The stock option grant was not issued under the Company’s 2020 Stock Incentive Plan. The stock option agreement provides for certain registration rights and for accelerated vesting upon the occurrence of certain events, including early termination of the agreement that is not the result of his voluntary termination or termination for cause, a sale or change in control of the Company, or a sale, licensing or other disposition of all or substantially all of the assets of the Company. The total fair value of the stock options to purchase 350,000 shares of common stock, as calculated pursuant to the Black-Scholes option-pricing model, was determined to be $728,671 ($2.0819 per share), which will be charged to operations from July 3, 2025 through December 31, 2025.
Compensatory Arrangements for Members of the Board of Directors
Effective April 9, 2021, the Board of Directors approved a comprehensive cash and equity compensation program for the non-officer directors for their services on the Board of Directors, which was subsequently amended effective May 25, 2022, July 9, 2024, and March 21, 2025. Subsequent to June 30, 2025, the Board of Directors commenced a review of this compensation program and is considering further revisions.
Officers who also serve on the Board of Directors are not compensated separately for their service on the Board of Directors.
Cash compensation for directors, payable quarterly, is as follows:
Base director compensation - $20,000 per year (except for Dr. Bernards, who was paid an additional annual cash fee of $40,000, in lieu of the annual June 30 grant of stock option as described below, through March 31, 2024)
Chairman of audit committee – additional $10,000 per year
Chairman of any other committees – additional $5,000 per year
Member of audit committee – additional $5,000 per year
Member of any other committees – additional $2,500 per year
In conjunction with the Company’s efforts to preserve cash, the Board of Directors approved amendments to this compensation program, such that for the quarters ended June 30, 2024 through December 31, 2025, the non-officer directors (including Dr. Bernards) have received or will receive, in lieu of cash compensation, stock options exercisable for a period of five years, vesting immediately, to purchase common stock at an exercise price based on the closing market price upon issuance, with the amount of such stock options equal to the cash payment such director would otherwise have been entitled to receive for such quarter, divided by their quarterly value as determined pursuant to the Black-Scholes option-pricing model.
Equity compensation for directors is as follows:
Appointment of new directors – The Company grants options to purchase 25,000 shares of common stock, exercisable for a period of five years, at the closing market price on the date of grant, vesting 50% on the grant date and the remaining 50% vesting 12.5% on the last day of each calendar quarter beginning in the quarter immediately subsequent to the date of the grant until fully vested, subject to continued service. At the discretion of the Board of Directors, for a nominee to the Board of Directors who is restricted by their respective institution or employer from receiving equity-based compensation, in lieu of the grant of such stock options, the Company may elect to pay a one-time cash fee of $100,000 to such director, payable upfront.
|
|
Annual grant of options to directors – Effective on the last business day of the month of June, the Company grants options to purchase 10,000 shares of common stock, exercisable for a period of five years, at the closing market price on the date of grant, vesting 12.5% on the last day of each calendar quarter beginning in the quarter immediately subsequent to the date of grant until fully vested, subject to continued service. If any director has served for less than 12 full calendar months on the grant date, the amount of such stock option grant is prorated based on the length of service of such director. At the discretion of the Board of Directors, for a nominee to the Board of Directors who is restricted by their respective institution or employer from receiving equity-based compensation, in lieu of the grant of such stock options, the Company may elect to pay an annual cash fee of $40,000 to such director, payable quarterly.
Total cash compensation paid to non-officer directors was $0 and $0, respectively, for the three months ended June 30, 2025 and 2024. Total cash compensation paid to non-officer directors was $0 and $38,819, respectively, for the six months ended June 30, 2025 and 2024.
Stock-based compensation granted to members of the Company’s Board of Directors, officers and affiliates is described at Note 6.
A summary of related party costs, including compensation under employment and consulting agreements and fees paid to non-officer directors for their services on the Board of Directors, for the three months and six months ended June 30, 2025 and 2024, is presented below.
Summary of Related Party Costs
|
Three Months Ended June 30, |
Six Months Ended June 30, |
|||||||||||||||
| 2025 | 2024 | 2025 | 2024 | |||||||||||||
| Related party costs: | ||||||||||||||||
| Cash-based | $ | 123,285 | $ | 175,663 | $ | 232,016 | $ | 390,398 | ||||||||
| Stock-based | 267,999 | 130,691 | 367,737 | 233,618 | ||||||||||||
| Total | $ | 391,284 | $ | 306,354 | $ | 599,753 | $ | 624,016 | ||||||||
The Company periodically issues common stock and stock options as incentive compensation to directors and as compensation for the services of employees, contractors, and consultants of the Company.
On July 14, 2020, the Board of Directors of the Company adopted the 2020 Stock Incentive Plan (the “2020 Plan”), which was subsequently approved by the stockholders of the Company. The 2020 Plan provides for the granting of equity-based awards, consisting of stock options, restricted stock, restricted stock units, stock appreciation rights, and other stock-based awards to employees, officers, directors and consultants of the Company and its affiliates, initially for a total of 233,333 shares of the Company’s common stock, under terms and conditions as determined by the Company’s Board of Directors. On October 7, 2022, the stockholders of the Company approved an amendment to the 2020 Plan to increase the number of common shares issuable thereunder by 180,000 shares, to a total of 413,333 shares. On November 27, 2023, the stockholders of the Company approved an amendment to the 2020 Plan to increase the number of common shares issuable thereunder by 336,667 shares, to a total of 750,000 shares.
As of June 30, 2025, unexpired stock options for 699,309 shares were issued and outstanding under the 2020 Plan and 50,691 shares were available for issuance under the 2020 Plan.
|
|
The fair value of a stock option award is calculated on the grant date using the Black-Scholes option-pricing model. The risk-free interest rate is based on the U.S. Treasury yield curve in effect as of the grant date. The expected dividend yield assumption is based on the Company’s expectation of dividend payouts and is assumed to be zero. The estimated volatility is based on the historical volatility of the Company’s common stock, calculated utilizing a look-back period approximately equal to the contractual life of the stock option being granted. Unless sufficient historical exercise data is available, the expected life of the stock option is calculated as the mid-point between the vesting period and the contractual term (the “simplified method”). The fair market value of the common stock is determined by reference to the quoted market price of the common stock on the grant date.
Schedule of Fair Value of Each Option Award Estimated Assumption
| Risk-free interest rate | 3.80% to 3.950 | % | ||
| Expected dividend yield | 0 | % | ||
| Expected volatility | 128.78% to 130.70 | % | ||
| Expected life | 2.5 to 3.5 years | |||
For stock options requiring an assessment of value during the six months ended June 30, 2024, the fair value of each stock option award was estimated using the Black-Scholes option-pricing model with the following assumptions:
| Risk-free interest rate | 4.290 | % | ||
| Expected dividend yield | 0 | % | ||
| Expected volatility | 126.45 | % | ||
| Expected life | 2.5 to 3.5 years |
On June 17, 2022, the Board of Directors appointed Bas van der Baan to the Board of Directors. In connection with his appointment to the Board of Directors, and in accordance with the Company’s cash and equity compensation package for members of the Board of Directors, Mr. van der Baan was granted stock options to purchase 25,000 shares of the Company’s common stock, exercisable for a period of five years at an exercise price of $7.40 per share (the closing market price on the grant date), vesting 50% on the grant date and the remainder vesting 12.5% on the last day of each subsequent calendar quarter-end until fully vested, subject to continued service. The fair value of these stock options, as calculated pursuant to the Black-Scholes option-pricing model, was determined to be $158,525 ($6.341 per share), of which $79,263 was attributable to the portion of the stock options fully vested on June 17, 2022 and was therefore charged to operations on that date. The remaining unvested portion of the fair value of the stock options was charged to operations ratably from June 17, 2022 through June 30, 2024. During the three months and six months ended June 30, 2024, the Company recorded charges to general and administrative costs in the consolidated statement of operations of $9,695 and $19,390, respectively, with respect to these stock options.
On June 30, 2022, the Board of Directors, in accordance with the Company’s cash and equity compensation package for members of the Board of Directors, granted to each of the five non-officer directors of the Company stock options to purchase 10,000 shares (a total of 50,000 shares) of the Company’s common stock, exercisable for a period of five years at an exercise price of $7.40 per share (the closing market price on the grant date), vesting 12.5% on the last day of each subsequent calendar quarter-end until fully vested, subject to continued service. The fair value of these stock options, as calculated pursuant to the Black-Scholes option-pricing model, was determined to be $316,700 ($6.334 per share), which was charged to operations ratably from July 1, 2022 through June 30, 2024. During the three months and six months ended June 30, 2024, the Company recorded a charge to general and administrative costs in the consolidated statement of operations of $23,655 and $47,310, respectively, with respect to these stock options.
On November 6, 2022, the Board of Directors granted to each of the four officers of the Company stock options to purchase 20,000 shares (a total of 80,000 shares) of the Company’s common stock, exercisable for a period of five years at an exercise price of $20.00 per share, vesting 25% on issuance and 25% on each anniversary date thereafter until fully vested, subject to continued service. The total fair value of the 80,000 stock options, as calculated pursuant to the Black-Scholes option-pricing model, was determined to be $262,560 ($3.282 per share), which is being charged to operations ratably from November 6, 2022 through November 6, 2025. During the three months ended June 30, 2025 and 2024, the Company recorded charges to general and administrative costs in the consolidated statements of operations of $4,088 and $12,396, respectively, with respect to these stock options. During the six months ended June 30, 2025 and 2024, the Company recorded charges to general and administrative costs in the consolidated statements of operations of $8,131 and $24,660, respectively, with respect to these stock options.
|
|
On June 30, 2023, the Board of Directors, in accordance with the Company’s cash and equity compensation package for members of the Board of Directors, granted to each of the four non-officer directors of the Company stock options to purchase 10,000 shares (a total of 40,000 shares) of the Company’s common stock, exercisable for a period of five years at an exercise price of $5.88 per share (the closing market price on the grant date), vesting 12.5% on the last day of each subsequent calendar quarter-end until fully vested, subject to continued service. The fair value of these stock options, as calculated pursuant to the Black-Scholes option-pricing model, was determined to be $192,593 ($4.8131 per share), which was charged to operations ratably from July 1, 2023 through June 30, 2025. During the three months ended June 30, 2025 and 2024, the Company recorded charges to general and administrative costs in the consolidated statements of operations of $23,968 and $24,100, respectively, with respect to these stock options. During the six months ended June 30, 2025 and 2024, the Company recorded charges to general and administrative costs in the consolidated statements of operations of $47,672 and $48,068, respectively, with respect to these stock options.
On September 26, 2023, in connection with the employment agreement entered into with Bas van der Baan, Mr. van der Baan was granted a stock option to purchase 250,000 shares of the Company’s common stock. The stock option can be exercised on a cashless basis. The stock option is exercisable for a period of five years at an exercise price of $1.95 per share, which was equal to the closing market price of the Company’s common stock on the grant date. The stock option initially vested in equal increments quarterly over a three-year period commencing on the last day of each calendar quarter commencing October 1, 2023, subject to continued service. The fair value of this stock option, as calculated pursuant to the Black-Scholes option-pricing model, was determined to be $403,066 ($1.612 per share), which was being charged to operations ratably from September 26, 2023 through September 30, 2026. Effective June 16, 2025, in connection with an amendment to Mr. van der Baan’s employment agreement (see Note 5), the stock option was deemed fully vested and the remaining unamortized fair value was charged to operations on such date, and the time period for Mr. van der Baan to exercise this stock option at any time in the future that he is no longer providing services to the Company as a consultant, employee or otherwise was increased from ninety days to one year. During the three months ended June 30, 2025 and 2024, the Company recorded charges to general and administrative costs in the consolidated statements of operations of $200,805 and $33,345, respectively, with respect to this stock option. During the six months ended June 30, 2025 and 2024, the Company recorded charges to general and administrative costs in the consolidated statements of operations of $233,784 and $66,690, respectively, with respect to this stock option.
On June 30, 2024, the Board of Directors, in accordance with the Company’s cash and equity compensation package for members of the Board of Directors, granted to each of the four non-officer directors of the Company stock options to purchase 10,000 shares (a total of 40,000 shares) of the Company’s common stock, exercisable for a period of five years at an exercise price of $2.37 per share (the closing market price on the grant date), vesting 12.5% on the last day of each subsequent calendar quarter-end until fully vested, subject to continued service. The fair value of these stock options, as calculated pursuant to the Black-Scholes option-pricing model, was determined to be $73,976 ($1.8494 per share), which is being charged to operations ratably from July 1, 2024 through June 30, 2026. During the three months and six months ended June 30, 2025, the Company recorded charges to general and administrative costs in the consolidated statements of operations of $9,220 and $18,340, respectively, with respect to these stock options.
On June 30, 2024, the Board of Directors, in conjunction with the Company’s efforts to preserve cash, granted to the four non-officer directors of the Company a total of 16,598 stock options to purchase shares of the Company’s common stock, exercisable for a period of five years at an exercise price of $2.37 per share (the closing market price on the grant date) The stock options were granted in lieu of cash compensation, are exercisable for a period of five years and were immediately vested. The number of stock options granted to each of the four non-officer directors of the Company was equal to the cash payment such director would otherwise have been entitled to receive for the quarter ended June 30, 2024, divided by their grant date value as determined pursuant to the Black-Scholes option-pricing model, and was determined to be $27,500 ($1.6570 per share), which was charged to operations on June 30, 2024, the date on which the stock options were fully vested.
|
|
On July 1, 2024, in connection with the consulting agreement with Dr. Jan H.M. Schellens, M.D., Ph.D., Dr. Schellens was granted a stock option to purchase 15,000 shares of the Company’s common stock. The stock option can be exercised on a cashless basis. The stock option is exercisable for a period of five years at an exercise e price of $2.39 per share, which was equal to the closing market price of the Company’s common stock on the grant date. The stock option vested quarterly over a three-year period commencing on the last day of each calendar quarter commencing September 30, 2024. The fair value of this stock option, as calculated pursuant to the Black-Scholes option-pricing model, was determined to be $29,074 ($1.9382 per share), which is being charged to operations ratably from July 1, 2024 through June 30, 2027. During the three months and six months ended June 30, 2025, the Company recorded charges to general and administrative costs in the consolidated statements of operations of $2,418 and $4,810, respectively, with respect to this stock option. Effective as of July 31, 2025, the Company agreed to accept the resignation of Dr. Schellens and to terminate his consulting agreement.
On September 30, 2024, the Board of Directors, in conjunction with the Company’s efforts to preserve cash, granted to the four non-officer directors of the Company a total of 21,217 stock options to purchase shares of the Company’s common stock, exercisable for a period of five years at an exercise price of $1.87 per share (the closing market price on the grant date) The stock options were granted in lieu of cash compensation, are exercisable for a period of five years and were immediately vested. The number of stock options granted to each of the four non-officer directors of the Company was equal to the cash payment such director would otherwise have been entitled to receive for the quarter ended September 30, 2024, divided by their quarterly value as determined pursuant to the Black-Scholes option-pricing model, and was determined to be $27,500 ($1.2961 per share), which was charged to operations on September 30, 2024, the date on which the stock options were fully vested.
On January 20, 2025, the Board of Directors, in conjunction with the Company’s efforts to preserve cash, granted to the four non-officer directors of the Company a total of 16,665 stock options to purchase shares of the Company’s common stock, exercisable for a period of five years at an exercise price of $2.33 per share (the closing market price on the grant date) The stock options were granted in lieu of cash compensation, are exercisable for a period of five years and were immediately vested. The number of stock options granted to each of the four non-officer directors of the Company was equal to the cash payment such director would otherwise have been entitled to receive for the quarter ended December 31, 2024, divided by their grant date value as determined pursuant to the Black-Scholes option-pricing model, and was determined to be $27,500 ($1.65002 per share). The grant date value of the stock options of $27,500 was accrued at December 31, 2024 and charged to operations at that date. During the six months ended June 30, 2025, there was no expense charged to operations with respect to these stock options.
On March 31, 2025, the Board of Directors, in conjunction with the Company’s efforts to preserve cash, granted to the four non-officer directors of the Company a total of 32,181 stock options to purchase shares of the Company’s common stock, exercisable for a period of five years at an exercise price of $1.21 per share (the closing market price on the grant date) The stock options were granted in lieu of cash compensation, are exercisable for a period of five years and were immediately vested. The number of stock options granted to each of the four non-officer directors of the Company was equal to the cash payment such director would otherwise have been entitled to receive for the quarter ended March 31, 2025, divided by their grant date value as determined pursuant to the Black-Scholes option-pricing model, and was determined to be $27,500 ($0.8546 per share), which was charged to operations on March 31, 2025, the date on which the stock options were fully vested.
On June 30, 2025, the Board of Directors, in accordance with the Company’s cash and equity compensation package for members of the Board of Directors, granted to each of the four non-officer directors of the Company stock options to purchase 10,000 shares (a total of 40,000 shares) of the Company’s common stock, exercisable for a period of five years at an exercise price of $0.905 per share (the closing market price on the grant date), vesting 12.5% on the last day of each subsequent calendar quarter-end until fully vested, subject to continued service. The fair value of these stock options, as calculated pursuant to the Black-Scholes option-pricing model, was determined to be $28,700 ($0.7175 per share), which is being charged to operations ratably from July 1, 2025 through June 30, 2027. During the three months and six months ended June 30, 2025, the Company did not record a charge to operations with respect to these stock options.
|
|
On June 30, 2025, the Board of Directors, in conjunction with the Company’s efforts to preserve cash, granted to the four non-officer directors of the Company a total of 42,648 stock options to purchase shares of the Company’s common stock, exercisable for a period of five years at an exercise price of $0.905 per share (the closing market price on the grant date) The stock options were granted in lieu of cash compensation, are exercisable for a period of five years and were immediately vested. The number of stock options granted to each of the four non-officer directors of the Company was equal to the cash payment such director would otherwise have been entitled to receive for the quarter ended June 30, 2025, divided by their grant date value as determined pursuant to the Black-Scholes option-pricing model, and was determined to be $27,500 ($0.6448 per share), which was charged to operations on June 30, 2025, the date on which the stock options were fully vested.
Gil Schwartzberg, a former director of the Company, died on October 30, 2022. Dr. John S. Kovach, the Chairman of the Board of Directors and the Company’s President and Chief Executive Officer, and Chief Scientific Officer, died on October 5, 2023, the employment agreement of the Company’s Chief Medical Officer, Dr. James S. Miser expired on July 31, 2024, the employment agreement of the Company’s Vice President and Chief Operating Officer, Eric J. Forman, terminated upon his resignation from the Company on December 31, 2024, and the consulting agreement of the Company’s Chief Medical Officer, Dr. Jan Schellens, was terminated effective with his resignation on July 31, 2025. Accordingly, the unvested stock options for each such person ceased vesting effective as of the respective dates that their services to the Company terminated. Furthermore, the expiration date of all vested stock options owned by each such person contractually expire one year from the respective dates that their services to the Company terminated.
Summary of Stock-based Compensation Costs
| Three Months Ended | Six Months Ended | |||||||||||||||
| June 30, | June 30, | |||||||||||||||
| 2025 | 2024 | 2025 | 2024 | |||||||||||||
| Related parties | $ | 267,999 | $ | 130,691 | $ | 367,737 | $ | 233,618 | ||||||||
| Non-related parties | — | — | — | — | ||||||||||||
| Total stock-based compensation costs | $ | 267,999 | $ | 130,691 | $ | 367,737 | $ | 233,618 | ||||||||
Summary of Stock Option Activity Including Options Form of Warrants
| Number of Shares |
Weighted Average Exercise Price |
Weighted Average Remaining Contractual Life (in Years) |
||||||||||
| Stock options outstanding at December 31, 2024 | 613,232 | $ | 12.317 | |||||||||
| Granted | 131,494 | 1.160 | ||||||||||
| Exercised | — | — | ||||||||||
| Expired | — | — | ||||||||||
| Stock options outstanding at June 30, 2025 | 744,726 | $ | 10.347 | 2.99 | ||||||||
| Stock options exercisable at December 31, 2024 | 409,897 | $ | 17.100 | |||||||||
| Stock options exercisable at June 30, 2025 | 669,726 | $ | 11.196 | 2.87 | ||||||||
Total deferred compensation expense for the outstanding value of unvested stock options was approximately $91,000 at June 30, 2025, which will be recognized subsequent to June 30, 2025 over a weighted-average period of approximately 18 months.
|
|
Schedule of Exercise Prices of Common Stock Options Outstanding and Exercisable Including Options Form of Warrants
| Exercise Prices | Options Outstanding (Shares) |
Options Exercisable (Shares) |
||||||||
| $ | 0.905 | 82,648 | 42,648 | |||||||
| $ | 1.210 | 32,181 | 32,181 | |||||||
| $ | 1.870 | 21,217 | 21,217 | |||||||
| $ | 1.950 | 250,000 | 250,000 | |||||||
| $ | 2.330 | 16,665 | 16,665 | |||||||
| $ | 2.370 | 56,598 | 36,598 | |||||||
| $ | 2.390 | 15,000 | 5,000 | |||||||
| $ | 5.025 | 8,750 | 8,750 | |||||||
| $ | 5.880 | 40,000 | 40,000 | |||||||
| $ | 7.400 | 55,000 | 55,000 | |||||||
| $ | 20.000 | 45,000 | 40,000 | |||||||
| $ | 20.600 | 20,000 | 20,000 | |||||||
| $ | 28.000 | 25,000 | 25,000 | |||||||
| $ | 30.300 | 30,000 | 30,000 | |||||||
| $ | 32.100 | 10,000 | 10,000 | |||||||
| $ | 60.000 | 8,333 | 8,333 | |||||||
| $ | 71.400 | 20,000 | 20,000 | |||||||
| $ | 120.000 | 8,334 | 8,334 | |||||||
| 744,726 | 669,726 | |||||||||
Based on the closing fair market value of $0.905 per common share on June 30, 2025, there was no intrinsic value attributed to exercisable but unexercised common stock options at June 30, 2025.
Outstanding stock options to acquire 75,000 shares of the Company’s common stock had not vested at June 30, 2025.
Upon the exercise of such stock options, the Company expects to satisfy the related stock obligations through the issuance of authorized but unissued shares of common stock.
7. Income Taxes
During the three months and six months ended June 30, 2025 and 2024, the Company did not record any provision for income taxes, as the Company incurred losses during such periods. Deferred tax assets and liabilities reflect the net tax effect of temporary differences between the carrying amount of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. The Company has recorded a full valuation allowance against its deferred tax assets for all periods presented as the Company currently believes it is more likely than not that the deferred tax assets will not be realized.
8. Commitments and Contingencies
Legal Claims
The Company may be subject to legal claims and actions from time to time as part of its business activities. As of June 30, 2025 and December 31, 2024, the Company was not subject to any threatened or pending lawsuits, legal claims or legal proceedings.
|
|
Principal Commitments
Clinical Trial Agreements
At June 30, 2025, the Company’s remaining financial contractual commitments pursuant to clinical trial agreements and clinical trial monitoring agreements not yet incurred, as described below, aggregated $524,000, including clinical trial agreements of $293,000 and clinical trial monitoring agreements of $231,000, which, based on current estimates, are currently scheduled to be incurred through approximately December 31, 2027. The Company’s ability to conduct and fund these contractual commitments is subject to the timely availability of sufficient capital to fund such expenditures, as well as any changes in the allocation or reallocation of such funds to the Company’s current or future clinical trial programs. The Company expects that the full amount of these expenditures will be incurred only if such clinical trial programs are conducted as originally designed and their respective enrollments and duration are not modified or reduced. Clinical trial programs, such as the types that the Company is engaged in, can be highly variable and can frequently involve a series of changes and modifications over time as clinical data is obtained and analyzed, and is frequently modified, suspended or terminated, in part based on receipt or lack of receipt of an indication of clinical benefit or activity, before the clinical trial endpoint is reached. Accordingly, such contractual commitments as discussed herein should be considered as estimates only based on current clinical assumptions and conditions and are typically subject to significant modifications and revisions over time.
The following is a summary of the Company’s ongoing active contractual clinical trials described below as of June 30, 2025:
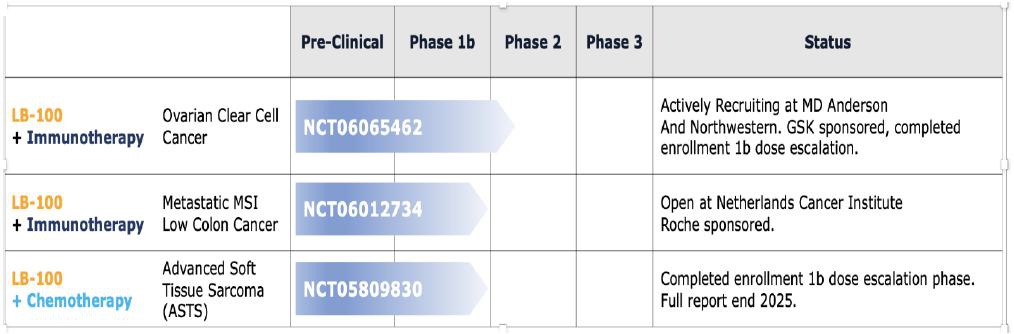
Schedule of Contractual Clinical Trials
| Description of Clinical Trial | Institution | Start Date | Projected End Date | Planned Number of Patients in Trial | Study Objective | Clinical Update | Expected Date of Preliminary Efficacy Signal | NCT No. | Remaining Financial Contractual Commitment | |||||||||||||||||||||||||||
| LB-100 combined with dostarlimab in ovarian clear cell carcinoma (Phase 1b/2) | MD Anderson | January 2024 | December 2027 | 21 | Determine the OS of patients with recurrent ovarian clear cell carcinoma | 16 patients entered | December 2026 | NCT06065462 | $ | -0- (1 ) | ||||||||||||||||||||||||||
| LB-100 combined with atezolizumab in microsatellite stable metastatic colorectal cancer (Phase 1b) | Netherlands Cancer Institute (NKI) | August 2024 | December 2026 | 37 | Determine RP2D with atezolizumab | First patient entered August 2024, in total two patients entered | June 2026 | NCT06012734 | -0- (1 ) | |||||||||||||||||||||||||||
| LB-100 combined with doxorubicin in advanced soft tissue sarcoma (Phase 1b) | GEIS | June 2023 | Recruitment completed September 2024 | 14 | Determine MTD and RP2D | Fourteen patients entered | December 2025 | NCT05809830 | 293,000 | |||||||||||||||||||||||||||
| Total | $ | 293,000 | ||||||||||||||||||||||||||||||||||
| (1) | The Company has no financial contractual commitments associated with these clinical trials at June 30, 2025. |
|
|
Netherlands Cancer Institute. Effective June 10, 2024, the Company entered into a Clinical Trial Agreement with the Netherlands Cancer Institute (“NKI”) (see Note 5) to conduct a Phase 1b clinical trial of the Company’s protein phosphatase inhibitor, LB-100, combined with atezolizumab, a PD-L1 inhibitor, the proprietary molecule of F. Hoffman-La Roche Ltd. (“Roche”), for patients with microsatellite stable metastatic colorectal cancer. Under the agreement, the Company will provide its lead compound, LB-100, and under a separate agreement between NKI and Roche, Roche will provide atezolizumab and financial support for the clinical trial. The Company has no obligation to and will not provide any reimbursement of clinical trial costs. Pursuant to the agreement and the protocol set forth in the agreement, the clinical trial will be conducted by NKI at NKI’s site in Amsterdam by principal investigator Neeltje Steeghs, MD, PhD, and NKI will be responsible for the recruitment of patients. The agreement provides for the protection of the respective intellectual property rights of each of the Company, NKI and Roche.
This Phase 1b clinical trial will evaluate safety, optimal dose and preliminary efficacy of LB-100 combined with atezolizumab for the treatment of patients with metastatic microsatellite stable colorectal cancer. Immunotherapy using monoclonal antibodies like atezolizumab can enhance the body’s immune response against cancer and hinder tumor growth and spread. LB-100 has been found to improve the effectiveness of anticancer drugs in killing cancer cells by inhibiting a protein called PP2A on cell surfaces. Blocking PP2A increases stress signals in tumor cells expressing the PP2A protein. Accordingly, combining atezolizumab with LB-100 may enhance treatment efficacy for metastatic colorectal cancer, as cancer cells with heightened stress signals are more vulnerable to immunotherapy.
This study comprises a dose escalation phase and a dose expansion phase. The objective of the dose escalation phase is to determine the recommended Phase 2 dose (RP2D) of LB-100 when combined with the standard dosage of atezolizumab. The dose expansion phase will further investigate the preliminary efficacy, safety, tolerability, and pharmacokinetics/dynamics of the LB-100 and atezolizumab combination. The clinical trial opened in August 2024 with the enrollment of the first patient. A total of two patients have been enrolled to date. Patient accrual is expected to take up to 24 months, with a maximum of 37 patients with advanced colorectal cancer to be enrolled in this study.
The principal investigator of the colorectal study testing LB-100 in combination with atezolizumab is currently investigating two Serious Adverse Events (“SAEs”) observed in the clinical trial. The Investigational Review Board (IRB) of NKI has requested additional information with respect to these SAEs and the study has been paused for enrollment until the IRB’s questions have been satisfactorily addressed (see “Specific Risks Associated with the Company’s Business Activities - Serious Adverse Events” below for additional information).
The Company has no financial contractual commitment associated with this clinical trial.
|
|
City of Hope. Effective January 18, 2021, the Company executed a Clinical Research Support Agreement (the “Agreement”) with the City of Hope National Medical Center, an NCI-designated comprehensive cancer center, and City of Hope Medical Foundation (collectively, “City of Hope”), to carry out a Phase 1b clinical trial of LB-100, the Company’s first-in-class protein phosphatase inhibitor, combined with an FDA-approved standard regimen for treatment of untreated extensive-stage disease small cell lung cancer (“ED-SCLC”). LB-100 was given in combination with carboplatin, etoposide and atezolizumab, an FDA-approved standard of care regimen, to previously untreated ED-SCLC patients. The LB-100 dose was to be escalated with the standard fixed doses of the 3-drug regimen to reach a recommended Phase 2 dose (“RP2D”). Patient entry was to be expanded so that a total of 12 patients would be evaluable at the RP2D to determine the safety of the LB-100 combination and to look for potential therapeutic activity as assessed by objective response rate, duration of overall response, progression-free survival, and overall survival.
The clinical trial was initiated on March 9, 2021, with patient accrual expected to take approximately two years to complete. Because patient accrual was slower than expected, effective March 6, 2023, the Company and City of Hope added the Sarah Cannon Research Institute (“SCRI”), Nashville, Tennessee, to the ongoing Phase 1b clinical trial. The Company and City of Hope continued efforts to increase patient accrual by adding additional sites and by modifying the protocol to increase the number of patients eligible for the clinical trial. The impact of these efforts to increase patient accrual and to decrease time to completion was evaluated in subsequent quarters.
After evaluating patient accrual through June 30, 2024, the Company and City of Hope agreed to close the clinical trial. Pursuant to the terms of the Agreement, the Company provided notice to City of Hope of the Company’s intent to terminate the Agreement effective as of July 8, 2024. Upon closure, the Company incurred a prorated charge of $207,004 for the cost of patients enrolled to date, which is included in accounts payable and accrued expenses at June 30, 2025 and December 31, 2024.
During the three months ended June 30, 2025 and 2024, the Company incurred costs of $0 and $78,015, respectively, pursuant to this Agreement. During the six months ended June 30, 2025 and 2024, the Company incurred costs of $0 and $78,015, respectively, pursuant to this Agreement. As of June 30, 2025, total costs of $732,532 had been incurred pursuant to this Agreement.
GEIS. Effective July 31, 2019, the Company entered into a Collaboration Agreement for an Investigator-Initiated Clinical Trial with the Spanish Sarcoma Group (Grupo Español de Investigación en Sarcomas or “GEIS”), Madrid, Spain, to carry out a study entitled “Randomized phase I/II trial of LB-100 plus doxorubicin vs. doxorubicin alone in first line of advanced soft tissue sarcoma”. The purpose of this clinical trial is to obtain information with respect to the efficacy and safety of LB-100 combined with doxorubicin in soft tissue sarcomas. Doxorubicin is the global standard for initial treatment of advanced soft tissue sarcomas (“ASTS”). Doxorubicin alone has been the mainstay of first line treatment of ASTS for over 40 years, with little improvement in survival from adding cytotoxic compounds to or substituting other cytotoxic compounds for doxorubicin. In animal models, LB-100 has consistently enhanced the anti-tumor activity of doxorubicin without apparent increases in toxicity.
GEIS has a network of referral centers in Spain and across Europe that have an impressive track record of efficiently conducting innovative studies in ASTS. The Company agreed to provide GEIS with a supply of LB-100 to be utilized in the conduct of this clinical trial, as well as to provide funding for the clinical trial. The goal is to enter approximately 150 to 170 patients in this clinical trial over a period of two to four years. The Phase 1 portion of the study began in the quarter ended June 30, 2023 to determine the recommended Phase 2 dose of the combination of doxorubicin and LB-100. As advanced sarcoma is a very aggressive disease, the design of the Phase 2 portion of the study assumes a median progression-free survival (“PFS”), no evidence of disease progression or death from any cause, of 4.5 months in the doxorubicin arm and an alternative median PFS of 7.5 months in the doxorubicin plus LB-100 arm to demonstrate a statistically significant decrease in relative risk of progression or death by adding LB-100. There is a planned interim analysis of the primary endpoint when approximately 50% of the 102 events required for final analysis is reached.
The Company had previously expected that this clinical trial would commence during the quarter ended June 30, 2020. However, during July 2020, the Spanish regulatory authority advised the Company that although it had approved the scientific and ethical basis of the protocol, it required that the Company manufacture new inventory of LB-100 under current Spanish pharmaceutical manufacturing standards. These standards were adopted subsequent to the production of the Company’s existing LB-100 inventory.
|
|
In order to manufacture a new inventory supply of LB-100 for the GEIS clinical trial, the Company engaged a number of vendors to carry out the multiple tasks needed to make and gain approval of a new clinical product for investigational study in Spain. These tasks included the synthesis under good manufacturing practice (GMP) of the active pharmaceutical ingredient (API), with documentation of each of the steps involved by an independent auditor. The API was then transferred to a vendor that prepares the clinical drug product, also under GMP conditions documented by an independent auditor. The clinical drug product was then sent to a vendor to test for purity and sterility, provide appropriate labels, store the drug, and distribute the drug to the clinical centers for use in the clinical trials. A formal application documenting all steps taken to prepare the clinical drug product for clinical use was submitted to the appropriate regulatory authorities for review and approval before being used in a clinical trial.
As of June 30, 2025, this program to provide new inventory of the clinical drug product for the Spanish Sarcoma Group study, and potentially for subsequent multiple trials within the European Union, had cost approximately $1,144,000.
On October 13, 2022, the Company announced that the Spanish Agency for Medicines and Health Products (Agencia Española de Medicamentos y Productos Sanitarios or “AEMPS”) had authorized a Phase 1b/randomized Phase 2 study of LB-100, the Company’s lead clinical compound, plus doxorubicin, versus doxorubicin alone, the global standard for initial treatment of ASTS. Consequently, this clinical trial commenced during the quarter ended June 30, 2023 and is expected to be completed and a report prepared by December 31, 2026. In April 2023, GEIS completed its first site initiation visit in preparation for the clinical trial at Fundación Jiménez Díaz University Hospital (Madrid). Up to 170 patents will be entered into the clinical trial. The recruitment for the Phase 1b portion of the protocol was extended with two patients and was completed during the quarter ended September 30, 2024. The Company expects to have data on toxicity and preliminary efficacy from this portion of the clinical trial during the quarter ending December 31, 2025.
Given the focus on the combination of LB-100 with immunotherapy in ovarian clear cell carcinoma and colorectal cancer and the availability of capital resources, the Company entered into Amendment No. 1 to the Collaboration Agreement effective March 11, 2025 that relieved the Company of the financial obligation to support the randomized Phase 2 portion of the clinical trial contemplated in the Collaboration Agreement of approximately $3,095,000. As a result, it is uncertain as to whether the Phase 2 portion of this clinical trial will proceed.
The Company’s agreement with GEIS provided for various payments based on achieving specific milestones over the term of the agreement. During the three months ended June 30, 2025 and 2024, the Company did not incur any costs pursuant to this agreement. During the six months ended June 30, 2025 and 2024, the Company did not incur any costs pursuant to this agreement. Through June 30, 2025, the Company has incurred charges of $685,107 for work done under this agreement through the fourth milestone.
The Company’s aggregate commitment pursuant to this agreement, less amounts previously paid to date, totaled approximately $293,000 for the Phase 1b portion of this clinical trial as of June 30, 2025, which is scheduled to be incurred through December 31, 2025. As the work is being conducted in Europe and is paid for in Euros, final costs are subject to foreign currency fluctuations between the United States Dollar and the Euro. Such fluctuations are recorded in the consolidated statements of operations as foreign currency gain or loss, as appropriate, and have not been significant.
MD Anderson Cancer Center Clinical Trial. On September 20, 2023, the Company announced an investigator-initiated Phase 1b/2 collaborative clinical trial to assess whether adding LB-100 to a human programmed death receptor-1 (“PD-1”) blocking antibody of GSK plc (“GSK”), dostarlimab-gxly, may enhance the effectiveness of immunotherapy in the treatment of ovarian clear cell carcinoma (“OCCC”). The study objective is to determine the overall survival (“OS”) of patients with OCCC. The clinical trial is being sponsored by The University of Texas MD Anderson Cancer Center (“MD Anderson”) and is being conducted at The University of Texas - MD Anderson Cancer Center. The Company is providing LB-100 and GSK is providing dostarlimab-gxly and financial support for the clinical trial. On January 29, 2024, the Company announced the entry of the first patient into this clinical trial. The Company currently expects that this clinical trial will be completed by December 31, 2027.
On February 25, 2025, the Company announced that it has added the Robert H. Lurie Comprehensive Cancer Center (Lurie Cancer Center) of Northwestern University as a second site in a clinical trial combining the Company’s proprietary compound LB-100 with GSK’s dostarlimab to treat ovarian clear cell cancer. Patient recruitment is underway, and the first patient has been dosed.
|
|
Clinical Trial Monitoring Agreements
MD Anderson Cancer Center Clinical Trial. On May 15, 2024, the Company signed a letter of intent with Theradex to monitor the MD Andersen investigator-initiated Phase 1b/2 collaborative clinical trial to assess whether adding LB-100 to a human programmed death receptor-1 (“PD-1”) blocking antibody of GSK plc (“GSK”), dostarlimab-gxly, may enhance the effectiveness of immunotherapy in the treatment of ovarian clear cell carcinoma (“OCCC”). On August 19, 2024, the Company signed a work order agreement with Theradex to monitor the MD Anderson clinical trial. The study oversight is expected to be completed by January 31, 2027.
Costs under this letter of intent and related work order agreement are estimated to be approximately $95,000. During the three months ended June 30, 2025 and 2024, the Company incurred costs of $4,614 and $8,228 pursuant to this letter of intent and subsequent work order. During the six months ended June 30, 2025 and 2024, the Company incurred costs of $11,892 and $8,228 pursuant to this letter of intent and subsequent work order. As of June 30, 2025, total costs of $38,655 have been incurred pursuant to this letter of intent and subsequent work order.
The Company’s aggregate commitment pursuant to this letter of intent, less amounts previously paid to date, totaled approximately $57,000 as of June 30, 2025, which is expected to be incurred through December 31, 2027.
City of Hope. On February 5, 2021, the Company signed a new work order agreement with Theradex to monitor the City of Hope investigator-initiated clinical trial in small cell lung cancer in accordance with FDA requirements for oversight by the sponsoring party. Costs under this work order agreement were estimated to be approximately $335,000. During the three months ended June 30, 2025 and 2024, the Company incurred costs of $0 and $4,500, respectively, pursuant to this work order. During the six months ended June 30, 2025 and 2024, the Company incurred costs of $0 and $9,000, respectively, pursuant to this work order. As of June 30, 2025, total costs of $87,823 had been incurred pursuant to this work order agreement.
As a result of the closure of the Agreement with City of Hope effective July 8, 2024 (see “Clinical Trial Agreements – City of Hope” above), the work order agreement with Theradex to monitor this clinical trial was concurrently terminated, although nominal oversight trailing costs subsequent to July 8, 2024 are expected to be incurred relating to the closure of this study.
GEIS. On June 22, 2023, the Company finalized a work order agreement with Theradex, to monitor the GEIS investigator-initiated clinical Phase I/II randomized trial of LB-100 plus doxorubicin vs. doxorubicin alone in first line of advanced soft tissue sarcoma. The study oversight is expected to be completed by December 31, 2026.
Costs under this work order agreement are estimated to be approximately $153,000, with such payments expected to be allocated approximately 72% to Theradex for services and approximately 28% for payments for pass-through software costs. During the three months ended June 30, 2025 and 2024, the Company incurred costs of $3,750 and $7,203, respectively, pursuant to this work order. During the six months ended June 30, 2025 and 2024, the Company incurred costs of $7,622 and $12,732, respectively, pursuant to this work order. As of June 30, 2025, total costs of $57,077 have been incurred pursuant to this work order agreement.
The Company’s aggregate commitment pursuant to this clinical trial monitoring agreement, less amounts previously paid to date, totaled approximately $95,000 as of June 30, 2025, which is expected to be incurred through December 31, 2026.
Netherlands Cancer Institute. On August 27, 2024, the Company finalized a work order agreement with Theradex, to monitor the NKI Phase 1b clinical trial of LB-100 combined with atezolizumab, a PD-L1 inhibitor, for patients with microsatellite stable metastatic colorectal cancer. The study oversight was expected to be completed by May 31, 2027.
|
|
Costs under this work order agreement were estimated to be approximately $106,380, with such payments expected to be allocated approximately 47% to Theradex for services and approximately 53% for payments for pass-through software costs. During three months and six months ended June 30, 2025, the Company incurred costs of $4,500 and $9,000, respectively, pursuant to this work order. As of June 30, 2025, total costs of $29,191 have been incurred pursuant to this work order agreement.
The Company’s aggregate commitment pursuant to this clinical trial monitoring agreement, less amounts previously paid to date, totaled approximately $79,000 as of June 30, 2025, which was expected to be incurred through May 31, 2027.
The Company was recently notified that the preparations for this clinical trial were suspended and the clinical trial is not expected commence. Accordingly, the Company expects that this agreement will be terminated and the Company will have no further financial commitment or cost.
Patent and License Agreements
National Institute of Health. Effective February 23, 2024, the Company entered into a Patent License Agreement (the “License Agreement”) with the National Institute of Neurological Disorders and Stroke (“NINDS”) and the National Cancer Institute (“NCI”), each an institute or center of the National Institute of Health (“NIH”). Pursuant to the License Agreement, the Company has licensed on an exclusive basis the NIH’s intellectual property rights claimed for a Cooperative Research and Development Agreement (“CRADA”) subject invention co-developed with the Company, and the licensed field of use, which focuses on promoting anti-cancer activity alone, or in combination with standard anti-cancer drugs. The scope of this clinical research extends to checkpoint inhibitors, immunotherapy, and radiation for the treatment of cancer. The License Agreement is effective, and shall extend, on a licensed product, licensed process, and country basis, until the expiration of the last-to-expire valid claim of the jointly owned licensed patent rights in each such country in the licensed territory, estimated at twenty years, unless sooner terminated.
The License Agreement contemplates that the Company will seek to work with pharmaceutical companies and clinical trial sites (including comprehensive cancer centers) to initiate clinical trials within timeframes that will meet certain benchmarks. Data from the clinical trials will be the subject of various regulatory filings for marketing approval in applicable countries in the licensed territories. Subject to the receipt of marketing approval, the Company would be expected to commercialize the licensed products in markets where regulatory approval has been obtained.
The Company is obligated to pay the NIH a non-creditable, non-refundable license issue royalty of $50,000 and a first minimum annual royalty within sixty days from the effective date of the Agreement. The first minimum annual royalty of $25,643 was prorated from the effective date of the License Agreement to the next subsequent January 1. Thereafter, the minimum annual royalty of $30,000 is due each January 1 and may be credited against any earned royalties due for sales made in that year. The license issue royalty of $50,000 and the first minimum annual royalty of $25,643 were paid in April 2024. The second minimum annual royalty for 2025 of $30,000 was paid in December 2024 and was included in other prepaid expenses in the consolidated balance sheet at December 31, 2024.
The Company is obligated to pay the NIH, on a country-by-country basis, earned royalties of 2% on net sales of each royalty-bearing product and process, subject to reduction by 50% under certain circumstances relating to royalties paid by the Company to third parties, but not less than 1%. The Company’s obligation to pay earned royalties under the License Agreement commences on the date of the first commercial sale of a royalty-bearing product or process and expires on the date on which the last valid claim of the licensed product or licensed process expires in such country.
The Company is obligated to pay the NIH benchmark royalties, on a one-time basis, within sixty days from the first achievement of each such benchmark. The License Agreement defines four such benchmarks, which the Company is required to pursue based on “commercially reasonable efforts” as defined in the License Agreement, with deadlines of October 1, 2024, 2027, 2029 and 2031, each with a different specified benchmark payment amount payable within thirty days of achieving such benchmark. The October 1, 2024 benchmark of $100,000 was defined as the dosing of the first patient with a licensed product in a Phase 2 clinical study of such licensed product in the licensed fields of use. The Company had not commenced a Phase 2 clinical study as of June 30, 2025. The total of all such benchmark payments is $1,225,000.
|
|
The Company is obligated to provide annual reports to the NIH on its progress toward the development and commercialization of products under the licensed patents. These reports, due within sixty days following the end of each calendar year, must include updates on research and development activities, regulatory submissions, manufacturing efforts, sublicensing, and sales initiatives. If any deviations from the established commercial development plan or agreed-upon benchmarks occur, the Company is obligated to provide explanation and may amend the commercial development plan and the benchmarks, which, subject to certain conditions, the NIH shall not unreasonably withhold, condition, or delay approval of any request of the Company to amend the commercial development plan and/or the benchmarks and to extend the time periods of the benchmarks.
The Company is obligated to pay the NIH sublicensing royalties of 5% on sublicensing revenue received for granting each sublicense within sixty days of receipt of such sublicensing revenue.
During the three months ended June 30, 2025 and 2024, the Company incurred costs of $7,397 and $7,455, respectively, in connection with its obligations under the License Agreement. During the six months ended June 30, 2025 and 2024, the Company incurred costs of $14,794 and $60,569, respectively, in connection with its obligations under the License Agreement. Such costs when incurred have been included in general and administrative costs in the Company’s consolidated statement of operations. As of June 30, 2025, total costs of $90,438 have been incurred pursuant to this agreement. The Company’s aggregate commitment pursuant to this agreement, less amounts previously paid to date, totaled approximately $1,765,000 as of June 30, 2025, which is expected to be incurred over approximately the next twenty years.
Other Significant Agreements and Contracts
NDA Consulting Corp. On December 24, 2013, the Company entered into a consulting agreement with NDA Consulting Corp. for consultation and advice in the field of oncology research and drug development. As part of the consulting agreement, NDA also agreed to have its president, Dr. Daniel D. Von Hoff, M.D., serve on the Company’s Scientific Advisory Committee during the term of such consulting agreement. The term of the consulting agreement was for one year and provided for a quarterly cash fee of $4,000. The consulting agreement had been automatically renewed for additional one-year terms on its anniversary date, most recently on December 24, 2023, but was subsequently terminated by mutual agreement effective September 30, 2024. Consulting and advisory fees charged to operations pursuant to this consulting agreement were $4,000 and $8,000 for the three months and six months ended June 30, 2024, respectively.
BioPharmaWorks. Effective September 14, 2015, the Company entered into a Collaboration Agreement with BioPharmaWorks, pursuant to which the Company engaged BioPharmaWorks to perform certain services for the Company. Those services included, among other things, assisting the Company to commercialize its products and strengthen its patent portfolio; identifying large pharmaceutical companies with a potential interest in the Company’s product pipeline; assisting in preparing technical presentations concerning the Company’s products; consultation in drug discovery and development; and identifying providers and overseeing tasks relating to clinical development of new compounds.
BioPharmaWorks was founded in 2015 by former Pfizer scientists with extensive multi-disciplinary research and development and drug development experience. The Collaboration Agreement was for an initial term of two years and automatically renews for subsequent annual periods unless terminated by a party not less than 60 days prior to the expiration of the applicable period. In connection with the Collaboration Agreement, the Company agreed to pay BioPharmaWorks a monthly fee of $10,000, subject to the right of the Company to pay a negotiated hourly rate in lieu of the monthly fee. Effective March 1, 2024, the compensation payable under the Collaboration Agreement was converted to an hourly rate structure.
The Company recorded charges to operations pursuant to this Collaboration Agreement of $10,800 and $7,200 during the three months ended June 30, 2025 and 2024, respectively, which were included in research and development costs in the consolidated statements of operations. The Company recorded charges to operations pursuant to this Collaboration Agreement of $24,800 and $27,200 during the six months ended June 30, 2025 and 2024, respectively, which were included in research and development costs in the consolidated statements of operations.
|
|
Netherlands Cancer Institute. On October 8, 2021, the Company entered into a Development Collaboration Agreement with the Netherlands Cancer Institute, Amsterdam (“NKI”) (see Note 5), one of the world’s leading comprehensive cancer centers, and Oncode Institute, Utrecht, a major independent cancer research center, for a term of three years. The Development Collaboration Agreement was subsequently modified by Amendment No. 1 thereto.
The Development Collaboration Agreement is a preclinical study intended to identify the most promising drugs to be combined with LB-100, and potentially LB-100 analogues, to be used to treat a range of cancers, as well as to identify the specific molecular mechanisms underlying the identified combinations. The Company agreed to fund the preclinical study, at an approximate cost of 391,000 Euros and provide a sufficient supply of LB-100 to conduct the preclinical study.
On October 3, 2023, the Company entered into Amendment No. 2 to the Development Collaboration Agreement with NKI, which provides for additional research activities, extends the termination date of the Development Collaboration Agreement by two years to October 8, 2026, and added 500,000 Euros to the operating budget being funded by the Company.
On October 4, 2024, the Company entered into Amendment No. 3 to the Development Collaboration Agreement with NKI, which suspended Amendment No. 2 and provided for a new study term of one year commencing upon the dosing of the first patient in the trial at a project cost of 100,000 Euros.
During the three months ended June 30, 2025 and 2024, the Company incurred charges of $0 and $67,119, respectively, with respect to this agreement, which amounts are included in research and development costs in the Company’s consolidated statements of operations. During the six months ended June 30, 2025 and 2024, the Company incurred charges of $0 and $134,084, respectively, with respect to this agreement, which amounts are included in research and development costs in the Company’s consolidated statements of operations. As of June 30, 2025, total costs of $695,918 have been incurred pursuant to this agreement.
The Company was recently notified that the preparations for this clinical trial were suspended and the clinical trial is not expected commence. Accordingly, the Company expects that this agreement will be terminated and the Company will have no further financial commitment or cost.
MRI Global. As amended, the Company has contracted with MRI Global for stability analysis, storage and distribution of LB-100 for clinical trials in the United States. During the three months ended June 30, 2025 and 2024, the Company incurred costs of $6,765 and $5,976, respectively, pursuant to this contract. During the six months ended June 30, 2025 and 2024, the Company incurred costs of $34,857 and $9,870, respectively, pursuant to this contract. As of June 30, 2025, total costs of $375,379 have been incurred pursuant to this contract.
The Company’s aggregate commitment pursuant to this contract, less amounts previously paid to date, totaled approximately $90,000 as of June 30, 2025.
Specific Risks Associated with the Company’s Business Activities
Serious Adverse Events
The Company’s lead drug candidate, LB-100, is currently undergoing various clinical trials, and there is a risk that one or more of these trials could be placed on hold by regulatory authorities due to serious adverse events (SAEs) related to the Company’s drug candidate or to another company’s drug used in combination in one of the Company’s clinical trials. It is possible that the SAEs could be attributable to the Company’s drug candidate and could include, but not be limited to, unexpected severe side effects, treatment-related deaths, or long-term health complications. A dose given could result in non-tolerable adverse events defined as dose-limiting toxicity (DLT). When two DLTs occur at the same dose-level that dose-level is considered too high and unsafe. Further treatment is only allowed at lower dose-levels that have previously been found safe.
|
|
If an SAE or a pattern of SAEs is observed during the course of a clinical trial involving the Company’s drug candidate, the U.S. Food and Drug Administration (FDA), European Medicines Agency (EMA), or other regulatory authorities may issue a clinical hold, requiring the Company to pause or discontinue further enrollment and dosing in the Company’s clinical trial. It is also possible that the clinical trial could be terminated. Any of these actions could delay or halt the development of the Company’s drug candidate, increase development costs, and negatively impact the Company’s ability to ultimately achieve regulatory approval. Additionally, if an SAE is confirmed to be drug-related, the Company may be required to conduct additional studies, modify the study design, or abandon further development of the drug candidate altogether, which could materially impact the Company’s business, financial condition, and prospects.
The occurrence of an SAE and any resulting clinical hold could also harm the Company’s reputation with patients, physicians, health institutions, and investors, diminish the Company’s ability to attract clinical trial participants, and damage the Company’s ability to interest investors and obtain financing in the future. There can be no assurances that the Company will not experience such SAEs in the future or that any related clinical hold will be lifted in a timely manner, or at all.
The principal investigator of the colorectal study testing LB-100 in combination with atezolizumab (Roche PD-L1 inhibitor) is currently investigating two SAEs observed in the clinical trial that was launched in August 2024. The Institutional Review Board (the “IRB”) of the Netherlands Cancer Institute (“NKI”) has put the colorectal cancer study on hold. The adverse reactions that developed in the two patients were dyspnea (shortness of breath) due to lung toxicity possibly or probably related to the combination of LB-100 and atezolizumab in one patient and fever and aphasia possibly or probably related to the combination of LB-100 and atezolizumab in the second patient. The patient who developed lung toxicity deceased due to the combination of lung metastases of colorectal cancer and dyspnea. The patient with fever and aphasia fully recovered from the adverse events with supportive medication.
Given the identified adverse events in the two patients in the clinical trial, the IRB requested from the principal investigator of the study at the NKI information as to whether the adverse events could have been caused by the combination of LB-100 and atezolizumab and information about the mode of action of the combination of LB-100 and atezolizumab. The principal investigator prepared a response to the IRB detailing the safety experience with LB-100 given alone and in combination with other cancer drugs, especially doxorubicin and dostarlimab. Doxorubicin is a well-known chemotherapy, and dostarlimab is a well-known immunotherapy of which the mode of action is closely related to that of atezolizumab.
The reported adverse events in the colorectal cancer study have not been seen in any other patients thus far treated with LB-100 alone or in combination with other cancer drugs. Through early July 2025, the Company has been informed that a total of 82 patients had received or were receiving experimental treatment with LB-100.
In May 2025, the Company updated the safety overview of LB-100 and delivered the updated version 5.0 of the Investigator’s Brochure (the “IB”), which contains all of the relevant preclinical, clinical and pharmacologic data with respect to the study of the LB-100 clinical compound in humans, to the investigators of all ongoing clinical trials. The investigators of the study in colorectal cancer (NCT06012734) submitted a detailed response to the IRB, including the updated IB. The Company is currently awaiting the outcome of the IRB review.
Other Business Risks
Covid-19 Virus. The global outbreak of the novel coronavirus (Covid-19) in early 2020 led to disruptions in general economic activities throughout the world as businesses and governments implemented broad actions to mitigate this public health crisis. Although the Covid-19 outbreak has subsided, the extent to which the coronavirus or any other pandemics may reappear and impact the Company’s clinical trial programs and capital raising efforts in the future is uncertain and cannot be predicted.
Inflation and Interest Rate Risk. The Company does not believe that inflation or increasing interest rates have had a material effect on its operations to date, other than their impact on the general economy. However, there is a risk that the Company’s operating costs could become subject to inflationary and interest rate pressures in the future, which would have the effect of increasing the Company’s operating costs, and which would put additional stress on the Company’s working capital resources.
|
|
Supply Chain Issues. The Company does not currently expect that supply chain issues will have a significant impact on its business activities, including its ongoing clinical trials.
Potential Recession. There have been some indications that the United States economy may be at risk of entering a recessionary period. Although it does not appear likely at this time, an economic recession could impact the general business environment and the capital markets, which could, in turn, affect the Company.
Geopolitical Risk. The geopolitical landscape poses inherent risks that could significantly impact the operations and financial performance of the Company. In the event of a military conflict, supply chain disruptions, geopolitical uncertainties, and economic repercussions may adversely affect the Company’s ability to conduct research, develop, test and manufacture products, and distribute them globally. This could lead to delays in product development, interruptions in the supply of critical materials, and delays in clinical trials, thereby impeding the Company’s clinical development and commercialization plans. Furthermore, the impact of a conflict on global financial markets may result in increased volatility and uncertainty in the capital markets, thereby affecting the valuation of the Company’s publicly-traded shares. Investor confidence, market sentiment, and access to capital could all be negatively influenced. Such geopolitical risks are outside the control of the Company, and the actual effects on the Company’s business, financial condition and results of operations may differ from current estimates.
Cybersecurity Risks. The Company has established policies and processes for assessing, identifying and managing material risk from cybersecurity threats, and has integrated these processes into its overall risk management systems and processes. The Company routinely assesses material risks from cybersecurity threats, including any potential unauthorized occurrence on or conducted through its information and email systems that may result in adverse effects on the confidentiality, integrity, or availability of the Company’s information and email systems or any information residing therein. The Company conducts periodic risk assessments to identify cybersecurity threats, as well as assessments in the event of a material change in the Company’s business practices that may affect information systems that are vulnerable to such cybersecurity threats. These risk assessments include identification of reasonably foreseeable internal and external risks, the likelihood and potential damage that could result from such risks, and the sufficiency of existing policies, procedures, systems and safeguards in place to manage such risks. The Company has not encountered any cybersecurity challenges to date that have materially impaired its operations or financial condition.
The Company is continuing to monitor these matters and will adjust its current business and financing plans as more information becomes available.
9. Subsequent Events
The Company performed an evaluation of subsequent events through the date of filing of these consolidated financial statements with the SEC. Other than as described below or elsewhere in the notes to the consolidated financial statements, there were no material subsequent events which affected, or could affect, the amounts or disclosures in the consolidated financial statements.
Sale of Common Stock, Preferred Stock, Pre-Funded Common Stock Purchase Warrants, and Common Stock Purchase Warrants; Exercise of Pre-Funded Common Stock Purchase Warrants
July 2, 2025 Equity Offering:
On June 30, 2025, the Company, entered into a Securities Purchase Agreement (the “Purchase Agreement”) with certain purchasers named therein (the “Purchasers”), pursuant to which the Company agreed to issue and sell, in a private placement (the “Offering”) 59,552 shares (the “Common Shares”) of the Company’s Common Stock, par value $0.0001 per share (the “Common Stock”); Pre-Funded Warrants (“Pre-Funded Warrants”) to purchase 2,322,532 shares of Common Stock; common stock warrants (the “Common Stock Warrants”) to purchase 6,355,214 shares of Common Stock; and 3,573,130 shares of the Company’s Series B Convertible Preferred Stock (the “Preferred Shares”). Each Preferred Share is convertible into one share of Common Stock, subject to standard adjustments such as stock splits and stock dividends. The Preferred Shares are non-voting, except that certain actions of the Company may not be taken except upon approval of holders who own a majority in stated value of the Preferred Shares. The Preferred Shares bear an 8% per annum cumulative dividend non-compounding and payable at conversion either in cash or, at the holder’s election, in shares of Common Stock valued at the then effective conversion rate. The holders of the Preferred Shares have the right to designate two members to the Company’s Board of Directors.
|
|
The Common Shares, Pre-Funded Warrants, the Preferred Shares, the Common Stock Warrants and the shares of Common Stock underlying the Common Stock Warrants, Pre-Funded Warrants and Preferred Shares have been registered under the Securities Act of 1933, as amended (the “Securities Act”) and were issued in reliance on an exemption from the registration requirements of the Securities Act afforded by Section 4(a)(2) thereof. The Company filed a registration statement on Form S-1 (the “Resale Registration Statement”) to cover the resale of the Common Shares and any shares of Common Stock underlying the Pre-Funded Warrants, the Common Stock Warrants, the Placement Agent Warrants and the Preferred Shares, which was declared effective by the Securities and Exchange Commission on July 15, 2025.
The Offering was priced at-the-market under Nasdaq rules at $0.8396 per common stock unit, with each unit consisting of one share of common stock at a price of $0.7146 and one common stock warrant at a price of $0.125 to acquire one share of common stock at an exercise price of $1.00 per share. The Offering resulted in gross proceeds of $5,050,000 before deducting the placement agent’s fees and related offering expenses of approximately $824,000. The initial Offering closed on July 2, 2025 with the Company receiving gross proceeds of approximately $4,050,000. The remaining $1,000,000 of gross proceeds were paid on July 18, 2025 upon the Resale Registration Statement having been declared effective.
Pursuant to a Placement Agent Agreement dated as of June 30, 2025, the Company engaged Spartan Capital Securities, LLC (the “Placement Agent”) to act as the Company’s exclusive placement agent in connection with the Offering. The Company paid the Placement Agent a cash fee equal to 8% of the aggregate gross proceeds raised in the Offering, a non-accountable expense allowance of 1.0% of the aggregate gross proceeds raised in the Offering, and $125,000 for its expenses including legal fees.
On the Closing Date, the Company issued to the Placement Agent warrants (the “Placement Agent’s Warrants”) to purchase up to 315,626 shares of Common Stock, which represented 5% of the Shares and Pre-Funded Warrants sold in the Offering. The Placement Agent’s Warrants had an exercise price of 125% of the offering price and otherwise had the same terms as the Common Stock Warrants. On July 15, 2025, the Placement Agent’s warrants were exercised on a cashless basis, resulting in the Placement Agent being issued 221,690 shares of the Company’s Common Stock.
During the period from July 2, 2025 through August 5, 2025, 658,455 pre-funded warrants exercisable at $0.00001 per share and sold in the private placement, were exercised, resulting in the issuance of 658,455 shares of Common Stock.
July 8, 2025 Equity Offering:
On July 3, 2025, the Company, entered into a Securities Purchase Agreement (the “Purchase Agreement”) with certain purchasers named therein (the “Purchasers”), pursuant to which the Company agreed to issue and sell, in a registered direct offering (the “Offering”) 210,675 shares (the “Common Shares”) of the Company’s Common Stock, par value $0.0001 per share (the “Common Stock”) and Pre-Funded Warrants (“Pre-Funded Warrants”) to purchase 763,351 shares of Common Stock, at an offering price of $1.54 per share.
The Offering resulted in gross proceeds of $1,500,000 before deducting placement agent’s fees and related offering expenses of $160,000. The Offering closed on July 8, 2025.
Pursuant to a Placement Agent Agreement dated as of July 3, 2025 (the “Placement Agent Agreement”), the Company engaged Spartan Capital Securities, LLC (the “Placement Agent”) to act as the Company’s exclusive placement agent in connection with the Offering. The Company paid the Placement Agent a cash fee equal to 8.0% of the aggregate gross proceeds raised in the Offering, and agreed to reimburse the Placement Agent $40,000 for its legal fees.
During the period from July 8, 2025 through August 5, 2025, 654,000 pre-funded warrants exercisable at $0.00001 per share and sold in the direct registered offering, were exercised, resulting in the issuance of 654,000 shares of Common Stock.
|
|
Resignation of Certain Directors and Officers; Appointment of New Directors
As described above, the Company entered into a Securities Purchase Agreement with certain purchasers named therein pursuant to which, among other things, the Company issued to the purchasers 3,573,190 shares of the Company’s Series B Preferred Stock (the “Preferred Shares”). The Certificate of Designation for the Preferred Shares grants to the holders the right to designate two members to the Company’s Board of Directors (the “Board”), and the holders designated Jason Sawyer and Dr. Michael Holloway as members of the Board. At a meeting of the Board on July 18, 2025, Mr. Sawyer and Dr. Holloway were appointed as independent members of the Board.
In connection with such appointment, Dr. Stephen Forman and Dr. Yun Yen resigned from the Board and were contemporaneously appointed to serve as members of the Company’s Scientific Advisory Committee. Mr. Sawyer will replace Dr. Yen as Chairman of the Compensation Committee and as a member of the Audit Committee. The compensation of Mr. Sawyer and Dr. Holloway will be determined by the Compensation Committee of the Board as part of an overall review of the Company’s compensation program for its independent directors.
Effective as of July 31, 2025, the Company agreed to accept the resignation of Dr. Jan Schellens, the Company’s Chief Medical Officer, and to terminate his consulting agreement dated as of May 31, 2024, to allow Dr. Schellens to pursue other employment opportunities.
Other Matters
Effective August 4, 2025, the Company entered into a Market Awareness Agreement (the “Agreement”) with MicroCap Advisory, LLC for a term of six months to develop a clear, impactful, and marketable corporate strategy to identify, reach and engage with potential investors. Following the initial 30 day term of the Agreement, either party may terminate it without cause by providing the other party with at least 15 days prior written notice. This corporate strategy is intended to serve as the foundation for a comprehensive investor communications program for the Company.
The Agreement provides for a one-time account set-up fee of $15,000 and a cash fee of $125,000 per month over a period of six months, subject to increase, depending on news, events, or other opportunities to amplify public awareness, which will be reviewed and approved by both parties. In addition, the Agreement provides for the issuance of 48,000 shares of the Company’s common stock to MicroCap Advisory, LLC. upon its signing. The Company has agreed to reimburse MicroCap Advisory, LLC for any pre-approved expenses incurred, including analyst reports and travel expenses.
|
|
ITEM 2. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
Forward-Looking Statements
This Quarterly Report on Form 10-Q of Lixte Biotechnology Holdings, Inc. (the “Company”) contains certain forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, and Section 21E of the Securities Exchange Act of 1934. These might include statements regarding the Company’s financial position, business strategy and other plans and objectives for future operations, and assumptions and predictions about future clinical trials and their timing and costs, product demand, supply, manufacturing costs, marketing and pricing factors are all forward-looking statements. These statements are generally accompanied by words such as “intend”, “anticipate”, “believe”, “estimate”, “potential(ly)”, “continue”, “forecast”, “predict”, “plan”, “may”, “will”, “could”, “would”, “should”, “expect” or the negative of such terms or other comparable terminology. The Company believes that the assumptions and expectations reflected in such forward-looking statements are reasonable, based on information available to it on the date hereof, but the Company cannot provide assurances that these assumptions and expectations will prove to have been correct or that the Company will take any action that the Company may presently be planning. These forward-looking statements are inherently subject to known and unknown risks and uncertainties. Actual results or experience may differ materially from those expected, anticipated or implied in the forward-looking statements. Factors that could cause or contribute to such differences include, but are not limited to, regulatory policies or changes thereto, available cash, research and development results, competition from other similar businesses, and market and general economic factors. This discussion should be read in conjunction with the condensed consolidated financial statements and notes thereto included in Item 1 of this Quarterly Report on Form 10-Q and the Company’s Annual Report on Form 10-K for the fiscal year ended December 31, 2024, including the section entitled “Item 1A. Risk Factors”. The Company does not intend to update or revise any forward-looking statements to reflect new information, future events or otherwise.
Overview
The Company is a clinical-stage biopharmaceutical company focused on identifying new targets for cancer drug development and developing and commercializing cancer therapies. The Company’s corporate office is located in Pasadena, California.
The Company’s product pipeline is primarily focused on inhibitors of protein phosphatase 2A, which is used to enhance cytotoxic agents, radiation, immune checkpoint blockers and other cancer therapies. The Company believes that inhibitors of protein phosphatases have significant therapeutic potential for a broad range of cancers. The Company is focusing on the clinical development of a specific protein phosphatase inhibitor, referred to as LB-100.
The Company’s activities are subject to significant risks and uncertainties, including the need for additional capital. The Company has not yet commenced any revenue-generating operations, does not have positive cash flows from operations, relies on stock-based compensation for a substantial portion of employee and consultant compensation, and is dependent on periodic access to equity capital to fund its operating requirements.
Recent Significant Developments
Sale of Common Stock, Preferred Stock, Pre-Funded Common Stock Purchase Warrants, and Common Stock Purchase Warrants
On July 2, 2025, the Company closed a private placement for gross proceeds of $5,050,000, consisting of shares of common stock, pre-funded warrants to purchase shares of common stock, warrants to purchase shares of common stock, and shares of Series B Convertible Preferred Stock.
On July 8, 2025, the Company closed a registered direct offering for gross proceeds of $1,500,000, consisting of shares of common stock and pre-funded warrants to purchase shares of common stock.
Information with respect to these equity financings is provided at Note 9 to the condensed consolidated financial statements for the three months and six months ended June 30, 2025 and 2024 included elsewhere in this document.
|
|
Resignation of Certain Directors and Officers; Appointment of New Directors
As described above, the Company entered into a Securities Purchase Agreement with certain purchasers named therein pursuant to which, among other things, the Company issued to the purchasers 3,573,190 shares of the Company’s Series B Preferred Stock (the “Preferred Shares”). The Certificate of Designation for the Preferred Shares grants to the holders the right to designate two members to the Company’s Board of Directors (the “Board”), and the holders designated Jason Sawyer and Dr. Michael Holloway as members of the Board. At a meeting of the Board on July 18, 2025, Mr. Sawyer and Dr. Holloway were appointed as independent members of the Board.
In connection with such appointment, Dr. Stephen Forman and Dr. Yun Yen resigned from the Board and were contemporaneously appointed to serve as members of the Company’s Scientific Advisory Committee. Mr. Sawyer will replace Dr. Yen as Chairman of the Compensation Committee and as a member of the Audit Committee. The compensation of Mr. Sawyer and Dr. Holloway will be determined by the Compensation Committee of the Board as part of an overall review of the Company’s compensation program for its independent directors.
Effective as of July 31, 2025, the Company agreed to accept the resignation of Dr. Jan Schellens, the Company’s Chief Medical Officer, and to terminate his related consulting agreement dated as of May 31, 2024, to allow Dr. Schellens to pursue other employment opportunities.
Summary of News Release
July 9, 2025 –
The Company issued a news release announcing that the Medical Journal Nature published findings by a team of physician scientists that validate the scientific premise underlying the Company’s ongoing clinical trials for Ovarian and Colorectal cancers
The team led by principal investigator Amir Jazaeri, MD, professor of Gynecologic Oncology and Reproductive Medicine at The University of Texas MD Anderson Cancer Center, studied survival outcomes of Ovarian Clear Cell Carcinoma (OCCC) patients treated with immune checkpoint blockade therapy (clinicaltrials.gov identifier: NCT03026062). The study showed that patients having tumors with inactivating mutations in PPP2R1A — the major scaffold subunit of protein phosphatase 2A (PP2A) — had significantly better overall survival, compared with patients who did not have this mutation in their tumors.
Inactivating mutations in PPP2R1A are known to reduce the enzymatic activity of PP2A, which is the target of the Company’s lead compound LB-100. Tumors with mutations in PPP2R1A were found to have increased the interferon gamma response pathway, which is known to be associated with improved immune checkpoint responses.
The Company is currently investigating the activity of LB-100 in combination with checkpoint immunotherapy in two clinical trials. The first is enrolling patients with OCCC, led by Dr. Jazaeri at MD Anderson Cancer Center, and also is open at Northwestern University. In this trial, the Company is collaborating with GSK to test LB-100 in combination with dostarlimab (anti PD1). In the second trial, at the Netherlands Cancer Institute, the Company is collaborating with Roche to test LB-100 in combination with atezolizumab (anti PDL1) in colon cancer patients.
Going Concern
For the six months ended June 30, 2025, the Company recorded a net loss of $1,485,228 and used cash in operations of $1,055,968. At June 30, 2025, the Company had cash of $887,212 available to fund its operations.
Because the Company is currently engaged in various early-stage clinical trials, it is expected that it will take a significant amount of time and resources to develop any product or intellectual property capable of generating sustainable revenues. Accordingly, the Company’s business is unlikely to generate any sustainable operating revenues in the next several years and may never do so. Even if the Company is able to generate revenues through licensing its technology, product sales or other commercial activities, there can be no assurance that the Company will be able to achieve and maintain positive earnings and operating cash flows. At June 30, 2025, the Company’s remaining financial contractual commitments pursuant to clinical trial agreements and clinical trial monitoring agreements not yet incurred aggregated approximately $524,000, which are currently scheduled to be incurred through approximately December 31, 2027.
|
|
The Company’s consolidated financial statements have been presented on the basis that it will continue as a going concern, which contemplates the realization of assets and satisfaction of liabilities in the normal course of business. The Company has no recurring source of revenues and has experienced negative operating cash flows since inception. The Company has financed its working capital requirements through the recurring sale of its equity securities. These factors raise substantial doubt about the Company’s ability to continue as a going concern within one year after the date the consolidated financial statements are issued. The consolidated financial statements also do not reflect any adjustments relating to the recoverability of assets and liabilities that might be necessary if the Company is unable to continue as a going concern
The Company’s ability to continue as a going concern is dependent upon its ability to raise additional equity capital to fund its research and development activities, including its ongoing clinical trials. The amount and timing of future cash requirements depends in substantial part on the pace, design and results of the Company’s clinical trial program, which, in turn, depends on the availability of operating capital to fund such activities.
Based on current operating plans, the Company estimates that its existing cash resources at June 30, 2025, together with the net proceeds from the July 2, 2025 private placement, and the July 8, 2025 registered direct offering, will provide sufficient working capital to fund the Company’s operations as currently configured, including its ongoing clinical trial program with respect to the development of the Company’s lead anti-cancer clinical compound LB-100, for at least the next 12 months. However, existing cash resources will not be sufficient to complete the development of and to obtain regulatory approval for the Company’s product candidate, which would require significant additional operating capital.
In addition, as a result of the appointment of a new Chairman and Chief Executive Officer in June 2025, the completion of the July 2025 equity financings, and other changes in senior management and the Board of Directors in July 2025, the Company’s operating strategies and business plans may change, including the incurrence of additional personnel and operating costs, which may require that the Company raise additional capital to fund operations. However, as market conditions present uncertainty as to the Company’s ability to secure additional funds, there can be no assurances that the Company will be able to secure additional financing on acceptable terms, as and when necessary, to continue to fund its operations.
The Company’s independent registered public accounting firm included an explanatory paragraph in their report with respect to this uncertainty that accompanied the Company’s audited consolidated financial statements as of and for the year ended December 31, 2024, in which they expressed substantial doubt about the Company’s ability to continue as a going concern. The Company’s consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
If cash resources are insufficient to satisfy the Company’s ongoing cash requirements, the Company would be required to scale back or discontinue its clinical trial program, as well as its licensing and patent prosecution efforts and its technology and product development efforts, or obtain funds, if available, through strategic alliances, joint ventures or other transaction structures that could require the Company to relinquish rights to and/or control of LB-100, or to curtail or discontinue operations entirely.
Nasdaq Compliance
The Company’s common stock and public warrants are traded on the Nasdaq Capital Market under the symbols “LIXT” and “LIXTW”, respectively.
|
|
On June 2, 2023, the Company effected a 1-for-10 reverse split of its outstanding shares of common stock in order to remain in compliance with the $1.00 minimum closing bid price requirement of the Nasdaq Stock Market LLC (“Nasdaq”).
On August 19, 2024, the Company received a letter from the Listing Qualifications Department (the “Staff”) of Nasdaq indicating that the Company was not in compliance with the minimum stockholders’ equity requirement of $2,500,000 for continued listing on the Nasdaq Capital Market under Listing Rule 5550(b)(1) (the “Stockholders’ Equity Requirement”).
On October 3, 2024, the Company submitted a plan to the Staff to regain compliance with the Stockholders’ Equity Requirement, which outlined the Company’s proposed initiatives to regain compliance by raising equity capital through various registered equity offerings.
On October 21, 2024, the Staff provided notice (the “Notice”) to the Company that it had granted an extension through February 18, 2025 to regain compliance with the Stockholders’ Equity Requirement, which required that the Company complete its capital raising initiatives and evidence compliance with the Stockholders’ Equity Requirement through filing a Current Report on Form 8-K with the SEC providing certain required information.
As of February 18, 2025, the Company had not regained compliance with the Stockholders’ Equity Requirement. On February 19, 2025, the Company received a Staff determination letter stating that the Company did not meet the terms of the extension because it did not complete its proposed financing initiatives to regain compliance. The Company timely requested a Hearing before a Nasdaq Hearings Panel (the “Panel”), which automatically stayed Nasdaq’s suspension or delisting of the Company’s common stock and public warrants pending the Panel’s decision.
On April 17, 2025, the Company received notice that the Panel had granted the Company an extension in which to regain compliance with all continued listing rules of the Nasdaq Capital Market. The Panel’s determination followed a hearing on April 3, 2025, at which the Panel considered the Company’s plan to regain compliance with the Stockholders’ Equity Requirement. As a result of the extension, the Panel granted the Company’s request for continued listing on the Nasdaq Capital Market, provided that the Company demonstrates compliance with the Stockholders’ Equity Requirement and all other continued listing requirements for the Nasdaq Capital Market by July 3, 2025.
On July 2, 2025, the Company closed a private placement for $5,050,000, consisting of shares of common stock, pre-funded warrants to purchase shares of common stock, warrants to purchase shares of common stock, and shares of Series B Convertible Preferred Stock, and on July 8, 2025, the Company closed a registered direct offering for $1,500,000, consisting of shares of common stock and pre-funded warrants to purchase shares of common stock.
On July 15, 2025, the Company received notice from Nasdaq that the Panel found that the Company was in compliance with the Stockholders’ Equity Requirement. The Company was also notified that it will remain subject to a “Panel Monitor”, as that term is defined in Nasdaq Listing Rule 5815(d)(4)(B), for a period of one year from the date of the Nasdaq notice, through July 15, 2026. If, during the term of the Panel Monitor, the Company does not continue to remain in compliance with the Stockholders’ Equity Requirement, the Company will not be provided with the opportunity to submit a compliance plan for review by the Listing Qualifications Staff and must instead request a hearing before the Panel to address the deficiency, with such request staying any further action with respect to the Company’s listing on Nasdaq pending completion of the hearing process.
The Company is undertaking measures to maintain compliance under Nasdaq’s continued listing requirements and to remain listed on the Nasdaq Capital Market. However, there can be no assurances that the Company will ultimately be able to maintain compliance with the Stockholders’ Equity Requirement, or be able to maintain compliance with all other applicable requirements for continued listing on the Nasdaq Capital Market. The Company’s failure to meet these requirements would result in the Company’s securities being delisted from the Nasdaq Capital Market.
|
|
Recent Accounting Pronouncements
Information with respect to recent accounting pronouncements is provided at Note 2 to the condensed consolidated financial statements for the three months and six months ended June 30, 2025 and 2024 included elsewhere in this document.
Concentration of Risk
Information with respect to concentration of risk is provided at Note 2 to the condensed consolidated financial statements for the three months and six months ended June 30, 2025 and 2024 included elsewhere in this document.
Critical Accounting Policies and Estimates
The preparation of financial statements in conformity with GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities at the date of the financial statements and the reported amounts of expenses during the reporting period. Some of those judgments can be subjective and complex, and therefore, actual results could differ materially from those estimates under different assumptions or conditions. Management bases its estimates on historical experience and on various assumptions that are believed to be reasonable in relation to the financial statements taken, as a whole, under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Management regularly evaluates the key factors and assumptions used to develop the estimates utilizing currently available information, changes in facts and circumstances, historical experience, and reasonable assumptions. After such evaluations, if deemed appropriate, those estimates are adjusted accordingly. Actual results could differ from those estimates. Significant estimates include those related to assumptions used in the calculation of accruals for clinical trial costs and other potential liabilities, and valuing equity instruments issued for services.
The following critical accounting policies affect the more significant judgements and estimates used in the preparation of the Company’s consolidated financial statements.
Cash
Cash is held in a cash bank deposit program maintained by Morgan Stanley Wealth Management, a division of Morgan Stanley Smith Barney LLC (“Morgan Stanley”). Morgan Stanley is a FINRA-regulated broker-dealer. The Company’s policy is to maintain its cash balances with financial institutions in the United States with high credit ratings and in accounts insured by the Federal Deposit Insurance Corporation (the “FDIC”) and/or by the Securities Investor Protection Corporation (the “SIPC”). The Company periodically has cash balances in financial institutions in excess of the FDIC and SIPC insurance limits of $250,000 and $500,000, respectively. Morgan Stanley Wealth Management also maintains supplemental insurance coverage for the cash balances of its customers. The Company has not experienced any losses to date resulting from this policy.
Segment Information
The Company’s Chief Executive Officer is the Company’s Chief Operating Decision Maker (“CODM”) and evaluates performance and makes operating decisions about allocating resources based on internal financial data presented on a consolidated basis. Because the CODM evaluates financial performance on a consolidated basis, the Company has determined that it operates in a single reportable segment, which consists of the development of a drug class called Protein Phosphatase 2A inhibitors, and is comprised of the consolidated financial results of the Company. The CODM uses consolidated net income (loss) as the sole measure of segment profit or loss.
In November 2023, the Financial Accounting Standards Board (“FASB”) issued Accounting Standards Update (“ASU”) 2023-07, Segment Reporting (Topic 280): Improvements to Reportable Segment Disclosure. ASU 2023-07 amends the FASB Accounting Standards Codification to require additional reportable segment disclosures of a public entity by requiring disclosure of significant segment expenses that are regularly provided to the chief operating decision maker, requiring other new disclosures, and requiring enhanced interim disclosures. ASU 2023-07 requires public entities with a single reportable segment to provide all the disclosures required by ASU 2023-07 and all existing segment disclosures in Topic 280 on an interim and annual basis. The Company adopted ASU 2023-07 effective January 1, 2024 for the 2024 annual period, including quarterly periods, on a retrospective basis.
|
|
Research and Development
Research and development costs consist primarily of fees paid to consultants and contractors, and other expenses relating to the negotiation, design, development, conduct and management of clinical trials with respect to the Company’s clinical compound and product candidate. Research and development costs also include the costs to manufacture compounds used in research and clinical trials, which are charged to operations as incurred. The Company’s inventory of LB-100 for clinical use has been manufactured separately in the United States and in the European Union in accordance with the laws and regulations of such jurisdictions.
Research and development costs are generally charged to operations ratably over the life of the underlying contracts, unless the achievement of milestones, the completion of contracted work, the termination of an agreement, or other information indicates that a different expensing schedule is more appropriate. However, payments for research and development costs that are contractually defined as non-refundable are charged to operations as incurred.
Obligations incurred with respect to mandatory scheduled payments under agreements with milestone provisions are recognized as charges to research and development costs in the Company’s consolidated statement of operations based on the achievement of such milestones, as specified in the respective agreement. Obligations incurred with respect to mandatory scheduled payments under agreements without milestone provisions are accounted for when due, are recognized ratably over the appropriate period, as specified in the respective agreement, and are recorded as liabilities in the Company’s consolidated balance sheet, with a corresponding charge to research and development costs in the Company’s consolidated statement of operations.
Payments made pursuant to contracts are initially recorded as advances on research and development contract services in the Company’s consolidated balance sheet and are then charged to research and development costs in the Company’s consolidated statement of operations as those contract services are performed. Expenses incurred under contracts in excess of amounts advanced are recorded as research and development contract liabilities in the Company’s consolidated balance sheet, with a corresponding charge to research and development costs in the Company’s consolidated statement of operations. The Company reviews the status of its various clinical trial and research and development contracts on a quarterly basis.
Patent and Licensing Legal and Filing Fees and Costs
Due to the significant uncertainty associated with the successful development of commercially viable products based on the Company’s research efforts and related patent applications, all patent and licensing legal and filing fees and costs related to the development and protection of the Company’s intellectual property are charged to operations as incurred. Patent and licensing legal and filing fees and costs are included in general and administrative costs in the Company’s consolidated statement of operations.
In September 2023, the Company appointed a new President and Chief Executive Officer, who, with the assistance of the Company’s management, Board of Directors and patent legal counsel, conducted a comprehensive review and analysis of the Company’s patent portfolio in order to implement a program to balance patent prosecution costs with intellectual property protection benefits. As a result of such review and analysis, the Company identified certain patent filings that it decided not to continue to support in 2024 and thereafter. In addition, the Company changed patent legal counsel in mid-2024. The Company expects that patent and licensing legal and filing fees and costs will continue to be a significant continuing cost in 2025 and thereafter as the Company continues to manage its patent portfolio related to the clinical development of LB-100.
As a result of such review and analysis, patent and licensing legal and filing fees and costs related to the development and protection of the Company’s intellectual property, primarily related to LB-100, decreased to $17,303 for the three months ended June 30, 2025, as compared to $63,612 for the three months ended June 30, 2024, a decrease of $46,309, or 72.8%. Patent and licensing legal and filing fees and costs related to the development and protection of the Company’s intellectual property, primarily related to LB-100, decreased to $73,386 for the six months ended June 30, 2025, as compared to $146,823 for the six months ended June 30, 2024, a decrease of $73,437, or 50.0%.
A descriptive summary of the patent portfolio for the Company’s most important clinical programs involving the development of LB-100, as well as a detailed listing of each domestic and international patent that has been issued, is presented at “ITEM 1. BUSINESS – Intellectual Property” in the Company’s Annual Report on Form 10-K for the fiscal year ended December 31, 2024.
|
|
Stock-Based Compensation
The Company periodically issues common stock and stock options to officers, directors, employees, contractors and consultants for services rendered. Options vest and expire according to terms established at the issuance date of each grant. Stock grants, which are generally time vested, are measured at the grant date fair value and charged to operations ratably over the vesting period.
The Company accounts for stock-based payments to officers, directors, employees, contractors, and consultants by measuring the cost of services received in exchange for equity awards utilizing the grant date fair value of the awards, with the cost recognized as compensation expense on the straight-line basis in the Company’s financial statements over the vesting period of the awards. Recognition of compensation expense for non-employees is in the same period and manner as if the Company had paid cash for the services.
The fair value of stock options granted as stock-based compensation is determined utilizing the Black-Scholes option-pricing model, and is affected by several variables, the most significant of which are the expected life of the stock option, the exercise price of the stock option as compared to the fair market value of the common stock on the grant date, and the estimated volatility of the common stock. Unless sufficient historical exercise data is available, the expected life of the stock option is calculated as the mid-point between the vesting period and the contractual term (the “simplified method”). The estimated volatility is based on the historical volatility of the Company’s common stock, calculated utilizing a look-back period approximately equal to the contractual life of the stock option being granted. The risk-free interest rate is based on the U.S. Treasury yield curve in effect at the time of grant. The fair market value of the common stock is determined by reference to the quoted market price of the Company’s common stock on the grant date. The expected dividend yield is based on the Company’s expectation of dividend payouts and is assumed to be zero.
The Company recognizes the fair value of stock-based compensation awards in general and administrative costs and in research and development costs, as appropriate, in the Company’s consolidated statements of operations. The Company issues new shares of common stock to satisfy stock option exercises.
Warrants
The Company accounts for warrants as either equity-classified or liability-classified instruments based on an assessment of the warrant’s specific terms and applicable authoritative guidance in Accounting Standards Codification (“ASC”) 480, Distinguishing Liabilities from Equity (“ASC 480”), and ASC 815, Derivatives and Hedging (“ASC 815”). The assessment considers whether the warrants are freestanding financial instruments pursuant to ASC 480, meet the definition of a liability pursuant to ASC 480, and whether the warrants meet all of the requirements for equity classification under ASC 815, including whether the warrants are indexed to the Company’s own common stock and whether the warrant holders could potentially require “net cash settlement” in a circumstance outside of the Company’s control, among other conditions for equity classification. The Company has determined that the warrants issued in the July 2023 equity financing, the February 2025 equity financing, and the July 2025 equity financings meet the requirements for equity classification. This assessment, which requires the use of professional judgment, is conducted when the warrants are issued and at the end each subsequent quarterly period while the warrants are outstanding. For issued or modified warrants that meet all of the criteria for equity classification, the warrants are required to be recorded as a component of additional paid-in capital at the time of issuance. For issued or modified warrants that do not meet all of the criteria for equity classification, the warrants are required to be liability-classified and recorded at their initial fair value on the date of issuance and remeasured at fair value at each balance sheet date thereafter. Changes in the estimated fair value of the warrants that are liability-classified are recognized as a non-cash gain or loss in the statement of operations at each balance sheet date. At June 30, 2025 and December 31, 2024, the Company did not have any liability-classified warrants.
|
|
Summary of Business Activities and Plans
Company Overview
The Company is a clinical-stage biopharmaceutical company focused on identifying new targets for cancer drug development and developing and commercializing cancer therapies. The Company’s product pipeline is primarily focused on inhibitors of protein phosphatase 2A, which is used to enhance cytotoxic agents, radiation, immune checkpoint blockers and other cancer therapies. The Company believes that inhibitors of protein phosphatases have significant therapeutic potential for a broad range of cancers. The Company is focusing on the clinical development of a specific protein phosphatase inhibitor, referred to as LB-100.
The Company believes that the mechanism by which LB-100 affects cancer cell growth is different from cancer agents currently approved for clinical use. LB-100 is currently being tested in clinical trials in Ovarian Clear Cell Carcinoma, Metastatic Colon Cancer, and Advanced Soft Tissue Sarcoma. LB-100 has shown anti-cancer activity in animal models of glioblastoma multiforme, neuroblastoma, and medulloblastoma, all cancers of neural tissue. LB-100 has also been shown to enhance the effectiveness of commonly used anti-cancer drugs in animal models of melanoma, breast cancer and sarcoma. The enhancement of anti-cancer activity of these anti-cancer drugs occurs at doses of LB-100 that do not significantly increase toxicity in animals. It is therefore hoped that, when combined with standard anti-cancer regimens against many tumor types, LB-100 will improve therapeutic benefit.
As a compound moves through the FDA-approval process, it becomes an increasingly valuable property, but at a cost of additional investment at each stage. As the potential effectiveness of LB-100 has been documented at the clinical trial level, the Company has allocated resources to manage its patent portfolio. The Company’s approach has been to operate with a minimum of overhead, moving compounds forward as efficiently and inexpensively as possible, and to raise funds to support each of these stages as certain milestones are reached. The Company’s longer-term objective is to secure one or more strategic partnerships or licensing agreements with pharmaceutical companies with major programs in cancer.
Specific Risks Associated with the Company’s Business Activities
Serious Adverse Events
The Company’s lead drug candidate, LB-100, is currently undergoing various clinical trials, and there is a risk that one or more of these trials could be placed on hold by regulatory authorities due to serious adverse events (SAEs) related to the Company’s drug candidate or to another company’s drug used in combination in one of the Company’s clinical trials. It is possible that the SAEs could be attributable to the Company’s drug candidate and could include, but not be limited to, unexpected severe side effects, treatment-related deaths, or long-term health complications. A dose given could result in non-tolerable adverse events defined as dose-limiting toxicity (DLT). When two DLTs occur at the same dose-level that dose-level is considered too high and unsafe. Further treatment is only allowed at lower dose-levels that have previously been found safe.
If an SAE or a pattern of SAEs is observed during the course of a clinical trial involving the Company’s drug candidate, the U.S. Food and Drug Administration (FDA), European Medicines Agency (EMA), or other regulatory authorities may issue a clinical hold, requiring the Company to pause or discontinue further enrollment and dosing in the Company’s clinical trial. It is also possible that the clinical trial could be terminated. Any of these actions could delay or halt the development of the Company’s drug candidate, increase development costs, and negatively impact the Company’s ability to ultimately achieve regulatory approval. Additionally, if an SAE is confirmed to be drug-related, the Company may be required to conduct additional studies, modify the study design, or abandon further development of the drug candidate altogether, which could materially impact the Company’s business, financial condition, and prospects.
The occurrence of an SAE and any resulting clinical hold could also harm the Company’s reputation with patients, physicians, health institutions, and investors, diminish the Company’s ability to attract clinical trial participants, and damage the Company’s ability to interest investors and obtain financing in the future. There can be no assurances that the Company will not experience such SAEs in the future or that any related clinical hold will be lifted in a timely manner, or at all.
|
|
The principal investigator of the colorectal study testing LB-100 in combination with atezolizumab (Roche PD-L1 inhibitor) is currently investigating two SAEs observed in the clinical trial that was launched in August 2024. The Institutional Review Board (the “IRB”) of the Netherlands Cancer Institute (“NKI”) has put the colorectal cancer study on hold. The adverse reactions that developed in the two patients were dyspnea (shortness of breath) due to lung toxicity possibly or probably related to the combination of LB-100 and atezolizumab in one patient and fever and aphasia possibly or probably related to the combination of LB-100 and atezolizumab in the second patient. The patient who developed lung toxicity deceased due to the combination of lung metastases of colorectal cancer and dyspnea. The patient with fever and aphasia fully recovered from the adverse events with supportive medication.
Given the identified adverse events in the two patients in the clinical trial, the IRB requested from the principal investigator of the study at the NKI information as to whether the adverse events could have been caused by the combination of LB-100 and atezolizumab and information about the mode of action of the combination of LB-100 and atezolizumab. The principal investigator prepared a response to the IRB detailing the safety experience with LB-100 given alone and in combination with other cancer drugs, especially doxorubicin and dostarlimab. Doxorubicin is a well-known chemotherapy, and dostarlimab is a well-known immunotherapy of which the mode of action is closely related to that of atezolizumab.
The reported adverse events in the colorectal cancer study have not been seen in any other patients thus far treated with LB-100 alone or in combination with other cancer drugs. Through early July 2025, the Company has been informed that a total of 82 patients had received or were receiving experimental treatment with LB-100.
In May 2025, the Company updated the safety overview of LB-100 and delivered the updated version 5.0 of the Investigator’s Brochure (the “IB”), which contains all of the relevant preclinical, clinical and pharmacologic data with respect to the study of the LB-100 clinical compound in humans, to the investigators of all ongoing clinical trials. The investigators of the study in colorectal cancer (NCT06012734) submitted a detailed response to the IRB, including the updated IB. The Company is currently awaiting the outcome of the IRB review.
External Risks Associated with the Company’s Business Activities
Covid-19 Virus. The global outbreak of the novel coronavirus (Covid-19) in early 2020 led to disruptions in general economic activities throughout the world as businesses and governments implemented broad actions to mitigate this public health crisis. Although Covid-19 outbreak has subsided, the extent to which the coronavirus pandemic may reappear and impact the Company’s clinical trial programs and capital raising efforts in the future is uncertain and cannot be predicted.
Inflation and Interest Rate Risk. The Company does not believe that inflation or increasing interest rates have had a material effect on its operations to date, other than their impact on the general economy. However, there is a risk that the Company’s operating costs could become subject to inflationary and interest rate pressures in the future, which would have the effect of increasing the Company’s operating costs, and which would put additional stress on the Company’s working capital resources.
Supply Chain Issues. The Company does not currently expect that supply chain issues will have a significant impact on its business activities, including its ongoing clinical trials.
Potential Recession. There have been some indications that the United States economy may be at risk of entering a recessionary period. Although it does not appear likely at this time, an economic recession could impact the general business environment and the capital markets, which could, in turn, affect the Company.
Geopolitical Risk. The geopolitical landscape poses inherent risks that could significantly impact the operations and financial performance of the Company. In the event of a military conflict, supply chain disruptions, geopolitical uncertainties, and economic repercussions may adversely affect the Company’s ability to conduct research, develop, test and manufacture products, and distribute them globally. This could lead to delays in product development, interruptions in the supply of critical materials, and delays in clinical trials, thereby impeding the Company’s clinical development and commercialization plans. Furthermore, the impact of a conflict on global financial markets may result in increased volatility and uncertainty in the capital markets, thereby affecting the valuation of the Company’s publicly-traded shares. Investor confidence, market sentiment, and access to capital could all be negatively influenced. Such geopolitical risks are outside the control of the Company, and the actual effects on the Company’s business, financial condition and results of operations may differ from current estimates.
|
|
Cybersecurity Risks. The Company has established policies and processes for assessing, identifying and managing material risk from cybersecurity threats, and has integrated these processes into its overall risk management systems and processes. The Company routinely assesses material risks from cybersecurity threats, including any potential unauthorized occurrence on or conducted through its information and email systems that may result in adverse effects on the confidentiality, integrity, or availability of the Company’s information and email systems or any information residing therein. The Company conducts periodic risk assessments to identify cybersecurity threats, as well as assessments in the event of a material change in the Company’s business practices that may affect information systems that are vulnerable to such cybersecurity threats. These risk assessments include identification of reasonably foreseeable internal and external risks, the likelihood and potential damage that could result from such risks, and the sufficiency of existing policies, procedures, systems and safeguards in place to manage such risks. The Company has not encountered any cybersecurity challenges to date that have materially impaired its operations or financial condition.
The Company is continuing to monitor these matters and will adjust its current business and financing plans as more information becomes available.
Results of Operations
At June 30, 2025, the Company had not yet commenced any revenue-generating operations, does not have any positive cash flows from operations, and is dependent on its ability to raise equity capital to fund its operating requirements.
The Company’s condensed consolidated statements of operations as discussed herein are presented below.
| Three Months Ended | Six Months Ended | |||||||||||||||
| June 30, | June 30, | |||||||||||||||
| 2025 | 2024 | 2025 | 2024 | |||||||||||||
| Revenues | $ | — | $ | — | $ | — | $ | — | ||||||||
| Costs and expenses: | ||||||||||||||||
| Research and development costs | 60,648 | 210,708 | 152,105 | 329,772 | ||||||||||||
| General and administrative costs | 714,161 | 798,448 | 1,329,644 | 1,646,263 | ||||||||||||
| Total costs and expenses | 774,809 | 1,009,156 | 1,481,749 | 1,976,035 | ||||||||||||
| Loss from operations | (774,809 | ) | (1,009,156 | ) | (1,481,749 | ) | (1,976,035 | ) | ||||||||
| Interest income | 365 | 2,233 | 806 | 5,092 | ||||||||||||
| Interest expense | (1,810 | ) | (4,154 | ) | (4,945 | ) | (11,340 | ) | ||||||||
| Foreign currency gain | 581 | 158 | 660 | 42 | ||||||||||||
| Net loss | $ | (775,673 | ) | $ | (1,010,919 | ) | $ | (1,485,228 | ) | $ | (1,982,241 | ) | ||||
| Net loss per common share – basic and diluted | $ | (0.29 | ) | $ | (0.45 | ) | $ | (0.57 | ) | $ | (0.88 | ) | ||||
| Weighted average common shares outstanding – basic and diluted | 2,720,533 | 2,249,290 | 2,596,509 | 2,249,290 | ||||||||||||
Three Months Ended June 30, 2025 and 2024
Revenues. The Company did not have any revenues for the three months ended June 30, 2025 and 2024.
Research and Development Costs. For the three months ended June 30, 2025, research and development costs were $60,648, which consisted of clinical and related oversight costs of $11,601, compound maintenance costs of $20,265, regulatory service costs of $1,190, and preclinical research focused on development of additional novel anti-cancer compounds to add to the Company’s clinical pipeline of $27,592.
|
|
For the three months ended June 30, 2024, research and development costs were $210,708, which consisted of clinical and related oversight costs of $97,947, compound maintenance costs of $5,976, regulatory service costs of $1,956, and preclinical research focused on development of additional novel anti-cancer compounds to add to the Company’s clinical pipeline of $104,829.
Included in preclinical research costs for the three months ended June 30, 2025 and 2024 were $0 and $67,119, respectively, of costs paid to the Netherlands Cancer Institute. On October 8, 2021, the Company entered into a Development Collaboration Agreement with the Netherlands Cancer Institute, Amsterdam, one of the world’s leading comprehensive cancer centers, and Oncode Institute, Utrecht, a major independent cancer research center, to identify the most promising drugs to be combined with LB-100, and potential LB-100 analogues, to be used to treat a range of cancers, as well as to identify the specific molecular mechanisms underlying the identified combinations.
On October 3, 2023, the Company entered into Amendment No. 2 to the Development Collaboration Agreement with the Netherlands Cancer Institute, which provided for additional research activities, extended the termination date of the Development Collaboration Agreement by two years to October 8, 2026, and added 500,000 Euros to the operating budget being funded by the Company.
On October 4, 2024, the Company entered into Amendment No. 3 to the Development Collaboration Agreement with NKI, which suspended Amendment No. 2 and provided for a new study term of one year commencing upon the dosing of the first patient in the clinical trial at a project cost of 100,000 Euros (see “Principal Commitments – Other Significant Agreements and Contracts – Netherlands Cancer Institute” below). The Company was recently notified that the preparations for this clinical trial were suspended and the clinical trial is not expected commence. Accordingly, the Company expects that this agreement will be terminated and the Company will have no further financial commitment or cost.
Research and development costs decreased by $150,060, or 71.2%, in 2025 as compared to 2024, primarily as a result of a decrease in clinical and related oversight costs of $86,346 and preclinical research focused on development of additional novel anti-cancer compounds to add to the Company’s clinical pipeline of $77,237, offset by an increase in compound maintenance costs of $14,289.
General and Administrative Costs. For the three months ended June 30, 2025, general and administrative costs were $714,161, which consisted of the fair value of vested stock options issued to directors and officers of $267,999 (including quarterly director and board committee fees of $27,500 and the acceleration of the vesting of stock options held by Bas van der Baan of $167,460 as a result of the amendment of his employment contract), patent and licensing legal and filing fees and costs of $17,303, other consulting and professional fees of $206,362, insurance expense of $64,277, officer compensation and related costs of $104,947, licensing and royalties of $7,397, shareholder reporting costs of $7,353, listing fees of $13,250, filing fees of $5,420, investor relations of $11,397, taxes and licenses of $5,056, and other operating costs of $3,400.
For the three months ended June 30, 2024, general and administrative costs were $798,448, which consisted of the fair value of vested stock options issued to directors and officers of $130,691 (including quarterly director and board committee fees of $27,500), patent and licensing legal and filing fees and costs of $63,612, other consulting and professional fees of $191,529, insurance expense of $126,873, officer compensation and related costs of $191,971, licensing and royalties of $7,455, shareholder reporting costs of $3,811, listing fees of $12,375, filing fees of $11,319, investor relations of $17,397, taxes and licenses of $15,406, rent of $4,230, conference fees of $14,475, and other operating costs of $7,304.
General and administrative costs decreased by $84,287, or 10.6%, in 2025 as compared to 2024, primarily as a result of decreases in patent and licensing legal and filing fees and costs of $46,309, insurance expense of $62,596, officer compensation and related costs of $87,024, investor relations of $6,000, taxes and licenses of $10,350, filing fees of $5,899, rent of $3,855, and conference fees of $14,475, offset by increases in fair value of vested stock options issued to directors and officers of $137,308, other consulting and professional fees of $14,833 and shareholder reporting of $3,542.
|
|
Interest Income. For the three months ended June 30, 2025, the Company had interest income of $365, as compared to interest income of $2,233 for the three months ended June 30, 2024, related to the investment of the Company’s cash resources.
Interest Expense. For the three months ended June 30, 2025, the Company had interest expense of $1,810, as compared to interest expense of $4,154 for the three months ended June 30, 2024, related to the financing of the premium for the Company’s directors and officers liability insurance policy.
Foreign Currency Gain. For the three months ended June 30, 2025, the Company had a foreign currency gain of $581, as compared to a foreign currency gain of $158 for the three months ended June 30, 2024, from foreign currency transactions.
Net Loss. For the three months ended June 30, 2025, the Company incurred a net loss of $775,673, as compared to a net loss of $1,010,919 for the three months ended June 30, 2024.
Six Months Ended June 30, 2025 and 2024
Revenues. The Company did not have any revenues for the six months ended June 30, 2025 and 2024.
Research and Development Costs. For the six months ended June 30, 2025, research and development costs were $152,105, which consisted of clinical and related oversight costs of $27,470, compound maintenance costs of $53,083, regulatory service costs of $1,190, and preclinical research focused on development of additional novel anti-cancer compounds to add to the Company’s clinical pipeline of $70,362.
For the six months ended June 30, 2024, research and development costs were $329,772, which consisted of clinical and related oversight costs of $107,977, compound maintenance costs of $9,870, regulatory service costs of $2,616, and preclinical research focused on development of additional novel anti-cancer compounds to add to the Company’s clinical pipeline of $209,309.
Included in preclinical research costs for the six months ended June 30, 2025 and 2024 were $0 and $134,084, respectively, of costs paid to the Netherlands Cancer Institute, On October 8, 2021, the Company entered into a Development Collaboration Agreement with the Netherlands Cancer Institute, Amsterdam, one of the world’s leading comprehensive cancer centers, and Oncode Institute, Utrecht, a major independent cancer research center, to identify the most promising drugs to be combined with LB-100, and potential LB-100 analogues, to be used to treat a range of cancers, as well as to identify the specific molecular mechanisms underlying the identified combinations.
On October 3, 2023, the Company entered into Amendment No. 2 to the Development Collaboration Agreement with the Netherlands Cancer Institute, which provided for additional research activities, extended the termination date of the Development Collaboration Agreement by two years to October 8, 2026, and added 500,000 Euros to the operating budget being funded by the Company.
On October 4, 2024, the Company entered into Amendment No. 3 to the Development Collaboration Agreement with NKI, which suspended Amendment No. 2 and provided for a new study term of one year commencing upon the dosing of the first patient in the clinical trial at a project cost of 100,000 Euros (see “Principal Commitments – Other Significant Agreements and Contracts – Netherlands Cancer Institute” below). The Company was recently notified that the preparations for this clinical trial were suspended and the clinical trial is not expected commence. Accordingly, the Company expects that this agreement will be terminated and the Company will have no further financial commitment or cost.
Research and development costs decreased by $177,667, or 53.9%, in 2025 as compared to 2024, primarily as a result of a decrease in clinical and related oversight costs of $80,507 and preclinical research focused on development of additional novel anti-cancer compounds to add to the Company’s clinical pipeline of $138,947, offset by an increase in compound maintenance costs of $43,213.
|
|
General and Administrative Costs. For the six months ended June 30, 2025, general and administrative costs were $1,329,644, which consisted of the fair value of vested stock options issued to directors and officers of $367,737 (including quarterly director and board committee fees of $55,000 and the acceleration of the vesting of stock options held by Bas van der Baan of $167,460 as a result of the amendment of his employment contract), patent and licensing legal and filing fees and costs of $73,386, other consulting and professional fees of $439,182, insurance expense of $128,553, officer compensation and related costs of $195,711, cash-based director and board committee fees of $0, licensing and royalties of $14,795, shareholder reporting costs of $12,164, listing fees of $46,500, filing fees of $15,170, investor relations of $22,794, taxes and licenses of $10,113, travel and entertainment of $435, and other operating costs of $5,587, offset by a rent refund of $2,483.
For the six months ended June 30, 2024, general and administrative costs were $1,646,263, which consisted of the fair value of vested stock options issued to directors and officers of $233,618 (including quarterly director and board committee fees of $27,500), patent and licensing legal and filing fees and costs of $146,823, other consulting and professional fees of $363,972, insurance expense of $253,727, officer compensation and related costs of $387,589, cash-based director and board committee fees of $38,819, licensing and royalties of $60,569, shareholder reporting costs of $12,749, listing fees of $24,750, filing fees of $19,053, investor relations of $34,794, taxes and licenses of $30,813, rent of $9,881, conference fees of $14,475, travel and entertainment of $9,725, and other operating costs of $4,906.
General and administrative costs decreased by $316,619, or 19.2%, in 2025 as compared to 2024, primarily as a result of decreases in patent and licensing legal and filing fees and costs of $73,437, insurance expense of $125,174, officer compensation and related costs of $191,878, cash-based director and board committee fees of $38,819, licensing and royalties of $45,774, investor relations of $12,000, taxes and licenses of $20,700, rent of $12,364, conference fees of $14,475, and travel and entertainment of $9,290, offset by increases in the fair value of vested stock options issued to directors and officers of $134,119, other consulting and professional fees of $75,210 and listing fees of $21,750.
Interest Income. For the six months ended June 30, 2025, the Company had interest income of $806, as compared to interest income of $5,092 for the six months ended June 30, 2024, related to the investment of the Company’s cash resources.
Interest Expense. For the six months ended June 30, 2025, the Company had interest expense of $4,945, as compared to interest expense of $11,340 for the six months ended June 30, 2024, related to the financing of the premium for the Company’s directors and officers liability insurance policy.
Foreign Currency Gain. For the six months ended June 30, 2025, the Company had a foreign currency gain of $660, as compared to a foreign currency gain of $42 for the six months ended June 30, 2024, from foreign currency transactions.
Net Loss. For the six months ended June 30, 2025, the Company incurred a net loss of $1,485,228, as compared to a net loss of $1,982,241 for the six months ended June 30, 2024.
Liquidity and Capital Resources – June 30, 2025
The Company’s condensed consolidated statements of cash flows as discussed herein are as follows:
| Six Months Ended June 30, | ||||||||
| 2025 | 2024 | |||||||
| Net cash used in operating activities | $ | (1,055,968 | ) | $ | (1,608,266 | ) | ||
| Net cash provided by (used in) investing activities | — | — | ||||||
| Net cash provided by financing activities | 904,228 | — | ||||||
| Net decrease in cash | $ | (151,740 | ) | $ | (1,608,266 | ) | ||
|
|
At June 30, 2025, the Company had working capital of $452,630, as compared to working capital of $827,219 at December 31, 2024, reflecting a net decrease in working capital of $374,589 for the six months ended June 30, 2025. The decrease in working capital during the six months ended June 30, 2025 was primarily the result of the level of continuing expenditures related to the Company’s ongoing operations, offset in part by the net proceeds of $914,228 from the sale of securities in a registered direct offering and concurrent private placement that closed on February 13, 2025. At June 30, 2025, the Company had cash of $887,212 available to fund its operations.
Going Concern
The Company’s consolidated financial statements have been presented on the basis that it will continue as a going concern, which contemplates the realization of assets and satisfaction of liabilities in the normal course of business. The Company has no recurring source of revenues and has experienced negative operating cash flows since inception. The Company has financed its working capital requirements through the recurring sale of its equity securities. These factors raise substantial doubt about the Company’s ability to continue as a going concern within one year after the date the consolidated financial statements are issued. The consolidated financial statements also do not reflect any adjustments relating to the recoverability of assets and liabilities that might be necessary if the Company is unable to continue as a going concern.
The Company’s ability to continue as a going concern is dependent upon its ability to raise additional equity capital to fund its research and development activities, including its ongoing clinical trials. The amount and timing of future cash requirements depends in substantial part on the pace, design and results of the Company’s clinical trial program, which, in turn, depends on the availability of operating capital to fund such activities.
Based on current operating plans, the Company estimates that its existing cash resources at June 30, 2025, together with the net proceeds from the July 2, 2025 private placement, and the July 8, 2025 registered direct offering, will provide sufficient working capital to fund the Company’s operations as currently configured, including its ongoing clinical trial program with respect to the development of the Company’s lead anti-cancer clinical compound LB-100, for at least the next 12 months. However, existing cash resources will not be sufficient to complete the development of and to obtain regulatory approval for the Company’s product candidate, which would require significant additional operating capital.
In addition, as a result of the appointment of a new Chairman and Chief Executive Officer in June 2025, the completion of the July 2025 equity financings, and other changes in senior management and the Board of Directors in July 2025, the Company’s operating strategies and business plans may change, including the incurrence of additional personnel and operating costs, which may require that the Company raise additional capital to fund operations. However, as market conditions present uncertainty as to the Company’s ability to secure additional funds, there can be no assurances that the Company will be able to secure additional financing on acceptable terms, as and when necessary, to continue to fund its operations.
If cash resources are insufficient to satisfy the Company’s ongoing cash requirements, the Company would be required to scale back or discontinue its clinical trial program, as well as its licensing and patent prosecution efforts and its technology and product development efforts, or obtain funds, if available, through strategic alliances, joint ventures or other transaction structures that could require the Company to relinquish rights to and/or control of LB-100, or to curtail or discontinue operations entirely.
At June 30, 2025, the Company’s remaining financial contractual commitments pursuant to clinical trial agreements and clinical trial monitoring agreements not yet incurred aggregated $524,000, which are currently scheduled to be incurred through approximately December 31, 2027.
At June 30, 2025, the Company did not have any transactions, obligations or relationships that could be considered off-balance sheet arrangements.
Operating Activities. For the six months ended June 30, 2025, operating activities utilized cash of $1,055,968, as compared to utilizing cash of $1,608,266 for the six months ended June 30, 2024, to fund the Company’s ongoing research and development activities and other operating expenses.
|
|
Investing Activities. For the six months ended June 30, 2025 and 2024, the Company did not have any investing activities.
Financing Activities. For the six months ended June 30, 2025, financing activities consisted of the gross proceeds from the sale of securities in the Company’s registered direct offering of $1,050,003, reduced by offering costs of $135,775, and the payment of deferred offering costs of $10,000. For the six months ended June 30, 2024, the Company had no financing activities.
Principal Commitments
Clinical Trial Agreements
At June 30, 2025, the Company’s remaining financial contractual commitments pursuant to clinical trial agreements and clinical trial monitoring agreements not yet incurred, as described below, aggregated $524,000, including clinical trial agreements of $293,000 and clinical trial monitoring agreements of $231,000, which, based on current estimates, are currently scheduled to be incurred through approximately December 31, 2027. The Company’s ability to conduct and fund these contractual commitments is subject to the timely availability of sufficient capital to fund such expenditures, as well as any changes in the allocation or reallocation of such funds to the Company’s current or future clinical trial programs. The Company expects that the full amount of these expenditures will be incurred only if such clinical trial programs are conducted as originally designed and their respective enrollments and duration are not modified or reduced. Clinical trial programs, such as the types that the Company is engaged in, can be highly variable and can frequently involve a series of changes and modifications over time as clinical data is obtained and analyzed, and is frequently modified, suspended or terminated, in part based on receipt or lack of receipt of an indication of clinical benefit or activity, before the clinical trial endpoint is reached. Accordingly, such contractual commitments as discussed herein should be considered as estimates only based on current clinical assumptions and conditions and are typically subject to significant modifications and revisions over time.
The following is a summary of the Company’s ongoing active contractual clinical trials described below as of June 30, 2025:
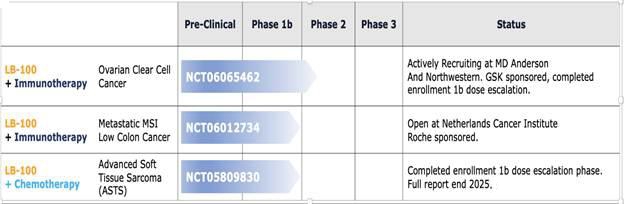
|
|
| Description of Clinical Trial | Institution | Start Date | Projected End Date |
Planned Number of Patients in Trial |
Study Objective | Clinical Update |
Expected Date of Preliminary Efficacy Signal |
NCT No. | Remaining Financial Contractual Commitment | |||||||||||||
| LB-100 combined with dostarlimab in ovarian clear cell carcinoma (Phase 1b/2) | MD Anderson | January 2024 | December 2027 | 21 | Determine the OS of patients with recurrent ovarian clear cell carcinoma | 16 patients entered | December 2026 | NCT06065462 | $ | -0- (1 | ) | |||||||||||
| LB-100 combined with atezolizumab in microsatellite stable metastatic colorectal cancer (Phase 1b) | Netherlands Cancer Institute (NKI) | August 2024 | December 2026 | 37 | Determine RP2D with atezolizumab | First patient entered August 2024, in total two patients entered | June 2026 | NCT06012734 | -0- (1) | |||||||||||||
| LB-100 combined with doxorubicin in advanced soft tissue sarcoma (Phase 1b) | GEIS | June 2023 | Recruitment completed September 2024 | 14 | Determine MTD and RP2D | Fourteen patients entered | December 2025 | NCT05809830 | 293,000 | |||||||||||||
| Total | $ | 293,000 | ||||||||||||||||||||
| (1) | The Company has no financial contractual commitments associated with these clinical trials at June 30, 2025. |
Netherlands Cancer Institute. Effective June 10, 2024, the Company entered into a Clinical Trial Agreement with the Netherlands Cancer Institute (“NKI”) (see Note 5) to conduct a Phase 1b clinical trial of the Company’s protein phosphatase inhibitor, LB-100, combined with atezolizumab, a PD-L1 inhibitor, the proprietary molecule of F. Hoffman-La Roche Ltd. (“Roche”), for patients with microsatellite stable metastatic colorectal cancer. Under the agreement, the Company will provide its lead compound, LB-100, and under a separate agreement between NKI and Roche, Roche will provide atezolizumab and financial support for the clinical trial. The Company has no obligation to and will not provide any reimbursement of clinical trial costs. Pursuant to the agreement and the protocol set forth in the agreement, the clinical trial will be conducted by NKI at NKI’s site in Amsterdam by principal investigator Neeltje Steeghs, MD, PhD, and NKI will be responsible for the recruitment of patients. The agreement provides for the protection of the respective intellectual property rights of each of the Company, NKI and Roche.
This Phase 1b clinical trial will evaluate safety, optimal dose and preliminary efficacy of LB-100 combined with atezolizumab for the treatment of patients with metastatic microsatellite stable colorectal cancer. Immunotherapy using monoclonal antibodies like atezolizumab can enhance the body’s immune response against cancer and hinder tumor growth and spread. LB-100 has been found to improve the effectiveness of anticancer drugs in killing cancer cells by inhibiting a protein called PP2A on cell surfaces. Blocking PP2A increases stress signals in tumor cells expressing the PP2A protein. Accordingly, combining atezolizumab with LB-100 may enhance treatment efficacy for metastatic colorectal cancer, as cancer cells with heightened stress signals are more vulnerable to immunotherapy.
|
|
This study comprises a dose escalation phase and a dose expansion phase. The objective of the dose escalation phase is to determine the recommended Phase 2 dose (RP2D) of LB-100 when combined with the standard dosage of atezolizumab. The dose expansion phase will further investigate the preliminary efficacy, safety, tolerability, and pharmacokinetics/dynamics of the LB-100 and atezolizumab combination. The clinical trial opened in August 2024 with the enrollment of the first patient. A total of two patients have been enrolled to date. Patient accrual is expected to take up to 24 months, with a maximum of 37 patients with advanced colorectal cancer to be enrolled in this study.
The principal investigator of the colorectal study testing LB-100 in combination with atezolizumab is currently investigating two Serious Adverse Events (“SAEs”) observed in the clinical trial. The Investigational Review Board (IRB) of NKI has requested additional information with respect to these SAEs and the study has been paused for enrollment until the IRB’s questions have been satisfactorily addressed (see “Specific Risks Associated with the Company’s Business Activities - Serious Adverse Events” below for additional information).
The Company has no financial contractual commitment associated with this clinical trial.
City of Hope. Effective January 18, 2021, the Company executed a Clinical Research Support Agreement (the “Agreement”) with the City of Hope National Medical Center, an NCI-designated comprehensive cancer center, and City of Hope Medical Foundation (collectively, “City of Hope”), to carry out a Phase 1b clinical trial of LB-100, the Company’s first-in-class protein phosphatase inhibitor, combined with an FDA-approved standard regimen for treatment of untreated extensive-stage disease small cell lung cancer (“ED-SCLC”). LB-100 was given in combination with carboplatin, etoposide and atezolizumab, an FDA-approved standard of care regimen, to previously untreated ED-SCLC patients. The LB-100 dose was to be escalated with the standard fixed doses of the 3-drug regimen to reach a recommended Phase 2 dose (“RP2D”). Patient entry was to be expanded so that a total of 12 patients would be evaluable at the RP2D to determine the safety of the LB-100 combination and to look for potential therapeutic activity as assessed by objective response rate, duration of overall response, progression-free survival, and overall survival.
The clinical trial was initiated on March 9, 2021, with patient accrual expected to take approximately two years to complete. Because patient accrual was slower than expected, effective March 6, 2023, the Company and City of Hope added the Sarah Cannon Research Institute (“SCRI”), Nashville, Tennessee, to the ongoing Phase 1b clinical trial. The Company and City of Hope continued efforts to increase patient accrual by adding additional sites and by modifying the protocol to increase the number of patients eligible for the clinical trial. The impact of these efforts to increase patient accrual and to decrease time to completion was evaluated in subsequent quarters.
After evaluating patient accrual through June 30, 2024, the Company and City of Hope agreed to close the clinical trial. Pursuant to the terms of the Agreement, the Company provided notice to City of Hope of the Company’s intent to terminate the Agreement effective as of July 8, 2024. Upon closure, the Company incurred a prorated charge of $207,004 for the cost of patients enrolled to date, which is included in accounts payable and accrued expenses at June 30, 2025 and December 31, 2024.
During the three months ended June 30, 2025 and 2024, the Company incurred costs of $0 and $78,015, respectively, pursuant to this Agreement. During the six months ended June 30, 2025 and 2024, the Company incurred costs of $0 and $78,015, respectively, pursuant to this Agreement. As of June 30, 2025, total costs of $732,532 had been incurred pursuant to this Agreement.
GEIS. Effective July 31, 2019, the Company entered into a Collaboration Agreement for an Investigator-Initiated Clinical Trial with the Spanish Sarcoma Group (Grupo Español de Investigación en Sarcomas or “GEIS”), Madrid, Spain, to carry out a study entitled “Randomized phase I/II trial of LB-100 plus doxorubicin vs. doxorubicin alone in first line of advanced soft tissue sarcoma”. The purpose of this clinical trial is to obtain information with respect to the efficacy and safety of LB-100 combined with doxorubicin in soft tissue sarcomas. Doxorubicin is the global standard for initial treatment of advanced soft tissue sarcomas (“ASTS”). Doxorubicin alone has been the mainstay of first line treatment of ASTS for over 40 years, with little improvement in survival from adding cytotoxic compounds to or substituting other cytotoxic compounds for doxorubicin. In animal models, LB-100 has consistently enhanced the anti-tumor activity of doxorubicin without apparent increases in toxicity.
GEIS has a network of referral centers in Spain and across Europe that have an impressive track record of efficiently conducting innovative studies in ASTS. The Company agreed to provide GEIS with a supply of LB-100 to be utilized in the conduct of this clinical trial, as well as to provide funding for the clinical trial. The goal is to enter approximately 150 to 170 patients in this clinical trial over a period of two to four years. The Phase 1 portion of the study began in the quarter ended June 30, 2023 to determine the recommended Phase 2 dose of the combination of doxorubicin and LB-100. As advanced sarcoma is a very aggressive disease, the design of the Phase 2 portion of the study assumes a median progression-free survival (“PFS”), no evidence of disease progression or death from any cause, of 4.5 months in the doxorubicin arm and an alternative median PFS of 7.5 months in the doxorubicin plus LB-100 arm to demonstrate a statistically significant decrease in relative risk of progression or death by adding LB-100. There is a planned interim analysis of the primary endpoint when approximately 50% of the 102 events required for final analysis is reached.
|
|
The Company had previously expected that this clinical trial would commence during the quarter ended June 30, 2020. However, during July 2020, the Spanish regulatory authority advised the Company that although it had approved the scientific and ethical basis of the protocol, it required that the Company manufacture new inventory of LB-100 under current Spanish pharmaceutical manufacturing standards. These standards were adopted subsequent to the production of the Company’s existing LB-100 inventory.
In order to manufacture a new inventory supply of LB-100 for the GEIS clinical trial, the Company engaged a number of vendors to carry out the multiple tasks needed to make and gain approval of a new clinical product for investigational study in Spain. These tasks included the synthesis under good manufacturing practice (GMP) of the active pharmaceutical ingredient (API), with documentation of each of the steps involved by an independent auditor. The API was then transferred to a vendor that prepares the clinical drug product, also under GMP conditions documented by an independent auditor. The clinical drug product was then sent to a vendor to test for purity and sterility, provide appropriate labels, store the drug, and distribute the drug to the clinical centers for use in the clinical trials. A formal application documenting all steps taken to prepare the clinical drug product for clinical use was submitted to the appropriate regulatory authorities for review and approval before being used in a clinical trial.
As of June 30, 2025, this program to provide new inventory of the clinical drug product for the Spanish Sarcoma Group study, and potentially for subsequent multiple trials within the European Union, had cost approximately $1,144,000.
On October 13, 2022, the Company announced that the Spanish Agency for Medicines and Health Products (Agencia Española de Medicamentos y Productos Sanitarios or “AEMPS”) had authorized a Phase 1b/randomized Phase 2 study of LB-100, the Company’s lead clinical compound, plus doxorubicin, versus doxorubicin alone, the global standard for initial treatment of ASTS. Consequently, this clinical trial commenced during the quarter ended June 30, 2023 and is expected to be completed and a report prepared by December 31, 2026. In April 2023, GEIS completed its first site initiation visit in preparation for the clinical trial at Fundación Jiménez Díaz University Hospital (Madrid). Up to 170 patents will be entered into the clinical trial. The recruitment for the Phase 1b portion of the protocol was extended with two patients and was completed during the quarter ended September 30, 2024. The Company expects to have data on toxicity and preliminary efficacy from this portion of the clinical trial during the quarter ending December 31, 2025.
Given the focus on the combination of LB-100 with immunotherapy in ovarian clear cell carcinoma and colorectal cancer and the availability of capital resources, the Company entered into Amendment No. 1 to the Collaboration Agreement effective March 11, 2025 that relieved the Company of the financial obligation to support the randomized Phase 2 portion of the clinical trial contemplated in the Collaboration Agreement of approximately $3,095,000. As a result, it is uncertain as to whether the Phase 2 portion of this clinical trial will proceed.
The Company’s agreement with GEIS provided for various payments based on achieving specific milestones over the term of the agreement. During the three months ended June 30, 2025 and 2024, the Company did not incur any costs pursuant to this agreement. During the six months ended June 30, 2025 and 2024, the Company did not incur any costs pursuant to this agreement. Through June 30, 2025, the Company has incurred charges of $685,107 for work done under this agreement through the fourth milestone.
The Company’s aggregate commitment pursuant to this agreement, less amounts previously paid to date, totaled approximately $293,000 for the Phase 1b portion of this clinical trial as of June 30, 2025, which is scheduled to be incurred through December 31, 2025. As the work is being conducted in Europe and is paid for in Euros, final costs are subject to foreign currency fluctuations between the United States Dollar and the Euro. Such fluctuations are recorded in the consolidated statements of operations as foreign currency gain or loss, as appropriate, and have not been significant.
|
|
MD Anderson Cancer Center Clinical Trial. On September 20, 2023, the Company announced an investigator-initiated Phase 1b/2 collaborative clinical trial to assess whether adding LB-100 to a human programmed death receptor-1 (“PD-1”) blocking antibody of GSK plc (“GSK”), dostarlimab-gxly, may enhance the effectiveness of immunotherapy in the treatment of ovarian clear cell carcinoma (“OCCC”). The study objective is to determine the overall survival (“OS”) of patients with OCCC. The clinical trial is being sponsored by The University of Texas MD Anderson Cancer Center (“MD Anderson”) and is being conducted at The University of Texas - MD Anderson Cancer Center. The Company is providing LB-100 and GSK is providing dostarlimab-gxly and financial support for the clinical trial. On January 29, 2024, the Company announced the entry of the first patient into this clinical trial. The Company currently expects that this clinical trial will be completed by December 31, 2027.
On February 25, 2025, the Company announced that it has added the Robert H. Lurie Comprehensive Cancer Center (Lurie Cancer Center) of Northwestern University as a second site in a clinical trial combining the Company’s proprietary compound LB-100 with GSK’s dostarlimab to treat ovarian clear cell cancer. Patient recruitment is underway, and the first patient has been dosed.
Clinical Trial Monitoring Agreements
MD Anderson Cancer Center Clinical Trial. On May 15, 2024, the Company signed a letter of intent with Theradex to monitor the MD Andersen investigator-initiated Phase 1b/2 collaborative clinical trial to assess whether adding LB-100 to a human programmed death receptor-1 (“PD-1”) blocking antibody of GSK plc (“GSK”), dostarlimab-gxly, may enhance the effectiveness of immunotherapy in the treatment of ovarian clear cell carcinoma (“OCCC”). On August 19, 2024, the Company signed a work order agreement with Theradex to monitor the MD Anderson clinical trial. The study oversight is expected to be completed by January 31, 2027.
Costs under this letter of intent and related work order agreement are estimated to be approximately $95,000. During the three months ended June 30, 2025 and 2024, the Company incurred costs of $4,614 and $8,228 pursuant to this letter of intent and subsequent work order. During the six months ended June 30, 2025 and 2024, the Company incurred costs of $11,892 and $8,228 pursuant to this letter of intent and subsequent work order. As of June 30, 2025, total costs of $38,655 have been incurred pursuant to this letter of intent and subsequent work order.
The Company’s aggregate commitment pursuant to this letter of intent, less amounts previously paid to date, totaled approximately $57,000 as of June 30, 2025, which is expected to be incurred through December 31, 2027.
City of Hope. On February 5, 2021, the Company signed a new work order agreement with Theradex to monitor the City of Hope investigator-initiated clinical trial in small cell lung cancer in accordance with FDA requirements for oversight by the sponsoring party. Costs under this work order agreement were estimated to be approximately $335,000. During the three months ended June 30, 2025 and 2024, the Company incurred costs of $0 and $4,500, respectively, pursuant to this work order. During the six months ended June 30, 2025 and 2024, the Company incurred costs of $0 and $9,000, respectively, pursuant to this work order. As of June 30, 2025, total costs of $87,823 had been incurred pursuant to this work order agreement.
As a result of the closure of the Agreement with City of Hope effective July 8, 2024 (see “Clinical Trial Agreements – City of Hope” above), the work order agreement with Theradex to monitor this clinical trial was concurrently terminated, although nominal oversight trailing costs subsequent to July 8, 2024 are expected to be incurred relating to the closure of this study.
GEIS. On June 22, 2023, the Company finalized a work order agreement with Theradex, to monitor the GEIS investigator-initiated clinical Phase I/II randomized trial of LB-100 plus doxorubicin vs. doxorubicin alone in first line of advanced soft tissue sarcoma. The study oversight is expected to be completed by December 31, 2026.
Costs under this work order agreement are estimated to be approximately $153,000, with such payments expected to be allocated approximately 72% to Theradex for services and approximately 28% for payments for pass-through software costs. During the three months ended June 30, 2025 and 2024, the Company incurred costs of $3,750 and $7,203, respectively, pursuant to this work order. During the six months ended June 30, 2025 and 2024, the Company incurred costs of $7,622 and $12,732, respectively, pursuant to this work order. As of June 30, 2025, total costs of $57,077 have been incurred pursuant to this work order agreement.
|
|
The Company’s aggregate commitment pursuant to this clinical trial monitoring agreement, less amounts previously paid to date, totaled approximately $95,000 as of June 30, 2025, which is expected to be incurred through December 31, 2026.
Netherlands Cancer Institute. On August 27, 2024, the Company finalized a work order agreement with Theradex, to monitor the NKI Phase 1b clinical trial of LB-100 combined with atezolizumab, a PD-L1 inhibitor, for patients with microsatellite stable metastatic colorectal cancer. The study oversight was expected to be completed by May 31, 2027.
Costs under this work order agreement were estimated to be approximately $106,380, with such payments expected to be allocated approximately 47% to Theradex for services and approximately 53% for payments for pass-through software costs. During three months and six months ended June 30, 2025, the Company incurred costs of $4,500 and $9,000, respectively, pursuant to this work order. As of June 30, 2025, total costs of $29,191 have been incurred pursuant to this work order agreement.
The Company’s aggregate commitment pursuant to this clinical trial monitoring agreement, less amounts previously paid to date, totaled approximately $79,000 as of June 30, 2025, which was expected to be incurred through May 31, 2027.
The Company was recently notified that the preparations for this clinical trial were suspended and the clinical trial is not expected commence. Accordingly, the Company expects that this agreement will be terminated and the Company will have no further financial commitment or cost.
Patent and License Agreements
National Institute of Health. Effective February 23, 2024, the Company entered into a Patent License Agreement (the “License Agreement”) with the National Institute of Neurological Disorders and Stroke (“NINDS”) and the National Cancer Institute (“NCI”), each an institute or center of the National Institute of Health (“NIH”). Pursuant to the License Agreement, the Company has licensed on an exclusive basis the NIH’s intellectual property rights claimed for a Cooperative Research and Development Agreement (“CRADA”) subject invention co-developed with the Company, and the licensed field of use, which focuses on promoting anti-cancer activity alone, or in combination with standard anti-cancer drugs. The scope of this clinical research extends to checkpoint inhibitors, immunotherapy, and radiation for the treatment of cancer. The License Agreement is effective, and shall extend, on a licensed product, licensed process, and country basis, until the expiration of the last-to-expire valid claim of the jointly owned licensed patent rights in each such country in the licensed territory, estimated at twenty years, unless sooner terminated.
The License Agreement contemplates that the Company will seek to work with pharmaceutical companies and clinical trial sites (including comprehensive cancer centers) to initiate clinical trials within timeframes that will meet certain benchmarks. Data from the clinical trials will be the subject of various regulatory filings for marketing approval in applicable countries in the licensed territories. Subject to the receipt of marketing approval, the Company would be expected to commercialize the licensed products in markets where regulatory approval has been obtained.
The Company is obligated to pay the NIH a non-creditable, non-refundable license issue royalty of $50,000 and a first minimum annual royalty within sixty days from the effective date of the Agreement. The first minimum annual royalty of $25,643 was prorated from the effective date of the License Agreement to the next subsequent January 1. Thereafter, the minimum annual royalty of $30,000 is due each January 1 and may be credited against any earned royalties due for sales made in that year. The license issue royalty of $50,000 and the first minimum annual royalty of $25,643 were paid in April 2024. The second minimum annual royalty for 2025 of $30,000 was paid in December 2024 and was included in other prepaid expenses in the consolidated balance sheet at December 31, 2024.
The Company is obligated to pay the NIH, on a country-by-country basis, earned royalties of 2% on net sales of each royalty-bearing product and process, subject to reduction by 50% under certain circumstances relating to royalties paid by the Company to third parties, but not less than 1%. The Company’s obligation to pay earned royalties under the License Agreement commences on the date of the first commercial sale of a royalty-bearing product or process and expires on the date on which the last valid claim of the licensed product or licensed process expires in such country.
|
|
The Company is obligated to pay the NIH benchmark royalties, on a one-time basis, within sixty days from the first achievement of each such benchmark. The License Agreement defines four such benchmarks, which the Company is required to pursue based on “commercially reasonable efforts” as defined in the License Agreement, with deadlines of October 1, 2024, 2027, 2029 and 2031, each with a different specified benchmark payment amount payable within thirty days of achieving such benchmark. The October 1, 2024 benchmark of $100,000 was defined as the dosing of the first patient with a licensed product in a Phase 2 clinical study of such licensed product in the licensed fields of use. The Company had not commenced a Phase 2 clinical study as of June 30, 2025. The total of all such benchmark payments is $1,225,000.
The Company is obligated to provide annual reports to the NIH on its progress toward the development and commercialization of products under the licensed patents. These reports, due within sixty days following the end of each calendar year, must include updates on research and development activities, regulatory submissions, manufacturing efforts, sublicensing, and sales initiatives. If any deviations from the established commercial development plan or agreed-upon benchmarks occur, the Company is obligated to provide explanation and may amend the commercial development plan and the benchmarks, which, subject to certain conditions, the NIH shall not unreasonably withhold, condition, or delay approval of any request of the Company to amend the commercial development plan and/or the benchmarks and to extend the time periods of the benchmarks.
The Company is obligated to pay the NIH sublicensing royalties of 5% on sublicensing revenue received for granting each sublicense within sixty days of receipt of such sublicensing revenue.
During the three months ended June 30, 2025 and 2024, the Company incurred costs of $7,397 and $7,455, respectively, in connection with its obligations under the License Agreement. During the six months ended June 30, 2025 and 2024, the Company incurred costs of $14,794 and $60,569, respectively, in connection with its obligations under the License Agreement. Such costs when incurred have been included in general and administrative costs in the Company’s consolidated statement of operations. As of June 30, 2025, total costs of $90,438 have been incurred pursuant to this agreement. The Company’s aggregate commitment pursuant to this agreement, less amounts previously paid to date, totaled approximately $1,765,000 as of June 30, 2025, which is expected to be incurred over approximately the next twenty years.
Other Significant Agreements and Contracts
NDA Consulting Corp. On December 24, 2013, the Company entered into a consulting agreement with NDA Consulting Corp. for consultation and advice in the field of oncology research and drug development. As part of the consulting agreement, NDA also agreed to have its president, Dr. Daniel D. Von Hoff, M.D., serve on the Company’s Scientific Advisory Committee during the term of such consulting agreement. The term of the consulting agreement was for one year and provided for a quarterly cash fee of $4,000. The consulting agreement had been automatically renewed for additional one-year terms on its anniversary date, most recently on December 24, 2023, but was subsequently terminated by mutual agreement effective September 30, 2024. Consulting and advisory fees charged to operations pursuant to this consulting agreement were $4,000 and $8,000 for the three months and six months ended June 30, 2024, respectively.
BioPharmaWorks. Effective September 14, 2015, the Company entered into a Collaboration Agreement with BioPharmaWorks, pursuant to which the Company engaged BioPharmaWorks to perform certain services for the Company. Those services included, among other things, assisting the Company to commercialize its products and strengthen its patent portfolio; identifying large pharmaceutical companies with a potential interest in the Company’s product pipeline; assisting in preparing technical presentations concerning the Company’s products; consultation in drug discovery and development; and identifying providers and overseeing tasks relating to clinical development of new compounds.
BioPharmaWorks was founded in 2015 by former Pfizer scientists with extensive multi-disciplinary research and development and drug development experience. The Collaboration Agreement was for an initial term of two years and automatically renews for subsequent annual periods unless terminated by a party not less than 60 days prior to the expiration of the applicable period. In connection with the Collaboration Agreement, the Company agreed to pay BioPharmaWorks a monthly fee of $10,000, subject to the right of the Company to pay a negotiated hourly rate in lieu of the monthly fee. Effective March 1, 2024, the compensation payable under the Collaboration Agreement was converted to an hourly rate structure.
|
|
The Company recorded charges to operations pursuant to this Collaboration Agreement of $10,800 and $7,200 during the three months ended June 30, 2025 and 2024, respectively, which were included in research and development costs in the consolidated statements of operations. The Company recorded charges to operations pursuant to this Collaboration Agreement of $24,800 and $27,200 during the six months ended June 30, 2025 and 2024, respectively, which were included in research and development costs in the consolidated statements of operations.
Netherlands Cancer Institute. On October 8, 2021, the Company entered into a Development Collaboration Agreement with the Netherlands Cancer Institute, Amsterdam (“NKI”) (see Note 5), one of the world’s leading comprehensive cancer centers, and Oncode Institute, Utrecht, a major independent cancer research center, for a term of three years. The Development Collaboration Agreement was subsequently modified by Amendment No. 1 thereto.
The Development Collaboration Agreement is a preclinical study intended to identify the most promising drugs to be combined with LB-100, and potentially LB-100 analogues, to be used to treat a range of cancers, as well as to identify the specific molecular mechanisms underlying the identified combinations. The Company agreed to fund the preclinical study, at an approximate cost of 391,000 Euros and provide a sufficient supply of LB-100 to conduct the preclinical study.
On October 3, 2023, the Company entered into Amendment No. 2 to the Development Collaboration Agreement with NKI, which provides for additional research activities, extends the termination date of the Development Collaboration Agreement by two years to October 8, 2026, and added 500,000 Euros to the operating budget being funded by the Company.
On October 4, 2024, the Company entered into Amendment No. 3 to the Development Collaboration Agreement with NKI, which suspended Amendment No. 2 and provided for a new study term of one year commencing upon the dosing of the first patient in the trial at a project cost of 100,000 Euros.
During the three months ended June 30, 2025 and 2024, the Company incurred charges of $0 and $67,119, respectively, with respect to this agreement, which amounts are included in research and development costs in the Company’s consolidated statements of operations. During the six months ended June 30, 2025 and 2024, the Company incurred charges of $0 and $134,084, respectively, with respect to this agreement, which amounts are included in research and development costs in the Company’s consolidated statements of operations. As of June 30, 2025, total costs of $695,918 have been incurred pursuant to this agreement.
The Company was recently notified that the preparations for this clinical trial were suspended and the clinical trial is not expected commence. Accordingly, the Company expects that this agreement will be terminated and the Company will have no further financial commitment or cost.
MRI Global. As amended, the Company has contracted with MRI Global for stability analysis, storage and distribution of LB-100 for clinical trials in the United States. During the three months ended June 30, 2025 and 2024, the Company incurred costs of $6,765 and $5,976, respectively, pursuant to this contract. During the six months ended June 30, 2025 and 2024, the Company incurred costs of $34,857 and $9,870, respectively, pursuant to this contract. As of June 30, 2025, total costs of $375,379 have been incurred pursuant to this contract.
The Company’s aggregate commitment pursuant to this contract, less amounts previously paid to date, totaled approximately $90,000 as of June 30, 2025.
Consideration of Strategic Alternatives
The Company will continue to evaluate various alternatives to be able to obtain the capital required to fund its operations and business development activities, and to maintain its listing on the Nasdaq Capital Market, including merger or acquisition opportunities (including reverse mergers and acquisitions) and funding transactions which could result in a change in control of the Company. There can be no assurances that the evaluation process will result in the identification of an appropriate transaction, the negotiation and execution of a definitive agreement to effect such a transaction, or that any such transaction will ultimately be approved by the Company’s stockholders and then be consummated. Even if such a strategic transaction is consummated, there can be no assurances that it would enhance stockholder value, and it may result in substantial dilution to existing stockholders. Any potential transaction would be dependent on a number of factors that may be outside of the control of the Company, including, among other things, market conditions, industry trends, the interest of third parties in a potential transaction with the Company, and the availability of appropriate financing for such a transaction.
|
|
Trends, Events and Uncertainties
Research and development of new pharmaceutical compounds by its nature is unpredictable. Although the Company undertakes research and development efforts with commercially reasonable diligence, there can be no assurance that the Company’s cash position will be sufficient to enable it to develop any pharmaceutical compound to the extent needed to create future sales to sustain operations as contemplated herein.
There can be no assurance that the Company’s pharmaceutical compound will obtain the regulatory approvals and market acceptance to achieve sustainable revenues sufficient to support the Company’s operations. Even if the Company is able to generate revenues, there can be no assurance that the Company will be able to achieve operating profitability or positive operating cash flows. There can be no assurance that the Company will be able to secure additional financing, to the extent required, on acceptable terms or at all. If cash resources are insufficient to satisfy the Company’s ongoing cash requirements, the Company would be required to reduce or discontinue its research and development programs, or attempt to obtain funds, if available, through strategic alliances, joint ventures or other transaction structures that could require the Company to relinquish rights to and/or control of LB-100, or to discontinue operations entirely.
Other than as discussed above, the Company is not currently aware of any trends, events or uncertainties that are likely to have a material effect on its financial condition in the near term, although it is possible that new trends or events may develop in the future that could have a material effect on the Company’s financial condition.
ITEM 3. QUANTITATIVE AND QUALITATIVE DISCLOSURE ABOUT MARKET RISK
Not applicable.
ITEM 4. CONTROLS AND PROCEDURES
Disclosure Controls and Procedures
The Company’s management is responsible for establishing and maintaining a system of disclosure controls and procedures (as defined in Rule 13a-15(e) and 15d-15(e) under the Securities Exchange Act of 1934, as amended (the “Exchange Act”)), that is designed to ensure that information required to be disclosed by the Company in the reports that it files or submits under the Exchange Act is recorded, processed, summarized, and reported, within the time periods specified in the rules and forms. Disclosure controls and procedures include, without limitation, controls and procedures designed to ensure that information required to be disclosed by an issuer in the reports that it files or submits under the Exchange Act is accumulated and communicated to the issuer’s management, including its principal executive officer and principal financial officer, or persons performing similar functions, as appropriate, to allow timely decisions regarding required disclosure.
In accordance with Exchange Act Rules 13a-15 and 15d-15, an evaluation was completed under the supervision and with the participation of the Company’s management, including its Chief Executive Officer and Chief Financial Officer, of the effectiveness of the design and operation of the Company’s disclosure controls and procedures as of June 30, 2025, the end of the most recent fiscal period covered by this report. Based on that evaluation, the Company’s management has concluded that the Company’s disclosure controls and procedures were effective in providing reasonable assurance that information required to be disclosed in the Company’s reports filed or submitted under the Exchange Act was recorded, processed, summarized, and reported within the time periods specified in the rules and forms of the Securities and Exchange Commission.
Limitations on Effectiveness of Disclosure Controls and Procedures
In designing and evaluating disclosure controls and procedures, management recognizes that any controls and procedures, no matter how well designed and operated, cannot provide absolute assurance that the objectives of the controls system are met, and no evaluation of controls can provide absolute assurance that all control issues and instances of fraud, if any, within a company have been detected. In addition, the design of disclosure controls and procedures must reflect that there are resource constraints and that management is required to apply judgment in evaluating the benefits of possible controls and procedures relative to their costs.
Changes in Internal Control Over Financial Reporting
The Company’s management, including its Chief Executive Officer and Chief Financial Officer, has determined that no change in the Company’s internal control over financial reporting (as that term is defined in Rules 13(a)-15(f) and 15(d)-15(f) of the Securities Exchange Act of 1934) occurred during the period ended June 30, 2025 that has materially affected, or is reasonably likely to materially affect, the Company’s internal control over financial reporting.
|
|
PART II - OTHER INFORMATION
ITEM 1. LEGAL PROCEEDINGS
The Company is not currently subject to any pending or threatened legal actions or claims.
ITEM 1A. RISK FACTORS
The Company’s business, financial condition, results of operations and cash flows may be impacted by a number of factors, many of which are beyond the Company’s control, including those set forth in the Company’s Annual Report on Form 10-K for the fiscal year ended December 31, 2024, as filed with the Securities and Exchange Commission on March 24, 2025 (the “2024 Form 10-K”).
The Risk Factors set forth in the 2024 Form 10-K should be read carefully in connection with evaluating the Company’s business and in connection with the forward-looking statements contained in this Quarterly Report on Form 10-Q. Any of the risks described in the 2024 Form 10-K could materially adversely affect the Company’s business, financial condition or future results, and the actual outcome of matters as to which forward-looking statements are made. These are not the only risks that the Company faces. Additional risks and uncertainties not currently known to the Company or that the Company currently deems to be immaterial also may materially adversely affect the Company’s business, financial condition and/or operating results.
As of the date of the filing of this document, except as disclosed elsewhere in this document, including Note 9. Subsequent Events, there have been no material changes to the Risk Factors previously disclosed in the Company’s 2024 Form 10-K.
ITEM 2. UNREGISTERED SALES OF EQUITY SECURITIES AND USE OF PROCEEDS
On May 16, 2025, the Company received a notice of conversion with respect to its 350,000 shares of Series A Convertible Preferred Stock outstanding, These shares of preferred stock were issued to an investor in 2015 and 2016 and were convertible into 72,917 shares of common stock on such date.
ITEM 3. DEFAULTS UPON SENIOR SECURITIES
Not applicable.
ITEM 4. MINE SAFETY DISCLOSURES
Not applicable.
ITEM 5. OTHER INFORMATION
During the six months ended June 30, 2025, no director or officer (as defined in Rule 16a-1(f) under the Exchange Act) of the Company adopted or terminated a “Rule 10b5-1 trading arrangement”, as such term is defined in Item 408(a) of Regulation S-K.
|
|
ITEM 6. EXHIBITS
The following documents are filed as part of this report:
* Filed herewith.
+ Indicates a management contract or any compensatory plan, contract or arrangement.
|
|
SIGNATURES
In accordance with the requirements of the Securities and Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned thereunto duly authorized.
| LIXTE BIOTECHNOLOGY HOLDINGS, INC. | ||
| (Registrant) | ||
| Date: August 7, 2025 | By: | /s/ GEORDAN PURSGLOVE |
| Geordan Pursglove | ||
| Chief Executive Officer | ||
| (Principal Executive Officer) | ||
| Date: August 7, 2025 | By: | /s/ ROBERT N. WEINGARTEN |
| Robert N. Weingarten | ||
| Vice President and Chief Financial Officer | ||
| (Principal Financial and Accounting Officer) | ||
|
|
Exhibit 10.3
AGREEMENT FOR GSK & LIXTE SUPPORTED COLLABORATIVE STUDY
| Institution: | The University of Texas M. D. Anderson Cancer Center | |
| Investigator: | Amir Jazaeri, MD | |
| GSK Investigational Product: | Dostarlimab | |
| LIXTE Investigational Product: | LB-100 | |
| Protocol Number and Title: | 219582 “Safety and Efficacy of Targeting PP2A in Ovarian Clear Cell Carcinoma (OCCC) using Dostarlimab and LB-100” | |
| Effective Date: | Last Signature Date |
THIS SUPPORTED COLLABORATIVE STUDY AGREEMENT (together with Appendices A, B, C, D, E, F and G (collectively “Agreement”) is made as of the Effective Date by and between GlaxoSmithKline LLC, with an office at 1000 Winter Street, Suite 3300, Waltham, MA 02451 (“GSK”), Lixte Biotechnology Holdings, Inc., having offices at 680 E. Colorado Boulevard, Suite 180, Pasadena, CA 91101 (“LIXTE”), and The University of Texas M. D. Anderson Cancer Center, a government agency of the State of Texas and a member institution of the University of Texas System (“System”) having an address at 1515 Holcombe Blvd, Houston, TX., 77030 (“Institution”) GSK, LIXTE and Institution collectively referred to as “Parties” or in the singular “Party,” under the following terms and conditions.
| 1. | Background. Dr. Amir Jazaeri, MD (“Investigator”), who is employed by the Institution, wishes to conduct at Institution and at Participating Site (as defined in Section 7.3), a clinical study of GSK’s proprietary drug known as Dostarlimab provided by GSK hereunder (the “GSK Investigational Product”) and LIXTE’s proprietary drug known as LB-100 provided by LIXTE hereunder (the “LIXTE Investigational Product”) each provided at no charge to Institution as necessary for the conduct of the clinical study , (collectively the “Investigational Products”) under the protocol number and title specified above (as may be amended from time to time, the “Protocol”) (such clinical study is hereinafter referred to as the “Study”). The Parties are interested in the development of scientific and medical knowledge concerning the Investigational Products and ovarian cancer, and GSK is therefore willing to supply Institution with quantities of the GSK Investigational Product as specified in Appendix A, and as outlined in Schedule 1, Appendix A and LIXTE is willing to supply Institution with the quantities of the LIXTE Investigational Product, as specified in Appendix A (“Delineation of GSK and LIXTE Investigational Product Responsibilities”). GSK is willing to provide funding to support the Study as specified in Section 3.4 below and further described in Appendix B (the “Funding”), however, the Parties agree and acknowledge LIXTE is not providing any funding or compensation to Institution or GSK for conducting the Study under this Agreement. Institution agrees to conduct the Study under the terms and conditions set forth in this Agreement. Insitution is the regulatory sponsor of the Study and is responsible for all sponsor and investigator regulatory obligations for the Study, if and to the extent applicable to the Study and if the Study is not exempt from such regulation. The Parties have identified responsibilities for this Study as indicated in Appendix C (“Task Responsibility Matrix”). Institution will notify GSK and LIXTE promptly in writing of any proposed change of the Investigator. Given that the Investigator is not a legal party to this Agreement, all references in this Agreement that purport to or arguably impose a legal obligation on the Investigator will be deemed to be a legal obligation on Institution, and Institution will be responsible for ensuring that the Investigator complies with such obligation. |
| Page |
| 2. | Compliance with Protocol/Law. |
| 2.1. | Institution will conduct the Study in accordance with (a) the Protocol (attached as Appendix D and acknowledged by the Parties as the confidential information of Institution); (b) this Agreement; (c) all applicable provisions of any and all federal, state and local laws, rules, regulations, orders and guidance relevant to the conduct of the Study including, (i) the United States Federal Food, Drug, and Cosmetic Act, as amended, and the applicable regulations promulgated under it from time to time, the Public Health Service Act, the Anti-Kickback Statute set forth at 42 U.S.C. § 1320a-7b(b), United States Code of Federal Regulations and applicable comparable state laws and regulations; (ii) the United States Health Insurance Portability and Accountability Act of 1996, as amended (“HIPAA”) and applicable comparable state laws and regulations; and (iii) publications of the International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use as adopted by the United States Food and Drug Administration (“FDA”), including current Good Clinical Practice (GCP) guidelines. Institution will be the regulatory sponsor of the Study and will fulfill, or cause the fulfillment of, all responsibilities of a regulatory sponsor (including postings on clinicaltrials.gov), if applicable to the Study and if the Study is not exempt from such regulation. If necessary, the Institution has filed or will file, and will maintain, an Investigational New Drug Application (“IND”) authorizing the Study with the FDA. Institution shall obtain and maintain, or shall cause Investigator to obtain and maintain, all other required authorizations for, and reviews of, the Study including approval of the IRB (as defined in Section 5 below) and proper oversight by all other applicable entities (e.g., ethics committees). Institution represents that the Investigator has the necessary licenses and has the expertise, time and resources to perform this Study. |
| 2.2. | Should there be any inconsistency between the Protocol and the terms of this Agreement, the language of this Agreement shall control over the Protocol or any amendments thereto, except that the terms of the Protocol (or any amendments thereto) shall govern with respect to medical, scientific and clinical issues of the Study. |
| 2.3. | The Parties will use reasonable efforts to conduct the Study in adherence with the Anticipated Timelines set out in Appendix E (“Anticipated Timelines”). Any material change, deviation, delay, modification, or reasonably expected delay to the Anticipated Timelines of which a Party becomes aware will be documented in writing to the other Parties for awareness and possible resolution. |
| 3. | GSK & LIXTE Support |
| 3.1. | Investigational Products Supply and Accountability. GSK and LIXTE each agree to provide Institution and Investigator with the quantities of GSK Investigational Product and LIXTE Investigational Product, respectively, as specified in Appendix A. For the avoidance of doubt, Institution and Investigator are performing the Study independently of GSK and LIXTE, and GSK and LIXTE have not determined the design of the Protocol and are not responsible for its implementation. Institution or its authorized designee will: (a) verify receipt of the GSK and LIXTE Investigational Products by signing the appropriate documentation provided by GSK and LIXTE or their respective designee; (b) handle and store all Investigational Products securely and in compliance with all applicable laws, rules and regulations and as specified in the Protocol, the applicable Investigational Products labeling and/or in writing by GSK or LIXTE, as the case may be; (c) be responsible for any additional labeling (e.g., for dispensing the Investigational Products for the Study) in compliance with and as required by applicable laws and the Protocol; (d) maintain complete and accurate records of use and disposition of the Investigational Products; (e) only dispense, or permit to be dispensed, the GSK Investigational Product and LIXTE Investigational Product supplied under this Agreement to Study subjects in accordance with the Protocol; and (f) after completion or early termination of the Study, at GSK’s or LIXTE’s respective expense, (i) return to GSK (or its designee) or destroy (as instructed by GSK) all unused GSK Investigational Product in accordance with the Protocol, applicable laws and regulations, and any reasonable written instructions provided by GSK to Institution, and, in the event of destruction of GSK Investigational Product, provide GSK, upon request, with a certificate/documentation of destruction to GSK’s reasonable satisfaction, and (ii) return to LIXTE (or its designee) or destroy (as instructed by LIXTE) all unused LIXTE Investigational Product in accordance with the Protocol, applicable laws and regulations, and any reasonable written instructions provided by LIXTE to Institution, and, in the event of destruction of LIXTE Investigational Product, provide LIXTE, upon request, with a certificate/documentation of destruction to LIXTE’s reasonable satisfaction. Notwithstanding the foregoing, Institution will have the right to destroy expired Investigational Products after thirty (30) days of the Investigational Products’ respective expiration date, unless otherwise agreed to by the respective Parties. Additionally, Institution and Investigator shall ensure that any Study subject clinical samples collected for purposes of the Study as required by the Protocol to be tested during and in the course of the Study are tested in accordance with the Protocol or are otherwise taken in accordance with standard of care. Investigational Products shall, at all times remain solely under the responsibility and overall control of the Institution until used in the Study, returned or destroyed. Institution and Investigator shall not supply or permit any other person to supply the Investigational Products to any third party, that is, any person who is not participating in the Study as a member of the Study Personnel (defined in Section 7.2 below) or a subject enrolled in the Study. GSK and LIXTE shall supply the GSK and LIXTE Investigational Product only after and as long as the Institution has obtained and holds the necessary authorizations and/or approvals for the Study in accordance with Section 5 below. |
| Page |
(a) Neither GSK nor LIXTE will be obligated to provide any quantity of the Investigational Products and/or Funding other than as specified in Appendix A and Appendix B, respectively, unless such obligation to provide additional Investigational Products and/or Funding is included in a written amendment to this Agreement signed by Institution, GSK and LIXTE.
(b) GSK & LIXTE will provide Institution and/or Investigator label proofs of such Investigational Products shipments for review, which are in compliance to FDA criteria, and understand that all Investigational Products should be shipped to Institution in accordance with Appendix G, provided that GSK and LIXTE are provided written details of shipping details for the Participating Sites from the Institution and/or Investigator so that GSK and LIXTE can ship Investigational Products directly to Participating Sites.
| 3.2. | Institution will not, and will ensure Investigator does not, and Institution will not (and will ensure Investigator does not) permit any third parties to, modify or reverse engineer, combine with another material, or formulate the Investigational Products or use the Investigational Products, in each case other than specified in this Agreement or the applicable Protocol without GSK’s and LIXTE’s prior written agreement for their respective Investigational Product. |
| 3.3. | Misconduct. If GSK or LIXTE reasonably believes with sufficient evidence there has been any research misconduct in relation to the Study, GSK or LIXTE will inform Institution and the Investigator and Institution will reasonably cooperate, and will cause Investigator to reasonably cooperate, to conduct a thorough investigation into any alleged research and, to the extent permitted by law, provide GSK or LIXTE with any applicable findings and/or outcomes. |
| 3.4. | Funding. GSK agrees to provide the Funding to Institution for the Study as set forth in Appendix B. Institution agrees that the amounts payable or otherwise provided by GSK under this Agreement represent amounts actually and reasonably required to enable the work to be performed by Institution and Investigator in connection with the Study and have not been determined in a manner that takes into account the volume or value of any referrals or business. Institution or its authorized designee will maintain complete and accurate records of the use and disposition of the Funding. Institution represents that any additional funding it may obtain to fund the Study and/or which is used in connection with the Study will not impose any obligations, restrictions and/or conditions on Institution, Investigator and/or any Participating Site or Sub-investigator that may conflict with the rights of GSK and LIXTE (including rights of GSK and LIXTE under Section 13 below and/or the obligations of Institution and Investigator set out in this Agreement). |
(i) Institution agrees and acknowledges that the Funding received from GSK will not be used to support any type of medical education programs (e.g., Continuing Medical Education, Independent Medical Education).
| Page |
(ii) No services by Institution or Investigator or any payments by GSK to Institution under this Agreement are related to or for any promotional or marketing activities.
(iii) Institution shall receive Funding from GSK on behalf of the Participating Site(s) and shall ensure that the Participating Site(s) are promptly and accurately reimbursed for their participation in this Study. GSK shall have no further responsibility to provide any further financial reimbursement to either Institution or any Participating Site(s) over and above that set out in this Agreement.
| 3.5. | Declaration of GSK and LIXTE Support. Subject to Section 11 below, Institution agrees to accurately describe GSK’s and LIXTE ‘s support for the Study in accordance with all applicable laws, regulations and institutional or publication policies applicable to the activities authorized by this Agreement. Institution agrees that: (a) all claims that Institution submits for reimbursement to any federal healthcare program or third party payor for any procedure that involves the Funding or that involves any Investigational Products provided by or on behalf of GSK and/or LIXTE at no cost to Institution will accurately reflect the provision of such Funding or supply by or on behalf of GSK and/or LIXTE; and (b) Institution will not seek reimbursement from any federal healthcare program or third party payor for any amounts paid, or Investigational Products supplied, by GSK and/or LIXTE under this Agreement. |
| 4. | Reports, Audits and Use of Study Data and Biological Samples. |
| 4.1. | Reports and Audits. Institution will maintain complete and up-to-date medical and other records relating to the Study and will keep GSK and LIXTE informed of the Study’s results and enrollment status through written reports to be provided monthly or as otherwise reasonably requested by GSK and/or LIXTE. The information to be provided in such reports will include the number of subjects enrolled in the Study and achievement of Study milestones. Subject to Section 11.1 below, Institution and/or Investigator will collect Study Data (as defined in Section 4.2 below) from the Participating Sites and will provide all Study Data (collected by Institution and Participating Sites), suitably de-identified, (as defined in HIPPA), to GSK and LIXTE (within such timeframe as is agreed in writing with GSK and LIXTE) after such Study Data is recorded in the Study database after final database freeze, or at such time that the Investigator provides the Study Data from the Study to a third party to the extent permitted hereunder, or upon termination of this Agreement as provided in Section 14.3, whichever is the earliest date. The Study Data will be provided in such format as GSK and LIXTE may reasonably request. Institution shall cause Investigator to provide each of GSK and LIXTE with a final Study report in the form of a draft manuscript, detailing the methodology, results and containing an analysis of the Study and drawing appropriate conclusions, conforming to International Committee of Medical Journal Editors guidelines and suitable for submission to a peer-reviewed journal within ninety (90) days after completion or early termination of the Study, whichever occurs first, and such manuscript will be submitted to GSK and LIXTE in accordance with Section 13 of this Agreement. At mutually agreeable times during normal administrative business hours and upon advance notice during the Study and for a period of one (1) year after completion of Study, Institution will give GSK and LIXTE and their respective designees access to all records and documentation (however stored) relating to the Study or to the care of Study subjects, in order for GSK and LIXTE to monitor the Study for source document verification and/or audit purposes. Institution and/or Investigator will also make those records and documents available for the purposes of any audit by a regulatory authority and agree not to destroy those records and documents without first giving GSK and LIXTE written notice and the opportunity to store them at GSK’s and/or LIXTE’s expense. Institution will ensure that all transactions under this Agreement are properly and accurately recorded in all material respects on its books and records and each document upon which entries in such books and records are based is complete and accurate in all material respects. Institution will maintain a system of internal accounting controls reasonably designed to ensure that it maintains no off-the-books accounts. GSK’s and/or LIXTE’s access rights under this Agreement shall be subject to GSK’s and/or LIXTE’s compliance, in connection with accessing Institution’s systems or at Institution’s facility, with Institution’s reasonable measures for purposes of confidentiality, safety, and security, and will be further subject to GSK’s and/or LIXTE’s compliance with Institution’s premises rules that are generally applicable to all persons at Institution’s facilities. Should GSK and/or LIXTE utilize one or more third party(s) in exercising its audit or inspection rights in this paragraph, GSK and/or LIXTE will ensure that such party(ies) shall be subject to an obligation of confidentiality consistent with the obligations of confidentiality required of GSK and/or LIXTE hereunder and such third party(ies) shall be subject to any and all conditions upon GSK’s and/or LIXTE’s access rights that are set forth under this Agreement. If GSK and/or LIXTE obtains, learns of, comes in contact with, or otherwise has access to any patient health and medical information in connection with the Study, GSK and/or LIXTE will keep such information confidential and will comply with all applicable laws regarding the confidentiality of such information, and GSK and/or LIXTE will not use or disclose such patient health and medical information in a manner that would violate any applicable law (including HIPAA) if such use or disclosure were made by Institution. References to “business days” in this Agreement means calendar days excluding Saturdays and Sundays and Study site holidays observed by Study site’s administrative staff, and “business hours” will refer to standard administrative work hours (generally between 8:00 am and 5:00 pm local time) on such business days. |
| Page |
| 4.2. | Use of Study Data. Institution shall own the Study Data and shall ensure that the Participating Site(s) and all Study Personnel, including Investigator and Sub-investigator(s), assign all right, title and interest in Study Data to Institution (or to Participating Site(s) for further assignment to Institution). Institution hereby grants to GSK, LIXTE, and their respective affiliates a fully sublicensable, royalty-free, fully paid-up, perpetual, irrevocable, transferable, worldwide right and non-exclusive license to use all data and information resulting from the Study, including any and all data and information arising from any analyses of Biological Samples (as defined below) conducted by or on behalf of Institution, Investigator and Sub-investigator(s) in accordance with the Protocol (collectively the “Study Data”), for any and all purposes, including for inclusion in GSK’s and LIXTE’s or their respective affiliates’ patent filings and regulatory submissions for their respective Investigational Products. All other data collected or created pursuant to or prepared in connection with the Protocol other than Study Data, including medical records, laboratory notebooks, and source documents, and all other primary data sources underlying data recorded on the case report forms (CRFs) (collectively “Source Records”) shall remain at Institution as property of Institution and shall be available for inspection in accordance with Section 4.1. Institution shall have the right to publish Study Data only in accordance with Section 12 below and following any such publication Institution shall have the right to use such published Study Data for any purpose. Institution shall then have the right to use any unpublished Study Data only for internal, non-commercial research purposes and/or in connection with Study subject care. Institution discloses the Study Data to GSK and LIXTE in confidence, and GSK and LIXTE should keep the Study Data confidential until the earlier of (i) publication or other public presentation of such Study Data by Institution in accordance with Section 12 of this Agreement, or (ii) twelve (12) months after the conclusion of the Study. Notwithstanding the foregoing, GSK and LIXTE and their respective affiliates may (a) perform analysis of such Study Data as GSK and LIXTE, respectively, deems appropriate, and GSK and LIXTE, respectively, may use such Study Data and analyses as it wishes for its internal purposes, which shall include, without limitation, disclosure to any third parties to the extent that they may be assisting GSK or LIXTE or their respective affiliates with any analysis, subject to that third party agreeing to keep such Study Data and analyses confidential on the same terms as provided herein, and (b), with the consent of the other Parties, not to be unreasonably withheld, include such Study Data in patent filings and regulatory submissions for their respective Investigational Products. Institution and/or Investigator will ensure that GSK and LIXTE are named in the ICF(s) (as defined in Section 5 below) and in the HIPAA authorization form(s) or analogous documents if signed separately from the ICF (“HIPAA Authorization(s)”) (each, a “Consent Document”), as parties to whom Study subjects’ protected health information (as that term is defined in HIPAA) (“PHI”) may be disclosed in connection with the Study, and that such Consent Document(s) will permit GSK, LIXTE and their respective designees access to Study subjects’ PHI and Source Records as may be necessary to audit the Study and to use the Study Data and Biological Samples (defined in Section 4.3 below), including for research and drug development purposes. Subject to Institution’s rights herein to publish Study Data and to use such published Study Data, Institution shall not and shall procure that the Participating Site(s) shall not, give access to the unpublished Study Data to any third party other than GSK (or GSK’s nominees) or LIXTE (or LIXTE’s nominees) during the term of this Agreement and for a period of five (5) years from the date of completion of the Study without the prior written consent of GSK and LIXTE. Institution shall notify all organizations to whom it grants access to the Study Data of its obligations under this Section and shall require such organizations to give similar undertakings for the benefit of GSK and LIXTE. Notwithstanding anything to the contrary herein, GSK and LIXTE shall access, disclose, and use any patient identifiable information provided to it/them or which it/they obtains or comes in contact with under this Agreement only as (1) authorized by an applicable Study subject’s ICF and/or HIPAA authorization; and (2) as permitted by applicable law and Institution’s IRB. |
| 4.3. | Biological Samples. |
| 4.3.1. | Definition. “Biological Samples” means blood, fluid and/or tissue samples collected from Study subjects for purposes of the Study as set forth in the Protocol, and tangible materials directly or indirectly derived from such samples. |
| 4.3.2. | Use of Biological Samples. Institution will collect, retain and/or use Biological Samples only as set forth in the Protocol. Institution and/or Investigator will provide GSK and LIXTE with quantities of Biological Samples as required by the Protocol. GSK and LIXTE may use such Biological Samples as specified in the Protocol and as permitted in the Consent Documents and by applicable law. The Consent Document(s) will permit GSK, LIXTE and their respective affiliates and designees access to Biological Samples, including for research and drug development purposes. |
| 4.3.3. | Stored Biological Samples. Institution and/or Investigator are permitted, at their own expense, to transfer Biological Samples to a licensed biorepository and store the Biological Samples at the licensed biorepository after completion or early termination of the Study (the “Stored Biological Samples”). During the Term of this Agreement, Institution and Investigator may use the Stored Biological Samples as specified in the Protocol and as permitted in the Consent Documents and by applicable law. All uses of Stored Biological Samples and any data or information arising therefrom are subject to the terms of this Agreement. |
| Page |
| 4.3.4. | Non-Permitted Use of Stored Biological Samples. Institution and/or Investigator will not use, and will ensure that the Participating Site(s) and other Study Personnel will not use, Biological Samples or Stored Biological Samples for any purpose other than as permitted under this Agreement and the Protocol. In the event that Institution, Participating Site(s) and/or any Study Personnel (including Sub-investigator(s)) and/or Investigator) use Biological Samples or Stored Biological Samples for any purpose other than as permitted under this Agreement or the Protocol (a “Non-Permitted Use”), Institution agrees that GSK shall be the sole and exclusive owner of any and all inventions made or invented (as such term is defined under U.S. Patent Law) in connection with the Non-Permitted Use that are solely related to GSK Confidential Information or GSK’s Investigational Product. Institution agrees that LIXTE shall be the sole and exclusive owner of any and all inventions made or invented (as such term is defined under U.S. Patent Law) in connection with the Non-Permitted Use that are solely related to LIXTE Confidential Information or LIXTE’s Investigational Product. Institution agrees that any and all inventions made or invented (as such term is defined under U.S. Patent Law) in connection with the Non-Permitted Use that are related to both GSK Confidential Information and LIXTE Confidential Information or both of GSK’s and LIXTE’s Investigational Products or that are directed to the combination therapy of the GSK Investigational Product with the LIXTE Investigational Product (“Combined Non-Permitted Use Inventions”) will be the joint property of GSK and LIXTE, and GSK and LIXTE will be the exclusive joint owners of any such Combined Non-Permitted Use Inventions. In the event of any Combined Non-Permitted Use Invention, GSK and LIXTE will negotiate in good faith an agreement that addresses each of GSK’s and LIXTE’s rights and obligations related to such Combined Non-Permitted Use Invention. Institution shall and hereby does assign any and all inventions described above in accordance with the foregoing and made as a result of a Non-Permitted Use to GSK and/or LIXTE (in accordance with the foregoing allocation of ownership) and shall execute and deliver, and shall cause its employees (including Investigator) to execute and deliver, any and all documents, including those for assignment and conveyance to memorialize such ownership of GSK and/or LIXTE in the invention and of all intellectual property rights therein consistent with Section 13 below. Notwithstanding the foregoing, any inventions made or invented by Institution personnel in connection with a Non-Permitted Use that are not owned by GSK and/or LIXTE in accordance with the foregoing shall be solely owned by Institution; provided that, Institution hereby grants to GSK, LIXTE, and their respective affiliates a non-exclusive, fully paid-up, worldwide, perpetual, irrevocable, royalty-free license to use such solely-owned Institution inventions for all purposes. For the avoidance of doubt, if any Participating Site or Sub-investigator is authorized under the Protocol, the Consent Documents and applicable law to use any Biological Samples or Stored Biological Samples, Institution shall ensure that the terms of this Section 4.3.4 are incorporated, mutatis mutandis, in the contract executed by Institution and the Participating Site in accordance with Section 7.4.1, such that GSK and LIXTE receive the same rights, assignments and licenses from Participating Site(s) as GSK and LIXTE would have received if Institution had performed the activities conducted by the Participating Site(s). |
| 5. | Institutional Review Board, Informed Consent Form, Review and Approvals. Initiation of the Study according to the Protocol shall not begin until the relevant Institutional Review Board (“IRB”) approval is obtained at Institution, and at Participating Site(s) for their Study initiation, and GSK and LIXTE have each been informed in writing of such approvals. Before submission to its IRB, Institution shall ensure Investigator supplies GSK and LIXTE with a copy of the informed consent form that is to be signed by all subjects enrolled in the Study (together with any amendments thereto, the “ICF”) for GSK’s and LIXTE ‘s review and approval. If a HIPAA Authorization form that is separate from the ICF will be used for the Study, then Institution shall ensure that Investigator also supplies GSK and LIXTE with a copy of such HIPAA Authorization form for GSK’s and LIXTE’s review and approval including, if applicable, prior to any submission to its IRB. Each Party will cooperate in the amendment of the HIPAA Authorization as may be necessary from time to time to comply with HIPAA to the extent HIPAA applies to such Party, and to ensure that the Study Data may be disclosed to and used by GSK and LIXTE and their respective affiliates and designees for the purposes contemplated by this Agreement and to the extent permitted by the IRB. Institution and/or Investigator shall inform GSK and LIXTE in writing of the IRB’s continuing reviews of the Study promptly after each such review takes place, which shall take place regularly as determined by the IRB of record and applicable law. If a Participating Site’s IRB requires material changes to its ICF and/or HIPAA Authorization that would impact the use of such site’s Study Data, Institution shall ensure that GSK and LIXTE review and approve such changes before that ICF and/or HIPAA Authorization are used at Participating Site. |
| 6. | Protocol and Informed Consent Form Changes. Institution and Investigator shall ensure that the Protocol is submitted to GSK for review and approval and submitted to LIXTE for review and approval. Institution and Investigator will not make any changes to the Protocol or the Consent Documents without first informing GSK and LIXTE of any such change and obtaining the written approval of the IRB, GSK, and LIXTE and, if necessary, making required amendments to the IND. Institution shall ensure that Investigator promptly incorporates into the ICF and Protocol any new core safety data of the Investigational Products provided by GSK and/or LIXTE and to promptly seek or procure approval of the IRB for such revised ICF. Institution, through Investigator, will be responsible for providing GSK and LIXTE with a copy of the final Protocol and Consent Documents approved by the IRB. The Protocol will be considered final after it is approved by the IRB. |
| Page |
| 7. | Personnel. |
| 7.1. | Institution and Investigator. Institution represents and certifies that Institution and Investigator have the necessary experience to conduct the Study and Institution is and shall remain throughout the Study, authorized to enter into this Agreement. Investigator will lead and direct the conduct of the Study as principal investigator at Institution. |
| 7.2. | Study Personnel. Study Personnel shall include any individual by way of example, engaged by Institution, Participating Site and/or Investigator who (a) participates in the conduct of the Study; (b) is privy to information related to the scientific elements of this Study that have the potential to give rise to any Inventions (defined in Section 13.2 below) or rights related to such Inventions; and/or (c) is privy to any other Confidential Information (defined below) and such other individuals set forth in the Study proposal and Study budget provided to GSK and/or LIXTE (“Study Personnel”). For avoidance of doubt, Sub-investigator(s) (as defined in Section 7.4 below) is/are Study Personnel. |
| 7.3. | Qualified Personnel. Institution will ensure (and will cause each Participating Site (as defined below) to ensure, with respect to each Sub-investigator) that all Study Personnel conducting the Study (a) are qualified to conduct the Study; (b) are subject to the terms under this Agreement or substantially similar to those outlined under this Agreement; (c) are obligated in writing or by the terms of their employment to give ownership to Institution (or to the applicable Participating Site, in the case of each Sub-investigator) of any rights they might have in the results of their work; and (d) will do so under the direction of the Investigator at Institution and under the direction of the Sub-investigator(s) at their respective Participating Site(s), if applicable, with the prior approval and ongoing oversight of relevant competent governmental authorities. “Participating Site” means such other institution and its facilities where a Sub-Investigator is employed or under contract with, as Institution may invite to participate, or which from time to time participates, in the Study and references to “Participating Sites” shall be to any one or more of such institutions. |
| 7.4. | Sub-investigators. Each of GSK and LIXTE hereby consents to the engagement by Institution of external sub-investigator(s) (“Sub-investigator(s)”) and their Participating Sites to participate in the conduct of the Study. Institution and Investigator will provide GSK and LIXTE with a list of the Sub-investigator (s) and their Participating Site(s) promptly upon request. Institution and Investigator will notify GSK and LIXTE promptly in writing of any subsequent changes to such list and will submit IRB approval letters to GSK and LIXTE promptly for any additional Participating Site(s). In addition, Institution will: |
| 7.4.1. | contract with Participating Site(s) with respect to the Study under the terms and conditions consistent with the terms under this Agreement such that GSK and LIXTE receive the same rights from Participating Site(s) as GSK and LIXTE would have received if Institution had performed the activities conducted by the Participating Site(s), including under the terms relating to confidentiality, intellectual property (including the assignments, licenses and option rights under Section 4.3.4 and Section 13), ICFs, publication, insurance, safety reporting, GSK’s and LIXTE’s right to audit, drug accountability, Biological Samples and Stored Biological Samples and compliance with laws and regulations; |
| 7.4.2. | ensure that the Sub-investigator(s) assign to their Participating Site(s), and shall cause the Participating Site(s) to assign to Institution, any and all an obligation to assign any intellectual property made by Sub-investigator(s) as a result of their participation in the conduct of the Study in order for Institution to fulfill its obligations to GSK and LIXTE regarding inventions under Section 4.3.4 and Inventions (as defined in Section 13.2 below) under this Agreement; and |
| Page |
| 7.4.3. | be solely responsible for providing shipping information to GSK and LIXTE for their distribution of Investigational Products to Sub-investigator(s), and for providing accountability information from Sub-investigator(s) for Investigational Products to the extent permitted by applicable law. |
| 8. | No Conflicts or Debarment. Institution will ensure that Institution, its trustees, officers and directors, Investigator, Participating Site(s), Sub-investigator(s), and Study Personnel: (a) are under no contractual or other obligation or restriction that is inconsistent with Institution’s and Investigator’s performance of or obligations under this Agreement; and (b) do not have a financial or other interest in GSK or LIXTE or the outcome of the Study that might interfere with their independent judgment. Institution will ensure that Institution, the Study Personnel and any and all Participating Site(s), and GSK and LIXTE, respectively, will ensure that it and any of its personnel who access Institution’s facilities, premises, or systems under this Agreement have not been, and are not under consideration to be (i) debarred from providing services pursuant to Section 306 of the United States Federal Food, Drug and Cosmetic Act 21 U.S.C. § 335a; (ii) excluded, debarred or suspended from, or otherwise ineligible to participate in any federal or state health care programs or federal procurement or non-procurement programs (as that term is defined in 42 U.S.C. § 1320a-7b(f)); (iii) disqualified by any government or regulatory agencies from performing specific services, and are not subject to a pending disqualification proceeding; or (iv) convicted of a criminal offense related to the provision of health care items or services, or under investigation or subject to any such action that is pending. During the Study and for a period of two (2) years following completion or early termination of the Study, each Party will notify the other Parties promptly if such party or any of its personnel involved in the conduct of the Study (or in the case of GSK and LIXTE any of their respective personnel who access Institution’s facilities, premises, or systems under this Agreement) are subject to the foregoing, or if any action, suit, claim, investigation, or proceeding relating to the foregoing is pending, or to the best of such Party’s knowledge, is threatened. |
| 9. | Safety Data Reporting. |
| 9.1. | Reporting. Institution is responsible for reporting all serious adverse events (SAE) (as defined in the Protocol) to regulatory authorities in the appropriate time frame as required by applicable law and as outlined in the Protocol and Appendix F. |
| 9.2. | Institution will promptly notify GSK and LIXTE of all SAEs, Special Situations (as defined in the Protocol), pregnancies, exposure via breastfeeding, and other safety information in accordance with the timelines and procedures specified in the Protocol and Appendix F. In addition, Institution will reasonably obtain and provide follow-up information as available, to GSK and LIXTE upon request. |
| 9.3. | Follow-Up Information. Institution and Investigator will assist GSK and LIXTE in investigating any SAE and will provide any follow-up information reasonably requested by GSK and LIXTE and consistent with applicable law. |
| 9.4. | Regulatory Reporting. Reporting an SAE to GSK and LIXTE does not relieve Investigator or Institution of responsibility for reporting it to the applicable regulatory authority, if and as required. |
| 10. | Communications with Regulatory Agencies. Institution shall ensure that the Investigator will |
(a) notify GSK and LIXTE of any communications from or to any regulatory authority received by the Investigator, including any safety reports, having an impact on the Investigational Products and the Study;
(b) include GSK and LIXTE in any discussions (other than immaterial discussions, such as for scheduling purposes) or meetings between Investigator and the FDA and/or other regulatory agencies regarding the Study unless prohibited by law or such regulatory agencies;
| Page |
(c) supply GSK and LIXTE with a copy of any correspondence from the FDA received by the Investigator regarding the Study, including any IND, approval letter, and any other IND-related correspondence unless prohibited by law or such regulatory agencies; and inform GSK and LIXTE (and provide a copy to GSK and LIXTE, if written) of (i) any communications from a regulatory and/or government agency regarding the performance of the Study, unless prohibited by law or such regulatory agencies; (ii)any request made by a regulatory authority to audit or inspect the Study Data/Participating Site(s) and/or the activities of the Investigator, relating to the Study; and/or (iii) the withdrawal or amendment of any IRB approval/regulatory authorization relating to the Study;
(d ) allow GSK and LIXTE a reasonable opportunity to comment on any correspondence being sent by the Investigator for submission to the FDA regarding the Study, including any submitted IND and IND annual reports.
Notwithstanding anything to the contrary herein, Institution does not provide GSK or LIXTE any records maintained by Institution’s Investigational New Drug Office or any access thereto.
| 11. | Confidentiality. |
| 11.1. | Medical Confidentiality. The Parties agree to adhere to the principles of medical confidentiality in relation to Study Subjects involved in the Study. Institution shall not and shall ensure that the Investigator does not disclose to GSK or LIXTE or their affiliates PHI pertaining to Study subjects, unless required directly or indirectly for the purpose of adverse event reporting or otherwise requested by GSK and/or LIXTE in conformance with the relevant Consent Document. GSK and LIXTE shall not disclose the identity of Study subjects disclosed to it/them for this purpose to third parties without prior written consent of the Study subject, except in accordance with the requirements of all applicable laws and statutes relating to data protection. |
| 11.2. | “Confidential Information” refers to non-public information of any kind related to the Study which is disclosed by GSK or LIXTE (each, a “Discloser”) to another Party (“Recipient”) for purposes of conducting the Study, either directly or indirectly, in writing, orally or by inspection of tangible objects, including, without limitation, information and materials regarding the Discloser’s technology, products, product candidates, research and development activities, results, compound designs or structures, manufacturing or other processes or methods, know-how, inventions or other intellectual property, the existence or content of licenses, the existence, status or content of licensing or collaboration negotiations, other agreements with third parties, information regarding facilities and financial and other business information, in each case whether or not identified or marked as “confidential” (if, by the nature of the information, it would reasonably constitute proprietary or confidential information) and as such are disclosed or made available to the Recipient. Discloser’s Confidential Information may also include information obtained by Discloser from its collaborators, customers, suppliers, vendors and other third parties who have entrusted their confidential information to Discloser. During the Study and for a period of seven (7) years after the termination or expiration of this Agreement, each Recipient agrees not to (a) publish, disseminate or otherwise disclose, deliver or make available the Confidential Information of any Discloser to any third party other than competent governmental authorities, Sub-investigator(s), Participating Site(s), other Study Personnel and GSK’s affiliates or designees (in the case of GSK’s Confidential Information) and LIXTE ‘s affiliates or designees (in the case of LIXTE’s Confidential Information), and then only for the purpose of conducting the Study; or (b) make use of any Confidential Information other than in the conduct of the Study or as permitted under this Agreement. |
| 11.3. | Exceptions to Confidential Information. The obligations of nondisclosure and limited use do not apply with respect to any of the Confidential Information that: |
| 11.3.1. | is or becomes public knowledge through no breach of this Agreement by Recipient; |
| 11.3.2. | is disclosed to Recipient by a third party entitled to disclose such information without an obligation of confidentiality to Discloser; |
| Page |
| 11.3.3. | is already known to Recipient prior to disclosure hereunder, or is independently developed by Recipient without use of Discloser’s Confidential Information, as shown by Recipient’s written records or competent documentation; |
| 11.3.4. | is necessary to obtain IRB approval of Study or required to be included in the written information summary provided to Study subject(s) and/or informed consent form or as necessary to be disclosed to a Study subject or potential subject in the informed consent process; |
| 11.3.5. | is released with the prior written consent of the Discloser; or |
| 11.3.6. | is required to support the medical care of a Study subject. |
| 11.4. | Recipient may disclose the Discloser’s Confidential Information to the extent that it is required to be produced pursuant to a requirement of applicable law, IRB, government agency, an order of a court of competent jurisdiction, or a facially valid administrative, Congressional, or other subpoena, provided that subject to the requirement, order, or subpoena the Discloser is notified if legally permissible and Recipient cooperates with Discloser’s efforts to limit the scope of the information to be provided or to obtain an order protecting the information from public disclosure. To the extent allowed under applicable law, Discloser may seek to limit the scope of such disclosure and/or seek to obtain a protective order. Recipient will disclose only the minimum amount of Confidential Information necessary to comply with such requirement, law, subpoena or court order as advised by Recipient’s legal counsel. |
| 11.5. | Upon Discloser’s written request, Recipient agrees to return all Confidential Information supplied to it by Discloser, at Discloser’s expense, pursuant to this Agreement except that Recipient may retain an archival copy of such Confidential Information in a secure location for purposes of identifying and satisfying its obligations and exercising its rights under this Agreement. |
| 11.6. | Any Party may disclose the existence and terms of this Agreement as and to the extent necessary to ensure compliance with applicable Federal, State and Institutional policies, regulations, and laws, and GSK and LIXTE may disclose the existence and terms of this Agreement to their respective actual or prospective licensees, sublicensees, collaborators, strategic partners and/or acquirers under confidentiality terms at least as stringent as those hereunder (but of commercially reasonable duration). |
| 11.7. | Institution agrees to not provide GSK’s Confidential Information to LIXTE and to not provide LIXTE’s Confidential Information to GSK without GSK’s and LIXTE’s respective written consent. Notwithstanding the foregoing, to the extent either GSK or LIXTE receives Confidential Information of the other in connection with this Agreement, the obligations of non-use and non-disclosure under this Section 11 will apply to it with respect to such Confidential Information of the other Party. |
| 12. | Publication. |
| 12.1. | GSK and LIXTE recognise that Institution and the Investigator intend that results of scientific interest arising from the Study will be appropriately published and disseminated. Institution and/or Investigator may publish or disclose the results of the Study; provided, that (a) a copy of any proposed disclosure, including any publication, abstract, manuscript, note, slides, oral presentation, poster, or any other document or material whether in written, electronic or printed form and, where publication is intended to be verbal presentation only, any other copy of the intended script, oral presentation, or poster that reports all or parts (interim or final) of the results of the Study or Study progress, is given to GSK and LIXTE for review at least thirty (30) days prior to the date of submission for publication (including abstracts) or of public disclosure; (b) if requested by GSK in writing during such review period, any reference to GSK’s Confidential Information is deleted, and if requested by LIXTE in writing during such review period, any reference to LIXTE’s Confidential Information is deleted; and (c) Institution and/or Investigator defer publication or disclosure for up to an additional sixty (60) days from the time GSK and/or LIXTE notifies Institution and/or Investigator in writing during such review period that GSK and/or LIXTE (i) desires patent application(s) to be filed on any Invention described in the proposed disclosure or (ii) desires to take any other measures as GSK or LIXTE considers necessary to establish, register, protect and/or preserve any intellectual property relating to the GSK Investigational Product and/or LIXTE Investigational Product. Institution and Investigator will ensure that any publication or public disclosure of the results of the Study proposed by a Sub-investigator(s) is/are made in accordance with this Section 12. |
| Page |
| 12.2. | Institution and/or Investigator agrees that all reasonable comments made by GSK or LIXTE in relation to a proposed publication by Institution and/or Investigator will be reasonably considered for incorporation by Institution and/or Investigator into the publication. Institution and/or Investigator shall ensure that any of GSK’s Confidential Information is removed from the proposed publication and any of LIXTE’s Confidential Information is removed from the proposed publication. |
| 13. | Intellectual Property. |
| 13.1. | Pre-Existing Intellectual Property. It is expressly agreed that no Party transfers to another Party by operation of this Agreement any patent right, copyright, or other proprietary or property right of any kind any Party owns as of the Effective Date. Nothing herein is intended to grant to any Party any license or other rights in and to such pre-existing or independently developed intellectual property of another Party, except for Institution’s limited right to use the Investigational Products, LIXTE Confidential Information, and GSK Confidential Information solely for the purposes of conducting the Study. |
| 13.2. | Inventions/Investigational Product Inventions. “Inventions” means any inventions, discoveries, know-how, and improvements (including new uses and improvements of the Investigational Products), and all intellectual property rights therein, whether or not protectable under patent or other intellectual property law, that are made or invented by Institution personnel, Investigator, Study Personnel (alone or with others), or any Participating Site(s) personnel, or Sub-investigator(s), (a) during and resulting from performance of the Study or the use of the GSK Investigational Product and/or the LIXTE Investigational Product; or (b) through the use of GSK’s Confidential Information and/or LIXTE’s Confidential Information under this Agreement during the Term of this Agreement and during the secrecy period described in Section 11.2 above. Each Party agrees that: |
13.2.1 any Invention that incorporates, is based on, refers to or relates solely to GSK Confidential Information or solely to the GSK Investigational Product, or the modification, use or manufacture thereof, (including Inventions that generically encompass within their scope the GSK Investigational Product, or Inventions relating to the use of the GSK Investigational Product), including any Invention that involves identification or use of biomarkers related to the safety, efficacy, or use of the GSK Investigational Product, but excluding Combined Therapy Inventions (as defined in Section 13.2.3 below) (“GSK Investigational Product Inventions”) shall be promptly disclosed by Institution to GSK in writing (and Institution shall ensure that any GSK Investigational Product Invention is promptly disclosed to Institution by all Study Personnel and Participating Site(s)) and shall be the sole and exclusive property of GSK. Institution shall assign and does hereby assign to GSK all right, title, and interest in the United States and throughout the world to all GSK Investigational Product Inventions (and, for the avoidance of doubt, Institution shall ensure that all Study Personnel, including Investigator and Sub-investigator(s), have assigned to Institution or Participating Site(s), as applicable, (and that Participating Site(s) have assigned to Institution) all right, title, and interest in the United States and throughout the world to all GSK Investigational Product Inventions). As agreed solely between GSK and LIXTE and without any obligation on the part of Institution, LIXTE will have no right and license to Study Data generated from the use of and related solely to GSK’s Investigational Product (for clarity, not in combination with LIXTE’s Investigational Product) or GSK Confidential Information, without GSK’s written consent.
| Page |
13.2.2 any Invention that incorporates, is based on, refers to or relates solely to LIXTE Confidential Information or solely to the LIXTE Investigational Product, or the modification, use or manufacture thereof, (including Inventions that generically encompass within their scope the LIXTE Investigational Product, or Inventions relating to the use of the LIXTE Investigational Product), including any Invention that involves identification or use of biomarkers related to the safety, efficacy, or use of the LIXTE Investigational Product, but excluding Combined Therapy Inventions (“LIXTE Investigational Product Inventions”) shall be promptly disclosed by Institution to LIXTE in writing (and Institution shall ensure that any LIXTE Investigational Product Invention is promptly disclosed to Institution by all Study Personnel and Participating Site(s)) and shall be the sole and exclusive property of LIXTE. Institution shall assign and does hereby assign to LIXTE all right, title, and interest in the United States and throughout the world to all LIXTE Investigational Product Inventions (and, for the avoidance of doubt, Institution shall ensure that all Study Personnel, including Investigator and Sub-investigator(s), have assigned to Institution or Participating Site(s), as applicable, (and that Participating Site(s) have assigned to Institution) all right, title, and interest in the United States and throughout the world to all LIXTE Investigational Product Inventions). As agreed solely between GSK and LIXTE and without any obligation on the part of Institution, GSK will have no right and license to Study Data generated from the use of and related solely to LIXTE’s Investigational Product (for clarity, not in combination with GSK’s Investigational Product) or LIXTE’s Confidential Information without LIXTE’s written consent.
13.2.3 any Invention that relates to both GSK Confidential Information and LIXTE Confidential Information or to both the GSK Investigational Product (or the modification, use or manufacture thereof) and the LIXTE Investigational Product (or the modification, use or manufacture thereof) as a combination therapy (“Combined Therapy Inventions”) will:
(i) be promptly disclosed by Institution to both GSK and LIXTE in writing (and Institution shall ensure that any Combined Therapy Invention is promptly disclosed to Institution by all Study Personnel and Participating Site(s)); and
(ii) shall be the joint property of GSK and LIXTE, and GSK and LIXTE shall be the exclusive joint owners of Combined Therapy Inventions. Institution shall assign and does hereby assign to GSK and LIXTE jointly all right, title, and interest in the United States and throughout the world to all Combined Therapy Inventions (and, for the avoidance of doubt, Institution shall ensure that all Study Personnel, including Investigator and Sub-investigator(s), have assigned to Institution or Participating Site(s), as applicable, (and that Participating Site(s) have assigned to Institution) all right, title, and interest in the United States and throughout the world to all Combined Therapy Inventions). In the event of any Combined Therapy Invention, GSK and LIXTE will negotiate in good faith an agreement that addresses each of GSK’s and LIXTE’s rights and obligations related to such Combined Therapy Invention.
| 13.3. | Institution will, and will cause Study Personnel (including Investigator), and will ensure that all Participating Site(s) will, and will cause the relevant Sub-investigator(s), to: |
(a) reasonably cooperate in the preparation of applications or registrations for patent rights and other proprietary protection for any and all patentable or protectable GSK Investigational Product Inventions, LIXTE Investigational Product Inventions, and Combined Therapy Inventions all in the name of (i) GSK for GSK Investigational Product Inventions, (ii) LIXTE for LIXTE Investigational Product Inventions, and (iii) GSK and LIXTE jointly for Combined Therapy Inventions, at
(x) GSK’s cost and expense for GSK Investigational Product Inventions;
(y) LIXTE’ cost and expense for LIXTE Investigational Product Inventions; and
(z) GSK’s and LIXTE’s cost and expense on a 50%/50% basis for Combined Therapy Inventions; and
(b) execute and deliver all requested applications, assignments, and other documents and take such other measures as GSK and/or LIXTE reasonably requests, that are necessary in order to perfect GSK’s and LIXTE’s respective rights in GSK Investigational Product Inventions, LIXTE Investigational Product Inventions, and Combined Therapy Inventions.
| Page |
| 13.4. | Other Inventions. Inventions (as defined in Section 13.2) that are not GSK Investigational Product Inventions, LIXTE Investigational Product Inventions or Combined Therapy Inventions (the “Other Inventions”) shall be owned in accordance with inventorship as determined under U.S. Patent Law. |
| 13.5. | License and Options. In the event Institution has an ownership interest in any Other Invention, Institution, on behalf of itself, all Participating Site(s), Investigator and all other Study Personnel (including each Sub-investigator(s)), hereby grants to each of GSK and LIXTE and their respective affiliates a non-exclusive, fully paid-up, worldwide, perpetual, royalty-free, irrevocable license to use Other Inventions for all purposes. In addition, Institution, on behalf of itself, all Participating Site(s), Investigator and all other Study Personnel (including each Sub-investigator(s)), hereby grants to each of GSK and LIXTE a first option to obtain a co-exclusive (exclusive to GSK and LIXTE) perpetual, irrevocable, transferable, royalty-bearing and sublicensable (through multiple tiers) license (“Co-Exclusive License”) to Institution’s, Investigator’s, Study Personnel’s or any Participating Site’s or Sub-investigator’s, interest in any Other Inventions to make, use and sell (and otherwise research, develop and commercialize) those inventions or any products that are covered by patent rights that claim or include those inventions. GSK’s and LIXTE’s Co-Exclusive License option may be exercised with respect to an Other Invention by notice in writing from GSK and/or LIXTE to each of the other Parties, at any time during a period of one-hundred & twenty (120) days (the “Option Period”) after the full written disclosure to GSK and LIXTE by Institution of each such Other Invention. Upon GSK and LIXTE both exercising their Co-Exclusive License with regard to any particular Other Invention, Institution, LIXTE and GSK will negotiate in good faith in an attempt to reach a license agreement satisfactory to all Parties (the “Negotiation Period”). Unless extended by the written consent of all of the Parties, the Option Period and the Negotiation Period shall not exceed two-hundred & forty (240) days in the aggregate. If either GSK or LIXTE declines to exercise the Co-Exclusive License option during an Option Period, then GSK or LIXTE, as the case may be, may exercise the Co-Exclusive License option alone which may result in an exclusive license to such Party. In the event that an Option Period lapses or, including any extensions, terminates, Institution may enter into negotiations to enter into a non-exclusive license agreement with a third party without further obligation to GSK or LIXTE under this Agreement with regard to Institution’s interest in such Other Inventions; provided, however, that the terms offered to a third party are not more favorable than last offered to GSK or LIXTE and provided further that the non-exclusive licenses to GSK and LIXTE will remain in full force and effect. |
| 13.6. | Notwithstanding anything to the contrary, Institution will retain a non-exclusive, royalty-free license to internally use any Invention to which Institution’s personnel or any Participating Site’s personnel makes an inventive contribution for Institution’s research, academic and patient care purposes, which Institution can sublicense only to the contributing Participating Site for such Participating Site’s research, academic and patient care purposes. |
| 14. | Term and Termination; Completion. |
| 14.1 | Term. This Agreement is effective as of the Effective Date and will continue in effect through completion of the Study, unless earlier terminated pursuant to this Section 14 (“Term”). |
| 14.2 | Termination. This Agreement may be terminated by any Party without cause upon thirty (30) days prior written notice to the other Parties. This Agreement may be terminated by any Party (a) immediately upon written notice to the other Parties if necessary to protect the safety, health or welfare of subjects enrolled in the Study or upon withdrawal of regulatory authorization for the Study; or (b) for a breach of a material provision hereof by a Party, which breach is not cured within thirty (30) days following receipt of written notice thereof; or (c) immediately upon written notice to the other Parties if Investigator is no longer able (for whatever reason) to act as Investigator and no replacement mutually acceptable to Institution, GSK and LIXTE can be found; or (d) if a Party becomes insolvent, or if an order is made or a resolution is passed for its winding up (except voluntarily for the purpose of solvent amalgamation or reconstruction), or if an administrator, administrative receiver or receiver is appointed over the whole or any part of its assets, or if it makes any arrangement with its creditors. Additionally, GSK or LIXTE shall be entitled to terminate this Agreement immediately on written notice to Institution and the other Party if Institution fails to perform its obligations in accordance with Section 17 below. Except for payment due to the Institution as detailed in this Agreement, including but not limited to Section 14.3 below and further set out in Appendix B, Institution shall have no claim against GSK or LIXTE for compensation for any loss of whatever nature by virtue of the termination of this Agreement in accordance with this Section 14.2. |
| Page |
| 14.3 | Effect of Termination/Completion of Study. Upon either an early termination under Section 14.2 or completion of the Study, (a) this Agreement will terminate; (b) Investigator will immediately stop enrolling subjects into the Study and determine the appropriate manner to cease conducting Study procedures and administration of the Investigational Products to subjects already entered into the Study; (c) Institution will provide GSK with a reconciliation of Study costs against budget as set forth in Appendix B; (d) Institution and/or Investigator will provide a copy of all Study Data to GSK and LIXTE and will return to GSK all of GSK’s Confidential Information and return to LIXTE all of LIXTE’s Confidential Information, except that Institution may retain one archival copy of each of GSK Confidential Information and LIXTE Confidential Information solely for purposes of determining future compliance with the terms of this Agreement and as necessary for legal, regulatory, compliance, or insurance purposes; (e) to the extent GSK and/or LIXTE received Confidential Information of another Party in connection with this Agreement, each of GSK and/or LIXTE, as applicable, will return the Confidential Information to the owning Party; and (f) GSK shall pay Institution all such amounts incurred or obligated by Institution in accordance with milestones as set out in Appendix B at the effective date of such termination. |
| 14.4 | Survival. No expiration or termination of this Agreement will release the Parties from their rights and obligations accrued prior to the effective date of expiration or termination. The rights and duties under Sections 3.1(f) (Investigational Products Supply and Accountability); 3.5 (Declaration of GSK and LIXTE Support); 4 (Reports, Audits and Use of Study Data and Biological Samples); 8 (No Conflicts or Debarment) (for the period of time set forth therein); 9 (Safety Data Reporting); 10 (Communications with Regulatory Agencies); 11 (Confidentiality) (for the period of time set forth therein), 12 (Publication), 13 (Intellectual Property); 14.3 (Effects of Termination); 14.4 (Survival); 15 (Indemnification, Remedies, Insurance and Study Subject Injury); 17.2 (Antibribery) and 18 (Miscellaneous) will survive the termination of this Agreement. |
| 15. | Indemnification, Remedies, Insurance and Study Subject Injury. |
| 15.1. | Indemnification by GSK. GSK will indemnify, defend and hold harmless Institution, System, and their Regents, directors, officers, employees (including Investigator), agents, Participating Site(s) and Sub-investigator(s), and any other third parties engaged at their discretion, (collectively, the “Institution Indemnitees”) and LIXTE, its directors, officers, employees and agents (collectively, the “LIXTE Indemnitees”) against any third party claims, including losses, liabilities, damages, claims, costs, charges, expenses and reasonable attorney’s fees for defending those claims (each, a “Claim”) to the extent a Claim arises out of or relates to (i) the personal injury (including death) or property damage arising out of or connected with GSK’s failure to manufacture and provide the GSK Investigational Product in accordance with current Good Manufacturing Practices (“GMP”), (ii) the use by GSK of the results of the Study, and (iii) the activities to be carried out by GSK under this Agreement. GSK’s obligations under this Section 15.1 will not apply to the extent that a Claim is indemnifiable by LIXTE or Institution under Section 15.2 or Section 15.3, respectively, or arises out of or relates to: (a) an Institution Indemnitee’s or a LIXTE Indemnitee’s respective (i) negligence, gross negligence or willful misconduct, or (ii) failure to adhere to the terms of this Agreement or any reasonable written instructions from GSK or its designee; or (b) an Institution Indemnitee’s (1) failure to conduct the Study in accordance with applicable laws or regulations, or (2) failure to adhere to the terms of the Protocol or any reasonable written instructions from GSK or its designees; or (c) any materials, equipment, device and/or drug products used in the Study that are not manufactured or provided by GSK. |
| Page |
| 15.2. | Indemnification by LIXTE. LIXTE will indemnify, defend, and hold harmless and the Institution Indemnitees and GSK, its directors, officers, employees and agents (collectively, the “GSK Indemnitees”) against any Claim to the extent such Claim arises out of or relates to (i) the personal injury (including death) or property damage arising out of or connected with LIXTE’s failure to manufacture and provide the LIXTE Investigational Product in accordance with GMP, (ii) the use by LIXTE of the results of the Study, and (iii) the activities to be carried out by LIXTE under this Agreement. LIXTE’s obligations under this Section 15.2 will not apply to the extent that a Claim is indemnifiable by GSK or Institution under Section 15.1 or Section 15.3, respectively, or arises out of or relates to: (a) an Institution Indemnitee’s or a GSK Indemnitee’s respective (i) negligence, gross negligence or willful misconduct, or (ii) failure to adhere to the terms of this Agreement or any reasonable written instructions from LIXTE or its designee; or (b) an Institution Indemnitee’s (1) failure to conduct the Study in accordance with applicable laws or regulations; or (2) failure to adhere to the terms of the Protocol or any reasonable written instructions from LIXTE or its designees; or (c) any materials, equipment, device and/or drug products used in the Study that are not manufactured or provided by LIXTE. |
| 15.3. | Indemnification by Institution. To the extent authorized under the Constitution and the laws of the State of Texas, Institution will indemnify, defend, and hold harmless the GSK Indemnitees and the LIXTE Indemnitees against any Claim to the extent such Claim arises out of or relates to (a) the performance of the Study by Institution or any Institution Indemnitees; or (b) any Institution Indemnitee’s: (i) negligence, gross negligence or willful misconduct, (ii) failure to adhere to the terms of the Protocol, this Agreement, or any reasonable written instructions from GSK or LIXTE or their respective designees, or (iii) failure to conduct the Study in accordance with applicable laws or regulations; provided, however, that deviations from the Protocol for health and safety reasons shall not constitute: failure to follow to the terms of the Protocol, negligence or willful misconduct by an Institution Indemnitee or a breach of this Agreement by an Institution Indemnitee. Institution’s obligations under this Section 15.3 will not apply to the extent that a Claim is indemnifiable by GSK or LIXTE under Section 15.1 or Section 15.2, respectively, or arises out of or relates to the negligence, gross negligence, or willful misconduct of GSK Indemnitees, LIXTE Indemnitees, or any person other than an Institution Indemnitee. |
| 15.4. | Indemnification Procedure. A Party (the “indemnified Party”) must use reasonable efforts to notify the indemnifying Party within thirty (30) days of receipt of any Claim for which such indemnifying Party might be liable under Section 15.1, 15.2, or 15.3, as the case may be; provided, however, the failure to promptly notify the indemnifying Party shall not relieve the indemnifying Party of its indemnification obligations unless the indemnifying Party is materially adversely affected by such failure. The indemnifying Party will have the sole right to defend, negotiate, and settle such Claim; provided, however, such settlements shall not require the indemnified Party to contribute to the settlement, admit fault or require the indemnified Party to change its operations or business practices, without the indemnified Party’s written consent. The indemnified Party will be entitled to participate in the defense of such matter and to employ counsel at its expense to assist in such defense; provided, however, that the indemnifying Party will have final decision-making authority regarding all aspects of the defense of the Claim. The indemnified Party seeking indemnification will provide the indemnifying Party with such information and assistance as the indemnifying Party may reasonably request, at the expense of the indemnifying Party. No indemnified Party will be responsible or bound by any settlement of any Claim or suit made without its prior written consent; provided, however, that the indemnified Party will not unreasonably withhold such consent. This Section 15 is subject to the statutory duties of the Texas Attorney General. |
| Page |
| 15.5. | Remedies. Institution agrees that (a) GSK and LIXTE may be irreparably injured by an impending or existing breach of this Agreement; (b) money damages may not be an adequate remedy for any such breach; and (c) GSK and LIXTE will be entitled to seek equitable relief, including injunctive relief and specific performance, without having to post a bond, as a remedy for any such breach. The provisions of this Section 15.5 are not intended to be exclusive and are without prejudice to the rights of GSK and/or LIXTE to seek any other right or remedy that it may have under this Agreement or otherwise. |
| 15.6. | Insurance. Each member of The University of Texas System is self-insured pursuant to The University of Texas Professional Medical Liability Benefit Plan under the authority of Chapter 59, Texas Education Code. Institution has and will maintain in force during the Term of this Agreement adequate insurance or financial resources to cover its indemnification obligations. Upon written request, Institution will provide written evidence of self-insurance or financial resources. |
| 15.7. | Study-Related Injury. If a Study subject is injured or becomes ill as a result of participating in the Study, Institution shall ensure that the Study subject has access to medical treatment necessary to treat such injury or illness. This Agreement does not obligate any of the Parties to provide medical treatment, except to the extent required by applicable law, nor does this Agreement obligate any Party to provide reimbursement for medical treatment if a Study subject requires medical treatment for physical illness or injury sustained as a direct result of the treatment of such Study subject in accordance with this Agreement and the Protocol. |
| 15.8. | Mitigation. Nothing in this Agreement shall relieve any party from its duty under applicable law to mitigate any loss or damage incurred by it as a result of any matter giving rise to a Claim. |
| 16. | Security Breaches and Data Privacy |
| 16.1 | Notification of Data Security Breaches. The Parties agree to notify each other in accordance with the below, without undue delay after discovery of a Security Breach (as defined below) and awareness that such Security Breach relates to this Agreement. |
| (i) | Notice of a Security Breach to GSK, will be sent via e-mail to csir@gsk.com. |
Notice of a Security Breach to LIXTE, will be sent via e-mail to: info@lixte.com
Notice of a Security Breach to Institution will be sent to: PrivacyCompliance@mdanderson.org
with a follow-up to: The University of Texas M. D. Anderson Cancer Center, Institutional Compliance Office, Unit 1640, P.O. Box 301407, Houston, Texas 77230-1407, Attn: Privacy Officer, Fax No. 713-563-4324.
| (ii) | In the course of notification to each other, the Parties will provide, as feasible, sufficient information for the Parties to jointly assess the Security Breach and, to the extent a Party or the Parties may be subject to an applicable legal requirement regarding disclosure to a competent governmental authority, make any required notification to any government authority within the timeline required by applicable laws. Such information may include, but is not necessarily limited to: |
| (a) | The nature of the Security Breach, the categories and approximate number of data subjects and records; |
| (b) | The likely consequences of the Security Breach, in so far as consequences are able to be determined; |
| (c) | Any measures taken to address or mitigate the incident. |
| (iii) | Following a breach of PHI, the Party (or Parties) who bear the regulatory duty to determine whether a Security Breach occurred will decide on the basis of all available information and applicable laws if the Security Breach will be considered a reportable Security Breach and arrange for notification to data subjects and/or government authorities, if required by applicable laws. Where the Parties decide that notification is required by applicable laws, the Party responsible for providing such notification under applicable data privacy laws shall be responsible for providing such notification. |
| Page |
| (iv) | Assistance in event of Security Breach. In the event of a Security Breach relating to the personal information of another Party’s personnel and/or GSK’s or LIXTE’s Confidential Information collected or received by a Party under this Agreement, each Party agrees to assist and fully cooperate with the other Party with respect to any internal or external investigation through the provision of access to information, employees, interviews, materials, databases, or any and all other items required to fully investigate and resolve any such incidents and provide information necessary to enable required notifications. The breached Party agrees to take such remedial actions as the Parties mutually agree is warranted. Notwithstanding the foregoing, the Parties will each comply with HIPAA, to the extent applicable, with regards to any breach that involves PHI. |
| (v) | No Party shall disclose, without the other Parties’ prior written approval, any information related to the suspected Security Breach to any third party other than a vendor hired to investigate/mitigate such Security Breach and bound by confidentiality obligations, except as required by applicable laws. |
| (vi) | “Security Breach” means any actual or suspected breach of security leading to the accidental or unlawful destruction, disclosure of, or access to, the Confidential Information or personal data of another Party’s personnel, as applicable, of any Party. |
| 17. | Anti-bribery and Corruption |
| 17.1 | Institution shall comply, and shall ensure Investigator complies, fully at all times with all applicable laws and regulations, including but not limited to anti-corruption laws, and that it has not, and covenants that it will not, in connection with the performance of this Agreement, directly or indirectly, make, promise, authorize, ratify or offer to make, or take any act in furtherance of any payment or transfer of anything of value for the purpose of influencing, inducing or rewarding any act, omission or decision to secure an improper advantage; or improperly assisting it or GSK or LIXTE in obtaining or retaining business, or in any way with the purpose or effect of public or commercial bribery, and warrants that it has taken reasonable measures to prevent subcontractors, agents or any other third parties, including Participating Site(s) and Sub-investigator(s), subject to its control or determining influence, from doing so. For the avoidance of doubt this includes facilitating payments, which are unofficial, improper, small payments or gifts offered or made to government officials to secure or expedite a routine or necessary action to which we are legally entitled. |
| 17.2 | Institution agrees that in the event that GSK or LIXTE believes that there has been a possible violation of the terms of this Section 17, GSK or LIXTE may make full disclosure of such belief and related information at any time and for any reason to any competent government bodies and their agencies, and, subject to the confidentiality obligations herein and with respect to any PHI, to the extent permitted by any applicable Consent Document and applicable law, to whomever GSK or LIXTE determines in good faith has a legitimate need to know. |
| 18. | Miscellaneous. |
| 18.1 | Independent Contractor. Institution, including Investigator, is an independent contractor and, as such, none of Institution, Institution’s employees, or Investigator will be entitled to any benefits applicable to employees of GSK and/or LIXTE. No Party is authorized or empowered to act as agent for any other Party for any purpose under this Agreement and will not, on behalf of another Party, enter into any contract, warranty or representation as to any matter herein. No Party will be bound by the acts or conduct of any other Party. Nothing in this Agreement and no action taken by the Parties under this Agreement shall constitute a partnership, association or other co-operative entity between any of the Parties or make any Party the agent of any other Party for any purpose. |
| Page |
| 18.2 | Use of Names; Publicity. Except to the extent required by applicable law or regulation or the rules of any stock exchange or listing agency, no Party will use the name of another Party in any form of advertising, promotion, social media or publicity or in any press release, without the prior written consent of that other Party. Institution agrees and will ensure that Study Personnel agree not to answer third party inquiries regarding the Study or the Investigational Products from financial analysts other than pursuant to a requirement of applicable law, IRB, government agency, an order of a court of competent jurisdiction, or a facially valid administrative, Congressional, or other subpoena and only in accordance with Section 11.4. |
| 18.3 | Sponsorship. Institution agrees that GSK or LIXTE will not be listed as the sponsor of the Study in any documentation related to the Study and will ensure that GSK or LIXTE is not identified as the sponsor for regulatory purposes nor referenced to have assumed any sponsor responsibilities for this Study. |
| 18.4 | Certain Disclosures and Transparency. Institution acknowledges that GSK and its affiliates and LIXTE and its affiliates are required to abide by applicable federal and state disclosure laws and GSK transparency policies governing their activities including providing reports to the government and to the public concerning financial or other relationships with healthcare providers and healthcare organizations. Institution agrees that GSK and its affiliates and LIXTE and its affiliates may, in their sole discretion, disclose information about this Agreement and about the Study as required by law, including relating to any transfers of value pursuant to this Agreement. Institution agrees to supply information reasonably requested by GSK and LIXTE for disclosure purposes, other than information that Institution previously provided to GSK. To the extent that Institution is independently obligated to disclose specific information concerning the Study, including relating to transfers of value from GSK or its affiliates pursuant to this Agreement, Institution will make timely and accurate required disclosures. Institution hereby represents that Investigator is aware of the possibility of such disclosure. |
(i) GSKs disclosure link is as follows: “https://www.gsk.com/en-gb/responsibility/ethical-standards/engaging-with-healthcare-professionals/#Disclosures”
| 18.5 | Assignment; Subcontracting. Except as expressly provided in this Agreement, Institution may not assign, delegate or transfer its obligations under this Agreement, in whole or in part, without the prior written consent of GSK and LIXTE, and any attempted assignment, delegation or transfer by Institution without such consent will be void. GSK and LIXTE may each, respectively, assign, delegate or transfer this Agreement to an affiliate or in connection with the acquisition, licensing, sale or purchase of any or all of its interests or its Investigational Product or its business or assets to which this Agreement relates, in whole or in part, without the consent of the other Parties; however, each of GSK and LIXTE will, as the case may be, provide notice to the other Parties if it assigns this Agreement. No assignment, delegation or transfer will relieve any Party of the performance of any accrued obligation that such Party may then have under this Agreement. With GSK’s and LIXTE’s prior written consent in each instance, Institution may subcontract the performance of certain of its activities under this Agreement to qualified third parties other than Sub-investigator(s) and Participating Site(s); provided, that (a) such permitted third parties perform such activities in a manner consistent with the terms and conditions in this Agreement; (b) Institution causes such permitted third parties to be bound by and comply with the terms of this Agreement, as applicable, including the obligations described in Section 7.4 above; (c) Institution remains liable for Institution’s obligations under this Agreement regardless of any delegations to such permitted third parties; and (d) other than as disclosed to GSK and LIXTE in writing, neither Investigator nor any Sub-investigator has any direct or indirect financial interest in any such permitted third parties. For the avoidance of doubt, and notwithstanding anything to the contrary herein, all permitted third parties used to perform the Study will be considered Study Personnel under this Agreement. |
| Page |
| 18.6 | Notice. All notices must be in writing and sent to the address for the recipient set forth below or at such other address as the recipient may specify in writing under this procedure. All notices must be given (a) by personal delivery, with receipt acknowledged; or (b) prepaid certified or registered mail, return receipt requested; or (c) by prepaid recognized express delivery service. Notices will be effective upon receipt or at a later date stated in the notice. |
| If to GSK: | GSK LLC | |
| 1000 Winter Street, Suite 3300, Waltham, MA 02451 | ||
| Attn: Tyler Lockard | ||
| Email: external.research@gsk.com; Fax: +1.339.309.5112 | ||
| With a copy to: | ||
| GSK LLC, Marc Harris, Contracting | ||
| 1250 South Collegeville Road, Bldg 4, 4th FL | ||
| Collegeville, PA., 19426 (marc.2.harris@gsk.com) | ||
| If to LIXTE: | LIXTE Biotechnology Holdings, Inc | |
| Attn: Eric J. Forman (eforman@lixte.com) | ||
| 680 E Colorado Blvd., Suite 180 | ||
| Pasadena, CA 91101 | ||
| With a copy to: | ||
| Cooley LLP | ||
| Attn: Matthew E. Langer (mlanger@cooley.com) | ||
| 55 Hudson Yards | ||
| New York, NY 10001-2157 | ||
| If to Institution: | ||
| The University of Texas M.D. Anderson Cancer Center | ||
| 7007 Bertner Avenue, 1MC11.3343 | ||
| Legal Services, Unit 1674 | ||
| Attn: Chief Legal Officer | ||
| Houston, TX 77030 | ||
| Phone: (713) 745-6633; Facsimile: (713) 745-6029 | ||
| With a copy to: | ||
| The University of Texas M.D. Anderson Cancer Center | ||
| 7007 Bertner Ave. | ||
| Office of Sponsored Programs, Unit 1676 | ||
| Attn: Associate VP, Research Administration | ||
| Houston, TX 77030 | ||
| Phone: (713) 792-3220; Facsimile: (713) 794-4595 | ||
| Investigator: | ||
| Vice Chair for Clinical Research | ||
| Director of the Gynecologic Cancer Immunotherapy Program | ||
| Professor, Department of Gynecologic Oncology and Reproductive Medicine | ||
| The University of Texas MD Anderson Cancer Center. | ||
| 1515 Holcombe Blvd., Unit 1362 | ||
| Houston, TX 77030 | ||
| Attn: Dr. Amir Jazaeri (aajazaeri@mdanderson.org) |
| Page |
| 18.7 | Entire Agreement; No Modification. This Agreement, including all Appendices which are incorporated into this Agreement, constitutes the entire agreement among the Parties with respect to the Study that is the specific subject matter of this Agreement and supersedes all prior agreements, oral or written, with respect to such subject matter. This Agreement may not be amended or modified except in a written instrument signed by an authorized representative of each of Institution, GSK and LIXTE, and acknowledged by Investigator. Any conflicts between any Appendices and this Agreement will be governed and controlled by provisions of the main text of this Agreement. |
| 18.8 | Severability; Reformation. Each provision in this Agreement is independent and severable from the others, and no provision will be rendered unenforceable because any other provision is found by a proper authority to be invalid or unenforceable in whole or in part. If any provision of this Agreement is found by such an authority to be invalid or unenforceable in whole or in part, such provision will be changed and interpreted so as to best accomplish the objectives of such unenforceable or invalid provision and the intent of the Parties, within the limits of applicable law. |
| 18.9 | Governing Law. Institution is an agency of the State of Texas and under the Constitution and the laws of the State of Texas possesses certain rights and privileges, is subject to certain limitations and restrictions, and only has such authority as is granted to it under the Constitution and laws of the State of Texas. This Agreement and any disputes arising out of or relating to this Agreement will be governed by, construed and interpreted in accordance with the Constitution and laws of the State of Texas, without regard to any choice of law principle that would require the application of the law of another jurisdiction. |
| 18.10 | Waivers. Any delay in enforcing a Party’s rights under this Agreement, or any waiver as to a particular default or other matter, will not constitute a waiver of such Party’s rights to the future enforcement of its rights under this Agreement, except with respect to an express written waiver relating to a particular matter for a particular period of time signed by an authorized representative of the waiving Party, as applicable. To clarify, any such waiver by GSK, LIXTE, or Institution must be evidenced by an instrument in writing executed by an officer of such party authorized to execute waivers. |
| 18.11 | Rights Cumulative. The rights and remedies contained in this Agreement are cumulative and not exclusive of any rights or remedies provided by law. |
| 18.12 | Party Rights. Except as specifically provided in this Agreement, nothing expressed or implied herein is intended, or will be construed, to confer upon or give any person other than the Parties hereto, and their successors or permitted assigns, any right, remedy, obligation or liability under or by reason of this Agreement, or result in such person being deemed a third-party beneficiary of this Agreement. |
| 18.13 | No Strict Construction; Headings; Interpretation. This Agreement has been prepared jointly and will not be strictly construed against any Party. This Agreement contains headings only for convenience and the headings do not constitute or form a part of this Agreement, and should not be used in the construction of this Agreement. The words “include,” “includes” and “including” when used in this Agreement are deemed to be followed by the phrase “but not limited to.” |
| 18.14 | Counterparts. This Agreement may be executed in any number of counterparts, each of which will be deemed to be an original and all of which together will constitute one and the same instrument. An executed counterpart of this Agreement (the entire Agreement, not just a signature page) may be delivered by e-mail (in PDF or another agreed format). |
| 18.15 | Force Majeure. No Party shall be liable to any other for any delay or non-performance of its obligations under this Agreement arising from any Force Majeure Event. “Force Majeure Event” means any act or event, in whole or in part, whether foreseen or unforeseen, that is beyond the reasonable control of a Party, but excludes economic hardship or insufficiency of funds. |
| Page |
| 18.16 | Academic Freedom. Subject to the obligations herein, nothing in this Agreement will limit or prohibit Institution or any of its personnel, including the Investigator, from conducting any research or from performing research for or with any entity or person, including any other outside sponsors. GSK and LIXTE acknowledges that this provision is intended to preserve the academic freedom and integrity of Institution and its faculty and to ensure that Institution and its faculty are not regarded as exclusive researchers for GSK or LIXTE. |
| 18.17 | Subordination to Applicable Law. The Parties will not be required to perform any act or to refrain from any act that would violate any law. This Agreement is subject to, and the Parties agree to comply with, all applicable laws. Any provision of any law, statute, rule or regulation that invalidates any provision of this Agreement, that is inconsistent with any provision of this Agreement, or that would cause one or any of the Parties hereto to be in violation of law will be deemed to have superseded the terms of this Agreement. The Parties, however, will use all reasonable endeavors to accommodate the terms and intent of this Agreement to the greatest extent possible consistent with the requirements of the law and will negotiate in good faith toward amendment of this Agreement in such respect. If the Parties cannot reach agreement on an appropriate amendment, then this Agreement may be immediately terminated by either Party. |
| 18.18 | Notice of Texas State Agency. Institution is an agency of the State of Texas and under the Constitution and the laws of the State of Texas possesses certain rights and privileges, is subject to certain limitations and restrictions, and only has such authority as is granted to it under the Constitution and laws of the State of Texas. Notwithstanding any provision hereof, nothing in this Agreement is intended to be, nor will it be construed to be, a waiver of the sovereign immunity of the State of Texas or a prospective waiver or restriction of any of the rights, remedies, claims, and privileges of the State of Texas. Moreover, notwithstanding the generality or specificity of any provision hereof, the provisions of this Agreement as they pertain to Institution are enforceable only to the extent authorized by the Constitution and laws of the State of Texas; accordingly, to the extent any provision hereof conflicts with the Constitution or laws of the State of Texas or exceeds the right, power or authority of Institution to agree to such provision, then that provision will not be enforceable against Institution or the State of Texas. |
[Signature page to follow]
| Page |
IN WITNESS WHEREOF, this Agreement is executed as of the Effective Date by a duly authorized representative of each of GSK, LIXTE and Institution.
| GLAXOSMITHKLINE LLC | Lixte Biotechnology Holdings. Inc. | |||
| By: | /s/ Marc Harris | By: | /s/ John S. Kovach | |
| Print Name: | Marc Harris | Print Name: | John S. Kovach, MD | |
| Title: | Assoc Director | Title: | CEO | |
| Date: | 18-Sep-2023 | Date: | Sep 18, 2023 | |
|
THE UNIVERSITY OF TEXAS M. D. ANDERSON CANCER CENTER |
||||
| By: | /s/ Amy M Moritz | |||
| Print Name: | Amy M Moritz | |||
| Title: | Assistant Director, ORA | |||
Date: |
8/28/2023 | |||
| Read and Acknowledged: | ||||
| INVESTIGATOR | ||||
| /s/ Amir Jazaeri | ||||
| Print Name: | Amir Jazaeri, MD | |||
| 8/28/2023 | ||||
| Page |
APPENDIX A
DELINEATION OF GSK AND LIXTE INVESTIGATIONAL PRODUCT (“IP”) RESPONSIBILITIES
| Institution Contact for IP | Investigational Pharmacy Services INVdrugs@mdanderson.org | |
|
GSK Contact for IP |
Tyler Lockard , Study Delivery Lead external.research@gsk.com | |
| LIXTE Contact for IP | Eric Forman eforman@lixte.com |
|
GSK IP LIXTE IP |
Investigational Dostarlimab (50mg/mL (10mLvial)) Investigational LB-100 (1 mg/mL (10mL vial) |
|
| Number of Study Subjects | Twenty-one (21) (includes Northwestern Study Subjects) | |
| Average Number of Cycles/Subject | 14 | |
| Participating Country | US | |
| Number of Participating Sites | 2 (two) | |
| Study Type |
☒ Open: Identity of the Investigational Product is not withheld from the investigator or subjects at the time of dispensing
☐ Blind: The investigator, pharmacist, and subjects are not able to distinguish between treatment groups at time of dispensing*
☐ Third-party Blind: The investigator and subjects are not able to distinguish between treatment groups, but pharmacy staff will have access to the identity of the IP at the time of dispensing |
| Page |
| Study Set Up |
Institution |
GSK or qualified delegate for GSK IP Only | LIXTE or qualified delegate for LIXTE IP Only | |||
| Description of IP provided to country regulatory authorities, and provision of cross-reference letter for use by Institution | X | X | ||||
| Submission to regulatory authority, as appropriate (Clinical Trial Application or IND. | X | |||||
| Supply of IP with appropriate labeling | X | X | ||||
| Additional labeling to comply with applicable local, legal and regulatory requirements, if necessary | X | |||||
| Retention samples of IP | X | X |
| IP Set Up |
Institution |
GSK or qualified delegate for GSK IP Only |
LIXTE or qualified delegate for LIXTE IP Only | |||
| Collection of regulatory approvals and submission of Institution’s and Participating Site’s initial shipment approval checklist to GSK and LIXTE | X |
| Shipment Strategy | Institution | GSK or qualified delegate for GSK IP Only | LIXTE or qualified delegate for LIXTE IP Only | |||
| Shipment direct to Institution and Participating Site(s) | X | X |
| IP Management | Institution | GSK or qualified delegate for GSK IP Only | LIXTE or qualified delegate for LIXTE IP Only | |||
| Provide Institution /Investigator with expiry date, storage conditions, and allowable excursions in a storage and handling manual | X | X | ||||
| Provide GSK & LIXTE with acknowledgement of receipt of all IP shipments to Participating Site(s) | X | |||||
| Monitor expiry date, comply with storage conditions, and report temperature excursions | X | |||||
| Assessment of potential quality issues which occur during shipment or storage of IP to Participating Site(s) | X | X | ||||
| Report inventory use and resupply IP requirements as specified by GSK or LIXTE, including real time use of IRT/RTSM system, as applicable. | X | |||||
| Decision to recall | X | X | ||||
| Execution of recall(s), if applicable, including communication of recall to Participating Site(s) | X | |||||
| Providing recall communication and reporting compliance of recall to GSK and LIXTE | X | |||||
| Destruction of IP (at end of study or recall) and written memo confirming destruction of IP at Participating Site(s) | X |
| Page |
SCHEDULE 1 TO APPENDIX A
SUPPLIES AGREEMENT FORM

| Page |

| Page |
| Page |

| Page |
APPENDIX B: FUNDING
| Institution: | The University of Texas MD Anderson Cancer Center | |
| Investigator: | Dr. Amir A.Jazaeri | |
| GSK Investigational Product: | Dostarlimab | |
| GSK Protocol # : | 219582 |
| 1. | Enrollment of Study Subjects |
Institution will enroll (which, for clarity, does not include any screening failures) a maximum of twenty-one (21) Study subjects (includes Study Subjects at Northwestern) on the Protocol and use reasonable efforts to achieve an expected rate of 2 Study subjects/month. GSK will pay Institution for Study subjects enrolled on the Protocol that receive at least one dose of GSK Investigational Product and LIXTE Investigational Product, in accordance with the schedule below.
| 2. | Payment Schedule |
GSK agrees to provide Funding in support of the conduct of the Study in the total amount of $1, 493,019.40 USD. Funding will be provided as follows:
| Milestone | $ Payment USD | |||
| Upon receipt by GSK of (i) the final and fully executed Agreement, (ii) all required documentation, and, (iii) confirmation that the summary Protocol has been posted on clinicaltrials.gov or other public register in accordance with the Agreement. | 160,345.04 | |||
| Upon enrollment of 1 (one) Study subject. | 172,345.08 | |||
| Upon enrollment of 4 additional (four) Study subjects (total 5). | 172,345.08 | |||
| Upon enrollment of 4 additional (four) Study Subjects (total 9). | 172,345.08 | |||
| Upon enrollment of 4 additional (four Study Subjects (total 13). | 172,345.08 | |||
| Upon enrollment of 4 additional (four) Study Subjects (total 17). | 172,345.08 | |||
| Upon enrollment of 4 additional (four) Study Subjects (total 21). | 172,345.08 | |||
| Upon receipt of documentation that (i) 21 Study Subjects have completed the Study; (ii) GSK’s receipt of the Final Report or draft manuscript in accordance the Communication of Data Section of this Agreement; (iii) confirmation that final Results summary has been posted to www.ClinicalTrials.gov or other public register in accordance with the Publication Section of this Agreement; and (iv) documentation of attempt of publication. If the Investigator and GSK agree that the Study Results do not support a publication, a written final study report may be accepted for final payment. | 298,603.88 | |||
TOTAL |
1,493,019.40 | |||
| Page |
| 3. | Invoicing |
To ensure timely payment, all invoices will be submitted to Tyler Lockard (external.research@gsk.com) (“SAP”) in accordance with the invoice instructions below. GSK will pay Institution within thirty (30) days from GSK receipt of invoice. Institution shall create and submit all its invoices and/or credit notes and sent through email to the SAP. Each Invoice will have to indicate the Purchase Order number, that will be communicated by GSK after contract execution, as well as the following information:
| i. | Invoice addressed to: GSK LLC, 1250 South Collegeville Road, Collegeville, PA., 19426 | |
| ii. | Institution Name/address: | |
| iii. | Study Title: 219582 | |
| iv. | CID: 574189 | |
| v. | Invoice number & date: | |
| vi. | Purchase Order Number: | |
| vii. | GSK Contact: Tyler Lockard | |
| viii. | VAT ID number | |
| ix. | Detailed description of services/milestone: | |
| x. | Bank account name/address & Sort Code |
Failure to provide the required information will delay approval and the invoice may be returned for revisions.
Payment Instructions:
Payments shall be made by Electronic Funds Transfer via the Automated Clearing House (ACH), which is Institution’s preferred method to receive payments.
FOR ACH DELIVERY
Bank Routing Number: 111000614; Account Number: 522292058
Account Name: Univ. of Texas MD Anderson Cancer Center-Office of Grants and Contracts
| 4. | Reconciliation |
In the event that the Study closes enrollment prior to achievement of the maximum number of Study subjects, Institution shall provide written notice to GSK of the date the Study was closed, the proposed funding reconciliation, the number of Study subjects that are eligible for payment in accordance with the terms in Section 1, and any other relevant documentation supporting the plan. GSK will review the proposed reconciliation and will respond with any objections.
The Payment Schedule described in Section 2 will be prorated up to a maximum of:
| Description | Per Unit Cost (USD) | # Units | $ Total USD | |||||||||
| One-time start-up fees | 160,345.04 | |||||||||||
| Per Study subject costs | 63,460.67 | 21 | 1,332,674.06 | |||||||||
| Total | 1,493,019.10 | |||||||||||
| Page |
Appendix C: TASK RESPONSIBILITY MATRIX
| TASK | GSK | LIXTE | Institution | |||
| Budget | ||||||
| Make payments according to milestones agreed in contract | X | |||||
| Develop itemised budget | X | |||||
| Study Start Up | ||||||
| Site/Investigator selection & training | X | |||||
| Scientific exchange on research proposal | X | X | X | |||
| Create study documents (Protocol, ICF, Statistical Plan, CRF, Diary Cards, and Laboratory Manual etc.) | X | |||||
|
Contribute to study documents (review and comment)
NB type of study documents to be detailed
Study Protocol, ICF(s), Pharmacy Manual, GSK /LIXTE Investigational Product Handling Instructions, Laboratory Manual
GSK /LIXTE also to contribute for comments received by CA / EC as required and especially in relation to the GSK Investigational Product and LIXTE Investigational Product |
X | X | X | |||
| Service Provider selection and contract, including laboratories if applicable | X | |||||
| Regulatory (e.g. IND application or amendment) | X | |||||
| Ethics Committee/Institutional Review Board Submission | X | |||||
| Protocol Summary posting on ClinicalTrials.gov | X | |||||
| Study Conduct | ||||||
| GSK and LIXTE to provide respective IP to Participating Site (s), upon Institution providing shipping details (address and name) to GSK and LIXTE | X | X | ||||
| Conduct study in adherence with applicable GxP and regulatory requirements | X | |||||
| Study site management (e.g., Monitoring visit, Investigator site file, etc.) | X | |||||
| Ensure sites use GSK’s IRT system in real-time to enroll patients, dispense drug and manage drug study | X | |||||
| Perform Quality Assurance site audits | X | |||||
| Updates to the Investigator’s Brochure for respective IP | X | X | ||||
| Data collection and data management | X | |||||
| Human Biological Sample Management | X | |||||
| Storage of samples as per Study Protocol for future research and development | X | |||||
| Report Clinical Safety Information (SAE & pregnancy initial and follow-up) to GSK and LIXTE | X | |||||
| Transfer of samples to GSK and/or LIXTE or preferred vendor for future research and development if required | x | |||||
| Submit safety report to ECs/IRBs/regulatory authorities | X | |||||
| Provide on a quarterly basis a line listing of all SAE and pregnancies received during a defined quarter to GSK. These listings should be sent to the following email address: PV.ICSRManagement@gsk.com | X | |||||
| Perform reconciliation and feedback to Institution on quarterly basis upon line listing receipt | X | X | ||||
| Perform audit activities of Institution activities | X | X | ||||
| STUDY CLOSURE & ARCHIVING | ||||||
| Conduct Participating Site Close out activities including product reconciliation | X | |||||
| Perform Data Management and Statistical Analysis | X | |||||
| Provide Data to GSK and to LIXTE | X | |||||
| Create Study Report and Manuscript | X | |||||
| Review Study Report and Manuscript | X | x | ||||
| Contribute (review and comment) to publications (abstracts, presentations, CSR, Manuscripts) | X | x | ||||
| Disclosure of Study Results Summary and submission of manuscripts | x | |||||
| Notification of Study end to regulatory authorities and ECs/IRBs | x | |||||
| Archiving of study files | x | |||||
| Page |
APPENDIX D
SAFETY AND EFFICACY OF TARGETING PP2A IN OVARIAN CLEAR CELL CARCINOMA (OCCC) USING DOSTARLIMAB AND LB-100
PROTOCOL PROVIDED SEPARATELY AND REFERRED TO IN THIS AGREEMENT AS IF SET FORTH IN FULL
| Page |
APPENDIX E
ANTICIPATED TIMELINES
| Milestone |
Target Date (dd/mon/year) |
|
| Final Protocol approved | 07/JUN/2023 | |
| Ethics Committee(EC) / Competent Authority (CA) submission | 21/JUN/2023 | |
| EC /CA approval | 01/AUG/2023 | |
| First Subject, First Visit | 01/OCT/2023 | |
| Last Subject, First Visit | 01/AUG/2025 | |
| Last Subject, Last Visit | 01/AUG/2026 | |
| Database Freeze | 01/AUG/2027 | |
| Final Report (or draft manuscript) delivered to GSK in accordance with Clause 4.1 | 01/JAN/2028 | |
| Manuscript in accordance with Clause 4.1 submitted for publication within 90 days of Study completion at all Study Site(s) | 01/APR/2028 |
| Page |
APPENDIX F:
SAFETY LANGUAGE FOR SUPPORTED STUDIES GSKInvestigational Product
Institution/Investigator Obligations
Under GCPs, applicable laws, and terms of this Agreement, Institution is responsible for and undertakes to assess all clinical safety information arising during the Study in order to generate all safety reports as required by applicable laws. Such safety reports will include, but may not be limited to, Individual Case Safety Reports (“ICSRs”) for Suspected Unexpected Serious Adverse Reactions (“SUSARs”) and, where applicable, Development Safety Update Report(s) (“DSURs”). Institution is responsible for submitting such reports to all concerned regulatory authorities, relevant Independent Ethics Committee(s) (“IEC”) or Institutional Review Board IRB(s) and individual Study investigator(s), as required, and within applicable timelines.
In the event that GSK maintains its own IB(s) for the GSK IMP being investigated under the Study, regardless of the indication under study, GSK will provide these IB(s), and any updates, and/or supplements to these IB(s) to Institution during the course of the Study for information purposes. Institution shall communicate the IB to the Investigator and Sub-investigators during the course of the Study for information purposes.
If any GSK IMP being investigated under the Study are marketed products, or become marketed products during the Study, Institution shall be responsible for providing to the Investigator, Participating Site(s), and Sub-investigator(s) access the current approved local country product information in respect of the marketed GSK IMP through whatever means available to health care professionals in the countries where the Study will be conducted (e.g., internet repositories, published compendia/formularies, etc.).
In the event that GSK produces any of its own DSURs in respect of the GSK IMP, GSK will provide to Institution on request and for the duration of the Study, copies of the executive summary and any line listings of serious adverse reactions extracted from approved GSK DSURs for information only and to assist Institution in the generation of its own DSUR(s), where applicable. Investigator and Institution agrees not to forward such GSK DSUR sections to any third party.
GSK will ensure that any urgent safety issues relating to the GSK IMP provided for the Study will be communicated to the Institution by whatever means that GSK, in its sole discretion, deems appropriate. The Institution shall communicate such issues to the Investigator, Participating Site(s), and Sub-investigator(s) during the course of the Study.
Forms
Institution will use its internal form for SAE reporting and GSKs forms for pregnancy reporting.
Case Exchange
| a. | Investigator shall report all SAEs arising during the Study in Study Subjects exposed to the GSK IMP (as defined by the Protocol), to GSK (as specified below) using an approved form within twenty-four (24) hours or latest one (1) business day of first becoming aware of the event, regardless of Investigator/designee causality assessments against GSK IMP. |
| Page |
Pregnancy Information
| b. | Investigator will report pregnancy information on any female Study Subject who becomes pregnant while participating in the Study and following exposure to a GSK IMP, to GSK (as specified below) using an approved form within 24 hours or latest one (1) business day of first becoming aware of the pregnancy. The Study Subject will also be followed to determine the outcome of the pregnancy (including any premature termination of the pregnancy). Information on the status of the mother and child will be forwarded to GSK. Generally, follow-up will be requested by GSK no longer than six (6) to eight (8) weeks following the estimated delivery date. |
Note:
There are two forms – initial and follow-up.
Reporting Clinical Safety Information to GSK
| c. | In this Study, there is the potential for the unsolicited reporting of GSK-product-related events, by a patient, to the Investigator, and/or there is the potential that the Investigator may read of GSK-product-related events in a patient’s medical records. Any events (AE, SAE, or pregnancies) considered to be related to a GSK product, that occur during the Study, need to be reported according to country regulatory guidelines. Study participation-related events should be collected and reported according to country regulatory guidelines. |
Reporting Period
| d. | The SAEs and pregnancy reports that are subject to the above reporting provisions are those that occur following the first dose of the GSK IMP as long as Protocol defines. |
Requesting Follow-up Information
| e. | Investigator will provide GSK with details of whom GSK shall address requests for follow up information on SAE and pregnancy reported from this Study, and further agrees to update such contact details as necessary. At the time of this Agreement, all such requests should be addressed to: |
Dr. Amir Jazaeri (aajazaeri@mdanderson.org)
Investigator shall submit to GSK (as specified above) such further detailed information relating thereto as GSK shall request within twenty-four (24) hours or latest one (1) business day of it becoming available.
Events Exempt from Reporting to GSK
| f. | Any blinded ICSRs or any unblinded reports for Study Subjects exposed only to placebo or a non-GSK comparator during the Study. |
Routing of Clinical Safety Data to GSK
| g. | Notwithstanding the Force Majeure Clause of this Agreement, such reports and information as outlined above, including Investigator causality assessments against all concerned GSK IMP and English translations where reporting is from a non-English speaking country, shall be sent via email to the below destination: |
Pharma:
OAX37649@GSK.com
| Page |
Reconciliations
If SCS: Quarterly reconciliations shall be performed.
Communication for reconciliation shall be sent via email to the below destination:
Reconciliation mailbox: Pharma: PV.ICSRManagement@gsk.com
Definitions
Adverse Event (AE) – Any untoward medical occurrence in a patient or clinical trial subject administered a medicinal product and which does not necessarily have a causal relationship with this treatment. An adverse event can therefore be any unfavourable and unintended sign (e.g., an abnormal laboratory finding), symptom, or disease temporally associated with the use of a medicinal product, whether or not considered related to the medicinal product.
Adverse Events of Special Interest (AESI) - An adverse event of special interest (serious or non-serious) is one of scientific and medical concern specific to the Institution’s product or program, for which ongoing monitoring and rapid communication by the investigator to the Institution can be appropriate. Such an event might warrant further investigation in order to characterize and understand it. Depending on the nature of the event, rapid communication by the trial Institution to other parties (e.g., regulators) might also be warranted.
Development Safety Update Report (DSUR) – A periodic reporting on drugs under development (see ICH-E2F Guideline, Volume 10 of the Rules Governing Medicinal Products in the EU).
Emerging Safety Issue - A safety issue considered by a marketing authorisation holder to require urgent attention by the competent authority because of the potential major impact on the risk-benefit balance of the medicinal product and/or on patients’ or public health and the potential need for prompt regulatory action and communication to patients and healthcare professionals. Examples include: major safety issues identified in the context of ongoing or newly completed studies, e.g., an unexpectedly increased rate of fatal or life-threatening adverse events; major safety issues identified through the spontaneous reporting system or publications in the scientific literature, which may lead to considering a contraindication, a restriction of use of a medicinal product or its withdrawal from the market; major safety-related regulatory actions outside the EU, e.g., a restriction of use of a medicinal product or its suspension.
Investigational New Drug Safety Report (INDSR) - Is a written safety report used by Institutions to notify FDA of any adverse experience associated with the use of the drug that is both serious and unexpected.
Investigator’s Brochure (IB) - A compilation of the clinical and nonclinical data on the investigational product(s) which is relevant to the study of the investigational product(s) in human subjects.
Pregnancy Cases - Cases originating from spontaneous or clinical trial sources. Pregnancy data is any abnormal pregnancy, normal pregnancy outcome or adverse event/special situation following direct exposure to a GSK product, via a patient’s partner, or via breast milk (lactation exposure).
| Page |
Pregnancy/Lactation Exposure - With or without any AEs related to the parent or child. Use of a product while pregnant and/or breastfeeding.
Reference Safety Information (RSI) - In periodic benefit-risk evaluation reports for medicinal products, all relevant safety information contained in the reference product information (e.g., the company core data sheet) prepared by the marketing authorisation holder and which the marketing authorisation holder requires to be listed in all countries where it markets the product, except when the local regulatory authority specifically requires a modification (see GVP Annex IV, ICH-E2C(R2) Guideline)”
Serious Adverse Event (SAE) - An untoward medical occurrence that at any dose: results in death, is life-threatening (NOTE: The term “life-threatening” in the definition of “serious” refers to an event in which the patient was at risk of death at the time of the event; it does not refer to an event which hypothetically might have caused death if it were more severe), requires inpatient hospitalisation or prolongation of existing hospitalization, results in persistent or significant disability/incapacity, is a congenital anomaly/birth defect, is a medically important event or reaction
Signal - Information arising from one or multiple sources, including observations and experiments, which suggests a new potentially causal association, or a new aspect of a known association between an intervention and an event or set of related events, either adverse or beneficial, that is judged to be of sufficient likelihood to justify verificatory action.
Suspected Unexpected Serious Adverse Reaction (SUSAR) - All suspected adverse reactions related to an IMP (the tested IMP and comparators) which occur in the concerned trial that are both unexpected and serious (SUSARs) are subject to expedited reporting.
Terms of Interest (TOI) - A group of MedDRA terms maintained in the Integrated Coding Dictionary System (ICDS). TOIs may be comprised of levels of the MedDRA hierarchy at the Preferred Term (PT) level or higher. TOIs may also be constructed from Standardized MedDRA Queries (SMQs), other TOIs in a ‘building-block’ manner or contain mixtures of TOIs and levels of the MedDRA hierarchy.
| Page |
APPENDIX G
Institution Investigational Pharmacy Services (IPS) Requirements
| 1. | Minimum Labeling Standards |
Product Labeling by GSK and LIXTE
| A. | GSK and LIXTE shall ensure that all immediate containers of their respective Investigational Product must, at a minimum, be labeled with the following information: |
| i. | Name of product | |
| ii. | Lot or batch number | |
| iii. | Storage conditions | |
| iv. | Quantity | |
| v. | Formulation | |
| vi. | Name and address of manufacturer or GSK or LIXTE, as applicable |
Note: Investigational Product that is shipped to Institution and not labeled as described above will be deemed unacceptable for use and will be destroyed or returned to GSK or LIXTE, as applicable, at its cost.
| 2. | Drug Expiration/Re-Test Dating Information |
GSK and LIXTE shall ensure that Re-test and/or expiration dating information shall be provided to Institution with each lot of their respective Investigational Product. Investigational Product may be quarantined by Institution until such information is provided.
| Page |
Exhibit 31.1
CERTIFICATION OF CHIEF EXECUTIVE OFFICER
UNDER SECTION 302 OF THE SARBANES-OXLEY ACT OF 2002
I, Geordan Pursglove, certify that:
| 1. | I have reviewed this Quarterly Report on Form 10-Q of Lixte Biotechnology Holdings, Inc.; |
| 2. | Based on my knowledge, this report does not contain any untrue statement of a material fact or omit to state a material fact necessary to make the statements made, in light of the circumstances under which such statements were made, not misleading with respect to the period covered by this report; |
| 3. | Based on my knowledge, the financial statements, and other financial information included in this report, fairly present in all material respects the financial condition, results of operations and cash flows of the registrant as of, and for, the periods presented in this report; |
| 4. | I am responsible for establishing and maintaining disclosure controls and procedures (as defined in Exchange Act Rules 13a-15(e) and 15d-15(e)) and internal control over financial reporting (as defined in Exchange Act Rules 13a-15(f) and 15d-15(f)), for the registrant and have: |
| (a) | Designed such disclosure controls and procedures, or caused such disclosure controls and procedures to be designed under my supervision, to ensure that material information relating to the registrant, including its consolidated subsidiaries, is made known to me by others within those entities, particularly during the period in which this report is being prepared; | |
| (b) | Designed such internal control over financial reporting, or caused such internal control over financial reporting to be designed under my supervision, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles; | |
| (c) | Evaluated the effectiveness of the registrant’s disclosure controls and procedures and presented in this report my conclusions about the effectiveness of the disclosure controls and procedures, as of the end of the period covered by this report based on such evaluation; and | |
| (d) | Disclosed in this report any change in the registrant’s internal control over financial reporting that occurred during the registrant’s most recent fiscal quarter (the registrant’s fourth fiscal quarter in the case of an annual report) that has materially affected, or is reasonably likely to materially affect, the registrant’s internal control over financial reporting; and |
| 5. | I have disclosed, based on my most recent evaluation of internal control over financial reporting, to the registrant’s auditors and the audit committee of registrant’s Board of Directors (or persons performing the equivalent functions): |
| (a) | All significant deficiencies and material weaknesses in the design or operation of internal control over financial reporting which are reasonably likely to adversely affect the registrant’s ability to record, process, summarize and report financial information; and | |
| (b) | Any fraud, whether or not material, that involves management or other employees who have a significant role in the registrant’s internal control over financial reporting. |
| Date: August 7, 2025 | By: | /s/ GEORDAN PURSGLOVE |
| Geordan Pursglove | ||
| Chief Executive Officer | ||
| (Principal Executive Officer) |
Exhibit 31.2
CERTIFICATION OF CHIEF FINANCIAL OFFICER
UNDER SECTION 302 OF THE SARBANES-OXLEY ACT OF 2002
I, Robert N. Weingarten, certify that:
| 1. | I have reviewed this Quarterly Report on Form 10-Q of Lixte Biotechnology Holdings, Inc.; | |
| 2. | Based on my knowledge, this report does not contain any untrue statement of a material fact or omit to state a material fact necessary to make the statements made, in light of the circumstances under which such statements were made, not misleading with respect to the period covered by this report; | |
| 3. | Based on my knowledge, the financial statements, and other financial information included in this report, fairly present in all material respects the financial condition, results of operations and cash flows of the registrant as of, and for, the periods presented in this report; | |
| 4. | I am responsible for establishing and maintaining disclosure controls and procedures (as defined in Exchange Act Rules 13a-15(e) and 15d-15(e)) and internal control over financial reporting (as defined in Exchange Act Rules 13a-15(f) and 15d-15(f)), for the registrant and have: | |
| (a) | Designed such disclosure controls and procedures, or caused such disclosure controls and procedures to be designed under my supervision, to ensure that material information relating to the registrant, including its consolidated subsidiaries, is made known to me by others within those entities, particularly during the period in which this report is being prepared; | |
| (b) | Designed such internal control over financial reporting, or caused such internal control over financial reporting to be designed under my supervision, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles; | |
| (c) | Evaluated the effectiveness of the registrant’s disclosure controls and procedures and presented in this report my conclusions about the effectiveness of the disclosure controls and procedures, as of the end of the period covered by this report based on such evaluation; and | |
| (d) | Disclosed in this report any change in the registrant’s internal control over financial reporting that occurred during the registrant’s most recent fiscal quarter (the registrant’s fourth fiscal quarter in the case of an annual report) that has materially affected, or is reasonably likely to materially affect, the registrant’s internal control over financial reporting; and | |
| 5. | I have disclosed, based on my most recent evaluation of internal control over financial reporting, to the registrant’s auditors and the audit committee of registrant’s Board of Directors (or persons performing the equivalent functions): | |
| (a) | All significant deficiencies and material weaknesses in the design or operation of internal control over financial reporting which are reasonably likely to adversely affect the registrant’s ability to record, process, summarize and report financial information; and | |
| (b) | Any fraud, whether or not material, that involves management or other employees who have a significant role in the registrant’s internal control over financial reporting. | |
| Date: August 7, 2025 | By: | /s/ ROBERT N. WEINGARTEN |
| Robert N. Weingarten | ||
| Vice President and Chief Financial Officer |
Exhibit 32.1
CERTIFICATIONS OF CHIEF EXECUTIVE OFFICER
UNDER SECTION 906 OF THE SARBANES-OXLEY ACT OF 2002
I, Geordan Pursglove, the Chief Executive Officer of Lixte Biotechnology Holdings, Inc. (the “Company”), certify, pursuant to Section 906 of the Sarbanes-Oxley Act of 2002, 18 U.S.C. Section 1350, that:
(i) The Quarterly Report on Form 10-Q of the Company for the quarterly period ended June 30, 2025 (the “Report”) fully complies with the requirements of Section 13(a) or Section 15(d) of the Securities Exchange Act of 1934; and
(ii) The information contained in the Report fairly presents, in all material respects, the financial condition and results of operations of the Company.
A signed original of this written statement required by Section 906 has been provided to the Company and will be retained by the Company and furnished to the Securities and Exchange Commission or its staff upon request.
| Date: August 7, 2025 | By: | /s/ GEORDAN PURSGLOVE |
| Geordan Pursglove | ||
| Chief Executive Officer | ||
| (Principal Executive Officer) |
Exhibit 32.2
CERTIFICATIONS OF CHIEF FINANCIAL OFFICER
UNDER SECTION 906 OF THE SARBANES-OXLEY ACT OF 2002
I, Robert N. Weingarten, the Chief Financial Officer of Lixte Biotechnology Holdings, Inc. (the “Company”), certify, pursuant to Section 906 of the Sarbanes-Oxley Act of 2002, 18 U.S.C. Section 1350, that:
(i) The Quarterly Report on Form 10-Q of the Company for the quarterly period ended June 30, 2025 (the “Report”) fully complies with the requirements of Section 13(a) or Section 15(d) of the Securities Exchange Act of 1934; and
(ii) The information contained in the Report fairly presents, in all material respects, the financial condition and results of operations of the Company.
A signed original of this written statement required by Section 906 has been provided to the Company and will be retained by the Company and furnished to the Securities and Exchange Commission or its staff upon request.
| Date: August 7, 2025 | By: | /s/ ROBERT N. WEINGARTEN |
| Robert N. Weingarten | ||
| Vice President and Chief Financial Officer | ||
| (Principal Financial and Accounting Officer) |