false2024FY0001771910P5Yhttp://fasb.org/us-gaap/2024#AccruedLiabilitiesCurrenthttp://fasb.org/us-gaap/2024#AccruedLiabilitiesCurrentP16M22DP28M22Dhttp://fasb.org/us-gaap/2024#CostsAndExpenseshttp://fasb.org/us-gaap/2024#CostsAndExpensesP2Yiso4217:USDxbrli:sharesiso4217:CHFxbrli:sharesiso4217:USDxbrli:sharesadc:subsidiaryadc:customerxbrli:pureadc:productadc:ageadc:participantiso4217:CHFadc:segment00017719102024-01-012024-12-3100017719102024-06-3000017719102025-03-0100017719102024-12-3100017719102023-12-310001771910us-gaap:ProductMember2024-01-012024-12-310001771910us-gaap:ProductMember2023-01-012023-12-310001771910adc:RoyaltyRevenueMember2024-01-012024-12-310001771910adc:RoyaltyRevenueMember2023-01-012023-12-3100017719102023-01-012023-12-310001771910us-gaap:CommonStockMember2022-12-310001771910us-gaap:AdditionalPaidInCapitalMember2022-12-310001771910us-gaap:TreasuryStockCommonMember2022-12-310001771910us-gaap:AccumulatedOtherComprehensiveIncomeMember2022-12-310001771910us-gaap:RetainedEarningsMember2022-12-3100017719102022-12-310001771910us-gaap:RetainedEarningsMember2023-01-012023-12-310001771910us-gaap:AccumulatedOtherComprehensiveIncomeMember2023-01-012023-12-310001771910us-gaap:AdditionalPaidInCapitalMember2023-01-012023-12-310001771910us-gaap:TreasuryStockCommonMember2023-01-012023-12-310001771910us-gaap:CommonStockMember2023-12-310001771910us-gaap:AdditionalPaidInCapitalMember2023-12-310001771910us-gaap:TreasuryStockCommonMember2023-12-310001771910us-gaap:AccumulatedOtherComprehensiveIncomeMember2023-12-310001771910us-gaap:RetainedEarningsMember2023-12-310001771910us-gaap:RetainedEarningsMember2024-01-012024-12-310001771910us-gaap:AccumulatedOtherComprehensiveIncomeMember2024-01-012024-12-310001771910us-gaap:CommonStockMember2024-01-012024-12-310001771910us-gaap:AdditionalPaidInCapitalMember2024-01-012024-12-310001771910us-gaap:TreasuryStockCommonMember2024-01-012024-12-310001771910us-gaap:CommonStockMember2024-12-310001771910us-gaap:AdditionalPaidInCapitalMember2024-12-310001771910us-gaap:TreasuryStockCommonMember2024-12-310001771910us-gaap:AccumulatedOtherComprehensiveIncomeMember2024-12-310001771910us-gaap:RetainedEarningsMember2024-12-310001771910adc:SeniorSecuredTermLoanMember2024-01-012024-12-310001771910adc:SeniorSecuredTermLoanMember2023-01-012023-12-310001771910adc:TopFourCustomersMemberus-gaap:CustomerConcentrationRiskMemberus-gaap:FinanceReceivablesMember2024-01-012024-12-310001771910adc:TopFourCustomersMemberus-gaap:CustomerConcentrationRiskMemberus-gaap:FinanceReceivablesMember2023-01-012023-12-310001771910currency:GBP2024-12-310001771910currency:GBP2023-12-310001771910currency:GBP2024-01-012024-12-310001771910currency:GBP2023-01-012023-12-310001771910srt:MinimumMember2024-01-012024-12-310001771910srt:MaximumMember2024-01-012024-12-310001771910srt:MinimumMemberus-gaap:LeaseholdImprovementsMember2024-12-310001771910srt:MaximumMemberus-gaap:LeaseholdImprovementsMember2024-12-310001771910adc:LaboratoryEquipmentMember2024-12-310001771910us-gaap:OfficeEquipmentMember2024-12-310001771910adc:HardwareAndSoftwareMember2024-12-310001771910country:CH2024-12-310001771910country:CH2023-12-310001771910country:GB2024-12-310001771910country:GB2023-12-310001771910country:US2024-12-310001771910country:US2023-12-310001771910us-gaap:FairValueInputsLevel1Member2024-12-310001771910us-gaap:FairValueInputsLevel2Member2024-12-310001771910us-gaap:FairValueInputsLevel3Member2024-12-310001771910us-gaap:FairValueInputsLevel1Member2023-12-310001771910us-gaap:FairValueInputsLevel2Member2023-12-310001771910us-gaap:FairValueInputsLevel3Member2023-12-310001771910us-gaap:LeaseholdImprovementsMember2024-12-310001771910us-gaap:LeaseholdImprovementsMember2023-12-310001771910adc:LaboratoryEquipmentMember2023-12-310001771910us-gaap:OfficeEquipmentMember2023-12-310001771910adc:HardwareAndSoftwareMember2023-12-310001771910country:CH2023-09-010001771910country:GB2023-01-300001771910adc:OverlandADCTBioPharmaCYLimitedMember2020-12-142020-12-140001771910adc:OverlandPharmaceuticalsMember2020-12-142020-12-140001771910adc:OverlandADCTBioPharmaMemberadc:OverlandPharmaceuticalsMember2020-12-140001771910adc:OverlandADCTBioPharmaMember2020-12-140001771910adc:OverlandADCTBioPharmaMember2020-12-140001771910adc:OverlandADCTBioPharmaCYLimitedMember2022-12-310001771910adc:OverlandADCTBioPharmaCYLimitedMember2023-01-012023-12-310001771910adc:OverlandADCTBioPharmaCYLimitedMember2023-12-310001771910adc:OverlandADCTBioPharmaCYLimitedMember2024-01-012024-12-310001771910adc:OverlandADCTBioPharmaCYLimitedMember2024-12-310001771910us-gaap:DomesticCountryMember2024-12-310001771910us-gaap:DomesticCountryMember2023-12-310001771910us-gaap:StateAndLocalJurisdictionMember2024-12-310001771910us-gaap:StateAndLocalJurisdictionMember2023-12-310001771910us-gaap:ResearchMember2024-12-310001771910us-gaap:ForeignCountryMember2024-12-310001771910adc:LoanAgreementMemberus-gaap:SecuredDebtMemberus-gaap:LineOfCreditMember2022-08-150001771910adc:LoanAgreementMemberus-gaap:SecuredDebtMemberus-gaap:LineOfCreditMember2022-08-152022-08-150001771910adc:LoanAgreementMemberus-gaap:SecuredDebtMemberus-gaap:LineOfCreditMemberus-gaap:SecuredOvernightFinancingRateSofrMemberadc:InterestRatePeriodOneMember2022-08-152022-08-150001771910adc:LoanAgreementMemberus-gaap:SecuredDebtMemberus-gaap:LineOfCreditMemberus-gaap:BaseRateMemberadc:InterestRatePeriodOneMember2022-08-152022-08-150001771910adc:LoanAgreementMemberus-gaap:SecuredDebtMemberus-gaap:LineOfCreditMemberus-gaap:SecuredOvernightFinancingRateSofrMemberadc:InterestRatePeriodTwoMember2022-08-152022-08-150001771910adc:LoanAgreementMemberus-gaap:SecuredDebtMemberus-gaap:LineOfCreditMemberus-gaap:BaseRateMemberadc:InterestRatePeriodTwoMember2022-08-152022-08-150001771910adc:LoanAgreementMemberus-gaap:SecuredDebtMemberus-gaap:LineOfCreditMemberus-gaap:SecuredOvernightFinancingRateSofrMember2022-08-150001771910adc:LoanAgreementMemberus-gaap:SecuredDebtMemberus-gaap:LineOfCreditMembersrt:MinimumMember2022-08-152022-08-150001771910adc:LoanAgreementMemberus-gaap:SecuredDebtMemberus-gaap:LineOfCreditMembersrt:MaximumMember2022-08-152022-08-150001771910adc:LoanAgreementWarrantsMember2022-08-1500017719102022-08-152022-08-1500017719102022-08-150001771910adc:LoanAgreementMember2024-01-012024-12-310001771910adc:LoanAgreementMemberus-gaap:SecuredDebtMemberus-gaap:LineOfCreditMember2023-01-012023-12-310001771910adc:LoanAgreementMemberus-gaap:SecuredDebtMemberus-gaap:LineOfCreditMember2024-12-310001771910adc:LoanAgreementMemberus-gaap:SecuredDebtMemberus-gaap:LineOfCreditMember2024-01-012024-12-310001771910adc:LoanAgreementMember2024-12-310001771910adc:DeerfieldWarrantsMember2022-08-152022-08-150001771910adc:WarrantsTrancheOneMemberadc:DeerfieldWarrantsMember2022-08-152022-08-150001771910adc:WarrantsTrancheOneMemberadc:DeerfieldWarrantsMember2022-08-150001771910adc:WarrantsTrancheTwoMemberadc:DeerfieldWarrantsMember2022-08-152022-08-150001771910adc:WarrantsTrancheTwoMemberadc:DeerfieldWarrantsMember2022-08-150001771910adc:DeerfieldWarrantsMember2022-08-150001771910adc:DeerfieldWarrantsMember2024-01-012024-12-310001771910adc:DeerfieldWarrantsMember2023-01-012023-12-310001771910adc:DeerfieldWarrantsMember2024-12-310001771910adc:DeerfieldWarrantsMember2023-12-310001771910adc:DeerfieldWarrantsMemberus-gaap:MeasurementInputExercisePriceMembersrt:MinimumMember2024-12-310001771910adc:DeerfieldWarrantsMemberus-gaap:MeasurementInputExercisePriceMembersrt:MaximumMember2024-12-310001771910adc:DeerfieldWarrantsMemberus-gaap:MeasurementInputExercisePriceMembersrt:MinimumMember2023-12-310001771910adc:DeerfieldWarrantsMemberus-gaap:MeasurementInputExercisePriceMembersrt:MaximumMember2023-12-310001771910adc:DeerfieldWarrantsMemberus-gaap:MeasurementInputSharePriceMember2024-12-310001771910adc:DeerfieldWarrantsMemberus-gaap:MeasurementInputSharePriceMember2023-12-310001771910adc:DeerfieldWarrantsMemberus-gaap:MeasurementInputRiskFreeInterestRateMember2024-12-310001771910adc:DeerfieldWarrantsMemberus-gaap:MeasurementInputRiskFreeInterestRateMember2023-12-310001771910adc:DeerfieldWarrantsMemberus-gaap:MeasurementInputPriceVolatilityMember2024-12-310001771910adc:DeerfieldWarrantsMemberus-gaap:MeasurementInputPriceVolatilityMember2023-12-310001771910us-gaap:MeasurementInputExpectedTermMember2024-12-310001771910us-gaap:MeasurementInputExpectedTermMember2023-12-310001771910adc:DeerfieldWarrantsMemberadc:MeasurementInputDividendYieldMember2024-12-310001771910adc:DeerfieldWarrantsMemberadc:MeasurementInputDividendYieldMember2023-12-310001771910adc:DeerfieldWarrantsMemberadc:MeasurementInputValueMembersrt:MinimumMember2024-12-310001771910adc:DeerfieldWarrantsMemberadc:MeasurementInputValueMembersrt:MaximumMember2024-12-310001771910adc:DeerfieldWarrantsMemberadc:MeasurementInputValueMembersrt:MinimumMember2023-12-310001771910adc:DeerfieldWarrantsMemberadc:MeasurementInputValueMembersrt:MaximumMember2023-12-310001771910adc:DeerfieldWarrantsMemberus-gaap:MeasurementInputExpectedTermMember2024-12-310001771910adc:DeerfieldWarrantsMemberus-gaap:MeasurementInputExpectedTermMember2023-12-3100017719102021-08-252021-08-250001771910adc:ZYNLONTAMember2021-08-252021-08-250001771910adc:CamiMember2021-08-252021-08-250001771910srt:MaximumMember2021-08-252021-08-250001771910srt:MaximumMember2023-01-012023-12-310001771910srt:MinimumMember2021-08-252021-08-250001771910srt:MinimumMember2023-01-012023-12-310001771910srt:MinimumMember2024-12-310001771910srt:MaximumMember2024-12-310001771910srt:MinimumMember2023-12-310001771910srt:MaximumMember2023-12-3100017719102022-01-012022-12-3100017719102021-01-012021-12-3100017719102021-08-250001771910country:US2024-01-012024-12-310001771910country:US2023-01-012023-12-310001771910us-gaap:ForeignPlanMember2024-01-012024-12-310001771910us-gaap:ForeignPlanMember2023-01-012023-12-310001771910srt:MedianMember2024-01-012024-12-310001771910adc:A2024EquityOfferingMember2024-05-012024-05-310001771910us-gaap:CommonStockMemberadc:A2024EquityOfferingMember2024-05-310001771910adc:A2024PreFundedWarrantMemberadc:A2024EquityOfferingMember2024-05-012024-05-310001771910adc:A2024EquityOfferingMember2024-05-3100017719102024-05-310001771910adc:A2024PreFundedWarrantMember2024-05-310001771910srt:MinimumMember2024-05-012024-05-310001771910srt:MaximumMember2024-05-012024-05-3100017719102024-05-012024-05-310001771910adc:PublicStockOfferingMember2024-01-012024-12-310001771910country:US2024-01-012024-12-310001771910country:US2023-01-012023-12-310001771910us-gaap:EMEAMember2024-01-012024-12-310001771910us-gaap:EMEAMember2023-01-012023-12-310001771910adc:DiscardedDrugRebateMember2022-12-310001771910adc:OtherAdjustmentsMember2022-12-310001771910adc:DiscardedDrugRebateMember2023-01-012023-12-310001771910adc:OtherAdjustmentsMember2023-01-012023-12-310001771910adc:DiscardedDrugRebateMember2023-12-310001771910adc:OtherAdjustmentsMember2023-12-310001771910adc:DiscardedDrugRebateMember2024-01-012024-12-310001771910adc:OtherAdjustmentsMember2024-01-012024-12-310001771910adc:DiscardedDrugRebateMember2024-12-310001771910adc:OtherAdjustmentsMember2024-12-310001771910adc:McKessonMemberus-gaap:CustomerConcentrationRiskMemberus-gaap:SalesRevenueNetMember2024-01-012024-12-310001771910adc:McKessonMemberus-gaap:CustomerConcentrationRiskMemberus-gaap:SalesRevenueNetMember2023-01-012023-12-310001771910adc:AmerisourceBergenCorporationMemberus-gaap:CustomerConcentrationRiskMemberus-gaap:SalesRevenueNetMember2024-01-012024-12-310001771910adc:AmerisourceBergenCorporationMemberus-gaap:CustomerConcentrationRiskMemberus-gaap:SalesRevenueNetMember2023-01-012023-12-310001771910adc:CardinalHealthMemberus-gaap:CustomerConcentrationRiskMemberus-gaap:SalesRevenueNetMember2024-01-012024-12-310001771910adc:CardinalHealthMemberus-gaap:CustomerConcentrationRiskMemberus-gaap:SalesRevenueNetMember2023-01-012023-12-310001771910adc:A2019EquityIncentivePlanMember2019-11-300001771910us-gaap:StockCompensationPlanMemberadc:A2019EquityIncentivePlanMember2024-12-310001771910adc:A2019EquityIncentivePlanMember2024-01-012024-12-310001771910adc:A2019EquityIncentivePlanMember2023-01-012023-12-310001771910adc:ConditionalShareCapitalPlanMember2019-11-300001771910us-gaap:RestrictedStockUnitsRSUMemberadc:ConditionalShareCapitalPlanMember2023-12-062023-12-060001771910us-gaap:StockCompensationPlanMemberadc:ConditionalShareCapitalPlanMember2023-12-062023-12-060001771910adc:ConditionalShareCapitalPlanMember2024-12-310001771910adc:ConditionalShareCapitalPlanMember2024-01-012024-12-310001771910adc:ConditionalShareCapitalPlanMember2023-01-012023-12-310001771910adc:InducementPlanMember2024-12-310001771910adc:InducementPlanMember2023-12-310001771910adc:InducementPlanMember2024-01-012024-12-310001771910adc:EquityExchangeProgramMember2023-03-062023-03-060001771910adc:EquityExchangeProgramMembersrt:MinimumMember2023-03-062023-03-060001771910adc:EquityExchangeProgramMembersrt:MaximumMember2023-03-062023-03-060001771910adc:EmployeeStockOptionExchangeMemberadc:EquityExchangeProgramMember2024-01-012024-12-310001771910adc:EmployeeStockOptionExchangeMemberadc:EquityExchangeProgramMember2023-03-062023-03-060001771910us-gaap:EmployeeStockOptionMember2019-11-012019-11-300001771910us-gaap:StockCompensationPlanMember2024-01-012024-12-310001771910us-gaap:StockCompensationPlanMember2023-01-012023-12-310001771910us-gaap:EmployeeStockOptionMember2024-01-012024-12-310001771910us-gaap:EmployeeStockOptionMembersrt:MinimumMember2024-12-310001771910us-gaap:EmployeeStockOptionMembersrt:MaximumMember2024-12-310001771910us-gaap:EmployeeStockOptionMembersrt:MinimumMember2023-12-310001771910us-gaap:EmployeeStockOptionMembersrt:MaximumMember2023-12-310001771910us-gaap:EmployeeStockOptionMembersrt:MinimumMember2024-01-012024-12-310001771910us-gaap:EmployeeStockOptionMembersrt:MaximumMember2024-01-012024-12-310001771910us-gaap:EmployeeStockOptionMembersrt:MinimumMember2023-01-012023-12-310001771910us-gaap:EmployeeStockOptionMembersrt:MaximumMember2023-01-012023-12-310001771910us-gaap:EmployeeStockOptionMember2023-01-012023-12-310001771910adc:EmployeeStockOptionExchangeMembersrt:MinimumMember2024-01-012024-12-310001771910adc:EmployeeStockOptionExchangeMembersrt:MaximumMember2024-01-012024-12-310001771910adc:EmployeeStockOptionExchangeMember2024-01-012024-12-310001771910adc:A2019EquityIncentivePlanMemberus-gaap:RestrictedStockUnitsRSUMembersrt:MinimumMember2019-11-012019-11-300001771910adc:A2019EquityIncentivePlanMemberus-gaap:RestrictedStockUnitsRSUMembersrt:MaximumMember2019-11-012019-11-300001771910us-gaap:RestrictedStockUnitsRSUMemberadc:A2019EquityIncentivePlanMember2024-01-012024-12-310001771910us-gaap:RestrictedStockUnitsRSUMemberadc:A2019EquityIncentivePlanMember2023-01-012023-12-310001771910us-gaap:RestrictedStockUnitsRSUMember2022-12-310001771910us-gaap:RestrictedStockUnitsRSUMember2023-01-012023-12-310001771910us-gaap:RestrictedStockUnitsRSUMember2023-12-310001771910us-gaap:RestrictedStockUnitsRSUMember2024-01-012024-12-310001771910us-gaap:RestrictedStockUnitsRSUMember2024-12-310001771910us-gaap:EmployeeStockMemberadc:A2022EmployeeStockPurchasePlanMember2022-06-012022-06-300001771910adc:A2022EmployeeStockPurchasePlanMember2024-01-012024-12-310001771910adc:A2022EmployeeStockPurchasePlanMember2023-01-012023-12-310001771910us-gaap:EmployeeStockOptionMemberadc:A2019EquityIncentivePlanMember2024-01-012024-12-310001771910us-gaap:EmployeeStockOptionMemberadc:A2019EquityIncentivePlanMember2023-01-012023-12-310001771910adc:InducementPlanMember2024-01-012024-12-310001771910adc:InducementPlanMember2023-01-012023-12-310001771910us-gaap:RestrictedStockUnitsRSUMemberadc:A2019EquityIncentivePlanMember2024-01-012024-12-310001771910us-gaap:RestrictedStockUnitsRSUMemberadc:A2019EquityIncentivePlanMember2023-01-012023-12-310001771910adc:ConditionalShareCapitalPlanMember2024-01-012024-12-310001771910adc:ConditionalShareCapitalPlanMember2023-01-012023-12-310001771910us-gaap:WarrantMember2024-01-012024-12-310001771910us-gaap:WarrantMember2023-01-012023-12-310001771910adc:A2022EmployeeStockPurchasePlanMember2024-01-012024-12-310001771910adc:A2022EmployeeStockPurchasePlanMember2023-01-012023-12-310001771910adc:ReportableSegmentMember2024-01-012024-12-310001771910adc:ReportableSegmentMember2023-01-012023-12-3100017719102024-10-012024-12-31
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
(Mark One)
|
|
|
|
|
|
| ☒ |
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
|
For the fiscal year ended December 31, 2024 |
|
OR |
| ☐ |
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
|
For the transition period from to |
Commission File Number: 001-39071
ADC Therapeutics SA
(Exact name of registrant as specified in its charter)
|
|
|
|
|
|
| Switzerland |
Not Applicable |
| (State or other jurisdiction of incorporation or organization) |
(I.R.S. Employer Identification No.) |
|
|
|
Biopôle
Route de la Corniche 3B
1066 Epalinges
Switzerland
(Address of principal executive offices) (Zip code)
|
+41 21 653 02 00
(Registrant’s telephone number)
|
Securities registered pursuant to Section 12(b) of the Act:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Title of each class |
|
Trading Symbol(s) |
|
Name of each exchange on which registered |
| Common Shares, par value CHF 0.08 per share |
|
ADCT |
|
The New York Stock Exchange |
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☐ No ☒
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ☐ No ☒
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports) and (2) has been subject to such filing requirements for the past 90 days. Yes ☒ No ☐
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files). Yes ☒ No ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Large accelerated filer |
|
☐ |
|
Accelerated filer |
|
☐ |
| Non-accelerated filer |
|
☒ |
|
Smaller reporting company |
|
☒ |
|
|
|
|
Emerging growth company |
|
☐ |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report. ☐
If securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant included in the filing reflect the correction of an error to previously issued financial statements. ☐
Indicate by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive-based compensation received by any of the registrant’s executive officers during the relevant recovery period pursuant to § 240.10D-1(b). ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ☐ No ☒
The aggregate market value of the voting and non-voting common equity held by non-affiliates, based on the closing price of the common shares on The New York Stock Exchange, as of the last business day of the registrant’s most recently completed second fiscal quarter was approximately $173.4 million.
As of March 17, 2025, the number of common shares outstanding was 99,083,838.
DOCUMENTS INCORPORATED BY REFERENCE:
ADC Therapeutics SA intends to file a definitive proxy statement pursuant to Regulation 14A relating to its 2025 Annual General Meeting within 120 days of the end of its fiscal year ended December 31, 2024. Portions of such definitive proxy statement are incorporated by reference into Part III of this Annual Report on Form 10-K to the extent stated herein.
Table of Contents
Unless otherwise indicated or the context otherwise requires, all references in this Annual Report to “ADC Therapeutics,” “ADCT,” the “Company,” “we,” “our,” “ours,” “us” or similar terms refer to ADC Therapeutics SA and its consolidated subsidiaries.
Trademarks
We own various trademark registrations and applications, and unregistered trademarks, including ADC Therapeutics, ADCT, ZYNLONTA and our corporate logo. All other trade names, trademarks and service marks of other companies appearing in this Annual Report are the property of their respective owners. Solely for convenience, the trademarks and trade names in this Annual Report may be referred to without the ® and ™ symbols, but such references should not be construed as any indicator that their respective owners will not assert, to the fullest extent under applicable law, their rights thereto. We do not intend to use or display other companies’ trademarks and trade names to imply a relationship with, or endorsement or sponsorship of us by, any other companies.
Market and Industry Data
This Annual Report contains industry, market and competitive position data that are based on general and industry publications, surveys and studies conducted by third parties, some of which may not be publicly available, and our own internal estimates and research. Third-party publications, surveys and studies generally state that they have obtained information from sources believed to be reliable, but do not guarantee the accuracy and completeness of such information. These data involve a number of assumptions and limitations and contain projections and estimates of the future performance of the industries in which we operate that are subject to a high degree of uncertainty.
FORWARD-LOOKING STATEMENTS
This Annual Report contains statements that constitute forward-looking statements. All statements other than statements of historical facts contained in this Annual Report, including statements regarding our future catalysts, results of operations and financial position, business and commercial strategy, market opportunities, products and product candidates, research pipeline, ongoing and planned preclinical studies and clinical trials, regulatory submissions and approvals, research and development costs, projected revenues and expenses and the timing of revenues and expenses, timing and likelihood of success, as well as plans and objectives of management for future operations are forward-looking statements. Many of the forward-looking statements contained in this Annual Report can be identified by the use of forward-looking words such as “anticipate,” “believe,” “could,” “expect,” “should,” “plan,” “intend,” “estimate,” “will” and “potential,” among others.
Forward-looking statements are based on our management’s beliefs and assumptions and on information available to our management at the time such statements are made. Such statements are subject to known and unknown risks and uncertainties, and actual results may differ materially from those expressed or implied in the forward-looking statements due to various factors, including, but not limited to:
•the substantial net losses that we have incurred since our inception, our expectation to continue to incur losses for the foreseeable future and our need to raise additional capital to fund our operations and execute our business plan;
•our indebtedness under the loan agreement and guaranty (the “Loan Agreement”) with certain affiliates and/or funds managed by each of Oaktree Capital Management, L.P. and Owl Rock Capital Advisors LLC, as lenders, and Blue Owl Opportunistic Master Fund I, L.P., as administrative agent, and the associated restrictive covenants thereunder;
•the purchase and sale agreement (the “HCR Agreement”) with certain entities managed by HealthCare Royalty Management, LLC (“HCR”) and its negative effect on the amount of cash that we are able to generate from sales of, and licensing agreements involving, ZYNLONTA and on our attractiveness as an acquisition target;
•our ability to complete clinical trials on expected timelines, if at all;
•the timing, outcome and results of ongoing or planned clinical trials and the sufficiency of such results;
•undesirable side effects or adverse events of our products and product candidates;
•our and our partners’ ability to obtain and maintain regulatory approval for our product and product candidates;
•our and our partners’ ability to successfully commercialize our products;
•the availability and scope of coverage and reimbursement for our products;
•the complexity and difficulty of manufacturing our products and product candidates;
•the substantial competition in our industry, including new technologies and therapies;
•our ability to continue to fund, or enter into collaborative or partnership arrangements for our research programs, and the timing, results and future clinical outcomes of such research programs;
•our reliance on third parties for preclinical studies and clinical trials and for the manufacture, production, storage and distribution of our products and product candidates and certain commercialization activities for our products;
•our ability to obtain, maintain and protect our intellectual property rights and our ability to operate our business without infringing on the intellectual property rights of others;
•our estimates regarding future revenue, expenses, liquidity, capital resources and needs for additional financing;
•the size and growth potential of the markets for our products and product candidates
•potential product liability lawsuits and product recalls for our products;
•and those identified in the “Item 1A. Risk Factors” section of this Annual Report and in our other reports filed with the U.S. Securities and Exchange Commission (the “SEC”), from time to time hereafter
Because forward-looking statements are inherently subject to risks and uncertainties, some of which cannot be predicted or quantified and some of which are beyond our control, you should not rely on these forward-looking statements as predictions of future events. Moreover, we operate in an evolving environment. New risk factors and uncertainties may emerge from time to time, and it is not possible for management to predict all risk factors and uncertainties. Except as required by applicable law, we do not plan to publicly update or revise any forward-looking statements, whether as a result of any new information, future events, changed circumstances or otherwise.
In addition, statements that “we believe” and similar statements reflect our beliefs and opinions on the relevant subject. These statements are based upon information available to us as of the date of this Annual Report, and while we believe such information forms a reasonable basis for such statements, such information may be limited or incomplete, and our statements should not be read to indicate that we have conducted an exhaustive inquiry into, or review of, all potentially available relevant information. These statements are inherently uncertain and investors are cautioned not to unduly rely upon these statements.
PART I
Item 1. Business
Overview
ADC Therapeutics is a commercial-stage global pioneer in the field of antibody drug conjugates (“ADCs”). The Company is advancing its proprietary ADC technology with the goal of transforming the cancer treatment paradigm for patients with hematologic malignancies and solid tumors. We have a validated and differentiated technology platform with multiple payloads, linkers and conjugation chemistry, enabling the design of next-generation potent ADCs with an enhanced therapeutic index. We are seeking to expand the label for our FDA-approved and marketed product, ZYNLONTA® (loncastuximab tesirine) into new indications and earlier lines of therapy while pursuing our early-stage solid tumor programs. We leverage our scientific and technical expertise and apply a disciplined approach to target selection to expand and advance our pipeline. Our portfolio of ADCs utilizes our highly potent pyrrolobenzodiazepine (“PBD”) payload, a differentiated exatecan-based payload with a novel hydrophilic linker and a next generation ADC toolbox.
In our hematology program, our flagship product, ZYNLONTA, a CD19-directed ADC, received accelerated approval from the U.S. Food and Drug Administration (“FDA”), conditional approval from the European Commission and conditional approval from the China National Medical Products Administration (“NMPA”) for the treatment of relapsed or refractory diffuse large B-cell lymphoma (“DLBCL”) after two or more lines of systemic therapy. We are seeking to continue expanding ZYNLONTA internationally, and into earlier lines of DLBCL and indolent lymphomas, including marginal zone lymphoma (“MZL”) and follicular lymphoma (“FL”), as a single agent and in combination through our LOTIS-5 confirmatory Phase 3 clinical trial and LOTIS-7 Phase 1b clinical trial as well as through investigator-initiated trials (“IITs”) at leading institutions. In addition, we are investigating a CD-22 targeted compound, ADCT-602, in collaboration with the MD Anderson Cancer Center in a Phase 1/2 IIT in relapsed or refractory B-cell acute lymphoblastic leukemia.
In solid tumors, we have early-stage preclinical research programs, including a portfolio of next-generation investigational ADCs targeting Claudin-6, PSMA, NaPi2b, and ASCT2, the most advanced of which are PSMA and Claudin-6. In addition, we are advancing research with a range of payloads, linkers and conjugation technologies against undisclosed targets. The Company is seeking strategic partnerships, collaborations or license arrangements to continue to fund these and other programs.
Strategy
Our goal is to be a leading ADC company that transforms the lives of those impacted by cancer. To achieve this, we are focused on unlocking the potential value of our robust ADC portfolio across two pillars of growth: maximizing the ZYNLONTA opportunity in hematology and pursuing our early-stage research portfolio in solid tumors.
We aim to expand our portfolio and accelerate the development of our pipeline through targeted investments and in collaboration with strategic partners. In this way, we plan to pursue multiple targets in parallel, enabling us to prioritize and ensure disciplined capital allocation strategy while advancing the most promising candidates in both hematology and solid tumors.
Hematology
In our hematology program, our strategy is to maximize and expand the ZYNLONTA opportunity in several ways:
•Maximize ZYNLONTA in third-line plus (“3L+”) DLBCL. We are driving utilization of ZYNLONTA in the 3L+ setting of DLBCL through increased awareness of ZYNLONTA’s efficacy and manageable safety profile and its suitability for use across treatment settings.
•Seek to expand ZYNLONTA into earlier lines of DLBCL and indolent lymphomas as a single agent and in combination with other therapies. We are studying the potential to move ZYNLONTA into earlier lines of therapy in combination with rituximab (LOTIS-5), glofitamab (LOTIS-7) and other novel combinations through our clinical trials and IITs. We believe these development efforts, if successful, will enable ZYNLONTA to move into earlier lines of treatment and potentially become a combination agent of choice in the 2L+ setting, increasing ZYNLONTA’s overall market opportunity.
•Continue to advance the development and commercialization of ZYNLONTA outside of the United States through strategic partnerships. We are committed to providing global access to ZYNLONTA for patients who may benefit from this treatment. In Europe, ZYNLONTA has received conditional approval from the European Commission for the treatment of relapsed or refractory DLBCL after two or more lines of systemic therapy. In China, we have received conditional approval for relapsed or refractory DLBCL after two or more lines of systemic therapy. We have entered into strategic agreements to maximize the commercial potential of ZYNLONTA, including an exclusive license agreement with Mitsubishi Tanabe Corporation (“MTPC”) in Japan, and a joint venture with Overland Pharmaceuticals in China (including Hong Kong and Macau), Singapore, and Taiwan, and an exclusive license agreement with Sobi for all other regions, excluding the U.S. In China, we and the joint venture are exploring commercialization options for ZYNLONTA. In Japan, MTPC is undertaking a Phase 1/2 bridging study and joined the LOTIS-5 confirmatory Phase 3 clinical trial.
We are also developing ADCT-602 targeting CD22 in collaboration with the MD Anderson Cancer Center for patients with relapsed or refractory acute lymphoblastic leukemia. We are currently in dose escalation with ADCT-602 and evaluating the 75 µg/kg dose.
Solid Tumors
In solid tumors, our strategy is to pursue multiple exatecan-based ADC pre-clinical candidates in parallel. We seek to maximize the value of our solid tumor program through strategic partnerships, collaborations and licenses for one or more research programs. Specifically, we seek to:
•Broaden and advance our ADC platform. We are advancing a portfolio of investigational ADCs including those targeting Claudin-6, PSMA, NaPi2b, and ASCT2, the most advanced of which are PSMA and Claudin-6. These candidates are based on an innovative proprietary approach that utilizes exatecan with a novel hydrophilic linker as a highly potent and differentiated payload. We are leveraging our decade-long expertise in the ADC field with multiple INDs and a proven track record of success to continue building this toolbox with new antibody formats, linkers and toxins while advancing a range of payloads, linkers and conjugation technologies against multiple targets to develop differentiated next-generation assets.
Our Competitive Capabilities
We are a pioneer and leader in the ADC field with specialized end-to-end capabilities for developing optimized ADCs. This includes a strong, integrated research & development organization and a validated technology platform with clinical-stage product candidates currently in the pipeline, multiple next-generation ADCs being developed and a proven executional track record that includes ZYNLONTA, the first PBD-based ADC receiving accelerated approval from the FDA, conditional approval from the European Commission and conditional approval from the NMPA in China for the treatment of relapsed or refractory DLBCL after two or more lines of systemic therapy.
Our executive leadership team is comprised of seasoned industry veterans with a history of proven expertise in biopharma from preclinical research and development to drug approval and commercialization.
Since the company was founded in 2011, ADC Therapeutics has significantly invested in all the core capabilities required for the design, preclinical and clinical development and manufacturing of novel ADCs. In the discovery stage, we utilize cutting-edge research to select optimal targeting moiety, linker and payload. The intersection of our technical capabilities, integrated organization and depth of experience allows us to move efficiently through preclinical development into the clinic in pursuit of therapeutic window, if needed in biomarker enriched patient populations. Further, our robust in-house chemistry, manufacturing and controls (“CMC”) capabilities utilize a highly experienced workforce to manage a top-tier external manufacturing network through third-party contract manufacturing organizations (“CMOs”) capable of manufacturing highly potent molecules and complex biologics.
Our Technology Platform and Key Strengths of ADCs
ADCs are an established therapeutic approach in oncology. ADCs selectively deliver potent cytotoxins directly to tumor cells, with the goal of maximizing activity in tumor cells while minimizing toxicity to healthy cells. ADCs are an important part of the cancer treatment paradigm for the following reasons:
•Selective Targeting. Traditional chemotherapies are unable to distinguish between healthy cells and tumor cells and therefore have a narrow therapeutic window (i.e., the dose range that can treat disease effectively without causing unacceptable toxic side effects). In contrast, ADCs, through their use of tumor-specific antibodies, target tumor cells with greater selectivity than chemotherapies. This selective targeting allows ADCs to use potent cytotoxins at dose levels that otherwise would not be tolerated and ADCs therefore represent a highly effective treatment approach while maintaining manageable side effects.
•Wide Addressable Patient Population. ADCs represent a treatment approach that expands the treatment options available to cancer patients. Many therapies are not appropriate for certain patient populations. For example, chemotherapy may not be appropriate when the patient is too sick to tolerate or does not respond to available chemotherapeutics, stem cell transplant may not be appropriate when the patient is frail, and some novel targeted therapies such as CAR-T (i.e., a type of treatment in which a patient’s T cells are modified in the laboratory so they will attack cancer cells) may not be appropriate when there is significant comorbidity. As a result of these limitations, there remains a significant unmet medical need for patients for whom other treatment options are inappropriate or ineffective.
•Potential in Relapsed or Refractory Patients. Traditional therapies typically have limited effectiveness for patients who exhibit relapsed (i.e., the cancer returns after an initial positive response to treatment) or refractory (i.e., the cancer is resistant to treatment) disease. In contrast, some ADCs have proven efficacious in such patient populations while maintaining a manageable safety profile. Therefore, ADCs represent an important part of the cancer treatment paradigm, expanding the treatment options available to patients suffering from relapsed or refractory disease.
ADC Design
An ADC consists of three components: (i) an antibody that selectively targets a distinct antigen preferentially expressed on tumor cells; (ii) a cytotoxic molecule, often referred to as the toxin or the warhead, that kills the target cell; and (iii) a chemical linker that joins together the antibody and the warhead. The warhead and the linker are together referred to as the payload. The figure below shows the three components of an ADC.
_______________
Schematic representation of an ADC, showing its three components.
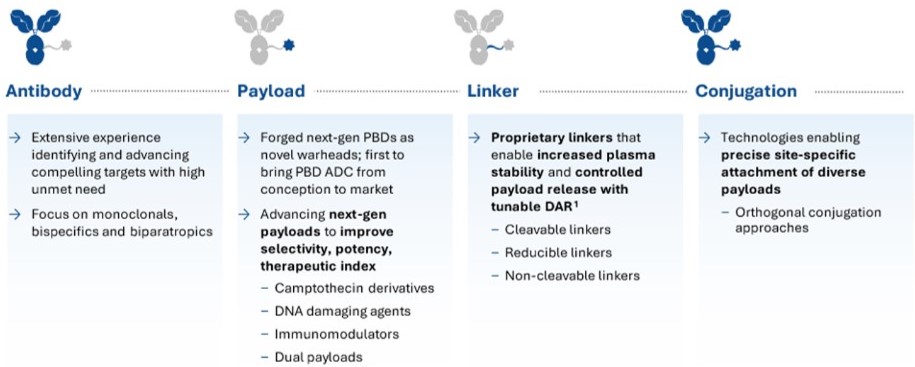
Within ADC Therapeutics, we have a strong focus on technology development with the goal of developing best-in-class ADC candidates with an optimal therapeutic window for any given tumor target. We have an expanding toolbox with a range of payloads, linkers and conjugation technologies.
Note: 1 DAR: Drug antibody ratio
Antibody
Different antibody technologies can be used to optimize targeting of the ADC to the tumor and/or to enhance uptake of the ADC into the tumor once it is bound to the target on the membrane of the tumor. Currently, while most of the ADCs in development are based on monoclonal antibody targeting, we are also exploring the use of bispecific and/or biparatopic antibodies in our ADC selection to enhance the uptake of the ADC into the tumor. We are also exploring the use of novel antibody formats in ADC design such conditionally binding antibodies. Conditionally binding antibodies bind stronger to target expressed in the more acidic local tumor environment and bind less strong to target expressed on healthy tissue which has neutral pH.
Toxins
Our current pipeline consists of multiple programs targeting hematological and solid tumor targets for which we have selected ADC candidates employing our proprietary exatecan platform or PBD dimer technology (under license from AstraZeneca). Exatecans belong to the family of camptothecins, which are naturally occurring pentacyclic quinoline alkaloids that bind to DNA topoisomerase I, inhibiting DNA relegation and finally causing apoptosis. Campothecins such as exatecan therefore possesses high cytotoxic activity against a variety of tumors. The potency of exatecan is slightly higher than DXd, the topoisomerase I inhibitor used in Enhertu® and other ADCs in clinical development such as ifinatamab deruxtecan, patritumab deruxtecan and raludotatug deruxtecan.
PBD dimers are highly potent and bind irreversibly to two guanines from opposite DNA strands in minor groove of DNA without distorting the double helix, potentially evading DNA repair mechanisms. The interstrand cross-links block DNA strand separation, disrupting essential DNA metabolic processes such as replication, and ultimately result in cell death. These interstrand cross-links persist in target cells and can lie dormant, potentially for weeks, which may contribute to the frequency and durability of responses in heavily pre-treated and primary refractory patients that we have observed in our clinical trials with PBD-based ADCs.
Both exatecans and PBD dimers cause a bystander effect, which occurs when a released warhead (from a target positive cells which has internalized and processed the ADC) is able to diffuse into and kill neighboring cells in the tumor microenvironment, irrespective of those cells’ antigen expression. Since exatecan and PBD dimers are cell-permeable, they may be able to diffuse into adjacent cells and kill them in an antigen-independent manner. Importantly, the bystander effect of exatecan has been observed to be considerably stronger than the other topoisomerase 1 inhibitor DXd. Both exatecan and PBD dimers also cause immunogenic cell death (ICD), whereby a cancer cell’s death expresses certain stress signals that induce the body’s anti-tumor immune response through the activation of T cells and antigen-presenting cells. This opens up the potential for combining our ADCs with other therapies, particularly with immuno-oncology therapies such as checkpoint inhibitors, that are specifically designed to activate the patient’s own immune system to combat cancer.
In addition to our proprietary exatecan platform and PBD dimer technology, we have access to another DNA alkylating cytotoxin which we can access under a license agreement. We are furthermore developing payloads based on immune modulators to develop immune stimulating antibody conjugates (ISACs) based on TLR7 agonists. Our ultimate objective is to have a variety of different toxins with orthogonal Mode of Action which can be used to design dual conjugate ADCs (i.e., an ADC to which two different toxins are conjugated).
Linkers
Our linker design focuses on different aspects. First, we focus on the spacer in the linker. Our proprietary exatecan platform is based on a novel hydrophilic spacer which allows conjugation of exatecan to antibodies at high drug to antibody ratio (“DAR”). Secondly, we have cleavable versus non-cleavable linker configurations. While cleavable linkers are the preferred choice if a bystander effect of the ADC is desired, in certain cases bystander activity should be avoided, which can be achieved using non-cleavable linkers. Finally, we have developed a set of branched linkers that allows us to increase the DAR or to generate ADCs with multiple toxins on a single conjugation site.
Conjugation Technologies
Depending on the desired DAR and other criteria such as the need for Fc gamma receptor binding, we can design the optimal ADC using a selection of different site-specific conjugation technologies, either enzymatic or non-enzymatic. We also have access to multiple conjugation chemistries such as classical maleimide and bio-orthogonal click chemistry and we are implementing additional proprietary approaches. We also have a variety of tools that allows us to design dual conjugate ADCs (i.e., an ADC to which two different toxins are conjugated).
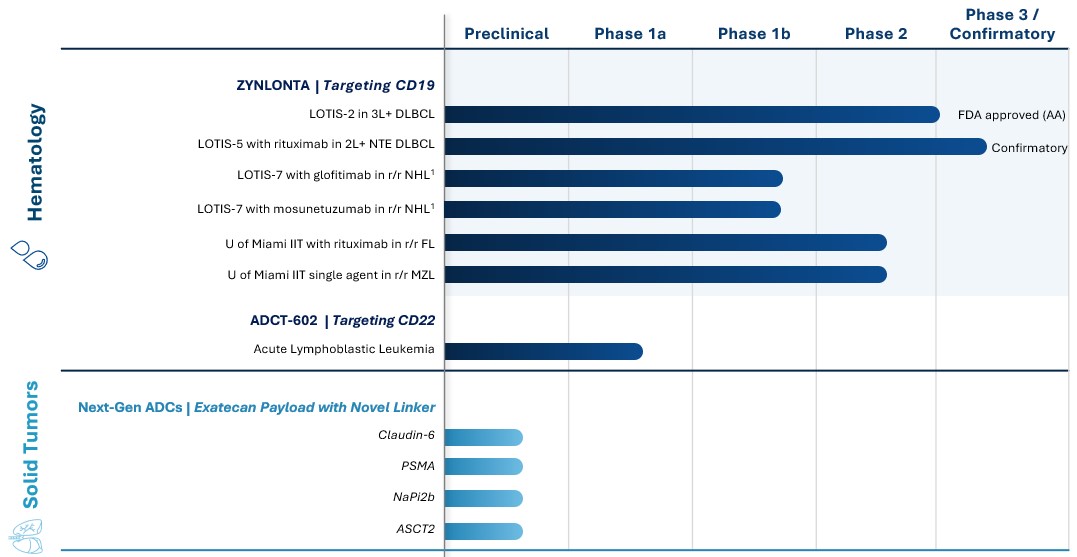
Our Portfolio and Pipeline
The following table provides an overview of our current product portfolio and research pipeline:
NTE: Non-Transplant Eligible. * The ADCT-602 trial is being led by the University of Texas MD Anderson Cancer Center.
Our Market Opportunity
Hematology
ZYNLONTA (loncastuximab tesirine): ADC Targeting CD19
The Lymphoma Disease Setting
We have and continue to develop ZYNLONTA for the treatment of B-cell lymphomas, including:
•DLBCL, the most common type of lymphoma. It is an aggressive form of non-Hodgkin lymphoma (“NHL”) and accounts for 30% of all NHL cases. Approximately 32,000 people in the United States are diagnosed with DLBCL each year and the five-year prevalence for DLBCL is an estimated 113,000 patients, with approximately 70% in the first-line setting, approximately 21% in the 2L setting and approximately 9% in the 3L setting.
•Marginal zone lymphoma (“MZL”) is a rare, indolent form of NHL. MZL is the third most common NHL subtype. In the United States, the five-year prevalence for MZL is an estimated 40,000 patients, with approximately 61% in the first-line setting, approximately 27% in the 2L setting and approximately 12% in the 3L setting.
•Follicular lymphoma (“FL”), is an indolent type of NHL. In the United States, the five-year prevalence for FL is an estimated 62,000 patients, with approximately 65% in the first-line setting, approximately 24% in the 2L setting and approximately 11% in the 3L setting.
Currently, ZYNLONTA is approved for the treatment of DLBCL in the 3L+ setting. We believe that our LOTIS-5 confirmatory Phase 3 clinical trial evaluating the efficacy of ZYNLONTA and rituximab, if successful, could allow this combination to be approved for use in the 2L setting for the treatment of DLBCL. In addition, our LOTIS-7 Phase 1b clinical trial is currently evaluating ZYNLONTA in combination with glofitamab in r/r DLBCL, which could provide initial data for the safety and efficacy of this novel combination in the 2L setting and support for future development of this combination.
Advancing ZYNLONTA Development
Our flagship product, ZYNLONTA (loncastuximab tesirine) is an ADC targeting CD19-expressing cancers. It received accelerated approval from the FDA, conditional approval from the European Commission and conditional approval from the NMPA in China for the treatment of relapsed or refractory DLBCL after two or more lines of systemic therapy. We are seeking to expand ZYNLONTA into international markets throughout the world, and into earlier lines of DLBCL and other indolent lymphomas as a single agent and in combination through our LOTIS-5 and LOTIS-7 clinical trials, as well as through IITs at leading institutions. ZYNLONTA as a monotherapy has a differentiated profile. Based on data from our LOTIS-2 trial, ZYNLONTA provides rapid responses with the median time to response of 1.5 months. Responses were durable for patients with a complete response and the median duration of complete response had not yet been reached at the 2-year follow-up. ZYNLONTA has a manageable safety profile with no cytokine release syndrome (“CRS”), no Risk Evaluation and Mitigation Strategies (“REMS”) or recommendation for in-patient stay. We believe ZYNLONTA’s profile is well-positioned for patients who do not have access to more complex therapies like CAR-T and bi-specific antibodies and for patients who have progressed post CAR-T and or bi-specific antibodies.
Structure and Mechanism of Action
ZYNLONTA is composed of a humanized monoclonal antibody (RB4v1.2) directed against human CD19 and conjugated through a cathepsin-cleavable linker to SG3199, a PBD dimer cytotoxin. Once bound to a CD19-expressing cell, it is internalized by the cell, following which the warhead is released. The warhead is designed to bind irreversibly to DNA to create highly potent interstrand cross-links that block DNA strand separation, thus disrupting essential DNA metabolic processes such as replication and ultimately resulting in cell death.
The human CD19 antigen is involved in the recognition, binding and adhesion processes of cells, mediating direct interactions between surfaces of different cell types and pathogen recognition. CD19 is expressed only on B cells (i.e., a type of white blood cell that plays a significant role in protecting the body from infection by producing antibodies) throughout all stages of B cell development and differentiation. Its expression is maintained at high levels in hematologic B cell malignancies, including NHL and certain types of leukemia.
Regulatory Approval
On April 23, 2021, ZYNLONTA received accelerated approval from the FDA for the treatment of adult patients with relapsed or refractory large B-cell lymphoma after two or more lines of systemic therapy, including DLBCL not otherwise specified, DLBCL arising from low-grade lymphoma and high-grade B-cell lymphoma. Continued approval for this indication is contingent upon verification and description of clinical benefit in a confirmatory trial, which we intend to satisfy through our LOTIS-5 confirmatory Phase 3 clinical trial.
On December 20, 2022, the European Commission granted conditional marketing authorization for the use of ZYNLONTA for the treatment of relapsed or refractory DLBCL. The decision is valid in all European Union Member States, Iceland, Norway, and Liechtenstein. Continued approval for this indication is contingent upon verification of clinical benefit in a confirmatory trial, which we intend to satisfy through our LOTIS-5 confirmatory Phase 3 clinical trial.
On December 06, 2024, ZYNLONTA received conditional approval in China from the NMPA for the treatment of adult patients with relapsed or refractory large B-cell lymphoma after two or more lines of systemic therapy. This conditional approval is based on overall response rate and duration of response results from a single-arm clinical trial. The full approval for this indication will be contingent upon the results of the subsequent confirmatory randomized controlled trial. We intend to satisfy this requirement through our LOTIS-5 Phase 3 clinical trial.
Commercialization
We continue to directly commercialize ZYNLONTA in the United States through a commercial organization comprised of cross-functional employees, including marketing, sales, market access, insights and analytics, and commercial operations functions. Our field sales team calls on healthcare providers across both the academic and community settings and has the potential to cover more than 90% of the DLBCL opportunity.
Outside of the United States, we have entered into strategic agreements to maximize the commercial potential of ZYNLONTA, including an exclusive license agreement with MTPC in Japan, and a joint venture with Overland Pharmaceuticals in China (including Hong Kong and Macau), Singapore, and Taiwan, and an exclusive license agreement with Sobi for all other regions, excluding the U.S. and Japan. In Europe, ZYNLONTA has received conditional approval from the European Commission for the treatment of relapsed or refractory DLBCL after two or more lines of systemic therapy. In China, we have received conditional approval from the NMPA for relapsed or refractory DLBCL after two or more lines of systemic therapy. In China, we and the joint venture are exploring commercialization options for ZYNLONTA. In Japan, MTPC is undertaking a Phase 1/2 bridging study and joined the LOTIS-5 confirmatory Phase 3 clinical trial.
Confirmatory Phase 3 Clinical Trial (LOTIS-5)
LOTIS-5 is a Phase 3, randomized, open-label, two-part, two-arm, multi-center clinical trial of ZYNLONTA combined with rituximab compared to immunochemotherapy in patients with relapsed or refractory DLBCL. We believe that this clinical trial, if successful, will support a supplemental biologics license application (“sBLA”) for ZYNLONTA to be used as a 2L therapy for the treatment of relapsed or refractory DLBCL in transplant-ineligible patients.
Clinical Trial Design
The primary objective of the clinical trial is to evaluate the efficacy of ZYNLONTA combined with rituximab compared to standard immunochemotherapy, as measured by progression-free survival (“PFS”). The secondary endpoints of the clinical trial are overall survival (“OS”), overall response rate (“ORR”), complete response (“CR”) rate and duration of response (“DoR”) as well as frequency and severity of adverse events as well as: (i) characterize the safety profile of ZYNLONTA combined with rituximab, (ii) characterize the pharmacokinetic profile of ZYNLONTA combined with rituximab, (iii) evaluate the immunogenicity of ZYNLONTA combined with rituximab and (iv) evaluate the impact of ZYNLONTA combined with rituximab treatment on treatment-related and disease-related symptoms, patient-reported functions and overall health status.
The clinical trial enrolled patients with pathologically confirmed relapsed or refractory DLBCL who are not considered by the investigator to be a candidate for stem cell transplantation (“SCT”) and who had failed at least one multi-agent systemic treatment regimen. Enrollment was completed in 2024.
The clinical trial is being conducted in two parts: In the safety run-in, the first 20 patients were non-randomly assigned to receive ZYNLONTA in combination with rituximab to compare the combination’s toxicity against historical safety data from monotherapy clinical trials of ZYNLONTA. The randomized part of the clinical trial was initiated after the last patient in the safety run-in completed the first treatment cycle and it was observed that there were no significant increases in toxicity of the combination as compared to historical safety data of ZYNLONTA used as a monotherapy.
Patients are randomly assigned 1:1 to receive either ZYNLONTA in combination with rituximab or rituximab in combination with gemcitabine and oxaliplatin.
In August 2023, we announced updated safety run-in results from the clinical trial. The 20 patients in the safety run-in were a median age of 74.5 years and had previously received a median of five cycles of ZYNLONTA in combination with rituximab and one previous therapy. As of the April 10, 2023, data cutoff:
•Seven patients completed treatment and five continue in follow-up.
•The ORR by central review was 16/20 (80%). A total of 10/20 (50%) and 6/20 (30%) patients attained complete and partial response, respectively.
•The median DoR was 8.0 months and the median PFS was 8.3 months.
•A total of 11 (55%) patients had Grade ≥3 TEAEs. The most common Grade ≥3 TEAEs were increased gamma-glutamyltransferase (five patients (25%) and neutropenia (three patients (15%)).
Pivotal Phase 2 Clinical Trial in Relapsed or Refractory Diffuse Large B-Cell Lymphoma (LOTIS-2)
We conducted a 145-patient Phase 2, multi-center, open-label, single-arm clinical trial to evaluate the safety and efficacy of ZYNLONTA in patients with relapsed or refractory DLBCL. This trial was used as the basis to obtain accelerated approval in the U.S. and conditional approvals in Europe and China.
Clinical Trial Design
The primary objective of the clinical trial was to evaluate the efficacy of ZYNLONTA in patients with relapsed or refractory DLBCL, measured by ORR based on the 2014 Lugano Classification Criteria. The secondary objectives were to (i) further evaluate the efficacy of ZYNLONTA measured by DoR, CR rate, PFS, relapse-free survival (“RFS”) and OS, (ii) characterize the safety profile of ZYNLONTA, (iii) characterize the pharmacokinetic profile of ZYNLONTA, (iv) evaluate the immunogenicity of ZYNLONTA and (v) evaluate the impact of ZYNLONTA treatment on health-related quality of life (“HRQoL”).
The clinical trial enrolled patients with pathologically confirmed relapsed or refractory DLBCL who have previously received two or more multi-agent systemic treatment regimens. The table below presents information about the patients’ characteristics.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Patient Characteristics |
|
|
|
n=145 |
|
|
| Age, median (minimum, maximum) |
|
|
|
66 |
|
(23, 94) |
| Histology, n (%) |
|
DLBCL Not otherwise specified |
|
128 |
|
(88.3) |
| |
|
HGBCL* |
|
10 |
|
(6.9) |
| |
|
PMBCL** |
|
7 |
|
(4.8) |
| Cancer characteristic, n (%) |
|
Double-hit or triple-hit disease*** |
|
15 |
|
(10.3) |
| |
|
Double/triple expressor |
|
20 |
|
(13.8) |
| |
|
Transformed disease**** |
|
29 |
|
(20.0) |
| Disease stage*****, n (%) |
|
I-II |
|
33 |
|
(22.8) |
| |
|
III-IV |
|
112 |
|
(77.2) |
| Number of previous systemic therapies received, median (minimum, maximum) |
|
|
|
3 |
|
(2, 7) |
| Response to first-line prior systemic therapy, n (%) |
|
Relapsed |
|
99 |
|
(68.3) |
| |
|
Refractory |
|
29 |
|
(20.0) |
| Response to most recent prior systemic therapy, n (%) |
|
Relapsed |
|
44 |
|
(30.3) |
| |
|
Refractory |
|
88 |
|
(60.7) |
| Refractory to all prior systemic therapies, n (%) |
|
Yes |
|
24 |
|
(16.6) |
| |
|
No |
|
115 |
|
(79.3) |
| Prior stem cell transplant, n (%) |
|
Autologous stem cell transplant |
|
21 |
|
(14.5) |
| |
|
Allogeneic stem cell transplant |
|
2 |
|
(1.4) |
| |
|
Both autologous and allogeneic stem cell transplant |
|
1 |
|
(0.7) |
| |
|
No |
|
121 |
|
(83.4) |
_______________
Information about the patients’ characteristics. *High-grade diffuse large B-cell lymphoma. **Primary mediastinal large B-cell lymphoma. ***Double-hit or triple-hit DLBCL are rare subtypes of DLBCL characterized by two or three recurrent chromosome translocations and are generally associated with poor prognosis. ****Transformed disease is recorded for patients who had another type of lymphoma that transformed to DLBCL. *****Disease stage is determined by the location of the tumor: Stage I means that the cancer is located in a single region, usually one lymph node and the surrounding area. Stage II means that the cancer is located in two separate regions, an affected lymph node or lymphatic organ and a second affected area, and that both affected areas are confined to one side of the diaphragm; Stage III means that the cancer has spread to both sides of the diaphragm, including one organ or area near the lymph nodes or the spleen; Stage IV means diffuse or disseminated involvement of one or more extralymphatic organs, including any involvement of the liver, bone marrow, or nodular involvement of the lungs.
Clinical Trial Results
The mean number of treatment cycles received was 4.6 and the maximum number of treatment cycles received was 26.
As of March 1, 2021, the main observed safety and tolerability findings were as follows:
•Grade ≥3 TEAEs were reported in 107 patients, or 73.8% of patients. The most common Grade ≥3 TEAEs that were reported in more than 10% of patients included neutropenia (reported in 26.2% of patients), thrombocytopenia (reported in 17.9% of patients), gamma-glutamyltransferase increased (reported in 17.2% of patients) and anemia (reported in 10.3% of patients).
•Treatment-related adverse events in 27 patients, or 18.6% of patients, led to treatment discontinuation. The most common of such adverse events that led to treatment discontinuation in more than 2% of patients included gamma-glutamyltransferase increased (led to treatment discontinuation in 11.7% of patients), peripheral edema (led to treatment discontinuation in 2.8% of patients) and localized edema (led to treatment discontinuation in 2.1% of patients).
•No increase in adverse events was observed in patients aged ≥65 years compared to younger patients.
The main observed efficacy findings were as follows:
•Thirty-six patients, or 24.8% of patients, achieved a complete response and another 34 patients, or 23.4% of patients, achieved a partial response, resulting in a 48.3% ORR. The median time to first response was 41.0 days.
•ZYNLONTA’s favorable clinical activity was observed across a broad patient population in this clinical trial, including transplant eligible and ineligible patients, patients who have not responded to first-line therapy or any prior therapy, double-hit and triple-hit disease and transformed disease and patients who had received prior CD19 therapies or SCT.
•The median DoR was 13.37 months for patients who achieved a response. The median DoR was not reached for patients who achieved a complete response and was 5.68 months for patients who achieved a partial response. The median DoR observed in subgroups at high risk of poor prognosis was comparable to that observed in the overall study population.
•Sixteen patients received CD-19 directed CAR-T after receiving treatment with ZYNLONTA, with an investigator-assessed ORR of 56.3% (eight complete response and one partial response). Eleven patients received SCT as consolidation after responding to treatment with ZYNLONTA.
•The median progression free survival was 4.93 months.
•The median overall survival was 9.53 months.
Other ZYNLONTA Clinical Trials
LOTIS-7
Clinical Trial Design
LOTIS-7 is a Phase 1b global multicenter, multi-arm study in patients with relapsed or refractory B-cell non-Hodgkin lymphoma (B-NHL), including Part 1 (dose escalation) and Part 2 (dose expansion). The three dosing arms include ZYNLONTA plus polatuzumab vedotin, ZYNLONTA plus glofitamab, and ZYNLONTA plus mosunetuzumab. Part 1 of the study uses a 3+3 dose escalation in third-line(“3L”)/3L+ heavily pre-treated patients with ZYNLONTA doses starting at 90 µg/kg and then proceeding to 120 µg/kg and 150 µg/kg. Part 2 currently includes dose expansion in second-line (“2L”)/second-line plus (“2L+”) large B-cell lymphoma in the ZYNLONTA plus glofitamab arm at dose levels determined from Part 1 (120 µg/kg and 150 µg/kg of ZYNLONTA plus the approved dosing of glofitamab). Primary endpoints of the study include safety and tolerability. Secondary efficacy endpoints include ORR, DOR, CRR, PFS, RFS, and OS as well as pharmacokinetics and immunogenicity.
The primary objective of the clinical trial is to characterize the safety and tolerability of loncastuximab tesirine in combination with polatuzumab vedotin, glofitamab, or mosunetuzumab, and to identify the maximum tolerated dose (“MTD”) and/or recommended dose for expansion (“RDE”) for any of the combinations.
The secondary objectives of the clinical trial are to evaluate the anti-cancer effect of loncastuximab tesirine in combination with polatuzumab vedotin, glofitamab, or mosunetuzumab, to characterize the pharmacokinetics (“PK”) profile of loncastuximab tesirine in combination with polatuzumab vedotin, glofitamab, or mosunetuzumab, and to evaluate the immunogenicity of loncastuximab tesirine, glofitamab, and mosunetuzumab, respectively.
The clinical trial is enrolling patients with relapsed or refractory B-cell NHL who have previously received two or more multi-agent systemic treatment regimens (in the dose escalation part) or who have previously received one or more multi-agent systemic treatment regimens (in the dose expansion part). The clinical trial is expected to enroll up to approximately 200 patients.
LOTIS-7 Initial Data
On December 11, 2024, we announced preliminary data from LOTIS-7, the ongoing Phase 1b global multicenter, multi-arm study in patients with relapsed or refractory B-NHL that includes Part 1 (dose escalation) and Part 2 (dose expansion). As of the November 20, 2024 cutoff date, a total of 29 B-NHL patients from Part 1 and Part 2 across all dose levels were treated and evaluated for safety and a total of 18 2L or later DLBCL patients who received dose levels of 120 μg/kg (n=9) or 150 μg/kg (n=9) of ZYNLONTA plus the bispecific antibody glofitamab (Arm E) were evaluated for efficacy. Patients had a median of two prior lines of systemic therapy.
Safety Data. No dose-limiting toxicities (“DLTs”) and no Grade 5 TEAEs were observed. The most common Grade ≥3 TEAEs occurring in 5% or more of patients were neutropenia (24% of patients), lymphopenia (7% of patients) and hypokalemia (7% of patients). No Grade ≥3 cytokine release syndrome CRS or Grade ≥3 immune effector cell-associated neurotoxicity syndrome (“ICANS”) were observed, although low-grade CRS was observed in 34.5% of patients and resolved with standard treatment and low-grade ICANS was observed in 7% of patients and fully resolved.
Efficacy Data. The ORR was 94% (17/18 patients), with 72% of patients (13/18 patients) achieving a CR and 22% of patients (4/18 patients) achieving a PR. Five patients with initial assessment of stable disease (“SD”) or partial response (“PR”) achieved CRs over time.
Of the patients who achieved a CR, 92% (12/13 patients) remained in CR as of the cutoff date. Of the patients who were treated with ZYNLONTA at the 150μg/kg dose — the FDA-approved dose for ZYNLONTA as a monotherapy for 3L or later DLBCL patients — the ORR was 100% (9/9 patients), with 78% (7/9 patients) achieving a CR and 22% (2/9 patients) achieving a PR. The encouraging anti-tumor activity was observed across patients with different numbers of lines and types of prior treatments and across different histologies.
For the LOTIS-7 clinical trial, we plan to fully enroll 20 patients per dosing arm in Part 2 of Arm E (dose levels of 120μg/kg (n=9) or 150 μg/kg (n=9) of ZYNLONTA plus glofitamab) in the first half of 2025.
Phase 2 Investor Initiated Trials (IITs)
On December 9, 2024, we announced updated data from a Phase 2 investigator-initiated clinical trial IIT evaluating ZYNLONTA in combination with rituximab for relapsed or refractory FL and data from a Phase 2 IIT evaluating ZYNLONTA for relapsed or refractory MZL were presented at the 66th American Society of Hematology (“ASH”) Annual Meeting and Exposition. Both IITs were conducted at the Sylvester Comprehensive Cancer Center at the University of Miami Miller School of Medicine.
Marginal Zone Lymphoma
The IIT evaluated ZYNLONTA as monotherapy in patients with relapsed or refractory MZL. As of the October 15, 2024 data cutoff, 23 patients were evaluated for safety and efficacy. Patients had a median of two prior lines of systemic therapy. According to the poster presentations at ASH:
Safety Data. No Grade 5 TEAEs were observed. The most common Grade ≥3 TEAEs occurring in 5% or more of patients were neutropenia (17% of patients), increased alkaline phosphatase (13% of patients) and increased alanine minotransferase (13% of patients).
Efficacy Data. The ORR was 91% (21/23 patients), with 70% of patients (16/23 patients) achieving a CR and 22% of patients (5/23 patients) achieving a PR. Of POD24 patients, 64% of such patients (7/11 patients) achieved a CR, including one patient who progressed after CAR-T. The 18-month duration of response DOR was 61%, and the estimated 12-month PFS was 92%. With a median follow-up duration of 11.5 months, of the patients who received a CR, all but one patient maintained a CR as of the cutoff date, and the median 18-month DOR was 67%.
Follicular Lymphoma
The IIT evaluated ZYNLONTA in combination with rituximab in patients with relapsed or refractory FL presenting high-disease burden as defined by GELF criteria or disease progression within 24 months (“POD24”) at enrollment. As of the September 13, 2024 data cutoff, the trial enrolled 39 patients, all of whom were evaluated for safety and 35 of whom were evaluated for efficacy. Patients had a median of one prior line of systemic therapy. According to the presentation at ASH:
Safety Data. No Grade 5 TEAEs were observed. The most common Grade ≥3 TEAEs occurring in 5% or more of patients were lymphopenia (21% of patients) and neutropenia (13% of patients). Four patients experienced treatment-related serious adverse events.
Efficacy Data. The ORR was 97% (38/39 patients), with 77% of patients (30/39 patients) achieving a complete response (“CR”) and 24% of patients (8/39 patients) achieving a PR. Of POD24 patients, 85% of such patients (17/20 patients) achieved a CR. After a median follow-up duration of 15.6 months, the median PFS was not reached and the 12-month PFS was 95%.
The data were selected for an oral presentation at ASH and simultaneously published in the December issue of The Lancet Haematology.
LOTIS-10
LOTIS-10 is a Phase 1b open-label, multi-center study to evaluate the safety, pharmacokinetics and anti-cancer activity of ZYNLONTA in patients with relapsed or refractory DLBCL or high-grade B-cell lymphoma (“HGBCL”) with hepatic impairment. The primary objective of the clinical trial is to determine the recommended dosing regimen of loncastuximab
tesirine in DLBCL or HGBCL patients with moderate and severe hepatic impairment. The secondary objectives of the clinical trial are to characterize the PK profile, safety and tolerability of loncastuximab tesirine in patients with hepatic impairment, and to evaluate the antitumor activity and the immunogenicity of loncastuximab tesirine in patients with hepatic impairment.
The clinical trial is enrolling patients with relapsed or refractory DLBCL or HGBCL with hepatic impairment. The clinical trial is expected to enroll approximately 56 patients.
Pediatric Trial
‘Glo-BNHL’ is an international multi-center, adaptive, platform trial of novel agents in pediatric and adolescent relapsed or refractory B-cell NHL. The primary objective of the clinical trial is to estimate the clinical efficacy of the specific treatment in patients with r/r B-NHL in either first relapse or subsequent relapse. The secondary objectives of the clinical trial are to assess the safety profile of the novel agent in children, adolescents, and young adults and to confirm the pharmacokinetics of the novel agent at the recommended trial dose in children, adolescents, and young adults, where relevant. The clinical trial is enrolling children, adolescents and young adults with relapsed or refractory B-cell NHL. The initial target sample size is 15 evaluable patients in each treatment arm or relevant sub-group, with additional patients to be added pending results from the no/no go decision from the initial 15 patients results.
The clinical trial consists of three arms: bispecific antibody (Arm A); ADC with standard chemotherapy (Arm B); and CAR-T (Arm C). Novel agents are selected for inclusion in the platform according to an overarching prioritization list and a robust systematic scientific assessment of each proposed asset. ZYNLONTA was selected for study in Arm B in combination with modified R-ICE (rituximab, ifosfamide, carboplatin and etoposide) chemotherapy to estimate the clinical efficacy of the combination in patients with relapsed or refractory B-cell NHL in first (only one prior line of therapy) or subsequent relapse (more than one prior line of therapy).
ADCT-602: PBD-Based ADC Targeting CD22
The Acute Lymphoblastic Leukemia Disease Setting
Acute lymphoblastic leukemia (“ALL”) is a rare form of blood cancer with an annual incidence of ~6,000. Adults make-up ~50% of ALL patients. The five-year survival rate is 71%; however, it declines significantly with age. Allogeneic transplant is the standard of care for patients who are relapsed or refractory to induction therapy. Achieving a response without toxicities which can prevent a patient’s ability to get to an allogeneic transplant is an area of unmet need. We are developing ADCT-602 (CD22) in this area of high unmet medical need.
Structure and Mechanism of Action
ADCT-602 (CD22) is composed of a humanized monoclonal antibody (hLL2-C220) directed against human CD22 and conjugated through a cathepsin-cleavable linker to SG3199, a PBD dimer cytotoxin. Once bound to a CD22-expressing cell, it is internalized by the cell, following which the warhead is released. The warhead is designed to bind irreversibly to DNA to create highly potent interstrand cross-links that block DNA strand separation, thus disrupting essential DNA metabolic processes such as replication and ultimately resulting in cell death. The human CD22 antigen plays a pivotal role in the recognition, binding and adhesion processes of cells. CD22 is only expressed on B cells throughout all stages of B cell development and differentiation. Its expression is maintained in high levels in hematological B cell malignancies, including in NHL and certain types of leukemia, including B-cell ALL.
Phase 1/2 Clinical Trial in Relapsed or Refractory Acute Lymphoblastic Leukemia
Pursuant to our collaboration agreement with MD Anderson Cancer Center, a Phase 1/2, open-label, dose escalation and dose expansion clinical trial of the safety and anti-tumor activity of ADCT-602 (CD22), used as monotherapy, in patients with relapsed or refractory ALL is progressing and additional clinical trial sites are being added to accelerate enrollment.
The primary objectives of the dose escalation stage are to (i) evaluate the safety and tolerability, and determine, as appropriate, the MTD of ADCT-602 (CD22) in patients with relapsed or refractory ALL and (ii) determine the recommended dose(s) of ADCT-602 (CD22) for the dose expansion stage. The primary objective of the dose expansion stage is to evaluate the efficacy of ADCT-602 (CD22) at the dose level(s) recommended from the results of the dose escalation stage. The secondary objectives of the clinical trial are to (i) evaluate the clinical activity of ADCT-602 (CD22), as measured by ORR, DoR, OS and PFS, (ii) characterize the pharmacokinetic profile of ADCT-602 and the free warhead SG3199, (iii) evaluate the immunogenicity of ADCT-602 (CD22) and (iv) characterize the effect of ADCT-602 (CD22) exposure on the QT interval.
The clinical trial is enrolling patients with pathologically confirmed relapsed or refractory B-ALL and patients with pathologically confirmed relapsed or refractory Ph+ ALL who have failed either first- or second-generation tyrosine kinase inhibitor. The clinical trial is expected to enroll approximately 65 patients.
As presented at ASH 2022 (data cutoff July 2022), 21 patients have been treated with ADCT-602 (CD22). We observed that ADCT-602 (CD22) was well tolerated with one DLT of prolonged myelosuppression. Four patients achieved MRD-negative remission, including two of six patients at the 50 µg/kg weekly dose level. One additional patient at 50 µg/kg weekly dose level had marrow blast clearance without count recovery.
Solid Tumors:
The Solid Tumor Disease Setting
There are many different types of solid tumors and they account for the majority of cancers. Some of the most commonly diagnosed solid tumor cancers include lung cancer and prostate cancer. There were an estimated ~200,000 new cases of non-small cell lung cancer and ~274,000 new cases of prostate cancer in the United States. The prognosis and treatment of solid tumor cancers vary based on the type of cancer.
Despite recent significant advances in the treatment of some solid tumor cancers, there remains a high medical need for novel therapies.
Research Strategy, Platform and Pipeline
Our solid tumor research strategy is focused on three key elements. First, we have identified areas of high unmet need that currently feature high use of chemotherapy. Second, we are pursuing targets within these tumor types that are amenable to an ADC approach. And third, we are utilizing our deep knowledge and broad toolkit to optimize the design of ADCs that fit with our first two criteria.
We have developed four lead candidates with differentiated profiles based on a novel, proprietary linker approach to tracelessly release exatecan and a high therapeutic index, which reflects the proprietary design of our ADCs: Claudin-6, PSMA, NaPi2b, and ASCT2, the most advanced of which are PSMA and Claudin-6. Details include:
Claudin-6 and NaPi2b: Preclinical studies demonstrated the Company’s novel, exatecan-based ADCs, targeting Claudin-6 and NaPi2b, were well tolerated with potent and specific in vitro and in vivo anti-tumor activity. Based on these results, we believe a Claudin-6 directed ADC and a NaPi2b directed ADC have the potential for high impact in platinum-resistant ovarian cancer and non-small cell lung cancer.
PSMA: With PSMA-PL2202, we are developing an optimized ADC directed against a validated target with high potential impact in metastatic castrate resistant prostate cancer.
ASCT2: And with ASCT2-PL2202, we are designing an optimized ADC directed against a novel target with high potential impact in many indications including - but not limited to - colorectal cancer and non-small cell lung cancer.
Beyond this, we are further diversifying our toolbox with drugs with other Mode of Actions, including other DNA damaging agents and immunomodulators and focus on conjugation technology to develop dual conjugate ADCs based on two different drugs with orthogonal modes of action.
Chemistry, Manufacturing and Controls
We believe that the manufacture of ADCs requires considerable expertise, know-how and resources. We are a pioneer and leader in the ADC field with specialized end-to-end capabilities for developing optimized manufacturing processes for ADCs. Our robust in-house CMC capabilities utilize a highly experienced workforce to manage a top-tier external manufacturing network through third-party CMOs. Our third-party CMO network is capable of manufacturing highly potent molecules and complex biologics.
We do not own or operate, and do not plan to own or operate, manufacturing infrastructure for the manufacture of clinical supply of our product candidates or commercial product. Instead, we contract with third-party current Good Manufacturing Process-compliant CMOs that have the facilities and capabilities to manufacture on our behalf the intermediate components and the final product candidates for use in clinical trials and commercial supply. We have sufficient commercial-grade drug product, ZYNLONTA, in stock and we believe we and our CMOs will be able to conduct additional manufacturing at the scheduled times.
Our in-house team oversees all aspects of the CMO manufacturing process, including defining the scope of work and monitoring all aspects of the manufacturing process, including conducting routine site visits and audits. We also contract with external specialist quality control and stability-testing organizations to monitor the quality of the materials manufactured by the CMOs.
Intellectual Property
Our success depends in part on our ability to obtain and maintain proprietary protection for our technology, programs and know-how related to our business, defend and enforce our intellectual property rights, in particular, our patent rights, preserve the confidentiality of our trade secrets, and operate without infringing valid and enforceable intellectual property rights of others. We seek to protect our proprietary position by, among other things, exclusively licensing and filing U.S. and foreign patent applications related to our technology, existing and planned programs and improvements that are important to the development of our business, where patent protection is available.
We also rely on trade secrets, know-how, continuing technological innovation and confidential information to develop and maintain our proprietary position and protect aspects of our business that are not amenable to, or that we do not consider appropriate for, patent protection. We seek to protect our proprietary technology and processes, in part, by confidentiality agreements with our employees, consultants, scientific advisors, and contractors. We also seek to preserve the integrity and confidentiality of our data and trade secrets by maintaining physical security of our premises and physical and electronic security of our information technology systems.
Notwithstanding these efforts, we cannot be sure that patents will be granted with respect to any patent applications we have licensed or filed or may license or file in the future, and we cannot be sure that any patents we have licensed or patents that may be licensed or granted to us in the future will not be challenged, invalidated, or circumvented or that such patents will be commercially useful in protecting our technology. Moreover, trade secrets can be difficult to protect. While we have confidence in the measures we take to protect and preserve our trade secrets, such measures can be breached, and we may not have adequate remedies for any such breach. In addition, our trade secrets may otherwise become known or be independently discovered by competitors.
In addition, we or our licensors may be subject to claims that former employees, collaborators, or other third parties have an interest in our owned or in-licensed patents or other intellectual property as an inventor or co-inventor. If we are unable to obtain an exclusive license to any such third-party co-owners’ interest in such patent applications, such co-owners may be able to license their rights to other third parties, including our competitors. In addition, we may need the cooperation of any such co-owners to enforce any patents that issue from such patent applications against third parties, and such cooperation may not be provided to us. For more information regarding the risks related to intellectual property, please see “Item 1A. Risk Factors—Risks Related to Intellectual Property.”
Patent Portfolio
The term of individual utility patents depends upon the countries in which they are granted. In most countries, including the United States, the utility patent term is generally 20 years from the earliest claimed filing date of a non-provisional utility patent application in the applicable country. United States provisional utility patent applications are not eligible to become issued patents until, among other things, non-provisional patent applications are filed within 12 months of the filing date of the applicable provisional patent applications and the failure to file such non-provisional patent applications within such timeline could cause us to lose the ability to obtain patent protection for the inventions disclosed in the associated provisional patent applications. In the United States, a patent’s term may, in certain cases, be lengthened by patent term adjustment, which compensates a patentee for administrative delays by the USPTO in examining and granting a patent, or may be shortened if a patent is terminally disclaimed over a commonly owned patent having an earlier expiration date. In certain circumstances, U.S. patents can also be eligible for patent term extension; for more information, see “—Government Regulation—Regulatory Approval in the United States—U.S. Patent Term Restoration and Marketing Exclusivity.” The expiration dates referred to below are without regard to potential patent term adjustment or extension that may be available to us.
In general, our licensed, owned or co-owned patents relate to our ADC products, the underlying antibodies, the warheads, such as PBD-based or Exatecan-based warheads, the linker used to connect such warheads to the antibodies to form an ADC, modifications of the antibodies to enhance efficacy, and the methods to use and administer or co-administer such ADCs. We typically file patent applications in the U.S. and other key foreign countries. We have over 350 patents issued in the U.S. and other countries with expirations ranging from 2031 to 2043 as well as numerous pending patent applications in the U.S. and other countries.
“PBD Warhead,” “PBD Warhead with Linker” Platform Patent Protection
As of December 31, 2024, with respect to the PBD-based warhead and ADC technology we use to develop our product candidates, we have exclusively licensed from MedImmune for particular target molecules, various patent families directed to different aspects of the chemistry of the PBD molecules and methods of using the molecules in the treatment of proliferative diseases. These families include approximately 7 issued U.S. utility patents. The issued utility patents, and any utility patents granted from the pending applications in these families, are expected to expire between 2023 and 2038.
Product-Specific Patent Protection
As of December 31, 2024, we co-own with MedImmune, and have exclusive rights to, approximately 12 patent families directed to ADCs with PBD warheads and targeting moieties that bind to specific target molecules, combinations of these ADCs with other therapeutic molecules and therapeutic uses of these ADCs. These families include approximately 13 issued U.S. utility patents. The issued utility patents, and any utility patents granted from the pending applications in these families, are expected to expire between 2033 and 2042. Further details in relation to particular marketed products are provided below. We also solely own eight patent families directed to antibodies, ADCs with Exatecan or other warheads, linker technology and therapeutic uses of these antibodies and ADCs. Any utility patents granted from the pending applications in these families are expected to expire between 2041 and 2044.
ZYNLONTA
The antibody for ZYNLONTA is in the public domain.
Patents more specifically directed to the ZYNLONTA ADC are co-owned by us and MedImmune, with us having the exclusive right to exploit the relevant patents during the term of our license and collaboration agreement with MedImmune.
As of December 31, 2024, there are six such patent families directed to the ADC product, methods of using the ADC as a single agent or in combination with other named molecules in the treatment of proliferative diseases, and dosing regimens. The issued utility patents, and any utility patents granted from the pending applications in these families, are expected to expire between 2033 and 2042.
For more information on the license and collaboration agreement with MedImmune, see “—Material Contracts—MedImmune License and Collaboration Agreement.”
Competition
The biotechnology industry, and the oncology subsector, are characterized by rapid evolution of technologies, fierce competition and strong defense of intellectual property. While we believe that our technology, intellectual property, know-how, scientific expertise and team provide us with certain competitive advantages, we face potential competition from many sources, including major pharmaceutical and biotechnology companies, academic institutions and public and private research organizations. Many competitors and potential competitors have substantially greater scientific, research and product development capabilities, as well as greater financial, marketing and human resources than we do.
Many companies are active in the oncology market and are developing or marketing products for the specific therapeutic markets that we target, including both antibody drug conjugate (“ADC”) and non-antibody drug conjugate therapies. Similarly, we also face competition from other companies and institutions that continue to invest in innovation in the ADC field including new payload classes, new conjugation approaches and new targeting moieties. Specifically, we are aware of multiple companies with ADC technologies that may be competitive with our product and product candidates, including, but not limited to, AbbVie, Inc., Daiichi Sankyo Company, Genmab, GlaxoSmithKline plc, Gilead Sciences, Inc., Johnson & Johnson, Mersana Therapeutics Inc., Sanofi S.A., Roche Holding AG, Pfizer Inc. and Zymeworks, Inc. There are hundreds of ADCs in development, the vast majority of which were being developed for the treatment of cancer.
In the relapsed or refractory DLBCL setting, for which we have developed ZYNLONTA, current 3L treatment options include CAR-T, allogeneic stem cell transplant, polatuzumab in combination with bendamustine and a rituximab product, selinexor, tafasitamab in combination with lenalidomide, chemotherapy using small molecules and bispecifics antibodies. If ZYNLONTA is approved for use as a 2L treatment for DLBCL patients, we will continue to compete with CAR-T, autologous stem cell transplant, rituximab in combination with chemotherapies, polatuzumab in combination with bendamustine and a rituximab product, and tafasitamab in combination with lenalidomide.
In addition, we expect potential future competition from bispecific antibodies such as glofitamab and epcoritamab alone or in combinations with chemotherapies or polatuzumab to gain approval in the 2L treatment of DLBCL.
Any products and product candidates that we successfully develop and commercialize may compete directly with approved therapies and any new therapies that may be approved in the future. Competition will be based on their safety and effectiveness, the timing and scope of marketing approvals, the availability and cost of supply, marketing and sales capabilities, reimbursement coverage, price levels and discounts offered, patent position and other factors. Our competitors may succeed in developing competing products before we do, obtaining marketing approval for products and gaining acceptance for such products in the same markets that we are targeting.
Material Contracts
The following descriptions of our material agreements are not complete and are qualified in their entirety by reference to the full text of such agreements, which are filed as exhibits to this Annual Report.
MedImmune License and Collaboration Agreement
In 2011, we (then operating under the name ADCT Sàrl) entered into a license and collaboration agreement with Spirogen (since renamed ADC Products UK Ltd.), pursuant to which Spirogen granted us access to its next-generation PBD-based warhead and linker technology. In connection with AstraZeneca plc’s acquisition of Spirogen Sàrl (which was at the time the direct parent company of Spirogen) and the transfer of certain of Spirogen’s intellectual property to Spirogen Sàrl, including its PBD-based warhead and linker technology, the agreement was subsequently amended and restated in October 2013 (with retroactive effect to September 2011), with Spirogen Sàrl also becoming a party to such agreement. Spirogen Sàrl subsequently transferred the PBD technology to MedImmune Limited, which, together with MedImmune LLC, is the global biologics research and development arm of AstraZeneca plc. Thereafter, Spirogen Sàrl transferred its rights and obligations under the agreement to MedImmune and the agreement was subsequently amended and restated again in May 2016 (with retroactive effect to September 2011), with MedImmune replacing Spirogen Sàrl as the licensor thereunder.
Under the terms of the agreement, MedImmune has granted us an exclusive, worldwide license under certain patent rights and related know-how to make, have made, use, sell, offer for sale and import product candidates in the field of human therapeutics and diagnostics that consist of (i) PBD-based molecules directly conjugated to an antibody (i.e., ADCs with a PBD-based warhead) that specifically bind to up to 11 approved targets (“ADC Targets”), and (ii) PBD-based molecules conjugated to a non-antibody (i.e., targeting-moiety conjugates with a PBD-based warhead) that specifically bind to up to ten approved targets (“XDC Targets”). As of the date hereof, there are 11 approved ADC Targets subject to the license, including CD19 (the target of ZYNLONTA), and ten approved XDC Targets subject to the license.
Under the terms of the agreement, we have the right to grant sublicenses to affiliates and, subject to MedImmune’s approval (not to be unreasonably withheld), third parties. In addition, with respect to each licensed target, we agreed to use commercially reasonable efforts to develop and commercialize at least one product and submit an IND application with the FDA (or its equivalent in another jurisdiction) for one product within 48 months after formal designation of the target as an approved target, which we have done with respect to CD19 and CD25 upon submitting the IND applications for ZYNLONTA and Cami, respectively.
As consideration for the rights granted to us under the agreement, in 2011 we paid Spirogen an up-front licensing fee of $2.5 million. No further payments in consideration for the grant of such rights are required to be paid to Spirogen or MedImmune under the agreement.
With respect to patent rights conceived during the course of our exercise of our rights under the agreement, rights are allocated as follows under the agreement: (i) we own any such patent that claims an antibody that binds to one of the ADC Targets approved under the agreement, (ii) we and MedImmune jointly own any such patent claiming any PBD-based ADC, with us owning the exclusive right to exploit such patent during the term of the agreement and (iii) MedImmune owns any such patent that claims a PBD or any PBD attached to an antibody that does not bind to one of the ADC Targets approved under the agreement. In addition, we have the right to prosecute and maintain all patents described in clauses (i) and (ii) above and are responsible for the costs of prosecuting and maintaining such patents. The ownership of any patent rights conceived during our exercise of our rights under the agreement in connection with non-ADC Targets will be determined under U.S. patent law.
Unless earlier terminated, the agreement terminates on the date of expiration of the last to expire licensed patent right that covers a product being exploited under the license. The agreement (including the licenses granted thereunder) will terminate upon our material breach of any provision of the agreement that is not cured within an applicable cure period.
Overland License and Collaboration Agreement
In December 2020, we entered into a joint venture with Overland Pharmaceuticals (“Overland”) to develop and commercialize ZYNLONTA, ADCT-602, ADCT-601 and ADCT-901 in greater China and Singapore. In connection with such joint venture, we entered into a license and collaboration agreement with the joint venture entity, Overland ADCT BioPharma (CY) Limited (“Overland ADCT BioPharma”) pursuant to which we granted Overland ADCT BioPharma an exclusive license or sublicense (as applicable) under all applicable patents and know-how now or in the future owned or controlled by us relating to ZYNLONTA, ADCT-602, ADCT-601 and ADCT-901 (collectively, the “Licensed Products”) in order to use, sell, offer for sale, import and commercialize such product candidates in China, Hong Kong, Macau, Taiwan and Singapore (the “Territory”). We also granted Overland ADCT BioPharma an exclusive right of first negotiation to obtain a license in the event we seek to grant to a third party a license in certain circumstances.
Overland ADCT BioPharma is responsible, at its sole cost, for the development, regulatory approval and commercialization of the Licensed Products in the Territory, and must use diligent efforts in order to obtain and maintain regulatory approval for and commercialize the products in each applicable jurisdiction. We maintain an exclusive option, on a product-by-product basis, to co-promote and participate in the detailing, promotion and marketing of the Licensed Products in the Territory. Upon any exercise by us of such option, we and Overland ADCT BioPharma will negotiate in good faith commercially reasonable terms for a co-promotion agreement. We maintain the right to control clinical trials for the Licensed Products conducted both in and out of the Territory, and the license agreement sets forth the division of costs between us and Overland ADCT BioPharma with respect to clinical trials depending on the territories within which such trials are conducted or relate to. We are required to use diligent efforts to manufacture and supply to Overland ADCT BioPharma (at our manufacturing cost), and Overland ADCT BioPharma must purchase from us, all of Overland ADCT BioPharma’s requirements of the Licensed Products for its development and commercialization activities.
The collaboration is managed by a joint steering committee (and other applicable committees and subcommittees) comprised of equal numbers of representatives from us and Overland ADCT BioPharma. In the event of a dispute that cannot be resolved by discussions between our CEO and Overland ADCT BioPharma’s CEO, Overland ADCT BioPharma shall have final decision-making authority with respect to matters that relate specifically to the development and commercialization of the Licensed Products in the Territory, except where such matters could also affect the products outside of the Territory and with respect to other specified matters (in which cases we shall have such authority).
As partial consideration for the rights granted to Overland ADCT BioPharma pursuant to the agreement, Overland ADCT BioPharma issued to us 44,590,000 Series A shares. We are also entitled to receive tiered quarterly royalties on Overland ADCT BioPharma’s net sales of the Licensed Products ranging from the low to mid-single digit percentages. Such royalties are payable, on a product-by-product and jurisdiction-by-jurisdiction basis, from the first commercial sale of a product in a jurisdiction until the latest of (i) the expiration of the last to expire claim in the licensed patent that covers such product or any components thereof in such jurisdiction, (ii) the last to expire regulatory exclusivity period for such product in such jurisdiction and (iii) a specified period after the first commercial sale of such product in such jurisdiction. Overland ADCT BioPharma must also reimburse us for any payments we are obligated to make to any of our licensors including in connection with the grant of a sublicense to Overland ADCT BioPharma under applicable intellectual property pursuant to this agreement, including any fees or payments directly resulting from or reasonably allocable to Overland ADCT BioPharma’s development and commercialization of the Licensed Products.
The license agreement remains in effect, on a product-by-product basis, for as long as Overland ADCT BioPharma continues to develop or commercialize such product. Either party may terminate the license agreement for a material breach by the other party, subject to specified notice and cure periods, or upon immediate written notice for an insolvency-related event experienced by the other party. We may also terminate the license agreement, subject to a specified notice and cure period, if Overland ADCT BioPharma commences a legal action challenging the validity, enforceability or scope of any patent owned or controlled by us that covers the applicable products.
In China, we and the joint venture are exploring commercialization options for ZYNLONTA.
Financing Agreement with HealthCare Royalty Partners
In August 2021, we entered into a royalty purchase agreement with certain entities managed by HCR for up to $325.0 million. Under the terms of the agreement, we received gross proceeds of $225.0 million upon closing and $75.0 million upon the first commercial sale of ZYNLONTA in Europe (collectively, the “Investment Amount”).
Under the agreement, we are obligated to pay to HCR (i) a 7% royalty on the worldwide (excluding China, Hong Kong, Macau, Taiwan, Singapore and South Korea) net sales of ZYNLONTA and any product that contains ZYNLONTA and on any upfront or milestone payments we receive from licenses that we grant to commercialize ZYNLONTA or any product that contains ZYNLONTA in any region other than China, Hong Kong, Macau, Taiwan, Singapore and South Korea, (ii) a 7% royalty on the worldwide net sales of Cami and any product that contains Cami and on any upfront or milestone payments we receive from licenses that we grant to commercialize Cami or any product that contains Cami in the United States and Europe, and (iii) outside the United States and Europe, a 7% share of any upfront or milestone payments derived from licenses that we grant to commercialize Cami or any product that contains Cami and, in lieu of the royalty on net sales under such licenses, a mid-teen percentage share of the net royalty we receive from such licenses. These royalty rates are subject to potential upward adjustment, up to a maximum of 10%, based on performance tests in 2026 and 2027. The 7% royalty rates described above are subject to adjustment to a potential high-single-digit percentage royalty rate after September 30, 2026 and/or a 10% royalty rate after September 30, 2027, if the aggregate net sales and license revenue subject to royalty obligations in the preceding twelve months do not exceed certain mid-nine-digit milestones by such dates. Our aggregate royalty obligations are capped at 2.50 times the amount paid by HCR under the agreement, or at 2.25 times the amount paid by HCR under the agreement if HCR receives royalty payments exceeding a mid-nine-digit amount on or prior to March 31, 2029 (the “Royalty Cap”). Once the Royalty Cap is reached, the royalty purchase agreement will terminate. Upon the occurrence of a change in control event, we are obligated to pay HCR an amount equal to the Royalty Cap, less any amounts we previously paid to HCR.
MTPC License Agreement
In January 2022, we entered into a license agreement with Mitsubishi Tanabe Pharma Corporation (“MTPC”) to develop and commercialize ZYNLONTA in Japan, pursuant to which we granted MPTC an exclusive license under all applicable patents and know-how relating to ZYNLONTA in order to use, sell, offer for sale, import and commercialize ZYNLONTA for all cancer indications in Japan. Under the agreement, we received an upfront payment of $30 million and are eligible to up to $205 million in regulatory and net sales-based milestones as well as royalties ranging from high teens to the low twenties based on net sales of ZYNLONTA in Japan. The royalties payable to us are subject to downward adjustment in certain circumstances, including the expiration of a patent covering ZYNLONTA, generic or biosimilar competition, and required royalty payments to third parties.
MTPC will conduct clinical studies and be responsible for the development and commercialization of ZYNLONTA in Japan and bear the associated costs. At MPTC’s option, it may also participate in any clinical studies of ZYNLONTA outside of Japan by bearing a portion of the costs of such studies. In addition, MPTC agreed that it will not engage in certain activities with respect to products that are similar to ZYNLONTA.
Unless terminated earlier, the agreement terminates upon the occurrence of the earliest of (i) the expiration of the last-to-expire patent covering ZYNLONTA in Japan, (ii) the expiration of exclusivity with respect to ZYNLONTA granted to MTPC by a regulatory authority, and (iii) ten years after the first commercial sale of ZYNLONTA in Japan. In addition, MTPC may terminate the agreement at its discretion upon 180 days’ notice to us, each party may terminate the agreement for the other party’s material breach or insolvency and we may terminate the agreement if MTPC engages in certain challenges of patents covering ZYNLONTA.
Sobi License Agreement
In July 2022, we entered into a license agreement with Swedish Orphan Biovitrum AB (publ) (“Sobi”) to develop and commercialize ZYNLONTA in all territories other than the United States, greater China, Singapore and Japan (the territories subject to the agreement, collectively, the “Covered Territory”). Pursuant to the agreement, we granted Sobi (i) an exclusive license under applicable patents and know-how relating to ZYNLONTA in order to use, develop, sell, offer for sale, distribute, import and commercialize ZYNLONTA for all human therapeutic and diagnostic uses in the Covered Territory, (ii) a non-exclusive license under applicable patents and know-how relating to ZYNLONTA in order to package and label ZYNLONTA for all human therapeutic and diagnostic uses in the Covered Territory and (iii) a non-exclusive license under applicable patents and know-how relating to ZYNLONTA in order to manufacture and have manufactured ZYNLONTA in any territory solely for the use and sale of ZYNLONTA for human therapeutic and diagnostic uses in the Covered Territory exercisable upon Sobi assuming responsibility to manufacture or have manufactured ZYNLONTA, in each case, with the right to sublicense to affiliates and, with our written consent, other third parties and the right to subcontract to third parties. The licenses are subject to certain retained rights as specified in the agreement.
Under the agreement, we received an upfront payment of $55 million in July 2022 and $50 million in February 2023 for the approval of a Marketing Authorization Application (“MAA”) by the European Commission for ZYNLONTA in 3L DLBCL, and are eligible for up to $332.5 million in other regulatory and net sales-based milestones, as well as tiered royalties ranging from the mid-teens to the mid-twenties based on net sales of ZYNLONTA in the Covered Territory. The royalties payable to us are subject to downward adjustment in certain circumstances, including the expiration of patents covering ZYNLONTA, generic or biosimilar competition and required payments to third parties. The royalty term with respect to a product in a given country begins upon the first commercial sale of the product in the country and terminates upon the latest of (x) 10 years after the first commercial sale of the product in the country, (y) the expiration of the last-to-expire valid patent claim covering the product in the country and (z) the expiration of regulatory exclusivity for the product in the country.
Sobi will conduct development and commercialization activities with respect to ZYNLONTA in the Covered Territory and bear the associated costs, other than the costs of global clinical studies that are intended to support regulatory approval in both the United States and the Covered Territory. In addition, Sobi will co-fund 25% of the costs of certain specified global clinical studies of ZYNLONTA and have the option to co-fund additional global clinical studies in exchange for use of the data generated from such additional studies, subject to an aggregate annual co-funding cap of $10 million.
Both we and Sobi agreed that neither party would (i) during the term of the agreement until the fifth anniversary of the first MAA approval of ZYNLONTA for the first indication in the first of Germany, France, United Kingdom, Spain or Italy, engage in the development (other than non-clinical and preclinical research activities) of any competitive product directed to CD19 for the treatment of DLBCL in the Covered Territory, or (ii) during the term of the agreement, commercialize any competitive product directed to CD19 for the treatment of DLBCL in the Covered Territory. If either party acquires or is acquired by a third party that has a competitive program, then such party may continue such competitive program as long as such party establishes firewalls to operate such competitive program separately from the ZYNLONTA program.
Loan Agreement
In August 2022, we, ADC Therapeutics (UK) Limited and ADC Therapeutics America, Inc. entered into a loan agreement and guaranty (as amended, the “Loan Agreement”) with certain affiliates and/or funds managed by each of Oaktree Capital Management, L.P. and Owl Rock Capital Advisors LLC, as lenders, and Owl Rock Opportunistic Master Fund I, L.P., as administrative agent and collateral agent, pursuant to which we borrowed $120.0 million principal amount of term loans. The secured term loans are scheduled to mature on August 15, 2029 and accrue interest at an annual rate of SOFR plus 7.50% per annum or a base rate plus 6.50% per annum for the first five years of the term loans, and thereafter, at an annual rate of SOFR plus 9.25% or a base rate plus 8.25%, in each case subject to a 1.00% per annum SOFR floor. At our election, for the first three years, we may choose to pay an amount of interest on the outstanding principal amount of term loans corresponding to up to 2.50% of the applicable interest rate in kind (in lieu of payment in cash). We are obligated to pay certain exit fees upon certain prepayments and repayments of the principal amount of the term loans. In addition, we have the right to prepay the term loans at any time subject to certain prepayment premiums applicable during the period commencing from the closing date until the fourth anniversary of the closing date. The Loan Agreement also contains certain prepayment provisions, including mandatory prepayments from the proceeds from certain asset sales, casualty events and from issuances or incurrences of debt, which may also be subject to prepayment premiums if made on or prior to the fourth anniversary of the closing date. The obligations under the Loan Agreement are secured by substantially all of our assets and those of certain of our subsidiaries and are guaranteed initially by our subsidiaries in the United States and the United Kingdom. The Loan Agreement contains customary covenants, including a covenant to maintain qualified cash of at least $60.0 million plus an amount equal to any accounts payable that remain unpaid more than ninety days after the date of the original invoice therefor, and negative covenants including limitations on indebtedness, liens, fundamental changes, asset sales, investments, dividends and other restricted payments and other matters customarily restricted in such agreements. In addition, the Loan Agreement contains a revenue covenant that, so long as the Company’s 30-day average market capitalization is less than $650 million, requires the Company achieve minimum levels of ZYNLONTA net sales in the United States, tested on a quarterly basis, which is subject to a customary cure right in favor of the Company that may be exercised by making certain prepayments and that, subject to certain limitations, may be exercised up to three times during the term of the Loan Agreement. The Loan Agreement also contains customary events of default, after which the term loan may become due and payable immediately, including payment defaults, material inaccuracy of representations and warranties, covenant defaults (including creation of any liens other than those that are expressly permitted), bankruptcy and insolvency proceedings, cross-defaults to certain other agreements, judgments against us and our subsidiaries and change in control.
Government Regulation
Government authorities in the United States at the federal, state and local level and in other countries and jurisdictions, including the European Union, extensively regulate, among other things, the research, development, testing, manufacture, quality control, approval, labeling, packaging, storage, record-keeping, promotion, advertising, distribution, post-approval monitoring and reporting, marketing and export and import of drug and biological products, such as our investigational medicines and any future investigational medicines. Generally, before a new drug or biologic can be marketed, considerable data demonstrating its quality, safety and efficacy must be obtained, organized into a format specific for each regulatory authority, submitted for review and approved by the regulatory authority.
Regulatory Approval in the United States
In the United States, pharmaceutical products are subject to extensive regulation by the FDA. The FDCA and other federal and state statutes and regulations govern, among other things, the research, development, testing, manufacture, storage, recordkeeping, approval, labeling, promotion and marketing, distribution, post-approval monitoring and reporting, sampling, and import and export of pharmaceutical products. Biological products used for the prevention, treatment or cure of a disease or condition of a human being are subject to regulation under the FDCA, except that the section of the FDCA that governs the approval of new drug applications (“NDAs”) does not apply to the approval of biological products. Biological products, such as our ADC product candidates, are approved for marketing under provisions of the Public Health Service Act (the “PHSA”), via a Biologics license application (“BLA”). However, the application process and requirements for approval of BLAs are very similar to those for NDAs, and biologics are associated with similar approval risks and costs as drugs. Failure to comply with applicable U.S. requirements may subject a company to a variety of administrative or judicial sanctions, such as clinical hold, FDA refusal to approve pending NDAs or BLAs, warning or untitled letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, civil penalties, and criminal prosecution.
Our investigational medicines and any future investigational medicines must be approved by the FDA pursuant to a BLA before they may be legally marketed in the United States. The process generally involves the following:
•completion of extensive preclinical laboratory and animal studies in accordance with applicable regulations, including studies conducted in accordance with good laboratory practice (GLP) requirements;
•submission to the FDA of an investigational new drug (“IND”) application, which must become effective before human clinical trials may begin;
•approval by an institutional review board (“IRB”) or independent ethics committee at each clinical trial site before each clinical trial may commence;
•performance of adequate and well-controlled human clinical trials in accordance with applicable IND regulations, good clinical practice (“GCP”) requirements and other clinical trial-related regulations to establish the safety and efficacy of the investigational product for each proposed indication;
•submission to the FDA of a BLA;
•a determination by the FDA within 60 days of its receipt of a BLA to file the submission for review;
•satisfactory completion of one or more FDA pre-approval inspections of the manufacturing facility or facilities where the biologic, or components thereof, will be produced to assess compliance with current good manufacturing practice (“cGMP”) requirements to assure that the facilities, methods and controls are adequate to preserve the biologic’s identity, strength, quality and purity;
•satisfactory completion of any potential FDA audits of the clinical trial sites that generated the data in support of the BLA to assure compliance with GCPs and integrity of the clinical data;
•payment of any user fees for FDA review of the BLA;
•FDA review and approval of the BLA, including consideration of the views of any FDA advisory committee; and
•compliance with any post-approval requirements, including REMS, where applicable, and post-approval studies required by the FDA as a condition of approval.
The preclinical and clinical testing and approval process requires substantial time, effort and financial resources, and we cannot be certain that any approvals for our product candidates will be granted on a timely basis, or at all.
Preclinical Studies
Before testing any biological product candidates in humans, the product candidate must undergo rigorous preclinical testing. Preclinical studies include laboratory evaluation of product chemistry and formulation, as well as in vitro and animal studies to assess the potential for adverse events and in some cases to establish a rationale for therapeutic use. The conduct of preclinical studies is subject to federal regulations and requirements, including GLP regulations for safety/toxicology studies. An IND sponsor must submit the results of the preclinical tests, together with manufacturing information, analytical data, any available clinical data or literature and plans for clinical studies, among other things, to the FDA as part of an IND. An IND is a request for authorization from the FDA to administer an investigational product to humans and must become effective before human clinical trials may begin. Some preclinical testing may continue after the IND is submitted. An IND automatically becomes effective 30 days after receipt by the FDA, unless before that time the FDA raises concerns or questions related to one or more proposed clinical trials and places the trial on clinical hold. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. As a result, submission of an IND may not result in the FDA allowing clinical trials to commence.
Clinical Trials
The clinical stage of development involves the administration of the investigational product to healthy volunteers or patients under the supervision of qualified investigators, generally physicians not employed by or under the trial sponsor’s control. Clinical trials must be conducted: (i) in compliance with federal regulations; (ii) in compliance with GCPs, an international standard meant to protect the rights and health of patients and to define the roles of clinical trial sponsors, administrators, and monitors; as well as (iii) under protocols detailing, among other things, the objectives of the trial, the parameters to be used in monitoring safety, and the effectiveness criteria to be evaluated in the trial. Each protocol involving testing on U.S. patients and subsequent protocol amendments must be submitted to the FDA as part of the IND. Furthermore, each clinical trial must be reviewed and approved by an IRB for each institution at which the clinical trial will be conducted to ensure that the risks to individuals participating in the clinical trials are minimized and are reasonable in relation to anticipated benefits. The IRB also approves the informed consent form that must be provided to each clinical trial subject or his or her legal representative and must monitor the clinical trial until completed.
There also are requirements governing the reporting of ongoing clinical trials and completed clinical trial results to public registries. Information about certain clinical trials, including clinical trial results, must be submitted within specific time frames for publication on the www.clinicaltrials.gov website. Information related to the product, patient population, phase of investigation, clinical trial sites and investigators, and other aspects of the clinical trial is then made public as part of the registration. Sponsors are also obligated to discuss the results of their clinical trials after completion. Disclosure of the results of these clinical trials can be delayed in certain circumstances for up to two years after the date of completion of the trial. Competitors may use this publicly available information to gain knowledge regarding the progress of development programs.
A sponsor who wishes to conduct a clinical trial outside of the United States may, but need not, obtain FDA authorization to conduct the clinical trial under an IND. If a foreign clinical trial is not conducted under an IND, the sponsor may submit data from the clinical trial to the FDA in support of a BLA. The FDA will accept a well-designed and well-conducted foreign clinical trial not conducted under an IND if the clinical trial was conducted in accordance with GCP requirements and the FDA is able to validate the data through an onsite inspection if deemed necessary.
Clinical trials are generally conducted in three sequential phases, known as Phase 1, Phase 2 and Phase 3:
•Phase 1 clinical trials generally involve a small number of healthy volunteers or disease-affected patients who are initially exposed to a single dose and then multiple doses of the product candidate. The primary purpose of these clinical trials is to assess the metabolism, pharmacokinetics, pharmacologic action, side effect tolerability, safety of the product candidate, and, if possible, early evidence of effectiveness. Phase 1 clinical trials may be designated as Phase 1a, which may involve dose escalation to determine the maximum tolerated dose, or Phase 1b, which may involve dose expansion at one or more dose levels to determine the recommended dose level for Phase 2 clinical trials.
•Phase 2 clinical trials generally involve studies in disease-affected patients to evaluate proof of concept and/or determine the dosing regimen(s) for subsequent investigations. At the same time, safety and further pharmacokinetic and pharmacodynamic information is collected, possible adverse effects and safety risks are identified, and a preliminary evaluation of efficacy is conducted.
•Phase 3 clinical trials generally involve a large number of patients at multiple sites and are designed to provide the data necessary to demonstrate the effectiveness of the product for its intended use, its safety in use and to establish the overall benefit/risk relationship of the product, and provide an adequate basis for product labeling. In most cases, the FDA requires two adequate and well-controlled Phase 3 clinical trials to demonstrate the efficacy of the biologic.
These Phases may overlap or be combined. For example, a Phase 1/2 clinical trial may contain both a dose-escalation stage and a dose-expansion stage, the latter of which may confirm tolerability at the recommended dose for expansion in future clinical trials (as in traditional Phase 1 clinical trials) and provide insight into the anti-tumor effects of the investigational therapy in selected subpopulation(s).
Typically, during the development of oncology therapies, all subjects enrolled in Phase 1 clinical trials are disease-affected patients and, as a result, considerably more information on clinical activity may be collected during such trials than during Phase 1 clinical trials for non-oncology therapies. A single adequate and well-controlled Phase 3 or Phase 2 trial may be sufficient in rare instances, including (1) where the trial is a large, multicenter trial demonstrating internal consistency and a statistically very persuasive finding of a clinically meaningful effect on mortality, irreversible morbidity or prevention of a disease with a potentially serious outcome and confirmation of the result in a second trial would be practically or ethically impossible or (2) when in conjunction with confirmatory evidence. Approval on the basis of a single adequate and well-controlled trial may be subject to the requirement of additional post-approval studies.
The manufacturer of an investigational drug in a Phase 2 or Phase 3 clinical trial for a serious or life-threatening disease is required to make available, such as by posting on its website, its policy on evaluating and responding to requests for expanded access to such investigational drug.
Phase 1, Phase 2, Phase 3 and other types of clinical trials may not be completed successfully within any specified period, if at all. The FDA, the IRB, or the sponsor may suspend or terminate a clinical trial at any time on various grounds, including non-compliance with regulatory requirements or a finding that the patients are being exposed to an unacceptable health risk. Similarly, an IRB can suspend or terminate approval of a clinical trial at its institution if the clinical trial is not being conducted in accordance with the IRB’s requirements or if the drug or biologic has been associated with unexpected serious harm to patients. Additionally, some clinical trials are overseen by an independent group of qualified experts organized by the clinical trial sponsor, known as a data safety monitoring board or committee. This group provides recommendations for whether a trial may move forward at designated checkpoints based on access to certain data from the trial.
Concurrent with clinical trials, companies usually complete additional animal studies and also must develop additional information about the chemistry and physical characteristics of the drug or biologic as well as finalize a process for manufacturing the product in commercial quantities in accordance with cGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the product and, among other things, companies must develop methods for testing the identity, strength, quality, potency, and purity of the final product. Additionally, appropriate packaging must be selected and tested, and stability studies must be conducted to demonstrate that the investigational medicines do not undergo unacceptable deterioration over their shelf life.
FDA Review Process
Following completion of the clinical trials, the results of preclinical studies and clinical trials are submitted to the FDA as part of a BLA, along with proposed labeling, chemistry and manufacturing information to ensure product quality and other relevant data. To support marketing approval, the data submitted must be sufficient in quality and quantity to establish the safety and efficacy of the investigational product to the satisfaction of the FDA. FDA approval of a BLA must be obtained before a biologic or drug may be marketed in the United States.
The cost of preparing and submitting a BLA is substantial. Under the Prescription Drug User Fee Act (“PDUFA”), each BLA must be accompanied by a substantial user fee. The FDA adjusts the PDUFA user fees on an annual basis. Fee waivers or reductions are available in certain circumstances, including a waiver of the application fee for the first application filed by a small business. Additionally, no user fees are assessed on BLAs for products designated as orphan drugs, unless the product also includes a non-orphan indication. The applicant under an approved BLA is also subject to an annual program fee.
The FDA reviews all submitted BLAs before it files them and may request additional information. The FDA must make a decision on filing a BLA within 60 days of receipt, and such decision could include a refusal to file by the FDA. Once the submission is filed, the FDA begins an in-depth review of the BLA. Under the goals and policies agreed to by the FDA under PDUFA, the FDA has 10 months, from the filing date, in which to complete its initial review of an original BLA for a new molecular entity and respond to the applicant, and six months from the filing date of an original BLA designated for priority review. The review process for both standard and priority review may be extended by the FDA for three additional months to consider certain late-submitted information or information intended to clarify information already provided in the submission. The FDA does not always meet its PDUFA goal dates for standard and priority BLAs, and the review process can be extended by FDA requests for additional information or clarification.
Before approving a BLA, the FDA will conduct a pre-approval inspection of the manufacturing facilities for the new product to determine whether they comply with cGMP requirements. The FDA will not approve the product unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications.
The FDA also may audit data from clinical trials to ensure compliance with GCP requirements and the integrity of the data supporting safety and efficacy. Additionally, the FDA may refer applications for novel products or products that present difficult questions of safety or efficacy to an advisory committee, typically a panel that includes clinicians and other experts, for review, evaluation and a recommendation as to whether the application should be approved and under what conditions, if any. The FDA is not bound by recommendations of an advisory committee, but it generally follows such recommendations when making decisions on approval. The FDA likely will reanalyze the clinical trial data, which could result in extensive discussions between the FDA and the applicant during the review process.
After the FDA evaluates a BLA, it will issue either an approval letter or a Complete Response Letter. An approval letter authorizes commercial marketing of the biologic with specific prescribing information for specific indications. A Complete Response Letter indicates that the review cycle of the application is complete, and the application will not be approved in its present form. A Complete Response Letter generally outlines the deficiencies in the BLA and may require additional clinical data, additional pivotal clinical trial(s), and/or other significant and time-consuming requirements related to clinical trials, preclinical studies or manufacturing in order for the FDA to reconsider the application. If a Complete Response Letter is issued, the applicant may either resubmit the BLA, addressing all of the deficiencies identified in the letter, or withdraw the application or request an opportunity for a hearing. The FDA has committed to reviewing such resubmissions in two or six months, depending on the type of information included. Even if such data and information are submitted, the FDA may decide that the BLA does not satisfy the criteria for approval.
As a condition of BLA approval, the FDA may require a risk evaluation and mitigation strategy (“REMS”) to help ensure that the benefits of the biologic outweigh the potential risks to patients. A REMS can include medication guides, communication plans for healthcare professionals, and elements to assure a product’s safe use (“ETASU”). An ETASU can include, but is not limited to, special training or certification for prescribing or dispensing the product, dispensing the product only under certain circumstances, special monitoring, and the use of patient-specific registries. The requirement for a REMS can materially affect the potential market and profitability of the product. Moreover, the FDA may require substantial post-approval testing and surveillance to monitor the product’s safety or efficacy.
Orphan Drug Designation
Under the Orphan Drug Act, the FDA may grant orphan designation to a drug or biological product intended to treat a rare disease or condition, which is generally a disease or condition that affects fewer than 200,000 individuals in the United States, or more than 200,000 individuals in the United States but for which there is no reasonable expectation that the cost of developing and making the product for this type of disease or condition will be recovered from sales of the product in the United States.
Orphan drug designation must be requested before submitting a BLA. After the FDA grants orphan drug designation, the identity of the therapeutic agent and its potential orphan use are disclosed publicly by the FDA. Orphan drug designation on its own does not convey any advantage in or shorten the duration of the regulatory review and approval process.
If a product that has orphan designation subsequently receives the first FDA approval for the disease or condition for which it has such designation, the product is entitled to orphan drug exclusivity, which means that the FDA may not approve any other applications to market the same product for the same indication for seven years from the date of such approval, except in limited circumstances, such as a showing of clinical superiority to the product with orphan exclusivity by means of greater effectiveness, greater safety, or providing a major contribution to patient care, or in instances of drug supply issues. Competitors, however, may receive approval of either a different product for the same indication or the same product for a different indication.
In the latter case, because healthcare professionals are free to prescribe products for off-label uses, the competitor’s product could be used for the orphan indication despite another product’s orphan exclusivity.
FDA’s determination of whether two ADCs are the same product for purposes of orphan drug exclusivity is based on a determination of sameness of the monoclonal antibody element and the functional element of the conjugated molecule. Two ADCs are deemed to be the same product if the complementarity determining region sequences of the antibody and the functional element of the conjugated molecule are the same. A difference in either of those two elements can result in a determination that the molecules are different.
Expedited Development and Review Programs
The FDA is authorized to designate certain products for expedited review if they are intended to address an unmet medical need in the treatment of a serious or life-threatening disease or condition.
Fast track designation may be granted for products that are intended to treat a serious or life-threatening disease or condition for which there is no effective treatment, and preclinical or clinical data demonstrate the potential to address unmet medical needs for the condition. Fast track designation applies to both the product and the specific indication for which it is being studied. The sponsor of a new biologic candidate can request the FDA to designate the candidate for a specific indication for fast track status concurrent with, or after, the submission of the IND for the candidate. The FDA must determine if the biologic candidate qualifies for fast track designation within 60 days of receipt of the sponsor’s request. For fast track products, sponsors may have greater interactions with the FDA and the FDA may initiate review of sections of a fast track product’s BLA before the application is complete. This “rolling review” is available if the FDA determines, after preliminary evaluation of clinical data submitted by the sponsor, that a fast track product may be effective. The sponsor must also provide, and the FDA must approve, a schedule for the submission of the remaining information and the sponsor must pay applicable user fees. Any product submitted to the FDA for marketing, including under a fast track program, may be eligible for other types of FDA programs intended to expedite development and review, such as priority review and accelerated approval.
Breakthrough therapy designation may be granted for products that are intended, alone or in combination with one or more other products, to treat a serious or life-threatening condition and preliminary clinical evidence indicates that the product may demonstrate substantial improvement over currently approved therapies on one or more clinically significant endpoints. Under the breakthrough therapy program, the sponsor of a new biologic candidate may request that the FDA designate the candidate for a specific indication as a breakthrough therapy concurrent with, or after, the submission of the IND for the biologic candidate. The FDA must determine if the biological product qualifies for breakthrough therapy designation within 60 days of receipt of the sponsor’s request. The FDA may take certain actions with respect to breakthrough therapies, including holding meetings with the sponsor throughout the development process, providing timely advice to the product sponsor regarding development and approval, involving more senior staff in the review process, assigning a cross-disciplinary project lead for the review team and taking other steps to design the clinical studies in an efficient manner.
Priority review may be granted for products that are intended to treat a serious or life-threatening condition and, if approved, would provide a significant improvement in safety and effectiveness compared to available therapies. The FDA will attempt to direct additional resources to the evaluation of an application designated for priority review in an effort to facilitate the review.
Accelerated approval may be granted for products that are intended to treat a serious or life-threatening condition and that generally provide a meaningful therapeutic advantage to patients over existing treatments. A product eligible for accelerated approval may be approved on the basis of either a surrogate endpoint that is reasonably likely to predict clinical benefit, or on a clinical endpoint that can be measured earlier than irreversible morbidity or mortality, that is reasonably likely to predict an effect on irreversible morbidity or mortality or other clinical benefit, taking into account the severity, rarity or prevalence of the condition and the availability or lack of alternative treatments. In clinical trials, a surrogate endpoint is a measurement of laboratory or clinical signs of a disease or condition that substitutes for a direct measurement of how a patient feels, functions, or survives. The accelerated approval pathway is most often used in settings in which the course of a disease is long and an extended period of time is required to measure the intended clinical benefit of a product, even if the effect on the surrogate or intermediate clinical endpoint occurs rapidly. Thus, accelerated approval has been used extensively in the development and approval of products for treatment of a variety of cancers in which the goal of therapy is generally to improve survival or decrease morbidity and the duration of the typical disease course requires lengthy and sometimes large studies to demonstrate a clinical or survival benefit. The accelerated approval pathway is contingent on a sponsor’s agreement to conduct additional post-approval confirmatory studies to verify and describe the product’s clinical benefit.
These confirmatory trials must be completed with due diligence, and, in some cases, the FDA may require that the trial be designed, initiated, and/or fully enrolled prior to approval. Failure to conduct required post-approval studies, or to confirm a clinical benefit during post-marketing studies, would allow the FDA to withdraw the product from the market on an expedited basis. All promotional materials for product candidates approved under accelerated approval are subject to prior review by the FDA. Pursuant to the Food and Drug Omnibus Reform Act (“FDORA”), the FDA is authorized to require a post-approval study to be underway prior to approval or within a specified time period following approval. FDORA also requires the FDA to specify conditions of any required post-approval study, which may include milestones such as a target date of study completion and requires sponsors to submit progress reports for required post-approval studies and any conditions required by FDA not later than 180 days following approval and not less frequently than every 180 days thereafter until completion or termination of the study. FDORA enables the FDA to initiate enforcement action for the failure to conduct with due diligence a required post-approval study, including a failure to meet any required conditions specified by the FDA or to submit timely reports.
Even if a product qualifies for one or more of these programs, the FDA may later decide that the product no longer meets the conditions for qualification or the time period for FDA review or approval may not be shortened. Furthermore, fast track designation, breakthrough therapy designation, priority review and accelerated approval do not change the standards for approval but may expedite the development or approval process.
Additional Controls for Biologics
To help reduce the increased risk of the introduction of adventitious agents, the PHSA emphasizes the importance of manufacturing controls for products whose attributes cannot be precisely defined. The PHSA also provides authority to the FDA to immediately suspend licenses in situations where there exists a danger to public health, to prepare or procure products in the event of shortages and critical public health needs, and to authorize the creation and enforcement of regulations to prevent the introduction or spread of communicable diseases in the United States and between states.
After a BLA is approved, the product may also be subject to official lot release as a condition of approval. As part of the manufacturing process, the manufacturer is required to perform certain tests on each lot of the product before it is released for distribution. If the product is subject to official release by the FDA, the manufacturer submits samples of each lot of product to the FDA together with a release protocol showing a summary of the history of manufacture of the lot and the results of all of the manufacturer’s tests performed on the lot. The FDA may also perform certain confirmatory tests on lots of some products, such as viral vaccines, before releasing the lots for distribution by the manufacturer. In addition, the FDA conducts laboratory research related to the regulatory standards on the safety, purity, potency, and effectiveness of biological products. As with drugs, after approval of biologics, manufacturers must address any safety issues that arise, are subject to recalls or a halt in manufacturing, and are subject to periodic inspection after approval.
Pediatric Information
Under the Pediatric Research Equity Act (“PREA), BLAs or supplements to BLAs must contain data to assess the safety and effectiveness of the biological product for the claimed indications in all relevant pediatric subpopulations and to support dosing and administration for each pediatric subpopulation for which the biological product is safe and effective. The FDA may grant full or partial waivers, or deferrals, for submission of data. Unless otherwise required by regulation, PREA generally does not apply to any biological product for an indication for which orphan designation has been granted. However, PREA applies to BLAs for orphan-designated biologics if the biologic is a molecularly targeted cancer product intended for the treatment of an adult cancer and is directed at a molecular target that FDA has determined is substantially relevant to the growth or progression of a pediatric cancer.
The Best Pharmaceuticals for Children Act (“BPCA”) provides a six-month extension of non-patent exclusivity for a biologic if certain conditions are met. Conditions for exclusivity include the FDA’s determination that information relating to the use of a new biologic in the pediatric population may produce health benefits in that population, the FDA making a written request for pediatric studies, and the applicant agreeing to perform, and reporting on, the requested studies within the statutory time frame. Applications under the BPCA are treated as priority applications, with all of the benefits that designation confers.
Post-Approval Requirements
Once a BLA is approved, a product will be subject to certain post-approval requirements. For instance, the FDA closely regulates the post-approval marketing and promotion of biologics, including standards and regulations for direct-to-consumer advertising, off-label promotion, industry-sponsored scientific and educational activities and promotional activities involving the internet. Biologics may be marketed only for the approved indications and in a manner consistent with the provisions of the approved labeling.
The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses, and a company that is found to have improperly promoted off-label uses may be subject to significant liability, including investigation by federal and state authorities.
Adverse event reporting and submission of periodic safety summary reports is required following FDA approval of a BLA. The FDA also may require post-marketing testing, known as Phase 4 testing, REMS, and surveillance to monitor the effects of an approved product, or the FDA may place conditions on an approval that could restrict the distribution or use of the product. In addition, quality control, biological product manufacture, packaging, and labeling procedures must continue to conform to cGMPs after approval. Biologic manufacturers and certain of their subcontractors are required to register their establishments with the FDA and certain state agencies. Registration with the FDA subjects entities to periodic unannounced inspections by the FDA, during which the agency inspects a biologic product’s manufacturing facilities to assess compliance with cGMPs. Accordingly, manufacturers must continue to expend time, money, and effort in the areas of production and quality-control to maintain compliance with cGMPs.
Once an approval is granted, the FDA may withdraw the approval or request product recalls if compliance with regulatory requirements and standards is not maintained or if problems occur after the product reaches the market. Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with manufacturing processes or failure to comply with regulatory requirements, may result in revisions to the approved labeling to add new safety information, imposition of post-market studies or clinical studies to assess new safety risks or imposition of distribution or other restrictions under a REMS program. Other potential consequences include, among other things:
•restrictions on the marketing or manufacturing of the product, suspension of the approval, complete withdrawal of the product from the market or product recalls;
•fines, warning or other enforcement-related letters or holds on post-approval clinical studies;
•refusal of the FDA to approve pending BLAs or supplements to approved BLAs, or suspension or revocation of product license approvals;
•product seizure or detention, or refusal to permit the import or export of products; or
•injunctions or the imposition of civil or criminal penalties.
Certain changes to an approved BLA or the conditions under which it was approved, including changes in its indications, safety information, labeling, or manufacturing processes or facilities, require submission and FDA approval of a new BLA or BLA supplement before the product can be marketed or distributed with those changes. A BLA supplement for a new indication typically requires clinical data similar to that in the original application. The FDA uses the same procedures and actions in reviewing BLA supplements as it does in reviewing original BLAs.
U.S. Patent Term Restoration, Marketing Exclusivity and Biosimilars
Depending upon the timing, duration, and specifics of FDA approval of our product candidates, some of our U.S. patents may be eligible for limited patent term extension under the Drug Price Competition and Patent Term Restoration Act of 1984 (the “Hatch-Waxman Amendments”). The Hatch Waxman Amendments provide for a patent term extension of up to five years as compensation for patent term lost during the FDA regulatory review process. Patent term extension, however, cannot extend the remaining term of a patent beyond a total of 14 years from the product’s approval date. The patent term extension period is generally one half the time between the effective date of an IND and the submission date of a BLA, plus the time between the submission date of a BLA and the approval of that application, up to five years. If the patent selected for extension was issued during the development or review period, the calculation begins from the date of patent issuance. The review period is reduced by any time during which the applicant failed to exercise due diligence. Only one patent applicable to an approved drug is eligible for such an extension, only those claims covering the approved drug, a method for using it, or a method for manufacturing it may be extended, and the application for the extension must be submitted prior to the expiration of the patent. Such application must be submitted within 60 days of approval. The USPTO, in consultation with the FDA, reviews and approves the application for any patent term extension or restoration. In the future, we or our licensors may apply for patent term extension for our owned or licensed patents to add patent life beyond their current expiration date, depending on the expected length of the clinical trials and other factors involved in the filing of the relevant BLA. However, an extension might not be granted because of, for example, failing to exercise due diligence during the testing phase or regulatory review process, failing to apply within applicable deadlines, failing to apply prior to expiration of relevant patents or otherwise failing to satisfy applicable eligibility or other requirements. Moreover, the extension awarded could be less than requested.
The BPCIA created an abbreviated approval pathway for biological products shown to be biosimilar to an FDA-licensed reference biological product. Biosimilarity, which requires that the biological product be highly similar to the reference product notwithstanding minor differences in clinically inactive components and that there be no clinically meaningful differences between the biological product and the reference product in terms of safety, purity and potency, can be shown through analytical studies, toxicity studies, and a clinical trial or trials. A biosimilar may be approved as interchangeable with its reference biological product. Interchangeability requires that a biological product both be biosimilar to the reference product and that the product can be expected to produce the same clinical results as the reference product in any given patient and, for products administered multiple times to an individual, that the product and the reference product may be alternated or switched after one has been previously administered without increasing safety risks or risks of diminished efficacy relative to exclusive use of the reference biological product without such alternation or switch. Under most state laws, interchangeable products may be used in place of the reference biological product.
A reference biological product is granted 12 years of data exclusivity from the time of first licensure of the product during which the FDA will not approve a biosimilar referencing the reference biological product, and the FDA will not accept an application for a biosimilar or interchangeable product based on the reference biological product until four years after the date of first licensure of the reference product. “First licensure” typically means the initial date the particular product at issue was licensed in the United States. Date of first licensure does not include the date of licensure of (and a new period of exclusivity is not available for) a biological product if the licensure is for a supplement for the biological product or for a subsequent application by the same sponsor or manufacturer of the biological product (or licensor, predecessor in interest, or other related entity) for a change that results in a new indication, route of administration, dosing schedule, dosage form, delivery system, delivery device or strength, or for a modification to the structure of the biological product that does not result in a change in safety, purity or potency.
Regulatory Approval in the European Union
The European Medicines Agency (the “EMA”) is a decentralized scientific agency of the European Union. It coordinates the evaluation and monitoring of centrally authorized medicinal products. It is responsible for the scientific evaluation of applications for EU marketing authorizations, as well as the development of technical guidance and the provision of scientific advice to sponsors. The EMA decentralizes its scientific assessment of medicines by working through a network of about 4,500 experts throughout the European Union, nominated by the member states. The EMA draws on resources of over 40 National Competent Authorities of European Union member states.
The process regarding approval of medicinal products in the European Union follows roughly the same lines as in the United States and likewise generally involves satisfactorily completing each of the following:
•preclinical laboratory tests, animal studies and formulation studies all performed in accordance with the applicable EU GLP regulations;
•submission to the relevant national authorities of a clinical trial application (“CTA”) for each trial in humans, which must be approved before the trial may begin in each country where patient enrollment is planned;
•performance of adequate and well-controlled clinical trials to establish the safety and efficacy of the product for each proposed indication;
•submission to the relevant competent authorities of an MAA, which includes the data supporting safety and efficacy as well as detailed information on the manufacture and composition of the product in clinical development and proposed labelling;
•satisfactory completion of an inspection by the relevant national authorities of the manufacturing facility or facilities, including those of third parties, at which the product is produced to assess compliance with strictly enforced cGMP;
•potential audits of the non-clinical and clinical trial sites that generated the data in support of the MAA; and
•review and approval by the relevant competent authority of the MAA before any commercial marketing, sale or shipment of the product.
Preclinical Studies
Preclinical tests include laboratory evaluations of product chemistry, formulation and stability, as well as studies to evaluate toxicity in animal studies, in order to assess the quality and potential safety and efficacy of the product. The conduct of the preclinical tests and formulation of the compounds for testing must comply with the relevant international, EU and national legislation, regulations and guidelines. The results of the preclinical tests, together with relevant manufacturing information and analytical data, are submitted as part of the CTA.
Clinical Trials
Pursuant to the Clinical Trials Directive 2001/20/EC, as amended (the “Clinical Trials Directive”), a system for the approval of clinical trials in the European Union has been implemented through national legislation of the member states. Under this system, approval must be obtained from the competent national authority of each European Union member state in which a clinical trial is planned to be conducted. To this end, a CTA is submitted, which must be supported by an investigational medicinal product dossier and further supporting information prescribed by the Clinical Trials Directive and other applicable guidance documents including, but not limited to, the clinical trial protocol. Furthermore, a clinical trial may only be started after a central ethics committee has issued a favorable opinion on the clinical trial application in that country.
Directive 2001/20/EC has been replaced by Regulation (EU) No. 536/2014, which became effective on January 31, 2022. The Regulation introduces an authorization procedure based on a single submission via a single EU portal, an assessment procedure leading to a single decision, as well as transparency requirements (the proactive publication of clinical trial data in the EU database). Since October 2016, based on its Policy 0070, the EMA has been publishing clinical data submitted by pharmaceutical companies to support their MAA for human medicines under this centralized procedure.
Manufacturing and import into the EU of investigational medicinal products is subject to the holding of appropriate authorizations and must be carried out in accordance with cGMP.
Review and Approval
Authorization to market a product in the European Union member states proceeds under one of four procedures: a centralized authorization procedure, a mutual recognition procedure, a decentralized procedure or a national procedure. Since our products by their virtue of being antibody-based biologics fall under the centralized procedure, only this procedure will be described here.
Certain drugs, including medicinal products developed by means of biotechnological processes, must be approved via the centralized authorization procedure for marketing authorization. A successful application under the centralized authorization procedure results in a marketing authorization from the European Commission, which is automatically valid in all European Union member states. The other European Economic Area member states (namely Norway, Iceland and Liechtenstein) are also obligated to recognize the European Commission decision. The EMA and the European Commission administer the centralized authorization procedure.
Under the centralized authorization procedure, the Committee for Medicinal Products for Human Use (the “CHMP”) serves as the scientific committee that renders opinions about the safety, efficacy and quality of human products on behalf of the EMA. The CHMP is composed of experts nominated by each member state’s national health authority, with one of them appointed to act as Rapporteur for the co-ordination of the evaluation with the assistance of a further member of the CHMP acting as a Co-Rapporteur. The CHMP is required to issue an opinion within 210 days of receipt of a valid application, though the clock is stopped if it is necessary to ask the applicant for clarification or further supporting data. The process is complex and involves extensive consultation with the regulatory authorities of member states and a number of experts. Once the procedure is completed, a European Public Assessment Report is produced. If the CHMP concludes that the quality, safety and efficacy of the medicinal product is sufficiently proven, it adopts a positive opinion. The CHMP’s opinion is sent to the European Commission, which uses the opinion as the basis for its decision whether or not to grant a marketing authorization. If the opinion is negative, information is given as to the grounds on which this conclusion was reached.
After a drug has been authorized and launched, it is a condition of maintaining the marketing authorization that all aspects relating to its quality, safety and efficacy must be kept under review. Sanctions may be imposed for failure to adhere to the conditions of the marketing authorization. In extreme cases, the authorization may be revoked, resulting in withdrawal of the product from sale.
Conditional Approval and Accelerated Assessment
As per Article 14(7) of Regulation (EC) 726/2004, a medicine that would fulfill an unmet medical need may, if its immediate availability is in the interest of public health, be granted a conditional marketing authorization on the basis of less complete clinical data than are normally required, subject to specific obligations being imposed on the authorization holder. These specific obligations are to be reviewed annually by the EMA. The list of these obligations shall be made publicly accessible. Such an authorization shall be valid for one year, on a renewable basis.
When an application is submitted for a marketing authorization in respect of a drug for human use which is of major interest from the point of view of public health and in particular from the viewpoint of therapeutic innovation, the applicant may request an accelerated assessment procedure pursuant to Article 14(9) of Regulation (EC) 726/2004. Under the accelerated assessment procedure, the CHMP is required to issue an opinion within 150 days of receipt of a valid application, subject to clock stops. We believe that some of the disease indications in which our product candidates are currently being or may be developed in the future qualify for this provision, and we will take advantage of this provision as appropriate.
Period of Authorization and Renewals
A full marketing authorization is initially valid for five years and may then be renewed on the basis of a re-evaluation of the risk-benefit balance by the EMA or by the competent authority of the authorizing member state. To this end, the marketing authorization holder shall provide the EMA or the competent authority with a consolidated version of the file in respect of quality, safety and efficacy, including all variants introduced since the marketing authorization was granted, at least nine months before the expiry date of the marketing authorization. Once renewed, the marketing authorization shall be valid for an unlimited period, unless the European Commission or the competent authority decides, on justified grounds relating to pharmacovigilance, to proceed with one additional five-year renewal. Any authorization which is not followed by the actual placing of the drug on the EU market (in case of centralized procedure) or on the market of the authorizing member state within three years after authorization shall cease to be valid (the so-called “sunset clause”).
Without prejudice to the law on the protection of industrial and commercial property, marketing authorizations for new medicinal products benefit from an 8+2+1 year period of regulatory protection. This regime consists of a regulatory data protection period of eight years plus a concurrent market exclusivity of 10 years plus an additional market exclusivity of one further year if, during the first eight years of those 10 years, the marketing approval holder obtains an approval for one or more new therapeutic indications which, during the scientific evaluation prior to their approval, are determined to bring a significant clinical benefit in comparison with existing therapies. Under the current rules, a third party may reference the preclinical and clinical data of the reference product beginning eight years after first approval, but the third party may market a generic version of the reference product after only 10 (or 11) years have lapsed.
Orphan Drug Designation
Regulation (EC) 141/2000 states that a drug shall be designated as an orphan drug if its sponsor can establish (i) that it is intended for the diagnosis, prevention or treatment of a life-threatening or chronically debilitating condition affecting not more than five in 10,000 persons in the European Union when the application is made, or that it is intended for the diagnosis, prevention or treatment of a life-threatening, seriously debilitating or serious and chronic condition in the European Union and that without incentives it is unlikely that the marketing of the drug in the European Union would generate sufficient return to justify the necessary investment; and (ii) that there exists no satisfactory method of diagnosis, prevention or treatment of the condition in question that has been authorized in the European Union or, if such method exists, the drug will be of significant benefit to those affected by that condition.
Regulation (EC) 847/2000 sets out criteria for the designation of orphan drugs. An application for designation as an orphan product can be made any time prior to the filing of an application for approval to market the product. Marketing authorization for an orphan drug leads to a 10-year period of market exclusivity, which means that no similar medicinal product can be authorized in the same indication. This period may, however, be reduced to six years if, at the end of the fifth year, it is established that the product no longer meets the criteria for orphan drug designation, for example because the product is sufficiently profitable not to justify continued market exclusivity. In addition, derogation from market exclusivity may be granted on an individual basis in very select cases, such as consent from the marketing authorization holder, inability to supply sufficient quantities of the product or demonstration of “clinically relevant superiority” by a similar medicinal product. Medicinal products designated as orphan drugs pursuant to Regulation (EC) 141/2000 are eligible for incentives made available by the European Union and by the member states to support research into, and the development and availability of, orphan drugs.
If the MAA of a medicinal product designated as an orphan drug pursuant to Regulation (EC) 141/2000 includes the results of all studies conducted in compliance with an agreed PIP, and a corresponding statement is subsequently included in the marketing authorization granted, the 10-year period of market exclusivity will be extended to 12 years.
European Data Collection and Processing
The collection, transfer, processing and other use of personal information, including health data, in the European Union is governed by the GDPR. This directive imposes several requirements relating to (i) obtaining, in some situations, the consent of the individuals to whom the personal data relates, (ii) the information provided to the individuals about how their personal information is used, (iii) ensuring the security and confidentiality of the personal data, (iv) the obligation to notify regulatory authorities and affected individuals of personal data breaches, (v) extensive internal privacy governance obligations and (vi) obligations to honor rights of individuals in relation to their personal data (for example, the right to access, correct and delete their data). The GDPR prohibits the transfer of personal data to countries outside the European Economic Area, such as the United States, which are not considered by the European Commission to provide an adequate level of data protection. Switzerland has adopted similar restrictions. Failure to comply with the requirements of the GDPR and the related national data protection laws of the European Union member states may result in fines and other administrative penalties. The GDPR introduces new data protection requirements in the EU and substantial fines for breaches of the data protection rules. The GDPR and related data protection laws may impose additional responsibility and liability in relation to personal data that we collect and process, and we may be required to put in place additional mechanisms ensuring compliance with such rules. This may be onerous and adversely affect our business, financial condition, results of operations, and prospects.
Marketing
Much like the Anti-Kickback Statute prohibition in the United States, the provision of benefits or advantages to physicians to induce or encourage the prescription, recommendation, endorsement, purchase, supply, order or use of medicinal products is also prohibited in the EU. The provision of benefits or advantages to physicians is governed by the national anti-bribery laws of European Union member states, such as the U.K. Bribery Act 2010. Infringement of these laws could result in substantial fines and imprisonment.
Payments made to physicians in certain European Union member states must be publicly disclosed. Moreover, agreements with physicians often must be the subject of prior notification and approval by the physician’s employer, his or her competent professional organization, and/or the regulatory authorities of the individual European Union member states. These requirements are provided in the national laws, industry codes or professional codes of conduct, applicable in the European Union member states. Failure to comply with these requirements could result in reputational risk, public reprimands, administrative penalties, fines or imprisonment.
International Regulation
In addition to regulations in the United States and Europe, a variety of foreign regulations govern clinical trials, commercial sales and distribution of product candidates. The approval process varies from country to country, and the time to approval may be longer or shorter than that required for FDA or European Commission approval.
Other Healthcare Laws and Regulations
For other material healthcare laws and regulations that affect our business, see “Item 1A. Risk Factors—Risks Related to Regulatory Approval and Government Regulation.”
Environmental, Health and Safety Laws and Regulations
We and our third-party contractors are subject to numerous environmental, health and safety laws and regulations, including those governing laboratory procedures and the use, generation, manufacture, distribution, storage, handling, treatment, remediation and disposal of hazardous materials and wastes. Hazardous chemicals, including flammable and biological materials, are involved in certain aspects of our business, and we cannot eliminate the risk of injury or contamination from the use, generation, manufacture, distribution, storage, handling, treatment or disposal of hazardous materials and wastes. In particular, our product candidates use PBDs, which are highly potent cytotoxins that require special handling by our and our contractors’ staff. In the event of contamination or injury, or failure to comply with environmental, health and safety laws and regulations, we could be held liable for any resulting damages, fines and penalties associated with such liability, which could exceed our assets and resources. Environmental, health and safety laws and regulations are becoming increasingly more stringent.
We may incur substantial costs in order to comply with current or future environmental, health and safety laws and regulations.
Government Pricing and Reimbursement Programs for Marketed Drugs in the United States
Medicaid, the 340B Drug Pricing Program, and Medicare
Federal law requires that a pharmaceutical manufacturer, as a condition of having its products receive federal reimbursement under Medicaid and Medicare Part B, must pay quarterly rebates to state Medicaid programs for all units of its covered outpatient drugs dispensed to Medicaid beneficiaries and paid for by a state Medicaid program under either a fee-for-service arrangement or through a managed care organization. This federal requirement is effectuated through a Medicaid drug rebate agreement between the manufacturer and the Secretary of U.S. Department of Health and Human Services (“HHS”). The Centers for Medicare & Medicaid Services (“CMS”) administers the Medicaid drug rebate agreements with manufacturers which provide, among other things, that the drug manufacturer will pay rebates to each state Medicaid agency on a quarterly basis and report certain price information on a monthly and quarterly basis. The rebates are based on the average manufacturer price (“AMP”) reported to CMS by manufacturers for their covered outpatient drugs and how those drugs are classified. For non-innovator products, generally generic drugs marketed under abbreviated new drug applications (“ANDAs”), the rebate amount is 13% of the AMP for the quarter. The AMP is the weighted average of prices paid to the manufacturer as defined by the applicable regulations. For innovator products (i.e., drugs that are marketed under NDAs or BLAs), the rebate amount is the greater of 23.1% of the AMP for the quarter or the difference between such AMP and the best price for that same quarter. The best price is essentially the lowest price available to non-governmental entities after accounting for discounts and rebates. Innovator products may also be subject to an additional rebate that is based on the amount, if any, by which the product’s AMP for a given quarter exceeds the inflation-adjusted baseline AMP, which for most drugs is the AMP for the first full quarter after launch. Since 2017, non-innovator products are also subject to an additional rebate for price increases that exceed inflation. Prior to January 1, 2024, the rebate amount for a drug was capped at 100% of the AMP; however, effective January 1, 2024, this cap was eliminated, which means that a manufacturer could pay a total rebate amount on a unit of the drug that is greater than the average price the manufacturer receives for the drug.
The terms of participation in the Medicaid drug rebate program impose an obligation to correct the prices reported in previous quarters, as may be necessary. Any such corrections could result in additional or lesser rebate liability, depending on the direction of the correction. A manufacturer may correct prices reported more than three years prior only in certain circumstances specified in regulations. In September 2024, CMS published a final rule that expanded its authority under one such circumstance, namely when a correction request addresses a manufacturer’s internal investigation. Under the final rule, CMS considers all change requests pursuant to an internal investigation to be possible violations of law and requires manufacturers to provide its investigation findings. Finally, the rule establishes a three-year limit for manufacturers to dispute, audit, or request a hearing on state utilization data.
If a manufacturer were found to have knowingly submitted false information to the government, or knowingly misclassified a drug, federal law provides for the collection of any outstanding rebates, as well as for civil monetary penalties for failing to provide required information, late submission of required information, and false information. The 2024 final rule expanded the definition of a “misclassification” to include any drug product information that is not supported by the statute or application regulations and allows CMS to suspend a Medicaid rebate agreement for a manufacturer’s failure to correct a misclassification, or a failure to submit the required pricing reports, within 90 calendar days receiving notice.
A manufacturer must also participate in a federal program known as the 340B drug pricing program for federal funds to be available to pay for the manufacturer’s drugs and biological products under Medicaid and Medicare Part B. Under this program, the participating manufacturer agrees to charge certain safety net healthcare providers no more than an established discounted price for its covered outpatient drugs. The formula for determining the discounted price is defined by statute and is based on the AMP and the unit rebate amount as calculated under the Medicaid drug rebate program, discussed above. Manufacturers are required to report pricing information to the Health Resources and Services Administration (“HRSA”) on a quarterly basis. HRSA has also issued regulations relating to the calculation of the ceiling price as well as imposition of civil monetary penalties for each instance of knowingly and intentionally overcharging a 340B covered entity. In November 2023, a federal district court decision broadened the types of patients that can purchase 340B discounted drugs from covered entities, which, if affirmed on appeal, will further grow this program. In addition, there is ongoing litigation that may restrict the number of third-party contract pharmacies that can dispense drugs that manufacturers sell to 340B covered entities and who qualify as patients of these 340B covered entities. The outcome of this litigation may change the scope of the 340B program in coming years.
Federal law also requires that manufacturers report data on a quarterly basis to CMS regarding the pricing of drugs that are separately reimbursable under Medicare Part B. These are generally drugs, such as injectable products, that are administered “incident to” a physician service and are not generally self-administered. The average selling price (“ASP”) reported by CMS manufacturers is the basis for reimbursement to providers for drugs covered under Medicare Part B. Under the Inflation Reduction Act (“IRA”), as of January 1, 2023, manufacturers are also required to provide quarterly rebates for certain single-source drugs and biologics (including biosimilars) covered under Medicare Part B with ASPs that increase faster than the rate of inflation. This requirement started on January 1, 2023, for drugs approved on or before December 1, 2020, and begins six quarters after a drug is first marketed for all other drugs. In November 2024, CMS finalized regulations for the Medicare Part B inflation rebates. As with the Medicaid drug rebate program, Federal law provides for civil monetary penalties for failing to provide required information, late submission of required information, and false information.
Manufacturers of certain single-source drugs (including biologics and biosimilars) separately paid for under Medicare Part B for at least 18 months and marketed in single-dose containers or packages (known as refundable single-dose container or single-use package drugs) are required to provide annual refunds for any portions of the dispensed drug that are unused and discarded if those unused or discarded portions exceed an applicable percentage defined by statute or regulation. Manufacturers will be subject to periodic audits and those that fail to pay refunds for their refundable single-dose container or single-use package drugs shall be subject to civil monetary penalties.
Medicare Part D provides prescription drug benefits for seniors and people with disabilities. Federal law requires that manufacturers with covered drugs under Medicare Part D pay a portion of the enrollee’s copay via a rebate to CMS. The Medicare Part D benefit design and the portion of the enrollee’s co-pay that manufacturers are required to pay has changed over time. The current manufacturer responsibility which began in 2019 is 70% of the coverage gap portion of the enrollee’s copay (amount between the initial coverage limit and start of catastrophic coverage). Medicare Part D enrollees once had a gap in their coverage (between the initial coverage limit and the point at which catastrophic coverage begins) where Medicare did not cover their prescription drug costs, known as the coverage gap. Beginning in 2025, the IRA eliminates the coverage gap under Medicare Part D by significantly lowering the enrollee maximum out-of-pocket cost and requiring manufacturers to enter into a new manufacturer discount program agreement and pay 10% of Part D enrollees’ prescription costs for brand drugs above a deductible and below the out-of-pocket maximum, and 20% once the out-of-pocket maximum has been reached. Although these discounts represent a lower percentage of enrollees’ costs than the current discounts required below the out-of-pocket maximum (that is, in the coverage gap phase of Part D coverage), the new manufacturer contribution required above the out-of-pocket maximum could be considerable for very high-cost patients and the total contributions by manufacturers to a Part D enrollee’s drug expenses may exceed those currently provided. Pursuant to the IRA, manufacturers are also required to provide annual Medicare Part D rebates for single-source drugs and biological products with prices that increase faster than the rate of inflation. As with the inflation rebates for Medicare Part B drugs, in November 2024, CMS finalized regulations for the Medicare Part B inflation rebates.
The IRA also allows HHS to directly negotiate the selling price of a statutorily specified number of drugs and biologics each year that CMS reimburses under Medicare Part B and Part D. Only high-expenditure single-source biologics that have been approved for at least 11 years (7 years for single-source drugs) can qualify for negotiation, with the negotiated price taking effect two years after the selection year. CMS selected the Medicare Part D products for negotiation in 2023, negotiations began in 2024, and the negotiated maximum fair price for each product has been announced. The price cap for each of these products, which cannot exceed a statutory ceiling price, will take with the negotiated price taking effect in 2026. Negotiations for Medicare Part B products begin in 2026 with the negotiated price taking effect in 2028. A drug or biological product that has an orphan drug designation for only one rare disease or condition will be excluded from the IRA's price negotiation requirements, but loses that exclusion if it has designations for more than one rare disease or condition, or if is approved for an indication that is not within that single designated rare disease or condition, unless such additional designation or such disqualifying approvals are withdrawn by the time CMS evaluates the drug for selection for negotiation.
U.S. Federal Contracting and Pricing Requirements
Manufacturers are also required to make their covered drugs, which are generally drugs approved under NDAs or BLAs, available to authorized users of the Federal Supply Schedule (“FSS”) of the General Services Administration. The law also requires manufacturers to offer deeply discounted FSS contract pricing for purchases of their covered drugs by the Department of Veterans Affairs, the Department of Defense, the Coast Guard, and the Public Health Service (including the Indian Health Service) in order for federal funding to be available for reimbursement or purchase of the manufacturer’s drugs under certain federal programs. FSS pricing to those four federal agencies for covered drugs must be no more than the Federal Ceiling Price (“FCP”), which is at least 24% below the Non-Federal Average Manufacturer Price (“Non-FAMP”) for the prior year.
The Non-FAMP is the average price for covered drugs sold to wholesalers or other middlemen, net of any price reductions.
The accuracy of a manufacturer’s reported Non-FAMPs, FCPs, or FSS contract prices may be audited by the government. Among the remedies available to the government for inaccuracies is recoupment of any overcharges to the four specified federal agencies based on those inaccuracies. If a manufacturer were found to have knowingly reported false prices, in addition to other penalties available to the government, the law provides for significant civil monetary penalties per incorrect item. Finally, manufacturers are required to disclose in FSS contract proposals all commercial pricing that is equal to or less than the proposed FSS pricing, and subsequent to award of an FSS contract, manufacturers are required to monitor certain commercial price reductions and extend commensurate price reductions to the government, under the terms of the FSS contract Price Reductions Clause. Among the remedies available to the government for any failure to properly disclose commercial pricing and/or to extend FSS contract price reductions is recoupment of any FSS overcharges that may result from such omissions.
Human Capital Resources
We believe that our success is largely dependent upon our ability to attract and retain qualified employees. We currently have 263 full-time employees and two part-time employees, of which 53 employees are based in our New Jersey office facility, 55 employees in our UK research facility, 24 employees in our Swiss corporate office, and 133 employees remote. Our workforce includes 60 employees directly involved in or supporting our commercial sales organization, 118 in research and development, 42 in manufacturing, CMC and quality and 45 in corporate and administrative functions. We are not party to any collective bargaining arrangements. We believe that we have been successful to date in attracting skilled and experienced scientific and business professionals.
We continue to focus on building a high performing organization with an engaging work culture and have established initiatives to support this strategic priority. We perform periodic employee engagement surveys, set and monitor retention goals, and offer training and leadership development to cultivate our organization. Additionally, we are committed to diversity and inclusion as a focus of our human capital strategy. We embrace differences, diversity and varying perspectives amongst our employee base, and are proud to be an equal opportunity employer. We do not discriminate based on race, religious creed, color, national origin, ancestry, physical disability, mental disability, medical condition, genetic information, marital status, sex, gender, gender identity, gender expression, age, military or veteran status, sexual orientation or any other protected characteristic established by federal, state or local laws. A diverse workforce, as well as an inclusive culture and work environment, are fundamentally important and strategic to us, beginning with our Board of Directors and extending to all levels of the organization. As of December 31, 2024, our total employee base was 57% diverse on the basis of gender and race.
We strongly believe that the success of ADCT depends, in part, on open and regular communication with employees to help foster a high performing and engaged workforce. To help ensure that employees fully understand the Company’s long-term strategy and annual goals, along with how their work contributes to the Company’s success, we use a variety of channels to facilitate open and direct communication, including: (i) regular CEO Town Hall meetings; (ii) regular ongoing update communications; and (iii) employee engagement surveys.
Talent management and leadership development is critical to our ability to execute on our long-term growth strategy. We seek to provide pay, benefits, and services that are competitive to market practice and create incentives to attract and retain employees. Our compensation package includes market-competitive base salary, discretionary broad-based stock grants and bonuses, health care and retirement benefits, paid time off and family leave. To help support the development and advancement of our high performing employees, we offer training and development programs encouraging advancement from within and continue to fill our team with strong and experienced management talent. We leverage both formal and informal programs to identify, foster, and retain top talent throughout the organization.
Our compensation philosophy is to pay for performance, and designed to support the Company’s business strategies, and offer competitive compensation arrangements to attract and retain key individuals. Our Board of directors have established a Compensation Committee to oversee and monitor our compensation practices. Consistent with this philosophy, the Compensation Committee considers the impact of our corporate performance in determining compensation for named executive officers, as well as each named executive officer’s individual performance, macroeconomic conditions, and data from peer group companies.
Availability of Information
We are subject to the informational requirements of the Exchange Act. Accordingly, we are required to file reports and other information with the SEC, including annual, quarterly and current reports and proxy and information statements. The SEC maintains an internet site at sec.gov that contains reports, proxy and information statements and other information we have filed electronically with the SEC.
Our website address is adctherapeutics.com. We make available free of charge on our website our Annual Reports on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K and amendments to those reports filed or furnished pursuant to Sections 13(a) and 15(d) of the Exchange Act, as soon as reasonably practicable after we file or furnish such materials with the SEC. Information on, or accessible through, our website is not part of this Annual Report or incorporated by reference in any of our filings with the SEC, except where we expressly incorporate such information.
Enforcement of Judgments
We are organized under the laws of Switzerland and our registered office and domicile is located in Epalinges, Switzerland. Moreover, a number of our directors and executive officers are not residents of the United States, and all or a substantial portion of the assets of such persons are located outside the United States. As a result, it may not be possible for investors to effect service of process within the United States upon us or upon such persons or to enforce against them judgments obtained in U.S. courts, including judgments in actions predicated upon the civil liability provisions of the federal securities laws of the United States. There is doubt as to the enforceability in Switzerland of original actions, or in actions for enforcement of judgments of U.S. courts, of civil liabilities to the extent solely predicated upon the federal and state securities laws of the United States. Original actions against persons in Switzerland based solely upon the U.S. federal or state securities laws are governed, among other things, by the principles set forth in the Swiss Federal Act on Private International Law (the “PILA”). The PILA provides that the application of provisions of non-Swiss law by the courts in Switzerland shall be precluded if the result would be incompatible with Swiss public policy. Also, mandatory provisions of Swiss law may be applicable regardless of any other law that would otherwise apply.
Switzerland and the United States do not have a treaty providing for reciprocal recognition of and enforcement of judgments in civil and commercial matters. The recognition and enforcement of a judgment of the courts of the United States in Switzerland is governed by the principles set forth in the PILA. The PILA provides in principle that a judgment rendered by a non-Swiss court may be enforced in Switzerland only if:
•the non-Swiss court had jurisdiction pursuant to the PILA;
•the judgment of such non-Swiss court has become final and non-appealable;
•the judgment does not contravene Swiss public policy;
•the court procedures and the service of documents leading to the judgment were in accordance with the due process of law; and
•no proceeding involving the same position and the same subject matter was first brought in Switzerland, or adjudicated in Switzerland, or was earlier adjudicated in a third state and this decision is recognizable in Switzerland.
Item 1A. Risk Factors
Our business faces significant risks and uncertainties. You should carefully consider all of the information set forth in this Annual Report and in other documents we file with or furnish to the SEC, including the following risk factors, before deciding to invest in or to maintain an investment in our securities. Our business, as well as our reputation, financial condition, results of operations, and share price, could be materially adversely affected by any of these risks, as well as other risks and uncertainties not currently known to us or not currently considered material.
Risk Factors Summary
Our ability to implement our business strategy is subject to numerous risks, as more fully described in this Annual Report and our other documents filed with the SEC. These risks include, among others:
•We have incurred substantial net losses since our inception, expect to continue to incur losses for the foreseeable future and may never achieve or sustain profitability. We will need to raise additional capital to fund our operations and execute our business plan.
•Our indebtedness under the Loan Agreement and the associated restrictive covenants thereunder could adversely affect our financial condition.
•The HCR Agreement reduces the amount of cash we are able to generate from sales of, and licensing agreements involving, ZYNLONTA and could make us a less attractive acquisition target.
•We may be unable to complete clinical trials on our expected timelines, if at all.
•Our products and product candidates may cause undesirable side effects or adverse events.
•We may be unable to obtain, or experience delays in obtaining, regulatory approval for our product candidates. We may be unable to maintain regulatory approval for any approved products.
•We or our partners may not be able to successfully commercialize our products.
•There can be no assurance regarding the outcome of ongoing or planned clinical trials or the sufficiency of results from such clinical trials.
•Coverage and reimbursement may be limited or unavailable for our products.
•Our products and product candidates are complex and difficult to manufacture.
•We face substantial competition, which may result in others discovering, developing or commercializing products, treatment methods or technologies before, or more successfully than, we do.
•We rely on third parties to conduct preclinical studies and clinical trials and for the manufacture, production, storage and distribution of our products and product candidates and certain commercialization activities for our products.
•If we are unable to obtain, maintain or protect our intellectual property rights in any products or technologies we develop, or if the scope of the intellectual property protection obtained is not sufficiently broad, third parties could develop and commercialize products and technology similar or identical to ours, and we may not be able to compete effectively in our market.
•We may become involved in lawsuits to protect or enforce our patents or other intellectual property, which could be expensive, time-consuming and unsuccessful, and our issued patents covering one or more of our products, product candidates or technologies or the technology we use in our products and product candidates, could be found invalid or unenforceable if challenged in court.
•We may be subject to claims by third parties asserting that our products infringe their intellectual property or that we or our employees, consultants or advisors have misappropriated their intellectual property, or claiming ownership of what we regard as our own intellectual property.
•Product liability lawsuits and product recalls could cause us to incur substantial liabilities and to limit development and commercialization of our products.
Risks Related to Our Financial Position, Capital Requirements and Ability to Raise Additional Capital
We have incurred substantial net losses since our inception, expect to continue to incur losses for the foreseeable future and may never achieve or sustain profitability. We will need to raise additional capital to fund our operations and execute our business plan and such additional capital could be dilutive, limit our ability to operate our business and adversely impact the price of our common shares.
We have incurred substantial net losses since our inception and expect to continue to incur losses for the foreseeable future. As of December 31, 2024, we had accumulated losses of $1,493 million. We expect to continue to incur net losses for the foreseeable future as we continue to devote substantial resources to research and development and marketing and commercialization efforts in both hematology and solid tumors, in particular to grow ZYNLONTA in the 3L+ DLBCL setting, continue to study and advance ZYNLONTA in earlier lines of therapy and in combinations to potentially expand our market opportunity and further develop our pipeline and our ADC platform. We are unable to accurately predict whether and when we will achieve profitability. Even if we achieve profitability, we may not be able to sustain profitability in subsequent periods. This risk is heightened as we only have one approved product, ZYNLONTA, at the present time and thus are heavily dependent on its commercial performance and its continued research and development.
As a result, we will need to raise additional capital to fund our operations and execute our business plan. We do not have any committed external source of funds, and additional funds may not be available when we need them or on terms that are acceptable to us. Our ability to raise additional funds will depend on financial, economic and market conditions and other factors, over which we may have no or limited control.
Further, as a Swiss company, we have less flexibility to raise capital, particularly in a quick and efficient manner, as compared to U.S. companies. See “—Risks Related to Our Common Shares—Our shareholders enjoy certain rights that may limit our flexibility to raise capital, issue dividends and otherwise manage ongoing capital needs.” The restrictions contained in our contractual agreements may also limit our ability to raise certain forms of capital. For example, subject to certain exceptions, the Loan Agreement restricts our ability to incur indebtedness and the HCR Agreement restricts our ability to sell, finance or loan any additional royalties on ZYNLONTA outside of China, Hong Kong, Macau, Taiwan, Singapore and South Korea or on Cami, and to incur indebtedness exceeding 20% of our market capitalization. If adequate funds are not available to us on a timely basis or on terms acceptable to us, we may be required to delay, limit, reduce or terminate our research and development, commercialization or growth efforts.
We may seek additional capital through a variety of means. If we raise additional capital through the sale of equity or convertible debt securities, your ownership interest will be diluted, and the terms of such equity or convertible debt securities may include liquidation or other preferences that are senior to or otherwise adversely affect your rights as a shareholder. If we raise additional capital through the sale of debt securities or through entering into credit or loan facilities, we may be restricted in our ability to take certain actions, such as incurring additional debt, making capital expenditures, acquiring or licensing intellectual property rights, declaring dividends or encumbering our assets to secure future indebtedness. If we raise additional capital through collaborations with third parties, we may be required to relinquish valuable rights to our intellectual property, products or product candidates or we may be required to grant licenses for our intellectual property, products or product candidates on unfavorable terms.
Our indebtedness under the Loan Agreement and the associated restrictive covenants thereunder could adversely affect our financial condition.
We have significant indebtedness outstanding under the Loan Agreement. Such indebtedness requires us to dedicate a substantial portion of our cash and cash equivalents to the payment of interest on, and principal of, the indebtedness, thereby reducing the amounts available to fund working capital, capital expenditures, research and development efforts, commercialization efforts and other general corporate purposes. Indebtedness under the Loan Agreement bears variable rates of interest based on the prevailing SOFR, thereby making us more vulnerable to rising interest rates.
The Loan Agreement contains certain restrictions on our activities and customary covenants, including a covenant to maintain qualified cash of at least $60.0 million plus an amount equal to any accounts payable that remain unpaid more than ninety days after the date of the original invoice therefor, and negative covenants including limitations on indebtedness, liens, fundamental changes, asset sales, investments, dividends and other restricted payments and other matters customarily restricted in such agreements. In addition, the Loan Agreement contains a revenue covenant that, so long as our 30-day average market capitalization is less than $650 million, requires us to achieve minimum levels of ZYNLONTA net sales in the United States, tested on a quarterly basis, which is subject to a customary cure right that may be exercised by making certain prepayments and that, subject to certain limitations, may be exercised up to three times during the term of the Loan Agreement. The obligations under the Loan Agreement are secured by substantially all of our assets and are guaranteed by certain of our subsidiaries. Such covenants could limit our flexibility in planning for, or reacting to, changes in our business and our industry; place us at a competitive disadvantage compared to our competitors who have less debt or competitors with comparable debt on more favorable terms; and limit our ability to borrow additional amounts.
Our ability to maintain compliance with the covenants imposed by our indebtedness and to repay the principal of, pay interest on and refinance our indebtedness depends on our future performance, which is subject to economic, financial, competitive and other factors, many of which are beyond our control. If we are unable to comply with the covenants imposed by our indebtedness or to generate sufficient cash flow to service or repay our indebtedness, we may be in default of the Loan Agreement and be required to adopt one or more alternatives, such as restructuring debt or obtaining additional financing on terms that may be unfavorable to us or highly dilutive.
The HCR Agreement reduces the amount of cash we are able to generate from sales of, and licensing agreements involving, ZYNLONTA and could make us a less attractive acquisition target.
Under the HCR Agreement, we are obligated to pay to HCR royalties representing a percentage of net sales of ZYNLONTA in certain jurisdictions, a percentage of any upfront or milestone payments we receive from licenses that we grant to commercialize ZYNLONTA in certain jurisdictions, and a percentage of any upfront or milestone payments (or on royalties) we receive from licenses that we grant to commercialize Cami. See “Item 1. Business—Material Contracts.” As a result, our ability to generate from sales of, and licensing agreements involving, ZYNLONTA is reduced, which could adversely affect our financial condition.
In addition, upon the occurrence of a change in control event, we are obligated to pay HCR an amount equal to 2.50 times the amount paid by HCR under the HCR Agreement, or at 2.25 times the amount paid by HCR under the agreement if HCR receives royalty payments exceeding a mid-nine-digit amount on or prior to March 31, 2029, less any amounts we previously paid to HCR. The foregoing provision may make us a less attractive acquisition target by reducing the benefit accruing to our shareholders in any change-of-control transaction.
Our consolidated statements of operations are subject to considerable non-cash charges and volatility due to factors that may be beyond our control.
The warrants that we have issued to Deerfield Partners, L.P. and Deerfield Private Design Fund IV, L.P. (the “Deerfield Warrants”) are presented in the audited consolidated balance sheets as a liability, which is remeasured to fair value at each reporting date. The fair value changes based on our share price and its expected volatility. Our obligation under the HCR Agreement is accounted for as a short-term and long-term debt obligation. To determine the accretion of the liability, we are required to estimate the total amount of future royalty payments and estimated timing of such payment to HCR based on our revenue projections as well as the achievement of certain milestones. Based on our periodic review, the amount and timing of repayment is likely to be different at each reporting period. To the extent the amount or timing of such payments is materially different than our initial estimates, we will record a cumulative catch-up adjustment. As a result, our Deerfield Warrants and obligations under the HCR Agreement could result in considerable non-cash charges to, and significant volatility in, our consolidated statements of operations.
Our ability to use tax loss carryforwards may be limited.
As of December 31, 2024, we reported $1,059 million in tax loss carryforwards for Swiss corporate income tax purposes. Such tax loss carryforwards and tax credits could, with certain limitations, be used to offset future taxable income. Swiss tax loss carryforwards generally expire seven years after the tax year in which they were incurred; U.S. federal and state tax credits generally expire after 20 years, although some state tax credits expire as quickly as seven years after the tax year in which they were incurred, and others do not expire. There can be no assurance that we will be able to generate sufficient income that allows us to use such tax loss carryforwards or tax credits before their expiration. We recognize U.S. federal and state credits in our consolidated financial statements based on our assessment of the value that we will be able to realize; however, such assessments are based on our projections of our future taxable income, which are subject to uncertainty and change based on numerous factors, including those described in this “Item 1A. Risk Factors” section. In addition, relevant tax authorities may not accept our claims of tax loss carryforwards or tax credits. Furthermore, changes in tax law, as well as interpretation of such tax laws, could reduce, eliminate, or otherwise impair our ability to use our tax loss carryforwards and U.S. federal and state tax credits.
Our inability to maintain our corporate tax structure may adversely impact our results of operations and financial condition.
We are subject to corporate taxation in Switzerland. We are also subject to taxation in other jurisdictions, in particular, the United States and the United Kingdom, where our two wholly-owned subsidiaries operate. We have entered into intercompany operating and transfer pricing arrangements among our various corporate entities. Changes in our intercompany arrangements, operational structure or practices, or in U.S. or foreign tax laws, regulations or rulings by U.S. tax authorities, could materially increase our tax liabilities and adversely affect our financial condition, results of operations and cash flows.
Exchange rate fluctuations may materially affect our results of operations and financial condition.
We operate internationally and are exposed to fluctuations in foreign exchange rates between the U.S. dollar and other currencies, particularly the British pound, the Euro and the Swiss franc. Our reporting currency is the U.S. dollar and, as a result, financial line items are converted into U.S. dollars at the applicable foreign exchange rates. As our business grows, we expect that at least some of our revenues and expenses will continue to be denominated in currencies other than the U.S. dollar. Therefore, unfavorable developments in the value of the U.S. dollar relative to other relevant currencies could adversely affect our business and financial condition.
We believe that we were a passive foreign investment company for U.S. federal income tax purposes for the 2023 taxable year, which could result in adverse U.S. tax consequences to certain U.S. investors.
Under the Internal Revenue Code of 1986, as amended (the “Code”), we will be a passive foreign investment company (“PFIC”), for any taxable year in which, after the application of certain look-through rules with respect to subsidiaries, either (i) 75% or more of our gross income consists of “passive income” or (ii) 50% or more of the average quarterly value of our assets consists of assets that produce, or are held for the production of, “passive income” (including cash). Passive income generally includes interest, dividends, certain non-active rents and royalties, and capital gains.
Cash is generally characterized as a passive asset for these purposes. Goodwill is generally characterized as a non-passive or passive asset based on the nature of the income produced in the activity to which the goodwill is attributable. The extent to which our goodwill should be characterized as a non-passive asset is not entirely clear. We hold a substantial amount of cash, and while this continues to be the case, our PFIC status for any taxable year depends largely on the value of our goodwill and the characterization of our goodwill as passive or non-passive. The value of our goodwill for any taxable year may be determined in large part by reference to the average of our market capitalization for that year. Because our market capitalization declined substantially during 2023, we believe we were a PFIC for our 2023 taxable year. We do not believe we were a PFIC for 2024, however there is a risk that we could be a PFIC in 2025 and possibly future taxable years. We have not obtained any valuation of our assets (including goodwill). A beneficial owner of our common shares who, for U.S. federal income tax purposes, is eligible for the benefits of the income tax treaty between Switzerland and the United States (the “Treaty”) and who is (i) a citizen or individual resident of the United States, (ii) a corporation, or other entity taxable as a corporation, created or organized in or under the laws of the United States, any state therein or the District of Columbia or (iii) an estate or trust the income of which is subject to U.S. federal income taxation regardless of its source (each, a “Holder”), should consult their tax advisers regarding the value and characterization of our assets for purposes of the PFIC rules, as they are subject to some uncertainties. In addition, our PFIC status is a factual annual determination that can be made only after the end of the relevant taxable year and will depend on the composition of our income and assets and the value of our assets from time to time. Accordingly, our PFIC status for any future taxable year is uncertain.
If we are a PFIC for any taxable year during which a U.S. Holder holds common shares, we generally will continue to be treated as a PFIC with respect to that U.S. Holder for all succeeding years during which the U.S. Holder holds common shares, even if we cease to meet the threshold requirements for PFIC status. Such a U.S. Holder may be subject to adverse U.S. federal income tax consequences, including (i) the treatment of all or a portion of any gain on disposition as ordinary income; (ii) the application of a deferred interest charge on such gain and the receipt of certain dividends; and (iii) compliance with certain reporting requirements. A “qualified electing fund” (“QEF”) election or, if our common shares are regularly traded on a qualified exchange, a “mark-to-market” election may be available that will alter the consequences of PFIC status.
Because we believe we were a PFIC for the 2023 taxable year, we have provided information necessary for our U.S. Holders to make a QEF election with respect to us for the 2023 taxable year and expect to provide such information for any subsequent year if we believe we are a PFIC, but there is no assurance that we will timely provide this information. There is also no assurance that we will have timely knowledge of our status as a PFIC in the future or of the information that a U.S. Holder would need in order to make a valid election. Any such information will be provided on our website.
Risks Related to Research and Development
We may expend our resources to pursue particular products or product candidates and fail to capitalize on those that may be more profitable or for which there is a greater likelihood of success.
Because we have limited financial resources and personnel, we may prioritize the research, development and commercialization of select products, product candidates and technologies and of products, product candidates and technologies in select indications or markets. As a result, we may forgo or delay the pursuit of other products, product candidates and technologies or of other indications and markets that later prove to have greater commercial potential. Decision-making about development and commercialization priorities involves inherent subjectivity and uncertainty, and there can be no assurance that we will pursue product candidates and technologies with the greatest likelihood of obtaining regulatory approval or products, product candidates and technologies with the greatest market potential. In addition, we may relinquish valuable rights to products, product candidates and technologies through partnering, licensing or other arrangements in cases in which it would have been more advantageous for us to retain sole development and commercialization rights to such products, product candidates and technologies.
We may be unable to complete clinical trials on our expected timelines, if at all.
Clinical trials are subject to the numerous risks described in this “Item 1A. Risk Factors” section and in our other filings with the SEC, and a failure, delay or termination of one or more clinical trials can occur at any stage of the clinical trial process. Events that could impede our ability to complete clinical trials on a timely basis include but are not limited to:
•delays in the timely commencement of clinical trials due to negative preclinical data, delays in receiving the required regulatory clearance from the appropriate regulatory authorities, delays in reaching an agreement on acceptable terms with prospective clinical research organizations (“CROs”) and clinical trial sites and difficulties in obtaining required IRB or ethics committee approval at each clinical trial site;
•challenges in recruiting and enrolling suitable patients that meet the study criteria to participate in clinical trials, which challenges may be heightened for clinical trials that seek to enroll patients with characteristics that are found in a small population and by the novel nature of our products and product candidates;
•competition from alternative clinical trials in a similar space or new treatments in similar indications which may limit or ability to recruit and enroll new subjects;
•difficulties in retaining and following up with subjects and subsequent censoring of patients;
•any failure by us or CROs, CMOs, and other third parties to adhere to applicable requirements, which risk may be heightened by our reliance on third parties, and which may result in delays, trial suspension or the imposition of clinical holds;
•safety issues, including occurrence of TEAEs and SAEs, which may result in trial suspension or the imposition of clinical holds;
•the inability to manufacture adequate quantities of a product or a product candidate or other materials necessary in accordance with cGMPs to conduct clinical trials, including, for example, quality issues and delays in the testing, validation, manufacturing delays or failures at our CROs and delivery of the product or product candidate to the clinical trial sites;
•the ability to obtain on a timely basis and on commercially reasonable terms an adequate supply of products or product candidates to be used in combination with our products and product candidates;
•changes in regulatory requirements and guidance;
•changes in the treatment landscape, such as new therapies or the withdrawal of a competing product; and
•lack of adequate funding to continue the clinical trial.
Any delays in the completion of clinical trials could increase costs, delay or prevent regulatory approval of our product candidates and impair our ability to maintain regulatory approval of and to commercialize any approved products.
There can be no assurance regarding the outcome of ongoing or planned clinical trials or the sufficiency of results from such clinical trials.
Drug research and clinical trials are inherently uncertain. There can be no assurance regarding the outcome of any ongoing or planned clinical trials, including whether such trials will meet their respective endpoints, whether severe adverse events will occur during the trials and whether the final results will ultimately be sufficient to support or maintain regulatory approval. For example, we are conducting a confirmatory Phase 3 trial of ZYNLONTA in combination with rituximab for the treatment of relapsed or refractory DLBCL. Despite ZYNLONTA having received accelerated approval from the FDA and conditional approval from the EMA and UK MHRA, ZYNLONTA may fail to achieve its endpoints in this clinical trial, which could result in our inability to maintain regulatory approval. Results from earlier-stage clinical trials are even more unpredictable due to the limited size of the clinical trials and number of unknown factors at such early stages.
Results from preclinical studies and early-stage clinical trials of a product candidate may not be predictive of results from late-stage clinical trials of that product candidate or of any other product or product candidate. In the past, despite promising results from preclinical studies and early-stage clinical trials, we have discontinued development of product candidates due to the results from late-stage clinical trials. In addition, positive and promising results from preclinical studies and clinical trials of a product or product candidate in one indication may not be predictive of results from clinical trials of that product or product candidate in other indications or in combination with other agents. There may be significant differences between clinical trials, including differences in inclusion and exclusion criteria, efficacy endpoints, dosing regimen and statistical design. For example, results from the pivotal Phase 2 clinical trial of ZYNLONTA for the treatment of relapsed or refractory DLBCL, or any other clinical trial of ZYNLONTA, may not be predictive of results from other clinical trials of ZYNLONTA, such as the confirmatory Phase 3 clinical trial, particularly those in which ZYNLONTA is used in combination with other agents and those involving different patient populations, such as the Phase 1 LOTIS-7 trial. If the results of our confirmatory trial for ZYNLONTA or the additional trials for ZYNLONTA in other indications do not meet their primary endpoints, then we may be unable to maintain regulatory approval for ZYNLONTA or obtain regulatory approval for expanded or new indications for ZYNLONTA.
Failure to maintain or obtain regulatory approval for ZYNLONTA could have an adverse impact on our ability to continue to generate and grow our revenue in the future.
From time to time, we may announce or publish preliminary data, such as we had done for the LOTIS-7 trial, but such data may not be predictive of future results for the next phase of the clinical program and are subject to the risk that one or more of the outcomes may materially change as more data become available. We also make assumptions, estimations, calculations and conclusions as part of our analyses of data, and we may not have received or had the opportunity to fully evaluate all data. Therefore, positive preliminary results in any ongoing clinical trial may not be predictive of results in the completed trial. Preliminary data also remain subject to audit and verification procedures that may result in the final data being materially different from the preliminary data we previously published. As a result, preliminary data should be viewed with caution until the final data are available.
Our products and product candidates may cause undesirable side effects or adverse events.
Undesirable side effects or adverse events caused by our products or product candidates could cause us or regulatory authorities to interrupt, delay or halt clinical trials, result in more restrictive labeling, boxed warnings, REMS or the denial or withdrawal of regulatory approval by the FDA, the EMA or other regulatory authorities, subject us to product liability claims or require us to issue product recalls. In addition, undesirable side effects or adverse events could impair our ability to market our products, limit patients’ and physicians’ willingness to use our products and make it more difficult for us to obtain adequate coverage and reimbursement for our products.
In our clinical trials, we have observed certain class toxicities associated with our warheads, including elevated liver enzymes, skin rash, and effusions and edema. The prescribing information for ZYNLONTA contains warnings and precautions for effusion and edema, myelosuppression, infections, cutaneous reactions and embryo-fetal toxicity.
Such information is based on adverse events observed in our clinical trials and post-marketing information. However, clinical trials by their nature utilize a sample of the potential patient population. With a limited number of subjects and limited duration of exposure, rare and severe side effects of our products or product candidates may only be uncovered with a significantly larger number of patients exposed to the drug. Therefore, there can be no assurance that ZYNLONTA will not cause side effects that are different or more severe in a greater proportion of patients when used by more patients as we commercialize the product. Similarly, as our other product candidates advance through late-stage clinical trials that involve more patients than earlier-stage clinical trials, these product candidates may cause side effects or adverse events that are different in nature, severity and frequency than observed in earlier-stage clinical trials.
We are also developing ZYNLONTA and certain of our product candidates in combination with other therapies, such as rituximab and bispecific antibodies. Combining therapies may cause additional, different or more severe side effects or adverse events than when a drug is used as a monotherapy. In addition, therapies used in combination may have common toxicities. When used in combination, the severity and frequency of such undesirable side effects or adverse events may be greater than the cumulative severity and frequency of such side effects or adverse events when the therapies are used as monotherapies.
In addition to our trials, there are several IITs studying Zynlonta, either as a monotherapy or in combination, and any new side effects or adverse events observed in these trials that are different in nature, severity and frequency than observed in earlier trials may require a change in our current approved labeling, impact ongoing trials, or impact our ability to maintain BLA approval for Zynlonta.
We may not be successful in our efforts to expand the market opportunity of ZYNLONTA, develop additional product candidates or build up our research pipeline.
ZYNLONTA is currently approved for the treatment of adult patients with relapsed or refractory large B-cell lymphoma after two or more lines of systemic therapy, including DLBCL not otherwise specified, DLBCL arising from low-grade lymphoma, and also high-grade B-cell lymphoma. We are undertaking clinical trials to potentially expand ZYNLONTA into other indications and into earlier lines of therapy. However, clinical development and regulatory review is inherently unpredictable and are subject to numerous risks and uncertainties described in this “Item 1A. Risk Factors” section. Failure to expand the indication(s) for ZYNLONTA could limit the market opportunity for ZYNLONTA and our potential future revenue which could have an adverse effect on our business and operations. There can be no assurance that we will succeed in expanding the market opportunity of ZYNLONTA.
A key element of our development strategy is to build a robust pipeline of ADCs targeting both novel and clinically validated cancer targets using a variety of technologies for the treatment of hematological malignancies and solid tumors. There can be no assurance that we will be able to identify suitable additional product candidates for clinical development or that our research and development efforts will yield safe, effective and commercially viable product candidates. If we are not successful in developing these new drugs, our future market opportunity and potential revenue may be negatively impacted, which could adversely impact our business and operations.
We do not control the conduct of current or any potential future investigator-initiated clinical trials, and the data from such trials is not subject to our review or quality control.
We have provided and may continue to provide clinical data from an investigator-initiated clinical trials (“IIT”). We do not control the design or administration of such trials, nor the submission, approval or maintenance of any regulatory and institutional filings required to conduct such trials. Furthermore, we have limited or no rights to audit, review or apply quality control procedures to the clinical data generated from such trials. As a result, we have no control over the conduct of such trials and the timing of any data releases from such trials and we cannot be certain that such trials are or will be conducted in accordance with applicable regulatory requirements or that the clinical data provided to us by the investigators of such trials are accurate, reliable or complete. There can be no assurance regarding the outcome or timing of any IITs, including whether such trials will meet their respective endpoint and whether severe adverse events will occur during the trials. In addition, positive preliminary results in any ongoing IIT may not be predictive of results in the completed trial.
Risks Related to Regulatory Approval and Government Regulation
We may be unable to obtain, or experience delays in obtaining, regulatory approval for our product candidates.
Our product candidates must be approved by the FDA in the United States, by the EMA in the European Union and by comparable regulatory authorities in other jurisdictions prior to commercialization. In order to obtain regulatory approval for the commercial sale of any product candidates, we must demonstrate through extensive preclinical studies and clinical trials that the product candidate is safe and effective for use in each target indication and that manufacturing of the product candidate is safe, robust and reproducible. The time and resources required to obtain regulatory approval is unpredictable, typically takes many years and significant investment following the commencement of clinical trials and depends upon numerous factors.
Regulatory authorities have substantial discretion in the approval process. They may refuse to accept any application or may decide that our data are insufficient for approval and require additional clinical trials or other studies. In this Annual Report and elsewhere in our public communications, we designate certain of our clinical trials as “pivotal” if we believe that these clinical trials, if successful, will support BLA submissions; however, there can be no assurance that any clinical trial that we designate as “pivotal” will be viewed as sufficient by the FDA, the EMA and other comparable regulatory authorities in other jurisdictions to support regulatory approval. If we are required to conduct additional clinical trials or other testing of any of our products and product candidates beyond those that are contemplated, we may incur significant additional costs and regulatory approval may be delayed or prevented.
Various regulatory programs in the United States, such as Breakthrough Therapy Designation, Fast Track Designation or Priority Review Designation, are designed to expedite the development and review of therapies to treat certain diseases. We may seek such designations, and comparable designations by foreign regulatory authorities, for one or more of our product candidates for the treatment of certain indications. However, regulatory authorities have broad discretion whether or not to grant such designations, and the receipt of such designations may not result in faster development, review or approval and does not guarantee regulatory approval.
We are developing certain of our products and product candidates in combination with other therapies. If we choose to develop a product or product candidate for use in combination with an approved therapy, we are subject to the risk that the FDA, the EMA or comparable regulatory authorities in other jurisdictions could revoke approval of, or that safety, efficacy, manufacturing or supply issues could arise with, the therapy used in combination with our product or product candidate. If the therapies we use in combination with our products and product candidates are replaced as the standard of care, the FDA, the EMA or comparable regulatory authorities in other jurisdictions may require us to conduct additional clinical trials. The occurrence of any of these risks could result in our products, if approved only for use in combination with another approved therapy, being removed from the market or being less successful commercially. Where we develop a product or product candidate for use in combination with a therapy that has not been approved by the FDA, the EMA or comparable regulatory authorities in other jurisdictions, we may not be able to market our product or product candidate for use in combination with such an unapproved therapy, unless and until the unapproved therapy receives regulatory approval.
Unapproved therapies face the same risks described with respect to our product candidates currently in development. In addition, other companies may also develop their products or product candidates in combination with the unapproved therapies with which we are developing our products and product candidates for use in combination. Any setbacks in these companies’ clinical trials, including the emergence of serious adverse effects, may delay or prevent the development and approval of our products and product candidates for use in combination with an approved therapy.
Furthermore, the process and time required to obtain regulatory approval differ by jurisdiction. Approval by one regulatory authority does not ensure approval by regulatory authorities in other jurisdictions. In particular, prior to regulatory approval, regulatory authorities may require additional clinical trials to be conducted with a local population. Moreover, in many countries outside the United States, a drug must be approved for reimbursement before it can be approved for sale in that country, which can take considerable time and be heavily impacted by political, economic and regulatory developments.
In addition, the approval policies or regulations of the FDA, the EMA or comparable regulatory authorities in other jurisdictions may change in a manner rendering our clinical data insufficient for approval. For example, the accelerated approval pathway has come under scrutiny within the FDA and by Congress. The FDA has put increased focus on ensuring that confirmatory studies are conducted with diligence and, ultimately, that such studies confirm the benefit. The Food and Drug Omnibus Reform Act (“FDORA”) included provisions related to the accelerated approval pathway and authorized the FDA to require a post-approval study to be underway prior to approval or within a specified time period following approval. Furthermore, the Oncology Center of Excellence within the FDA is advancing Project Optimus, which is an initiative to reform the dose optimization and dose selection paradigm in oncology drug development to emphasize selection of an optimal dose, which is a dose or doses that maximizes not only the efficacy of a drug but the safety and tolerability as well. This shift from the prior approach, which generally determined the maximum tolerated dose, may require sponsors to spend additional time and resources to further explore a product candidate’s dose-response relationship to facilitate optimum dose selection in a target population. These and other policies of the FDA, the EMA or comparable regulatory authorities in other jurisdictions may increase the time and costs associated with regulatory approval. Other recent Oncology Center of Excellence initiatives have included Project FrontRunner, a new initiative with a goal of developing a framework for identifying candidate drugs for initial clinical development in the earlier advanced setting rather than for treatment of patients who have received numerous prior lines of therapies or have exhausted available treatment options, and Project Equity, which is an initiative to ensure that the data submitted to the FDA for approval of oncology medical products adequately reflects the demographic representation of patients for whom the medical products are intended. More recently, as part of FDORA, sponsors will be required to submit Diversity Action Plans (“DAPs”) for Phase 3 studies or other pivotal studies of new drugs. DAPs must include the sponsor’s goals for enrollment for such studies, disaggregated by age group, sex, and racial and ethnic demographic characteristics of clinically relevant study populations; the sponsor’s rationale for such goals; and an explanation of how the sponsor intends to meet such goals. FDA is scheduled to issue final guidance on DAPs by late June 2025, and these requirements are scheduled to be implemented in late 2025. However, there remains significant uncertainty regarding the final details of these requirements and their impact on development programs.
We may be unable to maintain regulatory approval for any approved products.
As part of regulatory approval, we may be subject to a number of post-marketing requirements and commitments, such as post-marketing studies or clinical trials, surveillance to monitor the safety or efficacy of any approved product and risk evaluation and mitigation strategies. For example, our post-marketing obligations with respect to ZYNLONTA include a confirmatory trial to verify and describe the clinical benefit of ZYNLONTA, a deferred pediatric trial and a trial in patients with hepatic impairment. For ZYNLONTA and for any other products for which we receive accelerated approval from the FDA or conditional approval from the EMA or comparable regulatory authorities in other jurisdictions, we are required to complete confirmatory clinical trials. The FDA may withdraw approval of our products approved under the accelerated approval pathway if, for example, the clinical trial(s) required to verify the predicted clinical benefit of a product fails to verify such benefit or does not demonstrate sufficient clinical benefit to justify the risks associated with the product for any reason, including censoring of patients, other evidence demonstrates that a product is not shown to be safe or effective under the conditions of use, we fail to conduct any required post-marketing confirmatory clinical trial with due diligence or we disseminate false or misleading promotional materials relating to the relevant product. There can be no assurance that we will receive full approval or maintain the current accelerated approval for ZYNLONTA for the treatment of relapsed or refractory DLBCL or that we will receive full approval for ZYNLONTA in other indications or for any of our product candidates for which we receive accelerated approval. In addition, any products for which we receive regulatory approval in a particular jurisdiction and the activities associated with their commercialization, including testing, manufacture, recordkeeping, labeling, storage, approval, advertising, promotion, sale and distribution, will be subject to comprehensive regulation by the FDA, the EMA or comparable regulatory authorities in other jurisdictions.
These requirements include, without limitation, submissions of safety and other post-marketing information and reports, registration and listing requirements, the FDA’s cGMP requirements or comparable requirements in foreign jurisdictions, requirements relating to manufacturing, quality control, quality assurance and corresponding maintenance of records and documents, including periodic inspections by the FDA, the EMA or comparable regulatory authorities in other jurisdictions, requirements regarding the distribution of samples to physicians, tracking and reporting of payments to physicians and other healthcare providers and recordkeeping. If we are unable to complete the required confirmatory or post-marketing studies, if such studies fail to meet their safety and efficacy endpoints or if we otherwise fail to comply with post-marketing requirements and regulations, we may be unable to maintain regulatory approval for any approved products.
The policies of the FDA, the EMA and comparable regulatory authorities in other jurisdictions may change and additional regulations may be enacted. If we are slow or unable to adapt to changes in existing requirements or to the adoption of new requirements, or not able to maintain regulatory compliance, we may lose any regulatory approval that may have been obtained. We cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative action, either in the United States or abroad, as the regulatory environment changes rapidly.
We may not receive Orphan Drug Designation for our product candidates.
Regulatory authorities in some jurisdictions, including the United States and the European Union, may designate drugs for relatively small patient populations as orphan drugs. In the United States, orphan drug designation entitles a party to financial incentives such as tax advantages and user fee waivers and exemptions. In addition, if a product receives the first FDA approval for the condition for which it has orphan designation, the product is entitled to orphan drug exclusivity, which means the FDA may not approve any other application to market the same drug for the same condition for a period of seven years, except in limited circumstances, such as a showing of clinical superiority over the product with orphan exclusivity or where the manufacturer is unable to assure sufficient product quantity. In the European Union, orphan drug designation entitles a party to financial incentives such as reduction of fees or fee waivers and ten years of market exclusivity for the orphan indication following drug or biological product approval, provided that the criteria for orphan designation are still applicable at the time of the granting of the marketing authorization. This period may be reduced to six years if, at the end of the fifth year, the orphan drug designation criteria are no longer met, including where it is shown that the product is sufficiently profitable not to justify maintenance of market exclusivity. The respective orphan drug designation and exclusivity frameworks in the United States and in the European Union are subject to change, and any such changes may affect our ability to obtain, or the impact of obtaining, European Union or U.S. orphan designations in the future.
We may pursue orphan drug designation for one or more of our other product candidates. However, obtaining an orphan drug designation can be difficult, and we may not be successful in doing so. Even if we obtain orphan drug designation, we may not be able to maintain such designation. For example, in the process of seeking marketing authorization in the European Union, the Committee for Orphan Medicinal Products recommended to not uphold ZYNLONTA’s previously granted orphan drug designation. Orphan drug designation neither shortens the development time or regulatory review time of a product candidate nor gives the product candidate any advantage in the regulatory review or approval process. Even if we obtain orphan drug designation for our product candidates in specific conditions, we may not be the first to obtain regulatory approval of these product candidates for the orphan-designated condition and therefore we may not be eligible for orphan drug exclusivity in the U.S. In addition, exclusive marketing rights in the United States may not be awarded if we seek approval for an indication broader than the orphan-designated condition or, if awarded, may be lost if the FDA later determines that the request for designation was materially defective or if the manufacturer is unable to assure sufficient quantities of the product to meet the needs of patients with the rare disease or condition. Furthermore, even if we obtain orphan drug exclusivity for a product, that exclusivity may not effectively protect the product from competition because different ADCs with different monoclonal antibody elements or functional elements of the conjugated molecule can be approved for the same condition. Even after an orphan product is approved, the FDA can subsequently approve the same ADC with the same monoclonal antibody element and functional element of the conjugated molecule for the same condition if the FDA concludes that the later ADC is safer, more effective or makes a major contribution to patient care. Our inability to obtain orphan drug designation for any product candidates for the treatment of rare cancers and/or our inability to maintain that designation for the duration of the applicable exclusivity period, could reduce our ability to make sufficient sales of the applicable product candidate to balance our expenses incurred to develop it.
We may not receive the 12 years of data exclusivity from our anticipated Reference Product Exclusivity or data exclusivity in other jurisdictions.
We believe ZYNLONTA is the first loncastuximab tesirine product to have been licensed by the FDA and should be entitled to a period of 12 years of Reference Product Exclusivity (“RPE”). However, the FDA has not yet awarded ZYNLONTA such RPE, and the FDA may not do so for unknown reasons.
The Biologics Price Competition and Innovation Act of 2009 (the “BPCIA”) established an abbreviated pathway to licensure for follow-on biologics called biosimilars. Biosimilars are biological products approved under section 351(k) of the Public Health Act Service Act (“PHS Act”) relying on the FDA’s findings of safety, purity, and potency for a licensed biologic (“Reference Product”) submitted pursuant to section 351(k) of the PHS Act. A biosimilar is highly similar to its Reference Product, excluding minor differences in clinically inactive components for which there are no clinically meaningful differences between the proposed biological product and the Reference Product in safety, purity, or potency. Certain biosimilars may be substituted for the Reference Product in accordance with state law.
The BPCIA provides a 12-year period of RPE during which the FDA may not license a biosimilar application relying on the Reference Product; the applicant may not submit a biosimilar application relying on the Reference Product for the first 4 years of the 12-year RPE period. That RPE runs from the “date of first licensure,” which is the date that the FDA first licensed the Reference Product, and when such a period of RPE is awarded to a given Reference Product, it is listed in the FDA’s Database of Licensed Biological Products (the “Purple Book”) as a “Date of First Licensure.” The FDA historically has been slow to make these determinations and often does not do so until there is a biosimilar application pending.
RPE is available unless the putative Reference Product falls under one of several exclusions. Specifically, RPE is not available where licensure is for a supplement for the putative Reference Product or where the licensure is for a subsequent application filed by the same sponsor or manufacturer of the biological product for a change other than a modification to the structure of the biological product that results in a change in safety, purity, and potency. The “same sponsor” includes any licensor, predecessor in interest, or other related entity. For each putative Reference Product, the FDA assesses whether an application is considered a subsequent application filed by the same sponsor or manufacturer of the biological product and whether there is a modification to the structure of the biological product previously licensed by such an entity. If there is a structural modification, the FDA then determines whether such modification would result in a change in safety, purity, or potency.
ZYNLONTA is listed in the Purple Book, but the FDA has not yet listed a Date of First Licensure. Accordingly, it is unclear whether the FDA will award ZYNLONTA its 12 years of RPE. While we are not aware of any disqualifying factors, the FDA could determine that ZYNLONTA is not entitled to RPE if it determines that an entity related to us received licensure of a similar molecule in the past.
Even if ZYNLONTA does receive its 12 years of exclusivity, the value of RPE is limited. As data exclusivity, RPE would not preclude subsequent licensure of a similar or related product unless the application sought to rely on the FDA’s findings of safety, purity, and potency for ZYNLONTA in a biosimilar application filed pursuant to Section 351(k) of the PHS Act. Accordingly, the FDA could approve an identical loncastuximab tesirine product with full studies demonstrating safety, purity, and potency submitted under section 351(a) of the PHS Act. The FDA could also approve loncastuximab tesirine for a different indication or with a different route of administration or formulation despite any RPE for ZYNLONTA.
If we are found to have improperly promoted off-label use of our products, we may become subject to significant liability.
The FDA, the EMA and comparable regulatory authorities in other jurisdictions strictly regulate the promotional claims that may be made about prescription drug products, such as our products. While physicians, in the practice of medicine, may prescribe approved drugs for unapproved indications, a product may not be promoted for uses that are not approved by the applicable regulatory authority as reflected in the product’s approved labeling or for uses inconsistent with the product’s approved labeling. For example, despite ZYNLONTA being approved for the treatment of adult patients with relapsed or refractory large B-cell lymphoma after two or more lines of systemic therapy, including DLBCL not otherwise specified, DLBCL arising from low grade lymphoma and high-grade B-cell lymphoma, if our promotional materials and related activities are not consistent with the approved labeling or if physicians, in their professional medical judgment, nevertheless prescribe the drug product to their patients in a manner that is inconsistent with the approved labeling, we may be subject to claims that we promoted off-label use or otherwise violated applicable regulations. In addition, although we believe our warhead may provide for superior efficacy as compared to marketed ADCs, without head-to-head data, we will be unable to make comparative claims for our products. If we are found to have promoted such off-label use or made such unsubstantiated comparative claims, we may become subject to significant liability under the Federal Food, Drug, and Cosmetic Act (the “FDCA”) and other statutory authorities, such as laws prohibiting false claims for reimbursement.
Failure to comply with health and data protection laws and regulations could lead to government enforcement actions, private litigation and adverse publicity and could negatively affect our operating results and business.
We receive, generate and store significant and increasing volumes of sensitive information, such as employee and patient data. In addition, we actively seek access to medical information, including patient data, through research and development collaborations or otherwise. We and any potential collaborators may be subject to federal, state, local and foreign laws and regulations that apply to the collection, use, retention, protection, disclosure, transfer and other processing of personal data, including the Health Insurance Portability and Accountability Act of 1996 (“HIPAA”), as amended by the Health Information Technology for Economic and Clinical Health Act of 2009 (“HITECH”), the Regulation 2016/679, known as the General Data Protection Regulation (the “GDPR”), as well as European Union member state implementing legislations, the UK General Data Protection Regulation (“UK GDPR”) and the Swiss Federal Act on Data Protection. In addition, many state laws govern the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and may not have the same effect, and often are not pre-empted by HIPAA. For example, the California Consumer Privacy Act of 2018 (“CCPA”) as amended by the California Privacy Rights Act of 2020 (“CPRA”) on January 1, 2023, imposes obligations on businesses to which it applies, including, but not limited to, providing specific disclosures in privacy notices, affording California residents certain rights related to their personal data, although it exempts some data processed in the context of clinical trials, expanding consumers’ rights with respect to certain sensitive personal information, and creating a new state agency that is vested with authority to implement and enforce the CCPA and CPRA. Virginia’s Consumer Data Protection Act, which took effect on January 1, 2023, requires businesses subject to the legislation to conduct data protection assessments in certain circumstances and requires opt-in consent from consumers to acquire and process their sensitive personal information, which includes information revealing a consumer’s physical and mental health diagnosis and genetic and biometric information that can identify a consumer. In addition, Colorado enacted the Colorado Privacy Act, and Connecticut enacted the Connecticut Data Privacy Act, each of which took effect on July 1, 2023, and Utah enacted the Consumer Privacy Act, which became effective on December 31, 2023, and each of these laws may increase the complexity, variation in requirements, restrictions and potential legal risks, and could require increased compliance costs and changes in business practices and policies. Other states have also enacted, proposed, or are considering proposing, data privacy laws, which could further complicate compliance efforts, increase our potential liability and adversely affect our business. These laws and regulations are complex and change frequently, at times due to changes in political climate, and existing laws and regulations are subject to different and conflicting interpretations, which adds to the complexity of processing personal data from these jurisdictions. Compliance with U.S. and international data protection laws and regulations could require us to take on more onerous obligations in our contracts, restrict our ability to collect, use and disclose data, or in some cases, impact our ability to operate in certain jurisdictions. Failure to comply with these laws and regulations could result in government enforcement actions, which could include civil, criminal and administrative penalties, private litigation, and adverse publicity and could negatively affect our operating results and business. Moreover, clinical trial subjects, employees and other individuals about whom we or our potential collaborators obtain personal information, as well as the providers who share this information with us, may limit our ability to collect, use and disclose the information. Claims that we have violated individuals’ privacy rights, failed to comply with data protection laws, or breached our contractual obligations, even if we are not found liable, could be expensive and time-consuming to defend and could result in adverse publicity that could harm our business.
If we are unable to comply, or do not fully comply, with applicable fraud and abuse, transparency, government price reporting, privacy and security, and other healthcare laws, we could face substantial penalties.
Healthcare providers, physicians and third-party payors will play a primary role in the recommendation and prescription of our products for which we obtain marketing approval. Our operations, including any arrangements with healthcare providers, physicians, third-party payors and customers may expose us to broadly applicable fraud and abuse and other healthcare laws that may affect the business or financial arrangements and relationships through which we would market, sell and distribute our products. The healthcare laws that may affect our ability to operate include, but are not limited to:
•The federal Anti-Kickback Statute, which prohibits any person or entity from, among other things, knowingly and willfully soliciting, receiving, offering or paying any remuneration, directly or indirectly, overtly or covertly, in cash or in kind, to induce or reward either the referral of an individual for, or the purchase, order or recommendation of an item or service reimbursable, in whole or in part, under a federal healthcare program, such as the Medicare and Medicaid programs. The term “remuneration” has been broadly interpreted to include anything of value. The federal Anti-Kickback Statute has also been interpreted to apply to arrangements between pharmaceutical manufacturers, on the one hand, and prescribers, purchasers, and formulary managers, on the other hand. There are a number of statutory exceptions and regulatory safe harbors protecting some common activities from prosecution, but the exceptions and safe harbors are drawn narrowly and require strict compliance in order to offer protection.
•Federal civil and criminal false claims laws, such as the False Claims Act (“FCA”), which can be enforced by private citizens through civil qui tam actions, and the Civil Monetary Penalties Law prohibit individuals or entities from, among other things, knowingly presenting, or causing to be presented, false, fictitious or fraudulent claims for payment of federal funds, and knowingly making, using or causing to be made or used a false record or statement material to a false or fraudulent claim to avoid, decrease or conceal an obligation to pay money to the federal government. For example, pharmaceutical companies have been prosecuted under the FCA in connection with their alleged off-label promotion of drugs, purportedly concealing price concessions in the pricing information submitted to the government for government price reporting purposes, and allegedly providing free product to customers with the expectation that the customers would bill federal healthcare programs for the product. In addition, a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the FCA. As a result of a modification made by the Fraud Enforcement and Recovery Act of 2009, a claim includes “any request or demand” for money or property presented to the U.S. government. In addition, manufacturers can be held liable under the FCA even when they do not submit claims directly to government payors if they are deemed to “cause” the submission of false or fraudulent claims.
•HIPAA, which, among other things, imposes criminal liability for executing or attempting to execute a scheme to defraud any healthcare benefit program, including private third-party payors, knowingly and willfully embezzling or stealing from a healthcare benefit program, or willfully obstructing a criminal investigation of a healthcare offense, and creates federal criminal laws that prohibit knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement or representation, or making or using any false writing or document knowing the same to contain any materially false, fictitious or fraudulent statement or entry in connection with the delivery of or payment for healthcare benefits, items or services.
•HIPAA, as amended by HITECH, and its implementing regulations, which impose privacy, security and breach reporting obligations with respect to individually identifiable health information upon entities subject to the law, such as health plans, healthcare clearinghouses and certain healthcare providers, known as covered entities, and their respective business associates and covered subcontractors that perform services for them that involve individually identifiable health information. HITECH also created new tiers of civil monetary penalties, amended HIPAA to make civil and criminal penalties directly applicable to business associates, and gave state attorneys general new authority to file civil actions for damages or injunctions in U.S. federal courts to enforce HIPAA laws and seek attorneys’ fees and costs associated with pursuing federal civil actions.
•Federal and state consumer protection and unfair competition laws, which broadly regulate marketplace activities and activities that potentially harm consumers.
•The federal transparency requirements under the Physician Payments Sunshine Act, which requires, among other things, certain manufacturers of drugs, devices, biologics and medical supplies reimbursed under Medicare, Medicaid, or the Children’s Health Insurance Program to report annually to the Centers for Medicare & Medicaid Services (the “CMS”) information related to payments and other transfers of value provided to physicians, as defined by such law, certain other healthcare professionals, and teaching hospitals and physician ownership and investment interests, including such ownership and investment interests held by a physician’s immediate family members.
•State and foreign laws that are analogous to each of the above federal laws, such as anti-kickback and false claims laws, that may impose similar or more prohibitive restrictions, and may apply to items or services reimbursed by non-governmental third-party payors, including private insurers.
•State and foreign laws that require pharmaceutical companies to implement compliance programs, comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government, or to track and report gifts, compensation and other remuneration provided to physicians and other healthcare providers; state laws that require the reporting of marketing expenditures or drug pricing, including information pertaining to and justifying price increases; state and local laws that require the registration of pharmaceutical sales representatives; state laws that prohibit various marketing-related activities, such as the provision of certain kinds of gifts or meals; state laws that require the posting of information relating to clinical trials and their outcomes.
Ensuring that our operations and business arrangements with third parties comply with applicable healthcare laws and regulations is costly.
If our operations are found to be in violation of any of these laws or any other current or future healthcare laws that may apply to us, we may be subject to significant civil, criminal and administrative penalties, damages, fines, disgorgement, imprisonment, exclusion from government funded healthcare programs, such as Medicare and Medicaid, contractual damages, reputational harm, diminished profits and future earnings, additional reporting obligations and oversight if we become subject to a corporate integrity agreement or other agreement to resolve allegations of non-compliance with these laws, and the curtailment or restructuring of our operations, any of which could substantially disrupt our operations. Although effective compliance programs can mitigate the risk of investigation and prosecution for violations of these laws, these risks cannot be entirely eliminated. Any action against us for an alleged or suspected violation could cause us to incur significant legal expenses and could divert our management’s attention from the operation of our business, even if our defense is successful.
Healthcare reform legislation and other changes in the healthcare industry and in healthcare spending may adversely affect our business model.
Our revenues and revenue prospects could be affected by changes in healthcare spending and policies in the United States, the European Union and any other potential jurisdictions in which we or our collaborators may seek to commercialize our products. We operate in a highly regulated industry, and new laws, regulations and judicial decisions, or new interpretations of existing laws, regulations and decisions, related to healthcare availability, the method of delivery and payment for healthcare products and services could negatively affect our business, financial condition and prospects. There is significant interest in promoting healthcare reforms, and it is likely that federal and state legislatures within the United States and the governments of other countries will continue to consider changes to existing healthcare legislation. For example, there have been and continue to be a number of initiatives at the United States federal and state levels that seek to reduce healthcare and prescription drug costs, including the Budget Control Act (which, subject to certain sequestration periods, imposed 2% reductions in Medicare payments to providers per fiscal year) and the Infrastructure Investment and Jobs Act (which added a requirement for manufacturers of certain single-source drugs (including biologics and biosimilars) separately paid for under Medicare Part B for at least 18 months and marketed in single-dose containers or packages (known as refundable single-dose container or single-use package drugs), such as ZYNLONTA, to provide annual refunds for any portions of the dispensed drug that are unused and discarded if those unused or discarded portions exceed an applicable percentage defined by statute or regulation) which requirement has caused and we expect will continue to cause a significant adverse effect on ZYNLONTA net sales and thus our results of operations.
These initiatives culminated in the enactment of the Inflation Reduction Act (“IRA”), which, among other things, allows the U.S. Department of Health and Human Services (“HHS”) to directly negotiate the selling price of a statutorily specified number of drugs and biologics each year that the Centers for Medicare & Medicaid Services (“CMS”) reimburses under Medicare Part B and Part D. The negotiated price may not exceed a statutory ceiling price. Only high-expenditure single-source biologics that have been approved for at least 11 years (7 years for single-source drugs) can be selected by CMS for negotiations, with the negotiated price taking effect two years after the selection year. For 2026, the first year in which negotiated prices become effective, CMS selected 10 high-cost Medicare Part D products in 2023, negotiations began in 2024, and the negotiated maximum fair price for each product has been announced. CMS has selected 15 additional Medicare Part D drugs for negotiated maximum fair pricing in 2027. For 2028, an additional 15 drugs, which may be covered under either Medicare Part B or Part D, will be selected, and for 2029 and subsequent years, 20 Part B or Part D drugs will be selected. A drug or biological product that has an orphan drug designation for only one rare disease or condition will be excluded from the IRA’s price negotiations requirements, but loses that exclusion if it has designations for more than one rare disease or condition, or if is approved for an indication that is not within that single designated rare disease or condition, unless such additional designation or such disqualifying approvals are withdrawn by the time CMS evaluates the drug for selection for negotiation. The IRA also penalizes drug manufacturers that increase prices of Medicare Part B and Part D drugs at a rate greater than the rate of inflation, and in November 2024, CMS finalized regulations for these Medicare Part B and Part D inflation rebates. In addition, the IRA requires manufacturers that wish for their drugs to be covered by Medicare Part D to provide statutorily defined discounts to Part D enrollees. The IRA permits the Secretary of HHS to implement many of these provisions through guidance, as opposed to regulation, for the initial years. Manufacturers that fail to comply with the IRA may be subject to various penalties, some significant, including civil monetary penalties. The IRA also extends enhanced subsidies for individuals purchasing health insurance coverage in ACA marketplaces through plan year 2025. These provisions began taking effect progressively in 2023, although they may be subject to legal challenges. Thus, while it is unclear how some provisions of the IRA will be implemented, we are liable for Part B inflation rebates for ZYNLONTA under the IRA, which has and will continue to negatively impact our gross-to-net adjustment for ZYNLONTA sales.
Congress and CMS also continue to increase federal oversight on manufacturers participating in Medicaid and the Medicaid drug rebate program. In September 2024, CMS finalized a rule to implement certain provisions of the Medicaid Services Investment and Accountability Act of 2019 that increases the burden for manufacturers to report drug pricing information without error, and their potential liability if they fail to do so.
For example, CMS can, in certain circumstances, ask a manufacturer to share findings from an internal investigation if the manufacturer seeks to correct a price report submitted more than three years prior. The final rule also expands the circumstances in which CMS may penalize manufacturers for misclassifying a drug in its price reporting and, for the first time, allows CMS to suspend a Medicaid rebate agreement for a manufacturer’s failure to correct a misclassification, or a failure to submit the required pricing reports, within 90 calendar days receiving notice. These changes in the Medicaid program increase the burden for manufacturers to report drug pricing information without error, and their potential liability if they fail to do so. Finally, there is ongoing litigation in federal courts that may change the scope of the 340B program, a drug discount program for charge certain safety net healthcare providers, in incoming years.
Individual states in the United States have also become increasingly active in passing legislation and implementing regulations designed to control pharmaceutical product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing. In addition, regional healthcare authorities and individual hospitals are increasingly using bidding procedures to determine what pharmaceutical products and which suppliers will be included in their prescription drug and other healthcare programs.
We expect that additional state and federal healthcare reform measures will be adopted in the future. Any adopted healthcare reform measure could reduce the ultimate demand for our products or put pressure on our product pricing. It is likely that federal and state legislatures within the United States and foreign governments will continue to consider changes to existing healthcare legislation. We cannot predict the reform initiatives that may be adopted in the future or whether initiatives that have been adopted will be repealed or modified.
Risks Related to Commercialization and Manufacturing
We or our foreign commercialization partners may not be able to successfully commercialize our products.
To successfully commercialize our products, we must attract and retain qualified selling and marketing personnel and attain significant market acceptance of our products. We face significant competition for qualified personnel. See “—We face substantial competition, which may result in others discovering, developing or commercializing products, treatment methods or technologies before or more successfully than we do.” Establishing market acceptance of our products among physicians, patients, patient advocacy groups, third-party payors and the medical community is complex and resource intensive. The risk of our inability to establish market acceptance may be heightened as our products represent novel treatment methods and be influenced by factors beyond our control, including perceptions of ADC products generally or those of our competitors and coverage and reimbursement for our products. Further, changes to our commercialization strategy may result in disruptions to and adverse impacts on our commercialization efforts. If we do not successfully commercialize our products, we may not generate significant product revenues and may not receive a satisfactory return on our investment into the research and development of those products.
Alternatively, we have established collaborations with third parties to commercialize our product. See “Item 1. Business—Material Contracts.” In such collaborations, we depend on the performance of the contractual counterparty, over which we have limited control. Therefore, such collaborations may generate lower product revenues or profit than if we were to commercialize our products ourselves. We may wish to establish additional collaborations with third parties to commercialize our product. We may not be successful in entering into such marketing and distribution arrangements with third parties or in entering in such marketing and distribution arrangements with third parties on favorable terms. Moreover, such arrangements are complex and time-consuming to negotiate, document and implement and they may require substantial resources to maintain.
Coverage and reimbursement may be limited or unavailable for our products.
In both domestic and foreign markets, sales of our products will depend substantially on the extent to which the costs of our products will be covered by third-party payors, such as government health programs, commercial insurance and managed healthcare organizations. These third-party payors decide which products will be covered and establish reimbursement levels for those products. If coverage and adequate reimbursement are not available, or are available only to limited levels, we may not be able to successfully commercialize our products.
Obtaining coverage approval and reimbursement from a government or other third-party payor is a time-consuming and costly process that could require us to provide supporting scientific, clinical and cost-effectiveness data for the use of our products to the payor, which we may be unable to provide. In particular, there is significant uncertainty related to the insurance coverage and reimbursement of newly approved products. In the United States, there is no uniform policy for coverage and reimbursement and, as a result, coverage and reimbursement can differ significantly from payor to payor.
The principal decisions about reimbursement for new medicines are typically made by the CMS, which decides whether and to what extent a new medicine will be covered and reimbursed under Medicare. Private payors often, but not always, follow the CMS’s decisions regarding coverage and reimbursement. Further, coverage policies and third-party payor reimbursement rates may change at any time. It is difficult to predict what third-party payors will decide with respect to coverage and reimbursement for fundamentally novel products such as ours, as there is no body of established practices and precedents for these new products. Further, one payor’s determination to provide coverage and adequate reimbursement for a product does not assure that other payors will also provide coverage and adequate reimbursement for that product. In Europe, pricing and reimbursement schemes may be more restrictive than those in the United States and vary widely from country to country and may require additional clinical trials and additional cost-effectiveness assessments. Many foreign jurisdictions provide nationalized healthcare which may impact the ability to obtain coverage or the amount of reimbursement. In addition, countries may restrict the price of products through the use of nationalized tender processes, controls on the profitability of drug companies, guidance to physicians to limit prescriptions, reference pricing and parallel distribution. Furthermore, many countries have increased the amount of discounts required on pharmaceutical products. This risk may be heightened by our collaboration with Sobi, pursuant to which we do not control the commercialization of, including obtaining coverage and reimbursement for, ZYNLONTA. The downward pressure on healthcare costs in general, and prescription products in particular, has become increasingly intense.
Furthermore, the containment of healthcare costs has become a priority of governments and private third-party payors. Governments and private third-party payors have attempted to control costs by limiting coverage and the amount of reimbursement for particular medications. We also expect to experience pricing pressures due to the trend towards managed healthcare, the increasing influence of health maintenance organizations and additional legislative changes. In particular, we contract with group purchasing organizations, which increases our gross-to-net deductions. These and other cost-control initiatives could cause us to decrease the price we might establish for products, which could result in lower-than-anticipated product revenues. In addition, the publication of discounts by third-party payors or authorities may lead to further pressure on the prices or reimbursement levels within the country of publication and other countries. If pricing is set at unsatisfactory levels or if coverage and adequate reimbursement of our products is unavailable or limited in scope or amount, our revenues and the potential profitability of our products in those countries would be negatively affected.
Our products and product candidates are complex and difficult to manufacture.
Our products and product candidates are complex and difficult to manufacture. Problems with the manufacturing process, including even minor deviations from the normal process, could result in product defects or manufacturing failures that result in lot failures, product recalls, product liability claims, and insufficient inventory, negative impact on our sales and results of operations and make us a less attractive collaborator for potential partners. We may encounter problems achieving adequate quantities and quality of clinical-grade materials that meet FDA, EMA or other applicable standards or specifications with consistent and acceptable production yields and costs. In the past, we have received batches of certain of ZYNLONTA and our product candidates that did not meet our specifications. There can be no assurance that manufacturing issues will not occur in the future. We currently rely on third parties to manufacture all our raw materials, components and finished products, many of which are sole source suppliers, and this risk may be heightened by our reliance on CMOs to produce our products and product candidates. See “—Risks Related to Our Relationship with Third Parties.” In particular, our products, product candidates and research pipeline use highly potent cytotoxins and payloads and complex conjugation technology that require special manufacturing and handling, which may subject us to liability for any contamination or injury, or failure to comply with environmental, health and safety laws and regulations.
Increases in the costs and expenses of components or raw materials may also adversely influence our business, results of operations and financial condition. Supply sources could be interrupted from time to time and, if interrupted, it is not certain that supplies could be resumed, whether in part or in whole, within a reasonable time frame and at an acceptable cost, or at all. This risk is heightened by our use of sole source suppliers. The cost to manufacture our products could be significantly greater than we expect, which could limit the market acceptance of our products or reduce our potential profit on such product sales.
Furthermore, given the nature of biologics manufacturing, there is a risk of contamination during manufacturing. Any contamination could materially harm our ability to produce products and product candidates on schedule and could cause reputational damage. Some of the raw materials required in our manufacturing process are derived from biologic sources, which are difficult to procure and may be subject to contamination or recall. A material shortage, contamination, recall or restriction on the use of biologically derived substances in the manufacture of any products or product candidates could adversely impact or disrupt the commercial manufacturing or the production of clinical material, which could materially harm our development timelines and our business, financial condition, results of operations and prospects.
The market opportunities for our products and product candidates may be smaller than we estimate and any approval that we obtain may be based on a narrower definition of the patient population.
Our projections of the number of people who have the cancers we are targeting, the subset of people with these cancers in a position to receive a certain line of therapy and who have the potential to benefit from treatment with our products and product candidates, and the market opportunity for our products and product candidates, are based on estimates derived from a variety of sources, including scientific literature, surveys of clinicians and healthcare professionals and other forms of market research. These estimates may be inaccurate or based on imprecise data and are based on assumptions such as labeling, pricing, acceptance by HCPs and patients, patient access and pricing and reimbursement. The number of patients in the addressable markets may turn out to be lower than expected, new treatments may be approved in the future which may reduce our potential market opportunity, the pricing, coverage and reimbursement of our product candidates may be unfavorable, patients may not be otherwise amenable to treatment with our products and product candidates or new patients may become increasingly difficult to identify or gain access to, all of which could negatively impact our market opportunity estimate and materially adversely affect our business, financial condition, results of operations and prospects.
We face substantial competition, which may result in others discovering, developing or commercializing products, treatment methods or technologies before or more successfully than we do.
Our industry is characterized by rapidly advancing technologies, intense competition and a strong emphasis on proprietary products. We face competition with respect to our current products and product candidates and will face competition with respect to any products and product candidates that we may seek to develop or commercialize in the future. Our competitors include large pharmaceutical and biotechnology companies, academic institutions, government agencies and other public and private research organizations that conduct research, seek patent protection and establish collaborative arrangements for research, development, manufacturing and commercialization. Many of our competitors have significantly greater financial resources and capabilities in research and development, manufacturing, preclinical testing, conducting clinical trials, obtaining regulatory approval and marketing than we do. Furthermore, mergers and acquisitions in the biotechnology industry may result in even more resources being concentrated among a smaller number of our competitors.
Many companies are active in the oncology market and are developing or marketing products for the specific therapeutic markets that we target, including both ADC and non-antibody drug conjugate therapies. Similarly, we also face competition from other companies and institutions that continue to invest in innovation in the ADC field, including new payload classes, new conjugation approaches and new targeting moieties. Specifically, we are aware of multiple companies with ADC technologies that may be competitive with our product and product candidates, including, but not limited to, AbbVie, Inc., Daiichi Sankyo Company, Genmab, GlaxoSmithKline plc, Gilead Sciences, Inc., Johnson & Johnson, Mersana Therapeutics Inc., Sanofi S.A., Roche Holding AG, Pfizer Inc. and Zymeworks, Inc. There are hundreds of ADCs in development, the vast majority of which were being developed for the treatment of cancer.
In the relapsed or refractory DLBCL setting, for which we have developed ZYNLONTA, current 3L treatment options include CAR-T, allogeneic stem cell transplant, polatuzumab in combination with bendamustine and a rituximab product, selinexor, tafasitamab in combination with lenalidomide, chemotherapy using small molecules and bispecific antibodies. If ZYNLONTA is approved for use as a 2L treatment for DLBCL patients, we will continue to compete with CAR-T, autologous stem cell transplant, rituximab in combination with chemotherapies, polatuzumab in combination with bendamustine and a rituximab product, and tafasitamab in combination with lenalidomide. In addition, we expect potential future competition from bispecific antibodies such as glofitamab and epcoritamab alone or in combinations with chemotherapies or polatuzumab to gain approval in the 2L treatment of DLBCL.
Risks Related to Our Relationship with Third Parties
We rely on third parties to conduct preclinical studies and clinical trials and for the manufacture, production, storage and distribution of our products and product candidates and certain commercialization activities for our products.
We rely, and we expect that we will continue to rely, on CROs and other third parties to assist in managing, monitoring and otherwise carrying out preclinical studies and clinical trials of our products and product candidates and CMOs and other third parties for the manufacture, production, storage and distribution of our products and product candidates and certain commercialization activities for our products, including government pricing, reporting and chargeback and rebate processing, pharmacovigilance and adverse event reporting. We have less control over the activities of third parties than we would otherwise have if we relied entirely upon our own staff and we are exposed to different risks, including all the risks associated with such third parties’ businesses and financial condition, than if we performed such functions ourselves. There can be no assurance that these third parties will perform services for us in accordance with our timelines, standards and expectations.
If these third parties do not successfully carry out their duties under their agreements or otherwise fail to comply with regulatory requirements, we may experience delays in our research and development activities, be unable to obtain and maintain regulatory approval, be unable to commercialize our products and be required to issue product recalls. In addition, if any of our relationships with these third parties terminate, we may not be able to enter into alternative arrangements on a timely basis or on commercially reasonable terms, and even if successful in entering into alternative arrangements, we may experience significant delays during the transition. This risk may be heightened by our use of single-source supplier arrangements. Furthermore, if a CMO or other third-party manufacturer cannot maintain a compliance status acceptable to the FDA, or if the EMA or a comparable regulatory authority in another jurisdiction does not approve these facilities for the manufacture of our products and product candidates or if it withdraws any such approval in the future, we may need to find alternative manufacturing facilities, which would be time-consuming, costly and uncertain and significantly impact our ability to develop, obtain regulatory approval for, source adequate supply of or market our products and product candidates.
Our collaborators may not perform as expected, and we may be unable to maintain existing or establish additional collaborations for the development and commercialization of our products and product candidates.
We have entered into, and may in the future may enter into, collaboration agreements with third parties for the development and commercialization for products, product candidates and/or research programs. See “Item 1. Business—Material Contracts” for a description of such agreements that are material to us. There can be no assurance that we will be able to enter into additional collaboration agreements on favorable terms, or at all. Even if we are successful in our efforts to establish collaborations, we may not be able to maintain such collaborations if, for example, development or approval of a product or product candidate is delayed or sales of an approved product are disappointing. If we fail to establish and maintain collaborations, we could bear all of the risk and costs related to the development and commercialization of any such product or product candidate, which may require us to seek additional financing, hire additional employees and otherwise develop expertise for which we have not budgeted, and may have a detrimental effect on our financial position by reducing or eliminating the potential for us to receive technology access and license fees, milestones and royalties, and/or reimbursement of development costs.
In such collaborations, we will depend on the performance of our collaborators. Our collaborators may fail to perform their obligations under the collaboration agreements or may not perform their obligations in a timely manner. If conflicts arise between our collaborators and us, the other party may act in a manner adverse to us and could limit our ability to implement our strategies. Furthermore, our collaborators may not properly obtain, maintain, enforce or defend our intellectual property or proprietary rights or may use our proprietary information in such a way as to invite litigation that could jeopardize or invalidate our proprietary information or expose us to potential litigation. In addition, we cannot control the amount and timing of resources our collaborators may devote to our products and product candidates. They may separately pursue competing products, therapeutic approaches or technologies to develop treatments for the diseases targeted by us. Competing products, either developed by the collaborators or to which the collaborators have rights, may result in the withdrawal of support for our products and product candidates. Even if our collaborators continue their contributions to the strategic collaborations, they may nevertheless determine not to actively pursue the development or commercialization of any resulting products. Additionally, if our collaborators pursue different clinical or regulatory strategies with their product candidates based on similar technology as is used in our products and product candidates, adverse events with their product candidates could negatively affect our products and product candidates. Any of these developments could harm our development and commercialization efforts, which adversely impact our business and operations.
Risks Related to Intellectual Property
If we are unable to obtain, maintain or protect our intellectual property rights in any products or technologies we develop, or if the scope of the intellectual property protection obtained is not sufficiently broad, third parties could develop and commercialize products and technology similar or identical to ours, and we may not be able to compete effectively in our market.
Our success depends in significant part on our own and any of our licensors’ ability to obtain, maintain and protect patents and other intellectual property rights and operate without infringing, misappropriating, or otherwise violating the intellectual property rights of others. To protect our proprietary position, we have filed numerous patent applications both in the United States and in foreign jurisdictions to obtain patent rights to inventions we have developed that are important to our business. We have also licensed from third parties rights to patents and other intellectual property, including from MedImmune with respect to the PBD technology we use for our PBD-based ADCs and from other parties for some of our other product candidates and related technology. If we or our current or future licensors are unable to obtain or maintain patent protection with respect to such inventions and technology, our business, financial condition, results of operations and prospects could be materially harmed.
The patent prosecution process is expensive, time-consuming and complex and uncertain, and we and our current or future licensors may not be able to prepare, file, prosecute, maintain and enforce all necessary or desirable patent applications at a reasonable cost or in a timely manner. Patents may be invalidated and patent applications may not be granted for a number of reasons, including known and unknown prior art (including our own prior art), deficiencies in the patent applications or the lack of novelty of the underlying inventions or technology. It is also possible that we or our current and future licensors will fail to identify patentable aspects of inventions made in the course of research, development and commercialization activities in time to obtain patent protection. Although we enter into non-disclosure and confidentiality agreements with parties who have access to confidential or patentable aspects of our research, development and commercialization activities, such as our employees, corporate collaborators, outside scientific collaborators, CROs, consultants, advisors and other third parties, any of these parties may breach the agreements and disclose such activities before a patent application is filed, thereby jeopardizing our ability to seek patent protection. In addition, publications of discoveries in the scientific literature often lag behind actual discoveries, and patent applications in the United States and other jurisdictions are typically not published until eighteen months after filing, or in some cases not at all. Therefore, we cannot be certain that we or our current or future licensors were the first to make the inventions claimed in our owned or licensed patents or patent applications, or that we or our current or future licensors were the first to file for patent protection of such inventions.
Moreover, in some circumstances, we may not have the right to control the preparation, filing, prosecution, maintenance, enforcement and defense of patents and patent applications covering technology that we license from third parties, and are reliant on our licensors. For example, pursuant to our agreements with MedImmune, MedImmune retains control of the preparation, filing, prosecution, maintenance, enforcement and defense of certain of the patents and patent applications licensed to us. Therefore, these patents and applications may not be prepared, filed, prosecuted, maintained, enforced and defended in a manner consistent with the best interests of our business. If our current or future licensors fail to prosecute, maintain, enforce or defend such patents and other intellectual property rights, are not fully cooperative or disagree with us as to the prosecution, maintenance or enforcement of any patent rights, or lose rights to those patents or patent applications, the rights that we have licensed may be reduced or eliminated, and our right to develop and commercialize any of our products and product candidates that are the subject of such licensed rights could be adversely affected.
The patent position of biotechnology companies generally is highly uncertain, involves complex legal and factual questions and has, in recent years, been the subject of much litigation. As a result, the issuance, scope, validity, enforceability and commercial value of our and our current or future licensors’ patent rights are highly uncertain. Our owned and licensed pending and future patent applications may not result in patents being issued which protect the products or technologies we develop, in whole or in part, or which effectively prevent others from commercializing competitive technologies and products. Moreover, the patent examination process may require us or our current and future licensors to narrow the scope of the claims of our owned or licensed pending and future patent applications, which may limit the scope of patent protection that may be obtained. Additionally, the scope of patent protection can be reinterpreted after issuance. Even if our owned or licensed pending and future patent applications issue as patents, they may not issue in a form that will provide us with any meaningful protection, prevent competitors or other third parties from competing with us, or otherwise provide us with any competitive advantage. Any patents that we hold or in-license may be challenged, narrowed, circumvented or invalidated by third parties in court or in patent offices in the United States and abroad. Our owned or licensed patent applications cannot be enforced against third parties practicing the technology claimed in such applications unless and until a patent issues from such applications, and then, only to the extent the issued claims cover the technology. Our competitors or other third parties may also be able to circumvent our patents by developing similar or alternative technologies or products in a non-infringing manner.
We may be subject to a third-party pre-issuance submission of prior art to the United States Patent and Trademark Office (“USPTO”). We cannot assure you that all of the potentially relevant prior art relating to our patents and patent applications has been found. If such prior art exists, it can invalidate a patent or prevent a patent from issuing from a pending patent application. Even if patents do successfully issue and even if such patents cover our products and product candidates, third parties may initiate an opposition, interference, reexamination, post-grant review, inter partes review, nullification or derivation action in court or before patent offices, or other proceedings challenging the inventorship, validity, enforceability or scope of such patents, which may result in the patent claims being narrowed or invalidated. An adverse determination in any such proceeding or litigation could reduce the scope of, or invalidate, the patent rights we own or license, allow third parties to commercialize the products or technologies we develop and compete directly with us, without payment to us, or result in our inability to manufacture or commercialize products without infringing third-party patent rights. Such proceedings also may result in substantial cost and require significant time and attention from our scientific and management personnel, even if the eventual outcome is favorable to us.
Consequently, there can be no assurance that any product, product candidate or technology we develop will be protectable or remain protected by valid and enforceable patents. In addition, if the breadth or strength of protection provided by our patents or patent applications is threatened, regardless of the outcome, it could dissuade companies from collaborating with us to license, develop or commercialize current or future products and product candidates.
Because patent applications in the United States and most other countries are confidential for a period of time after filing, and some remain so until issued, we cannot be certain that we or our current and future licensors were the first to file any patent application related to a product or product candidate. Furthermore, if third parties have filed such patent applications on or before March 15, 2013, an interference proceeding in the United States can be initiated by such third parties to determine who was the first to invent any of the subject matter covered by the patent claims of our applications. If third parties have filed such applications after March 15, 2013, a derivation proceeding in the United States can be initiated by such third parties to determine whether our invention was derived from theirs. Even where we have a valid and enforceable patent, we may not be able to exclude others from practicing our invention where the other party can show that they used the invention in commerce before our filing date or the other party benefits from a compulsory license. Any of the foregoing could have a material adverse effect on our business, results of operations, financial condition and prospects.
We may become involved in lawsuits to protect or enforce our patents or other intellectual property, which could be expensive, time-consuming and unsuccessful, and our issued patents covering one or more of our products, product candidates or technologies or the technology we use in our products and product candidates, could be found invalid or unenforceable if challenged in court.
Competitors and other third parties may infringe, misappropriate or otherwise violate our issued patents or other intellectual property or the patents or other intellectual property of our licensors. To protect our competitive position, we or our licensors may, from time to time, resort to litigation in order to enforce or defend any patents or other intellectual property rights owned by or licensed to us, or to determine or challenge the scope or validity of patents or other intellectual property rights of third parties. Enforcement of intellectual property rights is difficult, unpredictable and expensive, and many of our or our licensors’ or collaboration partners’ adversaries in these proceedings may have the ability to dedicate substantially greater resources to prosecuting these legal actions than we or our licensors or collaboration partners can. Accordingly, despite our or our licensors’ or collaboration partners’ efforts, we or our licensors or collaboration partners may not prevent third parties from infringing upon, misappropriating or otherwise violating intellectual property rights we own or control, particularly in countries where the laws may not protect those rights as fully as in the United States and the European Union. Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation. We may fail in enforcing our rights, in which case third parties, including our competitors, may be permitted to use our technology without being required to pay us any license fees.
If we or one of our current or future licensors were to initiate legal proceedings against a third party to enforce a patent covering one of our products or product candidates, the defendant could counterclaim that such patent is invalid or unenforceable. In patent litigation in the United States and in Europe, defendant counterclaims alleging invalidity or unenforceability are commonplace. A claim for a validity challenge may be based on failure to meet any of several statutory requirements, for example, lack of novelty, obviousness or non-enablement. A claim for unenforceability could involve an allegation that someone connected with prosecution of the patent withheld relevant information from the USPTO or the European Patent Office or made a misleading statement during prosecution. Third parties may also raise similar claims before the USPTO or an equivalent foreign body, even outside the context of litigation. Potential proceedings include reexamination, post-grant review, inter partes review, interference proceedings, derivation proceedings and equivalent proceedings in foreign jurisdictions (e.g., opposition proceedings). Such proceedings could result in the revocation of, cancellation of, or amendment to our patents in such a way that they no longer cover our technology or any products or product candidates that we may develop. The outcome following legal assertions of invalidity and unenforceability during patent litigation is unpredictable. If a defendant were to prevail on a legal assertion of invalidity or unenforceability, we would lose at least part, and perhaps all, of the patent protection on one or more of our products or product candidates or certain aspects of the technology we use in our products and product candidates, and third parties, including our competitors, could compete directly with us, without payment to us. Such a loss of patent protection could have a material adverse impact on our business, financial condition, results of operations and prospects. Further, litigation could result in substantial costs and diversion of management resources, regardless of the outcome, and this could harm our business and financial results. Patents and other intellectual property rights also will not protect our technology if competitors design around our protected technology without infringing our patents or other intellectual property rights.
In addition, our patents or the patents of our licensors may become involved in inventorship or priority disputes. Interference proceedings provoked by third parties or brought by us or declared by the USPTO may be necessary to determine the priority of inventions with respect to our or our licensors’ patents or patent applications. If we or our licensors are unsuccessful in any interference proceedings to which we or they are subject, we may lose valuable intellectual property rights through the loss of one or more patents owned or licensed or our owned or licensed patent claims may be narrowed, invalidated or held unenforceable. If we or our licensors are unsuccessful in any interference proceeding or other priority or inventorship dispute, we may be required to obtain and maintain licenses from third parties, including parties involved in any such interference proceedings or other priority of inventorship disputes. Such licenses may not be available on commercially reasonable terms or at all, or may be non-exclusive. If we are unable to obtain and maintain such licenses, we may need to cease the development, manufacture and commercialization of one or more of the products and product candidates we may develop. The loss of exclusivity or narrowing of our owned or licensed patent claims could limit our ability to stop others from using or commercializing similar or identical technology and products. Any of the foregoing could have a material adverse effect on our business, results of operations, financial condition and prospects.
If we fail to comply with our obligations in the agreements under which we license intellectual property rights from third parties or otherwise experience disruptions to our business relationships with our licensors, we could lose the ability to continue the development and commercialization of our products and product candidates.
We are party to a number of intellectual property and technology licenses that are important to our business. For example, the PBD technology we use to generate our PBD-based ADCs was developed by, and is licensed on a target-exclusive basis from, MedImmune. If we fail to comply with our obligations under these or our other agreements, including payment and diligence terms, our current and future licensors may have the right to terminate these agreements, in which event we may not be able to develop, manufacture, market or sell any product that is covered by these agreements or may face other penalties under these agreements. Such an occurrence could adversely affect the value of the products and product candidates being developed under any such agreement. Termination of these agreements or reduction or elimination of our rights under these agreements may result in our having to negotiate new or reinstated agreements, which may not be available to us on equally favorable terms, or at all, or cause us to lose our rights under these agreements, including our rights to intellectual property or technology important to our development programs. Accordingly, termination of these agreements may require us to cease the development of our products and product candidates.
In addition, the agreements under which we license intellectual property or technology from third parties are generally complex, and certain provisions in such agreements may be susceptible to multiple interpretations. The resolution of any contract interpretation disagreement that may arise could narrow what we believe to be the scope of our rights to the relevant intellectual property or technology, or increase what we believe to be our financial or other obligations under the relevant agreements. Moreover, if disputes over intellectual property that we have licensed prevent or impair our ability to maintain our current licensing arrangements on commercially acceptable terms, we may be unable to successfully develop and commercialize the affected products and product candidates. Any of the foregoing could have a material adverse effect on our business, financial condition, results of operations and prospects.
We may not be successful in obtaining additional intellectual property rights necessary or required to further develop our products and product candidates.
A third party may hold intellectual property, including patent rights, that is important or necessary to the development of our products and product candidates. In order to avoid infringing these third-party patents, we may find it necessary or prudent to obtain licenses from such third-party intellectual property holders. Moreover, we may need to obtain additional licenses from our existing licensors and others to advance our research or allow commercialization of products and product candidates we may develop. In addition, many of our patents are co-owned with MedImmune, which licenses its interest in such patents to us. With respect to any patents we co-own with third parties, we may require licenses to such co-owners’ interest to such patents. In addition, we may need the cooperation of any co-owners of our patents in order to enforce such patents against third parties, and such cooperation may not be provided to us. We may be unable to secure such licenses or otherwise acquire or in-license any compositions, methods of use, processes or other intellectual property rights from third parties that we identify as necessary for products and product candidates we develop. The licensing or acquisition of third-party intellectual property rights is a competitive area, and more established companies may pursue strategies to license or acquire third-party intellectual property rights that we may consider attractive or necessary. These established companies may have a competitive advantage over us due to their size, capital resources and greater clinical development or commercialization capabilities. In addition, companies that perceive us to be a competitor may be unwilling to assign or license rights to us. As a result, we may be unable to obtain any such licenses at a reasonable cost or on reasonable terms, if at all. In that event, we may be required to expend significant time and resources to redesign our technology, products, product candidates or the methods for manufacturing them or to develop or license replacement technology, all of which may not be feasible on a technical or commercial basis.
If we are unable to do so, we may be unable to develop or commercialize the affected products and product candidates, which could significantly harm our business, financial condition, results of operations and prospects. In addition, even if we obtain a license, it may be non-exclusive, thereby giving third parties, including our competitors, access to the same technologies licensed to us. In addition, any license we obtain could require us to make substantial licensing and royalty payments. If we are unable to obtain an exclusive license to any third-party or co-owned patents or patent applications, such parties may be able to license their rights to other third parties, including our competitors, and such third parties could market competing products and technology. Any of the foregoing could have a material adverse effect on our business, results of operations, financial condition and prospects.
Third parties may initiate legal proceedings against us alleging that we infringe, misappropriate, or otherwise violate their intellectual property rights or we may initiate legal proceedings against third parties to challenge the validity or scope of intellectual property rights controlled by third parties, the outcome of which would be uncertain and could have an adverse effect on the success of our business.
Our commercial success depends upon our ability to develop, manufacture, market and sell our products and product candidates and use our and our current or future licensors’ proprietary technologies without infringing, misappropriating or otherwise violating the intellectual property rights of third parties. Third parties may initiate legal proceedings against us or our current and future licensors alleging that we or our current and future licensors infringe, misappropriate or otherwise violate their intellectual property rights. In addition, we and our licensors have initiated, and we and our current and future licensors may in the future initiate, legal proceedings against third parties to challenge the validity or scope of intellectual property rights controlled by third parties, including in oppositions, interferences, reexaminations, inter partes reviews or derivation proceedings in the United States or other jurisdictions. These proceedings can be expensive and time-consuming, and many of our or our current and future licensors’ adversaries in these proceedings may have the ability to dedicate substantially greater resources to prosecuting these legal actions than we or our current and future licensors. Numerous U.S.- and foreign-issued patents and pending patent applications which are owned by third parties exist in the fields in which we are pursuing our products and product candidates. As the biotechnology and pharmaceutical industries expand and more patents are issued, the risk increases that we may be subject to claims of infringement of the patent rights of third parties.
There are, and in the future, we may identify, other third-party patents or patent applications with claims to materials, formulations, methods of manufacture or methods for treatment related to the use or manufacture of one or more of our products and product candidates. Because patent applications can take many years to issue, there may be currently pending patent applications that may later result in issued patents that our products and product candidates may infringe. In addition, third parties may obtain patents in the future and claim that use of our technologies infringes upon these patents. Parties making infringement, misappropriation or other intellectual property claims against us may obtain injunctive or other equitable relief, which could effectively block our ability to further develop and commercialize one or more of our products and product candidates. Defense of these claims, regardless of their merit, would involve substantial litigation expense and would be a substantial diversion of management and employee resources. In addition, even if we believe any third-party intellectual property claims are without merit, there is no assurance that a court would find in our favor on questions of validity, enforceability, priority or non-infringement. A court of competent jurisdiction could hold that such third-party patents are valid, enforceable and infringed, which could materially and adversely affect our ability to commercialize any of our products, product candidates or technologies covered by the asserted third-party patents. In order to successfully challenge the validity of any such third-party U.S. patents in federal court, we would need to overcome a presumption of validity. As this burden is a high one requiring us to present clear and convincing evidence as to the invalidity of any such U.S. patent claim, there is no assurance that a court of competent jurisdiction would invalidate the claims of any such U.S. patent. An unfavorable outcome could require us or our current and future licensors to cease using the related technology or developing or commercializing our products and product candidates, or to attempt to license rights to it from the prevailing party. If we are not successful in defending a third-party claim of infringement, we may be enjoined from continuing to sell our products or our business could be harmed if the prevailing party does not offer us or our current and future licensors a license on commercially reasonable terms or at all. Even if we or our current and future licensors obtain a license, it may be non-exclusive, thereby giving our competitors access to the same technologies licensed to us or our current and future licensors, and it could require us to make substantial licensing and royalty payments. In addition, we could be found liable for monetary damages, including treble damages and attorneys’ fees, if we are found to have willfully infringed a patent. A finding of infringement, misappropriation or other violation of third-party intellectual property could prevent us from commercializing our products and product candidates or force us to cease some of our business operations, which could harm our business. Claims that we have misappropriated the confidential information or trade secrets of third parties could have a similar material adverse effect on our business, results of operations, financial condition and prospects. Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation or administrative proceedings, there is a risk that some of our confidential information could be compromised by disclosure.
We may be subject to claims by third parties asserting that we or our employees, consultants or advisors have misappropriated their intellectual property, or claiming ownership of what we regard as our own intellectual property.
Many of our employees, consultants, and advisors, including our senior management, were previously employed at other biopharmaceutical companies, including our competitors or potential competitors. Some of these employees executed proprietary rights, non-disclosure and/or non-competition agreements in connection with such previous employment. Although we try to ensure that our employees, consultants and advisors do not use the proprietary information or know-how of others in their work for us, we may be subject to claims that we or these individuals have used or disclosed confidential information or intellectual property, including trade secrets or other proprietary information, of any such individual’s current or former employer. Litigation may be necessary to defend against these claims. If we fail in defending against any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel. Such intellectual property rights could be awarded to a third party, and we could be required to obtain a license from such third party to commercialize our technology or products. Such a license may not be available on commercially reasonable terms, or at all.
In addition, while it is our policy to require our employees and contractors who may be involved in the conception or development of intellectual property to execute agreements assigning such intellectual property to us, we may be unsuccessful in executing such an agreement with each party who, in fact, conceives or develops intellectual property that we regard as our own. The assignment of intellectual property rights may not be self-executing, or the assignment agreements may be breached, and we may be forced to bring claims against third parties, or defend claims that they may bring against us, to determine the ownership of what we regard as our intellectual property. Such claims could have a material adverse effect on our business, results of operations, financial condition and prospects.
Changes in patent law could diminish the value of patents in general, thereby impairing our ability to protect our products and product candidates.
Obtaining and enforcing patents in the biopharmaceutical industry involves both technological and legal complexity and is therefore costly, time-consuming and inherently uncertain. Changes in either the patent laws or the interpretation of the patent laws in the United States or other jurisdictions could increase the uncertainties and costs surrounding the prosecution of patent applications and the enforcement or defense of issued patents. Depending on future actions by the U.S. Congress, the U.S. courts, the USPTO and the relevant law-making bodies in other countries, the laws and regulations governing patents could change in unpredictable ways that would weaken our ability to obtain new patents or to enforce our existing patents and patents that we might obtain in the future.
If we do not obtain patent term extension for any product candidates we may develop, our business may be materially harmed.
Patents have a limited lifespan. Due to the amount of time required for the development, testing and regulatory review of new product candidates, patents protecting such candidates might expire before or shortly after such candidates are commercialized. As a result, our owned and licensed patent portfolio may not provide us with sufficient rights to exclude others from commercializing products similar or identical to ours. In the United States, if all maintenance fees are timely paid, the natural expiration of a patent is generally 20 years from its earliest U.S. non-provisional filing date. Patent terms vary in other jurisdictions. Various extensions may be available, including under the Hatch-Waxman Amendments in the United States, but the life of a patent, and the protection it affords, is limited. Even if patents covering our product candidates are obtained, once the patent life has expired for a product, we may be open to competition from competitive medications, including biosimilar medications. At the time of the expiration of any relevant patents, the underlying technology covered by such patents can be used by any third party, including competitors.
We may not be able to protect our intellectual property rights throughout the world.
Filing, prosecuting, enforcing and defending patents on products and product candidates in all countries throughout the world would be prohibitively expensive, and our owned or licensed intellectual property rights may not exist in some countries outside the United States or may be less extensive in some countries than in the United States. In addition, the laws of some foreign countries do not protect intellectual property rights to the same extent as federal and state laws in the United States. For example, in some jurisdictions, including Europe, it is more difficult to obtain patents protecting a medical method of use, and any such patents we are able to obtain in such jurisdictions may issue with narrower scope than their U.S. counterparts. Many countries have compulsory licensing laws under which a patent owner may be compelled to grant licenses to third parties, and many countries limit the enforceability of patents against government agencies or government contractors.
In these countries, the patent owner may have limited remedies, which could materially diminish the value of such patent. Consequently, we and our current and future licensors may not be able to prevent third parties from practicing our owned or licensed inventions in all countries outside the United States, or from selling or importing products made using our owned or licensed inventions in and into the United States or other jurisdictions.
If we are unable to protect our confidential information and trade secrets, our business and competitive position would be harmed.
In addition to seeking patents for some of our technology, products and product candidates, we also rely on trade secrets, including unpatented know-how, technology and other proprietary information to maintain our competitive position. Trade secrets can be difficult to protect. We seek to protect these trade secrets, in part, by entering into non-disclosure, confidentiality and invention assignment agreements with parties who have access to them, such as our employees, corporate collaborators, outside scientific collaborators, CROs, contract manufacturers, consultants, advisors and other third parties. We also enter into confidentiality agreements with our employees and consultants. However, there can be no assurance that we have entered into such agreements with each party that may have or have had access to our trade secrets or proprietary technology and processes. Despite these efforts, any of these parties may breach the agreements and disclose our proprietary information, including our trade secrets, and we may not be able to obtain adequate remedies for such breaches. Misappropriation or unauthorized disclosure of our trade secrets could significantly affect our competitive position and may have a material adverse effect on our business.
Enforcing a claim that a party illegally disclosed or misappropriated a trade secret is difficult, expensive and time-consuming, and the outcome is unpredictable. Some courts both within and outside the United States may be less willing or unwilling to protect trade secrets. Furthermore, trade secret protection does not prevent competitors from independently developing substantially equivalent information and techniques, and we cannot guarantee that our competitors will not independently develop substantially equivalent information and techniques. If a competitor lawfully obtained or independently developed any of our trade secrets, we would have no right to prevent such competitor from using that technology or information to compete with us. Failure on our part to adequately protect our trade secrets or confidential information could have a material adverse effect on our business, results of operations, financial condition and prospects.
If our trademarks and trade names are not adequately protected, then we may not be able to build name recognition in our markets and our business may be adversely affected.
Our registered or unregistered trademarks or trade names may be challenged, circumvented, declared generic or determined to be infringing on other marks. There can be no assurance that competitors will not infringe our trademarks, that we will have adequate resources to enforce our trademarks or that any of our current or future trademark applications will be approved. During trademark registration proceedings, we may receive rejections and, although we are given an opportunity to respond, we may be unable to overcome such rejections. In addition, in proceedings before the USPTO and in proceedings before comparable agencies in many foreign jurisdictions, trademarks are examined for registrability against prior pending and registered third-party trademarks, and third parties are given an opportunity to oppose registration of pending trademark applications and/or to seek cancellation of registered trademarks. Applications to register our trademarks may be finally rejected, and opposition or cancellation proceedings may be filed against our trademarks, which may necessitate a change in branding strategy if such rejections and proceedings cannot be overcome or resolved. For example, in some jurisdictions the applicable trademark office has rejected our corporate name for registration, or a third party has objected to a published application for a product trademark, which, in some cases, has caused us to abandon or limit our applications, and rely more on the registration for our corporate logo.
Risks Related to Our Business and Industry
We may be unable to attract and retain senior management and qualified scientific personnel.
Our ability to compete in the highly competitive biotechnology industry depends upon our ability to attract, motivate and retain highly qualified managerial, scientific and medical personnel. The loss of the services of our senior management members, qualified employees and scientific and medical advisors could impede the achievement of our research, development and commercialization objectives. Members of our senior management are employed pursuant to employment agreements with no term and that require advance notice for termination, but these persons may terminate their employment with us at any time. In addition, laws and regulations on executive compensation, including legislation in our home country, Switzerland, may restrict our ability to attract, motivate and retain the required level of qualified personnel including those that (i) impose an annual binding shareholders’ “say-on-pay” vote with respect to the compensation of the members of the executive committee and the board of directors, (ii) prohibit severance, advances, transaction premiums and similar payments to the members of the executive committee and the board of directors, and (iii) require companies to specify various compensation-related matters in their articles of association, thus requiring them to be approved by a shareholders’ vote.
We do not maintain “key person” insurance for any of our executives or other employees. Further, we compensate our employees, in part, using share-based compensation, the effectiveness of which is influenced by the price of our common shares. If the price of our common shares continues to decrease or is subject to continued volatility, which may occur for various factors, including those beyond our control, we may be unable to attract or retain qualified personnel. Competition for skilled personnel is intense, particularly in the biotechnology industry. We face competition for personnel from other companies, universities, public and private research institutions and other organizations. This competition may limit our ability to hire and retain highly qualified personnel on acceptable terms, or at all. This possibility is further compounded by the novel nature of our product candidates, as fewer people are trained in or are experienced with product candidates of this type.
Our employees, agents, contractors or collaborators may engage in misconduct or other improper activities.
We cannot ensure that our compliance controls, policies and procedures will in every instance protect us from acts committed by our employees, agents, contractors or collaborators that would violate the laws or regulations of the jurisdictions in which we operate, including, without limitation, healthcare, employment, foreign corrupt practices, environmental, competition, and patient privacy and other privacy laws and regulations. In particular, because we operate globally and our business is heavily regulated and therefore involves significant interaction with public officials and because the healthcare providers and drug purchasers in certain countries are employed by their government, we face heightened risk with respect to compliance with the Foreign Corrupt Practices Act (the “FCPA”). There is no certainty that all of our employees, agents, contractors, or collaborators, or those of our affiliates, will comply with all applicable laws and regulations, particularly given the high level of complexity of these laws. We have provisions in our Code of Business Conduct and Ethics, an anti-corruption policy and certain controls and procedures in place that are designed to mitigate the risk of non-compliance with anti-corruption and anti-bribery laws. However, it is not always possible to identify and deter employee misconduct, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from government investigations or other actions stemming from a failure to comply with these laws or regulations. Violations of these laws and regulations could result in, among other things, significant administrative, civil and criminal fines and sanctions against us, our officers, or our employees, the closing of our facilities, exclusion from participation in federal healthcare programs including Medicare and Medicaid, implementation of compliance programs, integrity oversight and reporting obligations, and prohibitions on the conduct of our business.
Product liability lawsuits and product recalls could cause us to incur substantial liabilities and to limit development and commercialization of our products.
We face an inherent risk of product liability and product recalls as a result of the clinical testing of our product candidates in human clinical trials and as a result of the commercialization of approved products and their use by patients. Side effects or adverse events known or reported to be associated with, or manufacturing defects in, the products sold by us could exacerbate a patient’s condition or could result in serious injury or impairments or even death. This risk is heightened by our use of highly potent ADCs. If we cannot successfully defend ourselves against product liability claims, we may incur substantial liabilities or be required to limit the research and development and commercialization of our products and product candidates. Even a successful defense would require significant financial and management resources. We currently carry product and clinical trial liability insurance in an amount that we believe is appropriate for our business. Although we maintain such insurance, any claim that may be brought against us could result in a court judgment or settlement in an amount that is not covered, in whole or in part, by our insurance or that is in excess of the limits of our insurance coverage. Our insurance policies also have various exclusions, and we may be subject to a product liability claim for which we have no coverage. If we are unable to obtain or maintain sufficient insurance coverage at an acceptable cost or to otherwise protect against potential product liability claims, it could prevent or inhibit the development of our products and product candidates and the commercial production and sale of our products.
To the extent that a product fails to conform to its specifications or comply with the applicable laws or regulations, we or our partners may be required to or may decide to voluntarily recall the product or regulatory authorities may request or require that we recall a product even if there is no immediate potential harm to a patient. Recalls are costly and take time and effort to administer and damage our reputation and attractiveness as a collaborator. Even if a recall only initially relates to a single product, product batch, or a portion of a batch, recalls may later be expanded to additional products or batches or we or our partners may incur additional costs and need to dedicate additional efforts to investigate and rule out the potential for additional impacted products or batches. Moreover, if any of our partners recall a product due to an issue with a product or component that we supplied, they may claim that we are responsible for such issue and may seek to recover the costs related to such recall or be entitled to certain contractual remedies from us.
Recalls may further result in decreased demand for our or our partners’ products, could cause our partners or distributors to return products to us for which we may be required to provide refunds or replacement products, or could result in product shortages. Recalls may also require regulatory reporting and prompt regulators to conduct additional inspections of our or our partners’ or contractors’ facilities, which could result in findings of noncompliance and regulatory enforcement actions. A recall could also result in product liability claims by individuals and third-party payers and the suspension, variation, or withdrawal of regulatory approval.
Our internal computer systems, or those of our partners, third-party CROs or other contractors or consultants, may fail or suffer security incidents, which could result in a material disruption of our research and development and commercialization programs and significant monetary losses.
Despite the implementation of security measures, our internal computer systems and those of our current or future partners, third-party CROs and other contractors and consultants have been subject to attacks by, and may be vulnerable to damage from, various methods, including cybersecurity attacks, breaches, intentional or accidental mistakes or errors, or other technological failures which can include, among other things, computer viruses, malicious codes, employee theft or misuse, unauthorized copying of our website or its content, unauthorized access attempts including third parties gaining access to systems using stolen or inferred credentials, denial-of-service attacks, phishing attempts, service disruptions, natural disasters, fire, terrorism, war and telecommunication and electrical failures. As the cyber-threat landscape evolves, these attacks are growing in frequency, sophistication and intensity, and are becoming increasingly difficult to detect. If a failure, accident or security breach were to occur and cause interruptions in our, our partners’ or our CROs’ operations, it could result in a misappropriation of confidential information, including our intellectual property or financial information, a material disruption of our programs and significant monetary losses. In particular, because of our approach to running multiple clinical trials in parallel, any breach of our computer systems may result in a loss of data or compromised data integrity across many of our programs in many stages of development. Any such breach, loss or compromise of clinical trial participant personal data may also subject us to civil fines and penalties, including under the GDPR and relevant member state law in the European Union, the UK GDPR or the CCPA, HIPAA and other relevant state and federal privacy laws in the United States. Moreover, because we maintain sensitive company data on our computer networks, including our intellectual property and proprietary business information, any such security breach may compromise information stored on our networks and may result in significant data losses or theft of our intellectual property or proprietary business information. We currently carry cybersecurity liability insurance in an amount that we believe is appropriate for our business. However, our current cybersecurity liability insurance, and any such insurance that we may obtain in the future, may not cover the damages we would sustain based on any breach of our computer security protocols or other cybersecurity attack. To the extent that any disruption or security breach results in a loss of or damage to our data or applications or other data or applications relating to our technology, products or product candidates, or inappropriate disclosure of confidential or proprietary information, our reputation could be harmed and we could incur significant liabilities and the development and commercialization of our products and product candidates could be disrupted.
Our business is subject to economic, political, regulatory and other risks associated with conducting business internationally.
We are a global organization and thus subject to the risks associated with international operations, including inflationary pressures, economic weakness or political instability in particular non-U.S. economies and markets; global trends involving pharmaceutical pricing; differing regulatory requirements for drug approvals in non-U.S. countries; differing reimbursement, pricing and insurance regimes; potentially reduced protection for, and complexities and difficulties in obtaining, maintaining, protecting and enforcing, intellectual property rights; difficulties in compliance with non-U.S. laws and regulations; changes in non-U.S. regulations and customs, tariffs and trade barriers; changes in non-U.S. currency exchange rates and currency controls; changes in a specific country’s or region’s political or economic environment; trade protection measures, economic sanctions and embargoes, import or export licensing requirements or other restrictive actions by U.S. or non-U.S. governments; negative consequences from changes in tax laws; difficulties associated with staffing and managing international operations, including differing labor relations; production shortages resulting from any events affecting raw material supply or manufacturing capabilities abroad; business interruptions resulting from geopolitical actions and conflict, war and terrorism, including the recent conflict between Russia and the Ukraine and resulting sanctions, retaliatory measures, changes in the availability and price of various materials and effects on global financial markets; business interruptions resulting from natural disasters; and the impact of public health epidemics on employees and the global economy. In addition, as a result of the United Kingdom’s exit from the European Union, we may face increasingly divergent regulations in Europe, with which may be expensive and time-consuming for us to comply.
Our business could be adversely affected by the effects of health epidemics, pandemics and natural disasters.
Our business could be adversely affected by health epidemics, pandemics and natural disasters. To the extent any pandemic, epidemic or outbreak of an infectious disease adversely affects our business and financial results, it may also have the effect of heightening many of the other risks described in this “Item 1A. Risk Factors” section. In addition, any unplanned event, such as a flood, fire, explosion, earthquake, extreme weather condition, medical epidemic, power shortage, telecommunication failure or other natural or man-made accidents or incidents that result in us being unable to fully use our facilities, or the manufacturing facilities of our third-party contract manufacturers, may have a material and adverse effect on our ability to operate our business and have significant negative consequences on our financial and operating conditions. Certain of these events may become more frequent and severe as a result of the effects of climate change. Loss of access to these facilities may result in increased costs, reduced revenues, delays in the development of our products and product candidates or the interruption of our business operations for a substantial period of time. We maintain business continuity insurance coverage at levels that we believe are appropriate for our business. However, in the event of an accident or incident at these facilities, there can be no assurance that the amounts of insurance will be sufficient to satisfy any damages and losses. If our facilities, or the manufacturing facilities of our third-party contract manufacturers, are unable to operate because of an accident or incident or for any other reason, even for a short period of time, any or all of our research and development programs and commercialization efforts may be harmed.
If we fail to maintain an effective system of internal controls over financial reporting, we may not be able to accurately or timely report our financial condition or results of operations or prevent fraud.
Effective internal controls over financial reporting are necessary for us to provide reliable financial reports and, together with adequate disclosure controls and procedures, are designed to prevent fraud. Any failure to implement required new or improved controls, or difficulties encountered in their implementation could cause us to fail to meet our reporting obligations. Due to our filer status, our independent registered public accounting firm has not reported on the effectiveness of our internal control over financial reporting as of December 31, 2024. Any testing conducted by us in connection with Section 404 of the Sarbanes-Oxley Act, or any subsequent testing conducted by our independent registered public accounting firm, may reveal deficiencies in our internal controls over financial reporting that are deemed to be material weaknesses or that may require prospective or retroactive changes to our consolidated financial statements, or identify other areas for further attention or improvement. The failure to maintain controls compliant with Sarbanes-Oxley Act could also cause investors to lose confidence in our reported financial information, which could have a negative effect on the trading price of our shares.
Our corporate compliance program cannot guarantee that we are in compliance with all potentially applicable laws and regulations and we have incurred and will continue to incur costs relating to compliance with applicable laws and regulations.
As a biotechnology and pharmaceutical company, we are subject to a large body of legal and regulatory requirements, guidance, and recommendations from a variety of regulatory authorities, such as the FDA, the EMA, and HHS OIG. In addition, as a publicly traded company we are subject to significant regulations. While we have developed and instituted a corporate compliance program based on what we believe are the current best practices and continue to update the program in response to newly implemented regulatory requirements and guidance, we cannot ensure that we are or will be in compliance with all potentially applicable regulations. Failure to comply with all potentially applicable laws and regulations could lead to the imposition of fines, cause the value of our common shares to decline, and impede our ability to raise capital or list our securities on certain securities exchanges.
Risks Related to Our Common Shares
The market price of our common shares has been volatile.
The market price of shares of our common shares could be subject to wide fluctuations in response to many risk factors listed in this “Item 1A. Risk Factors” section, and others beyond our control such as actions by our shareholders (including if substantial amounts of common shares are sold in the public market or if the market perceives that such sales may occur), collaborators or competitors and general market and economic conditions. In particular, pharmaceutical, biotechnology and other life sciences company stocks have historically experienced significant volatility. As we operate in a single industry, we are especially vulnerable to these factors to the extent that they affect our industry. In the past, securities class action litigation has often been initiated against companies following periods of volatility in their stock price. This risk is especially relevant for biotechnology companies, which have experienced significant stock price volatility in recent years. Securities litigation could result in substantial costs and divert our management’s attention and resources, and could also require us to make substantial payments to satisfy judgments or to settle litigation.
Exercise of outstanding warrants will dilute existing shareholders’ ownership interest.
As of the date of this Annual Report, we have outstanding warrants to purchase an aggregate of 2,631,578 common shares at an exercise price of $24.70 per share (which are exercisable, on a cash or cashless basis, at the option of the holder at any time on or prior to May 19, 2025), warrants to purchase an aggregate of 1,781,262 common shares at an exercise price of $28.07 (which are exercisable, on a cash or cashless basis, at the option of the holder at any time on or prior to May 19, 2025), warrants to purchase an aggregate of 527,295 common shares at an exercise price of $8.30 per share (which are exercisable, on a cash or a cashless basis, at the option of the holder at any time on or prior to August 15, 2032) and pre-funded warrants to purchase 8,163,265 common shares at an exercise price of CHF 0.08 per share. The warrants also contain customary anti-dilution adjustments and will entitle holders to receive any dividends or other distributions paid on the underlying common shares prior to their expiration on an as-exercised basis. If our outstanding warrants are exercised into common shares, our existing shareholders’ ownership interest will be diluted.
We have never paid dividends and do not expect to pay any dividends in the foreseeable future.
We have not paid any cash dividends since our incorporation. Even if future operations lead to significant levels of distributable profits, we currently intend to reinvest any earnings in our business and do not anticipate declaring or paying any cash dividends until we have an established revenue stream to support continuing dividends. In addition, any proposal for the payment of future dividends will be at the discretion of our board of directors after taking into account various factors including our business prospects, liquidity requirements, financial performance and new product development. Furthermore, payment of future dividends is subject to certain limitations pursuant to our current and future debt instruments, Swiss law and our articles of association. In addition, the Loan Agreement limits our ability to pay dividends. See “—Risks Related to Our Financial Position and Capital Requirements.” Accordingly, investors cannot rely on dividend income from our common shares, and any returns on an investment in our common shares will likely depend entirely upon any future appreciation in the price of our common shares.
If securities or industry analysts do not continue to publish research, or publish inaccurate or unfavorable research, about our business, the price of our common shares and our trading volume could decline.
The trading market for our common shares depends, in part, on the research and reports that securities or industry analysts publish about us or our business. If one or more of the analysts who cover us downgrade our common shares or publish inaccurate or unfavorable research about our business, the price of our common shares would likely decline. In addition, if our operating results fail to meet the forecast of analysts, the price of our common shares would likely decline. If one or more of these analysts cease coverage of our company or fail to publish reports on us regularly, demand for our common shares could decrease, which might cause the price of our common shares and trading volume to decline.
The rights of our shareholders may be different from the rights of shareholders in companies governed by the laws of U.S. jurisdictions.
We are a Swiss corporation. Our corporate affairs are governed by our articles of association and by the laws governing companies incorporated in Switzerland. The rights of our shareholders and the responsibilities of members of our board of directors may be different from the rights and obligations of shareholders and directors of companies governed by the laws of U.S. jurisdictions. In particular, in the performance of its duties, our board of directors is required by Swiss law to consider the interests of our company, our shareholders, our employees and other stakeholders, in all cases with due observation of the principles of reasonableness and fairness. It is possible that some of these parties will have interests that are different from, or in addition to, shareholders’ interests. Swiss law limits the ability of our shareholders to challenge resolutions made or other actions taken by our board of directors in court. Our shareholders generally are not permitted to file a suit to reverse a decision or an action taken by our board of directors, but are instead only permitted to seek damages for breaches of fiduciary duty. As a matter of Swiss law, shareholder claims against a member of our board of directors for breach of fiduciary duty would have to be brought to the competent courts in Epalinges, Canton of Vaud, Switzerland, or where the relevant member of our board of directors is domiciled. In addition, under Swiss law, any claims by our shareholders against us must be brought exclusively to the competent courts in Epalinges, Canton of Vaud, Switzerland. For a further summary of applicable Swiss company law, see Exhibit 4.1 to this Annual Report. Accordingly, our shareholders do not have the same rights as those of a Delaware-incorporated company.
Our shareholders enjoy certain rights that may limit our flexibility to raise capital, issue dividends and otherwise manage ongoing capital needs.
Swiss law reserves for approval by shareholders certain corporate actions over which a board of directors would have authority in some other jurisdictions. For example, the payment of dividends must be approved by shareholders. Swiss law also requires that our shareholders themselves resolve to, or authorize our board of directors to, increase or decrease our share capital.
While our shareholders may authorize our board of directors to issue or cancel shares without additional shareholder approval, Swiss law limits this authorization to 50% of the issued share capital at the time of the authorization. The authorization, furthermore, has a limited duration of up to five years and must be renewed by the shareholders from time to time thereafter in order to be available for raising capital. Additionally, subject to specified exceptions, including exceptions described in our articles of association, Swiss law grants pre-emptive subscription rights to existing shareholders to subscribe for new issuances of shares. Swiss law also does not provide as much flexibility in the various rights and regulations that can attach to different categories of shares as do the laws of some other jurisdictions. These Swiss law requirements relating to our capital management may limit our flexibility, including our ability to raise capital, and situations may arise where greater flexibility would have provided benefits to our shareholders. See Exhibit 4.1 to this Annual Report.
Our shares are not listed in Switzerland, our home jurisdiction. As a result, our shareholders do not benefit from certain provisions of Swiss law that are designed to protect shareholders in a public takeover offer or a change-of-control transaction.
Because our common shares are listed exclusively on the NYSE and not in Switzerland, our shareholders do not benefit from the protection afforded by certain provisions of Swiss law that are designed to protect shareholders in the event of a public takeover offer or a change-of-control transaction. For example, Article 120 of the Swiss Financial Market Infrastructure Act and its implementing provisions require investors to disclose their interest in our company if they reach, exceed or fall below certain ownership thresholds. Similarly, the Swiss takeover regime imposes a duty on any person or group of persons who acquires more than one-third of a company’s voting rights to make a mandatory offer for all of the company’s outstanding listed equity securities. In addition, the Swiss takeover regime imposes certain restrictions and obligations on bidders in a voluntary public takeover offer that are designed to protect shareholders. However, these protections are applicable only to issuers that list their equity securities in Switzerland and, because our common shares are listed exclusively on the NYSE, are not be applicable to us. Furthermore, since Swiss law restricts our ability to implement rights plans or U.S.-style “poison pills,” our ability to resist an unsolicited takeover attempt or to protect minority shareholders in the event of a change of control transaction may be limited. Therefore, our shareholders may not be protected in the same degree in a public takeover offer or a change-of-control transaction as are shareholders in a Swiss company listed in Switzerland.
U.S. shareholders may not be able to obtain judgments or enforce civil liabilities against us or certain of our directors.
We are organized under the laws of Switzerland and our registered office and domicile is located in Epalinges, Canton of Vaud, Switzerland. Moreover, a number of our directors are not residents of the United States, and all or a substantial portion of the assets of such persons are located outside the United States. As a result, it may not be possible for investors to effect service of process within the United States upon us or upon such persons or to enforce against them judgments obtained in U.S. courts, including judgments in actions predicated upon the civil liability provisions of the federal securities laws of the United States. There is doubt as to the enforceability in Switzerland of original actions, or in actions for enforcement of judgments of U.S. courts, of civil liabilities to the extent solely predicated upon the federal and state securities laws of the United States. Original actions against persons in Switzerland based solely upon the U.S. federal or state securities laws are governed, among other things, by the principles set forth in the Swiss Federal Act on Private International Law (the “PILA”). The PILA provides that the application of provisions of non-Swiss law by the courts in Switzerland shall be precluded if the result is incompatible with Swiss public policy. Also, certain mandatory provisions of Swiss law may be applicable regardless of any other law that would otherwise apply.
Switzerland and the United States do not have a treaty providing for reciprocal recognition and enforcement of judgments in civil and commercial matters. The recognition and enforcement of a judgment of the courts of the United States in Switzerland is governed by the principles set forth in the PILA. The PILA provides in principle that a judgment rendered by a non-Swiss court may be enforced in Switzerland only if:
•the non-Swiss court had jurisdiction pursuant to the PILA;
•the judgment of such non-Swiss court has become final and non-appealable;
•the judgment does not contravene Swiss public policy;
•the court procedures and the service of documents leading to the judgment were in accordance with the due process of law; and
•no proceeding involving the same parties and the same subject matter was first brought in Switzerland, or adjudicated in Switzerland, or was earlier adjudicated in a third state, and this decision is recognizable in Switzerland.
Anti-takeover provisions in our articles of association could make an acquisition of us, which may be beneficial to our shareholders, more difficult.
Our articles of association contain provisions that may have the effect of discouraging, delaying or preventing a change in control of us that shareholders may consider favorable, including transactions in which our shareholders may receive a premium for their shares. Our articles of association include provisions that:
•in certain cases, allow our board of directors (i) to place up to 49,726,929 common shares, as well as any treasury shares that the Company may hold from time to time, and (ii) rights to acquire an additional 38,026,929 common shares with affiliates or third parties, without existing shareholders having statutory pre-emptive rights in relation to this share placement;
•allow our board of directors not to register any acquirer of common shares, or several acquirers acting in concert, in our share register as a shareholder with voting rights with respect to more than 15% of our share capital as set forth in the commercial register;
•limit the size of our board of directors to nine members;
•establish advance notice requirements for nominations for election to our board of directors or for proposing matters that can be acted upon by shareholders at shareholder meetings; and
•require two-thirds of the votes represented at a shareholder meeting for amending or repealing the above-mentioned voting and recording restrictions, for amending the provision setting a maximum board size or providing for indemnification of our directors and members of our executive committee and for removing the chairman or any member of the board of directors before the end of his or her term of office.
These and other provisions, alone or together, could delay or prevent takeovers and changes in control. See Exhibits 3.1 and 4.1 to this Annual Report. These provisions could also limit the price that investors might be willing to pay in the future for our common shares, thereby depressing the market price of our common shares.
Our articles of association provide that the competent court with jurisdiction over our registered office in Switzerland will be the exclusive forum for shareholder suits against us, members of our board of directors or members of our executive committee.
Our articles of association provide that the competent court with jurisdiction over our registered office in Switzerland will be the exclusive forum for shareholder suits against us, members of our board of directors or members of our executive committee; provided that the foregoing provision does not apply to claims brought to enforce a duty or liability created by the Securities Act or the Exchange Act or any claim for which the courts in the United States have exclusive jurisdiction. This forum selection provision may impose additional litigation costs on shareholders in pursuing any such claims, particularly if the shareholders do not reside in or near Switzerland and limit a shareholder’s ability to bring a claim in a judicial forum that it finds favorable for disputes with us, members of our board of directors or members of our executive committee, which may discourage lawsuits against us, members of our board of directors and members of our executive committee, although our shareholders will not be deemed to have waived our compliance with federal securities laws and the rules and regulations thereunder.
We are a “smaller reporting company” and the reduced disclosure requirements applicable to us may make our common shares less attractive to investors.
We are a “smaller reporting company,” which allows us to take advantage of certain provisions of the Exchange Act, including only being required to provide two years of audited consolidated financial statements and reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements. If some investors find our common shares less attractive as a result of our reliance on these reduced disclosure obligations, there may be a less active trading market for our common shares and our price of our common shares may be more volatile.
Item 1B. Unresolved Staff Comments
None.
Item 1C. Cybersecurity
Cybersecurity risk management is an integral part of our overall enterprise risk management program. Our cybersecurity risk management program is based on industry best practices and provides a framework for handling cybersecurity threats and incidents, including threats and incidents associated with the use of third-party service providers, and facilitates coordination across different departments of our company. This framework includes steps for assessing the severity of a cybersecurity threat, identifying the source of a cybersecurity threat including whether the cybersecurity threat is associated with a third-party service provider, implementing cybersecurity countermeasures and mitigation strategies and informing management and our board of directors of material cybersecurity threats and incidents. Our cybersecurity team also engages third-party security experts for risk assessment and system enhancements. Our cybersecurity team is responsible for assessing our cybersecurity risk management program. In addition, our cybersecurity team provides annual training to all employees.
Our board of directors has overall oversight responsibility for our risk management and has delegated cybersecurity risk management oversight to the audit committee. The audit committee is responsible for ensuring that management has processes in place designed to identify and evaluate cybersecurity risks to which the company is exposed and implement processes and programs to manage cybersecurity risks and mitigate cybersecurity incidents. The audit committee also reports material cybersecurity risks to our full board of directors. Management is responsible for identifying, considering and assessing material cybersecurity risks on an ongoing basis, establishing processes to ensure that such potential cybersecurity risk exposures are monitored, putting in place appropriate mitigation measures and maintaining cybersecurity programs. Our cybersecurity programs are under the direction of our Chief Information Officer (“CIO”), who receives reports from our cybersecurity team and monitors the prevention, detection, mitigation, and remediation of cybersecurity incidents. Our CIO has more than 25 years of leading technology specialist teams and leads a team of experienced information systems security professionals and information security managers. Management, together with the CIO, regularly present updates at standing audit committee meetings on the company’s cybersecurity programs, material cybersecurity risks and mitigation strategies and provide periodic cybersecurity reports that cover, among other topics, the company’s cybersecurity programs, developments in cybersecurity and updates to the company’s cybersecurity programs and mitigation strategies.
In 2024, we did not identify any cybersecurity threats that have materially affected or are reasonably likely to materially affect our business strategy, results of operations, or financial condition. However, despite our efforts, we cannot eliminate all risks from cybersecurity threats, or provide assurances that we have not experienced an undetected cybersecurity incident. See “Item 1A. Risk Factors.”
Item 2. Properties
We do not own any real property. The table below sets forth the sizes and uses of our leased facilities as of the date of this Annual Report:
|
|
|
|
|
|
|
|
|
| Location |
Primary Function |
Approximate Size |
Biopôle
Route de la Corniche 3B
1066 Epalinges
Switzerland |
Head office |
292 m2 |
430 Mountain Avenue, 4th Floor
New Providence, New Jersey 07974
United States |
Clinical, commercial and U.S. operations |
965 m2 |
84 Wood Lane
London, W12 0BZ
United Kingdom |
Research and preclinical development |
2,200 m2 |
|
|
|
We are not aware of any environmental issues or other constraints that would materially impact the intended use of our facilities.
Item 3. Legal Proceedings
From time to time, we may be subject to various legal proceedings and claims that arise in the ordinary course of our business activities. The results of litigation and claims cannot be predicted with certainty. As of the date of this Annual Report, we do not believe that we are party to any claim or litigation, the outcome of which would, individually or in the aggregate, be reasonably expected to have a material adverse effect on our business.
Item 4. Mine Safety Disclosure
Not applicable.
PART II
Item 5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities
Market Information
Our common shares are listed on the NYSE under the symbol “ADCT.”
Holders
As of March 17, 2025, we had 170 shareholders of record of our common shares. The actual number of shareholders is greater than this number of record holders and includes shareholders who are beneficial owners but whose shares are held in street name by brokers and other nominees or in trust or by other entities.
Dividends
We have never declared or paid cash dividends on our share capital. We intend to retain all available funds and any future earnings, if any, to fund the development and expansion of our business and we do not anticipate paying any cash dividends in the foreseeable future. In addition, the Loan Agreement limits our ability to pay dividends. Any future determination related to dividend policy will be made at the discretion of our board of directors and will depend upon, among other factors, our results of operations, financial condition, capital requirements, contractual restrictions, business prospects and other factors our board of directors may deem relevant.
Under Swiss law, any dividend must be approved by our shareholders. In addition, our auditors must confirm that the dividend proposal of our board of directors to the shareholders conforms to Swiss statutory law and our articles of association. A Swiss corporation may pay dividends only if it has sufficient distributable profits from the previous or current business year (bénéfice résultant du bilan) or brought forward from previous business years (report des bénéfices) or if it has distributable reserves (réserves à libre disposition), each as evidenced by its audited stand-alone statutory balance sheet prepared pursuant to Swiss law and after allocations to reserves required by Swiss law and its articles of association have been deducted. Distributable reserves are generally booked either as free reserves (réserves libres) or as reserves from capital contributions (apports de capital). Distributions out of share capital, which is the aggregate par value of a corporation’s issued shares, may be made only by way of a share capital reduction. See Exhibit 4.1 to this Annual Report.
Securities Authorized for Issuance under Equity Compensation Plans
The following table is a summary of the common shares authorized for issuance under equity compensation plans as of December 31, 2024:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Plan Category |
|
Number of common shares to be issued upon exercise of outstanding options, warrants and rights |
|
Weighted-average exercise price of outstanding options, warrants and rights |
|
Number of common shares remaining available for future issuance under equity compensation plans (excluding common shares to be issued upon exercise of outstanding options, warrants and rights) |
| Equity compensation plans approved by security holders: |
|
|
|
|
|
|
| 2022 Employee Stock Purchase Plan |
|
— |
|
— |
|
— * |
| Conditional Share Capital Plan |
|
|
|
|
|
|
| Options |
|
N/A |
|
N/A |
|
N/A |
| Restricted share units |
|
2,497,349 |
|
|
N/A |
|
N/A |
| Total for Conditional Share Capital Plan |
|
2,497,349 |
|
|
N/A |
|
3,355,883 |
|
| Equity compensation plans not approved by security holders: |
|
|
|
|
|
|
| 2019 Equity Incentive Plan: |
|
|
|
|
|
|
| Options |
|
10,054,588 |
|
|
$ |
9.02 |
|
|
N/A |
| Restricted share units |
|
608,961 |
|
|
N/A |
|
N/A |
| Total for 2019 Equity Incentive Plan |
|
10,663,549 |
|
|
N/A |
|
4,330,311 |
|
| Inducement Plan: |
|
|
|
|
|
|
| Options |
|
642,700 |
|
|
3.63 |
|
|
N/A |
| Restricted share units |
|
N/A |
|
N/A |
|
N/A |
| Total for Inducement Plan |
|
642,700 |
|
|
N/A |
|
357,300 |
|
*The aggregate number of shares that may be issued pursuant to rights granted under the 2022 Employee Stock Purchase Plan is equal to 1% of our common share capital at the plan’s adoption. In addition to the foregoing, on the first day of each calendar year beginning on January 1, 2024 and ending on and including January 1, 2033, the number of common shares available for issuance under the 2022 Employee Stock Purchase Plan is increased by that number of common shares equal to the least of (a) 1% of the common shares outstanding on the final day of the immediately preceding calendar year and (b) such smaller number of common shares as determined by the board of directors. The number of shares reported in this column represents the number of common shares available for future issuance as of December 31, 2024.
The material features of the equity compensation plans adopted without shareholder approval are described below. Any such material plans under which awards in Company shares may currently be granted are included as exhibits to this Annual Report.
2019 Equity Incentive Plan
Plan Administration. The 2019 Equity Incentive Plan is administered by the compensation committee of our board of directors, subject to the board of directors’ discretion to administer or appoint another committee to administer it.
Eligible Participants. The administrator is able to offer equity awards at its discretion under the 2019 Equity Incentive Plan to: (1) any employees of us or any of our subsidiaries; (2) any non-employee directors serving on our board of directors; and (3) any consultants or other advisors to us or any of our subsidiaries. The administrator of the plan may determine that an award for the benefit of a non-employee director will be granted to an affiliate of such director, but only to the extent consistent with the registration of shares offered under the plan on Form S-8 under the Securities Act.
Awards. The maximum number of common shares in respect of which awards may be granted under the 2019 Equity Incentive Plan is 17,741,355 common shares (including share-based equity awards granted to date, less awards forfeited), subject to adjustment in the event of certain corporate transactions or events if necessary to prevent dilution or enlargement of the benefits made available under the plan. Equity incentive awards under the 2019 Equity Incentive Plan may be granted in the form of options, share appreciation rights, restricted shares, restricted share units, performance awards or other share-based awards but not “incentive stock options” for purposes of U.S. tax laws. Options and share appreciation rights will have an exercise price determined by the administrator but will not be less than fair market value of the underlying common shares on the date of grant.
Vesting. The vesting conditions for grants under the equity incentive awards under the 2019 Equity Incentive Plan are set forth in the applicable award documentation.
Termination of Service and Change in Control. In the event of a participant’s termination of employment, the compensation committee may, in its discretion, determine the extent to which an equity incentive award may be exercised, settled, vested, paid or forfeited. In the event of our termination of a participant’s employment without cause or a participant’s resignation for good reason (as defined in the 2019 Equity Incentive Plan) upon or within 18 months following a change in control of the company (as defined in the 2019 Equity Incentive Plan), any awards outstanding to the participant (unless otherwise provided in the award agreement) will immediately vest and settle, and options and share appreciation rights will become fully exercisable.
In the event of a change in control that involves a merger, acquisition or other corporate transaction, any outstanding award not assumed, substituted, replaced or continued in connection with the transaction will immediately vest and settle, and options and share appreciation rights will become fully exercisable. In connection with a change of control, the compensation committee may, in its discretion, take any one or more of the following actions with respect to outstanding awards: (i) cancel any such award, in exchange for a payment in cash, securities or other property or any combination thereof with a value equal to the value of such award based on the per share value of common shares received or to be received by other shareholders in the event (or without payment of consideration if the committee determines that no amount would have been realized upon the exercise of the award or other realization of the participant’s rights); (ii) require the exercise of any outstanding option; (iii) provide for the assumption, substitution, replacement or continuation of any award by the successor or surviving corporation, along with appropriate adjustments with respect to the number and type of securities (or other consideration) of the successor or surviving corporation, subject to any replacement awards, the terms and conditions of the replacement awards (including performance targets) and the grant, exercise or purchase price per share for the replacement awards; (iv) make any other adjustments in the number and type of securities (or other consideration) subject to (a) such awards and in the terms and conditions of such awards in order to prevent the dilution or enlargement of benefits intended to be made available under the 2019 Equity Plan and (b) awards that may be granted in the future; (v) provide that any such award shall be accelerated and become exercisable, payable and/or fully vested with respect to all shares covered thereby or (vi) provide that any award shall not vest, be exercised or become payable as a result of such event.
Termination and Amendment. Unless terminated earlier, the 2019 Equity Incentive Plan will continue for a term of ten years. Our board of directors has the authority to amend or terminate the 2019 Equity Incentive Plan subject to shareholder approval with respect to certain amendments. However, no such action may impair the rights of the recipient of any options unless agreed to by the recipient.
Inducement Plan
Plan Administration. The Inducement Plan is administered by the compensation committee of our board of directors, subject to the board of directors’ discretion to administer or appoint another committee to administer it.
Eligible Participants. The administrator is able to offer equity awards at its discretion under the Inducement Plan to any employee who is eligible to receive an employment inducement grant in accordance with NYSE Listed Company Manual 303A.08.
Awards. The maximum number of common shares in respect of which awards may be granted under the Inducement Plan is 1,000,000 common shares (including share-based equity awards granted to date, less awards forfeited), subject to adjustment in the event of certain corporate transactions or events if necessary to prevent dilution or enlargement of the benefits made available under the plan. Equity incentive awards under the Inducement Plan may be granted in the form of options, share appreciation rights, restricted shares, restricted share units, performance awards or other share-based awards but not “incentive stock options” for purposes of U.S. tax laws. Options and share appreciation rights will have an exercise price determined by the administrator but will not be less than fair market value of the underlying common shares on the date of grant.
Vesting. The vesting conditions for grants under the equity incentive awards under the Inducement Plan are set forth in the applicable award documentation.
Termination of Service and Change in Control. In the event of a participant’s termination of employment, the compensation committee may, in its discretion, determine the extent to which an equity incentive award may be exercised, settled, vested, paid or forfeited. In the event of our termination of a participant’s employment without cause or a participant’s resignation for good reason (as defined in the Inducement Plan) upon or within 18 months following a change in control of the company (as defined in the Inducement Plan), any awards outstanding to the participant (unless otherwise provided in the award agreement) will immediately vest and settle, and options and share appreciation rights will become fully exercisable. In the event of a change in control that involves a merger, acquisition or other corporate transaction, any outstanding award not assumed, substituted, replaced or continued in connection with the transaction will immediately vest and settle, and options and share appreciation rights will become fully exercisable.
In connection with a change of control, the compensation committee may, in its discretion, take any one or more of the following actions with respect to outstanding awards: (i) cancel any such award, in exchange for a payment in cash, securities or other property or any combination thereof with a value equal to the value of such award based on the per share value of common shares received or to be received by other shareholders in the event (or without payment of consideration if the committee determines that no amount would have been realized upon the exercise of the award or other realization of the participant’s rights); (ii) require the exercise of any outstanding option; (iii) provide for the assumption, substitution, replacement or continuation of any award by the successor or surviving corporation, along with appropriate adjustments with respect to the number and type of securities (or other consideration) of the successor or surviving corporation, subject to any replacement awards, the terms and conditions of the replacement awards (including performance targets) and the grant, exercise or purchase price per share for the replacement awards; (iv) make any other adjustments in the number and type of securities (or other consideration) subject to (a) such awards and in the terms and conditions of such awards in order to prevent the dilution or enlargement of benefits intended to be made available under the Inducement Plan and (b) awards that may be granted in the future; (v) provide that any such award shall be accelerated and become exercisable, payable and/or fully vested with respect to all shares covered thereby or (vi) provide that any award shall not vest, be exercised or become payable as a result of such event.
Termination and Amendment. Unless terminated earlier, the Inducement Plan will continue for a term of ten years. Our board of directors has the authority to amend or terminate the Inducement Plan. However, no such action may impair the rights of the recipient of any options unless agreed to by the recipient.
Recent Sales of Unregistered Securities
There were no sales of unregistered equity securities during the period covered by this report.
Purchase of Equity Securities
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Period |
|
Total number of shares purchased |
|
Average price paid per share |
|
Total number of shares purchased as part of publicly announced plans or programs |
|
Maximum number (or approximate dollar value) of shares that may yet be purchased under the plans or programs |
| October 1, 2024 to October 31, 2024 |
|
— |
|
|
N/A |
|
— |
|
|
— |
|
| November 1, 2024 to November 30, 2024 |
|
— |
|
|
N/A |
|
— |
|
|
— |
|
| December 1, 2024 to December 31, 2024 |
|
— |
|
|
N/A |
|
— |
|
|
— |
|
Item 6. Reserved
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations
You should read the following discussion and analysis of our financial condition and results of operations together with our audited consolidated financial statements, including the notes thereto, included in this Annual Report. The following discussion includes forward-looking statements that involve risks, uncertainties and assumptions. Our actual results may differ materially from those anticipated in these forward-looking statements. See “Forward-Looking Statements.”
Overview
ADC Therapeutics is a commercial-stage global pioneer in the field of antibody drug conjugates (“ADCs”). The Company is advancing its proprietary ADC technology to transform the treatment paradigm for patients with hematologic malignancies and solid tumors. We have a validated and differentiated technology platform with multiple payloads, linkers and conjugation chemistry, enabling the design of next-generation potent ADCs with an enhanced therapeutic index. Our strategy is focused on expanding and maximizing the ZYNLONTA opportunity in hematology and pursuing our early-stage research portfolio in solid tumors. We are a pioneer and leader in the ADC field with specialized end-to-end capabilities for developing optimized ADCs. This includes a strong, integrated research & development organization and a validated technology platform with clinical-stage product candidates currently in the pipeline, multiple next-generation ADCs being developed and a proven executional track record that includes ZYNLONTA, the first PBD-based ADC receiving accelerated approval from the FDA, conditional approval from the European Commission and conditional approval from the NMPA in China for the treatment of relapsed or refractory DLBCL after two or more lines of systemic therapy.
In our hematology program, our flagship product, ZYNLONTA, a CD19-directed ADC, received accelerated approval from the U.S. Food and Drug Administration (“FDA”) conditional approval from the European Commission and conditional approval from the NMPA for the treatment of relapsed or refractory DLBCL after two or more lines of systemic therapy.
We are seeking to continue expanding ZYNLONTA internationally, and into earlier lines of DLBCL and indolent lymphomas, including marginal zone lymphoma (“MZL”) and follicular lymphoma (”FL”), as a single agent and in combination through our LOTIS-5 confirmatory Phase 3 clinical trial and LOTIS-7 Phase 1b clinical trial as well as through investigator-initiated trials (“IITs”) at leading institutions. In addition, we are investigating a CD-22 targeted compound, ADCT-602, in collaboration with the MD Anderson Cancer Center in a Phase 1/2 IIT in relapsed or refractory B-cell acute lymphoblastic leukemia.
In our solid tumor program, we have early stage preclinical research programs, including a portfolio of next-generation investigational ADCs targeting Claudin-6, PSMA, NaPi2b, and ASCT2, the most advanced of which are PSMA and Claudin-6. In addition, we are advancing research with a range of payloads, linkers and conjugation technologies against undisclosed targets. The Company is seeking to maximize the value of its solid tumor program through strategic partnerships, collaborations and license arrangements for one or more of its research programs.
Results of Operations
The following table summarizes our results of operations for the years ended December 31, 2024 and 2023:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended December 31, |
| (in thousands, except percentages and per share) |
2024 |
|
2023 |
|
Change |
|
% Change |
| Revenue |
|
|
|
|
|
|
|
| Product revenues, net |
$ |
69,280 |
|
|
$ |
69,060 |
|
|
$ |
220 |
|
|
0.3 |
% |
| License revenues and royalties |
1,557 |
|
|
498 |
|
|
1,059 |
|
|
212.7 |
% |
| Total revenue, net |
70,837 |
|
|
69,558 |
|
|
1,279 |
|
|
1.8 |
% |
|
|
|
|
|
|
|
|
| Operating expense |
|
|
|
|
|
|
|
| Cost of product sales |
(5,949) |
|
|
(2,529) |
|
|
(3,420) |
|
|
135.2 |
% |
| Research and development |
(109,633) |
|
|
(127,127) |
|
|
17,494 |
|
|
(13.8) |
% |
| Selling and marketing |
(44,015) |
|
|
(57,464) |
|
|
13,449 |
|
|
(23.4) |
% |
| General and administrative |
(41,894) |
|
|
(48,424) |
|
|
6,530 |
|
|
(13.5) |
% |
| Total operating expense |
(201,491) |
|
|
(235,544) |
|
|
34,053 |
|
|
(14.5) |
% |
| Loss from operations |
(130,654) |
|
|
(165,986) |
|
|
35,332 |
|
|
(21.3) |
% |
|
|
|
|
|
|
|
|
| Other income (expense) |
|
|
|
|
|
|
|
| Interest income |
12,272 |
|
|
10,540 |
|
|
1,732 |
|
|
16.4 |
% |
| Interest expense |
(50,211) |
|
|
(46,325) |
|
|
(3,886) |
|
|
8.4 |
% |
|
|
|
|
|
|
|
|
| Other, net |
12,457 |
|
|
6,352 |
|
|
6,105 |
|
|
96.1 |
% |
| Total other expense, net |
(25,482) |
|
|
(29,433) |
|
|
3,951 |
|
|
(13.4) |
% |
|
|
|
|
|
|
|
|
| Loss before income taxes |
(156,136) |
|
|
(195,419) |
|
|
39,283 |
|
|
(20.1) |
% |
| Income tax expense |
(166) |
|
|
(39,106) |
|
|
38,940 |
|
|
(99.6) |
% |
| Loss before equity in net losses of joint venture |
(156,302) |
|
|
(234,525) |
|
|
78,223 |
|
|
(33.4) |
% |
| Equity in net losses of joint venture |
(1,544) |
|
|
(5,528) |
|
|
3,984 |
|
|
(72.1) |
% |
| Net loss |
$ |
(157,846) |
|
|
$ |
(240,053) |
|
|
$ |
82,207 |
|
|
(34.2) |
% |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Net loss per share, basic and diluted |
$ |
(1.62) |
|
|
$ |
(2.94) |
|
|
$ |
1.32 |
|
|
(44.9) |
% |
Revenue
Product Revenues, net
We generate product revenue through the sale of ZYNLONTA in the United States. Revenue is recognized when control is transferred to the customer at the net selling price, which includes reductions for gross-to-net (“GTN”) sales adjustments such as government rebates, chargebacks, distributor service fees, other rebates and administrative fees, sales returns and allowances and sales discounts. In the long term, we expect that our product revenue will increase as we execute our business strategy, although our product revenue may fluctuate from period to period based on a number of factors, including patient demand, as well as the timing, dose and duration, of patient therapy and customers’ buying patterns and GTN deductions. We expect a relatively consistent level of GTN sales adjustments as a percentage of gross sales, but may also experience variability in GTN sales adjustments due to additional information and actual experience such as actual rebate and return rates.
Product revenues, net, were $69.3 million for the year ended December 31, 2024 as compared to $69.1 million for the year ended December 31, 2023, an increase of $0.2 million, or 0.3%. The increase is primarily attributable to a higher selling price and favorability in prior period GTN sales adjustments, partially offset by lower sales volume.
License Revenue and Royalties
We generate license revenue and royalties from our strategic agreements for the development and commercialization of ZYNLONTA and other product candidates outside of the United States. Under these agreements, we receive upfront payments and are eligible for certain milestone payments and royalties. See “Item 1. Business—Material Contracts.” We are unable to predict the timing and amounts of license revenue and royalties as meeting milestones is subject to many factors outside of our control and we have limited control over our partners’ commercialization efforts.
License revenues and royalties were $1.6 million for the year ended December 31, 2024 as compared to $0.5 million for the year ended December 31, 2023, an increase of $1.1 million. The increase was attributable to increased royalty revenue from our exclusive license agreement with SOBI to develop and commercialize ZYNLONTA in all territories other than the United States, greater China, Singapore and Japan.
Cost of Product Sales
Cost of product sales primarily includes direct and indirect costs relating to the third-party manufacture and distribution of ZYNLONTA, royalties payable to a collaboration partner based on net product sales of ZYNLONTA and inventory write-downs. We expect that cost of product sales will increase over time as we sell through pre-approval inventory that was previously expensed prior to commercialization under U.S. GAAP. Factors such as inflation may increase our cost of product sales as a percentage of product revenue if we are not able to increase the price at which we sell ZYNLONTA to offset such increases in our cost of product sales.
Cost of product sales were $5.9 million for the year ended December 31, 2024 as compared to $2.5 million for the year ended December 31, 2023, an increase of $3.4 million, or 135.2%. The increase is primarily attributable to higher stability, shipping and storage costs of $1.8 million, a $1.1 million batch cancellation fee and $0.6 million of commercial inventory used for the validation at a new CMO facility which was expensed as a period cost.
Research and Development Expenses
The following table summarizes our research and development expenses for our major development programs for the years ended December 31, 2024 and 2023:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended December 31, |
| (in thousands) |
2024 |
|
2023 |
Change |
| ZYNLONTA |
$ |
58,311 |
|
|
$ |
68,461 |
|
$ |
(10,150) |
|
ADCT-601(1) |
17,624 |
|
|
10,755 |
|
6,869 |
|
| Preclinical product candidates and research pipeline |
17,340 |
|
|
12,830 |
|
4,510 |
|
| ADCT-602 |
1,170 |
|
|
1,851 |
|
(681) |
|
Discontinued programs(2) |
5,056 |
|
|
21,707 |
|
(16,651) |
|
Not allocated to specific programs(3) |
8,211 |
|
|
7,572 |
|
639 |
|
| Share-based compensation |
1,921 |
|
|
3,951 |
|
(2,030) |
|
| Research and development expenses |
$ |
109,633 |
|
|
$ |
127,127 |
|
$ |
(17,494) |
|
(1) ADCT-601 was discontinued in November 2024.
(2) As of December 31, 2024, Cami, ADCT-901 and ADCT-212 were included in Discontinued programs. For the year ended December 31, 2023 these programs were separately presented as major development programs. Prior periods have been recast to conform to the current period presentation.
(3) Includes third-party contracting and employee expenses, as well as expense for preclinical research, storage, shipping and lab consumables that span multiple programs.
Research and development expense consists primarily of employee related expenses, including share-based compensation expense; costs for production of preclinical and clinical-stage product candidates by CMOs; fees and other costs paid to contract research organizations in connection with the performance of preclinical studies and clinical trials; costs of related facilities, materials and equipment; external costs associated with obtaining intellectual property; depreciation; and upfront fees and achieved milestone payments associated with R&D collaboration arrangements.
Our research and development expense may fluctuate from period to period based on a number of factors, including the timing, progress and stage of clinical trials, costs associated with regulatory approval processes and manufacturing costs associated with commercialization activities prior to the receipt of regulatory approval.
Our R&D expenses were $109.6 million for the year ended December 31, 2024 as compared to $127.1 million for the year ended December 31, 2023, a decrease of $17.5 million, or 13.8%, as driven by the following programs and activities:
ZYNLONTA
Research and development expenses for ZYNLONTA were $58.3 million for the year ended December 31, 2024 as compared to $68.5 million for the year ended December 31, 2023, a decrease of $10.2 million, or 14.8%. The overall decrease was primarily due to a net decrease in external clinical trial costs of $5.1 million (decrease in costs associated with LOTIS 5 and other trials offset by an increase in LOTIS 7), lower professional fees of $2.4 million, lower employee expenses of $1.6 million and lower CMC costs of $0.7 million as a result of the implementation of productivity initiatives and focused investment in prioritized development programs.
ADCT-601
Research and development expenses for ADCT-601 were $17.6 million for the year ended December 31, 2024 as
compared to $10.8 million for the year ended December 31, 2023, an increase of $6.9 million, or 63.9%. The increase is
primarily attributable to higher patient enrollment and progress towards the completion of the study. The ADCT-601
program was discontinued in November 2024.
Preclinical product candidates and research pipeline
Research and development expenses associated with our preclinical product candidates and research pipeline were $17.3 million for the year ended December 31, 2024 as compared to $12.8 million for the year ended December 31, 2023, an increase of $4.5 million, or 35.2%. The increase is primarily attributable to increased spending on our research strategy, platform and pipeline initiatives including PSMA and ASCT2.
Discontinued programs
Research and development expenses associated with our discontinued programs including Cami, ADCT-901 and ADCT-212 have decreased to $5.1 million for the year ended December 31, 2024 from $21.7 million, a decrease of $16.7 million, or 76.7%. The decrease was attributable to decreased spending on Cami of $8.7 million, ADCT-212 of $4.6 million and ADCT-901 of $3.4 million.
Share-based compensation
Share-based compensation was $1.9 million for the year ended December 31, 2024 as compared to $4.0 million for the year ended December 31, 2023, a decrease of $2.0 million, or 51.4%. The decrease was driven by fluctuations in our share price as well as forfeitures of awards in connection with employee terminations.
Selling and Marketing Expenses
The following table summarizes our selling and marketing expenses for the year ended December 31, 2024 and 2023:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended December 31, |
| (in thousands) |
2024 |
|
2023 |
|
Change |
| External costs and overhead |
$ |
21,442 |
|
|
$ |
33,006 |
|
|
$ |
(11,564) |
|
Employee expenses(1) |
22,269 |
|
|
24,780 |
|
|
(2,511) |
|
| Share-based compensation expense (reversal) |
304 |
|
|
(322) |
|
|
626 |
|
| Selling and marketing expenses |
$ |
44,015 |
|
|
$ |
57,464 |
|
|
$ |
(13,449) |
|
(1)Excludes share-based compensation expense (reversal).
Selling and marketing costs (“S&M”) are expensed as incurred and are primarily attributable to commercialization of ZYNLONTA in the United States. S&M includes employee costs and share-based compensation expense for commercial employees and external costs related to commercialization (including professional fees, communication costs and IT costs, travel expenses and depreciation of property and equipment).
Selling and marketing expenses were $44.0 million for the year ended December 31, 2024 as compared to $57.5 million for the year ended December 31, 2023, a decrease of $13.4 million, or 23.4%. The net decrease in external costs and overhead was primarily attributable to a reduction of $12.0 million in marketing and advertising expenses as a result of cost cutting initiatives. The decrease in employee expenses was primarily due to lower wages and benefits of $2.3 million primarily due to decreased headcount, as well as lower recruitment costs of $0.2 million. The increase in share-based compensation expense of $0.6 million was primarily due to forfeitures of awards in connection with prior year employee terminations.
General and Administrative Expenses
The following table summarizes our general and administrative expenses for the year ended December 31, 2024 and 2023:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended December 31, |
| (in thousands) |
2024 |
|
2023 |
|
Change |
| External costs and overhead |
$ |
17,683 |
|
|
$ |
20,542 |
|
|
$ |
(2,859) |
|
Employee expenses(1) |
18,705 |
|
|
18,017 |
|
|
688 |
|
| Share-based compensation expense |
5,506 |
|
|
9,865 |
|
|
(4,359) |
|
| General and administrative expenses |
$ |
41,894 |
|
|
$ |
48,424 |
|
|
$ |
(6,530) |
|
(1)Excludes share-based compensation expense.
General and administrative expense includes employee related costs (including wages, benefits and share-based compensation expense) for general and administrative employees, external costs (including, in particular, professional fees, legal fees and costs associated with maintaining patents and other intellectual property, communications costs and IT costs, facility expenses and travel expenses) and depreciation of property and equipment and right-of-use assets.
General and administrative expenses were $41.9 million for the year ended December 31, 2024 as compared to $48.4 million for the year ended December 31, 2023, an overall decrease of $6.5 million, or 13.5%. The decrease in external costs and overhead of $2.9 million was primarily related to lower professional fees of $1.5 million and lower insurance and IT costs of $1.3 million. The increase in employee expenses was primarily due to higher wages and benefits of $0.4 million and higher recruitment costs. The decrease in share-based compensation expense was primarily due to fluctuations in our share price as well as forfeitures of awards in connection with employee terminations.
Other Income (Expense)
Interest Income
Interest income includes interest received from banks on our cash balances. Our policy is to invest funds in a variety of capital preservation instruments, which may include all or a combination of cash and cash equivalents, short-term and long-term interest-bearing instruments, investment-grade securities, and direct or guaranteed obligations of the U.S. government.
Interest income was $12.3 million for the year ended December 31, 2024 as compared to $10.5 million for the year ended December 31, 2023, an increase of $1.7 million, or 16.4%. The increase was primarily due to higher yields received on our cash deposits.
Interest Expense
Interest expense is primarily related to the accretion of our deferred royalty obligation with HCR and the senior secured term loan facility. Interest expense was $50.2 million for the year ended December 31, 2024 as compared to $46.3 million for the year ended December 31, 2023, an increase of $3.9 million, or 8.4%. The increase was related to higher accretion of our deferred royalty obligation with HCR of $5.7 million as a result of the $73.1 million, net of transaction costs, received in June 2023 upon the first commercial sale of ZYNLONTA in the United Kingdom or any European Union country, which increased the liability. This was partially offset by lower interest on our senior secured term loan facility of $1.8 million as a result of a lower effective interest rate.
Other, net
Other, net consists primarily of cumulative catch-up adjustments related to our deferred royalty obligation, changes in the fair value (gains or losses) of the Deerfield warrant obligation and the R&D tax credit from our UK operations.
Other, net as of December 31, 2024 and 2023 included the following:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended December 31, |
| (in thousands) |
2024 |
|
2023 |
|
Change |
|
|
|
|
|
|
| Cumulative catch-up adjustment income, deferred royalty obligation |
$ |
11,178 |
|
|
$ |
4,972 |
|
|
$ |
6,206 |
|
| Deerfield warrant obligation, change in fair value income |
296 |
|
|
497 |
|
|
(201) |
|
| Exchange differences loss |
(80) |
|
|
(52) |
|
|
(28) |
|
| R&D tax credit |
1,063 |
|
|
935 |
|
|
128 |
|
| Total |
$ |
12,457 |
|
|
$ |
6,352 |
|
|
$ |
6,105 |
|
Cumulative catch-up adjustment income, deferred royalty obligation
We periodically assess the expected payments to HCR based on our underlying revenue projections and to the extent the amount or timing of such payments is materially different than our initial estimates we will record a cumulative catch-up adjustment to the deferred royalty obligation. The adjustment to the carrying amount is recognized in Other, net as an adjustment in the period in which the change in estimate occurred. The cumulative catch-up adjustment income was $11.2 million for the year ended December 31, 2024 as compared to $5.0 million for the year ended December 31, 2023, a change of $6.2 million. The change was primarily due to revised revenue forecasts incorporated into the valuation model in 2024 having a greater effect on the expected payments to HCR relative to the 2023 revised revenue forecasts. Revisions in both years were primarily attributable to changes in assumptions in the Company’s updated strategic and development plans, revenue projections and associated timing thereof.
Income Tax Expense
We are subject to corporate taxation in Switzerland. We are also subject to taxation in other jurisdictions in which we operate, in particular, the United States and the United Kingdom, where our two wholly-owned subsidiaries are incorporated. We are entitled under Swiss laws to carry forward any losses incurred for a period of seven years, which could be used to offset future taxable income. We are also entitled under U.S. tax law to carry forward R&D tax credits for a period of up to 20 years, which could be used to offset future taxable income.
We recorded an income tax expense of $0.2 million for the year ended December 31, 2024 as compared to $39.1 million for the year ended December 31, 2023, primarily driven by our U.S. operations and the full valuation allowance recognized on our deferred tax assets.
Income tax expense associated with our U.S. and UK operations was $0.2 million for the year ended December 31, 2024 driven by current period income tax expense of $0.5 million and partially offset by US and UK tax returns true-up benefit of $0.3 million. Generally, current income tax is primarily due to our internal arrangements to reimburse our foreign subsidiaries in the U.S. and the United Kingdom for the services they render to our parent company in Switzerland. Commercial sales in the U.S. also contributed to the current period income tax expense. Ultimately, the net profit at each subsidiary is subject to local income tax. During the year ended December 31, 2024, with respect to our U.S. operations, current income tax expense of $0.3 million was recorded and no deferred tax expense was recorded due to full valuation allowance on deferred tax assets.
Comparatively, our income tax expense of $39.1 million recorded during the year ended December 31, 2023 was driven by the recognition of a $47.8 million valuation allowance on our deferred tax assets due to a change in our intercompany operating and transfer pricing model and estimates of future taxable income and losses. During the year ended December 31, 2023, with respect to our U.S. operations, a deferred tax expense of $37.1 million and current income tax expense of $1.5 million was recorded.
Equity in Net Losses of Joint Venture
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended December 31, |
| (in thousands) |
2024 |
|
2023 |
|
Change |
| Share of Overland ADCT BioPharma net loss |
$ |
(1,544) |
|
|
$ |
(5,528) |
|
|
$ |
(3,984) |
|
We recorded our proportionate share of Overland ADCT BioPharma’s net loss of $1.5 million and $5.5 million for the years ended December 31, 2024 and 2023, respectively. For the year ended December 31, 2024, we recorded our share of Overland ADCT BioPharma’s net loss up until the point at which our share of losses exceeded our interest in Overland ADCT BioPharma. Losses were not recognized in excess of our total investment, as we have not incurred legal or constructive obligations or committed to additional funding on behalf of the joint venture.
Liquidity and Capital Resources
As of December 31, 2024, we had cash and cash equivalents of $250.9 million and believe that our current cash position and capital resources are sufficient to fund our operation and meet capital requirements for at least the next twelve months from the date of filing this Annual Report on Form 10-K.
We plan to continue to fund our operating needs through our existing cash and cash equivalents, revenues from sales of ZYNLONTA, potential milestone and royalty payments under our licensing agreements and additional equity financings, debt financings and/or other forms of financing, as well as funds provided by collaborations. We are continuously exploring strategic collaborations, business combinations, licensing opportunities or similar strategies for our early-stage research pipeline and for clinical development and commercialization of ZYNLONTA and/or our product candidates. However, we may be unable to obtain such financing, licensing and collaboration arrangements on favorable terms, if at all, and if so we may need to prioritize our portfolio and reduce our investment in early stage research and development activities.
Sources of Liquidity and Capital Resources
To date, we have financed our operations primarily through equity financings, convertible debt and senior secured term loan financings, and additional funds provided by collaborations and royalty financings and sales of ZYNLONTA in the United States. For a description of the Loan Agreement, HCR Agreement and other license and collaboration agreements, see “Item 1. Business - Material Contracts.”
In May 2024, we completed an underwritten offering which resulted in net proceeds of approximately $97.4 million. In August 2024, we filed a prospectus relating to an at-the-market offering program, pursuant to which we may offer and sell our common shares from time to time with an aggregate offering price of $100 million, subject to share limitations, through Jefferies LLC acting as sales agent. To date, we have not sold any shares under the program.
Uses of Capital Resources
Our primary uses of capital are, and we expect will continue to be, research and development expenses, selling and marketing expenses, compensation and related expenses, interest and principal payments on debt obligations and other operating expenses. We expect to incur substantial expenses as we continue to devote substantial resources to research and development and marketing and commercialization efforts, in particular to grow ZYNLONTA in the 3L+ DLBCL setting, continue to study and advance ZYNLONTA in earlier lines of therapy and in combinations to potentially expand our market opportunity and further develop our pipeline and our ADC platform. Cash used to fund operating expenses is impacted by the timing of when we pay expenses, as reflected in the change in our outstanding accounts payable and accrued expenses, as well as the timing of collecting receivables from the sale of ZYNLONTA and paying royalties related to our deferred royalty obligation.
Contractual Obligations and Commitments
Our contractual obligations relate to our outstanding indebtedness under the Loan Agreement, as described above, and our lease agreements. For information relating to our scheduled maturities with respect to our lease liabilities and long-term debt see Note 6, “Leases” and Note 10, “Senior secured term loan facility and warrants”, respectively, included in the Notes to our audited consolidated financial statements.
We have future royalty obligations to HCR, under our royalty purchase agreement, which royalty payment amounts and timing is dependent on the future sales results of ZYNLONTA. See Note 12, “Deferred royalty obligation”, included in the Notes to our audited consolidated financial statements for further information.
For information relating to our non-cancelable obligations under third party manufacturing agreements see Note 14, “Commitments and contingencies”, included in the Notes to our audited consolidated financial statements.
The Company has entered into various collaborations with development partners, including in-licensing and manufacturing agreements. These agreements provide for the Company to make potential future milestone and royalty payments that are conditional on success, and that are spread over various stages of development and commercialization, including filing an IND application, commencing or completing multiple clinical development stages, obtaining regulatory approval in multiple countries, and achieving various levels of commercial sales. Due to the nature of these arrangements, the future potential payments related to the attainment of the specified milestones are inherently uncertain, and accordingly, no amounts have been recorded for these future potential payments in the Company’s consolidated balance sheets as of December 31, 2024 and 2023. The aggregate amount of such potential milestone payments (excluding royalty payments), under all such collaboration agreements, was $212.2 million, including approximately $79.3 million contingent on the achievement of various research, development and regulatory approval milestones and approximately $132.9 million in sales-based milestones.
Cash Flows
The following table summarizes our cash flows for the years ended December 31, 2024 and 2023:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended December 31, |
| (in thousands) |
2024 |
|
2023 |
|
Change |
| Net cash (used in) provided by: |
|
|
|
|
|
| Operating activities |
$ |
(123,835) |
|
|
$ |
(118,686) |
|
|
$ |
(5,149) |
|
| Investing activities |
(867) |
|
|
(3,216) |
|
|
2,349 |
|
| Financing activities |
97,054 |
|
|
73,875 |
|
|
23,179 |
|
| Net change in cash and cash equivalents |
$ |
(27,648) |
|
|
$ |
(48,027) |
|
|
$ |
20,379 |
|
Net Cash Used in Operating Activities
Net cash used in operating activities increased to $123.8 million for the year ended December 31, 2024 from $118.7 million for the year ended December 31, 2023, an increase of $5.1 million. The increase in cash used in operating activities on a period over period basis was primarily due to the receipt of the $50.0 million in Sobi license milestone during the year ended December 31, 2023 which was recognized in revenue in December 2022 upon approval of the Marketing Authorisation Application by the European Commission for ZYNLONTA in 3L DLBCL as well as a decrease in deferred income taxes of $37.1 million, partially offset by the lower net loss for the period of $82.2 million attributable to a decrease in operating expenses and the timing of cash payments and receipts.
Net Cash Used in Investing Activities
Net cash used in investing activities decreased to $0.9 million for the year ended December 31, 2024 from $3.2 million for the year ended December 31, 2023, a decrease of $2.3 million. The decrease in net cash used in investing activities primarily relates to the timing of property and equipment purchases.
Net Cash Provided by Financing Activities
Net cash provided by financing activities was $97.1 million for the year ended December 31, 2024 and primarily related to the net proceeds received from the completion of the Company’s 2024 Equity Offering in May 2024. Net cash provided by financing activities was $73.9 million for the year ended December 31, 2023 and primarily related to the proceeds received under the deferred royalty obligation with HCR upon the first commercial sale of ZYNLONTA in the United Kingdom or any European Union country.
Off-Balance Sheet Arrangements
During the periods presented, we did not have, and we do not currently have, any off-balance sheet arrangements.
Critical Accounting Estimates
A summary of the significant accounting policies is provided in Note 2 “Summary of significant accounting policies,” included in the notes to our audited consolidated financial statements.
The preparation of financial statements in accordance with generally accepted accounting principles, or GAAP, requires us to make estimates, assumptions and judgments that affect the reported amounts of assets, liabilities, revenues and expenses, and related disclosures of contingent assets and liabilities.
We evaluate our estimates on an ongoing basis. We base our estimates on historical experience and on various other assumptions that we believe to be reasonable under the circumstances, the results of which form our basis for making judgments about the carrying values of assets and liabilities and the reported amounts of revenues and expenses that are not readily apparent from other sources. Actual results may differ from those estimates under different assumptions and conditions.
Management considers an accounting estimate to be critical if:
•it requires a significant level of estimation uncertainty; and
•changes in the estimate are reasonably likely to have a material effect on our financial condition or results of operations.
We believe the following critical accounting policies and estimates describe the more significant judgments and estimates used in the preparation of our consolidated financial statements.
Product revenues, net
We generate revenue from sales of ZYNLONTA in the U.S. for the treatment of relapsed or refractory DLBCL. Revenue is recognized when control is transferred to the customer at the net selling price, which includes reductions for gross-to-net (“GTN”) sales adjustments such as government rebates, chargebacks, distributor service fees, other rebates and administrative fees, sales returns and allowances and sales discounts.
GTN sales adjustments involve significant estimates and judgment after considering factors including legal interpretations of applicable laws and regulations, historical experience and drug product analogs in the absence of Company experience, payer channel mix, current contract prices under applicable programs, unbilled claims and processing time lags and inventory levels in the distribution channel. We also use information from external sources to identify prescription trends, patient demand, average selling prices, discarded volumes and sales return and allowance data for the Company and analog drug products. Our estimates are subject to inherent limitations of estimates that rely on third-party information, as certain third-party information was itself in the form of estimates and reflect other limitations including lags between the date as of which third-party information is generated and the date on which we receive third-party information. Estimates will be assessed each period and adjusted as required to revise information or actual experience. In particular, the following rebate requires a substantial degree of judgement.
Discarded Drug Rebate
The Infrastructure Investment and Jobs Act requires manufacturers of certain single-source drugs separately paid for under Medicare Part B and marketed in single-dose containers or packages to provide annual refunds (“discarded drug rebate”), if those portions of the dispensed drug that are unused and discarded exceed an applicable percentage defined by statute or regulation. The Centers for Medicare & Medicaid Services (the “CMS”) finalized regulations to implement this section on November 18, 2022, and the provision went into effect on January 1, 2023. In particular, the estimate for the discarded drug rebate requires a substantial degree of judgement.
We began estimating and recording a provision for the discarded drug rebate as a GTN sales adjustment beginning in the first quarter of 2023. The provision is recorded to Other current liabilities or Other long-term liabilities depending on when the annual refunds are expected to come due. The significant assumptions used to estimate the discarded drug rebate include legal interpretations of applicable laws and regulations, historical experience with discarded volumes and time lags in the processing of claims and invoicing from CMS. We use a number of factors to estimate the discarded drug rebate, including information from external sources to identify the Company’s discarded volumes and information from CMS on discarded volumes. We have now received from CMS the first annual report and invoice for 2023, the payment of which has been paid in the first quarter of 2025, and was generally consistent with our estimate and no significant prior period adjustments were made. We will continue to rely on projection methodologies and expect annual reports to be received from CMS. Given the annual nature of the proposed reporting schedule we will continue to estimate periodically discarded drug rebate liabilities.
Deferred royalty obligation
On August 25, 2021, we entered into a royalty purchase agreement with certain entities managed by Healthcare Royalty Partners (“HCR”). We accounted for the initial cash received as debt, less transaction costs and will subsequently account for the value of the debt at amortized cost. The amount received by us will be accreted to the total estimated royalty payments over the life of the agreement which will be recorded as interest expense. The carrying value of the debt will decrease for royalty payments made to HCR based on actual net sales and licensing revenue.
To determine the accretion of the liability related to the deferred royalty obligation, we are required to estimate the total amount of future royalty payments and estimated timing of such payment to HCR based on our revenue projections. The Company uses a third party valuation firm to assist in determining the total amount of future royalty payments and estimated timing of such payment to HCR using an option pricing Monte Carlo simulation model.
The significant assumptions used to estimate the HCR deferred royalty obligation accretion of the liability include the revenue projections and timing of payments. At each reporting period, we assess the expected payments to HCR based on its underlying revenue projections and to the extent the amount or timing of such payments is materially different than its initial estimates we will record a cumulative catch-up adjustment to the deferred royalty obligation. The adjustment to the carrying amount is recognized in earnings as an adjustment to Other, net in the period in which the change in estimate occurred.
The exact amount and timing of repayment is likely to be different each reporting period as compared to those estimated based on our revenue projections. A significant increase or decrease in actual net sales of ZYNLONTA compared to the Company’s revenue projections, as well as ZYNLONTA in other indications as well as licensing revenue could change the royalty rate and royalty cap due to HCR, which could materially impact the debt obligation as well as interest expense associated with the royalty purchase agreement. Also, our total obligation to HCR can vary depending on the achievement of the sales milestones as well as the timing of a change in control event.
Recently Issued and Adopted Accounting Pronouncements
Refer to Note 2, “Summary of significant accounting policies” to our audited consolidated financial statements for recently adopted accounting pronouncements and recently issued accounting pronouncements not yet adopted as of the date of this Annual Report.
Item 7A. Quantitative and Qualitative Disclosures About Market Risk
We are not required to provide the information required by this Item 7A as we are a smaller reporting company.
Item 8. Financial Statements and Supplementary Data
INDEX TO AUDITED CONSOLIDATED FINANCIAL STATEMENTS
|
|
|
|
|
|
| Audited Consolidated Financial Statements — ADC Therapeutics SA |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Report of Independent Registered Public Accounting Firm
To the Board of Directors and Shareholders of ADC Therapeutics SA
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of ADC Therapeutics SA and its subsidiaries (the “Company”) as of December 31, 2024 and 2023, and the related consolidated statements of operations, comprehensive loss, changes in shareholders’ equity (deficit) and cash flows for the years then ended, including the related notes (collectively referred to as the “consolidated financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2024 and 2023, and the results of its operations and its cash flows for the years then ended in conformity with accounting principles generally accepted in the United States of America.
Basis for Opinion
These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s consolidated financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits of these consolidated financial statements in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company's internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. We believe that our audits provide a reasonable basis for our opinion.
Critical Audit Matters
The critical audit matters communicated below are matters arising from the current period audit of the consolidated financial statements that were communicated or required to be communicated to the audit committee and that (i) relate to accounts or disclosures that are material to the consolidated financial statements and (ii) involved our especially challenging, subjective, or complex judgments. The communication of critical audit matters does not alter in any way our opinion on the consolidated financial statements, taken as a whole, and we are not, by communicating the critical audit matters below, providing separate opinions on the critical audit matters or on the accounts or disclosures to which they relate.
Deferred royalty obligation with HealthCare Royalty Partners
As described in Note 12 to the consolidated financial statements, on August 25, 2021, the Company entered into a royalty purchase agreement with certain entities managed by HealthCare Royalty Partners (HCR) for up to $325.0 million. Under the terms of the agreement, the Company received gross proceeds of $225.0 million upon closing and received an additional $75.0 million during the year ended December 31, 2023. The Company’s aggregate royalty obligations are capped at 2.50 times the amount paid by HCR under the agreement or at 2.25 times the amount paid by HCR under the agreement if HCR receives royalty payments exceeding a mid-nine-digit amount on or prior to March 31, 2029 (the “Royalty Cap”). Once the Royalty Cap is reached, the royalty purchase agreement will terminate.
The Company evaluated the terms of the royalty purchase agreement and concluded that the features of the investment amount are similar to those of a debt instrument. Accordingly, the Company recorded a liability relating to the initial gross proceeds received as debt less transaction costs in August 2021, and increased the liability in June 2023 for the eligible amount received as debt less transaction costs upon first commercial sale of ZYNLONTA in Europe. The Company accounts for the value of the debt at amortized cost. The amounts received by the Company will be accreted to the total estimated amount of the royalty payments necessary to extinguish the Company’s obligation under the agreement, which will be recognized as interest expense. The carrying value of the debt decreases for royalty payments made to HCR based on actual net sales and licensing revenue. To determine the accretion of the liability related to the deferred royalty obligation, the Company is required to estimate the total amount of future royalty payments and estimate the timing of such payments to HCR based on the Company's revenue projections. The Company uses a third party valuation firm to assist in determining the total amount of future royalty payments and estimates timing of such payment to HCR using an option pricing Monte Carlo simulation model. At each reporting period, the Company assesses the expected payments to HCR based on its underlying revenue projections and to the extent the amount or timing of such payments is materially different than its initial estimates, the Company will record a cumulative catch-up adjustment to the deferred royalty obligation. The Company’s deferred royalty obligation recognized within liabilities was $326.7 million as of December 31, 2024. The significant assumptions used to estimate the HCR deferred royalty obligation accretion of the liability include the revenue projections and timing of payments.
The principal considerations for our determination that performing procedures relating to the deferred royalty obligation with HCR is a critical audit matter are (i) the significant judgment by management when determining the value of the deferred royalty obligation; (ii) a high degree of auditor judgment, subjectivity, and effort in performing procedures and evaluating the audit evidence obtained related to management’s assumptions related to the revenue projections used to determine the timing of expected cash outflows through the Monte Carlo simulation model; and (iii) the audit effort involved the use of professionals with specialized skills and knowledge.
Addressing the matter involved performing procedures and evaluating audit evidence in connection with forming our overall opinion on the consolidated financial statements. These procedures included, among others, (i) testing management’s process for developing the deferred royalty obligation; (ii) evaluating the appropriateness of the Monte Carlo simulation model; (iii) testing the completeness and accuracy of underlying data used in the model; and (iv) evaluating the reasonableness of the significant assumptions used by management related to future revenue projections and the timing of payments considering external market and industry data. Professionals with specialized skill and knowledge were used to assist in evaluating (i) the appropriateness of the Monte Carlo simulation model and (ii) reasonableness of the revenue projections.
Product Revenue – Gross-to-net (GTN) sales adjustment – Discarded Drug Rebate
As described in Notes 2 and 16 to the consolidated financial statements, product revenue is recognized at the net selling price, which includes reductions for gross-to-net (“GTN”) sales adjustments such as government rebates, chargebacks, distributor service fees, other rebates and administrative fees, sales returns and allowances and sales discounts. Government rebates include the discarded drug rebate. The Infrastructure Investment and Jobs Act requires manufacturers of certain single-source drugs separately paid for under Medicare Part B and marketed in single-dose containers to refund the dispensed drug unused and discarded exceeding an applicable percentage defined by statute or regulation. The accrual for such rebate recognized within accrued expenses and other current liabilities was $15.1 million as of December 31, 2024. The significant assumptions used to estimate the discarded drug rebate include historical experience with discarded volumes considering time lags in the processing of claims and invoicing from the Centers for Medicare & Medicaid Services as well as legal interpretations of applicable laws and regulations used in the rebate calculation.
The principal considerations for our determination that performing procedures relating to the discarded drug rebate GTN sales adjustment is a critical audit matter are (i) the significant judgment made by management when determining the discarded drug rebate sales adjustment; and (ii) a high degree of auditor judgment, subjectivity, and effort in performing procedures and evaluating the audit evidence obtained related to the valuation of the discarded drug rebate and management’s assumptions related to historical experience with discarded volumes as well as legal interpretations of applicable laws and regulations used in the rebate calculation.
Addressing the matter involved performing procedures and evaluating audit evidence in connection with forming our overall opinion on the consolidated financial statements. These procedures included, among others, (i) testing management’s process for developing the discarded drug rebate estimates; (ii) testing the accuracy of the rebate calculation; (iii) testing the completeness and accuracy of inputs underlying data used in the calculation of the rebate; and (iv) evaluating the reasonableness of significant assumptions used by management related to the discarded volumes considering historical experience as well as legal interpretations of applicable laws and regulations used in the rebate calculation, and whether assumptions were consistent with evidence obtained in other areas of the audit.
/s/ PricewaterhouseCoopers SA
Pully, Switzerland
March 27, 2025
We have served as the Company's auditor since 2015.
CONSOLIDATED BALANCE SHEETS
(in thousands, except share amounts)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
As of December 31, |
|
|
|
2024 |
|
2023 |
| ASSETS |
|
|
|
|
|
| Current assets |
|
|
|
|
|
Cash and cash equivalents |
|
|
$ |
250,867 |
|
|
$ |
278,598 |
|
| Accounts receivable, net |
|
|
20,316 |
|
|
25,182 |
|
| Inventory |
|
|
18,387 |
|
|
16,177 |
|
Prepaid expenses |
|
|
8,370 |
|
|
10,344 |
|
| Other current assets |
|
|
9,450 |
|
|
5,990 |
|
Total current assets |
|
|
307,390 |
|
|
336,291 |
|
|
|
|
|
|
|
Property and equipment, net |
|
|
5,075 |
|
|
5,622 |
|
Operating lease right-of-use assets |
|
|
8,354 |
|
|
10,511 |
|
| Interest in joint venture |
|
|
— |
|
|
1,647 |
|
|
|
|
|
|
|
Other long-term assets |
|
|
1,161 |
|
|
711 |
|
|
|
|
|
|
|
Total assets |
|
|
$ |
321,980 |
|
|
$ |
354,782 |
|
|
|
|
|
|
|
| LIABILITIES AND SHAREHOLDERS’ (DEFICIT) EQUITY |
|
|
|
|
|
| Current liabilities |
|
|
|
|
|
Accounts payable |
|
|
$ |
18,029 |
|
|
$ |
15,569 |
|
Accrued expenses and other current liabilities |
|
|
62,440 |
|
|
52,101 |
|
|
|
|
|
|
|
|
|
|
|
|
|
Total current liabilities |
|
|
80,469 |
|
|
67,670 |
|
|
|
|
|
|
|
| Deferred royalty obligation, long-term |
|
|
320,093 |
|
|
303,572 |
|
| Senior secured term loans |
|
|
113,632 |
|
|
112,730 |
|
Operating lease liabilities, long-term |
|
|
7,995 |
|
|
10,180 |
|
| Other long-term liabilities |
|
|
2,433 |
|
|
8,879 |
|
Total liabilities |
|
|
524,622 |
|
|
503,031 |
|
Commitments and contingencies (Note 14) |
|
|
|
|
|
| Shareholders’ (deficit) equity |
|
|
|
|
|
Common shares, at CHF 0.08 par value |
|
|
8,425 |
|
|
7,312 |
|
Issued shares: 101,606,376 at December 31, 2024 and 89,041,946 at December 31, 2023; outstanding shares: 98,861,402 at December 31, 2024 and 82,293,137 at December 31, 2023 |
|
|
|
|
|
Additional paid-in capital |
|
|
1,283,892 |
|
|
1,180,545 |
|
Treasury shares |
|
|
(220) |
|
|
(541) |
|
At December 31, 2024: 2,744,974 and December 31, 2023: 6,748,809 |
|
|
|
|
|
Accumulated other comprehensive loss |
|
|
(1,421) |
|
|
(93) |
|
Accumulated deficit |
|
|
(1,493,318) |
|
|
(1,335,472) |
|
Total shareholders’ (deficit) equity |
|
|
(202,642) |
|
|
(148,249) |
|
|
|
|
|
|
|
Total liabilities and shareholders’ (deficit) equity |
|
|
$ |
321,980 |
|
|
$ |
354,782 |
|
The accompanying notes are an integral part of these consolidated financial statements.
CONSOLIDATED STATEMENTS OF OPERATIONS
(in thousands, except share and per share amounts)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
For the Years Ended December 31, |
|
|
|
2024 |
|
2023 |
|
|
| Revenue |
|
|
|
|
|
|
|
| Product revenues, net |
|
|
$ |
69,280 |
|
|
$ |
69,060 |
|
|
|
| License revenues and royalties |
|
|
1,557 |
|
|
498 |
|
|
|
| Total revenue, net |
|
|
70,837 |
|
|
69,558 |
|
|
|
| Operating expense |
|
|
|
|
|
|
|
| Cost of product sales |
|
|
(5,949) |
|
|
(2,529) |
|
|
|
| Research and development |
|
|
(109,633) |
|
|
(127,127) |
|
|
|
| Selling and marketing |
|
|
(44,015) |
|
|
(57,464) |
|
|
|
| General and administrative |
|
|
(41,894) |
|
|
(48,424) |
|
|
|
| Total operating expense |
|
|
(201,491) |
|
|
(235,544) |
|
|
|
| Loss from operations |
|
|
(130,654) |
|
|
(165,986) |
|
|
|
| Other income (expense) |
|
|
|
|
|
|
|
| Interest income |
|
|
12,272 |
|
|
10,540 |
|
|
|
| Interest expense |
|
|
(50,211) |
|
|
(46,325) |
|
|
|
|
|
|
|
|
|
|
|
| Other, net |
|
|
12,457 |
|
|
6,352 |
|
|
|
|
|
|
|
|
|
|
|
| Total other expense, net |
|
|
(25,482) |
|
|
(29,433) |
|
|
|
Loss before income taxes |
|
|
(156,136) |
|
|
(195,419) |
|
|
|
| Income tax expense |
|
|
(166) |
|
|
(39,106) |
|
|
|
| Loss before equity in net losses of joint venture |
|
|
(156,302) |
|
|
(234,525) |
|
|
|
| Equity in net losses of joint venture |
|
|
(1,544) |
|
|
(5,528) |
|
|
|
| Net loss |
|
|
$ |
(157,846) |
|
|
$ |
(240,053) |
|
|
|
|
|
|
|
|
|
|
|
Net loss per share |
|
|
|
|
|
|
|
Net loss per share, basic and diluted |
|
|
$ |
(1.62) |
|
|
$ |
(2.94) |
|
|
|
Weighted average shares outstanding, basic and diluted |
|
|
97,159,966 |
|
81,712,166 |
|
|
The accompanying notes are an integral part of these consolidated financial statements.
CONSOLIDATED STATEMENTS OF COMPREHENSIVE LOSS
(in thousands)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
For the Years Ended December 31, |
|
|
|
2024 |
|
2023 |
|
|
Net loss |
|
|
$ |
(157,846) |
|
|
$ |
(240,053) |
|
|
|
|
|
|
|
|
|
|
|
| Other comprehensive (loss) income: |
|
|
|
|
|
|
|
| Remeasurement of defined benefit pension liability |
|
|
(1,044) |
|
|
(1,854) |
|
|
|
Foreign currency translation adjustment |
|
|
(185) |
|
|
376 |
|
|
|
| Other comprehensive loss before share of other comprehensive loss in joint venture |
|
|
(1,229) |
|
|
(1,478) |
|
|
|
| Share of other comprehensive loss in joint venture |
|
|
(103) |
|
|
(438) |
|
|
|
| Other comprehensive loss |
|
|
(1,332) |
|
|
(1,916) |
|
|
|
|
|
|
|
|
|
|
|
| Total comprehensive loss |
|
|
$ |
(159,178) |
|
|
$ |
(241,969) |
|
|
|
The accompanying notes are an integral part of these consolidated financial statements.
CONSOLIDATED STATEMENTS OF CHANGES IN SHAREHOLDERS’ (DEFICIT) EQUITY
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| (in thousands, except share amounts) |
|
|
Number of shares |
|
Common shares, par value |
|
Additional paid-in capital |
|
Number of shares (held or received)/delivered |
|
Treasury
shares |
|
Accumulated other comprehensive
(loss) income |
|
Accumulated
deficit |
|
Total |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| December 31, 2022 |
|
|
89,041,946 |
|
|
$ |
7,312 |
|
|
$ |
1,166,414 |
|
|
(8,399,419) |
|
|
$ |
(679) |
|
|
$ |
1,823 |
|
|
$ |
(1,095,419) |
|
|
$ |
79,451 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Loss for the period |
|
|
— |
|
|
$ |
— |
|
|
$ |
— |
|
|
— |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
(240,053) |
|
|
$ |
(240,053) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Remeasurement of defined benefit pension liability |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
(1,854) |
|
|
— |
|
|
(1,854) |
|
|
| Foreign currency translation adjustment |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
376 |
|
|
— |
|
|
376 |
|
|
| Other comprehensive loss before share of other comprehensive loss in joint venture |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
(1,478) |
|
|
— |
|
|
(1,478) |
|
|
| Share of other comprehensive loss in joint venture |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
(438) |
|
|
— |
|
|
(438) |
|
|
| Total other comprehensive loss |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
(1,916) |
|
|
— |
|
|
(1,916) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Total comprehensive loss for the period |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
(1,916) |
|
|
(240,053) |
|
|
(241,969) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Vestings of RSUs |
|
|
— |
|
|
— |
|
|
(111) |
|
|
1,330,081 |
|
|
111 |
|
|
— |
|
|
— |
|
|
— |
|
|
| Issuance of shares, 2022 Employee Stock Purchase Plan |
|
|
— |
|
|
— |
|
|
747 |
|
|
320,529 |
|
|
27 |
|
|
— |
|
|
— |
|
|
774 |
|
|
| Share-based compensation expense |
|
|
— |
|
|
— |
|
|
13,495 |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
13,495 |
|
|
|
|
|
— |
|
|
— |
|
|
14,131 |
|
|
1,650,610 |
|
|
138 |
|
|
— |
|
|
— |
|
|
14,269 |
|
|
| December 31, 2023 |
|
|
89,041,946 |
|
|
$ |
7,312 |
|
|
$ |
1,180,545 |
|
|
(6,748,809) |
|
|
$ |
(541) |
|
|
$ |
(93) |
|
|
$ |
(1,335,472) |
|
|
$ |
(148,249) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Loss for the period |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
(157,846) |
|
|
(157,846) |
|
|
| Remeasurement of defined benefit pension liability |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
(1,044) |
|
|
— |
|
|
(1,044) |
|
|
| Foreign currency translation adjustment |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
(185) |
|
|
— |
|
|
(185) |
|
|
| Other comprehensive loss before share of other comprehensive loss in joint venture |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
(1,229) |
|
|
— |
|
|
(1,229) |
|
|
| Share of other comprehensive loss in joint venture |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
(103) |
|
|
— |
|
|
(103) |
|
|
| Total other comprehensive loss |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
(1,332) |
|
|
— |
|
|
(1,332) |
|
|
| Total comprehensive loss for the period |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
(1,332) |
|
|
(157,846) |
|
|
(159,178) |
|
|
| Vestings of RSUs |
|
|
2,146,768 |
|
|
192 |
|
|
(236) |
|
|
502,929 |
|
|
40 |
|
|
4 |
|
|
— |
|
|
— |
|
|
| Exercise of options |
|
|
5,750 |
|
|
— |
|
|
219 |
|
|
108,721 |
|
|
9 |
|
|
— |
|
|
— |
|
|
228 |
|
|
| Issuance of shares, 2022 Employee Stock Purchase Plan |
|
|
— |
|
|
— |
|
|
526 |
|
|
392,185 |
|
|
32 |
|
|
— |
|
|
— |
|
|
558 |
|
|
| Issuance of shares, underwritten offering, net of transaction costs |
|
|
10,411,912 |
|
|
921 |
|
|
59,345 |
|
|
3,000,000 |
|
|
240 |
|
|
— |
|
|
— |
|
|
60,506 |
|
|
| Issuance of warrants, underwritten offering, net of transaction costs |
|
|
— |
|
|
— |
|
|
36,925 |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
36,925 |
|
|
| Share-based compensation expense |
|
|
— |
|
|
— |
|
|
6,568 |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
6,568 |
|
|
|
|
|
12,564,430 |
|
|
1,113 |
|
|
103,347 |
|
|
4,003,835 |
|
|
321 |
|
|
4 |
|
|
— |
|
|
104,785 |
|
|
| December 31, 2024 |
|
|
101,606,376 |
|
|
$ |
8,425 |
|
|
$ |
1,283,892 |
|
|
(2,744,974) |
|
|
$ |
(220) |
|
|
$ |
(1,421) |
|
|
$ |
(1,493,318) |
|
|
$ |
(202,642) |
|
|
The accompanying notes are an integral part of these consolidated financial statements.
CONSOLIDATED STATEMENTS OF CASH FLOWS
(in thousands)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
For the Years Ended December 31, |
| |
|
|
2024 |
|
2023 |
|
|
| Cash used in operating activities |
|
|
|
|
|
|
|
| Net loss |
|
|
$ |
(157,846) |
|
|
$ |
(240,053) |
|
|
|
| Adjustments to reconcile net loss to net cash used in operations: |
|
|
|
|
|
|
|
| Deferred income taxes |
|
|
— |
|
|
37,104 |
|
|
|
| Share-based compensation expense |
|
|
6,568 |
|
|
13,495 |
|
|
|
| Accretion expense of deferred royalty obligation |
|
|
28,224 |
|
|
19,207 |
|
|
|
| Cumulative catch-up adjustment, deferred royalty obligation |
|
|
(11,178) |
|
|
(4,972) |
|
|
|
| Write-downs of inventory |
|
|
1,518 |
|
|
1,608 |
|
|
|
| Depreciation |
|
|
1,334 |
|
|
1,187 |
|
|
|
|
|
|
|
|
|
|
|
| Amortization of operating lease right-of-use assets |
|
|
1,942 |
|
|
2,080 |
|
|
|
|
|
|
|
|
|
|
|
| Share of results in joint venture |
|
|
1,544 |
|
|
5,528 |
|
|
|
|
|
|
|
|
|
|
|
| Change in defined benefit pension liabilities |
|
|
(186) |
|
|
(712) |
|
|
|
| Warrant obligations, decrease in fair value |
|
|
(296) |
|
|
(497) |
|
|
|
| Amortization of debt discount, senior secured term loan |
|
|
902 |
|
|
3,016 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Other |
|
|
(187) |
|
|
52 |
|
|
|
|
|
|
|
|
|
|
|
| Changes in operating assets and liabilities: |
|
|
|
|
|
|
|
| Accounts receivable, net |
|
|
4,771 |
|
|
47,789 |
|
|
|
| Inventory |
|
|
(3,728) |
|
|
(5,712) |
|
|
|
| Other current assets |
|
|
(1,480) |
|
|
7,289 |
|
|
|
| Other long-term assets |
|
|
(456) |
|
|
200 |
|
|
|
| Accounts payable |
|
|
2,462 |
|
|
3,173 |
|
|
|
|
|
|
|
|
|
|
|
| Accrued expenses and other short-term liabilities |
|
|
11,140 |
|
|
(14,143) |
|
|
|
|
|
|
|
|
|
|
|
| Operating lease liabilities |
|
|
(1,958) |
|
|
(1,716) |
|
|
|
| Other long-term liabilities |
|
|
(6,925) |
|
|
7,391 |
|
|
|
| Net cash used in operating activities |
|
|
(123,835) |
|
|
(118,686) |
|
|
|
| Cash flows used in investing activities |
|
|
|
|
|
|
|
| Payment for purchases of property and equipment |
|
|
(867) |
|
|
(3,216) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Net cash used in investing activities |
|
|
(867) |
|
|
(3,216) |
|
|
|
|
|
|
|
|
|
|
|
| Cash flows provided by (used in) financing activities |
|
|
|
|
|
|
|
| Proceeds from common shares, 2024 Equity Offering, net of transaction costs |
|
|
60,506 |
|
|
— |
|
|
|
| Proceeds from 2024 Pre-Funded Warrants, net of transaction costs |
|
|
36,925 |
|
|
— |
|
|
|
| Proceeds from deferred royalty transaction, net of transaction costs |
|
|
— |
|
|
73,102 |
|
|
|
| Proceeds from share issuance under stock purchase plan |
|
|
558 |
|
|
773 |
|
|
|
| Proceeds from the exercise of options |
|
|
228 |
|
|
— |
|
|
|
| Taxes paid related to net settled equity awards |
|
|
(1,163) |
|
|
— |
|
|
|
| Net cash provided by financing activities |
|
|
97,054 |
|
|
73,875 |
|
|
|
| Net decrease in cash and cash equivalents |
|
|
(27,648) |
|
|
(48,027) |
|
|
|
| Exchange (losses)/gains on cash and cash equivalents |
|
|
(83) |
|
|
184 |
|
|
|
| Cash and cash equivalents at beginning of year |
|
|
278,598 |
|
|
326,441 |
|
|
|
| Cash and cash equivalents at end of year |
|
|
$ |
250,867 |
|
|
$ |
278,598 |
|
|
|
| Supplemental Cash Flow Information: |
|
|
|
|
|
|
|
| Interest paid |
|
|
$ |
15,702 |
|
|
$ |
15,387 |
|
|
|
| Interest received |
|
|
13,707 |
|
|
9,725 |
|
|
|
| Payments made under royalty financing transaction |
|
|
5,384 |
|
|
8,709 |
|
|
|
|
|
|
|
|
|
|
|
| Supplemental Non-Cash Investing Activities: |
|
|
|
|
|
|
|
| Capital expenditures recorded in Accounts payable and Accrued expenses and other current liabilities |
|
|
— |
|
|
65 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The accompanying notes are an integral part of these consolidated financial statements.
|
|
|
|
| ADC Therapeutics SA |
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS |
| (in thousands, except per share amounts) |
1.Description of Business and Organization
ADC Therapeutics is a commercial-stage global pioneer in the field of antibody drug conjugates (“ADCs”) committed to advancing its proprietary ADC technology platform to transform the cancer treatment paradigm for patients with hematologic malignancies and solid tumors.
Since its inception, the Company has devoted its resources to developing a validated and differentiated technology platform with multiple payloads, linkers and conjugation chemistry, enabling the design of next-generation potent ADCs with an enhanced therapeutic index. The Company generates sales from its flagship product, ZYNLONTA, which is currently approved in the U.S. for the treatment of relapsed or refractory diffuse large B-cell lymphoma (“DLBCL”) in the 3L setting and has also been granted conditional approval from the European Commission and conditional approval from the China National Medical Products Administration. Additionally, the Company is seeking to expand ZYNLONTA into new indications and earlier lines of therapy while pursuing our early-stage solid tumor programs.
The Company was incorporated on June 6, 2011 under the laws of Switzerland, with its registered office located at Route de la Corniche 3B, 1066 Epalinges, Switzerland. The Company has two wholly-owned subsidiaries: ADC Therapeutics America, Inc. (“ADCT America”), which was incorporated in Delaware, USA on December 10, 2014 and ADC Therapeutics (UK) Ltd (“ADCT UK”), incorporated in England on December 12, 2014. The Company and its two subsidiaries form the ADCT Group (the “Group”). ADC Therapeutics (NL) B.V., previously a wholly-owned subsidiary of the Company, was dissolved effective October 4, 2024.
All references to “ADC Therapeutics,” “the Company", “we,” “us,” and “our” refer to ADC Therapeutics SA and its consolidated subsidiaries unless otherwise indicated.
2.Summary of Significant Accounting Policies
Basis of preparation and principles of consolidation
The accompanying consolidated financial statements have been prepared in accordance with generally accepted accounting principles in the United States (“U.S. GAAP”) and include the accounts of the Company and its wholly-owned subsidiaries. Intercompany transactions and balances have been eliminated in consolidation.
Use of Estimates
The preparation of financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the reported amounts of assets, liabilities, revenues and expenses, and related disclosures in the consolidated financial statements and accompanying notes. Management bases its estimates on historical experience and on assumptions believed to be reasonable under the circumstances. Actual results could differ materially from those estimates.
Risks and Uncertainties
The Company is subject to risks and uncertainties common to companies in the global biotechnology and pharmaceutical industries, including, but not limited to, risks of failure or unsatisfactory results of its research and development efforts and clinical studies, the need for significant capital to fund the continued development of its products and pipeline, the need to obtain and maintain marketing approval for its product and candidates, the need to successfully commercialize and gain market acceptance of any of its product candidates that obtain regulatory approval, dependence on strategic relationships with collaboration and commercialization partners and on key personnel, securing and protecting proprietary technology, compliance with government regulations, competition, and dependence on third-party service providers such as contract research organizations (“CROs”), contract manufacturing organizations (“CMOs”), other suppliers, and third-party logistics providers.
Concentrations of Risk
Foreign exchange risk
The Company operates internationally and is exposed to foreign exchange risk arising from various currency exposures, primarily with respect to British pounds, Euros and Swiss francs. Transaction exposure arises because the amount of local currency paid or received in transactions denominated in foreign currencies may vary due to changes in exchange rates. Foreign exchange risk arises from:
–forecast costs denominated in a currency other than the entity’s functional currency;
–recognized assets and liabilities denominated in a currency other than the entity's functional currency; and
–net investments in foreign operations.
Management believes that foreign exchange risk is minimal, as the Company pays invoices mainly in U.S. dollars and holds cash principally in U.S. dollars.
Interest rate risk
Interest rate risk arises from movements in interest rates which could have adverse effects on the Company's net loss or financial position. Changes in interest rates cause variations in interest income and expenses on interest-bearing assets and liabilities, and on the value of the net defined benefit pension obligation. In relation to the royalty purchase agreement with HCR, the Company is obligated to pay interest in the form of royalties in connection with certain net sales and licensing revenue. As the effective interest rate (“EIR”) on the deferred royalty obligation does not depend on market performance, the exposure to interest rate and market risk is deemed low. See Note 12, “Deferred royalty obligation” for further information. In regards to the senior secured term loans, the interest rate is variable and dependent upon market factors. The Company will update the EIR at the end of each reporting period for changes in the rate. See Note 10, "Senior secured term loan facility and warrants" for further information.
Credit risk
Credit risk is the risk that a counterparty will not meet its obligations under a financial instrument or customer contract, leading to a financial loss. The Company is exposed to credit risk from its operating activities and from its financing activities including deposits with banks and other financial institutions. The Company’s cash and cash equivalents accounts are maintained with well established, highly rated financial institutions. The Company’s wholly-owned subsidiaries are solvent, are managed on a cost-plus service provider basis, and are supported by the Company as the parent.
To date, the Company’s only source of product revenue, which commenced during May 2021, has been sales of ZYNLONTA only in the U.S., which is sold primarily through wholesale distributors. In addition, the Company earns license revenues and royalties through its license agreements with third parties. See Note 16 “Revenue” for further information. We continuously monitor the creditworthiness of our customers and have internal policies regarding customer credit limits. When determining customer allowances for estimated credit losses, the Company analyzes accounts that are past due, the creditworthiness of its customers, current economic conditions and, when sufficient historical data becomes available, actual credit losses incurred by the Company. As of December 31, 2024, and 2023, the Company did not record an allowance for expected credit losses as it was considered immaterial.
Liquidity risk
Liquidity risk is the risk that the Company may not be able to generate sufficient cash resources to settle its obligations in full as they fall due or can do so only on terms that are materially disadvantageous. Prudent liquidity risk management implies maintaining sufficient cash to cover working capital requirements. Cash is monitored by the Company’s management.
Funding and liquidity risks are reviewed regularly by management and the Board of Directors. The Board of Directors reviews the Company’s ongoing liquidity risks quarterly as part of the financial review process and on an ad hoc basis as necessary.
To date, the Company has funded its capital requirements through capital raises, including the issuance of the Company’s common shares, the issuance of convertible loans, the issuance of term loans, partnering of its programs and royalty financings. The Company may need to raise additional capital in the future, and that such financing may not be available on acceptable terms, or at all.
Other Concentrations of Risk
We depend on single source suppliers for certain components of our inventory, and our production, warehousing and distribution operations are outsourced to third-party suppliers and CMOs where a significant portion of our inventory is located. Disruption of supply from key vendors or third-party suppliers may have a material adverse impact on our operations and financial results.
Our primary source of revenue is from sales of ZYNLONTA. Historically, we have not experienced significant credit losses on our accounts receivable and as of December 31, 2024 and 2023, allowances on receivables were not material. Four customers accounted for 100% and 96% of gross accounts receivable as of December 31, 2024, and 2023, respectively.
Foreign currency translation
Functional and presentation currency
Items included in the financial statements of each of the Company’s entities are measured using the currency of the primary economic environment in which the entity operates (“the functional currency”). The consolidated financial statements are presented in US dollars (“$” or “USD”), which is the Company’s functional and Group’s reporting currency.
Transactions and balances
Foreign currency transactions are translated into the functional currency using the exchange rates prevailing at the dates of the transactions. Foreign exchange gains and losses resulting from the settlement of such transactions and from the translation at year-end exchange rates of monetary assets and liabilities denominated in foreign currencies are recognized within “Other, net” in the consolidated statements of operations.
Wholly-owned subsidiaries
The results and financial position of all the Company’s entities that have a functional currency different from the reporting currency are translated into the reporting currency as follows:
(i)assets and liabilities for each balance sheet presented are translated at the closing rate at the date of that balance sheet;
(ii)income and expenses for each consolidated statement of operations are translated at monthly average exchange rates; and
(iii)all resulting exchange differences are recognized in other comprehensive loss, under “Foreign currency translation adjustment.”
Monetary assets and liabilities are translated at exchange rates in effect at the balance sheet date while non-monetary assets and liabilities are translated at historical exchange rates. Exchange gains and losses resulting from remeasurement adjustments are recorded within general and administration costs in the consolidated statements of operations.
Foreign currency exchange rates
The following exchange rates have been used for the translation of the financial statements of ADCT UK, the functional currency of which is the British pound:
|
|
|
|
|
|
|
|
|
|
|
|
|
Year ended December 31, |
|
2024 |
|
2023 |
| US $ / GBP |
|
|
|
| Closing rate, GBP 1 |
1.2536 |
|
|
1.2731 |
|
| Weighted average exchange rate, GBP 1 |
1.2743 |
|
|
1.2431 |
|
Cash and cash equivalents
Cash and cash equivalents include demand deposits held at financial institutions and other short-term highly liquid investments with original maturities of three months or less that are readily convertible to cash.
Fair value measurements
Financial assets and liabilities are required to be measured and reported at fair value at each reporting period. Fair value is defined as the exchange price that would be received for an asset or paid to transfer a liability, an exit price, in the principal or most advantageous market for the asset or liability in an orderly transaction between market participants on the measurement date. Valuation techniques used to measure fair value must maximize the use of observable inputs and minimize the use of unobservable inputs. The three-tier fair value hierarchy, which prioritizes the inputs used in measuring fair value includes:
a.Level 1: quoted prices (unadjusted) in active markets for identical assets or liabilities;
b.Level 2: inputs other than quoted prices that are observable for the asset or liability, either directly (for example, as prices) or indirectly (for example, derived from prices);
c.Level 3: inputs for the asset or liability that are not based on observable market data.
Accounts receivable
Trade accounts receivable represent amounts due from customers from product sales and are stated net of customer sales allowances for chargebacks, product returns and estimated credit losses. The Company’s payment terms range from 30 to 90 days. When determining customer allowances for estimated credit losses, the Company analyzes accounts that are past due, the creditworthiness of its customers, current economic conditions and, when sufficient historical data becomes available, actual credit losses incurred by the Company. As of December 31, 2024, and 2023, the Company did not record an allowance for expected credit losses as it was considered immaterial.
License revenue and royalties receivable, as well as other amounts due from the Company’s partners, are included in accounts receivable and are typically payable to us within 45 to 60 days after the end of each quarter in which they were earned.
Inventory
Inventory is stated at the lower of cost or net realizable value with costs determined on a first-in, first-out basis. Reserves for potentially excess, dated or obsolete inventories are established as a write-down to inventory and a charge to cost of product sales based on forecasted product demand estimates and the likelihood of consumption in the normal course of business, considering the expiration dates of the inventories on hand, planned production volumes and required production lead times.
Property and equipment
All property and equipment is stated at historical cost less accumulated depreciation. Historical cost includes expenditure that is directly attributable to the acquisition of the items.
Depreciation is calculated using the straight-line method to reduce the cost of each asset to its residual value over its estimated useful life, as follows:
|
|
|
|
|
|
|
|
|
Leasehold improvements |
|
2 to 10 years |
Laboratory equipment |
|
5 years |
Office equipment |
|
5 years |
Hardware and computer software |
|
3 years |
Leases
The Company recognizes operating lease right-of-use (“ROU”) assets and operating lease liabilities when the Company obtains the right to control the asset under a leasing arrangement with an initial term greater than twelve months. The Company evaluates the nature of each lease at the inception of an arrangement to determine whether it is an operating or financing lease and recognizes the operating lease ROU asset and operating lease liability based on the present value of future minimum lease payments over the expected lease term. The lease payments are discounted using the interest rate implicit in the lease. If that rate cannot be determined, the Company uses the incremental borrowing rate we would expect to pay to borrow on a similar collateralized basis over a similar term in order to determine the present value of our lease payments. Operating lease ROU assets are comprised of the lease liability plus any lease payments made and excludes lease incentives. Certain lease arrangements contain renewal or termination options that have been included in the determination of the lease term if the options are reasonably certain of being exercised. For contracts that contain lease and non-lease components, the Company accounts for both components as a single lease component. Lease expense for operating leases is recognized on a straight-line basis over the lease term.
Investments in joint venture
The Company has an investment in a joint venture in which we own less than 50% and do not control the investee. The investment is accounted for using the equity method given our ability to exercise significant influence over the operating and financial decisions of the investee. The Company recognized its share of the investee’s profit or losses, other comprehensive income or losses and capital transactions as an adjustment to the carrying value of its investment in joint venture. The Company’s carrying value of its investment in a joint venture increases or decreases in relation to the Company’s proportionate share of comprehensive income or loss of the joint venture. When the Company’s share of losses of a joint venture exceeds the Company’s interest in that joint venture, the Company ceases to recognize its share of further losses. Additional losses are recognized only to the extent that the Company has incurred legal or constructive obligations or made payments on behalf of the joint venture. The Company’s Investment in joint venture is assessed for impairment when events or circumstances indicate the carrying value of the investment may be impaired based on qualitative factors.
Long-lived assets
Long-lived assets by geographic area are as follows:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| (in thousands) |
|
|
|
|
| Country |
|
December 31, 2024 |
|
December 31, 2023 |
Switzerland |
|
$ |
1,460 |
|
|
$ |
1,364 |
|
United Kingdom |
|
12,133 |
|
|
13,707 |
|
United States |
|
997 |
|
|
1,773 |
|
|
|
$ |
14,590 |
|
|
$ |
16,844 |
|
The Company reviews its long-lived assets for impairment whenever events or changes in circumstances indicate that the carrying value of an asset may not be recoverable. If such circumstances are determined to exist, an estimate of undiscounted future cash flows produced by the long-lived asset, including its eventual residual value, is compared to the carrying value to determine whether impairment exists. In the event that such cash flows are not expected to be sufficient to recover the carrying amount of the assets, the assets are written-down to their estimated fair values. For the years ended December 31, 2024 and 2023, there were no material impairments of the value of long-lived assets.
Loans
Loans are initially recognized at fair value, net of transaction costs incurred. Loans are subsequently measured at amortized cost using an effective interest rate (“EIR”). Loans are presented as a financial liability in the consolidated balance sheets. Debt is classified as a current liability when due within 12 months after the end of the reporting period. The remainder of the amount is presented as a long-term liability.
Warrants
The Company accounts for warrants as either equity-classified or liability-classified instruments based on an assessment of the warrant’s specific terms and applicable authoritative guidance included in Financial Accounting Standards Board (“FASB”) Accounting Standards Codification (“ASC”) 480, Distinguishing Liabilities from Equity (“ASC 480”), ASC 815, Derivatives and Hedging (“ASC 815”) and Accounting for Convertible Instruments and Contracts in an Entity’s Own Equity (“ASU 2020-06”). The assessment considers whether the warrants are freestanding financial instruments pursuant to ASC 480, whether the warrants meet the definition of a liability pursuant to ASC 480, and whether the warrants meet all of the requirements for equity classification under ASC 815, including whether the warrants are indexed to the Company’s own common stock and whether the warrant holders could potentially require “net cash settlement” in a circumstance outside of the Company’s control, among other conditions for equity classification. This assessment, which requires the use of professional judgment, is conducted at the time of warrant issuance and as of each subsequent quarterly period end date while the warrants are outstanding. For warrants that are classified as liabilities, the Company records the fair value of the warrants at each balance sheet date and records changes in the estimated fair value as a gain or loss in Other, net.
Employee benefits
Employee Pension Plans
The Company’s wholly-owned subsidiaries operate defined benefit and defined contribution pension plans in accordance with the local conditions and practices in the countries in which they operate. Certain employees of the UK subsidiary are covered by local defined contribution plans. The defined benefit schemes are generally funded through payments to insurance companies or trustee-administered funds, determined by periodic actuarial calculations. A defined contribution plan is a plan that provides an individual account for each participant and provides benefits that are based on all of the following: amounts contributed to the participant’s account by the employer or employee; investment experience; and any forfeitures allocated to the account, less any administrative expenses charged to the plan. A defined benefit plan is a pension plan that is not a defined contribution plan. Typically, defined benefit plans define an amount of pension benefit that an employee will receive on retirement, usually dependent on one or more factors such as age, years of service and compensation. However, as is the case with many Swiss pension plans, although the amount of ultimate pension benefit is not defined, certain legal obligations of the plan nevertheless create constructive obligations on the employer to pay further contributions to fund an eventual deficit. This results in the plan being accounted for as a defined benefit plan.
The liability recognized in the consolidated balance sheets in respect of defined benefit pension plans is the present value of the defined benefit obligation at the end of the reporting period less the fair value of plan assets. The defined benefit obligation is calculated annually by a third party using the projected unit credit method. The present value of the defined benefit obligation is determined by discounting the estimated future cash outflows using interest rates of high-quality corporate bonds that are denominated in the currency in which the benefits will be paid, and that have terms to maturity that approximate the terms of the related pension obligation.
The current service cost of the defined benefit plan is recognized in the consolidated statements of operations in employee benefit expenses, except where included in the cost of an asset, and reflects the increase in the defined benefit obligation resulting from employee service in the current year.
Past service costs, resulting from a plan amendment or curtailment, are recognized in Other comprehensive (loss) income and amortized to the consolidated statements of operations and comprehensive loss over the average remaining service period of active employees.
The net interest cost is calculated by applying the discount rate to the net balance of the present value of the defined benefit obligation and the fair value of plan assets. This cost is included in employee benefit expenses within research and development and general and administrative expenses in the consolidated statements of operations.
Actuarial gains and losses arising from experience adjustments and changes in actuarial assumptions are recognized in Other comprehensive (loss) income in the period in which they arise and amortized to the consolidated statements of operations and comprehensive loss in subsequent periods using the corridor approach.
For defined contribution plans, the company pays contributions to publicly or privately administered pension insurance plans on a mandatory, contractual or voluntary basis. Once the contributions have been paid, the company has no further payment obligations. The contributions are recognized as employee benefit expenses in the consolidated statements of operations. Prepaid contributions are recognized as an asset to the extent that a cash refund or a reduction in the future payments is available.
Share-based Compensation
The Company grants share-based awards. The fair value of awards expected to vest are recognized as an employee share-based compensation expense using the graded accelerated vesting method over the requisite service period of the award less actual forfeitures. The fair value of each stock option is estimated on the date of grant using the Black-Scholes option valuation model, that requires the use of assumptions including the expected volatility of the Company’s stock price, expected term, risk-free rate and the fair value of the underlying common stock. We estimate the fair value of restricted stock units granted based on the closing market price of our common stock on the date of grant. Actual forfeitures are recognized as they occur.
Employee Stock Purchase Plan
The fair value of purchase rights granted under the employee stock purchase plan is recognized as an employee share-based compensation expense with a corresponding increase in Additional paid-in capital. The total amount to be expensed is determined by reference to the fair value of the purchase rights granted.
The total expense is recognized over the offering period, which is the period over which all of the specified vesting conditions are to be satisfied. Participants that voluntarily withdraw from the plan are accounted for as a cancellation and total share-based compensation is recorded in the period in which the participant withdraws. Terminations are accounted for as forfeitures and any share-based compensation expense is reversed in the period the participant terminates. Accumulated payroll deductions are recorded within Accrued expenses in other current liabilities until the shares are purchased by the participant at the end of the offering period.
Revenue Recognition
The Company recognizes revenue when or as control of promised goods or services is transferred to its customers in an amount that reflects the consideration to which the Company expects to receive in exchange for those goods or services. The Company follows a five-step model: (i) identify the customer contract; (ii) identify the contract’s performance obligation; (iii) determine the transaction price; (iv) allocate the transaction price to the performance obligations; and (v) recognize revenue when, or as, a performance obligation is satisfied.
Product revenue
The Company generates revenue from sales of ZYNLONTA in the U.S. for the treatment of relapsed or refractory DLBCL. Revenue is recognized when control is transferred to the customer at the net selling price, which includes reductions for gross-to-net (“GTN”) sales adjustments such as government rebates, chargebacks, distributor service fees, other rebates and administrative fees, sales returns and allowances and sales discounts.
GTN sales adjustments involve significant estimates and judgment after considering factors including legal interpretations of applicable laws and regulations, historical experience and drug product analogs in the absence of Company experience, payer channel mix, current contract prices under applicable programs, unbilled claims and processing time lags and inventory levels in the distribution channel. Management also uses information from external sources to identify prescription trends, patient demand, average selling prices, discarded volumes and sales return and allowance data for the Company and analog drug products.
The Company’s estimates are subject to inherent limitations of estimates that rely on third-party information, as certain third-party information was itself in the form of estimates, and reflect other limitations including lags between the date as of which third-party information is generated and the date on which the Company receives third-party information. Estimates will be assessed each period and adjusted as required to revise information or actual experience. In particular, the following rebate requires a substantial degree of judgement.
Discarded Drug Rebate
The Infrastructure Investment and Jobs Act requires manufacturers of certain single-source drugs separately paid for under Medicare Part B and marketed in single-dose containers or packages to provide annual refunds (“discarded drug rebate”), if those portions of the dispensed drug that are unused and discarded exceed an applicable percentage defined by statute or regulation. The Centers for Medicare & Medicaid Services (the “CMS”) finalized regulations to implement this section on November 18, 2022, and the provision went into effect on January 1, 2023. In particular, the estimate for the discarded drug rebate requires a substantial degree of judgement.
The Company began estimating and recording a provision for the discarded drug rebate as a GTN sales adjustment beginning in the first quarter of 2023. The significant assumptions used to estimate the discarded drug rebate include legal interpretations of applicable laws and regulations, historical experience with discarded volumes and time lags in the processing of claims and invoicing from CMS. Management uses a number of factors to estimate the discarded drug rebate, including information from external sources to identify the Company’s discarded volumes and information from CMS on discarded volumes.
License arrangements
The Company recognizes revenues from license fees for intellectual property (IP) either at a point in time or over time. The Company must make an assessment as to whether such a license represents a right-to-use the IP (at a point in time) or a right to access the IP (over time). The Company recognizes revenue for a right-to-use license immediately if the licensee can begin to use and benefit from the IP upon commencement of the license term and the Company has no further obligations in the context of the IP. A license is considered a right to access the IP when the Company undertakes activities during the license term that may significantly affect the IP, which directly exposes the customer to any positive or negative effects arising from such activities. These activities do not result in the immediate transfer of a good or service to the customer. As such, revenues from the right to access the IP are recognized over time.
The Company may enter into agreements with multiple performance obligations. Performance obligations are identified and separated when the other party can benefit from the license on its own or together with other resources that are readily available, and the license is separately identifiable from other goods or services in the contract.
Transaction prices for out-license arrangements may include fixed up-front amounts as well as variable consideration such as contingent development and regulatory milestones, sales-based milestones and royalties. The most likely amount method is used to estimate contingent development and regulatory milestones because the ultimate outcomes are binary in nature. Variable consideration is included in the transaction price only to the extent that it is probable that a significant reversal in the amount of cumulative revenue will not occur when the uncertainty associated with the variable consideration is subsequently resolved. To the extent arrangements include multiple performance obligations that are distinct, the transaction price assigned to each distinct performance obligation is reflective of the relative stand-alone selling price when sold separately or estimated stand-alone selling price on the basis of comparable transactions with other customers when such goods or services are not sold separately. The residual approach is the method used to estimate a stand-alone selling price when the selling price for a good or service is highly variable or uncertain.
In determining the transaction prices for licenses of intellectual property only or when a license of intellectual property is the predominant item, both sales milestones and royalties are excluded from the variable consideration guidance and recognized at the later of when the subsequent sales transaction occurs, or the satisfaction or partial satisfaction of the performance obligation to which some or all of the royalty has been allocated.
Cost of product sales
Cost of product sales primarily includes direct and indirect costs relating to the manufacture of ZYNLONTA from third-party providers of manufacturing, distribution and logistics, and royalties to a collaboration partner based on net product sales of ZYNLONTA. Inventory amounts written down as a result of excess or obsolescence are charged to Cost of product sales.
Research and Development (“R&D”) expenses
R&D costs are expensed as incurred, and consist of salaries and benefits of employees, share-based compensation costs, fees paid to CROs and CMOs, upfront fees and achieved milestone payments associated with R&D collaboration arrangements, supplies, facilities costs and allocated overhead expenditures. Clinical study and certain research costs are recognized over the service periods specified in the contracts and adjusted as necessary based upon an ongoing review of the level of effort and costs actually incurred. The Company is required to estimate its expenses resulting from its obligation under contracts with vendors and consultants and clinical site agreements in connection with its R&D efforts. Although the Company does not expect its estimates to be materially different from amounts actually incurred, its understanding of the status and timing of services performed relative to the actual status and timing of services performed may vary, and will be assessed each period and adjusted as required to reflect amounts actually incurred. Research and development costs are presented net of reimbursements from development partners.
Income taxes
Income taxes are accounted for under the asset and liability method. Under this method, deferred tax assets and liabilities are recognized for the future tax consequences attributable to differences between the financial statement carrying amounts of assets and liabilities and their respective tax bases and for operating loss and tax credit carryforwards. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be reversed. The effect on deferred tax assets and liabilities of a change in tax rates is recognized in income in the period that includes the enactment date. A valuation allowance is provided when it is more-likely-than-not that some portion or all of a deferred tax asset will not be realized. Recognized income tax positions are measured at the largest amount that is greater than 50% likely of being realized upon settlement. Changes in recognition or measurement are reflected in the period in which the change in judgment occurs. Interest and penalties related to an underpayment of income taxes are included in the income tax expense and classified with the related liability on the consolidated balance sheets. The uncertain tax position is presented as a reduction to a deferred tax asset for credit carryforward.
Loss per share
Basic loss per share is calculated by dividing the net loss attributable to shareholders by the weighted average number of common shares in issue during the year, excluding common shares owned by the Company and held as treasury shares.
Diluted loss per share adjusts the shares used in the determination of basic loss per share to take into account potentially dilutive common shares, if applicable, and the weighted average number of ordinary shares that would have been outstanding assuming the conversion of all potentially dilutive common shares (share option plans, employee stock purchase plan and outstanding warrants). The Company has incurred a loss for the years ended December 31, 2024 and 2023 and therefore potentially dilutive common shares have been excluded from the calculation of diluted loss per share because the effect would be anti-dilutive.
Contingencies
From time to time, we may become involved in claims and other legal matters arising in the ordinary course of business. We record accruals for loss contingencies to the extent that we conclude that it is probable that a liability has been incurred and the amount of the related loss can be reasonably estimated. Legal fees and other expenses related to litigation are expensed as incurred and included in selling, general and administrative expenses.
Reclassification
During 2024, amounts previously presented on the consolidated balance sheets as "Prepaid expenses and other current assets" are now presented individually as "Prepaid expenses" and "Other current assets". Prior period amounts have been reclassified to conform to the current period presentation.
Recent Accounting Pronouncements
New accounting pronouncements which have been adopted
We adopted FASB ASU 2023-07, Segment Reporting (Topic 280): Improvements to Reportable Segment Disclosures, effective for fiscal years beginning in January 2024, which requires that a public entity disclose significant segment expenses regularly reviewed by the chief operating decision maker (“CODM”), including public entities with a single reportable segment. Adoption of this standard did not have a material impact on our financial results, but resulted in enhanced disclosures as included in Note 20, “Segment Reporting..”
Issued but not yet adopted
In December 2023, the FASB issued ASU 2023-09, Income Taxes (Topic 740): Improvements to Income Tax Disclosures. The ASU requires the annual financial statements to include consistent categories and greater disaggregation of information in the rate reconciliation, and income taxes paid disaggregated by jurisdiction. ASU 2023-09 is effective for the Company’s annual reporting periods beginning in January 2025. Adoption is either with a prospective method or a fully retrospective method of transition. Early adoption is permitted. The Company is currently evaluating the effect that adoption of ASU 2023-09 will have on its consolidated financial statements.
In November 2024, the FASB issued ASU 2024-03, Income Statement—Reporting Comprehensive Income—Expense Disaggregation Disclosures (Subtopic 220-40): Disaggregation of Income Statement Expenses Disclosure. The ASU requires disaggregation, in the notes to the financial statements, of certain cost and expense captions presented on the face of the Company’s interim and annual financial statements. ASU 2023-09 is effective for fiscal years beginning in January 2027 and interim periods beginning January 2028, with early adoption permitted. Adoption may be applied prospectively or retrospectively. The Company is currently evaluating the effect that adoption of ASU 2024-03 will have on its consolidated financial statements.
3.Fair value measurements
The carrying amount of Cash and cash equivalents, Accounts Receivable, net and Accounts payable is a reasonable approximation of fair value due to the short-term nature of these assets and liabilities. Financial liabilities that are not measured at fair value on a recurring basis include our senior secured term loan and deferred royalty financing obligation. The carrying value of our senior secured term loan approximates fair value as these borrowings are based on variable market rates. The fair value of our deferred royalty obligation was $312.0 million and $272.9 million as of December 31, 2024 and 2023, respectively, and is based on our current estimates of future royalties expected to be paid over the estimated life of the royalty purchase agreement which are level 3 inputs.
The Deerfield warrants, which are measured at fair value on a recurring basis, were as follows for the years ended December 31, 2024 and 2023:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| (in thousands) |
|
Total |
|
Quoted prices in active markets for identical assets and liabilities (Level 1) |
|
Other observable inputs (Level 2) |
|
Significant unobservable inputs (Level 3) |
| December 31, 2024: |
|
|
|
|
|
|
|
|
| Deerfield warrant obligation |
|
$ |
— |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
— |
|
|
|
|
|
|
|
|
|
|
| Total |
|
$ |
— |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
— |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| (in thousands) |
|
Total |
|
Quoted prices in active markets for identical assets and liabilities (Level 1) |
|
Other observable inputs (Level 2) |
|
Significant unobservable inputs (Level 3) |
| December 31, 2023: |
|
|
|
|
|
|
|
|
| Deerfield warrant obligation |
|
$ |
296 |
|
|
$ |
— |
|
|
$ |
296 |
|
|
$ |
— |
|
|
|
|
|
|
|
|
|
|
| Total |
|
$ |
296 |
|
|
$ |
— |
|
|
$ |
296 |
|
|
$ |
— |
|
Fair values must be estimated at the end of each reporting period with regard to the Deerfield warrants. The approach to valuation follows the fair value principle, and the key input factors are described for the Deerfield warrants in Note 11, "Deerfield warrants." A Black-Scholes model was used to calculate the fair values.
There were no transfers between the respective levels during the period.
4.Inventory
As of December 31, 2024 and December 31, 2023 inventory consisted of the following:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| (in thousands) |
|
December 31, 2024 |
|
December 31, 2023 |
|
|
|
|
|
| Work in progress |
|
$ |
18,338 |
|
|
$ |
16,095 |
|
| Finished goods |
|
49 |
|
|
82 |
|
| Total inventory, net |
|
$ |
18,387 |
|
|
$ |
16,177 |
|
Inventory write-downs of $1,518 and $1,608 were recognized and charged to cost of product sales in the Company’s consolidated statements of operations for the years ended December 31, 2024 and 2023, respectively.
5.Property and equipment
Property and equipment as of December 31, 2024 and December 31, 2023 consisted of the following:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| (in thousands) |
|
December 31, 2024 |
|
December 31, 2023 |
Leasehold improvements |
|
$ |
3,873 |
|
|
$ |
3,953 |
|
| Laboratory equipment |
|
4,415 |
|
|
3,652 |
|
| Office equipment |
|
970 |
|
|
1,119 |
|
Hardware and computer software |
|
1,137 |
|
|
1,173 |
|
|
|
|
|
|
|
|
10,395 |
|
|
9,897 |
|
| Less: accumulated depreciation |
|
(5,320) |
|
|
(4,275) |
|
| Property and equipment, net |
|
$ |
5,075 |
|
|
$ |
5,622 |
|
Depreciation expense for the years ended December 31, 2024 and 2023 was $1,334 and $1,187, respectively.
6.Leases
The Company leases space for its corporate offices and research and development facilities under non-cancellable operating leases.
During the third quarter of 2024, the Company modified the terms of its existing lease for its office in New Jersey, USA. The existing lease contract was scheduled to expire on November 30, 2024, including an extension option for three additional years, which at inception, the Company believed was reasonably likely to be exercised. The Company extended the term by seven months commencing on December 1, 2024 and removed the three-year extension option. Accordingly, the Company remeasured its lease liability and adjusted its right-of-use asset based on the revised lease term.
On September 1, 2023, the Company modified the terms of its existing lease for its office in Switzerland. The existing lease contract was originally scheduled to expire on June 15, 2024 and has been cancelled and replaced by the new lease. The new lease also includes a reduction in the amount of office space being occupied. The modified lease commenced on September 1, 2023 and expires November 30, 2028, and includes a renewal option for five additional years through November 2033. The Company is reasonably certain it will exercise the extension option and therefore has accounted for the modified lease as a ten-year lease term.
On January 30, 2023, the Company expanded the square footage of its existing lease related to its U.K. office. The lease commenced on January 30, 2023 and expires on January 27, 2031, and includes an option to terminate early on January 26, 2026. The Company is reasonably certain it will not terminate the lease early and therefore will account for the lease using an eight-year lease term.
Non-cash operating lease ROU assets obtained in exchange for operating lease obligations were $0.6 million and $4.9 million for the years ended December 31, 2024 and 2023, respectively.
Lease costs for the years ended December 31, 2024 and 2023 are $1,942 and $2,080, respectively.
Maturities of lease liabilities are as follows:
|
|
|
|
|
|
|
|
|
| Year ending December 31, 2025 |
|
$ |
1,780 |
|
| Year ending December 31, 2026 |
|
1,681 |
|
| Year ending December 31, 2027 |
|
1,718 |
|
| Year ending December 31, 2028 |
|
1,758 |
|
| Year ending December 31, 2029 |
|
1,799 |
|
| Thereafter |
|
2,096 |
|
| Total lease payments |
|
10,832 |
|
| Less: imputed interest |
|
1,466 |
|
| Present value of lease liabilities |
|
$ |
9,366 |
|
Other supplemental information related to leases is summarized below:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
As of December 31, |
|
|
2024 |
|
2023 |
|
|
| Weighted average remaining lease term (years) |
|
6.3 |
|
7.1 |
|
|
|
|
|
|
|
|
|
| Weighted average discount rate |
|
4.6 |
% |
|
4.4 |
% |
|
|
7.Interest in joint venture
On December 14, 2020, the Company formed a new joint venture company, Overland ADCT BioPharma, with Overland Pharmaceuticals (“Overland”) to develop and commercialize ZYNLONTA, and three of the Company’s ADC product candidates, ADCT-601, ADCT-602 and ADCT-901 (collectively, the “Licensed Products”), in greater China and Singapore (the “Territory”). The Company agreed to supply product to Overland ADCT BioPharma for its drug development and commercialization under a supply agreement entered into between the parties.
Under the terms of the license agreement between the Company and Overland ADCT BioPharma, the Company licensed exclusive development and commercialization rights to the Licensed Products (the “Licensed IP”) in the Territory to Overland ADCT BioPharma. Overland invested $50.0 million in Overland ADCT BioPharma, and is obligated to pay the Company potential development milestone payments related to ADCT-601, ADCT-602 and ADCT-901, for a 51% equity interest. The Company received a 49% equity interest in exchange for contribution of the Licensed IP. The Company and Overland have appointed an equal number of nominees to the board of directors of Overland ADCT BioPharma which includes the Chief Executive Officer of Overland ADCT BioPharma. Pursuant to the license agreement, the Company may also earn low to mid-single digit royalties on net sales of the Licensed Products.
In addition, Overland ADCT BioPharma elected to participate in the Company’s global clinical trials. The Company also received an option, which it may exercise at its sole discretion, to exchange any or all of its equity interest in Overland ADCT BioPharma into an equity interest in Overland upon an initial public offering of Overland. Given the uncertainty of an initial public offering of Overland, the Company did not assign any value to the option.
In connection with the formation of Overland ADCT BioPharma, the Company determined the fair value of its equity interest by implying a total equity value of Overland ADCT BioPharma using Overland’s investment of $50.0 million and the fair value of the contingent milestone consideration for Overland’s 51% equity interest. The fair value of the contingent consideration was determined to be nominal due to the high uncertainty related to achieving certain conditions associated with the contingent consideration as of the closing date.
The table below provides a rollforward of the Company’s interest in Overland ADCT BioPharma as of December 31, 2024 and 2023.
|
|
|
|
|
|
|
|
|
| (in thousands) |
|
|
| Interest in joint venture |
|
|
| January 1, 2023 |
|
$ |
7,613 |
|
| Share of comprehensive loss in joint venture |
|
(5,966) |
|
| December 31, 2023 |
|
1,647 |
|
| Share of comprehensive loss in joint venture |
|
(1,647) |
|
| December 31, 2024 |
|
$ |
— |
|
The Company recognized the proportionate share of Overland ADCT BioPharma’s net loss up to the investment
carrying amount. The equity method investment in Overland ADCT Biopharma was reduced to zero during the year ended December 31, 2024 and no further losses will be recorded in the Company's consolidated financial statements as the Company has not incurred legal or constructive obligations or committed to additional funding on behalf of the joint venture. The Company will begin recognizing its share of net income only when it is greater than the cumulative net losses not recognized during the period the equity method was suspended.
8.Income taxes
For the years ended December 31, 2024 and 2023, loss before taxes consists of the following:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended December 31, |
| (in thousands) |
|
2024 |
|
2023 |
|
|
US operations |
|
$ |
(44,722) |
|
|
$ |
(7,888) |
|
|
|
Swiss and other jurisdictions |
|
(111,414) |
|
|
(187,531) |
|
|
|
| Total |
|
$ |
(156,136) |
|
|
$ |
(195,419) |
|
|
|
Income tax expense (benefit) attributable to income consists of:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| (in thousands) |
|
Current |
|
Deferred |
|
|
| Year ended December 31, 2024: |
|
|
|
|
|
|
US federal and state |
|
$ |
314 |
|
|
$ |
— |
|
|
|
Swiss and other jurisdictions |
|
(148) |
|
|
— |
|
|
|
| Total |
|
$ |
166 |
|
|
$ |
— |
|
|
|
|
|
|
|
|
|
|
| Year ended December 31, 2023: |
|
|
|
|
|
|
| US federal and state |
|
$ |
1,479 |
|
|
$ |
37,104 |
|
|
|
| Swiss and other jurisdictions |
|
523 |
|
|
— |
|
|
|
| Total |
|
$ |
2,002 |
|
|
$ |
37,104 |
|
|
|
Tax Rate Reconciliation
Income tax expense attributable to income was $166 and $39,106 for the years ended December 31, 2024 and 2023, respectively. The increase in pre-tax book loss in the U.S. is due to internal transfer pricing restructuring started in October 2023. The income tax expense differed from the amounts computed by applying the Swiss income tax rate of 13.66% for both periods to pretax income from continuing operations as a result of the following:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
For the years ended December 31, |
|
|
| (in thousands) |
|
2024 |
|
|
|
2023 |
|
|
|
|
| Loss before income taxes |
|
$ |
(156,136) |
|
|
|
|
$ |
(195,419) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Rate reconciliation: |
|
|
|
|
|
|
|
|
|
|
Computed “expected” tax benefit |
|
(21,328) |
|
|
|
|
(26,694) |
|
|
|
|
|
Increase (reduction) in income taxes resulting from: |
|
|
|
|
|
|
|
|
|
|
| Differences in overseas taxation rates |
|
(2,991) |
|
|
|
|
(343) |
|
|
|
|
|
| US, state and local income taxes, net of federal income tax benefit |
|
(1,453) |
|
|
|
|
(1,022) |
|
|
|
|
|
| US Research & development tax credit |
|
(7,524) |
|
|
|
|
(5,800) |
|
|
|
|
|
| Stock compensation |
|
692 |
|
|
|
|
2,858 |
|
|
|
|
|
| Re-assessments of prior year estimates |
|
(1,111) |
|
|
|
|
(4,745) |
|
|
|
|
|
| Non-deductible expenses |
|
1,446 |
|
|
|
|
1,576 |
|
|
|
|
|
| Change in valuation allowance |
|
16,779 |
|
|
|
|
66,989 |
|
|
|
|
|
Expired net operating loss carryforward(1) |
|
14,839 |
|
|
|
|
5,638 |
|
|
|
|
|
Other(1) |
|
817 |
|
|
|
|
649 |
|
|
|
|
|
| Income tax expense |
|
$ |
166 |
|
|
|
|
$ |
39,106 |
|
|
|
|
|
(1) For the year ended December 31, 2023 Expired net operating loss carryforward was included in Other. The prior period has been recast to conform to the current period presentation.
In the table above, the Company used ADCT SA’s statutory tax rate as the starting point for the reconciliation since it is the parent entity of the business. The rate reconciliation begins with the blended Swiss Federal and Canton tax rate, rather than solely the Swiss Federal rate, as it more accurately reflects the statutory tax obligations applicable to the Company. Given the structure of the Swiss tax system, where both federal and canton tax components contribute significantly to the overall tax rate, using a blended rate provides a more meaningful basis for reconciliation to the effective tax rate. Effective tax rate varies from the statutory tax rate primarily due to valuation allowance recorded in the current losses in Switzerland and U.S. deferred tax assets.
Deferred Tax Assets
The income tax effect of each type of temporary difference comprising the net deferred tax asset as of December 31, 2024 and 2023 is as follows:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year ended December 31, |
|
|
| (in thousands) |
|
|
|
2024 |
|
2023 |
|
|
|
|
|
|
|
|
|
|
|
| Deferred tax assets: |
|
|
|
|
|
|
|
|
Net operating loss carryforwards |
|
|
|
$ |
149,925 |
|
|
$ |
162,877 |
|
|
|
| Federal R&D credit (US) |
|
|
|
28,999 |
|
|
21,606 |
|
|
|
| State R&D credits (US) |
|
|
|
6,310 |
|
|
6,417 |
|
|
|
| Share-based compensation |
|
|
|
14,880 |
|
|
14,873 |
|
|
|
| Capitalized research and development |
|
|
|
13,958 |
|
|
2,546 |
|
|
|
| Accrued Rebate Reserve |
|
|
|
1,880 |
|
|
1,843 |
|
|
|
| Other deferred tax assets |
|
|
|
5,796 |
|
|
3,326 |
|
|
|
| Total gross deferred tax assets |
|
|
|
221,748 |
|
|
213,488 |
|
|
|
| Less: Valuation allowance |
|
|
|
(219,836) |
|
|
(212,156) |
|
|
|
| Net deferred tax assets |
|
|
|
$ |
1,912 |
|
|
$ |
1,332 |
|
|
|
| Deferred tax liabilities: |
|
|
|
|
|
|
|
|
| Interest in joint venture |
|
|
|
— |
|
|
(225) |
|
|
|
| Other deferred tax liabilities |
|
|
|
(1,912) |
|
|
(1,107) |
|
|
|
| Total gross deferred tax liabilities |
|
|
|
(1,912) |
|
|
(1,332) |
|
|
|
| Net deferred tax liability |
|
|
|
$ |
(1,912) |
|
|
$ |
(1,332) |
|
|
|
| Total deferred tax |
|
|
|
$ |
— |
|
|
$ |
— |
|
|
|
The valuation allowance at December 31, 2024 was primarily related to Swiss NOL, Federal R&D and Orphan Drug credits, Capitalized Research & Development, and Share Based Compensation that, in the judgment of management, may not be realized. The valuation allowance increased by $7.7 million in 2024. The change in valuation allowance from 2023 was related to increases in U.S. capitalized R&D, share-based compensation, orphan drug credit deferred tax assets and Swiss NOL. In assessing the realizability of deferred tax assets, management considers whether it is more likely than not that all or some portion of the deferred tax assets will not be realized. The ultimate realization of deferred tax assets is dependent upon the generation of future taxable income during the periods in which those temporary differences become deductible. Management considers the scheduled reversal of deferred tax liabilities (including the effect of available carryback and carryforward periods), projected future taxable income, and tax-planning strategies in making this assessment. In order to fully realize the deferred tax asset, the Company will need to generate significant future taxable income prior to the expiration of R&D credit carryforwards in 2044.
At December 31, 2024, the Company has R&D credit for federal and state income tax purposes of $35.3 million which are available to offset partial future federal taxable income, if any, through 2044. The Company has not recognized the entire R&D carryforward due to the reasons stated above.
At December 31, 2024, the Company has Swiss Net Operating Loss Carryforwards of $1,059 million, with expiration dates ranging from 2025 to 2031. The Company has not recognized deferred tax assets on these NOLs due to the reasons stated above.
Uncertain Tax Positions
The Company recorded uncertain tax positions without interest and penalties of $7.5 million and $6.6 million as of December 31, 2024 and 2023, respectively. The increase of uncertain tax positions from tax year 2023 is related to current year positions. The uncertain tax position is recorded in the deduction of deferred tax assets.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| (in thousands) |
|
December 31, 2024 |
|
December 31, 2023 |
Balance at January 1, |
|
$ |
6,617 |
|
|
$ |
5,184 |
|
|
|
|
|
|
| Decreases related to prior year tax positions |
|
(1) |
|
|
— |
|
| Increase related to current year tax positions |
|
1,314 |
|
|
1,463 |
|
|
|
|
|
|
| Lapse of statute of limitations |
|
(420) |
|
|
(30) |
|
|
|
|
|
|
Balance at December 31, |
|
$ |
7,510 |
|
|
$ |
6,617 |
|
The Company files income tax returns in Switzerland, United States, and United Kingdom. The Company remains subject to tax examinations in the following jurisdictions as of December 31, 2024:
|
|
|
|
|
|
|
|
|
|
|
Tax Year |
| United States - Federal |
|
2021-2024 |
United States - States |
|
2020-2024 |
| Switzerland |
|
2018-2024 |
9.Accrued expenses and other current liabilities
Accrued expenses and other current liabilities consist of the following:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| (in thousands) |
|
December 31, 2024 |
|
December 31, 2023 |
| Accrued R&D costs |
|
$ |
20,523 |
|
|
$ |
24,902 |
|
| Accrued payroll and benefits |
|
13,600 |
|
|
12,693 |
|
| Gross-to-net sales adjustments, short-term |
|
15,697 |
|
|
1,543 |
|
| Operating lease liabilities, short-term |
|
1,371 |
|
|
1,467 |
|
| Other |
|
11,249 |
|
|
11,496 |
|
|
|
$ |
62,440 |
|
|
$ |
52,101 |
|
10.Senior secured term loan facility and warrants
On August 15, 2022, the Company, ADCT UK and ADCT America entered into the Loan Agreement, pursuant to which the Company could borrow up to $175.0 million principal amount of secured term loans, including (i) a First Tranche and (ii) Future Tranches. On August 15, 2022, the Company borrowed $120.0 million principal amount of term loans under the Loan Agreement. The secured term loans mature on August 15, 2029 and accrue interest at an annual rate of secured overnight financing rate (SOFR) plus 7.50% per annum (with respect to SOFR loans) or a base rate plus 6.50% per annum (with respect to alternative base rate ("ABR") loans) for the first five years of the term loans, and thereafter, at an annual rate of SOFR plus 9.25% (with respect to SOFR loans) or a base rate plus 8.25% (with respect to ABR loans), in each case subject to a 1.00% per annum SOFR floor. The secured term loans require the payment of interest only through June 30, 2026 and quarterly payments of principal, interest and exit fees thereafter until the maturity date of August 15, 2029. The Company has the option to elect for the loans to be either a SOFR loan or ABR loan and has elected the First Tranche of the secured term loan to be a SOFR loan. Interest is paid on the last business day of each quarter.
The Company is obligated to pay certain exit fees upon certain prepayments and repayments of the principal amount of the term loans in an amount ranging from zero to 4.0% of the amount of the loan so paid. In addition, the Company has the right to prepay the term loans at any time subject to certain prepayment premiums applicable until August 15, 2026. The Loan Agreement also contains certain prepayment provisions, including mandatory prepayments from the proceeds from certain asset sales, casualty events and from issuances or incurrences of debt, which may also be subject to prepayment premiums if made on or prior to August 15, 2026. The obligations under the Loan Agreement are secured by substantially all of the Company's assets and those of certain of the Company's subsidiaries and are guaranteed initially by the Company's subsidiaries in the US and the UK. The Loan Agreement contains customary covenants, including a covenant to maintain a balance at the end of each quarter of at least $60.0 million in cash and cash equivalents plus an amount equal to any accounts payable that remain unpaid more than ninety days after the original invoice therefore, and negative covenants including limitations on indebtedness, liens, fundamental changes, asset sales, investments, dividends and other restricted payments and other matters customarily restricted in such agreements. In addition, the Loan Agreement contains a revenue covenant that, so long as the Company’s 30-day average market capitalization is less than $650 million, requires the Company achieve minimum levels of ZYNLONTA net sales in the United States, tested on a quarterly basis, which is subject to a customary cure right in favor of the Company that may be exercised by making certain prepayments and that, subject to certain limitations, may be exercised up to three times during the term of the Loan Agreement. The Loan Agreement also contains customary events of default, after which the term loan may become due and payable immediately, including payment defaults, material inaccuracy of representations and warranties, covenant defaults (including creation of any liens other than those that are expressly permitted), bankruptcy and insolvency proceedings, cross-defaults to certain other agreements, judgments against the Company and its subsidiaries and change in control.
On August 15, 2022, the Company also issued to the lenders under the Loan Agreement warrants to purchase an aggregate of 527,295 common shares, which warrants have an exercise price of $8.30 per share. Each warrant is exercisable, on a cash or a cashless basis, at the option of the holder at any time on or prior to August 15, 2032. The warrants contain customary anti-dilution adjustments and will entitle holders to receive any dividends or other distributions paid on the underlying common shares prior to their expiration on an as-exercised basis. On August 15, 2022, the Company also entered into the Share Purchase Agreement with the lenders under the Loan Agreement to purchase 733,568 common shares of the Company.
The Company has accounted for the First Tranche of the senior secured term loans, the warrants and the common shares described above each as freestanding financial instruments.
The warrants are freestanding financial instruments that are indexed to the Company’s common stock and meet all other conditions for equity classification under ASC 480 and ASC 815. Accordingly, these warrants were recognized in equity and accounted for as a component of additional paid-in capital at the time of issuance.
The proceeds received were allocated to the loan, the warrants and the common shares based on the relative fair value method, resulting in a discount on the loan. The loan was recorded at $116.0 million on August 15, 2022. The loan is subsequently measured at its amortized cost. See further illustration of the allocation of proceeds in the table below:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(in thousands) |
|
Common shares |
|
Warrants |
|
Loan |
|
Total |
| Proceeds received |
|
$ |
6,250 |
|
|
$ |
— |
|
|
$ |
120,000 |
|
|
$ |
126,250 |
|
| Allocation of proceeds |
|
$ |
6,250 |
|
|
$ |
3,957 |
|
|
$ |
116,043 |
|
|
$ |
126,250 |
|
Transaction costs were allocated to the loan, the warrants and the common shares based on the relative fair value method. Transaction costs associated to the warrants and common shares have been deducted from the respective instrument in equity, while transaction costs associated to the loan have been deducted from the loan and amortized using the effective interest method over the expected life of the loan. See further illustration of the allocation of transaction costs in table below:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(in thousands) |
|
Common shares |
|
Warrants |
|
Loan |
|
Total |
| Allocation of proceeds |
|
$ |
6,250 |
|
|
$ |
3,957 |
|
|
$ |
116,043 |
|
|
$ |
126,250 |
|
| Transaction costs |
|
$ |
(120) |
|
|
$ |
(244) |
|
|
(7,187) |
|
|
$ |
(7,551) |
|
|
|
|
|
|
|
$ |
108,856 |
|
|
|
As illustrated in the table above, the transaction costs of the loan (net of the transaction costs allocated to the warrant and common shares) were deducted from the loan to determine the carrying value as of August 15, 2022. The implied EIR that would be needed to increase the book value of the loan to cover all future expected outflows, taking into account the deduction of transaction costs from the initial loan balance, and based on a 360-day year for a SOFR loan, was computed at inception at 14.99%. Given the interest rate in the senior secured term loans is variable and dependent upon market factors, the Company will update the EIR at the end of each reporting period for changes in the rate. For the years ended December 31, 2024 and December 31, 2023, the Company recorded interest expense on the senior secured term loan in the amount of $16.6 million and $18.4 million, respectively, which was recorded in interest expense in the consolidated statements of operations. The EIR at December 31, 2024 was 16.12%.
The following table provides a summary of the interest expense for the Company’s senior secured term loan for the years ended December 31, 2024 and December 31, 2023:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year ended December 31, |
|
|
2024 |
|
2023 |
| Contractual interest expense |
|
$ |
15,700 |
|
|
$ |
15,382 |
|
| Amortization of debt discount |
|
902 |
|
|
3,016 |
|
| Total |
|
$ |
16,602 |
|
|
$ |
18,398 |
|
The amount at which the senior secured term loan is presented as a liability in the consolidated balance sheets represents the net present value of all future cash outflows associated with the loan discounted at the EIR. The carrying value of the senior secured term loan is $113.6 million and $112.7 million as of December 31, 2024 and 2023, respectively.
Contractual payments due under our senior secured term loans, including exit fees are as follows (in thousands):
|
|
|
|
|
|
|
|
|
| 2025 |
|
$ |
— |
|
| 2026 |
|
3,090 |
|
| 2027 |
|
9,330 |
|
| 2028 |
|
12,480 |
|
| 2029 |
|
99,840 |
|
|
|
|
| Total |
|
$ |
124,740 |
|
11.Deerfield warrants
Pursuant to the Exchange Agreement with Deerfield entered into on August 15, 2022, the Company issued warrants to purchase an aggregate of 4,412,840 common shares. The warrants consist of warrants to purchase an aggregate of 2,631,578 common shares at an exercise price of $24.70 per share and warrants to purchase an aggregate of 1,781,262 common shares at an exercise price of $28.07 per share. Each warrant is exercisable, on a cash or a cashless basis, at the option of the holder, at any time on or prior to May 19, 2025. The warrants contain customary anti-dilution adjustments and entitle holders to receive any dividends or other distributions paid on the underlying common shares prior to their expiration on an as-exercised basis. Each holder also may require the Company to repurchase the warrants for their Black Scholes-based fair value in connection with certain transformative transactions or change of control of the Company that occur prior to their expiration.
These warrants have been recognized as a warrant obligation and presented in the consolidated balance sheets as a liability given the warrants may be settled through a cash or cashless exercise by the warrant holder. The liability was initially measured at fair value and was determined to approximate the fair value of the existing embedded conversion option features immediately prior to the consummation of the Exchange Agreement as the terms of the warrants are reflective of the terms of the embedded conversion option features of the Deerfield Facility Agreement prior to the Exchange Agreement. As such, the warrant obligation was recorded at an initial fair value of $12,297 on August 15, 2022. Subsequent to issuance, the warrant obligation is remeasured to fair value at the end of each reporting period. Changes in the fair value (gains or losses) of the warrant obligation at the end of each period are recorded in the consolidated statements of operations.
During the year ended December 31, 2024 and December 31, 2023, the Company recognized income of $296 and $497, respectively, as a result of changes in the fair value of the warrant obligation. The fair value of the warrant obligation as of December 31, 2024 and December 31, 2023 was $0 and $296, respectively. The decreases in fair value of the warrant obligation from December 31, 2023 to December 31, 2024 was primarily due to decreases in the expected term and expected volatility during the period. These amounts were recorded to Other, net in the consolidated statements of operations. See Note 17, "Other income (expense)" for further information.
The Company calculated the fair value of the Deerfield warrant obligation, using the Black-Scholes option-pricing model. Key inputs for the valuation of the warrant obligation as of December 31, 2024 and December 31, 2023 were as follows:
|
|
|
|
|
|
|
|
|
|
|
|
|
As of |
|
As of |
|
December 31, 2024 |
|
December 31, 2023 |
| Exercise price in $ |
24.70 and 28.07 |
|
24.70 and 28.07 |
| Share price in $ |
1.99 |
|
|
1.66 |
|
| Risk-free interest rate |
4.3 |
% |
|
4.6 |
% |
| Expected volatility |
83 |
% |
|
116 |
% |
| Expected term (years) |
0.39 years |
|
1.39 years |
| Dividend yield |
— |
|
|
— |
|
| Black-Scholes value in $ |
nil and nil |
|
0.07 and 0.06 |
12.Deferred royalty obligation
On August 25, 2021, the Company entered into a royalty purchase agreement with certain entities managed by HCR for up to $325.0 million. Under the terms of the agreement, the Company received gross proceeds of $225.0 million upon closing (the “First Investment Amount”) and received an additional $75.0 million during the year ended December 31, 2023 upon the first commercial sale of ZYNLONTA in the United Kingdom or any European Union country (the “Second Investment Amount”) and together with the First Investment Amount, the “Investment Amount”).
Under the agreement, the Company is obligated to pay to HCR (i) a 7% royalty on the worldwide (excluding China, Hong Kong, Macau, Taiwan, Singapore and South Korea) net sales of ZYNLONTA and any product that contains ZYNLONTA and on any upfront or milestone payments the Company receives from licenses that it grants to commercialize ZYNLONTA or any product that contains ZYNLONTA in any region other than China, Hong Kong, Macau, Taiwan, Singapore and South Korea, (ii) a 7% royalty on the worldwide net sales of Cami and any product that contains Cami and on any upfront or milestone payments the Company receives from licenses that it grants to commercialize Cami or any product that contains Cami in the United States and Europe, and (iii) outside the United States and Europe, a 7% share of any upfront or milestone payments derived from licenses that the Company grants to commercialize Cami or any product that contains Cami and, in lieu of the royalty on net sales under such licenses, a mid-teen percentage share of the net royalty the Company receives from such licenses. These royalty rates are subject to potential upward adjustment, up to a maximum of 10%, based on performance tests in 2026 and 2027. The 7% royalty rates described above are subject to adjustment to a potential high-single-digit percentage royalty rate after September 30, 2026 and/or a 10% royalty rate after September 30, 2027, if the aggregate net sales and license revenue subject to royalty obligations in the preceding twelve months do not exceed certain mid-nine-digit milestones by such dates. The Company’s aggregate royalty obligations are capped at 2.50 times the amount paid by HCR under the agreement ($750.0 million as of December 31, 2024 and December 31, 2023), or at 2.25 times the amount paid by HCR under the agreement ($675.0 million as of December 31, 2024 and December 31, 2023) if HCR receives royalty payments exceeding a mid-nine-digit amount on or prior to March 31, 2029 (the “Royalty Cap”). Once the Royalty Cap is reached, the royalty purchase agreement will terminate.
Upon the occurrence of a change in control event, the Company is obligated to pay HCR an amount equal to the Royalty Cap, less any amounts the Company previously paid to HCR. If the change in control event occurs prior to the 36-month anniversary of the closing of the royalty purchase agreement, the Company is obligated to pay HCR an amount equal to 2.0 times the amount paid by HCR, less any amounts the Company previously paid to HCR pursuant to the agreement ($574.7 million and $600.0 million as of December 31, 2024 and $580.1 million and $600.0 million as of December 31, 2023). In addition, the Company retains the right, at any time after the 27-month anniversary of the closing of the royalty purchase agreement, to terminate the remaining royalty obligations under the agreement by paying HCR an amount equal to the Royalty Cap, less any amounts the Company previously paid to HCR pursuant to the agreement (such amount, the “Buyout Amount”), provided that HCR may instead elect to receive 50% of the Buyout Amount and continue to receive 50% of the royalty payments under the agreement but with the Royalty Cap reduced to reflect the Company’s payment of 50% of the Buyout Amount. During the year ended December 31, 2021, the Company received gross proceeds of $225.0 million before deducting transaction costs of $7.0 million, all of which were paid during 2021, which resulted in net proceeds of $218.0 million. During the year ended December 31, 2023 the Company received an additional $75.0 million upon the first commercial sale of ZYNLONTA in the United Kingdom or any European Union country, which resulted in net proceeds of $73.1 million, net of transaction costs of $1.9 million.
The table below provides a rollforward of the Company’s debt obligation relating to the royalty purchase agreement.
|
|
|
|
|
|
|
|
|
| (in thousands) |
|
|
| Liability balance at January 1, 2023 |
|
$ |
222,277 |
|
| Plus: Additional proceeds from the sale of future royalties |
|
75,000 |
|
| Less: Transaction costs |
|
1,898 |
|
| Less: royalty payments |
|
8,709 |
|
| Plus: interest expense |
|
27,915 |
|
| Less: cumulative catch-up adjustment, Other, net |
|
4,972 |
|
| Liability balance at December 31, 2023 |
|
309,613 |
|
| Less: royalty payments |
|
5,384 |
|
| Plus: interest expense |
|
33,608 |
|
| Less: cumulative catch-up adjustment, Other, net |
|
11,178 |
|
| Liability balance at December 31, 2024 |
|
$ |
326,659 |
|
The Company evaluated the terms of the royalty purchase agreement and concluded that the features of the investment amount are similar to those of a debt instrument. Accordingly, the Company recorded a liability relating to the initial gross proceeds received as debt less transaction costs in August 2021, and increased the liability in June 2023 for the eligible amount received as debt upon the first commercial sale of ZYNLONTA in the United Kingdom or any European Union country less transaction costs. The Company accounts for the value of the debt at amortized cost. The amounts received by the Company will be accreted to the total estimated amount of the royalty payments necessary to extinguish the Company’s obligation under the agreement, which will be recognized as interest expense recorded in Interest expense within the Company’s consolidated statements of operations.
The carrying value of the debt will decrease for royalty payments made to HCR based on actual net sales and licensing revenue. The Company must periodically assess the expected payments to HCR based on its underlying revenue projections and to the extent the amount or timing of such payments is materially different than its initial estimates will record a cumulative catch-up adjustment to the deferred royalty obligation. The adjustment to the carrying amount is recognized in earnings as an adjustment to Other, net in the period in which the change in estimate occurred. The debt is classified as short-term and long-term which are recorded within Other current liabilities and Deferred royalty obligation, long-term, respectively, within the Company’s consolidated balance sheets.
To determine the accretion of the liability related to the deferred royalty obligation, the Company is required to estimate the total amount of future royalty payments and estimated timing of such payment to HCR based on the Company's revenue projections. Management uses a third party valuation firm to assist in determining the total amount of future royalty payments and estimated timing of such payment to HCR using an option pricing Monte Carlo simulation model. The amount ultimately received by the Company is accreted to the total amount of the royalty payments necessary to extinguish the Company’s obligation under the agreement, which is recorded as interest expense over the life of the royalty purchase agreement. The initial estimate of this total interest expense resulted in an EIR of 10%. As royalty payments are made to HCR, the balance of the debt obligation is effectively repaid over the life of the royalty purchase agreement. During the year ended December 31, 2024 and December 31, 2023, the Company made royalty payments to HCR of $5,384 and $8,709, respectively.
The exact amount and timing of repayment is likely to be different each reporting period as compared to those estimated based on the Company's revenue projections. A significant increase or decrease in actual net sales of ZYNLONTA compared to the Company’s revenue projections, and regulatory approval and commercialization of Cami, as well as ZYNLONTA in other indications as well as licensing revenue could change the royalty rate and royalty cap due to HCR, which could materially impact the debt obligation as well as interest expense associated with the royalty purchase agreement. Also, the Company’s total obligation to HCR can vary depending on the achievement of the sales milestones as well as the timing of a change in control event. At each reporting period, Management will assess the expected payments to HCR based on its underlying revenue projections and to the extent the amount or timing of such payments is materially different than its initial estimates it will record a cumulative catch-up adjustment.
The Company recorded a total cumulative catch-up adjustment of $11.2 million and $5.0 million recorded in Other, net for the years ended December 31, 2024 and 2023, respectively. The total cumulative catch-up adjustments were based on revised revenue forecasts used in the valuation models, which revisions were primarily attributable to updates made for the Company’s long range plans, including updated development plans, actual revenue results and the transaction costs related to the eligible amount received in June 2023. Under the cumulative catch-up method, the EIR is not revised when actual or estimated net sales differ from those estimated as of the inception of the debt obligation. Instead, the carrying amount of the debt obligation is adjusted to an amount equal to the present value of the estimated remaining future payments, discounted by using the original EIR, 10%, as of the date on which the estimate changes.
13.Pension and post-retirement benefit obligations
Defined contribution plans
For U.S. employees the Company maintains a defined contribution plan under section 401(k) of the Internal Revenue Code, whereby the Company provides a matching contribution of each employee’s eligible compensation. Total Company matching contributions to this plan were $2.1 million and $2.8 million for the years ended December 31, 2024 and 2023, respectively.
Employees of the UK subsidiary are covered by local defined contribution plans. Pension costs for these plans are charged to the consolidated statements of operations when incurred and were $278 and $280 for the years ended December 31, 2024 and 2023, respectively.
Defined benefit pension plan
The pension plan for Swiss employees is a defined benefit pension plan. The Company contracted with the Swiss Life Collective BVG Foundation based in Zurich for the provision of occupational benefits. All benefits in accordance with the regulations are reinsured in their entirety with Swiss Life SA within the framework of the corresponding contract. This pension solution fully reinsures the risks of disability, death and longevity with Swiss Life. Swiss Life invests the vested pension capital and provides a 100% capital and interest guarantee.
The pension plan is entitled to an annual bonus from Swiss Life comprising the effective savings, risk and cost results.
Although, as is the case with many Swiss pension plans, the amount of ultimate pension benefit is not defined, certain legal obligations of the plan create constructive obligations on the employer to pay further contributions to fund an eventual deficit; this results in the plan nevertheless being accounted for as a defined benefit plan.
In 2024, the guaranteed interest to be credited to employees' savings was 1.25% for mandatory retirement savings and 0.50% for supplementary retirement savings. The rate for converting mandatory savings to an annuity at age 65 for male employees will decrease from 5.9% in 2024 to 5.65% in 2025 and 5.40% in 2026 and for female employees age 65 will decrease from 6.0294% in 2024 to 5.8111% in 2025 and 5.5671% in 2026. The rate for converting supplementary savings to an annuity remains at 4.4855% from 2024 to 2025 for male age 65 and 4.6744% for female employees age 65 from 2024 to 2025.
The Swiss defined benefit plan scheme is valued by third-party actuaries every year using the projected unit credit method. The latest actuarial valuation was carried out as at December 31, 2024.
The movements in the projected benefit obligation and plan assets for the years ended December 31, 2024 and 2023 are as follows:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| (in thousands) |
|
2024 |
|
2023 |
| Projected benefit obligation: |
|
|
|
|
| January 1, |
|
$ |
(11,101) |
|
|
$ |
(12,685) |
|
| Current service cost |
|
(614) |
|
|
(736) |
|
| Interest cost |
|
(118) |
|
|
(254) |
|
| Actuarial losses (gains) & change in financial assumptions |
|
(973) |
|
|
(1,445) |
|
| Benefit payments (credits) |
|
(1,195) |
|
|
99 |
|
| Employer contributions |
|
(321) |
|
|
(421) |
|
| Settlement and curtailment |
|
3,177 |
|
|
5,482 |
|
| Plan amendment |
|
31 |
|
|
33 |
|
| Currency exchange difference |
|
778 |
|
|
(1,174) |
|
| December 31, |
|
$ |
(10,336) |
|
|
$ |
(11,101) |
|
|
|
|
|
|
| Fair value of plan assets: |
|
|
|
|
| January 1, |
|
$ |
9,909 |
|
|
$ |
12,640 |
|
| Actual return on plan assets |
|
174 |
|
|
58 |
|
| Employer contributions |
|
627 |
|
|
700 |
|
| Employee contributions |
|
321 |
|
|
421 |
|
| Benefit payments (credits) |
|
1,195 |
|
|
(100) |
|
| Settlement |
|
(3,177) |
|
|
(5,201) |
|
| Currency exchange difference |
|
(680) |
|
|
1,391 |
|
| December 31, |
|
$ |
8,369 |
|
|
$ |
9,909 |
|
|
|
|
|
|
| Defined benefit pension liability |
|
$ |
(1,967) |
|
|
$ |
(1,192) |
|
The defined benefit pension liability is recorded in Other long-term liabilities on the Company’s consolidated balance sheets. The present value of the defined benefit obligation related to 25 active employees based in Switzerland (2023: 24 active employees).
The net periodic benefit cost for the years ended December 31, 2024 and 2023 is as follows:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| (in thousands) |
|
2024 |
|
2023 |
| Net periodic benefit cost: |
|
|
|
|
| Service cost |
|
$ |
(614) |
|
|
$ |
(736) |
|
| Interest cost |
|
(118) |
|
|
(254) |
|
| Expected return on plan assets |
|
182 |
|
|
298 |
|
| Settlement loss |
|
(70) |
|
|
— |
|
| Amortization of prior service cost |
|
165 |
|
|
160 |
|
|
|
|
|
|
| Amortization due to settlement and benefit payments |
|
— |
|
|
79 |
|
| Amortization of prior service cost due to reduction in service years |
|
— |
|
|
244 |
|
| Net periodic benefit cost |
|
$ |
(455) |
|
|
$ |
(209) |
|
The components of net periodic benefit cost are included in operating expense on the consolidated statements of operations.
The principal actuarial assumptions used for accounting purposes are as follows for the years ending December 31, respectively:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2024 |
|
2023 |
Discount rate |
|
0.95 |
% |
|
1.45 |
% |
Interest credited on savings accounts |
|
1.25 |
% |
|
1.45 |
% |
Future salary increases |
|
1.50 |
% |
|
1.50 |
% |
Future pension increases |
|
0.00 |
% |
|
0.00 |
% |
| Expected rate of return |
|
3.03 |
% |
|
2.56 |
% |
Expected employer contributions for 2025 to the defined benefit plan for the year ending December 31, 2024 amounts to $675.
The weighted average duration of the defined benefit obligation is 18.7 years (2023: 18.0 years).
Estimated benefit payments for the next ten years are as follows:
|
|
|
|
|
|
|
|
|
| (in thousands) |
|
|
| 2025 |
|
$ |
444 |
|
| 2026 |
|
459 |
|
| 2027 |
|
478 |
|
| 2028 |
|
474 |
|
| 2029 |
|
501 |
|
| 2030-2034 |
|
2,246 |
|
| Total expected benefit payments |
|
$ |
4,602 |
|
Plan assets
The assets are invested by the pension plan, to which many companies contribute, in a diversified portfolio that respects the requirements of the Swiss BVG. Therefore, disaggregation of the pension assets and presentation of plan assets in classes that distinguish the nature and risks of those assets is not possible. The Company’s pension plan benefits from the economies of scale and diversification of risk available through the affiliations.
Comprehensive (loss) income
The movements in “Other comprehensive (loss) income” are as follows:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| (in thousands) |
|
2024 |
|
2023 |
January 1, |
|
$ |
325 |
|
|
$ |
2,179 |
|
Net gains |
|
(910) |
|
|
(1,483) |
|
| Prior service cost |
|
(134) |
|
|
(371) |
|
| Remeasurement of Defined Benefit Pension Liability |
|
(1,044) |
|
|
(1,854) |
|
| December 31, |
|
$ |
(719) |
|
|
$ |
325 |
|
Prior service cost for the years ended December 31, 2024 and 2023 includes a credit of $31 and $33, respectively, related to plan amendments.
14.Commitments and contingencies
Manufacturing Commitments
As of December 31, 2024, the Company has non-cancelable obligations under third party manufacturing agreements related to the supply of ZYNLONTA and the Company’s product candidates totaling $3.1 million, which will be paid in 2025.
Contingent liabilities
From time to time, we may be involved in various legal matters generally incidental to our business. Although the results of litigation and claims cannot be predicted with certainty, after discussion with legal counsel, we are not aware of any matters for which the likelihood of a loss is probable and reasonably estimable and which could have a material impact on our consolidated financial condition, liquidity, or results of operations.
15.Shareholders’ equity
Capital Range
The Board of Directors is authorized to increase or decrease the share capital at any time until November 11, 2029, by a maximum amount of CHF 12,106,664 (upper limit) and 8,128,510 (lower limit), by, among other things, issuing or cancelling common shares, fully paid up, with a par value of CHF 0.08 each. An increase of the share capital in partial amounts is permissible.
Conditional Share Capital
Conditional Share Capital for Financing Acquisitions and Other Purposes
The Company’s nominal share capital may be increased, including to prevent takeovers and changes in control, by a maximum aggregate amount of CHF 3,042,154 through the issuance of not more than 38,026,929 common shares, which would have to be fully paid-in, each with a par value of CHF 0.08 per share, by the exercise of option and conversion rights granted in connection with warrants, convertible bonds or similar instruments of the Company or one of its subsidiaries. Shareholders will not have pre-emptive subscription rights in such circumstances, but may have advance subscription rights to subscribe for such warrants, convertible bonds or similar instruments. The holders of warrants, convertible bonds or similar instruments are entitled to the new shares upon the occurrence of the applicable conversion feature.
Conditional Share Capital for Equity Incentive Plans
The Company’s nominal share capital may, to the exclusion of the pre-emptive subscription rights and advance subscription rights of shareholders, be increased by a maximum aggregate amount of CHF 763,799 through the issuance of not more than 9,547,482 common shares, which would have to be fully paid-in, each with a par value of CHF 0.08 per share, by the exercise of options, other rights to receive shares or conversion rights that have been granted to employees, members of the board of directors, contractors or consultants of the Company or of one of its subsidiaries or other persons providing services to the Company or to a subsidiary through one or more equity incentive plans created by the board of directors.
2024 Equity Offering
In May 2024, the Company completed an underwritten offering of equity securities in which 13,411,912 of the Company’s common shares were sold to public investors at a price of $4.90 per share and pre-funded warrants (the“2024 Pre-Funded Warrants”) to purchase 8,163,265 of the Company’s common shares were sold to public investors at a price of $4.81 per pre-funded warrant, which equals the per share 2024 Equity Offering price, less the CHF 0.08 exercise price for each pre-funded warrant (collectively, the “2024 Equity Offering”). Gross proceeds from the 2024 Equity Offering were approximately $105.0 million, and, after giving effect to $7.6 million of transaction costs related to the offering, net proceeds were approximately $97.4 million.
The 2024 Pre-Funded Warrants are exercisable, on a cash basis (or, if there is no registration statement and current prospectus covering the issuance of the shares upon exercise, then on a cashless basis), at the option of the holder after the date of issuance until the tenth anniversary of their original issuance. At any time during the last 90 days of the term, the holder may exchange the Pre-Funded Warrant for, and we will issue, a new pre-funded warrant for the number of common shares then remaining under the Pre-Funded Warrant. The Pre-Funded Warrants have certain limitations on exercise, including (i) any exercise must be for at least 50,000 common shares (or, if less, the remaining common shares available for purchase under the Pre-Funded Warrants), (ii) a holder cannot exercise for any amount that would cause such holder’s beneficial ownership of our common shares to exceed 9.99% (or 19.99% with 61-days’ notice to us), and (iii) cashless exercise is not available in certain circumstances as specified in the Pre-Funded Warrants. The warrants contain customary anti-dilution adjustments and will entitle holders to receive any dividends or other distributions paid on the underlying common shares prior to their expiration on an as-exercised basis.
Accounting for the 2024 Equity Offering and Pre-funded Warrants
The Company has accounted for the common shares and 2024 Pre-funded Warrants described above each as freestanding financial instruments.
The common shares for the underwritten offering of equity securities were (i) issued in part from the Company’s capital range and (ii) in part sourced from treasury shares. The common shares were recorded as $60.5 million to equity for the issuance of the common shares, net of transaction costs accrued and paid, and an increase in cash and cash equivalents.
The warrants are freestanding financial instruments that are indexed to the Company’s common stock and meet all other conditions for equity classification under ASC 480 and ASC 815, including the warrant holders cannot require “net cash settlement” in a circumstance outside of the Company’s control, and there is sufficient authorized and unissued shares to settle the warrants. Accordingly, these warrants are recognized as $36.9 million in equity and accounted for as a component of additional paid-in capital at the time of issuance, net of transaction costs paid, and an increase in cash and cash equivalents.
Transaction costs have been allocated to the warrants and the common shares based on the relative fair value method. Transaction costs associated to the warrants and common shares have been deducted from the respective instrument in equity.
2024 at-the-market offering program
In August 2024, we filed a prospectus relating to an at-the-market offering program, pursuant to which we may offer and sell our common shares from time to time with an aggregate offering price of $100.0 million through Jefferies LLC acting as sales agent. For the year ended December 31, 2024, we did not sell any shares under the program.
16.Revenue
The table below provides a disaggregation of revenues by type and customer location for the years ended December 31, 2024 and December 31, 2023:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| (in thousands) |
|
2024 |
|
2023 |
| Types of goods and services |
|
|
|
|
| Product revenue, net |
|
$ |
69,280 |
|
|
$ |
69,060 |
|
| Royalties |
|
1,557 |
|
|
498 |
|
| Total revenue |
|
$ |
70,837 |
|
|
$ |
69,558 |
|
|
|
|
|
|
| Customer Location |
|
|
|
|
| U.S. |
|
$ |
69,280 |
|
|
$ |
69,060 |
|
EMEA(1) |
|
1,557 |
|
|
498 |
|
|
|
|
|
|
| Total revenue |
|
$ |
70,837 |
|
|
$ |
69,558 |
|
(1) Europe, the Middle East and Africa
Product revenue, net
The table below provides a rollforward of the Company’s accruals related to the GTN sales adjustments for the years ended December 31, 2024 and December 31, 2023.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| (in thousands) |
|
Discarded Drug Rebate |
|
Other Adjustments |
|
Total |
| Balance as of December 31, 2022 |
|
$ |
— |
|
|
$ |
3,746 |
|
|
$ |
3,746 |
|
| GTN accruals for current period |
|
7,391 |
|
|
17,163 |
|
|
24,554 |
|
| Prior period adjustments |
|
— |
|
|
(1,028) |
|
|
(1,028) |
|
| Credits, payments and reclassifications |
|
— |
|
|
(15,935) |
|
|
(15,935) |
|
| Balance as of December 31, 2023 |
|
$ |
7,391 |
|
|
$ |
3,946 |
|
|
$ |
11,337 |
|
| GTN accruals for current period |
|
7,756 |
|
|
16,218 |
|
|
23,974 |
|
| Prior period adjustments |
|
(44) |
|
|
(1,971) |
|
|
(2,015) |
|
| Credits, payments and reclassifications |
|
— |
|
|
(15,807) |
|
|
(15,807) |
|
| Balance as of December 31, 2024 |
|
$ |
15,103 |
|
|
$ |
2,386 |
|
|
$ |
17,489 |
|
The table below provides the classification of the accruals related to the GTN sales adjustment included in the Company’s consolidated balance sheets as of December 31, 2024 and December 31, 2023.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| (in thousands) |
|
December 31, 2024 |
|
December 31, 2023 |
| Accounts receivable, net |
|
$ |
1,792 |
|
|
$ |
2,403 |
|
| Other current and non-current liabilities |
|
15,697 |
|
|
8,934 |
|
|
|
$ |
17,489 |
|
|
$ |
11,337 |
|
Customers from which we derive more than 10% of our total product revenues are as follows:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Years ended December 31, |
|
|
2024 |
|
2023 |
| McKesson |
|
41.0 |
% |
|
40.0 |
% |
AmerisourceBergen Corporation(1) |
|
35.0 |
% |
|
38.0 |
% |
| Cardinal Health |
|
24.0 |
% |
|
22.0 |
% |
(1) AmerisourceBergen also operates under the name Cencora
17.Other income (expense)
Interest Income
Interest income includes interest received from banks on our cash balances. Interest income was $12.3 million and $10.5 million for the years ended December 31, 2024 and December 31, 2023, respectively.
Interest Expense
The components of Interest expense for the years ended December 31, 2024 and December 31, 2023 are as follows:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| (in thousands) |
|
|
|
|
|
|
|
2024 |
|
2023 |
|
|
|
|
|
|
|
|
|
|
|
| Deferred royalty obligation interest expense |
|
|
|
|
|
|
|
$ |
33,608 |
|
|
$ |
27,915 |
|
| Effective interest expense on senior secured term loan facility |
|
|
|
|
|
|
|
16,602 |
|
|
18,398 |
|
| Other interest expense |
|
|
|
|
|
|
|
1 |
|
|
12 |
|
| Interest expense |
|
|
|
|
|
|
|
$ |
50,211 |
|
|
$ |
46,325 |
|
Other, net
The components of other, net for the years ended December 31, 2024 and December 31, 2023 are as follows:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| (in thousands) |
|
|
|
2024 |
|
2023 |
|
|
|
|
|
|
|
| Cumulative catch-up adjustment, deferred royalty obligation |
|
|
|
$ |
11,178 |
|
|
$ |
4,972 |
|
| Deerfield warrant obligation, change in fair value income |
|
|
|
296 |
|
|
497 |
|
| Exchange differences loss |
|
|
|
(80) |
|
|
(52) |
|
| R&D tax credit |
|
|
|
1,063 |
|
|
935 |
|
| Other, net |
|
|
|
$ |
12,457 |
|
|
$ |
6,352 |
|
Cumulative catch-up adjustment, deferred royalty obligation
We periodically assess the expected payments to HCR based on our underlying revenue projections and to the extent the amount or timing of such payments is materially different than our initial estimates we will record a cumulative catch-up adjustment to the deferred royalty obligation.
18.Share-based compensation
The Company has adopted various share-based compensation plans. Under these plans the Company may at its discretion grant to the plan participants, such as directors, certain employees, and service providers awards in the form of restricted shares and restricted share units (“RSUs”), share options, share appreciation rights, performance awards and other share-based awards. The 2019 Equity Incentive Plan was adopted in November 2019 while the Conditional Share Capital Plan and the Inducement Plan were adopted in December 2023.
2019 Equity Incentive Plan
In November 2019, the Company adopted the 2019 Equity Incentive Plan. Under the 2019 Equity Incentive Plan, the Company may at its discretion grant to plan participants, such as directors, certain employees and service providers, awards in the form of restricted shares and RSUs, share options, share appreciation rights, performance awards and other share-based awards. The Company has reserved 17,741,355 common shares for future issuance under the 2019 Equity Incentive Plan (including share-based equity awards granted to date less awards forfeited). As of December 31, 2024, the Company has 4,330,311 common shares available for the future issuance of share-based equity awards.
As of December 31, 2024 and 2023, the cumulative amount recorded as a net increase to additional paid-in capital within equity on the consolidated balance sheets in respect of the 2019 Equity Incentive Plan was $160,945 and $157,906. The amounts of expense for all awards recognized for services received during the years ended December 31, 2024 and December 31, 2023 were $3,039 and $13,123, respectively.
Conditional Share Capital Plan
In December 2023, the Company adopted the Conditional Share Capital Plan. Under the Conditional Share Capital Plan, the Company may at its discretion grant to plan participants, such as directors, certain employees and service providers, awards in the form of restricted shares and RSUs, share options, share appreciation rights, performance awards and other share-based awards. The Company has reserved 8,000,000 common shares for future issuance under this plan. On December 6, 2023, the Company issued its 2024 annual equity award under the Conditional Share Capital Plan, which was approved by the Compensation Committee of the Board of Directors and by the Board of Directors for the senior management team, consisted of 5,596,166 RSUs. No share options were issued in connection with this award. Accordingly, as of December 31, 2024, the Company has 3,355,883 common shares available for the future issuance of share-based equity awards.
As of December 31, 2024 and 2023, the cumulative amount recorded as a net increase to additional paid-in capital within equity on the consolidated balance sheets in respect of the Conditional Share Capital Plan was $2,964 and $319. The amounts of expense for all awards recognized for services received during the years ended December 31, 2024 and December 31, 2023 was $3,808 and $319.
Inducement Plan
In December 2023, the Company adopted the Inducement Plan. Under the Inducement Plan, the Company may at its discretion grant to any employee who is eligible to receive an employment inducement grant in accordance with NYSE Listed Company Manual 303A.08. The maximum number of common shares in respect of which awards may be granted under the Inducement Plan is 1,000,000 common shares (including share-based equity awards granted to date, less awards forfeited), subject to adjustment in the event of certain corporate transactions or events if necessary to prevent dilution or enlargement of the benefits made available under the plan. Equity incentive awards under the Inducement Plan may be granted in the form of options, share appreciation rights, restricted shares, restricted share units, performance awards or other share-based awards but not “incentive stock options” for purposes of U.S. tax laws. As of December 31, 2024, the Company has 357,300 common shares available for the future issuance of share-based equity awards under this plan. There were no awards issued under this plan during the year ended December 31, 2023.
As of December 31, 2024, the cumulative amount recorded as a net increase to additional paid-in capital within equity on the unaudited condensed consolidated balance sheets in respect of the Inducement Plan was $554. The amount of expense for all awards recognized for services received during the years ended December 31, 2024 was $554.
Equity Exchange Program
On March 6, 2023, the Company commenced a tender offer with employees to exchange some or all of their eligible stock options based on a pre-determined exchange ratio for new options as detailed in our Schedule TO filed March 6, 2023 with the Securities and Exchange Commission (the “Exchange Offer”), to, among other things, further align employee incentives with the current market conditions. The Exchange Offer expired on April 3, 2023 and new options were granted on April 4, 2023. Employees holding stock options to purchase 2.2 million common shares, with exercise prices ranging from $8.12 per share to $48.77 per share, participated in the Exchange Offer, and 0.9 million new options were granted based on the exchange ratios set forth in the Exchange Offer. The new options have an exercise price of $2.06 per share, which is equal to the closing price of the Company’s common shares as reported on the NYSE on April 4, 2023. The new options include additional vesting conditions. Any previously held options that were vested at the time of exchange would become fully vested on April 4, 2024. With respect to any options held that were unvested at the time of grant, a portion of the new options will vest on the first anniversary date with additional portions vesting monthly thereafter until the new options are fully vested five years after the original grant date.
Under U.S. GAAP, the incremental compensation expense of a modified award is measured as the excess of the fair value of each award of new options granted to participants in this Exchange Offer, measured as of the date the new options are granted, over the fair value of the eligible options replaced in exchange for the new options, measured immediately prior to the replacement. The Company utilized a binomial valuation model and determined there was no incremental share-based compensation expense associated with the new options granted under this Exchange Offer. The Company will continue to recognize share-based compensation expense equal to the grant date fair value of the exchanged options.
Share Options
Pursuant to the 2019 Equity Incentive Plan, the Conditional Share Capital Plan and Inducement Plan (the “Share-based Compensation Plans”) the Company may grant share options to its directors, certain employees and service providers working for the benefit of the Company at the time. The exercise price per share option is set by the Company at the fair market value of the underlying common shares on the date of grant, as determined by the Company, which is generally the closing share price of the Company’s common shares traded on the NYSE. The awards generally vest 25% on the first anniversary of the date of grant, and thereafter evenly on a monthly basis over the subsequent three years. The contractual term of each share option award granted is ten years. Under the grant, the options may be settled only in common shares of the Company. Therefore, the grants of share options under the 2019 Equity Incentive Plan and Inducement Plan have been accounted for as equity-settled under US GAAP. As such, the Company records a charge for the vested portion of award grants and for partially earned but non-vested portions of award grants. This results in a front-loaded charge to the Company’s consolidated statements of operations and a corresponding increase to additional paid-in capital within equity on the consolidated balance sheets.
The expense recognized for services received during the years ended December 31, 2024 and December 31, 2023 is $2,129 and $6,530, respectively.
Movements in the number of awards outstanding under the Plans described above and their related weighted average strike prices are as follows:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted average strike price per share (in $ per share) |
|
Number of
awards
|
|
Weighted average remaining life in years |
|
Aggregate Intrinsic Value (in $ thousands) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Outstanding as of January 1, 2023 |
|
$ |
18.30 |
|
|
10,755,494 |
|
|
8.46 |
|
$ |
— |
|
| Granted |
|
2.20 |
|
|
3,900,341 |
|
|
|
|
|
| Option Exchange - Granted |
|
2.06 |
|
|
898,585 |
|
|
|
|
|
| Forfeited |
|
12.58 |
|
(1,668,740) |
|
|
|
|
|
| Option Exchange - Forfeited |
|
22.55 |
|
(2,197,458) |
|
|
|
|
|
| Expired |
|
22.09 |
|
(943,816) |
|
|
|
|
|
| Outstanding as of December 31, 2023 |
|
11.00 |
|
|
10,744,406 |
|
|
8.14 |
|
$ |
— |
|
| Granted |
|
3.37 |
|
|
1,646,400 |
|
|
|
|
|
| Forfeited |
|
9.15 |
|
(610,083) |
|
|
|
|
|
| Expired |
|
30.84 |
|
(968,964) |
|
|
|
|
|
| Exercised |
|
2.15 |
|
(114,471) |
|
|
|
|
|
| Outstanding as of December 31, 2024 |
|
$8.69 |
|
10,697,288 |
|
|
7.56 |
|
$ |
— |
|
Of the total awards outstanding as of December 31, 2024, there were 5,875,127 awards are vested and exercisable. As of December 31, 2024, the weighted average strike price and weighted average remaining life for vested and exercisable awards is $12.57 and 6.81 years, respectively. Awards outstanding as of December 31, 2024 have expiration dates through 2034. The weighted average grant date fair value of the awards granted during the year ended December 31, 2024 and 2023 was $2.66 and $1.53, respectively. The aggregate intrinsic value of vested and exercisable options was zero. As of December 31, 2024, the unrecognized compensation cost related to 4,822,161 unvested share options expected to vest was $8.5 million. This unrecognized cost will be recognized over an estimated weighted-average amortization period of 1.56.
The fair values of the options were determined on the date of the grant using the Black-Scholes option-pricing model using the following assumptions:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year ended December 31, 2024 |
|
Year ended December 31, 2023 |
|
|
Share price, in $ |
|
|
|
1.69-4.86 |
|
0.67-5.45 |
|
|
Strike price, in $ |
|
|
|
1.69-4.86 |
|
0.67-5.45 |
|
|
Expected volatility, in % |
|
|
|
95 |
|
75-90 |
|
|
Award life, in years |
|
|
|
6.08 |
|
6.08 |
|
|
Expected dividends |
|
|
|
— |
|
|
— |
|
|
|
Risk-free interest rate, in % |
|
|
|
3.56-4.54 |
|
3.39-4.62 |
|
|
During the six months ended June 30, 2023, the expected volatility was based on the Company’s historical volatility and selected volatility determined by median values observed among other comparable public companies. Beginning in the third quarter of 2023, the Company’s expected volatility is no longer determined by values observed among other comparable companies and is now based solely on the Company’s historical volatility. The award life for options granted was based on the time interval between the date of grant and the date during the ten-year life after which, when making the grant, the Company expected on average that participants would exercise their options.
The fair value of the new options granted under the Equity Exchange program was estimated at the date of grant using a binomial model with the following assumptions: Share price of $2.06, expected volatility of 77% - 79%, expected risk-free interest rate of 3.29% - 3.31%, expected dividends of 0% and expected term was derived based on the contractual term of the options, the expected exercise behavior and expected post-vesting forfeiture rates. The Company used a third party valuation firm to assist in calculating the fair value of the new award grants per participant.
RSUs
Pursuant to the Share-based Compensation Plans, the Company may grant RSUs to its directors, certain employees and service providers working for the benefit of the Company at the time. The awards generally vest annually over a period of two to three years commencing on the first anniversary of the date of grant. The RSUs may be settled only in common shares of the Company. Therefore, the grant of RSUs under the 2019 Equity Incentive Plan and Conditional Share Capital Plan have been accounted for as equity-settled under US GAAP. As such, the Company records a charge for the vested portion of award grants and for partially earned but non-vested portions of award grants. This results in a front-loaded charge to the Company’s consolidated statements of operations and a corresponding increase to additional paid-in capital within equity on the consolidated balance sheets. The expense recognized for services received during the years ended December 31, 2024 and December 31, 2023 is $5,272 and 6,593, respectively. An amount of $1,163 was withheld for tax withholding obligations during the year ended December 31, 2024.
|
|
|
|
|
|
|
|
|
|
|
|
|
Number of awards |
|
Weighted average grant date fair value (in $ per share) |
| January 1, 2023 |
1,585,877 |
|
|
$ |
13.26 |
|
| Granted |
6,575,846 |
|
|
1.23 |
| Vested |
(1,330,081) |
|
|
10.13 |
| Forfeited |
(297,799) |
|
|
7.96 |
| December 31, 2023 |
6,533,843 |
|
|
2.03 |
| Granted |
280,000 |
|
|
3.16 |
Vested (1) |
(3,021,221) |
|
|
2.42 |
| Forfeited |
(686,312) |
|
|
1.78 |
| December 31, 2024 |
3,106,310 |
|
|
1.81 |
(1) Includes 371,524 shares withheld to cover tax withholding obligations in connection with the vesting of RSUs previously granted.
The total fair value of RSU awards vested (as measured on the date of vesting) during the years ended December 31, 2024 and December 31, 2023 was $10.0 million and $3.1 million, respectively.
Employee Stock Purchase Plan
In June 2022, the Company adopted the 2022 Employee Stock Purchase Plan (“ESPP”), which allows eligible employees to purchase designated shares of the Company's common shares at a discount, over a series of offering periods through accumulated payroll deductions. The Company generally offers the ESPP to employees twice a year with each having a six-month offering period. The first offering period is generally from January 1st through June 30th and the second offering period is from July 1st through December 31st. The grant date is the first day of each offering period.
The expense recognized related to the ESPP during the years ended December 31, 2024 and December 31, 2023 is $330 and $372, respectively.
19.Loss per share
The basic loss per share is calculated by dividing the net loss attributable to shareholders by the weighted average number of shares in issue during the period, excluding common shares owned by the Company and held as treasury shares, as follows:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
For the Years Ended December 31, |
| (in thousands, except per share amounts) |
|
2024 |
|
2023 |
|
|
Net loss |
|
$ |
(157,846) |
|
|
$ |
(240,053) |
|
|
|
Weighted average number of shares outstanding |
|
97,159,966 |
|
|
81,712,166 |
|
|
|
Basic and diluted loss per share |
|
$ |
(1.62) |
|
|
$ |
(2.94) |
|
|
|
For the year ended December 31, 2024 and December 31, 2023, basic and diluted loss per share are calculated on the weighted average number of shares issued and outstanding and also include the Pre-funded warrants issued in connection with the 2024 Equity Offering.
The calculation excludes shares to be issued under the Share-based Compensation Plans, the Company’s Deerfield warrants and shares to be issued under the 2022 ESPP as the effect of including those shares would be anti-dilutive. See Note 10, “Senior secured term loan facility and warrants,” Note 11, “Deerfield warrants”, Note 15, “Shareholders’ Equity” and Note 18, “Share-based compensation,” for further information.
Potentially dilutive securities that were not included in the diluted per share calculations because the effect of including them would be anti-dilutive were as follows:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
For the Years Ended December 31, |
|
|
2024 |
|
2023 |
|
|
| 2019 Equity Incentive Plan - Share Options |
|
10,054,588 |
|
|
10,744,406 |
|
|
|
| Inducement Plan - Share Options |
|
642,700 |
|
|
— |
|
|
|
| 2019 Equity Incentive Plan - RSUs |
|
608,961 |
|
|
937,677 |
|
|
|
| Conditional Share Capital Plan - RSUs |
|
2,497,349 |
|
|
5,596,166 |
|
|
|
| Outstanding warrants |
|
4,940,135 |
|
|
4,940,135 |
|
|
|
| 2022 ESPP |
|
163,576 |
|
|
229,675 |
|
|
|
|
|
18,907,309 |
|
|
22,448,059 |
|
|
|
20.Segment Reporting
The Company is managed and operated as one reportable segment, which includes all global activities related to the development and commercialization of targeted ADC cancer therapies. The determination of a single reportable segment is consistent with the consolidated financial information regularly provided to the Company’s CODM, which is its chief executive officer. The CODM uses loss from operations and consolidated net loss for purposes of assessing performance, making operating decisions, allocating resources and planning and forecasting for future periods. The CODM allocates resources and plans for the long-term growth of the Company based on the Company's available cash resources, forecasted cash flow, and expenditures on a consolidated basis, as well as an assessment of the probability of success of its research and development activities. Resource allocation and long-term growth decisions are informed by budgeted and forecasted expense information, along with actual expenses incurred to date. The measure of segment assets is reported on the consolidated balance sheets as total assets.
The following table is the summary of the segment profit or loss information, including the significant expense categories for the years ended December 31, 2024 and 2023, respectively:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
For the Years Ended December 31, |
|
|
2024 |
|
2023 |
|
|
| Total revenues, net |
|
$ |
70,837 |
|
|
$ |
69,558 |
|
|
|
| Significant operating expenses: |
|
|
|
|
|
|
| Cost of product sales |
|
(5,949) |
|
|
(2,529) |
|
|
|
Research and development(1) |
|
(107,712) |
|
|
(123,176) |
|
|
|
Selling and marketing(1) |
|
(43,711) |
|
|
(57,786) |
|
|
|
General and administrative(1) |
|
(36,388) |
|
|
(38,559) |
|
|
|
| Share-based compensation |
|
(7,731) |
|
|
(13,494) |
|
|
|
| Total operating expenses |
|
(201,491) |
|
|
(235,544) |
|
|
|
| Loss from operations |
|
(130,654) |
|
|
(165,986) |
|
|
|
Other segment items(2) |
|
(27,192) |
|
|
(74,067) |
|
|
|
| Net loss |
|
$ |
(157,846) |
|
|
$ |
(240,053) |
|
|
|
(1)Excludes share-based compensation
(2)Includes Other expense, net, Income tax expense and Equity in net losses of joint venture
Item 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure
None.
Item 9A. Controls and Procedures
Disclosure Controls and Procedures
Our management, under the supervision and with the participation of our Chief Executive Officer and Chief Financial Officer, has performed an evaluation of our disclosure controls and procedures (as defined in Rule 13a-15(e) under the Exchange Act) as of the end of the period covered by this report, as required by Rule 13a-15(b) under the Exchange Act. Based upon this evaluation, our Chief Executive Officer and Chief Financial Officer, has concluded that, as of the end of the period covered by this Annual Report, our disclosure controls and procedures were effective in ensuring that the information required to be disclosed by us in the reports that we file or submit under the Exchange Act is recorded, processed, summarized and reported within the time periods specified by the SEC’s rules and forms, and that the information required to be disclosed by us in the reports that we file or submit under the Exchange Act is accumulated and communicated to our management, including our Chief Executive Officer and Chief Financial Officer, as appropriate, to allow timely decisions regarding required disclosure.
Management’s Annual Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Rule 13a-15(f) under the Exchange Act. Our internal control over financial reporting is a process designed by or under the supervision of the Chief Executive Officer and Chief Financial Officer, and effected by our board of directors, management and other personnel, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of our consolidated financial statements for external reporting purposes in accordance with generally accepted accounting principles.
Our management conducted an assessment of the effectiveness of the Company’s internal control over financial reporting based on the criteria established in Internal Control – Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (“COSO”). Based on this assessment, our management has determined that the Company’s internal control over financial reporting as of December 31, 2024 is effective.
Our internal control over financial reporting includes policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect transactions and dispositions of our assets; (2) provide reasonable assurances that our transactions are recorded as necessary to permit preparation of financial statements in accordance with U.S. GAAP, and that our receipts and expenditures are being made only in accordance with authorizations of management; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of our assets that could have a material effect on our consolidated financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Due to our filer status, our independent registered public accounting firm has not reported on the effectiveness of our internal control over financial reporting as of December 31, 2024.
Changes in Internal Control Over Financial Reporting
There were no changes to internal control over financial reporting during the quarter ended December 31, 2024 that would have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
Item 9B. Other Information
Insider Trading Arrangements and Policies
There were no Rule 10b5-1 trading arrangements adopted or terminated by our officers or directors during the three months ended December 31, 2024.
We have adopted insider trading policies and procedures governing the purchase, sale and other dispositions of our securities by our directors, senior management and employees that are reasonably designed to promote compliance with applicable insider trading laws, rules and regulations and applicable listing standards. Our Insider Trading Policy and Rule 10b5-1 Plan Policy are filed as Exhibits 19.1 and 19.2, respectively, of this Annual Report. In addition, it is our policy to comply with applicable securities laws when we engage in transactions in our securities.
Item 9C. Disclosure Regarding Foreign Jurisdictions that Prevent Inspections
Not applicable.
PART III
Item 10. Directors, Executive Officers and Corporate Governance
The information required by this Item 10 will be included in our Definitive Proxy Statement to be filed with the SEC with respect to our 2025 Annual General Meeting and is incorporated herein by reference.
Item 11. Executive Compensation
The information required by this Item 11 will be included in our Definitive Proxy Statement to be filed with the SEC with respect to our 2025 Annual General Meeting and is incorporated herein by reference.
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters
The information required by this Item 12 will be included in our Definitive Proxy Statement to be filed with the SEC with respect to our 2025 Annual General Meeting and is incorporated herein by reference.
Item 13. Certain Relationships and Related Transactions, and Director Independence
The information required by this Item 13 will be included in our Definitive Proxy Statement to be filed with the SEC with respect to our 2025 Annual General Meeting and is incorporated herein by reference.
Item 14. Principal Accounting Fees and Services
The information required by this Item 14 will be included in our Definitive Proxy Statement to be filed with the SEC with respect to our 2025 Annual General Meeting and is incorporated herein by reference.
PART IV
Item 15. Exhibits, Financial Statement Schedules
Consolidated Financial Statements
For a list of the financial statements included in this Annual Report, see Index to the Consolidated Financial Statements on page 78 of this Annual Report, incorporated by reference in response to this Item.
Consolidated Financial Statement Schedules
All financial statement schedules have been omitted because they are either not required or not applicable or the information is included in the consolidated financial statements or the notes thereto.
Exhibits
The exhibits listed below are filed with or incorporated by reference into this Annual Report.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Incorporation by Reference |
| Exhibit No. |
Description |
|
Form |
File No. |
Exhibit No. |
Filing Date |
| 3.1* |
|
|
|
|
|
|
| 4.1* |
|
|
|
|
|
|
| 4.2 |
|
|
6-K |
001-39071 |
99.2 |
August 15, 2022 |
| 4.3 |
Registration Rights Agreement, dated August 15, 2022, between ADC Therapeutics SA and OR Opportunistic DL (C), L.P., Owl Rock Opportunistic Master Fund II, L.P., Oaktree LSL Holdings EURRC S.à r.l., Oaktree Specialty Lending Corporation, Oaktree AZ Strategic Lending Fund, L.P., Oaktree Strategic Credit Fund, Oaktree Diversified Income Fund, Inc., and Oaktree Loan Acquisition Fund, L.P. |
|
6-K |
001-39071 |
99.5 |
August 15, 2022 |
| 4.4 |
Registration Rights Agreement, dated February 6, 2023, between ADC Therapeutics SA and Oaktree Fund Administration LLC, OCM Strategic Credit Investments S.à r.l., OCM Strategic Credit Investments 2 S.à.r.l., Oaktree Gilead Investment Fund AIF (Delaware), L.P., Oaktree Huntington-GCF Investment Fund (Direct Lending AIF), L.P., Oaktree Specialty Lending Corporation, and Pathway Strategic Credit Fund III, L.P. |
|
6-K |
001-39071 |
4.1 |
February 6, 2023 |
| 4.5 |
|
|
8-K |
001-39071 |
10.1 |
January 24, 2024 |
| 10.1# |
|
|
F-1 |
333-237841 |
10.1 |
April 24, 2020 |
| 10.1.1# |
|
|
F-1 |
333-237841 |
10.2 |
April 24, 2020 |
| 10.2#† |
|
|
20-F |
001-39071 |
4.4 |
March 18, 2021 |
| 10.3#† |
|
|
20-F |
001-39071 |
4.5 |
March 18, 2021 |
| 10.4#† |
|
|
20-F |
001-39071 |
4.6 |
March 18, 2021 |
| 10.5#† |
|
|
6-K |
001-39071 |
99.1 |
August 26, 2021 |
| 10.6#† |
|
|
20-F |
001-39071 |
4.15 |
March 17, 2022 |
| 10.7#† |
|
|
20-F |
001-39071 |
4.9 |
March 15, 2023 |
| 10.8† |
|
|
6-K |
001-39071 |
99.3 |
August 15, 2022 |
| 10.8.1† |
|
|
8-K |
001-39071 |
10.1 |
January 19, 2024 |
| 10.8.2 |
|
|
10-K |
001-39071 |
10.8.2 |
March 13, 2024 |
| 10.8.3 |
|
|
10-Q |
001-39071 |
10.1 |
August 6, 2024 |
| 10.9 |
|
|
6-K |
001-39071 |
99.1 |
August 15, 2022 |
| 10.10 |
|
|
6-K |
001-39071 |
99.4 |
August 15, 2022 |
| 10.11 |
|
|
8-K |
0001-39071 |
4.1 |
May 8, 2024 |
| 10.12§ |
|
|
F-1 |
333-237841 |
10.12 |
April 24, 2020 |
| 10.13§ |
|
|
S-8 |
001-39071 |
99.1 |
March 15, 2023 |
| 10.13.1§ |
|
|
10-K |
001-39071 |
10.12.1 |
March 13, 2024 |
| 10.14§ |
|
|
S-8 |
333-265917 |
99.1 |
June 30, 2022 |
| 10.15§ |
|
|
S-8 |
333-275882 |
99.1 |
December 4, 2023 |
| 10.15.1§ |
|
|
10-K |
001-39071 |
10.14.1 |
March 13, 2024 |
| 10.16§ |
|
|
S-8 |
333-275882 |
99.2 |
December 4, 2023 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 10.17§ |
|
|
8-K |
001-39071 |
10.1 |
February 29, 2024 |
| 10.18§ |
|
|
10-K |
001-39071 |
10.17 |
March 13, 2024 |
| 10.18.1§ |
|
|
10-K |
001-39071 |
10.18 |
March 13, 2024 |
| 10.18.2§ |
|
|
10-K |
001-39071 |
10.19 |
March 13, 2024 |
| 10.19§ |
|
|
10-K |
001-39071 |
10.20 |
March 13, 2024 |
| 10.19.1§ |
|
|
10-K |
001-39071 |
10.21 |
March 13, 2024 |
| 10.20§ |
|
|
10-K |
001-39071 |
10.22 |
March 13, 2024 |
| 10.21§ |
|
|
10-K |
001-39071 |
10.23 |
March 13, 2024 |
| 10.21.1§ |
|
|
10-K |
001-39071 |
10.24 |
March 13, 2024 |
| 10.22§ |
|
|
10-K |
001-39071 |
10.28 |
March 13, 2024 |
| 19.1* |
|
|
|
|
|
|
| 19.2* |
|
|
|
|
|
|
| 21.1* |
|
|
|
|
|
|
| 23.1* |
|
|
|
|
|
|
| 31.1* |
|
|
|
|
|
|
| 31.2* |
|
|
|
|
|
|
| 32.1* |
|
|
|
|
|
|
| 32.2* |
|
|
|
|
|
|
| 97.1 |
|
|
20-F |
001-39071 |
97.1 |
March 17, 2022 |
| 101.SCH |
XBRL Taxonomy Extension Schema Document |
|
|
|
|
|
| 101.CAL |
XBRL Taxonomy Extension Calculation Linkbase Document |
|
|
|
|
|
| 101.DEF |
XBRL Taxonomy Extension Definition Linkbase Document |
|
|
|
|
|
| 101.LAB |
XBRL Taxonomy Extension Label Linkbase Document |
|
|
|
|
|
| 101.PRE |
XBRL Taxonomy Extension Presentation Linkbase Document |
|
|
|
|
|
| 104 |
Cover Page Interactive Data File (embedded with the Inline XBRL document) |
|
|
|
|
|
* Filed herewith.
# Portions of this exhibit have been omitted because they are both (i) not material and (ii) customarily and actually treated by the Company as private or confidential
† Certain schedules to this exhibit have been omitted pursuant to Item 601(a)(5) of Regulation S-K. A copy of any omitted schedule will be furnished supplementally to the SEC upon request; provided, however, that the parties may request confidential treatment pursuant to Rule 24b-2 of the Exchange Act for any document so furnished.
§ Management contract, compensatory plan or arrangement.
Item 16. Form 10-K Summary
None.
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, as amended, the Registrant has duly caused this Annual Report to be signed on its behalf by the undersigned, thereunto duly authorized.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
ADC Therapeutics SA |
|
|
|
/s/ Ameet Mallik |
Date: March 27, 2025 |
|
By: |
Ameet Mallik |
|
|
|
Chief Executive Officer |
|
|
|
|
Pursuant to the requirements of the Securities Exchange Act of 1934, as amended, this Annual Report has been signed below by the following persons on behalf of the Registrant in the capacities indicated on March 27, 2025.
|
|
|
|
|
|
|
|
|
| Name |
|
Title |
|
|
|
| /s/ Ameet Mallik |
|
Chief Executive Officer and Director
(Principal Executive Officer)
|
| Ameet Mallik |
|
|
|
|
| /s/ Jose Carmona |
|
Chief Financial Officer
(Principal Financial Officer)
|
| Jose Carmona |
|
|
|
|
| /s/ Lisa Kallebo |
|
Corporate Controller and Chief Accounting Officer
(Principal Accounting Officer)
|
| Lisa Kallebo |
|
|
|
|
| /s/ Ron Squarer |
|
Chairman of the Board of Directors |
| Ron Squarer |
|
|
|
|
| /s/ Robert Azelby |
|
Director |
| Robert Azelby |
|
|
|
|
| /s/ Jean-Pierre Bizzari |
|
Director |
| Jean-Pierre Bizzari |
|
|
|
|
| /s/ Peter Hug |
|
Director |
| Peter Hug |
|
|
|
|
| /s/ Viviane Monges |
|
Director |
| Viviane Monges |
|
|
|
|
| /s/ Thomas Pfisterer |
|
Director |
| Thomas Pfisterer |
|
|
|
|
| /s/ Tyrell Rivers |
|
Director |
| Tyrell Rivers |
|
|
|
|
| /s/ Victor Sandor |
|
Director |
| Victor Sandor |
|
EX-3.1
2
ex31adct-articlesofassocia.htm
EX-3.1
Document
EXHIBIT 3.1
Articles of Association
of ADC Therapeutics SA
(ADC Therapeutics AG)
(ADC Therapeutics Ltd)
Statuts
de ADC Therapeutics SA
(ADC Therapeutics AG)
(ADC Therapeutics Ltd)
Articles of Association of ADC Therapeutics SA
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Section 1
Name, Registered Office, Purpose and Duration of the Company |
|
|
Section 1
Raison sociale, siège, but de la société, durée |
|
|
|
|
|
|
Article 1 |
|
|
Article 1 |
Name, Place of Incorporation |
Under the name ADC Therapeutics SA (ADC Therapeutics AG) (ADC Therapeutics Ltd) (the Company) shall exist a corporation with its registered office in Epalinges, canton of Vaud. |
|
Raison sociale, siège |
Sous la raison sociale ADC Therapeutics SA (ADC Therapeutics AG), (ADC Therapeutics Ltd) (la Société) existe une société anonyme avec siège à Epalinges, canton de Vaud. |
|
|
|
|
|
|
Article 2 |
|
|
Article 2 |
Purpose |
1 The Company's purpose is to research, develop, produce and sell products in the fields of biotechnology, pharmaceutics, medical technology, diagnosis and therapy as well as to purchase, sell and use patents and licenses in these fields. The Company may engage in all types of transactions that appear appropriate to promote the purpose of the Company or that are related thereto. |
|
But |
1 La Société a pour but la recherche, le développement, la production et la vente de produits dans les domaines de la biotechnologie, de la pharmaceutique, de la technologie médicale, du diagnostic et de la thérapie ainsi que l’acquisition, la vente et l'utilisation de brevets et de licences dans ces domaines. La Société peut exercer toutes activités aptes à favoriser son but ou en rapport avec ce dernier. |
|
2 The Company may open branch offices and subsidiaries in Switzerland and abroad. It may also acquire participations or otherwise invest in other companies in Switzerland and abroad. |
|
|
2 La Société peut constituer des succursales et des filiales en Suisse et à l'étranger et participer à ou investir autrement dans d'autres entreprises en Suisse et à l'étranger. |
|
3 The Company may acquire, hold, manage, mortgage, exploit and sell real estate and intellectual property rights in Switzerland and abroad and may also finance other companies. |
|
|
3 La Société peut acquérir, détenir, gérer, gager, mettre en valeur et aliéner des immeubles et des droits de propriété intellectuelle en Suisse et à l'étranger, ainsi que financer d'autres sociétés. |
|
|
|
|
|
|
Article 3 |
|
|
Article 3 |
Duration |
The duration of the Company shall be unlimited. |
|
Durée |
La durée de la Société est illimitée. |
|
|
|
|
|
Articles of Association of ADC Therapeutics SA
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Section 2
Share Capital, Shares, Restrictions of Transferability
|
|
|
Section 2
Capital-actions, actions et restrictions à la transmissibilité
|
|
|
|
|
|
|
Article 4 |
|
|
Article 4 |
Share Capital |
The share capital of the Company is CHF 8,128,510.08 and is divided into 101,606,376 fully paid in registered shares with a par value of CHF 0.08 each. |
|
Capital-actions |
Le capital-actions de la Société s'élève à
CHF 8'128’510.08 et est divisé en 101'606’376 actions nominatives entièrement libérées d'une valeur nominale de CHF 0.08 chacune. |
|
|
|
|
|
|
Article 4a |
|
|
Article 4a |
Capital Range |
1 The Company has a capital range ranging from CHF 8,128,510.08 (lower limit) to CHF 12,106,664.40 (upper limit). The Board of Directors shall be authorized within the capital range to increase or reduce the share capital once or several times and in any amounts or to acquire or dispose of shares directly or indirectly, until November 11, 2029 or until an earlier expiry of the capital range. The capital increase or reduction may be effected by issuing fully paid-in registered shares with a par value of CHF 0.08 each and cancelling registered shares with a par value of CHF 0.08 each, as applicable, or by increasing or reducing the par value of the existing shares within the limits of the capital range or by simultaneous reduction and re-increase of the share capital. |
|
Marge de fluctuation du capital |
1 La Société dispose d'une marge de fluctuation du capital allant de CHF8’128'510.08 (limite inférieure) à CHF 12'106'664.40 (limite supérieure). Le Conseil d'administration peut, dans les limites définies de la marge de fluctuation, et ce jusqu'au 11 Novembre 2029 ou jusqu'à l'expiration anticipée de la marge de fluctuation, augmenter ou réduire le capital-actions en une ou plusieurs fois, de quelque montant que ce soit, ou acquérir ou aliéner des actions directement ou indirectement. L'augmentation ou la réduction du capital peut se faire par l'émission d'actions nominatives d'une valeur nominale de CHF 0.08 chacune, qui doivent être intégralement libérées, respectivement l'annulation d'actions nominatives d'une valeur nominale de CHF 0.08 chacune, ou par une augmentation ou une réduction, dans les limites de la marge de fluctuation, de la valeur nominale des actions nominatives existantes ou encore par une réduction et une nouvelle augmentation simultanées. |
Articles of Association of ADC Therapeutics SA
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2 In the event of an issue of shares, the subscription and acquisition of the new shares as well as any subsequent transfer of the shares shall be subject to the restrictions pursuant to Article 6 of these articles of association. |
|
|
2 En cas d'émission d'actions, la souscription et l'acquisition des nouvelles actions ainsi que tout transfert ultérieur des actions sont assujettis aux restrictions à la transmissibilité conformément à l'article 6 des présents statuts. |
|
3 In the event of a capital increase within the capital range, the Board of Directors shall, to the extent necessary, determine the issue price, the type of contribution (including cash contributions, contributions in kind, set-off and conversion of reserves or of profit carried forward into share capital), the date of issue, the conditions for the exercise of pre-emptive rights and the beginning date for dividend entitlement. In this regard, the Board of Directors may issue new shares by means of a firm underwriting through a financial institution, a syndicate of financial institutions or another third party and a subsequent offer of these shares to the existing shareholders or third parties (if the pre-emptive rights of the existing shareholders have been withdrawn or have not been duly exercised). The Board of Directors is entitled to permit, to restrict or to exclude the trading of pre-emptive rights. It may permit the expiration of pre-emptive rights that have not been duly exercised, or it may place such rights or shares as to which pre-emptive rights have been granted, but not duly exercised, at market conditions or may use them otherwise in the interest of the Company. |
|
|
3 En cas d'augmentation du capital-actions dans le cadre de la marge de fluctuation, le Conseil d'administration détermine, le cas échéant, le prix d'émission, la nature des apports (y compris la libération en espèces, les apports en nature, la compensation et la conversion de réserves ou de bénéfice reporté en capital-actions), le moment de l'émission, les conditions de l'exercice du droit de souscription préférentiel et le moment à partir duquel les actions donneront droit à des dividendes. A cet effet, le Conseil d'administration peut émettre des nouvelles actions par voie de prise ferme par un établissement financier, un consortium bancaire ou un tiers et d'offre subséquente de ces actions aux actionnaires actuels ou à des tiers (si les droits de souscription préférentiels des actionnaires actuels ont été supprimés ou qu'ils n'ont pas été valablement exercés). Le Conseil d'administration est en droit d'autoriser, de limiter ou d'exclure le négoce des droits de souscription préférentiels. Le Conseil d'administration peut laisser s'éteindre les droits de souscription préférentiels non exercés valablement; il peut aussi aliéner ceux-ci, respectivement les actions pour lesquelles des droits de souscription ont été accordés sans toutefois être valablement exercés, aux conditions du marché ou les utiliser autrement dans l'intérêt de la Société. |
|
4 In the event of an issue of shares, the Board of Directors is further authorized to withdraw or restrict pre-emptive rights of existing shareholders and to allocate such rights to third parties, the Company or any of its group companies: |
|
|
4 En cas d'émission d'actions, le Conseil d'administration peut aussi exclure ou limiter les droits de souscription préférentiels des actionnaires actuels et les attribuer à des tiers, à la Société ou à une des sociétés du groupe: |
Articles of Association of ADC Therapeutics SA
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(a) if the issue price of the new shares is determined by reference to the market price; or |
|
|
(a) si le prix d'émission des nouvelles actions est déterminé en fonction du prix du marché; ou |
|
(b) for raising equity capital in a fast and flexible manner, which would not be possible, or might only be possible with great difficulty or delays or at significantly less favorable conditions, without the exclusion of the pre-emptive rights of existing shareholders; or |
|
|
(b) pour créer des fonds propres de manière rapide et flexible, ce qui ne serait pas possible ou possible qu'avec difficulté ou tardivement ou à des conditions nettement plus défavorables sans l'exclusion des droits de souscription préférentiels des actionnaires actuels; ou |
|
(c) for the acquisition of companies, part(s) of companies or participations, for the acquisition of products, intellectual property or licenses by or for investment projects of the Company or any of its group companies, or for the financing or refinancing of any of such transactions through a placement of shares; or |
|
|
(c) pour l'acquisition de sociétés, de partie(s) de sociétés ou de participations, pour l'acquisition de produits, de droits de propriété intellectuelle, ou licences par ou pour des projets d'investissement de la Société ou de l'une des sociétés du groupe, ou pour le financement ou le refinancement de telles transactions par le placement d'actions; ou |
|
(d) for purposes of broadening the shareholder constituency of the Company in certain geographic, financial or investor markets, for purposes of the participation of strategic partners including financial investors, or in connection with the listing of new shares on domestic or foreign stock exchanges; or |
|
|
(d) pour élargir le cercle des actionnaires de la Société dans certains marchés géographiques, financiers ou d'investisseurs, pour permettre la participation de partenaires stratégiques y compris d'investisseurs financiers, ou en relation avec la cotation de nouvelles actions sur des bourses nationales ou étrangères; ou |
|
(e) for purposes of granting an over-allotment option (Greenshoe) or an option to subscribe for additional shares in a placement or sale of shares to the respective initial purchaser(s) or underwriter(s); or |
|
|
(e) pour octroyer une option de surallocation (Greenshoe) ou une option de souscription d'actions supplémentaires lors d'un placement ou de la vente d'actions à un ou plusieurs acheteurs initiaux ou souscripteurs; ou |
|
(f) for the participation of members of the Board of Directors, members of the executive management, employees, contractors, consultants or other persons performing services for the benefit of the Company or any of its group companies; or |
|
|
(f) pour la participation de membres du Conseil d'administration, de membres de la direction, d'employés, de co-contractants, de consultants ou d'autres personnes exerçant des services au bénéfice de la Société ou de l'une des sociétés du groupe; ou |
Articles of Association of ADC Therapeutics SA
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(g) following a shareholder or a group of shareholders acting in concert having accumulated shareholdings in excess of 20% of the share capital registered in the commercial register without having submitted to all other shareholders a takeover offer recommended by the Board of Directors; or |
|
|
(g) si un actionnaire ou un groupe d'actionnaires agissant de concert a acquis ou réuni une participation de plus de 20% du capital-actions inscrit au registre du commerce sans avoir présenté à tous les autres actionnaires une offre publique d'achat dont l'acceptation a été recommandée par le Conseil d'administration; ou |
|
(h) for the defense of an actual, threatened or potential takeover bid that the Board of Directors, upon consultation with an independent financial adviser retained by it, has not recommended or will not recommend to the shareholders to accept on the basis that the Board of Directors does not find such takeover bid to be financially fair to the shareholders or not to be in the Company's interest. |
|
|
(h) pour se défendre contre une offre publique d'achat hostile présentée, menaçante ou potentielle dont le rejet est, respectivement sera, recommandé par le Conseil d'administration, après consultation d'un conseiller financier indépendant qu'il aura choisi, dans la mesure où le Conseil d'administration estime que l'offre publique d'achat n'est pas équitable d'un point de vue financier vis-à-vis des actionnaires ou n'est pas dans l'intérêt de la Société. |
|
5 After a change of the par value, new shares shall be issued within the capital range with the same par value as the existing shares. |
|
|
5 En cas de modification de valeur nominale, les nouvelles actions émises dans le cadre de la marge de fluctuation du capital doivent être émises avec la même valeur nominale que les actions nominatives existantes. |
|
6 If the share capital increases as a result of an increase from conditional capital pursuant to Article 4b of these articles of association, the upper and lower limits of the capital range shall increase in an amount corresponding to such increase in the share capital. |
|
|
6 En cas d'augmentation du capital-actions en raison d'une augmentation conditionnelle du capital-actions conformément à l'article 4b, les limites supérieure et inférieure de la marge de fluctuation augmentent en fonction du montant de l'augmentation du capital-actions. |
|
7 In the event of a reduction of the share capital within the capital range, the Board of Directors shall, to the extent necessary, determine the use of the reduction amount. |
|
|
7 En cas de réduction du capital-actions dans le cadre de la marge de fluctuation, le Conseil d'administration détermine, si nécessaire, l'affectation du montant de la réduction. |
|
|
|
|
|
|
Article 4b |
|
|
Article 4b |
Articles of Association of ADC Therapeutics SA
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Conditional Share Capital for Employee Participation |
1 The share capital may be increased in an amount not to exceed CHF 763,798.56 through the issuance of up to 9,547,482 fully paid in registered shares with a par value of CHF 0.08 per share through the direct or indirect issuance of shares, or through the exercise or mandatory exercise of rights to acquire shares or through obligations to acquire shares, which were granted to or imposed on members of the Board of Directors, members of the executive management, employees, contractors or consultants of the Company or its group companies, or other persons providing services to the Company or its group companies. |
|
Capital-actions conditionnel pour la participation des employés |
1 Le capital-actions peut être augmenté d'un montant maximum de CHF 763’798.56 par l'émission de 9'547’482 actions nominatives au plus, d'une valeur nominale de CHF 0.08 chacune, qui doivent être intégralement libérées, par l'émission directe ou indirecte d'actions, ou par l'exercice ou l'exercice obligatoire de droits de souscription d'actions ou par des obligations d'acquisition d'actions accordées ou imposées aux membres du Conseil d'administration, aux membres de la direction, ou aux employés, co-contractants ou consultants de la Société ou de l'une des sociétés du groupe, ou d'autres personnes exerçant des services au bénéfice de la Société ou de l'une des sociétés du groupe. |
|
2 The pre-emptive rights and advance subscription rights of the shareholders of the Company shall be excluded in connection with the issuance of any shares, options, other rights to receive shares, or subscription rights therefor. Shares, options, other rights to receive shares, or subscription rights therefor shall be issued pursuant to one or more regulations to be issued by the Board of Directors or, to the extent delegated to it, the Compensation Committee, and to the extent applicable, taking into account the compensation principles pursuant to Article 28 of these articles of association. Shares, options, other rights to receive shares, or subscription rights therefor may be issued at a price or with an exercise price lower than the market price. |
|
|
2 Le droit de souscription préférentiel ainsi que le droit de souscription préalable des actionnaires de la Société sont exclus en relation avec l'émission de toutes actions, options, autres droits à recevoir des actions ou des droits de souscription qui y sont attachés. L'émission d'actions, d'options, d'autres droits à recevoir des actions ou des droits de souscription qui y sont attachés est faite selon un ou plusieurs règlements adoptés par le Conseil d'administration ou le Comité de rémunération, dans la mesure où cette compétence lui a été déléguée, et le cas échéant en tenant compte des principes de rémunération selon l'article 28 des présents statuts. L'émission d'actions, d'options, d'autres droits à recevoir des actions ou des droits de souscription qui y sont attachés peut se faire à un prix ou avec un prix d'exercice en-dessous du prix du marché. |
|
3 The direct or indirect acquisition of the new shares by persons listed in paragraph 1 in connection with an employee participation program and any subsequent transfer of such shares shall be subject to the restrictions of Article 6 of these articles of association. |
|
|
3 L'acquisition directe ou indirecte de nouvelles actions par des personnes mentionnées à l'alinéa 1 dans le cadre d'un programme de participation des collaborateurs ainsi que le transfert subséquent de ces actions sont assujettis aux restrictions à la transmissibilité conformément à l'article 6 des présents statuts. |
|
|
|
|
|
|
4 The declaration of acquisition of the shares based on this Article 4b shall refer to this Article 4b and be made in a form that allows proof by text. A waiver of the right to acquire shares based on this Article 4b may also occur informally or by lapse of time; this also applies to the waiver of the exercise and forfeiture of this right. |
|
|
4 La déclaration concernant l'acquisition d'actions fondée sur le présent article 4b doit faire référence à cet article 4b et doit être faite sous une forme permettant d'en établir la preuve par texte. La renonciation à un droit d'acquisition d'actions fondé sur le présent article 4b peut également avoir lieu de manière informelle ou par l'écoulement du temps; cela vaut également pour la renonciation à l'exercice et la déchéance de ce droit. |
Articles of Association of ADC Therapeutics SA
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Article 4c |
|
|
Article 4c |
Conditional Share Capital for Financing, Acquisitions and other Purposes |
1 The share capital may be increased including in connection with an intended takeover in an amount not to exceed CHF 3,042,154.32 through the issuance of up to 38,026,929 fully paid in registered shares with a par value of CHF 0.08 per share through the exercise or mandatory exercise of conversion, exchange, option, warrant or similar rights or obligations for the subscription of shares granted to shareholders or third parties on a stand-alone basis or in connection with bonds, notes, options, warrants or other securities or contractual obligations of the Company or any of its group companies, including without limitation a convertible debenture to be entered into by the Company, as may be amended or novated from time to time (hereinafter collectively, the Financial Instruments). |
|
Capital-actions conditionnel aux fins de financement, acquisitions ou d'autres buts |
1 Le capital-actions peut être augmenté, y compris en lien avec une future offre publique d'acquisition, d'un montant maximum de CHF 3'042'154.32 par l'émission de 38'026'929 actions nominatives au plus, d'une valeur nominale de CHF 0.08 chacune, qui doivent être intégralement libérées par l'exercice ou l'exercice obligatoire de droits de conversion, d'échange, d'option, de warrant ou d'autres droits ou obligations similaires pour la souscription d'actions octroyés aux actionnaires ou à des tiers de manière autonome ou en rapport avec des obligations, effets, options, warrants ou autres instruments financiers ou obligations contractuelles de la Société ou de l'une des sociétés du groupe, y compris, mais sans s'y limiter, une débenture convertible de la société, laquelle peut être amenée à être modifiée ou actualisée (ci-après désignés collectivement les Instruments Financiers). |
|
2 The pre-emptive rights of shareholders shall be excluded for the exercise of any Financial Instruments in connection with the issuance of shares. The then-current owners of such Financial Instruments shall be entitled to acquire the new shares issued upon conversion, exchange or exercise of any Financial Instruments. The key conditions of the Financial Instruments shall be determined by the Board of Directors. |
|
|
2 Le droit de souscription préférentiel des actionnaires est exclu en relation avec l'émission d'actions à l'occasion de l'exercice d'Instruments Financiers. Les personnes qui détiendront alors de tels Instruments Financiers seront en droit d'acquérir les nouvelles actions émises à l'occasion de la conversion, de l'échange ou de l'exercice d'Instruments Financiers. Le Conseil d'administration détermine les principales conditions des Instruments Financiers. |
|
3 The Board of Directors shall be authorized to restrict or withdraw advance subscription rights of shareholders in connection with the issuance of Financial Instruments by the Company or one of its group companies (1) if the issuance is for purposes of financing or refinancing, or the payment for, the acquisition of companies, parts of a company, participations, intellectual property rights, licenses or investments, (2) if the issuance occurs in domestic or international capital markets or through a private placement, (3) following a shareholder or a group of shareholders acting in concert having accumulated shareholdings in excess of 20% of the share capital registered in the commercial register without having submitted to all other shareholders a takeover offer recommended by the Board of Directors, (4) for the defense of an actual, threatened or potential takeover bid that the Board of Directors, upon consultation with an independent financial adviser retained by it, has not recommended or will not recommend to the shareholders to accept on the basis that the Board of Directors does not find such takeover bid to be financially fair to the shareholders or not in the Company's interest, or (5) if the Financial Instruments are issued on appropriate terms. If the advance subscription rights are neither granted directly nor indirectly by the Board of Directors, the following shall apply: |
|
|
3 Le Conseil d'administration est autorisé à limiter ou retirer le droit de souscription préalable des actionnaires en relation avec l'émission d'Instruments Financiers par la Société ou une des sociétés du groupe (1) si l'émission a pour but le financement, le refinancement ou le paiement de l'acquisition d'entreprises, de parties d'une entreprise, de participations, de droits de propriété intellectuelle, de licences ou d'investissements, (2) si l'émission a lieu sur les marchés de capitaux nationaux ou internationaux ou par le biais d'un placement privé, (3) si un actionnaire ou un groupe d'actionnaires agissant de concert a acquis ou réuni une participation de plus de 20% du capital-actions inscrit au registre du commerce sans avoir présenté à tous les autres actionnaires une offre publique d'achat dont l'acceptation a été recommandée par le Conseil d'administration, (4) pour se défendre contre une offre publique d'achat hostile présentée, menaçante ou potentielle dont le rejet est, respectivement sera, recommandé par le Conseil d'administration, après consultation d'un conseiller financier indépendant qu'il aura choisi, dans la mesure où le Conseil d'administration estime que l'offre publique d'achat n'est pas équitable d'un point de vue financier vis-à-vis des actionnaires ou n'est pas dans l'intérêt de la Société, ou (5) si les Instruments Financiers sont émis à des conditions appropriées. Si le droit de souscription préalable n'est pas accordé, de manière directe ou indirecte, par le Conseil d'administration, les règles suivantes s'appliquent: |
|
(a) the Financial Instruments may be converted, exchanged or exercised during a maximum period of 15 years from the date of issuance or contract conclusion; and |
|
|
(a) les Instruments Financiers peuvent être convertis, échangés ou exercés durant une période maximale de 15 ans suivant la date de l'émission ou de la conclusion du contrat; et |
Articles of Association of ADC Therapeutics SA
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(b) the conversion, exchange or exercise price of the Financial Instruments shall be set with reference to, and/or shall be subject to change based upon, the valuation of the Company's equity and/or market conditions at the time the Financial Instruments are issued. |
|
|
(b) le prix de conversion, d'échange ou d'exercice des Instruments Financiers est fixé en prenant en compte, et/ou peut être modifié en fonction, de la valorisation des fonds propres de la société et/ou des conditions du marché lors de l'émission des Instruments Financiers. |
|
4 The direct or indirect acquisition of the new shares acquired through the exercise of Financial Instruments and any subsequent transfer of such shares shall be subject to the restrictions of Article 6 of these articles of association. |
|
|
4 L'acquisition de nouvelles actions acquises directement ou indirectement par l'exercice d'Instruments Financiers ainsi que le transfert subséquent de ces actions sont assujettis aux restrictions à la transmissibilité conformément à l'article 6 des présents statuts. |
|
5 The declaration of acquisition of the shares based on this Article 4c shall refer to this Article 4c and be made in a form that allows proof by text. A waiver of the right to acquire shares based on this Article 4c may also occur informally or by lapse of time; this also applies to the waiver of the exercise and forfeiture of this right. |
|
|
5 La déclaration concernant l'acquisition d'actions fondée sur le présent article 4c doit faire référence à cet article 4c et doit être faite sous une forme permettant d'en établir la preuve par texte. La renonciation à un droit d'acquisition d'actions fondé sur le présent article 4c peut également avoir lieu de manière informelle ou par l'écoulement du temps; cela vaut également pour la renonciation à l'exercice et la déchéance de ce droit. |
|
Article 5 |
|
|
Article 5 |
Share Certificates and Intermediated Securities |
1 The Company may issue its registered shares in the form of single certificates, global certificates and uncertificated securities. Subject to applicable law, the Company may convert its registered shares from one form into another form at any time and without the approval of the shareholders. The Company shall bear the cost associated with any such conversion. |
|
Certificats d'actions et titres intermédiés |
1 La Société émet ses actions nominatives sous forme de certificats individuels, de certificats globaux ou de droits-valeurs. La Société est libre, dans les limites du droit applicable, en tout temps et sans l'approbation des actionnaires, de convertir ses actions nominatives émises sous l'une des formes ci-dessus, en une autre forme. La Société supporte les coûts d'une telle conversion. |
|
2 A shareholder has no right to request a conversion of the registered shares issued in one form into another form. Each shareholder may, however, at any time request from the Company a written confirmation of the registered shares held by such shareholder, as reflected in the share register. |
|
|
2 Un actionnaire n'a pas le droit de réclamer la conversion d'actions nominatives émises sous une certaine forme en une autre forme. Chaque actionnaire peut toutefois exiger en tout temps que la Société établisse une attestation relative aux actions nominatives qu'il détient selon le registre des actions. |
Articles of Association of ADC Therapeutics SA
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
3 Intermediated securities based on registered shares of the Company cannot be transferred by way of assignment. A security interest in any such intermediated securities also cannot be granted by way of assignment. |
|
|
3 Les titres intermédiés fondés sur des actions nominatives de la Société ne peuvent pas être transférés par cession. Il ne peut pas non plus être constitué de sûretés par cession sur ces titres intermédiés. |
|
|
|
|
|
|
Article 6 |
|
|
Article 6 |
Share Register, Restrictions on Registration, Nominees |
1 The Company shall maintain, itself or through a third party, a share register for the registered shares that lists the surname and name (the name of the company in case of a legal entity), the address and domicile (the registered office in case of a legal entity) of the shareholders or usufructuaries. A person registered in the share register shall notify the share registrar of any change of address. Until such notification has occurred, all written communications from the Company to persons registered in the share register shall be deemed to have validly been made if sent to the address previously recorded in the share register. |
|
Registre des actions, limitations à l'inscription, Nominees |
1 La Société ou un tiers mandaté par elle tient un registre des actions qui mentionne le nom et le prénom (la raison sociale pour les personnes morales), l'adresse et le domicile (le siège pour les personnes morales) des propriétaires et des usufruitiers. Si une personne inscrite au registre des actions change d'adresse, elle doit le communiquer à la personne en charge de la tenue du registre. Aussi longtemps que cette communication n'a pas eu lieu, toutes les communications écrites de la Société aux personnes inscrites au registre des actions seront valablement envoyées à l'adresse inscrite au registre des actions. |
Articles of Association of ADC Therapeutics SA
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2 Persons acquiring shares shall be registered in the share register as shareholders with voting rights upon their request if they expressly declare to have acquired these shares in their own name and for their own account, that there is no agreement on the redemption of the relevant shares and that they bear the economic risk associated with the shares. Subject to paragraph 4 of this Article 6 and article 685d para. 3 of the Swiss Code of Obligations, no person or entity shall be registered in the share register as a shareholder with voting rights for, and no person or entity may directly or indirectly, formally, constructively or beneficially own, or otherwise control alone or together with third parties voting rights (whether exercisable or not) with respect to more than 15% of the share capital as set forth in the commercial register as a shareholder with voting rights. This restriction shall also apply to persons or entities who hold some or all of their shares through Nominees (as defined in paragraph 4 of this Article 6). |
|
|
2 Les personnes qui acquièrent des actions sont inscrites dans le registre des actions, à leur demande, comme actionnaires avec droit de vote, pour autant qu'ils déclarent expressément avoir acquis les actions en leur nom et pour leur propre compte, qu'aucun contrat sur la reprise ou la restitution desdites actions n'a été conclu et qu'ils supportent le risque économique lié aux actions. Sous réserve de l'alinéa 4 du présent article 6 et de l'article 685d al. 3 du Code des obligations, aucune personne physique ou morale ne peut être inscrite au registre des actions comme actionnaire avec droit de vote et aucune personne physique ou morale ne peut détenir, directement ou indirectement, formellement, de fait ou comme ayant droit économique, ou contrôler autrement, seul ou avec des tiers, des droits de vote (exerçables ou non), par rapport à plus de 15% du capital-actions inscrit au registre du commerce en tant qu'actionnaire avec droit de vote. Cette restriction s'applique également aux personnes ou entités qui détiennent tout ou partie de leurs actions par l'intermédiaire de Nominees (tels que définis à l'alinéa 4 du présent article 6). |
|
3 Subject to Art. 652b para. 3 of the Swiss Code of Obligations, this transfer restriction also applies in the case of the acquisition of shares by the exercise of subscription, option and conversion rights. The transfer restriction does not apply to acquisitions by inheritance, division of an estate or matrimonial property law. |
|
|
3 Sous réserve de l'art. 652b al. 3 CO, les restrictions au transfert s'appliquent également lors de l'acquisition d'actions dans le cadre de l'exercice d'un droit de souscription, d'option ou de conversion. Les restrictions au transfert ne s'appliquent pas lors d'acquisitions par succession, partage successoral ou en vertu du droit matrimonial. |
Articles of Association of ADC Therapeutics SA
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
4 The Board of Directors may, in its own discretion, register persons who declare in the registration application that they hold the shares as nominees (each a Nominee) on behalf of third party beneficiaries (each a Beneficial Owner) in the share register as shareholders with voting rights. If, however, any Beneficial Owner should as a result of such registration being made or upheld, directly or indirectly, formally, constructively or beneficially own, or otherwise control or direct, alone or together with third parties, voting rights (whether exercisable or not) with respect to more than 15% of the share capital as set forth in the commercial register, the Board of Directors may cancel the registration of the Nominee holding shares for the account of such Beneficial Owner with respect to any shares in excess of such limit. The Board of Directors may make the registration with voting rights of the shares held by a Nominee subject to conditions, limitations and reporting requirements or may impose or adjust such conditions, limitations and requirements once registered. |
|
|
4 Le Conseil d'administration peut, à son entière discretion inscrire les personnes qui déclarent dans leur requête d'inscription qu'elles détiennent les actions en tant que nominees (chacun un Nominee) pour le compte de tiers ayants droit économiques (chacun un Ayant Droit Economique) en tant qu'actionnaires avec droit de vote. Toutefois, si suite à l'inscription ou à la confirmation de l'inscription, un Ayant Droit Economique détient directement ou indirectement, formellement, de fait ou comme ayant droit économique, ou contrôle ou dirige autrement, seul ou avec des tiers, des droits de votes (exerçables ou non) par rapport à plus de 15% du capital-actions inscrit au registre du commerce, le Conseil d'administration peut annuler l'inscription du Nominee détenant les actions pour le compte d'un tel Ayant Droit Economique pour les actions dépassant cette limite. Le Conseil d'administration peut soumettre l'inscription avec droit de vote des actions détenues par un Nominee à des conditions, limitations, exigences de rapports ou peut imposer de telles conditions, limitiations ou exigences suite à l'inscription. |
|
5 Legal entities and partnerships or other groups of persons or joint owners who are interrelated to one another through capital ownership, voting rights, uniform management or are otherwise linked, as well as individuals or legal entities or partnerships who act in concert or otherwise act in a coordinated manner or acquire shares indirectly, thereby circumventing the restrictions or limits pursuant to paragraph 2 or 4 of this article 6 shall be treated as one single person, entity, Nominee or as a person acquiring shares, as applicable, for purposes of paragraphs 2 and 4 of this article 6. |
|
|
5 Les personnes morales et communautés de personnes ou autres groupes de personnes ou de copropriétaires qui sont liés par le capital, les droits de vote, la gestion commune ou de toute autre manière, de même que les personnes physiques ou morales ou communautés de personnes qui agissent de concert ou de manière coordonnée ou acquièrent indirectement des actions, et contournent ainsi les restrictions ou limites visées aux alinéas 2 ou 4 du présent article 6 sont traités comme une seule personne, personne morale, Nominee ou comme une personne acquérant des actions, selon le cas, aux fins des alinéas 2 et 4 du présent article 6. |
Articles of Association of ADC Therapeutics SA
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
6 The Board of Directors may grant exceptions from the restrictions or limits pursuant to paragraph 2 or 4 of this article 6 for justified reasons with the majority vote of two thirds of all its members. A justified reason may include the situation where a person extends an offer to purchase with respect to all other shares of the Company, which the Board of Directors, after having consulted an independent financial advisor, recommends to the shareholders. Shareholders, other than Nominees, already being registered directly or through a Nominee with more than 15% at the time that this article takes effect remain registered with voting rights for such shares. |
|
|
6 Le Conseil d'administration peut octroyer des dérogations aux restrictions et limites mentionnées aux alinéas 2 ou 4 du présent article 6 pour des raisons justifiées, à la majorité des deux tiers de l'ensemble de ses membres. Peut aussi être considérée comme une raison justifiée le fait qu'une personne étende une offre d'achat par rapport à l'ensemble des autres actions de la Société et que le Conseil d'administration, après avoir consulté un conseiller financier indépendant, recommande aux actionnaires d'accepter cette offre. Les actionnaires autres que les Nominees déjà inscrits directement ou par l'intermédiaire d'un Nominee pour plus de 15% au moment où le présent article entre en vigueur demeurent enregistrés avec droit de vote pour ces actions. |
|
7 After hearing the registered shareholder or Nominee, the Board of Directors may cancel such person's registration in the share register with retroactive effect as of the date of registration if such registration was made based on false or misleading information or if such information becomes untrue or misleading. The relevant shareholder or Nominee shall be promptly informed of the cancellation. |
|
|
7 Le Conseil d'administration peut, après avoir entendu l'actionnaire ou le Nominee, radier du registre des actions l'inscription qui a été faite sur la base d'informations fausses ou trompeuses données par l'acquéreur, ou si les informations deviennent fausses ou trompeuses. L'actionnaire ou le Nominee doit être informé immédiatement de la radiation. |
|
8 The Board of Directors shall regulate all details and issue the instructions necessary to ensure compliance with the preceding provisions. The Board of Directors may delegate its duties. |
|
|
8 Le Conseil d'administration règle les détails et prend les mesures nécessaires au respect des dispositions ci-dessus. Le Conseil d'administration peut déléguer ses tâches. |
|
|
|
|
|
|
Article 7 |
|
|
Article 7 |
Exercise of Rights |
1 The Company shall only accept one representative per share. |
|
Exercice des droits |
1 La Société ne reconnaît qu'un représentant par action. |
Articles of Association of ADC Therapeutics SA
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2 The voting right and the rights associated therewith may be exercised vis-à-vis the Company by a shareholder, usufructuary or Nominee only to the extent that such person is registered in the share register with voting rights. |
|
|
2 Le droit de vote et les droits y relatifs ne peuvent être exercés à l'égard de la Société que par un actionnaire, un usufruitier ou un Nominee uniquement dans la mesure où celui-ci est inscrit avec droit de vote au registre des actions. |
|
|
|
|
|
|
|
|
|
|
|
Section 3
Corporate Bodies
|
|
|
Section 3
Organes
|
|
A. The General Meeting of Shareholders |
|
|
A. L'Assemblée générale |
|
|
|
|
|
|
Article 8 |
|
|
Article 8 |
Powers of the General Meeting of Shareholders |
1 The General Meeting of Shareholders is the supreme corporate body of the Company. |
|
Pouvoirs de l'Assemblée générale |
1 L'Assemblée générale est l'organe suprême de la Société. |
2 The General Meeting of Shareholders shall have the following inalienable powers: |
|
2 L'Assemblée générale a le droit inaliénable: |
1. the adoption and amendment of these articles of association; |
|
1. d'adopter et de modifier les présents statuts; |
2. the election of the members of the Board of Directors, the Chairperson of the Board of Directors and the members of the Compensation Committee; |
|
2. de nommer les membres du Conseil d'administration, le ou la président(e) du Conseil d'administration et les membres du Comité de rémunération; |
|
3. the election of the Auditors; |
|
|
3. de nommer l'organe de révision; |
|
4. the election of the independent voting rights representative; |
|
|
4. de nommer le représentant indépendant; |
|
5. the approval of the annual management report and the consolidated financial statements; |
|
|
5. d’approuver le rapport annuel et les comptes consolidés; |
Articles of Association of ADC Therapeutics SA
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
6. the approval of the annual financial statements as well as the resolution on the allocation of profit shown on the balance sheet, in particular the determination of dividends; |
|
|
6. d'approuver les comptes annuels et de déterminer l'emploi du bénéfice résultant du bilan, en particulier de fixer le dividende; |
|
7. the determination of interim dividends and the approval of the interim financial statements required for this purpose; |
|
|
7. de fixer le dividende intérimaire et d'approuver les comptes intermédiaires nécessaires à cet effet; |
|
8. the resolution on the repayment of the statutory capital reserve; |
|
|
8. de décider du remboursement de la réserve légale issue du capital; |
|
9. the discharge from liability of the members of the Board of Directors and the persons entrusted with management; |
|
|
9. de donner décharge aux membres du Conseil d'administration et aux personnes chargées de la gestion; |
|
10. the approval of the compensation of the Board of Directors and of the Executive Committee pursuant to Article 26 of these articles of association; |
|
|
10. d'approuver la rémunération du Conseil d'administration et de la Direction exécutive selon l'article 26 des présents statuts; et |
|
11. the delisting of the Company's equity securities; |
|
|
11. de procéder à la décotation des titres de participation de la société; |
|
12. the approval of the report on non-financial matters pursuant to article 964c CO (if applicable); and |
|
|
12. d'approuver le rapport sur les questions non finan-cières selon l'article 964c CO (si applicable); et |
|
13. the adoption of resolutions on matters that are reserved to the General Meeting of Shareholders by law or these articles of association or that are, subject to article 716a CO, submitted to the General Meeting of Shareholders by the Board of Directors. |
|
|
13. de prendre toutes les décisions qui lui sont réservées par la loi ou les présents statuts ou qui lui sont soumises par le Conseil d’administration, sous réserve de l’article 716a CO. |
|
|
|
|
|
|
Article 9 |
|
|
Article 9 |
Ordinary and Extraordinary General Meetings of Shareholders |
1 The Ordinary General Meeting of Shareholders shall be held each year within six months of the close of the financial year of the Company. |
|
Assemblées générales ordinaires et extraordinaires |
1 L'Assemblée générale ordinaire a lieu chaque année dans les six mois qui suivent la clôture de l'exercice de la Société. |
Articles of Association of ADC Therapeutics SA
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2 Extraordinary General Meetings of Shareholders shall be held if |
|
|
2 Des Assemblées générales extraordinaires ont lieu lorsque |
|
(a) the Board of Directors or the Auditors deem it necessary; |
|
|
(a) le Conseil d'administration ou l'organe de révision l'estime nécessaire; |
|
(b) so resolved by a General Meeting of Shareholders; or |
|
|
(b) une Assemblée générale le décide; ou |
|
(c) shareholders who hold, alone or together, shares representing at least 5% of the share capital or votes so request in writing, indicating the matters to be discussed and the corresponding proposals and, in case of elections, the names of the nominated candidates. |
|
|
(c) des actionnaires représentant seuls ou ensemble 5% au moins du capital-actions ou des voix le requièrent par écrit en indiquant les objets de discussion et les propositions, et en cas d'élections, les noms des candidats proposés. |
|
|
|
|
|
|
Article 10 |
|
|
Article 10 |
Notice |
1 Notice of a General Meeting of Shareholders shall be given by the Board of Directors or, if necessary, by the Auditors, no later than 20 calendar days prior to the date of the meeting. Liquidators and representatives of bond-holders are also entitled to call a General Meeting of Shareholders. |
|
Convocation |
1 L'Assemblée générale est convoquée par le Conseil d'administration ou, si nécessaire, par l'organe de révision au plus tard 20 jours calendaires avant le jour de l'assemblée. Les liquidateurs et les représentants de détenteurs d'obligations ont également le droit de convoquer l'Assemblée générale. |
|
2 Notice of the General Meeting of Shareholders shall be given by way of a single announcement in the official means of publication of the Company pursuant to Article 36 of these articles of association or by including the notice in the proxy statement pursuant to the rules of the United States Securities and Exchange Commission. |
|
|
2 La convocation à l'Assemblée générale a lieu par une annonce unique dans l'organe de publication de la Société selon l'article 36 des présents statuts ou en incluant la convocation dans le proxy statement conformément aux règles de la Securities and Exchange Commission des États-Unis.. |
|
3 The annual report, the compensation report, the Auditors' reports and any other reports required by law shall be made available to the shareholders no later than 20 calendar days prior to the Ordinary General Meeting of Shareholders.. |
|
|
3 Le rapport de gestion, le rapport de rémunération les rapports de révision et tout autre rapport exigés par la loi sont mis à la disposition des actionnaires au plus tard 20 jours calendaires avant l'Assemblée générale ordinaire. |
|
4 The notice shall include:
1.date, beginning, ending, mode and venue of the Shareholders' Meeting;
2.the agenda;
3.the proposals of the Board of Directors together with a brief statement of the reasons;
4.proposals of the shareholders, if any, together with a brief statement of the reasons; and
5.name and address of the independent voting rights representative.
|
|
|
4 La convocation doit mentionner:
1.la date, l'heure de début et de fin, le lieu et la forme de l'Assemblée générale;
2.les objets portés à l'ordre du jour;
3.les propositions du Conseil d'administration accompagnées d'une motivation succincte;
4.le cas échéant, les propositions des actionnaires, accompagnées d'une motivation succincte; et
5.le nom et l'adresse du représentant indépendant.
|
Articles of Association of ADC Therapeutics SA
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Article 11 |
|
|
Article 11 |
Agenda |
1 Shareholders who, alone or together, hold at least 0.5% of the share capital or the votes may request that an item be included on the agenda or that a proposal relating to an agenda item be included in the notice convening the General Meeting of Shareholders. Such request must be received by the Company at least 90 calendar days prior to the General Meeting of Shareholders, specifying the agenda item and the proposals of the shareholders. |
|
Objets à l'ordre du jour |
1 Des actionnaires qui représentent seuls ou ensemble détiennent au moins 0.5% du capital-actions ou des voix peuvent requérir l'inscription d'un objet à l'ordre du jour, ainsi que l'inscription dans la convocation à l'Assemblée générale de propositions concernant les objets portés à l'ordre du jour. La demande doit être reçue par la Société au moins 90 jours calendaires avant l'Assemblée générale avec indication des objets à l'ordre du jour et des propositions des actionnaires. |
|
2 No resolutions may be passed at a General Meeting of Shareholders on proposals concerning agenda items for which proper notice was not given. This provision shall not apply, however, to proposals made during a General Meeting of Shareholders to convene an Extraordinary General Meeting of Shareholders or to initiate a special audit. Each request for inclusion of an item on the agenda shall include (i) a brief description of the agenda item and the reason for which it is to be discussed at the meeting; (ii) the motions regarding the agenda item; (iii) the name and address, as they appear on the Company’s register of shareholders, of the shareholder proposing such business; (iv) the number of shares of the Company which are beneficially owned by such shareholder; (v) the dates upon which the shareholder acquired such shares; (vi) documentary support for any claim of beneficial ownership; (vii) any material interest of such shareholder in including the item in the agenda; (viii) a statement in support of the matter; and (ix) all other information required under applicable law and stock exchange rules. |
|
|
2 Aucune décision ne peut être prise par l'Assemblée générale sur des objets qui n'ont pas été dûment portés à l'ordre du jour, à l'exception des propositions de convoquer une Assemblée générale extraordinaire et d'instituer un contrôle spécial. Toute requête visant l'inscription d'un objet à l'ordre du jour doit inclure (i) une brève description de cet objet et la raison pour laquelle il doit être discuté lors de l'Asemblée générale; (ii) les propositions relatives à cet objet; (iii) le nom et l'adresse, tels qu'ils apparaissent dans le registre des actions, de l'actionnaire proposant un tel objet; (iv) le nombre d'actions de la Société dont cet actionnaire est ayant droit économique; (v) les dates auxquelles l'actionnaire a acquis ces actions; (vi) les pièces justificatives démontrant le statut d'ayant droit économique; (vii) l'intérêt important de l'actionnaire à l'inscription de l'objet à l'ordre du jour; (viii) une déclaration à l'appui de la requête; et (ix) toute autre information requise par la loi ou les règles boursières applicables. |
|
3 No prior notice is required to bring motions related to items already on the agenda or for the discussion of matters on which no resolution is to be taken. |
|
|
3 En revanche, il n'est pas nécessaire d'annoncer à l'avance les propositions entrant dans le cadre des objets portés à l'ordre du jour ni les délibérations qui ne doivent pas être suivies d'un vote. |
|
Article 11a |
|
|
Article 11a |
Venue |
1 The Board of Directors shall determine the venue of the General Meeting of Shareholders, which may be held in Switzerland or abroad. |
|
Lieu de réunion |
1 Le Conseil d'administration détermine le lieu de l'Assemblée générale, qui peut se tenir en Suisse ou à l'étranger. |
Articles of Association of ADC Therapeutics SA
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2 The Board of Directors can determine that the General Meeting of Shareholders be held simultaneously at different locations, provided that the contributions of the participants are transmitted directly in video and audio to all venues and that shareholders who are not present at the venue(s) of the General Meeting of Shareholders may exercise their rights by electronic means. |
|
|
2 Le Conseil d'administration peut décider que l'Assemblée générale se tiendra simultanément en plusieurs lieux, à condition que les votes des participants soient transmis directement par l'image et le son à tous les lieux de réunion et que les actionnaires qui ne sont pas présents au(x) lieu(x) de l'Assemblée générale puissent exercer leurs droits par voie électronique. |
|
3 Alternatively, the Board of Directors may provide that the General Meeting of Shareholders will be held by electronic means without a venue. |
|
|
3 Alternativement, le Conseil d'administration peut prévoir que l'Assemblée générale se déroule par voie électronique sans lieu de réunion. |
|
|
|
|
|
|
Article 12 |
|
|
Article 12 |
Chairperson, Vote Counters, Minutes |
1 The Chairperson of the Board of Directors shall chair the General Meeting of Shareholders. In his absence, the Vice-Chairperson of the Board of Directors, another member or a person designated by the Board of Directors shall chair the General Meeting of Shareholders. If no member of the Board of Directors is available and no other person has been designated by the Board of Directors, the acting chair shall be elected by the General Meeting of Shareholders. |
|
Présidence, scrutateurs, procès-verbal |
1 Le ou la président(e) du Conseil d'administration préside l'Assemblée générale. En son absence, le ou la vice-président(e) du Conseil d'administration, un autre membre ou une personne désignée par le Conseil d'administration préside l'Assemblée générale. Si aucun membre du Conseil d'administration n'est disponible et aucune personne n'a été désignée par le Conseil d'administration, l'Assemblée générale élit son président. |
|
2 The acting chair of the General Meeting of Shareholders shall appoint the secretary and the vote counter(s), none of whom need be shareholders. The minutes shall be signed by the acting chair of the General Meeting of Shareholders and the secretary. |
|
|
2 Le président de l'Assemblée générale désigne un rédacteur du procès-verbal et le ou les scrutateurs, qui ne doivent pas nécessairement être des actionnaires. Le procès-verbal doit être signé par le président de l'Assemblée générale et le secrétaire. |
|
3 The acting chair of the General Meeting of Shareholders shall have all powers and authority necessary and appropriate to ensure the orderly conduct of the General Meeting of Shareholders. |
|
|
3 Le président de l'Assemblée générale a tous les pouvoirs nécessaires et appropriés pour s'assurer de la conduite régulière de l'Assemblée générale. |
Articles of Association of ADC Therapeutics SA
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
4 The resolutions and election results shall be made available electronically within 15 calendar days after the General Meeting of Shareholders, stating the exact proportion of votes; each shareholder may request that the minutes be made available to him within 30 calendar days after the General Meeting of Shareholders. |
|
|
4 Les décisions et le résultat des élections, avec indication de la répartition exacte des voix, doivent être rendus accessibles par voie électronique dans les 15 jours calendaires qui suivent l'Assemblée générale; chaque actionnaire peut exiger que le procès-verbal soit mis à sa disposition dans les 30 jours calendaires qui suivent l'Assemblée générale. |
|
|
|
|
|
|
Article 13 |
|
|
Article 13 |
Voting Rights, Representation |
1 Each share shall convey the right to one vote. The voting rights are subject to the conditions of Articles 6 and 7 of these articles of association. |
|
Droit de vote, représentation |
1 Chaque action donne droit à une voix. Les droits de vote sont soumis aux conditions des articles 6 et 7 des présents statuts. |
2 The Board of Directors shall issue the rules regarding the participation in and representation at the General Meeting of Shareholders and determine the requirements as to proxies and instructions. A shareholder may be represented at the General Meeting of Shareholders by the independent voting rights representative, its legal representative or, by means of a written proxy, any other proxy who need not be a shareholder. All shares held by a shareholder may only be represented by one person. |
|
|
2 Le Conseil d'administration prend les dispositions relatives à la participation et à la représentation à l'Assemblée générale et détermine les exigences applicables aux procurations et instructions. Un actionnaire peut être représenté à l'Assemblée générale par le représentant indépendant, par son représentant légal ou, au moyen d'une procuration écrite, partout autre mandataire qui ne doit pas nécessairement être un actionnaire . Toutes les actions détenues par un actionnaire ne peuvent être représentées que par une seule personne. |
|
3 The General Meeting of Shareholders shall elect the independent voting rights representative for a term of office until completion of the next Ordinary General Meeting of Shareholders. Re-election is possible. |
|
|
3 L'Assemblée générale nomme le représentant indépendant pour une durée de fonctions s’achevant à la fin de l’Assemblée générale ordinaire suivante. La réélection est possible. |
|
4 If the Company does not have an independent voting rights representative, the Board of Directors shall appoint the independent voting rights representative for the next General Meeting of Shareholders. |
|
|
4 Dans le cas où la Société n'a pas de représentant indépendant, le Conseil d'administration nommera le représentant indépendant pour l'Assemblée générale suivante. |
|
|
|
|
|
|
Article 14 |
|
|
Article 14 |
Articles of Association of ADC Therapeutics SA
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Resolutions, Elections |
1 The General Meeting of Shareholders shall pass its resolutions and decide its elections by the majority of the votes attached to the shares represented, unless required otherwise by law or these articles of association. In the event of a tie, the resolution shall be deemed refused. |
|
Décisions, élections |
1 Les décisions de l'Assemblée générale sont prises à la majorité des voix attribuées aux actions représentées, à moins que la loi ou les présents statuts n'en disposent autrement. En cas d'égalité de voix, la décision est refusée. |
|
2 Two thirds of the votes represented and the majority of the par value of shares represented shall be required for the General Meeting of Shareholders to adopt resolutions on the following matters: |
|
|
2 Une décision de l'Assemblée générale recueillant au moins les deux tiers des voix attribuées aux actions représentées et la majorité des valeurs nominales représentées est nécessaire pour: |
|
1. the amendment of the purpose of the Company; |
|
|
1. la modification du but social de la Société; |
|
2. the creation of shares with privileged voting rights; |
|
|
2. l'introduction d'actions à droit de vote privilégié; |
|
3. the restriction on the transferability of registered shares or their registration with voting rights and the cancelation of such a restriction; |
|
|
3. la restriction de la transmissibilité des actions nominatives ou leur inscription avec droit de vote ainsi que la suppression d'une telle restriction; |
|
4. the introduction of conditional share capital or the introduction of a capital range; |
|
|
4. la création d'un capital conditionnel ou l'institution d'une marge de fluctuation du capital; |
|
5. an increase in share capital through the conversion of equity surplus, against contributions in kind, by set-off against a claim, or the granting of special benefits; |
|
|
5. l'augmentation du capital-actions au moyen de la conversion de fonds propres, contre apport en nature, par compensation de créance, ainsi que l'octroi d'avantages particuliers; |
|
6. the limitation or withdrawal of pre-emptive rights; |
|
|
6. la limitation ou la suppression du droit de souscription préférentiel; |
|
7. the change of currency of the share capital; |
|
|
7. le changement de la monnaie dans laquelle le capital-actions est fixé; |
|
8. the introduction of a casting vote for the person chairing the General Meeting of Shareholders; |
|
|
8. l'introduction de la voix prépondérante du ou de la président(e) à l'Assemblée générale; |
|
9. the relocation of the registered office of the Company; |
|
|
9. le transfert du siège de la Société; |
|
10. the delisting of the Company's equity securities; |
|
|
10. la décotation des titres de participation de la Société; |
|
11. the dissolution of the Company; |
|
|
11. la dissolution de la Société; |
Articles of Association of ADC Therapeutics SA
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
12. the introduction of an arbitration clause in the articles of association; |
|
|
12. l'introduction d'une clause d'arbitrage dans les statuts; |
|
13. mergers, demergers and conversions pursuant to the Merger Act; |
|
|
13. une fusion, scission ou transformation conformément à la Loi sur la fusion; |
|
14. the conversion of registered shares into bearer shares; |
|
|
14. la conversion d'actions nominatives en actions au porteur; |
|
15. the combination of shares; |
|
|
15. la réunion d'actions; |
|
16. the removal of any member of the Board of Directors or of its Chairperson before the end of his or her term of office; and |
|
|
16. la révocation de tout membre du Conseil d'administration ou de son président avant la fin de son mandat; et |
|
17. the amendment or repeal of the following provisions of these articles of association, with the exception of editorial amendments that do not effectively change their content:
(i) article 6;
(ii) article 14;
(iii) article 15;
(iv) article 18; and
(v) article 35a.
|
|
|
17. la modification ou la suppression des dispositions suivantes des présents statuts, à l'exception des modifications rédactionnelles qui ne modifient pas effectivement leur contenu:
(i) article 6;
(ii) article 14;
(iii) article 15;
(iv) article 18; et
(v) article 35a.
|
|
3 Resolutions and elections shall be decided by open ballot, unless the acting chair of the General Meeting of Shareholders decides that a secret ballot be held or that it be voted by electronic means. The acting chair may at any time order that a resolution or election be repeated if he considers the vote to be in doubt. The resolution or election previously held shall then be deemed not to have taken place. |
|
|
3 Les décisions et élections ont lieu à main levée, à moins qu'un vote à bulletins secrets ou électronique ne soit ordonné par le président de l'Assemblée générale. Le président peut en tout temps ordonner qu'une décision ou élection soit répétée s'il estime qu'il existe des doutes sur le résultat. Dans ce cas, la décision ou l'élection précédente est réputée ne pas avoir eu lieu. |
|
|
|
|
|
|
B. The Board of Directors |
|
|
B. Le Conseil d'administration |
|
|
|
|
|
Articles of Association of ADC Therapeutics SA
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Article 15 |
|
|
Article 15 |
Number of Directors |
The Board of Directors shall consist of not less than 3 and no more than 9 members. |
|
Nombre de membres |
Le Conseil d'administration se compose de 3 membres au moins et de 9 membres au plus. |
|
|
|
|
|
|
Article 16 |
|
|
Article 16 |
Election and Term of Office |
1 The General Meeting of Shareholders shall elect the members of the Board of Directors and the Chairperson of the Board of Directors individually and for a term of office until the completion of the next Ordinary General Meeting of Shareholders. Re-election is possible. |
|
Élection et durée des fonctions |
1 Les membres du Conseil d’administration et le ou la président(e) du Conseil d’administration sont élus individuellement par l’Assemblée générale pour une durée de fonctions s’achevant à la fin de l’Assemblée générale ordinaire suivante. La réélection est possible. |
|
2 If the office of the Chairperson of the Board of Directors is vacant, the Board of Directors shall appoint a new Chairperson from among its members for a term of office extending until completion of the next Ordinary General Meeting of Shareholders. |
|
|
2 Lorsque la fonction de président(e) du Conseil d’administration est vacante, le Conseil d’administration désigne un nouveau ou une nouvelle président(e) parmi ses membres pour une durée de fonctions s’achevant à la fin de l’Assemblée générale ordinaire suivante. |
|
|
|
|
|
|
Article 17 |
|
|
Article 17 |
Organization of the Board of Directors |
1 Except for the election of the Chairperson of the Board of Directors and the members of the Compensation Committee by the General Meeting of Shareholders, the Board of Directors shall constitute itself. The Board of Directors may elect one or several Vice-Chairperson. The Board of Directors shall further appoint a secretary who need not be member of the Board of Directors. |
|
Organisation du Conseil d'administration |
1 A l'exception de l'élection par l'Assemblée générale du de la président(e) du Conseil d'administration et des membres du Comité de rémunération, le Conseil d'administration se constitue lui-même. Il peut désigner au besoin, un(e) ou plusieurs vice-président(e)s. Le Conseil d'administration désigne en outre un secrétaire, qui ne doit pas nécessairement être membre du Conseil d'administration. |
|
2 Subject to these articles of association, the Board of Directors shall regulate its organization and the adoption of resolutions in the organizational regulations. |
|
|
2 Le Conseil d'administration règle en outre son organisation et la manière de prendre des décisions dans un règlement d'organisation, sous réserve des présents statuts. |
|
|
|
|
|
|
Article 18 |
|
|
Article 18 |
Articles of Association of ADC Therapeutics SA
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Reimbursement of Expenses, Indemnification |
1 The members of the Board of Directors shall be entitled to the reimbursement of all expenses incurred in the interest of the Company. |
|
Remboursement des frais, indemnisation |
1 Les membres du Conseil d'administration ont droit au remboursement de tous les frais engagés dans l'intérêt de la Société. |
2 To the extent not included in insurance coverage or paid by third parties, the Company shall indemnify and hold harmless, to the extent permitted by law, the existing and former members of the Board of Directors and Executive Committee, and their heirs, executors and administrators, out of the assets of the Company from and against all threatened, pending or completed actions, suits or proceedings – whether civil, criminal, administrative or investigative – and all costs, charges, losses, damages, and expenses which they or any of them, their heirs, executors or administrators, shall or may incur or sustain by or by reason of any actual or alleged actions, consents or omissions in or about the execution of their duty, or alleged duty, or by reason of the fact that he is or was a member of the Board of Directors or Executive Committee of the Company or one of its subsidiaries, or, while serving as a member of the Board of Directors or Executive Committee of the Company, is or was serving at the request of the Company as a director, member of the executive management, employee or agent of another corporation, partnership, joint venture, trust or other enterprise; provided, however, that this indemnity shall not extend to any matter in which any of said persons is found, in a final judgment or decree of a court or governmental or administrative authority of competent jurisdiction not subject to appeal, to have committed an intentional or grossly negligent breach of his statutory duties as a member of the Board of Directors or Executive Committee. |
|
2 Dans la mesure où la loi le permet, la Société indemnisera, à concurrence de la portion non couverte par une assurance ou payée par un tiers, sur ses propres biens les membres actuels et passés du Conseil d'administration et de la Direction exécutive ainsi que leurs héritiers, masse en faillite ou masse successorale contre toutes actions, procès ou poursuites, menaçants, en cours ou terminés, de nature civile, pénale, administrative ou autre, et tous les coûts, dépenses, pertes, dommages et frais qu'ils (ou leurs héritiers, masse en faillite ou masse successorale) subiraient ou pourraient subir en raison d'actions, consentements ou omissions, effectifs ou présumés, en relation avec l'exercice de leurs fonctions, leurs fonctions supposées ou en raison du fait d'être ou d'avoir été membres du Conseil d'administration ou de la Direction exécutive de la Société ou de l'une de ses filiales ou, sur instruction de la Société en tant que membres du Conseil d'administration ou de la Direction exécutive, en raison du fait d'être ou d'avoir été administrateur, membre de la direction, employé ou mandataire d'une autre société, entreprise, coentreprise, personne morale dénuée de la personnalité ou trust. L'obligation d'indemnisation s'éteint dès qu'un jugement définitif et exécutoire d'un tribunal ou d'une autorité compétente a décidé que la personne en question a violé, volontairement ou par grave négligence, ses devoirs de membre du Conseil d'administration ou de la Direction exécutive. |
|
3 Without limiting the foregoing paragraph 2 of this Article 18, the Company shall advance costs and expenses idemnifiable thereunder to the existing and former members of the Board of Directors and Executive Committee to the extent not included in insurance coverage or advanced by third parties. The Company may however recover such advanced costs if any of said persons is found, in a final judgment or decree of a court or governmental or administrative authority of competent jurisdiction not subject to appeal, to have committed an intentional or grossly negligent breach of his statutory duties as a member of the Board of Directors or Executive Committee. |
|
|
3 Sans préjudice de l'alinéa 2 du présent article 18, la Société avancera les frais et les coûts indemnisables en vertu de la disposition précitée aux membres actuels et passés du Conseil d'administration et de la Direction exécutive, à concurrence de la portion non couverte par une assurance ou payée par un tiers. La Société peut cependant recouvrer ces avances de frais si l'une de ces personnes a été reconnue coupable de violation intentionnelle ou par négligence grave de ses devoirs de membre du Conseil d'administration ou de la Direction exécutive par un jugement ou une décision final et exécutoire d'un tribunal ou d'une autorité gouvernementale ou administrative compétente. |
|
|
|
|
|
|
Article 19 |
|
|
Article 19 |
Articles of Association of ADC Therapeutics SA
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Convening of Meetings, Resolutions, Minutes |
1 The Board of Directors shall meet at the invitation of its Chairperson or, if not available, of the Vice-Chairperson or of another member of the Board of Directors as often as the business of the Company shall require or if a member requests it in writing or electronically, indicating the reasons. |
|
Convocation, décisions, procès-verbal |
1 Le Conseil d'administration est convoqué par son ou sa président(e) ou, en cas d'empêchement de ce dernier, par son ou sa vice-président(e) ou par un autre membre du Conseil d'administration, aussi souvent que cela apparaît nécessaire ou lorsqu'un membre du Conseil d'administration le demande par écrit ou par voie électronique, avec indication des motifs. |
|
2 Unless the organizational regulations adopted by the Board of Directors or a board resolution taken with the applicable attendance quorum provide otherwise, the Board of Directors shall only have a quorum if a majority of the members of the Board of Directors is present. No attendance quorum shall be required for resolutions of the Board of Directors providing for the amendment and ascertainment of capital changes or a change in the currency of the share capital. |
|
|
2 A moins que le contraire ne résulte d'une disposition du règlement d'organisation adopté par le Conseil d'administration ou d'une décision du Conseil d'administration prise conformément aux dispositions applicables au quorum de présence, la majorité des membres du Conseil d'administration doivent être présents afin de pouvoir prendre une décision. Ce quorum de présence n'est pas nécessaire pour les décisions de modification et de constatation du Conseil d'administration en lien avec les modifications du capital-actions ou de changement de la monnaie du capital-actions. |
|
3 Unless the organizational regulations adopted by the Board of Directors provide otherwise, the Board of Directors shall adopt its resolutions by a majority of votes cast. In the case of a tie, the Chairperson of the Board of Directors shall have the casting vote. |
|
|
3 Sauf si le règlement d'organisation adopté par le Conseil d'administration en dispose autrement, les décisions du Conseil d'administration sont prises à la majorité des voix exprimées. En cas d'égalité des voix, la voix du président du Conseil d'administration prévaut. |
|
4 Resolutions may also be adopted by way of written consent or electronically, unless a member requests discussion thereof. |
|
|
4 Les décisions du Conseil d'administration peuvent également être prises par voie de circulation ou par voie électronique, à moins qu'une discussion ne soit requise par l'un des membres du Conseil d'administration. |
|
5 The decisions of the Board of Directors shall be recorded in minutes to be signed by the acting chair and the secretary. |
|
|
5 Les décisions du Conseil d'administration sont consignées dans un procès-verbal signé par le président et par le secrétaire. |
|
|
|
|
|
|
Article 20 |
|
|
Article 20 |
Articles of Association of ADC Therapeutics SA
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Powers of the Board of Directors |
1 The Board of Directors may pass resolutions with respect to all matters which are not delegated to another corporate body of the Company by law, by these articles of association or by regulations. |
|
Attributions du Conseil d'administration |
1 Le Conseil d'administration peut prendre des décisions sur toutes les affaires qui ne sont pas attribuées à un autre organe de la Société par la loi, les présents statuts ou un règlement. |
|
2 It shall have the following non-transferable and inalienable duties: |
|
|
2 Il a les attributions intransmissibles et inaliénables suivantes: |
|
1. the ultimate management of the Company and the issuance of necessary instructions; |
|
|
1. exercer la haute direction de la Société et établir les instructions nécessaires; |
|
2. the determination of the organization of the Company; |
|
|
2. fixer l'organisation de la Société; |
|
3. the structuring of the accounting system, of the financial controls and of the financial planning; |
|
|
3. fixer les principes de la comptabilité et du contrôle financier ainsi que le plan financier; |
|
4. the appointment and dismissal of the persons entrusted with management and representation of the Company, and issuance of rules on the signature authority; |
|
|
4. nommer et révoquer les personnes chargées de la gestion et de la représentation de la Société et réglementer le droit de signature; |
|
5. the ultimate supervision of the persons entrusted with management, in particular in view of compliance with the law, these articles of association, regulations and directives; |
|
|
5. exercer la haute surveillance sur les personnes chargées de la gestion pour s'assurer notamment qu'elles observent la loi, les présents statuts, les règlements et les instructions données; |
|
6. the preparation of the annual report, the compensation report and, if applicable, the report on non-financial matters pursuant to article 964c CO and other reports as required by law, if any; |
|
|
6. établir le rapport de gestion, le rapport de rémunération, et, le cas échéant, le rapport sur les questions non financières selon l'article 964c CO et, le cas échéant, d'autres rapports exigés par la loi; |
|
7. the preparation of the General Meeting of Shareholders and the implementation of its resolutions; |
|
|
7. préparer l'Assemblée générale et exécuter ses décisions; |
Articles of Association of ADC Therapeutics SA
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
8. the adoption of resolutions on the change of the share capital to the extent that such power is vested in the Board of Directors, the ascertainment of capital changes , the preparation of the report on the capital increase, and the respective amendments of the articles of association (including deletions); |
|
|
8. prendre les décisions relatives aux modifications du capital-actions, dans la mesure où elles sont de la compétence du Conseil d'administration, ainsi que les décisions relatives à la constatation des modifications de capital, à l'établissement du rapport d'augmentation du capital-actions et aux modifications des statuts qui en résultent (radiation comprise); |
|
9. the non-transferable and inalienable duties and powers of the Board of Directors pursuant to the Merger Act; |
|
|
9. les attributions et compétences intransmissibles et inaliénables du Conseil d'administration selon la Loi sur la fusion; |
|
10. the submission of a petition for debt-restructuring moratorium and the notification of the court in case of over-indebtedness; and |
|
|
10. le dépôt d'une demande de sursis concordataire et l'avis au tribunal en cas de surendettement; et |
|
11. other powers and duties reserved to the Board of Directors by law or these articles of association. |
|
|
11. d'autres attributions et compétences réservées au Conseil d'administration par la loi ou les présents statuts. |
|
3 In all other respects, the Board of Directors may delegate in whole or in part the management and the representation of the Company within the framework set forth by these articles of association and the law to one or several of its members or to third parties by means of organizational regulations. |
|
|
3 En outre, le Conseil d'administration peut déléguer en tout ou en partie la gestion ainsi que la représentation de la Société, dans le cadre des présents statuts et de la loi, à un ou plusieurs de ses membres ou à des tiers conformément au règlement d'organisation. |
|
|
|
|
|
|
C. The Compensation Committee |
|
|
C. Le Comité de rémunération |
|
|
|
|
|
|
Article 21 |
|
|
Article 21 |
Number of Members |
The Compensation Committee shall consist of at least two members of the Board of Directors. |
|
Nombre de membres |
Le Comité de rémunération se compose d'au moins deux membres du Conseil d'administration. |
|
|
|
|
|
|
Article 22 |
|
|
Article 22 |
Articles of Association of ADC Therapeutics SA
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Election and Term of Office |
1 The General Meeting of Shareholders shall elect the members of the Compensation Committee individually for a term of office until the completion of the subsequent Ordinary General Meeting of Shareholders. Only members of the Board of Directors may be elected. Re-election is possible. |
|
Election et durée de fonctions |
1 L'Assemblée générale élit individuellement les membres du Comité de rémunération pour une durée de fonctions s'achevant à la fin de l'Assemblée générale ordinaire suivante. Seuls des membres du Conseil d'administration sont éligibles. La réélection est possible. |
|
2 If there are vacancies on the Compensation Committee, the Board of Directors may appoint substitute members from among its members for a term of office extending until completion of the next Ordinary General Meeting of Shareholders. |
|
|
2 En cas de vacance au sein du Comité de rémunération, le Conseil d'administration peut désigner des substituts parmi ses membres pour une durée de fonctions s'achevant à la fin de l'Assemblée générale ordinaire suivante. |
|
|
|
|
|
|
Article 23 |
|
|
Article 23 |
Organization of the Compensation Committee |
1 The Compensation Committee shall constitute itself. Unless the organizational regulations provide otherwise, the Board of Directors shall elect a chairperson from among the Compensation Committee's members. |
|
Organisation du Comité de rémunération |
1 Le Comité de rémunération se constitue lui-même. A moins que le règlement d'organisation n'en dispose autrement, le Conseil d'administration élit le ou la Président(e) du Comité de rémunération parmi les membres du Comité de rémunération. |
2 The Board of Directors shall issue regulations establishing the organization and decision-making process of the Compensation Committee, which may be part of the organizational regulations. |
|
2 Le Conseil d'administration établit un règlement concernant l'organisation et le processus de décision du Comité de rémunération, qui peut être intégré au règlement d'organisation. |
|
|
|
|
|
|
Article 24 |
|
|
Article 24 |
Duties and Powers |
1 The Compensation Committee shall support the Board of Directors in establishing and reviewing the compensation strategy and guidelines as well as in preparing the proposals to the General Meeting of Shareholders regarding the compensation of the Board of Directors and the Executive Committee. It may submit proposals to the Board of Directors in other compensation-related issues. |
|
Attributions |
1 Le Comité de rémunération assiste le Conseil d'administration dans l'établissement et la révision de la stratégie et des directives de rémunération, ainsi que dans la préparation des propositions à soumettre à l'Assemblée générale concernant la rémunération du Conseil d'administration et de la Direction exécutive. Il peut soumettre au Conseil d'administration des propositions en toutes autres matières relatives à la rémunération. |
Articles of Association of ADC Therapeutics SA
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2 The Board of Directors shall determine in regulations for which positions of the Board of Directors, the Executive Committee and other member of management (if any) the Compensation Committee shall submit proposals for the performance metrics, target values and/or the compensation of the members of the Board of Directors and the Executive Committee, and for which positions it shall itself determine, in accordance with these articles of association and the compensation guidelines established by the Board of Directors, such performance metrics, target values and/or the compensation. |
|
|
2 Le Conseil d'administration détermine dans un règlement pour quelles fonctions du Conseil d'administration, de la Direction exécutive et d'autres membres de la direction (si applicable) le Comité de rémunération proposera au Conseil d'administration les mesures de performances, les valeurs cibles et/ou la rémunération des membres du Conseil d'administration et de la Direction exécutive, et pour quelles fonctions il aura la compétence de déterminer de son propre chef, en accord avec les statuts et les directives de rémunération établies par le Conseil d'administration, les mesures de performances, les valeurs cibles et/ou la rémunération. |
|
3 The Board of Directors may delegate further tasks to the Compensation Committee. |
|
|
3 Le Conseil d’administration peut déléguer d'autres tâches au Comité de rémunération. |
|
|
|
|
|
|
D. The Auditors |
|
|
D. L'organe de révision |
|
|
|
|
|
|
Article 25 |
|
|
Article 25 |
|
1 The General Meeting of Shareholders shall elect the Auditors for a term of office until the completion of the next Ordinary General Meeting of Shareholders. Re-election is possible. |
|
|
1 L'Assemblée générale élit l'organe de révision pour une durée de fonctions s’achevant à la fin de l’Assemblée générale ordinaire suivante. La réélection est possible. |
|
2 The Auditors shall have the powers and duties vested in them by law. |
|
|
2 L'organe de révision a les pouvoirs et obligations que lui confère la loi. |
|
3 The Board of Directors may mandate the Auditors at any time to perform special investigations, in particular interim audits, and to prepare a report on their findings. |
|
|
3 Le Conseil d'administration peut en tout temps charger l'organe de révision de procéder à des contrôles spéciaux, notamment des révisions intermédiaires, et de lui en soumettre un rapport. |
|
|
|
|
|
|
|
|
|
|
Articles of Association of ADC Therapeutics SA
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Section 4
Compensation of the Members of the Board of Directors and the Executive Committee and Related Matters |
|
|
Section 4
Rémunération des membres du Conseil d'administration et de la Direction exécutive et affaires connexes |
|
|
|
|
|
|
Article 26 |
|
|
Article 26 |
Approval of the Compensation by the General Meeting of Shareholders |
1 The General Meeting of Shareholders shall approve the proposals of the Board of Directors in relation to the aggregate amounts of: |
|
Approbation de la rémunération par l'Assemblée générale |
1 L'Assemblée générale approuve les propositions du Conseil d'administration en relation avec les montants maximaux suivants: |
1. the maximum compensation of the Board of Directors until the completion of the next Ordinary General Meeting of Shareholders; |
|
1. la rémunération maximale du Conseil d'administration jusqu'à la fin de l'Assemblée générale ordinaire des actionnaires suivante; |
|
2. the maximum fixed compensation of the Executive Committee for the following financial year; and |
|
|
2. la rémunération fixe maximale de la Direction exécutive pour l'année comptable suivante; et |
|
3. the maximum variable compensation of the Executive Committee for the current financial year. |
|
|
3. la rémunération variable maximale de la Direction exécutive pour l'exercice en cours. |
|
2 The Board of Directors may submit for approval by the General Meeting of Shareholders deviating, additional or conditional proposals relating to the maximum aggregate amount or maximum partial amounts for the same or different periods and/or specific compensation components and/or in relation to additional amounts for specific compensation components. |
|
|
2 Le Conseil d'administration peut soumettre à l'approbation de l'Assemblée générale des propositions divergentes, supplémentaires ou conditionnelles concernant le montant maximal total ou les montants maximaux partiels pour les mêmes périodes ou des périodes différentes et/ou des éléments de rémunération spécifiques et/ou en relation avec des montants additionnels pour des éléments de rémunération spécifiques. |
|
3 In the event that the General Meeting of Shareholders does not approve a proposal of the Board of Directors, the Board of Directors shall determine, taking into account all relevant factors, the respective (maximum) aggregate amount or (maximum) partial amounts, and submit the amount(s) so determined for approval by a General Meeting of Shareholders. |
|
|
3 Si l'Assemblée générale n'approuve pas une proposition du Conseil d'administration, le Conseil d'administration détermine, en prenant en compte tous les critères pertinents, le montant (maximal) total ou des montants (maximaux) partiels respectifs, et soumet le(s) montant(s) ainsi déterminé(s) à l'approbation d'une Assemblée générale. |
Articles of Association of ADC Therapeutics SA
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
4 The Company or companies controlled by it may pay or grant compensation prior to approval by the General Meeting of Shareholders, subject to subsequent approval. |
|
|
4 La rémunération peut être versée ou octroyée par la Société ou les sociétés qu'elle contrôle avant l'approbation de l'Assemblée générale, sous réserve d'une approbation ultérieure. |
|
5 If variable compensation is approved prospectively, the Board of Directors shall submit the compensation report to the General Meeting of Shareholders for a consultative vote. |
|
|
5 Si des rémunérations variables sont approuvées de manière prospective, le Conseil d'administration soumet le rapport de rémunération au vote consultatif de l'Assemblée générale. |
|
|
|
|
|
|
Article 27 |
|
|
Article 27 |
Supplementary Amount for Changes to the Executive Committee |
If the maximum aggregate amount of compensation already approved by the General Meeting of Shareholders is not sufficient to also cover the compensation of one or more persons who become members of the Executive Committee after the General Meeting of Shareholders has approved the compensation of the Executive Committee for the relevant period, then the Company or companies controlled by it shall be authorized to pay such member(s) a supplementary amount during the compensation period(s) already approved. The supplementary amount per compensation period per member shall not exceed 100% of the aggregate amount of (maximum) compensation of the Executive Committee last approved. |
|
Montant complémentaire en cas de changements au sein de la Direction exécutive |
Si le montant global maximal de la rémunération déjà approuvé par l'Assemblée générale n'est pas suffisant pour couvrir également la rémunération d'une ou plusieurs personnes devenant membre(s) de la Direction exécutive après que l'Assemblée générale a approuvé la rémunération de la Direction exécutive pour la période visée, la Société ou toute autre société qu'elle contrôle est alors autorisée à verser à ce(s) membre(s) un montant complémentaire au cours de la (ou les) période(s) de rémunération déjà approuvée(s). Le montant complémentaire par période de compensation par membre ne doit pas dépasser 100% du montant global de la rémunération (maximale) de la Direction exécutive approuvée en dernier. |
|
|
|
|
|
|
Article 28 |
|
|
Article 28 |
Articles of Association of ADC Therapeutics SA
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
General Compensation Principles |
1 The compensation of the non-executive members of the Board of Directors may consist of fixed and variable compensation elements. Total compensation shall take into account the position and level of responsibility of the recipient. |
|
Principes généraux de rémunération |
1 La rémunération des membres non-exécutifs du Conseil d'administration peut être constituée d'éléments de rémunérations fixes et variables. La rémunération totale prend en compte la position et le niveau de responsabilité du bénéficiaire. |
2 The compensation of the members of the Executive Committee may consist of fixed and variable compensation elements. Fixed compensation comprises the base salary and may consist of other compensation elements. Variable compensation may take into account the achievement of specific performance targets. Total compensation shall take into account the position and level of responsibility of the recipient. |
|
2 La rémunération des membres de la Direction exécutive peut être constituée d'éléments de rémunération fixes et variables. La rémunération fixe comprend le salaire de base et peut être constituée d'autres éléments de rémunération. La rémunération variable peut prendre en compte l'accomplissement d'objectifs de performance spécifiques. La rémunération totale prend en compte la position et le niveau de responsabilité du bénéficiaire. |
|
3 The performance targets may include individual targets, targets of the Company, group or parts thereof or targets in relation to the market, other companies or comparable benchmarks, taking into account the position and level of responsibility of the recipient. The Board of Directors or, to the extent delegated to it, the Compensation Committee shall determine the relative weight of the performance targets and the respective target values as well as their achievement. |
|
|
3 Les objectifs de performance peuvent comprendre des objectifs personnels, des objectifs liés à la performance de la Société ou de tout ou partie du groupe ou des buts en relation avec le marché, d'autres sociétés ou d'autres repères comparables, prenant en compte la position et le niveau de responsabilité du bénéficiaire. Le Conseil d'administration ou le Comité de rémunération, dans la mesure où cette compétence lui est déléguée, détermine le poids relatif des objectifs de performance et les valeurs cibles respectives ainsi que leur accomplissement. |
Articles of Association of ADC Therapeutics SA
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
4 Compensation may be paid in the form of cash, shares, options or other share-based instruments or units, or in the form of other types of benefits. The Board of Directors or, to the extent delegated to it, the Compensation Committee shall determine grant, vesting, exercise, restriction and forfeiture conditions and periods. In particular, they may provide for continuation, acceleration or removal of vesting, exercise, restriction and forfeiture conditions and periods, for payment or grant of compensation based upon assumed target achievement, or for forfeiture, in each case in the event of pre-determined events such as a change of control or termination of an employment or mandate agreement. The Company may procure the required shares or other securities through purchases in the market, from treasury shares or by using conditional or authorized share capital. |
|
|
4 La rémunération peut être versée en espèces, sous forme d'actions, d'options ou d'instruments ou unités sur base d'actions ou d'autres types de prestations. Le Conseil d'administration ou le Comité de rémunération, dans la mesure où cette compétence lui a été déléguée, détermine les conditions et périodes d'octroi, d'acquisition (vesting), d'exercice, de restriction et de péremption. Ils peuvent en particulier prévoir la continuation, l'accélération ou la suppression des conditions ou périodes d'acquisition (vesting), d'exercice, de restriction et de péremption, le versement ou l'octroi d'une rémunération supposant l'atteinte des objectifs ou encore la déchéance des droits, dans chaque cas lors d'événements prédéterminés tels que, notamment, un changement de contrôle ou la fin d'un contrat de travail ou de mandat. La Société peut se procurer les actions ou autres instruments des marchés financiers requis par le biais d'achats sur le marché ou d'actions propres, ou en utilisant son capital-actions conditionnel ou autorisé. |
|
5 Compensation may be paid by the Company or companies controlled by it. |
|
|
5 La rémunération peut être versée par la Société ou tout autre société qu'elle contrôle. |
|
|
|
|
|
|
Article 29 |
|
|
Article 29 |
Agreements with Members of the Board of Directors and the Executive Committee |
1 The Company or companies controlled by it may enter into agreements with non-executive members of the Board of Directors relating to their compensation for a fixed term or for an indefinite term. The duration and termination are subject to the term of office and the law. |
|
Contrats avec les membres du Conseil d'administration et de la Direction exécutive |
1 La Société, ou toute société qu'elle contrôle, peut conclure des contrats de durée déterminée ou indéterminée avec les membres non-exécutifs du Conseil d'administration en relation avec leur rémunération. La durée et la résiliation doivent être conformes avec la durée des fonctions ainsi qu'avec les dispositions légales applicables. |
Articles of Association of ADC Therapeutics SA
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2 The Company or companies controlled by it may enter into employment agreements with executive members of the Board of Directors and other members of the Executive Committee for a fixed term or for an indefinite term. Fixed term agreements may have a maximum duration of one year; renewal is possible. Agreements for an indefinite term may have a notice period of maximum twelve months. |
|
|
2 La Société, ou toute société qu'elle contrôle, peut conclure des contrats de travail de durée déterminée ou indéterminée avec les membres exécutifs du conseil d'administration et les autres membres de la Direction exécutive. Les contrats de durée déterminée peuvent avoir une durée maximale d'une année; le renouvellement est possible. Les contrats de durée indéterminée peuvent prévoir un délai de congé d'au maximum douze mois. |
|
3 The Company or companies controlled by it may enter into non-compete agreements with members of the Executive Committee for the time after termination of employment. Their duration shall not exceed two years, and consideration paid per year for such non-compete undertaking shall not exceed the average of the compensation of the last three financial years. |
|
|
3 La Société, ou toute société qu'elle contrôle, peut conclure des accords de non-concurrence avec les membres de la Direction exécutive pour la période suivant la fin des rapports de travail. Leur durée ne peut excéder deux ans, et l'indemnisation par an versée en contrepartie d'un tel accord de non concurrence ne peuten aucun cas dépasser la moyenne des rémunérations des trois derniers exercices. |
|
|
|
|
|
|
Article 30 |
|
|
Article 30 |
Mandates Outside of the Group |
1 The number of mandates on the board of directors the executive committee or comparable functions at other enterprises with an economic purpose is limited: |
|
Mandats en dehors du groupe |
1 Le nombre de mandats d'administrateur, au sein de la Direction exécutive ou de fonctions similaires auprès d'autres entreprises poursuivant un but économique est limité: |
|
(a) for members of the Executive Committee, to seven mandates, of which no more than two in a listed company; and |
|
|
(a) pour les membres de la Direction exécutive, à sept mandats, dont pas plus de deux au sein de sociétés cotées; et |
|
(b) for members of the Board of Directors, to fifteen mandates, of which no more than five in listed companies. |
|
|
(b) pour les membres du Conseil d'administration à quinze mandats, dont pas plus de cinq au sein de sociétés cotées. |
|
2 Mandates in different legal entities being part of the same group or for the same group are deemed to be one mandate. |
|
|
2 Les mandats dans différentes entités juridiques appartenant au même groupe ou assumés pour le même groupe sont considérés comme un mandat. |
Articles of Association of ADC Therapeutics SA
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
3 Mandates in associations, charitable organizations, family trusts and foundations relating to post-retirement benefits as well as mandates held at the request of the Company or companies controlled by it are not subject to the above limitations. No member of the Board of Directors or the Executive Committee shall hold more than 10 such mandates. |
|
|
3 Les mandats dans des associations, organisations caritatives, fondations de famille et fondations de prévoyance professionnelle ainsi que les mandats exercés à la demande de la Société ou des sociétés qu'elle contrôle ne sont pas soumis aux limites mentionnées ci-dessus. Aucun membre du Conseil d'administration ou de la Direction exécutive ne peut exercer plus de 10 mandats de ce genre. |
|
|
|
|
|
|
Article 31 |
|
|
Article 31 |
Post-Retirement Benefits |
The Company or companies controlled by it may grant to members of the Board of Directors and the Executive Committee post-retirement benefits beyond the occupational benefit schemes which do not exceed the annual compensation of the respective member of the Board of Directors or the Executive Committee last paid or payable for the first time. |
|
Prestations de retraite |
La Société ou toute société qu'elle contrôle peut octroyer aux membres du Conseil d'administration et de la Direction exécutive des prestations de retraite allant au-delà du régime de prévoyance professionnelle n'excédant pas la rémunération annuelle du membre du Conseil d'administration ou de la Direction exécutive concerné versée ou à verser pour la première fois. |
|
|
|
|
|
|
|
|
|
|
|
Section 5
Financial Year, Profit Allocation |
|
|
Section 5
Exercice, répartition du bénéfice |
|
|
|
|
|
|
Article 32 |
|
|
Article 32 |
Financial Year, Annual and Compensation Report |
1 The Company's financial year shall be determined by the Board of Directors. |
|
Exercice social, rapport de gestion et de rémunération |
1 L'exercice est fixé par le Conseil d'administration.
|
2 The Board of Directors shall prepare an annual report for each financial year, comprising the annual financial statements, if required, the management report and any other report required by law and the consolidated financial statements, as well as a compensation report. |
|
2 Le Conseil d'administration établit pour chaque exercice un rapport de gestion, qui se compose des comptes annuels et, cas échéant, du rapport annuel et des comptes de groupe, ainsi qu'un rapport de rémunération et tout autre rapport requis par la loi. |
|
|
|
|
|
|
Article 33 |
|
|
Article 33 |
Articles of Association of ADC Therapeutics SA
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Allocation of Profit Shown on the Balance Sheet, Reserves |
1 The General Meeting of Shareholders shall resolve on the allocation of the profit as shown on the balance sheet in accordance with applicable law. The Board of Directors shall submit its proposals to the General Meeting of Shareholders. |
|
Utilisation du bénéfice résultant du bilan, réserves |
1 L'Assemblée générale détermine l'emploi du bénéfice résultant du bilan, sous réserve des prescriptions légales concernant la répartition du bénéfice. Le Conseil d'administration lui soumet ses propositions. |
2 In addition to the reserves required by law, the General Meeting of Shareholders may create other reserves. |
|
2 En sus des réserves légales, l'Assemblée générale peut constituer des réserves supplémentaires. |
|
3 Dividends that have not been collected within five years after their payment date shall inure to the Company and be allocated to the general statutory reserves. |
|
|
3 Les dividendes qui n'ont pas été perçus dans un délai de cinq ans après leur date de paiement sont prescrits et sont alloués aux réserves statutaires de la Société. |
|
|
|
|
|
|
|
|
|
|
|
Section 6
Dissolution, Liquidation |
|
|
Section 6
Dissolution, liquidation |
|
|
|
|
|
|
Article 34 |
|
|
Article 34 |
Dissolution, Liquidation |
1 The General Meeting of Shareholders may at any time resolve to dissolve and liquidate the Company in accordance with the law and the provisions set forth in these articles of association. |
|
Dissolution, liquidation |
1 L'Assemblée générale peut décider en tout temps de la dissolution et de la liquidation de la Société en conformité avec les prescriptions légales et statutaires. |
|
2 The liquidation shall be effected by the Board of Directors, unless the General Meeting of Shareholders appoints other persons as liquidators. |
|
|
2 La liquidation a lieu par les soins du Conseil d'administration, à moins que l'Assemblée générale ne désigne d'autres liquidateurs. |
|
3 The liquidation of the Company shall be effected pursuant to applicable law. The liquidators shall be entitled to sell assets (real estate included) in private transactions. |
|
|
3 La liquidation de la Société s'effectue conformément au droit applicable. Les liquidateurs sont autorisés à vendre des actifs (immeubles y compris) de gré à gré. |
|
4 Upon discharge of all liabilities of the Company, the assets shall be distributed to the shareholders in proportion to the share capital, unless these articles of association provide otherwise. |
|
|
4 Après paiement des dettes de la Société, l'actif est réparti entre les actionnaires au prorata du capital-actions, à moins que les présents statuts n'en disposent autrement. |
|
|
|
|
|
Articles of Association of ADC Therapeutics SA
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Section 7
Means of Publication, Communications |
|
|
Section 7
Organe de publication, communications |
|
|
|
|
|
|
Article 35 |
|
|
Article 35 |
Notices, Communications |
1 The official means of publication of the Company shall be the Swiss Official Gazette of Commerce. |
|
Communications, organe de publication |
1 L'organe de publication de la Société est la Feuille officielle suisse du commerce. |
2 In particular cases, the Board of Directors may specify additional means of publication. |
|
2 Le Conseil d'administration peut désigner d'autres organes de publication dans certains cas particuliers. |
|
3 Notices by the Company to the shareholders may, at the election of the Board of Directors, be validly given by publication in the Swiss Official Gazette of Commerce or in a form that allows proof by text, including notice through publication of a proxy statement filed pursuant to the rules of the United States Securities and Exchange Commission. |
|
|
3 Les communications aux actionnaires peuvent, au choix du Conseil d'administration, être valablement effectuées par publication dans la Feuille officielle suisse du commerce ou sous une forme permettant d'en établir la preuve par texte, y compris la communication par la publication d'un proxy statement soumise conformément aux règles de la Securities and Exchange Commission des États-Unis. |
|
|
|
|
|
|
Section 7a
Jurisdiction |
|
|
Section 7a
For |
|
|
|
|
|
|
Article 35a |
|
|
Article 35a |
Articles of Association of ADC Therapeutics SA
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Jurisdiction |
The exclusive place of jurisdiction for any disputes arising under, out of or in connection with or related to the corporate relationship shall be at the registered office of the Company; provided that this provision does not apply to claims brought to enforce a duty or liability created by the U.S. Securities Act of 1933, as amended, or the U.S. Securities Exchange Act of 1934, as amended, or any claim for which the courts in the United States have exclusive jurisdiction. |
|
For exclusif |
Le for exclusif pour tout litige découlant de ou en rapport avec la Société se situe au siège de la Société; à condition que cette disposition ne s'applique pas aux plaintes déposées pour faire respecter une obligation ou une responsabilité créée par la loi américaine sur les valeurs mobilières de 1933, telle que modifiée, ou la loi américaine sur l'échange de valeurs mobilières de 1934, telle que modifiée, ou toute plainte pour laquelle les tribunaux des États-Unis ont une compétence exclusive. |
|
|
|
|
|
|
Section 8
Authoritative Language |
|
|
Section 8
Langue faisant foi |
|
|
|
|
|
|
Article 36 |
|
|
Article 36 |
Authoritative Language |
In the event of discrepancies between the French and English version of these articles of association, the French version shall prevail. |
|
Langue faisant foi |
En cas de désaccord entre la version française et la version anglaise, la version française des présents statuts prévaut. |
Lausanne, le 6 décembre 2024
EX-4.1
3
exhibit41descriptionofsecu.htm
EX-4.1
Document
EXHIBIT 4.1
DESCRIPTION OF SHARE CAPITAL AND ARTICLES OF ASSOCIATION
The Company
We are a Swiss stock corporation (société anonyme) organized under the laws of Switzerland. We were incorporated as a Swiss limited liability company (société à responsabilité limitée) on June 6, 2011 with our registered office and domicile in Epalinges, Canton of Vaud, Switzerland. We converted to a Swiss stock corporation under the laws of Switzerland on October 13, 2015. Our domicile is in Epalinges, Canton of Vaud, Switzerland. Our registered office and head office is currently located at Biopôle, Route de la Corniche 3B, 1066 Epalinges, Switzerland.
Share Capital
As of December 31, 2024, our share capital as registered with the commercial register of the Canton of Vaud, Switzerland (the “Commercial Register”) amounted to 101,606,376 common shares with a par value of CHF 0.08 per share, 98,861,402 of which were outstanding.
Registration Rights
On August 15, 2022, we entered into a registration rights agreement with Deerfield Partners, L.P. and Deerfield Private Design Fund IV, L.P. that provides them with certain registration rights with respect to the common shares issuable to them upon the exercise of their warrants. We have filed a registration statement as required by the registration rights agreement. We are required to keep such registration statement effective until all such common shares have been sold, are eligible to be immediately sold to the public without registration or restriction, are no longer outstanding, are no longer held by persons entitled to registration rights or until the date that is one month following expiry of their warrants.
On August 15, 2022, we entered into a registration rights agreement with OR Opportunistic DL (C), L.P., Owl Rock Opportunistic Master Fund II, L.P., Oaktree LSL Holdings EURRC S.à r.l., Oaktree Specialty Lending Corporation, Oaktree AZ Strategic Lending Fund, L.P., Oaktree Strategic Credit Fund, Oaktree Diversified Income Fund, Inc. and Oaktree Loan Acquisition Fund, L.P.
that provides them with certain registration rights with respect to the common shares issuable upon the exercise of their warrants and the common shares issued to OR Opportunistic DL (C), L.P. and Owl Rock Opportunistic Master Fund II, L.P. pursuant to a share purchase agreement. We have filed a registration statement as required by the registration rights agreement. We are required to keep such registration statement effective until all such common shares have been sold, are eligible to be immediately sold to the public without registration or restriction, are no longer outstanding, are no longer held by persons entitled to registration rights or the date that is three years from the initial effective date of the registration statement. The registration rights agreement includes certain demand registration rights and piggyback registration rights exercisable if we do not maintain current public information for Rule 144 purposes.
On February 6, 2023, we entered into a registration rights agreement with Oaktree Fund Administration LLC, OCM Strategic Credit Investments S.à r.l., OCM Strategic Credit Investments 2 S.à.r.l., OCM Strategic Credit Investments 3 S.à r.l., Oaktree Gilead Investment Fund AIF (Delaware), L.P., Oaktree Huntington-GCF Investment Fund (Direct Lending AIF), L.P., Oaktree Specialty Lending Corporation and Pathway Strategic Credit Fund III, L.P. that provides them with certain registration rights with respect to the common shares received after a default by A.T. Holdings II Sarl and in the event of a foreclosure or other exercise of remedies under their lending arrangements with A.T. Holdings II Sàrl. If requested by such parties, we are required to file a registration statement with respect to such common shares. We are required to keep such registration statement effective until all such common shares have been sold, are no longer outstanding, are no longer held by persons entitled to registration rights or the date that is three years from the initial effective date of the registration statement.
Articles of Association
Ordinary Capital Increase, Capital Range and Conditional Share Capital
Under Swiss law, we may increase our share capital (capital-actions) with a resolution of the general meeting of shareholders (ordinary capital increase) that must be carried out by the board of directors within six months of the respective general meeting in order to become effective. Under our articles of association and Swiss law, in the case of subscription and increase against payment of contributions in cash, a resolution passed by a majority of the shares represented at the general meeting of shareholders is required. In the case of subscription and increase against contributions in kind or to fund acquisitions in kind, when shareholders’ statutory pre-emptive subscription rights or advance subscription rights are limited or withdrawn or where transformation of freely disposable equity into share capital is involved, a resolution passed by two-thirds of the shares represented at a general meeting of shareholders and the majority of the par value of the shares represented is required.
Under the Swiss Code of Obligations (Code des obligations) (the “CO”), our shareholders, by a resolution passed by two-thirds of the shares represented at a general meeting of shareholders and the majority of the par value of the shares represented, can:
▪adopt conditional share capital (capital-actions conditionnel) in the aggregate amount of up to 50% of the share capital for the purpose of issuing shares in connection with, among other things, option and conversion rights granted to shareholders, the creditors of bonds and similar debt instruments, employees, members of the board of directors of the Company or of any group company, or to any third parties; and
▪in the form of capital range (marge de fluctuation du capital), empower our board of directors to increase and/or decrease our share capital by up to 50% of the share capital, by issuing or canceling shares, or by increasing or decreasing the par value of shares, including through the creation of conditional share capital; such capital range is to be utilized by the board of directors within a period determined by the shareholders but not exceeding five years from the date of the shareholder approval.
Pre-Emptive and Advance Subscription Rights
Pursuant to the CO, shareholders have pre-emptive subscription rights (droits de souscription préférentiels) to subscribe for new issuances of shares. With respect to conditional capital, shareholders have (i) pre-emptive subscription rights for the subscription of option rights and (ii) advance subscription rights (droit de souscription préalable) for the subscription of bonds and similar debt instruments to which option or conversion rights are attached.
A resolution passed at a general meeting of shareholders by two-thirds of the shares represented and the majority of the par value of the shares represented may authorize our board of directors to withdraw or limit pre-emptive subscription rights or advance subscription rights in certain circumstances.
If pre-emptive subscription rights are granted, but not exercised, the board of directors may allocate the unexercised pre-emptive subscription rights at its discretion.
Our Capital Range
Under our articles of association, we have a capital range ranging from CHF 8,128,510.08 (lower limit) to CHF 12,106,664.40 (upper limit). Our board of directors is authorized within the capital range to increase or decrease our share capital once or several times and in any amounts and to acquire or dispose of shares, directly or indirectly, until November 11, 2029, or until an earlier expiry of the capital range. The capital increase or reduction may be effected by issuing fully paid-in registered shares with a par value of CHF 0.08 each and cancelling registered shares with a par value of CHF 0.08 each, as applicable, or by increasing or reducing the par value of the existing shares within the limits of the capital range, or by simultaneous reduction and re-increase of the share capital. If our share capital increases as a result of a share issue from conditional capital (see next subsection), the upper and lower limits of the capital range will increase in an amount corresponding to such increase.
In the event of a capital increase within the capital range, the board of directors has to determine the type of contributions, the issue price and the date on which the dividend entitlement starts. In the event of a capital reduction within the capital range, the board of directors has to determine the use of the reduction amount, to the extent necessary.
In a capital increase within the capital range, the board of directors is authorized by our articles of association to withdraw or to limit the pre-emptive subscription rights of shareholders, and to allocate them to third parties or to us, in the event that the newly issued shares are issued under the following circumstances:
▪if the issue price of the new registered shares is determined by reference to the market price;
▪for raising of equity capital (including private placements) in a fast and flexible manner, which would not be possible, or might only be possible with great difficulty or delays or at significantly less favorable conditions, without the exclusion of the statutory pre-emptive subscription rights of the existing shareholders;
▪for the acquisition of an enterprise, parts of an enterprise or participations, for the acquisition of products, intellectual property or licenses by or for investment projects of the Company or any of its group companies, or for the financing or refinancing of any of such transactions through a placement of shares;
▪for purposes of broadening the shareholder constituency of the Company in certain geographic, financial or investor markets, for purposes of the participation of strategic partners, including financial investors, or in connection with the listing of new shares on domestic or foreign stock exchanges;
▪for purposes of granting an over-allotment option or an option to purchase additional shares in a placement or sale of shares to the respective initial purchaser(s) or underwriter(s); ▪for the participation of members of the board of directors, members of the executive committee, employees, contractors, consultants or other persons performing services for the benefit of the Company or any of its group companies;
▪following a shareholder or a group of shareholders acting in concert having accumulated shareholdings in excess of 20% of our share capital registered in the Commercial Register without having submitted to all other shareholders a takeover offer recommended by the board of directors;
▪for the defense of an actual, threatened or potential takeover bid, that the board of directors, upon consultation with an independent financial adviser retained by it, has not recommended to the shareholders acceptance on the basis that the board of directors has not found the takeover bid to be financially fair to the shareholders or not to be in the Company’s interest; or
▪for other valid grounds in the sense of Article 652b para. 2 of the CO.
This authorization to withdraw or to limit the pre-emptive subscription of shareholders is exclusively linked to our capital range. If the capital range lapses for any reasons, such as if an ordinary capital increase is completed, the authorization to withdraw or to limit the pre-emptive subscription rights lapses simultaneously with the capital range.
We have agreed not to use the foregoing authorization to withdraw or to limit the pre-emptive subscription rights of shareholders, and to allocate them to third parties or to us, in certain circumstances. Specifically, we will not restrict the preemptive rights of Redmile Group, LLC (“Redmile”) or its affiliates based on Article 4a(4)(g) of the articles of association or restrict the advance subscription rights of Redmile or its affiliates based on Article 4c(3) of the articles of association as long as (i) Redmile (including its affiliates and any other person or entity forming a “group” (as defined in Rule 13d-5 under the Exchange Act)) does not directly or indirectly control, own or have the right to control or own, collectively, shares representing more than 20% of the Company’s share capital or (ii) Redmile (including its affiliates and any other person or entity forming a “group” (as defined in Rule 13d-5 under the Exchange Act)) directly or indirectly controls, owns or has the right to control or own, collectively, shares representing more than 20% of the Company’s share capital but the board of directors determines that Redmile does not have an intent to effect a change of control of the Company.
Our Conditional Share Capital
Conditional Share Capital for Warrants and Convertible Bonds
Our nominal share capital may be increased, including to prevent takeovers and changes in control, by a maximum aggregate amount of CHF 3,042,154.32 through the issuance of not more than 38,026,929 common shares, which would have to be fully paid-in, each with a par value of CHF 0.08 per share, by the exercise of option and conversion rights granted in connection with warrants, convertible bonds or similar instruments of the Company or one of our subsidiaries. Shareholders will not have pre-emptive subscription rights in such circumstances, but will have advance subscription rights to subscribe for such warrants, convertible bonds or similar instruments. The holders of warrants, convertible bonds or similar instruments are entitled to the new shares upon the occurrence of the applicable conversion feature.
When issuing convertible bonds, warrants or similar instruments, the board of directors is authorized to withdraw or to limit the advance subscription right of shareholders:
▪for the purpose of financing or refinancing, or the payment for, the acquisition of enterprises, parts of enterprises, participations, intellectual property rights, licenses or investments;
▪if the issuance occurs in domestic or international capital markets, including private placements;
▪following a shareholder or a group of shareholders acting in concert having accumulated shareholdings in excess of 20% of the share capital registered in the Commercial Register without having submitted to all other shareholders a takeover offer recommended by the board of directors; ▪for the defense of an actual, threatened or potential takeover bid that the board of directors, upon consultation with an independent financial adviser retained by it, has not recommended to the shareholders to accept on the basis that the board of directors has not found the takeover bid to be financially fair to the shareholders or not to be in the Company’s interest; or
▪if the convertible bonds, warrants or similar instruments are issued on appropriate terms.
To the extent that the advance subscription rights are withdrawn or limited, (i) the term to exercise the convertible bonds, warrants or similar instruments may not exceed fifteen years from the date of issue of the respective instrument and (ii) the conversion, exchange or exercise price of the convertible bonds, warrants or similar instruments has to be set with reference to or be subject to change based upon the valuation of the Company’s equity or market conditions.
Conditional Share Capital for Equity Incentive Plans
Our nominal share capital may, to the exclusion of the pre-emptive subscription rights and advance subscription rights of shareholders, be increased by a maximum aggregate amount of CHF 763,798.56 through the (direct or indirect) issuance of not more than 9,547,482 common shares, which would have to be fully paid-in, each with a par value of CHF 0.08 per share, by the exercise or mandatory exercise of options, other rights to receive or acquire shares or conversion rights that have been granted to or imposed on employees, members of the board of directors, contractors or consultants of the Company or of one of our subsidiaries or other persons providing services to the Company or to a subsidiary through one or more equity incentive plans created by the board of directors.
Uncertificated Securities
Our shares are in the form of uncertificated securities (droits-valeurs, within the meaning of Article 973c of the CO). In accordance with Article 973c of the CO, we maintain a non-public register of uncertificated securities (registre des droits-valeurs). We may at any time convert uncertificated securities into share certificates (including global certificates), one kind of certificate into another, or share certificates (including global certificates) into uncertificated securities. Following entry in the share register, a shareholder may at any time request from us a written confirmation in respect of his or her shares. Shareholders are not entitled, however, to request the conversion and/or printing and delivery of share certificates. We may print and deliver certificates for shares at any time.
General Meeting of Shareholders
Ordinary/Extraordinary Meetings, Powers
The general meeting of shareholders is our supreme corporate body. Under Swiss law, an annual general meeting of shareholders must be held annually within six months after the end of a corporation’s financial year. In our case, this generally means on or before June 30. In addition, extraordinary general meetings of shareholders may be held.
A general meeting of shareholders may take place in or outside Switzerland and at different places simultaneously if the votes of the participants are immediately transmitted to all meeting venues (multilocal shareholders’ meeting). The board of directors may allow shareholders that are not present at the meeting venue of the general meeting of shareholders to participate and exercise their rights electronically (“hybrid shareholder meeting”). Our articles of association also allow for general meetings of shareholders without a physical meeting venue but that takes place using electronic means (“virtual shareholder meeting”).
According to our articles of association, the following powers are vested exclusively in the general meeting of shareholders:
▪adopting and amending the articles of association, including the change of a company’s purpose or domicile;
▪electing the members of the board of directors, the chairperson of the board of directors, the members of the compensation committee, the auditors and the independent proxy; ▪approving the annual management report, the annual statutory and consolidated financial statements, and deciding on the allocation of profits as shown on the balance sheet, in particular with regard to dividends;
▪determining the interim dividend and approving the requisite interim financial statements;
▪resolving on the repayment of the statutory capital reserve (réserve légale issue du capital);
▪approving the aggregate amount of compensation of members of the board of directors and the executive committee;
▪discharging the members of the board of directors and the executive committee from liability with respect to their conduct of business;
▪resolving on the delisting of the company's equity securities;
▪approving the report on non-financial matters (if applicable);
▪dissolving a company with or without liquidation; and
▪deciding matters reserved to the general meeting of shareholders by law or the articles of association or submitted to it by the board of directors.
An extraordinary general meeting of shareholders may be called by a resolution of the board of directors or the general meeting of shareholders or, under certain circumstances, by a company’s auditors, liquidator or the representatives of bondholders, if any. In addition, our articles of association require the board of directors to convene an extraordinary general meeting of shareholders if shareholders representing at least 5% of our share capital or voting rights request such general meeting of shareholders in writing. A request for an extraordinary general meeting of shareholders must set forth the items to be discussed and the proposals to be acted upon. Further, the board of directors must convene an extraordinary general meeting of shareholders and propose financial restructuring measures if, based on our stand-alone annual statutory balance sheet, half of our share capital and statutory reserves are not covered by our assets and a contemplated restructuring measure falls within the competence of the general meeting of shareholders.
Voting and Quorum Requirements
Shareholder resolutions and elections (including elections of members of the board of directors) require the affirmative vote of the majority of shares represented at the general meeting of shareholders, unless otherwise stipulated by law or our articles of association.
Under our articles of association, a resolution of the general meeting of shareholders passed by two-thirds of the votes and the majority of the par value of the shares, each as represented at the meeting, is required for:
▪amending the Company’s corporate purpose;
▪creating shares with preference rights;
▪cancelling or amending the transfer restrictions of shares;
▪creating conditional share capital or a capital range;
▪increasing share capital out of equity, against contributions in-kind, by set-off against a claim or granting of specific benefits;
▪limiting or withdrawing shareholder’s pre-emptive subscription rights;
▪changing the currency of the share capital; ▪introducing a casting vote of the chairperson at the general meeting of shareholders;
▪changing the Company’s domicile;
▪delisting the Company's equity securities;
▪introducing an arbitration clause in the articles of association;
▪resolving on the consolidation of shares (reverse split);
▪amending or repealing the voting and recording restrictions, the provision setting a maximum board size, the indemnification provision for the board of directors and the executive committee or the forum selection clause set forth in our articles of association;
▪converting registered shares into bearer shares;
▪removing the chairperson or any member of the board of directors before the end of his or her term of office; and
▪dissolving or liquidating the Company.
The same voting requirements apply to resolutions regarding transactions among corporations based on Switzerland’s Federal Act on Mergers, Demergers, Transformations and the Transfer of Assets of 2003, as amended (the “Swiss Merger Act”). See “—Articles of Association—Compulsory Acquisitions; Appraisal Rights.”
In accordance with Swiss law and generally accepted business practices, our articles of association do not provide for quorum requirements generally applicable to general meetings of shareholders. To this extent, our practice varies from NYSE listing standards, which require an issuer to provide in its bylaws for a generally applicable quorum, and that such quorum may not be less than one-third of the outstanding voting shares.
Notice
General meetings of shareholders must be convened by the board of directors at least 20 days before the date of the meeting. The general meeting of shareholders is convened by way of a notice appearing in our official publication medium, currently the Swiss Official Gazette of Commerce, or by including the notice in the proxy statement. Registered shareholders may also be informed in a form that allows proof by text. The notice of a general meeting of shareholders must state the date, the starting and end time, the form and location of the meeting, the items on the agenda, the motions of the board of directors or of any shareholders including a short explanation, as well as the name and address of the independent representative. A resolution on a matter which is not on the agenda may not be passed at a general meeting of shareholders, except for motions to convene an extraordinary general meeting of shareholders or to initiate a special investigation, on which the general meeting of shareholders may vote at any time. No previous notification is required for motions concerning items included in the agenda or for debates that do not result in a vote.
All owners or representatives of our shares may, if no objection is raised, hold a general meeting of shareholders without complying with the formal requirements for convening general meetings of shareholders (a universal meeting). This universal meeting of shareholders may discuss and pass binding resolutions on all matters within the purview of the general meeting of shareholders, provided that the owners or representatives of all the shares are present at the meeting.
Agenda Requests
Pursuant to our articles of association, one or more shareholders whose combined shareholdings represent at least 0.5% of the share capital or the voting rights may request that an item be included in the agenda for a general meeting of shareholders or that a proposal relating to an agenda item be included in the notice convening the general meeting of shareholders.
To be timely, the shareholder’s request must be received by us generally at least 90 calendar days in advance of the meeting. The request must be made in writing and contain, for each of the agenda items, the following information:
▪a brief description of the business desired to be brought before the general meeting of shareholders and the reasons for conducting such business at the general meeting of shareholders;
▪the motions regarding the agenda item;
▪the name and address, as they appear in the share register, of the shareholder proposing such business;
▪the number of shares which are beneficially owned by such shareholder (including documentary support of such beneficial ownership); ▪the dates upon which the shareholder acquired such shares;
▪any material interest of the proposing shareholder in the proposed business;
▪a statement in support of the matter; and
▪all other information required under the applicable laws and stock exchange rules.
In addition, if the shareholder intends to solicit proxies from the shareholders of a company, such shareholder shall notify the company of this intent in accordance with applicable SEC rules.
Our business report, the compensation report and the auditor’s report must be published or otherwise made accessible to our shareholders no later than 20 days prior to the general meeting of shareholders. Shareholders of record may be notified of this in writing.
Minutes
We are required to make available the resolutions and election results of our general meeting of shareholders electronically within 15 calendar days after the meeting. In addition, each shareholder may request that the minutes be made available to them within 30 calendar days after the meeting.
Voting Rights
Each of our common shares entitles a holder to one vote. The common shares are not divisible. The right to vote and the other rights of share ownership may only be exercised by shareholders (including any nominees) or usufructuaries who are entered in the share register at a cut-off date determined by the board of directors. Those entitled to vote in the general meeting of shareholders may be represented by the independent proxy holder
(annually elected by the general meeting of shareholders), by its legal representative or by any other person with written authorization to act as proxy. The chairperson has the power to decide whether to recognize a power of attorney.
Our articles of association contain provisions that prevent investors from acquiring voting rights exceeding 15% of our issued share capital. Specifically, if an individual or legal entity acquires common shares and, as a result, directly or indirectly, has voting rights with respect to more than 15% of the registered share capital recorded in the Commercial Register, the registered shares exceeding the limit of 15% shall be entered in the share register as shares without voting rights (limitation à l’inscription). This restriction applies equally to parties acting in concert and to shares held or acquired via a nominee, including via Cede & Co., New York (or any successor), as the nominee of The Depository Trust Company (“DTC”), New York, acting in its capacity as clearing nominee. Specifically, if shares are being held by a nominee for third-party beneficiaries, which control (alone or together with third parties) voting rights with respect to more than 15% of the share capital recorded in the Commercial Register, our articles of association provide that the board of directors may cancel the registration of the shares with voting rights held by such nominee in excess of the limit of 15%. Furthermore, our articles of association contain provisions that allow
the board of directors to make the registration with voting rights of shares held by a nominee subject to conditions, limitations and reporting requirements or to impose or adjust such conditions, limitations and requirements once registered. However, any shareholders who held more than 15% prior to our initial public offering remain registered with voting rights for such shares. Furthermore, the board of directors may in special cases approve exceptions to these restrictions.
Dividends and Other Distributions
Our board of directors may propose to shareholders that a dividend or interim dividend or other distribution be paid but cannot itself authorize the distribution. Dividend and interim dividend payments require a resolution passed by a majority of the shares represented at a general meeting of shareholders. In addition, our auditors must confirm that the dividend proposal of our board of directors conforms to Swiss statutory law and our articles of association.
Under Swiss law, we may pay dividends only if we have sufficient distributable profits from the previous or current business year (bénéfice résultant du bilan) or if we have distributable capital reserves (réserve légale issue du capital), each as evidenced by audited stand-alone statutory annual or interim financial statements prepared pursuant to Swiss law, and after allocations to reserves required by Swiss law and by the articles of association have been deducted..
Under the CO at least 5% of our annual profit must be retained as statutory profit reserve (réserve légale). If there is a loss carried forward, such loss must be eliminated before allocation to the statutory profit reserve. The statutory profit reserve shall be accumulated until it reaches, together with the statutory capital reserve, 50% of our share capital recorded in the Commercial Register. In addition, we have to allocate, among other things, the net
proceeds of share issuances to the statutory capital reserve. The CO permits us to accrue additional reserves. Further, a purchase of our own shares (whether by us or a subsidiary) reduces the distributable reserves in an amount corresponding to the purchase price of such own shares. Finally, the CO under certain circumstances requires the creation of revaluation reserves which are not distributable.
Distributions out of issued share capital (i.e., the aggregate par value of our issued shares) are not allowed and may be made only by way of an ordinary capital reduction or within a capital range that (also) allows for a capital reduction (see “Description of Share Capital and Articles of Association—Articles of Association—Ordinary Capital Increase, Capital Range and Conditional Share Capital”). An ordinary capital reduction requires a resolution passed by a majority of the shares represented at a general meeting of shareholders. The board of directors must publish a call to creditors in the Swiss Official Gazette of Commerce in which creditors are advised that they may request, subject to certain conditions, security for their claims within 30 days of the publication of the creditor call. A licensed audit expert must then confirm, based on the results of the call to creditors, that the claims of the creditors remain fully covered despite the reduction in our share capital recorded in the Commercial Register. If all requirements for an ordinary capital reduction have been met, the board of directors has to amend the articles of association in a public deed. Our share capital may be reduced to a level below CHF 100,000 only if and to the extent that at the same time the statutory minimum share capital of CHF 100,000 is reestablished by sufficient new fully paid-up capital. An ordinary capital reduction must be completed within six months after the resolution of the general meeting of shareholders.
Our board of directors determines the date on which the dividend entitlement starts. Dividends are usually due and payable shortly after the shareholders have passed the resolution approving the payment, but shareholders may also resolve at the annual general meeting of shareholders to pay dividends in quarterly or other installments.
Transfer of Shares
Shares in uncertificated form (droits-valeurs) may only be transferred by way of assignment. Shares or the beneficial interest in shares, as applicable, credited in a securities account may only be transferred when a credit of the relevant intermediated securities to the acquirer’s securities account is made in accordance with applicable rules. Our articles of association provide that in the case of securities held with an intermediary such as a registrar, transfer agent, trust corporation, bank or similar entity, any transfer, grant of a security interest or usufructuary right in such intermediated securities and the appurtenant rights associated therewith requires the cooperation of the intermediary in order for such transfer, grant of a security interest or usufructuary right to be valid against us.
Voting rights may be exercised only after a shareholder has been entered in the share register (registre des actions) with his or her name and address (in the case of legal entities, the registered office) as a shareholder with voting rights. For a discussion of the restrictions applicable to the control and exercise of voting rights, see “Description of Share Capital and Articles of Association—Articles of Association—Voting Rights.”
Inspection of Books and Records
Under the CO, a shareholder has a right to inspect the share register with respect to his or her own shares and otherwise to the extent necessary to exercise his or her shareholder rights. No other person has a right to inspect the share register. Shareholders holding in the aggregate at least 5% of our nominal share capital or of our voting rights have the right to inspect our books and correspondence, subject to the safeguarding of our business secrets and other legitimate interests. Our board of directors is required to decide on an inspection request within four months after receipt of such request. Denial of the request will need to be justified in writing. If an inspection request is denied by the board of directors, shareholders may request the order of an inspection by the court within thirty days.
See “Comparison of Swiss Law and Delaware Law—Inspection of books and records.”
Special Investigation
If a shareholder has exercised its information or inspection rights, such shareholder may propose to the general meeting of shareholders that specific facts be examined by a special examiner in a special investigation. If the general meeting of shareholders approves the proposal, we or any shareholder may, within 30 calendar days after the general meeting of shareholders, request a court at our registered office (currently Epalinges, Canton of Vaud, Switzerland) to appoint a special examiner. If the general meeting of shareholders rejects the request, one or more shareholders representing at least 5% of our share capital or voting rights may request that the court appoint a
special examiner. The court will issue such an order if the petitioners can demonstrate that members of the board of directors or our executive committee infringed the law or our articles of association and that such violation is suitable to cause a damage to the Company or the shareholders. The costs of the investigation would generally be allocated to us and only in exceptional cases to the petitioners.
Compulsory Acquisitions; Appraisal Rights
Business combinations and other transactions that are governed by the Swiss Merger Act (i.e., mergers, demergers, transformations and certain asset transfers) are binding on all shareholders. A statutory merger or demerger requires approval of two-thirds of the shares represented at a general meeting of shareholders and the majority of the par value of the shares represented.
If a transaction under the Swiss Merger Act receives all of the necessary consents, all shareholders are compelled to participate in such transaction.
Swiss corporations may be acquired by an acquirer through the direct acquisition of the shares of the Swiss corporation. The Swiss Merger Act provides for the possibility of a so-called “cash-out” or “squeeze-out” merger with the approval of holders of 90% of the issued shares. In these limited circumstances, minority shareholders of the corporation being acquired may be compensated in a form other than through shares of the acquiring corporation (for instance, through cash or securities of a parent corporation of the acquiring corporation or of another corporation). For business combinations effected in the form of a statutory merger or demerger and subject to Swiss law, the Swiss Merger Act provides that if equity rights have not been adequately preserved or compensation payments in the transaction are unreasonable, a shareholder may request the competent court to determine a reasonable amount of compensation.
In addition, under Swiss law, the sale of “all or substantially all of our assets” by us may require the approval of two-thirds of the number of shares represented at a general meeting of shareholders and the majority of the par value of the shares represented. Whether a shareholder resolution is required depends on the particular transaction, including whether the following test is satisfied:
▪a core part of our business is sold without which it is economically impracticable or unreasonable to continue to operate the remaining business;
▪our assets, after the divestment, are not invested in accordance with our corporate purpose as set forth in the articles of association; and
▪the proceeds of the divestment are not earmarked for reinvestment in accordance with our corporate purpose but, instead, are intended for distribution to our shareholders or for financial investments unrelated to our corporate purpose.
A shareholder of a Swiss corporation participating in certain major corporate transactions may, under certain circumstances, be entitled to appraisal rights. As a result, such shareholder may, in addition to the consideration (be it in shares or in cash) receive an additional amount to ensure that the shareholder receives the fair value of the shares held by the shareholder. Following a statutory merger or demerger, pursuant to the Swiss Merger Act, shareholders can file an appraisal action against the surviving company. If the consideration is deemed inadequate, the court will determine an adequate compensation payment.
Board of Directors
Our articles of association provide that the board of directors shall consist of at least three and not more than nine members.
The members of the board of directors and the chairperson are elected annually by the general meeting of shareholders for a period until the completion of the subsequent annual general meeting of shareholders and are eligible for re-election. Each member of the board of directors must be elected individually.
Powers
According to our articles of association, the board of directors has the following non-delegable and inalienable powers and duties:
▪the ultimate direction of the business of the Company and issuing of the relevant directives;
▪laying down the organization of the Company;
▪formulating accounting procedures, financial controls and financial planning;
▪nominating and removing persons entrusted with the management and representation of the Company and regulating the power to sign for the Company;
▪the ultimate supervision of those persons entrusted with management of the Company, with particular regard to adherence to law, our articles of association, and regulations and directives of the Company;
▪preparing the annual report, the compensation report and, if applicable, the report on non-financial matters and other reports as required by law;
▪preparing for the general meeting of shareholders and carrying out its resolutions; and
▪submitting a petition for debt-restructuring moratorium and informing the court in case of over-indebtedness.
The board of directors may, while retaining such non-delegable and inalienable powers and duties, delegate some of its powers, in particular direct management, to a single or to several of its members, committees or to third parties (such as executive officers) who need be neither members of the board of directors nor shareholders.
Pursuant to Swiss law and our articles of association, details of the delegation and other procedural rules such as quorum requirements have been set in the organizational rules established by the board of directors.
Indemnification of Executive Officers and Directors
Subject to Swiss law, our articles of association provide for indemnification of the existing and former members of the board of directors and the executive committee and their heirs, executors and administrators, against liabilities arising in connection with the performance of their duties in such capacity, and permits us to advance the expenses of defending any act, suit or proceeding to our directors and executive officers to the extent not included in
insurance coverage or advanced by third parties.
In addition, under general principles of Swiss employment law, an employer may be required to indemnify an employee against losses and expenses incurred by such employee in the proper execution of his or her duties under the employment agreement with the employer. See “Comparison of Swiss Law and Delaware Law— Indemnification of directors and executive officers and limitation of liability.”
We have entered into indemnification agreements with each of the members of our board of directors and executive officers.
Conflicts of Interest, Management Transactions
The members of the board of directors and the executive committee are required to immediately and fully inform the board of directors about conflicts of interests concerning them. The board of directors is furthermore required to take measures in order to protect the interests of the company. More generally, the CO requires our directors and executive officers to safeguard the Company’s interests and imposes a duty of loyalty and duty of care on our directors and executive officers. This rule is generally understood to disqualify directors and executive officers from participation in decisions that directly affect them. Our directors and executive officers are personally liable to us for breaches of these obligations. In addition, Swiss law contains provisions under which directors and all persons engaged in the Company’s management are liable to the Company, each shareholder and the Company’s creditors for damages caused by an intentional or negligent violation of their duties. Furthermore, Swiss law contains a provision under which payments made to any of the Company’s shareholders or directors or any person related to any such shareholder or director, other than payments made at arm’s length, must be repaid to the Company if such shareholder or director acted in bad faith.
Our board of directors has adopted a Code of Business Conduct and Ethics and other policies that cover a broad range of matters, including the handling of conflicts of interest.
Principles of the Compensation of the Board of Directors and the Executive Committee
Pursuant to Swiss law, the aggregate amount of compensation of the board of directors and the persons whom the board of directors has, fully or partially, entrusted with the management (which we refer to as our “executive committee”) of the Company has to be submitted to our shareholders for approval each year. Our executive committee currently comprises the Chief Executive Officer, the Chief Financial Officer, the Chief Legal & Compliance Officer and the Chief Medical Officer.
The board of directors must issue, on an annual basis, a written compensation report that must be reviewed by our auditors. The compensation report must disclose, among other things, all compensation granted by the Company, directly or indirectly, to current members of the board of directors and the executive committee and, to the extent related to their former role within the Company or not on customary market terms, to former members of the board of directors and former executive officers.
The disclosure concerning compensation, loans and other forms of indebtedness must include the aggregate amount for the board of directors and the executive committee, respectively, as well as the particular amount for each member of the board of directors and for the highest-paid executive officer, specifying the name and function of each of these persons. If variable compensation is approved prospectively, as is currently the case with the Company, our board of directors must submit the compensation report to a non-binding vote of the general meeting of shareholders.
We are prohibited from granting certain forms of compensation to members of our board of directors and executive committee, such as:
▪severance payments (compensation due until the termination of a contractual relationship does not qualify as severance payment);
▪advance compensation;
▪incentive fees for the acquisition or transfer of companies, or parts thereof, by the Company or by companies being, directly or indirectly, controlled by us;
▪loans, other forms of indebtedness, pension benefits not based on occupational pension schemes and performance-based compensation not provided for in the articles of association; and
▪equity-based compensation not provided for in the articles of association.
Compensation to members of the board of directors and the executive committee for activities in entities that are, directly or indirectly, controlled by the Company is prohibited if (i) the compensation would be prohibited if it were paid directly by the Company, (ii) the articles of association do not provide for it, or (iii) the compensation has not been approved by the general meeting of shareholders.
Every year, the general meeting of shareholders has to vote on the proposals of the board of directors with respect to:
▪the maximum aggregate amount of compensation of the board of directors for the term of office until the next annual general meeting of shareholders; and
▪the maximum aggregate amount of fixed compensation of the executive committee for the following financial year; and
▪the maximum aggregate amount of variable compensation of the executive committee for the current financial year.
The board of directors may submit for approval at the general meeting of shareholders deviating or additional proposals relating to the same or different periods.
If, at the general meeting of shareholders, the shareholders do not approve a compensation proposal of the board of directors, the board of directors must prepare a new proposal, taking into account all relevant factors, and submit the new proposal for approval by the same general meeting of shareholders, at a subsequent extraordinary general meeting of shareholders or the next annual general meeting of shareholders.
If we appoint new members of the executive committee after the general meeting of shareholders has approved the compensation of the executive committee for the relevant period and such compensation is insufficient to also cover the new members' compensation, our articles of association allow us to pay each new member an amount not exceeding the aggregate amount of (maximum) compensation of the executive committee last approved.
In addition to fixed compensation, members of the board of directors and the executive committee may be paid variable compensation, depending on the achievement of certain performance criteria. The performance criteria may include individual targets, targets of the Company or parts thereof and targets in relation to the market, other companies or comparable benchmarks, taking into account the position and level of responsibility of the recipient of the variable compensation. The board of directors or, where delegated to it, the compensation committee shall determine the relative weight of the performance criteria and the respective target values as well as their achievement.
Compensation may be paid or granted in the form of cash, shares, financial instruments, in kind, or in the form of other types of benefits. The board of directors or, where delegated to it, the compensation committee shall determine grant, vesting, exercise and forfeiture conditions.
Borrowing Powers
Neither Swiss law nor our articles of association restricts our power to borrow and raise funds. The decision to borrow funds is made by or under the direction of our board of directors, and no approval by the shareholders is required in relation to any such borrowing.
Repurchases of Shares and Purchases of Own Shares
The CO limits our ability to repurchase and hold our own shares. We and our subsidiaries may repurchase shares only to the extent that (i) we have freely distributable reserves in the amount of the purchase price; and (ii) the aggregate par value of all shares held by us does not exceed 10% of our share capital. Pursuant to Swiss law, where shares are acquired in connection with a transfer restriction set out in the articles of association, the foregoing upper limit is 20%. If we own shares that exceed the threshold of 10% of our share capital, the excess must be sold or cancelled by means of a capital reduction within two years.
Shares held by us or our subsidiaries are not entitled to vote at the general meeting of shareholders but are entitled to the economic benefits applicable to the shares generally, including dividends and pre-emptive subscription rights in the case of share capital increases.
In addition, selective share repurchases are only permitted under certain circumstances. Within these limitations, as is customary for Swiss corporations, we may, subject to applicable law, purchase and sell our own shares from time to time in order to meet imbalances of supply and demand, to provide liquidity and to even out variances in the market price of shares.
Notification and Disclosure of Substantial Share Interests
The disclosure obligations generally applicable to shareholders of Swiss corporations under the Federal Act on Financial Market Infrastructures and Market Conduct in Securities and Derivatives Trading, or the Financial Market Infrastructure Act (the “FMIA”), do not apply to us since our shares are not listed on a Swiss exchange.
Mandatory Bid Rules
The obligation of any person or group of persons that acquires more than one-third of a company’s voting rights to submit a cash offer for all the outstanding listed equity securities of the relevant company at a minimum price pursuant to the FMIA does not apply to us since our shares are not listed on a Swiss exchange.
Nonresident or Foreign Owners
Other than limitations that apply to all holders of our common shares, there are no limitations on the right of nonresident or foreign owners of our common shares from holdings or voting such common shares imposed by Swiss law or our articles of association.
Exchange Controls
Other than sanctions against specific countries, individuals, and organizations, there are no governmental laws, decrees, regulations or other legislation in Switzerland affecting the remittance of dividends, interest and other payments to nonresident holders of our common shares.
Stock Exchange Listing
Our common shares are listed on the NYSE under the symbol “ADCT.”
The Depository Trust Company
Each person owning a beneficial interest in common shares held through DTC must rely on the procedures thereof and on institutions that have accounts therewith to exercise any rights of a holder of the shares.
Transfer Agent and Registrar of Shares
Our share register is kept by Computershare Trust Company, N.A., which acts as transfer agent and registrar. The share register reflects only record owners of our shares. Swiss law does not recognize fractional share interests.
Comparison of Swiss Law and Delaware Law
The Swiss laws applicable to Swiss corporations and their shareholders differ from laws applicable to U.S. corporations and their shareholders. The following table summarizes significant differences in shareholder rights pursuant to the provisions of the CO, by which our Company is governed, and the Delaware General Corporation Law applicable to companies incorporated in Delaware and their shareholders. Please note that this is only a general summary of certain provisions applicable to companies in Delaware. Certain Delaware companies may be permitted to exclude certain of the provisions summarized below in their charter documents.
|
|
|
|
|
|
| DELAWARE CORPORATE LAW |
SWISS CORPORATE LAW |
| Mergers and similar arrangements |
| Under the Delaware General Corporation Law, with certain exceptions, a merger, consolidation, sale, lease or transfer of all or substantially all of the assets of a corporation must be approved by the board of directors and a majority of the outstanding shares entitled to vote thereon. A shareholder of a Delaware corporation participating in certain major corporate transactions may, under certain circumstances, be entitled to appraisal rights pursuant to which such shareholder may receive cash in the amount of the fair value of the shares held by such shareholder (as determined by a court) in lieu of the consideration such shareholder would otherwise receive in the transaction. The Delaware General Corporation Law also provides that a parent corporation, by resolution of its board of directors, may merge with any subsidiary, of which it owns at least 90.0% of each class of capital stock without a vote by the shareholders of such subsidiary. Upon any such merger, dissenting shareholders of the subsidiary would have appraisal rights. |
Under Swiss law, with certain exceptions, a merger or a demerger of the corporation or a sale of all or substantially all of the assets of a corporation must be approved by two-thirds of the voting rights represented at the respective general meeting of shareholders as well as the majority of the par value of shares represented at such general meeting of shareholders. A shareholder of a Swiss corporation participating in a statutory merger or demerger pursuant to the Swiss Merger Act (Loi sur la fusion) can file a lawsuit against the surviving company. If the consideration is deemed “inadequate,” such shareholder may, in addition to the consideration (be it in shares or in cash) receive an additional amount to ensure that such shareholder receives the fair value of the shares held by such shareholder. Swiss law also provides that if the merger agreement provides only for a compensation payment, at least 90% of all members in the transferring legal entity who are entitled to vote shall approve the merger agreement. |
| Shareholders’ suits |
|
|
|
|
|
|
| Class actions and derivative actions generally are available to shareholders of a Delaware corporation for, among other things, breach of fiduciary duty, corporate waste and actions not taken in accordance with applicable law. In such actions, the court has discretion to permit the winning party to recover attorneys’ fees incurred in connection with such action. |
Class actions and derivative actions as such are not available under Swiss law. Nevertheless, certain actions may have a similar effect. A shareholder is entitled to bring suit against directors, officers or liquidators for breach of their duties and claim the payment of the company’s losses or damages to the corporation and, in some cases, to the individual shareholder. Likewise, an appraisal lawsuit won by a shareholder may indirectly compensate all shareholders. In addition, to the extent that U.S. laws and regulations provide a basis for liability and U.S. courts have jurisdiction, a class action may be available. |
|
Under Swiss law, the winning party is generally entitled to recover a limited amount of attorneys’ fees incurred in connection with such action. The court has discretion to permit the shareholder who lost the lawsuit to recover attorneys’ fees incurred to the extent that he or she acted in good faith. |
| Shareholder vote on board and management compensation |
| Under the Delaware General Corporation Law, the board of directors has the authority to fix the compensation of directors, unless otherwise restricted by the certificate of incorporation or bylaws. |
Pursuant to Swiss law, the general meeting of shareholders has the non-transferable right, amongst others, to vote separately and bindingly on the aggregate amount of compensation of the members of the board of directors, of the executive committee and of the advisory boards (if any). If variable compensation is approved for a future period rather than for a past period, the compensation report is subject to a non-binding vote of the general meeting of shareholders. |
| Annual vote on board renewal |
|
Unless directors are elected by written consent in lieu of an annual meeting, directors are elected in an annual meeting of shareholders on a date and at a time designated by or in the manner provided in the bylaws. Re-election is possible.
Classified boards are permitted.
|
The general meeting of shareholders elects the members of the board of directors, the chairperson of the board of directors and the members of the compensation committee individually and annually for a term of office until the end of the following general meeting of shareholders. Re-election is possible. |
| Indemnification of directors and executive officers and limitation of liability |
|
|
|
|
|
|
|
The Delaware General Corporation Law provides that a certificate of incorporation may contain a provision eliminating or limiting the personal liability of directors and officers (but not other controlling persons) of the corporation for monetary damages for breach of a fiduciary duty as a director, except no provision in the certificate of incorporation may eliminate or limit liability of:
•a director or officer for any breach of the duty of loyalty to the corporation or its shareholders;
•a director or officer for acts or omissions not in good faith or which involve intentional misconduct or a knowing violation of law;
•a director for statutory liability for unlawful payment of dividends or unlawful share purchase or redemption;
•a director or officer for any transaction from which the director or officer derived an improper personal benefit; or
•an officer in any action by or in right of the corporation.
A Delaware corporation may indemnify any person who was or is a party or is threatened to be made a party to any proceeding, other than an action by or on behalf of the corporation, because the person is or was a director or officer, against liability incurred in connection with the proceeding if the director or officer acted in good faith and in a manner reasonably believed to be in, or not opposed to, the best interests of the corporation, and the director or officer, with respect to any criminal action or proceeding, had no reasonable cause to believe his or her conduct was unlawful.
|
Under Swiss corporate law, an indemnification by the corporation of a director or member of the executive committee in relation to potential personal liability is not effective to the extent the director or member of the executive committee intentionally or negligently violated his or her corporate duties towards the corporation (certain views advocate that at least a grossly negligent violation is required to exclude the indemnification). Furthermore, the general meeting of shareholders may discharge (release) the directors and members of the executive committee from liability for their conduct to the extent the respective facts are known to shareholders. Such discharge is effective only with respect to claims of the company and of those shareholders who approved the discharge or who have since acquired their shares in full knowledge of the discharge. Most violations of corporate law are regarded as violations of duties towards the corporation rather than towards the shareholders. In addition, indemnification of other controlling persons is not permitted under Swiss corporate law, including shareholders of the corporation.
The articles of association of a Swiss corporation may also set forth that the corporation shall indemnify and hold harmless, to the extent permitted by the law, the directors and executive managers out of assets of the corporation against threatened, pending or completed actions.
Also, a corporation may enter into and pay for directors’ and officers’ liability insurance, which may cover negligent acts as well.
|
|
Unless ordered by a court, any foregoing indemnification is subject to a determination that the director or officer has met the applicable standard of conduct:
•by a majority vote of the directors who are not parties to the proceeding, even though less than a quorum;
•by a committee of directors designated by a majority vote of the eligible directors, even though less than a quorum;
•by independent legal counsel in a written opinion if there are no eligible directors, or if the eligible directors so direct; or
•by the shareholders.
Moreover, a Delaware corporation may not indemnify a director or officer in connection with any proceeding in which the director or officer has been adjudged to be liable to the corporation unless and only to the extent that the court determines that, despite the adjudication of liability but in view of all the circumstances of the case, the director or officer is fairly and reasonably entitled to indemnity for those expenses which the court deems proper.
|
|
| Directors’ fiduciary duties |
|
|
|
|
|
|
|
A director of a Delaware corporation has a fiduciary duty to the corporation and its shareholders. This duty has two components:
•the duty of care; and
•the duty of loyalty.
The duty of care requires that a director act in good faith, with the care that an ordinarily prudent person would exercise under similar circumstances. Under this duty, a director must inform himself or herself of, and disclose to shareholders, all material information reasonably available regarding a significant transaction.
The duty of loyalty requires that a director act in a manner he or she reasonably believes to be in the best interests of the corporation. He or she must not use his or her corporate position for personal gain or advantage. This duty prohibits self-dealing by a director and mandates that the best interest of the corporation and its shareholders take precedence over any interest possessed by a director, officer or controlling shareholder and not shared by the shareholders generally. In general, actions of a director are presumed to have been made on an informed basis, in good faith and in the honest belief that the action taken was in the best interests of the corporation. However, this presumption may be rebutted by evidence of a breach of one of the fiduciary duties.
Should such evidence be presented concerning a transaction by a director, a director must prove the procedural fairness of the transaction, and that the transaction was of fair value to the corporation.
|
The board of directors of a Swiss corporation manages the business of the corporation, unless responsibility for such management has been duly delegated to the executive committee based on organizational rules. However, there are several non-transferable duties of the board of directors:
•the overall management of the corporation and the issuing of all necessary directives;
•determination of the corporation’s organization;
•the organization of the accounting, financial control and financial planning systems as required for management of the corporation;
•the appointment and dismissal of persons entrusted with managing and representing the corporation;
•the overall supervision of the persons entrusted with managing the corporation, in particular with regard to compliance with the law, articles of association, operational regulations and directives;
•the compilation of the annual report, the compensation report, the report on non-financial matters and any other reports required by law, the preparation for the general meeting of the shareholders and implementation of its resolutions; and
•the filing of an application for a debt restructuring moratorium and notification of the court in the event that the company is over-indebted.
|
|
The members of the board of directors must perform their duties with all due diligence and safeguard the interests of the corporation in good faith. They must afford the shareholders equal treatment in equal circumstances.
The duty of care requires that a director act in good faith, with the care that an ordinarily prudent director would exercise under like circumstances.
The members of the board of directors and the executive committee are required to immediately and fully inform the board of directors about conflicts of interests concerning them. The board of directors is furthermore required to take measures in order to protect the interests of the company.
The duty of loyalty requires that a director safeguard the interests of the corporation and requires that directors act in the interest of the corporation and, if necessary, put aside their own interests. If there is a risk of a conflict of interest, the board of directors must take appropriate measures to ensure that the interests of the company are duly taken into account.
The burden of proof for a violation of these duties is with the corporation or with the shareholder bringing a suit against the director.
The Swiss Federal Supreme Court has established a doctrine that restricts its review of a business decision if the decision has been taken following proper preparation, on an informed basis and without conflicts of interest.
|
| Shareholder action by written consent |
| A Delaware corporation may, in its certificate of incorporation, eliminate the right of shareholders to act by written consent. |
Shareholders of a Swiss corporation may exercise their voting rights in a general meeting of shareholders. Shareholders can only act by written consents if no shareholder requests a general meeting of shareholders. The articles of association must allow for (independent) proxies to be present at a general meeting of shareholders. The instruction of such (independent) proxies may occur in writing or electronically. |
| Shareholder proposals |
|
|
|
|
|
|
| A shareholder of a Delaware corporation has the right to put any proposal before the annual meeting of shareholders, provided it complies with the notice provisions in the governing documents. A special meeting may be called by the board of directors or any other person authorized to do so in the governing documents, but shareholders may be precluded from calling special meetings. |
At any general meeting of shareholders, any shareholder may put proposals to the meeting if the proposal is part of an agenda item. No resolution may be taken on proposals relating to the agenda items that were not duly notified. Unless the articles of association provide for a lower threshold or for additional shareholders’ rights:
•shareholders together representing at least 5% of the share capital or voting rights may demand that a general meeting of shareholders be called for specific agenda items and specific proposals; and
•shareholders together representing shares with a par value of at least 0.5% of the share capital or the voting rights may demand that an agenda item including a specific proposal, or a proposal with respect to an existing agenda item, be put on the agenda for a scheduled general meeting of shareholders, provided such request is made with appropriate lead time.
Any shareholder can propose candidates for election as directors or make other proposals within the scope of an agenda item without prior written notice.
|
|
In addition, any shareholder is entitled, at a general meeting of shareholders and without advance notice, to (i) request information from the board of directors on the affairs of the company (note, however, that the right to obtain such information is limited), (ii) request information from the auditors on the methods and results of their audit, (iii) request that the general meeting of shareholders resolve to convene an extraordinary general meeting, or (iv) request that the general meeting of shareholders resolve to appoint an examiner to carry out a special examination (“examen spécial”). |
| Cumulative voting |
|
|
|
|
|
|
| Under the Delaware General Corporation Law, cumulative voting for elections of directors is not permitted unless the corporation’s certificate of incorporation provides for it. |
Cumulative voting is not permitted under Swiss corporate law. Pursuant to Swiss law, shareholders can vote for each proposed candidate, but they are not allowed to cumulate their votes for single candidates. An annual individual election of (i) all members of the board of directors, (ii) the chairperson of the board of directors, (iii) the members of the compensation committee, (iv) the election of the independent proxy for a term of office of one year (i.e., until the following annual general meeting of shareholders), as well as the vote on the aggregate amount of compensation of the members of the board of directors, of the executive committee and of the members of any advisory board, is mandatory for listed companies. Re-election is permitted. |
| Removal of directors |
| A Delaware corporation with a classified board may be removed only for cause with the approval of a majority of the outstanding shares entitled to vote, unless the certificate of incorporation provides otherwise. |
A Swiss corporation may remove, with or without cause, any director at any time with a resolution passed by a majority of the shares represented at a general meeting of shareholders. The articles of association may require the approval by a supermajority of the shares represented at a meeting for the removal of a director. |
| Transactions with interested shareholders |
| The Delaware General Corporation Law generally prohibits a Delaware corporation from engaging in certain business combinations with an “interested shareholder” for three years following the date that such person becomes an interested shareholder. An interested shareholder generally is a person or group who or which owns or owned 15.0% or more of the corporation’s outstanding voting shares within the past three years. |
No such rule applies to a Swiss corporation. |
| Dissolution; winding up |
| Unless the board of directors of a Delaware corporation approves the proposal to dissolve, dissolution must be approved by shareholders holding 100.0% of the total voting power of the corporation. Only if the dissolution is initiated by the board of directors may it be approved by a simple majority of the corporation’s outstanding shares. Delaware law allows a Delaware corporation to include in its certificate of incorporation a supermajority voting requirement in connection with dissolutions initiated by the board. |
A dissolution of a Swiss corporation requires the approval by two-thirds of the voting rights represented at the respective general meeting of shareholders as well as the majority of the par value of shares represented at such general meeting of shareholders. The articles of association may increase the voting thresholds required for such a resolution. |
| Variation of rights of shares |
|
|
|
|
|
|
| A Delaware corporation may vary the rights of a class of shares with the approval of a majority of the outstanding shares of such class, unless the certificate of incorporation provides otherwise. |
The general meeting of shareholders of a Swiss corporation may resolve that preference shares be issued or that existing shares be converted into preference shares with a resolution passed by a majority of the shares represented at the general meeting of shareholders. Where a company has issued preference shares, further preference shares conferring preferential rights over the existing preference shares may be issued only with the consent of both a special meeting of the adversely affected holders of the existing preference shares and of a general meeting of all shareholders, unless otherwise provided in the articles of association.
Shares with preferential voting rights are not regarded as preference shares for these purposes.
|
| Amendment of governing documents |
| A Delaware corporation’s governing documents may be amended with the approval of a majority of the outstanding shares entitled to vote, unless the certificate of incorporation provides otherwise. |
The articles of association of a Swiss corporation may be amended with a resolution passed by a majority of the shares represented at a general meeting of shareholders, unless otherwise provided in the articles of association.
There are a number of resolutions, such as an amendment of the stated purpose of the corporation, the introduction of a capital range and conditional capital and the introduction of shares with preferential voting rights that require the approval by two-thirds of the votes and a majority of the par value of the shares represented at such general meeting of shareholders. The articles of association may increase these voting thresholds.
|
| Inspection of books and records |
| Shareholders of a Delaware corporation, upon written demand under oath stating the purpose thereof, have the right during the usual hours for business to inspect for any proper purpose, and to obtain copies of list(s) of shareholders and other books and records of the corporation and its subsidiaries, if any, to the extent the books and records of such subsidiaries are available to the corporation. |
Shareholders of a Swiss corporation holding in the aggregate at least 5% of the nominal share capital or voting rights have the right to inspect books and records, subject to the safeguarding of the company’s business secrets and other interests warranting protection. A shareholder is only entitled to receive information to the extent required to exercise his or her rights as a shareholder. The board of directors has to decide on an inspection request within four months after receipt of such request. Denial of the request will need to be justified in writing. If the board of directors denies an inspection request, shareholders may request the order of an inspection by the court within 30 days.
A shareholder’s right to inspect the share register is limited to the right to inspect his or her own entry in the share register.
|
| Payment of dividends |
|
|
|
|
|
|
| The board of directors may approve a dividend without shareholder approval. Subject to any restrictions contained in its certificate of incorporation, the board may declare and pay dividends upon the shares of its capital stock either: |
Dividend (including interim dividend) payments are subject to the approval of the general meeting of shareholders. The board of directors may propose to shareholders that a dividend shall be paid but cannot itself authorize the distribution. |
|
•out of its surplus, or
•in case there is no such surplus, out of its net profits for the fiscal year in which the dividend is declared and/or the preceding fiscal year.
Shareholder approval is required to authorize capital stock in excess of that provided in the charter. Directors may issue authorized shares without shareholder approval.
|
Payments out of a corporation’s share capital (in other words, the aggregate par value of the corporation’s shares) in the form of dividends are not allowed and may be made only by way of a share capital reduction. Dividends may be paid only from the profits of the previous or current business year or brought forward from previous business years or if the corporation has distributable reserves, each as evidenced by the corporation’s audited stand-alone statutory balance sheet prepared pursuant to Swiss law and after allocations to reserves required by Swiss law and the articles of association have been deducted. |
| Creation and issuance of new shares |
| All creation of shares require the board of directors to adopt a resolution or resolutions, pursuant to authority expressly vested in the board of directors by the provisions of the company’s certificate of incorporation. |
All creation of shares require a shareholders’ resolution. The creation of a capital range or conditional share capital requires at least two-thirds of the voting rights represented at the general meeting of shareholders and a majority of the par value of shares represented at such meeting. The board of directors may issue or cancel shares out of the capital range during a period of up to five years by a maximum amount of 50% of the current share capital. Shares are created and issued out of conditional share capital through the exercise of options or of conversion rights that the board of directors may grant to shareholders, creditors of bonds or similar debt instruments, employees, contractors or consultants, directors of the company or another group company or third parties. |
EX-19.1
4
ex1912024insidertradingpol.htm
EX-19.1
Document
EXHIBIT 19.1
Effective January 1, 2024
ADC Therapeutics
Insider Trading Policy
Table of Contents
The Board of Directors of ADC Therapeutics (together with its subsidiaries, the “Company”) has adopted this Insider Trading Policy (this “Policy”) to promote compliance with federal and state securities laws by directors, officers, employees and consultants of the Company. This Policy is designed to prevent insider trading, as well as the appearance or allegations of insider trading. Your strict adherence to this Policy will help safeguard the Company’s reputation and will further ensure that the Company conducts its business in accordance with the highest ethical standards. You are responsible for the consequences of your actions. You also are responsible for understanding and complying with this Policy.
Federal and state securities laws generally prohibit the purchase and sale of a company’s securities by anyone who is aware of material information about that company that is not generally known or available to the public. These laws also generally prohibit anyone who is aware of material non-public information from disclosing this information to others who may trade on it.
It is important that you understand the breadth of activities that constitute illegal insider trading and the consequences, which can be severe. Both the U.S. Securities and Exchange Commission (“SEC”) and the Financial Industry Regulatory Authority (“FINRA”) investigate and are very effective at detecting insider trading. Both the SEC and the U.S. Department of Justice also pursue insider trading violations vigorously.
As discussed below, most of this Policy applies to everyone, and prohibits trading in the Company’s and other companies’ securities in certain circumstances. Another part of the Policy applies only to directors, officers and certain employees or consultants (called “Company Insiders”), who typically have access to financial and other highly sensitive information regarding the Company’s business, and imposes additional restrictions on those individuals with respect to trading in the Company’s securities.
What Is Insider Trading?
The federal securities laws prohibit so-called “insider trading.” Simply stated, insider trading occurs when a person uses material non-public information obtained through involvement with a company to make decisions to purchase, sell, give away or otherwise transact in the company’s securities or to provide that information to others. The prohibitions against insider trading apply to trades, tips and recommendations by virtually any person, including all persons associated with the company, if the information involved is “material” and “non-public.” The prohibitions would apply to any director, officer or employee who buys or sells a company’s stock on the basis of material non-public information that he or she obtained about the company, its customers, its suppliers, or other companies with which the company has contractual relationships or may be negotiating transactions.
1.Applicability
This Policy applies to all trading or other transactions in the Company’s securities, including common shares, options, preferred shares, notes, bonds, convertible securities and any other securities that the Company may issue, as well as to derivative securities relating to any of the foregoing, whether or not issued by the Company (each of the foregoing, a “Company Security”).
This Policy applies to members of the Board of Directors, all officers of the Company, all employees of the Company (including contractors, consultants and temporary employees) and their respective immediate family members or other persons with respect to whom they exercise control of securities trading decisions (together, “Covered Persons”).
2.General Policy: No Trading or Tipping While in Possession of Material Non-Public Information
No Covered Person may purchase or sell, offer to purchase or sell or otherwise transact in (including making a gift of) any Company Security while in possession of material non-public information about the Company. (The terms “material” and “non-public” are defined below.)
No Covered Person who knows of any material non-public information about the Company may communicate that information to “tip” any other person, including family members and friends, or otherwise disclose this information without the Company’s authorization.
The Company’s directors, officers and certain other individuals (together, “Company Insiders”) are presumed by the nature of their jobs to have access to material, non-public information regarding the Company. Company Insiders are subject to additional trading restrictions described in Section 5 below. The Chief Legal Officer or his designee maintains a list of Company Insiders, and may at any time determine that an individual is or is not a Company Insider. For some individuals, this may be a temporary designation based on an upcoming event or a special project they may be working on. Other individuals, by nature of their job functions, will always be Company Insiders. If you are determined to be a Company Insider, you will be informed by the Chief Legal Officer or his designee.
3.Definitions
“Material information” is any information for which there is a substantial likelihood that a reasonable investor would consider important in a decision to buy, sell or hold securities. Any information that could reasonably be expected to affect the price of the securities is likely to be considered material.
While it is not possible to identify every type of information that could be deemed “material,” depending on the facts and circumstances, information that could be considered material includes, but is not limited to:
•significant changes in the Company’s prospects;
•important clinical data related to safety or efficacy results;
•certain material discussions with or actions by the Food and Drug Administration or other regulatory agencies;
•developments regarding significant litigation or government agency investigations;
•actual, anticipated or targeted earnings and dividends and other financial information;
•financial, sales and other significant internal business forecasts, or a change in previously released estimates;
•award or loss of a significant contract, changes in earnings estimates or unusual gains or losses;
•major changes in the Company’s management or the Board of Directors;
•offerings of Company Securities;
•extraordinary borrowings;
•major changes in accounting methods or policies;
•cybersecurity risks and incidents, including vulnerabilities and breaches;
•changes in debt ratings;
•proposals, plans or agreements, even if preliminary in nature, involving mergers, acquisitions, divestitures, recapitalizations, strategic alliances, licensing arrangements, or purchases or sales of substantial assets; and
•significant write-downs in assets or increases in reserves.
Material information is not limited to historical facts. It may also include projections and forecasts. With respect to a future event, such as the launch of a new product, a clinical trial, a licensing arrangement or a merger, determining the point at which negotiations or product developments related to such event become material involves weighing the probability that the event will occur against the effect the event would have on the Company’s operations or stock price. Information concerning an event that would have a large effect on stock price may be material even if the possibility that the event will occur is relatively small. When in doubt about whether particular non-public information is material, you should presume that it is material. You should not transact in or recommend Company Securities nor discuss the information outside of the Company until you discuss your concerns with the Chief Legal Officer and determine whether the information is material and whether it has been publicly disclosed through proper channels.
“Non-public information.” Insider trading prohibitions come into play only when you possess information that is material and also “non-public.” The fact that information has been disclosed to a few members of the public does not make it public for insider trading purposes.
To be “public,” the information must have been disseminated in a manner designed to reach investors generally, and the investors must be given the opportunity to absorb the information. Even after public disclosure of information about the Company, you must wait until the close of business on the second trading day after the information is publicly disclosed before you can treat the information as public. When in doubt about whether particular information is non-public, you should presume that it is non-public unless you can point to a Company-issued press release or SEC filing that includes the information. With respect to questions of materiality, if you are not sure whether information is considered public, you should either consult with the Chief Legal Officer or assume that the information is non-public and treat it as confidential.
4.Trading Prohibitions During “Blackout Periods”
In furtherance of the general policy described above, Covered Persons may only trade or otherwise transact in Company Securities when no “Blackout Period” is in effect, provided, however, even during an open trading window, a Covered Person who is in possession of any material non-public information must not trade or otherwise transact in Company Securities until the information has been made publicly available or is no longer material. There are two types of Blackout Periods:
•Regular Blackout Periods, which apply to all Covered Persons, occur each quarter, beginning two calendar weeks prior to the last day of the fiscal quarter and continuing through the end of the second trading day following the public release of the Company’s financial results for that quarter. If the Company announces earnings before the market opens, this counts as one trading day.
•Ad-hoc Blackout Periods, which may apply to all Covered Persons or only certain designated persons, may be declared from time to time by the Chief Legal Officer or his designee, beginning and ending as determined by the Chief Legal Officer or his designee. Ad-hoc Blackout Periods may be declared when certain persons or the Company is in possession of material non-public information or for other reasons, at the discretion of the Chief Legal Officer or his designee.
Illustration – Regular Blackout Period and Trading Window
The Company ends its second fiscal quarter on June 30. A Blackout Period would be in effect from June 16 (two calendar weeks prior to June 30) until the end of the second full trading day following earnings release (if the Company announces earnings before the market opens, this counts as one trading day). Trading would then be permitted during the window beginning on the third trading day following announcement until two weeks before the close of the next quarter (i.e., September 16—two weeks prior to September 30), so long as the Covered Person is certain he or she possesses no material, non-public information.
5.Additional Trading Restrictions for Company Insiders
Trading and other transactions in Company Securities by Company Insiders is generally prohibited (subject to the exceptions referenced in Section 9 below), unless (a) the Company is in an “open trading window” and (b) the Company Insider has obtained pre-approval to trade or transact from the Chief Legal Officer or his designee. For administrative convenience, pre-approval requests should be submitted to the Chief Legal Officer via email and must certify that the Company Insider is not in possession of material non-public information regarding the Company. Unless revoked or otherwise conditioned, a grant of pre-approval to trade or transact will normally remain valid until the close of trading five trading days following the day on which it was granted.
However, if you become aware of material non-public information before the transaction is executed, the prior approval shall be void and the transaction must not be completed. If the transaction does not occur during the five-day period, prior approval of the transaction must be re-requested.
6.Post-Termination Transactions
If you are subject to a Blackout Period at the time your employment with, or service to, the Company ends, you must continue to follow this Policy and not transact in Company Securities until such Blackout Period has ended. The Company may advise of an additional period of time during which you should not transact in Company Securities. However, you are nonetheless required to comply with securities laws and refrain from insider trading while you are in possession of material non-public information about the Company, notwithstanding the Company not applying or the lapse of any trading prohibitions.
7.Other Prohibited Transactions
Covered Persons are prohibited from engaging in the following transactions in Company Securities unless advance approval is obtained from the Chief Legal Officer or his designee:
•Short sales. Covered Persons are prohibited from selling Company Securities short. A short sale has occurred if the seller (1) does not own the securities sold or (2) does own the securities sold but does not deliver them within 20 days after the sale or deposit them in the mail or other usual channels of transportation within five days after the sale.
•Trading on margin or pledging. Pledged securities may be sold by the pledgee without the pledgor’s consent under certain conditions. For example, securities held in a margin account may be sold by a broker without the customer’s consent if the customer fails to meet a margin call. Because such a sale may occur at a time when a Covered Person has material non-public information about the Company or is otherwise not permitted to trade in Company Securities, Covered Persons are prohibited from pledging Company Securities, including by purchasing Company Securities on margin or holding Company Securities in a margin account, unless the Board of Directors has authorized the pledge.
•Derivatives. Covered Persons are prohibited from engaging in any derivative transactions (including transactions involving options, puts, calls, prepaid variable forward contracts, equity swaps, collars and exchange funds or other derivatives) that are designed to hedge or speculate on any change in the market value of Company Securities. If the SEC or NYSE were to notice active options trading by Covered Persons prior to an announcement, it would investigate. Such an investigation could be embarrassing (as well as expensive) for the Company, and could result in severe penalties and expense for the persons involved. For all of these reasons, the Company prohibits Covered Persons from trading in options or other derivatives involving the Company’s shares.
•Short-term trading. Directors and officers of the Company (but not other Covered Persons) who purchase or sell, as applicable, Company Securities may not sell or purchase, as applicable, any Company Securities of the same class for at least six months after the purchase or sale, as applicable.
8.Other Companies’ Securities
Covered Persons may not purchase, sell or otherwise transact in, or recommend that another person purchase, sell or otherwise transact in, the securities of another company, if the Covered Person learns in the course of his or her employment material, non-public information about the other company that is likely to affect the value of those securities. For example, it would be a violation of this Policy and the securities laws if a Covered Person learned through Company sources that the Company intended to purchase assets from another company or was considering cancelling a large contract with another company, and then placed an order to buy or sell shares in that other company because of the likely increase or decrease in the value of those shares. It would equally be a violation if the Covered Person tipped off a third party that they should buy or sell shares in the other company based on such material, non-public information.
9.Exceptions
•Options: Exercising stock options granted under the Company’s 2019 Equity Incentive Plan by paying the exercise price in cash is always permitted. However, the sale of any shares issued on the exercise of Company-granted stock options, and any cashless exercise of Company-granted stock options, are subject to the trading restrictions under this Policy.
•10b5-1 Plans: The trading restrictions of this Policy do not apply to transactions under a trading plan or arrangement adopted and effected in compliance with the Company’s Rule 10b5-1 Plan Policy.
10. Consequences of Violating Insider Trading Laws
Penalties for trading on or communicating material non-public information can be severe, and may include prison terms, criminal fines, civil penalties and injunctions. Given the severity of the potential penalties, compliance with this Policy is absolutely mandatory.
•Legal Penalties. A person who violates insider trading laws by engaging in transactions in a company’s securities when he or she has material non-public information can be sentenced to a substantial prison term and required to pay a criminal penalty of several times the amount of profits gained or losses avoided.
In addition, a person who tips others may also be liable for transactions by the tippee. Tippers can be subject to the same penalties and sanctions as the tippee, and the SEC has imposed large penalties even when the tipper did not profit from the transaction.
The SEC can also seek substantial civil penalties from any person who, at the time of an insider trading violation, “directly or indirectly controlled the person who committed such violation,” which could apply to the Company or management and supervisory personnel. These control persons may be held liable for up to the greater of $1 million or three times the amount of the profits gained or losses avoided. Even for violations that result in little or no profit, the SEC can seek penalties from a company or its management and supervisory personnel as control persons.
•Company-Imposed Penalties. Employees who violate this Policy may be subject to disciplinary action by the Company, including dismissal for cause. Any exceptions to this Policy, if permitted, may only be granted by the Chief Legal Officer or his designee and must be provided before any activity contrary to the above requirements takes place.
11. Section 16 and Section 13 Filings
•Section 16(a) of the Exchange Act requires directors, executive officers, certain other officers and certain shareholders to publicly disclose any changes in their beneficial ownership of the Company’s equity securities (including as a result of receipt of options, RSUs, PSUs and other derivative securities). Those subject to Section 16(a)’s reporting requirements are responsible for complying with all filing requirements thereunder, including filing of Form 3 (which must be filed within ten calendar days of becoming subject to Section 16(a)’s reporting requirements), Forms 4 (which must be filed before the end of the second business day after any change in beneficial ownership) and Forms 5 (which must be filed before the end of the forty-fifth day after the end of the Company’s fiscal year), in a timely manner. As a convenience, the Company may assist directors and officers with these required filings, but ultimately such persons bear legal responsibility for complying with these requirements.
•Section 13 of the Exchange Act requires the filing of beneficial ownership statements by any person or group that acquires beneficial ownership of more than five percent of a class of equity securities registered under the Exchange Act. Persons required to file statements under Section 13 must do so in a timely manner and bear legal responsibility for complying with these requirements.
12. Rule 144 Limitations on Public Resales of Restricted Securities and Control Securities
•Rule 144 is an exemption available for the public resales of “restricted securities” (i.e., securities acquired in a private offering or sale) and “control securities” (i.e., securities held by any of the Company’s affiliates). Rule 144’s requirements include holding period, current public information regarding the Company, volume limitations (applicable to affiliates), manner of sale requirements (applicable to affiliates) and Form 144 filings (applicable to affiliates).
•Each person relying on Rule 144 for the public resale of restricted or control securities (including through Rule 10b5-1 plans) is responsible for complying with Rule 144’s requirements, including timely Form 144 filings.
13. Additional Securities Law Considerations and Restrictions
In addition to the above, Covered Persons are responsible for complying with all securities laws when trading or otherwise transacting in the Company Securities, including, but not limited to:
•Regulation M, which generally restricts the Company or any of its affiliates from buying Company shares in the open market during certain periods while a distribution, such as a public offering, is taking place. You should consult with the Chief Legal Officer if you desire to make purchases of Company shares during any period in which the Company is conducting an offering. Similar considerations may apply during period when the Company is conducting or has announced a tender offer.
•Section 16(b), which generally requires insiders to disgorge to the Company any “profit” resulting from “short-swing” trades, and Section 16(c), which prohibits insiders from engaging in short sales.
14. Inquiries
If securities transactions ever become the subject of scrutiny, they are likely to be viewed after-the-fact with the benefit of hindsight. As a result, before engaging in any transaction you should carefully consider how the transaction may be construed in the bright light of hindsight. If you have any questions or uncertainties about this Policy or a proposed transaction, you should ask the Chief Legal Officer.
EX-19.2
5
ex1922024rule10b5-1planpol.htm
EX-19.2
Document
EXHIBIT 19.2
Effective January 1, 2024
ADC Therapeutics
Rule 10b5-1 Plan Policy
Table of Contents
1
1Purpose
This Rule 10b5-1 Trading Plan Policy (the “Policy”) describes the requirements for how directors, officers and eligible employees of ADC Therapeutics SA (together with its subsidiaries, the “Company”) can establish trading plans meeting the requirements of Rule 10b5-1 under the Securities Exchange Act of 1934 (each such plan, a “Trading Plan”). A Trading Plan enables directors, officers and employees to sell or buy a predetermined number of shares of the Company or exercise a predetermined number of stock options at a predetermined time, including during blackout periods, while providing an affirmative defense to insider trading for transactions made under the Trading Plan.
2Eligible Persons
Directors, officers and employees who are subject to the Company’s regular quarterly blackout periods (“Eligible Persons”) may enter into Trading Plans.
3Pre-Clearance and Pre-Approval Requirements
Any employee of the Company may submit a good faith complaint regarding financial statement or other disclosures, accounting, internal accounting or disclosure controls, auditing matters or violations of law or violations of the Code of Business Conduct and Ethics without fear of dismissal or retaliation of any kind.
4
5Prescribed Requirements for Trading Plans
5.1Adoption Date
An Eligible Person may enter into a Trading Plan only (1) during an “open trading window” when the Company has not imposed a regular or ad hoc blackout period and (2) at a time when he/she does not otherwise possess any material non-public information regarding the Company.
5.2Specifications
A Trading Plan must (1) specify the amount of securities to be purchased or sold and the price at which and the date on which the securities are to be purchased or sold or include an objective formula or algorithm for determining the amount of securities to be purchased or sold and the price at which and date on which the securities are to be purchased or sold and (2) not permit the Eligible Person to exercise any subsequent influence over how, when or whether to affect purchases or sales.
5.3Duration
Trading Plans may not have a duration of less than six months or more than two years.
5.4Commencement of Transactions
For directors and officers, transactions may not commence under a Trading Plan until the Trading Plan is approved by the Company and a cooling-off period has passed, which is the later of (x) 90 days following the adoption of the Trading Plan and (y) two business days following the filing of the Company’s Form 10-K or 10-Q covering the period in which the Trading Plan is adopted, subject to a maximum of 120 days after adoption.
For Eligible Persons other than directors and officers, transactions may not commence under a Trading Plan until a cooling-off period has passed, which is 30 days following the adoption of the Trading Plan.
5.5Suspensions
A Trading Plan must provide for suspension of trades under such plan if legal, regulatory or contractual restrictions are imposed on the Eligible Person, or if these guidelines are amended, or other events occur, that would prohibit sales under such Trading Plan.
5.6Required Disclosures
Each Trading Plan must contain an explicit acknowledgement by the Eligible Person that all filings required by the Exchange Act or the Securities Act, as a result of or in connection with trades under such Trading Plan, are the sole obligation of the Eligible Person and not the Company.
To the extent that sales made under a Trading Plan are made pursuant to Rule 144 under the Securities Act, the Trading Plan must provide for specific procedures to comply with Rule 144, including the filing of Forms 144. To the extent that the Eligible Person is subject to Section 16 filing requirements, the Trading Plan must provide the broker will provide notice of any trades under the Trading Plan to the Eligible Person and the Company’s Stock Administrator in sufficient time to allow for the person to make timely filings under the Exchange Act.
6Modifications to and Terminations of Trading Plans
6.1
6.1Modifications
Trading Plans may be modified only (1) for Company Insiders, if pre-approval for such modification is obtained from the Company’s Chief Legal Officer, (2) during an open trading window and (3) when the Eligible Person does not possess any material non-public information regarding the Company.
Modification of one or more key terms of the Trading Plan, including the amount, price or timing of transactions under the Trading Plan, constitutes a termination of the Trading Plan. The Trading Plan cannot resume after a modification until after the same cooling-off period described above, as if a new Trading Plan is being adopted.
6.2Cancellations
Eligible Persons are prohibited from cancelling specific transactions under a Trading Plan.
6.3Terminations
An Eligible Person should enter into a Trading Plan with the expectation that it will not be terminated prior to its stated term. Company Insiders must obtain pre-approval from the Company’s Chief Legal Officer prior to terminating a Trading Plan. An Eligible Person who terminates a Trading Plan may not make transactions in Company stock or sell shares obtained through the exercise of stock options until 30 days following the termination date of the Trading Plan (and any such transactions must be made during an open window and pursuant to the Company’s regular pre-clearance process as described in the Company’s Insider Trading Policy).
If a Trading Plan is terminated early and another Trading Plan is already in place, the first trade under the later-commencing plan must not be scheduled to occur until after the end of the effective cooling-off period following the termination of the earlier plan.
If a Trading Plan expires according to its terms (either because all designated shares have been sold or the duration of the Trading Plan has lapsed), an Eligible Person may enter into a new Trading Plan after obtaining pre-clearance and pre-approval, as applicable and as described in this Policy.
7Limitations and Prohibited Practices
7.1Multiple Trading Plans
An Eligible Person may have only one Trading Plan in effect at any time; provided that (x) an Eligible Person may adopt a new Trading Plan so long as transactions under the later-commencing Trading Plan is not authorized to begin until all transactions under the earlier-commencing Trading Plan are completed or expire without execution (and not modified or terminated) and the cooling-off period has lapsed after the adoption of the later-commencing plan and (y) a Trading Plan will not count towards this limit if it is a “sell-to-cover plan” that (i) only authorizes sales of securities as necessary to satisfy tax withholding obligations arising solely from the vesting of limited types of compensatory awards (not including options), (ii) does not permit the Eligible Person to exercise control over the timing of sales under such Trading Plan, (iii) was adopted during an open trading window and when the Eligible Person does not possess any material non-public information regarding the Company and (iv) was adopted in good faith and not as part of a plan to evade the prohibition against illegal insider trading.
One Trading Plan may include multiple brokers so long as the Trading Plan acts as a single Trading Plan.
7.2Single-Trade Plans
In any 12-month period, an Eligible Person can have no more than one Trading Plan that is designed to effect the open market purchase or sale of the total amount of securities subject to the Trading Plan as one single transaction (“single-trade plan”); provided that the following do not constitute single-trade plans: (1) a Trading Plan that gives discretion to an agent over whether to execute the Trading Plan as a single transaction or that provides the agent’s future acts depend on facts not known at the time the Trading Plan’s adoption and might reasonably result in multiple transactions and (2) a sell-to-cover plan described above.
7.3Hedging
As described in the Company’s Insider Trading Policy, hedging or similar transactions designed to decrease the risks associated with holding Company securities are prohibited. Further to this end, an Eligible Person adopting a Trading Plan may not have entered into or altered a corresponding or hedging transaction or position with respect to the securities subject to the Trading Plan and must agree not to enter into any such transaction while the Trading Plan is in effect.
8Required Disclosures
For Eligible Persons who are required to file Forms 4 and 5 with the Securities and Exchange Commission, footnote disclosure indicating that reported transactions are pursuant to a Trading Plan will be included on Forms 4 prepared by the Company.
The Company will also disclose in its Form 10-K and Form 10-Q information regarding Trading Plans adopted or terminated (which includes modifications) by directors and officers as required by SEC rules, including the identity of the person, the date of adoption or termination, the duration of the trading arrangement and the aggregate number of securities under the Trading Plan.
EX-21.1
6
exhibit211listofsubsidiari.htm
EX-21.1
Document
Exhibit 21.1
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Subsidiaries of ADC Therapeutics SA |
|
|
|
|
|
| Name of Subsidiary |
|
Jurisdiction of Organization |
|
|
|
|
|
| ADC Therapeutics America, Inc. |
|
United States |
|
|
|
|
|
| ADC Therapeutics (UK) Limited |
|
England |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
EX-23.1
7
exhibit231adct_consentaudi.htm
EX-23.1
Document
Exhibit 23.1
CONSENT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
We hereby consent to the incorporation by reference in the Registration Statements on Form S-3 (No. 333-267295, No. 333-267293, and No. 333-281288) and Form S-8 (No. 333-238287, No. 333-255831, No. 333-265917, No. 333-270565, No. 333-275882, and No. 333-276110) of ADC Therapeutics SA of our report dated March 27, 2025 relating to the financial statements, which appears in this Form 10-K.
/s/ PricewaterhouseCoopers SA
Pully, Switzerland
March 27, 2025
EX-31.1
8
ex311-ceocertification302q.htm
EX-31.1
Document
Exhibit 31.1
CERTIFICATION UNDER SECTION 302
I, Ameet Mallik, certify that:
1.I have reviewed this annual report on Form 10-K of ADC Therapeutics SA;
2.Based on my knowledge, this report does not contain any untrue statement of a material fact or omit to state a material fact necessary to make the statements made, in light of the circumstances under which such statements were made, not misleading with respect to the period covered by this report;
3.Based on my knowledge, the financial statements, and other financial information included in this report, fairly present in all material respects the financial condition, results of operations and cash flows of the company as of, and for, the periods presented in this report;
4.The company’s other certifying officer(s) and I are responsible for establishing and maintaining disclosure controls and procedures (as defined in Exchange Act Rules 13a-15(e) and 15d-15(e)) and internal control over financial reporting (as defined in Exchange Act Rules 13a-15(f) and 15(d)-15(f)) for the company and have:
a.Designed such disclosure controls and procedures, or caused such disclosure controls and procedures to be designed under our supervision, to ensure that material information relating to the company, including its consolidated subsidiaries, is made known to us by others within those entities, particularly during the period in which this report is being prepared;
b.Designed such internal control over financial reporting, or caused such internal control over financial reporting to be designed under our supervision, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles;
c.Evaluated the effectiveness of the company’s disclosure controls and procedures and presented in this report our conclusions about the effectiveness of the disclosure controls and procedures, as of the end of the period covered by this report based on such evaluation; and
d.Disclosed in this report any change in the company’s internal control over financial reporting that occurred during the period covered by the annual report that has materially affected, or is reasonably likely to materially affect, the company’s internal control over financial reporting; and
5. The company’s other certifying officer(s) and I have disclosed, based on our most recent evaluation of internal control over financial reporting, to the company’s auditors and the audit committee of the company’s board of directors (or persons performing the equivalent functions):
a.All significant deficiencies and material weaknesses in the design or operation of internal control over financial reporting which are reasonably likely to adversely affect the company’s ability to record, process, summarize and report financial information; and
b.Any fraud, whether or not material, that involves management or other employees who have a significant role in the company’s internal control over financial reporting.
|
|
|
|
|
|
|
|
|
Date: |
March 27, 2025 |
/s/ Ameet Mallik
Ameet Mallik
Chief Executive Officer
|
EX-31.2
9
ex312-cfocertification302q.htm
EX-31.2
Document
Exhibit 31.2
CERTIFICATION UNDER SECTION 302
I, Jose Carmona, certify that:
1.I have reviewed this annual report on Form 10-K of ADC Therapeutics SA;
2.Based on my knowledge, this report does not contain any untrue statement of a material fact or omit to state a material fact necessary to make the statements made, in light of the circumstances under which such statements were made, not misleading with respect to the period covered by this report;
3.Based on my knowledge, the financial statements, and other financial information included in this report, fairly present in all material respects the financial condition, results of operations and cash flows of the company as of, and for, the periods presented in this report;
4.The company’s other certifying officer(s) and I are responsible for establishing and maintaining disclosure controls and procedures (as defined in Exchange Act Rules 13a-15(e) and 15d-15(e)) and internal control over financial reporting (as defined in Exchange Act Rules 13a-15(f) and 15(d)-15(f)) for the company and have:
a.Designed such disclosure controls and procedures, or caused such disclosure controls and procedures to be designed under our supervision, to ensure that material information relating to the company, including its consolidated subsidiaries, is made known to us by others within those entities, particularly during the period in which this report is being prepared;
b.Designed such internal control over financial reporting, or caused such internal control over financial reporting to be designed under our supervision, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles;
c.Evaluated the effectiveness of the company’s disclosure controls and procedures and presented in this report our conclusions about the effectiveness of the disclosure controls and procedures, as of the end of the period covered by this report based on such evaluation; and
d.Disclosed in this report any change in the company’s internal control over financial reporting that occurred during the period covered by the annual report that has materially affected, or is reasonably likely to materially affect, the company’s internal control over financial reporting; and
5.The company’s other certifying officer(s) and I have disclosed, based on our most recent evaluation of internal control over financial reporting, to the company’s auditors and the audit committee of the company’s board of directors (or persons performing the equivalent functions):
a.All significant deficiencies and material weaknesses in the design or operation of internal control over financial reporting which are reasonably likely to adversely affect the company’s ability to record, process, summarize and report financial information; and
b.Any fraud, whether or not material, that involves management or other employees who have a significant role in the company’s internal control over financial reporting.
|
|
|
|
|
|
|
|
|
|
|
|
Date: |
March 27, 2025 |
/s/ Jose Carmona
Jose Carmona
Chief Financial Officer
|
EX-32.1
10
ex321-ceocertification906q.htm
EX-32.1
Document
Exhibit 32.1
CERTIFICATION UNDER SECTION 906
The certification set forth below is being submitted in connection with ADC Therapeutics SA’s annual report on Form 10-K for the year ended December 31, 2024 (the “Report”) for the purpose of complying with Rule 13a-14(b) or Rule 15d-14(b) of the Securities Exchange Act of 1934 (the “Exchange Act”) and Section 1350 of Chapter 63 of Title 18 of the United States Code.
I, Ameet Mallik, the Chief Executive Officer of ADC Therapeutics SA, certify that:
1.the Report fully complies with the requirements of Section 13(a) or 15(d) of the Exchange Act; and
2.the information contained in the Report fairly presents, in all material respects, the financial condition and results of operations of ADC Therapeutics SA.
|
|
|
|
|
|
|
|
|
Date: |
March 27, 2025 |
/s/ Ameet Mallik
Ameet Mallik
Chief Executive Officer
|
EX-32.2
11
ex322-cfocertification906q.htm
EX-32.2
Document
Exhibit 32.2
CERTIFICATION UNDER SECTION 906
The certification set forth below is being submitted in connection with ADC Therapeutics SA’s annual report on Form 10-K for the year ended December 31, 2024 (the “Report”) for the purpose of complying with Rule 13a-14(b) or Rule 15d-14(b) of the Securities Exchange Act of 1934 (the “Exchange Act”) and Section 1350 of Chapter 63 of Title 18 of the United States Code.
I, Jose Carmona, the Chief Financial Officer of ADC Therapeutics SA, certify that:
1.the Report fully complies with the requirements of Section 13(a) or 15(d) of the Exchange Act; and
2.the information contained in the Report fairly presents, in all material respects, the financial condition and results of operations of ADC Therapeutics SA.
|
|
|
|
|
|
|
|
|
Date: |
March 27, 2025 |
/s/ Jose Carmona
Jose Carmona
Chief Financial Officer
|