UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
(Mark One)
☒ |
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2024
OR
☐ |
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to
Commission file number: 001-39130
TELA Bio, Inc.
(Exact name of registrant as specified in its charter)
|
Delaware (State or other jurisdiction of |
|
45-5320061 (I.R.S. Employer |
|
|
|
|
1 Great Valley Parkway, Suite 24 Malvern, Pennsylvania (Address of principal executive offices) |
|
19355 (Zip Code) |
(484) 320-2930
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
Title of each class: |
|
Trading Symbol |
|
Name of each exchange on which registered: |
Common Stock, $0.001 par value per share |
TELA |
The Nasdaq Global Market |
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. ◻ Yes ⌧ No
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. ◻ Yes ⌧ No
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. ⌧ Yes ◻ No
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files). ⌧ Yes ◻ No
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
Large accelerated filer ◻ |
|
Accelerated filer ◻ |
|
|
Smaller reporting company ⌧ |
Non-accelerated filer ⌧ |
|
Emerging growth company ◻ |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ◻
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C.7262(b)) by the registered public accounting firm that prepared or issued its audit report. ☐
If securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant included in the filing reflect the correction of an error to previously issued financial statements. ☐
Indicate by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive-based compensation received by any of the registrant’s executive officers during the relevant recovery period pursuant to § 240.10D-1(b). ◻
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ◻ No ⌧
As of June 28, 2024 (the last business day of the registrant’s most recently completed second fiscal quarter), the aggregate market value of the registrant’s common stock held by non-affiliates was approximately $114.5 million based on the closing price of the common stock as reported on the NASDAQ Global Market on June 28, 2024.
As of March 14, 2025, the registrant had 39,551,098 shares of Common Stock, $0.001 par value per share, outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the definitive proxy statement to be filed with the U.S. Securities and Exchange Commission (the “SEC”) for TELA Bio’s 2025 annual meeting of stockholders are incorporated by reference into Part III of this Form 10-K.
TABLE OF CONTENTS
Item No. |
|
Page No. |
7 |
||
40 |
||
80 |
||
80 |
||
81 |
||
81 |
||
81 |
||
|
|
|
82 |
||
82 |
||
MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS |
83 |
|
93 |
||
95 |
||
CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE |
95 |
|
95 |
||
96 |
||
DISCLOSURE REGARDING FOREIGN JURISDICTIONS THAT PREVENT INSPECTIONS |
96 |
|
|
|
|
96 |
||
97 |
||
SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS |
97 |
|
CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE |
97 |
|
97 |
||
|
|
|
97 |
||
98 |
||
|
|
|
99 |
||
102 |
||
2
CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS
This Annual Report on Form 10-K (this “Annual Report”) and the documents incorporated by reference herein contain “forward-looking statements” within the meaning of the Private Securities Litigation Reform Act of 1995. In addition, we may, through our officers and other authorized representatives, make certain forward-looking statements in publicly released materials, both written and oral, including statements contained in filings with the Securities and Exchange Commission, press releases, and our communications with our stockholders.
Forward-looking statements are neither statements of historical facts nor assurances of future performance, but instead discuss the future of our business, operations, future financial performance, future financial condition, plans, anticipated growth strategies, anticipated or perceived trends in our business, the industry in which we operate or the broader economy, and other objectives of management. In some cases, you can identify forward-looking statements by terminology such as “aim,” “anticipate,” “assume,” “believe,” “contemplate,” “continue,” “could,” “design,” “due,” “estimate,” “expect,” “goal,” “intend,” “may,” “objective,” “plan,” “predict,” “positioned,” “potential,” “seek,” “should,” “target,” “will,” “would,” the negative of such terms, and other similar expressions although not all forward-looking statements contain these identifying words.
You should understand that the following important factors could affect our future results and could cause those results or other outcomes to differ materially from those expressed or implied in our forward-looking statements:
| ● | estimates regarding future results of operations, financial position, research and development costs, capital requirements and our needs for additional financing; |
| ● | the commercial success and degree of market acceptance of our products; |
| ● | the introduction of new products or product enhancements by us or others in our industry, including new products which may be perceived to negatively impact the demand for our products now or in the future; |
| ● | our ability to expand, manage and maintain our direct sales and marketing organization and to market and sell our products in the U.S. and Europe; |
| ● | the performance of our exclusive contract manufacturer for our OviTex and OviTex PRS products, Aroa Biosurgery Ltd. (“Aroa”), in connection with the supply of product and in the development of additional products and product configurations within these products; |
| ● | our ability to maintain our supply chain integrity and expand our supply chain to manage increased demand for our products; |
| ● | our ability to compete successfully with larger competitors in our highly competitive industry; |
| ● | our ability to achieve and maintain adequate levels of coverage or reimbursement for our current products and any future products we may seek to commercialize; |
| ● | our ability to enhance our products, expand our indications and develop and commercialize additional products; |
| ● | the development, regulatory approval, efficacy and commercialization of competing products; |
| ● | our business model and strategic plans for our products, technologies and business, including our implementation thereof; |
| ● | the size of the markets for our current and future products; |
| ● | our ability to recruit and retain senior management and other highly qualified personnel; |
| ● | our ability to obtain additional capital to finance our planned operations; |
| ● | our ability to maintain regulatory approval for our products; |
| ● | our ability to commercialize or obtain regulatory approvals for our future products, or the effect of delays in commercializing or obtaining regulatory approvals; |
| ● | decreasing selling prices and pricing pressures; |
| ● | regulatory developments in the U.S. , including regulatory developments due to changes in the U.S. presidential administration and European markets; |
| ● | the potential impact of healthcare reform in the U.S., including the Inflation Reduction Act of 2022, and measures being taken worldwide designed to reduce healthcare costs; |
| ● | any decrease in frequency of surgical procedures using our products, whether through outbreak of illness or disease, cybersecurity events impacting hospital operations, labor and hospital staffing shortages, supply chain disruptions to critical surgical and hospital supplies, and any applicable adverse healthcare economic factors; |
3
| ● | the volatility of capital markets and other adverse macroeconomic factors, including due to inflationary pressures, interest rate and currency rate fluctuations, economic slowdown or recession, banking instability, monetary policy changes, changes in trade policies (including tariffs that have been or may in the future be imposed by the U.S. or other countries), geopolitical tensions or the outbreak of hostilities or war, including from the ongoing Russia-Ukraine conflict, the current conflicts in the Middle East (including any escalation or expansion) and increasing tensions between China and Taiwan; |
| ● | our ability to develop and maintain our corporate infrastructure, including our internal controls; |
| ● | our ability to establish and maintain intellectual property protection for our products, as well as our ability to operate our business without infringing the intellectual property rights of others; |
| ● | our expectations regarding the use of proceeds from recent and any future financings, if any; |
| ● | the occurrence of adverse safety events, restrictions on use with our products or product liability claims; and |
| ● | other risks and uncertainties, including those listed under the caption “Risk Factors.” |
These forward-looking statements are based on management’s current expectations, estimates, forecasts and projections about our business and the industry in which we operate, and management’s beliefs and assumptions are not guarantees of future performance or development and involve known and unknown risks, uncertainties and other factors that are in some cases beyond our control. In light of the significant uncertainties in these forward-looking statements, you should not rely upon forward-looking statements as predictions of future events. Although we believe the expectations reflected in the forward-looking statements are reasonable, the future results, levels of activity, performance or events and circumstances reflected in the forward-looking statements may not be achieved or occur at all.
You should refer to the section titled “Risk Factors” in this Annual Report for a discussion of important factors that may cause our actual results to differ materially from those expressed or implied by our forward-looking statements. Furthermore, if our forward-looking statements prove to be inaccurate, the inaccuracy may be material. Except as required by law, we undertake no obligation to publicly update any forward-looking statements, whether as a result of new information, future events or otherwise.
4
SUMMARY RISK FACTORS
We are providing the following summary of the risk factors contained in our Form 10-K to enhance the readability and accessibility of our risk factor disclosures. Additional discussion of the risks and uncertainties summarized in this risk factor summary, as well as other risks and uncertainties that we face, can be found under “Cautionary Note Regarding Forward-Looking Statements” and “Risk Factors” in this Annual Report. The below summary is qualified in its entirety by those more complete discussions of such risks and uncertainties.
Risks Related to Achieving or Sustaining Profitability, Financial Position and Capital Requirements
| ● | We have incurred significant operating losses since inception, we expect to incur operating losses in the future, and we may not be able to achieve or sustain profitability. |
| ● | Our indebtedness may limit our flexibility in operating our business and adversely affect our financial health and competitive position. |
| ● | We may require substantial additional capital to finance our planned operations, which may not be available to us on acceptable terms or at all. |
| ● | If we are unable to expand, manage and maintain our direct sales and marketing organizations, we may not be able to generate anticipated revenue. |
| ● | Macroeconomic conditions, including those placing financial strain on hospital systems and their ability to perform the procedures in which our products are used may negatively impact certain aspects of our business, our prospects, results of operations and financial condition. |
| ● | Rising inflation rates could negatively impact our revenues and profitability if increases in the prices of our product or a decrease in consumer spending results in lower volumes of elective surgeries. In addition, if our costs increase and we are not able to pass along these price increases, our profitability would be adversely affected, and the adverse impact may be material. |
Risks Related to the Commercialization of our Products
| ● | To date, the vast majority of our revenue has been generated from sales of our OviTex products, and we therefore are highly dependent on the commercial success of our OviTex products. |
| ● | The commercial success of our products will largely depend upon attaining significant market acceptance. |
| ● | Even if we are able to attain significant market acceptance of our products, the commercial success of our products is not guaranteed. |
| ● | The misuse or off-label use of our products may harm our reputation in the marketplace, result in injuries that lead to product liability suits or result in costly investigations, fines or sanctions by regulatory bodies if we are deemed to have engaged in the promotion of our products for these uses. |
| ● | If we are unable to achieve and maintain adequate levels of coverage or reimbursement for our OviTex and OviTex PRS products we may commercialize in the future, our commercial success may be hindered. |
| ● | Our long-term growth may depend on our ability to enhance our product offerings. |
| ● | In the future our products may become obsolete, which would negatively affect operations and financial condition. |
| ● | To successfully market and sell our products in markets outside of the U.S., we must address many international business risks with which we have limited experience. |
Risks Related to Our Reliance on Third Parties
| ● | We are highly dependent upon Aroa as the exclusive contract manufacturer of our OviTex and OviTex PRS products. |
| ● | We, or our partners, may experience development or manufacturing problems, capacity constraints, or delays in the production of our products that could limit the potential growth of our revenue or increase our losses. |
| ● | Our products contain materials derived from animal sources and may become subject to additional regulation. |
| ● | Our supply of ovine rumen for use in manufacturing our products may be vulnerable to disruption due to natural disaster, disease or other events. |
5
Risks Related to Intellectual Property Matters
| ● | We may need to license intellectual property from third parties, and such licenses may not be available or may not be available on commercially reasonable terms. |
| ● | If we fail to comply with our obligations under any license, collaboration or other agreements, we could lose intellectual property rights that are necessary for developing and protecting our products. |
| ● | If we are unable to adequately protect our intellectual property rights, or if we are accused of infringing on the intellectual property rights of others, our competitive position could be harmed, or we could be required to incur significant expenses to enforce or defend our rights. |
| ● | Litigation or other proceedings or third-party claims of intellectual property infringement could require us to spend significant time and money, enter into license agreements for disputed intellectual property and could prevent us from selling our products. |
| ● | If we are unable to protect the confidentiality of our trade secrets, our business and competitive position could be harmed. |
Risks Related to Government Regulation
| ● | Our products and operations are subject to extensive government regulation and oversight both in the U.S. and internationally. |
| ● | We may not receive, or may be significantly delayed in receiving, the necessary clearances or approvals for our future products and modifications to our current products may require new 510(k) clearances or premarket approval, and may require us to cease marketing or recall the modified products until clearances or approvals are obtained. |
| ● | Although we have obtained regulatory clearance for our products, they will remain subject to extensive regulatory scrutiny. |
| ● | If guidelines for soft-tissue reconstruction surgery change or the standard of care evolves, we may need to redesign and seek new marketing authorization from the U.S. Food and Drug Administration for our OviTex and OviTex PRS products or other products we may commercialize in the future. |
Risks Related to Our Business and Products
| ● | Our financial results may fluctuate significantly and may not fully reflect the underlying performance of our business. |
| ● | We may be unable to renew existing or obtain additional contract positions with major group purchasing organizations and integrated delivery networks for our products, and even if we are able to do so, such contracts may not generate sufficient sales of our products. |
| ● | We have limited data and experience regarding the safety and efficacy of certain of our products. Results of earlier studies may not be predictive of future clinical trial results, or the safety or efficacy profile for such products. |
| ● | Interim or preliminary data from our clinical trials that we announce or publish from time to time may change as more patient data become available and are subject to audit and verification procedures that could result in material changes in the final data. |
Risks Related to Our Securities
| ● | The trading price of the shares of our common stock has been and could in the future be highly volatile. |
| ● | Our directors, officers and principal stockholders have significant voting power and may take actions that may not be in the best interests of our other stockholders. |
| ● | Provisions in our corporate charter documents and under Delaware law could discourage another company from acquiring us and may prevent attempts by our stockholders to replace or remove our current management. |
6
PART I
ITEM 1.BUSINESS
Overview
We are a commercial-stage medical technology company focused on providing innovative soft-tissue reconstruction solutions that optimize clinical outcomes by prioritizing the preservation and restoration of the patient’s own anatomy. Our growing product portfolio is purposefully designed to leverage the patient’s natural healing response while minimizing long-term exposure to permanent synthetic materials. We are committed to delivering our advanced technologies with a strong economic value proposition to assist surgeons and institutions in providing next-generation soft-tissue repair solutions to more patients worldwide.
We are dedicated to building true partnerships with surgeons and healthcare providers to deliver solutions that provide both clinical and economic improvements. We believe that genuine collaboration with surgeons and healthcare providers results in the development of new solutions that empower patient care and addresses unmet needs within the soft tissue reconstruction market.
Our first portfolio of products, the OviTex Reinforced Tissue Matrix (“OviTex”) which we first commercialized in the U.S. in July 2016 and in Europe in February 2019, addresses unmet needs in hernia repair and abdominal wall reconstruction by combining the benefits of biologic matrices and polymer materials while minimizing their shortcomings, at a cost-effective price.
Hernia repair is one of the most common surgeries performed in the U.S., representing approximately 1.2 million procedures annually. Based on the volume weighted average selling price of our OviTex products, we estimate the annual U.S. total addressable market opportunity for our OviTex products to be approximately $1.8 billion.
Our OviTex portfolio consists of multiple product configurations intended to address various surgical procedures within hernia repair and abdominal wall reconstruction, including ventral, inguinal, and hiatal hernia repair. In addition, we have also designed an OviTex product specifically for use in laparoscopic and robotic-assisted hernia repair, which we market as OviTex LPR and began commercializing in November 2018. In February 2023, we launched two larger configurations of OviTex LPR, designed for ventral and incisional hernias. In April 2024, we launched OviTex IHR Reinforced Tissue Matrix, a new OviTex configuration specifically designed to address inguinal hernia procedures performed robotically and laparoscopically.
We have also focused on evaluating and publishing clinical data on the effectiveness and safety of our OviTex products. To date, there have been over forty published or presented works relating to these clinical findings, either by us or a third-party evaluating one or more product configurations in our OviTex portfolio. In October 2022, the 24-month results of our single arm, multicenter post-market clinical study, which we refer to as our BRAVO study, were published in the Annals of Medicine and Surgery. The BRAVO study was designed to evaluate the clinical performance of OviTex for primary or recurrent ventral hernias using open, laparoscopic, or robotic techniques in 92 enrolled patients. The recurrence rate at the 24-month time point was 2.6%, and surgical site occurrences (“SSOs”), were observed in 38% of the study population. Of the enrolled patients, 78% were characterized as high risk for experiencing an SSO based on at least one known risk factor, which included obesity, active smoking, chronic obstructive pulmonary disease (“COPD”), diabetes mellitus, coronary artery disease, or advanced age (≥75 years). The results also indicated that BRAVO patients experienced statistically significant and clinically meaningful improvements in their quality of life and perceived health based on patient responses to the EuroQol-5 Dimension (EQ-5D) health assessment and the validated 12-question Hernia-Related Quality of Life survey (HerQLes). In addition to the BRAVO study, we have also initiated other clinical data collection initiatives evaluating the use of OviTex across a variety of hernia and abdominal wall reconstruction procedures. Among these other initiatives, we continue to enroll patients for our BRAVO II study, a prospective study evaluating the use of OviTex in robot-assisted ventral and inguinal hernia repairs.
7
Our second portfolio of products, the OviTex PRS Reinforced Tissue Matrix, (“OviTex PRS”) which we first commercialized in the U.S. in May 2019, addresses unmet needs in plastic and reconstructive surgery. OviTex PRS is indicated for use in implantation to reinforce soft-tissue where weakness exists in patients requiring soft-tissue repair or reinforcement in plastic and reconstructive surgery. Our OviTex PRS portfolio consists of three product configurations with two or three layers of high-quality tissue derived from ovine rumen, which is reinforced with either permanent or resorbable polymer for added strength, stabilization, and controlled stretch. These products are designed to improve outcomes by facilitating functional tissue remodeling while controlling the degree and direction of stretch. OviTex PRS Long-Term Resorbable, our most recent product configuration, launched in August 2023, and was designed to enhance the OviTex PRS portfolio with specific design features including bi-directional stretch and a fully resorbable, long-term polymer for reinforcement.
Our OviTex PRS portfolio is supported by non-human primate data that demonstrated more rapid tissue integration and tissue remodeling compared to the market leading biologic matrix used in this indication. In addition, there have been a growing number of published or presented works evaluating the use of OviTex PRS in plastic and reconstruction applications. We also continue to enroll patients in our OPERA study, a retrospective-prospective trial evaluating the safety profile of OviTex PRS in previous pre-pectoral and sub-pectoral implant-based breast reconstructions. Based on the current sales of biologic matrices in the U.S., we estimate the annual U.S. current addressable market opportunity for our OviTex PRS products to be approximately $800 million.
Our OviTex products have received 510(k) clearances from the U.S. Food and Drug Administration, (“FDA”) which clearances were obtained and are currently held by our exclusive contract manufacturer of these products, Aroa. In April 2019, our first OviTex PRS products received 510(k) clearance from the FDA, which clearance was initially obtained by Aroa and is currently held by us. In March 2023, we received an additional 510(k) clearance for our OviTex PRS Long-Term Resorbable device, which is currently held by us. In May 2024, we received clearance of a Special 510(k) related to minor changes to our OviTex PRS Permanent and Short-Term Resorbable devices. In October 2024, we received approval from the FDA for our investigational device exemption application relating to the study of the safety and effectiveness of our OviTex PRS product in implant-based breast reconstruction. We continue to evaluate and finalize the clinical study protocol and anticipate additional FDA interactions related to such to support a pre-market application to obtain approval for an indication for OviTex PRS for use in breast reconstruction.
Historically, we have sought to expand our service offerings beyond our OviTex and OviTex PRS products through commercial partnerships to distribute complimentary soft tissue preservation and restoration solutions. Some additional product offerings include or have included atraumatic mesh fixation devices or surgical wound management and infection control solutions. In September 2023, we entered into a distribution agreement with Advanced Medical Solutions Limited, a company registered in England, to distribute their LiquiFix Hernia Mesh Fixation Devices (LIQUIFIX FIX8™ and LIQUIFIX Precision™). In March 2024, we announced the full commercial launch of LiquiFix in the U.S. We previously co-developed and commercialized the NIVIS Fibrillar Collagen Pack, (“NIVIS”) an absorbent matrix of Type I and Type III bovine collagen designed to manage moderately to heavily exudating wounds and to control minor bleeding, in partnership with Regenity Biosciences. In March 2024, we sold our distribution rights to MiMedx Group, Inc. in exchange for an initial $5.0 million payment and additional future payments aggregating between a minimum of $3.0 million and a maximum of $7.0 million based on net sales of NIVIS (now marketed as HELIOGEN) during the first two years following its launch by MiMedx Group, Inc. We may assess additional strategic partnerships with medical device companies whereby we may enter into distribution, product development and/or licensing agreements for additional products complimentary to, or related to, existing and future products in our distribution channel, which could result in the payment by us of single digit percentage royalties or other product acquisition costs
We have a broad portfolio of intellectual property protecting our products that we believe, when combined with the proprietary manufacturing processes associated with our products and our know-how, provides significant barriers to entry. Our intellectual property applies to our differentiated product construction and materials. In addition, we believe our exclusive manufacturing and long-term supply and license agreement with Aroa (the “Aroa License”) creates a competitive advantage by allowing us to secure an exclusive supply of ovine rumen at a low cost. Ovine rumen, the forestomach of a sheep, is the source of the biologic material used in both of our OviTex and OviTex PRS products.
8
We use biologic material from ovine rumen because of its plentiful supply, optimal biomechanical profile and open collagen architecture that allows for rapid cellular infiltration. Our OviTex and OviTex PRS products are manufactured by Aroa at their FDA registered and ISO 13485 compliant facility in Auckland, New Zealand. We purchase product from Aroa at a fixed transfer cost as a percentage of Aroa’s cost of goods sold, and subject to a true-up adjustment, resulting in an amount equal to 27% of our net sales of our OviTex and OviTex PRS products, with the exception of OviTex IHR product configurations, for which we pay the greater of the initial fixed transfer cost or 27% of our net sales of OviTex IHR. This revenue sharing arrangement allows us to competitively price our products and pass along cost-savings to our customers.
We primarily market our products through a single direct sales force, predominantly in the U.S., with a small number of sales representatives in the United Kingdom and European Union, and also utilize a smaller number of independent contractors and distributors in the United States and certain European countries. We have invested in our direct sales and marketing infrastructure to expand our presence and to promote awareness and adoption of our products. As of December 31, 2024, we had 75 sales territories in the U.S. and 13 sales territories in Europe. We believe we can enhance the productivity of our sales force by improving customer segmentation and targeting, implementing and further refining our proprietary training programs, leveraging support from our medical education and medical affairs functions to drive physician awareness, education and clinical understanding of our products, and utilizing engagement analytics to support further product development and enhancement opportunities. Additionally, we have contracted with three national group purchasing organizations (“GPOs”) in the United States covering our OviTex and OviTex PRS products and plan to continue to contract with additional GPOs and other integrated delivery networks (“IDNs”) to increase access to and penetration of hospital accounts for all products we commercialize.
We are currently devoting research and development resources to develop additional variations of our OviTex and OviTex PRS products, including larger versions of our current OviTex PRS product configurations, the development of OviTex configurations with longer-acting resorbable polymers and other potential product and packaging enhancements to extend the shelf life of our products. In addition, we also continue to explore the development of lower-cost, higher-margin resorbable polymer-based devices targeting our current indications. We are also exploring additional technologies that may complement our existing products, or expand the number of our products, in each case within the hernia, plastic and reconstruction, and broader soft-tissue reconstruction market. We intend to continue to make investments in research and development efforts to develop improvements and enhancements to our product portfolio.
Our revenue for the years ended December 31, 2024 and 2023 was $69.3 million and $58.5 million, respectively, which represents an increase of $10.8 million, or 19% for the year ended December 31, 2024. Our net loss for the same time periods was $37.8 million and $46.7 million, respectively, which represents a decrease of $8.8 million, or 19% for the year ended December 31, 2024 inclusive of the gain recognized of $7.6 million on the sale of NIVIS to the MiMedx Group, Inc. As of December 31, 2024, we had an accumulated deficit of $358.7 million. The vast majority of our revenue to date has been generated from sales of our OviTex and OviTex PRS products in the U.S., with the remainder generated from sales of our OviTex products in Europe and the sale of other products.
Market Opportunity
OviTex
Hernia repair is one of the most common surgeries performed in the U.S. There are an estimated 1.2 million hernia repairs annually in the U.S. including recurrences, which we categorize as approximately (i) 105,000 complex/moderate ventral hernia repairs and abdominal wall reconstructions, (ii) 395,000 simple ventral hernia repairs and (iii) 645,000 inguinal hernia repairs, and (iv) 42,000 hiatal hernia repairs.
The healthcare burden of hernia disease to patients, insurers and employers is significant. For the patient, a hernia may cause an increasing level of pain when lifting, straining during urination or a bowel movement, or sitting or standing for long periods of time. Increased pain from the hernia is the most common reason that a patient who is deferring surgical hernia repair will ultimately elect repair surgery. Following surgical hernia repair, convalescence has a significant socioeconomic impact.
9
Absence from work during this period can range from approximately five to 14 days according to one study. Pain is the most common cause of delay in returning to work, followed by wound problems. Long-term pain or discomfort at the hernia repair site is one of the most serious complications of hernia surgery and may, in some cases, persist for years.
Given the limitations of and lack of innovation in existing hernia repair products, we believe a significant market opportunity exists for our portfolio of OviTex products. Based on the volume weighted average selling price of our OviTex products, we estimate the annual U.S. total addressable market opportunity for our OviTex products to be approximately $1.8 billion.
|
|
Approximate |
|
|
|
|
|
|
|
Number of |
|
|
|
|
|
|
|
Annual |
|
|
|
|
|
|
|
U.S. Hernia |
|
Estimated |
|
|
|
|
|
Procedures |
|
Annual |
|
|
|
|
|
Using |
|
U.S. Total |
|
|
|
|
|
Tissue |
|
Addressable |
|
Traditional |
|
|
|
Reinforcement |
|
Market |
|
Products |
|
|
|
Material |
|
Opportunity |
|
Utilized |
|
Complex/Moderate Ventral Repair /Abdominal Wall Reconstruction |
|
105,000 |
|
$ |
630 |
million |
Biologic Matrices and Resorbable Synthetic Mesh |
Simple Ventral Hernia Repair |
|
395,000 |
|
$ |
590 |
million |
Permanent Synthetic Mesh |
Inguinal Hernia Repair |
|
645,000 |
|
$ |
540 |
million |
Permanent Synthetic Mesh |
Hiatal Hernia Repair |
|
42,000 |
|
$ |
42 |
million |
Biologic Matrices and Resorbable Synthetic Mesh |
Total |
|
1,187,000 |
|
$ |
1.8 |
billion |
|
OviTex PRS
Modern advances in tissue engineering have transformed the plastic and reconstructive surgeon’s management strategies across a wide variety of applications. Because biologic matrices incorporate into host tissues and enable revascularization and functional tissue remodeling, surgeons have realized multiple applications for their use, with techniques tailored to the specific requirements of the surgery. There is growing clinical literature validating the use of biologic matrices in head and neck surgery and reconstructions of the chest wall, pelvic region, extremities and breast.
In head and neck surgery, biologic matrices are used for both aesthetic and reconstructive purposes that include: surgery of the nose to change its shape or improve its function, referred to as rhinoplasty; lip augmentation; repair of perforations of the cartilage and thin bone separating the nostrils referred to as the nasal septum; complex reconstruction of the oral and oropharynx cavities after oncologic resection; cleft palate repair; upper and lower eyelid reconstruction; scalp defects and defects of the fibrous membrane covering the brain and spinal cord referred to as dura. In chest wall reconstruction, biologic matrices are used to repair defects from oncologic resections. In pelvic reconstruction, biologic matrices are utilized as an adjunct in the reconstruction of acquired pelvic defects caused by resections for colorectal, gynecologic and urologic malignancies. In extremities reconstruction, biologic matrices are used in the upper extremity for repair of the donor site following the harvest of a radial forearm free flap, a procedure used to harvest tissue and replace it in the head and neck after cancer has been resected. In breast reconstruction, biologic matrices are utilized for prosthetic based reconstruction following the removal of cancerous breast tissue.
Based on the current sales of biologic matrices in the U.S., we estimate the annual U.S. current addressable market opportunity for our OviTex PRS products to be approximately $800 million. Given the limitations of and lack of innovation in existing biologic matrices for plastic and reconstructive surgical procedures, we believe a significant market opportunity exists for our OviTex PRS products.
10
Current Materials Used in Hernia Repair and Abdominal Wall Reconstruction and Their Limitations
Hernia Repair and Abdominal Wall Reconstruction
The vast majority of hernias are treated with surgical repair. Surgical hernia repair is performed either through open repair, which uses a single incision to open the abdomen or groin across the hernia, or minimally invasive repair, which involves laparoscopic or robotic-assisted techniques. Laparoscopic surgery is a minimally invasive surgical technique performed in the abdomen or groin through small incisions. Surgical instruments and devices, such as mesh products, are then delivered to the surgical site through a trocar, which is an access port to the patient’s abdomen or groin. Robotic-assisted surgery is also performed using small incisions in the patient’s abdomen or groin and a trocar, but the surgeon sits at a console in the operating room and operates the robotic instruments remotely.
At the advent of hernia repair, all procedures were performed using an open surgical technique in which an incision is made through the body to access and repair the hernia. Due to the amount of healthy soft-tissue disruption required for an open procedure, there is a high risk of wound-related complications and seroma formation. In the early 1990s, surgeons began using a laparoscopic approach for hernia repair because it provided the benefits of lower wound complication rates, lower patient morbidity and decreased length of stay for patients. Despite these benefits, laparoscopic surgery presents surgeons with challenges, primarily due to restricted instrument dexterity that makes it difficult to achieve primary closure of the hernia defect, in which the connective tissue layer is sutured closed, and leads to a bridged repair. In a bridged repair, the tissue reinforcement material spans a portion of the hernia defect without any connective tissue layer above it to provide additional reinforcement. This leads to increased risk of bulging of the material or hernia recurrence. Robotic-assisted hernia repair addresses this issue while still providing the benefits of a laparoscopic repair. In robotic-assisted repair, the surgeon enjoys greater instrument dexterity and precision, and is able to achieve primary closure of the hernia defect. This has contributed to a significant increase in the number of robotic-assisted hernia repairs over the last several years.
It is estimated that about 90% of hernia repairs today use a form of reconstruction material to provide long-term support at the repair site. Reconstruction materials include synthetic mesh, which can be either permanent or resorbable, and biologic matrices made from tissue material.
In October 2020, we surveyed a group of 71 surgeons to better understand their receptivity to natural repair solutions, their technique preferences across their hernia practice and their views on the risks associated with plastic mesh. Feedback was gathered across inguinal hernia, simple ventral, moderate-to-complex ventral and hiatal hernia repair. Included in the group were 43 general surgeons (61%), 19 plastic reconstructive surgeons (27%) and the remainder were colorectal and trauma surgeons. These surgeons indicated they believe there is a role for natural repair products across all hernia segments and they expect to increase their usage of those products in the next 24 months. Almost 60% of surgeons stated that they are aware of the risks associated with plastic mesh and reported approximately 20% of their hernia patients have voiced concern about the use of plastic mesh within the past 12 months.
In May 2023, we commissioned a consumer survey of 1,152 consumers on consumer awareness, preferences and doctor expectations regarding hernia repair options. The results of this survey indicated a preference for more natural hernia repair options (57%), particularly among those who have previously had a repair using permanent synthetic mesh (77%). The majority of respondents also expressed a reliance on primary care physicians and healthcare professionals for guidance, emphasizing the importance of shared decision-making.
Permanent Synthetic Mesh
Permanent synthetic mesh, the oldest category of hernia repair materials, is made of plastic materials that are also used in industrial and consumer products. These products have gained popularity with surgeons because they are relatively inert, can be readily sterilized, exhibit biomechanical strength and durability and are available at relatively low upfront cost. Limitations of permanent synthetic mesh products may include:
| ● | significant persistent foreign body inflammatory response that can result in encapsulation of the implant by fibrotic tissue or contraction of the mesh; |
11
| ● | chronic post-operative pain; |
| ● | scar tissue formation and lack of regeneration of soft-tissue; |
| ● | permanent susceptibility to mesh infection; |
| ● | significant cost associated with subsequent repairs or failed and infected mesh; |
| ● | compromised abdominal wall anatomy due to damaged and eroded tissue rendering subsequent surgical repairs challenging; and |
| ● | migration of the permanent synthetic mesh which can result in organ erosion or perforation. |
Many of these complications caused by permanent synthetic mesh require additional surgical intervention, including, explantation of the mesh or repair of hernia recurrence or of the abdominal wall. Based on longitudinal data from the Danish Hernia Database, in an analysis of approximately 2,900 patients who received a hernia repair using a permanent synthetic mesh, the observed rate of surgical intervention due to either recurrence or mesh-related complications at five years post operatively was approximately 17%. As a result of these complications and litigation involving these complications, the number of adverse events reported to the FDA for synthetic mesh hernia repairs has climbed from over 2,400 reported events in 2016, to over 21,000 in 2019, while remaining in excess of 8,000 reported events per year in each of 2023 and 2024. Synthetic mesh products have been the subject of a significant number of lawsuits over this time period, with approximately 15,000 cases outstanding in federal and state courts across the U.S. as of November 2024, and not inclusive of more than 40,000 cases that have been settled or dismissed in the prior three-year period.
Biologic Matrices
The complications associated with permanent synthetic mesh prompted the development of biologic matrices as a second category of hernia repair materials. Biologic matrices are derived from human or animal dermis, pericardium or intestinal submucosa, which allows them to become replaced entirely by the patient’s own tissue over time, a process known as remodeling. The goal behind these biologic materials was to lower the foreign body inflammatory response and biomechanical requirements of the repair, while providing a matrix upon which tissue remodeling could occur. Compared to permanent synthetic mesh, biologic matrices are less likely to induce this inflammatory response and become infected; however, they may have the following limitations:
| ● | lack strength or durability as compared to synthetic mesh products; |
| ● | prone to laxity and stretching; |
| ● | difficult to handle, leading to longer operating times as compared to synthetic mesh products; |
| ● | inability to be placed in a patient through a trocar in laparoscopic or robotic-assisted surgery; and |
| ● | considerably more expensive upfront costs than permanent synthetic mesh, typically limiting their use to complex hernia repairs or abdominal wall reconstructions. |
Though hernia recurrence occurs with the use of all types of soft-tissue reconstruction, biologic matrices have the highest rates of recurrence, partly due to common use in complex hernia repairs or abdominal wall reconstructions. The RICH study, a multicenter, prospective study sponsored by LifeCell Corporation (“LifeCell”) that evaluated the performance of Strattice, the industry leader for biological tissue matrices in complex abdominal wall reconstruction, in open ventral incisional hernia repair in contaminated abdominal wall defects, demonstrated post-operative hernia recurrence rates of 19% and 28% at 12-months and 24-months follow-up, respectively.
12
Resorbable Synthetic Mesh
Resorbable synthetic mesh, including biologically-derived synthetic mesh, was introduced as a third category of hernia repair materials and as an alternative to permanent synthetic mesh and biologic matrices. Resorbable synthetic mesh was designed with the intended benefits of full degradation over several months, a moderately lower cost than biologic matrices and gradual transfer of strength from synthetic mesh to native tissue over time. Resorbable synthetic mesh is polymer-based and does not include biologic material to promote tissue remodeling and healing. Despite improvements compared to the use of permanent synthetic mesh or biologic matrices, current limitations of resorbable synthetic mesh may include:
| ● | significant foreign body inflammatory response that can result in encapsulation or contraction of the mesh until resorbed; |
| ● | scar tissue formation and lack of remodeling of soft-tissue; |
| ● | mesh infection until resorbed; |
| ● | migration of the mesh until resorbed which can result in organ erosion or perforation; and |
| ● | lack of mid-term and long-term soft-tissue reinforcement as resorption progresses. |
Many of these complications can require additional surgical intervention including explantation of the resorbable synthetic mesh or repair of hernia recurrence or the abdominal wall. Data from a published, multicenter, prospective study sponsored by C.R. Bard, Inc. (now a subsidiary of Becton, Dickinson and Company) that evaluated the performance of Phasix, the current market-leading resorbable synthetic mesh, in CDC Class I, high risk ventral and incisional hernia repair, showed a post-operative hernia recurrence rate of 9% at 18-months follow-up and 18% at 36-month follow-up.
Current Materials Used in Plastic and Reconstructive Surgery and Their Limitations
Biologic matrices are most commonly used in plastic and reconstructive surgery, including surgery of the nose to change its shape or improve its function, referred to as rhinoplasty, lip augmentation, repair of perforations of cartilage and thin bone separating the nostrils, complex reconstruction of the oral and oropharynx cavities after oncologic resection, cleft palate repair, upper and lower eyelid reconstruction, scalp defects, and defects of the fibrous membrane covering the brain and spinal cord, called the dura, because of their ability to define shape and position, improve tissue quality, reinforce existing soft-tissue and reduce the rate of complications associated with a foreign body inflammatory response, however they are prone to excessive stretching over time and difficult for surgeons to handle. These limitations may lead to undesirable results requiring additional surgical intervention. Additionally, biologic matrices are typically expensive to source.
Our Solution
We have created a new category of reinforced tissue matrices that were purposefully designed in close collaboration with more than 100 surgeons to address the unmet clinical needs in soft-tissue reconstruction. Our portfolio of products, generally designed with over approximately 95% biologic material, combines the benefits of both biologic and polymer materials while addressing their limitations by interweaving polymer fibers through layers of a minimally-processed biologic material. These products are priced competitively and designed for use with a range of surgical techniques, allowing the benefits of an advanced biologic repair to be available to more patients for use in accordance with the products’ 510(k) clearances and instructions for use.
The biologic material serves as the natural building block from which we can fabricate devices that meet specific clinical and surgical handling requirements. This material consists of an intact, minimally-processed extracellular matrix derived from ovine rumen, which is the forestomach of a sheep. Polymer fibers are interwoven through the layers of biologic material in unique embroidered patterns and contribute to approximately 5% of the overall device by mass.
13
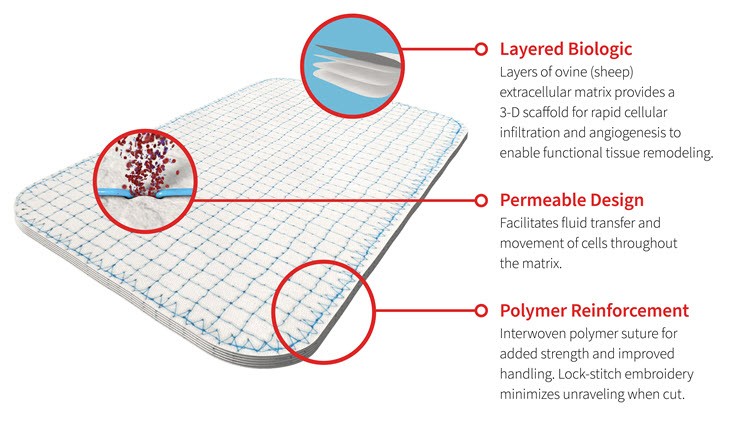
The interwoven polymer utilized can be either permanent, made from polypropylene, or resorbable, made from polyglycolic acid (“PGA”) or polylactic-co-glycolic acid (“PLGA”). The embroidering pattern varies between our OviTex and OviTex PRS products to impart different biomechanical properties tailored for their respective intended clinical applications. Our OviTex products are designed with a lockstitch embroidery pattern that is sewn in a grid pattern to minimize unraveling (when cut). Our OviTex PRS products are designed with a patented corner-lock stitch pattern designed to resist deformation and to control the degree and direction of stretching of the product.
Our capabilities in polymer science, biologics, textile engineering and analytical testing enable us to quickly design innovative products for development and manufacture. These competencies also allow our technical team to tailor the degree of stretch, direction of stretch, overall strength, handling properties, permeability, thickness, texture, size and shape of each reinforced tissue matrix to suit the needs of particular clinical applications and surgical techniques. This expertise has been utilized in the development of our OviTex and OviTex PRS products, including our OviTex LPR and OviTex IHR configurations and is currently being leveraged in the development of our additional configurations within product pipelines seeking to enhance product features for various applications within our indications.
Our reinforced tissue matrices are designed to improve the outcomes of soft-tissue reconstructions by reinforcing tissue while allowing rapid tissue integration, revascularization and biomechanical control. In addition to overall strength, a key property that we engineer into our products is the degree to which they stretch, which we refer to as compliance. Each of our products is designed to exhibit a degree of compliance appropriate for its intended clinical application.
The graphics below illustrate the key features of our OviTex and OviTex PRS products:
OviTex
14
OviTex PRS
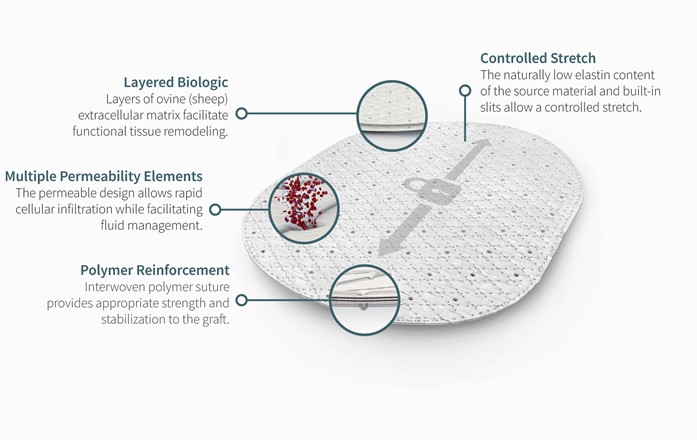
We believe the principal benefits of our reinforced tissue matrices are:
| ● | Reduced foreign body inflammatory response. The biologic material utilized in our reinforced tissue matrices is designed to minimize the body’s inflammatory response to the device. Our unique embroidered patterns create a macroporous grid within the biologic material. In our non-human primate study in which we compared our OviTex products to several commercially available synthetic mesh and biologic matrix products, at 24 weeks, our OviTex products demonstrated a minimal foreign body inflammatory response similar to that of biologic matrices, and less foreign body inflammatory response than all of the synthetic mesh tested. |
| ● | Enhanced remodeling of soft-tissue and rate of healing. Our reinforced tissue matrices are constructed to provide increased surface area and permeability, allowing for rapid absorption of wound fluids and blood during implantation and enabling oxygen supply, cellular infiltration, migration, and repopulation for revascularization and functional tissue remodeling during healing. In our non-human primate comparative study, at 24 weeks the pattern of collagen formation in our OviTex products resembled connective tissue as opposed to the random fibers typical of scar tissue that were seen adjacent to the synthetic mesh. By contrast, the synthetic mesh showed no signs of remodeling of soft-tissue and exhibited a high level of mesh contraction. |
| ● | Highly engineered biomechanical properties supported by clinical evidence. Our reinforced tissue matrices are reinforced with interwoven polymer fibers to provide mid-term and long-term support. The interwoven polymer increases the strength of our OviTex products by approximately 25% compared to the biologic material alone. When tensile forces are applied, this design allows for load sharing between the biologic material and the polymer during the remodeling process. Data from our strength testing demonstrated that our OviTex products meet or exceed that of published data from market-leading permanent and resorbable synthetic mesh. In our BRAVO study, the recurrence rate at the 24-month time point was 2.6%, and SSOs were observed in 38% of the study population. Of the enrolled patients, 78% |
15
| were characterized as high risk for experiencing an SSO based on at least one known risk factor, which included obesity, active smoking, COPD, diabetes mellitus, coronary artery disease, or advanced age (≥75 years). We believe based on a review of available literature that the BRAVO recurrence rate is among the lowest reported rate in any published study, including our biologic or resorbable synthetic mesh competitors evaluating product use in this procedural setting and with a similar cohort of high-risk patients. The addition of polymer to our reinforced tissue matrices allows each product to maintain its physiologic compliance properties, while resisting stretching and elongation. In our non-human primate comparative study, our OviTex devices best preserved their original shape, experiencing less contraction compared to biologic and synthetic mesh. |
| ● | Enhanced surgeon handling and satisfaction. Each of our embroidery patterns was designed specifically to allow the surgeon to trim and shape the product while minimizing the potential for unraveling of the polymer. Based upon our survey of approximately 50 surgeons, our OviTex products conform readily to the contours of surgical sites and are easy to handle, trim, suture and tack in all surgical approaches. In addition, in our BRAVO study, 32 of the 92 enrolled subjects received minimally invasive surgery, of whom 12 received laparoscopic repair and 20 received robotic repair. Of the surgeons who performed minimally invasive surgery, all reported at the time of surgery that the product was easy or very easy to place. The average surgeon satisfaction with the product was 9.7/10 at 30 days for the minimally invasive cohort and remained consistent over 24 months of follow-up. We are also actively enrolling patients in our BRAVO II study, a prospective study evaluating robot-assisted ventral and inguinal hernia repairs with OviTex, including our OviTex LPR, OviTex Core Permanent and OviTex 1S Permanent configurations. |
| ● | Lower upfront cost products. Our reinforced tissue matrices provide our customers with meaningful cost savings over leading competitive products across a range of clinical uses so that more patients can experience the benefits of an advanced biologic repair solution. We price our OviTex products competitively, and on average, our customers realize 20% to 40% cost savings over leading biologic matrices and resorbable synthetic mesh. Our OviTex PRS portfolio is priced below leading biologic matrices. |
Our Strengths
We are focused on developing and commercializing a new category of reinforced tissue matrix for surgeons and patients that aim to address the shortcomings of existing products. We believe the following strengths will allow us to build our business and potentially increase our market penetration:
| ● | Innovative and broad portfolio of products. Our OviTex and OviTex PRS products are the only FDA-cleared products to incorporate polymer fibers interwoven through layers of biologic material in a lockstitch pattern creating an embroidered construction. The biologic matrix is derived from ovine rumen and utilizes a patented process to create a reinforced tissue matrix that is optimized for soft-tissue reconstruction. Our OviTex and OviTex PRS products are available in resorbable and permanent polymer versions in a variety of configurations and sizes. For example, our OviTex devices are currently available in sizes ranging from 4 × 8 cm to 25 × 40 cm, and our OviTex LPR devices are designed with specific thickness, handling properties and shapes optimized for use in laparoscopic and robotic-assisted surgery. |
| ● | Disruptive technology supported by compelling pre-clinical and clinical evidence. OviTex product technology is supported by extensive pre-clinical research, including bench testing, in-vitro and in-vivo studies. These studies have demonstrated appropriate physiologic strength for the repair, compliance within the physiologic range of the human abdominal wall, retention of extracellular matrix proteins which may aid in tissue remodeling and porosity and permeability to promote fluid transfer. Our in-vivo non-human primate data demonstrated that use of our OviTex products resulted in more rapid tissue integration and revascularization compared to pure biologic matrices, as well as lower inflammatory response and better functional tissue remodeling compared to permanent and resorbable synthetic mesh. This preclinical data is supported by our compelling clinical evidence showing the safety and efficacy of our OviTex products in published data on over 1,200 hernia patients. |
16
| ● | Long-term supply agreement that provides pricing flexibility. Our Aroa License provides for the exclusive supply of ovine rumen and manufacture of our OviTex and OviTex PRS products, which gives us a low and fixed cost of raw materials. We purchase product from Aroa at a fixed transfer cost as a percentage of Aroa’s cost of goods sold, and subject to a true-up adjustment, resulting in an amount equal to 27% of our net sales of our OviTex and OviTex PRS products, with the exception of OviTex IHR product configurations, for which we pay the greater of the initial fixed transfer cost or 27% of our net sales of OviTex IHR. |
| ● | Potential cost savings to healthcare systems and hospitals. Our pricing flexibility allows us to sell our OviTex and OviTex PRS products to hospitals and healthcare systems at prices substantially below competitive products based on national average competitive pricing. Our OviTex products are sold at prices approximately 20% to 40% lower than other biologic matrices and resorbable synthetic mesh. We believe our pricing flexibility will continue to drive greater adoption of our products. Our OviTex PRS products are priced below leading biologic matrices, and as we further commercialize our OviTex PRS portfolio, we anticipate that our customers will realize cost savings over biologic matrices based on national average competitive pricing. We believe that the average selling prices across our products will provide financial benefits to our customers in addition to improving clinical outcomes. |
| ● | Established reimbursement pathway for hernia repair. The implantation of biologic matrices and synthetic mesh for hernia repair is coded using an established fixed procedure payment system known as a MS-DRG that consists of a lump sum payment rate that varies based on the degree of complications and comorbidities of each hernia. In addition, surgeons receive payment for their services depending on the coding associated with the procedure. The MS-DRG-based reimbursement system encourages hospitals to become more efficient in treating patients due to its fixed per-patient reimbursement nature. |
| ● | Broad intellectual property portfolio. Our products are covered by intellectual property that broadly covers changing a biologic matrix’s biomechanical properties by interweaving a polymer thread through the biologic matrix. Specifically, our patents claim the ability to tailor stretch resistance. The ability to predictably control the biomechanical properties of a biologic matrix is the cornerstone of our product portfolio. Our intellectual property also covers the development of extracellular matrix derived from ovine rumen, methods for isolating these scaffolds from ovine rumen, layering multiple sheets of these ovine rumen matrices together, sewing in an anti-adhesive layer into a matrix, and adding unique patterns sewn or embroidered into these matrices using different polymers to impart reinforcing strength. Our portfolio also includes patents covering implants with gripping strands, and implants with multivesicular liposomes that may be used to deliver drugs. Through the Aroa License and our issued or allowed patents and patent applications, we have a broad portfolio of intellectual property that is leveraged in all of our reinforced tissue matrix products. In addition, we believe that the trade secrets developed with Aroa create additional barriers to entry. |
| ● | Highly accomplished executive team with proven track record. Our executive team consists of seasoned medical device professionals with deep industry experience, and a broad network of relationships within the industry and the medical community. Our executive team has led and managed companies through significant growth and introduction and commercialization of multiple new products, including driving surgeon adoption of biologic and biosurgery technologies. Members of our team have held leading positions with medical technology companies such as Orthovita Inc., Stryker Corporation, OraSure Technologies, Inc., LifeCell and Medtronic plc. We believe this team is well-positioned to lead us through the commercial expansion of our products and development and launch of future products. |
17
Our Growth Strategy
Our goal is to become the leading provider of soft-tissue reconstruction products. The key elements of our strategy include:
| ● | Successfully deploy our U.S. commercial organization to support our growth. We primarily sell our products through a single direct sales organization in the U.S. As of December 31, 2024, we had 75 sales territories in the U.S. which are supported by 133 employees in our U.S. based commercial organization. We plan to hire additional territory managers and field-based support employees to support and service new accounts for soft-tissue reconstruction procedures. We believe we can also enhance the productivity of our sales force by improving customer segmentation and targeting, implementing and further refining our proprietary training programs, leveraging support from our medical education and medical affairs functions to drive physician awareness and education on our products, and utilizing engagement analytics to support product development. |
| ● | Promote awareness of our products to drive surgeon use. We educate surgeons regarding the value proposition of our products through presentations and exhibits at industry conferences, medical education symposia, direct training and education, webinars and publishing additional clinical data demonstrating the benefits of our products and establishing online peer-to-peer communities. We plan to continue to drive awareness of our products through in-person and virtual versions of these programs, while expanding their geographic reach and increasing the number of surgeon interactions. We will continue to increase our digital marketing efforts as well to build brand awareness with event marketing engagement, targeted ads and emails, various social media efforts and patient education and outreach efforts. |
| ● | Drive utilization through existing GPO and IDN contracts and secure additional contracts. We are focused on partnering with our existing GPO- and IDN-contracted customers to promote implementation of our contracts, increase our access to surgeon customers, broaden awareness of products and our economic messaging and help drive utilization of our products within associated hospitals and healthcare systems. To date, we have contracted with three national GPOs covering our OviTex and OviTex PRS products. In addition, we continue to pursue contracts with additional GPOs and IDNs. GPO and IDN contracts enable greater access to geographies with high procedural volumes and provide prioritized status within hospital procurement systems. |
| ● | Continue to build upon clinical evidence of the effectiveness and safety of our products. We are committed to evidence-based medicine and investing in clinical data to support the use of our products. In our BRAVO study, the recurrence rate at the 24-month time point was 2.6%, and SSOs were observed in 38% of the study population. 78% of all enrolled patients were characterized as high risk for experiencing an SSO based on at least one known risk factor, which included obesity, active smoking, COPD, diabetes mellitus, coronary artery disease, or advanced age (≥75 years). Our analysis of patients in the BRAVO study reaching 24-month follow-up was published in the Annals of Medicine and Surgery in October 2022. We have begun our next post-market prospective study, BRAVO II, which evaluates OviTex LPR, OviTex Core Permanent and OviTex 1S Permanent in the robotic repair of ventral and inguinal hernias over 24 months. With respect to OviTex PRS, in addition to independent, third-party publications evaluating the use of the product in various soft tissue applications, we also continue to enroll patients in our OPERA study, a retrospective-prospective trial evaluating the safety profile of OviTex PRS in previous pre-pectoral and sub-pectoral implant-based breast reconstructions. Following receipt of our investigational device exemption application in October 2024 relating to the study of the safety and effectiveness of our OviTex PRS product in implant-based breast reconstruction, we continue to evaluate and finalize the clinical study protocol to eventually support a pre-market application to obtain approval for an indication for OviTex PRS for use in breast reconstruction. |
| ● | Advance our portfolio of reinforced tissue matrices with the introduction of new product features and designs. We plan to continue to expand our product offerings and the treatment capabilities of our products to address a broader patient base within soft-tissue reconstruction. As we innovate and develop our |
18
| products, the new features and improved surgical techniques expand the clinical applications for soft-tissue reinforcement. Areas of focus include enhanced surgical handling, larger product configurations, increased permeability, and longer-acting resorbable polymers. Improving the surgical handling and implementation of our devices benefits both the clinician and patient. We believe that increasing the size of our product configurations will support utilization in new surgical applications or with certain patient populations. Increasing product permeability encourages a more-natural healing response. Longer-acting polymers can provide additional support for patients that need more time to heal. We believe these technology enhancements will continue to bolster our portfolio and expand the successful use of our products. |
| ● | Expand our service offerings and diversify our supplier base to create a broader soft tissue preservation and restoration portfolio. We plan to continue assessing internal development strategies and strategic partnerships with medical device companies whereby we may enter into distribution, product development and/or licensing agreements for new soft tissue preservation and restoration products complimentary to, or related to, existing and future products in our distribution channel. For example, in September 2023, we entered into a distribution agreement with Advanced Medical Solutions Limited, a company registered in England, to be their exclusive distributor of certain hernia mesh fixation devices in the U.S. In March 2024, we announced the full commercial launch of the LiquiFix Hernia Mesh Fixation Devices (LiquiFix FIX8™ and LiquiFix Precision™) in the U.S. Similarly, we continue to evaluate additional product opportunities that address patient health and unmet needs within the indications in which we operate. |
Our Products
Our Technology Platform
Our advanced reinforced tissue matrix technology consists of multiple layers of minimally-processed, decellularized extracellular matrix derived from ovine rumen with interwoven polymer fibers in a unique embroidered pattern. The extracellular matrix is the collagen component of the rumen that is retained following removal of the epithelium, muscle and cellular content, and has an optimal biomechanical profile and open collagen architecture that allows for rapid cellular infiltration. These thin, strong layers of ovine rumen are plentiful in supply and serve as building blocks from which we can construct multilayered devices to customize products to adapt to clinical needs and surgeon preferences. The layers of extracellular matrix provide a high degree of surface area for tissue remodeling. We strengthen these reinforced tissue matrix layers with interwoven polymers, that are either permanent (polypropylene), or resorbable (PGA or PLGA). These polymers were selected because they are well characterized suture materials with a history of significant clinical use and recognized safety profiles. Polypropylene has a high tensile strength and a low inflammatory response in small quantities. PGA is the fastest resorbing polymer and within three months it tends to be fully absorbed into the body, whereas using PLGA in our products provides a slower absorption option of approximately six months.
Our highly specialized and customizable textile engineering capability allows us to tailor the degree and direction of stretch, overall strength, handling properties, permeability, thickness, texture, size and shape of each reinforced tissue matrix to suit the needs of particular clinical applications and surgical techniques. Our textile engineering utilizes a computer-controlled fabrication method that is scalable, reproducible, efficient and customizable. This embroidery process creates hundreds of micro-channels to allow the multi-directional passage of the patients’ native cells and fluids throughout the product. The interwoven polymers are embroidered using a lockstitch pattern, which allows for the device to be trimmed while minimizing unraveling (when cut), and we use a patented corner-lock pattern, which creates a stable polymer fabric within the biologic material. We manipulate the polymer thread patterns to control the degree and stretch of our products. Denser grid patterns increase the amount of reinforcement and less dense patterns of different geometry allow for greater stretch. We are also able to manufacture products with smooth external layers that minimize the amount of exposed polymer such that the product can be placed in contact with the viscera.
OviTex Reinforced Tissue Matrix
Our OviTex Reinforced Tissue Matrix has received 510(k) clearance from the FDA, which clearance was obtained and is currently held by Aroa and is intended for use as a surgical mesh to reinforce and/or repair soft-tissue where weakness exists. Indications for use include the repair of hernias and/or abdominal wall defects that require the use of reinforcing or bridging material to obtain the desired surgical outcome.
19
Our OviTex products can be used in a variety of hernia repairs, including simple and complex ventral, inguinal and hiatal hernias, as well as abdominal wall reconstructions.
Our OviTex products are sterile reinforced tissue matrices derived from ovine rumen with either polypropylene or PGA. The product is provided in a dry, hydratable form and packaged in a double pouched configuration. The product can be stored at room temperature and only needs five minutes of rehydration for use. To be used in surgery our OviTex product is placed in a sterile dish, rehydrated with sterile saline for five minutes, trimmed to fit the site, if needed, and then positioned to achieve maximum contact between the device and the surrounding tissue. The device may be sutured, stapled or tacked into place.
All of our OviTex products were designed to minimize the amount of polymer material implanted in patients. The synthetic material in our OviTex products comprise less than 5% of our final product or approximately 12% in our OviTex LPR devices or approximately 15% in our OviTex IHR devices. Depending on the configuration selected, the amount of polymer is approximately 75% less than the polymer content of the most widely implanted permanent synthetic mesh, thereby reducing the patient’s foreign body inflammatory response to the polymer.
We market a variety of OviTex products in a range of sizes, thicknesses and degrees of reinforcement in order to suit surgeon preference and desired surgical technique. Our OviTex portfolio is designed to allow surgeons to select a device appropriate for any abdominal tissue plane. Generally, surgeons may place the reinforced tissue matrix in direct contact with internal organs, known as intraperitoneal placement, or away from these internal organs in a variety of tissue planes, known as pre-peritoneal placement. When selecting a product for intraperitoneal placement, surgeons require a surface that minimizes the risk of tissue attachment, whereas when selecting a product for pre-peritoneal placement, surgeons are able to use a product with polymer exposure on both sides. Surgeons may select the most appropriate product from our OviTex portfolio based on the size of the defect, necessity or surgeon preference for internal organ contact, use of a minimally invasive or open surgical technique and risk of infection.
OviTex Configurations for Laparoscopic and Robotic Procedures
Our OviTex LPR product was specifically designed for use in laparoscopic and robotic-assisted hernia surgical repairs. OviTex LPR was designed for use with a trocar and requires the same rehydration and fixation as our other OviTex products. This product includes design elements to improve surgical handling, including two extra embroidered lines of blue colored polypropylene fibers (ellipse shapes) to enhance endoscopic orientation and alignment. This product can be introduced into the patient’s body through various sized trocar ports. Based on surgeon feedback, OviTex LPR was designed in an elliptical or circular shape to minimize trimming.
Our OviTex IHR product was specifically designed for use in laparoscopic and robotic-assisted inguinal hernia repair and is available in anatomical and rectangle shapes.
20
OviTex Portfolio
|
|
OviTex |
|
OviTex 1S |
|
OviTex 2S |
|
OviTex LPR |
|
OviTex IHR |
|
|
|
|
|
|
|
|
|
|
|
Size and Shape |
|
4x8 cm to 25x40 cm* (Rectangle or Square) |
|
4x8 cm to 25x40 cm* (Rectangle or Square) |
|
4x8 cm to 25x40 cm* (Rectangle or Square) |
|
12x18 cm to 15x25 cm* (Ellipse); 9cm to 15cm (Round) |
|
10x17 cm (Anatomical); 13x17 cm (Rectangle) |
|
|
|
|
|
|
|
|
|
|
|
Strength |
|
+ |
|
++ |
|
+++ |
|
+ |
|
+ |
|
|
|
|
|
|
|
|
|
|
|
Layers of Ovine Rumen |
|
Four |
|
Six |
|
Eight |
|
Four |
|
Three or Four (anatomical); Three (Rectangle) |
|
|
|
|
|
|
|
|
|
|
|
Common Procedures |
|
Moderate ventral hernia (pre-peritoneal placement), inguinal hernia, hiatal hernia |
|
Moderate to complex ventral hernia, can be placed intraperitoneally |
|
Complex ventral hernia and abdominal wall reconstruction and can be used for bridging, can be placed intraperitoneally |
|
Laparoscopic or Robotic-assisted surgery |
|
Laparoscopic or Robotic-assisted inguinal hernia repair |
|
|
|
|
|
|
|
|
|
|
|
Polymer |
|
Resorbable (PGA) or Permanent (Polypropylene) |
|
Resorbable (PGA) or Permanent (Polypropylene) |
|
Resorbable (PGA) or Permanent (Polypropylene) |
|
Permanent (Polypropylene) |
|
Permanent (Polypropylene) |
|
|
|
|
|
|
|
|
|
|
|
Shelf Life |
|
Resorbable‑18 months |
|
Resorbable‑18 months |
|
Resorbable‑18 months |
|
36 months |
|
36 months |
|
|
|
|
|
|
|
|
|
|
|
Configuration |
|
Exposed polymer on both sides |
|
Exposed polymer on one side, and one smooth side |
|
Two smooth sides |
|
Exposed polymer on one side, and one smooth side |
|
Exposed polymer on both sides |
|
|
|
|
|
|
|
|
|
|
|
Commercial Availability |
|
U.S. |
|
U.S. |
|
U.S. |
|
U.S. |
|
U.S. |
*25 x 30 cm and 25 x 40 cm sizes currently only available with permanent (polypropylene) polymer.
+Denotes relative level of strength.
OviTex Plastic and Reconstructive Surgery — OviTex PRS
OviTex PRS, has received 510(k) clearance from the FDA, which clearance was obtained by Aroa and is held by us, and is indicated for use in implantation to reinforce soft-tissue where weakness exists in patients requiring soft-tissue repair or reinforcement in plastic and reconstructive surgery. In March 2023, we received an additional 510(k) clearance, which expands the OviTex PRS portfolio to include OviTex PRS Long-Term Resorbable. Our OviTex PRS portfolio can be stored at room temperature and comes in the same packaging and requires the same rehydration and fixation as our OviTex products.
Our OviTex PRS portfolio is a sterile reconstructive reinforced tissue matrix that comes in three different options. The short-term resorbable and permanent PRS options are composed of two or three layers of ovine rumen joined by a patented corner-lock embroidered diamond patterned polymer (PGA or polypropylene) that allows the product to stretch uni-directionally while also maintaining its shape. Machine punched regularly spaced fenestrations, or holes and die-cut slits in the product facilitate fluid management, allow for rapid cellular infiltration and create a directional bias to the stretch. The third option, the long-term resorbable PRS, provides bi-directional stretch and longer resorption profile utilizing PLGA. Our OviTex PRS product is available in arced rectangle, contour and oval shapes in a range of sizes (4 × 16 cm through 20 × 25 cm) to suit surgeon preference and nature of the soft-tissue repair in plastic and reconstructive surgery.
21
The device may be trimmed to a desired shape to further accommodate individual anatomy. The current shelf life of permanent OviTex PRS is 36 months, the current shelf life of short-term resorbable OviTex PRS is 12 months and the current shelf life of the long-term resorbable OviTex PRS is 18 months.
OviTex PRS

Product Pipeline and Research and Development
We continue to advance our product pipeline to broaden our treatment capabilities for soft-tissue reinforcement. As we innovate and develop our products, the new features and improved surgical techniques expand the clinical applications for soft-tissue reinforcement. Areas of focus include enhanced surgical handling, larger product configurations, increased permeability, and longer-acting resorbable polymers. Improving the surgical handling and implementation of our devices benefits both the clinician and patient. We believe that increasing the size of our product configurations will support utilization in new surgical applications or with certain patient populations. Increasing product permeability encourages a more-natural healing response. Longer-acting polymers can provide additional support for patients that need more time to heal. In addition, we continue to explore the development of lower-cost, higher margin resorbable polymer-based devices targeting our current indications. We believe these technology enhancements and new product alternatives will continue to bolster our portfolio and expand the successful use of our products across a variety of soft-tissue surgical applications.
Scientific Evidence
Overview of Preclinical and Clinical Programs
One of our key strategies is to continuously obtain evidence to support the safety and effectiveness of our products, which we believe will differentiate us from our competitors. As part of our strategy to gather and analyze high-quality data, we seek to ensure rigorous and reliable data collection and reporting. The data from our preclinical and clinical studies strengthens our ability to raise surgeon awareness and drive adoption of our products as a new category of soft-tissue reconstruction products. We expect our clinical evidence will provide surgeons with safety and efficacy data on the appropriate use of our products and we plan to obtain further clinical evidence to support additional regulatory clearances or approvals of our reinforced tissue matrices for additional indications for use in the future.
22
Preclinical Programs
Our pre-clinical program is paramount in the design of our products. Our program starts with bench performance characterization to ensure proper strength and compliance for the indication, followed by in-vitro and in-vivo studies to ensure proper biological performance to help promote remodeling of the repair site. We have developed an extensive pre-clinical research library on our devices, as well as on competitor devices. We continue to evaluate new and existing technologies for safety and biocompatibility as part of our product development process.
We believe we have completed the largest collection of non-human primate preclinical studies conducted in soft-tissue reconstruction surgery. In these studies, we compared our OviTex and OviTex PRS products to market leading competitive materials. The results showed our reinforced tissue matrices exhibited a minimal inflammatory response, rapid cellular infiltration and revascularization and demonstrated early and complete remodeling into functional tissue. The OviTex results have been published in the peer-reviewed journal Hernia (https://doi.org/10.1007/s10029-019-02119-z). The OviTex PRS results have been published in the peer-reviewed journal ePlasty (ePlasty 2022;22:e43).
Clinical Programs
We are committed to obtaining evidence to support the safety and efficacy of our products across their indications. Clinical data has been published on over 1,200 patients treated with OviTex in ventral hernia, inguinal hernia, hiatal hernia, and abdominal wall reconstruction. As part of our clinical research program, we have developed two post-market studies, BRAVO and BRAVO II. This commitment to generating clinical data through controlled prospective studies with 24-month follow-up will allow us to understand the short- and long-term benefits of using OviTex in hernia repair.
In October 2022, the 24-month results of our BRAVO study were published in the Annals of Medicine and Surgery. The BRAVO study was designed to evaluate the clinical performance of OviTex for primary or recurrent ventral hernias using open, laparoscopic, or robotic techniques in 92 enrolled patients. The recurrence rate at the 24-month time point was 2.6%, and SSOs were observed in 38% of the study population. 78% of all enrolled patients were characterized as high risk for experiencing an SSO based on at least one known risk factor, which included obesity, active smoking, COPD, diabetes mellitus, coronary artery disease, or advanced age (≥75 years). The results also indicated that BRAVO patients experienced statistically significant and clinically meaningful improvements in their quality of life and perceived health.
Surgeons continue to use our OviTex PRS reinforced tissue matrices in their surgeries and, in addition to a potential IDE study, we have also commenced our OPERA study, a retrospective-prospective trial evaluating the safety profile of OviTex PRS in previous pre-pectoral and sub-pectoral implant-based breast reconstructions.
Intellectual Property
Our success depends in part on our ability to obtain, maintain, protect and enforce our proprietary technology and intellectual property rights, in particular, our patent and trademark rights, preserving the confidentiality of our trade secrets, and operating without infringing the valid and enforceable patents and other proprietary rights of third parties. We rely on a combination of patent, trademark, trade secret and other intellectual property rights and measures to protect the intellectual property rights that we consider important to our business. We also rely on know-how and continuing technological innovation to develop and maintain our competitive position.
Aroa License
In August 2012, we entered into the Aroa License, which was amended and restated in July 2015, pursuant to which we obtained an exclusive license to certain patents and know-how to develop, commercialize and sell bovine and ovine extracellular matrix products for hernia repair, abdominal wall and breast reconstruction in North America and Europe, which we refer to as the Licensed Territory. In addition, under the Aroa License, Aroa is our exclusive manufacturer and supplier for the development of our bovine and ovine extracellular matrix products.
23
Pursuant to the terms of the Aroa License, we made upfront payments to Aroa totaling $2.3 million and granted Aroa 74,316 newly issued shares of our restricted common stock. We have made additional payments in the aggregate of $2.0 million to Aroa following the achievement of certain regulatory and operational milestones, including FDA 510(k) clearance of our OviTex products, which clearance was obtained and is currently held by Aroa, for use in surgical soft-tissue reinforcement and the receipt of the first CE mark for sale of our products in the European Economic Area for use in abdominal wall reconstruction and hernia repair and our acceptance of certain supply quantities manufactured by Aroa for our commercial launch in Europe. In addition, we paid Aroa $4.0 million in revenue-based milestone payments upon our achievement of certain net sales thresholds for sales of our products within the Licensed Territory. We have satisfied all milestone payment obligations under the Aroa License.
We are responsible for commercializing the products manufactured for us by Aroa. We pay Aroa for the supply and manufacturing of our products through a revenue sharing agreement. Pursuant to the Aroa License, we purchase product from Aroa at a fixed transfer cost as a percentage of Aroa’s cost of goods, and subject to a true-up adjustment, resulting in an amount equal to 27% of our net sales of our OviTex and OviTex PRS products, with the exception of OviTex IHR product configurations, for which we pay the greater of the initial fixed transfer cost or 27% of our net sales of OviTex IHR. If at any point during the term of the Aroa License we and Aroa determine that our anticipated product needs exceed Aroa’s manufacturing capabilities, we and Aroa will mutually approve an expansion and equally share the cost of such expansion. Our share of such expansion costs may be offset by us against future revenue share payments.
The initial term of the Aroa License terminates on the expiration of the last patent covering the OviTex and OviTex PRS products, currently March 9, 2031, with an option to extend for an additional ten-year period. Either party may terminate the Aroa License upon the other party’s material breach, subject to a ninety-day notice and cure period or upon thirty-days written notice in the event of bankruptcy. We may terminate manufacture and production of a specific product upon thirty-days prior written notice upon (i) a reasonable determination that such product infringes the intellectual property rights of a third party, (ii) an uncured supply failure by Aroa or (iii) such product proves unfeasible, and immediately upon written notice from a regulatory authority that such product must be withdrawn from the market. If we materially breach the Aroa License in one of the Licensed Territories, Aroa may terminate the Aroa License solely with respect to the Licensed Territory in which the breach occurred. Upon termination of the Aroa License, we have the right to purchase all or any part of the unsold portion of any completed products from Aroa and the right to continue to sell all products remaining in our inventory.
The Aroa License also contains customary representations and warranties, confidentiality, insurance, audit, indemnification and non-competition provisions.
Patents
As of December 31, 2024, we exclusively license two issued U.S. patents that will expire in 2029 and 2031. We own twenty-three U.S. issued or allowed patents which will expire between 2035 and 2041 and twelve pending U.S. patent applications, which subject to issuance, are projected to expire between 2035 and 2045, without taking into account potential patent term extensions or adjustments. In addition to our U.S. intellectual property, we also own eight issued non-U.S. patents and seven pending non-U.S. patent applications, including six applications under the Patent Cooperation Treaty (“PCT”), which, subject to issuance, would be projected to expire between 2036 and 2044 and have exclusively licensed issued patents in Europe and Canada that will expire in 2029.
Our patents and patent applications cover, among other things, our corner-lock embroidery pattern, the use of adhesion barriers sewn into soft-tissue and compliance associated with stretching.
Although the term of individual patents varies depending upon the country in which they were granted, in most countries, including the U.S., the patent term is 20 years from the earliest claimed filing date of a non-provisional patent application in the applicable country. In the U.S., a patent’s term may, in certain cases, be lengthened by patent term adjustment, which compensates a patentee for administrative delays by the U.S. Patent and Trademark Office in examining and granting a patent, or may be shortened if a patent is terminally disclaimed over a commonly owned patent or a patent naming a common inventor and having an earlier expiration date.
24
We cannot be sure that our pending patent applications that we have filed or may file in the future will result in issued patents, and we can give no assurance that any patents that have been issued or might issue in the future will protect our current or future products, will provide us with any competitive advantage, and will not be challenged, invalidated, or circumvented.
Trade Secrets
We seek to protect our proprietary rights through a variety of methods, including confidentiality agreements and proprietary information agreements with suppliers, employees, consultants and others who may have access to our proprietary information. However, trade secrets and proprietary information can be difficult to protect. While we have confidence in the measures we take to protect and preserve our trade secrets and proprietary information, such measures can be breached, and we may not have adequate remedies for any such breach. In addition, our trade secrets and proprietary information may otherwise become known or be independently discovered by competitors.
Trademarks
We also rely on trademarks and trade designs to develop and maintain our competitive position. TELA Bio®, OviTex®, the TELA Bio logo OviTex®, Minimize the Foreign Body Footprint®, and A More Natural Hernia Repair® are registered trademarks of ours in the U.S. and TELA Bio® and OviTex® are registered trademarks in the foreign jurisdictions in which we conduct our business.
For more information regarding the risks related to our intellectual property, please see the section titled “Risk Factors — Risks Related to Intellectual Property Matters.”
Research and Development
We invest in research and development to advance our reinforced tissue matrix products and to develop complimentary soft tissue preservation and restoration products, with the goal of improving upon and supplementing our existing product offerings. We believe our ability to rapidly develop new products and product configurations is attributable to the dynamic product innovation process that we have implemented, the versatility and leveragability of our core technology and the management philosophy behind that process. We have recruited and retained engineers and scientists with significant experience in the development of polymer science, biologics, textile engineering and analytical testing. We have a number of design improvements for our reinforced tissue matrices in various stages of development that are expected to enhance our current products and increase surgeon adoption of our products. In October 2024, we received approval from the FDA for our investigational device exemption application relating to the study of the safety and effectiveness of our OviTex PRS product in implant-based breast reconstruction. We continue to evaluate and finalize the clinical study protocol and anticipate additional FDA interactions related to such to support a pre-market application to obtain approval for an indication for OviTex PRS for use in breast reconstruction. Our research and development efforts are based at our facility in Malvern, Pennsylvania.
Commercial Strategy
Our commercial efforts are predominantly focused on the U.S. market where we have established strong relationships with key constituencies, including hospitals, ambulatory surgery centers, GPOs, IDN, third-party payors and other key clinical and economic decision makers by offering a unique high quality, cost-effective product. As part of our overall commercial strategy, we intend to contract with GPOs and IDNs to increase access and penetration with hospital accounts. To date, we have contracted with three national GPOs for coverage of our OviTex and OviTex PRS products. We have invested in our direct sales and marketing infrastructure in order to expand our presence to promote awareness and adoption of our products.
We market our products to hospitals, ambulatory surgery centers, surgeons, GPOs, IDNs and medical device supply chain participants primarily through our direct sales force. Our sales representatives and sales managers have substantial medical device experience. As of December 31, 2024, we had 133 employees in our U.S. based commercial organization in 75 sales territories, which includes sales management, territory managers, marketing and administrative and field-based support staff.
25
We plan to hire additional territory managers and field-based support employees to support and service new accounts for soft-tissue reconstruction procedures.
Manufacturing
The majority of our raw materials are sourced through and manufactured by Aroa in their Auckland, New Zealand facility under the terms of the Aroa License. Aroa’s facility is approximately 40,000 square feet of which approximately 25,000 square feet is dedicated to manufacturing, including an additional 15,000 square feet of additional manufacturing space in a neighboring facility. The Auckland facility is FDA registered and ISO 13485 certified. We believe that Aroa will be capable of providing sufficient quantities of our products to meet anticipated customer demands. In the event of an uncured supply failure by Aroa, we have the right to, directly or through a third-party, step in and operate the Aroa Auckland facility to manufacture our products on behalf of Aroa.
The proprietary ovine rumen used in the manufacturing of our products is obtained from sheep raised for human consumption in New Zealand and is currently sourced by Aroa from two abattoirs, or slaughterhouses. Although only two abattoirs are currently qualified, there are more than 30 additional abattoirs in New Zealand that could be used to source the ovine rumen. New Zealand cattle and sheep are considered by the USDA to be free of prion disease (progressive neurodegenerative disorders, including scrapie). The sheep receive veterinary inspection prior to slaughter and then each carcass is inspected post-mortem for the presence of disease according to USDA approved standards. Only sheep which pass full inspection can be used as a raw tissue source for our products and all the ovine rumen is processed in compliance with the FDA’s regulations for Medical Devices Containing Materials Derived from Animal Sources. Once the ovine rumen is procured, our reinforced tissue matrix products are then manufactured by Aroa at its facility in Auckland, New Zealand.
Distribution
The majority of our products are shipped directly from Auckland, New Zealand to our headquarters in Malvern, Pennsylvania. We sell our products directly to our customers, which are hospitals and ambulatory surgery centers. Outside of Europe, we do not use stocking distributors to sell our products.
Competition
The medical device industry is intensely competitive, subject to change and significantly affected by new product introductions and other market activities of industry participants.
In the hernia repair market, our primary competitors are Bard, a subsidiary of Becton, Dickinson and Company, which produces Phasix and Ventralight ST, and Allergan, a subsidiary of AbbVie, which produces Strattice. In the plastic and reconstructive surgery market, our primary competitors are Allergan, a subsidiary of AbbVie, which produces AlloDerm, MTF Biologics, which produces FlexHD, Novadaq, which produces DermACell, RTI Surgical, which produces Cortiva, Bard, which produces GalaFLEX, and Integra Lifesciences, which produces SurgiMend and DuraSorb.
Many of these competitors are large, well-capitalized companies with significantly greater market share and resources than we have, selling products that have been on the market prior to the commercialization of our products. As a consequence, they are able to spend more on product development, marketing, sales and other product initiatives than we can, while also benefiting from greater brand awareness. We also compete with smaller medical device companies that have single products or a limited range of products. Some of our competitors have:
| ● | significantly greater name recognition; |
| ● | broader or deeper relations with healthcare professionals, customers and third-party payors; |
| ● | more established distribution networks; |
26
| ● | greater experience in conducting research and development, manufacturing, clinical trials, marketing and obtaining regulatory clearance or approval for products; |
| ● | greater financial and human resources for product development, sales and marketing and patent prosecution; and |
| ● | more established, wider-ranging and deeper contractual relationships with GPO and IDNs that can be leveraged to drive greater utilization of their products. |
We believe that our continued ability to compete favorably depends on:
| ● | successfully deploying our commercial operations; |
| ● | continuing to innovate and maintain scientifically-advanced technology; |
| ● | attracting and retaining skilled personnel; |
| ● | maintaining and obtaining intellectual property protection for our products; and |
| ● | conducting clinical studies and obtaining and maintaining regulatory approvals. |
Government Regulation
Our products and operations are subject to extensive and rigorous regulation by the FDA and other federal, state and local authorities, as well as foreign regulatory authorities. The FDA regulates, among other things, the research, development, testing, design, manufacturing, approval, labeling, storage, recordkeeping, advertising, promotion and marketing, distribution, post-approval monitoring and reporting and import and export of medical devices in the U.S. to assure the safety and effectiveness of medical products for their intended use. The Federal Trade Commission also regulates the advertising of our products in the U.S. Further, we are subject to laws directed at preventing fraud and abuse, which subject our sales and marketing, training and other practices to government scrutiny.
Regulatory System for Medical Devices in the U.S.
All of our medical devices sold in the U.S. are subject to the Federal Food, Drug, and Cosmetic Act (“FDCA”) as implemented and enforced by the FDA.
Unless an exemption applies, each new or significantly modified medical device we seek to commercially distribute in the U.S. will require either a premarket notification to the FDA requesting permission for commercial distribution under Section 510(k) of the FDCA also referred to as a 510(k) clearance, or approval from the FDA of a PMA application. Both the 510(k) clearance and PMA processes can be resource intensive, expensive, and lengthy, and require payment of significant user fees, unless an exemption is available.
Device Classification
Under the FDCA, medical devices are classified into one of three classes — Class I, Class II or Class III — depending on the degree of risk associated with each medical device and the extent of control needed to provide reasonable assurances with respect to safety and effectiveness.
Class I includes devices with the lowest risk to the patient and are those for which safety and effectiveness can be reasonably assured by adherence to a set of FDA regulations, referred to as the General Controls for Medical Devices, which require compliance with the applicable portions of the Quality Systems Regulations, or QSR, facility registration and product listing, reporting of adverse events and malfunctions, and appropriate, truthful and non-misleading labeling and promotional materials. Some Class I devices, also called Class I reserved devices, also require premarket clearance by the FDA through the 510(k) premarket notification process described below.
27
Most Class I products are exempt from the premarket notification requirements.
Class II devices are those that are subject to the General Controls, and special controls as deemed necessary by the FDA to ensure the safety and effectiveness of the device. These special controls can include performance standards, patient registries, FDA guidance documents and post-market surveillance. Most Class II devices are subject to premarket review and clearance by the FDA. Premarket review and clearance by the FDA for Class II devices is accomplished through the 510(k) premarket notification process.
Class III devices include devices deemed by the FDA to pose the greatest risk such as life-supporting or life-sustaining devices, or implantable devices, in addition to those deemed novel and not substantially equivalent to a medical device cleared through the 510(k) process. The safety and effectiveness of Class III devices cannot be reasonably assured solely by the General Controls and special controls described above. Therefore, these devices are subject to the PMA application process, which is generally more costly and time consuming than the 510(k) process. Through the PMA application process, the applicant must submit data and information demonstrating reasonable assurance of the safety and effectiveness of the device for its intended use to the FDA’s satisfaction. Accordingly, a PMA application typically includes, but is not limited to, extensive technical information regarding device design and development, preclinical and clinical trial data, manufacturing information, labeling and financial disclosure information for the clinical investigators in device studies. The PMA application must provide valid scientific evidence that demonstrates to the FDA’s satisfaction a reasonable assurance of the safety and effectiveness of the device for its intended use.
510(k) Clearance Pathway
Our current products are subject to premarket notification and clearance under section 510(k) of the FDCA.
When a 510(k) clearance is required, we must submit a premarket notification to the FDA demonstrating that our proposed device is substantially equivalent to a predicate device, which is a previously cleared and legally marketed 510(k) device or a device that was in commercial distribution before May 28, 1976 (pre-amendments device) and for which a PMA is not required, a device that has been reclassified from Class III to Class II or I, or a device that was found substantially equivalent through the 510(k) process. By regulation, a premarket notification must be submitted to the FDA at least 90 days before we intend to distribute a device. As a practical matter, clearance often takes nine to twelve months, but may take significantly longer. To demonstrate substantial equivalence, the manufacturer must show that the proposed device has the same intended use as the predicate device, and it either has the same technological characteristics, or different technological characteristics and the information in the premarket notification demonstrates that the device is as safe and effective as the predicate device and does not raise different questions of safety and effectiveness. The FDA may require further information, including clinical data, to make a determination regarding substantial equivalence.
If the FDA agrees that the device is substantially equivalent to a predicate device currently on the market, it will grant 510(k) clearance to commercially market the device. If the FDA determines that the device is “not substantially equivalent” to a previously cleared device, the device is automatically designated as a Class III device. The device sponsor must then fulfill more rigorous PMA requirements, or can request a risk-based classification determination for the device in accordance with the de novo classification procedure, which is a route to market for novel medical devices that are low to moderate risk and are not substantially equivalent to a predicate device.
After a device receives 510(k) marketing clearance, any modification that could significantly affect its safety or effectiveness, or that would constitute a major change or modification in its intended use, will require a new 510(k) marketing clearance or, depending on the modification, a de novo classification or PMA approval. The FDA requires each manufacturer to determine whether the proposed change requires a premarket submission in the first instance, but the FDA can review any such decision and disagree with a manufacturer’s determination.
Many minor modifications today are accomplished by a manufacturer documenting the change in an internal letter-to-file. The letter-to-file is in lieu of submitting a new 510(k) to obtain clearance for every change. The FDA can always review these letters-to-file in an inspection. If the FDA disagrees with a manufacturer’s determination, the FDA can require the manufacturer to cease marketing and/or request the recall of the modified device until marketing authorization is obtained.
28
Also, in these circumstances, we may be subject to significant regulatory fines or penalties.
De Novo Classification
Medical device types that the FDA has not previously classified as Class I, II or III are automatically classified into Class III regardless of the level of risk they pose. The Food and Drug Administration Modernization Act of 1997, or FDAMA, established a new route to market for low to moderate risk medical devices that are automatically placed into Class III due to the absence of a predicate device, called the “Request for Evaluation of Automatic Class III Designation,” or the de novo classification procedure.
This procedure allows a manufacturer whose novel device is automatically classified into Class III to request down-classification of its medical device into Class I or Class II on the basis that the device presents low or moderate risk, rather than requiring the submission and approval of a PMA application. Prior to the enactment of the Food and Drug Administration Safety and Innovation Act of 2012, or FDASIA, a medical device could only be eligible for de novo classification if the manufacturer first submitted a 510(k) premarket notification and received a determination from the FDA that the device was not substantially equivalent to a predicate device. FDASIA streamlined the de novo classification pathway by permitting manufacturers to request de novo classification directly without first submitting a 510(k) premarket notification to the FDA and receiving a not substantially equivalent determination. Under FDASIA, the FDA is required to classify the device within 120 days following receipt of the de novo application, although the review of an application can occur over a significantly longer period of time. If the manufacturer seeks reclassification into Class II, the manufacturer must include a draft proposal for special controls that are necessary to provide a reasonable assurance of the safety and effectiveness of the medical device. In addition, the FDA may reject the reclassification petition if it identifies a legally marketed predicate device that would support a 510(k) or determines that the device is not low to moderate risk or that general controls would be inadequate to control the risks and special controls cannot be developed.
The PMA Approval Process
Class III devices require PMA approval before they can be marketed although some pre-amendment Class III devices for which the FDA has not yet required a PMA are cleared through the 510(k) process. The PMA process is more demanding than the 510(k) premarket notification process. In a PMA, the manufacturer must demonstrate that the device is safe and effective, and the PMA must be supported by extensive data, including data from preclinical studies and human clinical trials. The PMA must also contain a full description of the device and its components, a full description of the methods, facilities and controls used for manufacturing, and proposed labeling. While our current products are subject to the 510(k) clearance pathway, any future products or modifications to our existing products that we plan to develop for a breast reconstruction indication would be subject to the PMA approval process.
Following receipt of a PMA application, the FDA determines whether the application is sufficiently complete to permit a substantive review. If it is not, the agency will refuse to file the PMA. If it is, the FDA will accept the application for filing and begin the review. The FDA has 180 days to review a filed PMA application, although the review of an application can occur over a significantly longer period of time, and can take up to several years. During this review period, the FDA may request additional information or clarification of information already provided, or the FDA may issue a major deficiency letter to the applicant, requesting the applicant’s response to deficiencies communicated by the FDA. The FDA considers a PMA or PMA supplement to have been voluntarily withdrawn if an applicant fails to respond to an FDA request for information (e.g., a major deficiency letter) within 360 days. Before approving or denying a PMA, an FDA advisory committee may review the PMA at a public meeting and provide the FDA with the committee’s recommendation on whether the FDA should approve the submission, approve it with specific conditions, or not approve it. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions.
Prior to approval of a PMA, the FDA may conduct inspections of the clinical trial data and clinical trial sites, as well as inspections of the manufacturing facility and processes. Overall, the FDA review of a PMA application generally takes between one and three years, but may take significantly longer.
29
The FDA will approve the new device for commercial distribution if it determines that the data and information in the PMA constitute valid scientific evidence and that there is reasonable assurance that the device is safe and effective for its intended use(s).
If the FDA evaluation of a PMA is favorable, the FDA will issue either an approval letter, or an approvable letter, the latter of which usually contains a number of conditions that must be met in order to secure final approval of the PMA. When and if those conditions have been fulfilled to the satisfaction of the FDA, the agency will issue a PMA approval letter authorizing commercial marketing of the device, subject to the conditions of approval and the limitations established in the approval letter. If the FDA’s evaluation of a PMA application or manufacturing facilities is not favorable, the FDA will deny approval of the PMA or issue a not approvable letter. The FDA also may determine that additional tests or clinical trials are necessary, in which case the PMA approval may be delayed for several months or years while the trials are conducted and data is submitted in an amendment to the PMA, or the PMA is withdrawn and resubmitted when the data are available. The FDA may condition PMA approval on some form of post-market surveillance when deemed necessary to protect the public health or to provide additional safety and efficacy data for the device in a larger population or for a longer period of use. In such cases, the manufacturer might be required to follow certain patient groups for a number of years and to make periodic reports to the FDA on the clinical status of those patients. Failure to comply with the conditions of approval can result in material adverse enforcement action, including withdrawal of the approval.
New PMA applications or PMA supplements are required for changes to an approved device, such as modifications to the manufacturing process, equipment or facility, quality control procedures, sterilization, packaging, expiration date, labeling, device specifications, ingredients, materials or design. PMA supplements often require submission of the same type of information as an initial PMA application, except that the supplement is limited to information needed to support any changes from the device covered by the approved PMA application and may or may not require extensive technical or clinical data or the convening of an advisory committee, depending on the nature of the proposed change.
In approving a PMA application, as a condition of approval, the FDA may also require some form of post-approval study or post-market surveillance, whereby the applicant conducts a follow-up study or follows certain patient groups for a number of years and makes periodic reports to the FDA on the clinical status of those patients when necessary to protect the public health or to provide additional or longer term safety and effectiveness data for the device. The FDA may also require post-market surveillance for certain devices cleared under a 510(k) notification, such as implants or life-supporting or life-sustaining devices. The FDA may also approve a PMA application with other post-approval conditions intended to ensure the safety and effectiveness of the device, such as, among other things, restrictions on labeling, promotion, sale, distribution and use.
The Investigational Device Process
Clinical trials are almost always required to support a PMA and are sometimes required to support a 510(k) submission. All clinical investigations of investigational devices to determine safety and effectiveness must be conducted in accordance with the FDA’s IDE regulations which govern investigational device labeling, prohibit promotion of the investigational device, and specify an array of recordkeeping, reporting and monitoring responsibilities of study sponsors and study investigators. Some types of studies deemed to present a “non-significant risk” are deemed to have an approved IDE once certain requirements are addressed and Institutional Review Board, or IRB approval is obtained. If the device presents a “significant risk” to human health, as defined by the FDA, the sponsor must submit an IDE application to the FDA and obtain IDE approval prior to commencing the human clinical trials. The IDE will automatically become effective 30 days after receipt by the FDA unless the FDA notifies the company that the investigation may not begin. If the FDA determines that there are deficiencies or other concerns with an IDE for which it requires modification, the FDA may permit a clinical trial to proceed under an approval with conditions. The IDE application must be supported by appropriate data, such as animal and laboratory testing results, showing that it is safe to test the device in humans and that the testing protocol is scientifically sound. Generally, clinical trials for a significant risk device may begin once the IDE application is approved by the FDA and the study protocol and informed consent are approved by an appropriate IRB. There can be no assurance that submission of an IDE will result in the ability to commence clinical trials, and although the FDA’s approval of an IDE allows clinical testing to go forward for a specified number of subjects, it does not bind the FDA to accept the results of the trial as sufficient to prove the product’s safety and efficacy, even if the trial meets its intended success criteria.
30
During a study, the sponsor is required to comply with the applicable FDA requirements, including, for example, trial monitoring, selecting clinical investigators and providing them with the investigational plan, ensuring IRB review, adverse event reporting, record keeping and prohibitions on the promotion of investigational devices or on making safety or effectiveness claims for them. The clinical investigators in the clinical study are also subject to FDA good clinical practice regulations and must obtain patient informed consent, rigorously follow the investigational plan and study protocol, control the disposition of the investigational device, and comply with all reporting and recordkeeping requirements. Additionally, after a trial begins, we, the FDA or the IRB could suspend or terminate a clinical trial at any time for various reasons, including a belief that the risks to study subjects outweigh the anticipated benefits. The results of clinical testing may be unfavorable, or, even if the intended safety and efficacy success criteria are achieved, may not be considered sufficient for the FDA to grant marketing approval or clearance of a product.
Pervasive and Continuing FDA Regulation
After the FDA permits a device to enter commercial distribution, numerous and pervasive regulatory requirements continue to apply to our business operations, products and technologies. These include:
| ● | the FDA’s Quality Systems Regulations (“QSR”), which requires manufacturers, including third party manufacturers, to follow stringent design, testing, production, control, supplier/contractor selection, complaint handling, documentation and other quality assurance procedures during all aspects of the manufacturing process; |
| ● | labeling and marketing regulations which require that promotion is truthful, not misleading, fairly balanced and provides adequate directions for use and that all claims are substantiated; |
| ● | complying with requirements for Unique Device Identifiers on devices and also requiring the submission of certain information about each device to the FDA’s Global Unique Device Identification Database; |
| ● | advertising and promotion requirements, including FDA prohibitions against the promotion of products for uncleared, unapproved or off-label uses and FDA guidance on off-label dissemination of information and responding to unsolicited requests for information; |
| ● | restrictions on sale, distribution or use of a device; |
| ● | device establishment, registration and listing requirements and annual reporting requirements; |
| ● | approval or clearance of modifications to 510(k)-cleared devices that could significantly affect safety or effectiveness or that would constitute a major change in intended use of one of our cleared devices; |
| ● | medical device reporting regulations, which require that manufacturers report to the FDA if their device may have caused or contributed to a death or serious injury or malfunctioned in a way that would likely cause or contribute to a death or serious injury if the malfunction were to recur; |
| ● | medical device correction, removal and recall reporting regulations, which require that manufacturers report to the FDA field corrections and product recalls or removals if undertaken to reduce a risk to health posed by the device or to remedy a violation of the FDCA that may present a risk to health; |
| ● | recall requirements, including a mandatory recall if there is a reasonable probability that the device would cause serious adverse health consequences or death; |
| ● | an order of repair, replacement or refund; |
31
| ● | device tracking requirements; and |
| ● | post-market surveillance activities and regulations, which apply when necessary to protect the public health or to provide additional safety and effectiveness data for the device. |
The FDA has broad post-market and regulatory enforcement powers. Medical device manufacturers are subject to unannounced inspections by the FDA and other state, local and foreign regulatory authorities to assess compliance with the QSR and other applicable regulations, and these inspections may include the manufacturing facilities of any suppliers.
Failure to comply with applicable regulatory requirements can result in enforcement action by the FDA, which may include any of the following sanctions:
| ● | warning letters, untitled letters, Form 483s, fines, injunctions, consent decrees and civil penalties; |
| ● | recall or seizure of products; |
| ● | operating restrictions, partial suspension or total shutdown of production; |
| ● | the FDA’s refusal of requests for 510(k) clearance or premarket approval of new products, new intended uses or modifications to existing products; |
| ● | the FDA’s refusal to issue certificates to foreign governments needed to export products for sale in other countries; |
| ● | withdrawing approvals that have already been granted; and |
| ● | criminal prosecution. |
Regulatory System for Medical Devices in Europe
The European Union (“EU”) and the European Economic Area (“EEA”) (which is comprised of the 27 Member States of the EU plus Norway, Liechtenstein and Iceland) has a coordinated system for the authorization of medical devices. Until May 25, 2021, medical devices were regulated by the Council Directive 93/42/EEC, or the Medical Devices Directive (“MDD”), which has been repealed and replaced by Regulation (EU) No 2017/745, or the Medical Devices Regulation (“MDR”). There is a transition period during which certificates issued under the MDD remain valid, subject to compliance with certain requirements under the MDR (e.g. having put in place a quality management system in accordance with the MDR by May 26, 2024).
The MDR went into effect on May 26, 2021, and it:
| ● | strengthens the rules on placing devices on the market and reinforces surveillance once they are available; |
| ● | establishes explicit provisions on manufacturers’ responsibilities for the follow-up of the quality, performance and safety of devices placed on the market; |
| ● | improves the traceability of medical devices throughout the supply chain to the end-user or patient through a unique identification number; |
| ● | sets up a central database (Eudamed) to provide patients, healthcare professionals and the public with comprehensive information on products available in the EU; and |
32
| ● | strengthens rules for the assessment of certain high-risk devices, such as implants, which may have to undergo an additional check by experts before they are placed on the market. |
Under the MDR, the system of regulating medical devices operates by way of a certification for each medical device, which confirms that the device meets the relevant general safety and performance requirements laid down in Annex I of the MDR. Each certificated device is marked with a CE mark which shows that the device has a certificat de conformité, also referred to as a certificate of conformity. The means for achieving the requirements for a CE mark varies according to the nature of the device. Devices are classified in accordance with their perceived risks, similarly to the U.S. system. The class of a product determines the requirements to be fulfilled in accordance with the MDR before a CE mark can be placed on a product. The procedure by which a device is assessed to confirm if it complies with the applicable safety and performance requirements is known as a conformity assessment. Conformity assessment procedures require an assessment of available clinical evidence, literature data for the product, and post-market experience in respect of similar products already marketed. Specifically, a manufacturer must demonstrate that the device achieves its intended performance during normal conditions of use, that the known and foreseeable risks, and any adverse events, are minimized and acceptable when weighed against the benefits of its intended performance, and that any claims made about the performance and safety of the device are supported by suitable evidence. Except for low-risk medical devices (Class I non-sterile, non-measuring devices), where the manufacturer can self-certify compliance with the MDR based on a self-assessment of the conformity of its products with the applicable requirements of the MDR, a conformity assessment procedure requires the intervention of an independent organization accredited by a member state of the EEA to conduct conformity assessments, known as a notified body. If satisfied that the relevant product conforms to the relevant general safety and performance requirements, the notified body issues a certificate of conformity, which the manufacturer uses as a basis for its own declaration of conformity. The manufacturer may then apply the CE mark to the device, which allows the device to be placed on the market throughout the EEA.
The MDR requires that before placing a device, other than a custom-made device, on the market, manufacturers (as well as other economic operators such as authorized representatives and importers) must register by submitting identification information to the electronic system (Eudamed), unless they have already registered, and manufacturers must assign a unique identifier to the device and provide it along with other core data to the unique device identifier, or UDI, database. These new requirements aim at ensuring better identification and traceability of the devices. Manufacturers are responsible for entering the necessary data on Eudamed, which includes the UDI database, and for keeping it up to date. Eudamed is not yet fully functional and will be gradually rolled out. Use of a particular module of Eudamed (e.g. the UDI/device registration module) will become mandatory six months after the publication in the OJEU of the notice confirming the functionality of such module. The Medical Device Coordination Group (MDCG) has published guidance (in November 2024) on the gradual roll-out of Eudamed.
The United Kingdom formally left the EU on January 31, 2020. In respect of medical devices, since the end of the Brexit transitional period on January 1, 2021, medical devices must be registered with the Medicines and Healthcare products Regulatory Agency (“MHRA”) (the UK medicines and medical devices regulator) before being placed on the Great Britain market. If a manufacturer of a device placed on the market in Great Britain is based outside of the United Kingdom, the manufacturer must appoint a UK responsible person with a registered place of business in the United Kingdom to act on the manufacturer’s behalf in respect of certain activities (e.g. device registration). CE marks issued by EU notified bodies to place medical devices on the market in the EU will remain valid in the United Kingdom up until, at the latest, June 30, 2028 (for CE marks issued under the EU MDD) or June 30, 2030 (for CE marks issued under the EU MDR), following which a UK Conformity Assessed (“UKCA”) mark will be required to place a device on the Great Britain market. Manufacturers may choose to use the UKCA mark on a voluntary basis prior to such dates. UCKA marking will, however, not be recognized in the EU. The EU regulatory framework on medical devices continues to apply in Northern Ireland under the Windsor Framework and medical devices in Northern Ireland may either carry an EU CE mark or a UK and Northern Ireland CE mark (“CE UK(NI)”), although devices bearing the CE UK(NI) marking will not be accepted on the EU market.
Following a public consultation, the UK government is implementing changes to the medical devices legislation. The first piece of legislation will come into force on June 16, 2025, and implements changes to the post-market surveillance requirements for medical devices in Great Britain, with the aim of facilitating greater traceability of incidents. Further legislation will be put in place in 2025 and 2026 to introduce new pre-market requirements, including an international reliance procedure for approval of certain medical devices for the Great Britain market.
33
Privacy and Security Laws
There are numerous U.S. federal and state laws and regulations related to the privacy and security of personal information, including health information. Among others, the federal Health Insurance Portability and Accountability Act of 1996, as amended by the Health Information Technology for Economic and Clinical Health Act and their implementing regulations (collectively referred to as “HIPAA”) establish privacy and security standards that limit the use and disclosure of protected health information (“PHI”) and require covered entities and business associates to implement administrative, physical, and technical safeguards to ensure the confidentiality, integrity and availability of individually identifiable health information in electronic form, among other requirements.
Violations of HIPAA may result in civil and criminal penalties. Companies subject to HIPAA must also comply with HIPAA’s breach notification rule which requires notification of affected patients and the U.S. Department of Health and Human Services (“HHS”) and in certain cases of media outlets, in the case of a breach of unsecured PHI. The regulations also require business associates of covered entities to notify the covered entity of breaches by the business associate. State attorneys general also have the right to prosecute HIPAA violations committed against residents of their states, and HIPAA standards have been used as the basis for the duty of care in state civil suits, such as those for negligence or recklessness in misusing personal information. In addition, HIPAA mandates that HHS conduct periodic compliance audits of HIPAA covered entities and their business associates for compliance.
Many states have laws that protect the privacy and security of sensitive and personal information, including health information, to which we are subject. These laws may be similar to or even more protective than HIPAA and other federal privacy laws. For example, the California Consumer Privacy Act (“CCPA”) is a comprehensive privacy law that created individual privacy rights for California residents and increased the privacy and security obligations of entities handling certain personal data, including sensitive personal information.
We may be subject to other state and federal privacy laws, including laws that prohibit unfair privacy and security practices and deceptive statements about privacy and security, laws that place specific requirements on certain types of activities, such as data security and texting, and laws requiring holders of personal information to maintain safeguards and to take certain actions in response to a data breach.
Foreign data protection laws, including the General Data Protection Regulation, (“GDPR”) may also apply to health-related and other personal information belonging to individuals who reside outside of the U.S. The GDPR went into effect in the European Union in May 2018 and introduced strict requirements for processing the personal data of data subjects residing in the European Economic Area. Companies that must comply with the GDPR face increased compliance obligations and risk, including more robust regulatory enforcement of data protection requirements and potential fines for noncompliance of up to €20 million or 4% of the annual global revenues of the noncompliant company, whichever is greater. Among other requirements, the GDPR regulates cross-border transfers of personal data and requires transferee countries to have protections equivalent to protections available in the EU. In July 2023, the EU adopted the EU-U.S. Data Privacy Framework (“DPF”) to facilitate cross-border transfers of data from the EU to the U.S. A company may participate under the DPF by self-certifying and publicly committing to comply with the applicable DPF principles.
Further, the United Kingdom’s exit from the European Union, referred to as Brexit, has created uncertainty regarding data protection regulation in the United Kingdom. The United Kingdom has transposed the GDPR into domestic law with a United Kingdom version of the GDPR that took effect in January 2021 (“UK GDPR”). Currently, the GDPR and UK GDPR remain largely aligned, but the United Kingdom announced plans to reform the country’s data protection legal framework in its Data Reform Bill, which failed in the UK legislative process. A new Data (Use and Access) Bill, or UK Bill, has been introduced into parliament. If passed, the final version of the UK Bill may have the effect of further altering the similarities between the UK and EU data protection regime and threaten the UK adequacy decision from the EU Commission. This may lead to divergence between the GDPR and UK GDPR.
34
EU member states have introduced national laws implementing the GDPR which impose additional requirements; this adds to the complexity of processing personal data in or from the EEA or United Kingdom. Guidance on implementation and compliance practices are often updated or otherwise revised.
Anti-Kickback Statutes
The federal Anti-Kickback Statute prohibits persons from (among other things) knowingly and willfully soliciting, offering, receiving or providing remuneration, directly or indirectly, in exchange for or to induce the referral of an individual, or the recommending, furnishing or arranging for a good or service, for which payment may be made under a federal healthcare program such as Medicare or Medicaid.
Courts have interpreted the Anti-Kickback Statute quite broadly, holding that the statute will be violated if even one purpose of a payment — though not its sole or primary purpose — is to induce an act prohibited by the statute with a willful intent to act improperly. The statute prohibits many arrangements and practices that are otherwise lawful in businesses outside of the healthcare industry. A person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation. Prosecutors may infer intent from the surrounding circumstances and, because courts have interpreted the statute to be violated if even one purpose of a payment is to induce the purchase of items or services paid for by federal healthcare programs, prosecutors have broad discretion in choosing arrangements to prosecute under the statute. There are statutory exceptions and regulatory “safe harbors” available to protect certain appropriately structured arrangements that otherwise would implicate the Anti-Kickback Statute and those who structure their business arrangements to satisfy all of the criteria of a safe harbor are protected from liability under the statute. Our business is subject to these laws.
Many states have adopted anti-kickback and self-referral laws similar to the Anti-Kickback Statute; however, some of these state prohibitions are broader in scope and apply to arrangements involving healthcare items or services reimbursed by any source, and not only by Medicare, Medicaid or another federal healthcare program. These state laws do not always have the same exceptions or safe harbors as the federal Anti-Kickback Statute.
False Claims Laws
The federal False Claims Act imposes liability on any individual or entity that, among other things, knowingly presents, or causes to be presented, a false or fraudulent claim for payment by a federal healthcare program. The qui tam or “whistleblower” provisions of the False Claims Act allow a private individual to bring actions on behalf of the federal government alleging that the defendant has violated the False Claims Act and to share in any monetary recovery. In recent years, the number of lawsuits brought against healthcare industry participants by private individuals has increased dramatically.
There are many potential bases for liability under the False Claims Act. Liability arises, primarily, when an entity knowingly submits, or causes another to submit, a false claim for reimbursement to the federal government, but also may arise when an entity knowingly makes a false statement material to an obligation to pay or transmit money or property to the federal government or knowingly conceals or knowingly and improperly avoids or decreases an obligation to pay or transmit money or property to the federal government. Various states have also enacted false claims and insurance fraud laws that are analogous to the federal False Claims Act. Many of these state laws apply to claims submitted to any third-party payor and are not limited to claims submitted to a federal healthcare program. The scope of these laws and the interpretations of them vary from state to state and are enforced by state courts and regulatory authorities, each with broad discretion. A determination of liability under such laws could result in fines and penalties and restrictions on a company’s ability to operate in these jurisdictions.
Transparency Laws
35
The federal Physician Payments Sunshine Act (“Sunshine Act”) which was enacted as part of the Patient Protection and Affordable Care Act (“PPACA”) generally requires certain manufacturers of a drug, device, biologic or other medical supply that is covered by Medicare, Medicaid or the Children’s Health Insurance Program and applicable GPOs to report on an annual basis: (i) certain payments and other transfers of value given to certain healthcare professionals and teaching hospitals and (ii) any ownership or investment interest that U.S. physicians, or their immediate family members, have in their company. The payments required to be reported include the cost of meals provided to a healthcare professional, travel reimbursements and other transfers of value, including those provided as part of contracted services such as speaker programs, advisory boards, consultation services and clinical trial services. Under the statute, the federal government makes reported information available to the public. Failure to comply with the reporting requirements can result in significant civil monetary penalties or criminal penalties if an entity intentionally makes false statements in the reports.
There has been a recent trend of separate state regulation of payments and transfers of value by manufacturers of medical devices to healthcare professionals and entities, however, and some state transparency laws apply more broadly than the federal Sunshine Act. There are also an increasing number of analogous state laws that require manufacturers to file reports with states on pricing and marketing information. Many of these laws contain ambiguities as to what is required to comply with the laws. For example, several states have enacted legislation requiring manufacturers to, among other things, establish and implement commercial compliance programs, file periodic reports with the state, make periodic public disclosures on sales, marketing, pricing, clinical trials and other activities and/or register their sales representatives. Certain state laws also regulate manufacturers’ use of physician and patient identifiable data. These laws may affect our sales, marketing and other promotional activities by imposing administrative and compliance burdens. In addition, given the lack of clarity with respect to these laws and their implementation, our reporting actions could be subject to the penalty provisions of the pertinent state and federal authorities. All of our activities are also potentially subject to federal and state consumer protection and unfair competition.
Other Federal Healthcare Fraud and Abuse Laws
We may also be subject to other federal healthcare fraud and abuse laws, including provisions of HIPAA, which imposes criminal liability and amends provisions on the reporting, investigation, enforcement, and penalizing of civil liability for, among other things, knowingly and recklessly executing a scheme or artifice to defraud any healthcare benefit program, including private payors, as well as knowingly and willfully falsifying, concealing or covering up a material fact by any trick, scheme or device or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for healthcare benefits, items or services. A violation of this statute is a felony and may result in fines, imprisonment or exclusion from government-sponsored programs. As with the federal Anti-Kickback Statute, a person or entity no longer needs to have actual knowledge of this statute or specific intent to violate it in order to have committed a violation.
Foreign Corrupt Practices Act
The Foreign Corrupt Practices Act (“FCPA”) prohibits U.S. businesses and their representatives from offering to pay, paying, promising to pay or authorizing the payment of money or anything of value to a foreign official in order to influence any act or decision of the foreign official in his or her official capacity or to secure any other improper advantage in order to obtain or retain business. The FCPA also obligates companies whose securities are listed in the U.S. to comply with accounting provisions requiring us to maintain books and records, which in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the corporation, including international subsidiaries, if any, and to devise and maintain a system of internal accounting controls sufficient to provide reasonable assurances regarding the reliability of financial reporting and the preparation of financial statements. Our industry is heavily regulated and therefore involves significant interaction with public officials, including officials of non-U.S. governments. Additionally, in many other countries, the health care providers who prescribe pharmaceuticals are employed by their government, and the purchasers of pharmaceuticals are government entities; therefore, our dealings with these prescribers and purchasers are subject to regulation under the FCPA. Recently, the SEC and Department of Justice have increased their FCPA enforcement activities with respect to pharmaceutical companies. Violations could result in fines, criminal sanctions against us, our officers, or our employees, the closing down of our facilities, requirements to obtain export licenses, cessation of business activities in sanctioned countries, implementation of compliance programs, and prohibitions on the conduct of our business. Enforcement actions may be brought by the Department of Justice or the SEC, and recent enacted legislation has expanded the SEC’s power to seek disgorgement in all FCPA cases filed in federal court and extended the statute of limitations in SEC enforcement actions in intent-based claims such as those under the FCPA from five years to ten years.
36
International Laws
In Europe, and throughout the world, other countries have enacted anti-bribery laws and/or regulations similar to the FCPA. Violations of any of these anti-bribery laws, or allegations of such violations, could have a negative impact on our business, results of operations and reputation.
There are also international privacy laws that impose restrictions on the access, use, and disclosure of health information. All of these laws may impact our business. Our failure to comply with these privacy laws or significant changes in the laws restricting our ability to obtain required patient information could significantly impact our business and our future business plans.
U.S. Healthcare Reform
The U.S. and many foreign jurisdictions have enacted or proposed legislative and regulatory changes affecting the healthcare system. The U.S. government, state legislatures and foreign governments also have shown significant interest in implementing cost-containment programs to limit the growth of government-paid healthcare costs, including price controls and restrictions on reimbursement.
In the U.S., the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act, or collectively the “Affordable Care Act”, substantially changed the way healthcare is financed by both governmental and private insurers and significantly impacts the healthcare industry. The Affordable Care Act was intended to broaden access to health insurance, reduce or constrain the growth of healthcare spending, enhance remedies against healthcare fraud and abuse, add new transparency requirements for healthcare and health insurance industries, impose new fees on pharmaceutical and medical device manufacturers, and impose additional health policy reforms.
There have been significant ongoing judicial, administrative, executive and legislative efforts to modify or eliminate the Affordable Care Act.
Other legislative changes have been proposed and adopted since passage of the Affordable Care Act. The Budget Control Act of 2011, among other things, included aggregate reductions to Medicare payments to healthcare providers of up to 2.0% per fiscal year, and will last through 2031 unless additional Congressional action is taken. The American Taxpayer Relief Act of 2012 reduced Medicare payments to several types of providers, including hospitals, imaging centers and cancer treatment centers, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years. Due to the Statutory Pay-As-You-Go Act of 2010, estimated budget deficit increases resulting from the American Rescue Plan Act of 2021 and subsequent legislation, Medicare payments to providers were further reduced starting on January 1, 2025; however, legislation has been introduced (but not passed) in the U.S. Congress that would, if enacted, reverse these payment reductions.
The Affordable Care Act has also been subject to challenges in the courts since it was enacted. On June 17, 2021, for example, the Supreme Court dismissed the most recent challenge to the Affordable Care Act, ruling that the plaintiffs lacked standing to challenge the law as they had not alleged personal injury traceable to the allegedly unlawful conduct. As a result, the Supreme Court did not rule on the constitutionality of the Affordable Care Act or any of its provisions.
Further changes to and under the Affordable Care Act remain possible, but it is unknown what form any such changes or any law proposed to replace or revise the Affordable Care Act would take, and how or whether it may affect our business in the future.
At the state level, legislatures have increasingly passed legislation and implemented regulations designed to control healthcare costs, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing.
37
We expect that additional federal, state and foreign healthcare reform measures will be adopted in the future, any of which could limit the amounts that federal and state governments will pay for healthcare products and services, which could result in limited coverage and reimbursement and reduced demand for our products, once approved, or additional pricing pressures.
Coverage and Reimbursement
In the U.S. and markets in other countries, sales of any products for which we receive regulatory approval for commercial sale will depend in part on the availability of reimbursement from third party payors. Third party payors include government health administrative authorities, managed care providers, private health insurers, and other organizations. These third-party payors are increasingly challenging the price and examining the cost-effectiveness of medical products and services. In addition, significant uncertainty exists as to the reimbursement status of newly approved healthcare products, and there are continuing legislative and regulatory efforts by the federal government and the states to reduce the cost of medical products and services overall. We may need to conduct expensive studies in order to demonstrate the cost-effectiveness of our products. Our product candidates may not be considered cost-effective. Decisions regarding the extent of coverage and amount of reimbursement to be provided are made on a plan-by-plan basis. One third-party payor’s decision to cover a particular product or procedure using the product does not ensure that other payors will also provide coverage for the product. Adequate third-party reimbursement may not be available to enable us to maintain price levels sufficient to realize appropriate revenue levels. Future legislation could limit payments for medical devices, including our products and our future products.
The U.S. government, state legislatures and foreign governments have shown significant interest in implementing cost containment programs to limit the growth of government-paid health care costs, including price controls, restrictions on reimbursement and requirements for substitution of less costly products. Adoption of government controls and measures, and tightening of restrictive policies in jurisdictions with existing controls and measures, could limit payments for our products. The marketability of any products for which we receive regulatory approval for commercial sale may suffer if the government and third-party payors fail to provide adequate coverage and reimbursement. In addition, an increasing emphasis on managed care in the U.S. has increased and will continue to increase the pressure on medical product and service pricing.
Human Capital Resources
As of December 31, 2024, we had 209 employees worldwide. None of our employees are represented by a collective bargaining agreement and we have never experienced a work stoppage. We believe we have good relationships with our employees.
The success of our business is fundamentally connected to the well-being of our employees. Accordingly, we are committed to their health, safety and wellness. We provide our employees and their families with access to a variety of flexible and convenient health and wellness programs, including benefits that provide protection and security so they can have peace of mind concerning events that may require time away from work or that impact their financial well-being; that support their physical and mental health by providing tools and resources to help them improve or maintain their health status and encourage engagement in healthy behaviors; and that offer choice where possible so they can customize their benefits to meet their needs and the needs of their families.
We strive to provide a competitive mix of pay, benefits and services that help meet the needs of our employees. In addition to salaries, these programs include variable incentive compensation plans, potential annual discretionary bonuses, stock awards, a 401(k) Plan, healthcare and insurance benefits, health savings and flexible spending accounts, paid time off, family leave, and flexible work schedules, among others. In addition to our broad-based equity award programs, we have used targeted equity-based grants with vesting conditions to enhance retention of personnel.
Corporate Information
We were incorporated on April 17, 2012.
38
Our primary executive offices are located at 1 Great Valley Parkway, Suite 24, Malvern, Pennsylvania 19355 and our telephone number is (484) 320-2930. Our website address is www.telabio.com. The information contained in, or that can be accessed through, our website is not part of this Annual Report. We make available, free of charge and through our website, our Annual Reports on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K, and any amendments to any such reports filed or furnished pursuant to Section 13(a) or 15(d) of the Exchange Act as soon as reasonably practicable after they are electronically filed with or furnished to the SEC.
39
ITEM 1A.RISK FACTORS
You should carefully consider the following risks described below, together with all of the other information in this Annual Report, including our consolidated financial statements and related notes. While we believe that the risks and uncertainties described below are the material risks facing our business, additional risks that we do not know of or that we currently think are immaterial may also arise and materially affect our business. The realization of any of these risks could have a material adverse effect on our business, financial condition, results of operations, and our ability to accomplish our strategic objectives.
Risks Related to Achieving or Sustaining Profitability, Financial Position and Capital Requirements
We have incurred significant operating losses since inception, we expect to incur operating losses in the future, and we may not be able to achieve or sustain profitability.
We have incurred net losses since our incorporation on April 17, 2012. For the years ended December 31, 2024, 2023 and 2022, we had net losses of $37.8 million, $46.7 million and $44.3 million, respectively. As of December 31, 2024, we had an accumulated deficit of $358.7 million.
We expect to continue to incur significant sales and marketing, research and clinical development, regulatory and other expenses as we expand our sales and marketing efforts to increase adoption of our products, expand existing relationships with our customers, obtain regulatory clearances or approvals for our planned or future products, conduct clinical trials on our existing and planned or future products, develop, acquire or license complimentary products for our product portfolio, or add new features to our existing products. As a result, we expect to continue to incur operating losses for the foreseeable future and may never achieve profitability. Even if we do achieve profitability, we may not be able to sustain or increase profitability on an ongoing basis. If we do not achieve or sustain profitability, it will be more difficult for us to finance our business and accomplish our strategic objectives, either of which would have a material adverse effect on our business, financial condition and results of operations and may cause the market price of our common stock to decline.
Our indebtedness may limit our flexibility in operating our business and adversely affect our financial health and competitive position.
As of December 31, 2024, we had $40.0 million of indebtedness outstanding under our credit facility with MidCap Financial Trust (“MidCap”) that matures in May 2027.
To service this indebtedness and any additional indebtedness we may incur in the future, we need to generate cash from our operating activities. Our ability to generate cash is subject, in part, to our ability to successfully execute our business strategy, as well as general economic, financial, competitive, regulatory, and other factors beyond our control. We cannot assure you that our business will be able to generate sufficient cash flow from operations or that future borrowings or other financings will be available to us in an amount sufficient to enable us to service our indebtedness and fund our other liquidity needs. To the extent we are required to use cash from operations or the proceeds of any future financing to service our indebtedness, our ability to plan for, or react to, changes in our business, industry and the economy generally will be limited.
In addition, the MidCap credit facility contains certain covenants that limit our ability to engage in certain transactions that may be in our long-term best interests, including the incurrence of additional indebtedness, effecting certain corporate changes, making certain investments, acquisitions or dispositions and paying dividends.
We have not previously breached and are not currently in breach of these or any of the other covenants; however, there can be no guarantee that we will not breach these covenants in the future. In the event that we breach one or more covenants, our lender may choose to declare an event of default and require that we immediately repay all amounts outstanding, terminate any commitment to extend further credit and foreclose on the collateral granted to it to collateralize such indebtedness. The occurrence of any of these events could have a material adverse effect on our business, financial condition and results of operations.
40
We may require substantial additional capital to finance our planned operations, which may not be available to us on acceptable terms or at all.
If needed, any future funding requirements will depend on many factors, including:
| ● | surgeon and market acceptance of our products; |
| ● | the cost of our research and development activities; |
| ● | the cost and timing of obtaining regulatory clearances or approvals; |
| ● | the cost and timing of establishing additional sales and marketing capabilities; |
| ● | the cost and timing of clinical trials that we are currently conducting or may conduct in the future; |
| ● | costs associated with any product recall that may occur; |
| ● | the effect of competing products in our markets or competing technologies; |
| ● | the extent to which we acquire or invest in products, technologies and businesses, although we currently have no commitments or agreements relating to any of these types of transactions; |
| ● | the cost of filing and prosecuting patent applications and defending and enforcing our patent or other intellectual property rights; and |
| ● | the cost of defending, in litigation or otherwise, any claims that we infringe third-party patents or other intellectual property rights. |
Any additional equity or debt financing that we raise may contain terms that are not favorable to us or our stockholders. In addition, any future debt financing into which we enter may impose upon us additional covenants that restrict our operations, including limitations on our ability to incur liens or additional debt, pay dividends, repurchase our common stock, make certain investments or engage in certain merger, consolidation or asset sale transactions. If we raise additional funds through collaboration and licensing arrangements with third-parties, it may be necessary to relinquish some rights to our technologies or our products, or grant licenses on terms that are not favorable to us.
Furthermore, we cannot be certain that additional funding will be available on acceptable terms, if at all. If we do not have, or are not able to obtain, sufficient funds, we may have to delay development or commercialization of our products or license to third-parties the rights to commercialize products or technologies that we would otherwise seek to commercialize. We also may have to reduce marketing, customer support or other resources devoted to our products or cease operations. Any of these factors could harm our business, financial condition and results of operations.
If we are unable to expand, manage and maintain our direct sales and marketing organizations, we may not be able to generate anticipated revenue.
Building the requisite sales, marketing and distribution capabilities to successfully market and sell our products continues to be expensive and time-consuming and requires significant attention from our leadership team to manage. Any failure or delay in the expansion of our sales, marketing or distribution capabilities would adversely impact the commercialization of our products. Additionally, we may choose to collaborate, either globally or on a territory-by-territory basis, with third parties on the commercialization of our products. If we are unable to enter into such arrangements on acceptable terms or at all, we may not be able to successfully commercialize our products.
As of December 31, 2024, our commercial organization consisted of 133 employees in the U.S. and 16 employees in Europe. To generate future revenue growth, we plan to continue to expand the size and geographic scope of our direct sales organization.
41
This growth may require us to split or adjust existing sales territories, which may adversely affect our ability to retain customers in those territories. Additionally, our future success will depend largely on our ability to continue to hire, train, retain and motivate skilled sales and marketing personnel with significant industry experience and technical knowledge of medical devices and related products. The competition for talented individuals experienced in selling and marketing medical device products is intense, and we cannot assure you that we can assemble or maintain an effective team. We cannot assure you that we will be able to hire and retain additional personnel on favorable or commercially reasonable terms, if at all. Our operating results are directly dependent upon the sales and marketing efforts of our employees. Failure to hire or retain qualified sales and marketing personnel would prevent us from expanding our business and generating revenue. If we are unable to expand our sales and marketing capabilities, we may not be able to effectively commercialize our products, which could have an adverse effect on our business, financial condition and results of operations.
Macroeconomic conditions, including those placing financial strain on hospital systems and their ability to perform the procedures in which our products are used, may negatively impact certain aspects of our business, our prospects, results of operations and financial condition.
Macroeconomic conditions, including those placing financial strain on hospital systems and their ability to perform the procedures in which our products are used, or those stemming from external cybersecurity events, supply chain disruptions of critical surgical supplies, inflationary pressures, tariffs, geopolitical conflict or other macroeconomic events, may adversely impact our business, financial condition and prospects. These financial and resource strains on the healthcare system, including those first arising in response to the COVID-19 pandemic, may further impair labor and staffing in the hospital sector, and in turn hospital capacity for elective procedures. Any prolonged delays in normalized levels of elective surgeries by governmental, hospital or payor actions would continue to impair net sales of our products.
General supply chain disruptions, initially arising from COVID-19, have in the wake of severe weather events and geopolitical turmoil, such as the ongoing Russia-Ukraine conflict and the current conflict in the Middle East (including any escalation or expansion) continue to threaten trade globally and weaken supply systems. We currently rely on Aroa, which is headquartered in New Zealand, for supply of our products. While there have been minimal disruptions to our supply chain to date, there is a risk that in the future supplies of our products could be disrupted or delayed based on competition within the supply chain or otherwise affected by substantial inflationary pressures from other underlying macroeconomic conditions. There can be no assurance that we would be able to timely implement any mitigation plans relating to our supply chain.
Continued concerns about the systemic impact of potential economic slowdown or recession, liquidity constraints, failures and instability in the U.S. and international financial banking systems, and geopolitical turmoil, including the ongoing Russia-Ukraine conflict and the current conflict in the Middle East (including any escalation or expansion), have contributed to increased market volatility and diminished expectations for economic growth in the world. These conditions may lead to continued volatility in the future, which could result in a decline in our stock price, high inflation, increase our cost of capital and adversely affect our ability to access the capital markets in the future even after local conditions improve.
Market acceptance of our medical products in the U.S. and other countries is dependent upon the procurement practices of our customers, patient need for our products and procedures and the reimbursement of patients’ medical expenses by government healthcare programs and third-party payors. The continuing uncertainty surrounding global economic conditions and financial markets may adversely affect demand for our products and procedures and result in lower reimbursement rates or coverage for our products, resulting in lower sales volume and downward pricing pressure on our products and slower adoption of new products.
In addition, the sale of our medical products is correlated to the frequency of surgical procedural volumes at current and prospective hospital accounts. During the second quarter of 2024, we became aware of multiple cybersecurity events that impacted our customers, including ransomware attacks and other similar system disruptions and outages, in the U.S. and Europe that adversely impacted the procedural volumes at current customer accounts, including those affiliated across one of our GPOs. To the extent current or future cybersecurity events continue to impact the hospital systems we serve, or otherwise affect third-party payors or other vendors within the healthcare industry critical to the patient care, we may experience additional reductions in procedural volumes that lead to lower sales volume for our products or could have lasting impact resulting in slower rates of adoption for our products in these accounts.
42
The full extent to which these macroeconomic factors, will further, directly or indirectly, impact our business, results of operations and financial condition, including our sales, expenses, manufacturing capability, supply chain integrity, research and development activities, and employee-related matters, will depend on future developments that are highly uncertain.
Information pertaining to the impact of these macroeconomic pressures on our operations to date can be found in “Management’s Discussion and Analysis of Financial Position and Results of Operations” in this Annual Report on Form 10-K.
Rising inflation rates could negatively impact our revenues and profitability if increases in the prices of our product or a decrease in consumer spending results in lower volumes of elective surgeries. In addition, if our costs increase and we are not able to pass along these price increases, our profitability would be adversely affected, and the adverse impact may be material.
Inflation rates, particularly in the U.S., have increased recently to levels not seen in years. Increased inflation may result in decreased demand for our products, increased operating costs (including our labor costs), reduced liquidity, and limitations on our ability to access credit or otherwise raise debt and equity capital. In addition, the United States Federal Reserve has raised, and may in the future raise, interest rates in response to concerns about inflation. Increases in interest rates, especially if coupled with reduced government spending and volatility in financial markets, may have the effect of further increasing economic uncertainty and heightening these risks. In an inflationary environment, we may be unable to raise the prices of our products at or above the rate at which our costs increase, which could/would reduce our profit margins and have a material adverse effect on our financial results. We also may experience lower than expected sales and potential adverse impacts on our competitive position if there is a decrease in consumer spending or a negative reaction to our pricing. A reduction in our revenue would be detrimental to our profitability and financial condition and could also have an adverse impact on our future growth.
Adverse developments affecting the financial services industry, such as actual events or concerns involving liquidity, defaults or non-performance by financial institutions or transactional counterparties, could adversely affect our current and projected business operations and financial condition and results of operations.
Events involving limited liquidity, defaults, non-performance or other adverse developments that affect financial institutions, transactional counterparties or other companies in the financial services industry or the financial services industry generally, or concerns or rumors about any events of these kinds or other similar risks, have in the past and may in the future lead to market-wide liquidity problems. For example, in early 2023, several financial institutions closed and were taken into receivership by the Federal Deposit Insurance Corporation. Even though we assess our banking and customer relationships as we believe necessary or appropriate, our access to funding sources and other credit arrangements in amounts adequate to finance or capitalize our current and projected future business operations could be significantly impaired by factors that affect us, the financial services industry or economy in general. These factors could include, among others, events such as liquidity constraints or failures, the ability to perform obligations under various types of financial, credit or liquidity agreements or arrangements, disruptions or instability in the financial services industry or financial markets, or concerns or negative expectations about the prospects for companies in the financial services industry.
In addition, investor concerns regarding the U.S. or international financial systems could result in less favorable commercial financing terms, including higher interest rates or costs and tighter financial and operating covenants, or systemic limitations on access to credit and liquidity sources, thereby making it more difficult for us to acquire financing on acceptable terms or at all. Any decline in available funding or access to our cash and liquidity resources could, among other risks, adversely impact our ability to meet our operating expenses, financial obligations or fulfill our other obligations, result in breaches of our contractual obligations or result in violations of federal or state wage and hour laws. Any of these impacts, or any other impacts resulting from the factors described above or other related or similar factors not described above, could have material adverse impacts on our liquidity and our business, financial condition or results of operations.
43
Risks Related to the Commercialization of our Products
To date, the vast majority of our revenue has been generated from sales of our OviTex products, and we therefore are highly dependent on the commercial success of the OviTex product line.
Sales of our OviTex products accounted for 66%, 67% and 70% of total revenue for the years ended December 31, 2024, 2023 and 2022, respectively. We first commercialized OviTex products in the U.S. in 2016 and have subsequently launched our OviTex products in Europe, introduced our larger sized OviTex products, as well as OviTex LPR and OviTex IHR product configurations for deeper penetration into laparoscopic and robotic-assisted hernia surgical repairs. In addition to our OviTex products, we have also commercialized our OviTex PRS products for use in surgery for soft-tissue repair or reinforcement in plastic and reconstructive procedures and most recently the LIQUIFIX Hernia Mesh Fixation Devices (LIQUIFIX FIX8™ and LIQUIFIX Precision™) pursuant to our distribution agreement with Advanced Medical Solutions Limited. While we continue to diversify our portfolio and revenue sources, we expect that sales of our OviTex products will account for the majority of our revenue for the foreseeable future while we continue to grow market share for our OviTex PRS products, LIQUIFIX and any complementary products that we may develop or distribute from time to time. Our failure to successfully increase sales of these products or any other event impeding our ability to sell these products would result in a material adverse effect on our business, financial condition and results of operations.
The commercial success of our products will largely depend upon attaining significant market acceptance.
Our ability to execute our growth strategy, achieve commercial success and become profitable will depend upon the adoption by inpatient and outpatient hospitals, surgeons, and medical device supply chain participants of our reinforced tissue matrix products. We cannot predict how quickly, if at all, surgeons will accept our products or, if accepted, how frequently they will be used. Our products and planned or future products we may develop or market may never gain broad market acceptance among surgeons and the medical community for some or all of our indications. Some surgeons may have prior history with or a preference for other soft-tissue reinforcement products, such as permanent synthetic mesh, resorbable synthetic mesh, or other biologic matrices, or may be reluctant to alter their practice patterns to treat patients with our reinforced tissue matrix products. The degree of market acceptance of any of our products will depend on a number of factors, including:
| ● | whether surgeons and others in the medical community consider our products to be safe, effective and cost effective; |
| ● | the potential and perceived advantages of our products over alternative products; |
| ● | the effectiveness of our sales and marketing efforts for our products; |
| ● | the prevalence and severity of any complications associated with using our products; |
| ● | the convenience and ease of use of our products relative to competing products; |
| ● | product labeling or product insert requirements by regulatory authorities; |
| ● | the competitive pricing of our products; |
| ● | the quality of our products meeting patient and surgeon expectations; |
| ● | the results of clinical trials and post-market clinical studies relating to the use of our products; |
44
| ● | pricing pressure, including from GPOs and government payors; |
| ● | obtaining favorable contract treatment with GPOs and other third-party payors to enable growing adoption of our products across hernia procedures; |
| ● | the availability of coverage and adequate reimbursement for procedures using our products from third-party payors, including government authorities; |
| ● | the willingness of patients to pay out-of-pocket for our products in the absence of coverage and adequate reimbursement by third-party payors, including government authorities; and |
| ● | our ability to provide incremental clinical and economic data that show the safety, clinical efficacy and cost effectiveness, and patient benefits from, our products. |
Additionally, even if our products achieve market acceptance, they may not maintain that market acceptance over time if competing products or technologies, which are more cost effective or received more favorably, are introduced. Failure to achieve or maintain market acceptance and/or market share would limit our ability to generate revenue and would have a material adverse effect on our business, financial condition and results of operations.
Even if we are able to attain significant market acceptance of our products, the commercial success of our products is not guaranteed.
Our future financial success will depend substantially on our ability to effectively and profitably market and sell our products. Even if we are able to attain significant market acceptance of our products, the commercial success of our products and any of our planned or future products is dependent on a number of additional factors, including the results of clinical trials relating to the use of our products and our ability to obtain and maintain regulatory approval or clearance to market our products and maintain compliance with applicable regulatory requirements. Successful growth of our sales and marketing efforts will depend on the strength of our marketing and distribution infrastructure and the effectiveness of our marketing and sales efforts, including our efforts to expand our direct sales force, while our ability to satisfy demand for our products driven by our sales and marketing efforts will be largely dependent on the ability of Aroa to maintain a commercially viable manufacturing process that is compliant with regulatory standards. If we fail to successfully market and sell our products, we will not be able to achieve profitability, which will have a material adverse effect on our business, financial condition and results of operations.
Our ability to grow our revenue in future periods will depend on our ability to increase sales of our OviTex in hernia and abdominal wall reconstruction and OviTex PRS products in plastic and reconstructive procedures and any new product or product indications that we introduce, which will, in turn, depend in part on our success in expanding our customer base and driving increased use of our products. New products or product indications may also need to be approved or cleared by the FDA and comparable non-U.S. regulatory agencies to drive revenue growth. If we cannot achieve revenue growth, it could have a material adverse effect on our business, financial condition and results of operations.
The misuse or off-label use of our products may harm our reputation in the marketplace, result in injuries that lead to product liability suits or result in costly investigations, fines or sanctions by regulatory bodies if we are deemed to have engaged in the promotion of our products for these uses.
Surgeons and other medical professionals may misuse our reinforced tissue matrix products or use improper techniques if they are not adequately trained, potentially leading to injury and an increased risk of product liability. If our products are misused or used with improper technique, we may become subject to costly litigation by our customers or their patients. Product liability claims could divert management’s attention from our core business, be expensive to defend and result in sizeable damage awards against us that may not be covered by insurance. In addition, any of the events described above could harm our business.
45
The products we commercialize have been cleared by the FDA and other regulatory authorities for specific indications. Our OviTex products are reinforced tissue matrices designed for use as a surgical mesh to reinforce and/or repair soft-tissue where weakness exists and indications for use of our OviTex products include the repair of hernia and/or abdominal wall defects which require the use of reinforcing or bridging material to obtain the desired surgical outcome. Our OviTex PRS products are reconstructive reinforced tissue matrices designed for implantation to reinforce soft-tissue where weakness exists in patients requiring soft tissue repair or reinforcement in plastic and reconstructive surgery. In connection with the March 2019 meeting of the General and Plastic Surgery Devices Panel of the Medical Devices Advisory Committee, the FDA stated that no surgical mesh device has been cleared or approved for use in breast surgery, and that to obtain such indication, the product sponsor must obtain an approved PMA. This statement applies to our OviTex PRS products as they are not cleared or approved for use in breast surgery and thus, we are prohibited from marketing them for that use. OviTex PRS or any other product we may develop for use in breast surgery will need to be approved specifically for that indication and there can be no guarantee that it will be approved. In October 2024, we received approval from the FDA for our investigational device exemption application relating to the study of the safety and effectiveness of our OviTex PRS product in implant-based breast reconstruction, but anticipate additional FDA interactions related to identification of an adequate clinical protocol that would be sufficient to support a pre-market application to obtain approval for an indication for OviTex PRS for use in breast reconstruction. There can be no assurance that we will be able to secure a PMA approval in a timely manner, or at all. Any marketing for OviTex PRS or any other product for a use in breast reconstruction surgery would be deemed off-label promotion of that product if it has been cleared for a general indication of use to reinforce or repair soft-tissue and has not received an approval specifically for use in breast surgery. We train our marketing personnel and direct sales force to not promote our OviTex or OviTex PRS products for uses outside of the FDA-cleared indications for use, known as “off-label uses.” We cannot, however, prevent a surgeon or medical professional from using our OviTex or OviTex PRS products or other products we may commercialize in the future for off-label uses.
Although we train our direct sales force not to promote our products for off-label uses, and our instructions for use in all markets specify that our products are not intended for use outside of those indications cleared or approved for use, the FDA or another regulatory authority could conclude that we have engaged in off-label promotion. If the FDA determines that our promotional or training materials constitute promotion of an off-label use, or make claims that are not supported by the available clinical data, it could request that we modify our training or promotional materials or subject us to regulatory or enforcement actions. It is also possible that other federal, state or non-U.S. enforcement authorities might take action under other regulatory authority if they consider our business activities to constitute promotion of an off-label use, or are otherwise objectionable, which could result in significant penalties, including, but not limited to, criminal, civil and administrative penalties, damages, fines, disgorgement, exclusion from participation in government healthcare programs and the curtailment of our operations.
Even if surgeons or medical professionals use our OviTex and OviTex PRS products only for their approved indications, a failure by such surgeons and medical professionals to employ proper surgical techniques to handle and use our products in accordance with proper instructions for use could result in product liability lawsuits, costly investigations and potentially affect our ability to achieve sufficient market penetration for our OviTex and OviTex PRS products. In those possible events, our reputation could be damaged and adoption of the products would be impaired. We may also be required to reassess the training, written instructions and product warnings or other labeling information we provide our customers. This process could require us to expend significant time and capital and could have a material adverse effect on our business, financial condition and results of operations and impair our ability to grow our business.
If we are unable to achieve and maintain adequate levels of coverage or reimbursement for our OviTex, OviTex PRS or other products we may commercialize in the future, our commercial success may be hindered.
Our ability to successfully commercialize and achieve market acceptance of our products depends, in significant part, on the availability of adequate financial coverage and reimbursement from third-party payors, including governmental payors (such as the Medicare and Medicaid programs in the U.S.), managed care organizations and private health insurers. The primary customers for our products are hospitals and ambulatory surgery centers who will then seek reimbursement from third-party payors for the procedures performed using our products. While some third-party payors currently cover and provide reimbursement for procedures using our currently cleared or approved products, we can give no assurance that these third-party payors will continue to provide coverage and adequate reimbursement for the procedures using our products, to permit hospitals and surgeons to offer procedures using our products to patients requiring treatment, or that current reimbursement levels for procedures using our products will continue.
46
Additionally, no uniform policy for coverage and reimbursement exists in the U.S. and coverage and reimbursement can differ significantly from payor to payor. If third-party payors reverse or limit their coverage for the procedures using our currently cleared or approved products in the future, this could have a material adverse effect on our business. If we are forced to lower the price we charge for our products, this could have a material adverse effect on our business, financial condition and results of operations and impair our ability to grow our business. See the section of this Annual Report titled “Coverage and Reimbursement” for more information.
Healthcare costs have risen significantly over the past decade, which has resulted in or led to numerous cost reform initiatives. Third-party payors, whether U.S. or non-U.S., or governmental or commercial, are developing increasingly sophisticated methods of controlling healthcare costs, including examining the cost effectiveness of procedures, in addition to their safety and efficacy, when making coverage and payment decisions. Payors continually review new and existing technologies for possible coverage and can, without notice, deny or reverse coverage or alter pre-authorization requirements for new or existing procedures. We cannot provide assurance that we will be successful in any efforts we may potentially undertake to reverse such non-coverage decisions. If we are not successful in reversing non-coverage policies, or if third-party payors that currently cover or reimburse certain procedures reverse or limit their coverage of such procedures in the future, or if other third-party payors issue similar policies, our business could be adversely impacted.
Our long-term growth may depend on our ability to enhance our product offerings.
It is important to our business that we continue to enhance our OviTex and OviTex PRS products and develop and introduce new reinforced tissue matrix products and complementary soft -tissue reconstruction solutions. Developing products is expensive and time-consuming and could divert management’s attention away from other aspects of our business. The success of any new product offerings or product enhancements to our OviTex and OviTex PRS products will depend on several factors, including our ability to:
| ● | properly identify and anticipate surgeon and patient needs; |
| ● | develop and introduce new products and product enhancements in a timely manner; |
| ● | avoid infringing upon the intellectual property rights of third parties; |
| ● | ensure the quality, manufacture and supply of new products by Aroa or other third-party manufacturers we engage; |
| ● | demonstrate, if required, the safety and efficacy of new products with data from preclinical studies, clinical trials and post-market clinical studies; |
| ● | obtain the necessary regulatory clearances or approvals for expanded indications, new products or product modifications; |
| ● | be fully FDA-compliant with marketing of new devices or products; |
| ● | provide adequate training to potential users of our new products; |
| ● | receive adequate coverage and reimbursement for procedures performed with our new products; and |
| ● | develop and expand an effective and dedicated sales and marketing team. |
47
If we are not successful in introducing new product indications and developing and commercializing new products and product enhancements, our ability to increase our revenue may be impaired, which could have a material adverse effect on our business, financial condition and results of operations.
In the future our products may become obsolete, which would negatively affect operations and financial condition.
The medical device industry is characterized by rapid and significant change. There can be no assurance that other companies will not succeed in developing or marketing devices and products that are more effective than our reinforced tissue matrix products or that would render our reinforced tissue matrix products obsolete or noncompetitive. Additionally, new surgical procedures, medications and other therapies could be developed that replace or reduce the importance of our products. Accordingly, our success will depend in part on our ability to respond quickly to medical and other changes through the development and introduction of new products. Our reinforced tissue matrix products have a limited shelf life and will expire if not timely used. Product development involves a high degree of risk, and there can be no assurance that our new product development efforts will result in any commercially successful products.
To successfully market and sell our products in markets outside of the U.S., we must address many international business risks with which we have limited experience.
Approximately 15%, 10% and 8% of our revenue for the years ended December 31, 2024, 2023 and 2022, respectively, came from sales in markets outside of the U.S. Part of our sales strategy is to maintain our European presence. European sales are subject to a number of risks, including:
| ● | difficulties in staffing and managing international operations; |
| ● | increased competition as a result of more products and procedures receiving regulatory approval in international markets; |
| ● | longer accounts receivable payment cycles and difficulties in collecting accounts receivable; |
| ● | fluctuations in currency exchange rates; |
| ● | non-U.S. certification and regulatory clearance or approval requirements; |
| ● | difficulties in developing effective marketing campaigns in unfamiliar countries; |
| ● | customs clearance and shipping delays; |
| ● | complexities associated with managing multiple payor reimbursement regimes, government payors or patient self-pay systems; |
| ● | political, social, and economic instability abroad, terrorist attacks, and security concerns in general; |
| ● | the impact of the macroeconomic factors, including pandemics, epidemics and other public health outbreaks, inflationary pressures and geopolitical conflicts, such as the ongoing Russia-Ukraine conflict and the current conflict in the Middle East (including any escalation or expansion); |
| ● | natural disasters and pandemics, epidemics or public health outbreaks, which result in lock-downs, travel restrictions and other restrictions on our ability to operate internationally; |
| ● | preference for locally produced products; |
| ● | potentially adverse tax consequences, including the complexities of non-U.S. value-added tax systems, tax inefficiencies related to our corporate structure, and restrictions on the repatriation of earnings; |
48
| ● | the burdens of complying with a wide variety of non-U.S. laws and different legal standards; and |
| ● | increased financial accounting and reporting burdens and complexities. |
If one or more of these risks are realized, our business, financial condition and results of operations could be adversely affected.
Risks Related to Our Reliance on Third Parties
We are highly dependent upon Aroa, as the exclusive manufacturer and supplier of our OviTex and OviTex PRS products.
In August 2012, we entered into our Aroa License which was amended and restated in July 2015. The Aroa License grants us an exclusive license in North America, the EU, United Kingdom, Norway, Switzerland, Russia and former Soviet satellite countries to certain intellectual property rights, including patents relating to the use of bovine and ovine rumen as a source of extracellular matrix. Under the Aroa License, Aroa is our exclusive manufacturer and supplier of our OviTex and OviTex PRS products.
We are reliant upon the intellectual property we license from Aroa for the development and commercialization of our products. Under the Aroa License, we hold an exclusive license to certain intellectual and technology rights to develop, commercialize and sell certain endoform regenerative template products derived from cows and sheep. The Aroa License also provides for cooperative development of our products utilizing the licensed intellectual property and all of our products rely on intellectual property owned by Aroa and licensed to us under the Aroa License. The Aroa License imposes various developmental and regulatory requirements upon us along with requiring us to make milestone payments upon the achievement of certain commercial and regulatory milestones. If we fail to comply with our obligations under the Aroa License, Aroa will have the right to terminate the Aroa License, in which event we would not be able to develop and market our products.
Aroa is required under the Aroa License to manufacture all of our OviTex and OviTex PRS products at its manufacturing and warehousing facility in Auckland, New Zealand. The production of all of our OviTex and OviTex PRS products in a single location exposes us to the risk of Aroa’s facility being harmed or rendered inoperable by natural or man-made disasters or pandemics, which may render it difficult or impossible for Aroa to perform its manufacturing and assembly activities for some time. Although we and Aroa intend to establish redundant production facilities to lessen the risk of production disruptions, we will need to ensure that any manufacturing facility complies with our quality expectations and applicable regulatory requirements. If we are unable to establish redundant manufacturing facilities in a timely manner, any disruption in the manufacture of our OviTex and OviTex PRS products at Aroa’s manufacturing and warehouse facility, the continued commercialization of our OviTex and OviTex PRS products, the supply of our OviTex and OviTex PRS products to customers and the development of any new reinforced tissue matrix products will be delayed, limited or prevented, which could have material adverse effect on our business, financial condition and results of operations.
Under the Aroa License, Aroa is responsible for supplying all of the raw materials and components used in the manufacture and assembly of our OviTex and OviTex PRS products. If Aroa is unable to supply the raw materials and components or to manufacture and assemble our OviTex and OviTex PRS products reliably and at the levels we anticipate or that are required by the market, we may be unable to acquire a substitute supply of raw materials and components on a timely basis, if at all.
Under the Aroa License Aroa also holds the FDA clearances under which we commercialize our OviTex products, including OviTex LPR and OviTex IHR, and maintains ultimate responsibility for all regulatory interactions with FDA relating to these OviTex products and decisions made with respect to changing or updating those clearances. If Aroa fails to comply with all applicable regulatory requirements and maintain the FDA clearances related to our OviTex products, we may be unable to commercialize our OviTex products on a timely basis, or at all. Our ability to supply our OviTex and OviTex PRS products commercially and to develop any future products depends, in part, on our ability to obtain these materials, components and products in accordance with regulatory requirements and in sufficient quantities for commercialization and clinical testing.
49
While Aroa has historically met our demand for its products and services on a timely basis in the past, we cannot guarantee that it will always be able to meet our demand for its products. If Aroa fails to meet demand or notifies us that it believes it will fail to meet demand for our OviTex and OviTex PRS products, we are required under the Aroa License to work with Aroa to cure its supply failure and may, only in certain circumstances and on a temporary basis, engage a replacement contract manufacturer to mitigate a failure by Aroa to meet demand for our OviTex and OviTex PRS products. As such, we are highly dependent upon Aroa’s continued ability to supply our OviTex and OviTex PRS products at the levels we require and any production shortfall that impairs the supply of our OviTex and OviTex PRS products could have a material adverse effect on our business, financial condition and results of operations and adversely affect our ability to satisfy demand for our OviTex and OviTex PRS products, which could adversely affect our product sales and operating results materially.
We, or our partners, may experience development or manufacturing problems, capacity constraints, or delays in the production of our products that could limit the potential growth of our revenue or increase our losses.
We may encounter unforeseen situations in Aroa’s manufacturing and assembly of our OviTex and OviTex PRS products that would result in delays or shortfalls in its production. For example, Aroa was unable to supply us with our products from September 2017 to December 2017 due to a quality testing process failure identified by Aroa. Any personnel shortages and reduced manufacturing capacity may also result in a disruption in production.
Based upon our current planned market adoption we believe we will reach our capacity limitations in the Aroa facility. Aroa expanded its manufacturing capacity, with approximately 15,000 square feet of additional manufacturing space being constructed in a neighboring facility, in 2022. If we are unable to successfully expand capacity, we may not be able to meet the demand for our products. In addition, Aroa’s production processes and assembly methods may have to change in order to accommodate any significant future expansion of its manufacturing capacity, which may increase our manufacturing costs, delay production of our products and adversely impact our business. Conversely, if demand for our OviTex and OviTex PRS products shifts such that Aroa’s manufacturing facility is operated below its capacity for an extended period, it may adjust its manufacturing operations to reduce fixed costs, which could lead to uncertainty and delays in manufacturing times and quality during any transition period.
If Aroa’s manufacturing activities are adversely impacted or if it is otherwise unable to keep up with demand for our OviTex and OviTex PRS products by successfully manufacturing, assembling, testing and shipping our OviTex and OviTex PRS products in a timely manner, our revenue could be impaired, market acceptance for our products could be adversely affected and our customers might instead purchase our competitors’ products, which would have a material adverse effect on our business, financial condition and results of operations.
Our products contain materials derived from animal sources and may become subject to additional regulation.
Our products are manufactured using ovine rumen. Products that contain materials derived from animal sources are increasingly subject to scrutiny in the media and by regulatory authorities. Regulatory authorities are concerned about the potential for the transmission of disease, particularly progressive neurodegenerative disorders, from animals to humans via those materials. In addition, the COVID-19 pandemic heightened public awareness of animals and animal products as a disease vector. Products that contain materials derived from animals, including our products, may become subject to additional regulation, or even be banned in certain countries, because of concern over the potential for the transmission of infectious agents. Significant new regulation, or a ban of our products, could impair our current business or our ability to expand our business, and in the case of a ban or suspension, could have a material adverse effect on our business, financial condition and results of operations.
Our supply of ovine rumen for use in manufacturing our products may be vulnerable to disruption due to natural disaster, disease or other events.
The ovine rumen used in the manufacturing of our products is sourced through Aroa in New Zealand. Although Aroa obtains its supply of ovine rumen from jurisdictions with sheep that are not currently known to carry any prion disease (progressive neurodegenerative disorders, including scrapie disease), there can be no assurance that these flocks will remain prion disease-free or that a future outbreak or presence of other unintended and potentially hazardous agents would not adversely affect our products or patients that may receive them.
50
The geographic concentration of our supply chain increases our vulnerability to disruption due to natural disasters, disease or other events. If there is a disruption in the supply of ovine rumen to our manufacturer and supplier, we may be unable to fulfill customer orders or delay the commercialization of new products.
We may also be prohibited from importing our products into the U.S. in the event of disease outbreak or other event impacting the sheep population in New Zealand. Any disruption in our supply lines could have a material adverse effect on our business, financial condition and results of operations.
Performance issues, service interruptions or price increases by our shipping carriers could adversely affect our business and harm our reputation and ability to provide our products on a timely basis.
Expedited, reliable shipping is essential to our operations. We rely heavily on providers of transport services for reliable and secure point-to-point transport of our OviTex portfolio products (and would rely heavily on such providers for any other products we may commercialize and ship in the future) to our customers and for tracking of these shipments. Should a carrier encounter delivery performance issues such as loss, damage or destruction of any of our products, it would be costly to replace such products in a timely manner and such occurrences may damage our reputation and lead to decreased demand for our OviTex portfolio products (or any other products we commercialize in the future) and increased cost and expense to our business. In addition, any significant increase in shipping rates could adversely affect our operating margins and results of operations. Similarly, strikes, severe weather, natural disasters, disease or other service interruptions affecting delivery services we use would adversely affect our ability to deliver our OviTex and OviTex PRS products (or any other products we commercialize in the future) on a timely basis. For example, disruptions to transportation infrastructure as a result of macroeconomic conditions may impact our ability to provide our products to our customers.
Risks Related to Intellectual Property Matters
We may need to license intellectual property from third parties, and such licenses may not be available or may not be available on commercially reasonable terms.
We may need to obtain licenses from third parties to advance our research or allow commercialization of our products, and we cannot provide any assurances that third-party patents do not exist which might be enforced against our products in the absence of such a license. The licensing and acquisition of third-party intellectual property rights is a competitive practice and companies that may be more established, or have greater resources than we do, may also be pursuing strategies to license or acquire third-party intellectual property rights that we may consider necessary or attractive in order to commercialize our products. We may fail to obtain any of these licenses on commercially reasonable terms, if at all. Even if we are able to obtain a license, it may be non-exclusive, thereby giving our competitors access to the same technologies licensed to us. In that event, we may be required to expend significant time and resources to develop or license replacement technology. If we are unable to do so, we may be unable to develop or commercialize the affected products, which could materially harm our business and the third parties owning such intellectual property rights could seek either an injunction prohibiting our sales, or, with respect to our sales, an obligation on our part to pay royalties and/or other forms of compensation. Licensing of intellectual property is of critical importance to our business and involves complex legal, business and scientific issues. If disputes over intellectual property that we have licensed prevent or impair our ability to maintain our current licensing arrangements on acceptable terms, we may not be able to successfully develop and commercialize the affected products, which would have a material adverse effect on our business.
If we fail to comply with our obligations under any license, collaboration or other agreements, we could lose intellectual property rights that are necessary for developing and protecting our products.
We have licensed certain intellectual property rights covering our current products from third parties, including Aroa. We are heavily dependent on our agreements with such third parties for our current products. If, for any reason, one or more of our agreements is terminated or we otherwise lose those rights, it could harm our business. Our license and other agreements impose, and any future collaboration agreements or license agreements we enter into are likely to impose various development, commercialization, funding, milestone, royalty, diligence, sublicensing, insurance, patent prosecution and enforcement or other obligations on us.
51
If we breach any material obligations, or use the intellectual property licensed to us in an unauthorized manner, we may be required to pay damages and the licensor may have the right to terminate the license, which could result in us being unable to develop, manufacture and sell products that are covered by the licensed technology, having to negotiate new or reinstated licenses on less favorable terms, or enabling a competitor to gain access to the licensed technology.
If we are unable to adequately protect our intellectual property rights, or if we are accused of infringing on the intellectual property rights of others, our competitive position could be harmed or we could be required to incur significant expenses to enforce or defend our rights.
Our commercial success will depend in part on our success in obtaining and maintaining issued patents, trademarks and other intellectual property rights in the U.S. and elsewhere and protecting our proprietary technology. If we do not adequately protect our intellectual property and proprietary technology, competitors may be able to use our technologies or the goodwill we have acquired in the marketplace and erode or negate any competitive advantage we may have, which could harm our business and ability to achieve profitability.
We own twenty-three issued or allowed U.S. patents and have twelve pending U.S. patent applications. As of December 31, 2024, we had rights, whether through ownership or licensing, to twenty-five issued or allowed U.S. patents, twelve pending U.S. patent applications, eight issued non-U.S. patents and seven pending non-U.S. patent applications., including six applications under the Patent Cooperation Treaty (“PCT”). Our issued U.S. patents will expire between 2035 and 2041. The licensed patents will expire between 2029 and 2031.
Our ability to enforce our patent rights depends on our ability to detect infringement. It may be difficult to detect infringers who do not advertise the components that are used in their products. Moreover, it may be difficult or impossible to obtain evidence of infringement in a competitor’s or potential competitor’s product. We may not prevail in any lawsuits that we initiate and the damages or other remedies awarded if we were to prevail may not be commercially meaningful.
The patent prosecution process is expensive and time-consuming, and we may not be able to file and prosecute all necessary or desirable patent applications at a reasonable cost or in a timely manner. It is also possible that we will fail to identify patentable aspects of our research and development output before it is too late to obtain patent protection. We cannot provide any assurances that any of our patents, or patents to which we have ownership rights through licensing agreements, have, or that any of our pending patent applications that mature into issued patents will include, claims with a scope sufficient to protect our OviTex and OviTex PRS products, any additional features we develop for our OviTex and OviTex PRS products or any new products we seek to develop in the future. Other parties may have developed technologies that may be related or competitive to our OviTex and OviTex PRS products, may have filed or may file patent applications and may have received or may receive patents that overlap or conflict with our patent applications, either by claiming the same methods or devices or by claiming subject matter that could dominate our patent position. The patent positions of medical device companies, including our patent position, may involve complex legal, scientific and factual questions, and, therefore, the issuance, scope, validity and enforceability of any patent claims that we may obtain cannot be predicted with certainty. Patents, if issued, may be challenged, deemed unenforceable, invalidated or circumvented. Proceedings challenging our patents could result in either loss of the patent or denial of the patent application or loss or reduction in the scope of one or more of the claims of the patent or patent application. In addition, such proceedings may be costly. Thus, any patents that we may own, or to which we have ownership rights through licensing agreements, may not provide any protection against competitors. Furthermore, an adverse decision in a judicial or administrative proceeding can result in a third party receiving the patent right sought by us, which in turn could affect our ability to commercialize our products.
Patents covering our products could be found invalid or unenforceable if challenged in court or before administrative bodies in the U.S. or abroad.
Although an issued patent is presumed valid and enforceable, its issuance is not conclusive as to its validity or its enforceability and it may not provide us with adequate proprietary protection or competitive advantages against competitors with similar products.
52
Competitors could purchase our OviTex or OviTex PRS products and attempt to replicate the competitive advantages we derive from our development efforts, willfully infringe our intellectual property rights, design around the relevant patents, or develop and obtain patent protection for more effective technologies, designs or methods. We may be unable to prevent the unauthorized disclosure or use of our technical knowledge or trade secrets by consultants, suppliers, vendors, former employees and current employees. The laws of some non-U.S. countries do not protect our proprietary rights to the same extent as the laws of the U.S., and we may encounter significant problems in protecting our proprietary rights in these countries.
In addition, proceedings to enforce or defend our patents, or patents to which we have ownership rights through licensing agreements, could put those patents at risk of being invalidated, held unenforceable or interpreted narrowly. Such proceedings could also provoke third parties to assert claims against us, including that some or all of the claims in one or more of those patents are invalid or otherwise unenforceable. If any of the patents covering our OviTex and OviTex PRS products are invalidated or found unenforceable, or if a court found that valid, enforceable patents held by third parties covered one or more of our products, our competitive position could be harmed or we could be required to incur significant expenses to enforce or defend our rights.
Third parties may assert ownership or commercial rights to inventions we develop.
Third parties may in the future make claims challenging the inventorship or ownership of our intellectual property. In addition, we may face claims by third parties that our agreements with employees, contractors or consultants obligating them to assign intellectual property to us are ineffective or in conflict with prior or competing contractual obligations of assignment, which could result in ownership disputes regarding intellectual property we have developed or will develop and interfere with our ability to capture the commercial value of such intellectual property. Litigation may be necessary to resolve an ownership dispute, and if we are not successful, we may be precluded from using certain intellectual property or may lose our exclusive rights in such intellectual property. Either outcome could harm our business and competitive position.
Litigation or other proceedings or third-party claims of intellectual property infringement could require us to spend significant time and money, enter into license agreements for disputed intellectual property and could prevent us from selling our products.
Our commercial success will depend in part on not infringing the patents or violating other proprietary rights of others. Significant litigation regarding patent rights occurs in our industry. Our competitors may have applied for or obtained, or may in the future apply for and obtain, patents that will prevent, limit or otherwise interfere with our ability to make, use and sell our products. We do not always conduct independent reviews of patents issued to third parties. In addition, patent applications in the U.S. and elsewhere can be pending for many years before issuance, or unintentionally abandoned patents or applications can be revived, so there may be applications of others now pending or recently revived patents of which we are unaware. Patent applications in the U.S., the EU and elsewhere are published approximately 18 months after the earliest filing for which priority is claimed, with such earliest filing date being commonly referred to as the priority date. These applications may later result in issued patents, or the revival of previously abandoned patents, that will prevent, limit or otherwise interfere with our ability to develop and market our products. Third parties may assert claims that we are employing their proprietary technology without authorization, including claims from competitors or from nonpracticing entities that have no relevant product revenue and against whom our own patent portfolio may have no deterrent effect.
As we continue to commercialize our products in their current or updated forms, launch new products and enter new markets, we expect competitors may claim that one or more of our products infringe their intellectual property rights as a strategy to impede our commercialization and entry into new markets. The large number of patents, the rapid rate of new patent applications and issuances, the complexities of the technologies involved, and the uncertainty of litigation may increase the risk of business resources and management’s attention being diverted to patent litigation. We have received, and we may in the future receive, letters or other threats or claims from third parties inviting us to take licenses under, or alleging that we infringe, their patents.
53
Moreover, we may become party to adversarial proceedings regarding our or third-party patent portfolios. Such proceedings could include supplemental examination or contested post-grant proceedings such as review, reexamination, inter partes review, interference or derivation proceedings before the U.S. Patent and Trademark Office (“USPTO”) and challenges in U.S. District Courts. Patents may be subjected to opposition, post-grant review or comparable proceedings lodged in various foreign, both national and regional, patent offices. The legal threshold for initiating litigation or contested proceedings may be low, so that even lawsuits or proceedings with a low probability of success might be initiated. Litigation and contested proceedings can also be expensive and time-consuming, and our adversaries in these proceedings may have the ability to dedicate substantially greater resources to prosecuting these legal actions than we can. We may also occasionally use these proceedings to challenge the patent rights of others. We cannot be certain that any particular challenge will be successful in limiting or eliminating the challenged patent rights of the third party.
Any lawsuits resulting from such allegations could subject us to significant liability for damages and/ or invalidate our proprietary rights. Any potential intellectual property litigation also could force us to do one or more of the following:
| ● | stop making, selling or using products or technologies that allegedly infringe the asserted intellectual property; |
| ● | lose the opportunity to license our technology to others or to collect royalty payments; |
| ● | incur significant legal expenses, including, in some cases, the attorney’s fees and costs of litigation to the party whose intellectual property rights we may be found to be infringing; |
| ● | pay substantial damages (possibly treble damages) or royalties to the party whose intellectual property rights on which we may be found to be infringing; |
| ● | redesign products that contain the allegedly infringing intellectual property; and |
| ● | attempt to obtain a license to the relevant intellectual property from third parties, which may not be available on reasonable terms or at all. |
Any litigation or claim against us, even those without merit, may cause us to incur substantial costs, and could place a significant strain on our financial resources, divert the attention of management from our business and harm our reputation. If we are found to infringe the intellectual property rights of third parties, we could be required to pay substantial damages (which may be increased up to three times of awarded damages) and/or substantial royalties and could be prevented from selling our products unless we obtain a license or are able to redesign our products to avoid infringement. In addition, we may choose to seek, or be required to seek, a license from a third party, which may not be available on acceptable terms, if at all. Even if a license can be obtained on acceptable terms, the rights may be non-exclusive, which could give any competitors access to the same technology or intellectual property rights license to us. Any such license may not be available on reasonable terms, if at all, and there can be no assurance that we would be able to redesign our products in a technically feasible way that would not infringe the intellectual property rights of others. We could encounter delays in product introductions while we attempt to develop alternative methods or products. If we fail to obtain a required license, the holders of any such patents may be able to block us, our licenses or our collaborators from marketing products based on the disputed technology until such patents expire, which could limit our ability to generate revenue or achieve profitability and possibly prevent us from generating revenue sufficient to sustain our operations.
Even if we were ultimately to prevail, any of these events could require us to divert substantial financial and management resources that we would otherwise be able to devote to our business. Intellectual property litigation, regardless of its outcome, may cause negative publicity, adversely impact prospective customers, cause product shipment delays, or prohibit us from manufacturing, importing, marketing or otherwise commercializing our products, services and technology. In addition, if the breadth or strength of protection provided the patents and patent applications we own or in-license is threatened, it could dissuade companies from collaborating with us to license, develop or commercialize current or future products. In addition, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation.
54
There could also be public announcements of the results of hearings, motions or other interim proceedings or developments, and if securities analysts or investors view these announcements in a negative light, the price of our common stock could be adversely affected.
In addition, we generally indemnify our customers with respect to infringement by our products of the proprietary rights of third parties. Third parties may assert infringement claims against our customers. These claims may require us to initiate or defend protracted and costly litigation on behalf of our customers, regardless of the merits of these claims. If any of these claims succeed or settle, we may be forced to pay damages or settlement payments on behalf of our customers or may be required to obtain licenses for the products they use. If we cannot obtain all necessary licenses on commercially reasonable terms, our customers may be forced to stop using our products.
If we are unable to protect the confidentiality of our trade secrets, our business and competitive position could be harmed.
We also rely upon copyright and trade secret protection, as well as non-disclosure agreements and invention assignment agreements with our employees, consultants and third parties, to protect our confidential and proprietary information.
In addition to contractual measures, we try to protect the confidential nature of our proprietary information using commonly accepted physical and technological security measures. Such measures may not provide adequate protection for our proprietary information. Our security measures may not prevent an employee or consultant from misappropriating our trade secrets and providing them to a competitor, and recourse we take against such misconduct may not provide an adequate remedy to protect our interests fully. Unauthorized parties may also attempt to copy or reverse engineer certain aspects of our products that we consider proprietary. Enforcing a claim that a party illegally disclosed or misappropriated a trade secret can be difficult, expensive and time-consuming, and the outcome of any such claim is unpredictable. Trade secret violations are often a matter of state law, and the criteria for protection of trade secrets can vary among different jurisdictions. In addition, trade secrets may be independently developed or reverse engineered by others in a manner that could prevent legal recourse by us. If any of our confidential or proprietary information, such as our trade secrets, were to be disclosed or misappropriated, or if any such information was independently developed by a competitor, our business and competitive position could be harmed.
If our trademarks and trade names are not adequately protected, then we may not be able to build name recognition in our markets of interest.
If our trademarks and trade names are not adequately protected, then we may not be able to build name recognition in our target markets and our business may be adversely affected. At times, competitors may adopt trade names or trademarks similar to ours, thereby impeding our ability to build brand identity, possibly leading to market confusion and potentially requiring us to pursue legal action. In addition, there could be potential trade name or trademark infringement claims brought by owners of other registered trademarks or trademarks that incorporate variations of our unregistered trademarks or trade names. If we are unable to successfully register our trademarks and trade names and establish name recognition based on our trademarks and trade names, then we may not be able to compete effectively and our business may be adversely affected. Our efforts to enforce or protect our proprietary rights related to trademarks, trade secrets, domain names, copyrights or other intellectual property may be ineffective and could result in substantial costs and diversion of resources and could adversely impact our financial condition or results of operations.
We may be unable to enforce our intellectual property rights throughout the world.
Filing, prosecuting and defending patents covering our products in all countries throughout the world would be prohibitively expensive, and the laws of some foreign countries do not protect intellectual property rights to the same extent as the laws of the U.S. Many companies have encountered significant problems in protecting and defending intellectual property rights in certain foreign jurisdictions. This could make it difficult for us to stop infringement of our foreign patents, if obtained, or the misappropriation of our other intellectual property rights. For example, some foreign countries have compulsory licensing laws under which a patent owner must grant licenses to third parties. In addition, some countries limit the enforceability of patents against third parties, including government agencies or government contractors. In these countries, patents may provide limited or no benefit. Patent protection must ultimately be sought on a country-by-country basis, which is an expensive and time-consuming process with uncertain outcomes.
55
Accordingly, we may choose not to seek patent protection in certain countries, and we will not have the benefit of patent protection in such countries. Additionally, in the event that our trademarks are successfully challenged, we could be forced to rebrand our products, which could result in loss of brand recognition and could require us to devote resources to advertising and marketing new brands. Our competitors may infringe our trademarks, and we may not have adequate resources to enforce our trademarks.
Proceedings to enforce our patent or trademark rights in foreign jurisdictions could result in substantial costs and divert our efforts and attention from other aspects of our business. Accordingly, our efforts to protect our intellectual property rights in such countries may be inadequate.
Third parties may assert that our employees or consultants have wrongfully used or disclosed confidential information or misappropriated trade secrets.
We employ individuals who previously worked with other companies, including our competitors. Although we try to ensure that our employees and consultants do not use the proprietary information or know-how of others in their work for us, we may be subject to claims that we or our employees, consultants or independent contractors have inadvertently or otherwise used or disclosed intellectual property or personal data, including trade secrets or other proprietary information, of a former employer or other third party. Litigation may be necessary to defend against these claims. If we fail in defending any such claims or settling those claims, in addition to paying monetary damages or a settlement payment, we may lose valuable intellectual property rights or personnel. Even if we are successful in defending against such claims, litigation could result in substantial costs and be a distraction to management and other employees.
Recent changes in U.S. patent laws may limit our ability to obtain, defend and/or enforce our patents.
The U.S. has recently enacted and implemented wide ranging patent reform legislation. The U.S. Supreme Court has ruled on several patent cases in recent years, either narrowing the scope of patent protection available in certain circumstances or weakening the rights of patent owners in certain situations. In addition to increasing uncertainty with regard to our ability to obtain patents in the future, this combination of events has created uncertainty with respect to the value of patents, once obtained. Depending on actions by the U.S. Congress, the U.S. federal courts, and the USPTO, the laws and regulations governing patents could change in unpredictable ways that could weaken our ability to obtain new patents or to enforce patents that we have licensed or that we might obtain in the future. Similarly, changes in patent law and regulations in other countries or jurisdictions, changes in the governmental bodies that enforce them or changes in how the relevant governmental authority enforces patent laws or regulations may weaken our ability to obtain new patents or to enforce patents that we have licensed or that we may obtain in the future.
Obtaining and maintaining our patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements.
The USPTO and other patent agencies require compliance with a number of procedural, documentary, fee payment and other similar provisions during the patent application process. In addition, periodic maintenance and annuity fees on any issued patent are due to be paid to the USPTO and other patent agencies over the lifetime of the patent. While an inadvertent failure to make payment of such fees or to comply with such provisions can in many cases be cured by additional payment of a late fee or by other means in accordance with the applicable rules, there are situations in which non-compliance with such provisions will result in the abandonment or lapse of the patent or patent application, and the partial or complete loss of patent rights in the relevant jurisdiction. Non-compliance events that could result in abandonment or lapse of a patent or patent application include failure to respond to official actions within prescribed time limits, non-payment of fees and failure to properly legalize and submit formal documents within prescribed time limits. If we or our licensors fail to maintain the patents and patent applications covering our product or if we or our licensors otherwise allow our patents or patent applications to be abandoned or lapse, it can create opportunities for competitors to enter the market, which would hurt our competitive position and could impair our ability to successfully commercialize our products.
56
Patent terms may be inadequate to protect our competitive position on our product candidates for an adequate amount of time.
The term of any individual patent depends on applicable law in the country where the patent is granted. In the U.S., provided all maintenance fees are timely paid, a patent generally has a term of 20 years from its application filing date or earliest claimed non-provisional filing date. Extensions may be available under certain circumstances, but the life of a patent and, correspondingly, the protection it affords is limited. Even if we or our licensors obtain patents covering our products, when the terms of all patents covering a product expire, our business may become subject to competition from products identical or similar to ours. As a result, our owned and licensed patent portfolio may not provide us with sufficient rights to exclude others from commercializing products similar or identical to ours.
We may be unable to obtain a patent term extension in the U.S. under the Hatch-Waxman Act and in foreign countries under similar legislation.
In the U.S., a patent that covers a drug product or medical device approved by the FDA may be eligible for a term extension designed to restore the period of the patent term that is lost during the premarket regulatory review process conducted by the FDA. Depending upon the timing, duration and conditions of FDA marketing approval of our products, one or more of our U.S. patents may be eligible for limited patent term extension under the Drug Price Competition and Patent Term Restoration Act of 1984, or the Hatch-Waxman Act, which permits a patent term extension of up to five years for a patent covering an approved product as compensation for effective patent term lost during product development and the FDA regulatory review process. A patent term extension cannot extend the remaining term of a patent beyond a total of 14 years from the date of product approval, and only claims covering such approved drug product, a method for using it or a method for manufacturing it may be extended. In the European Union, our product candidates may be eligible for term extensions based on similar legislation. In either jurisdiction, however, we may not receive an extension if we fail to apply within applicable deadlines, fail to apply prior to expiration of relevant patents or otherwise fail to satisfy applicable requirements. Even if we are granted such extension, the duration of such extension may be less than our request. If we are unable to obtain a patent term extension, or if the term of any such extension is less than our request, the period during which we can enforce our patent rights for that product will be in effect shortened and our competitors may obtain approval to market competing products sooner. The resulting reduction of years of revenue from applicable products could be substantial.
Intellectual property rights do not necessarily address all potential threats.
The degree of future protection afforded by our intellectual property rights is uncertain because intellectual property rights have limitations and may not adequately protect our business or permit us to maintain our competitive advantage. For example:
| ● | others may be able to make products that are similar to our products or utilize similar technology but that are not covered by the claims of our patents or that incorporate certain technology in our products that is in the public domain; |
| ● | we, or our future licensors or collaborators, might not have been the first to make the inventions covered by the applicable issued patent or pending patent application that we own now or may own or license in the future; |
| ● | we, or our future licensors or collaborators, might not have been the first to file patent applications covering certain of our or their inventions; |
| ● | we may not be able to successfully commercialize our products before our relevant patents we may have, or to which we have ownership rights through licensing agreements, expire; |
| ● | others may independently develop similar or alternative technologies or duplicate any of our technologies without infringing our intellectual property rights; |
57
| ● | it is possible that our current or future pending patent applications will not lead to issued patents; |
| ● | issued patents that we hold rights to may be held invalid or unenforceable, including as a result of legal challenges by our competitors or other third parties; |
| ● | our competitors or other third parties might conduct research and development activities in countries where we do not have patent rights and then use the information learned from such activities to develop competitive products for sale in our major commercial markets; |
| ● | we may not develop additional proprietary technologies that are patentable; |
| ● | the patents of others may harm our business; and |
| ● | we may choose not to file a patent in order to maintain certain trade secrets or know-how, and a third party may subsequently file a patent covering such intellectual property. |
Any of the foregoing could have a material adverse effect on our business, financial condition and results of operations.
Risks Related to Government Regulation
Our products and operations are subject to extensive government regulation and oversight both in the U.S. and internationally.
Our products are regulated as medical devices. We and our products are subject to extensive regulation in the U.S. and internationally including by the FDA and competent authorities of the EU member states. The FDA and other foreign equivalents regulate, among other things, with respect to medical devices: design, development and manufacturing; testing, labeling, content and language of instructions for use and storage; clinical trials; product safety; establishment registration and device listing; marketing, sales and distribution; premarket clearance and approval; record keeping procedures; advertising and promotion; recalls and field safety corrective actions; post-market surveillance, including reporting of deaths or serious injuries and malfunctions that, if they were to recur, could lead to death or serious injury; post-market approval studies; and product import and export.
The regulations to which we are subject are complex, have become more stringent over time and are subject to further change. Failure to comply with applicable regulations could jeopardize our ability to sell our products and result in enforcement actions such as: warning letters; untitled letters; Form 483s; fines; injunctions; civil penalties; termination of distribution; recalls or seizures of products; delays in the introduction of products into the market; total or partial suspension of production; refusal to grant future clearances or approvals; withdrawals or suspensions of current approvals, resulting in prohibitions on sales of our products; and in the most serious cases, criminal penalties.
We may not receive, or may be significantly delayed in receiving, the necessary clearances or approvals for our future products and modifications to our current products may require new 510(k) clearances or PMA approvals, and may require us to cease marketing or recall the modified products until clearances or approvals are obtained.
An element of our strategy is to continue to add new features and expand the indications and uses for our current products. In the U.S., before we can market a new medical device, or a new use of, new claim for or significant modification to an existing product, we must first receive marketing authorization, such as either clearance under Section 510(k) of the FDCA or approval of a PMA from the FDA, unless an exemption applies. Our products are cleared with the FDA, through clearances obtained and, with the exception of the clearances relating to our OviTex PRS products, held by Aroa, under Section 510(k) of the FDCA, which permits marketing of a device if it is “substantially equivalent” to an already legally-marketed “predicate” device, which includes a device that has been previously cleared through the 510(k) process, a device that was legally marketed prior to May 28, 1976 (preamendments device), a device that was originally on the U.S. market pursuant to an approved PMA and later downclassified, or a 510(k)-exempt device.
58
To be “substantially equivalent,” the proposed device must have the same intended use as the predicate device, and either have the same technological characteristics as the predicate device or have different technological characteristics and the information in the premarket notification demonstrates that the device is as safe and effective and does not raise different questions of safety or effectiveness than the predicate device. Clinical data are sometimes required to support substantial equivalence. In the PMA process, the FDA must determine that a proposed device is safe and effective for its intended use based, in part, on extensive data, including, but not limited to, technical, preclinical, clinical trial, manufacturing and labeling data. The PMA process is typically required for devices that are deemed to pose the greatest risk, such as life-sustaining, life-supporting or implantable devices. To date, our products have been the subject of cleared 510(k)s, obtained and, with the exception of the clearances relating to our OviTex PRS products, held by Aroa. For more information regarding the regulation of our products, see “Business — Government Regulation.”
Modifications to products that are approved through a PMA application generally require FDA approval. Similarly, certain modifications made to products cleared through a 510(k) may require a new 510(k) clearance. Both the PMA approval and the 510(k) clearance process can be expensive, lengthy and uncertain. The FDA’s 510(k) clearance process usually takes from three to 12 months, but can last longer. The process of obtaining a PMA is much more costly and uncertain than the 510(k) clearance process and generally takes from one to three years, or even longer, from the time the application is filed with the FDA. In addition, a PMA generally requires the performance of one or more clinical trials. Despite the time, effort and cost, we cannot assure you that any particular device will be approved or cleared by the FDA. Any delay or failure to obtain necessary regulatory clearances or approvals could harm our business.
In the U.S., Aroa has obtained and holds 510(k) clearances from the FDA to market our OviTex products and obtained the 510(k) clearances from the FDA held by us for our first two OviTex PRS products, while we obtained and hold the 510(k) clearance for our OviTex PRS Long-Term Resorbable product. An element of our strategy is to continue to upgrade our reinforced tissue matrix products. We expect that any such modifications may require new 510(k) clearances; however, future modifications may be subject to the substantially more costly, time-consuming and uncertain PMA process. The FDA will require a PMA, rather than a 510(k) clearance for the use of OviTex PRS in breast reconstruction. If the FDA requires us to go through a lengthier, more rigorous examination for future products or modifications to existing products than we had expected, product introductions or modifications could be delayed or canceled, which could cause our sales to decline.
The FDA can delay, limit or deny clearance or approval of a device for many reasons, including:
| ● | we may not be able to demonstrate to the FDA’s satisfaction that the product or modification is substantially equivalent to the proposed predicate device or safe and effective for its intended use; |
| ● | the data from our preclinical studies and clinical trials may be insufficient to support clearance or approval, where required; and |
| ● | the manufacturing process or facilities we use may not meet applicable requirements. |
In addition, the FDA may change its clearance and approval policies, adopt additional regulations or revise existing regulations, or take other actions which may prevent or delay approval or clearance of our future products under development.
Even after we have obtained the proper regulatory clearance or approval to market a product, we have ongoing responsibilities under FDA regulations. The failure to comply with applicable regulations could jeopardize our ability to sell our reinforced tissue matrix products and result in enforcement actions such as:
| ● | warning letters, untitled letters or Form 483s; |
| ● | fines; |
| ● | injunctions; |
59
| ● | civil penalties; |
| ● | termination of distribution; |
| ● | recalls or seizures of products; |
| ● | delays in the introduction of products into the market; |
| ● | total or partial suspension of production; |
| ● | refusal to grant future clearances or approvals; |
| ● | withdrawals or suspensions of current clearances or approvals, resulting in prohibitions on sales of our products; and |
| ● | in the most serious cases, criminal penalties. |
Any of these sanctions could result in higher than anticipated costs or lower than anticipated sales and harm our reputation, business, financial condition and results of operations.
In addition, regulators may determine that our financial relationships with our principal investigators resulted in a perceived or actual conflict of interest that may have affected the interpretation of a study. Principal investigators for our clinical trials may serve as speakers or consultants to us from time to time and receive compensation in connection with such services. Under certain circumstances, we may be required to report some of these relationships to the FDA or other regulatory authority. The FDA or other regulatory authority may conclude that a financial relationship between us and a principal investigator has created a conflict of interest or otherwise affected interpretation of the study. The FDA or other regulatory authority may therefore question the integrity of the data generated at the applicable clinical trial site and the utility of the clinical trial itself may be jeopardized. This could result in a delay in approval, or rejection, of our marketing applications by the FDA or other regulatory authority, as the case may be, and may ultimately lead to the denial of marketing approval of one or more of our future products.
To sell our products in member countries of the EEA our products must comply with the general safety and performance requirements of the EU MDR, which became effective on May 26, 2021. Compliance with the new MDR requirements is a prerequisite to be able to affix the Conformité Européenne, or CE, mark to our products, without which they cannot be sold or marketed in the EEA. In the EEA, we have obtained the CE mark for our OviTex products. For more information regarding regulation of our products, see “Business—Government Regulation.”
An element of our strategy is to continue to add new features and expand the indications and uses for our current products. Any modification to a 510(k)-cleared device that could significantly affect its safety or effectiveness, or that would constitute a major change in its intended use, design or manufacture, requires a new 510(k) clearance or, possibly, approval of a PMA. The FDA requires every manufacturer to make this determination in the first instance, but the FDA may review any manufacturer’s decision. The FDA may not agree with our decisions regarding whether new clearances or approvals are necessary. Such modifications can be expensive and uncertain in time and outcome. We may not be able to obtain additional 510(k) clearances or PMAs for new products or for modifications to, or additional indications for, our products in a timely fashion, or at all. Delays in obtaining required future clearances or approvals would adversely affect our ability to introduce new or enhanced products in a timely manner, which in turn would harm our future growth. We have made modifications to our products in the past and expect to make additional modifications in the future that we believe do not or will not require additional clearances or approvals. If the FDA disagrees and requires new clearances or approvals for these modifications, we may be required to recall and to stop selling or marketing such products as modified until we obtain clearance or approval, which could harm our operating results and require us to redesign such products. In these circumstances, we may be subject to significant enforcement actions, including significant fines or penalties.
60
International regulatory approval processes may take more or less time than the FDA clearance or approval process. If we fail to comply with applicable FDA and comparable non-U.S. regulatory requirements, we may not receive regulatory clearances or approvals or may be subject to FDA or comparable non-U.S. enforcement actions.
We may be unable to obtain future regulatory clearance or approval in a timely manner, or at all, especially if existing regulations are changed or new regulations are adopted. For example, the FDA clearance or approval process can take longer than anticipated due to requests for additional clinical data and changes in regulatory requirements. A failure or delay in obtaining necessary regulatory clearances or approvals would materially adversely affect our business, financial condition and results of operations.
Although we have obtained regulatory clearance for our products, they will remain subject to extensive regulatory scrutiny.
We are subject to ongoing and pervasive regulatory requirements governing, among other things, the manufacturing, marketing, advertising, medical device reporting, selling and promoting our products. For example, we must submit periodic reports to the FDA as a condition of our clearance under Section 510(k). These reports include safety and effectiveness information about the device after its clearance. Failure to submit such reports, or failure to submit the reports in a timely manner, could result in enforcement action by the FDA.
Even after we have obtained the proper regulatory approval to market our products, they will be subject to ongoing regulatory requirements for design, development, manufacturing, testing, labeling, packaging, storage, advertising, promotion, sampling, record-keeping, recalls and field safety corrective actions, conduct of post-marketing studies and submission of safety, effectiveness and other post-market information, including both federal and state requirements in the U.S. and requirements of comparable non-U.S. regulatory authorities. Our failure to comply with applicable regulatory requirements could result in enforcement action by the FDA and applicable state regulatory authorities, which may include any of the following sanctions:
| ● | issue warning or untitled letters that would result in adverse publicity or may require corrective advertising; |
| ● | fines, injunctions, consent decrees and civil penalties; |
| ● | recalls, termination of distribution, administrative detention, or seizure of our products; |
| ● | customer notifications or repair, replacement or refunds; |
| ● | operating restrictions or partial suspension or total shutdown of production; |
| ● | delays in or refusal to grant our requests for future clearances under Section 510(k) or premarket approvals or EU regulatory approvals of new products, new intended uses, or modifications to existing products; |
| ● | withdrawal or suspension of regulatory clearances or approvals; |
| ● | FDA refusal to issue certificates to non-U.S. governments needed to export products for sale in other countries; and |
| ● | criminal prosecution. |
Any government investigation of alleged violations of law could require us to expend significant time and resources in response, and could generate negative publicity. Any failure to comply with ongoing regulatory requirements may significantly and adversely affect our ability to commercialize and generate revenue from our products. If regulatory sanctions are applied or if regulatory clearance or approval is withdrawn, it would have a material adverse effect on our business, financial condition and results of operations.
61
Our products must be manufactured in accordance with federal and state regulations, and we could be forced to recall our products or terminate production if we fail to comply with these regulations.
The methods used in, and the facilities used for, the manufacture of our products must comply with the FDA’s QSR which is a complex regulatory scheme that covers the procedures and documentation of the design, testing, production, process controls, quality assurance, labeling, packaging, handling, storage, distribution, installation, servicing and shipping of medical devices. Furthermore, Aroa must maintain facilities, procedures and operations that comply with our quality standards and applicable regulatory requirements. The FDA enforces the QSR through periodic announced or unannounced inspections of medical device manufacturing facilities, which may include the facilities of subcontractors. Our products are also subject to similar state regulations and various EU laws and regulations governing manufacturing.
Aroa may not take the necessary steps to comply with applicable regulations, which could cause delays in the delivery of our products. For example, following an inspection in March 2017, Aroa received an FDA Form 483 that contained multiple observations related to its manufacturing processes and procedures. In addition, failure to comply with applicable FDA requirements or later discovery of previously unknown problems with our products or manufacturing processes could result in, among other things: untitled letters or warning letters; fines, injunctions or civil penalties; suspension or withdrawal of approvals; seizures or recalls of our products; total or partial suspension of production or distribution; administrative or judicially imposed sanctions; the FDA’s refusal to grant pending or future clearances or approvals for our products; clinical holds; refusal to permit the import or export of our products; and criminal prosecution of us or our employees.
Any of these actions could significantly and negatively affect the supply of our products. If any of these events occurs, our reputation could be harmed, we could be exposed to product liability claims and we could lose customers and experience reduced sales and increased costs.
If guidelines for soft-tissue reconstruction surgery change or the standard of care evolves, we may need to redesign and seek new marketing authorization from the FDA for our OviTex and OviTex PRS products or other products we may commercialize in the future.
If guidelines for soft-tissue reconstruction surgery change or the standard of care for reconstructing tissue evolves, we may need to redesign the applicable product and seek new approvals from the FDA. Our clearances under Section 510(k) of the FDCA are based on current soft-tissue reconstruction surgery guidelines. If the guidelines change so that different surgeries or products become desirable, the clinical utility of one or more of our OviTex and OviTex PRS products or other products we may commercialize in the future could be diminished and our business could be adversely affected.
If any of our products cause or contribute to a death, serious injury, or other adverse medical events, or malfunction in certain ways, we will be required to report these events to FDA and other comparable regulatory authorities under applicable medical device reporting regulations, which can result in voluntary corrective actions or agency enforcement actions. If we fail to comply with our reporting obligations, we would be subject to sanctions that could harm our reputation, business, financial condition and results of operations. The discovery of serious safety issues with our products, or a recall of our products either voluntarily or at the direction of the FDA or another governmental authority, could have a negative impact on us.
We are subject to the FDA’s medical device reporting regulations and similar EU and other foreign regulations, which require us to report to the FDA when we receive or become aware of information that reasonably suggests that one or more of our products may have caused or contributed to a death or serious injury or malfunctioned in a way that, if the malfunction were to recur, could cause or contribute to a death or serious injury. The timing of our obligation to report is triggered by the date we become aware of the adverse event as well as the nature of the event. We may fail to report adverse events of which we become aware within the prescribed timeframe. We may also fail to recognize that we have become aware of a reportable adverse event, especially if it is not reported to us as an adverse event or if it is an adverse event that is unexpected or removed in time from the use of the product. If we fail to comply with our reporting obligations, the FDA could take action, including untitled letters, warning letters, administrative actions, criminal prosecution, imposition of civil monetary penalties, revocation of related approvals, seizure of our products or delay in clearance or approval of future products.
62
The FDA and foreign regulatory agencies have the authority to require the recall of commercialized products in the event of material deficiencies or defects in design or manufacture of a product or in the event that a product poses an unacceptable risk to health. The FDA’s authority to require a recall must be based on a finding that there is reasonable probability that the device could cause serious injury or death. We may also choose to voluntarily recall a product if any material deficiency is found. For example, in April 2018, Aroa, as the product manufacturer, issued a voluntary recall of our resorbable OviTex products due to a reduction in the labeled shelf life of such products from 24 months to 18 months. The recall included a total of 1,974 units from 48 manufacturing lots and was ultimately terminated in April 2019. A government-mandated or voluntary recall by us could also occur as a result of an unacceptable risk to health, component failures, malfunctions, manufacturing defects, labeling or design deficiencies, packaging defects or other deficiencies or failures to comply with applicable regulations. Product defects or other errors may occur in the future.
Depending on the corrective action we take to redress a product’s deficiencies or defects, the FDA may require, or we may decide, that we will need to obtain new clearances or approvals for the device before we may market or distribute the corrected device. Seeking such approvals may delay our ability to replace the recalled devices in a timely manner. Moreover, if we do not adequately address problems associated with our devices, we may face additional regulatory enforcement action, including FDA warning letters, product seizure, injunctions, administrative penalties or civil or criminal fines.
Companies are required to maintain certain records of recalls and corrections, even if they are not reportable to the FDA. We may initiate voluntary withdrawals or corrections for our products in the future that we determine do not require notification of the FDA. If the FDA disagrees with our determinations, it could require us to report those actions as recalls and we may be subject to enforcement action. A future recall announcement could harm our reputation with customers, potentially lead to product liability claims against us and negatively affect our sales. Any corrective action, whether voluntary or involuntary, as well as defending ourselves in a lawsuit, will require the dedication of our time and capital, distract management from operating our business and may harm our reputation and financial results.
Legislative or regulatory reforms may make it more difficult and costly for us to obtain regulatory clearances or approvals for our products or to manufacture, market or distribute our products after clearance or approval is obtained.
From time to time, legislation is drafted and introduced in Congress that could significantly change the statutory provisions governing the regulation of medical devices, or the FDA may change its clearance and approval policies, adopt additional regulations or revise existing regulations, or take other actions which may prevent or delay approval or clearance of our future products under development. In addition, FDA regulations and guidance are often revised or reinterpreted by the FDA in ways that may significantly affect our business and our products. Any new statutes, regulations or revisions or reinterpretations of existing regulations may impose additional costs or lengthen review times of any future products or make it more difficult to obtain clearance of or approval for, manufacture, market or distribute our products. We cannot determine what effect changes in regulations, statutes, legal interpretation or policies, when and if promulgated, enacted or adopted may have on our business in the future. Such changes could, among other things, require: additional testing prior to obtaining clearance or approval; changes to manufacturing methods; recall, replacement or discontinuance of our products; or additional record keeping.
The FDA’s and other regulatory authorities’ policies may change and additional government regulations may be enacted that could prevent, limit or delay regulatory approval of our product candidates. We cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative action, either in the U.S. or abroad. If we are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we are not able to maintain regulatory compliance, we may lose any marketing approval that we may have obtained and we may not achieve or sustain profitability.
63
In the EU, the MDR repealed and replaced the MDD on May 26, 2021. There is a transition period during which certificates issued under the MDD remain valid, subject to compliance with certain requirements under the MDR (e.g. having put in place a quality management system in accordance with the MDR by May 26, 2024). The Medical Devices Regulation is intended to, among other things, establish a uniform, transparent, predictable and sustainable regulatory framework across the EEA for medical devices and ensure a high level of safety and health while supporting innovation.
The MDR introduces new regulations which, among other things:
| ● | strengthen the rules on placing devices on the market and reinforce surveillance once they are available; |
| ● | establish explicit provisions on manufacturers’ responsibilities for the follow-up of the quality, performance and safety of devices placed on the market; |
| ● | improve the traceability of medical devices throughout the supply chain to the end-user or patient through a unique identification number; |
| ● | establish a central database to provide patients, healthcare professionals and the public with comprehensive information on products available in the EU; and |
| ● | strengthen rules for the assessment of certain high-risk devices, which may have to undergo an additional check by experts before they are placed on the market. |
Failure to comply with these regulations may harm our business.
Existing regulatory policies may change, and additional government regulations may be enacted that could prevent, limit or delay regulatory approval of our product candidates.
In June 2024, the U.S. Supreme Court overruled the Chevron doctrine, which gives deference to regulatory agencies’ statutory interpretations in litigation against federal government agencies, such as the FDA, where the law is ambiguous. This decision may result in more lawsuits against the FDA to challenge longstanding decisions and policies of the FDA, which could undermine the FDA’s authority, lead to uncertainties in the industry, and disrupt the FDA’s normal operations, any of which could delay the FDA’s review of our regulatory submissions. We cannot predict the full impact of this decision, future judicial challenges brought against the FDA, or the nature or extent of government regulation that may arise from future legislation or administrative action. If we are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we are not able to maintain regulatory compliance, we may lose any marketing approval that we may have obtained, and we may not achieve or sustain profitability.
Changes in funding for the FDA and other government agencies could hinder their ability to hire and retain key leadership and other personnel, or otherwise prevent new products and services from being developed or commercialized in a timely manner.
The ability of the FDA to review and approve new products can be affected by a variety of factors, including government budget and funding levels, ability to hire and retain key personnel and accept the payment of user fees, and statutory, regulatory, and policy changes. Average review times at the FDA have fluctuated in recent years as a result. In addition, government funding of other government agencies that fund research and development activities is subject to the political process, which is inherently fluid and unpredictable.
Disruptions at the FDA and other agencies may also slow the time necessary for new devices to be reviewed and/or approved or cleared by necessary government agencies, which would adversely affect our business. For example, over the last several years, the U.S. government has shut down several times and certain regulatory agencies, such as the FDA, have had to furlough critical FDA employees and stop critical activities. Currently, federal agencies in the U.S. are operating under a continuing resolution that is set to expire at the end of September 2025. A prolonged government shutdown, significant leadership, personnel, and/or policy changes, or other substantial modification in agency activities could significantly impact the ability of the FDA to timely review and process our regulatory submissions, which could have a material adverse effect on our business.
64
The U.S. Congress, the Trump administration, or any new administration may make substantial changes to fiscal, tax, and other federal policies that may adversely affect our business.
In 2017, the U.S. Congress and the Trump administration made substantial changes to U.S. policies, which included comprehensive corporate and individual tax reform. In addition, the Trump administration called for significant changes to U.S. trade, healthcare, immigration and government regulatory policy. With the transition to the Biden administration in early 2021, changes to U. S. policy occurred and since the start of the Trump Administration in 2025, U.S. policy changes have been implemented at a rapid pace and additional changes are likely. Changes to U.S. policy implemented by the U.S. Congress, the Trump administration or any new administration have impacted and may in the future impact, among other things, the U.S. and global economy, international trade relations, unemployment, immigration, healthcare, taxation, the U.S. regulatory environment, inflation and other areas. Although we cannot predict the impact, if any, of these changes to our business, they could adversely affect our business. Until we know what policy changes are made, whether those policy changes are challenged and subsequently upheld by the court system and how those changes impact our business and the business of our competitors over the long term, we will not know if, overall, we will benefit from them or be negatively affected by them.
Our relationships with surgeons, patients and payors in the U.S. are subject to applicable anti-kickback, fraud and abuse laws and regulations.
Our current and future operations with respect to the commercialization of our products are subject to various U.S. federal and state healthcare laws and regulations. These laws impact, among other things, our proposed sales, marketing, support and education programs and constrain our business and financial arrangements and relationships with third-party payors, surgeons and other healthcare professionals. For more information, see the sections entitled “Business – Government Regulation – Anti-Kickback Statutes, – False Claims Laws; – Transparency Laws; and – Other Federal Healthcare Fraud and Abuse Laws” in this Annual Report.
The shifting commercial compliance environment and the need to build and maintain robust and expandable systems to comply with different compliance or reporting requirements in multiple jurisdictions increase the possibility that a healthcare or medical device company may fail to comply fully with one or more of these requirements. Efforts to ensure that our business arrangements with third parties will comply with applicable healthcare laws and regulations may involve substantial costs. Certain physicians who influence the ordering or use of our products in procedures they perform have ownership interests in us and/or receive compensation for consulting services provided to us. It is possible that governmental authorities will conclude that our business practices do not comply with applicable fraud and abuse or other healthcare laws and regulations or guidance.
To enforce compliance with healthcare regulatory laws, certain enforcement bodies have recently increased their scrutiny of interactions between healthcare companies and healthcare providers, which has led to a number of investigations, prosecutions, convictions and settlements in the healthcare industry. Responding to investigations can be time- and resource-consuming and can divert management’s attention from the business. Additionally, as a result of these investigations, healthcare providers and entities may also have to agree to additional compliance and reporting requirements as part of a consent decree or corporate integrity agreement. Any such investigation or settlements could increase our costs or otherwise have an adverse effect on our business. Even an unsuccessful challenge or investigation into our practices could cause adverse publicity and be costly to respond to.
If our operations are found to be in violation of any of these laws or any other governmental regulations that may apply to us, we may be subject to significant civil, criminal and administrative penalties, damages, fines, imprisonment, exclusion from government funded healthcare programs, such as Medicare and Medicaid, additional oversight and reporting requirements if we become subject to a corporate integrity agreement to resolve allegations of non-compliance with these laws and the curtailment or restructuring of our operations. If any of the physicians or other providers or entities with whom we expect to do business is found not to be in compliance with applicable laws, they may be subject to the same criminal, civil or administrative sanctions, including exclusions from government funded healthcare programs.
65
We are subject to anti-bribery, anti-corruption, and anti-money laundering laws, including the U.S. Foreign Corrupt Practices Act, in which violations of these laws could result in substantial penalties and prosecution.
We are exposed to trade and economic sanctions and other restrictions imposed by the U.S. and other governments and organizations. The U.S. Departments of Justice, Commerce, State and Treasury and other federal agencies and authorities have a broad range of civil and criminal penalties they may seek to impose against corporations and individuals for violations of economic sanctions laws, export control laws, the U.S. Foreign Corrupt Practices Act, or the FCPA, and other federal statutes and regulations, including those established by the Office of Foreign Assets Control. In addition, the U.K. Bribery Act of 2010 (“Bribery Act”), prohibits both domestic and international bribery, as well as bribery across both private and public sectors. An organization that “fails to prevent bribery” by anyone associated with the organization can be charged under the Bribery Act unless the organization can establish the defense of having implemented “adequate procedures” to prevent bribery. Under these laws and regulations, as well as other anti-corruption laws, anti-money laundering laws, export control laws, customs laws, sanctions laws and other laws governing our operations, various government agencies may require export licenses, may seek to impose modifications to business practices, including cessation of business activities in sanctioned countries or with sanctioned persons or entities and modifications to compliance programs, which may increase compliance costs, and may subject us to fines, penalties and other sanctions. A violation of these laws or regulations would negatively affect our business, financial condition and results of operations.
We face risks related to our collection and use of data, which could result in investigations, inquiries, litigation, fines, legislative and regulatory action and negative press about our privacy and data protection practices.
Our business processes personal data, including some data related to health. When conducting clinical trials, we face risks associated with collecting trial participants’ data, especially health data, in a manner consistent with applicable laws and regulations. We also face risks inherent in handling large volumes of data and in protecting the security of such data. We could be subject to attacks on our systems by outside parties or fraudulent or inappropriate behavior by our service providers or employees. Third parties may also gain access to users’ accounts using stolen or inferred credentials, computer malware, viruses, spamming, phishing attacks or other means, and may use such access to obtain users’ personal data or prevent use of their accounts. Further, our general liability insurance and corporate risk program may not cover all potential claims to which we are exposed and may not be adequate to indemnify us for all liability that may be imposed.
As our operations and business grow, we may become subject to or affected by new or additional data protection laws and regulations and face increased scrutiny or attention from regulatory authorities. In the U.S., HIPAA imposes, among other things, certain standards relating to the privacy, security, transmission and breach reporting of individually identifiable health information. Certain states have also adopted comparable privacy and security laws and regulations, some of which may be more stringent than HIPAA. Such laws and regulations will be subject to interpretation by various courts and other governmental authorities, thus creating potentially complex compliance issues for us and our future customers and strategic partners. Further, led by California, with its CCPA, which created individual privacy rights for California residents and increased the privacy and security obligations of entities handling certain personal data, a great number of states have passed comprehensive privacy laws. These laws may increase our compliance costs and potential liability. Further, similar laws have been proposed in numerous other states and privacy-related laws have also been proposed at the federal level. There are also states that are specifically regulating health information. For example, Washington’s My Health My Data Act, which went into effect in March 2024, requires regulated entities to obtain consent to collect health information, grants consumers certain rights, including to request deletion, and provides for robust enforcement mechanisms, including enforcement by the state attorney-general and by litigants through a private right of action for consumer claims. These current and future data privacy laws and regulations may require us to modify our data collection or processing practices and policies, incur substantial costs and expenses in an effort to comply and increase our potential exposure to regulatory enforcement, reputational damage, and/or litigation.
66
A failure to comply with these current or future federal and state laws and regulations and industry standards relating to data privacy and security could lead to investigatory or regulatory action, private litigation or class actions that could result in exposure to civil or criminal penalties, monetary or statutory damages, attorney fee awards and/or exposure to adverse publicity that could negatively affect our operating results and business.
This risk is enhanced in certain jurisdictions as we expand our operations internationally. The EU’s GDPR became effective in May 2018. The GDPR applies extraterritorially and imposes several stringent requirements for controllers and processors of personal data, of data subjects residing in the European Economic Area. For example, the GDPR imposes higher standards for obtaining consent from individuals to process their personal data (where consent is required), more robust disclosures to individuals and a strengthened individual data rights regime, shortened timelines for data breach notifications, limitations on retention of information, increased requirements pertaining to special categories of personal data and pseudonymised (i.e., key-coded) data and additional obligations when we contract third-party processors in connection with the processing of the personal data. This risk is increased because EU member states have made their own laws and regulations limiting the processing of personal data, including special categories of data (e.g., racial or ethnic origin, political opinions, religious or philosophical beliefs) and profiling and automated individual decision-making of individuals, which limits our ability to process personal data or other data and could cause our compliance costs and liability risks to increase, harming our business and financial condition.
Further, the United Kingdom’s exit from the European Union, referred to as Brexit, has created uncertainty regarding data protection regulation in the United Kingdom. The United Kingdom has transposed the GDPR into domestic law with a United Kingdom version of the GDPR that took effect in January 2021 (UK GDPR). Currently, the EU GDPR and UK GDPR remain largely aligned, but the UK announced plans to reform the country’s data protection legal framework in its Data Reform Bill, which failed in the legislative process. A new Data (Use and Access) Bill, or UK Bill, has been introduced into parliament. If passed, the final version of the UK Bill may have the effect of further altering the similarities between the UK and EU data protection regime and threaten the UK adequacy decision from the EU Commission. This may lead to additional compliance costs and could increase our overall risk exposure as we may no longer be able to take a unified approach across the EEA and the UK, and we will need to amend our processes and procedures to align with the new framework. Non-compliance with GDPR, and UK GDPR, is subject to significant penalties, including fines of up to €20.0 million (£17.5 million under UK GDPR) or 4% of total worldwide revenue, whichever is greater. The implementation and enforcement of the GDPR (and UK GDPR) may subject us to enforcement risk and requirements to change certain of our data collection, processing and other policies and practices. We could incur significant costs investigating and defending such claims and, if we are found liable, significant damages. If any of these events were to occur, our business and financial results could be adversely affected. Other jurisdictions outside the EU and the United Kingdom are similarly introducing or enhancing laws and regulations relating to privacy and data security, which enhances risks relating to compliance with such laws.
The GDPR also regulates cross-border transfers of personal data and requires transferee countries to have protections equivalent to protections available in the EU. The GDPR imposes strict rules on the transfer of personal data to countries outside the EEA, Switzerland or the United Kingdom, including the United States, to other countries in respect of which the European Commission or the United Kingdom government has not issued a so-called “adequacy decision” or “ adequacy regulation” (known as “third countries”), unless the parties to the transfer have implemented specific safeguards to protect the transferred personal data. This includes putting in place the European Commission’s Standard Contractual Clauses (SCCs) for transfers outside of the EEA and a similar transfer mechanism for transfers of personal data outside of the United Kingdom, the International Data Transfer Agreement or Addendum (IDTA). Under both the GDPR and the UK GDPR, exporters are also required to assess the risk of the data transfer on a case-by-case basis, including conducting an analysis of the laws in the destination country. Finalizing the implementation of the updated SCCs and UK IDTA, and conducting the required risk assessments, may continue to necessitate significant contractual overhaul of our data transfer arrangements with customers, sub-processors and vendors. On June 28, 2021, the European Commission published its decision recognizing the United Kingdom as having adequate laws to the protect the rights and freedoms of data subjects such that personal data may transfer to from the EU to the United Kingdom without an approved transfer mechanism. The United Kingdom Government also confirmed that data transfers to the EU remain free flowing.
67
Compliance with U.S. federal and state laws and foreign data protection laws and regulations could require us to take on more onerous obligations in our contracts, restrict our ability to collect, use and disclose data, or in some cases, impact our ability to operate in certain jurisdictions. Failure to comply with United States and foreign data protection laws and regulations could result in government enforcement actions (which could include civil or criminal penalties), private litigation, and/or adverse publicity and could negatively affect our operating results and business. And claims that we have violated individuals’ privacy rights, failed to comply with data protection laws or breached our contractual obligations, even if we are not found liable, could be expensive and time-consuming to defend, could result in adverse publicity and could have a material adverse effect on our business, financial condition, results of operations, and prospects.
The Affordable Care Act and any changes in healthcare law may increase the difficulty and cost for us to successfully commercialize our products and affect the prices we may obtain.
Changes in regulations, statutes or the interpretation of existing regulations could impact our business in the future by requiring, for example: (i) changes to our manufacturing arrangements; (ii) additions or modifications to product labeling; (iii) the recall or discontinuation of our products; or (iv) additional record-keeping requirements. If any such changes were to be imposed, they could adversely affect the operation of our business. For more information, see the section entitled “Business – Government Regulation – U.S. Healthcare Reform” in this Annual Report.
In the U.S., for example, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act, or collectively the Affordable Care Act, substantially changed the way healthcare is financed by both governmental and private insurers, and significantly impacts the healthcare industry. The Affordable Care Act is intended to broaden access to health insurance, reduce or constrain the growth of healthcare spending, enhance remedies against healthcare fraud and abuse, add new transparency requirements for healthcare and health insurance industries, impose new taxes and fees on pharmaceutical and medical device manufacturers, and impose additional health policy reforms.
At the state level, legislatures have increasingly passed legislation and implemented regulations designed to control healthcare costs, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing.
We expect that additional federal, state and foreign healthcare reform measures will be adopted in the future, any of which could limit the amounts that federal and state governments will pay for healthcare products and services, which could result in limited coverage and reimbursement and reduced demand for our products, once approved, or additional pricing pressures. and could seriously harm our future revenues. Any reduction in reimbursement from Medicare, Medicaid, or other government programs may result in a similar reduction in payments from private payers. The implementation of cost containment measures or other healthcare reforms may prevent us from being able to generate revenue, attain and maintain profitability of our product and product candidates, if approved.
Our business involves the use of hazardous materials and we and Aroa must comply with environmental laws and regulations, which may be expensive and restrict how we do business.
Aroa’s activities in manufacturing our products may involve the controlled storage, use and disposal of hazardous materials. Aroa is or may be subject to federal, state, local and non-U.S. laws and regulations governing the use, generation, manufacture, storage, handling and disposal of these hazardous materials. We currently carry no insurance specifically covering environmental claims relating to the use of hazardous materials.
Although we believe that Aroa’s safety procedures for handling and disposing of these materials and waste products comply with the standards prescribed by these laws and regulations, we cannot eliminate the risk of accidental injury or contamination from the use, storage, handling or disposal of hazardous materials. In the event of an accident, federal, state or other applicable authorities may curtail Aroa’s use of these materials and interrupt their business operations which could adversely affect our business.
68
Compliance with environmental laws and regulations may be expensive and non-compliance could result in substantial liabilities, fines and penalties, personal injury and third-party property damage claims and substantial investigation and remediation costs. Environmental laws and regulations could become more stringent over time, imposing greater compliance costs and increasing risks and penalties associated with violations. We cannot assure you that violations of these laws and regulations will not occur in the future or have not occurred in the past as a result of human error, accidents, equipment failure or other causes. The expense associated with environmental regulation and remediation could harm our financial condition and results of operations.
Our business may be impacted by political, trade or regulatory developments in the jurisdictions in which we sell our products.
Significant political, trade, or regulatory developments in the jurisdictions in which we sell our products, such as those stemming from the change in U.S. federal administration, are difficult to predict and may have a material adverse effect on us. Similarly, changes in U.S. federal policy that affect the geopolitical landscape could give rise to circumstances outside our control that could have negative impacts on our business operations. For example in March 2025, the U.S. initially imposed a 25% tariff on imports from Canada and Mexico, and imposed a 20% tariff on imports from China. The U.S. largely reversed course and goods eligible for treatment under the 2020 United States-Mexico-Canada Agreement (“USMCA”) can enter the U.S. tariff free until April 2, 2025. Historically, tariffs have led to increased trade and political tensions. In response to tariffs, other countries have implemented retaliatory tariffs on U.S. goods. In retaliation to the recent U.S. imposed tariffs, China imposed tariffs up to 15% on a wide array of U.S. farm exports, and Canada and Mexico have stated they will impose tariffs on the U.S. Political tensions as a result of trade policies could reduce trade volume, investment, technological exchange and other economic activities between major international economies, resulting in a material adverse effect on global economic conditions and the stability of global financial markets. Any changes in political, trade, regulatory, and economic conditions, including U.S. trade policies, could have a material adverse effect on our financial condition, results of operations or our industry. The impact of these potential tariffs on our financial condition, results of operations or industry, if any, is subject to a number of factors that are not yet known, including any countermeasures that the target countries may take in response to such tariffs. In light of these uncertainties, we can provide no assurance that any mitigating actions that may become available to us, such as our ability to pass along some or all of the costs of any tariffs to some or all of our customers, will be successful.
Risks Related to Our Business and Products
Our financial results may fluctuate significantly and may not fully reflect the underlying performance of our business.
Our quarterly and annual results of operations may vary significantly in the future, and period-to-period comparisons of our operating results may not be meaningful. Accordingly, the results of any one quarter or period should not be relied upon as an indication of future performance. Our quarterly and annual financial results may fluctuate as a result of a variety of factors, many of which are outside our control.
Factors that may cause fluctuations in our quarterly and annual results include:
| ● | surgeon and patient adoption of our products; |
| ● | timing of new product offerings, acquisitions, licenses or other significant events by us or our competitors; |
| ● | changes in coverage policies by third-party payors that affect the reimbursement of procedures in which our products are used; |
| ● | unanticipated pricing pressure; |
| ● | established relationships or product purchase-level commitments of GPOs, IDNs and other third-party payors with whom we and our competitors contract; |
69
| ● | our ability to obtain and maintain regulatory clearance or approval for any products in development or for our current products for additional indications or in additional jurisdictions; |
| ● | the hiring, retention and continued productivity of our sales representatives; |
| ● | our ability to expand the geographic reach of our sales and marketing efforts; |
| ● | results of clinical research and trials on our existing products and products in development; |
| ● | delays in, or failure of, component and raw material deliveries by Aroa; |
| ● | recalls or other field safety corrective actions by Aroa; |
| ● | business interruptions resulting from geopolitical actions, including war and terrorism, or natural disasters such as earthquakes, floods or public health emergencies such as the COVID-19 pandemic; and |
| ● | positive or negative coverage in the media or clinical publications of our products or products of our competitors or our industry. |
Because our quarterly and annual results may fluctuate, period-to-period comparisons may not be the best indication of the underlying results of our business. These fluctuations may also increase the likelihood that we will not meet our forecasted performance, which could negatively affect the market price for our common stock.
We may be unable to compete successfully with larger competitors in our highly competitive industry.
The medical device industry is intensely competitive, subject to rapid change and significantly affected by new product introductions and other market activities of industry participants. Our competitors also compete with us in recruiting and retaining qualified scientific, management and commercial personnel, as well as in acquiring technologies complementary to, or necessary for, our products. Because of the complex and technical nature of our products and the dynamic market in which we compete, any failure to attract and retain a sufficient number of qualified employees could materially harm our ability to develop and commercialize our products, which would have a material adverse effect on our business, financial condition and results of operations.
In the U.S., we currently compete with Allergan, a subsidiary of AbbVie, C.R. Bard, a subsidiary of Becton, Dickinson and Company, MTF Biologics, RTI Surgical and Integra Life Sciences, which produce, among other things, soft-tissue reconstruction surgery products, including Strattice and Alloderm, Phasix, FlexHD, Cortiva,and SurgiMend and DuraSorb, respectively. In the EEA, we compete with Bard, who produces other soft-tissue reinforcement products. Many of these competitors are large, well-capitalized companies with significantly greater market share and contracting power than us, selling products that have been on the market prior to the commercialization of our products. As a consequence, they are able to spend more on product development, marketing, sales and other product initiatives than we can, while benefiting from greater brand awareness. We believe other emerging businesses are in the early stages of developing similar products designed for soft-tissue reconstruction surgery. Although we are the only ovine-derived implantable product designed for soft-tissue reconstruction surgery, there are other soft tissue reconstruction surgery products derived solely, or in part, from other biological sources.
Most of the other soft-tissue reconstruction surgery products currently have a greater penetration into the soft tissue reconstruction surgery market. Often, other soft-tissue reconstruction surgery products with which our products compete are marketed as part of a bundled product line, which may provide our potential customers a better price-per-product than we could offer. If we are unable to penetrate the soft-tissue reconstruction surgery market or offer competitive pricing on our products compared with products sold as part of a bundled product line, it could have a material adverse effect on our business, financial condition and results of operations.
70
In addition, competitors with greater financial resources could acquire other companies to gain enhanced name recognition and market share, as well as new technologies or products that could effectively compete with our existing products, which may cause our revenue to decline and would harm our business.
We may be unable to renew existing contracts with GPOs or obtain additional contract positions with major GPOs and IDNs, for our products, and even if we are able to do so, such contracts may not generate sufficient sales of our products.
Many existing and potential customers for our products within the U.S. are members of GPOs and IDNs, including accountable care organizations or public-based purchasing organizations, and our business strategy is focused on entering into major contracts with these organizations. Our products can be contracted under national tenders or with larger hospital GPOs. GPOs and IDNs typically award contracts on a category-by-category basis through a competitive bidding process. We are currently responding to bids and negotiating a number of GPO and IDN agreements.
We may not be able to renew existing contracts with GPOs or IDNs and due to the highly competitive nature of the bidding process and the GPO and IDN contracting processes in the U.S., we may not be able to obtain additional contract positions with major GPOs and IDNs for our products. If we are unable to renew existing contracts with GPOs or IDNs, our net sales and results of operations may be materially and adversely affected. In addition, while having a contract with a major purchaser for a given product category can facilitate sales, sales volumes of those products may not be maintained or may be limited based on preferential economic terms that can be offered by larger competitors across product categories. Further, we may fail to obtain a contract in an appropriate product category that will enable us to more effectively compete against competitive products within the same product category. For example, GPOs and IDNs are increasingly awarding contracts to multiple suppliers for the same product category. Even if we are the sole contracted supplier of a GPO or IDN for our product category, members of the GPO or IDN generally are free to purchase from other suppliers. Furthermore, GPO and IDN contracts typically are terminable without cause upon 60 to 90 days’ notice.
Supply chain disruptions could adversely impact our operations and financial condition.
Global supply chains have been impacted because of severe weather, recent geopolitical tensions such as the ongoing Russia-Ukraine conflict and the current conflict in the Middle East (including any escalation or expansion) and other factors, and this may impact the availability of raw materials and components used in the manufacture of our products. Additionally, even when we and our suppliers are able to source such materials and components, they may cost more and may only be available on a delayed basis. Higher materials and component costs could adversely affect our margins if we are unable to pass such costs along to customers in the form of price increases. Delays in receipt of materials and components could also interrupt our production and cause us to go into backorder on certain of our products, further exacerbating the effect of the global supply chain disruption.
We face the risk of product liability claims that could be expensive, divert management’s attention and harm our reputation and business.
Our business exposes us to the risk of product liability claims that are inherent in the testing, manufacturing and marketing of medical devices. This risk exists even if a product is cleared or approved for commercial sale by the FDA, and manufactured in facilities licensed and regulated by the FDA. Any side effects, manufacturing defects or misuse associated with our products could result in patient injury or death. The industry in which we operate has historically been subject to extensive litigation over product liability claims, and we cannot offer any assurance that we will not face product liability suits. We may be subject to product liability claims if our products cause, or merely appear to have caused, patient injury or death. In addition, an injury that is caused by the activities of Aroa may be the basis for a claim against us. Product liability claims may be brought against us by patients, healthcare providers or others selling or otherwise coming into contact with our products. If we cannot successfully defend ourselves against product liability claims, we will incur substantial liabilities and reputational harm. In addition, regardless of merit or eventual outcome, product liability claims may result in substantial litigation costs, product recalls or market withdrawals, decreased sales and demand for our products and damage to our reputation.
71
While we may attempt to manage our product liability exposure by proactively recalling or withdrawing from the market any defective products, any recall or market withdrawal of our products may delay the supply of those products to our customers and may impact our reputation. We can provide no assurance that we will be successful in initiating appropriate market recall or market withdrawal efforts that may be required in the future or that these efforts will have the intended effect of preventing product malfunctions and the accompanying product liability that may result. Such recalls and withdrawals may also be used by our competitors to harm our reputation for safety or be perceived by patients as a safety risk when considering the use of our products, either of which could have a material adverse effect on our business, financial condition and results of operations.
Although we have product liability insurance that we believe is appropriate, this insurance is subject to deductibles and coverage limitations. In addition, our current product liability insurance may not continue to be available to us on acceptable terms, if at all, and, if available, coverage may not be adequate to protect us against any future product liability claims. A product liability claim, recall or other claim with respect to uninsured liabilities or for amounts in excess of insured liabilities could have a material adverse effect on our business, financial condition and results of operations.
The continuing development of our products depends upon our maintaining strong working relationships with surgeons.
The research, development, marketing and sale of our current and future products and any future product indications for which we receive regulatory clearance or approval depend upon our maintaining working relationships with surgeons. We rely on these professionals to provide us with considerable knowledge and experience regarding the development, marketing and sale of our products. Surgeons assist us in clinical trials and in marketing, and as researchers, product consultants and public speakers. If we cannot maintain our strong working relationships with these professionals and continue to receive their advice and input, the development and marketing of our products could suffer, which could have a material adverse effect on our business, financial condition and results of operations. At the same time, the medical device industry’s relationship with surgeons is under increasing scrutiny by the U.S. Department of Health and Human Services Office of Inspector General (“OIG”), the U.S. Department of Justice (“DOJ”), the state attorneys general and other foreign and domestic government agencies. Our failure to comply with requirements governing the industry’s relationships with surgeons or an investigation into our compliance by the OIG, the DOJ, state attorneys general and other government agencies, could have a material adverse effect on our business, financial condition and results of operations. Additional information regarding the laws impacting our relationships with surgeons and other healthcare professionals can be found above under “Risks Related to Government Regulation.”
We have limited data and experience regarding the safety and efficacy of certain of our products. Results of earlier studies may not be predictive of future clinical trial results, or the safety or efficacy profile for such products.
Our single arm multicenter post-market clinical study, which we refer to as our BRAVO study, was fully enrolled at 92 patients. We conducted this study to support the marketing of our OviTex products for their cleared indicated uses, and do not currently have any clinical data for use of our OviTex PRS products in patients. The long-term effects of using certain of our products in a large number of patients have not been studied and the results of short-term clinical use of such products do not necessarily predict long-term clinical benefits or reveal long-term adverse effects. The results of preclinical studies and clinical studies of our products conducted to date and ongoing or future studies and trials of our current, planned or future products may not be predictive of the results of later clinical trials, and interim results of a clinical trial do not necessarily predict final results. Our interpretation of data and results from our clinical trials do not ensure that we will achieve similar results in future clinical trials in other patient populations. In addition, preclinical and clinical data are often susceptible to various interpretations and analyses, and many companies that have believed their products performed satisfactorily in preclinical studies and earlier clinical trials have nonetheless failed to replicate results in later clinical trials. Products in later stages of clinical trials may fail to show the desired safety and efficacy despite having progressed through nonclinical studies and earlier clinical trials.
72
Interim or preliminary data from our clinical trials that we announce or publish from time to time may change as more patient data become available and are subject to audit and verification procedures that could result in material changes in the final data.
From time to time, we may publicly disclose interim or preliminary data from our clinical studies, which is based on a preliminary analysis of then-available data, and the results and related findings and conclusions are subject to change following a full analyses of all data related to the particular trial. We also make assumptions, estimations, calculations and conclusions as part of our analyses of data, and we may not have received or had the opportunity to fully and carefully evaluate all data. As a result, the interim results that we report may differ from future results of the same trials, or different conclusions or considerations may qualify such results, once additional data have been received and fully evaluated. Interim or preliminary data also remain subject to audit and verification procedures that may result in the final data being materially different from the preliminary data we previously published. As a result, interim or preliminary data should be viewed with caution until the final data are available. We may also disclose interim data from our clinical trials. Interim data from clinical trials that we may complete are subject to the risk that one or more of the clinical outcomes may materially change as patient enrollment continues and more patient data become available. Adverse differences between preliminary or interim data and final data could significantly harm our business prospects.
Further, others, including regulatory agencies, may not accept or agree with our assumptions, estimates, calculations, conclusions or analyses or may interpret or weigh the importance of data differently, which could impact the value of the particular program, the approvability or commercialization of the particular product candidate or product and our business in general. In addition, the information we choose to publicly disclose regarding a particular study or clinical trial is based on what is typically extensive information, and you or others may not agree with what we determine is the material or otherwise appropriate information to include in our disclosure, and any information we determine not to disclose may ultimately be deemed significant with respect to future decisions, conclusions, views, activities or otherwise regarding a particular drug, product candidate or our business. If the interim or preliminary data that we report differ from actual results, or if others, including regulatory authorities, disagree with the conclusions reached, our ability to use such results to support the marketing of our products may be jeopardized.
The sizes of the markets for our current and future products have not been established with precision, and may be smaller than we estimate.
Our estimates of the annual total addressable markets for our current products and products under development are based on a number of internal and third-party estimates, including, without limitation, the number of hernia and soft-tissue reconstruction surgery patients and overall market and the assumed prices at which we can sell our products. While we believe our assumptions and the data underlying our estimates are reasonable, these assumptions and estimates may not be correct and the conditions supporting our assumptions or estimates may change at any time, thereby reducing the predictive accuracy of these underlying factors. As a result, our estimates of the annual total addressable market for our products may prove to be incorrect. If the price at which we can sell future products, or the annual total addressable market for our products is smaller than we have estimated, it may impair our sales growth and have an adverse impact on our business.
Our results of operations could be materially harmed if we are unable to accurately forecast customer demand for our products and manage our inventory.
Our reinforced tissue matrix products have a limited shelf life and will expire if not timely used. To ensure adequate inventory supply, we must forecast inventory needs and place orders with Aroa based on our estimates of future demand for our reinforced tissue matrix products. Our ability to accurately forecast demand for such products could be negatively affected by many factors, including:
| ● | product introductions by competitors; |
| ● | an increase or decrease in surgeon demand for our products or for products of our competitors; |
| ● | our failure to accurately manage our expansion strategy; |
73
| ● | our failure to accurately forecast surgeon acceptance of new products; |
| ● | our failure to obtain contracts with a significant number of GPOs and IDNs; |
| ● | unanticipated changes in general market conditions or regulatory matters; |
| ● | the severity and duration of market disruptions as a result of the COVID-19 outbreak; and |
| ● | weakening of economic conditions or consumer confidence. |
Inventory levels in excess of customer demand may result in inventory write-downs or write-offs, which would cause our gross margin to be adversely affected and could impair the strength of our brand. Additionally, we are subject to the risk that a portion of our inventory will expire, which could have a material adverse effect on our earnings and cash flows due to the resulting costs associated with the inventory impairment charges and costs required to replace such inventory. Conversely, if we underestimate customer demand for our products, Aroa may not be able to deliver products to meet our requirements, and this could result in damage to our reputation and customer relationships. In addition, if we experience a significant increase in demand, additional supplies of raw materials or additional manufacturing capacity may not be available when required on terms that are acceptable to us, or at all, or Aroa may not be able to allocate sufficient capacity to meet our increased requirements, which could have an adverse effect on our ability to meet customer demand for our products and our results of operations.
We rely on our own direct sales force for our products, which may result in higher fixed costs than our competitors and may slow our ability to reduce costs.
We rely on our own direct sales force, which as of December 31, 2024 consisted of 91 representatives in the U.S. and 12 representatives in Europe, to market and sell our products. A direct sales force may subject us to higher fixed costs than those of companies that market competing products through independent third parties, due to the costs that we will bear associated with employee benefits, training and managing sales personnel. As a result, we may be at a competitive disadvantage. Additionally, these fixed costs may slow our ability to reduce costs in the face of a sudden decline in demand for our products, which could have a material adverse effect on our business, financial condition and results of operations.
Our employees, independent contractors, consultants, commercial partners, distributors and vendors may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements.
We are exposed to the risk that our employees, independent contractors, consultants, commercial partners and vendors may engage in fraudulent or illegal activity. Misconduct by these parties could include intentional, reckless and/or negligent conduct or disclosure of unauthorized activities to us that violates: (i) the rules of the FDA and other similar foreign regulatory bodies; (ii) manufacturing standards; (iii) healthcare fraud and abuse laws in the U.S. and similar foreign fraudulent misconduct laws; (iv) data privacy laws and other similar non-U.S. laws; or (v) laws that require the true, complete and accurate reporting of financial information or data. These laws may impact, among other things, future sales, marketing and education programs.
It is not always possible to identify and deter misconduct by our employees and other third parties, and the precautions we take to detect and prevent these activities may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to be in compliance with such laws or regulations. In addition, we are subject to the risk that a person or government could allege such fraud or other misconduct, even if none occurred. If any such actions are instituted against us and we are not successful in defending ourselves or asserting our rights, those actions could result in the imposition of significant fines or other sanctions, including the imposition of civil, criminal and administrative penalties, additional integrity reporting and oversight obligations and possible exclusion from participation in Medicare, Medicaid and other federal healthcare programs, any of which could adversely affect our ability to operate our business and our results of operations. Whether or not we are successful in defending against any such actions or investigations, we could incur substantial costs, including legal fees, and divert the attention of management in defending ourselves against any of these claims or investigations, which could have a material adverse effect on our business, financial condition and results of operations.
74
We could be adversely affected by any interruption to our ability to conduct business at our current location.
We do not have redundant facilities. We perform substantially all of our research and development and back-office activity and maintain all our finished goods inventory in a single location in Malvern, Pennsylvania. Our facility, equipment and inventory would be costly to replace and could require substantial lead time to repair or replace. The facility may be harmed or rendered inoperable by natural or man-made disasters, including, but not limited to, tornadoes, flooding, fire, public health emergencies such as pandemics and power outages, which may render it difficult or impossible for us to perform our customer service research, development and commercialization activities for some period of time. The inability to perform those activities, combined with the time it may take to rebuild our inventory of finished product, may result in the loss of customers or harm to our reputation. Although we possess insurance for damage to our property and the disruption of our business, this insurance may not be sufficient to cover all of our potential losses and this insurance may not continue to be available to us on acceptable terms, or at all.
If we or our vendors experience a cybersecurity incident, significant disruption or a compromise of our information technology systems, our business could be adversely affected.
We rely extensively on information technology systems to conduct our business. These systems affect, among other things, ordering and managing products, shipping products to customers, processing transactions, summarizing and reporting results of operations, complying with regulatory, legal and tax requirements, data security and other processes necessary to manage our business. Our information systems require an ongoing commitment of significant resources to maintain, protect, and enhance existing systems and develop new systems to keep pace with continuing changes in information processing technology, evolving systems and regulatory standards, the increasing need to protect patient and customer information, changing customer patterns and an evolving threat landscape. If our systems are damaged or cease to function properly due to any number of causes, ranging from catastrophic events to power outages to security incidents, we may experience interruptions in our operations or security breaches, which could have an adverse effect on our business.
If we fail to maintain or protect our information systems and data integrity effectively, we could lose existing customers, have difficulty attracting new customers, suffer backlash from negative public relations, have regulatory sanctions or penalties imposed, have increases in operating expenses, incur expenses or lose revenues as a result of a data privacy breach, or suffer other adverse consequences. Furthermore, any compromise in our information technology systems could lead to the unauthorized access, disclosure and use of non-public information from our patient registry or other patient information which is protected by HIPAA and other laws. Any such access, disclosure, or other loss of information could require us to notify impacted stakeholders(including affected individuals, regulators and investors) and result in legal claims or proceedings, liability under laws that protect the privacy of personal information and damage to our reputation.
Although we develop and maintain systems and controls designed to prevent these events from occurring, there can be no assurance that our internal information technology systems or those of our third-party vendors will be sufficient to protect against breakdowns, service disruption, data deterioration or loss in the event of a system malfunction, or prevent data from being stolen or corrupted in the event of a cyberattack, security incident, industrial espionage attacks, ransomware, or insider threat attacks. Like other companies in our industry, we have experienced and may in the future experience, threats and cybersecurity incidents relating to our, our third-party vendors’, and our customers’ information systems.
If a material security incident related to our information technology systems or those of our vendors occurs, the market perception of the effectiveness of our cybersecurity measures could be harmed and our reputation and credibility could be damaged. We could be required to expend significant amounts of money and other resources to repair or replace information systems or networks, including costs to deploy additional personnel and protection technologies, train employees, engage third-party experts and consultants, and identify replacement vendors if necessary, which could materially and adversely affect our business, financial condition and results of operations.
75
We cannot be sure that our cyber insurance coverage will be adequate or sufficient to protect us from or to mitigate liabilities arising out of any such disruption in, or failure or security incident or breach of, our systems or third-party systems where information important to our business operations or commercial development is stored, that such coverage will continue to be available on commercially reasonable terms or at all, or that such coverage will pay future claims. In addition, we could be subject to regulatory actions and/or claims made by individuals and groups in private litigation involving privacy issues related to data collection and use practices and other data privacy laws and regulations, including claims for misuse or inappropriate disclosure of data, as well as unfair or deceptive practices. Our contracts may not contain limitations of liability, and even where they do, there can be no assurance that limitations of liability in our contracts are sufficient to protect us from liabilities, damages, or claims related to our privacy and data security obligations.
If we become profitable, our ability to use our net operating loss carryforwards and other tax attributes to offset future taxable income or taxes may be subject to limitations.
As of December 31, 2024, we had federal and state net operating loss carry forwards (“NOLs”) of approximately $266.7 million and $218.9 million, respectively. The federal carry forwards for losses incurred prior to 2018 will begin expiring in 2032 for federal purposes. Federal net operating losses incurred in 2018 and onward have an indefinite expiration under the 2017 Tax Cut & Jobs Act. The state carry forwards will begin expiring in 2026. An allowance for the majority of the NOLs which relate to the U.S. is provided for in our audited financial statements for the year of December 31, 2024 included in this Annual Report on Form 10-K. We cannot guarantee what the ultimate outcome or amount of the benefit we may receive from the NOLs, if any, will be. If we become profitable in the future, our ability to use net operating loss carryforwards and other tax attributes to offset future taxable income or reduce taxes may be subject to limitations.
Risks Related to Our Securities
The trading price of the shares of our common stock has been and could in the future be highly volatile.
The price of our common stock has been and may continue to be volatile. Even though our common stock is listed on the Nasdaq Global Market (“Nasdaq”), an active trading market for our common stock may not be sustained. The lack of an active trading market may impair the value of your shares and your ability to sell your shares at the time you wish to sell them. An inactive trading market may also impair our ability to raise capital by selling shares of our common stock and enter into strategic partnerships or acquire other complementary products, technologies or businesses by using shares of our common stock as consideration. Furthermore, there can be no guarantee that we will continue to satisfy the continued listing standards of Nasdaq. If we fail to satisfy the continued listing standards, we could be de-listed, which would have a negative effect on the price of our common stock.
We cannot predict the prices at which our shares of common stock may trade. The market price of our common stock is likely to be highly volatile and may fluctuate substantially due to many factors, including:
| ● | the volume and timing of sales of our products; |
| ● | the introduction of new products or product enhancements by us or others in our industry; |
| ● | disputes or other developments with respect to our or others’ intellectual property rights; |
| ● | our ability to develop, obtain regulatory clearance for, and market new and enhanced products on a timely basis; |
| ● | product liability claims or other litigation; |
| ● | quarterly variations in our results of operations or those of others in our industry; |
| ● | media exposure of our products or of those of others in our industry; |
76
| ● | changes in governmental regulations or in reimbursement; |
| ● | changes in earnings estimates or recommendations by securities analysts; |
| ● | broad trends impacting companies within the pharmaceutical, biotechnology and medical technology industries; and |
| ● | general market conditions and other factors, including factors unrelated to our operating performance or the operating performance of our competitors, including global pandemic such as the COVID-19 pandemic, or macroeconomic factors such as geopolitical tensions, tariffs, or the outbreak or escalation of hostilities or war. |
In recent years, the stock markets generally have experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of those companies. Broad market and industry factors may significantly affect the market price of our common stock, regardless of our actual operating performance.
In addition, in the past, class action litigation has often been instituted against companies whose securities have experienced periods of volatility in market price. Securities litigation brought against us following volatility in our stock price, regardless of the merit or ultimate results of such litigation, could result in substantial costs, which would hurt our financial condition and operating results and divert management’s attention and resources from our business.
We do not intend to pay cash dividends on our common stock for the foreseeable future.
We currently anticipate that we will retain future earnings for the development, operation and expansion of our business and do not anticipate declaring or paying any cash dividends for the foreseeable future. In addition, the agreement governing our credit facility precludes, and any future debt agreements may preclude us from paying cash dividends. Any future determination to declare dividends will be made at the discretion of our board of directors and will depend on, among other factors, our financial condition, operating results, capital requirements, general business conditions and other factors that our board of directors may deem relevant. Any return to stockholders will therefore be limited to the appreciation in the value of their stock, if any.
Our directors, officers and principal stockholders have significant voting power and may take actions that may not be in the best interests of our other stockholders.
Our officers, directors and principal stockholders each holding more than 5% of our common stock, collectively, control approximately 49% of our outstanding common stock. As a result, these stockholders, if they act together, will be able to significantly influence our management and affairs and most matters requiring stockholder approval, including the election of directors and approval of significant corporate transactions. The interests of these stockholders may not be the same as or may even conflict with your interests. For example, these stockholders could attempt to delay or prevent a change in control, even if such change in control would benefit our other stockholders, which could deprive our stockholders of an opportunity to receive a premium for their common stock as part of a sale of our capital stock or our assets, and might affect the prevailing market price of our common stock due to investors’ perceptions that conflicts of interest may exist or arise. As a result, this concentration of ownership may not be in the best interests of our other stockholders.
We are at risk of securities class action litigation.
In the past, securities class action litigation has often been brought against a company following a decline in the market price of its securities. This risk is especially relevant for us because medical device companies have experienced significant stock price volatility in recent years. If we face such litigation, it could result in substantial costs and a diversion of management’s attention and resources, which could harm our business.
77
If securities or industry analysts do not publish research or publish inaccurate or unfavorable research about our business, our stock price and trading volume could decline.
The trading market for our common stock depends in part on the research and reports that industry or financial analysts publish about us or our business. If one or more of the analysts who cover us downgrade our stock or publish inaccurate or unfavorable research about our business, the price of our stock could decline. If one or more of these analysts cease coverage of us or fail to publish reports covering us regularly, we could lose visibility in the market, which in turn could cause our stock price to decline.
Provisions in our corporate charter documents and under Delaware law could discourage another company from acquiring us and may prevent attempts by our stockholders to replace or remove our current management.
Provisions in our fourth amended and restated certificate of incorporation and our third amended and restated bylaws may discourage, delay or prevent a merger, acquisition or other change in control of us that stockholders may consider favorable, including transactions in which stockholders might otherwise receive a premium for their shares. These provisions could also limit the price that investors might be willing to pay in the future for shares of our common stock, thereby depressing the market price of our common stock. In addition, these provisions may frustrate or prevent any attempts by our stockholders to replace or remove our current management by making it more difficult for stockholders to replace members of our board of directors. As our board of directors is responsible for appointing the members of our management team, these provisions could in turn affect any attempt by our stockholders to replace current members of our management team. These provisions provide, among other things, that:
| ● | our board of directors has the exclusive right to expand the size of our board of directors and to elect directors to fill a vacancy created by the expansion of the board of directors or the resignation, death or removal of a director, which prevents stockholders from being able to fill vacancies on our board of directors; |
| ● | our board of directors is divided into three classes, Class I, Class II and Class III, with each class serving staggered three-year terms, which may delay the ability of stockholders to change the membership of a majority of our board of directors; |
| ● | our stockholders may not act by written consent, which forces stockholder action to be taken at an annual or special meeting of our stockholders; |
| ● | a special meeting of stockholders may be called only by the chair of our board of directors, our chief executive officer (or president, in the absence of a chief executive officer) or a majority of our board of directors, which may delay the ability of our stockholders to force consideration of a proposal or to take action, including the removal of directors; |
| ● | our fourth amended and restated certificate of incorporation prohibits cumulative voting in the election of directors, which limits the ability of minority stockholders to elect director candidates; |
| ● | our board of directors may alter certain provisions of our third amended and restated bylaws without obtaining stockholder approval; |
| ● | the approval of the holders of at least two-thirds of our shares entitled to vote at an election of our board of directors is required to adopt, amend or repeal our third amended and restated bylaws or repeal the provisions of our fourth amended and restated certificate of incorporation regarding the election and removal of directors; |
| ● | stockholders must provide advance notice and additional disclosures to nominate individuals for election to the board of directors or to propose matters that can be acted upon at a stockholders’ meeting, which may |
78
| discourage or deter a potential acquirer from conducting a solicitation of proxies to elect the acquirer’s own slate of directors or otherwise attempting to obtain voting control of our shares; and |
| ● | our board of directors is authorized to issue shares of preferred stock and to determine the terms of those shares, including preferences and voting rights, without stockholder approval, which could be used to significantly dilute the ownership of a hostile acquirer. |
Moreover, because we are incorporated in Delaware, we are governed by the provisions of Section 203 of the General Corporation Law of the State of Delaware (“DGCL”) which prohibits a person who owns in excess of 15% of our outstanding voting stock from merging or combining with us for a period of three years after the date of the transaction in which the person acquired in excess of 15% of our outstanding voting stock, unless the merger or combination is approved in a prescribed manner.
Our fourth amended and restated certificate of incorporation provides that the Court of Chancery of the State of Delaware will be the exclusive forum for substantially all disputes between us and our stockholders, which could limit our stockholders’ ability to obtain a favorable judicial forum for disputes with us or our directors, officers or employees.
Our fourth amended and restated certificate of incorporation provides that the Court of Chancery of the State of Delaware (or, if the Court of Chancery does not have jurisdiction, the United State District Court for the District of Delaware) is the exclusive forum, to the fullest extent permitted by law, for (i) any derivative action or proceeding brought on our behalf, (ii) any action asserting a claim of breach of a fiduciary duty or other wrongdoing by any of our directors, officers, employees or agents to us or our stockholders, (iii) any action asserting a claim arising pursuant to any provision of the DGCL or our fourth amended and restated certificate of incorporation or third amended and restated bylaws or (iv) any action asserting a claim governed by the internal affairs doctrine, except, in each case, (A) any claim as to which such court determines that there is an indispensable party not subject to the jurisdiction of such court (and the indispensable party does not consent to the personal jurisdiction of such court within 10 days following such determination), (B) which is vested in the exclusive jurisdiction of a court or forum other than such court, or (C) for which such court does not have subject matter jurisdiction, in all cases subject to the courts having jurisdiction over indispensable parties named as defendants. This provision may limit a stockholder’s ability to bring a claim in a judicial forum that it finds favorable for disputes with us or our directors, officers or other employees, which may discourage such lawsuits against us and our directors, officers and other employees. For example, stockholders who do bring a claim in the Court of Chancery could face additional litigations costs in pursuing any such claim, particularly if they do not reside in or near the State of Delaware. The Court of Chancery may also reach different judgments or results than would other courts, including courts where a stockholder considering an action may be located or would otherwise choose to bring the action, and such judgments or results may be more favorable to us than to our stockholders. The enforceability of similar choice of forum provisions in other companies’ certificates of incorporation has been challenged in legal proceedings, and it is possible that, in connection with any applicable action brought against us, a court could find the choice of forum provisions contained in our fourth amended and restated certificate of incorporation to be inapplicable or unenforceable in such action. Alternatively, if a court were to find the choice of forum provision contained in our fourth amended and restated certificate of incorporation to be inapplicable or unenforceable in an action, we may incur additional costs associated with resolving such action in other jurisdictions. This provision will not apply to actions arising under the Securities Act or Exchange Act. Our fourth amended and restated certificate of incorporation and third amended and restated bylaws further provide that the federal district courts of the U.S. will be the exclusive forum for resolving any complaint asserting a cause of action arising under the Securities Act. Section 22 of the Securities Act, however, creates concurrent jurisdiction for federal and state courts over all suits brought to enforce any duty or liability created by the Securities Act or the rules and regulations thereunder. Accordingly, there is uncertainty as to whether a court would enforce such a forum selection provision as written in connection with claims arising under the Securities Act.
79
General Risk Factors
Our ability to maintain our competitive position depends on our ability to attract and retain senior management and other highly qualified personnel.
We are highly dependent on our senior management and other key personnel. Our success depends in part on our continued ability to attract, retain and motivate highly qualified senior management and attract, retain and motivate qualified employees, including sales and marketing professionals, clinical specialists and other highly skilled personnel. Competition for skilled personnel in our market is intense and may limit our ability to hire and retain highly qualified personnel on acceptable terms, or at all. If we are not successful in attracting and retaining highly qualified personnel, it would have a material adverse effect on our business, financial condition and results of operations. The loss of highly qualified employees could result in delays in product development and commercialization and harm our business.
Although we have entered into employment agreements with all of our executive officers, each of them may terminate their employment with us at any time. The replacement of any of our key personnel likely would involve significant time and costs and may significantly delay or prevent the achievement of our business objectives and could therefore have an adverse effect on our business. We also do not maintain “key man” insurance policies on the lives of these individuals or the lives of any of our other employees.
Our disclosure controls and procedures may not prevent or detect all errors or acts of fraud.
We are subject to the periodic reporting requirements of the Exchange Act. We designed our disclosure controls and procedures to provide reasonable assurance that information we must disclose in reports we file or submit under the Exchange Act is accumulated and communicated to management, and recorded, processed, summarized and reported within the time periods specified in the rules and forms of the SEC. We believe that any disclosure controls and procedures, no matter how well those controls and procedures are conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met.
These inherent limitations include the realities that judgments in decision-making can be faulty, and that breakdowns can occur because of simple error or mistake. Additionally, controls can be circumvented by the individual acts of some persons, by collusion of two or more people or by an unauthorized override of the controls. Accordingly, because of the inherent limitations in our control system, misstatements due to error or fraud may occur and not be detected.
ITEM 1B.UNRESOLVED STAFF COMMENTS
None.
ITEM 1C.CYBERSECURITY
Cyber Risk Management and Strategy
Under the oversight of our board of directors and the Audit Committee of the board, we have adopted cybersecurity risk management processes that take a risk-based approach to assessing, identifying, and managing risks from cybersecurity threats. Management of cybersecurity risks is part of our overall risk management strategy.
We engage third-party service providers to assist us with our cybersecurity risk management, including for network monitoring, antivirus protection, and managing IT environments. We have also engaged third party advisors and consultants to conduct periodic testing of our processes and systems. Before contracting with certain third parties, such as those that have access to our IT networks, we have a process to conduct diligence on those third parties, which includes a security assessment. We have also implemented a process for employees to undergo cybersecurity training during onboarding, and thereafter, on an annual basis as part of our larger compliance training program.
We have established monitoring procedures in our effort to mitigate risks related to cybersecurity incidents. As part of our cybersecurity risk management, we have adopted a business continuity and incident response plan, which is designed to establish our processes for identifying and responding to significant events that may lead to a business disruption or crisis, including those arising from or related to cybersecurity threats.
80
Governance
Our board of directors holds oversight responsibility over our strategy and risk management, including risks related to cybersecurity. The board’s oversight of cybersecurity risk management is supported by the Audit Committee, which has responsibility for discussing with management significant cybersecurity risks and the measures we have implemented to monitor and control such cyber risk exposures. The Audit Committee receives quarterly updates from our Vice President, Information Technology (“IT Officer”) relating to IT and cybersecurity matters, including cybersecurity risks and threats. The Audit Committee provides periodic updates to our board of directors on cybersecurity matters discussed at such meetings. Our IT Officer also provides these and similar reports to the full board of directors on a biannual basis.
Our IT Officer oversees the day-to-day management of the Company’s cybersecurity risk management program. Our IT Officer has over 15 years of experience in IT leadership and has managed IT for our company for approximately 10 years. Our IT Officer reports to our Chief Operating Officer and Chief Financial Officer and is a member of our Compliance Committee. Our IT Officer coordinates with our legal department and relevant third parties, such as consultants and external legal advisors, to assess and manage material risks from cybersecurity threats. Our IT Officer is also supported by a cross-functional incident response team, which is empowered to review, assess, report, monitor and take action to mitigate or remedy any cybersecurity incidents pursuant to our business continuity and incident response plan. Our IT department further supports and has dedicated resources to assist our IT Officer in monitoring, preventing, detecting, mitigating, and remediating any cybersecurity incidents pursuant to our policies and procedures.
We have also established a Disclosure Committee, which regularly reviews relevant information related to potential public disclosure of critical business risks and material events.
We have not identified any cybersecurity incidents or threats that have materially affected our information or system or are reasonably likely to materially affect our information and systems, including our business strategy, results of operations, or financial condition. However, like other companies in our industry, we and our third-party vendors have from time to time experienced threats and security incidents that could affect our information or systems. For more information, please refer to Item 1A, “Risk Factors,” in this Form 10-K.
ITEM 2.PROPERTIES
Our products are manufactured by our exclusive manufacturer and supplier of our products, Aroa, at their facility in Auckland, New Zealand which currently totals approximately 40,000 square feet.
We lease our corporate headquarters in Malvern, Pennsylvania, which houses our research and development operations, controlled environment room, and office space, and currently totals approximately 41,000 square feet until June 30, 2025, after which we will relinquish approximately 5,000 square feet of excess office and warehouse space.
We believe that our current facilities meet our current and future anticipated needs, although we may seek to negotiate new leases or evaluate additional or alternate space for our operations. We believe appropriate office space will be readily available on commercially reasonable terms.
ITEM 3.LEGAL PROCEEDINGS
We may be subject to other legal proceedings and claims in the ordinary course of business. We cannot predict the results of any such disputes, and despite the potential outcomes, the existence thereof may have an adverse material impact on us due to diversion of management time and attention as well as the financial costs related to resolving such disputes.
ITEM 4.MINE SAFETY DISCLOSURES
81
Not applicable.
PART II
ITEM 5. MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Market Information
Our common stock has been publicly traded on the Nasdaq Global Market under the symbol “TELA” since November 8,
2019.
Holders
As of March 14, 2025, the Company had approximately 58 record holders of its common stock.
Dividends
The Company has not declared or paid any dividends since its inception nor does it expect to pay dividends in the
foreseeable future.
Securities Authorized for Issuance Under Equity Compensation Plans
The information under the heading “Securities Authorized for Issuance Under Equity Compensation Plans” will be filed in the Company’s definitive proxy statement for the 2025 annual meeting of stockholders and is incorporated herein by reference.
Recent Sales of Unregistered Securities
None.
Issuer Purchases of Equity Securities
None.
ITEM 6.RESERVED
82
ITEM 7. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
You should read the following discussion and analysis of our financial condition and results of operations and the consolidated financial statements and the related notes included elsewhere in this Annual Report. In addition to historical financial information, the following discussion contains forward-looking statements based upon our current plans, expectations and beliefs that involve risks, uncertainties and assumptions. Our actual results may differ materially from those described in or implied by these forward-looking statements as a result of many factors, including those set forth under the section titled “Risk Factors” and in other parts of this Annual Report.
We are a commercial-stage medical technology company focused on providing innovative soft-tissue reconstruction solutions that optimize clinical outcomes by prioritizing the preservation and restoration of the patient’s own anatomy. Our growing product portfolio is purposefully designed to leverage the patient’s natural healing response while minimizing long-term exposure to permanent synthetic materials. We are committed to delivering our advanced technologies with a strong economic value proposition to assist surgeons and institutions in providing next-generation soft-tissue repair solutions to more patients worldwide.
We are dedicated to building true partnerships with surgeons and healthcare providers to deliver solutions that provide both clinical and economic improvements. We believe that genuine collaboration with surgeons and healthcare providers results in the development of new solutions that empower patient care and addresses unmet needs within the soft tissue reconstruction market.
Our first portfolio of products, the OviTex Reinforced Tissue Matrix (“OviTex”) which we first commercialized in the U.S. in July 2016 and in Europe in February 2019, addresses unmet needs in hernia repair and abdominal wall reconstruction by combining the benefits of biologic matrices and polymer materials while minimizing their shortcomings, at a cost-effective price.
Hernia repair is one of the most common surgeries performed in the U.S., representing approximately 1.2 million procedures annually. Based on the volume weighted average selling price of our OviTex products, we estimate the annual U.S. total addressable market opportunity for our OviTex products to be approximately $1.8 billion.
Our OviTex portfolio consists of multiple product configurations intended to address various surgical procedures within hernia repair and abdominal wall reconstruction, including ventral, inguinal, and hiatal hernia repair. In addition, we have also designed an OviTex product specifically for use in laparoscopic and robotic-assisted hernia repair, which we market as OviTex LPR and began commercializing in November 2018. In February 2023, we launched two larger configurations of OviTex LPR, designed for ventral and incisional hernias. In April 2024, we launched OviTex IHR Reinforced Tissue Matrix, a new OviTex configuration specifically designed to address inguinal hernia procedures performed robotically and laparoscopically.
We have also focused on evaluating and publishing clinical data on the effectiveness and safety of our OviTex products. To date, there have been over forty published or presented works relating to these clinical findings, either by us or a third-party evaluating one or more product configurations in our OviTex portfolio. In October 2022, the 24-month results of our single arm, multicenter post-market clinical study, which we refer to as our BRAVO study, were published in the Annals of Medicine and Surgery. The BRAVO study was designed to evaluate the clinical performance of OviTex for primary or recurrent ventral hernias using open, laparoscopic, or robotic techniques in 92 enrolled patients. The recurrence rate at the 24-month time point was 2.6%, and surgical site occurrences (“SSOs”), were observed in 38% of the study population. Of the enrolled patients, 78% were characterized as high risk for experiencing an SSO based on at least one known risk factor, which included obesity, active smoking, chronic obstructive pulmonary disease (“COPD”), diabetes mellitus, coronary artery disease, or advanced age (≥75 years). The results also indicated that BRAVO patients experienced statistically significant and clinically meaningful improvements in their quality of life and perceived health based on patient responses to the EuroQol-5 Dimension (EQ-5D) health assessment and the validated 12-question Hernia-Related Quality of Life survey (HerQLes). In addition to the BRAVO study, we have also initiated other clinical data collection initiatives evaluating the use of OviTex across a variety of hernia and abdominal wall reconstruction procedures.
83
Among these other initiatives, we continue to enroll patients for our BRAVO II study, a prospective study evaluating the use of OviTex in robot-assisted ventral and inguinal hernia repairs.
Our second portfolio of products, the OviTex PRS Reinforced Tissue Matrix, (“OviTex PRS”) which we first commercialized in the U.S. in May 2019, addresses unmet needs in plastic and reconstructive surgery. OviTex PRS is indicated for use in implantation to reinforce soft-tissue where weakness exists in patients requiring soft-tissue repair or reinforcement in plastic and reconstructive surgery. Our OviTex PRS portfolio consists of three product configurations with two or three layers of high-quality tissue derived from ovine rumen, which is reinforced with either permanent or resorbable polymer for added strength, stabilization, and controlled stretch. These products are designed to improve outcomes by facilitating functional tissue remodeling while controlling the degree and direction of stretch. OviTex PRS Long-Term Resorbable, our most recent product configuration, launched in August 2023, and was designed to enhance the OviTex PRS portfolio with specific design features including bi-directional stretch and a fully resorbable, long-term polymer for reinforcement.
Our OviTex PRS portfolio is supported by non-human primate data that demonstrated more rapid tissue integration and tissue remodeling compared to the market leading biologic matrix used in this indication. In addition, there have been a growing number of published or presented works evaluating the use of OviTex PRS in plastic and reconstruction applications. We also continue to enroll patients in our OPERA study, a retrospective-prospective trial evaluating the safety profile of OviTex PRS in previous pre-pectoral and sub-pectoral implant-based breast reconstructions. Based on the current sales of biologic matrices in the U.S., we estimate the annual U.S. current addressable market opportunity for our OviTex PRS products to be approximately $800 million.
Our OviTex products have received 510(k) clearances from the U.S. Food and Drug Administration, (“FDA”) which clearances were obtained and are currently held by our exclusive contract manufacturer of these products, Aroa. In April 2019, our first OviTex PRS products received 510(k) clearance from the FDA, which clearance was initially obtained by Aroa and is currently held by us. In March 2023, we received an additional 510(k) clearance for our OviTex PRS Long-Term Resorbable device, which is currently held by us. In May 2024, we received clearance of a Special 510(k) related to minor changes to our OviTex PRS Permanent and Short-Term Resorbable devices. In October 2024, we received approval from the FDA for our investigational device exemption application relating to the study of the safety and effectiveness of our OviTex PRS product in implant-based breast reconstruction. We continue to evaluate and finalize the clinical study protocol and anticipate additional FDA interactions related to such to support a pre-market application to obtain approval for an indication for OviTex PRS for use in breast reconstruction.
Historically, we have sought to expand our service offerings beyond our OviTex and OviTex PRS products through commercial partnerships to distribute complimentary soft tissue preservation and restoration solutions. Some additional product offerings include or have included atraumatic mesh fixation devices or surgical wound management and infection control solutions. In September 2023, we entered into a distribution agreement with Advanced Medical Solutions Limited, a company registered in England, to distribute their LiquiFix Hernia Mesh Fixation Devices (LIQUIFIX FIX8™ and LIQUIFIX Precision™). In March 2024, we announced the full commercial launch of LiquiFix in the U.S. We previously co-developed and commercialized the NIVIS Fibrillar Collagen Pack, (“NIVIS”) an absorbent matrix of Type I and Type III bovine collagen designed to manage moderately to heavily exudating wounds and to control minor bleeding, in partnership with Regenity Biosciences. In March 2024, we sold our distribution rights to MiMedx Group, Inc. in exchange for an initial $5.0 million payment and additional future payments aggregating between a minimum of $3.0 million and a maximum of $7.0 million based on net sales of NIVIS (now marketed as HELIOGEN) during the first two years following its launch by MiMedx Group, Inc. We may assess additional strategic partnerships with medical device companies whereby we may enter into distribution, product development and/or licensing agreements for additional products complimentary to, or related to, existing and future products in our distribution channel, which could result in the payment by us of single digit percentage royalties or other product acquisition costs
We have a broad portfolio of intellectual property protecting our products that we believe, when combined with the proprietary manufacturing processes associated with our products and our know-how, provides significant barriers to entry. Our intellectual property applies to our differentiated product construction and materials. In addition, we believe our exclusive manufacturing and long-term supply and license agreement with Aroa (the “Aroa License”) creates a competitive advantage by allowing us to secure an exclusive supply of ovine rumen at a low cost.
84
Ovine rumen, the forestomach of a sheep, is the source of the biologic material used in both of our OviTex and OviTex PRS products. We use biologic material from ovine rumen because of its plentiful supply, optimal biomechanical profile and open collagen architecture that allows for rapid cellular infiltration. Our OviTex and OviTex PRS products are manufactured by Aroa at their FDA registered and ISO 13485 compliant facility in Auckland, New Zealand. We purchase product from Aroa at a fixed transfer cost as a percentage of Aroa’s cost of goods sold, and subject to a true-up adjustment, resulting in an amount equal to 27% of our net sales of our OviTex and OviTex PRS products, with the exception of OviTex IHR product configurations, for which we pay the greater of the initial fixed transfer cost or 27% of our net sales of OviTex IHR. This revenue sharing arrangement allows us to competitively price our products and pass along cost-savings to our customers.
We primarily market our products through a single direct sales force, predominantly in the U.S., with a small number of sales representatives in the United Kingdom and European Union, and also utilize a smaller number of independent contractors and distributors in the United States and certain European countries. We have invested in our direct sales and marketing infrastructure to expand our presence and to promote awareness and adoption of our products. As of December 31, 2024, we had 75 sales territories in the U.S. and 13 sales territories in Europe. We believe we can enhance the productivity of our sales force by improving customer segmentation and targeting, implementing and further refining our proprietary training programs, leveraging support from our medical education and medical affairs functions to drive physician awareness, education and clinical understanding of our products, and utilizing engagement analytics to support further product development and enhancement opportunities. Additionally, we have contracted with three national group purchasing organizations (“GPOs”) in the United States covering our OviTex and OviTex PRS products and plan to continue to contract with additional GPOs and other integrated delivery networks (“IDNs”) to increase access to and penetration of hospital accounts for all products we commercialize.
We are currently devoting research and development resources to develop additional variations of our OviTex and OviTex PRS products, including larger versions of our current OviTex PRS product configurations, the development of OviTex configurations with longer-acting resorbable polymers and other potential product and packaging enhancements to extend the shelf life of our products. In addition, we also continue to explore the development of lower-cost, higher-margin resorbable polymer-based devices targeting our current indications. We are also exploring additional technologies that may complement our existing products, or expand the number of our products, in each case within the hernia, plastic and reconstruction, and broader soft-tissue reconstruction market. We intend to continue to make investments in research and development efforts to develop improvements and enhancements to our product portfolio.
Our revenue for the years ended December 31, 2024 and 2023 was $69.3 million and $58.5 million, respectively, which represents an increase of $10.8 million, or 19% for the year ended December 31, 2024. Our net loss for the same time periods was $37.8 million and $46.7 million, respectively, which represents a decrease of $8.8 million, or 19% for the year ended December 31, 2024 inclusive of the gain recognized of $7.6 million on the sale of NIVIS to the MiMedx Group, Inc. As of December 31, 2024, we had an accumulated deficit of $358.7 million. The vast majority of our revenue to date has been generated from sales of our OviTex and OviTex PRS products in the U.S., with the remainder generated from sales of our OviTex products in Europe and the sale of other products.
Business Update Regarding Macroeconomic Conditions
Our business, results of operations and commercial operations have been, and may continue to be impacted by macroeconomic conditions outside of our control, including general economic uncertainty, external cybersecurity events impacting our customers, disruptions in supply of critical surgical supplies for procedures utilizing our products, inflationary pressures, tariffs, regulatory changes in the market in which we operate, fluctuations in foreign currency in the jurisdictions in which we operate, banking instability, monetary policy changes and geopolitical conflicts. These factors have and may continue to impact us in the following ways:
85
General Economic Uncertainty: Continued concerns about the systemic impact of a potential economic downturn or recession, increasing interest rates, further economic downturn or banking instability, monetary policy including the imposition of tariffs, changes and geopolitical issues, including the ongoing Russia-Ukraine conflict, the current conflict in the Middle East (including any escalation or expansion) and increasing tensions between China and Taiwan, have contributed to increased market volatility and diminished expectations for economic growth in the world. Due to this uncertainty and other factors, we have experienced high volatility in our stock price over the prior year. Continued uncertainty, perception of worsening market conditions and the introduction of new products which may, or may be perceived to, negatively impact the demand for our products now or in the future could result in a decline in our stock price, high inflation, an increase in our cost of capital and an adverse effect on our ability to access the capital markets in the future on terms acceptable to us or at all.
External Cybersecurity Events: The sale of our medical products is correlated to the frequency of surgical procedural volumes at current and prospective hospital accounts. During the second quarter of 2024, we became aware of multiple cybersecurity events, including ransomware attacks and other similar system disruptions and outages, in the U.S. and Europe that adversely impacted the procedural volumes at current customer accounts, including those affiliated across one of our GPOs. To the extent current or future cybersecurity events continue to impact the hospital systems we serve, or otherwise affect third-party payors or other vendors within the healthcare industry critical to the patient care, we may experience additional reductions in procedural volumes that lead to lower sales volume for our products.
External Supply Constraints for Critical Surgical Supplies: Any disruptions to the supply of critical surgical supplies, including, for example, IV fluids, could lead to deferrals of elective surgical procedures, including those utilizing our products. To the extent that our current and prospective hospital customers experience significant shortages of these critical supplies, whether due to extreme weather events, labor or work stoppages, or other supply chain disruptions, we may experience reductions in procedural volumes that lead to lower sales volume for our products.
Financial Strain: Market acceptance of our medical products in the U.S. and other countries is dependent upon the procurement practices of our customers, patient need for our products and procedures and the reimbursement of patients’ medical expenses by government healthcare programs and third-party payors. The continuing uncertainty surrounding macroeconomic conditions and financial markets, including the financial strain suffered by hospital customers first arising in response to the COVID-19 pandemic, may adversely affect demand for our products and procedures and result in lower reimbursement rates or coverage for our products, resulting in lower sales volume and downward pricing pressure on our products and slower adoption of new products.
Components of Our Results of Operations
Revenue
The majority of our revenue consists of direct sales of our products to hospital accounts in the U.S. Depending on the terms of our agreements with our customers, we recognize revenue related to product sales when control transfers, which generally occurs when the product is shipped to the customer, or when the product is utilized in a surgical procedure in the case of consignment agreements. Fees charged to customers for shipping are recognized as revenue. Recent revenue growth has been driven by increasing revenue from product sales due to our expanding customer base and deeper penetration across procedures in existing customer accounts, although macroeconomic pressures described in this Annual Report may impair our ability to continue to generate revenue, expand our customer base, and increase utilization of our products in existing customer accounts at historic rates.
86
Cost of Revenue
Cost of revenue primarily consists of the costs of licensed products, charges related to excess and obsolete inventory adjustments, royalties and costs related to shipping. We purchase product from Aroa at a fixed transfer cost as a percentage of Aroa’s cost of goods, which, subject to a true-up adjustment, results in an amount equal to 27% of our net sales of our OviTex and OviTex PRS products, with the exception of OviTex IHR product configurations, for which we pay the greater of the initial fixed transfer cost or 27% of our net sales of OviTex IHR. The initial term of our Aroa License terminates on the expiration of the last patent covering bovine and ovine products, with an option to extend for an additional ten-year period. We expect our cost of revenue to increase in absolute dollars as, and to the extent, our sales volume grows. Any delay in volume growth, whether due to macroeconomic pressures or otherwise, could lead to additional charges to excess and obsolete inventory.
Amortization of Intangible Assets
Amortization of intangible assets relates to the amortization of capitalized milestone amounts paid to Aroa related to license fees or commercialization rights after future economic benefit has been established for a product. These capitalized milestone amounts relate to regulatory clearances, the receipt of certain supply quantities of product, and amounts based upon aggregate net sales thresholds within a specified territory, and are amortized over the remaining useful life of the intellectual property.
Gross Profit and Gross Margin
Our gross profit is calculated by subtracting our cost of revenue and amortization of intangible assets from our revenue. We calculate our gross margin percentage as our gross profit divided by our revenue. Our gross margin has been, and we expect it will continue to be, affected by a variety of factors, including sales volume, royalties and inventory excess and obsolescence costs. Our gross profit may increase to the extent our revenue grows.
Sales and Marketing Expenses
Sales and marketing expenses consist of commercial activities related to the sale of our products, along with the salaries and related benefits, including sales commissions and stock-based compensation for employees focused on these efforts. Other significant sales and marketing expenses include costs incurred with post-market clinical studies, conferences and trade shows, promotional and marketing activities, market research, as well as travel and training expenses.
We expect future sales and marketing expenses will primarily depend on our ability to drive operational leverage and efficiencies from our commercial organization. We expect our sales and marketing expenses to continue to decrease as a percentage of revenue, as and to the extent, our revenue grows.
General and Administrative Expenses
General and administrative expenses consist primarily of salaries and related benefits, including stock-based compensation for personnel in executive, finance, information technology and administrative functions. General and administrative expenses also include professional service fees for legal, accounting, consulting, investor and public relations, insurance costs and direct and allocated facility-related costs.
We expect future general and administrative expenses will primarily depend on our ability to efficiently execute on our growth initiatives. We expect our general and administrative expenses to decrease as a percentage of revenue primarily as, and to the extent, our revenue grows.
Research and Development Expenses
Research and development expenses consist primarily of product research, engineering, product development, regulatory compliance and clinical development. These expenses include salaries and related benefits including stock-based compensation, for employees focused on these efforts, consulting services, costs associated with our preclinical studies and clinical studies undertaken to obtain regulatory clearance for new or expanded product indications, costs incurred with our manufacturing partner under development agreements related to technology transfer, costs incurred from license agreements with no alternative future uses, laboratory materials and supplies and an allocation of related facilities costs.
87
We expense research and development costs as they are incurred.
We expect future research and development expenses will primarily depend on our ability to efficiently develop new products, enhance existing products and conduct research to generate clinical data in support of new or expanded indications for our products. We expect research and development expenses as a percentage of revenue to vary over time depending on the level and timing of new product development and clinical trial initiatives.
Gain on Sale of Product Line
In March 2024, we entered into an asset purchase agreement with MiMedx Group, Inc. to sell certain assets related to NIVIS. These assets mainly included our existing inventory of NIVIS, with a net carrying value of $0.8 million, and certain intellectual property rights to sell NIVIS, with no carrying value. We transferred control of the nonfinancial asset group in March 2024 and recognized a gain of $7.6 million on the consolidated statement of operations and comprehensive loss during the year ended December 31, 2024. At each reporting date, we assess the constraint of variable consideration and record increases in the transaction price in the period that the estimate of variable consideration changes.
Interest Expense
Interest expense consists of cash interest under our credit facilities and non-cash interest attributable to the amortization of final payment fees and the amortization of deferred financing costs related to our indebtedness.
Other Income
Other income consists primarily of income earned on our cash and cash equivalents offset by miscellaneous tax expenses and foreign currency exchange gains and losses.
88
Results of Operations
Comparison of the Year Ended December 31, 2024 and 2023
|
Year Ended December 31, |
|
Change |
|
|||||||
|
2024 |
|
2023 |
|
Dollar |
|
Percentage |
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
Revenue |
$ |
69,300 |
|
$ |
58,453 |
|
$ |
10,847 |
|
19 |
% |
Cost of revenue (excluding amortization of intangible assets) |
|
22,432 |
|
|
17,961 |
|
|
4,471 |
|
25 |
|
Amortization of intangible assets |
|
380 |
|
|
380 |
|
|
— |
|
— |
|
Gross profit |
|
46,488 |
|
|
40,112 |
|
|
6,376 |
|
16 |
|
Gross margin |
|
67 |
% |
|
69 |
% |
|
|
|
|
|
Operating expenses: |
|
|
|
|
|
|
|
|
|
|
|
Sales and marketing |
|
64,648 |
|
|
59,681 |
|
|
4,967 |
|
8 |
|
General and administrative |
|
14,722 |
|
|
14,887 |
|
|
(165) |
|
(1) |
|
Research and development |
|
8,813 |
|
|
9,619 |
|
|
(806) |
|
(8) |
|
Total operating expenses |
|
88,183 |
|
|
84,187 |
|
|
3,996 |
|
5 |
|
Other operating income: |
|
|
|
|
|
|
|
|
|
|
|
Gain on sale of product line |
|
7,580 |
|
|
— |
|
|
7,580 |
|
NA |
|
Loss from operations |
|
(34,115) |
|
|
(44,075) |
|
|
9,960 |
|
(23) |
|
Other (expense) income: |
|
|
|
|
|
|
|
|
|
|
|
Interest expense |
|
(5,290) |
|
|
(5,223) |
|
|
(67) |
|
1 |
|
Other income |
|
1,420 |
|
|
2,634 |
|
|
(1,214) |
|
(46) |
|
Total other expense |
|
(3,870) |
|
|
(2,589) |
|
|
(1,281) |
|
49 |
|
Loss before income tax benefit |
|
(37,985) |
|
|
(46,664) |
|
|
8,679 |
|
(19) |
|
Income tax benefit |
|
144 |
|
|
— |
|
|
144 |
|
NA |
|
Net loss |
$ |
(37,841) |
|
$ |
(46,664) |
|
$ |
8,823 |
|
(19) |
% |
Revenue
Revenue increased by $10.8 million, or 19%, to $69.3 million for the year ended December 31, 2024 from $58.5 million for the year ended December 31, 2023. The increase in revenue was primarily driven by an increase in unit sales of our products due to the addition of new customers and growing international sales. This growth was partially offset by a decrease in average selling prices caused by product mix as the share of smaller-sized units increased. During the year ended December 31, 2024, we sold 18,121 units of OviTex compared to 13,675 units of OviTex during the year ended December 31, 2023, a 33% increase in unit sales volume. Additionally, we sold 4,645 units of OviTex PRS compared to 3,544 units during the year ended December 31, 2023, a 31% increase in unit sales volume.
Cost of Revenue
Cost of revenue (excluding amortization of intangible assets) increased by $4.5 million, or 25%, to $22.4 million for the year ended December 31, 2024 from $18.0 million for the year ended December 31, 2023. The increase in cost of revenue was primarily the result of an increase in products purchased to support demand from our higher unit sales and a higher charge for excess and obsolete inventory.
Amortization of Intangible Assets
Amortization of intangible assets was $0.4 million for both the years ended December 31, 2024 and 2023.
Gross Margin
Gross margin decreased to 67% for the year ended December 31, 2024 from 69% for the year ended December 31, 2023. The decrease was primarily due to higher expense recognized for excess and obsolete inventory adjustments as a percentage of revenue which resulted from the introduction of newer generation products.
89
Sales and Marketing
Sales and marketing expenses increased by $5.0 million, or 8%, to $64.6 million for the year ended December 31, 2024 from $59.7 million for the year ended December 31, 2023. The increase was primarily due to higher compensation costs, primarily from commissions on an increased revenue base and severance costs, increased travel and consulting expense, and additional selling-related expenses related to product sampling and meeting expenses, which were partially offset by decreased marketing expenses.
General and Administrative
General and administrative expenses decreased by $0.2 million, or 1%, to $14.7 million for the year ended December 31, 2024 from $14.9 million for the year ended December 31, 2023. The decrease was primarily due to decreases in professional fees, bad debt and insurance expense, partially offset by higher compensation costs.
Research and Development
Research and development expenses decreased by $0.8 million, or 8%, to $8.8 million for the year ended December 31, 2024 from $9.6 million for the year ended December 31, 2023. The decrease was primarily due to reduced clinical and preclinical study costs, including associated consulting expense, partially offset by higher compensation costs.
Gain on Sale of Product Line
In March 2024, we entered into an asset purchase agreement with MiMedx Group, Inc. to sell certain assets related to NIVIS. These assets mainly included our existing inventory of NIVIS, with a net carrying value of $0.8 million, and certain intellectual property rights to sell NIVIS, with no carrying value. We transferred control of the nonfinancial asset group in March 2024 and recognized a gain of $7.6 million during the year ended December 31, 2024.
Interest Expense
Interest expense increased by $0.1 million, or 1%, to $5.3 million for the year ended December 31, 2024 from $5.2 million for the year ended December 31, 2023 due to increases in the variable component of our interest rate.
Other Income (Expense)
Other income (expense) decreased by $1.2 million primarily due to lower interest income on cash balances and foreign currency translation adjustments.
Income Tax Benefit
We recorded a tax benefit of $0.1 million related to our foreign jurisdiction as we released a valuation allowance against our net operating loss tax asset.
Liquidity and Capital Resources
Overview
As of December 31, 2024, we had cash and cash equivalents of $52.7 million, working capital of $62.5 million and an accumulated deficit of $358.9 million. As of December 31, 2023, we had cash and cash equivalents of $46.7 million, working capital of $54.8 million and an accumulated deficit of $320.9 million.
On October 24, 2024, we completed an underwritten public offering of 14,670,000 shares of our common stock, including the exercise in full of the underwriters’ overallotment option to purchase additional shares of common stock, at a price to the public of $2.25 per share and, in lieu of common stock to investors who so chose, pre-funded warrants to purchase 5,800,000 shares of common stock at a public offering price of $2.2499 per pre-funded warrant, which represents the per share public offering price for the shares of common stock less the $0.0001 per share exercise price for each pre-funded warrant.
90
The offering resulted in net proceeds of $42.9 million, after deducting underwriting discounts and commissions and other estimated offering expenses and assuming no subsequent exercise of the pre-funded warrants. The exercise of the pre-funded warrants, if any, is not expected to provide significant additional funding to the Company.
In March 2024, we sold our distribution rights to MiMedx Group, Inc. in exchange for an initial $5.0 million payment and additional future payments aggregating between a minimum of $3.0 million and a maximum of $7.0 million based on net sales of NIVIS (now marketed as HELIOGEN) over the subsequent two years.
We have incurred operating losses since our inception, and we anticipate that our operating losses will continue in the near term as we seek to invest in our sales and marketing initiatives to support our growth in existing and new markets and in additional research and development activities. As of December 31, 2024, we had $40.0 million of borrowings outstanding under our Credit and Security Agreement (the “MidCap Credit Agreement”) with MidCap Financial Trust, as agent and certain lender parties thereto. The MidCap Credit Agreement matures in May 2027. Upon closing, we used a portion of the proceeds to repay borrowings under a previous credit facility and intend to use the remaining proceeds to fund operations and other general corporate purposes.
Based on our current business plan, we believe that our existing cash resources will be sufficient to meet our capital requirements and fund our operations for at least the next 12 months from the issuance of this Annual Report. If these sources are insufficient to satisfy our liquidity requirements, we may seek to sell common or preferred equity or debt securities or enter into a new credit facility. In November 2023, we entered into a new Equity Distribution Agreement (the “2023 Equity Agreement”) with Piper Sandler & Co, (“Piper”) in connection with the establishment of an at-the-market offering program under which we may sell shares of our common stock, from time to time through Piper as sales agent, in an initial amount of up to $50 million. No sales were made under the 2023 Equity Agreement or during the year ended December 31, 2024. If we raise additional funds by issuing equity or equity-linked securities, our stockholders would experience dilution and any new equity securities could have rights, preferences and privileges superior to those of holders of our common stock. Debt financing, if available, may involve covenants restricting our operations or our ability to incur additional debt. We cannot be assured that additional equity, equity-linked or debt financing will be available on terms favorable to us or our stockholders, or at all, including as a result of market volatility stemming from macroeconomic conditions, including those related to banking instability, increasing interest rates or other factors. If we are unable to obtain adequate financing, we may be required to delay or reduce the current development, commercialization and marketing plans for our products.
Cash Flows
The following table summarizes our sources and uses of cash for each of the periods presented:
|
Year Ended December 31, |
|||||||
(in thousands) |
2024 |
|
2023 |
|
2022 |
|||
Cash used in operating activities |
$ |
(41,595) |
|
$ |
(40,857) |
|
$ |
(40,748) |
Cash provided by (used in) investing activities |
|
4,451 |
|
|
(599) |
|
|
(1,872) |
Cash provided by financing activities |
|
43,057 |
|
|
46,267 |
|
|
40,852 |
Effect of exchange rate changes on cash and cash equivalents |
|
28 |
|
|
164 |
|
|
(144) |
Net increase (decrease) in cash and cash equivalents and restricted cash |
$ |
5,941 |
|
$ |
4,975 |
|
$ |
(1,912) |
Operating Activities
During the year ended December 31, 2024, we used $41.6 million of cash in operating activities, resulting from our net loss of $37.8 million and the change in operating assets and liabilities of $4.9 million, offset by non-cash items of $1.1 million. Our non-cash items were comprised of the gain on sale of NIVIS of $7.6 million offset by stock-based compensation expense of $4.4 million, our excess and obsolete inventory charge of $3.0 million, depreciation and amortization expense of $1.0 million and noncash interest expense of $0.6 million. The change in our operating assets and liabilities was primarily related to increases in accounts receivable and inventory, partially offset by increases in accrued expenses.
91
During the year ended December 31, 2023, we used $40.9 million of cash in operating activities, resulting from our net loss of $46.7 million and the change in operating assets and liabilities of $2.0 million, offset by non-cash items of $7.8 million. Our non-cash items were comprised of stock-based compensation expense of $5.0 million, our excess and obsolete inventory charge of $1.4 million, depreciation and amortization expense of $0.8 million and noncash interest expense of $0.6 million. The change in our operating assets and liabilities was primarily related to increases in accounts receivable and inventory, partially offset by increases in accrued expenses and other current and long-term liabilities.
During the year ended December 31, 2022, we used $40.7 million of cash in operating activities, resulting from our net loss of $44.3 million and the change in operating assets and liabilities of $5.3 million, offset by non-cash items of $8.9 million. Our non-cash items were comprised of stock-based compensation expense of $4.0 million, our excess and obsolete inventory charge of $1.9 million, loss on extinguishment of debt of $1.2 million, depreciation and amortization expense of $1.2 million and noncash interest expense of $0.7 million. The change in our operating assets and liabilities was primarily related to an increase in our inventory and accounts receivable, partially offset by increases in accrued expenses and other current and long-term liabilities.
Investing Activities
During the year ended December 31, 2024, cash provided by investing activities was $4.5 million, consisting of proceeds received from the sale of NIVIS of $5.4 million, partially offset by $1.0 million in purchases of property and equipment.
During the year ended December 31, 2023, cash used in investing activities was $0.6 million consisting of purchases of property and equipment.
During the year ended December 31, 2022, cash used in investing activities was $1.9 million consisting of a $1.0 million payment made for our intangible asset and purchases of property and equipment.
Financing Activities
During the year ended December 31, 2024, cash provided by financing activities was $43.1 million, consisting primarily of $42.9 million in proceeds received from the sale of our common stock and pre-funded warrants. $0.3 million of proceeds received from the issuance of stock under the employee stock purchase plan and $0.2 million of proceeds received from the exercise of stock options, partially offset by the payment of withholding taxes related to stock-based compensation to employees.
During the year ended December 31, 2023, cash provided by financing activities was $46.3 million, consisting primarily of $46.3 million in proceeds received from the sale of our common stock and $0.1 million of proceeds received from the exercise of stock options, partially offset by the payment of withholding taxes related to stock-based compensation to employees.
During the year ended December 31, 2022, cash provided by financing activities was $40.9 million, consisting primarily of $34.4 million in proceeds from an underwritten public offering, $40.0 million in proceeds received from the issuance of long-term debt, partially offset by $30.0 million in repayments of long-term debt and $3.5 million in payments of issuance costs.
Indebtedness
On May 26, 2022, we entered into the MidCap Credit Agreement with MidCap Financial Trust, as agent and certain lender parties thereto. The MidCap Credit Agreement provides for up to $40.0 million in MidCap Term Loans. Upon closing, we used a portion of the proceeds to fully repay borrowings under the OrbiMed Credit Facility and intend to use the remaining proceeds to fund operations and other general corporate purposes.
92
Pursuant to the MidCap Credit Agreement, we provided a first priority security interest in all existing and future acquired assets, including intellectual property, owned by us. The MidCap Credit Agreement contains certain covenants that limit our ability to engage in certain transactions that may be in our long-term best interests, including the incurrence of additional indebtedness, effecting certain corporate changes, making certain investments, acquisitions or dispositions and paying dividends.
The MidCap Credit Agreement also contains customary indemnification obligations and customary events of default, including, among other things, (i) non-payment, (ii) breach of warranty, (iii) non-performance of covenants and obligations, (iv) default on other indebtedness, (v) judgments, (vi) change of control, (vii) bankruptcy and insolvency, (viii) impairment of security, (ix) key permit events, (x) termination of a pension plan, (xi) regulatory matters, (xii) material adverse effect and (xiii) breach of material contracts.
In addition, we must maintain minimum net revenue levels tested quarterly. In the event of default under the MidCap Credit Agreement, we would be required to pay interest on principal and all other due and unpaid obligations at the current rate in effect plus 2%.
The MidCap Term Loans mature on May 1, 2027 and bear interest at a rate equal to 6.25% plus the greater of one-month Term SOFR (as defined in the MidCap Credit Agreement) or 1.0%. We are required to make 36 monthly interest payments beginning on June 1, 2022 (the “Interest-Only Period”). If we are in covenant compliance at the end of the Interest-Only Period, we will have the option to extend the Interest-Only Period by 12 months to 48 monthly interest payments, followed by 12 months of straight-line amortization, with the entire principal payment due at maturity. If we are not in covenant compliance at the end of the Interest-Only Period, we are required to make 24 months of straight-line amortization payments, with the entire principal amount due at maturity.
Subject to certain limitations, the MidCap Term Loans have a prepayment fee equal to 1.0% of the prepaid principal. We are also required to pay an exit fee at the time of maturity or prepayment event equal to 5% of all principal borrowings (or in the event of a prepayment event, the amount of principal being prepaid).
Contractual Obligations and Commitments
The following table summarizes our contractual obligations as of December 31, 2024 and the effects that such obligations are expected to have on our liquidity and cash flows in future periods:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Payments due by Period |
|||||||||||||
|
|
|
|
|
Less than |
|
|
|
|
|
|
|
|
||
(in thousands) |
|
Total |
|
1 year |
|
1 to 3 years |
|
3 to 5 years |
|
Thereafter |
|||||
Principal payments on long-term debt(1) |
|
$ |
40,000 |
|
$ |
— |
|
$ |
40,000 |
|
$ |
— |
|
$ |
— |
Interest and end of term charge on long-term debt(2) |
|
|
12,188 |
|
|
4,361 |
|
|
7,827 |
|
|
— |
|
|
— |
Operating lease commitments(3) |
|
|
3,135 |
|
|
580 |
|
|
1,127 |
|
|
1,178 |
|
|
250 |
Total |
|
$ |
55,323 |
|
$ |
4,941 |
|
$ |
48,954 |
|
$ |
1,178 |
|
$ |
250 |
| (1) | Assumes extension of Interest-Only Period to 48 months under the MidCap Credit Facility. |
| (2) | Interest payable reflects the rate in effect as of December 31, 2024. The interest rate on borrowings under the MidCap Credit Facility is variable and resets monthly. End of term fee reflects final payment fee due at maturity. |
| (3) | Reflects payments due for our lease of office and laboratory space in Malvern, Pennsylvania under an operating lease agreement that expires in 2030. |
ITEM 7A.QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
Our cash is held on deposit in demand accounts at high-credit-quality financial institutions in amounts in excess of the Federal Deposit Insurance Corporation (“FDIC”) insurance coverage limit of $250,000 per depositor, per FDIC-insured bank, per ownership category. Following the events relating to Silicon Valley Bank in 2023, we established a redundant account at a high-credit-quality financial institution to mitigate liquidity risk to our cash and cash equivalents from any further instability in the financial industry.
93
We have reviewed the consolidated financial statements of these financial institutions and believe they have sufficient assets and liquidity to conduct their operations in the ordinary course of business with little or no credit risk to us.
Financial instruments that potentially subject us to concentrations of credit risk principally consist of cash equivalents and accounts receivable. We limit our credit risk associated with cash equivalents by placing investments in highly-rated money market funds. We limit our credit risk with respect to accounts receivable by performing credit evaluations when deemed necessary, but we do not require collateral to secure amounts owed to us by our customers.
As discussed above in the section of this Annual Report entitled “Liquidity and Capital Resources — Indebtedness,” the MidCap Credit Facility bears interest at a floating rate of interest, which resets monthly and is equal to 6.25% plus the greater of one-month Term SOFR or 1.0%. As a result, we are exposed to risks from changes in interest rates. A 1% increase in interest rates would have resulted in a $0.4 million increase to our interest expense for the year ended December 31, 2024.
Inflationary factors, such as increases in our cost of revenue and operating expenses, may adversely affect our operating results. Although we do not believe inflation has had a material impact on our financial condition, results of operations or cash flows to date, a high rate of inflation in the future may have an adverse effect on our ability to maintain and increase our gross margin or decrease our operating expenses as a percentage of our revenue if our selling prices of our products do not increase as much or more than our costs increase.
We do not currently have any material exposure to foreign currency fluctuations and do not engage in any hedging activities as part of our normal course of business.
Critical Accounting Policies and Significant Judgments and Estimates
Our consolidated financial statements are prepared in accordance with generally accepted accounting principles in the U.S. (“GAAP”). The preparation of our consolidated financial statements and related disclosures requires us to make estimates and judgments that affect the reported amounts of assets and liabilities, the disclosure of contingent assets and liabilities at the date of the consolidated financial statements and the reported amount of revenue and expenses during the reporting period. We base our estimates on historical experience, known trends and events, and various other factors that we believe are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. We evaluate our estimates and assumptions on an ongoing basis. Our actual results may differ from these estimates under different assumptions or conditions.
While our significant accounting policies are described in more detail in Note 3 to our consolidated financial statements appearing elsewhere in this Annual Report, we believe that the following accounting policies are those most critical to the judgments and estimates used in the preparation of our consolidated financial statements.
Revenue Recognition
We account for revenue in accordance with Accounting Standards Codification Topic 606, Revenue from Contracts with Customers (“ASC 606”). Under ASC 606, we recognize revenue when our customer obtains control of our promised good, in an amount that reflects the consideration that the entity expects to be entitled in exchange for those goods.
Inventory Valuation
Inventory is stated at the lower of cost or net realizable value, with cost determined using the first-in-first-out method. Inventory, which consists primarily of our OviTex and OviTex PRS products held on consignment or held in our warehouse, is considered finished goods and is purchased from a third party.
94
We evaluate the carrying value of our inventory in relation to the estimated forecast of product demand, which takes into consideration the expiration date of the products. A significant decrease in demand could result in an increase in the amount of excess inventory on hand, which could lead to additional charges for excess and obsolete inventory. The need to maintain substantial levels of inventory impacts our estimates for excess and obsolete inventory. In addition, we continue to introduce new products and sizes, which we believe will increase our revenue. As a result, we may be required to take additional charges for excess and obsolete inventory in the future if the purchased units do not align with sales.
Recently Issued and Adopted Accounting Pronouncements
A description of recently issued accounting pronouncements that may potentially impact our financial position and results of operations is disclosed in Note 3 to our consolidated financial statements appearing elsewhere in this Annual Report.
ITEM 8.FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
The information required by this Item is set forth on pages F-1 through F-28 hereto.
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
None.
ITEM 9A. CONTROLS AND PROCEDURES
Evaluation of Disclosure Controls and Procedures
The term “disclosure controls and procedures,” as defined in Rules 13a-15(e) and 15d-15(e) of the Exchange Act, refers to controls and procedures that are designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Exchange Act is recorded, processed, summarized, and reported within the time periods specified in the SEC’s rules and forms. Disclosure controls and procedures include, without limitation, controls and procedures designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Exchange Act is accumulated and communicated to the company’s management, including its principal executive officer and principal financial officer, as appropriate to allow timely decisions regarding required disclosure. As required by Rules 13a-15(b) and 15d-15(b) of the Exchange Act, our management, with the participation of our Chief Executive Officer and Chief Operating Officer and Chief Financial Officer, evaluated the effectiveness of our disclosure controls and procedures as of the end of the period covered by this Annual Report on Form 10-K. Based on that evaluation, our Chief Executive Officer and our Chief Operating Officer and Chief Financial Officer concluded that our disclosure controls and procedures were effective as of December 31, 2024.
Management’s Report on Internal Control Over Financial Reporting
Internal control over financial reporting refers to the process designed by, or under the supervision of, our Chief Executive Officer and Chief Operating Officer and Chief Financial Officer, and effected by our board of directors, management and other personnel, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles, and includes those policies and procedures that: (1) pertain to the maintenance of records that in reasonable detail accurately and fairly reflect the transactions and dispositions of our assets; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that our receipts and expenditures are being made only in accordance with authorizations of our management and directors; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of the company’s assets that could have a material effect on the financial statements.
95
Internal control over financial reporting may not prevent or detect all errors and all fraud. A control system, no matter how well conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are achieved. Further, the design of a control system must be balanced against resource constraints, and therefore the benefits of controls must be considered relative to their costs. Given the inherent limitations in all systems of controls, no evaluation of controls can provide absolute assurance all control issues and instances of fraud, if any, within a company have been detected. These inherent limitations include the realities that judgments in decision making can be faulty and that breakdowns can occur because of a simple error or mistake. Additionally, controls can be circumvented by the individual acts of some persons, by collusion of two or more people or by management override of the controls. The design of any system of controls is also based in part upon certain assumptions about the likelihood of future events, and there can be no assurance that any design will succeed in achieving its stated goals under all potential future conditions; over time, controls may become inadequate because of changes in conditions or the degree of compliance with policies or procedures may deteriorate. Accordingly, given the inherent limitations in a cost-effective system of internal control, financial statement misstatements due to error or fraud may occur and may not be detected. Our disclosure controls and procedures are designed to provide reasonable, not absolute, assurance of achieving their objectives. We conduct periodic evaluations of our systems of controls to enhance, where necessary, our control policies and procedures.
Management is responsible for establishing and maintaining adequate internal control over our financial reporting, as such term is defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act. Under the supervision and with the participation of our management, including our Chief Executive Officer and Chief Operating Officer and Chief Financial Officer, we conducted an evaluation of the effectiveness of our internal control over financial reporting. Management has used the framework set forth in the report entitled “Internal Control—Integrated Framework (2013)” published by the Committee of Sponsoring Organizations of the Treadway Commission to evaluate the effectiveness of our internal control over financial reporting. Based on its evaluation, management has concluded that our internal control over financial reporting was effective as of December 31, 2024.
Changes in Internal Control over Financial Reporting
During the fourth quarter ended December 31, 2024, there were no changes in our internal control over financial reporting (as defined in Rule 13a-15(f) of the Exchange Act) which materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
ITEM 9B. OTHER INFORMATION
Rule 10b5-1 and Non-Rule 10b5-1 Trading Arrangements
During the three months ended December 31, 2024, none of our directors or officers adopted, terminated or modified a “Rule 10b5-1 trading arrangement” or “non-Rule 10b5-1 trading arrangement,” as defined in Item 408(a) of Regulation S-K of the Exchange Act.
ITEM 9C. DISCLOSURE REGARDING FOREIGN JURISDICTIONS THAT PREVENT INSPECTIONS
Not applicable.
PART III
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
The information concerning directors and compliance with Section 16(a) of the Exchange Act and our Code of Conduct that applies to our principal executive officer, principal financial officer, principal accounting officer or controller called for by Item 10 of Form 10-K will be set forth in our definitive proxy statement for the 2025 annual meeting of stockholders, to be filed within 120 days after the end of the fiscal year covered by this annual report on Form 10-K, and is incorporated herein by reference.
96
We have adopted insider trading policies and procedures governing the purchase, sale, and other dispositions of our securities by directors, officers, and employees that we believe are reasonably designed to promote compliance with insider trading laws, rules and regulations, and applicable Nasdaq listing standards. Our insider trading policy states, among other things, that our directors, officers, and employees are prohibited from trading in such securities while in possession of material, nonpublic information. The foregoing summary of our insider trading policies and procedures does not purport to be complete and is qualified by reference to our Insider Trading Policy filed as an exhibit to this Annual Report on Form 10-K. In addition, with regard to the Company’s trading in its own securities, it is our policy to comply with the federal securities laws and the applicable exchange listing requirements.
ITEM 11. EXECUTIVE COMPENSATION
The information required by Item 11 of Form 10-K is incorporated by reference to the information contained in our definitive proxy statement for the 2025 annual meeting of stockholders.
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
The information required by Item 12 of Form 10-K is incorporated by reference to the information contained in our definitive proxy statement for the 2025 annual meeting of stockholders.
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
The information required by Item 13 of Form 10-K is incorporated by reference to the information contained in our definitive proxy statement for the 2025 annual meeting of stockholders.
ITEM 14. PRINCIPAL ACCOUNTANT FEES AND SERVICES
Our independent registered public accounting firm is KPMG LLP, Philadelphia, PA, Auditor Firm ID: 185.
The information required by Item 14 of Form 10-K is incorporated by reference to the information contained in our definitive proxy statement for the 2025 annual meeting of stockholders.
PART IV
ITEM 15. EXHIBITS AND FINANCIAL STATEMENT SCHEDULES
(a) 1. Financial Statements
See Index to the Consolidated Financial Statements on page F-1 of this Annual Report.
2. Financial Statement Schedules
None, as all information required in these schedules is included in the Notes to the Consolidated Financial Statements.
3. Exhibits
Reference is made to the Exhibit Index on page 99 of this Annual Report for a list of exhibits required by Item 601 of Regulation S-K to be filed as part of this Annual Report.
97
TELA Bio, Inc.
Index to Consolidated Financial Statements
|
Page |
|
|
F-2 |
|
F-4 |
|
Consolidated Statements of Operations and Comprehensive Loss |
F-5 |
F-6 |
|
F-7 |
|
F-8 |
F-1
Report of Independent Registered Public Accounting Firm
To the Stockholders and the Board of Directors
TELA Bio, Inc.:
Opinion on the Consolidated Financial Statements
We have audited the accompanying consolidated balance sheets of TELA Bio, Inc. and subsidiaries (the Company) as of December 31, 2024 and 2023, the related consolidated statements of operations and comprehensive loss, stockholders’ equity, and cash flows for each of the years in the three-year period ended December 31, 2024, and the related notes (collectively, the consolidated financial statements). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2024 and 2023, and the results of its operations and its cash flows for each of the years in the three-year period ended December 31, 2024, in conformity with U.S. generally accepted accounting principles.
Basis for Opinion
These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these consolidated financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits, we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. We believe that our audits provide a reasonable basis for our opinion.
Critical Audit Matter
The critical audit matter communicated below is a matter arising from the current period audit of the consolidated financial statements that was communicated or required to be communicated to the audit committee and that: (1) relates to accounts or disclosures that are material to the consolidated financial statements and (2) involved our especially challenging, subjective, or complex judgments. The communication of a critical audit matter does not alter in any way our opinion on the consolidated financial statements, taken as a whole, and we are not, by communicating the critical audit matter below, providing a separate opinion on the critical audit matter or on the accounts or disclosures to which it relates.
Sufficiency of audit evidence over the existence of inventory
As discussed in Note 3 to the consolidated financial statements, the value of inventory was $12.8 million as of December 31, 2024. To facilitate the delivery of its products to customers, the Company maintains inventory at its headquarters and several field locations throughout the country, which includes finished goods inventory consigned to others and held by sales representatives. As of December 31, 2024, the Company had $3.2 million in finished goods consigned to others.
F-2
We identified the assessment of the sufficiency of audit evidence over the existence of inventory as a critical audit matter. The geographical dispersion of inventory required subjective auditor judgment in determining the nature and extent of procedures performed over the existence of inventory, including the determination of physical locations to observe physical inventory counts.
The following are the primary procedures we performed to address this critical audit matter. We obtained an understanding over the Company’s inventory process by inquiring with management and observing inventory counts for certain locations and determined where we would perform procedures. We applied auditor judgment to determine the nature and extent of procedures to be performed over the existence of inventory by evaluating:
• homogeneity of the locations
• historical inventory locations we have visited and results of prior physical counts
• amounts of inventory on-hand by location.
We evaluated the existence of inventory by performing independent test counts for all items at a certain location and comparing our counts to the Company’s records. We evaluated the sufficiency of audit evidence obtained by assessing the results of the procedures performed.
/s/ KPMG LLP
We have served as the Company’s auditor since 2013.
Philadelphia, Pennsylvania
March 21, 2025
F-3
TELA Bio, Inc.
Consolidated Balance Sheets
(In thousands, except share and per share amounts)
|
|
December 31, |
||||
|
|
2024 |
|
2023 |
||
Assets |
|
|
|
|
|
|
Current assets: |
|
|
|
|
|
|
Cash and cash equivalents |
|
$ |
52,670 |
|
$ |
46,729 |
Accounts receivable, net of allowances of $275 and $416 |
|
|
10,098 |
|
|
9,737 |
Inventory |
|
|
12,781 |
|
|
13,162 |
Prepaid expenses and other current assets |
|
|
2,522 |
|
|
2,098 |
Total current assets |
|
|
78,071 |
|
|
71,726 |
Property and equipment, net |
|
|
2,341 |
|
|
1,984 |
Intangible assets, net |
|
|
1,739 |
|
|
2,119 |
Right-of-use assets |
|
|
1,738 |
|
|
1,954 |
Other long-term assets |
|
|
2,276 |
|
|
— |
Deferred tax asset, net |
|
|
140 |
|
|
— |
Restricted cash |
|
|
265 |
|
|
265 |
Total assets |
|
$ |
86,570 |
|
$ |
78,048 |
|
|
|
|
|
|
|
Liabilities and stockholders’ equity |
|
|
|
|
|
|
Current liabilities: |
|
|
|
|
|
|
Accounts payable |
|
$ |
2,147 |
|
$ |
1,667 |
Accrued expenses and other current liabilities |
|
|
13,451 |
|
|
15,300 |
Total current liabilities |
|
|
15,598 |
|
|
16,967 |
Long‑term debt |
|
|
41,124 |
|
|
40,515 |
Other long‑term liabilities |
|
|
1,390 |
|
|
1,685 |
Total liabilities |
|
|
58,112 |
|
|
59,167 |
|
|
|
|
|
|
|
Commitments and contingencies (Note 12) |
|
|
|
|
|
|
|
|
|
|
|
|
|
Stockholders’ equity: |
|
|
|
|
|
|
Preferred stock; $0.001 par value: 10,000,000 shares authorized; no shares issued and outstanding |
|
|
— |
|
|
— |
Common stock; $0.001 par value: 200,000,000 shares authorized; 39,395,712 and 24,494,675 shares issued and outstanding at December 31, 2024 and December 31, 2023, respectively |
|
|
39 |
|
|
24 |
Additional paid-in capital |
|
|
387,059 |
|
|
339,655 |
Accumulated other comprehensive income |
|
|
90 |
|
|
91 |
Accumulated deficit |
|
|
(358,730) |
|
|
(320,889) |
Total stockholders’ equity |
|
|
28,458 |
|
|
18,881 |
Total liabilities and stockholders’ equity |
|
$ |
86,570 |
|
$ |
78,048 |
See accompanying notes to consolidated financial statements.
F-4
TELA Bio, Inc.
Consolidated Statements of Operations and Comprehensive Loss
(In thousands, except share and per share amounts)
|
|
Year ended December 31, |
|||||||
|
|
2024 |
|
2023 |
|
2022 |
|||
Revenue |
|
$ |
69,300 |
|
$ |
58,453 |
|
$ |
41,418 |
Cost of revenue (excluding amortization of intangible assets) |
|
|
22,432 |
|
|
17,961 |
|
|
13,570 |
Amortization of intangible assets |
|
|
380 |
|
|
380 |
|
|
804 |
Gross profit |
|
|
46,488 |
|
|
40,112 |
|
|
27,044 |
Operating expenses: |
|
|
|
|
|
|
|
|
|
Sales and marketing |
|
|
64,648 |
|
|
59,681 |
|
|
43,252 |
General and administrative |
|
|
14,722 |
|
|
14,887 |
|
|
13,862 |
Research and development |
|
|
8,813 |
|
|
9,619 |
|
|
8,937 |
Total operating expenses |
|
|
88,183 |
|
|
84,187 |
|
|
66,051 |
Other operating income: |
|
|
|
|
|
|
|
|
|
Gain on sale of product line |
|
|
7,580 |
|
|
— |
|
|
— |
Loss from operations |
|
|
(34,115) |
|
|
(44,075) |
|
|
(39,007) |
Other (expense) income: |
|
|
|
|
|
|
|
|
|
Interest expense |
|
|
(5,290) |
|
|
(5,223) |
|
|
(4,051) |
Loss on extinguishment of debt |
|
|
— |
|
|
— |
|
|
(1,228) |
Other income |
|
|
1,420 |
|
|
2,634 |
|
|
(10) |
Total other expense, net |
|
|
(3,870) |
|
|
(2,589) |
|
|
(5,289) |
Loss before income tax benefit |
|
$ |
(37,985) |
|
$ |
(46,664) |
|
$ |
(44,296) |
Income tax benefit |
|
|
144 |
|
|
— |
|
|
— |
Net loss |
|
|
(37,841) |
|
|
(46,664) |
|
|
(44,296) |
Net loss per common share, basic and diluted |
|
$ |
(1.33) |
|
$ |
(2.04) |
|
$ |
(2.72) |
Weighted average common shares outstanding, basic and diluted |
|
|
28,526,441 |
|
|
22,868,663 |
|
|
16,267,678 |
Comprehensive loss: |
|
|
|
|
|
|
|
|
|
Net loss |
|
$ |
(37,841) |
|
$ |
(46,664) |
|
$ |
(44,296) |
Foreign currency translation adjustment |
|
|
(1) |
|
|
(59) |
|
|
202 |
Comprehensive loss |
|
$ |
(37,842) |
|
$ |
(46,723) |
|
$ |
(44,094) |
See accompanying notes to consolidated financial statements.
F-5
TELA Bio, Inc.
Consolidated Statements of Stockholders’ Equity
(In thousands, except share amounts)
|
|
|
|
|
|
|
|
|
|
Accumulated |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Additional |
|
other |
|
|
|
|
|
|
||
|
|
Common stock |
|
paid‑in |
|
comprehensive |
|
Accumulated |
|
|
|
||||||
|
|
Shares |
|
Amount |
|
capital |
|
income (loss) |
|
deficit |
|
Total |
|||||
Balance at January 1, 2022 |
|
14,529,577 |
|
$ |
15 |
|
$ |
250,064 |
|
$ |
(52) |
|
$ |
(229,929) |
|
$ |
20,098 |
Vesting of common stock previously subject to repurchase |
|
29 |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
Vesting of restricted stock units and exercise of stock options |
|
44,346 |
|
|
— |
|
|
19 |
|
|
— |
|
|
— |
|
|
19 |
Issuance of common stock under the employee stock purchase plan |
|
4,523 |
|
|
— |
|
|
50 |
|
|
— |
|
|
— |
|
|
50 |
Shares withheld for employee taxes |
|
(13,448) |
|
|
|
|
|
(157) |
|
|
|
|
|
|
|
|
(157) |
Foreign currency translation adjustment |
|
— |
|
|
— |
|
|
— |
|
|
202 |
|
|
— |
|
|
202 |
Stock‑based compensation expense |
|
— |
|
|
— |
|
|
3,989 |
|
|
— |
|
|
— |
|
|
3,989 |
Sale of common stock, net of underwriting discounts, commissions and offering costs |
|
4,600,000 |
|
|
4 |
|
|
34,396 |
|
|
|
|
|
|
|
|
34,400 |
Net loss |
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
(44,296) |
|
|
(44,296) |
Balance at December 31, 2022 |
|
19,165,027 |
|
|
19 |
|
|
288,361 |
|
|
150 |
|
|
(274,225) |
|
|
14,305 |
Vesting of restricted stock units and exercise of stock options |
|
126,987 |
|
|
— |
|
|
127 |
|
|
— |
|
|
— |
|
|
127 |
Issuance of common stock under the employee stock purchase plan |
|
10,602 |
|
|
— |
|
|
88 |
|
|
— |
|
|
— |
|
|
88 |
Shares withheld for employee taxes |
|
(27,131) |
|
|
— |
|
|
(289) |
|
|
— |
|
|
— |
|
|
(289) |
Foreign currency translation adjustment |
|
— |
|
|
— |
|
|
— |
|
|
(59) |
|
|
— |
|
|
(59) |
Stock‑based compensation expense |
|
— |
|
|
— |
|
|
5,032 |
|
|
— |
|
|
— |
|
|
5,032 |
Sale of common stock, net of underwriting discounts, commissions and offering costs |
|
5,219,190 |
|
|
5 |
|
|
46,336 |
|
|
— |
|
|
— |
|
|
46,341 |
Net loss |
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
(46,664) |
|
|
(46,664) |
Balance at December 31, 2023 |
|
24,494,675 |
|
|
24 |
|
|
339,655 |
|
|
91 |
|
|
(320,889) |
— |
|
18,881 |
Vesting of restricted stock units and exercise of stock options |
|
229,606 |
|
|
1 |
|
|
225 |
|
|
— |
|
|
— |
|
|
226 |
Issuance of common stock under the employee stock purchase plan |
|
58,994 |
|
|
— |
|
|
281 |
|
|
— |
|
|
— |
|
|
281 |
Shares withheld for employee taxes |
|
(57,563) |
|
|
— |
|
|
(369) |
|
|
— |
|
|
— |
|
|
(369) |
Foreign currency translation adjustment |
|
— |
|
|
— |
|
|
— |
|
|
(1) |
|
|
— |
|
|
(1) |
Stock‑based compensation expense |
|
— |
|
|
— |
|
|
4,362 |
|
|
— |
|
|
— |
|
|
4,362 |
Sale of common stock and pre-funded warrants, net of underwriting discounts, commissions and offering costs |
|
14,670,000 |
|
|
14 |
|
|
42,905 |
|
|
— |
|
|
— |
|
|
42,919 |
Net loss |
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
(37,841) |
|
|
(37,841) |
Balance at December 31, 2024 |
|
39,395,712 |
|
$ |
39 |
|
$ |
387,059 |
|
$ |
90 |
|
$ |
(358,730) |
|
$ |
28,458 |
See accompanying notes to consolidated financial statements.
F-6
TELA Bio, Inc.
Consolidated Statements of Cash Flows
(In thousands)
|
|
Year ended December 31, |
|||||||
|
|
2024 |
|
2023 |
|
2022 |
|||
Cash flows from operating activities: |
|
|
|
|
|
|
|
|
|
Net loss |
|
$ |
(37,841) |
|
$ |
(46,664) |
|
$ |
(44,296) |
Adjustments to reconcile net loss to net cash used in operating activities: |
|
|
|
|
|
|
|
|
|
Depreciation expense |
|
|
632 |
|
|
428 |
|
|
383 |
Noncash interest expense |
|
|
609 |
|
|
599 |
|
|
657 |
Noncash loss on extinguishment of debt |
|
|
— |
|
|
— |
|
|
1,228 |
Amortization of intangible assets |
|
|
380 |
|
|
380 |
|
|
804 |
Net changes in operating lease ROU assets and liabilities |
|
|
(99) |
|
|
(49) |
|
|
(36) |
Inventory excess and obsolescence charge |
|
|
2,955 |
|
|
1,414 |
|
|
1,866 |
Stock‑based compensation expense |
|
|
4,362 |
|
|
5,032 |
|
|
3,989 |
Deferred income tax benefit |
|
|
(144) |
|
|
— |
|
|
— |
Gain on disposal of fixed assets |
|
|
— |
|
|
(12) |
|
|
— |
Gain on sale of product line |
|
|
(7,580) |
|
|
— |
|
|
— |
Change in operating assets and liabilities: |
|
|
|
|
|
|
|
|
|
Accounts receivable, net |
|
|
(762) |
|
|
(3,058) |
|
|
(2,421) |
Inventory |
|
|
(2,972) |
|
|
(2,718) |
|
|
(6,073) |
Prepaid expenses and other current assets |
|
|
227 |
|
|
(81) |
|
|
1,216 |
Accounts payable |
|
|
482 |
|
|
11 |
|
|
(884) |
Accrued expenses and other current and long-term liabilities |
|
|
(1,809) |
|
|
4,177 |
|
|
2,399 |
Foreign currency transaction (gain) loss |
|
|
(35) |
|
|
(316) |
|
|
420 |
Net cash used in operating activities |
|
|
(41,595) |
|
|
(40,857) |
|
|
(40,748) |
Cash flows from investing activities: |
|
|
|
|
|
|
|
|
|
Payment for intangible asset |
|
|
— |
|
|
— |
|
|
(1,000) |
Purchase of property and equipment |
|
|
(989) |
|
|
(611) |
|
|
(872) |
Proceeds from the sale of product line |
|
|
5,440 |
|
|
— |
|
|
— |
Proceeds from the sale of property and equipment |
|
|
— |
|
|
12 |
|
|
— |
Net cash provided by (used in) investing activities |
|
|
4,451 |
|
|
(599) |
|
|
(1,872) |
Cash flows from financing activities: |
|
|
|
|
|
|
|
|
|
Proceeds from sale of common stock and pre-funded warrants, net |
|
|
42,919 |
|
|
46,341 |
|
|
34,400 |
Proceeds from issuance of long-term debt |
|
|
— |
|
|
— |
|
|
40,000 |
Repayment of long-term debt |
|
|
— |
|
|
— |
|
|
(30,000) |
Payment of debt financing costs |
|
|
— |
|
|
— |
|
|
(3,460) |
Proceeds from exercise of stock options |
|
|
226 |
|
|
127 |
|
|
19 |
Payment of withholding taxes related to stock-based compensation to employees |
|
|
(369) |
|
|
(289) |
|
|
(157) |
Proceeds from issuance of common stock under the employee stock purchase plan |
|
|
281 |
|
|
88 |
|
|
50 |
Net cash provided by financing activities |
|
|
43,057 |
|
|
46,267 |
|
|
40,852 |
Effect of exchange rate on cash and cash equivalents |
|
|
28 |
|
|
164 |
|
|
(144) |
Net increase (decrease) in cash and cash equivalents and restricted cash |
|
|
5,941 |
|
|
4,975 |
|
|
(1,912) |
Cash and cash equivalents and restricted cash, beginning of year |
|
|
46,994 |
|
|
42,019 |
|
|
43,931 |
Cash and cash equivalents and restricted cash, end of year |
|
$ |
52,935 |
|
$ |
46,994 |
|
$ |
42,019 |
Supplemental disclosure of cash flow information: |
|
|
|
|
|
|
|
|
|
Cash paid during the period for interest |
|
$ |
4,071 |
|
$ |
4,624 |
|
$ |
3,394 |
Supplemental disclosures of noncash investing and financing activities: |
|
|
|
|
|
|
|
|
|
Property and equipment in accounts payable and accrued expenses and other current liabilities |
|
$ |
— |
|
$ |
119 |
|
$ |
7 |
Operating lease ROU asset exchanged for operating lease liabilities |
|
$ |
— |
|
$ |
895 |
|
$ |
1,376 |
Tenant improvement and deferred rent reclassified to operating lease liabilities |
|
$ |
— |
|
$ |
— |
|
$ |
380 |
Operating lease liabilities assumed for operating lease ROU assets |
|
$ |
— |
|
$ |
— |
|
$ |
1,756 |
See accompanying notes to consolidated financial statements.
F-7
TELA Bio, Inc.
Notes to Consolidated Financial Statements
(1) Background
TELA Bio, Inc. (the “Company”) was incorporated in the state of Delaware on April 17, 2012 and wholly owns TELA Bio Limited, a company incorporated in the United Kingdom and is the ultimate parent of TELA Bio GmbH, a company incorporated in Germany through TELA Bio Limited. The Company is a commercial-stage medical technology company focused on providing innovative soft-tissue reconstruction solutions that optimize clinical outcomes by prioritizing the preservation and restoration of the patient’s own anatomy. OviTex Reinforced Tissue Matrix (“OviTex”), the Company’s first portfolio of products, addresses unmet needs in hernia repair and abdominal wall reconstruction by combining the benefits of biologic matrices and polymer materials while minimizing their shortcomings, at a cost-effective price. OviTex PRS Reinforced Tissue Matrix (“OviTex PRS”), the Company’s second portfolio of products, addresses unmet needs in plastic and reconstructive surgery. The Company’s principal corporate office and research facility is located in Malvern, Pennsylvania.
(2) Risks and Liquidity
The Company’s operations to date have focused on commercializing products, developing and acquiring technology and assets, business planning, raising capital and organization and staffing. The Company has incurred recurring losses and negative cash flows from operations since inception and has an accumulated deficit of $358.7 million as of December 31, 2024. The Company anticipates incurring additional losses until such time, if ever, it can generate sufficient revenue from its products to cover its expenses.
In March 2024, the Company sold its distribution rights for NIVIS Fibrillar Collagen Pack to MiMedx Group, Inc. in exchange for an initial $5.0 million payment and additional future payments aggregating between a minimum of $3.0 million and a maximum of $7.0 million based on net sales of NIVIS (now marketed as HELIOGEN) over the subsequent two years.
On October 24, 2024, the Company completed an underwritten public offering of 14,670,000 shares of its common stock, including the exercise in full of the underwriters’ overallotment option to purchase additional shares of common stock, at a price to the public of $2.25 per share and, in lieu of common stock to investors who so chose, pre-funded warrants to purchase 5,800,000 shares of common stock at a public offering price of $2.2499 per pre-funded warrant, which represents the per share public offering price for the shares of common stock less the $0.0001 per share exercise price for each pre-funded warrant. The offering resulted in net proceeds of $42.9 million, after deducting underwriting discounts and commissions and other estimated offering expenses and assuming no subsequent exercise of the pre-funded warrants. The exercise of the pre-funded warrants, if any, is not expected to provide significant additional funding to the Company.
The operations of the Company are subject to certain risks and uncertainties including, among others, the uncertainty of product development, the impact of macroeconomic conditions, including, general economic uncertainty, inflationary pressures and the measures undertaken by various governments to address them, banking instability, monetary policy changes (including tariffs that have been or may in the future be imposed by the U.S. or other countries), geopolitical factors such as the ongoing Russia-Ukraine conflict, the current conflicts in the Middle East (including any escalation or expansion) and increasing tensions between China and Taiwan, cybersecurity events affecting or disrupting normal hospital operations, constraints on the supply of critical surgical and hospital supplies necessary to facilitate the surgical procedures in which our products are utilized, technological uncertainty, commercial acceptance of any developed products, alternative competing technologies, dependence on collaborative partners, uncertainty regarding patents and proprietary rights, comprehensive government regulations, and dependence on key personnel.
F-8
(3) Summary of Significant Accounting Policies
Basis of Presentation and Principles of Consolidation
The accompanying consolidated financial statements have been prepared in accordance with accounting principles generally accepted in the United States (“GAAP”). Any reference in these notes to applicable guidance is meant to refer to GAAP as found in the Accounting Standards Codification (“ASC”) and Accounting Standards Updates (“ASU”) promulgated by the Financial Accounting Standards Board (“FASB”). The consolidated financial statements include the accounts of TELA Bio, Inc. and its wholly owned subsidiaries TELA Bio Limited and TELA Bio GmbH. All intercompany accounts and transactions have been eliminated in consolidation.
Use of Estimates
The preparation of consolidated financial statements in conformity with GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and contingent liabilities at the date of the consolidated financial statements and the reported amounts of revenue and expenses during the reporting period. The most significant judgments are employed in estimates used to determine the recoverability of the carrying value of the Company’s inventory. As future events and their effects cannot be determined with precision, actual results may differ significantly from these estimates.
Segments
Operating segments are defined as components of an enterprise about which separate discrete information is available for evaluation by the chief operating decision maker, or decision-making group, in deciding how to allocate resources in assessing performance. The Company has one reportable segment which is focused on providing innovative soft-tissue reconstruction solutions that optimize clinical outcomes by prioritizing the preservation and restoration of the patient’s own anatomy. The Company’s chief operating decision maker (“CODM”) is the chief executive officer.
The accounting policies of its segment are the same as those described in the summary of significant accounting policies. The CODM uses budget to actual forecasts and net income in assessing entity-wide operating results and deciding how to invest in the Company. The CODM is regularly provided with net loss and consolidated assets, which are reported on the consolidated statement of operations and comprehensive loss and consolidated balance sheet, respectively.
The tables below summarizes the items included within net loss regularly provided to the CODM for the years ended December 31, 2024, 2023 and 2022:
F-9
|
|
Year ended December 31, |
|||||||
|
|
2024 |
|
2023 |
|
2022 |
|||
Revenue |
|
$ |
69,300 |
|
$ |
58,453 |
|
$ |
41,418 |
Cost of revenue (excluding amortization of intangible assets) |
|
|
22,432 |
|
|
17,961 |
|
|
13,570 |
Amortization of intangible assets |
|
|
380 |
|
|
380 |
|
|
804 |
Gross profit |
|
|
46,488 |
|
|
40,112 |
|
|
27,044 |
Sales and marketing: |
|
|
|
|
|
|
|
|
|
Sales and sales management |
|
|
44,132 |
|
|
35,469 |
|
|
13,657 |
International |
|
|
5,072 |
|
|
3,916 |
|
|
1,191 |
Other sales and marketing (a) |
|
|
15,444 |
|
|
20,296 |
|
|
28,404 |
Total sales and marketing |
|
|
64,648 |
|
|
59,681 |
|
|
43,252 |
General and Administrative: |
|
|
|
|
|
|
|
|
|
Finance and Legal |
|
|
7,503 |
|
|
7,715 |
|
|
6,461 |
Other General and administrative (b) |
|
|
7,219 |
|
|
7,172 |
|
|
7,401 |
Total general and administrative |
|
|
14,722 |
|
|
14,887 |
|
|
13,862 |
Research and Development: |
|
|
|
|
|
|
|
|
|
Clinical |
|
|
4,068 |
|
|
3,891 |
|
|
1,064 |
Regulatory and quality |
|
|
1,452 |
|
|
2,189 |
|
|
1,493 |
Other research and development (c) |
|
|
3,293 |
|
|
3,539 |
|
|
6,380 |
Total research and development |
|
|
8,813 |
|
|
9,619 |
|
|
8,937 |
Gain on sale of product line |
|
|
7,580 |
|
|
— |
|
|
— |
Other segment items (d) |
|
|
(3,726) |
|
|
(2,589) |
|
|
(5,289) |
Net loss |
|
$ |
(37,841) |
|
$ |
(46,664) |
|
$ |
(44,296) |
|
|
|
|
|
|
|
|
|
|
(a) Other sales and marketing includes strategy, analytics and allocated facility expenses. | |||||||||
(b) Other general and administrative includes executive, human resources, information technology and allocated facility expenses. | |||||||||
(c) Other research and development includes engineering and allocated facility expenses. | |||||||||
(d) Other segment items include other operating income and other expenses as disclosed in the consolidated statements of operations and comprehensive loss; interest expense, loss on extinguishment of debt, other income and income tax benefit. | |||||||||
Concentration of Risk
Financial instruments that potentially subject the Company to concentrations of credit risk consist primarily of cash and cash equivalents. The Company places its cash with high-credit-quality financial institutions and primarily invests in money market funds. The Company has established guidelines relative to credit ratings and maturities that seek to maintain safety and liquidity.
Following the events relating to Silicon Valley Bank in 2023, the Company established an additional redundant account with another high-credit-quality financial institution to mitigate liquidity risk to our cash and cash equivalents from any further instability in the financial industry.
As described in Note 12, the Company has licensed patents and other intellectual property from Aroa Biosurgery Ltd. (“Aroa”). As part of this agreement, Aroa is also the exclusive contract manufacturer of the Company’s OviTex portfolio of products. The inability of Aroa to fulfill supply requirements of the Company could materially impact future operating results. A change in the relationship with Aroa, or an adverse change in their business, could materially impact future operating results.
F-10
Cash and Cash Equivalents
The Company considers cash equivalents to be highly-liquid investments with maturities of three months or less from the date of purchase. Cash equivalents consist of investments in a money market fund. The Company’s cash and cash equivalents are carried at fair value.
Restricted Cash
Restricted cash represents an amount held in an escrow deposit account, securing a letter of credit for the Company’s office lease.
The following table presents a reconciliation of all captions of cash, cash equivalents and restricted cash reported on the balance sheets that sum to the total of those same amounts shown in the statements of cash flows.
|
|
December 31, |
||||
|
|
2024 |
|
2023 |
||
Cash and cash equivalents |
|
$ |
52,670 |
|
$ |
46,729 |
Restricted cash |
|
|
265 |
|
|
265 |
Total cash and cash equivalents and restricted cash shown in Statement of cash flows |
|
$ |
52,935 |
|
$ |
46,994 |
Inventory
Inventory consists of purchased materials, primarily finished goods and is identified and tracked by lot and stated at the lower of cost or net realizable value, with cost being determined on a first-in, first-out basis. Inventories consisted of the following (in thousands):
|
|
December 31, |
||||
|
|
2024 |
|
2023 |
||
Finished goods |
|
$ |
12,645 |
|
$ |
13,102 |
Raw materials |
|
|
136 |
|
|
60 |
Total inventory |
|
$ |
12,781 |
|
$ |
13,162 |
The Company periodically analyzes its inventory levels and writes down inventory that has become obsolete or that has a cost basis in excess of its expected net realizable value based on expected customer demand. To facilitate the delivery of its products to customers, the Company maintains inventory at its headquarters and several field locations throughout the country, which includes finished goods inventory consigned to others and held by sales representatives. As of December 31, 2024 and 2023, the Company had $3.2 million and $3.0 million, respectively, in finished goods consigned to others.
Property and Equipment
Property and equipment are stated at the aggregate cost incurred to acquire and place the asset in service. Expenditures for routine maintenance and repairs are charged to expense as incurred and costs of improvements and renewals are capitalized. Depreciation is provided over the estimated useful lives of the assets using the straight-line method.
Intangible Assets
Upfront payments and milestone payments due related to licenses or commercialization rights prior to future economic benefit being established are recorded as research and development expenses. Milestone payments due related to licenses or commercialization rights after future economic benefit is established are recorded as intangible assets. In 2024, 2023 and 2022, the Company recorded $0.4 million, $0.4 million and $0.8 million of amortization expense, respectively, related to intangible assets.
F-11
At December 31, 2024, the remaining life of intangible assets was 4.6 years. The Company anticipates recognizing amortization expense of $0.4 million in each of the next four years and $0.2 million thereafter.
Leases
The Company determines if an arrangement is a lease at contract inception. A lease exists when a contract conveys to the customer the right to control the use of identified property, plant, or equipment for a period of time in exchange for consideration. The definition of a lease embodies two conditions: (1) there is an identified asset in the contract that is land or a depreciable asset (i.e., property, plant, and equipment), and (2) the customer has the right to control the use of the identified asset.
Operating leases are included as a right-of-use (“ROU”) asset and a corresponding lease liability on the balance sheet for all leases with terms longer than 12 months.
Long-Lived Assets
Long-lived assets, such as property and equipment and intangible assets, are reviewed for impairment whenever events or changes in circumstances indicate the carrying amount of an asset may not be recoverable. If circumstances require a long-lived asset or asset group be tested for possible impairment, the Company first compares undiscounted cash flows expected to be generated by such asset or asset group to its carrying value. If the carrying value of the long-lived asset or asset group exceeds the undiscounted cash flows, an impairment is recognized to the extent the carrying value exceeds its fair value. Fair value is determined using various valuation techniques, including discounted cash flow models, quoted market values, and third-party independent appraisals, as considered necessary. No impairment losses were recognized during the years ended December 31, 2024, 2023 or 2022.
Debt Issuance Costs
Debt issuance costs incurred in connection with debt (Note 6) are amortized to interest expense over the term of the respective financing arrangement using the effective-interest method. Debt issuance costs, net of related amortization are deducted from the carrying amount of the related debt.
Revenue Recognition
Under ASC Topic 606, Revenue from Contracts with Customers, (“ASC 606”), an entity recognizes revenue when its customer obtains control of the promised good, in an amount that reflects the consideration that the entity expects to be entitled in exchange for those goods. The Company performs the following five steps to recognize revenue under ASC 606: (i) identify the contract(s) with a customer, (ii) identify the performance obligations in the contract, (iii) determine the transaction price, (iv) allocate the transaction price to the performance obligations in the contract, and (v) recognize revenue when (or as) the entity satisfies a performance obligation. The Company only recognizes revenue when it is probable that it will collect the consideration to which it is entitled in exchange for the goods or services that will be transferred to the customer.
A significant portion of the Company’s revenue is generated from product shipped to a customer or from consigned inventory maintained at hospitals or other surgical facilities. Revenue from the sale of consigned products is recognized when control is transferred to the customer, which occurs at the time the product is used in a surgical procedure. For product that is not held on consignment, the Company recognizes revenue when control transfers to the customer which occurs at the time the product is shipped or delivered. For all of the Company’s customer contracts, the only identified performance obligation is providing the product to the customer.
F-12
Revenue is recognized at the estimated net sales price, which includes estimates of variable consideration. The Company enters into contracts with certain third-party payors for the payment of rebates with respect to the utilization of its products. These rebates are based on contractual percentages. The Company estimates and records these rebates in the same period the related revenue is recognized, resulting in a reduction of product revenue.
Payment terms with customers do not exceed one year and, therefore, the Company does not account for a financing component in these arrangements. There are no incremental costs of obtaining a contract that would rise to or enhance an asset other than product costs, which are a component of inventory. The Company expenses incremental costs of obtaining a contract with a customer (e.g., sales commissions) when incurred as the period of benefit is less than one year. Fees charged to customers for shipping are recognized as revenue.
The following table presents revenue disaggregated (in thousands):
|
|
Year ended December 31, |
|||||||
|
|
2024 |
|
2023 |
|
2022 |
|||
OviTex |
|
$ |
45,925 |
|
$ |
39,416 |
|
$ |
28,879 |
OviTex PRS |
|
|
22,745 |
|
|
18,736 |
|
|
12,431 |
Other |
|
|
630 |
|
|
301 |
|
|
108 |
Total revenue |
|
$ |
69,300 |
|
$ |
58,453 |
|
$ |
41,418 |
Sales outside of the U.S. were $10.3 million, or 15%, of total revenue for the year ended December 31, 2024, $6.1 million or 10% of total revenue for the year ended December 31, 2023 and $3.2 million or 8% of total revenue for the year ended December 31, 2022.
Research and Development
Research and development costs are charged to expense as incurred and consist primarily of salaries, benefits, and other related costs, including stock-based compensation for personnel serving in the research and development functions as well as costs incurred with Aroa under development agreements related to technology transfer, laboratory materials and supplies. At the end of the reporting period, the Company compares payments made to third-party service providers to the estimated progress toward completion of the research or development objectives. Such estimates are subject to change as additional information becomes available. Depending on the timing of payments to the service providers and the progress that the Company estimates has been made as a result of the service provided, the Company may record net prepaid or accrued expense relating to these costs. Costs incurred in obtaining patent and other intellectual property licenses or milestone payments from license agreements for which there are no alternative future uses are charged to expense as incurred.
Stock-Based Compensation
The Company accounts for stock-based awards in accordance with provisions of ASC Topic 718, Compensation—Stock Compensation, under which the Company recognizes the grant-date fair value of stock-based awards issued to employees and nonemployee board members as compensation expense on a straight-line basis over the vesting period of the award while awards containing a performance condition are recognized as expense when the achievement of the performance criteria is considered probable. The Company uses the Black-Scholes option pricing model to determine the grant-date fair value of stock options. The Company estimates forfeitures that it expects will occur and adjusts expense for actual forfeitures in the periods they occur.
F-13
Warrants
The Company accounts for issued warrants either as a liability or equity in accordance with ASC Topic 480-10, Accounting for Certain Financial Instruments with Characteristics of both Liabilities and Equity (“ASC 480-10”) or ASC Topic 815-40, Accounting for Derivative Financial Instruments Indexed to, and Potentially Settled in, a Company’s Own Stock (“ASC 815-40”). The assessment considers whether the warrants are freestanding financial instruments pursuant to ASC 480, meet the definition of a liability pursuant to ASC 480, and whether the warrants meet all of the requirements for equity classification under ASC 815, including whether the warrants are indexed to the company’s own stock and whether the warrant holders could potentially require “net cash settlement” in a circumstance outside of the company’s control, among other conditions for equity classification. This assessment, which requires the use of professional judgment, is conducted at the time of warrant issuance and as of each subsequent quarterly period end date while the warrants are outstanding.
Warrants that are equity-classified instruments and recorded in additional paid-in capital at issuance are not subject to remeasurement. The Company periodically evaluates changes in facts and circumstances that could impact the classification of warrants.
Income Taxes
Income taxes are accounted for under the asset-and-liability method as required by ASC Topic 740, Income Taxes (“ASC 740”). Deferred tax assets and liabilities are recognized for the future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases and operating loss and tax credit carryforwards. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. The effect on deferred tax assets and liabilities of a change in tax rates is recognized in income in the period corresponding to the enactment date. Under ASC 740, a valuation allowance is required when it is more likely than not all or some portion of the deferred tax assets will not be realized through generating sufficient future taxable income.
ASC Subtopic 740-10, Accounting for Uncertainty of Income Taxes (“ASC 740-10”), defines the criterion an individual tax position must meet for any part of the benefit of the tax position to be recognized in consolidated financial statements prepared in conformity with GAAP. The Company may recognize the tax benefit from an uncertain tax position only if it is more likely than not such tax position will be sustained on examination by the taxing authorities, based solely on the technical merits of the respective tax position. The tax benefits recognized in the consolidated financial statements from such a tax position should be measured based on the largest benefit having a greater than 50% likelihood of being realized upon ultimate settlement with the tax authority. In accordance with the disclosure requirements of ASC 740-10, the Company’s policy on income statement classification of interest and penalties related to income tax obligations is to include such items as part of income tax expense.
Fair value of financial instruments
Fair value is the price that could be received to sell an asset or paid to transfer a liability in an orderly transaction among market participants. Fair value determination in accordance with applicable accounting guidance requires that a number of significant judgments are made. Additionally, fair value is used on a nonrecurring basis to evaluate assets for impairment or as required for disclosure purposes by applicable accounting guidance on disclosures about fair value of financial instruments. Depending on the nature of the assets and liabilities, various valuation techniques and assumptions are used when estimating fair value. The carrying amounts of certain of the Company’s financial instruments, including cash and cash equivalents, accounts receivable, other assets, and accounts payable are shown at cost, which approximates fair value due to the short-term nature of these instruments.
F-14
The carrying amounts of the Company’s Credit and Security Agreement approximates fair value due to its variable interest rate.
The Company follows the provisions of ASC Topic 820, Fair Value Measurement, for financial assets and liabilities measured on a recurring basis. The guidance requires fair value measurements be classified and disclosed in one of the following three categories:
| ● | Level 1: Unadjusted quoted prices in active markets that are accessible at the measurement date for identical, unrestricted assets or liabilities. |
| ● | Level 2: Quoted prices in markets that are not active, or inputs which are observable, either directly or indirectly, for substantially the full term of the asset or liabilities. |
| ● | Level 3: Prices or valuation techniques that require inputs that are both significant to the fair value measurement and unobservable (i.e., supported by little or no market activity). |
The following fair value hierarchy table presents information about each major category of the Company’s financial assets and liabilities measured at fair value on a recurring basis (in thousands):
|
|
Fair value measurement at reporting date using |
|||||||
|
|
Quoted prices in |
|
|
|
|
|
|
|
|
|
active markets |
|
Significant other |
|
Significant |
|||
|
|
for identical |
|
observable |
|
unobservable |
|||
|
|
assets |
|
inputs |
|
inputs |
|||
|
|
(Level 1) |
|
(Level 2) |
|
(Level 3) |
|||
December 31, 2024: |
|
|
|
|
|
|
|
|
|
Cash equivalents – money market fund |
|
$ |
48,131 |
|
$ |
— |
|
$ |
— |
|
|
|
|
|
|
|
|
|
|
December 31, 2023: |
|
|
|
|
|
|
|
|
|
Cash equivalents – money market fund |
|
$ |
41,561 |
|
$ |
— |
|
$ |
— |
Allowance for credit losses
The following table presents a rollforward of the allowance of credit losses (in thousands):
|
|
|
Balance at Beginning of Period |
|
Bad Debt Expense Recognized |
|
Write-offs of Uncollectible Balances |
|
|
Balance at End of Period |
Year ended December 31, 2022 |
|
$ |
(52) |
|
(116) |
|
25 |
|
$ |
(143) |
Year ended December 31, 2023 |
|
$ |
(143) |
|
(306) |
|
33 |
|
$ |
(416) |
Year ended December 31, 2024 |
|
$ |
(416) |
|
(65) |
|
206 |
|
$ |
(275) |
Net loss per share
Basic and diluted net loss per common share is determined by dividing net loss by the weighted-average shares of common stock outstanding during the reporting period. In periods in which the Company reports a net loss, diluted net loss per share is the same as basic net loss per share since dilutive shares are not assumed to have been issued if their effect is antidilutive. Therefore, the weighted-average shares used to calculate both basic and diluted loss per share are the same.
F-15
The following potentially dilutive securities have been excluded from the computation of diluted weighted-average shares outstanding, as they would be antidilutive.
|
|
Year ended December 31, |
||||
|
|
2024 |
|
2023 |
|
2022 |
Stock options |
|
2,119,183 |
|
2,162,453 |
|
2,071,848 |
Unvested restricted stock units |
|
948,788 |
|
907,203 |
|
311,991 |
Common stock warrants |
|
88,556 |
|
88,556 |
|
88,556 |
Total |
|
3,156,527 |
|
3,158,212 |
|
2,472,395 |
Due to their nominal exercise price of $0.0001 per share, the outstanding pre-funded warrants are considered common stock equivalents and are included in the calculation of weighted-average shares of common stock outstanding from the October 24, 2024 closing date.
Recently Issued Accounting Pronouncements
In August 2020, the FASB issued ASU No. 2020-06, Debt with Conversion and Other Options and Derivatives and Hedging - Contracts in Entity’s Own Equity (“ASU 2020-06”). ASU 2020-06 eliminates the beneficial conversion and cash conversion accounting models for convertible instruments. It also amends the accounting for certain contracts in an entity’s own equity that are currently accounted for as derivatives because of specific settlement provisions. The new guidance also modifies how particular convertible instruments and certain contracts that may be settled in cash or shares impact the diluted EPS computation. ASU 2020-06 is effective for fiscal years beginning after December 15, 2023, including interim periods within those annual periods. The adoption of this guidance did not have a significant impact on the consolidated financial statements and related disclosures.
In November 2023, the FASB issued ASU 2023-07, Improvements to Reportable Segment Disclosures, which expands public entities’ segment disclosures by requiring disclosure of significant segment expenses that are regularly provided to the chief operating decision maker and included within each reported measure of segment profit or loss, an amount and description of its composition for other segment items, and interim disclosures of a reportable segment’s profit or loss and assets. This guidance is effective for annual periods beginning after December 15, 2023, and interim periods within annual periods beginning after December 15, 2024, with early adoption permitted, including adoption in any interim period. See note 3 for additional disclosures related to the adoption of this ASU.
In December 2023, the FASB issued ASU 2023-09, Improvements to Income Tax Disclosures, requiring entities to provide additional information in the income tax rate reconciliation and additional disclosures about income taxes paid. The new accounting guidance requires entities to disclose in their rate reconciliation table additional categories of information about federal, state and foreign income taxes and to provide more details about the reconciling items in some categories if the items meet a quantitative threshold. This guidance is effective for annual periods beginning after December 15, 2024, and should be applied prospectively, but entities have the option to apply it retrospectively for each period presented. Early adoption is permitted for annual financial statements that have not yet been issued or made available for issuance. The Company is currently evaluating the expected impact that the standard could have on its consolidated financial statements and related disclosures.
In November 2024, the FASB issued ASU 2024-03, Disaggregation of Income Statement Expenses. ASU 2024-03 requires additional disclosure of specific types of expenses included in the expense captions presented on the face of the income statement as well as disclosures about selling expenses. ASU 2024-03 is effective for fiscal years beginning after December 15, 2026, and interim periods beginning after December 15, 2027, with early adoption permitted. The requirements will be applied prospectively with the option for retrospective application.
F-16
The Company is currently evaluating the impact that the adoption of ASU 2024-03 will have on its consolidated financial statements and disclosures.
(4) Property and Equipment
Property and equipment consisted of the following (in thousands):
|
|
|
|
December 31, |
||||
Asset description |
|
Estimated useful lives |
|
2024 |
|
2023 |
||
Lab equipment |
|
5 Years |
|
$ |
2,968 |
|
$ |
2,883 |
Furniture and fixtures |
|
5 Years |
|
|
366 |
|
|
284 |
Computer equipment and software |
|
3 Years |
|
|
733 |
|
|
645 |
Leasehold improvements |
|
Lesser of useful life or lease term |
|
|
2,934 |
|
|
2,507 |
Total |
|
|
|
|
7,001 |
|
|
6,319 |
Less accumulated depreciation and amortization |
|
|
|
|
(4,660) |
|
|
(4,335) |
Property and equipment, net |
|
|
|
$ |
2,341 |
|
$ |
1,984 |
The cost of property and equipment at both December 31, 2024 and 2023 includes $0.2 million of equipment located at Aroa. Depreciation expense was $0.6 million, $0.4 million and $0.4 million for the years ended December 31, 2024, 2023 and 2022, respectively.
(5) Accrued Expenses and Other Current Liabilities
Accrued expenses and other current liabilities consisted of the following (in thousands):
|
|
December 31, |
||||
|
|
2024 |
|
2023 |
||
Compensation and related benefits |
|
$ |
7,343 |
|
$ |
9,216 |
Third-party and professional fees |
|
|
2,493 |
|
|
2,828 |
Amounts due to contract manufacturer |
|
|
2,095 |
|
|
2,024 |
Current portion of operating lease liabilities |
|
|
545 |
|
|
565 |
Research and development expenses |
|
|
20 |
|
|
140 |
Other |
|
|
955 |
|
|
527 |
Total accrued expenses and other current liabilities |
|
$ |
13,451 |
|
$ |
15,300 |
F-17
(6) Debt
Long-term debt consisted of the following (in thousands):
|
|
December 31, |
||||
|
|
2024 |
|
2023 |
||
MidCap term loan |
|
$ |
40,000 |
|
$ |
40,000 |
Exit fee |
|
|
2,000 |
|
|
2,000 |
Unamortized exit fee and issuance costs |
|
|
(876) |
|
|
(1,485) |
Long-term debt |
|
$ |
41,124 |
|
$ |
40,515 |
MidCap Term Loan
On May 26, 2022, the Company entered into the Credit and Security Agreement (the “MidCap Credit Agreement”) with MidCap Financial Trust, as agent, and certain lender parties thereto. The MidCap Credit Agreement consists of $40.0 million in a term loan. Upon closing, the Company used a portion of the proceeds to repay borrowings under a previous credit facility.
Pursuant to the MidCap Credit Agreement, the Company provided a first priority security interest in all existing and future acquired assets, including intellectual property, owned by the Company. The MidCap Credit Agreement contains certain covenants that limit the Company’s ability to engage in certain transactions that may be in the Company’s long-term best interests, including the incurrence of additional indebtedness, effecting certain corporate changes, making certain investments, acquisitions or dispositions and paying dividends.
The MidCap Credit Agreement also contains customary indemnification obligations and customary events of default, including, among other things, (i) non-payment, (ii) breach of warranty, (iii) non-performance of covenants and obligations, (iv) default on other indebtedness, (v) judgments, (vi) change of control, (vii) bankruptcy and insolvency, (viii) impairment of security, (ix) key permit events, (x) termination of a pension plan, (xi) regulatory matters, (xii) material adverse effect and (xiii) breach of material contracts.
In addition, the Company must maintain minimum net revenue levels tested quarterly. In the event of default under the MidCap Credit Agreement, the Company would be required to pay interest on principal and all other due and unpaid obligations at the current rate in effect plus 2%.
The MidCap term loan matures on May 1, 2027 and bears interest at a rate equal to 6.25% plus the greater of one-month Term SOFR (as defined in the MidCap Credit Agreement) or 1.0%. The Company is required to make 36 monthly interest payments beginning on June 1, 2022 (the “Interest-Only Period”). If the Company is in covenant compliance at the end of the Interest-Only Period, the Company will have the option to extend the Interest-Only Period by 12 months to 48 monthly interest payments, followed by 12 months of straight-line amortization, with the entire principal payment due at maturity. If the Company is not in covenant compliance at the end of the Interest-Only Period, the Company is required to make 24 months of straight-line amortization payments, with the entire principal amount due at maturity.
Subject to certain limitations, the MidCap term loan has a prepayment fee equal to 1.0% of the prepaid principal amount. The Company is also required to pay an exit fee at the time of maturity or prepayment event equal to 5% of all principal borrowings (the “End of Term Charge”) (or in the event of a prepayment event, the amount of principal being prepaid). The exit fee has been accounted for as an additional debt issuance cost and is being amortized to interest expense over the term of the MidCap term loan. Interest expense associated with the MidCap Credit Facility recorded for the year ended December 31, 2024 was $5.3 million, of which $0.6 million was related to the amortization of debt issuance costs. Interest expense associated with the MidCap Credit Facility recorded for the year ended December 31, 2023 was $5.2 million, of which $0.6 million was related to the amortization of debt issuance costs. Interest expense associated with the MidCap Credit Facility recorded for the year ended December 31, 2022 was $2.6 million, of which $0.4 million was related to the amortization of debt issuance costs.
F-18
OrbiMed Term Loan
In November 2018, the Company entered into the OrbiMed Credit Facility with OrbiMed, a related party as the lender is affiliated with a stockholder of the Company, which consisted of up to $35.0 million in term loans (the “OrbiMed Term Loans”). The OrbiMed Term Loans consisted of two tranches, a $30.0 million Tranche 1 (“First Tranche”) and a $5.0 million Tranche 2 (“Second Tranche”). In November 2018, the Company borrowed $30.0 million of the First Tranche. The Company elected not to borrow the Second Tranche prior to its expiration on December 31, 2019. On May 26, 2022, the Company entered into the MidCap Credit Agreement and upon closing used a portion of the proceeds to repay all borrowings under the OrbiMed Credit Facility.
The OrbiMed Term Loan bore interest at a rate equal to 7.75% plus the greater of one-month LIBOR or 2.0% until the aggregate principal, interest and End of Term Charge of $3.0 million were paid with part of the proceeds received from the MidCap Credit Agreement. As a result of these payments, a $1.2 million loss on extinguishment was recorded during the year ended December 31, 2022. Interest expense associated with the OrbiMed Credit Facility recorded for the year ended December 31, 2022 was $1.5 million, of which $0.3 million was related to the amortization of debt issuance costs.
(7) Stockholders’ Equity
Public Stock Offerings
In November 2023, the Company entered into a new Equity Distribution Agreement (the “2023 Equity Agreement”) with Piper Sandler & Co, (“Piper”) in connection with the establishment of an at-the-market offering program under which the Company may sell shares of its common stock, from time to time through Piper as sales agent, in an initial amount of up to $50 million. The 2023 Equity Agreement supersedes and replaces the Company’s previous Equity Distribution Agreement with Piper dated December 18, 2020 (the “2020 Equity Agreement”), which is no longer effective. No sales were made under the 2023 Equity Agreement or the 2020 Equity Agreement during the years ended December 31, 2023, 2022 or 2021.
On October 24, 2024, the Company completed an underwritten public offering of 14,670,000 shares of its common stock, including the exercise in full of the underwriters’ overallotment option to purchase additional shares of common stock, at a price to the public of $2.25 per share and, in lieu of common stock to investors who so chose, pre-funded warrants to purchase 5,800,000 shares of common stock at a public offering price of $2.2499 per pre-funded warrant, which represents the per share public offering price for the shares of common stock less the $0.0001 per share exercise price for each pre-funded warrant. The offering resulted in net proceeds of $42.9 million, after deducting underwriting discounts and commissions and other estimated offering expenses and assuming no subsequent exercise of the pre-funded warrants. The exercise of the pre-funded warrants, if any, is not expected to provide significant additional funding to the Company.
In April 2023, the Company completed an underwritten public offering in which the Company issued and sold 5,219,190 shares of its common stock (including 469,190 shares sold pursuant to the underwriters’ overallotment option in May 2023) at a public offering price of $9.50 per share. The Company received net proceeds of approximately $46.3 million after deducting underwriting discounts, commissions and other offering expenses.
In August 2022, the Company completed an underwritten public offering in which the Company issued and sold 4,600,000 shares of its common stock at a public offering price of $8.00 per share. The Company received net proceeds of $34.4 million after deducting underwriting discounts, commissions and other offering expenses.
F-19
Warrants
There have been no exercises or cancellations of warrants during the year ended December 31, 2024. The Company had the following warrants outstanding at December 31, 2024:
|
|
|
|
|
Exercise |
|
Expiration |
|
|
Outstanding |
|
|
price |
|
dates |
Common stock warrants |
|
8,379 |
|
$ |
28.65 |
|
2028 |
Common stock warrants |
|
80,177 |
|
|
28.65 |
|
2027 |
Pre-funded common stock warrants |
|
5,800,000 |
|
|
0.0001 |
|
NA |
|
|
5,888,556 |
|
|
|
|
|
On October 24, 2024, in connection with the underwritten public offering, the Company granted pre-funded warrants to purchase 5,800,000 shares of common stock at a public offering price of $2.2499 per pre-funded warrant, which represents the per share public offering price for the shares of common stock less the $0.0001 per share exercise price for each pre-funded warrant. The common stock and pre-funded warrants each met the criteria for equity classification. Accordingly, the amount allocated to the pre-funded warrants was recorded as a component of stockholders’ equity within additional paid-in capital.
(8) Sale of Product Line
In March 2024, the Company entered into an Asset Purchase Agreement (“APA”) with MiMedx Group, Inc. (“MDXG”) to sell certain assets (the “Transaction”) related to NIVIS Fibrillar Collagen Pack Device (“NIVIS”). These assets mainly included the Company’s existing inventory of NIVIS, with a net carrying value of $0.8 million, and certain intellectual property rights to sell NIVIS, with no carrying value. MDXG assumed the Company’s existing supply agreements, including the minimum obligations for NIVIS that the Company entered into in 2022 ahead of the initial sales of NIVIS. In exchange for entering into the Transaction, the Company received an initial $5.0 million upfront payment and is entitled to receive future revenue-sharing payments based on the net sales of NIVIS (now marketed as HELIOGEN) during the first two years following its launch by MDXG, which revenue-sharing payments would range from a minimum of $3.0 million to a maximum of $7.0 million in the aggregate. In addition, $0.4 million of consideration was received for existing NIVIS inventory on-hand. Any consideration in excess of $3.0 million up to $7.0 million is considered variable consideration that is fully constrained.
The Company accounted for the Transaction as a sale of a nonfinancial asset group in accordance with ASC 610-20 and followed the principals of ASC 606 to determine the consideration of $8.4 million related to the Transaction which includes the consideration for the existing inventory. The Company transferred control of the nonfinancial asset group in March 2024 and recognized a gain of $7.6 million in the consolidated statement of operations and comprehensive loss during the year ended December 31, 2024. The $8.4 million transaction price included the minimum revenue-share payment of $3.0 million, which was recorded as a receivable when the deal closed. Revenue-share payments commenced after the third quarter of 2024. At December 31, 2024, $0.1 million of this amount had been collected. The remaining receivable included $0.6 million recorded as the current portion in prepaid expenses and other assets in the consolidated balance sheet and $2.3 million recorded as the long-term portion in other long-term assets in the consolidated balance sheet. At each reporting date, the Company assesses the constraint of variable consideration and records increases in the transaction price in the period that the estimate of variable consideration changes. For the year ended December 31, 2024, no changes were made to the variable consideration.
(9) Stock-Based Compensation
The Company has two equity incentive plans: the 2012 Stock Incentive Plan and the Amended and Restated 2019 Equity Incentive Plan. New awards can only be granted under the Amended and Restated 2019 Equity Incentive Plan (the “Plan”). At December 31, 2024, 1,345,582 shares of common stock were available for future issuances under the Plan.
F-20
The Plan is subject to an annual increase, subject to prior approval by the Company’s board of directors, equal to the lesser of (i) 432,442 shares, (ii) 4% of the shares outstanding on the last day of the immediately preceding fiscal year and (iii) such smaller number of shares as determined by the board of directors. The Plan provides for the grant of incentive stock options, nonqualified stock options, restricted stock awards, restricted stock units and/or stock appreciation rights to employees, directors, and other persons, as determined by the Company’s board of directors. The Company’s stock options vest based on the terms in each award agreements and generally vest over four years and have a term of 10 years. The Company estimates forfeitures that it expects will occur and adjusts expense for actual forfeitures in the periods they occur.
The Company measures employee and nonemployee stock-based awards at grant-date fair value and records compensation expense ratably over the vesting period of the award. The Company recorded stock-based compensation expense in the following expense categories of its accompanying consolidated statements of operations and comprehensive loss (in thousands):
|
|
Year ended December 31, |
|||||||
|
|
2024 |
|
2023 |
|
2022 |
|||
Sales and marketing |
|
$ |
1,293 |
|
$ |
1,824 |
|
$ |
1,373 |
General and administrative |
|
|
2,429 |
|
|
2,478 |
|
|
2,029 |
Research and development |
|
|
640 |
|
|
730 |
|
|
587 |
Total stock‑based compensation |
|
$ |
4,362 |
|
$ |
5,032 |
|
$ |
3,989 |
The following table summarizes stock option activity for the Plan:
|
|
|
|
|
|
|
Weighted |
|
|
|
|
|
|
|
average |
|
|
|
|
Weighted |
|
remaining |
|
|
|
Number of |
|
average exercise |
|
contractual term |
|
|
|
shares |
|
price per share |
|
(years) |
|
Outstanding at January 1, 2022 |
|
1,706,409 |
|
$ |
11.88 |
|
|
Granted |
|
450,410 |
|
|
10.24 |
|
|
Exercised |
|
(3,563) |
|
|
5.51 |
|
|
Canceled/forfeited |
|
(81,408) |
|
|
13.13 |
|
|
Outstanding at December 31, 2022 |
|
2,071,848 |
|
$ |
11.49 |
|
|
Granted |
|
212,960 |
|
|
10.50 |
|
|
Exercised |
|
(25,428) |
|
|
5.00 |
|
|
Canceled/forfeited |
|
(96,927) |
|
|
11.25 |
|
|
Outstanding at December 31, 2023 |
|
2,162,453 |
|
$ |
11.48 |
|
|
Granted |
|
259,900 |
|
|
6.86 |
|
|
Exercised |
|
(38,431) |
|
|
5.88 |
|
|
Canceled/forfeited |
|
(264,739) |
|
|
12.24 |
|
|
Outstanding at December 31, 2024 |
|
2,119,183 |
|
$ |
10.92 |
|
5.87 |
Vested and expected to vest at December 31, 2024 |
|
2,096,063 |
|
$ |
10.95 |
|
5.84 |
Exercisable at December 31, 2024 |
|
1,653,664 |
|
$ |
11.48 |
|
5.16 |
Included in outstanding options at December 31, 2024, were 313,149 stock options granted outside of the Plan. These grants were made pursuant to the Nasdaq inducement grant exception in accordance with Nasdaq listing rule 5635(c)(4). At December 31, 2024, the aggregate intrinsic value of both outstanding options and exercisable options was $0.
F-21
The weighted average grant-date fair value per share of options granted was $4.67, $7.19 and $6.55 for the years ended December 31, 2024, 2023 and 2022, respectively. The aggregate intrinsic value of options exercised was $41,000, $0.1 million and $16,000 for the years ended December 31, 2024, 2023 and 2022, respectively. As of December 31, 2024, the total unrecognized compensation expense related to unvested employee and nonemployee stock option awards was $2.3 million, which is expected to be recognized in expense over a weighted-average period of approximately 2.0 years.
Estimating Fair Value of Stock Options
The fair value of each grant of stock options was determined by the Company using the methods and assumptions discussed below. Certain of these inputs are subjective and generally require judgment to determine.
Expected term – The expected term of stock options represents the weighted average period the stock options are expected to be outstanding. The Company uses the simplified method for estimating the expected term as provided by the Securities and Exchange Commission. The simplified method calculates the expected term as the average time to vesting and the contractual life of the options.
Expected volatility – Due to the Company’s limited operating history and lack of sufficient company-specific historical or implied volatility, the expected volatility assumption was determined by examining the historical volatilities of a group of industry peers, including the Company, whose share prices are publicly available.
Risk-free interest rate – The risk-free rate assumption is based on the U.S. Treasury instruments, the terms of which were consistent with the expected term of the Company’s stock options.
Expected dividend – The Company has not paid and does not intend to pay dividends.
The fair value of each option was estimated on the date of grant using the weighted average assumptions in the table below:
|
|
Year ended December 31, |
|
||||
|
|
2024 |
|
2023 |
|
2022 |
|
Expected dividend yield |
|
— |
|
— |
|
— |
|
Expected volatility |
|
73.2 |
% |
74.3 |
% |
69.6 |
% |
Risk‑free interest rate |
|
4.29 |
% |
3.99 |
% |
2.55 |
% |
Expected term (in years) |
|
6.14 |
|
6.15 |
|
6.20 |
|
Restricted Stock Units
The Company has issued service-based and performance-based restricted stock units (“RSUs”). Vesting of the service-based RSUs is based on the terms in each award agreement and is generally over four years. Vesting of the performance-based RSUs is subject to continued service through 2026 and the achievement of certain performance milestones for fiscal year 2026. The amount of performance-based RSUs that will vest can range from 0% to 110% of the original number of RSUs granted. Expense for the performance-based RSUs is not recognized until the performance conditions are deemed probable of achievement. The Company has not recorded any expense related to the performance-based RSUs as the performance conditions are not deemed to be probable of achievement.
F-22
The following table summarizes the service-based RSUs for the Plan:
|
|
Number of |
|
|
shares |
Outstanding at January 1, 2022 |
|
163,043 |
Granted |
|
197,950 |
Vested |
|
(40,783) |
Canceled/forfeited |
|
(8,219) |
Outstanding at December 31, 2022 |
|
311,991 |
Granted |
|
479,585 |
Vested |
|
(101,559) |
Canceled/forfeited |
|
(32,963) |
Outstanding at December 31, 2023 |
|
657,054 |
Granted |
|
421,725 |
Vested |
|
(191,175) |
Canceled/forfeited |
|
(155,316) |
Outstanding at December 31, 2024 |
|
732,288 |
The following table summarizes the performance-based RSUs for the Plan:
|
|
Number of |
|
|
shares |
Outstanding at January 1, 2023 |
|
— |
Granted |
|
250,149 |
Vested |
|
— |
Canceled/forfeited |
|
— |
Outstanding at December 31, 2023 |
|
250,149 |
Granted |
|
— |
Vested |
|
— |
Canceled/forfeited |
|
(33,649) |
Outstanding at December 31, 2024 |
|
216,500 |
Included in outstanding RSUs at December 31, 2024, were 97,409 RSUs granted outside of the Plan. These grants were made pursuant to the Nasdaq inducement grant exception in accordance with Nasdaq listing rule 5635(c)(4). The weighted average grant-date fair value per RSU granted was $6.54, $10.25 and $11.21 during the years ended December 31, 2024, 2023 and 2022, respectively. The aggregate intrinsic value of RSUs outstanding was $2.9 million, $6.0 million and $3.6 million at December 31, 2024, 2023 and 2022, respectively. The total unrecognized compensation expense at December 31, 2024 related to RSUs was $4.0 million, which is expected to be recognized in expense over a weighted-average period of approximately 2.4 years.
(10) Employee Benefit Plans
401(k) Defined Contribution Plan
The Company sponsors a 401(k) defined-contribution plan covering all employees. Participants are permitted to contribute up to 100% of their eligible annual pretax compensation up to an established federal limit on aggregate participant contributions. Discretionary contributions made by the Company, if any, are determined annually by the board of directors. The Company matches 50% of employees’ contributions up to 6%, subject to a maximum annual amount. The Company’s contributions were $0.6 million, $0.5 million and $0.4 million for the years ended December 31, 2024, 2023 and 2022, respectively.
F-23
Participants are immediately vested in their own contributions to the plan and are fully vested in discretionary profit sharing made by the Company after three years of service.
2019 Employee Stock Purchase Plan
In November 2019, the Company adopted the 2019 Employee Stock Purchase Plan (the “ESPP”). At December 31, 2024, 567,243 shares were available for future issuance under the ESPP. The ESPP is subject to an annual increase, subject to prior approval by the Company’s board of directors, equal to the least of (i) 107,887 shares of common stock, (ii) 1% of the shares outstanding on the final day of the immediately preceding calendar year, and (iii) such smaller number of shares as determined by the board of directors. The ESPP provides the opportunity to purchase the Company’s common stock at a 15% discount to the market price through payroll deductions. As of December 31, 2024, 2023 and 2022, 58,994, 10,602 and 4,523 shares, respectively, were issued under the ESPP.
(11) Income Taxes
The Company has incurred losses since inception. Deferred tax assets and liabilities are determined based on the differences between the financial statement carrying amounts and tax bases of assets and liabilities using enacted tax rates in effect for years in which differences are expected to reverse.
Components of the Company’s current and deferred income tax expense or benefit for the period consisted of a tax
benefit of $0.1 million recorded related to its foreign jurisdiction recognized in the year ended December 31, 2024. No
benefit or expense was recognized during the years ended December 31, 2023 or 2022.
Significant components of the Company’s deferred tax assets for federal income taxes consisted of the following (in thousands):
|
|
December 31, |
||||
|
|
2024 |
|
2023 |
||
Deferred tax assets |
|
|
|
|
|
|
Net operating loss carryforwards |
|
$ |
66,265 |
|
$ |
61,427 |
Capitalized research and development expenses |
|
|
6,185 |
|
|
4,845 |
Stock-based compensation |
|
|
1,604 |
|
|
2,300 |
Accrued expenses and other |
|
|
1,193 |
|
|
1,235 |
Lease liability |
|
|
478 |
|
|
557 |
Research and development credits |
|
|
830 |
|
|
830 |
Inventory reserve |
|
|
432 |
|
|
289 |
Interest expense carryforward |
|
|
691 |
|
|
523 |
Gross deferred tax asset before valuation allowance |
|
|
77,678 |
|
|
72,006 |
Less: valuation allowance |
|
|
(76,782) |
|
|
(71,086) |
Total deferred tax asset |
|
|
896 |
|
|
920 |
Deferred tax liabilities |
|
|
|
|
|
|
Depreciation and amortization |
|
|
(326) |
|
|
(436) |
Right of use asset |
|
|
(430) |
|
|
(484) |
Gross deferred tax liability |
|
|
(756) |
|
|
(920) |
Net deferred tax asset |
|
$ |
140 |
|
$ |
— |
The Company does not have unrecognized tax benefits as of December 31, 2024 and 2023. The Company recognizes interest and penalties accrued on any unrecognized tax benefits as a component of income tax expense.
F-24
The Company’s net operating loss (“NOL”) carryforwards for federal and state income tax purposes consisted of the following (in thousands):
|
|
December 31, |
||||
|
|
2024 |
|
2023 |
||
NOL carryforwards |
|
|
|
|
|
|
Federal |
|
$ |
266,655 |
|
$ |
239,417 |
State |
|
|
218,944 |
|
|
196,132 |
The NOL carryforwards begin expiring in 2032 for federal purposes and in 2026 for state income tax purposes yet $187.3 million of the federal NOL carryforwards have no expiration. The Company recorded a valuation allowance on the majority of its deferred tax assets as of December 31, 2024 and 2023 because of the uncertainty of their realization. The valuation allowance increased by $5.7 million and $10.9 million for the years ended December 31, 2024 and 2023, respectively, mainly due to losses incurred.
Utilization of the net operating losses and general business tax credits carryforwards may be subject to a substantial limitation under Sections 382 and 383 of the Internal Revenue Code of 1986, as amended, if changes in ownership of the company have occurred previously or occur in the future. Ownership changes may limit the amount of net operating losses and general business tax credits carryforwards that can be utilized annually to offset future taxable income and tax, respectively. In general, an ownership change, as defined by Section 382, results from transactions increasing the ownership of 5% shareholders in the stock of a corporation by more than 50 percentage points over a three-year period. If the Company experiences a Section 382 ownership change, the tax benefits related to the NOL carryforwards may be further limited or lost. The Company has not performed an analysis under Section 382 and cannot predict or otherwise determine whether there would be any limitation to the amount of net operating losses and general business tax credits carryforwards that can be utilized.
A reconciliation of income tax benefit at the statutory federal income tax rate and as reflected in the consolidated financial statements is as follows:
|
|
Year ended December 31, |
|||||
|
|
2024 |
|
2023 |
|
2022 |
|
Rate reconciliation |
|
|
|
|
|
|
|
Federal tax benefit at statutory rate |
|
(21.0) |
% |
(21.0) |
% |
(21.0) |
% |
State rate, net of federal benefit |
|
2.0 |
|
(3.1) |
|
(3.1) |
|
Permanent differences |
|
0.9 |
|
0.7 |
|
0.6 |
|
Research and development |
|
— |
|
(0.4) |
|
— |
|
Change in valuation allowance |
|
15.0 |
|
23.4 |
|
23.4 |
|
Stock compensation true-up |
|
3.1 |
|
— |
|
— |
|
Other |
|
(0.4) |
|
0.4 |
|
0.1 |
|
Total tax provision |
|
(0.4) |
% |
— |
% |
— |
% |
The Company files income tax returns in the U.S. federal jurisdiction, various state jurisdictions and the United Kingdom. Tax years 2021 and forward remain open for examination for federal and the Company’s more significant state tax jurisdictions. Carryforward attributes from prior years may be adjusted upon examination by taxing authorities if used in an open period.
F-25
(12) Commitments and Contingencies
Legal Proceedings
From time to time, the Company may be a party to lawsuits, claims, and other legal proceedings that arise in the ordinary course of its business. While the outcomes of these matters are uncertain, management does not expect that the ultimate costs to resolve these matters will have a material adverse effect on the Company’s consolidated financial position, results of operations, or cash flows.
Agreements with Aroa
In August 2012, the Company entered into a License, Product Development, and Supply Umbrella Agreement (“Aroa Agreement”) with Aroa. The Aroa Agreement provides the Company a license to patent rights and other intellectual property related to Aroa’s products and technologies for use in certain indications and expires on the expiration of the last patent covering the products (currently March 9, 2031). The Company has the right to extend the term of the agreement by an additional 10 years following the expiration of the last patent covering the products on commercially reasonable terms to be negotiated by the parties. This agreement initially limited the Company’s license rights to the U.S. but was subsequently amended in March 2013 to include certain countries in Europe including the United Kingdom and members of the European Union and certain former Union of Soviet Socialist Republic satellite nations. The Aroa Agreement required payments aggregating up to $4.0 million upon the achievement of U.S. and European cumulative product sales targets. All amounts have been paid.
Other key terms of the amended Aroa agreement in addition to those disclosed above are as follows:
| ● | We purchase product from Aroa at a fixed transfer cost as a percentage of Aroa’s cost of goods, which, subject to a true-up adjustment, results in an amount equal to 27% of our net sales of our OviTex and OviTex PRS products, with the exception of OviTex inguinal hernia repair (“IHR”) product configurations, for which we pay the greater of the initial fixed transfer cost or 27% of our net sales of OviTex IHR. |
| ● | The transfer price and the quarterly true-up amount continued to equal 27% of Company’s net sales of licensed products, with the exception of the IHR products, where the total amount payable to Aroa will at least equal the aggregate transfer pricing paid to Aroa for such products during the applicable calendar year. |
| ● | Provisions exist for the Company to step in and operate Aroa’s plant if a supply failure occurs and is not cured within a set timeframe. Under the amended agreement, the criteria for a supply failure was modified to mean a failure by Aroa to timely supply, during any consecutive 60-day period, at least 75% of the products ordered by the Company under binding purchase orders. During the period that the Company steps in and assumes manufacturing responsibility, the Company shall pay a royalty of 6% of net sales in lieu of 27% of net sales of the licensed products. |
Research and Development Agreements
As of December 31, 2024, the Company had $1.8 million in milestone payments related to certain research and development arrangements which are currently deemed not probable as the timing and likelihood of such payments are not known with certainty.
F-26
Employment Agreements
The Company entered into employment agreements with key personnel providing for compensation and severance in certain circumstances, as defined in the respective employment agreements.
Leases
The Company leases office and laboratory space in Malvern, Pennsylvania under a noncancelable lease (the “Malvern Lease”). The Malvern Lease, which was concluded to be an operating lease, was amended in October 2023 to extend the term of the lease from May 2028 to May 2030 (the “Lease Amendment”). Pursuant to the Lease Amendment, the Company leased an additional 15,881 square feet at the Company’s corporate headquarters which commenced on December 1, 2023 (the “Expansion Premises”) and will relinquish 4,652 square feet of non-contiguous space currently subject to the lease agreement on June 30, 2025 (the “Relinquished Space”). The Expansion Premises increased the Company’s total leased square footage in the building from 24,725 square feet to 40,606 square feet, which will be subsequently reduced to 35,954 square feet as of June 30, 2025 following removal of the Relinquished Space. The modification of the lease terms for the Company’s existing space was not treated as a separate contract; however, the Company notes that the Expansion Premises is being treated as a new ROU asset. The Lease Amendment required the Company to pay an additional security deposit of $0.3 million. The Malvern Lease has annual scheduled payment increases and provides the Company with a renewal option for an additional term of 60 months at the end of the lease term. The Company evaluates renewal options at lease inception and on an ongoing basis and includes renewal options that it is reasonably certain to exercise in its expected lease terms when classifying leases and measuring lease liabilities. As the Company is not reasonably certain to exercise the renewal option, the additional 60-month term has been excluded.
Operating lease leasehold improvements are depreciated over the lesser of the useful lives of the leasehold improvements or the lease term.
The Company determined that the rate implicit in its lease is not readily determinable, and therefore, the Company uses its incremental borrowing rate as the discount rate when measuring operating lease liabilities. The incremental borrowing rate represents an estimate of the interest rate the Company would incur at lease commencement to borrow an amount equal to the lease payments on a collateralized basis over the term of a lease. The Company used an incremental borrowing rate of 11.66% to discount the Malvern Lease payments included in the operating lease liabilities recognized.
The Company recognized $0.5 million of lease cost during the year ended December 31, 2024 and $0.3 million of lease cost during both of the years ended December 31, 2023 and 2022. Cash paid for amounts included in the measurement of operating lease liabilities was $0.6 million, $0.4 million and $0.3 million for the years ended December 31, 2024, 2023 and 2022, respectively, and these amounts are included in operating activities in the consolidated statements of cash flows. As of December 31, 2024, the remaining lease term for the Malvern Lease is 5.4 years.
F-27
The following table reconciles the undiscounted future minimum lease payments (displayed in aggregate by year) under non-cancelable operating leases with terms of more than one year to the total operating lease liabilities recognized on the consolidated balance sheets as of December 31, 2024 (in thousands):
2025 |
$ |
580 |
2026 |
|
557 |
2027 |
|
570 |
2028 |
|
583 |
2029 |
|
595 |
Thereafter |
|
250 |
Total undiscounted future minimum lease payments |
$ |
3,135 |
Less imputed interest |
|
(1,200) |
Total operating lease liabilities |
$ |
1,935 |
As of December 31, 2024, $0.5 million representing the current portion of operating lease liabilities is included in accrued expenses and other current liabilities in the consolidated balance sheets and $1.4 million representing the long-term portion of operating lease liabilities is included in other long-term liabilities in the consolidated balance sheets.
F-28
99
10.14 |
|
|
10.15 |
|
Amendment No. 1 to TELA Bio, Inc. 2019 Employee Stock Purchase Plan (incorporated by reference to exhibit 10.15 to the Company’s Annual Report on Form 10-K, filed on March 23, 2023). |
10.16 |
|
|
10.17 |
|
|
10.18 |
|
TELA Bio, Inc. Amended and Restated Non-Employee Director Compensation Policy (filed herewith). |
10.19 |
|
|
10.20 |
|
|
10.21 |
|
|
10.22 |
|
|
10.23 |
|
|
10.24 |
|
|
10.25 |
|
|
10.26 |
|
|
10.27* |
|
|
10.28* |
|
|
10.29* |
|
|
10.30* |
|
|
10.31* |
|
|
10.32* |
|
|
10.33* |
|
100
10.34 |
|
|
10.35 |
|
|
10.36 |
|
|
10.37 |
|
|
10.38 |
|
|
10.39 |
|
|
19.1 |
|
|
21.1 |
|
|
23.1 |
|
|
|
31.1 |
|
|
|
31.2 |
|
|
|
32.1 |
|
|
|
32.2 |
|
|
97.1 |
|
|
101 INS |
|
Inline XBRL Instance Document (filed herewith). |
101 SCH |
|
Inline XBRL Taxonomy Extension Schema Document (filed herewith). |
101 CAL |
|
Inline XBRL Taxonomy Extension Calculation Linkbase Document (filed herewith). |
101 DEF |
|
Inline XBRL Taxonomy Extension Definition Linkbase Document (filed herewith). |
101 LAB |
|
Inline XBRL Taxonomy Extension Label Linkbase Document (filed herewith). |
101 PRE |
|
Inline XBRL Taxonomy Extension Presentation Linkbase Document (filed herewith). |
104 |
|
Cover Page Interactive Data File (formatted as inline XBRL and contained in Exhibit 101). |
|
|
|
* |
|
Certain confidential portions (indicated by brackets and asterisks) have been omitted from this exhibit. |
101
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
|
TELA BIO, INC. |
|
|
|
|
|
By: |
/s/ ANTONY KOBLISH |
|
|
Name: Antony Koblish |
|
|
Title: President, Chief Executive Officer and Director Date: March 21, 2025 |
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
Signature |
|
Title |
|
Date |
|
|
|
|
|
/s/ ANTONY KOBLISH |
|
President, Chief Executive Officer and Director (Principal Executive Officer) |
|
March 21, 2025 |
Antony Koblish |
|
|
||
|
|
|
|
|
/s/ ROBERTO CUCA |
|
Chief Operating Officer and Chief Financial Officer |
|
March 21, 2025 |
Roberto Cuca |
|
|
||
|
|
|
|
|
/s/ MEGAN SMEYKAL |
|
Chief Accounting Officer and Controller (Principal Accounting Officer) |
|
March 21, 2025 |
Megan Smeykal |
|
|
||
|
|
|
|
|
/s/ DOUG EVANS |
|
Chairman, Board of Directors |
|
March 21, 2025 |
Doug Evans |
|
|
|
|
|
|
|
|
|
/s/ KURT AZARBARZIN |
|
Director |
|
March 21, 2025 |
Kurt Azarbarzin |
|
|
|
|
|
|
|
|
|
/s/ JEFFREY BLIZARD |
|
Director |
|
March 21, 2025 |
Jeffrey Blizard |
|
|
|
|
|
|
|
|
|
/s/ VINCE BURGESS |
|
Director |
|
March 21, 2025 |
Vince Burgess |
|
|
|
|
|
|
|
|
|
/s/ LISA COLLERAN |
|
Director |
|
March 21, 2025 |
Lisa Colleran |
|
|
|
|
|
|
|
|
|
/s/ FEDERICA O’BRIEN |
|
Director |
|
March 21, 2025 |
Federica O’Brien |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
102
Exhibit 10.18
AMENDED AND RESTATED
NON-EMPLOYEE DIRECTOR COMPENSATION POLICY
Non-employee members of the board of directors (the “Board”) of TELA Bio, Inc. (the “Company”) shall be eligible to receive cash and equity compensation as set forth in this Amended and Restated Non-Employee Director Compensation Policy (this “Policy”). The cash and equity compensation described in this Policy shall be paid or granted, as applicable, automatically and without further action of the Board, to each member of the Board who is not an employee of the Company or any parent or subsidiary of the Company (each, a “Non-Employee Director”), unless such Non-Employee Director declines the receipt of such cash or equity compensation by written notice to the Company. This Policy shall become effective as of January 1, 2025 (the “Effective Time”) and shall remain in effect until it is revised or rescinded by further action of the Board. This Policy may be amended, modified or terminated by the Board at any time in its sole discretion. The terms and conditions of this Policy shall supersede any prior cash and/or equity compensation arrangements for service as a member of the Board between the Company and any of its Non-Employee Directors and between any subsidiary of the Company and any of its non-employee directors.
(1) |
Cash Compensation. |
(a)Annual Retainers. Each Non-Employee Director shall receive an annual retainer of $45,000 for service on the Board.
(b)Additional Annual Retainers. In addition, a Non-Employee Director shall receive the following annual retainers:
(i)Chairperson of the Board. A Non-Employee Director serving as Chairperson of the Board shall receive an additional annual retainer of $35,000 for such service.
(ii)Audit Committee. A Non-Employee Director serving as Chairperson of the Audit Committee shall receive an additional annual retainer of $20,000 for such service. A Non-Employee Director serving as a member of the Audit Committee (other than the Chairperson) shall receive an additional annual retainer of $10,000 for such service.
(iii)Compensation Committee. A Non-Employee Director serving as Chairperson of the Compensation Committee shall receive an additional annual retainer of $15,000 for such service. A Non-Employee Director serving as a member of the Compensation Committee (other than the Chairperson) shall receive an additional annual retainer of $7,500 for such service.
(iv)Nominating and Corporate Governance Committee. A Non-Employee Director serving as Chairperson of the Nominating and Corporate Governance Committee shall receive an additional annual retainer of $10,000 for such service. A Non-Employee Director serving as a member of the Nominating and Corporate Governance Committee (other than the Chairperson) shall receive an additional annual retainer of $5,000 for such service.
(c)Payment of Retainers.
(i)Timing. The annual retainers described in Sections 1(a) and 1(b) shall be earned on a quarterly basis based on a calendar quarter and shall be paid by the Company in arrears not later than the fifteenth day following the end of each calendar quarter.
(ii)Form. The annual retainers shall be paid in the form of cash; provided that the Board may, in its discretion, permit a Non-Employee Director to elect to receive any portion of the annual retainer in the form of shares of common stock of the Company (“Common Stock”) in lieu of cash. If such an election is permitted by the Board and made by a Non-Employee Director, the number of shares of Common Stock to be paid shall be determined by dividing the portion of the annual retainer payable in the form of Common Stock by the Fair Market Value (as defined in the Company’s Amended and Restated 2019 Equity Incentive Plan or any other applicable Company equity plan then maintained by the Company (such plan, as may be amended from time to time, the “Equity Plan”)) per share of Common Stock on the date the annual retainer is payable.
Shares issued in lieu of cash shall be fully vested and unrestricted shares of Common Stock. Any election by a Non-Employee Director to receive a portion of the annual retainer in shares of Common Stock must be made prior to the applicable payment date for such portion of the annual retainer and pursuant to an election form to be provided by the Company. An election must comply with all rules established from time to time by the Board, including any insider trading policy or similar policy. A Non-Employee Director may not make an election pursuant to this Section 1(c)(ii) during a Company blackout period or when the Non-Employee Director is otherwise in possession of material non-public information.
(iii)Termination of Service. In the event a Non-Employee Director does not serve as a Non-Employee Director, or in the applicable positions described in Section 1(b), for an entire calendar quarter, such Non-Employee Director shall receive a prorated portion of the retainer(s) otherwise payable to such Non-Employee Director for such calendar quarter pursuant to Section 1(b), with such prorated portion determined by multiplying such otherwise payable retainer(s) by a fraction, the numerator of which is the number of days during which the Non-Employee Director serves as a Non-Employee Director or in the applicable positions described in Section 1(b) during the applicable calendar quarter and the denominator of which is the number of days in the applicable calendar quarter.
(2)Equity Compensation. Non-Employee Directors shall be granted the equity awards described below (collectively, the “Awards”). The Awards shall be granted under and shall be subject to the terms and provisions of the Equity Plan and shall be granted subject to the execution and delivery of award agreements in substantially the forms approved by the Board. The approval of this Policy by the Board is intended to be effective for all purposes, including for purposes of satisfying Rule 16b-3(d)(1) of the Securities Exchange Act of 1934, as amended, in respect of each award issued hereunder.
(a)Initial Awards. Upon a Non-Employee Director’s initial appointment or election to the Board after the Effective Time, he or she will be granted: (i) an option to purchase 17,550 shares of Common Stock at a per-share exercise price equal to the closing price per share of Common Stock on the date of such appointment or election (or on the last preceding trading day, if the date of such appointment or election is not a trading day), and (ii) a restricted stock unit award with respect to 11,925 shares of Common Stock. The Awards described in this Section 2(a) shall be referred to as “Initial Awards.”
(b)Annual Awards. Each Non-Employee Director who serves on the Board as of the date of any annual meeting of the Company’s stockholders (an “Annual Meeting”) after the Effective Time, and will continue to serve as a Non-Employee Director immediately following such Annual Meeting, shall be automatically granted on the date of such Annual Meeting: (i) an option to purchase 11,700 shares of Common Stock at a per-share exercise price equal to the closing price per share of Common Stock on the date of such Annual Meeting (or on the last preceding trading day, if the date of the Annual Meeting is not a trading day), and (ii) a restricted stock unit award with respect to 7,950 shares of Common Stock. The Awards described in this Section 2(b) shall be referred to as “Annual Awards.”
(c)Vesting of Awards. Awards will vest as follows, in each case subject to the continued service of the grantee to the Company through the applicable vesting date or event:
(i)Initial Awards described in Section 2(a)(i) will vest in 36 equal monthly installments, on the monthly anniversary of the date of grant over the 36 calendar months commencing after the grantee’s initial appointment or election to the Board.
(ii)Initial Awards described in Section 2(a)(ii) will vest in three equal annual installments, on the first three anniversaries of the grantee’s initial appointment or election to the Board.
(iii)Annual Awards will vest on the earlier of (A) the first anniversary of the date of grant, and (B) the date of the subsequent Annual Meeting following the date of grant.
(iv)In addition, any otherwise unvested Awards will vest and become exercisable in full immediately prior to and contingent upon the occurrence of a Change in Control (as defined in the Equity Plan).
Exhibit 19.1
TELA BIO, INC.
INSIDER TRADING POLICY
Effective February 23, 2023
I.Purpose
TELA Bio, Inc. (the “Company”) has adopted this Insider Trading Policy (this “Policy”) to satisfy the Company’s obligation to prevent insider trading and to help the Company’s personnel and its external advisors avoid violating insider trading laws.
II.Persons Subject to the Policy
This Policy applies to (i) all officers, directors, and employees of the Company and any of its subsidiaries, (ii) immediate family members (as defined below) and any persons that reside in the same household as any of the foregoing persons and (iii) any other person whose transactions in Company Securities (as defined below) are directed by, or subject to influence or control by the foregoing persons, and any trust, partnership, corporation, or other entity over which such persons have investment control (collectively, “Insiders”). Individuals subject to this Policy are responsible for ensuring that members of their households also comply with this Policy and therefore should make such members of their households aware of the need to confer with them before they trade in Company Securities (as defined below) and should treat all such transactions for the purposes of this Policy and applicable securities laws as if the transactions were for your own account.
This Policy does not, however, apply to personal securities transactions of your immediate family members (as defined below) where the purchase or sale decision is made by a third party not controlled by, influenced by or related to you or your immediate family members (as defined below).
For purposes of this Policy, “immediate family member” means any spouse, child, stepchild, grandchild, parent, stepparent, grandparent, sibling, mother or father-in-law, son or daughter-in-law, or brother or sister-in-law (as well as other adoptive relationships), whether or not sharing the same household as the persons described in item (i) above.
All consultants and outside advisors assisting the Company on sensitive matters are expected to abide by the Policy, although the Company assumes no responsibility with respect to the actions of persons who are not under its direct control.
Persons in possession of material non-public information related to, affecting or regarding the Company or its subsidiaries (“Inside Information”) when their employment or service terminates may not trade in Company Securities (as defined below) until that information has become public or is no longer material.
Transactions Subject to the Policy
This Policy applies to all transactions in securities of the Company (collectively referred to in this Policy as “Company Securities”), including common stock, options to purchase common stock, preferred stock, convertible debt and warrants, or any other type of securities that the Company has or may issue, as well as derivative securities that are not issued by the Company, such as exchange-traded put or call options or swaps relating to the Company Securities.
III.General Policy
No Insider who is in possession of Inside Information may, either directly or indirectly, (i) purchase or sell Company Securities, (ii) engage in any other action to take advantage of Inside Information, or (iii) without the consent of the Company, provide Inside Information to any other person outside of the Company, including family and friends.
In addition, Insiders may not purchase or sell any securities of any other company, such as a joint venture partner, possible acquisition target, customer, or competitor of the Company, when in possession of material non-public information concerning any such other company obtained during his or her employment with, or service to, the Company or any of its subsidiaries.
Insiders may not disclose, convey or “tip” Inside Information to any person by providing them with Inside Information other than to disclose on a “need to know” basis to directors, officers and employees of the Company or outside advisors in the course of performing their duties for the Company. When sharing Inside Information with other directors, officers and employees of the Company or outside advisors, or other persons involved in the business and affairs of the Company, such information should be confined to as small a group as possible. Unlawful tipping includes passing on Inside Information to friends, family members or acquaintances under circumstances that suggest that persons subject to this Policy were trying to help the recipients of such information to make a profit or avoid a loss by trading in Company Securities based on such information.
IV.Definition of Inside Information
Material Information. Information is considered “material” if a reasonable investor would consider that information important in making a decision to buy, hold, or sell Company Securities or the securities of another public company. Any information that could be expected to affect the Company’s stock price, whether it is positive or negative, should be considered material. Determining whether information is material is not always straightforward; rather, materiality is based on an assessment of all of the facts and circumstances, and is often evaluated by enforcement authorities with the benefit of hindsight. When doubt exists as to whether information would be considered “material,” the information should be presumed to be material. While it is not possible to identify in advance all information that will be deemed to be material, some examples of such information would include the following:
· |
merger, acquisition, joint venture, partnerships, strategic alliances, collaborations, or investment proposals; |
· |
annual or quarterly financial statements; |
-2-
· |
projections of future earnings or losses, or other earnings guidance; |
· |
changes to operational or financial guidance; |
· |
significant expansion or curtailment of operations; |
· |
material information regarding an existing or potential customer or supplier; |
· |
unusual borrowings; |
· |
public or private securities offerings; |
· |
litigation (pending or threatened); |
· |
changes in senior management or members of the Board of Directors; |
· |
information concerning clinical trials and their results, intellectual property, regulatory approvals or other developments (positive or negative), product or technological plans, developments or agreements; |
· |
significant communications to or from regulatory agencies; |
· |
new product launches or the introduction of new business strategies; |
· |
the status of the Company’s progress toward achieving significant Company goals; |
· |
listing on or delisting from a stock exchange; |
· |
new major contracts, customers, distributors or suppliers, or the loss of any of the foregoing; |
· |
material changes in the Company’s pricing or cost structure for its products; |
· |
a company restructuring; |
· |
significant related party transactions; or |
· |
similar information concerning a significant subsidiary, business unit or investment. |
Non-Public Information. Information that has not been widely disseminated to the public is generally considered to be non-public information. Information generally becomes available to the public when it has been disclosed by the Company or third parties in a press release or other authorized public statement, including any filing with the Securities and Exchange Commission (the “SEC”). Once information is widely disseminated, it is still necessary to afford the investing public with sufficient time to absorb the information. As a general rule, information should not be considered fully absorbed by the marketplace until after the second full trading day after the information is released. If, for example, the Company were to make an announcement prior to the start of trading on a Monday, a person covered by this Policy should not trade in Company Securities until the start of trading on Wednesday. Depending on the particular circumstances, the Company may determine that a longer or shorter period should apply to the release of specific material non-public information.
If you are unsure whether you are in possession of Inside Information, you should consult with the General Counsel prior to engaging in, or entering into an agreement, understanding or arrangement to engage in, a purchase or sale transaction of any Company Securities. However, you are responsible for determining whether you are in possession of Inside Information and any action on the part of the Company, the General Counsel, or any other employee or director pursuant to this Policy or otherwise does not in any way constitute legal advice or insulate you from liability under applicable securities laws. If your securities transactions become the subject of scrutiny, they will be viewed after-the-fact with the benefit of hindsight. As a result, before engaging in any transaction you should carefully consider how regulators and others might view your transaction in hindsight.
-3-
VI.Special and Prohibited Transactions
In addition to the other restrictions set forth in this Policy, the following transactions are strictly prohibited at all times:
· |
trading in call or put options involving Company Securities and other derivative securities; |
· |
engaging in short sales of Company Securities; |
· |
holding Company Securities in a margin account; |
· |
standing and limit orders involving the Company’s Securities (other than in connection with an approved 10b5-1 trading plan as discussed below) if such standing or limit order is not limited to a short period of time and has not otherwise been approved pursuant to the pre-clearance procedures set forth in Section VIII herein; |
· |
all forms of hedging or monetization transactions, such as zero-cost collars and forward sale contracts; and |
· |
pledging Company Securities to secure margin or other loans. |
If you are unsure whether a particular transaction is prohibited under this Policy, you should consult with the General Counsel prior to engaging in, or entering into, an agreement, understanding, or arrangement to engage in, such transaction.
VII. Transactions Not Subject to Trading Restrictions Under the Policy
The trading restriction prohibitions in this Policy do not apply to:
· |
the granting of options or other equity awards or the purchase or sale of Company Securities from or to the Company, as applicable; |
· |
bona fide gifts of Company Securities; |
· |
purchases or sales of Company Securities made pursuant to any binding contract, specific instruction, or written plan entered into outside of a restricted period and while the purchaser or seller, as applicable, was unaware of any material, nonpublic information and which contract, instruction or plan (i) meets all of the requirements of the affirmative defense provided by Rule 10b5-1 promulgated under the Securities Exchange Act of 1934, as amended (a “10b5-1 trading plan”), (ii) was pre-cleared in advance pursuant to this Policy, and (iii) has not been amended or modified in any respect after such initial pre-clearance without such amendment or modification being pre-cleared in advance pursuant to this Policy; or |
· |
transactions between Insiders and the Company with respect to grants under its equity incentive plan (or, to the extent applicable, granted outside such plan), including the exercise of stock options for cash; the vesting of restricted stock or restricted stock units (“RSUs”), or the exercise of a tax withholding right pursuant to which a person has elected to have the Company withhold shares subject to an |
-4-
option to satisfy tax withholding upon the exercise of stock options or the vesting of any restricted stock or RSUs.
Consequently, restrictions contained in this Policy would apply to the sale of Company Securities in the open market to pay the exercise price of an option, any taxes due upon the vesting of restricted stock or an RSU, and to the “cashless exercise” effected through a broker or “same day sale” of an option, which generally entail the sale of a portion of the underlying stock on the market to cover the costs of exercise or the resulting taxes. In addition, any sale of the underlying securities acquired upon the exercise of an option or RSU is subject to the Policy.
VIII. Restricted Periods
Quarterly Restricted Periods. All Insiders are prohibited from trading in Company Securities during the period beginning on the 16th day of the last month of each fiscal quarter and ending on the beginning of the second trading day following public disclosure of the financial results for that quarter or the full year. The quarterly restricted periods have been established due to an increased likelihood of access to Inside Information at this time, where trading in the Company’s Securities may be subject to additional scrutiny. For illustration, a list of the restricted period for each quarter is attached hereto as Annex A.
Special Restricted Periods. In addition, from time to time, the Company may impose special restricted periods on Insiders if, in the judgment of the Chief Executive Officer, Chief Operating Officer and Chief Financial Officer, or the General Counsel, it is likely that such person or persons have become aware of significant corporate developments that have not yet been disclosed to the public, even when trading otherwise may be permitted. If certain Insiders become subject to a special restricted period, such persons are prohibited from (i) trading in Company Securities and (ii) without the consent of the Company, disclosing to others the fact that they are subject to such special restricted period. These special restricted periods may vary in length and may or may not be broadly communicated to Insiders. Unless otherwise specified, the Company will re-open trading at the beginning of the second day following the date of public disclosure of such significant corporate developments, or after the termination of any pending development, if applicable.
IX. Pre-Clearance Procedures for Restricted Persons
Certain Insiders may be designated as “Restricted Persons” and subject to additional restrictions on their ability to engage in purchase or sale transactions involving Company Securities. Any designation as a Restricted Person is intended to reflect a greater likelihood of access to Inside Information of the Company due to such person’s position or affiliation (e.g., as a director, officer, member of the Company’s legal, IT, or finance department), where trades in the Company’s Securities are more likely be subject to greater scrutiny. A list of current Restricted Persons is attached hereto as Annex B, and may be updated from time to time by the Chief Executive Officer, Chief Operating Officer and Chief Financial Officer, or the General Counsel.
A Restricted Person must obtain prior clearance from the General Counsel, or such person’s designee, by submitting (in writing or via email) the information contained in the Request for Clearance to Trade as set forth on Annex C attached hereto, before such Restricted Person makes any purchases, sales or gifts of Company Securities, regardless of whether a restricted period is then in effect.
-5-
In evaluating each proposed transaction, the General Counsel or such person’s designee will consult as necessary with senior management and outside counsel before clearing any proposed trade. The General Counsel is under no obligation to approve a transaction submitted for pre-clearance and may determine not to permit the transaction. If a person seeks pre-clearance and permission to engage in the transaction is denied, then he or she should refrain from initiating any transaction in Company Securities. Clearance of a transaction is valid for no more than the 5-business day period (or such shorter period as may be prescribed in the pre-clearance form) immediately following receipt by the Restricted Person of such clearance. Restricted Persons do not need to receive pre-clearance for trades pursuant to an approved 10b5-1 trading plan, but must receive prior approval before implementing, terminating or amending such a plan by the General Counsel, or such person’s designee.
For the avoidance of doubt, any restrictions of this Policy relating to Restricted Persons shall also apply to trades proposed by such person’s immediate family members or other persons sharing the same household, as well as other entities over which such person has investment control.
X.Rule 10b5-1 Plans
The Company permits all directors, officers, and other employees to adopt a 10b5-1 trading plan pursuant to the Company’s procedure for adopting such a trading plan. All Restricted Persons must obtain pre-clearance prior to entering into, modifying or terminating a 10b5-1 plan. No director, officer, or employee may enter into, modify or terminate a 10b5-1 plan during any restricted period as set forth in this policy, or during any period when such director, officer or employee is otherwise in possession of Inside Information. No director, officer, or employee may have more than one 10b5-1 trading plan at any time. Following the adoption or modification of a 10b5-1 trading plan, no transactions may be initiated until the later of (i) ninety (90) days following the adoption or modification of such 10b5-1 trading plan or (ii) two business days following the Company’s filing of the applicable Form 10-Q or Form 10-K for the fiscal quarter in which such 10b5-1 plan was adopted or modified (the “Cooling-Off Period”). Notwithstanding the foregoing, in no event shall the Cooling-Off Period exceed one hundred and twenty (120) days for a director or officer or thirty (30) days for any other employee of the Company. The restrictions on trading set forth in this Policy shall not apply to trades made pursuant to an approved 10b5-1 trading plan. For questions regarding the establishment, implementation, modification, or termination of a 10b5-1 plan, please seek additional guidance from the General Counsel.
XI.Consequences of Violations
The purchase or sale of Company Securities while aware of Inside Information, or the disclosure of Inside Information to others who then trade in Company Securities, is prohibited by federal and state securities laws. Insider trading violations are pursued vigorously by the SEC, U.S. Attorneys, and state enforcement authorities as well as the laws of foreign jurisdictions. Punishment for insider trading violations is severe and could include significant fines and imprisonment. While the regulatory authorities concentrate their efforts on the individuals who trade, or who tip inside information to others who trade, the federal securities laws also impose potential liability on companies and other “controlling persons” within the organization if they fail to take reasonable steps to prevent insider trading by company personnel.
-6-
In addition, an individual’s failure to comply with this Policy may subject the individual to Company-imposed sanctions, including dismissal for cause, whether or not the employee’s failure to comply results in a violation of law. A violation of law, or even an SEC investigation that does not result in prosecution, can tarnish a person’s reputation and irreparably damage a career.
XII.Administration of the Policy
The Company’s General Counsel, or in such person’s absence, an employee designated by the Chief Operating Officer and Chief Financial Officer in consultation with legal counsel, shall be responsible for administration of this Policy. All determinations and interpretations by the General Counsel (or his or her designees) shall be final and not subject to further review.
Any person who has a question about this Policy or its application to any proposed transaction may obtain additional guidance from the General Counsel.
XIII.Certification
You must sign, date and return the Certification set forth on Annex D attached hereto (or such other certification as the General Counsel may deem appropriate) stating that you have received, read, understand, and agree to comply with this Policy. The Company may require you to sign such a Certification on an annual basis, which Certification may be in electronic format. Please note that you are bound by this Policy whether or not you sign the Certification.
-7-
Annex A
Quarterly Restricted Periods
-8-
Annex B
Restricted Persons
All directors, executive officers (as designated by the board of directors), and any other vice president of the Company.
All members of the Company’s Legal, IT, Commercial Operations, Business Analytics and Finance departments.
All senior members of the Company’s Commercial function (e.g., Senior Directors, Area Sales Directors and Regional Sales Managers).
-9-
Annex C
Request for Clearance to Trade
To: TELA Bio, Inc. |
|
Attention: General Counsel |
||
1 Great Valley Parkway, Suite 24 |
|
Phone Number: (484) 320-2930 |
||
Malvern, PA 19355 |
|
E-mail: tocasio@telabio.com |
||
|
|
|
||
Name: |
|
|
Title: |
|
I hereby request clearance for myself (or a member of my immediate family or household) to execute the following transaction relating to the securities of TELA Bio, Inc.
Type of Transaction: |
|
Expiration Date: |
|
☐ I wish to purchase shares of common stock. Number of shares of common stock to be purchased: | |
| |
☐ I wish to sell shares of common stock. Number of shares of common stock to be sold: | |
| |
☐ I wish to gift shares of common stock. Number of shares of common stock to be gifted: | |
| |
Other: |
|
If the request is for a member of my immediate family or household:
Name of Person: |
|
|
Relationship: |
|
I hereby represent that I am not aware of any material, non-public information concerning TELA Bio, Inc. or its subsidiaries at the time of submitting this request and I agree that should I become aware of any material, non-public information concerning TELA Bio, Inc. or its subsidiaries prior to consummating the approved transaction, I will not consummate such transaction.
I understand that once approved, the authorization is valid on the date of approval and during the remaining term of the trading window in which it is approved. I further understand that the approval will lapse if, in the judgment of the General Counsel, I am likely to be aware of material, non-public information or at the expiration of the trading window in which approval is granted, whichever is the first to occur.
|
|
|
Date |
|
Signature |
|
|
|
Approved: |
|
|
|
|
|
General Counsel (or designee) |
|
Date |
-10-
Annex D
Certification
I hereby certify that:
1. |
I have read and understand TELA Bio, Inc.’s Insider Trading Policy (the “Policy”). I understand that the General Counsel is available to answer any questions I have regarding the Policy. |
2. |
Since I have been affiliated with the Company, I have complied with the Policy. |
3. |
I will continue to comply with the Policy for as long as I am subject to the Policy. |
Print name: |
|
|
|
|
|
|
|
Signature: |
|
|
|
|
|
|
|
Date: |
|
|
|
-11-
Exhibit 21.1
LIST OF SUBSIDIARIES
Subsidiary |
|
Ownership Percentage |
|
Jurisdiction of Incorporation or Organization |
TELA Bio, Limited |
|
100% |
|
England and Wales |
TELA Bio GmbH |
|
100% by TELA Bio, Limited |
|
Berlin |
Exhibit 23.1
Consent of Independent Registered Public Accounting Firm
We consent to the incorporation by reference in the registration statements (No. 333-275511) on Form S-3 and (Nos. 333-278183, 333-270803, 333-263797, 333-245707, and 333-235241) on Form S-8 of our report dated March 21, 2024, with respect to the consolidated financial statements of TELA Bio, Inc. and subsidiaries.
/s/ KPMG LLP
Philadelphia, Pennsylvania
March 21, 2025
EXHIBIT 31.1
CERTIFICATION
Pursuant to Rule 13a-14(a) under the Securities Exchange Act of 1934,
as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002
I, Antony Koblish, certify that:
1. I have reviewed this Form 10-K of TELA Bio, Inc.;
2. Based on my knowledge, this report does not contain any untrue statement of a material fact or omit to state a material fact necessary to make the statements made, in light of the circumstances under which such statements were made, not misleading with respect to the period covered by this report;
3. Based on my knowledge, the financial statements, and other financial information included in this report, fairly present in all material respects the financial condition, results of operations and cash flows of the registrant as of, and for, the periods presented in this report;
4. The registrant's other certifying officer and I are responsible for establishing and maintaining disclosure controls and procedures (as defined in Exchange Act Rules 13a-15(e) and 15d-15(e)) and internal control over financial reporting (as defined in Exchange Act Rules 13a-15(f) and 15d-15(f)) for the registrant and have:
(a) Designed such disclosure controls and procedures, or caused such disclosure controls and procedures to be designed under our supervision, to ensure that material information relating to the registrant, including its consolidated subsidiaries, is made known to us by others within those entities, particularly during the period in which this report is being prepared;
(b) Designed such internal control over financial reporting, or caused such internal control over financial reporting to be designed under our supervision, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles;
(c) Evaluated the effectiveness of the registrant's disclosure controls and procedures and presented in this report our conclusions about the effectiveness of the disclosure controls and procedures, as of the end of the period covered by this report based on such evaluation; and
(d) Disclosed in this report any change in the registrant's internal control over financial reporting that occurred during the registrant's most recent fiscal quarter (the registrant's fourth fiscal quarter in the case of an annual report) that has materially affected, or is reasonably likely to materially affect, the registrant's internal control over financial reporting; and
5. The registrant's other certifying officer and I have disclosed, based on our most recent evaluation of internal control over financial reporting, to the registrant's auditors and the audit committee of the registrant's board of directors (or persons performing the equivalent functions):
(a) All significant deficiencies and material weaknesses in the design or operation of internal control over financial reporting which are reasonably likely to adversely affect the registrant's ability to record, process, summarize and report financial information; and
(b) Any fraud, whether or not material, that involves management or other employees who have a significant role in the registrant's internal control over financial reporting.
Date: March 21, 2025 |
|
|
|
|
|
|
|
|
|
/s/ Antony Koblish |
|
|
Antony Koblish |
|
|
President and Chief Executive Officer |
|
|
(Principal Executive Officer) |
|
EXHIBIT 31.2
CERTIFICATION
Pursuant to Rule 13a-14(a) under the Securities Exchange Act of 1934,
as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002
I, Roberto Cuca, certify that:
1. I have reviewed this Form 10-K of TELA Bio, Inc.;
2. Based on my knowledge, this report does not contain any untrue statement of a material fact or omit to state a material fact necessary to make the statements made, in light of the circumstances under which such statements were made, not misleading with respect to the period covered by this report;
3. Based on my knowledge, the financial statements, and other financial information included in this report, fairly present in all material respects the financial condition, results of operations and cash flows of the registrant as of, and for, the periods presented in this report;
4. The registrant's other certifying officer and I are responsible for establishing and maintaining disclosure controls and procedures (as defined in Exchange Act Rules 13a-15(e) and 15d-15(e)) and internal control over financial reporting (as defined in Exchange Act Rules 13a-15(f) and 15d-15(f)) for the registrant and have:
(a) Designed such disclosure controls and procedures, or caused such disclosure controls and procedures to be designed under our supervision, to ensure that material information relating to the registrant, including its consolidated subsidiaries, is made known to us by others within those entities, particularly during the period in which this report is being prepared;
(b) Designed such internal control over financial reporting, or caused such internal control over financial reporting to be designed under our supervision, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles;
(c) Evaluated the effectiveness of the registrant's disclosure controls and procedures and presented in this report our conclusions about the effectiveness of the disclosure controls and procedures, as of the end of the period covered by this report based on such evaluation; and
(d) Disclosed in this report any change in the registrant's internal control over financial reporting that occurred during the registrant's most recent fiscal quarter (the registrant's fourth fiscal quarter in the case of an annual report) that has materially affected, or is reasonably likely to materially affect, the registrant's internal control over financial reporting; and
5. The registrant's other certifying officer and I have disclosed, based on our most recent evaluation of internal control over financial reporting, to the registrant's auditors and the audit committee of the registrant's board of directors (or persons performing the equivalent functions):
(a) All significant deficiencies and material weaknesses in the design or operation of internal control over financial reporting which are reasonably likely to adversely affect the registrant's ability to record, process, summarize and report financial information; and
(b) Any fraud, whether or not material, that involves management or other employees who have a significant role in the registrant's internal control over financial reporting.
Date: March 21, 2025 |
|
|
|
|
|
|
|
|
|
/s/ Roberto Cuca |
|
|
Roberto Cuca |
|
|
Chief Operating Officer and Chief Financial Officer |
|
|
(Principal Financial Officer) |
|
EXHIBIT 32.1
CERTIFICATION
Pursuant to 18 U.S.C. Section 1350,
as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002
Pursuant to the requirement set forth in Rule 13a-14(b) of the Securities Exchange Act of 1934, as amended (the “Exchange Act”), and Section 1350 of Chapter 63 of Title 18 of the United States Code (18 U.S.C. §1350), that: Antony Koblish, Chief Executive Officer of TELA Bio, Inc. (the “Company”), hereby certifies that, to the best of his knowledge:
(1) The Company’s Annual Report on Form 10-K for the period ended December 31, 2024, to which this Certification is attached as Exhibit 32.1 (the “Periodic Report”), fully complies with the requirements of Section 13(a) or Section 15(d) of the Exchange Act; and
(2) The information contained in the Periodic Report fairly presents, in all material respects, the financial condition and results of operations of the Company.
|
Date: March 21, 2025 |
|
|
|
|
|
/s/ Antony Koblish |
|
Antony Koblish |
|
President and Chief Executive Officer |
|
(Principal Executive Officer) |
EXHIBIT 32.2
CERTIFICATION
Pursuant to 18 U.S.C. Section 1350,
as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002
Pursuant to the requirement set forth in Rule 13a-14(b) of the Securities Exchange Act of 1934, as amended (the “Exchange Act”), and Section 1350 of Chapter 63 of Title 18 of the United States Code (18 U.S.C. §1350), that: Roberto Cuca, Chief Operating Officer and Chief Financial Officer of TELA Bio, Inc. (the “Company”), hereby certifies that, to the best of his knowledge:
(1) The Company’s Annual Report on Form 10-K for the period ended December 31, 2024, to which this Certification is attached as Exhibit 32.2 (the “Periodic Report”), fully complies with the requirements of Section 13(a) or Section 15(d) of the Exchange Act; and
(2) The information contained in the Periodic Report fairly presents, in all material respects, the financial condition and results of operations of the Company.
|
Date: March 21, 2025 |
|
|
|
|
|
/s/ Roberto Cuca |
|
Roberto Cuca |
|
Chief Operating Officer and Chief Financial Officer |
|
(Principal Financial Officer) |