UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
(Mark One)
☒ ANNUAL REPORT UNDER SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934.
FOR THE FISCAL YEAR ENDED JUNE 30, 2024
☐ TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the transition period from ____________to _____________
Commission File Number: 001-39015
BIOVIE INC.
(Exact name of registrant as specified in its charter)
| Nevada | 46-2510769 | |
| (State or other jurisdiction of incorporation or organization) | (I.R.S. Employer Identification Number) |
| 680 W Nye Lane Suite 204 | ||
| Carson City, NV 89703 | ||
| (Address of principal executive offices, Zip Code) | ||
| (775)-888-3162 | ||
| (Registrant’s telephone number, including area code) |
Securities registered pursuant to Section 12(b) of the Act:
| Title of each class | Trading Symbol(s) | Name of each exchange on which registered |
| Class A Common Stock, $0.0001 par value per share | BIVI | The NASDAQ Stock Market, LLC |
Securities registered pursuant to Section 12(g) of the Act:
None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act.
Yes ☐ No ☒
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act
Yes ☐ No ☒
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Exchange Act during the past 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days.
Yes ☒ No ☐
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files).
Yes ☒ No ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
| Large Accelerated Filer | ☐ | Accelerated Filer | ☐ |
| Non-Accelerated Filer | ☒ | Smaller reporting company | ☒ |
| Emerging growth company | ☐ | ||
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark if the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7362(b)) by the registered public accounting firm that prepared or issued its audit report.
Yes ☐ No ☒
If securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant included in the filing reflect the correction of an error to previously issued financial statements. ☐
Indicate by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive-based compensation received by any of the registrant’s executive officers during the relevant recovery period pursuant to §240.10D-1(b). ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act).
Yes ☐ No ☒
The aggregate market value of the voting and non-voting common equity held by non-affiliates computed by reference to the price at which the common equity was last sold, or the average bid and asked price of such common equity, as of the last business day of the registrant’s most recently completed second fiscal quarter was $20,590,705.
There were 7,956,660 shares of the Registrant’s Class A Common Stock, $0.0001 par value per share, outstanding as of September 25, 2024
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the Registrant’s definitive proxy statement relating to its 2024 annual meeting of stockholders (the “2024 Proxy Statement”) are incorporated by reference into Part III of this Annual Report on Form 10-K where indicated. The 2024 Proxy Statement was filed with the U.S. Securities and Exchange Commission on September 27, 2024.
BIOVIE INC.
FORM 10-K INDEX
| ( |
BIOVIE INC.
FORWARD-LOOKING STATEMENTS
This report contains forward-looking statements within the meaning of Section 21E of the Securities Exchange Act of 1934, and Section 27A of the Securities Act of 1933, as amended (the “Securities Act”). Any statements contained in this report that are not statements of historical fact may be forward-looking statements. When we use the words “intends,” “estimates,” “predicts,” “potential,” “continues,” “anticipates,” “plans,” “expects,” “believes,” “should,” “could,” “may,” “will” or the negative of these terms or other comparable terminology, we are identifying forward-looking statements. Forward-looking statements involve risks and uncertainties, which may cause our actual results, performance or achievements to be materially different from those expressed or implied by forward-looking statements. These factors include our research and development activities, distributor channel; compliance with regulatory impositions; and our capital needs. Although we believe that the expectations reflected in the forward-looking statements are reasonable, we cannot guarantee future results, levels of activity, performance or achievements.
Except as may be required by applicable law, we do not undertake or intend to update or revise our forward-looking statements, and we assume no obligation to update any forward-looking statements contained in this report as a result of new information or future events or developments. Thus, you should not assume that our silence over time means that actual events are bearing out as expressed or implied in such forward-looking statements. You should carefully review and consider the various disclosures we make in this report and our other reports filed with the Securities and Exchange Commission (the “Commission”) that attempt to advise interested parties of the risks, uncertainties and other factors that may affect our business.
All statements other than statements of historical fact are statements that could be deemed forward-looking statements. The Company assumes no obligation and does not intend to update these forward-looking statements, except as required by law. When used in this report, the terms “BioVie”, “Company”, “we”, “our”, and “us” refer to BioVie, Inc.
| ( |
PART I
| ITEM 1. | BUSINESS |
Overview
BioVie Inc. (the “Company” or “we” or “our”) is a clinical-stage company developing innovative drug therapies for the treatment of neurological and neurodegenerative disorders and advanced liver disease.
Neurodegenerative Disease Program
The Company acquired the biopharmaceutical assets of NeurMedix, Inc. (“NeurMedix”) a privately held clinical-stage pharmaceutical company and a related party in June 2021. The acquired assets included NE3107. In April 2024, the Company announced that the United States Adopted Names Council, and the World Health Organization International Nonproprietary Names expert committee had approved “bezisterim” as the non-proprietary (generic) name for NE3107. Bezisterim (NE3107) is an investigational, novel, orally administered small molecule that is thought to inhibit inflammation-driven insulin resistance and major pathological inflammatory cascades with a novel mechanism of action. There is emerging scientific consensus that both inflammation and insulin resistance may play fundamental roles in the development of Alzheimer’s disease (“AD”) and Parkinson’s disease (“PD”), and bezisterim (NE3107) could, if approved by U.S. Food and Drug Administration (“FDA”), represent an entirely new medical approach to treating these devastating conditions affecting an estimated 6 million Americans suffering from AD and 1 million Americans suffering from PD.
In neurodegenerative disease, bezisterim (NE3107) inhibits activation of inflammatory extracellular signal-regulated kinase (“ERK”) and nuclear factor kappa-light-chain-enhancer of activated B cells (“NFκB”) (including interactions with tumor necrosis factor (“TNF”) signaling and other relevant inflammatory pathways) that lead to neuroinflammation and insulin resistance. Bezisterim (NE3107) does not interfere with their homeostatic functions (e.g., insulin signaling and neuron growth and survival). Both inflammation and insulin resistance are drivers of AD and PD.
About Inflammation and Bezisterim’s (NE3107’s) Mechanism of Action
Neuroinflammation, insulin resistance, and oxidative stress are common features in the major neurodegenerative diseases, including AD, PD frontotemporal lobar dementia, and Amyotrophic lateral sclerosis (“ALS”). Bezisterim (NE3107) is an orally bioavailable, blood-brain permeable, small molecule, with potential anti-inflammatory, insulin sensitizing, and ERK-binding properties that may allow it to selectively inhibit ERK-, NFκB- and TNF-stimulated inflammation. Bezisterim’s (NE3107’s) potential to inhibit neuroinflammation and insulin resistance forms the basis for the Company’s work testing the molecule in AD and PD patients.
Parallels exist between AD and PD, among them activated microglia driving inflammation, involvement of TNFα, oxidative stress, protein misfolding, mitochondrial dysfunction, and insulin resistance. In preclinical and clinical studies, bezisterim (NE3107) reduced inflammation and enhanced insulin sensitivity, both of which are important to PD pathology. Preclinical studies in marmoset monkeys have shown bezisterim (NE3107) administered alone to be as pro-motoric as levodopa, underscoring the apparently critical role of inflammation in expression of PD motor symptoms. When bezisterim (NE3107) was administered with levodopa, the combination improved motor control better than either drug alone. Furthermore, in the marmoset study, bezisterim (NE3107) reduced the severity of levodopa induced dyskinesia (“LID”) concurrent with pro-motoric benefit and decreased neurodegeneration, preserving twice as many dopaminergic neurons compared to control.
Alzheimer’s Disease
AD Pathophysiology and Bezisterim (NE3107) Treatment Rationale
Alzheimer’s disease, which affects an estimated 6 million Americans, is a neuroinflammatory and neurodegenerative condition characterized by progressive deterioration of cognitive function and loss of short-term memory and executive function. Cognitive tests quantifying AD severity have been exhaustively developed. Formal diagnosis of AD has historically been dependent on the presence of extraneuronal amyloid beta (“Aβ”) plaques, which can only be observed at autopsy or with the aid of sophisticated radioimaging techniques. However, diagnostic methods have recently been approved that quantify Aβ in peripheral blood and correlate well with imaging results. Aβ plaques can also be found in people without apparent AD symptoms, which has cast doubt about the role of Aβ as the central mediator of disease pathology.
|
|
Scientific investigations in the past twenty years have provided strong evidence that inflammation, type 2 diabetes (“T2D”), and inflammation-driven insulin resistance are drivers of AD through interplay with the major inflammation signaling node, NFkB, and the cytokine, TNF, the activities of which are modulated by bezisterim (NE3107). The link between inflammation, T2D, and inflammation-driven insulin resistance and cognitive impairment are described by relatively new terms, type 3 diabetes and metabolic-cognitive syndrome.
Inflammation, insulin resistance, and associated metabolic dysregulation in the brain contribute to Aβ oligomerization and aggregation, phospho-tau formation, reduced neuron survival stimulus, and a forward-feeding cycle of neuronal energy deficit and oxidative stress, causing neuronal dysfunction (cognitive impairment) and neurodegeneration.
Insulin has a major role in metabolic regulation and neuron survival, while insulin resistance and T2D are closely linked to AD pathology. Insulin signaling is involved in synaptic plasticity, learning, and memory. Exogenous insulin enhances cognition in normal and cognitively impaired subjects. Insulin resistance is linked to cognitive impairment and senescence in the central nervous system (“CNS”).
Systemic inflammation from inflamed adipose tissue and associated mononuclear cells promotes CNS inflammation and is linked to cognitive decline and neurodegeneration. In addition to the afore mentioned factors contributing to AD pathophysiology, there is an extensive literature on the complex role of adipose tissue inflammation in systemic inflammation, insulin resistance, hypothalamus-pituitary-adrenal axis (“HPA”) dysregulation and chronic cortisol excess in cognitive impairment in AD. Obesity and inflammation are closely linked in expanding adipose tissue, where the production of inflammatory cytokines and increased cortisol are driven though up-regulation of 11β-hydroxysteroid dehydrogenase type 1 and adipocyte mineralocorticoid receptor activation. Inflamed adipose tissue interacts with the HPA axis and hippocampus to increase systemic cortisol, and promote hippocampal inflammation through chronically elevated cortisol, which freely penetrates the blood-brain barrier. Hyperglycemia (secondary to insulin resistance) exacerbates adrenal cortisol production and promotes forward feeding of inflammation and HPA-hippocampal dysregulation.
Bezisterim (NE3107) is believed to inhibit ERK/NFkB activation and TNF production stimulated by inflammatory stimuli, which includes oxidative stress. Inhibition of NFkB activation and TNF production from this type of stimulation has broad potential implications for reduction of pathological peripheral and CNS inflammatory signaling in AD, which includes reduction of inflammation-driven insulin resistance, decreased inflammatory cell infiltration into the CNS, and decreased microglia activation. Reduction of systemic inflammation and inflammation-driven insulin resistance are also predicted to have beneficial effects on HPA axis dysregulation and hippocampal dysregulation of cortisol secretion that are consequences of adipose inflammation and insulin resistance, and as described above, are known to promote cognitive impairment and forward-feeding insulin resistance. We believe bezisterim’s (NE3107’s) combination of anti-inflammatory and insulin sensitizing activity has the potential to disrupt this forward-feeding cycle of AD pathology. The multifactorial influence of insulin signaling on neuron survival and cognition suggests that correction of insulin signaling deficits with bezisterim (NE3107) in the target population may provide significant benefits on both cognition and disease progression.
Company’s Progress with Alzheimer’s Disease Clinical Trial
On November 29, 2023, the Company announced topline efficacy data from its Phase 3 clinical trial (NCT04669028) of bezisterim (NE3107) in the treatment of mild to moderate AD. The study had co-primary endpoints looking at cognition using the Alzheimer’s Disease Assessment Scale-Cognitive Scale (ADAS-Cog 12) and function using the Clinical Dementia Rating-Sum of Boxes. Patients were randomly assigned, 1:1 versus placebo, to receive sequentially 5 mg of bezisterim (NE3107) orally twice a day for 14 days, then 10 mg orally twice a day for 14 days, followed by 26 weeks of 20 mg orally twice daily.
Upon trial completion, as the Company began the process of analyzing the trial data, the Company found significant deviation from protocol and current good clinical practices (“cGCPs”) violations at 15 study sites (virtually all of which were from one geographic area). This highly unusual level of suspected improprieties led the Company to exclude all patients from these sites and to refer the sites to the FDA’s Office of Scientific Investigations (“OSI”) for potential action.
After the patient exclusions, 81 patients remained in the Modified Intent-to-Treat population, 57 of whom were in the Per-Protocol population which included those who completed the trial and were verified to take study drug based on pharmacokinetic data. The trial was originally designed to be 80% powered with 125 patients in each of the treatment and placebo arms. The unplanned exclusion of so many patients left the trial underpowered for its primary endpoints.
|
|
In the Per-Protocol population, which includes those patients who completed the trial and who were further verified to have taken the study drug (based on pharmacokinetics data), an observed but not statistically significant change from baseline appeared to suggest a slowing of cognitive loss; these same patients experienced an advantage in age deceleration vs. placebo as measured by deoxyribonucleic acid (“DNA”) epigenetic change. Age deceleration is used by longevity researchers to measure the difference between the patient’s biological age, in this case as measured by the Horvath DNA methylation Skin Blood Clock, relative to the patient’s actual chronological age. This test was a non-primary/secondary endpoint, other-outcome measure, done via blood test collected at week 30 (end of study).
Based on the efficacy signal seen in this trial, the Company is exploring (1) a discussion with the FDA to potentially employ the adaptive trial feature of the protocol to continue enrolling patients to achieve statistical significance; and/or (2) the design of a new Phase 3 study of bezisterim (NE3107) that leverages the most recent data and understanding of the potential effects bezisterim (NE3107) may have in persons with AD.
Parkinson’s Disease
Parkinson’s disease (PD), which affects an estimated 1 million Americans, is driven in large part by neuroinflammation and activation of brain microglia, leading to increased proinflammatory cytokines (particularly TNF). Multiple daily administrations of levodopa (converted to dopamine in the brain) is the current standard of care treatment for this movement disorder, but levodopa effectiveness diminishes over time necessitating increased dosage and prolonged daily administration leads to side effects of uncontrolled movements called levodopa-induced dyskinesia, commonly referred to as LID, which is exacerbated by high dose levodopa. Although levodopa provides symptomatic benefit, it does not slow PD progression.
The Company’s Phase 2 study of bezisterim (NE3107) for the treatment of PD (NCT05083260), completed in January 2023, was a double-blind, placebo-controlled, safety, tolerability, and pharmacokinetics study in PD participants treated with carbidopa/levodopa and NE3107. Forty-five patients with a defined L-dopa “off state” were randomized 1:1 to placebo or bezisterim (NE3107) 20 mg twice daily for 28 days. This trial was launched with two design objectives: (1) the primary objective was safety and drug-drug interaction, as requested by the FDA, to assess the potential for adverse interactions between bezisterim (NE3107) and carbidopa/ levodopa; and (2) the secondary objective was to determine if preclinical indications of promotoric activity and apparent enhancement of levodopa activity could be seen in humans. Both objectives were met. Results of the study include:
| ● | Five (26%) of the 19 patients treated with NE3107 vs zero of 19 placebo treated patients, experienced a morning ON state prior to receiving their initial morning C/L medications at the end of the study (day 28); this difference was statistically significant (p=0.046). | |
| ● | Patients treated with NE3107 + C/L experienced greater improvements in their Motor Disease Society- Unified Parkinson’s Disease Rating Scale (MDS UPDRS) Part III score than patients treated with placebo + C/L at the 2- and 3-hour marks after administration of the first daily dose of C/L. | |
| ● | Patients <70 years old treated with NE3107 + C/L experienced improvements that were ~6 points better than those who received placebo + C/L. | |
| ● | The study met its endpoints; investigators concluded that NE3107 + C/L combination treatment was associated with clinically meaningful and superior improvements (3+ points) on the motor examination part (Part III) of the MDS UPDRS. | |
| ● | NE3107 produced statistically significant improvements in nonmotor symptoms scale assessments (NMSS) for fatigue (Q4) p=0.02, urge to move legs (Q6) p=0.0036, and saliva dribbling (Q19) p= 0.0395. |
To extend this Phase 2 data in progressed patients, the Company has designed a new Phase 2 study of bezisterim (NE3107) as a potential first line therapy to treat patients with new onset PD. In July 2024, the Company submitted the protocol for this new study to the FDA for regulatory review.
|
|
Bezisterim (NE3107) may have the potential to become a non-dopaminergic alternative to PD patients. There are numerous scientific reports that support the critical role of inflammation in the manifestation of PD symptoms in addition to the essential role of inflammation in driving disease progression. We have shown in a mouse model of PD that bezisterim (NE3107) decreases inflammation and TNF in the brain and increases neuron survival (Nicoletti, 2012 Parkinson’s Disease 969418). In this neurotoxin induced model, bezisterim (NE3107) decreased clinical signs of disease and neuronal death compared to placebo treated mice. An unpublished study of a neurotoxin induced marmoset model of PD reported that administration of bezisterim (NE3107) decreased movement abnormalities that are the clinical signs of the disease. In the same study, bezisterim (NE3107) in combination with levodopa had a stronger effect on clinical signs of disease than levodopa or bezisterim (NE3107) alone, while marmosets treated with bezisterim (NE3107) developed less LID. Bezisterim (NE3107)-treated monkeys also exhibited neuroprotective activity that promoted the survival of twice as many neurons in the substantia nigra (primary region of the brain that degenerates to cause parkinsonism) as monkeys treated with placebo. The results from the marmoset study suggest that bezisterim (NE3107) may decrease clinical signs of disease in humans (improve motor function), which if true could enable a straightforward clinical development strategy to test bezisterim (NE3107) in PD patients needing promotoric therapy. If approved as a promotoric agent, NE3107 would provide a non-dopaminergic alternative to Parkinson’s patients, and an opportunity to significantly delay the need to start levodopa therapy. This could represent a first step toward supplanting levodopa as the primary PD therapy, and in addition to delaying the emergence of LID, could also slow disease progression, the most important and still unmet objective of PD drug development.
Long COVID Program
In April 2024, the Company announced the grant of a clinical trial award of up to $13.1 million from the U.S. Department of Defense (“DOD”), awarded through the Peer Reviewed Medical Research Program of the Congressionally Directed Medical Research Programs. In August 2024, U.S. Army Medical Research and Development Command, Office of Human Research Oversight (“OHRO”) approved the Company’s plan to evaluate bezisterim (NE3107) for the treatment of neurological symptoms that are associated with long COVID. The FDA had previously reviewed and approved the study as “Safe to Proceed” in August 2024. The approval form OHRO is the last scientific review milestone needed for the Company to receive the additional $12.6 million of the aggregate $13.1 million in grant funding from the DOD. The award can provide up to 2 years of non-dilutive funding for a Phase 2 clinical trial that will assess bezisterim (NE3107) for the treatment of neurological symptoms that are associated with long COVID. The Company anticipates the trial to commence by early 2025. The study protocol was finalized and submitted to the FDA for regulatory review in July 2024 and on August 22, 2024 the FDA authorized our IND application for Bezisterim (NE3107) allowing us to study a novel, anti-inflammatory approach or the treatment of the debilitating neurocognitive symptoms associated with long covid.
Long COVID is a condition in which symptoms of COVID-19, the acute respiratory disease caused by the SARS-CoV-2 virus, persist for an extended period of time, generally three months or more. The Centers for Disease Control recently reported that 6.8% of adults in the United States (more than 17 million individuals) currently or previously had long COVID. Symptoms, which include fatigue, cognitive dysfunction and sleep disturbances, are debilitating. The loss in quality of life and earnings and increased medical costs has an enormous economic impact estimated to be 3.7 trillion dollars. To date there are no therapies proven effective for treatment.
Chronic inflammation is one of the main hypotheses that researchers have proposed to explain the persistence of symptoms in long COVID. Specifically in individuals with “brain fog,” sustained systemic inflammation and persistent localized blood-brain-barrier (“BBB”) dysfunction are key physiological features. Bezisterim (NE3107) permeates the BBB and has been shown to modulate inflammation via the inhibition of NF-kB activation, thus representing a novel oral treatment targeting an underlying cause of long COVID symptoms.
Chronic neuroinflammation, insulin resistance, and oxidative stress are common features in the major neurodegenerative diseases, including AD, PD, frontotemporal lobar dementia, and ALS. Bezisterim (NE3107) is an investigational oral small molecule, blood-brain permeable, compound with potential anti-inflammatory, insulin sensitizing, and ERK-binding properties that may allow it to selectively inhibit ERK-, NFκB- and TNF-stimulated inflammation. Bezisterim’s (NE3107) potential to inhibit neuroinflammation and insulin resistance forms the basis for the Company’s work testing the molecule in AD, PD, and long COVID patients. Bezisterim (NE3107) is patented in the United States, Australia, Canada, Europe and South Korea.
Liver Cirrhosis Program
In liver disease, our investigational drug candidate BIV201 (continuous infusion terlipressin), which has been granted both FDA Fast Track designation status and FDA Orphan Drug status, is being evaluated and discussed after receiving guidance from the FDA regarding the design of Phase 3 clinical testing for the treatment of ascites due to chronic liver cirrhosis. BIV201 is administered as a patent-pending liquid formulation.
|
|
Ascites is a common complication of advanced liver cirrhosis involving the accumulation of large volumes of fluid in the abdomen, often exceeding five liters, due to liver and kidney dysfunction. The FDA has never approved a drug to treat ascites, and once patients reach the refractory stage the estimated one-year survival rate is only approximately 50%[10]. BIV201 is a continuous infusion of terlipressin, a drug used in over 40 countries to treat related complications of liver cirrhosis (Type 1 hepatorenal syndrome and bleeding esophageal varices) that was recently approved in the U.S. but is not approved in Japan. With the novel room temperature stable formulation in a pre-filled syringe, BIV201 could potentially provide a superior terlipressin drug delivery system throughout the world. The goal of BIV201 therapy is to interrupt the ascites disease pathway, thereby halting the cycle of accelerated fluid generation in ascites patients.
In June 2021, the Company initiated a Phase 2 study (NCT04112199) designed to evaluate the efficacy of BIV201 (terlipressin, administered by continuous infusion for two 28-day treatment cycles) combined with standard-of-care (“SOC”), compared to SOC alone, for the treatment of refractory ascites. The primary endpoints of the study are the incidence of ascites-related complications and change in ascites fluid accumulation during treatment compared to a pre-treatment period.
In March 2023, the Company announced enrollment was paused and that data from the first 15 patients treated with BIV201 plus SOC appeared to show at least a 30% reduction in ascites fluid during the 28 days after treatment initiation compared to the 28 days prior to treatment. The change in ascites volume was significantly different from those patients receiving SOC treatment. Patients who completed the treatment with BIV201 experienced a 53% reduction in ascites fluid, which was sustained (43% reduction) during the three months after treatment initiation as compared to the three-month pre-treatment period.
In June 2023, the Company requested and subsequently received guidance from the FDA regarding the design and endpoints for definitive clinical testing of BIV201 for the treatment of ascites due to chronic liver cirrhosis. The Company is currently finalizing protocol designs for the Phase 3 study of BIV201 for the treatment of ascites due to chronic liver cirrhosis.
While the active agent, terlipressin, is approved in the U.S. and in about 40 countries for related complications of advanced liver cirrhosis, treatment of ascites is not included in these authorizations. Patients with refractory ascites suffer from frequent life-threatening complications, generate more than $5 billion in annual treatment costs, and have an estimated 50% mortality rate within 6 to 12 months. The FDA has not approved any drug to treat refractory ascites.
Our proprietary novel liquid formulation of terlipressin is designed to improve convenience for outpatient administration and avoid potential formulation errors when pharmacists reconstitute the current powder version of terlipressin. To date, analytical testing results have confirmed room temperature stability of the prefilled syringe in storage for 18 months, with the potential for up two years stability. Room temperature storage presents a key product differentiation versus terlipressin products in countries where the drug is approved. To the best of the Company’s knowledge, all other terlipressin products sold globally must be stored under refrigeration and there is no prefilled syringe format of terlipressin available for treating patients in these countries. BioVie has also filed a Patent Cooperation Treaty (“PCT”) application covering our novel liquid formulations of terlipressin (international patent application PCT/US2020/034269, published as WO2020/237170) and we are seeking patent protection in at least the U.S., Europe, China, Japan and other jurisdictions.
BIV201 (continuous infusion terlipressin) has the potential to improve the health of thousands of patients suffering from life-threatening complications of liver cirrhosis due to hepatitis, nonalcoholic steatohepatitis, and alcoholism. The FDA has granted Fast-Track status and Orphan Drug designation for the most common of these complications, ascites, which represents a significant unmet medical need. Patients with cirrhosis and ascites account for an estimated 116,000 U.S. hospital discharges annually, with frequent early readmissions. According to the HCUP Nationwide Readmissions Database 2016, those requiring paracentesis (removal of ascites fluid) experience an average hospital stay lasting eight days incurring over $86,000 in medical costs. This translates into a total potentially addressable ascites market size for BIV201 therapy exceeding $650 million based on Company estimates. The FDA has never approved any drug specifically for treating ascites. For patients with refractory ascites the mean one-year survival rate is only 50% (Bureau et al. 2017). BIV201 has also received Orphan Drug designation for hepatorenal syndrome (“HRS”). Patients with refractory ascites often progress to HRS which is the onset of kidney failure and requires emergency hospitalization.
The BIV201 development program began at LAT Pharma LLC. On April 11, 2016, we acquired LAT Pharma LLC and the rights to its BIV201 development program and currently own all development and marketing rights to the product candidate. We and PharmaIN, LAT Pharma’s former partner focused on the development of new modified product candidates in the same therapeutic field but not including BIV201, have agreed to pay royalties equal to less than 1% of future net sales of each company’s ascites drug development programs, or if such program is licensed to a third party, less than 5% of each company’s net license revenues. On December 24, 2018, we returned our partial ownership rights to the PharmaIN modified terlipressin development program and simultaneously paid the remaining balance due on a related debt. PharmaIN’s rights to our program remain unchanged.
| 10 | Bureau et al. 2017 |
|
|
About Ascites and Liver Cirrhosis
Cirrhosis is a leading cause of death in the U.S. The condition results primarily from hepatitis, alcoholism, and fatty liver disease linked to obesity. Ascites is a common complication of advanced liver cirrhosis, involving kidney dysfunction and the accumulation of large amounts of fluid in the abdominal cavity.
The Need for an Ascites Therapy
With no medications approved by the FDA specifically for treating ascites, an estimated 40% of patients die within two years of diagnosis. Certain drugs approved for other uses such as diuretics may provide initial relief, but patients may fail to respond to treatment as ascites worsens. This represents a critical unmet medical need, reflected by the Fast Track designation granted to BIV201 by the FDA as a treatment for ascites refractory to or intolerant of diuretic therapy. U.S. treatment costs for liver cirrhosis, including ascites and other complications, are estimated at more than $5 billion annually.
The Ascites Development Pathway
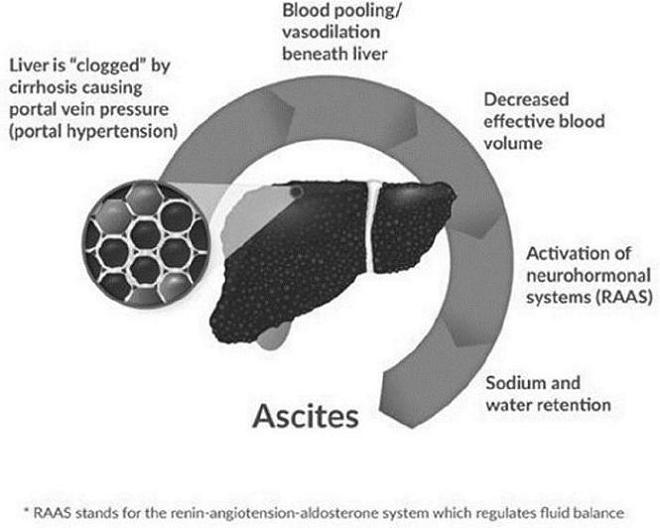
Most experts agree that ascites develops through a sequence of events illustrated by the above diagram. High blood pressure in the vein that supplies blood to the liver, called “portal hypertension,” occurs as increasing liver damage (fibrosis) impedes blood flow through the liver. This causes vasodilation and blood pooling in the central or “splanchnic” region of the body and low blood volume in the arteries. The decrease in effective blood volume activates a signaling pathway (“neurohormonal systems”) which tells the kidneys to retain large amounts of salt and water in an effort to increase blood volume. Ultimately the retention of excess sodium and water leads to the formation of ascites as these substances “weep” from the liver and lymph system and collect in the patient’s abdomen.
The BIV201 Proposed Mechanism of Action
BIV201 is being developed with the goal of alleviating portal hypertension and correcting splanchnic vasodilation, thereby increasing effective blood volume and reducing the signals to the kidneys to retain excess salt and water. If successful, BIV201 could halt the cycle of accelerating fluid generation in ascites patients and reduce the need for the frequent and painful paracentesis procedures many of these patients currently require.
|
|
Future Possible BIV201 Indications
Based on international investigative studies of the active agent in BIV201, terlipressin, we believe our drug candidate has potential future applications in other life-threatening conditions due to liver cirrhosis. Securing marketing approvals for any of these new uses will require well-controlled clinical trials to satisfy the FDA and/or other countries’ regulatory requirements, none of which have commenced at this time. The Company continues to evaluate other indications for the use of terlipressin continuous infusion. BioVie will discuss such indications if and when selected for testing.
Intellectual Property
BIV201
BioVie relies on a combination of patent, trade secret, other intellectual property laws (such as FDA data exclusivity), nondisclosure agreements, and other measures to protect our proposed products. We require our employees, consultants, and advisors to execute confidentiality agreements and to agree to disclose and assign to us all inventions conceived during the workday, using our property, or which relate to our business. Despite any measures taken to protect our intellectual property (IP), unauthorized parties may attempt to copy aspects of our products or to obtain and use information that we regard as proprietary.
BIV201 was awarded Orphan Drug Designations in the U.S. for the treatment of hepatorenal syndrome on November 21, 2018 and treatment of ascites due to all etiologies except cancer on September 8, 2016. We also filed a PCT application covering our novel liquid formulations of terlipressin (international patent application PCT/US2020/034269, published as WO2020/237170) and are seeking patent protection in U.S., Europe, China, Japan and other jurisdictions. To date patents have been granted in India (Patent No. 540813) and Chile (Patent No. 68965). Also, we own U.S. Patent 11,364,277, and European patent EP3347032, which is directed to a method of treating ascites with BIV201, and we are pursuing additional patent coverage in U.S., Japan, Europe, and China.
Bezisterim (NE3107) and related compounds
As of August 15, 2024, we have twelve (12) issued U.S. patents, six (6) pending U.S. patent applications, three (3) pending U.S. PCT applications, six (6) issued foreign patents, and six (6) pending foreign patent applications directed to protecting NE3107 and related compounds and methods of making and using thereof. The U.S. patents and pending patent applications and their projected expiration dates are provided below.
| Title | Patent Application Number |
Patent Number |
Expiration Date |
|||||
| Unsaturated Steroid Compounds | 13/030,326 | 8,586,770 | 6/2/2026 | |||||
| Solid State Forms of a Pharmaceutical | 12/418,559 | 8,252,947 | * | 4/18/2030 | ||||
| Crystalline Anhydrate Forms of a Pharmaceutical | 14/459,528 15/348,107 16/598,694 17/240,728 |
9,555,046 9,850,271 10,995,112 pending |
4/3/2029 4/3/2029 4/3/2029 — |
|||||
| Pharmaceutical Solid State Forms | 12/370,510 | 8,518,922 | 9/24/2031 | |||||
| Methods of Preparing Pharmaceutical Solid State Forms | 13/919,593 | 9,314,471 | 6/28/2029 | |||||
| Steroid Tetrol Solid State Forms | 12/272,767 | 8,486,926 | 1/10/2030 | |||||
| Drug Identification and Treatment Method | 11/941,936 | 8,354,396 | 7/7/2031 | |||||
| Method For Preparing Substituted 3,7-Dihydroxy Steroids | 13/664,304 14/886,738 |
9,163,059** 9,994,608 |
6/5/2029 6/5/2029 |
|||||
| Treatment Methods Using Pharmaceutical Solid State Forms | 14/459,493 | 9,877,972 | 4/3/2029 | |||||
| Compositions for Treatment of Neurodegenerative Conditions | 18/511,027 | pending | — | |||||
| Methods of Treating Long COVID | 63/621,280 | pending | — | |||||
| Modified C19 Steroids and Methods of Using the Same | 63/610,915 | pending | — | |||||
| Compositions and Methods for the Treatment of Diseases and Conditions Associated with Amyloid Beta Peptides | 63/592,364 | pending | — | |||||
| Methods for the Treatment of Biological Aging | 63/561,157 | pending | — | |||||
| * | Foreign counterparts issued in Australia, Canada, Europe and South Korea projected to expire 4/3/2029. |
| ** | Foreign counterparts issued in Europe and Japan projected to expire 6/5/2029. |
|
|
Government Regulation
Government authorities in the United States, at the federal, state and local level, and in other countries extensively regulate, among other things, the research, development, testing, manufacture, quality control, approval, labeling, packaging, storage, record-keeping, promotion, advertising, distribution, post-approval monitoring and reporting, marketing and export and import of products such as those we are developing. Any pharmaceutical candidate that we develop must be approved by the FDA before it may be legally marketed in the United States and by the appropriate foreign regulatory agency before it may be legally marketed in foreign countries.
United States Drug Development Process
In the United States, the FDA regulates drugs under the Federal Food, Drug and Cosmetic Act (“FDCA”), and implements regulations. Drugs are also subject to other federal, state and local statutes and regulations. Biologics are subject to regulation by the FDA under the FDCA, the Public Health Service Act (the “PHSA”), and related regulations, and other federal, state and local statutes and regulations. Biological products include, among other things, viruses, therapeutic serums, vaccines and most protein products. The process of obtaining regulatory approvals and the subsequent compliance with appropriate federal, state, local and foreign statutes and regulations require the expenditure of substantial time and financial resources. Failure to comply with the applicable United States requirements at any time during the product development process, approval process or after approval, may subject an applicant to administrative or judicial sanctions. FDA sanctions could include refusal to approve pending applications, withdrawal of an approval, a clinical hold, warning letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, refusals of government contracts, restitution, disgorgement or civil or criminal penalties. Any agency or judicial enforcement action could have a material adverse effect on us.
The process required by the FDA before a drug or biological product may be marketed in the United States generally involves the following:
| ● | Completion of preclinical laboratory tests, animal studies and formulation studies according to Good Laboratory Practices or other applicable regulations; |
| ● | Submission to the FDA of an Investigational New Drug Application (“IND”), which must become effective before human clinical trials may begin; |
| ● | Performance of adequate and well-controlled human clinical trials according to the FDA’s GCPs, to establish the safety and efficacy of the proposed drug or biologic for its intended use; |
| ● | Submission to the FDA of a New Drug Application (an “NDA”), for a new drug product, or a Biologics License Application (a “BLA”), for a new biological product; |
| ● | Satisfactory completion of an FDA inspection of the manufacturing facility or facilities where the drug or biologic is to be produced to assess compliance with the FDA’s current good manufacturing practice standards, or cGMP, to assure that the facilities, methods and controls are adequate to preserve the drug’s or biologic’s identity, strength, quality and purity; |
| ● | Potential FDA audit of the nonclinical and clinical trial sites that generated the data in support of the NDA or BLA; and |
| ● | FDA review and approval of the NDA or BLA. |
The lengthy process of seeking required approvals and the continuing need for compliance with applicable statutes and regulations require the expenditure of substantial resources. There can be no certainty that approvals will be granted.
Clinical trials involve the administration of the drug or biological candidate to healthy volunteers or patients having the disease being studied under the supervision of qualified investigators, generally physicians not employed by or under the trial sponsor’s control. Clinical trials are conducted under protocols detailing, among other things, the objectives of the clinical trial, dosing procedures, subject selection and exclusion criteria, and the parameters to be used to monitor subject safety. Each protocol must be submitted to the FDA as part of the IND. Clinical trials must be conducted in accordance with the FDA’s cGCP requirements. Further, each clinical trial must be reviewed and approved by an independent institutional review board (“IRB”), at or servicing each institution at which the clinical trial will be conducted. An IRB is charged with protecting the welfare and rights of trial participants and considers such items as whether the risks to individuals participating in the clinical trials are minimized and are reasonable in relation to anticipated benefits. The IRB also approves the informed consent form that must be provided to each clinical trial subject or his or her legal representative and must monitor the clinical trial until it is completed.
|
|
Human clinical trials prior to approval are typically conducted in three sequential phases that may overlap or be combined:
| ● | Phase 1. The drug or biologic is initially introduced into healthy human subjects and tested for safety, dosage tolerance, absorption, metabolism, distribution and excretion. In the case of some products for severe or life-threatening diseases, especially when the product may be too inherently toxic to ethically administer to healthy volunteers, the initial human testing is often conducted in patients having the specific disease. |
| ● | Phase 2. The drug or biologic is evaluated in a limited patient population to identify possible adverse effects and safety risks, to preliminarily evaluate the efficacy of the product for specific targeted diseases and to determine optimal dosage and dosing schedule for patients having the specific disease. |
| ● | Phase 3. Clinical trials are undertaken to further evaluate dosage, clinical efficacy and safety in an expanded patient population at geographically dispersed clinical trial sites. These clinical trials, which usually involve more subjects than earlier trials, are intended to establish the overall risk/benefit ratio of the product and provide an adequate basis for product labeling. Generally, two adequate and well-controlled Phase 3 clinical trials are required by the FDA for approval of an NDA or BLA. |
Post-approval studies, or Phase 4 clinical trials, may be conducted after initial marketing approval. These studies are used to gain additional experience from the treatment of patients in the intended therapeutic indication and may be required by the FDA as part of the approval process.
Progress reports detailing the results of the clinical trials must be submitted at least annually to the FDA and written IND safety reports must be submitted to the FDA by the investigators for serious and unexpected adverse events or any finding from tests in laboratory animals that suggests a significant risk for human subjects. Phase 1, Phase 2 and Phase 3 clinical trials may not be completed successfully within any specified period, if at all. The FDA or the sponsor or its data safety monitoring board may suspend a clinical trial at any time on various grounds, including a finding that the research subjects or patients are being exposed to an unacceptable health risk. Similarly, an IRB can suspend or terminate approval of a clinical trial at its institution if the clinical trial is not being conducted in accordance with the IRB’s requirements or if the drug or biologic has been associated with unexpected serious harm to patients.
Concurrent with clinical trials, companies usually complete additional animal studies and develop additional information about the chemistry and physical characteristics of the drug or biologic as well as finalize a process for manufacturing the product in commercial quantities in accordance with cGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the drug or biological candidate and, among other things, must include methods for testing the identity, strength, quality and purity of the final drug or biologic. Additionally, appropriate packaging must be selected and tested and stability studies must be conducted to demonstrate that the drug or biological candidate does not undergo unacceptable deterioration over its shelf life.
U.S. Review and Approval Processes
The results of product development, preclinical studies and clinical trials, along with descriptions of the manufacturing process, analytical tests conducted on the chemistry of the drug or biologic, proposed labeling and other relevant information are submitted to the FDA as part of an NDA or BLA requesting approval to market the product. The submission of an NDA or BLA is subject to the payment of substantial user fees; a waiver of such fees may be obtained under certain limited circumstances.
The FDA reviews all NDAs and BLAs submitted before it accepts them for filing and may request additional information rather than accepting an NDA or BLA for filing. Once the submission is accepted for filing, the FDA begins an in-depth review of the NDA or BLA.
After the NDA or BLA submission is accepted for filing, the FDA reviews the NDA to determine, among other things, whether the proposed product is safe and effective for its intended use, and whether the product is being manufactured in accordance with cGMP to assure and preserve the product’s identity, strength, quality and purity. The FDA reviews a BLA to determine, among other things, whether the product is safe, pure and potent and the facility in which it is manufactured, processed, packaged or held meets standards designed to assure the product’s continued safety, purity and potency. In addition to its own review, the FDA may refer applications for novel drug or biological products or drug or biological products which present difficult questions of safety or efficacy to an advisory committee, typically a panel that includes clinicians and other experts, for review, evaluation and a recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions. During the approval process, the FDA also will determine whether a risk evaluation and mitigation strategy, or REMS, is necessary to assure the safe use of the drug or biologic. If the FDA concludes that a REMS is needed, the sponsor of the NDA or BLA must submit a proposed REMS; the FDA will not approve the NDA or BLA without a REMS, if required.
|
|
Before approving an NDA or BLA, the FDA will inspect the facilities at which the product is to be manufactured. The FDA will not approve the product unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications. Additionally, before approving an NDA or BLA, the FDA will typically inspect one or more clinical sites to assure compliance with cGMP. If the FDA determines the application, manufacturing process or manufacturing facilities are not acceptable it will outline the deficiencies in the submission and often will request additional testing or information.
The NDA or BLA review and approval process is lengthy and difficult and the FDA may refuse to approve an NDA or BLA if the applicable regulatory criteria are not satisfied or may require additional clinical data or other information. Even if such data and information is submitted, the FDA may ultimately decide that the NDA or BLA does not satisfy the criteria for approval. Data obtained from clinical trials are not always conclusive and may be susceptible to varying interpretations, which could delay, limit or prevent regulatory approval. The FDA will issue a “complete response” letter if the agency decides not to approve the NDA or BLA. The complete response letter describes all of the specific deficiencies in the NDA or BLA identified by the FDA. The deficiencies identified may be minor, for example, requiring labeling changes, or major, for example, requiring additional clinical trials. Additionally, the complete response letter may include recommended actions that the applicant might take to place the application in a condition for approval. If a complete response letter is issued, the applicant may either resubmit the NDA or BLA, addressing all of the deficiencies identified in the letter, or withdraw the application.
If a product receives regulatory approval, the approval may be limited to specific diseases and dosages or the indications for use may otherwise be limited, which could restrict the commercial value of the product. Further, the FDA may require that certain contraindications, warnings or precautions be included in the product labeling. In addition, the FDA may require Phase 4 testing which involves clinical trials designed to further assess a product’s safety and effectiveness and may require testing and surveillance programs to monitor the safety of approved products that have been commercialized.
Orphan Drug Designation
Under the Orphan Drug Act, the FDA may grant orphan designation to a drug or biological product intended to treat a rare disease or condition, which is generally a disease or condition that affects fewer than 200,000 individuals in the United States, or more than 200,000 individuals in the United States and for which there is no reasonable expectation that the cost of developing and making a drug or biological product available in the United States for this type of disease or condition will be recovered from sales of the product. Orphan product designation must be requested before submitting an NDA or BLA. After the FDA grants orphan product designation, the identity of the therapeutic agent and its potential orphan use are disclosed publicly by the FDA. Orphan product designation does not convey any advantage in or shorten the duration of the regulatory review and approval process.
If a product that has Orphan designation subsequently receives the first FDA approval for the disease or condition for which it has such designation, the product is entitled to orphan product exclusivity, which means that the FDA may not approve any other applications to market the same drug or biological product for the same indication for seven years, except in limited circumstances, such as a showing of clinical superiority to the product with orphan exclusivity. Competitors, however, may receive approval of different products for the indication for which the Orphan product has exclusivity or obtain approval for the same product but for a different indication for which the Orphan product has exclusivity. Orphan product exclusivity also could block the approval of one of our products for seven years if a competitor obtains approval of the same drug or biological product as defined by the FDA or if our drug or biological candidate is determined to be contained within the competitor’s product for the same indication or disease. If a drug or biological product designated as an orphan product receives marketing approval for an indication broader than what is designated, it may not be entitled to orphan product exclusivity. Orphan Drug status in the European Union has similar but not identical benefits in the European Union.
Expedited Development and Review Programs
The FDA has a Fast Track program that is intended to expedite or facilitate the process for reviewing new drug and biological products that meet certain criteria. Specifically, new drug and biological products are eligible for Fast Track designation if they are intended to treat a serious or life-threatening condition and demonstrate the potential to address unmet medical needs for the condition. Fast Track designation applies to the combination of the product and the specific indication for which it is being studied. Unique to a Fast Track product, the FDA may consider for review sections of the NDA or BLA on a rolling basis before the complete application is submitted, if the sponsor provides a schedule for the submission of the sections of the NDA or BLA, the FDA agrees to accept sections of the NDA or BLA and determines that the schedule is acceptable, and the sponsor pays any required user fees upon submission of the first section of the NDA or BLA.
|
|
Any product submitted to the FDA for marketing approval, including those submitted to a Fast Track program, may also be eligible for other types of FDA programs intended to expedite development and review, such as priority review and accelerated approval. Any product is eligible for priority review if it has the potential to provide safe and effective therapy where no satisfactory alternative therapy exists or a significant improvement in the treatment, diagnosis or prevention of a disease compared with marketed products. The FDA will attempt to direct additional resources to the evaluation of an application for a new drug or biological product designated for priority review in an effort to facilitate the review. Additionally, a product may be eligible for accelerated approval. Drug or biological products studied for their safety and effectiveness in treating serious or life-threatening illnesses and that provide meaningful therapeutic benefit over existing treatments may receive accelerated approval, which means that they may be approved on the basis of adequate and well-controlled clinical studies establishing that the product has an effect on a surrogate endpoint that is reasonably likely to predict a clinical benefit, or on the basis of an effect on a clinical endpoint other than survival or irreversible morbidity. As a condition of approval, the FDA generally requires that a sponsor of a drug or biological product receiving accelerated approval perform adequate and well-controlled post-marketing clinical studies to establish safety and efficacy for the approved indication. Failure to conduct such studies or conducting such studies that do not establish the required safety and efficacy may result in revocation of the original approval. In addition, the FDA currently requires as a condition for accelerated approval pre-approval of promotional materials, which could adversely impact the timing of the commercial launch or subsequent marketing of the product. Fast Track designation, priority review and accelerated approval do not change the standards for approval but may expedite the development or approval process.
Post-Approval Requirements
Any drug or biological products for which we receive FDA approvals are subject to continuing regulation by the FDA, including, among other things, record-keeping requirements, reporting of adverse experiences with the product, providing the FDA with updated safety and efficacy information on an annual basis or as required more frequently for specific events, product sampling and distribution requirements, complying with certain electronic records and signature requirements and complying with FDA promotion and advertising requirements, which include, among others, standards for direct-to-consumer advertising, prohibitions against promoting drugs and biologics for uses or in patient populations that are not described in the drug’s or biologic’s approved labeling (known as “off-label use”), rules for conducting industry-sponsored scientific and educational activities, and promotional activities involving the internet. Failure to comply with FDA requirements can have negative consequences, including the immediate discontinuation of noncomplying materials, adverse publicity, enforcement letters from the FDA, mandated corrective advertising or communications with doctors, and civil or criminal penalties. Although physicians may prescribe legally available drugs and biologics for off-label uses, manufacturers may not market or promote such off-label uses.
We will need to rely on third parties for the production of our product candidates. Manufacturers of our product candidates are required to comply with applicable FDA manufacturing requirements contained in the FDA’s cGMP regulations. cGMP regulations require among other things, quality control and quality assurance as well as the corresponding maintenance of comprehensive records and documentation. Drug and biologic manufacturers and other entities involved in the manufacture and distribution of approved drugs and biologics are also required to register their establishments and list any products made there with the FDA and comply with related requirements in certain states, and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with cGMP and other laws. Accordingly, manufacturers must continue to expend time, money and effort in the area of production and quality control to maintain cGMP compliance. Discovery of problems with a product after approval may result in serious and extensive restrictions on a product, manufacturer, or holder of an approved NDA or BLA, including suspension of a product until the FDA is assured that quality standards can be met, continuing oversight of manufacturing by the FDA under a “consent decree,” which frequently includes the imposition of costs and continuing inspections over a period of many years, and possible withdrawal of the product from the market. In addition, changes to the manufacturing process generally require prior FDA approval before being implemented and other types of changes to the approved product, such as adding new indications and additional labeling claims, are also subject to further FDA review and approval.
The FDA also may require post-marketing testing, known as Phase 4 testing, risk minimization action plans and surveillance to monitor the effects of an approved product or place conditions on an approval that could otherwise restrict the distribution or use of the product.
Employees
Our business is managed by our officers who consist of Mr. Cuong Do, Chief Executive Officer & President; Dr. Joseph M Columbo, Executive Vice President -Chief Medical Officer; and Wendy Kim, our Chief Financial Officer and Corporate Secretary. These individuals devote their full-time efforts to the Company activities. The Company has 14 employees which are all full time. We also rely on a team of highly experienced scientific, medical, and regulatory consultants to conduct product development activities.
|
|
| ITEM 1A. | RISK FACTORS |
Our business, financial condition, operating results and prospects are subject to the following risks. Additional risks and uncertainties not presently foreseeable to us may also impair our business operations. If any of the following risks or the risks described elsewhere in this report actually occurs, our business, financial condition or operating results could be materially adversely affected. In such case, the trading price of our Company’s Class A Common Stock, par value $0.0001 (“Common Stock”) Common Stock could decline, and our stockholders may lose all or part of their investment in the shares of our Common Stock.
This Form 10-K contains forward-looking statements that involve risks and uncertainties. These statements can be identified by the use of forward-looking terminology such as “believes,” “expects,” “intends,” “plans,” “may,” “will,” “should,” “predict” or “anticipation” or the negative thereof or other variations thereon or comparable terminology. Actual results could differ materially from those discussed in the forward- looking statements as a result of certain factors, including those set forth below and elsewhere in this Form 10-K.
Risk Factor Summary
Our business operations are subject to numerous risks, factors and uncertainties, including those outside of our control, which could cause our actual results to be harmed, including risks regarding the following:
Risks Relating to Our Business and Industry
| · | If these third parties contractors do not successfully carry out their contractual duties or meet expected deadlines or do not successfully perform and comply with regulatory requirements, we may not be able to obtain regulatory approval of or commercialize our product candidates. |
| · | Successful development of biopharmaceuticals is highly uncertain and is dependent on numerous factors, many of which are beyond our control. |
| · | The concentration of our assets within certain financial institutions could have a material adverse effect on its business, financial condition and results of operations. |
| · | We are currently subject to securities class action litigation and may be subject to similar or other litigation in the future, which may have a material adverse effect on our business. |
| · | We have no products approved for commercial sale, have never generated any revenues, and may never achieve revenues or profitability, which could cause us to cease operations. |
| · | We are a development stage company with a limited operating history, making it difficult for you to evaluate our business and your investment. |
| · | If the FDA or comparable foreign regulatory authorities approve generic versions of any of our product candidates that receive marketing approval, or such authorities do not grant our products sufficient, or any, periods of exclusivity before approving generic versions of our products, the sales of our products could be adversely affected. |
| · | If we fail to obtain or maintain Orphan Drug exclusivity for BIV201, we will have to rely on other potential marketing exclusivity and on our intellectual property rights. |
| · | We will need to raise substantial additional capital in the future to fund our operations, which could have a materially adverse effect on our business. |
| · | We have limited experience in drug development and may not be able to successfully develop any drugs, which would cause us to cease operations. |
| · | Development of pharmaceutical products is a time-consuming process, subject to a number of risks, many of which are outside of our control. |
| · | We may expend our limited resources to pursue a particular drug candidate or indication and fail to capitalize on drug candidates or indications that may be more profitable or for which there is a greater likelihood of success. |
| · | We have no manufacturing experience, and the failure to comply with all applicable manufacturing regulations and requirements could have a materially adverse effect on our business. |
| · | We do not currently have the sales and marketing personnel necessary to sell products, and the failure to hire and retain such staff could have a materially adverse effect on our business. |
| · | Even if we were to successfully develop approvable drugs, we will not be able to sell these drugs if we or our third-party manufacturers fail to comply with manufacturing regulations. |
| · | We must comply with significant and complex government regulations, compliance with which may delay or prevent the commercialization of our product candidates. |
| · | We may face business disruption and related risks if there is another pandemic. |
|
|
| · | The loss or unavailability of our management could put us at a competitive disadvantage. |
| · | We may not be able to attract and retain highly skilled personnel. |
| · | We may be unable to compete with enterprises in the highly competitive biotechnology and biopharmaceutical industries and those equipped with more substantial resources than us. |
| · | There may be conflicts of interest among our officers, directors and stockholders. |
| · | We indemnify our officers and directors against liability to us and our security holders, and such indemnification could increase our operating costs. |
Risks Relating to Our Intellectual Property
| · | We may be unable to obtain or protect intellectual property rights relating to our product candidates. |
| · | If we fail to comply with our obligations in the licensing and collaboration agreements, our competitive position, business, financial condition, results of operations and prospects could be harmed. |
| · | Compliance with federal regulations such as “march-in” rights may limit our exclusive rights and our ability to contract with non-U.S. manufacturers. |
| · | Patent terms may be inadequate to establish our competitive position on our drug candidates for an adequate amount of time. |
| · | We may not be able to protect our intellectual property rights throughout the world. |
| · | Changes in patent law could diminish the value of our patents and impair our ability to protect our drug candidate. |
| · | We may be involved in lawsuits to protect or enforce our patents or other intellectual property, which could be expensive, time-consuming and unsuccessful, and our patents could be found invalid or unenforceable. |
| · | Our failure to identify relevant third-party patents or correctly interpret the relevance, scope or expiration of patents, we may be subject to infringement claims or may not be able to develop our drug candidates. |
| · | Third parties may initiate legal proceedings alleging that we are infringing, misappropriating or otherwise violating their intellectual property rights. |
| · | We may be subject to claims by third parties asserting that we or our employees have infringed, misappropriated or otherwise violated their intellectual property rights, or claiming ownership of what we regard as our own intellectual property. |
| · | We may be subject to claims challenging the inventorship of our patents and other intellectual property. |
| · | Intellectual property rights do not necessarily address all potential threats. |
| · | Intellectual property litigation may lead to unfavorable publicity that harms our reputation and causes the market price of shares of our Common Stock to decline. |
Risks Relating to Our Common Stock
| · | Our stock price is and may continue to be volatile and you may not be able to resell our Common Stock at or above the price you paid. |
| · | You may experience future dilution as a result of future equity offerings or if we issue shares subject to options, warrants, stock awards or other arrangements. |
| · | Certain stockholder of the Company may have significant control over our Company. |
| · | The reverse stock split effected on August 6, 2024 may not result in positive outcomes. |
| · | The market price and trading volume of our Common Stock may be volatile. |
| · | The large number of restricted shares outstanding may reduce the market price of our Common Stock. |
| · | Any failure to maintain effective internal control over financial reporting could harm us. |
| · | Limited trading market for our Common Stock could make it difficult to liquidate an investment. |
| · | The lack of public company experience of our management team could negatively affect our business. |
| · | Investors may be less attracted to our Common Stock because we are as a smaller reporting company. |
| · | Additional audit and legal costs associated with periodic reporting requirements of the Securities Exchange Act of 1934, as amended (the “Exchange Act”) will negatively affect our ability to earn a profit. |
| · | Because we do not intend to pay any cash dividends on our Common Stock, our stockholders will not be able to receive a return on their shares unless they sell them. |
| · | We are authorized to issue “blank check” preferred stock without stockholder approval, which could adversely impact the rights of holders of our securities. |
| · | Provisions in our Articles of Incorporation, our Bylaws, and Nevada law might discourage, delay or prevent a change in control of our company or changes in our management and, therefore, depress the trading price of our Common Stock. |
|
|
Risks Relating to Our Business and Industry
We rely and will continue to rely on third parties to conduct our clinical trials. If these third parties do not successfully carry out their contractual duties or meet expected deadlines or do not successfully perform and comply with regulatory requirements, we may not be able to obtain regulatory approval of or commercialize our product candidates.
We depend, and will continue to depend, on third parties, including, but not limited to, contract research organizations (“CROs”), clinical trial sites and clinical trial principal investigators, contract laboratories, IRBs, manufacturers, suppliers, and other third parties to conduct our clinical trials, including those for our drug candidates bezisterim (NE3107) and BIV201. We rely heavily on these third parties over the course of our clinical trials, and we control only certain aspects of their activities. Nevertheless, we retain ultimate responsibility for ensuring that each of our studies is conducted in accordance with the protocol and applicable legal, regulatory, and scientific standards and regulations, and our reliance on third parties does not relieve us of our regulatory responsibilities. We and these third parties are required to comply with cGCPs, which are regulations and guidelines enforced by the FDA and comparable foreign regulatory authorities for the conduct of clinical trials on product candidates in clinical development. Regulatory authorities enforce cGCPs through periodic inspections and for-cause inspections of clinical trial principal investigators and trial sites. If, due to the failure of either the Company or a third party, a clinical trial fails to comply with applicable cGCPs, FDA’s IND requirements, other applicable regulatory requirements, or requirements set forth in the applicable IRB-approved protocol, the Company may be required to conduct additional clinical trials to support our marketing applications, which would delay the regulatory approval process. For example, our drug product candidate bezisterim (NE3107) was cleared by FDA for use in a Phase 3, randomized, double blind, placebo controlled, parallel group, multicenter study in subjects who have mild to moderate AD. Enrollment in that trial began in August 2021, with a planned primary completion in late 2022/early 2023. On November 29, 2023, the Company announced topline efficacy data from its Phase 3 clinical trial (NCT04669028) of bezisterim (NE3107) in the treatment of mild to moderate AD. Upon trial completion, as the Company began the process of analyzing the trial data, the Company found significant deviations from the protocol and cGCP violations at 15 study sites (virtually all of which were from one geographic area). This highly unusual level of suspected improprieties led the Company to exclude all patients from these sites. We subsequently notified FDA’s OSI of such significant deviations from study protocol, the suspected improprieties, and the study sites involved. The identification of significant deviations from study protocol and numerous GCP violations at multiple study sites raised questions regarding the validity and robustness of data from these study sites. The unplanned exclusion of so many patients left the trial underpowered for its primary endpoints. However, based on the remaining dataset from those other sites determined to be in compliance with the protocol and GCP’s, a preliminary signal of efficacy was detected. The Company is considering: (1) employing the adaptive trial feature of the protocol to continue enrolling patients to achieve statistical significance; and/or (2) designing a new Phase 3 study of bezisterim (NE3107) that leverages the most recent scientific literature relating to AD along with the company's understanding regarding the effects of bezisterim (NE3107) in persons with mild-moderate AD.
Although we design the clinical trials for our product candidates, our CROs are tasked with facilitating and monitoring these trials. As a result, many aspects of our clinical development programs, including site and investigator selection, and the conduct, timing, and monitoring of the study, is outside our direct control, either partially or in whole. Our reliance on third parties to conduct clinical trials also results in less direct control over the collection, management, and quality of data developed through clinical trials than would be the case if we were relying entirely upon our own employees. Communicating with third parties can also be challenging, potentially leading to mistakes as well as difficulties in coordinating activities. Our business may be impacted if any of these third parties violates applicable federal, state, or foreign laws and/or regulations, including but not limited to FDA’s IND regulations, cGCPs, fraud and abuse or false claims laws, healthcare privacy and data security laws, or provide us or government agencies with inaccurate, misleading, or incomplete data.
Successful development of biopharmaceuticals is highly uncertain and is dependent on numerous factors, many of which are beyond our control.
Product candidates that appear promising in the early phases of development may fail to reach the market for several reasons. Pre-clinical study results may show the product candidate to be less effective than desired (e.g., the study failed to meet its primary endpoints) or to have harmful or problematic side effects. Product candidates may fail to receive the necessary regulatory approvals or may be delayed in receiving such approvals. Among other things, such delays may be caused by slow enrollment in clinical studies; length of time to achieve study endpoints; additional time requirements for data analysis; IND and later new drug application preparation; discussions with the FDA; an FDA request for additional pre-clinical or clinical data; unexpected safety or manufacturing issues; manufacturing costs; pricing or reimbursement issues; clinical sites deviating from the trial protocol, committing scientific misconduct, or other violations of regulatory requirements - which can render data from those sites unusable in support of regulatory approval; or other factors that make the product not economical. Proprietary rights of others and their competing products and technologies may also prevent the product from being commercialized.
|
|
Success in pre-clinical and early clinical studies does not ensure that large-scale clinical studies will be successful. Clinical results are frequently susceptible to varying interpretations that may delay, limit or prevent regulatory approvals. The length of time necessary to complete clinical studies and to submit an application for marketing approval for a final decision by a regulatory authority varies significantly from one product to the next, and may be difficult to predict. There can be no assurance that any of our products will develop successfully, and the failure to develop our products will have a materially adverse effect on our business and will cause you to lose all of your investment.
The concentration of our assets within a certain financial institution could have a material adverse effect on its business, financial condition and results of operations.
As of August 30, 2024, the Company had cash deposited in a certain financial institution in excess of federally insured levels. The Company regularly monitors the financial stability of these financial institutions and believes that it is not exposed to any significant credit risk in cash and cash equivalents. Bank failures, events involving limited liquidity, defaults, non-performance, or other adverse developments that affect financial institutions, or concerns or rumors about such events, may lead to liquidity constraints. In 2023, certain U.S. government banking regulators took steps to intervene in the operations of certain financial institutions due to liquidity concerns, which caused general heightened uncertainties in financial markets. While previous bank failures have not had a material direct impact on the Company’s operations, if further liquidity and financial stability concerns arise with respect to banks and financial institutions, either nationally or in specific regions, the Company’s ability to access cash or enter into new financing arrangements may be threatened, which could have a material adverse effect on its business, financial condition and results of operations.
We are currently subject to securities class action litigation and may be subject to similar or other litigation in the future, all of which will require significant management time and attention, result in significant legal expenses and may result in unfavorable outcomes, which may have a material adverse effect on our business, operating results and financial condition, and negatively affect the price of our Common Stock.
We are, and may in the future become, subject to various legal proceedings and claims that arise in or outside the ordinary course of business. For example, On January 19, 2024, a purported shareholder class action complaint, captioned Eric Olmstead v. BioVie Inc. et al., No. 3:24-cv-00035, was filed in the U.S. District Court for the District of Nevada, naming the Company and certain of its officers as defendants. On February 22, 2024, a second, related putative securities class action was filed in the same court asserting similar claims against the same defendants, captioned Way v. BioVie Inc. et al., No. 2:24-cv-00361. On April 15, 2024, the court consolidated these two actions under the caption In re BioVie Inc. Securities Litigation, No. 3:24-cv-00035, appointed the lead plaintiff, and approved selection of the lead counsel. On June 21, 2024, the lead plaintiff filed an amended complaint, alleging that the defendants made material misrepresentations and/or omissions of material fact relating to the Company’s business, operations, compliance, and prospects, including information related to the NM101 Phase 3 study and trial of bezisterim (NE3107) in mild to moderate probable Alzheimer’s Disease, in violation of Sections 10(b) and 20(a) of the Exchange Act, and Rule 10b-5 promulgated thereunder. The class action is on behalf of purchasers of the Company’s securities during the period from December 7, 2022 through November 28, 2023 and seeks unspecified monetary damages on behalf of the putative class and an award of costs and expenses, including attorney’s fees. The defendants filed a motion to dismiss the amended complaint on August 21, 2024. The defendants believe that the claims are without merit and intend to defend vigorously against them, but there can be no assurances as to the outcome.
It is possible that additional lawsuits will be filed, or allegations received from stockholders, with respect to these same or other matters and also naming us and/or our officers and directors as defendants. Such lawsuits and any other related lawsuits are subject to inherent uncertainties, and the actual defense and disposition costs will depend upon many unknown factors. The outcome of such lawsuits is necessarily uncertain. We could be forced to expend significant resources in the defense of the pending lawsuit and any additional lawsuits, and we may not prevail. In addition, we may incur substantial legal fees and costs in connection with such lawsuits. We currently are not able to estimate the possible cost to us from this matter, as the pending lawsuit is currently at an early stage, and we cannot be certain how long it may take to resolve the pending lawsuit or the possible amount of any damages that we may be required to pay. Monitoring, initiating and defending against legal actions is time-consuming for our management, is likely to be expensive and may detract from our ability to fully focus our internal resources on our business activities. We could be forced to expend significant resources in the settlement or defense of the pending lawsuit and any potential future lawsuits, and we may not prevail in such lawsuits.
|
|
Although we have insurance coverage that we believe applies to these actions, the coverage is subject to a $2 million deductible. That means that we are responsible for the first $2 million of loss arising from these actions, which includes both defense costs and damages, before any insurance coverage will apply. Furthermore, our insurance coverage may be insufficient, and our assets may be insufficient to cover any amounts that exceed our insurance coverage, and we may have to pay damage awards or otherwise may enter into a settlement arrangement in connection with such claim. A decision adverse to our interests in the pending lawsuit, or in similar or related litigation, could result in the payment of substantial damages, or possibly fines, and could have a material adverse effect on our business, our stock price, cash flow, results of operations and financial condition. We have not established any reserve for any potential liability relating to the pending lawsuit or any potential future lawsuits. Any such payments or settlement arrangements in current or future litigation could have a material adverse effect on our business, operating results or financial condition. In addition, such lawsuits may make it more difficult to finance our operations and affect our ability to make payments for damages.
We have no products approved for commercial sale, have never generated any revenues and may never achieve revenues or profitability, which could cause us to cease operations.
We have no products approved for commercial sale and, to date, we have not generated any revenue. Our ability to generate revenue depends heavily on (a) successful completion of one or more development programs demonstrating in human clinical trials that BIV201 and bezisterim (NE3107), our product candidates, are safe and effective; (b) our ability to seek and obtain regulatory approvals, including, without limitation, with respect to the indications we are seeking; (c) successful commercialization of our product candidates; and (d) market acceptance of our products. There are no assurances that we will achieve any of the forgoing objectives. Furthermore, our product candidates are in the development stage, and have not been fully evaluated in human clinical trials. If we do not successfully develop and commercialize our product candidates we will not achieve revenues or profitability in the foreseeable future, if at all. If we are unable to generate revenues or achieve profitability, we may be unable to continue our operations.
We are a development stage company with a limited operating history, making it difficult for you to evaluate our business and your investment.
Although our Company was incorporated on April 10, 2013, we are a development stage biopharmaceutical company with potential therapies that have not been fully evaluated in clinical trials, and our operations are subject to all of the risks inherent in the establishment of a new business enterprise, including but not limited to the absence of an operating history, the lack of commercialized products, insufficient capital, expected substantial and continual losses for the foreseeable future, limited experience in dealing with regulatory issues, the lack of manufacturing experience and limited marketing experience, possible reliance on third parties for the development and commercialization of our proposed products, a competitive environment characterized by numerous, well-established and well capitalized competitors and reliance on key personnel.
Since inception, we have not established any revenues or operations that would provide financial stability in the long term, and there can be no assurance that we will realize our plans on our projected timetable in order to reach sustainable or profitable operations.
Investors are subject to all the risks incident to the creation and development of a new business and each investor should be prepared to withstand a complete loss of his, her or its investment. Furthermore, the accompanying financial statements have been prepared assuming that we will continue as a going concern. We have not emerged from the development stage, and may be unable to raise further equity. These factors raise substantial doubt about our ability to continue as a going concern. The financial statements included elsewhere in this Form 10-K do not include any adjustments that might result from the outcome of this uncertainty.
Because we are subject to these risks, you may have a difficult time evaluating our business and your investment in our Company. Our ability to become profitable depends primarily on our ability to develop drugs, to obtain approval for such drugs, and if approved, to successfully commercialize our drugs, our research and development (“R&D”) efforts, including the timing and cost of clinical trials; and our ability to enter into favorable alliances with third-parties who can provide substantial capabilities in clinical development, regulatory affairs, sales, marketing and distribution.
Even if we successfully develop and market BIV201 and/or bezisterim (NE3107), we may not generate sufficient or sustainable revenue to achieve or sustain profitability, which could cause us to cease operations and cause you to lose all of your investment.
If the FDA or comparable foreign regulatory authorities approve generic versions of any of our product candidates that receive marketing approval, or such authorities do not grant our products sufficient, or any, periods of exclusivity before approving generic versions of our products, the sales of our products could be adversely affected.
|
|
Once a NDA is approved, the product covered thereby becomes a “reference listed drug” (“RLD”), in the FDA’s publication, “Approved Drug Products with Therapeutic Equivalence Evaluations,” commonly known as the Orange Book. Other manufacturers may seek approval of generic versions of reference listed drugs through submission of abbreviated new drug applications (“ANDAs”) in the United States. In support of an ANDA, a generic manufacturer need not conduct clinical trials. Rather, the applicant generally must show that its product has the same active ingredient(s), dosage form, strength, route of administration and conditions of use or labeling as the reference listed drug and that the generic version is bioequivalent to the reference listed drug, meaning it is absorbed in the body at the same rate and to the same extent as the RLD. Generic products may be significantly less costly to bring to market than the reference listed drug and companies that produce generic products are generally able to offer them at lower prices. Moreover, generic versions of RLDs are often automatically substituted for the RLD by pharmacies when dispensing a prescription written for the RLD. Thus, following the introduction of a generic drug, a significant percentage of the sales of any branded product or reference listed drug is typically lost to the generic product.
The FDA may not approve an ANDA for a generic product until any applicable period of non-patent exclusivity for the reference listed drug has expired. The FDCA provides a period of five years of non-patent exclusivity for a new drug containing a new chemical entity (“NCE”). An NCE is an active ingredient that has not previously been approved by FDA in any other NDA. Specifically, in cases where such exclusivity has been granted, an ANDA may not be submitted to the FDA until the expiration of five years unless the submission is accompanied by a Paragraph IV certification that a patent covering the reference listed drug is either invalid or will not be infringed by the generic product, in which case the applicant may submit its application four years following approval of the reference listed drug. If an ANDA is submitted to FDA with a Paragraph IV Certification, the generic applicant must also provide a “Paragraph IV Notification” to the holder of the NDA for the RLD and to the owner of the listed patent(s) being challenged by the ANDA applicant, providing a detailed written statement of the basis for the ANDA applicant’s position that the relevant patent(s) is invalid or would not be infringed. If the patent owner brings a patent infringement lawsuit against the ANDA applicant within 45 days of the Paragraph IV Notification, FDA approval of the ANDA will be automatically stayed for 30 months, or until 7-1/2 years after the NDA approval if the generic application was filed between 4 years and 5 years after the NDA approval. Any such stay will be terminated earlier if the court rules that the patent is invalid or would not be infringed.
Competition that our products may face from generic versions of our products could materially and adversely impact our future revenue, profitability and cash flows and substantially limit our ability to obtain a return on the investments we have made in those product candidates.
If we fail to obtain or maintain Orphan Drug exclusivity for BIV201, we will have to rely on other potential marketing exclusivity, and on our intellectual property rights, which may reduce the length of time that we can prevent competitors from selling generic versions of BIV201.
We have obtained Orphan Drug Designation for BIV201 (terlipressin) in the U.S. for the treatment of hepatorenal syndrome on November 21, 2018 and treatment of ascites due to all etiologies except cancer on September 8, 2016. Under the Orphan Drug Act, the FDA may designate a product as an Orphan Drug if it is a drug intended to treat a rare disease or condition, defined, in part, as a patient population of fewer than 200,000 in the U.S. In the European Union (“EU”), Orphan Drug designation may be granted to drugs intended to treat, diagnose or prevent a life-threatening or chronically debilitating disease having a prevalence of no more than five in 10,000 people in the EU, and which meet other specified criteria. The company that first obtains FDA approval for a designated Orphan Drug for the associated rare disease may receive a seven-year period of marketing exclusivity during which time FDA may not approve another application for the same drug for the same orphan disease or condition. Orphan Drug Exclusivity does not prevent FDA approval of another application for the same drug for a different disease or condition, or of an application for a different drug for the same rare disease or condition. Orphan Drug exclusive marketing rights may be lost under several circumstances, including a later determination by the FDA that the request for designation was materially defective or if the manufacturer is unable to assure sufficient quantity of the drug. Similar regulations are available in the EU with a ten-year period of market exclusivity.
Even though BioVie has obtained two Orphan Drug Designations for its lead product candidate, terlipressin, for treatment of ascites and for treatment of hepatorenal syndrome, and may seek other Orphan Drug Designations for BIV201, and Orphan Drug Designation for other product candidates, there is no assurance that BioVie will be the first to obtain marketing approval for any particular rare indication. Further, even though BioVie has obtained Orphan Drug Designations for its lead product candidate, or even if BioVie obtains Orphan Drug Designation for other potential product candidates, such designation may not effectively protect BioVie from competition because different drugs can be approved for the same condition and the same drug can be approved for different conditions and potentially used off-label in the Orphan indication. Even after an Orphan Drug is approved, the FDA can subsequently approve another competing drug with the same active ingredient for the same condition for several reasons, including, if the FDA concludes that the later drug is clinically superior due to being safer or more effective or because it makes a major contribution to patient care. Orphan Drug Designation neither shortens the development time or regulatory review time of a drug, nor gives the drug any advantage in the regulatory review or approval process.
|
|
In addition, other companies have received Orphan Drug designations for terlipressin. Mallinckrodt Hospital Products IP Limited received Orphan Drug designation in 2004 for terlipressin for the treatment of Hepatorenal Syndrome. Mallinckrodt has already gained FDA approval for its product, lyophilized terlipressin acetate for bolus intravenous administration for the treatment of hepatorenal syndrome Type 1 in September 2022. PharmaIN Corporation received Orphan Drug Designation in 2012 for PGC-C12E-terlipressin for treatment of ascites due to all etiologies except cancer. In addition, Ferring Pharmaceuticals Inc. received Orphan Drug designation in 1986 for terlipressin for the treatment of bleeding esophageal varices. If one of those or any other company with Orphan Drug Designation for the same drug as ours for the same proposed disease or condition receives FDA approval and Orphan Drug Exclusivity before our product is approved, approval of our drug(s) for the orphan indication may be blocked for seven years by the other company’s Orphan Exclusivity and they may obtain a competitive advantage even after the exclusivity period expires associated with being the first to market.
We will need to raise substantial additional capital in the future to fund our operations and we may be unable to raise such funds when needed and on acceptable terms, which could have a materially adverse effect on our business.
Developing biopharmaceutical products, including conducting pre-clinical studies and clinical trials and establishing manufacturing capabilities, requires substantial funding. Additional financing will be required to fund the research and development of our product candidates. We have not generated any product revenues, and do not expect to generate any revenues until, and only if, we develop, and receive approval to sell our product candidates from the FDA and other regulatory authorities for our product candidates.
We may not have the resources to complete the development and commercialization of any of our proposed product candidates. We will require additional financing to further the clinical development of our product candidates. In the event that we cannot obtain the required financing, we will be unable to complete the development necessary to file an NDA with the FDA for BIV201 or bezisterim (NE3107). This will delay or require termination of research and development programs, preclinical studies and clinical trials, material characterization studies, regulatory processes, the establishment of our own laboratory or a search for third party marketing partners to market our products for us, which could have a materially adverse effect on our business.
The amount of capital we may need will depend on many factors, including the progress, timing and scope of our research and development programs, the progress, timing and scope of our preclinical studies and clinical trials, the time and cost necessary to obtain regulatory approvals, the time and cost necessary to establish our own marketing capabilities or to seek marketing partners, the time and cost necessary to respond to technological and market developments, changes made or new developments in our existing collaborative, licensing and other commercial relationships, and new collaborative, licensing and other commercial relationships that we may establish.
Until we can generate a sufficient amount of product revenue, if ever, we expect to finance future cash needs through public or private equity offerings, debt financings, or corporate collaboration and licensing arrangements. Additional funds may not be available when we need them on terms that are acceptable to us, or at all. If adequate funds are not available, we may be required to delay, reduce the scope of, or eliminate one or more of our research or development programs or our commercialization efforts. In addition, we could be forced to discontinue product development and reduce or forego attractive business opportunities. To the extent that we raise additional funds by issuing equity securities, our stockholders may experience additional significant dilution, and debt financing, if available, may involve restrictive covenants. To the extent that we raise additional funds through collaboration and licensing arrangements, it may be necessary to relinquish some rights to our technologies or our product candidates or grant licenses on terms that may not be favorable to us. We may seek to access the public or private capital markets whenever conditions are favorable, even if we do not have an immediate need for additional capital at that time.
Our fixed expenses, such as rent and other contractual commitments, will likely increase in the future, as we may enter into leases for new facilities and capital equipment and/or enter into additional licenses and collaborative agreements. Therefore, if we fail to raise substantial additional capital to fund these expenses, we could be forced to cease operations, which could cause you to lose all of your investment.
We have limited experience in drug development and may not be able to successfully develop any drugs, which would cause us to cease operations.
|
|
We have never successfully developed a new drug and brought it to market. Our management and clinical teams have experience in drug development but they may not be able to successfully develop any drugs. Our ability to achieve revenues and profitability in our business will depend on, among other things, our ability to develop products internally or to obtain rights to them from others on favorable terms; complete laboratory testing and human studies; obtain and maintain necessary intellectual property rights to our products; successfully complete regulatory review to obtain requisite governmental agency approvals; enter into arrangements with third parties to manufacture our products on our behalf; and enter into arrangements with third parties to provide sales and marketing functions. If we are unable to achieve these objectives we will be forced to cease operations and you will lose all of your investment.
Development of pharmaceutical products is a time-consuming process, subject to a number of risks, many of which are outside of our control. Consequently, we can provide no assurance that our product candidates will obtain regulatory approval, and if we are unsuccessful or fail to timely develop new drugs, we could be forced to discontinue our operations.
Development and extensive testing will be required to determine the technical feasibility and commercial viability of BIV201 and bezisterim (NE3107). Our success will depend on our ability to achieve scientific and technological advances and to translate such advances into reliable, commercially competitive drugs on a timely basis. Drugs that we may develop are not likely to be commercially available, at a minimum, for several years, if ever. Our drug product candidate, BIV201 (continuous infusion terlipressin), was cleared by the FDA to undergo testing in a mid-stage (Phase 2b) clinical trial for the treatment of refractory ascites due to cirrhosis. On June 24, 2021, we announced that the first patient has been enrolled in this study. In March 2023, the open-label trial was stopped after 15 of the planned 30 patients were enrolled, and an evaluation of those completed patients assessed. Encouraging data from these patients appeared to show that treatment with BIV201 plus SOC resulted in a reduction in ascites fluid accumulation during treatment versus pre-treatment. In June 2023, the Company requested and subsequently received guidance from the FDA regarding the design and endpoints for definitive clinical testing of BIV201 for the treatment of ascites due to chronic liver cirrhosis. Over three years since the initial enrollment of this clinical trial, the Company is continuing to finalize protocol designs for the Phase 3 study of BIV201 for the treatment of ascites due to chronic liver cirrhosis.
The proposed development schedules for our product candidates may be affected by a variety of factors, including technological difficulties, proprietary technology of others, and changes in government regulation, many of which will not be within our control. In June 2021, FDA approved the drug aducanumab for treatment of Alzheimer’s despite a strong recommendation against approval from an FDA advisory committee. That FDA approval has generated significant medical and political controversy, including a Congressional investigation, announced on June 25, 2021, into the basis for FDA’s approval decision. That investigation, other potential investigations, and negative publicity of FDA’s approval decision could adversely impact the agency’s oversight of our clinical development program, how the agency may view and act upon any NDA we may file for bezisterim (NE3107), and the commercial viability of bezisterim (NE3107) if it were to be approved and marketed.
Any delay or further delay in the development, introduction or marketing of our product candidates could result either in such drugs being marketed at a time when their cost and performance characteristics would not be competitive in the marketplace or in the shortening of their commercial lives. In light of the long-term nature of our projects and other risk factors described elsewhere in this document, we may not be able to successfully complete the development or marketing of any drugs, which could cause us to cease operations.
From time to time, the FDA may have feedback on our clinical trial designs, including for example certain of our endpoints and outcome measures. As a result, we may consider revisions to our protocols which may delay progress in implementing our trials. We may fail to successfully develop and commercialize our product candidate(s) if it is found to be unsafe or ineffective in clinical trials; does not receive necessary approval from the FDA or foreign regulatory agencies; fails to conform to a changing standard of care for the disease it seeks to treat; or is less effective or more expensive than current or alternative treatment methods.
|
|
Drug development failure can occur at any stage of clinical trials and as a result of many factors, there can be no assurance that we or our collaborators will reach our anticipated clinical targets. Even if the trials are successfully completed, clinical data are often susceptible to varying interpretations and analyses, and we cannot guarantee that the FDA or comparable foreign regulatory authorities will interpret the results as we do, and more trials could be required before we submit our product candidates for approval. We cannot guarantee that the FDA or comparable foreign regulatory authorities will view our product candidates as having efficacy even if positive results are observed in clinical trials. In some instances, there can be significant variability in safety or efficacy results between different clinical trials of the same product candidate due to numerous factors, including changes in trial procedures set forth in protocols, differences in the size and type of the patient populations, changes in and adherence to the clinical trial protocols, and the rate of dropout among clinical trial participants. If the results of our ongoing or future clinical trials are inconclusive with respect to the efficacy of our product candidates, if we do not meet the clinical endpoints with statistical and clinically meaningful significance, or if there are safety concerns associated with our product candidates, we may be delayed in obtaining marketing approval, if at all. Additionally, any safety concerns observed in any one of our clinical trials in our targeted indications could limit the prospects for regulatory approval of our product candidates in those and other indications. We also do not know what the long-term effects of exposure to our product candidates will be. Furthermore, our product candidates may be used in combination with other treatments and there can be no assurance that such use will not lead to unique or unexpected safety issues.
Failure to complete clinical trials or to prove that our product candidates are safe and effective would have a material adverse effect on our ability to generate revenue and could require us to reduce the scope of or discontinue our operations, which could cause you to lose all of your investment.
We may expend our limited resources to pursue a particular drug candidate or indication and fail to capitalize on drug candidates or indications that may be more profitable or for which there is a greater likelihood of success.
Because we have limited human capital and financial resources, we focus on research programs and drug candidates that we identify for specific indications. As a result, we may forego or delay pursuit of opportunities with other drug candidates or for other indications that later prove to have greater commercial potential. Our resource allocation decisions may cause us to fail to capitalize on viable commercial drugs or profitable market opportunities. Our spending on current and future research and development programs and drug candidates for specific indications may not yield any commercially viable drugs. If we do not accurately evaluate the commercial potential or target market for a particular drug candidate, we may relinquish valuable rights to that drug candidate through collaboration, licensing or other royalty arrangements in cases in which it would have been more advantageous for us to retain sole development and commercialization rights to such drug candidate.
At any time and for any reason, we may determine that one or more of our discovery programs or preclinical or clinical drug candidates or programs does not have sufficient potential to warrant the allocation of resources toward such program or drug candidate. Accordingly, we may choose not to develop a potential drug candidate or elect to suspend, deprioritize or terminate one or more of our discovery programs or preclinical or clinical drug candidates or programs. For example, BIV201 has received Orphan Drug designation for HRS. On June 23, 2021, we announced that FDA has provided guidance on our planned Phase 3 clinical trial of BIV201 in (HRS-AKI) and have since reached agreement on the key elements of the trial design. Thereafter, we deprioritized HRS-AKI program to focus on bezisterim (NE3107). When we suspend, deprioritize or terminate a program or drug candidate in which we have invested significant resources, we will have expended resources on a program that will not provide a full return on our investment and may have missed the opportunity to have allocated those resources to potentially more productive uses, including existing or future programs or drug candidates.
We have no manufacturing experience, and the failure to comply with all applicable manufacturing regulations and requirements could have a materially adverse effect on our business.
We have never manufactured products in the highly regulated environment of pharmaceutical manufacturing, and our team has limited experience in the manufacture of drug therapies. There are numerous regulations and requirements that must be maintained to obtain licensure and permitting required prior to the commencement of manufacturing, as well as additional requirements to continue manufacturing pharmaceutical products. We currently do not own or lease facilities that could be used to manufacture any products that might be developed by us, and have contracted with an experienced Contract Manufacturing Organization (“CMO”) to perform the manufacturing of our new product candidates BIV201 and bezisterim (NE3107). In addition, we do not have the resources at this time to acquire or lease suitable facilities. If we or our CMO fail to comply with regulations, to obtain the necessary licenses and knowhow or to obtain the requisite financing in order to comply with all applicable regulations and to own or lease the required facilities in order to manufacture our products, we could be forced to cease operations, which would cause you to lose all of your investment.
|
|
In addition, the FDA and other regulatory authorities require that product candidates and drug products be manufactured according to cGMP. Any failure by our third-party manufacturers to comply with cGMP could lead to a shortage of BIV201 and NE3107. In addition, such failure could be the basis for action by the FDA to withdraw approval, if granted to us, and for other regulatory enforcement action, including Warning Letters, product seizure, injunction or other civil or criminal penalties.
BIV201 and bezisterim (NE3107) and any other product candidates that we develop may have to compete with other products and product candidates for access to manufacturing facilities. There are a limited number of manufacturers that operate under cGMP regulations and that are both capable of manufacturing for us and willing to do so. If we need to find another source of drug substance or drug product manufacturing for BIV201 and bezisterim (NE3107), we may not be able to identify, or reach agreement with, commercial-scale manufacturers on commercially reasonably terms, or at all. If we are unable to do so, we will need to develop our own commercial-scale manufacturing capabilities, which would: impact commercialization of BIV201 and bezisterim (NE3107) in the U.S. and other countries where it may be approved; require a capital investment by us that could be quite costly; and increase our operating expenses.
If our existing third-party manufacturers, or the third parties that we engage in the future to manufacture a product for commercial sale or for our clinical trials, should cease to continue to do so for any reason, we likely would experience significant delays in obtaining sufficient quantities of product for us to meet commercial demand or to advance our clinical trials while we identify and qualify replacement suppliers. If for any reason we are unable to obtain adequate supplies of BIV201 or any other product candidate that we develop, or the drug substances used to manufacture it, it will be more difficult for us to compete effectively, generate revenue, and further develop our products. In addition, if we are unable to assure a sufficient quantity of the drug for patients with rare diseases or conditions, we may lose any Orphan Drug exclusivity to which the product otherwise would be entitled.
We do not currently have the sales and marketing personnel necessary to sell products, and the failure to hire and retain such staff could have a materially adverse effect on our business.
We are an early stage development company with limited resources. Even if we had products available for sale, which we currently do not, we have not secured sales and marketing staff at this early stage of operations to sell products. We cannot generate sales without sales or marketing staff and must rely on others to provide any sales or marketing services until such personnel are secured, if ever. If we fail to hire and retain the requisite expertise in order to market and sell our products or fail to raise sufficient capital in order to afford to pay such sales or marketing staff, then we could be forced to cease operations and you could lose all of your investment.
Even if we were to successfully develop approvable drugs, we will not be able to sell these drugs if we or our third-party manufacturers fail to comply with manufacturing regulations, which could have a materially adverse effect on our business.
If we were to successfully develop approvable drugs, before we can begin selling these drugs, we must obtain regulatory approval of our manufacturing facility and process or the manufacturing facility and process of the third party or parties with whom we may outsource our manufacturing activities. In addition, the manufacture of our products must comply with the FDA’s current Good Manufacturing Practices regulations, commonly known as GMP regulations. The GMP regulations govern quality control and documentation policies and procedures. Our manufacturing facilities, if any in the future, and the manufacturing facilities of our third-party manufacturers will be continually subject to inspection by the FDA and other state, local and foreign regulatory authorities, before and after product approval. We cannot guarantee that we, or any potential third-party manufacturer of our products, will be able to comply with the GMP regulations or other applicable manufacturing regulations. The failure to comply with all necessary regulations would have a materially adverse effect on our business and could force us to cease operations and you could lose all of your investment.
We must comply with significant and complex government regulations, compliance with which may delay or prevent the commercialization of our product candidates, which could have a materially adverse effect on our business.
The R&D, manufacture and marketing of drug product candidates are subject to regulation, primarily by the FDA in the United States and by comparable authorities in other countries. These national agencies and other federal, state, local and foreign entities regulate, among other things, R&D activities (including testing in animals and in humans) and the testing, manufacturing, handling, labeling, storage, record keeping, approval, advertising and promotion of the product that we are developing. Noncompliance with applicable requirements can result in various adverse consequences, including approval delays or refusals to approve drug licenses or other applications, suspension or termination of clinical investigations, revocation of approvals previously granted, warning letters, fines, criminal prosecution, recalls or seizures of products, injunctions against shipping drugs and total or partial suspension of production and/or refusal to allow a company to enter into governmental supply contracts.
|
|
The process of obtaining FDA approval is costly and time consuming. Current FDA requirements for a new human drug or biological product to be marketed in the United States include, among other things: (a) the successful conclusion of pre-clinical laboratory and animal tests, if appropriate, to gain preliminary information on the product’s safety; (b) filing with the FDA of an IND application to conduct human clinical trials for drugs or biologics; (c) the successful completion of adequate and well-controlled human clinical investigations to establish the safety and efficacy of the product for its recommended use; and (d) filing by a company and acceptance and approval by the FDA of a NDA for a drug product or a BLA for a biological product to allow commercial distribution of the drug or biologic. A delay in one or more of the procedural steps outlined above could be harmful to us in terms of getting our product candidates through clinical testing and to market, which could have a materially adverse effect on our business.
The FDA, clinical investigators, Data Safety Monitoring Boards, and IRBs review the ongoing conduct of, and emerging safety information from, clinical trials and may order the temporary or permanent discontinuation of clinical trials at any time if it believes the product candidate exposes clinical subjects to an unacceptable health risk. Investigational drugs used in clinical studies must be produced in compliance with cGMP rules pursuant to FDA regulations.
Development, approval, and sales outside the United States of products that we develop will also be subject to regulatory requirements governing human clinical trials and marketing for drugs and biological products and devices. The requirements vary widely from country to country, but typically the registration and approval process takes several years and requires significant resources.
If we experience delays or discontinuations of our clinical trials by the FDA or comparable authorities in other countries, or if we fail to obtain registration or other approvals of our products or devices then we could be forced to cease our operations and you will lose all of your investment.
Even if we are successful in developing BIV201 and bezisterim (NE3107), our product candidates, we have limited experience in conducting or supervising clinical trials that must be performed to obtain data to submit in concert with applications for approval by the FDA. The regulatory process to obtain approval for drugs for commercial sale involves numerous steps. Drugs are subjected to clinical trials that allow development of case studies to examine safety, efficacy, and other issues to ensure that sale of drugs meets the requirements set forth by various governmental agencies, including the FDA. In the event that our protocols do not meet standards set forth by the FDA, or that our data is not sufficient to allow such trials to validate our drugs in the face of such examination, we might not be able to meet the requirements that allow our drugs to be approved for sale which could have a materially adverse effect on our business.
We depend upon our management and their loss or unavailability could put us at a competitive disadvantage which could have a material adverse effect on our business.
We currently depend upon the efforts and abilities of our executive management team of Cuong Do, our Chief Executive Officer–President; Wendy Kim, our Chief Financial Officer; Dr Joseph Palumbo, our Executive Vice President–Chief Medical Officer; Penelope Markham, our Senior Vice Ascites Programs & Strategic Initiatives–; Chris Reading, our Senior Vice President–Alzheimer’s Disease Program; Clarence Ahlem, our Senior Vice President – Operations President–Operations, Discovery and Parkinson’s Disease Program; ; and David Morse, our Senior Vice President–Chief Regulatory Officer; who all serve the Company full-time. The loss or unavailability of the services of any of these individuals for any significant period of time could have a material adverse effect on our business, prospects, financial condition and results of operations which may cause you to lose all of your investment. We have not obtained, do not own, nor are we the beneficiary of key-person life insurance.
We may not be able to attract and retain highly skilled personnel, which could have a materially adverse effect on our business.
Our ability to attract and retain highly skilled personnel is critical to our operations and expansion. We face competition for these types of personnel from other pharmaceutical companies and more established organizations, many of which have significantly larger operations and greater financial, technical, human and other resources than us. We may not be successful in attracting and retaining qualified personnel on a timely basis, on competitive terms, or at all. If we are not successful in attracting and retaining these personnel, our business, prospects, financial condition and results of operations will be materially and adversely affected.
The biotechnology and biopharmaceutical industries are characterized by rapid technological developments and a high degree of competition. We may be unable to compete with enterprises equipped with more substantial resources than us, which could cause us to curtail or cease operations.
|
|
The biotechnology and biopharmaceutical industries are characterized by rapid technological developments and a high degree of competition based primarily on scientific and technological factors. These factors include the availability of patent and other protection for technology and products, the ability to commercialize technological developments and the ability to obtain government approval for testing, manufacturing and marketing.
We compete with biopharmaceutical firms in the United States, Europe and elsewhere, as well as a growing number of large pharmaceutical companies that are applying biotechnology to their operations. Many biopharmaceutical companies have focused their development efforts in the human therapeutics area. Many major pharmaceutical companies have developed or acquired internal biotechnology capabilities or made commercial arrangements with other biopharmaceutical companies. These companies, as well as academic institutions, government agencies and private research organizations, also compete with us in recruiting and retaining highly qualified scientific personnel and consultants. Our ability to compete successfully with other companies in the pharmaceutical field will also depend to a considerable degree on the continuing availability of capital to us.
Although there are not currently any therapies approved by the FDA specifically for the treatment of ascites due to liver cirrhosis, we still face significant competitive and market risk. Other companies, such as Ocelot Bio, are developing therapies for severe complications of advanced liver cirrhosis, which may in the future be developed for the treatment of ascites, and these therapies could compete indirectly or directly with our product candidate. Similarly, other companies, such as Biogen and Eli Lilly, are developing treatments for AD and PD, which could compete indirectly or directly with our product candidate. There may be other competitive development programs of which we are unaware. Even if our product candidates are ultimately approved by the FDA, there is no guarantee that once it is on the market doctors will adopt them in favor of current ascites treatment procedures such as diuretics and paracentesis with respect to BIV201 and AD and PD with respect to bezisterim (NE3107). These competitive and market risks could have a material adverse effect on our business, prospects, financial condition and results of operations which may cause you to lose all of your investment.
Our competition will be determined in part by the potential indications for which drugs are developed and ultimately approved by regulatory authorities. Additionally, the timing of the market introduction of some of our potential product candidate or of competitors’ products may be an important competitive factor. Accordingly, the relative speed with which we can develop drugs, complete pre-clinical testing, clinical trials, approval processes and supply commercial quantities to market are important competitive factors. We expect that competition among drugs approved for sale will be based on various factors, including product efficacy, safety, reliability, availability, price and patent protection.
The successful development of biopharmaceuticals is highly uncertain. A variety of factors including, pre-clinical study results or regulatory approvals, could cause us to abandon the development of our product candidates.
There may be conflicts of interest among our officers, directors and stockholders.
Certain of our executive officers and directors and their affiliates are engaged in other activities and have interests in other entities on their own behalf or on behalf of other persons. Neither we nor any of our shareholders will have any rights in these ventures or their income or profits. In particular, our executive officers or directors or their affiliates may have an economic interest in or other business relationship with partner companies that invest in us or are engaged in competing drug development. Our executive officers or directors may have conflicting fiduciary duties to us and third parties. The terms of transactions with third parties may not be subject to arm’s length negotiations and therefore may be on terms less favorable to us than those that could be procured through arm’s length negotiations. Although we have established an audit committee comprised solely of independent directors to oversee transactions between us and our insiders, we do not have any formal policies in place to deal with such conflicting fiduciary duties should such a conflict arise.
We indemnify our officers and directors against liability to us and our security holders, and such indemnification could increase our operating costs.
Our Articles of Incorporation and Bylaws require us to indemnify our officers and directors against claims associated with carrying out the duties of their offices. We are also required to advance the costs of certain legal defenses upon the indemnitee undertaking to repay such expenses to the extent it is determined that such person was not entitled to indemnification of such expenses. Insofar as indemnification for liabilities arising under the Securities Act may be permitted to our officers, directors, or control persons, the Commission has advised that such indemnification is against public policy and is therefore unenforceable.
|
|
Risks Relating to Our Intellectual Property
We may be unable to obtain or protect intellectual property rights relating to our product candidates, which could have a materially adverse effect on our business.
Our ability to compete effectively will depend on our ability to maintain the proprietary nature of our technologies. We cannot assure investors that we will continue to innovate and file new patent applications, or that if filed any future patent applications will result in granted patents with respect to the technology owned by us or licensed to us. Further, we cannot predict how long it will take for such patents to issue, if at all. The patent position of pharmaceutical or biotechnology companies, including ours, is generally uncertain and involves complex legal and factual considerations and, therefore, validity and enforceability cannot be predicted with certainty. Patents may be challenged, deemed unenforceable, invalidated or circumvented.
We have pending patent applications for our liquid formulations of terlipressen the following jurisdictions which claim priority to PCT/US2020/034269 filed on May 22, 2020 and published as WO2020/237170: US, Europe, China, and Japan and 6 other jurisdictions. In two jurisdictions, we have patents for our liquid formulations of terlipressen which claim priority to PCT/US2020/034269 filed on May 22, 2020 and published as WO2020/237170. We also have thirteen (13) issued U.S. patents, six (6) pending U.S. applications, three (3) pending Patent Cooperation Treaty applications, six (6) issued foreign patents, and six (6) pending foreign patent applications directed to protecting bezisterm (NE3107) and related compounds and methods of making and using thereof. However, there can be no assurance that our pending patent applications will result in issued patents, or that any issued patent claims from pending or future patent applications will be sufficiently broad to protect BIV201, bezisterim (NE3107), or any other product candidates or to provide us with competitive advantages.
We can provide no assurance that any issued patents will provide us with any competitive advantage. We cannot be certain that there is no invalidating prior art of which we and the patent examiner are unaware or that our interpretation of the relevance of prior art is correct. If a third-party patent or patent application is determined to have an earlier priority date, it may prevent our patent applications from issuing at all or issuing in a form that provides any competitive advantage for our drug candidates. Failure to obtain additional issued patents could have a material adverse effect on our ability to develop and commercialize our drug candidates. Even if our patent applications do issue as patents, third parties may be able to challenge the validity and enforceability of our patents on a variety of grounds, including that such third party’s patents and patent applications have an earlier priority date, and if such challenges are successful, we may be required to obtain one or more licenses from such third parties, if available on commercially reasonable terms, or be prohibited from commercializing our drug candidates.
We seek to protect our proprietary positions by, among other things, filing patent applications in the United States and abroad related to our current drug candidates and other drug candidates that we may identify. Obtaining, maintaining, defending and enforcing pharmaceutical patents is costly, time-consuming and complex, and we may not be able to file and prosecute all necessary or desirable patent applications, or maintain, enforce and license any patents that may issue from such patent applications, at a reasonable cost or in a timely manner. It is also possible that we will fail to identify patentable aspects of our research and development output before it is too late to obtain patent protection. Moreover, under certain of our license or collaboration agreements, we may not have the right to control the preparation, filing, prosecution and maintenance of patent applications, or to maintain the rights to patents licensed to or from third parties.
We currently are the assignee of a number of U.S. provisional patent applications. U.S. provisional patent applications are not eligible to become issued patents until, among other things, we file a non-provisional patent application within 12 months of filing one or more of our related provisional patent applications. With regard to such U.S. provisional patent applications, if we do not timely file any non-provisional patent applications, we may lose our priority dates with respect to our provisional patent applications and any patent protection on the inventions disclosed in our provisional patent applications. Further, in the event that we do timely file non-provisional patent applications relating to our provisional patent applications, we cannot predict whether any such patent applications will result in the issuance of patents or if such issued patents will provide us with any competitive advantage.
As to our material inventions, trade secrets, and intellectual property, our employees, consultants, and advisors execute confidentiality agreements and agree to disclose and assign to us all inventions conceived during the workday, using our property, or which relate to our business. However, any of these parties may breach these agreements and disclose such output before a patent application is filed, thereby jeopardizing our ability to seek patent protection. Further, we may not be aware of all third-party intellectual property rights potentially relating to our drug candidates. Publications of discoveries in the scientific literature often lag behind the actual discoveries, and patent applications in the United States and other jurisdictions are typically not published until 18 months after filing or, in some cases, not at all. Therefore, we cannot know with certainty whether we were the first to make the inventions claimed in our patents or pending patent applications, or that we were the first to file for patent protection of such inventions.
|
|
The patent position of pharmaceutical companies generally is highly uncertain, involves complex legal, technological and factual questions and has, in recent years, been the subject of much debate and litigation throughout the world. In addition, the laws of foreign countries may not protect our rights to the same extent as the laws of the United States, or vice versa. The standards that the United States Patent and Trademark Office (the “USPTO”) (and foreign countries) use to grant patents are not always applied predictably or uniformly and can change. There is also no uniform, worldwide policy regarding the subject matter and scope of claims granted or allowable in pharmaceutical or biotechnology patents. Accordingly, we do not know the degree of future protection for our proprietary rights or the breadth of claims that will be allowed in any patents issued to us or to others. The issuance, scope, validity, enforceability, and commercial value of our patent rights are highly uncertain. The subject matter claimed in a patent application can be significantly reduced or eliminated before the patent issues, if at all, and its scope can be reinterpreted or narrowed after issuance. Therefore, our pending and future patent applications may not result in patents being issued in relevant jurisdictions that protect our drug candidates, in whole or in part, or that effectively prevent others from commercializing competitive drug candidates, and even if our patent applications issue as patents in relevant jurisdictions, they may not issue in a form that will provide us with any meaningful protection for our drug candidates or technology, prevent competitors from competing with us or otherwise provide us with any competitive advantage. Additionally, our competitors may be able to circumvent our patents by challenging their validity or by developing similar or alternative drug candidates or technologies in a non-infringing manner.
The issuance of a patent is not conclusive as to its inventorship, scope, validity or enforceability, and our patents may be challenged in the courts or patent offices in the United States and abroad. We may be subject to a third-party preissuance submission of prior art to the USPTO, or become involved in opposition, derivation, reexamination, inter partes review, post-grant review or interference proceedings challenging our patent rights or the patent rights of others, or other proceedings in the USPTO or applicable foreign offices that challenge priority of invention or other features of patentability. An adverse determination in any such submission, proceeding or litigation could result in loss of exclusivity or ability to sell our products free from infringing the patents of third parties, patent claims being narrowed, invalidated or held unenforceable, in whole or in part, and limitation of the scope or duration of the patents directed to our drug candidates, all of which could limit our ability to stop others from using or commercializing similar or identical drug candidates or technology to compete directly with us, without payment to us, or result in our inability to manufacture or commercialize drug candidates or approved products (if any) without infringing third-party patent rights. In addition, if the breadth or strength of the claims of our patents and patent applications is threatened, regardless of the outcome, it could dissuade companies from collaborating with us to license, develop or commercialize current or future drug candidates, or could have a material adverse effect on our ability to raise funds necessary to continue our research programs or clinical trials. Such proceedings also may result in substantial cost and require significant time from our scientists and management, even if the eventual outcome is favorable to us.
In addition, given the amount of time required for the development, testing and regulatory review of new drug candidates, patents protecting such candidates might expire before or shortly after such candidates are commercialized. As a result, our patent portfolio may not provide us with sufficient rights to exclude others from commercializing products or technology similar or identical to ours for a meaningful amount of time, or at all. Moreover, some of our licensed patents and owned or licensed patent applications may in the future be co-owned with third parties. If we are unable to obtain exclusive licenses to any such co-owners’ interest in such patents or patent applications, such co-owners may be able to license their rights to other third parties, including our competitors, and our competitors could market competing products and technology. In addition, we may need the cooperation of any such co-owners in order to enforce such patents against third parties, and such cooperation may not be provided to us. Any of the foregoing could harm our competitive position, business, financial condition, results of operations and prospects.
Further, we rely on a combination of trade secrets, know-how, technology and nondisclosure, and other contractual agreements and technical measures to protect our rights in the technology. If any trade secret, know-how or other technology not protected by a patent were to be disclosed to or independently developed by a competitor, our business and financial condition could be materially and adversely affected. The laws of some foreign countries do not protect our proprietary rights to the same extent as the laws of the U.S., and we may encounter significant problems in protecting our proprietary rights in these countries.
Our success depends in significant part on our ability to obtain, maintain, enforce and defend patents and other intellectual property rights with respect to our drug candidates and technology and to operate our business without infringing, misappropriating, or otherwise violating the intellectual property rights of others. If we are unable to obtain and maintain sufficient intellectual property protection for our drug candidates or other drug candidates that we may identify, or if the scope of the intellectual property protection obtained is not sufficiently broad, our competitors and other third parties could develop and commercialize drug candidates similar or identical to ours, and our ability to successfully commercialize our drug candidates and other drug candidates that we may pursue may be impaired.
|
|
Confidentiality agreements with employees and others may not adequately prevent disclosure of trade secrets and other proprietary information and disclosure of our trade secrets or proprietary information could compromise any competitive advantage that we have, which could have a materially adverse effect on our business.
Our success depends, in part, on our ability to protect our proprietary rights to the technologies used in our product candidates. We depend heavily upon confidentiality agreements with our officers, employees, consultants and subcontractors to maintain the proprietary nature of our technology. These measures may not afford us complete or even sufficient protection, and may not afford an adequate remedy in the event of an unauthorized disclosure of confidential information. If we fail to protect and/or maintain our intellectual property, third parties may be able to compete more effectively against us, we may lose our technological or competitive advantage, and/or we may incur substantial litigation costs in our attempts to recover or restrict use of our intellectual property. In addition, others may independently develop technology similar to ours, otherwise avoiding the confidentiality agreements, or produce patents that would materially and adversely affect our business, prospects, financial condition and results of operations, in which event you could lose all of your investment.
We may enter into licensing and collaboration agreements with third parties. If we fail to comply with our obligations in the agreements under which we license intellectual property rights to or from third parties, or these agreements are terminated, or we otherwise experience disruptions to our business relationships with our licensors or licensees, our competitive position, business, financial condition, results of operations and prospects could be harmed.
It may be necessary for us to use the patented or proprietary technology of third parties to commercialize our products (if approved), in which case we would be required to obtain a license from these third parties. The licensing of third-party intellectual property rights is a competitive area, and more established companies may pursue strategies to license or acquire third-party intellectual property rights that we may consider attractive or necessary. More established companies may have a competitive advantage over us due to their size, capital resources and greater clinical development and commercialization capabilities. In addition, companies that perceive us to be a competitor may be unwilling to assign or license rights to us. We also may be unable to license or acquire third-party intellectual property rights on terms that would allow us to make an appropriate return on our investment or at all. If we are unable to license such technology, or if we are forced to license such technology on unfavorable terms, our business could be materially harmed.
We may fail to obtain any of these licenses or intellectual property rights on commercially reasonable terms. Even if we are able to obtain a license, it may be non-exclusive, thereby giving our competitors access to the same technologies licensed to us. Licenses may not provide us with exclusive rights to use the applicable intellectual property and technology in all relevant fields of use and in all territories in which we may wish to develop or commercialize our drug candidates, products (if approved) and technology in the future. In that event, we may be required to expend significant time and resources to develop or license replacement technology. If we are unable to do so, we may be unable to develop or commercialize the affected products, which could materially harm our business and the third parties owning such intellectual property rights could seek either an injunction prohibiting our sales, or, with respect to our sales, an obligation on our part to pay royalties and/or other forms of compensation. Conversely, we may not always be able to successfully pursue our claims against others that infringe upon our technology. Thus, the proprietary nature of our technology or technology licensed by us may not provide adequate protection against competitors, and we may not be able to prevent competitors from developing and commercializing competitive products or technologies.
In addition, in some circumstances, we may not have the right to control the preparation, filing and prosecution of patent applications or to maintain, defend and enforce the patents that we license to or from third parties, and we may have to rely on our partners to fulfill these responsibilities. If our current or future licensors, licensees or collaborators fail to prepare, file, prosecute, maintain, enforce, and defend licensed patents and other intellectual property rights, such rights may be reduced or eliminated, and our right to develop and commercialize any of our drug candidates or technology that are the subject of such licensed rights could be adversely affected. In addition, our licensors may own or control intellectual property that has not been licensed to us and, as a result, we may be subject to claims, regardless of their merit, that we are infringing or otherwise violating the licensor’s rights.
|
|
If we fail to comply with our obligations, including the obligation to make various milestone payments and royalty payments, under any of the agreements under which we license intellectual property rights from third parties, the licensor may have the right to terminate the license. If any of our license agreements is terminated, the underlying licensed patents fail to provide the intended exclusivity or we otherwise experience disruptions to our business relationships with our licensors, we could lose intellectual property rights that are important to our business or be prevented from developing and commercializing our drug candidates, and competitors could have the freedom to seek regulatory approval of, and to market, products identical to ours. Termination of these agreements or reduction or elimination of our rights under these agreements may also result in our having to negotiate new or reinstated agreements with less favorable terms, cause us to lose our rights under these agreements, including our rights to important intellectual property or technology, or impede, delay or prohibit the further development or commercialization of one or more drug candidates that rely on such agreements. It is possible that we may be unable to obtain any additional licenses at a reasonable cost or on reasonable terms, if at all. In that event, we may be required to expend significant time and resources to redesign our drug candidates or the methods for manufacturing them or to develop or license replacement technology, all of which may not be feasible on a technical or commercial basis
Licensing of intellectual property is of critical importance to our business and involves complex legal, business and scientific issues and certain provisions in intellectual property license agreements may be susceptible to multiple interpretations. Disputes may arise between us and our licensing partners regarding intellectual property subject to a license agreement, including:
| · | the scope of rights granted under the license agreement and other interpretation-related issues; |
| · | whether and the extent to which technology and processes of one party infringe intellectual property of the other party that are not subject to the licensing agreement; |
| · | rights to sublicense patent and other rights to third parties; |
| · | any diligence obligations with respect to the use of the licensed technology in relation to development and commercialization of our drug candidates, and what activities satisfy those diligence obligations; |
| · | the ownership of inventions and know-how resulting from the joint creation or use of intellectual property; |
| · | rights to transfer or assign the license; and |
| · | the effects of termination. |
The resolution of any contract interpretation disagreement that may arise could narrow what we believe to be the scope of our rights to the relevant intellectual property or technology, or increase what we believe to be our financial or other obligations under the relevant agreement, either of which could harm our business, financial condition, results of operations and prospects. If disputes over intellectual property that we have licensed prevent or impair our ability to maintain our current licensing arrangements on acceptable terms or at all, we may be unable to successfully develop and commercialize the affected drug candidates. Moreover, any dispute or disagreement with our licensing partners may result in the delay or termination of the research, development or commercialization of our drug candidates or any future drug candidates, and may result in costly litigation or arbitration that diverts management attention and resources away from our day-to-day activities, which may adversely affect our business, financial condition, results of operations and prospects.
Furthermore, current and future collaborators or strategic partners may develop, either alone or with others, products in related fields that are competitive with the products or potential products that are the subject of these collaborations. Competing products, either developed by our collaborators or strategic partners or to which the collaborators or strategic partners have rights, may result in the withdrawal of partner support for our drug candidates. Any of these developments could harm our product development efforts.
In addition, if our licensors fail to abide by the terms of the license, if the licensors fail to prevent infringement by third parties or if the licensed patents or other rights are found to be invalid or unenforceable, our business, competitive position, financial condition, results of operations and prospects could be materially harmed.
Some of our intellectual property may be subject to federal regulations such as “march-in” rights, certain reporting requirements and a preference for U.S.-based companies if it is determined that our intellectual property has been discovered through government-funded programs. Compliance with such regulations may limit our exclusive rights, and limit our ability to contract with non-U.S. manufacturers.
|
|
Some of the intellectual property rights we have acquired or licensed or may acquire or license in the future may have been generated through the use of U.S. government funding and may therefore be subject to certain federal regulations. These U.S. government rights include a non-exclusive, non-transferable, irrevocable worldwide license to use inventions for any governmental purpose. In addition, the U.S. government has the right, under certain limited circumstances, to require us to grant exclusive, partially exclusive, or non-exclusive licenses to any of these inventions to a third party if it determines that: (i) adequate steps have not been taken to commercialize the invention; (ii) government action is necessary to meet public health or safety needs; or (iii) government action is necessary to meet requirements for public use under federal regulations (also referred to as “march-in rights”). The U.S. government also has the right to take title to these inventions if the grant recipient fails to disclose the invention to the government or fails to file an application to register the intellectual property within specified time limits. Intellectual property generated under a government funded program is also subject to certain reporting requirements, compliance with which may require us to expend substantial resources. In addition, the U.S. government requires that any products embodying any of these inventions or produced through the use of any of these inventions be manufactured substantially in the United States. This preference for U.S. industry may be waived by the federal agency that provided the funding if the owner or assignee of the intellectual property can show that reasonable but unsuccessful efforts have been made to grant licenses on similar terms to potential licensees that would be likely to manufacture substantially in the United States or that under the circumstances domestic manufacture is not commercially feasible. This preference for U.S. industry may limit our ability to contract with non-U.S. product manufacturers for products relating to such intellectual property. To the extent any of our future intellectual property is also generated through the use of U.S. government funding, the provisions of the Bayh-Dole Act may similarly apply.
Patent terms may be inadequate to establish our competitive position on our drug candidates for an adequate amount of time.
Patents have a limited lifespan. In the United States, if all maintenance fees are timely paid, the natural expiration of a patent is generally 20 years from its earliest U.S. non-provisional filing date. Various extensions may be available, but the life of a patent, and the protection it affords, is limited. Even if patents directed to our drug candidates are obtained, once the patent life has expired for a drug candidate, we may be open to competition from competitive medications, including generic versions. Given the amount of time required for the development, testing and regulatory review of new drug candidates, patents directed towards such drug candidates might expire before or shortly after such drug candidates are commercialized. As a result, our owned and licensed patent portfolio may not provide us with sufficient rights to exclude others from commercializing drug candidates similar or identical to ours for a meaningful amount of time, or at all.
Depending upon the timing, duration and conditions of any FDA marketing approval of our drug candidates, one or more of our owned or licensed U.S. patents may be eligible for limited patent term extension under the Hatch-Waxman Act, and similar legislation in the EU and certain other countries. The Hatch-Waxman Act permits a patent term extension of up to five years for a patent covering an approved product as compensation for effective patent term lost during product development and the FDA regulatory review process. However, we may not receive an extension if we fail to exercise due diligence during the testing phase or regulatory review process, fail to apply within applicable deadlines, fail to apply prior to expiration of relevant patents or otherwise fail to satisfy applicable requirements. Moreover, the length of the extension could be less than we request. Only one patent per approved product can be extended, the extension cannot extend the total patent term beyond 14 years from approval and only those claims for the approved drug, a method for using it or a method for manufacturing it may be extended. If we are unable to obtain patent term extension or the term of any such extension is less than we request, the period during which we can enforce our patent rights for the applicable drug candidate will be shortened and our competitors may obtain approval to market competing products sooner. As a result, our revenue from applicable products could be reduced. Further, if this occurs, our competitors may take advantage of our investment in development and trials by referencing our clinical and nonclinical data and launch their product earlier than might otherwise be the case, and our competitive position, business, financial condition, results of operations and prospects could be materially harmed.
We may not be able to protect our intellectual property rights throughout the world.
Filing, prosecuting, maintaining, defending and enforcing patents on our drug candidates in all countries throughout the world would be prohibitively expensive, and consequently our intellectual property rights in some countries outside the United States may be less extensive than those in the United States. In addition, the laws of some foreign countries do not protect intellectual property rights to the same extent as federal and state laws in the United States. Consequently, we may not be able to prevent third parties from practicing our inventions in all countries outside the United States, or from selling or importing products made using our inventions in and into the United States or other jurisdictions. Competitors may use our technologies in jurisdictions where we have not obtained patents to develop their own products and may export otherwise infringing products to territories where we have patents, but enforcement rights are not as strong as those in the United States. These products may compete with our drug candidates and our patents or other intellectual property rights may not be effective or sufficient to prevent them from competing.
|
|
Many companies have encountered significant problems in protecting and defending intellectual property rights in foreign jurisdictions. The legal systems of some countries do not favor the enforcement or protection of patents, trade secrets and other intellectual property, which could make it difficult for us to stop the infringement of our patents or marketing of competing products in violation of our intellectual property and proprietary rights generally. Proceedings to enforce our intellectual property rights in foreign jurisdictions could result in substantial costs and divert our efforts and attention from other aspects of our business, could put our patents at risk of being invalidated or interpreted narrowly and our patent applications at risk of not issuing and could provoke third parties to assert claims against us. We may not prevail in any lawsuits that we initiate, and the damages or other remedies awarded, if any, may not be commercially meaningful.
Many foreign countries, including some EU countries, India, Japan and China, have compulsory licensing laws under which a patent owner may be compelled under specified circumstances to grant licenses to third parties. In addition, many countries limit the enforceability of patents against government agencies or government contractors. In those countries, we may have limited remedies if patents are infringed or if we are compelled to grant a license to a third party, which could materially diminish the value of the applicable patents and limit our potential revenue opportunities. Accordingly, our efforts to enforce our intellectual property rights around the world may be inadequate to obtain a significant commercial advantage from the intellectual property that we develop or license, which could adversely affect our business, financial condition, results of operations and prospects.
In 2012, the European Patent Package, or EU Patent Package, regulations were passed with the goal of providing a single pan-European Unitary Patent and a new European Unified Patent Court (“UPC”), for litigation involving European patents. Implementation of the EU Patent Package occurred in 2023. Under the UPC, all European patents, including those issued prior to ratification of the European Patent Package, will by default automatically fall under the jurisdiction of the UPC. The UPC will provide our competitors with a new forum to centrally revoke our European patents, and allow for the possibility of a competitor to obtain pan-European injunctions. It will be several years before we will understand the scope of patent rights that will be recognized and the strength of patent remedies that will be provided by the UPC. Under the EU Patent Package as currently proposed, we will have the right to opt our patents out of the UPC over the first seven years of the court’s existence, but doing so may preclude us from realizing the benefits of the new unified court.
Changes in patent law could diminish the value of patents in general, thereby impairing our ability to protect our drug candidates.
Obtaining and enforcing patents in the pharmaceutical industry is inherently uncertain, due in part to ongoing changes in the patent laws. For example, in the United States, depending on decisions by Congress, the federal courts, and the USPTO, the laws and regulations governing patents, and interpretation thereof, could change in unpredictable ways that could weaken our and our collaborators’ or licensors’ ability to obtain new patents or to enforce existing or future patents. For example, the U.S. Supreme Court has ruled on several patent cases in recent years, either narrowing the scope of patent protection available in certain circumstances or weakening the rights of patent owners in certain situations. Therefore, there is increased uncertainty with regard to our and our collaborators’ or licensors’ ability to obtain patents in the future, as well as uncertainty with respect to the value of patents once obtained.
Patent reform legislation could increase the uncertainties and costs surrounding the prosecution of our and our collaborators’ or licensors’ patent applications and the enforcement or defense of our or our collaborators’ or licensors’ issued patents. For example, assuming that other requirements for patentability are met, prior to March 2013, in the United States, the first to invent the claimed invention was entitled to the patent, while outside the United States, the first to file a patent application was entitled to the patent. After March 2013, under the Leahy-Smith America Invents Act (the “Leahy-Smith Act”), enacted in September 2011, the United States transitioned to a first inventor to file system in which, assuming that other requirements for patentability are met, the first inventor to file a patent application will be entitled to the patent on an invention regardless of whether a third party was the first to invent the claimed invention. The Leahy-Smith Act also includes a number of significant changes that affect the way patent applications are prosecuted and may also affect patent litigation. These include allowing third-party submission of prior art to the USPTO during patent prosecution and additional procedures to challenge the validity of a patent by USPTO-administered post-grant proceedings, including post-grant review, inter partes review and derivation proceedings. The USPTO has developed regulations and procedures to govern administration of the Leahy-Smith Act, and many of the substantive changes to patent law associated with the Leahy-Smith Act, particularly the first inventor-to-file provisions. Accordingly, it is not clear what, if any, impact the Leahy-Smith Act will have on the operation of our business. However, the Leahy-Smith Act and its implementation could increase the uncertainties and costs surrounding the prosecution of our or our licensors’ patent applications and the enforcement or defense of our or our licensors’ issued patents. Similarly, statutory or judicial changes to the patent laws of other countries may increase the uncertainties and costs surrounding the prosecution of patent applications and the enforcement or defense of issued patents. Any of the foregoing could harm our business, financial condition, results of operations and prospects.
We may become involved in lawsuits to protect or enforce our patents or other intellectual property, which could be expensive, time-consuming and unsuccessful, and issued patents directed towards our technology and drug candidates could be found invalid or unenforceable if challenged.
|
|
We are not aware that our patents directed to either BIV201 or bezisterim (NE3107), the product candidates we are currently developing, are infringed by third parties. However, there can be no assurance that our patents will not be found in the future to be infringed by others. Any patents we do obtain may be challenged by reexamination or otherwise invalidated or eventually found unenforceable. Both the patent application process and the process of managing patent disputes can be time-consuming and expensive.
Significantly, our pending patent applications cannot be enforced against third parties practicing the technology claimed in such applications unless and until a patent issues from such applications. Our ability to enforce patent rights also depends on our ability to identify infringement. It may be difficult to identify infringers who do not advertise the components or methods that are used in connection with their products and services. Moreover, it may be difficult or impossible to obtain evidence of infringement in a competitor’s or potential competitor’s product or service. Any claims we assert against perceived infringers could provoke these parties to assert counterclaims against us alleging that we infringe their patents or that our patents are invalid or unenforceable. In a patent infringement proceeding, a court may decide that a patent of ours is invalid or unenforceable, in whole or in part, construe the patent’s claims narrowly or refuse to stop the other party from using the technology at issue on the grounds that our patents do not cover the technology. An adverse result in any litigation proceeding could put one or more of our owned or licensed patents at risk of being invalidated, held unenforceable or interpreted narrowly. We may find it impractical or undesirable to enforce our intellectual property against some third parties.
If we were to initiate legal proceedings against a third party to enforce a patent directed to our drug candidates, or one of our future drug candidates, the defendant could counterclaim that our patent is invalid or unenforceable. In patent litigation in the United States, defendant counterclaims alleging invalidity or unenforceability are commonplace. Grounds for a validity challenge could be an alleged failure to meet any of several statutory requirements, including lack of novelty, obviousness, non-enablement or insufficient written description. Grounds for a presentability assertion could be an allegation that someone connected with prosecution of the patent withheld material information from the USPTO or made a misleading statement during prosecution. Third parties may also raise similar claims before the USPTO or an equivalent foreign body, even outside the context of litigation. Potential proceedings include reexamination, post-grant review, inter partes review, interference proceedings, derivation proceedings and equivalent proceedings in foreign jurisdictions (e.g., opposition proceedings). Such proceedings could result in the revocation of, cancellation of, or amendment to our patents in such a way that they no longer cover our technology or any drug candidates that we may develop. The outcome following legal assertions of invalidity and unenforceability is unpredictable. With respect to the validity question, for example, we cannot be certain that there is no invalidating prior art of which we and the patent examiner were unaware during prosecution. These assertions may also be based on information known to us or the USPTO. If a defendant were to prevail on a legal assertion of invalidity or unenforceability, we would lose at least part, and perhaps all, of the patent rights directed towards the applicable drug candidates or technology related to the patent rendered invalid or unenforceable. Such a loss of patent rights would materially harm our business, financial condition, results of operations and prospects.
Interference proceedings provoked by third parties or brought by us or declared by the USPTO may be necessary to determine the priority of inventions with respect to our patents or patent applications. An unfavorable outcome could require us to cease using the related technology or to attempt to license rights to it from the prevailing party. Our business could be materially harmed if the prevailing party does not offer us a license on commercially reasonable terms or at all.
Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation.
The pharmaceutical industry is characterized by extensive litigation regarding patents and other intellectual property rights. Moreover, the cost to us of any litigation or other proceeding relating to our patents and other intellectual property rights, even if resolved in our favor, could be substantial, and the litigation would divert our management’s efforts. We may not have sufficient resources to bring any such action to a successful conclusion. Uncertainties resulting from the initiation and continuation of any litigation could limit our ability to continue our operations and you could lose all of your investment.
Some of our competitors are larger than we are and have substantially greater resources. They are, therefore, likely to be able to sustain the costs of complex patent litigation or proceedings more effectively than we can because of their greater financial resources and more mature and developed intellectual property portfolios. Accordingly, despite our efforts, we may not be able to prevent third parties from infringing, misappropriating or otherwise violating our intellectual property. Even if resolved in our favor, litigation or other legal proceedings relating to intellectual property claims could result in substantial costs and diversion of management resources, which could harm our business. In addition, the uncertainties associated with litigation could compromise our ability to raise the funds necessary to continue our clinical trials, continue our internal research programs, or in-license needed technology or other drug candidates. There could also be public announcements of the results of the hearing, motions, or other interim proceedings or developments. If securities analysts or investors perceive those results to be negative, it could cause the price of shares of our Common Stock to decline. Any of the foregoing events could harm our business, financial condition, results of operation and prospects.
|
|
We may not identify relevant third-party patents or may incorrectly interpret the relevance, scope or expiration of a third-party patent, which might subject us to infringement claims or adversely affect our ability to develop and market our drug candidates.
We cannot guarantee that any of our or our licensors’ patent searches or analyses, including the identification of relevant patents, the scope of patent claims or the expiration of relevant patents, are complete or thorough, nor can we be certain that we have identified each and every third-party patent and pending patent application in the United States and abroad that is relevant to or necessary for the commercialization of our drug candidates in any jurisdiction. For example, U.S. patent applications filed before November 29, 2000 and certain U.S. patent applications filed after that date that will not be filed outside the United States remain confidential until patents issue. As mentioned above, patent applications in the United States and elsewhere are published approximately 18 months after the earliest filing for which priority is claimed, with such earliest filing date being commonly referred to as the priority date. Therefore, patent applications covering our drug candidates could have been filed by third parties without our knowledge. Additionally, pending patent applications that have been published can, subject to certain limitations, be later amended in a manner that could cover our drug candidates or the use of our drug candidates. The scope of a patent claim is determined by an interpretation of the law, the written disclosure in a patent and the patent’s prosecution history. Our interpretation of the relevance or the scope of a patent or a pending application may be incorrect, which may negatively impact our ability to market our drug candidates. We may incorrectly determine that our drug candidates are not covered by a third-party patent or may incorrectly predict whether a third party’s pending application will issue with claims of relevant scope. Our determination of the expiration date of any patent in the United States or abroad that we consider relevant may be incorrect, which may negatively impact our ability to develop and market our drug candidates. Our failure to identify and correctly interpret relevant patents may negatively impact our ability to develop and market our drug candidates.
In addition, if we fail to identify and correctly interpret relevant patents, we may be subject to infringement claims. We cannot guarantee that we will be able to successfully settle or otherwise resolve such infringement claims. If we fail in any such dispute, in addition to being forced to pay damages, which may be significant, we may be temporarily or permanently prohibited from commercializing any of our drug candidates that are held to be infringing. We might, if possible, also be forced to redesign drug candidates so that they no longer infringe the third-party intellectual property rights. Any of these events, even if we were ultimately to prevail, could require us to divert substantial financial and management resources that we would otherwise be able to devote to our business and could adversely affect our business, financial condition, results of operations and prospects.
Third parties may initiate legal proceedings alleging that we are infringing, misappropriating or otherwise violating their intellectual property rights, the outcome of which would be uncertain and could negatively impact the success of our business.
Our commercial success depends upon our ability to develop, manufacture, market and sell our drug candidates and use our proprietary technologies without infringing, misappropriating or otherwise violating the intellectual property and other proprietary rights of third parties. There is considerable intellectual property litigation in the pharmaceutical industry. We may become party to, or be threatened with, future adversarial proceedings or litigation regarding intellectual property rights with respect to our drug candidates and their manufacture and our other technology, including reexamination, interference, post-grant review, inter partes review or derivation proceedings before the USPTO or an equivalent foreign body. Numerous U.S.- and foreign-issued patents and pending patent applications owned by third parties exist in the fields in which we are developing our drug candidates. Third parties may assert infringement claims against us based on existing patents or patents that may be granted in the future, regardless of their merit.
We do not believe that either BIV201 or bezisterim (NE3107), the product candidates we are currently developing, infringe the patents of any third parties. However, there can be no assurance that our technology will not be found in the future to infringe the patents of others. Moreover, patent applications are in some cases maintained in secrecy until patents are issued. The publication of discoveries in the scientific or patent literature frequently occurs substantially later than the date on which the underlying discoveries were made and patent applications were filed. Because patents can take many years to issue, there may be currently pending applications of which we are unaware that may later result in issued patents that our products or product candidates infringe. For example, pending applications may exist that provide support or can be amended to provide support for a claim that results in an issued patent that our product infringes.
Even if we believe third-party intellectual property claims are without merit, there is no assurance that a court would find in our favor on questions of claim scope, infringement, validity, enforceability or priority. A court of competent jurisdiction could hold that third-party patents asserted against us are valid, enforceable and infringed, which could materially and adversely affect our ability to commercialize any drug candidates we may develop and any other drug candidates or technologies covered by the asserted third-party patents. In order to successfully challenge the validity of any such U.S. patent in federal court, we would need to overcome a presumption of validity. As this burden is a high one requiring us to present clear and convincing evidence as to the invalidity of any such U.S. patent claim, there is no assurance that a court of competent jurisdiction would invalidate the claims of any such U.S. patent.
|
|
If we are found to infringe, misappropriate or otherwise violate a third party’s intellectual property rights, and we are unsuccessful in demonstrating that such rights are invalid or unenforceable, we could be required to obtain a license from such a third party in order to continue developing and marketing our products and technology. However, we may not be able to obtain any required license on commercially reasonable terms or at all. Even if we were able to obtain a license, it could be or may become non-exclusive, thereby giving our competitors access to the same technologies licensed to us. We could be forced, including by court order, to cease commercializing the infringing technology or product. A finding of infringement could prevent us from commercializing our drug candidates or force us to cease some of our business operations. In the event of a successful claim of infringement against us, we may have to pay substantial damages, including treble damages and attorneys’ fees for willful infringement, pay royalties and other fees, redesign our infringing drug candidate or obtain one or more licenses from third parties, which may be impossible or require substantial time and monetary expenditure. Claims that we have misappropriated the confidential information or trade secrets of third parties could have a similar negative impact on our business. Any of the foregoing events would harm our business, financial condition, results of operations and prospects.
We may be subject to claims by third parties asserting that we or our employees have infringed, misappropriated or otherwise violated their intellectual property rights, or claiming ownership of what we regard as our own intellectual property.
Many of our employees were previously employed at other biotechnology or pharmaceutical companies. Although we try to ensure that our employees, consultants and advisors do not use the proprietary information or know-how of others in their work for us, we may be subject to claims that we or these individuals have used or disclosed intellectual property, including trade secrets or other proprietary information, of any such individual’s former employer. We may also be subject to claims that patents and applications we have filed to protect inventions made on our behalf by our employees, consultants and advisors, even those related to one or more of our drug candidates, are rightfully owned by their former or concurrent employer. Litigation may be necessary to defend against these claims
If we fail in prosecuting or defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel. Even if we are successful in prosecuting or defending against such claims, litigation could result in substantial costs, delay development of our drug candidates and be a distraction to management. Any of the foregoing events would harm our business, financial condition, results of operations and prospects.
We may be subject to claims challenging the inventorship of our patents and other intellectual property.
We or our licensors may be subject to claims that former employees, collaborators or other third parties have an interest (including co-ownership or ownership) in our owned or in-licensed patents, trade secrets, or other intellectual property as an inventor or co-inventor. For example, we or our licensors or collaborators may have inventorship disputes arising from conflicting obligations of employees, consultants or others who are involved in developing our drug candidates. While it is our policy to require our employees and contractors who may be involved in the development of intellectual property to execute agreements assigning such intellectual property to us, we may be unsuccessful in executing such an agreement with each party who in fact develops intellectual property that we regard as our own. Our and their assignment agreements may not be self-executing or may be breached, and litigation may be necessary to defend against these and other claims challenging inventorship or our or our licensors’ or collaborators’ ownership of our owned or in-licensed patents, trade secrets or other intellectual property. If we or our licensors or collaborators fail in defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights, such as exclusive ownership of, or right to use, intellectual property that is important to our drug candidates. Even if we are successful in defending against such claims, these claims may create considerable distraction to management and other employees of the company. Any of the foregoing could have a material adverse effect on our business, financial condition, results of operations and prospects.
|
|
Intellectual property rights do not necessarily address all potential threats.
The degree of future protection, if any, afforded by our intellectual property rights is uncertain because intellectual property rights have limitations and may not adequately protect our business or permit us to maintain our competitive advantage. For example:
| · | others may be able to make products that are similar to any drug candidates we may develop or utilize similar technology but that are not covered by the claims of the patents that we license or may own in the future; |
| · | we, or our current or future licensors or collaborators, might not have been the first to make the inventions covered by the issued patent or pending patent application that we license or may own in the future; |
| · | we, or our current or future licensors or collaborators might not have been the first to file patent applications covering certain of our or their inventions; |
| · | others may independently develop similar or alternative technologies or duplicate any of our technologies without infringing our owned or licensed intellectual property rights; |
| · | it is possible that our pending owned or licensed patent applications or those that we may own or license in the future will not lead to issued patents; |
| · | issued patents that we hold rights to may be held invalid or unenforceable, including as a result of legal challenges by our competitors; |
| · | our competitors might conduct research and development activities in countries where we do not have patent rights and then use the information learned from such activities to develop competitive products for sale in our major commercial markets; |
| · | we may not develop additional proprietary technologies that are patentable; |
| · | the intellectual property rights of others may harm our business; and |
| · | we may choose not to file a patent in order to maintain certain trade secrets or know-how, and a third party may subsequently file a patent directed to such intellectual property. |
Should any of these events occur, they could harm our business, financial condition, results of operations and prospects.
Intellectual property litigation may lead to unfavorable publicity that harms our reputation and causes the market price of shares of our Common Stock to decline.
During the course of any intellectual property litigation, there could be public announcements of the initiation of the litigation as well as results of hearings, rulings on motions, and other interim proceedings in the litigation. If securities analysts or investors regard these announcements as negative, the perceived value of our existing products, programs or intellectual property could be diminished. Accordingly, the market price of shares of our Common Stock may decline. Such announcements could also harm our reputation or the market for our future products, which could have a material adverse effect on our business.
|
|
Risks Relating to Our Common Stock
Our stock price is and may continue to be volatile and you may not be able to resell our Common Stock at or above the price you paid.
The market price for our Common Stock is volatile and may fluctuate significantly in response to a number of factors, many of which we cannot control, such as quarterly fluctuations in financial results, the timing and our ability to advance the development of our product candidates or changes in securities analysts’ recommendations could cause the price of our stock to fluctuate substantially. In addition, stock markets generally have recently experienced volatility. Our stock price is likely to experience significant volatility in the future. The price of our Common Stock may decline and the value of any investment in our Common Stock may be reduced regardless of our performance. Further, the daily trading volume of our Common Stock has historically been relatively low. As a result of the historically low volume, our shareholders may be unable to sell significant quantities of Common Stock in the public trading markets without a significant reduction in the price of our shares of Common Stock. Each of these factors, among others, could harm your investment in our Common Stock and could result in your being unable to resell the shares of our Common Stock that you purchase at a price equal to or above the price you paid.
In the past, when the market price of a stock has been volatile, holders of that stock have sometimes instituted securities class action litigation against the issuer. If any of our stockholders were to bring such a lawsuit against us, we could incur substantial costs defending the lawsuit and the attention of our management would be diverted from the operation of our business.
You may experience future dilution as a result of future equity offerings or if we issue shares subject to options, warrants, stock awards or other arrangements.
As of June 30, 2024, our Articles of Incorporation, as amended, authorize the issuance of 800,000,000 shares of Common Stock, and we had 6,216,398 shares of Common Stock issued and 6,190,072 issued and outstanding. Accordingly, we may issue up to an additional 793,783,602 shares of Common Stock. The future issuance of Common Stock may result in substantial dilution in the percentage of our Common Stock held by our then existing stockholders. We may value any Common Stock in the future on an arbitrary basis. The issuance of Common Stock for future services or acquisitions or other corporate actions may have the effect of diluting the value of the shares held by our investors, might have an adverse effect on any trading market for our Common Stock and could impair our ability to raise capital in the future through the sale of equity securities.
In order to raise additional capital, we may in the future offer additional shares of our Common Stock or other securities convertible into or exchangeable for our Common Stock, including under the Controlled Equity Offering Sales Agreement (the “ATM Agreement”), dated as of August 31, 2022, between the Company and Cantor Fitzgerald & Co. (the “Cantor”), pursuant to which the Company may issue and sell from time to time shares of our Common Stock through Cantor. We may sell shares or other securities in any other offering at a price per share that is less than the current market price of our securities, and investors purchasing shares or other securities in the future could have rights superior to existing stockholders. The sale of additional shares of our Common Stock or other securities convertible into or exchangeable for our Common Stock would dilute all of our stockholders, and if such sales of convertible securities into or exchangeable into our Common Stock occur at a deemed issuance price that is lower than the current exercise price of our outstanding warrants sold to Acuitas Group Holdings, LLC (“Acuitas”) in August 2022, the exercise price for those warrants would adjust downward to the deemed issuance price pursuant to price adjustment protection contained within those warrants.
As of June 30, 2024, there were warrants outstanding to purchase an aggregate of 1,932,029 shares of our Common Stock at exercise prices ranging from $10.00 to $125.00 per share and 518,076 shares issuable upon exercise of outstanding options at exercise prices ranging from $4.74 to $420.90 per share and restricted stock units totaling 40,291. In addition, pursuant to the Loan and Security Agreement and the Supplement to the Loan and Security Agreement, each entered into on November 30, 2021, with Avenue Venture Opportunities Fund II, L.P. and Avenue Venture Opportunities Fund, L.P., the lenders have the option to convert up to $5 million of the outstanding loan amount into shares of our Common Stock at a conversion price of $58.20 per share. We may also grant additional options, warrants or equity awards. To the extent such shares are issued, the interest of holders of our Common Stock will be diluted.
Moreover, we are obligated to issue shares of our Common Stock upon achievement of certain clinical, regulatory and commercial milestones with respect to certain of our drug candidates (i.e., bezisterim (NE3107), NE3291, NE3413, and NE3789) pursuant to the asset purchase agreement, dated April 27, 2021, by and among the Company, NeurMedix and Acuitas, as amended on May 9, 2021. The achievement of these milestones could result in the issuance of up to 1.8 million shares of our Common Stock, further diluting the interest of holders of our Common Stock.
|
|
Certain stockholder of the Company may have significant control over our Company.
As of August 30, 2024, Acuitas beneficially owns 3,050,397 shares of our Common Stock, which includes warrants to purchase 727,273 shares of our Common Stock and options to purchase 6,500 shares of our Common Stock that are exercisable within 60 days of August 30, 2024 and currently constitutes 43.2% of our issued and outstanding Common Stock. As a result, Acuitas has substantially influence over the management and affairs of our Company, as well as the ability to control the outcome of matters submitted to our stockholders for approval, including the election of directors, the approval of significant corporate transactions, including any merger, consolidation or sale of all or substantially all of our assets, the issuance or redemption of equity interests in certain circumstances, and any other significant transaction. The interests of Acuitas may not always align with, and in some cases may conflict with, our interests or the interests of our other stockholders. For instance, this concentration of ownership may have the effect of delaying or preventing a change of control otherwise favored by our other stockholders and could deprive our other stockholders of an opportunity to receive a premium for their Common Stock. This concentration of ownership may also negatively affect the prevailing market price of our Common Stock due to investors’ perceptions that conflicts of interest may exist or arise. As a result, this concentration of ownership may not be in your best interests.
We effected a reverse stock split on August 6, 2024, and we cannot predict the effect that such reverse stock split will have on the market price for shares of our Common Stock.
Our board of directors approved a one-for-ten (1:10) reverse stock split of our Common Stock, which became effective at 12:01 a.m. Eastern Time on August 6, 2024. We cannot predict the effect that the reverse stock split will have on the market price for shares of our Common Stock, and the history of similar reverse stock splits for companies in like circumstances has varied. Some investors may have a negative view of a reverse stock split. Even if the reverse stock split has a positive effect on the market price for shares of our Common Stock, performance of our business and financial results, general economic conditions and the market perception of our business, and other adverse factors which may not be in our control could lead to a decrease in the price of our Common Stock following the reverse stock split.
Furthermore, even if the reverse stock split does result in an increased market price per share of our Common Stock, the market price per share following the reverse stock split may not increase in proportion to the reduction of the number of shares of our Common Stock outstanding before the implementation of the reverse stock split. Accordingly, even with an increased market price per share, the total market capitalization of shares of our Common Stock after a reverse stock split could be lower than the total market capitalization before the reverse stock split. Also, even if there is an initial increase in the market price per share of our Common Stock after a reverse stock split, the market price many not remain at that level.
If the market price of shares of our Common Stock declines following the reverse stock split, the percentage decline as an absolute number and as a percentage of our overall market capitalization may be greater than would occur in the absence of the reverse stock split due to decreased liquidity in the market for our Common Stock. Accordingly, the total market capitalization of our Common Stock following the reverse stock split could be lower than the total market capitalization before the reverse stock split.
The market price and trading volume of our Common Stock may be volatile.
The market price and trading volume of our Common Stock has been volatile. We expect that the market price of our Common Stock will continue to fluctuate significantly for many reasons, including in response to the risk factors described in this prospectus or for reasons unrelated to our specific performance. In recent years, the stock market has experienced extreme price and volume fluctuations. This volatility has affected the market prices of securities issued by many companies for reasons unrelated to their operating performance and may adversely affect the market price and trading volume of our Common Stock. Prices for our Common Stock may also be influenced by the depth and liquidity of the market for our Common Stock, investor perceptions about us and our business, our future financial results, the absence of cash dividends on our Common Stock and general economic and market conditions. In the past, securities class action litigation has often been instituted against companies following periods of volatility in their stock price. This type of litigation could result in substantial costs and could divert our management and other resources.
Any failure to maintain effective internal control over financial reporting could harm us.
Our management is responsible for establishing and maintaining adequate internal control over financial reporting. Internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements in accordance with U.S. generally accepted accounting principles (“GAAP”). Under standards established by the Public Company Accounting Oversight Board (“PCAOB”), a deficiency in internal control over financial reporting exists when the design or operation of a control does not allow management or personnel, in the normal course of performing their assigned functions, to prevent or detect misstatements on a timely basis. The PCAOB defines a material weakness as a deficiency, or combination of deficiencies, in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of annual or interim financial statements will not be prevented, or detected and corrected, on a timely basis.
|
|
If we are unable to assert that our internal control over financial reporting is effective, or when required in the future, if our independent registered public accounting firm is unable to express an unqualified opinion as to the effectiveness of our internal control over financial reporting, investors may lose confidence in the accuracy and completeness of our financial reports, the market price of our Common Stock could be adversely affected and we could become subject to litigation or investigations by the stock exchange on which our securities are listed, the Commission or other regulatory authorities, which could require additional financial and management resources.
There is a limited trading market for our Common Stock, which could make it difficult to liquidate an investment in our Common Stock, in a timely manner.
Our Common Stock is currently traded on the Nasdaq Capital Market. Because there is a limited public market for our Common Stock, investors may not be able to liquidate their investment whenever desired. We cannot assure that there will be an active trading market for our Common Stock and the lack of an active public trading market could mean that investors may be exposed to increased risk. In addition, if we failed to meet the criteria set forth in the regulations of the Commission, various requirements would be imposed by law on broker dealers who sell our securities to persons other than established customers and accredited investors. Consequently, such regulations may deter broker-dealers from recommending or selling our Common Stock, which may further affect its liquidity.
The lack of public company experience of our management team could adversely impact our ability to comply with the reporting requirements of U.S. securities laws, which could have a materially adverse effect on our business.
Our officers have limited public company experience, which could impair our ability to comply with legal and regulatory requirements such as those imposed by Sarbanes-Oxley Act of 2002. Such responsibilities include complying with federal securities laws and making required disclosures on a timely basis. Any such deficiencies, weaknesses or lack of compliance could have a materially adverse effect on our ability to comply with the reporting requirements of the Exchange Act, which is necessary to maintain our public company status. If we were to fail to fulfill those obligations, our ability to continue as a U.S. public company would be in jeopardy in which event you could lose your entire investment in our Company.
We are considered a smaller reporting company that is exempt from certain disclosure requirements, which could make our stock less attractive to potential investors.
Rule 12b-2 of the Exchange Act defines a “smaller reporting company” as an issuer that is not an investment company, an asset-backed issuer, or a majority-owned subsidiary of a parent that is not a smaller reporting company and that:
| ● | Had a public float of less than $250 million as of the last business day of its most recently completed fiscal quarter, computed by multiplying the aggregate number of worldwide number of shares of its voting and non-voting common equity held by non-affiliates by the price at which the common equity was last sold, or the average of the bid and asked prices of common equity, in the principle market for the common equity; or |
| ● | In the case of an initial registration statement under the Securities Act or the Exchange Act for shares of its common equity, had a public float of less than $250 million as of a date within 30 days of the date of the filing of the registration statement, computed by multiplying the aggregate worldwide number of such shares held by non-affiliates before the registration plus, in the case of a Securities Act registration statement, the number of such shares included in the registration statement by the estimated public offering price of the shares; or |
| ● | In the case of an issuer who had annual revenue of less than $100 million during the most recently completed fiscal year for which audit financial statements are available, had a public float as calculated under paragraph (1) or (2) of this definition that was either zero or less than $700 million. |
As a “smaller reporting company” we are not required and may not include a Compensation Discussion and Analysis section in our proxy statements; we provide only 3 years of business development information; and have other “scaled” disclosure requirements that are less comprehensive than issuers that are not “smaller reporting companies” which could make our stock less attractive to potential investors, which could make it more difficult for you to sell your shares.
|
|
We are subject to the periodic reporting requirements of the Exchange Act, which require us to incur audit fees and legal fees in connection with the preparation of such reports. These additional costs will negatively affect our ability to earn a profit.
We are required to file periodic reports with the Commission pursuant to the Exchange Act and the rules and regulations thereunder. In order to comply with such requirements, our independent registered auditors have to review our financial statements on a quarterly basis and audit our financial statements on an annual basis. Moreover, our legal counsel has to review and assist in the preparation of such reports. Factors such as the number and type of transactions that we engage in and the complexity of our reports cannot accurately be determined at this time and may have a major negative effect on the cost and amount of time to be spent by our auditors and attorneys. However, the incurrence of such costs is an expense to our operations and thus has a negative effect on our ability to meet our overhead requirements and earn a profit.
Because we do not intend to pay any cash dividends on our Common Stock, our stockholders will not be able to receive a return on their shares unless they sell them.
We intend to retain any future earnings to finance the development and expansion of our business. We do not anticipate paying any cash dividends on our Common Stock in the foreseeable future. Unless we pay dividends, our stockholders will not be able to receive a return on their shares unless they sell them. There is no assurance that stockholders will be able to sell shares when desired.
We are authorized to issue “blank check” preferred stock without stockholder approval, which could adversely impact the rights of holders of our securities.
Our Articles of Incorporation authorize us to issue up to 10,000,000 shares of blank check preferred stock. Any preferred stock that we issue in the future may rank ahead of our Common Stock in terms of dividend priority or liquidation premiums and may have greater voting rights than our Common Stock. Any preferred stock issued may contain provisions allowing those shares to be converted into shares of Common Stock, which could dilute the value of our Common Stock to current stockholders and could adversely affect the market price, if any, of our Common Stock. The preferred stock could be utilized, under certain circumstances, as a method of discouraging, delaying, or preventing a change in control of our company. Although we have no present intention to issue any shares of our authorized preferred stock, there can be no assurance that we will not do so in the future.
Provisions in our Articles of Incorporation, our Bylaws, and Nevada law might discourage, delay or prevent a change in control of our company or changes in our management and, therefore, depress the trading price of our Common Stock.
Provisions of our Articles of Incorporation, our Bylaws, and Nevada law may have the effect of deterring unsolicited takeovers or delaying or preventing a change in control of our company or changes in our management, including transactions in which our stockholders might otherwise receive a premium for their shares over then current market prices. In addition, these provisions may limit the ability of stockholders to approve transactions that they may deem to be in their best interests. These provisions include:
| ● | the inability of stockholders to call special meetings; |
| ● | the “business combinations” and “control share acquisitions” provisions of Nevada law, to the extent applicable, could discourage attempts to acquire our stockholders stock even on terms above the prevailing market price; and |
| ● | the ability of our board of directors to designate the terms of and issue new series of preferred stock without stockholder approval, which could include the right to approve an acquisition or other change in our control or could be used to institute a rights plan, also known as a poison pill, that would dilute the stock ownership of a potential hostile acquirer, likely preventing acquisitions that have not been approved by our board of directors. |
The existence of the forgoing provisions and anti-takeover measures could limit the price that investors might be willing to pay in the future for shares of our Common Stock. They could also deter potential acquirers of our company, thereby reducing the likelihood that you could receive a premium for your Common Stock in an acquisition.
|
|
| ITEM 1B. | UNRESOLVED STAFF COMMENTS |
None.
| ITEM 1C. | CYBERSECURITY |
We maintain an information technology and security program appropriate for a company our size, taking into account our operations and risks. The Company recognizes the critical importance of maintaining the trust and confidence of our investors, employees, and vendors. The Company's cybersecurity policies and processes are integrated into the Company's enterprise risk management program and are informed by recognized frameworks established by the National Institute of Standards and Technology, and other applicable industry standards.
In the ordinary course of our business, we collect, use, store, and transmit digitally confidential, sensitive, proprietary, and personal information. The secure maintenance of this information and our information technology systems is important to our operations and business strategy. To this end, we have implemented processes designed to assess, identify, and manage risks from potential unauthorized occurrences on or through our information technology systems that may result in adverse effects on the confidentiality, integrity, and availability of these systems and the data residing therein. These processes are managed and monitored by an outside information technology vendor under the supervision of our Chief Financial Officer, and include mechanisms, controls, technologies, systems, and other processes designed to prevent or mitigate data loss, theft, misuse, or other security incidents or vulnerabilities affecting the data and maintain a stable and secure information technology environment. For example, we conduct ongoing monitoring of critical systems for any compromised or potentially compromised accounts. We conduct regular trainings on cyber and information security, along with phishing simulations, among other topics. In addition, we consult with our outside information technology vendor on a regular basis to assist with assessing, identifying, and managing cybersecurity risks, including to anticipate future threats and trends, and their impact on the Company’s risk environment.
In the last fiscal year, we have not identified any risks from known cybersecurity threats, including as a result of any prior cybersecurity incidents, that have materially affected us, including our operations, business strategy, results of operations, or financial condition. If we were to experience a material cybersecurity incident in the future, such incidents are reasonably likely to materially affect us, including our operations, business strategy, results of operations, or financial condition. The Company's Board of Directors, as a whole and at the committee level, has oversight for the most significant risks facing us and for our processes to identify, prioritize, assess, manage, and mitigate those risks. The Audit Committee, which is composed solely of independent directors, has been designated by our Board to oversee cybersecurity risks. The Board receives updates from the Company's management on cybersecurity risks on at least an annual basis.
| ITEM 2. | PROPERTIES |
The Company pays an annual rent of $2,200 for its headquarters at 680 W Nye Lane, Suite 201, Carson City Nevada 89703. The rental agreement was for a one-year term commenced on October 1, 2022 and has been subsequently renewed for another year at the same rate.
On February 26, 2022, the Company’s San Diego office relocated to Suite 206 at 5090 Shoreham Place, San Diego, CA 92122. The amended lease entered into on February 12, 2024, increased the base rate to $9,685 per month for the larger space, with an annual increase of 4%; and a new term of 60 months.
|
|
| ITEM 3. | LEGAL PROCEEDINGS |
To our knowledge, other than described below, neither the Company nor any of our officers or directors is a party to any material legal proceeding or litigation and such persons know of no material legal proceeding or contemplated or threatened litigation, other than as described below. There are no judgments against us or our officers or directors. None of our officers or directors has been convicted of a felony or misdemeanor relating to securities or performance in corporate office.
On January 19, 2024, a purported shareholder class action complaint, captioned Eric Olmstead v. BioVie Inc. et al., No. 3:24-cv-00035, was filed in the U.S. District Court for the District of Nevada, naming the Company and certain of its officers as defendants. On February 22, 2024, a second, related putative securities class action was filed in the same court asserting similar claims against the same defendants, captioned Way v. BioVie Inc. et al., No. 2:24-cv-00361. On April 15, 2024, the court consolidated these two actions under the caption In re BioVie Inc. Securities Litigation, No. 3:24-cv-00035, appointed the lead plaintiff, and approved selection of the lead counsel. On June 21, 2024, the lead plaintiff filed an amended complaint, alleging that the defendants made material misrepresentations and/or omissions of material fact relating to the Company’s business, operations, compliance, and prospects, including information related to the NM101 Phase 3 study and trial of bezisterim (NE3107) in mild to moderate probable AD, in violation of Sections 10(b) and 20(a) of the Exchange Act, and Rule 10b-5 promulgated thereunder. The class action is on behalf of purchasers of the Company’s securities during the period from December 7, 2022 through November 28, 2023 and seeks unspecified monetary damages on behalf of the putative class and an award of costs and expenses, including attorney’s fees. The defendants filed a motion to dismiss the amended complaint on August 21, 2024. The defendants believe that the claims are without merit and intend to defend vigorously against them, but there can be no assurances as to the outcome.
| ITEM 4. | MINE SAFETY DISCLOSURES |
None.
|
|
PART II
| ITEM 5. | MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES |
Unregistered Sales of Securities
All sales of unregistered securities during the year ended June 30, 2024 were previously disclosed in a Quarterly Report on Form 10-Q or Current Report on Form 8-K.
Issuer Purchases of Common Stock
During the year ended June 30, 2024, there were no issuer repurchases of shares of Common Stock.
| ITEM 6. | [Reserved] |
| ITEM 7. | MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS |
The following discussion of the Company’s financial condition and the results of operations should be read in conjunction with the Financial Statements and Notes thereto appearing elsewhere in this report.
Overview
We are a clinical-stage company developing innovative drug therapies to treat chronic debilitating conditions including liver disease and neurological and neuro-degenerative disorders.
Neurodegenerative Disease Program
The Company acquired the biopharmaceutical assets of NeurMedix a privately held clinical-stage pharmaceutical company and a related party in June 2021. The acquired assets included NE3107. In April 2024, the Company announced that the United States Adopted Names Council, and the World Health Organization International Nonproprietary Names expert committee had approved “bezisterim” as the non-proprietary (generic) name for NE3107. Bezisterim (NE3107) is an investigational, novel, orally administered small molecule that is thought to inhibit inflammation-driven insulin resistance and major pathological inflammatory cascades with a novel mechanism of action. There is emerging scientific consensus that both inflammation and insulin resistance may play fundamental roles in the development of AD and PD, and bezisterim (NE3107) could, if approved by FDA, represent an entirely new medical approach to treating these devastating conditions affecting an estimated 6 million Americans suffering from AD and 1 million Americans suffering from PD.
In neurodegenerative disease, bezisterim (NE3107) inhibits activation of inflammatory ERK and nuclear factor kappa-light-chain-enhancer of activated B cells (“NFκB”) (including interactions with TNF signaling and other relevant inflammatory pathways) that lead to neuroinflammation and insulin resistance. Bezisterim (NE3107) does not interfere with their homeostatic functions (e.g., insulin signaling and neuron growth and survival). Both inflammation and insulin resistance are drivers of AD and PD.
About Inflammation and Bezisterim’s (NE3107’s) Mechanism of Action
Neuroinflammation, insulin resistance, and oxidative stress are common features in the major neurodegenerative diseases, including AD, PD, frontotemporal lobar dementia, and ALS. Bezisterim (NE3107) is an orally bioavailable, blood-brain permeable, small molecule, with potential anti-inflammatory, insulin sensitizing, and ERK-binding properties that may allow it to selectively inhibit ERK-, NFκB- and TNF-stimulated inflammation. Bezisterim’s (NE3107’s) potential to inhibit neuroinflammation and insulin resistance forms the basis for the Company’s work testing the molecule in AD and PD patients.
Parallels exist between AD and PD, among them activated microglia driving inflammation, involvement of TNFα, oxidative stress, protein misfolding, mitochondrial dysfunction, and insulin resistance. In preclinical and clinical studies, bezisterim (NE3107) reduced inflammation and enhanced insulin sensitivity, both of which are important to PD pathology. Preclinical studies in marmoset monkeys have shown bezisterim (NE3107) administered alone to be as pro-motoric as levodopa, underscoring the apparently critical role of inflammation in expression of PD motor symptoms. When bezisterim (NE3107) was administered with levodopa, the combination improved motor control better than either drug alone. Furthermore, in the marmoset study, bezisterim (NE3107) reduced the severity of LID concurrent with pro-motoric benefit and decreased neurodegeneration, preserving twice as many dopaminergic neurons compared to control.
|
|
Parkinson’s Disease
Parkinson’s disease (PD), which affects an estimated 1 million Americans, is driven in large part by neuroinflammation and activation of brain microglia, leading to increased proinflammatory cytokines (particularly TNF). Multiple daily administrations of levodopa (converted to dopamine in the brain) is the current standard of care treatment for this movement disorder, but levodopa effectiveness diminishes over time necessitating increased dosage and prolonged daily administration leads to side effects of uncontrolled movements called levodopa-induced dyskinesia, commonly referred to as LID, which is exacerbated by high dose levodopa. Although levodopa provides symptomatic benefit, it does not slow PD progression.
Long COVID Program
In April 2024, the Company announced the grant of a clinical trial award of up to $13.1 million from the U.S. Department of Defense, awarded through the Peer Reviewed Medical Research Program of the Congressionally Directed Medical Research Programs. The award can provide up to 2 years of non-dilutive funding for a Phase 2 clinical trial that will assess bezisterim (NE3107) for the treatment of neurological symptoms that are associated with long COVID. The Company anticipates the trial to commence by early 2025. The study protocol has been finalized and submitted to the FDA for regulatory review in July 2024. The FDA notified the Company that the study can proceed on August 22, 2024.
Long COVID is a condition in which symptoms of COVID-19, the acute respiratory disease caused by the SARS-CoV-2 virus, persist for an extended period of time, generally three months or more. The Centers for Disease Control recently reported that 6.8% of adults in the United States (more than 17 million individuals) currently or previously had long COVID. Symptoms, which include fatigue, cognitive dysfunction and sleep disturbances, are debilitating. The loss in quality of life and earnings and increased medical costs has an enormous economic impact estimated to be 3.7 trillion dollars. To date there are no therapies proven effective for treatment.
Chronic inflammation is one of the main hypotheses that researchers have proposed to explain the persistence of symptoms in long COVID. Specifically in individuals with “brain fog,” sustained systemic inflammation and persistent localized blood-brain-barrier (“BBB”) dysfunction are key physiological features. Bezisterim (NE3107) permeates the BBB and has been shown to modulate inflammation via the inhibition of NF-kB activation, thus representing a novel oral treatment targeting an underlying cause of long COVID symptoms.
Chronic neuroinflammation, insulin resistance, and oxidative stress are common features in the major neurodegenerative diseases, including AD, PD, frontotemporal lobar dementia, and Amyotrophic lateral sclerosis. Bezisterim (NE3107) is an investigational oral small molecule, blood-brain permeable, compound with potential anti-inflammatory, insulin sensitizing, and ERK-binding properties that may allow it to selectively inhibit ERK-, NFκB- and TNF-stimulated inflammation. Bezisterim’s (NE3107) potential to inhibit neuroinflammation and insulin resistance forms the basis for the Company’s work testing the molecule in AD, PD, and long COVID patients. Bezisterim (NE3107) is patented in the United States, Australia, Canada, Europe and South Korea.
|
|
Results of Operations
Comparison of the Year Ended June 30, 2024 to the Year Ended June 30, 2023
Net loss
The net loss was approximately $33.0 million and $50.3 million for the years ended June 30, 2024 and 2023, respectively. The decrease in net loss of approximately $17.3 million was comprised of a net decrease in research and development expenses of approximately $10.2 million and selling, general and administrative expenses of approximately $2.7 million, and further reduced by an increase in interest income of approximately $574,000, a reduction in interest expense of approximately $1.4 million and the reduction in the fair value of derivative liabilities of approximately $3.3 million.
Total operating expenses were approximately $32.2 million and $45.1 million for the year ended June 30, 2024, and 2023, respectively. The net decrease of approximately $12.9 million for the year ended June 30, 2024, was comprised of a decrease in research and development expenses of approximately $10.2 million and a decrease in selling, general and administrative expenses of approximately $2.7 million.
Research and Development Expenses
Research and development expenses were approximately $23.1 million and $33.3 million for the year ended June 30, 2024, and 2023, respectively. The net decrease of approximately $10.2 million for the year ended June 30, 2024, was primarily attributed to a reduction in expenses totaling approximately $13.8 million due to the completion of clinical studies, offset by increased expenses of approximately $3.6 million primarily comprised of the planning and development of new studies of approximately $836,000 which included the development of Sunrise 1 PD PH2b and Radiance 1 AD MCI Ph2b/3 studies which have been put on hold in the first quarter of the fiscal year ended June 30,2024 pending financing and were designed to be large potentially registrational studies. an increase in Chemistry, Manufacturing and Controls (“CMC”) expense of approximately $1.0 million for drug production and development, an increase in clinical team compensation from the clinical team expansion and the use of consultants totaling approximately $732,000 and $474,000, respectively, and other increases in regulatory and other consultants of approximately $362,000 and publications and travel of approximately $187,000. The decreased expenses from completed clinical studies of approximately $13.8 million were comprised of approximately $2.7 million from Ascites BIV201 Phase 2b study, approximately $2.1 million from the PD Phase 2 study, both studies were completed in the prior fiscal year ended June 30, 2023, approximately $238,000 from the Investigator-Initiated Trial in MCI and Mild Alzheimer’s Disease, and approximately $8.8 million from the AD pivotal Phase 3 clinical study that completed on December 31, 2023.
The following summarizes the expenses incurred during the fiscal years ending June 30, 2024 and 2023 for new developmental studies and the completed studies:
| 2024 | 2023 | Increase (Decrease) |
||||||||||
| New Development | ||||||||||||
| Sunrise PD Phase 2 | $ | 181,000 | $ | - | $ | 181,000 | ||||||
| Long Covid Program | 106,000 | - | 106,000 | |||||||||
| Sunrise 1 PD PH2b (On hold) | 822,000 | 600,000 | 222,000 | |||||||||
| Radiance 1 AD MCI-301 Ph2b/3 (On hold) | 778,000 | 451,000 | 327,000 | |||||||||
| $ | 1,887,000 | $ | 1,051,000 | $ | 836,000 | |||||||
| Completed Studies | ||||||||||||
| Ascites BIV201 Phase 2b | $ | 672,000 | $ | 3,377,000 | $ | (2,705,000 | ) | |||||
| AD mild to moderate pivotal Phase 3 | 7,888,000 | 16,630,000 | (8,742,000 | ) | ||||||||
| PD Phase 2 | 612,000 | 2,747,000 | (2,135,000 | ) | ||||||||
| Investigator-Initiated studies | 73,000 | 311,000 | (238,000 | ) | ||||||||
| $ | 9,245,000 | $ | 23,065,000 | $ | (13,820,000 | ) | ||||||
Selling, General and Administrative Expenses
Selling, general and administrative expenses were approximately $8.8 million and $11.6 million for the year ended June 30, 2024 and 2023, respectively. The net decrease of approximately $2.7 million was primarily attributed to decreased stock and cash compensation of approximately $2.8 million of the executives and directors, and a decline in investor relations fees of approximately $396,000, offset by insurance expense of approximately $248,000 and legal fees of approximately $306,000.
Other Income and Expense
Other income, net was approximately $59,000 compared to other expense, net of $5.2 million, for the year ended June 30, 2024 and 2023, respectively. The net increase in other income of approximately $5.2 million was primarily driven by the change in fair value of the derivative liabilities of approximately $3.3 million, reduction in interest expense of approximately $1.4 million due to amortization and accretion of the financing costs, unearned discount, and premium relating to the note payable, and an increase in interest income of approximately $574,000 from the investment in U.S. Treasury Bills.
Capital Resources and Liquidity
As of June 30, 2024, the Company had working capital of approximately $14.7 million, cash and cash equivalents of approximately $23.8 million, stockholders’ equity of approximately $15.5 million, and an accumulated deficit of approximately $334.2 million. Additionally, the Company had a net loss of approximately $32.1 million and net cash used in operating activities of approximately $27.9 million during the year ended June 30, 2024. In addition, the Company has not generated any revenues to date and no revenues are expected in the foreseeable future. The Company’s future operations are dependent on the success of the Company’s ongoing development and commercialization efforts, as well as its ability to secure additional financing as needed. Projected cash flows could be extended if further measures are taken to delay planned expenditures in our research protocols and slow the progress in the Company’s development and launch of next phase clinical programs.
|
|
The future viability of the Company is largely dependent upon its ability to raise additional capital to finance its operations. Management expects that future sources of funding may include sales of equity, obtaining loans, or other strategic transactions.
Although management continues to pursue the Company’s strategic plans, there is no assurance that the Company will be successful in obtaining sufficient financing on terms acceptable to the Company, if at all, to fund continuing operations. These circumstances raise substantial doubt on the Company’s ability to continue as a going concern. The financial statements included elsewhere in this Form 10-K do not include any adjustments that might result from the outcome of this uncertainty.
At-The-Market Facility
On August 31, 2022, the Company entered into a Sales Agreement with Cantor, pursuant to which the Company may issue and sell from time-to-time shares of the Company’s Common Stock through Cantor, subject to the terms and conditions of the Sales Agreement. During the year ended June 30, 2024, the Company sold 333,749 shares of its Common Stock under the ATM Agreement with Cantor for total net proceeds of approximately $9.3 million after 3% commissions and cost totaling approximately $377,000.
Underwritten Offering
On March 6, 2024, the Company closed the best efforts public offering (the “March 2024 Offering”) of 1,500,000 shares (the “Shares”) of Common Stock, pre-funded warrants (the “Pre-funded Warrants”) to purchase 600,000 shares of Common Stock, and warrants to purchase up to 1,050,000 shares of Common Stock (the “Common Warrants”) (CUSIP 09074F132) at a combined public offering price of $10.00 per Share, or Pre-funded Warrant, and the associated Common Warrant. The gross proceeds to the Company from the March 2024 Offering were approximately $21 million, before deducting placement agent fees and offering expenses of approximately $2.5 million. Upon closing of the March 2024 Offering, the Company issued the placement agent a warrant (“Placement Agent’s Warrant”) to purchase 105,000 shares of Common Stock exercisable at a per share price of $12.50, which was equal to 125% of the public offering price per Share. The Placement Agent’s Warrant is exercisable during a five-year period commencing 180 days from March 6, 2024.
Off-Balance Sheet Arrangements
The term “off-balance sheet arrangement” generally means any transaction, agreement or other contractual arrangement to which an entity unconsolidated with the Company is a party, under which the Company has (i) any obligation arising under a guarantee contract, derivative instrument or variable interest; or (ii) a retained or contingent interest in assets transferred to such entity or similar arrangement that serves as credit, liquidity or market risk support for such assets. The Company has no off-balance sheet arrangements that have or are reasonably likely to have a current or future effect or change on the Company’s financial condition, revenues or expenses, results of operations, liquidity, capital expenditures or capital resources that are material to investors.
Critical Accounting Policies and Estimates
Cash and cash equivalents
Cash and cash equivalents consisted of cash deposits and money market funds held at a bank and funds held in a brokerage account which included a U.S. treasury money market fund and U.S. Treasury Bills with original maturities of three months or less.
Concentration of Credit Risk in the Financial Service Industry
As of June 30, 2024, the Company had cash deposited in certain financial institutions in excess of federally insured levels. The Company regularly monitors the financial stability of these financial institutions and believes that it is not exposed to any significant credit risk in cash and cash equivalents. However, in March and April 2023, certain U.S. government banking regulators took steps to intervene in the operations of certain financial institutions due to liquidity concerns, which caused general heightened uncertainties in financial markets. While these events have not had a material direct impact on the Company’s operations, if further liquidity and financial stability concerns arise with respect to banks and financial institutions, either nationally or in specific regions, the Company’s ability to access cash or enter into new financing arrangements may be threatened, which could have a material adverse effect on its business, financial condition and results of operations.
Investments in U.S. Treasury Bills
Investments in U.S. Treasury Bills with maturities greater than three months, are accounted for as available for sale and are recorded at fair value. Unrealized gains were included in other comprehensive income in the accompanying statements of operations and comprehensive loss.
|
|
Research and Development
Research and development expenses consist primarily of costs associated with the preclinical and/or clinical trials of drug candidates, compensation and other expenses for research and development, personnel, supplies and development materials, costs for consultants and related contract research and facility costs.
Accounting for Stock-based Compensation
The Company follows the provision of Accounting Standards Codification (“ASC”) Topic 718 - Stock Compensation (“ASC 718”), which requires the measurement of compensation expense for all share-based payment awards made to employees and non-employee director, including employee stock options. Share-based compensation expense is based on the grant date fair value estimated in accordance with the provisions of ASC 718 and is generally recognized as an expense over the requisite service period, net of forfeitures which are recorded as they occur.
Fair value measurement of assets and liabilities
We determine the fair values of our financial instruments based on the fair value hierarchy, which requires an entity to maximize the use of observable inputs and minimize the use of unobservable inputs when measuring fair value. Fair value is defined as the price that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date. The fair value assumes that the transaction to sell the asset or transfer the liability occurs in the principal or most advantageous market for the asset or liability and establishes that the fair value of an asset or liability shall be determined based on the assumptions that market participants would use in pricing the asset or liability. The classification of a financial asset or liability within the hierarchy is based upon the lowest level input that is significant to the fair value measurement. The fair value hierarchy prioritizes the inputs into three levels that may be used to measure fair value:
Level 1 - Inputs are unadjusted quoted prices in active markets for identical assets or liabilities.
Level 2 - Inputs are quoted prices for similar assets and liabilities in active markets or inputs that are observable for the asset or liability, either directly or indirectly through market corroboration, for substantially the full term of the financial instrument.
Level 3 - Inputs are unobservable inputs based on our assumptions.
| ITEM 7A. | QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK |
Not applicable.
| ITEM 8. | FINANCIAL STATEMENTS |
Our financial information required to be filed hereunder are indexed under Item 15 of this report and are incorporated herein by reference.
| ITEM 9. | CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURES |
Not applicable.
| ITEM 9A. | CONTROLS AND PROCEDURES |
Evaluation of Disclosure Controls and Procedures
We have evaluated, with the participation of our principal executive and our principle financial officer, the effectiveness of our disclosure controls and procedures as defined in Rules 13a-15(e) and 15(d)-15(e) under the Exchange Act as of the end of the period covered by this Annual Report on Form 10-K. Based on this evaluation, our principal executive officer and our principal financial officer have concluded that our disclosure controls and procedures were effective to ensure that information required to be disclosed by us in the reports that we file or submit under the Exchange Act is recorded, processed, summarized and reported, within the time periods specified in the Commission’s rules and forms, and is accumulated and communicated to our management, including our principal executive and principal financial officers, or persons performing similar functions, as appropriate to allow timely decisions regarding required disclosure.
|
|
Management’s Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting as defined in Rule 13a-15(f) and 15d-15(f) under the Exchange Act. Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Projections of any evaluation of the effectiveness of internal control to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with policies or procedures may deteriorate. Under the supervision and with the participation of our management, including our Chief Executive Officer and Chief Financial Officer, we conducted an evaluation of the effectiveness of our internal control over financial reporting as of June 30, 2024 using the criteria established in Internal Control Integrated Framework issued by the Committee of Sponsoring Organization of the Treadway Commission. Based on our evaluation using those criteria, our management has concluded that, as of June 30, 2024, our internal control over financial reporting was effective to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles for the reasons discussed above.
Changes in Internal Control Over Financial Reporting
There were no changes in our internal controls over financial reporting during the quarter ended June 30, 2024, that materially affected, or are reasonably likely to materially affect our internal controls over financial reporting.
| ITEM 9B. | OTHER INFORMATION |
None.
| ITEM 9C. | DISCLOSURE REGARDING FOREIGN JURISDICTIONS THAT PREVENT INSPECTIONS |
Not applicable.
|
|
PART III.
| ITEM 10. | DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE |
The information required by this item is included in our 2024 Proxy Statement and is incorporated herein by reference.
| ITEM 11. | EXECUTIVE COMPENSATION |
The information required by this item is included in our 2024 Proxy Statement and is incorporated herein by reference.
| ITEM 12. | SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS |
The information required by this item is included in our 2024 Proxy Statement and is incorporated herein by reference.
| ITEM 13. | CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE |
The information required by this item is included in our 2024 Proxy Statement and is incorporated herein by reference.
| ITEM 14. | PRINCIPAL ACCOUNTANT FEES AND SERVICES |
The information required by this item is included in our 2024 Proxy Statement and is incorporated herein by reference.
|
|
PART IV
| ITEM 15. | EXHIBITS AND FINANCIAL STATEMENT SCHEDULES |
(a)(1),(2) Financial Statements
The Financial Statements listed on page F-1 of this document are filed as part of this filing.
(a)(3) Exhibits
The following is a list of exhibits filed as a part of this report:
|
|
# Indicates a management contract or compensatory plan or arrangement
* Filed herewith.
** Furnished herewith.
|
|
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this Report to be signed on its behalf by the undersigned, thereunto duly authorized.
| Date: September 30, 2024 | BIOVIE INC. | ||
| By: | /s/ Cuong Do | ||
| Name: | Cuong Do | ||
| Title: | Chief Executive Officer (Principal Executive Officer) |
||
POWER OF ATTORNEY
Each person whose signature appears below constitutes and appoints Cuong Do and Joanne Wendy Kim, and each of them acting individually and without the other, as his or her true and lawful attorneys-in-fact and agents, with full power of substitution and re-substitution, for him or her and in his or her name, place, and stead, in any and all capacities, to sign any and all amendments (including post-effective amendments, exhibits thereto and other documents in connection therewith) to this Report, and to file the same, with all exhibits thereto, and other documents in connection therewith, with the Commission, granting unto said attorneys-in-fact and agents, and each of them, full power and authority to do and perform each and every act and thing requisite and necessary to be done in and about the premises, as fully to all intents and purposes as he or she might or could do in person, hereby ratifying and confirming all that said attorneys-in-fact and agents, or either of them individually, or their or his substitute or substitutes, may lawfully do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed by the following persons in the capacities and on the dates indicated.
| Person | Capacity | Date | ||
| /s/ Cuong Do | Chief Executive Officer | September 30, 2024 | ||
| Cuong Do | (Principal Executive Officer) | |||
| /s/ Joanne Wendy Kim | Chief Financial Officer | September 30, 2024 | ||
| Joanne Wendy Kim | (Principal Financial Officer) | |||
| /s/ Jim Lang | Director | September 30, 2024 | ||
| Jim Lang | ||||
| /s/ Michael Sherman | Director | September 30, 2024 | ||
| Michael Sherman | ||||
| /s/ Richard J. Berman | Director | September 30, 2024 | ||
| Richard J. Berman | ||||
| /s/ Robert Hariri | Director | September 30, 2024 | ||
| Robert Hariri | ||||
| /s/ Sigmund Rogich | Director | September 30, 2024 | ||
| Sigmund Rogich |
|
|
BioVie Inc.
Index to Financial Statements
| F- |
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders of
BioVie, Inc.
Opinion on the Financial Statements
We have audited the accompanying balance sheets of BioVie, Inc. (the “Company”) as of June 30, 2024 and 2023, and the related statements of operations and comprehensive loss, changes in stockholders’ equity, and cash flows for each of the years then ended, and the related notes (collectively referred to as the “financial statements”). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of June 30, 2024 and 2023, and the results of its operations and its cash flows for of the years then ended, in conformity with accounting principles generally accepted in the United States of America.
Going Concern
The accompanying financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 2 to the financial statements, the Company‘s recurring losses from operations and negative cash flows from operating activities raise substantial doubt about its ability to continue as a going concern. Management’s plans regarding these matters are also described in Note 2. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Basis for Opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (“PCAOB”) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits, we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
Critical Audit Matter
The critical audit matter communicated below is a matter arising from the current period audit of the financial statements that was communicated or required to be communicated to the audit committee and that: (1) relates to accounts or disclosures that are material to the financial statements and (2) involved our especially challenging, subjective, or complex judgments. The communication of the critical audit matter does not alter in any way our opinion on the financial statements, taken as a whole, and we are not, by communicating the critical audit matter below, providing a separate opinion on the critical audit matter or on the accounts or disclosures to which it relates.
Research and development expenses and related accruals
As described in Note 3 to the accompanying financial statements, research and development expenses consist primarily of costs associated with the preclinical and/or clinical trials of drug candidates, compensation and other expenses for research and development, personnel, supplies and development materials, costs for consultants and related contract research and third-party facility costs. The amounts recorded for clinical trial expenses represent the Company’s estimates of clinical trial expenses based on facts and circumstances known to the Company at that time, and are dependent upon the timely and accurate reporting of contract research organizations and other third-party vendors.
| F- |
We identified the accounting for the research and development expenses and related accruals to be a critical audit matter due to the degree of management judgement in ensuring they are complete, accurate and classified correctly, their significance, and the risk of material misstatement due to the nature and timing of these costs and accruals. This in turn led to a high degree of auditor judgment, subjectivity, and effort in applying the procedures related to their accounting.
Addressing the matter involved performing procedures and evaluating audit evidence in connection with forming our overall opinion on the financial statements. These procedures included, obtaining an understanding of management’s process and evaluating the design of controls over research and development expense classification and the completeness and accuracy of related accruals, independently researching vendors, testing a selection of research and development expense transactions to determine, based on the underlying supporting documents, the mathematical accuracy of the expense and the appropriateness of the expense classification. In addition, we made inquiries of management and reviewed subsequent payments, invoices and agreements relating to certain research and development expenses to ensure that accruals were properly recorded as of June 30, 2024.
/s/ EisnerAmper LLP
We have served as the Company’s auditor since 2019.
EISNERAMPER LLP
Iselin, New Jersey
September 30, 2024
| F- |
BioVie Inc.
Balance Sheets
| June 30, | June 30, | |||||||
| 2024 | 2023 | |||||||
| ASSETS | ||||||||
| CURRENT ASSETS: | ||||||||
| Cash and cash equivalents | $ | 23,843,798 | $ | 19,460,883 | ||||
| Investments in U.S. Treasury Bills (available-for-sale) | - | 14,477,726 | ||||||
| Prepaid and other current assets | 204,392 | 102,526 | ||||||
| Total current assets | 24,048,190 | 34,041,135 | ||||||
| Operating lease right-of-use assets, net | 406,726 | 80,789 | ||||||
| Intangible assets, net | 407,718 | 637,095 | ||||||
| Goodwill | 345,711 | 345,711 | ||||||
| TOTAL ASSETS | $ | 25,208,345 | $ | 35,104,730 | ||||
| LIABILITIES AND STOCKHOLDERS' EQUITY | ||||||||
| CURRENT LIABILITIES: | ||||||||
| Accounts payable and accrued expenses | $ | 3,586,912 | $ | 3,476,259 | ||||
| Other current liabilities | - | 48,385 | ||||||
| Current portion of operating lease liabilities | 60,343 | 44,909 | ||||||
| Current portion of note payable, net of financing cost, unearned premium and discount of $701,210 at June 30, 2024 and $894,926 at June 30, 2023 | 5,701,210 | 9,105,074 | ||||||
| Warrant liability | 3,771 | 894,280 | ||||||
| Embedded derivative liability | - | 925,762 | ||||||
| Total current liabilities | 9,352,236 | 14,494,669 | ||||||
| Operating lease liabilities, net of current portion | 349,894 | 42,505 | ||||||
| Note payable, net of current portion, financing cost, unearned premium and discount of $0 and $227,268 at June 30, 2024 and June 30, 2023, respectively. | - | 5,227,270 | ||||||
| TOTAL LIABILITIES | 9,702,130 | 19,764,444 | ||||||
| Commitments and contingencies (Note 11) | ||||||||
| STOCKHOLDERS' EQUITY : | ||||||||
| Preferred stock; $0.001 par value; 10,000,000 shares authorized; 0 shares issued and outstanding | - | - | ||||||
| Common stock, $0.0001 par value; 800,000,000 shares authorized at June 30, 2024 and June 30, 2023, respectively; 6,216,398 shares issued of which 6,190,072 shares are outstanding at June 30, 2024; and 3,645,183 shares issued of which 3,642,895 shares outstanding at June 30, 2023 | 6,229 | 3,643 | ||||||
| Additional paid in capital | 349,732,674 | 316,385,759 | ||||||
| Accumulated other comprehensive income | - | 176,591 | ||||||
| Accumulated deficit | (334,232,661 | ) | (301,225,705 | ) | ||||
| Treasury stock | (27 | ) | (2 | ) | ||||
| Total stockholders' equity | 15,506,215 | 15,340,286 | ||||||
| TOTAL LIABILITIES AND STOCKHOLDERS' EQUITY | $ | 25,208,345 | $ | 35,104,730 | ||||
The accompanying notes are an integral part of the financial statements.
| F- |
BioVie Inc.
Statements of Operations and Comprehensive Loss
| Year ended | Year ended | |||||||
| June 30, 2024 | June 30, 2023 | |||||||
| OPERATING EXPENSES: | ||||||||
| Amortization of intangible assets | $ | 229,377 | $ | 229,377 | ||||
| Research and development expenses | 23,100,394 | 33,299,503 | ||||||
| Selling, general and administrative expenses | 8,849,814 | 11,551,568 | ||||||
| TOTAL OPERATING EXPENSES | 32,179,585 | 45,080,448 | ||||||
| LOSS FROM OPERATIONS | (32,179,585 | ) | (45,080,448 | ) | ||||
| OTHER (INCOME) EXPENSE: | ||||||||
| Change in fair value of derivative liabilities | (1,816,271 | ) | 1,437,481 | |||||
| Interest expense | 2,893,922 | 4,300,150 | ||||||
| Interest income | (1,136,703 | ) | (562,264 | ) | ||||
| TOTAL OTHER (INCOME) EXPENSE, NET | (59,052 | ) | 5,175,367 | |||||
| NET LOSS | $ | (32,120,533 | ) | $ | (50,255,815 | ) | ||
| Deemed dividend related to ratchet adjustment to warrants | 886,423 | - | ||||||
| NET LOSS ATTRIBUTABLE TO COMMON STOCKHOLDERS | $ | (33,006,956 | ) | $ | (50,255,815 | ) | ||
| NET LOSS PER COMMON SHARE | ||||||||
| - Basic | $ | (7.30 | ) | $ | (15.47 | ) | ||
| - Diluted | $ | (7.30 | ) | $ | (15.47 | ) | ||
| WEIGHTED AVERAGE NUMBER OF COMMON SHARES OUTSTANDING | ||||||||
| - Basic | 4,518,533 | 3,248,349 | ||||||
| - Diluted | 4,518,533 | 3,248,349 | ||||||
| NET LOSS ATTRIBUTABLE TO COMMON STOCKHOLDERS | $ | (33,006,956 | ) | $ | (50,255,815 | ) | ||
| Other comprehensive (loss) income | ||||||||
| Unrealized gain on available-for-sale investments | - | 176,591 | ||||||
| Reclassification of unrealized gains on available-for-sale investments upon settlement | (176,591 | ) | - | |||||
| Total other comprehensive (loss) income | (176,591 | ) | 176,591 | |||||
| Comprehensive loss | $ | (33,183,547 | ) | $ | (50,079,224 | ) | ||
The accompanying notes are an integral part of the financial statements.
| F- |
BioVie Inc.
Statements of Changes in Stockholders’ Equity
For the Years Ended June 30, 2024 and 2023
| Accumulated | ||||||||||||||||||||||||||||||||
| Additional | Other | Total | ||||||||||||||||||||||||||||||
| Common Stock | Common Stock | Paid in | Treasury Stock | Treasury Stock | Comprehensive | Accumulated | Stockholders' | |||||||||||||||||||||||||
| Shares | Amount | Capital | Shares | Amount | Income | Deficit | Equity | |||||||||||||||||||||||||
| Balance, June 30, 2022 | 2,498,408 | $ | 2,496 | $ | 254,638,329 | - | $ | - | $ | - | $ | (250,969,890 | ) | 3,670,935 | ||||||||||||||||||
| Stock option-based compensation | - | - | 4,222,845 | - | - | - | - | 4,222,845 | ||||||||||||||||||||||||
| Stock-based compensation - restricted stock units | 21,518 | 21 | 1,780,028 | (2,288 | ) | (2 | ) | - | - | 1,780,047 | ||||||||||||||||||||||
| Stock-based compensation - issuance of common stock for services rendered | 5,000 | 5 | 372,495 | - | - | - | - | 372,500 | ||||||||||||||||||||||||
| - | ||||||||||||||||||||||||||||||||
| Cashless exercise of options | 2,256 | 3 | (3 | ) | - | - | - | - | - | |||||||||||||||||||||||
| - | ||||||||||||||||||||||||||||||||
| Cashless exercise of warrants | 359 | - | - | - | - | - | - | - | ||||||||||||||||||||||||
| - | ||||||||||||||||||||||||||||||||
| Proceeds from exercise of options | 80 | - | 2,240 | - | - | - | - | 2,240 | ||||||||||||||||||||||||
| Proceeds from issuance of common stock, net costs of $2,008,898 | 753,925 | 754 | 49,464,349 | - | - | - | - | 49,465,103 | ||||||||||||||||||||||||
| Proceeds from issuance of common stock, net of costs of $94,160 - Related Party | 363,636 | 364 | 5,905,476 | - | - | - | - | 5,905,840 | ||||||||||||||||||||||||
| Unrealized gain on available-for-sale investments | - | - | - | - | - | 176,591 | - | 176,591 | ||||||||||||||||||||||||
| Net loss | - | - | - | - | - | - | (50,255,815 | ) | (50,255,815 | ) | ||||||||||||||||||||||
| Balance, June 30, 2023 | 3,645,183 | 3,643 | 316,385,759 | (2,288 | ) | (2 | ) | 176,591 | (301,225,705 | ) | 15,340,286 | |||||||||||||||||||||
| Stock - based compensation - stock options | - | - | 2,823,764 | - | - | - | - | 2,823,764 | ||||||||||||||||||||||||
| Stock-based compensation - restricted stock units | - | - | 1,763,450 | - | - | - | - | 1,763,450 | ||||||||||||||||||||||||
| Proceeds from issuance of common stock, net of costs of $2,908,141 | 2,433,749 | 2,449 | 27,800,490 | - | - | - | - | 27,802,939 | ||||||||||||||||||||||||
| Issuance of common stock from vesting of restricted stock units | 122,395 | 122 | (97 | ) | (24,038 | ) | (25 | ) | - | - | - | |||||||||||||||||||||
| Stock-based compensation - issuance of common stock for services rendered | 15,000 | 15 | 72,885 | - | - | - | - | 72,900 | ||||||||||||||||||||||||
| Deemed dividend for ratchet adjustment to warrants | - | - | 886,423 | - | - | - | (886,423 | ) | - | |||||||||||||||||||||||
| Relcassification of unrealized gains on available-for-sale investments upon settlement | - | - | - | - | - | (176,591 | ) | - | (176,591 | ) | ||||||||||||||||||||||
| Issuance of additional shares for fractional shares effected by the reverse split | 71 | - | - | - | - | - | - | - | ||||||||||||||||||||||||
| Net Loss | - | - | - | - | - | - | (32,120,533 | ) | (32,120,533 | ) | ||||||||||||||||||||||
| Balance, June 30, 2024 | 6,216,398 | $ | 6,229 | $ | 349,732,674 | (26,326 | ) | $ | (27 | ) | $ | - | $ | (334,232,661 | ) | $ | 15,506,215 | |||||||||||||||
The accompanying notes are an integral part of the financial statements.
| F- |
BioVie Inc.
Statements of Cash Flows
| Year ended | Year ended | |||||||
| June 30, 2024 | June 30, 2023 | |||||||
| CASH FLOWS FROM OPERATING ACTIVITIES: | ||||||||
| Net loss | $ | (32,120,533 | ) | $ | (50,255,815 | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||
| Amortization of intangible assets | 229,377 | 229,377 | ||||||
| Stock based compensation - restricted stock units | 1,763,450 | 1,780,047 | ||||||
| Stock based compensation expense - stock options | 2,823,764 | 4,222,845 | ||||||
| Stock based compensation expense - stock issued | 72,900 | 372,500 | ||||||
| Amortization of financing costs | 108,751 | 170,219 | ||||||
| Accretion of unearned loan discount | 1,023,145 | 1,601,445 | ||||||
| Accretion of loan premium | 236,970 | 421,994 | ||||||
| Realized gain on maturity of available-for sale | (223,865 | ) | - | |||||
| Non-cash lease expense from right-of-use assets | 49,346 | 37,465 | ||||||
| Gain on termination of operating lease | (5,215 | ) | - | |||||
| Change in fair value of derivative liabilities | (1,816,271 | ) | 1,437,481 | |||||
| Changes in operating assets and liabilities: | ||||||||
| Prepaid and other assets | (101,866 | ) | 39,915 | |||||
| Accounts payable and accrued expenses | 110,653 | 1,033,455 | ||||||
| Operating lease liabilities | (47,245 | ) | (38,884 | ) | ||||
| Other current liabilities | (48,385 | ) | (1,304,925 | ) | ||||
| Net cash used in operating activities | (27,945,024 | ) | (40,252,881 | ) | ||||
| CASH FLOWS FROM INVESTING ACTIVITIES: | ||||||||
| Proceeds from (purchases of) U.S. Treasury Bills (available-for-sale) | 14,525,000 | (14,301,135 | ) | |||||
| Net cash provided by (used in) investing activities | 14,525,000 | (14,301,135 | ) | |||||
| CASH FLOWS FROM FINANCING ACTIVITIES: | ||||||||
| Net proceeds from issuance of common stock | 27,802,939 | 49,465,103 | ||||||
| Payments of note payable | (10,000,000 | ) | - | |||||
| Proceeds from exercise of stock options | - | 2,240 | ||||||
| Net proceeds from issuance of common stock - Related Party | - | 5,905,840 | ||||||
| Net cash provided by financing activities | 17,802,939 | 55,373,183 | ||||||
| Net increase in cash and cash equivalents | 4,382,915 | 819,167 | ||||||
| Cash and cash equivalents, beginning of period | 19,460,883 | 18,641,716 | ||||||
| Cash and cash equivalents, end of period | $ | 23,843,798 | $ | 19,460,883 | ||||
| SUPPLEMENTAL CASH FLOW INFORMATION: | ||||||||
| Cash paid for interest | $ | 1,525,056 | $ | 2,106,491 | ||||
| SUPPLEMENTAL DISCLOSURE OF NONCASH INVESTING ACTIVITIES: | ||||||||
| Right-of-use assets obtained in exchange for lease obligations | $ | 432,192 | $ | - | ||||
| Unrealized gain on U.S. Treasury Bills (available-for-sale) | $ | - | $ | 176,591 | ||||
| Reclassification of unrealized gains on U.S. Treasury Bills (available-for-sale investments) upon settlement | $ | 176,591 | $ | - | ||||
| Deemed dividend of ratchet adjustment to warrants | $ | 886,423 | $ | - | ||||
The accompanying notes are an integral part of the financial statements.
| F- |
BioVie Inc.
Notes to Financial Statements
| 1. | Background Information |
BioVie Inc. (the “Company” or “we” or “our”) is a clinical-stage company developing innovative drug therapies to treat chronic debilitating conditions including neurological and neuro-degenerative disorders and liver disease.
The Company acquired the biopharmaceutical assets of NeurMedix, Inc. (“NeurMedix”) a privately held clinical-stage pharmaceutical company and a related party in June 2021. The acquired assets included NE3107. NE3107 is an investigational, novel, orally administered small molecule that is thought to inhibit inflammation-driven insulin resistance and major pathological inflammatory cascades with a novel mechanism of action. There is emerging scientific consensus that both inflammation and insulin resistance may play fundamental roles in the development of Alzheimer’s disease (“AD”) and Parkinson’s disease (“PD”), and NE3107 could, if approved by the U.S. Food and Drug Administration (“FDA”), represent an entirely new medical approach to treating these devastating conditions affecting an estimated 6 million Americans suffering from AD and 1 million Americans suffering from PD.
Neurodengenerative Disease Program
In neurodegenerative disease, the Company’s drug candidate NE3107 inhibits activation of inflammatory actions extracellular single-regulated kinase (“ERK”) and nuclear factor kappa-light-chain-enhancer of activated B cells (“NFκB”) (including interactions with tumor necrosis factor (“TNF”) signaling and other relevant inflammatory pathways) that lead to neuroinflammation and insulin resistance. NE3107 does not interfere with their homeostatic functions (e.g., insulin signaling and neuron growth and survival). Both inflammation and insulin resistance are drivers of AD and PD.
Alzheimer’s Disease (NCT05083260)
On November 29, 2023, the Company announced the analysis of its unblinded, topline efficacy data from its Phase 3 clinical trial (NCT04669028) of NE3107 in the treatment of mild to moderate AD. The study has co-primary endpoints looking at cognition using the Alzheimer’s Disease Assessment Scale-Cognitive Scale (ADAS-Cog 12) and function using the Clinical Dementia Rating-Sum of Boxes (CDR-SB). Patients were randomly assigned, 1:1 versus placebo, to receive sequentially 5 mg of NE3107 orally twice a day for 14 days, then 10 mg orally twice a day for 14 days, followed by 26 weeks of 20 mg orally twice daily.
Upon trial completion, as the Company began the process of unblinding the trial data, the Company found significant deviation from protocol and current good clinical practices (“cGCPs”) violations at 15 study sites (virtually all of which were from one geographic area). This highly unusual level of suspected improprieties led the Company to exclude all patients from these sites and to refer the sites to the FDA Office of Scientific Investigations (“OSI”) for potential further action. After the patient exclusions, 81 patients remained in the Modified Intent to Treat population, 57 of whom were in the Per-Protocol population which included those who completed the trial and were verified to take study drug from pharmacokinetic data.
The trial was originally designed to be 80% powered with 125 patients in each of the treatment and placebo arms. The unplanned exclusion of so many patients has left the trial underpowered for the primary endpoints. In the Per-Protocol population, which included those patients who completed the trial and who were further verified to have taken the study drug (based on pharmacokinetic data), an observed descriptive change from baseline appeared to suggest a slowing of cognitive loss; these same patients experienced an advantage in age deceleration vs. placebo as measured by DNA epigenetic change. Age deceleration is used by longevity researchers to measure the difference between the patient’s biological age, in this case as measured by the Horvath DNA methylation Skin Blood Clock, relative to the patient’s actual chronological age. This test was a non-primary/secondary endpoint, other-outcome measure, done via blood test collected at week 30 (end of study). Additional DNA methylation data continues to be collected and analyzed.
Parkinson’s Disease (NCT05083260)
The Phase 2 study of bezisterim (NE3107) for the treatment of PD (NCT05083260), completed in December 2022, was a double-blind, placebo-controlled, safety, tolerability, and pharmacokinetics study in PD participants treated with carbidopa/levodopa and bezisterim (NE3107). Forty-five patients with a defined L-dopa “off state” were randomized 1:1 to placebo: bezisterim (NE3107) 20 mg twice daily for 28 days. This trial was launched with two design objectives: 1) the primary objective was safety and a drug-drug interaction study as requested by the FDA to measure the potential for adverse interactions of bezisterim (NE3107) with carbidopa/ levodopa; and 2) the secondary objective was to determine if preclinical indications of promotoric activity and apparent enhancement of levodopa activity could be seen in humans. Both objectives were met.
| F- |
| 1. | Background Information (continued) |
Long COVID Program
In April 2024, the Company announced the grant of a clinical trial award of up to $13.1 million from the DOD, awarded through the Peer Reviewed Medical Research Program (“PRMRP”) of the Congressionally Directed Medical Research Programs (“CDMRP”). The award can provide up to 2 years of non-dilutive funding for a Phase 2b clinical trial that will assess bezisterim (NE3107) for the treatment of neurological symptoms that are associated with long COVID. The Company anticipates the trial to commence by early 2025.
Liver Disease Program
In liver disease, our investigational drug candidate BIV201 (continuous infusion terlipressin), which has been granted both FDA Fast Track designation status and FDA Orphan Drug status, is being evaluated and discussed after receiving guidance from the FDA regarding the design of Phase 3 clinical testing of BIV201 for the treatment of ascites due to chronic liver cirrhosis. BIV201 is administered as a patent-pending liquid formulation.
In June 2021, the Company initiated a Phase 2 study (NCT04112199) designed to evaluate the efficacy of BIV201 (terlipressin, administered by continuous infusion for two 28-day treatment cycles) combined with standard-of-care (“SOC”), compared to SOC alone, for the treatment of refractory ascites. The primary endpoints of the study are the incidence of ascites-related complications and change in ascites fluid accumulation during treatment compared to a pre-treatment period.
In March 2023, the Company announced enrollment was paused and that data from the first 15 patients treated with BIV201 plus SOC appeared to show at least a 30% reduction in ascites fluid during the 28 days after treatment initiation compared to the 28 days prior to treatment. The change in ascites volume was significantly different from those patients receiving SOC treatment. Patients who completed the treatment with BIV201 experienced a 53% reduction in ascites fluid, which was sustained (43% reduction) during the three months after treatment initiation as compared to the three-month pre-treatment period.
In June 2023, the Company requested and subsequently received guidance from the FDA regarding the design and endpoints for definitive clinical testing of BIV201 for the treatment of ascites due to chronic liver cirrhosis. The Company is currently finalizing protocol designs for the Phase 3 study of BIV201 for the treatment of ascites due to chronic liver cirrhosis.
The BIV201 development program was initiated by LAT Pharma LLC. On April 11, 2016, the Company acquired LAT Pharma LLC and the rights to its BIV201 development program. The Company currently owns all development and marketing rights to this drug candidate. Pursuant to the Agreement and Plan of Merger entered into on April 11, 2016, between our predecessor entities, LAT Pharma LLC and NanoAntibiotics, Inc., BioVie is obligated to pay a low single digit royalty on net sales of BIV201 (continuous infusion terlipressin). to be shared among LAT Pharma Members, PharmaIn Corporation, and The Barrett Edge, Inc.
| 2. | Liquidity and Going Concern |
The Company’s operations are subject to a number of factors that can affect its operating results and financial conditions. Such factors include, but are not limited to: the results of clinical testing and trial activities of the Company’s products, the Company’s ability to obtain regulatory approval to market its products; competition from products manufactured and sold or being developed by other companies; the price of, and demand for, Company products; the Company’s ability to negotiate favorable licensing or other manufacturing and marketing agreements for its products; and the Company’s ability to raise capital. The Company’s financial statements have been prepared assuming the Company will continue as a going concern, which contemplates the realization of assets and the satisfaction of liabilities in the normal course of business. As of June 30, 2024, the Company had working capital of approximately $14.7 million, cash and cash equivalents of approximately $23.8 million, stockholders’ equity of approximately $15.5 million, and an accumulated deficit of approximately $334.2 million. The Company is in the pre-revenue stage and no revenues are expected in the foreseeable future. The Company’s future operations are dependent on the success of the Company’s ongoing development and commercialization efforts, as well as its ability to secure additional financing as needed. Projected cash flows could be extended if further measures are taken to delay planned expenditures in our research protocols and slow the progress in the Company’s development and launch of next phase clinical programs.
The future viability of the Company is largely dependent upon its ability to raise additional capital to finance its operations. Management expects that future sources of funding may include sales of equity, obtaining loans, or other strategic transactions.
| F- |
| 2. | Liquidity and Going Concern (continued) |
Although management continues to pursue the Company’s strategic plans, there is no assurance that the Company will be successful in obtaining sufficient financing on terms acceptable to the Company, if at all, to fund continuing operations. These circumstances raise substantial doubt on the Company’s ability to continue as a going concern. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
| 3. | Significant Accounting Policies |
Basis of Presentation
The Company’s financial statements have been prepared in accordance with accounting principles generally accepted in the United States (“GAAP”) and include all adjustments necessary for the fair presentation of the Company’s financial position for the periods presented.
Use of Estimates
The preparation of financial statements in conformity with GAAP requires management to make estimates and assumptions that affect the amounts reported in the financial statements and accompanying notes. The Company bases its estimates on historical experience and on various assumptions that are believed to be reasonable under the circumstances. The amounts of assets and liabilities reported in the Company’s balance sheets and the amounts of expenses reported for each of the periods presented in the statements of operations and comprehensive loss are affected by estimates and assumptions, which are used for, but not limited to, accounting for clinical accruals, share-based compensation, accounting for derivatives, assumptions used in recording leases, the inputs used in the valuation of goodwill and intangible assets in connection with impairment testing and accounting for income taxes. Actual results could differ from those estimates.
Cash and cash equivalents
Cash and cash equivalents consisted of cash deposits and money market funds held at a bank and funds held in a brokerage account which included a U.S. treasury money market fund and U.S. Treasury Bills with original maturities of three months or less.
Investments in U.S. Treasury Bills
Investments in U.S. Treasury Bills with maturities greater than three months, are accounted for as available-for-sale and are recorded at fair value. Unrealized gains were included in other comprehensive income in the accompanying statements of operations and comprehensive loss.
Concentration of Credit Risk in the Financial Service Industry
As of June 30, 2024, the Company had cash deposited in certain financial institutions in excess of federally insured levels. The Company regularly monitors the financial stability of these financial institutions and believes that it is not exposed to any significant credit risk in cash and cash equivalents. However, in March and April 2023, certain U.S. government banking regulators took steps to intervene in the operations of certain financial institutions due to liquidity concerns, which caused general heightened uncertainties in financial markets. While these events have not had a material direct impact on the Company’s operations, if further liquidity and financial stability concerns arise with respect to banks and financial institutions, either nationally or in specific regions, the Company’s ability to access cash or enter into new financing arrangements may be threatened, which could have a material adverse effect on its business, financial condition and results of operations.
| F- |
| 3. | Significant Accounting Policies (continued) |
Fair value measurement of assets and liabilities
We determine the fair values of our financial instruments based on the fair value hierarchy, which requires an entity to maximize the use of observable inputs and minimize the use of unobservable inputs when measuring fair value. Fair value is defined as the price that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date. The fair value assumes that the transaction to sell the asset or transfer the liability occurs in the principal or most advantageous market for the asset or liability and establishes that the fair value of an asset or liability shall be determined based on the assumptions that market participants would use in pricing the asset or liability. The classification of a financial asset or liability within the hierarchy is based upon the lowest level input that is significant to the fair value measurement. The fair value hierarchy prioritizes the inputs into three levels that may be used to measure fair value:
Level 1 - Inputs are unadjusted quoted prices in active markets for identical assets or liabilities.
Level 2 - Inputs are quoted prices for similar assets and liabilities in active markets or inputs that are observable for the asset or liability, either directly or indirectly through market corroboration, for substantially the full term of the financial instrument.
Level 3 - Inputs are unobservable inputs based on our assumptions.
The Company’s financial instruments include cash, accounts payable, the carrying value of the operating lease liabilities and notes payable. The carrying amounts of cash and accounts payable approximate their fair value, due to the short-term nature of these items. The carrying amounts of notes payable and operating lease liabilities approximate their fair values since they bear interest at rates which approximate market rates for similar debt instruments.
Prepaid and other assets
Prepaid and other assets consist of prepayments of certain expenses and a security deposit paid in connection with a lease agreement.
Leases
The Company determines whether an arrangement contains a lease at inception. Operating leases are included in operating lease right-of-use (“ROU”) assets, current portion of operating lease liabilities, and operating lease liabilities, net of current portion on our balance sheets. ROU assets represent the Company’s right to use an underlying asset for the lease term and lease liabilities represent an obligation to make lease payments arising from the lease. ROU assets and lease liabilities are recognized based on the present value of the future minimum lease payments over the lease term at the commencement date. As the Company’s leases do not provide an implicit rate, an incremental borrowing rate is used based on the information available at the commencement date in determining the present value of lease payments. The Company does not include options to extend or terminate the lease term in its calculation unless it is reasonably certain that the Company will exercise any such options. Rent expense is recognized under the operating leases on a straight-line basis. The Company does not recognize right-of-use assets or lease liabilities for short-term leases, which have a lease term of 12 months or less at inception, and instead will recognize lease payments as expense on a straight-line basis over the lease term.
Research and Development
Research and development expenses consist primarily of costs associated with the preclinical and/or clinical trials of drug candidates, compensation and other expenses for research and development, personnel, supplies and development materials, costs for consultants and related contract research and facility costs.
Income Taxes
The Company uses the asset and liability method of accounting for deferred income taxes. Deferred income taxes are measured by applying enacted statutory rates to net operating loss carryforwards and to the differences between the financial reporting and tax bases of assets and liabilities. Deferred tax assets are reduced, by a valuation allowance if it is more likely than not that some portion or all of the deferred tax assets will not be realized. The Company decided to apply a full valuation allowance against its deferred tax assets due to the continuing losses.
| F- |
| 3. | Significant Accounting Policies (continued) |
The Company recognizes uncertainty in income taxes in the financial statements using a recognition threshold and measurement attribute of a tax position taken or expected to be taken in a tax return. The Company applies the “more-likely-than-not” recognition threshold to all tax positions, commencing at the adoption date of the applicable accounting guidance, which resulted in no unrecognized tax benefits as of such date. Additionally, there have been no unrecognized tax benefits subsequent to adoption. The Company has opted to classify interest and penalties that would accrue, if any, according to the provisions of relevant tax law as general and administrative expenses, in the Statements of Operations and Comprehensive Loss. For the years ended June 30, 2024 and 2023, there was no such interest or penalty.
Basic net loss per common share is computed by dividing the net loss attributable to Common Stockholders by the weighted average number of shares of Common Stock outstanding during the period. Diluted net loss per common share is computed by dividing the net loss attributable to Common Stockholders by the weighted average number of shares of Common Stock outstanding and potentially outstanding shares of Common Stock during the period to reflect the potential dilution that could occur from common shares issuable through stock options, warrants, and convertible debentures. For the years ended June 30, 2024 and 2023, such amounts were excluded from the diluted loss since their effect was considered anti-dilutive due to the net loss for the periods presented.
The table below shows the potential shares of common stock, presented based on amounts outstanding at each year end, that were excluded from the computation of diluted net loss per share attributable to common stockholders because including them would have had an anti-dilutive effect:
| Schedule of dilutive securities were excluded from the computation of diluted loss per share | ||||||||
| June 30, 2024 | June 30, 2023 | |||||||
| Number of Shares | Number of Shares | |||||||
| Stock Options | 518,076 | 395,286 | ||||||
| Warrants | 1,932,029 | 777,029 | ||||||
| Restricted Stock Units | 40,291 | 59,646 | ||||||
Notes payable conversion option |
71,633 | 71,633 | ||||||
| Total | 2,562,029 | 1,303,594 | ||||||
Stock-based Compensation
The Company has accounted for stock-based compensation under the provisions of Accounting Standards Codification (“ASC”) Topic 718 – “Stock Compensation” (“ASC 718”) which requires the use of the fair-value based method to determine compensation for all arrangements under which employees and others receive shares of stock or equity instruments (stock options and Common Stock purchase warrants). For employees and non-employees awards, the fair value of each stock option award is estimated on the date of grant using the Black-Scholes valuation model that uses assumptions for expected volatility, expected dividends, expected term, and the risk-free interest rate. For non-employees, the Company utilizes the graded vesting attribution method under which the entity treats each separately vesting portion (tranche) as a separate award and recognizes compensation cost for each tranche over its separate vesting schedule. Expected volatilities are based on historical volatility of peer companies and other factors estimated over the expected term of the stock options. For employee and non-employee awards, the expected term of options granted is derived using the “simplified method” which computes expected term as the average of the sum of the vesting term plus the contract term. The risk-free rate is based on the U.S. Treasury yield curve in effect at the time of grant for the period of the expected term. The Company recognizes forfeitures as they occur.
Goodwill
Goodwill is recorded when the purchase price paid for an acquisition exceeds the fair value of the net identified tangible and intangible assets acquired. The Company performs an annual impairment test of goodwill and further periodic tests to the extent indicators of impairment develop between annual impairment tests. The Company’s impairment review process compares the fair value of the reporting unit to its carrying value, including the goodwill related to the reporting unit. To determine the fair value of the reporting unit, the Company may use various approaches including an asset or cost approach, market approach or income approach or any combination thereof. These approaches may require the Company to make certain estimates and assumptions including future cash flows, revenue and expenses. These estimates and assumptions are reviewed each time the Company tests goodwill for impairment and are typically developed as part of the Company’s routine business planning and forecasting process. While the Company believes its estimates and assumptions are reasonable, variations from those estimates could produce materially different results. The Company did not recognize any goodwill impairments for the years ended June 30, 2024 and 2023.
| F- |
| 3. | Significant Accounting Policies (continued) |
Impairment of Long-Lived Assets
Long-lived assets, including intangible assets, are reviewed for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. Recoverability of assets to be held and used is measured by a comparison of the carrying amount of an asset to estimated undiscounted future cash flows expected to be generated by the asset.
If the carrying amount of an asset exceeds its undiscounted estimated future cash flows, an impairment review is performed. An impairment charge is recognized in the amount by which the carrying amount of the asset exceeds the fair value of the asset. Generally, fair value is determined using valuation techniques such as expected discounted cash flows or appraisals, as appropriate. Assets to be disposed of would be separately presented in the balance sheet and reported at the lower of the carrying amount or fair value less costs to sell, and are no longer depreciated or amortized. The assets and liabilities of a disposed group classified as held for sale would be presented separately in the appropriate asset and liability sections of the balance sheets. The Company did not recognize any long-lived asset impairments for the years ended June 30, 2024 and 2023.
Reverse stock split up
The company effected a 1:10 reverse split of the issued and outstanding shares of its Class A commons stock which was approved by the board of director after the approval obtained from shareholders at a special meeting on July 29, 2024 which became effective on Nasdaq on August 6, 2024, 5 trading days after the shareholders’ approval was obtained. All historical share and earnings per share amounts have been retroactively adjusted to reflect the split.
Recent Accounting Pronouncements
The Company considers the applicability and impact of all Accounting Standards Updates (“ASU’s”). There were no recent ASU’s that are expected to have a material impact on our balance sheets or statements of operations and comprehensive loss.
In June 2016, the Financial Accounting Standards Board (“FASB”) issued ASU No. 2016-13, “Financial Instruments - Credit Losses (Topic 326), Measurement of Credit Losses on Financial Instruments” (“ASU 2016-13”). This amendment replaces the incurred loss impairment methodology in current GAAP with a methodology that reflects expected credit losses on instruments within its scope, including trade receivables. This update is intended to provide financial statement users with more decision-useful information about the expected credit losses. The Company adopted ASU 2016-13 effective July 1, 2023 and the adoption had an insignificant impact on the accompanying financial statements.
In November 2023, the FASB issued Accounting Standards Update (ASU) 2023-07, "Segment Reporting (Topic 280): Improvements to Reportable Segment Disclosures," to enhance disclosures for significant segment expenses for all public entities required to report segment information in accordance with ASC 280. The standard did not change the definition of a segment, the method for determining segments or the criteria for aggregating operating segments into reportable segments. The amendments are effective for fiscal years beginning after December 15, 2023, and interim periods within fiscal years beginning after December 15, 2024. Retrospective adoption is required for all prior periods presented in the financial statements. The adoption is not expected to have a material impact to our financial statements or disclosures.
In December 2023, the FASB issued ASU 2023-09, "Income Taxes (Topic 740): Improvements in Income Tax Disclosures" to enhance the transparency and decision usefulness of income tax disclosures. This amendment requires public companies to disclose specific categories in the rate reconciliation and provide additional information for reconciling items that meet a quantitative threshold. Additionally, under the amendment entities are required to disclose the amount of income taxes paid disaggregated by federal, state and foreign taxes, as well as disaggregated by material individual jurisdictions. Finally, the amendment requires entities to disclose income from continuing operations before income tax expense disaggregated between domestic and foreign and income tax expense from continuing operations disaggregated by federal, state and foreign. The new rules are effective for annual periods beginning after December 15, 2024. We will adopt this standard on a prospective basis as allowed by the standard. The adoption of this standard is not expected to have a material impact on our financial statements.
| F- |
4. Investments in U.S. Treasury Bills available-for-sale
The following is a summary of the U.S. Treasury Bills held at June 30, 2023:
| Schedule of U.S. treasury bills held | ||||||||||||||||||||
| Amortized Cost Basis | Gross Unrealized Gain | Gross Unrealized loss | Fair Value | Total Accumulated Other Comprehensive Income | ||||||||||||||||
| U.S. Treasury Bills due in 3 - 6 months | $ | 14,301,136 | $ | 176,591 | $ | — | $ | 14,477,726 | $ | 176,591 | ||||||||||
During the fiscal year ended June 30, 2023, the Company purchased a total of approximately $46 million of U.S. Treasury Bills. All outstanding investments in U.S. Treasury Bills available-for-sale held at June 30, 2023 matured during the three months ended September 30, 2023 and were settled, resulting in a realized gain of $223,865 recorded as a component of interest income on the accompanying statement of operations and comprehensive loss.
| 5. | Intangible Assets |
The Company’s intangible assets consist of intellectual property acquired from LAT Pharma, Inc. and are amortized over their estimated useful lives. The following is a summary of the intangible assets as of June 30, 2024 and 2023:
| Schedule of intangible assets | ||||||||
| June 30, 2024 | June 30, 2023 | |||||||
| Intellectual Property | $ | 2,293,770 | $ | 2,293,770 | ||||
| Less: Accumulated Amortization | (1,886,052 | ) | (1,656,675 | ) | ||||
| Intellectual Property, Net | $ | 407,718 | $ | 637,095 | ||||
Amortization expense amounted to $229,377 for each of the years ended June 30, 2024 and 2023, respectively. The Company amortizes intellectual property over the expected original useful lives of 10 years.
Estimated future amortization expense is as follows:
| Schedule of future amortization expense | ||||
| Year ending June 30, | ||||
| 2025 | $ | 229,377 | ||
| 2026 | 178,341 | |||
| Finite lived intangible assets, net | $ | 407,718 | ||
| 6. | Related Party Transactions |
Equity Transactions with Acuitas
On July 15, 2022, the Company entered into a securities purchase agreement with Acuitas Group Holdings, LLC (“Acuitas”), the Company’s largest stockholder, pursuant to which Acuitas agreed to purchase from the Company, in a private placement, (i) an aggregate of 363,636 shares of the Company’s Common Stock, at a price of $16.50 per share (the “PIPE Shares”), and (ii) a warrant to purchase 727,273 shares of Common Stock (“PIPE Warrant Shares”), at an exercise price of $18.20, with a term of exercise of five years. The down round feature reduced the exercise price of the PIPE Warrant Shares to $10.00 per share on March 6, 2024 in connection with the offering further described in Note 9 as the Company sold stock at a price lower than its initial exercise price. The Company calculated the difference in fair value of the PIPE Warrant Shares between the stated exercise price and the reduced exercise price and recorded $886,423 as a deemed dividend. The fair value of the PIPE Warrant Shares were estimated using the Black Scholes Method with the following inputs, the stock price of $10.65, exercise price of $18.20 and $10.00, remaining term of 3.5 years, risk free rate of 4.4% and volatility of 95.0%.
On August 15, 2022, the Company received net proceeds of approximately $5.9 million, net of costs of approximately $94,000, and entered into an amended and restated registration agreement with Acuitas, which amended and restated that certain registration rights agreement, dated as of June 10, 2021, by and between the Company and Acuitas (the “Existing Registration Rights Agreement”), to amend the definition of “Registrable Securities” in the Existing Registration Rights Agreement to include the PIPE Shares and the PIPE Warrant Shares as Registrable Securities thereunder.
| F- |
| 7. | Notes Payable |
On November 30, 2021 (the “Closing Date”), the Company entered into a Loan and Security Agreement and the Supplement to the Loan and Security Agreement and Promissory Notes (together, the “Loan Agreement”) with Avenue Venture Opportunities Fund, L.P. (“AVOPI”) and Avenue Venture Opportunities Fund II, L.P. (“AVOPII,” and together with AVOPI, “Avenue”) for growth capital loans in an aggregate commitment amount of up to $20 million (the “Loan”). On the Closing Date, $15 million of the Loan was funded (“Tranche 1”). The Loan provided for an additional $5 million to be available to the Company on or prior to September 15, 2022, subject to the Company’s achievement of certain milestones with respect to certain of its ongoing clinical trials, which were not achieved. The Loan bears interest at an annual rate equal to the greater of (a) the sum of 7.00% plus the prime rate as reported in The Wall Street Journal and (b) 10.75%. The prime rate at June 30, 2024 was 8.50%. The Loan is secured by a lien upon and security interest in all of the Company’s assets, including intellectual property, subject to agreed exceptions. The maturity date of the Loan is December 1, 2024.
The Loan Agreement required monthly interest-only payments during the first eighteen months of the term of the Loan. Following the interest-only period, on July 1, 2023, the Company pays equal monthly payments of principal, plus accrued interest, until the Loan’s maturity date when all remaining principal and accrued interest is due. If the Company prepays the Loan, it will be required to pay (a) a prepayment fee in an amount equal to 3.0% of the principal amount of the Loan that is prepaid during the interest-only period; and (b) a prepayment fee in an amount equal to 1.0% of the principal amount of the Loan that is prepaid after the interest-only period. At the Loan’s maturity date, or on the date of the prepayment of the Loan, the Company will be obligated to pay a final payment equal to 4.25% of the Loan commitment amount, the sum of Tranche 1 and Tranche 2, which amounts to $850,000 (the “Loan Premium”).
The Loan Agreement includes a conversion option to convert up to $5.0 million of the principal amount of the Loan outstanding at the option of Avenue, into shares of the Company’s Common Stock at a conversion price of $69.80 per share (the “Conversion Option”).
On the Closing Date, the Company issued to Avenue warrants to purchase 36,101 shares of Common Stock of the Company (the “Avenue Warrants”) at an exercise price per share equal to $58.20. The Avenue Warrants are exercisable until November 30, 2026.
The amount of the carrying value of the notes payable was determined by allocating portions of the outstanding principal of the notes, approximately $1.4 million, to the fair value of the Avenue Warrants, and approximately $2.2 million to the fair value of the embedded Conversion Option. Accordingly, the total amount of unearned discount of approximately $3.6 million, the total direct financing cost of approximately $390,000 and the Loan Premium of $850,000 are being amortized using the effective interest method over the term of the Loan. The adjusted effective interest rate is 27%.
Total interest expense for the year ended June 30, 2024 was approximately $2.9 million on the accompanying statement of operations and comprehensive loss. Interest expense was comprised of interest incurred on the outstanding principal of the loan of approximately $1.5 million, amortization of financing costs of approximately $109,000, amortization of the unearned discount of $1.0 million, and the accretion of the Loan Premium of approximately $237,000.
Total interest expense for the year ended June 30, 2023 was approximately $4.3 million on the accompanying statement of operations and comprehensive loss. Interest expense was comprised of interest incurred on the outstanding principal of the loan of approximately $2.1 million, amortization of financing costs of approximately $170,000, amortization of the unearned discount of $1.6 million, and the accretion of the Loan Premium of approximately $422,000.
As of June 30, 2024, the remaining principal balance of $5.0 million under the Loan is payable in 6 monthly equal installments. For the year ended June 30, 2024, the Company paid back $10 million, of the original loan of $15 million.
| F- |
| 7. | Notes Payable (continued) |
The following is a summary of the Notes Payable as of June 30, 2024 and 2023:
Current portion of Notes Payable
| Schedule of note payable | ||||||||
| June 30, 2024 | June 30, 2023 | |||||||
| Current portion of Notes Payable | $ | 5,000,000 | $ | 10,000,000 | ||||
| Less: debt financing costs | (11,820 | ) | (108,751 | ) | ||||
| Less: unearned discount | (111,212 | ) | (1,023,145 | ) | ||||
| Plus: accretion of Loan Premium | 824,242 | 236,970 | ||||||
| Current portion of Notes Payable, net of financing costs, unearned premium and discount | $ | 5,701,210 | $ | 9,105,074 | ||||
Non-current portion of Notes Payable
| June 30, 2024 | June 30, 2023 | |||||||
| Notes Payable | $ | - | $ | 5,000,000 | ||||
| Less: debt financing costs | - | (11,820 | ) | |||||
| Less: unearned discount | - | (111,212 | ) | |||||
| Plus: accretion of Loan Premium | - | 350,302 | ||||||
| Notes Payable, net of the current portion financing costs, unearned premium and discount | $ | - | $ | 5,227,270 | ||||
Estimated future amortization expense and accretion of Loan Premium is as follows:
| Schedule of estimated future amortization expense and accretion of premium | ||||||||||||
| Unearned Discount | Debt Financing Costs | Loan Premium | ||||||||||
| Year ending June 30, | ||||||||||||
| 2025 | $ | 111,212 | $ | 11,820 | $ | 25,758 | ||||||
| Total | $ | 111,212 | $ | 11,820 | $ | 25,758 | ||||||
| F- |
| 8. | Fair Value Measurements |
At June 30, 2024 and 2023, the estimated fair value of derivative liabilities measured on a recurring basis are as follows:
| Schedule of derivative liabilities at fair value | ||||||||||||||||
| Fair Value Measurements at | ||||||||||||||||
| June 30, 2024 | ||||||||||||||||
| Level 1 | Level 2 | Level 3 | Total | |||||||||||||
| Derivative liability - Warrants | $ | - | $ | - | $ | 3,771 | $ | 3,771 | ||||||||
| Derivative liability - Conversion Option | - | - | - | - | ||||||||||||
| Total derivative liabilities | $ | - | $ | - | $ | 3,771 | $ | 3,771 | ||||||||
| Fair Value Measurements at | ||||||||||||||||
| June 30, 2023 | ||||||||||||||||
| Level 1 | Level 2 | Level 3 | Total | |||||||||||||
| Derivative liability - Warrants | $ | - | $ | - | $ | 894,280 | $ | 894,280 | ||||||||
| Derivative liability - Conversion option | - | - | 925,762 | 925,762 | ||||||||||||
| Total derivative liabilities | $ | - | $ | - | $ | 1,820,042 | $ | 1,820,042 | ||||||||
The following table presents the activity for level 3 liabilities measured at fair value using unobservable inputs for the years ended June 30, 2024 and 2023:
| Fair value, liabilities measured on recurring basis | ||||||||
| Derivative liability - Warrants | Derivative liability - Conversion Option | |||||||
| Balance at June 30, 2022 | $ | 194,531 | $ | 188,030 | ||||
| Additions to level 3 liabilities | - | - | ||||||
| Change in in fair value of level 3 liabilities | 699,749 | 737,732 | ||||||
| Transfer in and/or out of level 3 | - | - | ||||||
| Balance at June 30, 2023 | $ | 894,280 | $ | 925,762 | ||||
| Additions to level 3 liabilities | - | - | ||||||
| Change in in fair value of level 3 liabilities | (890,509 | ) | (925,762 | ) | ||||
| Transfer in and/or out of level 3 | - | - | ||||||
| Balance at June 30, 2024 | $ | 3,771 | $ | - | ||||
The fair values of derivative liabilities for the Avenue Warrants and Conversion Option at June 30, 2024 in the accompanying balance sheet, were approximately $3,800 and approximately zero, respectively. The total change in the fair value of the derivative liabilities totaled approximately $(1.8) million and $1.4 million for the years ended June 30, 2024, and 2023, respectively; and accordingly, was recorded in the accompanying statements of operations and comprehensive loss. The assumptions used in the Black Scholes model to value the derivative liabilities at June 30, 2024 included the closing stock price of $4.00 per share; for the Avenue Warrants, the exercise price of $58.20, remaining term 2.4 year, risk free rate of 4.6% and volatility of 82.0%; and for the Conversion Option, the conversion price of $69.80; remaining term of 5 months, risk free rate of 5.38% and volatility of 91.0%.
Derivative liability – Avenue Warrants
The Avenue Warrants were not considered to be indexed to the Company’s own stock, and accordingly, were recorded as a derivative liability at fair value in the accompanying balance sheets at June 30, 2024 and 2023.
The Black Scholes model was used to calculate the fair value of the warrant derivative to bifurcate the warrant derivative amount from the Avenue Loan amount funded. The Avenue Warrants are recorded at their fair values at the date of issuance and remeasured at each subsequent reporting period end date.
| F- |
| 8. | Fair Value Measurements (continued) |
Embedded derivative liability – Conversion Option
The Conversion Option is accounted for as an embedded derivative liability and required bifurcation from the Loan amount. The Black Scholes model was used to calculate the fair value of the Conversion Option to bifurcate it from the Loan.
Financial assets
As of June 30, 2024, investments in U.S. Treasury Bills were valued through use of quoted prices and are classified as Level 1. The following table presents information about our assets that are measured at fair value on a recurring basis using the above input categories.
| Measured at fair value on a recurring basis | ||||||||||||||||
| Fair Value Measurements at | ||||||||||||||||
| June 30, 2024 | ||||||||||||||||
| Level 1 | Level 2 | Level 3 | Total | |||||||||||||
| Cash | $ | 12,763,941 | $ | - | $ | - | $ | 12,763,941 | ||||||||
| U.S. Treasury Bills due in 3 months or less at purchase | 11,079,857 | - | - | 11,079,857 | ||||||||||||
| Total | $ | 23,843,798 | $ | - | $ | - | $ | 23,843,798 | ||||||||
| Fair Value Measurements at | ||||||||||||||||
| June 30, 2023 | ||||||||||||||||
| Level 1 | Level 2 | Level 3 | Total | |||||||||||||
| Cash | $ | 6,304,543 | $ | - | $ | - | $ | 6,304,543 | ||||||||
| U.S. Treasury Bills due in 3 months or less at purchase | 13,156,340 | - | - | 13,156,340 | ||||||||||||
| U.S. Treasury Bills due in 3 - 6 months at purchase | 14,477,726 | - | - | 14,477,726 | ||||||||||||
| Total | $ | 33,938,609 | $ | - | $ | - | $ | 33,938,609 | ||||||||
| 9. | Equity Transactions |
Issuance of common stock for cash
On August 31, 2022, the Company entered into a Controlled Equity Offering Sales Agreement (the “Sales Agreement”) with Cantor Fitzgerald & Co. and B. Riley Securities, Inc. (collectively, the “Agents”), pursuant to which the Company may issue and sell from time-to-time shares of the Company’s common stock through the Agents, subject to the terms and conditions of the Sales Agreement. On April 6, 2023, the Company and B. Riley Securities, Inc. mutually agreed to terminate B. Riley Securities, Inc.’s role as a sales agent under the Sales Agreement. During the year ended June 30, 2024, the Company sold 333,749 shares of common stock under the Sales Agreement for total net proceeds of approximately $9.3 million after deducting 3% commissions and expenses of approximately $377,000. During the year ended June 30, 2023, the Company sold 753,925 shares of common stock under the Sales Agreement for total net proceeds of approximately $49.5 million after 3% commissions and expenses of approximately $2.0 million.
On March 6, 2024, the Company closed a best efforts public offering (the “Offering”) of 1,500,000 shares (the “Shares”) of its common stock, par value $0.001 per share (the “Common Stock”), pre-funded warrants (the “Pre-funded Warrants”) to purchase 600,000 shares of Common Stock, and warrants to purchase up to 1,050,000 shares of Common Stock (the “Common Warrants”) at a combined public offering price of $10.00 per Share, or Pre-funded Warrant, and the associated Common Warrant. The Common Warrants have an exercise price of $15.00 per share and are immediately exercisable upon issuance for a period of five years following the date of issuance. The gross proceeds to the Company from the Offering were approximately $21.0 million, before deducting placement agent fees and offering expenses of approximately $2.5 million, resulting in net proceeds of approximately $18.5 million. Additionally, upon closing the Company issued the placement agent warrants (“Placement Agent’s warrants”) to purchase 105,000 shares of Common Stock exercisable at a per share price of $12.50, which was equal to 125% of the public offering price per share. The Placement Agent’s Warrants are exercisable during a five-year period commencing 180 days from March 6, 2024. The Pre-Funded Warrants were exercised shortly after issuance and the 600,000 shares of Common Stock were issued during the year ended June 30, 2024.
| F- |
| 9. | Equity Transactions (continued) |
Issuance of common stock for services
On April 6, 2023, the Company awarded 5,000 shares of Common Stock to a vendor as part of their fees in exchange for services. The fair value of the Common Stock at the date of issuance was $74.50 per share. The stock-based compensation expense related to this Common Stock issuance was $372,500.
On May 10, 2024, the Company awarded 15,000 shares of Common Stock to a vendor as part of their fees in exchange for services. The fair value of the Common Stock at the date of issuance was $4.86 per share. The stock-based compensation expense related to this Common Stock issuance was $72,900.
Stock Options
The following table summarizes the activity relating to the Company’s stock options for the years ended June 30, 2024 and 2023:
| Schedule of summarizes the activity relating to the Company’s stock options | ||||||||||||||||
| Options | Weighted-Average Exercise Price | Weighted Remaining Average Contractual Term | Aggregate Intrinsic Value | |||||||||||||
| Outstanding at June 30, 2022 | 339,876 | $ | 74.20 | 5.5 | $ | - | ||||||||||
| Granted | 71,467 | 59.00 | 8.6 | 38,610 | ||||||||||||
| Options Expired | (1,000 | ) | 286.90 | - | - | |||||||||||
| Options Canceled | (4,967 | ) | 77.40 | |||||||||||||
| Options Exercised | (10,090 | ) | 81.20 | - | - | |||||||||||
| Outstanding at June 30, 2023 | 395,286 | 71.00 | 6.3 | 1,067,966 | ||||||||||||
| Granted | 155,242 | 11.70 | 9.8 | - | ||||||||||||
| Options Expired | (640 | ) | 46.09 | - | - | |||||||||||
| Options Canceled | (31,812 | ) | 57.19 | - | - | |||||||||||
| Outstanding at June 30, 2024 | 518,076 | $ | 54.11 | 6.1 | $ | - | ||||||||||
| Exercisable at June 30, 2024 | 296,934 | $ | 66.65 | 4.9 | $ | - | ||||||||||
The fair value of each option grant on the date of grant is estimated using the Black-Scholes model. The following weighted-average assumptions were utilized for the years ended:
| Schedule of assumptions used | |||
| June 30, 2024 | June 30, 2023 | ||
| Expected life of options (in years) | 6 | 6 | |
| Expected volatility | 86.28% | 81.65% | |
| Risk free interest rate | 4.40% | 3.82% | |
| Dividend Yield | 0% | 0% |
The Company recorded stock based compensation expense relating to the vesting of stock options of approximately $2.8 million and $4.2 million for the years ended June 30, 2024 and 2023, respectively.
Issuance and modification of restricted stock units and options:
On June 21, 2022, the Company awarded 12,452 restricted stock units (“RSUs”) to the President and CEO under the Company’s 2019 Omnibus Plan. Each RSU awarded to the CEO entitles him to receive one share of Common Stock upon vesting. The RSUs vest in three equal annual installments beginning on the first anniversary grant date. 4,151 and 4,151 RSUs vested in June 2023 and 2024, respectively.
On November 23, 2022, the Company awarded 38,198 RSUs to certain employees and a consultant, with a grant date fair value of $61.20 per share. 25% of these RSUs vested on the grant date and the remaining RSUs vest in three equal installments over three years beginning on the first anniversary of the grant date. During the year ended June 30, 2023, 9,550 of these RSUs vested, of which 2,288 shares were withheld in Treasury stock in exchange for payment of withholding tax on behalf of the employees.
| F- |
| 9. | Equity Transactions (continued) |
On November 23, 2022, the Company issued equity awards for the board of directors’ annual compensation. Four directors received 15,564 RSUs with a grant date fair value of $61.20 per share. In addition, three directors received stock options to purchase 19,500 shares of common stock at an exercise price of $61.20 per share with a grant date fair value of $40.60 per share. The equity awards vest quarterly on February 23, 2023, May 23, 2023, August 23, 2023 and earlier of November 23, 2023 or the next annual shareholders’ meeting. During the year ended June 30, 2024, 7,746 of these RSUs vested. These RSUs and options contain certain contractual vesting terms where the vesting can be accelerated outside the Company’s control and as a result, for accounting purposes, are assumed to have been fully vested on the grant date, and accordingly, the Company recognized the total compensation cost of $1,744,192 on November 23, 2022.
On November 9, 2023, the Company issued equity awards for the board of directors’ annual compensation. Four directors received 18,270 RSUs with a grant date fair value of $30.10 per share. In addition, two directors received stock options to purchase 18,325 shares of common stock at an exercise price of $30.10 per share with a grant date fair value of $18.30 per share. The equity awards vest quarterly on February 9, 2024, May 9, 2024, August 9, 2024 and earlier of November 9, 2024 or the next annual shareholders’ meeting. During the year ended June 30, 2024, 4,568 of these RSUs vested.
In December 2023, the Company terminated five employees and as part of their severance agreement modified their equity awards that had been granted pursuant to the 2019 Omnibus Plan. The modifications included the acceleration of certain stock option awards to purchase a total of 5,623 shares of common stock (“Accelerated Options”), effective on the December Separation Date, as defined in severance agreement (“Separation Date”), and extended the expiration date for one year from the Separation Date for both the Accelerated Options and any vested and unexercised stock options held by the terminated employees as of the Separation Date. Accordingly, the Company remeasured the Accelerated Options based on the stock price of $15.40 per share at the close on the Separation Date and a one-year extension of the term. The net adjustment for the modification was a net credit of $127,199 and was recognized as an adjustment to stock compensation expense during the year ended June 30, 2024.
Additionally, 1,030 vesting RSUs were accelerated as of the Separation date. The modified RSUs were remeasured based on the stock price of $15.40 per share at close on the Separation Date and $15,865, was recorded to additional in stock-based compensation for the year ended June 30, 2024 as a result of the modification.
In connection with the separation, the Company canceled 18,396 unvested stock options and 1,030 unvested RSUs. Additionally, the Company canceled an additional 13,416 unvested stock options for employees that voluntarily left the company.
In June 2023, the Company issued 14,950 RSUs with a grant date fair value of $41.10 per share to the President and CEO under the Company’s 2019 Omnibus Plan. The RSUs vest in three equal annual installments beginning on the first anniversary grant date. 4,983 RSUs vested in June 2024.
In June 2024, the Company issued 85,800 RSUs to employees, with a grant date fair value of $4.74 per share. The RSUs vested on the grant date. The Company delivered the vested portion of the RSU’s and issued 85,800 shares of Common Stock, of which 21,450 shares were withheld in Treasury stock in exchange for payment of withholding tax on behalf of the employees.
The following table summarizes vesting of restricted stock units:
| Schedule of vesting of restricted common stock | ||||||||
| Number of Shares | Weighted Average Grant Date Fair Value Per Share | |||||||
| Unvested at June 30, 2022 | 12,452 | $ | 16.90 | |||||
| Granted | 68,711 | 58.87 | ||||||
| Vested | (21,518 | ) | 52.70 | |||||
| Unvested at June 30, 2023 | 59,646 | $ | 52.40 | |||||
| Issued | 104,070 | 9.16 | ||||||
| Vested | (122,395 | ) | 16.94 | |||||
| Canceled | (1,030 | ) | 61.20 | |||||
| Unvested at June 30, 2024 | 40,291 | $ | 44.59 | |||||
| F- |
| 9. | Equity Transactions (continued) |
The total stock-based compensation expense from restricted stock units for the year ended June 30, 2024 and 2023 was approximately $1.8 million and $1.8 million, respectively.
Issuance of Common Stock through exercise of Stock Options and Warrants
In December 2022, the Company issued 2,209 shares of Common Stock pursuant to a cashless exercise of stock options to purchase 9,930 shares at an average exercise price of $76.40.
In November 2022, the Company issued 80 shares of Common Stock pursuant to a cash exercise of stock options to purchase 80 shares at an average exercise price of $28.00 per share.
In October 2022, the Company issued 359 shares of Common Stock pursuant to a cashless exercise of warrants to purchase 800 shares at an average exercise price of $22.50.
In May 2023, the Company issued 48 shares of Common Stock pursuant to a cashless exercise of stock options to purchase 80 shares at an average exercise price of $31.30.
Issuance of Stock Options under the 2019 Omnibus Plan.
Pursuant to a former employee’s Separation Agreement, dated April 11, 2022, the Company modified their stock option award granted on August 20, 2021, pursuant to the 2019 Omnibus Plan (“2021 Options Grant”). Pursuant to the terms of the Separation Agreement, effective July 8, 2022 (the “Separation Date”), the Company accelerated the vesting of options scheduled to vest on the first and second anniversary of the grant date as deemed vested (“Accelerated Options”) and after giving effect to the Accelerated Options, extended the exercise period of the total vested outstanding and unexercised options (totaling 7,450 options) to one year following the Separation Date. The unvested portion of the 2021 Option Grant (totaling 4,967 options) was canceled. The modification was remeasured as of July 8, 2022, and the incremental difference in fair value resulted in a net credit to stock based compensation expense of $181,154, due to the original exercise price of $77.40 being greater than the stock price of $18.00 on the remeasurement date, and accordingly was recognized on July 8, 2022.
On June 7, 2023, the Company granted stock options to purchase 14,800 shares of Common Stock to certain employees. 20% of the shares underlying the options awarded vested on the grant date, and the remaining 80% will vest in four equal annual installments beginning, on the first grant date anniversary. The exercise price of the options is $57.80 per share, the grant date fair value and the options terminate on the earlier of the tenth grant date anniversary or the date of which the options are fully exercised.
During the fiscal year ended June 30, 2023, the Company granted stock options to purchase a total of 28,617 shares of Common Stock in connection with compensation packages of three new employees. The exercise prices were set at the grant date fair value with vesting terms over a five year period and the options terminate on the earlier of tenth grant date anniversary or the date of which the options are fully exercised.
On October 3, 2023, the Company granted stock options to purchase 21,117 shares of Common Stock to new hire employees. 20% of the shares underlying the options awarded vest on the one-year anniversary of the grant date, and the remaining 80% will vest in equal monthly installments over 48 months each month thereafter. The exercise price of the options is $34.10, the grant date fair value, and the options terminate on the earlier of the tenth grant date anniversary or the date of which the options are fully exercised.
In June 2024, the Company granted stock options to purchase 115,800 shares of Common Stock to employees. 33% of the shares underlying the options awarded vest on the grant date, and the remaining 67% will vest over 2 years on first and second anniversary of the grant date. The exercise price of the options is $4.70, the grant date fair value, and the options terminate on the earlier of the tenth grant date anniversary or the date of which the options are fully exercised.
| F- |
| 9. | Equity Transactions (continued) |
Stock Warrants
The following table summarizes the warrants activity during the years ended June 30, 2024 and 2023:
| Summary of warrants activity | ||||||||||||||||
| Number of Shares | Weighted Average Exercise Price | Weighted Average Remaining Life (Years) | Aggregate Intrinsic Value | |||||||||||||
| Outstanding and exercisable at June 30, 2022 | 51,037 | $ | 61.70 | 3.8 | $ | - | ||||||||||
| Granted | 727,273 | 18.20 | 5.0 | - | ||||||||||||
| Expired | (482 | ) | 750.00 | - | - | |||||||||||
| Exercised | (800 | ) | 22.50 | - | - | |||||||||||
| Outstanding and exercisable at June 30, 2023 | 777,029 | 20.60 | 4.0 | 18,318,954 | ||||||||||||
| Granted | 1,755,000 | 13.14 | 5.0 | - | ||||||||||||
| Exercised | (600,000 | ) | 10.00 | - | - | |||||||||||
| Outstanding and exercisable at June 30, 2024 | 1,932,029 | $ | 14.03 | 4.0 | $ | - | ||||||||||
Of the above warrants outstanding at June 30, 2024, 10,138 expire in the fiscal year ending June 30, 2025, 3,518 expire in the fiscal year ending June 30, 2026, 763,373 expire in the fiscal year ending June 30, 2027 and 1,155,000 expire in the fiscal year ending June 30, 2029.
| 10. | Leases |
Office Leases
The Company pays an annual rent of $2,200 for its headquarters at 680 W Nye Lane, Suite 201, Carson City Nevada 89703. The rental agreement was for a one-year term, commenced on October 1, 2022 and has been subsequently renewed for another year at the same rate.
The Company’s San Diego office lease at 5090 Shoreham Place Suite 212, San Diego, CA 92122 which commenced on March 1, 2022, was for a term of 38 months with a base monthly rate of $4,300, and annual increases of three percent. In February 2024, the Company amended the lease agreement which allowed the Company to vacate the then current space and move to a larger space at Suite 206. The current monthly base rate for the new office space is $9,685, with an annual increase of four percent. The term for the new office lease is 60 months and commenced on February 12, 2024. The lease that was in place for the 5090 Shoreham Place Suite 212 office was effectively extinguished upon the commencement of the new office space lease on February 12, 2024, resulting in the write off of the corresponding remaining right-of-use asset and operating lease liability of $56,909 and $62,124, respectively, and a gain to selling, general and administrative expenses of $5,215 for the year ended June 30, 2024.
Total operating lease expense for the years ended June 30, 2024 and 2023 of approximately $78,000 and $52,000, respectively were included in the accompanying statements of operations and comprehensive loss as a component of selling, general and administrative expenses.
The right-of-use asset, net and current and non-current portion of the operating lease liabilities included in the accompanying balance sheets are as follows:
| F- |
| 10. | Leases (continued) |
| Schedule of deferred tax assets | ||||||||
| June 30, 2024 | June 30, 2023 | |||||||
| Assets | ||||||||
| Operating lease, right-of-use asset, net | $ | 406,726 | $ | 80,789 | ||||
| Liabilities | ||||||||
| Current portion of operating lease liability | $ | 60,343 | $ | 44,909 | ||||
| Operating lease liability, net of current portion | 349,894 | 42,505 | ||||||
| Total operating lease liability | $ | 410,237 | $ | 87,414 | ||||
At June 30, 2024, the future estimated minimum lease payments under non-cancelable operating leases are as follows:
| Schedule of future estimated minimum lease payments under non-cancelable operating leases | ||||
| Year ending June 30, | ||||
| 2025 | $ | 117,915 | ||
| 2026 | 122,042 | |||
| 2027 | 126,313 | |||
| 2028 | 130,734 | |||
| 2029 | 77,796 | |||
| Total minimum lease payments | 574,800 | |||
| Less amount representing interest | (164,563 | ) | ||
| Present value of future minimum lease payments | 410,237 | |||
| Less current portion of operating lease liability | (60,343 | ) | ||
| Operating lease liability, net of current portion | $ | 349,894 | ||
Total cash paid for amounts included in the measurement of lease liabilities were $83,910 and $50,600 for the years ended June 30, 2024 and 2023, respectively.
The weighted average remaining lease term and discount rate as of June 30, 2024 and 2023 were as follows:
| Schedule of weighted average remaining lease term and discount rate | ||||||||
| June 30, 2024 | June 30, 2023 | |||||||
| Weighted average remaining lease term (Years) | ||||||||
| Operating lease | 4.6 | 1.8 | ||||||
| Weighted average discount rate | ||||||||
| Operating lease | 15.00 | % | 10.75 | % | ||||
| F- |
| 11. | Commitments and Contingencies |
Royalty Agreements
Pursuant to the Agreement and Plan of Merger entered into on April 11, 2016, by and between our predecessor entities, LAT Pharma and NanoAntibiotics, Inc., the Company is obligated to pay a low single digit royalty on net sales of BIV201 (continuous infusion terlipressin) to be shared by the members of LAT Pharma Members, PharmaIn Corporation, and The Barrett Edge, Inc.
Pursuant to the Technology Transfer Agreement entered into on July 25, 2016, by and between the Company and the University of Padova (Italy), the Company is obligated to pay a low single digit royalty on net sales of all terlipressin products covered by US patent no. 9,655,645 and any future foreign issuances, capped at a maximum of $200,000 per year.
Shareholder class action complaint
On January 19, 2024, a purported shareholder class action complaint, captioned Eric Olmstead v. BioVie Inc. et al., No. 3:24-cv-00035, was filed in the U.S. District Court for the District of Nevada, naming the Company and certain of its officers as defendants. On February 22, 2024, a second, related putative securities class action was filed in the same court asserting similar claims against the same defendants, captioned Way v. BioVie Inc. et al., No. 2:24-cv-00361. On April 15, 2024, the court consolidated these two actions under the caption In re BioVie Inc. Securities Litigation, No. 3:24-cv-00035, appointed the lead plaintiff, and approved selection of the lead counsel. On June 21, 2024, the lead plaintiff filed an amended complaint, alleging that the defendants made material misrepresentations and/or omissions of material fact relating to the Company’s business, operations, compliance, and prospects, including information related to the NM101 Phase 3 study and trial of bezisterim (NE3107) in mild to moderate probable AD, in violation of Sections 10(b) and 20(a) of the Exchange Act, and Rule 10b-5 promulgated thereunder. The class action is on behalf of purchasers of the Company’s securities during the period from December 7, 2022 through November 28, 2023 and seeks unspecified monetary damages on behalf of the putative class and an award of costs and expenses, including attorney’s fees. The defendants filed a motion to dismiss the amended complaint on August 21, 2024.
The Company believes the lawsuit is without merit and intends to defend the case vigorously. At this early stage of the proceedings, the Company is unable to make any prediction regarding the outcome of the litigation. No adjustment or accruals have been reflected in the accompanying financial statements.
| 12. | Employee Benefit Plan |
On August 1, 2021, the Company began sponsoring an employee benefit plan subject to Section 401(K) of the Internal Revenue Service Code (the “401K Plan”) pursuant to which, all employees meeting eligibility requirements are able to participate.
Subject to certain limitations in the Internal Revenue Code, eligible employees are permitted to make contributions to the 401K Plan on a pre-tax salary reduction basis and the Company will match 5% of the first 5% of an employee’s contributions to the 401K Plan. The Company made contributions into the plan of approximately $153,200 and $171,900, for the years ended June 30, 2024 and 2023, respectively.
| F- |
| 13. | Income Taxes |
Significant components of the Company’s deferred tax assets (liabilities) are as follows:
| Schedule of deferred tax assets | ||||||||
| June 30, 2024 | June 30, 2023 | |||||||
| Deferred tax assets (liabilities): | ||||||||
| Tax loss carryforward | $ | 51,429,074 | $ | 48,080,664 | ||||
| Intangible assets | (114,161 | ) | (189,854 | ) | ||||
| Stock based compensation | 5,860,272 | 4,575,852 | ||||||
| R&D capitalized | 12,467,969 | 8,171,276 | ||||||
| Valuation Allowance | (69,643,154 | ) | (60,637,938 | ) | ||||
| Net deferred tax assets | $ | - | $ | - | ||||
At June 30, 2024 and 2023, the Company has recorded a full valuation against its net deferred tax assets of approximately $69.6 million and $60.6 million, respectively, since in the judgement of management, these assets are not more than likely to be realized. The increase in the valuation allowance during the year ended June 30, 2024 was approximately $9.0 million.
At June 30, 2024, the Company had a Net Operating Loss (“NOL”) carryforward of approximately $184 million. NOL’s generated prior to 2018 have expiration dates ranging from 2032 to 2037.
The Company has no current tax expense due to its net losses and a full valuation allowance.
Reconciliation of the differences between income tax benefit computed at the federal and state statutory tax rates and the provision for income tax benefit for the years ended June 30, 2024 and 2023 is as follows:
| Schedule of effective income tax rate reconciliation | ||||||||
| 2024 | 2023 | |||||||
| Income tax expense at federal statutory rate | 21 | % | 21 | % | ||||
| State taxes, net of federal benefit | 7 | % | 7 | % | ||||
| Change in valuation allowance | (28 | )% | (28 | )% | ||||
| Effective tax rate | - | - | ||||||
| 14. | Subsequent Events |
On September 25, 2024, the Company closed a best efforts public offering (the “September 2024 Offering”) of 1,360,800 shares of its common stock, par value $0.0001 per share, pre-funded warrants (the “September Pre-funded Warrants”) to purchase 600,000 shares of Common Stock, and warrants to purchase up to 1,960,800 shares of Common Stock (the “September Common Warrants”) at a combined public offering price of $1.53 per Share, or September Pre-funded Warrant, and the associated September Common Warrant. The September Common Warrants have an exercise price of $1.53 per share and are immediately exercisable upon issuance and will expire on the fifth anniversary date of the original issuance date. The gross proceeds to the Company from the September 2024 Offering were approximately $3.0 million, before deducting placement agent fees and offering expenses of approximately $560,000. Additionally, upon closing the Company issued the placement agent warrants (“September Placement Agent’s Warrants”) to purchase 98,040 shares of Common Stock exercisable at a per share price of $1.91, which was equal to 125% of the public offering price per share. The September Placement Agent’s Warrants are exercisable during a five-year period commencing 180 days from September 25, 2024.
| F- |
Exhibit 23.1
CONSENT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
We consent to the incorporation by reference in the Registration Statements of BioVie Inc. on Form S-3 (Nos. 333-274083 and 333-271054) and Form S-8 (No. 333-260019), of our report dated September 30, 2024, on our audits of the financial statements as of June 30, 2024 and 2023, and for each of the years then ended, which report is included in this Annual Report on Form 10-K to be filed on or about September 30, 2024. Our report includes an explanatory paragraph about the existence of substantial doubt concerning the Company’s ability to continue as a going concern.
/s/ EisnerAmper LLP
EISNERAMPER LLP
Iselin, New Jersey
September 30, 2024
Exhibit 31.1
CERTIFICATION PURSUANT TO
RULE 13-a-14(a) and 15d-14(a)
AS ADOPTED PURSUANT TO
SECTION 302 OF THE SARBANES OXLEY ACT OF 2002
| I, Cuong Do, certify that: | ||
| 1. | I have reviewed this annual report on Form 10-K of Biovie Inc.; | |
| 2. | Based on my knowledge, this report does not contain any untrue statement of a material fact or omit to state a material fact necessary to make the statements made, in light of the circumstances under which such statements were made, not misleading with respect to the period covered by this report; | |
| 3. | Based on my knowledge, the financial statements, and other financial information included in this report, fairly present in all material respects the financial condition, results of operations and cash flows of the registrant as of, and for, the periods presented in this report; | |
| 4. | The registrant’s other certifying officer(s) and I are responsible for establishing and maintaining disclosure controls and procedures (as defined in Exchange Act Rules 13a-15(e) and 15d-15(e)) and internal control over financial reporting (as defined in Exchange Act Rules 13a – 15(f) and 15d – 15(f)) for the registrant and have: | |
| a) | Designed such disclosure controls and procedures, or caused such disclosure controls and procedures to be designed under our supervision, to ensure that material information relating to the registrant, including its consolidated subsidiaries, is made known to us by others within those entities, particularly during the period in which this report is being prepared; | |
| b) | Designed such internal control over financial reporting, or caused such internal control over financial reporting to be designed under our supervision, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles; | |
| c) | Evaluated the effectiveness of the registrant’s disclosure controls and procedures and presented in this report our conclusions about the effectiveness of the disclosure controls and procedures, as of the end of the period covered by this report based on such evaluation; and | |
| d) | Disclosed in this report any change in the registrant’s internal control over financial reporting that occurred during the registrant’s most recent fiscal quarter (the registrant’s fourth fiscal quarter in the case of the annual report) that has materially affected, or is reasonably likely to materially affect, the registrant’s internal control over financial reporting; and | |
| 5. | The registrant’s other certifying officer(s) and I have disclosed, based on our most recent evaluation of internal control over financial reporting, to the registrant’s auditors and the audit committee of the registrant’s board of directors (or persons performing the equivalent functions): | |
| a) | All significant deficiencies and material weaknesses in the design or operation of internal control over financial reporting which are reasonably likely to adversely affect the registrant’s ability to record, process, summarize and report financial information; and | |
| b) | Any fraud, whether or not material, that involves management or other employees who have a significant role in the registrant’s internal control over financial reporting. | |
Date: September 30, 2024
| /s/ Cuong Do | ||||||||
|
Cuong Do Chief Executive Officer (Principal Executive Officer) |
Exhibit 31.2
CERTIFICATION PURSUANT TO
RULE 13-a-14(a) and 15d-14(a)
AS ADOPTED PURSUANT TO
SECTION 302 OF THE SARBANES OXLEY ACT OF 2002
| I, Joanne Wendy Kim, certify that: | ||
| 1. | I have reviewed this annual report on Form 10-K of Biovie Inc.; | |
| 2. | Based on my knowledge, this report does not contain any untrue statement of a material fact or omit to state a material fact necessary to make the statements made, in light of the circumstances under which such statements were made, not misleading with respect to the period covered by this report; | |
| 3. | Based on my knowledge, the financial statements, and other financial information included in this report, fairly present in all material respects the financial condition, results of operations and cash flows of the registrant as of, and for, the periods presented in this report; | |
| 4. | The registrant’s other certifying officer(s) and I are responsible for establishing and maintaining disclosure controls and procedures (as defined in Exchange Act Rules 13a-15(e) and 15d-15(e)) and internal control over financial reporting (as defined in Exchange Act Rules 13a – 15(f) and 15d – 15(f)) for the registrant and have: | |
| a) | Designed such disclosure controls and procedures, or caused such disclosure controls and procedures to be designed under our supervision, to ensure that material information relating to the registrant, including its consolidated subsidiaries, is made known to us by others within those entities, particularly during the period in which this report is being prepared; | |
| b) | Designed such internal control over financial reporting, or caused such internal control over financial reporting to be designed under our supervision, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles; | |
| c) | Evaluated the effectiveness of the registrant’s disclosure controls and procedures and presented in this report our conclusions about the effectiveness of the disclosure controls and procedures, as of the end of the period covered by this report based on such evaluation; and | |
| d) | Disclosed in this report any change in the registrant’s internal control over financial reporting that occurred during the registrant’s most recent fiscal quarter (the registrant’s fourth fiscal quarter in the case of the annual report) that has materially affected, or is reasonably likely to materially affect, the registrant’s internal control over financial reporting; and | |
| 5. | The registrant’s other certifying officer(s) and I have disclosed, based on our most recent evaluation of internal control over financial reporting, to the registrant’s auditors and the audit committee of the registrant’s board of directors (or persons performing the equivalent functions): | |
| a) | All significant deficiencies and material weaknesses in the design or operation of internal control over financial reporting which are reasonably likely to adversely affect the registrant’s ability to record, process, summarize and report financial information; and | |
| b) | Any fraud, whether or not material, that involves management or other employees who have a significant role in the registrant’s internal control over financial reporting. | |
Date: September 30, 2024
| /s/Joanne Wendy Kim | ||||||||
|
Joanne Wendy Kim Chief Financial Officer (Principal Financial and Accounting Officer) |
Exhibit 32.1
CERTIFICATION PURSUANT TO 18 U.S. C. SECTION 1350 AS ADOPTED PURSUANT TO
SECTION 906 OF THE SARBANES-OXLEY ACT OF 2002
In connection with the Annual Report of Biovie Inc., (the “Company”) on Form 10-K for the year ended June 30, 2024, as filed with the Securities and Exchange Commission on the date hereof (the “Report”), I, Cuong Do, Chief Executive Officer and Chairman of the Board of the Company, certify, pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes Oxley Act of 2002, that:
| (1) | The Report fully complies with the requirements of Section 13 (a) or 15 (d) of the Securities Exchange Act of 1934 (15 U.S.C. 78m (a) or 78o(d)); and |
| (2) | The information contained in the Report fairly presents, in all material respects, the financial condition and results of operations of the Company. |
Date: September 30, 2024
| /s/ Cuong Do | ||||||||
| Cuong Do Chief Executive Officer (Principal Executive Officer) |
Exhibit 32.2
CERTIFICATION PURSUANT TO 18 U.S. C. SECTION 1350 AS ADOPTED PURSUANT TO
SECTION 906 OF THE SARBANES-OXLEY ACT OF 2002
In connection with the Annual Report of Biovie Inc., (the “Company”) on Form 10-K for the year ended June 30, 2024, as filed with the Securities and Exchange Commission on the date hereof (the “Report”), I, Joanne Wendy Kim, Chief Financial Officer of the Company, certify, pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes Oxley Act of 2002, that, to my knowledge:
| (1) | The Report fully complies with the requirements of Section 13 (a) or 15 (d) of the Securities Exchange Act of 1934 (15 U.S.C. 78m (a) or 78o(d)); and |
| (2) | The information contained in the Report fairly presents, in all material respects, the financial condition and results of operations of the Company. |
Date: September 30, 2024
| /s/ Joanne Wendy Kim | ||||||||
|
Joanne Wendy Kim Chief Financial Officer |