UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, DC 20549
FORM 10-K
(Mark One)
| ☒ | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
FOR THE FISCAL YEAR ENDED MARCH 31, 2023
OR
| ☐ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
Commission file number: 001-38892
BEYOND AIR, INC.
(Exact name of registrant as specified in its charter)
| Delaware | 47-3812456 | |
|
(State or other jurisdiction of incorporation or organization) |
(I.R.S. Employer Identification No.) |
|
900 Stewart Avenue, Suite 301 Garden City, NY |
11530 | |
| (Address of principal executive offices) | (Zip Code) |
516-665-8200
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
| Title of each class: | Trading Symbol | Name of each exchange on which registered: | ||
| Common Stock, par value $0.0001 per share | XAIR | The Nasdaq Stock Market LLC |
Securities registered pursuant to Section 12(g) of the Act:
None
Indicate by a check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act.
| Yes ☐ | No ☒ |
Indicate by a check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Securities Exchange Act of 1934.
| Yes ☐ | No ☒ |
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports) and (2) has been subject to such filing requirements for the past 90 days.
| Yes ☒ | No ☐ |
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files).
| Yes ☒ | No ☐ |
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer | ☐ | Accelerated filer | ☐ | |
| Non-accelerated filer | ☒ | Smaller reporting company | ☒ | |
| Emerging growth company | ☐ |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report. ☐
If securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant included in the filing reflect the correction of an error to previously issued financial statements. ☐
Indicate by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive-based compensation received by any of the registrant’s executive officers during the relevant recovery period pursuant to §240.10D-1(b). ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act).
| Yes ☐ | No ☒ |
As of September 30, 2022, the last business day of the registrant’s most recently completed second fiscal quarter, the aggregate market value of the registrant’s voting stock held by non-affiliates was approximately $201.2 million based on the last reported sale price of the registrant’s common stock on the Nasdaq Capital Market.
There were 31,675,521 shares of common stock outstanding as of June 22, 2023.
DOCUMENTS INCORPORATED BY REFERENCE
None.
Beyond Air, Inc.
TABLE OF CONTENTS
FORM 10-K
For the Year Ended March 31, 2023
INDEX
|
|
References in this Annual Report on Form 10-K (this “Annual Report”) to the “Company,” “Beyond Air,” “we,” “our,” or “us” mean Beyond Air, Inc. and its subsidiaries except where the context otherwise requires.
FORWARD-LOOKING STATEMENTS AND MARKET DATA
This Annual Report contains forward-looking statements. We intend such forward-looking statements to be covered by the safe harbor provisions for forward-looking statements contained in Section 27A of the Securities Act of 1933 (the “Securities Act”) and Section 21E of the Securities Exchange Act of 1934 (the “Exchange Act”). All statements other than statements of historical facts contained in this Annual Report, including statements regarding our future results of operations and financial position, business strategy, approved product and product candidates, certifications or approvals, timing of our clinical development activities, research and development costs, our commercialization plans and the expected timing thereof, timing and likelihood of success, and the plans and objectives of management for future operations and future results of anticipated products are forward-looking statements. These statements involve known and unknown risks, uncertainties and other important factors that may cause our actual results, performance or achievements to be materially different from any future results, performance or achievements expressed or implied by the forward-looking statements.
In some cases, you can identify forward-looking statements by terms such as “may,” “will,” “should,” “expect,” “plan,” “anticipate,” “expect,” “could,” “intend,” “target,” “project,” “contemplate,” “believe,” “estimate,” “predict,” “potential,” or “continue” or the negative of these terms or other similar conditional expressions. The forward-looking statements in this Annual Report are only predictions. We have based these forward-looking statements largely on our current expectations and projections about future events and financial trends that we believe may affect our business, financial condition and results of operations. These forward-looking statements speak only as of the date of this Annual Report and are subject to a number of important factors that could cause actual results to differ materially from those in the forward-looking statements, including the factors described under the sections in this Annual Report titled “Risk Factors” and “Management’s Discussion and Analysis of Financial Condition and Results of Operations” as well as the following:
- our ability to successfully commercialize our LungFit® PH system in the U.S.;
- our ability to obtain a CE Certificate of Conformity to CE mark LungFit® in the European Union (the “EU”);
- our expectation to incur losses for the next few years;
- our ability to predict accurately the demand for our products, and products under development and to develop strategies to address markets successfully;
- the possibility that products may contain undetected errors or defects or otherwise not perform as anticipated;
- the anticipated development of markets we sell our products into and the success of our products in these markets;
- our future capital needs and our need to raise additional funds;
- our ability to build a pipeline of product candidates and develop and commercialize our approved products;
- our ability to enroll patients in clinical trials, timely and successfully complete those trials and receive necessary certifications or regulatory approvals;
- our ability to maintain our existing or future collaborations or licenses;
- our ability to protect and enforce our intellectual property rights;
- federal, state, and foreign regulatory requirements, including the U.S. Food and Drug Administration (“FDA”) regulation of our approved product and product candidates;
- our ability to obtain and retain key executives and attract and retain qualified personnel; and
- our ability to successfully manage our growth, including as a commercial-stage company.
Moreover, we operate in an evolving environment. New risk factors and uncertainties may emerge from time to time, and it is not possible for management to predict all risk factors and uncertainties.
You should read this Annual Report and the documents that we reference in this Annual Report completely and with the understanding that our actual future results may be materially different from what we expect. We qualify all of our forward-looking statements by these cautionary statements. Except as required by applicable law, we do not plan to publicly update or revise any forward-looking statements contained herein, whether as a result of any new information, future events, changed circumstances or otherwise.
Beyond Air, Inc., the Beyond Air logo, and other trademarks or service marks of Beyond Air, Inc. appearing in this Annual Report are the property of Beyond Air, Inc. This Annual Report also includes trademarks, tradenames and service marks that are the property of other organizations. Solely for convenience, trademarks and tradenames referred to in this Annual Report may appear without the ® and ™ symbols, but those references are not intended to indicate, in any way, that we will not assert, to the fullest extent under applicable law, our rights, or that the applicable owner will not assert its rights, to these trademarks and tradenames.
|
|
MARKET, INDUSTRY AND OTHER DATA
This Annual Report contains estimates, projections, market research and other information concerning our industry, our business, markets for LungFit® PH and our product candidates and the size of those markets, the prevalence of certain medical conditions, LungFit® PH market access, prescription data and other physician, patient and payor data. Unless otherwise expressly stated, we obtain this information from reports, research surveys, studies and similar data prepared by market research firms and other third parties, industry, medical and general publications, government data and similar sources as well as from our own internal estimates and research and from publications, research, surveys and studies conducted by third parties on our behalf. Information that is based on estimates, projections, market research or similar methodologies is inherently subject to uncertainties and actual events or circumstances may differ materially from events and circumstances that are reflected in this information. As a result, you are cautioned not to give undue weight to such information.
SUMMARY OF PRINCIPAL RISK FACTORS
This summary briefly lists the principal risks and uncertainties facing our business, which are only a select portion of those risks. A more complete discussion of those risks and uncertainties is set forth in Part I, Item 1A of this Annual Report, entitled Risk Factors. Additional risks not presently known to us or that we currently deem immaterial may also affect us. If any of these risks occur, our business, financial condition or results of operations could be materially and adversely affected.
Our business is subject to the following principal risks and uncertainties:
Risks Related to our Financial Position and Capital Requirements
| ● | Numerous factors, including the incurring of significant losses, are relevant to our financial success and any or all such factors could have a disproportionate impact on our bottom line. |
| ● | It is highly likely that we will need to raise additional capital to meet our business requirements in the future, and such capital raising may be costly or difficult to obtain, and could dilute current stockholders’ ownership interests. |
| ● | Our failure to comply with the covenants or other terms of the Loan Agreement (as defined below), including as a result of events beyond our control, could result in a default under the Loan Agreement that could materially and adversely affect the ongoing viability of our business. |
Risks Related to Commercialization of our Approved Product or Product Candidates
| ● | If the market opportunities for our approved product or product candidates are smaller than we believe they are, our revenue may be adversely affected, and our business may suffer. |
| ● | Intense competition and rapid technological changes may adversely affect our ability to successfully commercialize our approved product or product candidates. |
| ● | If we are unable to scale sales and marketing capabilities or enter into agreements with third parties to market and sell our approved product or product candidates, we may be unable to generate revenue. |
| ● | Our approved product and any future product candidates that we may bring to market may fail to achieve the degree of market acceptance by physicians, patients, third-party payors and others in the medical community necessary for commercial success, and the market opportunity for our approved product or product candidates may be smaller than we estimate. |
| ● | If we fail to properly manage our anticipated growth, our business could suffer. |
| ● | Pricing pressure from our competitors and our customers may impact our ability to sell our products at prices necessary to support our current business strategies. |
| ● | Many of our current and potential competitors have substantially greater financial, technical and marketing resources than we do, and they may succeed in developing products that would render our approved product and any future product candidates obsolete or noncompetitive. |
| ● | We may be subject to enforcement action if we engage in improper marketing or promotion of our products. |
| ● | Healthcare legislative or regulatory reform measures may have a negative impact on our business and results of operations. |
| ● | Cybersecurity risks and the failure to maintain the confidentiality, integrity, and availability of our computer hardware, software, and Internet applications and related tools and functions could result in harm to our business and/or subject us to costs, fines or lawsuits. |
| ● | Failure to comply with applicable privacy, data and security regulations could result in substantial penalties, liability and negative publicity which could adversely affect our business operations. |
|
|
Risks Related to the Discovery and Development of Our Product Candidates
| ● | We cannot give any assurance that any of our product candidates will receive certification or regulatory approvals, which is necessary before they can be commercialized. |
| ● | We may incur additional costs or experience delays in completing, or ultimately be unable to complete, the development of our product candidates. |
| ● | We have conducted, and may in the future conduct, clinical trials for certain of our product candidates at sites outside the United States, and the FDA may not accept data from trials conducted in such locations. |
| ● | If we experience any of a number of possible unforeseen events in connection with clinical trials of our product candidates, potential certification or marketing approval or commercialization of our product candidates could be delayed or prevented. |
| ● | Our subsidiaries are exploring novel therapeutic processes with NO, and their product candidates are likely to be classified as pharmaceutical drugs by the FDA and thus face stringent regulation. |
| ● | We may experience delays or difficulties in the enrollment of patients in clinical trials. |
| ● | Serious adverse events (“SAEs”), or undesirable side effects of our product candidates may be identified. |
| ● | Even if we obtain certification or regulatory approvals for our product candidates, we will still face extensive, ongoing regulatory requirements and review, and our products may face future development and regulatory difficulties. |
Risks Related to our Reliance on Third Parties
| ● | We rely on third parties to conduct our clinical trials, and those third parties may not perform satisfactorily. |
| ● | Any failure by a third-party supplier or manufacturer to produce or deliver supplies for us or to provide necessary servicing may delay or impair our ability to complete our clinical trials or commercialize our approved product or product candidates. |
| ● | We may seek to enter into collaborations with third parties for the development and commercialization of our product candidates. If we fail to enter into such collaborations, or such collaborations are not successful, we may not be able to capitalize on the market potential of our approved product or product candidates. |
| ● | The third-party manufacturing facilities on which we rely are subject to significant regulation and may not continue to meet regulatory requirements. |
| ● | Our reliance on third parties necessitates the sharing of trade secrets, which increases the possibility of their improper disclosure or appropriation. |
Risks Related to our Intellectual Property
| ● | If we fail to obtain and maintain sufficiently broad patent protection of our technology, our competitors could develop and commercialize technology and products similar or identical to ours, and our ability to successfully commercialize our technology and products may be impaired. |
| ● | We may become involved in lawsuits to protect or enforce our patents or other intellectual property, which could be expensive, time-consuming and unsuccessful. |
| ● | We may be subject to claims by third parties asserting that we or our employees have misappropriated their intellectual property, or claiming ownership of what we regard as our own intellectual property. |
| ● | Patent policy and rule changes could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents. |
| ● | We may not be successful in obtaining or maintaining necessary intellectual or proprietary rights to our approved product or product candidates including, but not limited to acquisitions and in-licenses. |
|
|
Risks Related to our Business Operations
| ● | We manage our business through a small number of employees and key consultants. |
| ● | We may need to expand our operations and increase the size of our Company, which may expose us to difficulties in managing growth as well as additional regulatory operational and financial risks. |
| ● | Our international operations and plans to expand such operations present challenges and risks related to doing business internationally. |
| ● | The use of our product or any of our product candidates could result in product liability or similar claims that could be expensive, damage our reputation and harm our business. |
| ● | Natural disasters, geopolitical unrest, war, terrorism, public health issues or other catastrophic events could disrupt the supply, delivery or demand of products, which could negatively affect our operations and performance. |
| ● | We may continue to face business disruption and related risks resulting from the COVID-19 pandemic. |
| ● | We are dependent on information technology and our systems and infrastructure face certain risks, including from cybersecurity breaches and data leakage. |
Risks Related to the Ownership of our Common Stock
| ● | Stockholder disputes may be limited by the terms of our amended and restated certificate of incorporation. |
| ● | Trading in our common stock has been volatile and may continue to be volatile in the future. |
| ● | Anti-takeover provisions in our amended and restated certificate of incorporation and bylaws, as well as Delaware law, may discourage, delay, or prevent a change of control of our Company. |
Risks Related to Employee Matters
| ● | Our business could suffer if we lose the services of key members of our senior management, key advisors or personnel. |
| ● | Our employees may engage in misconduct or other non-compliant activities. |
| ● | We are subject to various healthcare laws; failure to comply with such laws may result in substantial penalties. |
| ● | Employee litigation and unfavorable publicity could negatively affect our future business. |
| ● | We may not be able to enforce covenants not to compete and therefore may be unable to prevent our competitors from benefiting from the expertise of some of our former employees. |
General Risk Factors
| ● | The increasing use and relevance of social media platforms may present new risks. |
| ● | Our business may be affected by unfavorable global or U.S. economic conditions. |
|
|
PART I
ITEM 1. BUSINESS
Business Overview
We are a commercial-stage medical device and biopharmaceutical company developing a platform of nitric oxide (“NO”) generators and delivery systems (the “LungFit® platform”) capable of generating NO from ambient air. Our first device, LungFit® PH received premarket approval (“PMA”) approval from the FDA in June 2022. The NO generated by the LungFit® PH system is indicated to improve oxygenation and reduce the need for extracorporeal membrane oxygenation in term and near-term (>34 weeks gestation) neonates with hypoxic respiratory failure associated with clinical or echocardiographic evidence of pulmonary hypertension in conjunction with ventilatory support and other appropriate agents. This condition is commonly referred to as persistent pulmonary hypertension of the newborn or “PPHN”. The LungFit® platform can generate NO up to 400 parts per million (“ppm”) for delivery to a patient’s lungs directly or via a ventilator. LungFit® can deliver NO either continuously or for a fixed amount of time at various flow rates and has the ability to either titrate dose on demand or maintain a constant dose. In July 2022, the Company commenced marketing LungFit® PH in the United States for PPHN as a medical device.
LungFit® can be used to treat patients on ventilators that require NO, as well as patients with chronic or acute severe lung infections via delivery through a breathing mask or similar apparatus. Furthermore, we believe that there is a high unmet medical need for patients suffering from certain severe lung infections that the LungFit® platform can potentially address. Our current areas of focus with LungFit® are PPHN, viral community-acquired pneumonia (“VCAP”) including COVID-19, bronchiolitis (“BRO”), nontuberculous mycobacteria (“NTM”) lung infection and those with various severe lung infections with underlying chronic obstructive pulmonary disease (“COPD”). The Company’s current product candidates will be subject to premarket reviews and approvals by the FDA, certification through the conduct of a conformity assessment by a notified body in the EU for the product to be CE marked, as well as comparable foreign regulatory authorities.
With Beyond Air’s focus on NO and its effect on the human condition, there are two additional programs that do not utilize our LungFit® system. Through our majority-owned affiliate Beyond Cancer, Ltd. (“Beyond Cancer”), NO is used to target solid tumors. The LungFit® platform is not utilized for the solid tumor indication due to need for ultra-high concentrations of gaseous nitric oxide (“UNO”). A proprietary delivery system has been developed that can safely deliver UNO in excess of 10,000 ppm directly to a solid tumor. This program has advanced to a phase 1 human study.
The second program which does not utilize the LungFit® platform, partially inhibits neuronal nitric oxide synthase (“nNOS”) in the brain to treat neurological conditions. The first target indication is autism spectrum disorder (“ASD”). ASD is a serious neurodevelopmental and behavioral disorder, and one of the most disabling conditions and chronic illnesses in children. ASD includes a wide range of developmental disorders that share a core of neurobehavioral deficits manifested by abnormalities in social interactions, deficits in communication, restricted interests, and repetitive behaviors. In 2023, the CDC reported that approximately 1 in 36 children in the U.S. is diagnosed with an ASD. The cost of caring for Americans with autism had reached $268 billion in 2015 and would rise to $461 billion by 2025 in the absence of more-effective interventions and support across the life span. We expect this program to progress from pre-clinical to a phase 1 first-in-human study by early 2025.
|
|
Our approved product and active pipeline of product candidates is shown in the table below:
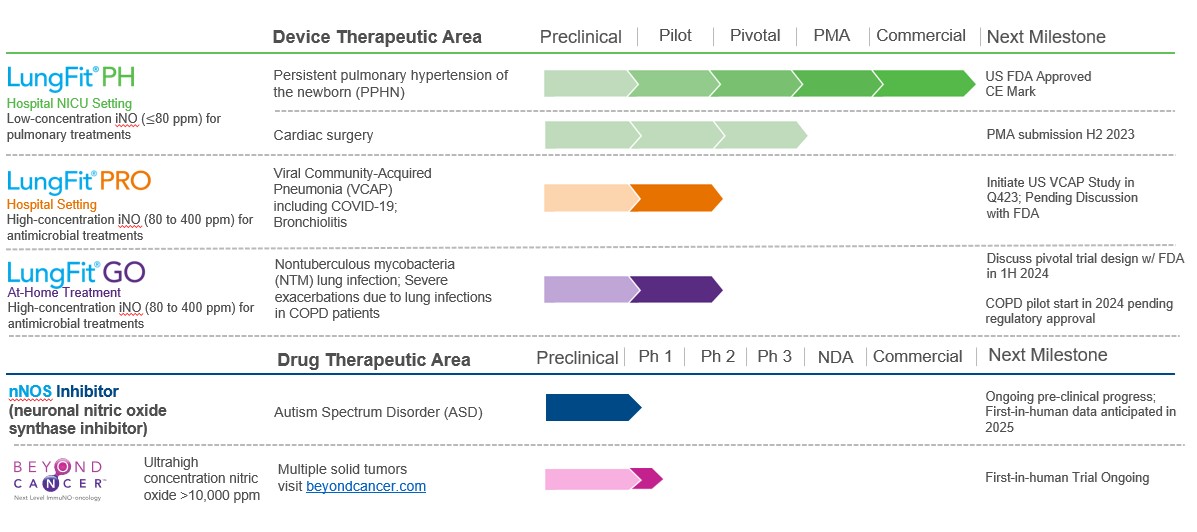
| (a) | All dates are calendar year, and based on projections and appropriate financing, anticipated first launch on a global basis pending appropriate regulatory approvals |
Our programs represent large market opportunities:

All figures are Company estimates for peak year sales: Global sales potential includes U.S. sales potential.
|
|
LungFit® PH is the first FDA approved system using our patented ionizer technology to generate on-demand NO from ambient air and, regardless of dose or flow, deliver it to a ventilator circuit. The device uses a medical air compressor to drive room air through a plasma chamber in the center of the unit where pulses of electrical discharge are created between two electrodes. The system uses the power equivalent to a 60-watt lightbulb to ionize the nitrogen and oxygen molecules, which then combine as NO with low levels of nitrogen dioxide (“NO2”) created as a byproduct. The products are then passed through a Smart Filter, which removes the toxic NO2 from the internal circuit. With respect to PPHN, the novel LungFit® PH is designed to deliver a dosage of NO to the lungs that is consistent with current guidelines for delivery of 20 ppm NO with a range of 0.5 ppm – 80 ppm (low concentration NO) for ventilated patients.
We believe the ability of LungFit® PH to generate NO from ambient air provides us with many competitive advantages over the current standard of NO delivery systems in the U.S., the EU, Japan and other markets. For example, LungFit® PH does not require the use of a high-pressure cylinder, does not require cumbersome purging procedures and places less burden on hospital staff in carrying out safety procedures.
Our novel LungFit® platform can also deliver a high concentration (>150 ppm) of NO directly to the lungs, which we believe has the potential to eliminate microbial infections including bacteria, fungi and viruses, among others. We believe that current FDA-approved NO vasodilation treatments would have limited success in treating microbial infections given the low concentrations of NO being delivered (<100 ppm). Given that NO is produced naturally by the body as an innate immunity mechanism, at a concentration of 200 ppm, supplemental high dose NO should aid in the body’s fight against infection. Based on our preclinical and clinical studies, we believe that 150 ppm is the minimum therapeutic dose to achieve the desired pulmonary antimicrobial effect of NO. To date, neither the FDA nor comparable foreign regulatory agencies in other countries or regions have approved any NO formulation and/or delivery system for >80 ppm NO.
LungFit® PH for the treatment of Persistent Pulmonary Hypertension of the Newborn (PPHN)
In June 2022, the FDA approved LungFit® PH to improve oxygenation and reduce the need for extracorporeal membrane oxygenation in term and near-term (>34 weeks gestation) neonates with hypoxic respiratory failure associated with clinical or echocardiographic evidence of pulmonary hypertension in conjunction with ventilatory support and other appropriate agents. LungFit® PH is the inaugural device from the LungFit® platform of NO generators that use patented ionizer technology and is the first FDA-approved product for Beyond Air.
We also expect to be CE Mark certified under the EU Medical Device Regulation (“EU MDR”) in the EU in the second half of calendar year 2023. According to the most recent year-end report from Mallinckrodt Pharmaceuticals (“Mallinckrodt”), sales of NO were $339.7 million in 2022 (down from $448.5 million in 2021) for the United States, Canada, Japan, Mexico and Australia, with ~90% in the United States. Outside of the U.S. there are multiple market participants which translates to considerably lower sales than in the U.S. We believe the U.S. sales potential of LungFit® PH in PPHN to be approximately $400 million and worldwide sales potential to be greater than $700 million. We initiated the first phase of our commercial launch in June 2022, and entered into phase 2 with an expanded commercial presence during the spring of 2023 in the U.S. and will continue to work toward a potential launch in the EU and globally in 2023 and beyond.
LungFit® PRO for the treatment of viral lung infections in hospitalized patients
Viral Community-Acquired Pneumonia (including COVID-19)
Viral pneumonia in adults is most commonly caused by rhinovirus, respiratory syncytial virus (“RSV”) and influenza virus. However, newly emerging viruses (including SARS-CoV-1, SARS-CoV-2, avian influenza A, and H1N1 viruses) have been identified as pathogens contributing to the overall burden of adult viral pneumonia. COVID-19 is an infectious disease caused by SARS-CoV-2, that resulted in a global pandemic, causing millions of hospitalizations and over 6.6 million deaths worldwide as of January 2023 according to the World Health Organization. Excluding the pandemic, there are approximately 350,000 annual viral pneumonia hospitalizations in the U.S., and up to 16 million annual viral pneumonia hospitalizations globally. For the broader annual viral pneumonia hospitalizations, we believe U.S. market potential to be greater than $1.5 billion and worldwide market potential to be greater than $3 billion.
We initiated a pilot study in late 2020 using our novel LungFit® PRO system at 150 ppm to treat patients with VCAP. The trial was a multi-center, open-label, randomized clinical trial in Israel, including patients infected with COVID-19. Patients were randomized in a 1:1 ratio to receive either inhalations of 150 ppm NO given intermittently for 40 minutes four times per day for up to seven days in addition to standard supportive treatment (“NO+SST”) or standard supportive treatment alone (“SST”). Endpoints related to safety (primary endpoint), oxygen saturation and ICU admission, among others, were assessed.
|
|
We presented results from the pilot study at the 32nd European Congress of Clinical Microbiology & Infectious Diseases (ECCMID 2022), which took place from April 23, 2022 through April 26, 2022 as a hybrid event both onsite in Lisbon, Portugal and online. At the time of the data cut off, the trial enrolled a total of 40 subjects hospitalized for VCAP (SARS-CoV-2, n=39; other viruses n=1). The ITT population included 35 subjects with 16 subjects in the inhaled NO group and 19 subjects in the control group. The primary COVID-19 treatments used during the study were Remdesivir (>30%) and Dexamethasone (>65%). Safety data from the study show that inhaled NO treatment was well tolerated overall with no treatment related adverse events as assessed by the investigators. There were two SAEs reported in the group receiving inhaled NO along with SST, which were determined to be related to underlying conditions and unrelated to study drug/device. From an efficacy perspective, results show a trend of shortening length of stay (“LOS”) by a factor 1.8 in favor of inhaled NO treatment. Duration of oxygen support, measured in-hospital and at home, was significantly shorter (p=0.0339) for inhaled NO treated subjects. Subjects with unstable oxygen saturation during hospitalization, 66.7% of the inhaled NO treatment group, reached stable saturation of ≥93% during hospital stay as compared to 26.7% in the SST group.
Following completion of the study and the 180-day follow-up period, incremental data were provided in a poster presentation at IDWeek 2022 held from October 19, 2022, through October 23, 2022 in Washington, D.C. In addition to the positive clinical results provided at ECCMID 2022, the poster showed a larger decline in c-reactive protein (“CRP”) from baseline for subjects treated with NO + SST compared to the control group. Analysis of the data provides compelling evidence that high concentration NO delivery with the LungFit® PRO generator and delivery system can be a powerful tool against any type of pneumonia, especially COVID-19. The Company anticipates commencing a study in calendar year 2023 following discussions with the FDA.
Bronchiolitis (BRO)
Bronchiolitis is the leading cause of hospital admission in children less than 1 year of age. The incidence is estimated to be 150 million new cases a year worldwide, with 2-3% (over 3 million) of them severe enough to require hospitalization. Worldwide, 95% of all cases occur in developing countries. In the U.S., there are approximately 120,000 annual bronchiolitis hospitalizations and approximately 3.2 million annual child hospitalizations globally. Currently, there is no approved treatment for bronchiolitis. The treatment for acute viral lung infections that cause bronchiolitis in infants is largely supportive care and is based primarily on prolonged hospitalization during which the infant receives a constant flow of oxygen to treat hypoxemia, a reduced concentration of oxygen in the blood. In addition, systemic steroids and inhalation with bronchodilators are sometimes utilized until recovery, but we believe that these treatments do not successfully reduce hospital length of stay. We believe the U.S. market potential for bronchiolitis to be greater than $500 million and worldwide market potential to be greater than $1.2 billion.
The pivotal study for bronchiolitis was originally set to be performed in the winter of 2020/21 but was delayed due to the pandemic. We have completed three successful pilot studies for bronchiolitis. A further analysis of the three previously reported pilot studies was presented at the ATS International Conference 2021, which was held virtually from May 14, 2021 through May 19, 2021. Analysis across the studies (n=198 infants, mean age 3.9 months) showed that 150 ppm – 160 ppm NO administered intermittently was generally safe and well tolerated with adverse event rates similar among treatment groups with no reported treatment-related serious adverse events. The short course of treatments with intermittent high concentration inhaled NO was effective in shortening hospital length of stay and accelerating time to fit for discharge – a composite endpoint of clinical signs and symptoms to indicate readiness to be evaluated for hospital discharge. This treatment was also effective in accelerating time to stable oxygen saturation – measured as SpO2 ≥ 92% in room air. Additionally, NO at a dose of 85 ppm NO showed no difference compared to control for all efficacy endpoints, while 150 ppm NO showed statistical significance when compared to control.
|
|
Additionally, long-term safety data for high concentration inhaled NO in bronchiolitis was presented at the Pediatric Academic Societies Meeting 2022 (PAS 22), which was held in Denver, Colorado from April 21, 2022 through April 25, 2022. A total of 101 infants from the three prior pilot studies for bronchiolitis (n=198) participated in the long-term follow-up study. Study endpoints for the long-term safety study included percentage of subjects re-hospitalized for bronchiolitis related reasons, such reasons included wheezing episodes, pneumonia, and asthma and the percentage of subjects re-hospitalized for any reason. Data from the study showed the re-hospitalization rate per 100 Patient Exposure Years (PEY) due to bronchiolitis related reasons trended favorably for the inhaled NO group. In addition, the long-term subject re-hospitalization rate for any reason was similar between inhaled NO and control groups. As such, the study concluded that the treatment of hospitalized infants with acute bronchiolitis by intermittent high dose inhaled NO show a favorable long-term safety profile.
We believe that the entirety of data at 150 ppm - 160 ppm NO in both adult and infant patient populations supports further development of LungFit® PRO in a pivotal study for patients hospitalized with VCAP or bronchiolitis.
LungFit® GO for the treatment of Nontuberculous mycobacteria (NTM)
NTM lung infection is a rare and serious pulmonary disease associated with increased morbidity and mortality. Patients with NTM lung disease may experience a multitude of symptoms such as fever, weight loss, cough, lack of appetite, night sweats, blood in the sputum and fatigue. Patients with NTM lung disease, specifically Mycobacterium abscessus (M. abscessus) representing 20% - 25% of all NTM and other forms of NTM that are refractory to antibiotic therapy, frequently require lengthy and repeated hospital stays to manage their condition. There are no treatments specifically indicated for the treatment of M. abscessus lung disease in North America, Europe or Japan.
There are approximately 50,000 to 90,000 people with NTM infections in the U.S. In Asia, the number of patients suffering from NTM surpasses what is seen in the U.S. There is one inhaled antibiotic approved for the treatment of refractory Mycobacterium avium complex (“MAC”). Current guideline-based approaches to treat NTM lung disease involve multi-drug regimens of antibiotics that may cause severe, long lasting side effects, and treatment can be longer than 18 months. Median survival for NTM MAC patients is approximately 13 years while median survival for patients with other variations of NTM is typically 4.6 years. The prevalence of human disease attributable to NTM has increased over the past two decades. In a study conducted between 2007 and 2016, researchers found that the prevalence of NTM in the U.S. is increasing at approximately 7.5% per year. M. abscessus treatment costs are estimated to be more than double that of MAC. A 2015 publication by co-authors from several U.S. government departments stated that cases in 2014 alone cost the U.S. healthcare system approximately $1.7 billion. For this indication, we believe U.S. sales potential to be greater than $1 billion and worldwide sales potential to be greater than $2.5 billion.
In December 2020 we began a 12-week, multi-center, open-label clinical trial in Australia intended to enroll approximately 20 adult patients with chronic refractory NTM lung disease. We received a grant of up to $2.17 million from the Cystic Fibrosis Foundation (“CFF”) to fund this study and advance the clinical development of inhaled NO to treat NTM pulmonary disease. The trial enrolled both cystic fibrosis (“CF”) and non-CF patients infected with MAC, M. abscessus or any strain of NTM. The study consisted of a run-in period followed by two treatment phases. The run-in period provided a baseline for the efficacy endpoints. The first treatment phase took place over a two-week period and began in the hospital setting where patients were titrated from 150 ppm NO up to 250 ppm NO over several days. During this phase patients received NO for 40 minutes, four times per day while Methemoglobin (“MetHb”) levels were monitored. Patients were also trained to use LungFit® GO and subsequently discharged to complete the remaining portion of the two-week treatment period at their home at the highest tolerated NO concentration. For the second treatment phase, a 10-week maintenance phase, the administration was twice daily. The study evaluated safety, quality of life, physical function, and bacterial load among other parameters.
At the American Thoracic Society International Conference 2022 (ATS 2022), which was held in San Francisco from May 13, 2022 through May 18, 2022, we presented positive interim data from the ongoing study. At the time of data cutoff on April 4, 2022, a total of 15 subjects were enrolled in the pilot study. The mean age of subjects was 62.1 years (range: 22 – 82 years) with the majority female (80%), a distribution consistent with real-world NTM disease. All 15 subjects were successfully titrated to 250 ppm NO in the hospital setting, and no patients required dose reductions during the subsequent at-home portion of the study. Patients were followed up for 12 weeks after the 12-week treatment period was completed.
|
|
After completion of the study, we presented positive results at the American College of Chest Physicians (“CHEST”) annual meeting, held from October 16, 2022 through October 19, 2022, further supporting development of intermittent high dose NO for the treatment of NTM. The study demonstrated that high dose NO treatment was well-tolerated in both the home and hospital settings. During the 10-week at-home treatment period of the study, a total of 2,492 inhalations were self-administered with overall high treatment compliance (>90%). There were no SAEs related to treatment discontinuations reported over the 12-week treatment or 12-week follow up periods. Key efficacy endpoints showed strong results with improvement seen in the majority of quality-of-life domains. Respiratory function and physical function were maintained during treatment and follow-up. Trends in the reduction of microbial load were observed and one subject achieved culture conversion with three consecutive negative sputum samples. We anticipate commencing a pivotal study in the first half of calendar year 2025 following discussions with the FDA.
Our program in COPD is in the preclinical stage and, subject to obtaining additional financing, is expected to enter clinical trials in calendar year 2024.
Ultra-High Concentration NO (UNO) in solid tumors through majority-owned affiliate Beyond Cancer, Ltd.
In the fourth calendar quarter of 2021, Beyond Cancer, our majority-owned affiliate, raised $30 million in a private placement of common shares. The investors purchased a 20% equity ownership in Beyond Cancer, while Beyond Air maintained 80%. The funding is being used to accelerate ongoing preclinical work, including the completion of IND-enabling studies, completion of a Phase 1 study, expansion of preclinical programs for combination studies, hiring of additional Beyond Cancer team members, and optimization of the delivery system, as well as for general corporate purposes.
Beyond Cancer will benefit from Beyond Air’s NO expertise, IP portfolio, preclinical oncology team, and regulatory progress, and will pay Beyond Air a single-digit royalty on all future revenues. Beyond Cancer is being led by a seasoned leadership team with experience in emerging healthcare companies and clinical oncology.
Selena Chaisson, MD, currently serves as Beyond Cancer’s Chief Executive Officer. Previously, Dr. Chaisson was the Director of Healthcare Investments at Bailard, where she spent 16 years focusing on highly specialized, emerging healthcare opportunities with more than one-third of her portfolio dedicated to investing in oncology companies. Prior to Bailard, Dr. Chaisson held senior executive roles at RCM Capital Management and Tiger Management. RCM Capital Management was acquired by Dresdner Bank in 1996 and then subsequently acquired by Allianz Global Investors U.S. in 2001. Dr. Chaisson received a BS in microbiology in 1987 from Louisiana State University in Baton Rouge, LA, where she graduated summa cum laude. She earned her MBA and MD from Stanford University in 1992 and 1993, respectively.
The Beyond Cancer Board of Directors consists of six members:
| ● | Steve Lisi, Chairman of the Board, and CEO and Chairman of the Board of Beyond Air | |
| ● | Selena Chaisson, MD, Director, and Chief Executive Officer of Beyond Cancer | |
| ● | Amir Avniel, Executive Director, and Chief Business Officer and Co-Founder of Beyond Air | |
| ● | Robert Carey, Director, and Board Member of Beyond Air | |
| ● | David Dvorak, Director | |
| ● | Gregory Berk, MD, Director |
|
|
UNO has shown anticancer properties in preclinical trials by eliciting an immune response from the host. We have released preclinical data at several medical/scientific conferences showing the promise of delivering NO directly to tumors at concentrations of 20,000 ppm – 200,000 ppm. Results showed that local tumor ablation with NO conveyed anti-tumor immunity to the host. In April 2022, we presented in vivo and in vitro preclinical data at the American Association for Cancer Research (“AACR”) 2022 annual meeting. The in vivo study assessed the mode of action following a single 5-minute gaseous NO (“gNO”) treatment which provided data showing an effect on the primary tumor 14 days post-treatment. These data showed that intratumoral injections of concentrations of gNO at 20,000 and 50,000 ppm led to increased recruitment of T cells, B cells, macrophages, and dendrocytes to the primary tumor. An elevated number of T cells and B cells were also detected in the spleen and blood 21 days following gNO treatment. In addition, at the same time point, a marked reduction in the number of myeloid-derived suppressor cells was observed in the spleen. Results from the in vitro study showed that exposure of six different cancer cell lines – including human ovarian and pancreatic and mouse lung, melanoma, colon, and breast – to UNO ranging from 10,000 ppm to 100,000 ppm for up to 10 minutes resulted in a dose-dependent cytotoxic response. The higher concentration doses of gNO led to near-instant cell death, while the lower concentration doses required a longer exposure period to elicit cell death. Cell viability was assessed using two assays: XTT and clonogenic assay. After one minute of exposure to 25,000 ppm gNO, less than 10% viability was observed in all cell lines.
In March 2022 we received approval from the IMOH (Israeli Ministry of Health) to do a Phase 1 study.
The second half of calendar year 2022 was a time of significant progress for Beyond Cancer. On August 23, 2022, we announced that the first patient was treated in a first-in-human Phase 1 study to assess the safety and immune biomarkers of UNO therapy. In November, at the annual meeting of the Society for Immunotherapy of Cancer (“SITC”), we presented new in vivo combination data that support the potential of the Company’s novel UNO therapy to treat various types of solid tumors in combination with immune checkpoint inhibitor (“ICI”) therapies, including anti-PD-1. The data presented at SITC appears to indicate that UNO in combination with anti-PD-1 treatment may lead to higher tumor regression rates and prolonged survival. Also in 2022, on December 13, we announced the publication of preclinical data in the peer-reviewed journal Cancer Cell International (CCI), which showed that our proprietary tumor ablation technology utilizing UNO induced a potent innate and adaptive immune response that prevented metastases and resulted in a statistically significant survival benefit.
Calendar year 2023 began with the announcement of Beyond Cancer’s entry into a sponsored research agreement with Stanford School of Medicine and the appointment of Frederick M. Dirbas, MD, Associate Professor of Surgery, Division of Surgical Oncology, Stanford School of Medicine, and Mark D. Pegram, MD, the Suzy Yuan-Huey Hung Endowed Professor of Medical Oncology at the Stanford School of Medicine, to the Beyond Cancer Scientific Advisory Board (“SAB”). In addition to the research agreement, Dr. Dirbas was named as Chair of the SAB, which provides guidance for ongoing preclinical studies as well as ongoing and planned future clinical trials in the use of UNO to treat solid tumors. The newly appointed members of the SAB will work to provide input on the clinical development of Beyond Cancer’s UNO technology, particularly as it relates to the U.S. regulatory submission.
In April 2023, Beyond Cancer presented additional preclinical data for UNO therapy in solid tumors during the AACR 2023 annual meeting. Data showed a statistically significant survival benefit for repeat dosing of UNO compared to anti-mCTLA-4 as monotherapy and repeat doses of UNO prolonged survival in combination with anti-PD-1 compared to gNO alone. With regard to tumor volume, statistically significant reductions were observed with repeat dosing of UNO versus anti-mPD-1 as a monotherapy and in combination with anti-CTLA-4 versus anti-CTLA-4 alone. Additionally, the data shows that short exposures between 10 seconds to one minute of tumor cells to UNO at increasing concentrations of 25,000 ppm to 100,000 ppm NO significantly upregulate mPD-L1 expression in a dose and time-dependent manner. Also, in vivo experiments exhibited a statistically significant day 1 increase in M1 macrophages, decrease in Tregs, and reduction in tumor cell viability was directionally maintained through day 5. We believe that together with the known ability of NO to activate and recruit the immune system, the data presented at this year’s AACR annual meeting appears to indicate that repeat dosing of UNO is feasible and may be effective even in difficult-to-treat, non-immunogenic tumor types.
The Company expects to present top-line data from the first-in-human Phase 1 trial before the end of calendar year 2023.
Selective neuronal nitric oxide synthase (nNOS) inhibitor for the treatment of neurological conditions in collaboration with Hebrew University of Jerusalem
On June 15, 2023, the Company announced that it had entered into an agreement with Yissum Research Development Company of the Hebrew University of Jerusalem, LTD. to acquire the commercial rights for neuronal nitric oxide synthase (nNOS) inhibitors being developed for the treatment of autism spectrum disorder (“ASD”) and other neurological conditions. Currently, there are no FDA approved therapies utilizing nNOS inhibitors specifically for the treatment of ASD. Under the terms of the agreement, Beyond Air will make payments to the University over the next two years for preclinical work. Also, the Company will pay a low single digit royalty on net sales and certain one-time payments based on clinical, regulatory and sales milestones.
Work is currently being done by the University in a preclinical setting. We expect the program to progress into a phase 1 first-in-human study early in 2025.
|
|
Background and NO Mechanism of Action
NO is recognized as a vital molecule involved in many physiological and pathological processes. NO is naturally produced by the body’s immune system to provide a first line of defense against invading pathogens. It is a powerful molecule with a short half-life of a few seconds in the blood, enabling it to be cleared rapidly from the body. NO has been shown to play a critical role in the function of several body systems. For example, as vasodilator of smooth muscles, NO enhances blood flow and circulation. In addition, NO is involved in regulation of wound healing and immune responses to infection. The pharmacology, toxicity and other data for NO in humans is generally well known, and its use has been approved by the FDA as a vasodilator. The precise effect of inhaled NO is dependent on concentration, oxidation state and type of pathogen.
NO has multiple immunoregulatory and antimicrobial functions that are likely to be of relevance to inhaled NO therapy. In vitro studies suggest that NO possesses anti-microbial activity against common bacteria, gram positive and gram negative, as well as mycobacteria, fungi, yeast, parasites and helminths. It has the potential to eliminate multi-drug resistant strains of the above. Anti-viral activity covers respiratory viruses such as influenza, corona viruses, RSV and others. In healthy humans, NO has been shown to stimulate mucociliary clearance, and low levels of nasal NO correlate with impaired mucociliary function in the human upper airway. Unlike other inhaled drugs, NO is also a smooth muscle relaxant and avoids the concomitant bronchial constriction often associated with inhaled antibiotics and mucolytics. A potential benefit of these multiple mechanisms may be that in addition to treating lung infections in CF patients, this suggests that NO may be useful in directly treating the mucus caused by CF, which is the principal manifestation of the disease.
Nitric Oxide and Infection
NO possesses broad-spectrum anti-microbial activity acting against bacteria, fungi and viruses. NO is produced at high output as part of the innate immune response. NO and its by-products (for example, reactive nitrogen species (“RNS”)) are responsible for the process of killing microorganisms within white blood cells called macrophages and in organs such as the lungs and other mucolytic tissues.
|
|
More than a decade ago, several research groups showed that NO and RNS possess anti-viral activity and affect several viruses including coxsackievirus, RSV, influenza, severe acute respiratory syndrome (SARS), coronavirus, rhinovirus, herpes simplex virus, Epstein-Barr virus (EBV), and others. NO has also been shown to be useful in preventing bacterial growth on surfaces.
Continuous exposure to 150 ppm NO and above, especially in the lungs, may have side effects and cause damage to host cells. Intermittent exposure to NO in cycles retains NO anti-microbial activity both in vitro and in animal model of infection. Exposure of bacteria to concomitant 30-minute treatments with 160 ppm NO resulted in a significant reduction in bacterial load. A similar dose has been shown to reduce viruses (common influenza) by 30-100% in a canine kidney infection model. In vivo, in a pneumonia model in rats, inhaled 160 ppm NO, for 30 minutes, every 4 hours, resulted in significant reduction in bacteria counts in the lungs, without affecting the body’s defense mechanisms, and without any other adverse effect. In addition, we believe a daily dose of 160 ppm of NO can treat bovine respiratory disease (BRD) in cattle.
Importantly, several studies report synergy between NO and antibiotic drugs. Adjunctive treatment combining NO together with inhaled tobramycin antibiotics or other anti-microbial agents has been shown to greatly enhance the efficacy of the antibiotics in dispersing P. aeruginosa biofilms and to increase their ability to elicit anti-microbial activity. These studies suggest that adjuvant treatment combining NO with antibiotics might have a beneficial role by reducing bacterial infectivity, and therefore reduce the dependency on antibiotics.
Beyond Air Technology
We have developed the Beyond Air NO generator and delivery system which we call LungFit®, a novel and precise delivery system that uses NO generated from ambient air with a novel NO generator, the ionizer. Our system provides continuous monitoring and control of the gaseous content administered during intermittent and continuous NO inhalation treatments, as well as a precise and reliable monitoring system that is able to monitor patient status and alert medical staff to any adverse effects.
The LungFit® system is innovatively designed to provide patients with a gaseous dose of NO (ranging from 0.5 ppm up to 400 ppm) combined with ambient air. The gaseous blend is supplied to the patient via a ventilator, face mask, or similar apparatus. LungFit® is designed to minimize the time that NO is mixed with oxygen and air. The system is also designed to continuously monitor inhaled NO concentration, NO2 concentration and oxygen. A dedicated screen allows for monitoring of the gas mixture. Further, our approved product and product candidates resemble other inhalation systems, making them user friendly, with operation and maintenance that we believe will be immediately familiar to medical staff. Our LungFit® system has been manufactured at commercial scale with a contract manufacturer.
When programmed for lung infections, the LungFit®, is designed to specifically deliver a NO dosage of 150 ppm and higher. We believe that the LungFit® has a number of advantages over other NO formulation delivery systems. For example, it is:
| ● | optimized to deliver 150 ppm and higher of NO, whereas existing NO delivery systems on the market consist of a maximum deliverable NO concentration of 80 ppm; | |
| ● | equipped with a monitoring system that continuously monitors system parameters (e.g., NO, NO2 and inhaled fraction of inspired oxygen (“FiO2”) concentrations); | |
| ● | capable of providing constant flow of NO, which we believe allows it to adequately cover the surface area of the lung to eliminate bacteria, viruses, fungi and other microbes; | |
| ● | programmable and able to deliver different dosage regimens for a wide range of lung infections; | |
| ● | able to generate NO from ambient air, eliminating the need for the use of high-pressure cylinders; | |
| ● | designed to be used by the patient, thus convenient and portable; and | |
| ● | administered non-invasively through a facial mask, which has the potential to address severe infections in large, underserved chronic-care markets, such as CF and COPD. |
|
|
We believe that our solution has the potential for a number of additional benefits and opportunities, as follows:
| ● | The antimicrobial and multiple other properties of the NO molecule delivered to the lungs suggest the potential for application in a wide range of respiratory diseases. In contrast to the often arduous and slow drug discovery process for small molecules, proteins, and peptides, the use of NO in medicine is well-known, and therefore the identification of conditions where NO provides benefits has been, and we expect will continue to be, much simpler, quicker and less costly. | |
| ● | The FDA approved the use of NO as an inhaled drug for the treatment of PPHN in 1999. More than 20 years of clinical experience in the delivery, monitoring and understanding of NO in the clinical environment for vascular uses has been documented. | |
| ● | NO is naturally produced by the immune system and acts as a first line of defense against infectious diseases. We believe therapeutic use of NO for viral and bacterial co-infections would potentially improve the success of antimicrobial and anti-viral treatments by mimicking the body’s natural defense mechanism and thereby directly reduce viral infectivity, as well as antibiotic drug resistant bacteria. | |
| ● | NO is used naturally by the body for vasodilation and we believe that the benefits to patients with various medical conditions will be seen via vasodilation when delivered with our system. |
NitricGen License
On January 31, 2018, we entered into a definitive agreement to acquire a global, exclusive, perpetual, transferable license to the eNOGenerator and associated critical assets including intellectual property, know-how, trade secrets and confidential information (the “License”) from NitricGen Inc. (“NitricGen”). The eNOGenerator is a novel and precise delivery system that uses NO generated from ambient air with a novel NO generator.
The Beyond Air LungFit® system, which incorporates the eNOGenerator, has been designated as a medical device by the FDA. The eNOGenerator can generate NO on demand for delivery to the lungs at concentrations ranging from 0.5 to 400 ppm. With the License, we expect that we will be able to target all conditions requiring NO at any concentration, regardless of the need for intermittent or continuous dosing.
Under the terms of the License, we agreed to pay NitricGen an aggregate of $2 million in up-front, clinical, and regulatory milestone payments, with the majority pertaining to regulatory milestones, as well as royalties on net sales of the delivery system containing the eNOGenerator at a percentage in the low-single digits. As partial consideration for the License, we issued to NitricGen warrants to purchase 100,000 shares of our common stock at an exercise price of $6.90 per share. To date, $1.5 million has been paid for milestones that were earned.
Cystic Fibrosis Foundation Agreement
On February 10, 2021, we received a grant for up to $2.17 million from the CFF to advance the clinical development of high concentration NO for the treatment of nontuberculous mycobacteria pulmonary disease, which disproportionally affects CF patients. Under the terms of the agreement, the funding will be allocated to the ongoing LungFit® GO NTM pilot study. The Company met the third milestone and received a reimbursement of $0.4 million in the fiscal year ended March 31, 2023. The Company has received a total of $1.3 million from the CFF since February 2021.
|
|
Strategies
Our objective is to build a leading medical device and biopharmaceutical company that develops and commercializes patented and proprietary products for the treatment of respiratory infections and diseases, with an initial focus on the treatment of PPHN, AVP, BRO, NTM and severe infections in COPD patients, among others. We are exploring and testing the effects of NO on solid tumors through our subsidiary, Beyond Cancer. Additionally, we are exploring the development of nNOS inhibitors for the treatment of ASD and other neurological conditions. If our clinical trials for our product candidates are successful, we expect to seek certification or marketing approval from the FDA and other worldwide authorities and notified regulatory bodies.
Our Clinical Results to Date
We have conducted several clinical trials to assess our 150-250 ppm NO inhalation-treatment in various indications. These trials include:
| Date | Study | Indication | Primary | Results |
| 2011 | Pilot Safety (n=10) | All comers | Safety | No SAEs |
| 2013 – 2014 | Proof of Concept (“POC”) double blind randomized (n=43) | Bronchiolitis (due to any virus) | Safety & Efficacy | No SAEs; 24 hour reduction in hospital length of stay |
| 2013 – 2014 | Pilot open label (n=9) | Cystic Fibrosis (CF) | Safety & Efficacy | No SAEs; Lowered bacterial load |
| 2016 | Compassionate use investigator sponsored research (“ISR”) (n=2) | NTM abscessus (CF) | Safety & Efficacy | No SAEs; clinical & surrogate endpoints improved |
| 2017 | Compassionate use National Institute of Health, U.S. (n=1) | NTM abscessus (CF) | Safety & Efficacy | No SAEs; Improvements in clinical endpoints |
| 2017 | Pilot open label (n=9) | NTM abscessus | Safety & Efficacy | No SAEs; clinical & surrogate endpoints improved |
| 2017-2018 | Pilot; double blind randomized (n=68) | Bronchiolitis (due to any virus) | Safety & Efficacy | No SAEs; 27 hour reduction in hospital length of stay |
| 2018 | Compassionate use ISR (n=1) | NTM abscessus (CF) | Safety | No SAEs at 250 ppm NO dose |
| 2019-2020 | Pilot; double blind randomized (n=87) | Bronchiolitis (due to any virus) | Safety & Efficacy | No SAEs; 150 ppm treatment showed statistically significant improvements in primary and key secondary endpoints compared to both 85 ppm and control |
| 2020-2022 | Pilot; randomized, controlled (n=35) | VCAP (due to any virus, including COVID-19) | Safety & Efficacy | No SAEs related to treatment; 150 ppm NO showed statistically significant improvement in duration of oxygen use |
| 2020-2022 | Pilot open label (n=15) | NTM (all strains) | Safety & Efficacy | All patients titrated to 250 ppm NO with no treatment related discontinuations during 11+ weeks of self-administration at home; Improvements in QoL |
| 2021-2022 | Pilot; randomized, controlled (n=101) | Bronchiolitis Long Term Follow-up | Safety | Favorable long term safety profile based on re-hospitalization rate |
|
|
Cystic Fibrosis and NTM Clinical Development
In 2011, a prospective, open label, controlled, single-center pilot safety study was conducted on ten healthy adults between 20 and 62 years of age. The data were published in the Journal of Cystic Fibrosis in 2012. Subjects received 160 ppm NO for 30 minutes, five times a day, for five consecutive days via direct inhalation to the lungs using a prototype delivery system. The primary objective of the study was to determine the effect of inhaled 160 ppm NO on pulmonary function tests and characterize the relationship between high-concentration NO administration and MetHb – a form of hemoglobin that is a biproduct of NO and hemoglobin that cannot bind oxygen – and establish a MetHb safety threshold level to assess adverse events associated with the treatment. Secondary objectives of the study were to assess the changes in cytokine levels. Multiple safety markers were continuously monitored including: NO levels, NO2 (a biproduct of NO and O2 that can be toxic at high concentrations), FiO2, as well as MetHb and oxygen saturation (“SaO2”). Vital signs, lung function, blood chemistry (including nitrite/nitrates), hematology, prothrombin time, inflammatory cytokine/chemokines levels and endothelial activation (angiopoietin ratio) were also closely monitored. All individuals tolerated the NO formulation treatment courses well. No SAEs occurred. The maximum amount of air one can forcefully exhale in one second, known as forced expiratory volume in one second (“FEV1”) and other lung function parameters, serum nitrites/nitrates, prothrombin, pro-inflammatory cytokine and chemokine levels did not differ between baseline and day 5, while MetHb increased during the study period by an average of 0.9%, as expected. These data suggest that inhalation of 160 ppm NO for 30 minutes, five times a day, for five consecutive days is well tolerated in healthy individuals.
In 2014, we completed a pilot open label, multi-center study in nine CF patients (≥10 years old). Patients received intermittent (30 minutes, three times a day) inhalation of 160 ppm NO formulation, five days a week, over a two-week period. The study was performed in two centers, Soroka Medical Center and Schneider Children’s Medical Center of Israel. The primary endpoints of the study were to determine the MetHb percentage, adverse events associated with inhaled NO and the percentage of subjects who prematurely discontinued the study due to adverse events (“AEs”) and/or SAEs, or for any other reason. AEs were reported by five (55.5%) subjects. There were no SAEs related to NO therapy, no treatment-related withdrawals due to AEs, and no deaths. AEs considered by the investigator as possibly or probably related to treatment were reported for two (22.2%) subjects. There were no AEs of MetHb elevation >5% or NO 2 elevation >5 ppm (study safety threshold of MetHb and NO2, respectively). In total, seven cases of hemoptysis were reported in two subjects and all events were mild in severity. There was no cumulative effect of MetHb exposure during the study. The maximum MetHb level reported was 4.6%. Several secondary efficacy analyses were conducted in this study, and though the study was not powered for efficacy, results show various positive effects of the treatment regime. Bacterial and fungal sputum load analysis results were highly variable, though marked reductions of Methicillin-sensitive Staphylococcus aureus (“MSSA”), Achromabacter, P. aeruginosa, and Aspergillus were seen in several subjects. These results suggest non-specific targeting of bacteria and fungi that commonly manifest in CF patients. In subjects with systemic inflammation (CRP >5 mg/mL) at baseline, CRP levels decreased over the treatment period, showing the effect of NO in the reduction of systemic inflammation. There were no statistically significant or clinically relevant changes in FEV1 over time, and lung function indices also remained relatively constant throughout the study duration.
In 2016, Rambam healthcare campus in Israel conducted a compassionate use treatment for two patients with CF who suffer from M, abscessus lung infections. The data were published in the Pediatric Infectious Disease Journal in 2017. The NO treatment regime, as well as the device for this treatment, was supplied by BA Ltd., our wholly owned subsidiary. Patients received intermittent 30-minute treatments of 160 ppm NO, with two different regimes including hospitalization (5 times a day) and ambulatory treatment (2-3 inhalations a day). Treatment was well tolerated with no evidence of any serious side effects. We observed significant improvement in sputum production (up to 5-10 times more sputum), and subjective improvement in the well-being of both patients. Significant reduction in systemic inflammation was observed in the first patient, as observed by reduction of CRP (C-reactive protein, a systemic inflammation marker that rises in response to inflammation) levels during treatment. In addition, the first patient had a 2 log (100-fold) reduction in M. abscessus during treatment (an effect that was lost after the treatment regime changed to ambulatory). The second patient showed a significant increase in the 6-minute walk (“6MW”) test and the sputum culture became negative, which is consistent with eradication of M. abscessus. Further information is needed, but we believe these results suggest that the treatment of M. abscessus with high-concentration inhaled NO is effective.
In 2017, we treated one patient with CF who suffered from NTM infections (specifically, M. abscessus) under compassionate use in the United Sates at the National Heart, Lung and Blood Institute with our generator based NO delivery system. The patient saw improvements in 6MW, FEV1, most Quality-of-Life measures and had no SAEs. The bacteria was not eradicated. The patient requested to be treated again and this treatment commenced in February 2018. A total of 38 treatments were administered over 8 days, 29 of them at a concentration of 240 ppm, with no SAEs believed to be related to NO reported.
Additionally in 2017, we completed a single-arm, open-label Pilot trial in nine patients with M. abcessus lung disease, who were refractory to standard-of-care (“SOC”). The patients were treated with inhaled NO at a concentration of 160 ppm for 30 minutes, in addition to treatment with SOC. Our inhaled NO treatment was administered intermittently five times per day over a 14-day period, followed by a seven-day period with three treatments per day. The primary endpoint of safety, as measured by NO-related SAEs, over the 21-day treatment period was met with no SAEs reported. Secondary endpoints of a 6MW test FEV1, Quality of Life and M. abscessus load in sputum all trended positively. 6MW showed an increase of >40 meters at the end of treatment at day 21 versus baseline and an increase of >25 meters on day 81 (60 days after the cessation of therapy). The mean percentage change in FEV1 at day 21 and day 51 (30 days after the cessation of treatment) was > 3.5% with FEV1 returning to baseline at day 81 (60 days after the cessation of therapy). At day 81 (60 days after the cessation of therapy) bacterial load was 65% lower than baseline. 1 of 9 patients saw culture conversion. This study was published in the Journal of Cystic Fibrosis in 2019.
In 2018, an additional CF patient infected with M. abscessus was treated over a 4-week period with 76 of 84 treatments at 250 ppm NO in Israel at Soroka Medical Center. The patient saw improvements in 6MW, FEV1 and most Quality-of-Life measures. The bacteria were not eradicated. Importantly, there were no SAE’s reported and all treatments were completed without incident.
In December 2020 we began a 12-week, multi-center, open-label clinical trial in Australia intended to enroll approximately 20 adult patients with chronic refractory NTM lung disease. We received a grant of up to $2.17 million from the CFF to fund this study and advance the clinical development of inhaled NO to treat NTM pulmonary disease. The trial enrolled both CF and non-CF patients infected with MAC, M. abscessus or any strain of NTM. The study consisted of a run-in period followed by two treatment phases. The run-in period provided a baseline for the efficacy endpoints. The first treatment phase took place over a two-week period and begun in the hospital setting where patients were titrated from 150 ppm NO up to 250 ppm NO over several days. During this phase patients received NO for 40 minutes, four times per day while MetHb levels were monitored. Patients were also trained to use LungFit® GO and subsequently discharged to complete the remaining portion of the two-week treatment period at their home at the highest tolerated NO concentration. For the second treatment phase, a 10-week maintenance phase, the administration was twice daily. The study was evaluating safety, quality of life, physical function, and bacterial load among other parameters. The mean age of subjects was 62.1 years (range: 22 – 82 years) with the majority female (80%), a distribution consistent with real-world NTM disease. All 15 subjects were successfully titrated to 250 ppm NO in the hospital setting, and no patients required dose reductions during the subsequent at-home portion of the study. Patients were followed up for 12 weeks after the 12-week treatment period was completed.
|
|
We presented positive results at the 2022 CHEST annual meeting, held from October 16, 2022 through October 19, 2022, further supporting development of intermittent high dose NO for the treatment of NTM. The study demonstrated that high dose NO treatment was safe and well-tolerated in both the home and hospital settings. During the 10-week at-home treatment period of the study, a total of 2,492 inhalations were self-administered with overall high treatment compliance (>90%). There were no SAEs related to treatment discontinuations reported over the 12-week treatment or 12-week follow up periods. Key efficacy endpoints showed strong results with improvement seen in the majority of quality-of-life domains. Respiratory function and physical function were maintained during treatment and follow-up. Trends in the reduction of microbial load were observed and one subject achieved culture conversion with three consecutive negative sputum samples.
In addition to further supporting development of intermittent high dose NO for the treatment of NTM, we believe this study breaks new ground in the development of NO therapy by showing the potential for the Company’s at-home generator-based system to be used safely and consistently by this patient population in a real-world setting.
VCAP and BRO Clinical Development
In 2014, we completed a double blind, randomized Pilot study for infants with bronchiolitis (n=43) for which the data were published in the Pediatric Pulmonology Journal in 2017. The study was performed at Soroka University Medical Center in Israel. Forty-three infants between the ages of two to 12 months diagnosed with bronchiolitis were randomly assigned to either the treatment group or the control group. The treatment group comprised 21 subjects who received intermittent (30 minutes, five times a day) inhalation of 160 ppm NO formulation, in addition to supportive O2 treatment for up to five days. The control group, 22 subjects, received ongoing inhalation of the supportive O2 treatment. Primary endpoints included determination of the MetHb levels, adverse events associated with the inhaled NO formulation and proportion of subjects who prematurely discontinued the study. Baseline clinical score, indicating disease severity at screening, was similar between treatment groups (~8). Results were encouraging, with similar overall incidence of AEs between the treatment groups. Out of 43 patients, 39 (~90%) completed the study per protocol (“PP”), with similar percentages (90%) for both the control and the treatment groups, individually. Only one subject from the treatment group discontinued treatment due to an adverse event, namely – repeated MetHb levels above 5%. Adverse events were reported by 23 (53.5%) subjects overall, with ten (47.6%) subjects in the NO group reporting a total of 22 AEs, and 13 (59.1%) subjects in the control group reporting a total of 22 AEs. SAEs were reported by four (19.0%) subjects in the NO group and four (18.2%) in the standard treatment group. There were no treatment-related SAEs in the NO treatment group.
In the NO group, six (28.6%) subjects had any MetHb measurement >5% during the study treatment period, and three of these subjects had more than one MetHb >5%. The maximum MetHb level was 5.6% in one subject in the NO group. There was no cumulative effect of MetHb exposure during the study. The MetHb levels in this study were defined to <5% as a safety measure, though previous findings have shown that higher levels (6.4%) are non-toxic in children. Secondary and exploratory analyses were performed, and results show positive impact of the treatment regime. In a subgroup of subjects that stayed at the hospital at least 24 hours (LOS >24 hours), a statistically significant treatment benefit of NO versus standard treatment was demonstrated. Mean results for subjects with LOS > 24 hours show that LOS was shortened by approximately 34% in the NO group compared to the standard treatment group, with a one-day difference between the groups (PP, N=24). Time to normal oxygenation (SaO2 of 92%) was shortened by approximately 44% (27.75 hours) in the NO group compared to the standard treatment group (PP, N=24). An 80% improvement in time to clinical score (indicating improvement in disease severity) and time to normal oxygenation (92%) was observed in favor of the NO group (PP, N=24).
|
|
In 2018 we completed a second pilot study in bronchiolitis in 6 centers in Israel. The data were published in Nature in 2020. The prospective, randomized, double blind, controlled pilot study enrolled 67 patients, aged 0-12 months, who were hospitalized due to bronchiolitis. The patients received either SOC (typically oxygen and hydration) or SOC plus inhaled NO at a concentration of 160 ppm for 30 minutes 5 times per day for up to 5 days. The primary endpoint of hospital LOS was met with a 26.7-hour reduction in hospital length of stay demonstrated (p=0.04). Secondary endpoints of time required to achieve a clinical score of 5 or less on the modified Tal score and time required to achieve SaO2of 92% or greater showed improvement versus the SOC. There were no issues with NO2 or MetHb and no SAEs were recorded.
In 2020 we completed a third pilot study in bronchiolitis in 8 centers in Israel and presented the data at 2020 CHEST annual meeting. The prospective, randomized, double blind, controlled pilot study enrolled 89 patients (ITT n=87), aged 0-12 months, who were hospitalized due to bronchiolitis. The patients were randomized 1:1:1 to receive either SOC (typically oxygen and hydration) or SOC plus inhaled NO at 85 ppm or SOC plus inhaled NO at 150 ppm for 40 minutes 4 times per day for up to 5 days. There were no SAEs related to NO therapy. Efficacy results are shown in the table below.
|
150 ppm vs. 85 ppm Hazard Ratio (p-value) |
150 ppm vs. SST Hazard Ratio (p-value) |
|||
| Fit for Discharge | 2.11 (0.041) | 2.32 (0.049) | ||
| Hospital Length of Stay (LOS) | 2.01 (0.046) | 2.28 (0.043) | ||
| Oxygen Saturation of > 92% | 2.15 (0.056) | 2.62 (0.039) |
We plan to seek certification or regulatory approval for our remaining current product candidates and, if approved, we expect they will be marketed as medical devices.
If we reach the commercialization stage for any of our remaining product candidates, we expect that we will collaborate with companies outside the U.S. for all indications. We are still determining whether to attempt to collaborate for any indication in the U.S. We are commercializing LungFit® PH in the U.S. ourselves and expect to partner with third parties to commercialize LungFit® PH outside the U.S.
Our PreClinical Results to Date for LungFit®
We have completed 4 separate toxicology studies in animals.
| ● | Rats: 30 days of intermittent treatments with LungFit® at 400 ppm NO showed no observations (differences) between control rats and treated rats on observation during the treatment period prior to sacrifice and no observations on histopathology | |
| ● | Rats: 12 weeks of intermittent treatments with LungFit® at 250 ppm NO showed no observations (differences) between control rats and treated rats on observation during the treatment period prior to sacrifice and no observations on histopathology | |
| ● | Dogs: 12 weeks of intermittent treatments with LungFit® at 250 ppm NO showed no observations (differences) between control dogs and treated dogs on observation during the treatment period prior to sacrifice and no observations on histopathology | |
| ● | Rats: Geno toxicology study of intermittent with LungFit® NO at 200 – 400 ppm showed a non-genotoxic response at all concentrations |
|
|
Additional Programs
Beyond Cancer’s research data has shown that UNO has anticancer properties and elicits an immune response from the host. Beyond Cancer utilizes an intratumoral UNO technology as a gas delivery of NO at high concentrations to tumors to induce an immune response. Gas based intratumoral therapies for the treatment of cancer are considered novel and new medical science. Beyond Cancer’s efforts are currently also focused on utilizing UNO in combination with Keytruda, a PD-1 inhibitor or other PD-1 or PDL-1 inhibitors as a treatment for cancers. To date, no such gas-based therapy has been approved for commercialization by the FDA or other regulatory agencies.
We have entered into an agreement with Yissum Research Development Company of the Hebrew University of Jerusalem, LTD. to acquire the commercial rights for neuronal nitric oxide synthase (nNOS) inhibitors being developed for the treatment of autism spectrum disorder (“ASD”) and other neurological conditions. Currently, there are currently no FDA approved therapies utilizing nNOS inhibitors specifically for the treatment of ASD.
Competition
The biotechnology, pharmaceutical and medical device industries are highly competitive. There are many pharmaceutical companies, biotechnology companies, medical device companies, public and private universities and research organizations actively engaged in the research and development (“R&D”) of products that may be similar to our approved product or product candidates. We are aware of several companies currently developing and selling NO therapies for various indications such as hypoxic respiratory failure (HRF). For example, Mallinckrodt commercializes INOMAX® (nitric oxide) for inhalation, which is approved for use to treat newborns suffering from hypoxic respiratory failure (“HRF”)-PPHN, in the U.S., Canada, Australia, Mexico and Japan. Linde Group markets a generic version of the Mallinckrodt offering with their delivery system called NOxBOX®. The Linde Group has marketing rights to INOMAX® in Europe. Air Liquide sells a similar product in Europe, called KINOX™, together with their delivery platform called SoKINOX™, for the treatment of pulmonary hypertension of the newborn. In Europe, EKU, International Biomedical, Cahouet and ITC each have a device that delivers nitric oxide. Bellerophon Therapeutics is developing an NO delivery system for patients with chronic pulmonary diseases such as COPD, PH-sarcoidosis, or fibrotic interstitial lung disease. VERO Biotech LLC (formerly known as Geno LLC) received FDA approval for their delivery system GENOSYL DS for HRF associated with PPHN in 2019, and received FDA approval for a third generation of that delivery system in 2023. In addition, other companies may be developing inhaled NO delivery systems at various concentrations. Novan Inc. has recently received approval for a nitric oxide-based prescription treatment called berdazimer for molluscum, a contagious skin infection. SaNOtize has an NO nasal spray that has received approval in India, Israel and eight other countries for preventing COVID-19 after exposure. Third Pole has reported the development of an NO generator and delivery system, but we are not aware of any display of any product at any medical/scientific conference in recent years. Our patents surrounding LungFit® have priority date over those of Third Pole.
Some of our competitors, either alone or through their strategic partners, might have substantially greater name recognition and financial, technical, manufacturing, marketing and human resources than we do and greater experience and infrastructure in the research and clinical development of pharmaceutical products, obtaining FDA and other regulatory approvals of those products and commercializing those products around the world.
As it relates to Beyond Cancer’s programs, no such gas-based therapies have been approved for commercialization by the FDA or other regulatory agencies to date. Similarly, there are currently no FDA approved therapies utilizing nNOS inhibitors specifically for the treatment of ASD.
Manufacturing and Distribution
We have contracted with third-party contract manufacturers, Spartronics LLC (“Spartronics”), and Medisize Ireland Limited (“Medisize”) who have completed a substantial portion of the commercial manufacturing process for our LungFit® PH system. In addition, we will be reliant on our partners for commercial manufacture of our systems for both clinical studies and commercial supply. In the twelve months ended March 31, 2023, the Company purchased approximately 80% of our materials from these two third-party vendors, with these vendors representing 67% and 13%, respectively.
We have a central warehouse in Atlanta, GA for the staging of products to be deployed to forward-stocking locations (“FSL”) in major cities close to our potential customers. We contract with third party logistics providers to manage both the central warehouse and the FSLs. FSLs allow us to supply LungFit® devices and consumables in a timely manner to customers.
We market directly to our customers through our field sales team. A separate team of clinical specialists provides the training necessary to use the devices. Invoicing and cash collection are managed from our headquarters location.
Intellectual Property
We own or have exclusively licensed patents, pending patent applications, know-how and trade secrets that relate to our NO generator, NO2 filtration, delivery systems, devices configured for delivering NO to patients by inhalation, methods of exposing patients to inhalation of NO, and methods for treating subjects in need of NO inhalation.
We are party to a global, exclusive, transferable license agreement with NitricGen for the eNOGenerator, its components, and all associated patents and know how related thereto. Additionally, we have a broad intellectual property portfolio directed to our product candidates and mode of delivery, monitoring parameters and methods of treating specific disease indications. Our intellectual property portfolio consists of issued patents and pending applications, which includes patents we acquired pursuant to the exercise of an option in 2017 granted to us by Pulmonox Technologies Corporation (“Pulmonox”).
|
|
On August 31, 2015, we entered into an agreement with Pulmonox (the “Option Agreement”) whereby we acquired the option to purchase certain intellectual property assets, including Pulmonox’s rights in 17 issued U.S. patents, including eight patents jointly owned with CareFusion which are directed to:
| ● | devices and methods for delivering NO formulations to a patient at steady and alternating concentrations (80-400 ppm), including intermittent delivery of NO; | |
| ● | a device and methods for treatment of surface infections; and | |
| ● | use of NO as a mucolytic agent and for treatment and disinfection of biofilms. |
We exercised the Option in January 2017, acquiring Pulmonox’s rights in the patents described above. Upon exercise of the Option, we became obligated to make certain one-time development and sales milestone payments to Pulmonox, commencing with the date on which we receive regulatory approval for the commercial sale of the first product candidate qualifying under the agreement. These milestone payments are almost entirely sales-related and are capped at a total of $87 million across three separate and distinct indications that fall under the agreement with the majority of them, approximately $83 million, being sales-related based on cumulative sales milestones for each of the three products. In addition, the Company issued a fully vested warrant to purchase up to 178,570 shares of our common stock at an exercise price of $4.80 per share for each share of common stock. On May 10, 2018, we issued to Pulmonox, an additional fully vested warrant to purchase up to 29,763 shares of our common stock at an exercise price of $4.80 per share.
Patent Applications. We have over 50 pending patent applications worldwide, including U.S., foreign and Patent Corporation Treaty (“PCT”) patent applications.
A PCT patent application is a filing under the Patent Cooperation Treaty to which the U.S. and a number of other countries are a party. It provides a unified procedure for filing a single patent application to protect inventions in those countries. A search with respect to the application is conducted by the International Searching Authority, accompanied by a written opinion regarding the patentability of the invention. A PCT application does not itself result in the grant of a patent, and the grant of patent is a prerogative of each national or regional authority where the PCT application is filed during national phase filings.
Settlement Agreement
On January 23, 2019, we entered into an agreement for commercial rights (the “Circassia Agreement”) with Circassia Limited and its affiliates (collectively, “Circassia”) for PPHN and future related indications at concentrations of < 80 ppm in the hospital setting in the United States and China. On December 18, 2019, we terminated the Circassia Agreement. Circassia contended that the termination was wrongful.
On May 25, 2021, we and Circassia Limited entered into a Settlement Agreement resolving all claims by and between both parties and mutually terminating the Circassia agreement disclosed in Note 10 of the consolidated financial statements included in this Annual Report. Pursuant to the terms of the Settlement Agreement, we agreed to pay Circassia $10.5 million in three installments, the first being a payment of $2.5 million to Circassia within fifteen (15) days following FDA approval of the LungFit® PH (the “Initial Payment Due Date”). Thereafter, we shall pay $3.5 million to Circassia on the first anniversary of the Initial Payment Due Date and $4.5 million on the second anniversary of the Initial Payment Due Date. Additionally, beginning in year three post-approval, Circassia will receive a quarterly royalty payment equal to 5% of LungFit® PH net sales in the U.S. This royalty will terminate once the aggregate payment reaches $6 million. $10.5 million was accrued as a liability as of March 31, 2022 when management deemed the approval of LungFit® PH by the FDA was probable. FDA approval was received on June 28, 2022. $2.5 million was paid to Circassia Limited in July 2022 and $8.0 million remains as an accrued liability as of March 31, 2023. $3.5 million will be paid in July 2023 and the final $4.5 million will be paid in July 2024.
|
|
Government Regulation
U.S. Regulation. In the U.S., the FDA regulates drug and medical device products under the Federal Food, Drug, and Cosmetic Act (“FD&C Act”), and its implementing regulations. Our products have been designated as devices by the FDA and will be regulated by the Center for Devices and Radiological Health (CDRH). Given that currently approved NO products and delivery systems were approved either separately (NO drug approval and NO delivery systems cleared as devices) or as drug-device combinations in the United States, we expect our device to not only be reviewed by CDRH, but also have input from the Center for Drug Evaluation and Research (“CDER”).
FDA Premarket Clearance and Approval Requirements for Medical Devices. Unless an exemption applies, each medical device commercially distributed in the United States requires either FDA clearance of a 510(k) premarket notification, authorization of a de novo application, or approval of a PMA application. Under the FD&C Act, medical devices are classified into one of three classes—Class I, Class II or Class III—depending on the degree of risk associated with each medical device and the extent of manufacturer and regulatory control needed to ensure its safety and effectiveness. Class I includes devices with the lowest risk to the patient and are those for which safety and effectiveness can be assured by adherence to the FDA’s General Controls for medical devices, which include compliance with the applicable portions of the Quality System Regulation (“QSR”) facility registration and product listing, reporting of adverse medical events, and truthful and non-misleading labeling, advertising, and promotional materials. Class II devices are subject to the FDA’s General Controls, and special controls as deemed necessary by the FDA to ensure the safety and effectiveness of the device. These special controls can include performance standards, post-market surveillance, patient registries and FDA guidance documents.
While most Class I devices are exempt from the 510(k) premarket notification requirement, manufacturers of most Class II devices are required to submit to the FDA a premarket notification under Section 510(k) of the FFDCA requesting permission to commercially distribute the device. The FDA’s permission to commercially distribute a device subject to a 510(k) premarket notification is generally known as 510(k) clearance. Devices deemed by the FDA to pose the greatest risks, such as life sustaining, life supporting or some implantable devices, or devices that have a new intended use, or use advanced technology that is not substantially equivalent to that of a legally marketed device, are placed in Class III, requiring approval of a PMA. Some pre-amendment devices are unclassified, but are subject to FDA’s premarket notification and clearance process in order to be commercially distributed.
510(k) Clearance Marketing Pathway. To obtain 510(k) clearance, a company must submit to the FDA a premarket notification submission demonstrating that the proposed device is “substantially equivalent” to a predicate device already on the market. A predicate device is a legally marketed device that is not subject to PMA, i.e., a device that was legally marketed prior to May 28, 1976 (pre-amendments device) and for which a PMA is not required, a device that has been reclassified from Class III to Class II or I, or a device that was found substantially equivalent through the 510(k) process. The FDA’s 510(k) clearance process usually takes from three to twelve months, but often takes longer. The FDA may require additional information, including clinical data, to make a determination regarding substantial equivalence. In addition, the FDA collects user fees for certain medical device submissions and annual fees for medical device establishments.
If the FDA agrees that the device is substantially equivalent to a predicate device currently on the market, it will grant 510(k) clearance to commercially market the device. If the FDA determines that the device is “not substantially equivalent” to a previously cleared device, the device is automatically designated as a Class III device. The device sponsor must then fulfill more rigorous PMA requirements, or can request a risk-based classification determination for the device in accordance with the “de novo” process, which is a route to market for novel medical devices that are low to moderate risk and are not substantially equivalent to a predicate device.
After a device receives 510(k) marketing clearance, any modification that could significantly affect its safety or effectiveness, or that would constitute a major change or modification in its intended use, will require a new 510(k) clearance or, depending on the modification, PMA approval. The FDA requires each manufacturer to determine whether the proposed change requires submission of a 510(k) or a PMA in the first instance, but the FDA can review any such decision and disagree with a manufacturer’s determination. If the FDA disagrees with a manufacturer’s determination, the FDA can require the manufacturer to cease marketing and/or request the recall of the modified device until 510(k) marketing clearance, authorization of a de novo application, or PMA approval is obtained. Also, in these circumstances, the manufacturer may be subject to significant regulatory fines or penalties.
|
|
PMA Approval Pathway. Class III devices require approval of a PMA before they can be marketed, although some pre-amendment Class III devices for which the FDA has not yet required a PMA are cleared through the 510(k) process. The PMA process is more demanding than the 510(k) premarket notification process. In a PMA application, the manufacturer must demonstrate that the device is safe and effective, and the PMA application must be supported by extensive data, including data from preclinical studies and human clinical trials. The PMA application must also contain a full description of the device and its components, a full description of the methods, facilities, and controls used for manufacturing, and proposed labeling. Following receipt of a PMA application, the FDA determines whether the application is sufficiently complete to permit a substantive review. If the FDA accepts the application for review, it has 180 days under the FFDCA to complete its review of a PMA application, although in practice, the FDA’s review often takes significantly longer, and can take up to several years. An advisory panel of experts from outside the FDA may be convened to review and evaluate the application and provide recommendations to the FDA as to the approvability of the device. The FDA may or may not accept the panel’s recommendation. In addition, the FDA will generally conduct a pre-approval inspection of the applicant or its third-party manufacturers’ or suppliers’ manufacturing facility or facilities to ensure compliance with the QSR. PMA devices are also subject to the payment of user fees.
The FDA will approve the new device for commercial distribution if it determines that the data and information in the PMA application constitute valid scientific evidence and that there is reasonable assurance that the device is safe and effective for its intended use(s). A PMA may include post-approval conditions intended to ensure the safety and effectiveness of the device, including, among other things, restrictions on labeling, promotion, sale and distribution, and collection of long-term follow-up data from patients in the clinical study that supported the PMA or requirements to conduct additional clinical studies post-approval. The FDA may condition PMA approval on some form of post-market surveillance when deemed necessary to protect the public health or to provide additional safety and efficacy data for the device in a larger population or for a longer period of use. In such cases, the manufacturer might be required to follow certain patient groups for a number of years and to make periodic reports to the FDA on the clinical status of those patients. Failure to comply with the conditions of approval can result in material adverse enforcement action, including withdrawal of the approval.
Certain changes to an approved device, such as changes in manufacturing facilities, methods, or quality control procedures, or changes in the design performance specifications, which affect the safety or effectiveness of the device, require submission of a PMA supplement. PMA supplements often require submission of the same type of information as a PMA, except that the supplement is limited to information needed to support any changes from the device covered by the original PMA and may not require as extensive clinical data or the convening of an advisory panel. Certain other changes to an approved device require the submission of a new PMA, such as when the design change causes a different intended use, mode of operation, and technical basis of operation, or when the design change is so significant that a new generation of the device will be developed, and the data that were submitted with the original PMA are not applicable for the change in demonstrating a reasonable assurance of safety and effectiveness. None of our products are currently marketed pursuant to a PMA.
On November 10, 2020 we submitted a PMA application to the FDA for the use of LungFit® PH in PPHN and subsequently received PMA approval in June 2022.
De-Novo Pathway. Another pathway, known as de-novo down-classification also can be used for lower risk devices for which there is no existing product code or predicate device. The Food and Drug Administration Modernization Act of 1997 established the de-novo down-classification procedure as a new route to market for low to moderate risk medical devices that automatically require a PMA due to the absence of a predicate device. This procedure allows a manufacturer whose novel device automatically requires a PMA to request down-classification of its medical device (to allow clearance through the 510(k) pathway) on the basis that the device presents low or moderate risk, rather than requiring the submission and approval of a PMA application. Manufacturers can request de-novo down-classification directly without first submitting a 510(k) premarket notification to the FDA and receiving a “not substantially equivalent” determination. Under this pathway, the FDA is required to classify the device within 120 days following receipt of the de-novo application. If the manufacturer seeks reclassification into Class II, the manufacturer must include a draft proposal for special controls that are necessary to provide a reasonable assurance of the safety and effectiveness of the medical device. In addition, the FDA may reject the reclassification petition if it identifies a legally marketed predicate device that would be appropriate for a 510(k) or determines that the device is not low to moderate risk or that general controls would be inadequate to control the risks and special controls cannot be developed.
|
|
Clinical Trials. Clinical trials are almost always required to support a PMA and are sometimes required to support a 510(k) submission. All clinical investigations of devices to determine safety and effectiveness must be conducted in accordance with the FDA’s Investigational Device Exemption (“IDE”) regulations which govern investigational device labeling, prohibit promotion of the investigational device, and specify an array of recordkeeping, reporting and monitoring responsibilities of study sponsors and study investigators. If the device presents a “significant risk,” to human health, as defined by the FDA, the FDA requires the device sponsor to submit an IDE application to the FDA, which must become effective prior to commencing human clinical trials. A significant risk device is one that presents a potential for serious risk to the health, safety or welfare of a patient and either is implanted, used in supporting or sustaining human life, substantially important in diagnosing, curing, mitigating or treating disease or otherwise preventing impairment of human health, or otherwise presents a potential for serious risk to a subject. An IDE application must be supported by appropriate data, such as animal and laboratory test results, showing that it is safe to test the device in humans and that the testing protocol is scientifically sound. The IDE will automatically become effective 30 days after receipt by the FDA unless the FDA notifies the company that the investigation may not begin. If the FDA determines that there are deficiencies or other concerns with an IDE for which it requires modification, the FDA may permit a clinical trial to proceed under a conditional approval.
In addition, the study must be approved by, and conducted under the oversight of, an Institutional Review Board (“IRB”) for each clinical site. The IRB is responsible for the initial and continuing review of the IDE study, and may pose additional requirements for the conduct of the study. If an IDE application is approved by the FDA and one or more IRBs, human clinical trials may begin at a specific number of investigational sites with a specific number of patients, as approved by the FDA. If the device presents a non-significant risk to the patient, a sponsor may begin the clinical trial after obtaining approval for the trial by one or more IRBs without separate approval from the FDA, but must still follow abbreviated IDE requirements, such as monitoring the investigation, ensuring that the investigators obtain informed consent, and labeling and record-keeping requirements. Acceptance of an IDE application for review does not guarantee that the FDA will allow the IDE to become effective and, if it does become effective, the FDA may or may not determine that the data derived from the trials support the safety and effectiveness of the device or warrant the continuation of clinical trials. An IDE supplement must be submitted to, and approved by, the FDA before a sponsor or investigator may make a change to the investigational plan that may affect its scientific soundness, study plan or the rights, safety or welfare of human subjects.
During a study, the sponsor is required to comply with the applicable FDA requirements, including, for example, trial monitoring, selecting clinical investigators and providing them with the investigational plan, ensuring IRB review, adverse event reporting, record keeping and prohibitions on the promotion of investigational devices or on making safety or effectiveness claims for them. The clinical investigators in the clinical study are also subject to FDA regulations and must obtain patient informed consent, rigorously follow the investigational plan and study protocol, control the disposition of the investigational device, and comply with all reporting and recordkeeping requirements. Additionally, after a trial begins, we, the FDA or the IRB could suspend or terminate a clinical trial at any time for various reasons, including a belief that the risks to study subjects outweigh the anticipated benefits.
Post-market Regulation. After a device is cleared or approved for marketing, numerous and pervasive regulatory requirements continue to apply. These include:
| ● | establishment registration and device listing with the FDA; | |
| ● | QSR requirements, which require manufacturers, including third-party manufacturers, to follow stringent design, testing, control, documentation and other quality assurance procedures during all aspects of the design and manufacturing process; | |
| ● | labeling regulations and FDA prohibitions against the promotion of investigational products, or the promotion of off-label (as defined below) uses of cleared or approved products; | |
| ● | requirements related to promotional activities; |
|
|
| ● | clearance or approval of product modifications to 510(k)-cleared devices that could significantly affect safety or effectiveness or that would constitute a major change in intended use of one of our cleared devices, or approval of certain modifications to PMA-approved devices; | |
| ● | medical device reporting regulations which require that a manufacturer report to the FDA if a device it markets may have caused or contributed to death or serious injury, or has malfunctioned and the device or a similar device that it markets would be likely to cause or contribute to a death or serious injury, if the malfunction were to recur. | |
| ● | correction, removal and recall reporting regulations, which require that manufacturers report to the FDA field corrections and product recalls or removals if undertaken to reduce a risk to health posed by the device or to remedy a violation of the FFDCA that may present a risk to health; | |
| ● | the FDA’s recall authority, whereby the agency can order device manufacturers to recall from the market a product that is in violation of governing laws and regulations; and | |
| ● | post-market surveillance activities and regulations, which apply when deemed by the FDA to be necessary to protect the public health or to provide additional safety and effectiveness data for the device. |
The manufacturing processes for medical devices are required to comply with the applicable portions of the QSR, which cover the methods and the facilities and controls for the design, manufacture, testing, production, processes, controls, quality assurance, labeling, packaging, distribution, installation and servicing of finished devices intended for human use. The QSR also requires, among other things, maintenance of a device master file, device history file, and complaint files. These requirements impose certain procedural and documentation requirements upon us and our third-party manufacturers related to the methods used in and the facilities and controls used for designing, manufacturing, packaging, labeling, storing, medical devices. As a manufacturer, we are subject to periodic scheduled or unscheduled inspections by the FDA. Following these inspections, the FDA may assert noncompliance with QSR requirements on a Form 483, which is a report of observations from an inspection, or by way of “untitled letters” or “warning letters” that could cause us or any third-party manufacturers to modify certain activities. A Form 483 notice, if issued at the conclusion of an FDA inspection, can list conditions the FDA investigators believe may have violated QSR or other FDA requirements. We cannot be certain that we or our present or any future third-party manufacturers or suppliers will be able to comply with QSR or other FDA regulatory requirements to the agency’s satisfaction. Failure to comply with these obligations may lead to possible legal or regulatory enforcement action by the FDA.
The FDA has broad regulatory compliance and enforcement powers. If the FDA determines that we failed to comply with applicable regulatory requirements, it can take a variety of compliance or enforcement actions, which may result in any of the following sanctions:
| ● | untitled letters, warning letters, fines, injunctions, consent decrees and civil penalties; | |
| ● | unanticipated expenditures to address or defend such actions; | |
| ● | customer notifications or repair, replacement, refunds, recall, detention or seizure of our products; | |
| ● | operating restrictions, partial suspension or total shutdown of production; | |
| ● | refusing or delaying our requests for regulatory approvals or clearances of new products or modified products; | |
| ● | withdrawing a PMA that has already been granted; | |
| ● | refusal to grant export approval for our products; or | |
| ● | criminal prosecution. |
Advertising and Promotion. The FDA and comparable foreign regulatory authorities closely regulate the post-approval marketing and promotion of medical devices, including standards and regulations for direct-to-consumer advertising, communications about unapproved uses, industry- sponsored scientific and educational activities and promotional activities involving the internet. Devices may be marketed only for the approved or cleared indications and in accordance with the provisions of the approved or cleared label.
Healthcare providers are permitted to prescribe approved devices for “off-label” uses—that is, uses not approved by the FDA and therefore not described in the product’s labeling. These off-label uses are common across medical specialties. Physicians may believe that such off-label uses are the best treatment for many patients in varied circumstances. The FDA does not regulate the behavior of physicians in their choice of treatments. The FDA does, however, impose stringent restrictions on manufacturers’ communications regarding off-label use. Thus, we may market our products, if approved by the FDA, only for their approved indications, but under certain conditions may engage in non-promotional, balanced communication regarding off-label uses. Failure to comply with applicable FDA requirements and restrictions in this area may subject us to adverse publicity and a variety of sanctions, which could harm our business and financial condition.
|
|
Combination Products. A combination product is the combination of two or more regulated components, i.e., drug/device, biologic/device, drug/biologic, or drug/device/biologic, that are combined or mixed and produced as a single entity; packaged together in a single package or as a unit; or a drug, device, or biological product packaged separately that according to its investigational plan or proposed labeling is intended for use only with an approved individually specified drug, device, or biological product where both are required to achieve the intended use, indication, or effect.
To determine which FDA center or centers will review a combination product candidate submission, companies may submit a request for assignment to the FDA. Those requests may be handled formally or informally. In some cases, jurisdiction may be determined informally based on FDA experience with similar products. However, informal jurisdictional determinations are not binding on the FDA. Companies also may submit a formal Request for Designation to the FDA Office of Combination Products. The Office of Combination Products will review the request and make its jurisdictional determination within 60 days of receiving a Request for Designation.
FDA will determine which center or centers within the FDA will review the product candidate and under what legal authority the product candidate will be reviewed. Depending on how the FDA views the product candidates that are developed, the FDA may have aspects of the product candidate reviewed by the Center for Biologics Evaluation and Research (“CBER”), CDRH, or CDER, though one center will be designated as the center with primary jurisdiction, based on the product candidate’s primary mode of action. The FDA determines the primary mode of action based on the single mode of action that provides the most important therapeutic action of the combination product candidate – the mode of action expected to make the greatest contribution to the overall intended therapeutic effects of the combination product candidate. The review of such combination product candidates is often complex and time-consuming, as the FDA may select the combination product candidate to be reviewed and regulated by one or multiple of the FDA centers identified above, which could affect the path to regulatory clearance or approval. Furthermore, the FDA may also require submission of separate applications to multiple centers.
The post-market requirements that apply to the cleared or approved product will largely be aligned with the agency center determined to have primary jurisdiction over the product candidate and that provided marketing authorization, but manufacturers must also comply with certain post-market requirements with respect to the constituent parts of combination products. In April 2019, FDA published a final guidance document entitled Compliance Policy for Combination Product Post-Marketing Safety Reporting, which is intended to assist manufacturers of combination products comply with reporting requirements applicable to such products.
After issuing marketing authorizations, the FDA has discretion in determining post-approval compliance requirements for combination products. The FDA has also promulgated regulations pertaining to compliance with certain current good manufacturing practices (“cGMP”) requirements for drug components as well as QSR requirements for device constituents of a combination product. Other post-market requirements in the same vein as those described above for medical devices and drugs will also apply, depending on the application type and center overseeing regulation of the combination product, including:
| ● | Other record-keeping requirements; | |
| ● | Post-market adverse event and Medical Device Reporting requirements; | |
| ● | Labeling regulations and FDA prohibitions against the promotion of products for uncleared, unapproved or off-label uses; | |
| ● | Advertising and promotion requirements; | |
| ● | Restrictions on sale, distribution or use of the product; | |
| ● | Requirements for recalls being conducted and recall reporting; | |
| ● | An order of repair, replacement or refund; | |
| ● | Product tracking requirements; and | |
| ● | Post-market surveillance or clinical trials. |
|
|
Coverage and Reimbursement. Coverage and reimbursement for medical devices in the U.S. is determined by third-party payors, including Medicare and Medicaid, commercial health insurers, and managed care organizations. Each payor has a unique process for determining whether to cover a device for a particular indication and how to set reimbursement rates for the device. A payor can decide to cover a device yet not provide adequate reimbursement to ensure access to the device. New devices often face significant uncertainty about coverage and reimbursement. Payors may require additional evidence, beyond the data required for FDA approval, to demonstrate that a device should be covered for a particular indication or that it should be reimbursed at a higher rate than other technologies. In addition, health care spending continues to be a concern for federal and state governments, as well as for commercial payors. Governments continue to debate methods of controlling health care costs, including reductions in reimbursement or additional controls on utilization of new technologies in Medicare and Medicaid, and commercial payors may similarly seek to limit spending on new devices. Restrictions on coverage and reimbursement could harm our future revenues and ability to realize an appropriate return on our investment.
Orphan Drug Designation and Exclusivity. Under the Orphan Drug Act, the FDA may grant orphan drug designation to products that are intended to treat rare diseases or conditions (i.e., those affecting fewer than 200,000 individuals in the U.S.), or diseases or conditions that affect more than 200,000 individuals in the U.S. but there is no reasonable expectation that the cost of developing and making the drug product would be recovered from sales in the U.S. Although orphan drug designation does not convey any advantage in the regulatory review and approval process, it can provide certain tax benefits and access to certain grants. Additionally, FDA user fees, which can be substantial, are waived for products that obtain orphan drug designation. Further, if a product with orphan drug designation subsequently receives FDA approval for the designated disease or condition, the product is generally granted seven years of orphan drug exclusivity, which (with certain limited exceptions) blocks for seven years FDA approval of another product with the same active ingredient for the same indication. Orphan drug exclusivity does not prevent the FDA from approving a different drug for the same disease or condition, or the same drug for a different disease or condition.
Healthcare Fraud and Abuse Laws. In addition to the FDA’s ongoing post-approval regulation of devices discussed above, manufacturers are also subject to several other types of laws and regulations, subject to differing enforcement regimes. In recent years, marketing and promotional activities regarding FDA-regulated products have come under intense scrutiny and have been the subject of enforcement action brought by the U.S. Department of Justice and the Office of Inspector General of the Department of Health and Human Services, as well as state authorities and even private individuals.
Healthcare providers, physicians and third-party payors play a primary role in the recommendation and selection of medical devices for patients. Arrangements with third-party payors and customers are subject to broadly applicable fraud and abuse and other healthcare laws and regulations. Such restrictions under applicable federal and state healthcare laws and regulations include the following:
| ● | The federal health care program Anti-Kickback Statute (“AKS”) prohibits, among other things, persons from knowingly and willfully soliciting, offering, receiving or providing remuneration, directly or indirectly, in cash or in kind, to induce or in return for purchasing, leasing, ordering or arranging for the purchase, lease or order of any good or service, for which payment may be made, in whole or in part, under a federal healthcare program such as Medicare and Medicaid. This statute has been interpreted to apply to arrangements between pharmaceutical or device manufacturers, on the one hand, and prescribers, purchasers and formulary managers and others on the other. The term “remuneration” has been broadly interpreted to apply to anything of value including, for example, gifts, cash payments, donations, waivers of payment, ownership interests, and providing any item, service, or compensation for something other than fair market value. Liability under the AKS may be established without proving actual knowledge of the statute or specific intent to violate it. Although there are a number of statutory exceptions and regulatory safe harbors to the AKS protecting certain common business arrangements and activities from prosecution or regulatory sanctions, the exceptions and safe harbors are drawn narrowly. Practices that involve remuneration to those who prescribe, purchase, or recommend medical device products, including certain discounts, or engaging such individuals as consultants, advisors and speakers, may be subject to scrutiny if they do not fit squarely within an exception or safe harbor. Moreover, there are no safe harbors for many common practices, such as educational grants and reimbursement support programs. Violations are punishable by up to 10 years in prison, criminal fines, administrative civil monetary penalties and exclusion from participation in federal healthcare programs. Any sales or marketing practices that involve remuneration intended to induce prescribing, purchases or recommendations may be subject to scrutiny under the AKS; |
|
|
| ● | the federal civil False Claims Act (“FCA”) imposes liability on individuals or entities for, among other things, knowingly presenting, or causing to be presented, false or fraudulent, claims for payment of government funds, knowingly making, using, or causing to be made or used a false statement or record material to an obligation to pay money to the government, or knowingly concealing or knowingly and improperly avoiding, decreasing or concealing an obligation to pay money to the federal government. A claim including items or services resulting from a violation of the AKS constitutes a false or fraudulent claim for purposes of the FCA. Actions under the FCA may be brought by the government or as a qui tam action by a private individual in the name of the government, who may also share in any monetary recovery. Qui tam actions are filed under seal and impose a mandatory duty on the U.S. Department of Justice to investigate such allegations. Manufacturers have faced liability under the FCA for providing inaccurate billing or coding information to customers or promoting a product off-label. FCA liability is potentially significant in the healthcare industry because the statute provides for treble damages and significant mandatory penalties per false or fraudulent claim or statement for violations, as well as exclusion from participation in federal healthcare programs; | |
| ● | the federal Health Insurance Portability and Accountability Act of 1996, as amended by the Health Information Technology for Economic and Clinical Health Act, and their respective implementing regulations (collectively, “HIPAA”), imposes criminal and civil liability for, among other things, knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program, including private third-party payors, or knowingly and willfully falsifying, concealing, or covering up a material fact or making any materially false, fictitious, or fraudulent statement or representation, or using any false writing or document knowing the same to contain any materially false, fictitious, or fraudulent statement or entry, in connection with the delivery of or payment for healthcare benefits, items, or services; | |
| ● | the federal Physician Payments Sunshine Act (the “Sunshine Act”) requires applicable manufacturers of devices, biologics and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program (with certain exceptions) to report annually to CMS information related to payments and other transfers of value to physicians, physician assistants, nurse practitioners, clinical nurse specialists, certified nurse anesthetists, and certified nurse midwives, and teaching hospitals, as well as ownership and investment interests held by physicians and their immediate family members. | |
| ● | analogous state and foreign laws and regulations, such as state anti-kickback and false claims laws, may apply to sales or marketing arrangements and claims involving healthcare items or services reimbursed by non-governmental third-party payors, including private insurers. Several states have enacted legislation requiring medical device manufacturers to, among other things, establish marketing compliance programs; file periodic reports with the state, including reports on gifts and payments to individual health care providers; and/or register their sales representatives. Some states prohibit certain sales and marketing practices, including the provision of gifts, meals, or other items to health care providers. |
Additionally, other laws such as the federal Lanham Act and similar state laws allow competitors and others to initiate litigation relating to advertising claims. If we sell our device outside the United States, it must comply with the Foreign Corrupt Practices Act (“FCPA”) and local laws of other countries. FCPA is a complex patchwork of laws can change rapidly with relatively short notice.
|
|
Environmental Laws. Elements of our potential products may be classified as hazardous materials, subject to regulation by the Department of Transportation, the International Air Transportation Association, the International Maritime Organization, the Environmental Protection Agency and the Occupational Safety and Health Administration, which may impose various requirements pertaining to the way we manufacture, transport, store, handle and dispose of our products.
European Regulation of Medical Devices. In the European Economic Area (“EEA”), we expect our products to be regulated as a medical device product falling within the scope of EU MDR.
In the EEA, medical devices must currently comply with the General Safety and Performance Requirements laid down in Annex I to the EU MDR. Compliance with these requirements is a prerequisite to be able to affix the CE mark on products, without which they cannot be marketed or sold in the EEA. To demonstrate compliance with the General Safety and Performance Requirements of the EU MDR and obtain the right to affix the CE mark, medical devices manufacturers must undergo a conformity assessment procedure, which varies according to the type of medical device and its classification. Apart from low risk medical devices (Class I with no measuring function and which are not sterile), in relation to which the manufacturer may issue an EC Declaration of Conformity based on a self-assessment of the conformity of its products with the General Safety and Performance Requirements, a conformity assessment procedure requires the intervention of a notified body, which is an organization designated by a Competent Authority of an EEA country to conduct conformity assessments. Depending on the relevant conformity assessment procedure, the notified body would audit and examine the technical documentation and the quality system for the manufacture, design and final inspection of the medical devices. The notified body issues a CE Certificate of Conformity following successful completion of a conformity assessment procedure conducted in relation to the medical device and its manufacturer and their conformity with the General Safety and Performance Requirements. This Certificate and the related conformity assessment process entitles the manufacturer to affix the CE mark to its medical devices after having prepared and signed a related EC Declaration of Conformity. Notified bodies must be accredited by the EEA countries’ accreditation bodies to conduct assessment procedures for medical devices in accordance with the EU MDR. There are currently a relatively small number of notified bodies that have been accredited to conduct these assessments. This may delay conformity assessment procedures in the future in the EU.
As a general rule, demonstration of conformity of medical devices and their manufacturers with the General Safety and Performance Requirements must be based, among other things, on the evaluation of clinical data supporting the safety and performance of the products during normal conditions of use. Specifically, a manufacturer must demonstrate that the device achieves its intended performance during normal conditions of use and that the known and foreseeable risks, and any adverse events, are minimized and acceptable when weighed against the benefits of its intended performance, and that any claims made about the performance and safety of the device (e.g., product labeling and instructions for use) are supported by suitable evidence. This assessment must be based on clinical data, which can be obtained from (1) clinical studies conducted on the devices being assessed, (2) scientific literature from similar devices whose equivalence with the assessed device can be demonstrated or (3) both clinical studies and scientific literature. The conduct of clinical studies in the EEA is governed by detailed regulatory obligations. These may include the requirement of prior authorization by the competent authorities of the country in which the study takes place and the requirement to obtain a positive opinion from a competent Ethics Committee. This process can be expensive and time-consuming.
The EU MDR repeals and replaces the EU Medical Devices Directive 93/42/EEC. Unlike directives, which must be implemented into the national laws of the EEA countries, the regulations is directly applicable, i.e., without the need for adoption of EEA country laws implementing them, in all countries and are intended to eliminate current differences in the regulation of medical devices among EEA countries. The EU MDR, among other things, establishes a uniform, transparent, predictable and sustainable regulatory framework across the EEA for medical devices and ensures a high level of safety and health while supporting innovation. The EU MDR entered into application on May 26, 2021, and among others things:
| ● | strengthens the rules on placing devices on the market and reinforce surveillance once they are available; | |
| ● | establishes explicit provisions on manufacturers’ responsibilities for the follow-up of the quality, performance and safety of devices placed on the market; | |
| ● | improves the traceability of medical devices throughout the supply chain to the end-user or patient through a unique identification number; | |
| ● | sets up a central database to provide patients, healthcare professionals and the public with comprehensive information on products available in the EU; | |
| ● | strengthens rules for the assessment of certain high-risk devices which may have to undergo an additional check by experts before they are placed on the market. |
|
|
Continuing Regulation. As in the U.S., manufacturers of medical devices are subject to comprehensive regulatory oversight by notified bodies and the competent authorities of the EEA countries. This oversight applies both before and after certification. It includes control of compliance with the EU MDR General Safety and Performance Requirements and post-market surveillance.
In the EEA, the advertising and promotion of our products will also be subject to EEA countries national laws implementing Directive 2006/114/EC concerning misleading and comparative advertising, and Directive 2005/29/EC on unfair commercial practices, as well as other national legislation of individual EEA countries governing the advertising and promotion of medical devices. EEA countries’ legislation may also restrict or impose limitations on our ability to advertise our products directly to the general public. In addition, voluntary EU and national Codes of Conduct provide guidelines on the advertising and promotion of our products to the general public and may impose limitations on our promotional activities with healthcare professionals. Violations of the rules governing the promotion of medical devices in the EEA could be penalized by administrative measures, fines and imprisonment.
Data Privacy Regulation. The collection and use of personal health data in the EEA is governed by the data protection laws and regulations adopted by the EEA countries and the EU General Data Protection Regulation (“GDPR”). The GDPR became applicable on May 25, 2018 and repealed the EU Data Protection Directive. The GDPR is directly applicable in each EEA country and imposes several requirements on companies that process personal data, strict rules on the transfer of personal data out of the EEA, including to the U.S., and fines and penalties for failure to comply with the requirements of the GDPR and the related national data protection laws of the EAA countries. The GDPR confers a private right of action on data subjects and consumer associations to lodge complaints with supervisory authorities, seek judicial remedies, and obtain compensation for damages resulting from violations of the GDPR. Failure to comply with the requirements of GDPR may result in fines of up to 20,000,000 Euros or up to 4% of the total worldwide annual turnover of the preceding financial year, whichever is higher, and other administrative penalties.
Orphan Designation and Exclusivity. In the EU, the Committee for Medicinal Products for Human Use grants orphan drug designation to promote the development of products that are intended for the diagnosis, prevention or treatment of life-threatening or chronically debilitating conditions affecting not more than five in 10,000 persons in the EU Community and for which no satisfactory method of diagnosis, prevention or treatment has been authorized (or the product would be a significant benefit to those affected). Additionally, designation is granted for products intended for the diagnosis, prevention or treatment of a life-threatening, seriously debilitating or serious and chronic condition and when, without incentives, it is unlikely that sales of the drug in the EU would be sufficient to justify the necessary investment in developing the medicinal product.
In the EU, orphan drug designation entitles a party to financial incentives such as reduction of fees or fee waivers and ten years of market exclusivity is granted following medicinal product approval. This period may be reduced to six years if the orphan drug designation criteria are no longer met, including where it is shown that the product is sufficiently profitable not to justify maintenance of market exclusivity.
Orphan drug designation must be requested before submitting an application for marketing approval. Orphan drug designation does not convey any advantage in, or shorten the duration of, the regulatory review and approval process.
Exceptional Circumstances/Conditional Approval. Orphan medicinal product or products for unmet medical needs may be eligible for EU approval under exceptional circumstances or with conditional approval. Approval under exceptional circumstances is applicable to orphan products and is used when an applicant is unable to provide comprehensive data on the efficacy and safety under normal conditions of use because the indication for which the product is intended is encountered so rarely that the applicant cannot reasonably be expected to provide comprehensive evidence, when the present state of scientific knowledge does not allow comprehensive information to be provided, or when it is medically unethical to collect such information. Conditional marketing authorization is applicable to orphan medicinal products, medicinal products for seriously debilitating or life- threatening diseases or medicinal products to be used in emergency situations in response to recognized public threats. Conditional marketing authorization can be granted on the basis of less complete data than is normally required in order to meet unmet medical needs and in the interest of public health, provided the risk-benefit balance is positive, it is likely that the applicant will be able to provide the comprehensive clinical data, and unmet medical needs will be fulfilled.
|
|
Conditional marketing authorization is subject to certain specific obligations to be reviewed annually.
Other Regulations. We are also subject to numerous federal, state and local laws relating to such matters as safe working conditions, manufacturing practices, environmental protection, fire hazard control and disposal of hazardous or potentially hazardous substances. We may incur significant costs to comply with such laws and regulations now or in the future.
Regulation in Israel. In order to conduct clinical testing on humans in the State of Israel, special authorization must first be obtained from the ethics committee and general manager of the institution in which the clinical studies are scheduled to be conducted, as required under the Guidelines for Clinical Trials in Human Subjects implemented pursuant to the Israeli Public Health Regulations (Clinical Trials in Human Subjects), as amended from time to time, and other applicable legislation. These regulations require authorization by the institutional ethics committee and general manager as well as from the Israeli Ministry of Health, except in certain circumstances, and in the case of genetic trials, special fertility trials and complex clinical trials, an additional authorization of the Ministry of Health’s overseeing ethics committee. The institutional ethics committee must, among other things, evaluate the anticipated benefits that are likely to be derived from the project to determine if it justifies the risks and inconvenience to be inflicted on the human subjects, and the committee must ensure that adequate protection exists for the rights and safety of the participants as well as the accuracy of the information gathered in the course of the clinical testing. Since we perform a portion of the clinical studies on certain of our therapeutic candidates in Israel, we are required to obtain authorization from the ethics committee and general manager of each institution in which we intend to conduct our clinical trials, and in most cases, from the Israeli Ministry of Health.
Corporate History
We were incorporated on April 24, 2015. On June 25, 2019, our name was changed to Beyond Air, Inc. from AIT Therapeutics, Inc.
We have the following wholly owned subsidiaries:
| - | Beyond Air Ltd. (“BA Ltd.”), incorporated in Israel on May 1, 2011. | |
| - | Advanced Inhalation Therapies (“AIT”), a wholly-owned subsidiary of BA Ltd., incorporated on August 29, 2014, in Delaware. AIT was dissolved on March 1, 2021. | |
| - | Beyond Air Australia Pty Ltd., incorporated on December 17, 2019 in Australia. | |
| - | Beyond Air Ireland Limited, incorporated on March 5, 2020 in Ireland. | |
| - | Beyond Air Cyprus Limited, incorporated on October 13, 2021 in Cyprus. |
We also have 80% ownership in the following entity:
| - | Beyond Air Bermuda Limited, incorporated on August 13, 2021 in Bermuda. In September 2022, its name was changed to Beyond Cancer Bermuda Limited. |
Beyond Air Bermuda has the following wholly owned subsidiaries:
| - | XAIR Israel Ltd, incorporated on October 3, 2021 in Israel. | |
| - | Beyond Cancer U.S., Inc., incorporated on March 17, 2022 in Delaware. |
|
|
Emerging Growth Company Status
As of April 1, 2022, we ceased to be an “emerging growth company” (“EGC”) as defined in the Jumpstart our Business Startups Act of 2012. As such, the Company became subject to new accounting pronouncement effective dates for non-EGCs for fiscal year 2023. As a result, we have experienced, and expect to continue to experience, additional costs associated with being a public company. We may also experience additional costs associated with compliance with the requirements of Section 404 of the Sarbanes-Oxley Act, the adoption of certain Accounting Standards Updates upon losing such status, and additional disclosure requirements.
Available Information
We file electronically with the Securities and Exchange Commission (the “SEC”) our annual reports on Form 10-K, quarterly reports on Form 10-Q and current reports on Form 8-K, and amendments related thereto, pursuant to Section 13(a) or 15(d) of the Exchange Act. We make available on our website at www.beyondair.net free of charge, copies of these reports, as soon as reasonably practicable after we electronically file such material with, or furnish it to, the SEC. Reports filed with the SEC may be viewed at www.sec.gov. The information in or accessible through the SEC and our website are not incorporated into, and are not considered part of, this filing. Further, our references to the URLs for these websites are intended to be inactive textual references only.
Human Capital
As of March 31, 2023, we had 98 employees globally, all of whom were full time employees. None of our employees are represented by a labor union and we consider our employee relations to be good.
Our workforce is highly educated and diverse, which we believe is important for our continued success as a leading innovator in the medical device market. We employ a number of strategies to best enable us to attract, retain, and engage our team members. Our human capital resources objectives include, as applicable, identifying, recruiting, retaining, and incentivizing our management team and our clinical, scientific and other employees and consultants. The principal purposes of our equity and cash incentive plans are to attract, retain and motivate personnel through the granting of stock-based and cash-based compensation awards, in order to align our interests and the interests of our stockholders with those of our employees and consultants.
|
|
ITEM 1A. RISK FACTORS
Investing in our common stock involves a high degree of risk. You should consider carefully the risks described below, together with the other information included or incorporated by reference in this Annual Report. If any of the following risks occur, our business, financial condition, results of operations and future growth prospects could be materially and adversely affected. In these circumstances, the market price of our common stock could decline. Other events that we do not currently anticipate or that we currently deem immaterial may also affect our business, prospects, financial condition and results of operations.
Risks Related to Our Financial Position and Capital Requirements
Numerous factors, including the incurring of significant losses, are relevant to our financial success and any or all such factors could have a disproportionate impact on our bottom line.
Our ability to implement our business strategy is subject to numerous risks that you should be aware of before making an investment decision. These are not the only risks we face. These risks include, among others, that:
| ● | we are a medical device and biopharmaceutical company with only one FDA-approved product and a limited operating history on which to assess our business, have incurred significant losses since our inception and incurred a net cash used in operating activities for the year ended March 31, 2023 of approximately $33.0 million. As of March 31, 2023, we have an accumulated deficit of approximately $179.5 million and we anticipate to continue to incur significant losses for the foreseeable future; | |
| ● | we are unable to predict the extent of future losses or when we will become profitable based on the sale of any product, if at all. Even if we succeed in scaling the commercialization of our approved product and succeed in developing and commercializing our product candidates, we may never generate revenue to sustain profitability; | |
| ● | we have only one FDA-approved product, and we expect that we will need to raise additional funding before we can expect to become profitable from sales of our products; | |
| ● | we are heavily dependent upon the success of our approved product and product candidates (which are in various stages of clinical development), and we cannot provide any assurance that the FDA or comparable foreign regulatory authorities will allow us to conduct further clinical trials; | |
| ● | we are in the process of further developing our proprietary NO delivery system, and unexpected delays will adversely impact the timing of our U.S.-based clinical trials and approvals; | |
| ● | we might be unable to develop product candidates that will achieve commercial success in a timely and cost-effective manner, or ever; | |
| ● | our competitors may develop or commercialize products faster or more successfully than us; | |
| ● | because some of the target patient populations of our approved product or product candidates are small, we must be able to successfully identify patients and achieve a significant market share to maintain profitability and growth; | |
| ● | we rely on third parties to help conduct our preclinical studies, clinical trials and commercial scale manufacturing; |
| ● | if we are unable to obtain and maintain effective intellectual property rights for our technologies, approved product or current or future product candidates, we may not be able to compete effectively in our markets; and | |
| ● | our future success depends in part upon our ability to retain our executive and scientific teams, and to attract, retain and motivate other qualified personnel. |
Because of the numerous risks and uncertainties associated with product development and commercialization, we are unable to accurately predict the timing or amount of expenses or when, or if, we will be able to achieve profitability. If we are required by regulatory authorities to perform studies in addition to those expected or if there are any delays in the initiation and completion of our clinical trials or the development of any of our product candidates, our expenses could increase.
|
|
It is highly likely that we will need to raise additional capital to meet our business requirements in the future, and such capital raising may be costly or difficult to obtain, and could dilute current stockholders’ ownership interests.
Our future capital requirements will depend on many factors, including the success and costs of our commercialization activities, including product marketing, sales, and distribution progress, the results of our clinical trials, the timing and outcome of regulatory review of our product candidates, commercial manufacturing success, and the number and development requirements of product candidates that we pursue. Because of the numerous risks and uncertainties associated with the development and commercialization of our approved product and product candidates, we are unable to reasonably estimate the amounts of additional capital outlays and operating expenditures that our business will require. It is likely that we will need to raise additional funds through public or private debt or equity financings to meet various objectives including, but not limited to:
| ● | expanding commercialization of our approved product; | |
| ● | clinical trials for our product candidates; | |
| ● | researching and developing new products; | |
| ● | pursuing growth opportunities, including more rapid expansion; | |
| ● | acquiring complementary businesses or technologies; | |
| ● | making capital improvements to improve our infrastructure; | |
| ● | hiring qualified management and key employees; | |
| ● | responding to competitive pressures; | |
| ● | complying with regulatory requirements; and | |
| ● | maintaining compliance with applicable laws. |
Any additional capital raised through the sale of equity or equity-linked securities may dilute our current stockholders’ ownership in us and could also result in a decrease in the market price of our common stock. The terms of those securities issued by us in future capital transactions may be more favorable to new investors and may include preferences, superior voting rights and the issuance of warrants or other derivative securities, which may have a further dilutive effect.
Furthermore, any debt or equity financing that we may need may not be available on terms favorable to us, or at all.
Additionally, we may incur substantial costs in pursuing future capital financing, including investment banking fees, legal fees, accounting fees, securities law compliance fees, printing and distribution expenses and other costs. We may also be required to recognize non-cash expenses in connection with certain securities we issue, such as convertible notes and warrants, which may adversely impact our financial condition.
If we are unable to obtain required additional capital, we may have to curtail our growth plans or cut back on existing business, and we may not be able to continue operating if we do not generate sufficient revenues from operations needed to stay in business.
Our failure to comply with the covenants or other terms of the Loan Agreement, including as a result of events beyond our control, could result in a default under the Loan Agreement that could materially and adversely affect the ongoing viability of our business.
Subsequent to the fiscal year ended March 31, 2023, on June 15, 2023 (the “Closing Date”), we and our wholly owned subsidiary, Beyond Air Ltd., entered into a Loan and Security Agreement (the “Agreement”) with Avenue Capital Management II, L.P., as administrative agent and collateral agent (the “Agent”), Avenue Venture Opportunities Fund, L.P., a Delaware limited partnership (“Avenue”), and Avenue Venture Opportunities Fund II, L.P, a Delaware limited partnership (“Avenue 2” and, together with Avenue, the “Lenders”). Also on June 15, 2023, the Company entered into a Supplement to the Agreement (collectively with the Agreement, the “Loan Agreement”) with the Agent and the Lenders. The Loan Agreement provides for senior secured term loans (the “Loans”) in an aggregate principal amount up to $40 million, with (i) $17.5 million advanced on the Closing Date (“Tranche 1”), (ii) up to $10 million which may be advanced upon the request of the Company between April 1, 2024 and September 30, 2024, subject to the Company having achieved total revenue derived from the sale of LungFit® PH (other than licensing revenue) (“Product Revenue”) for the three-month period prior to funding of not less than 85% of projected Product Revenue for such period (“Tranche 2”), and (iii) up to $12.5 million which may be advanced after April 1, 2024 (the “Discretionary Tranche”), subject to (a) the Agent and Lenders having received investment committee approval and (b) the Company and Lenders having mutually agreed to draw and fund, respectively, such amount. The Loans are due and payable on June 1, 2027 (the “Maturity Date”). The Loan principal is repayable in equal monthly installments beginning on January 1, 2025, with the possibility of deferring principal payments an additional 6 to 18 months contingent upon the Company’s achievement of at least $40 million of Product Revenue in the fiscal year ending March 31, 2025 and whether the Company has fully drawn Tranche 2. The Loans bear interest at a rate per annum (subject to increase during an event of default) equal to the greater of (i) the prime rate, as published by the Wall Street Journal from time to time, plus 3.75% and (ii) 12.00%. The Company may, subject to certain parameters, voluntarily prepay the Loans, in whole or in part, at any time. If prepayment occurs on or before the one-year anniversary of the Closing Date, the Company is required to pay a fee equal to the principal amount of the Loans prepaid multiplied by 3.00%; if prepayment occurs after the one-year anniversary of the Closing Date and on or before the two-year anniversary of the Closing Date, the Company is required to pay a fee equal to the principal amount of the Loans prepaid multiplied by 2.00%; if prepayment occurs after the two-year anniversary and on or before the three-year anniversary of the Closing Date, the Company is required to pay a fee equal to the principal amount of the Loans prepaid multiplied by 1.50%; and if prepayment occurs after the three-year anniversary of the Closing Date and before the Maturity Date, the Company is required to pay a fee equal to the principal amount of the Loans prepaid multiplied by 1.00%. A final payment fee of 3.50% of the principal amount of the Tranche 1 and Tranche 2 Loans is also due upon the Maturity Date or any earlier date of prepayment (in the case of any partial prepayment, solely with respect to the principal amount being prepaid). The Loans are guaranteed by the Company’s subsidiaries, Beyond Air Ltd. and Beyond Air Ireland Limited, and certain of the Company’s future subsidiaries (collectively, the “Guarantors”). The Company’s obligations under the Loan Agreement and the guarantee of such obligations are secured by a pledge of substantially all of the Company’s assets and will be secured by a pledge of substantially all of the assets of the Guarantors.
Pursuant to the Loan Agreement, the Company is subject to a financial covenant requiring the Company to maintain at all times $5,000,000 in unrestricted cash. The Loan Agreement also contains affirmative and negative covenants customary for financings of this type that, among other things, limit the ability of the Company and its subsidiaries to (i) incur additional debt, guarantees or liens; (ii) pay any dividends; (iii) enter into certain change of control transactions; (iv) sell, transfer, lease, license, or otherwise dispose of certain assets; (v) make certain investments or loans; and (vi) engage in certain transactions with related persons, in each case, subject to certain exceptions.
The Loan Agreement also includes events of default customary for financings of this type, in certain cases subject to customary periods to cure, following which the Agent may accelerate all amounts outstanding under the Loans. Events of default include, among other things:
| ● | our failure to pay any principal or interest under the Loan Agreement or any note in connection therewith; | |
| ● | any representation or warranty made shall prove to have been false or misleading in any material respect; | |
| ● | the occurrence of circumstances that could reasonably be expected to have a material adverse effect; | |
| ● | our default in any obligation under any other agreement involving indebtedness exceeding $250,000 that (i) results in the acceleration of such indebtedness or (ii) enables the holder of such indebtedness to accelerate such indebtedness; | |
| ● | judicial or administrative actions by governmental or regulatory authorities that could reasonably be expected to have a material adverse effect; | |
| ● | our breach of the restrictive covenants, reporting obligations or other terms of the Loan Agreement; and | |
| ● | certain specified insolvency and bankruptcy-related events. |
Subject to any applicable cure period set forth in the Loan Agreement, all amounts outstanding with respect to the Loans (principal and accrued interest), as well as any applicable prepayment premiums, final payment fees and other obligations and amounts owing under the loan documents, would become due and payable at the option of the Agent at the direction of the Lenders. Our assets or cash flow may not be sufficient to fully repay our obligations under the Loans if the obligations thereunder are accelerated upon any events of default. The duration and magnitude of any negative impact from a potential resurgence of the COVID-19 pandemic on LungFit® PH commercialization, development or net revenues could also affect our ability to meet the requirements to draw on one or more of the tranches and to remain in compliance with our liquidity financial covenant. Further, if we are unable to repay, refinance or restructure our obligations under the Loans, the Agent on behalf of the Lenders could proceed to protect and enforce their rights under the Agreement and the other loan documents by exercising such remedies (including foreclosure on the assets securing our obligations under the Agreement and the other loan documents) as are available to the Agent and the Lenders and in respect thereof under applicable law, either by suit in equity or by action at law, or both, whether for specific performance of any covenant or other agreement contained in the Agreement or the other loan documents or in aid of the exercise of any power granted in the Agreement or other loan documents. The foregoing would materially and adversely affect the ongoing viability of our business.
If we are unable to satisfy certain conditions in the Loan Agreement, we will be unable to draw down the remaining tranches of the Loans.
Pursuant to the Loan Agreement, we must satisfy certain conditions to be eligible to draw down the Tranche 2 of $10 million and the Discretionary Tranche of $12.5 million. Tranche 2 may be advanced upon the request of the Company between April 1, 2024 and September 30, 2024, subject to the Company having achieved Product Revenue for the three-month period prior to funding of not less than 85% of projected Product Revenue for such period. The uncommitted Discretionary Tranche may be advanced after April 1, 2024 subject to (i) the Agent and Lenders having received investment committee approval and (ii) the Company and Lenders having mutually agreed to draw and fund, respectively, such amount. If we are unable to satisfy the conditions for the draw-down of Tranche 2 or the Lenders do not agree to fund the Discretionary Tranche, we would not be able to draw down such tranches and may not be able to obtain alternative financing on commercially reasonable terms or at all.
Our Loan Agreement contains restrictions that limit our flexibility in operating our business.
The Loan Agreement contains various covenants that limit our ability to engage in specified types of transactions without the prior consent of the Agent and the Lenders. These covenants limit our ability to, among other things:
| ● | sell, transfer, lease or dispose of our assets; | |
| ● | create, incur or assume additional indebtedness; | |
| ● | encumber or permit liens on certain of our assets; | |
| ● | make restricted payments, including paying dividends on, repurchasing or making distributions with respect to our common stock; | |
| ● | make specified investments (including loans, guaranties and advances); | |
| ● | consolidate, merge, sell or otherwise dispose of all or substantially all of our assets; | |
| ● | enter into certain transactions with our affiliates; and | |
| ● | permit our unrestricted cash held in certain deposit accounts to at any time be less than $5,000,000. |
The covenants in the Loan Agreement may limit our ability to take certain actions that may be in our long-term best interests. In the event that we breach one or more covenants, the Agent at the direction of the Lenders may choose to declare an event of default and require that we immediately repay all amounts outstanding under the Loan Agreement, plus penalties and interest, terminate the Lenders’ commitments to fund any undrawn tranches and foreclose on the collateral granted to them to secure the obligations under the Agreement and the other loan documents. Such repayment could have a material adverse effect on our business, operating results and financial condition.
Risks Related to Commercialization of Our Approved Product or Product Candidates
If the market opportunities for our approved product or product candidates are smaller than we believe they are, our revenue may be adversely affected, and our business may suffer.
Our projections of both the number of people who have our target diseases, as well as the subset of people with these diseases who have the potential to benefit from treatment with LungFit® PH and our product candidates, are based on our beliefs and estimates. These estimates have been derived from a variety of sources, including scientific literature, surveys of clinics, patient foundations or market research and may prove to be incorrect. Further, new studies may change the estimated incidence or prevalence of these diseases. The number of patients may turn out to be lower than expected. The effort to identify patients with diseases we seek to treat is in early stages, and we cannot accurately predict the number of patients for whom treatment might be possible. Additionally, the potentially addressable patient population for LungFit® PH and each of our product candidates may be limited or may not be amenable to treatment with LungFit® PH or our product candidates, and new patients may become increasingly difficult to identify or gain access to, which would adversely affect our results of operations and our business.
|
|
The insurance coverage and reimbursement status of newly-approved products is uncertain. Failure to obtain or maintain adequate coverage and reimbursement for new or current products could limit our ability to market those products and decrease our ability to generate revenue.
The pricing, coverage and reimbursement of our approved product and product candidates, if approved, must be adequate to support our commercial infrastructure. Our per-patient prices must be sufficient to recover our development and manufacturing costs and potentially achieve profitability. Accordingly, the availability and adequacy of coverage and reimbursement by governmental and private payors are essential for most patients to be able to afford expensive treatments such as ours, assuming certification or approval. Sales of our approved product and product candidates will depend substantially, both domestically and abroad, on the extent to which the costs of our approved product and product candidates will be paid for by health maintenance, managed care, pharmacy benefit and similar healthcare management organizations, or reimbursed by government authorities, private health insurers and other third-party payors. If coverage and reimbursement are not available, or are available only to limited levels, we may not be able to successfully commercialize our approved product and product candidates. Even if coverage is provided, the approved reimbursement amount may not be high enough to allow us to establish or maintain pricing sufficient to realize a return on our investment.
There is significant uncertainty related to the insurance coverage and reimbursement of newly approved products. In the U.S., there is no uniform system among payors for making coverage and reimbursement decisions. In addition, the process for determining whether a payor will provide coverage for a product or service may be separate from the process for setting the price or reimbursement rate that the payor will pay for the product or service once coverage is approved. Payors may limit coverage to specific products or services on an approved list, or formulary, which might not include all of the FDA-approved or -cleared products for a particular indication. In the U.S., the principal decisions about Medicare coverage and reimbursement for new medical devices are typically made by the Centers for Medicare & Medicaid Services (“CMS”), an agency within the U.S. Department of Health and Human Services, or Medicare contractors that process and pay claims, as CMS and/or the contractors decide whether and to what extent a new device will be covered and reimbursed under Medicare. Private payors tend to, but are not required to, follow the coverage and reimbursement policies established by CMS to a substantial degree. It is difficult to predict what CMS or the Medicare contractors will decide with respect to reimbursement for products such as ours.
Outside the U.S., international operations are generally subject to extensive governmental price controls and other market regulations, and we believe the increasing emphasis on cost-containment initiatives in the EEA, Canada and other countries has and will continue to put pressure on the pricing and usage of our approved product or product candidates. In many countries, the prices of medical products are subject to varying price control mechanisms as part of national health systems. In general, the prices of medical devices under such systems are substantially lower than in the U.S. Other countries allow companies to fix their own prices for medical products, but monitor and control company profits. Additional foreign price controls or other changes in pricing regulation could restrict the amount that we are able to charge for our approved product or product candidates. Accordingly, in markets outside the U.S., the reimbursement for our products may be reduced compared with the U.S. and may be insufficient to generate commercially reasonable revenue and profits.
Moreover, increasing efforts by governmental and third-party payors in the U.S. and abroad to cap or reduce healthcare costs may cause such organizations to limit both coverage and the level of reimbursement for new products approved and, as a result, they may not cover or provide adequate payment for our approved product and product candidates. We expect to experience pricing pressures in connection with the sale of any of our approved product and product candidates due to the trend toward managed healthcare, the increasing influence of health maintenance organizations and additional legislative changes. The downward pressure on healthcare costs in general, particularly prescription drugs and surgical procedures and other treatments, has become very intense. As a result, increasingly high barriers are being erected to the entry of new products.
Accordingly, the certification or approval of our product and product candidates for insurance coverage and reimbursement by governmental and private payors may impact our ability to generate revenues.
|
|
We face intense competition and rapid technological change and the possibility that our competitors may discover, develop or commercialize therapies that are similar, more advanced or more effective than ours, which may adversely affect our financial condition and our ability to successfully commercialize LungFit® PH and our product candidates.
We are working on PPHN which is a highly competitive market. A delivery system with a generator of NO has never been commercialized anywhere in the world, and market acceptance at an appropriate price is proving to be a somewhat difficult and lengthy process. The biotechnology, pharmaceutical and medical device industries are highly competitive. There are many pharmaceutical companies, biotechnology companies, medical device companies, public and private universities and research organizations actively engaged in the research and development of products that may be similar to our approved product or our product candidates. We are aware of several companies currently developing and selling NO therapies for various indications such as hypoxic respiratory failure (HRF). For example, Mallinckrodt commercializes INOMAX® (nitric oxide) for inhalation, which is approved for use to treat newborns suffering from HRF-PPHN in the U.S., Canada, Australia, Mexico and Japan. Linde Group markets a generic version of the Mallinckrodt offering with their delivery system called NOxBOX®. The Linde Group has marketing rights to INOMAX® in Europe. Air Liquide sells a similar product in Europe, called KINOX™, together with their delivery platform called SoKINOX™, for the treatment of pulmonary hypertension of the newborn. In Europe, EKU, International Biomedical, Cahouet and ITC each have a device that delivers nitric oxide. Bellerophon Therapeutics is developing an NO delivery system for patients with chronic pulmonary diseases such as COPD, PH-sarcoidosis, or fibrotic interstitial lung disease. VERO Biotech LLC (formerly known as Geno LLC) received FDA approval for their delivery system GENOSYL DS for HRF associated with PPHN in 2019, and received FDA approval for a third generation of that delivery system in 2023. In addition, other companies may be developing inhaled NO delivery systems at various concentrations. Novan Inc. has recently received approval for a nitric oxide-based prescription treatment called berdazimer for molluscum, a contagious skin infection. SaNOtize has an NO nasal spray that has received approval in India, Israel, and eight other countries for preventing COVID-19 after exposure. Third Pole has reported the development of an NO generator and delivery system, but we are not aware of any display of any product at any medical/scientific conference in recent years. Our patents surrounding LungFit® have priority date over those of Third Pole.
In addition to NO treatments currently available or under development, we also face competition from non-NO-based drugs and therapies. For example, the successful development of immunizations for bronchiolitis may render useless any product we develop for that indication. Also, antibiotic treatments for infections associated with CF and other underlying lung conditions may be preferred over any product that we develop. Even if we successfully develop our product candidates, and obtain certification or approval for them, other treatments may be preferred and we may not be successful in commercializing our product candidates.
Some of our competitors have substantially greater financial, technical and other resources, such as larger research and development staff and experienced marketing and manufacturing organizations. Additional mergers and acquisitions in the medical device, biotechnology and pharmaceutical industries may result in even more resources being concentrated in our competitors. As a result, these companies may obtain certification or regulatory approval more rapidly than we are able to and may be more effective in selling and marketing their products as well. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large, established companies. Competition may increase further as a result of advances in the commercial applicability of technologies and greater availability of capital for investment in these industries. Our competitors may succeed in developing, acquiring or licensing on an exclusive basis, products that are more effective or less costly than LungFit® PH or any product candidate that we may develop, or achieve earlier patent protection, certification or regulatory approval, product commercialization and market penetration than we do. Additionally, technologies developed by our competitors may render LungFit® PH or our potential product candidates uneconomical or obsolete, and we may not be successful in marketing LungFit® PH or our product candidates against competitors.
|
|
We currently have a limited marketing and sales organization. If we are unable to scale sales and marketing capabilities or enter into agreements with third parties to market and sell LungFit® PH or our product candidates, we may be unable to generate revenue.
Although some of our employees may have sold other similar products in the past while employed at other companies, we as a company have limited experience selling and marketing our product candidates and we currently have a nascent marketing and sales organization. To successfully commercialize LungFit® PH or any other products that may result from our development programs, we will need to further develop these capabilities, either on our own or with others. We continue to refine our commercialization efforts for LungFit® PH and intend to establish a more complete sales and marketing organization with technical expertise to potentially reach all U.S. hospitals using or capable of using NO. This will be an expensive, difficult and time-consuming endeavor. Any failure or delay in the development of our internal sales, marketing and distribution capabilities would adversely impact the commercialization of our products.
Further, given our limited experience in marketing and selling medical device products, our estimate of the size of the required sales force may be materially more or less than the size of the sales force actually required to effectively commercialize LungFit® PH and our product candidates. As such, we may be required to hire substantially more sales representatives to adequately support the commercialization of LungFit® PH and our product candidates, or we may incur excess costs as a result of hiring more sales representatives than necessary. With respect to certain geographical markets, we may enter into collaborations with other entities to utilize their local marketing and distribution capabilities, but we may be unable to enter into such agreements on favorable terms, if at all. If our future collaborators do not commit sufficient resources to commercialize our future products, if any, and we are unable to develop the necessary marketing capabilities on our own, we will be unable to generate sufficient product revenue to sustain our business. We may be competing with companies that currently have extensive and well-funded marketing and sales operations. Without an internal team or the support of a third party to perform marketing and sales functions, we may be unable to compete successfully against these more established companies.
The commercial success of LungFit® PH and any current or future product candidate will depend upon the degree of market acceptance by physicians, patients, third-party payors and others in the medical community.
Even with approval from the FDA and potential future certification or approvals from comparable foreign regulatory authorities, the commercial success of LungFit® PH and our product candidates will depend in part on the medical community, patients and third-party payors accepting LungFit® PH and our product candidates as medically useful, cost-effective and safe. Any product that we bring to the market may not gain market acceptance by physicians, patients, third-party payors and others in the medical community. The degree of market acceptance of LungFit® PH and any of our product candidates that become approved for commercial sale will depend, in part, on a number of factors, including:
| ● | the safety and efficacy of the product(s) as demonstrated in clinical trials and potential advantages over competing treatments; | |
| ● | the prevalence and severity of any side effects, including any limitations or warnings contained in a product’s approved labeling; | |
| ● | the clinical indications for which certification or approval is granted; | |
| ● |
relative convenience and ease of administration;
|
|
| ● | familiarity of group purchasing organizations with our products; | |
| ● | the cost of treatment, particularly in relation to competing treatments; | |
| ● | the willingness of the target patient population to try new therapies and of physicians to prescribe these therapies; | |
| ● | the strength of marketing and distribution support and timing of market introduction of competitive products; | |
| ● | publicity concerning our products or competing products and treatments; and | |
| ● | sufficient third-party insurance coverage and reimbursement. |
Even if a potential product displays a favorable efficacy and safety profile in preclinical studies and clinical trials, market acceptance of the product will not be fully known until after it is launched. Our efforts to educate the medical community and third-party payors on the benefits of the product candidates may require significant resources and may never be successful. If the LungFit® PH or our product candidates are approved for commercialization but fail to achieve an adequate level of acceptance by physicians, patients, third-party payors and others in the medical community, we will not be able to generate sufficient revenue to become or remain profitable.
|
|
If we fail to properly manage our anticipated growth, our business could suffer.
Our rapid growth has placed, and will continue to place, a significant strain on our management and on our operational and financial resources and systems. Failure to manage our growth effectively could cause us to over-invest or under-invest in infrastructure, and result in losses or weaknesses in our infrastructure, which could materially adversely affect us. Additionally, our anticipated growth will increase the demands placed on our suppliers, resulting in an increased need for us to carefully monitor for quality assurance. Any failure by us to manage our growth effectively could have an adverse effect on our ability to achieve our development and commercialization goals.
Pricing pressure from our competitors and our customers may impact our ability to sell our products at prices necessary to support our current business strategies.
The industry in which we operate is characterized by intense competition, and the market continues to attract numerous new companies and technologies, which has encouraged more established companies to intensify competitive pricing pressure. As a result of this increased competition, as well as the challenges of third-party coverage and reimbursement practices, we believe there will be continued pricing pressure in the future. If competitive forces drive down the prices we are able to charge for our products, our profit margins will shrink, which will adversely affect our ability to maintain our profitability and to invest in and grow our business.
Cybersecurity risks and the failure to maintain the confidentiality, integrity, and availability of our computer hardware, software, and Internet applications and related tools and functions could result in harm to our business and/or subject us to costs, fines or lawsuits.
We rely on sophisticated information technology systems and network infrastructure to operate and manage our business. We also maintain personally identifiable information (“PII”) about our employees, and given the nature of our business, we have access to protected health information (“PHI”). Our business therefore depends on the continuous, effective, reliable, and secure operation of our computer hardware, software, networks, Internet servers, and related infrastructure. To the extent that our hardware or software malfunctions or access to our data by internal personnel, suppliers or customers through the Internet is interrupted or compromised, our business could suffer.
The integrity and protection of our customer, personnel, financial, research and development, and other confidential data is critical to our business, and our customers and employees have a high expectation that we will adequately protect their personal information. The regulatory environment governing information, security and privacy laws is increasingly demanding and continues to evolve and a number of states have adopted laws and regulations that may affect our privacy and data security practices regarding the use, disclosure and protection of PII. For example, the California Consumer Privacy Act (“the CCPA”), among other things, creates individual privacy rights and imposes increased obligations on companies handling PII.
Although our computer and communications hardware are protected through physical and software safeguards, they are still vulnerable to system malfunction, computer viruses, malware and ransomware, and other cybersecurity threats such as phishing and social engineering attacks. These events could lead to the unauthorized access of our information technology systems and result in financial loss and the misappropriation or unauthorized disclosure of confidential information belonging to us, our employees, partners, customers, or suppliers. The techniques used by criminal elements to attack computer systems are sophisticated, change frequently and may originate from less regulated and remote areas of the world. As a result, we may not be able to address these techniques proactively or implement adequate preventative measures. If our information technology systems are compromised, we could be subject to fines, damages, litigation and enforcement actions, incur financial losses, suffer reputational damage, and lose trade secrets or other confidential information, each of which could significantly harm our business.
Healthcare legislative or regulatory reform measures, including government restrictions on pricing and reimbursement, may have a negative impact on our business and results of operations.
In the U.S., there have been and continue to be a number of legislative and regulatory changes and proposed changes to contain healthcare costs. For example, in March 2010, the Patient Protection and Affordable Care Act (“ACA”) was enacted, which, among other things, substantially changes the way health care is financed by both governmental and private insurers, and significantly impacts the U.S. medical device industry. Some of the provisions of the ACA have been subject to judicial challenges as well as efforts to modify them or alter their interpretation or implementation. For example, the Tax Cuts and Jobs Act of 2017 (“Tax Act”), includes a provision that eliminated the tax-based shared responsibility payment imposed by the ACA on certain individuals who fail to maintain qualifying health coverage for all or part of a year, commonly referred to as the “individual mandate,” effective January 1, 2019. It is unclear how efforts to modify or invalidate the ACA or its implementing regulations, or portions thereof, will affect our business. Additional legislative changes, regulatory changes and judicial challenges related to the ACA remain possible. We cannot predict what effect further changes related to the ACA would have on our business.
We cannot be sure whether additional legislative changes will be enacted, or whether government regulations, guidance or interpretations will be changed, or what the impact of such changes would be on the certification or marketing approvals, sales, pricing, or reimbursement of our approved product or product candidates, if any, may be. We expect that any such healthcare reform measures that may be adopted in the future, may result in more rigorous coverage criteria and in additional downward pressure on the price that we receive for any approved product. Any reduction in reimbursement from Medicare or other government programs may result in a similar reduction in payments from private payors. The implementation of cost containment measures or other healthcare reforms may prevent us from being able to generate revenue, attain profitability, or commercialize our approved product or product candidates.
|
|
Moreover, in order to obtain reimbursement for our products in some EEA countries, including some EU Member States, we may be required to compile additional data comparing the cost-effectiveness of our products to other available therapies. Health Technology Assessment (“HTA”) of both medicinal products and medical devices is becoming an increasingly common part of the pricing and reimbursement procedures in some EU Member States, including those representing the larger markets. The HTA process, which is currently governed by national laws in each EU Member State, is the procedure to assess therapeutic, economic and societal impact of a given medical product in the national healthcare systems of the individual country. The outcome of an HTA will often influence the pricing and reimbursement status granted to these medical products by the competent authorities of the respective EU Member State. The extent to which pricing and reimbursement decisions are influenced by the HTA of the specific medical product currently varies between EU Member States. On December 13, 2021, the EU adopted a new HTA Regulation which entered into force on January 11, 2022 and will become applicable to all EU Member States from January 12, 2025. The new EU HTA regulation aims to harmonize the clinical benefit assessment of HTA across the EU and provides the basis for permanent and sustainable cooperation at the EU level for joint clinical assessments in these areas.
We are subject to additional federal and state laws and regulations relating to our business, and our failure to comply with those laws could have a material adverse effect on our results of operations and financial conditions.
We are subject to additional health care regulation and enforcement by the federal government and the states in which we conduct our business. Of note, regulations that may relate to our operations include the following:
| ● | the federal health care program AKS prohibits, among other things, persons from knowingly and willfully soliciting, receiving, offering, or providing remuneration, directly or indirectly, in cash or in kind, to induce or reward either the referral of an individual for, or the purchase, lease, or order or arranging for the purchase, lease or order of any good or service, for which payment may be made, in whole or in part, under federal health care programs such as Medicare and Medicaid. This statute has been interpreted to apply to arrangements between pharmaceutical or device manufacturers, on the one hand, and prescribers, purchasers and formulary managers and others on the other. The term “remuneration” has been broadly interpreted to apply to anything of value including, for example, gifts, cash payments, donations, waivers of payment, ownership interests, and providing any item, service, or compensation for something other than fair market value. Liability under the AKS may be established without proving actual knowledge of the statute or specific intent to violate it. Although there are a number of statutory exceptions and regulatory safe harbors to the AKS protecting certain common business arrangements and activities from prosecution or regulatory sanctions, the exceptions and safe harbors are drawn narrowly. Practices that involve remuneration to those who prescribe, purchase, or recommend medical device products, including certain discounts, or engaging such individuals as consultants, advisors and speakers, may be subject to scrutiny if they do not fit squarely within an exception or safe harbor. Moreover, there are no safe harbors for many common practices, such as educational grants and reimbursement support programs; | |
| ● | the federal civil FCA that prohibits, among other things, individuals or entities from knowingly presenting, or causing to be presented, false or fraudulent, claims for payment of government funds, knowingly making, using or causing to be made or used a false statement or record material to an obligation to pay money to the government, or knowingly concealing or knowingly and improperly avoiding, decreasing or concealing an obligation to pay money to the federal government. A claim including items or services resulting from a violation of the AKS constitutes a false or fraudulent claim for purposes of the FCA. Actions under the FCA may be brought by the government or as a qui tam action by a private individual in the name of the government, who may also share in any monetary recovery. Qui tam actions are filed under seal and impose a mandatory duty on the U.S. Department of Justice to investigate such allegations. Manufacturers have faced liability under the FCA for providing inaccurate billing or coding information to customers or promoting a product off-label. FCA liability is potentially significant in the healthcare industry because the statute provides for treble damages and significant mandatory penalties per false or fraudulent claim or statement for violations, as well as exclusion from participation in federal healthcare programs; | |
| ● | HIPAA imposes criminal and civil liability for, among other things, knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program, including private third-party payors, or knowingly and willfully falsifying, concealing, or covering up a material fact or making any materially false, fictitious, or fraudulent statement or representation, or using any false writing or document knowing the same to contain any materially false, fictitious, or fraudulent statement or entry, in connection with the delivery of or payment for healthcare benefits, items, or services; | |
| ● | the federal Sunshine Act requires applicable manufacturers of devices, biologics and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program (with certain exceptions) to report annually to CMS information related to payments and other transfers of value to physicians, physician assistants, nurse practitioners, clinical nurse specialists, certified nurse anesthetists, and certified nurse midwives, and teaching hospitals, as well as ownership and investment interests held by physicians and their immediate family members; and | |
| ● | analogous state and foreign laws and regulations, such as state anti-kickback and false claims laws, may apply to sales or marketing arrangements and claims involving healthcare items or services reimbursed by non-governmental third-party payors, including private insurers. Several states have enacted legislation requiring medical device manufacturers to, among other things, establish marketing compliance programs; file periodic reports with the state, including reports on gifts and payments to individual health care providers; and/or register their sales representatives. Some states prohibit certain sales and marketing practices, including the provision of gifts, meals, or other items to health care providers. |
Efforts to ensure that our business arrangements with third parties will comply with applicable healthcare laws and regulations will involve substantial costs. Because of the breadth of these laws and the narrowness of the statutory exceptions and safe harbors available, it is possible that some of our business activities could be subject to challenge under one or more of such laws. The scope and enforcement of these laws is uncertain and subject to change in the current environment of health care reform. We cannot predict the impact on our business of any changes in these laws. Federal or state regulatory authorities may challenge our current or future activities under these laws. Any such challenge, even if we are able to successfully defend against it, could have a material adverse effect on our reputation, business, results of operations, and financial condition. Any state or federal regulatory review of us, regardless of the outcome, would be costly and time-consuming. If our operations are found to be in violation of any of the laws described above or any other governmental regulations that apply to us, we may be subject to penalties, including civil and criminal penalties, damages, fines, exclusion from participation in government health care programs, such as Medicare and Medicaid, imprisonment and the curtailment or restructuring of our operations, any of which could adversely affect our ability to operate our business and our results of operations.
|
|
If we fail to comply with applicable privacy, data protection and data security laws and regulations, we could face substantial penalties, liability and adverse publicity and our business, operations and financial condition could be adversely affected.
We are subject to various laws and regulations globally regarding privacy and data protection, including laws and regulations relating to the collection, storage, handling, use, disclosure, transfer and security of personal data. The restrictions under applicable privacy, data protection and data security laws and regulations that may affect our ability to operate include but are not limited to:
| ● | HIPAA governs the use, disclosure, and security of protected health information by HIPAA “covered entities” and their “business associates.” Covered entities are health plans, health care clearinghouses and health care providers that engage in specific types of electronic transactions. A business associate is any person or entity (other than members of a covered entity’s workforce) that performs a service for or on behalf of a covered entity involving the use or disclosure of protected health information. Most healthcare providers who prescribe our products and from whom we obtain patient health information are subject to privacy and security requirements under HIPAA, as are we in certain circumstances. The U.S. Department of Health and Human Services (through the Office for Civil Rights) has direct enforcement authority against covered entities and business associates with regard to compliance with HIPAA regulations. We also could be subject to criminal penalties if we knowingly obtain individually identifiable health information from a covered entity in a manner that is not authorized or permitted by HIPAA or for aiding and abetting and/or conspiring to commit a violation of HIPAA. We are unable to predict whether our actions could be subject to prosecution in the event of an impermissible disclosure of health information to us; | |
| ● | numerous U.S. federal and state laws and regulations, including state data breach notification laws, state health information privacy laws and federal and state consumer protection laws, govern the collection, use, disclosure and protection of personal information. These laws may impose a number of compliance obligations on us, including requiring that we obtain consent before we collect, use, or disclose personal information, implement certain security protections to safeguard personal information, and notify individuals or regulators in the event of a breach; | |
| ● | other countries also have, or are developing, laws governing the collection, use, disclosure and protection of personal information. The GDPR, for example, imposes restrictions on the processing (e.g., collection, use, disclosure) of personal data in the EEA and also imposes strict restrictions on the transfer of personal data out of the EU to the U.S. Our business could be adversely impacted if our ability to transfer personal data outside of the EEA or Switzerland is restricted, which could adversely impact our operating results. For example, in July 2020, the Court of Justice of the European Union (the CJEU) declared the EU-U.S. Privacy Shield framework between the EU and U.S. to be invalid and raised concerns about other data transfer mechanisms in a case known colloquially as “Schrems II”, which could adversely impact our ability to transfer personal data from the EU to the U.S or otherwise may cause us to incur significant costs to do so legally. At present, there are few viable alternatives to the EU-U.S. Privacy Shield and the Standard Contractual Clauses (“SCCs”). If the level of protection in the U.S. or any other importing country is called into question under the SCCs, this could further impact our ability to transfer data outside of the EEA or Switzerland. Furthermore, following the Brexit and the UK’s exit from the EU, the UK became a third country to the EU in terms of personal data transfers. The European Commission has adopted an Adequacy Decision concerning the level of personal data protection in the UK under which personal data may now flow freely from the EU to the UK. However, personal data transfers from the EU to the UK may nevertheless be at a greater risk than before because the Adequacy Decision could in theory someday be suspended. On December 13, 2022, the European Commission adopted a draft adequacy decision for the EU-U.S. Data Privacy Framework. This draft decision follows the signature of a U.S. Executive Order by President Biden on October 7, 2022, along with the regulations issued by the U.S. Attorney General Merrick Garland. These two instruments implemented into U.S. law the agreement in principle announced by President von der Leyen and President Biden in March 2022. The draft decision concludes that the U.S. ensures an adequate level of protection for personal data transferred from the EU to U.S. companies. On February 14, 2023, the European Parliament’s Committee on Civil Liberties, Justice and Home Affairs published its draft motion for a resolution regarding the adequacy of the protection of personal data. On February 28, 2023, the European Data Protection Committee (EDPB) published its Opinion 5/2023 on the European Commission’s draft adequacy decision. The two sides are now expected to finalize the details of this agreement and translate it into legal texts that will form the basis of a draft adequacy decision to be proposed by the European Commission; and | |
| ● | the legislative and regulatory landscape for privacy and data security continues to evolve, and there has been an increasing amount of focus on privacy and data security issues with the potential to affect our business. For example, the CCPA, as amended by the California Privacy Rights Act (CPRA), contains disclosure obligations for businesses that collect personal information about California residents and affords those individuals new rights relating to their personal information that may affect our ability to use personal information. Other states, including Virginia, Colorado, Utah, Connecticut, Indiana, Iowa, Tennessee, Utah, and others have enacted privacy laws similar to the CCPA that impose new obligations or limitations in areas affecting our business and we continue to assess the impact of these state legislation, on our business as additional information and guidance becomes available. The federal government has also considered similar privacy laws that could impose new obligations or limitations in areas affecting our business. |
|
|
These privacy and data security laws and regulations could increase our cost of doing business, and failure to comply with these laws and regulations could result in government enforcement actions (which could include civil or criminal penalties), private litigation and/or adverse publicity and could materially and negatively affect our operating results and business. Although a thorough privacy compliance program could mitigate the risk of investigation and prosecution for violations of these laws and regulations, the risks cannot be entirely eliminated. Any action against us for violation of these laws or regulations, even if we successfully defend against it, could cause us to incur significant legal expenses and divert our management’s attention from the operation of our business. Moreover, achieving and sustaining compliance with applicable federal, state, and foreign privacy and data security laws and regulations may prove costly.
If we or our suppliers fail to comply with ongoing FDA or other foreign regulatory authority requirements, or if we experience unanticipated problems with our products, these products could be subject to restrictions or withdrawal from the market.
Any product for which we obtain FDA certification, clearance or approval, and the manufacturing processes, post-market surveillance, post-certification/approval clinical data and promotional activities for such product, remain subject to continued regulatory review, oversight, requirements, and periodic inspections by the FDA and other domestic and foreign regulatory authorities and notified bodies. For such products, we must also comply with equivalent standards in non-U.S. countries should we choose to engage in comparable activity in those jurisdictions. These obligations extend to our third-party suppliers as well.
In particular, we and our suppliers are required to comply with the FDA’s QSR in the U.S. and other regulations enforced outside the United States which cover the manufacture of our products and the methods and documentation of the design, testing, production, control, quality assurance, labeling, packaging, storage and shipping of medical devices. Regulatory authorities, such as the FDA, and notified bodies enforce the QSR in the U.S. and other regulations through periodic inspections. The failure by us or one of our suppliers to comply with applicable statutes and regulations administered by the FDA, or the failure to timely and adequately respond to any adverse inspectional observations or product safety issues, could result in, among other things, any of the following enforcement actions:
| ● | untitled letters, warning letters, fines, injunctions, consent decrees and civil penalties; | |
| ● | unanticipated expenditures to address or defend such actions; | |
| ● | customer notifications for repair, replacement, refunds; | |
| ● | recall, detention or seizure of our products; | |
| ● | operating restrictions or partial suspension or total shutdown of production; | |
| ● | refusing or delaying our requests for 510(k) clearance , de novo authorization or PMA approval of new products or modified products; | |
| ● | operating restrictions; | |
| ● | withdrawal of 510(k) clearances on PMA approvals that have already been granted; | |
| ● | refusal to grant export approval for our products; or | |
| ● | criminal prosecution. |
If any of these actions were to occur, it would harm our reputation and cause our product sales and profitability to suffer and may prevent us from generating revenue. Furthermore, our key component suppliers may not currently be or may not continue to be in compliance with all applicable regulatory requirements which could result in our failure to produce our products on a timely basis and in the required quantities, if at all.
In addition, we are required to conduct costly post-market testing and surveillance to monitor the safety or effectiveness of our approved products, and we must comply with medical device reporting requirements, including the reporting of adverse events and malfunctions related to our products. Later discovery of previously unknown problems with such products, including unanticipated adverse events or adverse events of unanticipated severity or frequency, manufacturing problems, or failure to comply with regulatory requirements such as QSR, may result in changes to labeling, restrictions on such products or manufacturing processes, withdrawal of the products from the market, voluntary or mandatory recalls, a requirement to repair, replace or refund the cost of any medical device we manufacture or distribute, fines, suspension of certification or regulatory approval, product seizures, injunctions or the imposition of civil or criminal penalties which would adversely affect our business, operating results and prospects.
|
|
Moreover, we may be required to conduct costly post-market testing and surveillance to monitor the safety or effectiveness of our products in the EEA. We must comply with medical device reporting requirements, including the reporting of adverse events and malfunctions related to our products. Later discovery of previously unknown problems with our products, including unanticipated adverse events or adverse events of unanticipated severity or frequency, manufacturing problems, or failure to comply with regulatory requirements may result in changes to labeling, restrictions on such products or manufacturing processes, withdrawal of the products from the market, voluntary or mandatory recalls, a requirement to repair, replace or refund the cost of any medical device we manufacture or distribute, fines, suspension of certification, regulatory clearances or approvals, product seizures, injunctions or the imposition of civil or criminal penalties which would adversely affect our business, operating results and prospects.
Our products may cause or contribute to adverse medical events or be subject to failures or malfunctions that we are required to report to the FDA or comparable foreign regulatory authorities, and if we fail to do so, we would be subject to sanctions that could harm our reputation, business, financial condition and results of operations. The discovery of serious safety issues with our products, or a recall of our products either voluntarily or at the direction of the FDA or comparable foreign regulatory authorities, could have a negative impact on us.
As a commercial-stage company, we are subject to the FDA’s medical device reporting regulations and similar foreign regulations, which require us to report to the FDA when we receive or become aware of information that reasonably suggests that one or more of our products may have caused or contributed to a death or serious injury or malfunctioned in a way that, if the malfunction were to recur, it could cause or contribute to a death or serious injury. The timing of our obligation to report is triggered by the date we become aware of the adverse event as well as the nature of the event. We may fail to report adverse events of which we become aware within the prescribed timeframe. We may also fail to recognize that we have become aware of a reportable adverse event, especially if it is not reported to us as an adverse event or if it is an adverse event that is unexpected or removed in time from the use of the product. If we fail to comply with our reporting obligations, the FDA could take action, including warning letters, untitled letters, administrative actions, criminal prosecution, imposition of civil monetary penalties, revocation of our device clearance or approval, seizure of our products or delay in clearance or approval of future products.
The FDA and comparable foreign regulatory authorities have the authority to require the recall of commercialized products in the event of material deficiencies or defects in design or manufacture of a product or in the event that a product poses an unacceptable risk to health. The FDA’s authority to require a recall must be based on a finding that there is reasonable probability that the device could cause serious injury or death. We may also choose to voluntarily recall a product if any material deficiency is found. A government-mandated or voluntary recall by us could occur as a result of an unacceptable risk to health, component failures, malfunctions, manufacturing defects, labeling or design deficiencies, packaging defects or other deficiencies or failures to comply with applicable regulations. Product defects or other errors may occur in the future.
Depending on the corrective action we take to redress a product’s deficiencies or defects, the FDA may require, or we may decide, that we will need to obtain new clearances or approvals for the device before we may market or distribute the corrected device. Seeking such clearances or approvals may delay our ability to replace the recalled devices in a timely manner. Moreover, if we do not adequately address problems associated with our devices, we may face additional regulatory enforcement action, including FDA warning letters, product seizure, injunctions, administrative penalties or civil or criminal fines.
Companies are required to maintain certain records of recalls and corrections, even if they are not reportable to the FDA. We may initiate voluntary withdrawals or corrections for our products in the future that we determine do not require notification of the FDA. If the FDA disagrees with our determinations, it could require us to report those actions as recalls and we may be subject to enforcement action. A future recall announcement could harm our reputation with customers, potentially lead to product liability claims against us and negatively affect our sales. Any corrective action, whether voluntary or involuntary, as well as defending ourselves in a lawsuit, will require the dedication of our time and capital, will distract management from operating our business and may harm our reputation and financial results.
|
|
All manufacturers placing medical devices on the market in the EEA are legally bound to report to the relevant competent authorities (a) any serious incident involving devices made available on the EEA market, except expected side-effects which are clearly documented in the product information and quantified in the technical documentation and are subject to trend reporting, and (b) any field safety corrective action in respect of devices made available on the EEA market, including any field safety corrective action undertaken in a third country in relation to a device which is also legally made available on the EEA market, if the reason for the field safety corrective action is not limited to the device made available in the third country. Reports should be submitted through the electronic system set up and managed by the European Commission in collaboration with EEA countries. Reports of serious incidents will be automatically transmitted to the competent authority of the EEA country in which the incident occurred and reports on field safety corrections actions will be automatically transmitted to the competent authority of the EEA country in which the field safety corrective action is being or is to be undertaken and the EEA country in which the manufacturer has its registered place of business.
Under the EU MDR, a “serious incident” means any incident that directly or indirectly led, might have led or might lead to any of the following: (a) the death of a patient, user or other person; (b) the temporary or permanent serious deterioration of a patient’s, user’s or other person’s state of health; or (c) a serious public health threat. A “field safety corrective action” means corrective action taken by a manufacturer for technical or medical reasons to prevent or reduce the risk of a serious incident in relation to a device made available on the market.
Malfunction of our products could result in future voluntary corrective actions, such as recalls, including corrections, or customer notifications, or agency action, such as inspection or enforcement actions. If malfunctions do occur, we may be unable to correct the malfunctions adequately or prevent further malfunctions, in which case we may need to cease manufacture and distribution of the affected products, initiate voluntary recalls, and redesign the products. Regulatory authorities may also take actions against us, such as ordering recalls, imposing fines, or seizing the affected products. Any corrective action, whether voluntary or involuntary, will require the dedication of our time and capital, distract management from operating our business, and may harm our reputation and financial results.
Our approved product or product candidates may in the future be subject to product recalls that could harm our reputation, business and financial results.
Medical devices can experience performance problems in the field that require review and possible corrective action. The occurrence of component failures, manufacturing errors, software errors, design defects or labeling inadequacies affecting a medical device could lead to a government-mandated or voluntary recall by the device manufacturer, in particular when such deficiencies may endanger health. The FDA requires that certain classifications of recalls be reported to the FDA within 10 working days after the recall is initiated. Comparable foreign regulatory authorities impose similar deadlines. Companies are required to maintain certain records of recalls, even if they are not reportable to the FDA or to comparable foreign regulatory authorities. We may initiate voluntary recalls involving our products in the future that we determine do not require notification of the FDA. If the FDA disagrees with our determinations, they could require us to report those actions as recalls. Product recalls may divert management attention and financial resources, expose us to product liability or other claims, harm our reputation with customers and adversely impact our business, financial condition and results of operations.
We may be subject to regulatory or enforcement actions if we engage in improper marketing or promotion of our approved product or product candidates.
Our educational and promotional activities and training methods must comply with FDA and other applicable laws, including the prohibition of the promotion of a medical device for a use that has not been cleared or approved by the FDA. Use of a device outside of its cleared or approved indications is known as “off-label” use. Physicians may use our products off-label in their professional medical judgment, as the FDA does not restrict or regulate a physician’s choice of treatment within the practice of medicine. However, if the FDA determines that our educational and promotional activities or training constitutes promotion of an off-label use, it could request that we modify our training or promotional materials or subject us to regulatory or enforcement actions, including the issuance of warning letters, untitled letters, fines, penalties, injunctions, or seizures, any of which could have an adverse impact on our reputation and financial results.
|
|
It is also possible that other federal, state or comparable foreign regulatory authorities might take action if they consider our educational and promotional activities or training methods to constitute promotion of an off-label use, which could result in significant fines or penalties under other statutory authorities, such as laws prohibiting false claims for reimbursement. In that event, our reputation could be damaged, and adoption of the products could be impaired. Although our policy is to refrain from statements that could be considered off-label promotion of our products, the FDA or comparable foreign regulatory authorities could disagree and conclude that we have engaged in off-label promotion. It is also possible that other federal, state or comparable foreign regulatory authorities might take action, including, but not limited to, through a whistleblower action under the FCA, if they consider our business activities constitute promotion of an off-label use, which could result in significant penalties, including, but not limited to, criminal, civil or administrative penalties, treble damages, fines, disgorgement, exclusion from participation in government healthcare programs, reporting requirements and compliance oversight if we become subject to a corporate integrity agreement or similar agreement to resolve allegations of non-compliance with these laws, and the curtailment or restructuring of our operations. In addition, the off-label use of our products may increase the risk of product liability claims. Product liability claims are expensive to defend and could divert our management’s attention, result in substantial damage awards against us, and harm our reputation.
The advertising and promotion of our products in the EEA is subject to EEA countries’ national laws implementing Directive 2006/114/EC concerning misleading and comparative advertising, and Directive 2005/29/EC on unfair commercial practices, as well as other national legislation of individual EEA country governing the advertising and promotion of medical devices. EEA country legislation may also restrict or impose limitations on our ability to advertise our products directly to the general public. In addition, voluntary EU and national Codes of Conduct provide guidelines on the advertising and promotion of our products to the general public and may impose limitations on our promotional activities with healthcare professionals.
We face extensive, ongoing regulatory requirements and review, and our products may face future development and regulatory difficulties.
The holder of an approved PMA, de novo authorization, or cleared 510(k) is subject to obligations to monitor and report adverse events and instances of the failure of a product to meet the specifications in the marketing application. Application holders must submit new or supplemental applications and obtain FDA approval for certain changes to the approved product, product labeling, or manufacturing process. Legal requirements have also been enacted to require disclosure of clinical trial results on publicly available databases.
In addition, manufacturers of FDA regulated products and their facilities are subject to continual review and periodic inspections by the FDA and comparable foreign regulatory authorities for compliance with the FDA’s QSR and, as applicable, cGMP regulations. Our relationships with healthcare providers, physicians and third-party payors must comply with FDA laws and regulations, the AKS, the FCA, HIPAA, various transparency laws, and similar state and foreign laws. If products are made available to authorized users of the Federal Supply Schedule of the General Services Administration and to low-income patients of certain hospitals, additional laws and requirements apply. Our activities are also potentially subject to federal and state consumer protection and unfair competition laws. If we or our third-party collaborators fail to comply with applicable regulatory requirements, a regulatory authority may take any of the following actions:
| ● | conduct an investigation into our practices and any alleged violation of law; | |
| ● | issue warning letters or untitled letters asserting that we are in violation of the law; | |
| ● | seek an injunction or impose civil or criminal penalties or monetary fines; | |
| ● | suspend or withdraw certification or regulatory approval; | |
| ● | require that we suspend or terminate any ongoing clinical trials; |
|
|
| ● | refuse to approve pending applications or supplements to applications filed by us; | |
| ● | suspend or impose restrictions on operations, including costly new manufacturing requirements; | |
| ● | seize or detain products, refuse to permit the import or export of products, or require us to initiate a product recall; or | |
| ● | exclude us from providing our products to those participating in government health care programs, such as Medicare and Medicaid, and refuse to allow us to enter into supply contracts, including government contracts. |
The occurrence of any of the foregoing events or penalties may force us to expend significant amounts of time and money and may significantly inhibit our ability to bring to market or continue to market our products and generate revenue. Similar regulations apply in foreign jurisdictions.
Risks Related to the Discovery and Development of Our Product Candidates
We are heavily dependent on the success of our product candidates, which are in various stages of clinical development. We cannot give any assurance that any of our product candidates will receive certification or regulatory approval, which is necessary before they can be commercialized.
To date, we have invested substantially all of our efforts and financial resources to design and develop our product candidates, including conducting clinical trials and providing general and administrative support for these operations. Our future success is dependent on our ability to successfully develop, obtain regulatory certification or approval for, and then successfully commercialize one or more product candidates.
Several of our product candidates are in the early stages of development and will require additional clinical development (and in some cases additional preclinical development), management of nonclinical, clinical and manufacturing activities, certification or regulatory approval, obtaining adequate manufacturing supply, building of a commercial organization and significant marketing efforts before we generate any revenue from product sales. To date, we have conducted 3 pilot clinical trials involving 198 patients with bronchiolitis (mainly caused by RSV) and a pilot clinical trial in nine patients with CF. In addition, Rambam healthcare campus in Israel conducted a compassionate treatment for two patients with CF who suffer from NTM infections (specifically M. abscessus). Additionally, two pilot clinical trials were completed in 2022, one in viral pneumonia and one in NTM lung infection. Both of these studies were using our LungFit® system (PRO and GO, respectively). Although the results of these trials demonstrated improvements in various endpoints and clinical outcomes which we believe support our efforts towards obtaining FDA approval, these trials were small and conducted outside the US, so it is unlikely that the FDA will view them as significant because of their size and scope. Therefore, we intend to conduct larger clinical trials aiming for statistically and clinically significant favorable results, or we will not be able to obtain certification or regulatory approval to market such product candidates. It may be some time before a pivotal trial is initiated, if at all, for such product candidates. Before a medical device clinical trial can be undertaken in the U.S., the sponsor of the trial must submit an IDE application for a medical device and the FDA must permit the trial to go forward. We cannot assure that we will obtain such agency acquiescence in a timely manner, or at all. In addition to our respiratory program using the LungFit® device, we have programs in Cancer and Autism which require significant further development before submission to the FDA.
Although we received approval of LungFit® PH from the FDA, we can make no assurances as to what any other comparable foreign regulatory authorities and notified bodies where we are seeking certification or regulatory approval will do. We are expending significant resources to commercialize LungFit® PH in the U.S. and we can make no assurances that our efforts will be successful. We cannot be certain that any of our product candidates will be successful in clinical trials or receive certification or regulatory approval. Further, our product candidates may not receive certification or regulatory approval even if they are successful in clinical trials. If we do not receive certification or regulatory approvals for our other product candidates, we may not be able to continue our operations.
|
|
We generally plan to seek certification or regulatory approval to commercialize our approved product and product candidates in the U.S., the EU and in additional foreign countries, as applicable. To obtain certification or regulatory approvals we must comply with the numerous and varying regulatory requirements of such countries regarding safety, efficacy, chemistry, manufacturing and controls, clinical trials, commercial sales, pricing and distribution of our approved product and product candidates. Even if we are successful in obtaining marketing certification or regulatory approval in one jurisdiction, we cannot ensure that we will obtain certification or regulatory approval in any other jurisdictions. If we are unable to obtain certification, clearance or approval for our product candidates in multiple jurisdictions, our revenue and results of operations would be negatively affected.
Some of our product candidates may be considered a drug/device combination and the process for obtaining regulatory approval in the U.S. on our product candidates will require compliance with complex procedures because concordance between two centers of the FDA (CDRH and CDER) is necessary for approval of this combination product. A change in the FDA’s prior determination that CDRH would lead the review of a marketing application for our product candidates would adversely impact our development timeline and significantly raise our costs to complete clinical development and obtain regulatory approvals.
The success of our business may also depend upon our ability to identify, license or discover additional product candidates.
Although a substantial amount of our effort will focus on the continued clinical testing, potential certification, regulatory approval and commercialization of LungFit® PH and our existing product candidates, the success of our business may also depend upon our ability to identify, license or discover additional product candidates. Our research programs or licensing efforts may fail to yield additional product candidates for clinical development for a number of reasons, including but not limited to the following:
| ● | our research or business development methodology or search criteria and process may be unsuccessful in identifying potential product candidates; | |
| ● | we may not be able or willing to assemble sufficient resources to acquire or discover additional product candidates; | |
| ● | our product candidates may not succeed in preclinical or clinical testing; | |
| ● | our potential product candidates may be shown to have harmful side effects or may have other characteristics that may make the product candidates unmarketable or unlikely to receive certification or marketing approval; | |
| ● | competitors may develop alternatives that render our product candidates obsolete or less attractive; | |
| ● | product candidates we develop may be covered by third parties’ patents or other exclusive rights; | |
| ● | the market for a product candidate may change during our program so that such a product may become unreasonable to continue to develop; | |
| ● | a product candidate may not be capable of being produced in commercial quantities at an acceptable cost, or at all; and | |
| ● | a product candidate may not be accepted as safe and effective by patients, the medical community or third-party payors. |
If any of these events occur, we may be forced to abandon our development efforts for a program or programs, or we may not be able to identify, license or discover additional product candidates, which would have a material adverse effect on our business and could potentially cause us to cease operations. Research programs to identify new product candidates require substantial technical, financial and human resources. We may focus our efforts and resources on potential programs or product candidates that ultimately prove to be unsuccessful.
|
|
The certification or regulatory approval processes of the FDA and comparable foreign regulatory authorities and notified bodies are lengthy, time-consuming and inherently unpredictable. If we are ultimately unable to obtain certification or regulatory approval for our product candidates, our business will be substantially harmed.
The time required to obtain certification or regulatory approval by the FDA or notified bodies in the EU is unpredictable, typically takes many years following the commencement of clinical trials and depends upon numerous factors. In addition, certification or regulatory approval policies, regulations or the type and amount of clinical data necessary to gain certification or regulatory approval may change during the course of a product candidate’s clinical development and may vary among jurisdictions, which may cause delays in the certification or regulatory approval or the decision not to certify or approve an application. We have not obtained certification or regulatory approval for any product other than LungFit® PH, and it is possible that none of our existing product candidates or any product candidates we may seek to develop in the future will ever obtain certification or regulatory approval.
The process required by the FDA before a new medical device may be marketed in the U.S. generally involves the following:
| ● | completion of or reference to extensive preclinical laboratory tests and preclinical animal studies, all performed in accordance with the FDA’s Good Laboratory Practice (“GLP”); | |
| ● | performance of adequate and well-controlled human clinical trials to establish the safety and efficacy of the medical device candidate for each proposed indication; and | |
| ● | submission to the FDA of a 510(k), de novo application, or PMA, after completion of all pivotal clinical trials. |
Applications for our product candidates could fail to receive regulatory approval for many reasons, including but not limited to the following:
| ● | the FDA or comparable foreign regulatory authorities may disagree with the design or implementation of our clinical trials; | |
| ● | we may be unable to demonstrate to the FDA or comparable foreign regulatory authorities that a product candidate’s risk-benefit ratio for its proposed indication is acceptable; | |
| ● | the FDA may determine that the population studied in the clinical program was not sufficiently broad or representative to assure safety in the full population for which we seek approval; | |
| ● | the FDA may disagree with our interpretation of data from preclinical studies or clinical trials; | |
| ● | the data collected from clinical trials of our product candidates may not be sufficient to support the submission of a PMA in the U.S. or elsewhere; | |
| ● | the FDA or comparable foreign regulatory authorities may fail to approve the manufacturing processes, test procedures and specifications or facilities of third-party manufacturers with which we contract for clinical and commercial supplies; | |
| ● | the approval policies or regulations of the FDA or comparable foreign regulatory authorities and notified bodies may significantly change in a manner rendering our clinical data insufficient for certification or approval; and |
This lengthy certification or regulatory approval process, as well as the unpredictability of the results of clinical trials, may result in our failing to obtain certification or regulatory approval to market any of our product candidates, which would significantly harm our business, results of operations and prospects.
|
|
Our business and eventual sale of our approved product and product candidates are subject to extensive regulatory requirements, including compliance with labelling, manufacturing and reporting controls. If we fail or are unable to timely obtain the necessary 510(k) clearances, de-novo authorizations, or PMA approvals for new products, or equivalent steps in third countries including the EEA, our ability to generate revenue could be materially harmed.
Our approved product and product candidates are classified as medical devices and are subject to extensive regulation in the United States by the FDA and other federal, state and local authorities and by comparable foreign regulatory authorities. The FDA can delay, limit or deny 510(k) clearance, authorization of a de novo application, or PMA approval of a device for many reasons, including:
| ● | we may not be able to demonstrate to the FDA’s satisfaction that our systems are safe and effective for its intended use; |
| ● | the data from our preclinical studies and clinical trials may be insufficient to support clearance or approval, where required; |
| ● | the manufacturing process or facilities we use or contract to use may not meet applicable requirements; and |
| ● | disruptions at the FDA caused by funding shortages or global health concerns, including the COVID-19 pandemic. |
The FDA may refuse our requests for 510(k) clearance, de-novo or PMA of new products, new intended uses or modifications to existing products.
From time to time, legislation is drafted and introduced in the United States that could significantly change the statutory provisions governing any regulatory approval or clearance that we receive in the United States. In addition, the FDA may change its clearance and approval policies, adopt additional regulations or revise existing regulations, or take other actions which may prevent or delay approval or clearance of our test kits under development or impact our ability to modify our currently approved or cleared test kits on a timely basis.
Our products are also subject to approval, certification and regulation by foreign regulatory and safety agencies. For example, the EU has adopted the EU MDR, which imposes stricter requirements for the marketing and sale of medical devices, including in the area of clinical evaluation requirements, quality systems and post-market surveillance. Complying with the requirements of the EU MDR may require us to incur significant expenditures. Failure to meet these requirements could adversely impact our business in the EEA and other regions that tie their product registrations to the EU requirements.
Once commercialized, modifications to our marketed products may require new 510(k) clearances or approval of PMA supplements, or equivalent steps in other countries or regions including the EEA, or may require us to cease marketing or recall the modified products until certification, clearances or regulatory approvals are obtained.
Modifications to any of our products once they are commercialized may require new regulatory approvals or clearances, including 510(k) clearances or approval of PMA supplements, or require us to recall or cease marketing the modified systems until these clearances or approvals are obtained. The FDA requires device manufacturers to initially make and document a determination of whether or not a modification requires a new approval, supplement or clearance. A manufacturer may determine that a modification could not affect safety or efficacy and does not represent a major change in its intended use, so that no new clearance or approval is necessary. However, the FDA can review a manufacturer’s decision and may disagree. The FDA may also on its own initiative determine that a new clearance or approval of a PMA Supplement is required. We may make modifications in the future that we believe do not or will not require additional clearances or approvals. If the FDA disagrees and requires new clearances or approvals for the modifications, we may be required to recall and to stop marketing our products as modified, which could require us to redesign our products and/or seek new marketing authorizations and harm our operating results. In these circumstances, we may be subject to significant enforcement actions.
|
|
For example, if a manufacturer determines that a modification to a PMA approved device could affect its safety or effectiveness or would constitute a major change in its intended use, then the manufacturer must file for a new a new PMA or approval of a PMA supplement. Where we determine that modifications to our products require a new PMA approval, we may not be able to obtain those additional approvals for the modifications or additional indications in a timely manner, or at all. Obtaining new approvals can be a time-consuming process, and delays in obtaining required future approvals would adversely affect our ability to introduce new or enhanced products in a timely manner, which in turn would harm our future growth.
For those products sold in the EEA, we must notify our EU notified body if significant changes are made to the products or if there are substantial changes to our quality assurance systems affecting those products. Obtaining certification can be a time-consuming process, and delays in obtaining required future clearances or approvals would adversely affect our ability to introduce new or enhanced products in a timely manner, which in turn would harm our future growth.
Clinical development involves a lengthy and expensive process with an uncertain outcome, and results of earlier studies may not be predictive of future study results.
Clinical testing is expensive and can take many years to complete, and its outcome is inherently uncertain. Failure can occur at any time during the clinical trial process. The results of preclinical studies and early clinical trials of our product candidates may not be predictive of the results of later-stage clinical trials. Product candidates that have shown promising results in early-stage clinical trials may still suffer significant setbacks in subsequent advanced clinical trials. There is a high failure rate for product candidates proceeding through clinical trials, and product candidates in later stages of clinical trials may fail to show the desired safety and efficacy traits despite having progressed satisfactorily through preclinical studies and initial clinical trials. A number of companies in the medical device and biopharmaceutical industry have suffered significant setbacks in advanced clinical trials due to lack of efficacy or adverse safety profiles, notwithstanding promising results in earlier studies. Moreover, preclinical and clinical data are often susceptible to varying interpretations and analyses. We do not know whether any pivotal clinical trials we may conduct will demonstrate consistent or adequate efficacy and safety sufficient to obtain certification or regulatory approval to market our product candidates. Nor do we know whether the FDA will permit us to proceed directly to pivotal trials without performing pilot trials in the U.S. using the same delivery system that we will seek approval by the agency.
Legislative or regulatory reforms may make it more difficult and costly for us to obtain certification, regulatory clearance or approval of any future products and to manufacture, market and distribute our products after certification, clearance or approval is obtained.
From time to time, legislation is drafted and introduced in Congress that could significantly change the statutory provisions governing the regulatory approval, manufacture and marketing of regulated products or the reimbursement thereof. In addition, the FDA may change its clearance and approval policies, adopt additional regulations or revise existing regulations, or take other actions, which may prevent or delay approval or clearance of our future products under development or impact our ability to modify our currently cleared products on a timely basis. Any new regulations or revisions or reinterpretations of existing regulations may impose additional costs or lengthen review times of planned or future products. It is impossible to predict whether legislative changes will be enacted or FDA regulations, guidance or interpretations changed, and what the impact of such changes, if any, may be.
FDA regulations and guidance are often revised or reinterpreted by the FDA in ways that may significantly affect our business and our products. Any new statutes, regulations or revisions or reinterpretations of existing regulations may impose additional costs or lengthen review times of any future products or make it more difficult to obtain clearance or approval for, manufacture, market or distribute our products. We cannot determine what effect changes in regulations, statutes, legal interpretation or policies, when and if promulgated, enacted or adopted may have on our business in the future. Such changes could, among other things, require: additional testing prior to obtaining clearance or approval; changes to manufacturing methods; recall, replacement or discontinuance of our products; or additional record keeping.
|
|
The FDA’s and comparable foreign regulatory authorities’ policies may change and additional government regulations may be promulgated that could prevent, limit or delay certification, regulatory clearance or approval of our product candidates. We cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative action, either in the U.S. or abroad. For example, the results of the upcoming mid-term Congressional elections may impact our business and industry. Any change in the laws or regulations that govern the clearance and approval processes relating to our current, planned and future products could make it more difficult and costly to obtain clearance or approval for new products or to produce, market and distribute existing products. Significant delays in receiving clearance or approval or the failure to receive clearance or approval for any new products would have an adverse effect on our ability to expand our business. If we are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we are not able to maintain regulatory compliance, we may lose any marketing clearance that we may have obtained and we may not achieve or sustain profitability.
In addition, on May 25, 2017, the new EU MDR entered into force for medical devices marketed in the EEA. Implementation of the EU MDR was delayed by one year due to the COVID-19 pandemic. Following its entry into application on May 26, 2021, the EU MDR introduced substantial changes to the obligations with which medical device manufacturers must comply in the EEA. High risk medical devices are subject to additional scrutiny during the conformity assessment procedure. Specifically, the EU MDR repeals and replaces the EU Medical Devices Directive. Unlike directives, which must be implemented into the national laws of the EEA countries, the regulations is directly applicable, i.e., without the need for adoption of EEA country laws implementing them, in all EEA countries and are intended to eliminate current differences in regulation of medical devices among EEA countries. The EU MDR, among other things, is intended to establish a uniform, transparent, predictable and sustainable regulatory framework across the EEA for medical devices to ensure a high level of safety and health while supporting innovation. The EU MDR entered into application on May 26, 2021 and among other things:
| ● | strengthens the rules on placing devices on the market and reinforce surveillance once they are available; | |
| ● | establishes explicit provisions on manufacturers’ responsibilities for the follow-up of the quality, performance and safety of devices placed on the market; | |
| ● | improves the traceability of medical devices throughout the supply chain to the end-user or patient through a unique identification number; | |
| ● | sets up a central database to provide patients, healthcare professionals and the public with comprehensive information on products available in the EEA; and | |
| ● | strengthens rules for the assessment of certain high-risk devices which may have to undergo an additional check by experts before they are placed on the market. |
The EU MDR imposes a number of new requirements on manufacturers of medical devices. Notified bodies need to be accredited by the EU Member States’ accreditation bodies to conduct assessment procedures for medical devices in accordance with the EU MDR. There are currently a relatively small number of notified bodies that have been accredited to conduct these assessments and their capacity to deal with new applications is currently limited. In addition, the timeline to go through an EU MDR conformity assessment is substantially longer than under the previous Directive (currently between 13 and 18 months in average). This may delay conformity assessment procedures in the future in the EEA. This may impact any of our future activities in the EEA and the UK, the renewal of our existing CE Certificates of Conformity and conformity assessment related to future bodies.
Further, the EU MDR imposes increased compliance obligations for us to access the EEA market. Our failure to comply with applicable foreign regulatory requirements, including those administered by authorities of the EEA countries, could result in enforcement actions against us, including refusal, suspension, variation, or withdrawal of any CE Certificates of Conformity by the applicable EU notified body, which could impair our ability to market products in the EEA. Any changes to the membership of the EU, such as the departure of the United Kingdom (Brexit), may impact the regulatory requirements for the impacted countries and impair our business operations and our ability to market products in such countries.
Brexit has created significant uncertainty concerning the future relationship between the UK and the EU. On 24 December 2020, the EU and UK reached an agreement in principle on the framework for their future relationship, the EU-UK Trade and Cooperation Agreement (the “Trade Agreement”), which took effect on May 1, 2021. The Trade Agreement primarily focuses on ensuring free trade between the EU and the UK in relation to goods. The Trade Agreement does not however, specifically address medical devices. The Trade Agreement seeks to ensure that the parties ensure “regulatory cooperation”. Among the changes that will now occur are that Great Britain (England, Scotland and Wales) will be treated as a third country. Northern Ireland will, with regard to EU regulations, continue to follow the EU regulatory rules. In light of the fact that the CE marking process is set out in EU law, which no longer applies in the UK, the UK has devised a new route to market culminating in a UK Conformity Assessed (“UKCA”) mark to replace the CE mark. Northern Ireland will, however, continue to be covered by the regulations governing CE marks. As part of the Trade Agreement, the EU and the UK have agreed to continue to recognize declarations of conformity based on a self-assessment in the other territory. The UK Medical Device Regulations 2002 (SI 2002 No 618, as amended) (“UK MDR”) currently provide that the acceptance of CE marked medical devices on the Great Britain market will end on June 30, 2023. However, the UK government intends to put in place legislation to extend the acceptance of CE marked medical devices on the Great Britain market. Subject to Parliamentary approval, the UK government intends to introduce legislation before June 30, 2023 which will provide that medical devices CE marked under the EU MDR may be placed on the Great Britain market up until June 30, 2030. Given the lack of comparable precedent to Brexit, it is unclear what the financial, regulatory, and legal implications of Brexit will be and how it will affect us and our commercialization plans in Europe. However, potentially changing regulatory schemes and tariffs engendered by Brexit may add additional complexity, cost and delays in marketing or selling our products in the United Kingdom.
|
|
We are working on NTM lung infection which is very rare.
NTM lung infection is a very rare disease and only a small number of people suffer from this condition. As a result of these small numbers, we may not be able to complete the study related to NTM or, even if approved, the device for that indication may never be profitable.
We are working on bronchiolitis that usually is caused by the RSV virus and any related trials we conduct are dependent on a number of factors outside of our control, which may lead to varied trial results and possibly delays in our plans.
RSV is a seasonal virus (only in the winter). For any RSV related clinical trial that we pursue, we are heavily dependent on the occurrence and the severity of this virus. Treating for RSV is highly reliant on the weather conditions in winter. The weather in the winter is not predictable. For example, if the winter is warm or short, or the RSV infection was not severe enough when we conducted our trial, or the length of stay in the hospital at the year that trial was conducted was different from previous seasons, then we might miss the optimal trial season or the results can be significantly different between two seasons or between different countries or even between different sites. Due to these factors, it may take us longer than anticipated to obtain data from RSV related trials.
Our subsidiaries are exploring novel therapeutic processes with NO and once they put forth product candidates, those product candidates are likely to be classified as pharmaceutical drugs by the FDA; pharmaceutical regulation is more stringent than the standards to which our current approved product is subject, and as such, as our subsidiaries’ research and results expand, the Company’s regulatory and related costs will increase, which may affect our financial results, in particular, to the extent that these therapies remain investigative and do not lead to the outcomes anticipated.
We expect the novel nature of our subsidiaries’ product candidates to create challenges in obtaining regulatory approval, and we anticipate that they may likely be classified as drug product candidates based on the therapeutic processes being created. The FDA has limited experience with the commercial development of NO-related drug therapies for cancer and autism, respectively. Accordingly, the regulatory approval pathway for such future product candidates may be uncertain, and complex, in addition to being expensive and lengthy, and approval may not be obtained. Beyond Cancer’s research data has shown that UNO has anticancer properties and elicits an immune response from the host. Beyond Cancer utilizes an intratumoral UNO technology as a gas delivery of NO at high concentrations to tumors to induce an immune response. Gas based intratumoral therapies for the treatment of cancer are considered novel and new medical science. Beyond Cancer’s efforts are currently also focused on utilizing UNO in combination with Keytruda, a PD-1 inhibitor or other PD-1 or PDL-1 inhibitors as a treatment for cancers. To date, no such gas-based therapy has been approved for commercialization by the FDA or other regulatory agencies. As a result, the processes and requirements imposed by relevant regulatory authorities in multiple jurisdictions for these future pharmaceutical product candidates may cause delays and additional costs in obtaining approvals for marketing authorization. Likewise, pharmaceutical regulation as opposed to medical device regulation, requires different clearance standards than those the Company is currently subject to, in order to conduct business. As a result, once our subsidiaries go through initial preclinical research and expand into clinical trials, they are likely to face additional cost burdens in order to get a product candidate to market.
Clinical trials are necessary to support our future product submissions to the FDA and such trials involve regulatory complexity, are lengthy, , iterative and involve working with third parties, including CROs and patients for enrollment. These and other factors may affect our ability to complete clinical trials and may lead to delays or failures that would affect our business and financial prospects.
Initiating and completing clinical trials necessary to support any future PMAs, and additional safety and efficacy data beyond that typically required for a 510(k) clearance, for our possible future product candidates, will be time-consuming and expensive and the outcome uncertain. Moreover, the results of early clinical trials are not necessarily predictive of future results, and any product we advance into clinical trials may not have favorable results in later clinical trials. The results of preclinical studies and clinical trials of our products and product candidates conducted to date and ongoing or future studies and trials of our current, planned or future products may not be predictive of the results of later clinical trials, and interim results of a clinical trial do not necessarily predict final results. Our interpretation of data and results from our clinical trials do not ensure that we will achieve similar results in future clinical trials. In addition, preclinical and clinical data are often susceptible to various interpretations and analyses, and many companies that have believed their products performed satisfactorily in preclinical studies and earlier clinical trials have nonetheless failed to replicate results in later clinical trials. Products in later stages of clinical trials may fail to show the desired safety and efficacy despite having progressed through nonclinical studies and earlier clinical trials. Failure can occur at any stage of clinical testing. Our clinical trials may produce negative or inconclusive results, and we may decide, or regulators may require us, to conduct additional clinical and non-clinical testing in addition to those we have planned.
|
|
The initiation and completion of our clinical trials may be prevented, delayed, or halted for numerous reasons. We may experience delays in our ongoing clinical trials for a number of reasons, which could adversely affect the costs, timing or successful completion of our clinical trials, including related to the following:
| ● | we may be required to submit an IDE application to the FDA, which must become effective prior to commencing certain human clinical trials of medical devices, and the FDA may reject our IDE application and notify us that we may not begin clinical trials; | |
| ● | regulators and other comparable foreign regulatory authorities may disagree as to the design or implementation of our clinical trials; | |
| ● | regulators and/or an IRB, or other reviewing bodies may not authorize us or our investigators to commence a clinical trial, or to conduct or continue a clinical trial at a prospective or specific trial site; | |
| ● | we may not reach agreement on acceptable terms with prospective contract research organizations (“CROs”) and clinical trial sites, the terms of which can be subject to extensive negotiation and may vary significantly among different CROs and trial sites; | |
| ● | clinical trials may produce negative or inconclusive results, and we may decide, or regulators may require us, to conduct additional clinical trials or abandon product development programs; | |
| ● | the number of subjects or patients required for clinical trials may be larger than we anticipate, enrollment in these clinical trials may be insufficient or slower than we anticipate, and the number of clinical trials being conducted at any given time may be high and result in fewer available patients for any given clinical trial, or patients may drop out of these clinical trials at a higher rate than we anticipate; | |
| ● | our third-party contractors, including those manufacturing products or conducting clinical trials on our behalf, may fail to comply with regulatory requirements or meet their contractual obligations to us in a timely manner, or at all; | |
| ● | we might have to suspend or terminate clinical trials for various reasons, including a finding that the subjects are being exposed to unacceptable health risks; | |
| ● | we may have to amend clinical trial protocols or conduct additional studies to reflect changes in regulatory requirements or guidance, which we may be required to submit to an IRB and/or regulatory authorities for re-examination; | |
| ● | regulators, IRBs, or other parties may require or recommend that we or our investigators suspend or terminate clinical research for various reasons, including safety signals or noncompliance with regulatory requirements; | |
| ● | the cost of clinical trials may be greater than we anticipate; | |
| ● | clinical sites may not adhere to the clinical protocol or may drop out of a clinical trial; | |
| ● | we may be unable to recruit a sufficient number of clinical trial sites; | |
| ● | regulators, IRBs, or other reviewing bodies may fail to approve or subsequently find fault with our manufacturing processes or facilities of third-party manufacturers with which we enter into agreement for clinical and commercial supplies, the supply of devices or other materials necessary to conduct clinical trials may be insufficient, inadequate or not available at an acceptable cost, or we may experience interruptions in supply; | |
| ● | approval policies or regulations of the FDA or comparable foreign regulatory authorities may change in a manner rendering our clinical data insufficient for certification or approval; | |
| ● | our current or future products may have undesirable side effects or other unexpected characteristics; and | |
| ● | impacts of regional or global public health crises could adversely affect any clinical trials we are conducting or plan to conduct, including delays or difficulties in enrolling or onboarding patients, initiating clinical sites, or obtaining the requisite certification or regulatory approvals, interruption of key clinical trial activities, or supply chain disruptions that delay or make it more difficult or costly to obtain the supplies and materials we need for clinical trials. |
|
|
Any of these occurrences may significantly harm our business, financial condition and prospects. In addition, many of the factors that cause, or lead to, a delay in the commencement or completion of clinical trials may also ultimately lead to the denial of certification or regulatory approval of our product candidates.
Clinical trials must be conducted in accordance with the laws and regulations of the FDA and other comparable foreign regulatory authorities’ legal requirements, regulations or guidelines, and are subject to oversight by these governmental authorities and IRBs at the medical institutions where the clinical trials are conducted. Conducting successful clinical trials will require the enrollment of large numbers of patients, and suitable patients may be difficult to identify and recruit. Patient enrollment in clinical trials and completion of patient participation and follow-up depends on many factors, including the size of the patient population, the nature of the trial protocol, the attractiveness of, or the discomforts and risks associated with, the treatments received by enrolled subjects, the availability of appropriate clinical trial investigators, support staff, and proximity of patients to clinical sites and able to comply with the eligibility and exclusion criteria for participation in the clinical trial and patient compliance. For example, patients may be discouraged from enrolling in our clinical trials if the trial protocol requires them to undergo extensive post-treatment procedures or follow-up to assess the safety and effectiveness of our products or if they determine that the treatments received under the trial protocols are not attractive or involve unacceptable risks or discomforts.
We depend on our collaborators and on medical institutions and CROs to conduct our clinical trials in compliance with Good Clinical Practice (“GCP”) requirements. To the extent our collaborators or the CROs fail to enroll participants for our clinical trials, fail to conduct the study to GCP standards or are delayed for a significant time in the execution of trials, including achieving full enrollment, we may be affected by increased costs, program delays or both. In addition, clinical trials that are conducted in countries outside the United States may subject us to further delays and expenses as a result of increased shipment costs, additional regulatory requirements and the engagement of non-U.S. CROs, as well as expose us to risks associated with clinical investigators who are unknown to the FDA, and different standards of diagnosis, screening and medical care.
Development of sufficient and appropriate clinical protocols to demonstrate safety and efficacy are required and we may not adequately develop such protocols to support clearance and approval. Further, the FDA may require us to submit data on a greater number of patients than we originally anticipated and/or for a longer follow-up period or change the data collection requirements or data analysis applicable to our clinical trials. Delays in patient enrollment or failure of patients to continue to participate in a clinical trial may cause an increase in costs and delays in the approval and attempted commercialization of our products or result in the failure of the clinical trial. In addition, despite considerable time and expense invested in our clinical trials, the FDA may not consider our data adequate to demonstrate safety and efficacy. Such increased costs and delays or failures could adversely affect our business, operating results and prospects.
Even if our products are approved or cleared in the United States and CE marked in the EEA in the future, comparable regulatory authorities of additional foreign countries must also approve the manufacturing and marketing of our products in those countries, should we desire to access those markets. Certification, approval and clearance procedures vary among jurisdictions and can involve requirements and administrative review periods different from, and greater than, those in the United States or the EEA, including additional preclinical studies or clinical trials. Any of these occurrences may harm our business, financial condition and prospects significantly.
We may experience delays in obtaining a CE mark in the EEA for our approved product due to possible EU MDR regulatory classification that differs from the FDA’s regulatory paradigm.
In the EEA, we expect that, if approved, our products would be classified as a medical device. However, competent regulatory authorities in EEA countries or notified bodies could disagree and consider our products to be a drug-delivery combination product composed of a medical device and a medicinal product. In the EEA, drug-delivery systems can fall within the scope of the medical device legislation or the pharmaceutical legislation depending on their combination with the relevant medicinal substance.
If our device is considered as being intended to administer a medicinal product and our device and the medicinal product are placed on the market in such a way that they form a single integral product which is intended exclusively for use in the given combination and which is not reusable, that single integral product shall be governed by Directive 2001/83/EC and be subject to a marketing authorization. The medical device part of the drug-delivery combination product would not need to be CE marked. However, the relevant general safety and performance requirements set out in Annex I to the EU MDR would apply as far as the safety and performance of the device part of the single integral product are concerned. As a result, we would need to pursue different regulatory pathways for placing our product on the EEA market which may lead to additional costs and time.
|
|
We may find it difficult to enroll patients in our clinical trials. Difficulty in enrolling patients could delay or prevent clinical trials of our product candidates.
Identifying and qualifying patients to participate in clinical trials of our product candidates is critical to our success. The timing of our clinical trials depends in part on the speed at which we can recruit patients to participate in testing our product candidates, and we may experience delays in our clinical trials if we encounter difficulties in enrollment.
Some of the conditions for which we plan to evaluate our current product candidates are for rare diseases. For example, we estimate that 15,000 patients suffer from refractory NTM lung infection in the U.S. Accordingly, there is a limited patient pool from which to draw for clinical trials. Further, the eligibility criteria of our clinical trials will further limit the pool of available study participants as we will require that patients have specific characteristics that we can measure or to assure their disease is either severe enough or not too advanced to include them in a study.
Additionally, the process of finding patients may prove costly. We also may not be able to identify, recruit and enroll a sufficient number of patients to complete our clinical trials because of the perceived risks and benefits of the product candidate under study, particularly the toxicity of NO in certain doses, the availability and efficacy of competing therapies and clinical trials, the proximity and availability of clinical trial sites for prospective patients and the patient referral practices of physicians. If patients are unwilling to participate in our studies for any reason, the timeline for recruiting patients, conducting studies and obtaining certification or regulatory approval of potential products will be delayed.
If we experience delays in the completion or termination of any clinical trial of our product candidates, the commercial prospects of our product candidates will be harmed, and our ability to generate product revenue from any of these product candidates could be delayed or prevented. In addition, any delays in completing our clinical trials will increase our costs, slow down our product candidate development and certification or approval process and jeopardize our ability to commence product sales and generate revenue. Any of these occurrences may harm our business, financial condition and prospects significantly. In addition, many of the factors that cause, or lead to, a delay in the commencement or completion of clinical trials may also ultimately lead to the denial of certification or regulatory approval of our product candidates.
We may encounter substantial delays in our clinical trials, or we may fail to demonstrate safety and efficacy to the satisfaction of applicable regulatory authorities.
Before obtaining certification or marketing approval from regulatory authorities and notified bodies for the sale of our product candidates, we must conduct extensive clinical trials to demonstrate the safety and efficacy of the product candidates in humans. Clinical testing is expensive, time-consuming and uncertain as to outcome. We cannot guarantee that any clinical trials will be conducted as planned or completed on schedule, if at all. Our clinical trials involve infants, children, and adults and, before we are permitted to enroll them in clinical trials, we must demonstrate that although the research may pose a risk to the subjects, there is a prospect of direct benefit to each patient. We must do so to the satisfaction of each research site’s IRB. If we fail to adequately demonstrate this to the satisfaction of the relevant IRB, it will decline to approve the research, which could have significant adverse consequences for us.
|
|
A failure of one or more clinical trials can occur at any stage of testing, and our future clinical trials may not be successful. Events that may prevent successful or timely completion of clinical development include but are not limited to:
| ● | inability to generate sufficient preclinical, toxicology or other in vivo or in vitro data to support the initiation of human clinical trials; | |
| ● | delays in reaching a consensus with regulatory authorities on study design; | |
| ● | delays in reaching agreement on acceptable terms with prospective CROs and clinical trial sites, the terms of which can be subject to extensive negotiation and may vary significantly among different CROs and clinical trial sites; | |
| ● | delays in obtaining required IRB approval at each clinical trial site; | |
| ● | imposition of a clinical hold by regulatory authorities, after review of an IDE application or equivalent application, or an inspection of our clinical trial operations or study sites; | |
| ● | delays in recruiting suitable patients to participate in our clinical trials; | |
| ● | difficulty collaborating with patient groups and investigators; | |
| ● | failure by our CROs, other third parties or us to adhere to clinical trial requirements; | |
| ● | failure to perform in accordance with the FDA’s GCP requirements, or applicable regulatory guidelines in other foreign countries; | |
| ● | delays in having patients complete participation in a study or return for post-treatment follow-up; | |
| ● | occurrence of serious adverse events associated with the product candidate that are viewed to outweigh its potential benefits; | |
| ● | changes in regulatory requirements and guidance that require amending or submitting new clinical protocols; | |
| ● | the cost of clinical trials of our product candidates being greater than we anticipate; | |
| ● | clinical trials of our product candidates producing negative or inconclusive results, which may result in us deciding, or regulators requiring us, to conduct additional clinical trials or abandon product development programs; and | |
| ● | delays in manufacturing, testing, releasing, validating or importing/exporting sufficient stable quantities of our product candidates for use in clinical trials or the inability to do any of the foregoing. |
Any inability to successfully complete preclinical and clinical development could result in additional costs to us or impair our ability to generate revenue. We may also be required to conduct additional safety, efficacy and comparability studies before we will be allowed to start clinical trials. Clinical trial delays could also shorten any periods during which our products have patent protection and may allow our competitors to bring products to market before we do, which could impair our ability to successfully commercialize our product candidates and may harm our business and results of operations.
Our product candidates may cause undesirable side effects or have other properties that could delay or prevent their certification or regulatory approval, limit the commercial profile of an approved label or result in significant negative consequences following marketing approval, if any.
Undesirable side effects caused by our product candidates could cause us or regulatory authorities to interrupt, delay or halt clinical trials and could result in a more restrictive marketing label or the delay or denial of certification or regulatory approval by the FDA or other comparable foreign regulatory authorities. There is currently limited data regarding possible side effects for an antimicrobial dosage of NO treatments, such as our product candidates. Potential side effects of NO treatments may include high MetHb, NO2 toxicity, nose bleeding and low blood pressure. Results of our studies may identify unacceptable severity and prevalence of these or other side effects. In such an event, our studies could be suspended or terminated, and the FDA or comparable foreign regulatory authorities or notified bodies could order us to cease further development of or deny certification or approval of our product candidates for any or all targeted indications.
|
|
NO-related side effects could affect patient recruitment, the ability of enrolled patients to complete the study or result in potential product liability claims.
Additionally, if our product candidates receive certification or marketing approval, and we or others later identify undesirable side effects caused by such products, a number of potentially significant negative consequences could result, including but not limited to:
| ● | regulatory authorities and notified bodies may withdraw certification or approvals of such product; | |
| ● | regulatory authorities may require additional warnings on the label; | |
| ● | we could be sued and held liable for harm caused to patients; and | |
| ● | our reputation may suffer. |
Any of these events could prevent us from achieving or maintaining market acceptance of the particular product candidate, if approved, and could significantly harm our business, results of operations and prospects.
Risks Related to our Reliance on Third Parties
We rely on third parties to conduct our preclinical studies and clinical trials and perform other tasks for us. If these third parties do not successfully carry out their contractual duties, meet expected deadlines or comply with regulatory requirements, we may not be able to obtain certification or regulatory approval for or commercialize our approved product or product candidates and our business could be substantially harmed.
We have relied on and plan to continue to rely on third-party CROs to monitor and manage data for our ongoing preclinical and clinical programs. We rely on these parties for execution of our preclinical studies and clinical trials, and we directly control only certain aspects of their activities, although from a regulatory perspective we are responsible for their actions. We are responsible for ensuring that each of our studies is conducted in accordance with the applicable protocol, legal, regulatory and scientific standards and our reliance on the CROs does not relieve us of our regulatory responsibilities. We and our CROs and other vendors are required to comply with GCP, QSR and GLP, which are regulations and guidelines enforced by the FDA, the competent authorities of the EEA countries, and comparable foreign regulatory authorities for all of our product candidates in clinical development. Regulatory authorities enforce these regulations through periodic inspections of study sponsors, principal investigators, study sites and other contractors. If we or any of our CROs or vendors fail to comply with applicable regulations, the clinical data generated in our clinical trials may be deemed unreliable and the FDA or comparable foreign regulatory authorities may require us to perform additional clinical trials before approving our marketing applications. We cannot assure you that upon inspection by a given regulatory authority, such regulatory authority will determine that any of our clinical trials comply with GCP regulations. In addition, our clinical trials must be conducted with products that are produced under QSR regulations. Our failure to comply with these regulations may require us to repeat clinical trials, which would delay the certification or regulatory approval process, or have other adverse consequences.
If any of our relationships with these third-party CROs terminate, we may not be able to enter into arrangements with alternative CROs or do so on commercially reasonable terms. In addition, our CROs are not our employees, and except for remedies available to us under our agreements with such CROs, we cannot control whether they devote sufficient time and resources to our on-going clinical, nonclinical and preclinical programs. If CROs do not successfully carry out their contractual duties or obligations or meet expected deadlines, if they need to be replaced or if the quality or accuracy of the clinical data they obtain is compromised due to the failure to adhere to our clinical protocols, regulatory requirements or for other reasons, our clinical trials may be extended, delayed or terminated and we may not be able to obtain certification or regulatory approval for or successfully commercialize our approved product or product candidates. CROs may also generate higher costs than anticipated. As a consequence, our results of operations and the commercial prospects for our approved product or product candidates would be harmed, our costs could increase and our ability to generate revenue could be delayed.
Switching or adding additional CROs involves additional cost and requires management time and focus. In addition, there is a natural transition period when a new CRO commences work. As a result, delays may occur, which could materially impact our ability to meet our desired clinical development timelines. Though we carefully manage our relationships with our CROs, there can be no assurance that we will not encounter similar challenges or delays in the future or that these delays or challenges will not have a material adverse impact on our business, financial condition and prospects.
|
|
We rely on third parties to manufacture our NO generator and delivery system. Our business could be harmed if those third parties fail to provide us with sufficient quantities of our needed supplies, or fail to do so at acceptable quality levels or prices.
We do not currently have the infrastructure or capability internally to manufacture the components of our NO generator and delivery system, and we lack the resources and the capability to manufacture our approved product or any of our product candidates on a clinical or commercial scale. We rely on third parties for such supplies. There are a limited number of manufacturers who have the ability to produce our delivery system, and there may be a need to identify alternate manufacturers to prevent a possible disruption of our clinical trials. Any significant delay or discontinuity in the supply of these components could considerably delay commercialization of our approved product, completion of our clinical trials, product testing and potential certification or regulatory approval of our product candidates, which could harm our business and results of operations.
We and our collaborators and contract manufacturers are subject to significant regulation with respect to manufacturing our approved product or product candidates. The manufacturing facilities on which we rely may not continue to meet regulatory requirements and have limited capacity.
All entities involved in the preparation of medical devices for clinical trials or commercial sale, including our existing contract manufacturers for our approved product and product candidates, are subject to extensive regulation. Components of a finished medical device product approved for commercial sale or used in late-stage clinical trials must be manufactured in accordance with QSR in the U.S., and similar requirements in foreign countries. These regulations govern manufacturing processes and procedures (including record keeping) and the implementation and operation of quality systems to control and assure the quality of investigational products and products approved for sale. Poor control of production processes can lead to the introduction of contaminants or to inadvertent changes in the properties or stability of our approved product or product candidates that may not be detectable in final product testing. We, our collaborators or our contract manufacturers must supply all necessary documentation in support of any marketing application on a timely basis and must adhere to GLP and QSR regulations enforced by the FDA and comparable foreign regulatory authorities through their facilities inspection program. The facilities and quality systems of some or all of our collaborators and third-party contractors must pass a pre-approval inspection for compliance with the applicable regulations as a condition of certification or regulatory approval of our product candidates or any of our other potential products. In addition, the regulatory authorities may, at any time, audit or inspect a manufacturing facility involved with the preparation of our approved product, product candidates or our other potential products or the associated quality systems for compliance with the regulations applicable to the activities being conducted. We do not control the manufacturing process of, and are completely dependent on, our contract manufacturing partners for compliance with the regulatory requirements. If these facilities do not pass a pre-approval plant inspection, certification or regulatory approval of the products may not be granted or may be substantially delayed until any violations are corrected to the satisfaction of the regulatory authority, if ever.
The regulatory authorities also may, at any time following certification or approval of a product for sale, audit the manufacturing facilities of our collaborators and third-party contractors. If any such inspection or audit identifies a failure to comply with applicable regulations or if a violation of our product specifications or applicable regulations occurs independent of such an inspection or audit, we or the relevant regulatory authority may require remedial measures that may be costly and/or time-consuming for us or a third party to implement, and that may include the temporary or permanent suspension of a clinical trial or commercial sales, or the temporary or permanent closure of a facility. Any such remedial measures imposed upon us or third parties with whom we contract could materially harm our business. If we, our collaborators, or any of our third-party manufacturers fail to maintain regulatory compliance, the FDA or comparable foreign regulatory authorities can impose regulatory sanctions including, among other things, refuse to approve a pending application for a new product, withdrawal of a certification or approval, suspend production, suspend clinical trials, require a recall or suspension of production. As a result, our business, financial condition and results of operations may be materially harmed.
|
|
Additionally, if supply from one approved manufacturer is interrupted, an alternative manufacturer would need to be qualified through a PMA Supplement or Marketing Authorization Application amendment, or equivalent foreign regulatory filing, which could result in further delays. The regulatory authorities may also require additional studies if a new manufacturer is relied on for commercial production. Switching manufacturers may involve substantial costs and is likely to result in a delay in our desired clinical and commercial timelines.
These factors could cause us to incur higher costs and could cause the delay or termination of clinical trials, regulatory submissions, required certification or approvals or commercialization of our approved product or product candidates. Furthermore, if our suppliers fail to meet contractual requirements and we are unable to secure one or more replacement suppliers capable of production at a substantially equivalent cost, our clinical trials may be delayed or we could lose potential revenue.
If we encounter issues with our contract manufacturers or suppliers, we may need to qualify alternative manufacturers or suppliers, which could impair our ability to sufficiently and timely manufacture and supply LungFit® PH.
We currently depend on contract manufacturers and suppliers for LungFit® PH and its components. Although we could obtain each of these components from other third-party suppliers, we would need to qualify and obtain FDA approval for another contract manufacturer or supplier as an alternative source for each such component, which could be costly and cause significant delays. Each of our current commercial manufacturing and supply agreements include limitations on our ability to utilize alternative manufacturers or suppliers for these components above certain specified thresholds during the terms of the agreements, which impairs our ability to fully implement any future manufacturing strategies to prevent supply shortages or quality issues.
In addition, some of our suppliers and contract manufacturers, including Spartronics and Medisize, conduct their manufacturing operations for us at a single facility. Unless and until we qualify additional facilities, we may face limitations in our ability to respond to manufacturing and supply issues. For example, if regulatory, manufacturing or other problems require one of these manufacturers or suppliers to discontinue production at their respective facility, or if the equipment used for the production of LungFit® PH in these facilities is significantly damaged or destroyed by fire, flood, earthquake, power loss or similar events, the ability of such manufacturer or supplier to provide components needed for LungFit® PH, or to manufacture LungFit® PH may be significantly impaired. In the event that these parties suffer a temporary or protracted loss of its facility or equipment, we would still be required to obtain FDA approval to qualify a new manufacturer or supplier, as applicable, as an alternate manufacturer or source for the respective component before any components manufactured by such manufacturer or by such supplier could be sold or used.
Any production shortfall that impairs the supply of LungFit® PH or any of these components could have a material adverse effect on our business, financial condition and results of operations and adversely affect our ability to satisfy demand for LungFit® PH, which could adversely affect our product sales and operating results materially.
We depend on third-party manufacturers, including sole source suppliers, to manufacture LungFit® PH and our product candidates and the materials we require for our clinical trials. We may not be able to maintain these relationships and could experience supply disruptions outside of our control.
We rely on a network of third-party manufacturers to manufacture and supply LungFit® PH for commercial sale and post-certification/approval clinical trials, and our product candidates for clinical trials and any commercial sales if they are approved. As a result of our reliance on these third-party manufacturers and suppliers, including sole source suppliers of certain components of LungFit® PH and our product candidates, we could be subject to significant supply disruptions, in particular, should any issues occur with our sole source suppliers. Our supply chain for sourcing raw materials and manufacturing our products ready for distribution is a multi-step endeavor. In some cases, third-party contract manufacturers supply us with raw materials, and contract manufacturers in the United States convert these raw materials into substances from which we need to test our product’s final dosage. Establishing and managing this supply chain requires a significant financial commitment and the creation and maintenance of numerous third-party contractual relationships. Although we attempt to effectively manage the business relationships with companies in our supply chain, we do not have control over their operations.
|
|
We require a supply of LungFit® PH for sale in the United States, and we will require a supply of LungFit® PH for sale in international markets if we obtain certification or marketing approvals outside of the United States. We currently rely, and expect to continue to rely, on sole source third party manufacturers to produce starting materials, substance, and final product, and to package and label LungFit® PH and our product candidates. While we have identified and expect to qualify and engage back-up third party manufacturers as additional or alternative suppliers for the commercial supply of LungFit® PH, we currently do not have such arrangements in place. Moreover, some of these alternative manufacturers will have to be approved by the FDA before we can use them for manufacturing LungFit® PH. It is also possible that supplies of materials that cannot be second-sourced can be managed with inventory planning. There can be no assurance, however, that failure of any of our original sole source third-party manufacturers to meet our commercial demands for LungFit® PH in a timely manner, or our failure to engage qualified additional or back-up suppliers for the commercial supply of LungFit® PH, would not have a material adverse effect on commercialization of LungFit® and our business.
Supply disruptions may result from a number of factors, including shortages in product raw materials, labor or technical difficulties, regulatory inspections or restrictions, shipping or customs delays or any other performance failure by any third-party manufacturer on which we rely. Any supply disruptions could disrupt sales of LungFit® PH and/or the timing of our clinical trials, which could have a material adverse impact on our business. Furthermore, we may be required to modify our production methods to permit us to economically manufacture our product for sale and our product candidates for clinical trials. These modifications may require us to re-evaluate our resources and the resources of our third-party manufacturers, which could result in abrupt changes in our production methods and supplies.
In the course of providing its services, a contract manufacturer may develop process technology related to the manufacture of our products or product candidates that the manufacturer owns, either independently or jointly with us. This would increase our reliance on that manufacturer or require us to obtain a license from that manufacturer in order to have LungFit® PH or our product candidates manufactured by other suppliers utilizing the same process.
In the twelve months ended March 31, 2023, we Company purchased approximately 80% of our materials from two third-party vendors, with these vendors representing 67% and 13%, respectively. In the twelve months ended March 31, 2022, we had no significant supplier concentration.
The failure of our third-party manufacturers to meet our commercial demands for LungFit® PH in a timely manner, or our failure to engage qualified additional or back-up suppliers for the commercial supply of LungFit® PH, would have a material adverse effect on our business, results of operations and financial position.
Our reliance on third parties may require us to share our trade secrets, which increases the possibility that a competitor will discover them or that our trade secrets will be misappropriated or disclosed.
Because we rely on third parties to develop and manufacture LungFit® PH and our product candidates, we must, at times, share trade secrets with them. We seek to protect our proprietary technology in part by entering into confidentiality agreements and, if applicable, material transfer agreements, collaborative research agreements, consulting agreements or other similar agreements with our collaborators, advisors, employees and consultants prior to beginning research or disclosing proprietary information. These agreements typically limit the rights of the third parties to use or disclose our confidential information, such as trade secrets. Despite the contractual provisions employed when working with third parties, the need to share trade secrets and other confidential information increases the risk that such trade secrets become known by our competitors, are inadvertently incorporated into the technology of others, or are disclosed or used in violation of these agreements. Given that our proprietary position is based, in part, on our know-how and trade secrets, a competitor’s discovery of our trade secrets or other unauthorized use or disclosure would impair our competitive position and may have a material adverse effect on our business.
|
|
Risks Related to Our Intellectual Property
If we are unable to obtain and maintain effective patent rights for LungFit® PH, our product candidates or any future product candidates, we may not be able to compete effectively in our markets.
We rely on a combination of patents, trade secret protection and confidentiality agreements to protect the intellectual property related to our technologies, approved product and product candidates. Our success depends in large part on our and our licensors’ ability to obtain and maintain intellectual property protection in the U.S. and in other countries with respect to our proprietary technology and products.
We have sought to protect our proprietary position by filing patent applications in the U.S. and abroad related to our novel technologies and products that are important to our business. This process is expensive and time-consuming, and we may not be able to file and prosecute all necessary or desirable patent applications at a reasonable cost or in a timely manner. We may also fail to identify patentable aspects of our research and development output before it is too late to obtain patent protection.
The patent position of medical device, biotechnology and pharmaceutical companies generally is highly uncertain and involves complex legal and factual questions for which legal principles remain unsolved. The patent applications that we own or in-license may fail to result in issued patents with claims that cover our approved product or product candidates in the U.S. or in other foreign countries. There is no assurance that all potentially relevant prior art relating to our patents and patent applications has been found, which can invalidate a patent or prevent a patent from issuing from a pending patent application. Even if patents do successfully issue, and even if such patents cover our approved product or product candidates, third parties may challenge their validity, enforceability or scope, which may result in such patents being narrowed, found unenforceable or invalidated. Furthermore, even if they are unchallenged, our patents and patent applications may not adequately protect our intellectual property, provide exclusivity for our approved product or product candidates or prevent others from designing around our claims. Any of these outcomes could impair our ability to prevent competition from third parties, which may have an adverse impact on our business.
We have filed several patent applications directed to various aspects of our approved product and product candidates. We cannot offer any assurances about which, if any, patents will issue, the breadth of any such patent or whether any issued patents will be found invalid and unenforceable or will be threatened by third parties. Any successful opposition to these patents or any other patents owned by or licensed to us after patent issuance could deprive us of rights necessary for the successful commercialization of our approved product or any product candidates that we may develop. Further, if we encounter delays in certification or regulatory approvals, the period of time during which we could market a product candidate under patent protection could be reduced. In addition, some or all of our patent applications may not result in issued patents.
If we cannot obtain and maintain effective patent rights for our approved product or product candidates, we may not be able to compete effectively and our business and results of operations would be harmed.
Intellectual property rights of third parties could adversely affect our ability to commercialize our approved product or product candidates, and we might be required to litigate or obtain licenses from third parties in order to develop or market our approved product or product candidates. Such litigation or licenses could be costly or not available on commercially reasonable terms.
Given the number of companies developing various types of NO devices, it is difficult to conclusively assess our freedom to operate without infringing on third-party rights. There are numerous companies that have pending patent applications and issued patents in the field of therapeutic NO delivery. Our competitive position may suffer if patents issued to third parties or other third-party intellectual property rights cover our products or elements thereof, or our manufacture or uses relevant to our development plans. In such cases, we may not be in a position to develop or commercialize our approved product or product candidates unless we successfully pursue litigation to nullify or invalidate the third-party intellectual property right concerned, or enter into a license agreement with the intellectual property right holder, if available on commercially reasonable terms. There may be pending patent applications of which we are not aware, that if they result in issued patents, could be alleged to be infringed by our approved product or product candidates. If such an infringement claim should be brought and be successful, we may be required to pay substantial damages, be forced to abandon our approved product or product candidates or seek a license from any patent holders. No assurances can be given that a license will be available on commercially reasonable terms, if at all.
|
|
It is also possible that we have failed to identify relevant third-party patents or applications. Patent applications in the U.S. and elsewhere are published approximately 18 months after the earliest filing for which priority is claimed, with such earliest filing date being commonly referred to as the priority date. Therefore, patent applications covering our approved product, product candidates or platform technology could have been filed by others without our knowledge. Additionally, pending patent applications which have been published can, subject to certain limitations, be later amended in a manner that could cover our platform technologies, our approved product or product candidates or the use of our approved product or product candidates. Third-party intellectual property right holders may also actively bring infringement claims against us. We cannot guarantee that we will be able to successfully settle or otherwise resolve such infringement claims. If we are unable to successfully settle future claims on terms acceptable to us, we may be required to engage in or continue costly, unpredictable and time-consuming litigation and may be prevented from or experience substantial delays in pursuing the development of and/or marketing our approved product or product candidate. If we fail in any such dispute, in addition to being forced to pay damages, we may be temporarily or permanently prohibited from commercializing our approved product or product candidate that is held to be infringing. We might, if possible, also be forced to redesign our approved product or product candidate so that we no longer infringe the third-party intellectual property rights. Any of these events, even if we were ultimately to prevail, could require us to divert substantial financial and management resources that we would otherwise be able to devote to our business.
Patent terms are limited and we may not be able to effectively protect our products and business.
Patents have a limited lifespan. In the U.S., the natural expiration of a patent is generally 20 years after it is filed. Although various extensions may be available, the life of a patent, and the protection it affords, is limited.
In addition, upon issuance in the U.S., the patent term may be extended based on certain delays caused by the applicant(s) or the U.S. Patent and Trademark Office (“USPTO”). Even if we obtain effective patent rights for our approved product or product candidates, we may not have sufficient patent terms or regulatory exclusivity to protect our products, and our business and results of operations would be adversely affected.
Patent policy and rule changes could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents.
Changes in either the patent laws or interpretation of the patent laws in the U.S. and other countries may diminish the value of our patents or narrow the scope of our patent protection. The laws of foreign countries may not protect our rights to the same extent as the laws of the U.S. Publications of discoveries in the scientific literature often lag behind the actual discoveries, and patent applications in the U.S. and other jurisdictions are typically not published until 18 months after filing, or in some cases not at all. We therefore cannot be certain that we or our licensor were the first to make the invention claimed in our owned and licensed patents or pending applications, or that we or our licensor were the first to file for patent protection of such inventions. Assuming the other requirements for patentability are met, in the U.S. prior to March 15, 2013, the first to invent the claimed invention is entitled to the patent, while outside the U.S., the first to file a patent application is entitled to the patent. After March 15, 2013, under the Leahy-Smith America Invents Act (“Leahy-Smith Act”), enacted on September 16, 2011, the U.S. moved to a first-to-file system. The Leahy-Smith Act also includes a number of significant changes that affect the way patent applications will be prosecuted and may also affect patent litigation. The effects of these changes are currently unclear as the USPTO must still implement various regulations, the courts have yet to address these provisions and the applicability of the act and new regulations on specific patents discussed herein have not been determined and would need to be reviewed. In general, the Leahy-Smith Act and its implementation could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents, all of which could have a material adverse effect on our business and financial condition.
If we are unable to maintain effective proprietary rights for our approved product, product candidates or any future product candidates, we may not be able to compete effectively in our markets.
In addition to the protection afforded by patents, we rely on trade secret protection and confidentiality agreements to protect proprietary know-how that is not patentable or that we elect not to patent, processes for which patents are difficult to enforce and any other elements of our product candidate discovery and development processes that involve proprietary know-how, information or technology that is not covered by patents. However, trade secrets can be difficult to protect. We seek to protect our proprietary technology and processes, in part, by entering into confidentiality agreements with our employees, consultants, scientific advisors, vendors, collaborators and contractors. We also seek to preserve the integrity and confidentiality of our data and trade secrets by maintaining physical security of our premises and physical and electronic security of our information technology systems. While we have confidence in these individuals, organizations and systems, agreements or security measures may be breached, and we may not have adequate remedies for any breach. In addition, our trade secrets may otherwise become known or be independently discovered by competitors.
|
|
All of our employees, consultants, advisors and any third parties who have access to our proprietary know-how, information or technology enter into confidentiality agreements and we expect they will assign all rights in their inventions to us pursuant to the terms of such agreements; however, we cannot provide any assurances that all such agreements have been duly executed or that our trade secrets and other confidential proprietary information will not be disclosed or that competitors will not otherwise gain access to our trade secrets or independently develop substantially equivalent information and techniques. Misappropriation or unauthorized disclosure of our trade secrets could impair our competitive position and may have a material adverse effect on our business. Additionally, if the steps taken to maintain our trade secrets are deemed inadequate, we may have insufficient recourse against third parties for misappropriating the trade secret.
Third-party claims of intellectual property infringement may prevent or delay our development and commercialization efforts.
Our commercial success depends in part on our avoiding infringement of the patents and proprietary rights of third parties. There have been many lawsuits and other proceedings involving patent and other intellectual property rights in the biotechnology and pharmaceutical industries, including with respect to NO delivery systems and formulations, including patent infringement lawsuits, interferences, oppositions and reexamination proceedings before the USPTO and corresponding foreign patent offices. Numerous U.S. and foreign issued patents and pending patent applications, which are owned by third parties, exist in the fields in which we are developing product candidates. As the biotechnology and pharmaceutical industries expand and more patents are issued, the risk increases that our approved product or product candidates may be subject to claims of infringement of the patent rights of third parties.
Third parties may assert that we are employing their proprietary technology without authorization. There may be third-party patents or patent applications with claims to materials, formulations, methods of manufacture or methods for treatment related to the use or manufacture of our approved product or product candidates. We do not know whether there are any third-party patents that would impair our ability to commercialize our approved product or such product candidates. We also cannot be sure that we have identified each and every patent and pending patent application in the U.S. and abroad that is relevant or necessary to the commercialization of our approved product and product candidates. Because patent applications can take many years to issue, there may be currently pending patent applications that may later result in issued patents that our approved product or product candidates may infringe. In addition, third parties may obtain patents in the future and claim that use of our technologies infringes upon these patents. If any third-party patents were held by a court of competent jurisdiction to cover the manufacturing process of our approved product or any of our product candidates, any molecules formed during the manufacturing process or any final product itself, the holders of any such patents may be able to block our ability to continue commercializing our approved product or such product candidates unless we obtained a license under the applicable patents, or until such patents expire or are finally determined to be invalid or unenforceable.
Similarly, if any third-party patents were held by a court of competent jurisdiction to cover aspects of our formulations, processes for manufacture or methods of use, the holders of any such patents may be able to block our ability to develop and commercialize our approved product or the applicable product candidate unless we obtained a license or until such patent expires or is finally determined to be invalid or unenforceable. In either case, such a license may not be available on commercially reasonable terms or at all.
Parties making claims against us may obtain injunctive or other equitable relief, which could effectively block our ability to further develop and further commercialize our approved product or one or more of our product candidates. Defense of these claims, regardless of their merit, would involve substantial litigation expense and would be a substantial diversion of employee resources from our business. In the event of a successful claim of infringement against us, we may have to pay substantial damages, including treble damages and attorneys’ fees for willful infringement, pay royalties, redesign our “infringing” products or obtain one or more licenses from third parties, which may be impossible or require substantial time and monetary expenditure.
|
|
We may not be successful in obtaining or maintaining necessary rights to our approved product or product candidates through acquisitions and in-licenses.
We currently own and have in-licensed rights to intellectual property through licenses from third parties and under patents that we own, to develop our approved product and product candidates. Because our programs may require the use of proprietary rights held by third parties, the growth of our business will likely depend in part on our ability to acquire, in-license or use these proprietary rights. In addition, our approved product or product candidates may require specific formulations to work effectively and efficiently and the rights to these formulations may be held by others. We may be unable to acquire or in-license any compositions, methods of use, processes or other third-party intellectual property rights from third parties that we identify as necessary for our approved product or product candidates. The licensing and acquisition of third-party intellectual property rights is a competitive area, and a number of more established companies are also pursuing strategies to license or acquire third-party intellectual property rights that we may consider attractive. These established companies may have a competitive advantage over us due to their size, cash resources and greater clinical development and commercialization capabilities.
For example, we sometimes collaborate with U.S. and foreign academic institutions to accelerate our preclinical research or development underwritten agreements with these institutions. Typically, these institutions provide us with an option to negotiate a license to any of the institution’s rights in technology resulting from the collaboration. Regardless of such option, we may be unable to negotiate a license within the specified timeframe or under terms that are acceptable to us. If we are unable to do so, the institution may offer the intellectual property rights to other parties, potentially blocking our ability to pursue our program.
In addition, companies that perceive us to be a competitor may be unwilling to assign or license rights to us. We also may be unable to license or acquire third-party intellectual property rights on terms that would allow us to make an appropriate return on our investment. If we are unable to successfully obtain rights to required third-party intellectual property rights, we may have to abandon development of that program and our business and financial condition could suffer.
If we fail to comply with our obligations in the agreements under which we license intellectual property and other rights from third parties or otherwise experience disruptions to our business relationships with our licensors, we could lose license rights that are important to our business.
We are party to intellectual property license agreements that are important to our business, and we may enter into additional license agreements in the future. Our existing license agreements impose, and we expect that future license agreements will impose, various diligence, milestone payment, royalty and other obligations on us.
Licensing of intellectual property is of critical importance to our business and involves complex legal, business and scientific issues. Disputes may arise regarding intellectual property subject to a licensing agreement, including but not limited to:
| ● | the scope of rights granted under the license agreement and other interpretation-related issues; | |
| ● | the extent to which our technology and processes infringe on intellectual property of the licensor that is not subject to the licensing agreement; | |
| ● | the sublicensing of patent and other rights; | |
| ● | our diligence obligations under the license agreement and what activities satisfy those diligence obligations; | |
| ● | the ownership of inventions and know-how resulting from the joint creation or use of intellectual property by our licensors and us and our collaborators; and | |
| ● | the priority of invention of patented technology. |
If disputes over intellectual property and other rights that we have licensed prevent or impair our ability to maintain our current licensing arrangements on acceptable terms, we may be unable to successfully develop and commercialize the affected approved product or product candidates and this may affect our financial performance.
|
|
We may be involved in lawsuits or post-grant proceedings to protect or enforce our patents or the patents of our licensor, which could be expensive, time-consuming and unsuccessful.
We may face risk if our competitors infringe the patents of any licensor with whom we may be involved. If any such licensing partner were to initiate legal proceedings against a third party to enforce a patent covering one of our product candidates, the defendant could counterclaim that the patent covering our product candidate is invalid and/or unenforceable. In patent litigation in the U.S., defendant counterclaims alleging invalidity and/or unenforceability are commonplace. Grounds for a validity challenge could be an alleged failure to meet any of several statutory requirements, including lack of novelty, obviousness or non-enablement. Grounds for an unenforceability assertion could be an allegation that someone connected with prosecution of the patent withheld relevant information from the USPTO, or made a misleading statement, during prosecution. The outcome following legal assertions of invalidity and unenforceability is unpredictable.
Pending patent applications may be subject to third-party pre-issuance submission of prior art to the USPTO, and any patents issuing thereon may become involved in derivation, reexamination, inter parties review, post grant review, interference proceedings or other patent office proceedings in the U.S. challenging our patent rights.
Proceedings provoked by third parties or brought by us or declared by the USPTO may be necessary to determine the priority of inventions with respect to our patents or patent applications or those of our licensor. An unfavorable outcome could require us to cease using the related technology or to attempt to license rights to it from the prevailing party. Our business could be harmed if the prevailing party does not offer us a license on commercially reasonable terms. Our defense of litigation or proceedings may fail and, even if successful, may result in substantial costs and distract our management and other employees. In addition, the uncertainties associated with litigation could have a material adverse effect on our ability to raise the funds necessary to continue our clinical trials, continue our research programs, license necessary technology from third parties or enter into development partnerships that would help us bring our approved product and product candidates to market.
Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation. There could also be public announcements of the results of hearings, motions or other interim proceedings or developments. If securities analysts or investors perceive these results to be negative, it could have a material adverse effect on the price of our common stock.
We may be subject to claims that our employees, consultants or independent contractors have wrongfully used or disclosed confidential information of third parties or that our employees have wrongfully used or disclosed alleged trade secrets of their former employers.
We employ individuals who were previously employed at universities or other biotechnology or pharmaceutical companies, including our competitors or potential competitors. Although we try to ensure that our employees, consultants and independent contractors do not use the proprietary information or know-how of others in their work for us, we may be subject to claims that we or our employees, consultants or independent contractors have inadvertently or otherwise used or disclosed intellectual property, including trade secrets or other proprietary information, of any of our employee’s former employer or other third parties. Litigation may be necessary to defend against these claims. If we fail in defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel, which could adversely impact our business. Even if we are successful in defending against such claims, litigation could result in substantial costs and be a distraction to management and other employees.
|
|
We may be subject to claims challenging the inventorship of our patents and other intellectual property.
We may be subject to claims that former employees, collaborators or other third parties have an interest in or right to compensation with respect to our patents or other intellectual property as an inventor or co-inventor. For example, we may have inventorship disputes arise from conflicting obligations of consultants or others who are involved in developing our approved product or product candidates. Litigation may be necessary to defend against these and other claims challenging inventorship or claiming the right to compensation. If we fail in defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights, such as exclusive ownership of, or right to use, valuable intellectual property. Such an outcome could have a material adverse effect on our business. Even if we are successful in defending against such claims, litigation could result in substantial costs and be a distraction to management and other employees. To the extent that our employees have not effectively waived the right to compensation with respect to inventions that they helped create, they may be able to assert claims for compensation with respect to our future revenue may be successful. As a result, we may receive less revenue from future products if such claims are successful which in turn could impact our future profitability.
Changes in U.S. patent law could diminish the value of patents in general, thereby impairing our ability to protect our products.
As is the case with other biopharmaceutical companies, our success is heavily dependent on intellectual property, particularly patents. Obtaining and enforcing patents in the biotechnology industry involves both technological and legal complexity. Therefore, obtaining and enforcing biotechnology patents is costly, time-consuming and inherently uncertain. In addition, the U.S. has recently enacted and is currently implementing wide-ranging patent reform legislation. Recent U.S. Supreme Court rulings have narrowed the scope of patent protection available in certain circumstances and weakened the rights of patent owners in certain situations. In addition to increasing uncertainty with regard to our ability to obtain patents in the future, this combination of events has created uncertainty with respect to the value of patents, once obtained. Depending on future actions by the U.S. Congress, the federal courts and the USPTO, the laws and regulations governing patents could change in unpredictable ways that would weaken our ability to obtain new patents or to enforce our existing patents and patents that we might obtain in the future.
We may not be able to protect our intellectual property rights throughout the world.
Filing, prosecuting and defending patents on our approved product or product candidates in all countries throughout the world would be prohibitively expensive, and our intellectual property rights in some countries outside the U.S. can be less extensive than those in the U.S. In addition, the laws of some foreign countries do not protect intellectual property rights to the same extent as federal and state laws in the U.S.
Competitors may use our technologies in jurisdictions where we have not obtained patent protection to develop their own products and may also export otherwise infringing products to territories where we have patent protection, but enforcement is not as strong as that in the U.S. These products may compete with our products and our patents or other intellectual property rights may not be effective or sufficient to prevent them from competing.
Many companies have encountered significant problems in protecting and defending intellectual property rights in foreign jurisdictions. The legal systems of certain countries, particularly certain developing countries, do not favor the enforcement of patents, trade secrets and other intellectual property protection, particularly those relating to biotechnology products, which could make it difficult for us to stop the infringement of our patents or marketing of competing products in violation of our proprietary rights generally. Proceedings to enforce our patent rights in foreign jurisdictions, whether or not successful, could result in substantial costs and divert our efforts and attention from other aspects of our business, could put our patents at risk of being invalidated or interpreted narrowly and our patent applications at risk of not issuing and could provoke third parties to assert claims against us. We may not prevail in any lawsuits that we initiate and the damages or other remedies awarded, if any, may not be commercially meaningful. Accordingly, our efforts to enforce our intellectual property rights around the world may be inadequate to obtain a significant commercial advantage from the intellectual property that we develop or license.
|
|
Risks Related to Our Business Operations
We manage our business through a small number of employees and key consultants.
As of March 31, 2023, we had a total of 98 full-time employees between us and our subsidiaries and a number of dedicated consultants, all of whom work for us on a part-time basis. In addition, any of our employees and consultants may leave the Company at any time, subject to certain notice periods. The loss of the services of any of our executive officers or any key employees or consultants would adversely affect our ability to execute our business plan and harm our operating results.
We do not currently carry “key person” insurance on the lives of members of management.
We may need to expand our organization and we may experience difficulties in recruiting needed additional employees and consultants, which could disrupt our operations.
As our development and commercialization plans and strategies develop, we may need additional managerial, operational, sales, marketing, financial, legal and other resources. The competition for qualified personnel in the life sciences field is intense. Due to this intense competition, we may be unable to attract and retain qualified personnel necessary for the development of our business or to recruit suitable replacement personnel. We may experience difficulty retaining and motivating existing employees and attracting qualified personnel to fill key positions. In addition, labor shortages and employee mobility may make it more difficult to hire and retain employees.
Our management may need to divert a disproportionate amount of its attention away from our day-to-day activities and devote a substantial amount of time to managing these growth activities. We may not be able to effectively manage the expansion of our operations, which may result in weaknesses in our infrastructure, operational mistakes, loss of business opportunities, loss of employees and reduced productivity among remaining employees. Our expected growth could require significant capital expenditures and may divert financial resources from other projects, such as the development of additional product candidates. If our management is unable to effectively manage our growth, our expenses may increase more than expected, our ability to generate and/or grow revenue could be reduced and we may not be able to implement our business strategy. Our future financial performance and our ability to commercialize our approved product and compete effectively will depend, in part, on our ability to effectively manage any future growth.
Our international operations and plans to expand such operations present challenges and risks related to doing business internationally.
International expansion of our business further exposes us to business, regulatory, political, operational, financial and economic risks associated with doing business outside of the U.S., the EEA or Israel.
Other than our operations that are located in the EEA and Israel (as further described below), we currently have limited international operations, but our business strategy incorporates potentially significant international expansion, particularly in anticipation of certification or regulatory approval of our product candidates. We plan to maintain non-commercial infrastructure and conduct physician and patient association outreach activities, as well as clinical trials, outside of the U.S., the EEA and Israel. Doing business internationally involves a number of risks, including but not limited to:
| ● | multiple, conflicting and changing laws and regulations such as privacy regulations, tax laws, export and import restrictions, employment laws, regulatory requirements and other governmental certification, approvals, permits and licenses; | |
| ● | failure by us to obtain certification or regulatory approvals for the use of our products in various countries; | |
| ● | additional potentially relevant third-party patent rights; | |
| ● | complexities and difficulties in obtaining protection and enforcing our intellectual property; | |
| ● | difficulties in staffing and managing foreign operations; | |
| ● | complexities associated with managing multiple payor reimbursement regimes, government payors or patient self-pay systems; | |
| ● | limits on our ability to penetrate international markets; |
|
|
| ● | financial risks, such as longer payment cycles, difficulty collecting accounts receivable, the impact of local and regional financial crises on demand and payment for our products and exposure to foreign currency exchange rate fluctuations; | |
| ● | natural disasters, political and economic instability, including wars, terrorism and political unrest, outbreak of disease, boycotts, curtailment of trade and other business restrictions; | |
| ● | certain expenses including, among others, expenses for travel, translation and insurance; and | |
| ● | regulatory and compliance risks that relate to maintaining accurate information and control over sales and activities that may fall within the purview of the FCPA, its books and records provisions or its anti-bribery provisions. |
Any of these factors could significantly harm our future international expansion and operations and, consequently, our results of operations.
The use of any of our products could result in product liability or similar claims that could be expensive, damage our reputation and harm our business.
Our business exposes us to an inherent risk of potential product liability or similar claims. The medical device industry has historically been litigious, and we face financial exposure to product liability or similar claims if the use of any of our products were to cause or contribute to injury or death. There is also the possibility that defects in the design or manufacture of any of our products might necessitate a product recall. Although we maintain product liability insurance, the coverage limits of these policies may not be adequate to cover future claims. In the future, we may be unable to maintain product liability insurance on acceptable terms or at reasonable costs and such insurance may not provide us with adequate coverage against potential liabilities. A product liability claim, regardless of merit or ultimate outcome, or any product recall could result in substantial costs to us, damage to our reputation, customer dissatisfaction and frustration and a substantial diversion of management attention. A successful claim brought against us in excess of, or outside of, our insurance coverage could have a material adverse effect on our business, financial condition and results of operations.
Natural disasters, geopolitical unrest, war, terrorism, public health issues or other catastrophic events could disrupt the supply, delivery or demand of products, which could negatively affect our operations and performance.
We are subject to the risk of disruption by earthquakes, floods and other natural disasters, fire, power shortages, geopolitical unrest, war, terrorist attacks and other hostile acts, public health issues, epidemics or pandemics and other events beyond our control and the control of the third parties on which we depend. Any of these catastrophic events, whether in the United States, Europe or abroad, may have a strong negative impact on the global economy, our employees, facilities, partners, suppliers, distributors or customers, and could decrease demand for our products, create delays and inefficiencies in our supply chain and make it difficult or impossible for us to deliver products to our customers.
We may continue to face business disruption and related risks resulting from the COVID-19 pandemic, which continue to impact our business plans.
COVID-19 impacted our business plans and ability to conduct preclinical studies and clinical trials as well as on our reliance on third-party manufacturing and our supply chain. The Company experienced significant delays in the supply chain for LungFit® PH due to the redundancy in parts and suppliers for ventilator manufacturing, which has since been remedied. The development of our product candidates and the commercialization of our approved product could be further disrupted and adversely affected by a resurgence of the COVID-19 pandemic. We continuously assess the impact that COVID-19 may have on our business plans and our ability to conduct preclinical studies and clinical trials as well as on our reliance on third-party manufacturing and our supply chain. There can be no assurance that we will be able to avoid part or all of any impact from COVID-19 or its consequences if a resurgence occurs. The extent to which the effects of COVID-19 will have on our operations will depend on future developments outside of our control.
We are dependent on information technology and our systems and infrastructure face certain risks, including from cybersecurity breaches and data leakage.
We rely to a large extent upon sophisticated information technology systems to operate our businesses, some of which are managed, hosted provided and/or used for third parties or their vendors. We may collect, store and transmit large amounts of confidential information (including personal information and pseudonymized information), and we deploy and operate an array of technical and procedural controls to maintain the confidentiality and integrity of such confidential information. A significant breakdown, invasion, corruption, destruction, interruption, or unavailability of critical information technology systems or infrastructure, by our workforce, others with authorized access to our systems or unauthorized persons could negatively impact operations. Hardware, software, or applications we develop or obtain from third parties may contain defects in design or manufacture or other supply chain problems that could unexpectedly compromise our information and network security.
The ever-increasing use and evolution of technology, including cloud-based computing, creates opportunities for the unintentional dissemination or intentional destruction of confidential information stored in our or our third-party providers’ systems, portable media or storage devices. We could also experience a business interruption, theft of confidential information or reputational damage from industrial espionage attacks, malware or other cyber-attacks (including ransomware), which may compromise our system infrastructure or lead to data leakage, either internally or at our third-party providers. While we have invested in the protection of data and information technology, there can be no assurance that our efforts will prevent service interruptions or security breaches. Any such interruption or breach of our systems could adversely affect our business operations and/or result in the loss of critical or sensitive confidential information or intellectual property, and could result in financial, legal, business and reputational harm to us. In addition, as the regulatory environment related to information security, data collection and use, and privacy becomes increasingly rigorous, with new and constantly changing requirements applicable to our business, compliance with those requirements could also result in additional costs.
|
|
Risks Related to the Ownership of our Common Stock
Our amended and restated certificate of incorporation provides that the Court of Chancery of the State of Delaware will be the exclusive forum for substantially all disputes between us and our stockholders, which could limit our stockholders’ ability to obtain a favorable judicial forum for disputes with us or our directors, officers or employees.
Our certificate of incorporation provides that the Court of Chancery of the State of Delaware is the exclusive forum for (A) any derivative action or proceeding brought on behalf of us; (B) any action asserting a claim of breach of a fiduciary duty owed by any of our directors, officers or other employees to us or our stockholders; (C) any action asserting a claim against us arising pursuant to any provision of the Delaware General Corporation Law, our Amended and Restated Certificate of Incorporation or our Bylaws; or (D) any action asserting a claim against us governed by the internal affairs doctrine. Section 27 of the Exchange Act creates exclusive federal jurisdiction over all suits brought to enforce any duty or liability created by the Exchange Act or the rules and regulations thereunder. As a result, the exclusive forum provision will not apply to suits brought to enforce any duty or liability created by the Exchange Act or any other claim for which the federal courts have exclusive jurisdiction. In addition, Section 22 of the Securities Act creates concurrent jurisdiction for federal and state courts over all suits brought to enforce any duty or liability created by the Securities Act or the rules and regulations thereunder. As a result, the exclusive forum provision will not apply to suits brought to enforce any duty or liability created by the Securities Act or any other claim for which the federal and state courts have concurrent jurisdiction.
The choice of forum provision may limit a stockholder’s ability to bring a claim in a judicial forum that it finds favorable for disputes with us or our directors, officers or other employees, which may discourage such lawsuits against us and our directors, officers and other employees. Alternatively, if a court were to find the choice of forum provision contained in our certificate of incorporation to be inapplicable or unenforceable in an action, we may incur additional costs associated with resolving such action in other jurisdictions, which could adversely affect our business and financial condition.
Trading in our common stock has been volatile and may continue to be volatile in the future.
The stock market in general has experienced extreme price and volume fluctuations. The market prices of the securities of biotechnology and specialty pharmaceutical companies, particularly companies like ours without product revenues and earnings, have been highly volatile and may continue to be highly volatile in the future. This volatility has often been unrelated to the operating performance of particular companies.
The following factors, in addition to other risk factors described in this section, may have a significant impact on the market price of our common stock:
● announcements of technological innovations or new products by us or our competitors;
● announcement of FDA approval, disapproval or delay of approval of our product candidates or other product-related actions;
● developments involving our discovery efforts and clinical trials;
● developments or disputes concerning patents or proprietary rights, including announcements of infringement, interference or other litigation against us or our potential licensees;
● developments involving our efforts to commercialize our products, including developments impacting the timing of commercialization;
● announcements concerning our competitors, or the biotechnology, pharmaceutical or drug delivery industry in general;
● public concerns as to the safety or efficacy of our approved product or product candidates or our competitors’ products;
● changes in government regulation of the pharmaceutical or medical industry;
● changes in the reimbursement policies of third-party insurance companies or government agencies;
● actual or anticipated fluctuations in our operating results;
● changes in financial estimates or recommendations by securities analysts;
● developments involving corporate collaborators, if any;
● changes in accounting principles; and
● the loss of any of our key scientific or management personnel.
In the past, securities class action litigation has often been brought against companies that experience volatility in the market price of their securities. Whether or not meritorious, litigation brought against us could result in substantial costs and a diversion of management’s attention and resources, which could adversely affect our business, operating results and financial condition.
We cannot assure you that our common stock price and volume will remain at current levels in which case investors may sustain large losses.
In addition, the stock market in general, and the stocks of small-cap biotechnology companies in particular, have experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of these companies. Broad market and industry factors may negatively affect the market price of our common stock, regardless of our actual operating performance. The realization of any of the above risks or any of a broad range of other risks, including those described in these “Risk Factors,” could have a dramatic and material adverse impact on the market price of our common stock.
|
|
Anti-takeover provisions in our amended and restated certificate of incorporation and our amended and restated bylaws, as well as provisions of Delaware law, might discourage, delay or prevent a change in control of the Company or changes in our Board of Directors or management and, therefore, depress the trading price of our common stock.
Our amended and restated certificate of incorporation, amended and restated bylaws and Delaware law contain provisions that may depress the market price of our common stock by acting to discourage, delay or prevent a merger, acquisition or other change in control that stockholders may consider favorable, including transactions in which you might otherwise receive a premium for your shares of our common stock. These provisions may also prevent or frustrate attempts by our stockholders to replace or remove members of our Board of Directors or our management. Our corporate governance documents include provisions:
| ● | providing that directors may be removed by stockholders with or without cause; | |
| ● | limiting the ability of our stockholders to call and bring business before special meetings and to take action by written consent in lieu of a meeting; | |
| ● | requiring advance notice of stockholder proposals for business to be conducted at meetings of our stockholders and for nominations of candidates for election to our Board of Directors; | |
| ● | authorizing blank check preferred stock, which could be issued with voting, liquidation, dividend and other rights superior to our common stock; and | |
| ● | limiting the liability of, and providing indemnification to, our directors and officers. |
As a Delaware corporation, we are also subject to provisions of Delaware law, including Section 203 of the Delaware General Corporation Law, which limits the ability of stockholders owning in excess of 15% of our outstanding voting stock from engaging in certain business combinations with us. Any provision of our amended and restated certificate of incorporation, amended and restated bylaws or Delaware law that has the effect of delaying or deterring a change in control could limit the opportunity for our stockholders to receive a premium for their shares of our common stock, and could also affect the price that some investors are willing to pay for our common stock.
The existence of the foregoing provisions and anti-takeover measures could limit the price that investors might be willing to pay in the future for shares of our common stock. They could also deter potential acquirers of the Company, thereby reducing the likelihood that you could receive a premium for your common stock in an acquisition.
Risks Related to Employee Matters
Our business could suffer if we lose the services of key members of our senior management, key advisors or personnel.
We are dependent upon the continued services of key members of our senior management and a limited number of key advisors and personnel. The loss of any one of these individuals could disrupt our operations or our strategic plans. Additionally, our future success will depend on, among other things, our ability to continue to hire and retain the necessary qualified scientific, technical and managerial personnel, for whom we compete with numerous other companies, academic institutions and organizations. The loss of members of our management team, key advisors or personnel, or our inability to attract or retain other qualified personnel or advisors, could have a material adverse effect on our business, results of operations and financial condition. Though members of our sales force generally enter into noncompetition agreements that restrict their ability to compete with us, most of the members of our executive management team are not subject to such agreements. Accordingly, the adverse effect resulting from the loss of certain executives could be compounded by our inability to prevent them from competing with us.
Our employees may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements and insider trading.
We are exposed to the risk of employee fraud or other misconduct. Misconduct by employees could include intentional failures to comply with FDA regulations, to provide accurate information to the FDA, to comply with federal and state healthcare fraud and abuse laws and regulations, to report financial information or data accurately, to disclose unauthorized activities to us or to comply with our code of business conduct and ethics. In particular, sales, marketing and business arrangements in the healthcare industry are subject to extensive laws and regulations intended to prevent fraud, kickbacks, false claims, inappropriate promotion, self-dealing and other abusive practices. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, customer incentive programs and other business arrangements. Employee misconduct could also involve the improper use of information obtained in the course of clinical trials, which could result in regulatory sanctions and serious harm to our reputation. The precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to be in compliance with such laws or regulations. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition of significant fines or other sanctions.
|
|
We are subject to applicable fraud and abuse laws, including anti-kickback and false claims, transparency, health information privacy and security and other healthcare laws. Failure to comply with such laws may result in substantial penalties.
We are subject to broadly applicable healthcare laws and regulations that may constrain the business or financial arrangements and relationships through which we conduct research, market, sell and distribute any product candidates for which we obtain marketing approval. The healthcare laws that may affect us include: the federal fraud and abuse laws, including the federal anti-kickback, and false claims and civil monetary penalties laws; federal data privacy and security laws; and federal transparency laws related to ownership and investment interests and payments and/or other transfers of value made to or held by physicians (including doctors, dentists, optometrists, podiatrists and chiropractors) and teaching hospitals and, information regarding payments and transfers of value provided to and other healthcare professionals during the previous year. In addition, many states have similar laws and regulations that may differ from each other and federal law in significant ways, thus complicating compliance efforts. Moreover, several states require biopharmaceutical companies to comply with the biopharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government and may require medical device manufacturers to report information related to payments and other transfers of value to physicians and other healthcare providers or marketing expenditures. Additionally, some state and local laws require the registration of biopharmaceutical sales representatives in the jurisdiction.
Ensuring that our operations and future business arrangements with third parties comply with applicable healthcare laws and regulations will involve substantial costs. It is possible that governmental authorities will conclude that our business practices, including our relationships with physicians and other healthcare providers, some of whom are compensated in the form of stock options for consulting services provided, may not comply with current or future statutes, regulations, agency guidance or case law involving applicable fraud and abuse or other healthcare laws and regulations. If our operations are found to be in violation of any of the laws described above or any other governmental laws and regulations that may apply to us, we may be subject to significant civil, criminal and administrative penalties, damages, disgorgement, fines, imprisonment, exclusion of products from government funded healthcare programs, such as Medicare and Medicaid, additional reporting requirements and/or oversight if a corporate integrity agreement or similar agreement is executed to resolve allegations of non-compliance with these laws and the curtailment or restructuring of operations. In addition, violations may also result in reputational harm, diminished profits and future earnings.
Employee litigation and unfavorable publicity could negatively affect our future business.
Our employees may, from time to time, bring lawsuits against us regarding injury, creating a hostile work place, discrimination, wage and hour disputes, sexual harassment, or other employment issues. In recent years, there has been an increase in the number of discrimination and harassment claims generally. Coupled with the expansion of social media platforms and similar devices that allow individuals access to a broad audience, these claims have had a significant negative impact on some businesses. Certain companies that have faced employment- or harassment-related lawsuits have had to terminate management or other key personnel, and have suffered reputational harm that has negatively impacted their business. If we were to face any employment-related claims, our business could be negatively affected.
Under applicable employment laws, such as in Israel, we may not be able to enforce covenants not to compete and therefore may be unable to prevent our competitors from benefiting from the expertise of some of our former employees.
We generally enter into non-competition agreements with our employees and certain key consultants. These agreements prohibit our employees and certain key consultants, if they cease working for us, from competing directly with us or working for our competitors or clients for a limited period of time. We may be unable to enforce these agreements under the laws of the jurisdictions in which our employees work and it may be difficult for us to restrict our competitors from benefitting from the expertise our former employees or consultants developed while working for us. For example, Israeli courts have required employers seeking to enforce non-compete undertakings of a former employee to demonstrate that the competitive activities of the former employee will harm one of a limited number of material interests of the employer which have been recognized by the courts, such as the secrecy of a company’s confidential commercial information or the protection of its intellectual property. If we cannot demonstrate that such interests will be harmed, we may be unable to prevent our competitors from benefiting from the expertise of our former employees or consultants and our ability to remain competitive may be diminished.
General Risk Factors
The increasing use of social media platforms presents new risks and challenges.
Social media is increasingly being used to communicate about our research, development candidates, investigational medicines, and the diseases our development candidates and investigational medicines are being developed to treat. Social media practices in the biopharmaceutical industry continue to evolve and regulations relating to such use are not always clear. This evolution creates uncertainty and risk of noncompliance with regulations applicable to our business, resulting in potential regulatory actions against us. For example, subjects may use social media channels to comment on their experience in an ongoing blinded clinical trial or to report an alleged adverse event. When such disclosures occur, there is a risk that we fail to monitor and comply with applicable adverse event reporting obligations or we may not be able to defend our business or the public’s legitimate interests in the face of the political and market pressures generated by social media due to restrictions on what we may say about our development candidates and investigational medicines. There is also a risk of inappropriate disclosure of sensitive information or negative or inaccurate posts or comments about us on any social networking website. If any of these events were to occur or we otherwise fail to comply with applicable regulations, we could incur liability, face regulatory actions, or incur other harm to our business.
Unfavorable U.S. or global economic conditions could adversely affect our business, financial condition, or results of operations.
Our results of operations could be adversely affected by general conditions in the global economy and financial markets, including global pandemics, recent geopolitical events, unfavorable changes related to interest rates and rising inflation. The most recent global financial crisis caused extreme volatility and disruptions in the capital and credit markets. A severe or prolonged economic downturn, such as the most recent global financial crisis, could result in a variety of risks to our business, including weakened demand for our investigational medicines and our ability to raise additional capital when needed on favorable terms, if at all. A weak or declining economy could strain our suppliers, possibly resulting in supply disruption, or cause delays in payments for our services by third-party payors or our collaborators. Any of the foregoing could harm our business and we cannot anticipate all of the ways in which the current economic climate and financial market conditions could adversely impact our business.
|
|
ITEM 1B. UNRESOLVED STAFF COMMENTS
None.
ITEM 2. PROPERTIES
As of March 31, 2023, the Company leased facilities for corporate and R&D purposes at locations throughout the United States and in various locations outside of the United States. Our executive office is located at 900 Stewart Avenue, Suite 310, Garden City, NY 11530 under a lease that expires in June 2031. We also lease office space in Garden City, New York under a lease that expires in June 2025, an office in Atlanta, Georgia under a lease that expires in September 2026, an office in Dublin, Ireland under a lease that expires in September 2028 and office spaces in Rehovot, Israel under several lease agreements that expire over the course of the next fiscal year. The Company has a research and development facility in Madison, Wisconsin under a lease that expires in May 2026. The Company believes the existing facilities which are used by all reportable segments and are in good operating condition and are suitable for the conduct of its business.
ITEM 3. LEGAL PROCEEDINGS
On March 23, 2023, the Supreme Court of the State of New York, Appellate Division, First Judicial Department (the “Appellate Court”), rendered its opinion in Empery Asset Master, Ltd., et. al. vs. AIT Therapeutics, Inc. (the “Empery Suit”). The Appellate Court opinion affirmed the judgment by the Supreme Court of the State of New York against Beyond Air, Inc. In connection with the appeal, the Company had used approximately $7.4 million of cash as collateral to secure a supersedeas bond for the full amount of damages and interest in case it was unsuccessful in its appeal. Accordingly, the Company does not believe that the outcome from this appeal will materially impact its available liquidity or financial position. In April 2023, as a subsequent event to the fiscal year ended March 31, 2023, the Company paid $7.6 million, including releasing the funds held in the supersedeas bond, in satisfaction of judgment.
On December 28, 2021 Hudson Bay Master Fund (“Hudson”) filed a lawsuit in the Supreme Court of the State of New York against us relating to the notice of adjustment of the exercise price of and the number of warrant shares issuable under warrants issued to Hudson in January 2017. Hudson received 83,334 warrants in connection with the January 2017 offering. Hudson’s complaint alleges breach of contract and that Hudson is entitled to damages and interest estimated at approximately $2.6 million as a result of certain adjustments to the exercise price and number of warrant shares issuable following a February 2018 financing transaction. The Company accrued a total of $2.7 million as of March 31, 2023, reflecting management’s estimate of most likely outcome of the lawsuit.
ITEM 4. MINE SAFETY DISCLOSURES
Not applicable.
|
|
PART II
ITEM 5. MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Market Information
Our common stock has been listed under the symbol “XAIR” on the Nasdaq Capital Market since May 7, 2019. From August 28, 2018 until May 6, 2019, our common stock was quoted on the OTC Pink.
Stockholders
As of June 22, 2023, there were approximately 101 holders of record for shares of our common stock. This does not reflect beneficial stockholders who held their common stock in “street” or nominee name through brokerage firms.
Securities Authorized for Issuance Under Equity Compensation Plans
Information regarding securities authorized for issuance under our equity compensation plans is contained in Part III, Item 12 of this Annual Report.
Dividend Policy
We have never declared or paid any cash dividends on our capital stock. We currently intend to retain all available funds and any future earnings to support our operations and finance the growth and development of our business. We do not intend to pay cash dividends on our common stock for the foreseeable future.
Unregistered Sales of Equity Securities
(a) Sales of Unregistered Securities
None.
(b) Use of Proceeds
None.
(c) Issuer Purchases of Equity Securities
None.
ITEM 6. [RESERVED]
|
|
ITEM 7. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
The following discussion and analysis of our financial condition and results of operations should be read in conjunction with our consolidated financial statements and related notes appearing elsewhere in this Annual Report on Form 10-K. This discussion and other parts of this Annual Report contain forward-looking statements that involve risks and uncertainties, such as statements regarding our plans, objectives, expectations, intentions and projections. Our actual results could differ materially from those discussed in these forward-looking statements. Factors that could cause or contribute to such differences include, but are not limited to, those discussed in Item 1A “Risk Factors.”
Introduction
We are a commercial stage medical device and biopharmaceutical company developing the LungFit® platform of NO generators and delivery systems capable of generating NO from ambient air. Our first device, LungFit® PH received FDA PMA approval and is indicated to improve oxygenation and reduce the need for extracorporeal membrane oxygenation in term and near-term (>34 weeks gestation) neonates with hypoxic respiratory failure associated with clinical or echocardiographic evidence of pulmonary hypertension in conjunction with ventilatory support and other appropriate agents. This condition is commonly referred to as persistent pulmonary hypertension of the newborn or “PPHN”. LungFit® can deliver NO either continuously or for a fixed amount of time at various flow rates and has the ability to either titrate dose on demand or maintain a constant dose. In July 2022, the Company commenced marketing LungFit® PH in the United States for PPHN as a medical device.
We believe that LungFit® can be used to treat patients on ventilators that require NO, as well as patients with chronic or acute severe lung infections via delivery through a breathing mask or similar apparatus. Furthermore, we believe that there is a high unmet medical need for patients suffering from certain severe lung infections that the LungFit® platform can potentially address. Our current areas of focus with LungFit® is PPHN, VCAP including COVID-19, BRO, NTM lung infection and those with various severe lung infections with underlying COPD. Additionally, The Company has ongoing clinical programs in Cancer and Autism which are nitric oxide focused, but do not utilize the LungFit® device. Our current product candidates will be subject to premarket reviews and certifications or regulatory approvals by the FDA, as well as comparable foreign regulatory authorities in other countries or regions.
We expect to be certified under the EU MDR in the second half of calendar year 2023. We also expect to make certain regulatory filings outside of the U.S. this year. If certifications or regulatory approvals are obtained, we anticipate to launch LungFit® outside of the U.S. in late 2023.
In addition to the above-mentioned programs, we have two subsidiaries that are currently engaging in novel preclinical stage pharmaceutical research, Beyond Cancer, Ltd. and Beyond Air Cyprus.
Financial Operations Overview
Critical Accounting Policies
Research and Development
Research and development expenses are charged to the consolidated statement of operations as incurred. Research and development expenses include salaries, benefits, stock-based compensation and costs incurred by outside laboratories, manufacturers, clinical research organizations, consultants, and accredited facilities in connection with clinical trials and preclinical studies. Research and development expenses are partially offset by the benefit of tax incentive payments for qualified research and development expenditures from the Australian tax authority (“AU Tax Rebates”).
|
|
Stock-Based Compensation
We measure the cost of employee and non-employee services received in exchange for an award of equity instruments based on the grant date fair value of the award. Fair value for restricted stock awards is valued using the closing price of our common stock on the date of grant. The grant date fair value is recognized over the period during which an employee and non-employee is required to provide service in exchange for the award – the requisite service period. The grant date fair value of employee share options is estimated using the Black-Scholes option pricing model. The risk-free interest rate assumptions were based upon the observed interest rates appropriate for the expected term of the equity instruments. The expected dividend yield was assumed to be zero as we have not paid any dividends since our inception and do not anticipate paying dividends in the foreseeable future from either Beyond Air or Beyond Cancer. In 2023, we began using Beyond Air’s historic volatility as the input for expected volatility in the fair value calculations for Beyond Air. Due to its lack of marketability, we utilize an implied volatility for Beyond Cancer based on an aggregate of guideline companies. The peer companies selected have similar characteristics, including industry and market capitalization. We routinely review our calculation of volatility based on our life cycle, our peer group, and other factors. We use the simplified method to estimate the expected term.
Contingencies
On March 23, 2023, the Appellate Court rendered its opinion in the Empery Suit. The Appellate Court opinion affirmed the judgment by the Supreme Court of the State of New York against the Company. In connection with the appeal, the Company had used approximately $7.4 million of cash as collateral to secure a supersedeas bond for the full amount of damages and interest in case it was unsuccessful in its appeal. The Company accrued a total of $7.6 million for damages and interest as of March 31, 2023. As a subsequent event to the fiscal year ended March 31, 2023, the Company paid a total of $7.6 million including damages and interest in satisfaction of judgment.
On December 28, 2021 Hudson filed a lawsuit against us related to the notice of adjustment of the exercise price of and the number of warrant shares issuable under warrants issued to Hudson in January 2017. Hudson received 83,334 warrants in connection with the January 2017 offering. Hudson’s complaint alleges breach of contract and that Hudson is entitled to damages and interest estimated at approximately $2.6 million as a result of certain adjustments to the exercise price and number of warrant shares issuable following a February 2018 financing transaction. In consultation with outside legal counsel, we believe that we have several meritorious defenses against Hudson’s claims. The Company accrued a total of $2.7 million as of March 31, 2023, reflecting management’s estimate of most likely outcome of the lawsuit.
|
|
Results of Operations and Other Comprehensive Income (Loss)
| (in thousands, except number of shares and loss per share) |
Year Ended March 31, 2023 |
Year Ended March 31, 2022 |
||||||
| Revenue | $ | - | $ | - | ||||
| Cost of revenue | (555) | - | ||||||
| Gross loss | (555) | - | ||||||
| Research and development | (16,810) | (11,802 | ) | |||||
| General and administrative | (34,694) | (18,408 | ) | |||||
| Milestone expenses incurred upon regulatory approval | - | (10,500) | ||||||
| Total operating expenses | (51,504) | (40,710) | ||||||
| Loss from operations | (52,059) | (40,710 | ) | |||||
| Estimated contingent loss | (7,863) | (2,435) | ||||||
| Dividend and interest income | 656 | 4 | ||||||
| Interest expense and financing expense | (30) | (775 | ) | |||||
| Foreign exchange loss (gain) | (105) | (144 | ) | |||||
| Total other loss | (7,342) | (3,350 | ) | |||||
| Net loss before income taxes | (59,401) | (44,060 | ) | |||||
| Provision for income taxes | - | - | ||||||
| Net loss | $ | (59,401) | $ | (44,060 | ) | |||
| Less : Net loss attributable to non-controlling interests | (3,585) | (882) | ||||||
| Net loss attributed to Beyond Air, Inc. | $ | (55,816) | $ | (43,177 | ) | |||
| Other comprehensive income: | ||||||||
| Foreign currency translation gain / (loss) | (43) | 96 | ||||||
| Comprehensive loss attributable to Beyond Air, Inc. | (55,859) | (43,081 | ) | |||||
| Net loss per share – basic and diluted | $ | (1.86) | $ | (1.68 | ) | |||
| Weighted average number of shares of common stock outstanding – basic and diluted | 29,973,639 | 25,668,230 | ||||||
|
|
Comparison of the year ended March 31, 2023 to the year ended March 31, 2022
Revenue and Cost of Revenue
On June 28, 2022 the FDA approved LungFit® PH to treat PPHN. No revenue was recognized for the years ended March 31, 2023 and 2022. Cost of revenue for the year ended March 31, 2023 was $0.6 million, primarily driven by the costs to implement our supply chain infrastructure in select geographies across the U.S. as part of our commercial launch. There was no Cost of Revenue for the year ended March 31, 2022.
Research and Development
Research and development expenses for the year ended March 31, 2023 were $16.8 million, as compared to $11.8 million for the year ended March 31, 2022. The increase of $5.0 million was attributed primarily to an increase in development costs in Cancer research ($4.0 million), Autism ($0.8 million) and preparing for the approval of PPHN ($0.5 million), partially offset by a decrease in spend on NTM ($1.0 million) and BRO ($0.3 million). The Company also made further increases in R&D staff, with an increase in stock-based compensation, salaries and travel (for a total of $1.0 million).
General and Administrative Expenses
General and administrative expense for the years ended March 31, 2023 and March 31, 2022 were $34.7 million and $18.4 million, respectively. The increase of $16.3 million was attributed primarily to an increase in stock-based compensation ($7.3 million in Beyond Cancer and $2.1 million in Beyond Air), salaries ($2.6 million in Beyond Air and $1.1 million in Beyond Cancer) and travel ($0.7 million, mainly in Beyond Air) driven by an increase of 16 positions globally, legal and professional fees ($0.7 million), leases and related office costs ($1.3 million), IT costs ($0.1 million) and increased insurance costs ($0.2 million).
Other Operating Expenses
Other operating expenses for the year ended March 31, 2023 were $0, versus $10.5 million for the year ended March 31, 2022, which was attributed to the recognition of liabilities that were contingent on FDA approval of LungFit® PH with Circassia $10.5 million.
Other Income and Expense
Net other expense for the year ended March 31, 2023 and March 31, 2022 were $7.3 million and $3.4 million, respectively. The increase of $4.0 million was attributed primarily to an increase in the accrual for the Empery Suit of $5.1 million versus $2.4 million in the prior year and an estimated liability for a contingent loss on the lawsuit with Hudson Bay ($2.7 million), partially offset by $0.7 million of interest and dividend income from our investments in marketable securities and a reduction of interest expense and financing costs of $0.8 million.
Net Loss Attributable to Non-controlling Interests
Net loss attributed to non-controlling interests for the year ended March 31, 2023, was $3.6 million, compared to $0.9 million for the year ended March 31, 2022. Non-controlling interests represent 20% of the net loss of our Beyond Cancer subsidiary which was established in November 2021 and the increase in net loss is reflective of the increase in spend in Beyond Cancer since its inception in late 2021.
Net Loss Attributed to Common Stockholders
Net loss attributed to common stockholders for the year ended March 31, 2023, was $55.8 million or a loss of $1.86 per share, basic and diluted, as a result of the foregoing. Our net loss attributed to common stockholders for the year ended March 31, 2022, was $43.2 million or a loss of $1.68 per share, basic and diluted.
|
|
Liquidity and Capital Resources
We started generating costs related to the commercial launch since obtaining regulatory approval for the LungFit® PH at the end of the first quarter of fiscal 2023 and expect to generate revenue commencing in the first fiscal quarter of 2024. We used cash flow in operations of $33.0 million for the year ended March 31, 2023 and we have experienced an accumulated deficit of $179.5 million since inception through March 31, 2023. As of March 31, 2023, we had cash, cash equivalents and marketable securities of $45.9 million and $10.1 million in restricted cash. We believe that our cash and cash equivalents as of March 31, 2023 in addition to $5.6 million raised through our ATM in the first fiscal quarter of 2024 and the funds available through the debt with the Avenue Capital Group (as described below) will enable us to fund our operating expenses and capital expenditure requirements through at least one year from the date these consolidated financial statements were filed.
Our future capital needs and the adequacy of its available funds beyond one year from the date of filing these financial statements will depend on many factors, including, but not necessarily limited to, the cost and time necessary for the development, clinical studies and certification or regulatory approval of our other medical devices, indications as well as the commercial success of our approved product and any product candidates that receive marketing approval by the FDA. We may be required to raise additional funds through sale of equity or debt securities or through strategic collaborations and/or licensing agreements in order to fund operations until we are able to generate enough product or royalty revenues, if any. Financing may not be available on acceptable terms, or at all, and our failure to raise capital when needed could have a material adverse effect on our strategic objectives, results of operations and financial condition.
On May 25, 2021, we and Circassia Limited entered into the Settlement Agreement resolving all claims by and between both parties and mutually terminating the Circassia Agreement disclosed in Note 10 of the consolidated financial statements included in this Annual Report. Pursuant to the terms of the Settlement Agreement, we agreed to pay Circassia $10.5 million in three installments, the first being a payment of $2.5 million on the Initial Payment Due Date. Thereafter, we shall pay $3.5 million to Circassia on the first anniversary of the Initial Payment Due Date and $4.5 million on the second anniversary of the Initial Payment Due Date. Additionally, beginning in year three post-approval, Circassia will receive a quarterly royalty payment equal to 5% of LungFit® PH net sales in the U.S. This royalty will terminate once the aggregate payment reaches $6 million. The Initial Payment of $2.5 million was made in July 2022 and $8.0 million has been recorded as an accrued liability for the year ended March 31, 2023.
On February 4, 2022, we entered into an At-The-Market Equity Offering Sales Agreement with Truist Securities, Inc. and Oppenheimer & Co, Inc. (the “2022 ATM”). Under the 2022 ATM, we may sell shares of our common stock having aggregate sales proceeds of up to $50 million, from time to time and at various prices. If shares of our common stock are sold, there is a 3% fee paid to the sales agent. As of March 31, 2023, there was a balance of $46.3 million available under the 2022 ATM.
As a subsequent event to the fiscal year ended March 31, 2023, on June 15, 2023, we entered into a Loan Agreement with Avenue Capital providing for senior secured term loans of up to $40 million, with (i) $17.5 million advanced on the Closing Date, (ii) up to $10 million between April 1, 2024 and September 30, 2024, subject to the Company achieving revenue milestones, and (iii) up to $12.5 million after April 1, 2024, subject to mutual agreement. The Loans are due and payable on June 1, 2027. The Loan principal is repayable beginning on January 1, 2025, with the possibility of deferring principal payments an additional 6 to 18 months. The Loans bear interest at a rate per annum equal to the greater of (i) the prime rate, as published by the Wall Street Journal from time to time, plus 3.75% and (ii) 12.00%. A final payment fee of 3.50% of the principal amount of the Tranche 1 and Tranche 2 Loans is also due upon repayment of the principle.
The Company is subject to a financial covenant requiring the Company to maintain $5,000,000 in unrestricted cash. The Loan Agreement also contains affirmative and negative covenants customary for financing of this type.
The Loan Agreement also includes events of default customary for financings of this type, in certain cases subject to customary periods to cure, following which the Agent may accelerate all amounts outstanding under the Loans. We granted Avenue Capital Group warrants to purchase 233,843 shares of common stock at an exercise price of the lesser of $5.88 or the price per share of our next bona fide round of equity financing before June 30, 2024.
We received $15.7 million in net proceeds on June 15, 2023 after all fees and advanced interest had been deducted.
Our ability to continue to operate beyond the first fiscal quarter of 2025 will be largely dependent upon the successful commercial launch of LungFit® PH, as well as obtaining partners in other parts of the world, and raising additional funds to finance our activities until we are generating cash flow from operations. Further, there are no assurances that we will be successful in obtaining an adequate level of financing for the development and commercialization of our other product candidates.
|
|
There are numerous risks and uncertainties associated with the development of our NO delivery system and we are unable to estimate the amounts of increased capital outlays and operating expenses associated with completing the research and development of our product candidates.
Our future capital requirements will depend on many factors, including:
| ● | the progress and costs of our preclinical studies, clinical trials and other research and development activities; | |
| ● | the costs of commercializing the LungFit® system; | |
| ● | the scope, prioritization and number of our clinical trials and other research and development programs; | |
| ● | the costs and timing of obtaining certification or regulatory approval for our product candidates; | |
| ● | the costs of filing, prosecuting, enforcing and defending patent claims and other intellectual property rights; | |
| ● | the costs of, and timing for, strengthening our manufacturing agreements for production of sufficient clinical quantities of our product candidate; | |
| ● | the potential costs of contracting with third parties to provide marketing and distribution services for us or for building such capacities internally; | |
| ● | the costs of acquiring or undertaking the development and commercialization efforts for additional, future therapeutic applications of our product candidate; | |
| ● | the magnitude of our general and administrative expenses; and | |
| ● | any cost that we may incur under current and future in-licensing and out-licensing arrangements relating to our product candidate. |
Cash Flows
Below is a summary of the statements of cash flows for the years ended March 31, 2023 and March 31, 2022.
| (in thousands) |
For The Year Ended March 31, 2023 |
For The Year Ended March 31, 2022 |
||||||
| Net cash provided by (used in): | ||||||||
| Operating activities | $ | (33,009 | ) | $ | (23,134 | ) | ||
| Investing activities | $ | (20,587 | ) | $ | (1,450 | ) | ||
| Financing activities | $ | 2,696 | $ | 79,450 | ||||
| Effect of exchange rate changes on cash and cash equivalents | $ | (43 | ) | $ | 96 | |||
| Net increase (decrease) in cash, cash equivalents and restricted cash | $ | (50,944 | ) | $ | 54,962 | |||
Comparison between Fiscal Years Ended March 31, 2023 and March 31, 2022
Operating Activities
For the year ended March 31, 2023, net cash used by operating activities was $33.0 million which was primarily due to our net loss of $59.4 million, which included non-cash stock based compensation of $19.6 million and an increase in accrued liabilities of $4.8 million including an increase in liabilities related to lawsuits of $7.8 million, partially offset by the payment of the first tranche of the Circassia settlement of $2.5 million. For the year ended March 31, 2022, net cash used by operating activities was $23.1 million which was primarily due to our net loss of $44.1 million, which included non-cash stock based compensation of $7.8 million and an increase in liabilities for the recognition of contingent liabilities of $10.5 million and a contingent liability for a lawsuit of $2.5 million.
|
|
Investing Activities
For the year ended March 31, 2023, cash used in investing activities was $20.6 million which was primarily from investments in marketable securities for a net movement of $16.7 million in the fiscal year, and the purchase of property and equipment for $3.9 million. For the year ended March 31, 2022, cash used in investing activities was $1.4 million which was from the purchase of property and equipment.
Financing Activities
For the year ended March 31, 2023, net cash provided by financing activities was $2.7 million which was primarily from the issuance of common stock in connection with our 2022 ATM for $3.7 million, partially offset by loan payments of $1.0 million. For the year ended March 31, 2022, net cash provided by financing activities was $79.5 million which was primarily from the issuance of common stock in connection with a Stock Purchase Agreement with Lincoln Park Capital Fund, LLC, that has since expired ($11.1 million) and 2020 ATM ($36.5 million), issuance of common stock in connection with the exercise of warrants and stock options ($6.0 million and $0.3 million, respectively), and proceeds from the sale of common stock of Beyond Cancer for $30.0 million less ($4.8) million from lenders in our retired loan facility.
ITEM 7A. Quantitative and Qualitative Disclosure about Market Risk
We are exposed to market risks in the ordinary course of our business. Market risk represents the risk of loss that may impact our financial position due to adverse changes in financial market prices and rates. Our market risk exposure is primarily a result of foreign currency exchange rates.
Foreign Currency Exchange Risk
Our results of operations and cash flow are subject to fluctuations due to changes in foreign currency exchange rates. Certain of our expenses are denominated in New Israeli Shekels (“NIS”), the Euro and the Australian Dollar. Our results of operations and cash flow are, therefore, subject to fluctuations due to changes in foreign currency exchange rates and may be adversely affected in the future due to changes in foreign exchange rates. We do not hedge our foreign currency exchange risk. In the future, we may enter into formal currency hedging transactions to decrease the risk of financial exposure from fluctuations in the exchange rates of our principal operating currencies. These measures, however, may not adequately protect us from significant changes in such fluctuations.
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
The consolidated financial statements together with the report of our independent registered public accounting firm, required to be filed pursuant to this Item 8 are appended to this Annual Report. An index of those consolidated financial statements is found in Item 15 of this Annual Report.
|
|
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
Based on information provided by Friedman LLP (“Friedman”), effective September 1, 2022, Friedman, which has served as our independent registered public accounting firm since 2019, combined with Marcum LLP (“Marcum”), an independent registered public accounting firm. As a result, the Audit Committee of the Company has approved the dismissal of Friedman and the engagement of Marcum of East Hanover, New Jersey on October 6, 2022 to serve as our independent registered public accounting firm for the fiscal year ending March 31, 2023.
Friedman’s reports regarding our financial statements for the years ended March 31, 2022 and March 31, 2021 did not contain any adverse opinion or disclaimer of opinion and were not qualified or modified as to uncertainty, audit scope or accounting principles.
During our two most recent fiscal years and the interim period from the end of the most recently completed year through October 6, 2022, the date of Marcum’s engagement, there were no (i) disagreements with Friedman on any matter of accounting principles or practices, financial statement disclosure or auditing scope or procedures which, if not resolved to Friedman’s satisfaction, would have caused it to make reference to the subject matter of the disagreement in connection with its reports, or (ii) “reportable events” as defined in Item 304(a)(1)(v) of Regulation S-K.
ITEM 9A. CONTROLS AND PROCEDURES
(a) Disclosure Controls and Procedures
We performed an evaluation of the effectiveness of our disclosure controls and procedures that are designed to ensure that information required to be disclosed in this Annual Report and filed with the SEC is recorded, processed, summarized and reported timely within the time period specified in the SEC’s rules and forms. Disclosure controls and procedures include, without limitation, controls and procedures designed to ensure that information required to be disclosed by an issuer in the reports that it files or submits under the Exchange Act, is accumulated and communicated to the issuer’s management, including its principal executive and principal financial officers, or persons performing similar functions, as appropriate to allow timely decisions regarding required disclosure. There can be no assurance that our disclosure controls and procedures will detect or uncover all failures of persons within the Company to disclose information otherwise required to be set forth in our reports. Nevertheless, our disclosure controls and procedures are designed to provide reasonable assurance of achieving the desired control objectives. Based on our evaluation, our Chief Executive Officer (our principal executive officer) and Chief Financial Officer (our principal financial officer) have concluded that our disclosure controls and procedures (as defined in Rules 13a-15(e) and 15(d)-15(e) of the Exchange Act) were effective at such reasonable assurance level as of March 31, 2023.
(b) Management’s Annual Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act. Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Our management, with the participation of our Chief Executive Officer (our principal executive officer) and Chief Financial Officer (our principal financial officer), conducted an evaluation of the effectiveness of our internal control over financial reporting as of March 31, 2023, based on the framework in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework). Based on this evaluation, management concluded that our internal control over financial reporting was effective as of March 31, 2023.
(c) Attestation Report of Registered Public Accounting Firm
This report does not include an attestation report of our registered public accounting firm as we are not an accelerated filer or a large accelerated filer.
(d) Changes in Internal Control over Financial Reporting
We continued to deploy our ERP system across our subsidiaries during the year ended March 31, 2023 and have completed deployment to all entities where we have employees and finance professionals. During the fiscal year we added enhanced controls on our banking platforms and expanded our Information Technologies team to help detect fraud and prevent threats to our critical systems.
ITEM 9B. OTHER INFORMATION
None.
ITEM 9C. DISCLOSURE REGARDING FOREIGN JURISDICTIONS THAT PREVENT INSPECTIONS
Not applicable.
|
|
PART III
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
Directors and Executive Officers
The table below sets forth the name, age and position of each of our directors and executive officers and as of the date of this Annual Report on Form 10-K.
| Name | Age | Position | ||
| Steven A. Lisi | 52 | Chief Executive Officer and Chairman of the Board of Directors | ||
| Amir Avniel | 49 | President, Chief Business Officer and Director | ||
| Douglas Q. Larson | 53 | Chief Financial Officer | ||
| Michael Gaul | 69 | Chief Operating Officer | ||
| Jeff Myers | 58 | Chief Medical Officer | ||
| Ron Bentsur | 57 | Director | ||
| Erick J. Lucera | 55 | Director | ||
| Yoori Lee | 50 | Director | ||
| Dr. William Forbes | 61 | Director | ||
| Robert F. Carey | 64 | Director |
Steven A. Lisi, Chief Executive Officer and Chairman of the Board
Steven Lisi has served on our Board of Directors since January 13, 2017, and has served on the Board of Directors of BA Ltd., our wholly-owned subsidiary, since June 2016. Mr. Lisi has served as our Chief Executive Officer since June 14, 2017.
Mr. Lisi was previously Senior Vice President of Business and Corporate Development at Avadel Pharmaceuticals (AVDL) where he was instrumental in restructuring the company, raising over $125 million and transforming it from a $100 million enterprise value to $1 billion in three years. Prior to his position at Avadel, Mr. Lisi spent 18 years investing in the global healthcare industry at Deerfield Management, Millennium Management and SAC Capital, amongst others. Mr. Lisi serves as Chairman of the Board of Beyond Cancer, an NO based immuno-oncology company targeting solid tumors. He received his master’s degree in International Business from Pepperdine University.
Our Board of Directors believes that Mr. Lisi’s experience and perspective as our Chief Executive Officer, as well as his depth of operating and senior management experience and specific skills in the areas of general operations and financial operations, provide him with the qualifications and skills to serve as a director.
Amir Avniel, President, Chief Business Officer and Director
Amir Avniel has served on BA Ltd.’s Board since 2011 and became BA Ltd.’s Chief Executive Officer in August 2014. He has served on our Board of Directors since January 2017, as our President since June 2017 and as Beyond Air’s Chief Business Officer since July 2022. Mr Avniel also served as our Chief Operating Officer between June 2017 and June 2022 and as our Chief Executive Officer from January, 2017 to June, 2017. He has more than ten years of management experience in the biotechnology industry. From 2013 through 2014, Mr. Avniel served as Strategy and Business Development of A.B. Seeds, a wholly owned subsidiary of Monsanto Company. Mr. Avniel served as the Chief Executive Officer of Rosetta Green Ltd. from 2010 through 2013 and led Rosetta Green in its acquisition by Monsanto. Mr. Avniel also served as the President and the Chief Executive Officer of Rosetta Genomics from 2006 to 2009, and he is a named inventor in over 20 patent applications. He studied computer science at the Academic College of Tel Aviv - Jaffa Israel and earned a Bachelor’s degree in Social Sciences and Humanities from Open University in Israel. Prior to his academic studies, he served as an officer in the Israel Defense Force, where he was awarded four commendations for excellence.
Our Board of Directors believes that Mr. Avniel’s experience and perspective as our President and Chief Business Officer, as well as his depth of operating and senior management experience in the biotechnology industry, provide him with the qualifications and skills to serve as a director.
|
|
Douglas Larson, Chief Financial Officer
Douglas Larson has been our Chief Financial Officer since September, 2021. Mr. Larson joined the Company with over 20 years of international and operational financial leadership experience. Most recently, he served as an independent consultant providing operational and financial consulting services from February 2021 to August 2021. Prior to that, he served as Vice President, Finance and Head of Global Controlling at DBV Technologies, Inc. (Nasdaq: DBVT) (“DBV”), a global clinical stage biopharmaceutical company headquartered in France, from June 2017 to September 2020. Prior to DBV, Mr. Larson served as the Chief Financial Officer of The Scotts Miracle-Gro Company’s (NYSE: SMG) International division, based in Lyon, France from January 2001 to May 2015. Mr. Larson graduated from the Certified General Accountant’s Association of Canada in 2001 and HEC Paris’s Executive MBA program in 2015.
Michael Gaul, Chief Operating Officer
Michael Gaul has been our Chief Operating Officer since July 1, 2022. Mr. Gaul joined the Company in May 2020 as Senior VP, Operations, a title he held until June 2022. Mr. Gaul has led teams in operations, sales and marketing, supply chain, distribution, quality, regulatory, human resources, and finance. He has had P&L experience with multi-plant responsibility in both the U.S. and Asia, including roles with Sparton Corporation (Group VP, Manufacturing & Design Services Business Unit; Group VP, Medical Business Unit), a defense contractor for maritime defense, from September 2011 to February 2020, SynCardia Systems (COO; VP Operations), a manufacturer and provider of the commercially approved Total Artificial Heart, from June 2005 to February 2011, Ventana Medical Systems (now known as Roche Tissue Diagnostics) (VP & General Manager, Operations), a medical device company, from September 2003 to April 2005, and Robotic Vision Systems, Inc. (General Manager, Vanguard Division; VP Operations, Systemation Division), a maker of machine vision systems, from September 1997 to June 2003. He also currently serves on the Board of Directors of the Ohio Life Sciences Association to support and promote Ohio’s bioscience industry. Mr. Gaul has a BS, Business Administration from Delaware Valley University, and an MBA from Florida Institute of Technology.
Jeff Myers, Chief Medical Officer
Jeff Myers, MD joined Beyond Air as our Chief Medical Officer on March 27, 2023 with nearly 15 years of leadership experience as a biopharmaceutical executive overseeing clinical development, clinical operations, and regulatory affairs. Prior to industry, Dr. Myers was a cardiothoracic surgeon for nine years, most recently at Massachusetts General Hospital. Dr. Myers’ previous leadership responsibilities include overseeing clinical development, clinical operations, business development, medical affairs, and implementing regulatory strategies in the U.S. and abroad. Previously, he was the Chief Medical Officer for Revolo Biotherapeutics, initiating clinical trials in the U.S. and Europe before leaving to become the CEO of Bioceptive where he continues to serve as a member of the Board of Directors. Dr. Myers also served as the Chief Medical Officer for Portola Pharmaceuticals where he was instrumental in the acquisition by Alexion Pharmaceuticals, and Vice President, Medical and Regulatory Affairs, at SteadyMed Therapeutics. Prior to beginning his career in biotechnology, he was a practicing congenital cardiac surgeon and served as the Chief of Pediatric Cardiac Surgery at Tulane University and Massachusetts General Hospital with appointments to Tulane and Harvard Medical Schools. He is passionate about developing novel, first-in-class therapies that significantly improve the lives of patients. Dr. Myers began his work with inhaled NO in pursuit of his PhD at Georgetown University.
Ron Bentsur, Director
Ron Bentsur joined BA Ltd. in August 2015 and has served on our Board of Directors since January 2017. Mr. Bentsur has served as Chief Executive Officer, President, and Chairman of the board of directors of Nuvectis Pharma, Inc., (“Nuvectis”) since 2021. Prior to Nuvectis, Mr. Bentsur served as Chief Executive Officer and on the board of directors of UroGen Pharma, Ltd. between August 2015 and January 2019. From 2009 to April 2015, Mr. Bentsur served as Chief Executive Officer and Director of Keryx Biopharmaceuticals, Inc. Mr. Bentsur’s tenure as CEO of Keryx Biopharmaceuticals culminated in the September 2014 FDA approval of Auryxia TM (ferric citrate) and its December 2014 U.S. launch. Prior to joining Keryx Biopharmaceuticals, Inc., from 2006 to 2009, Mr. Bentsur served as Chief Executive Officer of XTL Biopharmaceuticals, Ltd. Prior to that, Mr. Bentsur served as Vice President Finance and Chief Financial Officer of Keryx Biopharmaceuticals, Inc., as Director of Technology Investment Banking at Leumi Underwriters, where he was responsible for all technology and biotechnology private placement and advisory transactions, and as a New York City-based investment banker, primarily at ING Barings Furman Selz. Mr. Bentsur holds a B.A. in Economics and Business Administration with distinction from the Hebrew University of Jerusalem and an M.B.A., magna cum laude, from New York University’s Stern Graduate School of Business. Mr. Bentsur also serves on the Board of Directors of Stemline Therapeutics, Inc., an oncology therapeutics company.
Our Board of Directors believes that Mr. Bentsur’s experience and perspective advising the Company and other life sciences companies, as well as his depth of operating and senior management experience in the biopharma industry, provide him with the qualifications and skills to serve as a director.
Yoori Lee, Director
Yoori Lee joined our Board of Directors in January 2018. She has served as Co-founder and President of Trio Health Advisory Group, Inc. since 2013. Trio Health’s mission is to improve the quality of care in patient outcomes through coordinating the efforts of all patient care stakeholders. Prior to Trio Health, Ms. Lee spent over 15 years at Leerink Partners LLC, a leading healthcare investment bank, where she was Managing Director, and Director of MEDACorp Services. Additionally, she helped found the MEDACorp network, a cadre of experts including more than 35,000 healthcare professionals in diverse areas of practice such as clinical medicine, biomedical research, regulatory affairs, public policy, healthcare administration and healthcare information technology.
Our Board of Directors believes that Ms. Lee’s experience and perspective advising the Company as well as her experience with Leerink Partners LLC and MEDACorp. provide her with the qualifications and skills to serve as a director.
|
|
Dr. William Forbes, Director
Dr. William Forbes joined our Board of Directors in August 2018. He brings to our Board of Directors more than 30 years of pharmaceutical product development experience and, working with health authorities in the U.S. and Europe, has contributed to numerous marketing approvals spanning a diverse range of therapeutic areas. Dr. Forbes has served as the Chief Development Officer of Trevi Therapeutics, a clinical-stage pharmaceutical company focused on serious neurologically mediated diseases since February 2021. Prior to joining Trevi, Dr. Forbes was at Salix Pharmaceuticals as the Chief Development Officer and also Head of Medical and R&D from February 2015 to June 2015. Prior to Salix, Dr. Forbes spent 15 years in Clinical Development & Regulatory Affairs and Clinical Research at a number of global pharmaceutical companies.
Our Board of Directors believes that Dr. Forbes’ experience and perspective advising the Company, as well as his depth of operating and senior management experience in our industry, provide him with the qualifications and skills to serve as a director.
Robert F. Carey, Director
Robert Carey joined our Board of Directors in February 2019. He has an extensive track record of accomplishment within the biopharmaceutical and healthcare investment banking industry. He has assisted biotech and specialty pharma companies raise more than $10 billion in initial public offerings, follow-on offerings, debt offerings, and private placements. He has served as a financial advisor on mergers, acquisitions, and strategic alliance transactions with a total deal value of more than $10 billion. From July 2020 until December 2023, Mr. Carey was co-founder and President of ACELYRIN, INC., a biopharmaceutical company developing life-changing drug therapies. Mr. Carey previously served as Executive Vice President and Chief Business Officer at Horizon Therapeutics plc from March 2014 to September 2019, during which Horizon Therapeutics deployed in excess of $3.5 billion to acquire or license eight commercial products and three products in development and grew net sales from $74 million in 2013 to approximately $1.2 billion in 2018, a compound annual growth rate of 75%. Before Horizon, he spent more than 11 years as Managing Director and Head of the Life Sciences Investment Banking Group at JMP Securities. Mr. Carey was also Managing Director in the healthcare groups at Dresdner Kleinwort Wasserstein and Vector Securities. He received his B.B.A. in Accounting from the University of Notre Dame. Mr. Carey currently serves on the Board of Beyond Cancer Ltd.and Sangamo Therapeutics, Inc.
Our Board of Directors believes that Mr. Carey’s experience and perspective advising the Company and other life sciences companies in connections with financings and strategic transactions, as well as his depth of operating and senior management experience in our industry, provide him with the qualifications and skills to serve as a director.
Erick J. Lucera, Director
Erick J. Lucera joined our Board of Directors in August 2017 and serves on our audit committee. He was appointed Executive Vice President and Chief Financial Officer of Editas Medicine, Inc., a leading gene editing company focused on developing CRISPR medicines for people with serious diseases, in May 2023. Mr. Lucera previously served as Chief Financial Officer of AVEO Pharmaceuticals, Inc., a commercial-stage biopharmaceutical company focused on targeted medicines for oncology and other unmet medical needs, from January 2020 until March 2023, following its acquisition by LG Chem, Ltd. Since April 2023, Mr. Lucera has served on the board of directors of SAB Biotherapeutics, Inc., a public clinical-stage biopharmaceutical company . Mr. Lucera was the Chief Financial Officer of Valeritas Holdings, Inc., a U.S. Nasdaq traded commercial stage company developing new technology for diabetes, from 2016 to 2019. Mr. Lucera served as Chief Financial Officer, Treasurer and Secretary of Viventia Bio from 2015 to 2016. From 2012 to 2015, he was Vice President, Corporate Development at Aratana Therapeutics, a veterinary biopharmaceutical company. While at Aratana, he helped grow the company’s product pipeline through a series of acquisitions and in licensing transactions financed through five public and private offerings of nearly $250 million. Before his career as a healthcare company executive, Mr. Lucera spent over 15 years in investment management as a healthcare analyst at Eaton Vance, the portfolio manager of the Triathlon Life Sciences Fund at Intrepid Capital and as head of the healthcare research team at Independence Investments. He has served on the board of directors of Bone Biologics, a Nasdaq traded orthobiologics company, since October 2021. He holds a CPH from Harvard University, an MS in quantitative finance from Boston College, an MBA from Indiana University Bloomington, and a BS in accounting from the University of Delaware. Mr. Lucera has obtained CFA, CMA, and CPA designations.
Our Board of Directors believes that Mr. Lucera’s experience and perspective advising the Company and other life sciences companies on strategic transactions and financings, as well as his depth of operating and senior management experience in our industry, provide him with the qualifications and skills to serve as a director.
|
|
Term of Office of Directors
Our directors are elected at each annual meeting of stockholders and serve until their successors are elected and qualified at the next annual meeting of stockholders, or until their prior death, resignation or removal.
Family Relationships
There are no family relationships among any of our current or former directors or executive officers.
Involvement in Certain Legal Proceedings
Erick J. Lucera was the Chief Financial Officer of Valeritas Holdings, Inc. until January 3, 2020. On February 9, 2020, Valeritas Holdings, Inc. filed a voluntary petition for bankruptcy protection under Chapter 11 of Title 11 of the U.S. Bankruptcy Code in the United States Bankruptcy Court for the District of Delaware in order to facilitate its sale to a Denmark-based biotechnology company. The plan of liquidation was approved on June 8, 2020 and became effective on June 30, 2020.
Except as set forth above, none of our directors, executive officers, significant employees, promoters or control persons has been involved in any legal proceeding in the past ten years that would require disclosure under Item 401(f) of Regulation S-K promulgated under the Securities Act.
Board Committees
Our Board of Directors has established four standing committees: the audit committee, the compensation committee, the nominating committee and the compliance committee. The current members of our audit committee are Erick Lucera, Ron Bentsur and Robert F. Carey with Erick Lucera serving as chairperson. The current members of our compensation committee are Yoori Lee, Erick J. Lucera, and Ron Bentsur with Yoori Lee serving as chairperson. The current members of our nominating committee are Erick Lucera, Yoori Lee and Dr. William Forbes, with Erick Lucera serving as chairperson. The compliance committee currently has five members, of which one member serves on our Board of Directors.
Our Board of Directors has determined that Erick Lucera, Ron Bentsur and Robert F. Carey meet the additional test for independence for audit committee members imposed by SEC regulations and Section 5605(c)(2)(A) of the Nasdaq Stock Market listing rules and that Erick J. Lucera, Yoori Lee and Ron Bentsur meet the additional test for independence for compensation committee members imposed by Section 5605(d)(2)(A) of the Nasdaq Stock Market listing rules.
Audit Committee
The primary purpose of our audit committee is to assist the Board of Directors in the oversight of the integrity of our accounting and financial reporting process, the audits of our consolidated financial statements, and our compliance with legal and regulatory requirements. It also oversees internal controls and discusses Company policies related to risk assessment and risk management. Our audit committee met four times during the fiscal year ended March 31, 2023. The functions of our audit committee include, among other things:
| ● | hiring the independent registered public accounting firm to conduct the annual audit of our consolidated financial statements and monitoring its independence and performance; | |
| ● | reviewing and approving the planned scope of the annual audit and the results of the annual audit; |
|
|
| ● | pre-approving all audit services and permissible non-audit services provided by our independent registered public accounting firm; | |
| ● | reviewing the significant accounting and reporting principles to understand their impact on our consolidated financial statements; | |
| ● | reviewing our internal financial, operating and accounting controls with management, our independent registered public accounting firm and our internal audit provider; | |
| ● | reviewing with management and our independent registered public accounting firm, as appropriate, our financial reports, earnings announcements and our compliance with legal and regulatory requirements; | |
| ● | periodically reviewing and discussing with management the effectiveness and adequacy of our system of internal controls; | |
| ● | in consultation with management and the independent auditors, reviewing the integrity of our financial reporting process and adequacy of disclosure controls; | |
| ● | reviewing potential conflicts of interest under and violations of our code of conduct; | |
| ● | establishing procedures for the treatment of complaints received by us regarding accounting, internal accounting controls or auditing matters and confidential submissions by our employees of concerns regarding questionable accounting or auditing matters; | |
| ● | reviewing and approving related-party transactions; and | |
| ● | reviewing and evaluating, at least annually, our audit committee’s charter. |
With respect to reviewing and approving related-party transactions, our audit committee will review related-party transactions for potential conflicts of interests or other improprieties. Under SEC rules, related-party transactions are those transactions to which we are or may be a party in which the amount involved exceeds the lesser of $120,000 or 1% of the average of our total assets at year-end for the last two completed fiscal years, and in which any of our directors or executive officers or any other related person had or will have a direct or indirect material interest, excluding, among other things, compensation arrangements with respect to employment and Board of Directors membership. Our audit committee could approve a related-party transaction if it determines that the transaction is in our best interests. Our directors are required to disclose to this committee or the full Board of Directors any potential conflict of interest, or personal interest in a transaction that our Board of Directors is considering. Our executive officers are required to disclose any related-party transaction to the audit committee. We also poll our directors on an annual basis with respect to related-party transactions and their service as an officer or director of other entities. Any director involved in a related-party transaction that is being reviewed or approved must recuse himself or herself from participation in any related deliberation or decision. Whenever possible, the transaction should be approved in advance and if not approved in advance, must be submitted for ratification as promptly as practical. The Company has written policies and procedures with respect to related person transactions for managing such situations.
The financial literacy requirements of the SEC require that each member of our audit committee be able to read and understand fundamental financial statements. In addition, at least one member of our audit committee must qualify as an audit committee financial expert, as defined in Item 407(d)(5) of Regulation S-K promulgated under the Securities Act, and have financial sophistication in accordance with the Nasdaq Stock Market listing rules. Our Board of Directors has determined that Erick Lucera qualifies as an audit committee financial expert.
Both our independent registered public accounting firm and management periodically meet privately with our audit committee.
|
|
Compensation Committee
The primary purpose of our compensation committee is to assist our Board of Directors in exercising its responsibilities relating to compensation of our executive officers and employees and to administer our equity compensation and other benefit plans. In carrying out these responsibilities, this committee reviews all components of executive officer and employee compensation for consistency with its compensation philosophy, as in effect from time to time. Our compensation committee met one time during the year ended March 31, 2023. The functions of our compensation committee include, among other things:
| ● | designing and implementing competitive compensation, retention and severance policies to attract and retain key personnel; | |
| ● | reviewing and formulating policy and determining the compensation of our Chief Executive Officer, our other executive officers and certain employees; | |
| ● | reviewing and recommending to our Board of Directors the compensation of our non-employee directors; | |
| ● | reviewing and evaluating our compensation risk policies and procedures; | |
| ● | administering our equity incentive plans and granting equity awards to our employees, consultants and directors under these plans; | |
| ● | administering our performance bonus plans and granting bonus opportunities to our employees, consultants and non-employee directors under these plans; | |
| ● | if required from time to time, preparing the analysis or reports on executive officer compensation required to be included in our annual proxy statement; | |
| ● | engaging compensation consultants or other advisors it deems appropriate to assist with its duties; and | |
| ● | reviewing and evaluating, at least annually, our compensation committee’s charter. |
The compensation committee retains sole authority to hire any compensation consultant, approve such consultant’s compensation, determine the nature and scope of its services, evaluate its performance, and terminate its engagement.
The compensation committee reviews our compensation policies and practices for all employees, including our named executive officers, as they relate to risk management practices and risk-taking incentives to assess and determine that there are no risks arising from these policies and practices that are reasonably likely to have a material adverse effect on us.
Nominating Committee
The primary purpose of our nominating committee is to assist our Board of Directors in promoting the best interest of the Company and our stockholders through the implementation of sound corporate governance principles and practices. Our nominating committee met one time during the fiscal year ended March 31, 2023. The functions of our nominating committee include, among other things:
| ● | identifying, reviewing and evaluating candidates to serve on our Board of Directors; | |
| ● | determining the minimum qualifications for service on our Board of Directors; | |
| ● | developing and recommending to our Board of Directors an annual self-evaluation process for our Board of Directors and overseeing the annual self-evaluation process; | |
| ● | developing, as appropriate, a set of corporate governance principles, and reviewing and recommending to our Board of Directors any changes to such principles; and | |
| ● | periodically reviewing and evaluating our nominating committee’s charter. |
Compliance Committee
The primary purpose of our compliance committee is to assist our Board of Directors with oversight of healthcare compliance risk management arising from the FDA, Office of Inspector General, the U.S. Department of Justice and other federal, state or foreign corporate compliance requirements related to medical devices that specifically govern the AKS, FCA, FD&C Act, FCPA, Sunshine Act and similar state laws, HIPAA, the AdvaMed Code, and similar applicable state and local regulations and (when applicable) equivalent global requirements. Our compliance committee met one time during the fiscal year ended March 31, 2023.
Director Candidates
Our Board of Directors has a critical role in guiding our strategic direction and overseeing the management of our business, and accordingly, we seek to attract and retain highly qualified directors who have sufficient time to engage in the activities of our Board of Directors and to understand and enhance their knowledge of our industry and business plans. In evaluating the suitability of individual candidates, our Board of Directors, in approving (and, in the case of vacancies, appointing) such candidates, may take into account many factors, including: personal and professional integrity; ethics and values; experience in corporate management, such as serving as an officer or former officer of a publicly held company; strong finance experience; experience relevant to our industry; experience as a board member or executive officer of another publicly held company; relevant academic expertise or other proficiency in an area of our operations; diversity of expertise and experience in substantive matters pertaining to our business relative to other board members; diversity of background and perspective, including, but not limited to, with respect to age, gender, race, place of residence and specialized experience; practical and mature business judgment, including, but not limited to, the ability to make independent analytical inquiries; and any other relevant qualifications, attributes or skills. The core competencies of directors should address accounting or finance experience, market familiarity, business or management experience, industry knowledge, customer-base experience or perspective, crisis response, leadership, and/or strategic planning. Our Board of Directors evaluates each individual in the context of the Board as a whole, with the objective of assembling a group that can best perpetuate the success of the business and represent stockholder interests through the exercise of sound judgment using its diversity of experience in these various areas.
|
|
Stockholder Communications
Although we do not have a formal policy regarding stockholder communications with our Board of Directors, stockholders may communicate with our Board of Directors, or any individual director on our Board of Directors, by writing to us at the address of our principal executive offices, addressing the communication to the attention of our Chief Executive Officer, and specifying the Board of Directors or, if applicable, the individual member thereof as the intended recipient of the communication. Our Corporate Secretary will forward to the directors all communications that, in his judgment, are appropriate for consideration by the directors. Examples of communications that would not be appropriate for consideration by the directors include commercial solicitations and matters not relevant to the stockholders, to the functioning of the Board of Directors or to the affairs of our Company. Any correspondence received that is addressed generically to the Board of Directors will be forwarded to the Chairman of the Board of Directors.
Board Leadership Structure and Role in Risk Oversight
The Board of Directors does not have a formal policy on whether or not the roles of Chairman of the Board and Chief Executive Officer should be separate and believes that it should retain the flexibility to make this determination in the manner it believes will provide the most appropriate leadership for the Company from time to time. Currently, Steven A. Lisi serves as Chairman of the Board and Chief Executive Officer, working closely with former CEO and present CBO and President, Amir Avniel. We do not have a lead independent director. Mr. Lisi sets the strategic direction for the Company and provides day-to-day leadership. As Chairman of the Board of Directors, Mr. Lisi further oversees the agenda for board meetings in collaboration with the other board members. Our Board believes that it is in the best interest of the Company and its stockholders for Mr. Lisi to serve in both roles at this time given his knowledge of the Company and industry. We believe that this structure provides appropriate leadership and oversight of the Company and facilitates effective functioning of both management and our Board of Directors. Our Board of Directors will continue to reassess the structure to determine what is in the best interests of the Company and stockholders.
The Board of Directors oversees our exposure to risk through its interaction with management and receipt from management of periodic reports outlining matters related to financial, operational, regulatory, legal and strategic risks. Risk assessment and oversight are an integral part of our governance and management processes. Our Board of Directors encourages management to promote a culture that incorporates risk management into our corporate strategy and day-to-day business operations. Management discusses strategic and operational risks at regular management meetings and conducts specific strategic planning and review sessions during the year that include a focused discussion and analysis of the risks facing us. Throughout the year, senior management reviews these risks with the Board of Directors at regular board meetings as part of management presentations that focus on particular business functions, operations or strategies and presents the steps taken by management to mitigate or eliminate such risks.
Code of Business Conduct and Ethics
We have adopted a Code of Business Conduct and Ethics that applies to all our directors, officers (including our Chief Executive Officer, Chief Financial Officer and any person performing similar functions) and employees. We have made our Code of Business Conduct and Ethics available on our website at www.beyondair.net under “Investors—Governance—Governance Documents”. We expect that any future amendments to our Code of Business Conduct and Ethics or any waivers of its requirements will be disclosed on our website.
Delinquent Section 16(a) Reports
Section 16(a) of the Exchange Act requires our directors and executive officers, and persons who own more than 10% of a registered class of our equity securities, to file with the SEC initial reports of ownership and reports of changes in ownership of our common stock and other equity securities. Executive officers, directors and greater than 10% stockholders are required by SEC regulations to furnish us with copies of all Section 16(a) forms they file.
SEC regulations require us to identify in this Annual Report anyone who filed a required report late during the most recent fiscal year. To our knowledge, based solely on a review of the copies of such reports furnished to us and written representations that no other reports were required, during the year ended March 31, 2023, all of our officers, directors and greater than 10% beneficial owners have complied with Section 16(a) filing requirements on a timely basis, other than set forth below:
| Filer | Number of Late Reports | Number of Transactions not Reported Timely | ||
| Amir Avniel | One Form 4 | One |
ITEM 11. EXECUTIVE COMPENSATION
Processes and Procedures for Compensation Decisions
Our compensation committee is responsible for the executive compensation programs for our executive officers and reports to our Board of Directors on its discussions, decisions and other actions. Our compensation committee reviews and approves corporate goals and objectives relating to the compensation of our Chief Executive Officer, evaluates the performance of our Chief Executive Officer in light of those goals and objectives and determines and approves the compensation of our Chief Executive Officer based on such evaluation. Our compensation committee has the sole authority to determine our Chief Executive Officer’s compensation. In addition, our compensation committee, in consultation with our Chief Executive Officer, reviews and approves all compensation for other officers, as well as the directors.
|
|
The compensation committee is authorized to retain the services of one or more executive compensation and benefits consultants or other outside experts or advisors as it sees fit, in connection with the establishment of our compensation programs and related policies.
The compensation committee has full authority to form and delegate authority to one or more subcommittees consisting solely of one or more members of the compensation committee as it deems appropriate from time to time. The compensation committee may delegate to the Chief Executive Officer or any other executive officer the authority to grant equity awards to employees of the Company who are not directors or officers of the Company, on such terms and subject to such limitations as the compensation committee may determine in compliance with Delaware corporate law.
Summary Compensation Table
The following table provides information regarding the compensation earned by our named executive officers for the years ended March 31, 2023 and March 31, 2022.
|
Name and Principal Position |
Year | Salary Cost | Restricted Stock Awards (A)(C) |
Option Awards (B)(C) |
Total | ||||||||||||||
| Steven A. Lisi | 2023 | $ | 650,000 | $ | - | $ | 8,278,000 | $ | 8,928,000 | ||||||||||
| Chief Executive Officer and Chairman of the Board | 2022 | $ | 450,000 | $ | 2,000,800 | $ | 6,942,000 | $ | 9,392,800 | ||||||||||
| Amir Avniel | 2023 | $ | 400,000 | $ | 1,507,200 | $ | 2,040,000 | $ | 3,947,200 | ||||||||||
| President, Chief Business Officer and Director | 2022 | $ | 400,000 | $ | 1,000,400 | $ | 2,921,000 | $ | 4,321,400 | ||||||||||
| Mike Gaul | 2023 | $ | 350,000 | $ | - | $ | 782,000 | $ | 1,132,000 | ||||||||||
| Chief Operating Officer | 2022 | $ | 250,000 | $ | 335,975 | $ | 360,500 | $ | 946,475 | ||||||||||
| (A) | The fair market value of the restricted stock units for stock-based expense is equal to the closing price of the Company’s stock at the date of grant based upon the total award. The restricted stock units vest over five years. |
| (B) | This column represents the grant date fair value of awards from both Beyond Air and Beyond Cancer in accordance with stock-based compensation rules under Accounting Standards Codification (“ASC”) Topic 718. For a more detailed discussion of the valuation model and assumptions used to calculate the fair value of each restricted stock award and option award, refer to Note 5. |
| (C) | The respective agreements include a change of control provision that would automatically vest any unvested restricted stock units or unvested stock options if triggered. |
|
|
Employment Agreements with Named Executive Officers
Our employment agreements with our named executive officers contain provisions standard for a company in our industry regarding non-competition, confidentiality of information and assignment of inventions.
Employment Agreement with Steven Lisi
On June 30, 2018, we entered into an employment agreement with Mr. Lisi to serve as our Chief Executive Officer with an annual base salary of $450,000, subject to review of the compensation committee at least annually. On April 1, 2022, Mr. Lisi’s annual salary was increased to $650,000. In addition to his base salary, Mr. Lisi is eligible to receive a short-term incentive bonus equal to a percentage of his base salary in effect at the end of the fiscal year, based partially on performance weighted bonus objectives established for Mr. Lisi by the Board of Directors (which includes both corporate objectives and individual objectives) for the fiscal year, with such objectives to be discussed with Mr. Lisi prior to being established, and partially based on the discretion of the Board of Directors. The target bonus percentage each fiscal year is an amount equal to 60% of Mr. Lisi’s base salary in effect at the end of each fiscal year. However, the actual short-term incentive bonus as determined by the Board of Directors may range from 0% to higher than 100% of the base salary. Any short-term incentive bonus shall be paid on or before April 15 of the following year and may include cash, stock options and restricted stock awards. If paid in stock options or restricted stock awards, the short-term incentive bonus must be paid separately from, and independently of, any long-term equity incentive award. Pursuant to the employment agreement, Mr. Lisi is also eligible to receive awards of stock options or restricted stock grants as may be determined from time to time by the Board of Directors or the compensation committee of the Board of Directors. Pursuant to the terms and conditions of employment, Mr. Lisi received options to purchase 400,000 shares of our common stock at an exercise price of $4.25 per share. 25% of the options vested on June 30, 2018 and thereafter an additional 25% vested on December 31, 2018 and December 31st of each of the two ensuing years thereafter until the options vested in full. The options expire on the tenth anniversary of the date of grant and were fully vested as of March 31, 2021.
In the event of Mr. Lisi’s termination without “cause” or his resignation for “good reason”, as such terms are defined in his employment agreement, Mr. Lisi, subject to his execution and non-revocation of a release of claims and compliance with the restrictive covenants set forth in his employment agreement, will be entitled to (i) severance equal to twenty-four months of base salary, payable in a lump sum, (ii) a lump sum payment equal to 1.5 times that of the most recent earned short-term incentive award, (iii) all outstanding options and restricted common stock awards held by Mr. Lisi would automatically vest and (iv) provided Mr. Lisi timely elects to continue health care coverage under the Consolidated Omnibus Reconciliation Act of 1985 (“COBRA”), continued participation by Mr. Lisi and his eligible dependents in our standard group medical and dental plans until the earlier of (a) the end of the 18th month following Mr. Lisi’s termination and (b) the date Mr. Lisi secures subsequent employment with medical and dental coverage.
In the event of Mr. Lisi’s termination without “cause” or his resignation for “good reason”, in each case within three months prior to a “change of control”, as such term is defined in Mr. Lisi’s employment agreement, or within 18 months following a “change of control”, Mr. Lisi, subject to his execution and non-revocation of a release of claims and compliance with the restrictive covenants set forth in his employment agreement, will be entitled to (i) a one-time grant of 650,000 shares of our common stock, (ii) all outstanding options and restricted common stock awards held by Mr. Lisi would automatically vest and (iii) provided Mr. Lisi timely elects to continue health care coverage under COBRA, continued participation by Mr. Lisi and his eligible dependents in our standard group medical and dental plans until the earlier of (a) the end of the 24th month following Mr. Lisi’s termination and (b) the date Mr. Lisi secures subsequent employment with medical and dental coverage.
Mr. Lisi’s employment agreement contains restrictive covenants relating to non-disclosure of confidential information, assignment of inventions, and non-solicitation of employees and customers that runs for a period of one year following his termination of employment for any reason.
|
|
Employment Agreement with Amir Avniel
On June 30, 2018, we entered into an employment agreement with Mr. Avniel to serve as our President and Chief Operating Officer with an annual base salary of $400,000, subject to review of the compensation committee at least annually. In addition to his base salary, Mr. Avniel is eligible to receive a short-term incentive bonus equal to a percentage of his base salary in effect at the end of the fiscal year, based partially on performance weighted bonus objectives established for Mr. Avniel by the Board of Directors (which includes both corporate objectives and individual objectives) for the fiscal year, with such objectives to be discussed with Mr. Avniel prior to being established, and partially based on the discretion of the Board of Directors. The target bonus percentage each fiscal year is an amount equal to 60% of Mr. Avniel’s base salary in effect at the end of each fiscal year. However, the actual short-term incentive bonus as determined by the Board of Directors may range from 0% to higher than 100% of the base salary. Any short-term incentive bonus shall be paid on or before April 15 of the following year and may include cash, stock options and restricted stock awards. If paid in stock options or restricted stock awards, the short-term incentive bonus must be paid separately from, and independently of, any long-term equity incentive award. Pursuant to the employment agreement, Mr. Avniel is also eligible to receive awards of stock options or restricted stock grants as may be determined from time to time by the Board of Directors or the compensation committee of the Board of Directors. Pursuant to the terms and conditions of employment, Mr. Avniel received options to purchase 250,000 shares of our common stock at an exercise price of $4.25 per share. 25% of the options vested on June 30, 2018 and thereafter an additional 25% vested on December 31, 2018 and December 31st of each of the two ensuing years thereafter until the options vested in full. The options expire on the tenth anniversary of the date of grant and the options were fully vested as of March 31, 2021. On July 2, 2022, Mr. Avniel stepped down as Chief Operating Officer and assumed the role of Chief Business Officer. Notwithstanding this titular change, the other terms of his employment agreement with us, did not change.
In the event of Mr. Avniel’s termination without “cause” or his resignation for “good reason”, as such terms are defined in his employment agreement, Mr. Avniel, subject to his execution and non-revocation of a release of claims and compliance with the restrictive covenants set forth in his employment agreement, will be entitled to (i) severance equal to twenty-four months of base salary, payable in a lump sum, (ii) a lump sum payment equal to 1.5 times that of the most recent earned short-term incentive award, (iii) all outstanding options and restricted common stock awards held by Mr. Avniel would automatically vest and (iv) provided Mr. Avniel timely elects to continue health care coverage under COBRA, continued participation by Mr. Avniel and his eligible dependents in our standard group medical and dental plans until the earliest of (a) the end of the 18th month following Mr. Avniel’s termination and (b) the date Mr. Avniel secures subsequent employment with medical and dental coverage.
In the event of Mr. Avniel’s termination without “cause” or his resignation for “good reason”, in each case within three months prior to a “change of control”, as such term is defined in Mr. Avniel’s employment agreement, or within 18 months following a “change of control”, Mr. Avniel, subject to his execution and non-revocation of a release of claims and compliance with the restrictive covenants set forth in his employment agreement, will be entitled to (i) a one-time grant of 350,000 shares of our common stock, (ii) all outstanding options and restricted common stock awards held by Mr. Avniel would automatically vest and (iii) provided Mr. Avniel timely elects to continue health care coverage under COBRA, continued participation by Mr. Avniel and his eligible dependents in our standard group medical and dental plans until the earliest of (a) the end of the 24th month following Mr. Avniel’s termination and (b) the date Mr. Avniel secures subsequent employment with medical and dental coverage.
Mr. Avniel’s employment agreement contains restrictive covenants relating to non-disclosure of confidential information, assignment of inventions, and non-solicitation of employees and customers that runs for a period of one year following his termination of employment for any reason.
|
|
Employment Agreement with Mike Gaul
On April 24, 2020, we entered into an Employment Agreement with Michael Gaul to serve as Senior VP, Operations of the Company, effective May 4, 2020. Mr. Gaul’s Employment Agreement provides that his employment will continue until either the Company or Mr. Gaul terminates his employment in accordance with the terms of the Employment Agreement. Pursuant to the employment Agreement, Mr. Gaul is entitled to receive an annual base salary of $250,000, which is subject to adjustment pursuant to the Company’s employee compensation practices. In April 2022, Mr. Gaul’s base salary was increased to $350,000. In addition, pursuant to the Employment Agreement, Mr. Gaul is eligible to be considered for an incentive bonus for each fiscal year of the Company, which will be awarded based on objective or subjective criteria established by the Chief Executive Officer and approved by the Board or a committee thereof. As of his start date, the Company also granted Mr. Gaul a stock option award, subject to the Company’s 2013 Equity Incentive Plan, for the purchase of 10,000 shares of the Company’s common stock, at an exercise price of $5.32. This option vests over a four-year period, with 25% of the shares underlying the stock option award vesting on the first anniversary of the date of grant after one-year of continuous service and annually thereafter in three equal installments. This option is in addition to those granted previously to Mr. Gaul pursuant to that certain Consulting Agreement, dated February 26, 2020. On July 1, 2022, Mr. Gaul stepped down as Senior VP, Operations of the Company and assumed the role of Chief Operating Officer. Notwithstanding this titular change, the other terms of his Employment Agreement with us did not change.
Under the Employment Agreement, termination of Mr. Gaul’s employment by the Company without cause, or by Mr. Gaul for “Good Reason” (as such term is defined in the Employment Agreement), will require the Company to pay severance to Mr. Gaul. Upon any such termination, and subject to the terms of the Employment Agreement and the Company’s policies, Mr. Gaul will be entitled to receive his base salary for a period of six months, plus an additional month of severance payments for every two months of employment, with such additional amounts to be cumulative and not to exceed a total of 12 months of severance payments. The Company will also continue to contribute to Mr. Gaul’s health and dental benefits in the same proportion as during employment for the same duration as severance payments are made. In the event of a Change of Control (as defined in the Employment Agreement), Mr. Gaul will receive severance payments equal to six months’ base salary and the Company will continue to provide health and dental benefits in the same proportion as during employment for six months. Mr. Gaul will also receive an additional month of severance for every two months of employment, with such amounts to be cumulative and not to exceed a total of 12 months of severance payments. In addition, all of Mr. Gaul’s options to acquire Company stock and restricted common stock awards which have not vested as of the date of termination will become immediately vested as of the date of termination.
The Employment Agreement also contains customary non-solicitation and non-competition covenants, which covenants remain in effect for 1 year following any cessation of employment with respect to Mr. Gaul.
Option Awards Granted During the Year Ended March 31, 2023
On November 3, 2022, Messrs. Lisi and Avniel were granted options to purchase 550,000 and 200,000 shares of the common stock of Beyond Cancer, respectively, with an exercise price of $2.25 per share. The options vest annually over four years commencing on December 31, 2023.
On March 29, 2023, Messrs. Lisi, Avniel and Gaul were granted options to purchase 800,000, 80,000 and 170,000 shares of our common stock, respectively, with an exercise price of $6.28 per share, which was equal to the closing price of our common stock on the date of grant. The options vest in equal annual installments over four years commencing on December 31, 2023.
On March 29, 2023, Mr. Avniel was granted restricted stock units to receive 240,000 shares of our common stock, respectively. The restricted stock units vest in equal annual installments over five years commencing on December 15, 2023.
|
|
Outstanding Equity Awards as of March 31, 2023
| Equity awards | ||||||||||||||||||||||||||||||
| Name | Date
of Grant |
Number
of securities underlying unexercised options (#) exercisable |
Number
of securities underlying unexercised options (#) unexercisable |
Equity incentive plan awards: Number of securities underlying unexercised unearned options (#) |
Option exercise price ($) |
Option expiration date |
Number
of shares or units of stock that have not vested (#) |
Market (6) |
||||||||||||||||||||||
| Steven A. Lisi | 08/31/2018 | 400,000 | (1) | - | - | 4.25 | 08/13/2029 | - | - | |||||||||||||||||||||
| 03/31/2019 | 250,000 | (2) | - | - | 4.80 | 03/31/2029 | - | - | ||||||||||||||||||||||
| 03/11/2020 | 52,500 | (2) | 17,500 | - | 5.32 | 03/11/2030 | - | 118,125 | ||||||||||||||||||||||
| 03/04/2021 | 100,000 | (2) | 100,000 | - | 5.45 | 03/04/2031 | - | 675,000 | ||||||||||||||||||||||
| 03/03/2022 | 70,000 | (2) | 210,000 | - | 6.87 | 03/03/2032 | - | 1,417,500 | ||||||||||||||||||||||
| 03/29/2023 | (2) | 800,000 | - | 6.28 | 03/29/2033 | - | 5,400,000 | |||||||||||||||||||||||
| 12/31/2018 | - | (3) | - | - | - | - | 17,600 | 118,800 | ||||||||||||||||||||||
| 01/01/2019 | - | (3) | - | - | - | - | 2,400 | 16,200 | ||||||||||||||||||||||
| 12/31/2019 | - | (3) | - | - | - | - | 41,800 | 282,150 | ||||||||||||||||||||||
| 01/04/2020 | - | (3) | - | - | - | - | 6,200 | 41,850 | ||||||||||||||||||||||
| 10/05/2021 | - | (4) | - | - | - | - | 60,000 | 405,000 | ||||||||||||||||||||||
| 03/03/2022 | - | (4) | - | - | - | - | 112,000 | 765,000 | ||||||||||||||||||||||
| Amir Avniel | 02/20/2017 | 100,000 | (1) | - | - | 4.25 | 02/20/2027 | - | - | |||||||||||||||||||||
| 08/31/2018 | 250,000 | (1) | - | - | 4.25 | 08/13/2029 | - | - | ||||||||||||||||||||||
| 03/31/2019 | 140,000 | (2) | - | - | 4.80 | 03/31/2029 | - | - | ||||||||||||||||||||||
| 03/11/2020 | 30,000 | (2) | 10,000 | - | 5.32 | 03/11/2030 | - | 67,500 | ||||||||||||||||||||||
| 03/04/2021 | 50,000 | (2) | 50,000 | - | 5.45 | 03/04/2031 | - | 337,500 | ||||||||||||||||||||||
| 03/03/2022 | 35,000 | (2) | 105,000 | - | 6.87 | 03/03/2032 | - | 708,750 | ||||||||||||||||||||||
| 03/29/2023 | - | (2) | 80,000 | - | 6.28 | 03/29/2033 | - | 540,000 | ||||||||||||||||||||||
| 12/31/2018 | - | (3) | - | - | - | - | 9,000 | 120,240 | ||||||||||||||||||||||
| 01/01/2019 | - | (3) | - | - | - | - | 2,400 | 32,064 | ||||||||||||||||||||||
| 12/31/2019 | - | (3) | - | - | - | - | 20,200 | 202,404 | ||||||||||||||||||||||
| 01/04/2020 | - | (3) | - | - | - | - | 5,800 | 58,116 | ||||||||||||||||||||||
| 10/05/2021 | - | (4) | - | - | - | - | 30,000 | 267,200 | ||||||||||||||||||||||
| 03/03/2022 | - | (4) | - | - | - | - | 56,000 | 467,600 | ||||||||||||||||||||||
| 03/28/2023 | - | (2) | - | - | - | - | 240,000 | 1,620,000 | ||||||||||||||||||||||
| Mike Gaul | 03/11/2020 | 30,000 | (2) | 10,000 | - | 5.32 | 03/11/2030 | - | 67,500 | |||||||||||||||||||||
| 05/04/2020 | 5,000 | (5) | 5,000 | - | 5.32 | 05/04/2030 | - | 33,750 | ||||||||||||||||||||||
| 03/04/2021 | 12,500 | (2) | 12,500 | - | 5.45 | 03/04/2031 | - | 84,375 | ||||||||||||||||||||||
| 03/03/2022 | 17,500 | (2) | 52,500 | - | 6.87 | 03/03/2032 | - | 354,375 | ||||||||||||||||||||||
| 03/29/2023 | - | (2) | 170,000 | - | 6.28 | 03/29/2033 | - | 1,147,500 | ||||||||||||||||||||||
| 10/05/2021 | - | (4) | - | - | - | - | 7,500 | 50,625 | ||||||||||||||||||||||
| 03/03/2022 | - | (4) | - | - | - | - | 24,000 | 162,000 | ||||||||||||||||||||||
| (1) | 25% options vests immediately, 25% vests December 31, 2018, 25% vests each following December 31. | |
| (2) | 25% options vests in December of the year of grant, 25% vests each following December. | |
| (3) | Restricted stock units vest 20% per year at the date of grant. | |
| (4) | Restricted stock units vest 20% per year, with the first tranche vesting in December in the year of grant. | |
| (5) | 25% options vests on the anniversary date of the grant, 25% each following year on the anniversary date. | |
| (6) | Market value was calculated based upon the stock price at the last trading day of the fiscal year ended March 31, 2023 ($6.75). |
|
|
Director Compensation
Persons serving as both an Officer and a Director of the Company are only included in the Summary Compensation Table above for the year ended March 31, 2023.
| Name | Fees earned or paid in cash ($) |
Stock awards ($) |
Option awards ($) (1) (2) |
Non-equity incentive plan compensation ($) |
Nonqualified deferred compensation earnings ($) |
All Other Compensation ($) |
Total ($) |
|||||||||||||||||||||
| Dr. William Forbes | - | - | 199,200 | - | - | - | 199,200 | |||||||||||||||||||||
| Ron Bentsur | - | - | 199,200 | - | - | - | 199,200 | |||||||||||||||||||||
| Erick J. Lucera | - | - | 199,200 | - | - | - | 199,200 | |||||||||||||||||||||
| Yoori Lee | - | - | 199,200 | - | - | - | 199,200 | |||||||||||||||||||||
| Robert F. Carey | - | - | 324,600 | - | - | - | 324,600 | |||||||||||||||||||||
| (1) | During the year ended March 31, 2023, each director on the Board of Directors received options to purchase 40,000 shares of Beyond Air’s common stock and each option expires in ten years from the date of grant. Mr. Carey also serves on the Board of Directors of Beyond Cancer and received options to purchase 15,000 shares of Beyond Cancer’s common stock. Compensation expense was based upon the grant date fair value of the award in accordance with stock-based compensation rules under ASC Topic 718. As of March 31, 2023, the aggregate number of options to purchase Beyond Air’s common stock held by each director on the Board of Directors was as follows: (i) 153,000 by Dr. Forbes; (ii) 144,000 by Mr. Bentsur; (iii) 165,000 by Mr. Lucera; (iv) 160,000 by Ms. Lee; and (v) 146,000 by Mr. Carey. As of March 31, 2023, Mr. Carey additionally held an aggregate of 65,000 options to purchase Beyond Cancer’s common stock. |
| (2) | The respective agreements include a change of control provision that would automatically vest any unvested stock options if triggered. |
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
The table below sets forth information with respect to the beneficial ownership of our common stock by each person known by us to beneficially own more than 5.0% of any class of our voting securities together with:
| ● | each of our directors; | |
| ● | each of our named executive officers; and | |
| ● | all of our directors and executive officers as a group. |
The percentages of common stock beneficially owned are reported on the basis of regulations of the SEC governing the determination of beneficial ownership of securities. Except as indicated in the footnotes to this table, each beneficial owner named in the table below has sole voting and sole investment power with respect to all shares beneficially owned. Percentage computations are based on 31,438,506 shares of our common stock outstanding as of June 22, 2023.
|
|
Under the terms of the warrants issued by us to Charles Mosseri Marlio, he may not exercise a warrant to the extent such exercise would cause him, together with his affiliates and any other persons acting as a group with him or any of his affiliates, to have acquired a number of shares of common stock which would exceed 9.985% (subject to an increase of such percentage to 9.99% on 61 days’ notice by the holder to the Company) of our then outstanding common stock, excluding for purposes of such determination shares of common stock issuable upon exercise of warrants that have not been exercised. We refer to the foregoing limitation applicable to Mr. Mosseri Marlio as the “Ownership Cap.” The share numbers in the table below do not reflect the Ownership Cap, but the figures contained in the “Percentage of Outstanding Shares” column reflect the Ownership Cap applicable to Mr. Mosseri Marlio.
| Name and Address of Beneficial Owner (1) | Number
of Shares |
Percentage
of Outstanding Shares % (2) |
||||||
| 5% Owners | ||||||||
| Charles Mosseri Marlio | 3,018,707 | (3) | 9.6 | |||||
| Executive Officers and Directors | ||||||||
| Steven A. Lisi | 2,246,349 | (4) | 6.6 | |||||
| Amir Avniel | 957,894 | (5) | 3.0 | |||||
| Ron Bentsur | 207,486 | (6) | * | |||||
| Dr. William Forbes | 86,605 | (7) | * | |||||
| Robert F. Carey | 951,246 | (8) | 2.9 | |||||
| Erick Lucera | 88,092 | (9) | * | |||||
| Yoori Lee | 91,404 | (10) | * | |||||
| Michael Gaul | 109,150 | (11) | * | |||||
| Executive Officers and Directors as a Group (Ten persons) | 4,766,323 | 13.7 | ||||||
| * | Less than one percent (1.0%). |
| (1) | The address of these persons, unless otherwise noted, is c/o Beyond Air, Inc., 900 Stewart Avenue, Suite 301 Garden City, New York, 11530. |
| (2) | Shares of common stock beneficially owned and, except as limited by the Ownership Cap, the respective percentages of beneficial ownership of common stock includes for each person or entity shares issuable on the exercise of all options and warrants and the conversion of other convertible securities beneficially owned by such person or entity that are currently exercisable or will become exercisable or convertible within 60 days following June 22, 2023. Such shares, however, are not included for the purpose of computing the percentage ownership of any other person. |
| (3) | Based, in part, on information provided on Schedule 13G/A filed with the SEC on February 14, 2023. Includes 108,816 shares of common stock issuable upon exercise of the warrants issued to Mr. Mosseri Marlio in connection with Facility Agreement in March 2020. |
| (4) | Includes 872,500 vested options to purchase shares of common stock. |
| (5) | Includes 605,000 vested options to purchase common stock and 32,666 shares of common stock held by Dandelion Investments Ltd., over which Mr. Avniel has sole voting and dispositive power. |
| (6) | Includes 62,750 vested options to purchase shares of common stock. |
| (7) | Includes 71,750 vested options to purchase shares of common stock. |
| (8) | Includes 64,750 vested options to purchase shares of common stock. |
| (9) | Includes 83,750 vested options to purchase shares of common stock. |
| (10) | Includes 78,750 vested options to purchase shares of common stock. |
| (11) | Includes 67,500 vested options to purchase shares of common stock. |
|
|
Equity Compensation Plan Information
We maintain the Fifth Amended and Restated 2013 Beyond Air Equity Incentive Plan (the “2013 BA Plan”). The 2013 BA Plan provides for the grant of incentive stock options, non-statutory stock options, restricted stock awards, restricted stock unit awards, stock appreciation rights, performance share awards, and other stock-based awards (collectively, the “stock awards”). Stock awards may be granted under the 2013 BA Plan to our employees, directors and consultants, other than incentive stock options which may only be granted to employees of the Company. The maximum number of shares of common stock available for issuance under the 2013 BA Plan is 10,600,000 shares.
The 2013 BA Plan is scheduled to terminate on August 13, 2028. No stock awards shall be granted pursuant to the 2013 BA Plan after such date, but awards theretofore granted may extend beyond that date. The Board may suspend or terminate the 2013 BA Plan at any earlier date. No stock awards may be granted under the 2013 BA Plan while the Plan is suspended or after it is terminated.
The following table summarizes the total number of outstanding options and shares available for other future issuances of options under the 2013 BA Plan and the 2021 Employee Stock Purchase Plan as of March 31, 2023:
| Plan Category | Number of Shares to be Issued Upon Exercise of Outstanding Options, Warrants and Rights |
Weighted- Average Exercise Price of Outstanding Options, Warrants and Rights |
Number of Shares Remaining Available for Future Issuance Under the Equity Compensation Plan (Excluding Shares in First Column) |
|||||||||
| Equity compensation plans approved by stockholders (1) | 6,812,000 | $ | 6.09 | 1,133,221 | (3) | |||||||
| Equity compensation plans not approved by stockholders (2) | 1,386,881 | $ | 4.67 | - | ||||||||
| Total | 8,198,881 | $ | 5.85 | 1,133,221 | ||||||||
(1) Represents shares of common stock reserved for future issuance upon exercise of outstanding stock options under the 2013 BA Plan that were approved by our stockholders.
(2) Represents shares of common stock issuable upon exercise of outstanding stock options under the 2013 BA Plan that were not approved by our stockholders including 125,000 options issued as inducement awards.
(3) Represents 383,221 shares of common stock reserved for future issuance under the 2013 BA Plan and 750,000 shares of common stock reserved for future issuance under the 2021 Employee Stock Purchase Plan.
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
Transactions with Related Persons
Under SEC rules, related-party transactions are those transactions to which we are or may be a party in which the amount involved exceeds the lesser of $120 thousand or 1% of the average of total assets at year-end for the last two completed fiscal years, and in which any of our directors or executive officers or any other related person had or will have a direct or indirect material interest, excluding, among other things, compensation arrangements with respect to employment and Board of Directors membership. Our audit committee could approve a related-party transaction if it determines that the transaction is in our best interests. Our directors are required to disclose to this committee or the full Board of Directors any potential conflict of interest, or personal interest in a transaction that our Board of Directors is considering. Our executive officers are required to disclose any related-party transaction to the audit committee. We also poll our directors on an annual basis with respect to related-party transactions and their service as an officer or director of other entities. Any director involved in a related-party transaction that is being reviewed or approved must recuse himself or herself from participation in any related deliberation or decision. Whenever possible, the transaction should be approved in advance and if not approved in advance, must be submitted for ratification as promptly as practical.
|
|
Since April 1, 2021, there were no transactions to which we have been a party in which the amount involved exceeded or will exceed the lesser of $120,000 or one percent of the average of the company’s total assets at year-end for the last two completed fiscal years, and in which any of our directors, executive officers or beneficial owners of more than 5% of our capital stock, or any members of their immediate family, had or will have a direct or indirect material interest, other than compensation arrangements that are described in the “Executive Compensation” and “Director Compensation” sections of this Annual Report .
Director Independence
Our Board of Directors has determined that each of Ron Bentsur, Erick Lucera, Yoori Lee, William Forbes and Robert F. Carey is independent within the meaning of Rule 5605(a)(2) of the Nasdaq Listing Rules and the rules and regulations promulgated by the SEC. In making its independence determinations, the Board of Directors sought to identify and analyze all of the facts and circumstances related to any relationship between a director, his immediate family and the Company and our affiliates and did not rely on categorical standards other than those contained in the Nasdaq rule referenced above.
ITEM 14. PRINCIPAL ACCOUNTANT FEES AND SERVICES
Audit Fees
In September 2022, Marcum LLP acquired certain assets of Friedman LLP, at which point the Company’s auditor became Marcum LLP. The aggregate fees billed for (i) the fiscal year ended March 31, 2022 for professional services rendered by Friedman LLP for the audit of our annual financial statements provided by Friedman LLP, and (ii) the fiscal year ended March 31, 2023 for professional services rendered in part by Friedman LLP and in part by Marcum LLP, in connection with statutory and regulatory filings or engagements for this fiscal period were as follows:
|
Year Ended March 31, 2023 |
Year Ended March 31, 2022 |
|||||||
| Audit Fees | $ | 267,580 | $ | 215,800 | ||||
| Audit Related Fees | $ | - | $ | - | ||||
| Tax Fees | $ | - | $ | - | ||||
| All Other Fees | $ | 39,140 | $ | - | ||||
| Total | $ | 306,720 | $ | 215,800 | ||||
In the above table, “audit fees” are fees billed by our independent registered public accounting firm for services provided in auditing our annual financial statements for the subject year. Audit fees also include professional services performed for filing of our registration statement on Form S-3 for equity offerings, Form S-8 for shares of our common stock underlying our 2013 Equity Incentive Option Plan and for the resale of certain shares of our common stock and other filings. “Audit-related fees” are fees not included in audit fees that are billed by the independent registered public accounting firm for assurance and related services that are reasonably related to the performance of the audit review of our financial statements. “Tax fees” are fees billed by the independent registered public accounting firm for professional services rendered for tax compliance, tax advice and tax planning. “All other fees” are fees billed by the independent registered public accounting firm for products and services not included in the foregoing categories.
Policy on Pre-Approval by Audit Committee of Services Performed by Independent Auditors
The audit committee pre-approves all services provided by our independent registered public accounting firm. All of the above services and fees were reviewed and approved by the audit committee before the respective services were rendered.
The Board of Directors has considered the nature and amount of fees billed by Marcum LLP and believes that the provision of services for activities unrelated to the audit, if any, is compatible with maintaining Marcum LLP’s independence.
|
|
PART IV
ITEM 15. EXHIBITS AND FINANCIAL STATEMENT SCHEDULES
| 1. | Financial Statements. |
See Index to Consolidated Financial Statements on page F-1.
2. Finance Statement Schedules.
All schedules are omitted because they are not applicable or the required information is shown in the financial statements or notes thereto.
3. Exhibits
|
|
|
|
|
|
+ Management contract or compensation plan arrangement
* Pursuant to Item 601(b)(10) of Regulation S-K, portions of this exhibit have been omitted as the registrant has determined that the omitted information is not material and is the type that registrant treats as private or confidential.
** Filed herewith
*** Furnished herewith.
Item 16. Form 10-K Summary
Information with respect to this item is not required and has been omitted at the Company’s option.
|
|
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
Date: June 22, 2023
| BEYOND AIR, INC. | ||
| By: | /s/ Steven Lisi | |
|
Steven Lisi Chairman and Chief Executive Officer (Principal Executive Officer) |
||
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| Name | Title | Date | ||
| /s/ Steven Lisi | Chairman and Chief Executive Officer | June 22, 2023 | ||
| Steven Lisi | (Principal Executive Officer) | |||
| /s/ Douglas Larson | Chief Financial Officer (Principal Financial | June 22, 2023 | ||
| Douglas Larson | Officer and Principal Accounting Officer) | |||
| /s/ Amir Avniel | Chief Business Officer, President and Director | June 22, 2023 | ||
| Amir Avniel | ||||
| /s/ Erick Lucera | Director | June 22, 2023 | ||
| Erick Lucera | ||||
| /s/ Yoori Lee | Director | June 22, 2023 | ||
| Yoori Lee | ||||
| /s/ William Forbes | Director | June 22, 2023 | ||
| William Forbes | ||||
| /s/ Ron Bentsur | Director | June 22, 2023 | ||
| Ron Bentsur | ||||
| /s/ Robert Carey | Director | June 22, 2023 | ||
| Robert Carey |
|
|
BEYOND AIR, INC. AND SUBSIDIARIES
CONSOLIDATED FINANCIAL STATEMENTS
AS OF MARCH 31, 2023
INDEX
| Page | |
| Reports of Independent Registered Public Accounting Firms Marcum LLP (PCAOB ID:688) and Friedman LLP (PCAOB ID:711) | F-2 |
| Consolidated Balance Sheets | F-4 |
| Consolidated Statements of Operations and Comprehensive Loss | F-5 |
| Consolidated Statements of Changes in Stockholders’ Equity | F-6 |
| Consolidated Statements of Cash Flows | F-8 |
| Notes to Consolidated Financial Statements | F-9 – F-25 |
| F- |
To the Stockholders and Board of Directors of
Beyond Air, Inc.
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheet of Beyond Air, Inc. (the “Company”) as of March 31, 2023, the related consolidated statements of operations and comprehensive loss, stockholders’ equity and cash flows for the year ended March 31, 2023, and the related notes (collectively referred to as the “financial statements”). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of March 31, 2023, and the results of its operations and its cash flows for the year ended March 31, 2023, in conformity with accounting principles generally accepted in the United States of America.
Basis for Opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s financial statements based on our audit. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (“PCAOB”) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audit we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audit included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audit also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audit provides a reasonable basis for our opinion.
Critical Audit Matter
The critical audit matter communicated below is a matter arising from the current period audit of the financial statements that were communicated or required to be communicated to the audit committee and that: (1) relate to accounts or disclosures that are material to the financial statements and (2) involved our especially challenging, subjective, or complex judgments. The communication of the critical audit matter does not alter in any way our opinion on the financial statements, taken as a whole, and we are not, by communicating the critical audit matter below, providing a separate opinion on the critical audit matter or on the accounts or disclosures to which it relates.
Going concern – Assessing the probability of the Company’s ability to continue as a going concern
|
Critical Audit Matter Description |
As described in Note 2 of the financial statements, the Company believes it has adequate cash on hand in addition to cash provided from debt and equity financings subsequent to March 31, 2023, which will provide sufficient liquidity to finance the operating activities of the Company at its current level of operations for at least the next twelve months from the issuance date of these financial statements. We determined the Company’s ability to continue as a going concern is a critical audit matter due to the estimation uncertainty regarding the Company’s future cash flows and the risk of bias in management’s judgments and assumptions in estimating these cash flows. |
|
How We Addressed the Matter in Our Audit
|
Our audit procedures related to the Company’s assertion on its ability to continue as a going concern included the following, among others; we reviewed the design and underlying factors relating to the preparation of forecasted information and considerations of the Company’s obligations; testing the reasonableness of the forecasted operating expenses, as well as the sources of cash used in management’s assessment as to whether the Company has sufficient liquidity to fund operations for at least one year from the financial statement issuance date. This testing included inquiries with management, comparison of prior period forecasts to actual results, a sensitivity analysis, consideration of positive and negative evidence impacting management’s forecasts, and the Company’s financing arrangements in place as of the report date. |
/s/ Marcum llp
We have served as the Company’s auditor since 2019 (such date considers the acquisition of certain assets of Friedman LLP by Marcum LLP effective September 1, 2022)
East Hanover, New Jersey
June 22, 2023
| F- |
To the Board of Directors and
Stockholders of Beyond Air, Inc. and Subsidiaries
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheet of Beyond Air, Inc. and Subsidiaries (the “Company”) as of March 31, 2022, and the related consolidated statements of operations and comprehensive loss, stockholders’ equity and cash flows for the year ended March 31, 2022, and the related notes (collectively referred to as the “financial statements”). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of March 31, 2022, and the results of its operations and its cash flows for the year ended March 31, 2022, in conformity with accounting principles generally accepted in the United States of America.
Basis for Opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s financial statements based on our audit. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (“PCAOB”) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audit we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audit included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audit also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audit provides a reasonable basis for our opinion.
/s/ Friedman LLP
We have served as the Company’s auditor from 2019 through 2022
East Hanover, New Jersey
June 28, 2022
| F- |
BEYOND AIR, INC. AND SUBSIDIARIES
CONSOLIDATED BALANCE SHEETS
(in thousands, except share data)
| March 31, 2023 | March 31, 2022 | |||||||
| ASSETS | ||||||||
| Current assets | ||||||||
| Cash and cash equivalents | $ | 29,158 | $ | 80,242 | ||||
| Marketable securities | 16,724 | - | ||||||
| Restricted cash | 10,129 | 9,988 | ||||||
| Inventory, net | 1,129 | 350 | ||||||
| Grant receivable | 420 | 322 | ||||||
| Other current assets and prepaid expenses | 1,850 | 2,044 | ||||||
| Total current assets | 59,410 | 92,945 | ||||||
| Licensed right to use technology | 1,632 | 1,837 | ||||||
| Right-of-use lease assets | 2,493 | 2,216 | ||||||
| Property and equipment, net | 5,003 | 1,995 | ||||||
| Other assets | 212 | 207 | ||||||
| TOTAL ASSETS | $ | 68,749 | $ | 99,199 | ||||
| LIABILITIES AND STOCKHOLDERS’ EQUITY | ||||||||
| Current liabilities | ||||||||
| Accounts payable | $ | 2,016 | $ | 1,129 | ||||
| Accrued expenses | 16,613 | 8,374 | ||||||
| Operating lease liability, current portion | 376 | 281 | ||||||
| Loans payable, current portion | 775 | 927 | ||||||
| Total current liabilities | 19,780 | 10,712 | ||||||
| Operating lease liability, net | 2,321 | 2,079 | ||||||
| Long-term debt, net | 120 | 200 | ||||||
| Other long-term liabilities | 4,500 | 8,000 | ||||||
| Total liabilities | 26,721 | 20,990 | ||||||
| Stockholders’ equity | ||||||||
| Preferred Stock, $0.0001 par value per share: 10,000,000 shares authorized, 0 shares issued and outstanding | - | - | ||||||
| Common Stock, $0.0001 par value per share: 100,000,000 shares authorized, 30,738,585 and 29,886,173 shares issued and outstanding as of March 31, 2023 and 2022, respectively | 3 | 3 | ||||||
| Treasury stock | (25 | ) | (25 | ) | ||||
| Additional paid-in capital | 217,339 | 196,269 | ||||||
| Accumulated deficit | (179,455 | ) | (123,639 | ) | ||||
| Accumulated other comprehensive income | 53 | 96 | ||||||
| Total stockholders’ equity attributable to Beyond Air, Inc. | 37,915 | 72,704 | ||||||
| Non-Controlling Interests | 4,113 | 5,505 | ||||||
| Total equity | 42,028 | 78,209 | ||||||
| TOTAL LIABILITIES AND STOCKHOLDERS’ EQUITY | $ | 68,749 | $ | 99,199 | ||||
The accompanying notes are an integral part of these consolidated financial statements.
| F- |
BEYOND AIR, INC. AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF OPERATIONS AND COMPREHENSIVE LOSS
(in thousands except share data)
|
Year Ended March 31, 2023 |
Year Ended March 31, 2022 |
|||||||
| Revenue | $ | - | $ | - | ||||
| Cost of revenue | (555 | ) | - | |||||
| Gross loss | (555 | ) | - | |||||
| Operating expenses | ||||||||
| Research and development | (16,810 | ) | (11,802 | ) | ||||
| General and administrative | (34,694 | ) | (18,408 | ) | ||||
| Milestone expenses incurred upon regulatory approval | - | (10,500 | ) | |||||
| Total operating expenses | (51,504 | ) | (40,710 | ) | ||||
| Loss from operations | $ | (52,059 | ) | $ | (40,710 | ) | ||
| Other income (expense) | ||||||||
| Estimated contingent loss | (7,863 | ) | (2,435 | ) | ||||
| Dividend and interest income | 656 | 4 | ||||||
| Interest and finance expense | (30 | ) | (775 | ) | ||||
| Foreign exchange loss | (105 | ) | (144 | ) | ||||
| Total other loss / expense | (7,342 | ) | (3,350 | ) | ||||
| Net loss before income taxes | (59,401 | ) | (44,060 | ) | ||||
| Provision for income taxes | - | - | ||||||
| Net loss | $ | (59,401 | ) | $ | (44,060 | ) | ||
| Less: net loss attributable to non-controlling interests | (3,585 | ) | (882 | ) | ||||
| Net loss attributable to Beyond Air, Inc. | $ | (55,816 | ) | $ | (43,177 | ) | ||
| Other comprehensive loss, net of tax: | ||||||||
| Foreign currency translation gain (loss) | (43 | ) | 96 | |||||
| Comprehensive loss attributable to Beyond Air, Inc. | $ | (55,859 | ) | $ | (43,081 | ) | ||
| Net basic and diluted loss per share attributable to Beyond Air, Inc | $ | (1.86 | ) | $ | (1.68 | ) | ||
| Weighted average number of shares of common stock outstanding – basic and diluted | 29,973,639 | 25,668,230 | ||||||
The accompanying notes are an integral part of these consolidated financial statements.
| F- |
BEYOND AIR, INC. AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF CHANGES IN STOCKHOLDERS’ EQUITY
FOR THE YEAR ENDED MARCH 31, 2022
(in thousands except share data)
| Common Stock | Treasury | Additional Paid-in |
Accumulated |
Accumulated Other |
Non-Controlling | Total | ||||||||||||||||||||||||||
| Number | Amount | Stock | Capital | Deficit | Income | Interest | Equity | |||||||||||||||||||||||||
| Balance as of April 1, 2021 | 21,828,244 | $ | 2 | $ | (25 | ) | $ | 110,948 | $ | (80,462 | ) | $ | - | $ | - | $ | 30,464 | |||||||||||||||
| Issuance of common stock – At The Market equity offering | 4,053,424 | - | 36,489 | 36,489 | ||||||||||||||||||||||||||||
| Issuance of common stock pursuant to a Purchase Agreement, net | 1,100,000 | - | 11,138 | 11,138 | ||||||||||||||||||||||||||||
| Issuance of common stock upon the exercise of warrants – for cash | 1,630,002 | - | 251 | 251 | ||||||||||||||||||||||||||||
| Issuance of common stock upon excise of warrants – cashless | 894,823 | - | - | - | ||||||||||||||||||||||||||||
| Issuance of common stock upon exercise of options – for cash | 55,875 | - | 5,966 | 5,966 | ||||||||||||||||||||||||||||
| Issuance of common stock upon exercise of options – cashless | 121,205 | - | - | - | ||||||||||||||||||||||||||||
| Vested restricted stock | 202,600 | - | - | - | ||||||||||||||||||||||||||||
| Beyond Cancer issuance of stock | 24,000 | 6,000 | 30,000 | |||||||||||||||||||||||||||||
| Net recovery of short swing profits | - | - | 33 | 33 | ||||||||||||||||||||||||||||
| Stock-based compensation | - | - | 7,444 | 387 | 7,831 | |||||||||||||||||||||||||||
| Other comprehensive income | 96 | 96 | ||||||||||||||||||||||||||||||
| Net loss | - | - | - | - | (43,177 | ) | (882 | ) | (44,060 | ) | ||||||||||||||||||||||
| Balance as of March 31, 2022 | 29,886,173 | $ | 3 | $ | (25 | ) | $ | 196,269 | $ | (123,639 | ) | $ | 96 | $ | 5,505 | $ | 78,209 | |||||||||||||||
The accompanying notes are an integral part of these consolidated financial statements.
| F- |
BEYOND AIR, INC. AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF CHANGES IN STOCKHOLDERS’ EQUITY
FOR THE YEAR ENDED MARCH 31, 2023
(in thousands except share data)
| Common Stock | Treasury | Additional Paid-in |
Accumulated |
Accumulated Other |
Non-Controlling | Total | ||||||||||||||||||||||||||
| Number | Amount | Stock | Capital | Deficit | Income | Interest | Equity | |||||||||||||||||||||||||
| Balance as of April 1, 2022 | 29,886,173 | $ | 3 | $ | (25 | ) | $ | 196,269 | $ | (123,639 | ) | $ | 96 | $ | 5,505 | $ | 78,209 | |||||||||||||||
| Issuance of common stock – At The Market equity offering | 572,078 | - | - | 3,705 | 3,705 | |||||||||||||||||||||||||||
| Issuance of common stock upon exercise of options – cashless | 5,734 | - | - | - | - | - | - | - | ||||||||||||||||||||||||
| Vested restricted stock | 274,600 | - | - | - | - | - | - | - | ||||||||||||||||||||||||
| Stock-based compensation | - | - | 17,365 | - | - | 2,194 | 19,559 | |||||||||||||||||||||||||
| Other comprehensive loss | (43 | ) | (43 | ) | ||||||||||||||||||||||||||||
| Net loss | - | - | (55,816 | ) | - | (3,585 | ) | (59,401 | ) | |||||||||||||||||||||||
| Balance as of March 31, 2023 | 30,738,585 | $ | 3 | $ | (25 | ) | $ | 217,339 | $ | (179,455 | ) | $ | 53 | $ | 4,113 | $ | 42,028 | |||||||||||||||
The accompanying notes are an integral part of these consolidated financial statements.
| F- |
BEYOND AIR, INC. AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF CASH FLOWS
(in thousands except share data)
| For The Year Ended March 31, 2023 |
For The Year Ended March 31, 2022 |
|||||||
| Cash flows from operating activities | ||||||||
| Net loss | $ | (59,401 | ) | $ | (44,060 | ) | ||
| Adjustments to reconcile net loss used in provided by operating activities | ||||||||
| Depreciation | 634 | 314 | ||||||
| Stock-based compensation | 19,558 | 7,831 | ||||||
| Amortization of debt discount and accretion of debt issuance costs | - | 528 | ||||||
| Amortization of licensed right to use technology | 205 | 38 | ||||||
| Amortization of operating lease assets | 331 | 236 | ||||||
| Unrealized gain on marketable securities | (12 | ) | - |
|||||
| Foreign currency adjustments | - | 144 | ||||||
| Disposal of clinical and medical equipment | 235 | - | ||||||
| Provision for inventory losses | 49 | - | ||||||
| Changes in: | ||||||||
| Grant receivable | (98 | ) | 103 | |||||
| Inventory | (781 | ) | - | |||||
| Other current assets and prepaid expenses | 918 | (816 | ) | |||||
| Accounts payable | 838 | (327 | ) | |||||
| Accrued expenses | 4,791 | 4,874 | ||||||
| Operating lease liabilities | (275 | ) | - | |||||
| Long-term accrued liabilities | - | 8,000 | ||||||
| Net cash used in operating activities | (33,009 | ) | (23,134 | ) | ||||
| Cash flows from investing activities | ||||||||
| Purchase of marketable securities | (35,656 | ) | - |
|||||
| Proceeds from sale of marketable securities | 18,945 | - | ||||||
| Security deposits made on operating leases | 1 | (69 | ) | |||||
| Purchase of property and equipment | (3,877 | ) | (1,380 | ) | ||||
| Net cash used in investing activities | (20,587 | ) | (1,450 | ) | ||||
| Cash flows from financing activities | ||||||||
| Proceeds from issuance of common stock through at the market offerings | 3,705 | 36,489 | ||||||
| Proceeds from issuance of common stock through exercise of warrants | - | 5,966 | ||||||
| Proceeds from issuance of common stock through exercise of stock options | - | 251 | ||||||
| Proceeds from the sale of common stock of Beyond Cancer | - | 25,200 | ||||||
| Proceeds from issuance of common stock in connection with a purchase agreement | - | 11,138 | ||||||
| Proceeds from the net recovery of short-swing profits from related parties | - | 33 | ||||||
| Proceeds from loan | - | 1,029 | ||||||
| Payment of loan | (1,009 | ) | (658 | ) | ||||
| Net cash provided by financing activities | 2,696 | 79,450 | ||||||
| Effect of exchange rate changes on cash and cash equivalents | (43 | ) | 96 | |||||
| Increase (decrease) in cash, cash equivalents and restricted cash | (50,944 | ) | 54,962 | |||||
| Cash, cash equivalents and restricted cash at beginning of year | 90,230 | 35,268 | ||||||
| Cash, cash equivalents and restricted cash at end of year | $ | 39,287 | $ | 90,230 | ||||
| Supplemental disclosure of non-cash investing and financing activities: | ||||||||
| Right-of-use lease assets | $ | 609 | $ | 693 | ||||
| Operating lease liability | $ | 609 | $ | 693 | ||||
| Accelerated amortization of debt discount costs on retirement of debt | $ | - | $ | 390 | ||||
| Increase in Licensed Right to Use Technology from NitricGen agreement | $ | - | $ | 1,500 | ||||
| NitricGen accrued liability due to FDA approval | $ | - | $ | 1,500 | ||||
| Supplemental disclosure of cash flow items: | ||||||||
| Interest paid | $ | 36 | $ | 340 | ||||
| Income taxes paid | $ | - | $ | - | ||||
The accompanying notes are an integral part of these consolidated financial statements.
| F- |
BEYOND AIR, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 1 ORGANIZATION AND BUSINESS
Beyond Air, Inc. (together with its subsidiaries, “Beyond Air” or the “Company”) was incorporated on April 28, 2015 under Delaware law. On June 25, 2019, the Company’s name was changed to Beyond Air, Inc. from AIT Therapeutics, Inc.
The Company is a commercial stage medical device and biopharmaceutical company developing a platform of nitric oxide (“NO”) generators and delivery systems (the “LungFit® platform”) capable of generating NO from ambient air. The Company’s first device, LungFit® PH (“LungFit® PH”) received premarket approval (“PMA”) approval from the U.S. Food and Drug Administration (“FDA”) in June 2022. The NO generated by the LungFit® PH system is indicated to improve oxygenation and reduce the need for extracorporeal membrane oxygenation in term and near-term (>34 weeks gestation) neonates with hypoxic respiratory failure associated with clinical or echocardiographic evidence of pulmonary hypertension in conjunction with ventilatory support and other appropriate agents. This condition is commonly referred to as persistent pulmonary hypertension of the newborn or “PPHN”. The LungFit® platform can generate NO up to 400 parts per million (“ppm”) for delivery to a patient’s lungs directly or via a ventilator. LungFit® can deliver NO either continuously or for a fixed amount of time at various flow rates and has the ability to either titrate dose on demand or maintain a constant dose. In July 2022, the Company commenced marketing LungFit® PH in the United States for PPHN as a medical device.
LungFit® can be used to treat patients on ventilators that require NO, as well as patients with chronic or acute severe lung infections via delivery through a breathing mask or similar apparatus. Furthermore, the Company believes that there is a high unmet medical need for patients suffering from certain severe lung infections that the LungFit® platform can potentially address. The Company’s other areas of focus with the LungFit® platform beyond PPHN are viral community-acquired pneumonia (“VCAP”) including COVID-19, bronchiolitis (“BRO”), nontuberculous mycobacteria (“NTM”) lung infection and those with various severe lung infections with underlying chronic obstructive pulmonary disease (“COPD”).
With Beyond Air’s focus on NO and its effect on the human condition, the Company has two additional programs that do not utilize the LungFit® system. Through the Company’s majority-owned affiliate Beyond Cancer, Ltd. (“Beyond Cancer”) NO is used to target solid tumors. The LungFit® platform is not utilized for the solid tumor indication due to need for ultra-high concentrations of gaseous nitric oxide (“UNO”). A proprietary delivery system has been developed that can safely deliver UNO in excess of 10,000 ppm directly to a solid tumor. This program has advanced to a phase 1 human study.
On November 4, 2021, Beyond Air reorganized its oncology business into a new private company called Beyond Cancer. Beyond Air’s preclinical oncology team and the exclusive right to the intellectual property portfolio utilizing ultra-high concentration of gaseous nitric oxide (“UNO”) for the treatment of solid tumors now reside with Beyond Cancer. The new subsidiary secured $30 million in private placement of common shares, including $4.8 million in conjunction with the retirement of long-term debt, providing investors with 20% equity ownership in Beyond Cancer. Beyond Air retained 80% ownership in Beyond Cancer (see Note 16).
The second program which does not utilize the LungFit® platform partially inhibits neuronal nitric oxide synthase (nNOS) in the brain to treat neurological conditions. The first target indication is autism spectrum disorder (ASD). On June 15, 2023, the Company announced that it has entered into an agreement with Yissum Research Development Company of the Hebrew University of Jerusalem, LTD. to acquire the commercial rights for neuronal nitric oxide synthase (nNOS) inhibitors being developed for the treatment of autism spectrum disorder (ASD) and other neurological conditions. Currently, there are no FDA approved therapies specifically for the treatment of ASD. Under the terms of the agreement, Beyond Air will make payments to the University over the next two years for pre-clinical work. Also, the Company will pay a low single digit royalty on net sales and certain one-time payments based on clinical, regulatory and sales milestones. The Company expects this program to progress from pre-clinical to phase 1 human study by early 2025.
The Company’s current product candidates will be subject to premarket reviews and approvals by the FDA, certification through the conduct of a conformity assessment by a notified body in the European Union (the “EU”), as well as comparable foreign regulatory authorities’ reviews or approvals in other countries or regions.
| F- |
BEYOND AIR, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 2 SIGNIFICANT ACCOUNTING POLICIES
Principles of Consolidation
These consolidated financial statements include the accounts of the Company and the accounts of all of the Company’s subsidiaries and a variable interest entity (“VIE”) for which the Company is the primary beneficiary. As the Company has both the power to direct activities of Beyond Cancer that most significantly impact Beyond Cancer’s economic performance and the right to receive benefits and losses that may potentially be significant, these financial statements are fully consolidated with those of the Company. The non-controlling owners’ 20% interest in Beyond Cancer’s net assets and result of operations is reported as “non-controlling interest” on the Company’s consolidated balance sheets and as “net loss attributable to non-controlling interest” in the Company’s consolidated statements of operations and comprehensive loss. All intercompany balances and transactions have been eliminated in the accompanying consolidated financial statements.
Reclassifications
Certain prior period amounts have been reclassified to conform to the current period presentation. These reclassifications had no effect on the reported results of operations.
Use of Estimates
The preparation of financial statements in conformity with generally accepted accounting principles in the United States (“U.S. GAAP”) requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses for the reporting period. Actual results could significantly differ from those estimates. On an ongoing basis, the Company evaluates its significant estimates and assumptions including expense recognition and accrual assumptions under consulting and clinical trial agreements, stock-based compensation, impairment assessments, accounting for licensed rights to use technologies and other long-lived assets, contingency recognition and accruals and the determination of valuation allowance requirements on deferred tax attributes.
Liquidity Risks and Uncertainties
The Company used cash in operating activities of $33.0 million for the year ended March 31, 2023, and has accumulated deficit attributable to the stockholders of Beyond Air of $179.5 million. The Company had cash and cash equivalents and marketable securities of $45.9 million as of March 31, 2023. Based on management’s current business plan, after taking into consideration the proceeds received from the debt financing subsequent to the end of the fiscal year, the Company estimates that its cash and liquidity is sufficient to finance its operating requirements for at least one year from the date these consolidated financial statements were filed.
The Company’s future capital needs and the adequacy of its available funds will depend on many factors, including, but not necessarily limited to the success and costs of commercialization of the Company’s approved product and the actual cost and time necessary for current and anticipated preclinical studies, clinical trials and other actions needed to obtain certification or regulatory approval of the Company’s product candidates.
The Company entered into an At-The-Market Offering Sales Agreement, dated February 4, 2022 (the “2022 ATM”) for $50 million (see Note 5). The Company has $46.3 million available under this agreement as of March 31, 2023.
The Company may be required to raise additional funds through equity or debt securities offerings or strategic collaboration and/or licensing agreements in order to fund operations if it is unable to generate enough product or royalty revenues, if any. Such financing may not be available on acceptable terms, or at all, and the Company’s failure to raise capital when needed could have a material adverse effect on its strategic objectives, results of operations and financial condition.
Other Risks and Uncertainties
The Company is subject to risks common to development and early-stage medical device companies including, but not limited to, new technological innovations, certifications or regulatory approval, dependence on key personnel, protection of proprietary technology, compliance with government regulations, product liability, uncertainty of market acceptance of approved products and the potential need to obtain additional financing. The Company is also dependent on third-party suppliers and, in some cases single-source suppliers.
The Company’s products require approval or clearance from the FDA prior to commencement of commercial sales in the United States. There can be no assurance that the Company’s products beyond LungFit® PH in the U.S. will receive the required approvals or clearances. Certifications, approvals or clearances are also required in foreign jurisdictions in which the Company may license or sell its products. If the Company is denied such certifications or approvals or clearances or such certifications, approvals or clearances are delayed, such denial or delay may have a material adverse impact on the Company’s results of operations, financial position and liquidity. Further, there can be no assurance that the Company’s product will be accepted in the marketplace, nor can there be any assurance that any future products can be developed or manufactured at an acceptable cost and with appropriate performance characteristics, or that such products will be successfully marketed, if at all.
| F- |
BEYOND AIR, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 2 SIGNIFICANT ACCOUNTING POLICIES (continued)
Cash and Cash Equivalents, Short-Term Investments and Restricted Cash
The Company considers all highly liquid investments with original maturities of three months or less at the date of purchase and an investment in a U.S. government money market fund to be cash equivalents. The Company maintains its cash and cash equivalents in highly rated financial institutions in Australia, Israel, Ireland and the U.S., the balances of which, at times, may exceed federally insured limits.
Marketable securities include investment in fixed income bonds and U.S. Treasury securities that are considered to be highly liquid and easily tradeable. The marketable securities are considered trading securities and are measured at fair value and are accounted for in accordance with ASC 320. The marketable securities are valued using inputs observable in active markets for identical securities and are therefore classified as Level 1 within the Company’s fair value hierarchy.
As of March 31, 2023 and March 31, 2022, restricted cash included approximately $2.5 million and $2.6 million designated for a contract manufacturer, respectively. This cash is expected to be used for materials and parts that require long lead times. As of March 31, 2023 and March 31, 2022, restricted cash included $7.4 million for a supersedeas bond held as collateral pending the outcome of the appeal on the Empery lawsuit. Subsequent to March 31, 2023, restricted cash declined by $7.4 million as the bond was released in satisfaction of judgement in April 2023 (see Note 15).
The following table is the reconciliation of the presentation and disclosure of cash, cash equivalents, marketable securities by major security type and restricted cash as shown on the Company’s consolidated statements of cash flows for:
SCHEDULE OF CASH AND CASH EQUIVALENTS AND RESTRICTED CASH
| (in thousands) | March 31, 2023 | March 31, 2022 | ||||||
| Cash and cash equivalents | $ | 29,158 | $ | 80,242 | ||||
| Restricted cash | 10,129 | 9,988 | ||||||
| Total cash, cash equivalents and restricted cash | $ | 39,287 | $ | 90,230 | ||||
| Marketable securities: | ||||||||
| Marketable debt securities | ||||||||
| Corporate debt securities | 1,597 | - | ||||||
| U.S. government securities | 1,013 | - | ||||||
| Mutual fund (Ultra-Short-Term Income) | 14,114 | - | ||||||
| Total marketable securities | 16,724 | - | ||||||
| Total cash, cash equivalents, marketable securities and restricted cash | $ | 56,011 | $ | 90,230 | ||||
The following table summarizes our short-term marketable securities with unrealized gains and losses as of March 31, 2023 aggregated by major security type:
SUMMARY OF SHORT-TERM MARKETABLE SECURITIES WITH UNREALIZED GAINS AND LOSSES
| (in thousands) | Fair Value |
Unrealized (Losses) |
||||||
| Corporate debt securities | 1,597 | 2 | ||||||
| US government securities | 1,013 | 10 | ||||||
| Mutual fund (Ultra-Short-Term Income) | 14,114 | - | ||||||
| Total short-term marketable securities | $ | 16,724 | $ | 12 | ||||
All marketable securities are A- or higher rated. No marketable securities have maturities greater than 12 months. All investments are level 1 investments.
Revenue Recognition
The Company recognizes revenue when it transfers promised goods or services to customers in an amount that reflects the consideration to which the Company expects to be entitled in exchange for those goods or services. To determine revenue recognition for contracts with customers, the Company performs the following five steps: (i) identify the contract(s) with a customer, (ii) identify the performance obligation(s) in the contract, (iii) determine the transaction price, (iv) allocate the transaction price to the performance obligation(s) in the contract and (v) recognize revenue when (or as) the Company satisfies the performance obligation(s). At contract inception, the Company assesses the goods or services promised within each contract, assesses whether each promised good or service is distinct and identifies those promised goods or services that are performance obligations.
| F- |
BEYOND AIR, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 2 SIGNIFICANT ACCOUNTING POLICIES (continued)
The Company will be required to use judgment to determine (a) the number of performance obligations based on the determination under step (ii) above and whether those performance obligations are distinct from other performance obligations in the contract (b) the transaction price under step (iii) above and (c) the stand-alone selling price for each performance obligation identified in the contract for the allocation of the transaction price in step (iv) above. The Company will also be required to use judgment to determine whether variable consideration should be included in the transaction price. The transaction price is allocated to each performance obligation on an estimated stand-alone selling price basis, for which the Company recognizes revenue as or when the performance obligations under contract are satisfied.
Inventories
Raw materials, work in progress and finished goods are stated at the lower of cost or net realizable value. Cost comprises direct materials, third-party manufacturing costs and shipping costs incurred to deliver goods to the Company’s central warehouse location.
Costs are assigned to individual items of inventory on the basis of weighted average costs. Inventory items are tracked by batch/lot number for specific identification whenever possible. The Company uses judgement to determine the proportion of inventory items held which will be provided to a hospital as part of its service delivery versus the inventory items that will be provided free of charge on hospital evaluations. Inventory items held for hospital evaluations have no realizable value.
Leases
Operating lease assets are included within operating lease right-of-use assets, and the corresponding operating lease obligation on the consolidated balance sheets as of March 31, 2023 and March 31, 2022 in accordance with ASC 842, Leases. The Company has elected not to present short-term leases as these leases have a lease term of 12 months or less at lease inception and do not contain purchase options or renewal terms that the Company is reasonably certain to exercise. All other lease assets and lease liabilities are recognized based on the present value of lease payments over the lease term at commencement date. Because most of the Company’s leases do not provide an implicit rate of return, the Company used an incremental borrowing rate based on the information available at adoption date in determining the present value of lease payments.
Grant receivable
Under a collaboration arrangement with the Cystic Fibrosis Foundation (“CFF”), grant milestones are achieved subject to certain performance steps and requirements under a development program. Grant milestones are recorded as reimbursements against the applicable portion of the Company’s research and development expenses. Such reimbursements are reflected as a reduction of research and development expenses in the Company’s consolidated statements of operations and comprehensive income (loss), as the performance of research and development services for reimbursement is not considered to be an ongoing component or central to the Company’s operations. See Note 11.
Segment Reporting
Commencing with the creation of Beyond Cancer in November 2021 (see Note 16), the Company’s operations became classified into two segments, Beyond Air and Beyond Cancer. Each segment has its own Management team, Board of Directors, Corporate Officers and legal entities. As of March 31, 2023, Beyond Air, Inc. owns 80% of the common stock of Beyond Cancer. The segment reporting is based on the manner in which the Company’s CEO as chief operating decision maker assesses performance and allocates resources across the organization. The Beyond Air segment includes unallocated corporate expenses associated with the public company fees as well as all corporate related assets and liabilities.
| F- |
BEYOND AIR, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 2 SIGNIFICANT ACCOUNTING POLICIES (continued)
Research and Development
Research and development expenses are charged to the consolidated statements of operations as incurred. Research and development expenses include salaries, benefits, stock-based compensation and costs incurred by outside laboratories, manufacturers, clinical research organizations, consultants, and accredited facilities in connection with preclinical studies and clinical trials. Research and development expenses are partially offset by the benefit of tax incentive payments for qualified research and development expenditures from the Australian tax authority (“AU Tax Rebates”). The Company does not record AU Tax Rebates until payment is received due to the uncertainty of receipt. During the years ended March 31, 2023, and March 31, 2022, the Company received $0.5 million and $0, respectively, in AU Tax Rebates.
Foreign Exchange Transactions
The Company’s subsidiaries transact in U.S. dollars, Euros, New Israeli Shekels and Australian dollars. The Company’s main operations are in the United States and the U.S. dollar is the currency of the primary economic environment in which the Company operates and expects to continue to operate in the foreseeable future. The Company translated its non-U.S. operations’ assets and liabilities denominated in foreign currencies into U.S. dollars at current rates of exchange as of the balance sheet date and income and expense items at the average exchange rate for the reporting period. Gains or losses from foreign currency transactions are included in other income (expense) in the Consolidated Statements of operations as foreign currency exchange gain/(loss).
In consolidating international subsidiaries, balance sheet currency effects are recorded as a component of accumulated other comprehensive income. The accumulated other comprehensive income account includes the cumulative results of translating certain balance sheet assets and liabilities at current exchange rates and some accounts at historical rates. For the year ended March 31, 2023, the Company recorded a loss of $43 thousand in accumulated other comprehensive income.
The Company measures the cost of employee and non-employee services received in exchange for an award of equity instruments based on the grant date fair value of the award. Fair value for restricted stock unit awards is valued using the closing price of the Company’s common stock on the date of grant. The grant date fair value is recognized over the requisite service period during which an employee and non-employee is required to provide service in exchange for the award, using the accelerated method with each tranche being expensed over its vesting period. The grant date fair value of employee and non-employee share options is estimated using the Black-Scholes option pricing model. The risk-free interest rate assumptions were based upon the observed interest rates appropriate for the expected term of the equity instruments. The expected dividend yield was assumed to be zero as the Company has not paid any dividends since its inception and does not anticipate paying dividends in the foreseeable future. Starting in 2023, Beyond Air used its own historical volatility as an input for expected volatility, but due to the Beyond Cancer’s lack of marketability, the Company utilizes the implied volatility based on an aggregate of guideline companies for expected volatility. The Company uses the simplified method to estimate the expected term.
Property and Equipment
Property and equipment are stated at cost less accumulated depreciation and accumulated amortization. Depreciation and amortization are calculated using the straight-line method over the estimated useful life of the assets as follows:
SCHEDULE OF PROPERTY AND EQUIPMENT USEFUL LIFE OF ASSETS
| Computer equipment | Three years |
| Furniture and fixtures | Five years |
| Clinical and medical equipment | Five years |
| Equipment deployable as part of a service offering | Five years |
| Leasehold improvements | Shorter of term of lease or estimated useful life of the asset |
Licensed Right to Use Technology
Licensed right to use technology that is considered platform technology with alternative future uses is recorded as an intangible asset and is amortized on a straight-line method over its estimated useful life, determined to be thirteen years (see Note 15).
The expected amortization expense for the next five years and thereafter is as follows for the year ended March 31 (in thousands):
SCHEDULE OF FUTURE EXPECTED AMORTIZATION EXPENSE
| 2024 | $ | 205 | ||
| 2025 | 205 | |||
| 2026 | 205 | |||
| 2027 | 205 | |||
| 2028 | 205 | |||
| Thereafter | 607 | |||
| Total | $ | 1,632 |
Long-Lived Assets
The Company assesses the impairment of long-lived assets on an ongoing basis and whenever events or changes in circumstances indicate that the carrying value may not be recoverable. Factors that the Company considers as potential triggers of an impairment review include the following:
| ● | significant underperformance relative to expected historical or projected future operating results, |
| ● | significant changes in the manner of the Company’s use of the acquired assets or the strategy for its overall business, |
| ● | significant negative regulatory or economic trends, and |
| ● | significant technological changes, which would render the platform technology, equipment, and manufacturing processes obsolete. |
| F- |
BEYOND AIR, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 2 SIGNIFICANT ACCOUNTING POLICIES (continued)
Recoverability of assets that will continue to be used in the Company’s operations is measured by comparing the carrying value to the future net undiscounted cash flows expected to be generated by the asset or asset group. Future undiscounted cash flows include estimates of future revenues, driven by market growth rates, and estimates of future costs. There were no events during the reporting periods that were deemed to be a triggering event that would require an impairment assessment.
Income Taxes
The Company accounts for income taxes using the asset and liability method. Accordingly, deferred tax assets and liabilities are recognized for the future tax consequences attributable to differences between financial statement carrying amounts of existing assets and liabilities and their respective tax bases. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. The effect on deferred tax assets and liabilities of a change in the tax rate is recognized in income or expense in the period that the change is effective. Tax benefits are recognized when it is probable that the deduction will be sustained. A valuation allowance is established when it is more likely than not that all or a portion of a deferred tax asset will either expire before the Company is able to realize the benefit, or that future deductibility is uncertain. As of March 31, 2023, the Company recorded a valuation allowance to the full extent of the Company’s net deferred tax assets since the likelihood of realization of the benefit does not meet the more-likely-than-not threshold.
The Company’s reserves related to taxes are based on a determination of whether and how much of a tax benefit taken by the Company in its tax filings or positions is more likely than not to be realized following resolution of any potential contingencies present related to the tax benefit. As of March 31, 2023, the Company had no unrecognized tax benefits or related interest and penalties accrued. The Company has not, as yet, conducted a study of research and development (R&D) credit carryforwards. This study may result in an adjustment to the Company’s R&D credit carryforwards; however, until a study is completed and any adjustment is known, no amounts are being presented as an uncertain tax position. A full valuation allowance has been provided against the Company’s R&D credits and, if an adjustment is required, this adjustment would be offset by an adjustment to the valuation allowance. Thus, there would be no impact to the consolidated balance sheet, statement of operations or comprehensive loss if an adjustment were required. The Company would recognize both accrued interest and penalties related to unrecognized benefits in income tax expense. The Company’s uncertain tax positions are related to years that remain subject to examination by relevant tax authorities. Since the Company is in a loss carryforward position, the Company is generally subject to examination by the U.S. federal, state and local income tax authorities for all tax years in which a loss carryforward is available.
Basic and diluted net loss per share attributable to common stockholders is computed by dividing the net loss attributable to Beyond Air, Inc., by the weighted average number of shares of common stock outstanding for the period. The dilutive effect of outstanding options, warrants, restricted stock and other stock-based compensation awards is reflected in diluted net loss per share by application of the treasury stock method. The calculation of diluted net loss attributed to common stockholders per share excludes all anti-dilutive shares of common stock. For periods in which the Company has reported net losses, diluted net loss per share attributable to common stockholders is the same as basic net loss per share attributable to common stockholders, because such shares of common stock are not assumed to have been issued if their effect is anti-dilutive, see Note 9.
Recently Issued Accounting Standards Adopted
In September 2016, the FASB issued ASU 2016-13, “Financial Instruments – Credit Losses (Topic 326)”. The ASU sets forth a “current expected credit loss” (CECL) model which requires the Company to measure all expected credit losses for financial instruments held at the reporting date based on historical experience, current conditions, and reasonable supportable forecasts. This replaces the existing incurred loss model and is applicable to the measurement of credit losses on financial assets measured at amortized cost and applies to some off-balance sheet credit exposures. In February 2020, the FASB issued ASU 2020-02, “Financial Instruments – Credit Losses (Topic 326)”, which amends the effective date of the original pronouncement for smaller reporting companies. ASU 2016-13 and its amendments became effective for the Company for interim and annual periods for the fiscal period ending December 31, 2022. ASU 2016-13 did not have a material impact on the Company’s consolidated financial statements or disclosures.
| F- |
BEYOND AIR, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 3 FAIR VALUE MEASUREMENT
The Company’s financial instruments primarily include cash, cash equivalents, restricted cash, accounts payable, and a short-term loan. Due to the short-term nature of these financial instruments, the carrying amounts of these assets and liabilities approximate their fair value. The long-term debt approximates fair value due to the prevailing market conditions for similar debt with remaining maturity and terms.
Fair value is defined as the price that would be received to sell an asset or paid to transfer a liability (an exit price) in an orderly transaction between market participants at the reporting date. The accounting guidance establishes a three-tiered hierarchy, which prioritizes the inputs used in the valuation methodologies in measuring fair value. A fair value hierarchy has been established for valuation inputs that give the highest priority to quoted prices in active markets for identical assets or liabilities and the lowest priority to unobservable inputs. The fair value hierarchy is as follows:
| Level 1 - | quoted prices in active markets for identical assets or liabilities; | |
| Level 2 - | inputs other than Level 1 that are observable, either directly or indirectly, such as quoted prices in active markets for similar assets or liabilities, quoted prices for identical or similar assets or liabilities in markets that are not active, or other inputs that are observable or can be corroborated by observable market data for substantially the full term of the assets or liabilities; or | |
| Level 3 - | unobservable inputs that are supported by little or no market activity and that are significant to the fair value of the assets or liabilities. |
NOTE 4 PROPERTY AND EQUIPMENT
Property and equipment consist of the following:
SCHEDULE OF PROPERTY AND EQUIPMENT
| (in thousands) | March 31, 2023 | March 31, 2022 | ||||||
| Clinical and medical equipment | $ | 1,365 | $ | 1,682 | ||||
| Equipment deployable as part of a service offering | 3,027 | - | ||||||
| Computer equipment | 779 | 364 | ||||||
| Furniture and fixtures | 505 | 311 | ||||||
| Leasehold improvements | 581 | 404 | ||||||
| Property and Equipment, gross | 6,256 | 2,762 | ||||||
| Accumulated depreciation and amortization | (1,254 | ) | (767 | ) | ||||
| Property and equipment , net | $ | 5,003 | $ | 1,995 | ||||
Depreciation and amortization expense for the years ended March 31, 2023 and March 31, 2022 was $634 thousand and $314 thousand, respectively. In the year ended March 31, 2023, upon retirement of clinical equipment determined to have no remaining useful life, $382 thousand of clinical equipment was removed less $147 thousand of accumulated depreciation and the charge was recorded in R&D in the accompanying consolidated statement of operations.
NOTE 5 STOCKHOLDERS’ EQUITY
On April 2, 2020, the Company entered into an At-The-Market Equity Offering Sales Agreement (the “2020 ATM”), allowing the Company to sell its common stock for aggregate sales proceeds of up to $50 million from time to time and at various prices, subject to the conditions and limitations set forth in the sales agreement. When shares of the Company’s common stock were sold, there was a 3% fee paid to the sales agent. For the year ended March 31, 2022, the Company received net proceeds of $36.5 million from the sale of 4,053,424 shares of the Company’s common stock. There were no remaining funds available under the 2020 ATM as of March 31, 2022.
On May 14, 2020, the Company entered into the Stock Purchase Agreement with Lincoln Park Capital Fund, LLC (“LPC”) (the “New Stock Purchase Agreement”), which provides for the issuance of up to $40 million of its common stock which the Company may sell from time to time in its sole discretion to LPC over 36 months, provided that the closing price of the Company’s common stock is not below $0.25 per share and subject to certain other conditions and limitations set forth in the New Stock Purchase Agreement. For the twelve months ended March 31, 2022, the Company received net proceeds of $11.1 million from the sale of 1,100,000 shares of common stock. No proceeds were received in the twelve months ended March 31, 2023. The New Stock Purchase Agreement expired on May 14, 2023 and no additional proceeds had been received prior to that date.
On February 4, 2022, the Company entered into a new At-The-Market Equity Offering Sales Agreement with Truist Securities, Inc. and Oppenheimer & Co, Inc. (the “2022 ATM”), allowing the Company to sell its common stock for aggregate sales proceeds of up to $50 million from time to time and at various prices, subject to the conditions and limitations set forth in the 2022 ATM. If shares of the Company’s common stock are sold, there is a 3% fee paid to the sales agent.
For the twelve months ended March 31, 2023, the Company received net proceeds of $3.7 million from the sale of 572,078 shares of common stock. There were no proceeds received from the 2022 ATM in the twelve months ended March 31, 2022. As of March 31, 2023, there were $46.3 million in funds available under the 2022 ATM.
| F- |
BEYOND AIR, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 5 STOCKHOLDERS’ EQUITY (continued)
Stock Option Plans
The Company’s Fifth Amended and Restated 2013 Beyond Air Equity Incentive Plan (the “2013 BA Plan”) allows for awards to officers, directors, employees, and consultants of stock options, restricted stock units and restricted shares of the Company’s common stock. On January 9, 2023, the Company’s Board of Directors approved an amendment to the 2013 BA Plan to increase the number of shares in the 2013 BA Plan by 3,000,000, which was approved by the Company’s stockholders at the 2023 annual stockholder meeting on March 9, 2023. The 2013 BA Plan has 10,600,000 shares authorized for issuance. As of March 31, 2023, 383,221 shares were available under the 2013 BA Plan.
Restricted Stock Units
The fair value for the restricted stock unit awards was valued at the closing price of the Company’s common stock on the date of grant. Restricted stock units vest annually over five years.
A summary of the Company’s restricted stock unit awards for the years ended March 31, 2023 and March 31, 2022 is as follows:
SCHEDULE OF RESTRICTED STOCK AWARDS
| Number Of Shares |
Weighted Average Grant Date Fair Value |
|||||||
| Unvested as of March 31, 2021 | 554,200 | $ | 5.07 | |||||
| Granted | 615,000 | 8.37 | ||||||
| Vested | (202,600 | ) | 6.32 | |||||
| Forfeited | (17,000 | ) | 5.23 | |||||
| Unvested as of March 31, 2022 | 949,600 | $ | 6.92 | |||||
| Granted | 434,500 | 6.27 | ||||||
| Vested | (274,600 | ) | 6.50 | |||||
| Forfeited | (8,400 | ) | 5.06 | |||||
| Unvested as of March 31, 2023 | 1,101,100 | $ | 6.78 | |||||
Stock-based compensation expense related to these grants for the years ended March 31, 2023 and March 31, 2022 was $2.4 million and $1.7 million, respectively.
As of March 31, 2023, the Company had unrecognized stock-based compensation expense for the restricted stock unit awards in the 2013 BA Plan of approximately $5.0 million which is expected to be expensed over the weighted average remaining service period of 2.1 years. For the years ended March 31, 2023 and March 31, 2022, the weighted average fair value of options granted was $6.27 and $8.37 per share, respectively.
As of March 31, 2023, all vested shares had been issued.
Stock Options
The vesting terms of the options issued under the 2013 BA Plan are generally four years and expire ten years from the grant date.
SCHEDULE OF OPTION ACTIVITY
| Number Of Options |
Weighted Average Exercise Price–- Options |
Weighted Average Remaining Contractual Life- Options |
Aggregate (thousands) |
|||||||||||||
| Options outstanding as of March 31, 2021 | 4,195,097 | $ | 4.91 | 8.4 | $ | 2,609 | ||||||||||
| Granted | 1,673,500 | 7.14 | ||||||||||||||
| Exercise | (259,904 | ) | 4.97 | |||||||||||||
| Forfeited | (100,062 | ) | 5.11 | |||||||||||||
| Options outstanding as of March 31, 2022 | 5,508,631 | $ | 5.60 | 8.1 | $ | 6,831 | ||||||||||
| Granted | 2,805,500 | 6.32 | ||||||||||||||
| Exercise | (15,000 | ) | 5.21 | |||||||||||||
| Forfeited | (100,250 | ) | 6.38 | |||||||||||||
| Outstanding as of March 31, 2023 | 8,198,881 | $ | 5.83 | 8.4 | $ | 8,306 | ||||||||||
| Exercisable as of March 31, 2023 | 3,481,943 | $ | 5.85 | 6.4 | $ | 6,110 | ||||||||||
| F- |
BEYOND AIR, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 5 STOCKHOLDERS’ EQUITY (continued)
The Company’s 2021 Beyond Cancer Ltd Equity Incentive Plan (the “2021 BC Plan”) allows for awards to officers, directors, employees, and consultants of stock options, restricted stock units and restricted shares of Beyond Cancer Ltd.’s common stock. The vesting terms of the options issued under the 2021 BC Plan are generally four years and they expire ten years from the grant date. On November 3, 2022, the Company’s Board of Directors approved an amendment to reserve for issuance an additional 2,000,000 shares of common stock. The 2021 BC Plan has 4,000,000 shares authorized for issuance. As of March 31, 2023, 183,000 shares were available under the 2021 BC Plan.
A summary of the change in stock options for Beyond Cancer for the years ended March 31, 2023 and March 31, 2022 is as follows:
| Number Of Options |
Weighted Average Exercise Price–- Options |
Weighted Average Remaining Contractual Life- Options |
Aggregate (thousands) |
|||||||||||||
| Options outstanding as of March 31, 2021 | - | $ | - | - | $ | - | ||||||||||
| Granted | 1,763,500 | 2.76 | ||||||||||||||
| Options outstanding as of March 31, 2022 | 1,763,500 | $ | 2.76 | 9.3 | $ | 12,768 | ||||||||||
| Granted | 2,076,500 | 3.01 | ||||||||||||||
| Exercised | - | - | - | |||||||||||||
| Forfeited | (23,000 | ) | 5.91 | - | ||||||||||||
| Outstanding as of March 31, 2023 | 3,817,000 | $ | 2.88 | 9.2 | $ | 23,486 | ||||||||||
| Exercisable as of March 31, 2023 | 457,375 | $ | 3.08 | 8.8 | $ | 2,728 | ||||||||||
As of March 31, 2023, the Company had unrecognized stock-based compensation expense for the stock options in the 2013 BA Plan of approximately $17.5 million which is expected to be expensed over the weighted average remaining service period of 2.1 years. For the years ended March 31, 2023 and March 31, 2022, the weighted average fair value of options granted was $4.68 and $5.58 per share, respectively.
As of March 31, 2023, The Company had unrecognized stock-based compensation expense for the stock options in the 2021 BC Plan of approximately $20.2 million which is expected to be expensed over the weighted average remaining service period of 2.0 years. For the years ended March 31, 2023 and March 31, 2022, the weighted average fair value of options granted was $8.32 and $9.08 per share, respectively.
SCHEDULE OF FAIR VALUE OF OPTION
| March 31, 2023 | March 31, 2022 | |||||||
| Risk -free interest rate | 2.5% - 4.3% | 0.9% - 2.2% | ||||||
| Expected volatility (Beyond Air) | 82.8% - 96.1% | 89.3 – 91.1% | ||||||
| Expected volatility (Beyond Cancer) | 103.0% - 107.9% | 101.8% - 103.1% | ||||||
| Dividend yield | 0% | 0% | ||||||
| Expected terms (in years) | 6.25 | 6.25 | ||||||
The Company determined the fair value per share of Beyond Cancer’s common stock to be $9.00 and $10.00 in fiscal year 2023 and 2022 based on a third-party valuation. See Note 16.
SCHEDULE OF STOCK-BASED COMPENSATION EXPENSE
| Year Ended | ||||||||
| March 31, | ||||||||
| 2023 | 2022 | |||||||
| Research and development | $ | 4,181 | $ | 1,820 | ||||
| General and administrative | 15,378 | 6,011 | ||||||
| Total stock-based compensation expense | $ | 19,558 | $ | 7,831 | ||||
ESPP
On March 4, 2021, the stockholders approved the 2021 Employee Stock Purchase Plan (“ESPP”). The purpose of the ESPP is to encourage and to enable eligible employees of the Company, through after-tax payroll deductions, to acquire proprietary interests in the Company through the purchase and ownership of shares of Stock. The ESPP is intended to benefit the Company and its stockholders by (a) incentivizing participants to contribute to the success of the Company and to operate and manage the Company’s business in a manner that will provide for the Company’s long-term growth and profitability and that will benefit its stockholders and other important stakeholders and (b) encouraging participants to remain in the employ of the Company. As of March 31, 2023 and March 31, 2022, no shares were issued under the ESPP.
| F- |
BEYOND AIR, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 5 STOCKHOLDERS’ EQUITY (continued)
Warrants
A summary of the Company’s outstanding warrants as of March 31, 2023 are as follows:
SUMMARY OF COMPANY’S OUTSTANDING WARRANTS
| Warrant Holders |
Number Of Warrants |
Exercise Price |
Intrinsic Value (thousands) |
Date of Expiration |
||||||||||
| Third-party license agreement | 208,333 | $ | 4.80 | $ | 406 | January 2024 | ||||||||
| March 2020 loan (see Note 12) | 172,187 | $ | 7.26 | - | March 2025 | |||||||||
| NitricGen Agreement | 80,000 | $ | 6.90 | - | January 2028 | |||||||||
| Total | 460,520 | $ | 6.08 | $ | 406 | |||||||||
For the years ended March 31, 2023 and March 31, 2022, 0 and 1,630,002 warrants were exercised into common shares from the proceeds of $0 and $6.0 million, respectively. For the years ended March 31, 2023 and 2022, warrant holders exercised 0 and 1,358,896 warrant shares on a cashless basis for 0 and 894,823 shares of common stock, respectively.
NOTE 6 OTHER CURRENT ASSETS AND PREPAID EXPENSES
A summary of current assets and prepaid expenses is as follows (in thousands):
SCHEDULE OF CURRENT ASSETS AND PREPAID EXPENSES
| March 31, 2023 | March 31, 2022 | |||||||
| Research and development | $ | 128 | $ | 216 | ||||
| Insurance | 908 | 1,037 | ||||||
| Value added tax receivable | 231 | 282 | ||||||
| Demonstration materials | 245 | - | ||||||
| Other | 337 | 508 | ||||||
| Total | $ | 1,850 | $ | 2,044 | ||||
NOTE 7 ACCRUED EXPENSES
A summary of the accrued expenses is as follows (in thousands):
SUMMARY OF ACCRUED EXPENSES
March 31, 2023 |
March 31, 2022 |
|||||||
| Research and development | $ | 426 | $ | 1,006 | ||||
| Professional fees | 1,221 | 442 | ||||||
| Employee salaries and benefits | 985 | 409 | ||||||
| Contingent litigation and settlements (Note 15) | 10,298 | 2,435 | ||||||
| Circassia settlement – current portion (Note 10) | 3,500 | 2,500 | ||||||
| NitricGen agreement (Note 15) | - | 1,500 | ||||||
| Other | 184 | 82 | ||||||
| Total short-term accrued expenses | $ | 16,613 | $ | 8,374 | ||||
| Circassia settlement - long-term portion (Note 10) | $ | 4,500 | $ | 8,000 | ||||
| Total other long-term liabilities | $ | 4,500 | $ | 8,000 | ||||
| F- |
BEYOND AIR, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 8 LEASES
Lessees are required to recognize operating and financing lease liabilities and corresponding right-of-use assets on the balance sheet and provide disclosures surrounding the amount, timing and uncertainty of cash flows arising from leasing arrangements. During the year ended March 31, 2023, the Company entered into one new lease and two lease extensions, which resulted in the recognition of operating lease liabilities and right-of-use assets of same amount of $609 thousand. The right-of use assets and operating lease liability are as follows (in thousands):
SCHEDULE OF OPERATING LEASE LIABILITY
| March 31, 2023 | March 31, 2022 | |||||||
| Right-of-use assets | $ | 2,493 | $ | 2,216 | ||||
| Operating lease liability short-term | $ | 376 | $ | 281 | ||||
| Operating lease liability long-term | 2,321 | 2,079 | ||||||
| Total | $ | 2,697 | $ | 2,361 | ||||
Operating lease liabilities and their corresponding right-of-use assets are recorded based on the present value of lease payments over the expected remaining lease term. Certain adjustments to the right-of-use asset may be required for items such as prepaid or accrued rent. The interest rate implicit in the Company’s leases is typically not readily determinable. As a result, the Company utilizes its incremental borrowing rate, which reflects the fixed rate at which the Company could borrow on a collateralized basis the amount of the lease payments in the same currency, for a similar term, in a similar economic environment. Operating lease expense is recognized on a straight-line basis over the lease term and is included in general and administrative and research development expenses. The Company has other operating lease agreements with commitments of less than one year or that are not significant. The Company elected the practical expedient option and as such these lease payments are expensed as incurred. For the year ended March 31, 2023, the Company recognized $0.5 million of lease expense in general and administrative costs and $0.1 million in research and development. For the year ended March 31, 2022, the Company recognized $0.5 million of lease expense in general and administrative costs and $0.1 million in research and development.
SCHEDULE OF LEASE OTHER INFORMATION
| Other Information For The Year Ended March 31, 2023 | ||||
| Cash paid for amounts included in the measurement of lease liabilities: | ||||
| Cash paid | $ | 500 | ||
| Right-of-use assets obtained in exchange for new operating lease liabilities: | ||||
| Weighted-average remaining lease term — operating leases | 6.5 | |||
| Weighted-average discount rate — operating leases | 8.5 | % | ||
SCHEDULE OF MATURITY OF LEASE LIABILITIES
| Maturity of Lease Liabilities | Operating Leases | |||
| Payments remaining for the year ended March 31: | ||||
| 2024 | $ | 582 | ||
| 2025 | 593 | |||
| 2026 | 551 | |||
| 2027 | 452 | |||
| 2028 | 404 | |||
| Thereafter | 916 | |||
| Total lease payments | 3,497 | |||
| Less: interest | (800 | ) | ||
| Present value of lease liabilities | $ | 2,697 | ||
SCHEDULE OF POTENTIAL ANTI-DILUTIVE SECURITIES
| March 31, 2023 | March 31, 2022 | |||||||
| Common stock warrants | 460,520 | 460,520 | ||||||
| Common stock options | 8,198,881 | 5,508,631 | ||||||
| Restricted shares | 1,101,100 | 949,600 | ||||||
| Total | 9,760,501 | 6,918,751 | ||||||
| F- |
BEYOND AIR, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 10 CIRCASSIA AGREEMENT
On January 23, 2019, the Company entered into an agreement for commercial rights (the “Circassia Agreement”) with Circassia Limited and its affiliates (collectively, “Circassia”) for PPHN and future related indications at concentrations of < 80 ppm in the hospital setting in the United States and China. On December 18, 2019, the Company terminated the Circassia Agreement.
On May 25, 2021, the Company and Circassia entered into a Settlement Agreement resolving all claims by and between both parties and mutually terminating the Circassia Agreement. Pursuant to the terms of the Settlement Agreement, the Company agreed to pay Circassia $10.5 million in three installments, the first payment of $2.5 million was triggered upon FDA approval (fixing the “Initial Payment Due Date” at July 28, 2022). Thereafter, the Company will pay $3.5 million to Circassia on the first anniversary of the Initial Payment Due Date and $4.5 million on the second anniversary of the Initial Payment Due Date. Additionally, beginning in year three post-approval, Circassia will receive a quarterly royalty payment equal to 5% of LungFit® PH net sales in the U.S. This royalty will terminate once the aggregate payment reaches $6 million. $2.5 million was paid to Circassia Limited in July 2022 and $8.0 million remains as an accrued liability as of March 31, 2023. $3.5 million will be paid in July 2023 and the final $4.5 million will be paid in July 2024.
NOTE 11 GRANT COLLABORATON AGREEMENT
On February 10, 2021, the Company received a grant for up to $2.2 million from the CFF to advance the clinical development of high concentration NO for the treatment of NTM pulmonary disease, which disproportionally affects Cystic Fibrosis patients. Under the terms of the agreement, the funding will be allocated to the ongoing LungFit® GO NTM pilot study. The grant provides milestones based upon achieving performance steps and requirements under a development program. The grant provides for royalty payments to CFF upon the commercialization of any product developed under the grant program at a rate of 10% of net sales. The royalties are capped at four times the grant actually paid to the Company. For the years ended March 31, 2023 and March 31, 2022, the Company recorded $0.5 million and $0.7 million as a reduction to R&D expense, respectively. A total of $1.7 million has been recognized as a reduction of R&D costs from this grant to date. Since the beginning of the pilot study, the Company has received milestone payments totaling $1.3 million and accrued an additional $0.4 million as a grant receivable as of March 31, 2023.
NOTE 12 LONG-TERM LOAN
On March 17, 2020, the Company entered into a Facility Agreement (the “Facility Agreement”) with certain lenders for up to $25.0 million in five tranches of $5.0 million per tranche. The Company received proceeds from the first tranche in fiscal year 2020. During October 2021, the Company amended the Facility Agreement to offer the lenders the ability to accept redemption of all amounts outstanding from the first tranche of $5.0 million and to terminate the Facility Agreement without penalty. The Facility Agreement was terminated on November 10, 2021.
In connection with the first tranche, the Company issued, in March 2020, warrants to the lenders for the purchase of 172,826 shares of the Company’s common stock at $7.26 per share. The warrants expire in five years. The Company allocated the fair market value of the warrants at the date of grant to stockholders’ equity and reflected a debt discount of $595 thousand. Debt discount and debt issuance costs are amortized over the life of the loan. Upon termination of the Facility Agreement, the Company accelerated amortization of debt discount and debt issuance costs and have recognized the full amount as of March 31, 2022.
A lender who is an over 5% stockholder of the Company loaned the Company $3.2 million of the first tranche and, as such, related party interest expense for the twelve months ended March 31, 2022 was $206 thousand (not including amortization of debt discount and deferred offering costs), respectively. See Note 17.
In connection with the termination of the Facility Agreement, on November 8, 2021, the Company entered into a modification of the Facility Agreement for one lender to allow for repayment of $200 thousand on unchanged payment terms. The loan is unsecured with interest at 10% per year which is to be paid quarterly. The loan shall be repaid in installments commencing on June 15, 2023 with all outstanding amounts due on March 17, 2025.
SCHEDULE OF MATURITY OF LONG-TERM LOAN
| Maturity of Term Loan (thousands) | March 31, 2023 | |||
| 2024 | $ | 80 | ||
| 2025 | 120 | |||
| Total | $ | 200 | ||
| F- |
BEYOND AIR, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 13 LOAN PAYABLE
As of March 31, 2023 and March 31, 2022 in connection with the Company’s insurance policy, a loan was used to finance part of the premium. The details concerning the loan are as follows:
SCHEDULE OF LOAN PAYABLE
| March 31, 2023 | March 31, 2022 | |||||||
| Amount outstanding | $ | 695 | $ | 927 | ||||
| Monthly payments | $ | 88 | $ | 104 | ||||
| Number of monthly payments remaining | 8 | 10 | ||||||
| Interest rate | 4.85 | % | 1.3 | % | ||||
| Due date | November 2023 | December 2022 | ||||||
NOTE 14 INCOME TAXES
As of March 31, 2023, the Company has approximately $86.4 million, $18.1 million, $3.4 million and $0.9 million of net operating losses (NOL) carryforwards for U.S. federal, Israeli, Irish and Cypriot tax purposes, respectively. The U.S. federal NOL carryforwards of approximately $1.4 million, which were generated prior to March 2018 expire through 2037. The NOL of approximately $85.0 million can be carried forward indefinitely but limited to offset 80% of taxable income. The entire NOL for Israel, Ireland and Australia can be carried forward indefinitely. The Company also has state NOL carryforwards in the amount of approximately $82.3 million expiring during the years from 2036 to 2042. The Tax Cuts and Jobs Act of 2017 (TCJA) has modified the IRC 174 expenses related to research and development for tax years beginning after December 31, 2021. Under the TCJA, the Company must now capitalize the expenditures related to research and development activities and amortize over five years for U.S. activities and 15 years for non-U.S. activities using a mid-year convention. Therefore, the capitalization of research and development costs in accordance with IRC 174 resulted in a gross deferred tax asset of $8.0 million.
Pursuant to Section 382 of the Internal Revenue Code, changes in the Company’s ownership may limit the amount of its NOL carryforwards that could be utilized annually to offset future taxable income, if any. This limitation would generally apply in the event of a cumulative change in ownership of the Company of more than 50% within a three-year period. The Company has performed a study as of March 31, 2021 and determined that on or around February 15, 2017 and February 15, 2020 ownership changes for purposes of Section 382 have occurred. The annual limitations caused by these prior ownership changes will no longer impact the utilizations of NOL’s after March 31, 2022. The Company has not updated the study since March 31, 2021 and therefore has not determined if any other NOL limitations exist.
The components of net loss before the provision for income taxes are as follows (in thousands):
SCHEDULE OF PROFIT (LOSS) BEFORE TAXES
|
For the Year Ended March 31, 2023 |
For the Year Ended March 31, 2022 |
|||||||
| Domestic | $ | (49,720 | ) | $ | (39,148 | ) | ||
| Foreign | (9,681 | ) | (4,912 | ) | ||||
| Total | $ | (59,401 | ) | $ | (44,060 | ) | ||
| F- |
BEYOND AIR, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 14 INCOME TAXES (continued)
The tax effects of temporary differences that gave rise to significant portions of the deferred tax assets were as follows (in thousands):
SCHEDULE OF DEFERRED TAX ASSET/LIABILITY
| March 31, 2023 | March 31, 2022 | |||||||
| Net operating loss carryforward | $ | 28,240 | $ | 22,952 | ||||
| Research and development tax credits | 1,624 | 1,226 | ||||||
| Research and development tax credit capitalization | 2,158 | |||||||
| Other | 23 | (162 | ) | |||||
| Depreciation | (1,228 | ) | - | |||||
| Stock-based compensation | 6,731 | 3,268 | ||||||
| Capital loss carryforward | 1,559 | 1,559 | ||||||
| Reserves and Accruals | 5,291 | 3,580 | ||||||
| Right-of-use asset | (484 | ) | (464 | ) | ||||
| Lease liability | 524 | 495 | ||||||
| Net deferred tax | 44,438 | 32,454 | ||||||
| Valuation allowance | (44,438 | ) | (32,454 | ) | ||||
| Net deferred tax asset | $ | - | $ | - | ||||
A reconciliation of income tax expense calculated at the federal enacted statutory rate of 21% is as follows:
SCHEDULE OF STATUTORY US FEDERAL EFFECTIVE RATE
| March 31, 2023 | March 31, 2022 | |||||||
| Federal income tax at statutory rate | (21.00 | )% | (21.00 | )% | ||||
| State income tax, net of federal benefit | (4.35 | ) | (6.21 | ) | ||||
| Permanent items | 0.08 | (0.86 | ) | |||||
| Change in valuation allowance | 20.17 | 29.31 | ||||||
| Research and development tax credits | (0.67 | ) | (0.65 | ) | ||||
| Foreign tax rate differential | 2.81 | 0.00 | ||||||
| Other | 2.96 | (0.59 | ) | |||||
| Effective income tax expense rate | 0.00 | % | 0.00 | % | ||||
NOTE 15 COMMITMENTS AND CONTINGENCIES
License Agreements
In August 2015, BA Ltd. entered into an Option Agreement (the “Option Agreement”) with Pulmonox whereby BA Ltd. acquired the option (the “Option”) to purchase certain intellectual property assets and rights. On January 13, 2017, the Company exercised the Option and paid $500 thousand to Pulmonox. The Company becomes obligated to make certain one-time development and sales milestone payments to Pulmonox, commencing with the date on which the Company receive regulatory approval for the commercial sale of the first product candidate qualifying under the Option Agreement. These milestone payments are capped at a total of $87 million across three separate and distinct indications that fall under the agreement, with the majority of them, approximately $83 million, being sales-related based on cumulative sales milestones for each of the three products. The Company is not currently developing any qualifying products.
| F- |
BEYOND AIR, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 15 COMMITMENTS AND CONTINGENCIES (continued)
On January 31, 2018, the Company entered into an agreement (the “NitricGen Agreement”) with NitricGen, Inc. (“NitricGen”) to acquire a global, exclusive, transferable license and associated assets including intellectual property, know-how, trade secrets and confidential information from NitricGen related to the LungFit®. The Company acquired the licensing right to use the technology and agreed to pay NitricGen a total of $2.0 million in future payments based upon achieving certain milestones, as defined in the NitricGen Agreement, and single-digit royalties on sales of the LungFit®. The Company paid NitricGen $100 thousand upon the execution of the NitricGen Agreement, $100 thousand upon achieving the next milestone, accrued $1.5 million in March 2022 (which was paid in January 2023, six months after approval of the LungFit® by the FDA) and issued 100,000 warrants to purchase the Company’s common stock valued at $295 thousand upon executing the NitricGen Agreement. The remaining future milestone payments are $0.3 million.
Supply Agreement and Purchase Order
In August 2020, the Company entered into a supply agreement expiring on December 31, 2024. The agreement will renew automatically for successive three-year periods unless and until the Company provides twelve months’ notice of intent not to renew. The Company has opened several non-cancellable purchase orders and the outstanding amount remaining under the purchase order as of March 31, 2023 was approximately $4.8 million with this supplier.
Contingencies
On March 23, 2023, the Supreme Court of the State of New York, Appellate Division, First Judicial Department (the “Appellate Court”), rendered its opinion in Empery Asset Master, Ltd., et. al. vs. AIT Therapeutics, Inc. (the “Empery Suit”). The Appellate Court opinion affirmed the judgment by the Supreme Court of the State of New York against the Company. In connection with the appeal, the Company had used approximately $7.4 million of cash as collateral to secure a supersedeas bond for the full amount of damages and interest in case it was unsuccessful in its appeal. The Company accrued a total of $7.6 million for damages and interest as of March 31, 2023. As a subsequent event to the fiscal year ended March 31, 2023, the Company paid a total of $7.6 million including damages and interest in satisfaction of judgment.
On December 28, 2021 Hudson Bay Master Fund (“Hudson”) filed a lawsuit against the Company related to the notice of adjustment of the exercise price of and the number of warrant shares issuable under warrants issued to Hudson in January 2017. Hudson received 83,334 warrants in connection with the January 2017 offering. Hudson’s complaint alleges breach of contract and that Hudson is entitled to damages estimated at approximately $2.6 million as a result of certain adjustments to the exercise price and number of warrant shares issuable following a February 2018 financing transaction. The Company accrued a total of $2.7 million as of March 31, 2023, reflecting management’s estimate of most likely outcome of the lawsuit.
| F- |
BEYOND AIR, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 16 BEYOND CANCER VARIABLE INTEREST ENTITY
On November 4, 2021, the Company announced that Beyond Air and Beyond Cancer, Ltd agreed to terms to which the Company, through its subsidiaries would be licensing certain intellectual property and other assets related to, or necessary for the development, commercialization, manufacture and distribution of certain cancer treatment products and/or technologies to a subsidiary of the Company (the “Transaction”). In connection and concurrently with the closing of the Transaction, Beyond Cancer issued and sold common shares, par value $1.00 to certain investors pursuant to a subscription agreement (the “Offering”). The Offering consisted of an aggregate of 3 million common shares of Beyond Cancer at a purchase price of $10.00 per share. On November 18, 2021, the Company announced that the maximum amount of shares offered had been purchased for a total of $30 million (including $4.8 million from the terminated Loan Facility and $1.1 million from related parties) for 20% of the equity in Beyond Cancer. The Company retained 80% ownership of Beyond Cancer, which will have exclusive right to the intellectual property portfolio utilizing UNO for the treatment of solid tumors. Beyond Cancer will pay Beyond Air a single digit royalty on all future revenues.
Members of the Board of Directors of Beyond Air who are also members of the Board of Directors of Beyond Cancer, and their families, are considered related parties to the Offering. Related parties invested $1.1 million in the Offering. See Note 17.
The carrying amount of net assets of the VIE included in the consolidated financial statements, after the elimination of intercompany balances and transactions, was $20.6 million (including $20.5 million of cash) at March 31, 2023, compared with $27.5 million (including $27.7 million of cash) at March 31, 2022. Beyond Cancer generated $17.9 million of losses (before elimination of intercompany amounts) for the year ended March 31, 2023. The Company’s attributed losses as the primary beneficiary was proportional to its equity interest in Beyond Cancer (80%) for the year ended March 31, 2023.
The following table summarizes segment financial information by business segment for the year ended and at March 31, 2023:
SCHEDULE OF SEGMENT FINANCIAL INFORMATION BY BUSINESS SEGMENT
| (in thousands) | Beyond Air | Beyond Cancer | Total | |||||||||
| Cash, cash equivalents and marketable securities | $ | 25,388 | $ | 20,494 | $ | 45,882 | ||||||
| All other assets | 22,310 | 557 | 22,867 | |||||||||
| Total assets | $ | 47,699 | $ | 21,051 | $ | 68,749 | ||||||
| Total liabilities | (26,201 | ) | (520 | ) | (26,721 | ) | ||||||
| Net assets | $ | 21,498 | $ | 20,530 | $ | 42,028 | ||||||
| Non-controlling interests | $ | - | $ | 4,113 | $ | 4,113 | ||||||
| (in thousands) | ||||||||||||
| Net loss for the year ended March 31, 2023 | $ | (41,475 | ) | $ | (17,927 | ) | $ | (59,401 | ) | |||
| Operating activities included in net loss: | ||||||||||||
| Depreciation and amortization | $ | 1,107 | $ | 63 | $ | 1,170 | ||||||
| Stock-based compensation expense | $ | 8,590 | $ | 10,969 | $ | 19,559 | ||||||
| Cash used in operations | $ | (25,864 | ) | $ | (7,145 | ) | $ | (33,009 | ) | |||
| Cash used in investing | (20,482 | ) | (106 | ) | (20,587 | ) | ||||||
| Cash from financing | 2,696 | - | 2,696 | |||||||||
| Impact of exchange rates | (2 | ) | (42 | ) | (43 | ) | ||||||
| Net change for the period | $ | (43,651 | ) | $ | (7,292 | ) | $ | (50,944 | ) | |||
The following table summarizes segment financial information by business segment for the year ended and at March 31, 2022:
| (in thousands) | Beyond Air | Beyond Cancer | Total | |||||||||
| Cash and cash equivalents | $ | 52,515 | $ | 27,727 | $ | 80,242 | ||||||
| All other assets | 18,674 | 283 | 18,957 | |||||||||
| Total assets | $ | 71,189 | $ | 28,010 | $ | 99,199 | ||||||
| Total liabilities | (20,505 | ) | (485 | ) | (20,990 | ) | ||||||
| Net assets | $ | 50,684 | $ | 27,525 | $ | 78,209 | ||||||
| Non-controlling interests | $ | - | $ | 5,505 | $ | 5,505 | ||||||
| (in thousands) | ||||||||||||
| Net loss for the year ended March 31, 2022 | $ | (39,150 | ) | $ | (4,910 | ) | $ | (44,060 | ) | |||
| Operating activities included in net loss: | ||||||||||||
| Depreciation and amortization | $ | 312 | $ | 2 | $ | 314 | ||||||
| Stock-based compensation expense | $ | 5,813 | $ | 2,018 | $ | 7,831 | ||||||
| Cash used in operations | $ | (20,975 | ) | $ | (2,159 | ) | $ | (23,134 | ) | |||
| Cash used in investing | (1,334 | ) | (116 | ) | (1,450 | ) | ||||||
| Cash from financing | 49,450 | 30,000 | 79,450 | |||||||||
| Impact of exchange rates | 96 | - | 96 | |||||||||
| Net change for the period | $ | 27,171 | $ | 27,792 | $ | 54,962 | ||||||
| F- |
BEYOND AIR, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 17 RELATED PARTY TRANSACTIONS
In March 2020, as part of the first tranche of the Facility Agreement, a lender who is an over 5% stockholder of the Company loaned the Company $3.2 million and, as such, related party interest expense for the year ended March 31, 2022 was $0.2 million. See Note 12.
Members of the Board of Directors of Beyond Air who are also members of the Board of Directors of Beyond Cancer, and their families, are considered related parties to this transaction. Related parties invested $1.1 million in the Offering in November 2021. See Note 16.
In the fourth quarter of fiscal year 2022, the Company recorded approximately $37 thousand related to the net recovery of short-swing profits from one of the Company’s shareholders under Section 16(b) of the Securities Exchange Act of 1934, as amended. The Company recognized these related party proceeds, net of $4 thousand related legal fees, as an increase to additional paid-in capital as of March 31, 2022 as well as cash proceeds of approximately $33 thousand as cash provided by financing activities in the consolidated statement of cash flows for the fiscal quarter ended March 31, 2022.
There were no related party transactions in the fiscal year ended March 31, 2023.
NOTE 18 SUBSEQENT EVENTS
On April 12, 2023 the Company paid $7.6 million in satisfaction of judgment on the Empery lawsuit, including releasing the funds on the supersedeas bond that had been recorded as restricted cash as of March 31, 2023.
On June 15, 2023 (the “Closing Date”), The Company entered into a Loan and Security Agreement (the “Agreement”) with Avenue Capital Management II, L.P., as administrative agent and collateral agent (the “Agent”), Avenue Venture Opportunities Fund, L.P., a Delaware limited partnership (“Avenue”), and Avenue Venture Opportunities Fund II, L.P, a Delaware limited partnership (“Avenue 2” and, together with Avenue, the “Lenders”). Also on June 15, 2023, the Company entered into a Supplement to the Agreement (collectively with the Agreement, the “Loan Agreement”) with the Agent and the Lenders. The Loan Agreement provides for senior secured term loans (the “Loans”) in an aggregate principal amount up to $40 million, with (i) $17.5 million advanced on the Closing Date (“Tranche 1”), (ii) up to $10 million which may be advanced upon the request of the Company between April 1, 2024 and September 30, 2024, subject to the Company having achieved total revenue derived from the sale of LungFit® PH (other than licensing revenue) (“Product Revenue”) for the three-month period prior to funding of not less than 85% of projected Product Revenue for such period (“Tranche 2”), and (iii) up to $12.5 million which may be advanced after April 1, 2024 (the “Discretionary Tranche”), subject to (a) the Agent and Lenders having received investment committee approval and (b) the Company and Lenders having mutually agreed to draw and fund, respectively, such amount. The Loans are due and payable on June 1, 2027 (the “Maturity Date”). The Loan principal is repayable in equal monthly installments beginning on January 1, 2025, with the possibility of deferring principal payments an additional 6 to 18 months contingent upon the Company’s achievement of at least $40 million of Product Revenue in the fiscal year ending March 31, 2025 and whether the Company has fully drawn Tranche 2. The Loans bear interest at a rate per annum (subject to increase during an event of default) equal to the greater of (i) the prime rate, as published by the Wall Street Journal from time to time, plus 3.75% and (ii) 12.00%. The Company may, subject to certain parameters, voluntarily prepay the Loans, in whole or in part, at any time. If prepayment occurs on or before the one-year anniversary of the Closing Date, the Company is required to pay a fee equal to the principal amount of the Loans prepaid multiplied by 3.00%; if prepayment occurs after the one-year anniversary of the Closing Date and on or before the two-year anniversary of the Closing Date, the Company is required to pay a fee equal to the principal amount of the Loans prepaid multiplied by 2.00%; if prepayment occurs after the two-year anniversary and on or before the three-year anniversary of the Closing Date, the Company is required to pay a fee equal to the principal amount of the Loans prepaid multiplied by 1.50%; and if prepayment occurs after the three-year anniversary of the Closing Date and before the Maturity Date, the Company is required to pay a fee equal to the principal amount of the Loans prepaid multiplied by 1.00%. A final payment fee of 3.50% of the principal amount of the Tranche 1 and Tranche 2 Loans is also due upon the Maturity Date or any earlier date of prepayment (in the case of any partial prepayment, solely with respect to the principal amount being prepaid). The Loans are guaranteed by the Company’s subsidiaries, Beyond Air Ltd. and Beyond Air Ireland Limited, and certain of the Company’s future subsidiaries (collectively, the “Guarantors”). The Company’s obligations under the Loan Agreement and the guarantee of such obligations are secured by a pledge of substantially all of the Company’s assets and will be secured by a pledge of substantially all of the assets of the Guarantors.
Pursuant to the Loan Agreement, the Company is subject to a financial covenant requiring the Company to maintain at all times $5,000,000 in unrestricted cash. The Loan Agreement also contains affirmative and negative covenants customary for financings of this type that, among other things, limit the ability of the Company and its subsidiaries to (i) incur additional debt, guarantees or liens; (ii) pay any dividends; (iii) enter into certain change of control transactions; (iv) sell, transfer, lease, license, or otherwise dispose of certain assets; (v) make certain investments or loans; and (vi) engage in certain transactions with related persons, in each case, subject to certain exceptions.
The Loan Agreement also includes events of default customary for financings of this type, in certain cases subject to customary periods to cure, following which the Agent may accelerate all amounts outstanding under the Loans. The Company granted Avenue Capital Group warrants to purchase 233,843 shares of common stock at an exercise price of the lesser of $5.88 or the price per share of our next bona fide round of equity financing before June 30, 2024.
The Company received $15.7 million in net proceeds on June 15, 2023 after all fees and advanced interest had been deducted.
| F- |
Exhibit 10.29
EMPLOYMENT AGREEMENT
EMPLOYMENT AGREEMENT (“Agreement”) is effective on March 27th, 2023 between Beyond Air, Inc. (the “Company”), a Delaware Company, and Jeff L. Myers, M.D., Ph.D. (the “Employee”), residing at 1001 Julia Street, New Orleans, LA 70113.
Recital:
The parties desire to enter into this Agreement so as to provide for the employment of the Employee by the Company and for certain other matters in connection with such employment, all as set forth more fully in this Agreement.
NOW, THEREFORE, in consideration of the premises and covenants set forth herein, and intending to be legally bound hereby, the parties to this Agreement hereby agree as follows:
1. Duties. The Company agrees that the Employee shall be employed by the Company to serve as the Chief Medical Officer of the Company. The Employee shall report to the Chief Executive Officer of the Company (the “CEO”). The Employee agrees to be so employed by the Company and to devote his best efforts and full working time to advance the interests of the Company and to perform the duties customarily incident to the position of Chief Medical Officer and such other duties assigned to the Employee by the CEO and the Board of Directors of the Company (the “Board”), provided such other duties are commensurate with the Employee’s employment level at the Company.
2. Term. The Employee’s employment under this Agreement shall continue in effect until terminated pursuant to Section 4 of this Agreement.
3. Compensation.
(a) Salary. During the term of the Employee’s employment under this Agreement, the Employee shall be paid an annual salary at the rate of not less than $425,000 (the “Base Salary”). The Base Salary may be increased from time to time by the Board in its sole discretion. The Base Salary shall be paid in accordance with the Company’s regular payroll practices.
(b) Annual Bonus. At the end of each fiscal year of the Company that ends during the term of this Agreement, the Board or its Compensation Committee in its sole discretion shall consider whether to award a bonus to the Employee. Any bonus, if awarded, shall be paid within two and one-half months after the close of each such year and Employee must be employed on the date of the bonus to be eligible for the bonus. A bonus in one year does not guarantee a bonus in any subsequent year.
(c) Equity Incentive Awards. On the first date of employment, the Employee will be granted an inducement stock option award exercisable for the purchase of 50,000 shares of the Company’s Common Stock , subject to the terms and conditions of the Company’s 2013 Equity Incentive Plan, as amended, and the applicable award agreement. The exercise price of the stock option will be equal to the last reported sale price on the Nasdaq on the date of grant. The stock option will vest 25% on the first anniversary and annually thereafter in 3 equal installments, provided that, no portion of the stock option that is not exercisable at the time of the Employee’s termination of employment for any reason shall thereafter become exercisable.
In addition, on the first date of employment, the Employee will be granted an inducement restricted stock unit award, subject to the terms and conditions of the Company’s 2013 Equity Incentive Plan, as amended, and the applicable award agreement. The RSUs will vest 20% annually beginning on December 15, 2023 until fully vested.
The Employee shall be eligible to participate in equity incentive programs established by the Company from time to time to provide stock options and other equity-based incentives to key employees of the Company in accordance with the terms of those programs.
(d) Vacation and Fringe Benefits. Based on the Employee’s level in the Company, the Employee shall be eligible to take paid time off during the year, provided that the Employee schedules and takes time off in a manner that ensures that all Company needs are met and the Employee maintains satisfactory performance and productivity levels. Paid time off is additional to bank holidays and days the Company does not operate. The Employee shall be entitled to participate in all insurance and other fringe benefit programs of the Company to the extent and on the same terms and conditions as are accorded to other officers and key employees of the Company.
(e) Reimbursement of Expenses. The Employee shall be reimbursed for all normal items of travel, entertainment and miscellaneous business expenses reasonably incurred by the Employee on behalf of the Company, provided that such expenses are documented and submitted in accordance with the reimbursement policies of the Company as in effect from time to time.
4. Termination.
(a) Death. This Agreement and Employee’s employment shall automatically terminate effective as of the date of the Employee’s death, in which event the Company shall not have any further obligation or liability under this Agreement except that the Company shall pay to the Employee’s estate: (i) any portion of the Employee’s Base Salary for the period up to the Employee’s date of death that has been earned but remains unpaid; and (ii) any benefits that have accrued to the Employee under the terms of the employee benefit plans of the Company, which benefits shall be paid in accordance with the terms of those plans.
(b) Total Disability. The Company may terminate the employment of the Employee immediately upon written notice to the Employee in the event of the Disability (as that term is hereinafter defined) of the Employee, in which event, the Company shall not have any further obligation or liability under this Agreement except that the Company shall pay to the Employee: (i) any portion of the Employee’s Base Salary for the period up to the date of termination that has been earned but remains unpaid; and (ii) any benefits that have accrued to the Employee under the terms of the employee benefit plans of the Company, which benefits shall be paid in accordance with the terms of those plans. For purposes of this Agreement, the term “Disability” shall mean an illness, incapacity or a mental or physical condition that renders the Employee unable or incompetent to carry out the job responsibilities that the Employee held or the tasks that the Employee was assigned at the time the disability commenced for 120 consecutive days or 180 days in a one-year period, as determined by the Board and supported by the opinion of a physician. The Employee shall fully cooperate with the physician retained to furnish such opinion, including submitting to such examinations and tests as may be requested by the physician.
(c) Termination by the Company for Cause. The Company may terminate the Employee’s employment hereunder and this Agreement immediately (unless stated otherwise) upon written notice to the Employee for “Cause.” For purposes of this Agreement, Cause shall be defined as: (i) the Employee’s use of alcoholic beverages, controlled substances or other narcotics, which use has had or is reasonably likely to have a material adverse effect on the business or financial affairs of the Company or the reputation of the Company; (ii) failure by the Employee to cooperate with the Company in any investigation or formal proceeding; (iii) the commission by the Employee of, or a plea by the Employee of guilty or nolo contendere with respect to, or conviction of the Employee for, a felony (or any lesser included offense or crime in exchange for withdrawal of a felony indictment or charged crime that might result in a penalty of incarceration), a crime involving moral turpitude, or any other offense that results in or could result in any prison sentence; (iv) adjudication of Employee as an incompetent; (v) a breach by the Employee of any material term of this Agreement, including the Employee’s failure to faithfully, diligently and adequately perform the Employee’s duties under this Agreement, that is not corrected within ten days after written notice from the Company, which notice shall set forth the nature of the breach; (vi) violation by Employee in any material respect of any of the Company’s rules, regulations or policies; (vii) gross insubordination by the Employee in the performance of the Employee’s duties under this Agreement; (viii) any conduct, action or behavior by Employee that, in the reasonable opinion of the Company, has had or is likely to have a material adverse effect on the reputation of the Company or the Employee; (ix) any continued or repeated absence of Employee from the Company, unless the absence is approved or excused by the CEO or the result of the Employee’s illness, disability or incapacity (in which event the provisions of Section 4(b) hereof shall control); or (x) misappropriation by Employee of any funds or property of the Company, theft, embezzlement or fraud. For the avoidance of doubt, “Cause” shall not mean a failure to achieve scientific goals, financial goals or forecasted timelines. In the event that the Company shall discharge the Employee pursuant to this Section 4(c), the Company shall not have any further obligation or liability under this Agreement, except that the Company shall pay to the Employee: (i) any portion of the Employee’s Base Salary for the period up to the date of termination that has been earned but remains unpaid; and (ii) any benefits that have accrued to the Employee under the terms of the employee benefit plans of the Company, which benefits shall be paid in accordance with the terms of those plans.
(d) Other Termination by the Company. The Company may terminate the employment of the Employee and this Agreement for any reason other than one specified in Section 4(b) or 4(c) hereof immediately upon written notice to the Employee, in which event the Employee shall be entitled to receive: (i) any portion of the Employee’s Base Salary for the period up to the date of termination that has been earned but remains unpaid; (ii) any benefits that have accrued to the Employee under the terms of any employee benefit plans of the Company, which benefits shall be paid in accordance with the terms of those plans; and (iii) subject to the satisfaction of the provisions of Section 4(g) and the compliance by the Employee with all terms and provisions of this Agreement that survive the termination of the Employee’s employment by the Company, (A) the Employee’s Base Salary for a period of one (1) month for every two months of employment to a maximum of six (6) months, less applicable taxes and withholdings, payable in accordance with the Company’s regular payroll practices, with an accelerated payment of any balance upon the occurrence of a Change in Control; provided, however, that if such termination of employment shall occur within three months before or within twelve months after the occurrence of a Change in Control (such period being referred to herein as the “Change of Control Period”), the severance payable to the Employee shall be increased to an amount equal to the Employee’s Base Salary for a period of twelve (12) months and be payable in a single lump sum payment, less applicable taxes and withholdings; and (B) payment or reimbursement (upon presentation of proof of payment) of the Employee’s medical insurance premiums at the same level as was in effect on the termination date for a period equal to the number of months of salary continuance which period shall increase to eighteen months (18) if such termination of employment shall occur within the Change in Control Period. Any payments due hereunder shall commence as soon as administratively feasible within 60 days after the date of the Employee’s termination of employment provided the Employee has timely executed and returned the Release referred to in Section 4(g) and, if a revocation period is applicable, the Employee has not revoked the Release; provided, however, that if the 60-day period begins in one calendar year and ends in a second calendar year, the severance payments shall begin to be paid in the second calendar year. On the date that severance payments commence, the Company will pay the Employee in a single lump sum payment, less applicable taxes and withholding, the Severance payments that the Employee would have received on or prior to such date but for the delay imposed by the immediately preceding sentence, with the balance of the severance payments to be paid as originally scheduled.
(e) Termination by the Employee for Good Reason. The Employee may terminate the Employee’s employment by providing written notice to the Company of a breach constituting Good Reason. “Good Reason” shall be deemed to exist with respect to any termination of employment by the Employee for any of the following reasons: (i) any material failure by the Company to comply with any material term of this Agreement; (ii) the sustained demotion of the Employee to a lesser position than described in Section 1 hereof or a substantial diminution of the Employee’s authority, duties or responsibilities as in effect on the date of this Agreement or as hereafter increased; or (iv) a material diminution of the Executive’s Base Salary and benefits, in the aggregate, unless such reduction is part of a Company-wide reduction in compensation and/or benefits for all of its senior executives. If the Employee shall terminate the Employee’s employment hereunder for Good Reason, the Employee shall be entitled to receive the same payments and benefits on the same terms and conditions (including satisfaction of the provisions of Section 4(g)) as would be applicable upon a termination of the Employee’s employment by the Company without Cause, as provided in Section 4(d) and subject to the satisfaction of the other provisions of this Section 4(e). The Employee may not resign with Good Reason pursuant to this Section 4(e), and shall not be considered to have done so for any purpose of this Agreement, unless (A) the Employee, within 60 days after the initial existence of the act or failure to act by the Company that the Employee believes constitutes “Good Reason” within the meaning of this Agreement, provides the Company with written notice that describes, in particular detail, the act or failure to act that the Employee believes to constitute “Good Reason” and identifies the particular clause of this Section 4(e) that the Employee contends is applicable to such act or failure to act; (B) the Company, within 30 days after its receipt of such notice, fails or refuses to cure the act(s) or failure(s) that the Employee claims to be Good Reason (the “Cure Period”), and (C) the Employee actually resigns from the employ of the Company on or before that date that is 30 days after the Cure Period ends with the Company not having cured the act or failure that the Employee claims to be Good Reason. If the requirements of the preceding sentence are not fully satisfied on a timely basis, then the resignation by the Employee from the employ of the Company shall not be deemed to have been for “Good Reason,” the Employee shall not be entitled to any of the benefits to which the Employee would have been entitled if the Employee had resigned from the employ of the Company for “Good Reason,” and the Company shall not be required to pay any amount or provide any benefit that would otherwise have been due to the Employee under this Section 4(e) had the Employee resigned with “Good Reason.”
(f) Other Termination by the Employee. The Employee may terminate the Employee’s employment and this Agreement for any reason other than one specified in Section 4(e) upon at least 30 days’ prior written notice to the Company, which notice shall specify the effective date of the termination. In the event the Employee shall terminate the Employee’s employment pursuant to this Section 4(f), the Company shall not have any further obligation or liability under this Agreement, except that the Company shall pay to the Employee: (i) any portion of the Employee’s Base Salary for the period up to the date of termination that has been earned but remains unpaid; and (ii) any benefits that have accrued to the Employee under the terms of the employee benefit plans of the Company, which benefits shall be paid in accordance with the terms of those plans.
(g) Execution of Release. The Employee shall not be entitled to any payments or benefits under Sections 4(d) or 4(e) unless the Employee executes and does not revoke a Release and Agreement (the “Release”), as drafted by the Company at the time of the Employee’s termination of employment, which shall include, but not limited to the following terms:
(i) an unconditional release of all rights to any claims, charges, complaints, grievances, known or unknown to the Employee, against the Company, its affiliates or assigns, through the date of the Employee’s termination from employment other than post-termination payments and benefits pursuant to this Agreement;
(ii) a representation and warranty that the Employee has not filed or assigned any claims, charges, complaints, or grievances against the Company, its affiliates, or assigns; (iii) an agreement not to use, disclose or make copies of any confidential information of the Company, as well as to return any such confidential information and property to the Company upon execution of the Release;
(iv) a mutual agreement to maintain the confidentiality of the Release or disclose the reasons for any termination of employment;
(v) an agreement not to disparage the Company or its affiliates, or its or their respective officers, directors, stockholders, products or business;
(vi) an agreement to continue to comply with the terms of Section 5 herein; and
(vii) an agreement to indemnify the Company, or its affiliates or assigns, in the event that the Employee breaches any portion of this Agreement or the Release.
Notwithstanding any provision of this Agreement to the contrary, in no event shall the timing of the Employee’s execution of the Release, directly or indirectly, result in the Employee designating the calendar year of payment, and if a payment that is subject to execution of the Release could be made in more than one taxable year, payment shall be made in the later taxable year.
(h) Definition of Change in Control. As used in this Agreement, the term “Change in Control” shall mean the definition set forth in the Company’s 2013 Equity Incentive Plan, as amended.
(i) Base Salary Continuation. The Base Salary continuation set forth in Sections 4(c) and (e) above shall be intended either (i) to satisfy the safe harbor set forth in the regulations issued under section 409A of the Internal Revenue Code of 1986, as amended (the “Code”) (Treas. Regs. 1.409A-1(n)(2)(ii)) or (ii) be treated as a Short-term Deferral as that term is defined under Code section 409A (Treas. Regs. 1.409A-1(b)(4)). To the extent such continuation payments exceed the applicable safe harbor amount or do not constitute a Short-term Deferral, the excess amount shall be treated as deferred compensation under Code section 409A and as such shall be payable pursuant to the following schedule: such excess amount shall be paid via standard payroll in periodic installments in accordance with the Company’s usual practice for its senior executives. Solely for purposes of Code section 409A, each installment payment is considered a separate payment. Notwithstanding any provision in this Agreement to the contrary, in the event that the Employee is a “specified employee” as defined in Section 409A, any continuation payment, continuation benefits or other amounts payable under this Agreement that would be subject to the special rule regarding payments to “specified employees” under Section 409A(a)(2)(B) of the Code shall not be paid before the expiration of a period of six months following the date of the Employee’s termination of employment or before the date of the Employee’s death, if earlier.
(j) Parachute Provisions. Notwithstanding any provisions of this Agreement to the contrary:
(i) If any of the payments or benefits received or to be received by the Employee in connection with the Employee’s termination of employment in respect of a Change in Control, whether pursuant to the terms of this Agreement or any other plan, arrangement or agreement with the Company (all such payments and benefits, being hereinafter referred to as the “Total Payments”), would be subject to the excise tax (the “Excise Tax”) imposed under Section 4999 of the Code, the Employee shall receive the Total Payments and be responsible for the Excise Tax; provided, however that the Employee shall not receive the Total Payments and the Total Payments shall be reduced to the Safe Harbor Amount (defined below) if (A) the net amount of such Total Payments, as so reduced to the Safe Harbor Amount (and after subtracting the net amount of federal, state and local income taxes on such reduced Total Payments) is greater than or equal to (B) the net amount of such Total Payment without such reduction (but after subtracting the net amount of federal, state and local income taxes on such Total Payments and the amount of Excise Tax to which the Employee would be subject in respect of such unreduced Total Payments). The “Safe Harbor Amount” is the amount to which the Total Payments would hypothetically have to be reduced so that no portion of the Total Payments would be subject to the Excise Tax.
(ii) For purposes of determining whether any of the Total Payments will be subject to the Excise Tax and the amount of such Excise Tax, (A) all of the Total Payments shall be treated as “parachute payments” (within the meaning of Section 280G(b)(2) of the Code) unless, in the opinion of tax counsel (“Tax Counsel”) selected by the accounting firm that was, immediately prior to the Change in Control, the Company’s independent auditor (the “Auditor”), such payments or benefits (in whole or in part) do not constitute parachute payments, including by reason of Section 280G(b)(4)(A) of the Code, (B) all “excess parachute payments” within the meaning of Section 280G(b)(1) of the Code shall be treated as subject to the Excise Tax unless, in the opinion of Tax Counsel, such excess parachute payments (in whole or in part) represent reasonable compensation for services actually rendered (within the meaning of Section 280G(b)(4)(B) of the Code) in excess of the base amount (within the meaning of Section 280G(b)(3) of the Code) allocable to such reasonable compensation, or are otherwise not subject to the Excise Tax, and (C) the value of any noncash benefits or any deferred payment or benefit shall be determined by the Auditor in accordance with the principles of Sections 280G(d)(3) and (4) of the Code. If the Auditor is prohibited by applicable law or regulation from performing the duties assigned to it hereunder, then a different auditor, acceptable to both the Company and Employee, shall be selected. The fees and expenses of Tax Counsel and the Auditor shall be paid by the Company.
(iii) In the event it is determined that the Safe Harbor Amount is payable to Employee, then the severance payments provided under this Agreement that are cash shall first be reduced on a pro rata basis, and the non-cash severance payments shall thereafter be reduced on a pro rata basis, to the extent necessary so that no portion of the Total Payments is subject to the Excise Tax.
5. Non-Disclosure and Non-Competition.
(a) Non-Disclosure. The Employee acknowledges that in the course of performing services for the Company, the Employee will obtain knowledge of the Company’s business plans, products, processes, software, know-how, trade secrets, formulas, methods, models, prototypes, discoveries, inventions, improvements, disclosures, customers, clients, vendors, pricing, names and positions of employees and/or other proprietary and/or confidential information (collectively the “Confidential Information”). The Employee agrees to keep the Confidential Information secret and confidential and not to publish, disclose or divulge to any other party, and the Employee agrees not to use any of the Confidential Information for any purpose other than in the lawful course of his employment with the Company and in furtherance of the Company’s interests, whether or not such Confidential Information was discovered or developed by the Employee. The Employee also agrees not to divulge, publish or use any proprietary and/or confidential information of others that the Company is obligated to maintain in confidence.
(b) Non-Competition. The Employee agrees that during the Employee’s employment by the Company hereunder and for an additional period of twelve (12) months after the termination of the Employee’s employment hereunder for any reason (the “Restricted Period”), the Employee will (i) not anywhere in the world where the Company markets or sells its products, engage or assist others in engaging in any business or enterprise (whether as owner, partner, officer, director, employee, consultant, investor or otherwise) that is competitive with the Company’s business, including but not limited to any business or enterprise that develops, manufactures, markets, licenses, sells or provides any product that competes with any product developed, manufactured, marketed, licensed, sold or provided, or planned to be developed, manufactured, marketed, licensed sold or provided, by the Company (“Competitive Business”) while Employee was employed by the Company; or (ii) solicit, hire, contract for services or otherwise employ, directly or indirectly, anyone who is or was employed by the Company within the six-month period prior to Employee’s termination of employment with the Company. The foregoing prohibition shall not prevent any employment or engagement of the Employee, after termination of employment with the Company, by any company or business organization not substantially engaged in a Competitive Business as long as the activities of any such employment or engagement, in any capacity, do not involve work on matters related to any product or service being developed, manufactured, marketed, distributed or planned in writing by the Company at the time of termination of Employee’s employment with the Company. The Employee’s ownership of no more than 5% of the outstanding voting stock of a publicly traded company shall not constitute a violation of this Section 5(b). The Employee is entering into this covenant not to compete in consideration of the additional agreements of the Company in this Agreement, including but not limited to the rights of the Employee set forth in Sections 4(d) and 4(e). The Restricted Period shall be tolled for any period of time during which the Employee is in breach of any obligations under this Section 5(b).
(c) Blue Pencil. In the event that the terms of this Section 5 shall be determined by any court of competent jurisdiction to be unenforceable in whole or in part by reason of their extending for too great a period of time or over too great a geographical area or by reason of their being too extensive in any other respect, the parties intend for such court to interpret such terms to extend only over the maximum period of time for which they may be enforceable, over the maximum geographical area as to which they may be enforceable, or to the maximum extent in all other respects as to which they may be enforceable.
(d) Permitted Disclosures. Notwithstanding anything to the contrary herein, the Employee understands that nothing in this Agreement restricts or prohibits the Employee from initiating communications directly with, responding to any inquiries from, providing testimony before, providing confidential information to, reporting possible violations of law or regulation to, or from filing a claim or assisting with an investigation directly with a self-regulatory authority or a government agency or entity, or from making other disclosures that are protected under the whistleblower provisions of state or federal law or regulation, and pursuant to 18 USC § 1833(b), an individual may not be held liable under any criminal or civil federal or state trade secret law for disclosure of a trade secret: (i) made in confidence to a government official, either directly or indirectly, or to an attorney, solely for the purpose of reporting or investigating a suspected violation of law or (ii) in a complaint or other document filed in a lawsuit or other proceeding, if such filing is made under seal. Additionally, an individual suing an entity for retaliation based on the reporting of a suspected violation of law may disclose a trade secret to the individual’s attorney and use the trade secret information in the court proceeding, so long as any document containing the trade secret is filed under seal and the individual does not disclose the trade secret except pursuant to court order. Nothing in this Agreement is intended to conflict with 18 USC § 1833(b) or create liability for disclosures of trade secrets that are expressly allowed by 18 USC § 1833(b).
6. Inventions and Discoveries.
(a) Disclosure. The Employee shall promptly and fully disclose to the Company, with all necessary detail, all developments, know-how, discoveries, inventions, improvements, concepts, ideas, formulae, processes and methods (whether copyrightable, patentable or otherwise) made, received, conceived, acquired or written by the Employee (whether or not at the request or upon the suggestion of the Company, solely or jointly with others), during the period of the Employee’s employment with the Company that (i) result from, arise out of, or relate to any work, assignment or task performed by the Employee on behalf of the Company, whether undertaken voluntarily or assigned to the Employee within the scope of the Employee’s responsibilities to the Company, or (ii) were developed using the Company’s facilities or other resources or in Company time, or (iii) result from the Employee’s use or knowledge of the Company’s Confidential Information, or (iv) relate to the Company’s business or any of the products or services being developed, manufactured or sold by the Company or that may be used in relation therewith (collectively referred to as “Inventions”). The Employee hereby acknowledges that all original works of authorship that are made by the Employee (solely or jointly with others) within the above terms and that are protectable by copyright are “works made for hire,” as that term is defined in the United States Copyright Act. The Employee understands and hereby agrees that the decision whether or not to commercialize or market any Invention developed by the Employee solely or jointly with others is within the Company’s sole discretion and for the Company’s sole benefit and that no royalty shall be due to the Employee as a result of the Company’s efforts to commercialize or market any such Invention.
(b) Assignment and Transfer. The Employee agrees to assign and hereby does irrevocably assign to the Company all of the Employee’s right, title and interest in and to the Inventions, and the Employee further agrees to deliver to the Company any and all drawings, notes, specifications and data relating to the Inventions, and to sign, acknowledge and deliver all such further papers, including applications for and assignments of copyrights and patents, and all renewals thereof, as may be necessary to obtain copyrights and patents for any Inventions in any and all countries and to vest title thereto in the Company and its successors and assigns and to otherwise protect the Company’s interests therein. The Employee shall not charge the Company for time spent in complying with these obligations. If the Company is unable because of the Employee’s mental or physical incapacity or for any other reason to secure the Employee’s signature to apply for or to pursue any application for any United States or foreign patents or copyright registrations covering Inventions or original works of authorship assigned to the Company as above, then the Employee hereby irrevocably designates and appoints the Company and its duly authorized officers and agents as the Employee’s agent and attorney in fact, to act for and in the Employee’s behalf and stead to execute and file any such applications and to do all other lawfully permitted acts to further the prosecution and issuance of letters patent or copyright registrations thereon with the same legal force and effect as if executed by the Employee.
(c) Company Documentation. The Employee shall hold in a fiduciary capacity for the benefit of the Company all documentation, disks, programs, data, records, drawings, manuals, reports, sketches, blueprints, letters, notes, notebooks and all other writings, electronic data, graphics and tangible information and materials of a secret, confidential or proprietary information nature relating to the Company or the Company’s business that are in the possession or under the control of the Employee. The Employee agrees that in connection with any research, development or other services performed for the Company, the Employee will maintain careful, adequate and contemporaneous written records of all Inventions, which records shall be the property of the Company.
7. Injunctive Relief. The Employee acknowledges that the Employee’s compliance with the agreements in Sections 5 and 6 hereof is necessary to protect the good will and other proprietary interests of the Company and that the Employee is one of the principal executives of the Company and conversant with its affairs, its trade secrets and other proprietary information. The Employee acknowledges that a breach of any of the Employee’s agreements in Sections 5 and 6 hereof will result in irreparable and continuing damage to the Company for which there will be no adequate remedy at law; and the Employee agrees that in the event of any breach of the aforesaid agreements, the Company and its successors and assigns shall be entitled to injunctive relief in any court of competent jurisdiction, without being required to post bond, and to such other and further relief as may be proper.
8. Full Agreement. This Agreement constitutes the entire agreement of the parties concerning its subject matter and supersedes all other oral or written understandings, discussions, and agreements, and may be modified only in a writing signed by both parties. The parties acknowledge that they have read and fully understand the contents of this Agreement and execute it after having an opportunity to consult with legal counsel.
9. Amendments. Any amendment to this Agreement shall be made in writing and signed by the parties hereto.
10. Enforceability. If any provision of this Agreement shall be invalid or unenforceable, in whole or in part, then such provision shall be deemed to be modified or restricted to the extent and in the manner necessary to render the same valid and enforceable, or shall be deemed excised from this Agreement, as the case may require, and this Agreement shall be construed and enforced to the maximum extent permitted by law as if such provision had been originally incorporated herein as so modified or restricted or as if such provision had not been originally incorporated herein, as the case may be.
11. Construction. This Agreement shall be construed and interpreted in accordance with the internal laws of the State of New York.
12. Assignment.
(a) By the Company. The rights and obligations of the Company under this Agreement shall inure to the benefit of, and shall be binding upon, the successors and assigns of the Company. This Agreement may be assigned by the Company without the consent of the Employee.
(b) By the Employee. This Agreement and the obligations created hereunder may not be assigned by the Employee, but all rights of the Employee hereunder shall inure to the benefit of and be enforceable by the Employee’s heirs, devisees, legatees, executors, administrators and personal representatives.
13. Notices. All notices required or permitted to be given hereunder shall be in writing and shall be deemed to have been given when mailed by certified mail, return receipt requested, or delivered by a national overnight delivery service addressed to the intended recipient as follows:
If to the Company:
Beyond Air, Inc.
900 Stewart Ave
Suite 301
Garden City, New York, 11565
Attention: Chief Executive Officer
If to the Employee, to address stated on the signature page to this Agreement.
Any party may from time to time change its address for the purpose of notices to that party by a similar notice specifying a new address, but no such change shall be deemed to have been given until it is actually received by the party sought to be charged with its contents.
14. Waivers. No claim or right arising out of a breach or default under this Agreement shall be discharged in whole or in part by a waiver of that claim or right unless the waiver is supported by consideration and is in writing and executed by the aggrieved party hereto or such party’s duly authorized agent. A waiver by any party hereto of a breach or default by the other party hereto of any provision of this Agreement shall not be deemed a waiver of future compliance therewith, and such provisions shall remain in full force and effect.
15. Section 409A. It is intended that this Agreement be drafted and administered in compliance with section 409A of the Code, including, but not limited to, any future amendments to Code section 409A, and any other Internal Revenue Service or other governmental rulings or interpretations (together, “Section 409A”) issued pursuant to Section 409A so as not to subject the Employee to payment of interest or any additional tax under Code section 409A. The parties intend for any payments under this Agreement to either satisfy the requirements of Section 409A or to be exempt from the application of Section 409A, and this Agreement shall be construed and interpreted accordingly. In furtherance thereof, if payment or provision of any amount or benefit hereunder that is subject to Section 409A at the time specified herein would subject such amount or benefit to any additional tax under Section 409A, the payment or provision of such amount or benefit shall be postponed to the earliest commencement date on which the payment or provision of such amount or benefit could be made without incurring such additional tax. In addition, to the extent that any Internal Revenue Service guidance issued under Section 409A would result in the Employee being subject to the payment of interest or any additional tax under Section 409A, the parties agree, to the extent reasonably possible, to amend this Agreement in order to avoid the imposition of any such interest or additional tax under Section 409A, which amendment shall have the minimum economic effect necessary and be reasonably determined in good faith by the Company and the Employee.
16. Survival of Covenants. The provisions of Sections 4, 5, 6 and 7 hereof shall survive the termination of this Agreement. Furthermore, each other provision of this Agreement that, by its terms, is intended to continue beyond the termination of the Employee’s employment shall continue in effect thereafter.
(Signature
page follows.)
IN WITNESS WHEREOF, this Agreement has been executed by the parties.
| BEYOND AIR INC. | ||
| By: | /s/ Steve Lisi | |
| Steve Lisi | ||
| Chief Executive Officer | ||
| Date: February 17, 2023 | ||
| /s/ Jeff Myers | ||
| By: Jeff Myers | ||
| 1001 Julia Street | ||
| New Orleans, LA 70113 | ||
| Date: February 28, 2023 | ||
Exhibit 21.1
| Subsidiary | Jurisdiction of Incorporation |
|
| Beyond Air Ltd. | Israel | |
| Beyond Air Ireland Limited | Ireland | |
| Beyond Air Australia Pty. Ltd. | Australia | |
| Beyond Cancer Bermuda Limited | Bermuda | |
| Beyond Cancer U.S., Inc. | Delaware | |
| XAIR Israel Ltd | Israel | |
| Beyond Cancer Cyprus Limited | Cyprus |
Exhibit 23.1
INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM’S CONSENT
We consent to the incorporation by reference in the Registration Statements of Beyond Air, Inc. on Form S-3 (File No.’s 333-231416, 333-233283, and 333-237958) and S-8 (File No.’s 333-227697, 333-238239, 333-257562 and 333-269861) of our report dated June 22, 2023, with respect to our audit of the consolidated financial statements of Beyond Air, Inc. as of and for the year ended March 31, 2023, which report is included in this Annual Report on Form 10-K of Beyond Air, Inc. for the year ended March 31, 2023.
/s/ Marcum llp
Marcum llp
East Hanover, New Jersey
June 22, 2023
Exhibit 23.2
INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM’S CONSENT
We consent to the incorporation by reference in the Registration Statements of Beyond Air, Inc. on Form S-3 (File No.’s 333-231416, 333-233283, and 333-237958), and Form S-8 (File No.’s 333-269861, 333-227697, 333-238239, and 333-257562) of our report dated June 28, 2022, with respect to our audit of the consolidated financial statements as of and for the year ended March 31, 2022, which report is included in this Annual Report on Form 10-K of Beyond Air, Inc. for the year ended March 31, 2023. We were dismissed as auditors on October 6, 2022 and, accordingly, we have not performed any audit or review procedures with respect to any financial statements incorporated by reference for the periods after the date of our dismissal.
/s/ Friedman LLP
FRIEDMAN LLP
East Hanover, NJ
June 22, 2023
Exhibit 31.1
CERTIFICATIONS
I, Steven A. Lisi, certify that:
| 1. | I have reviewed this Annual Report on Form 10-K of Beyond Air, Inc.; |
| 2. | Based on my knowledge, this report does not contain any untrue statement of a material fact or omit to state a material fact necessary to make the statements made, in light of the circumstances under which such statements were made, not misleading with respect to the period covered by this report; |
| 3. | Based on my knowledge, the financial statements, and other financial information included in this report, fairly present in all material respects the financial condition, results of operations and cash flows of the registrant as of, and for, the periods presented in this report; |
| 4. | The registrant’s other certifying officer and I are responsible for establishing and maintaining disclosure controls and procedures (as defined in Exchange Act Rules 13a-15(e) and 15d-15(e)) and internal control over financial reporting (as defined in Exchange Act Rules 13a-15(f) and 15d-15(f)) for the registrant and have: |
| (a) | Designed such disclosure controls and procedures, or caused such disclosure controls and procedures to be designed under our supervision, to ensure that material information relating to the registrant, including its consolidated subsidiaries, is made known to us by others within those entities, particularly during the period in which this report is being prepared; | |
| (b) | Designed such internal control over financial reporting, or caused such internal control over financial reporting to be designed under our supervision, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles; | |
| (c) | Evaluated the effectiveness of the registrant’s disclosure controls and procedures and presented in this report our conclusions about the effectiveness of the disclosure controls and procedures, as of the end of the period covered by this report based on such evaluation; and | |
| (d) | Disclosed in this report any change in the registrant’s internal control over financial reporting that occurred during the registrant’s most recent fiscal quarter (the registrant’s fourth fiscal quarter in the case of an annual report) that has materially affected, or is reasonably likely to materially affect, the registrant’s internal control over financial reporting; and |
| 5. | The registrant’s other certifying officer and I have disclosed, based on our most recent evaluation of internal control over financial reporting, to the registrant’s auditors and the audit committee of the registrant’s board of directors (or persons performing the equivalent functions): |
| (a) | All significant deficiencies and material weaknesses in the design or operation of internal control over financial reporting which are reasonably likely to adversely affect the registrant’s ability to record, process, summarize and report financial information; and | |
| (b) | Any fraud, whether or not material, that involves management or other employees who have a significant role in the registrant’s internal control over financial reporting. |
| June 22, 2023 | /s/ Steven A. Lisi |
| Steven A. Lisi | |
|
Chairman and Chief Executive Officer (Principal Executive Officer) |
Exhibit 31.2
CERTIFICATIONS
I, Douglas Larson, certify that:
| 1. | I have reviewed this Annual Report on Form 10-K of Beyond Air, Inc.; |
| 2. | Based on my knowledge, this report does not contain any untrue statement of a material fact or omit to state a material fact necessary to make the statements made, in light of the circumstances under which such statements were made, not misleading with respect to the period covered by this report; |
| 3. | Based on my knowledge, the financial statements, and other financial information included in this report, fairly present in all material respects the financial condition, results of operations and cash flows of the registrant as of, and for, the periods presented in this report; |
| 4. | The registrant’s other certifying officer and I are responsible for establishing and maintaining disclosure controls and procedures (as defined in Exchange Act Rules 13a-15(e) and 15d-15(e)) and internal control over financial reporting (as defined in Exchange Act Rules 13a-15(f) and 15d-15(f)) for the registrant and have: |
| (a) | Designed such disclosure controls and procedures, or caused such disclosure controls and procedures to be designed under our supervision, to ensure that material information relating to the registrant, including its consolidated subsidiaries, is made known to us by others within those entities, particularly during the period in which this report is being prepared; | |
| (b) | Designed such internal control over financial reporting, or caused such internal control over financial reporting to be designed under our supervision, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles; | |
| (c) | Evaluated the effectiveness of the registrant’s disclosure controls and procedures and presented in this report our conclusions about the effectiveness of the disclosure controls and procedures, as of the end of the period covered by this report based on such evaluation; and | |
| (d) | Disclosed in this report any change in the registrant’s internal control over financial reporting that occurred during the registrant’s most recent fiscal quarter that has materially affected, or is reasonably likely to materially affect, the registrant’s internal control over financial reporting; and |
| 5. | The registrant’s other certifying officer and I have disclosed, based on our most recent evaluation of internal control over financial reporting, to the registrant’s auditors and the audit committee of the registrant’s board of directors (or persons performing the equivalent functions): |
| (a) | All significant deficiencies and material weaknesses in the design or operation of internal control over financial reporting which are reasonably likely to adversely affect the registrant’s ability to record, process, summarize and report financial information; and | |
| (b) | Any fraud, whether or not material, that involves management or other employees who have a significant role in the registrant’s internal control over financial reporting. |
| June 22, 2023 | /s/ Douglas Larson |
| Douglas Larson | |
|
Chief Financial Officer (Principal Financial Officer and Principal Accounting Officer) |
Exhibit 32.1
CERTIFICATION PURSUANT TO
18 U.S.C. SECTION 1350,
AS ADOPTED PURSUANT TO
SECTION 906 OF THE SARBANES-OXLEY ACT OF 2002
I, Steven A. Lisi, Chairman and Chief Executive Officer of Beyond Air, Inc. (the “Company”), hereby certify, pursuant to 18 U.S.C. §1350, as adopted pursuant to §906 of the Sarbanes-Oxley Act of 2002, that, to the best of my knowledge:
| 1. | The Annual Report on Form 10-K of the Company for the year ended March 31, 2023 (the “Report”) fully complies with the requirements of Section 13(a) or 15(d) of the Securities Exchange Act of 1934, as amended; and |
| 2. | The information contained in the Report fairly presents, in all material respects, the financial condition and results of operations of the Company. |
| June 22, 2023 | /s/ Steven A. Lisi |
| Steven A. Lisi | |
|
Chairman and Chief Executive Officer (Principal Executive Officer) |
Exhibit 32.2
CERTIFICATION PURSUANT TO
18 U.S.C. SECTION 1350,
AS ADOPTED PURSUANT TO
SECTION 906 OF THE SARBANES-OXLEY ACT OF 2002
I, Douglas Larson, Chief Financial Officer of Beyond Air, Inc. (the “Company”), hereby certify, pursuant to 18 U.S.C. §1350, as adopted pursuant to §906 of the Sarbanes-Oxley Act of 2002, that, to the best of my knowledge:
| 1. | The Annual Report on Form 10-K of the Company for the year ended March 31, 2023 (the “Report”) fully complies with the requirements of Section 13(a) or 15(d) of the Securities Exchange Act of 1934, as amended; and |
| 2. | The information contained in the Report fairly presents, in all material respects, the financial condition and results of operations of the Company. |
| June 22, 2023 | /s/ Douglas Larson |
| Douglas Larson | |
|
Chief Financial Officer (Principal Financial Officer and Principal Accounting Officer) |