UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
(Mark One)
| ☒ |
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2023
or
| ☐ |
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from ___________ to
Commission file number: 001-34475
OMEROS CORPORATION
(Exact name of registrant as specified in its charter)
| Washington |
91-1663741 |
| (State or other jurisdiction of |
(I.R.S. Employer |
| 201 Elliott Avenue West | |
| (Address of principal executive offices and zip code) | |
(206) 676-5000
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
| Title of each class |
Trading Symbol |
Name of each exchange on which registered |
| Common Stock, par value $0.01 per share |
OMER |
The Nasdaq Stock Market LLC |
Securities registered pursuant to Section 12(g) of the Act:
None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☐ No ☒
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ☐ No ☒
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934, as amended, during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ☒ No ☐
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files). Yes ☒ No ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer |
☐ |
Accelerated filer |
☐ | ||
| Non-accelerated filer |
☒ | Smaller reporting company |
☒ |
||
| Emerging growth company |
☐ |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report. ☐
If securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant included in the filing reflect the correction of an error to previously issued financial statements. ☐
Indicate by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive-based compensation received by any of the registrant’s executive officers during the relevant recovery period pursuant to §240.10D-1(b). ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ☐ No ☒
The aggregate market value of the voting and non-voting common stock held by non-affiliates of the registrant as of the last business day of the registrant’s most recently completed second fiscal quarter was $328,153,747.
As of March 28, 2024, the number of outstanding shares of the registrant’s common stock, par value $0.01 per share, was 57,942,695.
DOCUMENTS INCORPORATED BY REFERENCE
Specified portions of the registrant’s proxy statement with respect to the 2024 Annual Meeting of Shareholders, which is to be filed pursuant to Regulation 14A within 120 days after the end of the registrant’s fiscal year ended December 31, 2023, are incorporated by reference into Part III of this Form 10-K.
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
This Annual Report on Form 10-K contains forward-looking statements within the meaning of Section 27A of the Securities Act of 1933 (the “Securities Act”) and Section 21E of the Securities Exchange Act of 1934 (the “Exchange Act”), which are subject to the “safe harbor” created by those sections for such statements. Forward-looking statements are based on our management’s beliefs and assumptions and on currently available information. All statements other than statements of historical fact are “forward-looking statements.” Terms such as “anticipate,” “believe,” “could,” “estimate,” “expect,” “goal,” “intend,” “likely,” “may,” “plan,” “possible,” “potential,” “predict,” “project,” “should,” “target,” “will,” “would,” and similar expressions and variations thereof are intended to identify forward-looking statements, but these terms are not the exclusive means of identifying such statements. Examples of these statements include, but are not limited to, statements regarding:
● our estimates of future operating expenses and projections regarding how long our existing cash, cash equivalents and short-term investments will fund our anticipated operating expenses, capital expenditures and debt service obligations;
● our ability to raise additional capital through the capital markets or one or more future equity offerings, debt financings, industry collaborations, licensing arrangements, asset sales or other means;
● our expectations regarding amounts potentially payable to us based on sales of our former commercial ophthalmology product OMIDRIA®;
● our expectations regarding clinical plans and anticipated or potential paths to regulatory approval of narsoplimab by the U.S. Food and Drug Administration (“FDA”) and the European Medicines Agency (“EMA”) in hematopoietic stem cell transplant-associated thrombotic microangiopathy (“TA-TMA”), COVID-19 or any other indication;
● whether and when our biologics license application (“BLA”) for narsoplimab in TA-TMA may be resubmitted to FDA, whether and when a marketing authorization application (“MAA”) may be submitted to the EMA for narsoplimab in any indication, and whether and when the FDA, the EMA or any other regulatory authority will grant approval for narsoplimab in any indication;
● our plans for the commercial launch of narsoplimab following any regulatory approval and our estimates and expectations regarding coverage and reimbursement for any approved products;
● our expectation that our contract manufacturer will manufacture narsoplimab when needed to support any regulatory filing and, if approved, to support commercial sale;
● our expectations regarding the clinical, therapeutic and competitive benefits and importance of our drug candidates;
● our ability to design, initiate and/or successfully complete clinical trials and other studies for our drug candidates and our plans and expectations regarding our ongoing or planned clinical trials;
● our expectations regarding: our ability to recruit and enroll patients in any ongoing or planned clinical trial; whether we can capitalize on the financial and regulatory incentives provided by orphan drug designations granted by the FDA, the European Commission (“EC”), or the EMA; and whether we can utilize the opportunities for expedited development and review that may be provided by fast-track or breakthrough therapy designations granted by FDA; ● our expectations about the commercial competition that our drug candidates, if commercialized, face or may face;
● our involvement in existing or potential claims, legal proceedings and administrative actions, and the merits, potential outcomes and effects of both existing and potential claims, legal proceedings and administrative actions, as well as regulatory determinations, on our business, prospects, financial condition and results of operations;
● the extent of protection that our patents provide and that our pending patent applications will provide, if patents are issued from such applications, for our technologies, programs, and drug candidates;
● the factors on which we base our estimates for accounting purposes and our expectations regarding the effect of changes in accounting guidance or standards on our operating results; and
● our expected financial position, performance, revenues, growth, costs and expenses, magnitude of net losses and the availability of resources.
Our actual results could differ materially from those anticipated in these forward-looking statements for many reasons, including the risks, uncertainties and other factors described in Item 1A of Part I of this Annual Report on Form 10-K under the heading “Risk Factors” and in Item 7 of Part II under the heading “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and in our other filings with the Securities and Exchange Commission (“SEC”). Given these risks, uncertainties and other factors, actual results or anticipated developments may not be realized or, even if substantially realized, they may not have the expected consequences to or effects on our company, business or operations. Accordingly, you should not place undue reliance on these forward-looking statements, which represent our estimates and assumptions only as of the date of the filing of this Annual Report on Form 10-K. You should read this Annual Report on Form 10-K completely and with the understanding that our actual results in subsequent periods may materially differ from current expectations. Except as required by applicable law, including the securities laws of the United States and the rules and regulations of the SEC, we assume no obligation to update or revise any forward-looking statements contained herein, whether as a result of any new information, future events or otherwise.
SUMMARY RISK FACTORS
The risk factors described below are a summary of the principal risk factors associated with an investment in our company. These are not the only risks we face. You should carefully consider the risk factors discussed in this summary, as well as the risk factors described in Item 1A. of this Annual Report on Form 10-K.
Risks related to our drug candidates, programs and operations include, but are not limited to, the following:
● inability to raise capital when needed;
● that our drug candidates may not successfully complete clinical development or be suitable for successful commercialization or generation of revenue;
● failure to obtain and maintain regulatory approval for marketing of future commercial products in the U.S. or in foreign jurisdictions;
● lack of adequate coverage or reimbursement from government and/or private payers for OMIDRIA or any of our drug candidates that we commercialize in the future;
● whether and to what extent future royalty and milestone payments that we are eligible to receive based on net sales of OMIDRIA by Rayner Surgical Inc. (“Rayner”) will become payable;
● unpredictability of our operating results;
● any failure to comply with current or future government regulations;
● lack of internal manufacturing capacity and reliance on third parties;
● inability to acquire ingredients, excipients, test kits and other materials to manufacture our drug candidates on commercially reasonable terms;
● delays, suspensions or terminations of our clinical trials or clinical protocols;
● substantial costs as a result of commercial disputes, claims, litigation or other legal proceedings;
● inability to protect our intellectual property and proprietary technologies;
● our indebtedness and liabilities, which could limit the cash flow available for our operations;
● products developed by our competitors, which may diminish or eliminate the success of any products that we may commercialize;
● reliance on members of our management team and our ability to recruit and retain key personnel;
● reliance on third parties to conduct portions of our preclinical research and clinical trials; and
● the impact of our share repurchase program on our stock price.
General risks related to our business include the following:
● cyber-attacks or failures in telecommunications or other information technology systems;
● volatility of our stock price;
● dilution to our existing shareholders if we issue additional shares of our common stock or other securities that may be convertible into, or exercisable for, our common stock;
● adverse effects of natural disasters or other events on us or the third parties on whom we rely; and
● the impact of anti-takeover provisions in our charter documents and under Washington law on potential acquisitions of our company.
ANNUAL REPORT ON FORM 10-K FOR THE YEAR ENDED DECEMBER 31, 2023
INDEX
This Annual Report on Form 10-K contains forward-looking statements reflecting our current expectations that involve risks and uncertainties. Actual results may differ materially from those discussed in these forward-looking statements due to a number of factors, including those set forth in the section entitled “Risk Factors” and elsewhere in this Annual Report. Please refer to the special note regarding forward-looking statements at the beginning of this Annual Report on Form 10-K for further information.
Overview
Omeros Corporation (“Omeros,” the “Company” or “we”) is a clinical-stage biopharmaceutical company committed to discovering, developing and commercializing small-molecule and protein therapeutics for large-market as well as orphan indications targeting immunologic diseases, including complement-mediated diseases and cancers related to dysfunction of the immune system, as well as addictive and compulsive disorders.
Complement-targeted Therapeutic Development Programs
We are advancing multiple development programs focused on diseases and disorders associated with the complement system, a group of specialized proteins that protect against invasive pathogens as well as damaged cells inside the body and comprise an important part of the body’s immune system. When triggered, the various components of complement cooperate to generate an immune response that fights infection and clears damaged or dead cells, maintaining healthy function of the body’s systems. However, dysregulation of the complement system (i.e., over- or under-activation) can be harmful and is associated with increased vulnerability to infections and non-infectious diseases, including autoimmune disorders, chronic inflammation, thrombotic microangiopathy, and cancer.
There are three distinct pathways of complement, each activatedvia one or more unique mechanisms:
| ● |
Classical pathway: activated by antigen-antibody complexes |
| ● |
Lectin pathway: activated by lectin binding of carbohydrate patterns on the surfaces of damaged cells and microbes |
| ● |
Alternative pathway: constitutively active and amplifies classical and lectin pathway activation |
Our complement-targeted therapeutic development programs are primarily focused on diseases and disorders associated with the lectin and/or alternative pathways of complement. Our lectin pathway program includes inhibitors of mannan-binding lectin-associated serine protease 2 (“MASP-2”) and our alternative pathway program includes inhibitors of mannan-binding lectin-associated serine protease 3 (“MASP-3”).
Narsoplimab (OMS721), the lead candidate in our lectin pathway program, is a proprietary, patented human monoclonal antibody inhibitor of MASP-2, the key effector enzyme of the lectin pathway. Clinical development of narsoplimab is currently focused primarily on hematopoietic stem cell transplant-associated thrombotic microangiopathy (“TA-TMA”). We are also developing OMS1029, a long-acting, next-generation antibody and an orally administered small molecule targeting MASP-2 and the lectin pathway.
The lead drug candidate in our development program focused on the alternative pathway of complement is OMS906, a proprietary, patented monoclonal antibody targeting MASP-3. MASP-3 is the key activator of the alternative pathway of complement. We believe OMS906 has the potential to treat a wide range of alternative pathway-related diseases and that its attributes favorably differentiate OMS906 from other marketed and in-development alternative pathway inhibitors. Clinical development of OMS906 is currently ongoing in multiple alternative pathway-related disorders, including complement 3 glomerulopathy (“C3G”), a rare chronic kidney disease, and paroxysmal nocturnal hemoglobinuria (“PNH”), a rare and life-threatening hemolytic blood disorder. An orally administered small molecule MASP-3 inhibitor is also in development.
Other Development Programs
Our development pipeline also includes OMS527, our phosphodiesterase 7 (“PDE7”) inhibitor program focused on addiction and movement disorders. We also have a diverse group of preclinical programs, including five immuno-oncology platforms directed to development of novel adoptive T cell/CAR-T therapies, immunomodulators, immunotoxins and cancer vaccines.
OMIDRIA Sale and Royalty Monetization Transactions
We previously developed and commercialized OMIDRIA® (phenylephrine and ketorolac intraocular solution) 1%/0.3%, which is approved for use during cataract surgery or intraocular lens (“IOL”) replacement to maintain pupil size by preventing intraoperative miosis (pupil constriction) and to reduce postoperative ocular pain. We marketed OMIDRIA in the United States (the “U.S.”) from the time of its commercial launch in 2015 until December 2021.
On December 23, 2021, we sold OMIDRIA and certain related assets, including inventory and prepaid expenses to Rayner Surgical Inc. (“Rayner”) pursuant to an Asset Purchase Agreement, dated December 1, 2021 (the “Asset Purchase Agreement”). Under the Asset Purchase Agreement, Rayner paid us $126.0 million in cash at the closing and we retained all outstanding accounts receivable, accounts payable, and accrued expenses as of the closing date. Rayner is also obligated under the Asset Purchase Agreement to pay us royalties based on Rayner’s net sales of OMIDRIA for a term that extends for the life of the patents covering OMIDRIA in the relevant jurisdiction. The latest expiration of a patent covering OMIDRIA in the United States is currently in 2035. Also pursuant to the Asset Purchase Agreement, we were entitled to receive a milestone payment of $200.0 million (the “Milestone Payment”) within 30 days following an event (the “Milestone Event”) that establishes separate payment for OMIDRIA for a continuous period of at least four years when furnished in the ambulatory surgery center (“ASC”) setting. The Milestone Event occurred in December 2022 and we recorded a $200.0 million milestone receivable. We received the Milestone Payment together with accrued interest in February 2023.
On September 30, 2022, we entered into a Royalty Purchase Agreement (the “Original Agreement”) with DRI Healthcare Acquisitions LP (“DRI”) under which we received $125.0 million in cash in exchange for a portion of our royalties on global net sales of OMIDRIA payable by Rayner between September 1, 2022 and December 31, 2030, subject to certain annual caps on the royalty amounts payable to DRI, with Omeros entitled to receive all royalties paid in excess of the applicable caps.
On February 1, 2024, we entered into an amended and restated royalty purchase agreement (the “Amendment”) with DRI to effect the sale to DRI of an expanded interest in the OMIDRIA royalties. The Amendment eliminated the annual caps on royalty payments to which DRI is entitled and provides that DRI will now receive all royalties on U.S. net sales of OMIDRIA payable between January 1, 2024 and December 31, 2031. We received $115.5 million in cash upon closing of the Amendment. Additionally, we are eligible under the Amendment to receive two milestone payments of up to $27.5 million each, payable in January 2026 and January 2028, respectively, based on achievement of certain thresholds for U.S. net sales of OMIDRIA. DRI is entitled to payment only to the extent of royalty payments that are payable on U.S. net sales of OMIDRIA on or before December 31, 2031 and DRI has no recourse to our assets other than its interest in the OMIDRIA royalties. Omeros retains the right to receive all royalties payable by Rayner on any net sales of OMIDRIA outside the U.S. payable from and after January 1, 2024, as well as all royalties on global net sales of OMIDRIA payable from and after December 31, 2031.
As discussed above, the term for royalty payments under the Asset Purchase Agreement expires based on the last-expiring OMIDRIA-related patent in the relevant country, which in the United States currently extends into 2035. The royalty rate payable by Rayner on net sales of OMIDRIA is currently 30% in the United States and 15% outside the U.S. These royalty rates are subject to reduction upon the occurrence of certain events described in the Asset Purchase Agreement. For example, the applicable U.S. royalty rate would be reduced to 10% during any specific period in which OMIDRIA is no longer eligible for separate payment (i.e., included in the packaged payment rate for the surgical procedure) under Medicare Part B. Pursuant to legislation enacted in late 2022, we expect separate payment for OMIDRIA under Medicare Part B to extend until at least January 1, 2028.
Our Drug Candidates and Development Programs
Our clinical drug candidates consist of the following:
| Drug Candidate/Program |
Targeted Disease(s) |
Development Status |
Next Expected Milestone |
|||
| Narsoplimab (MASP-2 / Lectin Pathway) |
Hematopoietic stem-cell transplant-associated thrombotic microangiopathy (TA-TMA) | Pivotal trial complete; CRL received; working with FDA on BLA resubmission | BLA resubmission | |||
| Narsoplimab (MASP-2 / Lectin Pathway) |
Severe COVID-19, post-acute sequelae of SARS-CoV-2 infection (PASC, i.e., long COVID) and other causes of acute respiratory distress syndrome (ARDS) | Phase 2 trial in severe COVID-19 completed |
Continue development of narsoplimab and diagnostic for lectin pathway hyperactivation for COVID-19/ARDS and related indications |
|||
| OMS1029 (MASP-2 / Lectin Pathway)
|
Long-acting second-generation antibody targeting lectin pathway disorders |
Phase 1 trial nearly complete (dosing completed and follow-up ongoing) |
Select indication for Phase 2 development | |||
| OMS906 (MASP-3 / Alternative Pathway) |
Paroxysmal nocturnal hemoglobinuria (PNH), complement 3 glomerulopathy (C3G) and other alternative pathway disorders |
Phase 2 |
Complete Phase 2 trials with treated patients moving to extension study of long-term efficacy and finalize dose selections; Initiate pivotal Phase 3 clinical trials. | |||
| OMS527 (PDE7) |
Addictions and compulsive disorders; movement |
Phase 1 |
Continue NIDA-funded research through completion of a Phase 1 cocaine interaction study and Phase 2 clinical trial in patients with cocaine use disorder; Determine whether and how best to continue development of OMS527 in levodopa-induced dyskinesia (LID). |
Our pipeline of preclinical development programs includes the following:
| Preclinical Program |
Targeted Disease(s) |
Development Status |
Next Expected Milestone |
|||
| MASP-2: small-molecule inhibitors |
Lectin pathway disorders | Preclinical |
Continue IND-enabling studies of current drug development candidate | |||
| MASP-3: small-molecule inhibitors |
Alternative pathway disorders |
Preclinical |
Identify drug development candidate for clinical trials |
|||
| Adoptive T-Cell and Chimeric Antigen Receptor (CAR) T-Cell Therapies |
Wide range of cancers |
Preclinical |
Complete preclinical proof of concept studies and evaluate data | |||
| Immunomodulators/Immunotoxins/Cancer Vaccines |
Wide range of cancers |
Preclinical |
Complete preclinical proof of concept studies and evaluate data |
Complement Inhibitor Programs
The complement system plays a role in the body’s inflammatory response and becomes activated as a result of tissue damage or trauma or microbial pathogen invasion. Inappropriate or uncontrolled activation of the complement system can cause diseases characterized by serious tissue injury. Three main pathways can activate the complement system: classical, lectin, and alternative. Omeros is focused on development of therapeutics to treat diseases associated with the lectin and/or alternative pathways of complement.
MASP-2 Program - Lectin Pathway Disorders
MASP-2, a novel pro-inflammatory protein target, is the effector enzyme of the lectin pathway and is required for the function of this pathway. Omeros is developing antibodies and small-molecule inhibitors of MASP-2 as potential therapeutics for diseases in which the lectin pathway has been shown to contribute to significant tissue injury and pathology. When not treated, these diseases are typically characterized by significant end-organ damage, such as kidney or central nervous system injury. Importantly, inhibition of MASP-2 has been demonstrated not to interfere with the antibody-dependent classical complement activation pathway, a critical component of the acquired immune response to infection. In addition to our clinical programs evaluating narsoplimab, we have generated positive preclinical data from MASP-2 inhibition in in vivo models of AMD, myocardial infarction, diabetic neuropathy, stroke, ischemia-reperfusion injury, and other diseases and disorders. We own or hold worldwide exclusive licenses to rights related to MASP-2, the antibodies targeting MASP-2 and the therapeutic applications for those antibodies.
Narsoplimab (OMS721)
The lead candidate in our MASP-2 inhibitor program is narsoplimab (OMS721), a proprietary, patented human monoclonal antibody targeting MASP-2. Narsoplimab is in clinical development for several indications.
Hematopoietic stem-cell transplant-associated thrombotic microangiopathy (“TA-TMA”): We successfully completed a pivotal clinical trial for narsoplimab in TA-TMA and previously submitted to the FDA a biologics license application (“BLA”) seeking marketing approval for narsoplimab in this indication. In late 2021, FDA issued a complete response letter (“CRL”) with respect to the BLA in which the agency indicated that additional information would be needed to support regulatory approval. We appealed FDA’s decision to issue the CRL through a formal dispute resolution process that concluded in late 2022. Although our appeal was denied, the decision identified potential paths for resubmission of the BLA based on both response data and survival data from the completed pivotal trial versus a historical control group, with or without an independent literature analysis or based on survival data alone. Consistent with subsequent interactions with FDA’s review division, we submitted to FDA in the fall of 2023 an analysis plan to assess already existing clinical trial data, existing data from a historical control population available from an external source, data from the narsoplimab expanded access program, and data directed to the mechanism of action of narsoplimab. We are having ongoing discussions with the agency regarding the proposed analysis plan. As a result, we are currently unable to estimate when we will submit the BLA or, subsequently, FDA’s timing for a decision regarding approval. There can be no guarantee that FDA's specific recommendations for resubmission will be acceptable to Omeros in terms of the time and/or expenditure required or that any resubmission of the BLA will result in approval of narsoplimab for TA-TMA.
In the U.S., FDA has granted narsoplimab (1) breakthrough therapy designation in patients who have persistent TMA despite modification of immunosuppressive therapy, (2) orphan drug designation for the prevention (inhibition) of complement-mediated TMAs, and (3) orphan drug designation for the treatment of TA-TMA. The European Commission (the “EC”) also granted narsoplimab designation as an orphan medicinal product for treatment in hematopoietic stem cell transplantation.
In Europe, the European Medicines Agency (“EMA”) has confirmed narsoplimab’s eligibility for the EMA’s centralized review of a single marketing authorization application (“MAA”) that, if approved, authorizes the product to be marketed in all EU member states and European Economic Area countries. We expect to complete our MAA submission following the resubmission of our BLA to FDA.
COVID-19 and Acute Respiratory Distress Syndrome (“ARDS”): There is strong and increasingly well-established evidence of the central role of the lectin pathway in COVID-19 and ARDS and we have developed both mechanistic and proof-of-concept clinical data indicating that narsoplimab may be an effective therapeutic for COVID-19, ARDS and/or related indications. We also continue to explore the mounting evidence that MASP-2 and the lectin pathway are important drivers of post-acute sequelae SARS-CoV-2 (“PASC”), commonly known as long COVID, as well as potential approaches to identify acute COVID-19 patients at high-risk for hospitalization and mortality, to identify those PASC patients with hyperactive lectin pathway-driven disease, and to monitor response to treatment with MASP-2 inhibitors.
Narsoplimab has been administered under compassionate use to treat COVID-19 patients in Italy and in the U.S., achieving encouraging results. Additionally, narsoplimab was the only complement inhibitor included in the I-SPY COVID-19 trial, a nationwide, late-stage adaptive platform trial sponsored by Quantum Leap Healthcare Collaborative (“Quantum Leap”), that evaluated multiple agents as potential treatments for severe COVID-19 requiring mechanical ventilation. Results of the I-SPY COVID-19 trial were reported in September 2022. The narsoplimab treatment arm was terminated prior to accrual of the maximum of 125 patients on the basis of analysis in a pre-consented patient population in which substantial bias was detected. Although narsoplimab was not observed in this study to shorten the time to recovery in critically ill patients with COVID-19, analysis in the randomized patient population showed that the addition of narsoplimab to treatment of critically ill patients with COVID-19 reduces the mortality risk (hazard ratio [HR]=0.81, with probability [HR <1] equal to 0.77). In approximately half of the patients who died in the narsoplimab group, narsoplimab was not given or was prematurely stopped, with those patients dying 9 to 35 days later. Neither the trial’s futility nor graduation criteria had been met in the analysis of the randomized population at the time the narsoplimab arm was terminated. Narsoplimab demonstrated the greatest reported survival benefit of all therapeutics evaluated in the I-SPY platform trial.
We have also developed an assay platform to identify hyperactivation of the lectin pathway. Lectin pathway hyperactivation is correlated with COVID-19-related-ARDS and may be involved in the pathogenesis of other forms of ARDS and/or PASC. As such, the assay may be useful to identify patients who are at greatest risk of hospitalization and/or mortality as well as those who are particularly amenable to lectin pathway inhibition therapy for the treatment of one or more of these conditions. We continue to validate the clinical correlation of lectin pathway hyperactivation with COVID-19, ARDS and PASC. We continue to engage in discussions with potential partners as well as with representatives of the U.S. government regarding potential opportunities to obtain funding and advance development of our potential diagnostic and/or therapeutic product candidates for COVID-19, PASC or other infectious diseases.
IgA Nephropathy: In October 2023, we announced the results of a pre-specified interim analysis in ARTEMIS-IGAN, our Phase 3 clinical trial evaluating narsoplimab for the treatment of immunoglobulin A (“IgA”) nephropathy. Topline results of the interim analysis showed that narsoplimab did not achieve statistical significance on the primary endpoint of reduction in proteinuria from baseline compared to placebo. Based on the absence of statistical significance and as previously agreed with FDA, we determined not to submit an application for approval of narsoplimab in this indication and the ARTEMIS-IGAN clinical trial has been discontinued.
OMS1029
Our lectin pathway program also includes OMS1029, our long-acting antibody targeting MASP-2. This next-generation MASP-2 inhibitor is intended to be complementary to narsoplimab, enabling us to pursue chronic indications in which dosing convenience would be of significant benefit to patients. Dosing of all cohorts in a single-ascending dose Phase 1 clinical trial of OMS1029 was successfully completed in early 2023. Pharmacokinetic (“PK”) and pharmacodynamic (“PD”) data show dose-proportional exposure and sustained lectin pathway inhibition, consistent with dosing of OMS1029 once quarterly, either intravenously or subcutaneously. Dosing has also been completed in both of two planned cohorts of our ongoing Phase 1 multiple-ascending-dose study of OMS1029 in healthy volunteers and we expect the study to conclude in mid-2024. OMS1029 has been well tolerated to date with no safety concerns identified. We continue to evaluate several potential indications for Phase 2 clinical development for OMS1029.
MASP-3 Program - Alternative Pathway Disorders
As part of our program to develop complement-targeted therapeutics, we have identified MASP-3, which has been shown to be the key activator of the complement system’s alternative pathway (“APC”), and we believe that we are the first to make this and related discoveries associated with the APC. The complement system is part of the immune system’s innate response, and the APC is considered the amplification loop within the complement system. MASP-3 is responsible for the conversion of pro-factor D to mature factor D; which is necessary for the activation of the APC.
We believe that MASP-3 inhibitors have the potential to treat patients suffering from a wide range of diseases and conditions including: PNH; C3G; multiple sclerosis; neuromyelitis optica; age-related macular degeneration; Alzheimer’s disease; systemic lupus erythematosus; diabetic retinopathy; chronic obstructive pulmonary disease; antineutrophil cytoplasmic antibody-associated vasculitis; anti-phospholipid syndrome; atherosclerosis; myasthenia gravis and others. Several of these indications have been clinically validated by other agents targeting the APC. Our MASP-3 program has also generated positive data in a well-established animal model of arthritis.
OMS906
The lead candidate in our MASP-3 inhibitor program is OMS906, a proprietary, patented human monoclonal antibody targeting MASP-3. Clinical development of OMS906 is currently focused on rapidly advancing to Phase 3 clinical trials in multiple APC-related disorders, including PNH and C3G. OMS906 received designation from FDA as an orphan drug for the treatment of PNH in July 2022.
Paroxysmal nocturnal hemoglobinuria (“PNH”): We have three ongoing Phase 2 clinical trials evaluating OMS906 for PNH. One in PNH patients who have not previously been treated with a complement inhibitor, and the second in PNH patients who have had an unsatisfactory response to ravulizumab, an inhibitor of complement component 5 (“C5”). The third Phase 2 clinical trial is an open-label extension study to assess the long-term efficacy and safety of OMS906 in patients who have completed either of the other two PNH Phase 2 clinical trials.
In December 2023, updated results from a pre-specified interim analysis of our ongoing Phase 2 clinical trial of OMS906 in complement-inhibitor-naïve adults with PNH were featured in a podium presentation at the annual meeting of the American Society of Hematology. The interim analysis results showed statistically significant and clinically meaningful improvements in all measured markers of hemolysis, including hemoglobin and lactate dehydrogenase. No patients were reported to have had a clinical breakthrough of PNH or a thrombotic event, and none were reported to require a transfusion while receiving OMS906 treatment.
Enrollment is complete and dosing is ongoing in our Phase 2 trial evaluating OMS906 in PNH patients who have had an unsatisfactory response to the C5 inhibitor ravulizumab. Utilizing a “switch-over” design, this study enrolls PNH patients receiving ravulizumab, adds OMS906 to provide combination therapy with ravulizumab for 24 weeks, and then, in those patients who demonstrate a hemoglobin response with the combination therapy, switches to OMS906 monotherapy. Data from a pre-specified interim analysis showed that the addition of OMS906 therapy to ravulizumab treatment resulted in statistically significant and clinical meaningful improvements in both mean hemoglobin levels and absolute reticulocyte counts by week 4 of combination therapy, with a sustained response demonstrated through week 24 (the latest assessment prior to the interim analysis cutoff). Full details from the interim analysis are expected to be presented at a major hematology conference in mid-2024. Interim analysis data from the monotherapy portion of the trial is expected to be available in late 2024.
We have initiated an open-label extension study to assess the long-term efficacy and safety of OMS906 in patients with PNH. In the extension study, PNH patients who have completed a previous study evaluating OMS906 roll directly into the extension study without a break in OMS906 treatment. Data from this study will contribute to a planned BLA for OMS906 in the treatment of PNH. Based on PK data from a successful Phase 1 single-ascending-dose study of OMS906 in healthy subjects and interim data from our ongoing clinical trials in PNH patients, we are exploring two different dosing frequencies - once every eight weeks and once every 12 weeks - for the Phase 3 studies and commercialization.
In February 2024 we met with FDA to discuss our development program for OMS906 in PNH. We presented clinical and nonclinical data and requested input on expectations for Phase 3 studies and BLA submission. FDA confirmed that the scope of our nonclinical program is sufficient to support Phase 3 clinical studies and provided input on dosing and design of the proposed Phase 3 program to support a BLA in PNH. We expect to meet again with FDA later this year to discuss further details of the design of our Phase 3 studies. Phase 3 clinical trials evaluating OMS906 in PNH are targeted to begin in late 2024.
Complement 3 glomerulopathy (“C3G”): We also have an ongoing Phase 2 clinical program evaluating OMS906 for the treatment of C3G, a rare and debilitating renal disease driven by complement dysregulation. Notably, the relevance of the alternative pathway to C3G has been clinically validated in a Phase 3 trial with another inhibitor of the alternative pathway that reported positive results in the treatment of C3G. Although a protocol amendment to modify the OMS906 dose based on information learned from our PNH program delayed initiation of the study, sites are now open in multiple countries and patients are being screened for enrollment. We are targeting to initiate Phase 3 development for C3G in the first part of 2025, after Phase 2 results are available and discussions occur with regulators.
Preclinical Complement Inhibitor Programs
We have also directed efforts to development of small-molecule inhibitors of MASP-2 and MASP-3 designed for oral administration. In our MASP-2 small molecule inhibitor program we continue to advance testing to enable the filing of an investigational new drug application. Our MASP-3 small-molecule inhibitor is advancing rapidly toward selection of a drug development candidate.
Other Clinical Programs
PDE7 Inhibitor Programs - OMS527
Our PDE7 inhibitor programs, which we refer to as OMS527, comprise multiple PDE7 inhibitor compounds and are based on our discoveries of previously unknown links between PDE7 and any addiction or compulsive disorder, and between PDE7 and any movement disorders. PDE7 appears to modulate the dopaminergic system, which plays a significant role in regulating both addiction and movement. We believe that PDE7 inhibitors could be effective therapeutics for the treatment of addictions and compulsions as well as for movement disorders. Data generated in preclinical studies support the use of PDE7 inhibitors in both of these therapeutic areas.
Cocaine Use Disorder (“CUD”): In April 2023, we were awarded a grant from the National Institute on Drug Abuse, part of the National Institutes of Health, to develop our lead orally administered PDE7 inhibitor compound, for which we have successfully completed a Phase 1 study, for the treatment of CUD. The grant amount, a total of $6.69 million over three years, is intended to support preclinical cocaine interaction/toxicology studies to assess safety of the therapeutic candidate in the presence of concomitant cocaine administration, as well as an in-patient, placebo-controlled clinical study evaluating the safety and effectiveness of OMS527 in adults with CUD who receive concurrent intravenous cocaine. The preclinical study is intended to provide the toxicology data necessary to support the human study of OMS527 in CUD. That study is underway and is expected to be completed in late 2024.
Levodopa-induced dyskinesia (“LID”): With investigators at Emory University, we are also evaluating OMS527 as a potential treatment for LID, which are involuntary and often crippling movements in patients with Parkinson’s disease that are caused by prolonged treatment with levodopa, the most prescribed therapy for Parkinson’s disease. More than 10 million patients are living with Parkinson’s disease worldwide. Reportedly 50 percent or more of levodopa-treated patients with Parkinson’s disease suffer from LID.
We hold an exclusive license to certain PDE7 inhibitors claimed in patents and pending patent applications owned by Daiichi Sankyo Co., Ltd. (“Daiichi Sankyo”), as successor-in-interest to Asubio Pharma Co., Ltd. for use in the treatment of movement, addiction and compulsive disorders as well as other specified indications. For a more detailed description of our agreement with Daiichi Sankyo, see “License and Development Agreements” below.
PPARγ Program - OMS405
In our peroxisome proliferator-activated receptor gamma (“PPARγ”) program, we have engaged in development of proprietary compositions that include PPARγ agonists for the treatment and prevention of addiction to substances of abuse, which may include opioids, nicotine and alcohol. We believe that Omeros is the first to demonstrate a link between PPARγ and addiction disorders. Data from clinical studies and from animal models of addiction suggest that PPARγ agonists could be efficacious in the treatment of a wide range of addictions.
Our collaborators at The New York State Psychiatric Institute have completed two Phase 2 clinical trials related to our PPARγ program. These studies evaluated a PPARγ agonist, alone or in combination with other agents, for treatment of addiction to heroin and to nicotine. The published results of the heroin study demonstrated that, although not altering the reinforcing or positive subjective effects of heroin, the PPARγ agonist significantly reduced heroin craving and overall anxiety. The National Institute on Drug Abuse (“NIDA”) provided substantially all of the funding for these clinical trials and solely oversaw the conduct of these trials. We have the right or expect to be able to reference the data obtained from these studies for any future FDA submission and continue to retain all other rights in connection with the PPARγ program.
We have also reported positive results (i.e., decreased cravings and protection of brain white matter) from a Phase 2 clinical trial conducted by an independent investigator evaluating the effects of a PPARγ agonist in patients with cocaine use disorder. An investigator-sponsored study evaluating the effects of a PPARγ agonist on the prevention of relapse following treatment of cocaine use disorder is ongoing. The study is funded by NIDA.
We own patents, patent applications and other intellectual property rights related to our PPARγ program, as described under “Intellectual Property” below.
Preclinical Programs and Platforms
Immuno-Oncology Platform
We have five immuno-oncology (I-O) platforms in preclinical development – adoptive T-cell therapy, CART-T, signaling-driven immunomodulators, antigen-driven immunomodulators that function both as therapeutics and vaccines, and oncotoxins. To date, in vitro, ex vivo and animal studies using human cellular components have been positive with high response rates. These data collectively reinforce the scientific basis for each platform, confirming our rationale for their design and development. The data from our studies to date have demonstrated a number of potential advantages of our immuno-oncology franchise over other I-O approaches.
Our I-O franchise should be applicable to cancers broadly. Rather than targeting only cell-surface antigens, our I-O franchise is designed to target both cell-surface and intracellular cancer targets, significantly broadening the range of indications. Unlike other therapeutic approaches that affect either CD4 or CD8 levels, we have designed and demonstrated the ability of our technologies to increase levels of both CD4 and CD8 cells against a given cancer, both of which are necessary to kill tumor cells. By increasing both the CD4 and CD8 cells, we should also be able to mitigate the “treatment exhaustion” – or the wearing-off of the treatment effect – seen with many currently available therapies. This would allow repeated treatment, providing a sustained and better anti-tumor response.
Our cellular platforms do not require modification or engineering. Instead, cells from the patient are simply treated outside the body and administered back to the patient. We expect this simplicity in process, if achieved, to represent a major advance over currently available T-cell therapies, greatly decreasing both preparation time and cost. When injected into the body, our novel biologic molecules should result not only in elimination of tumor cells but, importantly, in immune memory against future cancer relapse. Our core technology is also amenable potentially to cancer prevention broadly.
We believe that all five platforms are entirely novel and proprietary. We continue to confirm our results and to generate new data, all of which contribute to our intellectual property position.
Sales and Marketing
We have retained all worldwide marketing and distribution rights to our drug candidates and our development programs. As such, we will be able to market any drug candidate that is approved in the future independently, through arrangements with third parties, or via some combination of these approaches.
We maintained internal marketing and sales capabilities with respect to OMIDRIA until the completion of the divestiture of that product on December 23, 2021. As part of the divestiture, substantially all of our OMIDRIA sales and marketing team members accepted employment with Rayner and were separated from their employment at Omeros, effective as of December 31, 2021.
Manufacturing, Supply and Commercial Operations
We currently do not own or operate manufacturing facilities. We utilized contract manufacturers to produce, store and distribute OMIDRIA and currently rely on third parties to produce sufficient quantities of our drug candidates for use in pre-clinical and clinical studies and for the manufacture of narsoplimab for commercial use following potential regulatory approval.
OMIDRIA. We assigned or otherwise transitioned to Rayner our agreements with the third parties that produced, stored and distributed OMIDRIA. We required manufacturers that produced active pharmaceutical ingredients (“APIs”) and finished drug products to operate in accordance with current Good Manufacturing Practices (“cGMPs”) and all other applicable laws and regulations.
In the U.S., we sold OMIDRIA through a limited number of wholesalers that distributed the product to ASCs and hospitals. Title transferred upon delivery of OMIDRIA to the wholesaler. For additional information, see Part II, Item 8, “Note 7 – Discontinued Operations – Sale of OMIDRIA” to our Consolidated Financial Statements in this Annual Report on Form 10-K.
Drug Candidates. We have laboratories in-house for analytical method development, bioanalytical testing, formulation, stability testing and small-scale compounding of laboratory supplies of drug candidates. We utilize contract manufacturers to produce sufficient quantities of drug candidates for use in preclinical and clinical studies and to store and distribute our drug candidates. We require manufacturers that produce bulk drug substance and finished drug products for clinical use to operate in accordance with cGMPs and all other applicable laws and regulations. We anticipate that we will rely on contract manufacturers to develop and manufacture our drug candidates for commercial sale. We maintain agreements with potential and existing manufacturers that include confidentiality and intellectual property provisions to protect our proprietary rights related to our drug candidates.
In July 2019, we entered into a master services agreement with Lonza Biologics Tuas Pte. Ltd. (“Lonza”) for the commercial production of narsoplimab and for certain regulatory support and related services to be provided by Lonza from time to time. Under the agreement Lonza will manufacture narsoplimab pursuant to purchase orders issued in accordance with certain forecast and confirmation procedures specified in the contract. We will purchase narsoplimab that meets agreed specifications in batches, with the price per batch varying according to the total number of batches ordered for serial production in a single manufacturing campaign. We are obligated to purchase a minimum number of batches annually beginning on a specified anniversary of the first commercial sale of narsoplimab in either the U.S. or EU. We may be obligated to pay certain fees to Lonza upon cancellation of purchase orders.
The initial term of the agreement expires five years after the first commercial sale of narsoplimab in either the U.S. or EU and is subject to automatic renewal for an additional four-year term unless we provide notice of non-renewal at least three years prior to the end of the initial term. In addition, either party may terminate the agreement, subject to applicable notice and cure periods under certain circumstances. Other than our agreement for commercial supply of narsoplimab, we have not yet entered into a commercial supply agreement for any of our drug candidates.
If approved for commercial sale in the U.S., we expect to utilize one or more wholesalers for distribution of narsoplimab.
License and Development Agreements
MASP-3. In August 2020, we entered into a technology license agreement with Xencor, Inc., pursuant to which we received an exclusive license to apply Xencor’s Xtend Fc technology to OMS906 and options to access exclusive licenses to apply Xtend Fc technology to additional antibodies (the “Xencor Agreement”). Exercise of an option to access additional licenses would require payment of a $3.0 million upfront license fee. With respect to each antibody for which we license the Xencor technology we are obligated to make milestone payments of up to $65.0 million, comprised of $15.0 million in development milestones, $25.0 million in regulatory milestones and $25.0 million in sales milestones. In August 2023, we paid $5.0 million to Xencor in connection with the achievement of a development milestone in our OMS906 program. We are obligated on a product-by-product and country-by-country basis to pay Xencor royalties in the mid-single digit percentage range on net sales of any product covered by the license so long as there is a valid, subsisting and enforceable claim in any patents or patent applications covering the licensed technology. Thereafter, the royalty rate is reduced to the low single-digit percentage range, if the applicable licensed product is covered by Xencor know-how, or to zero, if the applicable licensed product is not covered by Xencor know-how. The term of the Xencor Agreement continues on a product-by-product basis until the later of (i) expiration for the last-to-expire patent covering the licensed technology or (ii) five years from the date of first commercial sale of the applicable product.
PDE7. Under an agreement with Daiichi Sankyo, we hold an exclusive worldwide license to PDE7 inhibitors claimed in certain patents and pending patent applications owned by Daiichi Sankyo for use in the treatment of (1) movement disorders and other specified indications, (2) addiction and compulsive disorders and (3) all other diseases except those related to dermatologic conditions. Under the agreement, we agreed to make milestone payments to Daiichi Sankyo of up to an aggregate total of $33.5 million upon the achievement of certain events in each of these three fields; however, if only one of the three indications is advanced through the milestones, the total milestone payments would be $23.5 million. The milestone payment events include successful completion of preclinical toxicology studies; dosing of human subjects in Phase 1, 2 and 3 clinical trials; receipt of marketing approval of a PDE7 inhibitor drug candidate; and reaching specified sales milestones. In addition, Daiichi Sankyo is entitled to receive from us a low single-digit percentage royalty of any net sales of a PDE7 inhibitor licensed under the agreement by us and/or our sublicensee(s) provided that, if the sales are made by a sublicensee, then the amount payable by us to Daiichi Sankyo is capped at an amount equal to a low double-digit percentage of all royalty and specified milestone payments received by us from the sublicensee.
The term of the agreement with Daiichi Sankyo continues so long as there is a valid, subsisting and enforceable claim in any patents covered by the agreement. The agreement may be terminated sooner by us, with or without cause, upon 90 days advance written notice or by either party following a material breach of the agreement by the other party that has not been cured within 90 days or immediately if the other party is insolvent or bankrupt. Daiichi Sankyo also has the right to terminate the agreement if we and our sublicensee(s) cease to conduct all research, development and/or commercialization activities for a PDE7 inhibitor covered by the agreement for a period of six consecutive months, in which case all rights held by us under Daiichi Sankyo’s patents will revert to Daiichi Sankyo.
Competition
Overview. The pharmaceutical and biotechnology industry is highly competitive and characterized by a number of established, large pharmaceutical and biotechnology companies as well as smaller companies like ours. We expect to compete with other pharmaceutical and biotechnology companies, and our competitors may:
● develop and market products that are less expensive, more effective or safer than our future products;
● commercialize competing products before we can launch our products;
● operate larger research and development programs, possess greater manufacturing capabilities or have substantially greater financial resources than we do;
● initiate or withstand substantial price competition more successfully than we can;
● have greater success in recruiting skilled technical and scientific workers from the limited pool of available talent;
● more effectively negotiate third-party licenses and strategic relationships; and
● take advantage of acquisition or other opportunities more readily than we can.
We expect to compete for market share against large pharmaceutical and biotechnology companies, smaller companies that are collaborating with larger pharmaceutical companies, new companies, academic institutions, government agencies and other public and private research organizations. In addition, the pharmaceutical and biotechnology industry is characterized by rapid technological change. Because our research approach integrates many technologies, it may be difficult for us to remain current with the rapid changes in each technology. Further, our competitors may render our technologies obsolete by advancing their existing technological approaches or developing new or different approaches. If we fail to stay at the forefront of technological change, we may be unable to compete effectively.
Drug Candidates, Development Programs and Platforms. There are a number of complement-targeted therapeutics that are in advanced stages of clinical development, or which have been approved for commercial use. These include Soliris® (eculizumab), Ultomiris® (ravulizumab-cwvz), Empaveli® (pegcetacoplan), Tavneos® (avocopan) and Fabhalta® (iptacopan). Narsoplimab, OMS1029 and/or OMS906 will face competition from one or more of these products if approved for any indication(s) for which one or more of these potentially competitive products are also approved or for which a potentially competitive product is used off-label to treat a relevant condition.
Intellectual Property
We have retained control of all worldwide manufacturing, marketing and distribution rights for each of our drug candidates and programs. Some of our drug candidates and programs are based on inventions and other intellectual property rights that we acquired through assignments, exclusive licenses or acquisitions described in further detail under “License and Development Agreements” above.
As of February 15, 2024, we owned or held worldwide exclusive licenses to a total of 78 issued patents and 60 pending patent applications in the U.S. and 1,334 issued patents and 580 pending patent applications in foreign markets directed to therapeutic compositions and methods and other technologies related to our research and development programs. For each program, our decision to seek patent protection in specific foreign markets, in addition to the U.S., is based on many factors, including one or more of the following: our available resources, the size of the commercial market, the presence of a potential competitor or a contract manufacturer in the market and whether the legal authorities in the market effectively enforce patent rights.
● MASP-2 Program - Narsoplimab (OMS721) and OMS1029. We own and hold worldwide exclusive licenses to rights in connection with MASP-2, antibodies targeting MASP-2, small-molecule MASP-2 inhibitors, and related therapeutic applications. As of February 15, 2024, we exclusively controlled 38 issued patents and 32 pending patent applications in the U.S., and 793 issued patents and 435 pending patent applications in foreign markets, related to our MASP-2 program, including narsoplimab and our second-generation MASP-2 antibody OMS1029. Our MASP-2-related patents have terms that will expire as late as 2038 and, if currently pending patent applications are issued, as late as 2043.
● MASP-3 Program - OMS906. We own and exclusively control rights in connection with MASP-3, antibodies targeting MASP-3 and related therapeutic applications. We also hold an exclusive license from Xencor, Inc. for the application of certain antibody technology to OMS906, as well as the option to obtain additional licenses to such technology for exclusive application to additional antibodies that we may select. As of February 15, 2024, we exclusively controlled four issued patents and eight pending patent applications in the U.S. and 188 issued and 104 pending patent applications in foreign markets that are related to our MASP-3 program. Our MASP-3-related patents have terms that will expire as late as 2037 and, if currently pending patent applications are issued, as late as 2043.
● PPARγ Program - OMS405. As of February 15, 2024, we owned three issued patents and one pending patent application in the U.S., and 37 issued patents and two pending patent applications in foreign markets, directed to our discoveries linking PPARγ and addictive disorders. Our PPARγ-related patents have terms that will expire as late as 2030.
● PDE7 Program - OMS527. As of February 15, 2024, we owned two issued patents and two pending patent application in the U.S., and 61 issued patents and two pending patent applications in foreign markets directed to our discoveries linking PDE7 to movement disorders, as well as three issued patent and two pending patent applications in the U.S., and 54 issued patents and three pending patent applications in foreign markets directed to the link between PDE7 and addiction and compulsive disorders. Additionally, under a license from Daiichi Sankyo, we exclusively control rights to two issued U.S. patents and 53 issued patents in foreign markets that are directed to proprietary PDE7 inhibitors. Our PDE7-related patents have terms that will expire as late as 2031 and, if currently pending patent applications are issued, as late as 2043. For a more detailed description of our agreement with Daiichi Sankyo, see “License and Development Agreements” below.
● Immuno-oncology Program. Our Immuno-oncology program includes five proprietary platforms relating to potential therapies for cancer. As of February 15, 2024, we owned two pending patent applications in foreign markets directed to potential cancer therapies.
All of our employees enter into our standard employee proprietary information and inventions agreement, which includes confidentiality provisions and provides us ownership of all inventions and other intellectual property made by our employees that pertain to our business or that relate to our employees’ work for us or that result from the use of our resources. Our commercial success will depend in part on obtaining and maintaining patent protection and trade secret protection of the use, formulation and structure of our drug candidates and the methods used to manufacture them, as well as on our ability to defend successfully these patents against third-party challenges. Our ability to protect our drug candidates from unauthorized making, using, selling, offering to sell or importing by third parties is dependent on the extent to which we have rights under valid and enforceable patents that cover these activities.
The patent positions of pharmaceutical, biotechnology and other life sciences companies can be highly uncertain and involve complex legal and factual questions for which important legal principles remain unresolved. No consistent policy regarding the breadth of claims allowed in biotechnology patents has emerged to date in the U.S., and tests used for determining the patentability of patent claims in all technologies are in flux. The pharmaceutical, biotechnology and other life sciences patent situation outside the U.S. is even more uncertain. Changes in either the patent laws or in interpretations of patent laws in the U.S. and other countries may diminish the value of our intellectual property. Accordingly, we cannot predict the breadth of claims that may be allowed or enforced in the patents that we own or have licensed or in third-party patents.
We have registered, and intend to maintain, the trademark “OMEROS” within the U.S. Patent and Trademark Office in connection with the products and services we offer. We are not aware of any material claims of infringement or other challenges to our right to use the “OMEROS” trademark in the U.S.
Government Regulation
Government authorities in the U.S., the EU and other countries extensively regulate the research, development, testing, manufacture, labeling, promotion, advertising, distribution, marketing, and export and import of drug and biologic products including the drug candidates that we are developing. Failure to comply with applicable requirements, both before and after receipt of regulatory approval, may subject us, our third-party manufacturers, and other partners to administrative and judicial sanctions, such as warning letters, product recalls, product seizures, a delay in approving or refusal to approve pending applications, civil and other monetary penalties, total or partial suspension of production or distribution, injunctions, and/or criminal prosecutions.
In the U.S., our drug candidates are regulated by FDA as drugs or biologics under the Federal Food, Drug, and Cosmetic Act (“FDCA”) and implementing regulations and under the Public Health Service Act (“PHSA”). In the EU, our drug candidates are regulated by the EMA and national medicines regulators under the rules governing medicinal products in the EU as well as national regulations in individual countries. Our drug candidates are in various stages of testing and none of our drug candidates has received marketing approval from FDA or the applicable regulatory authorities in the EU.
The steps required before a product may be approved for marketing by FDA, or the applicable regulatory authorities outside of the U.S., typically include the following:
● formulation development and manufacturing process development;
● preclinical laboratory and animal testing;
● submission to FDA of an Investigational New Drug application (“IND”) for human clinical testing, which must become effective before human clinical trials may begin; and in countries outside the U.S., a Clinical Trial Application (“CTA”), is filed according to the country’s local regulations;
● adequate and well-controlled human clinical trials to establish the efficacy and safety of the product for each indication for which approval is sought;
● adequate assessment of drug product stability to determine shelf life/expiry dating;
● in the U.S., submission to FDA of a New Drug Application (“NDA”), in the case of a drug product, or a BLA in the case of a biologic product and, in Europe, submission to the EMA or a national regulatory authority of an MAA;
● satisfactory completion of inspections of one or more clinical sites at which clinical trials with the product were carried out and of the manufacturing facility or facilities at which the product is produced to assess compliance with Good Clinical Practices (“GCPs”), and cGMPs; and
● FDA review and approval of an NDA or BLA, or review and approval of an MAA by the applicable regulatory authorities in the EU.
Manufacturing. Manufacturing of drug products for use in clinical trials must be conducted according to relevant national and international guidelines, for example, cGMP. Process and formulation development are undertaken to design suitable routes to manufacture the drug substance and the drug product for administration to animals or humans. Analytical development is undertaken to obtain methods to quantify the potency, purity and stability of the drug substance and drug product as well as to measure the amount of the drug substance and its metabolites in biological fluids, such as blood.
Preclinical Tests. Preclinical tests include laboratory evaluations and animal studies to assess efficacy, toxicity and pharmacokinetics. The results of the preclinical tests, together with manufacturing information, analytical data, clinical development plan, and other available information are submitted as part of an IND or CTA.
The IND/CTA Process. An IND or CTA must become effective before human clinical trials may begin. INDs are extensive submissions including, among other things, the results of the preclinical tests, together with manufacturing information and analytical data. In addition to including the results of the preclinical studies, the IND will also include one or more protocols for proposed clinical trials detailing, among other things, the objectives of the clinical trial, the parameters to be used in monitoring safety and the effectiveness criteria to be evaluated. An IND will become effective 30 days after receipt by FDA unless, before that time, FDA raises concerns or questions and imposes a clinical hold. In that event, the IND sponsor and FDA must resolve any outstanding FDA concerns or questions before the clinical hold is lifted and clinical trials can proceed. Similarly, a CTA must be cleared by the local independent ethics committee and competent authority prior to conducting a clinical trial in the country in which it was submitted. There can be no assurance that submission of an IND or CTA will result in authorization to commence clinical trials. Once an IND or CTA is in effect, there are certain reporting requirements.
Clinical Trials. Clinical trials involve the administration of the investigational product to human subjects under the supervision of qualified personnel and must be conducted in accordance with local regulations and GCPs. Clinical trials are conducted under protocols detailing, for example, the parameters to be used in monitoring patient safety and the efficacy criteria, or endpoints, to be evaluated. Each trial must be reviewed and approved by an independent institutional review board or ethics committee for each clinical site at which the trial will be conducted before it can begin. Clinical trials are typically conducted in three defined phases, but the phases may overlap or be combined:
● Phase 1 usually involves the initial administration of the investigational product to human subjects, who may or may not have the disease or condition for which the product is being developed, to evaluate the safety, dosage tolerance, pharmacodynamics and, if possible, to gain an early indication of the effectiveness of the product.
● Phase 2 usually involves trials in a limited patient population with the disease or condition for which the product is being developed to evaluate appropriate dosage, to identify possible adverse side effects and safety risks, and to evaluate preliminarily the effectiveness of the product for specific indications.
● Phase 3 clinical trials usually further evaluate and confirm effectiveness and test further for safety by administering the product in its final form in an expanded patient population.
We, our product development partners, institutional review boards or ethics committees, FDA or other regulatory authorities may suspend or terminate clinical trials at any time on various grounds, including a belief that the subjects are being exposed to an unacceptable health risk.
Disclosure of Clinical Trial Information. Sponsors of clinical trials of certain FDA-regulated products, including prescription drugs, are required to register and disclose certain clinical trial information on a public website maintained by the U.S. National Institutes of Health. Information related to the product, patient population, phase of investigation, study sites and investigator, and other aspects of the clinical trial is made public as part of the registration. Sponsors are also obligated to disclose the results of these trials after completion. Disclosure of the results of these trials can be delayed for up to two years if the sponsor certifies that it is seeking approval of an unapproved product or that it will file an application for approval of a new indication for an approved product within one year. Clinical trials conducted in European countries are required to be registered at a similar public database maintained and overseen by European health authorities. Competitors may use this publicly available information to gain knowledge regarding the design and progress of our development programs.
The Application Process. If the necessary clinical trials are successfully completed, the results of the preclinical trials and the clinical trials, together with other detailed information, including information on the manufacture and composition of the product, are submitted to FDA in the form of an NDA or a BLA, as applicable, and to the EMA or national regulators in the form of an MAA, requesting approval to market the product for a specified indication. In the EU, an MAA may be submitted to the EMA for review and, if the EMA gives a positive opinion, the EC may grant a marketing authorization that is valid across the EU (centralized procedure). Alternatively, an MAA may be submitted to one or more national regulators in the EU according to one of several national or decentralized procedures. The type of submission in Europe depends on various factors and must be cleared by the appropriate authority prior to submission. For most of our drug candidates, the centralized procedure will be either mandatory or available as an option.
If the regulatory authority determines that the application is not acceptable, it may refuse to accept the application for filing and review, outlining the deficiencies in the application and specifying additional information needed to file the application. Notwithstanding the submission of any requested additional testing or information, the regulatory authority ultimately may decide that the proposed product is not safe or effective, or that the application does not otherwise satisfy the criteria for approval. In the U.S., to support an approval an NDA must demonstrate, among other things, that the proposed drug product is safe and effective, has a favorable benefit-risk profile, is manufactured in a way that preserves its identity, strength, purity and potency, and that its labeling is adequate and not false or misleading. A similar standard exists for BLAs. Before approving an NDA or BLA, or an MAA, FDA or the EMA, respectively, may inspect one or more of the clinical sites at which the clinical studies were conducted to ensure that GCPs were followed and may inspect facilities at which the product is manufactured to ensure satisfactory compliance with cGMP. The FDA may refer the NDA or BLA to an advisory committee for review and recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendation of an advisory committee, but it generally follows such recommendation. In addition, even if a drug candidate satisfied its endpoints with statistical significance during clinical trials, FDA could determine that the overall balance of risks and benefits for the drug candidate is not adequate to support approval, or only justifies approval for a narrow set of clinical uses and/or subject to restricted distribution or other burdensome post-approval requirements or limitations. If approval is obtained changes to the approved product such as adding new indications, manufacturing changes, or additional labeling claims will require submission of a supplemental application, referred to as a variation in the EU, or, in some instances, a new application, for further review and approval. The testing and approval process requires substantial time, effort, and financial resources, and we cannot be sure that any future approval will be granted on a timely basis, if at all.
Some of our drug candidates, such as those from our MASP-2 and MASP-3 programs, are considered biologics because they are derived from natural sources as opposed to being chemically synthesized. The added complexity associated with manufacturing biologics may result in additional monitoring of the manufacturing process and product changes.
In addition, we, our suppliers and our contract manufacturers are required to comply with extensive regulatory requirements both before and after approval. For example, we must establish a pharmacovigilance system and are required to report adverse reactions and production problems, if any, to the regulatory authorities. If any of our drug candidates are approved, we will be required to also comply with certain requirements concerning advertising and promotion for our products. The regulatory authorities may impose specific obligations as a condition of the marketing authorization, such as additional safety monitoring, or the conduct of additional clinical trials or post-marketing safety studies, or the imposition of a Risk Evaluation and Mitigation Strategy (“REMS”), which could include significant restrictions on distribution or use of the product. Also, quality control and manufacturing procedures must continue to conform to cGMPs after approval. Accordingly, manufacturers must continue to expend time, money, and effort in all areas of regulatory compliance, including production and quality control to comply with cGMPs. In addition, discovery of problems such as safety issues may result in changes in labeling or restrictions on a product manufacturer or marketing authorization holder, including removal of the product from the market.
Fast-Track and Priority Review Designations. Section 506(b) of the FDCA provides for the designation of a drug as a fast-track product if it is intended, whether alone or in combination with one or more other drugs, for the treatment of a serious or life-threatening disease or condition, and it demonstrates the potential to address unmet medical needs for such a disease or condition. A program with fast-track status is afforded greater access to FDA for the purpose of expediting the product’s development, review and potential approval. Many products that receive fast-track designation are also considered appropriate to receive priority review, and their respective applications may be accepted by FDA as a rolling submission in which portions of an NDA or BLA are reviewed before the complete application is submitted. Together, these may reduce time of development and FDA review time. In Europe, products that are considered to be of major public health interest are eligible for accelerated assessment, which shortens the review period. The grant of fast-track status, priority review or accelerated assessment does not alter the standard regulatory requirements for obtaining marketing approval.
Breakthrough Therapy Designation. In 2012, Congress enacted the Food and Drug Administration Safety and Innovation Act. This law established a regulatory process allowing for increased interactions with FDA with the goal of expediting development and review of products designated as “breakthrough therapies.” A product may be designated as a breakthrough therapy if it is intended, either alone or in combination with one or more other products, to treat a serious or life-threatening disease or condition and preliminary clinical evidence indicates that the product may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development. The FDA may take certain actions with respect to breakthrough therapies, including holding meetings with the sponsor throughout the development process; providing timely advice to the product sponsor regarding development and approval; involving more senior staff in the review process; assigning a cross-disciplinary project lead for the review team; and taking other steps to design the clinical trials in an efficient manner.
Accelerated Approval. The FDA may grant accelerated approval to a product for a serious or life-threatening condition that provides a meaningful therapeutic advantage to patients over existing treatments based upon a determination that the product has an effect on a surrogate endpoint that is reasonably likely to predict clinical benefit. The FDA may also grant accelerated approval for such a condition when the product has an effect on an intermediate clinical endpoint that can be measured earlier than an effect on irreversible morbidity or mortality and that is reasonably likely to predict an effect on irreversible morbidity or mortality or other clinical benefit. In both cases, FDA must take into account the severity, rarity, or prevalence of the condition and the availability or lack of alternative treatments. Studies that are conducted to demonstrate a drug’s effect on a surrogate or intermediate clinical endpoint for accelerated approval must be adequate and well-controlled as required by the FDCA.
Following accelerated approval, FDA requires that the company provide confirmatory evidence, which may include certain adequate and well-controlled post-marketing clinical studies to verify the clinical benefit of the product, and FDA may impose restrictions on distribution to assure safe use. Pursuant to new statutory authority under the Food and Drug Omnibus Reform Act of 2022, FDA can require confirmatory studies to be underway at the time of the accelerated approval. If the required confirmatory studies fail to verify the clinical benefit of the drug, or if the applicant fails to perform the required confirmatory studies with due diligence, FDA may withdraw approval of the drug under streamlined procedures in accordance with FDA’s regulations. FDA may also withdraw approval of a drug if, among other things, other evidence demonstrates that the drug product is not shown to be safe or effective under its conditions of use.
The EU also has accelerated approval programs. In the EU, a marketing authorization may be granted on the basis of less complete data than are normally required in certain “exceptional circumstances,” such as when the product’s indication is encountered so rarely that the applicant cannot reasonably be expected to provide comprehensive data. Alternatively, a conditional marketing authorization may be granted prior to obtaining the comprehensive clinical data required for a full MAA if a product fulfills an unmet medical need and the benefit to public health of the product’s immediate availability outweighs the risk inherent in the incomplete data.
Orphan Drug Designation. Under the Orphan Drug Act (“ODA”), FDA may grant orphan drug designation to drugs or biologics intended to treat a rare disease or condition that affects fewer than 200,000 individuals in the U.S. or more than 200,000 individuals in the U.S. for which the cost of developing and making the product available in the U.S. for this type of disease or condition is not likely to be recovered from U.S. sales for that product. The granting of orphan designation does not alter the standard regulatory requirements (other than payment of certain fees and the applicability of certain pediatric assessment requirements), nor does it alter the standards or process for obtaining marketing approval. The sponsor of a product that has an orphan drug designation qualifies for various development incentives specified in the ODA, including a tax credit of up to 25% of expenditures on qualified clinical testing for the orphan drug. Furthermore, if the orphan designated product subsequently receives the first FDA approval for the orphan indication, the product is entitled to an orphan drug exclusivity period, which means that FDA may not grant approval to any other application to market the same drug for the same indication for a period of seven years except in limited circumstances, such as a showing of clinical superiority to the product with orphan drug exclusivity for the protected indication. Orphan drug exclusivity does not prevent FDA from approving a different drug for the same disease or condition, or the same drug for a different disease or condition. The EU has a similar Orphan Drug program to that of the U.S., and it is administered through the EMA’s Committee for Orphan Medicinal Products.
Pediatric Testing and Exclusivity. In the U.S., NDAs and BLAs are subject to both mandatory pediatric testing requirements and voluntary pediatric testing incentives in the form of exclusivity. An additional six months of exclusivity in the U.S. may be granted to a sponsor of an NDA or BLA if the sponsor conducts certain pediatric studies, which studies are conducted pursuant to a written request from FDA. This process is initiated when FDA issues a Written Request for pediatric studies to determine if the drug or biologic could have meaningful pediatric health benefits. If FDA determines that the sponsor has conducted the requested pediatric studies in accordance with the written request, then an additional six months of exclusivity may attach in the case of a drug to any other regulatory exclusivity or patent protection applicable to the drug and, in the case of a biologic, to any other regulatory exclusivity applicable to the biologic. The EU has a similar requirement and incentive for the conduct of pediatric studies according to the pediatric investigation plan, which must be adopted by the EMA before an MAA may be submitted.
Expanded Access. “Expanded access” refers to the use of an investigational drug where the primary purpose is to diagnose, monitor, or treat a patient’s disease or condition rather than to collect information about the safety or effectiveness of a drug. There are three FDA-recognized categories of expanded access trials: expanded access for individual patients, including for emergency use; expanded access for intermediate-size patient populations; and expanded access for large patient populations under a treatment IND or treatment protocol. For all types of expanded access, FDA must determine prior to authorizing expanded access that: (1) the patient or patients to be treated have a serious or life-threatening disease or condition and there is no comparable or satisfactory alternative therapy; (2) the potential patient benefit justifies the potential risks of use and that the potential risks are not unreasonable in the context of the disease or condition to be treated; and (3) granting the expanded access will not interfere with the initiation, conduct, or completion of clinical studies in support of the drug’s approval. Only a licensed physician or the drug’s manufacturer may apply for expanded access. Manufacturers are not required to supply the investigational product for expanded access. The FDA has established streamlined processes for physicians to request individual patient expanded access whereby physicians can submit a single patient IND. In cases of individual patient emergency expanded access, physicians can receive FDA approval for access by phone and follow up with the abbreviated form. In addition, the sponsor of an expanded access IND must submit IND safety reports and, in the cases of protocols continuing for one year or longer, annual reports to FDA.
U.S. Labeling, Marketing and Promotion. The FDA closely regulates the labeling, marketing and promotion of drugs. In general, our labeling and promotion must not be false or misleading in any particular, and claims that we make must be adequately substantiated. In addition, our approved labeling must include adequate directions to physicians for each intended use of our products. Failure to comply with these requirements can result in adverse publicity, warning letters, corrective advertising, injunctions and potential civil and criminal penalties.
In addition to regulation by FDA, the research, manufacturing, distribution, sale and promotion of drug products in the U.S. are subject to regulation by various federal, state and local authorities, including CMS, other divisions of the U.S. Department of Health and Human Services (e.g., the Office of Inspector General), the U.S. Department of Justice, state Attorneys General, and other state and local government agencies. All of these activities are also potentially subject to federal and state consumer protection and unfair competition laws. Violations of these laws are punishable by prison sentences, criminal fines, administrative civil money penalties, and exclusion from participation in federal healthcare programs.
There are also an increasing number of state laws that require manufacturers to make reports to states on pricing and marketing information or impose other special requirements for the sale and marketing of drug products. Many of these laws contain ambiguities as to what is required to comply with the laws. In addition, federal and state “transparency laws” require manufacturers to track and report certain payments made to health care providers and, under some state laws, other information concerning our products. These laws may affect our sales, marketing and other promotional activities by imposing administrative and compliance burdens on us. In addition, our reporting actions could be subject to the penalty provisions of the pertinent state and federal authorities.
Drug Supply Chain Security Act. Title II (the Drug Supply Chain Security Act (the “DSCSA”)), of the Drug Quality and Security Act imposes on manufacturers of certain pharmaceutical products new obligations related to product tracking and tracing, among others, which began a several-year phase-in process in 2015. Among the requirements of this legislation, manufacturers subject to the DSCSA are required to provide certain documentation regarding the drug product to trading partners to which product ownership is transferred, label drug product with a product identifier (i.e., serialize), respond to verification requests from trading partners, provide transaction documentation upon request by federal or state government entities, and keep certain records regarding the drug product. The transfer of information to subsequent product owners by manufacturers must be done electronically. For products and transactions falling within DSCSA’s scope, manufacturers are required to verify that purchasers of the manufacturers’ products are appropriately licensed. Further, under the DSCSA, covered manufacturers have drug product investigation, quarantine, disposition, and notification responsibilities for product that is reasonably believed or that credible evidence shows to be counterfeit, diverted, stolen, intentionally adulterated such that the product would result in serious adverse health consequences or death, the subject of fraudulent transactions or otherwise unfit for distribution such that they would be reasonably likely to result in serious health consequences or death. Anti-counterfeiting and serialization requirements similar to those under the DSCSA have also been adopted in the EU and became effective in February 2019.
Foreign Regulatory Requirements. Outside of the U.S., our ability to conduct clinical trials or market our products will also depend on receiving the requisite authorizations from the appropriate regulatory authorities. The foreign regulatory approval processes include similar requirements and many of the risks associated with FDA and/or the EU approval process described above, although the precise requirements may vary from country to country.
Hatch-Waxman Act. In seeking approval for a drug through an NDA, applicants are required to list with FDA each patent with claims that cover the applicant’s drug or an approved method of use of the drug. Upon approval of a drug, each of the patents listed in the application for the drug is then published in FDA’s Approved Drug Products with Therapeutic Equivalence Evaluations, commonly known as the Orange Book. Drugs listed in the Orange Book can, in turn, be cited by potential competitors in support of approval of an Abbreviated New Drug Application (“ANDA”) or a 505(b)(2) application. In this case the original NDA, i.e., the pioneer drug, is known as the “listed” drug or “reference-listed” drug. An ANDA provides for marketing of a drug that has the same active ingredients and, in some cases, also the same inactive ingredients, in the same strengths, route of administration and dosage form as the listed drug and has been shown through testing to be bioequivalent to the listed drug or receives a waiver from bioequivalence testing. ANDA applicants are generally not required to conduct or submit results of preclinical or clinical tests to prove the safety or effectiveness of their drug, other than the requirement for bioequivalence testing. Drugs approved in this way are considered therapeutically equivalent, and are commonly referred to as “generic equivalents” to the listed drug. These drugs then generally can be substituted by pharmacists under prescriptions written for the original listed drug.
The ANDA or 505(b)(2) applicant is required to certify to FDA concerning any patents listed for the referenced approved drug in FDA’s Orange Book. Specifically, for each listed patent, the applicant must certify that: (1) the required patent information has not been filed; (2) the listed patent has expired; (3) the listed patent has not expired but will expire on a particular date and approval is sought after patent expiration; or (4) the listed patent is invalid, unenforceable or will not be infringed by the new drug. A certification that the new drug will not infringe the already approved drug’s listed patents or that such patents are invalid or unenforceable is called a Paragraph IV certification. If the ANDA or 505(b)(2) applicant does not include a Paragraph IV certification, the ANDA or 505(b)(2) application will not be approved until all of the listed patents claiming the referenced drug have expired, except for any listed patents that only apply to uses of the drug not being sought by the ANDA or 505(b)(2) applicant.
If the ANDA or 505(b)(2) applicant has made a Paragraph IV certification, the applicant must also send a Paragraph IV Notice Letter to the NDA and patent holders once the ANDA or 505(b)(2) application has been accepted for filing by FDA. The NDA and patent holders may then initiate a patent infringement lawsuit in response to the Paragraph IV Notice Letter. The filing of a patent infringement lawsuit within 45 days of the receipt of a Paragraph IV Notice Letter automatically prevents FDA from approving the ANDA until the earlier of 30 months, expiration of the patent, settlement of the lawsuit, modification by a court or a decision in the infringement case that is favorable to the ANDA or 505(b)(2) applicant.
The ANDA or 505(b)(2) application also will not be approved until any applicable non-patent exclusivity, such as exclusivity for obtaining approval of a new chemical entity, listed in the Orange Book for the reference-listed drug has expired. The U.S. Drug Price Competition and Patent Term Restoration Act of 1984, more commonly known as the Hatch-Waxman Act, provides a period of five years following approval of a drug containing no previously approved active moiety, during which ANDAs for generic versions of those drugs and 505(b)(2) applications referencing those drugs cannot be submitted unless the submission contains a Paragraph IV challenge to a listed patent, in which case the submission may be made four years following the original drug approval. The Hatch-Waxman Act also provides for a period of three years of exclusivity following approval of a listed drug that contains previously approved active ingredients but is approved in a new dosage form, route of administration or combination, or for a new use, the approval of which was supported by new clinical trials other than bioavailability studies that were essential to the approval and conducted by or for the sponsor. During those three years of exclusivity, FDA cannot grant approval of an ANDA or 505(b)(2) application for the protected dosage form, route of administration or combination, or use of that listed drug.
In December 2019, the Creating and Restoring Equal Access to Equivalent Samples Act of 2019 (“CREATES Act”) was signed into law. The legislation is intended to address the concern that some brand manufacturers have improperly denied generic and biosimilar product developers access to samples of brand products. The CREATES Act establishes a private cause of action that permits a generic or biosimilar product developer to sue the brand manufacturer to compel it to furnish the necessary samples on commercially reasonable, market-based terms. If the developer prevails, the court may grant the developer a monetary award up to the brand product’s revenue for the period of delay in providing samples.
Biosimilars. The enactment of federal healthcare reform legislation in March 2010 provided a new pathway for approval of follow-on biologics (i.e., biosimilars) under the PHSA. FDA licensure of a biosimilar is dependent upon many factors, including a showing that the proposed biosimilar is “highly similar” to the reference product, notwithstanding minor differences in clinically inactive components, and has no clinically meaningful differences from the reference product in terms of safety, purity, and potency. The types of data ordinarily required in a biosimilar application to show high similarity include analytical data, animal studies (including toxicity studies), and clinical studies (including immunogenicity and pharmacokinetic/pharmacodynamic studies). A biosimilar must seek licensure for a condition of use for which the reference-listed product is licensed.
Furthermore, the PHSA provides that for a biosimilar to be considered “interchangeable” (i.e., the biological product may be substituted for the reference product without the intervention of the health care provider who prescribed the reference product), the applicant must make an additional showing that the biosimilar can be expected to produce the same clinical result as the reference product in any given patient, and if the product is administered more than once to a patient, that risks in terms of safety or diminished efficacy of alternating or switching between the biological product and the reference product is no greater than the risk of using the reference product without switching. Although FDA has provided guidance on what information and data an applicant should submit to enable an interchangeability determination, thus far FDA has not licensed any biologic as being interchangeable with its reference product.
The PHSA also provides a period of exclusivity for pioneer biologics. Specifically, FDA may not accept a biosimilar application referencing data from a pioneer biologic (i.e., one approved through a full BLA) until four years have elapsed from the date of first licensure of the pioneer biologic. FDA may not approve a biosimilar application referencing data from a pioneer biologic until 12 years have elapsed since the date of first licensure of the pioneer biologic. There are certain restrictions and limitations on the types of BLAs that are eligible for biologics exclusivity as well as what constitutes the date of first licensure for a pioneer biologic.
In the EU, a pathway for the approval of biosimilars has existed since 2005.
Healthcare compliance laws. In the U.S., commercialization of our drug candidates, if approved, is subject to regulation and enforcement under a number of federal and state healthcare compliance laws administered and enforced by various agencies. These include, but are not limited to, the following:
● the federal Anti-Kickback Statute, which prohibits offering or paying anything of value to a person or entity to induce or reward referrals for goods or services reimbursed by a federal healthcare program such as Medicare or Medicaid;
● the federal False Claims Act, which prohibits presenting or causing to be presented a false claim for payment by a federal healthcare program, and which has been interpreted to also include claims caused by improper drug-manufacturer product promotion or the payment of kickbacks;
● a variety of governmental pricing, price reporting, and rebate requirements, including those under Medicaid and the Veterans Health Care Act; and ● the so-called Sunshine Act and certain provisions of the Affordable Care Act, which require that we report to the federal government information on certain financial payments and other transfers of value made to certain health care providers and institutions, as well as certain information regarding our distribution of drug samples.
In addition to these federal law requirements, several U.S. states have enacted similar laws requiring periodic reporting and/or disclosure related to our marketing, sales and other activities, or regulating certain sales and marketing activities, such as provision of meals to certain health care providers. We may also be subject to federal or state privacy laws if we receive protected patient health information.
Similar requirements apply to our operations outside of the U.S. Laws in the U.S. such as the Foreign Corrupt Practices Act prohibit the offering or payment of bribes or inducements to foreign public officials for business, including physicians or other medical professionals who are employees of public healthcare entities. In addition, many non-U.S. jurisdictions in which we operate, or may operate in the future, have their own laws similar to the healthcare compliance laws that exist in the U.S.
Pharmaceutical Pricing and Reimbursement
Overview. In both U.S. and foreign markets, our ability to commercialize our drug candidates successfully, and to attract commercialization partners for our drug candidates, depends in significant part on the availability of adequate financial coverage and reimbursement from third-party payers including, in the U.S., managed care organizations and other private health insurers as well as governmental payers such as the Medicare and Medicaid programs. Reimbursement by a third-party payer may depend on a number of factors, including the payer’s determination that use of a product is:
● a covered benefit under its health plan;
● safe, effective and medically necessary;
● appropriate for the specific patient;
● cost-effective; and
● neither experimental nor investigational.
Reimbursement by government payers is based on statutory authorizations and complex regulations that may change with annual or more frequent rulemaking, as well as legislative reform measures.
Third-party private and governmental payers are increasingly challenging the prices charged for medicines and examining their cost-effectiveness in addition to their safety and efficacy. We may need to conduct expensive pharmacoeconomic studies in order to demonstrate the cost effectiveness of our products or drug candidates. Even with the availability of such studies, third-party private and/or governmental payers may not provide coverage and reimbursement for our drug candidates, in whole or in part.
United States. Political, economic and regulatory influences are subjecting the healthcare industry in the U.S. to fundamental changes. There have been, and we expect there will continue to be, legislative and regulatory proposals to change the healthcare system in ways that could significantly affect our business. For example, the 2010 Affordable Care Act (the “ACA”), is intended to broaden access to health insurance, reduce or constrain the growth of healthcare spending, enhance remedies against fraud and abuse, add transparency requirements for the healthcare and health insurance industries, impose new taxes and fees on the health industry and impose additional health policy reforms. Other legislative changes included a two percent across-the-board reduction to Medicare payments to providers, effective April 1, 2013, which, due to subsequent legislative amendments, will begin to increase gradually starting in April 2030, reaching 4 percent in April 2031 and continuing until the reduction ends in October 2031, unless additional congressional action is taken. The American Taxpayer Relief Act of 2012, among other things, reduced Medicare payments to several providers, and increased the period for the government to recover overpayments to providers from three to five years. In December 2017, portions of the ACA dealing with the individual mandate insurance requirement were effectively repealed by the Tax Cuts and Jobs Act of 2017.
Containment of healthcare costs has been a priority of federal, state, and foreign governments, and the prices of drug products have been a focus of this effort. Governments have shown significant interest in implementing cost-containment programs. This interest has resulted in significant proposed and enacted reform measures affecting healthcare reimbursement and drug pricing, including the enactment in August 2022 of significant changes to potential Medicare drug product reimbursement through government negotiation of certain drug prices, as well as manufacturer discount and inflation rebate obligations under the Inflation Reduction Act (the “IRA”).
We are unable to predict what additional legislation, regulations, policies or court orders, if any, relating to the healthcare industry or coverage and reimbursement may be enacted or imposed in the future or what effect such legislation, regulations, policies or court orders would have on our business. Any cost-containment measures, including those listed above, or other healthcare system reforms that are adopted could have a material adverse effect on our business prospects and financial operations.
Europe. Governments in the various member states of the EU influence or control the price of medicinal products in their countries through their pricing and reimbursement rules and control of national healthcare systems that fund a large part of the cost of those products to consumers. To obtain reimbursement or pricing approval, some of these countries may require the completion of clinical trials or pharmacoeconomic studies that assess the cost-effectiveness of a product or drug candidate relative to currently available therapies or relative to a specified standard. The downward pressure on healthcare costs in general, and prescription medicines in particular, has become very intense and is creating increasingly high barriers to the entry of new products in these markets.
Research and Development
We have built a research and development organization that includes expertise in discovery research, preclinical development, product formulation, analytical and medicinal chemistry, manufacturing, clinical development and regulatory and quality assurance. We operate cross-functionally and are led by an experienced management team. We strive to make disciplined strategic decisions regarding our research and development programs and to limit the risk profile of our product pipeline. We also access relevant market information and key opinion leaders in creating target product profiles and, when appropriate, as we advance our programs to commercialization. We engage third parties on a limited basis to conduct portions of our preclinical research; however, we are not substantially dependent on any third parties for our preclinical research nor do any of these third parties conduct a major portion of our preclinical research. We also engage multiple clinical sites to conduct our clinical trials and rely on third-party contract research organizations (“CROs”) to coordinate and execute aspects of clinical trial operations. None of these CROs or clinical sites are responsible for the major portion of our clinical trials and we are not substantially dependent on any one of them.
Employees
As of December 31, 2023, we had 198 full-time employees, 132 of whom are in research and development, 19 of whom are in sales and marketing and 47 of whom are in finance, legal, business development and administration. Our full-time employees include six with M.Ds and 40 with Ph.Ds., of whom five and 39, respectively, are in research and development. None of our employees are represented by a labor union, and we consider our employee relations to be good.
Information about Our Executive Officers and Significant Employees
The following table provides information regarding our executive officers and significant employees as of April 1, 2024:
| Name |
Age |
Position(s) |
||
| Executive Officers: |
||||
| Gregory A. Demopulos, M.D. |
65 |
President, Chief Executive Officer and Chairman of the Board of Directors |
||
| Michael A. Jacobsen |
65 |
Vice President, Finance, Chief Accounting Officer and Treasurer |
||
| Peter B. Cancelmo, J.D. |
45 |
Vice President, General Counsel and Secretary |
||
| Significant Employees: |
||||
| Nadia Dac |
54 |
Vice President, Chief Commercial Officer |
||
| Mariana N. Dimitrova, Ph.D. |
58 |
Vice President, Chemistry, Manufacturing and Controls |
||
| George A. Gaitanaris, M.D., Ph.D. |
67 |
Vice President, Science and Chief Scientific Officer |
||
| Andreas Grauer, M.D. |
63 |
Vice President, Chief Medical Officer |
||
| Catherine A. Melfi, Ph.D. |
65 |
Vice President, Regulatory Affairs & Quality Systems and Chief Regulatory Officer |
||
| J. Steven Whitaker, M.D., J.D. |
68 |
Vice President, Clinical Development |
||
| Peter W. Williams |
56 |
Vice President, Human Resources |
Gregory A. Demopulos, M.D. founded our company and has served as our president, chief executive officer and chairman of the board of directors since June 1994. He also served as our chief financial officer and treasurer from January 2009 to October 2013 in an interim capacity and as our chief medical officer from June 1994 to March 2010. Prior to founding Omeros, Dr. Demopulos completed his residency in orthopedic surgery at Stanford University and his fellowship training in hand and microvascular surgery at Duke University. Dr. Demopulos currently serves on the board of trustees of the Smead Funds Trust, an open-end mutual fund company registered under the Investment Company Act of 1940. Dr. Demopulos received his M.D. from the Stanford University School of Medicine and his B.S. from Stanford University. Dr. Demopulos is the brother of Peter A. Demopulos, M.D., a member of our board of directors.
Michael A. Jacobsen has served as our vice president, finance, chief accounting officer and treasurer since October 2013. Prior to joining Omeros, Mr. Jacobsen served as vice president of finance of Sarepta Therapeutics, Inc. from September 2011 to May 2013 and as its chief accounting officer from September 2011 to December 2012. From April 2007 to August 2011, Mr. Jacobsen was vice president and chief accounting officer at ZymoGenetics, Inc. Prior to his service with ZymoGenetics, Mr. Jacobsen held various roles at ICOS Corporation, including senior director of finance and corporate controller. From April 1995 to October 2001, Mr. Jacobsen held vice president of finance or chief financial officer roles at three companies in the software, computer hardware and internet retailing industries, two of which were publicly traded. Mr. Jacobsen is a certified public accountant and received his bachelor’s degree in accounting from Idaho State University.
Peter B. Cancelmo, J.D. has served as our vice president, general counsel and secretary since June 2019. He joined Omeros as deputy general counsel in January 2019. Prior to joining Omeros, Mr. Cancelmo was a principal and shareholder at Garvey Schubert Barer, P.C., where he represented clients in the life sciences and other technology industries in mergers, acquisitions, strategic alliances, public and private securities offerings, and a range of other corporate, commercial and financial transactions. He served as chair of the firm’s business practice group from 2016 until his departure in December 2018. Mr. Cancelmo previously practiced corporate and transactional law at Davies, Ward, Philips and Vineberg LLP, in New York, and Choate, Hall & Stewart LLP, in Boston. Mr. Cancelmo received his J.D. from Boston University and his B.A. from Saint Michael’s College.
Nadia Dac has served as our chief commercial officer since January 2021. Ms. Dac brings nearly three decades of international experience as a strategic commercial leader at large and small biopharmaceutical companies. Prior to joining Omeros, Ms. Dac served as the chief commercial officer at Alder Pharmaceuticals, Inc. (acquired in 2019 by Lundbeck) from April 2019 until June 2020 and as vice president of global specialty commercial development at AbbVie, Inc. from December 2014 to March 2019. She previously served as vice president of marketing at Auxilium Pharmaceuticals, Inc. from May 2013 to September 2014, when the company was acquired by Endo International plc. From 2009 to 2013, Ms. Dac held several roles of increasing responsibility at Novartis AG, including global vice president of neuroscience professional relations prior to her role as vice president of Novartis’ multiple sclerosis franchise, and at Biogen Inc., Johnson & Johnson, and Eli Lilly and Company. She holds a B.S. in Marketing from Rutgers University.
Mariana N. Dimitrova, Ph.D., has served as our vice president chemistry, manufacturing, and controls (“CMC”) since October 2022. Prior to joining Omeros in this role, Dr. Dimitrova had 20 years of pharmaceutical experience with CMC leadership spanning formulation development, drug product and device development, drug delivery and Human Factors engineering, analytical sciences, process development, and clinical manufacturing. In her career, Dr. Dimitrova contributed to the development of a number of monoclonal antibodies, Fc-fusion proteins, PEG-proteins, bispecific molecules, cytokines, DNA, peptides, and small molecules at Amgen Inc., MedImmune (Astra Zeneca), Biogen, and Jazz Pharmaceuticals. Dr. Dimitrova contributed to the commercialization of nine patient-convenient drug/device combination products for the treatment of autoimmune, respiratory, neurodegenerative, hematology, and infectious diseases. Most recently, from May 2019 to September 2022, Dr. Dimitrova was vice president of product and device development at Akero Therapeutics, developing Fc-FGF21 fusion protein for treatment of NASH. Prior to her industry work, Dr. Dimitrova spent five years in academia, including at the National Heart, Lung, and Blood Institute at the National Institutes of Health and the National Institute of Advanced Industrial Science and Technology (AIST) in Japan. Dr. Dimitrova holds a Ph.D. in Biophysics and Biological Sciences from the Bulgarian Academy of Sciences and the AIST, and a M.S. in Chemistry from Kliment Ohridski University in Bulgaria.
George A. Gaitanaris, M.D., Ph.D. has served as our vice president, science since August 2006 and as our chief scientific officer since January 2012. From August 2003 until our acquisition of nura, inc., in August 2006, Dr. Gaitanaris served as the chief scientific officer of nura, a company that he co-founded, and that developed treatments for central nervous system disorders. From 2000 to 2003, Dr. Gaitanaris served as president and chief scientific officer of Primal, Inc., a biotechnology company that was acquired by nura in 2003. Prior to co-founding Primal, Dr. Gaitanaris served as staff scientist at the National Cancer Institute. Dr. Gaitanaris received his Ph.D. in cellular, molecular and biophysical studies and his M.Ph. and M.A. from Columbia University and his M.D. from the Aristotelian University of Greece.
Andreas Grauer, M.D. has served as our chief medical officer since October 2023. Prior to joining Omeros, Dr. Grauer served as chief medical officer at Federation Bio from October 2021, where he led all clinical activities with a focus on hyperoxaluria and immuno-oncology. From March 2019 to August 2021, Dr. Grauer was chief medical officer of Corcept Therapeutics, Inc., leading its global development organization in the design and execution of clinical programs directed to oncology, neurology, endocrinology, and metabolism indications. From December 2007 to December 2018, Dr. Grauer held several roles of increasing responsibility at Amgen, most recently serving as vice president of global development, therapeutic area head, and co-chair of the franchise steering committee for bone, nephrology and inflammation. Earlier in his career, Dr. Grauer was at Proctor and Gamble Pharmaceuticals where he held roles as global executive medical director for bone and for new technology development. Dr. Grauer received his M.D. from the University of Heidelberg Medical School in Germany, where he also completed his clinical training in internal medicine and endocrinology. He did research in molecular and cellular endocrinology both there and during a post-doctoral fellowship at Baylor College of Medicine. He holds an active associate professorship of medicine at the University of Heidelberg Medical School.
Catherine A. Melfi, Ph.D. has served as our vice president, regulatory affairs and quality systems since October 2012 and has served as our chief regulatory officer since April 2016. Dr. Melfi previously served from January 1996 to September 2012 at Eli Lilly and Company, where she held technical and leadership roles of increasing scope and responsibility, including as senior director and scientific director in global health outcomes and regulatory affairs, respectively. Prior to joining Eli Lilly, Dr. Melfi held various faculty and research positions at Indiana University, including appointments in its Economics Department, in the School of Public and Environmental Affairs, and in the Indiana University School of Medicine. Dr. Melfi received her Ph.D. in Economics from the University of North Carolina - Chapel Hill and B.S. in Economics from John Carroll University.
J. Steven Whitaker, M.D., J.D. has served as our vice president, clinical development since joining Omeros in 2010, and served as our chief medical officer from March 2010 to August 2018 and from November 2019 to October 2023. From May 2008 to March 2010, Dr. Whitaker served as the chief medical officer, vice president of clinical development at Allon Therapeutics, Inc., a biotechnology company focused on developing drugs for neurodegenerative diseases. From August 2007 to May 2008, he served as a medical consultant to Accelerator Corporation, a biotechnology-company investor and incubator. From May 1994 to May 2007, Dr. Whitaker served at ICOS Corporation, which was acquired by Eli Lilly and Company in 2007. At ICOS, he held roles of increasing responsibility in clinical research and medical affairs, most recently as divisional vice president, clinical research as well as medical director of the Cialis® global product team. Dr. Whitaker received his M.D. from the Indiana University School of Medicine, his J.D. from the University of Washington and his B.S. from Butler University.
Peter W. Williams has served as our vice president, human resources since June 2020. Prior to joining Omeros, Mr. Williams served as the senior vice president of human resources at Redbox Automated Retail, LLC from 2016 to 2019, where he led human resources and internal communications functions. From 2013 to 2016, Mr. Williams served as the vice president, HR operations at Outerwall Inc. (Coinstar) and before that he held human resources leadership roles at Coinstar from 2009 to 2013. Prior to 2009, Mr. Williams held human resources leadership roles at various technology and consumer focused companies, including Washington Mutual, Inc., Sterling Commerce, Inc., Expedia, Inc., and Verio, Inc. Mr. Williams received a B.A. in Business Administration and a B.A. in English from the University of Washington.
Corporate Information
We were incorporated in 1994 as a Washington corporation. Our principal executive offices are located at 201 Elliott Avenue West, Seattle, Washington, 98119, and our telephone number is (206) 676-5000. Our website address is www.omeros.com. We make available, free of charge through our investor relations website at investor.omeros.com, our annual report on Form 10-K, our quarterly reports on Form 10-Q, our current reports on Form 8-K and amendments to those reports filed or furnished pursuant to Section 13(a) or 15(d) of the Exchange Act, including exhibits to those reports, as soon as reasonably practicable after we electronically file such material with, or furnish it to, the SEC. Our websites and the information contained therein or incorporated therein are not intended to be incorporated into this Annual Report on Form 10-K. The SEC maintains a website that contains reports, proxy and information statements, and other information regarding reports that we file or furnish electronically with them at www.sec.gov.
The risks and uncertainties described below may have a material adverse effect on our business, prospects, financial condition or operating results. In addition, we may be adversely affected by risks that we currently deem immaterial or by other risks that are not currently known to us. You should carefully consider these risks before making an investment decision. The trading price of our common stock could decline due to any of these risks and you may lose all or part of your investment. In assessing the risks described below, you should also refer to the other information contained in this Annual Report on Form 10-K.
Risks Related to Our Products, Programs and Operations
● continue our research and development in our programs;
● make principal, interest and fee payments as required under our 5.25% Convertible Senior Notes due 2026 (the “2026 Notes”); and
● commercialize and launch drug candidates for which we may receive regulatory approval.
Failure to obtain and maintain regulatory approval in the U.S. or in foreign jurisdictions would prevent us from commercializing and marketing our drug candidates.
The regulatory process is subject to substantial agency discretion and risks, including those described herein and elsewhere in these “Risk Factors.” In October 2021, we received a CRL from FDA regarding our BLA for narsoplimab for the treatment of TA-TMA. In the CRL, FDA expressed difficulty in estimating the treatment effect of narsoplimab in TA-TMA and asserted that additional information would be needed to support regulatory approval. We appealed FDA’s decision to issue the CRL through a formal dispute resolution process that concluded in late 2022. Although our appeal was denied, the decision identified a potential path for resubmission of the BLA based on inclusion of certain additional information and analyses. Consistent with subsequent interactions with FDA’s review division regarding resubmission of the BLA,we submitted to FDA in the fall of 2023 an analysis plan to assess already existing clinical trial data, existing data from an historical control population available from an external source, data from the narsoplimab expanded access program, and data directed to the mechanism of action of narsoplimab. We are having ongoing discussions with the agency regarding the proposed analysis plan and are currently unable to estimate when we will submit the BLA. Additionally, the requirements for resubmission of our BLA may be costly, require significant time and may not result in approval. Ultimately, we cannot guarantee that FDA will ever approve narsoplimab for the treatment of TA-TMA or any other indication.
We also intend to market outside the U.S. any of our drug candidates that are approved in the future. In order to market our products in non-U.S. jurisdictions, we or our partners must obtain separate regulatory approvals and comply with numerous and varying regulatory requirements. The regulatory approval procedure varies among countries and can involve additional testing and data review. The requirements governing marketing authorization, the conduct of clinical trials, pricing and reimbursement vary from country to country. Approval by FDA does not ensure approval by the EMA, and approval by one foreign regulatory authority does not ensure approval by regulatory agencies in other foreign countries or by FDA. The time required to obtain regulatory approval outside the U.S. and EU may differ from that required to obtain FDA or EU approval. The foreign regulatory approval process may include all of the risks associated with obtaining FDA approval discussed in these “Risk Factors” and we may not obtain foreign regulatory approvals on a timely basis, or at all. In addition, even if we were able to obtain regulatory approval for a product in one or more foreign jurisdictions, we may need to complete additional requirements to maintain that approval and our ability to market the product in the applicable jurisdiction.
If any product that we develop and commercialize does not receive adequate coverage or reimbursement from governments and/or private payers our prospects for revenue and profitability would suffer.
The success of any product that we or our third-party business partners commercialize in the future will depend heavily on the pricing, availability and duration of adequate coverage or reimbursement for any such product from government, private and other third-party payers, both in the U.S. and in other countries.
There may be significant delays in obtaining coverage or reimbursement for newly approved products, and we may not be able to provide data sufficient to be granted adequate coverage or reimbursement. Even when a payer determines that a product is eligible for reimbursement, coverage may be limited to the uses of a product that are either approved by FDA (or, in other countries, the relevant country’s regulatory agency) and/or appear in a recognized drug compendium, or other conditions may apply. Moreover, eligibility for coverage does not mean that any product will be reimbursed at a rate that allows us to make a profit or at a rate that covers our costs, including research, development, manufacturing, sales and distribution. Increasingly, government and private third-party payers that reimburse for healthcare services and products are requiring that companies provide them with predetermined discounts from list prices and are challenging the prices charged for medical products, which could adversely impact the pricing of our products. Any reduction in reimbursement from Medicare, including as a result of the Inflation Reduction Act, or other government programs may result in a similar reduction in payments from private payers. Pricing may also be adversely affected by changes in the terms, scope and/or complexity of government pricing requirements. Even if we achieve coverage or reimbursement for a product, the initial rate or method at which the product will be reimbursed could become unfavorable to us at the time reimbursement is initiated or in the future or may be of a limited duration. In addition, obtaining acceptable coverage and reimbursement from one payer does not guarantee that we will obtain similar acceptable coverage or reimbursement from another payer.
In non-U.S. jurisdictions, we must obtain separate reimbursement approvals and comply with related foreign legal and regulatory requirements. In some countries, including those in the EU, our products may be subject to government price controls. Pricing negotiations with governmental authorities can take a considerable amount of time and expenditure of resources after the receipt of marketing approval for a product. We provide no assurances that the price of any product in one or more of these countries or regions will allow us to make a profit or cover our costs, including research, development, manufacturing, sales and distribution, and as a result we may decide to delay, potentially indefinitely, initiating sales in the particular country or region.
If the reimbursement or pricing that we are able to obtain and maintain for any product that we develop and commercialize is inadequate, is significantly delayed or is subject to overly restrictive conditions, our ability to generate revenue, attain profitability and/or commercialize our drug candidates may be impaired and there could be a material adverse effect on our business, financial condition, results of operations and growth prospects and the trading price of our stock could decline.
Our ability to meet our future capital requirements is partially dependent on certain milestone and royalty payments that we are eligible to receive based on Rayner’s sales of OMIDRIA, and, if sales of OMIDRIA are less than anticipated and/or Rayner is unable to expand sales of OMIDRIA outside the U.S., our financial condition and results of operations may be materially adversely affected, the price of our common stock may decline and we may be unable to access needed capital on favorable terms, or at all.
In February 2024, we sold to DRI an expanded interest in OMIDRIA royalties payable by Rayner. Pursuant to the Amendment with DRI, DRI is entitled to receive all royalties on U.S. net sales of OMIDRIA between January 1, 2024 and December 31, 2031. Omeros retains the right to receive all royalties payable by Rayner on any net sales of OMIDRIA outside the U.S. payable from and after January 1, 2024, as well as all royalties on global net sales of OMIDRIA payable from and after December 31, 2031. We received $115.5 million in cash upon closing of the Amendment. Additionally, we are eligible under the Amendment to receive two milestone payments of up to $27.5 million each, payable in January 2026 and January 2028, respectively, based on achievement of certain thresholds for U.S. net sales of OMIDRIA.
The royalty rate payable by Rayner on net sales of OMIDRIA is currently 30% in the United States and 15% outside the U.S. The royalty rate is subject to further reduction to 10% of U.S. net sales upon the occurrence of certain events, including during any specific period in which OMIDRIA is no longer eligible for separate payment. The availability of royalties from Rayner and/or milestone payments from DRI is dependent on Rayner’s net sales of OMIDRIA and may be of lesser magnitude than anticipated or may not become payable at all. We cannot provide assurance that royalty income from Rayner and/or milestone payments from DRI, if they become payable, will be a meaningful source of capital in the future. Sales-based royalty income and milestone payments may be affected by any number of factors, including:
● Rayner’s ability to successfully market and sell OMIDRIA in the U.S.;
● whether, and to what extent, Rayner is able to expand sales of OMIDRIA outside the U.S.;
● pricing, coverage and reimbursement policies of government and private payers such as Medicare, Medicaid, the U.S. Department of Veterans Affairs, group purchasing organizations, insurance companies, health maintenance organizations and other plan administrators;
● a lack of acceptance by physicians, patients and other members of the healthcare community;
● interruptions in the supply of OMIDRIA;
● the availability, relative price and efficacy of the product as compared to alternative treatment options or branded, compounded or generic competing products;
● an unknown safety risk; and
● changed or increased regulatory restrictions in the U.S., EU and/or other foreign territories.
Our operating results are unpredictable and may fluctuate.
Our operating results are difficult to predict and will likely fluctuate from quarter to quarter and year to year. We believe that our quarterly and annual results of operations may be affected by a variety of factors, including:
● the extent and magnitude of certain payments to which we may be entitled based on Rayner’s net sales of OMIDRIA may be affected by the extent of coverage and reimbursement for OMIDRIA, market acceptance of the product and Rayner’s ability to execute an effective sales strategy;
● the extent of any payments received from any collaboration agreements or development funding arrangements that we may enter into from time to time, as well as the extent of any payments that we are required to make under existing or future collaboration and license agreements, which may include sales-based royalties and milestone payments based on the achievement of development, regulatory and sales milestones and may vary significantly from quarter to quarter;
● the timing, cost and level of investment in our research and development activities as well as expenditures we may incur to acquire or develop additional technologies, drug candidates, or in preparation for potential commercialization of our drug candidates; and
● whether we are able to obtain marketing approval for any of our drug candidates, the extent and timing of revenue from sales of any such approved product and the magnitude and timing of expenses associated with the manufacturing and sale of any such approved product.
Any of these risk factors, should one or more occur, could adversely affect our results of operations and financial condition and cause the trading price of our stock to decline.
We are subject to extensive government regulation and the failure to comply with these regulations may have a material adverse effect on our operations and business.
Both before and after approval of any product, we and our suppliers, contract manufacturers and clinical investigators are subject to extensive regulation by governmental authorities in the U.S. and other countries, covering, among other things, testing, manufacturing, quality control, clinical trials, post-marketing studies, reporting, risk management plans, labeling, advertising, promotion, distribution, import and export, governmental pricing, price reporting and rebate requirements. Failure to comply with applicable requirements could result in one or more of the following actions: warning letters; unanticipated expenditures; delays in approval or refusal to approve a drug candidate; product recall or seizure; interruption of manufacturing or clinical trials; operating or marketing restrictions; injunctions; criminal prosecution and civil or criminal penalties including fines and other monetary penalties; adverse publicity; and disruptions to our business. Further, government investigations into potential violations of these laws would require us to expend considerable resources and face adverse publicity and the potential disruption of our business even if we are ultimately found not to have committed a violation.
Obtaining FDA approval of our drug candidates requires substantial time, effort and financial resources and may be subject to both expected and unforeseen delays, and there can be no assurance that any approval will be granted on any of our drug candidates on a timely basis, if at all. As was the case with our BLA for narsoplimab in TA-TMA, with respect to which FDA issued a CRL, even after collaborating closely with FDA or regulators with corollary responsibilities in jurisdictions outside the U.S. regarding the contents of a marketing application a regulator may decide that the design of our clinical trials or clinical data collection protocols as actually run, or our resulting data, are insufficient for approval of our drug candidates. FDA or other regulators may require us to run additional preclinical, clinical or other studies or perform additional work related to chemistry, manufacturing and controls. In addition, we, FDA or an independent institutional review board or ethics committee may suspend or terminate human clinical trials at any time on various grounds, including a finding that the patients are or would be exposed to an unacceptable health risk or because of the way in which the investigators on whom we rely carry out the trials. We are subject to extensive government regulation of the testing of our investigational products, including the requirement that we conduct all of our clinical trials in accordance with FDA’s GCP requirements and similar requirements outside of the U.S. If we are unable to comply with these requirements, if we are required to conduct additional trials or to conduct other testing of our drug candidates beyond that which we currently contemplate for regulatory approval, if we are unable to complete our clinical trials or other testing successfully, or if the results of these and other trials or tests fail to demonstrate efficacy or raise safety concerns, we may face substantial additional expenses, be delayed in obtaining marketing approval for our drug candidates or may never obtain marketing approval.
We are also required to comply with extensive governmental regulatory requirements after a product has received marketing authorization. Governing regulatory authorities may require post-marketing studies that may negatively impact the commercial viability of a product. Once on the market, a product may become associated with previously undetected adverse effects and/or may develop manufacturing difficulties. We are required to comply with other post-marketing requirements including cGMPs, advertising and promotion restrictions, pharmacovigilance requirements including risk management activities, reporting and recordkeeping obligations, and other requirements. As a result of any of these or other problems or failure to comply with our regulatory obligations, a product’s regulatory approval could be withdrawn, which could harm our business and operating results. In addition, we must maintain an effective healthcare compliance program in order to comply with U.S. and other laws applicable to marketed drug products and, in particular, laws (such as the Anti-Kickback Statute, the False Claims Act and the Sunshine Act) applicable when drug products are reimbursed by a federal or state healthcare program. U.S. laws such as the Foreign Corrupt Practices Act prohibit the offering or payment of bribes or inducements to foreign public officials, including potentially physicians or other medical professionals who are employees of public healthcare entities in jurisdictions outside the U.S. In addition, many countries have their own laws similar to the healthcare compliance laws that exist in the U.S. Implementing and maintaining an effective compliance program requires the expenditure of significant time and resources. If we are found to be in violation of any of these laws, we may be subject to significant penalties, including but not limited to civil or criminal penalties, damages and fines as well as exclusion from government healthcare programs.
We may face difficulties from changes to current regulations as well as future legislation.
Existing regulatory policies may change and additional government regulations may be enacted that could prevent, limit or delay regulatory approval of our drug candidates. We cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative action, either in the U.S. or abroad. If we are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we are not able to maintain regulatory compliance, we may lose any marketing approval that we may have obtained and we may not achieve or sustain profitability.
Any reduction in reimbursement from Medicare resulting from the IRA or other legislative or policy changes or from other government programs may result in a similar reduction in payments from private payers. These healthcare reforms and the implementation of any future cost containment measures or other reforms may prevent us from being able to generate sufficient revenue, attain and/or maintain profitability or commercialize our drug candidates. We cannot be sure whether additional legislative changes will be enacted, whether existing legislation will be implemented, interpreted or enforced as anticipated or whether FDA regulations, guidance or interpretations will be changed, or what the impact of such changes on our drug candidates, if any, may be.
We have no internal capacity to manufacture commercial or clinical supplies of our drug candidates and intend to continue to rely solely on third-party manufacturers, which could significantly limit or delay our clinical trials or regulatory submissions and may negatively impact our financial conditions and results of operations. If we are unable to establish relationships with contract manufacturers that have sufficient manufacturing capacity available to meet our needs, or if the contract manufacturers that we rely on experience difficulties manufacturing and supplying our drug candidates, or fail FDA or other regulatory inspections, then our clinical trials or regulatory submissions may be significantly limited or delayed or we may have inadequate supply to meet demand for any product that we commercialize in the future.
We rely and intend to continue to rely on third-party manufacturers to produce quantities of clinical drug supplies of our drug candidates that are needed for clinical trials and to support NDAs, BLAs, or similar applications to regulatory authorities seeking marketing approval for our drug candidates, as well as to produce inventory of our drug candidates for commercial use in anticipation of marketing approval. Global demand for contract manufacturing is volatile and the available supply of contract manufacturing capacity is limited and unpredictable. We cannot provide any assurance that we will be able to enter into or maintain these types of arrangements on commercially reasonable terms, or at all, or that manufacturing arrangements will meet our requirements. Our contract manufacturers previously have and may in the future require us to place orders or make other financial commitments several years in advance of manufacturing commencement based on forecasts of our long-term commercial supply requirements for drug candidates that have not yet received, and may never receive, regulatory approval. We may be required to pay significant cancellation fees or other financial penalties in connection with the withdrawal or cancellation of any binding order for manufacturing that we later determine is not needed. The fees or other financial obligations that we may incur in connection with withdrawn or cancelled orders may be material and any such financial penalty would negatively impact our financial condition and results of operations.
If we or one of our manufacturers were to terminate one of these arrangements early, or the manufacturer was unable to supply product quantities sufficient to meet our requirements, we would be required to transfer manufacturing to an approved alternative facility and/or establish additional manufacturing and supply arrangements. We may also need to establish additional or replacement manufacturers, potentially with little or no notice, in the event that one of our manufacturers fails to comply with FDA and/or other pharmaceutical manufacturing regulatory requirements. Even if we are able to establish additional or replacement manufacturers, identifying these sources and entering into definitive supply agreements and obtaining regulatory approvals may require a substantial amount of time and cost and may create a shortage of the product. It can take several years to qualify and validate a new contract manufacturer, and we cannot guarantee that we would be able to complete in a successful and timely manner the appropriate validation processes or obtain the necessary regulatory approvals for one or more additional or replacement manufacturers. Such alternate supply arrangements may not be available on commercially reasonable terms, or at all. Additionally, if we are unable to engage multiple suppliers to manufacture our products, we may have inadequate supply to meet demand for our product.
In addition, narsoplimab, OMS906 and OMS1029 are biologic drug products and other drug candidates from certain of our programs, including but not limited to MASP-2 and MASP-3, could be biologic drug products. We do not have the internal capability to produce biologics for use in clinical trials or on a commercial scale. There are only a limited number of manufacturers of biologic drug products and we may be unable to enter into agreements on commercially reasonable terms with a sufficient number of them to meet clinical or commercial demand, if at all. The regulatory requirements for commercial supply are more stringent than for clinical supply and we cannot guarantee that a contract manufacturer producing drug product for clinical trials will be able to complete successfully the appropriate validation processes or obtain the necessary regulatory approvals for marketing approval and commercial supply in a timely manner or at all.
Our contract manufacturers may encounter difficulties with formulation, manufacturing, supply chain and/or release processes that could result in delays in clinical trials and/or regulatory submissions or that could impact adversely the commercialization of our products or drug candidates, as well as in the initiation of enforcement actions by FDA and other regulatory authorities. For example, our manufacturers are required to comply with FDA’s GMP requirements and are subject to periodic inspections by FDA. If our manufacturers are unable to comply with FDA requirements, they may be unable to meet our supply needs. These difficulties also could result in the recall or withdrawal of a product from the market or a failure to have adequate supplies to meet market demand. If the safety or manufacturing quality of any drug candidate supplied by contract manufacturers is compromised due to one or more of those contract manufacturers’ failure to adhere to applicable laws or for other reasons, we may not be able to maintain regulatory approval to run clinical trials or to obtain and maintain regulatory approval for one or more of our drug candidates, which would harm our business and prospects significantly.
Any significant delays in the manufacture and/or supply of clinical or commercial supplies could materially harm our business, financial condition, results of operations and prospects.
Ingredients, excipients, test kits and other materials necessary to manufacture our drug candidates may not be available on commercially reasonable terms, or at all, which may adversely affect the development and commercialization of our drug candidates.
We and our third-party manufacturers must obtain from third-party suppliers the APIs, excipients, and/or other raw materials plus primary and secondary packaging materials necessary for our contract manufacturers to produce our drug candidates for our clinical trials and, to the extent approved or commercialized, for commercial distribution. Although we have entered or intend to enter into agreements with third-party suppliers that will guarantee the availability and timely delivery of APIs, excipients, test kits and materials for our drug candidates, we have not entered into agreements for the supply of all such ingredients, excipients, test kits or materials, and we may be unable to secure all such supply agreements or guarantees on commercially reasonable terms, if at all. Even if we were able to secure such agreements or guarantees, our suppliers may be unable or choose not to provide us the ingredients, excipients, test kits or materials in a timely manner or in the quantities required. Further, if we or our third-party manufacturers are unable to obtain APIs, excipients, test kits and materials as necessary for our clinical trials or for the manufacture of commercial supplies of our drug candidates, if approved, potential regulatory approval or commercialization would be delayed, which would materially and adversely affect our ability to generate revenue from the sale of our drug candidates.
We may be unable to advance clinical development of narsoplimab for treatment of COVID-19 and, even if successful, we may be unable to manufacture narsoplimab in sufficient quantities.
Narsoplimab has been used to treat critically ill COVID-19 patients under our compassionate use program with highly positive results and, in an analysis of the randomized population in the narsoplimab treatment arm of I-SPY COVID-19 trial, the addition of narsoplimab to standard-of-care treatment of critically ill COVID-19 patients resulted in a mortality benefit. Notwithstanding these results, we may determine not to continue clinical development of narsoplimab for COVID-19 and/or further clinical evaluation of narsoplimab for the treatment of COVID-19 may not be feasible as a result of a number of factors, including decreasing rates of severe illness in patients with COVID-19 and the availability of alternative preventive or therapeutic agents for COVID-19. Additionally, the results of the I-SPY-COVID-19 trial may be not be viewed by regulators, government officials and others as strong evidence of narsoplimab’s efficacy in the treatment of severe COVID-19 because the narsoplimab treatment arm of the I-SPY-COVID-19 trial was terminated prior to accrual of the maximum of 125 patients on the basis of analysis in a pre-consented population in which substantial bias was detected. Also, contract manufacturing capacity and supplies of raw materials necessary for the production of narsoplimab are limited and we may be unable to secure the large-scale manufacturing capacity from third parties necessary to manufacture narsoplimab in sufficient quantities to enable broad availability of narsoplimab for COVID-19 patients. These risks could limit our ability to develop or commercialize a therapeutic for COVID-19.
If our clinical trials or clinical protocols are delayed, suspended or terminated, we may be unable to develop our drug candidates on a timely basis, which would adversely affect our ability to obtain regulatory approvals, increase our development costs and delay or prevent commercialization of approved products.
We cannot predict whether we will encounter problems with any of our completed, ongoing or planned clinical trials or clinical data collection protocols that will cause regulatory agencies, institutional review boards or ethics committees, or us to delay our clinical trials or suspend or delay the analysis of the data from those trials. Clinical trials and clinical data protocols have been, and in the future can be, delayed for a variety of reasons, including:
● discussions with FDA, the EMA or other foreign authorities regarding the scope or design of our clinical trials or clinical data collection protocols;
● delays or the inability to obtain required approvals from institutional review boards, ethics committees or other responsible entities at clinical sites selected for participation in our clinical trials;
● delays in enrolling patients into clinical trials, collecting data from enrolled patients or collecting historical control data for any reason including disease severity, trial or data collection protocol design, study eligibility criteria, patient population size (e.g., for orphan diseases or for some pediatric indications), proximity and/or availability of clinical trial sites for prospective patients, availability of competing therapies and clinical trials, regional differences in diagnosis and treatment, perceived risks and benefits of the product or drug candidate, disruptions due to external events or conditions affecting the localities or regions in which our clinical trials are conducted, such as terrorism, political crises, natural disasters, war and wartime conditions, such as those in Ukraine, which has affected the operation of our clinical trials of OMS906, or outbreaks of contagious disease such as the COVID-19 pandemic, which previously slowed enrollment in our clinical trials of narsoplimab;
● lower than anticipated retention rates of patients in clinical trials;
● the need to repeat or conduct additional clinical trials as a result of inconclusive or negative results, failure to replicate positive early clinical data in subsequent clinical trials, failure to deliver an efficacious dose of a drug candidate, poorly executed testing, a failure of a clinical site to adhere to the clinical protocol or to follow GCPs or other study requirements, an unacceptable study design or other problems;
● adverse findings in clinical or nonclinical studies related to the safety of our drug candidates in humans;
● an insufficient supply of drug candidate materials or other materials necessary to conduct our clinical trials;
● the need to qualify new suppliers of drug candidate materials for FDA and foreign regulatory approval;
● an unfavorable inspection or review by FDA or other regulatory authority of a clinical trial site or records of any clinical investigation;
● the occurrence of unacceptable drug-related side effects or adverse events experienced by participants in our clinical trials;
● the suspension by a regulatory agency of a trial by imposing a clinical hold; or
● the amendment of clinical trial or data collection protocols to reflect changes in regulatory requirements and guidance or other reasons as well as subsequent re-examination of amendments to clinical trial or data collection protocols by regulatory agencies, institutional review boards or ethics committees.
In particular, because PNH and C3G, the indications for which our ongoing clinical trials are evaluating OMS906, are rare conditions, we have opened and expect to continue opening clinical sites in Ukraine and other countries that may be affected by armed conflict or political instability or that have not been traditionally established as centers for clinical research. Like Ukraine, some of these areas have been, and may continue to be, affected by such conflict, instability and/or health infrastructure challenges. Enrollment and retention of patients in, or the ability to receive results from, these clinical trials could be disrupted by the existing conditions in these areas or other geopolitical or macroeconomic developments. If patients withdraw from our trials, miss scheduled doses or follow-up visits or otherwise fail to follow trial protocols, if we are unable to resupply the drugs to clinical sites on schedule, or if our trial results are otherwise disrupted or disputed due to such conditions and developments, the integrity of data from our trials may be compromised or not accepted by FDA or other regulatory authorities, which would represent a significant setback for the development of this drug candidate.
In addition, our clinical trial or development programs have been, and in the future may be, suspended or terminated by us, FDA or other regulatory authorities, or institutional review boards or ethics committees due to a number of factors, including:
● failure to conduct the clinical trial in accordance with regulatory requirements or our clinical protocols;
● inspection of the clinical trial operations or trial sites by FDA or other regulatory authorities resulting in the imposition of a clinical hold;
● our failure to comply with our regulatory obligations as a sponsor of clinical research, such as adverse event reporting, control of study drug, adequate study monitoring, and other obligations;
● the failure to remove a clinical hold in a timely manner, if at all;
● unforeseen safety issues or any determination that a trial presents unacceptable health risks;
● inability to deliver an efficacious dose of a drug candidate; or
● lack of adequate funding to continue the clinical trial or development program, including as a result of unforeseen costs due to enrollment delays, requirements to conduct additional trials and studies and/or increased expenses associated with the services of our contract research organizations (“CROs”), or other third parties.
If the results of our clinical trials are not available when we expect or if we encounter any delay in the analysis of data from our clinical trials, we may be unable to file for regulatory approval or conduct additional clinical trials on the schedule we currently anticipate. Many of the factors that cause, or lead to, a delay in the commencement or completion of clinical trials may also ultimately lead to the denial of regulatory approval of a drug candidate. Any delays in completing our clinical trials could increase our development costs, could slow down our product development and regulatory submission process, could delay our receipt of product revenue and could make it difficult to raise additional capital. In addition, significant clinical trial delays also could allow our competitors to bring products to market before we do and impair our ability to commercialize our future products, potentially harming our business.
Because we have a number of drug candidates and development programs, we may expend our limited resources to pursue a particular drug candidate or indication and fail to capitalize on drug candidates or indications for which there is a greater likelihood of obtaining regulatory approval and that may be more profitable, if approved.
We have limited resources and must focus on the drug candidates and clinical and preclinical development programs that we believe are the most promising. As a result, we may forgo or delay the pursuit of opportunities with other drug candidates or other indications that later prove to have greater commercial potential and may not be able to progress development programs as rapidly as otherwise possible. Further, if we do not accurately evaluate the commercial potential or target market for a particular drug candidate, we may relinquish valuable rights to that drug through collaboration, license or other royalty arrangements in cases in which it would have been advantageous for us to retain sole development and commercialization rights.
Our drug candidates may not successfully complete clinical development or be suitable for successful commercialization or generation of revenue through partnerships, and our preclinical programs may not produce drug candidates that are suitable for clinical trials.
We must successfully complete preclinical testing, which may include demonstrating efficacy and the lack of toxicity in established animal models, before commencing clinical trials for any drug candidate. Many pharmaceutical and biological drug candidates do not successfully complete preclinical testing. There can be no assurance that positive results from preclinical studies will be predictive of results obtained from subsequent preclinical studies or clinical trials.
Even if preclinical testing is successfully completed, we cannot be certain that any drug candidates that do advance into clinical trials will successfully demonstrate safety and efficacy in clinical trials. Even if we achieve positive results in early clinical trials, they may not be predictive of the results in later trials, and safety and/or efficacy outcomes of early clinical trials may not be consistent with outcomes of subsequent clinical trials. There can be no assurance that we will be able to successfully commercialize our current or future drug candidates or to meet our expectations with respect to revenues or profits from such products.
We may incur substantial costs as a result of commercial disputes, claims, litigation or other legal proceedings relating to our business operations, especially with regard to patent and other intellectual property rights, and such costs or an adverse outcome in such a proceeding may adversely affect our financial condition, results of operations and/or stock price.
Our business involves numerous commercial contractual arrangements, important intellectual property rights, potential product liability, uncertainties with respect to clinical development, manufacture and regulatory approvals and other aspects that create heightened risks of disputes, claims and legal proceedings. These include claims that may be faced in one or more jurisdictions related to the safety of our drug candidates, the development of our drug candidates, our ability to obtain regulatory approval for our drug candidates, our expectations regarding product development and regulatory approval, sales and marketing practices, commercial disputes including with contract manufacturers, competition, environmental matters, employment matters and other matters. These matters could consume significant time and resources, even if we are successful. Many of our competitors and contractual counterparties are significantly larger than we are and, as a result, may be able to sustain the costs of complex litigation more effectively than we can because they have substantially greater resources. In addition, we may pay damage awards or settlements or become subject to equitable remedies that could, individually or in the aggregate, have a material negative effect on our financial condition, results of operations or stock price. Any uncertainties resulting from the initiation and continuation of any litigation also could have a material adverse effect on our ability to raise the capital necessary to continue our operations.
We may initiate or become subject to litigation regarding patents and other intellectual property rights. Patent infringement litigation involves many complex technical and legal issues and its outcome is often difficult to predict and the risk involved in doing so can be substantial. Manufacturers of generic or biosimilar drugs could seek approval to market a generic or biosimilar version of our products or challenge our intellectual property rights with respect to our drug candidates.
Further, our industry has produced a large number of patents and it is not always clear which patents cover various types of products or methods of use. A third party may claim that we or our contract manufacturers are using inventions covered by the third party’s patent rights and may go to court to stop us from engaging in the alleged infringing activity, including making, using or selling our drug candidates. These lawsuits are costly and could affect our results of operations and divert the attention of managerial and technical personnel. There is a risk that a court would decide that we, or our contract manufacturers, are infringing the third party’s patents and would order us or our contractors to stop the activities covered by the patents. In addition, if we or our contract manufacturers are found to have violated a third party’s patent, we or our contract manufacturers could be ordered to pay damages to the other party. We have agreed or may in the future agree to indemnify our contract manufacturers against certain patent infringement claims and thus may be responsible for any of their costs associated with such claims and actions. If we were sued for patent infringement, we would need to demonstrate that our drug candidates or methods of use either do not infringe the patent claims of the relevant patent or that the patent claims are invalid, and we might be unable to do this. Proving invalidity, in particular, is difficult since it requires clear and convincing evidence to overcome the presumption of validity enjoyed by issued patents.
It is difficult and costly to protect our intellectual property and our proprietary technologies, and we may not be able to ensure their protection.
Our commercial success will depend in part on obtaining and maintaining patent protection and trade secret protection for the use, formulation and structure of our drug candidates, the methods used to manufacture them, the related therapeutic targets and associated methods of treatment as well as on successfully defending these patents against potential third-party challenges. Our ability to protect our drug candidates from unauthorized making, using, selling, offering to sell or importing by third parties is dependent on the extent to which we have rights under valid and enforceable patents that cover these activities.
The patent positions of pharmaceutical, biotechnology and other life sciences companies can be highly uncertain and involve complex legal and factual questions for which important legal principles remain unresolved. Changes in either the patent laws or in interpretations of patent laws in the U.S. and other countries may diminish the value of our intellectual property. Further, the determination that a patent application or patent claim meets all of the requirements for patentability is a subjective determination based on the application of law and jurisprudence. The ultimate determination by the U.S. Patent and Trademark Office or by a court or other trier of fact in the U.S., or corresponding foreign national patent offices or courts, on whether a claim meets all requirements of patentability cannot be assured. Although we have conducted searches for third-party publications, patents and other information that may affect the patentability of claims in our various patent applications and patents, we cannot be certain that all relevant information has been identified. Accordingly, we cannot predict the breadth of claims that may be allowed or enforced in our patents or patent applications, in our licensed patents or patent applications or in third-party patents.
We cannot provide assurances that any of our patent applications will be found to be patentable, including over our own prior art patents, or will issue as patents. Neither can we make assurances as to the scope of any claims that may issue from our pending and future patent applications nor to the outcome of any proceedings by any potential third parties that could challenge the patentability, validity or enforceability of our patents and patent applications in the U.S. or foreign jurisdictions. Any such challenge, if successful, could limit patent protection for our drug candidates and/or materially harm our business.
The degree of future protection for our proprietary rights is uncertain because legal means afford only limited protection and may not adequately protect our rights or permit us to gain or keep our competitive advantage. In addition, to the extent that we are unable to obtain and maintain patent protection for one of our drug candidates or in the event that such patent protection expires or is limited to method of use patent protection, it may no longer be cost-effective to extend our portfolio by pursuing additional development of a product or drug candidate for follow-on indications.
We also may rely on trade secrets to protect our technologies or drug candidates, especially where we do not believe patent protection is appropriate or obtainable. Although we use reasonable efforts to protect our trade secrets, our employees, consultants, contractors, outside scientific collaborators and other advisers may unintentionally or willfully disclose our information to competitors. Enforcing a claim that a third-party entity illegally obtained and is using any of our trade secrets is expensive and time-consuming, and the outcome is unpredictable. In addition, courts outside the U.S. are sometimes less willing to protect trade secrets. Moreover, our competitors may independently develop equivalent knowledge, methods and know-how.
Our indebtedness and liabilities could limit the cash flow available for our operations and expose us to risks that could adversely affect our business, financial condition and results of operations.
As of December 31, 2023, we had $213.2 million total aggregate principal amount of our 2026 Notes outstanding, and we had approximately $1.3 million of outstanding finance lease obligations. We may incur additional indebtedness to meet future financing needs. Our existing and future indebtedness could have significant negative consequences for our security holders and our business, results of operations and financial condition by, among other things:
● requiring a substantial portion of our cash flow from operations to service and repay our indebtedness, which will reduce the amount of cash available for other purposes;
● limiting our ability to obtain additional financing;
● limiting our flexibility to plan for, or react to, changes in our business; ● diluting the interests of our existing stockholders as a result of issuing shares of our common stock upon any conversion of the Convertible Notes;
● placing us at a possible competitive disadvantage with competitors that are less leveraged than we are or have better access to capital; and
● increasing our vulnerability to adverse economic and industry conditions.
Our ability to make scheduled payments of the principal of, to pay interest on, or to refinance our indebtedness, including the Convertible Notes, depends on our future performance, which is subject to many factors, including, economic, financial, competitive and other circumstances beyond our control. Our business may not generate sufficient funds, and we may otherwise be unable to maintain sufficient cash reserves, to pay amounts due under our indebtedness, including the Convertible Notes, and our cash needs may increase in the future. In addition, future indebtedness that we may incur may contain financial and other restrictive covenants that limit our ability to operate our business, raise capital or make payments under our other indebtedness. If we fail to comply with these covenants or to make payments under our indebtedness when due, then we would be in default under that indebtedness, which could, in turn, result in that and our other indebtedness becoming immediately payable in full.
Competitors may develop products that are less expensive, safer or more effective, or which may otherwise diminish or eliminate the success of any products that we may commercialize.
We may not achieve commercial success if our competitors, many of which have significantly more resources and experience than we have, market products that are safer, more effective, less expensive or faster to reach the market than any products that we may develop and commercialize. Our competitors also may market a product that proves to be unsafe or ineffective, which may affect the market for future product we are developing, regardless of the safety or efficacy of our product. The failure of any future product that we may market to compete effectively with products marketed by our competitors would impair our ability to generate revenue, which would have a material adverse effect on our future business, our financial condition and our results of operations.
The loss of members of our management team could substantially disrupt our business operations.
Our success depends to a significant degree on the continued individual and collective contributions of our management team. The members of our management team are at-will employees, and we do not maintain any key-person life insurance policies other than on the life of Gregory A. Demopulos, M.D., our president, chief executive officer and chairman of the board of directors. Losing the services of any key member of our management team, whether from death or disability, retirement, competing offers or other causes, without having a readily available and appropriate replacement could delay the execution of our business strategy, cause us to lose a strategic partner, or otherwise materially affect our operations.
We rely on highly skilled personnel and, if we are unable to retain or motivate key personnel or hire qualified personnel, we may not be able to maintain our operations or grow effectively.
Our performance is largely dependent on the talents and efforts of highly skilled individuals, many of whom possess specialized expertise that may be difficult to replace. Our future success depends on our continuing ability to identify, hire, develop, motivate and retain highly skilled personnel for all areas of our organization. If we are unable to hire and train a sufficient number of qualified employees for any reason, we may not be able to implement our current initiatives or grow effectively. We maintain a rigorous, highly selective and time-consuming hiring process. We believe that our approach to hiring has significantly contributed to our success to date. If we do not succeed in attracting qualified personnel and retaining and motivating existing personnel, our existing operations may suffer and we may be unable to grow effectively.
We may encounter difficulties managing our growth, which could delay our business plans or adversely affect our results of operations.
To manage our future growth, we must continue to implement and improve our managerial, operational and financial systems and continue to recruit, train and retain qualified personnel. We may not be able to implement necessary business processes and systems, recruit, train and retain additional qualified personnel and otherwise manage the growth of our enterprise due to factors such as limited financial resources and competition for qualified personnel within local, national and international markets. The expansion of our operations may lead to significant costs and may divert our management and business development resources. Any inability to manage growth could delay the execution of our business plans or disrupt our operations. Additionally, our inability to manage growth effectively could cause our operating costs to exceed our forecasts.
Product liability claims may damage our reputation and, if insurance proves inadequate, these claims may harm our business.
We may be exposed to the risk of product liability claims that is inherent in the biopharmaceutical industry. A product liability claim may damage our reputation by raising questions about our product’s safety and efficacy and could limit our ability to sell one or more products by preventing or interfering with commercialization of our drug candidates. In addition, product liability insurance for the biopharmaceutical industry is generally expensive to the extent it is available at all. There can be no assurance that we will be able to obtain or maintain such insurance on acceptable terms for any product we bring to market. Further, our product liability insurance coverage may not provide coverage for or may be insufficient to reimburse us for any or all expenses or losses we may suffer. A successful claim against us with respect to uninsured liabilities or in excess of insurance coverage could have a material adverse effect on our business, financial condition and results of operations.
We rely on third parties to conduct portions of our preclinical research and clinical trials. If these third parties do not perform as contractually required or otherwise expected, or if we fail to adequately supervise or monitor these parties, we may not be able to obtain regulatory approval for or commercialize our drug candidates.
We rely on third parties, such as CROs, medical and research institutions and clinical investigators, to conduct a portion of our preclinical research, assist us in conducting our clinical trials or to conduct third party-sponsored clinical trials of our drug candidates. Nonetheless, we are responsible for confirming that our preclinical research and clinical trials are conducted in accordance with applicable regulations, the relevant trial protocol and within the context of approvals by an institutional review board or ethics committee, and we may not always be successful in ensuring such compliance. Our reliance on these third parties does not relieve us of responsibility for ensuring compliance with FDA and other regulations and standards for conducting, monitoring, recording and reporting the results of preclinical research and clinical trials to assure that data and reported results are credible and accurate and that the trial participants are adequately protected. If these third parties do not successfully carry out their contractual duties or regulatory obligations or meet expected deadlines, if the third parties need to be replaced or if the quality or accuracy of the data they obtain is compromised due to their failure to adhere to our clinical protocols or regulatory requirements or for other reasons, our preclinical and clinical development processes may be extended, delayed, suspended or terminated, and we may not be able to commercialize or obtain regulatory approval for our drug candidates.
If we are unable to obtain licenses from third parties on commercially reasonable terms or fail to comply with our obligations under such agreements, our business could be harmed.
It may be necessary for us to use the patented or proprietary technology of third parties to commercialize our products, in which case we would be required to obtain a license from these third parties. If we are unable to license such technology, or if we are forced to license such technology on unfavorable terms, our business could be materially harmed. If we are unable to obtain a necessary license, we may be unable to develop or commercialize the affected products or product candidates, which could materially harm our business and the third parties owning such intellectual property rights could seek either an injunction prohibiting our sales, or, with respect to our sales, an obligation on our part to pay royalties and/or other forms of compensation. Even if we are able to obtain a license, it may be non-exclusive, which could enable our competitors to obtain access to the same technologies licensed to us.
If we fail to comply with our obligations under license agreements, our counterparties may have the right to terminate these agreements, in which event we might not be able to develop, manufacture or market, or may be forced to cease developing, manufacturing or marketing, any product that is covered by these agreements or may face other penalties under such agreements. Such an occurrence could materially adversely affect the value of the product or product candidate being developed under any such agreement. Termination of these agreements or reduction or elimination of our rights under these agreements may result in our having to negotiate new or reinstated agreements with less favorable terms, cause us to lose our rights under these agreements, including our rights to important intellectual property or technology, or impede, delay or prohibit the further development or commercialization of one or more product candidates that rely on such agreements.
As a non-accelerated filer, we are no longer required to comply with the auditor attestation requirements of the Sarbanes-Oxley Act.
As of December 31, 2023, we are a non-accelerated filer under the Exchange Act and, therefore, we are no longer required to comply with the auditor attestation requirements of Section 404(b) of the Sarbanes-Oxley Act. Therefore, our internal controls over financial reporting will not receive the level of review provided by the process relating to the auditor attestation included in annual reports of issuers that are subject to the auditor attestation requirements. In addition, we cannot predict if investors will find our common stock less attractive because we are not required to comply with the auditor attestation requirements. If some investors find our common stock less attractive as a result, there may be a less active trading market for our common stock and the trading price for our common stock may be negatively affected.
Our share repurchase program could affect the price of our common stock and increase volatility and may be suspended or terminated at any time, which may result in a decrease in the trading price of our common stock.
In November 2023, our board of directors authorized a share repurchase program to repurchase, from time to time, up to $50.0 million of our outstanding shares of common stock in the open market, including under trading plans established pursuant to Rule 10b5-1 and Rule 10b-18 under the Exchange Act, or in privately negotiated transactions. The share repurchase program does not have a fixed expiration date, may be suspended or discontinued at any time, and does not obligate us to acquire any amount of our common stock. The timing, manner, price, and amount of any repurchases may be determined by us at our discretion and will depend on a variety of factors, including business, economic and market conditions, prevailing stock prices, corporate and regulatory requirements, and other considerations. As of March 26, 2024, approximately $33.8 million remained available to repurchase of our outstanding shares of common stock under the share repurchase program.
Repurchases pursuant to our share repurchase program could affect our stock price and increase its volatility. The existence of a share repurchase program could also cause our stock price to be higher than it would be in the absence of such a program and could potentially reduce the market liquidity for our common stock. There can be no assurance that any repurchases will enhance shareholder value, because the market price of our common stock may decline below the levels at which we repurchased our common stock. Although our share repurchase program is intended to enhance long-term shareholder value, short-term stock price fluctuations could reduce the share repurchase program’s effectiveness.
General Risk Factors Related to our Business
Cyber-attacks or other failures in telecommunications or information technology systems could result in information theft, data corruption and significant disruption of our business operations.
We utilize information technology systems and networks to process, transmit and store electronic information in connection with our business activities. As use of digital technologies has increased, cyber incidents, including deliberate attacks and attempts to gain unauthorized access to computer systems and networks, have increased in frequency and sophistication. These threats pose a risk to the security of our systems and networks, the confidentiality and the availability and integrity of our data. There can be no assurance that we will be successful in preventing cyber-attacks or mitigating their effects. Similarly, there can be no assurance that our collaborators, CROs, third-party logistics providers, distributors and other contractors and consultants will be successful in protecting our clinical and other data that is stored on their systems. While we have not experienced any previous cybersecurity incidents that have had a material adverse effect on or company, we cannot provide assurance that a future cybersecurity incident will not occur or that it would not materially affect our business, results of operations or financial condition. In addition, we may suffer reputational harm or face litigation or adverse regulatory action as a result of cyber-attacks or other data security breaches and may incur significant additional expense to implement further data protection measures.
Our stock price has been and may continue to be volatile, and the value of an investment in our common stock may decline.
During the 12-month period ended December 31, 2023, the closing price of our stock ranged from as high as $7.57 per share and as low as $1.08 per share. The trading price of our common stock is likely to continue to be highly volatile and could be subject to wide fluctuations in response to numerous factors, many of which are beyond our control. In addition, the stock market has experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of publicly traded companies. Broad market and industry factors may seriously affect the market price of companies’ stock, including ours, regardless of actual operating performance. These fluctuations may be even more pronounced in the trading market for our stock. In addition, in the past, following periods of volatility in the overall market and the market price of a particular company’s securities, securities class action litigation has often been instituted against these companies. This litigation, if instituted against us, could result in substantial costs and a diversion of our management’s attention and resources.
If we issue additional shares of our common stock or other securities that may be convertible into, or exercisable or exchangeable for, our common stock, our existing shareholders would experience further dilution.
To the extent that we raise additional funds in the future by issuing equity securities, our shareholders would experience dilution, which may be significant and could cause the market price of our common stock to decline significantly. In addition, approximately 15.3 million shares of common stock were subject to outstanding options as of December 31, 2023 and may become eligible for sale in the public market to the extent permitted by the provisions of various vesting agreements. As of December 31, 2023, we also had approximately 8.8 million additional shares of common stock reserved for future issuance under our employee benefit plans that are not subject to outstanding options. Further, to the extent we issue common stock upon conversion of the Convertible Notes, such conversion would dilute the ownership interests of existing stockholders despite the expected reduction of such dilution as a result of the capped call transactions that we entered into in connection with the original issuances of the Convertible Notes. If the holders of outstanding options or warrants elect to exercise some or all of them, or if the shares subject to our employee benefit plans are issued and become eligible for sale in the public market, or we issue common stock upon conversion of the Convertible Notes, our shareholders would experience dilution and the market price of our common stock could decline.
If we or the third parties upon whom we rely are adversely affected by natural disasters or other events, our business continuity and disaster recovery plans may not adequately protect us from such interruptions.
Any unplanned event, such as flood, fire, explosion, earthquake, tsunami, extreme weather condition, power shortage, power outage, telecommunication failure, or other natural or man-made accidents or incidents could disrupt our operations. If a natural disaster or other event were to occur that prevents us from using all or a significant portion of our headquarters, that damages critical infrastructure, such as the manufacturing facilities of our third-party manufacturers, or that otherwise disrupts operations, it may be difficult or, in certain cases, impossible for us to continue our business for a substantial period of time. We may not carry sufficient business interruption insurance to compensate us for all losses that may occur. The disaster recovery and business continuity plans we have in place may not be adequate in the event of a serious disaster or similar event. We may incur substantial expenses as a result of a natural disaster or other event, which could have a material adverse effect on our business, and we could potentially lose valuable data and other items. The occurrence of any of the foregoing could have a material adverse effect on our business, financial condition and results of operations.
Anti-takeover provisions in our charter documents and under Washington law could make an acquisition of us, which may be beneficial to our shareholders, difficult and prevent attempts by our shareholders to replace or remove our current management.
Provisions in our articles of incorporation and bylaws and under Washington law may delay or prevent an acquisition of us or a change in our management. These provisions include a classified board of directors, a prohibition on shareholder actions by less than unanimous written consent, restrictions on the ability of shareholders to fill board vacancies and the ability of our board of directors to issue preferred stock without shareholder approval. In addition, because we are incorporated in Washington, we are governed by the provisions of Chapter 23B.19 of the Washington Business Corporation Act, which, among other things, restricts the ability of shareholders owning 10% or more of our outstanding voting stock from merging or combining with us. Although we believe these provisions collectively provide for an opportunity to receive higher bids by requiring potential acquirers to negotiate with our board of directors, they would apply even if an offer may be considered beneficial by some shareholders. In addition, these provisions may frustrate or prevent any attempts by our shareholders to replace or remove our current management by making it difficult for shareholders to replace members of our board of directors, which is responsible for appointing the members of our management.
We have never declared or paid dividends on our capital stock, and we do not anticipate paying dividends in the foreseeable future.
Our business requires significant funding. We currently plan to invest all available funds and future earnings, if any, in the development and growth of our business. Therefore, we currently do not anticipate paying any cash dividends on our common stock in the foreseeable future. As a result, a rise in the market price of our common stock, which is uncertain and unpredictable, will be the sole source of potential gain for shareholders in the foreseeable future, and an investment in our common stock for dividend income should not be relied upon.
ITEM 1B. UNRESOLVED STAFF COMMENTS
None.
Risk Management and Strategy
Omeros maintains a cybersecurity risk management program that is designed to assess, identify, manage and respond to risks from cybersecurity threats in a robust manner. This program shares certain common methodologies, reporting channels and governance processes applicable to our management of other risk areas, such as legal, compliance, strategic, operational and financial risk.
We utilize a range of internal and external resources to assess and identify cybersecurity threats and vulnerabilities. We access and utilize information drawn from a range of publications, reports and services to assess our cybersecurity risk profile, develop awareness of emerging cybersecurity threats and threat actors and identify risk factors that are particularly relevant to the biotechnology and pharmaceutical sector and to our company. We also work with third parties that assist us to identify, assess and manage cybersecurity risks, including external auditors, consulting firms, managed security service providers and penetration testing firms.
We have implemented and maintain various technical, physical and organizational measures, processes, standards and/or policies designed to manage and mitigate material risks from cybersecurity threats. These include data encryption, network security controls, access controls, physical security, asset management, system hardening, vulnerability management and patching and continuous monitoring of information technology systems and network telemetry data using a variety of manual and automated tools and systems designed to detect and respond to suspicious or unusual activity. We maintain systems and plans for business continuity and disaster recovery and have implemented tools and procedures for cybersecurity incident detection and response. We also operate a cybersecurity training program for employees that includes required webinars and deployment of simulated phishing and similar attacks in which threat actors utilize social engineering to gain access to company systems.
We take a risk-weighted approach to mitigation of cybersecurity risks associated with use of third-party service providers. Based on an assessment of the cybersecurity risks presented by a given third-party service provider, we seek to minimize third-party cybersecurity risk on a case-by-case basis, generally through a combination of due diligence in the selection of qualified vendors and the imposition of contractual terms requiring the vendor to maintain specified cybersecurity safeguards and/or to accept financial responsibility for security breaches occurring within the vendor’s area of responsibility.
We are not aware of any specific risks from specific cybersecurity threats, and have not experienced any previous cybersecurity incidents, that have materially affected or are reasonably likely to materially affect our company, including our business strategy, results of operations or financial condition. While we continue to invest in the security and resiliency of our information technology systems and to enhance our cybersecurity controls and processes, we cannot provide assurance that a future cybersecurity incident will not occur or that it would not materially affect our company. Please see Item 1A of Part I of this Annual Report under the heading “Risk Factors” for additional discussion about risks related to cybersecurity.
Governance
Cybersecurity is an important part of our risk management processes and an area of focus for our board of directors and management. Pursuant to its charter, the audit committee of our board of directors is responsible for the oversight of management’s efforts to address cybersecurity risk. Management reports to the audit committee on cybersecurity risk matters periodically, typically twice annually. These reports normally address matters such as: the evolving cybersecurity risk environment and the emergence of new threats; outcomes and learnings from penetration testing, security audits or vulnerability assessments; evaluation of existing controls, tools and procedures and progress on implementation of any new initiatives to manage and mitigate cybersecurity risk. In addition, members of our board of directors regularly engage in discussions with management on cybersecurity-related news events and discuss any updates to our cybersecurity risk management and strategy programs.
Our cybersecurity risk management program is managed by our Director of Information Technology (the “IT Director”), whose team is responsible for leading enterprise-wide cybersecurity strategy, policy, standards, architecture and processes. The IT Director has been with the organization since 2007, has a post-graduate degree in Information Security, and is a member of InfraGard, a partnership between the Federal Bureau of Investigation and members of the private sector for the protection of U.S. critical infrastructure. The IT Director is informed about and monitors prevention, detection, mitigation and remediation of cybersecurity risks and incidents through various means, which may include, among other things, briefings with dedicated internal security personnel, threat intelligence and other information obtained from governmental, public or private sources, including external consultants engaged by us, and alerts and reports produced by security tools deployed in our information technology environment. The IT Director provides periodic reports on cybersecurity risk to the audit committee of our board of directors, as well as our chief executive officer and other members of our senior management as appropriate.
We lease approximately 113,060 square feet for our principal office and laboratory space in the building located at 201 Elliott Avenue West, Seattle, Washington (“the Omeros Building”), which includes 7,245 square feet of laboratory space that we are subleasing to third parties. The lease term for our space is through November 2027. We also have two options to extend the lease term, each by five years. The annual base rent due under the lease for our principal office and laboratory space is $7.0 million for 2024, $7.1 million for 2025, $6.9 million for 2026, and $5.9 million for 2027. In addition, we are responsible for paying our proportionate share of the building’s utilities, taxes, insurance and maintenance as well as a property management fee.
We believe that our facilities are sufficient for our anticipated near-term needs.
From time to time, in the ordinary course of business, we may be involved in various claims, lawsuits and other proceedings. As of the date of filing of this Annual Report on Form 10-K, we were not involved in any material legal proceedings.
ITEM 4. MINE SAFETY DISCLOSURES Our common stock is traded on The Nasdaq Global Market under the symbol “OMER.”
Not applicable.
ITEM 5. MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED SHAREHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Market Information
Holders
As of March 28, 2024, there were approximately 57,942,695 shares of our common stock outstanding, which were held by 84 holders of record.
Dividends
We have never declared or paid any cash dividends on our capital stock. We expect to retain all available funds and future earnings to fund the development and growth of our business and we do not anticipate paying any cash dividends in the foreseeable future.
Recent Sales of Unregistered Securities
We did not sell any equity securities that were not registered under the Securities Act during the three fiscal years ended December 31, 2023.
Stock Performance Graph
The following graph compares the cumulative total shareholder return for our common stock (OMER), the Nasdaq Biotechnology Index (NBI) and the Nasdaq U.S. Benchmark TR Index (NQUSBT) for the period beginning December 31, 2018 and ending December 31, 2023. This graph assumes that $100 was invested on December 31, 2018 in our common stock, the Nasdaq Biotechnology Index and the Nasdaq U.S. Benchmark TR Index. It also assumes that any dividends were reinvested. The data shown in the following graph are not necessarily indicative of future stock price performance.
Comparison of 5 Year Cumulative Return
Assumes Initial Investment of $100
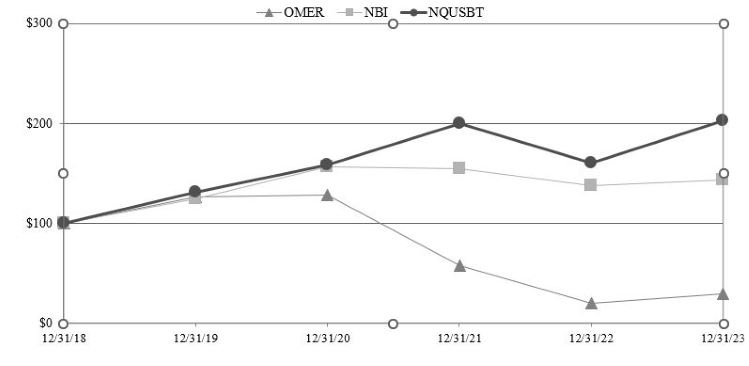
The foregoing information shall not be deemed to be “soliciting material” or to be “filed” for purposes of Section 18 of the Exchange Act or otherwise subject to liability under that Section. In addition, the foregoing information shall not be deemed to be incorporated by reference into any of our filings under the Exchange Act or the Securities Act, except to the extent that we specifically incorporate this information by reference.
Issuer Purchases of Equity Securities
The following table provides information regarding our repurchases of our common stock during the quarter ended December 31, 2023:
| Period |
Total Number of Shares Purchased |
Average Price Paid per Share |
Total Number of Shares Purchased as Part of Publicly Announced Plans or Programs (1) |
Maximum Approximate Dollar Value of Shares That May Yet Be Purchased Under the Plans or Programs (1) |
||||||||||||
| (In thousands) |
||||||||||||||||
| 10/01/23 – 10/31/23 |
— | $ | — | — | $ | — | ||||||||||
| 11/01/23 – 11/30/23 |
579,387 | 1.98 | 579,387 | 48,852 | ||||||||||||
| 12/01/23 – 12/31/23 |
1,224,757 | 2.80 | 1,224,757 | 45,424 | ||||||||||||
| Total |
1,804,144 | $ | 2.54 | 1,804,144 | ||||||||||||
____________
| (1) |
On November 9, 2023, our board of directors approved an indefinite term share repurchase program under which we may repurchase from time to time up to $50.0 million of our common stock in the open market, including under trading plans established pursuant to Rule 10b5-1 and Rule 10b-18 under the Exchange Act, or in privately negotiated transactions. As of March 26, 2024, approximately $33.8 million remained available for repurchase of our outstanding shares of common stock under the share repurchase program. |
ITEM 7. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
The following discussion and analysis should be read in conjunction with the audited annual consolidated financial statements and the related notes thereto included elsewhere in this Annual Report on Form 10-K. This discussion contains forward-looking statements reflecting our current expectations that involve risks and uncertainties. Actual results may differ materially from those discussed in these forward-looking statements due to a number of factors, including those set forth in the section entitled “Risk Factors” and elsewhere in this Annual Report on Form 10-K. For further information regarding forward-looking statements, please refer to the special note regarding forward-looking statements at the beginning of this Annual Report on Form 10-K. Throughout this discussion, unless the context specifies or implies otherwise, the terms “Company,” “we,” “us” and “our” refer to Omeros Corporation and our wholly owned subsidiaries.
Overview
We are a clinical-stage biopharmaceutical company committed to discovering, developing and commercializing small-molecule and protein therapeutics for large-market as well as orphan indications targeting immunologic disorders including complement-mediated diseases, cancers related to the dysfunction of the immune system, and addictive and compulsive disorders.
Complement Inhibitor Programs
The complement system plays a role in the body’s inflammatory response and becomes activated as a result of tissue damage or trauma or microbial pathogen invasion. Inappropriate or uncontrolled activation of the complement system can cause diseases characterized by serious tissue injury. Three main pathways can activate the complement system: classical, lectin, and alternative. Omeros is focused on development of therapeutics to treat diseases associated with the lectin and/or alternative pathways of complement. Omeros is developing antibodies as well as small-molecule inhibitors of key enzymes known to be centrally involved in the in activation of the targeted pathway of complement.
Lectin Pathway / MASP 2
MASP-2, is a novel pro-inflammatory protein target that is the effector enzyme of the lectin pathway and is required for the function of this pathway. Omeros is developing antibodies and small-molecule inhibitors of MASP-2 as potential therapeutics for diseases in which the lectin pathway has been shown to contribute to significant tissue injury and pathology. When not treated, these diseases are typically characterized by significant end-organ damage, such as kidney or central nervous system injury. Importantly, inhibition of MASP-2 has been demonstrated not to interfere with the antibody-dependent classical complement activation pathway, a critical component of the acquired immune response to infection.
The lead drug candidate in our MASP-2 inhibitor program is narsoplimab (OMS721), a proprietary, patented human monoclonal antibody targeting MASP-2, the effector enzyme of the lectin pathway of complement. Clinical development of narsoplimab is currently focused primarily on TA-TMA and development efforts are also directed to COVID-19, ARDS and PASC. We are also developing OMS1029, a long-acting, next-generation antibody targeting MASP-2 and the lectin pathway which we expect will be well-suited to indications requiring long-term, chronic administration. In addition, we are advancing our orally administered small-molecule MASP-2 inhibitor through IND-enabling studies. For more information, see Part I, Item 1 in this Annual Report on Form 10-K under the heading “Complement Inhibitor Programs: MASP-2 Program – Lectin Pathway Disorders”.
Alternative Pathway / MASP-3
Our pipeline of clinical-stage complement-targeted therapeutic candidates also includes OMS906, a proprietary, patented monoclonal antibody targeting MASP-3, the key activator of the alternative pathway of complement. We believe OMS906 has the potential to treat a wide range of alternative pathway-related diseases and that its attributes favorably differentiate OMS906 from other marketed and in-development alternative pathway inhibitors. Clinical development of OMS906 is currently focused on rapidly advancing to Phase 3 clinical trials in multiple alternative pathway-related disorders, including PNH and C3G. We have multiple ongoing Phase 2 clinical trials evaluating OMS906 in these indications. For more information, see Part I, Item 1 in this Annual Report on Form 10-K under the heading “Complement Inhibitor Programs: MASP-3 Program – Alternative Pathway Disorders”.
PDE7 Inhibitor Programs
Our PDE7 inhibitor program, which we refer to as OMS527, comprises multiple PDE7 inhibitor compounds and is based on our discoveries of previously unknown links between PDE7 and any addiction or compulsive disorder, and between PDE7 and any movement disorders. In April 2023, we were awarded a grant from the National Institute on Drug Abuse, part of the National Institutes of Health, to develop our lead orally administered PDE7 inhibitor compound, for which we have successfully completed a Phase 1 study, for the treatment of cocaine use disorder (“CUD”). The grant amount, a total of $6.69 million over three years, is intended to support preclinical cocaine interaction/toxicology studies to assess safety of the therapeutic candidate in the presence of concomitant cocaine administration, as well as an in-patient, placebo-controlled clinical study evaluating the safety and effectiveness of OMS527 in adults with CUD who receive concurrent intravenous cocaine. The preclinical study is intended to provide the toxicology data necessary to support the human study of OMS527 in CUD. The toxicology study is underway and is expected to be completed in late 2024. Additionally, with investigators at Emory University, we are also evaluating OMS527 as a potential treatment for levodopa-induced dyskinesia, a common and debilitating side effect of long-term levodopa dosing in patients with Parkinson’s disease. For more information, see Part I, Item 1 in this Annual Report on Form 10-K under the heading “Other Clinical Programs: PDE7 Inhibitor Programs – OMS527”.
Pre-clinical Programs
We are advancing preclinical research on potential molecular and cellular therapies for cancer. On the molecular front, we have developed novel biologic platforms to target cancer cells specifically and kill them directly or indirectly through the potentiation of the immune system. Our novel molecules combine tumor antigens with a potent adjuvant and show high levels of killing in cancer cells. We believe that some of these molecules could function as therapeutic vaccines against a broad range of tumors, potentially transforming treatment of both solid tumors and hematologic cancers. On the cellular front, we are evaluating novel approaches for both adoptive T cell and CAR T therapies. We have identified specific T cell signaling pathways, which, once inhibited, significantly and preferentially enhance the expansion of memory T cells that distinctively recognize and efficiently kill tumor cells. We continue to develop and validate our novel approach, which we believe could improve response rates for patients receiving either engineered or native T cell therapies for liquid or solid tumors.
OMIDRIA Sale and Royalty Monetization Transactions
We previously developed and commercialized OMIDRIA® (phenylephrine and ketorolac intraocular solutions) 1%/0.3%, which is approved by FDA for use during cataract surgery or intraocular ("IOL") replacement to maintain pupil size by preventing intraoperative miosis (pupil constriction) and to reduce postoperative ocular pain. We marketed OMIDRIA in the U.S. from the time of its commercial launch in 2015 until December 2021.
On December 23, 2021, we sold our commercial product OMIDRIA and certain related assets, including inventory and prepaid expenses, to Rayner. Rayner paid us $126.0 million in cash at the closing and we retained all outstanding accounts receivable, accounts payable, and accrued expenses as of the closing date.
Under the Asset Purchase Agreement, we were entitled to receive a $200.0 million Milestone Payment within 30 days following an event (the "Milestone Event") that establishes separate payment for OMIDRIA for a continuous period of at least four years when furnished in the ambulatory surgery center ("ASC") setting. The Milestone Event occurred in December 2022 and we recorded a $200.0 milestone receivable. We received the Milestone Payment together with accrued interest in February 2023.
Under the Asset Purchase Agreement, the occurrence of the Milestone Event in December 2022 triggered a reduction in the U.S. royalty rate from 50% to 30% on OMIDRIA net sales until the expiration or termination of the last issued and unexpired U.S. patent, which we expect to occur no earlier than 2035. Upon the occurrence of certain events described in the Asset Purchase Agreement, including during any specific period in which OMIDRIA is no longer eligible for certain separate payment (i.e., becomes included in the packaged payment rate for the surgical procedure) under Medicare Part B, the U.S. base royalty rate would be reduced to 10%. Pursuant to legislation enacted in late 2022, we expect separate payment for OMIDRIA under Medicare Part B to extend until at least January 1, 2028.
As a result of the OMIDRIA divestiture, the results of OMIDRIA operations have been reclassified to net income from discontinued operations, net of tax in our consolidated statements of operations and comprehensive income (loss) and excluded from continuing operations for all periods presented.
On September 30, 2022, we sold to DRI an interest in a portion of our future OMIDRIA royalty receipts and received $125.0 million in cash consideration which we recorded as an OMIDRIA royalty obligation on our consolidated balance sheet. Interest expense is recorded as a component of continuing operations. The aggregate amount of royalties to which DRI is entitled under this arrangement is capped at $188.4 million.
On February 1, 2024, we sold to DRI an expanded interest in the OMIDRIA royalties pursuant to the terms of an amended and restated royalty purchase agreement dated February 1, 2024 (the “Amendment”). We received $115.5 million in cash upon closing of the Amendment. The Amendment eliminated the caps on royalty payments effective beginning in the first quarter of 2024, and provides that DRI will now receive all royalties on U.S. net sales of OMIDRIA payable between January 1, 2024 and December 31, 2031. DRI is entitled to payment only to the extent of royalty payments that are payable on U.S. net sales of OMIDRIA on or before December 31, 2031 and DRI has no recourse to our assets other than its interest in the OMIDRIA royalties. Omeros retains the right to receive all royalties payable by Rayner on any net sales of OMIDRIA outside the U.S. payable from and after January 1, 2024, as well as all royalties on global net sales of OMIDRIA payable from and after December 31, 2031. In addition to the cash consideration received at closing, the Amendment also entitles us to receive a milestone payment ranging between $10.0 million and $27.5 million if U.S. net sales of OMIDRIA reach applicable thresholds ranging between a total of $156.0 million and $160.0 million for any period of four consecutive quarters ending prior to January 1, 2026 as well as a separate milestone payment ranging between $8.0 million and $27.5 million if U.S. net sales of OMIDRIA reach applicable thresholds ranging between a total of $181.0 million and $185.0 million for any period of four consecutive quarters ending prior to January 1, 2028. See Part II, Item 8, “Note 8 – OMIDRIA Royalty Obligation” to our Consolidated Financial Statements in this Annual Report on Form 10-K for additional information.
As of December 31, 2023, we had cash, cash equivalents and short-term investments of $171.8 million and, in February 2024, we received $115.5 million from DRI.
Results of Operations
Research and Development Expenses
Our research and development expenses can be divided into three categories: direct external expenses, which include clinical research and development and preclinical research and development activities; internal, overhead and other expenses; and stock-based compensation expense. Direct external expenses consist primarily of expenses incurred pursuant to agreements with third-party manufacturing organizations prior to receiving regulatory approval for a drug candidate, CROs, clinical trial sites, collaborators, licensors and consultants. Pre-clinical research and development includes costs prior to beginning Phase 1 studies in human subjects. Internal, overhead and other expenses primarily consist of costs for personnel, overhead, rent, utilities and depreciation. The discontinued operations of OMIDRIA relates to the costs of drug manufacturing stability, quality control testing and costs of employees and consultants. The following table illustrates our expenses associated with these activities:
| Year Ended |
||||||||||||
| Year Ended December 31, |
||||||||||||
| 2023 |
2022 |
2021 |
||||||||||
| (In thousands) |
||||||||||||
| Continuing research and development expenses: |
||||||||||||
| Direct external expenses: |
||||||||||||
| Clinical research and development: |
||||||||||||
| MASP-2 program - OMS721 (narsoplimab) |
$ | 35,352 | $ | 50,408 | $ | 48,806 | ||||||
| MASP-3 program - OMS906 |
22,853 | 6,304 | 7,005 | |||||||||
| MASP-2 program - OMS1029 |
6,249 | 2,687 | — | |||||||||
| Other |
153 | 442 | 555 | |||||||||
| Total clinical research and development |
64,607 | 59,841 | 56,366 | |||||||||
| Preclinical research and development |
5,172 | 7,254 | 15,031 | |||||||||
| Total direct external expenses |
69,779 | 67,095 | 71,397 | |||||||||
| Internal, overhead and other expenses |
40,337 | 39,503 | 40,587 | |||||||||
| Stock-based compensation expenses |
4,754 | 6,123 | 6,791 | |||||||||
| Total continuing research and development expenses |
114,870 | 112,721 | 118,775 | |||||||||
| Discontinued research and development expenses |
— | — | 3,839 | |||||||||
| Total research and development expenses |
$ | 114,870 | $ | 112,721 | $ | 122,614 | ||||||
Clinical research and development expenses increased $4.8 million between 2023 and 2022. The $16.5 million increase in OMS906 development costs was due to an increase in manufacturing and Phase 2 clinical trial costs and a $5.0 million development milestone paid in 2023 under a technology license agreement. The $3.6 million increase in OMS1029 expense was primarily due to costs associated with initiation of human trials and other clinical development costs, i.e. the transition from preclinical to clinical development status in the third quarter of 2022. These increases were offset by decreased narsoplimab manufacturing costs during 2023.
The $3.5 million increase in clinical research and development costs between 2022 and 2021 was primarily due to the advancement of OMS1029 from preclinical status to clinical research and development status on initiation of the Phase 1 clinical trial in the third quarter of 2022. Additionally, we incurred increased narsoplimab drug manufacturing costs in 2022 compared to the prior year. These costs were partially offset by reduced costs in our OMS906 program resulting from the completion of toxicology study work in the second quarter of 2022.
Preclinical research and development expenses decreased $2.1 million in 2023 compared to 2022, primarily due to the migration of OMS1029 from preclinical to clinical research and development status during the third quarter of 2022, offset by an increase in preclinical oncology research costs during 2023.The $7.8 million decrease in 2022 over 2021 in preclinical research and development expenses was primarily due to the migration of OMS1029 from preclinical to clinical research and development status during the third quarter of 2022.
The changes in stock-based compensation expense between the three covered years were due to the valuations of employee stock options.
We expect our overall research and development costs in 2024 to be similar to 2023, driven by commercial narsoplimab manufacturing costs expected to be incurred prior to FDA approval of TA-TMA, increases in OMS906 clinical and manufacturing costs, and decreases in OMS721 clinical costs. Our accounting policy is to expense all manufacturing costs related to drug candidates until regulatory approval is reasonably assured in either the U.S. or Europe.
At this time, we are unable to estimate with certainty the longer-term costs we will incur in the continued development of our drug candidates due to the inherently unpredictable nature of our preclinical and clinical development activities. Clinical development timelines, the probability of success and development costs can differ materially as new data become available and as expectations change. Our future research and development expenses will depend, in part, on the preclinical or clinical success of each drug candidate as well as ongoing assessments of each program’s commercial potential. In addition, we cannot forecast with precision which drug candidates, if any, may be subject to future collaborations, when such arrangements will be secured, if at all, and to what degree such arrangements would affect our development plans and capital requirements.
We are required to expend substantial resources in the development of our drug candidates due to the lengthy process of completing clinical trials and seeking regulatory approval. Any failure or delay in completing clinical trials, or in obtaining regulatory approvals, could delay our generation of product revenue and increase our research and development expenses.
Selling, General and Administrative Expenses
Our selling, general and administrative expenses are comprised primarily of salaries, benefits and stock-based compensation costs for sales, marketing and administrative personnel who are not directly engaged in research and development. Costs also include marketing and selling expenses, professional and legal services, general corporate costs and an allocation of our occupancy costs.
| Year Ended December 31, |
||||||||||||
| 2023 |
2022 |
2021 |
||||||||||
| (In thousands) |
||||||||||||
| Continuing selling, general and administrative expenses: |
||||||||||||
| Selling, general and administrative expenses, excluding stock-based compensation expense |
$ | 42,520 | $ | 42,626 | $ | 46,688 | ||||||
| Stock-based compensation expense |
7,140 | 8,042 | 8,154 | |||||||||
| Total continuing selling, general and administrative expenses |
49,660 | 50,668 | 54,842 | |||||||||
| Discontinued selling, general and administrative expenses |
— | — | 25,428 | |||||||||
| Total selling, general and administrative expenses |
$ | 49,660 | $ | 50,668 | $ | 80,270 | ||||||
Continuing selling, general and administrative expenses, excluding stock-based compensation expense, decreased $4.1 million between 2022 and 2021 primarily related to reduced spending on pre-commercialization sales and marketing activities which were higher in 2021 as we prepared for the then anticipated approval and commercial launch of narsoplimab for the treatment of TA-TMA.
The changes in stock-based compensation expense between the three covered years were due to the valuations of employee stock options.
Our selling, general and administrative expenses for 2024 will be highly dependent on whether narsoplimab receives U.S. marketing approval for treatment of TA-TMA. If TA-TMA is approved in 2024, we expect to hire a field sales force and initiate commercial launch activities which will increase our selling, general and administrative expenses. If narsoplimab is not approved in 2024, our selling, general and administrative expenses are expected to decrease in 2024.
Interest Expense
| Year Ended December 31, |
||||||||||||
| 2023 |
2022 |
2021 |
||||||||||
| (In thousands) |
||||||||||||
| Interest expense |
$ | 30,844 | $ | 22,702 | $ | 19,669 | ||||||
Interest expense is primarily comprised of interest and amortization of debt discount and issuance costs related to our convertible senior notes and interest on our DRI royalty obligation (see Part II, Item 8, "Note 6 – Convertible Senior Notes" and "Note 8 – OMIDRIA Royalty Obligation” to our Consolidated Financial Statements in this Annual Report on Form 10-K for additional information).
Interest expense increased $8.1 million in 2023 compared to 2022 primarily due to incurring interest from our DRI royalty obligation for the full year. Interest expense increased $3.0 million in 2022 compared to 2021 primarily due to interest incurred from our DRI royalty obligation only in the fourth quarter of 2022.
Interest and Other Income
| Year Ended December 31, |
||||||||||||
| 2023 |
2022 |
2021 |
||||||||||
| (In thousands) |
||||||||||||
| Interest and other income |
$ | 16,342 | $ | 4,062 | $ | 1,740 | ||||||
The $12.3 million increase in interest and other income between 2023 and 2022 was primarily due to holding higher average cash and investment balances than in the prior year as a result of receiving a $200.0 million Milestone Payment from Rayner in February 2023. The $2.3 million increase in interest and other income between 2022 and 2021 was primarily attributable to obtaining significantly higher interest rates on our cash and investments in 2022.
We expect interest and other income in 2024 to be less than 2023 primarily due to lower average cash and investment balances during 2024.
Gain on Early Extinguishment of Convertible Senior Notes
| Year Ended December 31, |
||||||||||||
| 2023 |
2022 |
2021 |
||||||||||
| (In thousands) |
||||||||||||
| Gain on early extinguishment of convertible senior notes |
$ | 4,112 | $ | — | $ | — | ||||||
In December 2023, we repurchased $9.1 million par value of our 2026 Notes at a discount, realizing a $4.1 million non-cash gain on extinguishment.
Net Income from Discontinued Operations, Net of Tax
On December 23, 2021, we sold our commercial drug, OMIDRIA, to Rayner. As a result of the OMIDRIA divestiture, the results of OMIDRIA operations have been reclassified to discontinued operations for all periods presented.
Net income from OMIDRIA discontinued operations, net of tax is shown below:
| Year Ended December 31, |
||||||||||||
| 2023 |
2022 |
2021 |
||||||||||
| (In thousands) |
||||||||||||
| Product sales, net |
$ | — | $ | — | $ | 110,735 | ||||||
| Costs and expenses |
— | — | 30,631 | |||||||||
| Gross margin |
— | — | 80,104 | |||||||||
| Gain on sale of OMIDRIA |
— | — | 305,648 | |||||||||
| Milestone income |
— | 200,000 | — | |||||||||
| Interest on OMIDRIA contract royalty asset |
15,315 | 18,634 | — | |||||||||
| Remeasurement adjustments |
41,167 | 14,457 | — | |||||||||
| Other income |
1,087 | 307 | 1,035 | |||||||||
| Income before income tax |
57,569 | 233,398 | 386,787 | |||||||||
| Income tax expense (1) |
(462 | ) | (3,952 | ) | (1,006 | ) | ||||||
| Net income from discontinued operations, net of tax |
$ | 57,107 | $ | 229,446 | $ | 385,781 | ||||||
(1) For further discussion of income tax expense, please refer to Part II, Item 8, “Note 13 – Income Taxes” to our Consolidated Financial Statements in this Annual Report on Form 10-K.
Gain on the Sale of OMIDRIA
On December 23, 2021, we completed the sale of OMIDRIA to Rayner and received $126.0 million in cash at the closing. Additionally, we recorded an OMIDRIA contract royalty asset of $184.6 million for the rights to receive future royalties from Rayner on OMIDRIA net sales. The sale of OMIDRIA qualified as an asset sale under GAAP.
Rayner’s U.S. net sales of OMIDRIA for the years ended December 31, 2023 and 2022 were $135.3 million and $130.9 million, respectively. We earned royalties of $40.6 million and $65.4 million on OMIDRIA net sales for the years ended December 31, 2023 and 2022, respectively, which we recorded as a reduction from the OMIDRIA contract royalty asset. The decrease in royalty earnings between the years ended December 31, 2023 and 2022 was due to a reduction of our royalty rate on U.S. net sales of OMIDRIA from 50% to 30% upon achievement of the $200.0 million Milestone Event. (For further discussion of discontinued operations, please refer to Part II, Item 8, “Note 7 – Discontinued Operations – Sale of OMIDRIA” to our Consolidated Financial Statements in this Annual Report on Form 10-K).
Milestone Income
The Milestone Event occurred in December 2022, entitling us to receive a Milestone Payment of $200.0 million from Rayner. We received the Milestone Payment together with accrued interest in February 2023.
Interest Income
During the years ended December 31, 2023 and 2022, we recorded $15.3 million and $18.6 million, respectively, of income in discontinued operations, representing interest income on the outstanding OMIDRIA contract royalty asset at an implied interest rate of 11.0%.
Remeasurement Adjustments
During the years ended December 31, 2023 and 2022, we recorded $41.2 million and $14.5 million, respectively, of remeasurement adjustments. The $26.7 million increase in 2023 was primarily attributable to assigning a greater probability of achieving higher royalty earnings on net sales of OMIDRIA as supported by our most recent transaction with DRI, which closed on February 1, 2024.
Income Tax Expense
For the years ended December 31, 2023, 2022 and 2021, we recorded state income tax expense of $0.5 million, $4.0 million and $1.0 million, respectively, which could not be offset by prior period net operating losses and tax credit carryforwards.
Financial Condition - Liquidity and Capital Resources
As of December 31, 2023, we had cash, cash equivalents and short-term investments of $171.8 million.For the year ended December 31, 2023, our cash provided by operations was $74.7 million and our net loss was $117.8 million. In February 2024, we received $115.5 million upon the sale to DRI of our U.S. OMIDRIA royalty receipts payable between January 1, 2024 and December 31, 2031.
Historically, we have incurred net losses from continuing operations and negative operating cash flows. We have not yet established an ongoing source of revenue sufficient to cover our operating costs; therefore, we potentially need to continue to raise additional capital to accomplish our business plan and to retire our outstanding convertible senior notes due in 2026. We plan to continue to fund our operations for at least the next twelve months with our existing cash and investments and the $115.5 million we received in February 2024 from DRI. We have a sales agreement to sell shares of our common stock, from time to time, in an “at the market” equity offering facility through which we may offer and sell shares of our common stock equaling an aggregate amount up to $150.0 million. Should it be determined to be strategically advantageous, we could pursue debt financings as well as public and private offerings of our equity securities, similar to those we have previously completed, or other strategic transactions, which may include licensing a portion of our existing technology. Should it be necessary to manage our operating expenses, we could also reduce our projected cash requirements by delaying clinical trials, reducing selected research and development efforts, or implementing other restructuring activities.
Cash Flow Data
| Year Ended December 31, |
||||||||||||
| 2023 |
2022 |
2021 |
||||||||||
| (In thousands) |
||||||||||||
| Selected cash flow data |
||||||||||||
| Cash provided by (used in): |
||||||||||||
| Operating activities |
$ | 74,726 | $ | (86,483 | ) | $ | (109,722 | ) | ||||
| Investing activities |
$ | 27,454 | $ | (127,564 | ) | $ | 193,710 | |||||
| Financing activities |
$ | (106,084 | ) | $ | 124,248 | $ | 6,319 | |||||
Operating Activities. Net cash provided by operating activities for the year ended December 31, 2023 increased by $161.2 million compared to the same period in 2022. This increase was primarily due to collecting the $200.00 million Milestone Payment from Rayner in the current year and a $15.3 increase in accounts payable and accrued expenses. These increases were offset by a $26.7 million change in the remeasurement of the OMIDRIA contract royalty asset, $8.7 million related to the accretion of interest on U.S. government treasury bills and a $4.1 million gain on the early extinguishment of a portion of our 2026 Notes.
Net cash used in operating activities for the year ended December 31, 2022 decreased by $23.2 million compared to the same period in 2021. This change was primarily due to a decrease in net income of $146.8 million as we recognized $310.6 million of non-cash gain from the sale of OMIDRIA in the prior year and a change in cash collections of $124.7 million through accounts receivables and royalty earnings. This was offset by a $200.0 million milestone receivable recognized in 2022 as well as $35.6 million in non-cash charges and $29.7 million of accounts payable, accrued expenses and other.
Investing Activities. Net cash provided by investing activities increased $155.0 million during 2023 compared to 2022 driven by net proceeds from the purchase and sale of investments.
Net cash provided by investing activities decreased $321.3 million during 2022 compared to 2021. This was driven by a $194.5 million decrease in net proceeds from the purchase and sale of investments and recognizing $126.0 million in proceeds from the sale of OMIDRIA in 2021.
Financing Activities. Net cash used in financing activities decreased $230.3 million during 2023 compared to the prior year. The decrease was primarily due to receiving $125.0 million in 2022 in connection with selling a portion of our OMIDRIA royalties to DRI and extinguishing $95.0 million of our 6.25% convertible senior notes (the "2023 Notes"). In addition, we paid $4.9 million to retire $9.1 million par value of our 2026 Notes and repurchased $4.7 million of our common stock through a stock repurchase program in 2023.
Net cash provided by financing activities increased $117.9 million during 2022 compared to the prior year. The increase was primarily due to receiving cash proceeds of $125.0 million in connection with the sale of a portion of our OMIDRIA royalties to DRI, which was partially offset by a reduction in stock option exercises of $8.0 million during 2022.
Contractual Obligations and Commitments
Operating Leases
We lease our office and laboratory space in The Omeros Building under a lease agreement with BMR - 201 Elliott Avenue LLC. The initial term of the lease ends in November 2027 and we have two options to extend the lease term, each by five years. As of December 31, 2023, the remaining aggregate non-cancelable rent payable under the initial term of the lease, excluding common area maintenance and related operating expenses, was $26.9 million.
We have finance leases for certain laboratory and office equipment that have lease terms expiring through November 2026.
Convertible Notes
For more information regarding the convertible senior notes extinguished in mid-November 2023 and convertible senior notes due in February 2026, see Part II, Item 8, “Note 6 - Convertible Senior Notes”.
OMIDRIA Royalty Obligation
For more information regarding the OMIDRIA Royalty Obligation, see Part II, Item 8, “Note 8 - OMIDRIA Royalty Obligation”.
Goods & Services
We have certain non-cancelable obligations under other agreements for the acquisitions of goods and services associated with the manufacturing of our drug candidates, which contain firm commitments. As of December 31, 2023, our aggregate firm commitments were $25.8 million.
We may be required, in connection with in-licensing or asset acquisition agreements, to make certain royalty and milestone payments and we cannot, at this time, determine when or if the related milestones will be achieved or whether the events triggering the commencement of payment obligations will occur. Therefore, such payments are not included in the table above. For information regarding agreements that include these royalty and milestone payment obligations, see Part II, Item 8, “Note 10 - Commitments and Contingencies” to our Consolidated Financial Statements in this Annual Report on Form 10-K.
Critical Accounting Policies and Significant Judgments and Estimates
The preparation of our consolidated financial statements in conformity with U.S. generally accepted accounting principles (“GAAP”) requires management to make estimates and assumptions that affect the amounts reported in the financial statements and accompanying notes. We base our estimates on historical experience and on various other factors that we believe are reasonable under the circumstances; however, actual results could differ from those estimates. An accounting policy is considered critical if it is important to a company’s financial condition and results of operations and if it requires the exercise of significant judgment and the use of estimates on the part of management in its application. Although we believe that our judgments and estimates are appropriate, actual results may differ materially from our estimates. For a summary of our critical accounting policies, see Part II, Item 8, “Note 2 - Significant Accounting Policies” to our Consolidated Financial Statements in this Annual Report on Form 10-K.
We believe the following to be our critical accounting policies because they are both important to the portrayal of our financial condition and results of operations and they require critical judgment by management and estimates about matters that are uncertain:
● revenue recognition;
● OMIDRIA royalties and contract asset accounting;
● OMIDRIA royalty obligation accounting;
● research and development expenses related to clinical trials;
● accounting for convertible debt issuances, primarily related to fair valuing debt and issuance costs; and
● stock-based compensation, primarily related to our fair value assumptions.
If actual results or events differ materially from those contemplated by us in making these estimates, our reported financial condition and results of operations for future periods could be materially affected.
Product Revenue Recognition
Prior to the December 23, 2021 sale of OMIDRIA to Rayner, we recorded revenue from product sales when the product was delivered to our wholesalers and title for the product was transferred. Product sales were recorded net of wholesaler distribution fees and estimated chargebacks, rebates, returns and purchase-volume discounts. Accruals or allowances were established for these deductions in the same period when revenue was recognized, and actual amounts incurred were offset against the applicable accruals or allowances. We reflected each of these accruals or allowances as either a reduction in the related accounts receivable or as an accrued liability depending on how the amount was expected to be settled.
OMIDRIA Royalties, Milestones and Contract Royalty Assets
We have rights to receive future royalties from Rayner on OMIDRIA net sales at royalty rates that vary based on geography and certain regulatory contingencies. Therefore, future OMIDRIA royalties are treated as variable consideration. To measure the OMIDRIA contract royalty asset, we used the expected value approach which is the discounted sum of probability-weighted royalty payments, we would receive using a range of potential outcomes, to the extent that it is probable that a significant reversal in the amount of cumulative income recognized will not occur. Our calculations take the net present value of the sum to arrive at the OMIDRIA contract royalty asset stated on the balance sheet. We revalued the contract royalty asset to reduce the applicable royalty percentage from 50% to 30%, as required under the Asset Purchase Agreement following the occurrence of the December 2022 event triggering the $200.0 million Milestone Payment. Royalties earned will be recorded as a reduction to the OMIDRIA contract royalty asset. The amount recorded in discontinued operations in future periods will reflect interest earned on the outstanding OMIDRIA contract royalty asset and any amounts received different from the expected royalties recorded at closing. The OMIDRIA contract royalty asset is subject to changes in net sales of OMIDRIA. All else being equal, a 10% decrease or increase in net sales results in a $16.8 million change in value of the OMIDRIA contract royalty asset, resulting in a potential contract royalty asset valued within the range of $151.3 million to $184.9 million. Changes in net sales could occur due to various risks such as competitors entering the market, changes in the standard of care for cataract patients and loss of separate payment status for OMIDRIA. In determining the value of the OMIDRIA contract royalty asset, we have considered all of these factors. The OMIDRIA contract royalty asset will be re-measured periodically using the expected value approach based on actual results and future expectations. Any required adjustment to the OMIDRIA contract royalty asset will be recorded in discontinued operations.
We receive monthly royalty payments based on Rayner’s OMIDRIA product sales in accordance with the Asset Purchase Agreement. Upon the closing of the Asset Purchase Agreement, we determined the expected minimum net present value of future OMIDRIA royalty receipts and recognized the amount as a gain on the sale of OMIDRIA in discontinued operations on our income statement and as an OMIDRIA contract royalty asset on our balance sheet. To determine the OMIDRIA contract royalty asset, we used the expected value approach which is based on the sum of probability-weighted payments we would receive using a range of potential outcomes at an effective interest rate of 11%. The contract royalty asset excludes any revenue which potentially may be reversed in the event of an over estimation.
OMIDRIA Royalty Obligations
The sale of any portion of our OMIDRIA royalty receipts is treated as a liability on our consolidated balance sheet to the extent that any of our royalties are capped, as this does not result in the transfer of a participating interest. We amortize royalty obligation liabilities over the term of the arrangement using the effective interest method and classify interest expense as a component of continuing operations.
To the extent our estimates of future royalties are less than previous estimates, we will adjust the carrying amount of the royalty obligation to the present value of the revised estimated cash flows, discounted at the original effective interest rate utilizing the cumulative catch-up method. The adjustment would be recognized as a component of net income (loss) from continuing operations.
Research and Development Expenses
Research and development costs are comprised primarily of:
● contracted research and manufacturing costs;
● clinical study costs;
● costs of personnel, including salaries, benefits and stock compensation;
● consulting arrangements;
● depreciation and an allocation of our occupancy costs; and
● other expenses incurred to sustain our overall research and development programs.
Contracted research and manufacturing costs are primarily incurred in the development and production of our drug candidates. Prior to approval, our estimates are based on the timing of services provided. We record accrued expenses equal to our estimated expense in excess of amount invoiced by the suppliers.
Clinical trial expenses are estimated on a cost per patient that varies depending on the clinical trial site. As actual costs become known to us, we adjust our estimates; these changes in estimates may result in understated or overstated expenses at any given point in time.
Convertible Debt Issuances
On January 1, 2021, we adopted Accounting Standards Update (“ASU”) 2020-06, Debt—Debt with Conversion Options (Subtopic 470.20 and Derivatives and Hedging—Contracts in Entity’s Own Equity (Subtopic 815-40) on a modified retrospective basis. ASU 2020-06 removes the separate liability and equity accounting for our convertible senior notes. As of January 1, 2021, we account for our convertible senior notes wholly as debt. Prior to January 1, 2021, we accounted for convertible debt that may be settled wholly or partially in cash upon conversion as having both a liability component (debt) and an equity component (conversion option). The cash conversion guidance applies as the embedded conversion features meet the requirements for a derivative scope exception for instruments that are both indexed to an entity’s own stock and classified in stockholders’ equity in the balance sheet. Principal cash proceeds from the instrument are allocated first to the liability component based on the fair value of non-convertible debt using the income and market-based approaches to determine an effective interest rate for present valuing the cash proceeds. For the income-based approach, we use a convertible bond pricing model that includes several assumptions such as volatility and a risk-free rate. For the market-based approach, we observe the price of derivative price instruments purchased in conjunction with our convertible senior note issuances or evaluate issuances of convertible debt securities by other companies with similar credit risk ratings at the time of issuance. The amount of the equity component is then calculated by deducting the fair value of the liability component from the principal amount of the instrument. Issuance costs from the instrument are then allocated to the liability and equity components in the same proportion as the proceeds. The equity component of the cash principal proceeds and the liability component of the issuance costs represent a debt discount.
Transactions involving contemporaneous exchanges of cash between the same debtor and creditor in connection with the issuance of a new debt obligation and satisfaction of an existing debt obligation by the debtor are evaluated as a modification or an exchange transaction depending on whether the exchange is determined to have substantially different terms. We extinguished the 2023 Notes at maturity. The partial repurchase of the 2026 Notes was deemed to be a modification which we accounted for as a debt extinguishment.
Stock-Based Compensation
Stock-based compensation expense is recognized for all share-based payments made to employees, directors and non-employees based on estimated fair values. The fair value of our stock options is calculated using the Black-Scholes valuation model, which requires assumptions regarding volatility, risk-free rates, forfeiture rates and expected option life. We estimate forfeitures for expense recognition based on our historical experience. Groups of employees that have similar historical forfeiture behavior are considered separately. If any of the assumptions used in the Black-Scholes model change significantly, stock-based compensation expense for new awards may differ materially from that recorded for existing awards and stock-based compensation for non-employees will vary as the awards are re-measured over the vesting term.
Recent Accounting Pronouncements
Please refer to Part II, Item 8, “Note 2 - Significant Accounting Policies” to our Consolidated Financial Statements in this Annual Report on Form 10-K for information regarding recent accounting pronouncements.
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
Our exposure to market risk is primarily confined to our investment securities. The primary objective of our investment activities is to preserve our capital to fund operations, and we do not enter into financial instruments for trading or speculative purposes. We also seek to maximize income from our investments without assuming significant risk. To achieve our objectives, we maintain a portfolio of investments in high-credit-quality securities. As of December 31, 2023, we had cash, cash equivalents and short-term investments of $171.8 million. In accordance with our investment policy, we invest funds in highly liquid, investment-grade securities. The securities in our investment portfolio are not leveraged and are classified as held-to-maturity. We currently do not hedge interest rate exposure. Because of the short-term maturities of our investments, we do not believe that an increase in market rates would have a material negative effect on the realized value of our investment portfolio. We actively monitor changes in interest rates and, with our current portfolio of short-term investments which we intend to hold to maturity, we are not exposed to significant loss due to changes in interest rates.
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
Index to Consolidated Financial Statements
| Page |
|
| Report of Independent Registered Public Accounting Firm (PCAOB ID: 42) |
|
| Consolidated Statements of Operations and Comprehensive Income (Loss) |
|
Report of Independent Registered Public Accounting Firm
To the Shareholders and the Board of Directors Omeros Corporation
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of Omeros Corporation (the Company) as of December 31, 2023 and 2022, the related consolidated statements of operations and comprehensive income (loss), shareholders' equity (deficit) and cash flows for each of the three years in the period ended December 31, 2023, and the related notes (collectively referred to as the “consolidated financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company at December 31, 2023 and 2022, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2023, in conformity with U.S. generally accepted accounting principles.
Basis for Opinion
These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits we are required to obtain an understanding of internal control over financial reporting but not for the purpose of an expressing an opinion on the effectiveness of the Company's internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
Critical Audit Matter
The critical audit matter communicated below is a matter arising from the current period audit of the financial statements that was communicated or required to be communicated to the audit committee and that: (1) relates to accounts or disclosures that are material to the financial statements and (2) involved our especially challenging, subjective, or complex judgments. The communication of the critical audit matter does not alter in any way our opinion on the consolidated financial statements, taken as a whole, and we are not, by communicating the critical audit matter below, providing a separate opinion on the critical audit matter or on the account or disclosures to which it relates.
| OMIDRIA Contract Royalty Asset | |
| Description of the Matter |
As more fully described in Note 2 of the financial statements, the Company recorded a contract royalty asset in connection with its sale of OMIDRIA to Rayner Surgical, Inc. on December 23, 2021. To measure that contract royalty asset, the Company used the expected value approach, which is the discounted sum of the probability-weighted royalty payments using a range of potential outcomes, to the extent that it is probable that a significant reversal in the amount of cumulative income recognized will not occur.
Auditing management’s forecasts is complex and requires judgment due to the level of estimation uncertainty and the sensitivity of the asset’s value to changes in assumptions. In particular, the value of the OMIDRIA contract royalty asset is sensitive to changes in significant assumptions such as forecasted royalties due from Rayner Surgical, Inc. in various scenarios and the probability-weighting of those scenarios, which are affected by expectations about future market and regulatory conditions. |
| How We Addressed the Matter in Our Audit |
To test the measurement of the OMIDRIA contract royalty asset, we performed audit procedures that included, among others, evaluating (1) the estimated future royalties in various scenarios, and (2) management’s relative weighting of those scenarios. We compared estimated future royalties to the Company’s historical revenues and royalty rates in the asset purchase agreement. We evaluated the appropriateness and likelihood of occurrence of the various scenarios included in management’s calculation, given the Company’s experience and industry trends, and verified the clerical accuracy of the calculation. We also evaluated the Company’s disclosures in the consolidated financial statements related to these matters. |
/s/Ernst & Young LLP
We have served as the Company’s auditor since 1998.
Seattle, Washington
April 1, 2024
OMEROS CORPORATION
(In thousands, except share and per share data)
| December 31, |
December 31, |
||||||
| 2023 |
2022 |
||||||
| Assets |
|||||||
| Current assets: |
|||||||
| Cash and cash equivalents |
$ | 7,105 | $ | 11,009 | |||
| Short-term investments |
164,743 | 183,909 | |||||
| OMIDRIA contract royalty asset, short-term |
29,373 | 28,797 | |||||
| Receivables |
8,096 | 213,221 | |||||
| Prepaid expense and other assets |
8,581 | 6,300 | |||||
| Total current assets |
217,898 | 443,236 | |||||
| OMIDRIA contract royalty asset |
138,736 | 123,425 | |||||
| Right of use assets |
18,631 | 21,762 | |||||
| Property and equipment, net |
1,950 | 1,492 | |||||
| Restricted investments |
1,054 | 1,054 | |||||
| Total assets |
$ | 378,269 | $ | 590,969 | |||
| Liabilities and shareholders’ equity (deficit) |
|||||||
| Current liabilities: |
|||||||
| Accounts payable |
$ | 7,712 | $ | 5,989 | |||
| Accrued expenses |
31,868 | 30,551 | |||||
| Current portion of convertible senior notes, net |
— | 94,381 | |||||
| Current portion of OMIDRIA royalty obligation |
8,576 | 1,152 | |||||
| Current portion of lease liabilities |
5,160 | 4,310 | |||||
| Total current liabilities |
53,316 | 136,383 | |||||
| Convertible senior notes, net |
213,155 | 220,906 | |||||
| OMIDRIA royalty obligation |
116,550 | 125,126 | |||||
| Lease liabilities, non-current |
18,143 | 22,426 | |||||
| Other accrued liabilities - noncurrent |
2,088 | 444 | |||||
| Commitments and contingencies (Note 10) |
|||||||
| Shareholders’ equity (deficit): |
|||||||
| Preferred stock, par value $0.01 per share, 20,000,000 shares authorized; none issued and outstanding at December 31, 2023 and December 31, 2022 |
— | — | |||||
| Common stock, par value $0.01 per share, 150,000,000 shares authorized at December 31, 2023 and December 31, 2022; 61,128,597 and 62,828,765 shares issued and outstanding at December 31, 2023 and December 31, 2022, respectively. |
611 | 628 | |||||
| Additional paid-in capital |
727,936 | 720,773 | |||||
| Accumulated deficit |
(753,530 | ) | (635,717 | ) | |||
| Total shareholders’ equity (deficit) |
(24,983 | ) | 85,684 | ||||
| Total liabilities and shareholders’ equity (deficit) |
$ | 378,269 | $ | 590,969 | |||
See accompanying Notes to Consolidated Financial Statements
OMEROS CORPORATION
CONSOLIDATED STATEMENTS OF OPERATIONS AND COMPREHENSIVE INCOME (LOSS)
(In thousands, except share and per share data)
| Year Ended December 31, |
||||||||||||
| 2023 |
2022 |
2021 |
||||||||||
| Costs and expenses: |
||||||||||||
| Research and development |
$ | 114,870 | $ | 112,721 | $ | 118,775 | ||||||
| Selling, general and administrative |
49,660 | 50,668 | 54,842 | |||||||||
| Total costs and expenses |
164,530 | 163,389 | 173,617 | |||||||||
| Loss from operations |
(164,530 | ) | (163,389 | ) | (173,617 | ) | ||||||
| Interest expense |
(30,844 | ) | (22,702 | ) | (19,669 | ) | ||||||
| Interest and other income |
16,342 | 4,062 | 1,740 | |||||||||
| Gain on early extinguishment of convertible senior notes |
4,112 | — | — | |||||||||
| Net loss from continuing operations |
(174,920 | ) | (182,029 | ) | (191,546 | ) | ||||||
| Net income from discontinued operations, net of tax |
57,107 | 229,446 | 385,781 | |||||||||
| Net income (loss) |
$ | (117,813 | ) | $ | 47,417 | $ | 194,235 | |||||
| Basic and diluted net income (loss) per share: |
||||||||||||
| Net loss from continuing operations |
$ | (2.79 | ) | $ | (2.90 | ) | $ | (3.07 | ) | |||
| Net income from discontinued operations |
0.91 | 3.66 | 6.19 | |||||||||
| Net income (loss) |
$ | (1.88 | ) | $ | 0.76 | $ | 3.12 | |||||
| Weighted-average shares used to compute basic and diluted net income (loss) per share |
62,739,227 | 62,737,091 | 62,344,100 | |||||||||
See accompanying Notes to Consolidated Financial Statements
OMEROS CORPORATION
CONSOLIDATED STATEMENTS OF SHAREHOLDERS’ EQUITY (DEFICIT)
(In thousands, except share data)
| Additional |
Total |
|||||||||||||||
| Common Stock |
Paid-in |
Accumulated |
Shareholders’ |
|||||||||||||
| Shares |
Amount |
Capital |
Deficit |
Equity/(Deficit) |
||||||||||||
| Balance at December 31, 2020 |
61,671,231 | $ | 616 | $ | 751,304 | $ | (872,672 | ) | $ | (120,752 | ) | |||||
| Issuance of common stock upon exercise of stock options |
945,924 | 10 | 8,372 | — | 8,382 | |||||||||||
| Issuance of common stock upon grant of restricted stock awards |
11,700 | — | 91 | — | 91 | |||||||||||
| At the market offering fees |
— | — | (241 | ) | — | (241 | ) | |||||||||
| Stock-based compensation |
— | — | 17,539 | — | 17,539 | |||||||||||
| Cumulative effect of adopting ASU 2020-06 |
— | — | (70,777 | ) | (4,697 | ) | (75,474 | ) | ||||||||
| Net income |
— | — | — | 194,235 | 194,235 | |||||||||||
| Balance at December 31, 2021 |
62,628,855 | 626 | 706,288 | (683,134 | ) | 23,780 | ||||||||||
| Issuance of common stock upon exercise of stock options |
101,160 | 1 | 414 | — | 415 | |||||||||||
| Issuance of common stock upon vesting of restricted stock units |
98,750 | 1 | (1 | ) | — | — | ||||||||||
| Stock-based compensation |
— | — | 14,072 | — | 14,072 | |||||||||||
| Net income |
— | — | — | 47,417 | 47,417 | |||||||||||
| Balance at December 31, 2022 |
62,828,765 | 628 | 720,773 | (635,717 | ) | 85,684 | ||||||||||
| Issuance of common stock upon exercise of stock options |
36,726 | — | 150 | — | 150 | |||||||||||
| Issuance of common stock upon vesting of restricted stock units |
67,250 | 1 | (1 | ) | — | — | ||||||||||
| Repurchases of common stock |
(1,804,144 | ) | (18 | ) | (4,636 | ) | — | (4,654 | ) | |||||||
| Stock-based compensation |
— | — | 11,650 | — | 11,650 | |||||||||||
| Net loss |
— | — | — | (117,813 | ) | (117,813 | ) | |||||||||
| Balance at December 31, 2023 |
61,128,597 | $ | 611 | $ | 727,936 | $ | (753,530 | ) | $ | (24,983 | ) | |||||
See accompanying Notes to Consolidated Financial Statements
OMEROS CORPORATION
CONSOLIDATED STATEMENTS OF CASH FLOWS
(In thousands)
| Year Ended December 31, |
||||||||||||
| 2023 |
2022 |
2021 |
||||||||||
| Operating activities: |
||||||||||||
| Net income (loss) |
$ | (117,813 | ) | $ | 47,417 | $ | 194,235 | |||||
| Adjustments to reconcile net income (loss) to net cash provided by (used in) operating activities: |
||||||||||||
| Stock-based compensation expense |
11,650 | 14,072 | 17,630 | |||||||||
| Non-cash interest expense on convertible senior notes |
1,853 | 1,830 | 1,696 | |||||||||
| Depreciation and amortization |
920 | 952 | 1,386 | |||||||||
| Remeasurement on OMIDRIA contract royalty asset |
(41,167 | ) | (14,457 | ) | — | |||||||
| Interest on OMIDRIA contract royalty asset |
(15,315 | ) | (18,634 | ) | — | |||||||
| Accretion on U.S. government treasury bills, net |
(8,714 | ) | — | — | ||||||||
| Gain on early extinguishment of convertible senior notes |
(4,112 | ) | — | — | ||||||||
| Gain on sale of OMIDRIA, gross |
— | — | (310,563 | ) | ||||||||
| Non-cash interest expense on future royalty obligation |
— | 1,695 | — | |||||||||
| Changes in operating assets and liabilities: |
||||||||||||
| Receivables |
205,125 | (175,066 | ) | (34,314 | ) | |||||||
| OMIDRIA contract royalty asset |
40,595 | 65,439 | — | |||||||||
| Accounts payable and accrued expense |
4,682 | (10,665 | ) | 14,640 | ||||||||
| Prepaid expenses and other |
(2,978 | ) | 934 | 5,568 | ||||||||
| Net cash provided by (used in) operating activities |
74,726 | (86,483 | ) | (109,722 | ) | |||||||
| Investing activities: |
||||||||||||
| Purchases of investments |
(1,018,602 | ) | (429,045 | ) | (32,006 | ) | ||||||
| Proceeds from the sale and maturities of investments |
1,046,482 | 301,594 | 100,000 | |||||||||
| Purchases of property and equipment |
(426 | ) | (113 | ) | (277 | ) | ||||||
| Cash proceeds on sale of OMIDRIA |
— | — | 125,993 | |||||||||
| Net cash provided by (used in) investing activities |
27,454 | (127,564 | ) | 193,710 | ||||||||
| Financing activities: |
||||||||||||
| Payments on convertible senior notes |
(99,873 | ) | — | — | ||||||||
| Repurchases on common stock |
(4,654 | ) | — | — | ||||||||
| Principal payments on OMIDRIA royalty obligation |
(1,152 | ) | (417 | ) | — | |||||||
| Payments on finance lease obligations |
(555 | ) | (750 | ) | (1,823 | ) | ||||||
| Proceeds upon exercise of stock options |
150 | 415 | 8,383 | |||||||||
| Proceeds upon entering into OMIDRIA royalty obligation |
— | 125,000 | — | |||||||||
| At the market offering costs |
— | — | (241 | ) | ||||||||
| Net cash provided by (used in) financing activities |
(106,084 | ) | 124,248 | 6,319 | ||||||||
| Net increase (decrease) in cash and cash equivalents |
(3,904 | ) | (89,799 | ) | 90,307 | |||||||
| Cash and cash equivalents at beginning of period |
11,009 | 100,808 | 10,501 | |||||||||
| Cash and cash equivalents at end of period |
$ | 7,105 | $ | 11,009 | $ | 100,808 | ||||||
| Supplemental cash flow information |
||||||||||||
| Cash paid for interest |
$ | 29,923 | $ | 19,178 | $ | 17,876 | ||||||
| Equipment acquired under finance lease |
$ | 952 | $ | 40 | $ | 289 | ||||||
See accompanying Notes to Consolidated Financial Statements
OMEROS CORPORATION
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Note 1—Organization and Basis of Presentation
General
Omeros Corporation (“Omeros,” the “Company” or “we”) is a clinical-stage biopharmaceutical company committed to discovering, developing and commercializing small-molecule and protein therapeutics for large-market as well as orphan indications targeting immunologic disorders including complement-mediated diseases, cancers, and addictive and compulsive disorders. We marketed our first drug product OMIDRIA® (phenylephrine and ketorolac intraocular solution) 1% / 0.3% for use during cataract surgery or intraocular lens replacement in the United States (the “U.S.”) until we sold OMIDRIA and related business assets on December 23, 2021 (see “Sale of OMIDRIA Assets” below for additional information).
Our pipeline of clinical-stage development programs includes: narsoplimab, our antibody targeting mannan-binding lectin-associated serine protease 2 ("MASP-2"), the effector enzyme of the lectin pathway of complement; OMS1029, our long-acting antibody targeting MASP-2; OMS906, our antibody targeting mannan-binding lectin-associated serine protease-3 ("MASP-3"), the key activator of the alternative pathway of complement; and OMS527, our phosphodiesterase 7 ("PDE7") inhibitor program.
Clinical development of narsoplimab is currently focused primarily on hematopoietic stem cell transplant-associated thrombotic microangiopathy ("TA-TMA"). Our Biologics License Application ("BLA") for narsoplimab in TA-TMA is anticipated to be resubmitted with additional information to support potential approval of narsoplimab in this indication. In October 2023, we announced the results of a pre-specified interim analysis of our Phase 3 ARTEMIS-IGAN trial evaluating narsoplimab for the treatment of immunoglobulin A ("IgA") nephropathy. Topline results showed that narsoplimab did not reach statistically significant improvement over placebo on the primary endpoint of reduction in proteinuria. Based on this result, we have discontinued the ARTEMIS-IGAN clinical trial.
Phase 1 and Phase 2 clinical programs are underway in our other clinical-stage assets.
Sale of OMIDRIA Assets
On December 23, 2021, we closed on an Asset Purchase Agreement (the “Asset Purchase Agreement”) with Rayner Surgical Inc. (“Rayner”) for the sale of our commercial product OMIDRIA. Rayner paid us $126.0 million in cash at closing, and we retained all outstanding accounts receivable, accounts payable and accrued expenses as of the closing date. Additionally, we are entitled to future royalty payments on net sales of OMIDRIA.
Under the Asset Purchase Agreement, Omeros is entitled to receive a milestone payment of $200.0 million (the “Milestone Payment”) following an event (the "Milestone Event") that establishes separate payment for OMIDRIA for a continuous period of at least four years when furnished in the ambulatory surgery center (“ASC”) setting. In December 2022, the Milestone Event occurred and we recorded a $200.0 million milestone receivable. We received the Milestone Payment in February 2023.
As a result of the divestiture, the results of OMIDRIA operations (e.g., revenues and operating costs) have been reclassified to discontinued operations in our consolidated statements of operations and comprehensive income (loss) and excluded from continuing operations for all periods presented (See“Note 7 – Discontinued Operations – Sale of OMIDRIA ”).
Basis of Presentation
Our consolidated financial statements include the financial position and results of operations of Omeros and our wholly owned subsidiaries. All inter-company transactions have been eliminated. The accompanying consolidated financial statements have been prepared in accordance with U.S. generally accepted accounting principles (“GAAP”).
Liquidity and Capital Resources
As of December 31, 2023, we had cash, cash equivalents and short-term investments of $171.8 million. Our cash provided by operations for the year ended December 31, 2023 was $74.7 million and included our 2023 net loss for the year of $117.8 million and collection of the $200.0 million Milestone Payment in the first quarter of 2023. We extinguished $95.0 million outstanding of convertible senior notes at maturity in November 2023. In February 2024, we received $115.5 million upon the sale to DRI Healthcare Acquisition LP ("DRI") of substantially all of our expected remaining U.S.-only Rayner OMIDRIA royalty receipts payable through December 31, 2031 (see “Note 8 - OMIDRIA Royalty Obligation”).
Historically, we have incurred net losses from continuing operations and negative operating cash flows. We have not yet established an ongoing source of revenue sufficient to cover our operating costs; therefore, we potentially need to continue to raise additional capital to accomplish our business plan and to retire our outstanding convertible senior notes due in 2026. We plan to continue to fund our operations for at least the next twelve months with our existing cash and investments and the $115.5 million we received in February 2024 from DRI. We have a sales agreement to sell shares of our common stock, from time to time, in an “at the market” equity offering facility through which we may offer and sell shares of our common stock equaling an aggregate amount up to $150.0 million. Should it be determined to be strategically advantageous, we could pursue debt financings as well as public and private offerings of our equity securities, similar to those we have previously completed, or other strategic transactions, which may include licensing a portion of our existing technology. Should it be necessary to manage our operating expenses, we could also reduce our projected cash requirements by delaying clinical trials, reducing selected research and development efforts, or implementing other restructuring activities.
Segments
We operate in one segment. Management uses cash flow as the primary measure to manage our business and does not segment our business for internal reporting or decision-making.
Use of Estimates
The preparation of financial statements in conformity with GAAP requires management to make estimates and assumptions that affect the amounts reported in the financial statements and accompanying notes. Significant items subject to such estimates include OMIDRIA contract royalty asset valuation, stock-based compensation expense, and accruals for clinical trials and manufacturing of drug product. We base our estimates on historical experience and on various other factors that we believe are reasonable under the circumstances; however, actual results could differ from these estimates.
Note 2—Significant Accounting Policies
Discontinued Operations
We review the presentation of planned or completed business dispositions in the consolidated financial statements based on the available information and events that have occurred. The review consists of evaluating whether the business meets the definition of a component for which the operations and cash flows are clearly distinguishable from the other components of the business and, if so, whether it is anticipated that after the disposal the cash flows of the component would be eliminated from continuing operations and whether the disposition represents a strategic shift that has a major effect on operations and financial results.
Planned or completed business dispositions are presented as discontinued operations when all the criteria described above are met. For those divestitures that qualify as discontinued operations, all comparative periods presented are reclassified in the consolidated balance sheets. Additionally, the results of operations of a discontinued operation are reclassified to income from discontinued operations, net of tax, for all periods presented in the consolidated statements of operations and comprehensive income (loss). Results of discontinued operations include all revenues and expenses directly derived from such businesses. General corporate overhead is not allocated to discontinued operations. The OMIDRIA asset sale to Rayner qualifies as a discontinued operation and has been presented as such for all reporting periods presented. The Company included information regarding cash flows from discontinued operations (see “Note 7 – Discontinued Operations – Sale of OMIDRIA”).
OMIDRIA Royalties, Milestones and Contract Royalty Assets
We have rights to receive future royalties from Rayner on OMIDRIA net sales at royalty rates that vary based on geography and certain regulatory contingencies. Therefore, future OMIDRIA royalties are treated as variable consideration. The sale of OMIDRIA qualified as an asset sale under GAAP. To measure the OMIDRIA contract royalty asset, we used the expected value approach which is the sum of the discounted probability-weighted royalty payments, we would receive using a range of potential outcomes, to the extent that it is probable that a significant reversal in the amount of cumulative income recognized will not occur. As contemplated by the Asset Purchase Agreement, the royalty rate applicable to U.S. net sales of OMIDRIA was reduced from 50% to 30% upon the occurrence, in December 2022, of the event triggering the $200.0 million Milestone Payment. The reduction in our royalty rate to 30% continues until the expiration or termination of the last issued and unexpired U.S. patent, which we expect to occur no earlier than 2035. Consequently, we revalued the OMIDRIA contract royalty asset using the 30% royalty rate on U.S. net sales and adjusted the probability weighted outcomes to reflect the occurrence of the Milestone Event. Royalties earned are recorded as a reduction to the OMIDRIA contract royalty asset. The amount recorded in discontinued operations in future periods will reflect interest earned on the outstanding OMIDRIA contract royalty asset at 11.0% and any amounts we receive that are different from the expected royalties. The OMIDRIA contract royalty asset is re-measured periodically using the expected value approach based on actual results and future expectations. Any required adjustment to the OMIDRIA contract royalty asset is recorded in discontinued operations.
OMIDRIA Royalty Obligation
On September 30, 2022, we sold to DRI an interest in a portion of our future OMIDRIA royalty receipts for a purchase price of $125.0 million and recorded as an “OMIDRIA royalty obligation” on our consolidated balance sheet. The liability is amortized over the term of the arrangement using the implied effective interest rate of 9.4%. Interest expense is recorded as a component of continuing operations.
To the extent our estimates of future royalties are less than previous estimates, we will adjust the carrying amount of the OMIDRIA royalty obligation to the present value of the revised estimated cash flows, discounted at the effective interest rate utilizing the cumulative catch-up method. The adjustment would be recognized as a component of net income (loss) from continuing operations (see “Note 8 - OMIDRIA Royalty Obligation”).
Cash and Cash Equivalents, Short-Term Investments and Restricted Investments
Cash and cash equivalents include highly liquid investments with a maturity of three months or less on the date of purchase which can be easily converted into cash without a significant impact to their value. Short-term investment securities are classified as held-to-maturity. Investments classified as held-to-maturity are carried at cost. Amortization, accretion, interest, and dividends, realized gains and losses and declines in value judged to be other-than-temporary are included in other income. The cost of securities sold is based on the specific-identification method. Investments with maturities of less than one year, or those for which management intends to use the investments to fund current operations, are included in current assets. We evaluate whether an investment is other-than-temporarily impaired based on the specific facts and circumstances. Factors that are considered in determining whether an other-than-temporary decline in value has occurred include: the market value of the security in relation to its cost basis; the financial condition of the investee; and the intent and ability to retain the investment for a sufficient period of time to allow for recovery in the market value of the investment. Restricted investments held in money-market funds include security deposits held by our landlord.
Investment income, which is included as a component of other income, consists primarily of interest earned.
Inventory
We expense inventory costs related to product candidates as research and development expenses until regulatory approval is reasonably assured in the U.S. or the European Union (“EU”). Once approval is reasonably assured, costs, including amounts related to third-party manufacturing, transportation and internal labor and overhead, will be capitalized.
Receivables
Receivables at December 31, 2023 primarily consist of royalties receivable from Rayner. Receivables at December 31, 2022 also included the $200.0 million Milestone Payment which we received in February 2023. Considering the nature of our receivables, we concluded an allowance for doubtful accounts was not necessary as of December 31, 2023 and 2022, respectively.
Property and Equipment, Net
Property and equipment are stated at cost, and depreciation is calculated using the straight-line method over the estimated useful life of the assets, which is generally three to 10 years. Equipment acquired through finance leases is recorded as property and equipment and is amortized over the shorter of the useful lives of the related assets or the lease term. Expenditures for repairs and maintenance are expensed as incurred.
Convertible Senior Notes
On January 1, 2021, we adopted Accounting Standards Update (“ASU”) 2020-06, Debt—Debt with Conversion Options (Subtopic 470.20 and Derivatives and Hedging—Contracts in Entity’s Own Equity (Subtopic 815-40) on a modified retrospective basis. ASU 2020-06 removed the separate liability and equity accounting for our convertible senior notes that was required under previous guidance and allows us to account for our convertible senior notes wholly as debt. Upon adoption, we removed the equity component allocated to debt issuance costs.
Transactions involving contemporaneous exchanges of cash between the same debtor and creditor in connection with the issuance of a new debt obligation and satisfaction of an existing debt obligation are evaluated as a modification or an extinguishment depending on whether the exchange is determined to have substantially different terms. We extinguished the 6.25% convertible senior notes (the “2023 Notes”) at par upon maturity on November 15, 2023. In December 2023, we repurchased $9.1 million par value of our 5.25% convertible senior notes (“2026 Notes”) at a discount, realizing a $4.1 million non-cash gain on extinguishment.
Impairment of Long-Lived Assets
We assess the impairment of long-lived assets, whenever events or changes in circumstances indicate that the carrying value of an asset may not be recoverable. Recoverability of these assets is measured by comparing the carrying value to future undiscounted cash flows that the asset is expected to generate. If the asset is impaired, the amount of any impairment will be reflected in the results of operations in the period of impairment. We have not recognized any impairment losses for the years ended December 31, 2023, 2022 and 2021.
Revenue Recognition
When we enter into a customer contract, we perform the following five steps: (i) identify the contract with a customer; (ii) identify the performance obligations in the contract; (iii) determine the transaction price; (iv) allocate the transaction price to the performance obligations in the contract; and (v) recognize revenue when (or as) we satisfy a performance obligation.
Prior to the sale of OMIDRIA to Rayner, we recorded product sales as revenue when the product was delivered to our wholesalers and title for the product was transferred. Product sales were recorded net of wholesaler distribution fees and estimated chargebacks, rebates, returns and purchase-volume discounts.
Research and Development
Research and development expenses are comprised primarily of contracted research, clinical trial study and manufacturing costs prior to approval; consulting services; contract milestones; materials and supplies; costs for personnel, including salaries, benefits and stock compensation; depreciation; an allocation of our occupancy costs; and other expenses incurred to sustain our overall research and development programs. Advance payments for goods or services that will be used for future research and development activities are deferred and then recognized as an expense as the related goods are delivered or the services are performed. All other research and development costs are expensed as incurred.
Selling, General and Administrative
Selling, general and administrative expenses are comprised primarily of marketing and selling expenses; professional and legal services; patent costs; and salaries, benefits, and stock-compensation costs for sales, marketing, and other personnel not directly engaged in research and development. Additionally, selling, general and administrative expenses include depreciation; an allocation of our occupancy costs; and other general corporate expenses. Advertising costs are expensed as incurred. We had no advertising costs during the years ended December 31, 2023 and 2022. For the year ended December 31, 2021, we incurred $0.8 million in advertising costs related to our sales of OMIDRIA.
Income Taxes
Deferred tax assets and liabilities are recognized for the future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their tax bases. Deferred tax assets and liabilities are measured using enacted tax rates applied to taxable income in the years in which those temporary differences are expected to be recovered or settled. We recognize the effect of income tax positions only if those positions are more likely than not of being sustained upon an examination. A valuation allowance is established when it is more likely than not that the deferred tax assets will not be realized.
Stock-Based Compensation
Stock-based compensation expense is recognized for all share-based payments, including grants of stock option awards and restricted stock units (“RSU”) based on estimated fair values. The fair value of our stock is calculated using the Black-Scholes option-pricing model, which requires judgmental assumptions around volatility, forfeiture rates, risk-free rate and expected term. Compensation expense is recognized over the requisite service periods, which is generally the vesting period, using the straight-line method. Forfeiture expense is estimated at the time of grant and revised in subsequent periods if actual forfeitures differ from those estimates.
Common Stock Repurchases
We may repurchase shares of our common stock from time to time under authorization made by our Board of Directors. Under applicable Washington State law, repurchased shares are retired and not presented separately as treasury stock on the consolidated financial statements.
Accumulated Other Comprehensive Income (Loss)
Accumulated other comprehensive income (loss) is comprised of net income (loss) and certain changes in equity that are excluded from net income (loss). There was no difference between comprehensive income (loss) and net income (loss) for the years ended December 31, 2023, 2022 and 2021.
Financial Instruments and Concentrations of Credit Risk
Cash and cash equivalents, receivables, accounts payable and accrued liabilities, which are recorded at invoiced amount or cost, approximate fair value based on the short-term nature of these financial instruments. The fair value of short-term investments is based on quoted market prices. Financial instruments that potentially subject us to concentrations of credit risk consist primarily of cash and cash equivalents, short-term investments and receivables. Cash and cash equivalents are held by financial institutions and are federally insured up to certain limits. At times, our cash and cash equivalents balance held at a financial institution may exceeds the federally insured limits. To limit the credit risk, we invest our excess cash in high-quality securities such as money market mutual funds, certificates of deposit and U.S. treasury bills.
Recent Accounting Pronouncements
In December 2023, the Financial Accounting Standards Board issued ASU 2023-09, Income Taxes - Improvements to Income Tax Disclosure (Topic 740), to enhance the transparency of income tax disclosures. ASU 2023-09 provides enhancements to the income tax disclosures related to the rate reconciliation and income taxes paid information. ASU 2023-09 is effective for fiscal years after December 15, 2025 and applied prospectively. The Company is evaluating the impact of this pronouncement on its consolidated financial statements.
Note 3—Net Income (Loss) Per Share
Basic net income (loss) per share ("Basic EPS") is computed by dividing net income (loss) by the weighted average number of common shares outstanding during the period. Diluted net income (loss) per share (“Diluted EPS”) is computed by dividing net income (loss) by the weighted average number of common shares and potentially dilutive common shares outstanding during the period. Our potentially dilutive securities include common shares related to our stock options, RSUs and convertible senior notes calculated using the treasury stock method. In periods where we have a net loss from continuing operations but overall net income, we do not compute Diluted EPS. Potentially dilutive securities excluded from Diluted EPS are as follows:
| Year Ended December 31, |
||||||||||||
| 2023 |
2022 |
2021 |
||||||||||
| 2026 Notes convertible to common stock (1) |
11,132,366 | 12,172,008 | 12,172,008 | |||||||||
| 2023 Notes convertible to common stock (1)(2) |
4,318,944 | 4,941,739 | 4,941,739 | |||||||||
| Outstanding options to purchase common stock |
38,462 | 9,488 | 1,707,371 | |||||||||
| Outstanding restricted stock units |
— | 98,750 | 2,642 | |||||||||
| Total dilutive shares excluded from net income (loss) per share |
15,489,772 | 17,221,985 | 18,823,760 | |||||||||
(1) The 2023 Notes were, and the 2026 Notes are subject to a capped call arrangement that potentially reduces the dilutive effect as described in “Note 6 - Convertible Senior Notes”. Any potential impact of the capped call arrangement is excluded from this table.
(2) The 2023 Notes were fully extinguished on November 15, 2023.
Note 4—Fair-Value Measurements
All of our investments are held in our name and are classified as short-term and held-to-maturity. Interest income from investments for the years ended December 31, 2023 and December 31, 2022 were $14.7 million and $2.2 million, respectively.
The following tables summarize our investments:
| |
December 31, 2023 |
|||||||||||
| |
Gross Unrealized |
|||||||||||
| Amortized Cost |
Gains/(Losses) |
Estimated Fair Value |
||||||||||
| |
(In thousands) |
|||||||||||
| |
|
|
|
|||||||||
| U.S. government securities classified as short-term investments |
$ | 102,100 | $ | 19 | $ | 102,119 | ||||||
| Money-market funds classified as short-term investments |
62,643 | — | 62,643 | |||||||||
| Total short-term investments |
164,743 | 19 | 164,762 | |||||||||
| Certificate of deposit classified as non-current restricted investments |
1,054 | — | 1,054 | |||||||||
| Total investments |
$ | 165,797 | $ | 19 | $ | 165,816 | ||||||
| |
December 31, 2022 |
|||||||||||
| |
Gross Unrealized |
|||||||||||
| Amortized Cost |
Gains/(Losses) |
Estimated Fair Value |
||||||||||
| |
(In thousands) |
|||||||||||
| |
|
|
|
|||||||||
| U.S. government securities classified as short-term investments |
$ | 99,027 | $ | 22 | $ | 99,049 | ||||||
| Money-market funds classified as short-term investments |
84,882 | — | 84,882 | |||||||||
| Total short-term investments |
183,909 | 22 | 183,931 | |||||||||
| Certificate of deposit classified as non-current restricted investments |
1,054 | — | 1,054 | |||||||||
| Total investments |
$ | 184,963 | $ | 22 | $ | 184,985 | ||||||
Fair value is defined as the exchange price that would be received for an asset or paid to transfer a liability, an exit price, in the principal or most advantageous market for the asset or liability in an orderly transaction between market participants on the measurement date. The accounting standard establishes a fair value hierarchy that requires an entity to maximize the use of observable inputs, where available. The following summarizes the three levels of inputs required:
Level 1—Observable inputs for identical assets or liabilities, such as quoted prices in active markets;
Level 2—Inputs other than quoted prices in active markets that are either directly or indirectly observable; and Level 3—Unobservable inputs in which little or no market data exists, therefore they are developed using estimates and assumptions developed by us, which reflect those that a market participant would use.
Our fair-value hierarchy for our financial assets are as follows:
| December 31, 2023 |
||||||||||||||||
| Level 1 |
Level 2 |
Level 3 |
Total |
|||||||||||||
| (In thousands) |
||||||||||||||||
| U.S. government securities classified as short-term investments |
$ | — | $ | 102,119 | $ | — | $ | 102,119 | ||||||||
| Money-market funds classified as short-term investments |
62,643 | — | — | 62,643 | ||||||||||||
| Total short-term investments |
62,643 | 102,119 | — | 164,762 | ||||||||||||
| Money-market funds classified as non-current restricted investments |
1,054 | — | — | 1,054 | ||||||||||||
| Total investments |
$ | 63,697 | $ | 102,119 | $ | — | $ | 165,816 | ||||||||
| December 31, 2022 |
||||||||||||||||
| Level 1 |
Level 2 |
Level 3 |
Total |
|||||||||||||
| (In thousands) |
||||||||||||||||
| U.S. government treasury bills classified as short-term investments |
$ | — | $ | 99,049 | $ | — | $ | 99,049 | ||||||||
| Money-market funds classified as short-term investments |
84,882 | — | — | 84,882 | ||||||||||||
| Total short-term investments |
84,882 | 99,049 | — | 183,931 | ||||||||||||
| Money-market funds classified as non-current restricted investments |
1,054 | — | — | 1,054 | ||||||||||||
| Total investments |
$ | 85,936 | $ | 99,049 | $ | — | $ | 184,985 | ||||||||
Unrealized gains and losses on our short-term investments were not material for either period presented. Cash held in demand deposit accounts of $7.1 million and $11.0 million is excluded from our fair-value hierarchy disclosure as of December 31, 2023 and 2022, respectively. The carrying amounts for receivables, accounts payable and accrued liabilities, and other current monetary assets and liabilities, including lease financing obligations, approximate fair value.
See “Note 6 - Convertible Senior Notes” and “Note 8 – OMIDRIA Royalty Obligation” for the carrying amount and estimated fair value of our 2023 Notes, 2026 Notes and the OMIDRIA royalty obligation.
Note 5—Certain Balance Sheet Accounts
Receivables
Receivables consists of the following:
| December 31, |
December 31, |
|||||||
| 2023 |
2022 |
|||||||
| (In thousands) |
||||||||
| OMIDRIA milestone receivable |
$ | — | $ | 200,000 | ||||
| OMIDRIA royalty receivables |
6,724 | 12,966 | ||||||
| Other receivables |
1,372 | 255 | ||||||
| Total receivables |
$ | 8,096 | $ | 213,221 | ||||
Property and Equipment, Net
Property and equipment, net consists of the following:
| December 31, |
December 31, |
|||||||
| 2023 |
2022 |
|||||||
| (In thousands) |
||||||||
| Equipment under finance leases |
$ | 6,929 | $ | 6,204 | ||||
| Laboratory equipment |
3,525 | 3,135 | ||||||
| Computer equipment |
1,113 | 1,076 | ||||||
| Office equipment and furniture |
624 | 625 | ||||||
| Total cost |
12,191 | 11,040 | ||||||
| Less accumulated depreciation and amortization |
(10,241 | ) | (9,548 | ) | ||||
| Total property and equipment, net |
$ | 1,950 | $ | 1,492 | ||||
For the years ended December 31, 2023, 2022 and 2021, depreciation and amortization expenses were $0.9 million, $1.0 million and $1.4 million, respectively.
Accrued Expenses
Accrued expenses consist of the following:
| December 31, |
December 31, |
|||||||
| 2023 |
2022 |
|||||||
| (In thousands) |
||||||||
| Clinical trials |
$ | 10,168 | $ | 5,536 | ||||
| Employee compensation |
7,380 | 6,665 | ||||||
| Contract research and development |
6,223 | 3,209 | ||||||
| Interest payable |
4,242 | 5,172 | ||||||
| Consulting and professional fees |
3,539 | 4,425 | ||||||
| Other accrued expenses |
316 | 5,544 | ||||||
| Total accrued expenses |
$ | 31,868 | $ | 30,551 | ||||
Note 6—Convertible Senior Notes
On January 1, 2021, we adopted ASU 2020-06, Debt—Debt with Conversion Options (Subtopic 470-20) and Derivatives and Hedging—Contracts in Entity’s Own Equity (Subtopic 815-40) on a modified retrospective basis. ASU 2020-06 removes the separate liability and equity accounting for our convertible senior notes. Consequently, we now account for our convertible senior notes wholly as debt. Upon adoption, we removed the equity component allocated to debt issuance costs increasing convertible senior notes and shareholders’ equity by $75.5 million.
In December 2023, we repurchased $9.1 million par value of our 2026 Notes realizing a non-cash gain on debt extinguishment of $4.1 million to our consolidated statement of operations and comprehensive loss in the current year. On November 15, 2023, we also extinguished at par the $95.0 million outstanding principal amount on our 2023 Notes.
Convertible senior notes outstanding at December 31, 2023 and 2022, respectively, are as follows:
| Balance as of December 31, 2023 |
||||||||||||
| 2023 Notes |
2026 Notes |
Total |
||||||||||
| (In thousands) |
||||||||||||
| Principal amount |
$ | — | $ | 215,924 | $ | 215,924 | ||||||
| Unamortized debt issuance costs |
— | (2,769 | ) | (2,769 | ) | |||||||
| Total convertible senior notes, net |
$ | — | $ | 213,155 | $ | 213,155 | ||||||
| Fair value of outstanding convertible senior notes (1) |
$ | — | $ | 131,444 | ||||||||
| Balance as of December 31, 2022 |
||||||||||||
| 2023 Notes |
2026 Notes |
Total |
||||||||||
| (In thousands) |
||||||||||||
| Principal amount |
$ | 95,000 | $ | 225,030 | $ | 320,030 | ||||||
| Unamortized debt issuance costs |
(619 | ) | (4,124 | ) | (4,743 | ) | ||||||
| Total convertible senior notes, net |
$ | 94,381 | $ | 220,906 | $ | 315,287 | ||||||
| Fair value of outstanding convertible senior notes (1) |
$ | 92,031 | $ | 118,141 | ||||||||
(1) The fair value is classified as Level 3 due to the limited trading activity for the convertible senior notes.
2023 Convertible Senior Notes
The 2023 Notes accrued interest at an annual rate of 6.25% per annum. The 2023 Notes matured on November 15, 2023, and the $95.0 million outstanding principal and related accrued interest were paid at that time.
The following table sets forth total interest expense recognized in connection with the 2023 Notes:
| Year Ended December 31, |
||||||||||||
| 2023 |
2022 |
2021 |
||||||||||
| (In thousands) |
||||||||||||
| Contractual interest expense |
$ | 5,195 | $ | 5,938 | $ | 5,938 | ||||||
| Amortization of debt issuance costs |
619 | 663 | 618 | |||||||||
| Total interest expense |
$ | 5,814 | $ | 6,601 | $ | 6,556 | ||||||
2026 Convertible Senior Notes
The 2026 Notes are unsecured and accrue interest at an annual rate of 5.25% per annum, payable semi-annually in arrears on February 15 and August 15 of each year. The 2026 Notes mature on February 15, 2026, unless earlier purchased, redeemed or converted in accordance with their terms.
The initial conversion rate is 54.0906 shares of our common stock per $1,000 of note principal (equivalent to an initial conversion price of approximately $18.4875 per share of common stock), which equals approximately 12.2 million shares issuable upon conversion, subject to adjustment in certain circumstances.
The 2026 Notes are convertible at the option of the holders on or after November 15, 2025 at any time prior to the close of business on February 12, 2026, the second scheduled trading day immediately before the stated maturity date of February 15, 2026. Additionally, holders may convert their 2026 Notes at their option at specified times prior to the maturity date only if:
(1) during any calendar quarter, beginning after September 30, 2020, that the last reported sale price per share of our common stock exceeds 130% of the conversion price of the 2026 Notes for each of at least 20 trading days in the period of 30 consecutive trading days ending on, and including, the last trading day of the immediately preceding calendar quarter;
(2) during the five consecutive business days immediately after any five-consecutive-trading-day period (such five-consecutive-trading-day period, the “measurement period”) in which the trading price per $1,000 principal amount of 2026 Notes for each trading day of the measurement period was less than 98% of the product of the last reported sale price per share of our common stock on such trading day and the conversion rate on such trading day;
(3) there is an occurrence of one or more certain corporate events or distributions of our common stock; or
(4) we call the 2026 Notes for redemption.
We will settle any conversions by paying or delivering, as applicable, cash, shares of our common stock or a combination of cash and shares of our common stock, at our election, based on the applicable conversion rate(s).
Subject to the satisfaction of certain conditions, we may redeem in whole or in part the 2026 Notes at our option beginning August 15, 2023 through the 50th scheduled trading day immediately before the maturity date at a cash redemption price equal to the principal amount of the 2026 Notes to be redeemed plus any accrued and unpaid interest to, but excluding, the redemption date. The 2026 Notes are subject to redemption only if certain requirements are satisfied, including that the last reported sale price per share of our common stock exceeds 130% of the conversion price on (i) each of at least 20 trading days, whether or not consecutive, during the 30 consecutive trading days ending on, and including, the trading day immediately before the date we send the related redemption notice and (ii) the trading day immediately before the date we send such notice.
In order to reduce the dilutive impact or potential cash expenditure associated with the conversion of the 2026 Notes, we entered into capped call transactions in connection with the issuances of the 2026 Notes (the "2026 Capped Call"). The 2026 Capped Call will cover, subject to anti-dilution adjustments substantially similar to those applicable to the 2026 Notes, the number of shares of common stock underlying the 2026 Notes when our common stock is trading within the range of approximately $18.49 and $26.10. However, should the market price of our common stock exceed the $26.10 cap, then the conversion of the 2026 Notes would have an additional dilutive impact or may require a cash expenditure to the extent the market price exceeds the cap price. The 2026 Capped Call will expire on various dates over the 50-trading-day period ranging from December 2, 2025 to February 12, 2026, if not exercised earlier. The 2026 Capped Call is a separate transaction and not part of the terms of the 2026 Notes and was executed separately from the issuance of the 2026 Notes. The amount paid for the 2026 Capped Call was recorded as a reduction to additional paid-in capital in the consolidated balance sheet. As of December 31, 2023, approximately 12.2 million shares remained outstanding under the 2026 Capped Call.
Further, we concluded the 2026 Capped Call qualifies for a derivative scope exception for instruments that are both indexed to an entity’s own stock and classified in stockholders’ equity in its balance sheet. Consequently, the fair value of the 2026 Capped Call of $23.2 million is classified as equity, not accounted for as derivatives, and will not be subsequently remeasured.
The unamortized debt issuance costs of $2.8 million as of December 31, 2023 will be amortized to interest expense at an effective interest rate of 5.9% over the remaining term.
The following table sets forth interest expense recognized related to the 2026 Notes:
| Year Ended December 31, |
||||||||||||
| 2023 |
2022 |
2021 |
||||||||||
| (In thousands) |
||||||||||||
| Contractual interest expense |
$ | 11,774 | $ | 11,814 | $ | 11,814 | ||||||
| Amortization of debt issuance costs |
1,355 | 1,167 | 1,078 | |||||||||
| Total interest expense |
$ | 13,129 | $ | 12,981 | $ | 12,892 | ||||||
Note 7—Discontinued Operations - Sale of OMIDRIA
On December 23, 2021, we closed the sale of OMIDRIA and related assets, which is reported as discontinued operations in our consolidated statements of operations and comprehensive income. Upon closing, we received an up-front cash payment from Rayner of $126.0 million, and we retained the outstanding receivables and liabilities related to OMIDRIA as of the closing date.
The year ended December 31, 2021, included a gain on the sale of OMIDRIA comprised as follows (in thousands):
| Cash proceeds |
$ | 125,993 | ||
| OMIDRIA contract royalty asset |
184,570 | |||
| Gain on sale of OMIDRIA, gross |
310,563 | |||
| Transaction and closing costs |
(1,972 | ) | ||
| RSUs granted to transferred employees |
(1,419 | ) | ||
| Prepaid assets and inventory at cost |
(1,524 | ) | ||
| Gain on sale of OMIDRIA |
$ | 305,648 |
In December 2022, the achievement of the Milestone Event triggered a $200.0 million Milestone Payment from Rayner which we received in February 2023. The Milestone Event also resulted in a reduction in the U.S. royalty rate from 50% to 30% on OMIDRIA net sales.
The results of operations for OMIDRIA are recorded as income from discontinued operations for all periods presented in the consolidated statements of operations and comprehensive income (loss).
| Year Ended December 31, |
||||||||||||
| 2023 |
2022 |
2021 |
||||||||||
| (In thousands) |
||||||||||||
| Product sales, net |
$ | — | $ | — | $ | 110,735 | ||||||
| Costs and expenses |
— | — | 30,631 | |||||||||
| Gross margin |
— | — | 80,104 | |||||||||
| Gain on sale of OMIDRIA |
— | — | 305,648 | |||||||||
| Milestone income |
— | 200,000 | — | |||||||||
| Interest on OMIDRIA contract royalty asset |
15,315 | 18,634 | — | |||||||||
| Remeasurement adjustments |
41,167 | 14,457 | — | |||||||||
| Other income |
1,087 | 307 | 1,035 | |||||||||
| Income before income tax |
57,569 | 233,398 | 386,787 | |||||||||
| Income tax expense (1) |
(462 | ) | (3,952 | ) | (1,006 | ) | ||||||
| Net income from discontinued operations, net of tax |
$ | 57,107 | $ | 229,446 | $ | 385,781 | ||||||
(1) For further discussion of income tax expense refer to “Note 13 – Income Taxes”.
The following schedule is a rollforward of the OMIDRIA contract royalty asset (in thousands):
| Balance at December 31, 2021 |
$ | 184,570 | ||
| Royalties earned |
(65,439 | ) | ||
| Interest on OMIDRIA contract royalty asset |
18,634 | |||
| Remeasurement adjustments |
14,457 | |||
| Balance at December 31, 2022 |
152,222 | |||
| Royalties earned |
(40,595 | ) | ||
| Interest on OMIDRIA contract royalty asset |
15,315 | |||
| Remeasurement adjustments |
41,167 | |||
| Balance at December 31, 2023 |
$ | 168,109 |
Cash flow from discontinued operations is as follows:
| Year Ended December 31, |
||||||||||||
| 2023 |
2022 |
2021 |
||||||||||
| (In thousands) |
||||||||||||
| Net cash provided by discontinued operations from operating activities |
$ | 241,317 | $ | 78,082 | $ | 55,380 | ||||||
| Net cash provided by discontinued operations from investing activities |
$ | — | $ | — | $ | 125,993 | ||||||
Note 8—OMIDRIA Royalty Obligation
In September 2022, we sold to DRI an interest in our future OMIDRIA royalty receipts and received $125.0 million in cash consideration which was recorded as an OMIDRIA royalty obligation on our consolidated balance sheet. DRI is entitled to receive royalties on OMIDRIA net sales between September 1, 2022 and December 31, 2030, subject to annual caps. DRI receives their prorated monthly cap amount before we receive any royalty proceeds. DRI is not entitled to carry-forward nor recoup any shortfall if the royalties paid by Rayner for an annual period are less than the cap amount applicable to each discrete calendar year. Additionally, DRI has no recourse to or security interest in our assets other than our OMIDRIA royalty receipts.
The changes in the OMIDRIA royalty obligation during the year ended December 31, 2023 are as follows (in thousands):
| Balance at December 31, 2022 |
$ | 126,278 | ||
| Principal payments |
(1,152 | ) | ||
| Balance at December 31, 2023 |
$ | 125,126 |
The OMIDRIA royalty obligation is classified as a Level 3 liability as its valuation requires substantial judgment and estimation of factors that are not currently observable in the market. As of December 31, 2023, the approximate fair value of our obligation was $116.3 million.
For the years ended December 31, 2023 and December 31, 2022, we incurred interest expense of $11.8 million and $2.9 million, respectively, on the OMIDRIA royalty obligation.
As of December 31, 2023, the maximum scheduled principal and interest payments (based on an implied effective interest rate of 9.4%) are as follows:
| Total |
||||||||||||
| Principal |
Interest |
Annual Cap |
||||||||||
| (In thousands) |
||||||||||||
| 2024 |
$ | 8,576 | $ | 11,424 | $ | 20,000 | ||||||
| 2025 |
14,641 | 10,359 | 25,000 | |||||||||
| 2026 |
16,081 | 8,919 | 25,000 | |||||||||
| 2027 |
17,664 | 7,336 | 25,000 | |||||||||
| 2028 |
19,402 | 5,598 | 25,000 | |||||||||
| Thereafter |
48,762 | 4,988 | 53,750 | |||||||||
| Total scheduled payments |
$ | 125,126 | $ | 48,624 | $ | 173,750 | ||||||
Subsequent Event
In February 2024, Omeros and DRI expanded their royalty purchase agreement, resulting in Omeros receiving $115.5 million in cash consideration from DRI upon closing. The Amended and Restated Royalty Purchase Agreement ("RPA") eliminated the caps on royalty payments effective in the first quarter of 2024 and provides that DRI will now receive all royalties on U.S. net sales of OMIDRIA payable between January 1, 2024 and December 31, 2031. DRI is entitled to payment only to the extent of royalty payments that are payable in the respect of U.S. net sales of OMIDRIA on or before December 31, 2031 and DRI has no recourse to our assets other than its interest in OMIDRIA royalties. Omeros retains the right to receive all royalties payable by Rayner on any net sales of OMIDRIA outside the U.S. payable from and after January 1, 2024, as well as royalties on global net sales of OMIDRIA payable from and after December 31, 2031. To date, international royalties have not been significant. We are also entitled to receive a milestone ranging between $10.0 million and $27.5 million if U.S. net sales of OMIDRIA reach applicable thresholds ranging between a total of $156.0 million and $160.0 million for any period of four consecutive quarters prior to January 1, 2026. In addition, we are entitled to receive a separate milestone ranging between $8.0 million and $27.5 million if U.S. net sales of OMIDRIA reach applicable thresholds ranging between a total of $181.0 million and $185.0 million for any period of four consecutive quarters prior to January 1, 2028.
Note 9—Lease Liabilities
We have operating leases related to our office and laboratory space. The initial term of the leases is through November 2027 and we have two options to extend the lease term, each by five years. We have finance leases for certain laboratory and office equipment that have lease terms expiring through November 2026.
Lease-related assets and liabilities recorded on our consolidated balance sheet are as follows:
| December 31, |
December 31, |
|||||||
| 2023 |
2022 |
|||||||
| (In thousands) |
||||||||
| Assets |
||||||||
| Operating lease assets |
$ | 18,631 | $ | 21,762 | ||||
| Finance lease assets, net |
1,220 | 945 | ||||||
| Total lease assets |
$ | 19,851 | $ | 22,707 | ||||
| Liabilities |
||||||||
| Current: |
||||||||
| Operating leases |
$ | 4,590 | $ | 3,888 | ||||
| Finance leases |
570 | 422 | ||||||
| Non-current: |
||||||||
| Operating leases |
17,424 | 21,971 | ||||||
| Finance leases |
719 | 455 | ||||||
| Total lease liabilities |
$ | 23,303 | $ | 26,736 | ||||
| Weighted-average remaining lease term |
||||||||
| Operating leases (years) |
3.8 | 4.8 | ||||||
| Finance leases (years) |
2.3 | 2.3 | ||||||
| Weighted-average discount rate |
||||||||
| Operating leases |
12.81 | % | 12.81 | % | ||||
| Finance leases |
8.57 | % | 10.44 | % | ||||
The components of total lease costs are as follows:
| Year Ended |
||||||||
| December 31, |
||||||||
| 2023 |
2022 |
|||||||
| (In thousands) |
||||||||
| Lease cost |
||||||||
| Operating lease cost |
$ | 6,464 | $ | 6,152 | ||||
| Finance lease cost: |
||||||||
| Amortization |
677 | 812 | ||||||
| Interest |
174 | 174 | ||||||
| Variable lease cost |
3,160 | 3,191 | ||||||
| Sublease income |
(1,500 | ) | (1,755 | ) | ||||
| Net lease cost |
$ | 8,975 | $ | 8,574 | ||||
The supplemental cash flow information related to leases is as follows:
| Year Ended |
||||||||
| December 31, |
||||||||
| 2023 |
2022 |
|||||||
| (In thousands) |
||||||||
| Cash paid for amounts included in the measurement of lease liabilities |
||||||||
| Cash payments for operating leases |
$ | 7,144 | $ | 7,072 | ||||
| Cash payments for financing leases |
$ | 655 | $ | 790 | ||||
The future maturities of our lease liabilities as of December 31, 2023 are as follows:
| Operating |
Finance |
|||||||||||
| Leases |
Leases |
Total |
||||||||||
| (In thousands) |
||||||||||||
| 2024 |
$ | 8,528 | $ | 684 | $ | 9,212 | ||||||
| 2025 |
7,088 | 517 | 7,605 | |||||||||
| 2026 |
6,870 | 258 | 7,128 | |||||||||
| 2027 |
5,950 | — | 5,950 | |||||||||
| Total undiscounted lease payments |
28,436 | 1,459 | 29,895 | |||||||||
| Less interest |
(6,422 | ) | (170 | ) | (6,592 | ) | ||||||
| Total lease liabilities |
$ | 22,014 | $ | 1,289 | $ | 23,303 | ||||||
Note 10—Commitments and Contingencies
Contracts
We have various agreements with third parties that collectively require payment of termination fees totaling $25.8 million as of December 31, 2023 if we cancel the work within specific time frames, either prior to commencing or during performance of the contracted services.
Development Milestones and Product Royalties
We have entered a variety of development, collaboration, licensing or similar agreements with third parties under which we have accessed technology or services in connection with our development assets and programs. Some of these agreements require milestone payments based on achievements of development, regulatory or sales milestones, and/or low-single to low-double digit royalties on net income or net sales of the relevant product. For the years ended December 31, 2023, 2022 and 2021, we paid $5.0 million, $0.3 million and $0.5 million, respectively in development milestones.
Note 11—Shareholders’ Equity (Deficit)
Common Stock
As of December 31, 2023, we had reserved shares of common stock under our equity plans as follows:
| Stock options outstanding |
15,255,154 | |||
| Awards available to issue under the 2017 Plan |
8,802,249 | |||
| Total shares reserved |
24,057,403 |
At the Market Sales Agreement – We have a sales agreement to sell shares of our common stock having an aggregate offering price of up to $150.0 million, from time to time, through an “at the market” equity offering program.
Amendment of 2017 Omnibus Incentive Compensation Plan - At our June 23, 2023 annual meeting, our shareholders approved a 5,000,000 share increase in the number of shares of common stock available for grant under the 2017 Omnibus Incentive Compensation Plan, as amended and restated.
Share Repurchase Program - On November 9, 2023, the Board of Directors approved an indefinite-term share repurchase program under which we may repurchase from time to time up to $50.0 million of our common stock in the open market or through privately negotiated transactions. For the year ended December 31, 2023, we repurchased and retired 1.8 million shares of common stock at an average share price of $2.54, for an aggregate repurchase price of $4.7 million.
Note 12—Stock-Based Compensation
Our equity plans provide for the grant of incentive and non-statutory stock options, stock appreciation rights, restricted stock awards, restricted stock units, performance units, performance shares and other stock and cash awards to employees and consultants. Stock options are granted with an exercise price not less than the fair market value of Omeros’ common stock on the date of the grant. Any unexercised options expire 10 years from grant date, and any unvested stock options granted which are subsequently canceled become available for future reissuance.
Vesting schedules for our equity plans generally are as follows:
| Grant Type |
Vesting Schedule |
| Employee initial options grants |
25% at one-year anniversary, 1/48 monthly thereafter |
| Employee recurring options grants |
1/48 monthly |
| Non-employee consultant options grants |
1/12 or 1/48 monthly |
| Employee RSUs |
50% after one year, 50% after two years |
Stock-based compensation expense is as follows:
| Year Ended December 31, |
||||||||||||
| 2023 |
2022 |
2021 |
||||||||||
| (In thousands) |
||||||||||||
| Continuing operations: |
||||||||||||
| Research and development |
$ | 4,754 | $ | 6,123 | $ | 6,791 | ||||||
| Selling, general and administrative |
7,140 | 8,042 | 8,154 | |||||||||
| Total stock-based compensation in continuing operations |
11,894 | 14,165 | 14,945 | |||||||||
| Discontinued operations |
(244 | ) | (93 | ) | 2,685 | |||||||
| Total stock-based compensation |
$ | 11,650 | $ | 14,072 | $ | 17,630 | ||||||
The fair value of each option grant is estimated on the date of grant using the Black-Scholes option-pricing model. The following assumptions were applied to stock option grants during the periods ended:
| Year Ended December 31, |
||||||||||||
| 2023 |
2022 |
2021 |
||||||||||
| Estimated weighted-average fair value |
$ | 2.44 | $ | 2.94 | $ | 10.54 | ||||||
| Weighted-average assumptions: |
||||||||||||
| Expected volatility |
93 | % | 90 | % | 81 | % | ||||||
| Expected life, in years |
7.2 | 6.0 | 6.0 | |||||||||
| Risk-free interest rate |
3.97 | % | 2.83 | % | 1.06 | % | ||||||
| Expected dividend yield |
— | % | — | % | — | % | ||||||
Expected volatility is based on the historical volatility of our stock price weighted by grant issuances over the reporting period. We estimated the expected life of the stock options granted using the historical exercise behavior of option holders. The risk-free interest rate is based on the U.S. Treasury yield curve in effect at the time of grant. Forfeiture expense is estimated at the time of grant and revised in subsequent periods if actual forfeitures differ from those estimates.
Stock option activity for all stock option plans is as follows:
| Options Outstanding |
Weighted- Average Exercise Price per Share | Remaining Contractual Life (In years) |
Aggregate Intrinsic Value (In thousands) |
||||||||||
| Balance at December 31, 2022 |
13,872,973 | $ | 11.02 | ||||||||||
| Granted |
3,153,200 | 3.01 | |||||||||||
| Exercised |
(36,726 | ) | 4.10 | ||||||||||
| Forfeited |
(1,734,293 | ) | 9.96 | ||||||||||
| Balance at December 31, 2023 |
15,255,154 | $ | 9.50 | 6.2 | $ | 1,388 | |||||||
| Vested and expected to vest at December 31, 2023 |
14,762,090 | $ | 9.65 | 6.0 | $ | 1,272 | |||||||
| Exercisable at December 31, 2023 |
10,554,140 | $ | 11.50 | 4.7 | $ | 217 | |||||||
Of the 15.3 million common stock options outstanding as of December 31, 2023, 12.3 million have an exercise price above the $3.27 closing price of our stock on the Nasdaq exchange on December 31, 2023. The total intrinsic value of stock options exercised during the years ended December 31, 2023, 2022 and 2021 was $0.1 million, $0.2 million and $7.8 million, respectively.
At December 31, 2023, there were 4.7 million unvested stock options outstanding that vest over a weighted-average period of 2.1 years. The remaining estimated compensation expense to be recognized in connection with these unvested stock options is $14.5 million.
RSU activity for all stock plans is as follows:
| RSUs Outstanding |
Weighted- Average Grant Date Fair Value Per Share |
||||||
| Balance at December 31, 2022 |
98,750 | $ | 7.53 | ||||
| Vested and released |
(67,250 | ) | 7.53 | ||||
| Forfeited |
(31,500 | ) | 7.53 | ||||
| Balance at December 31, 2023 |
— | $ | — | ||||
Note 13—Income Taxes
The components of income tax benefit from continuing and discontinued operations were as follows:
| December 31, |
||||||||||||
| 2023 |
2022 |
2021 |
||||||||||
| (In thousands) |
||||||||||||
| Continuing operations: |
||||||||||||
| Current income tax expense: |
||||||||||||
| Federal |
$ | — | $ | — | $ | — | ||||||
| State |
— | — | — | |||||||||
| Total current income tax expense |
— | — | — | |||||||||
| Deferred income tax benefit: |
||||||||||||
| Federal |
— | — | — | |||||||||
| State |
— | — | — | |||||||||
| Total deferred income tax benefit |
— | — | — | |||||||||
| Income tax benefit in continuing operations |
$ | — | $ | — | $ | — | ||||||
| Income tax expense as a component of discontinued operations |
$ | 462 | $ | 3,952 | $ | 1,006 | ||||||
For the years ended December 31, 2023, 2022 and 2021, for federal and state income tax purposes, we had net losses from continuing operations and net income from discontinued operations, which resulted in an overall tax loss. At December 31, 2023, 2022 and 2021, we had federal net operating loss ("NOL") carryforwards of approximately $398.6 million, $361.4 million and $630.6 million, respectively, for all periods. At December 31, 2023, 2022 and 2021, we had state NOL carryforwards of approximately $245.8 million, $226.3 million and $245.1 million, respectively. In 2023, we had a net loss for federal income tax purposes and in 2022 and 2021, we utilized existing net operating loss carryforwards of $268.6 million and $245.1 million, respectively to fully offset our federal tax liability for both periods. We recorded state income tax expense of $0.5 million, $4.0 million and $1.0 million in discontinued operations in 2023, 2022 and 2021, respectively as we did not have adequate net operating losses and tax credits to fully offset our state tax liability.
Deferred income tax assets and liabilities reflect the tax effect of net operating loss and tax credit carryforwards and the net temporary difference between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes.
Significant components of deferred income taxes were as follows:
| December 31, |
||||||||
| 2023 |
2022 |
|||||||
| (In thousands) |
||||||||
| Deferred tax assets: |
||||||||
| Net operating loss carryforwards |
$ | 95,183 | $ | 85,887 | ||||
| Research and development tax credits |
92,837 | 78,992 | ||||||
| Capitalized research and development |
39,318 | 21,864 | ||||||
| OMIDRIA royalty obligation |
28,903 | 28,938 | ||||||
| Stock-based compensation |
10,132 | 12,517 | ||||||
| Lease liability |
5,085 | 5,926 | ||||||
| Other |
10,283 | 9,234 | ||||||
| Total deferred tax assets |
281,741 | 243,358 | ||||||
| Deferred tax liabilities: |
||||||||
| OMIDRIA contract royalty asset |
(38,832 | ) | (34,883 | ) | ||||
| Right of use assets |
(4,304 | ) | (4,987 | ) | ||||
| Property and equipment |
(122 | ) | (288 | ) | ||||
| Total deferred tax liabilities |
(43,258 | ) | (40,158 | ) | ||||
| Net deferred tax assets before valuation allowance |
238,483 | 203,200 | ||||||
| Less valuation allowance |
(238,483 | ) | (203,200 | ) | ||||
| Net deferred tax liabilities |
$ | — | $ | — | ||||
As of December 31, 2023, we had federal net operating loss carryforwards of approximately $398.6 million and state net operating loss carryforwards of approximately $245.8 million. Pre-2018 federal net operating losses of $109.8 million expire between 2035 and 2037. Post-2018 federal net operating losses of $288.8 million do not expire. Research and development tax credit carryforwards of $93.0 million expire between 2024 and 2043.
The Tax Cuts and Jobs Act was enacted on December 22, 2017 and includes the requirement to capitalize and amortize research and experimental expenditures beginning in 2022. Prior to 2022, we expensed these costs as incurred for tax purposes.
Reconciliation of income tax computed at federal statutory rates to the reported provisions for income taxes from continuing operations are as follows:
| Year ended December 31, |
||||||||||||
| 2023 |
2022 |
2021 |
||||||||||
| U.S. Federal statutory rate on net loss |
(21.0 | )% | (21.0 | )% | (21.0 | )% | ||||||
| State tax, net of federal tax benefit |
(2.1 | )% | (1.7 | )% | (0.6 | )% | ||||||
| Change in valuation allowance |
27.7 | % | 28.3 | % | 26.9 | % | ||||||
| Tax credits |
(8.0 | )% | (6.8 | )% | (5.5 | )% | ||||||
| Stock compensation |
1.5 | % | 1.4 | % | 0.3 | % | ||||||
| Other |
1.9 | % | (0.2 | )% | (0.1 | )% | ||||||
| Effective tax rate |
0.0 | % | 0.0 | % | 0.0 | % | ||||||
We file federal and certain state income tax returns, which provides varying statutes of limitations on assessments. However, because of net operating loss carryforwards, substantially all our tax years remain open to federal and state tax examination.
As of December 31, 2023 and 2022, the total amount of gross unrecognized tax benefits was $2.0 million and $0.2 million, respectively. We recognized $0.3 million of interest and penalties at December 31, 2023 as an unrecognized tax benefit. As of December 31, 2023, $1.8 million of the total unrecognized tax benefits, if recognized, would have an impact on our effective tax rate. We estimate that there will be no material changes in this uncertain tax position for the next 12 months. Our policy is to recognize interest and penalties accrued on any unrecognized tax benefits as a component of income tax expense.
The following table summarizes the activities related to our gross unrecognized tax benefits (in thousands):
| Balance at December 31, 2022 |
$ | 212 | ||
| Increase in balance related to tax positions taken during prior years |
1,796 |
|||
| Decrease in balance related to tax positions during prior years |
(30 | ) | ||
| Decrease in balance as a result of a lapse of the applicable statute of limitations |
(12 | ) | ||
| Balance at December 31, 2023 |
$ | 1,966 |
Note 14—401(k) Retirement Plan
Our 401(k) retirement plan provides for an annual company discretionary match on employee contributions. For the years-ended December 31, 2023, 2022 and 2021, Omeros' 401(k) match expense was $0.6 million, $0.6 million and $0.8 million, respectively. We match up to 4.0% of each participating employee’s eligible earnings, with a maximum company match of $4,000 per employee per year. All employees are eligible to participate.
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
None.
ITEM 9A. CONTROLS AND PROCEDURES
Disclosure Controls and Procedures
Our management, with the participation of our principal executive officer and principal financial officer, evaluated the effectiveness of our disclosure controls and procedures, as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act, as of December 31, 2023. Management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving their objectives, and management necessarily applies its judgment in evaluating the cost-benefit relationship of possible controls and procedures. Based on the evaluation of our disclosure controls and procedures as of December 31, 2023, our principal executive and principal financial officers concluded that, as of such date, our disclosure controls and procedures were effective at the reasonable assurance level.
Management’s Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Exchange Act Rules 13a-15(f) and 15d-15(f). Internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles.
Internal control over financial reporting cannot provide absolute assurance of achieving financial reporting objectives because of its inherent limitations. Internal control over financial reporting is a process that involves human diligence and compliance and is subject to lapses in judgment and breakdowns resulting from human failures. Internal control over financial reporting also can be circumvented by collusion or improper management override. Because of such limitations, there is a risk that material misstatements may not be prevented or detected on a timely basis by internal control over financial reporting. However, these inherent limitations are known features of the financial reporting process. Therefore, it is possible to design into the process safeguards to reduce, though not eliminate, this risk.
Our management, with the participation of our principal executive and principal financial officers, conducted an assessment of the effectiveness of our internal control over financial reporting as of December 31, 2023. In making this assessment, management used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission in Internal Control-Integrated Framework (2013 framework). Based on the results of this assessment and on those criteria, our management concluded that our internal control over financial reporting was effective as of December 31, 2023.
There was no change in our internal control over financial reporting identified in connection with the evaluation required by Rules 13a-15(d) and 15d-15(d) of the Exchange Act that occurred during our fourth fiscal quarter of 2023 that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
(a) None.
(b) During the three months ended December 31, 2023, none of our directors or officers adopted or terminated any contract, instruction or written plan for the purchase or sale of our securities that was intended to satisfy the affirmative defense conditions of Rule 10b5-1(c) under the Exchange Act or any “non-Rule 10b5-1 trading arrangement” (as defined in Item 408(c) of Regulation S-K).
ITEM 9C. DISCLOSURE REGARDING FOREIGN JURISDICTIONS THAT PREVENT INSPECTIONS
Not applicable.
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
The information required by this item will be contained in our definitive proxy statement issued in connection with the 2024 Annual Meeting of Shareholders and is incorporated herein by reference. Certain information required by this item concerning executive officers is set forth in Part I of this Annual Report on Form 10-K under the heading “Business - Information About Our Executive Officers and Significant Employees.”
ITEM 11. EXECUTIVE COMPENSATION
The information required by this item will be contained in our definitive proxy statement issued in connection with the 2024 Annual Meeting of Shareholders and is incorporated herein by reference.
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED SHAREHOLDER MATTERS
Except for the information set forth below, the information required by this item will be contained in our definitive proxy statement issued in connection with the 2024 Annual Meeting of Shareholders and is incorporated herein by reference.
Securities Authorized for Issuance Under Equity Compensation Plans
The following table provides certain information regarding our equity compensation plans in effect as of December 31, 2023:
| Number of Securities to be Issued Upon Exercise of Outstanding Options, Warrants and Rights |
Weighted-Average Exercise Price of Outstanding Options, Warrants and Rights |
Number of Securities Remaining Available for Future Issuance Under Equity Compensation Plans |
||||||||||
| Equity compensation plans approved by security holders: |
||||||||||||
| 2017 Omnibus Incentive Compensation Plan (1) |
11,201,040 | $ | 8.82 | 8,802,249 | ||||||||
| 2008 Equity Incentive Plan (2) |
4,054,114 | $ | 11.37 | — | ||||||||
| Total |
15,255,154 | $ | 9.50 | 8,802,249 | ||||||||
(1) Our 2017 Plan provides for the grant of incentive and non-statutory stock options, restricted stock, restricted stock units, stock appreciation rights, performance units and performance shares to employees, directors and consultants and subsidiary corporations’ employees and consultants. The 2017 Plan replaced the 2008 Plan, and as a result we will not grant any new awards under the 2008 Plan. Any stock option awards granted under the 2008 Plan that were outstanding as of the effective date of the 2017 Plan remained in effect pursuant to their terms and, if the award is canceled or is repurchased, the shares underlying such award become available for grant under the 2017 Plan.
(2) The 2008 Plan provided for the grant of incentive and non-statutory stock options, restricted stock, stock appreciation rights, performance units and performance shares to employees, directors and consultants and subsidiary corporations’ employees and consultants.
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE ITEM 14.
The information required by this item will be contained in our definitive proxy statement issued in connection with the 2024 Annual Meeting of Shareholders and is incorporated herein by reference.
PRINCIPAL ACCOUNTING FEES AND SERVICES
The information required by this item will be contained in our definitive proxy statement issued in connection with the 2024 Annual Meeting of Shareholders and is incorporated herein by reference.
ITEM 15. EXHIBITS, FINANCIAL STATEMENT SCHEDULES
1. Financial Statements
See the Index to Consolidated Financial Statements in Part II, Item 8 of this Form 10-K.
2. Financial Statement Schedules
All schedules have been omitted as the required information is either not required, not applicable or otherwise included in the Financial Statements and notes thereto.
3. Exhibits
The following list of exhibits includes exhibits submitted with this Form 10-K as filed with the SEC and those incorporated by reference to other filings.
EXHIBIT INDEX
| Incorporated by Reference | ||||||||||||
| Exhibit |
Exhibit Description |
Form |
File No. |
Exhibit |
Filing Date |
Filed |
||||||
| 1.1 |
Sales Agreement, dated March 1, 2021, between Omeros Corporation and Cantor Fitzgerald & Co. |
10-K |
001-34475 |
1.1 |
03/01/2021 |
|||||||
| 3.1 |
Amended and Restated Articles of Incorporation of Omeros Corporation |
10-K |
001-34475 |
3.1 |
03/31/2010 |
|||||||
| 3.2 |
10-K |
001-34475 |
3.2 |
03/31/2010 |
||||||||
| 4.1 |
10-K |
001-34475 |
4.1 |
03/01/2021 |
||||||||
| 4.2 |
S-1/A |
333-148572 |
4.1 |
10/02/2009 |
||||||||
| 4.3 |
8-K |
001-34475 |
4.1 |
08/14/2020 |
||||||||
| 4.4 |
8-K |
001-34475 |
4.2 |
08/14/2020 |
||||||||
| 10.1* |
S-1 |
333-148572 |
10.1 |
01/09/2008 |
||||||||
| 10.2* |
10-K |
001-34475 |
10.6 |
03/16/2017 |
||||||||
| 10.3* |
Form of Stock Option Award Agreement under the 2008 Equity Incentive Plan |
10-Q |
001-34475 |
10.2 |
11/07/2013 |
|||||||
| 10.4* |
2017 Omnibus Incentive Compensation Plan (as amended and restated effective as of June 23, 2023) |
8-K |
001-34475 |
10.1 |
06/28/2023 |
|||||||
| 10.5* |
Form of Stock Option Award Agreement under the 2017 Omnibus Incentive Compensation Plan |
S-8 |
333-218882 |
4.4 |
06/21/2017 |
| 10.6* |
8-K |
001-34475 |
10.1 |
04/12/2010 |
||||||||
| 10.7* |
Omeros Corporation Non-Employee Director Compensation Policy |
10-K | 001-34475 | 10.11 | 03/13/2023 |
|
||||||
| 10.8 |
Lease dated January 27, 2012 between Omeros Corporation and BMR-201 Elliott Avenue LLC |
8-K |
001-34475 |
10.1 |
02/01/2012 |
|||||||
| 10.9 |
10-Q |
001-34475 |
10.2 |
11/09/2012 |
||||||||
| 10.10 |
10-K |
001-34475 |
10.18 |
03/18/2013 |
||||||||
| 10.11 |
10-K |
001-34475 |
10.18 |
03/13/2014 |
||||||||
| 10.12 |
10-Q |
001-34475 |
10.3 |
11/09/2015 |
||||||||
| 10.13 |
10-Q |
001-34475 |
10.1 |
05/10/2017 |
||||||||
| 10.14 |
10-K |
001-34475 |
10.19 |
03/01/2019 |
| 10.15 |
10-Q |
001-34475 |
10.1 |
08/08/2019 |
||||||||
| 10.16 |
10-K |
001-34475 |
10.20 |
03/02/2020 |
||||||||
| 10.17 |
10-Q |
001-34475 |
10.1 |
05/11/2020 |
||||||||
| 10.18 |
10-Q |
001-34475 |
10.1 |
11/09/2020 |
||||||||
| 10.19 |
10-K |
001-34475 |
10.23 |
03/01/2021 |
||||||||
| 10.20 |
10-K |
001-34475 |
10.24 |
03/01/2021 |
||||||||
| 10.21 |
10-Q |
001-34475 |
10.1 |
08/09/2021 |
||||||||
| 10.22 | Fourteenth Amendment to Lease dated January 14, 2022 between Omeros Corporation and BMR-201 Elliott Avenue LLC | 10-Q | 001-34475 | 10.1 | 05/10/2022 |
| 10.23† |
X | |||||||||||
| 10.24† |
X | |||||||||||
| 10.25† |
X | |||||||||||
| 10.26 |
8-K |
001-34475 |
10.1 |
08/14/2020 |
||||||||
| 10.27† |
10-Q |
001-34475 |
10.1 |
11/12/2019 |
||||||||
| 10.28† | Technology License Agreement, effective August 28, 2020 between Omeros Corporation and Xencor, Inc. | 10-K | 001-34475 | 10.1 | 03/13/2023 | |||||||
| 10.29† | Asset Purchase Agreement, dated as of December 1, 2021 among Omeros Corporation, Rayner Surgical Inc. and Rayner Surgical Group, Limited, as Parent Guarantor | 10-K | 001-34475 | 10.1 | 03/01/2022 | |||||||
| 10.30† | Amended and Restated Royalty Purchase Agreement between Omeros Corporation and DRI Healthcare Acquisitions LP dated February 1, 2024 | X | ||||||||||
| 23.1 |
X |
|||||||||||
| 31.1 |
X |
|||||||||||
| 31.2 |
X |
|||||||||||
| 32.1 |
X |
|||||||||||
| 32.2 |
X |
|||||||||||
| 97.1 | Omeros Corporation Compensation Clawback Policy | X | ||||||||||
| 101.INS |
Inline XBRL Instance Document |
X |
||||||||||
| 101.SCH |
Inline XBRL Taxonomy Extension Schema Document |
X |
||||||||||
| 101.CAL |
Inline XBRL Taxonomy Extension Calculation Linkbase Document |
X |
||||||||||
| 101.DEF |
Inline XBRL Taxonomy Extension Definition Linkbase Document |
X |
||||||||||
| 101.LAB |
Inline XBRL Taxonomy Extension Labels Linkbase Document |
X |
| 101.PRE |
Inline XBRL Taxonomy Extension Presentation Linkbase Document |
X |
||||||||||
| 104.1 |
Cover Page Interactive Data File, formatted in Inline XBRL (included in Exhibit 101) |
X |
| * |
Indicates management contract or compensatory plan or arrangement. |
| † |
Certain identified information has been excluded from the exhibit because it both (A) is not material and (B) would be competitively harmful if publicly disclosed. |
ITEM 16. FORM 10-K SUMMARY Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
Not included.
| OMEROS CORPORATION |
|
| /s/ GREGORY A. DEMOPULOS, M.D. |
|
| Gregory A. Demopulos, M.D. |
|
| President, Chief Executive Officer |
Dated: April 1, 2024
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| Signature |
Title |
Date |
||
| /s/ GREGORY A. DEMOPULOS, M.D. |
President, Chief Executive Officer and Chairman of the Board of Directors (Principal Executive Officer) |
April 1, 2024 | ||
| Gregory A. Demopulos, M.D. |
||||
| /s/ MICHAEL A. JACOBSEN |
Vice President, Finance, Chief Accounting Officer and Treasurer (Principal Financial Officer and Principal Accounting Officer) |
April 1, 2024 | ||
| Michael A. Jacobsen |
||||
| /s/ THOMAS F. BUMOL, PH.D. |
Director |
April 1, 2024 | ||
| Thomas F. Bumol, Ph.D. |
||||
| /s/ THOMAS J. CABLE |
Director |
April 1, 2024 | ||
| Thomas J. Cable |
||||
| /s/ PETER A. DEMOPULOS, M.D. |
Director |
April 1, 2024 | ||
| Peter A. Demopulos, M.D. |
||||
| /s/ ARNOLD C. HANISH |
Director |
April 1, 2024 | ||
| Arnold C. Hanish |
||||
| /s/ LEROY E. HOOD, M.D., PH.D. |
Director |
April 1, 2024 | ||
| Leroy E. Hood, M.D., Ph.D. |
||||
| /s/ DIANA PERKINSON, M.D. |
Director |
April 1, 2024 | ||
| Diana Perkinson, M.D. |
||||
|
|
Director |
April 1, 2024 | ||
| Rajiv Shah, M.D. |
Exhibit 10.23
[***] CERTAIN IDENTIFIED INFORMATION HAS BEEN EXCLUDED FROM THE EXHIBIT BECAUSE IT BOTH (A) IS NOT MATERIAL AND (B) WOULD BE COMPETITIVELY HARMFUL IF PUBLICLY DISCLOSED.
LICENSE AGREEMENT
between
ASUBIO PHARMA CO., LTD. and OMEROS CORPORATION
This license agreement (this “Agreement”) is made effective the 3rd day of March 2010 (the “Effective Date”) between Asubio Pharma Co., Ltd., a Japanese Corporation having a place of business at 9-11 Akasaka 2-Chome, Minato-Ku, Tokyo 107-8541 Japan (“Asubio”), and Omeros Corporation, a Washington corporation having a principal place of business at 1420 Fifth Avenue, Suite 2600, Seattle, WA 98101 USA (“Omeros”).
WHEREAS Asubio owns rights to certain phosphodiesterase-7 (“PDE7”) inhibitors and derivatives thereof, claimed in certain related patents and pending patent applications owned by Asubio;
WHEREAS Asubio and Omeros entered into a Mutual Confidential Disclosure Agreement executed on June 6, 2008 and amended on June 18, 2009 (the “Mutual CDA”) and a Material Transfer Agreement executed on October 20, 2008 (the “MTA”) to permit Omeros to evaluate certain of Asubio’s PDE7 inhibitors and related confidential information;
WHEREAS Omeros owns rights to certain pending patent applications directed to the use of PDE7 inhibitors for the treatment of movement disorders;
WHEREAS Omeros wishes to undertake an exclusive license to Asubio’s rights in certain of Asubio’s PDE7 inhibitors under Asubio’s related patents for development and commercialization by Omeros for the treatment of movement disorders [***]; and
WHEREAS Asubio wishes to grant Omeros an exclusive license to such inhibitors and related patents and patent applications in the field of movement disorders [***] in consideration of the milestone and royalty payments set forth in this Agreement;
NOW THEREFORE, in consideration for the mutual covenants and obligations set forth herein as well as other good and valuable consideration, the parties hereby agree as follows:
|
1 |
Key Definitions |
|
|
1.1 |
“Affiliate” as used herein shall include any affiliate, subsidiary or parent of either party and in each case shall mean any corporation or other entity directly or indirectly controlled by, controlling or under common control with the party, and for such purposes “control” shall mean the direct or indirect ownership of more than fifty percent (50%) of the voting interest in such other corporation or other entity, or the power to direct the management of such other corporation or other entity. |
|
|
1.2 |
“Asubio Patents” means the patents and patent applications owned by Asubio that are listed on Schedule A attached to this Agreement, as well as all foreign and national counterparts, all continuations, divisionals, reissues and reexaminations corresponding thereto or claiming priority therefrom, and all patents, inventor certificates and utility models issuing therefrom. |
|
|
1.3 |
“Asubio Know-How” means Asubio’s data and information listed in Schedule B |
|
[***] |
CERTAIN IDENTIFIED INFORMATION HAS BEEN EXCLUDED FROM THE EXHIBIT BECAUSE IT BOTH (A) IS NOT MATERIAL AND (B) WOULD BE COMPETITIVELY HARMFUL IF PUBLICLY DISCLOSED. |
|
attached to this Agreement, which data was disclosed by Asubio to Omeros under the Mutual CDA and/or the MTA, and any additional data, information and records disclosed from Asubio to Omeros in accordance with this Agreement. |
||
|
1.4 |
“Compounds” means the Parent Compounds, samples of some of which were previously supplied by Asubio to Omeros in accordance with the terms of the MTA, and any Improved Compounds. Asubio and Omeros agree that the Excluded Compounds are not included in the Compounds. |
|
|
1.5 |
“Compound Improvement Patents” mean all patents and patent applications claiming substances that are new improvements (including any structural derivatives or analogs), variations, updates, modifications, and enhancements to the Parent Compounds made by or for Omeros at any time commencing upon execution of the MTA and ending upon the termination of this Agreement, but excluding any patent or patent application claims to new uses or methods of use of the Compounds. |
|
|
1.6 |
“Excluded Compounds” means the compounds described in Schedule D attached to this Agreement. |
|
|
1.7 |
“Field” means all movement disorders described in WHO ICD-10 (G20-G26) and/or in Omeros’ published International PCT Patent Application WO 2008/119057 A2, including, without limitation, Parkinson’s Disease, Restless Legs Syndrome, Post-encephalitic Parkinsonism, Dopamine-Responsive Dystonia, Shy-Drager Syndrome, Periodic Limb Movement Disorder, Periodic Limb Movements in Sleep, Tourette’s Syndrome, all other movement disorders treatable with a dopamine receptor agonist or a precursor of a dopamine receptor agonist, [***]. |
|
|
1.8 |
“Field Improvement Patents” means all patents and patent applications claiming new uses or methods of use of the Compounds solely in the Field made by or for Omeros at any time commencing upon execution of the MTA and ending upon the termination of this Agreement, but excluding any claim to the chemical structure of the Compounds and excluding any claim to uses or methods of use relating to treatment of diseases in the dermatology and dermatologic affections defined as any diseases of the skin, hair/scalp or nails. |
|
|
1.9 |
“Improved Compounds” means any compound, other than the Parent Compounds, encompassed by the claims of the Asubio Patents and/or Compound Improvement Patents, including new improvements (including any structural derivatives or analogs), variations, updates, modifications, and enhancements to the Parent Compounds, made by or for Omeros at any time commencing upon execution of the MTA and ending upon the termination of this Agreement. Asubio and Omeros agree that the Excluded Compounds are not included in the Improved Compounds. Improved Compounds shall include Improved Compounds that are encompassed only by valid claim(s) of the Asubio Patents (“Minor Improved Compounds”), Improved Compounds that are encompassed by valid claim(s) of both the Asubio Patents and the Compound Improvement Patents (“Major Improved Compounds”) and Improved Compounds that are encompassed only by valid claim(s) of the Compound Improvement Patents (“Other Improved Compounds”). |
|
[***] |
CERTAIN IDENTIFIED INFORMATION HAS BEEN EXCLUDED FROM THE EXHIBIT BECAUSE IT BOTH (A) IS NOT MATERIAL AND (B) WOULD BE COMPETITIVELY HARMFUL IF PUBLICLY DISCLOSED. |
|
1.10 |
“Measure Date” means any given point in time when a determination is being made. |
|
|
1.11 |
“Net Sales” means (a) the gross total of the monetary amounts collected by Omeros, when Omeros is the initial seller and distributor, and by Omeros’ sublicensee(s), when such sublicensee(s) are the initial seller(s) and distributor(s), for the sale or distribution of the Products to independent third parties, less (b) the sum of the following actual and customary deductions where applicable: cash, trade, or quantity discounts; sales, use, tariff, import/export duties or other excise taxes, and any other governmental taxes imposed on particular sales; transportation charges and allowances; sales commissions to third parties (but excluding sales commissions to Omeros’ employees); wholesale charge backs; distributor fees; Medicare/Medicaid rebates; customer rebates; refunds for recalls; and allowances or credits to customers because of rejections or returns, provided such deductions are documented. |
|
|
1.12 |
“Parent Compounds” means the seven compounds listed in Schedule C attached to this Agreement that are claimed in one or more of the Asubio Patents. |
|
|
1.13 |
“Product” or “Products” means all drug product(s) and drug product candidate(s) containing one or more Compounds that are encompassed by any valid and subsisting claim(s) of any issued patent within the Asubio Patents or the Compound Improvement Patents in the country or countries in which such products are offered for sale, sold, manufactured or used. Notwithstanding the foregoing, any certain drug product or drug product candidate that initially meets the definition of a Product set forth in the foregoing sentence of this Subsection 1.13 shall still be deemed to be a Product after the expiration of all claim(s) of all issued patent within the Asubio Patents or the Compound Improvement Patents covering the Compound contained in such product if, and only if, the use of such Compound is encompassed by any valid and subsisting claim(s) of any issued patent within the Field Improvement Patents in the country or countries in which such drug product or drug product candidate is used. |
|
|
1.14 |
“Third Party Marketing and Distribution Agreement” means any agreement conveying a sublicense of the marketing and distribution rights for a Product to a third party entity other than any Affiliate of either party. |
|
|
2 |
Grant of License |
|
|
2.1 |
Asubio hereby grants to Omeros for the term of this Agreement a royalty-bearing, exclusive worldwide license in the Field under the Asubio Patents and the Asubio Know-How, including the right to grant sublicenses, to use (including, without limitation, manufacture, formulate, preclinical and clinical research and development and commercialization), apply for approval, sell, offer for sale, market, distribute, import and export the Compounds and the Products (the “License”). |
|
|
2.2 |
If requested by Omeros, Asubio will consider in good faith expanding the License in the Field, on terms consistent with this Agreement, to include additional PDE7 inhibitors created by Asubio, other than the Compounds, that are claimed in the Asubio Patents and that are not reasonably expected, based on a preclinical assessment, to have a clinically meaningful immunologic function. |
|
|
|
|
2.3 |
If requested by Omeros, Asubio will consider in good faith expanding the Field to include other central nervous system diseases and disorders that do not involve meaningful immunologic dysfunction. |
|
|
2.4 |
If, in accordance with Omeros’ right to grant sublicenses under the License, Omeros elects to convey to any third party other than an Affiliate of Omeros any rights to offer for sale, sell, and market a Product in any ex-U.S. country in the world, Omeros will consider in good faith the exclusive licensing of such Product to Asubio or its Affiliates in such country. Omeros shall give Asubio prompt notification of the identity of each third party who is conveyed such sublicense rights. Omeros shall ensure that such third parties are bound by the same obligations, to the extent practicable and applicable, as those set forth in this Agreement, and shall be responsible to Asubio for the acts and omissions of such third party. |
|
|
2.5 |
Asubio shall, as part of the Asubio Know-How, disclose and provide to Omeros any and all additional data, information and records it may have or may develop or obtain during the term of this Agreement that would facilitate Omeros’ development, manufacture, approval for marketing and commercialization of any of the Parent Compounds, and Omeros shall have the right to reference the Asubio Know-How in any regulatory submission. |
|
|
3 |
Milestone Payments |
|
|
3.1 |
Omeros shall pay Asubio the following one-time milestone fees (each a “Milestone Fee”) in U.S. dollars following the satisfaction of the following corresponding milestone events (each a “Milestone”): |
|
|
3.1.1 |
Upon execution of this Agreement, Omeros shall pay Asubio a Milestone Fee of [***]. |
|
|
3.1.2 |
Upon Omeros’ or its sublicensee(s)’ receipt of positive data from completed toxicology studies, each of three-months minimum duration, of a first Product in a rodent species and in a non-rodent species, which studies have been conducted in conformance with current good laboratory practice guidance (“GLP”) promulgated by the U.S. Food and Drug Administration (“USFDA”), which data and studies are sufficient to support the submission by Omeros or its sublicensee(s) to USFDA of an Investigational New Drug Application (“IND”), Omeros shall pay Asubio a Milestone Fee of [***]. |
|
|
3.1.3 |
Upon the first dosing of a human subject in the first Phase 1 clinical study sponsored or authorized by Omeros or its sublicensee(s) of a first Product anywhere in the world, which study has been cleared by USFDA or a corresponding foreign regulatory agency, Omeros shall pay Asubio a Milestone Fee of [***]. |
|
|
3.1.4 |
Upon the first dosing of a human subject in the first Phase 2 clinical study sponsored or authorized by Omeros or its sublicensee(s) of a first Product anywhere in the world, which study has been cleared by USFDA or a corresponding foreign regulatory agency, Omeros shall pay Asubio a Milestone Fee of [***]. |
|
[***] |
CERTAIN IDENTIFIED INFORMATION HAS BEEN EXCLUDED FROM THE EXHIBIT BECAUSE IT BOTH (A) IS NOT MATERIAL AND (B) WOULD BE COMPETITIVELY HARMFUL IF PUBLICLY DISCLOSED. |
|
3.1.5 |
Upon the first dosing of a human subject in the first Phase 3 clinical study sponsored or authorized by Omeros or its sublicensee(s) of a first Product anywhere in the world, which study has been cleared by USFDA or a corresponding foreign regulatory agency, Omeros shall pay Asubio a Milestone Fee of [***]. |
|
|
3.1.6 |
Upon receipt of the first new drug application (“NDA”) marketing approval for a first Product obtained by or on behalf of Omeros or its sublicensee(s) from USFDA, Omeros shall pay Asubio a Milestone Fee of [***]. |
|
|
3.1.7 |
Upon receipt of the first marketing authorization for a first Product obtained by or on behalf of Omeros or its sublicensee(s) from an ex-U.S. regulatory authority corresponding to USFDA, Omeros shall pay Asubio a Milestone Fee of [***]. |
|
|
3.1.8 |
Upon reaching an aggregate of all Net Sales of [***], Omeros shall pay Asubio a Milestone Fee of [***]. |
|
|
3.1.9 |
Upon reaching an aggregate of all Net Sales of [***], Omeros shall pay Asubio a Milestone Fee of [***]. |
|
|
3.2 |
If any Milestone above is achieved with respect to a particular Product before a prior Milestone has been achieved, then all prior Milestones that have not previously been paid with respect to that Product shall be deemed achieved upon achievement of the subsequent Milestone, and the corresponding payment shall become payable, provided, however, that the NDA approval Milestone set forth in Subsection 3.1.6 shall not be treated as a “prior Milestone” when the ex-U.S. marketing authorization Milestone set forth in Subsection 3.1.7 is achieved, and the ex-U.S. marketing authorization Milestone set forth in Subsection 3.1.7 shall not be treated as a “prior Milestone” when the NDA approval Milestone set forth in Subsection 3.1.6 is achieved. |
|
|
4 |
Royalty Payments |
|
|
4.1 |
Omeros shall pay Asubio a royalty (the “Royalty”) as a percentage of Net Sales. The Royalty shall be computed in accordance with the applicable one of the following subsections: |
|
|
4.1.1 |
For Products containing one or more Parent Compounds, the Royalty shall be either (a) prior to the expiration of all applicable valid claims of the Asubio Patents, [***] of Net Sales of such Products, and then (b) after the expiration of all applicable valid claims of the Asubio Patents, [***] of Net Sales of such Products if the use of such Products is encompassed by valid claim(s) in the Field Improvement Patents in the countries in which such Products are used; or |
|
|
4.1.2 |
For Products containing one or more Minor Improved Compounds, the Royalty shall be either (a) prior to the expiration of all applicable valid claims of the Asubio Patents, [***] of Net Sales of such Products, and then (b) after the expiration of all applicable valid |
|
[***] |
CERTAIN IDENTIFIED INFORMATION HAS BEEN EXCLUDED FROM THE EXHIBIT BECAUSE IT BOTH (A) IS NOT MATERIAL AND (B) WOULD BE COMPETITIVELY HARMFUL IF PUBLICLY DISCLOSED. |
|
claims of the Asubio Patents, [***] of Net Sales of such Products if the use of such Products is encompassed by valid claim(s) in the Field Improvement Patents in the countries in which such Products are used; or |
||
|
4.1.3 |
For Products containing one or more Major Improved Compounds, the Royalty shall be (a) prior to the expiration of all applicable valid claims of the Asubio Patents, [***] of Net Sales of such Products, and then (b) after the expiration of all applicable valid claims of the Asubio Patents and prior to the expiration of all applicable valid claims of the Compound Improvement Patents, [***] of Net Sales of such Products, and then (c) after the expiration of all applicable valid claims of the Asubio Patents and the Compound Improvement Patents, [***] of Net Sales of such Products if the use of such Products is encompassed by valid claim(s) in the Field Improvement Patents in the countries in which such Products are used; or |
|
|
4.1.4 |
For Products containing one or more Other Improved Compounds, the Royalty shall be either (a) prior to the expiration of all applicable valid claims of the Compound Improvement Patents, [***] of Net Sales of such Products, and then (b) after the expiration of all applicable valid claims of the Compound Improvement Patents, [***] of Net Sales of such Products if the use of such Products is encompassed by valid claim(s) in the Field Improvement Patents in the countries in which such Products are used. |
|
|
4.2 |
In the event that the provisions of multiple Subsections 4.1.1 through 4.1.4 apply to any Product at any Measure Date, the Subsection that provides the highest Royalty shall be utilized for only so long as the conditions set forth in the corresponding Subsection apply. |
|
|
4.3 |
Notwithstanding the royalty provisions of Subsections 4.1 and 4.2, in the event that Omeros enters into a Third Party Marketing and Distribution Agreement for a Product, then at any Measure Date the sum of all Royalty payments paid or payable over the life of this Agreement up to the Measure Date based on the Net Sales collected by the sublicensee under such Third Party Marketing and Distribution Agreement (the “Summed Royalties”) shall not exceed [***] of the gross total of the monetary amounts collected up to the Measure Date by Omeros in the form of royalty payments and milestone payments under the Third Party Marketing and Distribution Agreement; provided, that such gross total shall exclude (i) any Net Sales independent of the Third Party Marketing and Distribution Agreement, (ii) any amounts received as funding for further research and development activities; (iii) any amounts received in connection with the conveyance of other rights not specifically and directly pertaining to such Product; (iv) any amounts received subject to a repayment obligation by Omeros; and (v) any amounts reasonably received as consideration for the purchase of equity of Omeros and not as consideration for sublicense from Omeros, (the “Adjusted Gross Partnering Revenue”). If on any Measure Date the Summed Royalties exceeds [***] of the Adjusted Gross Partnering Revenue, then no further Royalty shall be paid under Section 4 until the Summed Royalties on a subsequent Measure Date falls below [***] of the Adjusted Gross Partnering Revenue for that Measure Date, provided, however, that Asubio shall not be required to refund any Royalty payments that have already been paid to Asubio by Omeros. |
|
[***] |
CERTAIN IDENTIFIED INFORMATION HAS BEEN EXCLUDED FROM THE EXHIBIT BECAUSE IT BOTH (A) IS NOT MATERIAL AND (B) WOULD BE COMPETITIVELY HARMFUL IF PUBLICLY DISCLOSED. |
|
5 |
Payment Procedures |
|
|
5.1 |
Omeros shall promptly notify Asubio of the achievement of each Milestone, and Asubio shall then invoice Omeros for the corresponding Milestone Fee. All Milestone Fees shall be paid within thirty (30) days of receipt of the corresponding invoice. |
|
|
5.2 |
Omeros shall pay Asubio Royalty payments on a quarterly basis for Net Sales realized during each respective quarter. Royalty payments for each quarter shall be made within sixty (60) days of the end of the quarter. If Omeros is required to use any estimated figures to meet this royalty payment time frame, Omeros shall note such figures as estimated in an accompanying report and shall adjust the Royalty paid the next quarter when actual figures are available for the prior quarter. Net Sales and Royalty payments shall be computed based on a conversion from any other denomination to U.S. Dollars for any revenues received or costs and expenses incurred by Omeros during the relevant quarter, as provided herein, using the exchange rate published in The Wall Street Journal, West Coast edition, on the last business day of the applicable calendar quarter. Each quarterly Royalty payment shall be accompanied by (a) a report specifying the source and amount of the Royalty itemized by Product-by-Product basis and country-by-country basis including information reasonably necessary and sufficient for Asubio to calculate the Adjusted Gross Partnering Revenue under any Third Party Marketing and Distribution Agreements for the Products, (b) the total of all discounts, returns, credits and commissions deducted from gross proceeds to determine Net Sales, and (c) other information as Asubio may reasonably request from time to time and as Omeros may agree. |
|
|
5.3 |
Milestone Fees and Royalty payments shall be made in U.S. Dollars by wire transfer in accordance with payment instructions to be provided by Asubio in writing. Asubio shall be responsible for updating its payment instructions as may be required. Any and all charges from Omeros’ bank and similar fees incurred by Omeros in processing such payments shall be borne by Omeros. If any of the payments made or to be made by Omeros to Asubio become subject to withholding taxes under any applicable law, then Omeros shall withhold the amount of such taxes for the account of Asubio to the extent required by such applicable laws, and shall pay the amounts of such taxes to the proper governmental authorities in a timely manner and promptly transmit to Asubio an official tax certificate or other evidence of such tax obligations together with proof of payment from the relevant governmental authorities of all amounts withheld sufficient to enable Asubio to claim such payment of taxes. Omeros will provide Asubio with reasonable assistance (not including professional advice or representation) to enable Asubio to recover such taxes as permitted by applicable laws. Any other taxes levied on Omeros arising out of or in connection with Omeros’ activities hereunder shall be borne by Omeros. |
|
|
5.4 |
Asubio reserves the right to employ a certified public accountant to review and reconcile the directly relevant accounting records and procedures of Omeros as they relate to the determination of Royalty payments during reasonable business hours and no more than |
|
|
|
|
|
|
twice a year, and Omeros agrees to make available at Omeros’ place of business all such directly relevant accounting records for that purpose within 30 (thirty) days of written request by Asubio. The cost of such review shall be borne by Asubio, unless it is found that Omeros under-paid a quarterly Royalty for any quarter by an amount of [***] or greater, in which case the cost of such review shall be borne by Omeros. |
||
|
5.5 |
In the event that any Milestone Fees or Royalty payments are not timely paid by Omeros when due, Omeros shall pay to Asubio interest charges on such late payments at a rate of [***] per annum. |
|
|
5.6 |
Notwithstanding anything to the contrary herein, Omeros shall have no obligation to pay any Royalty or Milestone Fee for any Product based on any patent claim that has been declared invalid or unenforceable by a court or governmental body of competent jurisdiction or based on any patent claim that is not enforceable in the jurisdiction(s) where such Product is manufactured, used, sold, offered for sale, imported or distributed. |
|
|
6 |
Progress Reports and Reversion Rights |
|
|
6.1 |
Omeros shall, commencing on the one-year anniversary of the Effective Date of this Agreement and annually thereafter, deliver to Asubio a written progress report summarizing the status of Omeros’ efforts to develop and commercialize one or more Products. |
|
|
6.2 |
Asubio shall have the right to terminate the License and this Agreement, at its discretion, if Omeros and each of its sublicensee(s) for a period of at least six (6) consecutive months ceases to conduct, or to cause to have conducted, all research, development and/or commercialization activities for all Products, including without limitation the cessation for such period of time all medicinal chemistry efforts, all formulation activities, all chemistry, manufacturing and control activities, all preclinical research and development, all clinical research and development, and all regulatory, patent and business partnering activities concerning all Products, and Asubio sends Omeros a notice of termination prior to recommencement by Omeros of such activities; provided, however, that any cessation or delay of such activities due to a regulatory process, availability of compounds, materials or necessary processes, the procurement of intellectual property rights, any dispute or legal proceeding concerning third party intellectual property rights that are necessary to the research, development and/or commercialization of a Product, or any other material factor not reasonably within Omeros’ or its sublicensee’s control (e.g., strikes, terrorism, natural disasters, war), shall be excused. In the event of such a termination under this provision, the License and other rights held by Omeros under the Asubio Patents shall revert to Asubio, and Omeros and Asubio shall be relieved of all further obligations under this Agreement except for the surviving clauses as set forth in Section 11.4 below. |
|
|
7 |
Patent Prosecution and Enforcement |
|
|
7.1 |
Asubio shall retain ownership of the Asubio Patents, which Asubio Patents cannot be assigned to a third party other than the Affiliate of Asubio without Omeros’ advance |
|
[***] |
CERTAIN IDENTIFIED INFORMATION HAS BEEN EXCLUDED FROM THE EXHIBIT BECAUSE IT BOTH (A) IS NOT MATERIAL AND (B) WOULD BE COMPETITIVELY HARMFUL IF PUBLICLY DISCLOSED. |
|
written consent (not to be unreasonably withheld); provided, however, that this provision shall not act to prohibit Asubio from granting other licenses under the Asubio Patents consistent with the provisions of Section 2 above. Asubio shall also retain the sole right and the obligation to diligently use commercially reasonable efforts to file, prosecute and maintain the patent applications and patents included in the Asubio Patents that include claims encompassing the Compounds, including the filing of continuation applications, divisional applications, appeals, reissues and reexaminations where reasonably warranted. |
||
|
7.2 |
Asubio shall keep Omeros timely informed of all actions reasonably considered to be important, including, without limitation, all application filings, search reports, examination reports, office actions, responses, amendments and appeal proceedings, that are taken in the filing, prosecution and maintenance of any patent or patent application included in the Asubio Patents that include claims encompassing the Compounds. Omeros shall cooperate with Asubio in the course of the procedure for extension of the Asubio Patents, including any supplemental protection certificates. Should Asubio determine not to proceed with or to abandon the filing, prosecution or maintenance of any patent or patent application included in the Asubio Patents that claims any of the Compounds, Asubio shall provide Omeros timely advance notice of its determination and Omeros shall be entitled at its discretion and upon written notice to Asubio to assume the right to file, prosecute and maintain such patent or patent application, at Omeros’ sole expense, which such patent or patent application shall thereafter be excluded from the basis for payment of any Milestone Fee or Royalty to Asubio. |
|
|
7.3 |
Omeros shall own and retain ownership of the Compound Improvement Patents and the Field Improvement Patents, which Compound Improvement Patents and Field Improvement Patents cannot be assigned to a third party other than an Affiliate of Omeros without Asubio’s advance written consent (not to be unreasonably withheld). Omeros shall also retain the sole right and the obligation to diligently use commercially reasonable efforts to file, prosecute and maintain the patent applications and patents included in the Compound Improvement Patents and the Field Improvement Patents, including the filing of continuation applications, divisional applications, appeals, reissues and reexaminations where reasonably warranted. |
|
|
7.4 |
Should Omeros determine not to proceed with or to abandon the filing, prosecution or maintenance of any patent or patent application included in the Compound Improvement Patents or the Field Improvement Patents, Omeros shall provide Asubio timely advance notice of its determination and Asubio shall be entitled at its discretion and upon written notice to Omeros to assume the right to file, prosecute and maintain such patent or patent application, at Asubio’s sole expense. |
|
|
7.5 |
Whenever either party becomes aware of the possible infringement of the Asubio Patents, the Compound Improvement Patents or the Field Improvement Patents by a third party, such party shall promptly notify the other party of any such infringement and provide such other party with any available evidence of such infringement. |
|
|
7.6 |
Asubio shall have the first right, but not the obligation, to bring any suit or action for |
|
|
|
infringement of the Asubio Patents. Any infringement action brought by Asubio shall be solely at Asubio’s expense and in such actions in which Omeros has not elected to participate and share in the expenses, Asubio shall have no duty to account to Omeros for any award, settlement or any other recovery resulting from such enforcement action. Omeros shall provide reasonable assistance at Asubio’s reasonable expense in the prosecution of such suit or action. Omeros shall have the right, but not the obligation, at its cost to join as a party in any infringement action brought by Asubio. In the event that monetary damages are awarded or obtained by Asubio whether by judgment, award, decree, settlement or otherwise, as a result of such enforcement action brought by ASB in which Omeros joins as a party, the money actually received shall be divided appropriately between Asubio and Omeros with reference to the relative monetary injury suffered by the party hereto by reason of the infringement, after first deducting the expenses incurred by Asubio and Omeros in filing, prosecuting, and maintaining such suit or action. Asubio shall not settle any such action in any manner that conflicts with Omeros’ rights in the Asubio Patents, without the prior written consent of Omeros (which shall not be unreasonably withheld). |
||
|
7.7 |
In the event that Asubio fails to or elects not to commence any infringement suit or action under Subsection 7.6, Omeros shall have the sole right in its discretion to enforce, in its name or Asubio’s name, the Asubio Patents against any third party that infringes one or more claims of the Asubio Patent by the use, manufacture, offering for sale or sale of a product that competes with a Product in the Field or that is a generic or reformulated version of a Product. Any such enforcement action in accordance with this Subsection 7.7 shall be undertaken at Omeros’ sole cost and Omeros shall have no duty to account to Asubio for any award, settlement or any other recovery resulting from such enforcement action. Asubio shall provide reasonable assistance requested by Omeros in connection with such enforcement action at Omeros’ reasonable expense. Asubio shall have the right, but not the obligation, at its cost to join as a party in any infringement action brought by Omeros. Omeros shall not settle such action in any manner that conflicts with Asubio’s rights in the Asubio Patents without the prior written consent of Asubio (which shall not be unreasonably withheld). |
|
|
7.8 |
Except as expressly set forth in Section 2 or elsewhere in this Agreement, neither party grants any license under its preexisting or independently created or obtained intellectual property rights to the other party. |
|
|
7.9 |
Each party shall execute and cause its employees and agents to execute any assignment, declaration or other document required to effectuate the patent ownership, application, prosecution and enforcement provisions of this Section 7. |
|
|
8 |
Representations, Warranties |
|
|
8.1 |
Each party represents and warrants that it has the requisite corporate power and authority and the legal right to enter into this Agreement and to perform its obligations hereunder. |
|
|
8.2 |
Asubio warrants, as of the Effective Date, to Asubio’s knowledge that: the Asubio Patents include valid issued claims and/or patentable pending claims that encompass each |
|
|
|
of the Parent Compounds and the Parent Compounds do not infringe the intellectual property of any third party; and Asubio has complied with its duty of disclosure of prior art and other material information, where applicable, to national and regional patent offices with respect to the Asubio Patents. Asubio also warrants that Asubio has not granted any other license, right, security interest or lien, or undertaken any other obligation, that limits its ability to grant the License; and Asubio will diligently use all reasonable efforts to apply for, prosecute, maintain and enforce (except as provided for Omeros in Section 7 above) all patents and patent applications in the Asubio Patents. |
||
|
8.3 |
Prior to Omeros’ marketing of any Product or making any Product available for use in any human patients, Omeros will obtain and maintain reasonably adequate product liability insurance. |
|
|
8.4 |
EXCEPT AS OTHERWISE EXPRESSLY SET FORTH IN THIS AGREEMENT, NEITHER PARTY MAKES ANY REPRESENTATIONS OR EXTENDS ANY WARRANTIES OF ANY KIND, EITHER EXPRESS OR IMPLIED, INCLUDING, BUT NOT LIMITED TO WARRANTIES OF MERCHANTABILITY, FITNESS FOR A PARTICULAR PURPOSE, OR NON-INFRINGEMENT. |
|
|
9 |
Confidentiality |
|
|
9.1 |
Asubio and Omeros hereby affirm and incorporate by reference the terms of the Mutual CDA, a copy of which is attached hereto as Exhibit A, except to the extent that the terms of the Mutual CDA may conflict with the terms of this Agreement, in which case the terms of this Agreement shall prevail. The parties further agree that all Confidential Information (as defined in the Mutual CDA) disclosed by either party to the other party during the term of this Agreement shall be subject to the terms of the Mutual CDA, and that the mutual obligations of nondisclosure and non-use set forth in the Mutual CDA shall subsist for a period of five (5) years after the termination of this Agreement. |
|
|
9.2 |
The terms of this Agreement shall be maintained in strict confidence by both Asubio and Omeros, and may not be disclosed by either party without the consent of the other party, except to each party’s Affiliates, employees, directors, auditors, counsel, financial advisers, consultants, shareholders, investors, as part of due-diligence reviews by prospective corporate partners, financers and acquirers, and as may be required under a court order or decree or as required to comply with any governmental law, rule or regulation. Asubio also acknowledges and agrees that Omeros will be legally required and shall be permitted to disclose this Agreement and its terms in filings with the U.S. Securities and Exchange Commission. |
|
|
10 |
Indemnification |
|
|
10.1 |
Each party (the “Indemnifying Party”) shall indemnify, hold harmless and defend the other party and its Affiliates, and their employees, officers, directors, consultants and agents (the “Indemnified Party”) against any and all claims, suits, losses, liabilities, damages, costs, fees, and expenses (“Claims”) resulting from or arising directly out of the Indemnifying Party’s breach of any representation, warranty or obligation under this |
|
|
|
Agreement, or the Indemnifying Party’s exercise of the rights and obligations under the License or any sublicense, except that such obligation to indemnify, hold harmless and defend shall not extend to any Claims to the extent such Claims result from or arise directly from the negligence or misconduct of the Indemnified Party. Neither party shall be liable to the other party under this Agreement for any indirect, incidental, consequential or special damages. |
||
|
11 |
Term and Termination |
|
|
11.1 |
Unless terminated earlier as set forth in Subsections 6.2, 11.2 or 11.3, this Agreement and the License shall remain in full force and effect so long as there is a valid, subsisting and enforceable claim in any patent included within the Asubio Patents, the Compound Improvement Patents or the Field Improvement Patents or any patentable claim included in any pending patent application included in the Asubio Patents, the Compound Improvement Patents or the Field Improvement Patents. |
|
|
11.2 |
Omeros may terminate this Agreement by providing ninety (90) days advance written notice of termination under this Subsection 11.2 to Asubio, with or without cause; provided that if Omeros terminates this Agreement under this Subsection then Omeros shall thereafter not make, use, offer for sale, sell or sublicense any Product that is encompassed by one or more unexpired, valid and enforceable claim(s) of the Asubio Patents, the Compound Improvement Patents or the Field Improvement Patents. |
|
|
11.3 |
Either party may terminate this Agreement at any time in the event that the other party (a) breaches any material obligation of this Agreement by first submitting written notice of breach to the breaching party, which breach is not substantially cured within ninety (90) days of the receipt of such notice, followed by written notice of termination then being sent to the breaching party, or (b) declares or is adjudged by a court of competent jurisdiction to be insolvent, bankrupt or in receivership, and such insolvency, bankruptcy or receivership materially limits such party’s ability to perform its obligation under this Agreement, excluding reorganizations entered into by such party with the consent of the other party, which consent shall not be unreasonably withheld. |
|
|
11.4 |
The provisions of Sections and Subsections 7.1 (limited to the patent ownership provisions), 7.3 (limited to the patent ownership provisions), 8 (Representations, Warranties, but excluding continued obligations regarding patent prosecution and maintenance), 9 (Confidentiality), 10 (Indemnification), 12 (Use of Names) and 13 (Miscellaneous) above shall survive expiration or termination of this Agreement for the period set forth therein or, if no period is set forth therein, then indefinitely. |
|
|
11.5 |
Termination of this Agreement for any reason shall not release any party hereto from any liability which at the time of such termination has already accrued to the other party or which is attributable to a period prior to such termination, nor preclude either party from pursuing all rights and remedies it may have hereunder or at law or in equity with respect to any breach of this Agreement. |
|
|
|
12 |
Use of Names |
|
|
12.1 |
Nothing contained in this Agreement confers any right to either party to use in advertising, publicity, or other promotional activities any name, trade name, trademark, or other designation of the other party hereto, and neither party shall make such use without the prior written consent of the other party. The parties agree not to make any press release or public announcement, written or oral, relating to this Agreement without the prior written approval of the other party. Asubio acknowledges that Omeros intends to issue a press release legally required or advised by its legal counsel concerning this Agreement concurrent with making a legally required filing of a Form 8-K disclosing the signing of this Agreement with the U.S. Securities and Exchange Commission, and shall provide Asubio with a draft of such press release for approval, which approval shall not be unreasonably withheld. When filing of a copy of this Agreement with the U.S. Securities and Exchange Commission, which may be subsequent to the filing of the Form 8-K disclosure of the signing of this Agreement, Omeros shall use reasonable efforts to redact commercially sensitive information in a confidential treatment request, shall provide Asubio a copy of such request for approval and comment, and shall incorporate all timely received reasonable comments to the extent legally permissible. |
|
|
13 |
Miscellaneous |
|
|
13.1 |
This Agreement including appended Schedules A-D and appended Exhibit A constitutes the entire understanding of the parties hereto regarding the subject matter of this Agreement, and no other representation, agreement, promise or undertaking altering, modifying, taking from or adding to the terms of this Agreement shall have any effect unless the same is reduced to writing and duly executed by the parties hereto. In the event of any conflict between the main body of this Agreement and any attachments thereto or documents incorporated by reference therein, the provisions of the main body of this Agreement shall control. This Agreement expressly supersedes the MTA. |
|
|
13.2 |
Either party’s failure to enforce any provision of this Agreement will not be considered a waiver of future enforcement of that or any other provision. |
|
|
13.3 |
The laws of the state of Delaware, United States, without regard to its conflict-of-laws provisions, shall govern this Agreement, its interpretation and its enforcement, and any disputes arising out of or related to this Agreement. |
|
|
13.4 |
The parties agree that the U.S. Federal Courts located in the state of Delaware, United States will have sole and exclusive jurisdiction over any disputes arising under this Agreement, and each party hereby consents to the jurisdiction and venue of such courts for such purposes. |
|
|
13.5 |
In the event that it is necessary for either party of this Agreement to take legal action to enforce any of the terms, conditions or rights contained herein, or to defend any such action, then the prevailing party in such action shall be entitled to recover from the other party all reasonable costs and expenses, including attorneys fees, related to such legal action. |
|
|
|
13.6 |
In the event that any portion of this Agreement is held invalid or unenforceable by a court of law, that provision will be construed and reformed to permit enforcement of the provision to the maximum extent permissible consistent with the parties’ original intent, and if such construction is not possible, such provision shall be struck from this Agreement, and the remainder of the Agreement shall remain in full force and effect as if such provision had never been part of this Agreement. |
||
|
13.7 |
For the purposes of this Agreement, the parties hereto are independent contractors, and nothing in this Agreement shall be construed to place them in the relationship of partners, principal and agent, employer/employee or joint venturers. Except as provided expressly herein, each party agrees that it shall have no authority to bind or obligate the other party, nor shall any party hold itself out as having such authority. |
||
|
13.8 |
Neither party will be liable for failure or delay in performing any obligation under this Agreement, or will be considered in breach of this Agreement, if such failure or delay is due to a natural disaster or any cause reasonably beyond such party’s control, provided that such party resumes performance as soon as possible following the end of the event that caused such delay or failure of performance. |
||
|
13.9 |
This Agreement including all right and obligations hereunder shall not be assignable by either party without the prior written consent of the other party except in connection with any acquisition or merger of such party or sale of all or substantially all of its assets; provided, however, that this Agreement shall be assignable by either party to its Affiliates. This Section shall not be construed in any way to limit Omeros’ rights to grant, at Omeros’ sole discretion, sublicenses under the License. Subject to these restrictions, this Agreement will be binding upon and will inure to the benefit of the parties’ permitted successors and assignees, including each party’s successor-Affiliates. |
||
|
13.10 |
Either party shall be permitted to cause its Affiliates to perform any and all obligation under this Agreement on behalf of such party. Such party shall guarantee and be responsible all obligations and performances undertaken by its Affiliates. |
||
|
13.11 |
Any notice required or permitted to be given hereunder by either party shall be in writing and shall be (a) delivered personally, (b) sent by an internationally recognized courier service, charges prepaid, or (c) delivered by facsimile (with the original promptly sent by any of the foregoing manners) to the addresses or facsimile numbers of the other party set forth below, or at such other addresses as may from time to time be furnished by similar notice by either party. The effective date of any notice hereunder shall be the date of receipt by the receiving party. |
||
|
If to Omeros: |
If to Asubio: |
|
|
Omeros Corporation |
Asubio Pharma Co., Ltd. |
|
|
1420 Fifth Avenue, Suite 2600 |
9-11, Akasaka 2-Chome |
|
|
Seattle, WA 98101 |
Minato-Ku, Tokyo 107-8541 |
|
|
U.S.A. |
Japan |
|
|
|
Attention: Gregory A. Demopulos, M.D., |
Attention: General Manager |
|
|
Chairman & CEO |
Intellectual Property and |
|
|
|
Licensing Department |
|
|
And copy to: Marcia S. Kelbon, |
||
|
Patent & General Counsel |
||
|
Fax: (206) 676.5005 |
Fax: +81-3-3588-9602 |
|
|
Phone: (206) 676.5000 |
Phone: +81-3-3588-9710 |
|
13.12 |
This Agreement may be executed in one or more counterparts, each of which will be considered an original, and all of which will constitute the same instrument. |
IN WITNESS WHEREOF, Omeros and Asubio have each acknowledged and accepted this Agreement by causing it to have been signed by their respective duly authorized officials.
|
OMEROS CORPORATION |
ASUBIO PHARMA CO., LTD. |
||
|
By: |
/s/ Gregory A. Demopulos |
By: |
/s/ Seiichi Yokoyama |
|
|
Name: Gregory A. Demopulos, M.D. |
Name: Seiichi Yokoyama |
|
|
|
Title: Chairman & CEO |
Title: President |
|
|
|
Date: 3/3/10 |
Date: 3/3/10 |
|
|
|
Schedule A
Asubio Pharma Co., Ltd. — Omeros Corporation
License Agreement
Asubio Patents
1) [***] having PDE7 inhibitory activity
International Publication Number: [***]
|
Country |
Application Date |
Application Number |
||
|
[***] |
[***] | [***] | ||
|
[***] |
[***] |
[***] |
||
|
[***] |
[***] |
[***] |
||
|
[***] |
[***] |
[***] |
||
|
[***] |
[***] |
[***] |
||
|
[***] |
[***] |
[***] |
||
|
[***] |
[***] |
[***] |
||
|
[***] |
[***] |
[***] |
||
|
[***] |
[***] |
[***] |
||
|
[***] |
[***] |
[***] |
||
|
[***] |
[***] |
[***] |
2) [***] having PDE7-inhibitory activity
International Publication Number: [***]
|
Country |
Application Date |
Application Number |
||
|
[***] |
[***] |
[***] |
||
|
[***] |
[***] |
[***] |
||
|
[***] |
[***] |
[***] |
||
|
[***] |
[***] |
[***] |
||
|
[***] |
[***] |
[***] |
||
|
[***] |
[***] |
[***] |
||
|
[***] |
[***] |
[***] |
||
|
[***] |
[***] |
[***] |
|
[***] |
CERTAIN IDENTIFIED INFORMATION HAS BEEN EXCLUDED FROM THE EXHIBIT BECAUSE IT BOTH (A) IS NOT MATERIAL AND (B) WOULD BE COMPETITIVELY HARMFUL IF PUBLICLY DISCLOSED. |
Schedule B
Asubio Pharma Co., Ltd. — Omeros Corporation
License Agreement
Asubio Know-How
|
Document/File Title |
Date of Disclosure |
|
|
[***] |
[***] |
|
|
[***] |
[***] |
|
|
[***] |
[***] |
|
|
[***] |
[***] |
|
|
[***] |
[***] |
|
|
[***] |
[***] |
|
|
[***] |
[***] |
|
|
[***] |
[***] |
|
|
[***] |
[***] |
|
|
[***] |
[***] |
|
|
[***] |
[***] |
|
|
[***] |
[***] |
|
|
[***] |
[***] |
|
[***] |
CERTAIN IDENTIFIED INFORMATION HAS BEEN EXCLUDED FROM THE EXHIBIT BECAUSE IT BOTH (A) IS NOT MATERIAL AND (B) WOULD BE COMPETITIVELY HARMFUL IF PUBLICLY DISCLOSED. |
Schedule C
Asubio Pharma Co., Ltd. — Omeros Corporation
License Agreement
Parent Compounds
|
Identification |
||||||
|
Number in |
||||||
|
Asubio Reference |
Applicable European |
|||||
|
Number |
Structure |
Molecular Weight |
Patent Application |
|||
|
[***] |
[***] |
[***] |
[***] |
|||
|
[***] |
[***] |
[***] |
[***] |
|||
|
[***] |
[***] |
[***] |
[***] |
|||
|
[***] |
[***] |
[***] |
[***] |
|||
|
[***] |
[***] |
[***] |
[***] |
|||
|
[***] |
[***] |
[***] |
[***] |
|||
|
[***] |
[***] |
[***] |
[***] |
|
|
|
|
[***] |
CERTAIN IDENTIFIED INFORMATION HAS BEEN EXCLUDED FROM THE EXHIBIT BECAUSE IT BOTH (A) IS NOT MATERIAL AND (B) WOULD BE COMPETITIVELY HARMFUL IF PUBLICLY DISCLOSED. |
Schedule D
Asubio Pharma Co., Ltd. — Omeros Corporation
License Agreement
Excluded Compounds
[***]
|
|
|
|
[***] |
CERTAIN IDENTIFIED INFORMATION HAS BEEN EXCLUDED FROM THE EXHIBIT BECAUSE IT BOTH (A) IS NOT MATERIAL AND (B) WOULD BE COMPETITIVELY HARMFUL IF PUBLICLY DISCLOSED. |
Exhibit A
Mutual Confidential Disclosure Agreement of June 6, 2008 and
Amendment of June 18, 2009
OMEROS CORPORATION
MUTUAL CONFIDENTIAL DISCLOSURE AGREEMENT
This Confidential Disclosure Agreement (“Agreement”) is entered into as of June 6, 2008 by and between OMEROS CORPORATION (“Omeros”) and ASUBIO PHARMA CO., LTD. (“Asubio”). In the course of business negotiations and transactions between the parties hereto, either or both parties and agents thereof (including without limitation, attorneys and consultants representing the parties) may disclose certain confidential and proprietary information for the sole purpose of evaluating a potential business relationship and/or performing in accordance with any separate agreement that may be reached between the parties that does not supersede this Agreement (“Purpose”). The parties want to provide for the protection of any such confidential and proprietary information disclosed by one party (the “disclosing party”) to which the other party receiving the information (the “recipient”) may have access. Omeros and Asubio agree that this Agreement shall be binding on each company’s affiliates. For purposes of this Agreement, the term “affiliates” shall include each party’s subsidiary corporations, other corporations or business entities for which the party owns or controls at least a majority interest.
In consideration of continuing negotiations for or entering into business transactions, the parties agree:
|
1. |
Covenant Not to Disclose. For a period of five (5) years from the date of last disclosure hereunder, the recipient of any Confidential Information (defined in Section 2) will not at any time disclose or otherwise make known or available to any person, firm, corporation (including, without limitation, any parent corporation) or other entity, or use for its own account or for any purpose other than the Purpose, any Confidential Information prior to or during the term of this Agreement, without the express prior written consent of the disclosing party. The recipient shall utilize reasonable procedures to safeguard Confidential Information, including releasing Confidential Information only to employees or consultants who have agreed to abide by the recipient’s obligations hereunder on a “need-to-know” basis. All Confidential Information shall be disclosed in writing or, if first disclosed orally or visually, shall be summarized in writing and then provided to the recipient within thirty (30) days of initial disclosure. |
|
|
2. |
Confidential Information. |
|
|
2.1 |
For information disclosed by Omeros, “Confidential Information” means any and all information relating to Omeros’ programs concerning agents, compositions and therapeutic methods targeting phosphodiesterase 7 (“PDE7”), and includes, without limitation, research and development information, know-how, inventions, trade secrets, patent applications, technical data, targets (genes or proteins), knock-out and knock-in mouse strains, gene expression profiles, behavioral and physiological assays, phenotypes, cell lines, cellular, biochemical and chemical assays, chemical structures, chemical structure-activity relationships, formulae, treatment methods, clinical trial design criteria, protocols, investigators’ brochures, drawings, designs, models, samples, processes, chemistry, manufacturing and controls information, regulatory information, and any type |
|
|
|
of product development, business or marketing plans or strategies, financial information, customer lists or other customer information. |
||
|
For information disclosed by Asubio, “Confidential Information” means any and all information relating to Asubio’s compounds and programs targeting PDE7, and includes, without limitation, research and development information, know-how, inventions, trade secrets, patent applications, technical data, targets (genes or proteins), knock-out and knock-in mouse strains, gene expression profiles, behavioral and physiological assays, phenotypes, cell lines, cellular, biochemical and chemical assays, chemical structures, chemical structure-activity relationships, formulae, treatment methods, clinical trial design criteria, protocols, investigators’ brochures, drawings, designs, models, samples, processes, chemistry, manufacturing and controls information, regulatory information, and any type of product development, business or marketing plans or strategies, financial information, customer lists or other customer information. |
||
|
2.2 |
Notwithstanding the foregoing, Confidential Information does not include any information concerning any agents, compositions or therapeutic methods for the treatment of diseases that are currently classified as immune diseases or skin diseases. In addition, Confidential Information does not include information that the recipient can establish: |
|
|
2.2.1 |
is or becomes generally available to the public other than as a result of a disclosure by the recipient; |
|
|
2.2.2 |
was in the possession of the recipient prior to its being furnished to the recipient under this Agreement, provided that the source of such information was not known to the recipient to be bound by a confidentiality agreement with, or other contractual, legal, or fiduciary obligation of confidentiality to the disclosing party or any other party with respect to such information and that such prior possession can reasonably be proven by the recipient by written records; |
|
|
2.2.3 |
becomes available to the recipient on a non-confidential basis from a source other than the disclosing party, provided that such source is not bound by a confidentiality agreement with, or other contractual, legal, or fiduciary obligation of confidentiality to the disclosing party or any other party with respect to such information; or |
|
|
2.2.4 |
is independently developed by the recipient without reference to the Confidential Information, provided that such independent development can reasonably be proven by the recipient by written records. |
|
|
2.3 |
If the recipient is required by order of a court of law, administrative agency, or other governmental body to disclose any of the Confidential Information, the recipient will promptly provide the disclosing party with reasonable advance written notice if at all possible to enable the disclosing party the opportunity to seek a protective order or to otherwise prevent or limit such legally required disclosure, will use reasonable efforts to cooperate with the disclosing party to obtain such protection, and will disclose only the legally required portion of the Confidential Information. Any such legally required disclosure will not relieve recipient from its obligations under this Agreement to otherwise limit the disclosure and use of such information as Confidential Information. |
|
|
3. |
Limitations on Use. In further recognition of the value of Confidential Information, the recipient acknowledges that it shall not engage in the reproduction of Confidential Information through the techniques of “reverse engineering”. The recipient shall not make any use, either directly or indirectly, of any Confidential Information to which the recipient has been, is or will be exposed, except in the ordinary course of business |
|
|
|
pursuant to this Agreement for the Purpose or as may be expressly authorized in a separate specific written agreement between the parties. Nothing in this Agreement shall be construed as giving recipient any license or other right under any intellectual property of the disclosing party. Neither party shall disclose the existence and nature of this Agreement or the fact that it is evaluating the other party’s Information, except that such disclosure to a party’s present and potential employees, consultants, officers, directors, shareholders and investors is permitted, and neither party shall use the name of the other party in any publicity or advertising without that party’s prior written approval. |
||
|
4. |
Return of Confidential Information. When requested by the disclosing party or at the termination of the relationship giving rise to this Agreement, whichever first occurs, the recipient immediately shall deliver all Confidential Information and all copies thereof in its possession or in the possession of its employees, provided that the recipient’s legal counsel may retain one archival copy of the Confidential Information solely for purposes of ensuring compliance with this Agreement. |
|
|
5. |
Specific Performance. The parties acknowledge that (a) the covenants set forth in Sections 1, 3 and 4 are essential elements of the transactions contemplated in this Agreement and that, but for the agreement to comply with such covenants, the parties would not have entered into such transactions, and that the parties have consulted with, or have had the opportunity to consult with, counsel and have been advised in all respects concerning the reasonableness of such covenants as to scope and limit of time; (b) the disclosing party will not have any adequate remedy at law if the recipient violates the terms of Sections 1, 3 or 4 fails to perform any of its other obligations hereunder; and (c) the disclosing party shall have the right, in addition to any other rights it may have, to obtain in any court of competent jurisdiction temporary, preliminary and permanent injunctive relief to restrain any breach, threatened breach, or otherwise to specifically enforce any of such covenants or any other obligations of the recipient if the recipient fails to perform any of its obligations under this Agreement. |
|
|
6. |
Term. This Agreement and the obligations of nondisclosure and nonuse set forth herein shall terminate five (5) years after the date of the last disclosure of Confidential Information under this Agreement, provided that the obligations concerning improvements of Section 5 of this Agreement shall survive termination of the Agreement. Prior to termination of this Agreement, either party may deliver written notice to the other party that it no longer wishes to receive Confidential Information under this Agreement, after receipt of which any information subsequently sent in writing or orally disclosed by either party shall be deemed non-confidential. |
|
|
7. |
Miscellaneous. This Agreement shall be binding upon and inure to the benefit of the parties’ successors and assigns. The waiver of any breach of any provision of this Agreement or failure to enforce any provision hereof shall not operate or be construed as a waiver of any subsequent breach by any party. The invalidity of all or any part of any section of this Agreement shall not render invalid the remainder of this Agreement or the remainder of such section. If any provision of this Agreement is so broad as to be unenforceable, such provision shall be interpreted to be only so broad as is enforceable. In any litigation or disputes arising out of this Agreement, the substantially prevailing party will be entitled to recover all reasonable costs and attorneys’ fees, including costs and fees on appeal. The provisions of this Agreement shall not be construed as limiting any rights or remedies that either party may otherwise have under the applicable law. |
|
|
|
8. |
Governing Law. This Agreement shall be governed by and construed in accordance with the laws of the State of New York, USA. |
||
|
OMEROS CORPORATION |
|||
|
By: |
/s/ Marcia S. Kelbon |
||
|
Printed name Marcia S. Kelbon |
|||
|
Its VP, Patent & General Counsel |
|||
|
ASUBIO PHARMA CO., LTD. |
|||
|
By |
/s/ Keijiro Sugimura |
||
|
Printed name Keijiro Sugimura, Ph. D. |
|||
|
Its General Manager, Intellectual Property & Licensing Department |
|||
|
|
AMENDMENT TO MUTUAL CONFIDENTIAL DISCLOSURE AGREEMENT
AMENDMENT TO MUTUAL CONFIDENTIAL DISCLOSURE AGREEMENT (this “Amendment”) is made and entered into this 18th day of June, 2009, by and between Asubio Pharma Co., Ltd., with an address at 9-11 Akasaka 2-Chome, Minato-Ku, Tokyo 107-8541 Japan (“Asubio”) and Omeros Corporation, with an address at 1420 Fifth Avenue, Suite 2600, Seattle. WA 98101, U. S. A. (“Omeros”),
WITNESSETH:
WHEREAS, Asubio and Omeros have entered into a mutual confidential disclosure agreement dated June 6, 2008 (the “CDA”); and
WHEREAS, Asubio and Omeros now wish to amend the CDA to provide for the disclosure of the Confidential Information to certain officers and employees of Asubio’s affiliates.
NOW, THEREFORE, the parties agree as follows:
1. Any initially capitalized terms not otherwise defined herein shall have the meanings given in the CDA.
2. Notwithstanding the Section 1 of the CDA, Asubio may disclose the Confidential Information to its Affiliates’ officers and employees who have a need to know for the Purpose, provided that Asubio has first advised such officers and employees of the confidential nature of the Confidential Information and ensures that such officer and employee is subject to confidentiality obligations substantially similar to those set forth in the CDA. For purposes of this Amendment, “Affiliates” shall mean any corporation or other entity directly or indirectly controlled by, controlling or under common control with Asubio, and for such purpose “control” shall mean the direct or indirect ownership of more than fifty percent (50%) of the voting interest in such corporation or other entity, or the power to direct the management of such corporation or other entity.
3. Except as expressly amended hereby, all terms of the CDA, shall remain unchanged and in full force and effect.
4. This Amendment may be executed in two (2) counterparts, each of which shall be deemed an original, but all of which together shall constitute one and the same instrument.
IN WITNESS HEREOF the parties hereto have executed this Amendment as of the day and year first above written.
|
ASUBIO PHARMA CO., LTD. |
OMEROS CORPORATION |
||||
|
By: |
/s/ Keijiro Sugimura |
By: |
/s/ Marcia S. Kelbon |
||
|
|
Name: Keijiro Sugimura, Ph. D. |
Name: Marcia S. Kelbon |
|||
|
|
Title: General Manager, Intellectual Property & Licensing Department |
Title: VP, Patent & General Counsel |
|||
|
|
Exhibit 10.24
[***] CERTAIN IDENTIFIED INFORMATION HAS BEEN EXCLUDED FROM THE EXHIBIT BECAUSE IT BOTH (A) IS NOT MATERIAL AND (B) WOULD BE COMPETITIVELY HARMFUL IF PUBLICLY DISCLOSED.
Amendment No. 1 to LICENSE AGREEMENT
This Amendment No. 1 to License Agreement (this “Amendment No. 1”) is made effective the 5th day of January 2011 (the “Effective Date”) between Daiichi Sankyo Company, Limited, a Japanese Corporation having a place of business at 5-1, Nihonbashi Honcho 3-Chome, Chuo-ku, Tokyo 103-8426 Japan (“DS”), and Omeros Corporation, a Washington corporation having a principal place of business at 1420 Fifth Avenue, Suite 2600, Seattle, WA 98101 USA (“Omeros”).
WHEREAS Asubio Pharma Co., Ltd. (“Asubio”) and Omeros entered into a license agreement dated February 26, 2010 (“Agreement”) under which Asubio grants Omeros an exclusive license to certain phosphodiesterase-7 (“PDE7”) inhibitors and related patents and patent applications in the field of movement disorders [***];
WHEREAS Asubio was acquired by DS on April 1, 2010, and DS succeeds all rights and obligations of Asubio under the Agreement in accordance with Section 13.9 thereof;
WHEREAS Omeros requests DS to expand the Field (as defined in the License Agreement) to include certain central nervous system diseases and disorders in accordance with Section 2.3 of the Agreement; and
WHEREAS DS wishes to accept such Omeros’ request in consideration of certain payment set forth in this Amendment No. 1.
NOW THEREFORE, in consideration for the mutual covenants and obligations set forth herein as well as other good and valuable consideration, the parties hereby agree as follows:
|
1 |
Definitions |
|
1.1 |
Unless otherwise set forth in this Amendment No. 1, the capitalized terms herein shall have the meaning as defined in the Agreement. |
|
1.2 |
“Asubio” or “Asubio Pharma Co., Ltd.” in the Agreement shall be amended to read “DS” or “Daiichi Sankyo Company, Limited” respectively. |
|
1.3 |
Section 1.7 of the Agreement is amended and restated in its entirety to read as follows: |
|
“ “Field” means (a) all movement disorders described in WHO ICD-10 (G20-G26) and/or in Omeros’ published International PCT Patent Application WO 2008/119057 A2, including, without limitation, Parkinson’s Disease, Restless Legs Syndrome, Post-encephalitic Parkinsonism, Dopamine-Responsive Dystonia, Shy-Drager Syndrome, Periodic Limb Movement Disorder, Periodic Limb Movements in Sleep, Tourette’s Syndrome, all other movement disorders treatable with a dopamine receptor agonist or a precursor of a dopamine receptor agonist [***] (collectively “Movement Disorder Indications”) and (b) all addiction and compulsive disorders described in WHO ICD-10 (F10-F19, F40-F48, F50-F59) and/or in Omeros’ pending U.S. Provisional Patent |
|
[***] |
CERTAIN IDENTIFIED INFORMATION HAS BEEN EXCLUDED FROM THE EXHIBIT BECAUSE IT BOTH (A) IS NOT MATERIAL AND (B) WOULD BE COMPETITIVELY HARMFUL IF PUBLICLY DISCLOSED. |
Application 61/411,437 (collectively “Addiction Indications”).”
|
2 |
Milestone Payments |
|
Sections 3.1 and 3.2 of the Agreement are amended and restated in their entirety to read as follows: |
|
|
3.1 |
Omeros shall pay DS the following one-time milestone fees (each a “Milestone Fee”) in U.S. dollars following the satisfaction of the following corresponding milestone events (each a “Milestone”). References below in this Section 3 to a “First Indication” shall mean the initial one of either a Movement Disorder Indication or an Addiction Indication to reach the corresponding Milestone, and “Second Indication” shall mean the other of a Movement Disorder Indication or an Addiction Indication, e.g., if the initial indication to reach a Phase 1 clinical Milestone is a Movement Disorder Indication, such Movement Disorder Indication shall trigger the First Indication Phase 1 Clinical Milestone Fee, and thereafter the Second Indication Phase 1 clinical Milestone Fee shall be triggered only upon an Addiction Indication reaching a Phase 1 clinical Milestone. |
|
3.1.1.1 |
Upon execution of this Agreement, Omeros shall pay DS a Milestone Fee of [***]. |
|
3.1.1.2 |
Upon execution of the Amendment No. 1, Omeros shall pay DS a Milestone Fee of [***]. |
|
3.1.2.1 |
Upon Omeros’ or its sublicensee(s)’ receipt of positive data from completed toxicology studies, each of three-months minimum duration, of a first Product in a rodent species and in a non-rodent species, which studies have been conducted in conformance with current good laboratory practice guidance (“GLP”) promulgated by the U.S. Food and Drug Administration (“USFDA”), which data and studies are sufficient to support the submission by Omeros or its sublicensee(s) to USFDA of an Investigational New Drug Application (“IND”) for a First Indication, Omeros shall pay DS a Milestone Fee of [***]. |
|
3.1.2.2 |
Should Omeros be required to conduct a second set of toxicology studies to support an IND for a Second Indication, then upon Omeros’ or its sublicensee(s)’ receipt of positive data from the completed second set of toxicology studies, each of three-months minimum duration, of a first Product in a rodent species and in a non-rodent species, which studies have been conducted in conformance with current GLP promulgated by the USFDA, which data and studies are sufficient to support the submission by Omeros or its sublicensee(s) to USFDA of an IND for a Second Indication, Omeros shall pay DS a Milestone Fee of [***]. If such second set of toxicology studies is not required, then this Milestone Fee is not payable to DS. If two sets of toxicology studies to support an IND for First Indication and Second Indication respectively are conducted and Milestones described in Section 3.1.2.1 and this Section 3.1.2.2 are achieved simultaneously, then Milestone Fees in Sections 3.1.2.1 and 3.1.2.2 are payable to DS. |
|
[***] |
CERTAIN IDENTIFIED INFORMATION HAS BEEN EXCLUDED FROM THE EXHIBIT BECAUSE IT BOTH (A) IS NOT MATERIAL AND (B) WOULD BE COMPETITIVELY HARMFUL IF PUBLICLY DISCLOSED. |
|
3.1.3.1 |
Upon the first dosing of a human subject in the first Phase 1 clinical study for a First Indication sponsored or authorized by Omeros or its sublicensee(s) of a first Product anywhere in the world, which study has been cleared by USFDA or a corresponding foreign regulatory agency, Omeros shall pay DS a Milestone Fee of [***]. |
|
3.1.3.2 |
Upon the first dosing of a human subject in the first Phase 1 clinical study for a Second Indication sponsored or authorized by Omeros or its sublicensee(s) of a first Product anywhere in the world, which study has been cleared by USFDA or a corresponding foreign regulatory agency, Omeros shall pay DS a Milestone Fee of [***]. |
|
3.1.4.1 |
Upon the first dosing of a human subject in the first Phase 2 clinical study for a First Indication sponsored or authorized by Omeros or its sublicensee(s) of a first Product anywhere in the world, which study has been cleared by USFDA or a corresponding foreign regulatory agency, Omeros shall pay DS a Milestone Fee of [***]. |
|
3.1.4.2 |
Upon the first dosing of a human subject in the first Phase 2 clinical study for a Second Indication sponsored or authorized by Omeros or its sublicensee(s) of a first Product anywhere in the world, which study has been cleared by USFDA or a corresponding foreign regulatory agency, Omeros shall pay DS a Milestone Fee of [***]. |
|
3.1.5.1 |
Upon the first dosing of a human subject in the first Phase 3 clinical study for a First Indication sponsored or authorized by Omeros or its sublicensee(s) of a first Product anywhere in the world, which study has been cleared by USFDA or a corresponding foreign regulatory agency, Omeros shall pay DS a Milestone Fee of [***]. |
|
3.1.5.2 |
Upon the first dosing of a human subject in the first Phase 3 clinical study for a Second Indication sponsored or authorized by Omeros or its sublicensee(s) of a first Product anywhere in the world, which study has been cleared by USFDA or a corresponding foreign regulatory agency, Omeros shall pay DS a Milestone Fee of [***]. |
|
3.1.6.1 |
Upon receipt of the first new drug application (“NDA”) marketing approval for a first Product for a First Indication obtained by or on behalf of Omeros or its sublicensee(s) from USFDA, Omeros shall pay DS a Milestone Fee of [***]. |
|
3.1.6.2 |
Upon receipt of the first NDA marketing approval for a first Product for a Second Indication obtained by or on behalf of Omeros or its sublicensee(s) from USFDA, Omeros shall pay DS a Milestone Fee of [***]. |
|
3.1.7.1 |
Upon receipt of the first marketing authorization for a first Product for a First Indication obtained by or on behalf of Omeros or its sublicensee(s) from an ex-U.S. regulatory authority corresponding to USFDA, Omeros shall pay DS a Milestone Fee of [***]. |
|
3.1.7.2 |
Upon receipt of the first marketing authorization for a first Product obtained for a Second Indication by or on behalf of Omeros or its sublicensee(s) from an ex-U.S. regulatory authority corresponding to USFDA, Omeros shall pay DS a Milestone Fee of [***]. |
|
[***] |
CERTAIN IDENTIFIED INFORMATION HAS BEEN EXCLUDED FROM THE EXHIBIT BECAUSE IT BOTH (A) IS NOT MATERIAL AND (B) WOULD BE COMPETITIVELY HARMFUL IF PUBLICLY DISCLOSED. |
|
3.1.8 |
Upon reaching an aggregate of all Net Sales of [***], Omeros shall pay DS a Milestone Fee of [***]. |
|
3.1.9 |
Upon reaching an aggregate of all Net Sales of [***], Omeros shall pay DS a Milestone Fee of [***]. |
|
3.2 |
If any Milestone above is achieved with respect to a particular Product for a particular First or Second Indication before a prior Milestone has been achieved for such First or Second Indication, then all prior Milestones for such First or Second Indication that have not previously been paid with respect to that Product shall be deemed achieved upon achievement of the subsequent Milestone, and the corresponding payment shall become payable; provided, however, that the NDA approval Milestone set forth in Subsections 3.1.6.1 and/or 3.1.6.2 shall not be treated as a “prior Milestone” when the ex-U.S. marketing authorization Milestone set forth in Subsections 3.1.7.1 and/or 3.1.7.2, respectively, is achieved, and the ex-U.S. marketing authorization Milestone set forth in Subsection 3.1.7.1 and/or Subsection 3.1.7.2 shall not be treated as a “prior Milestone” when the NDA approval Milestone set forth in Subsection 3.1.6.1 and/or 3.1.6.2, respectively, is achieved. It is understood by the parties that on a certain Measure Date, Movement Disorder Indication would be “a particular First Indication”, and on another Measure Date, Addiction Indication would be “a particular First Indication” depending on the progress of their development.” |
It is understood by the parties that Omeros has already paid and DS has already received the Milestone Fee of [***] as provided in Section 3.1.1.1 of the Agreement.
|
3 |
Term |
This Amendment No. 1 shall become effective as of the Effective Date and shall continue to be in effect as long as the Agreement is in effect.
|
4 |
Miscellaneous |
|
4.1 |
Section 13.11 of the Agreement is amended and restated in its entirety to read as follows: |
“Any notice required or permitted to be given under the Agreement by either party shall be in writing and shall be (a) delivered personally, (b) sent by an internationally recognized courier service, charges prepaid, or (c) delivered by facsimile (with the original promptly sent by any of the foregoing manners) to the addresses or facsimile numbers of the other party set forth below, or at such other addresses as may from time to time be furnished by similar notice by either party. The effective date of any notice hereunder shall be the date of receipt by the receiving party.
|
[***] |
CERTAIN IDENTIFIED INFORMATION HAS BEEN EXCLUDED FROM THE EXHIBIT BECAUSE IT BOTH (A) IS NOT MATERIAL AND (B) WOULD BE COMPETITIVELY HARMFUL IF PUBLICLY DISCLOSED. |
|
If to Omeros: Omeros Corporation 1420 Fifth Avenue, Suite 2600 Seattle, WA 98101 U.S.A. |
If to DS: Daiichi Sankyo Company, Limited 5-1, Nihonbashi Honcho 3-Chome Chuo-ku, Tokyo 103-8426 Japan |
||
|
Attention: |
Gregory A. Demopulos, M.D. |
Attention: Noriaki Ishida |
|
|
Chairman & CEO |
Corporate Officer, Vice President, |
||
|
Business Development & Licensing |
|||
|
Department |
|||
|
And copy to: |
Marcia S. Kelbon, |
||
|
Patent & General Counsel |
|||
|
Fax: (206) 676.5005 Phone: (206) 676.5000 |
Fax: +81-3-6225-1903 Phone: +81-3-6225-1008 |
||
|
4.2 |
This Amendment may be executed in one or more counterparts, each of which will be considered an original, and all of which will constitute the same instrument. |
|
4.3 |
Except as expressly amended by this Amendment No. 1, all terms and conditions of the Agreement shall continue to be in full force and effect. |
IN WITNESS WHEREOF, DS and Omeros have each acknowledged and accepted this Agreement by causing it to have been signed by their respective duly authorized officials.
|
DAIICHI SANKYO COMPANY, LIMITED |
OMEROS CORPORATION |
|||
|
By: |
/s/ Noriaki Ishida |
By: |
/s/ Gregory A. Demopulos |
|
|
Name: |
Noriaki Ishida |
Name: |
Gregory A. Demopulos, M.D. |
|
|
Title: Corporate Officer, Vice President, |
Title: Chairman & CEO |
|||
|
Business Development & Licensing Department |
||||
|
Date: |
January 31, 2011 |
Date: |
December 28, 2010 |
|
|
|
|
Exhibit 10.25
[***] CERTAIN IDENTIFIED INFORMATION HAS BEEN EXCLUDED FROM THE EXHIBIT BECAUSE IT BOTH (A) IS NOT MATERIAL AND (B) WOULD BE COMPETITIVELY HARMFUL IF PUBLICLY DISCLOSED.
Amendment No. 2 to LICENSE AGREEMENT
This Amendment No. 2 to License Agreement (this “Amendment No. 2”) is made effective the 25 day of January 2013 (the “Effective Date”) between Daiichi Sankyo Company, Limited, a Japanese Corporation having a place of business at 5-1, Nihonbashi Honcho 3-Chome, Chuo-ku, Tokyo 103-8426 Japan (“DS”), and Omeros Corporation, a Washington corporation having a principal place of business at 1420 Fifth Avenue, Suite 2600, Seattle, WA 98101 USA (“Omeros”).
WHEREAS Asubio Pharma Co., Ltd. (“Asubio”) and Omeros entered into a license agreement dated February 26, 2010 (“Agreement”) under which Asubio grants Omeros an exclusive license to certain phosphodiesterase-7 (“PDE7”) inhibitors and related patents and patent applications in the field of movement disorders and [***];
WHEREAS Asubio was acquired by DS on April 1, 2010, and DS succeeds all rights and obligations of Asubio under the Agreement in accordance with Section 13.9 thereof;
WHEREAS DS and Omeros entered into an amendment No. 1 to license agreement dated January 5, 2011 (“Amendment No. 1”) under which the parties agreed to expand the Field (as defined in the Agreement) to include certain central nervous system diseases and disorders in accordance with Section 2.3 of the Agreement; and
WHEREAS Omeros further requests DS to expand the Field to include additional diseases and disorders in accordance with Section 2.3 of the Agreement; and
WHEREAS DS wishes to accept such Omeros’ request in consideration of certain payment obligations set forth in this Amendment No. 2.
NOW THEREFORE, in consideration for the mutual covenants and obligations set forth herein as well as other good and valuable consideration, the parties hereby agree as follows:
|
1 |
Definitions |
|
1.1 |
Unless otherwise set forth in this Amendment No. 2, the capitalized terms herein shall have the meaning as defined in the Agreement. |
|
1.2 |
“Asubio” or “Asubio Pharma Co., Ltd.” in the Agreement shall be amended to read “DS” or “Daiichi Sankyo Company, Limited” respectively. |
|
1.3 |
Section 1.7 of the Agreement is amended and restated in its entirety to read as follows: |
|
“ “Field” means (a) all movement disorders described in WHO ICD-10 (G20-G26) and/or in Omeros’ published International PCT Patent Application WO 2008/119057 A2, including, without limitation, Parkinson’s Disease, Restless Legs Syndrome, Post-encephalitic Parkinsonism, Dopamine-Responsive Dystonia, Shy-Drager Syndrome, Periodic Limb Movement Disorder, Periodic Limb Movements in Sleep, Tourette’s Syndrome, all other movement disorders treatable with a dopamine receptor agonist or a precursor of a dopamine receptor agonist [***] (collectively “Movement Disorder Indications”), (b) all addiction and compulsive disorders described in WHO ICD-10 (F10-F19, F40-F48, F50-F59) and/or in Omeros’ published International PCT Patent Application WO 2012/064667A2 A2 (collectively “Addiction Indications”), and (c) all other diseases except (i) those described in above (a) and (b) and (ii) diseases in the dermatology and dermatologic affections defined as any disease of the skin, the hair/scalp and nails (collectively “Other Indications”).” |
|
[***] |
CERTAIN IDENTIFIED INFORMATION HAS BEEN EXCLUDED FROM THE EXHIBIT BECAUSE IT BOTH (A) IS NOT MATERIAL AND (B) WOULD BE COMPETITIVELY HARMFUL IF PUBLICLY DISCLOSED. |
|
2 |
Milestone Payments |
Sections 3.1 and 3.2 of the Agreement are amended and restated in their entirety to read as follows:
|
“3.1 |
Omeros shall pay DS the following one-time milestone fees (each a “Milestone Fee”) in U.S. dollars following the satisfaction of the following corresponding milestone events (each a “Milestone”). References below in this Section 3 to a “First Indication” shall mean the initial indication among a Movement Disorder Indication, an Addiction Indication and an Other Indication that reaches the corresponding Milestone, and “Second Indication” shall mean an indication among such three indications, excluding the one from which the First Indication was drawn, that secondly reaches the corresponding Milestone, and “Third Indication” shall mean the last indication among such three indications, excluding the two from which the First Indication and Second Indication were drawn, that reaches the corresponding Milestone, e.g., if the initial indication to reach a Phase 1 clinical Milestone is a Movement Disorder Indication, such Movement Disorder Indication shall trigger the First Indication Phase 1 Clinical Milestone Fee, if the indication to secondly reach a Phase 1 clinical Milestone is an Addiction Indication, such Addiction Indication shall trigger the Second Indication Phase 1 Clinical Milestone Fee, and thereafter the Third Indication Phase 1 clinical Milestone Fee shall be triggered if and when an Other Indication reaching a Phase 1 clinical Milestone. |
|
3.1.1.1 |
Upon execution of this Agreement, Omeros shall pay DS a Milestone Fee of [***]. |
|
3.1.1.2 |
Upon execution of the Amendment No. 1, Omeros shall pay DS a Milestone Fee of [***]. |
|
3.1.1.3 |
Upon execution of the Amendment No. 2, Omeros shall pay DS a Milestone Fee of [***]. |
|
3.1.2.1 |
Upon Omeros’ or its sublicensee(s)’ receipt of positive data from completed toxicology studies, each of three-months minimum duration, of a first Product in a rodent species and in a non-rodent species, which studies have been conducted in conformance with current good laboratory practice guidance (“GLP”) promulgated by the U.S. Food and Drug Administration (“USFDA”), which data and studies are sufficient to support the submission by Omeros or its sublicensee(s) to USFDA of an Investigational New Drug Application (“IND”) for a First Indication, Omeros shall pay DS a Milestone Fee of [***]. |
|
3.1.2.2 |
Should Omeros be required to conduct a second set of toxicology studies to support an IND for a Second Indication, then upon Omeros’ or its sublicensee(s)’ receipt of positive data from the completed second set of toxicology studies, each of three-months minimum duration, of a first Product in a rodent species and in a non-rodent species, which studies have been conducted in conformance with current GLP promulgated by the USFDA, which data and studies are sufficient to support the submission by Omeros or its sublicensee(s) to USFDA of an IND for a Second Indication, Omeros shall pay DS a Milestone Fee of [***]. If such second set of toxicology studies is not required, then this Milestone Fee is not payable to DS. If two sets of such toxicology studies to support an IND for a First Indication and a Second Indication, respectively, are conducted and Milestones described in Section 3.1.2.1 and this Section 3.1.2.2 are achieved simultaneously, then Milestone Fees in Sections 3.1.2.1 and 3.1.2.2 are payable to DS. |
|
3.1.2.3 |
Should Omeros be required to conduct a third set of toxicology studies to support an IND for a Third Indication, then upon Omeros’ or its sublicensee(s)’ receipt of positive data from the completed third set of toxicology studies, each of three-months minimum duration, of a first Product in a rodent species and in a non-rodent species, which studies have been conducted in conformance with current GLP promulgated by the USFDA, which data and studies are sufficient to support the submission by Omeros or its sublicensee(s) to USFDA of an IND for a Third Indication, Omeros shall pay DS a Milestone Fee of [***]. If such third set of toxicology studies is not required, then this Milestone Fee is not payable to DS. If three sets of such toxicology studies to support an IND for a First Indication, a Second Indication and a Third Indication, respectively, are conducted and Milestones described in Section 3.1.2.1, Section 3.1.2.2 and this Section 3.1.2.3 are achieved simultaneously, then Milestone Fees in Sections 3.1.2.1, 3.1.2.2 and 3.1.2.3 are payable to DS. |
|
3.1.3.1 |
Upon the first dosing of a human subject in the first Phase 1 clinical study for a First Indication sponsored or authorized by Omeros or its sublicensee(s) of a first Product anywhere in the world, which study has been cleared by USFDA or a corresponding foreign regulatory agency, Omeros shall pay DS a Milestone Fee of [***]. |
|
[***] |
CERTAIN IDENTIFIED INFORMATION HAS BEEN EXCLUDED FROM THE EXHIBIT BECAUSE IT BOTH (A) IS NOT MATERIAL AND (B) WOULD BE COMPETITIVELY HARMFUL IF PUBLICLY DISCLOSED. |
|
3.1.3.2 |
Upon the first dosing of a human subject in the first Phase 1 clinical study for a Second Indication sponsored or authorized by Omeros or its sublicensee(s) of a first Product anywhere in the world, which study has been cleared by USFDA or a corresponding foreign regulatory agency, Omeros shall pay DS a Milestone Fee of [***]. |
|
3.1.3.3 |
Upon the first dosing of a human subject in the first Phase 1 clinical study for a Third Indication sponsored or authorized by Omeros or its sublicensee(s) of a first Product anywhere in the world, which study has been cleared by USFDA or a corresponding foreign regulatory agency, Omeros shall pay DS a Milestone Fee of [***]. |
|
3.1.4.1 |
Upon the first dosing of a human subject in the first Phase 2 clinical study for a First Indication sponsored or authorized by Omeros or its sublicensee(s) of a first Product anywhere in the world, which study has been cleared by USFDA or a corresponding foreign regulatory agency, Omeros shall pay DS a Milestone Fee of [***]. |
|
3.1.4.2 |
Upon the first dosing of a human subject in the first Phase 2 clinical study for a Second Indication sponsored or authorized by Omeros or its sublicensee(s) of a first Product anywhere in the world, which study has been cleared by USFDA or a corresponding foreign regulatory agency, Omeros shall pay DS a Milestone Fee of [***]. |
|
3.1.4.3 |
Upon the first dosing of a human subject in the first Phase 2 clinical study for a Third Indication sponsored or authorized by Omeros or its sublicensee(s) of a first Product anywhere in the world, which study has been cleared by USFDA or a corresponding foreign regulatory agency, Omeros shall pay DS a Milestone Fee of [***]. |
|
3.1.5.1 |
Upon the first dosing of a human subject in the first Phase 3 clinical study for a First Indication sponsored or authorized by Omeros or its sublicensee(s) of a first Product anywhere in the world, which study has been cleared by USFDA or a corresponding foreign regulatory agency, Omeros shall pay DS a Milestone Fee of [***]. |
|
3.1.5.2 |
Upon the first dosing of a human subject in the first Phase 3 clinical study for a Second Indication sponsored or authorized by Omeros or its sublicensee(s) of a first Product anywhere in the world, which study has been cleared by USFDA or a corresponding foreign regulatory agency, Omeros shall pay DS a Milestone Fee of [***]. |
|
3.1.5.3 |
Upon the first dosing of a human subject in the first Phase 3 clinical study for a Third Indication sponsored or authorized by Omeros or its sublicensee(s) of a first Product anywhere in the world, which study has been cleared by USFDA or a corresponding foreign regulatory agency, Omeros shall pay DS a Milestone Fee of [***]. |
|
3.1.6.1 |
Upon receipt of the first new drug application (“NDA”) marketing approval for a first Product for a First Indication obtained by or on behalf of Omeros or its sublicensee(s) from USFDA, Omeros shall pay DS a Milestone Fee of [***]. |
|
3.1.6.2 |
Upon receipt of the first NDA marketing approval for a first Product for a Second Indication obtained by or on behalf of Omeros or its sublicensee(s) from USFDA, Omeros shall pay DS a Milestone Fee of [***]. |
|
3.1.6.3 |
Upon receipt of the first NDA marketing approval for a first Product for a Third Indication obtained by or on behalf of Omeros or its sublicensee(s) from USFDA, Omeros shall pay DS a Milestone Fee of [***]. |
|
3.1.7.1 |
Upon receipt of the first marketing authorization for a first Product for a First Indication obtained by or on behalf of Omeros or its sublicensee(s) from an ex-U.S. regulatory authority corresponding to USFDA, Omeros shall pay DS a Milestone Fee of [***]. |
|
[***] |
CERTAIN IDENTIFIED INFORMATION HAS BEEN EXCLUDED FROM THE EXHIBIT BECAUSE IT BOTH (A) IS NOT MATERIAL AND (B) WOULD BE COMPETITIVELY HARMFUL IF PUBLICLY DISCLOSED. |
|
3.1.7.2 |
Upon receipt of the first marketing authorization for a first Product obtained for a Second Indication by or on behalf of Omeros or its sublicensee(s) from an ex-U.S. regulatory authority corresponding to USFDA, Omeros shall pay DS a Milestone Fee of [***]. |
|
3.1.7.3 |
Upon receipt of the first marketing authorization for a first Product obtained for a Third Indication by or on behalf of Omeros or its sublicensee(s) from an ex-U.S. regulatory authority corresponding to USFDA, Omeros shall pay DS a Milestone Fee of [***]. |
|
3.1.8 |
Upon reaching an aggregate of all Net Sales of [***], Omeros shall pay DS a Milestone Fee of [***]. |
|
3.1.9 |
Upon reaching an aggregate of all Net Sales of [***], Omeros shall pay DS a Milestone Fee of [***]. |
|
3.2 |
If any Milestone above is achieved with respect to a given Product for a given First, Second or Third Indication before a prior Milestone has been achieved for such First, Second or Third Indication, the achieved milestone being the “Accelerated Milestone”, then all Milestones prior to the Accelerated Milestone for such First, Second or Third Indication that have not previously been paid with respect to that Product shall be deemed achieved upon achievement of the Accelerated Milestone, and the corresponding payment(s) shall become payable; provided, however, that the NDA approval Milestone set forth in Subsections 3.1.6.1, 3.1.6.2 and/or 3.1.6.3 shall not be treated as a “prior Milestone” when the ex-U.S. marketing authorization Milestone set forth in Subsections 3.1.7.1, 3.1.7.2 and/or 3.1.7.3, respectively, is achieved, and the ex-U.S. marketing authorization Milestone set forth in Subsection 3.1.7.1, Subsection 3.1.7.2 and/or 3.1.7.3 shall not be treated as a “prior Milestone” when the NDA approval Milestone set forth in Subsection 3.1.6.1, 3.1.6.2 and/or 3.1.6.3, respectively, is achieved. It is understood by the parties that, for example, for a given Milestone, a Movement Disorder Indication could be “a given First Indication”, and for another Milestone, an Addiction Indication could be “a given First Indication” depending on the progress of development of the first Product(s) for such indications.” |
It is understood by the parties that Omeros has already paid and DS has already received the Milestone Fees of [***] as provided in Section 3.1.1.1 of the Agreement and [***] as provided in Section 3.1.1.2 of the Agreement.
|
3 |
[*] |
The following paragraphs shall be added as Sections 2.6 and 2.7 of the Agreement:
|
“2.6 |
If Omeros retains [***] for a certain Product [***] at the time of the [***] for the First Indication for such Product (the “FPE”), Omeros shall [***] with Omeros for [***] of such Product in such [***] in which Omeros retains [***]. Such [***] shall commence upon Omeros’ delivery of written notice to DS of the FPE and shall expire [***] after delivery of such notice, during which [***] the parties agree to [***]. |
|
“2.7 |
If Omeros enters into a sublicense agreement with a third party pharmaceutical company (a “Third Party Partner”) for the commercialization of a certain Product (the “Partner Agreement”) in accordance with Omeros’ right to grant sublicenses under the License, and such Partner Agreement applies to any or all of [***], such Partner Agreement shall be [***]. Such [***] shall be commenced upon the delivery from the Third Party Partner to DS of a written notice of the entry into the Partner Agreement and shall expire [***] from delivery of such notice. In case DS realizes that the Third Party Partner does not deliver to DS such written notice after the execution of the Partner Agreement, DS shall have the right to require Omeros to have the Third Party Partner provide DS such written notice as soon as possible, and Omeros shall diligently attempt to require the Third Party Partners to do so. If the Third Party Partner should breach its obligations to provide such written notice to DS and/or [***], such breach shall not affect the validity of the License granted to Omeros under this Agreement and Omeros shall not be liable for the Third Party Partner’s breach as far as Omeros in good faith and over a period of at least [***] continues to attempt to cure such breach.” |
|
[***] |
CERTAIN IDENTIFIED INFORMATION HAS BEEN EXCLUDED FROM THE EXHIBIT BECAUSE IT BOTH (A) IS NOT MATERIAL AND (B) WOULD BE COMPETITIVELY HARMFUL IF PUBLICLY DISCLOSED. |
|
4 |
Term |
This Amendment No. 2 shall become effective as of the Effective Date and shall continue to be in effect as long as the Agreement is in effect. Amendment No. 1 shall be terminated on the Effective Date.
|
5 |
Miscellaneous |
|
5.1 |
Section 13.11of the Agreement is amended and restated in its entirety to read as follows: |
|
“Any notice required or permitted to be given under the Agreement by either party shall be in writing and shall be (a) delivered personally, (b) sent by an internationally recognized courier service, charges prepaid, or (c) delivered by facsimile (with the original promptly sent by any of the foregoing manners) to the addresses or facsimile numbers of the other party set forth below, or at such other addresses as may from time to time be furnished by similar notice by either party. The effective date of any notice hereunder shall be the date of receipt by the receiving party. |
|
If to Omeros: |
If to DS: |
|
|
Omeros Corporation |
Daiichi Sankyo Company, Limited |
|
|
1420 Fifth Avenue, Suite 2600 |
5-1, Nihonbashi Honcho 3-Chome |
|
|
Seattle, WA 98101 |
Chuo-ku, Tokyo 103-8426 |
|
|
U.S.A. |
Japan |
|
|
Attention: Gregory A. Demopulos, M.D. |
Attention: Kazuo Sato, Ph. D. |
|
|
Chairman & CEO |
Corporate Officer, Vice President, |
|
|
Business Development & Licensing |
||
|
Department |
||
|
And copy to: Marcia S. Kelbon, |
||
|
Patent & General Counsel |
||
|
Fax: (206) 676.5005 |
Fax: +81-3-6225-1903 |
|
|
Phone: (206) 676.5000 |
Phone: +81-3-6225-1008 |
|
5.2 |
This Amendment may be executed in one or more counterparts, each of which will be considered an original, and all of which will constitute the same instrument. |
|
5.3 |
Except as expressly amended by this Amendment No. 2, all terms and conditions of the Agreement shall continue to be in full force and effect. |
IN WITNESS WHEREOF, DS and Omeros have each acknowledged and accepted this Agreement by causing it to have been signed by their respective duly authorized officials.
|
DAIICHI SANKYO COMPANY, LIMITED |
OMEROS CORPORATION |
||
|
By: |
/s/ Kazuo Sato |
By: |
/s/ Gregory A. Demopulos |
|
Name: |
Kazuo Sato, Ph. D. |
Name: |
Gregory A. Demopulos, M.D. |
|
Title: |
Corporate Officer, Vice President, Business Development & Licensing |
Title: |
Chairman & CEO |
|
Department |
|||
|
Date: |
January 21, 2013 |
Date: |
January 25, 2013 |
Exhibit 10.30
[***] CERTAIN IDENTIFIED INFORMATION HAS BEEN EXCLUDED FROM THE EXHIBIT BECAUSE IT BOTH (A) IS NOT MATERIAL AND (B) WOULD BE COMPETITIVELY HARMFUL IF PUBLICLY DISCLOSED.
AMENDED AND RESTATED ROYALTY PURCHASE AGREEMENT
BETWEEN
OMEROS CORPORATION
AND
DRI HEALTHCARE ACQUISITIONS LP
Dated as of February 1, 2024
TABLE OF CONTENTS
|
Page |
||
|
ARTICLE I |
||
|
DEFINITIONS; INTERPRETATION |
||
|
Section 1.1. |
Definitions |
1 |
|
Section 1.2. |
Certain Interpretations |
9 |
|
ARTICLE II |
||
|
PURCHASE AND SALE OF PURCHASED ASSETS |
||
|
Section 2.1. |
Purchase and Sale of Purchased Assets |
10 |
|
Section 2.2. |
No Purchase or Sale of Excluded Assets |
12 |
|
Section 2.3. |
No Obligations Transferred |
12 |
|
Section 2.4. |
Sale of Purchased Assets; Back-Up Security Interest |
13 |
|
ARTICLE III |
||
|
CLOSING; DELIVERABLES |
||
|
Section 3.1. |
Closing |
14 |
|
Section 3.2. |
Payment of Additional Floor Purchase Price |
14 |
|
Section 3.3. |
Closing Certificates |
14 |
|
Section 3.4. |
New Bill of Sale and Assignment |
14 |
|
Section 3.5. |
Tax Forms |
14 |
|
Section 3.6. |
New Rayner Surgical Instruction Letter |
15 |
|
Section 3.7. |
Receipt |
15 |
|
ARTICLE IV |
||
|
SELLER’S REPRESENTATIONS AND WARRANTIES |
||
|
Section 4.1. |
Representations and Warranties |
15 |
|
Section 4.2. |
Representations and Warranties regarding the 2022 Royalty Purchase Agreement |
21 |
|
ARTICLE V |
||
|
PURCHASER’S REPRESENTATIONS AND WARRANTIES |
||
|
Section 5.1. |
Representations and Warranties |
21 |
|
Section 5.2. |
Representations and Warranties regarding the 2022 Royalty Purchase Agreement |
23 |
|
ARTICLE VI |
||
|
RECEIVABLES AND PAYMENTS |
||
|
Section 6.1. |
No Out-of-Pocket Payments by Seller for Purchased Receivables |
23 |
|
Section 6.2. |
Adjustments to Pre-2024 Royalty Payments |
23 |
|
Section 6.3. |
Treatment of Late Payment of Purchased Receivables |
24 |
|
ARTICLE VII |
||
|
COVENANTS |
||
|
Section 7.1. |
Misdirected Payments; Set-Offs by Rayner Surgical |
24 |
|
Section 7.2. |
Interest |
25 |
|
Section 7.3. |
Royalty Reports; Notices; Correspondence |
25 |
|
Section 7.4. |
Audits of Rayner Surgical |
25 |
|
Section 7.5. |
Performance of APA |
28 |
|
Section 7.6. |
Amendment of APA |
28 |
|
Section 7.7. |
Enforcement of APA |
29 |
|
Section 7.8. |
Termination of APA |
30 |
|
Section 7.9. |
Assignments of APA |
30 |
|
Section 7.10. |
Confidentiality |
31 |
|
Section 7.11. |
Public Announcements; Use of Names |
34 |
|
Section 7.12. |
Taxes |
35 |
|
Section 7.13. |
Further Actions |
35 |
|
Section 7.14. |
New Rayner Surgical Instruction Letter |
35 |
|
Section 7.15. |
Acknowledgment and Agreement by Purchaser; Limitation of Seller’s Duties and Obligations |
36 |
|
ARTICLE VIII |
||
|
INDEMNIFICATION |
||
|
Section 8.1. |
Obligation of Parties to Indemnify |
36 |
|
Section 8.2. |
Procedures Relating to Indemnification for Third Party Claims |
37 |
|
Section 8.3. |
Procedures Relating to Indemnification for Other Claims |
38 |
|
Section 8.4. |
Limitations on Indemnification |
38 |
|
Section 8.5. |
Survival of Representations and Warranties |
39 |
|
Section 8.6. |
Indemnification and Liability under the 2022 Royalty Purchase Agreement |
39 |
|
Section 8.7. |
No Implied Representations and Warranties |
39 |
|
Section 8.8. |
Exclusive Remedy |
40 |
|
Section 8.9. |
Equitable Remedies/Specific Performance |
40 |
|
Section 8.10. |
Limitations on Damages |
41 |
|
ARTICLE IX |
||
|
MISCELLANEOUS |
||
|
Section 9.1. |
Headings |
41 |
|
Section 9.2. |
Notices |
41 |
|
Section 9.3. |
No Personal Liability |
42 |
|
Section 9.4. |
Expenses |
43 |
|
Section 9.5. |
Assignment |
43 |
|
Section 9.6. |
Amendment and Waiver |
44 |
|
Section 9.7. |
Entire Agreement |
44 |
|
Section 9.8. |
No Partnership; Independent Contractors |
45 |
|
Section 9.9. |
No Third Party Beneficiaries |
45 |
|
Section 9.10. |
Governing Law |
45 |
|
Section 9.11. |
Jurisdiction; Venue; Service of Process; Waiver of Jury Trial |
45 |
|
Section 9.12. |
Severability |
45 |
|
Section 9.13. |
Counterparts; Electronic Signatures |
46 |
|
Section 9.14. |
Termination of Agreement |
46 |
List of Exhibits
A Updated Schedule of Exceptions to Seller’s Representations and Warranties
B Form of New Bill of Sale and Assignment
C Form of New Rayner Surgical Instruction Letter
List of Schedules
4.1 (i)(iii) Third Party Licenses
4.1 (i)(iv) Section 1.1(h)(B) of Seller Schedule to APA
4.1 (k)(i) APA; Royalty Reports; Material Notices (updated)
AMENDED AND RESTATED ROYALTY PURCHASE AGREEMENT
This Amended and Restated Royalty Purchase Agreement (this “Agreement”), dated as of February 1, 2024, between Omeros Corporation, a Washington corporation (“Seller”), and DRI Healthcare Acquisitions LP, a Delaware limited partnership (“Purchaser”) (each, a “Party” and together, the “Parties”), amends and restates in its entirety that certain Royalty Purchase Agreement (the “2022 Royalty Purchase Agreement”), dated as of September 30, 2022 (the “2022 Effective Date”), between Seller and Purchaser.
RECITALS
WHEREAS, Seller is a party to that certain Asset Purchase Agreement, by and among Seller and Rayner Surgical (as defined below) and solely for the purpose of Article V and Section 6.24 thereof, Rayner Surgical Group Limited, dated as of December 1, 2021, as supplemented by (i) the letter agreement between Seller and Rayner Surgical dated December 23, 2021 (“2021 Letter Agreement”) and (ii) the 2022 Letter Agreement (as defined below) (such Asset Purchase Agreement as so supplemented by the 2021 Letter Agreement and 2022 Letter Agreement, all of three of which are as set forth on Schedule 4.1(k)(i), and as may be further supplemented, amended or modified from time to time solely to the extent such supplement, amendment or modification is made in accordance therewith and in accordance with this Agreement (and it being understood, for the avoidance of doubt, that no such supplement, amendment or modification will have a retroactive effect unless such supplement, amendment or modification specifically states that it shall have a retroactive effect), the (“APA”).
WHEREAS, pursuant to the 2022 Royalty Purchase Agreement, Seller sold, transferred, assigned and conveyed to Purchaser, and Purchaser purchased acquired and accepted from Seller, all of Seller’s right, title and interest in and to the Initial Purchased Assets (as defined below), for the consideration and on the terms and subject to the conditions set forth in the 2022 Royalty Purchase Agreement.
WHEREAS, Seller and Purchaser desire to amend and restate the 2022 Royalty Purchase Agreement in its entirety on the terms and subject to the conditions set forth herein, pursuant to which, among other things, (i) Purchaser will sell, transfer, assign and convey back to Seller the Initial Purchased Non-U.S. Assets 2024-2030 (as defined below) and (ii) Seller will sell, transfer, assign and convey to Purchaser the Additional Purchased Assets (as defined below).
In consideration of the representations, warranties, covenants, and agreements set forth herein and for good and valuable consideration, the receipt and adequacy of which are hereby acknowledged, Seller and Purchaser hereby agree as follows:
ARTICLE I
DEFINITIONS; INTERPRETATION
Section 1.1. Definitions. As used in this Agreement, the following terms shall have the following meanings, and capitalized terms not otherwise defined in this Agreement shall have the definitions set forth in the APA. In the event a capitalized term used herein is defined in both this Agreement and the APA, the meaning given to such term in this Agreement shall control unless otherwise specified:
“2021 Letter Agreement” has the meaning set forth in the Recitals.
“2022 Letter Agreement” means the letter agreement among Seller, Rayner Surgical and Rayner Surgical Group Limited dated September 28, 2022.
“2022 Bill of Sale and Assignment” means that certain bill of sale and assignment entered into by Seller and Purchaser as of September 30, 2022, in connection with the 2022 Royalty Purchase Agreement.
“2022 Royalty Purchase Agreement” has the meaning set forth in the preamble.
“2022 Effective Date” has the meaning set forth in the preamble.
“Additional Floor Purchase Price” has the meaning set forth in Section 2.1(b)(ii).
“Additional Purchased Assets” means all Purchased Assets that are not Initial Purchased Assets.
“Affiliate” means, with respect to any Person, any other Person that directly, or indirectly through one or more intermediaries, Controls, or is Controlled by, or is under common Control with, such Person.
“Agreement” has the meaning set forth in the preamble hereto.
“APA” has the meaning set forth in the Recitals.
“APA Closing” means the closing of the transactions contemplated by the APA, which closing was consummated on December 23, 2021.
“APA Omidria Provisions” means the following provisions of the APA (and the definitions in the APA of the defined terms used in such provisions), in each case solely to the extent, and solely in the circumstances where, such provisions (and such definitions as used in such provisions) apply to the Purchased Receivables: Sections 2.7(c) through (g) (inclusive) (“Royalty Terms”), Sections 2.9 through 2.11 (inclusive) (“Payment Mechanics; Withholding and Indirect Taxes”), Section 6.27 (“Patents; Licensing; Royalty Buy-Out”), Article IX (“Indemnification; Survival”), Section 11.1 (“Assignment”) and Section 11.13 (“Equitable Remedies”). For clarity, and as an example, Sections 2.9 through 2.11 of the APA to the extent, and in the circumstances where, they are applied to the Milestone Payment (as defined in the APA) shall not constitute “APA Omidria Provisions”.
“Applicable Withholding Certificate” means (a) IRS Form W-9 (or any applicable successor form) if Purchaser (or, if Purchaser is a disregarded entity, Purchaser’s owner) is a “United States person” (as defined in Section 7701(a)(30) of the Internal Revenue Code of 1986, as amended) or (b) IRS Form W-8BEN-E (or any applicable successor form) if Purchaser (or, if Purchaser is a disregarded entity, Purchaser’s owner) is not a United States person (as defined in Section 7701(a)(30) of the Internal Revenue Code of 1986, as amended).
“Back-Up Security Interest” has the meaning set forth in Section 2.4(b).
“Business Day” means any day other than (a) a Saturday or Sunday or (b) a day on which banking institutions located in (i) the City of New York in the State of New York, (ii) the City of Seattle in the State of Washington or (iii) the City of Toronto in the Province of Ontario, Canada are permitted or required by applicable Law to remain closed.
“Closing” has the meaning set forth in Section 3.1.
“Closing Date” has the meaning set forth in Section 3.1.
“Confidential Information” has the meaning set forth in Section 7.10(b).
“Consent” means any consent, approval, license, permit, order, authorization, registration, declaration, filing or notice.
“Contingent Royalty Reduction” means a reduction in the Royalty as expressly permitted pursuant to Section 2.7(c)(ii) of the APA.
“Contract” means any legally binding agreement, arrangement, loan or credit agreement, note, bond, guaranty, mortgage, indenture, instrument, lease, sublease, license, deed of trust, undertaking, commitment or other oral or written contract or binding understanding.
“Control” means the possession, directly or indirectly, of the power to direct or cause the direction of the management and policies of a Person, whether through the ownership of voting securities or other voting interests, by contract or otherwise.
“Cumulative Purchase Price” means, collectively, as of any date, (i) the Initial Purchase Price, (ii) the Additional Floor Purchase Price and (iii) to the extent paid to Seller on or before such date, each Purchaser Milestone Payment.
“Escrow Agent” means Wilmington Trust, as escrow agent.
“Escrow Agreement” means that certain Escrow Agreement entered into by Seller, Purchaser, and the Escrow Agent as of September 30, 2022, in connection with the 2022 Royalty Purchase Agreement.
“Excluded Assets” means collectively: (a) the contractual right to payment under the APA of Retained Receivables; (b) the Retained Receivables; (c) any Royalty Payments payable by Rayner Surgical during the time prior to January 1, 2024 (other than the Specified Initial Receivables), (d) any Royalty Payments payable by Rayner Surgical from and after January 1, 2032; and (e) any reimbursements, milestone payments, fees, indemnification, damages, awards, settlement payments or any other payments, compensation or consideration of any kind pursuant to, under or in respect of the APA, excluding such consideration paid or payable to Purchaser pursuant to the terms of this Agreement relating to Purchased Receivables and Related Payment Provisions. For clarity, with respect to Net Revenue of Subject Products occurring in November 2031 and December 2031, each Royalty Payment in respect of such Net Revenue shall constitute Excluded Assets (because such Royalty Payments will be payable by Rayner Surgical during calendar year 2032).
“Ex-US Receivables” means, with respect to any Subject Product and any country other than the United States, each Royalty Payment to the extent attributable to Royalties payable by Rayner Surgical with respect to such Subject Product and such country.
“Financing Sources” means, with respect to any Person, (a) any commercial bank or other lender that provides non-convertible debt financing in the ordinary course of its business to such Person, (b) those Persons that provide financing (whether in the form of debt, equity or otherwise) to such Person in accordance with contractual relationships and not directly related to or entered into in contemplation of the transactions contemplated hereby and (c) rating agencies.
“First Payment” has the meaning set forth in Section 2.1(b)(ii)(y).
“Governmental Entity” means any federal, state, provincial, local or foreign government or any court of competent jurisdiction, arbitral body, administrative, judicial or agency, department, political subdivision, commission, bureau or tribunal or other governmental authority, domestic or foreign.
“Initial Purchased Assets” means those certain assets Seller sold, transferred, assigned and conveyed to Purchaser pursuant to the 2022 Royalty Purchase Agreement and the 2022 Bill of Sale and Assignment.
“Initial Purchase Price” has the meaning set forth in Section 2.1(b)(i).
“Initial Purchased Non-U.S. Assets 2024-2030” has the meaning set forth in Exhibit B.
“January 2024 Amount” has the meaning set forth in Section 2.1(b)(ii)(y).
“Judgment” means any judgment, order, injunction, consent, writ, decree or stipulation granted, issued or entered by a Governmental Entity.
“Knowledge of Seller” or “Knowledge”, when used with respect to Seller, means the current actual knowledge of (a) Gregory A. Demopulos, MD, Chief Executive Officer of Seller, (b) Michael A. Jacobsen, Chief Accounting Officer of Seller, and (c) Peter B. Cancelmo, JD, General Counsel of Seller.
“Law” means any provision of federal, state, provincial, local or foreign law (including common law), statute, rule, regulation, ordinance or code issued or promulgated by any Governmental Entity.
“Liens” means any mortgage, security interest, pledge, hypothecation, charge, adverse claims, easements, rights of first or last refusal, negotiation or similar priority, option, deed of trust, participation interest, deposit arrangement, title retention, conditional sale, financing lease or other security arrangement, or encumbrance, whether imposed by Contract, Law or otherwise.
“Milestone Event 1” means an event after January 1, 2024, and prior to January 1, 2026 where Net Revenue with respect to OMIDRIA within the United States (over a total of four (4) consecutive quarters) reaches an aggregate total equal to or greater than one hundred fifty six million dollars (U.S. $156,000,000).
“Milestone Event 2” means an event after January 1, 2024, and prior to January 1, 2028 where Net Revenue with respect to OMIDRIA within the United States (over a total of four (4) consecutive quarters) reaches an aggregate total equal to or greater than one hundred eighty one million dollars (U.S. $181,000,000).
“Net Revenue” has the meaning set forth in the APA.
“New Bill of Sale and Assignment” means that certain bill of sale and assignment, substantially in the form of Exhibit B attached hereto, entered into by Seller and Purchaser as of the date hereof.
“New Rayner Surgical Instruction Letter” means that certain letter of instruction, substantially in the form of Exhibit C attached hereto, from Seller to Rayner Surgical, and countersigned by Purchaser.
“OMIDRIA” means the Product and all Subject Products (as those terms are defined in the APA).
“Out-of-Scope Set-Off” means, in all cases, any Set-Off against Royalties by Rayner Surgical of any actual or alleged amount owing from Seller to Rayner Surgical in respect of any right of Rayner Surgical against Seller arising from or in connection with any matter other than the Purchased Receivables, whether such Set-Off is made pursuant to Section 2.7(c)(iii) of the APA or otherwise (it being understood and agreed that any dispute between Seller and Rayner Surgical that is unrelated to the Purchased Receivables constitutes a “matter other than the Purchased Receivables”). For clarity, (a) a Contingent Royalty Reduction is not an Out-of-Scope Set-Off, (b) any withholding or other Set-Off permitted pursuant to Section 2.10 or Section 2.11 of the APA is not an Out-of-Scope Set-Off and (c) a credit or other Set-Off against future Royalties by Rayner Surgical of amounts it may be owed pursuant to Section 2.7(e) of the APA for an overpayment of Purchased Receivables is not an Out-of-Scope Set-Off, provided, that, such Set-Off shall be subject to Section 7.4(b)(iii) and Section 7.4(b)(iv). For the avoidance of doubt, any Set-Off against Purchased Receivables by Rayner Surgical of amounts it may be owed pursuant to Section 2.7(e) of the APA for an overpayment of any Royalty Payment that was payable by Rayner Surgical prior to January 1, 2024 is an Out-of-Scope Set-Off and is subject to Section 7.1(c).
“Party” shall have the meaning set forth in the preamble.
“Permitted Liens” means, collectively:
(i) Liens expressly set forth in Section 2.4(b);
(ii) Liens expressly set forth in the Escrow Agreement;
(iii) the Escrow Agreement itself;
(iv) Liens created by, or in favor of, Purchaser;
(v) the deposit arrangement expressly set forth in Section 2.9(a) of the APA; and
(vi) the provisions set forth in Section 6.27(c) of the APA and in the 2021 Letter Agreement.
“Person” means any individual, firm, corporation, partnership, limited liability company, trust, joint venture, association, unincorporated organization, Governmental Entity or other entity or organization.
“Proceeds” means any amounts actually recovered from a Person (other than Purchaser) as a result of any litigations, settlement or resolution of actions, suits, proceedings, claims or disputes to the extent related to the Purchased Receivables or the APA Omidria Provisions.
“Product” has the meaning set forth in the APA.
“Purchased Assets” means the Purchased Receivables, together with the contractual right to payment under the APA to the Purchased Receivables.
“Purchased Receivables” means, with respect to any Subject Product and the United States, each Royalty Payment to the extent attributable to Royalties payable by Rayner Surgical with respect to such Subject Product and the United States during the Purchased Royalty Period applicable to such Subject Product and the United States. For clarity, the foregoing is without prejudice to the Related Payment Provisions, pursuant to which Purchaser may be paid certain of the Purchased Receivables and other amounts.
“Purchased Royalty Period” means with respect to any Subject Product and the United States, the period beginning on (and including) January 1, 2024 and ending on (and including) the earlier of (a) December 31, 2031, and (b) the last day of the calendar month that is two calendar months after the calendar month during which the Royalty Term (as defined in the APA) applicable to such Subject Product and the United States expires.
“Purchaser” has the meaning set forth in the preamble.
“Purchaser Material Adverse Effect” means any one or more of: (a) a material adverse effect on the ability of Purchaser to consummate the transactions contemplated by the Transaction Documents and perform its obligations under the Transaction Documents and (b) a material adverse effect on (i) the validity or enforceability of any Transaction Document against Purchaser or (ii) the rights of Seller thereunder.
“Purchaser Milestone Payment 1” means a one-time lump sum payment by Purchaser to Seller of (i) ten million dollars (U.S. $10,000,000) if Milestone Event 1 is equal to or greater than one hundred fifty six million dollars (U.S. $156,000,000) but less than [***] dollars (U.S. $[***]); (ii) [***] dollars (U.S. $[***]) if Milestone Event 1 is equal to or greater than [***] dollars (U.S. $[***]) but less than [***] dollars (U.S. $[***]); or (iii) twenty-seven million five hundred thousand dollars (U.S. $27,500,000) if Milestone Event 1 is equal to or greater than one hundred sixty million dollars (U.S. $160,000,000).
“Purchaser Milestone Payment 2” means a one-time lump sum payment by Purchaser to Seller of (i) eight million dollars (U.S. $8,000,000) if Milestone Event 2 is equal to or greater than one hundred eighty one million dollars (U.S. $181,000,000) but less than [***] dollars (U.S. $[***]); (ii) [***] dollars (U.S. $[***]) if Milestone Event 2 is equal to or greater than [***] dollars (U.S. $[***]) but less than [***] dollars (U.S. $[***]); or (iii) twenty-seven million five hundred thousand dollars (U.S. $27,500,000) if Milestone Event 2 is equal to or greater than one hundred eighty five million dollars (U.S. $185,000,000).
“Purchaser Milestone Payments” has the meaning set forth in Section 2.1(b)(ii).
“Purchaser Participated Audit” has the meaning set forth in Section 7.4(b)(ii).
“Rayner Surgical” means Rayner Surgical Inc. (including, for clarity, any successors or assigns, as applicable, solely to the extent permitted pursuant to the terms of this Agreement and the APA).
“Rayner Surgical Instruction Letter” means the letter of instruction delivered by Seller to Rayner Surgical on October 3, 2022, in connection with the 2022 Royalty Purchase Agreement.
“Receivables” means each Royalty Payment to the extent attributable to Royalties payable by Rayner Surgical with respect to any Subject Product and any country.
“Related Payment Provisions” means Section 7.1(a) (“Payments to Purchaser”), Section 7.2 (“Interest”), Section 7.4 (“Audits of Rayner Surgical”), Section 7.7 (“Enforcement of APA”) and Section 8.1(a) (“Indemnification by Seller”).
“Representatives” means, collectively, with respect to any Person, the managers, shareholders (provided, that, such Person does not have any equity shares that are publicly traded), partners, directors, officers, trustees, employees, agents, advisors or other representatives (including attorneys, accountants, consultants, scientists and financial advisors) of such Person.
“Responsible Employee of Seller” means any employee of Seller referred to in the definition of “Knowledge of Seller” and any successor to such employee.
“Retained Ex-US Receivables” means the Ex-US Receivables other than the Specified Purchased Ex-US Receivables.
[***] CERTAIN IDENTIFIED INFORMATION HAS BEEN EXCLUDED FROM THE EXHIBIT BECAUSE IT BOTH (A) IS NOT MATERIAL AND (B) WOULD BE COMPETITIVELY HARMFUL IF PUBLICLY DISCLOSED.
“Retained Receivables” means the portion of the Receivables that does not constitute Purchased Receivables. For the avoidance of doubt, “Retained Receivables” includes the Retained Ex-US Receivables.
“Royalty” has the meaning set forth in the APA. For clarity, “Royalty” does not include any of the following: the Milestone Payment, the Upfront Payment, any reimbursements for Taxes (each such defined term as defined in the APA) pursuant to Section 10.3 of the APA, or any payments pursuant to Sections 2.7(h), 2.7(i), 2.7(j) or 2.7(k) of the APA.
“Royalty Payment” means each payment by Rayner Surgical of Royalties pursuant to Sections 2.7(c)(i)-(ii) of the APA with respect to each Subject Product and each country (including after giving effect to the Contingent Royalty Reductions applicable thereto pursuant to Section 2.7(c)(ii) of the APA).
“Royalty Reports” means (a) the monthly and quarterly reports relating to Royalties required to be delivered by Rayner Surgical to Seller pursuant to Section 2.7(c)(ii)(D) of the APA and (b) the additional materials required to be delivered by Rayner Surgical to Seller pursuant to Section 2.7(c)(ii)(E) of the APA.
“Seller” shall have the meaning set forth in the preamble.
“Seller Material Adverse Effect” means any one or more of: (a) a material adverse effect on the ability of Seller to consummate the transactions contemplated by the Transaction Documents and perform its obligations under the Transaction Documents, (b) a material adverse effect on (i) the validity or enforceability of any Transaction Document against Seller or (ii) the rights of Purchaser thereunder, (c) a material adverse effect on the rights of Seller under the APA that could have a material adverse effect on the Purchased Receivables, or (d) a material adverse effect on the Purchased Assets (including the timing, amount or duration thereof).
“Seller Participated Audit” has the meaning set forth in Section 7.4(b)(i).
“Specified Financing Statement” has the meaning set forth in Section 2.4(c).
“Specified Initial Receivables” means the “Purchased Receivables” under and as defined in the 2022 Royalty Purchase Agreement, but excluding the Retained Ex-US Receivables.
“Specified Purchased Ex-US Receivables” means the portion of the Ex-US Receivables included in the Initial Purchased Assets to the extent such portion of the Ex-US Receivables are payable by Rayner Surgical during the period beginning on (and including) September 1, 2022 and ending on (and including) the earlier of (a) December 31, 2023, and (b) the last day of the calendar month that is two calendar months after the calendar month during which the Royalty Term (as defined in the APA) applicable to such Subject Product and such country expires.
“Specified Purchased Intellectual Property” means the United States issued Patents (as defined in the APA) and United States Patent applications included in the Purchased Intellectual Property (as defined in the APA) listed on Section 1.1(h)(B) of the Seller Schedule (as defined in the APA) other than any such United States issued Patents and United States Patent applications listed as “abandoned” thereon.
“Separate Payment” has the meaning set forth in Section 4.1(j).
“Set-Off” means any adjustments, modifications, offsets, set-offs, credits, deductions or reductions.
“Subject Product” has the meaning set forth in the APA.
“Transaction Documents” means this Agreement, the New Bill of Sale and Assignment, the Escrow Agreement and the New Rayner Surgical Instruction Letter.
“UCC” means the Uniform Commercial Code as in effect from time to time in the State of Washington.
“UCC Amendment” has the meaning set forth in Section 2.4(c).
Section 1.2. Certain Interpretations. Except where expressly stated otherwise in this Agreement, the following rules of interpretation apply to this Agreement:
(a)“ either” and “or” are not exclusive and “include”, “includes” and “including” shall be deemed to be followed by the words “without limitation”;
(b)“ extent” in the phrase “to the extent” means the degree to which a subject or other thing extends, and such phrase does not mean simply “if”;
(c)“ hereof”, “hereto”, “herein” and “hereunder” and words of similar import when used in this Agreement refer to this Agreement as a whole and not to any particular provision of this Agreement;
(d) references to a Contract mean such Contract as from time to time amended, amended and restated, supplemented or otherwise modified, in each case to the extent not prohibited by such Contract or this Agreement;
(e) references to a Person are also to its permitted successors and assigns;
(f) definitions are applicable to the singular as well as the plural forms of such terms;
(g) references to an “Article”, “Section”, “Exhibit” or “Schedule” refer to an Article or Section of, or an Exhibit or Schedule to, this Agreement;
(h) references to “$” or otherwise to dollar amounts refer to the lawful currency of the United States;
(i) references to a Law include any amendment or modification to such Law and any rules and regulations issued thereunder, whether such amendment or modification is made, or issuance of such rules and regulations occurs, before, on or after the date of this Agreement; and
(j) references to this “Agreement” shall include a reference to all Schedules and Exhibits attached to this Agreement (including the Updated Schedule of Exceptions attached hereto as Exhibit A), all of which constitute a part of this Agreement and are incorporated herein for all purposes.
ARTICLE II
PURCHASE AND SALE OF PURCHASED ASSETS
Section 2.1. Purchase and Sale of Purchased Assets.
(a) Purchase and Sale. Pursuant to the 2022 Royalty Purchase Agreement and the 2022 Bill of Sale and Assignment, Seller sold, transferred, assigned and conveyed to Purchaser, and Purchaser purchased, acquired and accepted from Seller, free and clear of all Liens (other than Permitted Liens), all of Seller’s right, title and interest in and to the Initial Purchased Assets. Upon the terms and subject to the conditions of this Agreement, at the Closing, (i) Seller shall sell, transfer, assign and convey to Purchaser, and Purchaser shall purchase, acquire and accept from Seller, free and clear of all Liens (other than Permitted Liens), all of Seller’s right, title and interest in and to the Additional Purchased Assets and (ii) Purchaser shall transfer, assign and convey to Seller, and Seller shall accept from Purchaser, free and clear of all Liens, all of Purchaser’s right, title and interest in and to the Initial Purchased Non-U.S. Assets 2024-2030. It is understood and agreed that Purchaser shall not, by its purchase of the Initial Purchased Assets and the Additional Purchased Assets (and by its transfer of the Initial Purchased Non-U.S. Assets 2024-2030 to Seller), acquire any assets or rights of Seller under, or relating to, the APA other than those specified in the immediately preceding two sentences or the rights as set forth in this Agreement with respect to the APA Omidria Provisions.
(b) Purchase Price.
(i) The purchase price for the Initial Purchased Assets was one hundred twenty-five million dollars (U.S. $125,000,000.00) minus any deduction by Purchaser in respect of any applicable withholding taxes (the “Initial Purchase Price”) (it being acknowledged and agreed by Purchaser that no such withholding or deduction was required and Seller delivered to Purchaser an executed IRS Form W-9 certifying that Seller is exempt from United States federal withholding tax).
(ii) The purchase price for the Additional Purchased Assets is:
(x) an upfront payment of one hundred fifteen million dollars (U.S. $115,000,000) to be paid by Purchaser to Seller on the Closing Date minus any deduction by Purchaser in respect of any applicable withholding taxes (the “Additional Floor Purchase Price”) (it being acknowledged and agreed by Purchaser that no such withholding or deduction shall be required, provided, that, Seller delivers to Purchaser an executed IRS Form W-9 certifying that Seller is exempt from United States federal withholding tax);
(y) five hundred twenty five thousand four hundred seventy dollars (U.S. $525,470) minus any deduction by Purchaser in respect of any applicable withholding taxes (the “January 2024 Amount”) (it being acknowledged and agreed by Purchaser that no such withholding or deduction shall be required, provided, that, Seller delivers to Purchaser the executed IRS Form W-9 pursuant to Section 2.1(b)(ii)(x)); it being understood and agreed that such January 2024 Amount will be deducted from the first payment of Purchased Receivables to be made to Purchaser from the Escrow Account after the Closing Date (the “First Payment”) and such January 2024 Amount will instead be paid to Seller from the Escrow Account at the same time the remaining amount (after the deduction of the January 2024 Amount) of the First Payment is paid from the Escrow Account to Purchaser (and Purchaser agrees that it shall appropriately reflect the foregoing allocation in the instructions that are furnished to the Escrow Agent pursuant to Section 1.3 of the Escrow Agreement with respect to the distribution of the First Payment); and
(z) the Purchaser Milestone Payment 1 and the Purchaser Milestone Payment 2, in each case to the extent the same become due and payable under Section 2.1(c), minus any deduction by Purchaser in respect of any applicable withholding taxes (collectively, the “Purchaser Milestone Payments”) (it being acknowledged and agreed by Purchaser that no such withholding or deduction shall be required, provided, that, at the time any Purchaser Milestone Payment is paid to Seller, (A) the executed IRS Form W-9 provided by Seller pursuant to Section 2.1(b)(ii)(x) remains accurate and complete or (B) Seller has provided Purchaser any additional tax forms or information establishing that Seller is exempt from United States federal withholding tax).
(c) Milestone Events; Milestone Payments.
(i) If the Milestone Event 1 is achieved, Purchaser shall pay the Purchaser Milestone Payment 1 to Seller on the later of (x) Thursday, January 15, 2026 and (y) the date that is two (2) Business Days after Seller notifies Purchaser in writing of the occurrence of the Milestone Event 1 (which notice shall include copies of the Royalty Reports in respect of the four (4) consecutive quarters that are used to satisfy Milestone Event 1), by wire transfer of immediately available funds to the account specified by Seller in writing to Purchaser prior to the date of such payment.
(ii) If the Milestone Event 2 is achieved, Purchaser shall pay the Purchaser Milestone Payment 2 to Seller on the later of (x) Friday, January 14, 2028 and (y) the date that is two (2) Business Days after Seller notifies Purchaser in writing of the occurrence of the Milestone Event 2 (which notice shall include copies of the Royalty Reports in respect of the four (4) consecutive quarters that are used to satisfy Milestone Event 2), by wire transfer of immediately available funds to the account specified by Seller in writing to Purchaser prior to the date of such payment.
(iii) It is understood and agreed that the same four (4) consecutive quarters ending before January 1, 2026 can be used to satisfy Milestone Event 1 and Milestone Event 2. For example, if aggregate Net Revenue with respect to OMIDRIA within the United States in Q3 2024, Q4 2024, Q1 2025 and Q2 2025 totals U.S. $184 million or more, then both Milestone Event 1 and Milestone Event 2 shall be achieved and (A) a Purchaser Milestone Payment 1 amount of U.S. $27.5 million shall be paid by Purchaser to Seller by the due date set forth in Section 2.1(c)(i) and (B) a Purchaser Milestone Payment 2 amount of at least U.S. $[***] shall be paid by Purchaser to Seller by the due date set forth in Section 2.1(c)(ii) (unless a higher Net Revenue threshold amount is reached with respect to Milestone Event 2 prior to January 1, 2028, in which case the higher corresponding Purchaser Milestone Payment 2 amount shall be paid by Purchaser to Seller by the due date set forth in Section 2.1(c)(ii)). For the avoidance of doubt, in no event shall Purchaser Milestone Payment 1 or Purchaser Milestone Payment 2 be paid more than once. It is further understood and agreed that any Purchaser Milestone Payment may become due and payable hereunder whether or not the other Purchaser Milestone Payment has become, or in the future becomes, due and payable hereunder.
(iv) Without limiting Purchaser’s obligations to pay the Purchaser Milestone Payments to Seller by the due dates set forth in Sections 2.1(c)(i) and 2.1(c)(ii), if Purchaser fails to timely pay any Purchaser Milestone Payment to Seller in accordance with such Section 2.1(c)(i) or 2.1(c)(ii), as applicable, Purchaser shall pay interest to Seller (payable on demand and compounded quarterly) for any such late payment at a per annum rate equal to the U.S. Prime Rate, as reported in the Wall Street Journal, Eastern Edition, for the first date on which such payment was to be paid to Seller under Section 2.1(c)(i) or Section 2.1(c)(ii), respectively, plus two percent (2%), from the first (1st) day the payment was to be paid to Seller under Section 2.1(c)(i) or Section 2.1(c)(ii), respectively, to the day of actual payment of the amount to Seller.
Section 2.2. No Purchase or Sale of Excluded Assets. Notwithstanding anything to the contrary contained in this Agreement, Seller shall retain all its right, title and interest in and to, and there shall be excluded from the sale, transfer, assignment and conveyance to Purchaser under this Agreement, all Excluded Assets.
Section 2.3. No Obligations Transferred. Notwithstanding anything to the contrary contained in this Agreement, (a) the sale, transfer, assignment and conveyance to Purchaser of the Purchased Assets pursuant to this Agreement (and the 2022 Royalty Purchase Agreement) shall not in any way subject Purchaser to, or transfer, novate, affect or modify, any obligation or liability of Seller under the APA and (b) Purchaser expressly does not assume or agree to become responsible for any obligation or liability of Seller whatsoever (it being understood and agreed that this clause (b) shall not serve to limit Purchaser’s obligations under Section 7.4 and Section 7.10).
[***] CERTAIN IDENTIFIED INFORMATION HAS BEEN EXCLUDED FROM THE EXHIBIT BECAUSE IT BOTH (A) IS NOT MATERIAL AND (B) WOULD BE COMPETITIVELY HARMFUL IF PUBLICLY DISCLOSED.
Section 2.4. Sale of Purchased Assets; Back-Up Security Interest.
(a) It is the intention of the Parties that the sale, transfer, assignment and conveyance contemplated by this Agreement (and by the 2022 Royalty Purchase Agreement) be, and is, a true, complete, absolute and irrevocable sale, transfer, assignment and conveyance by Seller to Purchaser of all of Seller’s right, title and interest in and to the Purchased Assets. Neither Seller nor Purchaser intends the transactions contemplated by this Agreement (and by the 2022 Royalty Purchase Agreement) to be, or for any purpose characterized as, a loan from Purchaser to Seller, or a pledge, security interest or other Lien, or a financing transaction or a borrowing. It is the intention of the Parties that the beneficial interest in and title to the Purchased Receivables and any “proceeds” (as such term is defined in the UCC) thereof shall not be part of Seller’s estate in the event of the filing of a petition by or against Seller under any U.S. bankruptcy Laws or similar state Laws relating to or affecting creditors’ rights generally. Each of Seller and Purchaser hereby waives, to the maximum extent permitted by applicable Law, any right to contest or otherwise assert that the sale contemplated by this Agreement (and by the 2022 Royalty Purchase Agreement) does not constitute a true, complete, absolute and irrevocable sale, transfer, assignment and conveyance by Seller to Purchaser of all of Seller’s right, title and interest in and to the Purchased Assets under applicable Law, which waiver shall, to the maximum extent permitted by applicable Law, be enforceable against Seller in any bankruptcy or insolvency proceeding relating to Seller.
(b) Accordingly, the Parties shall treat the sale, transfer, assignment and conveyance of the Purchased Assets as a sale of an “account” or a “payment intangible” (as appropriate) in accordance with the UCC and Seller does hereby authorize Purchaser, from and after the Closing, to file such financing statement (and continuation statements with respect to such financing statement when applicable) naming Seller as the seller and Purchaser as the purchaser of the Purchased Assets as may be necessary to perfect such sale. Not in derogation of the foregoing statement of the intent of the Parties in this regard, and for the purposes of providing additional assurance to Purchaser in the event that, despite the intent of the parties hereto, the sale, transfer, assignment and conveyance contemplated hereby (and by the 2022 Royalty Purchase Agreement) is hereafter held not to be a sale for any reason, Seller shall, and hereby does, grant to Purchaser a security interest in and to all right, title and interest in, to and under the Purchased Assets as continuing security to secure payment to Purchaser of amounts equal to the Purchased Receivables as they become due and payable under the APA. Seller hereby authorizes Purchaser, from and after the Closing, to file such financing statements (and continuation statements with respect to such financing statements when applicable) in such manner and such jurisdictions as are necessary or appropriate to perfect such security interest (the “Back-Up Security Interest”), and this Agreement shall constitute a security agreement for purposes of the UCC. The Parties agree that the Back-Up Security Interest is being granted as security for the payment of amounts to Purchaser equal to the Purchased Receivables as they become due and payable under the APA. Notwithstanding the foregoing, nothing in this Section 2.4 shall bind either Party regarding the reporting of the transactions contemplated by the Transaction Documents for accounting purposes, securities Law purposes or tax purposes.
(c) With respect to the Uniform Commercial Code financing statement naming Seller as seller/debtor and Purchaser as buyer/secured party in respect of the Initial Purchased Assets that was filed by Purchaser with the Department of Licensing of the State of Washington on October 3, 2022 in accordance with the 2022 Royalty Purchase Agreement (the “Specified Financing Statement”), Seller hereby authorizes Purchaser to file, and Purchaser hereby agrees to file, with the Department of Licensing of the State of Washington after the consummation of the Closing, a Uniform Commercial Code Financing Statement Amendment (Form UCC3) that restates the collateral description set forth in the Specified Financing Statement to instead cover the Purchased Assets, in form and substance reasonably satisfactory to Seller (the “UCC Amendment”).
ARTICLE III
CLOSING; DELIVERABLES
Section 3.1. Closing. The closing of this Agreement (the “Closing”) shall take place remotely on the date hereof, or at such place, time and date as the parties hereto may mutually agree. The date on which the Closing occurs is referred to in this Agreement as the “Closing Date”.
Section 3.2. Payment of Additional Floor Purchase Price. At the Closing, Purchaser shall deliver to Seller payment of the Additional Floor Purchase Price by wire transfer of immediately available funds to the account specified by Seller in writing to Purchaser prior to the Closing Date.
Section 3.3. Closing Certificates.
(a) Seller’s Closing Certificate. At the Closing, Seller shall deliver to Purchaser a certificate of the Secretary of Seller, dated the Closing Date, certifying as to (i) the incumbency of the officer (or officers) of Seller executing the Transaction Documents and (ii) the attached copies of Seller’s organizational documents and resolutions adopted by Seller’s Board of Directors authorizing the execution and delivery by Seller of the Transaction Documents and the consummation by Seller of the transactions contemplated thereby.
(b) Purchaser’s Closing Certificate. At the Closing, Purchaser shall deliver to Seller a certificate of an officer of Purchaser, dated the Closing Date, certifying as to (x) the incumbency of the officer of Purchaser executing the Transaction Documents and (y) the attached copies of relevant extracts from Purchaser’s organizational documents and resolutions adopted by Purchaser’s general partner authorizing the execution and delivery by Purchaser of the Transaction Documents and the consummation by Purchaser of the transactions contemplated thereby.
Section 3.4. New Bill of Sale and Assignment. At the Closing, Seller and Purchaser shall each deliver to the other Party hereto a duly executed counterpart to the New Bill of Sale and Assignment, evidencing (i) the assignment to Seller of the Initial Purchased Non-U.S. Assets 2024-2030 and (ii) the sale and assignment to Purchaser of the Additional Purchased Assets.
Section 3.5. Tax Forms. Prior to the Closing, (i) Purchaser shall have delivered or cause to be delivered to Seller a validly executed, true and complete Applicable Withholding Certificate certifying that Purchaser (or Purchaser’s owner, if Purchaser is a disregarded entity for United States federal income tax purposes) is not subject to United States federal withholding tax (including backup withholding) in respect of amounts payable to Purchaser hereunder, and (ii) Seller shall have delivered to Purchaser a validly executed, true and complete IRS Form W-9 certifying that Seller is not subject to United States federal withholding tax (including backup withholding) in respect of amounts payable to Seller hereunder.
Section 3.6. New Rayner Surgical Instruction Letter. As soon as practicable after the Closing, and in any event no later than two (2) Business Days after the Closing, Seller shall deliver to Rayner Surgical the duly executed New Rayner Surgical Instruction Letter (so long as Purchaser has delivered to Seller prior to such time a counterpart to the New Rayner Surgical Instruction Letter duly executed by Purchaser).
Section 3.7. Receipt. As soon as practicable after the Closing, and in any event no later than two (2) Business Days after the Closing, Seller shall deliver to Purchaser a duly executed receipt for the payment of the Additional Floor Purchase Price.
ARTICLE IV
SELLER’S REPRESENTATIONS AND WARRANTIES
Section 4.1. Representations and Warranties. Except as set forth on Exhibit A, Seller hereby represents and warrants to Purchaser as of the date hereof:
(a) Existence; Good Standing. Seller is a corporation duly incorporated, validly existing and in good standing under the Laws of the State of Washington. Seller is duly licensed or qualified to do business and is in corporate good standing in each jurisdiction in which the nature of the business conducted by it or the character or location of the properties and assets owned, leased or operated by it makes such licensing or qualification necessary, except where the failure to be so licensed or qualified and in corporate good standing has not and would not reasonably be expected to have, either individually or in the aggregate, a Seller Material Adverse Effect.
(b) Authorization. Seller has the requisite corporate power and authority to execute, deliver and perform the Transaction Documents and to consummate the transactions contemplated thereby. The execution, delivery and performance of the Transaction Documents, and the consummation of the transactions contemplated thereby, have been duly authorized by all necessary corporate action on the part of Seller.
(c) Enforceability. Each of the Transaction Documents has been duly executed and delivered by Seller, and constitutes a valid and binding obligation of Seller, enforceable against Seller in accordance with its terms, except as may be limited by general principles of equity (regardless of whether considered in a proceeding at law or in equity) and by applicable bankruptcy, insolvency, moratorium and other similar Laws of general application relating to or affecting creditors’ rights generally.
(d) Absence of Conflicts. The execution, delivery and performance by Seller of the Transaction Documents and the consummation of the transactions contemplated thereby do not constitute a breach of or default under any provision of (i) the organizational documents of Seller, (ii) any Law or Judgment applicable to Seller, (iii) the APA or (iv) any written Contract (other than the APA) to which Seller is a party or by which Seller is bound (or, to the Knowledge of Seller, any oral Contract to which Seller is a party or by which Seller is bound), except, in the case of clauses (ii) and (iv), for such breaches or defaults that, individually or in the aggregate, would not reasonably be expected to result in a Seller Material Adverse Effect.
(e) Consents. No Consent of any Governmental Entity or any other Person is required by or with respect to Seller in connection with (i) the execution and delivery by Seller of the Transaction Documents, (ii) the consummation of the transactions contemplated thereby or (iii) the performance by Seller of its obligations under this Agreement, except for (A) any filings required by federal securities Laws or stock exchange rules, (B) the Rayner Surgical Instruction Letter and the New Rayner Surgical Instruction Letter, (C) the filing of the financing statements referred to in Section 2.4(b), and the filing of the UCC Amendment, with the Department of Licensing of the State of Washington, (D) such Consents, the failure of which to be obtained or made, would not reasonably be expected to result, individually or in the aggregate, in a Seller Material Adverse Effect, and (E) such Consents as shall have been obtained on or prior to the date hereof.
(f) Litigation. No action, suit, proceeding or investigation before any Governmental Entity, court or arbitrator is pending, or, to the Knowledge of Seller, threatened in writing, against Seller relating to the APA or to the Product. Since the APA Closing, Seller has not received any written notice from Rayner Surgical, and otherwise has no Knowledge, of any action, suit, proceeding or investigation before any Governmental Entity, court or arbitrator that is pending against Rayner Surgical relating to the APA or to the Product.
(g) Compliance With Laws. With respect to the operation of the Business (as defined in the APA), Seller did not, during the three (3) year period immediately preceding December 1, 2021, violate any Laws (as defined in the APA) applicable to the Business or the Specified Purchased Intellectual Property (as defined below), that has had or would reasonably be expected to result, individually or in the aggregate, in a Seller Material Adverse Effect. Since the APA Closing, Seller has not received any written notice from Rayner Surgical, and otherwise has no Knowledge, that Rayner Surgical has violated any Laws (as defined in the APA) applicable to the development, manufacture, commercialization or use of the Product or the Specified Purchased Intellectual Property that has had or would reasonably be expected to result, individually or in the aggregate, in a Seller Material Adverse Effect.
(h) Brokers’ Fees. There is no investment banker, broker, finder, financial advisor or other Person who has been retained by or is authorized to act on behalf of Seller who is entitled to any fee or commission from Purchaser in connection with the transactions contemplated by this Agreement.
(i) Intellectual Property.
(i) All right, title and interest in and to the Specified Purchased Intellectual Property was owned solely and exclusively by Seller or Omeros Ireland Limited (“Omeros Ireland”) as of immediately prior to the APA Closing, and all such right, title and interest was assigned to Rayner Surgical as a result of the APA Closing.
(ii) As of immediately prior to the APA Closing, the Specified Purchased Intellectual Property was free of Liens.
(iii) As of December 1, 2021, except with respect to non-exclusive licenses and authorizations to use granted to or by third parties in the ordinary course of business or as otherwise contemplated by the APA, Schedule 4.1(i)(iii) attached hereto lists all of the written Contracts (as defined in the APA) pursuant to which Seller or Omeros Ireland had granted a third party a license to use or practice under any Patent (as defined in the APA) that is material to the operation of the Business (as defined in the APA) and included in the Specified Purchased Intellectual Property.
(iv) Attached hereto as Schedule 4.1(i)(iv) is a true and complete copy of Section 1.1(h)(B) of the Seller Schedule (as defined in the APA) as of the APA Closing. As of immediately prior to the APA Closing, Seller had not received any written notice challenging the validity, enforceability or good standing of any Specified Purchased Intellectual Property and all required maintenance fees, annuity fees or renewal fees for the Specified Purchased Intellectual Property that were due and payable prior to the APA Closing were paid prior to the APA Closing. Since the APA Closing, Seller has not received any written notice from Rayner Surgical, and otherwise has no Knowledge, that Rayner Surgical has not paid all required maintenance fees, annuity fees or renewal fees for the Specified Purchased Intellectual Property.
(v) As of immediately prior to the APA Closing, all Specified Purchased Intellectual Property and the rights to any inventions claimed or disclosed therein, were properly assigned to Seller or Omeros Ireland, and all such assignments were properly recorded in the United States Patent and Trademark Office as of immediately prior to the APA Closing.
(vi) As of December 1, 2021, there were no written (or, to the Knowledge of Seller, oral) third party allegations made to Seller by any Person alleging that the operation or conduct of the Business (as defined in the APA) infringes or misappropriates the Intellectual Property (as defined in the APA) of such Person in any material respect.
(vii) As of immediately prior to the APA Closing, none of the Patents (as defined in the APA) in the Specified Purchased Intellectual Property was involved in any interference, reissue, reexamination, derivation, supplemental examination, inter partes review, post-grant review, conflict, opposition, cancellation, litigation or other post-issuance proceeding. Since the APA Closing, Seller has not received any written notice from Rayner Surgical, and otherwise has no Knowledge, that any of the Patents (as defined in the APA) in the Specified Purchased Intellectual Property is involved in any interference, reissue, reexamination, derivation, supplemental examination, inter partes review, post-grant review, conflict, opposition, cancellation, litigation or other post-issuance proceeding.
(j) Separate Payments. Seller has not received any written notice from Rayner Surgical, and otherwise has no Knowledge, of any non-public written communications by any Governmental Entity that would materially affect the eligibility of the Product for separate payment by the Centers for Medicare and Medicaid Services (“Separate Payment”). Seller has not received any written notice from Rayner Surgical, and otherwise has no Knowledge, that Generic Entry (as defined in the APA) with respect to the Product has occurred.
(k) APA.
(i) APA; Royalty Reports; Material Notices. Attached hereto as Schedule 4.1(k)(i) are true, correct and complete copies of: (x) the APA; (y) the Royalty Reports received by Seller from Rayner Surgical prior to the date hereof; and (z) all material written notices (including any affecting the timing, amount or duration of Purchased Receivables) delivered to Rayner Surgical by Seller, or by Rayner Surgical to Seller, pursuant to the APA relating to, or involving, the Purchased Assets (including the APA Omidria Provisions), in each case since the date of the APA Closing (excluding, for the avoidance of doubt, (A) copies of, and notices to the extent related to, any of the Ancillary Agreements (as defined in the APA) (but including any such notices to the extent they relate to the Purchased Assets) and (B) drafts and prior versions of the 2022 Letter Agreement and any accompanying correspondence to the extent related to the 2022 Letter Agreement (but including any such correspondence to the extent it addresses additional matters (apart from the 2022 Letter Agreement) that are related to the Purchased Assets).
(ii) Validity and Enforceability of APA. The APA is in full force and effect with respect to Seller and, to the Knowledge of Seller, is in full force and effect with respect to Rayner Surgical, and the APA is a valid and binding obligation of Seller and, to the Knowledge of Seller, of Rayner Surgical, enforceable against each of Seller and, to the Knowledge of Seller, Rayner Surgical, in accordance with its terms, except as may be limited by general principles of equity (regardless of whether considered in a proceeding at law or in equity) and by applicable bankruptcy, insolvency, moratorium and other similar Laws of general application relating to or affecting creditors’ rights generally. Seller has not received any written notice from Rayner Surgical challenging the validity or enforceability of the APA or any obligation of Rayner Surgical to pay the Royalties thereunder (or setting forth any basis for, or any intention to mount, any such challenge), nor has Seller delivered any such notice to Rayner Surgical.
(iii) No Amendments, Waivers or Releases. Seller has not granted or agreed to any Modification (as defined below) of the APA, except, in each case, to the extent set forth in the APA or as have not had or would not reasonably be expected to result, individually or in the aggregate, in a Seller Material Adverse Effect. Other than the 2022 Letter Agreement and 2021 Letter Agreement, Seller has not received from Rayner Surgical any written (or, to the Knowledge of Seller, oral) proposal, and has not made any proposal to Rayner Surgical, to amend, waive or release any of the terms and conditions of the APA in any material respect.
(iv) No Termination, Force Majeure, etc. Seller has not (x) given Rayner Surgical any notice of termination with respect to the APA (in whole or in part) or any notice of force majeure under the APA or (y) received from Rayner Surgical any written notice of termination with respect to the APA or any written notice of force majeure under the APA. Seller does not have any rights under Section 8.1 of the APA to terminate the APA after the APA Closing without the consent of Rayner Surgical, and, to the Knowledge of Seller, Rayner Surgical does not have any rights under Section 8.1 of the APA to terminate the APA after the APA Closing without the consent of Seller. Seller has not received any written notice from Rayner Surgical, and has not delivered any notice to Rayner Surgical, expressing any intention or desire to terminate the APA. There is no agreement between Seller and Rayner Surgical to terminate the APA.
(v) No Breaches. Seller has not breached any provision of the APA in any respect, and, to the Knowledge of Seller, Rayner Surgical has not breached any provision of the APA in any respect, other than, in each case, any breaches that have not had and would not reasonably be expected to result, individually or in the aggregate, in a Seller Material Adverse Effect. Seller has not delivered any notice to Rayner Surgical, and Seller has not received any written notice from Rayner Surgical, alleging, inquiring about or seeking information relating to any actual or potential breach of the APA, other than, in each case, any breaches that have not had and would not reasonably be expected to result, individually or in the aggregate, in a Seller Material Adverse Effect.
(vi) Payments Made. Seller has received from Rayner Surgical (x) all Royalty Reports described in clause (a) of the definition of “Royalty Reports” required to be delivered under the APA since the APA Closing and prior to the date of this Agreement; and (y) the full amounts specified next to the heading “Royalty Due Omeros” in the Royalty Reports referred to in the immediately preceding clause (x) (except with respect to such Royalty Reports that were delivered during the period after September 30, 2022 and prior to the date hereof, in which case the full amounts specified therein next to the heading “Royalty Due Omeros”, the heading “Royalty to Omeros” or the heading “Estimated Royalties (30%)”, as the case may be, were paid by Rayner Surgical to the Escrow Account (as defined in the Escrow Agreement)).
(vii) No Set-Offs or Contingent Royalty Reduction. As of the APA Closing, the Contingent Royalty Reduction was not being taken against the Royalty Payments. To the Knowledge of Seller, the Contingent Royalty Reduction is not, as of the date hereof, being taken against the Royalty Payments. Seller has not received any written (or, to the Knowledge of Seller, oral) notice from Rayner Surgical expressing an intention for, or other consideration by, Rayner Surgical to take any Contingent Royalty Reduction from the Purchased Receivables or otherwise Set-Off from the Purchased Receivables, including because of any amount owed or claimed owed from Seller to Rayner Surgical. For the avoidance of doubt, it is understood and agreed by the Parties that the items specified in clauses (a) through (g) of the definition of “Net Revenue” in the APA do not constitute Contingent Royalty Reductions or other Set-Offs from the Purchased Receivables).
(viii) No Assignments. Seller has not consented to any assignment or other transfer or delegation (in whole or in part) by Rayner Surgical of, and, to the Knowledge of Seller, Rayner Surgical has not assigned, transferred or delegated, the APA (in whole or in part), other than (x) the assignment by Rayner Surgical Inc. (pursuant to Section 11.1 of the APA) to Rayner Surgical (Ireland) Limited (an Affiliate (as defined in the APA) of Rayner Surgical Inc.) of all of its rights under the APA, and the designation by Rayner Surgical Inc. (pursuant to Section 11.1 of the APA) of Rayner Surgical (Ireland) Limited to perform all of its obligations under the APA (which assignment and designation were effective as of May 18, 2023), and (y) the assignment by Rayner Surgical (Ireland) Limited (pursuant to Section 11.1 of the APA) to Rayner Intraocular Lenses Limited (an Affiliate (as defined in the APA) of Rayner Surgical (Ireland) Limited and Rayner Surgical Inc.) of all of its rights under the APA, and the designation by Rayner Surgical (Ireland) Limited (pursuant to Section 11.1 of the APA) of Rayner Intraocular Lenses Limited to perform all of its obligations under the APA (which assignment and designation were effective as of December 18, 2023). Except as contemplated by the 2022 Royalty Purchase Agreement, this Agreement and the Escrow Agreement (and except for (i) such Liens as shall have been released on or prior to the date hereof and (ii) Permitted Liens), Seller has not assigned, transferred or delegated, in whole or in part, and has not granted any Liens with respect to, the APA or the Purchased Receivables.
(ix) No Indemnification Claims. Seller has not given any notice to Rayner Surgical of, and is not in the process of evaluating or seeking, any claim for indemnification or equitable remedies pursuant to Article IX of the APA against Rayner Surgical. Rayner Surgical has not given any written notice to Seller of any claim for indemnification or equitable remedies pursuant to Article IX of the APA against Seller, nor has Rayner Surgical given any written notice to Seller expressing any intention to do so.
(x) Audits. Seller has not initiated, pursuant to Section 2.7(e) of the APA, any audit of Rayner Surgical.
(xi) No Other Agreements. Other than (x) the APA (and the Ancillary Agreements as defined therein) and (y) Contracts that are in the process of being assigned by Seller to Rayner Surgical pursuant to the APA (and the Ancillary Agreements as defined therein), there are no written Contracts between Seller, on the one hand, and Rayner Surgical or any other Person, on the other hand, that relate to the Product, the Royalties or the Purchased Receivables that would reasonably be expected to result in a Seller Material Adverse Effect. Other than written agreements assigned, transferred or novated as contemplated by the APA, Seller has not received any written notice from Rayner Surgical, and otherwise has no Knowledge, of any written agreements between Rayner Surgical and any Third Party pursuant to which rights to develop, manufacture or commercialize the Product in the United States have been granted or transferred to such Third Party.
(xii) Subject Product. The pharmaceutical product known as Omidria® is a Subject Product, and all Royalties from all Subject Product(s) (other than the Excluded Assets) are subject to this Agreement.
(xiii) No Disputes. Seller has not received any written notice of any dispute from, or given any notice of any dispute to, Rayner Surgical in connection with the APA.
(l) UCC Matters. Seller’s exact legal name is, and for the preceding ten (10) years has been, “Omeros Corporation”. Seller’s principal place of business is, and for the preceding ten (10) years has been, located in the State of Washington. Seller’s jurisdiction of organization is, and for the preceding ten (10) years has been, the State of Washington.
(m) Taxes. All material tax returns required under applicable Law to have been filed by or on behalf of Seller with respect to the Purchased Assets have been duly and timely filed, and all material taxes required to be paid under applicable Law by Seller with respect to the Purchased Assets have been paid. To the Knowledge of Seller, no deficiencies for taxes with respect to the Purchased Assets have been claimed, proposed, or assessed by any tax authority against the Seller.
(n) Title to Purchased Assets. Seller has good and valid title to the Additional Purchased Assets, free and clear of all Liens (other than Permitted Liens). Upon payment of the Additional Floor Purchase Price by Purchaser to Seller and the filing of the UCC Amendment with the Department of Licensing of the State of Washington on the Closing Date, and with effect from the Closing, Purchaser will have acquired good and valid title to the Additional Purchased Assets, free and clear of all Liens (other than Permitted Liens).
(o) Tax Status. Seller is acting as a principal, and not as an agent for any other Person, in connection with the execution, delivery and performance by Seller of the Transaction Documents and the consummation of the transactions contemplated thereby.
Section 4.2. Representations and Warranties regarding the 2022 Royalty Purchase Agreement. Seller hereby represents and warrants to Purchaser that, except as set forth on Exhibit C to the 2022 Royalty Purchase Agreement, the representations and warranties of Seller contained in Sections 4.1 through 4.6, Section 4.8 and Section 4.14 of the 2022 Royalty Purchase Agreement were true and correct when made as of the 2022 Effective Date (except to the extent such representations and warranties specifically relate to an earlier date, in which case such representations and warranties are true and correct as of such earlier date).
ARTICLE V
PURCHASER’S REPRESENTATIONS AND WARRANTIES
Section 5.1. Representations and Warranties. Purchaser represents and warrants to Seller that as of the date hereof:
(a) Existence. Purchaser is a limited partnership duly organized, validly existing and in good standing under the Laws of the State of Delaware, United States of America. Purchaser is duly licensed or qualified to do business and is in corporate good standing in each jurisdiction in which the nature of the business conducted by it or the character or location of the properties and assets owned, leased or operated by it makes such licensing or qualification necessary, except where the failure to be so licensed or qualified and in corporate good standing has not had and would not reasonably be expected to have, either individually or in the aggregate, a Purchaser Material Adverse Effect.
(b) Authorization. Purchaser has the requisite partnership power and authority to execute, deliver and perform the Transaction Documents and to consummate the transactions contemplated thereby. The execution, delivery and performance of the Transaction Documents, and the consummation of the transactions contemplated thereby, have been duly authorized by all necessary corporate action on the part of the general partner of Purchaser.
(c) Enforceability. Each of the Transaction Documents has been duly executed and delivered by Purchaser, and constitutes a valid and binding obligation of Purchaser, enforceable against Purchaser in accordance with its terms, except as may be limited by general principles of equity (regardless of whether considered in a proceeding at law or in equity) and by applicable bankruptcy, insolvency, moratorium and other similar Laws of general application relating to or affecting creditors’ rights generally.
(d) Absence of Conflicts. The execution, delivery and performance by Purchaser of the Transaction Documents and the consummation of the transactions contemplated thereby do not constitute a breach or default under any provision of (a) the organizational documents of Purchaser, (b) any Law or Judgment applicable to Purchaser or (c) any written Contract to which Purchaser is a party or by which Purchaser is bound, except, in the case of clauses (b) and (c), for such breaches or defaults that, individually or in the aggregate, would not reasonably be expected to result in a Purchaser Material Adverse Effect.
(e) Consents. No Consent of any Governmental Entity or any other Person is required by or with respect to Purchaser in connection with (a) the execution and delivery by Purchaser of the Transaction Documents, (b) the consummation of the transactions contemplated thereby or (c) the performance by Purchaser of its obligations under this Agreement, except for (x) such Consents, the failure of which to be obtained or made, would not reasonably be expected to result, individually or in the aggregate, in a Purchaser Material Adverse Effect, and (y) such Consents as shall have been obtained on or prior to the date hereof.
(f) Litigation. No action, suit, proceeding or investigation before any Governmental Entity, court or arbitrator is pending, or, to the knowledge of Purchaser, threatened, against Purchaser that, individually or in the aggregate, would reasonably be expected to result in a Purchaser Material Adverse Effect.
(g) Compliance With Laws. Purchaser has not violated, is not in violation of, has not been given notice that it has violated, and, to the knowledge of Purchaser, Purchaser is not under investigation with respect to its violation of, and has not been threatened to be charged with any violation of, any applicable Law or any Judgment of any Governmental Entity, which violation would reasonably be expected to result, individually or in the aggregate, in a Purchaser Material Adverse Effect.
(h) Brokers’ Fees. There is no investment banker, broker, finder, financial advisor or other Person who has been retained by or is authorized to act on behalf of Purchaser who is entitled to any fee or commission from or on behalf of Seller in connection with the transactions contemplated by this Agreement.
(i) Financing. Purchaser (x) has sufficient cash on hand or binding and enforceable commitments to provide it with funds sufficient to satisfy its obligation to pay the Additional Floor Purchase Price at Closing and (y) will have sufficient cash on hand or binding and enforceable commitments to provide it with funds sufficient to satisfy its obligations to pay the Purchaser Milestone Payment 1 and the Purchaser Milestone Payment 2 if and when they become due and payable in accordance with Section 2.1(c). Purchaser has no reason to believe, and has not been provided with any notice (whether written or otherwise), that any of the Persons providing the commitments referred to above are unable or are not required or do not intend, for any reason, to satisfy their obligations under such commitments. Purchaser acknowledges that its obligations under this Agreement are not contingent on obtaining financing.
(j) Tax Status. Purchaser is acting as a principal, and not as an agent for any other Person, in connection with the execution, delivery and performance by Purchaser of the Transaction Documents and the consummation of the transactions contemplated thereby. The Applicable Withholding Certificate of Purchaser delivered to Seller on the Closing Date is valid, true and properly executed. As of the date hereof, payments to be made hereunder are exempt from and not subject to United States withholding tax or, to the Purchaser’s knowledge, Irish withholding tax.
Section 5.2. Representations and Warranties regarding the 2022 Royalty Purchase Agreement. Purchaser hereby represents and warrants to Seller that the representations and warranties of Purchaser contained in Sections 5.1 through 5.10 of the 2022 Royalty Purchase Agreement were true and correct when made as of the 2022 Effective Date.
ARTICLE VI
RECEIVABLES AND PAYMENTS
Section 6.1. No Out-of-Pocket Payments by Seller for Purchased Receivables. The Parties agree and acknowledge that, subject to the Related Payment Provisions, subject to Sections 6.2(d) and 7.1(c) and subject to Purchaser’s remedies for breach of this Agreement, (a) Seller is obligated to pay to Purchaser the Purchased Receivables only out of the Royalty Payments to the extent attributable to Royalties paid by Rayner Surgical with respect any Subject Product and the United States during the Purchased Royalty Period applicable to such Subject Product and the United States and (b) Seller will not have any obligation to pay Purchaser the Purchased Receivables from any other sources of funds.
Section 6.2. Adjustments to Pre-2024 Royalty Payments. It is acknowledged and agreed that (a) Purchaser has heretofore received Specified Initial Receivables that were payable by Rayner Surgical during the partial calendar year from September 1, 2022 through December 31, 2022 in an amount equal to the Annual Purchased Receivables Cap (as defined in the 2022 Royalty Purchase Agreement) applicable to such partial calendar year, (b) Purchaser has heretofore received Specified Initial Receivables that were payable by Rayner Surgical during the calendar year from January 1, 2023 through December 31, 2023 in an amount equal to the Annual Purchased Receivables Cap (as defined in the 2022 Royalty Purchase Agreement) applicable to such calendar year, (c) Seller (and not Purchaser) is entitled to any and all payments made by Rayner Surgical after the date hereof in respect of Royalty Payments that were payable by Rayner Surgical prior to January 1, 2024 (whether such payments constitute corrections of underpayments or otherwise, including, without limitation, corrections of gross to net adjustments and other corrections of Net Revenue calculations) and (d) with respect to any overpayments by Rayner Surgical of any Royalty Payments that were payable by Rayner Surgical prior to January 1, 2024, Purchaser shall not be liable for any such overpayments, and Seller shall be responsible for any such overpayments.
Section 6.3. Treatment of Late Payment of Purchased Receivables. In the event of (a) any late payments by Rayner Surgical of any Purchased Receivables or (b) any late payments by Seller to Purchaser of misdirected payments of Purchased Receivables as described in Section 7.1(a), such Purchased Receivables will be allocated to the calendar month(s) in which such payments were originally due and payable under Section 2.7(c) of the APA, regardless of when actually paid.
ARTICLE VII
COVENANTS
Section 7.1. Misdirected Payments; Set-Offs by Rayner Surgical.
(a) Payments to Purchaser. If Seller shall, notwithstanding the provisions of the Rayner Surgical Instruction Letter, the New Rayner Surgical Instruction Letter and the Escrow Agreement, receive from Rayner Surgical, the Escrow Agent or any other Person any Purchased Receivables, Seller shall promptly, and in any event no later than five (5) Business Days, following a Responsible Employee of Seller becoming aware of the receipt by Seller of such Purchased Receivables, remit to Purchaser such Purchased Receivables.
(b) Payments to Seller. If Purchaser shall, notwithstanding the provisions of the Rayner Surgical Instruction Letter, the New Rayner Surgical Instruction Letter and the Escrow Agreement, receive from Rayner Surgical, the Escrow Agent or any other Person (i) any Royalty Payment that does not consist entirely of Purchased Receivables or (ii) any Excluded Asset, then Purchaser shall promptly, and in any event no later than five (5) Business Days, following the date Purchaser becomes aware of its receipt thereof, remit to Seller (X) such Royalty Payment, or portion thereof, that does not constitute Purchased Receivables or (Y) such Excluded Asset, as the case may be.
(c) Out-of-Scope Set-Offs. If, with respect to any Purchased Receivables that are paid by Rayner Surgical pursuant to monthly Royalty Payments, Rayner Surgical undertakes an Out-of-Scope Set-Off against such Purchased Receivables, then Seller shall promptly, and in any event no later than twenty (20) Business Days, following the date on which a Responsible Employee of Seller becomes aware of such Out-of-Scope Set-Off (including the amount and nature thereof), pay to Purchaser a sum equal to the amount of such Out-of-Scope Set-Off against the Purchased Receivables.
(d) Remittances. All remittances pursuant to this Section 7.1 shall be made (i) without set-off or deduction of any kind (except as required by applicable Law) and (ii) by wire transfer of immediately available funds to such account as the relevant payee may designate in writing to the other Party hereto (such designation to be made at least three Business Days prior to any such payment).
(e) Payments Held in Trust. Each Party hereto agrees that it shall hold any amounts received by it to which the other Party hereto is entitled under Section 7.1(a) or Section 7.1(b) in trust and agrees that it shall have no right, title or interest whatsoever in such amounts, shall make no use thereof and shall not impose, or permit the imposition of, any Liens on such amounts (other than, in the case of such amounts held by Seller to which Purchaser is entitled under Section 7.1(a), Permitted Liens).
Section 7.2. Interest. Without limiting Seller’s obligation to promptly remit Purchased Receivables to Purchaser pursuant to Section 7.1(a), if Seller fails to timely pay Purchaser in accordance with Section 7.1(a), Seller shall pay interest for any such late payment to Purchaser at a per annum rate equal to the U.S. Prime Rate, as reported in the Wall Street Journal, Eastern Edition, for the first date on which such payment was to be paid to Purchaser under Section 7.1(a), plus two percent (2%), from the first (1st) day the payment was to be paid to Purchaser under Section 7.1(a) to the day of actual payment of the amount. Without limiting Purchaser’s obligation to promptly remit amounts to Seller pursuant to Section 7.1(b), if Purchaser fails to timely pay Seller in accordance with Section 7.1(b), Purchaser shall pay interest for any such late payment to Seller at a per annum rate equal to the U.S. Prime Rate, as reported in the Wall Street Journal, Eastern Edition, for the first date on which such payment was to be paid to Seller under Section 7.1(b), plus two percent (2%), from the first (1st) day the payment was to be paid to Seller under Section 7.1(b) to the day of actual payment of the amount.
Section 7.3. Royalty Reports; Notices; Correspondence. Promptly, and in any event no later than five (5) Business Days, following the receipt by Seller of (i) a Royalty Report or (ii) any material written notice or material written correspondence pursuant to the APA relating to, or involving, the Purchased Receivables (including any relating to, or involving, any APA Omidria Provisions) Seller shall furnish full and complete copies of such Royalty Report or such written notice or written correspondence to Purchaser. Except for the Rayner Surgical Instruction Letter, the New Rayner Surgical Instruction Letter and notices and correspondence required to be given or made by Seller (x) under the APA or (y) by applicable Law, Seller shall not send any notice or correspondence to Rayner Surgical relating to, or involving, the Purchased Receivables, in each case, without the prior written consent of Purchaser (such consent not to be unreasonably withheld, conditioned or delayed), unless the sending of such notice or correspondence would not reasonably be expected to adversely affect (A) the Purchased Receivables (including any APA Omidria Provisions) in any material respect or (B) the timing, amount or duration of the Purchased Receivables in any material respect. If Purchaser does not provide consent to Seller within ten (10) Business Days after receipt of such proposed notice or correspondence, such consent will be deemed to have been given by Purchaser. Seller shall promptly, and in any event no later than five (5) Business Days, following the delivery thereof by Seller to Rayner Surgical, provide to Purchaser a copy of any material notice or material correspondence (including any notice or correspondence that (1) relates to, or involves, any APA Omidria Provisions in a material respect or (2) adversely effects the timing, amount or duration of the Purchased Receivables) sent by Seller to Rayner Surgical pursuant to the APA relating to, or involving, the Purchased Receivables (including the APA Omidria Provisions).
Section 7.4. Audits of Rayner Surgical.
(a) Consultation. Seller and Purchaser shall consult with each other regarding the timing, manner and conduct of any audit of Rayner Surgical with respect to the Royalties pursuant to Section 2.7(e) of the APA. Upon the termination of this Agreement pursuant to Section 9.14, the provisions of this Section 7.4 shall survive until the fifth (5th) anniversary of the effective date of such termination; provided, however, that (I) Purchaser’s right to receive amounts pursuant to, and in accordance with, Section 7.4(b)(v) and (II) the provisions of Sections 7.4(b)(iii) and 7.4(b)(iv), shall survive such termination indefinitely.
(b) Audits.
(i) Seller may, and if requested in writing by Purchaser, shall (as promptly as practicable following receipt of such written request from Purchaser), cause an independent certified public accountant of nationally recognized standing, to audit Rayner Surgical’s Financial Records (as such term is defined in the APA) relating to the Purchased Receivables (it being understood and agreed that Purchaser shall not be entitled to request an audit with respect to any matters other than matters in respect of the Purchased Receivables) pursuant to Section 2.7(e) of the APA (which limits Seller to one (1) audit per calendar year); provided, however, that Purchaser shall not be entitled to request such an audit if such an audit would contravene the provisions of Sections 2.7(d) or 2.7(e) of the APA. With respect to any such audit, Seller shall engage any independent certified public accountant as Seller shall determine for such purpose (as long as such independent certified public accountant is reasonably acceptable to (X) Rayner Surgical pursuant to Section 2.7(e) of the APA and (Y) Purchaser). Notwithstanding the foregoing, any such audit initiated at the request of Purchaser as described in this Section 7.4(b)(i) shall include such additional matters (whether or not related to the Purchased Receivables) as reasonably requested by Seller and subject to the provisions of Section 2.7(e) of the APA (such an audit, a “Seller Participated Audit”). Subject to Section 7.4(b)(vi), all of the out-of-pocket costs and expenses of any audit described in this Section 7.4(b)(i) (including the fees and expenses of any independent certified public accountant) that would otherwise be borne by Seller pursuant to the APA shall instead be borne (as such costs and expenses are incurred) as follows: (A) in the event such audit is solely with respect to Net Revenues for the period from January 1, 2024 through December 31, 2027 (and only if one or both of Milestone Event 1 and Milestone Event 2 have not yet occurred at the time of such audit), then twenty percent (20%) by Seller and eighty percent (80%) by Purchaser, and Purchaser shall promptly upon request reimburse Seller for Purchaser’s respective eighty percent (80%) of such out-of-pocket costs and expenses; (B) in the event such audit is a Seller Participated Audit, then twenty percent (20%) by Seller and eighty percent (80%) by Purchaser, and Purchaser shall promptly upon request reimburse Seller for Purchaser’s respective eighty percent (80%) of such out-of-pocket costs and expenses; and (C) in the event such audit is neither an audit described in the foregoing clause (A) nor an audit described in the foregoing clause (B), then one hundred percent (100%) by Purchaser, and Purchaser shall promptly upon request reimburse Seller for one hundred percent (100%) of such out-of-pocket costs and expenses. With respect to any audit described in this Section 7.4(b)(i), each Party shall provide to the other Party copies of the auditor’s results from such audit and the auditor’s work product from such audit received by such Party (subject to a reasonable and customary common interest agreement to protect attorney-client privilege, if applicable).
(ii) With respect to any audit of Rayner Surgical initiated by Seller pursuant to Section 2.7(e) of the APA solely with respect to matters unrelated to the Purchased Receivables, Seller shall give Purchaser at least ten (10) Business Days prior written notice before Seller initiates such audit. If, no later than five (5) Business Days after Purchaser receives such a notice, Purchaser reasonably requests (by written notice to Seller) that additional matters solely relating to the Purchased Receivables be included in such audit, then Seller shall include such additional matters subject to the provisions of Sections 2.7(d) and 2.7(e) of the APA (such an audit, a “Purchaser Participated Audit”). Subject to Section 7.4(b)(vi) and in deviation from Section 7.4(b)(i), all of the out-of-pocket costs and expenses of any audit described in this Section 7.4(b)(ii) (including the fees and expenses of any independent certified public accountant) that would otherwise be borne by Seller pursuant to the APA shall instead be borne (as such costs and expenses are incurred) as follows: (A) in the event such audit is a Purchaser Participated Audit, then twenty percent (20%) by Seller and eighty percent (80%) by Purchaser, and Purchaser shall promptly upon request reimburse Seller for Purchaser’s respective eighty percent (80%) of such out-of-pocket costs and expenses; and (B) in the event such audit is not a Purchaser Participated Audit, then one hundred percent (100%) by Seller. With respect to any audit described in this Section 7.4(b)(ii), each Party shall provide to the other Party copies of the auditor’s results from such audit and the auditor’s work product from such audit received by such Party (subject to a reasonable and customary common interest agreement to protect attorney-client privilege, if applicable).
(iii) If at any time (including following the completion of any audit under Section 2.7(e) of the APA), Seller is required to reimburse Rayner Surgical for an overpayment of Purchased Receivables, then Seller shall provide copies of the auditor’s results of such audit and the auditor’s work product of such audit to Purchaser (if not already provided) (subject to a reasonable and customary common interest agreement to protect attorney-client privilege, if applicable), and Purchaser shall promptly (and in compliance with the APA) reimburse such overpayment (to the extent such overpayment was received by Purchaser) (A) to Rayner Surgical on behalf of Seller or (B) if Seller has already reimbursed Rayner Surgical for the entirety of such amount (or if Rayner Surgical Sets-Off all or a portion of such amount against payments that Rayner Surgical owes to Seller in connection with any matter other than the Purchased Receivables), to Seller, and, in each case of clauses (A) and (B), Purchaser shall promptly (and in any event within five (5) Business Days) after making such payment provide evidence satisfactory to Seller that such payment was made.
(iv) If at any time (including following the completion of any audit under Section 2.7(e) of the APA), Rayner Surgical Sets-Off (in accordance with the last sentence of Section 2.7(e) of the APA) all or any portion of an overpayment of Purchased Receivables against future Royalties that constitute Excluded Assets (including Retained Receivables), then the amount of such Set-Off shall be borne one hundred percent (100%) by Purchaser (to the extent such overpayment was received by Purchaser), and Purchaser shall promptly upon request reimburse Seller for one hundred percent (100%) of such amount. If at any time (including following the completion of any audit under Section 2.7(e) of the APA), Rayner Surgical Sets-Off (in accordance with the last sentence of Section 2.7(e) of the APA) all or any portion of an overpayment of Purchased Receivables against future Royalties that constitute Purchased Receivables, then the amount of such Set-Off shall be borne one hundred percent (100%) by Purchaser (to the extent such overpayment was received by Purchaser).
(v) If, following the completion of any audit under Section 2.7(e) of the APA, Rayner Surgical is required to make additional payments to Seller for underpaid Purchased Receivables, then upon the payment thereof, the Parties shall allocate such payments as follows: (A) first, to reimburse the Parties for all out-of-pocket costs and expenses of any such audit (including the fees and expenses of any independent certified public accountant) borne by the Parties pursuant to Section 7.4(b)(i) or Section 7.4(b)(ii), as applicable (and that are not to be reimbursed pursuant to Section 7.4(b)(vi) below); and (B) second, to Purchaser.
(vi) If, following the completion of any audit under Section 2.7(e) of the APA, Rayner Surgical reimburses Seller for the out-of-pocket costs and expenses of such audit pursuant to Section 2.7(e) of the APA, Seller shall promptly (and in any event within five (5) Business Days of receipt by Seller of such reimbursement) reimburse Purchaser for the out-of-pocket costs and expenses paid by Purchaser pursuant to Section 7.4(b)(i) or Section 7.4(b)(ii), as applicable (and Purchaser shall provide reasonable documentation of such out-of-pocket costs and expenses).
Section 7.5. Performance of APA.
(a) Seller agrees that (i) it shall not breach the APA with respect to the Purchased Receivables if such breach would reasonably be expected to result in a Seller Material Adverse Effect, (ii) it shall promptly, and in any event no later than five (5) Business Days, following the date on which Seller becomes aware of a breach of the APA by Seller of the type described in the immediately preceding clause (i), provide written notice to Purchaser of such breach (including a reasonable description thereof), and (iii) after consultation with Purchaser and taking into account all reasonable comments from Purchaser, and without limiting its obligations under this Agreement, it shall use commercially reasonable efforts to promptly cure any such breach by Seller of the APA (and provide prompt written notice to Purchaser upon curing such breach).
(b) Seller will not exercise or fail to exercise any of its rights or perform or fail to perform its obligations under the APA in any manner that would result in a material adverse effect on the Purchased Receivables (including a material adverse effect on the timing, amount or duration of the Purchased Receivables); provided, that, the foregoing shall not limit Seller with respect to the research, development or commercialization of any biopharmaceutical product or technology that competes with OMIDRIA (to the extent otherwise permitted by the APA).
Section 7.6. Amendment of APA. Seller shall provide Purchaser a copy of any proposed amendment, supplement, modification, waiver, release or excuse (a “Modification”) of any provision of the APA as soon as practicable and in any event not less than five (5) Business Days prior to the date Seller proposes to execute such Modification. Seller shall not, without the prior written consent of Purchaser (such consent not to be unreasonably withheld, conditioned or delayed), execute or agree to execute any proposed Modification if such Modification would reasonably be expected to adversely affect in any material respect the Purchased Receivables (including the timing, amount or duration thereof) or the APA Omidria Provisions. Promptly, and in any event within five (5) Business Days following receipt by Seller of a fully executed Modification of the APA, Seller shall furnish a true and complete copy of such Modification to Purchaser.
Section 7.7. Enforcement of APA.
(a) Notice of Rayner Surgical Breaches. Promptly, and in any event within five (5) Business Days, following a Responsible Employee of Seller becoming aware of a breach of the APA by Rayner Surgical that would reasonably be expected to adversely affect in any material respect the Purchased Receivables (including the timing, amount or duration thereof) or the APA Omidria Provisions (such breach, a “Related Breach”), Seller shall provide notice of such Related Breach to Purchaser.
(b) Enforcement of APA. Upon receipt of notice of a Related Breach, Seller and Purchaser promptly shall consult and cooperate with each other regarding such Related Breach and as to the timing, manner and conduct of any enforcement of Rayner Surgical’s obligations under the APA relating thereto (including any indemnification obligations of Rayner Surgical under Article IX of the APA relating thereto); provided, however, that the Parties shall enter into a reasonable and customary common-interest agreement to protect attorney client privilege. Seller may, and if reasonably requested in writing by Purchaser within five (5) Business Days after receipt of notice of a Related Breach, shall, proceed, in consultation and cooperation with Purchaser, to use commercially reasonably efforts to enforce compliance by Rayner Surgical with the relevant provisions of the APA and to use commercially reasonable efforts to exercise such rights and remedies relating to such Related Breach as shall be available to Seller (including litigation of such matter), whether under the APA or by operation of applicable Law. In connection with any enforcement of Rayner Surgical’s obligations under the APA in respect of a Related Breach pursuant to this Section 7.7, Seller shall employ such lead counsel as Seller shall choose, in its sole discretion, for such purpose (as long as such counsel is reasonably acceptable to Purchaser). Nothing contained herein shall limit Seller or Purchaser from retaining, at its sole cost, separate outside counsel who shall be permitted, where reasonably practical (and subject to a reasonable and customary common interest agreement to protect attorney-client privilege), to consult with the lead counsel selected pursuant to the immediately preceding sentence for such enforcement.
(c) Allocation of Costs and of Proceeds from Enforcement.
(i) All out-of-pocket costs and expenses (including attorneys’ fees and expenses) incurred by Seller in connection with any enforcement of Rayner Surgical’s obligations under the APA in respect of a Related Breach pursuant to this Section 7.7 shall be borne (as such costs and expenses are incurred) one hundred percent (100%) by Purchaser, and Purchaser shall promptly upon request reimburse Seller for one hundred percent (100%) of such out-of-pocket costs and expenses. In furtherance of the foregoing, and subject to the remainder of this Section 7.7(c), any retainers or advances required by the lead counsel selected pursuant to the penultimate sentence of Section 7.7(b) for such enforcement (and that are incurred by Seller) shall be funded one hundred percent (100%) by Purchaser (such amounts to be credited or deducted from the actual amounts owed by Purchaser under the immediately preceding sentence), and Purchaser shall promptly upon request reimburse Seller for one hundred percent (100%) of such retainers and advances.
(ii) The Proceeds of any enforcement of Rayner Surgical’s obligations under the APA in respect of any Related Breach pursuant to this Section 7.7, shall be allocated (A) first, to reimburse the Parties for all out-of-pocket costs and expenses (including reasonable attorneys’ fees and expenses) borne by the Parties pursuant to Section 7.7(c)(i); and (B) second, to Purchaser.
(d) Other Matters. For clarity, (i) this Section 7.7 shall not apply to any enforcement of Rayner Surgical’s obligations under the APA other than in respect of a Related Breach (for example, this Section 7.7 shall not apply to any enforcement of Rayner Surgical’s obligations under the APA to the extent relating to any of the Retained Receivables), and (ii) except as otherwise provided in Article VIII hereof, the costs and expenses incurred by Seller in connection with any such enforcement described in the immediately preceding clause (i) shall be borne one hundred percent (100%) by Seller and the proceeds of any such enforcement shall inure one hundred percent (100%) to Seller.
Section 7.8. Termination of APA. Seller shall not terminate the APA or agree with Rayner Surgical to terminate the APA, except with the prior written consent of Purchaser (such consent not to be unreasonably withheld, conditioned or delayed), provided, that it will not be unreasonable for Purchaser to withhold consent if such proposed termination would adversely affect the timing, amount or duration of the Purchased Receivables in any material respect.
Section 7.9. Assignments of APA.
(a) Seller may not encumber, assign, delegate or otherwise transfer the APA, in whole or in part, without the prior written consent of Purchaser (such consent not to be unreasonably withheld, conditioned or delayed), and any such purported assignment without such consent shall be void ab initio and of no effect; provided, that, it will not be unreasonable for Purchaser to withhold consent if such proposed termination would adversely affect the timing, amount or duration of the Purchased Receivables in any material respect; provided, further, however, that, following the Closing, so long as none of the following encumbrances, assignments, delegations or other transfers, individually or in the aggregate, would reasonably be expected to result in a Seller Material Adverse Effect, no consent shall be required in connection with:
(i) Any (x) assignment, sale or other transfer (in whole or in part) of Seller’s right, title and interest in and to the Excluded Assets (including the Retained Receivables) or the delegation of any of Seller’s duties with respect to the Excluded Assets (including the Retained Receivables), or (y) buy-out of any Excluded Assets (including any Retained Receivables) by, or other transfer of any Excluded Assets (including any Retained Receivables) to, Rayner Surgical;
(ii) Any assignment that would not require consent of Rayner Surgical under Section 11.1 of the APA (so long as this Agreement is assigned in conjunction therewith); or
(iii) Any assignment of the APA to any successor by merger, operation of Law, or change of control of Seller (including as a result of any change, directly or indirectly, in the beneficial ownership of the voting securities of Seller) (so long as this Agreement is assigned in conjunction therewith);
provided, that, (1) in the case of the immediately preceding clauses (ii) and (iii), the assignee (if other than Seller) agrees in writing to perform all relevant obligations under, and to be bound by all the relevant provisions of, the APA as if such assignee were the “Seller” under the APA, and (2) for all cases such assignment shall not require Purchaser to enter into any agreement requiring Purchaser to undertake any obligations in favor of any party (directly or indirectly).
(b) Promptly, and in any event no later than five (5) Business Days, following Seller’s receipt of any executed assignment of the APA by Seller (other than any assignment, sale, transfer or delegation that is described in Section 7.9(a)(i) above), Seller shall furnish a copy of such assignment to Purchaser.
Section 7.10. Confidentiality.
(a) Confidentiality. Each Party shall keep confidential and not disclose to any Person (other than its Affiliates and its and its Affiliates’ Representatives and Financing Sources), and shall cause its Affiliates and its and its Affiliates’ Representatives and Financing Sources to keep confidential and not disclose to any Person, any Confidential Information (as defined below). Each Party shall, and shall cause its Affiliates and its and its Affiliates’ Representatives and Financing Sources to, use the Confidential Information solely in connection with such Party’s evaluation, administration and enforcement of the Transaction Documents (including in connection with its performance, exercise and enforcement of its rights under the Transaction Documents and the transactions contemplated thereby). The foregoing obligations shall continue until the date that is six years after the termination of this Agreement.
(b) Confidential Information. “Confidential Information” means, collectively, all information (whether written or oral, or in electronic or other form, and whether furnished before, on or after the date of this Agreement) disclosed to the other Party in connection with this Agreement to the extent concerning or relating to the following: (i) in the case of Confidential Information of either Party, that Party, its rights and interests relating to the Royalties, or its investment or investment performance pursuant to this Agreement; (ii) in the case of Confidential Information of both Parties, the Purchased Receivables, this Agreement, any Modifications, assignments, notices, requests, correspondence or other information furnished pursuant to this Agreement (including this Article VII) and any other reports, data, information, materials, notices, correspondence or documents of any kind relating to this Agreement; and (iii) in the case of Confidential Information of Seller, the APA or the Receivables, including (x) the APA and any license, sublicense or other agreements involving or relating to the Receivables or the intellectual property, compounds or products giving rise to the Receivables, whether or not such licenses, sublicenses or other agreements currently exist, are executed after the date hereof, or have been previously terminated, and including all terms and conditions hereof and thereof and the identities of the parties thereto, (y) any Royalty Reports, Modifications, assignments, notices, requests, correspondence, documents or other information furnished pursuant to this Agreement with respect to the APA (including this Article VII) and any other reports, data, information, materials, notices, correspondence or documents of any kind relating to Seller, Seller’s Affiliates, this Agreement, the APA, the Receivables, the Retained Receivables or the intellectual property, compounds or products giving rise to the Receivables, and including reports, data, information, materials, notices, correspondence or documents of any kind delivered pursuant to or under this Agreement or any of the other agreements referred to in the immediately preceding clause (x), and (z) any inventions, devices, improvements, formulations, discoveries, compositions, ingredients, patents, patent applications, know-how, processes, trial results, research, developments or other intellectual property, trade secrets or information to the extent involving or relating to the Receivables or the compounds or products giving rise to the Receivables. Notwithstanding the foregoing, “Confidential Information” shall not include any information that (A) was known, other than under an obligation of confidentiality, by the receiving Party at the time such information was disclosed to the receiving Party, its Affiliates or its or its Affiliates’ Representatives or Financing Sources in accordance herewith or in accordance with the Confidentiality Agreement (as defined below), as evidenced by its written records; (B) was or becomes part of the public domain (other than as a result of a disclosure by the receiving Party, its Affiliates or its or its Affiliates’ Representatives or Financing Sources in violation of this Agreement or the Confidentiality Agreement) prior to any disclosure of such information by the receiving Party, its Affiliates or its or its Affiliates ‘ Representatives or Financing Sources; (C) becomes known to the receiving Party on a non-confidential basis from a source other than the disclosing Party, its Affiliates and its and its Affiliates ‘ Representatives (and without any breach of this Agreement or the Confidentiality Agreement by the receiving Party, its Affiliates or its or its Affiliates’ Representatives or Financing Sources); provided, that, such source, to the knowledge of Purchaser, (x) had the right to disclose such information to the receiving Party (without breaching any legal, contractual or fiduciary obligation to the receiving Party or any of its Affiliates) and (y) did not obtain such information directly or indirectly from, or on behalf of, the disclosing Party, its Affiliates or its or its Affiliates ‘ Representatives; or (D) is or has been independently developed by the receiving Party, its Affiliates or its or its Affiliates’ Representatives without use of or reference to the Confidential Information, as evidenced by its written records.
(c) Permitted Disclosures. In the event that either Party or its Affiliates or any of its or its Affiliates’ Representatives or Financing Sources are requested by a governmental or regulatory or self-regulatory authority or required by applicable Law, regulation or legal process (including the regulations of a stock exchange or governmental or regulatory or self-regulatory authority or the order or ruling of a court, administrative agency or other government or regulatory body of competent jurisdiction) to disclose any Confidential Information, such Party shall promptly, to the extent permitted by Law, notify the disclosing Party in writing of such request or requirement so that the disclosing Party may seek an appropriate protective order or other appropriate remedy (and if the disclosing Party seeks such an order or other remedy, the receiving Party will provide such cooperation, at the disclosing party’s expense, as the disclosing Party shall reasonably request). If no such protective order or other remedy is obtained and the receiving Party or its Affiliates or its or its Affiliates’ Representatives or Financing Sources are, in the view of their respective counsel (which may include their respective internal counsel), legally required to disclose Confidential Information, the receiving Party or its Affiliates or its or its Affiliates’ Representatives or Financing Sources, as the case may be, shall only disclose that portion of the Confidential Information that their respective counsel advises that the receiving Party or its Affiliates or its or its Affiliates’ Representatives, as the case may be, are required to disclose and will exercise commercially reasonable efforts, at the disclosing Party ‘s expense, to obtain reliable assurance that confidential treatment will be accorded to that portion of the Confidential Information that is being disclosed. In any event, the receiving Party will not oppose action by the disclosing Party to obtain an appropriate protective order or other reliable assurance that confidential treatment will be accorded the Confidential Information. Notwithstanding the foregoing, notice to the disclosing Party shall not be required where disclosure is made (i) in response to a request by a governmental or regulatory authority having competent jurisdiction over the receiving Party, its Affiliates or its or its Affiliates ‘ Representatives or Financing Sources, as the case may be, or (ii) in connection with a routine examination by a regulatory or self-regulatory examiner, where in each case such request or examination does not expressly reference the disclosing Party, its Affiliates, the Receivables or this Agreement. Nothing provided herein limits any Party ‘s use or disclosure of its own Confidential Information if such Confidential Information is not also Confidential Information of the other Party.
(d) Purchaser Financial Statements. Notwithstanding anything herein to the contrary, nothing in this Section 7.10 (or in Section 7.11) shall or shall be construed to restrict Purchaser from (i) including disclosure of (A) the Initial Purchase Price, the Additional Floor Purchase Price, the January 2024 Amount, the Purchaser Milestone Payment 1 and the Purchaser Milestone Payment 2 and (B) the amount and nature of the Specified Initial Receivables and the amount and nature of the Purchased Receivables in the footnotes to Purchaser’s financial statements, to the extent so required or advised by Purchaser’s independent accountants, or including comparable or analogous disclosure in Purchaser’s unaudited quarterly financial statements, or (ii) providing copies of Confidential Information, including such audited annual and unaudited quarterly financial statements, to Purchaser’s existing or prospective lenders or direct or indirect beneficial owners, Affiliates or its or its Affiliates’ Representatives or Financing Sources, as long as such parties have agreed to be bound by the provisions of this Section 7.10 or are otherwise subject to reasonable restrictions of confidentiality. In addition, notwithstanding anything herein to the contrary, it is hereby agreed and confirmed that Purchaser (and its Affiliates) is permitted to disclose Confidential Information to the extent such disclosure is required by applicable Law (including the Securities Act of 1933, as amended, the Securities Exchange Act of 1934, as amended, and regulations promulgated by securities exchanges, or equivalent Canadian Law).
(e) Required Filings by Seller and Financial Statements. Notwithstanding anything herein to the contrary, nothing in this Section 7.10 (or in Section 7.11) shall or shall be construed to restrict Seller from (i) including disclosure of (A) the Initial Purchase Price, the Additional Floor Purchase Price, the January 2024 Amount, the Purchaser Milestone Payment 1 and the Purchaser Milestone Payment 2 and (B) the amount and nature of the Royalties, the amount and nature of the Receivables, and the amount and nature of the Purchased Receivables in the footnotes to Seller’s financial statements, to the extent so required or advised by Seller’s independent accountants, or including comparable or analogous disclosure in Sellers’s unaudited quarterly financial statements, or (ii) providing copies of Confidential Information, including such audited annual and unaudited quarterly financial statements, to Seller’s existing or prospective lenders, potential and actual buyers of Retained Receivables or other Excluded Assets, or direct or indirect beneficial owners, as long as such parties have agreed to be bound by the provisions of this Section 7.10 or are otherwise subject to reasonable restrictions of confidentiality. In addition, notwithstanding anything herein to the contrary, it is hereby agreed and confirmed that Seller (and its Affiliates) is permitted to disclose Confidential Information to the extent such disclosure is required by applicable Law (including the Securities Act of 1933, as amended, the Securities Exchange Act of 1934, as amended, and regulations promulgated by securities exchanges).
(f) Termination of Confidentiality Agreement. Effective upon the 2022 Effective Date, the Confidential Disclosure Agreement, dated April 12, 2022 (the “Confidentiality Agreement”), between DRI Capital Inc. (an Affiliate of Purchaser) and Seller shall terminate and be of no further force or effect, and shall be superseded by the provisions of this Section 7.10.
Section 7.11. Public Announcements; Use of Names.
(a) Neither Party shall, and each Party shall instruct its Affiliates and its and its Affiliates’ Representatives and Financing Sources not to, issue a press release or other public announcement or otherwise make any public disclosure with respect to this Agreement or the subject matter hereof without the prior consent of the other Party (which consent shall not be unreasonably withheld, conditioned or delayed), except as may be required by applicable Law (in which case the Party required by applicable Law to issue or make the press release, public announcement or other public disclosure shall allow the other Party reasonable time to comment on such press release, public announcement or other public disclosure in advance of such issuance or making thereof to the extent practicable and permitted by Law). Notwithstanding anything herein to the contrary, Seller and Purchaser hereby agree that (i) a joint press release issued by Seller and Purchaser jointly, or separate press releases issued by Seller and Purchaser separately, relating to the consummation of the transactions contemplated by this Agreement may be issued following the Closing in form(s) to be agreed by Purchaser and Seller (such press releases, the “Specified Press Releases”) and (ii) any Party may, without the consent of the other Party, make public disclosures of any information with respect to this Agreement or the subject matter hereof which is the same as the information that has already been publicly disclosed by such Party, or the other Party, in the Specified Press Release or otherwise in compliance with the foregoing provisions of this Section 7.11(a).
(b) Except as contemplated by the last sentence of Section 7.11(a), each Party hereto (the “First Party”) shall not, without the prior written consent of the other Party hereto, identify such other Party, its Affiliates or its or its Affiliates’ trustees, directors, managers, investors, owners, owner or officer family members, officers or employees in any advertising, sales literature or other promotional materials to be disseminated to any Person other than to such First Party, its Affiliates and its and its Affiliates’ Representatives, provided that each Party may refer to the name of the other Party in capital raising documentation that is governed by reasonable restrictions of confidentiality.
(c) Notwithstanding anything herein to the contrary, Seller may, without the consent of Purchaser, disclose (and nothing herein shall be construed to restrict Seller from disclosing) Purchaser’s identity, the Initial Purchase Price, the Additional Floor Purchase Price, the January 2024 Amount, the Purchaser Milestone Payment 1, the Purchaser Milestone Payment 2, the amount and nature of the Royalties, the amount and nature of the Receivables and the amount and nature of the Purchased Receivables (i) to potential and actual buyers of Retained Receivables or other Excluded Assets and (ii) to Rayner Surgical and its Affiliates. Notwithstanding anything herein to the contrary, each of Purchaser and Seller may without consent of the other Party disclose (and nothing herein shall be construed to restrict either Party from disclosing) the other Party’s name, the Initial Purchase Price, the Additional Floor Purchase Price, the January 2024 Amount, the Purchaser Milestone Payment 1, the Purchaser Milestone Payment 2 and the amount and nature of the Purchased Receivables in their respective annual and other periodic and current reports and financial statements and in those of their respective Affiliates (including, without limitation, any related press releases).
Section 7.12. Taxes. Seller and Purchaser agree that for United States federal income tax purposes, (x) any and all Purchased Receivables remitted by Seller to Purchaser pursuant to Section 7.1(a) or otherwise under this Agreement shall be treated as received by Seller as agent for Purchaser, and (y) any and all amounts remitted by Seller to Purchaser pursuant to Section 7.1(a) of this Agreement shall be treated as remittances of amounts collected by Seller on behalf of Purchaser. Purchaser agrees (i) to notify Seller, Rayner Surgical and the Escrow Agent immediately in writing if any Applicable Withholding Certificate, other tax form or information furnished in connection therewith or in connection with this Agreement that was previously delivered pursuant to this Agreement ceases to be accurate or complete, and (ii) to the extent it is legally eligible to do so, to provide to Seller, Rayner Surgical and the Escrow Agent any additional tax forms or information relating to any Applicable Withholding Certificate (A) upon reasonable request by Seller, Rayner Surgical or the Escrow Agent and (B) promptly upon any Applicable Withholding Certificate or other tax form previously delivered pursuant to this Agreement becoming obsolete. Purchaser agrees to notify each of Seller, Rayner Surgical and the Escrow Agent immediately if the statements in Section 5.1(j) (if made as of any date after the Closing Date) cease, or because of any change of Law or any act or omission planned, suffered or performed by Purchaser, would in the future cease, to be true. Seller shall be entitled to deduct (or cause to be deducted) from any amount payable hereunder (but for this sentence) to Purchaser any income, gross withholding or other tax that Seller determines that it is required to withhold under applicable Law with respect to such amount payable to Purchaser under this Agreement prior to remittance to Purchaser. Seller shall remit (or cause to be remitted) any amount withheld or deducted pursuant to this Section 7.12 to the relevant taxing authority, and any amount so remitted shall be treated as paid hereunder to Purchaser. Seller shall use commercially reasonable efforts to give or cause to be given to Purchaser such assistance and such information concerning the reasons for deduction as may be reasonably necessary to enable Purchaser to claim exemption therefrom, or credit therefor, and, in each case, shall furnish Purchaser with proper evidence of the taxes withheld and remitted to the relevant taxing authority.
Section 7.13. Further Actions. From and after the Closing, each of Purchaser and Seller shall, at the expense of the requesting party, execute and deliver such additional documents, certificates and instruments, and perform such additional acts, as may be reasonably requested and necessary or appropriate to carry out all of the provisions of this Agreement and to give full effect to and consummate the transactions contemplated by this Agreement.
Section 7.14. New Rayner Surgical Instruction Letter.
(a) Prior to the termination of this Agreement pursuant to Section 9.14 (and except for Seller’s delivery of the Rayner Surgical Instruction Letter and the New Rayner Surgical Instruction Letter to Rayner Surgical), Seller shall not, without Purchaser’s prior written consent (such consent not to be unreasonably withheld, delayed or conditioned), deliver any further directions to Rayner Surgical regarding the payment of the Purchased Receivables of the type referred to in paragraph no. 3 of the New Rayner Surgical Instruction Letter.
(b) As soon as practicable after the Closing, and in any event no later than two (2) Business Days after the Closing, Seller shall deliver to Rayner Surgical the duly executed New Rayner Surgical Instruction Letter (so long as Purchaser has delivered to Seller prior to such time a counterpart to the New Rayner Surgical Instruction Letter duly executed by Purchaser).
Section 7.15. Acknowledgment and Agreement by Purchaser; Limitation of Seller’s Duties and Obligations. Notwithstanding any provision of this Agreement (including other provisions of this Article VII) to the contrary, nothing contained in this Agreement shall obligate Seller to take any action, or omit to take any action, that (i) would conflict with, violate or cause a violation of, contravene or cause a default under, the APA or any applicable Law or any Judgment binding upon Seller, or (ii) would, or would involve any disclosure that would, result in the loss or waiver of any attorney-client privilege available to Seller; provided, that Seller shall use its commercially reasonable efforts to implement arrangements that would permit such action, omission or disclosure while preserving such privilege.
ARTICLE VIII
INDEMNIFICATION
Section 8.1. Obligation of Parties to Indemnify.
(a) Indemnification by Seller. Subject to the limitations set forth in this Article VIII, from and after the Closing, Seller shall indemnify Purchaser, DRC Management III LLC 2, DRI Healthcare LP, DRI Healthcare GP, LLC, DRI Healthcare ICAV, DRI Healthcare Trust, DRI Capital Inc., and DRI Capital (US), Inc. (collectively, the “DRI Entities”) against any and all losses, liabilities, costs, expenses (including reasonable attorneys’ fees and expenses in connection with any third party action, suit or proceeding) and damages (collectively, “Losses”) incurred by the DRI Entities or their directors, officers, employees, limited partners or agents (each, a “Purchaser Indemnified Party”), to the extent arising or resulting from any of the following:
(i) any breach of any representation or warranty made by Seller in this Agreement; and
(ii) any breach of any covenant or agreement of Seller contained in this Agreement or any other Transaction Document.
(b) Indemnification by Purchaser. Subject to the limitations set forth in this Article VIII, from and after the Closing, Purchaser shall indemnify Seller against any and all Losses incurred by Seller or its directors, officers, employees or agents (each, a “Seller Indemnified Party”), to the extent arising or resulting from any of the following:
(i) any breach of any representation or warranty made by Purchaser in this Agreement; and
(ii) any breach of any covenant or agreement of Purchaser contained in this Agreement or any other Transaction Document.
Section 8.2. Procedures Relating to Indemnification for Third Party Claims.
(a) Notice of Third Party Claim. In order for a Purchaser Indemnified Party, on the one hand, or a Seller Indemnified Party, on the other hand (such Purchaser Indemnified Party and Seller Indemnified Party being herein referred to as “Indemnified Party”) to be entitled to any indemnification under this Article VIII in respect of Losses arising out of or involving a claim or demand made by any Person other than Purchaser or Seller against a Purchaser Indemnified Party or a Seller Indemnified Party, as applicable (a “Third Party Claim”), the Indemnified Party must, promptly after its receipt of notice of the commencement of such Third Party Claim, notify the party from whom indemnification is sought under this Article VIII (the “Indemnifying Party”) in writing (including in such notice a brief description of such Third Party Claim, including damages sought or estimated, to the extent actually known or reasonably capable of estimation by the Indemnified Party); provided, however, that the failure to promptly provide such notice shall not affect the indemnification provided under this Article VIII except to the extent that the Indemnifying Party has been actually prejudiced as a result of such failure. Thereafter, the Indemnified Party shall deliver to the Indemnifying Party, promptly after the Indemnified Party’s receipt thereof, copies of all documents (including court papers) received by the Indemnified Party relating to such Third Party Claim.
(b) Defense of Third Party Claims. The Indemnifying Party shall be entitled to participate in the defense of any Third Party Claim and, if it so chooses, to assume the defense thereof, at its own expense, with counsel selected by the Indemnifying Party; provided, that, such counsel is not reasonably objected to by the Indemnified Party. If the Indemnifying Party elects to assume the defense of any Third Party Claim, the Indemnifying Party shall not be liable to the Indemnified Party for legal expenses subsequently incurred by the Indemnified Party in connection with the defense thereof, except that, if the Indemnifying Party and the Indemnified Party have conflicting interests or different defenses available with respect to such Third Party Claim, the Indemnified Party may hire its own separate counsel (provided, that, such counsel is not reasonably objected to by the Indemnifying Party) with respect to such Third Party Claim and the related action or suit, and the reasonable fees and expenses of such counsel shall be considered Losses for purposes of this Agreement. If the Indemnifying Party elects to assume the defense of any Third Party Claim, the Indemnifying Party shall permit the Indemnified Party to participate in, but not control, the defense of such Third Party Claim through counsel chosen by the Indemnified Party; provided, that, such counsel is not reasonably objected to by the Indemnifying Party and, except in the circumstances described in the immediately preceding sentence, the fees and expenses of such counsel shall be borne by the Indemnified Party. The Indemnifying Party shall be liable for the reasonable fees and expenses of counsel employed by the Indemnified Party in the defense of a Third Party Claim (which shall all be considered Losses for purposes of this Agreement) for any period during which the Indemnifying Party has not assumed the defense thereof (other than during the period prior to the time the Indemnified Party shall have notified the Indemnifying Party of such Third Party Claim).
(c) Cooperation. The parties hereto shall cooperate in the defense or prosecution of any Third Party Claim, with such cooperation to include (i) the retention of and the provision to the Indemnifying Party of records and information that are reasonably relevant to such Third Party Claim and (ii) the making available of employees and representatives on a mutually convenient basis for the purpose of providing factual information and explanation of any material provided hereunder. If the Indemnifying Party shall have assumed the defense of a Third Party Claim, the Indemnified Party shall agree to any settlement, compromise or discharge of such Third Party Claim that the Indemnifying Party may recommend and that by its terms obligates the Indemnifying Party to pay the full amount of the liability (if any) in connection with such Third Party Claim and which does not involve any non-monetary penalties and releases the Indemnified Party completely and unconditionally in connection with such Third Party Claim. Regardless of whether the Indemnifying Party shall have assumed the defense of a Third Party Claim, the Indemnified Party shall not be entitled to be indemnified or held harmless pursuant to this Article VIII if the Indemnified Party shall settle such Third Party Claim without the prior written consent of the Indemnifying Party (such consent not to be unreasonably withheld, conditioned or delayed).
Section 8.3. Procedures Relating to Indemnification for Other Claims. In order for an Indemnified Party to be entitled to any indemnification under this Article VIII in respect of Losses that do not arise out of or involve a Third Party Claim, the Indemnified Party must notify the Indemnifying Party promptly in writing (including in such notice a brief description of the claim for indemnification and the Loss, including damages sought or estimated, to the extent actually known or reasonably capable of estimation by the Indemnified Party); provided, however, that the failure to promptly provide such notice shall not affect the indemnification provided under this Article VIII except to the extent that the Indemnifying Party has been actually prejudiced as a result of such failure.
Section 8.4. Limitations on Indemnification.
(a) Seller. Notwithstanding anything in this Agreement to the contrary, Seller shall not have any liability under clause (i) of Section 8.1(a):
(i) with respect to any individual item (or any series of related items) if the Losses related thereto are less than U.S. $250,000 in the aggregate;
(ii) unless the aggregate liability for all Losses suffered by the Purchaser Indemnified Parties thereunder exceeds one percent (1%) of the Cumulative Purchase Price, and then only to the extent of such excess; or
(iii) in excess of the Cumulative Purchase Price, less (A) the aggregate amount of Purchased Receivables received by Purchaser on or prior to the day on which an indemnity claim under clause (i) of Section 8.1(a) is paid by Seller, (B) without duplication of clause (A), the aggregate amount of Specified Initial Receivables received by Purchaser on or prior to the day on which an indemnity claim under clause (i) of Section 8.1(a) is paid by Seller and (C) the aggregate amount of payments made by Seller under clause (i) of Section 8.1(a) on or prior to such day (provided, however, that the limitation under this clause (iii) will not be applicable with respect to instances of fraud and intentional misrepresentation).
(b) Purchaser. Notwithstanding anything in this Agreement to the contrary, Purchaser shall not have any liability under clause (i) of Section 8.1(b):
(i) with respect to any individual item (or any series of related items) if the Losses related thereto are less than U.S. $250,000 in the aggregate;
(ii) unless the aggregate liability for all Losses suffered by the Purchaser Indemnified Parties thereunder exceeds one percent (1%) of the Cumulative Purchase Price, and then only to the extent of such excess; or
(iii) in excess of the Cumulative Purchase Price, in the aggregate (provided, however, that the foregoing limitation will not be applicable with respect to instances of fraud and intentional misrepresentation).
Section 8.5. Survival of Representations and Warranties. The representations and warranties contained in this Agreement shall survive the Closing solely for purposes of Section 8.1 and shall terminate on the date that is the second (2nd) anniversary of the Closing Date; provided, however, that the representations and warranties contained in Sections 4.1(a), 4.1(b), 4.1(c), 4.1(e), 4.1(l), 5.1(a), 5.1(b), 5.1(c) and 5.1(e) shall terminate on the date that is the fourth (4th) anniversary of the Closing Date. No Party hereto shall have any liability or obligation of any nature with respect to any representation or warranty after the termination thereof, unless the other party hereto shall have delivered a notice to such Party, pursuant to Section 8.2(a) or Section 8.3, claiming such a liability or obligation under Section 8.1(a)(i) or 8.1(b)(i), prior to such termination.
Section 8.6. Indemnification and Liability under the 2022 Royalty Purchase Agreement. It is understood and agreed that: (i) except as otherwise specifically provided in Article IV, the representations and warranties made by Seller in the 2022 Royalty Purchase Agreement are hereby terminated and Seller shall not have any liability or obligation of any nature with respect to any such representation or warranty (and the indemnification obligations set forth in Section 8.1(a)(i) shall only apply to the representations and warranties made by Seller in this Agreement); (ii) except as otherwise specifically provided in Article V, the representations and warranties made by Purchaser in the 2022 Royalty Purchase Agreement are hereby terminated and Purchaser shall not have any liability or obligation of any nature with respect to any such representation or warranty (and the indemnification obligations set forth in Section 8.1(b)(i) shall only apply to the representations and warranties made by Purchaser in this Agreement); (iii) the covenants and agreements of Seller contained in the 2022 Royalty Purchase Agreement are hereby terminated and Seller shall not have any liability or obligation of any nature with respect to any such covenant or agreement (and the indemnification obligations set forth in Section 8.1(a)(ii) shall only apply to the covenants and agreements made by Seller in this Agreement and the other Transaction Documents); and (iv) the covenants and agreements of Purchaser contained in the 2022 Royalty Purchase Agreement are hereby terminated and Purchaser shall not have any liability or obligation of any nature with respect to any such covenant or agreement (and the indemnification obligations set forth in Section 8.1(b)(ii) shall only apply to the covenants and agreements made by Purchaser in this Agreement and the other Transaction Documents).
Section 8.7. No Implied Representations and Warranties.
Purchaser acknowledges and agrees that (x) other than the representations and warranties of Seller specifically contained in Article IV, there are no representations or warranties of Seller or any other Person either expressed or implied with respect to Seller (or any of its Affiliates), the Royalties, the Receivables, the Purchased Receivables, OMIDRIA, Separate Payments, the APA or the transactions contemplated by the Transaction Documents or the APA and (y) Purchaser does not rely on, and shall have no remedies in respect of, any representation or warranty not specifically set forth in Article IV. Without limiting the foregoing, Purchaser acknowledges and agrees that (a) Purchaser, together with its Affiliates and its and its Affiliates’ Representatives, have made their own investigation of Seller (and its Affiliates), the Royalties, the Receivables, the Purchased Receivables, OMIDRIA, Separate Payments, the APA and the transactions contemplated by the Transaction Documents and the APA and, except as expressly set forth in any representation or warranty in Article IV, are not relying on, and shall have no remedies in respect of, (i) any implied warranties, (ii) any representation or warranty whatsoever as to the future amount or potential amount of the Royalties, the Receivables and the Purchased Receivables or as to the creditworthiness of Rayner Surgical or Rayner Surgical Group Limited (or any of their respective Affiliates) or (iii) any representation or warranty whatsoever as to the availability, amount or likelihood of any Separate Payment and (b) except as expressly set forth in any representation or warranty in Article IV, Purchaser shall have no claim or right regarding losses or damages pursuant to this Article VIII (or otherwise) with respect to any information, documents, or materials relating to the transactions contemplated by the Transaction Documents or the APA furnished or made available to Purchaser or any of its Affiliates or its or its Affiliates’ Representatives by Seller or Seller’s Representatives. Purchaser further acknowledges and agrees that, without limiting the representations and warranties expressly set forth in Article IV, (A) as between the Parties hereto, Purchaser is assuming all market risk associated with OMIDRIA (including with respect to the Receivables and the Purchased Receivables) and, as such, shall have no recourse against Seller or any of Seller’s Affiliates based on the failure of the sales of OMIDRIA to meet its or any other Person’s projections and (B) neither Seller nor any of Seller’s Affiliates guarantees any obligations of Rayner Surgical or Rayner Surgical Group Limited under the APA.
Section 8.8. Exclusive Remedy. Other than for breaches of any covenants or agreements set forth in Section 7.10 or Article IX, the Parties hereto acknowledge and agree that, from and after the Closing, this Article VIII (including Section 8.4, Section 8.5 and Section 8.9) shall provide the Parties’ sole and exclusive remedy with respect to any matter or claim arising out of, relating to or in connection with any of the Transaction Documents, the 2022 Royalty Purchase Agreement (or any of the other Transaction Documents as defined in the 2022 Royalty Purchase Agreement) or any of the transactions contemplated thereby, except that any such claim or matter based upon fraud, deliberate or willful breach of covenant or willful misconduct shall not be subject to or limited by this Article VIII. For clarity, and without limiting the foregoing, the parties acknowledge and agree that rescission of this Agreement shall not be an available remedy for breach of or misrepresentation of any representation made by either Party under this Agreement, except in the case of fraud and intentional misrepresentation.
Section 8.9. Equitable Remedies/Specific Performance. Each Party acknowledges and agrees that the other Party may be damaged irreparably in the event any of the provisions of this Agreement are not performed in accordance with their specific terms or otherwise are breached or violated. Accordingly, each Party agrees that, without posting bond or other undertaking, the other Party shall be entitled to seek an injunction or injunctions to prevent breaches or violations of the provisions of this Agreement, or other equitable remedies, and to seek to enforce specifically this Agreement and the terms and provisions hereof in any action, suit or other proceeding instituted in any New York Court in addition to any other remedy to which it may be entitled, at law or in equity.
Section 8.10. Limitations on Damages. Notwithstanding anything to the contrary in this Agreement or any other Transaction Document, in no event shall either Party hereto be liable (including, without limitation, under Section 8.1) for any (i) special, indirect, incidental, exemplary, punitive, multiple or consequential damages (except to the extent of Losses for which an Indemnifying Party is liable under Article VIII that do constitute direct damages of the Indemnified Party, but that constitute indirect damages of such Indemnifying Party), or (ii) loss of use, business interruption, loss of any contract or other business opportunity or good will, in each case, of the other Party hereto (other than any such damages or losses occasioned by any breach of the covenants or agreements set forth in Section 7.10), whether or not caused by or resulting from the actions of such Party or the breach of its covenants, agreements, representations or warranties under any of the Transaction Documents (except as aforesaid) and whether in contract, tort or breach of statutory duty or otherwise, even if such Party has been advised of the possibility of such damages.
ARTICLE IX
MISCELLANEOUS
Section 9.1. Headings. The captions to the Articles, Sections and subsections hereof are not a part of this Agreement but are for convenience only and shall not be deemed to limit or otherwise affect the construction thereof.
Section 9.2. Notices. All notices and other communications under this Agreement shall be in writing and shall be sent by email with PDF attachment, an internationally recognized courier or personal delivery to the following addresses, or to such other addresses as shall be designated from time to time by a Party hereto in accordance with this Section 9.2:
If to Seller:
Omeros Corporation
201 Elliott Avenue West
Seattle, WA 98119
Attention: General Counsel
Email: generalcounsel@omeros.com
With a copy to (which shall not constitute service of process):
Foley Hoag LLP 1301 Avenue of the Americas New York, NY 10019 Attention: Rachel Beller Email: rbeller@foleyhoag.com DRI Capital Inc. First Canadian Place 100 King St. West, Suite 7250 PO Box 62 Toronto, ON M5X 1B1 Attention: Behzad Khosrowshahi Email: DRINotices@dricapital.com
If to Purchaser:
With a copy to (which shall not constitute service of process):
Cravath, Swaine & Moore LLP
Worldwide Plaza
825 Eighth Avenue
New York, NY 10019
Attention: David J. Kappos
Daniel J. Cerqueira
Email: dkappos@cravath.com
dcerqueira@cravath.com
All notices and communications under this Agreement shall be effective upon receipt by the addressee. Notwithstanding anything to the contrary in this Section 9.2, (a) all notices and communications under Sections 8.2(a) and 8.3 and all service of legal process shall be sent by an internationally recognized courier or by personal delivery, (b) if any notices or other documentation required to be delivered to Purchaser under Article VII is not sent by email, then in addition to any other method of notice permitted under this Section 9.2, Seller shall send a copy of such notices or other documentation to Purchaser by email with PDF attachment to DRINotices@dricapital.com and (c) if any notices or other documentation required to be delivered to Seller under this Agreement is not sent by email, then in addition to any other method of notice permitted under this Section 9.2, Purchaser shall send a copy of such notices or other documentation to Seller by email with PDF attachment to generalcounsel@omeros.com.
Section 9.3. No Personal Liability. It is expressly understood and agreed by Seller and Purchaser that:
(a) each of the representations, warranties, covenants and agreements in the Transaction Documents made on the part of either Party is made by such Party and is not intended to be nor is a personal representation, warranty, covenant or agreement of any other Person including any Representative of a Party or its Affiliates, and further including, with respect to Seller, those Persons named in the definition of “Knowledge of Seller” (the “Non-Warranting Parties”);
(b) other than Seller or Purchaser, respectively, no Person, including the Non-Warranting Parties, shall have any liability whatsoever for breach of any representation, warranty, covenant or agreement made in the Transaction Documents on the part of Seller or Purchaser or in respect of any claim or matter arising out of, relating to or in connection with the Transaction Documents or the transactions contemplated thereby; and
(c) the provisions of this Section 9.3 are intended to benefit each and every one of the Non-Warranting Parties and shall be enforceable by each and every one of them to the fullest extent permitted by Law.
Section 9.4. Expenses. Other than the fees, costs and expenses of the Escrow Agent payable to the Escrow Agent pursuant to the Escrow Agreement, all fees, costs and expenses (including any legal, accounting, financial advisory and banking fees) incurred in connection with the preparation, negotiation, execution and delivery of the Transaction Documents and to consummate the transactions contemplated thereby shall be paid by the party incurring such fees, costs and expenses. The fees, costs and expenses of the Escrow Agent payable to the Escrow Agent pursuant to the Escrow Agreement shall be borne and paid 100% by Purchaser.
Section 9.5. Assignment.
(a) By Purchaser. Purchaser may not encumber, assign, delegate, or otherwise transfer this Agreement, in whole or in part, without the prior written consent of Seller, and any such purported assignment, delegation or other transfer without such consent shall be void ab initio and of no effect; provided, however, that following the Closing, Purchaser may, without the prior written consent of Seller, assign, delegate, or otherwise transfer this Agreement, in whole or in part, only such that there are no more than two (2) assignees at any time, and only (i) as part of a sale of all or substantially all of Purchaser’s business; (ii) to an Affiliate of Purchaser; (iii) to a special purpose vehicle created to be bankruptcy remote and for financing purposes of Purchaser and its Affiliates; (iv) to any successor by merger, by operation of Law, or in the event of a change of control of Purchaser (including as a result of any change, directly or indirectly, in the beneficial ownership of the voting securities of Purchaser); or (v) by way of a grant of a security interest therein to a financial institution or other lender (with consent to foreclose thereon) subject to the conditions set forth in this Section 9.5(a). In the event of an assignment, delegation, or other transfer of Purchaser’s obligations under this Agreement pursuant to clauses (i) through (iv) (inclusive), the transferee under such assignment, delegation, or other transfer must (A) agree, in writing, for the benefit of Seller, to perform all such assigned obligations under this Agreement (and the corresponding obligations under the Escrow Agreement), and to be bound by all the provisions of this Agreement (and of the Escrow Agreement) relating to such assigned obligations, as if such transferee were the “Purchaser” under this Agreement (and under the Escrow Agreement) (and Purchaser shall deliver a copy of such writing to Seller within five (5) Business Days following the effectiveness of such assignment), and (B) such transferee must be subject to confidentiality and non-use obligations at least as stringent as those set forth in Section 7.10. In the event of an assignment, delegation, or other transfer by Purchaser permitted under clause (v) (i.e., by way of a grant of a security interest), Purchaser shall (1) notify the secured party that such secured party shall be bound by the applicable provisions of this Agreement (and of the Escrow Agreement) and (2) use its commercially reasonable best efforts to ensure compliance with clauses (A) and (B). In the event that, as a result of an assignment under this Section 9.5(a), there are two transferees as permitted under this Section 9.5(a), Purchaser (or the transferees, as applicable) shall designate one such transferee as the primary party with which Seller shall correspond for purposes of this Agreement.
(b) By Seller. Seller may not encumber assign, delegate or otherwise transfer this Agreement, in whole or in part, without the prior written consent of Purchaser, and any such purported assignment, delegation or other transfer without such consent shall be void ab initio and of no effect; provided, however, that following the Closing, Seller may assign, delegate or otherwise transfer this Agreement in whole or in part without the prior written consent of Purchaser, (i) in conjunction with any assignment, delegation or transfer of the APA by Seller that does not require the consent of Rayner Surgical under Section 11.1 of the APA; (ii) as part of a sale of all or substantially all of Seller’s business; (iii) to an Affiliate of Seller; or (iv) to any successor by merger, by operation of Law or in the event of a change of control of Seller (including as a result of any change, directly or indirectly, in the beneficial ownership of the voting securities of Seller), so long as (A) a corresponding assignment, delegation or transfer, as the case may be, by Seller of the APA (or the relevant provisions of the APA) occurs concurrently therewith, (B) if any obligations under this Agreement are assigned, delegated or transferred to the assignee, the assignee (if other than the same Seller legal entity) agrees in writing, for the benefit of Purchaser, to perform all such assigned obligations under this Agreement (and the corresponding obligations under the Escrow Agreement), and to be bound by all the provisions of this Agreement (and of the Escrow Agreement) relating to such assigned obligations, as if such assignee were the “Seller” under this Agreement (and under the Escrow Agreement) (and Seller shall deliver a copy of such writing to Purchaser within five (5) Business Days after the effectiveness of such assignment) and (C) the assignee is subject to confidentiality and non-use obligations at least as stringent as those set forth in Section 7.10.
(c) Successors and Assigns. Subject to the provisions of Section 9.5(a) and Section 9.5(b), this Agreement shall be binding upon, inure to the benefit of and be enforceable by, the Parties hereto and their respective permitted successors and assigns.
Section 9.6. Amendment and Waiver.
(a) This Agreement may be amended, modified or supplemented only in a written agreement signed by both Parties hereto. Any provision of this Agreement may be waived only in a written agreement, which agreement may be signed only by the Party granting such waiver.
(b) No failure or delay on the part of either Party hereto in exercising any right, power or remedy hereunder shall operate as a waiver thereof, nor shall any single or partial exercise of any such right, power or remedy preclude any other or further exercise thereof or the exercise of any other right, power or remedy. No course of dealing between the parties hereto shall be effective to amend, modify, supplement or waive any provision of this Agreement.
Section 9.7. Entire Agreement. This Agreement, including the Exhibits and Schedules attached to this Agreement, sets forth the entire agreement and understanding between the Parties hereto as to the subject matter hereof. All express or implied agreements, promises, assurances, arrangements, representations, warranties and understandings as to the subject matter hereof, whether oral or written, heretofore made are superseded by this Agreement. For the avoidance of doubt, the Parties also acknowledge, for purposes of this Section 9.7, the provisions of Sections 8.7, 8.8 and 8.10 of this Agreement.
Section 9.8. No Partnership; Independent Contractors. The parties hereto recognize and agree that each is operating as an independent contractor and not as an agent, partner or fiduciary of the other. Neither Party shall have the authority to bind, obligate or represent the other Party.
Section 9.9. No Third Party Beneficiaries. Except to the extent otherwise contemplated by Section 9.3, this Agreement is for the sole benefit of Seller and Purchaser and their respective permitted successors and assigns, and nothing herein expressed or implied shall give or be construed to give to any Person, other than the parties hereto and such successors and assigns, any legal or equitable rights hereunder. For the avoidance of doubt, indemnification under Article VIII in respect of Losses incurred by a Purchaser Indemnified Party or a Seller Indemnified Party may only be enforced by Purchaser or Seller, respectively, and not by any other Person.
Section 9.10. Governing Law. This Agreement shall be governed by and construed in accordance with the Laws of the State of New York, excluding any conflicts or choice of Law rule or principle that might otherwise refer construction or interpretation of this Agreement to the substantive Law of another jurisdiction.
Section 9.11. Jurisdiction; Venue; Service of Process; Waiver of Jury Trial. Each party hereto irrevocably submits to the exclusive jurisdiction of the Supreme Court of the State of New York for the County of New York, the United States District Court for the Southern District of New York and any appellate court from either of them (such courts, collectively, the “New York Courts”) for the purposes of any action, suit or other proceeding arising out of, relating to or in connection with this Agreement or any transaction contemplated hereby. Each Party hereto agrees to commence any action, suit or other proceeding arising out of, relating to or in connection with this Agreement or any transaction contemplated hereby in the New York Courts. Each Party hereto further agrees that service of any process, summons, notice or document by an internationally recognized courier or personal delivery in accordance with Section 9.2 shall be effective service of process for any action, suit or other proceeding in the New York Courts with respect to any matters to which it has submitted to jurisdiction in this Section 9.11. Nothing herein shall affect the right of a Party hereto to serve process on the other Party hereto in any other manner permitted by applicable Law. Each Party hereto irrevocably and unconditionally waives any objection to the laying of venue of any action, suit or other proceeding arising out of, relating to or in connection with this Agreement or any transaction contemplated hereby in the New York Courts, and hereby further irrevocably and unconditionally waives, and shall not assert by way of motion, defense, or otherwise, in any such action, suit or other proceeding, any claim that it is not subject personally to the jurisdiction of the New York Courts, that its property is exempt or immune from attachment or execution, that such action, suit or other proceeding is brought in an inconvenient forum, that the venue of such action, suit or other proceeding is improper, or that this Agreement or the transactions contemplated hereby may not be enforced in or by any of the New York Courts. Each Party hereto irrevocably and unconditionally waives any right to trial by jury with respect to any proceeding arising out of, relating to or in connection with this Agreement or any transaction contemplated hereby.
Section 9.12. Severability. If any term or provision of this Agreement is held to be invalid, illegal or unenforceable by a court or Governmental Entity of competent jurisdiction, such invalidity, illegality or unenforceability shall not affect any other term or provision of this Agreement, which shall remain in full force and effect, and the parties hereto shall replace such term or provision with a new term or provision permitted by applicable Law and having an economic effect as close as possible to the invalid, illegal or unenforceable term or provision. The holding of a term or provision to be invalid, illegal or unenforceable in a jurisdiction shall not have any effect on the application of the term or provision in any other jurisdiction.
Section 9.13. Counterparts; Electronic Signatures. This Agreement may be executed in any number of counterparts and by the Parties hereto in separate counterparts, each of which when so executed shall be deemed to be an original and all of which taken together shall constitute one and the same agreement. Counterparts may be (i) signed in person and delivered in person, via facsimile, or via other means of electronic delivery (including emailing an electronic copy thereof) and/or (ii) signed and delivered by means of employing electronic signature technology that complies with the Electronic Signatures in Global and National Commerce Act of 2000 (E-SIGN), or other applicable Law governing the execution and delivery of this Agreement through electronic means, and any counterpart so executed and delivered shall be deemed to have been duly and validly executed and delivered and be valid and enforceable for all purposes.
Section 9.14. Termination of Agreement.
(a) Subject to Section 9.14(b), this Agreement shall continue in full force and effect until the earlier of (i) December 31, 2031, and (ii) the date on which Purchaser has received the last payment of Purchased Receivables made pursuant to the APA. Immediately upon the effective date of termination of this Agreement pursuant to this Section 9.14(a), this Agreement shall automatically terminate (without the need for any action by any party hereto), save for any rights, obligations or claims of either party hereto which have accrued prior to such effective date of termination (along with any corresponding limitations of liability in respect thereof).
(b) The following provisions shall survive any termination of this Agreement pursuant to this Section 9.14: Article I (Definitions; Interpretation), Section 7.4 (Audits of Rayner Surgical) (provided that Section 7.4 shall terminate on the fifth (5th) anniversary of the effective date of termination of this Agreement; provided, further, that (I) Purchaser’s right to receive amounts pursuant to, and in accordance with, Section 7.4(b)(v) and (II) the provisions of Sections 7.4(b)(iii) and 7.4(b)(iv), shall survive such termination of this Agreement indefinitely), Section 7.10 (Confidentiality), Section 7.11 (Public Announcements; Use of Names), Section 7.15 (Acknowledgment and Agreement by Purchaser; Limitation of Seller’s Duties and Obligations) and Article IX (Miscellaneous).
(c) Immediately upon the effective date of termination of this Agreement, Purchaser shall execute and deliver to Seller all documents as Seller shall reasonably request to evidence the termination of this Agreement.
(d) Promptly following the effective date of termination of this Agreement, Purchaser and Seller shall execute and deliver a joint written direction to the Escrow Agent terminating the Escrow Agreement with respect to Purchaser.
[Signature Page Follows]
IN WITNESS WHEREOF, the Parties hereto have caused this Agreement to be executed by their respective representatives thereunto duly authorized as of the date first above written.
|
|
SELLER: |
|
|
| OMEROS CORPORATION | |||
|
|
|
|
|
|
|
|
|
|
|
|
By: |
/s/ Gregory A. Demopulos, M.D. |
|
|
|
|
Name: Gregory A. Demopulos, M.D. |
|
|
|
|
Title: Chairman of Board & CEO |
|
|
|
PURCHASER: |
|
|
| DRI HEALTHCARE ACQUISITIONS LP | |||
|
|
|
|
|
| By: | DRC Management III LLC 2 | ||
| Its: | General Partner | ||
|
|
|
|
|
|
|
By: |
/s/ Grant Cellier |
|
|
|
|
Name: Grant Cellier |
|
|
|
|
Title: Manager |
|
[Signature Page to Amended and Restated Royalty Purchase Agreement]
Exhibit 23.1
Consent of Independent Registered Public Accounting Firm
We consent to the incorporation by reference in the Registration Statements (Form S-8 Nos. 333-162732, 333-165861, 333-172905, 333-180216, 333-187344, 333-194693, 333-202788, 333-210219, 333-216749, 333-218882, 333-232071, 333-257148 and 333-273855) pertaining to the Omeros Corporation 2008 Equity Incentive Plan, the Omeros Corporation Second Amended and Restated 1998 Stock Option Plan, the nura, Inc. 2003 Stock Option Plan, the Omeros Corporation Stock Option Grant to Gregory A. Demopulos, M.D., the Omeros Corporation Stock Option Grant to Pamela Pierce Palmer, M.D., Ph.D., and the Omeros Corporation 2017 Omnibus Incentive Compensation Plan, and the Registration Statement (Form S-3 No. 333- 268269) and related Prospectus of Omeros Corporation pertaining to the registration of common stock, preferred stock, debt securities, depositary shares, warrants, subscription rights, and units, of our reports dated April 1, 2024, with respect to the consolidated financial statements of Omeros Corporation, included in this Annual Report (Form 10-K) of Omeros Corporation for the year ended December 31, 2023.
/s/ Ernst & Young LLP
Seattle, Washington
April 1, 2024
Exhibit 31.1
CERTIFICATION OF PRINCIPAL EXECUTIVE OFFICER PURSUANT TO RULE 13a-14(a)/15d-14(a) OF THE SECURITIES EXCHANGE ACT OF 1934, AS ADOPTED PURSUANT TO SECTION 302 OF THE SARBANES-OXLEY ACT OF 2002
I, Gregory A. Demopulos, M.D., certify that:
1. I have reviewed this annual report on Form 10-K of Omeros Corporation;
2. Based on my knowledge, this report does not contain any untrue statement of a material fact or omit to state a material fact necessary to make the statements made, in light of the circumstances under which such statements were made, not misleading with respect to the period covered by this report.
3. Based on my knowledge, the financial statements, and other financial information included in this report, fairly present in all material respects the financial condition, results of operations and cash flows of the registrant as of, and for, the periods presented in this report;
4. The registrant’s other certifying officer(s) and I are responsible for establishing and maintaining disclosure controls and procedures (as defined in Exchange Act Rules 13a-15(e) and 15d-15(e)) and internal control over financial reporting (as defined in Exchange Act Rules 13a-15(f) and 15d-15(f)) for the registrant and have:
a. Designed such disclosure controls and procedures, or caused such disclosure controls and procedures to be designed under our supervision, to ensure that material information relating to the registrant, including its consolidated subsidiaries, is made known to us by others within those entities, particularly during the period in which this report is being prepared;
b. Designed such internal control over financial reporting, or caused such internal control over financial reporting to be designed under our supervision, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles;
c. Evaluated the effectiveness of the registrant’s disclosure controls and procedures and presented in this report our conclusions about the effectiveness of the disclosure controls and procedures, as of the end of the period covered by this report based on such evaluation; and
d. Disclosed in this report any change in the registrant’s internal control over financial reporting that occurred during the registrant’s most recent fiscal quarter (the registrant’s fourth fiscal quarter in the case of an annual report) that has materially affected, or is reasonably likely to materially affect, the registrant’s internal control over financial reporting; and
5. The registrant’s other certifying officer(s) and I have disclosed, based on our most recent evaluation of internal control over financial reporting, to the registrant’s auditors and the audit committee of the registrant’s board of directors (or persons performing the equivalent functions):
a. All significant deficiencies and material weaknesses in the design or operation of internal control over financial reporting which are reasonably likely to adversely affect the registrant’s ability to record, process, summarize and report financial information; and
b. Any fraud, whether or not material, that involves management or other employees who have a significant role in the registrant’s internal control over financial reporting.
|
Dated: April 1, 2024 |
|
|
/s/ Gregory A. Demopulos |
|
|
Gregory A. Demopulos, M.D. |
Exhibit 31.2
CERTIFICATION OF PRINCIPAL FINANCIAL OFFICER PURSUANT TO RULE 13a-14(a)/15d-14(a) OF THE SECURITIES EXCHANGE ACT OF 1934, AS ADOPTED PURSUANT TO SECTION 302 OF THE SARBANES-OXLEY ACT OF 2002
I, Michael A. Jacobsen, certify that:
1. I have reviewed this annual report on Form 10-K of Omeros Corporation;
2. Based on my knowledge, this report does not contain any untrue statement of a material fact or omit to state a material fact necessary to make the statements made, in light of the circumstances under which such statements were made, not misleading with respect to the period covered by this report.
3. Based on my knowledge, the financial statements, and other financial information included in this report, fairly present in all material respects the financial condition, results of operations and cash flows of the registrant as of, and for, the periods presented in this report;
4. The registrant’s other certifying officer(s) and I are responsible for establishing and maintaining disclosure controls and procedures (as defined in Exchange Act Rules 13a-15(e) and 15d-15(e)) and internal control over financial reporting (as defined in Exchange Act Rules 13a-15(f) and 15d-15(f)) for the registrant and have:
a. Designed such disclosure controls and procedures, or caused such disclosure controls and procedures to be designed under our supervision, to ensure that material information relating to the registrant, including its consolidated subsidiaries, is made known to us by others within those entities, particularly during the period in which this report is being prepared;
b. Designed such internal control over financial reporting, or caused such internal control over financial reporting to be designed under our supervision, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles;
c. Evaluated the effectiveness of the registrant’s disclosure controls and procedures and presented in this report our conclusions about the effectiveness of the disclosure controls and procedures, as of the end of the period covered by this report based on such evaluation; and
d. Disclosed in this report any change in the registrant’s internal control over financial reporting that occurred during the registrant’s most recent fiscal quarter (the registrant’s fourth fiscal quarter in the case of an annual report) that has materially affected, or is reasonably likely to materially affect, the registrant’s internal control over financial reporting; and
5. The registrant’s other certifying officer(s) and I have disclosed, based on our most recent evaluation of internal control over financial reporting, to the registrant’s auditors and the audit committee of the registrant’s board of directors (or persons performing the equivalent functions):
a. All significant deficiencies and material weaknesses in the design or operation of internal control over financial reporting which are reasonably likely to adversely affect the registrant’s ability to record, process, summarize and report financial information; and
b. Any fraud, whether or not material, that involves management or other employees who have a significant role in the registrant’s internal control over financial reporting.
|
Dated: April 1, 2024 |
|
|
/s/ Michael A. Jacobsen |
|
|
Michael A. Jacobsen |
Exhibit 32.1
CERTIFICATION OF PRINCIPAL EXECUTIVE OFFICER PURSUANT TO 18 U.S.C. SECTION 1350, AS ADOPTED PURSUANT TO SECTION 906 OF THE SARBANES-OXLEY ACT OF 2002
In connection with the annual report on Form 10-K of Omeros Corporation (the “Company”) for the fiscal year ended December 31, 2023, as filed with the Securities and Exchange Commission on the date hereof (the “Report”), the undersigned officer of the Company certifies, pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002, that:
|
(1) |
The Report fully complies with the requirements of Section 13(a) or 15(d) of the Securities Exchange Act of 1934; and |
|
(2) |
The information contained in the Report fairly presents, in all material respects, the financial condition and results of operations of the Company. |
This certification accompanies the Report pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 and shall not, except to the extent required by the Sarbanes-Oxley Act of 2002, be deemed filed by the Company for purposes of Section 18 of the Securities Exchange Act of 1934, as amended (the “Exchange Act”), or incorporated by reference in any filing under the Securities Act of 1933, as amended, or the Exchange Act, except as may be expressly set forth by specific reference in such filing.
|
Dated: April 1, 2024 |
|
|
/s/ Gregory A. Demopulos |
|
|
Gregory A. Demopulos, M.D. |
Exhibit 32.2
CERTIFICATION OF PRINCIPAL FINANCIAL OFFICER PURSUANT TO 18 U.S.C. SECTION 1350, AS ADOPTED PURSUANT TO SECTION 906 OF THE SARBANES-OXLEY ACT OF 2002
In connection with the annual report on Form 10-K of Omeros Corporation (the “Company”) for the fiscal year ended December 31, 2023, as filed with the Securities and Exchange Commission on the date hereof (the “Report”), the undersigned officer of the Company certifies, pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002, that:
|
(1) |
The Report fully complies with the requirements of Section 13(a) or 15(d) of the Securities Exchange Act of 1934; and |
|
(2) |
The information contained in the Report fairly presents, in all material respects, the financial condition and results of operations of the Company. |
This certification accompanies the Report pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 and shall not, except to the extent required by the Sarbanes-Oxley Act of 2002, be deemed filed by the Company for purposes of Section 18 of the Securities Exchange Act of 1934, as amended (the “Exchange Act”), or incorporated by reference in any filing under the Securities Act of 1933, as amended, or the Exchange Act, except as may be expressly set forth by specific reference in such filing.
|
Dated: April 1, 2024 |
|
|
/s/ Michael A. Jacobsen |
|
|
Michael A. Jacobsen |
Exhibit 97.1
OMEROS CORPORATION COMPENSATION CLAWBACK POLICY
PURPOSE
The Board of Directors (the “Board”) of Omeros Corporation (the “Company”) has adopted this compensation clawback policy (the “Policy”), which provides for the recovery of incentive-based compensation in the event of an accounting restatement. This Policy is intended to comply with Section 10D of the Securities Exchange Act of 1934 (the “Act”), the rules promulgated thereunder by the Securities and Exchange Commission (the “SEC”), and the listing standards of The Nasdaq Stock Market LLC (“Nasdaq,” and such above-referenced Act, rules, and listing standards, collectively, the “Applicable Rules”) and will be interpreted consistent therewith.
EFFECTIVE DATE AND APPLICABILITY
This Policy is effective October 2, 2023 (the “Effective Date”) and is applicable to all Incentive- Based Compensation received by a Covered Executive (i) after beginning service as a Covered Executive,
(ii) who served as a Covered Executive at any time during the performance period for such Incentive-Based Compensation, and (iii) while the Company had a listed class of securities on a national securities exchange. Notwithstanding the foregoing, this Policy will not apply to Incentive-Based Compensation received by a Covered Executive before the Effective Date.
Each Covered Executive will be required to execute the acknowledgement and agreement in Appendix A of this Policy as soon as practicable after the later of (i) the Effective Date and (ii) the date on which the employee is designated as a Covered Executive; provided, however, that failure to execute such acknowledgement and agreement will have no impact on the enforceability of this Policy.
ADMINISTRATION
This Policy will be administered by the Board or, if so designated by the Board, a committee of the Board (the Board or such committee charged with administration of this Policy, the “Administrator”). The Administrator will have authority to (i) exercise all of the powers granted to it under the Policy, (ii) construe, interpret, and implement the Policy, and (iii) make all determinations necessary or advisable in administering the Policy other than as set forth herein. Any determination made by the Administrator under the Policy will be final and binding on all affected individuals and need not be uniform with respect to each individual covered by the Policy.
This Policy will be enforced and, if applicable, appropriate proxy disclosures and exhibit filings will be made in accordance with the Applicable Rules and any other applicable rules and regulations of the SEC and applicable Nasdaq listing standards.
RESTATEMENT CLAWBACK
In the event the Company is required to prepare an Accounting Restatement, the Company will recover reasonably promptly the amount of any Excess Compensation received by any Covered Executive during the Look-Back Period.
METHOD OF REPAYMENT, CONDITIONS FOR NON-RECOVERY
The Administrator will have sole discretion to determine the appropriate means of recovery of Excess Compensation, which may include, without limitation, (i) seeking reimbursement of all or part of any cash or equity-based award, (ii) cancelling prior cash or equity-based awards, whether vested or unvested or paid or unpaid, (iii) cancelling or offsetting against any planned salary increase or future cash or equity-based awards, and (iv) any other method authorized by applicable law or contract and determined by the Administrator to be advisable to achieve reasonably prompt recovery of Excess Compensation. At the direction of the Administrator and subject to compliance with any applicable law, the Company will take all actions reasonable and appropriate to recover Excess Compensation from any applicable Covered Executive in accordance with this Policy.
No recovery of Excess Compensation (or a portion thereof) is required if and to the extent that the Compensation Committee of the Board determines that recovery would be impracticable under any of the following conditions: (i) the direct expense paid to a third party to assist in enforcing this Policy would exceed the amount to be recovered, provided the Company has (A) made a reasonable attempt to recover such Excess Compensation, (B) documented such reasonable attempt, and (C) provided such documentation to Nasdaq; or (ii) recovery would likely cause an otherwise tax-qualified retirement plan, under which benefits are broadly available to employees of the Company, to fail to meet the requirements of 26 U.S.C. 401(a)(13) or 26 U.S.C. 411(a) and the regulations thereunder.
NO INDEMNIFICATION
Notwithstanding the terms of any indemnification or insurance policy or any contractual arrangement with any Covered Executive that may be interpreted to the contrary, the Company will not indemnify any Covered Executive against the loss of any Excess Compensation, including any payment or reimbursement for the cost of third-party insurance purchased by any Covered Executive to fund potential clawback obligations under this Policy.
AMENDMENT, TERMINATION, AND INTERPRETATION
The Board may amend, modify, supplement, rescind or replace all or any portion of this Policy at any time and from time to time in its discretion, and will amend this Policy as it deems necessary to comply with the Applicable Rules; provided, however, that any such revision to this Policy will not be effective if it would (after taking into account any actions taken by the Company contemporaneously with such revision) cause the Company to violate the Applicable Rules.
In the event of any conflict or inconsistency between this Policy and any other policies, plans, or other materials of the Company (including any agreement between the Company and any Covered Executive subject to this Policy), this Policy will govern.
DEFINITIONS
Capitalized terms used and not otherwise defined in this Policy have the meanings ascribed to them below:
“Accounting Restatement” means an accounting restatement due to the material noncompliance of the Company with any financial reporting requirement under securities laws, including any required accounting restatement to correct an error in previously issued financial statements that is material to the previously issued financial statements, or that corrects an error that is not material to previously issued financial statements but would result in a material misstatement if the error were corrected in the current period or left uncorrected in the current period.
Changes to financial statements that do not constitute an Accounting Restatement include retrospective (i) application of a change from one generally accepted accounting principle to another generally accepted accounting principle; (ii) revision to reportable segment information due to a change in the structure of the Company’s internal organization; (iii) reclassification due to a discontinued operation; (iv) application of a change in reporting entity, such as from a reorganization of entities under common control; and (v) revision for stock splits, reverse stock splits, stock dividends, or other changes in capital structure.
“Covered Executive” means each of the Company’s current and former executive officers, as determined by the Administrator in accordance with the definition of “executive officer” set forth in Rule 10D-1(d) under the Act.
“Excess Compensation” means the amount of Incentive-Based Compensation received by a Covered Executive after commencement of service as a Covered Executive that exceeds the amount of Incentive-Based Compensation that otherwise would have been received had it been determined based on the restated amounts, computed without regard to any taxes paid. For Incentive-Based Compensation based on stock price or total shareholder return, where the amount of Excess Compensation is not subject to mathematical recalculation directly from information in the Accounting Restatement, (i) the Administrator will determine the amount based on a reasonable estimate of the effect of the Accounting Restatement on the stock price or total shareholder return, as applicable, upon which the Incentive-Based Compensation was received, and (ii) the Company will maintain documentation of the determination of such estimate and provide such documentation to Nasdaq if so required by the Applicable Rules.
“Incentive-Based Compensation” means any compensation that is granted, earned, or vested based wholly or in part on the attainment of a financial reporting measure, including (i) stock price, (ii) total shareholder return, and/or (iii) any financial reporting measure(s) that are determined and presented in accordance with the accounting principles used in preparing the Company’s financial statements and any other measures that are derived in whole or in part from such measures. Incentive-Based Compensation is deemed “received” in the Company’s fiscal period during which the applicable financial reporting measure, stock price and/or total shareholder return measure upon which the Incentive-Based Compensation is based, is attained, even if the payment or grant of the Incentive-Based Compensation occurs after the end of that period.
Compensation that does not constitute “Incentive-Based Compensation” includes, but is not limited to, (i) salaries (except to the extent a salary increase is earned wholly or in part based on the attainment of a financial reporting measure performance goal, stock price and/or total shareholder return); (ii) bonuses paid solely at the discretion of the Compensation Committee or Board that are not paid from a “bonus pool” that is determined by satisfying a financial reporting measure performance goal, stock price and/or total shareholder return; (iii) bonuses paid solely upon satisfying one or more subjective standards and/or completion of a specified employment period; (iv) non-equity incentive plan awards earned solely upon satisfying one or more strategic measures or operation measures; and (v) equity awards for which the grant is not contingent upon achieving any financial reporting measure performance goal, stock price and/or total shareholder return and vesting is contingent solely upon completion of a specified employment period and/or attaining one or more nonfinancial reporting measures.
“Look-Back Period” means the three completed fiscal years immediately preceding the date that the Company is required to prepare an Accounting Restatement, as well as any transition period (that results from a change in the Company’s fiscal year) within or immediately following those three completed fiscal years (except that a transition period that comprises a period of at least nine months will count as a completed fiscal year). The “date that the Company is required to prepare an Accounting Restatement” is the earlier to occur of (i) the date the Board, the Audit Committee of the Board, or the officer or officers of the Company authorized to take such action if Board action is not required, concludes, or reasonably should have concluded, that the Company is required to prepare an Accounting Restatement or (ii) the date a court, regulator, or other legally authorized body directs the Company to prepare an Accounting Restatement, in each case regardless of if or when the restated financial statements are filed.
2
APPENDIX A:
ACKNOWLEDGMENT AND AGREEMENT
REGARDING OMEROS CORPORATION’S COMPENSATION CLAWBACK POLICY
I, the undersigned, agree and acknowledge that I am fully bound by, subject to, and will abide with, all of the terms and conditions of Omeros Corporation’s Compensation Clawback Policy (as may be amended, restated, supplemented or otherwise modified from time to time, the “Policy”). In the event of any inconsistency between the Policy and the terms of any employment agreement to which I am a party, or the terms of any compensation plan, program, or agreement under which any compensation has been granted, awarded, earned, or paid, the terms of the Policy shall govern. In the event it is determined by the Administrator that any amounts granted, awarded, earned, or paid to me must be forfeited or reimbursed to the Company under the Policy, I will promptly take any action determined by the Administrator under the Policy to effectuate such forfeiture and/or reimbursement. Any capitalized terms used in this Acknowledgment and Agreement without definition have the meaning set forth in the Policy.
|
|
|
|
|
|
|
|
|
|
|
|
|
Covered Executive’s Signature |
|
|
|
|
|
|
|
|
|
Covered Executive’s Name (printed) |
|
|
|
|
|
|
| Date | |||