UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM 10-K
(Mark One)
☒ ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the fiscal year ended June 30, 2024
OR
☐ TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the transition period from to
Commission file number: 001-39685
INMED PHARMACEUTICALS INC.
(Exact name of registrant as specified in its charter)
| British Columbia, Canada | 98-1428279 | |
| (State or other jurisdiction of incorporation or organization) | (IRS employer Identification number) |
|
| 1445, 885 West Georgia St., Vancouver, B.C., Canada | V6C 3E8 | |
| (Address of principal executive office) | (Zip Code) |
(604) 669-7207
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Exchange Act:
| Title of Each Class | Trading Symbol | Name of Each Exchange On Which Registered | ||
| Common Stock, no par value | INM | The Nasdaq Capital Market |
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☐ No ☒
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Exchange Act. Yes ☐ No ☒
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ☒ No ☐
Indicate by check mark whether the registrant has submitted electronically, every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit such files). Yes ☒ No ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer | ☐ | Accelerated filer | ☐ |
| Non-accelerated filer | ☒ | Smaller reporting company | ☒ |
| Emerging growth company | ☒ |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant has fi led a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting fi rm that prepared or issued its audit report. ☐
If securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant included in the filing reflect the correction of an error to previously issued financial statements. ☐
Indicate by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive-based compensation received by any of the registrant’s executive officers during the relevant recovery period pursuant to §240.10D-1(b). ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ☐ No ☒
As of December 31, 2023, the last business day of the Registrant’s most recently completed second fiscal quarter, the aggregate market value of the Company’s voting and non-voting common equity held by non-affiliates of the Registrant was $2,351,381.
On September 20, 2024, there were 13,340,245 shares of the registrant’s common shares, no par value, outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the registrant’s definitive proxy statement for the registrant’s 2024 Annual Meeting of Stockholders to be filed pursuant to Regulation 14A within 120 days of the registrant’s fiscal year ended June 30, 2024 are incorporated herein by reference into Part III of this Annual Report (as defined below).
InMed Pharmaceuticals Inc.
TABLE OF CONTENTS
PART I
Special Note Regarding Forward-Looking Statements
This Annual Report on Form 10-K (this “Annual Report”) contains “forward-looking statements” within the meaning of United States Private Securities Litigation Reform Act of 1995 and “forward-looking information” within the meaning of applicable Canadian securities law, which are included but are not limited to statements with respect to InMed Pharmaceuticals Inc.’s (the “Company” “InMed”, “we”, “our”, or “us”) anticipated results and progress of the Company’s operations, research and development in future periods, plans related to its business strategy, and other matters that may occur in the future. These statements relate to analyses and other information that are based on forecasts of future results, estimates of amounts not yet determinable and assumptions of management. We may, in some cases, use words such as “anticipate”, “believe”, “could”, “estimate”, “expect”, “intend”, “may”, “plan”, “predict”, “project”, “will”, “would”, “budget”, “possible”, “should”, “future”, and similar expressions that convey uncertainty of future events or outcomes to identify these forward-looking statements. These forward-looking statements reflect our current views with respect to future events and are based on assumptions and subject to risks and uncertainties. You should not place undue reliance on these forward-looking statements. Any statements contained herein that are not statements of historical facts may be deemed to be forward-looking statements. Our actual results could differ materially from those anticipated in these forward-looking statements. Among the factors that could cause actual results to differ materially are the risks and uncertainties described under “Item 1A. Risk Factors” of this Annual Report, “Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations” of this Annual Report, and the following:
| ● | The Company’s ability to stem operating losses and the Company’s ability to obtain additional financing to fund its operations. |
| ● | The revenues of BayMedica, LLC (“BayMedica”) and the commercial viability of its product portfolio; | |
| ● | The Company’s ability to effectively research, develop, manufacture and commercialize pharmaceutical drug candidates that will treat diseases with high unmet medical needs; |
| ● | The continued optimization of key, proprietary manufacturing approaches and technologies; |
| ● | Our ability to commercialize and, where required, register products in the pharmaceutical R&D programs (“Product Candidates”) and those targeted to the health and wellness sector (“Products”) in the United States and other jurisdictions; |
| ● | Our success in initiating discussions with potential partners for licensing various aspects of our Product Candidates; |
| ● | Our ability to successfully access existing manufacturing capacity via leases with third-parties or to transfer our manufacturing processes to contract manufacturing organizations; |
| ● | Our belief that our manufacturing approaches that we are developing are robust and effective and will result in commercially viable yields of cannabinoids and will be a significant improvement upon existing manufacturing platforms; |
| ● | Our ability to successfully scale up our IntegraSyn approach to cannabinoid manufacturing. InMed has created genetically engineered microbes that produce proprietary enzymes, which are then used to optimize subsequent biotransformation reactions or other cost-effective manufacturing approaches so that it will be commercial-scale ready after Phase 2 clinical trials are completed, after which time we may no longer need to source active pharmaceutical ingredients (“APIs”) from third-party API manufacturers; |
| ● | The success of the key next steps in our manufacturing approaches, including continuing efforts to diversify the number of products produced, scaling-up the processes to larger vessels and identifying external vendors to assist in the commercial scale-up of the process; |
| ● | Our ability to successfully make determinations as to which research and development programs to continue based on several strategic factors; |
| ● | Our ability to continue to outsource the majority of our research and development activities through scientific collaboration agreements and arrangements with various scientific collaborators, academic institutions and their personnel; |
| ● | The success of work to be conducted under the research and development collaboration between us and various contract development and manufacturing organizations (“CDMOs”); |
| ● | Our ability to develop our therapies through early human testing; |
| ● | Our ability to evaluate the financial returns on various commercialization approaches for our Product Candidates, such as a ‘go-it-alone’ commercialization effort, out-licensing to third parties, or co-promotion agreements with strategic collaborators; |
| ● | Our ability to find a partnership early in the development process for our various programs; |
| ● | Our ability to explore our manufacturing technologies as processes which may confer certain benefits, including cost, yield, speed, or all the above, when pursuing specific types of molecules, and filing a provisional patent application for same; |
| ● | Plans regarding our next steps, options, and targeted benefits of our manufacturing technologies; |
| ● | Our Products being bio-identical to the naturally occurring molecules, and offering superior ease, control and quality of manufacturing when compared to alternative methods; |
| ● | U.S. Food and Drug Administration (“FDA”) regulatory acceptance of Product Candidates for potential use in the pharmaceutical industry; |
| ● | Our ability to successfully file, prosecute and defend patent applications; | |
| ● | The potential for any of our patent applications to provide intellectual property protection for us; |
| ● | The termination or renegotiation of our supplier, technology and other material contracts, including the invoking of force majeure or termination clauses, and actual or threatened claims of our failure to comply with any obligations set forth under such contracts; | |
| ● | The adequacy of, or gaps in, insurance coverage upon the occurrence of a catastrophic or other material adverse event, as well as our ability to (i) expand our insurance coverage to include the commercial sale of Products and Product Candidates and (ii) secure insurance coverage for shipping and storage of Product Candidates, and clinical trial insurance; |
| ● | Developing patentable New Chemical Entities (“NCE”) which, if issued, will confer market exclusivity to us for the potential development into pharmaceutical Product Candidates, license, partner or sell to interested external parties; | |
| ● | Our ability to initiate discussions and conclude strategic partnerships to assist with development of certain programs; |
| ● | Our ability to position ourselves to achieve value-driving, near term milestones for our Product Candidates with limited investment; |
| ● | Our ability to effectively execute our business strategy; |
| ● | The sufficiency of our internal controls, including any exposure arising from the failure to (i) establish and maintain effective internal control over financial reporting in accordance with applicable regulatory requirements, and (ii) fully remediate any material weakness identified with respect to such internal controls; |
| ● | Epidemics, pandemics, global health crises, or other public health events and concerns, including any future resurgence of COVID-19, and the effectiveness of associated vaccinations and treatments; |
| ● | Consolidation of our competitors and suppliers; |
| ● | Effects of new products and new technology on the market, including with respect to automation and the use of artificial intelligence (“AI”); |
| ● | The impact of geopolitical, global, regional or local economic and financial market risks and challenges, applicability of foreign laws, including foreign labor and employment laws, foreign tax and customs regimes, and foreign currency exchange rate risk; |
| ● | Political disturbances, geopolitical instability and tensions, or terrorist attacks, and associated changes in global trade policies and economic sanctions, including, but not limited to, in connection with (i) the Russo-Ukrainian war and (ii) any impact, effect, damage, destruction and/or bodily harm directly or indirectly relating to the ongoing hostilities in the Middle East; and |
| ● | Our failure to satisfy any applicable listing standards, including compliance with the minimum bid price rule, and the actual or threatened delisting of our securities by Nasdaq. |
This list is not exhaustive of the factors, events, conditions and circumstances that may affect the “forward-looking statements” and “forward-looking information” contained in this Annual Report. Although we have attempted to identify important factors that could cause actual results to differ materially from those described in forward-looking statements, there may be other factors that cause results not to be as anticipated, estimated or intended. Should one or more of these risks or uncertainties materialize, or should underlying assumptions prove incorrect, actual results may vary materially from those anticipated, believed, estimated, or expected. We caution readers not to place undue reliance on any such forward-looking statements, which speak only as of the date made and are based only on the information available to us at that time. Except as required by law, we disclaim any obligation to subsequently revise any forward-looking statements to reflect events or circumstances after the date of such statements or to reflect the occurrence of anticipated or unanticipated events.
Overview
We are a pharmaceutical company developing a pipeline of proprietary small molecule drug candidates that are preferential signaling ligands of the endogenous cannabinoid 1 (“CB1”) and cannabinoid 2 (“CB2”) receptors as well as other receptor targets linked to human diseases. CB1 and CB2 receptors are each part of the endocannabinoid system that is found throughout the human body and is responsible for many homeostatic functions. CB1 receptors are primarily located in the brain and central nervous system, while CB2 receptors are involved in modulating neuroinflammation and immune responses. Our research efforts target the treatment of diseases with high unmet medical needs. Together with our wholly-owned subsidiary, BayMedica, we also have garnered significant know-how in developing proprietary manufacturing approaches to produce and sell bulk rare cannabinoids as ingredients for various market sectors (“Products”).
InMed has sought to focus on the research and development of preferential signaling ligands of CB1 and CB2 and has produced a library of novel, proprietary drug candidates (“Product Candidates’). These Product Candidates are patentable NCEs for pharmaceutical drug development, aimed at targeting diverse clinical indications. Our current pharmaceutical pipeline consists of three programs, with drug candidates targeting Alzheimer’s disease, dry age-related macular degeneration (“AMD”), and Epidermolysis Bullosa (“EB”).
InMed’s INM-901 is a proprietary small molecule, disease modifying drug candidate that is being developed as a potential treatment for Alzheimer’s disease. INM-901 has multiple potential mechanisms of action as a preferential signaling agonist for both CB1 and CB2 receptors, as well as impacting the PPAR signaling pathway. Combined, these mechanisms of action may offer a unique treatment approach targeting several biological pathways associated with Alzheimer’s disease.
Outcome from our ocular research, based on the proprietary small molecule INM-089, indicates potentially promising neuroprotective effects in the back of the eye, which may lead to the preservation of the retinal function. Neuroprotection in dry Aged-related Macular Degeneration (“dry AMD”) remains an unmet medical need and a new treatment option may help solve this multifactorial disease.
InMed has completed a Phase 2 clinical trial of INM-755 (cannabinol) cream studying its safety and efficacy in treating symptoms related to EB. Results from the Phase 2 clinical trial conducted during 2022 and 2023 showed a positive indication of enhanced anti-itch activity for INM-755 (cannabinol) cream versus the control cream alone in an exploratory clinical evaluation. The Company is also pursuing strategic partnership opportunities for INM-755 in EB and other itch-related skin conditions.
Together with our wholly owned subsidiary BayMedica, our manufacturing capabilities include traditional approaches such as chemical synthesis and biosynthesis, as well as a proprietary, integrated manufacturing approach called IntegraSyn. With several manufacturing approaches, InMed has sought to maintain enhanced flexibility to select the most cost-effective method to deliver high quality, high-purity Products and Product Candidates fit for their intended use. BayMedica’s commercial business specializes in the business-to-business (“B2B”) commercialization of bulk rare, non-intoxicating cannabinoids as raw materials for the ‘health and wellness’ sector that are bioidentical to those found in nature.
Corporate Information
We were originally incorporated in the Province of British Columbia, under the Business Corporations Act (British Columbia) (the “BCBCA”), on May 19, 1981 (the “Incorporation Date”), and we have undergone a number of executive management, corporate name and business sector changes since such Incorporation Date, ultimately changing our name to “InMed Pharmaceuticals Inc.” on October 6, 2014. Our principal executive offices are located at Suite 1445, 885 West Georgia Street, Vancouver, BC, V6C3E8 and our telephone number is +1-604-669-7207. Our website is https://www.inmedpharma.com/. The contents of our website are not incorporated, in whole or in part, into this Annual Report in any respect.
Employees and Human Capital
Our management team is comprised of highly experienced pharmaceutical and biotechnology executives with successful track records in researching, developing, gaining approval for and commercializing novel medicines to treat serious diseases. Each member of our management team has 20 to 30+ years of industry experience, including our Chief Executive Officer (“CEO”), Chief Operating Officer (“COO”), Chief Financial Officer (“CFO”), General Manager and VPs of Preclinical Drug Development, Discovery Research, Chemistry, Synthetic Biology and of Sales & Marketing. Together, this management team has covered the spectrum of pharmaceutical drug discovery, preclinical research, formulation development, manufacturing, human clinical trials, regulatory submissions and approval, and global commercialization of pharmaceutical and wellness products. Additionally, the management team has significant experience in company formation, capital raises, mergers and acquisitions, business development, and sales and marketing in the pharmaceutical and other industries. Our Board is constituted by individuals with significant experience in the pharmaceutical and biotechnology industries. As of September 20, 2024, inclusive of our management team, we had 13 full-time employees, and we also utilize the services of several consultants. None of our employees are represented by a collective bargaining agreement, nor have we experienced any work stoppage. We believe that we maintain strong relations with our employees.
We are committed to growing our business over the long-term. As a result of the competitive nature of the industry in which we operate, employees have significant career mobility and opportunity, and as a result, the competition for experienced employees is great. The existence of this competition, and the need for talented and experienced employees to realize our business objectives, underlies the design and implementation of our compensation programs. At the same time, we seek to keep our approach to compensation simple and streamlined to reflect the still relatively moderate size of the Company. We have therefore implemented compensation, leave and benefits programs necessary to attract and retain the talented and experienced employees necessary to develop our business, including what we believe to be competitive salaries, stock options awards to permanent employees (both upon initial hiring and on an annual basis thereafter), and pay annual bonuses to permanent employees contingent on the achievement of corporate and/or personal objectives. We have developed an Employee Handbook that contains all corporate policies and guidelines for professional behavior. Our policies and practices apply to all employees, regardless of title. These guidelines include, among others, our Code of Business Conduct as well as our policies for corporate disclosure, insider trading and whistle blower, all of which are posted on our website.
For all current and future pharmaceutical Product Candidates we intend to submit new drug applications (“NDAs”) (or their international equivalents) in most major jurisdictions, including the United States, either alone or with development/commercial partners.
Our Business Strategy
Our goal is to develop a pipeline of prescription-based Product Candidates targeting treatments for diseases with high unmet medical needs.
| ● | Develop and produce proprietary small molecule Product Candidates for use in our drug development programs |
| ● | Advance pharmaceutical drug Product Candidates through preclinical and clinical development, thereby establishing important human proof-of-concept in multiple therapeutic applications |
These activities are at various stages of development, including, with INM-901 (for the treatment of Alzheimer’s disease), INM-089 (for the treatment of dry AMD) and INM-755 (for the treatment of symptoms related to EB). We have the internal capabilities to design and execute, together with multiple external vendors, the preclinical experimentation and clinical studies required to advance pharmaceutical drug candidates towards commercialization.
| ● | Actively seek avenues to accelerate drug development via licensing, partnering or sale to external companies |
We do not currently have an internal organization for the sales, marketing and distribution of pharmaceutical Products. With respect to the commercialization of each Product Candidate, we may therefore rely on (i) a “go-it-alone” commercialization effort; (ii) out-licensing to third parties; or, (iii) co-promotion agreements with strategic collaborators for our Product Candidates. The decision to pursue a “go-it-alone” commercialization effort versus out-licensing to third parties will depend on various factors including, but not limited to, the complexity of the Product Candidate and process, the expertise required and related cost of building any such infrastructure for our Product Candidates. For INM-755 in EB, we are actively seeking development and commercial partnerships. The optimal commercial strategy for the INM-901 and INM-089 compounds will be evaluated in due course.
| ● | Expand portfolio and revenues of Products into the existing distribution network and to end-product manufacturers of specialty health and wellness products |
| ● | Develop cost effective and scalable manufacturing processes for high quality Products and Product Candidates as APIs for our core internal drug candidate pipeline and for licensing opportunities of non-core drug candidates. |
Our Strengths
We are a pharmaceutical drug development company as well as a developer and supplier of rare, naturally occurring cannabinoids that is focused on commercializing important \ medicines to treat diseases with high unmet medical needs. Our key strengths include the following:
Experienced executive team and board of directors with proven track records.
One key critical success factor in the field of pharmaceutical drug development is the experience and skill set of the individuals leading the company. We have been successful in attracting and retaining executive and directors with extensive experience in all facets of the pharmaceutical industry, including fundamental research and development, multiple manufacturing techniques, drug formulation, clinical trial execution, regulatory approvals, pharmaceutical commercialization, company and capital formation, business development, legal, and corporate governance. Our leadership team is well-poised to lead us through all facets of drug development and product commercialization, either internally or externally via partnerships. It is this group of individuals that will help optimize our chances for success.
Scope of research and robust pharmaceutical pipeline
Over several years of dedicated research, InMed has built a robust pipeline of drug development candidates, including two preclinical programs targeting Alzheimer’s (INM-901) and ocular diseases (INM-088 for glaucoma and INM-089 for AMD), as well as a completed Phase 2 study in dermatology (INM-755). The INM-089 and INM-901 preclinical programs offer a differentiated treatment approach using proprietary, disease-modifying small molecules that target the CB1 and CB2 receptors, which management believes is a key strength of the Company.
Multiple manufacturing approaches.
Our management team believes that the combined manufacturing technologies from InMed and BayMedica provide us with a competitive advantage to utilize the most cost-efficient methodology (i.e. chemical synthesis, biosynthesis and IntegraSyn) for the development and commercialization of new Products and Product Candidates to a wide spectrum of market opportunities.
Early mover status as a B2B supplier of rare cannabinoids to the health and wellness sector.
As demonstrated by the launch of several rare cannabinoids into the health and wellness sector, the team at BayMedica has substantial expertise in the commercial manufacturing scale-up to produce rare cannabinoids at large scale as well as extensive sales and marketing expertise. This know-how is important to establishing an early-mover status and to maintain cost leadership with regards to specific rare cannabinoids.
Diverse portfolio of patent applications covering a spectrum of commercial opportunities.
Success in pharmaceutical markets often rests with the strength of intellectual property, including patents, to protect our commercialization interests. We have filed several patents on our novel findings and expect to continue to do so. The acquisition of BayMedica brought several additional new patent families to bolster our manufacturing as well as drug development opportunities.
BayMedica’s chemical synthesis and biosynthesis technologies for the development and production of cannabinoids, their variants and analogs
BayMedica continues to develop manufacturing techniques that are ‘method agnostic’, utilizing the most practicable, expeditious and cost-effective means to produce any particular Product or novel Product Candidate.
Research and Development Pipeline of Therapeutic Drug Candidates
INM-901 for the Treatment of Alzheimer’s Disease (“AD”)
Traditionally, Alzheimer’s disease has been defined by the buildup of amyloid beta (“Aβ”) plaques and neurofibrillary, also referred to as tau protein, tangles within the brain, making it a central focus of neurological research for many years. However, more recently, other factors such as neuroprotection and synaptic dysfunction are being recognized as contributors to disease progression.
Our early research demonstrating the neuroprotective capabilities of CB1 and CB2 agonists in the eye led us to investigate how such molecules might play a role in protecting other neurons in the human body, potentially, impacting different diseases. To this end, we initiated research on the neurons that are associated with the brain and how our proprietary CB1 and CB2 agonist drug candidates could affect neurodegenerative diseases such as Alzheimer’s, Parkinson’s, and Huntington’s. In October 2023, InMed announced it had selected and would be advancing a lead AD drug candidate, named INM-901, following positive results from several proof-of-concept studies. INM-901 is a proprietary small molecule drug candidate. which, based on preclinical studies in well-characterized AD study models, may address multiple pathologies related to AD progression. In these preclinical study models, INM-901 demonstrated neuroprotective effects, reduced neuroinflammation, the ability to extend the length of neurites signifying enhanced neuronal function, and improvement in behavior, cognitive function and memory. These early studies show the potential of INM-901 to reverse neuronal damage from AD and potentially provide disease-modifying effects.
As a small molecule compound, INM-901 may offer various modes of administration including oral delivery, which could overcome several limitations associated with currently approved antibody therapies for AD, such as the high drug expenses, complicated and inconvenient drug administration and its associated compliance and accessibility challenges.
INM-901’s promising preclinical studies, multifactorial mechanism of action and small molecule profile offer a potentially attractive treatment option for AD.
Alzheimer’s Disease Prevalence and Impact – A Major Medical and Societal Burden
Alzheimer’s disease is a progressive neurodegenerative condition that predominantly afflicts the elderly, resulting in severe cognitive impairments. It is a subset of dementia that impacts the part of the brain that controls memory and language and leads to increased morbidity and mortality.
According to the U.S. Alzheimer’s Disease Association, AD accounts for 60-80% of dementia cases and is the fifth leading cause of death for people aged 65 and older. It’s estimated that 6.9 million Americans are living with AD, and it’s expected to grow to 12.7 million by 2050. About 1 in 9 people aged 65 and older has AD (10.7%), affecting 1 in 5 women and 1 in 10 men in their lifetime.
The disease has a major medical and societal burden with health and long-term care costs valued at $360 billion. In addition to the cost to the healthcare system, it’s estimated 11 million Americans are providing 18.4 billion hours of unpaid care valued at $350 billion for people living with AD or other dementias, making it one of the costliest diseases to society.
Additionally, the emotional and mental health burden on patients and their caregivers cannot be overstated.
Pathology of Alzheimer’s disease
Alzheimer’s disease is a complex neurodegenerative disease with multiple pathologies leading to its development and progression. Hallmarks of the disease point to the toxicity and disruption of proteostasis caused by misfolded amyloid beta protein and neurofibrillary tangles or tau tangles. Amyloid-beta is a naturally occurring protein in the brain, but when abnormal levels of amyloid-beta clump together to form plaques, it causes damage to neuronal cell function resulting in AD.
The focus of Alzheimer’s research has been traditionally centered around amyloid-beta plaques and tau protein, which play a crucial role in stabilizing microtubules within neurons, supporting their structure and function. Increased activity of enzymes called tau kinase causes the tau protein to misfold and clump, creating neurofibrillary tangles which disrupt the normal functioning of neurons. The stage and severity of AD is associated with an abundance of tau tangles.
In addition to these two aspects of Alzheimer’s disease, neuroinflammation and synaptic dysfunction are also recognized as contributors to AD progression. Microglia, the brain’s immune cells, are involved in the removal of amyloid-beta and has been a focus of research in neuroinflammation. Therapies targeting the modulation of microglial activity aim to reduce inflammation and protect neurons.
Current treatments in Neurodegenerative Diseases
| Brand | Company | Mechanism of Action | Status | |||
| Aducanumab (Aduhelm™) | Biogen | Anti-amyloid beta target both insoluble and soluble aggregates | Approved June 2021 | |||
| Lecanemab (Leqembi™) | Biogen/ Eisai | Anti-amyloid beta, electively binds to large, soluble Aβ protofibrils | Approved January 2023 | |||
| Gantenerumab | Roche | Anti-amyloid beta, target aggregated forms of AB including oligomers and plaques | Phase 3 failed November 2022 | |||
| Donanemab | Eli Lilly | Anti-amyloid beta, target pyroglutamated AB in plaques | Approved July 2024 | |||
| Semorinemab | Genentech | Anti-tau | Phase 2 Failed | |||
| HMTM | TauRx Therapeutics | Anti-Tau, tau aggregation inhibitor | Applied to UK MAA for market authorization |
Currently approved medications for AD fall into two main categories. The first category comprises drugs designed to address symptoms related to memory and cognitive function. While these medications cannot halt the damage that AD inflicts on brain cells, they can help alleviate or stabilize symptoms for a limited duration by influencing specific chemicals responsible for transmitting messages between nerve cells in the brain. Essentially, these medications are aimed at preserving neurotransmitters. However, they do not replace the deteriorating ones and thus do not impede the disease’s progression.
Until recently, cholinesterase inhibitors and glutamate regulators were the only treatments available to people living with AD. These drugs are intended to improve cognitive and behavioral symptoms and do not address the prevention or progression of the disease.
Over the past three decades, only four drugs have received approval for AD treatment, and while they can manage certain symptoms, they do not address the prevention or progression of the disease. These drugs, known as cholinesterase inhibitors and glutamate regulators, primarily target cognitive symptoms.
In recent years, there has been a growing emphasis on developing disease-modifying treatments that target the underlying biology of AD. One major focus of these research and development endeavors has centered on addressing the accumulation of amyloid plaques and the removal of both these plaques and tau proteins. This approach aligns with the long-standing amyloid hypothesis, which posits that AD is triggered by the buildup of (Aβ) in the brain. This accumulation leads to neuronal toxicity within the central nervous system, disrupting neuronal and synaptic function, ultimately culminating in neuronal degeneration and cell death.
Role of CB1 and CB2 Agonists in Alzheimer’s disease:
Numerous studies have indicated dysregulation of the Endocannabinoid System (“ECS”), which encompasses receptors, endocannabinoids, and synthesizing/metabolizing enzymes, in various neurodegenerative conditions, notably AD. These investigations have unveiled the potential of CB1 and CB2 agonists, both endogenous and synthetic, in mitigating the harmful effects of AD pathology. These CB1 and CB2 agonists have been suggested to diminish Aβ toxicity, reduce tau hyper-phosphorylation, and suppress neuroinflammatory responses while curbing the production of reactive oxygen species (“ROS”). As a result, they may enhance the survival of neurons in the aftermath of Aβ aggregation.
CB1 and CB2 agonists exert their biological effects through two primary membrane receptors, endogenous CB1 and CB2 receptors, which are widely distributed in the central nervous system and peripheral tissues. Activation of CB1 has demonstrated its ability to alleviate neurotoxicity in various AD models. Conversely, CB2 agonism and increased expression have been associated with the removal of Aβ by macrophages.
The precise molecular mechanisms responsible for safeguarding specific neuronal populations remain elusive. However, several observations support this concept:
a) CB1 and CB2 agonists possess a capacity to exert broad effects on multiple molecular targets, including critical brain structures and behavior;
b) CB1 and CB2 agonists act not only through ECS receptors but also interact with other non-ECS receptors such as transient receptor potential vanilloid 1, peroxisome proliferator-activated receptors (“PPARs”), and transcription factors such as nuclear factor kappa B (“NFkB”); and
c) CB1 and CB2 agonists exhibit anti-inflammatory properties, modulate neurotransmitter release, and limit oxidative stress, collectively contributing to the enhancement of neuronal viability.
AD is a progressive neurodegenerative condition primarily driven by the toxicity and disruption of proteostasis caused by misfolded Aβ protein. CB1 and CB2 agonists have emerged as promising agents capable of preserving neuronal integrity and functionality, offering a potential strategy to slow down disease progression and enhance the quality of life for affected individuals. Furthermore, CB1 and CB2 agonists exhibit the capacity to mitigate neuroinflammation, shield against beta-amyloid-induced neurotoxicity, and mitigate neurodegeneration in animal models of AD. Additionally, research has unveiled dysregulation of the ECS in the brains of AD patients, which could contribute to the cognitive and behavioral symptoms associated with the disease.
INM-901 is highly lipophilic (dissolves in fats, oils and lipids) and can easily cross the blood-brain barrier, a capability that renders it a promising candidate for pharmaceutical use in the treatment of neurological disorders.
The use of CB1 and CB2 agonists in AD treatment holds great promise; however, further research is needed to fully understand the mechanisms to develop safe and effective CB1 and CB2 agonists-based therapeutics.
INM-901: A Multi-factorial Approach to Treating Alzheimer’s disease
While progress has been made recently in the development of new treatments for AD, there are no treatments addressing the multiple aspects of this complex disease such as neuroinflammation, neuroprotection, synaptic dysfunction or the restoration of the damaged neurons – factors that may help to restore brain function loss or reverse the damage caused by AD.
Preclinical studies indicate that INM-901 may target multiple biological pathways. InMed has conducted several in vitro and in vivo studies to test the pharmacological effects of INM-901 in well-characterized AD preclinical models.
Figure 1. Neuroprotection of human neuronal cells

Phyto-cannabinoids (pCBx) promote neuroprotection. (A) Amyloid peptide (Aβ, 5µM) induces cytotoxicity in SHSY5Y cells. Aβ1-42 insult induced approximately ~45% cytotoxicity of the SH-SY5Y cells. (B) Concurrent exposure of Aβ with pCBx at 5 µM and 10 µM concentrations protected cells from Aβ induced toxicity in a dose-dependent manner. Cell viability was determined by MTT assay.
Figure 2. Neurogenesis of human neuronal cells

Cannabinoid (pCBx) promotes neuritogenesis. Tuj1 Tubulins are building blocks of microtubules. As such, Tuj1 expression can reveal the fine details of axonal structures and dendrites. Therefore, changes in Tuj1 expression can be directly correlated with neuronal health and communication. (A) Photomicrographs illustrating Tuj1 expression in control and pCBx (5 and 10µM) treated cells. The formation of extended neurites and arborization is evident upon pCBx treatment.
Key Results:
Preclinical studies in AD models demonstrated the following:
| ● | neuroprotective effects by reducing cell death in an amyloid-beta-induced cytotoxicity and attenuated increased Bax/Cas-3 expression in the presence of Aβ (5µM) in SH-SY5Y neuroblastoma cells; |
| ● | improves neuritogenesis in human neuroblastoma cells, enhanced expression of neurite marker MAP2 and Tuj1; |
| ● | reduces neuroinflammation; |
| ● | promotes neurite outgrowth, signifying the potential to improve neuronal function, a potential breakthrough in the treatment of AD; and |
| ● | improvement in cognitive function and memory, locomotor activity, anxiety-based behavior, sound awareness and neuronal function. |
INM-901 Interacts with Specific Receptors in the Brain
Studies of INM-901 demonstrate activity as a preferential signaling ligand for CB1 and CB2 and impacts the PPAR signaling pathway. Research indicates that activating CB1 and CB2 receptors may induce neuroprotective effects and may help to protect brain cells from damage and death. Enhancing the activity of these receptors may help to slow down the progression of the AD, in which neuronal cell death is a hallmark. Moreover, the activation of these receptors, along with other cellular receptors, has also been shown to have an impact on neuroinflammation. As neuroinflammation is believed to contribute to the progression of AD, targeting these receptors could help alleviate this inflammatory response.
INM-901 is a Proprietary Small Molecule Compound
INM-901 is a small molecule compound, one of several cannabinoid analogs developed by the Company’s scientists. Cannabinoids are small molecules known to be highly lipophilic and can safely cross the blood-brain barrier, enabling the potential therapeutic modulation of brain signaling and making them promising pharmaceutical targets for neurological diseases such as AD.
Small molecule drugs have several advantages that contribute to their widespread use. Those advantages include oral administration (making it convenient for patients to comply), good bioavailability (allowing these compounds to be efficiently absorbed), ability to cross the blood-brain barrier (enabling therapeutic modulation in brain signaling), stability in storage and transport (ease of drug handling and dose adjustment) and low-cost manufacturing.
INM-901 Next Steps
Research & Development
| ● | Assess long-term (7-month dosing) study in 5xFAD mice (on-going) |
| ● | Plan/execute study in PS19 Tau model |
| ● | Continue Chemistry/Manufacturing/Controls (“CMC”) activities for drug substance and drug product |
| ● | Continue studies of receptor interactions (MoA) and Distribution, Metabolism, Pharmacokinetics (“DMPK”) |
| ● | Plan to execute IND enabling toxicology studies |
Key Milestones
| ● |
November 3, 2021 — we announced the filing of an international patent application demonstrating neuroprotection and enhanced neuronal function using a rare cannabinoid for the potential treatment of neurodegenerative diseases such as Alzheimer’s Disease, Parkinson’s Disease, Huntington’s Disease and others. This Patent Cooperation Treaty (PCT) application, entitled “Compositions and Methods for Treating Neuronal Disorders with Cannabinoids”, specifies a rare cannabinoid that may inhibit or slow the progression of neurodegenerative diseases by providing neuroprotection in a population of affected neurons. Furthermore, the PCT application also demonstrates the subject cannabinoid compound can also be used to promote neurite outgrowth, signifying the potential to enhance neuronal function. The rare cannabinoid included in the PCT application is new to InMed’s portfolio. |
| ● | November 16, 2022 — we announced announces the launch of its neurodegenerative disease program (INM-900 series), investigating the effects of cannabinoid analogs in diseases such as Alzheimer’s, Huntington’s and Parkinson’s. In addition, Dr. Ujendra Kumar of the Faculty of Pharmaceuticals Sciences at UBC has been awarded an Alliance grant from NSERC, with InMed as the named industry partner. The funding will support the research and development studies of InMed’s cannabinoid pharmaceutical candidates, investigating their potential therapeutic effects in neurodegenerative diseases. The collaboration project is entitled “Pharmacological Characterization of Phytocannabinoids and the Endocannabinoid System”. |
| ● | June 1, 2023 — we announced that results from a neurodegenerative disease study was presented in a scientific poster at the Canadian Neuroscience Meeting in Montreal from May 28-31, 2023. The InMed sponsored research, entitled “Cannabinoids modulate cytotoxicity and neuritogenesis in Amyloid-beta-treated neuronal cells”, demonstrated the ability of a specific rare cannabinoid (“pCBx”) in our INM-900 series of potential candidates that reduces amyloid toxicity and tau protein expression while enhancing neuronal cell growth and neuritogenesis markers in vitro, all considered to be important targets in the potential treatment of neurodegenerative diseases such as Alzheimer’s. | |
| ● | October 2, 2023 — we announced the selection of a lead Alzheimer’s disease drug candidate, named INM-901, following positive results from several proof-of-concept studies in a validated Alzheimer’s disease treatment model. InMed will be advancing INM-901, a cannabinoid analog, in its pharmaceutical drug development program. In vitro Alzheimer’s disease studies showed that INM-901 treated groups display neuroprotection and extended neurite length, a potential marker for improved neuronal function. INM-901 treated groups in an in vivo Alzheimer’s disease model demonstrated improved behavioral, cognitive and memory outcomes in several Alzheimer’s proof-of-concept studies. | |
| ● | April 4, 2024 — we announced additional preclinical data demonstrating INM-901’s positive pharmacological effects in the potential treatment of Alzheimer’s disease (“AD”). Additionally, the studies demonstrated INM-901 is a preferential signaling agonist of the CB1and CB2 receptors and impacts the PPAR signaling pathway, reduced neuroinflammation and improved neuronal function, and that mRNA data supports the observations made in the previously released behavior studies in locomotor activity, cognition and memory. |
| ● | April 18, 2024 — we announced the addition of Dr. David G. Morgan, a renowned leader in neurodegenerative disease to its Scientific Advisory Board (“SAB”) reinforcing the Company’s commitment to advancing it’s INM-901 program in the treatment of Alzheimer’s disease. |
| ● | July 30, 2024 — we announced positive results from initial data sets from a long-term (7 months of dosing) in vivo preclinical Alzheimer’s Disease (“AD”) study of INM-901 which confirms previously reported findings from a short-term (3 months of dosing) pilot study. All assessments of the INM-901-treated AD groups showed a positive trend towards behaviour similar to the untreated disease-free group, with most assessments demonstrating a clear dose response. Furthermore, INM-901-treated AD groups achieved a statistically significant improvement in certain behavior criteria in comparison to the placebo-treated AD groups. These results not only supported but, in several instances, improved upon the prior short-term pilot study outcomes. |
|
| ● | August 20, 2024 — we announced the confirmation of INM-901 as an oral formulation that will be utilized in its development programs for Alzheimer’s disease. Recent preclinical studies have demonstrated that INM-901can be administered orally and achieve therapeutic levels in the brain comparable to those obtained through intraperitoneal (“IP”) injection, which is a common route of administration for preclinical investigation of neurodegenerative diseases. The data indicates the INM-901 formulation can be administered orally and maintains a similar drug exposure levels as IP delivery over a 24-hour period in the brain. This oral delivery method offers potential advantages such as reduction in treatment delivery costs versus intravenous delivery of current disease modifying large molecule antibody therapies. |
INM-089 for the Treatment of AMD
Introduction
While conducting the preclinical studies of a previous drug candidate, INM-088 in glaucoma, which involved comparing various naturally occurring compounds including InMed’s proprietary small molecule candidates, it was discovered that one of InMed’s candidates was demonstrating interesting pharmacological effects in the back of the eye. Further preclinical studies of this compound using AMD study models demonstrated significant functional and pathological improvements. InMed has selected drug candidate INM-089, a proprietary small molecule analog of INM-088, for further preclinical development in the potential treatment of dry AMD.
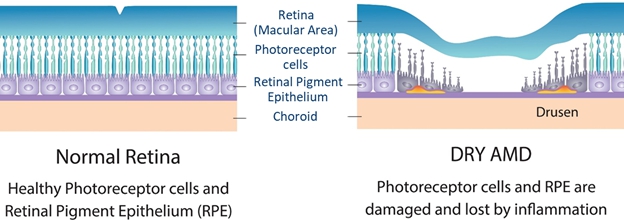
AMD is a progressive eye disease that causes damage to the macula which affects a person’s central vision. AMD is common amongst the elderly and is a leading cause of vision loss. Dry AMD is the most common form of AMD, accounting for 80% of AMD cases according to the American Academy of Ophthalmology.
Until recently, there were no approved pharmaceutical treatments for people with dry age-related macular degeneration. In 2023, the FDA approved two new treatments which are complement inhibitors for advanced stages of dry AMD (called geographic atrophy (“GA”)).
In vitro and in vivo studies of INM-089 have demonstrated neuroprotection of photoreceptors, improvement of the integrity of the retinal pigment epithelium, a reduction in extracellular autofluorescent deposits (a hallmark of AMD), preservation of the retinal function in the back of the eye and improvement in the thickness of the outer nuclear layer of the retina.
As a small molecule, INM-089 is likely deliverable via various modes of administration, such as a topical eye drop or intravitreal injection (“IVT”) formulation.
Pathology of Age-related Macular Degeneration
AMD is a progressive eye disease that causes damage to the macula which is part of the retina at the back of the eye. The macula controls the sharp vision straight ahead of you, and damage to the macular affects a person’s central vision.
There are two principal forms of AMD, atrophic (non-exudative) dry AMD and neovascular (exudative) wet AMD. Wet AMD constitutes about 10%-20% of all cases of AMD and occurs when an abnormal blood vessel grows in or under the retina leading to central vision loss. Dry AMD is the most common form affecting nearly 80%-90% of all patients with AMD. It is associated with the gradual loss of the outer nuclear layer (ONL) photoreceptors and the retinal pigment epithelium (RPE) thinning, formation of drusen deposits, and loss of the vessels in the retinal choriocapillaris. Advanced stage of dry AMD is characterized by geographic atrophy (GA) at the center of the macula extending through the outer neuroretina, RPE and choroid. GA is characterized by the atrophy of RPE, photoreceptors, choriocapillaris, and ONL. The loss of functional RPE and photoreceptors in GA is not endogenously replaced and can result in complete sight loss.
AMD is a leading cause of vision loss in adults
According to the World Health Organization, 196 million people worldwide live with age-related macular degeneration. An estimated 19.8M Americans aged 40+, about 12.6% of the population, suffer from AMD. While AMD does not cause complete vision loss, it affects central vision and impairs one’s ability to perform daily tasks such as cooking, reading and driving.
As the name suggests, aging is a strong risk factor for developing AMD. Adults aged 50 or older, smoke, have a diet of high saturated fat, have cardiovascular disease or have a family history of AMD are more at risk of developing AMD. People of European ancestry are more likely to develop AMD than Blacks, Hispanics or Asians. In addition, people with blue eyes have higher incidence rates of AMD than someone with brown eyes.
Early detection is key to slowing the progression of AMD. A sign of whether you might have AMD is when straight lines look wavy.
A Major Unmet Medical Need for New AMD Treatments
Until recently, there were no approved pharmaceutical treatments for people with dry age-related macular degeneration, which accounts for about 80%-90% of AMD cases.
In 2023, the FDA approved two new treatments which are complement inhibitors for advanced stages of dry AMD (or geographic atrophy). These complement inhibitors are injected directly into the eye every one to two months.
Syforvre® (pegcetacoplan), developed by Apellis Pharmaceuticals, was approved by the FDA in February of 2023 for the treatment of geography atrophy, the late stage of dry AMD. Syforvre®, a C3 complement inhibitor, is injected into each eye every 25-60 days and reduces the rate of lesion growth in the eye.
In August 2023, the FDA approved Iveric’s Izervay® (avacincaptad pegol), a complement C5 inhibitor, which aims to reduce an immune response that damages retinal cells. Similar to Syforvre®, the drug is approved for geographic atrophy and is administered via intravitreal injection every month. According to the Alzheimer’s Association, these new complement inhibitor drugs slow the development of GA by about 14%-20%, but do not improve eyesight, nor restore lost vision. Side effects of these new treatments include inflammation, bleeding beneath the clear lining of the eye, blurred vision and fluid pressure, and some patients develop wet AMD.
In addition to the new complement inhibitors, there are surgical implants and ongoing clinical drug trials. An ophthalmologist may recommend specific vitamins to slow the progression of AMD in its intermediate stage.
The approval of complement inhibitors offers hope to people living with dry AMD, however, the modest effect, inconvenient drug delivery and the increased risk of developing wet AMD may outweigh the benefit for some patients and their physicians. There remains a large unmet medical need for more effective and convenient treatments for the large patient population affected by dry AMD.
Treatments approved or in late-stage development for Geographic Atrophy (late-stage dry AMD)
| Brand | Company | Mechanism of Action | Status | |||
| Syforvre® | Apellis Pharmaceuticals | Complement C3 inhibitor | Approved February 2023 | |||
| Izervay® | Iveric | Complement C5 inhibitor | Approved August 2023 | |||
| Tinlarebant | Belite Bio | Targets retinol binding protein 4 (RBP4) | Phase 3 | |||
| ANX007 | Annexon Biosciences | Anti-C1q antibody | Initiating Phase 3 | |||
| JNJ-1887/HMR59 | Hemmera/ Janssen | Increase expression of soluble form of CD59 | Phase 2 | |||
| IONIS-FB-LRx / RG6299 | Ionis /Roche | Anti-sense complement factor B inhibitor | Phase 2 | |||
| Danicopan (ALXN2040) | Alexion | Complement Factor D inhibitor | Phase 2 |
Role of CB1 and CB2 agonists in ocular disease
Mounting scientific research is pointing to the neuroprotective effects of CB1 and CB2 agonists, supporting their therapeutic potential in ocular diseases such as AMD and glaucoma, in which neuroprotection is key to preserving the nerve cells in the eyes and potentially slowing or reversing eye damage. Several preclinical studies conducted by InMed in three of its drug development programs have consistently shown the neuroprotective effects of naturally occurring CB1 and CB2 agonists and their analogs in well-recognized study models.
In was during this research of INM-088 when InMed scientists observed the ability of a novel CB1 and CB2 agonist, now called INM-089, to proactively protect the nerve cells in the back of the eye. As a result of this discovery, InMed launched the INM-089 drug development program for the potential treatment of AMD.
INM-089: Small molecule compound acting as a selective dual CB1 / CB2 agonist
CB1 and CB2 receptors are both part of the endocannabinoid system and are found throughout the body and are responsible for many homeostatic functions. CB1 receptors are primarily located in the brain and central nervous system, while CB2 receptors are involved in modulating neuroinflammation and immune responses. Activation of CB1 and CB2 receptors has been shown to have neuroprotective effects and protect cells from damage and death.
INM-089 is a small molecule compound, one of several proprietary CB1and CB2 agonists discovered and developed by the Company’s team of scientists.
INM-089 in vitro and in vivo studies to date
Preclinical studies of INM-089 demonstrate significant functional and pathological improvements in an AMD disease study model. Results from several in vitro and in vivo studies demonstrate INM-089’s pharmacological effects in the potential treatment of dry AMD:
| ● | INM-089 provides neuroprotection of retinal cells |
| ● | INM-089 improves the integrity of the retinal pigment epithelium (“RPE”) |
| ● | INM-089 reduces extracellular autofluorescent (“AF”) deposits, including drusen, a hallmark of dry AMD |
| ● | INM-089 preserves photoreceptor function and retinal cells in the back of the eye |
| ● | INM-089 improves thickness of outer nuclear layer (“ONL”) of the retina where photoreceptors are located. |
Based on widely accepted ocular research, the thickness of the outer nuclear layer is strongly correlated with photoreceptor preservation and visual acuity.
INM-089 Study: Neuroprotective effects
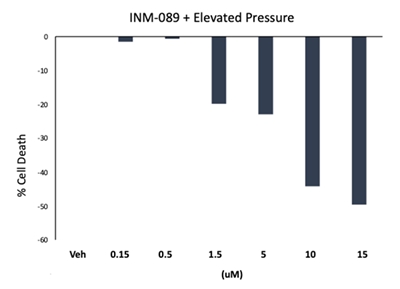
INM-089 demonstrates neuroprotective effects in pressure-induced toxicity in vitro model in retinal ganglion cells in a dose-dependent manner.
INM-089 Study: Photoreceptor Function Preservation
In a light-induced toxicity in vivo AMD model, a single intravitreal injection was performed at the back of the eye to deliver either INM-089, INM-088 or vehicle control. INM-089 outperforms INM-088 (‘CBN’ in the graph below) and vehicle control in preserving photo-receptor function.

INM-089 Study: Autofluorescent Extracellular Deposit Level
In a light-induced toxicity in vivo AMD model, a single intravitreal injection was performed at the back of the eye to deliver either INM-089 or vehicle control. INM-089 reduced build-up of autofluorescent extracellular deposits, which causes damage to the macular. The build-up of autofluorescent deposits such as drusen is a hallmark of dry AMD.

INM-089 Next Steps
Research & Development
| ● | Additional Protein and RNA analysis results are pending |
| ● | Continuing CMC activities for drug substance and drug product |
| ● | On-going studies of receptor interactions (MoA) and DMPK |
| ● | GLP studies to follow |
INM-755 for the Treatment of Epidermolysis Bullosa (‘EB’)
Introduction
INM-755 (cannabinol, or ‘CBN’) cream is a proprietary, topical product candidate intended as a therapy in dermatological diseases. The first clinical indication under development is treatment of symptoms related to EB. EB is a collective name for a group of genetic disorders of connective tissues characterized by skin fragility leading to extensive blistering and wounding. It affects skin and mucous membranes, particularly of the gastrointestinal tract, genitourinary and respiratory systems. EB is a debilitating disease affecting a small proportion of people in the United States, thus earning it an orphan-disease status. The disease has no definitive cure, and all current treatments are directed towards symptom relief. There are, however, a number of products, mainly gene therapies, currently in clinical trials, in which a cure is being explored, according to several recent scientific publications. Our preclinical research has identified a specific Product Candidate, CBN, that may prove beneficial to patients: first, by addressing certain key disease hallmarks (which may include wound healing, infection, pain, inflammation, and itch); and second, by regulating the expression of various proteins (keratins) that may compensate for reduced expression of others.
The active ingredient in INM-755, CBN, is an agonist for both CB 1 and CB2 receptors, with a higher affinity for CB2, which means it should have a greater effect on the immune system than on the central nervous system. The distribution of CB1 and CB2 receptors in sensory nerves and inflammatory cells in the skin make it an attractive pharmaceutical agent for dermal treatments in medical conditions characterized by inflammation and pain.
Summary of Completed Clinical Trials
Phase 1 Clinical Trials (Studies 755-101-HV and 755-102-HV)
A regulatory application to support our first Phase 1 clinical trial in healthy volunteers with INM-755 (755-101-HV) was submitted November 4, 2019 and approved December 6, 2019 in the Netherlands. The initial Phase 1 clinical trial evaluated the safety, tolerability, and pharmacokinetics of INM-755 cream in 22 healthy volunteers with normal, intact skin; the volunteers had cream applied once daily for a period of 14 days. All subjects in this first clinical trial completed treatment and evaluations by March 27, 2020. Database completion and data analyses were delayed by pandemic restrictions. Study results were reported November 25, 2020. A blinded interim safety review from the first 16 subjects in this Phase 1 clinical trial were included in a regulatory application that was approved April 17, 2020, for a second Phase 1 clinical trial of 8 healthy volunteers to test the local safety and tolerability of applying sterile INM-755 cream to small wounds once daily for 14 days. As with the initial Phase 1 trial, the second clinical trial (755-102-HV) was conducted with two different drug concentrations and a vehicle control. Enrollment began in early July 2020 and the clinical trial completed treatment and evaluations at the end of September 2020. Study results were reported January 8, 2021.
Phase 2 Clinical Trial (Study 755-201-EB)
Regulatory applications to support this global trial were filed for review by the National Competent Authorities and Ethics Committees in 8 countries for 13 clinical sites. Approvals were obtained in all countries (Austria, France, Germany, Greece, Israel, Italy, Serbia, and Spain) as of March 2022. Enrollment and patient treatment began in December 2021 and completed in April 2023.
The goal of the Phase 2 study was to obtain safety and preliminary efficacy of INM-755 cream in treating symptoms and wound healing in patients with EB, using a within-patient design in which matched index areas were randomized to INM-755 cream or vehicle (no drug) cream in a blinded manner. A target of up to 20 patients were to be enrolled with treatment for 28 days, the longest period supported by nonclinical toxicology studies.
No single primary endpoint was set for the trial to allow for possible variations in presenting symptoms in each patient. These include the presence of open wounds, wound pain associated with dressing changes, background wound pain, wound itch, and itch in non-wound areas. To this end, InMed’s goal was to harvest data from the trial to evaluate the ability of INM-755 to treat chronic non-wound itch and to heal wounds and treat associated pain and itch.
The Phase 2 Trial enrolled a total of 19 patients. Data from one patient were excluded from efficacy analyses due to a significant protocol deviation. Of the 18 remaining patients whose data were considered reliable for clinical review, 17 were treated for chronic non-wound itch and one patient was treated for wound-related itch. The remaining endpoints (pain, wound healing) could not be analyzed due to too few enrollees with such symptoms.
Of the 18 participants assessed, chronic itch improved by a clinically meaningful amount in 12 patients (66.7%), of whom:
| ● | 6 patients (33.3%) had the same level of itch improvement with INM-755 cream as with control cream; |
| ● | 5 patients (27.8%) treated with INM-755 showed meaningful anti-itch activity beyond that of the control cream; and |
| ● | 1 patient (5.6%) showed better itch reduction with the control cream. |
In summary, results from the Phase 2 clinical trial showed a positive indication of enhanced anti-itch activity for INM-755 cream versus the control cream alone in an exploratory clinical evaluation. The results for non-wound itch were not statistically significant in this small trial due, in part, to the clinically important anti-itch effect of the underlying control cream. We are, nevertheless, encouraged by and satisfied with the INM-755 clinical data for non-wound itch treatment. That the majority of the assessed patients in the trial showed clinically meaningful improvement in non-wound itch from the application of INM-755, be it with similar outcomes to the control cream or better than the control cream, can be considered impressive.
Based on the safety and efficacy data for treating non-wound itch in this EB study, as well as previous safety data from Phase 1 trials, we are now seeking R&D and commercial partnership opportunities for any continued development of INM-755 cream. Continued development of INM-755 cream will likely move beyond EB into broader indications involving chronic itch, with potentially much larger target populations and commercial opportunities than offered solely by the EB indication.
On average, it takes at least ten years to complete the development of an investigational drug from its initial discovery to the marketplace, with clinical trials alone taking six to seven years on average. It is not possible with any degree of certainty to estimate how long it will take to complete clinical trials and potentially obtain marketing approval for INM-755. To the extent that INM-755 may potentially be designated as either a Fast Track drug, a Breakthrough Therapy, or eligible for Priority/Accelerated Review, the timeline to any potential marketing approval may be shorter than might otherwise be the case.
Additional Indications for INM-755
Once a company has gone to the significant investments of bringing a new chemical entity into human clinical trials, the traditional approach is to investigate as many therapeutic uses as possible of that product in different indications, or specific diseases. We intend to pursue this strategy with a co-development partner as a way to leverage our knowledge of CBN and investment in the development of INM-755 as a topical skin cream. Under the assumption that we would use the same formulation for other dermatological indications, there should be no need for further Phase 1 safety studies allowing us to proceed directly to Phase 2 safety and preliminary efficacy studies in humans, since the toxicology and initial human safety studies have been completed; however, the adequacy of the nonclinical and human safety data to support new dermatologic indications will be determined by the appropriate health authority.
Key Milestones for the EB Program:
| ● | April 30, 2020 — we announced clinical trial application approval in the Netherlands for Study 755-102-HV, a randomized, double-blind, vehicle-controlled Phase 1 study designed to evaluate the safety and tolerability of INM-755 (two strengths) applied daily for 14 days on epidermal wounds in 8 healthy volunteers. |
| ● | November 25, 2020 — we announced the top-line results of Study 755-101-HV (“Study 101”). Study 101 was a randomized, vehicle-controlled, double-blind, Phase 1 trial, which examined the safety and tolerability of two strengths of INM-755 cream on intact skin in 22 healthy adult volunteers over a 14-day treatment period. The Study 101 results indicate that INM-755 was safe and well-tolerated on intact skin, caused no systemic or serious adverse effects. In addition, there were no subject withdrawals due to adverse events. Drug concentrations in the blood were very low, as expected. |
| ● | January 8, 2021 — we announced the top-line results of Study 755-102-HV (“Study 102”). Study 102 was a randomized, double-blind, vehicle controlled, single-center study, in 8 healthy adult volunteers to test the tolerability of 14 days of application of the INM-755 cream on epidermal wounds under treatment procedures designed to simulate wound care for Epidermolysis Bullosa (“EB”) patients with open wounds. Results of Study 102 indicate that INM-755 cream was safe and well-tolerated on induced open epidermal wounds, caused no systemic or serious adverse effects. In addition, there were no subject withdrawals due to adverse events. These data from Study 101 and Study 102 support moving forward into clinical trials in patients with EB. |
| ● | April 28, 2021 — we announced that we filed Clinical Trial Applications (“CTAs”) in Austria, Israel and Serbia as part of a Phase 2 clinical trial of INM-755 (cannabinol) cream in EB. Additional CTAs for 755-201-EB (the ‘201 study) will be submitted to National Competent Authorities (“NCAs”) and Ethics Committees (“ECs”) in France, Germany, Greece, and Italy in the coming weeks. |
| ● | September 30, 2021 — we announced commencement of a Phase 2 clinical trial, the 755-201-EB study, of INM-755 (cannabinol) cream in the treatment of EB, marking the first time cannabinol has advanced to a Phase 2 clinical trial to be studied as a therapeutic option to treat a disease. The 755-201-EB study is designed to enroll up to 20 patients. InMed will evaluate the safety of INM-755 (cannabinol) cream and its preliminary efficacy in treating symptoms and wound healing over a 28-day treatment period. All four subtypes of inherited EB; EB Simplex, Dystrophic EB, Junctional EB, and Kindler Syndrome are eligible for this study. | |
| ● | July 25, 2022 — we announced, based on the safety data of the first five adult patients who completed treatment with INM-755 CBN cream for the treatment of symptoms in the Phase 2 clinical trial, an independent Data Monitoring Committee agreed it was safe to allow the enrollment of adolescent patients, defined as persons aged twelve to seventeen. |
| ● | March 28, 2023 — we announced we had concluded enrollment of our Phase 2 clinical trial using investigational drug INM-755 cannabinol (“CBN”) cream for the treatment of patients with EB. The Phase 2 study enrolled 19 of its targeted 20 patients. |
| ● | June 22, 2023 — we announced safety and efficacy results from the Phase 2 clinical trial (755-201-EB) for the treatment of symptoms in patients with EB. |
Rare Cannabinoid Products in the Health and Wellness Sector
BayMedica has a revenue-generating commercial business unit that leverages our significant expertise in synthetic biology and chemistry to develop efficient, scalable, and proprietary manufacturing approaches to produce high quality, regulatory-compliant, non-intoxicating rare cannabinoids (“Products”) for consumer applications. BayMedica is currently commercializing Products as a B2B supplier to distributors and manufacturers in the health and wellness sector, including nutraceuticals, cosmetics, functional foods and beverages, as well as animal health markets. BayMedica currently has a robust portfolio of four different non-intoxicating Products including: cannabichromene (“CBC”), cannabidivarin (“CBDV”) tetrahydrocannabivarin (“THCV”) and cannabicitran (“CBT”).
Following the acquisition of BayMedica in 2021, a key priority in 2022 was accelerating commercial activities and building out a robust product portfolio as a supplier of Products to the health and wellness sector. While there was slower than expected revenue growth in 2022, we have seen increased demand through 2023 and the first half of 2024 due to better research of rare cannabinoids, companies and brands looking for product innovation and effects-based outcomes, and the ability of companies like ours to reliably supply high quality and consistent Products. During the financial year ended June 30, 2024, BayMedica had sales of approximately $4.6 million.
The increased sales in part resulted from expanded marketing efforts and increased demand for certain Products. BayMedica will continue to evaluate opportunities for potential structured supply arrangements and collaborations for the commercial business. Sales and marketing efforts will remain focused on Products that contribute highest margins where BayMedica continues to hold a strong competitive position.
Chemical Synthesis-Derived Cannabinoids Commercialized by BayMedica
Cannabichromene (CBC)
The high cost of goods for rare / minor cannabinoids (e.g. CBC) extracted and purified from the plant made adoption of these more difficult due to the cost of manufacture and cost to include in the final marketed product(s). Chemical synthesis has provided a consistent, scalable approach to produce CBC and other minor cannabinoids at a highly competitive cost of goods with consistent batch to batch variability and high purity of the cannabinoid. BayMedica has successfully manufactured and commercialized the rare cannabinoid CBC, for sale to distributors into the health and wellness industry. The development of a scalable process for the manufacturing of CBC began in 2018 using well established chemical synthesis protocols.
In 2019, a Material Services Agreement was completed with a multinational contract research, development and manufacturing organization (“Chemistry CDMO”) to facilitate the optimization and scale-up of BayMedica’s proprietary CBC manufacturing process using commercially available starting materials sourced from various manufacturers. We scaled to a batch size of greater than 1kg by calendar 2019 at which time we contracted a leading U.S. manufacturer to provide the final purification of CBC to greater than 95% purity. This manufacturer also operates a North American based toll-processing facility with the capability to process from 10kg to metric ton quantities of our crude CBC material under food-grade GMP conditions. By late 2019, our Chemistry CDMO had scaled the process to greater than 10kg, and by year end 2019 to almost 30kg with final purification at the NA contractor. We commenced commercial sales of CBC in November 2019.
Large scale manufacturing of crude CBC began at our Chemistry CDMO in 2020 at >40kg. The emergence of the Covid-19 pandemic significantly impacted sales beginning in calendar 2020. Large scale production continues with current batch sizes exceeding 100kg.
Cannabicitran (CBT)
We have developed a process for the efficient chemical synthesis of CBT through both in-house R&D efforts and via our CDMO. We began scaling this process and conducted downstream processing and purification trials in late calendar 2021. We received initial purchase orders and commenced commercial sales of CBT in calendar 2022.
Cannabidivarin (CBDV)
Beginning in early 2021, BayMedica worked internally and with external parties to access and develop manufacturing technologies for the chemical synthesis of the rare, non-intoxicating “varin” cannabinoid, CBDV. In calendar 2021, via a Chemistry CDMO, we successfully scaled CBDV synthesis to commercial quantities. In April 2022, we commenced B2B sales of CBDV to the health and wellness sector. As of fiscal 2023, a new source of CBDV was developed to provide a lower cost of goods replacement to the previously produced CBDV. Sales of this CBDV were initiated in the second quarter of calendar 2024.
Tetrahydrocannabivarin (THCV)
As part of the R&D into manufacturing techniques to synthesize and produce CBDV, we also began researching and developing processes to convert CBDV to the non-intoxicating rare cannabinoid THCV. In conjunction with our Chemistry CDMO and our in-house team, we developed a robust pilot-scale process that produces THCV. We have now developed a purification process to produce the finished THCV material. We began scale-up of our novel process with this CDMO in the first quarter of calendar 2022 and commenced sales in second half of calendar 2022. A new, more cost-effective, manufacturing approach for the supply of THCV was initiated in calendar 2023 with initial sales in the third quarter of calendar 2023. We will continue to assess further initiatives to reduce cost of goods for THCV.
Analogs of Cannabinoids / New Chemical Entities (“NCE”)
In addition to the natural cannabinoids above, we have leveraged our expertise in pharmaceutical chemistry and biosynthesis to produce a number of novel cannabinoid analogs and variants of pharmaceutical interest.
In the field of pharmaceutical drug development, the term analog is used to describe structural and functional similarity between an original (or parent) molecule and one that has been somewhat modified. While any company researching a naturally occurring compound, like cannabinoids, cannot own a patent on the molecule itself for commercial exclusivity, a modified molecule, which has certain structural and pharmacological similarities with the original compound, can be patented. As well, modifications of the original molecule (ie, the analog) can be designed to confer certain improvement in activity of the parent, such as an elevation of the desired physiological effects, a decrease in unwanted side effects, improvement in aspects related to drug delivery to targeted tissues, etc., or a combination of these targeted outcomes. We have filed patents covering numerous structural additions and modifications of the naturally occurring cannabinoids. Each individual modification to each individual cannabinoid represents a NCE which can be patented. If issued, this patent family will confer market exclusivity to us for the analogs that we intend to develop into pharmaceutical Product Candidates, license, partner or sell to interested external parties.
Notable Milestones:
| ● | On May 21, 2015 — we commenced the development of our biosynthesis process for the manufacturing of cannabinoids through a research collaboration with Dr. Vikramaditya Yadav from the Department of Biological and Chemical Engineering at the University of British Columbia under a project titled “The Metabolic Engineering of yeast and bacteria for synthesis of cannabinoids and Cannabis derived terpenoids”. On May 31, 2017, we signed a Technology Assignment Agreement with the University of British Columbia whereby we retain sole worldwide rights to all patents emergent from the technology under development in exchange for a royalty of less than 1% on sales revenues from products utilizing cannabinoids manufactured using the technology (the “1% royalty”) and a single digit royalty on sub-licensing revenues. On May 15, 2018, we extended our Collaborative Research Agreement with the University of British Columbia for an additional three years, which expired in 2021. Other than the 1% royalty, we do not have any ongoing financial commitments under these arrangements with the University of British Columbia. |
| ● | February 2019 — we entered into a separate process development collaboration by way of a Master Service Agreement with the Almac Group (UK)(“Almac”), a seasoned GMP pharmaceutical CDMO. Almac was initially tasked to develop a down-stream purification process to support the fermentation optimization activities at the National Research Council of Canada. In addition, we also engaged Almac to assist in the development of an “alternative” manufacturing process for cannabinoids which integrates the best available technologies across the spectrum of pharmaceutical drug production. This process is now referred to as IntegraSyn. In May 2020, we announced our working relationship with Almac on an integrated approach to augment current biosynthesis-based methods for cannabinoid production. The companies have been engaged in developing a streamlined cannabinoid manufacturing process, specifically optimizing the upstream cannabinoid assembly processes as well as downstream purification processes, to achieve cost-efficient, GMP-grade active pharmaceutical ingredients for prescription-based cannabinoid medications. Almac is an international, privately-owned organization which has grown organically over the past five decades now employing over 5,600 highly skilled personnel across 18 facilities including Europe, the United States and Asia. We retain all rights to this new process while Almac retains certain rights-of-first refusal on the production and supply of certain precursors, or starting materials, for this alternative process. |
Other Milestones Include:
| ● | September 2020 — we announced the filing of a PCT patent application as part of a growing portfolio of intellectual property related to the IntegraSyn manufacturing approach for producing low-cost, pharmaceutical-grade cannabinoids (refer to “Intellectual Property”, immediately below). |
| ● | April 2021 — we announced that the IntegraSyn cannabinoid manufacturing approach has achieved a level of 2g/L cannabinoid yield, a milestone that signals commercial viability and supports advancement to large-scale production in the coming months. Having achieved a 2g/L yield level, we will now focus on manufacturing scale-up to larger batch sizes while continuing process and enzyme optimization, targeting increased cannabinoid yield and further reducing the overall cost of goods. In parallel, we continue to prepare the manufacturing process to be Good Manufacturing Practice (“GMP”)-ready for pharmaceutical quality production. |
Intellectual Property
A patent is a monopoly granted by a government for a period of up to 20 years. A patent provides an enforceable legal right to prevent others from exploiting an invention being a product, device, system, substance, process or method in the country of grant. For an invention to be patentable, it must be novel, involve an inventive step and useful at the time of filing the initial patent application for that invention. At 18 months from the initial patent application, the detailed description of the invention is published. In order to secure patent protection, a patent application is filed with the patent office in each country of interest, the application is considered under the patent laws of that country, and a patent will issue if the application meets the patentability criteria of that country. After a patent expires or lapses, anyone can then use the invention.
The grant of a patent does not guarantee validity, and a patent may be challenged by third parties at a patent office by re-examination in some countries or through the courts by revocation proceedings. The grant of a valid patent does not mean that the invention may be exploited in a given country without infringing third party intellectual property rights in that country.
The owner of a patent has the exclusive right to prevent others from making, selling, importing or otherwise using the patented invention for the life of the patent. Patent infringement occurs when someone makes, hires, uses, imports or sells the patented invention, or a product made by a patented method, or offers to do these things, within the country covered by the patent without the permission of the owner of the patent.
Adequate protection of intellectual property is a means to ensure that we can commercialize our intellectual property and reduce the likelihood of imitation by competitors. We intend to utilize patents available to protect our IP wherever commercially realizable. In addition, we also rely on trade secrets and process know-how to protect our intellectual property. While we cannot patent the naturally occurring individual cannabinoids used in our Products and Product Candidates, there are a number of other approaches to protect our inventions. These include:
| ● | patents on individual or combinations of cannabinoids that provide novel methods for treating diseases; |
| ● | cannabinoid delivery technology, formulations designed specifically to increase the safety and efficacy of drug treatments; and |
| ● | manufacturing processes for cannabinoids. |
The patent methodologies listed above will be designed with the intention to maximize the protection of our multi-faceted approach to developing novel cannabinoid medicines. We typically file patent applications in US, Canada, EU and other selected commercially significant foreign jurisdictions.
InMed Patent Portfolio
| Subject Matter | Scope | Ownership/ Origin | Filing Status / Filing Date |
Patent Reference Number – Patent Nos. |
Earliest Patent |
Jurisdictions - Status | ||||||
| Metabolic engineering of E. coli for the biosynthesis of cannabinoid products | Manufacturing Process | InMed, UBC1 | PCT Application filed 09/05/2018 |
WO2019/046941 US (12,077,802) |
2038 | Pending: AU, CA, EP, JP, US (patent will issue on 9/3/24) |
||||||
| Compositions and methods for biosynthesis of terpenoids or cannabinoids in a heterologous system | Manufacturing Process | InMed, UBC1 | PCT Application filed 3/6/2020 |
WO2020/176998 | 2040 | Pending: AU, CA, EP, JP, SG, US | ||||||
| Ocular drug delivery formulation (Hydrogel) | Formulation, Use | InMed | PCT Application filed 05/08/2018 |
WO2018/205022 AU 2018266262 EP 3621656 IN 426267 JP 7323458 US 12083229 |
2038 | Granted: US, AU, EP, IN, JP Pending: CA, SG, |
||||||
| Compositions and methods for use of cannabinoids for neuroprotection | Use | InMed | PCT Application filed 04/24/2020 |
WO2020/215164 | 2040 | Pending: AU, CA, CN, EP, IL, JP, MX, SG, US, ZA | ||||||
| Topical formulations of cannabinoids and use thereof in the treatment of pain | Formulation, Use | InMed | PCT Application filed 09/21/2018 |
WO2019/056123 |
2038 | Pending: EP, US | ||||||
| Use of topical formulations of cannabinoids in the treatment of epidermolysis bullosa and related connective tissue disorders | Use | InMed | PCT Application filed 05/04/2017 |
WO2017/190249 AU 2017260706 IL 262702 JP 7054691 JP 7342183 US (12042479) |
2037 | Granted: AU, IL, JP, JP, US Pending: CA, EP, |
||||||
| Compositions and methods for treating neuronal disorders with cannabinoids | Use | InMed | PCT Application filed 10/21/2022 |
WO 2022/082313 |
2041 | Pending: AU, CA, CN, EP, IL, JP, MX, US | ||||||
| Compositions and Methods for Use of Cannabinol Compounds in Neuroprotection | New Chemical Entity, Composition, Use | InMed | PCT Application filed 5/8/2024 |
63/545361 |
2043 | Pending: PCT (National stage application filings due April/May 2026) |
PCT = Patent Cooperation Treaty. Members in this treaty includes over 150 countries including USA, Canada, Europe and others.
Patents typically expire 20 years from their filing dates, if granted, the patent expiry may be extended by patent agencies and/or health regulatory authorities.
| 1 | UBC is a co-inventor and has assigned all commercial rights to InMed in exchange for a royalty of less than 1% on sales revenues from products utilizing cannabinoids manufactured using the technology and a single digit royalty on any sub-licensing revenues. |
| BayMedica Patent Portfolio |
| Subject Matter | Scope | Ownership/ Origin | Filing Status / Filing Date |
Patent Reference Number |
Earliest Potential/ Patent |
Jurisdictions | ||||||
| Recombinant production systems for prenylated polyketides of the cannabinoid family | Manufacturing Process | BayMedica | PCT Application filed 05/10/2018 |
WO2018/209143 US 10837031 US11555211 MX 411852 |
2038 | Granted: US, US, MX Pending: AU, CA, CN, EP, IN, |
||||||
| Cannabinoid analogs and methods for their preparation | New Chemical Entity; Manufacturing Process | BayMedica | PCT Application filed 10/31/2019 |
WO2020/092823 |
2039 | Pending: AU, CA, CN, EP, IL, IN, IN, JP, MX, US | ||||||
| Preparation of cannabichromene and related cannabinoids | Manufacturing Process | BayMedica | PCT Application filed 12/23/2020 |
WO2021/133989 |
2040 | Pending: CA, CN, EP, IN, JP, US | ||||||
| Genetically modified yeast for the production of cannabigerolic acid, cannabichromenic acid and related cannabinoids | Manufacturing Process | BayMedica | PCT Application filed 01/20/2021 |
WO2021/150636 |
2041 | Pending: CA, CN, EP, IN, JP, US | ||||||
| Acyl activating enzymes for preparation of cannabinoids | Manufacturing Process | BayMedica | PCT Application filed 01/20/2022 |
WO2022/159589 |
2042 | Pending: CA, EP, IN, JP, US |
PCT = Patent Cooperation Treaty. Members in this treaty includes over 150 countries including USA, Canada, Europe and others.
Patents typically expire 20 years from their filing dates, if granted, the patent expiry may be extended by patent agencies and/or health regulatory authorities.
| 1 | UBC is a co-inventor and has assigned all commercial rights to InMed in exchange for a royalty of less than 1% on sales revenues from products utilizing cannabinoids manufactured using the technology and a single digit royalty on any sub-licensing revenues. |
| 2 | Patents typically expire 20 years from their filing dates, if granted, the patent expiry may be extended by patent agencies and/or health regulatory authorities. |
PCT = Patent Cooperation Treaty. Members in this treaty includes over 150 countries including USA, Canada, Europe and others.
As of August 2024, we have a total of thirteen patent families covering the following areas:
| ● | Four patent families covering novel methods for treating diseases including two for our INM-755 program (WO/2017/190249 and WO/2019/056123), one for our INM-088 program (WO/2020/215164) and one for treating neurodegenerative diseases (WO/2022/082313). If these patents applications are granted and all maintenance fees or annuities are paid, these patents are expected to expire in 2037-2042. In some situations, the patent may be eligible for adjustment or extension of the patent terms due to delay in the patent office during the prosecution phase. The expiration date above does not include the adjustments or extensions; |
| ● | Eight patent families covering manufacturing process for cannabinoids of interest (WO2019/046941, WO2020/176998, WO2018/209143, WO2020/092823, WO2020/102430, WO2021/133989, WO2021/150636 and WO2022/159589). If these patents applications are granted and all maintenance fees or annuities are paid, these patents are expected to expire in 2038-2042. In some situations, the patent may be eligible for adjustment or extension of the patent terms due to delay in the patent office during the prosecution phase. The expiration date above does not include the adjustments or extensions; and |
| ● | One patent family covering a delivery technology for the ocular program (WO/2018/205022). |
If these patents applications are granted and all maintenance fees or annuities are paid, these patents are expected to expire in 2038. In some situations, the patent may be eligible for adjustment or extension of the patent terms due to delay in the patent office during the prosecution phase. The expiration date above does not include the adjustments or extensions.
The Patent Cooperation Treaty (“PCT”), is an international patent law treaty, which provides a unified procedure for filing patent applications to protect inventions in each of its member states. There are 151 member countries within the PCT, enabling near-global patent coverage through successful patent prosecution in the U.S., Japan, Europe, Canada, Australia, New Zealand, China, Brazil, Russia, India and many other countries. We have several filed patent applications currently either in the provisional stage or PCT stage of review as shown above. None have been granted to date. We retain the full commercial rights to all of these patents with any exceptions noted in the above table.
Government Regulations
The research, development, testing, manufacture, quality control, packaging, labeling, storage, record-keeping, distribution, import, export, promotion, advertising, marketing, sale, and reimbursement of pharmaceutical products are extensively regulated by governmental authorities in the United States and other jurisdictions. The processes for obtaining regulatory approvals in the United States and in foreign countries and jurisdictions, along with compliance with applicable statutes and regulations and other requirements, both pre-approval and post-approval, require the expenditure of substantial time and financial resources. The regulatory requirements applicable to product development, approval and marketing are subject to change, and regulations and administrative guidance often are revised or reinterpreted by the agencies in ways that may have a significant impact on our business.
Licensure and Regulation of Biological Products in the United States
In the United States, the FDA regulates human drugs under the Federal Food, Drug, and Cosmetic Act (the “FDCA”) and, in the case of biological products, also under the Public Health Service Act, and their implementing regulations. The failure to comply with the applicable U.S. requirements may result in the FDA’s refusal to approve any pending applications or delays in development and may subject an applicant to administrative or judicial sanctions, such as issuance of warning letters, or the imposition of fines, civil penalties, product recalls, product seizures, total or partial suspension of production or distribution, and injunctions and/or civil or criminal prosecution brought by the FDA and the U.S. Department of Justice or other governmental entities. The FDA must approve all product candidates, including the Product Candidates, for therapeutic indications before they may be marketed in the United States.
Other U.S. Healthcare Laws and Regulations
In the United States, biopharmaceutical manufacturers and their products are subject to extensive regulation at the federal and state level, such as laws intended to prevent fraud and abuse in the healthcare industry. These laws, some of which apply only to approved products, include: (i) federal false claims, false statements, and civil monetary penalties laws prohibiting, among other things, any person from knowingly presenting, or causing to be presented, a false claim for payment of government funds or knowingly making, or causing to be made, a false statement to get a false claim paid; (ii) federal healthcare program anti-kickback law, which prohibits, among other things, persons from offering, soliciting, receiving, or providing remuneration, directly or indirectly, to induce either the referral of an individual for, or the purchasing or ordering of, a good or service for which payment may be made under federal healthcare programs such as Medicare and Medicaid; (iii) the federal Health Insurance Portability and Accountability Act of 1996 (“HIPAA”), which, in addition to privacy protections applicable to healthcare providers and other entities, prohibits executing a scheme to defraud any healthcare benefit program or making false statements relating to healthcare matters; (iv) the FDCA, which among other things, strictly regulates marketing, prohibits manufacturers from marketing such products prior to approval or for off-label use, and regulates the distribution of samples; (v) federal laws that require pharmaceutical manufacturers to report certain calculated product prices to the government or provide certain discounts or rebates to government authorities or private entities, often as a condition of reimbursement under government healthcare programs; (vi) federal transparency law, which requires pharmaceutical companies to report certain payments to healthcare providers; (vii) state laws and regulations analogous to the above; and (viii) laws and regulations prohibiting bribery and corruption such as the FCPA (as defined below), which, among other things, prohibits U.S. companies and their employees and agents from authorizing, promising, offering, or providing, directly or indirectly, corrupt or improper payments or anything else of value to foreign government officials, employees of public international organizations or foreign government-owned or affiliated entities, candidates for foreign public office, and foreign political parties or officials thereof. Violations of these laws are punishable by criminal and/or civil sanctions, including, in some instances, exclusion from participation in federal and state health care programs, such as Medicare and Medicaid. Ensuring compliance is time consuming and costly. Similar healthcare laws and regulations exist in the EU and other jurisdictions, including reporting requirements detailing interactions with and payments to healthcare providers and laws governing the privacy and security of personal information.
U.S. Privacy Law
In the U.S., there are numerous state and federal laws and regulations governing the security and privacy of personal information. Additionally, state and federal regulators have begun to pay more attention to companies’ data processing activities. At the state level, laws require companies to safeguard personal information and take action in the event of a data breach (e.g., notifying governmental authorities and data subjects). State attorney generals have been active in using their consumer protection authority to investigate companies’ data security practices. A number of states have passed laws governing data privacy and many others have similar legislation under consideration. Although many of these laws contain exceptions for certain health data, these exceptions are not comprehensive. All of these laws give rights to residents in their states and require businesses to take certain actions with respect to those rights (similar to the GDPR in effect in the EU, but with notable differences). At the federal level in the United States, the Federal Trade Commission has been active in using its Section 5 authority to bring enforcement actions against companies for deceptive or unreasonable data processing activities.
ITEM 1A. RISK FACTORS
Summary of Risk Factors
The following is a summary of material risks that could affect the Company. This summary may not contain all of our material risks, and it is qualified in its entirety by the more detailed risk factors set forth below.
| ● | Our prospects depend on the success of our Product Candidates, which are in the early stages of development with a statistically high probability of failure and are subject to lengthy, time-consuming and inherently unpredictable regulatory processes. |
| ● | If clinical trials of our Product Candidates fail to demonstrate safety and efficacy to the satisfaction of regulatory authorities or do not otherwise produce positive results, we would incur additional costs or experience delays in completing, or ultimately be unable to complete, the development and commercialization of our Product Candidates. | |
| ● | We intend to expend our limited resources to pursue our Product Candidates for certain indications and may fail to capitalize on other Product Candidates or other indications for our Product Candidates that may be more profitable or for which there is a greater likelihood of success. |
| ● |
Our Product Candidates contain compounds that may be classified as “controlled substances”, the use of which may generate public controversy and restrict their development or commercialization. |
| ● | Any actual or threatened delisting of our securities by Nasdaq due to our inability to satisfy applicable listing standards, including compliance with the minimum bid price rule, could have a material and adverse effect on our business, operations and financial condition, and the liquidity and value of our securities. |
| ● | Research restrictions, product shipment delays or prohibitions could have a material adverse effect on our business, results of operations and financial condition. |
| ● | Our relationships with customers and third-party payors are subject to applicable anti-kickback, fraud and abuse, and other healthcare laws and regulations, which could expose us to, among other things, sanctions, penalties, damages, reputational harm and diminished profits and future earnings. |
| ● | Our insurance may be insufficient to cover losses that may occur as a result of our operations. | |
| ● | There may be changes in laws, regulations and guidelines which are detrimental to our business. | |
| ● | Controlled substance legislation may differ in other jurisdictions and could restrict our ability to market our products internationally, which could materially and adversely affect our financial results. |
| ● | Failure to protect our information technology infrastructure against cyber-based attacks, network security breaches, service interruptions, or data corruption could significantly disrupt our operations and adversely affect our business and operating results. | |
| ● | Our failure to comply with data protection laws and regulations could lead to government enforcement actions and significant penalties against us, and adversely impact our operating results. |
| ● | The market prices for our common shares, no par value (the “Common Shares”), are volatile and are anticipated to fluctuate in the near term. |
| ● | Raising additional capital may cause dilution to our existing shareholders, restrict our operations or require us to relinquish rights to our technologies or Product Candidates. |
| ● | Future offerings of debt or equity securities may rank senior to our Common Shares. |
| ● | For as long as we are an “emerging growth company” we intend to take advantage of reduced disclosure and governance requirements applicable to emerging growth companies, which could result in our Common Shares being less attractive to investors and could make it more difficult for us to raise capital. | |
| ● | If we fail to maintain an effective system of internal control over financial reporting in the future, we may not be able to accurately report our financial condition, results of operations or cash flows, which may adversely affect investor confidence in us and, as a result, the value of our Common Shares. |
| ● | Our disclosure controls and procedures may not prevent or detect all errors or acts of fraud. |
| ● | Deficiencies in disclosure controls and procedures and internal control over financial reporting could result in a material misstatement in our financial statements, and our ability to identify and effectively remediate any such material weaknesses that could have a material and adverse effect. |
| ● | In connection with the audit of our financial statements as of and for the year ended June 30, 2023, a material weakness in our internal control over financial reporting was identified and we may identify additional material weaknesses in the future. |
| ● | Future sales and issuances of, and rights to purchase, our Common Shares, including by officers and directors could materially dilute the percentage ownership of our shareholders and may cause our share price to fall. |
| ● | We (i) have incurred significant losses since our inception and (ii) anticipate we will incur losses in the future, and our operating losses have raised substantial doubt regarding our ability to continue as a going concern | |
| ● | We will require additional capital to fund our operations and if we fail to obtain necessary financing, we will not be able to complete the development and commercialization of our Product Candidates. |
| ● | We currently have limited commercial revenue and may never become profitable. | |
| ● | Our success is largely dependent upon our patents, proprietary technology, and other intellectual property. | |
| ● | Obtaining and maintaining our patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements. | |
| ● | We may become subject to claims or become involved in lawsuits related to intellectual property. |
| ● | We may become involved in lawsuits to protect or enforce our intellectual property, which could be expensive, time consuming and unsuccessful, and materially and adversely effect our business. |
| ● | If we are not able to adequately prevent disclosure of trade secrets and other proprietary information, the value of our technology and products could be significantly diminished. | |
| ● | We may not be able to protect our intellectual property rights throughout the world. | |
| ● |
Patent terms may be inadequate to protect our competitive position on our Product Candidates. |
|
| ● | Intellectual property rights do not necessarily address all potential threats to our competitive advantage. | |
| ● | We rely heavily on contract manufacturers over whom we have limited control and our existing collaboration agreements and any that we may enter into in the future may not be successful. |
Risk Factors
Investing in our Common Shares involves a high degree of risk. Therefore, you should carefully consider each of the following risks, together with all other information set forth in this Annual Report, including the consolidated financial statements and the related notes, before making a decision to buy our Common Shares. If any of the following risks actually occurs, our business could be harmed. In that case, the trading price of our Common Shares could decline, and you may lose all or part of your investment.
Risks Related to our Business and Industry
Our prospects depend on the success of our Product Candidates which are at early stages of development with a statistically high probability of failure.
Given the Company’s early stage of development, we can make no assurance that our research and development programs will result in regulatory approval or commercially viable products. To achieve profitable operations, we, alone or with others, must successfully develop, gain regulatory approval, and market our future products. We currently have no products that have been approved by the FDA, HC, or any similar regulatory authority. To obtain regulatory approvals for our Product Candidates being developed and to achieve commercial success, clinical trials must demonstrate that the Product Candidates are safe for human use and that they demonstrate efficacy. We have no products or technologies which are currently in human clinical trials. Additionally, we have no products for commercial sale or licensed for commercial sale, nor do we expect to have any such products for the next several years.
Many potential pharmaceuticals products never reach the stage of clinical testing and even those that do have only a small chance of successfully completing clinical development and gaining regulatory approval. Our Product Candidates may fail for a number of reasons, including, but not limited to, being unsafe for human use or due to the failure to provide therapeutic benefits equal to or better than the standard of treatment at the time of testing. Positive results of early preclinical research may not be indicative of the results that will be obtained in later stages of preclinical or clinical research. Similarly, positive results from early-stage clinical trials may not be indicative of favorable outcomes in later-stage clinical trials. We can make no assurance that any future studies, if undertaken, will yield favorable results.
The early stage of our product development makes it particularly uncertain whether any of our product development efforts will prove to be successful and meet applicable regulatory requirements, and whether any of our Product Candidates will receive the requisite regulatory approvals, be capable of being manufactured at a reasonable cost or be successfully marketed. If we are successful in developing our current and future Product Candidates into approved products, we will still experience many potential obstacles, such as the need to develop or obtain manufacturing, marketing and distribution capabilities. If we are unable to successfully commercialize any of our products, our financial condition and results of operations may be materially and adversely affected.
Even if our Product Candidates advance through preclinical studies and clinical trials, we may experience difficulties in managing our growth and expanding our operations.
We have limited resources to carry out objectives for our current and future preclinical studies and clinical trials. Since our inception as a pharmaceutical company in October 2014, we have conducted numerous preclinical experiments and are currently conducting early-stage clinical trials, which is a time-consuming, expensive and uncertain process. In addition, while we have experienced management and expect to contract out many of the activities related to conducting these programs, we are a small company with less than 15 employees and, therefore, have limited internal resources both to conduct preclinical studies and clinical trials and to monitor third-party providers. As our Product Candidates advance through preclinical studies and clinical trials, we will need to expand our development, regulatory and manufacturing operations, either by expanding our internal capabilities or contracting with other organizations to provide these capabilities for us. In the future, we expect to have to manage additional relationships with collaborators or partners, suppliers and other organizations. Our ability to manage our operations and future growth will require us to continue to improve our operational, financial and management controls, reporting systems and procedures.
Any actual or threatened delisting of our securities by Nasdaq could have a material and adverse effect on our business, operations and financial condition, and could, among other things, limit investors’ ability to make transactions in our securities and subject us to additional trading restrictions.
As previously reported by the Company, on March 19, 2024, the Company received written notification from the Listing Qualifications Department of Nasdaq that the Company has been granted an additional 180-day compliance period, or until September 16, 2024 (the “Extended Compliance Period”), to regain compliance with Nasdaq’s minimum bid price requirement for the continued listing on the Nasdaq Capital Market, as set forth in Nasdaq Listing Rule 5550(a)(2) (the “Minimum Bid Price Rule”). Nasdaq’s determination was based on the Company meeting the continued listing requirement for market value of publicly held shares and all other applicable requirements for initial listing on the Nasdaq Capital Market, with the exception of the bid price requirement, and the Company’s written notice of its intention to consider all available options to regain compliance during the Extended Compliance Period, including, if necessary, effecting a reverse stock split. The Company was unable to regain compliance during the Extended Compliance Period and on September 17, 2024, the Company received an additional notification from the Listing Qualifications Department stating that due to the deficiency, the Company’s securities would be delisted from Nasdaq on September 26, 2024, unless the Company appealed Nasdaq’s determination to a Hearings Panel (the “Panel”). A hearing request would stay the suspension of the Company’s securities pending the Panel’s discussion. On September 17, 2024, the Company submitted the hearing request to appeal (the “Appeal Request”) Nasdaq’s determination before the Panel. The hearing will take place on October 31, 2024 and it is anticipated that the Panel’s decision will follow shortly thereafter. The pendency of the Appeal Request does not have an immediate effect on the listing of our Common Shares and our Common Shares will continue to trade on Nasdaq under the symbol “INM”.
While the Company has filed the Appeal Request, there can be no assurances, however, that we will be successful in regaining compliance with the continued listing requirements and maintaining the listing of our Common Shares on Nasdaq. Delisting from Nasdaq could materially and adversely affect our ability to raise additional financing through the public or private sale of equity securities, would significantly affect the ability of investors to trade our securities and would negatively affect the value and liquidity of our securities, including our Common Shares. The actual or threatened delisting of our securities could also have other material and adverse consequences, including the potential loss of confidence by employees and other stakeholders, the loss of institutional investor interest and fewer business development opportunities, limited availability of market quotations for our securities, reduced liquidity with respect to our securities, a determination that our Common Shares is “penny stock,” which will require brokers trading in shares of our Common Shares to adhere to more stringent rules, possibly resulting in a reduced level of trading activity in the secondary trading market for our shares of our Common Shares, and limited amount of news and analyst coverage of the Company. To the extent that our Common Shares became eligible to trade on the OTC Bulletin Board, another over-the-counter quotation system, or on the pink sheets, an investor may find it more difficult to dispose of their Common Shares or obtain accurate quotations as to the market value of our Common Shares.
Furthermore, the National Securities Markets Improvement Act of 1996, which is a federal statute, prevents or preempts the states from regulating the sale of certain securities, which are referred to as “covered securities.” Because our Common Shares are currently listed on Nasdaq, such securities will be deemed covered securities. Although the states will be preempted from regulating the sale of our securities, the federal statute does allow states to investigate companies if there is a suspicion of fraud and, if there is a finding of fraudulent activity, then the states can regulate or bar the sale of covered securities in a particular case. Additionally, if we were no longer listed on Nasdaq, our securities would not be covered securities and we would be subject to regulations in each state in which we offer our securities.
If we have difficulty enrolling patients in clinical trials, the completion of the trials may be delayed or cancelled.
As our Product Candidates advance from preclinical testing to clinical testing, and then through progressively larger and more complex clinical trials, we will need to enroll an increasing number of patients that meet the eligibility criteria for those trials. The factors that affect our ability to enroll patients are largely uncontrollable and include, but are not limited to, the following:
| ● | size and nature of the patient population; |
| ● | inclusion and exclusion criteria for the trial; |
| ● | design of the study protocol; |
| ● | competition with other companies for clinical sites or patients; |
| ● | the perceived risks and benefits of the product candidate under study; |
| ● | the patient referral practices of physicians; and |
| ● | the number, availability, location and accessibility of clinical trial sites. |
As a result of the foregoing factors, we may have difficulty enrolling or maintaining the enrollment of patients in any clinical trials conducted for our products, which may result in the delay or cancellation of such trials. The delay or cancellation of any clinical trials could shorten any periods during which we may have the exclusive right to commercialize our Product Candidates or allow our competitors to bring products to market before us, which would impair our ability to successfully commercialize our Product Candidates and may harm our financial condition, results of operations and prospects.
If clinical trials of our Product Candidates fail to demonstrate safety and efficacy to the satisfaction of regulatory authorities or do not otherwise produce positive results, we would incur additional costs or experience delays in completing, or ultimately be unable to complete, the development and commercialization of our Product Candidates.
Before obtaining marketing approval from regulatory authorities for the sale of our Product Candidates, we must conduct preclinical studies in animals and extensive clinical trials in humans to demonstrate the safety and efficacy of the Product Candidates. Clinical testing is expensive and difficult to design and implement, can take many years to complete and has uncertain outcomes. The outcome of preclinical studies and early clinical trials may not predict the success of later clinical trials and interim results of a clinical trial do not necessarily predict final results. A number of companies in the pharmaceutical and biotechnology industries have suffered significant setbacks in advanced clinical trials due to lack of efficacy or unacceptable safety profiles, notwithstanding promising results in earlier trials. We do not know whether the clinical trials we may conduct will demonstrate adequate efficacy and safety to result in regulatory approval to market any of our Product Candidates in any jurisdiction. A product candidate may fail for safety or efficacy reasons at any stage of the testing process. A major risk we face is the possibility that none of our Product Candidates under development will successfully gain market approval from the FDA or other regulatory authorities, resulting in us being unable to derive any commercial revenue from them after investing significant amounts of capital in multiple stages of preclinical and clinical testing.
If we experience delays in clinical testing, we will be delayed in commercializing our Product Candidates, and our business may be substantially harmed.
We cannot predict whether any clinical trials will begin as planned, will need to be restructured, or will be completed on schedule, or at all. Our product development costs will increase if we experience delays in clinical testing. Significant clinical trial delays could shorten any periods during which we may have the exclusive right to commercialize our Product Candidates or allow our competitors to bring products to market before us, which would impair our ability to successfully commercialize our Product Candidates and may harm our financial condition, results of operations and prospects. The commencement and completion of clinical trials for our products may be delayed for a number of reasons, including delays related, but not limited, to:
| ● | failure by regulatory authorities to grant permission to proceed or placing the clinical trial on hold; |
| ● | import/export and research restrictions for cannabinoid-based pharmaceuticals may delay or prevent clinical trials in various geographical jurisdictions; |
| ● | patients failing to enroll or remain in our trials at the rate we expect; |
| ● |
suspension or termination of clinical trials by regulators for many reasons, including concerns about patient safety or failure of our contract manufacturers to comply with current good manufacturing practice (“cGMP”) requirements; |
| ● | any changes to our manufacturing process that may be necessary or desired; |
| ● | delays or failure to obtain clinical supply from contract manufacturers of our products necessary to conduct clinical trials; |
| ● | Product Candidates demonstrating a lack of safety or efficacy during clinical trials; |
| ● | patients choosing an alternative treatment for the indications for which we are developing any of our Product Candidates or participating in competing clinical trials and/or scheduling conflicts with participating clinicians; |
| ● | patients failing to complete clinical trials due to dissatisfaction with the treatment, side effects or other reasons; |
| ● | reports of clinical testing on similar technologies and products raising safety and/or efficacy concerns; |
| ● | clinical investigators not performing our clinical trials on their anticipated schedule, dropping out of a trial, or employing methods not consistent with the clinical trial protocol, regulatory requirements or other third parties not performing data collection and analysis in a timely or accurate manner; |
| ● | failure of our CROs, to satisfy their contractual duties or meet expected deadlines; |
| ● | inspections of clinical trial sites by regulatory authorities or Institutional Review Boards (“IRBs”) or ethics committees finding regulatory violations that require us to undertake corrective action, resulting in suspension or termination of one or more sites or the imposition of a clinical hold on the entire study; |
| ● | one or more IRBs or ethics committees rejecting, suspending or terminating the study at an investigational site, precluding enrollment of additional subjects, or withdrawing its approval of the trial; or |
| ● | failure to reach agreement on acceptable terms with prospective clinical trial sites. |
Our product development costs will increase if we experience delays in testing or approval or if we need to perform more or larger clinical trials than planned. Additionally, changes in regulatory requirements and policies may occur, and we may need to amend study protocols to reflect these changes. Amendments may require us to resubmit our study protocols to regulatory authorities or IRBs or ethics committees for re-examination, which may impact the cost, timing or successful completion of that trial. Delays or increased product development costs may have a material adverse effect on our business, financial condition and prospects.
Our IntegraSyn manufacturing approach may prove unsuccessful in achieving yields and/or cost levels required to be economically competitive with alternative methods of manufacturing.
Given the early stage of development of the IntegraSyn program and the risks inherent in research and development, it is too early to project the commercial viability of cannabinoids produced via this process. Potential negative outcomes from this program include but are not limited to:
| ● | the technology fails to produce sufficient quantities of cannabinoids or ones for which we or others have a need; or |
| ● | the cost structure of the technology is such that it is not commercially competitive with alternate methods of cannabinoid manufacturing leading to the technology having no value proposition nor incremental value to the Company. |
Negative results from clinical trials or studies of others and adverse safety events involving the targets of our products may have an adverse impact on our future commercialization efforts.
From time to time, studies or clinical trials on various aspects of pharmaceutical products are conducted by academic researchers, competitors or others. The results of these studies or trials, when published, may have a significant effect on the market for the pharmaceutical product that is the subject of the study. The publication of negative results of studies or clinical trials or adverse safety events related to our Product Candidates, or the therapeutic areas in which our Product Candidates compete, could adversely affect the price of our Common Shares and our ability to finance future development of our Product Candidates, and our business and financial results could be materially and adversely affected.
We intend to expend our limited resources to pursue our Product Candidates for certain indications and may fail to capitalize on other Product Candidates or other indications for our Product Candidates that may be more profitable or for which there is a greater likelihood of success.
Because we have limited financial and managerial resources, we are focusing on research programs relating to our Product Candidates for certain indications, primarily for the treatment of EB, which concentrates the risk of product failure in the event our Product Candidates prove to be unsafe or ineffective or inadequate for clinical development or commercialization. As a result, we may forego or delay pursuit of opportunities with other Product Candidates or for other indications that could later prove to have greater commercial potential. We may also deem it advisable to refocus our clinical development programs based on clinical trial results.
The regulatory approval processes of the FDA, HC, the EMA and other comparable foreign regulatory authorities are lengthy, time-consuming and inherently unpredictable, and if we are ultimately unable to obtain regulatory approval for our Product Candidates, our business will be substantially harmed.
We are not permitted to market our Product Candidates in any jurisdiction until we receive formal approval from the appropriate regulatory authorities. For example, prior to submitting an NDA to the FDA or an MAA to the EMA for approval of our Product Candidates, we will need to complete our preclinical studies and clinical trials. Successfully completing our clinical program and obtaining approval of an application seeking commercialization approval is a complex, lengthy, expensive and uncertain process, and the regulatory authorities may delay, limit or deny approval of our Product Candidates for many reasons, including, among others, because:
| ● | we may not be able to demonstrate that our Product Candidates are safe and effective in treating patients to the satisfaction of the regulatory authorities such as the FDA, HC or EMA; |
| ● | the results of our clinical trials may not meet the level of statistical or clinical significance required by the regulatory authorities for marketing approval; |
| ● | the regulatory authorities may disagree with the number, design, size, conduct or implementation of our clinical trials; |
| ● | the regulatory authorities may require that we conduct additional clinical trials; |
| ● | the regulatory authorities or other applicable foreign regulatory authorities may not approve the formulation, labeling or specifications of our Product Candidates; |
| ● | the contract manufacturing organizations and other contractors that we may retain to conduct our clinical trials may take actions outside of our control that materially adversely impact our clinical trials; |
| ● | the regulatory authorities may find the data from clinical studies and clinical trials insufficient to demonstrate that our Product Candidates are safe and effective for their proposed indications; |
| ● | the regulatory authorities may disagree with our interpretation of data from our preclinical studies and clinical trials; |
| ● | the regulatory authorities may not accept data generated at our clinical trial sites or may disagree with us over whether to accept efficacy results from clinical trial sites outside the United States, Canada or outside the European Union, as applicable, where the standard of care is potentially different from that in the United States, Canada or in the European Union, as applicable; |
| ● | if our applications are submitted to the regulatory authorities, the regulatory authorities may have difficulties scheduling the necessary review meetings in a timely manner, may recommend against approval of our application or may recommend or require, as a condition of approval, additional preclinical studies or clinical trials, limitations on approved labeling or distribution and use restrictions; |
| ● | the FDA may require development of a Risk Evaluation and Mitigation Strategy which would use risk minimization strategies to ensure that the benefits of certain prescription drugs outweigh their risks, as a condition of approval or post-approval, and the EMA may grant only conditional marketing authorization or impose specific obligations as a condition for marketing authorization, or may require us to conduct post-authorization safety studies; |
| ● | the FDA, DEA, HC, EMA or other applicable foreign regulatory agencies may not approve the manufacturing processes or facilities of third-party manufacturers with which we contract or DEA or other applicable foreign regulatory agency quotas may limit the quantities of controlled substances available to our manufacturers; or |
| ● | the FDA, HC, EMA or other applicable foreign regulatory agencies may change their approval policies or adopt new regulations. |
In the United States, our activities are potentially subject to additional regulation by various federal, state and local authorities in addition to the FDA, including, among others, the Centers for Medicare and Medicaid Services, other divisions of the United States Department of Health and Human Services (“HHS”), (for example, the Office of Inspector General), the Department of Justice (“DOJ”), and individual United States Attorney offices within the DOJ, and state and local governments. Because of the breadth of these laws and the narrowness of available statutory and regulatory exemptions, it is possible that some of our business activities could be subject to challenge under one or more of such laws. If our operations are found to be in violation of any of the federal and state laws described above or any other governmental regulations that apply to us, we may be subject to penalties, including criminal and significant civil monetary penalties, damages, fines, imprisonment, exclusion from participation in government programs, injunctions, recall or seizure of products, total or partial suspension of production, denial or withdrawal of pre marketing product approvals, private “qui tam” actions brought by individual whistleblowers in the name of the government or refusal to allow us to enter into supply contracts, including government contracts, and the curtailment or restructuring of our operations, any of which could adversely affect our ability to operate our business and our results of operations. To the extent that any of our products are sold in a foreign country, we may be subject to similar foreign laws and regulations, which may include, for instance, applicable post-marketing requirements, including safety surveillance, anti-fraud and abuse laws, and implementation of corporate compliance programs and reporting of payments or transfers of value to healthcare professionals.
Any of these factors, many of which are beyond our control, could increase development costs, jeopardize our ability to obtain regulatory approval for and successfully market our Product Candidates and generate product revenue.
We intend to conduct clinical trials for our Product Candidates in several international jurisdictions, and acceptance by all regulatory authorities for such “international” data is not certain.
We intend to conduct clinical trials for our Product Candidates both inside and outside the United States. To date, all of our clinical development has been conducted outside of the United States. Ultimately, we plan to submit NDAs for our Product Candidates to the FDA and other regulatory authorities upon completion of all requisite clinical trials. As an example, although the FDA may accept data from clinical trials conducted outside the United States, acceptance of such study data by the FDA is subject to certain conditions. For example, the clinical trial must be conducted in accordance with FDA regulations relating governing human subject protection and the conduct of clinical trials, which are referred to as “Good Clinical Practice” (“GCP”) requirements and the FDA must be able to validate the data from the clinical trial through an onsite inspection if it deems such inspection necessary. Where data from foreign clinical trials are intended to serve as the sole basis for marketing approval in the United States, the FDA will not approve the application on the basis of foreign data alone unless those data are considered applicable to the U.S. patient population and U.S. medical practice, the clinical trials were performed by clinical investigators of recognized competence, and the data is considered valid without the need for an on-site inspection by the FDA or, if the FDA considers such an inspection to be necessary, the FDA is able to validate the data through an on-site inspection or other appropriate means. In addition, such clinical trials would be subject to the applicable local laws of the foreign jurisdictions where the clinical trials are conducted. There can be no assurance the FDA or any other regulatory authorities will accept data from clinical trials conducted outside of the United States or other international jurisdictions. If the FDA or any other regulatory authorities does not accept any such data, it would likely result in the need for additional clinical trials, which would be costly and time-consuming and delay aspects of our development plan.
In addition, the conduct of clinical trials outside the United States could have a significant impact on us. Risks inherent in conducting international clinical trials include:
| ● | foreign regulatory requirements that could burden or limit our ability to conduct our clinical trials; |
| ● | administrative burdens of conducting clinical trials under multiple foreign regulatory schema; |
| ● | foreign currency fluctuations which could negatively impact our financial condition since certain payments are paid in local currencies; |
| ● | manufacturing, customs, shipment and storage requirements; |
| ● | cultural differences in medical practice and clinical research; and |
| ● | diminished protection of intellectual property in some countries. |
Our Product Candidates contain compounds that may be classified as “controlled substances”, the use of which may generate public controversy and restrict their development or commercialization.
If a drug has a potential for abuse, the NDA or other regulatory submission must include a description and analysis of studies or information related to abuse of the drug, including a proposal for scheduling (for example, in the U.S. under the federal Controlled Substances Act (“CSA”). A description of any studies related to overdosage is also required, including information on dialysis, antidotes, or other treatments, if known. While we believe there would be relatively minimal abuse potential with our Product Candidates given the low drug concentration and topical route of administration, we could be incorrect or they may be perceived as having the potential for substance abuse. In either case, there may be a negative effect on our ability to successfully develop or commercialize our Product Candidates. Since our Product Candidates contain purified substances that are chemically identical to those occurring in nature, they may, therefore, be classified as “controlled substances”, and their regulatory approval may generate public controversy. Political and social pressures and adverse publicity could lead to delays in approval of, and increased expenses for, our Product Candidates. These pressures could also limit or restrict the introduction and marketing of our Product Candidates. Despite that fact that our APIs, which are the ingredients that give medicines their effects, are synthetically made and, therefore, we have no interaction with the Cannabis plant, adverse publicity from Cannabis misuse or adverse side effects from Cannabis or other cannabinoid products may adversely affect the commercial success or market penetration achievable for our Product Candidates. The nature of our business attracts a high level of public and media interest, and in the event of any resultant adverse publicity, our reputation may be harmed. Furthermore, if our Product Candidates are classified as “controlled substances”, they may be subject to import/export and research restrictions that could delay or prevent the development of our products in various geographical jurisdictions. The successful commercialization of our Product Candidates may require permits or approvals from regulatory bodies, such as the DEA, that regulate controlled substances.
If any of our Product Candidates receives marketing approval and we or others later identify undesirable side effects caused by the Product Candidate, our ability to market and derive revenue from the product candidates could be compromised.
In the event that any of our Product Candidates receive regulatory approval and we or others identify undesirable side effects caused by one of our products, any of the following adverse events could occur, which could result in the loss of significant revenue to us and materially and adversely affect our results of operations and business: (i) regulatory authorities may withdraw their approval of the product or seize the product; (ii) we may be required to recall the product or change the way the product is administered to patients; (iii) additional restrictions may be imposed on the marketing of the particular product or the manufacturing processes for the product or any component thereof; (iv) we may be subject to fines, injunctions or the imposition of civil or criminal penalties; (v) regulatory authorities may require the addition of labeling statements, such as a “black box” warning or a contraindication; (vi) we may be required to create a Medication Guide outlining the risks of such side effects for distribution to patients; (vii) we could be sued and held liable for harm caused to patients; (viii) the product may become less competitive; and (ix) our reputation being harmed.
Research restrictions, product shipment delays or prohibitions could have a material adverse effect on our business, results of operations and financial condition.
Research on and the shipment, import and export of our Product Candidates and the API used in our Product Candidates will require research permits, import and export licenses by many different authorities. For instance, in the United States, the FDA, U.S. Customs and Border Protection, and the DEA; in Canada, the Canada Border Services Agency, and HC; in Europe, the EMA and the European Commission; in Australia and New Zealand, the Australian Customs and Border Protection Service, the Therapeutic Goods Administration, the New Zealand Medicines and Medical Device Safety Authority and the New Zealand Customs Service; and in other countries, similar regulatory authorities, regulate the research on and import and export of pharmaceutical products that contain controlled substances. Specifically, the import and export process require the issuance of import and export licenses by the relevant controlled substance authority in both the importing and exporting country. We may not be granted, or if granted, maintain, such licenses from the authorities in certain countries. Even if we obtain the relevant licenses, shipments of API and our Product Candidates may be held up in transit, which could cause significant delays and may lead to product batches being stored outside required temperature ranges. Inappropriate storage may damage the product shipment resulting in delays in clinical trials or, upon commercialization, a partial or total loss of revenue from one or more shipments of API or our Product Candidates. Once shipment is complete, we or the research contractors we are working with may also suffer further delays or restrictions as a result of regulations governing research on cannabinoids. A delay in a clinical trial or, upon commercialization, a partial or total loss of revenue from one or more shipments of API or our Product Candidates could have a material adverse effect on our business, results of operations and financial condition. The aforementioned examples and lists of various authorities that may currently, or in the future, affect our ability to conduct research on or import or export our Product Candidates and/or API, should not be construed as exhaustive or comprehensive in any way.
Healthcare legislation, including potentially unfavorable pricing regulations or other healthcare reform initiatives, may increase the difficulty and cost for us to obtain marketing approval of and commercialize our Product Candidates.
Particularly in the United States but also in other jurisdictions, there have been a number of legislative and regulatory changes and proposed changes in recent years regarding the healthcare system that could prevent or delay marketing approval of our Product Candidates, restrict or regulate post-approval activities or affect our ability to profitably sell any Product Candidates for which we obtain marketing approval. Healthcare reform measures that have been and may be adopted in the future may result in more rigorous coverage criteria, new payment methodologies and additional downward pressure on the price that we receive for any approved product, and could seriously harm our future revenue. Any reduction in reimbursement from Medicare or other government programs may result in a similar reduction in payments from private payors. The implementation of cost containment measures or other healthcare reforms may compromise our ability to generate revenue, attain profitability or commercialize our products.
Any Product Candidates we develop may become subject to unfavorable pricing regulations, third-party reimbursement practices or healthcare reform initiatives, thereby materially and adversely impacting our business.
The regulations that govern marketing approvals, pricing and reimbursement for new drugs vary widely from country to country. Some countries require approval of the sale price of a drug before it can be marketed. In many countries, the pricing review period begins after marketing or product licensing approval is granted. In some foreign markets, drug pricing remains subject to continuing governmental control even after initial approval is granted. Although we intend to monitor these regulations, our programs are currently in the early stages of development and we will not be able to assess the impact of price regulations for a number of years. As a result, we might obtain regulatory approval for a product in a particular country, but then be subject to price regulations that delay our commercial launch of the product and negatively impact the revenues we are able to generate from the sale of the product in that country.
Our ability to commercialize any products also will depend in part on the extent to which reimbursement for these products and related treatments will be available from government health administration authorities, private health insurers and other organizations. Even if we succeed in bringing one or more products to the market, these products may not be considered cost-effective, and the amount reimbursed for any products may be insufficient to allow us to sell our products on a competitive basis. Because our programs are in the early stages of development, we are unable at this time to determine their cost effectiveness or the likely level or method of reimbursement. Increasingly, the third-party payors who reimburse patients or healthcare providers, such as government and private insurance plans, are requiring that drug companies provide them with predetermined discounts from list prices and are seeking to reduce the prices charged or the amounts reimbursed for pharmaceutical products. If the price we are able to charge for any products we develop, or the reimbursement provided for such products, is inadequate in light of our development and other costs, our return on investment could be adversely affected.
Increased scrutiny on drug pricing or changes in pricing regulations could restrict the amount that we are able to charge for our Product Candidates, which could adversely affect our revenue and results of operations.
Drug pricing by pharmaceutical companies is currently under increased scrutiny and is expected to continue to be the subject of intense political and public debate in the United States and other jurisdictions. Specifically, there have been several recent U.S. Congressional inquiries and hearings with respect to pharmaceutical drug pricing practices, including in connection with the investigation of specific price increases by several pharmaceutical companies. Additionally, several states have recently passed laws designed to, among other things, bring more transparency to drug pricing, and other states may pursue similar initiatives in the future. We cannot predict the extent to which our business may be affected by these or other potential future legislative or regulatory developments. However, increased scrutiny on drug pricing, negative publicity related to the pricing of pharmaceutical drugs generally, or changes in pricing regulations could restrict the amount that we are able to charge for our Product Candidates, which could have a material adverse effect on our revenue and results of operations.
Negative publicity may adversely affect us and our business.
Media coverage and public statements that insinuate improper actions by us, regardless of their factual accuracy or truthfulness, may result in negative publicity, litigation or governmental investigations by regulators. Addressing negative publicity and any resulting litigation or investigations may distract management, increase costs and divert resources. Negative publicity may have an adverse impact on our reputation and the morale of our employees, which could have a material adverse effect on our business, financial condition, results of operations and cash flows.
Even if we are able to commercialize our Product Candidates, they may not receive coverage and adequate reimbursement from third-party payors, which could harm our business.
The availability of reimbursement by governmental and private payors is essential for most patients to be able to afford their treatments. Sales of our Product Candidates, if approved, will depend substantially on the extent to which the costs of these Product Candidates will be paid by health maintenance, managed care, pharmacy benefit and similar healthcare management organizations, or reimbursed by government health administration authorities, private health coverage insurers and other third-party payors. If reimbursement is not available, or is available only to limited levels, we may not be able to successfully commercialize our Product Candidates. Even if coverage is provided, the approved reimbursement amount may not be high enough to allow us to establish or maintain pricing sufficient to realize a sufficient return on our investment.
In the United States, the Medicare Modernization Act, established the Medicare Part D program and provided authority for limiting the number of drugs that will be covered in any therapeutic class thereunder. The Medicare Modernization Act, including its cost reduction initiatives, could decrease the coverage available for any of our approved products. Furthermore, private payors often follow Medicare in setting their own coverage policies. Therefore, any reduction in coverage that results from the Medicare Modernization Act may result in a similar reduction from private payors.
There is significant uncertainty related to the insurance coverage and reimbursement of newly approved products. In the United States, the principal decisions about reimbursement for new medicines are typically made by the Centers for Medicare & Medicaid Services (the “CMS”), an agency within the HHS, as CMS decides whether and to what extent a new medicine will be covered and reimbursed under Medicare. Private payors tend to follow CMS to a substantial degree.
The intended use of a drug product by a physician can also affect pricing. For example, CMS could initiate a National Coverage Determination administrative procedure, by which the agency determines which uses of a therapeutic product would and would not be reimbursable under Medicare. This determination process can be lengthy, thereby creating a long period during which the future reimbursement for a particular product may be uncertain.
Outside the United States, particularly in EU Member States, the pricing of prescription drugs is subject to governmental control. In these countries, pricing negotiations or the successful completion of Health Technology Assessment (“HTA”) procedures with governmental authorities can take considerable time after receipt of marketing authorization for a product. In addition, there can be considerable pressure by governments and other stakeholders on prices and reimbursement levels, including as part of cost containment measures. Certain countries allow companies to fix their own prices for medicines but monitor and control company profits. Political, economic and regulatory developments may further complicate pricing negotiations, and pricing negotiations may continue after reimbursement has been obtained. Reference pricing used by various EU Member States and parallel distribution, or arbitrage between low-priced and high-priced EU member states, can further reduce net realized prices. In some countries, we or our collaborators may be required to conduct a clinical trial or other studies that compare the cost-effectiveness of our Product Candidates to other available therapies in order to obtain or maintain reimbursement or pricing approval. Publication of discounts by third-party payors or authorities may lead to further pressure on the prices or reimbursement levels within the country of publication and other countries. If reimbursement of any product candidate approved for marketing is unavailable or limited in scope or amount, or if pricing is set at unsatisfactory levels, our business, financial condition, results of operations or prospects could be adversely affected.
Our relationships with customers and third-party payors will be subject to applicable anti-kickback, fraud and abuse, federal exclusion or debarment, and other healthcare laws and regulations, which could expose us to criminal sanctions, civil penalties, contractual damages, reputational harm and diminished profits and future earnings.
Healthcare providers, physicians and third-party payors play a primary role in the recommendation and prescription of any Product Candidates for which we obtain marketing approval. Our future arrangements with third-party payors and customers may expose us to broadly applicable fraud and abuse and other healthcare laws and regulations that may constrain the business or financial arrangements and relationships through which we market, sell and distribute our products for which we obtain marketing approval. As a pharmaceutical company, even though we do not and will not control referrals of healthcare services or bill directly to Medicare, Medicaid or other third-party payors, certain federal and state healthcare laws and regulations pertaining to fraud and abuse and patients’ rights are and will be applicable to our business. Restrictions under applicable federal and state healthcare laws and regulations that may affect our ability to operate include the following:
| ● | the U.S. federal healthcare Anti-Kickback Statute impacts our marketing practices, educational programs, pricing policies and relationships with healthcare providers or other entities, by prohibiting, among other things, persons from knowingly and willfully soliciting, offering, receiving or providing remuneration, directly or indirectly, in cash or in kind, to induce or reward, or in return for, either the referral of an individual for, or the purchase, order or recommendation of, any good or service, for which payment may be made under a federal healthcare program such as Medicare and Medicaid; |
| ● | federal civil and criminal false claims laws and civil monetary penalty laws impose criminal and civil penalties, including through civil whistleblower or qui tam actions, against individuals or entities for, among other things, knowingly presenting, or causing to be presented, false or fraudulent claims for payment of government funds (including through reimbursement by Medicare or Medicaid or other federal health care programs), which has been applied to impermissible promotion of pharmaceutical products for off-label uses, or making a false statement or record to avoid, decrease or conceal an obligation to pay money to the federal government; |
| ● | the U.S. Health Insurance Portability and Accountability Act (“HIPPA”), as amended by the Health Information Technology for Economic and Clinical Health Act (“HITECH Act”), among other things, imposes criminal and civil liability for executing a scheme to defraud any healthcare benefit program and also prohibits knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement or representation, or making or using any false writing or document knowing the same to contain any materially false, fictitious or fraudulent statement or entry in connection with the delivery of or payment for healthcare benefits, items or services; |
| ● | the U.S. federal Physician Payment Sunshine Act, being implemented as the Open Payments Program, requires applicable manufacturers of covered drugs, devices, biologics and medical supplies to report annually to HHS information related to payments and other transfers of value to physicians and teaching hospitals, and ownership and investment interests held by physicians and their immediate family members; |
| ● | analogous state laws and regulations, such as state anti-kickback laws, false claims laws and privacy and security of health information laws, may apply to sales or marketing arrangements, claims involving healthcare items or services reimbursed by non-governmental third-party payors, including private insurers, or health information; and |
| ● | certain state laws require pharmaceutical companies to adopt codes of conduct consistent with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government; restrict certain marketing-related activities including the provision of gifts, meals, or other items to certain health care providers; and/or require drug manufacturers to report information related to payments and other transfers of value to physicians and certain other healthcare providers or marketing expenditures. |
Comparable laws and regulations exist in the countries within the European Economic Area (“EEA”). Although such laws are partially based upon European Union (“EU”), law, they may vary from country to country. Healthcare specific, as well as general EU and national laws, regulations and industry codes constrain, for example, our interactions with government officials and healthcare professionals, and the collection and processing of personal health data. Non-compliance with any of these laws or regulations could lead to criminal or civil liability.
Efforts to ensure that our business arrangements with third parties will comply with applicable healthcare laws and regulations will involve substantial costs. It is possible that governmental authorities will conclude that our business practices may not comply with current or future statutes, regulations or case law involving applicable fraud and abuse or other healthcare laws and regulations. If our operations are found to be in violation of any of these laws or any other governmental regulations that may apply to us, we may be subject to significant civil, criminal and administrative penalties, damages, fines, imprisonment, exclusion from government funded healthcare programs, such as Medicare and Medicaid, and the curtailment or restructuring of our operations. If any physicians or other healthcare providers or entities with whom we expect to do business are found to not be in compliance with applicable laws, they may be subject to criminal, civil or administrative sanctions, including exclusions from government funded healthcare programs.
Failure to comply with the FCPA, the CFPOA, and other global anti-corruption and anti-bribery laws could subject us to penalties and other adverse consequences.
The FCPA and the CFPOA, as well as any other applicable domestic or foreign anti-corruption or anti-bribery laws to which we are or may become subject generally prohibit corporations and individuals from engaging in certain activities to obtain or retain business or to influence a person working in an official capacity and requires companies to maintain accurate books and records and internal controls, including at foreign-controlled subsidiaries. It is illegal to pay, offer to pay or authorize the payment of anything of value to any foreign government official, government staff member, political party or political candidate in an attempt to obtain or retain business or to otherwise influence a person working in an official capacity.
Compliance with these anti-corruption laws and anti-bribery laws may be expensive and difficult, particularly in countries in which corruption is a recognized problem. In addition, these laws present particular challenges in the pharmaceutical industry, because, in many countries, hospitals are operated by the government, and physicians and other hospital employees are considered to be foreign officials. Certain payments by other companies to hospitals in connection with clinical trials and other work have been deemed to be improper payments to governmental officials and have led to FCPA enforcement actions.
Our internal control policies and procedures may not protect us from reckless or negligent acts committed by our employees, future distributors, licensees or agents. We are currently working to get policies and processes in place to monitor compliance with the FCPA and CFPOA. We can make no assurance that they will not engage in prohibited conduct, and we may be held liable for their acts under applicable anti-corruption and anti-bribery laws. Noncompliance with these laws could subject us to investigations, sanctions, settlements, prosecution, other enforcement actions, disgorgement of profits, significant fines, damages, other civil and criminal penalties or injunctions, suspension or debarment from contracting with certain persons, the loss of export privileges, whistleblower complaints, reputational harm, adverse media coverage, and other collateral consequences. Any investigations, actions or sanctions or other previously mentioned harm could have a material negative effect on our business, operating results and financial condition.
Any change in export or import controls, anti-corruption laws, economic sanctions or related legislation, or change in the countries, governments, persons, or technologies targeted by such restrictions or legislation, could result in decreased use of our products by customers or in our decreased ability to offer our products internationally, which would harm our business, operating results and financial condition. Furthermore, failure to comply with export or import controls or with anti-corruption or economic sanctions laws may expose us to government investigations, more onerous compliance requirements and significant penalties, which could harm our business, operating results and financial condition. In addition, responding to any action will likely result in a significant diversion of management’s attention and resources and an increase in professional fees. Enforcement actions and sanctions could harm our business, operating results and financial condition.
Trade wars and changes in international trade law and policies may have a material adverse effect on our business, financial condition and results of operations. In October 2022, the Biden administration issued a new set of export controls which (i) banned Chinese companies from buying advanced chips and chip-making equipment in the United States without a license, and (ii) restricted the ability of U.S. persons from providing support for the development or production of chips at certain manufacturing facilities in China. Moreover, in December 2022, the United States imposed new duties on imports from certain major solar panel makers in China after an investigation determined that such manufacturers were avoiding tariffs by finishing their products in Southeast Asian countries. More recently, President Biden signed an executive order that will make it more difficult for U.S. firms to invest in certain Chinese companies—citing national security concerns, the executive order prohibits investments in AI and quantum computing. In response to the foregoing, China implemented its own export controls on two rare elements, germanium and gallium, which the United States relies on to produce chips, fiber optics and solar panels.
The ongoing trade war between China and the United States and its potential escalation may have an adverse effect on our business operations and revenues.
Starting in April 2018, the United States imposed a 25% tariff on steel and a 10% tariff on aluminum imports from other countries. On July 6, 2018, the United States imposed 25% tariffs on $34 billion worth of Chinese goods. China instituted retaliatory tariffs on certain United States goods. In 2019, the United States and China implemented several rounds of tariff increases and retaliations. On January 15, 2020, the United States and China signed a Phase One trade deal pursuant to which, among other things, the United States will modify existing tariffs. In October 2022, the Biden administration issued a new set of export controls which (i) banned Chinese companies from buying advanced chips and chip-making equipment in the United States without a license, and (ii) restricted the ability of U.S. persons from providing support for the development or production of chips at certain manufacturing facilities in China. Moreover, in December 2022, the United States imposed new duties on imports from certain major solar panel makers in China after an investigation determined that such manufacturers were avoiding tariffs by finishing their products in Southeast Asian countries. More recently, President Biden signed an executive order that will make it more difficult for U.S. firms to invest in certain Chinese companies—citing national security concerns, the executive order prohibits investments in AI and quantum computing. In response to the foregoing, China implemented its own export controls on two rare elements, germanium and gallium, which the United States relies on to produce chips, fiber optics and solar panels.
Since we operate in the United States and deliver products and services to customers in the United States, the trade war could materially and adversely affect us, and especially if, when and to the extent escalated, may cause global economic turmoil and adversely impact the supply chain for our products, the cost of our products and the demand for our products and, thus, may have a material adverse effect on our business and results of operations.
Federal legislation and actions by state and local governments may permit reimportation of drugs from/to foreign countries where the drugs are sold at lower prices than in the country of origination, which could materially adversely affect our business and financial condition.
We may face competition for our Product Candidates, if approved, from cheaper generics and/or cannabinoid therapies sourced from foreign countries that have placed price controls on pharmaceutical products. This is referred to as parallel importation. For instance, the Medicare Modernization Act contains provisions that may change U.S. importation laws and expand pharmacists’ and wholesalers’ ability to import cheaper versions of an approved drug and competing products from Canada, where there are government price controls. These changes to U.S. importation laws will not take effect unless and until the Secretary of HHS certifies that the changes will pose no additional risk to the public’s health and safety and will result in a significant reduction in the cost of products to consumers. The Secretary of HHS has so far declined to approve a reimportation plan. Proponents of drug reimportation, including certain state legislatures, may attempt to pass legislation that would directly allow reimportation under certain circumstances. Legislation or regulations allowing the reimportation of drugs, if enacted, could decrease the price we receive for any products that we may develop, including our Product Candidates, and adversely affect our future revenues and prospects for profitability.
We are dependent upon our key personnel to achieve our business objectives.
We depend on key personnel, the loss of any of whom could harm our business. Our future performance and development will depend to a significant extent on the efforts and abilities of its executive officers, key employees, and consultants. The loss of the services of one or more of these individuals could harm our business. Our success will depend largely on our continuing ability to attract, develop and retain skilled employees and consultants in our business. Because of the specialized scientific and managerial nature of our business, we rely heavily on our ability to attract and retain qualified scientific, technical and managerial personnel. The competition for qualified personnel in our field is intense. Due to this intense competition, we may be unable to continue to attract and retain qualified personnel necessary for the development of our business or to recruit suitable replacement personnel. Any delay in replacing such persons, or an inability to replace them with persons of similar expertise, would have a material adverse effect on our business, financial condition and results of operations.
Our employees may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements, which could subject us to significant liability and harm our reputation.
We are exposed to the risk of employee fraud or other misconduct. Misconduct by employees could include intentional failures to comply with regulations of domestic or foreign regulatory authorities. In addition, misconduct by employees could include intentional failures to comply with certain development standards, to report financial information or data accurately, or to disclose unauthorized activities to us. Employee misconduct could also involve the improper use of information obtained in the course of clinical trials, which could result in regulatory sanctions and serious harm to our reputation. While prohibited, it is not always possible to identify and deter employee misconduct, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to be in compliance with such laws or regulations. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business and results of operations, including the imposition of significant fines or other sanctions.
Our internal computer systems, or those of our contractors or consultants, may fail or suffer security breaches, which could result in a material disruption of our product development programs.
Despite the implementation of cyber security measures, our internal computer systems and those of our contractors and consultants are vulnerable to damage from computer viruses, unauthorized access, natural disasters, terrorism, war and telecommunication and electrical failures. Such events could cause interruptions of our operations. For example, the loss of preclinical data or data from any future clinical trial involving our product candidates could result in delays in our development and regulatory filing efforts and significantly increase our costs. To the extent that any disruption or security breach were to result in a loss of, or damage to, our data, or inappropriate disclosure of confidential or proprietary information, we could incur liability and the development of our product candidates could be delayed.
Our insurance may be insufficient to cover losses that may occur as a result of our operations.
We currently maintain directors’ and officers’ liability insurance, clinical trial insurance and property and general liability insurance and intend in the future to obtain shipping and storage insurance for Product Candidates. This insurance may not remain available to us or be obtainable by us at commercially reasonable rates, and the amount of our coverage may not be adequate to cover any liability we incur. Future increases in insurance costs, coupled with the increase in deductibles, will result in higher operating costs and increased risk. If we were to incur substantial liability and such damages were not covered by insurance or were in excess of policy limits, or if we were to incur such liability at a time when we were not able to obtain liability insurance, our business, results of operations and financial condition could be materially adversely affected.
Our insurance costs may increase significantly, we may be unable to obtain the same level of insurance coverage and our insurance coverage may not be adequate to cover all possible losses we may suffer.
We generally renew our insurance policies annually. If the cost of coverage becomes too high or if we believe certain coverage becomes inapplicable, we may need to reduce our policy limits, increase retention amounts or agree to certain exclusions from our coverage to reduce the premiums to an acceptable amount or to otherwise reduce coverage for certain occurrences. On the other hand, we may determine that we either do not have certain coverage that would be prudent for our business and the risks associated with our business or that our current coverages are too low to adequately cover such risks. In either event, we may incur additional or higher premiums for such coverage than in prior years.
Among other factors, national security concerns, catastrophic events, pandemics such as the COVID-19 pandemic, or any changes in any applicable statutory requirement binding insurance carriers to offer certain types of coverage could also adversely affect available insurance coverage and result in, among other things, increased premiums on available coverage (which may cause us to elect to reduce our policy limits or not renew our coverage) and additional exclusions from coverage. As cyber incidents and threats continue to evolve, we may be required to expend additional, perhaps significant, resources to continue to update, modify or enhance our protective measures or to investigate and remediate any vulnerability to cyber incidents. Although we maintain and monitor our information technology systems and maintain coverage to indemnify us from losses arising from cyber-attacks, such systems and insurance coverage may not be sufficient to protect against or cover all the losses we may experience as a result of any cyberattacks.
We may suffer damage due to a casualty loss (such as fire, natural disasters, pandemics and acts of war or terrorism) or other losses, such as those related to labor, professional liability or certain actions or inactions by our management, directors, employees or others, that could severely disrupt its business or subject us to claims by third parties who are injured or harmed. Although we maintain insurance that we believe to be adequate, such insurance may be inadequate or unavailable to cover all the risks to which our business and assets may be exposed, including risks related to certain litigation. Should an uninsured loss (including a loss that is less than the applicable deductible or that is not covered by insurance) or loss in excess of insured limits occur, it could have a significant adverse impact on our business, results of operations or financial condition.
There may be changes in laws, regulations and guidelines which are material and detrimental to our business.
Our operations are subject to a variety of laws, regulations and guidelines relating to pharmacology, cannabinoids and drug delivery, as well as laws and regulations relating to health and safety, the conduct of operations, and the protection of the environment. While, to the knowledge of our management, we are currently in compliance with all such laws, changes to such laws, regulations and guidelines due to matters beyond our control may cause adverse effects to our operations and financial condition. These changes may require us to incur substantial costs associated with legal and compliance fees and ultimately require us to alter our business plan. In addition, if the governments of Canada or the United States were to enact or amend laws relating to our industry, it may decrease the size of, or eliminate entirely, the market for our Product Candidates, may introduce significant new competition into the market and may otherwise potentially materially and adversely affect our business, results of operations and financial condition.
If we do not comply with laws regulating the protection of the environment and health and human safety, our business could be adversely affected.
The research and development that we carry out either directly or through third parties involves, and may in the future involve, the use of potentially hazardous materials and chemicals. Our operations may produce hazardous waste products. Although we believe that our safety procedures for handling and disposing of these materials comply with the standards mandated by local, state and federal laws and regulations, the risk of accidental contamination or injury from these materials cannot be eliminated. If an accident occurs, we could be held liable for resulting damages, which could be substantial. We are also subject to numerous environmental, health and workplace safety laws and regulations and fire and building codes. Although we maintain workers’ compensation insurance as prescribed by the Province of British Columbia to cover us for costs and expenses, we may incur due to injuries to our employees, this insurance may not provide adequate coverage against potential liabilities. We do not maintain insurance for environmental liability or toxic tort claims that may be asserted against us. Additional federal, state and local laws and regulations affecting our operations may be adopted in the future. We may incur substantial costs to comply with, and substantial fines or penalties if we violate, any of these laws or regulations.
Our proprietary information, or that of our customers, suppliers and business partners, may be lost or we may suffer security breaches.
In the ordinary course of our business, we may collect and store sensitive data, including intellectual property, data from preclinical studies, clinical trial data, our proprietary business information and that of our customers, suppliers and business partners, and personally identifiable information of our customers, clinical trial subjects and employees, in our data centers and on our networks. The secure processing, maintenance and transmission of this information is critical to our operations. Despite our security measures, our information technology and infrastructure may be vulnerable to attacks by hackers or breached due to employee error, malfeasance or other disruptions. Although to our knowledge we have not experienced any such material security breach to date, any such breach could compromise our networks and the information stored there could be accessed, publicly disclosed, lost or stolen. Any such access, disclosure or other loss of information could result in legal claims or proceedings, liability under laws that protect the privacy of personal information, regulatory penalties, disrupt our operations, damage to our ability to obtain patent protection for our Product Candidates, damage to our reputation, and cause a loss of confidence in our products and our ability to conduct clinical trials, which could adversely affect our business and reputation and lead to delays in gaining regulatory approvals.
We expect to face intense competition, often from companies with greater resources and experience than we have.
The pharmaceutical industry is highly competitive and subject to rapid change. The industry continues to expand and evolve as an increasing number of competitors and potential competitors enter the market. Many of these competitors and potential competitors have substantially greater financial, technological, managerial and research and development resources and experience than we have. Some of these competitors and potential competitors have more experience than we have in the development of pharmaceutical products, including validation procedures and regulatory matters. Other companies researching in the same disease areas may develop products that are competitive or superior to our Product Candidates. Other companies working in cannabinoid research may develop products targeting the same diseases that we are focused on that are competitive or superior to our Product Candidates. In addition, there are non-FDA approved Cannabis / cannabinoid preparations being made available from companies in the so-called “medical marijuana” industry, which may be competitive to our products. If we are unable to compete successfully, our commercial opportunities will be reduced and our business, results of operations and financial conditions may be materially harmed.
Industry consolidation may lead to increased competition and costs, and may harm our operating results.
We rely on certain third parties to provide supplies and services necessary for our business. Any reduction in market participants and available suppliers and vendors, whether through transactions or consolidation, could result in fewer alternatives for sourcing key supplies and services. Such consolidation could result in a shortage of supplies and services thereby increasing the cost of such supplies and services, and potentially inhibit the ability of suppliers and vendors to deliver on time, if at all. Cost increases and delays in, or the unavailability of, critical supplies and services could have a material and adverse effect on our results of operations.
If we receive regulatory approvals, we intend to market our Product Candidates in multiple jurisdictions where we have limited or no operating experience and may be subject to increased business and economic risks that could affect our financial results.
If we receive regulatory approvals, we may plan to market our Product Candidates in jurisdictions where we have limited or no experience in marketing, developing and distributing our products. Certain markets have substantial legal and regulatory complexities that we may not have experience navigating. We are subject to a variety of risks inherent in doing business internationally, including risks related to the legal and regulatory environment in non-U.S. jurisdictions, including with respect to privacy and data security, trade control laws and unexpected changes in laws, regulatory requirements and enforcement, as well as risks related to fluctuations in currency exchange rates and political, social and economic instability in foreign countries. If we are unable to manage our international operations successfully, our financial results could be adversely affected.
Controlled substance legislation may differ in other jurisdictions and could restrict our ability to market our products internationally, which would result in increased business and economic risks that could affect our financial results.
Controlled substance legislation may differ in other jurisdictions and could restrict our ability to market our products internationally. Most countries are parties to the Single Convention on Narcotic Drugs 1961, which governs international trade and domestic control of narcotic substances, including Cannabis extracts. Countries may interpret and implement their treaty obligations in a way that creates a legal obstacle to our obtaining marketing approval for Product Candidates in those countries. These countries may not be willing or able to amend or otherwise modify their laws and regulations to permit our Product Candidates to be marketed or achieving such amendments to the laws and regulations may take a prolonged period of time. We would be unable to market our Product Candidates in countries with such obstacles in the near future or perhaps at all without modification to laws and regulations.
Product liability lawsuits against us could cause us to incur substantial liabilities.
Our use of our Product Candidates in clinical trials and the sale of our Product Candidates, if approved, exposes us to the risk of product liability claims. Product liability claims might be brought against us by patients, healthcare providers or others selling or otherwise coming into contact with our Product Candidates. For example, we may be sued if any product we develop allegedly causes injury or is alleged to be otherwise unsuitable during product testing, manufacturing, marketing or sale. Any such product liability claims may include allegations of defects in manufacturing, defects in design, a failure to warn of dangers inherent in the product, including as a result of interactions with alcohol or other drugs, negligence, strict liability, and a breach of warranties. Claims could also be asserted under local jurisdiction consumer protection acts. If we become subject to product liability claims and cannot successfully defend ourselves against them, we could incur substantial liabilities. In addition, regardless of merit or eventual outcome, product liability claims may result in, among other things:
| ● | withdrawal of patients from our clinical trials; |
| ● | substantial monetary awards to patients or other claimants; |
| ● | decreased demand for our Product Candidates following marketing approval, if obtained; |
| ● | damage to our reputation and exposure to adverse publicity; |
| ● | increased FDA warnings on product labels or increased warnings imposed by the EMA or other regulatory authorities; |
| ● | litigation costs; |
| ● | distraction of management’s attention from our primary business; |
| ● | loss of revenue; and |
| ● | the inability to successfully commercialize our Product Candidates, if approved. |
Our current clinical trial liability insurance coverage may not be sufficient to reimburse us for any expenses or losses we may suffer. Moreover, insurance coverage is becoming increasingly expensive and, in the future, we may not be able to maintain insurance coverage at a reasonable cost or in sufficient amounts to protect us against losses due to liability. If we obtain marketing approval for our Product Candidates, we intend to expand our insurance coverage to include the sale of commercial products; however, we may be unable to obtain product liability insurance on commercially reasonable terms or in adequate amounts. Large judgments have been awarded in class action lawsuits based on drugs that had unanticipated side effects. The cost of any product liability litigation or other proceedings, even if resolved in our favor, could be substantial, particularly in light of the size of our business and financial resources. A product liability claim or series of claims brought against us could cause our share price to decline and, if we are unsuccessful in defending such a claim or claims and the resulting judgments exceed our insurance coverage, our financial condition, results of operations, business and prospects could be materially adversely affected.
Failure to protect our information technology infrastructure against cyber-based attacks, network security breaches, service interruptions, or data corruption could significantly disrupt our operations and adversely affect our business and operating results.
We rely on information technology, telephone networks and systems, including the internet, to process and transmit sensitive electronic information and to manage or support a variety of business processes and activities. We use enterprise information technology systems to record, process and summarize financial information and results of operations for internal reporting purposes and to comply with regulatory, financial reporting, legal and tax requirements. Despite the implementation of security measures, our information technology systems, and those of our third-party contractors and consultants, are vulnerable to a cyber-attack, malicious intrusion, breakdown, destruction, loss of data privacy or other significant disruption. Any such successful attacks could result in the theft of intellectual property or other misappropriation of assets, or otherwise compromise our confidential or proprietary information and disrupt our operations. Cyber-attacks are becoming more sophisticated and frequent, and our systems could be the target of malware and other cyber-attacks. We have invested in our systems and the protection of our data to reduce the risk of an intrusion or interruption, and we monitor our systems on an ongoing basis for any current or potential threats. Nonetheless, our computer systems are subject to penetration and our data protection measures may not prevent unauthorized access. We can give no assurances that these measures and efforts will prevent interruptions or breakdowns. If we are unable to detect or prevent a security breach or cyber-attack or other disruption from occurring, then we could incur losses or damage to our data, or inappropriate disclosure of our confidential information or that of others; and we could sustain damage to our reputation, suffer disruptions to our research and development and incur increased operating costs including increased cybersecurity and other insurance premiums, costs to mitigate any damage caused and protect against future damage, and be exposed to additional regulatory scrutiny or penalties and to civil litigation and possible financial liability. For instance, the loss of preclinical or clinical data could result in delays in our development and regulatory filing efforts and significantly increase our costs.
Certain macroeconomic and geopolitical conditions, which are outside of our control, as well as the evolution of methods and techniques used by bad actors, may also make us more susceptible to a cybersecurity attack. For example, tensions between Russia and several western nations (and their respective allies) in connection with the Russia-Ukraine War could result in retaliatory actions being undertaken by supporters of Russia, including in the form of espionage, phishing campaigns and other forms of cyber-attacks. Moreover, pro-Russian ransomware cybercriminals and gangs have previously publicly threatened to augment their hacking efforts in response to the implementation of sanctions and other responsive actions taken by western countries (and their allies). Increasing costs associated with information security, such as increased investment in technology, the cost of compliance and costs resulting from consumer fraud could cause our business and results of operations to suffer materially. Likewise, within a few hours of the commencement of the Hamas-Israel conflict, activist hackers commenced cyberattacks against both Israeli and Palestinian websites, and in a short period, had targeted dozens of government websites and media outlets. Such cyber-intrusions included DDOS attacks, attempts to overload websites with ‘junk’ traffic and ultimately bring down the site.
The methods and techniques used by cyber threat actors to gain entry into our network and access our computer systems, software and data will become more advanced with the use of AI and may become increasingly difficult or impossible to detect and prevent. As these threats continue to evolve, we may be required to invest significant additional resources to modify and enhance our information security and controls or to investigate and remediate any security vulnerabilities. While our technology infrastructure is designed to safeguard and protect personal and business information, we have limited ability to monitor the implementation of similar safeguards by our vendors.
Any cyberattack, unauthorized intrusion, malicious software infiltration, network disruption, corruption of data, misuse or theft of private or other sensitive information, or inadvertent acts by our own employees, could result in the disclosure or misuse of confidential or proprietary information, which could have a material adverse effect on our business operations or that of our clients. If we experience a significant data security breach, fail to detect and appropriately respond to a significant data security breach, or fail to comply with the various cybersecurity regulations, including the California Consumer Privacy Act and the California Privacy Rights Act in the United States, we could be exposed to government enforcement actions and private litigation. These losses may exceed our insurance coverage for such incidents. In addition, our employees and clients could lose confidence in our ability to protect their personal and proprietary information, which could cause them to terminate their relationships with us. Any loss of confidence arising from a significant data security breach could hurt our reputation, further damaging our business.
Our failure to comply with data protection laws and regulations could lead to government enforcement actions and significant penalties against us, and adversely impact our operating results.
We are subject to various domestic and international data protection laws and regulations (i.e., laws and regulations that address privacy and data security). The legislative and regulatory landscape for data protection continues to evolve, and in recent years there has been an increasing focus on privacy and data security issues. Numerous laws, including data breach notification laws, health information privacy laws and consumer protection laws, govern the collection, use and disclosure of health-related and other personal information. In addition, we may obtain health information from third parties (e.g., healthcare providers who prescribe our products) that are subject to privacy and security requirements under HIPAA regulations.
EU Member States, Australia and other countries have also adopted data protection laws and regulations, which impose significant compliance obligations. For example, the collection and use of personal data in the EU is governed by the provisions of the General Data Protection Regulation (“GDPR”). The GDPR and the national implementing legislation of the EU Member States impose strict obligations and restrictions on the ability to collect, analyze and transfer personal data, including health data from clinical trials and adverse event reporting. In particular, these obligations and restrictions concern the consent of the individuals to whom the personal data relates, the information provided to the individuals, the rights of individuals to control personal data and the security and confidentiality of the personal data. The related UK GDPR and the UK Data Protection Act of 2018, which ensures that the United Kingdom has in effect the same high standards for data protection in place as under the GDPR, impose stringent operational requirements in the United Kingdom (including through restrictions on processing of personal data and cross-border transfers of personal data, and mandatory breach reporting to regulators and, under certain circumstances, to the individuals whose personal data was compromised in the breach). In addition, the Australian Privacy Act 1986 (Cth), and other laws in the states and territories in Australia where we conduct certain of our clinical trials, apply similar restrictions on our ability to collect, analyze and transfer medical records and other patient data.
Other new laws and regulations are rapidly coming into effect while existing legislation is quickly evolving. In the United States, the SEC adopted new rules requiring public companies to disclose information about a material cybersecurity incident, including any breach of personal data, within four business days of determining that it has experienced a material cybersecurity incident. Likewise, several privacy laws in the United States came into effect in 2023, including in California, Virginia, Colorado, Connecticut and Utah, and new state privacy laws that have or will come into effect in 2024, including in Montana, Oregon and Texas, all of which give new data privacy rights to their respective residents and impose significant obligations on controllers and processors of consumer data.
There is additionally increasing U.S. and foreign activity in the regulation of AI, and other similar uses of technology. For example, in Europe, there is a proposed regulation (the Artificial Intelligence Act) that, if adopted and approved, could impose onerous and substantial obligations related to the use of AI-related systems. Additionally, several states and localities in the United States have enacted measures related to the use of AI and machine learning in products and services. In October 2023, the President of the United States issued an executive order on the Safe, Secure and Trustworthy Development and Use of AI, emphasizing the need for transparency, accountability and fairness in the development and use of AI tools, and AI is the subject of evolving review by various governmental and regulatory agencies, including the SEC and the Federal Trade Commission. Depending on how these AI laws and regulations are interpreted, and to the extent that our business practices, products and services utilize AI, we could be subject to, and need to comply with, such obligations. Moreover, our development and use of AI, and the uncertain regulatory environment, could result in reputational harm, liability or other material and adverse consequences to our financial condition and business operations. The introduction of AI technologies into new or existing products may also result in new or enhanced governmental or regulatory scrutiny, litigation, confidentiality or security risks, ethical concerns, or other complications that could adversely affect our business, reputation, or financial results. The intellectual property ownership and license rights, including copyright, surrounding AI technologies has not been fully addressed by courts or national or local laws or regulations, and the use or adoption of third-party AI technologies into our products and services may result in exposure to claims of copyright infringement or other intellectual property misappropriation. Uncertainty around new and emerging AI technologies, such as generative AI, may require additional investment in the development and maintenance of proprietary datasets and machine learning models, development of new approaches and processes to provide attribution or remuneration to creators of training data, and development of appropriate protections and safeguards for handling the use of customer data with AI technologies, which may be costly and could impact our expenses if we decide to expand generative AI into our product offerings. AI technologies, including generative AI, may create content that appears correct but is factually inaccurate or flawed. Our customers or others may rely on or use this flawed content to their detriment, which may expose us to brand or reputational harm, competitive harm, and/or legal liability. The use of AI technologies presents emerging ethical and social issues, and if we enable or offer solutions that draw scrutiny or controversy due to their perceived or actual impact on customers or on society as a whole, we may experience brand or reputational harm, competitive harm, and/or legal liability.
Existing privacy-related laws and regulations in the United States and other countries are evolving and are subject to potentially differing interpretations, and various U.S. federal and state or other international legislative and regulatory bodies may expand or enact laws regarding privacy and data security-related matters. Due to the fact that privacy and information security laws and regulations are subject to change from time to time, our compliance with them may result in cost increases due to necessary systems changes and the development of new processes. Any new or modified laws and regulations may require that we modify our data processing practices and policies, and incur substantial costs and expenses in an effort to comply with such laws and regulations. These laws are complex and there is no ubiquitous approach to maintaining compliance. Requirements may be interpreted and applied in a manner that is inconsistent from one jurisdiction to another or may conflict with other rules or our practices. If we fail to comply with any of these laws and regulations, we could be subjected to legal risk and other adverse effects to our business and operations.
A claim or series of claims brought against us alleging a failure to comply with these laws, or changes in the way in which these laws are implemented, could lead to government enforcement actions and significant penalties against us, and adversely impact our operating results and could cause our share price to decline and, if we are unsuccessful in defending such a claim or claims and the resulting judgments exceed our insurance coverage, our financial condition, results of operations, business and prospects could be materially adversely affected.
Our results of operations could be materially and adversely affected if we cannot keep pace with technological changes impacting the development of our products and implementation of our business needs, including with respect to automation and the use of AI.
Our success depends on our ability to keep pace with rapid technological changes affecting the development of our products and implementation of our business needs. Emerging technological trends such as AI, machine learning and automation are impacting industries and business operations. If we do not sufficiently invest in new technology and industry developments, appropriately implement new technologies or evolve our business at sufficient speed and scale in response to such developments, or if we do not make the right strategic investments to respond to these developments, our products, results of operations and ability to develop and maintain our business could be negatively affected. Our competitors or other third parties may incorporate AI technologies into their services, products and business more quickly or more successfully than us, which could impair our ability to compete effectively and materially and adversely affect our results of operations and financial condition.
Growing emphasis by the investment community, regulators and other stakeholders on environmental, social and governance-related matters could impact our business and operations.
As members of the investment community have started to heavily factor in a company’s commitment to environmental, social and governance (“ESG”)-related initiatives and sustainability performance as part of their overall investment thesis and strategy, such investors could elect to eventually forego their investment in us to the extent we fail to satisfy such metrics. Moreover, the increased focus by investors, regulators and other stakeholders on ESG related practices and disclosures has created, and will likely create for the foreseeable future, increased pressure regarding the enhancement of, and modification to, our disclosure and governance practices. Recently, there has been a growing concern and emphasis by governmental agencies regarding the effects of climate change on the environment and the need to make disclosures to investors regarding a company’s environmental footprint. For example, on March 6, 2024, the SEC adopted a final rule requiring public companies to include certain climate-related disclosures in their respective registration statements and annual reports filed with the SEC, including climate-related financial statement metrics, greenhouse gas emissions and climate-related targets and goals, and management’s role in managing material climate-related risks. A number of state legislators and regulators, including California laws S.B. 253, S.B. 261 and A.B. 1305 in the State of California, as well as non-U.S. governmental agencies (such as the EU’s Corporate Sustainability Reporting Directive), have adopted or are currently considering proposing or adopting other rules, regulations, directives, initiatives and laws requiring ESG-related disclosures or limiting (or affirmatively requiring) certain ESG-related conduct. In the event that we were to become subject to any of the newly adopted climate change and/or ESG-related disclosure regimes, including in the United States and elsewhere, it could require us to, among other things, (i) restrict or limit our operating activities or other conduct, (ii) make material capital improvements and expend material capital resources in connection with such compliance efforts, and (iii) alter our business and operational strategy more generally. Furthermore, there continues to be a lack of consistent proposed climate change and ESG-related legislation, which creates regulatory and economic uncertainty. Separately, enhanced climate-related disclosure requirements and obligations could lead to reputational or other harm with customers, regulators, investors or other stakeholders and could also increase our litigation risks relating to statements alleged to have been made by us or others in our industry regarding climate change risks, or in connection with any future disclosures we may make regarding reported emissions, particularly given the inherent approximations, estimations and uncertainties with respect to calculating, determining and reporting greenhouse gas emissions. Additionally, governmental regulators, including the SEC, have also from time to time applied additional scrutiny to existing climate change-related assertions in public disclosures, increasing the potential for enforcement if any such governmental regulator were to allege that our climate change-related disclosures are misleading or deficient. As a result of the foregoing, we currently face, and are likely to continue to face, increasing pressure regarding our ESG-related disclosures, practices, initiatives and sustainability performance in the near- and long-term. We continue to monitor for these changes and their potential impact on our business, financial condition and industry at large, and seek to implement measures to comply with all such newly implemented requirements; however, given the rapidly changing nature of these rules, regulations, directives, initiatives and laws, and the heightened regulatory scrutiny being applied by governmental agencies across numerous jurisdictions, it is not possible to predict how such matters will ultimately impact our business or that of our critical counterparties at this time.
Climate change may have an impact on our business.
While we seek to mitigate our business-related risks associated with climate change, we recognize that there are inherent climate-related risks wherever business is conducted. Any of our locations may be vulnerable to the adverse effects of climate change. Furthermore, it is more difficult to mitigate the impact of these events on our employees while they work from home as a result of the COVID-19 pandemic. Changing market dynamics, global policy developments, and the increasing frequency and impact of extreme weather events on critical infrastructure in the United States, Canada and elsewhere have the potential to disrupt our business, the business of our suppliers, and the business of our customers, and may cause us to experience higher attrition, losses and additional costs to maintain or resume operations. In particular, we rely on data centers to deliver our solutions, which consume significant amounts of energy. To the extent that energy prices increase as a result of carbon pricing or other measures, this could affect our cost structure.
Our success depends on our ability to continue to innovate and create new products and enhancements to our existing products.
To keep pace with technological developments, satisfy increasingly sophisticated customer requirements and achieve market acceptance, we must enhance and improve existing products and continue to introduce new products and services. If we are unable to successfully develop new products, integrate acquired products or enhance and improve existing products or if we fail to position and/or price our products to meet market demand, our business and operating results will be adversely affected. Accelerated product introductions and short product life cycles require high levels of expenditures for research and development that could adversely affect our results of operations. Further, the introduction of new products could require long development and testing periods and may not be introduced in a timely manner or may not achieve the broad market acceptance necessary to generate significant revenue. Further, if a competitor develops a new, less expensive product using a different technological approach to delivering informational services over existing networks, our products would no longer be competitive. Conversely, even if we are successful in rapidly developing new products ahead of our competitors, if we do not cost-effectively manage our inventory levels of existing products when making the transition to new products, our financial results could be negatively affected by write-offs as a result of high levels of obsolete inventory. If any of the foregoing were to occur, our operating results could be materially and adversely impacted.
Risks Related to our Securities
The market prices for our Common Shares are volatile and will fluctuate.
The trading price of our Common Shares has been and could remain volatile, and the market price of our Common Shares may decrease. The market price of our Common Shares has historically experienced and may continue to experience significant volatility. From July 1, 2023 through June 30, 2024, the closing market price of our Common Shares has fluctuated from a high of $1.22 per share to a low of $0.22 per share. The volatile nature of our Common Share price may cause investment losses for our stockholders. In addition, the market price of stock in small capitalization biotech companies is often driven by investor sentiment, expectation, and perception, all of which may be independent of fundamental, objective, and intrinsic valuation metrics or traditional financial performance metrics, thereby exacerbating volatility.
The market price for our Common Shares is anticipated to be volatile and subject to wide fluctuations in response to numerous factors, many of which are beyond our control, including the following: (i) actual or anticipated fluctuations in our quarterly financial results; (ii) recommendations by securities research analysts; (iii) changes in the economic performance or market valuations of other issuers that investors deem comparable to ours; (iv) addition or departure of our executive officers or members of our Board and other key personnel; (v) release or expiration of lock-up or other transfer restrictions on outstanding Common Shares; (vi) sales or perceived sales of additional Common Shares; (vii) liquidity of the Common Shares; (viii) significant acquisitions or business combinations, strategic partnerships, joint ventures or capital commitments by or involving us or our competitors; and (ix) news reports relating to trends, concerns, technological or competitive developments, regulatory changes and other related issues in our industry or target markets. Financial markets often experience significant price and volume fluctuations that affect the market prices of equity securities of public entities and that are, in many cases, unrelated to the operating performance, underlying asset values or prospects of such entities. Accordingly, the market price of our Common Shares may decline even if our operating results, underlying asset values or prospects have not changed. Additionally, these factors, as well as other related factors, may cause decreases in asset values that are deemed to be other than temporary, which may result in impairment losses. As well, certain institutional investors may base their investment decisions on consideration of our environmental, governance and social practices and performance against such institutions’ respective investment guidelines and criteria, and failure to meet such criteria may result in limited or no investment in our Common Shares by those institutions, which could materially adversely affect the trading price of our Common Shares. There can be no assurance that continuing fluctuations in price and volume will not occur. If such increased levels of volatility and market turmoil continue for a protracted period of time, our operations could be materially adversely impacted and the trading price of our Common Shares may be materially adversely affected.
Raising additional capital may cause dilution to our existing shareholders, restrict our operations or require us to relinquish rights to our technologies or Product Candidates.
We will seek additional capital through a combination of private and public equity offerings, debt financings, strategic partnerships and alliances and licensing arrangements. To the extent that we raise additional capital through the sale of equity or convertible debt securities, existing ownership interests will be diluted and the terms of such financings may include liquidation or other preferences that adversely affect the rights of existing shareholders. Debt financings may be coupled with an equity component, such as warrants to purchase shares, which could also result in dilution of our existing shareholders’ ownership. The incurrence of indebtedness would result in increased fixed payment obligations and could also result in certain restrictive covenants, such as limitations on our ability to incur additional debt, limitations on our ability to acquire or license intellectual property rights and other operating restrictions that could adversely impact our ability to conduct our business and may result in liens being placed on our assets and intellectual property. If we were to default on such indebtedness, we could lose such assets and intellectual property. If we raise additional funds through strategic partnerships and alliances and licensing arrangements with third parties, we may have to relinquish valuable rights to our Product Candidates or grant licenses on terms that are not favorable to us.
Future offerings of debt or equity securities may rank senior to our Common Shares.
If we decide to issue debt or equity securities in the future ranking senior to our Common Shares or otherwise incur additional indebtedness, it is possible that these securities or indebtedness will be governed by an indenture or other instrument containing covenants restricting our operating flexibility and limiting our ability to pay dividends to shareholders. Additionally, any convertible or exchangeable securities that we issue in the future may have rights, preferences and privileges, including with respect to dividends, more favorable than those of Common Shares and may result in dilution to shareholders. Because our decision to issue debt or equity securities in any future offering or otherwise incur indebtedness will depend on market conditions and other factors beyond our control, we cannot predict or estimate the amount, timing or nature of our future offerings or financings, any of which could reduce the market price of our Common Shares and dilute their value.
More generally, our level of indebtedness could have significant and adverse effects on our business. For example, our level of indebtedness and the terms of our debt agreements could: (i) make it more difficult for us to satisfy our financial obligations under our indebtedness and our contractual and commercial commitments and increase the risk that we may default on our debt obligations; (ii) prevent us from raising the funds necessary to repurchase notes tendered to us if we undergo a change of control; (iii) require us to use a substantial portion of our cash flow from operations to pay interest and principal on the Second Amended and Restated Credit Agreement and other debt, which would reduce the funds available for working capital, capital expenditures and other general corporate purposes; (iv) limit our ability to obtain additional financing for working capital, capital expenditures, acquisitions and other investments, or general corporate purposes, which may limit our ability to execute our business strategy; (v) limit our ability to refinance our current or future indebtedness on terms that are commercially reasonable, if at all; (vi) heighten our vulnerability to downturns in our business, our industry or in the general economy, and restrict us from exploiting business opportunities or making acquisitions; (vii) place us at a competitive disadvantage compared to those of our competitors that may have proportionately less debt; (viii) limit management’s discretion in operating our business; and (ix) limit our flexibility in planning for, or reacting to, changes in our business, the industry in which we operate or the general economy. Each of these factors may have a material and adverse effect on our financial condition and viability. Our ability to satisfy our other debt obligations will depend on our future operating performance, which will be affected by prevailing economic conditions and financial, business and other factors affecting our company and industry, many of which are beyond our control.
Future sales of Common Shares by our officers, directors and affiliates may negatively impact the market price for our Common Shares.
Subject to compliance with applicable securities laws, our directors and officers and their affiliates may sell some or all of their Common Shares in the future. No prediction can be made as to the effect, if any, such future sales of Common Shares may have on the market price of the Common Shares prevailing from time to time. However, the future sale of a substantial number of Common Shares by our directors and officers and their affiliates, or the perception that such sales could occur, could adversely affect prevailing market prices for our Common Shares.
We do not currently pay dividends on our Common Shares and have no intention to pay dividends on our Common Shares for the foreseeable future.
No dividends on our Common Shares have been paid by us to date. We do not intend to declare or pay any cash dividends in the foreseeable future. Payment of any future dividends will be at the discretion of our Board, after taking into account a multitude of factors appropriate in the circumstances, including our operating results, financial condition and current and anticipated cash needs. In addition, the terms of any future debt or credit facility may preclude us from paying any dividends unless certain consents are obtained and certain conditions are met.
We are exposed to risks related to currency exchange rates.
We currently hold most of our cash, cash equivalents and short-term investments in U.S. dollars which is our functional currency. A portion of our current operations is conducted in Canadian dollars. Exchange rate fluctuations between other currencies and the U.S. dollar create risk in several ways, including the following:
| ● | weakening of the Canadian dollar may decrease the value of our Canadian dollar cash, cash equivalents and short-term investments; | |
| ● | weakening of the U.S. dollar may increase the cost of operations and products/services sourced in Canada; | |
| ● | the exchange rates on non-U.S. dollar transactions and cash deposits can distort our financial results; and | |
| ● | commercial product pricing and profit margins are affected by currency fluctuations. |
For as long as we are an “emerging growth company” we intend to take advantage of reduced disclosure and governance requirements applicable to emerging growth companies, which could result in our Common Shares being less attractive to investors and could make it more difficult for us to raise capital as and when we need it.
We are an “emerging growth company,” as defined in the JOBS Act, and we have taken advantage, and intend to continue to take advantage, of certain exemptions from various reporting requirements that are applicable to other public companies that are not emerging growth companies including, but not limited to, not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act, reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements, and exemptions from the requirements of holding a non-binding advisory vote on executive compensation and shareholder approval of any golden parachute payments not previously approved.
Investors may find our Common Shares less attractive because we rely on these exemptions, which could contribute to a less active trading market for our Common Shares or volatility in our share price. In addition, we may be less attractive to investors and it may be difficult for us to raise additional capital as and when we need it. Investors may be unable to compare our business with other companies in our industry if they believe that our financial accounting is not as transparent as other companies in our industry. If we are unable to raise additional capital as and when we need it, our financial condition and results of operations may be materially and adversely affected.
We may take advantage of these reporting exemptions until we are no longer an emerging growth company.
There remains increased focus from lawmakers and regulators on corporate ESG practices, including climate change and related ESG disclosure requirements. Expectations regarding voluntary ESG initiatives and disclosures may result in increased costs (including but not limited to increased costs related to compliance, stakeholder engagement, contracting and insurance), changes in demand for certain products, enhanced compliance or disclosure obligations, or other adverse impacts to our business, financial condition or results of operations. In addition, standards for tracking and reporting ESG matters continue to evolve, and our business may be impacted by new laws, regulations or investor criteria in the United States, Europe and around the world related to ESG. In March 2024, the SEC adopted new rules that will require registrants to provide certain climate-related information in their registration statements and annual reports. The rules require information about a registrant’s climate-related risks that are reasonably likely to have a material impact on its business, results of operations, or financial condition. The required information about climate-related risks will also include disclosure of a registrant’s greenhouse gas emissions. In addition, the rules will require registrants to present certain climate-related financial metrics in their audited financial statements. The SEC’s newly adopted climate-related disclosure rules may require us to incur significant additional costs to comply, including the implementation of significant additional internal controls processes and procedures regarding matters that have not been subject to such controls in the past and expanded data collection, analysis and certification with respect to greenhouse gas emissions reporting that may not be complete or accurate, and impose increased oversight obligations on our management and board of directors. These and other regulations, disclosure-related and otherwise, including California laws S.B. 253, S.B. 261 and A.B. 1305 and the EU’s Corporate Sustainability Reporting Directive, may increase our costs as well as increase scrutiny regarding our ESG efforts, which may enhance the risks discussed in this risk factor. These legal and regulatory requirements, as well as investor expectations related to ESG practices and disclosures are subject to change, can be unpredictable, and may be difficult and expensive for us to comply with. If we fail to adapt to or comply with all laws, regulations, policies and related interpretations, our business and reputation could be negatively impacted, and our share price and access to/cost of capital could be materially and adversely affected. Additionally, many of our customers and suppliers may be subject to similar expectations, which may augment or create additional risks, including risks that may not be known to us.
If we fail to maintain an effective system of internal control over financial reporting in the future, we may not be able to accurately report our financial condition, results of operations or cash flows, which may adversely affect investor confidence in us and, as a result, the value of our Common Shares.
We will be required, under Section 404 of the Sarbanes-Oxley Act, to furnish a report by management on, among other things, the effectiveness of our internal control over financial reporting. This assessment includes disclosure of any material weaknesses identified by our management in our internal control over financial reporting. A material weakness is a deficiency, or combination of deficiencies, in internal control over financial reporting that results in more than a reasonable possibility that a material misstatement of annual or interim financial statements will not be prevented or detected on a timely basis. Section 404 of the Sarbanes-Oxley Act also generally requires an attestation from our independent registered public accounting firm on the effectiveness of our internal control over financial reporting. However, for as long as we remain an emerging growth company as defined in the JOBS Act, we intend to take advantage of the exemption permitting us not to comply with the independent registered public accounting firm attestation requirement.
Our compliance with Section 404 will require that we incur substantial accounting expense and expend significant management efforts. We may not be able to complete our evaluation, testing and any required remediation in a timely fashion. During the evaluation and testing process, if we identify one or more material weaknesses in our internal control over financial reporting, we will be unable to assert that our internal control over financial reporting is effective. We cannot assure you that there will not be material weaknesses or significant deficiencies in our internal control over financial reporting in the future. Any failure to maintain internal control over financial reporting could severely inhibit our ability to accurately report our financial condition, results of operations or cash flows. This may expose us, including individual executives, to potential liability which could significantly affect our business. If we are unable to conclude that our internal control over financial reporting is effective, or if our independent registered public accounting firm determines we have a material weakness in our internal control over financial reporting once that firm begins its audits of internal control over financial reporting, we could lose investor confidence in the accuracy and completeness of our financial reports, the market price of our Common Shares could decline, and we could be subject to sanctions or investigations by Nasdaq, the SEC, or other regulatory authorities. Failure to remedy any material weakness in our internal control over financial reporting, or to implement or maintain other effective control systems required of public companies, could also restrict our future access to the capital markets.
Our disclosure controls and procedures may not prevent or detect all errors or acts of fraud.
Our disclosure controls and procedures are designed to reasonably assure that information required to be disclosed by us in reports we file or submit under the Securities Exchange Act of 1934 is accumulated and communicated to management, recorded, processed, summarized and reported within the time periods specified in the rules and forms of the SEC. We believe that any disclosure controls and procedures or internal controls and procedures, no matter how well conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met.
These inherent limitations include the realities that judgments in decision-making can be faulty, and that breakdowns can occur because of simple error or mistake. Additionally, controls can be circumvented by the individual acts of some persons, by collusion of two or more people or by an unauthorized override of the controls. Accordingly, because of the inherent limitations in our control system, misstatements or insufficient disclosures due to error or fraud may occur and not be detected.
Deficiencies in disclosure controls and procedures and internal control over financial reporting could result in a material misstatement in our financial statements.
We could be adversely affected if there are deficiencies in our disclosure controls and procedures or in our internal controls over financial reporting. The design and effectiveness of our disclosure controls and procedures and our internal controls over financial reporting may not prevent all errors, misstatements or misrepresentations. Consistent with other entities in similar stages of development, we have a limited number of employees currently in the accounting group, limiting our ability to provide for segregation of duties and secondary review. A lack of resources in the accounting group could lead to material misstatements resulting from undetected errors occurring from an individual performing primarily all areas of accounting with limited secondary review. Deficiencies in internal controls over financial reporting which may occur could result in material misstatements of our results of operations, restatements of financial statements, other required remediations, a decline in the price of our Common Shares, or otherwise materially adversely affect our business, reputation, results of operations, financial condition or liquidity.
In connection with the audit of our financial statements as of and for the year ended June 30, 2023, a material weaknesse in our internal control over financial reporting was identified and we may identify additional material weaknesses in the future.
In connection with the preparation and audits of our financial statements as of and for the year ended June 30, 2023, a material weakness, (as defined under the Exchange Act and by the auditing standards of the U.S. Public Company Accounting Oversight Board (“PCAOB”)), was identified in our internal control over financial reporting. A material weakness is a deficiency, or a combination of deficiencies, in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of our annual financial statements will not be prevented or detected on a timely basis. The identified control deficiencies arose from a lack of resources in our finance function, which rose to a material weakness due to segregation of duty issues.
In light of the identified material weakness, it is possible that, had we performed a formal assessment of our internal control over financial reporting or had our independent registered public accounting firm performed an audit of our internal control over financial reporting in accordance with PCAOB standards, additional control deficiencies may have been identified.
We subsequently designed and implemented controls to remediate the material weakness, including strengthening our finance function and addressing segregation of duty issues. While our management has determined that we have remediated such material weakness as of our fiscal year ended June 30, 2024, we cannot assure you that (i) the measures we have taken or will take will be entirely sufficient to remediate the material weakness or avoid the identification of additional material weaknesses in the future nor (ii) we will not experience flaws in our internal controls and procedures in the future. Our failure to implement and maintain effective internal control over financial reporting could result in the identification of additional errors in our consolidated financial statements that could result in a further restatement of our financial statements and could cause us to fail to meet our periodic reporting obligations, any of which could diminish investor confidence in us, cause a decline in the price of our Common Shares and subject us to litigation or regulatory enforcement actions.
We have incurred, and will continue to incur, increased costs as a result of operating as a public company, and our management has been required, and will continue to be required, to devote substantial time to new compliance initiatives.
As a public company, we have incurred and are continuing to incur significant legal, accounting and other expenses and these expenses may increase even more after we are no longer an “emerging growth company.” We are subject to the reporting requirements of the Exchange Act and the rules adopted, and to be adopted, by the SEC. Our management and other personnel devote a substantial amount of time to these compliance initiatives.
Moreover, these rules and regulations have substantially increased our legal and financial compliance costs and made some activities more time-consuming and costly. The increased costs have increased our net loss. These rules and regulations may make it more difficult and more expensive for us to maintain sufficient director’s and officer’s liability insurance coverage. We cannot predict or estimate the amount or timing of additional costs we may continue to incur to respond to these requirements. The ongoing impact of these requirements could also make it more difficult for us to attract and retain qualified persons to serve on our Board, our Board committees or as executive officers.
Future sales and issuances of our Common Shares or rights to purchase Common Shares pursuant to our equity incentive plan could result in additional dilution of the percentage ownership of our shareholders and may cause our share price to fall.
We expect that significant additional capital will be needed in the future to continue our planned operations. To raise capital, we may sell substantial amounts of Common Shares or securities convertible into or exchangeable for Common Shares. These future issuances of Common Shares or Common Share-related securities to purchase Common Shares, together with the exercise of outstanding options and any additional shares issued in connection with acquisitions, if any, may result in material dilution to our investors. Such sales may also result in material dilution to our existing shareholders, and new investors could gain rights, preferences and privileges senior to those of holders of our Common Shares.
Pursuant to our 2017 Amended and Restated Stock Option Plan, and as amended at our Annual General Meeting in November 2020, our compensation committee is authorized to grant equity-based incentive awards in the form of options to purchase common shares to our directors, executive officers and other employees and service providers. As of September 20, 2024, there were 179,293 options available for future allocation pursuant to the 20% of the issued and outstanding shares allowed to be issued according to the terms of the Plan. Future equity incentive grants under our stock option plan may result in material dilution to our shareholders and may have an adverse effect on the market price of our common shares.
Provisions in our corporate charter documents and certain Canadian laws could delay or deter a change of control.
Provisions in our articles and our by-laws, as well as certain provisions under the BCBCA and applicable Canadian securities laws, may discourage, delay or prevent a merger, acquisition, tender offer or other change in control of us that some shareholders may consider favorable. In addition, because our Board is responsible for appointing the members of our management team, these provisions may frustrate or prevent any attempts by our shareholders to replace or remove our current management by making it more difficult for shareholders to replace members of our Board. As well, our preferred shares are available for issuance from time to time at the discretion of our Board, without shareholder approval. Our articles allow our Board, without shareholder approval, to determine the special rights to be attached to our preferred shares, and such rights may be superior to those of our Common Shares.
In addition, limitations on the ability to acquire and hold our Common Shares may be imposed by the Competition Act in Canada. This legislation permits the Commissioner of Competition of Canada (the “Commissioner”), to review any acquisition of a significant interest in us. This legislation grants the Commissioner jurisdiction to challenge such an acquisition before the Canadian Competition Tribunal if the Commissioner believes that it would, or would be likely to, result in a substantial lessening or prevention of competition in any market in Canada. The Investment Canada Act subjects an acquisition of control of a company by a non-Canadian to government review if the value of our assets, as calculated pursuant to the legislation, exceeds a threshold amount. A reviewable acquisition may not proceed unless the relevant minister is satisfied that the investment is likely to result in a net benefit to Canada. Any of the foregoing could prevent or delay a change of control and may deprive or limit strategic opportunities for our shareholders to sell their shares.
If securities or industry analysts publish inaccurate or unfavorable research about our business, our share price and trading volume may decline.
The trading market for our Common Shares depends in part on the research and reports that securities or industry analysts publish about us or our business. If one or more of the analysts who cover us downgrade our shares or publish inaccurate or unfavorable research about our business, our shares price may decline. If one or more of these analysts cease coverage of our company or fail to publish reports on us regularly, demand for our shares may decrease, which may cause our shares price and trading volume to decline.
We are incorporated in Canada, with our assets and officers primarily located in Canada, with the result that it may be difficult for investors to enforce judgments obtained against us or some of our officers.
We are a company organized and existing under the laws of British Columbia, Canada. Many of our directors and officers and the experts named in this Annual Report are residents of Canada or otherwise reside outside the United States, and all or a substantial portion of their assets, and a substantial portion of our assets, are located outside the United States. It may be difficult for holders of Common Shares who reside in the United States to effect service within the United States upon those directors, officers and experts who are not residents of the United States. It may also be difficult for holders of securities who reside in the United States to realize in the United States upon judgments of courts of the United States predicated upon our civil liability and the civil liability of our directors, officers and experts under the U.S. federal securities laws. Our Canadian counsel has advised us that there is doubt as to the enforceability in Canada against us or against our directors, officers and experts who are not residents of the United States, in original actions or in actions for enforcement of judgments of courts of the United States, of liabilities predicated solely upon U.S. federal or state securities laws.
Conversely, some of our directors and officers reside outside Canada and some of our assets are also located outside Canada. Therefore, it may not be possible for you to enforce in Canada against our assets or those directors and officers residing outside Canada, judgments obtained in Canadian courts based upon the civil liability provisions of the Canadian securities laws or other laws of Canada.
Risks Related to our Financial Position and Capital Needs
Our operating losses have raised substantial doubt regarding our ability to continue as a going concern.
Our operating losses raise substantial doubt about our ability to continue as a going concern. As a result, our independent registered public accounting firm included an explanatory paragraph in its report on our financial statements as of and for the years ended June 30, 2024 and June 30, 2023 with respect to this uncertainty. The perception of our ability to continue as a going concern may make it more difficult for us to obtain financing for the continuation of our operations and could result in the loss of confidence by investors, suppliers and employees.
We have incurred significant losses since our inception and anticipate that we will continue to incur losses in the future.
Since our inception as a pharmaceutical company in October 2014, we have devoted substantially all of our resources to the development of our proprietary Product Candidates. We have generated significant operating losses since our inception with an accumulated deficit to June 30, 2024 of approximately $109.1 million. Our net loss for the fiscal years ended June 30, 2024 and 2023 was approximately $7.7 million and $8.0 million, respectively. Substantially all of our losses have resulted from expenses incurred in connection with our research and development programs and from general and administrative costs associated with our operations.
We expect to continue to incur significant expenses and operating losses for the foreseeable future. We anticipate these losses will increase as we continue the research and development of, and clinical trials for, our Product Candidates. In addition to budgeted expenses, we may encounter unforeseen expenses, difficulties, complications, delays and other unknown factors that may adversely affect our business. If our Product Candidates fail in preclinical or clinical trials, or do not gain regulatory approval, or even if approved, fail to achieve market acceptance, we may never become profitable. Even if we achieve profitability in the future, we may not be able to sustain profitability in subsequent periods.
Due to our limited operating history and history of losses, any predictions about our future success, performance or viability may not be accurate.
We will require additional capital to fund our operations and if we fail to obtain necessary financing, we will not be able to complete the development and commercialization of our Product Candidates.
Our operations have consumed substantial amounts of cash since inception. We expect to continue to spend substantial and increasing amounts to conduct further research and development, preclinical testing and clinical trials of our Product Candidates, to seek regulatory approvals and reimbursement for our Product Candidates and to launch and commercialize any Product Candidates for which we receive regulatory approval.
As of June 30, 2024, we had approximately $6.6 million in cash, cash equivalents and short-term investments, which, we currently estimate funds our operations to the end of the fourth quarter of calendar 2024 (being the second fiscal quarter of 2025), depending on the level and timing of realizing revenues from the sale of BayMedica inventory as well as the level and timing of the Company’s operating expenses. Our ability to develop our research and development programs is subject to accessing additional capital, including through the sale of equity, partnership revenues, and out-licensing activities. There is no assurance that we will be successful in these efforts.
The progress of our Product Candidates for both current and prospective target indication(s) is uncertain because it is difficult to predict our spending for our Product Candidates up to the time that we seek FDA approval due to numerous factors, including, without limitation, the rate of progress of clinical trials, the results of preclinical studies and clinical trials for such indication, the costs and timing of seeking and obtaining FDA and other regulatory approvals for clinical trials and FDA guidance regarding clinical trials for such indication. Moreover, changing circumstances may cause us to expend cash significantly faster than we currently anticipate, and we may need to spend more cash than currently expected because of circumstances beyond our control. For these reasons, we are unable to state unequivocally the actual funds we will require for development and any approved marketing and commercialization activities. Our future funding requirements, both near and long-term, will depend on many factors, including, but not limited to:
| ● | the initiation, progress, timing, costs and results of preclinical studies and clinical trials for our Product Candidates; |
| ● | any change in the clinical development plans or target indications for these Product Candidates; |
| ● | the number and characteristics of Product Candidates that we develop or may in-license; |
| ● | the terms of any collaboration agreements we may choose to execute; |
| ● | the outcome, timing and cost of meeting regulatory requirements established by the Drug Enforcement Administration (“DEA”), the FDA, the European Medicines Agency, Health Canada (“HC”), or other comparable foreign regulatory authorities; |
| ● | The cost of filing, prosecuting, defending and enforcing our patent claims and other intellectual property rights; |
| ● | the cost of defending intellectual property disputes, including patent infringement actions brought by third parties against us; |
| ● | the effect of competing product and market developments; |
| ● | the costs and timing of the implementation of commercial scale manufacturing activities; and |
| ● | the cost of establishing, or outsourcing, sales, marketing and distribution capabilities for any Product Candidates for which we may receive regulatory approval in regions where we choose to commercialize our products on our own. |
We cannot be certain that additional funding will be available on acceptable terms, or at all. If we are unable to raise additional capital in sufficient amounts or on terms acceptable to us, we may have to significantly delay, scale back or discontinue the development or commercialization of one or more of our Product Candidates or one or more of our other research and development initiatives.
Any doubt about our ability to continue as a going concern may materially and adversely affect the price of our Common Shares, and it may be more difficult for us to obtain financing. Any doubt about our ability to continue as a going concern may also adversely affect our relationships with current and future collaborators, contract manufacturers and investors, who may become concerned about our ability to meet our ongoing financial obligations. If potential collaborators decline to do business with us or potential investors decline to participate in any future financings due to such concerns, our ability to increase our financial resources may be limited. We have prepared our financial statements on a going concern basis, which assumes that we will be able to meet our commitments, realize our assets and discharge our liabilities in the normal course of business. Our consolidated financial statements do not include any adjustment to reflect the possible future effects on the recoverability and classification of assets or the amounts and classification of liabilities that may result from the outcome of this uncertainty.
We may have difficulties identifying, successfully completing or integrating acquisitions, or maintaining or growing our acquired businesses.
We remain committed to our growth strategy of organically growing our strategic portions of our business while assessing strategic acquisitions, dispositions, partnerships and other strategic transactions. While we believe we have the experience required to execute this strategy, we do not have control over the market conditions prevailing or likely to prevail in the future, which may impact the ability to execute this strategy. There can be no assurances that we will be able to identify suitable acquisition candidates available for sale at reasonable valuations, consummate any acquisition or successfully integrate any acquired business into our operations. Moreover, there can be no assurance that we will be able to access further financial resources for other suitable acquisition opportunities that may become available to us. We have and will likely continue to have competition for acquisition opportunities from other parties including those that have greater financial resources or are willing to pay higher valuation multiples. To the extent we were to pursue or engage in any transactions, including acquisitions and dispositions, there is no guarantee that such transactions will be successful or, even if consummated, improve our operating results and financial condition. We may incur costs, breakage fees or other expenses in connection with any such transactions, and any such transactions may ultimately have a material adverse effect on our operating results.
Acquisitions involve significant risks and uncertainties, including, but not limited to, the following:
| ● | unanticipated costs and liabilities; | |
| ● | difficulties in marketing and integrating new products, software, businesses, operations and technology infrastructure in an efficient, effective and secure manner, including the integration of businesses where a portion or all of the business is in an adjacent industry; | |
| ● | the inability to achieve synergy and cost reduction targets assumed at the time of acquisition; | |
| ● | difficulties in maintaining customer and key supplier relations, including changing contract manufacturers as a result of lower volumes of business; | |
| ● | the potential loss of key employees of the acquired businesses, including as a result of cultural differences between the acquired company and our own; | |
| ● | the diversion of the attention of our senior management from the operation of our daily business; | |
| ● | the potential adverse effect on our net debt and liquidity position as a result of all or a portion of an acquisition purchase price being paid in cash; | |
| ● | the potential significant increase of our interest expense, leverage and debt service requirements if we incur additional debt to pay for an acquisition; | |
| ● | the potential issuance of securities that would dilute our shareholders’ percentage ownership; | |
| ● | the potential to incur restructuring and other related expenses, including significant transaction costs that may be incurred regardless of whether a potential strategic acquisition or investment is completed; | |
| ● | use of resources that are needed in other areas of our business; | |
| ● | the inability to maintain uniform standards, controls, policies and procedures, including the inability to establish and maintain adequate internal controls over financial reporting, and remediate, in whole or in part, any material weaknesses or significant deficiencies identified with respect to internal controls over financial reporting; | |
| ● | difficulties in entering markets in which we have no or limited direct prior experience and where competitors in such markets have stronger market positions; | |
| ● | difficulties in securing required regulatory approvals or otherwise satisfy closing conditions for a proposed transaction in a timely manner, or at all; |
| ● | potential impairment charges on higher levels of goodwill and intangible assets as a result of impairment testing performed on a regular basis; | |
| ● | higher amortization expenses related to acquired definite-lived intangible assets; and | |
| ● | becoming subject to intellectual property or other litigation. |
We currently have limited commercial revenue and may never become profitable.
In addition to the limited revenues from our BayMedica Products, our ability to generate revenue and become profitable depends upon our ability to obtain regulatory approval for, and successfully commercialize, our Product Candidates that we may develop, in-license or acquire in the future.
Even if we are able to successfully achieve regulatory approval for these Product Candidates, we do not know what the reimbursement status of our Product Candidates will be or when any of these products will generate revenue for us, if at all. We have not generated, and do not expect to generate, any revenue from Product Candidates for the foreseeable future, and we expect to continue to incur significant operating losses for the foreseeable future due to the cost of research and development, preclinical studies and clinical trials and the regulatory approval process for our Product Candidates. The number of future losses is uncertain and will depend, in part, on the rate of growth of our expenses.
Our ability to generate revenue and become profitable depends upon a number of additional factors, including our ability to:
| ● | successfully complete development activities, including the remaining preclinical studies and ongoing and planned clinical trials for our Product Candidates; |
| ● | in-license or acquire in the future, Product Candidates and other potential lines of business that we may develop; |
| ● | complete and submit NDAs to the FDA and Marketing Authorization Applications (“MAAs”) to the EMA, and obtain regulatory approval for indications for which there is a commercial market; |
| ● | complete and submit applications to, and obtain regulatory approval from, other foreign regulatory authorities; |
| ● | manufacture any approved products in commercial quantities and on commercially reasonable terms; |
| ● | develop a commercial organization, or find suitable partners, to market, sell and distribute approved products in the markets in which we have retained commercialization rights; |
| ● | achieve acceptance among patients, clinicians and advocacy groups for any products we develop; |
| ● | obtain coverage and adequate reimbursement from third parties, including government payors; and |
| ● | set a commercially viable price for any products for which we may receive approval. |
We are unable to predict the timing or amount of increased expenses, or when or if we will be able to achieve or maintain profitability. Even if we are able to complete the processes described above, we anticipate incurring significant costs associated with commercializing our Product Candidates.
Changes in tax laws and unanticipated tax liabilities could adversely affect our effective income tax rate and ability to achieve profitability.
We are subject to income taxes in the United States and Canada. As our operations expand, we may become subject to income tax in jurisdictions outside of the United States and Canada. Our effective income tax rate in the future could be adversely affected by a number of factors including changes in the mix of earnings (losses) in countries with differing statutory tax rates, changes in the valuation of deferred tax assets and liabilities and changes in tax laws. We regularly assess all of these matters to determine the adequacy of our tax provision which is subject to discretion. If our assessments are incorrect, it could have an adverse effect on our business and financial condition. There can be no assurance that income tax laws and administrative policies with respect to the income tax consequences generally applicable to us or to our subsidiaries will not be changed in a manner which adversely affects our shareholders.
Our ability to use our net operating loss carryforwards and other tax attributes may be materially limited.
As of June 30, 2024, we had non-capital loss (“NOL”) carryforwards of approximately $80.2 million available to offset future taxable income in Canada and the United States. These NOL carry-forwards begin to expire in 2026.
Our NOL carryforwards could expire unused and be unavailable to offset future income tax liabilities. Under provisions in the Canadian Income Tax Act, and corresponding provisions of Canadian provincial law, if a corporation undergoes an “ownership change,” generally defined as a greater than 50% change, by value, the corporation’s ability to use its pre-change Canadian NOLs and other pre-change tax attributes, such as research and development tax credits, to offset its post-change income may be limited. Specifically, NOLs from a business before the change of control may be carried forward to taxation years after the change of control, but only if the same business is carried forward on after the change in control with a reasonable expectation of profit, and only to offset income from that business or a similar business. We have not performed any analyses under the applicable provisions in the Canadian Income Tax Act and cannot forecast or otherwise determine our ability to derive benefit from our various federal or provincial tax attribute carryforwards. As a result, if we earn net taxable income, our ability to use our pre-change NOL carryforwards to offset Canadian federal taxable income may be subject to limitations, which could potentially result in increased future tax liability to us. In addition, at the provincial level, there may be periods during which the use of NOLs is suspended or otherwise limited, which could accelerate or permanently increase provincial taxes owed.
In addition, we may experience ownership changes in the future as a result of subsequent shifts in our share ownership, including in any future offerings, some of which may be outside of our control. If we determine that an ownership change has occurred and our ability to use our NOL carryforwards is materially limited, it would harm our future operating results by effectively increasing our future tax obligations.
Changes to accounting standards may adversely impact the manner in which we report our financial position and operating results.
There are ongoing projects conducted by the Financial Accounting Standards Board in the United States that are expected to result in new pronouncements that continue to evolve, which could adversely impact the manner in which we report our financial position and operating results.
Natural disasters, public health crises, political crises, or other catastrophic events may adversely affect our business affairs, results of operations, financial condition, liquidity, availability of credit and foreign exchange exposure.
Changes in the global economic environment have created market uncertainty and volatility in recent years. The market and demand for metal commodities and related products has in recent years been adversely affected by global economic uncertainty, reduced confidence in financial markets, the COVID-19 pandemic, including any resurgence thereof, bank failures and credit availability concerns. These macro-economic events negatively affected the mining and minerals sectors in general. Global financial conditions remain subject to sudden and rapid destabilizations in response to economic shocks. A slowdown in the financial markets or other economic conditions, including but not limited to reduced consumer spending, decreased employment rates, adverse business conditions, high inflation, high fuel and energy costs, high consumer debt levels, a lack of available credit, the state of turmoil in the financial markets, high interest rates and/or tax rates, may adversely affect the Company’s growth and profitability. Future economic shocks may be precipitated by a number of causes, including the slowdown in the Chinese economy, a rise in the price of oil and other commodities, climate change disasters, geopolitical instability, including as a direct or indirect result of the Russo-Ukraine war and the ongoing Israel-Hamas conflict, further wars or acts of terrorism, the devaluation and volatility of global stock markets and natural disasters. Any sudden or rapid destabilization of global economic conditions could impact the Company’s ability to obtain equity or debt financing in the future on terms favorable to the Company or at all. In such an event, the Company’s operations and financial condition could be adversely impacted.
The Company assesses on a quarterly basis the carrying values of its assets. Should market conditions and commodity prices worsen and persist in a worsened state for a prolonged period of time, an assessment of the Company’s assets for impairment may be required.
The ongoing Russo-Ukraine War and the Israel-Hamas conflict, including the actual or perceived threat of an exacerbation of such conflicts, could have a material and adverse effect on our business, operations and financial condition.
Russia’s invasion of Ukraine in February 2022 has caused, and could continue to cause, increased volatility across the global financial markets, increased inflation, and turbulence in the markets in which we operate. In response to actions undertaken by Russia in Ukraine, several countries (including Canada, the United States and other western governments) have imposed stringent economic sanctions and export control measures, and may impose additional sanctions or export control measures in the near-term, which have included severe and complete restrictions on exports and other commerce and business dealings involving Russia, certain regions of Ukraine, Belarus and/or particular entities and individuals.
In May 2023, in coordination with the G7, Australia, and other partners, the United States imposed new sanctions on Russia. As part of these actions, the U.S. Department of State imposed sanctions on over 200 entities, individuals, vessels, and aircraft, as well as designated certain entities and individuals (i) across Russia’s defense and related materiel, technology, and metals and mining sectors and (ii) involved in expanding Russia’s future energy production and capacity. In December 2023, President Biden signed an executive order which seeks to strengthen U.S. sanctions authorities against financial facilitators of Russia’s war efforts, and additionally provided authority to broaden U.S. import bans on certain Russian goods. Likewise, in February 2024, the United States’ Treasury Department, State Department and Department of Commerce, collectively, imposed an extensive set of new sanctions on Russia, which specifically target Russia’s financial sector and military-industrial operations. Such sanctions seek to restrict Russia’s energy industry and limit the evasion of sanctions outside the United States, including by encompassing 500 additional persons associated with the ongoing Russo-Ukraine War. Moreover, the resulting impact of sanctions imposed by western nations against Russia, Russian-backed separatist regions in Ukraine, certain banks, companies, government officials, and other individuals in Russia and Belarus, could adversely impact our ability to access additional capital funding sources.
Likewise, the recent and ongoing conflict in the Middle East has impacted and could continue to impact the global economy for the foreseeable future, and is threatening to spread, and may in the future spread, into other Middle Eastern countries. The conflicts have caused, and could intensify, volatility in market prices, and the extent and duration of the military actions, sanctions and resulting market disruptions could be significant and could potentially have a substantial negative impact geopolitical stability and on our business for an unknown period of time.
In addition, any further changes in regulations or shifts in political conditions are beyond the control of the Company and may materially and adversely affect our business, or if significant enough, may significantly impede our ability to transact in certain countries. Operations may be affected in varying degrees by government regulations with respect to restrictions on production, price controls and foreign exchange restrictions.
While we do not have any direct significant exposure or connection to Russia, Ukraine, Belarus or the Middle East at large, it is uncertain as to how such events and any related economic sanctions could impact the global economy. Any negative developments in respect thereof could have a material and adverse effect on our business, operations, financial condition, and the value of our securities.
High rates of global inflation, the occurrence of a recession and higher interest rates could have a material and adverse impact on our business, results of operations and financial condition.
During 2022 and 2023, the global markets experienced, and continue to experience, higher rates of inflation as a result of several market factors, including in the form of increased costs pertaining to labor, materials and overhead. The U.S. capital markets in particular have experienced and continue to experience extreme volatility and disruption. Inflation rates in the U.S. significantly increased in 2022 and 2023 resulting in action by the U.S. federal government to increase interest rates, adversely affecting capital markets activity. Interest rates are sensitive to factors that are beyond our control, including domestic and international economic conditions, including inflation, and the policies of various governmental and regulatory agencies, including the Federal Reserve Board in the United States (the “Federal Reserve”). Interest rates may increase further, or they may remain at current levels for the near-term, and this new interest rate environment could materially and adversely affect our business, the counterparties with which we interact and the global economy at large
As a result of these inflationary pressures, governments in many countries have implemented tight monetary policies, which could slow the growth rate of local economies and restrict the availability of credit. In particular, the monetary policies of the Federal Reserve, implemented through open market operations, the federal funds rate (“Fed Funds Rate”) targets, and the discount rate for banking borrowings and reserve requirements, affect prevailing interest rates. A material change in any of these policies could have a material impact on us or our customers, and therefore on our results of operations. Beginning early in 2022 and continuing in 2023, in response to growing indications of inflation, the Federal Reserve increased interest rates rapidly and the Fed Funds Rate reached a 23-year high in 2023. While we experienced increases in the cost of labor and materials, we believe that our financial condition and results of operations have thus far not been materially impacted by inflationary pressures. However, to the extent the current rates of inflation and shifts in fiscal and monetary policy result in prolonged and slower growth or a recession, it could have a material and adverse effect on the demand for our products and services and, in the process, our business, results of operations and financial condition as a whole, including with respect to our ability to maintain current levels of gross margin and general and administrative expenses as a percentage of total revenue. Moreover, in the event that a global recession were to occur, it could adversely impact the critical counterparties that we engage, including in the form of a decrease in the products and services they seek to obtain from us. Additionally, due to our current level of debt, our business and results of operations have been, and may continue to be, impacted by a rise in interest rates, which may continue to rise. Relatively high interest rates will increase cost of capital and the cost of borrowings for any other corporate purpose. As a result, if we need or seek significant borrowings and interest rates remain elevated or increase, the cost of such borrowing to us could be significant, which may have a significant adverse impact on our financial condition and results of operations. We continue to monitor our operations and will seek to take appropriate actions to mitigate the potential impact of heightened inflation on our business. Nevertheless, there can be no assurances that we will be successful in doing so, if at all.
Material and adverse developments impacting the financial services industry at large, including the occurrence of actual (or widespread concerns regarding the potential occurrence of) defaults, illiquidity, operational failures and non-performance by financial institutions and critical counterparties, could have a material and adverse effect on our business, financial condition and results of operations.
The occurrence of actual (or widespread concerns regarding the potential occurrence of) illiquidity, operational failures, defaults, non-performance or other material and adverse developments that impact financial institutions and transactional counterparties, or other entities within the financial services industry at large, have previously caused, and could continue to cause, market-wide liquidity issues, bank-runs and general contagion across the global financial industry. For example, on March 10, 2023, Silicon Valley Bank (“SVB”) was closed by the California Department of Financial Protection and Innovation and the Federal Deposit Insurance Corporation (the “FDIC”) was subsequently appointed as a receiver. Similarly, on March 12, 2023, Signature Bank and Silvergate Capital Corp. were each placed into receivership. While the U.S. Federal Reserve Board, the FDIC and the U.S. Department of Treasury collectively agreed to guarantee all deposits, above and beyond the limit on insured deposits of $250,000 at these financial institutions, there can be no assurance that there will not be additional bank failures or issues in the broader financial system. Likewise, there is no guarantee that any of the U.S. Department of Treasury, the FDIC or the Federal Reserve Board will provide access to any additional uninsured funds in the future in the event of the closure or failure of any other banks or financial institutions, or that they would do so promptly or in a timely fashion. Additionally, substantial and rapid increases in interest rates and inflation have led to a decline in the trading value of previously issued government securities with interest rates below current market interest rates. While the U.S. Department of Treasury, Federal Reserve and the FDIC have announced a program to provide up to $25 billion of loans to financial institutions secured by certain of such government securities held by financial institutions to mitigate the risk of potential losses on the sale of such instruments, the liquidity needs of financial institutions, including as a result of widespread demands for customer withdrawals, may exceed the capacity of such program.
Furthermore, we and other parties with who we conduct business and engage commercially may be unable to access critical funds in deposit accounts or other accounts held with a closed or failing financial institution or pursuant to lending arrangements with such financial institutions. Accordingly, in such instance, our ability to pay our obligations, and any of our counterparties’ ability to pay their respective obligations, or enter into new commercial arrangements requiring additional payments, could be materially and adversely affected.
Risks Related to our Intellectual Property
Our success is largely dependent upon our patents, proprietary technology, and other intellectual property.
Our success will depend, in part, on our ability to obtain patents, protect our trade secrets and operate without infringing on the proprietary rights of others. Patents and other proprietary rights are essential to our business. We rely on trade secret, patent, copyright and trademark laws, and confidentiality and other agreements with employees and third parties, all of which offer only limited protection. Our general policy has been to file patent applications to protect our inventions and improvements to our inventions that are considered important to the development of our business. In certain cases, we have chosen to protect our intellectual property by treating it as confidential internal know-how. Our success will depend in part on our ability to obtain patents, defend patents, maintain internal know-how/trade secret protection and operate without infringing on the proprietary rights of others. Interpretation and evaluation of pharmaceutical patent claims present complex legal and factual questions. Further, patent protection may not be available for some of the products or technology we are developing. If we are placed in a position where we must spend significant time and money defending or enforcing our patents, designing around patents held by others or licensing patents or other proprietary rights held by others, our business, results of operations and financial condition may be harmed. In seeking to protect our inventions using patents it is important to note that we have no assurance that:
| ● | patent applications will result in the issuance of patents; |
| ● | additional proprietary products developed will be patentable; |
| ● | patents issued will provide adequate protection or any competitive advantages; |
| ● | patents issued will not be successfully challenged by third parties; |
| ● | commercial exploitation of our inventions does not infringe the patents or intellectual property of others; or |
| ● | we will be able to obtain any extensions of the patent term. |
A number of pharmaceutical, biotechnology and medical device companies and research and academic institutions have developed technologies, filed patent applications or received patents on various technologies that may be related to our business. Some of these technologies, applications or patents could limit the scope of the patents, if any, that we may be able to obtain. It is also possible that these technologies, applications or patents may preclude us from obtaining patent protection for our inventions. Further, there may be uncertainty as to whether we may be able to successfully defend any challenge to our patent portfolio. Moreover, we may have to participate in derivation proceedings, inter partes review proceedings, post-grant review proceedings, or opposition proceedings in the various jurisdictions around the world. An unfavorable outcome in a derivation proceeding, an inter partes review proceeding, a post-grant review proceeding, or an opposition proceeding could preclude us or our collaborators or licensees from making, using or selling products using the technology, or require us to obtain license rights from third parties. It is not known whether any prevailing party would offer a license on commercially acceptable terms, if at all. Further, any such license could require the expenditure of substantial time and resources and could harm our business. If such licenses are not available, we could encounter delays or prohibition of the development or introduction of our product. In the case of intellectual property where we have chosen to protect it by treating it as internal knowhow, there can be no assurance that others with greater expertise or access to greater resources do not develop similar or superior technology that impairs the competitive value of our internal know-how.
Moreover, a number of aspects of intellectual property protection in the field of AI are currently under development, and there is uncertainty and ongoing litigation in different jurisdictions as to the degree and extent of protection warranted for AI and machine learning systems, as well as relevant system input and outputs. If we fail to obtain protection for the intellectual property rights concerning our AI technologies, or later have our intellectual property rights invalidated or otherwise diminished, our competitors may be able to take advantage of our research and development efforts to develop competing products, and our business, financial condition and operations could be materially and adversely impacted.
Obtaining and maintaining our patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements.
The U.S. Patent and Trademark Office (“PTO”) and various foreign national or international patent agencies require compliance with a number of procedural, documentary, fee payment and other similar provisions during the patent application process. Periodic maintenance fees on any issued patent are due to be paid to the PTO and various foreign national or international patent agencies in several stages over the lifetime of the patent. While an inadvertent lapse can in many cases be cured by payment of a late fee or by other means in accordance with the applicable rules, there are situations in which noncompliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. Non-compliance events that could result in abandonment or lapse of patent rights include, but are not limited to, failure to timely file national and regional stage patent applications based on our international patent application, failure to respond to official actions within prescribed time limits, non-payment of fees and failure to properly legalize and submit formal documents. If we fail to maintain the patents and patent applications covering our Product Candidates, our competitors might be able to enter the market, which would have a material adverse effect on our business.
We may become subject to claims by third parties asserting that we or our employees have misappropriated their intellectual property or claiming ownership of what we regard as our own intellectual property.
Our commercial success depends upon our ability to develop, manufacture, market and sell our Product Candidates, and to use our related proprietary technologies without violating the intellectual property rights of others. We may become party to, or threatened with, future adversarial proceedings or litigation regarding intellectual property rights with respect to our Product Candidates, including interference or derivation proceedings before the PTO or other international patent offices. Third parties may assert infringement claims against us based on existing patents or patents that may be granted in the future. If we are found to infringe a third party’s intellectual property rights, we could be required to obtain a license from such third party to continue commercializing our Product Candidates. However, we may not be able to obtain any required license on commercially reasonable terms or at all. Under certain circumstances, we could be forced, including by court order, to cease commercializing the applicable product candidate. In addition, in any such proceeding or litigation, we could be found liable for monetary damages. A finding of infringement could prevent us from commercializing our Product Candidates or force us to cease some of our business operations, which could materially harm our business. Any claims by third parties that we have misappropriated their confidential information or trade secrets could have a similar negative impact on our business.
While our preclinical studies are ongoing, we believe that the use of our Product Candidates in these preclinical studies fall within the scope of the exemptions provided by 35 U.S.C. Section 271(e) in the United States, which exempts from patent infringement liability activities reasonably related to the development and submission of information to the FDA. As our Product Candidates progress toward clinical trials and, ultimately, commercialization, the possibility of a patent infringement claim against us increases. We attempt to ensure that our Product Candidates and the methods we employ to manufacture them, as well as the methods for their uses we intend to promote, do not infringe other parties’ patents and other proprietary rights. There can be no assurance they do not, however, and competitors or other parties may assert that we infringe their proprietary rights in any event.
We may become involved in lawsuits to protect or enforce our intellectual property, which could be expensive, time consuming and unsuccessful and have a material adverse effect on the success of our business.
Competitors may infringe our patents or misappropriate or otherwise violate our intellectual property rights. To counter infringement or unauthorized use, litigation may be necessary in the future to enforce or defend our intellectual property rights, to protect our trade secrets or to determine the validity and scope of our own intellectual property rights or the proprietary rights of others. Also, third parties may initiate legal proceedings against us to challenge the validity or scope of intellectual property rights we own. These proceedings can be expensive and time consuming. Many of our current and potential competitors have the ability to dedicate substantially greater resources to defend their intellectual property rights than we can. Accordingly, despite our efforts, we may not be able to prevent third parties from infringing upon or misappropriating our intellectual property. Litigation could result in substantial costs and diversion of management resources, which could harm our business and financial results. In addition, in an infringement proceeding, a court may decide that a patent owned by us is invalid or unenforceable or may refuse to stop the other party from using the technology at issue on the grounds that our patents do not cover the technology in question. An adverse result in any litigation proceeding could put one or more of our patents at risk of being invalidated, held unenforceable or interpreted narrowly. Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation. There could also be public announcements of the results of hearings, motions or other interim proceedings or developments. If securities analysts or investors perceive these results to be negative, it could have a material adverse effect on the price of our common shares.
If we are not able to adequately prevent disclosure of trade secrets and other proprietary information, the value of our technology and products could be significantly diminished.
We rely on trade secrets to protect our proprietary technologies, especially where we do not believe patent protection is appropriate or obtainable. However, trade secrets are difficult to protect. We rely in part on confidentiality agreements with our current and former employees, consultants, outside scientific collaborators, sponsored researchers, contract manufacturers, vendors and other advisors to protect our trade secrets and other proprietary information. These agreements may not effectively prevent disclosure of confidential information and may not provide an adequate remedy in the event of unauthorized disclosure of confidential information. In addition, we cannot guarantee that we have executed these agreements with each party that may have or have had access to our trade secrets. Any party with whom we or they have executed such an agreement may breach that agreement and disclose our proprietary information, including our trade secrets, and we may not be able to obtain adequate remedies for such breaches.
Enforcing a claim that a party illegally disclosed or misappropriated a trade secret is difficult, expensive and time-consuming, and the outcome is unpredictable. In addition, some courts are less willing or unwilling to protect trade secrets. If any of our trade secrets were to be lawfully obtained or independently developed by a competitor, we would have no right to prevent them, or those to whom they disclose such trade secrets, from using that technology or information to compete with us. If any of our trade secrets were to be disclosed to or independently developed by a competitor or other third-party, our competitive position would be harmed.
We may not be able to protect our intellectual property rights throughout the world.
Filing, prosecuting and defending patents on all of our Product Candidates throughout the world would be prohibitively expensive. Therefore, we have filed applications and/or obtained patents only in key markets such as the United States, Canada, Japan and Europe. Competitors may use our technologies in jurisdictions where we have not obtained patent protection to develop their own products and, further, may be able to export otherwise infringing products to territories where we have patent protection but where enforcement is not as strong as that in the United States. These products may compete with our products in jurisdictions where we do not have any issued patents and our patent claims or other intellectual property rights may not be effective or sufficient to prevent them from so competing.
Many companies have encountered significant problems in protecting and defending intellectual property rights in certain foreign jurisdictions. The legal systems of certain countries, particularly certain developing countries, do not favor the enforcement of patents and other intellectual property protection, particularly those relating to pharmaceuticals, which could make it difficult for us to stop the infringement of our patents or marketing of competing products in violation of our proprietary rights generally. For example, an April 2016 report from the Office of the United States Trade Representative identified a number of countries, including India and China, where challenges to the procurement and enforcement of patent rights have been reported. Several countries, including India and China, have been listed in the report every year since 1989. As a result, proceedings to enforce our patent rights in certain foreign jurisdictions could result in substantial cost and divert our efforts and attention from other aspects of our business and could be unsuccessful.
Patent terms may be inadequate to protect our competitive position on our Product Candidates for an adequate amount of time.
Given the amount of time required for the development, testing and regulatory review of new Product Candidates, patents protecting such candidates might expire before or shortly after such candidates are commercialized. We expect to seek extensions of patent terms in the United States and, if available, in other countries where we are prosecuting patents. In the United States, the Drug Price Competition and Patent Term Restoration Act of 1984 permits a patent term extension of up to five years beyond the normal expiration of the patent, which is limited to the approved indication (or any additional indications approved during the period of extension). However, the applicable authorities, including the FDA and the PTO, and any equivalent regulatory authorities in other countries, may not agree with our assessment of whether such extensions are available, and may refuse to grant extensions to our patents, or may grant more limited extensions than we request. If this occurs, our competitors may be able to take advantage of our investment in development and clinical trials by referencing our clinical and preclinical data and launch their product earlier than might otherwise be the case.
Intellectual property rights do not necessarily address all potential threats to our competitive advantage.
The degree of future protection afforded by our intellectual property rights is uncertain because intellectual property rights have limitations, and may not adequately protect our business, or permit us to maintain our competitive advantage. For example:
| ● | others may be able to make compounds that are the same as or similar to our Product Candidates but that are not covered by the claims of the patents that we own; |
| ● | we might not have been the first to make the inventions covered by the issued patents or pending patent applications that we own; |
| ● | we might not have been the first to file patent applications covering certain of our inventions; |
| ● | others may independently develop similar or alternative technologies or duplicate any of our technologies without infringing our intellectual property rights; |
| ● | it is possible that our pending patent applications will not lead to issued patents; |
| ● | issued patents that we own may not provide us with any competitive advantages, or may be held invalid or unenforceable as a result of legal challenges; |
| ● | our competitors might conduct research and development activities in the United States and other countries that provide a safe harbor from patent infringement claims for certain research and development activities, as well as in countries where we do not have patent rights and then use the information learned from such activities to develop competitive products for sale in our major commercial markets; or |
| ● | the patents of others may have an adverse effect on our business. |
Risks Related to our Third Parties
We rely heavily on contract manufacturers over whom we have limited control. If we are subject to quality, cost or delivery issues with the preclinical and clinical grade materials supplied by contract manufacturers, our business operations could suffer significant harm.
We currently have no manufacturing capabilities and rely on CDMOs to manufacture our Product Candidates for preclinical studies and clinical trials. We rely on CDMOs for manufacturing, filling, packaging, testing, storing and shipping of drug products in compliance with cGMP, regulations applicable to our products. The FDA and other regulatory agencies ensure the quality of drug products by carefully monitoring drug manufacturers’ compliance with cGMP regulations. The cGMP regulations for drugs contain minimum requirements for the methods, facilities and controls used in manufacturing, processing and packaging of a drug product. If our CDMOs increase their prices or fail to meet our quality standards, or those of regulatory agencies such as the FDA, and cannot be replaced by other acceptable CDMOs, our ability to obtain regulatory approval for and commercialize our Product Candidates may be materially adversely affected.
The APIs used in all of our Product Candidates are currently sourced from either contract manufacturers or, for smaller quantities, from research material suppliers, that typically utilize synthetic chemistry as their manufacturing method. This is intended to be an interim step to enable us to proceed with developing our formulation, execute preclinical toxicology studies and progress through Phase 1 and 2 clinical trials, after which time we anticipate that we will have been able to successfully scale-up our IntegraSyn manufacturing approach so that it will be GMP ready at pharmaceutical grade. Bridging studies consisting of chemical analysis and, possibly, animal studies may be required in order to switch our APIs from the current external manufacturing sources to our internally manufactured products. There is no guarantee that we will be successful in scaling up our IntegraSyn manufacturing process for cannabinoids, or successfully complete any required bridging studies, or be able to successfully transfer our IntegraSyn manufacturing process to a CDMO. The key risks and challenges associated with the development of the IntegraSyn process include: failure to continue optimization and development of the process manufacturing steps from the current scale while maintaining the same or greater output of the selected cannabinoid; equipment and techniques may not be able to be scaled up using existing commercial processing equipment; supply of the key starting materials for the process may not be secured to ensure stability and security of commercial supply; and, failure of the large scale process to consistently produce the selected cannabinoid within set specifications and meeting the process parameters and in process controls to enable the manufacturing process to be validated for GMP commercial production of an API, among others. Failing to accomplish these or other criteria for the IntegraSyn manufacturing process with a CDMO may mean that we are not able to produce certain cannabinoids in a cost-effective manner. This could result in us not being able to successfully commercialize or utilize our APIs in our Product Candidates, if any, that may obtain regulatory approval.
Our existing collaboration agreements and any that we may enter into in the future may not be successful.
We also have relationships with scientific collaborators at academic and other institutions, some of whom conduct research at our request or assist us in formulating our research and development strategies. These scientific collaborators are not our employees and may have commitments to, or consulting or advisory contracts with, companies that conflict in interests with and pose a competitive threat to us. Moreover, to the extent that we decide to enter into collaboration agreements, we will face significant competition in seeking appropriate collaborators. Collaboration arrangements are complex and time consuming to negotiate, document and implement. We may not be successful in our efforts to establish, implement and maintain collaborations or other alternative arrangements if we choose to enter into such arrangements and our selected partners may be given, and may exercise, a right to terminate their agreement with us without cause. Our Collaborative Research Agreement with the University of British Columbia may be terminated by either party upon 30 calendar days written notice. The terms of any collaboration or other arrangements that we may establish may not be favorable to us.
For all of the aforesaid reasons and others set forth in this Annual Report, an investment in Common Shares and any other securities that we may offer from time to time involves a high degree of risk. Any person considering an investment in our Common Shares or any other of our securities should be aware of these and other factors set forth in this Annual Report and should consult with his or her legal, tax and financial advisors prior to making an investment in our Common Shares or any other of our securities that may be offered from time to time. Our Common Shares and any other securities that we may offer from time to time should only be purchased by persons who can afford to lose all of their investment.
ITEM 1B. UNRESOLVED STAFF COMMENTS
None.
ITEM 1C. CYBERSECURITY
Risk Management and Strategy
We regularly assess, identify, and manage material risks from cybersecurity threats, and have integrated these processes into our overall risk management systems and processes. We routinely assess material risks from cybersecurity threats, including any potential unauthorized occurrence on or conducted through our information systems that may result in adverse effects on the confidentiality, integrity, or availability of our information systems or any information residing therein.
We conduct risk assessments at least annually to identify cybersecurity threats. These risk assessments include identifying reasonably foreseeable potential internal and external risks, the likelihood of occurrence and any potential damage that could result from such risks, and the sufficiency of existing policies, procedures, systems, controls and other safeguards we have put in place to manage such risks. Our risk management process also encompasses cybersecurity risks associated with the use of our major third-party vendors and service providers. Additionally, we maintain cyber coverage through our insurance carrier to mitigate risks associated with cybersecurity incidents, subject to customary terms and exclusions.
Following these risk assessments, we design, implement, and maintain reasonable safeguards to minimize the identified risks; reasonably address any identified gaps in existing safeguards; update existing safeguards as necessary; and monitor the effectiveness of our safeguards. Moreover, we remain committed to investing in the development and improvement of our security processes and controls, as well as maintaining our technology infrastructure. These processes include a plan for notifying, informing, consulting, analyzing, and communicating any risks or incidents to a range of internal stakeholders, including executive management and the Board of Directors, as well as external stakeholders, as deemed necessary and appropriate based on the circumstances. We believe we have allocated adequate resources related to our cybersecurity risk management processes and have designated our Chief Financial Officer with the responsibility of managing the cybersecurity risk assessment and mitigation process.
As part of our overall risk management program, we provide required training to employees in high risk areas on cybersecurity and will distribute standard operating procedures to all employees. For additional information regarding whether any risks from cybersecurity threats, including as a result of any previous cybersecurity incidents, have materially affected or are reasonably likely to materially affect the Company, including our business strategy, results of operations, or financial condition, please refer to Item 1A, “Risk Factors,” in this Annual Report.
Governance
One of the key functions of our Board of Directors is informed oversight of our risk management process, including risks arising from cybersecurity threats. Our Chief Financial Officer and Chief Operating Officer are primarily responsible for assessing and managing material risks from cybersecurity threats on a day to day basis. Our Board of Directors is responsible for monitoring and assessing strategic risk exposure, and our management team is additionally responsible for the day-to-day management of the material risks we face. Our Board of Directors administers its cybersecurity risk oversight function directly as a whole. The Company additionally utilizes the assistance of a third-party service provider, an information technology solutions service provider located in Richmond, BC, for purposes of broadly managing its cybersecurity risks.
ITEM 2. PROPERTIES
Our corporate headquarters are located at Suite 1445 - 885 West Georgia Street, Vancouver, British Columbia V6C 1B4, Canada. This new lease was signed in July 2024 and the office space occupies approximately 2,243 square feet with a monthly basic rental rate and operating charges of an estimated C$12,296 over the two-year term of the agreement.
In July 2019, InMed entered into a facility lease agreement for approximately 4,000 square feet of office space in Vancouver, BC, which served as our corporate headquarters until August 2024, when the lease expired. The Company did not take the option to renew for an additional three-year period.
In October 2023, BayMedica entered into an amended facility lease agreement for approximately 7,000 square feet of office space in San Francisco, California. The lease expires in April 2027.
We believe substantially all of our property and equipment is in good condition and that InMed has sufficient capacity to meet its current operational needs. We further believe that, should it be needed, suitable additional space is available to accommodate any expansion of our operations, but such space may not be available in the same building, if and when such space is needed.
ITEM 3. LEGAL PROCEEDINGS
From time to time, we are subject to various legal proceedings, claims and administrative proceedings that arise in the ordinary course of our business activities. Although the results of the litigation and claims cannot be predicted with certainty, as of the date of this report, we do not believe we are party to any claim, proceeding or litigation the outcome of which, if determined adversely to us, would individually or in the aggregate be reasonably expected to have a material adverse effect on our business. Regardless of the outcome, litigation can have an adverse impact on us because of defense and settlement costs, diversion of management resources and other factors. However, as of the date of this Annual Report, we are not involved in any material pending legal or governmental proceedings.
ITEM 4. MINE SAFETY DISCLOSURES
Not applicable.
PART II
ITEM 5. MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Market Information
The Company’s shares are listed on the on the Nasdaq Capital Market (“Nasdaq”) under the trading symbol “INM”).
There were approximately 11,764 holders of record of our Common Shares as of September 20, 2024. On September 20, 2024, the last reported sales price per share of our Common Shares was $0.26 per share.
Unregistered Sales of Equity Securities
None.
Repurchases of Equity Securities
None.
ITEM 6. [RESERVED]
ITEM 7. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
This discussion and analysis contains certain forward-looking statements within the meaning of Section 27A of the Securities Act and Section 21E of the Exchange Act, and is subject to the safe harbor created by those sections. For more information, see “Special Note Regarding Forward-Looking Statements.” When reviewing the discussion below, you should keep in mind the substantial risks and uncertainties that impact our business. In particular, we strongly encourage you to review the risks and uncertainties described in “Risk Factors” in this Annual Report, and other filings we make from time to time with the SEC. These risks and uncertainties could cause actual results to differ materially from those projected or implied by our forward-looking statements contained in this report. These forward-looking statements are made as of the date of this report, and we do not intend, and do not assume any obligation, to update these forward-looking statements, except as required by law.
The following discussion and analysis should be read in conjunction with our audited consolidated financial statements for the year ended June 30, 2024, and the related notes thereto, which have been prepared in accordance with U.S. GAAP. Additionally, the following discussion and analysis should be read in conjunction with our audited consolidated financial statements included in this Annual Report. Throughout this discussion, unless the context specifies or implies otherwise the terms “InMed,” “Company,” “we,” “us,” and “our” refer to InMed Pharmaceuticals Inc.
All dollar amounts stated herein are in U.S. dollars unless specified otherwise.
Overview
We are a clinical stage pharmaceutical company developing a pipeline of proprietary small molecule drug candidates that are preferential signaling ligands of the endogenous CB1and CB2 receptors as well as other receptor targets linked to human disease. CB1 and CB2 receptors are each part of the endocannabinoid system that is found throughout the human body and is responsible for many homeostatic functions. CB1 receptors are primarily located in the brain and central nervous system, while CB2 receptors are involved in modulating neuroinflammation and immune responses. Our research efforts target the treatment of diseases with high unmet medical needs. Together with BayMedica, we also have significant know-how in developing proprietary manufacturing approaches to produce and sell bulk rare cannabinoids as ingredients for various market sectors.
InMed has sought to focus on the research and development of preferential signaling ligands of CB1 and CB2 and has produced a library of novel, proprietary drug candidates (“Product Candidates”). These Product Candidates are patentable new chemical entities (“NCEs”) for pharmaceutical development, aimed at targeting diverse clinical indications. Our current pharmaceutical pipeline consists of three programs, with drug candidates targeting Alzheimer’s disease, dry age-related macular degeneration, and Epidermolysis Bullosa. InMed’s INM-901 is a proprietary small molecule, disease modifying drug candidate being developed as a potential treatment for Alzheimer’s disease. INM-901 has multiple potential mechanisms of action as a preferential signaling agonist for both CB1 and CB2 receptors, as well as impacting the peroxisome proliferator-activated receptor (“PPAR”) signaling pathway. Combined, these mechanisms of action may offer a unique treatment approach targeting several biological pathways associated with Alzheimer’s disease. Our ocular research, based on the proprietary small molecule INM-089, indicates potentially promising neuroprotective effects in the back of the eye, which may lead to the preservation of the retinal function. Neuroprotection in dry Aged-related Macular Degeneration (“dry AMD”) remains an unmet medical need and a new treatment option may help solve this multifactorial disease.
InMed has also completed a Phase 2 clinical trial of INM-755 (cannabinol) cream studying its safety and efficacy in treating symptoms related to Epidermolysis Bullosa (“EB”). Results from the Phase 2 clinical trial showed a positive indication of enhanced anti-itch activity for INM-755 cream versus the control cream alone in an exploratory clinical evaluation. The Company is also pursuing strategic partnership opportunities for INM-755 in epidermolysis bullosa and other itch-related skin conditions.
Together with BayMedica, our manufacturing capabilities include traditional approaches such as chemical synthesis and biosynthesis, as well as a proprietary, integrated manufacturing approach called IntegraSyn. With multiple manufacturing approaches, InMed has sought to maintain enhanced flexibility to select the most cost-effective method to deliver high quality, high purity Products and Product Candidates fit for their intended use. BayMedica’s commercial business specializes in the B2B commercialization of bulk rare, non-intoxicating cannabinoids as raw materials for the Health and Wellness sector that are bioidentical to those found in nature.
Recent Developments
NASDAQ Delisting Notice
As previously reported by the Company, on March 19, 2024, the Company received written notification from the Listing Qualifications Department of Nasdaq that the Company has been granted an additional 180-day compliance period, or until September 16, 2024 (the “Extended Compliance Period”), to regain compliance with Nasdaq’s minimum bid price requirement for the continued listing on the Nasdaq Capital Market, as set forth in Nasdaq Listing Rule 5550(a)(2) (the “Minimum Bid Price Rule”). Nasdaq’s determination was based on the Company meeting the continued listing requirement for market value of publicly held shares and all other applicable requirements for initial listing on the Nasdaq Capital Market, with the exception of the bid price requirement, and the Company’s written notice of its intention to consider all available options to regain compliance during the Extended Compliance Period, including, if necessary, effecting a reverse stock split. The Company was unable to regain compliance during the Extended Compliance Period and, on September 17, 2024, the Company received an additional notification from the Listing Qualifications Department stating that due to the deficiency, the Company’s securities would be delisted from Nasdaq on September 26, 2024, unless the Company appealed Nasdaq’s determination to a Hearings Panel (the “Panel”). A hearing request would stay the suspension of the Company’s securities pending the Panel’s discussion. On September 17, 2024, the Company submitted the hearing request to appeal (the “Appeal Request”) Nasdaq’s determination before the Panel. The hearing will take place on October 31, 2024 and it is anticipated that the Panel’s decision will follow shortly thereafter. The pendency of the Appeal Request does not have an immediate effect on the listing of our Common Shares and our Common Shares will continue to trade on Nasdaq under the symbol “INM”.
While the Company has filed the Appeal Request, there can be no assurances, however, that we will be successful in regaining compliance with the continued listing requirements and maintaining the listing of our Common Shares on Nasdaq. Delisting from Nasdaq could materially and adversely affect our ability to raise additional financing through the public or private sale of equity securities, would significantly affect the ability of investors to trade our securities and would negatively affect the value and liquidity of our securities, including our Common Shares. The actual or threatened delisting of our securities could also have other material and adverse consequences, including the potential loss of confidence by employees and other stakeholders, the loss of institutional investor interest and fewer business development opportunities, limited availability of market quotations for our securities, reduced liquidity with respect to our securities, a determination that our Common Shares is “penny stock,” which will require brokers trading in our Common Shares to adhere to more stringent rules, possibly resulting in a reduced level of trading activity in the secondary trading market for our Common Shares, and limited amount of news and analyst coverage of the Company. To the extent that our Common Shares became eligible to trade on the OTC Bulletin Board, another over-the-counter quotation system, or on the pink sheets, an investor may find it more difficult to dispose of their Common Shares or obtain accurate quotations as to the market value of our Common Shares.
Renewal of ATM Program
On June 27, 2024, the Company entered into an amendment (the “ATM Amendment”) to its At-the-Market Offering Agreement, dated April 7, 2022 (the “Original ATM Agreement” and together with the ATM Amendment, the “Amended ATM Agreement”), by and between the Company and H.C. Wainwright & Co., LLC (the “Agent”), as sales agent, pursuant to which the Company may offer and sell shares of our Common Shares, from time to time, in “at the market” offerings through the Agent. The Original ATM Agreement was previously filed with the Securities and Exchange Commission on April 7, 2022 on the Company’s Current Report on Form 8-K. The ATM Amendment amends the Original ATM Agreement to reflect, among other provisions, updates to certain sales settlement provisions and reimbursement terms, and to supplement the representations being made by the Company to the Agent. Our Common Shares sold under the Amended ATM Agreement will be offered and sold pursuant to the Company’s shelf registration statement on Form S-3, which was initially filed on February 4, 2022 and amended on February 9, 2022, and was declared effective by the SEC on February 11, 2022. The foregoing description of the terms of the ATM Amendment does not purport to be complete and is qualified in its entirety by reference to the full text of the ATM Amendment, a copy of which is filed as Exhibit 10.1 to the Company’s Current Report on Form 8-K filed with the SEC on June 28, 2024 and is incorporated herein by reference.
Appointments to the Scientific Advisory Board (“SAB”)
On September 5, 2024, the Company appointed Dr. Barry Greenberg to its Scientific Advisory Board (“SAB”). Dr. Greenberg is an Associate Professor in the Department of Neurology and Director of the Alzheimer’s Disease Translational Center at the Johns Hopkins University School of Medicine. He serves on several committees and advisory boards for NIA-funded initiatives focused on genetics, model development and clinical trials in AD, and has recently been selected as Editor-in-Chief of the journal “Alzheimer’s & Dementia: Translational Research and Clinical Interventions”
On April 18, 2024, the Company announced the addition of Dr. David G. Morgan, a renowned leader in neurodegenerative disease, to its SAB reinforcing the Company’s commitment to advancing it’s INM-901 program in the treatment of Alzheimer’s disease.
Ocular Research Program
On April 16, 2024, the Company announced additional preclinical data for INM-089 further demonstrating positive pharmacological effects targeting dry AMD. In vivo preclinical studies in AMD disease models demonstrated significant outcomes for INM-089 including neuroprotection of photoreceptors as well as improved photoreceptor’s function, improved integrity of retinal pigment epithelium and reduction in extracellular autofluorescent deposits, a hallmark of dry AMD. Additionally, data indicates that INM-089 may be more effective as a therapeutic treatment for dry AMD compared to neovascular, or wet, AMD. More specifically, data suggests INM-089 may be an important candidate for geographic atrophy (“GA”) which is common in more advanced cases of dry AMD, affecting the center of the macula.
The Company has strategically prioritized the utilization of its proprietary small molecule drug candidates in its drug development initiatives, resulting in the advancement of the INM-089 program in the treatment of dry AMD taking precedence over the INM-088 program in the treatment of glaucoma. Therefore, the Company will not be advancing INM-088 in the immediate future. Notably, the initial research and data from the INM-088 program have played an instrumental role in shaping the development of INM-089 program.
Additional Preclinical Data for INM-901’s Pharmacological Effects
On April 4, 2024, the Company announced additional preclinical data demonstrating INM-901’s positive pharmacological effects in the potential treatment of Alzheimer’s disease (“AD”). Several preclinical studies were conducted in well-characterized in vivo AD models demonstrating that INM-901 is a preferential signaling agonist of the CB1/CB2 receptors and impacts the PPAR signaling pathway, reduces neuroinflammation and improves neuronal function. Analysis of mRNA data supports the observations made in the previously released behavioral studies results showing improvement of locomotor activity, cognition and memory in diseased animals.
Other Personnel Matters
On February 20, 2024, Ms. Netta Jagpal joined the Company as Chief Financial Officer and Corporate Secretary. In conjunction with this appointment, Mr. Jonathan Tegge stepped down as interim Chief Financial Officer and currently holds the position of Corporate Controller.
On May 10, 2024, Ms. Alexandra D.J. Mancini, Senior Vice President, Clinical & Regulatory Affairs, provided notice to the Company and the Company’s Board of Directors of her intention to retire from her position, effective June 30, 2024. In connection with Ms. Mancini’s retirement and eventual departure, and to ensure a smooth transition, the Company retained Ms. Mancini under the terms of a Consulting Agreement (the “Consulting Agreement”), pursuant to which Ms. Mancini will provide certain consulting services to the Company for a period to be mutually agreed upon by both the Company, on the one hand, and Ms. Mancini, on the other. The foregoing description of the Consulting Agreement does not purport to be complete and is subject, and qualified by reference, to the full text of the Consulting Agreement, which has been filed as Exhibit 10.19 attached hereto.
Notice of Termination with Respect to the Technology Licensing Agreement
On May 10, 2024, the Company delivered a 90-day notice of termination to EyeCRO LLC with respect to the Technology Licensing Agreement, specifying an effective date of termination of August 8, 2024.
Components of Results of Operations
Revenue
Our revenue consists of manufacturing and distribution sales of bulk rare cannabinoid Products, which are generally recognized at a point in time. The Company recognizes revenue when control over the products have been transferred to the customer and the Company has a present right to payment.
Cost of Sales
Cost of sales consist primarily of the purchase price of goods and cost of services rendered, freight costs, warehousing costs, and purchasing costs. Cost of sales also includes production and labor costs for our manufacturing business.
Operating Expenses
Research and Development and Patent Expenses
Research and development and patent expenses represent costs incurred by us for the discovery, development, and manufacture of our Products and Product Candidates and include:
| ● | external research and development expenses incurred under agreements with contract research organizations (“CROs”), CDMOs and consultants; |
| ● | salaries, payroll taxes, employee benefits expenses for individuals involved in research and development efforts; | |
| ● | research supplies; and | |
| ● | legal and patent office fees related to patent and intellectual property matters. |
We expense research and development costs as incurred. We recognize expenses for certain development activities, such as preclinical studies and manufacturing, based on an evaluation of the progress to completion of specific tasks using data or other information provided to us by our vendors. Payments for these activities are based on the terms of the individual agreements, which may differ from the pattern of expenses incurred. Non-refundable advance payments for goods or services to be received in the future for use in research and development activities are recorded as prepaid expenses. These amounts are recognized as an expense as the goods are delivered or the related services are performed, or until it is no longer expected that the goods will be delivered, or the services rendered.
External costs represent a significant portion of our research and development expenses, which we track on a program-by-program basis following the nomination of a development candidate. Our internal research and development expenses consist primarily of personnel-related expenses, including salaries, benefits and stock-based compensation expense. We do not track our internal research and development expenses on a program-by-program basis as the resources are deployed across multiple projects.
The successful development of our Products and Product Candidates is highly uncertain. At this time, we cannot reasonably estimate or know the nature, timing, and estimated costs of the efforts that will be necessary to complete the remainder of the development of our Product Candidates or to develop and commercialize additional Products. We are also unable to predict when, if ever, material net cash inflows will commence from our Product Candidates, if approved. This is due to the numerous risks and uncertainties associated with development, including the uncertainty related to:
| ● | the timing and progress of preclinical and clinical development activities; |
| ● | the number and scope of preclinical and clinical programs we decide to pursue; |
| ● | our ability to raise additional funds necessary to complete preclinical and clinical development and commercialization of our Product Candidates, to further advance the development of our manufacturing technologies, and to develop and commercialize additional Products, if any; |
| ● | our ability to maintain our current research and development programs and to establish new ones; |
| ● | our ability to establish sales, licensing or collaboration arrangements; |
| ● | the progress of the development efforts of parties with whom we may enter into collaboration arrangements; |
| ● | the successful initiation and completion of clinical trials with safety, tolerability and efficacy profiles that are satisfactory to the FDA or any comparable foreign regulatory authority; |
| ● | the receipt and related terms of regulatory approvals from applicable regulatory authorities; |
| ● | the availability of materials for use in production of our Products and Product Candidates; |
| ● | our ability to secure manufacturing supply through relationships with third parties or establish and operate a manufacturing facility; |
| ● | our ability to consistently manufacture our Product Candidates in quantities sufficient for use in clinical trials; |
| ● | our ability to obtain and maintain intellectual property protection and regulatory exclusivity, both in the United States and internationally; |
| ● | our ability to maintain, enforce, defend and protect our rights in our intellectual property portfolio; |
| ● | the commercialization of our Product Candidates, if and when approved, and of new Products; |
| ● | our ability to obtain and maintain third-party payor coverage and adequate reimbursement for our Product Candidates, if approved; |
| ● | the acceptance of our Product Candidates, if approved, by patients, the medical community and third-party payors; |
| ● | competition with other products; and |
| ● | a continued acceptable safety profile of our Product Candidates following receipt of any regulatory approvals. |
A change in the outcome of any of these variables with respect to the development of any of our Products or Product Candidates would significantly change the costs and timing associated with the development of those Products or Product Candidates.
Research and development activities account for a significant portion of our operating expenses. Research and development expenses decreased in fiscal 2024 as compared to fiscal 2023, largely due to high start-up costs associated with the multicenter Phase 2 clinical trial in our INM-755 program during fiscal 2022. However, we expect our research and development expenses to increase significantly in future periods as we continue to implement our business strategy, which includes advancing our drug candidates and our manufacturing technologies into and through clinical development, expanding our research and development efforts, including hiring additional personnel to support our research and development efforts, ultimately seeking regulatory approvals for our drug candidates that successfully complete clinical trials, and further developing selected R&D and commercial BayMedica activities. In addition, drug candidates in later stages of clinical development generally incur higher development costs than those in earlier stages of clinical development, primarily due to the increased size and duration of later-stage clinical trials. Accordingly, although we expect our research and development expenses to increase as our drug candidates advance into later stages of clinical development, we do not believe that it is possible, at this time, to accurately project total program-specific expenses through to commercialization. There are numerous factors associated with the successful commercialization of any of our Product Candidates, including future trial design and various regulatory requirements, many of which cannot be determined with accuracy at this time based on our stage of development.
General and Administrative Expenses
General and administrative expenses consist of personnel-related costs, including salaries, benefits and stock-based compensation expense, for our personnel in executive, finance and accounting, human resources, business operations and other administrative functions, investor relations activities, legal fees related to corporate matters, fees paid for accounting and tax services, consulting fees and facility-related costs.
Amortization and Depreciation
Intangible assets are comprised of intellectual property that we acquired in 2014 and 2015 and trade secrets, product formulation knowledge, patents that we acquired in October 2021. The acquired intellectual property and patents are amortized on a straight-line basis based on their estimated useful lives. Equipment and leasehold improvements are depreciated using the straight-line method based on their estimated useful lives.
Share-based Payments
Share-based payments is the stock-based compensation expense related to our granting of stock options to employees and others. The fair value, at the grant date, of equity-settled share awards is charged to our loss over the period for which the benefits of employees and others providing similar services are expected to be received. The vesting components of graded vesting employee awards are measured separately and expensed over the related tranche’s vesting period. The amount recognized as an expense is adjusted to reflect the number of share options expected to vest. The fair value of awards is calculated using the Black-Scholes option pricing model, which considers the exercise price, current market price of the underlying shares, expected life of the award, risk-free interest rate, expected volatility and the dividend yield.
Other Income
Other income consists primarily of interest income earned on our cash, cash equivalents and short-term investments.
Results of Operations
The Company has two operating and reportable segments based on the management approach which designates the internal reporting used by the Chief Operating Decision Maker (“CODM”), which is the Company’s Chief Executive Officer and the senior management team, for making decisions and assessing performance as the source of the Company’s reportable segments. The CODM allocates resources and assesses the performance of each operating segment based on potential licensing opportunities, historical and potential future product sales, operating expenses, and operating income (loss) before interest and taxes. The Company has determined its reportable segments to be InMed Pharmaceuticals (“InMed Pharma”) and BayMedica Commercial based on the information used by the CODM.
Comparison of the year ended June 30, 2024 and 2023 for InMed Segment
| Year Ended June 30, |
||||||||||||||||
| 2024 | 2023 | Change | % Change | |||||||||||||
| (in thousands) | ||||||||||||||||
| Operating expenses: | ||||||||||||||||
| Research and development and patents | 3,627 | 3,579 | 48 | 1 | % | |||||||||||
| General and administrative | 4,495 | 4,997 | (502 | ) | (10 | )% | ||||||||||
| Amortization and depreciation | 217 | 201 | 16 | 8 | % | |||||||||||
| Foreign exchange loss | 62 | 48 | 14 | 29 | % | |||||||||||
| Total operating expenses | 8,401 | 8,825 | (424 | ) | (5 | )% | ||||||||||
| Interest and other income | 533 | 491 | 42 | 9 | % | |||||||||||
| Net loss | $ | (7,868 | ) | $ | (8,334 | ) | $ | 466 | (6 | )% | ||||||
Research and Development and Patents Expenses
Research and development and patents expenses increased by less than $0.1 million in our InMed segment, or 1%, for the year ended June 30, 2024 as compared to the year ended June 30, 2023. The increase in research and development and patents expenses was due primarily to an increase in patent fees and compensation. This was offset by a decrease in research supplies. However, we expect our research and development expenses to increase significantly in future periods as we continue to implement our business strategy.
General and administrative expenses
General and administrative expenses decreased by $0.5 million in our InMed segment, or 10%, for the year ended June 30, 2024 as compared to the year ended June 30, 2023. The decrease results primarily from a combination of changes including lower office and administrative expenses, investor relation expenses, and personnel expenses.
Foreign exchange loss
The Company’s functional currency is the US dollar and our foreign exchange loss is predominantly due to transactions with foreign currency. Foreign exchange loss increased by less than $0.1 million in our InMed segment, or 29% for the year ended June 30, 2024, as compared to the year ended June 30, 2023, as a consequence of holding non-US denominated assets and liabilities combined with fluctuations in foreign exchange rates.
Comparison of the year ended June 30, 2024 and 2023 for the BayMedica Segment
| Year Ended June 30, |
||||||||||||||||
| 2024 | 2023 | Change | % Change | |||||||||||||
| (in thousands) | ||||||||||||||||
| Sales | $ | 4,598 | $ | 4,136 | $ | 462 | 11 | % | ||||||||
| Cost of sales | 3,497 | 2,733 | 764 | 28 | % | |||||||||||
| Gross profit | 1,101 | 1,403 | (302 | ) | (22 | )% | ||||||||||
| Operating expenses: | ||||||||||||||||
| Research and development and patents | 138 | 153 | (15 | ) | (10 | )% | ||||||||||
| General and administrative | 756 | 851 | (95 | ) | (11 | )% | ||||||||||
| Amortization and depreciation | 2 | 2 | - | - | % | |||||||||||
| Total operating expenses | 896 | 1,006 | (412 | ) | (41 | )% | ||||||||||
| Interest and other income | (5 | ) | 2 | (7 | ) | (350 | )% | |||||||||
| Tax expense | (7 | ) | (13 | ) | 6 | (46 | )% | |||||||||
| Net Income | $ | 193 | $ | 386 | $ | (193 | ) | (50 | )% | |||||||
Sales
Sales increased by $0.5 million in our BayMedica segment, or 11%, for the year ended June 30, 2024 as compared to the year ended June 30, 2023. The increase in sales results from expanded marketing efforts and increased demand in certain cannabinoid products. BayMedica will continue to evaluate opportunities for potential structured supply arrangements and collaborations for the commercial business. Sales and marketing efforts will remain focused on products that contribute highest margins, where BayMedica continues to hold a strong competitive position.
Cost of Sales
Cost of goods sold increased by $0.8 million in our BayMedica segment, or 28%, for the year ended June 30, 2024 as compared to the year ended June 30, 2023. The increase in cost of goods sold is primarily the result of hiring a full-time resource to support the cost of goods function, leading to higher personnel costs, as well as an increase in sales mentioned above, during the year ended June 30, 2024.
Research and Development and Patents Expenses
Research and development and patents expenses decreased by less than $0.1 million in our BayMedica segment, or 10%, for the year ended June 30, 2024 as compared to the year ended June 30, 2023. The decrease in research and development and patents expenses was primarily due to research supplies. This was offset by an increase in external contractors.
General and administrative expenses
General and administrative expenses decreased by less than $0.1 million in our BayMedica segment, or 11%, for the year ended June 30, 2024 as compared to the year ended June 30, 2023. The decrease results primarily from a combination of changes including lower personnel expenses, accounting fees and, legal fees. This was offset by an increase in sales and marketing expenses.
Liquidity and Capital Resources
Since our inception, we have generated revenue from BayMedica product sales and no sales from any other sources and have incurred significant operating losses and negative cash flows from our operations. We have not yet commercialized any of our Product Candidates and we do not expect to generate revenue from sales of any Product Candidates for several years, if at all. We have funded our operations to date primarily with proceeds from the sale of Common Shares.
As of June 30, 2024, we had cash, cash equivalents and short-term investments of $6.6 million.
The following table summarizes our cash flows for each of the periods presented:
| (in thousands) | Year Ended June 30, 2024 |
Year Ended June 30, 2023 |
||||||
| Net cash (used in) operating activities | $ | (6,986 | ) | $ | (7,283 | ) | ||
| Net cash (used in) investing activities | (9 | ) | (662 | ) | ||||
| Net cash provided by financing activities | 4,654 | 10,681 | ||||||
| Net increase (decrease) in cash and cash equivalents | $ | (2,341 | ) | $ | 2,736 | |||
Operating Activities
During the year ended June 30, 2024, we used cash in operating activities of $7.0 million, primarily resulting from our net loss of $7.7 million combined with $0.4 million used in changes in our non-cash working capital, partially offset by non-cash share-based compensation expenses and inventory write-down.
During the year ended June 30, 2023, we used cash in operating activities of $7.3 million, primarily resulting from our net loss of $7.9 million combined with $0.6 million used in changes in our non-cash working capital, partially offset by non-cash share-based compensation expenses and inventory write-down.
Investing Activities
During the year ended June 30, 2024, cash used in investing activities of less than $0.01 million resulted from the purchases of property and equipment.
During the year ended June 30, 2023, cash used in investing activities of $0.7 million resulted from escrow payments made to BayMedica’s historical equity and convertible debt holders and purchase of property and equipment.
Financing Activities
During the year ended June 30, 2024, cash provided by financing activities of $4.7 million consisted of $5.2 million in gross proceeds derived from the 2023 Private Placement, offset by total transaction costs of $0.5 million.
During the year ended June 30, 2023, cash provided by financing activities of $10.7 million consisted of $12.0 million of gross proceeds from private placements of our Common Shares, offset by total transaction costs of $1.3 million.
Funding Requirements
We expect our expenses to increase substantially in connection with our ongoing research and development activities, particularly as we continue the research and development of and the clinical trials for our Product Candidates. In addition, we expect to incur additional costs associated with operating as a US-listed public company and associated with any required investment into BayMedica’s R&D efforts targeting cannabinoid analogs. As a result, we expect to incur substantial operating losses and negative operating cash flows for the foreseeable future.
In accordance with the Financial Accounting Standards Board (“FASB”) Accounting Standards Update (“ASU”) 2014-15, Disclosure of Uncertainties about an Entity’s Ability to Continue as a Going Concern (Subtopic 205-40), we have evaluated whether there are conditions and events, considered in the aggregate, that raise substantial doubt about the Company’s ability to continue as a going concern within one year after the date that the consolidated financial statements are issued.
Through June 30, 2024, we have funded our operations primarily with proceeds from the sale of our Common Shares. We have incurred recurring losses and negative cash flows from operations since its inception, including net losses of $7.7 million and $7.9 million for the years ended June 30, 2024 and 2023, respectively. In addition, we have an accumulated deficit of $109.1 million as of June 30, 2024.
As of the issuance date of these consolidated annual financial statements, the Company expects its cash, cash equivalents and short-term investments of $6.6 million as of June 30, 2024 will be sufficient to fund its operating expenses and capital expenditure requirements to the end of the fourth quarter of calendar 2024, depending on the level and timing of realizing BayMedica revenues from the sale of bulk rare cannabinoids in the health & wellness sector as well as the level and timing of the Company operating expenses. The future viability of the Company is dependent on its ability to raise additional capital to finance its operations. The Company has concluded that there is substantial doubt about its ability to continue as a going concern within one year after the date that the consolidated financial statements are issued.
We expect to continue to seek additional funding through equity financings, debt financings or other capital sources, including collaborations with other companies, government contracts or other strategic transactions. We may not be able to obtain financing on acceptable terms, or at all. The terms of any financing may adversely affect the holdings or the rights of our existing stockholders.
Our funding requirements and timing and amount of our operating expenditures will depend largely on:
| ● | the scope, progress, results and costs of discovery research, preclinical development, laboratory testing and clinical trials for our Product Candidates; |
| ● | the scope, progress, results and costs of development of our manufacturing technologies; |
| ● | the number of and development requirements for other Products and Product Candidates that we pursue; |
| ● | the costs, timing and outcome of regulatory review of our Product Candidates; |
| ● | our ability to enter into contract manufacturing arrangements for supply of materials and manufacture of our Products and Product Candidates and the terms of such arrangements; |
| ● | the impact of any acquired, or in-licensed, externally developed product(s) and/or technologies; |
| ● | our ability to establish and maintain strategic collaborations, licensing or other arrangements, including sales arrangements, and the financial terms of such arrangements; |
| ● | the sales, costs and timing of future commercialization activities, including product manufacturing, sales, marketing and distribution, for any of our Products and for Product Candidates for which we may receive marketing approval; |
| ● | the costs and timing of preparing, filing and prosecuting patent applications, maintaining and enforcing our intellectual property and proprietary rights and defending any intellectual property- related claims; |
| ● | expansion costs of our operational, financial and management systems and increases to our personnel, including personnel to support our clinical development, manufacturing and commercialization efforts and our operations as a dual listed company; |
| ● | the costs to obtain, maintain, expand and protect our intellectual property portfolio; and |
| ● | the level and timing of realizing revenues from the BayMedica commercial operations. |
A change in the outcome of any of these, or other variables with respect to the development of any of our Products and Product Candidates, could significantly change the costs and timing associated with their development. We will need to continue to rely on additional financing to achieve our business objectives.
In addition to the variables described above, if and when any of our Product Candidates successfully complete development, we will incur substantial additional costs associated with regulatory filings, marketing approval, post-marketing requirements, maintaining our intellectual property rights, and regulatory protection, in addition to other commercial costs. We cannot reasonably estimate these costs at this time.
Until such time, if ever, as we can generate substantial revenues from either our Products or Product Candidates, we expect to finance our cash needs through a combination of equity or debt financings and collaboration arrangements. We currently have no credit facility or committed sources of capital. To the extent that we raise additional capital through the future sale of equity securities, the ownership interests of our shareholders will be diluted, and the terms of these securities may include liquidation or other preferences that adversely affect the rights of our existing common shareholders. If we raise additional funds through the issuance of debt securities, these securities could contain covenants that would restrict our operations. We may require additional capital beyond our currently anticipated amounts, and additional capital may not be available on reasonable terms, or at all. If we raise additional funds through collaboration arrangements or other strategic transactions in the future, we may have to relinquish valuable rights to our technologies, future revenue streams, Products or Product Candidates, or grant licenses on terms that may not be favorable to us. If we are unable to raise additional funds through equity or debt financings when needed, we may be required to delay, limit, reduce or terminate development or future commercialization efforts or grant rights to develop and market Products or Product Candidates that we would otherwise prefer to develop and market ourselves.
Off-Balance Sheet Arrangements
During the periods presented, we did not have, and we do not currently have, any off-balance sheet arrangements, as defined in the rules and regulations promulgated by the SEC.
Critical Accounting Policies and Significant Judgments and Estimates
We periodically review our financial reporting and disclosure practices and accounting policies to ensure that they provide accurate and transparent information relative to the current economic and business environment. As part of this process, we have reviewed our selection, application and communication of critical accounting policies and financial disclosures. Management has discussed the development and selection of the critical accounting policies with the Audit Committee of the Board of Directors and the Audit Committee has reviewed the disclosure relating to critical accounting policies in this Management’s Discussion and Analysis.
This discussion and analysis of our financial condition and results of operations is based on our consolidated financial statements included as part of this report, which have been prepared in accordance with U.S. GAAP. The preparation of our consolidated financial statements requires us to make estimates and assumptions that affect the reported amounts of assets and liabilities and the revenue and expenses incurred during the reported periods. We base estimates on our historical experience, known trends and various other factors that we believe are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying value of assets and liabilities that are not apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions.
The full details of our accounting policies are presented in Note 2 of our audited consolidated financial statements for the year ended June 30, 2024. These policies are considered by management to be essential to understanding the processes and reasoning that go into the preparation of our consolidated financial statements and the uncertainties that could have a bearing on its financial results. The significant accounting policies that we believe to be most critical in fully understanding and evaluating our financial results are research and development costs and share based payments.
Use of Estimates
The preparation of financial statements in compliance with US GAAP requires management to make estimates and assumptions that affect the reported amount of assets and liabilities as of the balance sheet date, and the corresponding revenues and expenses for the periods reported. It also requires management to exercise judgment in applying the Company’s accounting policies. In the future, actual experience may differ from these estimates and assumptions. The areas involving a higher degree of judgment or complexity, or areas where assumptions and estimates are significant to these consolidated financial statements are the application of the going concern assumptions, determining the fair value of share-based payments, income tax provisions, write-down of inventories to net realizable value, warrant valuations, and the assumptions used in the determination of research & development accruals. Actual results could differ from those estimates.
Research & Development and Patents costs:
Research and development and patents costs is a critical accounting estimate due to the magnitude and nature of the assumptions that are required to calculate third-party accrued and prepaid research and development expenses. Research and development costs are charged to expense as incurred and include, but are not limited to, personnel compensation, including salaries and benefits, services provided by CROs that conduct preclinical and clinical studies, costs of filing and prosecuting patent applications, and lab supplies.
The amount of expenses recognized in a period related to service agreements is based on estimates of the work performed using an accrual basis of accounting. These estimates are based on services provided and goods delivered, contractual terms and experience with similar contracts. We monitor these factors and adjust our estimates accordingly.
Share-based payments:
The fair value, at the grant date, of equity share awards is charged to income or loss over the period for which the benefits of employees and others providing similar services are expected to be received, generally the vesting period. The corresponding accrued entitlement is recorded in contributed surplus. The amount recognized as an expense is adjusted to reflect the number of share options expected to vest. The fair value of awards is calculated using the Black-Scholes option pricing model which considers the following factors:
| ● | Exercise price; |
| ● | Current market price of the underlying shares; |
| ● | Expected life of the award; |
| ● | Risk-free interest rate; |
| ● | Expected volatility; and |
| ● | Dividend yield. |
Management determines costs for share-based payments using market-based valuation techniques. The fair value of the market-based and performance-based share awards are determined at the date of grant using generally accepted valuation techniques. Assumptions are made and judgment used in applying valuation techniques. These assumptions and judgments include estimating the future volatility of the stock price, expected dividend yield, forfeiture rates and corporate performance. For employee awards, we use the “simplified method” to determine the expected term of options. Under this method, the expected term represents the average of the vesting period and the contractual term. Such judgments and assumptions are inherently uncertain. Changes in these assumptions affect the fair value estimates. If we had made different judgments and assumptions than those described previously, the amount of our share-based payments expense, net loss and net loss per common shares amounts could have been materially different.
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISKS
We are a smaller reporting company as defined by Rule 12b-2 of the Exchange Act and are not required to provide the information required under this item.
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA

Consolidated Financial Statements of
InMed Pharmaceuticals Inc.
For the Years Ended June 30, 2024 and 2023
F-
InMed Pharmaceuticals Inc.
(Expressed in U.S. Dollars)
June 30, 2024
| INDEX | Page | |
| Consolidated Financial Statements | ||
| ● | Report of Independent Registered Public Accounting Firm (PCAOB Firm ID 688) | F-3 |
| ● | Consolidated Balance Sheets | F-4 |
| ● | Consolidated Statements of Operations | F-5 |
| ● | Consolidated Statements of Shareholders’ Equity | F-6 |
| ● | Consolidated Statements of Cash Flows | F-7 |
| ● | Notes to the Consolidated Financial Statements | F-8 |
F-
Report of Independent Registered Public Accounting Firm
To the Shareholders and Board of Directors of
InMed Pharmaceuticals Inc.
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of InMed Pharmaceuticals Inc. (the “Company”) as of June 30, 2024 and 2023, the related consolidated statements of operations, changes in shareholders’ equity and cash flows for each of the two years in the period ended June 30, 2024, and the related notes (collectively referred to as the “financial statements”). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of June 30, 2024 and 2023, and the results of its operations and its cash flows for each of the two years in the period ended June 30, 2024, in conformity with accounting principles generally accepted in the United States of America.
Explanatory Paragraph – Going Concern
The accompanying financial statements have been prepared assuming that the Company will continue as a going concern. As more fully described in Note 1, the Company has incurred recurring losses and negative cash flows and has an accumulated deficit that raise substantial doubt about the Company's ability to continue as a going concern. Management's plans in regard to these matters are also described in Note 1. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Basis for Opinion
These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on the Company's financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) ("PCAOB") and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits, we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company's internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
/s/ Marcum LLP
Marcum LLP
We have served as the Company’s auditor since 2023
New York, NY
September 27, 2024
F-
InMed Pharmaceuticals Inc.
CONSOLIDATED BALANCE SHEETS
Expressed in U.S. Dollars
| June 30, | June 30, | |||||||
| 2024 | 2023 | |||||||
| $ | $ | |||||||
| ASSETS | ||||||||
| Current | ||||||||
| Cash and cash equivalents | 6,571,610 | 8,912,517 | ||||||
| Short-term investments | 43,064 | 44,422 | ||||||
| Accounts receivable (less provision for credit losses of $nil and $66,775 in June 30, 2024 and 2023, respectively) | 352,838 | 260,399 | ||||||
| Inventories, net | 1,244,324 | 1,616,356 | ||||||
| Prepaids and other current assets | 477,749 | 498,033 | ||||||
| Total current assets | 8,689,585 | 11,331,727 | ||||||
| Non-Current | ||||||||
| Property, equipment and ROU assets, net | 1,249,999 | 723,426 | ||||||
| Intangible assets, net | 1,783,198 | 1,946,279 | ||||||
| Other assets | 100,000 | 104,908 | ||||||
| Total Assets | 11,822,782 | 14,106,340 | ||||||
| LIABILITIES AND SHAREHOLDERS’ EQUITY | ||||||||
| Current | ||||||||
| Accounts payable and accrued liabilities | 1,654,011 | 1,608,735 | ||||||
| Current portion of lease obligations | 317,797 | 375,713 | ||||||
| Deferred rent | 16,171 | |||||||
| Total current liabilities | 1,971,808 | 2,000,619 | ||||||
| Non-current | ||||||||
| Lease obligations, net of current portion | 644,865 | 15,994 | ||||||
| Total Liabilities | 2,616,673 | 2,016,613 | ||||||
| Commitments and Contingencies (Note 12) | ||||||||
| Shareholders’ Equity | ||||||||
| Common shares, no par value, unlimited authorized shares: 8,918,956 and 3,328,191 as of June 30, 2024 and 2023, respectively, issued and outstanding | 82,784,400 | 77,620,252 | ||||||
| Additional paid-in capital | 35,368,899 | 35,741,115 | ||||||
| Accumulated deficit | (109,075,759 | ) | (101,400,209 | ) | ||||
| Accumulated other comprehensive income | 128,569 | 128,569 | ||||||
| Total Shareholders’ Equity | 9,206,109 | 12,089,727 | ||||||
| Total Liabilities and Shareholders’ Equity | 11,822,782 | 14,106,340 | ||||||
| Related Party Transactions (Note 13) | ||||||||
The accompanying notes form an integral part of these consolidated financial statements.
F-
InMed Pharmaceuticals Inc.
CONSOLIDATED STATEMENTS OF OPERATIONS
Expressed in U.S. Dollars
| For the Year Ended | ||||||||
| June 30, | ||||||||
| 2024 | 2023 | |||||||
| $ | $ | |||||||
| Sales | 4,597,730 | 4,135,561 | ||||||
| Cost of sales | 3,496,817 | 2,732,525 | ||||||
| Gross profit | 1,100,913 | 1,403,036 | ||||||
| Operating Expenses | ||||||||
| Research and development and patents | 3,765,028 | 3,732,056 | ||||||
| General and administrative | 5,250,715 | 5,847,518 | ||||||
| Amortization and depreciation | 219,600 | 202,249 | ||||||
| Foreign Exchange Loss | 61,921 | 48,175 | ||||||
| Total operating expenses | 9,297,264 | 9,829,998 | ||||||
| Other Income (Expense) | ||||||||
| Interest and other income | 527,901 | 492,440 | ||||||
| Loss before income tax expense | (7,668,450 | ) | (7,934,522 | ) | ||||
| Income tax expense | (7,100 | ) | (13,100 | ) | ||||
| Net loss for the year | (7,675,550 | ) | (7,947,622 | ) | ||||
| Net loss per share for the year | ||||||||
| (1.01 | ) | (3.25 | ) | |||||
| Weighted average outstanding common shares | ||||||||
| 7,621,075 | 2,448,458 | |||||||
The accompanying notes form an integral part of these consolidated financial statements.
F-
InMed Pharmaceuticals Inc.
CONSOLIDATED STATEMENTS OF SHAREHOLDERS’ EQUITY
For the years ended June 30, 2024 and 2023
Expressed in U.S. Dollars
| Common Shares | Additional Paid-in Capital |
Accumulated Deficit | Accumulated Other Comprehensive Income | Total | ||||||||||||||||||||
| # | $ | $ | $ | $ | $ | |||||||||||||||||||
| Balance July 1, 2023 | 3,328,191 | 77,620,252 | 35,741,115 | (101,400,209 | ) | 128,569 | 12,089,727 | |||||||||||||||||
| Private placement | 3,272,733 | 3,240,006 | 1,976,188 | 5,216,194 | ||||||||||||||||||||
| Share issuance costs | (562,151 |
) |
(562,151 |
) | ||||||||||||||||||||
| Exercise of pre-funded warrants | 2,318,032 | 1,924,142 | (1,923,967 | ) | 175 | |||||||||||||||||||
| Loss for the period | - | (7,675,550 | ) | (7,675,550 | ) | |||||||||||||||||||
| Share-based compensation | - | 137,714 | 137,714 | |||||||||||||||||||||
| Balance June 30, 2024 | 8,918,956 | 82,784,400 | 35,368,899 | (109,075,759 | ) | 128,569 | 9,206,109 | |||||||||||||||||
| Common Shares | Additional Paid-in Capital |
Accumulated Deficit | Accumulated Other Comprehensive Income |
Total | ||||||||||||||||||||
| # | $ | $ | $ | $ | $ | |||||||||||||||||||
| Balance July 1, 2022 | 650,667 | 70,718,461 | 31,684,098 | (93,452,587 | ) | 128,569 | 9,078,541 | |||||||||||||||||
| Private placement | 240,000 | 673,748 | 11,326,042 | 11,999,790 | ||||||||||||||||||||
| Share issuance costs | - | (115,955 | ) | (1,895,311 | ) | (2,011,266 | ) | |||||||||||||||||
| Agents’ investment options | - | 691,483 | 691,483 | |||||||||||||||||||||
| Exercise of pre-funded warrants | 2,437,524 | 6,343,998 | (6,343,352 | ) | 646 | |||||||||||||||||||
| Loss for the period | - | (7,947,622 | ) | (7,947,622 | ) | |||||||||||||||||||
| Share-based compensation | - | 278,155 | 278,155 | |||||||||||||||||||||
| Balance June 30, 2023 | 3,328,191 | 77,620,252 | 35,741,115 | (101,400,209 | ) | 128,569 | 12,089,727 | |||||||||||||||||
The accompanying notes form an integral part of these consolidated financial statements.
F-
InMed Pharmaceuticals Inc.
CONSOLIDATED STATEMENTS OF CASH FLOWS
For the years ended June 30, 2024 and 2023
Expressed in U.S. Dollars
| 2024 | 2023 | |||||||
| $ | $ | |||||||
| Cash provided by (used in): | ||||||||
| Operating Activities | ||||||||
| Net loss | (7,675,550 | ) | (7,947,622 | ) | ||||
| Items not requiring cash: | ||||||||
| Amortization and depreciation | 219,600 | 202,249 | ||||||
| Share-based compensation | 137,714 | 278,155 | ||||||
| Amortization of right-of-use assets | 384,918 | 393,748 | ||||||
| Interest income received on short-term investments | (1,250 | ) | (803 | ) | ||||
| Unrealized foreign exchange loss | 12,262 | 1,183 | ||||||
| Inventory write-down | 305,812 | 308,937 | ||||||
| Credit losses | 46,775 | |||||||
| Changes in operating assets and liabilities: | ||||||||
| Inventories | 66,220 | 565,561 | ||||||
| Prepaids and other currents assets | 20,284 | 299,192 | ||||||
| Other non-current assets | 4,908 | 5,507 | ||||||
| Accounts receivable | (92,439 | ) | (219,147 | ) | ||||
| Accounts payable and accrued liabilities | 45,282 | (806,530 | ) | |||||
| Deferred rent | (16,171 | ) | 16,171 | |||||
| Lease obligations | (397,422 | ) | (426,575 | ) | ||||
| Total cash used in operating activities | (6,985,832 | ) | (7,283,199 | ) | ||||
| Investing Activities | ||||||||
| Payment of acquisition consideration | (500,000 | ) | ||||||
| Payment of deposit on equipment | (1,790 | ) | ||||||
| Purchase of property and equipment | (9,293 | ) | (160,014 | ) | ||||
| Sale of short-term investments | 42,082 | 42,268 | ||||||
| Purchase of short-term investments | (42,082 | ) | (42,268 | ) | ||||
| Total cash used in investing activities | (9,293 | ) | (661,804 | ) | ||||
| Financing Activities | ||||||||
| Proceeds from the exercise of pre-funded warrants | 175 | 646 |
||||||
| Proceeds from the private placement net of issuance costs | 4,654,043 | 10,680,008 | ||||||
| Total cash provided by financing activities | 4,654,218 | 10,680,654 | ||||||
| Increase (decrease) in cash during the year | (2,340,907 | ) | 2,735,651 | |||||
| Cash and cash equivalents beginning of the year | 8,912,517 | 6,176,866 | ||||||
| Cash and cash equivalents end of the year | 6,571,610 | 8,912,517 | ||||||
| SUPPLEMENTARY CASH FLOW INFORMATION: | ||||||||
| Cash Paid During the Year for: | ||||||||
| Income taxes | $ | 7,100 | $ | 13,100 | ||||
| Interest | $ | $ | ||||||
| SUPPLEMENTARY DISCLOSURE OF NON-CASH INVESTING AND FINANCING ACTIVITIES: | ||||||||
| Preferred investment options to its placement agent | $ | 325,699 | $ | 691,484 | ||||
| Fair value of warrant modification recorded as equity issuance costs | $ | 3,508,749 | $ | |||||
| Recognition of Right-of-use asset and corresponding operating lease | $ | 968,376 | $ | |||||
The accompanying notes form an integral part of these consolidated financial statements.
F-
| 1. | CORPORATE INFORMATION AND CONTINUING OPERATIONS |
Business
InMed Pharmaceuticals Inc. (“InMed” or the “Company”) was incorporated in the Province of British Columbia on May 19, 1981 under the Business Corporations Act of British Columbia. InMed is a clinical stage pharmaceutical company developing a pipeline of prescription-based products, including rare cannabinoids and novel cannabinoid analogs, targeting the treatment of diseases with high unmet medical needs as well as developing proprietary manufacturing technologies to produce rare cannabinoids for sale in the health and wellness industry.
The Company’s shares are listed on the Nasdaq Capital Market (“Nasdaq”) under the trading symbol “INM”. InMed’s office and principal place of business is located at Suite 1445– 885 West Georgia Street, Vancouver, B.C., Canada, V6C 1B4.
Going Concern
In accordance with the Financial Accounting Standards Board (“FASB”) Accounting Standards Update (“ASU”) 2014-15, Disclosure of Uncertainties about an Entity’s Ability to Continue as a Going Concern (Subtopic 205-40), the Company has evaluated whether there are conditions and events, considered in the aggregate, that raise substantial doubt about the Company’s ability to continue as a going concern within one year after the date that the consolidated financial statements are issued.
Through June 30, 2024, the Company has funded its operations primarily with proceeds from the sale of Common Shares. The Company has incurred recurring losses and negative cash flows from operations since its inception, including net losses of approximately $7.7 million and $7.9 million for the years ended June 30, 2024 and 2023, respectively. In addition, the Company had an accumulated deficit of approximately $109.1 million as of June 30, 2024. The Company expects to continue to generate operating losses for the foreseeable future.
As of the issuance date of these consolidated annual financial statements, the Company expects its cash, cash equivalents and short-term investments of $6.6 million as of June 30, 2024 will be sufficient to fund its operating expenses and capital expenditure requirements to the end of the fourth quarter of calendar 2024, depending on the level and timing of realizing BayMedica revenues from the sale of bulk rare cannabinoids in the health & wellness sector as well as the level and timing of the Company operating expenses. The future viability of the Company is dependent on its ability to raise additional capital to finance its operations. The Company has concluded that there is substantial doubt about its ability to continue as a going concern within one year after the date that the consolidated financial statements are issued.
The Company expects to continue to seek additional funding through equity financings, debt financings or other capital sources, including collaborations with other companies, government contracts or other strategic transactions. The Company may not be able to obtain financing on acceptable terms, or at all. The terms of any financing may adversely affect the holdings or the rights of the Company’s existing shareholders.
In connection with the Company’s assessment of going concern considerations in accordance with Subtopic 205-40, management has determined that the Company’s liquidity condition raises substantial doubt about the Company’s ability to continue as a going concern, which is considered to be for a period of one year from the issuance of these financial statements. These consolidated financial statements do not include any adjustments relating to recoverability and classification of recorded asset amounts or the amounts of classification of liabilities that might result from the outcome of this uncertainty. Such adjustments could be material.
F-
| 2. | SIGNIFICANT ACCOUNTING POLICIES |
Basis of Presentation
These consolidated financial statements have been prepared in accordance with generally accepted accounting principles as applied in the United States (“US GAAP”) and pursuant to the rules and regulations of the United States Securities and Exchange Commission (“SEC”) for financial information.
Reclassifications
Certain prior year amounts in the consolidated financial statements and the notes thereto have been reclassified where necessary to conform to the current year’s presentation. These reclassifications did not affect the prior period’s total assets, total liabilities, stockholders’ deficit, net loss or net cash used in operating activities. During the three months and nine months ended March 31, 2024, we adopted a change in presentation on our consolidated statements of operations in order to include foreign exchange loss in operating expenses. The Company has adopted ASU 2023-07 - Improvements to Reportable Segment Disclosures which has required prior period to reflect the change in presentation. Refer to discussion on Recent Accounting Pronouncements below.
Use of Estimates
The preparation of financial statements in compliance with US GAAP requires management to make estimates and assumptions that affect the reported amount of assets and liabilities as of the balance sheet date, and the corresponding revenues and expenses for the periods reported. It also requires management to exercise judgment in applying the Company’s accounting policies. In the future, actual experience may differ from these estimates and assumptions. The areas involving a higher degree of judgment or complexity, or areas where assumptions and estimates are significant to these consolidated financial statements are the application of the going concern assumptions, determining the fair value of share-based payments, income tax provisions, write-down of inventories to net realizable value, warrant valuations, and the assumptions used in the determination of research & development accruals.
Actual results could differ from those estimates.
Basis of Consolidation
These consolidated financial statements include the accounts of the Company and its subsidiaries, including subsidiaries: InMed Pharmaceutical Ltd., BayMedica, LLC, Biogen Sciences Inc., and Sweetnam Consulting Inc. A subsidiary is an entity that the Company controls, either directly or indirectly, where control is defined as the power to govern the financial and operating policies of an entity so as to obtain benefits from its activities. All inter-company transactions and balances including unrealized income and expenses arising from intercompany transactions are eliminated in preparing these consolidated financial statements.
Foreign Currency
The functional currency of the Company and its subsidiaries is the U.S. Dollar. These consolidated financial statements are presented in U.S. Dollars. References to “$” and “US$” are to United States (“U.S.”) dollars and references to “C$” are to Canadian dollars.
Business Combinations
Business combinations are accounted for using the acquisition method. The fair value of total purchase consideration is allocated to the fair values of identifiable tangible and intangible assets acquired and liabilities assumed, with the remaining amount being classified as goodwill. All assets, liabilities and contingent liabilities acquired or assumed in a business combination are recorded at their fair values at the date of acquisition. If the Company’s interest in the fair value of the acquiree’s net identifiable assets exceeds the cost of the acquisition, the excess is recognized in earnings. Transaction costs that are incurred in connection with a business combination, other than costs associated with the issuance of debt or equity securities, are expensed as incurred.
F-
Cash and Cash Equivalents
Cash and cash equivalents include cash-on-hand, demand deposits with financial institutions and other short-term, highly liquid investments with original maturities of three months or less when acquired that are readily convertible to known amounts of cash and subject to an insignificant risk of change in value. As of June 30, 2024 and 2023, the Company holds $1,939,482 and $1,478,487 respectively, of cash equivalents in a money market fund that is considered Level 1 in the financial instruments hierarchy due to the readily available quoted prices in active markets for identical instruments.
Short-term Investments
Short-term investments include fixed and variable rate guaranteed investment certificates, with terms greater than three months and less than twelve months. Due to the short-term nature of these investments the fair value of the investments approximates the current value. Guaranteed investment certificates are convertible to known amounts of cash and are subject to an insignificant risk of change in value.
Accounts Receivable
Accounts receivable are recorded at invoiced amounts, net of any credit losses. The allowance for doubtful accounts is the Company’s best estimate of the amount of probable credit losses in existing accounts receivable.
The Company evaluates the collectability of accounts receivable on a regular basis based upon various factors including the financial condition and payment history of customers, an overall review of collections experience on other accounts and economic factors or events expected to affect future collections experience. Expected credit losses on our accounts receivable were $0 and $66,775 as at June 30, 2024 and 2023 respectively. We had $66,775 and $0 of write-offs during the year ended June 30, 2024 and 2023, respectively.
Concentration of Credit Risk and Other Risks and Uncertainties
At times, cash balances may exceed the Federal Deposit Insurance Corporation (“FDIC”) or Canadian Deposit Insurance Corporation (CDIC) insurable limits. The Company has not experienced any losses related to these balances. The uninsured cash balance as of June 30, 2024, was $2.4 million. The Company does not believe it is exposed to significant credit risk on cash and cash equivalents.
The Company’s customers are primarily concentrated in the United States.
As of June 30, 2024, we had five customers with an accounts receivable balance representing 32%, 20%, 15%,15% and 14% of total accounts receivable.
For the year ended June 30, 2024, the Company had five customers that accounted for 34%, 18%, 14%, 12% and 10% of revenue. For the year ended June 30, 2023, the Company had four customers that accounted for 22%, 17%, 16% and 11% of revenue.
Inventories
Inventories are initially valued at weighted average cost and subsequently valued at the lower of weighted average cost and net realizable value. Costs included in inventories are the purchase price of goods and cost of services rendered, freight costs, warehousing costs, purchasing costs and production and labor costs related to manufacturing.
In determining any valuation allowances, the Company reviews inventory for obsolete, redundant, and slow-moving goods. As of June 30, 2024, the Company has $103,434 as a valuation allowance to reduce weighted average cost to net realizable value. As of June 30, 2023, the Company has $93,820 as a valuation allowance to reduce weighted average cost to net realizable value. During the year ended June 30, 2024 and 2023 the Company record an inventory write-down due to net realizable value of $103,136 and $308,937 respectively. During the year ended June 30, 2024 and 2023 the Company record an inventory write-down due to obsolescence of $208,737 and $nil, respectively.
F-
Property, Equipment and ROU Assets, Net
Computer equipment, lab equipment and furnishings are recorded at cost, less accumulated depreciation and accumulated impairment losses. The initial cost of computer equipment, lab equipment and furnishings comprises their purchase price. The computer equipment, lab equipment and furnishings are reviewed at least once per year for impairment. Equipment and furniture are depreciated using the straight-line method based on their estimated useful lives as follows:
| ● | Computer equipment — 5 years | |
| ● | Lab equipment — 6 - 10 years |
| ● | Furnishings — 5 years |
Computer equipment, lab equipment and furnishings, acquired or disposed of during the year, are depreciated proportionately for the period they are in use.
The right-of-use assets are initially measured based on the initial amount of the lease liability adjusted for any lease payments made at or before the commencement date, less any lease incentives received. The assets are amortized to the earlier of the end of the useful life of the right-of-use asset or the lease term using the straight-line method as this most closely reflects the expected pattern of consumption of the future economic benefits. The lease term includes periods covered by an option to extend if the Company is reasonably certain to exercise that option. In addition, the right-of-use assets are periodically reduced by impairment losses, if any, and adjusted for certain re-measurements of the lease liability (see Note 2 Lease (i)).
Intangible Assets, Net
Intangible assets are comprised of acquired intellectual property, which consists of certain patents and technical know-how. The intellectual property is recorded at cost and is amortized on a straight-line basis over an estimated useful life of 18 years net of any accumulated impairment losses. There is no impairment loss during the years ended June 30, 2024 and 2023.
In-Process R&D
In-process R&D (“IPR&D”) is classified as an indefinite-lived intangible asset and is not amortized. IPR&D becomes definite-lived upon the completion or abandonment of the associated research and development efforts. All research and development costs incurred subsequent to the acquisition of IPR&D are expensed as incurred. Indefinite-lived intangible assets are evaluated for impairment on an annual basis or more frequently if an indicator of impairment is present.
Impairment of Long-Lived Assets
The Company assesses the recoverability of its long-lived assets whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. Recoverability of the long-lived asset is measured by a comparison of the carrying amount of the asset to future undiscounted net cash flows expected to be generated by the asset or assets. If carrying value exceeds the sum of undiscounted cash flows, the Company then determines the fair value of the underlying asset. Any impairment to be recognized is measured as the amount by which the carrying amount of the asset group exceeds the estimated fair value of the asset group. Assets classified as held for sale are reported at the lower of the carrying amount or fair value, less costs to sell.
Fair Value Measurements
Financial Assets
Financial assets are initially recognized at fair value, plus transaction costs that are directly attributable to their acquisition or issue and subsequently carried at amortized cost, using the effective interest rate method, less any impairment losses. No financial assets are or elected to be carried at fair value through profit or loss or where changes in fair value are recognized in the consolidated statements of operations and comprehensive loss in other comprehensive loss.
F-
Short-term investments are subsequently recorded at cost plus accrued interest, which approximates fair value due to short-term nature. Accounts receivable are reported at outstanding amounts, net of credit losses.
Financial Liabilities
To determine the fair value of financial instruments, the Company uses the fair value hierarchy for inputs used to measure fair value of financial assets and liabilities. This hierarchy prioritizes the inputs to valuation techniques used to measure fair value into three levels: Level 1 (highest priority), Level 2, and Level 3 (lowest priority).
| Level 1 – | Unadjusted quoted prices in active markets for identical instruments. |
| Level 2 – | Inputs other than quoted prices included within Level 1 that are observable for the asset or liability, either directly or indirectly. Level 2 inputs include quoted prices for similar assets or liabilities in active markets, quoted prices for identical or similar assets or liabilities in markets that are not active, inputs other than quoted prices that are observable for the asset or liability (i.e., interest rates, yield curves, etc.), and inputs that are derived principally from or corroborated by observable market data by correlation or other means (market corroborated inputs). |
| Level 3 – | Inputs are unobservable and reflect the Company’s assumptions as to what market participants would use in pricing the asset or liability. The Company develops these inputs based on the best information available. Assets and liabilities are classified based on the lowest level of input that is significant to the fair value measurements. Changes in the observability of valuation inputs may result in a reclassification of levels for certain securities within the fair value hierarchy. |
The carrying value of cash and cash equivalents, short-term investments, accounts receivable, and accounts payable and accrued liabilities, approximate their carrying values as at June 30, 2024 and 2023 due to their immediate or short-term maturities.
Income Taxes
Deferred tax assets and liabilities are recognized for the future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases and operating loss and tax credit carry forwards. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. The effect on deferred tax assets and liabilities of a change in tax rates is recognized in income in the period that includes the enactment date. At June 30, 2024, and June 30, 2023, the Company had a full valuation allowance against its deferred tax assets.
Per FASB ASC 740-10, disclosure is not required of an uncertain tax position unless it is considered probable that a claim will be asserted and there is a more-likely-than-not possibility that the outcome will be unfavorable. Using this guidance, as of June 30, 2024, and 2023, the Company has no uncertain tax positions that qualify for either recognition or disclosure in the financial statements. The Company’s 2024, 2023, 2022, and 2021 United States and Canadian tax returns remain subject to examination by their respective taxing authorities. Neither of the Company’s tax returns are currently under examination.
Revenue Recognition
The Company recognizes revenue when the Company satisfies the performance obligations under the terms of a contract and control of its products and services is transferred to its customers in an amount that reflects the consideration the Company expects to receive from its customers in exchange for those products and services. ASC 606, Revenue from Contracts with Customers defines a five-step process to recognize revenue that requires judgment and estimates, including identifying the contract with the customer, identifying the performance obligations in the contract, determining the transaction price, allocating the transaction price to the performance obligations in the contract, and recognizing revenue when or as the performance obligation is satisfied.
F-
Revenue consists of manufacturing and distribution sales of bulk rare cannabinoids, which are generally recognized at a point in time. The Company recognizes revenue when control over the products has been transferred to the customer and the Company has a present right to payment. Sales and other taxes that are required to be remitted to regulatory authorities are recorded as liabilities and excluded from sales. Limited rights of return, for claims of damaged or non-compliant products, exist with the Company’s customers.
The Company has elected the practical expedient that allows it to recognize the incremental costs of obtaining a contract as an expense, when incurred, if the amortization period of the asset that the Company otherwise would have recognized is one year or less.
Revenues within the scope of ASC 606 do not include material amounts of variable consideration. Customer payments are generally due in advance of when control is transferred to the customer. Some of our larger customers with which we have history with are eligible for payment terms up to net 30.
Cost of Sales
Cost of sales consists primarily of the purchase price of goods and cost of services rendered, freight costs, warehousing costs, and purchasing costs. Cost of sales also includes production and labor costs for the Company’s manufacturing business.
Shipping and Handling
The Company records freight billed to customers within Net sales. Shipping and handling costs associated with inbound freight and goods shipped to customers are recorded in cost of sales. Other shipping and handling costs, such as for quality assurance, are recorded in operating expenses.
Earnings (Loss) Per Share
Basic earnings (loss) per common share (“EPS”) is computed by dividing the net income or loss applicable to common shares of the Company by the weighted average number of common shares outstanding for the relevant period. The Company has 694,017 pre-funded warrants included in the basic earnings (loss) per share. Diluted earnings (loss) per common share (“Diluted EPS”) is computed by dividing the net income or loss applicable to common shares by the sum of the weighted average number of common shares issued and outstanding and all additional common shares that would have been outstanding, if potentially dilutive instruments were converted. If the conversion of outstanding stock options and warrants into common share is anti-dilutive, then diluted EPS is not presented separately from EPS.
The following table sets forth the number of potential shares of common stock that have been excluded from diluted net income (loss) per because their effect was anti-dilutive:
| Year ended June 30, | ||||||||
| 2024 | 2023 | |||||||
| Options | 674,473 | 102,642 | ||||||
| Warrants | 10,192,044 | 3,516,529 | ||||||
| 10,866,517 | 3,619,171 | |||||||
Share-based Payments
The Company follows the requirements of FASB ASC 718-10-10, Share-Based Payments with regards to stock-based compensation issued to employees and non-employees. The Company has agreements and arrangements that call for stock to be awarded to the employees and consultants at various times as compensation and periodic bonuses. The expense for this stock-based compensation is equal to the fair value of the stock price on the day the stock was awarded multiplied by the number of shares awarded. The Company has a relatively low forfeiture rate of stock-based compensation and forfeitures are recognized as they occur.
F-
The valuation methodology used to determine the fair value of the options issued during the period is the Black-Scholes option-pricing model. The Black-Scholes model requires the use of a number of assumptions including the volatility of the stock price, the average risk-free interest rate, and the weighted average expected life of the options. Risk-free interest rates are calculated based on continuously compounded risk-free rates for the appropriate term. The dividend yield is assumed to be zero as the Company has never paid or declared any cash dividends on its Common Stock and does not intend to pay dividends on its Common Stock in the foreseeable future. The expected forfeiture rate is estimated based on management’s best assessment.
Estimated volatility is a measure of the amount by which InMed’s stock price is expected to fluctuate each year during the expected life of the award. The Company’s calculation of estimated volatility is based on historical stock prices over a period equal to the expected life of the awards.
Research and Development Costs
The Company conducts research and development programs and incurs costs related to these activities, including research and development personnel compensation, services provided by contract research organizations and lab supplies. Research and development costs are expensed in the periods in which they are incurred.
Patents and Intellectual Property Costs
The costs of filing for patents and of prosecuting and maintaining intellectual property rights are expensed as incurred due to the uncertainty surrounding the drug development process and the uncertainty of future benefits. Patents and intellectual property acquired from third parties for approved products or where there are alternative future uses are capitalized and amortized over the remaining life of the patent.
Segment reporting
The Company’s operations consist of two operating and reportable segments, the InMed Pharma segment and the BayMedica Commercial segment.
The InMed Pharma segment is largely organized around the research and development of small molecule pharmaceuticals drug candidates and the BayMedica Commercial segment is largely organized around manufacturing technologies to produce and commercialize bulk rare cannabinoids for sale as ingredients in the health and wellness industry (See Note 11).
Leases
At inception of a contract, the Company assesses whether a contract is, or contains, a lease based on whether the contract conveys the right to control the use of an identified asset for a period of time in exchange for consideration.
The lease liability is initially measured as the present value of future lease payments excluding payments made at the commencement date, discounted using the interest rate implicit in the lease or, if that rate cannot be readily determined, the Company’s incremental borrowing rate. Generally, the Company uses its incremental borrowing rate as the discount rate. The lease liability is measured at amortized cost using the effective interest method. It is re-measured when there is a change in future lease payments arising from a change in an index or rate, if there is a change in the Company’s estimate of the amount expected to be payable under a residual value guarantee, or if the Company changes its assessment of whether it will exercise a purchase, extension, or termination option. When the lease liability is re-measured in this way, a corresponding adjustment is made to the carrying amount of the right-of-use asset or is recorded in profit or loss if the carrying amount of the right-of-use asset has been reduced to nil.
The Company has lease arrangements that include both lease and non-lease components. The Company accounts for each separate lease component and its associated non-lease components as a single lease component for all of its asset classes.
F-
The Company has elected to apply the practical expedient to exclude initial direct costs such as annual operating costs from the measurement of the right-of-use asset at the date of initial application. The Company has elected to apply the practical expedient not to recognize right-of-use assets and lease liabilities for short-term leases that have a lease term of 12 months or less. The lease payments associated with these leases is recognized as an expense on a straight-line basis over the lease term.
Recent Accounting Pronouncements
The Company has reviewed recent accounting pronouncements and concluded that they are either not applicable to the Company or that there was no material impact or no material impact is expected in the consolidated financial statements as a result of future adoption.
In November 2023, the Financial Accounting Standards Board (“FASB”) issued ASU 2023-07, Segment Reporting (Topic 280): Improvements to Reportable Segment Disclosures, which enhances reportable segment disclosure requirements primarily through expanded disclosures around significant segment expenses. The amendments are effective for fiscal years beginning after December 15, 2023, and for interim periods within fiscal years beginning after December 15, 2024. The amendments should be applied retrospectively to all prior periods presented in the financial statements. The Company has early adopted this accounting pronouncement.
In December 2023, the FASB issued ASU 2023-09, Income Taxes (Topic 740): Improvements to Income Tax Disclosures, which requires disclosure of specific categories meeting a quantitative threshold within the income tax rate reconciliation, as well as disaggregation of income taxes paid by jurisdiction. This ASU, which can be applied either prospectively or retrospectively, is effective for annual periods beginning after December 15, 2024, with early adoption permitted. The Company is currently evaluating the impact of the ASU and expects to include updated income tax disclosures in its fiscal year 2026.
| 3. | INVENTORIES |
Inventories consisted of the following:
| June 30, 2024 |
June 30, 2023 |
|||||||
| $ | $ | |||||||
| Raw materials | 372,695 | 208,737 | ||||||
| Work in process | 30,817 | 514,113 | ||||||
| Finished goods | 840,812 | 893,506 | ||||||
| Inventories | 1,244,324 | 1,616,356 | ||||||
In determining any valuation allowances, the Company reviews inventory for obsolete, redundant, and slow-moving goods. During the year ended June 30, 2024 and 2023, the write-down of inventories to net realizable value was $9,614 and $308,937 respectively. Contributing factors to the decrease in net realizable value included lower demand and downward pricing pressure for certain products. As of June 30, 2024 and 2023, the Company has $103,434 and $93,820 respectively as a valuation allowance to reduce weighted average cost to new basis.
| 4. | PROPERTY, EQUIPMENT AND ROU ASSETS, NET |
Property, equipment and ROU assets consisted of the following:
| June 30, 2024 |
June 30, 2023 |
|||||||
| $ | $ | |||||||
| Right-of-Use Assets (leases) | 2,135,811 | 1,167,436 | ||||||
| Equipment | 429,090 | 440,902 | ||||||
| Furnishing | 40,409 | 40,409 | ||||||
| Property and equipment | 2,605,310 | 1,648,747 | ||||||
| Less: accumulated depreciation and amortization | (1,355,311 | ) | (925,321 | ) | ||||
| Property, equipment and ROU assets, net | 1,249,999 | 723,426 | ||||||
F-
Depreciation expense on computer equipment, lab equipment and furnishing for the year ended June 30, 2024 and 2023, was $47,742 and $39,613 respectively and was recorded in general and administrative expenses. Amortization expense related to the right-of-use assets for the year ended June 30, 2024 and 2023, was $384,918 and $369,239 respectively and was recorded in general and administrative expenses.
| 5. | INTANGIBLE ASSETS |
The following table summarizes the Companies intangible assets:
| June 30, 2024 |
June 30, 2023 |
|||||||
| $ | $ | |||||||
| Intellectual property | 1,736,420 | 1,736,420 | ||||||
| Patents | 1,191,000 | 1,191,000 | ||||||
| Intangible assets | 2,927,420 | 2,927,420 | ||||||
| Less: accumulated amortization | (1,144,222 | ) | (981,141 | ) | ||||
| Intangible assets, net | 1,783,198 | 1,946,279 | ||||||
Acquired intellectual property is recorded at cost and is amortized on a straight-line basis over 18 years. Acquired patents consist of patents related to the development of cannabinoid analogs. This intangible asset is being amortized over an estimated useful life of 18 years. As at June 30, 2024, the definite-lived intangible assets had a weighted average estimated remaining useful life of approximately 11 years.
Amortization expense on intangible assets for the year ended June 30, 2024 and 2023 was $171,858 and $159,228 respectively. The Company expects amortization expense to be incurred over the next five years as follows:
| Twelve months ending June 30, | $ | |||
| 2024 | 162,746 | |||
| 2025 | 162,746 | |||
| 2026 | 162,746 | |||
| 2027 | 162,746 | |||
| 2028 | 162,746 | |||
| Thereafter | 969,468 | |||
| Total | 1,783,198 | |||
| 6. | ACCOUNTS PAYABLE AND ACCRUED LIABILITIES |
Accounts payable and accrued liabilities consist of the following:
| June 30, 2024 |
June 30, 2023 |
|||||||
| $ | $ | |||||||
| Trade payables | 626,190 | 544,179 | ||||||
| Accrued research and development expenses | 242,066 | 164,587 | ||||||
| Inventory related accruals | 41,004 | |||||||
| Employee compensation, benefits and related accruals | 488,278 | 542,305 | ||||||
| Accrued general and administrative expenses | 256,473 | 357,664 | ||||||
| Accounts payable and accrued liabilities | 1,654,011 | 1,608,735 | ||||||
F-
| 7. | SHARE CAPITAL AND RESERVES |
Authorized
As of June 30, 2024, the Company’s authorized share structure consisted of: (i) an unlimited number of common shares without par value; and (ii) an unlimited number of preferred shares without par value. No preferred shares were issued and outstanding as of June 30, 2024 and 2023.
The Company may, from time to time, issue preferred shares and may, at the time of issuance, determine the rights, preferences and limitations pertaining to these shares. Holders of preferred shares may be entitled to receive a preference payment in the event of any liquidation, dissolution or winding up of the Company before any payment is made to the holders of common shares.
On June 27, 2024, the Company entered into an amendment (the “ATM Amendment”) to its At-the-Market Offering Agreement, dated April 7, 2022 (the “Original ATM Agreement” and together with the ATM Amendment, the “Amended ATM Agreement”), pursuant to which the Company may offer and sell shares of the Company’s common shares, no par value per share (the “Common Shares”), from time to time, in “at the market” offerings through the Agent. The Original ATM Agreement was previously filed with the Securities and Exchange Commission on April 7, 2022 on the Company’s Current Report on Form 8-K. The ATM Amendment amends the Original ATM Agreement to reflect, among other provisions, updates to certain sales settlement provisions and reimbursement terms, and to supplement the representations being made by the Company to the Agent.
On October 24, 2023, the Company entered into a securities purchase agreement (the “2023 Securities Purchase Agreement”) with two accredited institutional investors (the “Accredited Institutional Investors”) for the sale (the “2023 Private Placement”) of 3,012,049 pre-funded warrants of the Company’s common shares at a purchase price of $0.83 per share. The pre-funded warrants have an exercise price of $0.0001 and do not have an expiration date. The pre-funded warrants had a fair value of $1,248,376 at the time of issuance. As of June 30, 2024, there were 694,017 pre-funded warrants outstanding. In addition, the Company agreed, as part of the 2023 Private Placement, to issue to the purchasers unregistered preferred investment options to purchase up to an aggregate of 3,012,049 common shares. These preferred investment options have an exercise price of $0.83 and have a term of 5.5 years from issuance. The preferred investment options had a fair value of $1,251,449 at the time of their issuance.
Concurrently with the Company’s entry into the 2023 Securities Purchase Agreement, the Company also entered into an inducement offer letter agreement (the “Inducement Offer Letter”) with the holders of existing preferred investment options (the “Existing Holders”) to purchase up to an aggregate of 3,272,733 common shares issued to the Existing Holders on November 21, 2022. Pursuant to the Inducement Offer Letter, the Existing Holders agreed to exercise for cash their existing preferred investment options to purchase an aggregate of 3,272,733 common shares (at a reduced exercise price of $0.83 per share) in consideration of the Company’s agreement to issue new unregistered preferred investment options to purchase up to an aggregate of 6,545,466 shares of the Company’s common shares at an exercise price of $0.83 per share). Due to ownership limitations, the Accredited Institutional Investors had 1,796,552 common shares held in abeyance as of the closing of the 2023 Private Placement. The abeyance shares had a fair value of $1,491,138 and the common shares issued had a fair value of $1,225,230 on their respective issuance date. As of June 30, 2024, the Accredited Institutional Investors had drawn down 1,796,552 abeyance shares.
F-
The inducement contemplated by the Inducement Offer Letter (the “Inducement”) is considered a warrant modification due to the changing of the terms of the warrants. The modification had a fair value of $3.5 million as of the date of the Inducement, using a Black-Scholes model and is recognized as an equity issuance cost in accordance with ASC 718-20-35-3.
On October 26, 2023, the parties consummated the 2023 Private Placement and the other transactions contemplated by the 2023 Securities Purchase Agreement. In connection with such transactions, the Company (i) received gross proceeds of approximately $5.2 million and paid approximately $560,000 in cash fees and (ii) issued 408,511 warrants to our placement agent. These warrants have an exercise price of $1.0375 and a term of 5.5 years. The placement agent warrants had a fair value of $325,699 as of the date of their issuance, using a Black-Scholes model and were recorded as an equity issuance cost.
On September 13, 2022, the Company closed a private placement of its common shares and issued an aggregate of 90,000 common shares and 601,245 pre-funded warrants, for gross proceeds of $5,999,946. The pre-funded warrants were determined to be common stock equivalents. Each common share and each pre-funded warrant were sold in the offering with an investment option to purchase a common share. Transaction costs were allocated proportionally between common shares and investment options with $77,242 allocated to common shares and the balance of $1,052,101 allocated to additional paid-in capital and recorded as a component of shareholders’ equity in the consolidated balance sheet. As of June 30, 2023, there were no pre-funded warrants outstanding.
In connection with the September 13, 2022, private placement the company issued 1,382,490 warrants. These warrants were issued with an exercise price of $8.44 per share, were immediately exercisable upon issuance, and expire 7 years following the date of issuance. On November 21, 2022, these preferred investment options were surrendered to the Company for cancellation.
On November 21, 2022, the Company closed a private placement of its common shares and issued an aggregate of 150,000 common shares and 1,668,185 pre-funded warrants, for gross proceeds of $5,999,844. The pre-funded warrants were determined to be common stock equivalents. Each common share and each pre-funded warrant were sold in the offering with an investment option to purchase a common share. Transaction costs were allocated proportionally between common shares and investment options with $38,713 allocated to common shares and the balance of $831,292 allocated to additional paid-in capital and recorded as a component of shareholders’ equity in the consolidated balance sheet. As of June 30, 2023, there were no pre-funded warrants outstanding.
In connection with the November 21, 2022, private placement the company issued 3,272,733 warrants. These warrants were issued with an exercise price of $3.044 per share, were immediately exercisable upon issuance, and expire 7 years following the date of issuance. The allocated value of these investment options was recorded in additional paid-in capital.
Common Share Warrants
The assumptions used in the Black-Scholes model to value the new warrants issued during the years ended June 30, 2024 and 2023, are set forth in the table immediately below.
| June 30, 2024 |
||||
| Exercise price | $ | 0.83 – 1.04 | ||
| Risk-free interest rate | 4.82 | % | ||
| Volatility | 109 – 111 | % | ||
| Expected life (years) | 5.0 – 5.5 | |||
| Dividend yield | $ | 0 | % | |
| June 30, 2023 |
||||
| Exercise price | $ | 3.04 – 8.44 | ||
| Risk-free interest rate | 2.92 – 3.12 | % | ||
| Volatility | 114 - 117 | % | ||
| Expected life (years) | 7 Years | |||
| Dividend yield | $ | 0 | % | |
F-
The assumptions used in the Black-Scholes model to value the modification of warrants issued during the year ended June 30, 2024, are set forth in the table immediately below.
| 2024 | ||||
| Exercise price | $ | 0.83 – 3.04 | ||
| Risk-free interest rate | 0.56 – 4.82 | % | ||
| Volatility | 109 – 614 | % | ||
| Expected life (years) | 0 – 6.8 | |||
| Dividend yield | $ | 0 | % | |
A summary of the Company’s warrant activity and related information for the periods covered were as follows:
| Number of Shares Under Warrants |
Weighted Average Exercise Price |
|||||||
| Balance as at July 1, 2022 | 505,128 | $ | 31.92 | |||||
| Granted | 4,818,336 | 4.69 | ||||||
| Expired / Cancelled Exercised | (1,806,935 | ) | 10.75 | |||||
| Exercised | ||||||||
| Balance as at June 30, 2023 | 3,516,529 | $ | 5.37 | |||||
| Warrants Granted | 12,978,075 | 0.64 | ||||||
| Exercised | (5,590,765 | ) | 0.49 | |||||
| Expire/Cancelled | (17,778 | ) | 18.50 | |||||
| Warrants Outstanding at June 30, 2024 | 10,886,061 | $ | 1.06 | |||||
| Warrants Exercisable at June 30, 2024 | 10,886,061 | $ | 1.06 | |||||
As of June 30, 2024 and 2023, the warrants exercisable and outstanding have an intrinsic value of $184,539 and $0 respectively with a weighted average remaining life of 4 years and 6 years respectively.
| 8. | SHARE-BASED PAYMENTS |
| a) | Option Plan Details |
On March 24, 2017, and as amended on November 20, 2020, the Company’s shareholders approved: (i) the adoption of a new stock option plan (the “Plan”) pursuant to which the Company’s Board of Directors may, from time to time, in its discretion and in accordance with applicable regulatory requirements, grant to directors, officers, employees and consultants of the Company, non-transferable options to purchase common shares, provided that the number of common shares reserved for issuance will not exceed twenty percent (20%) of the issued and outstanding common shares at the date the options are granted (on a non-diluted and rolling basis); and (ii) the application of the Plan to all outstanding stock options of the Company that were granted prior to March 24, 2017 under the terms of the Company’s previous stock option plan. On December 19, 2023, the Company’s Board of Directors approved the reservation of an additional 700,000 common shares under the Plan (which common shares were registered on the Company’s Form S-8 previously filed with the SEC on December 22, 2023).
As of June 30, 2024 and 2023, there were 179,293 and 51,633 options, respectively, immediately available for future allocation pursuant to applicable regulatory requirements. The maximum number of options issuable under the terms of the Plan equates to 20% of the then issued and outstanding shares. The option price under each option shall not be less than the closing price on the day prior to the date of grant. All options vest upon terms as set by the Board of Directors, either over time, up to 36 months, or upon the achievement of certain corporate milestones.
On December 23, 2023, the Company issued 502,000 options to its employees and consultants pursuant to the Plan. The options have an exercise price of $0.37 with a term of 5 years. The options vest in equal installments monthly over three years.
On December 23, 2023, the Company additionally issued 28,400 options to members of the Company’s Board of Directors pursuant to the Plan. The options have an exercise price of $0.37 with a term of five years. The options vest on the earlier of (i) December 23, 2024 or (ii) immediately prior to the next Annual General Meeting.
F-
On February 20, 2024, the Company issued 50,000 options to its employees pursuant to the Plan. The options have an exercise price of $0.37 with a term of five years. The options vest in equal installments monthly over three years.
On August 10, 2022, the Company issued 560 options to a member of the Company’s Board of Directors pursuant to the Plan. The options have an exercise price of $9.75 with a term of five years. The options vest 1 year from issuance.
On December 16, 2022, the Company issued 57,800 options to its employees and consultants pursuant to the Plan. The options have an exercise price of $1.32 with a term of 5 years. The options vest in equal installments monthly over three years.
On December 16, 2023, the Company additionally issued 3,360 options to members of the Company’s Board of Directors pursuant to the Plan. The options have an exercise price of $1.32 with a term of five years. The options vest on the earlier of (i) December 16, 2023 or (ii) immediately prior to the next Annual General Meeting.
The assumptions used in the Black-Scholes model during the years ended June 30, 2024 and 2023, are set forth in the table immediately below:
| June 30, 2024 |
||||
| Exercise price | $ | 0.37 | ||
| Risk-free interest rate | 3.95 - 4.30 | % | ||
| Volatility | 116 - 203 | % | ||
| Expected life (years) | 3.5 - 3.6 | |||
| Dividend yield | $ | 0 | % | |
| June 30, 2023 | ||||
| Exercise price | $1.78 – 9.75 | |||
| Risk-free interest rate | 3.37 - 3.77 | % | ||
| Volatility | 92 - 125 | % | ||
| Expected life (years) | 3.1 - 5 | |||
| Dividend yield | $ | 0 | % | |
The following is a summary of changes in outstanding options from July 1, 2022 to June 30, 2024:
| Number | Weighted Average Exercise Price |
|||||||
| Balance as at July 1, 2022 | 55,603 | $ | 128.59 | |||||
| Granted | 61,720 | 1.85 | ||||||
| Expired/Forfeited | (14,681 | ) | 267.13 | |||||
| Balance as at June 30, 2023 | 102,642 | 31.28 | ||||||
| Granted | 580,400 | 0.37 | ||||||
| Expired/Forfeited | (8,569 | ) | 164.34 | |||||
| Balance as at June 30, 2024 | 674,473 | $ | 2.89 | |||||
| June 30, 2023: | ||||||||
| Vested and exercisable | 51,067 | $ | 57.44 | |||||
| Unvested | 51,575 | $ | 5.38 | |||||
| June 30, 2024: | ||||||||
| Vested and exercisable | 112,920 | $ | 14.90 | |||||
| Unvested | 561,553 | $ | 0.4 | |||||
Total expenses arising from share-based payment transactions recognized during the years ended June 30, 2024 and 2023 were $137,714 and $278,154, respectively, of which $80,513 and $162,200, respectively, was allocated to general and administrative expenses, $56,408 and $115,954, respectively, was allocated to research and development expenses, and $793 and $0, respectively, was allocated to Cost of Goods sold.
Unrecognized compensation cost at June 30, 2024 related to unvested options was $107,586 which will be recognized over a weighted-average vesting period of approximately 1.38 years.
F-
| 9. | LEASE OBLIGATIONS |
The Company is committed to minimum lease payments as follows:
| Maturity Analysis | June 30, 2024 |
|||
| $ | ||||
| Year 1 | 364,887 | |||
| Year 2 | 366,753 | |||
| Year 3 | 313,231 | |||
| Year 4 | ||||
| Year 5 | ||||
| More than five years | ||||
| Total undiscounted lease liabilities(1) | 1,044,871 | |||
| Less: imputed interest | (82,209 | ) | ||
| Present value of lease liabilities | 962,662 | |||
| Less: Current portion of lease liabilities | (317,797 | ) | ||
| Non-current portion of lease liabilities | (644,865 | ) | ||
In July 2019, InMed entered into a facility lease agreement for approximately 4,000 square feet of office space in Vancouver, BC, which served as our corporate headquarters until August 2024, when the lease expired. The Company did not take the option to renew for an additional three-year period. Subsequent to June 30 2024, the Company entered into a lease agreement for new office space. This office occupies approximately 2,243 square feet with a monthly basic rental rate and operating charges of an estimated C$12,296 for the two-year term of the agreement.
On October 5, 2023, BayMedica amended its lease located at 458 Carlton Court, Suite C, South San Francisco, California, in order to extend its lease to May 14, 2027. The Company is obligated to pay $1,295,759 over the three-year period unless terminated before the end of the period. The Company used an incremental borrowing rate of 6.15% and recognized a right-of-use asset and corresponding operating lease liability of $953,935.
| 10. | INCOME TAXES |
The following is a reconciliation of income taxes calculated at the combined Canadian federal and provincial income statutory corporate tax rate of 27.0% to the tax expense:
| 2024 | 2023 | |||||||
| $ | $ | |||||||
| US net loss before taxes | (1,756,965 | ) | (1,212,372 | ) | ||||
| Canada net loss before tax | (5,911,485 | ) | (6,735,250 | ) | ||||
| Net loss before taxes | (7,668,450 | ) | (7,947,622 | ) | ||||
| Income tax expense (recovery) at the statutory rate | (1,966,981 | ) | (2,073,116 | ) | ||||
| Increase (reduction) in income taxes resulting from: | ||||||||
| Change in valuation allowance | 1,625,998 | 2,532,867 | ||||||
| State taxes | (21,942 | ) | 9,638 | |||||
| Permanent differences | 39,252 | 76,605 | ||||||
| True up to the return | (303,595 | ) | ||||||
| State Rate Change | 2,949 | |||||||
| Foreign exchange differences | 618,907 | 417,194 | ||||||
| Share issuance cost capitalized in equity | (123,415 | ) | (553,877 | ) | ||||
| Other | 135,927 | (396,211 | ) | |||||
| Income tax expense (recovery) | 7,100 | 13,100 | ||||||
As of June 30, 2024, the Company has non-capital loss carry-forwards of approximately $72,724,671 (June 30, 2023 - $67,875,659) available to offset future taxable income in Canada. These non-capital loss carryforwards begin to expire in 2026. As of June 30, 2024, the Company has US Federal net operating losses of $7,451,316 and state net operating losses of $3,689,190. The US Federal NOLs have an indefinite carryforward period, and the state NOLs begin to expire in 2042.
F-
Deferred tax assets and liabilities are as follows:
| 2024 | 2023 | |||||||
| $ | $ | |||||||
| Non-capital losses | 21,501,476 | 19,490,270 | ||||||
| Financing costs | 861,867 | 1,265,542 | ||||||
| Accrued expenses | 12,831 | 11,638 | ||||||
| Intangible assets, net | 146,193 | 131,714 | ||||||
| Tax credits | 241,270 | 241,270 | ||||||
| Lease liability | 164,288 | 98,827 | ||||||
| 22,927,925 | 21,239,261 | |||||||
| Property and equipment, net | (116,231 | ) | (119,889 | ) | ||||
| Lease obligations | (157,701 | ) | (91,377 | ) | ||||
| (273,932 | ) | (211,266 | ) | |||||
| Net deferred tax asset | 22,653,993 | 21,027,995 | ||||||
| Valuation allowance | (22,653,993 | ) | (21,027,995 | ) | ||||
A full valuation allowance has been applied against the net deferred tax assets because it is more likely than not that future taxable income will be available against which the Company can utilize the benefits therefrom.
The Company recognizes tax benefits from an uncertain tax position only if it is more likely than not that the tax position will be sustained on examination by taxing authorities, based on the technical merits of the position. The tax benefits recognized in the consolidated financial statements from any such position would be measured based on the largest benefit that has a greater than fifty percent likelihood of being realized upon ultimate settlement. It is the Company’s policy to recognize interest and penalties accrued on any uncertain tax benefits as a component of income tax expense.
The Company files income tax returns in the U.S. federal jurisdiction, various state jurisdictions, and Canada. With few exceptions, the Company is no longer subject to U.S. federal and state tax examinations for fiscal years prior to 2021.
The Company is subject to taxation at the federal, state, and local levels in the United States and Canada.
| 11. | SEGMENT INFORMATION |
The Company reports segment information based on the management approach which designates the internal reporting used by the Chief Operating Decision Maker (“CODM”), which is the Company’s Chief Executive Officer and the senior management team, for making decisions and assessing performance as the source of the Company’s reportable segments. The CODM allocates resources and assesses the performance of each operating segment based on potential licensing opportunities, historical and potential future product sales, operating expenses, and operating income (loss) before interest and taxes. The Company has determined its reportable segments to be InMed Pharmaceuticals (‘InMed Pharma’) and BayMedica Commercial based on the information used by the CODM. As such, the pharmaceutical related research and development carried out at BayMedica is included with the InMed Pharma segment. Other than cash, cash equivalents and short-term investments (“Unrestricted cash”) balances, the CODM does not regularly review asset information by reportable segment and, therefore, the Company does not report asset information by reportable segment.
The InMed Pharma segment is largely organized around the research and development of small molecule pharmaceuticals drug candidates and the BayMedica Commercial segment is largely organized around manufacturing technologies to produce and commercialize bulk rare cannabinoids for sale as ingredients in the health and wellness industry. Total assets held in the InMed Pharma segment as of June 30, 2024 and 2023 are $9.2 million and $11.2 million, respectively. Total assets as of June 30, 2024 and 2023, held in the BayMedica segment are $2.6 million and $2.9 million, respectively.
F-
The following table presents information about the Company’s reportable segments for the year ended June 30, 2024 and 2023:
| Year Ended June 30, | ||||||||||||||||||||||||
| 2024 | 2023 | |||||||||||||||||||||||
| InMed | BayMedica | Total | InMed | BayMedica | Total | |||||||||||||||||||
| $ | $ | $ | $ | $ | $ | |||||||||||||||||||
| Sales | 4,597,730 | 4,597,730 | 4,135,561 | 4,135,561 | ||||||||||||||||||||
| Cost of sales | (3,496,817 | ) | (3,496,817 | ) | (2,732,525 | ) | (2,732,525 | ) | ||||||||||||||||
| Operating expenses | (8,400,411 | ) | (896,853 | ) | (9,297,264 | ) | (8,824,275 | ) | (1,005,723 | ) | (9,829,998 | ) | ||||||||||||
| Other income (expense) | 532,782 | (4,881 | ) | 527,901 | 490,753 | 1,687 | 492,440 | |||||||||||||||||
| (Loss) income before income taxes | (7,867,629 | ) | 199,179 | (7,668,450 | ) | (8,333,522 | ) | 399,000 | (7,934,522 | ) | ||||||||||||||
| Unrestricted cash | 5,669,113 | 902,497 | 6,571,610 | 8,036,714 | 875,803 | 8,912,517 | ||||||||||||||||||
| 12. | COMMITMENTS AND CONTINGENCIES |
Pursuant to the terms of agreements with various contract research organizations, as of June 30, 2024, the Company is committed for contract research services and materials at a cost of approximately $1.5 million, expected to occur in the twelve months following period.
Pursuant to the terms of agreements with various vendors, as of June 30, 2024, the Company is committed for contract materials and equipment at a cost of approximately $0.7 million, expected to occur in the twelve months following June 30, 2024.
Pursuant to the terms of a certain Technology Assignment Agreement, dated as of May 31, 2017 (the “Technology Agreement”), between the Company and the University of British Columbia (“UBC”), the Company is committed to pay royalties to UBC on certain licensing and royalty revenues received by the Company for biosynthesis of certain drug products that are covered by the Technology Agreement. To date, no payments have been required to be made.
Pursuant to the terms of a certain Collaborative Research Agreement, dated as of December 13, 2018, between the Company and UBC, pursuant to which the Company owns all rights, title and interests in and to any intellectual property, in addition to funding research at UBC, the Company is committed to make a one-time payment upon filing of any PCT patent application arising from the research. To date, one such payment has been made to UBC.
Pursuant to the terms of a certain Contribution Agreement, dated as of November 1, 2018, between the Company and National Research Council Canada, as represented by its Industrial Research Assistance Program (NRC-IRAP), under certain circumstances contributions received, including the disposition of the underlying intellectual property developed in part with NRC-IRAP contributions, may become repayable. As of June 30, 2024, there have been no triggering events to cause a repayment.
Short-term investments include guaranteed investment certificates, with one year terms, of $43,064 and $44,422 as of June 30, 2024 and 2023 respectively, that are pledged as security for a corporate credit card.
In addition to the foregoing, the Company has entered into certain agreements in the ordinary course of operations that may include indemnification provisions, which are common in such agreements. In some cases, the maximum amount of potential future indemnification is unlimited; however, the Company currently holds commercial general liability insurance. This insurance may limit the Company’s overall liability and may enable the Company to recover a portion of any future amounts paid. Historically, the Company has not made any indemnification payments under such agreements, and it believes that the fair value of these indemnification obligations is minimal. Accordingly, the Company has not recognized any liabilities relating to these obligations for any period presented.
Pursuant to a certain Technology Licensing Agreement, dated as of March 11, 2021, between the Company and EyeCRO, the Company is committed to issue, subject to regulatory approval, up to 700 warrants to purchase 700 Common Shares upon the achievement of certain milestones. The exercise price of the warrants will be equal to the five-day VWAP of our Common Shares prior to each milestone achievement and the warrants will be exercisable for a period of three years from the issuance date. On May 10, 2024, the Company delivered a 90-day notice of termination to EyeCRO LLC with respect to the Technology Licensing Agreement, specifying an effective date of termination of August 8, 2024 (see Note 14 – Subsequent Events).
F-
BayMedica entered into a patent license agreement (“Patent License Agreement”) with a third party (the “Licensor”) on February 15, 2021. The Company was required to begin making royalty payments to the Licensor based on net sales of licensed products in 2021 in order to maintain an exclusive license. In December 2021, the Company amended the Patent License Agreement, which amendment included the deferral of the 2021 minimum payments to 2022. As of June 30, 2023, the Company has paid $300,000 for the minimum payments due and payable under the Patent License Agreement. On February 10, 2023, BayMedica received a letter from the Licensor alleging a breach of the Patent License Agreement and asserting a right to monies due thereunder. On April 6, 2023, BayMedica sent a letter to the Licensor disputing the Licensor’s interpretation of the Patent License Agreement and asserted that the counterparty’s only remedy under the Patent License Agreement to be either (a) the conversion of an exclusive technology license into a non-exclusive license or (b) the termination of the Patent License Agreement.
On July 18, 2024, BayMedica received a letter from the Licensor alleging breach of the Patent License Agreement and asserting monies due thereunder. On August 7, 2024, BayMedica responded asserting that the counterparty’s interpretation of the Agreement is incorrect again and that BayMedica does not owe any funds under the Agreement.
To date, the Licensor has not initiated a lawsuit with respect to the foregoing matters. If a lawsuit is ultimately brought alleging a breach of the Patent License Agreement, the proceeding will be subject to final, binding and non-appealable arbitration under the Arbitration Act, 1991 (Ontario) and determined pursuant to Ontario law. BayMedica intends to vigorously defend its position. At this time, it is not possible to reasonably estimate a potential loss due to the terms of the Agreement, the nature of the legal theory advanced by the counterparty, and the ultimate outcome of any proceeding (including the interpretation by the arbitrator with respect to applicable requirements under Ontario law regarding contract formation).
| 13. | RELATED PARTY TRANSACTIONS |
On February 11, 2022, the Board of Directors appointed Janet Grove as a director of the Company. Ms. Grove is a Partner of Norton Rose Fulbright Canada LLP (“NRF”). During the years ended June 30, 2024 and 2023, NRF rendered legal services in the amount of $226,793 and $634,208, respectively, to the Company. These transactions were in the normal course of operations and were measured at the exchange amount which represented the amount of consideration established and agreed to by NRF. No legal services rendered by NRF were provided by Ms. Grove directly.
| 14. | SUBSEQUENT EVENTS |
The Company has evaluated subsequent events through the date of the filing of this Annual Report on Form 10-K and determined that there have been no events that have occurred that would require adjustments to our disclosures in the consolidated financial statements except for the matters described below.
On September 17, 2024, the Company received a notification (the “Delisting Notice”) from the Nasdaq Listing Qualifications Department stating that due to the deficiency in the Company’s compliance with Listing Rule 5550(a)(2) which required the Company to maintain a bid price of its listed securities over $1 for 30 consecutive days, the Company’s securities would be delisted from Nasdaq on September 26, 2024, unless the Company appealed Nasdaq’s determination to a Hearings Panel (the “Panel”). A hearing request will stay the suspension of the Company’s securities pending the Panel’s discussion. On September 17, 2024, the Company submitted the hearing request to appeal (the “Appeal Request”) Nasdaq’s determination before the Panel. The hearing will take place on October 31, 2024 and it is anticipated that the Panel’s decision will follow shortly thereafter. The pendency of the Appeal Request does not have an immediate effect on the listing of our Common Shares and our Common Shares will continue to trade on Nasdaq under the symbol “INM”. While the Company has filed the Appeal Request, there can be no assurances, however, that we will be successful in regaining compliance with the continued listing requirements and maintaining the listing of our Common Shares on Nasdaq. Delisting from Nasdaq could materially and adversely affect our ability to raise additional financing through the public or private sale of equity securities, would significantly affect the ability of investors to trade our securities and would negatively affect the value and liquidity of our securities, including our Common Shares. See Item 1A. “Risk Factors” of this Annual Report, including the risk that “any actual or threatened delisting of our securities by Nasdaq could have a material and adverse effect on our business, operations and financial condition, and could, among other things, limit investors’ ability to make transactions in our securities and subject us to additional trading restrictions,” for further information regarding the risks pertaining to the Delisting Notice and the Appeal Request.
Refer to note 9 – Lease Obligations with reference to a new lease agreement.
Refer to note 12 - Commitments and Contingencies with reference to the Patent License Agreement.
Under the Amended ATM Agreement, the Company issued 3,727,272 Common Shares for gross proceeds of $1,030,000 during the period of July 11, 2024 through July 29, 2024.
F-
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
None.
ITEM 9A. CONTROLS AND PROCEDURES
Evaluation of Disclosure Controls and Procedures
Our management, with the participation of our principal executive officer and principal financial officer, evaluated the effectiveness of our disclosure controls and procedures as of June 30, 2024. The term “disclosure controls and procedures,” as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act, means controls and other procedures of a company that are designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Exchange Act is recorded, processed, summarized and reported, within the time periods specified in the SEC’s rules and forms. Disclosure controls and procedures include, without limitation, controls and procedures designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Exchange Act is accumulated and communicated to its management, including its principal executive and principal financial officers, as appropriate to allow timely decisions regarding required disclosure. Management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving their objectives and management necessarily applies its judgment in evaluating the cost-benefit relationship of possible controls and procedures. Based on the evaluation of our disclosure controls and procedures as of June 30, 2024, our principal executive officer and principal financial officer concluded that, as of such date, our disclosure controls and procedures were effective.
Management’s Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting. Internal control over financial reporting is defined in Rules 13a-15(f) and 15d-15(f) under the Securities Exchange Act of 1934, as amended, as a process designed by, or under the supervision of, the Company’s principal executive and principal financial officers and effected by the company’s board of directors, management and other personnel to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles and includes those policies and procedures that:
| ● | Pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the Company; |
| ● | Provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures are being made only in accordance with authorizations of management and directors of the Company; and |
| ● | Provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of the Company’s assets that could have a material effect on the financial statements. |
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Therefore, even those systems determined to be effective can provide only reasonable assurance with respect to financial statement preparation and presentation. Projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Our management assessed the effectiveness of our internal control over financial reporting as of June 30, 2024. In making this assessment, management used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission in Internal Control-Integrated Framework (2013). Based on this evaluation, management has concluded our internal control over financial reporting as of June 30, 2024 was effective. The material weakness in the Company’s internal control over financial reporting for year ended June 30, 2023 has been remediated.
Remediation of Previously Reported Material Weakness
In connection with the audit of our consolidated financial statements as of and for the year ended June 30, 2023, our management identified a material weakness in the Company’s internal control over financial reporting, primarily the result of inadequate resources required to respond to financial reporting matters other than in the normal course of business. The material weakness was attributable to a lack of personnel and inadequate controls around segregation of duties in the accounting department resulting from employee turnover.
Management implemented a remediation plan to address the root cause that contributed to the material weakness and is committed to a strong Internal Control over Financial Reporting environment. Management engaged a third party that provides accounting and financial reporting services to provide a resource to serve as interim CFO to replace our former VP of Accounting and Controller, until a CFO could be hired. During the year, the Company hired a CFO and continued to retain the services of the interim CFO in the capacity of Corporate Controller. Both individuals have the requisite technical accounting knowledge. Furthermore, management also implemented review controls and processes to provide oversight over the Company’s period-end close and reporting processes. Management engaged a third party consultant to review and test its internal control processes. Once the applicable remedial controls operated for a sufficient period of time, they were tested and management has concluded that these controls are operating effectively.
Changes in Internal Control over Financial Reporting
Other than the actions we have taken to remediate the material weakness described above for the financial year ended June 30, 2023 there were no changes in our internal control over financial reporting identified in connection with the evaluation required by Rule 13a-15(d) and 15d-15(d) of the Exchange Act that occurred during the year ended June 30, 2024 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
ITEM 9B. OTHER INFORMATION
During the three months ended June 30, 2024, no director or officer of the Company adopted or terminated a “Rule 10b5-1 trading arrangement” or “non-Rule 10b5-1 trading arrangement,” as each term is defined in item 408(a) of Regulation S-K.
ITEM 9C. DISCLOSURE REGARDING FOREIGN JURISDICTIONS THAT PREVENTS INSPECTIONS
Not applicable
PART III
The information required by Part III is omitted from this report because we will file a definitive proxy statement (the “2024 Proxy Statement”) for our 2024 Annual Meeting of Stockholders within 120 days after the end of our 2024 fiscal year pursuant to Regulation 14A of the Exchange Act. If the 2024 Proxy Statement is not filed within 120 days after the end of the fiscal year covered by this Annual Report, the omitted information will be included in an amendment to this Annual Report filed not later than the end of such 120-day period.
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
The response to this item is incorporated by reference from the discussion responsive thereto under the captions “Management and Corporate Governance,” “Section 16(a) Beneficial Ownership Reporting Compliance,” and “Code of Business Conduct and Ethics” in the Company’s Proxy Statement for the 2024 Annual Meeting of Stockholders.
ITEM 11. EXECUTIVE COMPENSATION
The response to this item is incorporated by reference from the discussion responsive thereto under the caption “Executive Officer and Director Compensation” in the Company’s Proxy Statement for the 2024 Annual Meeting of Stockholders.
The Company’s Board of Directors has adopted a clawback policy in order to comply with U.S. federal securities laws and the listing requirements of Nasdaq. As such, we have adopted a clawback policy in which we may seek the recovery or forfeiture of incentive compensation paid by us, including cash, equity or equity-based compensation, in the event we restate our financial statements under certain circumstances. The clawback policy applies to our Section 16 officers, any employee who was eligible to receive incentive compensation and whose conduct contributed to the need for a restatement, and any other former Section 16 officer or other employee who contributed to the need for such restatement. A copy of the clawback policy has been filed as Exhibit 97.1 to this Annual Report.
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
The response to this item is incorporated by reference from the discussion responsive thereto under the captions “Security Ownership of Certain Beneficial Owners and Management,” and “Equity Compensation Plan Information” in the Company’s Proxy Statement for the 2024 Annual Meeting of Stockholders.
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
The response to this item is incorporated by reference from the discussion responsive thereto under the captions “Certain Relationships and Related Person Transactions” and “Management and Corporate Governance” in the Company’s Proxy Statement for the 2024 Annual Meeting of Stockholders.
ITEM 14. PRINCIPAL ACCOUNTING FEES AND SERVICES
Our independent registered public accounting firm is Marcum LLP PCAOB Auditor ID 688. The information required by this item will be included in our definitive proxy statement with respect to our 2024 Annual Meeting of Shareholders to be filed with the SEC, and is incorporated herein by reference.
PART IV
ITEM 15. EXHIBITS AND FINANCIAL STATEMENT SCHEDULES
The following documents are being filed as part of this report:
| (1) | The following financial statements of the Company and the report of Marcum LLP are included in Part II, Item 8: |
| (2) | All financial statement supporting schedules are omitted because the information is inapplicable or presented in the Notes to Consolidated Financial Statements. |
| (3) | A list of exhibits filed with this report or incorporated herein by reference is found in the Exhibit Index immediately following the signature page of this Annual Report. |
ITEM 16. 10-K SUMMARY
Not applicable.
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| INMED PHARMACEUTICALS INC. | ||
| (Registrant) | ||
| September 27, 2024 | By: | /s/ Netta Jagpal |
| Netta Jagpal | ||
| Chief Financial Officer | ||
Pursuant to the requirements of the Securities and Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| Signature | Title | Date | ||
| /s/ Eric A. Adams | President, Chief Executive Officer and Director | September 27, 2024 | ||
| Eric A. Adams | (Principal Executive Officer) | |||
| /s/ Netta Jagpal | Interim Chief Financial Officer | September 27, 2024 | ||
| Netta Jagpal | (Principal Financial Officer and Principal Accounting Officer) | |||
| /s/ Andrew Hull | Director | September 27, 2024 | ||
| Andrew Hull | (Chairman to the Board of Directors) | |||
| /s/ Janet Grove | Director | September 27, 2024 | ||
| Janet Grove | ||||
| /s/ Bryan Baldasare | Director | September 27, 2024 | ||
| Bryan Baldasare | ||||
| /s/ Nicole Lemerond | Director | September 27, 2024 | ||
| Nicole Lemerond |
84
Exhibit 4.13
Description of Registrant’s Securities
Registered under Section 12
of the Securities Exchange Act of 1934
The following description (this “Description”) of our Common Shares is a summary and does not purport to be complete. It is subject to, and qualified in its entirety by reference to, our Amended and Restated Articles (our “Articles”), which has been filed with the Securities and Exchange Commission. This Description also summarizes relevant provisions of the British Columbia Business Corporations Act (the “BCBCA”) and securities laws in the provinces and territories of Canada. We encourage you to read our Articles, the applicable provisions of the BCBCA and the applicable provisions of securities laws in the provinces and territories of Canada for additional information.
General
Our authorized share capital consists of an unlimited number of Common Shares without par value and an unlimited number of preferred shares without par value. As of the date of the Annual Report on Form 10-K for which this Description forms a part, no preferred shares were issued and outstanding.
Common Shares
Each common share entitles the holder thereof to one vote at all meetings of shareholders.
There are no limitations on the rights of non-Canadian owners to hold or vote Common Shares.
In the event of our liquidation, dissolution or winding-up, whether voluntary or involuntary, or other distribution of our assets among shareholders for the purpose of winding up our affairs, subject to the rights, privileges and restrictions attaching to any preferred shares that may then be outstanding, the shareholders shall be entitled to receive our remaining property.
The shareholders are entitled to receive dividends, as and when declared by our board of directors, subject to the rights, privileges and restrictions attaching to our securities, which may be paid in money, property or by the issue of fully paid shares in our capital.
Certain Takeover Bid Requirements
Unless such offer constitutes an exempt transaction, an offer made by a person to acquire outstanding shares of a Canadian entity that, when aggregated with the offeror’s holdings (and those of persons or companies acting jointly with the offeror), would constitute 20% or more of the outstanding shares, would be subject to the take-over provisions of Canadian securities laws. The foregoing is a limited and general summary of certain aspects of applicable securities law in the provinces and territories of Canada, all in effect as of the date of the Annual Report of which this Description forms a part.
In addition to the take-over bid requirements noted above, the acquisition of shares may trigger the application of additional statutory regimes including amongst others, the Investment Canada Act and the Competition Act.
This summary is not a comprehensive description of relevant or applicable considerations regarding such requirements and, accordingly, is not intended to be, and should not be interpreted as, legal advice to any existing or prospective investor and no representation with respect to such requirements to any existing or prospective investor is made. Existing and prospective investors should consult their own Canadian legal advisors with respect to any questions regarding securities law in the provinces and territories of Canada.
Actions Requiring a Special Majority
Under the BCBCA, unless otherwise stated in our Articles, certain corporate actions require the approval of a special majority of shareholders, meaning holders of shares representing 66 2/3% of those votes cast in respect of a shareholder vote addressing such matter. Those items requiring the approval of a special majority generally relate to fundamental changes with respect to our business, and include amongst others, resolutions: (i) removing a director prior to the expiry of his or her term; (ii) altering our Articles, (iii) approving an amalgamation; (iv) approving a plan of arrangement; and (v) providing for a sale of all or substantially all of our assets.
* * * * *
Exhibit 10.19
CONSULTING AGREEMENT
THIS AGREEMENT made as of the 17th day of May, 2024 with an effective date of 1st day of July, 2024 (the “Effective Date”).
BETWEEN:
InMed Pharmaceuticals Inc., a corporation registered in the Province of British Columbia and having its principal place of business at 310-815 W. Hastings St., Vancouver, BC, V6C 1B4,
(the “Company”)
| AND: | ALEXANDRA D. J. MANCINI, d.b.a TRUE NORTH SYNERGY INC, a corporation registered in the Province of British Columbia and having its principal place of business at [* * * * *] |
(the “Consultant”)
(each a “Party” and together, the “Parties”)
WHEREAS
A. The Company is a clinical stage pharmaceutical company that specializes in the research and development of novel, cannabinoid-based drug candidates and wishes to retain the consulting services of the Consultant;
B. The Consultant has relevant experience in matters related to drug development, clinical trials and regulatory affairs; and
C. The Company wishes to retain the Consultant and the Consultant wishes to be retained to provide consulting advice to the Company with respect to the advancement of its Business and other related matters on the terms and conditions set out in this Agreement.
NOW THEREFORE THIS AGREEMENT WITNESSES that for and in consideration of the premises and mutual covenants and agreements hereinafter contained, the Company and the Consultant agree as follows:
Section 1 - Interpretation
| 1.1 | In and for the purposes of this Agreement, unless there is something in the subject matter or context inconsistent therewith, each of the following words, phrases and expressions will have the meanings ascribed to them below: |
| (a) | “Business” means all activities of the Company and its affiliates (as affiliate is defined in the Company Act (British Columbia)); |
| (b) | “Company” includes any related or affiliated entity; |
| (c) | “Confidential Information” includes, but is not limited to, all information related to processes, formulae, research, development, financial and business information, trade secrets or other proprietary information in whatever form, concerning the past, present and planned future products, services, operations and marketing techniques and procedures of the Company, and further includes any information related to the past, present and prospective customers, suppliers, clients, distributors and employees of the Company, but does not include information which is in the public domain, without any fault or responsibility on the part of the Consultant. |
| (d) | “Effective Date” means 1 July 2024. |
| (e) | “Material” includes all documentation, work-in-progress, reports and other materials the Consultant produces in the course of providing the Services; |
| (f) | “Services” includes work related to various drug development, clinical and regulatory affairs, business development support related to INM-755and such further and other services as the Company may request of the Contractor from time to time and as agreed to between the parties ; and |
| (g) | “Term” means the time period from the date of this Agreement until this Agreement is terminated in accordance with Section 6. |
| 1.2 | For the purposes of this Agreement, the singular of any term includes the plural, and vice versa, the use of any term is generally applicable to either gender and, where applicable, to a corporation, the word “or” is not exclusive and the word “including” is not limiting whether or not non-limiting language (such as “without limitation” or “but not limited to” or words of similar import) is used with reference thereto. |
Section 2- Engagement
| 2.1 | This Agreement will commence on the Effective Date and will continue until such terminated in accordance with Section 6 of this Agreement (the “Term”). |
Section 3 - Services
| 3.1 | During the Term, the Consultant will provide the Services to the Company on a part-time basis, at times mutually compatible with the Parties’ schedules. |
| 3.2 | The Consultant will spend the hours necessary to competently, effectively and efficiently perform the Services. |
| 3.3 | The Consultant will determine the manner and procedure of the performance of the Services, subject to the deadlines, time requirements and other reasonable requirements of the Company. |
| 3.4 | The Consultant will perform the Services in a competent and professional manner and fully in accordance with all policies of the Company and all applicable laws and regulations. |
| 3.5 | The Consultant will not subcontract out any portion of the Services and will perform the Services personally unless otherwise agreed to by the Parties. |
Section 4 - Compensation
| 4.1 | Consulting Fees. During the Term, the Company will pay the Consultant USD $[* * * *] per hour. |
If the Company requires the Consultant to travel long distances on behalf of the Company, as determined by the Company in the Company’s sole discretion, the travel time (e.g., on airplane) will be paid at the full rate for time spent providing the Services and at a reduced rate of USD $[* * *] per hour otherwise.
The Consultant will submit an invoice to the Company, reflecting the Services provided during the previous month, on or before the 5th day of each month. The Invoice will provide a detailed breakdown of the Services provided, including, without limitation, the number of hours spent by the Consultant in providing the Services.
| 4.2 | Expenses. Subject to the following provisions: |
| (a) | Compliance with Company Policies. Subject to compliance by the Consultant with the Company’s expense and travel policies as may be in effect from time to time; and |
| (b) | Expense Reports. Provided that the Consultant provides the Company with written expense accounts including receipts |
the Company shall reimburse the Consultant for all reasonable pre-approved expenses incurred in the performance of this Agreement, if any.
Section 5 – Debarment
| 5.1 | The Consultant certifies that she is not under investigation by the United States Food and Drug Administration (the “FDA”) for debarment action and has not been debarred under and that she will not use in any capacity the services of any person or entity that is under investigation for debarment action under the Generic Drug Enforcement Act of 1992 (21 U.S.C. 301 et seq.) or has been so debarred, or who is otherwise restricted or disqualified from performing services relating to clinical trials, to perform any Services under this Agreement. If, during the course of this Agreement, the Consultant becomes aware that the Consultant is under investigation by the FDA or any health regulatory authority for debarment action or is debarred, or otherwise restricted or disqualified; |
| (a) | the Consultant shall promptly inform the Company of such event and, upon the Company’s request, will assist the Company in conducting an inquiry or audit regarding the Services performed by the Consultant for the Company; and |
| (b) | the Company may in its sole discretion elect to terminate this Agreement with immediate effect. |
Section 6 – Termination
| 6.1 | The following terms and conditions apply to a termination of the engagement of the Consultant pursuant to this Agreement: |
| (a) | either Party may terminate this Agreement at any time upon 7 days’ written notice to the other Party; |
| (b) | either Party may terminate this Agreement immediately without notice or any payment in lieu of notice in the event of a material breach by the other Party of any term of this Agreement. |
| 6.2 | The Parties agree that the amount of notice provided herein is sufficient and in full satisfaction of any and all rights and entitlements that may arise pursuant to the termination of their engagement for whatever reason. The waiver by the Company of any breach by the Consultant of any provision of this Agreement will not operate or be construed as a waiver of any subsequent breach by the Consultant. |
Section 7 – Confidentiality, Ownership of Material and Assignment of Intellectual Property Rights
| 7.1 | The Consultant agrees to enter into a Confidentiality and Assignment of Inventions Agreement with the Company as attached in Attachment A to this Consulting Agreement. |
Section 8- Non-Exclusive Services
| 8.1 | The Company acknowledges and agrees that the Consultant is providing Services to the Company during the Term of this Agreement on a non-exclusive basis. The Company acknowledges that the Consultant may provide services to other companies. |
Section 9 – Relationship between the Parties
| 9.1 | In the performance of the Services under this Agreement, the Consultant is an independent contractor. The Parties acknowledge and agree that this Agreement is not intended and does not constitute or create an employment, agency, partnership, joint venture or any other relationship between the Consultant and the Company. |
| 9.2 | The Consultant agrees and acknowledges that the Consultant will not have the authority to enter into contracts on behalf of the Company, nor to legally bind the Company in any way, except with the prior written consent of the Company. |
| 9.3 | The manner and means by which the Consultant will provide the Services are under their sole and exclusive control, provided, however, that the Services meet the Company’s standards regarding quality and timeliness. |
| 9.4 | The Consultant will be wholly responsible for all taxes and other fees levied on the fees and services under this Agreement. Without limitation, the Consultant will make all statutory deductions, contributions and remittances, including, without limitations, federal and provincial sales tax, any taxes pursuant to the Income Tax Act, employment insurance, workers’ compensation, other similar levies, and all fines and penalties levied for failure to make payment. |
| 9.5 | The Consultant will indemnify and hold the Company harmless from: |
| (a) | any and all claims or demands that may be made by Canada Revenue Agency requiring the Company to pay income tax, penalties, interest or other charges under the Income Tax Act (Canada), the Income Tax Act (BC), the Employment Insurance Act (Canada), or similar statute of British Columbia or Canada in respect of any payments made to the Consultant for the provision of the Services, or in respect of deductions or remittances unpaid to any statutory agency; and |
| (b) | all costs, charges, legal fees and other expenses reasonably incurred by the Company, or its related or subsidiary companies in connection with defending any civil, criminal or administrative action, proceeding or other remedy taken against them with respect to the claims or demands set out in (a) above. |
Section 10- General Provisions
| 10.1 | Severability. Each provision of this Agreement constitutes a separate and distinct obligation and if any provision of this Agreement is determined to be void or unenforceable, in whole or in part, it will be deemed not to affect or impair the validity of any other obligation or provision. |
| 10.2 | Entire Agreement. This Agreement constitutes the entire agreement between the Parties, and supersedes and replaces any and all other representations, understandings, negotiations, and previous agreements, written or oral, express or implied. The Parties do not rely upon or regard as being material any representations or other agreements not specifically incorporated into and made part of this Agreement. |
| 10.3 | Succession. This Agreement will enure to the benefit of and be binding upon each of the Company and the Consultant and their respective successors and assigns, and may not be assigned or transferred by either party except with the prior written consent of the other party. |
| 10.4 | Notices. Any notices to be given hereunder by either party to the other party may be effected in writing, either by personal delivery, by mail if sent certified, postage prepaid, with return receipt requested or by email upon acknowledgment of receipt. Mailed notices will be addressed to the parties at the address set out on the first page of this Agreement, or as otherwise specified from time to time. Mailed notice will be effective upon delivery. If emailed to the Consultant, at [* * * * *]or [* * * * *] and if emailed to the Company at [* * * * *]. |
| 10.5 | Amendments and Waivers. No amendment to this Agreement will be valid or binding unless set forth in writing and duly executed by all of the parties hereto. No waiver of any breach of any provision of this Agreement will be effective or binding unless made in writing and signed by the party purporting to give the same and, unless otherwise provided in the written waiver, will be limited to the specific breach waived. |
| 10.6 | Survival. Notwithstanding the expiration or early termination of this Agreement, Sections 1.1, 1.2, 4.2, 6, 7, 9.4, 9.5 and this Section 10 shall survive any termination of this Agreement. |
| 10.7 | Governing Law. This Agreement will be governed by and construed, enforced and interpreted exclusively in accordance with the laws of the Province of British Columbia and the applicable laws of Canada therein. |
| 10.8 | Independent Legal Advice. The Consultant specifically confirms that the Consultant has been provided with the opportunity to retain independent legal advice prior to entering into this Agreement. |
[Remainder of page intentionally left blank.]
IN WITNESS WHEREOF this Agreement has been executed by the parties hereto as of the Effective Date.
| SIGNED, SEALED AND DELIVERED by | |||
| Alexandra D. J. Mancini, President, TRUE NORTH | |||
| SYNERGY INC. in the presence of | /s/ Alexandra D. J. Mancini | ||
| ALEXANDRA D. J. MANCINI | |||
| Consultant | |||
| Witness Signature | |||
| Witness Name | |||
| Witness Address | |||
| Witness Occupation | |||
| INMED PHARMACEUTICALS INC. | |||
| by its authorized signatory: | |||
| Per: | |||
| /s/ Eric A. Adams | |||
| Name: | Eric A. Adams | ||
| Title: | President and CEO | ||
Attachment A
Confidentiality and Assignment of Inventions Agreement
Effective Date: 1 July 2024
| WHEREAS: |
| A. | In accordance with the terms of the Consulting Agreement between the Consultant and the Company dated 17 May 2024, the Consultant has agreed to execute this Confidentiality and Assignment of Inventions Agreement on the terms set out herein. |
NOW THEREFORE for good and valuable consideration, including the consideration contemplated by the Consulting Agreement, the receipt and sufficiency of which is acknowledged by the Consultant, the Consultant and the Company agree as follows:
| 1 | INTERPRETATION |
The capitalized terms have the meanings ascribed to them in the Consulting Agreement, and the following terms have the following meanings:
| (a) | “Affiliate” means, in respect of the Company, a company or other entity which directly or indirectly controls, is controlled by, or is under common control with, the Company. For the purposes of this definition, “control” means direct or indirect beneficial ownership of a greater than 50% interest in the income of such company or entity or such other relationship as, in fact, constitutes actual control. |
| (b) | “Business” or “Business of the Company” means: |
| (i) | researching, developing, commercializing, producing and marketing novel, cannabinoid-based and other pharmaceutical therapies to treat disease; or |
| (ii) | any other area in which the Company has an active research and development program on the date the Consultant’s engagement with the Company terminates and in connection with which the Consultant directly provided Services. |
| (c) | “Confidential Information” shall mean all information, knowledge, or data, whether in written, oral, electronic or other form, relating to the Business of the Company, whether or not conceived, originated, discovered or developed in whole or in part by the Consultant, that is not generally known to the public or to other persons who are not bound by obligations of confidentiality and: |
| (i) | from which the Company or its Affiliates derive economic value, actual or potential, from the information not being generally known; or |
| (ii) | in respect of which the Company or its Affiliates otherwise have a legitimate interest in maintaining secrecy; |
and which, without limiting the generality of the foregoing, shall include:
| (iii) | all proprietary information licensed to, acquired, used or developed by the Company and its Affiliates in its research and development activities (including but not restricted to the research and development of cannabinoid-based and other pharmaceutical therapeutics and delivery technology), other scientific strategies and concepts, designs, know-how, information, material, formulas, processes, research data and proprietary rights in the nature of copyrights, patents, trademarks, licenses and industrial designs; |
| (iv) | all information relating to the Business of the Company, and to all other aspects of the structure, personnel and operations of the Company and its Affiliates, including financial, clinical, regulatory, marketing, advertising and commercial information and strategies, customer lists, compilations, agreements and contractual records and correspondence; programs, devices, concepts, inventions, designs, methods, processes, data, know-how, unique combinations of separate items that is not generally known and items provided or disclosed to the Company or its Affiliates by third parties subject to restrictions on use or disclosure; |
| (v) | all know-how relating to the Business of the Company, including all biological, chemical, pharmacological, toxicological, pharmaceutical, physical and analytical, clinical, safety, manufacturing and quality control data and information, and all applications, registrations, licenses, authorizations, approvals and correspondence submitted to regulatory authorities; |
| (vi) | all information relating to the businesses of competitors of the Company or its Affiliates, including information relating to competitors’ research and development, intellectual property, operations, financial, clinical, regulatory, marketing, advertising and commercial strategies, that is not generally known; |
| (vii) | all information provided to the Company or its Affiliates by their agents, consultants, lawyers, contractors, licensors or licensees and relating to the Business of the Company. |
All Work Product shall be deemed to be the Company’s Confidential Information.
Notwithstanding the foregoing, “Confidential Information” does not include information which the Consultant can prove is information that was in the public domain at the date of disclosure to the Consultant, or thereafter entered the public domain through no fault of the Consultant (but only after it has entered the public domain) provided that any combination of information that is Confidential Information will not be included within the exception merely because parts of the information were within the public domain unless the whole of the combination itself was in the public domain.
| (d) | “Intellectual Property” is used in its broadest sense and means and includes any statutory, common law, equitable, contractual or proprietary rights or interests, recognized currently or in future, in and to any Inventions, including, without limitation, rights and interests in and to the following: |
| (i) | knowledge, know-how and its embodiments, including trade secret information; |
| (ii) | patents in inventions, and all applications therefor; |
| (iii) | copyrights in artistic, literary, dramatic, musical, and neighbouring works, copyrightable works of authorship including technical descriptions for products, user guides, illustrations, advertising materials, computer programs, source code and object code, and all applications therefor; |
| (iv) | trademarks, service marks, tradenames, business names and domain names and all applications therefor; |
| (v) | industrial designs and all other industrial or intellectual property and all applications therefor; and |
| (vi) | all goodwill connected with the foregoing. |
| (e) | “Inventions” shall mean any and all inventions, discoveries, developments, enhancements, improvements, concepts, formulas, designs, processes, ideas, writings and other works, whether or not reduced to practice, and whether or not protectable under patent, copyright, trade secret or similar laws. |
| (f) | “Work Product” shall mean any and all Inventions and possible Inventions relating to the Business of the Company and which the Consultant may make or conceive, alone or jointly with others, during their involvement in any capacity with the Company, whether during or outside their regular working hours, except those Inventions made or conceived by the Consultant entirely on their own time that do not relate to the Business of the Company and do not derive from any equipment, supplies, facilities, Confidential Information or other information, gained, directly or indirectly, from or through their involvement in any capacity with the Company. |
| 2 | CONFIDENTIALITY |
| (a) | Property of the Company. The Company shall exclusively own all right, title and interest in and to the Confidential Information, whether or not created or developed by the Consultant. |
| (b) | Prior Business Confidential Information. The Consultant represents and warrants to the Company that the Consultant has not brought or used, and the Consultant covenants and agrees that the Consultant will not use or bring to the Company any confidential information of any kind whatsoever of any prior party other than the Company (the “Prior Business”) with whom the Consultant was previously involved, whether such involvement was as an employee, director or officer of that Prior Business, an investor in that Prior Business, a partner in that Prior Business, a consultant to that Prior Business or other relationship to that Prior Business (the “Prior Involvement”). The Company and the Consultant acknowledge and agree that the Company is not engaging the Consultant to obtain confidential information relating to any Prior Involvement and the Consultant acknowledges that the Company has advised the Consultant to comply with any and all legal obligations the Consultant may have to such Prior Business. The Consultant covenants and agrees to indemnify and hold the Company harmless from any and all loss, claims, damages, expenses and costs (including legal costs on a solicitor-client basis) of any kind whatsoever that the Company may suffer related to of any breach by the Consultant of their obligations to such Prior Business in that regard. |
| (c) | Basic Obligation of Confidentiality. The Consultant hereby acknowledges and agrees that the Company has disclosed and will continue to disclose to the Consultant, and that the Consultant has had and will continue to have access to Confidential Information. The Consultant will receive and hold all Confidential Information on the terms and conditions set out in this Attachment A. Except as otherwise expressly set out in this Attachment A, the Consultant will keep strictly confidential all Confidential Information and all other information belonging to the Company that the Consultant acquires, observes or is informed of, directly or indirectly, in connection with the Consultant’s involvement, in any capacity, with the Company both during and after this engagement in any capacity with the Company. |
| (d) | Non-Disclosure. Except with the prior written consent of the Company or as may be expressly required in the course of performing the Services, the Consultant will not at any time, either during or after their engagement in any capacity with the Company; |
| (i) | use or copy any Confidential Information or recollections thereof for any purpose other than the performance of the Services for the benefit of the Company and its Affiliates; |
| (ii) | publish or disclose any Confidential Information or recollections thereof to any person other than to employees of the Company and its Affiliates who have a need to know such Confidential Information in the performance of their duties for the Company or its Affiliates; |
| (iii) | permit or cause any Confidential Information to be used, copied, published, disclosed, translated or adapted except as otherwise expressly permitted by this Agreement; or |
| (iv) | permit or cause any Confidential Information to be stored off the premises of the Company, including permitting or causing such Confidential Information to be stored in electronic format on personal computers, except in accordance with written procedures of the Company, as amended from time to time in writing. |
| (e) | Taking Precautions. The Consultant will take all reasonable precautions necessary or prudent to prevent material in his possession or control that contains or refers to Confidential Information from being discovered, used or copied by third parties. |
| (f) | Control of Confidential Information and Return of Information. All physical materials produced or prepared by the Consultant containing Confidential Information, including, without limitation, records, devices, computer files, data, notes, reports, proposals, lists, correspondence, specifications, drawings, plans, materials, accounts, reports, financial statements, estimates and all other materials prepared in the course of his responsibilities to or for the benefit of the Company or its Affiliates, together with all copies thereof (in whatever medium recorded), shall belong exclusively to the Company, and the Consultant will promptly turn over to the Company’s possession every original and copy of any and all such items in his possession or control upon request by the Company. If the material is such that it cannot reasonably be delivered, upon request from the Company, the Consultant will provide reasonable evidence that such materials have been destroyed, purged or erased. |
| (g) | Purpose of Use. The Consultant agrees that they will use Confidential Information only for purposes authorized or directed by the Company. |
| (h) | Exemptions. The obligations of confidentiality set out in this Section 2 will not apply to |
| (i) | information required by operation of law, court order or government agency to be disclosed, provided that: |
| (ii) | in the event that the Consultant is required to disclose such information or material, upon becoming aware of the obligation to disclose, unless prohibited by law the Consultant will provide to the Company prompt written notice so that the Company may seek a protective order or other appropriate remedy and/or waive compliance with the provisions of this Agreement; |
| (iii) | if the Company agrees that the disclosure is required by law, it will promptly give the Consultant written authorization to disclose the information for the required purposes only; |
| (iv) | if the Company promptly informs the Consultant that the Company does not agree that the disclosure is required by law, this Agreement will continue to apply, except to the extent that a Court of competent jurisdiction orders otherwise; and |
| (v) | if a protective order or other remedy is not obtained or if compliance with this Agreement is waived, the Consultant will furnish only that portion of the Confidential Information that is legally required and will exercise all reasonable efforts to obtain confidential treatment of such Confidential Information. |
| 3 | INTELLECTUAL PROPERTY RIGHTS |
| (a) | Property of the Company: All Inventions and Work Product will be the sole and exclusive property of the Company. |
| (b) | Notice of Invention. The Consultant will promptly and fully inform the Company of all Work Product, whether or not patentable, throughout the course of their involvement, in any capacity with the Company and from which there is a reasonable basis to believe that Intellectual Property may be derived therefrom, whether or not developed before or after execution of this Agreement. On their ceasing to be engaged by the Company for any reason whatsoever, the Consultant will immediately deliver up to the Company all Work Product. |
| (c) | Assignment of Rights. The Consultant will irrevocably assign, and does hereby irrevocably assign, to the Company or, at the option of the Company and upon notice from the Company, to the Company’s designee, all of their right, title and interest in and to all Work Product, including all Intellectual Property rights therein. To the extent that the Consultant retains or acquires legal title to any such Intellectual Property rights and interests, the Consultant hereby declares and confirms that such legal title is and will be held by them only as trustee and agent for the Company or the Company’s designee until such time as the Consultant is able to execute a binding assignment of such rights. The Consultant agrees that the Company’s rights hereunder shall attach to all Intellectual Property rights in their Work Product, notwithstanding that it may be perfected or reduced to specific form after they have terminated their relationship with the Company. The Consultant further agrees that the Company’s rights hereunder are worldwide rights and are not limited to Canada, but shall extend to every country of the world. |
| (d) | Moral Rights. Without limiting the foregoing, the Consultant hereby irrevocably waives any and all moral rights worldwide, including without limitation those arising under the Copyright Act (Canada), as amended, or any successor legislation of similar force and effect or similar legislation in other applicable jurisdictions or at common law that they may have with respect to all Work Product, and agrees never to assert any moral rights which they may have in the Work Product, including, without limitation, the right to the integrity of the Work Product, the right to be associated with the Work Product, the right to restrain or claim damages for any distortion, mutilation or other modification or enhancement of the Work Product and the right to restrain the use or reproduction of the Work Product in any context and in connection with any product, service, cause or institution, and the Consultant further confirms that the Company may use or alter any Work Product as the Company sees fits in its absolute discretion. |
| (e) | Goodwill. The Consultant hereby agrees that all goodwill that the Consultant has established or may establish with clients, customers, suppliers, principals, shareholders, investors, collaborators, strategic partners, licensees, contacts or prospects of the Company relating to the Business of the Company (or of its partners, subsidiaries or affiliates), both before and after the Effective Date, shall be and remain the property of the Company exclusively, for the Company to use, alter, vary, adapt and exploit as the Company shall determine in its discretion. |
| (f) | Assistance. The Consultant hereby agrees to reasonably assist the Company, at the Company’s request and expense, in: |
| (i) | making patent applications for all Work Product, including instructions to lawyers and/or patent agents as to the characteristics of the Work Product in sufficient detail to enable the preparation of a suitable patent specification, to execute all formal documentation incidental to an application for letters patent and to execute assignment documents in favour of the Company for such applications; |
| (ii) | making applications for all other forms of Intellectual Property registration relating to all Work Product; |
| (iii) | prosecuting and maintaining the patent applications and other Intellectual Property relating to all Work Product; and |
| (iv) | registering, maintaining and enforcing the patents and other Intellectual Property registrations relating to all Work Product. |
| (v) | If the Company is unable for any reason to secure the Consultant’s signature with respect to any Work Product including, without limitation, to apply for or to pursue any application for any patents or copyright registrations covering such Work Product, then the Consultant hereby irrevocably designates and appoints the Company and its duly authorized officers and agents as their agent and attorney-in-fact, to act for and in their behalf and stead to execute and file any papers, oaths and to do all other lawfully permitted acts with respect to such Work Product with the same legal force and effect as if executed by them. |
| (g) | Assistance with Proceedings. The Consultant will reasonably assist the Company, at the Company’s request and expense, in connection with any defence to an allegation of infringement of another person’s intellectual property rights, claim of invalidity of another person’s intellectual property rights, opposition to, or intervention regarding, an application for letters patent, copyright or trademark or other proceedings relating to Intellectual Property or applications for registration thereof. |
| (h) | Commercialization. The Consultant understands that the decision whether or not to commercialize or market any Work Product is within the Company’s sole discretion and for the Company’s sole benefit and that no royalty or other consideration will be due or payable to him as a result of the Company’s efforts to commercialize or market any such Work Product. |
| (i) | Prior Business Intellectual Property. The Consultant represents and warrants to the Company that they have not brought or used, and the Consultant covenants and agrees that they will not use or bring to the Company any Intellectual Property of any kind whatsoever of any Prior Business with whom the Consultant had a Prior Involvement or any Intellectual Property directly owned by the Consultant. The Company and the Consultant acknowledge and agree that the Company is not engaging the Consultant to obtain Intellectual Property relating to any Prior Involvement and the Consultant acknowledges that the Company has advised the Consultant to comply with any legal obligations the Consultant may have to such Prior Business. The Consultant covenants and agrees to indemnify and hold the Company harmless from any and all losses, claims, damages, expenses, and costs (including legal costs on a solicitor-client basis) of any kind whatsoever that the Company may suffer related to any breach by the Consultant of his obligations to such Prior Business in that regard. |
| 4 | PUBLICITY |
The Consultant shall not, without the prior written consent of the Company, make or give any public announcements, press releases or statements to the public or the press regarding any Work Product or any Confidential Information.
| 5 | FURTHER ASSURANCES |
The Parties will execute and deliver to each other such further instruments and assurances and do such further acts as may be required to give effect to this Attachment A.
| 6 | FIDUCIARY STATUS |
The provisions of this Attachment A are additional to and do not amend, replace or otherwise reduce the Consultant’s fiduciary obligations at law or equity.
| 7 | TERMINATION OF ENGAGEMENT / SURVIVAL |
| (a) | The covenants in this Attachment A apply regardless of which Party initiated the termination of the Consultant’s engagement or the reasons for the termination of the Consultant’s engagement. |
| (b) | If the engagement of the Consultant is terminated for any reason by the Consultant or the Company and there is any dispute with respect to whether any obligations have been breached or to what extent compensation or other entitlements are owing to the Consultant then despite the dispute and whether the Company is, or is later determined to be, otherwise in compliance with the terms and conditions of the Consultant’s engagement, the Consultant will at all times remain bound by the obligations set out in this Attachment A. |
| (c) | This Attachment A will survive the termination of engagement of the Consultant for any reason and will continue in full force and effect. |
| 8 | NO CONFLICTING OBLIGATIONS |
The Consultant hereby represents and warrants that the Consultant has no agreements with or obligations to any other person with respect to the matters covered by this Attachment A or concerning the Confidential Information that are in conflict with anything in this Attachment A.
| 9 | SEVERABILITY |
For the purposes of section 10.6 of the Consulting Agreement, each covenant or obligation set out in this Attachment A is a separate and distinct provision.
| 10 | INDEPENDENT LEGAL ADVICE |
The Consultant agrees that the Consultant has obtained or has had an opportunity to obtain independent legal advice in connection with this Attachment A, and further acknowledges that the Consultant has read, understands, and agrees to be bound by all of the terms and conditions contained herein.
| Agreed: | /s/ Alexandra D.J. Mancini | Date: 17 May 2024 | |
| Alexandra
D.J. Mancini, President, TRUE NORTH SYNERGY INC., Consultant |
|||
| Agreed: | Eric A. Adams | Date: 17 May 2024 | |
| Eric A. Adams | |||
| INMED PHARMACEUTICALS INC. | |||
14
Exhibit 10.20
SCIENTIFIC ADVISORY BOARD CONSULTING AGREEMENT
THIS AGREEMENT (this “Agreement”) dated for reference the 4th day of September 2024, (the “Effective Date”)
BETWEEN:
INMED PHARMACEUTICALS INC., a company incorporated under the laws of British Columbia having an office at Suite 1445, 885 W. Georgia Street, Vancouver, British Columbia, Canada V6C 3E8
(“InMed”)
AND:
Barry Greenberg, PhD, [* * * * *]
(“Advisor”)
WHEREAS:
A. InMed is a pharmaceutical company that is developing proprietary small molecule therapeutics targeting human disease;
B. InMed desires to retain distinguished scientists and other qualified individuals to advise InMed with respect to its technology strategy and to assist it in the research, development and analysis of its technology and products;
C. Advisor has the expertise and qualifications required to perform the services contemplated by this Agreement; and
D. InMed desires to retain Advisor as a member of its Scientific Advisory Board to perform the services described in this Agreement, and Advisor desires to perform such services, on the terms and conditions set out in this Agreement.
NOW THEREFORE THIS AGREEMENT WITNESSES THAT in consideration of the mutual covenants and agreements contained in this Agreement and for other good and valuable consideration, the receipt and sufficiency of which is hereby acknowledged, the parties agree as follows:
Article 1 - Interpretation
| 1.1 | Definitions |
In this Agreement:
| (a) | “Affiliate” means any Person which directly or indirectly controls, is controlled by, or is under common control with a party. |
| (b) | “Business Day” means a day that is not a Saturday or a Sunday or a statutory holiday in British Columbia or in the USA. |
| (c) | “Confidential Business Information” means all information, knowledge or data pertaining to the business and affairs of InMed, including, without limitation, its research and development activities, employees, customers, consultants, licensees, licensors, product development plans, supplier information, forecasts, strategies and financial plans, financial information, marketing and commercial strategies, the existence of this Agreement and the terms and conditions of this Agreement. |
| (d) | “Confidential Information” means the Confidential Business Information and the Proprietary Information, whether oral, written or any other form or medium, disclosed by InMed to Advisor before or after the Effective Date, including, without limitation, all analyses, compilations, data, studies, reports, copies or other documents prepared, based upon or including any such information, knowledge or data, but excluding any such information: |
| (i) | is or becomes available to the general public other than through a breach of this Agreement or another agreement of confidentiality with InMed; |
| (ii) | obtained by Advisor from a third party with a valid right to disclose it, provided that such third party is not under an obligation of confidentiality, directly or indirectly, to InMed; or |
| (iii) | that, in the reasonable opinion of Advisor’s legal counsel, is required to be disclosed by operation of law or the requirement of a governmental authority or stock exchange on which InMed’s securities are listed, provided that |
| (A) | Advisor shall promptly notify InMed prior to any such disclosure and InMed shall be given the opportunity to oppose such disclosure by seeking a protective order or other appropriate remedy, or to waive compliance with the provisions of this Agreement, |
| (B) | Advisor shall disclose only that portion of the information legally required to be disclosed, and |
| (C) | Advisor will exercise reasonable efforts to maintain the confidential treatment of the information disclosed. |
| (e) | “Contract Year” means each 12 month period during the Term, commencing on the Effective Date or anniversary thereof; |
| (f) | “Intellectual Property” means any patents, copyrights, trademarks and other forms of intellectual property, industrial and other designs, trade secrets, know-how or utility models, whether or not copyrighted or patented or registered or protected, or capable of such registration or protection. |
| (g) | “Person” means any individual, partnership, firm or corporation, governmental authority, regulatory body or agency, or other legal entity. |
-
| (h) | “Proprietary Information” means all information, knowledge or data of an intellectual, technical, scientific or industrial nature relating to compositions and formulations having immunomodulatory effects, methods of using such compositions and formulations, including targeted site-specific immunomodulation, in which InMed has a proprietary or ownership interest or has a legal duty to protect, including, without limitation, concepts, compositions, designs, formulas, specifications, biological or other materials, inventions and applications for registration of same, whether filed or not, manufacturing processes, methods, test procedures, results, databases and computer programs. |
| (i) | “Representative” of a party means any director, officer, employee, consultant, agent, lawyer, accountant or other professional advisor of such party. |
| (j) | “Services” the activities to be performed by Advisor, as specified in Section 2.1. |
| (k) | “Term” has the meaning set out in Section 9.1. |
| (l) | “Work Product” has the meaning set out in Section 5.1. |
Any words defined elsewhere in this Agreement shall have the particular meaning assigned to such words.
Article 2 - Appointment of Advisor
| 2.1 | Engagement |
Commencing on the Effective Date, InMed hereby engages Advisor, and Advisor hereby accepts such engagement, to serve as a member of InMed’s Scientific Advisory Board (the “SAB”) and to undertake and perform, during the Term, on the terms and conditions set out in this Agreement, the duties and responsibilities set out in Schedule A attached hereto (the “Duties and Responsibilities”). The parties acknowledge and agree that nothing in this agreement shall prevent the parties from entering into additional consulting agreements with each other or with the Institute for services beyond the scope of the services to be provided by Advisor under this Agreement, on such terms and conditions as may be mutually agreed by the parties.
| 2.2 | Performance of Services |
| (a) | Advisor shall perform the Services in a competent, workmanlike fashion and in accordance with customary industry standards and with at least the same level of care and attention used in performing similar work within biopharmaceutical industry. |
| (b) | Advisor will perform the Services at InMed’s location set out on the first page of this Agreement or at Advisor’s own premises, or at such other location as may be agreed between Advisor and InMed. |
| (c) | For each Contract Year during the Term, the Advisor shall provide issue-specific consultation to InMed, up to 12 times per Contract Year as further defined in Schedule A. Advisor acknowledges that the type of Services to be provided by Advisor under this Agreement will be dependent on InMed’s need for Advisor’s expertise, in light of the areas of research conducted by InMed in accordance with its corporate objectives and commercial strategies. |
-
| (d) | The Advisor may provide services to other companies or clients during the Term, so long as such services do not interfere with or conflict with the Advisor’s obligations under this Agreement, including the restrictive covenants set out in Article 10. |
| (e) | The Advisor shall be responsible for: |
| (i) | providing office space, equipment and technological needs as required to complete the Services, unless otherwise agreed to be provided by InMed; |
| (ii) | paying or remitting in a timely manner all relevant income or other taxes and any other required statutory payments that the Advisor is required to pay or remit in connection with the payments it receives from InMed for the provision of the Services; and |
| (iii) | providing InMed with proof of payment of the taxes and required statutory payments as and when requested. |
| (f) | If applicable, Advisor shall provide progress reports and/or other reports, information and work product in respect of the Services as and when directed by InMed. |
Article 3 - Compensation
| 3.1 | Compensation for Services |
In consideration of the Services performed by the Advisor and in recognition of the Advisor’s non-qualification to receive stock options, InMed shall compensate the Advisor as follows:
| (a) | InMed shall pay to Advisor, and Advisor shall accept from InMed, a fee of $[* * * * *] per Contract Year, payable in arrears, during the initial term of two (2) years; and |
| (b) | Advisor shall notify InMed for all reasonable out-of-pocket expenses actually and properly incurred by Advisor in performing the Services, provided that Advisor obtains the prior written approval of InMed as to the nature and quantum of such expenses, and submits to InMed with his payment notes under Section 3.2(a) reasonable documentation supporting his claims for reimbursement. |
| 3.2 | Payment |
| (a) | InMed will pay fees in equal quarterly installments, in arrears, during the Term as per the terms of this Agreement. Payments will be remitted directly to the Advisor’s bank account (details to be provided separately). |
| (b) | InMed will pay all undisputed invoices within fifteen (15) days of receipt by wire transfer of funds to Advisor’s bank account, or by cheque, at InMed’s discretion. |
| 3.3 | Taxes |
Advisor agrees to pay and be responsible for all deductions payable by Advisor in connection with the performance of the Services.
-
| 3.4 | Currency |
All references to money in this Agreement shall mean the lawful money of the United States, except as otherwise indicated.
| 3.5 | Records |
Advisor must keep complete, true, and accurate books of account and records of its performance of the Services for a period of at least three (3) years after the expiration of the Term.
Article 4 – Institutional Affiliations
| 4.1 | Advisor’s Obligations under Institutional Policies and Previous Commitments |
InMed acknowledges that Advisor is an Associate Professor in the Department of Neurology and Director, Alzheimer’s Disease Translational center at Johns Hopkins University School of Medicine (the “Institute”) and is subject to the Institute’s policies, including policies concerning consulting, conflicts of interest and intellectual property. InMed acknowledges that, to the extent that such policies conflict with the terms of this Agreement, Advisor’s obligations under the Institute’s policies take priority over the obligations of Advisor under this Agreement. The Advisor agrees to use reasonable efforts to avoid or minimize any such conflict. Advisor agrees that he will use his best efforts to avoid using any facilities or resources of the Institute in performing the Services hereunder.
| 4.2 | Disclosure of Institutional Policies |
Advisor agrees to provide to InMed copies of Institute’s policies or guidelines relating to Advisor’s obligations to the Institute and consulting services, if any, promptly upon request by InMed. If Advisor is required by the Institute, pursuant to applicable guidelines or policies, to make any disclosure or take any action that conflicts with the Services being provided by Advisor hereunder or is that contrary to the terms of this Agreement, Advisor will promptly notify InMed of such obligation, specifying the nature of such disclosure or action and identifying the applicable guideline or policy under which disclosure or action is required, prior to making such disclosure or taking such action.
Article 5 - Work Product
| 5.1 | Ownership of Work Product |
Advisor agrees that all inventions, discoveries, improvements, processes, technology and know-how (whether or not patentable and whether or not reduced to practice) and all works of authorship:
| (a) | arising directly or indirectly from the performance of the Services under this Agreement; or |
-
| (b) | which Advisor may conceive or make (either alone or jointly with others) during the Term with InMed, and which relate to the field in which the Services are to be performed under this Agreement and all Intellectual Property rights thereto (collectively, “Work Product”) shall be the sole and exclusive property and Proprietary Information of InMed, and that the Advisor will retain only the right to be listed as an inventor in any applicable patents, as determined by InMed in its sole discretion. Advisor will promptly and fully disclose in writing to InMed any and all Work Product resulting from the conduct of the Services. Advisor will assign, and hereby assigns all right, title and interest in and to all such Wok Product to InMed and will execute all documents necessary to confirm same. All original works of authorship which are made by Advisor (solely or jointly with others) in the course of Advisor’s performance of the Services under this Agreement and which are able to be protected by copyright are “works made for hire,” as that term is defined in the United States Copyright Act. |
| 5.2 | Further Assurances |
During and after the Term, Advisor agrees to assist InMed, at InMed’s request and expense, in executing patent applications and copyright registrations and such other documents considered necessary by InMed or its counsel to apply for and obtain letters, patents and copyright registrations in any or all countries, as InMed may deem advisable, or to otherwise protect for the benefit of InMed any Work Product. Advisor shall also make such assignments and execute such other instruments as may be necessary to convey to InMed the ownership and exclusive rights in and to such Work Product. Advisor further agrees, whether or not Advisor is still providing services to InMed, to cooperate to the extent and in the manner requested by InMed in the prosecution or defense of any such patent or copyright claims or any litigation or other proceeding involving any Work Product in any country of the world, at InMed’s cost and expense.
| 5.3 | Avoidance of Third Party Rights |
Except with InMed’s prior written consent, Advisor shall not engage in any activities, on its own or in collaboration with a third party or use any third party facilities or third party Intellectual Property in performing the Services which could result in any claims of any ownership interest in any Work Product being made by such third party.
Article 6 - Confidential Information
| 6.1 | Obligation of Confidentiality |
Advisor agrees:
| (a) | to keep and use all Confidential Information in strict confidence and to not, without InMed’s prior written consent, disclose any Confidential Information or recollections thereof to any Person; |
| (b) | to use the Confidential Information only for the purpose of performing the Services; |
-
| (c) | to clearly label as “confidential” all copies, duplicates, reproductions, translations or adaptations of the Confidential Information; and |
| (d) | to take all reasonable steps to prevent material in its possession or control that contains or refers to Confidential Information from being discovered, used or copied by third parties and to protect and safeguard such Confidential Information from loss, theft or destruction. |
| 6.2 | Acknowledgement of Confidentiality |
Advisor agrees that all communications and information received from InMed prior to the execution of this Agreement will be deemed to be InMed’s Confidential Information from the time of its receipt, subject to the exceptions set out in the definition of “Confidential Information”.
| 6.3 | Title to Confidential Information |
All right, title and interest in and to the Confidential Information will be owned by InMed. Advisor is not granted any licence or other rights to use any Confidential Information except as expressly set out in this Agreement. Advisor will hold all Confidential Information in trust for InMed.
Article 7 - Representations and Warranties
| 7.1 | Advisor’s Representations and Warranties |
Advisor represents and warrants to InMed that:
| (a) | the Services will be performed in a professional manner in accordance with industry standards and any quality standards set out in this Agreement; |
| (b) | Advisor will comply with all laws, regulations and ordinances, whether foreign, federal, provincial, municipal or otherwise with respect to the performance of the Services; and |
| (c) | to the best knowledge of Advisor, the performance of the Services and the use of the Work Product will not infringe any Intellectual Property right of any third party. |
| 7.2 | No Warranty |
All Confidential Information is provided to Advisor on an “AS IS” basis. InMed makes no representation or warranty of any kind, express or implied, to Advisor as to the accuracy, performance or completeness of any Confidential Information disclosed hereunder. Advisor will rely upon its own investigation, due diligence and analysis in evaluating and satisfying itself as to all matters relating to the Confidential Information. Neither InMed nor its Representatives will be liable to Advisor or any other Person, directly or indirectly, for any claims arising from the furnishing of any Confidential Information to Advisor or any use by Advisor of such Confidential Information.
-
| 7.3 | No Conflict with Advisor’s Other Obligations |
Advisor represents that Advisor is presently retained by [list any companies here] to advise on research that is not similar to that of InMed, and Advisor agrees that Advisor will not accept any new retention to provide advice that might conflict with InMed during the term of this Agreement without prior written approval of InMed. Advisor represents that Advisor has no agreements with or obligations to others with respect to the matters covered by this Agreement or concerning the Confidential Information that is in conflict with anything in this Agreement except as disclosed in Schedule B attached hereto. Advisor represents that Advisor’s performance of the terms of the Agreement does not and will not conflict with the terms of any agreement to keep in confidence proprietary information and trade secrets acquired in confidence or in trust prior to Advisor’s advisory relationship with InMed. Advisor will not disclose to InMed, or induce InMed to use, any confidential or proprietary information or material belonging to any third party. Advisor also represents to InMed that he is not subject to any pre-existing restrictive covenants that could be breached as a result of entering into this Agreement and/or performing the Services.
Article 8 - Indemnification
| 8.1 | Indemnification by InMed |
InMed shall indemnify, defend, and hold harmless Advisor from and against any and all claims, suits, losses, expenses (including reasonable attorneys’ fees and legal expenses), costs and damages of every kind and nature (including but not limiting the generality of the foregoing, in respect of death, injury, loss or damage to any person or property) (together, “Claims”) of any third party resulting from or arising out of the Advisor’s services and his role as an SAB member hereunder, except to the extent any such Claim is caused by the Advisor’s wilful misconduct.
Article 9 - Termination
| 9.1 | Term |
This Agreement shall commence on the Effective Date and shall remain in full force and effect for an initial term of two (2) years, unless and until terminated in accordance with Section 9.2 or extended as provided herein (the “Term”). At the end of such initial term, the Term will automatically be extended for an additional period of one (1) year, unless Advisor or InMed shall have given to the other written notice to the contrary at least thirty (30) days prior to the commencement of such additional period. Compensation for this extended period will be mutually agreed between the parties.
| 9.2 | Termination for Default and Convenience |
This Agreement may be terminated:
| (a) | upon 30 days prior written notice by either party to the other party; |
| (b) | by a party upon a material default in the performance or observance of the other party’s obligations under this Agreement, and failure of the defaulting party to remedy such default within 30 days after receiving written notice of the default from the non-defaulting party; or |
-
| (c) | immediately upon either party becoming bankrupt or making an assignment for the benefit of creditors, or upon a receiver or trustee in bankruptcy being appointed for either party, or upon any proceeding in bankruptcy, receivership, or liquidation being instituted against a party and continuing for 30 days without being dismissed, or upon a party otherwise ceasing to exist; |
provided that any right of termination set out above shall be in addition to all other rights and remedies available to the parties, if any, for default or wrong-doing by each other.
| 9.3 | Effect of Termination |
Upon termination of this Agreement:
| (a) | upon receipt of notice of termination, Advisor shall immediately cease performing the Services; |
| (b) | Advisor shall provide InMed with all Work Product, including data, whether or not complete, and all information relating to such Work Product, all in a form and with content reasonably satisfactory to InMed; and |
| (c) | except in the case of a breach of this Agreement by Advisor or the negligence or intentional misconduct of Advisor, Advisor shall invoice InMed for, and InMed shall pay to Advisor, within thirty (30) days after the date of the invoice any out-of-pocket expenses reasonably and properly incurred by Advisor prior to the effective date of termination. |
| 9.4 | Return of Confidential Information |
Upon termination of this Agreement, Advisor will cease all use of the Confidential Information. Advisor shall, within thirty (30) days after termination:
| (a) | return to InMed all original copies of the Confidential Information; and |
| (b) | destroy any and all copies and extracts thereof and all other documents, computer files, memoranda, notes and other writings prepared by or for Advisor based on the Confidential Information. |
Notwithstanding the foregoing, Advisor may retain one copy of the Confidential Information solely to determine its obligations hereunder and will not be required to destroy electronic information stored to electronic back-up systems in the ordinary course of business, provided that such electronic information must not be accessible or usable by any person except as provided in this section.
| 9.5 | Survival |
The terms and provisions, covenants and conditions contained in Article 5 (Work Product), Article 6 (Confidential Information), Article 7 (Representations and Warranties), Article 8 (Indemnification), Sections 9.2 through 9.5 (Termination), Article 10 (Restrictive Covenants) and Article 11 (General Provisions) shall remain in force, survive indefinitely (or otherwise in accordance with their terms) and be binding upon the parties, their successors and their permitted assigns notwithstanding any expiration or other termination of this Agreement for any reason whatsoever.
-
Article 10 - Restrictive Covenants
| 10.1 | Definitions |
In this Agreement:
| (a) | “Business” means: |
| (i) | the researching, developing, commercializing, producing and marketing of novel therapies, as such business grows and evolves during the Term; and |
| (ii) | any other material business carried on from time to time by InMed or any Affiliate of InMed. |
| (b) | “Competing Business” means any endeavor, activity or business which is competitive in any material way with the Business world-wide. |
| (c) | “Contact” means any person, firm, corporation or other entity that was a client, customer, supplier, principal, shareholder, investor, collaborator, strategic partner, licensee, contact or prospect of InMed (or of its partners, funders or Affiliates) with whom Advisor dealt or otherwise became aware of during the Term or Advisor’s relationship in any capacity with InMed. |
| (d) | “Restricted Period” means a period of twelve (12) months from the date of termination of this Agreement for any reason. |
| 10.2 | Reasonableness |
Advisor hereby acknowledges and agrees that:
| (a) | both before and since the Effective Date InMed has operated and competed and will operate and compete world-wide, with respect to the Business of InMed; |
| (b) | competitors of InMed and the Business are located worldwide; |
| (c) | in order to protect InMed adequately, any enjoinder of competition would have to apply to any country in which InMed, during the Term, had material business relationships; |
| (d) | during the Term, on behalf of InMed, Advisor will acquire knowledge of, and will come into contact with, initiate and establish relationships with, both existing and new clients, customers, suppliers, principals, contacts and prospects of InMed, and that in some circumstances Advisor may become the senior or sole representative of InMed dealing with such persons; and |
| (e) | in light of the foregoing, the provisions of this Article 10 are reasonable and necessary for the proper protection of the Business of InMed. |
-
| 10.3 | Restrictive Covenant |
| (a) | During the Term and for the Restricted Period, Advisor shall not, without the prior written consent of InMed’s Board of Directors, such consent to be granted or withheld in the sole discretion of InMed’s Board of Directors, within the geographic scope of any country in which InMed, during the Term, had material business relationships, including, without limitation, Canada, the United States, the United Kingdom and the countries of the European Union, carry on or be employed by or engaged in or have any financial or other interest in or be otherwise commercially involved in a Competing Business, whether legal or illegal, directly or indirectly, either individually or in partnership or jointly or in conjunction with any person, firm, corporation or other entity, as principal, agent, consultant, advisor, employee, shareholder or in any manner whatsoever. |
| (b) | Advisor shall not be in default of Section 10.3(a) by virtue of Advisor: |
| (i) | following the termination of this Agreement, holding, strictly for portfolio purposes and as a passive investor, no more than five percent (5%) of the issued and outstanding shares of, or any other interest in, any corporation or other entity which is listed on any recognized stock exchange, that is a Competing Business; or |
| (ii) | during the Term, holding, strictly for portfolio purposes and as a passive investor, issued and outstanding shares of, or any other interest in, any corporation or other entity, the business of which corporation or other entity is in the same Business as InMed provided such corporation is not a Competing Business, and provided further that Advisor first obtains InMed’s written consent, which consent will not be unreasonably withheld. |
If Advisor holds issued and outstanding shares or any other interest in a corporation or other entity pursuant to Section 10.3(b)(ii), and following the acquisition of such shares or other interest the business of the corporation or other entity becomes a Competing Business, Advisor will promptly dispose of his shares or other interest in such corporation or other entity.
| 10.4 | Non-Solicitation |
Advisor shall not, during the Term and for the Restricted Period, whether legal or illegal, either individually or in partnership or jointly or in conjunction with any person, firm, corporation or other entity, as principal, agent, consultant, advisor, employee, shareholder or in any manner whatsoever, without the prior written and informed consent of InMed, directly or indirectly:
| (a) | canvass or solicit the business of (or procure or assist the canvassing or soliciting of the business of) any Contact, or otherwise solicit, induce or encourage any Contact to curtail or cease its relationship with InMed, for any purpose which is competitive with the Business; or |
| (b) | accept (or procure or assist the acceptance of) any business from any Contact which business is competitive with the Business; or |
| (c) | be employed by or supply (or procure or assist the supply of) any goods or services to any Contact for any purpose which is competitive with the Business; or |
| (d) | employ, engage, offer employment or engagement to or solicit the employment or engagement of or otherwise entice away from or solicit, induce or encourage to leave the employment or engagement of InMed, any individual who is employed or engaged by InMed whether or not such individual would commit any breach of his contract or terms of employment or engagement by leaving the employ or the engagement of InMed, provided that Advisor shall be permitted, solely in a personal capacity, to provide letters of reference for individuals who are employed by InMed. |
-
| 10.5 | Validity |
Advisor expressly recognizes and acknowledges that it is the intent of the parties that his activities following the termination of this Agreement be restricted in the manner described in this Article 10, and acknowledges that good, valuable, and sufficient consideration has been provided in exchange for such restrictions. Advisor agrees that should any of the restrictions contained in this Article 10 be found to be unreasonable to any extent by a court of competent jurisdiction adjudicating upon the validity of the restriction, whether as to the scope of the restriction, the area of the restriction or the duration of the restriction, then such restriction shall be reduced to that which is in fact declared reasonable by such court, or a subsequent court of competent jurisdiction, requested to make such a declaration, in order to ensure that the intention of the parties is given the greatest possible effect.
Article 11 - General Provisions
| 11.1 | General Provisions |
| (a) | This Agreement may not be amended except in writing signed by both parties. |
| (b) | The following schedules attached hereto shall be deemed to form an integral part of this Agreement: |
Schedule A: Duties & Responsibilities
Schedule B: Advisor’s Conflicting Obligations
| (c) | Advisor shall not assign this Agreement nor delegate any of Advisor’s obligations hereunder, in whole or in part, without the prior written consent of InMed. InMed may assign this Agreement without consent to any assignee of all or substantially all of InMed’s business or assets. Any assignment not in accordance with this section shall be void. |
| (d) | This Agreement constitutes the entire agreement between the parties concerning the subject matter hereof and supersedes all prior agreements or understandings with respect thereto. |
| (e) | This Agreement shall enure to the benefit of and be binding upon the parties and their respective successors, heirs and permitted assigns. |
| (f) | The parties hereby undertake to do such further acts and take such steps as may be reasonably required to implement the intent of this Agreement. |
| (g) | This Agreement shall be governed by and construed in accordance with the laws of the Province of British Columbia, Canada. |
| (h) | Nothing in this Agreement shall form any partnership or joint venture between the parties. The parties shall, in relation to their respective obligations hereunder, act as independent contractors, and nothing in this Agreement shall be construed to give either party the power or authority to act for, bind or commit the other party in any way whatsoever. |
| (i) | Advisor agrees that InMed may be irreparably damaged if any provision of this Agreement is not performed by Advisor in accordance with its terms. Accordingly, InMed shall be entitled to apply for an injunction or injunctions to prevent breaches of any of the provisions of this Agreement and may specifically enforce such provisions by an action instituted in a court having jurisdiction. These specific remedies are in addition to any other remedy to which InMed may be entitled at law or in equity. |
-
| (j) | All notices or communications hereunder shall be sent in writing to the other party by hand delivery, registered mail, receipted commercial courier or receipted facsimile transmission, at the address provided below or to such other address as the parties may notify each other from time to time. Notices and communications sent by hand delivery, registered letter or commercial courier shall be deemed received upon actual receipt, and those sent by facsimile transmission shall be deemed received on the next Business Day. |
|
To the Advisor:
[* * * * *] |
To InMed:
InMed Pharmaceuticals Inc.
Attention: Chief Executive Officer
Tel: [* * * * *] |
| (k) | A waiver of any breach of any provision of this Agreement shall not be construed as a continuing waiver of other breaches of the same or other provisions of this Agreement. |
| (l) | No exercise of a specific right or remedy by either party precludes it or prejudices it in exercising another right or pursuing another remedy or maintaining an action to which it may otherwise be entitled either at law or in equity. |
| (m) | If any provision of this Agreement is held invalid, illegal or found to be unenforceable by a court of competent jurisdiction for any reason whatsoever, the unenforceability will not affect the validity, legality or enforceability of the remaining provisions of this Agreement, and the unenforceable, illegal or invalid provision or provisions will be severable from the remainder of this Agreement. |
| (n) | The obligations of confidentiality and non-use of Confidential Information under this Agreement are and shall be continuing obligations and shall be perpetual. |
| (o) | Except for disputes arising in respect of Article 10, all disputes arising out of or in connection with this Agreement and the relationship between the parties, are to be referred to and finally resolved by arbitration administered by the British Columbia International Commercial Arbitration Centre, pursuant to its Rules. The place of arbitration will be Vancouver, British Columbia. |
| (p) | The parties each acknowledge that they have not relied upon the other party to this Agreement for advice, whether legal or otherwise, in connection with this Agreement. Advisor agrees that he has obtained or has had an opportunity to obtain independent legal advice in connection with this Agreement, and further acknowledges that he has read, understands, and agrees to be bound by all of the terms and conditions contained herein. |
| (q) | This Agreement may be signed and delivered in counterparts with the same effect as if the parties had signed the same document and all counterparts shall be construed together and shall constitute one and the same agreement. A signed version of this Agreement scanned and delivered by facsimile or e-mail transmission shall have the same effect as a signed original. |
-
IN WITNESS WHEREOF this Agreement has been executed by the parties hereto as of the Effective Date.
|
SIGNED, SEALED AND DELIVERED in the presence of |
||
| /s/ Barry Greenberg | ||
| Witness Signature | Barry Greenberg, PhD | |
| Witness Name | ||
| Witness Address | ||
| Witness Occupation |
| INMED PHARMACEUTICALS INC. | |
| by its authorized signatory: |
| Per: | |
| /s/ Eric A. Adams | |
| Name: Eric A. Adams | |
| Title: President and CEO |
-
SCHEDULE A
Duties & Responsibilities
Advisor shall undertake and perform the following duties and responsibilities:
| (a) | serving expectedly for 20 hours per year as a scientific and technical advisor to InMed and its senior team members on specific topics with respect to the field of InMed proprietary small molecule drug candidate and their development in the context of neurodegenerative/neurological diseases (collectively, “InMed’s Product Development”), including the application of unique, special and extraordinary skills and knowledge that Advisor possesses in such field; |
| (b) | attending meetings of InMed’s SAB, if required, at InMed’s expense as outlined in the Agreement; and |
| (c) | providing issue-specific ad hoc consulting services to InMed at its request, within the expected amount of not more than 10 hours per year. |
-
SCHEDULE B
Advisor’s Conflicting Obligations
| a) | Advisor is an Associate Professor in the Department of Neurology and Director, Alzheimer’s Disease Translational center at Johns Hopkins University School of Medicine, [* * * * *], USA |
| b) | Advisor is a consultant to [* * * * *]. |
-16-
Exhibit 21.1
SUBSIDIARIES OF INMED PHARMACEUTICALS INC.
| Subsidiary | Jurisdiction | |
| Biogen Sciences Inc. | BC | |
| Sweetnam Consulting Inc. | BC | |
| InMed Pharmaceuticals Ltd. | Delaware | |
| BayMedica, LLC | Delaware |
Exhibit 23.1
INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM’S CONSENT
We consent to the incorporation by reference in the Registration Statement of InMed Pharmaceuticals Inc. on Forms S-1 [File Nos. 333-253925, 333-257858, 333-265731, 333-267831, 333-268700 and 333-275410], on Forms S-3 [File Nos. 333-262532, 333-262533 and 333-264187], and on Forms S-8 [File Nos. 333,253912, 333-260323, and 333-276212] of our report dated September 27, 2024, which includes an explanatory paragraph as to the Company’s ability to continue as a going concern, with respect to our audits of the consolidated financial statements of InMed Pharmaceuticals Inc. as of June 30, 2024 and 2023 and for the years ended June 30, 2024 and 2023, which report is included in this Annual Report on Form 10-K of InMed Pharmaceuticals Inc. for the year ended June 30, 2024.
/s/ Marcum llp
Marcum llp
New York, NY
September 27, 2024
Exhibit 31.1
Certification Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002
I, Eric A. Adams, certify that:
| 1. | I have reviewed this Annual Report on Form 10-K of InMed Pharmaceuticals Inc.; |
| 2. | Based on my knowledge, this report does not contain any untrue statement of a material fact or omit to state a material fact necessary to make the statements made, in light of the circumstances under which such statements were made, not misleading with respect to the period covered by this report; |
| 3. | Based on my knowledge, the financial statements, and other financial information included in this report, fairly present in all material respects the financial condition, results of operations and cash flows of the registrant as of, and for, the periods presented in this report; |
| 4. | The registrant’s other certifying officer and I are responsible for establishing and maintaining disclosure controls and procedures (as defined in Exchange Act Rules 13a- 15(e) and 15d-15(e)) and internal control over financial reporting (as defined in Exchange Act Rules 13a-15(f) and 15d-15(f)) for the registrant and have: |
| a) | Designed such disclosure controls and procedures, or caused such disclosure controls and procedures to be designed under our supervision, to ensure that material information relating to the registrant, including its consolidated subsidiaries, is made known to us by others within those entities, particularly during the period in which this report is being prepared; |
| b) | Designed such internal control over financial reporting, or caused such internal control over financial reporting to be designed under our supervision, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles; |
| c) | Evaluated the effectiveness of the registrant’s disclosure controls and procedures and presented in this report our conclusions about the effectiveness of the disclosure controls and procedures, as of the end of the period covered by this report based on such evaluation; and |
| d) | Disclosed in this report any change in the registrant’s internal control over financial reporting that occurred during the registrant’s fourth fiscal quarter that has materially affected, or is reasonably likely to materially affect, the registrant’s internal control over financial reporting; and |
| 5. | The registrant’s other certifying officer and I have disclosed, based on our most recent evaluation of internal control over financial reporting, to the registrant’s auditors and the audit committee of the registrant’s board of directors (or persons performing the equivalent functions): |
| a) | All significant deficiencies and material weaknesses in the design or operation of internal control over financial reporting which are reasonably likely to adversely affect the registrant’s ability to record, process, summarize and report financial information; and |
| b) | Any fraud, whether or not material, that involves management or other employees who have a significant role in the registrant’s internal control over financial reporting. |
Date: September 27, 2024
| /s/ Eric A. Adams | ||
| Name: | Eric A. Adams | |
| Title: | President and Chief Executive Officer | |
Exhibit 31.2
Certification Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002
I, Netta Jagpal, certify that:
| 1. | I have reviewed this Annual Report on Form 10-K of InMed Pharmaceuticals Inc.; |
| 2. | Based on my knowledge, this report does not contain any untrue statement of a material fact or omit to state a material fact necessary to make the statements made, in light of the circumstances under which such statements were made, not misleading with respect to the period covered by this report; |
| 3. | Based on my knowledge, the financial statements, and other financial information included in this report, fairly present in all material respects the financial condition, results of operations and cash flows of the registrant as of, and for, the periods presented in this report; |
| 4. | The registrant’s other certifying officer and I are responsible for establishing and maintaining disclosure controls and procedures (as defined in Exchange Act Rules 13a- 15(e) and 15d-15(e)) and internal control over financial reporting (as defined in Exchange Act Rules 13a-15(f) and 15d-15(f)) for the registrant and have: |
| a) | Designed such disclosure controls and procedures, or caused such disclosure controls and procedures to be designed under our supervision, to ensure that material information relating to the registrant, including its consolidated subsidiaries, is made known to us by others within those entities, particularly during the period in which this report is being prepared; |
| b) | Designed such internal control over financial reporting, or caused such internal control over financial reporting to be designed under our supervision, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles; |
| c) | Evaluated the effectiveness of the registrant’s disclosure controls and procedures and presented in this report our conclusions about the effectiveness of the disclosure controls and procedures, as of the end of the period covered by this report based on such evaluation; and |
| d) | Disclosed in this report any change in the registrant’s internal control over financial reporting that occurred during the registrant’s fourth fiscal quarter that has materially affected, or is reasonably likely to materially affect, the registrant’s internal control over financial reporting; and |
| 5. | The registrant’s other certifying officer and I have disclosed, based on our most recent evaluation of internal control over financial reporting, to the registrant’s auditors and the audit committee of the registrant’s board of directors (or persons performing the equivalent functions): |
| a) | All significant deficiencies and material weaknesses in the design or operation of internal control over financial reporting which are reasonably likely to adversely affect the registrant’s ability to record, process, summarize and report financial information; and |
| b) | Any fraud, whether or not material, that involves management or other employees who have a significant role in the registrant’s internal control over financial reporting. |
Date: September 27, 2024
| /s/ Netta Jagpal | ||
| Name: | Netta Jagpal | |
| Title: | Chief Financial Officer | |
Exhibit 32.1
Certification Pursuant to Section 906 of the Sarbanes-Oxley Act of 2002
Pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002, I, Eric A. Adams, the President and Chief Executive Officer of InMed Pharmaceuticals Inc. (the “Company”), hereby certify that, to my knowledge:
1. The Annual Report on Form 10-K for the year ended June 30, 2024 (the “Report”) of the Company fully complies with the requirements of Section 13(a) or Section 15(d) of the Securities Exchange Act of 1934; and
2. The information contained in the Report fairly presents, in all material respects, the financial condition and results of operations of the Company.
Date: September 27, 2024
| /s/ Eric A. Adams | ||
| Name: | Eric A. Adams | |
| Title: | President and Chief Executive Officer | |
Exhibit 32.2
Certification Pursuant to Section 906 of the Sarbanes-Oxley Act of 2002
Pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002, I, Netta Jagpal, the Chief Financial Officer of InMed Pharmaceuticals Inc. (the “Company”), hereby certify that, to my knowledge:
1. The Annual Report on Form 10-K for the year ended June 30, 2024 (the “Report”) of the Company fully complies with the requirements of Section 13(a) or Section 15(d) of the Securities Exchange Act of 1934; and
2. The information contained in the Report fairly presents, in all material respects, the financial condition and results of operations of the Company.
Date: September 27, 2024
| /s/ Netta Jagpal | ||
| Name: | Netta Jagpal | |
| Title: | Chief Financial Officer | |
Exhibit 97.1
InMed Pharmaceuticals Inc.
EXECUTIVE COMPENSATION CLAWBACK POLICY
Introduction
The Board of Directors (the “Board”) of InMed Pharmaceuticals Inc. (the “Company”) believes that it is in the best interests of the Company and its shareholders to create and maintain a culture that emphasizes integrity and accountability and that reinforces the Company’s pay-for-performance compensation philosophy. The Board has therefore adopted this policy which provides for the recoupment of certain executive compensation in the event of an accounting restatement resulting from material noncompliance with financial reporting requirements under the federal securities laws (the “Policy”). This Policy is designed to comply with Section 10D of the Securities Exchange Act of 1934 (the “Exchange Act”), Rule 10D-1 promulgated under the Exchange Act and Section 303A.14 of the New York Stock Exchange Listed Company Manual (the “Listing Standards”).
Administration
The Board has delegated administration of this Policy to the Compensation Committee of the Board (the “Committee”). Any determinations made by the Committee shall be final and binding on all affected individuals.
Covered Executives
This Policy applies to the Company’s current and former executive officers, as determined by the Committee in accordance with Section 10D of the Exchange Act and the Listing Standards, and such other senior executives or employees who may from time to time be deemed subject to the Policy by the Committee (“Covered Executives”). The following are examples of persons who may be deemed executive officers:
| ● | Chief Executive Officer; |
| ● | President; |
| ● | Chief Financial Officer or principal financial officer; |
| ● | Principal accounting officer or controller; |
| ● | Any vice president in charge of a principal business unit, division or function, such as sales administration or finance; |
| ● | Any other officer who performs a policy-making function; and |
| ● | Any other person (such as an executive officer of a subsidiary or parent entity) who performs similar policy-making functions for the company. |
Recoupment; Accounting Restatement
In the event the Company is required to prepare an accounting restatement of its financial statements due to the Company’s material noncompliance with any financial reporting requirement under the securities laws, the Committee will require reimbursement or forfeiture of any excess Incentive Compensation received by any Covered Executive during the three completed fiscal years immediately preceding the date on which the Company is required to prepare an accounting restatement. However, no reimbursement or forfeiture will apply to Incentive Compensation received by a Covered Executive before such Covered Executive began providing services as a Covered Executive.
Incentive Compensation
For purposes of this Policy, Incentive Compensation means any compensation that is granted, earned or vested based wholly or in part upon the attainment of a Financial Reporting Measure. Incentive Compensation is “received” for purposes of this Policy in the Company’s fiscal period during which the Financial Reporting Measure specified in the Incentive Compensation award is attained, even if the payment or grant of such Incentive Compensation occurs after the end of that period. The following are examples of Incentive Compensation that may be based on a Financial Reporting Measure:
| ● | Annual bonuses and other short- and long-term cash incentives. |
| ● | Stock options. |
| ● | Stock appreciation rights. |
| ● | Restricted stock. |
| ● | Restricted stock units. |
| ● | Performance shares. |
| ● | Performance units. |
A “Financial Reporting Measure” is any measure that is determined and presented in accordance with the accounting principles used in preparing the Company’s financial statements, and any measure that is derived wholly or in part from such measure. A Financial Reporting Measure need not be presented within the Company’s financial statements or included in a filing with the Securities Exchange Commission. Examples of Financial Reporting Measures may include:
| ● | Company stock price; |
| ● | Total shareholder return; |
| ● | Revenues; |
| ● | Net income; |
| ● | Earnings before interest, taxes, depreciation, and amortization (EBITDA); |
| ● | Funds from operations; |
| ● | Liquidity measures such as working capital or operating cash flow; |
| ● | Return measures such as return on invested capital or return on assets; or |
| ● | Earnings measures such as earnings per share. |
Excess Incentive Compensation: Amount Subject to Recovery
The amount to be recovered will be the excess of the Incentive Compensation paid to the Covered Executive based on the erroneous data over the Incentive Compensation that would have been paid to the Covered Executive had it been based on the restated results, as determined by the Committee.
If the Committee cannot determine the amount of excess Incentive Compensation received by the Covered Executive directly from the information in the accounting restatement, then it will make its determination based on a reasonable estimate of the effect of the accounting restatement.
Method of Recoupment
The Committee will determine, in its sole discretion, the method for recouping Incentive Compensation hereunder which may include, without limitation:
| ● | requiring reimbursement of cash Incentive Compensation previously paid; |
| ● | seeking recovery of any gain realized on the vesting, exercise, settlement, sale, transfer, or other disposition of any equity-based awards; |
| ● | offsetting the recouped amount from any compensation otherwise owed by the Company to the Covered Executive; |
| ● | (d)) cancelling outstanding vested or unvested equity awards; and |
| ● | taking any other remedial and recovery action permitted by law, as determined by the Committee. |
No Indemnification
The Company shall not indemnify any Covered Executives against the loss of any incorrectly awarded Incentive Compensation.
Interpretation
The Committee is authorized to interpret and construe this Policy and to make all determinations necessary, appropriate, or advisable for the administration of this Policy. It is intended that this Policy be interpreted in a manner that is consistent with the requirements of Section 10D of the Exchange Act and any applicable rules or standards adopted by the Securities and Exchange Commission or any national securities exchange on which the Company’s securities are listed.
Effective Date
This Policy has been adopted by the Committee effective as of December 1, 2023 (the “Effective Date”) and shall apply to Incentive Compensation that is approved, awarded or granted to Covered Executives on or after that date. This Policy amends and replaces in its entirety the Company’s former Executive Compensation Clawback Policy.
Amendment; Termination
The Committee may amend this Policy from time to time in its discretion and shall amend this Policy as it deems necessary to reflect further regulations adopted by the Securities and Exchange Commission under Section 10D of the Exchange Act or rules or interpretations promulgated thereunder and to comply with any Listing Standards. The Committee may terminate this Policy at any time.
Other Recoupment Rights
The Committee intends that this Policy will be applied to the fullest extent of the law. The Committee may require that any employment agreement, equity award agreement, or similar agreement entered into on or after the Effective Date shall, as a condition to the grant of any benefit thereunder, require a Covered Executive to agree to abide by the terms of this Policy. Any right of recoupment under this Policy is in addition to, and not in lieu of, any other remedies or rights of recoupment that may be available to the Company pursuant to the terms of any similar policy in any employment agreement, equity award agreement, or similar agreement and any other legal remedies available to the Company.
Impracticability
The Committee shall recover any excess Incentive Compensation in accordance with this Policy unless such recovery would be impracticable, as determined by the Committee in accordance with Rule 10D-1 of the Exchange Act and the Listing Standards.
Successors
This Policy shall be binding and enforceable against all Covered Executives and their beneficiaries, heirs, executors, administrators or other legal representatives.
Exhibit Filing Requirement
A copy of this Policy and any amendments thereto shall be posted on the Company’s website and filed as an exhibit to the Company’s annual report on Form 10-K.
4