UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, DC 20549
FORM 8-K
CURRENT REPORT
Pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934
Date of Report (Date of earliest event reported): June 25, 2024
Unicycive Therapeutics, Inc.
(Exact name of registrant as specified in its charter)
Delaware |
001-40582 | 81-3638692 | ||
|
(State or other jurisdiction of incorporation or organization) |
(Commission File Number) |
IRS Employer Identification No.) |
4300 El Camino Real, Suite 210
Los Alto, CA 94022
(Address of principal executive offices)
Registrant’s telephone number, including area code: (650) 351-4495
(Former name or former address, if changed since last report)
Securities registered pursuant to Section 12(b) of the Act:
| Title of each class: | Trading Symbol(s) | Name of each exchange on which registered: | ||
| Common Stock | UNCY | Nasdaq Capital Market |
Check the appropriate box below if the Form 8-K filing is intended to simultaneously satisfy the filing obligation of the registrant under any of the following provisions:
| ☐ | Written communication pursuant to Rule 425 under the Securities Act (17 CFR 230.425) |
| ☐ | Soliciting material pursuant to Rule 14a-12 under the Exchange Act (17 CFR 240.14a-12) |
| ☐ | Pre-commencement communications pursuant to Rule 14d-2(b) under the Exchange Act (17 CFR 240.14d-2(b)) |
| ☐ | Pre-commencement communications pursuant to Rule 13e-4(c) under the Exchange Act (17 CFR 240.13e-4(c)) |
Indicate by check mark whether the registrant is an emerging growth company as defined in as defined in Rule 405 of the Securities Act of 1933 (§230.405 of this chapter) or Rule 12b-2 of the Securities Exchange Act of 1934 (§240.12b-2 of this chapter).
Emerging growth company ☒
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Item 7.01 Regulation FD Disclosure.
On June 25, 2024, Unicycive Therapeutics, Inc. (the “Company”) issued a press release announcing positive results from the Oxylanthanum Carbonate (OLC) UNI-OLC-201 pivotal clinical trial including achievement of both the Primary and Secondary Endpoints. A copy of the press release is furnished as Exhibit 99.1 to this Form 8-K.
In addition, on June 25, 2024, the Company presented its clinical results from the pivotal clinical trial in a corporate presentation. A copy of the corporate presentation is furnished as Exhibit 99.2 to this Form 8-K.
The foregoing (including Exhibits 99.1 and 99.2) is being furnished pursuant to Item 7.01 and will not be deemed to be filed for purposes of Section 18 of the Securities Exchange Act of 1934, as amended (the “Exchange Act”), or otherwise be subject to the liabilities of that section, nor will it be deemed to be incorporated by reference in any filing under the Securities Act of 1933 (the “Securities Act”), as amended, or the Exchange Act.
Item 9.01. Financial Statements and Exhibits
(d) Exhibits.
| 99.1 |
Press Release of Unicycive Therapeutics, Inc. dated June 25, 2024. |
|
| 99.2 | Unicycive Therapeutics, Inc. Corporate Presentation | |
| 104 | Cover Page Interactive Data File (embedded within the Inline XBRL document). |
SIGNATURE
Pursuant to the requirements of the Securities Exchange Act of 1934, the Registrant has duly caused this report to be signed on its behalf by the undersigned hereunto duly authorized.
Dated: June 25, 2024
|
UNICYCIVE THERAPEUTICS, INC. | |
| By: | /s/ Shalabh Gupta | |
| Shalabh Gupta | ||
| Chief Executive Officer | ||
2
Exhibit 99.1

Unicycive Therapeutics Achieves Study Objective in Oxylanthanum Carbonate (OLC) Pivotal Clinical Trial
– Successfully Established Favorable Tolerability and Safety of OLC –
– New Drug Application (NDA) Submission Anticipated in Q3 2024 –
– Webcast and Conference Call Today at 8:30 A.M. ET –
LOS ALTOS, California, June 25, 2024 -- Unicycive Therapeutics, Inc. (Nasdaq: UNCY), a clinical-stage biotechnology company developing therapies for patients with kidney disease (the “Company or “Unicycive”), today announced positive results from the Oxylanthanum Carbonate (OLC) UNI-OLC-201 pivotal clinical trial with regard to both safety and tolerability endpoints. OLC is a next-generation lanthanum-based phosphate binding agent utilizing proprietary nanoparticle technology being developed for the treatment of hyperphosphatemia in patients with chronic kidney disease (CKD).
The study established promising tolerability of OLC at clinically effective doses in CKD patients on hemodialysis. In terms of tolerability, OLC had a low rate of discontinuation due to adverse events (AEs) with only 5/86 patients (6%) discontinuing from the Study. Of the 5 discontinuations, 3 were treatment-related and 2 were not related to treatment. Importantly, the Company believes the low discontinuation rate for OLC compares favorably to a discontinuation rate due to AEs of 14% for Fosrenol® from its U.S. Food and Drug Administration (FDA)-approved Package Insert.
The primary endpoint was defined as the rate of discontinuations due to treatment-related AEs leading to discontinuation in the maintenance period. In the UNI-OLC-201 trial, there was only 1 discontinuation due to a treatment-related AE in the Evaluable Population (n=71), a rate of 1.4%. In the full Safety Population, a total of 3 patients discontinued due to treatment-related AEs, a rate of 3.5%.
| Patient Population | n | Treatment-Related Discontinuations |
Percent | |||||
| Evaluable | 71 | 1 | 1.4 | % | ||||
| Safety | 86 | 3 | 3.5 | % | ||||
The secondary endpoint assessing safety was also favorable as most treatment-related AEs were mild to moderate in severity and there were no treatment-related serious adverse events (SAEs) reported in the Safety Population. The treatment-related AEs reported in ≥5% of patients were diarrhea (9%) and vomiting (6%) which also compares favorably to Fosrenol and other phosphate binders on the market.
While the study was not designed to evaluate efficacy, the trial enrolled patients on stable doses of approved hyperphosphatemia medications. At baseline approximately 59% of patients had phosphate levels ≤5.5 mg/dL, the level recommended by KDOQI guidelines. After washout from the prior phosphate binders, 90% of patients were able to achieve phosphate levels ≤5.5ng/dL at the end of titration with OLC.
“We are immensely pleased with the outcome of the UNI-OLC-201 clinical trial as the results demonstrate extremely promising tolerability results in real-world dialysis patients,” said Shalabh Gupta, MD, Chief Executive Officer of Unicycive. “The study was well received by investigators and patients as we were able to successfully over-enroll the study with 71 evaluable patients. In addition, we were able to obtain phosphate control in 90% of the Safety Population during titration and OLC proved to be highly potent at lower doses. We would like to thank our investigators, study coordinators, and the patients who dedicated their time and energy for our clinical trial.”
Dr. Gupta continued, “We believe these favorable results confirming tolerability for OLC are the final data component needed to support submission of a New Drug Application to the FDA utilizing the 505(b)(2) regulatory pathway. The submission package will also include the previously disclosed preclinical data and the data establishing bioequivalence to Fosrenol. The encouraging performance of OLC gives us a high degree of confidence and provides potential clinical validation of OLC’s best-in-class commercial promise for patients suffering from hyperphosphatemia.”
“Based on real world evidence, approximately 40% of patients on dialysis are unable to achieve adequate serum phosphate control as defined by established KDOQI treatment guidelines1. The UNI-OLC-201 study was representative of the U.S. dialysis patient population. Uncontrolled hyperphosphatemia is an important problem for patients and physicians because it can lead to other major complications including cardiovascular disease. I am encouraged by the results from the OLC pivotal trial, and I believe that a product like OLC that improves phosphate control and reduces the number of pills could have a meaningful impact on the overall care of CKD patients on dialysis,” added Pablo Pergola, MD, PhD, Research Director, Clinical Advancement Center, Renal Associates, P.A., and principal investigator for the UNI-OLC-201 trial
Conference Call & Webcast Details
Unicycive will host a webcast and conference call with accompanying slides today at 8:30 a.m. ET. The live and archived webcast may be accessed on the Unicycive website under the Investors section: Events and Presentations. The live call can be accessed by dialing +1 (646) 876-9923 with Meeting ID: 96518079674 and Passcode: 273069.
Presentation slides will be provided at the start of the conference call on the Unicycive website under the Investors section: Events and Presentations.
OLC-201 Data Summary
Overview
The results from the trial are focused on two patient populations: the full Safety Population and the Evaluable Population. The Safety Population (n=86) included all patients who entered titration and received at least one dose of OLC. The Evaluable Population (n=71) required a patient to have a serum phosphate level of ≤5.5 mg/dL at the end of titration and received at least one dose of OLC in the maintenance period.
Once patients were enrolled into the trial, they went through a washout period for two weeks to clear their current phosphate binder from the body. Participants were initially dosed at 500 mg of OLC three times a day (TID) and titrated to a clinically effective dose that is defined as the dose required to achieve a serum phosphate level of ≤5.5 mg/dL. The maximum dose of OLC tested was 3000 mg/day (1000 mg TID). As a reminder, all approved phosphate binders are administered on a dose titration schedule based on the control of serum phosphate. Once titrated to a clinically effective dose, patients were then treated in a maintenance period with OLC for four weeks to evaluate tolerability.
| 1 | KDOQI treatment guidelines |
Demographics and Enrollment Summary
In the study, 106 patients were enrolled, of which 86 patients entered titration and were followed as the Safety Population. Of the 86, 78 entered the maintenance period. Of the 78 patients that entered maintenance, 7 patients did not have phosphate control, leaving an Evaluable Population of 71 patients, exceeding the planned enrollment number of 60. Of the 86 patients, the trial enrolled 47 males and 39 females with a mean age of 62. Renvela® was the most prescribed phosphate binder for patients entering the study.
Primary Endpoint - Tolerability:
The objective of the OLC-201 trial was to evaluate the tolerability of clinically effective doses of OLC in CKD patients on dialysis. A clinically effective dose was established when a patient achieved a serum phosphate level ≤5.5 mg/dL. Tolerability was assessed based on the incidence of treatment-related AEs leading to discontinuation from the study in the maintenance period. In the OLC-201 trial, there was only 1 discontinuation due to a treatment-related AE in the Evaluable Population, a rate of 1.4%. In the Safety Population of 86 patients there were only 3 treatment-related discontinuations, a rate of 3.5%. In total, 5 patients discontinued due to AEs in the Safety Population, 3 were related to OLC and 2 were deemed unrelated to OLC.
Secondary Endpoint - Safety:
The secondary endpoint assessing safety was reported as the treatment-related AEs occurring in ≥5% of patients. The safety analysis covered all 86 patients in the Safety Population. Consistent with the AEs observed with other phosphate binders, the AEs were gastrointestinal related with diarrhea and vomiting being the most common at 9% and 6% respectively. There were no treatment-related serious adverse events (SAEs). Six patients experienced SAEs but those were deemed not related to OLC treatment. Most treatment-related AEs were mild to moderate in severity with only 2 AEs reported as severe.
Unicycive continues to assess the pharmacokinetics from this trial and those final data will be included in the NDA package.
Serum Phosphate Control
While the UNI-OLC-201 study was not designed to evaluate efficacy, the trial enrolled patients on stable doses of approved hyperphosphatemia medications. At baseline 59% of patients had phosphate levels ≤5.5 mg/dL, the level recommended by KDOQI guidelines. After washout from the prior phosphate binders, 90% of patients were able to achieve phosphate levels ≤5.5ng/dL at the end of titration with OLC. This includes the last serum phosphate levels from all patients including those that discontinued during titration: 77/86 (90%).
In addition, 69% of the 71 Evaluable Patients achieved a target serum phosphate level of ≤5.5 mg/dL at OLC doses of 1500 mg/day or lower.
About Oxylanthanum Carbonate (OLC)
Oxylanthanum carbonate is a next-generation lanthanum-based phosphate binding agent utilizing proprietary nanoparticle technology being developed for the treatment of hyperphosphatemia in patients with chronic kidney disease (CKD). OLC has over forty issued and granted patents globally. Its potential best-in-class profile may have meaningful patient adherence benefits over currently available treatment options as it requires a lower pill burden for patients in terms of number and size of pills per dose that are swallowed instead of chewed. Based on a survey conducted in 2022, Nephrologists stated that the greatest unmet need in the treatment of hyperphosphatemia with phosphate binders is a lower pill burden and better patient compliance.2 The global market opportunity for treating hyperphosphatemia is projected to be in excess of $2.5 billion in 2023, with the United States accounting for more than $1 billion of that total.3 Despite the availability of several FDA-cleared medications, 75 percent of U.S. dialysis patients fail to achieve the target phosphorus levels recommended by published medical guidelines.
Unicycive is seeking FDA approval of OLC via the 505(b)(2) regulatory pathway. As part of the clinical development program, two prior clinical studies were conducted in over 100 healthy volunteers. The first study was a dose-ranging Phase I study to determine safety and tolerability. The second study was a randomized, open-label, two-way crossover bioequivalence study to establish pharmacodynamic bioequivalence between OLC and Fosrenol. Based on the topline results of the bioequivalence study, pharmacodynamic (PD) bioequivalence of OLC to Fosrenol was established.
About Hyperphosphatemia
Hyperphosphatemia is a serious medical condition that occurs in nearly all patients with End Stage Renal Disease (ESRD). If left untreated, hyperphosphatemia leads to secondary hyperparathyroidism (SHPT), which then results in renal osteodystrophy (a condition similar to osteoporosis and associated with significant bone disease, fractures and bone pain); cardiovascular disease with associated hardening of arteries and atherosclerosis (due to deposition of excess calcium-phosphorus complexes in soft tissue). Importantly, hyperphosphatemia is independently associated with increased mortality for patients with chronic kidney disease on dialysis. Based on available clinical data to date, over 80% of patients show signs of cardiovascular calcification by the time they become dependent on dialysis.
| 2 | Reason Research, LLC 2022 company sponsored survey. Results here. |
| 3 | Fortune Business InsightsTM, Hyperphosphatemia Treatment Market, 2021-2028 |
Dialysis patients are already at an increased risk for cardiovascular disease (because of underlying diseases such as diabetes and hypertension), and hyperphosphatemia further exacerbates this. Treatment of hyperphosphatemia is aimed at lowering serum phosphate levels via two means: (1) restricting dietary phosphorus intake; and (2) using, on a daily basis, and with each meal, oral phosphate binding drugs that facilitate fecal elimination of dietary phosphate rather than its absorption from the gastrointestinal tract into the bloodstream.
About Unicycive Therapeutics
Unicycive Therapeutics is a biotechnology company developing novel treatments for kidney diseases. Unicycive’s lead drug candidate, oxylanthanum carbonate (OLC), is a novel investigational phosphate binding agent being developed for the treatment of hyperphosphatemia in chronic kidney disease patients on dialysis. UNI-494 is a patent-protected new chemical entity in clinical development for the treatment of conditions related to acute kidney injury. For more information, please visit Unicycive.com and follow us on LinkedIn, X, and YouTube.
Forward-looking statements
Certain statements in this press release are forward-looking within the meaning of the Private Securities Litigation Reform Act of 1995.
These statements may be identified using words such as “anticipate,” “believe,” “forecast,” “estimated”
and “intend” or other similar terms or expressions that concern Unicycive’s expectations, strategy, plans or intentions. These
forward-looking statements are based on Unicycive’s current expectations and actual results could differ materially. There are several
factors that could cause actual events to differ materially from those indicated by such forward-looking statements. These factors include,
but are not limited to, clinical trials involve a lengthy and expensive process with an uncertain outcome, and results of earlier studies
and trials may not be predictive of future trial results; our clinical trials may be suspended or discontinued due to unexpected side
effects or other safety risks that could preclude approval of our product candidates; risks related to business interruptions, which could
seriously harm our financial condition and increase our costs and expenses; dependence on key personnel; substantial competition; uncertainties
of patent protection and litigation; dependence upon third parties; and risks related to failure to obtain FDA clearances or approvals
and noncompliance with FDA regulations. Topline data from the Oxylanthanum carbonate (OLC) pivotal trial is preliminary and subject to
change based on further detailed analysis. Actual results may differ materially from those indicated by such forward-looking statements
as a result of various important factors, including: the uncertainties related to market conditions and other factors described more fully
in the section entitled ‘Risk Factors’ in Unicycive’s Annual Report on Form 10-K for the year ended December 31, 2023,
and other periodic reports filed with the Securities and Exchange Commission. Any forward-looking statements contained in this press release
speak only as of the date hereof, and Unicycive specifically disclaims any obligation to update any forward-looking statement, whether
as a result of new information, future events or otherwise.
Fosrenol® (lanthanum carbonate) is a registered trademark of Shire International Licensing BV.
Renvela® (sevelamer carbonate) is a registered trademark of Sanofi.
Investor Contact:
ir@unicycive.com
(650) 543-5470
SOURCE: Unicycive Therapeutics, Inc.
6
Exhibit 99.2

Oxylanthanum Carbonate (OLC) UNI - OLC - 201 Pivotal Trial Topline Data June 25, 2024 NASDAQ: UNCY

Forward Looking Statements This presentation contains certain “forward - looking” statements that are made pursuant to the safe harbor provisions of the Private Securities Litigation Reform Ac t of 1995. All statements, other than statements of historical or present facts, are forward - looking statements, including statements regarding our future financial condition, f uture revenues, projected costs, prospects, business strategy, and plans and objectives of management for future operations, including our plans for clinical trials and plans to submit for regulatory filings. In s ome cases, you can identify forward - looking statements by terminology such as “believe,” “will,” “may,” “might,” “estimate,” “continue,” “anticipate,” “intend,” “target,” “project,” “model,” “should,” “would,” “pla n,” “expect,” “predict,” “could,” “seek,” “goal,” “potential,” or the negative of these terms or other similar terms or expressions that concern our expectations, strategy, plans, or intentions. These statements are based on our intentions, beliefs, projections, outlook, analyses, or current expectations using currently available information, and are not guarantees of future performance, and involve certain risks and uncertainties. A lth ough we believe that the expectations reflected in these forward - looking statements are reasonable, we cannot assure you that our expectations will prove to be correct. Therefore, actual outcomes and results c oul d materially differ from what is expressed, implied, or forecasted in these statements. Any differences could be caused by a number of factors including but not limited to: our expectations regarding t he timing, costs, conduct, and outcome of our clinical trials, including statements regarding the timing of the initiation and availability of data from such trials; the timing and likelihood of regulatory fil ing s and approvals for our product candidates; whether regulatory authorities determine that additional trials or data are necessary in order to obtain approval; our ability to obtain funding for our operations, includ ing funding necessary to complete further development and commercialization of our product candidates; our plans to research, develop, and commercialize our product candidates; the commercialization of our product ca ndi dates, if approved; the rate and degree of market acceptance of our product candidates; our expectations regarding the potential market size and the size of the patient populations for our product cand ida tes, if approved for commercial use, and the potential market opportunities for commercializing our product candidates; the success of competing therapies that are or may become available; our expectations re garding our ability to obtain and maintain intellectual property protection for our product candidates; the ability to license additional intellectual property relating to our product candidates and to comply wit h our existing license agreements; our ability to maintain and establish relationships with third parties, such as contract research organizations, suppliers, and distributors; our ability to maintain and establish co lla borators with development, regulatory, and commercialization expertise; our ability to attract and retain key scientific or management personnel; our ability to grow our organization and increase the size of our fac ilities to meet our anticipated growth; the accuracy of our estimates regarding expenses, future revenue, capital requirements, and needs for additional financing; our expectations related to the use of ou r a vailable cash; our ability to develop, acquire, and advance product candidates into, and successfully complete, clinical trials; the initiation, timing, progress, and results of future preclinical studies and devel opm ents and projections relating to our competitors and our industry. Topline data from the Oxylanthanum carbonate (OLC) pivotal trial is preliminary and subject to change based on further detailed analysis. Additional factors that could cause actual results to differ materially from our expectations can be found in our Securities and Exchange Commission filings. Moreover, we operate in a very competitive and rapidly changing environment. New risk factors emerge from time to time, and it is not possible for our management to predict all ris k f actors, nor can we assess the effects of all factors on our business or the extent to which any factor, or combination of factors, may cause actual results to differ materially from those contained in, or implie d b y, any forward - looking statements. All forward - looking statements included in this presentation are expressly qualified in their entirety by these cautionary statements. The forward - looking statements speak only as of the date made and, other than as required by law, we undertake no obligation to publicly update or revise any forward - looking statements, whether as a result of new information, future events, or otherwise . The company obtained the industry, market and competitive position data used throughout this presentation from its own intern al estimates and research, as well as from industry and general publications, and research, surveys and studies conducted by third parties. Internal estimates are derived from publicly available information rel eased by industry analysts and third - party sources, the company’s internal research and our industry experience, and are based on assumptions made by the company based on such data and its knowledge of the ind ust ry and market, which the company believes to be reasonable. In addition, while the company believes the industry, market and competitive position data included in this presentation is reliable and b ase d on reasonable assumptions, the company has not independently verified any third - party information, and all such data involve risks and uncertainties and are subject to change based on various factors. This presentation shall not constitute an offer to sell or the solicitation of an offer to buy, nor shall there be any sale o f t hese securities in any state or other jurisdiction in which such offer, solicitation or sale would be unlawful prior to registration or qualification under the securities laws of any such state or other jurisdiction. 2 2

Pivotal Clinical Trial (UNI - OLC - 201) 3 Study Objective Achieved ▪ Demonstrated tolerability and safety of OLC in CKD patients on hemodialysis Tolerability / Safety ▪ Total discontinuations due to adverse events were 5/86 (6%) ▪ 3 treatment - related, 2 not related to treatment ▪ For reference*, Fosrenol ® FDA package insert lists AE discontinuation rate at 14% ▪ Treatment related adverse events reported in ≥ 5% of patients were diarrhea (9%) and vomiting (6%) ▪ OLC safety profile compares favorably to Fosrenol and other phosphate binders on the market Serum Phosphate Control ▪ 90% of OLC treated patients achieved effective serum phosphate control at the end of titration (n=86) ▪ 69% of patients controlled at OLC dose of ≤1,500 mg/day (n=71) Implications ▪ We believe that these results for OLC will support the demonstration of similarity to Fosrenol with regard to tolerability an d safety required for our 505(b)(2) NDA filing ▪ NDA filing next quarter (Q3, 2024) *UNI - OLC - 201 was not a comparator trial

Oxylanthanum carbonate (OLC) is an unapproved investigational new drug being developed under FDA’s 505(b)(2) regulatory pathway. If approved, OLC will share substantially the same product label and prescribing information as the reference - listed drug (RLD) Fosrenol (lanthanum carbonate) with the exception that OLC tablets are smaller in size and swallowed whole with water and not chewed OLC Background

Oxylanthanum Carbonate (OLC) Product Profile Overview ▪ Potential best - in - class product being developed under FDA’s 505(b)(2) regulatory pathway for the treatment of hyperphosphatemia ▪ If approved, OLC is expected to share substantially the same product label and prescribing information as the reference - listed drug Fosrenol (lanthanum carbonate) ▪ OLC advantages: (1) Potency : shares high phosphate binding capacity of lanthanum , (2) Pill Burden: smaller and fewer pills, ( 3 ) Palatability: swallowed whole with water and not chewed ▪ Proprietary Nanoparticle Technology ▪ UNICYCIVE has harnessed the phosphate binding potency of lanthanum to reduce the number and size of pills that patients must take to control hyperphosphatemia • Enhanced surface area • Lower molecular weight • Immediate release tablets Strong Global Intellectual Property ▪ Enables smaller pills ▪ Pills are swallowed (not chewed) 5

6RXUFH)'$DSSURYHGSDFNDJHLQVHUWV3LOOYROXPHV'DWDRQȅOH8QLF\FLYH7KHUDSHXWLFV3URGXFWLPDJHVDUHSURSRUWLRQDOO\V L]H G 5HQYHOD n LVDUHJLVWHUHGWUDGHPDUNRI6DQRȅ$XU\[LD n LVDUHJLVWHUHGWUDGHPDUNRI$NHELD7KHUDSHXWLFV_)RVUHQRO n LVDWUDGHPDUNRI 7DNHGD3KDUPDFHXWLFDO&RPSDQ\/LPLWHG3KRVOR n DQG9HOSKRUR n DUHUHJLVWHUHGWUDGHPDUNVRI9LIRU)UHVHQLXV Renvela ® sevelamer carbonate 800 mg 2 tablets 3 times per day, swallowed Volume: 6.5 cm 3 Phoslo ® calcium acetate 667 mg 2 tablets 3 times per day, swallowed Volume: 6.8 cm 3 Auryxia ® ferric citrate 210 mg 2 tablets 3 times per day, swallowed Volume: 5.5 cm 3 Velphoro ® sucroferric oxyhydroxide 500 mg 1 tablet three times per day, chewed Volume: 5.5 cm 3 Fosrenol ® lanthanum carbonate 500 mg 1 tablet 3 times per day, chewed Volume: 4.0 cm 3 Recommended Daily Starting Dose for Phosphate Binders MOST PRESCRIBED 2[\ODQWKDQXP&DUERQDWH2/& PJ WDEOHW WLPHVSHUGD\VZDOORZHG 9ROXPH FP 6 * Expected OLC recommended daily starting dose, if approved

Dialysis Patients Experience Excessive Pill Burden: Phosphate Binders Account for Half of the Problem The daily pill burden for maintenance dialysis patients is among the highest across various chronic disease states including HIV/AIDS, diabetes mellitus, and congestive heart failure 1 • 19 pills per day (median) • 49% of pill burden from phosphate binders • Higher pill burden was independently associated with lower quality of life scores (HR - QOL) • 62% of patients are non - adherent (self - reported) 7 1 Chiu YW, et al. Clin J Am Soc Nephrol. 2009

ZĞŐƵůĂƚŽƌLJWĂƚŚǁĂLJ

Studies to Support OLC NDA Filing 9 Demonstrate similar Efficacy to reference drug (Fosrenol) Demonstrate similar Safety / Tolerability to reference drug (Fosrenol) Bridge to Efficacy Bridge to Safety/Tolerability Bridge to Safety/Tolerability ŝŽĞƋƵŝǀĂůĞŶĐĞ ŽŶĨŝƌŵĞĚ Some quantitative variability observed – additional study needed Favorable Tolerability Profile Confirmed 2/&YV)RVUHQRO n 3KDUPDFRG\QDPLF %LRHTXLYDOHQFH7ULDO LQ+HDOWK\9ROXQWHHUV 1 2/&YV)RVUHQRO n 3UH &OLQLFDO6WXG\ 2 3LYRWDO&OLQLFDO 7ROHUDELOLW\([SRVXUH 7ULDOLQ'LDO\VLV 3DWLHQWV 3 505(b)(2) Requirement ^ƚƵĚŝĞƐŽŶĚƵĐƚĞĚΎ * All study designs agreed upon with FDA &RPSOHWHG Completed 6/2024 &RPSOHWHG

Study Overview UNI - OLC - 201 Topline data from the Oxylanthanum carbonate (OLC) pivotal trial is preliminary and subject to change based on further detailed analysis.

11 Secondary Endpoints Primary Endpoint 1) Safety: Safety is assessed based on the incidence of treatment - related adverse events 2) Pharmacokinetics: Pharmacokinetics of OLC Tolerability: Tolerability is assessed based on the incidence of treatment - related adverse events leading to discontinuation from the study in the Evaluable Population ^ƚƵĚLJKďũĞĐƚŝǀĞĂŶĚŶĚƉŽŝŶƚƐ Study Objective: To evaluate the tolerability of clinically effective doses (serum phosphate ≤5.5 mg/dL) of OLC in CKD patients on dialysis

12 WŝǀŽƚĂůůŝŶŝĐĂůdƌŝĂůĞƐŝŐŶ • Washout Period: Designed to clear existing binders from the circulation and ensure that serum phosphate was above 5.5 mg/dL • Washout Failure: Those who had phospha te levels ≤ 5.5 mg/dL were considered wash out failures and excluded from the study. • Titration: First 2 weeks of titration, all patients received 500 mg three times a day (TID) with meals. OLC was titrated every 2 weeks based on serum phosphate levels, up and down titration was allowed to a maximum dose 1000 mg TID. • Evaluable Patients: Patients with serum phosphate levels ≤5.5 mg/dL and received at least one dose of OLC during maintenance. 4 Weeks Up to 6 Weeks Up to 3 Weeks Up to 4 Weeks OLC Maintenance OLC Titration Washout 6FUHHQLQJ ≤5.5 Evaluable Population for Primary Endpoint ≤5.5 >5.5 to ≤10 ≥4 to ≤7.5 Target Serum Phosphate Level (mg/dL) Maximum Dose 1000 mg TID Maximum Dose 1000 mg TID Starting Dose 500 mg TID Design, endpoints and sample size (n=60 evaluable) agreed on with the FDA

Key Inclusion Criteria 1. Patient must be ≥18 years of age. 2. Patient has stable chronic kidney disease and is undergoing and compliant with hemodialysis treatment 3 times per week for at least 12 weeks prior to screening 3. Patient has serum phosphate levels of ≥ 4.0 mg/dL and ≤7.5 mg/dL on phosphate binders fo r at least 8 weeks prior to screening 4. Patients with a Kt/V of ≥1.2 during the most recent monthly evaluation Key Exclusion Criteria 1. Patient has had prior treatment with lanthanum - based binder (i.e., Fosrenol) within the past 6 months. 13 Key Eligibility Criteria
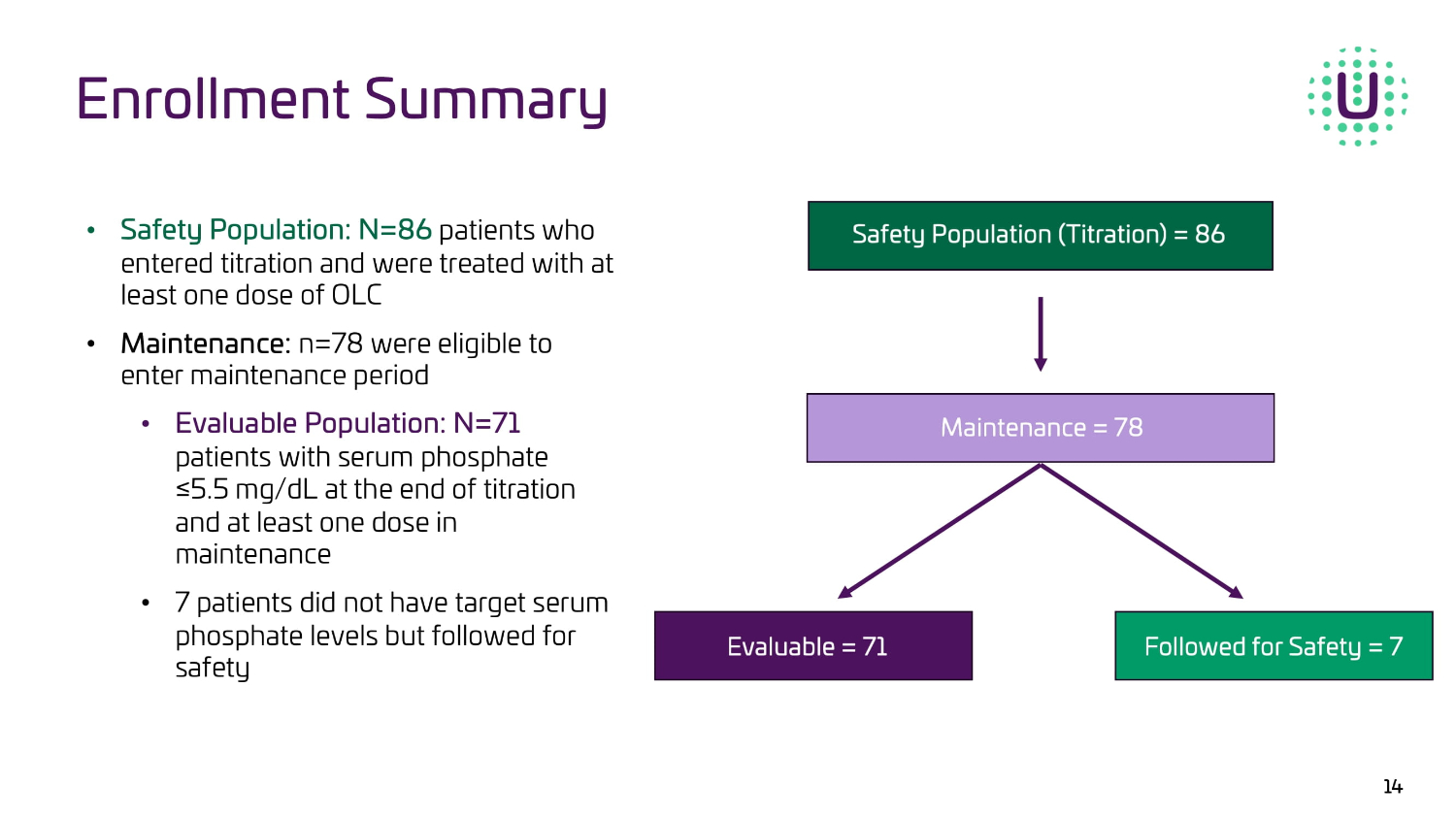
Enrollment Summary 14 ‡ ^ĂĨĞƚLJWŽƉƵůĂƚŝŽŶ͗Eс ϴϲ SDWLHQWVZKR HQWHUHGWLWUDWLRQDQGZHUHWUHDWHGZLWKDW OHDVWRQHGRVHRI2/& ‡ DĂŝŶƚĞŶĂŶĐĞ͗ Q ZHUHHOLJLEOHWRHQWHU PDLQWHQDQFHSHULRG ‡ ǀĂůƵĂďůĞWŽƉƵůĂƚŝŽŶ͗Eс ϳϭ SDWLHQWV ZLWKVHUXPSKRVSKDWH Ǫ PJG/DWWKHHQGRIWLWUDWLRQ DQGDWOHDVWRQHGRVHLQ PDLQWHQDQFH ‡ SDWLHQWVGLGQRWKDYHWDUJHWVHUXP SKRVSKDWHOHYHOVEXWIROORZHGIRU VDIHW\

15 OLC (N=86) n (%) Patient Demographics 62.4 (10.7) Age (years) mean (SD) 39 (45.3) 47 (54.7) Gender Female Male 57 (66.3) 18 (20.9) 8 (9.3) 2 (2.3) 1 (1.2) Ethnicity Caucasian African American American Indian or Alaska Native Hispanic Asian Prior Phosphate Binder and Patient Demographics n (%) Prior Phosphate Binder 43 (50%) 17 (20%) 13 (15% 12 (14%) 1 (1%) Renvela (sevelamer carbonate) Phoslo (calcium acetate) Auryxia (ferric citrate) Velphoro (sucroferric oxyhydroxide) Other

Results: Primary & Secondary Endpoints UNI - OLC - 201

17 Rate of Discontinuation Due to Treatment - Related Adverse Events during the Maintenance Period (Evaluable Patients) Primary Endpoint: Tolerability Percent Treatment - Related Discontinuations n WĂƚŝĞŶƚWŽƉƵůĂƚŝŽŶ 1 . 4% 1 71 Evaluable 3 . 5% 3 86 Safety Total discontinuation due to AEs was 5/86 patients (6%)

18 Primary Endpoint: Tolerability ● OLC - 201 is an open - label/non - comparator study design that was agreed upon with the FDA ● We believe that the low rate of OLC treatment - related discontinuations observed in this clinical trial compare favorably to the historical data that was submitted in the original Fosrenol NDA ○ Rate of discontinuations due to adverse events (treatment - related and non - treatment related) from Fosrenol Package Insert was 14% ○ OLC rate of discontinuations due to adverse events (treatment - related and non - treatment related) was 6% We believe that these results for OLC are sufficient to support the demonstration of similarity to Fosrenol with regard to tolerability required for our 505(b)(2) NDA filing

19 Secondary Endpoints: Safety & PK (N=86) n (%) Adverse Event 8 (9%) a Diarrhea 5 (6%) a Vomiting Treatment - Related Adverse Events in ≥5% Patients a) Two patients experienced both diarrhea and vomiting Safety • No treatment - related Serious Adverse Events (SAEs) • 6 patients had non - treatment - related SAEs • Most AEs were mild - to - moderate; only 2 patients with severe treatment - related AEs Pharmacokinetics • PK assessment is ongoing and will be submitted in the NDA • Prior Phase 1 clinical trial completed showed PK in healthy volunteers similar to Fosrenol

Adverse Event (AE) Profiles of Phosphate Lowering Therapies from FDA - Approved Product Labels Disclaimer: FDA cautions that because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in c lin ical trials of a drug cannot to directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in pr act ice. * 20 We believe that the AE profile observed in the OLC pivotal trial compares favorably with the historical clinical experience with Fosrenol and other phosphate binders and supports a similar safety profile required for our 505(b)(2) NDA filing

Serum Phosphate Control UNI - OLC - 201

Relative phosphate - binding coefficient per gram of binder (calcium carbonate with an index value of 1.0) Relative urinary phosphate - lowering effect 2.0 1.9 1.7 1.5 1.3 1.0 1.0 0.75 0.0 0.5 1.0 1.5 2.0 2.5 Renal failure rat model (5/6 nephrectomy). All binders dosed at 1000 mg/kg/day. Index Value Damment S. Renal Failure. 2011;33(2):217 – 224. 1 Carbonate, 2 hydroxide, 3 hydrate, 4 acetate. Daugirdas JT, et al. Seminars in Dialysis . 2011;24(1):41 – 49. Lanthanum Literature Reported Relative Phosphate Binder Potency 22
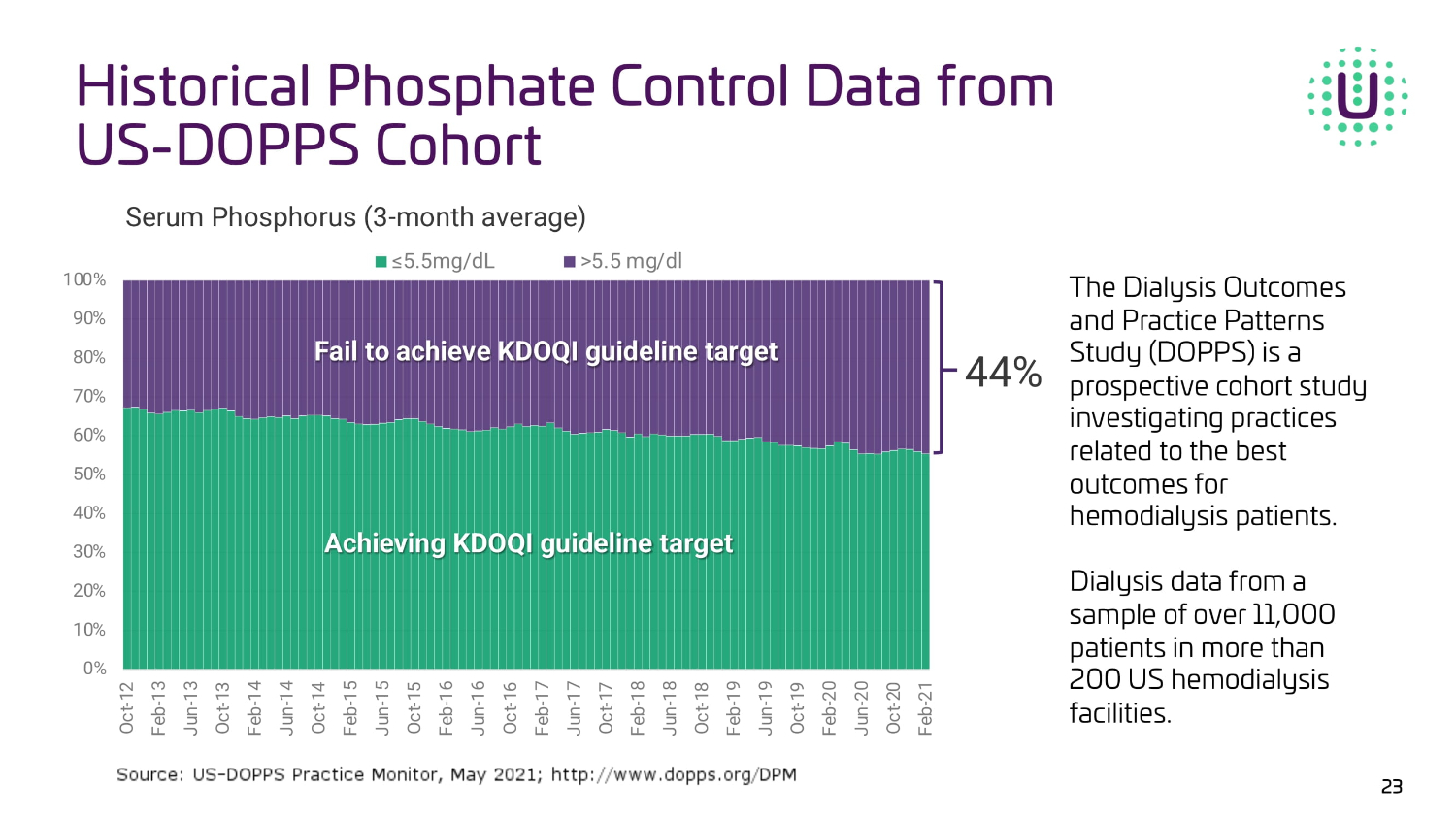
0% 10% 20% 30% 40% 50% 60% 70% 80% 90% 100% Oct-12 Feb-13 Jun-13 Oct-13 Feb-14 Jun-14 Oct-14 Feb-15 Jun-15 Oct-15 Feb-16 Jun-16 Oct-16 Feb-17 Jun-17 Oct-17 Feb-18 Jun-18 Oct-18 Feb-19 Jun-19 Oct-19 Feb-20 Jun-20 Oct-20 Feb-21 ≤5.5mg/dL >5.5 mg/dl Serum Phosphorus (3 - month average) |||| || ||||||| ||||| ||||||||| |||||| 44% ||||||||| ||||| ||||||||| |||||| The Dialysis Outcomes and Practice Patterns Study (DOPPS) is a prospective cohort study investigating practices related to the best outcomes for hemodialysis patients. Dialysis data from a sample of over 11,000 patients in more than 200 US hemodialysis facilities. Historical Phosphate Control Data from US - DOPPS Cohort 23

Phosphate Control in Safety Population (N=86) in UNI - OLC - 201 24 ≤5.5 mg/dL >5.5 mg/dL 59% 41% 10% 90% Baseline End of Titration 0 20 40 60 80 100 P e r c e n t o f P a t i e n t s Baseline - Serum phosphate levels at screening before washout End of Titration – includes last serum phosphate levels from all patients including those that discontinued during titration 77/ 86 (90%) / 9/86 (10%) Prior Phosphate Binder OLC Treatment

Of the 71 evaluable patients, 69% achieved a target serum phosphate level of ≤5.5 mg/dL at an OLC dose of ≤1500 mg/day 500 1000 1500 2000 2500 3000 0 20 40 60 Dose (mg/day) P e r c e n t o f P a t i e n t s 1% 14% 54% 6% 9% 17% End of Titration (mg/day) 69% 25 Phosphate Control in Evaluable Population (n=71) in UNI - OLC - 201

OLC Data Results Support NDA Filing Achieved the Study Objective demonstrating tolerability and safety of OLC Primary Endpoint: Tolerability ▪ Treatment - related discontinuation rate in Evaluable Population of 1.4% (1/71) ▪ Total discontinuations due to adverse events were 5/86 (6%) ▪ 3 treatment - related, 2 non - treatment related ▪ For reference, discontinuation rate due to AEs from Fosrenol FDA package insert is 14% Secondary Endpoint: Safety ▪ Diarrhea (9%) and vomiting (6%) occurred in ≥ 5% of patients; there were no treatment - related SAEs ▪ OLC safety profile compares favorably to Fosrenol and other phosphate binders on the market Serum Phosphate Control ▪ OLC demonstrated 90% serum phosphate control during titration in the Safety P opulation ▪ At the end of titration, 69% achieved target levels at an OLC dose of ≤1500 mg/day 26 These encouraging results in hemodialysis patients validate our confidence in OLC’s best - in - class potential and as an important new treatment option for patients with hyperphosphatemia, if approved.

Anticipated Milestones

OLC Expected Catalysts x Successful bioequivalence study in healthy volunteers x FDA alignment on regulatory path x Completed enrollment in pivotal clinical trial x Oral and Poster presentations at NKF & ERA (Q2 ‘24) x Pivotal trial readout (Q2 ‘24) □ NDA Filing (Q3 ‘24) □ NDA Acceptance & PDUFA date designated by FDA □ Buildout of commercial infrastructure □ FDA Approval (mid - year ‘25) □ TDAPA Designation 28

T: (650) 900 - 5470 ir@unicycive.com Investor Relations