UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM 8-K
CURRENT REPORT
Pursuant to Section 13 or 15(d)
of the Securities Exchange Act of 1934
Date of report (Date of earliest event reported): May 15, 2025
Invivyd, Inc.
(Exact Name of Registrant as Specified in its Charter)
| Delaware | 001-40703 | 85-1403134 | ||
| (State or Other Jurisdiction of Incorporation) |
(Commission File Number) |
(IRS Employer Identification No.) |
| 1601 Trapelo Road, Suite 178 Waltham, MA |
02451 | |
| (Address of Principal Executive Offices) | (Zip Code) |
Registrant’s telephone number, including area code: (781) 819-0080
Not applicable
(Former Name or Former Address, if Changed Since Last Report)
Check the appropriate box below if the Form 8-K filing is intended to simultaneously satisfy the filing obligation of the registrant under any of the following provisions:
| ☐ | Written communications pursuant to Rule 425 under the Securities Act (17 CFR 230.425) |
| ☐ | Soliciting material pursuant to Rule 14a-12 under the Exchange Act (17 CFR 240.14a-12) |
| ☐ | Pre-commencement communications pursuant to Rule 14d-2(b) under the Exchange Act (17 CFR 240.14d-2(b)) |
| ☐ | Pre-commencement communications pursuant to Rule 13e-4(c) under the Exchange Act (17 CFR 240.13e-4(c)) |
Securities registered pursuant to Section 12(b) of the Act:
| Title of each class |
Trading |
Name of each exchange on which registered |
||
| Common stock, par value $0.0001 per share | IVVD | The Nasdaq Stock Market LLC |
Indicate by check mark whether the registrant is an emerging growth company as defined in Rule 405 of the Securities Act of 1933 (§230.405 of this chapter) or Rule 12b-2 of the Securities Exchange Act of 1934 (§240.12b-2 of this chapter).
Emerging growth company ☒
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
| Item 2.02. | Results of Operations and Financial Condition. |
On May 15, 2025, Invivyd, Inc. (the “Company”) issued a press release announcing its financial results for the quarter ended March 31, 2025, and recent business highlights. A copy of the press release is furnished as Exhibit 99.1 to this Current Report on Form 8-K and is incorporated by reference into this Item 2.02.
The information furnished pursuant to this Item 2.02, including Exhibit 99.1, shall not be deemed to be “filed” for purposes of Section 18 of the Securities Exchange Act of 1934, as amended (the “Exchange Act”), or otherwise subject to the liabilities of that Section, nor shall it be deemed to be incorporated by reference into any of the Company’s filings with the Securities and Exchange Commission under the Exchange Act or the Securities Act of 1933, as amended, whether made before or after the date hereof, regardless of any general incorporation language in such a filing, except as expressly set forth by specific reference in such a filing.
| Item 8.01. | Other Events. |
On May 15, 2025, the Company posted an updated corporate presentation on its website at www.invivyd.com. A copy of the presentation is filed as Exhibit 99.2 to this Current Report on Form 8-K and is incorporated by reference into this Item 8.01.
| Item 9.01. | Financial Statements and Exhibits. |
(d) Exhibits
| Exhibit No. |
Description |
|
| 99.1 | Press Release, dated May 15, 2025 | |
| 99.2 | Corporate Presentation, dated May 15, 2025 | |
| 104 | Cover Page Interactive Data File (embedded within the Inline XBRL document) | |
SIGNATURES
Pursuant to the requirements of the Securities Exchange Act of 1934, as amended, the registrant has duly caused this report to be signed on its behalf by the undersigned hereunto duly authorized.
| INVIVYD, INC. | ||||||
| Date: May 15, 2025 | By: | /s/ Jill Andersen |
||||
| Jill Andersen | ||||||
| Chief Legal Officer and Corporate Secretary | ||||||
Exhibit 99.1

Invivyd Reports First Quarter 2025 Financial Results and Recent Business Highlights
| • | PEMGARDA™ (pemivibart) net product revenue of $11.3 million reported for Q1 2025, influenced by planned transition (Jan/Feb) from a contracted to an internalized sales force |
| • | PEMGARDA revenue re-acceleration observed in Q2 2025 to date |
| • | Invivyd continues to target near-term profitability (1H 2025) with existing cash and cash equivalents, anticipated growth of net product revenue, and continued reduction of operating expenses |
| • | Since Emergency Use Authorization (EUA) of PEMGARDA in March 2024, no documented cases of anaphylaxis reported, across thousands of post-authorization doses |
| • | VYD2311 Phase 1 clinical trial data read-out anticipated later in Q2 2025 |
WALTHAM, Mass., May 15, 2025 – Invivyd, Inc. (Nasdaq: IVVD) today announced financial results for the quarter ended March 31, 2025, and provided recent business highlights.
“To drive long-term topline growth, we made a strategic decision to internalize our sales force at the beginning of 2025,” said Bill Duke, Chief Financial Officer of Invivyd. “Although this shift created a short-term headwind, we’re now seeing positive momentum with a return to growth and early signs of acceleration in Q2 2025. Backed by a strong cash position and potential to access up to $30 million in non-dilutive funding through our term loan facility, we remain focused on disciplined financial execution and continue to target profitability by the end of the first half of 2025.”
Recent Business Highlights
| • | Commercial Execution |
| • | PEMGARDA™ (pemivibart) uptake continues to grow among healthcare providers caring for immunocompromised patients, supported by Invivyd’s in-house sales force and expanded field presence across key specialties. |
| • | Ongoing Variant Coverage and Safety Profile of PEMGARDA |
| • | In vitro neutralization data show sustained neutralizing activity of PEMGARDA against currently dominant SARS-CoV-2 variants, including LP.8.1 and XEC, consistent with expectations based on the observed stability of PEMGARDA target epitope and prior variant surveillance. LP.8.1 and XEC represent more than 75% of SARS-CoV-2 variants circulating in the U.S., according to the Centers for Disease Control. |
| • | PEMGARDA safety profile remains consistent with the Fact Sheet for Healthcare Providers; no additional documented cases of anaphylaxis reported since emergency use authorization (EUA) in March 2024. |
| • | Regulatory Developments |
| • | In February 2025, the U.S. Food and Drug Administration (FDA) declined Invivyd’s request to expand the existing EUA of PEMGARDA to include treatment of mild-to-moderate COVID-19 for certain immunocompromised patients who have no alternative therapeutic options. The FDA declination letter provides reasoning that may provide a near-term pathway for VYD2311. |
| • | Pipeline Expansion |
| • | Invivyd has initiated discovery efforts to assess pipeline expansion beyond SARS-CoV-2, including potential targets such as respiratory syncytial virus (RSV) and measles |
| • | These evaluations are focused on high-value unmet needs in which a best- in class or first-and-best in class antibody may offer an attractive alternative or complement to traditional vaccines, or a high-value treatment. |
| • | Corporate and Financial Updates |
| • | In April 2025, Invivyd secured a $30 million non-dilutive term loan facility with Silicon Valley Bank, a division of First Citizens Bank, supporting balance sheet optionality and providing potential additional runway for commercial and pipeline execution if certain conditions and milestones are met. |
Recent Pipeline Highlights
| • | VYD2311 Phase 1 clinical trial data read-out, including potency, half-life and full safety unblinding anticipated later in Q2 2025. |
First Quarter 2025 Financial Results:
| • | Revenue: Reported Q1 2025 PEMGARDA net product revenue of $11.3 million, as compared to $13.8 million in Q4 2024. There were no revenues reported during Q1 2024. |
| • | Cash Position: Cash and cash equivalents were $48.1 million as of March 31, 2025. |
| • | Research & Development (R&D) Expenses (including In-Process R&D): R&D expenses were $10.6 million for the quarter ended March 31, 2025, compared to $31.2 million for the comparable period of 2024. This decrease is primarily attributable to a decrease in commercial manufacturing costs of PEMGARDA, a decrease in clinical trial costs related to our CANOPY Phase 3 clinical trial and a decrease in personnel-related costs. |
| • | Selling, General & Administrative (SG&A) Expenses: SG&A expenses were $16.8 million for the quarter ended March 31, 2025, compared to $14.9 million for the comparable period of 2024. This increase is primarily attributable to sales and marketing costs related to PEMGARDA. |
| • | Net Loss and Net Loss per Share: Net loss was $16.3 million for the quarter ended March 31, 2025, compared to $43.5 million for the comparable period in 2024. Basic and diluted net loss per share was $0.14 for the quarter ended March 31, 2025, compared to $0.38 for the comparable period in 2024. |
Conference Call & Webcast
Listeners can register for the webcast via this link. Analysts wishing to participate in the question and answer session should use this link. A replay of the webcast will be available via the company’s investor website approximately two hours after the call’s conclusion. Those who plan on participating are advised to join 15 minutes prior to the start time.
About PEMGARDA
PEMGARDA™ (pemivibart) is a half-life extended investigational monoclonal antibody (mAb). PEMGARDA was engineered from adintrevimab, Invivyd’s investigational mAb that has a robust safety data package and provided evidence of clinical efficacy in global Phase 2/3 clinical trials for the prevention and treatment of COVID-19. PEMGARDA has demonstrated in vitro neutralizing activity against major SARS-CoV-2 variants, including JN.1, KP.3.1.1, XEC and LP.8.1. PEMGARDA targets the SARS-CoV-2 spike protein receptor binding domain (RBD), thereby inhibiting virus attachment to the human ACE2 receptor on host cells.
PEMGARDA (pemivibart) injection (4500 mg), for intravenous use is an investigational mAb that has not been approved, but has been authorized for emergency use by the U.S. FDA under an EUA for the pre-exposure prophylaxis (prevention) of COVID-19 in adults and adolescents (12 years of age and older weighing at least 40 kg) who have moderate-to-severe immune compromise due to certain medical conditions or receipt of certain immunosuppressive medications or treatments and are unlikely to mount an adequate immune response to COVID-19 vaccination. Recipients should not be currently infected with or have had a known recent exposure to an individual infected with SARS-CoV-2.
PEMGARDA is not authorized for use for treatment of COVID-19 or post-exposure prophylaxis of COVID-19. Pre-exposure prophylaxis with PEMGARDA is not a substitute for vaccination in individuals for whom COVID-19 vaccination is recommended. Individuals for whom COVID-19 vaccination is recommended, including individuals with moderate-to-severe immune compromise who may derive benefit from COVID-19 vaccinations, should receive COVID-19 vaccination. In individuals who have recently received a COVID-19 vaccine, PEMGARDA should be administered at least 2 weeks after vaccination.
Anaphylaxis has been observed with PEMGARDA and the PEMGARDA Fact Sheet for Healthcare Providers includes a boxed warning for anaphylaxis. The most common adverse reactions included systemic infusion-related reactions and hypersensitivity reactions, local infusion site reactions, and infusion site infiltration or extravasation. For additional information, please see the PEMGARDA full product Fact Sheet for Healthcare Providers, including important safety information and boxed warning.
To support the EUA for PEMGARDA, an immunobridging approach was used to determine if PEMGARDA may be effective for pre-exposure prophylaxis of COVID-19. Immunobridging is based on the serum virus neutralizing titer-efficacy relationships identified with other neutralizing human mAbs against SARS-CoV-2. This includes adintrevimab, the parent mAb of pemivibart, and other mAbs that were previously authorized for EUA. There are limitations of the data supporting the benefits of PEMGARDA. Evidence of clinical efficacy for other neutralizing human mAbs against SARS-CoV-2 was based on different populations and SARS-CoV-2 variants that are no longer circulating. Further, the variability associated with cell-based EC50 value determinations, along with limitations related to pharmacokinetic data and efficacy estimates for the mAbs in prior clinical trials, impact the ability to precisely estimate protective titer ranges. Additionally, certain SARS-CoV-2 viral variants may emerge that have substantially reduced susceptibility to PEMGARDA, and PEMGARDA may not be effective at preventing COVID-19 caused by these SARS-CoV-2 viral variants.
The emergency use of PEMGARDA is only authorized for the duration of the declaration that circumstances exist justifying the authorization of the emergency use of drugs and biological products during the COVID-19 pandemic under Section 564(b)(1) of the Federal Food, Drug, and Cosmetic Act, 21 U.S.C. § 360bbb-3(b)(1), unless the declaration is terminated or authorization revoked sooner. PEMGARDA is authorized for use only when the combined national frequency of variants with substantially reduced susceptibility to PEMGARDA is less than or equal to 90%, based on available information including variant susceptibility to PEMGARDA and national variant frequencies.
About CANOPY
The CANOPY Phase 3 clinical trial was designed to evaluate the safety and tolerability of pemivibart and to assess immunobridging from pemivibart to certain historical data from the company’s previous Phase 2/3 clinical trial of adintrevimab (ADG20) for the prevention of symptomatic COVID-19 (EVADE). Additionally, there were pre-specified exploratory endpoints through three, six and twelve months to evaluate clinical efficacy of pemivibart compared to placebo in the prevention of RT-PCR-confirmed symptomatic COVID-19. The latest analysis from the Phase 3 CANOPY clinical trial included 365-day data. The CANOPY clinical trial enrolled participants in two cohorts: Cohort A was a single-arm, open-label trial in adults with moderate-to-severe immune compromise including complex underlying medical conditions. Cohort B was a randomized, placebo-controlled cohort that enrolled adults without moderate-to-severe immune compromise at risk of acquiring COVID-19 due to regular unmasked face-to-face interactions in indoor settings.
About VYD2311
VYD2311 is a novel monoclonal antibody (mAb) candidate being developed for COVID-19 to continue to address the urgent need for new prophylactic and therapeutic options. The pharmacokinetic profile and antiviral potency of VYD2311 may offer the ability to deliver clinically meaningful titer levels through more patient-friendly means such as an intramuscular route of administration.
VYD2311 was engineered using Invivyd’s proprietary integrated technology platform and is the product of serial molecular evolution designed to generate an antibody optimized for neutralizing contemporary virus lineages. VYD2311 leverages the same antibody backbone as pemivibart, Invivyd’s investigational mAb granted emergency use authorization in the U.S. for the pre-exposure prophylaxis (PrEP) of symptomatic COVID-19 in certain immunocompromised patients, and adintrevimab, Invivyd’s investigational mAb that has a robust safety data package and demonstrated clinically meaningful results in global Phase 2/3 clinical trials for the prevention and treatment of COVID-19.
About Invivyd
Invivyd, Inc. (Nasdaq: IVVD) is a biopharmaceutical company devoted to delivering protection from serious viral infectious diseases, beginning with SARS-CoV-2. Invivyd deploys a proprietary integrated technology platform unique in the industry designed to assess, monitor, develop, and adapt to create best in class antibodies. In March 2024, Invivyd received emergency use authorization (EUA) from the U.S. FDA for a monoclonal antibody (mAb) in its pipeline of innovative antibody candidates. Visit https://invivyd.com/ to learn more.
Cautionary Note Regarding Forward-Looking Statements
This press release contains forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995. Words such as “anticipates,” “believes,” “could,” “expects,” “estimates,” “intends,” “potential,” “predicts,” “projects,” and “future” or similar expressions (as well as other words or expressions referencing future events, conditions or circumstances) are intended to identify forward-looking statements. Forward-looking statements include statements concerning, among other things, the company’s goal of near-term profitability; the company’s expectations regarding anticipated growth of net product revenue and continued reduction of operating expenses; expectations related to the company’s term loan facility; the company’s ongoing research and development activities, as well as future potential research and development efforts; the ongoing in vitro neutralizing activity of PEMGARDA against dominant SARS-CoV-2 variants; the potential of PEMGARDA as a mAb for pre-exposure prophylaxis (prevention) of COVID-19 in certain adults and adolescents who have moderate-to-severe immune compromise; the potential of VYD2311 as a novel mAb candidate that may be able to deliver clinically meaningful titer levels through more patient-friendly means, and potentially available regulatory pathways; the company’s devotion to delivering protection from serious viral infectious diseases, beginning with SARS-CoV-2; potential pipeline expansion beyond SARS-CoV-2, including potential targets such as RSV and measles; and other statements that are not historical fact. The company may not actually achieve the plans, intentions or expectations disclosed in the company’s forward-looking statements and you should not place undue reliance on the company’s forward-looking statements.
These forward-looking statements involve risks and uncertainties that could cause the company’s actual results to differ materially from the results described in or implied by the forward-looking statements, including, without limitation: uncertainties regarding the company’s expectations, projections and estimates regarding future costs and expenses, future revenue, capital requirements, and the availability of and the need for additional financing; whether the company’s cash and cash equivalents are sufficient to support its operating plan for as long as anticipated; uncertainties regarding market acceptance, payor coverage and reimbursement, or future revenue generated by PEMGARDA; how long the EUA granted by the FDA for PEMGARDA will remain in effect and whether the EUA is revised or revoked by the FDA; the ability to maintain a continued acceptable safety, tolerability and efficacy profile of any product candidate following regulatory authorization or approval; the success of the company’s in-house sales force, and company’s ability to maintain and expand sales, marketing and distribution capabilities to successfully commercialize PEMGARDA; changes in expected or existing competition; changes in the regulatory environment; the outcome of the company’s engagement with regulators; uncertainties related to the regulatory authorization or approval process, and available development and regulatory pathways; the timing, progress and results of the company’s discovery, preclinical and clinical development activities; unexpected safety or efficacy data observed during preclinical studies or clinical trials; the predictability of clinical success of the company’s product candidates based on neutralizing activity in nonclinical studies; the risk that results of nonclinical studies or clinical trials may not be predictive of future results, and interim data are subject to further analysis; potential variability in neutralizing activity of product candidates tested in different assays, such as pseudovirus assays and authentic assays; variability of results in models and methods used to predict activity against SARS-CoV-2 variants; whether the epitope that pemivibart and VYD2311 targets remains structurally intact; whether the company’s product candidates are able to demonstrate and sustain neutralizing activity against major SARS-CoV-2 variants, particularly in the face of viral evolution; whether the company’s integrated technology platform is able to produce mAbs with broad and durable viral protection along with improved drug properties; the company’s reliance on third parties; clinical trial site activation or enrollment rates; the complexities of manufacturing mAb therapies; macroeconomic and political uncertainties; the company’s ability to realize the anticipated benefits of its term loan facility; the company’s ability to continue as a going concern; and whether the company has adequate funding to meet future operating expenses and capital expenditure requirements. Other factors that may cause the company’s actual results to differ materially from those expressed or implied in the forward-looking statements in this press release are described under the heading “Risk Factors” in the company’s Annual Report on Form 10-K for the year ended December 31, 2024, as filed with the Securities and Exchange Commission (SEC), and in the company’s other filings with the SEC, and in its future reports to be filed with the SEC and available at www.sec.gov. Forward-looking statements contained in this press release are made as of this date, and Invivyd undertakes no duty to update such information whether as a result of new information, future events or otherwise, except as required under applicable law.
This press release contains hyperlinks to information that is not deemed to be incorporated by reference in this press release.
Contacts:
Media Relations
(781) 208-1747
media@invivyd.com
Investor Relations
(781) 208-1747
investors@invivyd.com
INVIVYD, INC.
CONDENSED CONSOLIDATED BALANCE SHEETS
(UNAUDITED)
(In thousands, except share and per share amounts)
| March 31, 2025 |
December 31, 2024 |
|||||||
| Assets |
||||||||
| Current assets: |
||||||||
| Cash and cash equivalents |
$ | 48,078 | $ | 69,349 | ||||
| Accounts receivable |
8,561 | 10,906 | ||||||
| Prepaid expenses and other current assets |
19,186 | 20,426 | ||||||
|
|
|
|
|
|||||
| Total current assets |
75,825 | 100,681 | ||||||
| Inventory |
25,419 | 25,907 | ||||||
| Property and equipment, net |
1,523 | 1,508 | ||||||
| Operating lease right-of-use assets |
953 | 1,385 | ||||||
| Other non-current assets |
24 | 34 | ||||||
|
|
|
|
|
|||||
| Total assets |
$ | 103,744 | $ | 129,515 | ||||
|
|
|
|
|
|||||
| Liabilities, Preferred Stock and Stockholders’ Equity |
||||||||
| Current liabilities: |
||||||||
| Accounts payable |
$ | 8,739 | $ | 10,448 | ||||
| Accrued expenses(1) |
39,928 | 50,197 | ||||||
| Operating lease liabilities |
894 | 1,304 | ||||||
| Other current liability |
34 | 27 | ||||||
|
|
|
|
|
|||||
| Total current liabilities |
49,595 | 61,976 | ||||||
|
|
|
|
|
|||||
| Total liabilities |
49,595 | 61,976 | ||||||
|
|
|
|
|
|||||
| Commitments and contingencies |
||||||||
| Stockholders’ equity: |
||||||||
| Preferred stock (undesignated), $0.0001 par value; 10,000,000 shares authorized and no shares issued and outstanding at March 31, 2025 and December 31, 2024 |
— | — | ||||||
| Common stock, $0.0001 par value; 1,000,000,000 shares authorized, 119,961,445 shares issued and outstanding at March 31, 2025; 119,835,162 shares issued and outstanding at December 31, 2024 |
12 | 12 | ||||||
| Additional paid-in capital |
972,433 | 969,526 | ||||||
| Accumulated other comprehensive loss |
(13 | ) | (5 | ) | ||||
| Accumulated deficit |
(918,283 | ) | (901,994 | ) | ||||
|
|
|
|
|
|||||
| Total stockholders’ equity |
54,149 | 67,539 | ||||||
|
|
|
|
|
|||||
| Total liabilities, preferred stock and stockholders’ equity |
$ | 103,744 | $ | 129,515 | ||||
|
|
|
|
|
|||||
| (1) | Includes related-party amounts of $456 and $1,274 as of March 31, 2025 and December 31, 2024, respectively. |
INVIVYD, INC.
CONDENSED CONSOLIDATED STATEMENTS OF OPERATIONS AND COMPREHENSIVE LOSS
(UNAUDITED)
(In thousands, except share and per share amounts)
| Three Months Ended March 31, 2025 |
Three Months Ended March 31, 2024 |
|||||||
| Revenue: |
||||||||
| Product revenue, net |
$ | 11,304 | $ | — | ||||
|
|
|
|
|
|||||
| Total revenue |
11,304 | — | ||||||
|
|
|
|
|
|||||
| Operating costs and expenses: |
||||||||
| Cost of product revenue(1) |
834 | — | ||||||
| Research and development (2) |
10,641 | 31,160 | ||||||
| Selling, general and administrative |
16,751 | 14,929 | ||||||
|
|
|
|
|
|||||
| Total operating costs and expenses |
28,226 | 46,089 | ||||||
|
|
|
|
|
|||||
| Loss from operations |
(16,922 | ) | (46,089 | ) | ||||
|
|
|
|
|
|||||
| Other Income: |
||||||||
| Other Income, net |
633 | 2,593 | ||||||
|
|
|
|
|
|||||
| Total other income, net |
633 | 2,593 | ||||||
|
|
|
|
|
|||||
| Net Loss |
(16,289 | ) | (43,496 | ) | ||||
|
|
|
|
|
|||||
| Other comprehensive income (loss) |
||||||||
| Unrealized (loss) gain, net of tax |
(8 | ) | 1 | |||||
| Comprehensive loss |
$ | (16,297 | ) | (43,495 | ) | |||
|
|
|
|
|
|||||
| Net loss per share attributable to common stockholders, basic and diluted |
$ | (0.14 | ) | $ | (0.38 | ) | ||
|
|
|
|
|
|||||
| Weighted-average common shares outstanding, basic and diluted |
119,883,479 | $ | 115,618,209 | |||||
|
|
|
|
|
|||||
| (1) | Includes related-party amounts of $452 for the three months ended March 31, 2025 and no related-party amounts for the three months ended March 31, 2024. |
| (2) | Includes related-party amounts of $1,128 and $1,135 for the three months ended March 31, 2025 and 2024, respectively. |

Exhibit 99.2 INVIVYD Q1 2025 FINANCIAL RESULTS & BUSINESS HIGHLIGHTS May 15, 2025 © 2025 Invivyd, Inc. Invivyd , Pemgarda , and the Ribbon logos are trademarks of Invivyd, Inc. All trademarks in this presentation are the property of their respective owners.

CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS This presentation contains forward-looking statements within the meaning of the U.S. Private Securities Litigation Reform Act of 1995. Statements in this presentation that are not statements of historical fact are forward-looking statements. Words such as “may,” “will,” “should,” “expect,” “plan,” “anticipate,” “seek,” “could,” “intend,” “target,” “aim,” “project,” “designed to,” “estimate,” “believe,” “predict,” “potential” or “continue” or the negative of these terms or other similar expressions are intended to identify forward-looking statements, though not all forward-looking statements contain these identifying words. Forward-looking statements include statements concerning, among other things, PEMGARDA (pemivibart) as a monoclonal antibody (mAb) for pre-exposure prophylaxis (PrEP) of COVID-19 in certain immunocompromised patients; the company’s plans, strategies, goals and expectations related to the commercialization of PEMGARDA, including key commercial metrics; the company’s aim for near-term profitability; the company’s belief that its existing cash and cash equivalents, anticipated growth of net product revenue, and continued reduction of operating expenses will be sufficient to fund operations through profitability; expectations related to the company’s loan facility; the company’s research and clinical development efforts, including statements regarding initiation or completion of studies or trials, the time-frame during which results may become available; the potential of VYD2311 as a novel mAb candidate that may be able to deliver clinically meaningful titer levels through more patient-friendly means, and potentially available regulatory pathways; the company’s discovery efforts, including for respiratory syncytial virus (RSV) and measles; the company’s expectations regarding the neutralization activity of pemivibart and VYD2311 against SARS-CoV-2 variants; the government and regulatory landscape; the company’s business strategies and objectives, and ability to execute on them; potential market opportunities; the company’s future prospects; and other statements that are not historical fact. The company may not actually achieve the plans, intentions or expectations disclosed in the company’s forward-looking statements and you should not place undue reliance on the company’s forward-looking statements. These forward-looking statements involve risks and uncertainties that could cause the company’s actual results to differ materially from the results described in or implied by the forward-looking statements, including, without limitation: uncertainties regarding the company’s expectations, projections and estimates regarding future costs and expenses, future revenue, capital requirements, and the availability of and the need for additional financing; whether the company’s cash and cash equivalents are sufficient to support its operating plan for as long as anticipated; uncertainties regarding market acceptance, payor coverage and reimbursement, or future revenue generated by PEMGARDA; how long the EUA granted by the U.S. Food & Drug Administration (FDA) for PEMGARDA for COVID-19 PrEP in certain immunocompromised patients will remain in effect and whether such EUA is revised or revoked by the FDA; the ability to maintain a continued acceptable safety, tolerability and efficacy profile of any product candidate following regulatory authorization or approval; the success of the company’s in-house sales force, and company’s ability to maintain and expand sales, marketing and distribution capabilities to successfully commercialize PEMGARDA; changes in expected or existing competition; changes in the regulatory environment; the outcome of the company’s engagement with regulators; uncertainties related to the regulatory authorization or approval process, and available development and regulatory pathways; the timing, progress and results of the company’s discovery, preclinical and clinical development activities; unexpected safety or efficacy data observed during preclinical studies or clinical trials; the predictability of clinical success of the company’s product candidates based on neutralizing activity in nonclinical studies; the risk that results of nonclinical studies or clinical trials may not be predictive of future results, and interim data are subject to further analysis; the company’s reliance on third parties; potential variability in neutralizing activity of product candidates tested in different assays, such as pseudovirus assays and authentic assays; variability of results in models and methods used to predict activity against SARS-CoV-2 variants; whether the epitope that pemivibart and VYD2311 targets remains structurally intact; whether the company’s product candidates are able to demonstrate and sustain neutralizing activity against major SARS-CoV-2 variants, particularly in the face of viral evolution; whether the company’s integrated technology platform is able to produce mAbs with broad and durable viral protection along with improved drug properties; the complexities of manufacturing mAb therapies; macroeconomic and political uncertainties; the company’s ability to realize the anticipated benefits of its loan facility; the company’s ability to continue as a going concern; and whether the company has adequate funding to meet future operating expenses and capital expenditure requirements. Other factors that may cause the company’s actual results to differ materially from those expressed or implied in the forward-looking statements in this presentation are described under the heading “Risk Factors” in the company’s Annual Report on Form 10-K for the year ended December 31, 2024, as filed with the Securities and Exchange Commission (SEC), and in the company’s other filings with the SEC, and in its future reports to be filed with the SEC and available at www.sec.gov. Forward- looking statements contained in this press release are made as of this date, and Invivyd undertakes no duty to update such information whether as a result of new information, future events or otherwise, except as required under applicable law. 2

u Executive Summary u Commercial Update u R&D Overview u Clinical & Regulatory Finance Q&A 3

INVIVYD FOCUS: GROWTH AND EVOLUTION • Executed a commercial field force changeover with expected growth disruption during Q1 • Key commercial metrics in Q2 encouraging; focus remains on break-even and beyond • Pemivibart epitope has remained stable across now incalculable virus evolution post- Omicron – with no meaningful change to neutralization activity anticipated • Secured loan facility with SVB in April, allowing for potential access to $30 million in capital • COVID-19 pipeline emerging with VYD2311 positioned as a therapeutic and vaccine- alternative pending engagement with new FDA • Discovery program updates anticipated on RSV and measles later in 2025 SVB = Silicon Valley Bank, a division of First Citizens Bank; RSV = Respiratory Syncytial Virus 4

GOVERNMENT / REGULATORY LANDSCAPE • New Administration carries views on infectious disease that align with Invivyd strategy o Seek RCT for vaccines in contemporary, seropositive Americans against contemporary, immune- evasive virus, and hopefully over the long term, as for monoclonal antibodies (mAbs) o Favorable view on mAbs o Focus on treatment of active disease o Focus on choice • All of the above represents a break from the previous Administration which prioritized mRNA vaccines over alternatives like mAbs • Invivyd engaging with FDA and HHS directly for pipeline and on strategy; in May 2025, Invivyd submitted Citizen Petition which should be available for viewing shortly RCT = Randomized Controlled Trial; FDA = U.S. Food and Drug Administration; HHS = U.S. Department of Health and Human Services 5

u Executive Summary u Commercial Update R&D Overview Clinical & Regulatory Finance Q&A 6
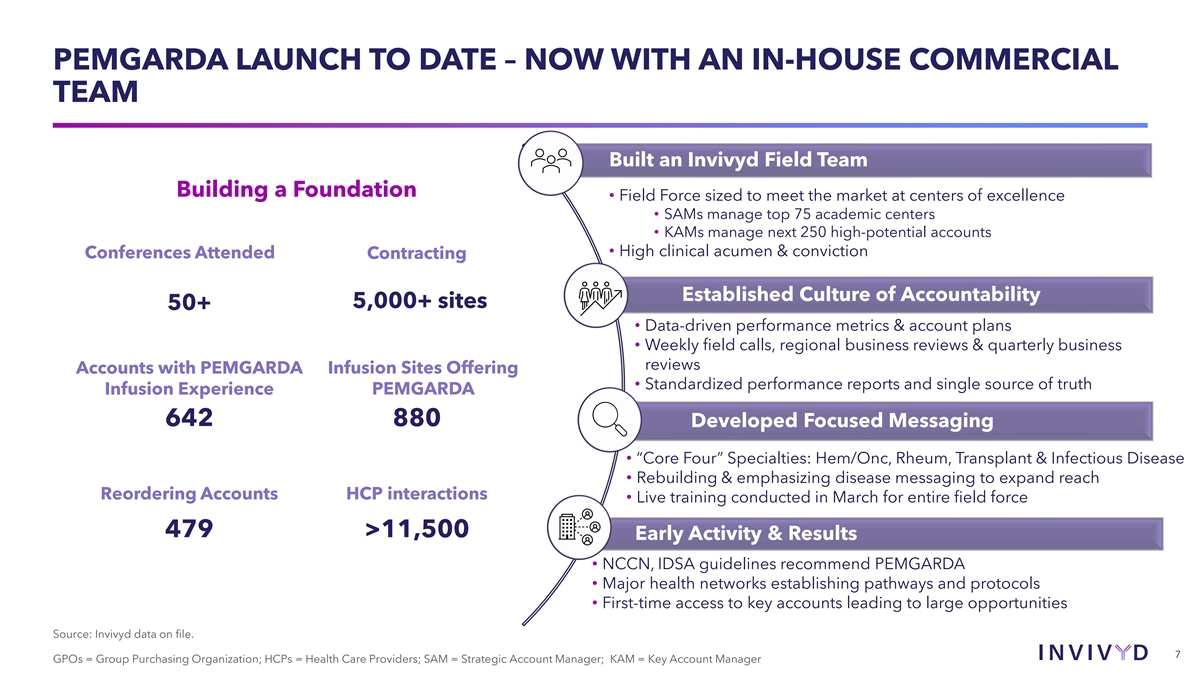
PEMGARDA LAUNCH TO DATE – NOW WITH AN IN-HOUSE COMMERCIAL TEAM Built an Invivyd Field Team Building a Foundation • Field Force sized to meet the market at centers of excellence • SAMs manage top 75 academic centers • KAMs manage next 250 high-potential accounts • High clinical acumen & conviction Conferences Attended Contracting Established Culture of Accountability 5,000+ sites 50+ • Data-driven performance metrics & account plans • Weekly field calls, regional business reviews & quarterly business reviews Accounts with PEMGARDA Infusion Sites Offering • Standardized performance reports and single source of truth Infusion Experience PEMGARDA 642 880 Developed Focused Messaging • “Core Four” Specialties: Hem/Onc, Rheum, Transplant & Infectious Disease • Rebuilding & emphasizing disease messaging to expand reach Reordering Accounts HCP interactions • Live training conducted in March for entire field force 479 >11,500 Early Activity & Results • NCCN, IDSA guidelines recommend PEMGARDA • Major health networks establishing pathways and protocols • First-time access to key accounts leading to large opportunities Source: Invivyd data on file. 7 7 GPOs = Group Purchasing Organization; HCPs = Health Care Providers; SAM = Strategic Account Manager; KAM = Key Account Manager
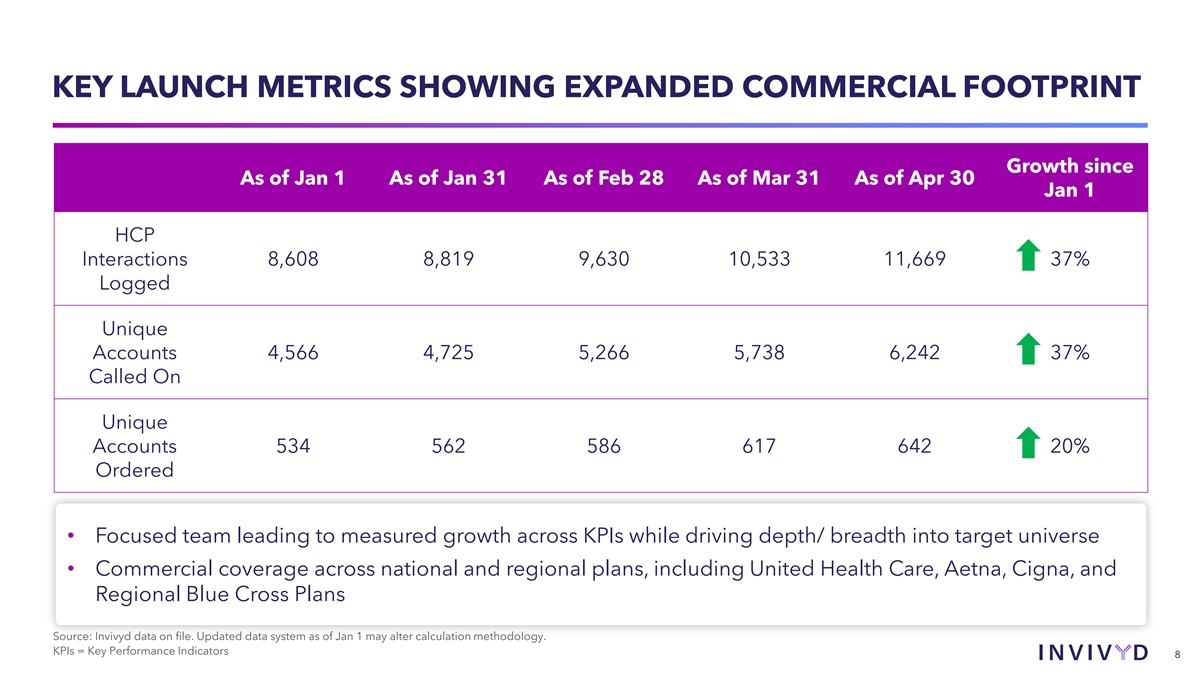
KEY LAUNCH METRICS SHOWING EXPANDED COMMERCIAL FOOTPRINT Growth since As of Jan 1 As of Jan 31 As of Feb 28 As of Mar 31 As of Apr 30 Jan 1 HCP Interactions 8,608 8,819 9,630 10,533 11,669 37% Logged Unique Accounts 4,566 4,725 5,266 5,738 6,242 37% Called On Unique Accounts 534 562 586 617 642 20% Ordered • Focused team leading to measured growth across KPIs while driving depth/ breadth into target universe • Commercial coverage across national and regional plans, including United Health Care, Aetna, Cigna, and Regional Blue Cross Plans Source: Invivyd data on file. Updated data system as of Jan 1 may alter calculation methodology. KPIs = Key Performance Indicators 8

PEMGARDA IS NOW ON SEVERAL SOCIETY GUIDELINES NCCN GUIDELINES FOR IDSA GUIDELINES B-CELL LYMPHOMAS Pemivibart has been added to the NCCN IDSA Guidelines recommend: Guidelines® for B-cell Lymphomas as a PEMGARDA for PrEP in moderately or recommended option for pre-exposure severely immunocompromised individuals prophylaxis of COVID-19 for individuals with 12 years or older at risk for COVID-19 moderate to severe immunocompromise IDSA Guidelines are endorsed by the Pediatric Infectious Diseases Society, the Society of Infectious Diseases ~76,000 new ~820,000 people Pharmacists, the Society for Healthcare patients diagnosed living with B-Cell Epidemiology of America, and the Lymphoma in US per year Society of Critical Care Medicine NCCN = National Comprehensive Cancer Network, NCCN Guidelines® = NCCN Clinical Practice Guidelines in Oncology, IDSA = Infectious Disease Society of America 9
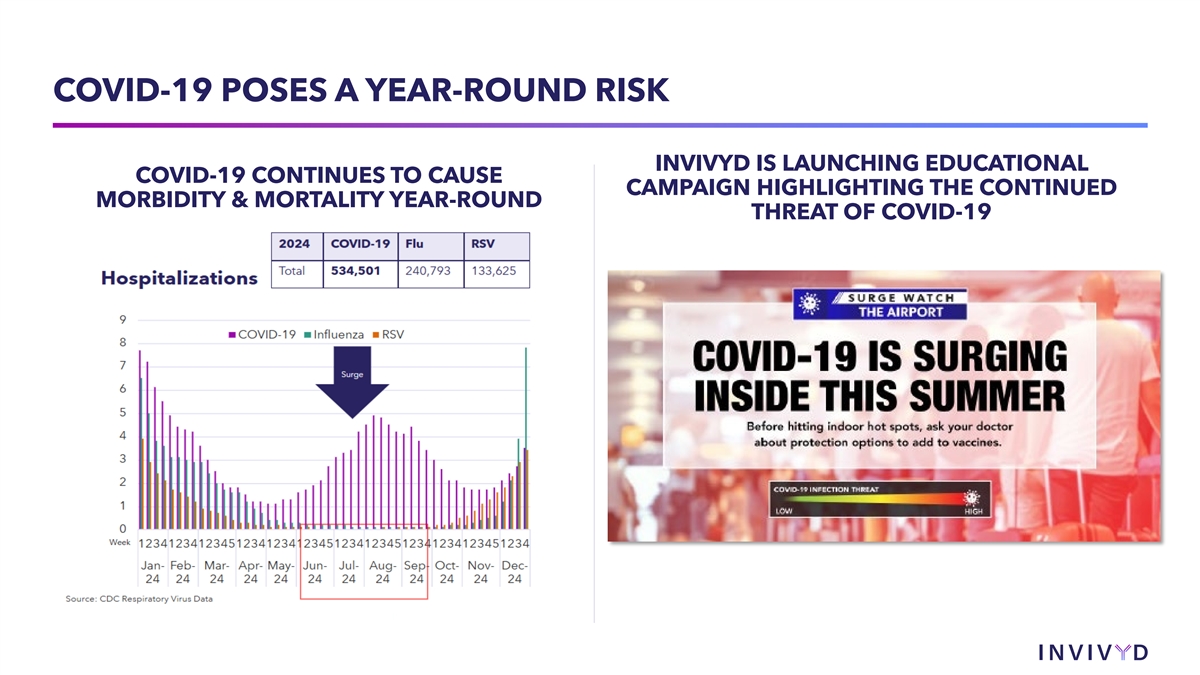
COVID-19 POSES A YEAR-ROUND RISK INVIVYD IS LAUNCHING EDUCATIONAL COVID-19 CONTINUES TO CAUSE CAMPAIGN HIGHLIGHTING THE CONTINUED MORBIDITY & MORTALITY YEAR-ROUND THREAT OF COVID-19
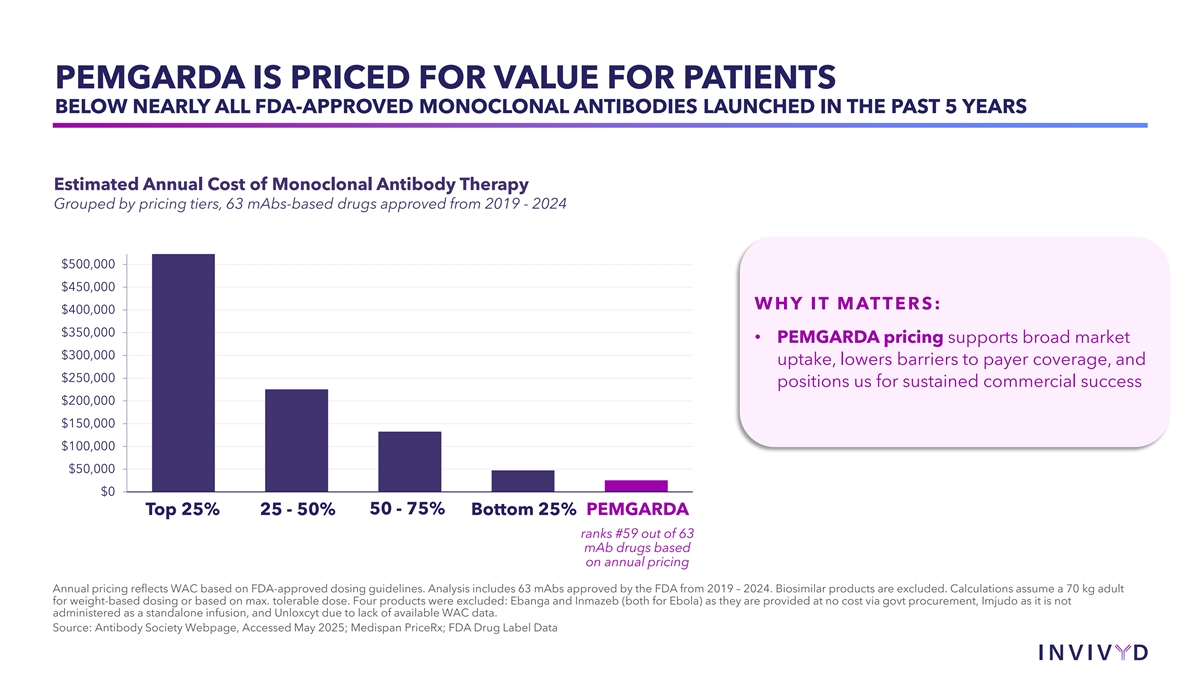
PEMGARDA IS PRICED FOR VALUE FOR PATIENTS BELOW NEARLY ALL FDA-APPROVED MONOCLONAL ANTIBODIES LAUNCHED IN THE PAST 5 YEARS Estimated Annual Cost of Monoclonal Antibody Therapy Grouped by pricing tiers, 63 mAbs-based drugs approved from 2019 - 2024 $500,000 $450,000 WHY IT MATTERS: $400,000 $350,000 • PEMGARDA pricing supports broad market $300,000 uptake, lowers barriers to payer coverage, and $250,000 positions us for sustained commercial success $200,000 $150,000 $100,000 $50,000 $0 Top 25% 25 - 50% 50 - 75% Bottom 25% PEMGARDA ranks #59 out of 63 mAb drugs based on annual pricing Annual pricing reflects WAC based on FDA-approved dosing guidelines. Analysis includes 63 mAbs approved by the FDA from 2019 – 2024. Biosimilar products are excluded. Calculations assume a 70 kg adult for weight-based dosing or based on max. tolerable dose. Four products were excluded: Ebanga and Inmazeb (both for Ebola) as they are provided at no cost via govt procurement, Imjudo as it is not administered as a standalone infusion, and Unloxcyt due to lack of available WAC data. Source: Antibody Society Webpage, Accessed May 2025; Medispan PriceRx; FDA Drug Label Data

PEMGARDA (PEMIVIBART) Please see below PEMGARDA has not been approved but has been authorized for emergency use by the FDA under an emergency use authorization (EUA), for pre-exposure prophylaxis of COVID-19 in certain adults and adolescents (12 years of age and older weighing at least 40 kg) with moderate-to-severe immune compromise. Pre-exposure prophylaxis with PEMGARDA is not a substitute for vaccination in individuals for whom COVID-19 vaccination is recommended. Individuals for whom COVID-19 vaccination is recommended, including individuals with moderate-to-severe immune compromise who may derive benefit from COVID-19 vaccinations, should receive COVID-19 vaccination. In individuals who have recently received a COVID-19 vaccine, PEMGARDA should be administered at least 2 weeks after vaccination. The emergency use of PEMGARDA is only authorized for the duration of the declaration that circumstances exist justifying the authorization of the emergency use of drugs and biological products during the COVID-19 pandemic under Section 564(b)(1) of the Federal Food, Drug, and Cosmetic Act, 21 U.S.C. § 360bbb-3(b)(1), unless the declaration is terminated or authorization is revoked sooner. PEMGARDA is authorized for use only when the combined national frequency of variants with substantially reduced susceptibility to PEMGARDA is less than or equal to 90%, based on available information including variant susceptibility to PEMGARDA and national variant frequencies. For additional information, please see the PEMGARDA full product Fact Sheet for Healthcare Providers, including Important Safety Information and Boxed Warning. COVID-19=coronavirus disease 2019; mAb=monoclonal antibody. Reference: PEMGARDA Fact Sheet for Healthcare Providers. Invivyd; February 2025. 12

u Executive Summary u Commercial Update u R&D Overview Clinical & Regulatory Finance Q&A 13

PEMIVIBART EPITOPE AND POTENCY REMAIN STABLE Virus Evolution • Pemivibart and Invivyd follow-on mAbs (e.g., VYD2311) are designed to target a highly conserved epitope • No significant structural change to the pemivibart epitope observed over an incalculably enormous quantum of virus variant exploration • Accordingly, no meaningful change to pemivibart or VYD2311 measured EC50 14
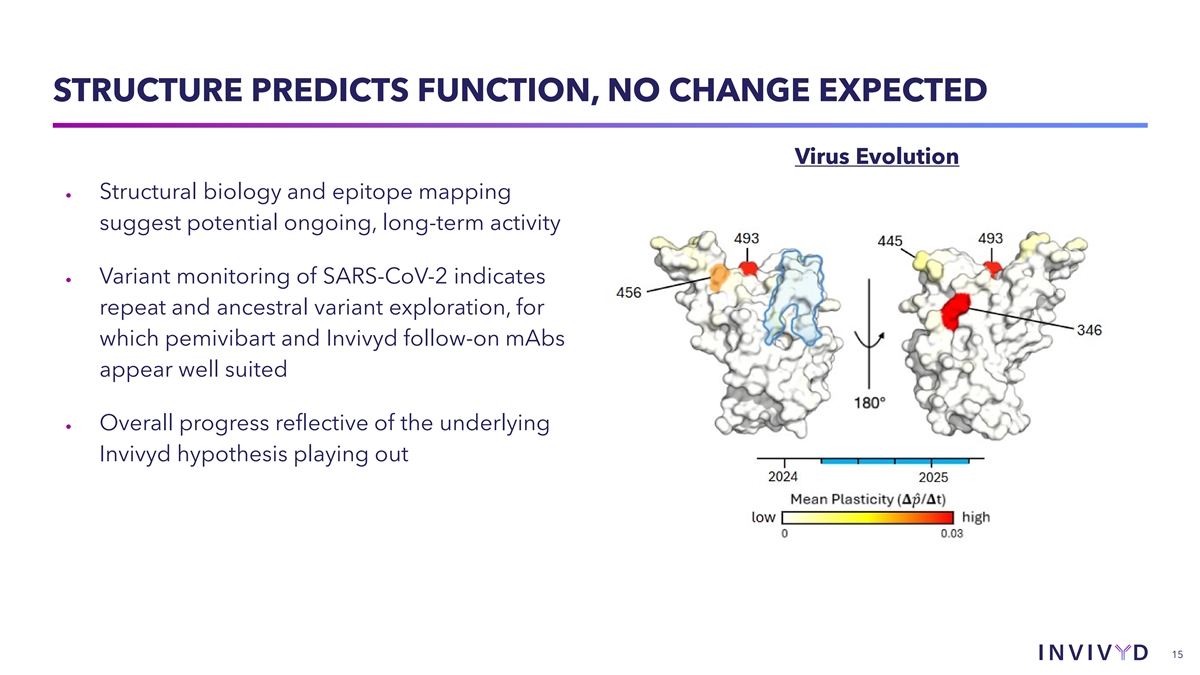
STRUCTURE PREDICTS FUNCTION, NO CHANGE EXPECTED Virus Evolution • Structural biology and epitope mapping suggest potential ongoing, long-term activity • Variant monitoring of SARS-CoV-2 indicates repeat and ancestral variant exploration, for which pemivibart and Invivyd follow-on mAbs appear well suited • Overall progress reflective of the underlying Invivyd hypothesis playing out 15
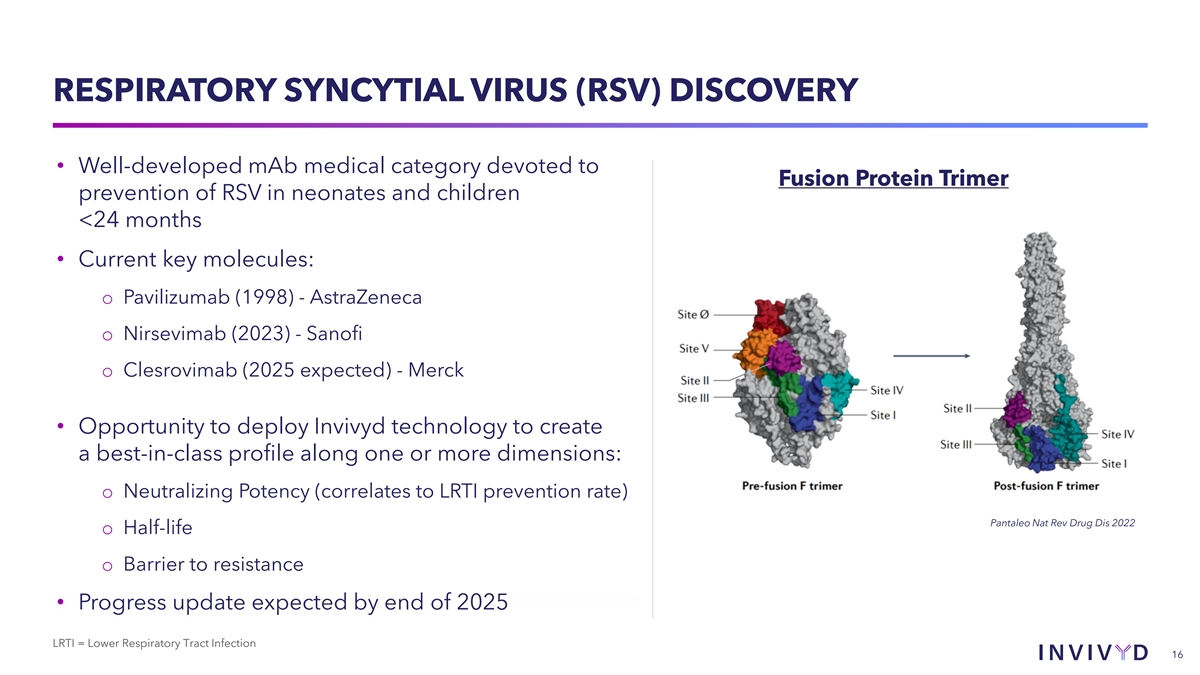
RESPIRATORY SYNCYTIAL VIRUS (RSV) DISCOVERY • Well-developed mAb medical category devoted to Fusion Protein Trimer prevention of RSV in neonates and children <24 months • Current key molecules: o Pavilizumab (1998) - AstraZeneca o Nirsevimab (2023) - Sanofi o Clesrovimab (2025 expected) - Merck • Opportunity to deploy Invivyd technology to create a best-in-class profile along one or more dimensions: o Neutralizing Potency (correlates to LRTI prevention rate) Pantaleo Nat Rev Drug Dis 2022 o Half-life o Barrier to resistance • Progress update expected by end of 2025 LRTI = Lower Respiratory Tract Infection 16
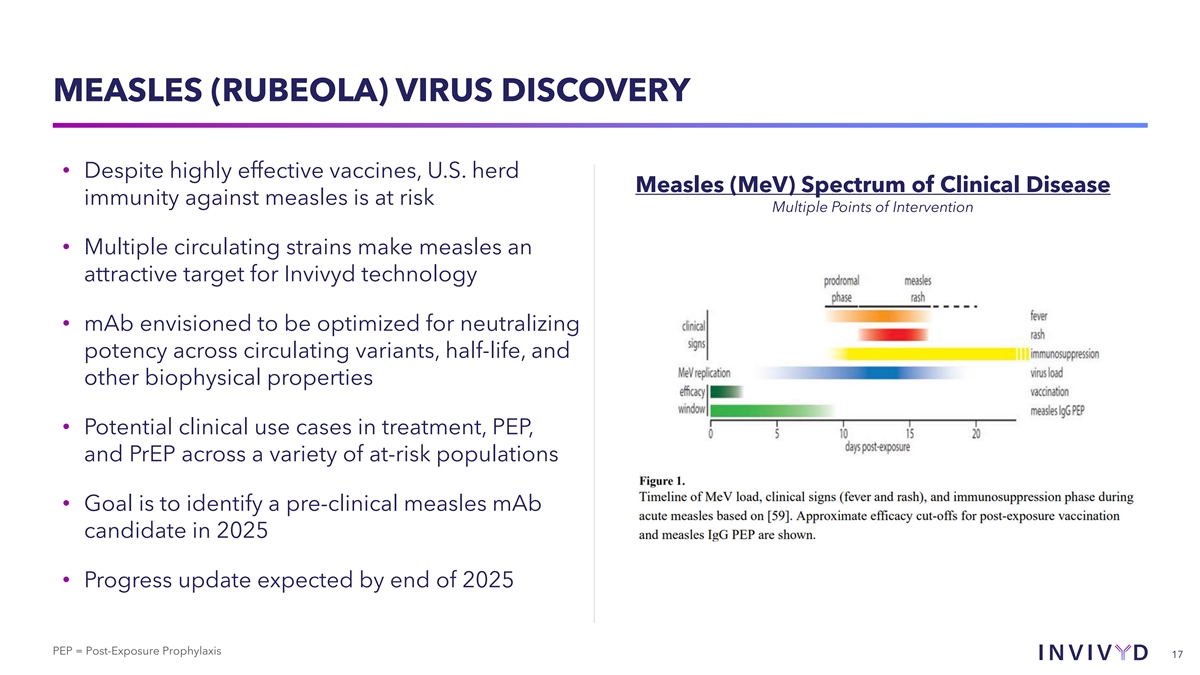
MEASLES (RUBEOLA) VIRUS DISCOVERY • Despite highly effective vaccines, U.S. herd Measles (MeV) Spectrum of Clinical Disease immunity against measles is at risk Multiple Points of Intervention • Multiple circulating strains make measles an attractive target for Invivyd technology • mAb envisioned to be optimized for neutralizing potency across circulating variants, half-life, and other biophysical properties • Potential clinical use cases in treatment, PEP, and PrEP across a variety of at-risk populations • Goal is to identify a pre-clinical measles mAb candidate in 2025 • Progress update expected by end of 2025 PEP = Post-Exposure Prophylaxis 17

u Executive Summary u Commercial Update u R&D Overview u Clinical & Regulatory Finance Q&A 18

VYD2311 OVERVIEW • VYD2311 is the third iteration of Invivyd’s platform approach targeting SARS-CoV-2 spike protein • Carries 99%+ sequence identity to predicate antibodies, ADG20 and pemivibart; approximately same or less structural change, version to version, as mRNA vaccine updates • Series of antibodies is honing access to a conserved epitope; goal is to refine antibody properties and substantially expand TAM with successive iterations TAM = Total Addressable Market 19
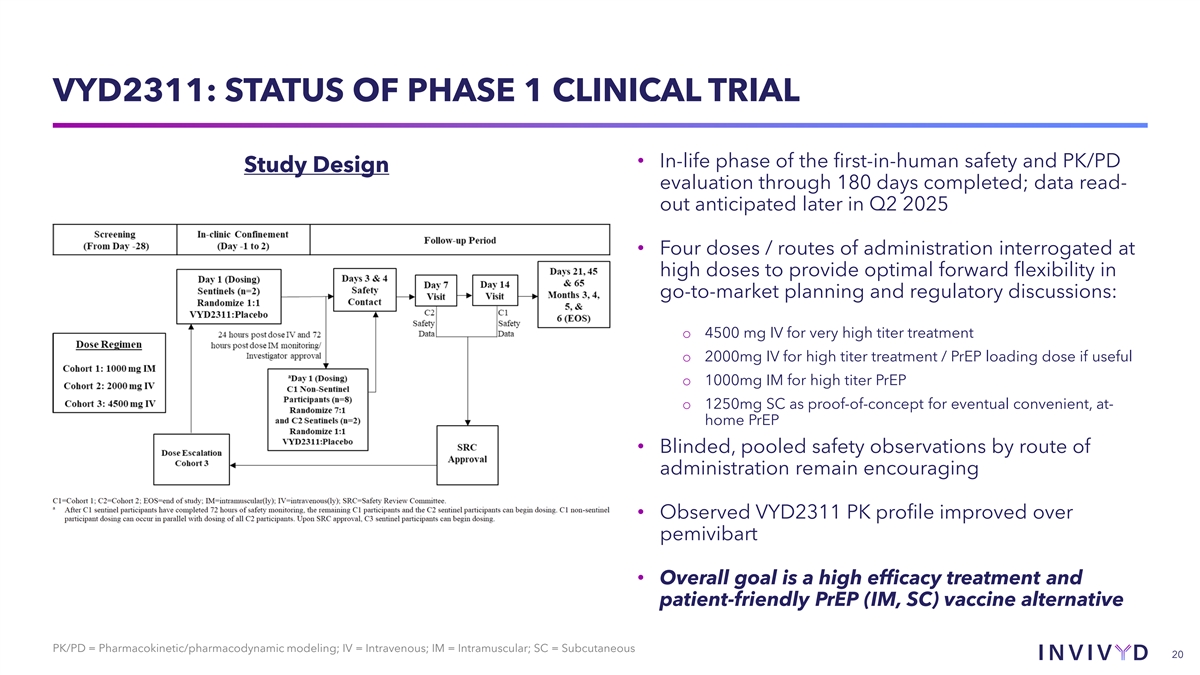
VYD2311: STATUS OF PHASE 1 CLINICAL TRIAL • In-life phase of the first-in-human safety and PK/PD Study Design evaluation through 180 days completed; data read- out anticipated later in Q2 2025 • Four doses / routes of administration interrogated at high doses to provide optimal forward flexibility in go-to-market planning and regulatory discussions: o 4500 mg IV for very high titer treatment o 2000mg IV for high titer treatment / PrEP loading dose if useful o 1000mg IM for high titer PrEP o 1250mg SC as proof-of-concept for eventual convenient, at- home PrEP • Blinded, pooled safety observations by route of administration remain encouraging • Observed VYD2311 PK profile improved over pemivibart • Overall goal is a high efficacy treatment and patient-friendly PrEP (IM, SC) vaccine alternative PK/PD = Pharmacokinetic/pharmacodynamic modeling; IV = Intravenous; IM = Intramuscular; SC = Subcutaneous 20
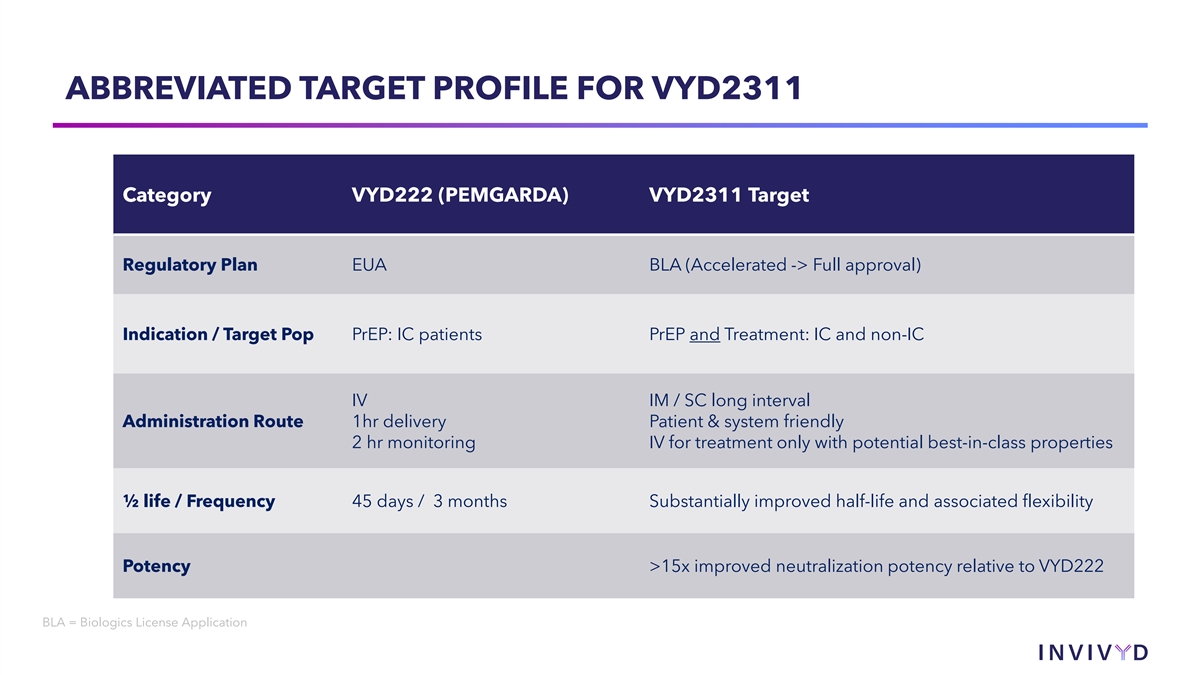
ABBREVIATED TARGET PROFILE FOR VYD2311 Category VYD222 (PEMGARDA) VYD2311 Target Regulatory Plan EUA BLA (Accelerated -> Full approval) Indication / Target Pop PrEP: IC patients PrEP and Treatment: IC and non-IC IV IM / SC long interval Administration Route 1hr delivery Patient & system friendly 2 hr monitoring IV for treatment only with potential best-in-class properties ½ life / Frequency 45 days / 3 months Substantially improved half-life and associated flexibility Potency >15x improved neutralization potency relative to VYD222 BLA = Biologics License Application

PEMGARDA TREATMENT EUA BACKGROUND: IMMUNOCOMPROMISED PATIENTS WITH NO OPTIONS • EUA was the preferred concept for the Biden Administration FDA for mAbs • FDA indicated openness to an immunobridging treatment EUA for pemivibart in 1H 2024 based primarily on comparison to adintrevimab; clear desire was for conservative (high) titers • Different potencies, half-lives, and routes of administration means, by definition, the sVNA titer curves would differ between adintrevimab, pemivibart and other COVID-19 mAbs; the key regulatory issue appeared to be the sVNA titer levels and shape of titer curves, and the associated basis of FDA assurance on potential clinical benefit • EUA request was for treatment of mild to moderate COVID-19” in certain immunocompromised patients “for whom alternative COVID-19 treatment options approved by FDA are not accessible or clinically appropriate EUA = Emergency Use Authorization; sVNA = Serum Virus Neutralizing Antibody 22

EXCERPT FROM FDA TREATMENT EUA DECLINATION LETTER: THE OVERALL FINDING Based on the totality of scientific evidence FDA provided four specific arguments against available, we are unable to reasonably authorization relative to the bridging and expected efficacy (benefits) in their conclusions, as follows: conclude that the known and potential • Immunobridging to adintrevimab (primary) benefits of pemivibart, when used for the • Meta-analysis (supportive) treatment of COVID-19 as described above, • Optimal dose for severely immunocompromised outweigh the known and potential risks… • Possible non-neutralization, non-effector functions of antibodies FDA Declination Letter (February 2025), Excerpt 23
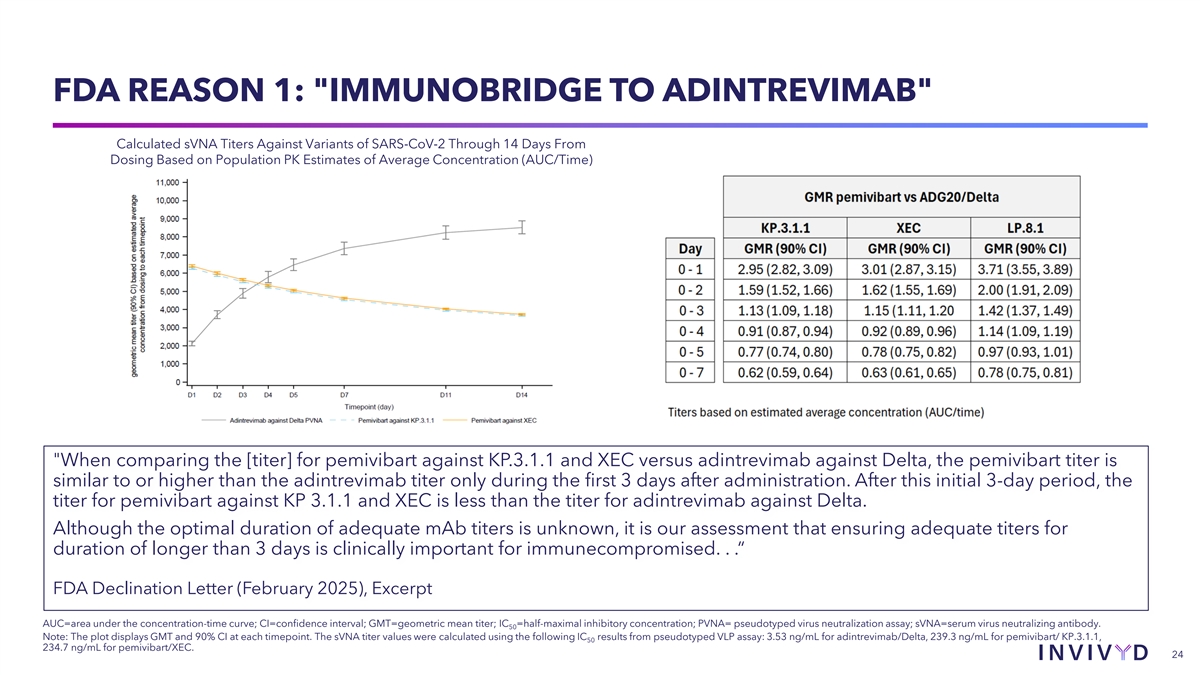
FDA REASON 1: IMMUNOBRIDGE TO ADINTREVIMAB Calculated sVNA Titers Against Variants of SARS-CoV-2 Through 14 Days From Dosing Based on Population PK Estimates of Average Concentration (AUC/Time) When comparing the [titer] for pemivibart against KP.3.1.1 and XEC versus adintrevimab against Delta, the pemivibart titer is similar to or higher than the adintrevimab titer only during the first 3 days after administration. After this initial 3-day period, the titer for pemivibart against KP 3.1.1 and XEC is less than the titer for adintrevimab against Delta. Although the optimal duration of adequate mAb titers is unknown, it is our assessment that ensuring adequate titers for duration of longer than 3 days is clinically important for immunecompromised. . .“ FDA Declination Letter (February 2025), Excerpt AUC=area under the concentration-time curve; CI=confidence interval; GMT=geometric mean titer; IC =half-maximal inhibitory concentration; PVNA= pseudotyped virus neutralization assay; sVNA=serum virus neutralizing antibody. 50 Note: The plot displays GMT and 90% CI at each timepoint. The sVNA titer values were calculated using the following IC results from pseudotyped VLP assay: 3.53 ng/mL for adintrevimab/Delta, 239.3 ng/mL for pemivibart/ KP.3.1.1, 50 234.7 ng/mL for pemivibart/XEC. 24
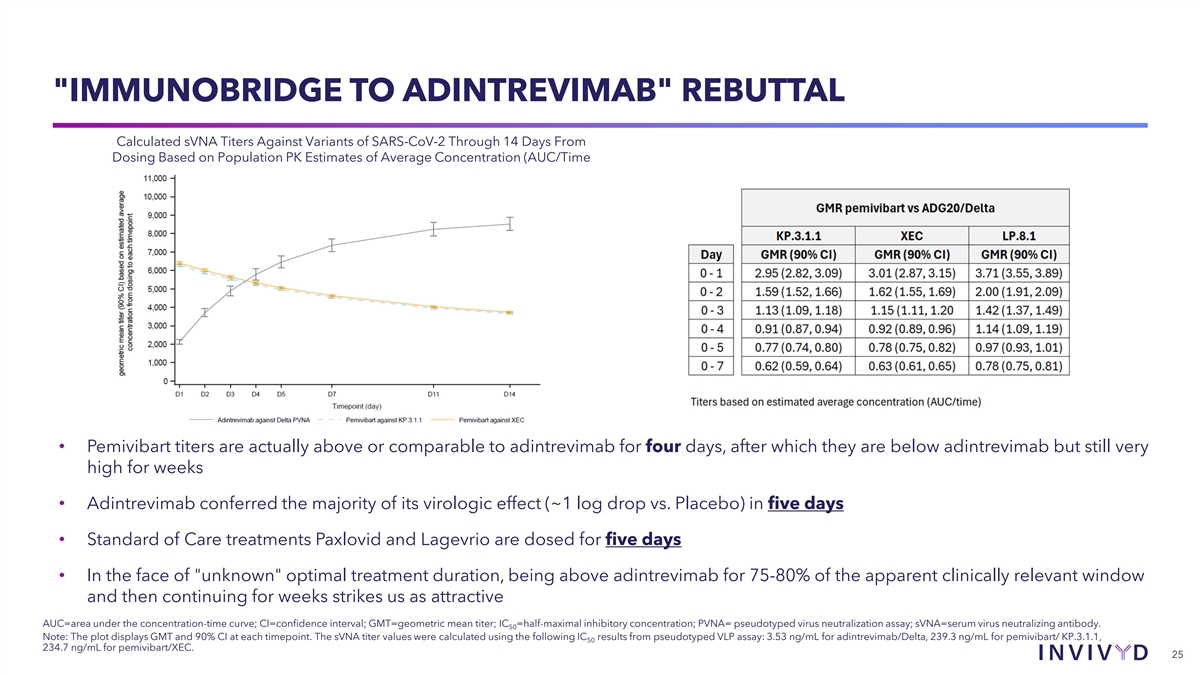
IMMUNOBRIDGE TO ADINTREVIMAB REBUTTAL Calculated sVNA Titers Against Variants of SARS-CoV-2 Through 14 Days From Dosing Based on Population PK Estimates of Average Concentration (AUC/Time • Pemivibart titers are actually above or comparable to adintrevimab for four days, after which they are below adintrevimab but still very high for weeks • Adintrevimab conferred the majority of its virologic effect (~1 log drop vs. Placebo) in five days • Standard of Care treatments Paxlovid and Lagevrio are dosed for five days • In the face of unknown optimal treatment duration, being above adintrevimab for 75-80% of the apparent clinically relevant window and then continuing for weeks strikes us as attractive AUC=area under the concentration-time curve; CI=confidence interval; GMT=geometric mean titer; IC =half-maximal inhibitory concentration; PVNA= pseudotyped virus neutralization assay; sVNA=serum virus neutralizing antibody. 50 Note: The plot displays GMT and 90% CI at each timepoint. The sVNA titer values were calculated using the following IC results from pseudotyped VLP assay: 3.53 ng/mL for adintrevimab/Delta, 239.3 ng/mL for pemivibart/ KP.3.1.1, 50 234.7 ng/mL for pemivibart/XEC. 25
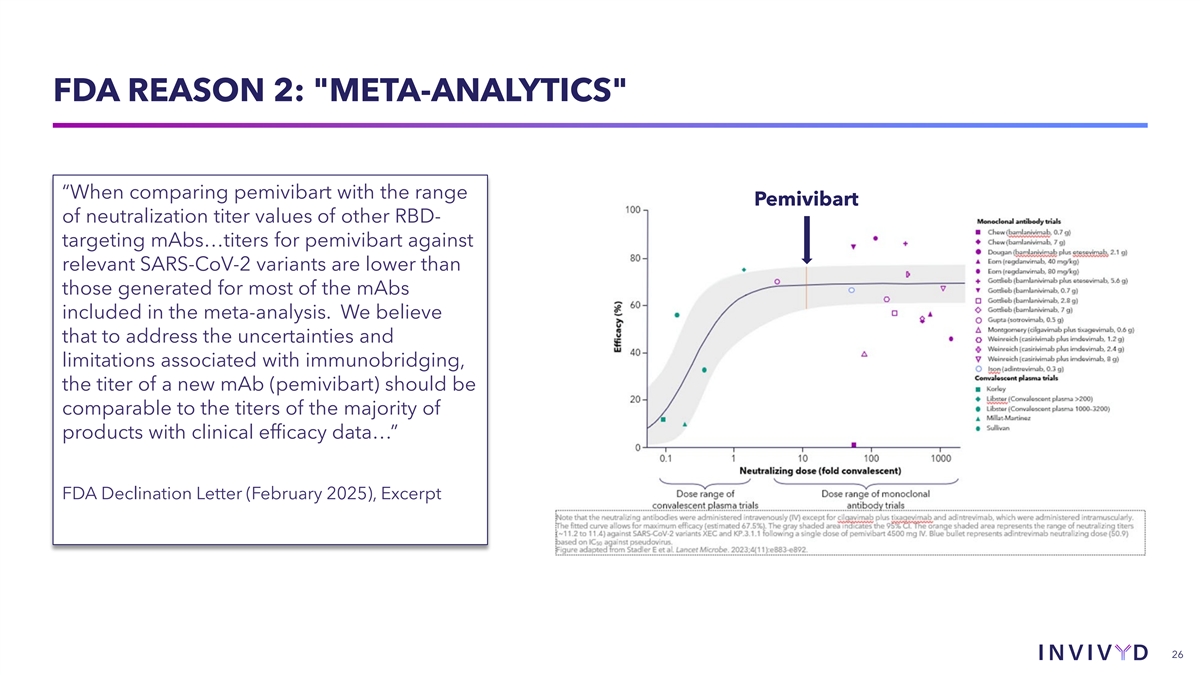
FDA REASON 2: META-ANALYTICS “When comparing pemivibart with the range Pemivibart of neutralization titer values of other RBD- targeting mAbs…titers for pemivibart against relevant SARS-CoV-2 variants are lower than those generated for most of the mAbs included in the meta-analysis. We believe that to address the uncertainties and limitations associated with immunobridging, the titer of a new mAb (pemivibart) should be comparable to the titers of the majority of products with clinical efficacy data…” FDA Declination Letter (February 2025), Excerpt 26
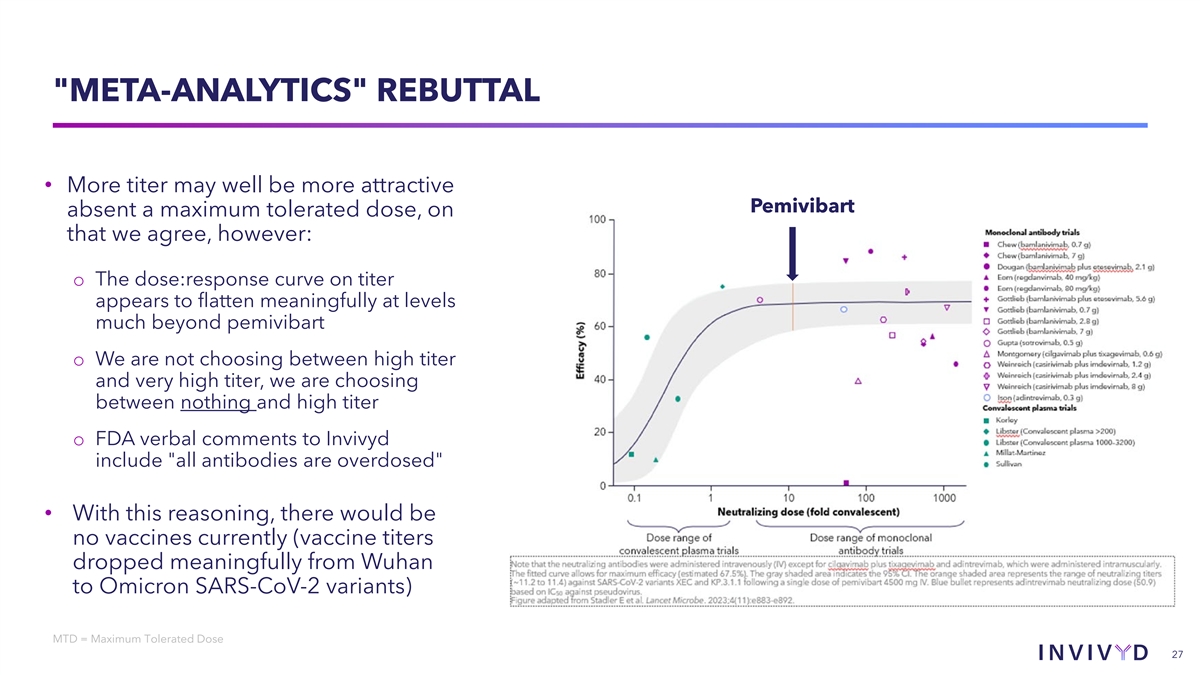
META-ANALYTICS REBUTTAL • More titer may well be more attractive Pemivibart absent a maximum tolerated dose, on that we agree, however: o The dose:response curve on titer appears to flatten meaningfully at levels much beyond pemivibart o We are not choosing between high titer and very high titer, we are choosing between nothing and high titer o FDA verbal comments to Invivyd include all antibodies are overdosed • With this reasoning, there would be no vaccines currently (vaccine titers dropped meaningfully from Wuhan to Omicron SARS-CoV-2 variants) MTD = Maximum Tolerated Dose 27

FDA REASON 3: OPTIMAL DOSE FOR SEVERELY IMMUNOCOMPROMISED Optimal drug concentrations / titers for successful treatment in immunocompetent individuals may differ substantially from those required in severely immunocompromised individuals who lack an adequate immunological response after infection is established. FDA Declination Letter (February 2025), Excerpt 28

OPTIMAL DOSE REBUTTAL So, if FDA does not know the optimal dose, which is itself an odd concept in drug development, the FDA would prefer that “severely immunocompromised” persons who lack an adequate immunological response after infection is established … … do their best to survive infection with no additional immune support at all. 29

FDA REASON 4: OTHER MAB ACTIVITIES Variability in antibody-mediated activities other than neutralization may contribute to differences in treatment effects between antibodies and may not be readily normalizable, thus limiting the ability to conclude comparable effectiveness based on upon similar neutralization titers. FDA Declination Letter (February 2025), Excerpt 30

REBUTTAL: OTHER MAB ACTIVITIES • Pemivibart and adintrevimab both retain effector function, arguing for enhanced activity in active treatment versus other antibodies • Fc effector function assessed and found comparable between adintrevimab and pemivibart; no surprise given structural identity 31

THE CURRENT STATE OF PLAY • The Biden Administration prioritized prevention over treatment; the new Administration appears to be reversing course • Pemivibart EUA treatment declination letter signatory at OND and leadership at CDER, OID has either been fired or resigned • Invivyd is planning to reengage with FDA on both pemivibart and VYD2311 for treatment; Citizen Petition regarding mAbs submitted to the Agency; VYD2311 briefing book to be sent shortly to the Agency CDER = Center for Drug Evaluation and Research; OID = Office of Infectious Diseases 32
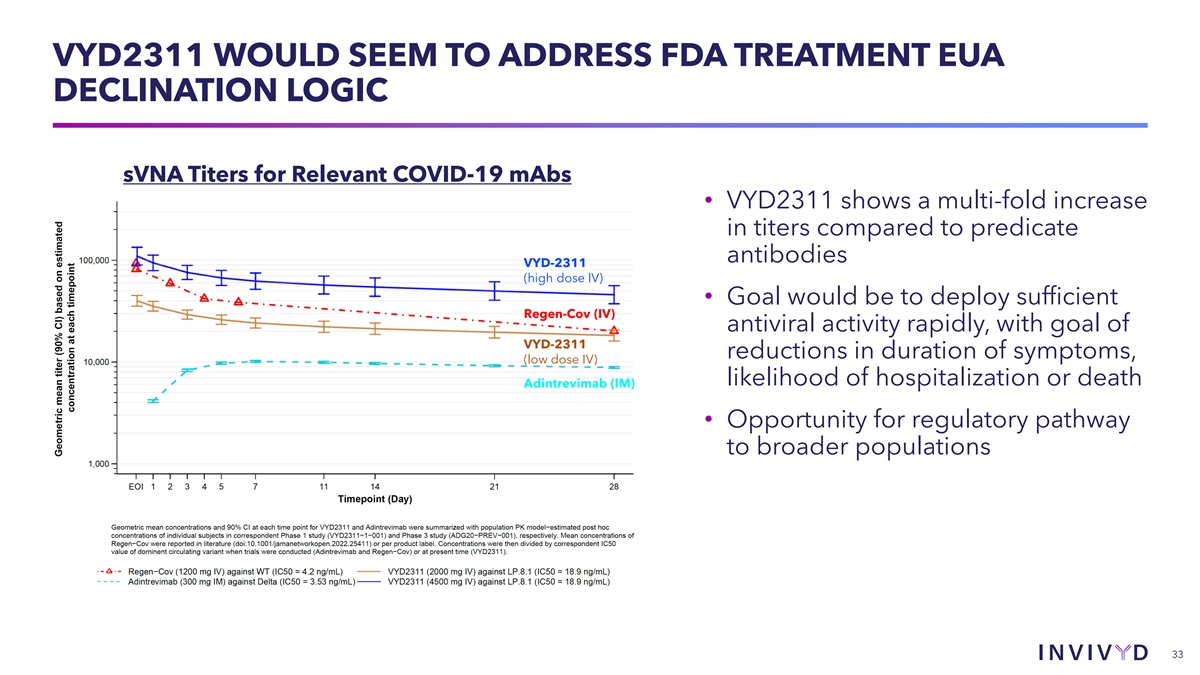
VYD2311 WOULD SEEM TO ADDRESS FDA TREATMENT EUA DECLINATION LOGIC sVNA Titers for Relevant COVID-19 mAbs • VYD2311 shows a multi-fold increase in titers compared to predicate antibodies VYD-2311 (high dose IV) • Goal would be to deploy sufficient Regen-Cov (IV) antiviral activity rapidly, with goal of VYD-2311 reductions in duration of symptoms, (low dose IV) likelihood of hospitalization or death Adintrevimab (IM) • Opportunity for regulatory pathway to broader populations 33

u Executive Summary u Commercial Update u R&D Overview u Clinical & Regulatory u Finance Q&A 34

FINANCIALS • Q1 2025 PEMGARDA (pemivibart) net product revenue of $11.3 million • Continued execution of financial discipline and reduction of operating expenses – $27.4 million in Q1 2025 vs. $32.3 million in Q4 2024 • Ended Q1 2025 with approximately $48.1 million in cash and cash equivalents; potential financial flexibility with $30 million loan facility secured in April 2025 • Targeting near-term profitability (1H 2025) with existing cash and cash equivalents, anticipated growth of net product revenue, and continued reduction of operating expenses • Well-insulated from potential tariffs and most-favored-nation impact on PEMGARDA, with commercial supply located in U.S. and not commercialized outside of U.S. • Continuing to evaluate multiple sources of additional capital 35 Source: Invivyd Data on File

u Executive Summary u Commercial Update u R&D Overview u Clinical and Regulatory u Finance u Q&A 36

APPENDIX: FDA DECLINATION LETTER (FEBRUARY 2025), EXCERPT 37 Source: Invivyd Data on File

APPENDIX: FDA DECLINATION LETTER (FEBRUARY 2025), EXCERPT 38 Source: Invivyd Data on File

APPENDIX: FDA DECLINATION LETTER (FEBRUARY 2025), EXCERPT * NB: The supportive meta-analysis for immunobridging was presented via two marginally different analytics, hence the FDA notes areas of uncertainty for all three methods * 39 Source: Invivyd Data on File