|
|
|
FORM 20-F
|
|
Israel
|
|
2 HaMa’ayan Street
Modi’in 7177871, Israel |
|
(Jurisdiction of incorporation or organization)
|
|
(Address of principal executive offices)
|
|
Title of each class
|
|
Name of each exchange on which registered
|
|
American Depositary Shares, each representing 600 ordinary shares, par value NIS 0.10 per share
|
|
Nasdaq Capital Market
|
|
|
|
|
|
Ordinary shares, par value NIS 0.10 per share
|
|
Nasdaq Capital Market*
|
|
|
|
|

|
Large accelerated filer ☐
|
Accelerated filer ☐
|
Non-accelerated filer ☒
|
Emerging growth company ☐
|
|
U.S. GAAP ☐
|
International Financial Reporting Standards as issued by the International Accounting Standards Board ☒
|
Other ☐
|
|
TABLE OF CONTENTS |
|
|
|
|
| 1 | |
| 1 | |
| 1 | |
| 1 | |
| 30 | |
| 60 | |
| 60 | |
| 71 | |
| 88 | |
| 91 | |
| 92 | |
| 93 | |
| 103 | |
| 104 | |
| 105 | |
| 105 | |
| 105 | |
| 106 | |
| 106 | |
| 106 | |
| 107 | |
| 107 | |
| 107 | |
| 107 | |
| 107 | |
| 108 | |
| 108 | |
| 109 | |
| 109 | |
| 110 | |
| 110 | |
| 110 | |
| 113 |
|
|
• |
the clinical development, commercialization and market acceptance of our therapeutic
candidates, including the degree and pace of market uptake of APHEXDA for the mobilization of hematopoietic stem cells for autologous
transplantation in multiple myeloma patients; |
|
|
• |
the initiation, timing, progress and results of our preclinical studies, clinical
trials and other therapeutic candidate development efforts; |
|
|
• |
our ability to advance our therapeutic candidates into clinical trials or to successfully
complete our preclinical studies or clinical trials; |
|
|
• |
whether the clinical trial results for APHEXDA will be predictive of real-world results;
|
|
|
• |
our receipt of regulatory approvals for our therapeutic candidates, and the timing
of other regulatory filings and approvals; |
|
|
|
|
|
|
• |
whether access to APHEXDA is achieved in a commercially viable manner and whether
APHEXDA receives adequate reimbursement from third-party payors; |
|
|
|
|
|
|
• |
our ability to establish, manage, and maintain corporate collaborations, as well as
the ability of our collaborators to execute on their development and commercialization plans; |
|
|
• |
our ability to integrate new therapeutic candidates and new personnel, as well as
new collaborations; |
|
|
• |
the interpretation of the properties and characteristics of our therapeutic candidates
and of the results obtained with our therapeutic candidates in preclinical studies or clinical trials; |
|
|
• |
the implementation of our business model and strategic plans for our business and
therapeutic candidates; |
|
|
• |
the scope of protection that we are able to establish and maintain for intellectual
property rights covering our therapeutic candidates and our ability to operate our business without infringing the intellectual property
rights of others; |
|
|
• |
estimates of our expenses, future revenues, capital requirements and our need for
and ability to access sufficient additional financing; |
|
|
• |
risks related to changes in healthcare laws, rules and regulations in the United States
or elsewhere; |
|
|
• |
competitive companies, technologies and our industry; |
|
|
• |
our ability to maintain the listing of our ADSs on Nasdaq; |
|
|
• |
statements as to the impact of the political and security situation in Israel on our
business, including the impact of Israel’s war with Hamas and other militant groups, which may exacerbate the magnitude of the factors
discussed above; and |
|
|
• |
those factors referred to in “Item 3.D. Risk Factors,” “Item 4.
Information on the Company,” and “Item 5. Operating and Financial Review and Prospects”, as well as in this Annual Report
on Form 20-F generally. |
|
|
• |
We have incurred significant losses since inception and expect to incur additional losses in the future
and may never be profitable. |
|
|
• |
We cannot assure investors that our existing cash and investment balances will be sufficient to meet our
future capital requirements. |
|
|
• |
If we default under our secured loan agreement with BlackRock EMEA Venture and Growth Lending (previously
Kreos Capital VII Aggregator SCSP), or BlackRock, all or a portion of our assets could be subject to forfeiture. |
|
|
• |
Management has concluded that there is substantial doubt about our ability to continue as a going concern,
which could prevent us from obtaining new financing on reasonable terms or at all. |
|
|
• |
We have earned limited commercialization revenues to date. We may never achieve profitability. |
|
|
• |
APHEXDA, or any other therapeutic candidate that may receive marketing approval in
the future, may fail to achieve the degree of market acceptance by physicians, patients, third-party payors and others in the medical
community necessary for commercial success and the market opportunity for APHEXDA or any other therapeutic candidate may be smaller than
our estimates. |
|
|
• |
If we or our collaborators are unable to obtain and/or maintain U.S. and/or foreign
regulatory approval for our therapeutic candidates, in a timely manner or at all, we will be unable to commercialize our therapeutic candidates.
|
|
|
• |
We and our collaborators may not obtain additional marketing approvals for motixafortide
in other indications or initial approval for any other therapeutic candidates we may develop in the future. |
|
|
• |
Clinical trials involve a lengthy and expensive process with an uncertain outcome,
and results of earlier studies and trials may not be predictive of future trial results. |
|
|
• |
Even if we obtain regulatory approvals, our therapeutic candidates will be subject
to ongoing regulatory review and if we fail to comply with continuing U.S. and applicable foreign regulations, we could lose those approvals
and our business would be seriously harmed. |
|
|
• |
We generally rely on third parties to conduct our preclinical studies and clinical
trials and to provide other services, and those third parties may not perform satisfactorily, including by failing to meet established
deadlines for the completion of such services. |
|
|
• |
We recently entered into and may in the future rely on out-licensing arrangements
for late-stage development, marketing or commercialization of our therapeutic candidates. |
|
|
• |
If we cannot meet requirements under our in-license agreements, we could lose the
rights to our therapeutic candidates, which could have a material adverse effect on our business. |
|
|
• |
We have partnered with and may seek to partner with third-party collaborators with
respect to the development and commercialization of motixafortide, and we may not succeed in establishing and maintaining collaborative
relationships, which may significantly limit our ability to develop and commercialize our therapeutic candidates successfully, if at all.
|
|
|
• |
If competitors develop and market therapeutics that are more effective, safer or less
expensive than our current or future therapeutic candidates, our prospects will be negatively impacted. |
|
|
• |
APHEXDA, or any other therapeutic candidate that we or our licensees are able to commercialize,
may become subject to unfavorable pricing regulations, third-party payor reimbursement practices or healthcare reform initiatives, any
of which could harm our business. |
|
|
• |
We rely upon third-party manufacturers to produce therapeutic supplies for the clinical
trials, and commercialization, of APHEXDA. If we manufacture any therapeutic candidates in the future, we will be required to incur significant
costs and devote significant efforts to establish and maintain manufacturing capabilities. |
|
|
• |
Healthcare reforms and related reductions in pharmaceutical pricing, reimbursement and coverage by government
authorities and third-party payors may adversely affect our business. |
|
|
• |
If third-party payors do not adequately reimburse customers for any of our therapeutic candidates that
are approved for marketing, they might not be purchased or used, and our revenues and profits will not develop or increase. |
|
|
• |
Our business has a substantial risk of clinical trial and product liability claims. If we are unable to
obtain and maintain appropriate levels of insurance, a claim could adversely affect our business. |
|
|
• |
Significant disruptions of our information technology systems or breaches of our data security could adversely
affect our business. |
|
|
• |
We deal with hazardous materials and must comply with environmental, health and safety laws and regulations,
which can be expensive and restrict how we do business. |
|
|
• |
We are currently party to, and may in the future, become subject to litigation or claims arising in or
outside the ordinary course of business that could negatively affect our business operations and financial condition. |
|
|
• |
Our access to most of the intellectual property associated with our therapeutic candidates
results from in-license agreements with biotechnology companies and a university, the termination of which would prevent us from commercializing
the associated therapeutic candidates. |
|
|
• |
Our business, operating results and growth rates may be adversely affected by current or future unfavorable
economic and market conditions and adverse developments with respect to financial institutions and associated liquidity risk. |
|
|
• |
The market prices of our ordinary shares and ADSs are subject to fluctuation, which could result in substantial
losses by our investors. |
|
|
• |
Future sales of our ordinary shares or ADSs could reduce the market price of our ordinary shares and ADSs.
|
|
|
• |
Raising additional capital by issuing securities may cause dilution to existing shareholders. |
|
|
• |
If we fail to comply with the continued listing requirements of the Nasdaq, our ADSs may be delisted and
the price of our ADSs and our ability to access the capital markets could be negatively impacted. |
|
|
• |
We conduct a substantial part of our operations in Israel and therefore our results may be adversely affected
by political, economic and military instability in Israel and its region. |
|
|
|
|
|
|
• |
Provisions of Israeli law may delay, prevent or otherwise impede a merger with, or an acquisition of, our
company, which could prevent a change of control, even when the terms of such a transaction are favorable to us and our shareholders.
|
|
|
• |
It may be difficult to enforce a U.S. judgment against us and our officers and directors in Israel or the
United States, or to serve process on our officers and directors. |
|
|
• |
Your rights and responsibilities as a shareholder will be governed by Israeli law, which may differ in
some respects from the rights and responsibilities of shareholders of U.S. companies. |
|
|
• |
the advantages of the treatment compared
to competitive therapies; |
|
• |
the number of competitors approved for similar
uses; |
|
• |
the relative promotional effort and marketing
success of us as compared with our competitors; |
|
• |
how the product is positioned in physician
treatment guidelines and pathways; |
|
• |
the prevalence and severity of any side
effects; |
|
• |
the efficacy and safety of the product;
|
|
• |
our ability to offer the product for sale
at competitive reimbursement; |
|
• |
the product’s tolerability, convenience
and ease of administration compared to alternative treatments; |
|
• |
the willingness of the target patient population
to try, and of physicians to prescribe, the product; |
|
• |
limitations or warnings, including use restrictions,
contained in the product’s approved labeling; |
|
• |
the strength of sales, marketing and distribution
support; |
|
• |
the timing of market introduction of our
approved products as well as competitive products; |
|
• |
adverse publicity about the product or favorable
publicity about competitive products; |
|
• |
potential product liability claims;
|
|
• |
changes in the standard of care for the
targeted indications of the product; and |
|
• |
availability and amount of coverage and
reimbursement from government payors, managed care plans and other third-party payors. |
|
• |
regulatory authorities may withdraw their
approval of the product or seize the product; |
|
• |
we, or any of our collaborators, may be
required to recall the product, change the way the product is administered or conduct additional clinical trials; |
|
• |
additional restrictions may be imposed on
the marketing of, or the manufacturing processes for, the particular product; |
|
• |
we, or any of our collaborators, may be
subject to fines, injunctions or the imposition of civil or criminal penalties; |
|
• |
regulatory authorities may require the addition
of labeling or warning statements, such as a “black box” warning or a contraindication; |
|
• |
we, or any of our collaborators, may be
required to create a Medication Guide outlining the risks of the previously unidentified side effects for distribution to physicians,
health care professionals and patients; |
|
• |
we could be sued and held liable for harm
caused to patients; |
|
• |
physicians and patients may stop using our
product; and |
|
• |
our reputation may suffer. |
|
• |
delays in securing clinical investigators
or trial sites for the clinical trials; |
|
• |
delays in obtaining institutional review
board and other regulatory approvals to commence a clinical trial; |
|
• |
slower-than-anticipated patient recruitment
and enrollment; |
|
• |
negative or inconclusive results from clinical
trials; |
|
• |
unforeseen safety issues; |
|
• |
uncertain dosing issues; |
|
• |
an inability to monitor patients adequately
during or after treatment; and |
|
• |
problems with investigator or patient compliance
with the trial protocols. |
|
• |
restrictions on such product, manufacturer
or manufacturing process; |
|
• |
warning letters from the FDA or other regulatory
authorities; |
|
• |
withdrawal of the product from the market;
|
|
• |
suspension or withdrawal of regulatory approvals;
|
|
• |
refusal to approve pending applications
or supplements to approved applications that we or our licensees submit; |
|
• |
voluntary or mandatory recall; |
|
• |
fines; |
|
• |
refusal to permit the import or export of
our products; |
|
• |
product seizure or detentions; |
|
• |
injunctions or the imposition of civil or
criminal penalties; or |
|
• |
adverse publicity. |
|
• |
we have limited control over the amount
and timing of resources that a licensee devotes to our therapeutic candidate; |
|
• |
a licensee may experience financial difficulties;
|
|
• |
a licensee may fail to secure adequate commercial
supplies of our therapeutic candidate upon marketing approval, if at all; |
|
• |
our future revenues depend heavily on the
efforts of a licensee; |
|
• |
business combinations or significant changes
in a licensee’s business strategy may adversely affect the licensee’s willingness or ability to complete its obligations under
any arrangement with us; |
|
• |
a licensee could move forward with a competing
therapeutic candidate developed either independently or in collaboration with others, including our competitors; and |
|
• |
out-licensing arrangements are often terminated
or allowed to expire, which would delay the development and may increase the development costs of our therapeutic candidates. |
|
• |
a collaboration partner may shift its priorities
and resources away from our therapeutic candidates due to a change in business strategies, or a merger, acquisition, sale or downsizing;
|
|
• |
a collaboration partner may seek to renegotiate
or terminate their relationships with us due to unsatisfactory clinical results, manufacturing issues, a change in business strategy,
a change of control or other reasons; |
|
• |
a collaboration partner may cease development
in therapeutic areas which are the subject of our strategic collaboration; |
|
• |
a collaboration partner may not devote sufficient
capital or resources towards our therapeutic candidates; |
|
• |
a collaboration partner may change the success
criteria for a therapeutic candidate, thereby delaying or ceasing development of such candidate; |
|
• |
a significant delay in initiation of certain
development activities by a collaboration partner will also delay payment of milestones tied to such activities, thereby impacting our
ability to fund our own activities; |
|
• |
a collaboration partner could develop a
product that competes, either directly or indirectly, with our therapeutic candidate; |
|
• |
a collaboration partner with commercialization
obligations may not commit sufficient financial or human resources to the marketing, distribution or sale of a product; |
|
• |
a collaboration partner with manufacturing
responsibilities may encounter regulatory, resource or quality issues and be unable to meet demand requirements; |
|
• |
a partner may exercise a contractual right
to terminate a strategic alliance; |
|
• |
a dispute may arise between us and a partner
concerning the research, development or commercialization of a therapeutic candidate resulting in a delay in milestones, royalty payments
or termination of an alliance and possibly resulting in costly litigation or arbitration which may divert management attention and resources;
and |
|
• |
a partner may use our products or technology
in such a way as to invite litigation from a third party. |
|
• |
reliance on the third party for regulatory
compliance and quality assurance; |
|
• |
limitations on supply availability resulting
from capacity and scheduling constraints of the third parties; |
|
• |
impact on our reputation in the marketplace
if manufacturers of our products, once commercialized, fail to meet customer demands; |
|
• |
the possible breach of the manufacturing
agreement by the third party because of factors beyond our control; and |
|
• |
the possible termination or nonrenewal of
the agreement by the third party, based on its own business priorities, at a time that is costly or inconvenient for us. |
|
• |
a covered benefit under its health plan;
|
|
• |
safe, effective and medically necessary;
|
|
• |
appropriate for the specific patient;
|
|
• |
cost-effective; and |
|
• |
neither experimental nor investigational.
|
|
• |
announcements of technological innovations
or new products by us or others; |
|
• |
announcements by us of significant acquisitions,
strategic partnerships, in-licensing, out-licensing, joint ventures or capital commitments; |
|
• |
expiration or terminations of licenses,
research contracts or other collaboration agreements; |
|
• |
public concern as to the safety of drugs
we, our licensees or others develop; |
|
• |
general market conditions; |
|
• |
the volatility of market prices for shares
of biotechnology companies generally; |
|
• |
success of research and development projects;
|
|
• |
departure of key personnel; |
|
• |
developments concerning intellectual property
rights or regulatory approvals; |
|
• |
variations in our and our competitors’
results of operations; |
|
• |
changes in earnings estimates or recommendations
by securities analysts, if our ordinary shares or ADSs are covered by analysts; |
|
• |
statements about the Company made in the
financial media or by bloggers on the Internet; |
|
• |
statements made about drug pricing and other
industry-related issues by government officials; |
|
• |
changes in government regulations or patent
decisions; |
|
• |
developments by our licensees; and
|
|
• |
general market conditions and other factors,
including factors unrelated to our operating performance. |
|
• |
the failure to obtain regulatory approval,
in a timely manner or at all, or achieve commercial success of our therapeutic candidates; |
|
• |
our success in effecting out-licensing arrangements
with third parties; |
|
• |
our success in establishing other out-licensing
or co-development arrangements; |
|
• |
the success of our licensees in selling
products that utilize our technologies; |
|
• |
the results of our preclinical studies and
clinical trials for our earlier stage therapeutic candidates, and any decisions to initiate clinical trials if supported by the preclinical
results; |
|
• |
the costs, timing and outcome of regulatory
review of our therapeutic candidates that progress to clinical trials; |
|
• |
the costs of establishing or acquiring specialty
sales, marketing and distribution capabilities, if any of our therapeutic candidates are approved, and we decide to commercialize them
ourselves; |
|
• |
the costs of preparing, filing and prosecuting
patent applications, maintaining and enforcing our issued patents and defending intellectual property-related claims; |
|
• |
the extent to which we acquire or invest
in businesses, products or technologies and other strategic relationships; and |
|
• |
the costs of financing unanticipated working
capital requirements and responding to competitive pressures. |
|
• |
Revenue sharing payments. These are payments to be made to licensors with respect
to revenue we receive from sub-licensing to third parties for further development and commercialization of our drug products. These payments
are generally fixed at a percentage of the total revenues we earn from these sublicenses. |
|
• |
Milestone payments. These payments are generally linked to the successful achievement
of milestones in the development and approval of drugs, such Phases 1, 2 and 3 of clinical trials and approvals of NDAs, and achievement
of sales milestones. |
|
• |
Royalty payments. To the extent we elect to complete the development, licensing and
marketing of a therapeutic candidate, we are generally required to pay our licensors royalties on the sales of the end drug product. These
royalty payments are generally based on the net revenue from these sales. In certain instances, the rate of the royalty payments decreases
upon the expiration of the drug’s underlying patent and its transition into a generic drug. |
|
• |
Additional payments. In addition to the above payments, certain of our in-license
agreements provide for a one-time or periodic payment that is not linked to milestones. Periodic payments may be paid until the commercialization
of the product, either by direct sales or sublicenses to third parties. Other agreements provide for the continuation of these payments
even following the commercialization of the licensed drug product. |
|
• |
The motixafortide drug product composition of matter and methods of manufacturing thereof are covered by
a granted U.S. patent and patent applications pending in the USA (two applications received notice of allowance). Israel, Europe, Japan,
Canada, Australia, China, India, Mexico, Brazil, Hong-Kong and Korea. The patents, if granted, will expire in December 2041, not including
any applicable patent term extension, which may add an additional term of up to five years for the U.S. patents. We also have an exclusive
license to a patent family that covers motixafortide combined with a PD1 antagonist for the treatment of cancer. Patents of this family
have been granted in the U.S., Israel, Australia, China, India, Mexico and Hong Kong; and member patent applications are pending in Australia,
Hong Kong, Europe, China, Canada, India, Israel and Brazil. The granted U.S. patent and patents to issue in the future based on pending
patent applications in this family will expire in 2036, not including any applicable patent term extension. In addition, we have an exclusive
license to nineteen other patent families pending or granted worldwide directed to methods of synthesis of motixafortide and methods of
use of motixafortide either alone or in combination with other drugs for the treatment of certain types of cancer and other indications.
Furthermore, we have Orphan Drug status for AML, pancreatic cancer and stem cell mobilization, as well as data exclusivity protection
afforded to motixafortide as an NCE. |
|
• |
With respect to BL-5010, we have an exclusive license to a patent family directed
to a novel applicator uniquely configured for applying the BL-5010 composition to targeted skin tissue safely and effectively. Patents
in this family have been granted in the U.S., Europe, Israel, Japan, China, Australia and New Zealand. The patents will expire in 2033-2034.
|
|
|
• |
preclinical laboratory tests, animal studies and formulation development; |
|
|
• |
submission to the FDA of an Investigational New Drug, or IND, application to conduct human clinical testing; |
|
|
• |
adequate and well controlled clinical trials to determine the safety and efficacy of the drug for each indication as well as to establish
the exposure levels; |
|
|
• |
submission to the FDA of an application for marketing approval; |
|
|
• |
satisfactory completion of an FDA inspection of the manufacturing facility or facilities at which the drug is manufactured; and
|
|
|
• |
FDA review and approval of the drug and drug labeling for marketing. |
|
|
• |
Consistent rules for conducting clinical trials throughout the EU; |
|
|
• |
Making information on the authorization, conduct and results of each clinical trial carried out in the EU publicly available;
|
|
|
• |
Harmonized electronic submission and assessment process for clinical trials conducted in multiple member states; |
|
|
• |
Improved collaboration, information sharing and decision-making between and within member states; |
|
|
• |
Increased transparency of information on clinical trials; and |
|
|
• |
Higher standards of safety for all participants in EU clinical trials. |
|
|
• |
the federal Anti-Kickback Statute, which prohibits, among other
things, persons and entities from knowingly and willfully soliciting, offering, receiving or providing remuneration (including any kickback,
bribe or rebate), directly or indirectly, in cash or in kind, to induce or reward either the referral of an individual for, or the purchase,
lease or order of, any good or service, for which payment may be made, in whole or in part, under a federal healthcare program such as
Medicare and Medicaid; |
|
|
• |
the federal civil and criminal false claims laws, including
the civil False Claims Act, and civil monetary penalties laws, which prohibit individuals or entities from, among other things, knowingly
presenting, or causing to be presented, to the federal government, claims for payment that are false or fraudulent or making a false statement
to avoid, decrease or conceal an obligation to pay money to the federal government; |
|
|
• |
the federal Health Insurance Portability and Accountability
Act of 1996, or HIPAA, which created additional federal criminal laws that prohibit, among other things, knowingly and willingly executing,
or attempting to execute, a scheme or making false statements in connection with the delivery of or payment for health care benefits,
items, or services; |
|
|
• |
HIPAA, as amended by the Health Information Technology for
Economic and Clinical Health Act and its implementing regulations, which also imposes obligations, including mandatory contractual terms,
with respect to safeguarding the privacy, security and transmission of individually identifiable health information on covered entities
and their business associates that associates that perform certain functions or activities that involve the use or disclosure of protected
health information on their behalf; |
|
|
• |
the Foreign Corrupt Practices Act, or FCPA, which prohibits
companies and their intermediaries from making, or offering or promising to make improper payments to non-U.S. officials for the purpose
of obtaining or retaining business or otherwise seeking favorable treatment; |
|
|
• |
the federal transparency requirements known as the federal
Physician Payments Sunshine Act, under the Patient Protection and Affordable Care Act, as amended by the Health Care Education Reconciliation
Act, or collectively the ACA, which requires certain manufacturers of drugs, devices, biologics and medical supplies for which payment
is available under Medicare, Medicaid, or the Children’s Health Insurance Program, with specific exceptions, to report annually
to the Centers for Medicare & Medicaid Services, or CMS, within HHS, information related to payments and other transfers of value
to certain healthcare providers and teaching hospitals and information regarding ownership and investment interests held by physicians
and their immediate family members; and |
|
|
• |
analogous state and foreign laws and regulations, such as state
anti-kickback and false claims laws, which may apply to healthcare items or services that are reimbursed by non-governmental third-party
payors, including private insurers. |
|
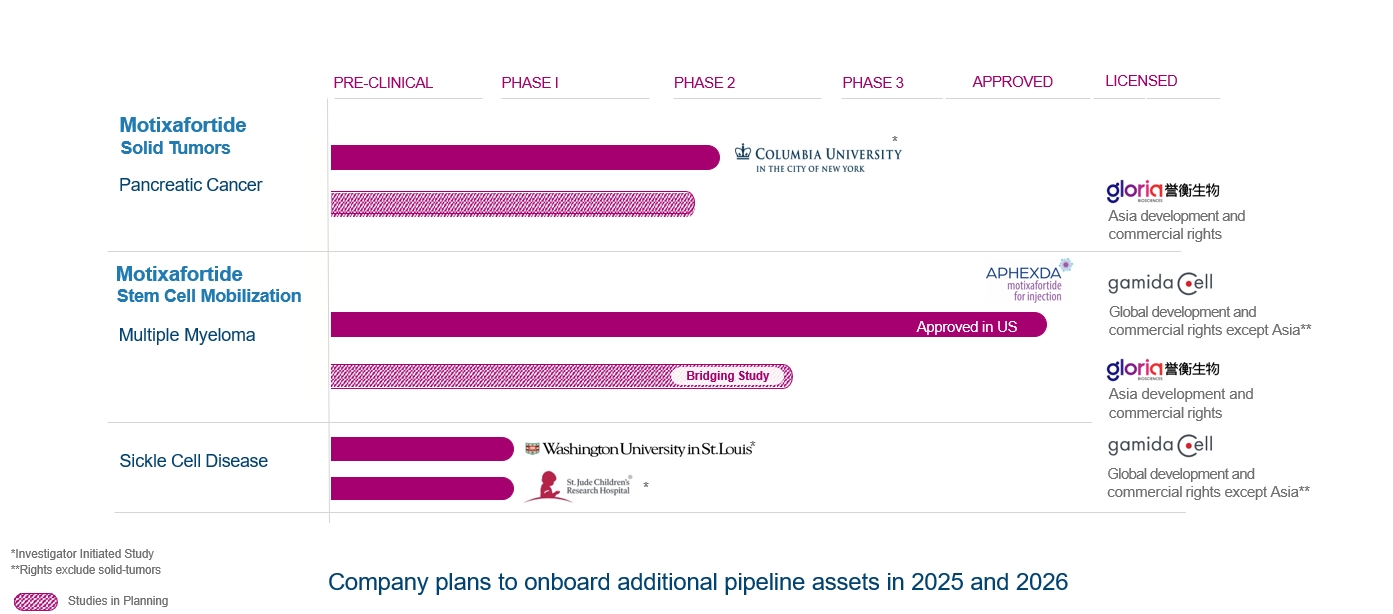
Project |
Status |
Expected Near Term Milestones |
||
|
motixafortide |
1. |
FDA approval received on September 8, 2023 for stem-cell mobilization
in multiple myeloma patients. |
1. |
Out-licensed to Ayrmid in November 2024; five-year long-term
follow-up of GENESIS patients ongoing |
|
2. |
Reported data from single-arm pilot phase of the investigator-initiated
Phase 2 combination trial in first-line PDAC. Of 11 patients with metastatic pancreatic cancer enrolled, 7 patients (64%) experienced
partial response (PR), of which 6 (55%) were confirmed PRs with one patient experiencing resolution of the hepatic (liver) metastatic
lesion. 3 patients (27%) experienced stable disease, resulting in a disease control rate of 91%. Based on these encouraging results, study
was substantially revised to a multi-institution, randomized Phase 2b trial of 108 patients |
2. |
First patient dosed in February 2024. Interim data expected
in 2026 and full enrollment projected for 2027* | |
|
3. |
Phase 1 study for gene therapies in SCD (with Washington University
School of Medicine in St. Louis)** |
3. |
First patient dosed in December 2023 and initial data from
the study released in November 2024. Final data planned in 2025* | |
|
4. |
Phase 1 study for gene therapies in SCD (with St. Jude Children’s
Research Hospital, Inc.)** |
4. |
First patient dosed in February 2025, with data planned in
2025/2026* | |
|
5. |
IND approved in China for initiation of pivotal bridging study
in SCM under license agreement with Gloria |
5. |
Initiation of the study is currently delayed*** |
|
|
6. |
Phase 2b randomized study in first-line PDAC in China under
license agreement with Gloria |
6. |
IND submission and protocol finalization is currently delayed***
| |
|
|
* |
These studies are investigator-initiated studies; therefore, the timelines are ultimately controlled by the independent investigators
and are subject to change. |
|
** |
Study to be continued under
the Ayrmid License Agreement |
|
|
*** |
Under the Gloria License Agreement, Gloria is late in the payment of $2.4 million to
us for the achievement of a specific milestone under the Gloria License Agreement and for certain product supply, which was due during
2024. In addition, the planned studies of motixafortide in China under the Gloria License Agreement are currently not advancing according
to schedule and it is unclear when such studies will be initiated, if at all. There can be no assurance that Gloria will meet its payment
obligations or any other obligations under the Gloria License Agreement. |
|
|
• |
the number of sites included in the clinical trials; |
|
|
• |
the length of time required to enroll suitable patients; |
|
|
|
|
|
|
• |
the number of patients that participate, and are eligible to participate, in the clinical
trials; |
|
|
• |
the duration of patient follow-up; |
|
|
|
|
|
|
• |
whether the patients require hospitalization or can be treated on an outpatient basis;
|
|
|
• |
the development stage of the therapeutic candidate; and |
|
|
• |
the efficacy and safety profile of the therapeutic candidate. |
|
|
• |
identify the contract with a customer; |
|
|
• |
identify the performance obligations in the contract; |
|
|
• |
determine the transaction price; |
|
|
• |
allocate the transaction price to the performance obligations in the contract; and
|
|
|
• |
recognize revenue when (or as) the entity satisfies a performance obligation.
|
|
|
• |
the progress and costs of our preclinical studies, clinical trials and other research
and development activities; |
|
|
• |
the scope, prioritization and number of our clinical trials and other research and
development programs; |
|
|
• |
the amount of revenues we receive, if any, under our collaboration or licensing arrangements;
|
|
|
|
|
|
|
• |
the costs of the development and expansion of our operational infrastructure;
|
|
|
• |
the costs and timing of obtaining regulatory approval of our therapeutic candidates;
|
|
|
• |
our success in effecting out-licensing arrangements with third parties; |
|
|
• |
the ability of our collaborators and licensees to achieve development milestones,
marketing approval and other events or developments under our collaboration and out-licensing agreements; |
|
|
• |
the costs of filing, prosecuting, enforcing and defending patent claims and other
intellectual property rights; |
|
|
• |
the costs and timing of securing manufacturing arrangements for clinical or commercial
production; |
|
|
• |
the costs of establishing sales and marketing capabilities or contracting with third
parties to provide these capabilities for us; |
|
|
• |
the costs of acquiring or undertaking development and commercialization efforts for
any future therapeutic candidates; |
|
|
• |
the magnitude of our general and administrative expenses; |
|
|
• |
interest and principal payments on the loan from BlackRock; |
|
|
• |
any cost that we may incur under current and future licensing arrangements relating
to our therapeutic candidates; and |
|
|
• |
market conditions. |
|
Name |
|
Age |
|
Position(s) |
|
|
|
|
|
|
|
Philip A. Serlin, CPA, MBA |
|
64 |
|
Chief Executive Officer |
|
Mali Zeevi, CPA |
|
49 |
|
Chief Financial Officer |
|
Ella Sorani, Ph.D. |
|
57 |
|
Chief Development Officer |
|
Aharon Schwartz, Ph.D. (1) |
|
82 |
|
Chairman of the Board of Directors, Class III Director |
|
Rami Dar, MBA (1)(2)(3)(4) |
|
68 |
|
Class I Director |
|
B.J. Bormann, Ph.D. (1)(3) |
|
66 |
|
Class II Director |
|
Raphael Hofstein, Ph.D. (1)(2)(3) |
|
75 |
|
Class II Director |
|
Avraham Molcho, M.D. (1)(2)(3) |
|
67 |
|
Class I Director |
|
Sandra Panem, Ph.D. (1)(4) |
|
78 |
|
Class III Director |
|
Shaoyu Yan, Ph.D. |
|
60 |
|
Class III Director |
|
Gal Cohen (1) |
|
52 |
|
Class I Director |
|
(1) |
Independent director under applicable Nasdaq Capital Market, as affirmatively determined by our board of
directors. | |
|
|
| |
|
(2) |
A member of our audit committee. | |
|
|
| |
|
(3) |
A member of our compensation committee. | |
|
|
| |
|
(4) |
A member of our investment monitoring committee |
|
|
Salaries, fees,
commissions
and bonuses |
Pension,
retirement,
options and
other similar benefits |
||||||
|
|
(in thousands of U.S. dollars) |
|||||||
|
All directors and senior management as a group, consisting of 13 persons |
1,912 |
899 |
||||||
|
Name and Position |
Salary |
Social Benefits(1)
|
Bonuses(2)
|
Value of
Share-Based
Compensation(3)
|
All Other
Compensation(4)
|
Total |
||||||||||||||||||
|
|
(in thousands of U.S. dollars) |
|||||||||||||||||||||||
|
Philip A. Serlin
Chief Executive Officer |
296 |
109 |
74 |
265 |
20 |
764 |
||||||||||||||||||
|
Mali Zeevi
Chief Financial Officer |
195 |
59 |
49 |
32 |
22 |
357 |
||||||||||||||||||
|
Ella Sorani
Chief Development Officer |
221 |
54 |
- |
32 |
20 |
328 |
||||||||||||||||||
|
Holly W. May
Former President of BioLineRx USA, Inc.* |
440 |
61 |
226 |
- |
220 |
947 |
||||||||||||||||||
|
(1) |
“Social Benefits” include payments to the National Insurance Institute,
advanced education funds, managers’ insurance and pension funds, vacation pay and recuperation pay as mandated by Israeli law.
|
|
(2) |
With the exception of Ms. May, does not include annual bonuses for 2024, which remain
subject to the approval of the Company’s compensation committee and board of directors. | |
|
(3) |
Consists of amounts recognized as share-based compensation expense on the Company’s
statement of comprehensive loss for the year ended December 31, 2024. |
|
(4) |
“All Other Compensation” includes automobile-related expenses pursuant to the Company’s
automobile leasing program, telephone, basic health insurance and holiday presents, as well as termination benefits to Ms. May.
|
|
|
• |
the Class I directors, consisting of Dr. Avraham Molcho, Mr. Rami Dar and Gal Cohen, will hold office until our annual general meeting
of shareholders to be held in 2027; |
|
|
• |
the Class II directors, consisting of Dr. B.J. Bormann and Dr. Raphael Hofstein, will hold office until our annual general meeting
of shareholders to be held in 2025; and |
|
|
• |
the Class III directors, consisting of Dr. Sandra Panem, Dr. Aharon Schwartz and Dr. Shaoyu Yan, will hold office until our annual
general meeting of shareholders to be held in 2026. |
|
|
• |
oversight of the company’s independent registered public accounting firm and
recommending the engagement, compensation or termination of engagement of our independent registered public accounting firm to our board
of directors in accordance with Israeli law; |
|
|
• |
recommending the engagement or termination of the office of our internal auditor;
and |
|
|
• |
reviewing and pre-approving the terms of audit and non-audit services provided by
our independent auditors. |
|
|
• |
to make recommendations to the board of directors for its approval of (i) a compensation
policy for office holders, (ii) once every three years whether to extend the then current compensation policy (approval of either a new
compensation policy or the continuation of an existing compensation policy must, in any case, occur every three years); and (iii) periodic
updates to the compensation policy which may be required from time to time. In addition, the compensation committee is required
to periodically examine the implementation of the compensation policy; and |
|
|
• |
to approve transactions relating to terms of office and employment of company office
holders that require the approval of the compensation committee pursuant to the Companies Law (including determining whether the compensation
terms of a candidate for chief executive officer of the company need not be brought to approval of the shareholders). |
|
|
• |
the majority of the votes voted in favor includes at least a majority of all the votes
of shareholders who are not controlling shareholders of the company and shareholders who do not have a personal interest in the compensation
policy, present and voting on the matter (excluding abstentions); or |
|
|
• |
the total of opposing votes from among the shareholders who are non-controlling shareholders
and shareholders who do not have a personal interest in the matter does not exceed 2% of all the voting rights in the company. |
|
|
• |
a person (or a relative of a person) who holds more than 5% of the company’s
shares; |
|
|
• |
a person (or a relative of a person) who has the power to appoint a director or the
general manager of the company; |
|
|
• |
an executive officer or director of the company (or a relative thereof); or
|
|
|
• |
a member of the company’s independent accounting firm. |
|
|
• |
information on the advisability of a given action brought for his or her approval
or performed by virtue of his or her position; and |
|
|
• |
all other important information pertaining to these actions. |
|
|
• |
refrain from any act involving a conflict of interest between the performance of his
or her duties in the company and his or her other duties or personal affairs; |
|
|
• |
refrain from any activity that is competitive with the business of the company;
|
|
|
• |
refrain from exploiting any business opportunity of the company for the purpose of
gaining a personal advantage for himself or herself or others; and |
|
|
• |
disclose to the company any information or documents relating to the company’s
affairs which the office holder received as a result of his or her position as an office holder. |
|
|
• |
a transaction other than in the ordinary course of business; |
|
|
• |
a transaction that is not on market terms; or |
|
|
• |
a transaction that may have a material impact on the company’s profitability, assets or liabilities.
|
|
|
• |
Executive officers other than the Chief Executive
Officer. The compensation of an office holder in a public company who is neither a director nor the chief executive officer
generally requires approval by the (i) compensation committee; and (ii) the board of directors. Approval of terms of office and
employment for such officers which do not comply with the compensation policy may nonetheless be approved, in special circumstances, subject
to two cumulative conditions: (i) the compensation committee and thereafter the board of directors, approved the terms after having taken
into account the various considerations and mandatory requirements set forth in the Companies Law with respect a compensation policy,
and (ii) the shareholders of the company have approved the terms by the Special Majority for Compensation . However, if the shareholders
do not approve a compensation arrangement with an executive officer that is inconsistent with the company’s compensation policy,
a company’s compensation committee and board of directors, may, in special circumstances approve the compensation despite shareholder
objection, provided that the compensation committee and thereafter the board of directors have determined to approve the compensation
based on detailed reasoning, after each having re- discussed the terms of compensation, and after examining the objection of the shareholders.
|
|
|
• |
Chief Executive Officer. The compensation
of a chief executive officer in a public company generally requires approval by the (i) compensation committee; (ii) the board of directors;
and (iii) the shareholders of the company by the Special Majority for Compensation. Approval of the compensation terms of a chief executive
officer which do not comply with the compensation policy may nonetheless be approved, in special circumstances, subject to two cumulative
conditions: (i) the compensation committee and thereafter the board of directors, approved the terms after having taken into account the
various considerations and mandatory requirements set forth in the Companies Law with respect to a compensation policy and (ii) the shareholders
of the company have approved the terms by means of the Special Majority for Compensation . However, a company’s compensation committee
and board of directors, may, in special circumstances approve the compensation of a chief executive officer (who is not a director) that
is not approved by shareholders despite shareholder objection, provided that the company’s compensation committee and thereafter
the board of directors have determined to approve the compensation, based on detailed reasoning, after each having re-discussed the terms
of office and employment, and after examining the objection of the shareholders. In addition, the compensation committee may exempt from
shareholder approval the compensation terms of a candidate for the office of chief executive officer where such officer has no prior business
relationship with the controlling shareholder or the company, if it has found, based on detailed reasons, that bringing the compensation
to the approval of the shareholders would impede the employment of such candidate by the company, provided that the terms of office and
employment are in accordance with the company’s compensation policy. |
|
|
• |
Directors. The compensation of a director
(who is not the chief executive officer) of a public company generally requires approval by the (i) compensation committee; (ii) the board
of directors; and (iii) unless exempted under regulations promulgated under the Companies Law, the shareholders of the company. Approval
of terms of the compensation of directors of a company which do not comply with the compensation policy may nonetheless be approved, in
special circumstances, subject to two cumulative conditions: (i) the compensation committee and thereafter the board of directors, approved
the terms after having taken into account the various considerations and mandatory requirements set forth in the Companies Law with respect
to a compensation policy and (ii) the shareholders of the company have approved the terms by means of the Special Majority for Compensation.
|
|
|
• |
at least a majority of the shares held by shareholders who have no personal interest in the matter who
are present and voting at the meeting must be voted in favor of approving the transaction, excluding abstentions; or |
|
|
• |
the shares voted by shareholders who have no personal interest in the matter who are present and vote against
the transaction represent no more than 2% of the voting rights in the company. |
|
|
• |
an amendment to the articles of association; |
|
|
• |
an increase in the company’s authorized share capital; |
|
|
• |
a merger; and |
|
|
• |
the approval of related party transactions and acts of office holders that require shareholder approval
under the Companies Law. |
|
|
• |
monetary liability imposed on him or her in favor of another person pursuant to a
judgment, including a settlement or arbitrator’s award approved by a court. However, if an undertaking to indemnify an office holder
with respect to such liability is provided in advance, then such an undertaking must be limited to events which, in the opinion of the
board of directors, can be foreseen based on the company’s activities when the undertaking to indemnify is given, and to an amount
or according to criteria determined by the board of directors as reasonable under the circumstances, and such undertaking shall detail
the abovementioned foreseen events and amount or criteria; |
|
|
• |
reasonable litigation expenses, including attorneys’ fees, incurred by the office
holder (i) as a result of an investigation or proceeding instituted against him or her by an authority authorized to conduct such investigation
or proceeding, provided that (1) no indictment was filed against such office holder as a result of such investigation or proceeding; and
(2) no financial liability was imposed upon him or her as a substitute for the criminal proceeding as a result of such investigation or
proceeding or, if such financial liability (such as a criminal penalty) was imposed, it was imposed with respect to an offense that does
not require proof of criminal intent and (ii) in connection with a monetary sanction; |
|
|
• |
a monetary liability imposed on an office holder in favor of an injured party at an Administrative Procedure
(as defined below) pursuant to Section 52(54)(a)(1)(a) of the Israeli Securities Law; |
|
|
• |
expenses incurred by an office holder or certain compensation payments made to an injured party that were
instituted against an office holder in connection with an Administrative Procedure under the Israeli Securities Law, including reasonable
litigation expenses and reasonable attorneys’ fees; and |
|
|
• |
reasonable litigation expenses, including attorneys’ fees, incurred by the office
holder or imposed by a court in proceedings instituted against him or her by the company, on its behalf or by a third party or in connection
with criminal proceedings in which the office holder was acquitted or as a result of a conviction for an offense that does not require
proof of criminal intent. |
|
|
• |
a breach of duty of loyalty to the company, provided that the office holder acted in good faith and had
a reasonable basis to believe that the act would not prejudice the company; |
|
|
• |
a breach of duty of care to the company or to a third party, including a breach arising out of the negligent
(but not intentional or reckless) conduct of the office holder; |
|
|
• |
a financial liability imposed on the office holder in favor of a third party; |
|
|
• |
a monetary liability imposed on the office holder in favor of an injured party in an Administrative Procedure
pursuant to Section 52(54)(a)(1)(a) of the Israeli Securities Law; and |
|
|
• |
expenses, including reasonable litigation expenses and reasonable attorneys’ fees, incurred by an
office holder in connection with an Administrative Procedure instituted against him or her pursuant to certain provisions of the Israeli
Securities Law. |
|
|
• |
a breach of duty of loyalty, except for indemnification and insurance for a breach of the duty of loyalty
to the company to the extent that the office holder acted in good faith and had a reasonable basis to believe that the act would not prejudice
the company; |
|
|
• |
a breach of duty of care committed intentionally or recklessly, excluding a breach arising out of the negligent
conduct of the office holder; |
|
|
• |
an act or omission committed with intent to derive illegal personal benefit; or |
|
|
• |
a fine, monetary sanction or forfeit levied against the office holder. |
|
|
December 31, |
|||||||||||
|
|
2022 |
2023 |
2024 |
|||||||||
|
|
||||||||||||
|
Management and administration |
12 |
12 |
8 |
|||||||||
|
Research and development |
29 |
29 |
19 |
|||||||||
|
Commercialization and business development |
8 |
38 |
1 |
|||||||||
|
Total |
49 |
79 |
28 |
|||||||||
|
|
● |
each of our directors and senior management; |
|
|
● |
all of our directors and senior management as a group; and |
|
|
● |
each person (or group of affiliated persons) known by us to be the beneficial owner
of 5% or more of the outstanding ordinary shares. |
|
|
Number of |
|||||||
|
|
Ordinary Shares |
|||||||
|
|
Beneficially |
Percent of |
||||||
|
|
Held |
Class |
||||||
|
|
||||||||
|
5% or Greater Shareholder |
||||||||
|
Hong Seng Technology Limited(1)
|
102,437,055 |
4.6 |
% | |||||
|
Intracoastal Capital LLC(2)
|
102,236,115 |
4.6 |
% | |||||
|
Directors |
||||||||
|
|
||||||||
|
Aharon Schwartz(3)
|
6,071,400 |
* |
||||||
|
B.J. Bormann(4) |
2,366,400 |
* |
||||||
|
Rami Dar(5) |
1,917,600 |
* |
||||||
|
Raphael Hofstein(6)
|
2,366,400 |
* |
||||||
|
Avraham Molcho(7)
|
2,366,400 |
* |
||||||
|
Sandra Panem(8) |
2,366,400 |
* |
||||||
|
Shaoyu Yan |
- |
|||||||
|
Gal Cohen (9) |
342,600 |
* |
||||||
|
|
||||||||
|
Executive officers |
||||||||
|
|
||||||||
|
Philip A. Serlin(10)
|
16,166,400 |
* |
||||||
|
Mali Zeevi(11) |
4,279,200 |
* |
||||||
|
Ella Sorani(12) |
4,176,000 |
* |
||||||
|
All directors and executive officers as a group (11 persons)(13)
|
42,418,800 |
1.1 |
% | |||||
|
(1) |
Based on Schedule 13D filed with the SEC on October 26, 2023. According to the Schedule
13D, includes 170,728 ADS, representing 102,437,055 ordinary shares held by Hong Seng Technology Limited. Lepu (Hong Kong) Co.,
Limited holds 66.67% equity interest of Hong Seng Technology Limited. Lepu Holdings Limited holds 99.5% equity interest of Lepu
(Hong Kong) Co., Limited. Lepu Medical (Europe) Cooperatief U.A. holds 100% equity interest of Lepu Holdings Limited. Lepu Medical
Technology (Beijing) Co., Ltd. holds 99.95% equity interest of Lepu Medical (Europe) Cooperatief U.A. Lepu Medical Technology (Beijing)
Co., Ltd. is a company publicly listed on Shenzhen Stock Exchange in the PRC (300003.SZ). |
|
(2) |
Based on Schedule 13G filed with the SEC on January 10, 2025.
According to the Schedule 13G, includes (i) 3,125 ADSs representing 1,875,000 ordinary shares and (ii) 45,394 ADSs representing 27,236,115
ordinary shares issuable upon exercise of a warrant issued in January 2025. Mitchell P. Kopin and Daniel B. Asher, each of whom
are managers of Intracoastal Capital LLC, or Intracoastal, have shared voting control and investment discretion over the securities reported
herein that are held by Intracoastal. As a result, each of Mr. Kopin and Mr. Asher may be deemed to have beneficial ownership (as determined
under Section 13(d) of the Exchange Act) of the securities reported herein that are held by Intracoastal. The warrant is subject to a
beneficial ownership limitation of 4.99%, which such limitation restricts the shareholder from exercising that portion of the warrant
that would result in the shareholder and its affiliates owning, after exercise, a number of shares in excess of the beneficial ownership
limitation. The principal business office of Intracoastal is 245 Palm Trail, Delray Beach, FL 33483. |
|
(3) |
Includes 3,705,000 ordinary shares and 2,366,400 ordinary shares issuable upon exercise
of outstanding options currently exercisable or exercisable within 60 days of March 26, 2025. Does not include 1,937,400 ordinary shares
issuable upon exercise of outstanding options that are not exercisable within 60 days of March 26, 2025. |
|
(4) |
Includes 2,366,400 ordinary shares issuable upon exercise of outstanding options currently exercisable
or exercisable within 60 days of March 26, 2025. Does not include 1,937,400 ordinary shares issuable upon exercise of outstanding options
that are not exercisable within 60 days of March 26, 2025. |
|
(5) |
Includes 1,917,600 ordinary shares issuable upon exercise of outstanding options currently
exercisable or exercisable within 60 days of March 26, 2025. Does not include 1,937,400 ordinary shares issuable upon exercise of outstanding
options that are not exercisable within 60 days of March 26, 2025. |
|
(6) |
Includes 2,366,400 ordinary shares issuable upon exercise of
outstanding options currently exercisable or exercisable within 60 days of March 26, 2025. Does not include 1,937,400 ordinary shares
issuable upon exercise of outstanding options that are not exercisable within 60 days of March 26, 2025. |
|
(7) |
Includes 2,366,400 ordinary
shares issuable upon exercise of outstanding options currently exercisable or exercisable within 60 days of March 26, 2025. Does
not include 1,937,400 ordinary shares issuable upon exercise of outstanding options that are not exercisable within 60 days of March 26,
2025. |
|
(8) |
Includes 2,366,400 ordinary shares issuable upon exercise of outstanding options currently exercisable
or exercisable within 60 days of March 26, 2025. Does not include 1,937,400 ordinary shares issuable upon exercise of outstanding options
that are not exercisable within 60 days of March 26, 2025. |
|
(9) |
Includes 342,600 ordinary shares issuable upon exercise of outstanding
options currently exercisable or exercisable within 60 days of March 26, 2025. Does not include 1,712,400 ordinary shares issuable upon
exercise of outstanding options that are not exercisable within 60 days of March 26, 2025. |
|
(10) |
Includes 171,600 ordinary shares and 15,994,800 ordinary
shares issuable upon exercise of outstanding options and PSUs currently exercisable or exercisable within 60 days of March 26,
2025. Does not include 6,006,000 ordinary shares issuable upon exercise of outstanding options and PSUs that are not exercisable within
60 days of March 26, 2025. |
|
(11) |
Includes 329,400 ordinary shares and 3,949,800 ordinary shares issuable upon exercise of outstanding options
and PSUs currently exercisable or exercisable within 60 days of March 26, 2025. Does not include 1,441,200 ordinary shares issuable upon
exercise of outstanding options and PSUs that are not exercisable within 60 days of March 26, 2025. |
|
(12) |
Includes 66,600 ordinary shares and 4,109,400 ordinary shares issuable upon exercise of outstanding options
and PSUs currently exercisable or exercisable within 60 days of March 26, 2025. Does not include 1,441,200 ordinary shares issuable upon
exercise of outstanding options and PSUs that are not exercisable within 60 days of March 26, 2025. |
|
(13) |
See footnotes (3)-(12) for certain information regarding beneficial ownership.
|
|
• |
amendments to our Articles of Association; |
|
• |
appointment, termination or the terms of service of our auditors; |
|
• |
appointment of external directors (if applicable); |
|
• |
approval of certain related party transactions; |
|
• |
increases or reductions of our authorized share capital; |
|
• |
a merger; and |
|
• |
the exercise of our board of directors’ powers by a general meeting, if our
board of directors is unable to exercise its powers and the exercise of any of its powers is required for our proper management.
|
|
|
• |
the excess distribution or gain would be allocated ratably over the Non-Electing U.S.
Investor’s holding period for the ordinary shares or ADSs; |
|
|
• |
the amount allocated to the current taxable year and any year prior to us becoming
a PFIC would be taxed as ordinary income; and |
|
|
• |
the amount allocated to each of the other taxable years would be subject to tax at
the highest rate of tax in effect for the applicable class of taxpayer for that year, and an interest charge for the deemed deferral benefit
would be imposed with respect to the resulting tax attributable to each such other taxable year. |
|
|
• |
taxes and other governmental charges; |
|
|
• |
any applicable transfer or registration fees; |
|
|
• |
certain cable, telex and facsimile transmission charges as provided in the deposit
agreement; |
|
|
• |
any expenses incurred in the conversion of foreign currency; |
|
|
• |
a fee of $5.00 or less per 100 ADSs (or a portion thereof) for the execution and delivery
of ADRs and the surrender of ADRs, including if the deposit agreement terminates; |
|
|
• |
a fee of $.01 or less per ADS (or portion thereof) for any cash distribution made
pursuant to the deposit agreement; |
|
|
• |
a fee for the distribution of securities pursuant to the deposit agreement;
|
|
|
• |
in addition to any fee charged for a cash distribution, a fee of $.01 or less per
ADS (or portion thereof) per annum for depositary services; |
|
|
• |
a fee for the distribution of proceeds of rights that the Depositary sells pursuant
to the deposit agreement; and |
|
|
• |
any other charges payable by the Depositary, any of the Depositary’s agents,
or the agents of the Depositary’s agents in connection with the servicing of ordinary shares or other Deposited Securities.
|
|
a. |
Disclosure Controls and Procedures
|
|
b. |
Management’s Annual Report on Internal
Control over Financial Reporting |
|
c. |
Attestation Report of Registered Public Accounting
Firm |
|
d. |
Changes in Internal Control over Financial
Reporting |
|
|
Year Ended December 31, |
|||||||
|
|
2023 |
2024 |
||||||
|
Services Rendered |
(in thousands of U.S. dollars) |
|||||||
|
|
||||||||
|
Audit Fees(1) |
130 |
160 |
||||||
|
Audit-Related Fees(2)
|
17 |
40 |
||||||
|
Tax Fees(3) |
52 |
41 |
||||||
|
Total |
199 |
211 |
||||||
|
(1) |
Audit fees consist of services that would normally be provided in connection with statutory and regulatory
filings or engagements, including services that generally only the independent accountant can reasonably provide. | |
|
|
| |
|
(2) |
Audit-related services relate to reports to the IIA and work regarding a public listing or offering.
| |
|
|
| |
|
(3) |
Tax fees relate to tax reports for BioLineRx USA, Inc., tax compliance, planning
and advice. |
|
|
• |
Distribution of periodic reports to shareholders.
Under Israeli law, a public company whose shares are traded on the TASE, is not required to distribute periodic reports directly to shareholders
and the generally accepted business practice in Israel is not to distribute such reports to shareholders but to make such reports publicly
available through a public website. We will only mail such reports to shareholders upon request. In addition, we make our audited financial
statements available to our shareholders at our offices. |
|
|
• |
Quorum. While the Nasdaq Rules require that
the quorum for purposes of any meeting of the holders of a listed company’s common voting stock, as specified in the company’s
bylaws, be no less than 33 1/3% of the company’s outstanding common voting stock, as permitted under the Companies Law, our Articles
of Association provide that a quorum of two or more shareholders holding at least 25% of the voting rights in person or by proxy is required
for commencement of business at a general meeting (and, with respect to an adjourned meeting, a quorum consists of any number of shareholders
present in person or by proxy). |
|
|
• |
Nomination of Directors. We follow Israeli
corporate governance practices instead of the requirements of the Nasdaq Rules with regard to the nomination committee and director nomination
procedures. Israeli law and practice does not require director nominations to be made by a nominating committee of our board of
directors consisting solely of independent directors, as required under the Nasdaq Rules. In accordance with Israeli law and practice,
directors are recommended by our board of directors for election by our shareholders (other than directors elected by our board of directors
to fill a vacancy), and certain of our shareholders may nominate candidates for election as directors by the general meeting of shareholders
in accordance with the Companies Law and our Articles of Association. |
|
|
• |
Compensation of Officers. We follow Israeli
law and practice with respect to the approval of officer compensation, pursuant to which transactions with office holders regarding their
terms of office and employment generally require the approval of the compensation committee, the board of directors and under certain
circumstances (such as if the officer is a director or controlling shareholder) the shareholders, either in accordance with our compensation
policy or, in special circumstances in deviation therefrom, taking into account certain considerations set forth in the Companies Law.
See “Item 6.C— Directors, Senior Management and Employees — Board Practices — Compensation Committee” for
information regarding the Compensation Committee, and “Item 6.C — Directors, Senior Management and Employees — Approval
of Related Party Transactions under Israeli Law” for information regarding the approvals required with respect to approval of terms
of office and employment of office holders, pursuant to the Companies Law. |
|
|
• |
Approval of Related Party Transactions.
We follow Israeli law and practice with respect to the approval of interested party acts and transactions, as set forth in sections 268
to 275 of the Companies Law, and the regulations promulgated thereunder, which generally require the approval of the audit committee,
the board of directors and, under certain circumstances (such as if the transaction is with a controlling shareholder or another party
in which the controlling shareholder has a personal interest) the shareholders, as may be applicable, for specified transactions. See
“Item 6.C— Directors, Senior Management and Employees —Board Practices — Approval of Related Party Transactions
under Israeli Law” for information regarding the approvals required with respect to approval of related party transactions pursuant
to the Companies Law. |
|
|
• |
Shareholder Approval. We intend to seek shareholder
approval for all corporate actions requiring such approval in accordance with the requirements of the Companies Law, which are different
or in addition to the requirements for seeking shareholder approval under Nasdaq Listing Rule 5635, rather than seeking approval for corporation
actions in accordance with such listing rules. |
|
|
• |
Equity Compensation Plans. We do not necessarily
seek shareholder approval for the establishment of, and amendments to, stock option or equity compensation plans (as set forth in Nasdaq
Listing Rule 5635(c)), as such matters are not subject to shareholder approval under Israeli law and practice. However, any equity-based
compensation arrangement with a director or the chief executive officer or the material amendment of such an arrangement must be approved
by our Compensation Committee, board of directors and shareholders, in that order. |
109
|
Exhibit
Number
|
|
Exhibit Description
|
|
101
|
The following financial information from BioLineRx Ltd.’s Annual Report on Form 20-F for the fiscal year ended December 31, 2023 formatted in Inline XBRL (Extensible Business Reporting Language): (i) Consolidated Statements of Financial Position at December 31, 2023 and 2022; (ii) Consolidated Statements of Comprehensive Loss for the years ended December 31, 2023, 2022 and 2021; (iii) Statements of Changes in Equity for the years ended December 31, 2023, 2022 and 2021; (iv) Consolidated Cash Flow Statements for the years ended December 31, 2023, 2022 and 2021; and (v) Notes to the Consolidated Financial Statements.
|
|
*
|
Filed herewith.
|
|
†
|
Portions of this exhibit have been omitted and filed separately with the Securities and Exchange Commission pursuant to a confidential treatment request.
|
|
(1)
|
Incorporated by reference to the Registrant’s Annual Report on Form 20-F filed on February 23, 2021.
|
|
(2)
|
Incorporated by reference to Exhibit 1 of the Registration Statement on Form F-6EF (No. 333-218969) filed by the Bank of New York Mellon on June 26, 2017 with respect to the Registrant’s American Depositary Shares.
|
|
(3)
|
Incorporated by reference to the Registrant’s Annual Report on Form 20-F filed on March 23, 2017.
|
|
(4)
|
Incorporated by reference to the Registrant’s Registration Statement on Form 20-F (No. 001-35223) filed on July 1, 2011.
|
|
(5)
|
Incorporated by reference to the Registrant’s Annual Report on Form 20-F filed on March 10, 2016.
|
|
(6)
|
Incorporated by reference to the Registrant’s Annual Report on Form 20-F/A filed on May 31, 2016.
|
|
(7)
|
Incorporated by reference to the Registrant’s Form 6-K filed on October 3, 2018.
|
|
(8)
|
Incorporated by reference to the Registrant’s Form 6-K filed on May 27, 2022.
|
|
(9)
|
Incorporated by reference to the Registrant’s Annual Report on Form 20-F filed on March 12, 2020.
|
|
(10)
|
Incorporated by reference to the Registrant’s Annual Report on Form 20-F filed on March 23, 2015.
|
|
(11)
|
Incorporated by reference to the Registrant’s Annual Report on Form 20-F/A filed on September 22, 2015.
|
|
(12)
|
Incorporated by reference to the Registrant’s Form 6-K filed on February 7, 2019.
|
|
(13)
|
Incorporated by reference to the Registrant’s Form 6-K filed on January 21, 2021.
|
|
(14)
|
Incorporated by reference to the Registrant’s Form 6-K filed on September 3, 2021.
|
|
(15)
|
Incorporated by reference to the Registrant’s Form 6-K filed on September 15, 2022.
|
|
(16)
|
Incorporated by reference to the Registrant’s Form 6-K filed on September 21, 2022.
|
|
(17)
|
Incorporated by reference to the Registrant’s Annual Report on Form 20-F filed on March 16, 2022.
|
|
(18)
|
Incorporated by reference to the Registrant’s Form 6-K filed on August 30, 2023.
|
|
(19)
|
Incorporated by reference to the Registrant’s Annual Report on Form 20-F filed on March 26, 2024.
|
|
(20)
|
Incorporated by reference to the Registrant’s Form 6-K filed on April 1, 2024.
|
|
(21)
|
Incorporated by reference to the Registrant’s Form 6-K filed on November 21, 2024.
|
|
(22)
|
Incorporated by reference to the Registrant’s Form 6-K filed on January 7, 2025.
|
|
|
BIOLINERX LTD.
|
|
|
|
|
|
|
|
|
|
By:
|
/s/ Philip A. Serlin
|
|
|
|
|
Philip A. Serlin
|
|
|
|
|
Chief Executive Officer
|
|
| Page | |
|
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM (PCAOB name: Kesselman & Kesselman C.P.A.s and PCAOB ID No. 1309)
|
F-2 |
|
CONSOLIDATED FINANCIAL STATEMENTS:
|
|
| F-4 | |
| F-5 | |
| F-6 | |
| F-7 | |
| F-9 |

|
/s/ Kesselman & Kesselman
|
|
Certified Public Accountants (Isr.)
|
|
A member firm of PricewaterhouseCoopers International Ltd.
|
|
Note
|
December 31,
|
||||||||||
|
2023
|
2024
|
||||||||||
|
in USD thousands
|
|||||||||||
|
Assets
|
|||||||||||
|
CURRENT ASSETS
|
|||||||||||
|
Cash and cash equivalents
|
5
|
4,255
|
10,436
|
||||||||
|
Short-term bank deposits
|
6
|
38,739
|
9,126
|
||||||||
|
Trade receivables
|
358
|
2,476
|
|||||||||
|
Prepaid expenses
|
1,048
|
443
|
|||||||||
|
Other receivables
|
20a
|
830
|
1,478
|
||||||||
|
Inventory
|
7
|
1,953
|
3,145
|
||||||||
|
Total current assets
|
47,183
|
27,104
|
|||||||||
|
NON-CURRENT ASSETS
|
|||||||||||
|
Property and equipment, net
|
8
|
473
|
386
|
||||||||
|
Right-of-use assets, net
|
10
|
1,415
|
967
|
||||||||
|
Intangible assets, net
|
9
|
14,854
|
10,449
|
||||||||
|
Total non-current assets
|
16,742
|
11,802
|
|||||||||
|
Total assets
|
63,925
|
38,906
|
|||||||||
|
Liabilities and equity
|
|||||||||||
|
CURRENT LIABILITIES
|
|||||||||||
|
Current maturities of long-term loan
|
11
|
3,145
|
4,479
|
||||||||
|
Contract liabilities
|
16
|
12,957
|
-
|
||||||||
|
Accounts payable and accruals:
|
|||||||||||
|
Trade
|
20b
|
10,869
|
5,583
|
||||||||
|
Other
|
20b
|
3,353
|
3,131
|
||||||||
|
Current maturities of lease liabilities
|
10
|
528
|
522
|
||||||||
|
Warrants
|
12c
|
11,932
|
1,691
|
||||||||
|
Total current liabilities
|
42,784
|
15,406
|
|||||||||
|
NON-CURRENT LIABILITIES
|
|||||||||||
|
Long-term loan, net of current maturities
|
11
|
6,628
|
8,958
|
||||||||
|
Lease liabilities
|
10
|
1,290
|
1,081
|
||||||||
|
Total non-current liabilities
|
7,918
|
10,039
|
|||||||||
|
COMMITMENTS AND CONTINGENT LIABILITIES
|
15
|
||||||||||
|
Total liabilities
|
50,702
|
25,445
|
|||||||||
|
EQUITY
|
12
|
||||||||||
|
Ordinary shares
|
31,355
|
38,097
|
|||||||||
|
Share premium
|
355,482
|
353,693
|
|||||||||
|
Warrants
|
1,408
|
5,367
|
|||||||||
|
Capital reserve
|
17,000
|
17,547
|
|||||||||
|
Other comprehensive loss
|
(1,416
|
)
|
(1,416
|
)
|
|||||||
|
Accumulated deficit
|
(390,606
|
)
|
(399,827
|
)
|
|||||||
|
Total equity
|
13,223
|
13,461
|
|||||||||
|
Total liabilities and equity
|
63,925
|
38,906
|
|||||||||
F - 4
|
Note
|
Year ended December 31,
|
||||||||||||||
|
2022
|
2023
|
2024
|
|||||||||||||
|
in USD thousands
|
|||||||||||||||
|
REVENUES:
|
|||||||||||||||
|
License revenues
|
16, 17
|
-
|
4,610
|
22,917
|
|||||||||||
|
Product sales, net
|
-
|
190
|
6,023
|
||||||||||||
|
Total revenues
|
-
|
4,800
|
28,940
|
||||||||||||
|
COST OF REVENUES
|
20c
|
-
|
(3,692
|
)
|
(9,263
|
)
|
|||||||||
|
GROSS PROFIT
|
-
|
1,108
|
19,677
|
||||||||||||
|
RESEARCH AND DEVELOPMENT EXPENSES
|
20d
|
(17,629
|
)
|
(12,519
|
)
|
(9,149
|
)
|
||||||||
|
SALES AND MARKETING EXPENSES
|
20e
|
(6,462
|
)
|
(25,270
|
)
|
(23,605
|
)
|
||||||||
|
GENERAL AND ADMINISTRATIVE EXPENSES
|
20f
|
(5,066
|
)
|
(6,310
|
)
|
(6,321
|
)
|
||||||||
|
IMPAIRMENT OF INTANGIBLE ASSETS
|
9
|
-
|
(6,703
|
)
|
(1,010
|
)
|
|||||||||
|
OPERATING LOSS
|
(29,157
|
)
|
(49,694
|
)
|
(20,408
|
)
|
|||||||||
|
NON-OPERATING INCOME (EXPENSES), NET
|
20g
|
5,670
|
(10,819
|
)
|
18,435
|
||||||||||
|
FINANCIAL INCOME
|
20h
|
694
|
2,068
|
1,871
|
|||||||||||
|
FINANCIAL EXPENSES
|
20i
|
(2,158
|
)
|
(2,169
|
)
|
(9,119
|
)
|
||||||||
|
LOSS AND COMPREHENSIVE LOSS
|
(24,951
|
)
|
(60,614
|
)
|
(9,221
|
)
|
|||||||||
|
in USD
|
|||||||||||||||
|
LOSS PER ORDINARY SHARE – BASIC AND DILUTED
|
14
|
(0.03
|
)
|
(0.06
|
)
|
(0.01
|
)
|
||||||||
|
WEIGHTED AVERAGE NUMBER OF SHARES USED IN CALCULATION OF LOSS PER ORDINARY SHARE
|
14
|
773,956,973
|
963,365,525
|
1,198,107,761
|
|||||||||||
F - 5
|
Ordinary
shares
|
Share
premium
|
Warrants
|
Capital
reserve
|
Other
comprehensive
loss
|
Accumulated
deficit
|
Total
|
||||||||||||||||||||||
|
in USD thousands
|
||||||||||||||||||||||||||||
|
BALANCE AT JANUARY 1, 2022
|
21,066
|
339,346
|
975
|
13,157
|
(1,416
|
)
|
(305,041
|
)
|
68,087
|
|||||||||||||||||||
|
CHANGES IN 2022:
|
||||||||||||||||||||||||||||
|
Issuance of share capital and warrants, net
|
6,029
|
(1,007
|
)
|
433
|
-
|
-
|
-
|
5,455
|
||||||||||||||||||||
|
Employee stock options exercised
|
5
|
14
|
-
|
(14
|
)
|
-
|
-
|
5
|
||||||||||||||||||||
|
Employee stock options expired
|
-
|
623
|
-
|
(623
|
)
|
-
|
-
|
-
|
||||||||||||||||||||
|
Share-based compensation
|
-
|
-
|
-
|
2,245
|
-
|
-
|
2,245
|
|||||||||||||||||||||
|
Comprehensive loss for the year
|
-
|
-
|
-
|
-
|
(24,951
|
)
|
(24,951
|
)
|
||||||||||||||||||||
|
BALANCE AT DECEMBER 31, 2022
|
27,100
|
338,976
|
1,408
|
14,765
|
(1,416
|
)
|
(329,992
|
)
|
50,841
|
|||||||||||||||||||
|
CHANGES IN 2023:
|
||||||||||||||||||||||||||||
|
Issuance of share capital, net
|
3,242
|
10,847
|
-
|
-
|
-
|
-
|
14,089
|
|||||||||||||||||||||
|
Warrants exercised
|
1,000
|
5,559
|
-
|
-
|
-
|
-
|
6,559
|
|||||||||||||||||||||
|
Employee stock options exercised
|
13
|
45
|
-
|
(31
|
)
|
-
|
-
|
27
|
||||||||||||||||||||
|
Employee stock options expired
|
-
|
55
|
-
|
(55
|
)
|
-
|
-
|
-
|
||||||||||||||||||||
|
Share-based compensation
|
-
|
-
|
-
|
2,321
|
-
|
-
|
2,321
|
|||||||||||||||||||||
|
Comprehensive loss for the year
|
-
|
-
|
-
|
-
|
-
|
(60,614
|
)
|
(60,614
|
)
|
|||||||||||||||||||
|
BALANCE AT DECEMBER 31, 2023
|
31,355
|
355,482
|
1,408
|
17,000
|
(1,416
|
)
|
(390,606
|
)
|
13,223
|
|||||||||||||||||||
|
CHANGES IN 2024:
|
||||||||||||||||||||||||||||
|
Issuance of share capital and warrants, net
|
4,712
|
(3,060
|
)
|
6,650
|
-
|
-
|
-
|
8,302
|
||||||||||||||||||||
|
Pre-funded warrants exercised
|
2,009
|
682
|
(2,691
|
)
|
-
|
-
|
-
|
-
|
||||||||||||||||||||
|
Employee stock options exercised
|
21
|
50
|
-
|
(49
|
)
|
-
|
-
|
22
|
||||||||||||||||||||
|
Employee stock options expired
|
-
|
539
|
-
|
(539
|
)
|
-
|
-
|
-
|
||||||||||||||||||||
|
Share-based compensation
|
-
|
-
|
-
|
1,135
|
-
|
-
|
1,135
|
|||||||||||||||||||||
|
Comprehensive loss for the year
|
-
|
-
|
-
|
-
|
-
|
(9,221
|
)
|
(9,221
|
)
|
|||||||||||||||||||
|
BALANCE AT DECEMBER 31, 2024
|
38,097
|
353,693
|
5,367
|
17,547
|
(1,416
|
)
|
(399,827
|
)
|
13,461
|
|||||||||||||||||||
F - 6
|
Year ended December 31,
|
||||||||||||
|
2022
|
2023
|
2024
|
||||||||||
|
in USD thousands
|
||||||||||||
|
CASH FLOWS - OPERATING ACTIVITIES
|
||||||||||||
|
Loss
|
(24,951
|
)
|
(60,614
|
)
|
(9,221
|
)
|
||||||
|
Adjustments required to reflect net cash used in operating activities (see appendix below)
|
(1,289
|
)
|
38,006
|
(34,652
|
)
|
|||||||
|
Net cash used in operating activities
|
(26,240
|
)
|
(22,608
|
)
|
(43,873
|
)
|
||||||
|
CASH FLOWS - INVESTING ACTIVITIES
|
||||||||||||
|
Investments in short-term deposits
|
(44,000
|
)
|
(47,588
|
)
|
(26,350
|
)
|
||||||
|
Maturities of short-term deposits
|
48,322
|
49,329
|
55,778
|
|||||||||
|
Purchase of property and equipment
|
(131
|
)
|
(116
|
)
|
(53
|
)
|
||||||
|
Purchase of intangible assets
|
(185
|
)
|
(181
|
)
|
(1
|
)
|
||||||
|
Net cash provided by investing activities
|
4,006
|
1,444
|
29,374
|
|||||||||
|
CASH FLOWS - FINANCING ACTIVITIES
|
||||||||||||
|
Issuance of share capital and warrants, net of issuance costs
|
14,359
|
14,089
|
16,357
|
|||||||||
|
Exercise of warrants
|
-
|
2,928
|
-
|
|||||||||
|
Employee stock options exercised
|
5
|
27
|
22
|
|||||||||
|
Proceeds from long-term loan, net of issuance costs
|
9,126
|
-
|
19,223
|
|||||||||
|
Repayments of loan
|
(2,832
|
)
|
(1,543
|
)
|
(14,433
|
)
|
||||||
|
Repayments of lease liabilities
|
(220
|
)
|
(445
|
)
|
(511
|
)
|
||||||
|
Net cash provided by financing activities
|
20,438
|
15,056
|
20,658
|
|||||||||
|
INCREASE (DECREASE) IN CASH AND CASH EQUIVALENTS
|
(1,796
|
)
|
(6,108
|
)
|
6,159
|
|||||||
|
CASH AND CASH EQUIVALENTS - BEGINNING
OF YEAR
|
12,990
|
10,587
|
4,255
|
|||||||||
|
EXCHANGE DIFFERENCES ON CASH AND CASH EQUIVALENTS
|
(607
|
)
|
(224
|
)
|
22
|
|||||||
|
CASH AND CASH EQUIVALENTS - END OF YEAR
|
10,587
|
4,255
|
10,436
|
|||||||||
F - 7
|
Year ended December 31,
|
||||||||||||
|
2022
|
2023
|
2024
|
||||||||||
|
in USD thousands
|
||||||||||||
|
APPENDIX
|
||||||||||||
|
Adjustments required to reflect net cash used in operating activities:
|
||||||||||||
|
Income and expenses not involving cash flows:
|
||||||||||||
|
Depreciation and amortization
|
654
|
1,384
|
4,065
|
|||||||||
|
Exchange differences on cash and cash equivalents
|
607
|
224
|
(22
|
)
|
||||||||
|
Fair value adjustments of warrants
|
(6,425
|
)
|
11,054
|
(18,965
|
)
|
|||||||
|
Share-based compensation
|
2,245
|
2,321
|
1,135
|
|||||||||
|
Interest and exchange differences on short-term deposits
|
(672
|
)
|
15
|
185
|
||||||||
|
Interest on loan
|
1,117
|
1,148
|
(1,126
|
)
|
||||||||
|
Warrant issuance costs
|
171
|
-
|
669
|
|||||||||
|
Exchange differences on lease liabilities
|
(224
|
)
|
(42
|
)
|
(31
|
)
|
||||||
|
Intangible assets impairment
|
-
|
6,703
|
1,010
|
|||||||||
|
Loss on abandonment of right-of-use asset
|
-
|
-
|
246
|
|||||||||
|
(2,527
|
)
|
22,807
|
(12,834
|
)
|
||||||||
|
Changes in operating asset and liability items:
|
||||||||||||
|
Increase in trade receivables
|
-
|
(358
|
)
|
(2,118
|
)
|
|||||||
|
Increase in inventory
|
-
|
(1,953
|
)
|
(1,192
|
)
|
|||||||
|
Increase in prepaid expenses and other receivables
|
(650
|
)
|
(959
|
)
|
(43
|
)
|
||||||
|
Increase (decrease) in accounts payable and accruals
|
1,888
|
5,512
|
(5,508
|
)
|
||||||||
|
Increase (decrease) in contract liabilities
|
-
|
12,957
|
(12,957
|
)
|
||||||||
|
1,238
|
15,199
|
(21,818
|
)
|
|||||||||
|
(1,289
|
)
|
38,006
|
(34,652
|
)
|
||||||||
|
Supplemental information on interest received in cash
|
342
|
2,020
|
1,992
|
|||||||||
|
Supplemental information on interest paid in cash
|
593
|
1,111
|
10,387
|
|||||||||
|
Supplemental information on non-cash transactions:
|
||||||||||||
|
Changes in right-of-use asset and lease liabilities
|
706
|
149
|
327
|
|||||||||
|
Warrant issuance costs
|
262
|
-
|
-
|
|||||||||
|
Purchase of property and equipment
|
28
|
-
|
-
|
|||||||||
|
Fair value of exercised warrants (portion related to accumulated fair value adjustments)
|
-
|
3,631
|
-
|
|||||||||
F - 8
| a. |
General
|
F - 9
BioLineRx Ltd.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
| b. |
War in Israel
|
F - 10
BioLineRx Ltd.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
| c. |
Going concern
|
| d. |
Change in ratio of ADSs
|
| e. |
Approval of consolidated financial statements
|
F - 11
BioLineRx Ltd.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
| a. |
Basis of presentation
|
| b. |
Functional and reporting currency
|
| c. |
Inventory
|
F - 12
BioLineRx Ltd.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
| d. |
Property and equipment
|
|
%
|
|
|
Computers and communications equipment
|
33
|
|
Office furniture and equipment
|
6
|
|
Laboratory equipment
|
15
|
| e. |
Intangible assets
|
| f. |
Impairment of non-financial assets
|
F - 13
BioLineRx Ltd.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
| g. |
Warrants
|
| h. |
Borrowings
|
F - 14
BioLineRx Ltd.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
| i. |
Revenues
|
| • |
identify the contract with a customer;
|
| • |
identify the performance obligations in the contract;
|
| • |
determine the transaction price;
|
| • |
allocate the transaction price to the performance obligations in the contract; and
|
| • |
recognize revenue when (or as) the entity satisfies a performance obligation.
|
| 1. |
Distribution fees - The Company pays distribution fees to its three main distributors. The distribution fees are paid based on contractually determined rates from the gross consideration. When the service is received and the products sold to distributors, it is recognized as a reduction of revenues in the period the related revenues from the sale of products are recognized.
|
| 2. |
Rebates and patient discount programs - The Company offers various rebate and patient discount programs, which result in discounted prescriptions to qualified patients. The Company estimates the allowance for these rebates, based on the estimated utilization of the rebate and discount programs, at the time the revenues are recognized. These estimates are recognized as a reduction of revenues.
|
| 3. |
Product returns - The Company offers customers a right of return as part of the distributor agreements. The Company estimates the amount of product sales that may be returned by its customers and records this estimate as a reduction of revenues at the time of sale, based on estimates of product returns based on its own sales information, its visibility into the inventory remaining in the distribution channel, and product dating.
|
F - 15
BioLineRx Ltd.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
NOTE 2 – MATERIAL ACCOUNTING POLICIES (cont.)
| i. |
Revenues (cont.)
|
|
| 1) |
Development milestones: Variable payments, contingent on achieving additional milestones, are included in the transaction price based on the most likely amount method. Amounts included in the transaction price are recognized only when it is highly probable that a material reversal of cumulative revenues will not occur, usually upon achievement of the specific milestone, in accordance with the relevant agreement.
|
| 2) |
Sales-based royalties and sales-based milestones are recognized as the related sale occurs, due to the specific exception of IFRS 15 for sales-based royalties from licensing of intellectual properties.
|
| j. |
Research and development expenses
|
F - 16
BioLineRx Ltd.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
| k. |
Share-based payments
|
| l. |
Loss per share
|
| 1) |
Basic
|
| 2) |
Diluted
|
F - 17
BioLineRx Ltd.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
NOTE 2 – MATERIAL ACCOUNTING POLICIES (cont.)
| m. |
Leases |
|
Years
|
|
|
Property
|
11
|
|
Motor vehicles
|
3
|
F - 18
BioLineRx Ltd.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
| n. |
New International Financial Reporting Standards, amendments to standards and new interpretations:
|
F - 19
BioLineRx Ltd.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
| a. |
Market risk
|
| 1) |
Concentration of currency risk
|
|
December 31, 2024
|
||||||||||||||||||||
|
Income (loss)
|
Value on
|
Income (loss)
|
||||||||||||||||||
|
Sensitive instrument
|
10% increase
|
5% increase
|
balance sheet
|
5% decrease
|
10% decrease
|
|||||||||||||||
|
in USD thousands
|
||||||||||||||||||||
|
NIS-linked balances:
|
||||||||||||||||||||
|
Cash and cash equivalents
|
(87
|
)
|
(45
|
)
|
955
|
50
|
106
|
|||||||||||||
|
Other receivables
|
(52
|
)
|
(27
|
)
|
568
|
30
|
63
|
|||||||||||||
|
Trade payables
|
115
|
60
|
(933
|
)
|
(67
|
)
|
(141
|
)
|
||||||||||||
|
Other payables
|
154
|
81
|
(1,495
|
)
|
(89
|
)
|
(188
|
)
|
||||||||||||
|
Total NIS-linked balances
|
130
|
69
|
(905
|
)
|
(76
|
)
|
(160
|
)
|
||||||||||||
|
Euro-linked trade payables
|
(83
|
)
|
(44
|
)
|
(919
|
)
|
48
|
102
|
||||||||||||
|
Total
|
47
|
25
|
(1,824
|
)
|
(28
|
)
|
(58
|
)
|
||||||||||||
|
December 31, 2023
|
||||||||||||||||||||
|
Income (loss)
|
Value on
|
Income (loss)
|
||||||||||||||||||
|
Sensitive instrument
|
10% increase
|
5% increase
|
balance sheet
|
5% decrease
|
10% decrease
|
|||||||||||||||
|
in USD thousands
|
||||||||||||||||||||
|
NIS-linked balances:
|
||||||||||||||||||||
|
Cash and cash equivalents
|
(132
|
)
|
(69
|
)
|
1,457
|
77
|
162
|
|||||||||||||
|
Short term deposit
|
(251
|
)
|
(131
|
)
|
2,758
|
145
|
306
|
|||||||||||||
|
Other receivables
|
(31
|
)
|
(16
|
)
|
344
|
18
|
38
|
|||||||||||||
|
Trade payables
|
243
|
127
|
(2,670
|
)
|
(141
|
)
|
(297
|
)
|
||||||||||||
|
Other payables
|
169
|
89
|
(1,853
|
)
|
(98
|
)
|
(207
|
)
|
||||||||||||
|
Total NIS-linked balances
|
(2
|
)
|
0
|
36
|
1
|
2
|
||||||||||||||
|
Euro-linked trade payables
|
(117
|
)
|
(61
|
)
|
(1,283
|
)
|
67
|
142
|
||||||||||||
|
Total
|
(119
|
)
|
(61
|
)
|
(1,247
|
)
|
68
|
144
|
||||||||||||
F - 20
BioLineRx Ltd.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
| a. |
Market risk (cont.)
|
| 1) |
Concentration of currency risk (cont.)
|
|
|
December 31, 2023
|
December 31, 2024
|
|||||||||||||||||||||||
|
Dollar
|
NIS
|
Other currencies
|
Dollar
|
NIS
|
Other Currencies
|
|||||||||||||||||||
|
USD in thousands
|
USD in thousands
|
|||||||||||||||||||||||
|
Assets:
|
||||||||||||||||||||||||
|
Current assets:
|
||||||||||||||||||||||||
|
Cash and cash equivalents
|
2,768
|
1,457
|
30
|
9,428
|
955
|
53
|
||||||||||||||||||
|
Short-term bank deposits
|
35,981
|
2,758
|
-
|
9,126
|
-
|
-
|
||||||||||||||||||
|
Other receivables
|
480
|
350
|
-
|
819
|
568
|
91
|
||||||||||||||||||
|
39,229
|
4,565
|
30
|
19,373
|
1,523
|
144
|
|||||||||||||||||||
|
Liabilities:
|
||||||||||||||||||||||||
|
Current liabilities:
|
||||||||||||||||||||||||
|
Current maturities of long-term loan
|
3,145
|
-
|
-
|
4,479
|
-
|
-
|
||||||||||||||||||
|
Accounts payable and accruals:
|
||||||||||||||||||||||||
|
Trade
|
6,663
|
2,670
|
1,536
|
3,549
|
933
|
1,101
|
||||||||||||||||||
|
Other
|
1,500
|
1,853
|
-
|
1,636
|
1,495
|
-
|
||||||||||||||||||
|
Non-current liabilities
|
||||||||||||||||||||||||
|
Long-term loan, net of current maturities
|
6,628
|
-
|
-
|
8,958
|
-
|
-
|
||||||||||||||||||
|
17,936
|
4,523
|
1,536
|
18,622
|
2,428
|
1,101
|
|||||||||||||||||||
|
Net balance
|
21,293
|
42
|
(1,506
|
)
|
751
|
(905
|
)
|
(957
|
)
|
|||||||||||||||
F - 21
BioLineRx Ltd.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
| a. |
Market risk (cont.)
|
|
| 2) |
Fair value of financial instruments
|
| 3) |
Exposure to market risk and management thereof
|
| 4) |
Interest rate risk
|
| b. |
Credit risk
|
|
December 31,
|
||||||||
|
2023
|
2024
|
|||||||
|
in USD thousands
|
||||||||
|
Assets:
|
||||||||
|
Cash and cash equivalents
|
4,255
|
10,436
|
||||||
|
Short-term bank deposits
|
38,739
|
9,126
|
||||||
|
Trade receivables
|
358
|
2,476
|
||||||
|
Other receivables
|
830
|
1,478
|
||||||
|
Total
|
44,182
|
23,516
|
||||||
F - 22
BioLineRx Ltd.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
| c. |
Liquidity risk
|
| d. |
Fair value of financial instruments
|
|
Level 1
|
Quoted prices (unadjusted) in active markets for identical assets or liabilities.
|
|
Level 2
|
Inputs, other than quoted prices included within level 1 that are observable for the asset or liability, either directly (as prices) or indirectly (derived from prices).
|
|
Level 3
|
Inputs for the asset or liability that are not based on observable market data (unobservable inputs).
|
F - 23
BioLineRx Ltd.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
| e. |
Changes in financial liabilities with cash flows included in financing activities
|
|
|
Long-term loan
|
Warrants
|
Total
|
||||||||||
|
in USD thousands
|
||||||||||||
|
Balance as of January 1, 2022
|
2,757
|
1,859
|
4,616
|
|||||||||
|
Changes during the year 2022:
|
||||||||||||
|
Net proceeds
|
9,126
|
9,075
|
18,201
|
|||||||||
|
Principal and interest payments
|
(3,177
|
)
|
-
|
(3,177
|
)
|
|||||||
|
Amounts recognized through profit and loss
|
1,462
|
(6,425
|
)
|
(4,963
|
)
|
|||||||
|
Balance as of December 31, 2022
|
10,168
|
4,509
|
14,677
|
|||||||||
|
Changes during the year 2023:
|
||||||||||||
|
Principal and interest payments
|
(1,543
|
)
|
-
|
(1,543
|
)
|
|||||||
|
Amounts recognized through profit and loss
|
1,148
|
11,054
|
12,202
|
|||||||||
|
Share premium resulting from exercise of warrants
|
-
|
(3,631
|
)
|
(3,631
|
)
|
|||||||
|
Balance as of December 31, 2023
|
9,773
|
11,932
|
21,705
|
|||||||||
|
Changes during the year 2024:
|
||||||||||||
|
Net proceeds
|
19,223
|
8,724
|
27,947
|
|||||||||
|
Principal and interest payments
|
(22,795
|
)
|
-
|
(22,795
|
)
|
|||||||
|
Amounts recognized through profit and loss
|
7,236
|
(18,965
|
)
|
(11,729
|
)
|
|||||||
|
Balance as of December 31, 2024
|
13,437
|
1,691
|
15,128
|
|||||||||
| f. |
Fair value measurement of warrants using significant unobservable inputs (level 3)
|
|
Warrants
|
||||
|
in USD thousands
|
||||
|
Balance as of January 1, 2022
|
1,859
|
|||
|
Changes during 2022:
|
||||
|
Issuances
|
9,075
|
|||
|
Changes in fair value through profit and loss
|
(6,425
|
)
|
||
|
Balance as of December 31, 2022
|
4,509
|
|||
|
Changes during 2023:
|
||||
|
Exercises
|
(3,631
|
)
|
||
|
Changes in fair value through profit and loss
|
11,054
|
|||
|
Balance as of December 31, 2023
|
11,932
|
|||
|
Changes during 2024:
|
||||
|
Issuances
|
8,724
|
|||
|
Changes in fair value through profit and loss
|
(18,965
|
) | ||
|
Balance as of December 31, 2024
|
1,691
|
|||
F - 24
BioLineRx Ltd.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
F - 25
BioLineRx Ltd.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
| • |
The allocation of consideration between the license agreement and the SPA, based on the fair value of the Company’s shares on the date considered as the closing date of the transaction
|
| • |
The estimated stand-alone, selling-price value between the contract components (i.e., between the main therapeutic areas covered by the contract), as well as the performance obligations relating to each of the components
|
| • |
The period of time over which revenue should be recognized for each component. The revenue recognition method is the ratio of support hours to the total hours expected to be incurred.
|
|
December 31,
|
||||||||
|
2023
|
2024
|
|||||||
|
in USD thousands
|
||||||||
|
Cash on hand and in bank
|
3,154
|
944
|
||||||
|
Short-term bank deposits
|
1,101
|
9,492
|
||||||
|
4,255
|
10,436
|
|||||||
F - 26
BioLineRx Ltd.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
|
December 31,
|
||||||||
|
2023
|
2024
|
|||||||
|
in USD thousands
|
||||||||
|
Raw materials
|
903
|
-
|
||||||
|
Work-in-progress
|
471
|
2,980
|
||||||
|
Finished goods
|
579
|
165
|
||||||
|
1,953
|
3,145
|
|||||||
F - 27
BioLineRx Ltd.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
|
Cost
|
Accumulated depreciation
|
|||||||||||||||||||||||||||||||||||||||
|
Balance at
|
Additions
|
Deletions
|
Balance at
|
Balance at
|
Additions
|
Deletions
|
Balance at
|
Net book value
|
||||||||||||||||||||||||||||||||
|
beginning
|
during
|
during
|
end of
|
beginning
|
during
|
during
|
end of
|
December 31,
|
||||||||||||||||||||||||||||||||
|
of year
|
year
|
year
|
year
|
of year
|
year
|
year
|
year
|
2023
|
2024
|
|||||||||||||||||||||||||||||||
|
in USD thousands
|
in USD thousands
|
in USD thousands
|
||||||||||||||||||||||||||||||||||||||
|
Composition in 2024
|
||||||||||||||||||||||||||||||||||||||||
|
Office furniture and equipment
|
252
|
-
|
1
|
251
|
165
|
27
|
1
|
191
|
87
|
60
|
||||||||||||||||||||||||||||||
|
Computers and communications equipment
|
1,062
|
17
|
612
|
467
|
820
|
83
|
612
|
291
|
242
|
176
|
||||||||||||||||||||||||||||||
|
Laboratory equipment
|
1,613
|
36
|
78
|
1,571
|
1,598
|
5
|
78
|
1,525
|
15
|
46
|
||||||||||||||||||||||||||||||
|
Leasehold improvements
|
2,036
|
-
|
-
|
2,036
|
1,907
|
25
|
-
|
1,932
|
129
|
104
|
||||||||||||||||||||||||||||||
|
4,963
|
53
|
691
|
4,325
|
4,490
|
140
|
691
|
3,939
|
473
|
386
|
|||||||||||||||||||||||||||||||
F - 28
BioLineRx Ltd.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
| - |
$6.7 million originally recorded as a result of the acquisition of Agalimmune, primarily related to its main drug candidate, AGI-134 (see Note 1a). In December 2023, the Company made a decision to terminate the development of AGI-134. Accordingly, the intellectual property related to AGI-134 was written-off in the 2023 financial statements.
|
| - |
$15.0 million associated with BL-8040 were recorded following an amendment to the in-licensing agreement with Biokine Therapeutics Ltd. (“Biokine”). This amendment reduced the payments owed by the Company on sublicense receipts (as defined in the license agreement) from 40% to 20%. This intellectual property is amortized proportionally with the revenues recognized from the licensing transaction with HST and Gloria in Asia (see Note 16), and with Ayrmid for the rest of the world (excluding solid tumors) (see Note 17). During the period of self-commercialization in the U.S. of motixafortide in 2024, this intellectual property was amortized in accordance with the lifespan of the patents in the U.S. In addition, as a result of an impairment review of its remaining rights to motixafortide in solid tumor indications in the fourth quarter of 2024, the Company recorded an impairment loss of $1.0 million. Sensitivity tests on impairment of the remaining rights to motixafortide in solid tumor indications were carried out as of December 31, 2024, by increasing the WACC by 0.5% and by decreasing the annual growth rate of new cases by 0.5%. Such changes, if realized, would result in additional impairment charges of $0.7 million and $0.6 million, respectively.
|
|
Cost
|
Accumulated depreciation and impairment
|
|||||||||||||||||||||||||||||||||||||||||||||||
|
Balance at
|
Additions
|
Deletions
|
Disposal
|
Balance at
|
Balance at
|
Additions
|
Deletions
|
Impairment
|
Balance at
|
Net book value
|
||||||||||||||||||||||||||||||||||||||
|
beginning
|
during
|
during
|
during
|
end of
|
beginning
|
during
|
during
|
during
|
end of
|
December 31,
|
||||||||||||||||||||||||||||||||||||||
|
of year
|
year
|
year
|
year
|
year
|
of year
|
year
|
year
|
year
|
year
|
2023
|
2024
|
|||||||||||||||||||||||||||||||||||||
|
in USD thousands
|
in USD thousands
|
in USD thousands
|
||||||||||||||||||||||||||||||||||||||||||||||
|
Composition in 2023
|
||||||||||||||||||||||||||||||||||||||||||||||||
|
Intellectual property
|
21,792
|
-
|
-
|
450
|
21,342
|
96
|
-
|
-
|
6,703
|
6,799
|
14,543
|
- | ||||||||||||||||||||||||||||||||||||
|
Computer software
|
801
|
181
|
-
|
-
|
982
|
612
|
59
|
-
|
-
|
671
|
311
|
- | ||||||||||||||||||||||||||||||||||||
|
22,593
|
181
|
-
|
450
|
22,324
|
708
|
59
|
-
|
6,703
|
7,470
|
14,854
|
- | |||||||||||||||||||||||||||||||||||||
|
Composition in 2024
|
||||||||||||||||||||||||||||||||||||||||||||||||
|
Intellectual property
|
21,342
|
-
|
- |
3,323
|
18,019
|
6,799
|
-
|
- |
1,010
|
7,809
|
14,543
|
10,210
|
||||||||||||||||||||||||||||||||||||
|
Computer software
|
982
|
1
|
185
|
-
|
798
|
671
|
73
|
185
|
-
|
559
|
311
|
239
|
||||||||||||||||||||||||||||||||||||
|
22,324
|
1
|
185
|
3,323
|
18,817
|
7,470
|
73
|
185
|
1,010
|
8,368
|
14,854
|
10,449
|
|||||||||||||||||||||||||||||||||||||
F - 29
BioLineRx Ltd.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
| A. |
Right-of-use assets
|
|
Cost
|
Accumulated depreciation
|
|||||||||||||||||||||||||||||||||||||||
|
Balance at
|
Additions
|
Deletions
|
Balance at
|
Balance at
|
Additions
|
Deletions
|
Balance at
|
Net book value
|
||||||||||||||||||||||||||||||||
|
beginning
|
during
|
during
|
end of
|
beginning
|
during
|
during
|
end of
|
December 31,
|
||||||||||||||||||||||||||||||||
|
of year
|
year
|
year
|
year
|
of year
|
year
|
year
|
year
|
2023
|
2024
|
|||||||||||||||||||||||||||||||
|
in USD thousands
|
in USD thousands
|
in USD thousands
|
||||||||||||||||||||||||||||||||||||||
|
Composition in 2024
|
||||||||||||||||||||||||||||||||||||||||
|
Property
|
2,097
|
239
|
785
|
1,551
|
970
|
379
|
539
|
810
|
1,127
|
741
|
||||||||||||||||||||||||||||||
|
Motor vehicles
|
368
|
88
|
-
|
456
|
80
|
150
|
-
|
230
|
288
|
226
|
||||||||||||||||||||||||||||||
|
2,465
|
327
|
785
|
2,007
|
1,050
|
529
|
539
|
1040
|
1,415
|
967
|
|||||||||||||||||||||||||||||||
| B. |
Lease liabilities
|
|
Balance at
|
Additions
|
Deletions
|
Interest expense
|
Exchange differences
|
Payments
|
Balance at
|
||||||||||||||||||||||
|
beginning
|
during
|
during
|
during
|
during
|
during
|
end of
|
||||||||||||||||||||||
|
of year
|
year
|
year
|
year
|
year
|
year
|
year
|
||||||||||||||||||||||
|
in USD thousands
|
||||||||||||||||||||||||||||
|
Composition in 2024
|
||||||||||||||||||||||||||||
|
Property
|
1,556
|
239
|
-
|
208
|
(23
|
)
|
571
|
1,409
|
||||||||||||||||||||
|
Motor vehicles
|
262
|
88
|
-
|
32
|
(8
|
)
|
180
|
194
|
||||||||||||||||||||
|
1,818
|
327
|
-
|
240
|
(31
|
)
|
751
|
1,603
|
|||||||||||||||||||||
F - 30
BioLineRx Ltd.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
| C. |
Additional disclosures
|
| 1) |
The Company leases two premises – its corporate headquarters and development facilities in Modi’in, Israel, and its U.S. commercial headquarters in Waltham, Massachusetts.
|
| a. |
The Company leases its premises in Israel under a lease agreement entered into in August 2014. Payments under the lease commenced in June 2015, and the initial term of the lease expired in June 2020. The lease agreement included three options to extend the lease through June 30, 2030 in the aggregate, each at a 5% increase to the preceding lease payment amount. The Company exercised its first option to extend the lease through June 30, 2025, and is currently making monthly lease payments of approximately $26,000. In December 2024, the Company notified the lessor of its intention to exercise the second lease extension option through June 30, 2028.
|
| b. |
The Company leases its premises in Boston under a lease agreement entered into and commencing in October 2022. The lease term will expire in December 2025. The monthly lease fee is approximately $24,000. Following the shutdown of US operations towards the end of 2024, the full amount of the lease commitment was accrued, and the related right-of-use asset, with a net value of $246,000, was written off in its entirety in the 2024 financial statements.
|
| 2) |
The Company has entered into lease agreements in connection with a number of vehicles. The lease periods are generally for three years. The annual lease fees, linked to the CPI, are approximately $244,000. To secure the terms of the lease agreements, the Company has prepaid two months of lease payments to the leasing companies.
|
| 3) |
As of December 31, 2024, minimum future rental payments (taking into consideration the aforementioned extension periods) under the leases are as follows:
|
|
Year
|
Property
|
Motor vehicles
|
Total
|
||||||||||
|
in USD thousands
|
|||||||||||||
|
2025
|
569
|
146
|
715
|
||||||||||
|
2026
|
286
|
61
|
347
|
||||||||||
|
2027
|
300
|
13
|
313
|
||||||||||
|
2028-2030
|
751
|
-
|
751
|
||||||||||
|
1,906
|
220
|
2,126
|
|||||||||||
F - 31
BioLineRx Ltd.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
F - 32
BioLineRx Ltd.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
| a. |
Share capital
|
|
Number of Ordinary Shares
|
||||||||
|
December 31,
|
||||||||
|
2023
|
2024
|
|||||||
|
Authorized share capital
|
2,500,000,000
|
5,000,000,000
|
||||||
|
Issued and paid-up share capital
|
1,086,589,165
|
1,336,670,575
|
||||||
|
In USD and NIS Amounts
|
||||||||
|
December 31,
|
||||||||
|
2023
|
2024
|
|||||||
|
Authorized share capital (in NIS)
|
250,000,000
|
500,000,000
|
||||||
|
Issued and paid-up share capital (in NIS)
|
108,658,916
|
133,667,057
|
||||||
|
Issued and paid-up share capital (in USD)
|
31,355,056
|
38,096,940
|
||||||
| b. |
Rights related to shares
|
F - 33
BioLineRx Ltd.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
| c. |
Changes in the Company’s equity
|
| 1) |
In September 2022, the Company completed a registered direct offering of 340,909 ADSs at a price of $44.00 per ADS. The Company also issued to investors in the offering unregistered warrants to purchase 340,909 ADSs. The warrants are exercisable immediately, expire five years from the date of issuance and have an exercise price of $46.00 per ADS. In addition, the Company granted to the placement agent in the offering, as part of the placement fee, warrants to purchase 17,045 ADSs. These warrants are exercisable immediately, expire five years from the date of issuance and have an exercise price of $55.00 per ADS. Gross proceeds from the offering totaled $15.0 million, with net proceeds of $13.5 million, after deducting fees and expenses. The offering consideration allocated to the placement agent warrants amounted to $0.4 million.
The warrants issued to the investors have been classified as a financial liability due to a net settlement provision. This liability was initially recognized at its fair value on the issuance date and is subsequently accounted for at fair value at each balance sheet date. The fair value changes are charged to non-operating income and expense in the statement of comprehensive loss.
The fair value of the warrants is computed using the Black-Scholes option pricing model. The fair value of the warrants upon issuance was computed based on the then-current price of an ADS, a risk-free interest rate of 3.62%, and an average standard deviation of 82.5%. The gross consideration initially allocated to the investor warrants amounted to $9.1 million, with total issuance costs initially allocated to the warrants amounting to $0.8 million.
The fair value of the warrants amounted to $478,000 as of December 31, 2024 (December 31, 2023 - $11,905,000), and was based on the then current price of an ADS, a risk-free interest rate of 4.27%, an average standard deviation of 89.61%, and on the remaining contractual life of the warrants.
The changes in fair value for the years ended December 31, 2023 and 2024 of ($11,033,000) and $11,427,000, respectively, have been recorded as non-operating income (expenses) in the statement of comprehensive loss.
As of December 31, 2024, 63,636 of these warrants had been exercised.
The placement agent warrants have been classified in shareholders’ equity, with initial recognition at fair value on the date issued, using the same assumptions as the investor warrants.
|
| 2) |
In August 2023, the Company entered into a securities purchase agreement, pursuant to which the Company agreed to sell in a private placement an aggregate of 170,728 ADSs of the Company, at a purchase price of $85.44 per ADS. Aggregate gross proceeds from the sale, which were received by the Company at closing, amounted to $14.6 million, with related issuance costs amounting to approximately $0.9 million. Pursuant to IFRS 15, approximately $12.0 million of gross proceeds and $0.7 million of issuance costs were recognized as equity (see Note 16).
|
|
F - 34
BioLineRx Ltd.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
| c. |
Changes in the Company’s equity (cont.)
|
|
| 3) |
In April 2024, the Company completed a registered direct offering of 187,500 ADSs at a price of $32.00 per ADS. The Company also issued to investors in the offering unregistered warrants to purchase 187,500 ADSs. The warrants are exercisable immediately, expire five years from the date of issuance and have an exercise price of $32.00 per ADS. Gross proceeds from the offering totaled $6.0 million, with net proceeds of $5.4 million, after deducting fees and expenses.
The warrants have been classified as a financial liability due to a net settlement provision. This liability was initially recognized at its fair value on the issuance date and is subsequently accounted for at fair value at each balance sheet date. The fair value changes are charged to non-operating income and expense in the statement of comprehensive loss.
The fair value of the warrants is computed using the Black-Scholes option pricing model and is determined by using a level 3 valuation technique. The fair value of the warrants upon issuance was computed based on the then-current price of an ADS, a risk-free interest rate of 4.21%, and an average standard deviation of 84.7%. The fair value initially allocated to the investor warrants amounted to $6,250,000, with total issuance costs initially allocated to the warrants amounting to $642,000.
Due to a difference between the fair value at initial recognition and the transaction price (“day 1 loss”), upon initial recognition, the fair value of the warrants was adjusted by the amount of $250,000, to reflect the unrecognized day 1 loss. Following initial recognition, the unrecognized day 1 loss of the warrants is being amortized over its contractual life.
The fair value of the warrants amounted to $679,000 as of December 31, 2024, and was based on the then current price of an ADS, a risk-free interest rate of 4.33%, an average standard deviation of 89.1%, and on the remaining contractual life of the warrants. The changes in fair value for the years ended December 31, 2024, amounting to $5,571,000, have been recorded as a non-operating income in the statement of comprehensive loss.
As of December 31, 2024, none of these warrants had been exercised.
|
|
F - 35
BioLineRx Ltd.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
| c. |
Changes in the Company’s equity (cont.)
|
| 4) |
In November 2024, the Company completed a registered direct offering to certain funds associated with Highbridge Capital Management LLC (“Highbridge”) of 103,037 ADSs and 308,749 pre-funded warrants to purchase ADSs. Each ADS and pre-funded warrant was sold at a purchase price of $21.86 and $21.85, respectively. The Company also issued to the investors unregistered ordinary warrants to purchase an aggregate of 205,893 ADSs. Gross proceeds from the Offering totaled $9.0 million, with net proceeds of $8.9 million, after deducting fees and expenses. See Note 17.
The pre-funded warrants are exercisable immediately, do not expire until exercised in full, and have an exercise price of $0.004 per ADS. The ordinary warrants are exercisable immediately, expire four years from the date of issuance, and have an exercise price of $23.60 per ADS.
A holder of the pre-funded or ordinary warrants cannot exercise such warrants if the holder, together with its affiliates, would beneficially own in excess of 4.99% of the outstanding share capital of the Company immediately after giving effect to such exercise.
The ordinary warrants have been classified as a financial liability due to a net settlement provision. This liability was initially recognized at its fair value on the issuance date and is subsequently accounted for at fair value at each balance sheet date. The fair value changes are charged to non-operating income and expense in the statement of comprehensive loss.
The pre-funded warrants have been classified in shareholders’ equity, with initial recognition at fair value on the date issued, using the same assumptions as the ordinary warrants.
The fair value of the ordinary warrants is computed using the Black-Scholes option pricing model. The fair value of the ordinary warrants upon issuance was computed based on the then-current price of an ADS, a risk-free interest rate of 4.19%, and an average standard deviation of 84.5%. The gross consideration initially allocated to ordinary warrants amounted to $2,721,000, with total issuance costs initially allocated to the ordinary warrants amounting to $27,000.
The fair value of the ordinary warrants amounted to $745,000 as of December 31, 2024, and was based on the then current price of an ADS, a risk-free interest rate of 4.33%, an average standard deviation of 85.5%, and on the remaining contractual life of the ordinary warrants.
The changes in fair value for the years ended December 31, 2024 of $1,976,000, have been recorded as non-operating income in the statement of comprehensive loss.
As of December 31, 2024, 125,034 of the pre-funded warrants had been exercised, and none of the ordinary warrants had been exercised.
|
F - 36
BioLineRx Ltd.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
| d. |
Share purchase agreement
|
| e. |
Share-based payments
|
| 1) |
Share Incentive plan – general
|
F - 37
BioLineRx Ltd.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
| e. |
Share-based payments (cont.)
|
|
| 1) |
Share Incentive plan – general (cont.)
|
| 2) |
Employee share incentive plan:
|
|
Year ended December 31,
|
||||||||||||||||||||||||
|
2022
|
2023
|
2024
|
||||||||||||||||||||||
|
Number
of options
|
Weighted average exercise price
(in NIS)
|
Number
of options
|
Weighted average exercise price
(in NIS)
|
Number
of options
|
Weighted average exercise price
(in NIS)
|
|||||||||||||||||||
|
Outstanding at beginning of year
|
40,956,214
|
0.70
|
89,871,858
|
0.44
|
150,429,825
|
0.32
|
||||||||||||||||||
|
Granted
|
53,696,305
|
0.27
|
64,855,380
|
0.18
|
23,190,000
|
0.20
|
||||||||||||||||||
|
Expired
|
(1,496,168
|
)
|
0.98
|
(1,379,640
|
)
|
0.68
|
(3,825,990
|
)
|
0.53
|
|||||||||||||||
|
Forfeited
|
(3,121,894
|
)
|
0.73
|
(2,424,535
|
)
|
0.21
|
(27,389,565
|
)
|
0.23
|
|||||||||||||||
|
Exercised
|
(162,599
|
)
|
0.10
|
(493,238
|
)
|
0.22
|
(770,025
|
)
|
0.14
|
|||||||||||||||
|
Outstanding at end of year*
|
89,871,858
|
0.44
|
150,429,825
|
0.32
|
141,634,245
|
0.32
|
||||||||||||||||||
|
Exercisable at end of year
|
26,663,961
|
0.78
|
51,970,635
|
0.54
|
88,080,600
|
0.40
|
||||||||||||||||||
| * |
As of December 31, 2022, 2023 and 2024, includes 10,482,277, 12,219,465, and 4,010,670 PSUs at an exercise price of 0.10 NIS (par value of ordinary shares), for which performance obligations have not been met.
|
F - 38
BioLineRx Ltd.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
| e. |
Share-based payments (cont.)
|
|
| 2) |
Employee share incentive plan (cont.):
|
|
As of December 31,
|
|||||||||||||||||
|
2023
|
2024
|
||||||||||||||||
|
Range of
exercise
prices
(in NIS)
|
Number
of options
outstanding
|
Weighted
average
remaining
contractual
life (in yrs.)
|
Number
of options
outstanding
|
Weighted
average
remaining
contractual
life (in yrs.)
|
|||||||||||||
|
Up to 0.49
|
120,593,415
|
8.8
|
114,352,425
|
8.07
|
|||||||||||||
|
0.5-0.99
|
16,147,110
|
6.9
|
15,104,100
|
6.54
|
|||||||||||||
|
1.00-2.00
|
13,149,390
|
4.7
|
11,727,795
|
4.57
|
|||||||||||||
|
2.01-3.4
|
539,910
|
3.2
|
449,925
|
3.18
|
|||||||||||||
|
150,429,825
|
8.2
|
141,634,245
|
7.60
|
||||||||||||||
|
2022
|
2023
|
2024
|
||||||||||
|
Expected dividend yield
|
0
|
%
|
0
|
%
|
0
|
%
|
||||||
|
Expected volatility
|
67
|
%
|
69
|
%
|
73
|
%
|
||||||
|
Risk-free interest rate
|
4
|
%
|
4
|
%
|
4
|
%
|
||||||
|
Expected life of options (in years)
|
6
|
6
|
6
|
|||||||||
F - 39
BioLineRx Ltd.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
| e. |
Share-based payments (cont.)
|
| 3) |
Stock options to consultants
|
| a. |
Corporate taxation
|
| b. |
Tax loss carryforwards
|
| c. |
Tax assessments
|
F - 40
BioLineRx Ltd.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
| d. |
Theoretical taxes
|
|
Year ended December 31,
|
||||||||||||||||||||||||
|
2022
|
2023
|
2024
|
||||||||||||||||||||||
|
in USD
|
in USD
|
in USD
|
||||||||||||||||||||||
|
thousands
|
thousands
|
thousands
|
||||||||||||||||||||||
|
Loss before taxes
|
23.0
|
%
|
(24,951
|
)
|
23.0
|
%
|
(60,614
|
)
|
23.0
|
%
|
(9,221
|
)
|
||||||||||||
|
Theoretical tax benefit
|
(5,739
|
)
|
(13,941
|
)
|
(2,121
|
)
|
||||||||||||||||||
|
Disallowed deductions (tax exempt income):
|
||||||||||||||||||||||||
|
Loss (gain) on adjustment of warrants to fair value
|
(1,478
|
) |
2,542
|
(4,056
|
)
|
|||||||||||||||||||
|
Share-based compensation
|
516
|
534
|
261
|
|||||||||||||||||||||
|
Impairment of intangible asset
|
-
|
1,542
|
232
|
|||||||||||||||||||||
|
Other
|
11
|
11
|
11
|
|||||||||||||||||||||
|
Increase in taxes for tax losses and timing differences incurred in the reporting year for which deferred taxes were not created
|
6,690
|
9,312
|
5,673
|
|||||||||||||||||||||
|
Taxes on income for the reported year
|
-
|
-
|
-
|
|||||||||||||||||||||
|
Year ended December 31,
|
||||||||||||
|
2022
|
2023
|
2024
|
||||||||||
|
in USD thousands
|
||||||||||||
|
Loss attributed to ordinary shares
|
(24,951
|
)
|
(60,614
|
)
|
(9,221
|
)
|
||||||
|
in thousands
|
||||||||||||
|
Number of shares used in basic calculation
|
773,957
|
963,366
|
1,198,108
|
|||||||||
|
in USD
|
||||||||||||
|
Basic and diluted loss per ordinary share
|
(0.03
|
)
|
(0.06
|
)
|
(0.01
|
)
|
||||||
F - 41
BioLineRx Ltd.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
| a. |
Commitments
|
| 1) |
Obligation to pay royalties to the State of Israel
|
| 2) |
Licensing agreements
|
F - 42
BioLineRx Ltd.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
| a. |
Commitments (cont.)
|
| 2) |
Licensing agreements (cont.)
|
|
F - 43
BioLineRx Ltd.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
| a. |
Commitments (cont.)
|
| 2) |
Licensing agreements (cont.)
|
|
| 3) |
Commitments in respect of Biokine
|
| 4) |
Purchase orders
|
F - 44
BioLineRx Ltd.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
| b. |
Guarantees
|
| c. |
Contingent liabilities
|
F - 45
BioLineRx Ltd.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
F - 46
BioLineRx Ltd.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
| a. |
Revenue for the SCM license was recognized in the fourth quarter of 2023, upon transfer of control over the license to the licensee, in the amount of $2.0 million.
|
| b. |
Revenue from providing the SCM support services has been recognized using the input method, which is based on costs incurred and labor hours expended, resulting in straight-line revenue recognition for the year ended December 31, 2024 totaling $0.1 million.
|
| c. |
Revenue from the PDAC performance obligation has been recognized over time, with the percentage of completion determined based on support hours incurred, with a total amount of $15.5 million recognized for the year ended December 31, 2024.
|
F - 47
BioLineRx Ltd.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
F - 48
BioLineRx Ltd.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
|
Year ended December 31,
|
||||||||||||
|
2022
|
2023
|
2024
|
||||||||||
|
in USD thousands
|
||||||||||||
|
Benefits to related parties:
|
||||||||||||
|
Compensation and benefits to senior management, including benefit component of equity instrument grants
|
2,968
|
3,155
|
2,396
|
|||||||||
|
Compensation and benefits to directors, including benefit component of equity instrument grants
|
507
|
587
|
415
|
|||||||||
|
Year ended December 31,
|
||||||||||||
|
2022
|
2023
|
2024
|
||||||||||
|
in USD thousands
|
||||||||||||
|
Salaries and other short-term employee benefits
|
2,298
|
2,425
|
2,018
|
|||||||||
|
Post-employment benefits
|
131
|
256
|
394
|
|||||||||
|
Other long-term benefits
|
35
|
31
|
29
|
|||||||||
|
Share-based compensation
|
1,011
|
1,030
|
370
|
|||||||||
|
3,475
|
3,742
|
2,811
|
||||||||||
|
Year ended December 31,
|
||||||||||||
|
2022
|
2023
|
2024
|
||||||||||
|
in USD thousands
|
||||||||||||
|
China
|
-
|
4,610
|
14,957
|
|||||||||
|
U.S.
|
-
|
190
|
13,983
|
|||||||||
|
-
|
4,800
|
28,940
|
||||||||||
F - 49
BioLineRx Ltd.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
| a. |
Other receivables
|
|
December 31,
|
||||||||
|
2023
|
2024
|
|||||||
|
in USD thousands
|
||||||||
|
Government institutions
|
245
|
455
|
||||||
|
Advance payments
|
480
|
91
|
||||||
|
Other
|
105
|
932
|
||||||
|
830
|
1,478
|
|||||||
| b. |
Accounts payable and accruals
|
|
December 31,
|
||||||||
|
2023
|
2024
|
|||||||
|
in USD thousands
|
||||||||
|
1) Trade:
|
||||||||
|
Accounts payable:
|
||||||||
|
Overseas
|
7,704
|
4,250
|
||||||
|
In Israel
|
3,165
|
1,333
|
||||||
|
10,869
|
5,583
|
|||||||
|
2) Other:
|
||||||||
|
Payroll and related expenses
|
2,184
|
1,993
|
||||||
|
Accrued expenses
|
662
|
707
|
||||||
|
Accrual for vacation and recreation pay
|
419
|
431
|
||||||
|
Other
|
88
|
-
|
||||||
|
3,353
|
3,131
|
|||||||
F - 50
BioLineRx Ltd.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
| c. |
Cost of revenues
|
|
Year ended December 31,
|
||||||||||||
|
2022
|
2023
|
2024
|
||||||||||
|
in USD thousands
|
||||||||||||
|
License fees and royalties payable to licensor
|
-
|
3,027
|
3,024
|
|||||||||
|
Direct costs related to license revenues
|
-
|
203
|
2,605
|
|||||||||
|
Amortization of intangible asset
|
-
|
450
|
3,323
|
|||||||||
|
Cost of product sales
|
-
|
12
|
311
|
|||||||||
|
-
|
3,692
|
9,263
|
||||||||||
| d. |
Research and development expenses
|
|
Year ended December 31,
|
||||||||||||
|
2022
|
2023
|
2024
|
||||||||||
|
in USD thousands
|
||||||||||||
|
Payroll and related expenses
|
4,495
|
4,452
|
3,524
|
|||||||||
|
Research and development services
|
9,296
|
4,603
|
3,170
|
|||||||||
|
Lab, occupancy and telephone
|
902
|
969
|
1,102
|
|||||||||
|
Professional fees
|
954
|
935
|
273
|
|||||||||
|
Share-based compensation
|
1,198
|
760
|
451
|
|||||||||
|
Depreciation and amortization
|
615
|
583
|
431
|
|||||||||
|
Other
|
169
|
217
|
198
|
|||||||||
|
17,629
|
12,519
|
9,149
|
||||||||||
| e. |
Sales and marketing expenses
|
|
Year ended December 31,
|
||||||||||||
|
2022
|
2023
|
2024
|
||||||||||
|
in USD thousands
|
||||||||||||
|
Payroll and related expenses
|
947
|
8,868
|
12,224
|
|||||||||
|
Medical Affairs
|
1,316
|
4,824
|
2,891
|
|||||||||
|
Marketing
|
1,805
|
4,091
|
2,041
|
|||||||||
|
Travel
|
84
|
986
|
1,506
|
|||||||||
|
Office-related expenses
|
519
|
1,923
|
1,477
|
|||||||||
|
Business Analytics
|
106
|
1,005
|
1,264
|
|||||||||
|
Market Access
|
1,023
|
1,606
|
701
|
|||||||||
|
Professional fees
|
521
|
745
|
627
|
|||||||||
|
Share-based compensation
|
112
|
751
|
98
|
|||||||||
|
Depreciation and amortization
|
-
|
314
|
330
|
|||||||||
|
Loss on abandonment of right-of-use asset
|
-
|
-
|
246
|
|||||||||
|
Other
|
29
|
157
|
200
|
|||||||||
|
6,462
|
25,270
|
23,605
|
||||||||||
F - 51
BioLineRx Ltd.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
| f. |
General and administrative expenses
|
|
Year ended December 31,
|
||||||||||||
|
2022
|
2023
|
2024
|
||||||||||
|
in USD thousands
|
||||||||||||
|
Payroll and related expenses
|
1,706
|
2,117
|
2,000
|
|||||||||
|
Professional fees
|
1,248
|
2,028
|
1,991
|
|||||||||
|
Provision for doubtful accounts receivable
|
-
|
-
|
800
|
|||||||||
|
Insurance
|
1,046
|
939
|
808
|
|||||||||
|
Share-based compensation
|
895
|
780
|
585
|
|||||||||
|
Depreciation
|
39
|
37
|
10
|
|||||||||
|
Other
|
132
|
409
|
127
|
|||||||||
|
5,066
|
6,310
|
6,321
|
||||||||||
| g. |
Non-operating income (expenses), net
|
|
Year ended December 31,
|
||||||||||||
|
2022
|
2023
|
2024
|
||||||||||
|
in USD thousands
|
||||||||||||
|
Changes in fair value of warrants
|
6,425
|
(11,054
|
)
|
18,965
|
||||||||
|
Issuance costs
|
(762
|
)
|
-
|
(669
|
)
|
|||||||
|
Other
|
7
|
235
|
139
|
|||||||||
|
5,670
|
(10,819
|
)
|
18,435
|
|||||||||
| h. |
Financial income
|
|
Year ended December 31,
|
||||||||||||
|
2022
|
2023
|
2024
|
||||||||||
|
in USD thousands
|
||||||||||||
|
Interest income
|
694
|
2,007
|
1,820
|
|||||||||
|
Exchange differences, net
|
-
|
61
|
51
|
|||||||||
|
694
|
2,068
|
1,871
|
||||||||||
| i. |
Financial expenses
|
|
Year ended December 31,
|
||||||||||||
|
2022
|
2023
|
2024
|
||||||||||
|
in USD thousands
|
||||||||||||
|
Interest expense
|
1,786
|
2,144
|
9,074
|
|||||||||
|
Bank commissions
|
26
|
25
|
45
|
|||||||||
|
Exchange differences, net
|
346
|
-
|
-
|
|||||||||
|
2,158
|
2,169
|
9,119
|
||||||||||
F - 52
BioLineRx Ltd.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
F - 53
| 1. |
Name of Company
|
| 2. |
Goals of the Company
|
| 3. |
Interpretation
|
|
|
3.1 |
Any statement in the singular shall also include the plural and vice versa; any statement in the masculine shall also include the feminine and vice versa.
|
|
|
3.2 |
Except insofar as these Articles include special definitions of certain terms, any word and expression in these Articles shall have the meaning attributed thereto in the Companies Law, 5759-1999 (in these Articles – “the Companies Law,”) unless this contradicts the written matter or the content thereof.
|
|
|
3.3 |
To prevent doubt it is clarified that regarding matters regulated in the Companies Law in such manner that the arrangements in these matters may be conditioned in the Articles, and in cases in which these Articles do not include
different provisions from those in the Companies Law, the provisions of the Companies Law shall apply.
|
|
|
3.4 |
It is hereby clarified that the provisions of the Articles of Association of the Company as detailed below are subject to the provisions of the Companies Law, the Securities Law, and any law.
|
| 4. |
The Share Capital of the Company and the Rights Attached to Shares
|
|
|
4.1 |
The registered capital of the Company is NIS 500,000,000, divided into 5,000,000,000 ordinary shares with a nominal value of NIS 0.10 each.
|
|
|
4.2 |
The ordinary shares shall entitle their owners to –
|
|
|
4.2.1 |
An equal right to participate in and vote at the general meetings of the Company, whether ordinary meetings or extraordinary meetings. Each of the shares in the Company shall entitle its owner present at the meeting and participating in
the vote in person, by proxy, or by means of a letter of voting, to one vote;
|
|
|
4.2.2 |
An equal right to participate in the distribution of dividends, whether in cash or in benefit shares, in the distribution of assets, or in any other distribution, according to the proportionate nominal value of the shares held thereby;
|
|
|
4.2.3 |
An equal right to participate in the distribution of the surplus assets of the Company in the event of its liquidation in accordance with the proportionate nominal value of the shares held thereby.
|
|
|
4.3 |
The Board of Directors is entitled to issue shares and other convertible securities or securities that may be realized as shares up to the limit of the Company’s registered capital. For the purpose of calculating the limit of the
registered capital, convertible securities or securities that may be realized as shares shall be considered to have been converted or realized as of their date of issue.
|
| 5. |
Limited Liability
|
| 6. |
Joint Shares and Share Certificates
|
|
|
6.1 |
The owner of a share registered in the registry of shareholders is entitled to receive from the Company, without payment and within a period of three months following the allocation or the registration of transfer, one share certificate
stamped with the Company’s stamp regarding all the shares registered in his name, which certificate shall detail the number of shares. In the event of a jointly owned share, the Company shall issue one share certificate for all the joint
owners of the share, and the delivery of such a certificate to one of the partners shall be considered delivery to them all.
|
|
|
6.2 |
A share certificate that has been defaced, destroyed, or lost may be renewed on the basis of such proof and guarantees as shall be required by the Company from time to time.
|
| 7. |
The Company’s Reliefs relating to Shares that Have Not Been Fully Paid
|
|
|
7.1 |
If any or all of the remuneration the shareholder undertook to pay the Company in return for his shares has not been paid by such date and on such conditions as established in the conditions for the allocation of his shares and/or in the
payment request as stated in section 7.2 below, the Company is entitled, by way of a decision of the Board of Directors, to forfeit the shares whose remuneration has not been fully paid. The forfeiture of shares shall take place provided
that the Company has sent the shareholder written warning of its intention to forfeit the shares after at least 7 days from the date of receipt of the warning, insofar as payment shall not be made during the period determined in the letter
of warning.
|
|
|
7.2 |
If, in accordance with the conditions of allocation of the shares, there is no fixed date for the payment of any part of the price to be paid on account thereof, the Board of Directors is entitled, from time to time, to present payment
requests to the shareholders on account of monies not yet removed for the shares they hold, and each shareholder shall be obliged to pay the Company the amount requested on the date determined as stated, provided that he shall receive prior
notice of 14 days of the date and place of payment (hereinafter – “the Payment Request.”) The notification shall specify that non-payment by or before the determined date and in the specified place
may lead to the forfeiture of the shares regarding which payment is requested. A Payment Request may be nullified or postponed to another date, all as shall be decided by the Board of Directors.
|
|
|
7.3 |
Unless otherwise determined in the conditions of allocations of the shares, a shareholder shall not be entitled to receive a dividend or to exercise any right as a shareholder on account of shares that have not yet been fully paid.
|
|
|
7.4 |
Persons who are the joint owners of a share shall be liable jointly and severally for payment of the amounts due to the Company on account of the share.
|
|
|
7.5 |
The content of this section shall not derogate from any other relief of the Company vis-à-vis a shareholder who fails to pay his debt to the Company on account of his shares.
|
| 8. |
Transfer of Shares
|
|
|
8.1 |
The Company’s shares are transferable.
|
|
|
8.2 |
The transfer of shares must be made in writing, and it shall be recorded only if –
|
|
|
8.2.1 |
A proper certificate for the transfer of shares, together with the certificates of the share intended for transfer, if such were issued, is delivered to the Company at its registered office. The certificate of transfer shall be signed by
the transferor and by a witness confirming the signature of the transferor. In the event of the transfer of shares that are not fully paid as of the date of transfer, the certificate of transfer shall also be signed by the recipient of the
share and by a witness testifying to the signature of the recipient; or
|
|
|
8.2.2 |
A court order for the amendment of the registration shall be delivered to the Company; or
|
|
|
8.2.3 |
It shall be proved to the Company that lawful conditions pertain for the transfer of the right to the share.
|
|
|
8.3 |
The transfer of shares that have not been fully paid requires the authorization of the Board of Directors, which is entitled to refuse to grant its authorization at its absolute discretion and without stating grounds therefore.
|
|
|
8.4 |
The recipient of the transfer shall be considered the shareholder regarding the transferred shares from the moment of the registration of his name in the registry of shareholders.
|
| 9. |
Changes in Capital
|
|
|
9.1 |
The general meeting is entitled to increase the Company’s registered share capital by creating new shares of an existing type or a new type, all as shall be determined in the decision of the general meeting.
|
|
|
9.2 |
The general meeting is entitled to nullify registered share capital that has not yet been allocated, provided that there is no commitment, including a conditioned commitment, by the Company to allocate the shares.
|
|
|
9.3 |
The general meeting shall be entitled, subject to the provisions of any law:
|
|
|
9.3.1 |
To unify and redivide its share capital, or any part thereof, into shares of a nominal value greater than the nominal value of the existing shares.
|
|
|
9.3.2 |
To divide, by way of the redivision of any or all of the existing shares, its share capital into shares of a nominal value smaller than the nominal value of the existing shares.
|
|
|
9.3.3 |
To reduce its share capital and any reserved fund for the repayment of capital in such manner and on such conditions and with the receipt of such authorization as shall be required by the Companies Law.
|
| 10. |
Changes in the Rights of Share Types
|
|
|
10.1 |
Unless otherwise stated in the conditions of issue of the shares, and subject to the provisions of any law, the rights of any share type may be changed following a decision of the Company’s Board of Directors, and with the authorization
of the general meeting of shareholders of that type, or with the written consent of all the shareholders of that type. The provisions of the Company’s Articles of Association regarding general meetings shall apply, mutatis mutandis, to a general meeting of type shareholders.
|
|
|
10.2 |
The rights granted to the holders of shares of a specific type issued with special rights shall not be considered to have been changed by virtue of the creation or issue of additional shares of equal grade, unless otherwise conditioned
in the conditions of issue of the said shares.
|
| 11. |
General Meetings
|
|
|
11.1 |
Company decisions on the following matters shall be taken at the general meeting –
|
|
|
11.1.1 |
Changes to the Articles;
|
|
|
11.1.2 |
Exercising the authorities of the Board of Directors in the event that the Board of Directors is unable to perform its function;
|
|
|
11.1.3 |
Appointment of the auditing accountant of the Company and the cessation of employment thereof;
|
|
|
11.1.4 |
Appointment of directors, including external directors;
|
|
|
11.1.5 |
Authorization of actions and transactions requiring the authorization of the general meeting in accordance with the provisions of the Companies Law and any other law;
|
|
|
11.1.6 |
Increasing and decreasing the registered share capital;
|
|
|
11.1.7 |
Merger as defined in the Companies Law.
|
|
|
11.2 |
Subject to the provisions of the law, the general meeting is entitled to assume authorities granted to another organ in the Company, including the Board of Directors, for a particular matter or for a given period of time.
|
| 12. |
Convening of General Meetings
|
|
|
12.1 |
General meetings shall be convened at least once a year at such a venue and on such a date as shall be determined by the Board of Directors, and subject to the provisions of the law, but not later than 15 months after the previous
general meeting. These general meetings shall be called “annual meetings.” The remaining meetings of the Company shall be called “extraordinary meetings.”
|
|
|
12.2 |
The agenda at the annual meeting shall include discussion of the report of the Board of Directors and financial statements as required by law. The annual meeting shall appoint an auditing accountant; shall appoint the directors in
accordance with these Articles; and shall discuss all other matters to be discussed at the annual meeting of the Company in accordance with these Articles or in accordance with the Companies Law, as well as any other matter as shall be
determined by the Board of Directors.
|
|
|
12.3 |
The Board of Directors is entitled to convene an extraordinary meeting in accordance with its decision, and must convene a general meeting if a written request is received from any of the following (hereinafter – “Request to Convene:”)
|
|
|
12.3.1 |
Two directors or one-fourth of the incumbent directors; and/or
|
|
|
12.3.2 |
One or more shareholders holding at least five percent of the issued capital and at least one percent of the voting rights in the Company; and/or
|
|
|
12.3.3 |
One or more shareholders holding at least five percent of the voting rights in the Company.
|
|
|
12.4 |
Any Request to Convene must specify the goals for whose purpose the meeting is to be convened, and shall be signed by those requesting the convening and delivered at the Company’s registered office. The request may consist of a number of
documents of identical format, each signed by one or more individuals making the request.
|
|
|
12.5 |
A Board of Directors required to convene an extraordinary meeting shall convene such meeting within twenty-one days from the date on which the Request to Convene was submitted thereto, for a date determined in an invitation in accordance
with section 12.6 below and subject to any law.
|
|
|
12.6 |
Notification of the members of the Company regarding the convening of a general meeting shall be published or delivered to all the shareholders registered in the registry of shareholders in the Company in accordance with the requirements
of the law. The notification shall include the agenda, the proposed decisions, and arrangements regarding voting in writing.
|
| 13. |
Discussion at General Meetings
|
|
|
13.1 |
The discussion at the general meeting shall be opened only if a legal quorum is present at the time the discussion begins. A legal quorum is the presence of at least two shareholders holding at least 25 percent of the voting rights
(including presence by means of proxy or through a letter of voting) within one half-hour from the time specified for the opening of the meeting.
|
|
|
13.2 |
If, at the end of one half-hour from the time specified for the opening of the meeting, no legal quorum is present, the meeting shall be postponed by one week, to the same day, the same hour, and the same venue, or to a later date, if
specified on the invitation to the meeting or in the notification of the meeting (hereinafter – “the Postponed Meeting.”) Notification and invitation regarding a Postponed Meeting postponed for a
period of not more than 21 days shall be made not later than seventy-two hours prior to the Postponed Meeting. Notification of a Postponed Meeting shall be made as stated in section 12.6, mutatis mutandis.
|
|
|
13.3 |
The legal quorum for commencing a Postponed Meeting shall be any number of participants.
|
|
|
13.4 |
The chairperson of the Board of Directors shall serve as the chairperson of the general meeting. If the chairperson of the Board of Directors is absent from the meeting after 15 minutes from the time specified for the meeting, or if he
refuses to serve as the chairperson of the meeting, the chairperson shall be elected by the general meeting.
|
|
|
13.5 |
A general meeting with a legal quorum is entitled to decide on the postponement of the meeting to another date and to such venue as shall be determined and, in this case, notifications and invitations to the Postponed Meeting shall be
made as stated in section 13.2 above.
|
| 14. |
Voting at a General Meeting
|
|
|
14.1 |
A shareholder in the Company shall be entitled to vote at general meetings in person or by means of a proxy or a letter of voting.
|
|
|
14.2 |
In any vote, each shareholder shall have a number of votes equivalent to the number of shares in their possession entitling the holder to a vote.
|
|
|
14.3 |
A decision at the general meeting shall be taken by an ordinary majority unless another majority is determined in the Companies Law or in these Articles.
|
|
|
14.4 |
The declaration by the chairperson of the meeting that a decision has been adopted unanimously or by a given majority, or rejected or not adopted by a given majority, shall constitute prima facie evidence of the content thereof.
|
|
|
14.5 |
If the votes at the meeting are equally divided, the chairperson of the meeting shall not have an additional or casting opinion and the decision presented for voting shall be rejected.
|
|
|
14.6 |
Subject to any law, the shareholders in the Company are entitled to vote in any matter on the agenda of a general meeting (including type meetings) by means of a letter of voting, provided that the Board of Directors, subject to any law,
has not negated in its decision to convene the general meeting the possibility of voting by means of a letter of voting on that matter.
|
|
|
14.7 |
A shareholder is entitled to state the manner of his vote in the letter of voting and to deliver this to the Company up to 48 hours prior to the time of commencement of the meeting. A letter of voting stating the manner of voting of the
shareholder reaching the Company at least 48 hours prior to the time of commencement of the meeting shall be considered tantamount to presence at the meeting, including for the matter of the presence of the legal quorum as stated in section
13.1 above.
|
|
|
14.8 |
Appointment of a proxy shall be in writing, signed by the appointer (hereinafter – “Power of Attorney.”) A corporation shall vote by means of its representatives, who shall be appointed in a
document signed properly by the corporation (hereinafter – “Letter of Appointment.”)
|
|
|
14.9 |
A vote in accordance with the conditions of a Power of Attorney shall be lawful even if the appointer dies before the voting, or becomes legally incompetent, is liquidated, becomes bankrupt, nullifies the Letter of Appointment, or
transfers the share regarding which it was given, unless written notification is received at the Company’s office prior to the meeting that the shareholder has died, become legally incompetent, been liquidated, become bankrupt, or has
nullified the Letter of Appointment or transferred the shares as stated.
|
|
|
14.10 |
The Letter of Appointment and the Power of Attorney, or a copy authorized by an attorney, shall be deposited at the Company’s registered offices at least forty eight (48) hours prior to the time determined for the meeting or for the
Postponed Meeting at which the person mentioned in the document intends to vote in accordance therewith.
|
|
|
14.11 |
A shareholder in the Company shall be entitled to vote at the Company’s meetings by means of several proxies appointed thereby, provided that each proxy shall be appointed on account of different sections of the shares held by the said
shareholder. There shall be no impediment to each proxy as stated voting in a different manner in the Company’s meetings.
|
|
|
14.12 |
If a shareholder is legally incompetent, he is entitled to vote by means of his trustees, the recipient of his assets, his natural guardian or other legal guardian, and these are entitled to vote in person or by proxy or a Letter of
Voting.
|
|
|
14.13 |
When two or more persons are the joint owners of a share, in a vote on any matter the vote of the person whose name is registered first in the registry of shareholders as the owner of that share shall be accepted, whether in person or by
proxy, and he is entitled to deliver Letters of Voting to the Company.
|
| 15. |
The Board of Directors
|
| 16. |
Appointment of the Board of Directors and Cessation of Office Thereof
|
|
|
16.1 |
The number of directors in the Company shall be determined from time to time by the annual general meeting, provided that this shall not be fewer than 5 and not more than 10 directors, including external directors (if any were elected).
The number of external directors in the Company shall not be less than the number determined in the Companies Law.
|
|
|
16.2
|
(a) |
Directors shall be elected at the annual general meeting by vote of a shareholders’ resolution. The directors, excluding the external directors (if any were elected), shall be classified, with respect to the term for which they each
severally hold office, into three classes, as nearly equal in number as practicable, hereby designated as Class I, Class II and Class III.
|
|
(i) The term of office of the initial Class I directors shall expire when their successors are elected and qualified at the first annual
general meeting to be held following the approval of this provision;
(ii) The term of office of the initial Class II directors shall expire when their successors are elected and qualified at the first
annual general meeting following the annual general meeting referred to in clause (i) above; and
(iii) The term of office of the initial Class III directors shall expire when their successors are elected and qualified at the first
annual general meeting following the annual general meeting referred to in clause (ii) above.
|
|
|
(b) |
At each annual general meeting, commencing with the annual general meeting to be held following the approval of this provision (anticipated to be in 2024), each of the successors elected to replace the directors of a Class whose term
shall have expired at such annual general meeting shall be elected to hold office until the third annual general meeting next succeeding his or her election (or re-election) and until his or her respective successor shall have been elected
and qualified. Notwithstanding anything to the contrary, each director shall serve subject to section 16 hereof until his or her successor is elected and qualified or until such earlier time as such director’s office is vacated, or his
earlier removal pursuant to this section 16.
|
|
|
(c) |
If the number of directors (excluding external directors, if any were elected) that comprise the Board of Directors is hereafter changed, any newly created directorships or decrease in directorships shall be so apportioned by the Board
of Directors among the classes as to make all classes as nearly equal in number as is practicable, provided that no decrease in the number of directors constituting the Board of Directors shall shorten the term of any incumbent director.
|
|
|
16.3 |
In addition to the content of section 16.2 above, the Board of Directors is entitled to appoint a director in place of a director whose position has become vacant and/or by way of an addition to the Board of Directors, subject to the
maximum number of directors on the Board of Directors as stated in section 16.1 above. The appointment of a director by the Board of Directors to fill any vacancy shall only be for the remaining period of time during which the director
whose service has ended would have held office, or in case of a vacancy due to the number of directors serving being less than the maximum number stated in section 16.1 above, the Board of Directors shall determine at the time of
appointment the class, pursuant to section 16.2, to which the additional director is assigned.
|
|
|
16.4 |
A director whose period of office has expired may be reelected, with the exception of an external director, who may be reelected for an additional period of office subject to the provisions of the law.
|
|
|
16.5 |
The office of a director shall commence on the date of his/her appointment by the annual meeting and/or the Board of Directors, or on a later date if this date is determined in the decision of appointment at the annual meeting and/or the
Board of Directors.
|
|
|
16.6 |
The Board of Directors shall elect one of its members as the chairperson of the Board of Directors. The elected chairperson shall run the meetings of the Board of Directors and shall sign the minutes of the discussion. If no chairperson
is elected, or if the chairperson of the Board of Directors is not present after 15 minutes from the time set for the meeting, the directors present shall choose one of their number to serve as the chairperson at that meeting, and the
chosen member shall run the meeting and sign the minutes of the discussion.
|
|
|
16.7 |
The general meeting is entitled to remove any director from their office prior to the end of the period of their office, inter alia whether the director was appointed thereby in accordance with section 16.2 above or was appointed by the
Board of Directors in accordance with section 16.3 above, provided that the director shall be given a reasonable opportunity to state their case before the general meeting.
|
|
|
16.8 |
Any director is entitled, with the agreement of the Board of Directors, to appoint a substitute for themselves (hereinafter – a “Substitute Director”),
provided that a person who is not competent shall not be appointed to serve as a Substitute Director, nor a person who has been appointed as a Substitute Director for another director and/or a person who is already serving as a director in
the Company.
|
|
|
16.9 |
The office of a director shall become vacant in any of the following cases:
|
|
|
16.9.1 |
The director resigns from office by means of a letter signed in his hand, submitted to the Company and detailing the reasons for his resignation;
|
|
|
16.9.2 |
The director is removed from office by the general meeting;
|
|
|
16.9.3 |
The director is convicted of an offense as stated in Article 232 of the Companies Law;
|
|
|
16.9.4 |
In accordance with a court decision as stated in Article 233 of the Companies Law;
|
|
|
16.9.5 |
The director is declared legally incompetent or died;
|
|
|
16.9.6 |
The director is declared bankrupt and, if the director is a corporation – it opted for voluntary liquidation or a liquidation order was issued against it.
|
|
|
16.10 |
In the event that the position of a director becomes vacant, the remaining directors shall be entitled to continue to act, provided the number of directors remaining shall not be less than the minimum number of directors as stated above
in section 16.1 above. If the number of directors falls below the above-mentioned minimum number, the remaining directors shall be entitled to act solely in order to fill the place of the director that has become vacant as stated in section
16.3 above, or in order to convene a general meeting of the Company, and pending the convening of the general meeting of the Company as stated they may act to manage the Company’s affairs solely in matters that cannot be delayed.
|
|
|
16.11 |
The terms and conditions of the office of the members of the Board of Directors shall be authorized in accordance with the provisions of the Companies Law.
|
|
|
16.12 |
Notwithstanding anything to the contrary herein, clauses 16.1, 16.2 and 16.3 above may only be amended, replaced or suspended by a resolution adopted at an annual general meeting by a majority of at least 65% of the ordinary shares
represented at such meeting (in person, by proxy, by voting instruction form or via the electronic voting system of the Israel Securities Authority) voting on the proposal.
|
|
|
16.13 |
Notwithstanding anything to the contrary in these Articles, the election, qualification, removal or dismissal of external directors, if so elected, shall be only in accordance with the applicable provisions set forth in the Companies
Law.
|
| 17. |
Meetings of the Board of Directors
|
|
|
17.1 |
The Board of Directors shall convene for a meeting in accordance with the needs of the Company, and at least once every three months.
|
|
|
17.2 |
The chairperson of the Board of Directors is entitled to convene the Board at any time. In addition, the Board of Directors shall hold a meeting on such subject as shall be specified in the following cases:
|
|
|
17.2.1 |
In accordance with the request of two directors; however, if at the time the Board of Directors comprises five directors or less – in accordance with the request of one director;
|
|
|
17.2.2 |
In accordance with the request of one director if, in his request to convene the Board, he states that he has learned of a matter in the Company ostensibly entailing a violation of the law or infringement of proper business practice;
|
|
|
17.2.3 |
If a general director has been appointed in the Company or if a notification or report by the general director require an action on the part of the Board of Directors;
|
|
|
17.2.4 |
If the auditing accountant has informed the chairperson of the Board of Directors – or, in the event that no chairperson was appointed for the Board of Directors, has informed the Board of Directors – of substantial defects in the
accounting control of the Company.
|
|
|
17.3 |
Notification of the meeting of the Board of Directors shall be delivered to all members of the Board at least three days prior to the date of convening of the Board, or with shorter prior notice insofar as the chairperson of the Board
decided that, in the circumstances of the matter, it is vital and reasonable to convene the Board of Directors with notice shorter than three days. Notification shall be delivered to the address of the director as forwarded to the Company
in advance, and shall stipulate the time of the meeting and the venue at which it shall convene, as well as reasonable detail of all subjects on the agenda.
|
|
|
17.4 |
The agenda of the meetings of the Board of Directors shall be determined by the chairperson of the Board and shall include: Subjects determined by the chairperson of the Board; subjects deriving from the report of the general director
and/or the auditing accountant; any subject a director of the general director have requested of the chairperson of the Board to include on the agenda, at least two days prior to the convening of the meeting of the Board.
|
|
|
17.5 |
The details of the subjects on the agenda as stated in section 17.4 above do not prevent discussion of a subject or subjects not mentioned in the notification of the meeting of the Board of Directors (hereinafter: “a New Subject.”)
|
|
|
17.6 |
The legal quorum for the commencement of a meeting of the Board of Directors shall be a majority of the members of the Board of Directors. If, at the end of one half-hour from the time set for the commencement of the meeting, no quorum
is present, the meeting shall be postponed to another date as decided by the chairperson of the Board, or, in his absence, by the directors present at the convened meeting, provided that prior notification of three days shall be given to
all directors regarding the date of the Postponed Meeting. The legal quorum for the opening of a Postponed Meeting shall be any number of participants.
|
|
|
17.7 |
The Board of Directors is entitled to hold meetings by use of any means of communication, providing that all the participating directors can hear each other simultaneously.
|
|
|
17.8 |
The Board of Directors is entitled to take decisions without actually convening, provided that all the directors entitled to participate in the discussion and to vote on the subject brought for decision agree thereto. If decisions are
made as stated in this section, the chairperson of the Board of Directors shall record minutes of the decisions stating the manner of voting of each director on the subjects brought for decision, as well as the fact that all the directors
agreed to take the decision without convening.
|
| 18. |
Voting on the Board of Directors
|
|
|
18.1 |
Each director shall have one vote when voting on the Board of Directors.
|
|
|
18.2 |
Decisions of the Board of Directors shall be taken by a majority vote. The chairperson of the Board of Directors shall not have any additional or casting opinion, and in the event of a tie vote, the decision brought for voting shall be
rejected.
|
| 19. |
Committees of the Board of Directors
|
|
|
19.1 |
The Board of Directors is entitled to establish committees and to appoint members thereto (hereinafter – “the Committees of the Board of Directors.”) If Committees of the Board of Directors are
established, the Board of Directors shall determine, in the conditions of empowerment thereof, whether specific authorities of the Board of Directors shall be delegated to the Committees of the Board of Directors, in such manner that the
decision of the Committee of the Board of Directors shall be considered tantamount to a decision of the Board of Directors, or whether the decision of the Committee of the Board of Directors shall merely constitute a recommendation, subject
to the authorization of the Board of Directors; provided that authorities to make decisions in the matters stated in Article 112 of the Companies Law shall not be delegated to a committee.
|
|
|
19.2 |
A person who is not a director shall not serve in a Committee of the Board of Directors to which the Board of Directors has delegated authorities. Persons who are not members of the Board of Directors may serve in a Committee of the
Board of Director whose function is merely to advise or submit recommendations to the Board of Directors.
|
|
|
19.3 |
The provisions included in these Articles relating to the meetings of the Board of Directors and voting therein shall apply, mutatis mutandis and subject to the decisions of the Board of
Directors regarding the procedures for the meetings of the committee (if any), to any Committee of the Board of Directors comprising two or more members.
|
| 20. |
Audit Committee
|
|
|
20.1 |
The Board of Directors of the Company shall appoint an audit committee from among its members. The number of members of the audit committee shall be not less than three, and any external director may be a member thereof. The chairperson
of the Board of Directors or any director employed by the Company, or providing it with services on a regular basis, or a controlling shareholder in the Company, or a relative thereof shall not be appointed to the committee.
|
|
|
20.2 |
The functions of the audit committee shall be –
|
|
|
20.2.1 |
To identify defects in the business management of the Company, inter alia through consultation with the internal auditor of the Company or the auditing accountant, and to propose methods to the Board of Directors for correcting these;
|
|
|
20.2.2 |
To decide whether to authorize actions and transactions requiring the authorization of the audit committee in accordance with the Companies Law.
|
| 21. |
General Director
|
| 22. |
Exemption, Insurance, and Indemnification
|
|
|
22.1 |
enter into a contract for the insurance of the liability, in whole or in part, of any of its “Office Holders” (as defined in the Companies Law) with respect to an obligation imposed on such Office Holder due to an act performed by the
Office Holder in the Office Holder’s capacity as an Office Holder of the Company arising from any of the following:
|
|
|
22.1.1 |
a breach of duty of care to the Company or to any other person;
|
| 22.1.2 | a breach of the duty of loyalty to the Company provided that the Office Holder acted in good faith and had reasonable grounds to assume that the act would not harm the interests of the Company; |
| 22.1.3 | a financial liability imposed on such Office Holder in favor of any other person: |
| 22.1.4 | reasonable litigation expenses, including attorneys fees, incurred by the Office Holder as a result of an ongoing administrative enforcement proceeding instituted against him in accordance with the Israeli Securities Law. Without derogating from the generality of the foregoing, such expenses will include, and the Company may procure insurance for, a payment imposed on the Office Holder in favor of an injured party as set forth in Section 52CIV(a)(1)(a) of the Israeli Securities Law and expenses that the Office Holder incurred in connection with a proceeding under Chapters VIII”3, VIII”4 or IX”1 of the Israeli Securities Law, including reasonable legal expenses, which term includes attorney fees; and |
|
|
22.1.5 | any other incident for which it is or shall be permitted to insure the liability of an officer. |
|
|
22.2 |
undertake, in advance to indemnify, or may indemnify retroactively, an Office Holder of the Company with respect to any of the following liabilities or expenses that arise from an act performed by the Office Holder by virtue of being an
Office Holder of the Company:
|
|
|
22.2.1 |
a financial liability imposed on an Office Holder in favor of another person by any judgment, including a judgment given as a result of a settlement or an arbitrator’s award which has been confirmed by a court;
|
|
|
22.2.2 |
reasonable litigation expenses including attorney’s fees, incurred by him as a result of an investigation or proceeding instituted against him by an authority empowered to conduct an investigation or proceedings, which are concluded
without the filing of an indictment against the Office Holder and without the levying of a monetary obligation in lieu of criminal proceedings upon the Office Holder, or which are concluded without the filing of an indictment against the
Office Holder but with levying a monetary obligation in substitute of such criminal proceedings upon the Office Holder for a crime that does not require proof of criminal intent; or in connection with an administrative enforcement
proceeding or a financial sanction. Without derogating from the generality of the foregoing, such expenses will include, and the Company may undertake to indemnify an Office Holder of the Company as aforesaid, for a payment imposed on the
Office Holder in favor of an injured party as set forth in Section 52LIV(a)(1)(a) of the Israeli Securities Law and expenses that the Office Holder incurred in connection with a proceeding under Chapters VIII”3, VIII”4 or IX’1 of the
Israeli Securities Law, including reasonable legal expenses, which term includes attorney fees; and
|
|
|
22.2.3 |
reasonable litigation expenses, including attorney’s fees, expended by an Office Holder or which were imposed on an Office Holder by a court in proceedings filed against the Office Holder by the Company or in its name or by any other
person or in a criminal charge on which the Office Holder was acquitted or in a criminal charge on which the Office Holder was convicted for an offense which did not require proof of criminal intent; and
|
|
|
22.2.4 |
any other obligation or expense for which it is or shall be permitted to indemnify an officer, provided however, that in the event the Company wishes to indemnify an Office Holder in advance for financial liabilities under Article 22
it may only do so if the undertaking to indemnify the Office Holder for such liabilities was restricted to those events that the Board may deem foreseeable in light of the Company’s actual activities, at the time of giving of such
undertaking, and to a specific sum or a reasonable criterion under such circumstances as determined by the Board.
|
| 23. |
Subject to the provisions of the Law and the Israeli Securities Law, the Company hereby releases, in advance, its Office Holders from liability to the Company for damage that arises from the breach of the
Office Holder’s duty of care to the Company.
|
| 24. | The provisions of Articles 22 and 23 are not intended, and shall not be interpreted, to restrict the Company in any manner in respect of the procurement of insurance or in respect of indemnification (i) in connection with any person who is not an Office Holder, including, without limitation, any employee, agent, consultant or contractor of the Company who is not an Office Holder, or (ii) in connection with any Office Holder to the extent that such insurance and/ or indemnification is not specifically prohibited under the Companies Law; provided that the procurement of any such insurance or the provision of any such indemnification shall be approved by the Board. Any modification of Articles 22 through 24, and any amendment to the Companies Law, the Israeli Securities Law or any other applicable law, shall be prospective in effect and shall not affect the Company’s obligation or ability to indemnify an Office Holder for any act or omission occurring prior to such modification or amendment, unless otherwise provided by the Companies Law, the Israeli Securities Law or such applicable law. |
| 25. |
Internal Auditor
|
|
|
25.1 |
The Board of Directors of the Company shall appoint an internal auditor in accordance with the proposal of the audit committee. A person who is an interested party in the Company, an office holder therein, or the relative or either of
the above, as well as the auditing accountant or any person on his behalf, shall not serve as an internal auditor in the Company.
|
|
|
25.2 |
The Board of Directors shall determine which office holder shall be organizationally accountable for the internal auditor and, in the absence of such determination; this shall be the chairperson of the Board of Directors.
|
|
|
25.3 |
The internal audit plan prepared by the auditor shall be submitted to the audit committee for authorization; however, the Board of Directors is permitted to determine that the plan shall be submitted to the Board of Directors for
authorization.
|
| 26. |
Auditing Accountant
|
|
|
26.1 |
The general meeting shall appoint an auditing accountant for the Company. The auditing accountant shall service in his office through the end of the following annual meeting, or for a longer period as determined by the annual meeting,
provided that the period of office shall not be extended beyond the end of the third annual meeting following that at which he was appointed.
|
|
|
26.2 |
The fee of the auditing accountant for the auditing operations shall be determined by the Board of Directors. The Board of Directors shall report to the annual meeting on the fee of the auditing accountant.
|
| 27. |
Signing in the Company’s Name
|
|
|
27.1 |
The rights to sign in the Company’s name shall be determined from time to time by the Board of Directors of the Company.
|
|
|
27.2 |
The Company’s authorized signatory shall do so together with the Company’s stamp, or alongside its printed name.
|
| 28. |
Dividend and Benefit Shares
|
| 28.1 | The decision by the Company to allocate a dividend and/or to allocate benefit shares shall be taken by the Company’s Board of Directors. |
|
|
28.2 |
Unless determined otherwise by the Board of Directors, it shall be permitted to pay any dividend by way of check or payment order to be sent by mail in accordance with the registered address of the shareholder or the personal eligible
thereto or, in the case of joint registered owners of the same share, to that shareholder whose name is mentioned first in the registry of shareholders with regard to the joint ownership. Any such check shall be made out to order of the
person to whom it is sent. A receipt from a person whose name, as of the date of declaration of the dividend, is registered in the registry of shareholders as the owner of any share or, in the case of joint owners, of one of the joint
owners, shall serve as authorization regarding all payments made in connection with that share and regarding which the receipt was received.
|
|
|
28.3 |
For the purpose of executing any decision in accordance with the provisions of this section, the Board of Directors is entitled to resolve as it sees fit any difficulty that emerges regarding distribution of the dividend and/or the
benefit shares, including determining the value for the purpose of the said division of certain assets, and to determine that payments in cash shall be made to members on the basis of the value so determined; to determine provisions
regarding fractions of shares; or to determine that sums of less than NIS 50 shall not be paid to a shareholder.
|
| 29. |
Redeemable Securities
|
| 30. |
Donations
|
| 31. |
Accounts
|
|
|
31.1 |
The Company shall maintain accounts and shall prepare financial statements in accordance with the Securities Law and in accordance with any law.
|
|
|
31.2 |
The account ledgers shall be held at the Company’s registered offices or in any other place as the directors shall see fit, and shall always be open for inspection by the directors.
|
| 32. |
Notifications
|
|
|
32.1 |
Subject to any law, a notification or any other document that shall be delivered by the Company, and which it is entitled or required to issue in accordance with the provisions of the Articles and/or the Companies Law, the Securities
Law, or any law, shall be delivered by the Company to any person in one of the following manners as decided by the Company in each individual case: (A) By dispatch by registered mail in a letter addressed in accordance with the registered
address of that shareholder in the registry of shareholders, or in accordance with such address as stated by the shareholder in a letter to the Company as the letter for the delivery of notifications or other documents; or (B) By dispatch
by facsimile in accordance with the number stated by the shareholder as the number for the delivery of facsimile notifications; or (C) By way of publication in two daily newspapers appearing in Israel; or (D) By way of publication in the
distribution site of the Securities Authority and the Tel Aviv Stock Exchange Ltd.
|
|
|
32.2 |
Any notification to be made to shareholders shall be made, regarding jointly owned shares, to that person whose name is mentioned first in the registry of shareholders as the holder of that share, and any notification made in this manner
shall be sufficient notification for the holders of that share.
|
|
|
32.3 |
Any notification or other document sent in accordance with the provisions of section 30.1 above shall be considered to have reached its destination: (A) Within 3 business days – if sent by registered mail in Israel; or (B) On the first
business day after its dispatch, if delivered by hand or sent by facsimile; or (C) On the date of publication, if published in a newspaper or on the distribution site of the Securities Authority and the Tel Aviv Stock Exchange Ltd.
|
|
|
32.4 |
Any record made in an ordinary manner in the company’s registry shall be considered prima facie evidence of dispatch as recorded in that registry.
|
|
|
32.5 |
When it is necessary to provide prior notification of a certain number of days, or when notification is valid for a certain period, the date of delivery shall be included in reckoning the number of days or the period.
|
|
•
|
taxes and other governmental charges;
|
|
|
•
|
any applicable transfer or registration fees;
|
|
|
•
|
certain cable, telex and facsimile transmission charges as provided in the deposit agreement;
|
|
|
•
|
any expenses incurred in the conversion of foreign currency;
|
|
|
•
|
a fee of $5.00 or less per 100 ADSs (or a portion thereof) for the execution and delivery of ADRs and the surrender of ADRs,
including if the deposit agreement terminates;
|
|
|
•
|
a fee of $.05 or less per ADS (or portion thereof) for any cash distribution made pursuant to the deposit agreement;
|
|
|
•
|
a fee for the distribution of securities pursuant to the deposit agreement;
|
|
|
•
|
in addition to any fee charged for a cash distribution, a fee of $.05 or less per ADS (or portion thereof) per annum for
depositary services;
|
|
|
•
|
a fee for the distribution of proceeds of rights that the Depositary sells pursuant to the deposit agreement; and
|
|
|
•
|
any other charges payable by the Depositary, any of the Depositary’s agents, or the agents of the Depositary’s agents in
connection with the servicing of ordinary shares or other Deposited Securities.
|
|
|
Number of ADSs:
|
Initial Exercise Date: January 7, 2025
|
|
|
d) |
Mechanics of Exercise.
|
|
BIOLINERX LTD.
|
|
|
By:__________________________________________
Name:
Title:
|
|
Name:
|
______________________________________
|
|
(Please Print)
|
|
|
Address:
|
______________________________________
|
|
Phone Number:
Email Address:
|
(Please Print)
______________________________________
______________________________________
|
|
Dated: _______________ __, ______
|
|
|
Holder’s Signature: ______________________________________
|
|
|
Holder’s Address: ______________________________________
|
|

|
Title: Securities Trades by BioLineRx Personnel
|
| 1. |
PURPOSE
|
| 2. |
SCOPE
|
| 3. |
RESPONSIBILITY
|
|
|
3.1. |
This Policy shall be administered by the “Policy Administrator,” who shall be the Chief Financial Officer. The Policy Administrator may, however, change from time to time and employees are encouraged to consult the copy of this Policy that
is included on the Company’s website to obtain current information concerning the Policy Administrator.
|
|
|
3.2. |
QA is responsible for ensuring the document control process is in accordance with Documentation Management (SOP-10-002).
|
| 4. |
OBJECTIVE
|
|
|
• |
to educate all Company personnel
|
|
|
• |
to set forth guidelines for courses of action
|
|
|
• |
to protect the Company and all of its personnel against legal liability
|
|
|
• |
to preserve the reputation of the Company and its personnel for integrity and ethical conduct.
|
| 5. |
DEFINITIONS AND/OR ABBREVIATIONS
|
| 6. |
EQUIPMENT AND MATERIALS
|
| 7. |
PROCEDURE
|
|
|
• |
a civil penalty of up to three times the profit gained or loss avoided;
|
|
|
• |
a criminal fine (no matter how small the profit) of up to $5 million; and
|
|
|
• |
a jail term of up to twenty years.
|
|
|
• |
a civil penalty of the greater of $1 million or three times the profit gained or loss avoided as a result of the employee’s violation; and
|
|
|
• |
a criminal penalty of up to $25 million.
|
|
|
• |
is not generally known to the public, and
|
|
|
• |
which, if publicly known, would likely affect either the market price of the Company’s securities or a person’s decision to buy, sell or hold the Company’s securities.
|
|
|
• |
quarterly or annual earnings results
|
|
|
• |
projections of future results or sales
|
|
|
• |
earnings or losses
|
|
|
• |
news of a pending or proposed merger, acquisition or tender offer
|
|
|
• |
an important financing transaction
|
|
|
• |
significant clinical or regulatory developments
|
|
|
• |
the entry into or termination of a significant collaboration, joint venture or strategic alliance
|
|
|
• |
changes in management
|
|
|
• |
significant new products or discoveries
|
|
|
• |
plans regarding strategy or significant capital investments
|
|
|
• |
impending bankruptcy or financial liquidity problems
|
|
|
• |
criminal charge or government investigations
|
|
|
• |
internal financial information which departs from what the market would expect
|
|
|
• |
the gain or loss of any significant contract or agreement.
|
|
|
• |
Don’t discuss material information in elevators, hallways, restaurants, airplanes, taxicabs or any place where you can be overheard.
|
|
|
• |
Don’t gossip about confidential information.
|
|
|
• |
Don’t read confidential documents in public places or discard them where they can be retrieved by others.
|
|
|
• |
Don’t carry confidential documents in elevators, hallways, etc. in an exposed manner.
|
|
|
• |
Beware of the carrying quality of conversations conducted on speaker telephones in offices, and the potential for eavesdropping on conversations conducted on car or airplane telephones, on marine radios etc.
|
|
|
• |
Don’t leave confidential documents in unattended conference rooms; don’t leave confidential documents behind when the conference is over.
|
|
|
• |
Cover confidential documents on you desk before you leave your room; don’t leave confidential papers lying where visitors can see them.
|
|
|
• |
Be careful when giving out the whereabouts of personnel not in the office or revealing the presence of specific visitors to the office. The mere fact of a meeting or the destination of a trip may reveal
something confidential.
|
|
|
• |
Under no circumstances are employees to provide confidential Company documents to third parties, without express consent of the supervisor. This includes, but is not limited to, any confidential Company
documents relating to customers, competitors or suppliers of the Company.
|
|
|
• |
Trading in the Company’s securities on a short-term basis. Any ordinary shares of the Company purchased in the open market should be held for a minimum of 60 days.
|
|
|
• |
Short sales of the Company’s securities.
|
|
|
• |
Use of the Company’s securities to secure a margin or other loan, except in limited cases with the prior approval of the Policy Administrator.
|
|
|
• |
Transactions in straddles, collars, or other similar risk reduction devices, except in limited cases with the prior approval of the Policy Administrator.
|
|
|
• |
Transactions in publicly-traded options relating to the Company’s securities (i.e., options that are not granted by the Company), except in limited cases with the prior approval of the Policy Administrator.
|
|
|
• |
the period starting on the 15th day after the close of each fiscal quarter and ending at the beginning of the second business day after the release of the Company’s financial results for each quarter and, in the case of the
fourth quarter, financial results for the year end; and
|
|
|
• |
any other periods as determined by the Company. You will be notified by e-mail when you may not trade in the Company’s securities during such periods, and you will also be notified when trading restrictions are lifted.
|
| 8. |
REFERENCES - NA
|
| 9. |
RELATED DOCUMENTS - NA
|
| 10. |
APPENDICES - NA
|
|
1.
|
I have reviewed this annual report on Form 20-F of BioLineRx Ltd.;
|
|
2.
|
Based on my knowledge, this report does not contain any untrue statement of a material fact or omit to
state a material fact necessary to make the statements made, in light of the circumstances under which such statements were made, not misleading with respect to the period covered by this report;
|
|
3.
|
Based on my knowledge, the financial statements, and other financial information included in this
report, fairly present in all material respects the financial condition, results of operations and cash flows of the company as of, and for, the periods presented in this report;
|
|
4.
|
The company’s other certifying officer(s) and I are responsible for establishing and maintaining
disclosure controls and procedures (as defined in Exchange Act Rules 13a-15(e) and 15d-15(e)) and internal control over financial reporting (as defined in Exchange Act Rules 13a-15(f) and 15d-15(f)) for the company and have:
|
|
|
a)
|
Designed such disclosure controls and procedures, or caused such disclosure controls and procedures to be
designed under our supervision, to ensure that material information relating to the company, including its consolidated subsidiaries, is made known to us by others within those entities, particularly during the period in which this
report is being prepared;
|
|
|
b)
|
Designed such internal control over financial reporting, or caused such internal control over financial
reporting to be designed under our supervision, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally
accepted accounting principles;
|
|
|
c)
|
Evaluated the effectiveness of the company’s disclosure controls and procedures and presented
in this report our conclusions about the effectiveness of the disclosure controls and procedures, as of the end of the period covered by this report based on such evaluation; and
|
|
|
d)
|
Disclosed in this report any change in the company’s internal control over financial
reporting that occurred during the period covered by the annual report that has materially affected, or is reasonably likely to materially affect, the company’s internal control over financial reporting; and
|
|
5.
|
The company’s other certifying officer(s) and I have disclosed, based on our most recent evaluation of internal control
over financial reporting, to the company’s auditors and the audit committee of the company’s board of directors (or persons performing the equivalent functions):
|
|
|
a)
|
All significant deficiencies and material weaknesses in the design or operation of internal control over
financial reporting which are reasonably likely to adversely affect the company’s ability to record, process, summarize and report financial information; and
|
|
|
b)
|
Any fraud, whether or not material, that involves management or other employees who have a
significant role in the company’s internal control over financial reporting.
|
|
|
1.
|
I have reviewed this annual report on Form 20-F of BioLineRx Ltd.;
|
|
|
2.
|
Based on my knowledge, this report does not contain any untrue statement of a material fact or omit to state a material
fact necessary to make the statements made, in light of the circumstances under which such statements were made, not misleading with respect to the period covered by this report;
|
|
|
3.
|
Based on my knowledge, the financial statements, and other financial information included in this report, fairly
present in all material respects the financial condition, results of operations and cash flows of the company as of, and for, the periods presented in this report;
|
|
|
4.
|
The company’s other certifying officer(s) and I are responsible for establishing and maintaining disclosure controls
and procedures (as defined in Exchange Act Rules 13a-15(e) and 15d-15(e)) and internal control over financial reporting (as defined in Exchange Act Rules 13a-15(f) and 15d-15(f)) for the company and have:
|
|
|
a)
|
Designed such disclosure controls and procedures, or caused such disclosure controls and procedures to be designed
under our supervision, to ensure that material information relating to the company, including its consolidated subsidiaries, is made known to us by others within those entities, particularly during the period in which this report is being
prepared;
|
|
|
b)
|
Designed such internal control over financial reporting, or caused such internal control over financial
reporting to be designed under our supervision, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted
accounting principles;
|
|
|
c)
|
Evaluated the effectiveness of the company’s disclosure controls and procedures and presented in this
report our conclusions about the effectiveness of the disclosure controls and procedures, as of the end of the period covered by this report based on such evaluation; and
|
|
|
d)
|
Disclosed in this report any change in the company’s internal control over financial reporting that
occurred during the period covered by the annual report that has materially affected, or is reasonably likely to materially affect, the company’s internal control over financial reporting; and
|
|
|
5.
|
The company’s other certifying officer(s) and I have disclosed, based on our most recent evaluation of internal control
over financial reporting, to the company’s auditors and the audit committee of the company’s board of directors (or persons performing the equivalent functions):
|
|
|
a)
|
All significant deficiencies and material weaknesses in the design or operation of internal control over
financial reporting which are reasonably likely to adversely affect the company’s ability to record, process, summarize and report financial information; and
|
|
|
b)
|
Any fraud, whether or not material, that involves management or other employees who have a significant
role in the company’s internal control over financial reporting.
|
|
(i)
|
the accompanying Annual Report on Form 20-F of the Company for the year ended December 31, 2024 (the “Report”) fully complies with the
requirements of Section 13(a) or Section 15(d), as applicable, of the Securities Exchange Act of 1934, as amended; and
|
|
(ii)
|
the information contained in the Report fairly presents, in all material respects, the financial condition and results of operations of
the Company.
|
|
(i)
|
the accompanying Annual Report on Form 20-F of the Company for the year ended December 31, 2024 (the “Report”) fully complies with the
requirements of Section 13(a) or Section 15(d), as applicable, of the Securities Exchange Act of 1934, as amended; and
|
|
(ii)
|
the information contained in the Report fairly presents, in all material respects, the financial condition and results of operations of
the Company.
|
|
Tel-Aviv, Israel
|
/s/Kesselman & Kesselman
|
|
March 31, 2025
|
Certified Public Accountants (Isr.)
|
|
|
A member firm of PricewaterhouseCoopers International Limited
|