Gyre Therapeutics’ Hydronidone Met the Primary Endpoint and Demonstrated Statistically Significant Fibrosis Regression in Pivotal Phase 3 Trial for the
Treatment of CHB-associated Liver Fibrosis in China
|
• |
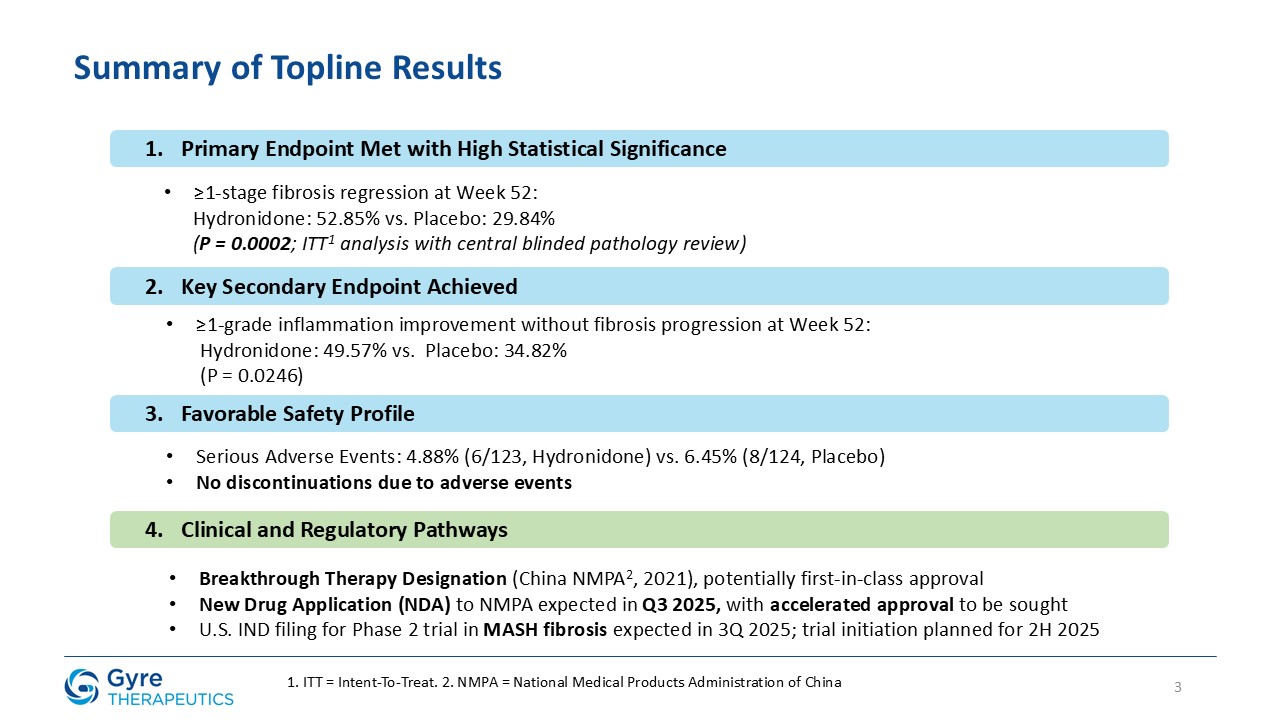
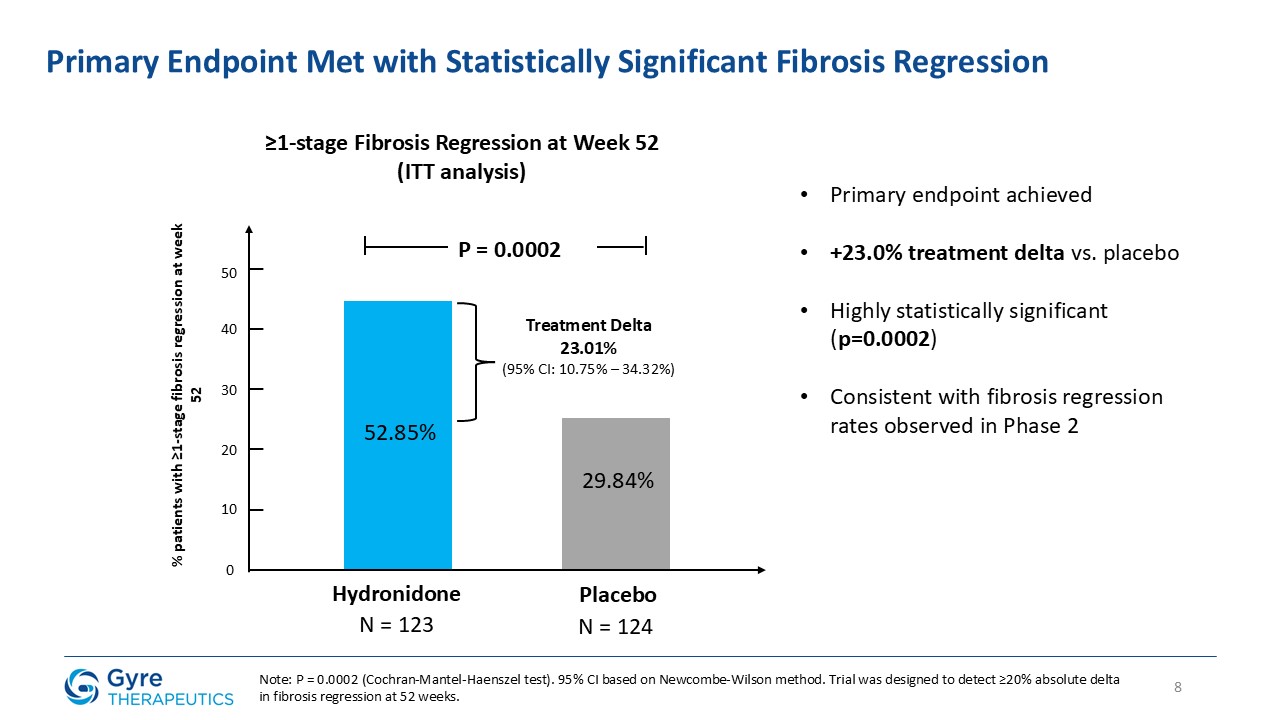
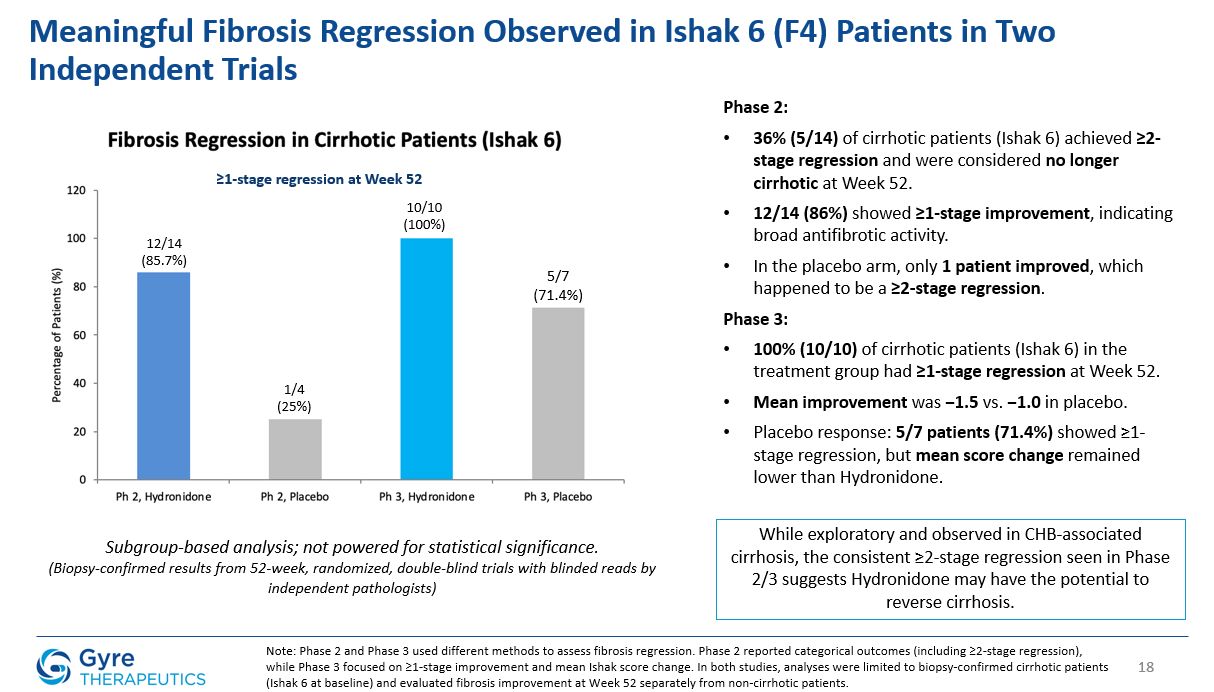
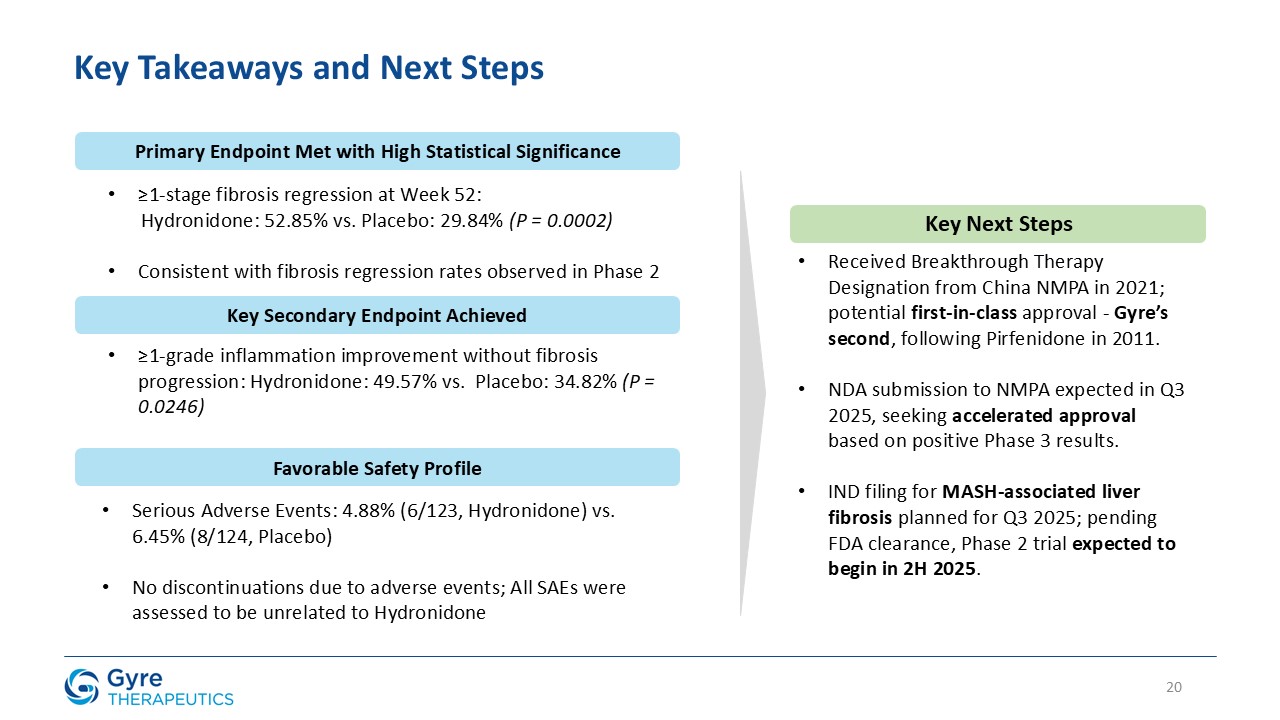
Achieved statistically significant ≥1-stage fibrosis regression at Week 52 vs. placebo (52.85% vs.
29.84%, P=0.0002).
|
|
• |
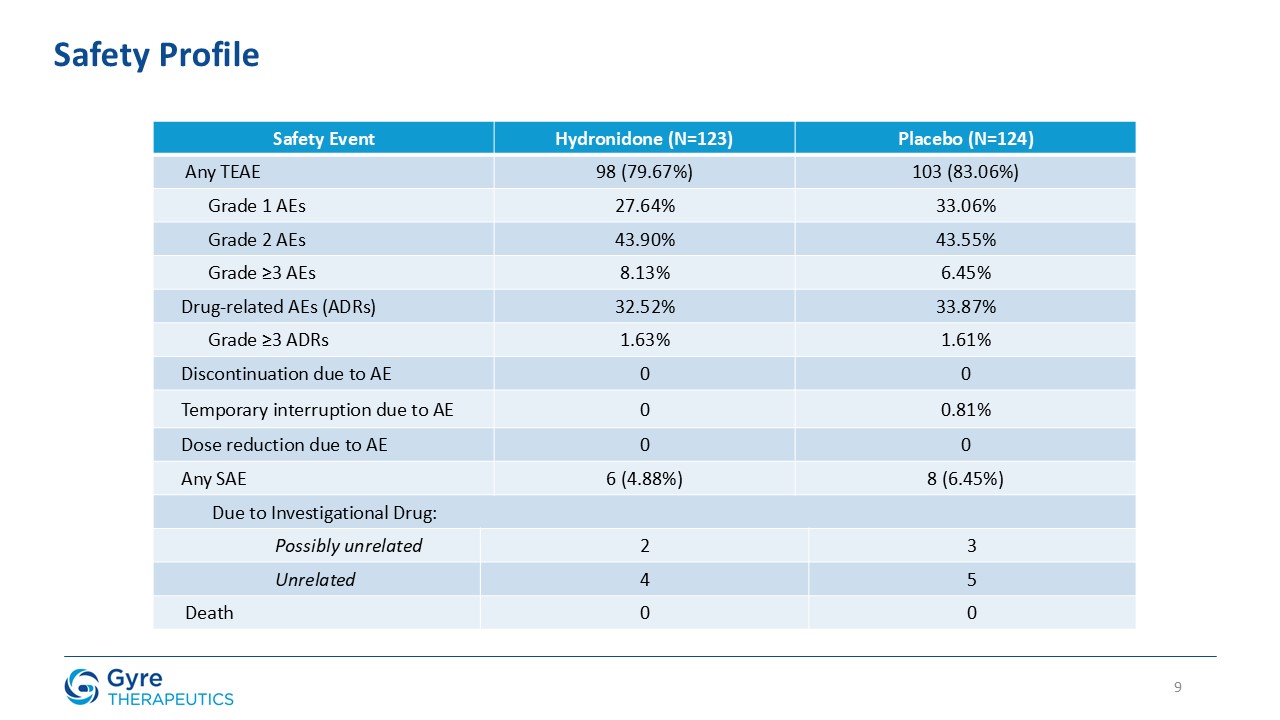
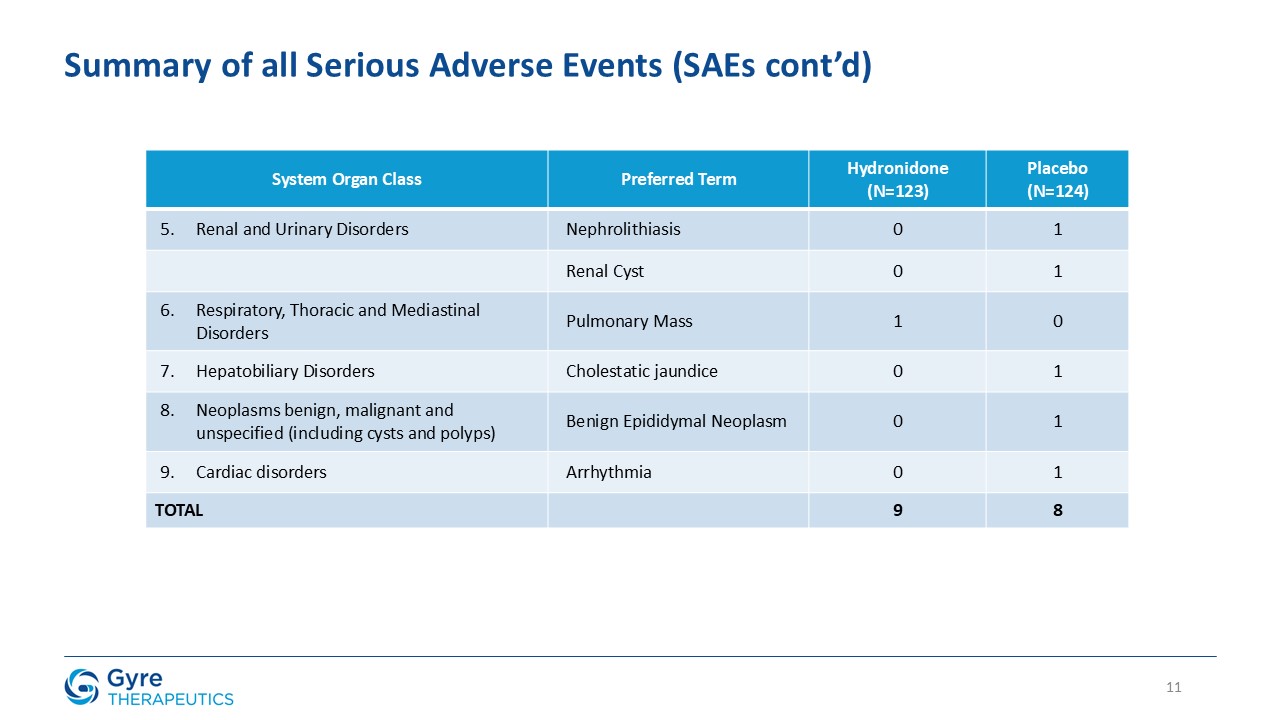
Demonstrated favorable safety and tolerability profile: 4.88% serious adverse events vs. 6.45% for placebo; zero discontinuations due to adverse events.
|
|
• |
Breakthrough Therapy Designation granted by China’s National Medical Products Administration (“NMPA”) in 2021 supports potential first-in-class approval in CHB-associated liver fibrosis (“CHB fibrosis”).
|
|
• |
Gyre intends to seek accelerated approval for Hydronidone in CHB fibrosis, with a New Drug Application (“NDA”) submission to the NMPA expected in Q3 2025.
|
|
• |
U.S. Phase 2 trial in MASH-associated liver fibrosis expected to begin in 2H2025.
|
SAN DIEGO, May 22, 2025 (GLOBE NEWSWIRE) – Gyre Therapeutics (“Gyre”) (Nasdaq: GYRE), an innovative, commercial-stage biotechnology company focused on organ fibrosis, today
announced that its lead compound, Hydronidone (F351), met the primary endpoint in a pivotal Phase 3 trial evaluating its efficacy and safety for the treatment of liver fibrosis in patients with chronic hepatitis B (“CHB”) in China.
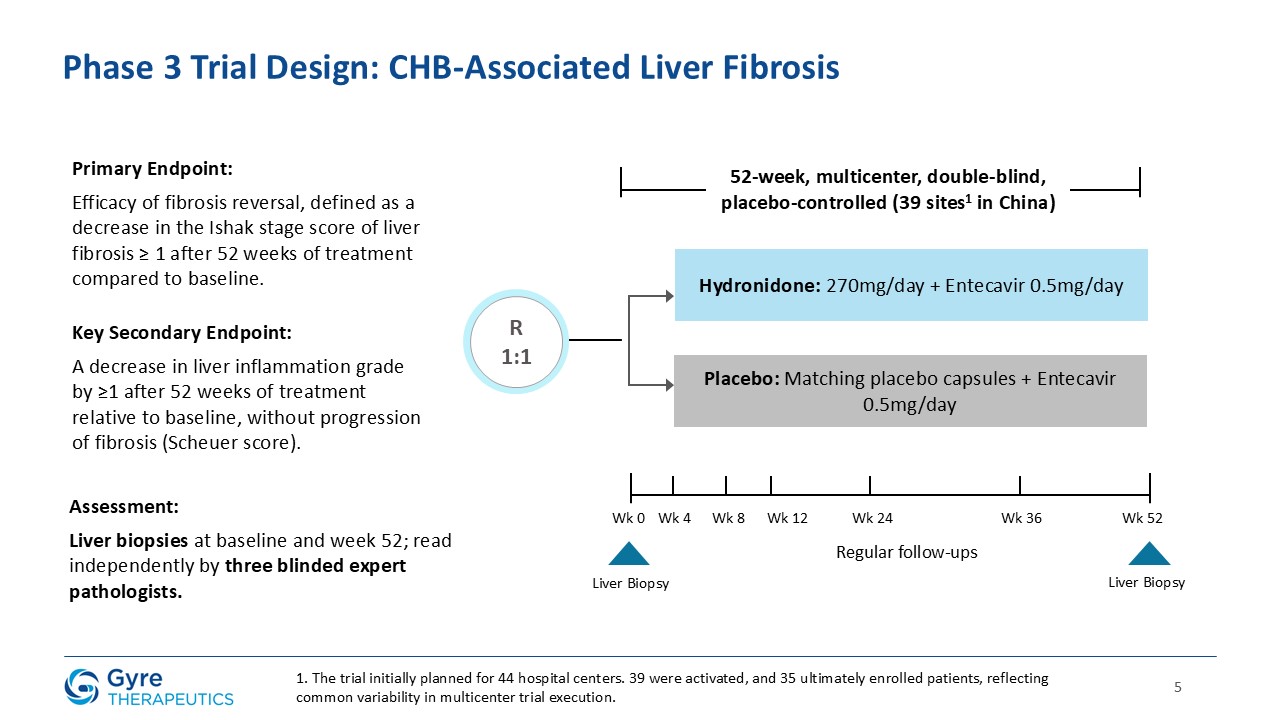
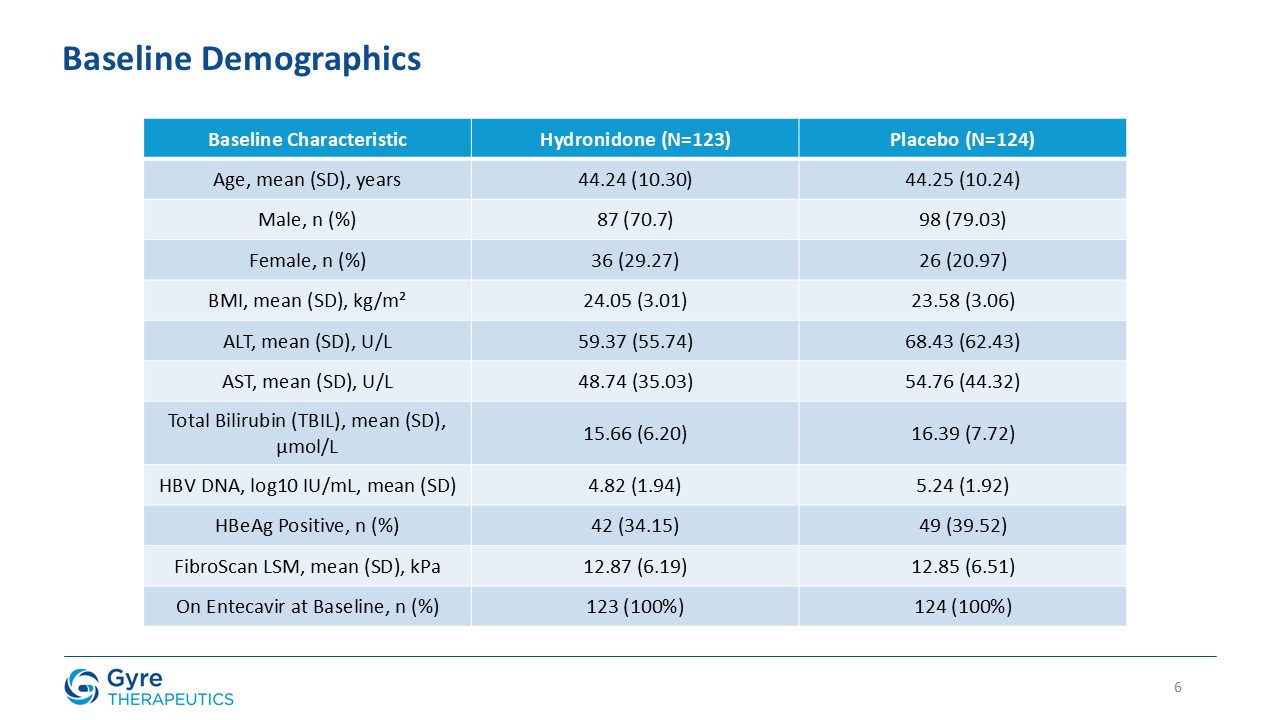
The 52-week, multicenter, double-blind, placebo-controlled trial enrolled 248 patients with CHB fibrosis across 39 hospitals in China. Patients were
randomized 1:1 to receive either Hydronidone (270 mg/day, orally) or placebo, in addition to background entecavir antiviral therapy. The trial met its primary endpoint, with a statistically significant proportion of patients receiving Hydronidone
achieving a ≥1-stage regression in liver fibrosis compared to placebo (P=0.0002). These results are consistent with the efficacy and safety outcomes observed in Gyre’s prior Phase 2 trial.
“These landmark Phase 3 results represent a major step forward for Gyre and for the millions of Chinese patients living with CHB fibrosis,” said Han Ying, Ph.D., CEO of
Gyre Therapeutics. “Pending regulatory approval in China, Hydronidone may become the first therapy specifically indicated for reversing liver fibrosis in CHB patients and the foundation for broader expansion into metabolic dysfunction-associated
steatohepatitis (“MASH”)-related fibrosis in the United States. We are deeply grateful to the patients and investigators who participated in this pivotal trial and made this important milestone possible.”
“The fibrosis regression in our trial marks a breakthrough in the treatment of CHB fibrosis,” said Prof. Lungen Lu, M.D., Dean of the Department of Gastroenterology at
Shanghai General Hospital, Shanghai Jiaotong University School of Medicine, and lead principal investigator. “With no approved anti-fibrotic therapies for this liver disease currently available, Hydronidone has the potential to transform the
treatment landscape and offer new hope to patients facing the serious risks of cirrhosis, liver failure, and hepatocellular carcinoma.”
Hydronidone Week 52 Results
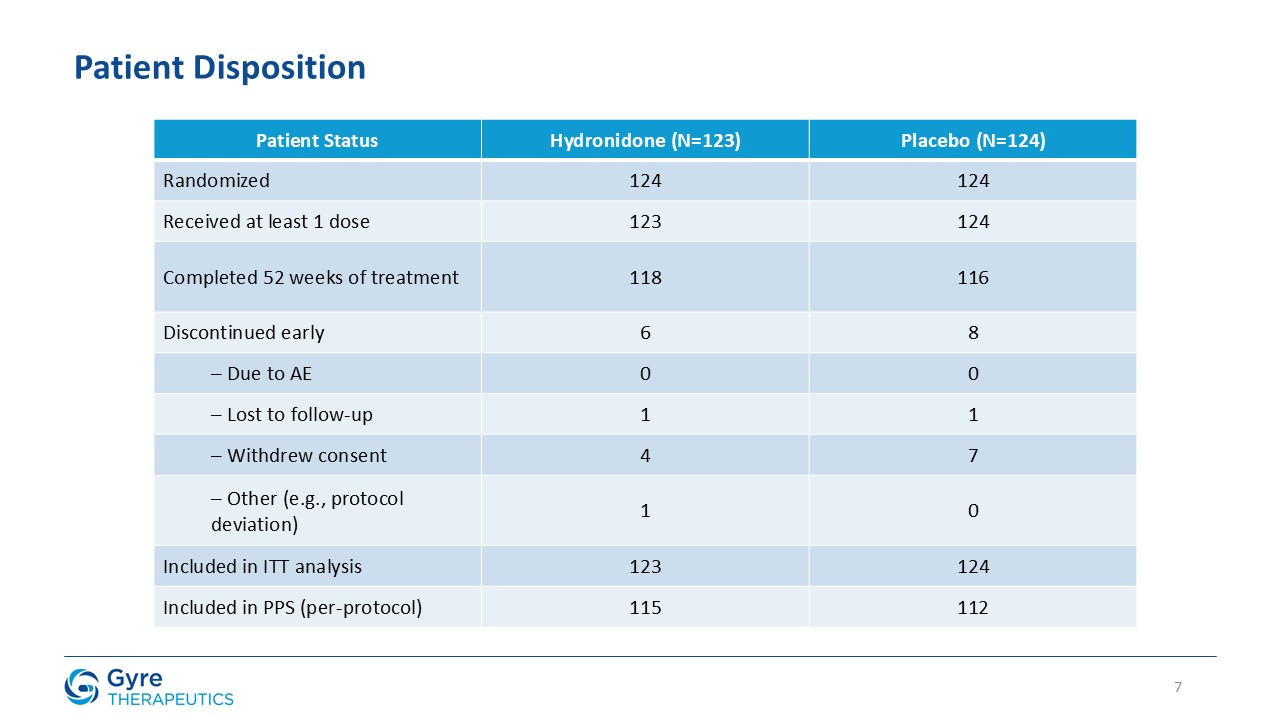
The efficacy analysis followed the intent-to-treat (“ITT”) principle. The ITT population comprised all randomized subjects who received at least one dose of study drug. One
randomized subject who did not receive any treatment was excluded. All biopsies were independently reviewed by three blinded central pathologists to ensure consistency and objectivity of fibrosis and inflammation assessments.
Efficacy Results
In the ITT population, Hydronidone demonstrated statistically significant regression in liver fibrosis compared to placebo.
| |
Primary Endpoint
|
Hydronidone 270mg
N=123
|
Placebo
N=124
|
P-value
|
| |
≥1-stage fibrosis regression (Ishak)
|
52.85%
|
29.84%
|
P=0.0002
|
| |
|
|
|
|
| |
Key Secondary Endpoints
|
|
|
|
| |
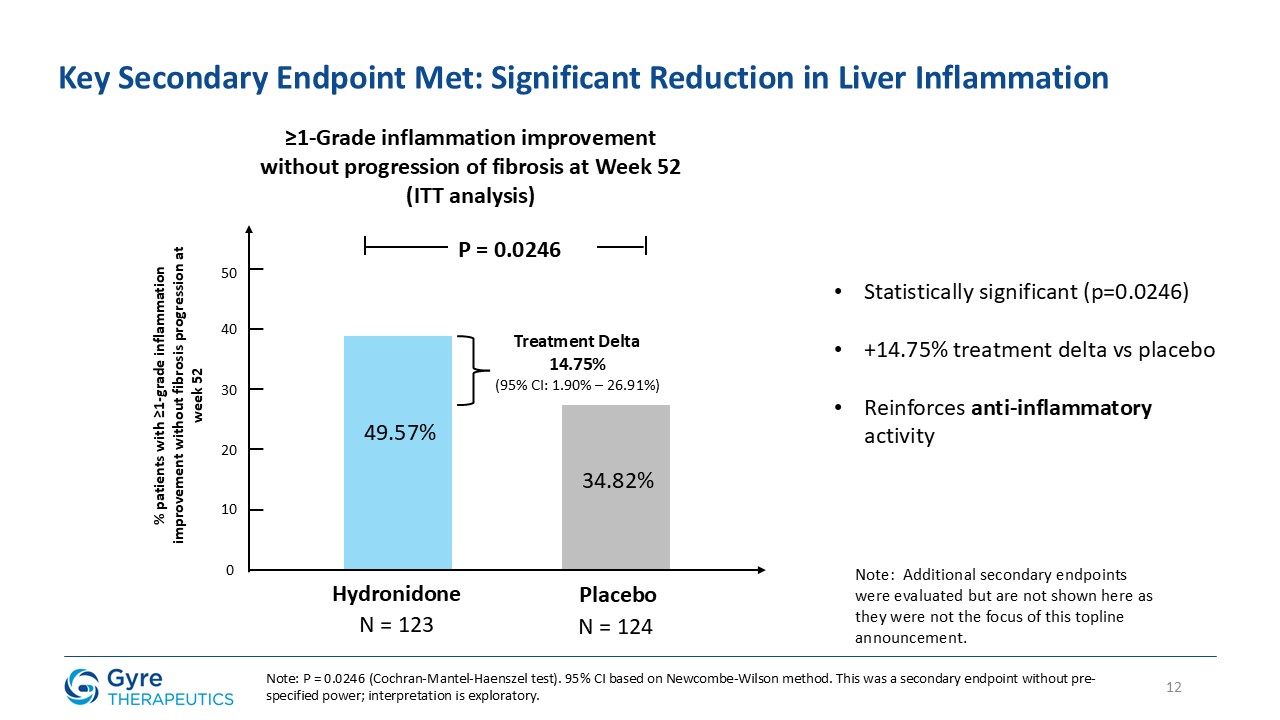
≥1-Grade inflammation improvement (Scheuer score) without progression of fibrosis
|
49.57%
|
34.82%
|
P=0.0246
|
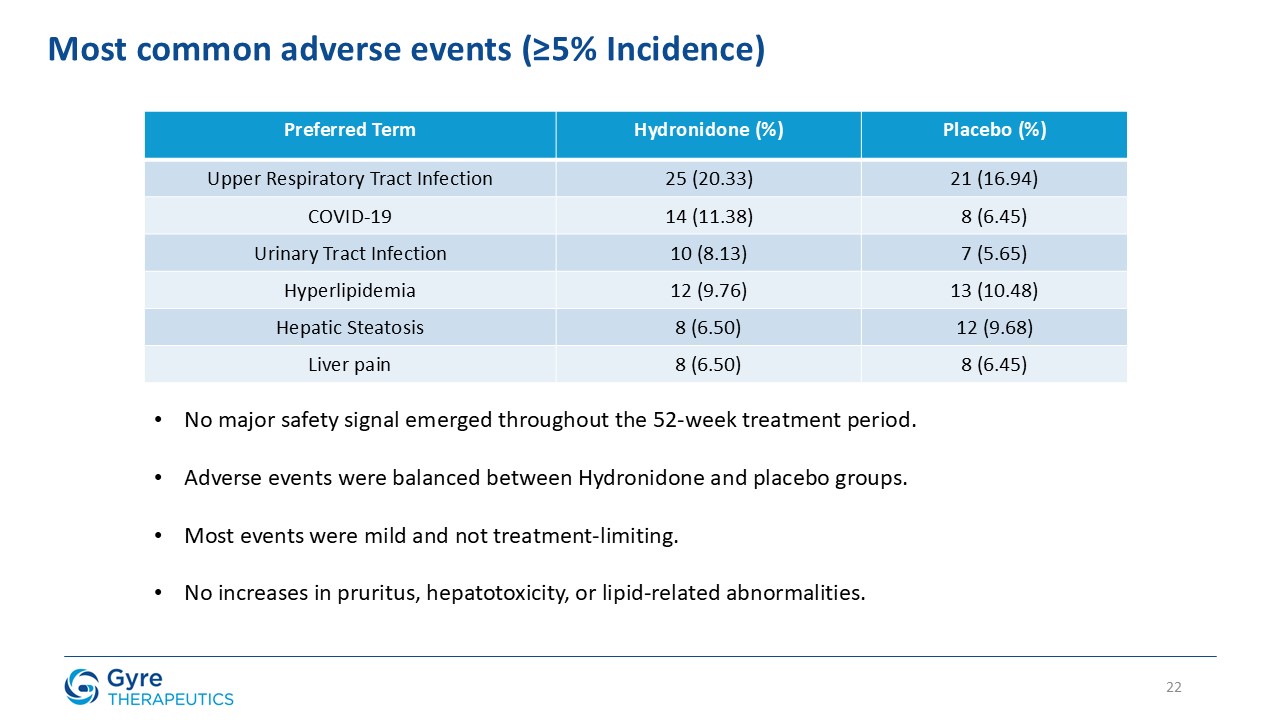
Safety Results
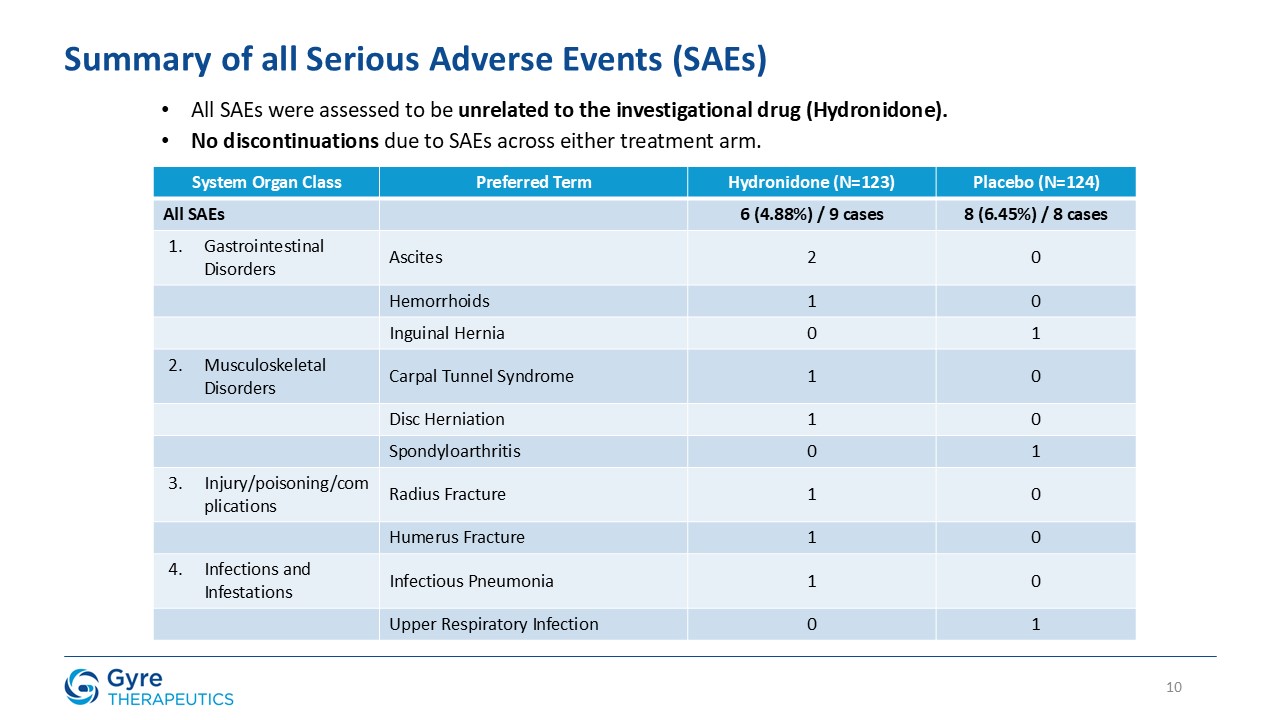
Hydronidone was well tolerated, with a comparable incidence of serious adverse events (4.88% vs. 6.45% in the placebo group) and no discontinuations due to adverse events
in either group. Most adverse events were mild or moderate and unrelated to Hydronidone, while a small number of severe adverse events occurred, none of which were considered related to the trial drug.
Our Path Forward
Gyre plans to submit primary results for publication in a peer-reviewed journal and present full trial results at a future medical congress.
Based on these positive results, Gyre plans to file an NDA with China’s NMPA in the third quarter of 2025. In parallel, the Company is actively preparing to file an
investigational new drug (“IND”) application in the third quarter of 2025 and, subject to IND clearance, plans to initiate a Phase 2 trial in the U.S. evaluating Hydronidone for the treatment of MASH-associated fibrosis in the
second half of 2025. A prior U.S. Phase 1 trial in healthy volunteers confirmed Hydronidone’s tolerability and demonstrated a pharmacokinetic profile consistent with the Chinese population.
About the Phase 3 Trial
The randomized, double-blind, placebo-controlled multicenter Phase 3 trial (NCT05115942) enrolled 248 patients with CHB fibrosis (Ishak fibrosis stage ≥3) across 39 hospitals
in China. Patients were randomized 1:1 to receive either 270 mg Hydronidone or placebo daily, in combination with entecavir. The primary endpoint of the trial was the efficacy of fibrosis regression, defined as a decrease in the Ishak stage score
of liver fibrosis ≥ 1 after 52 weeks of treatment compared to baseline. A key secondary endpoint was a ≥1-grade reduction in liver inflammation, as assessed by the Scheuer
scoring system, after 52 weeks of treatment compared to baseline, without progression of fibrosis.
Patient Population for CHB-Associated Liver Fibrosis
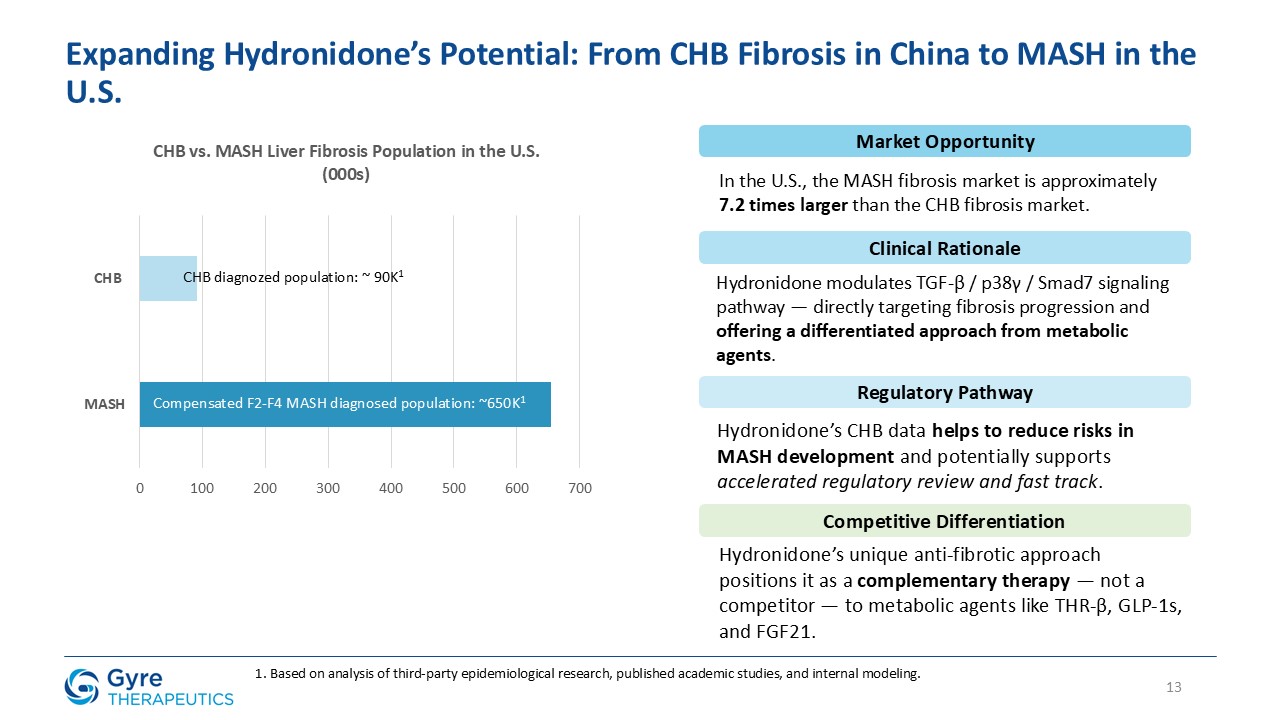
According to national serological surveys, approximately 75 million people in China are chronically infected with hepatitis B virus. A significant subset develops CHB with
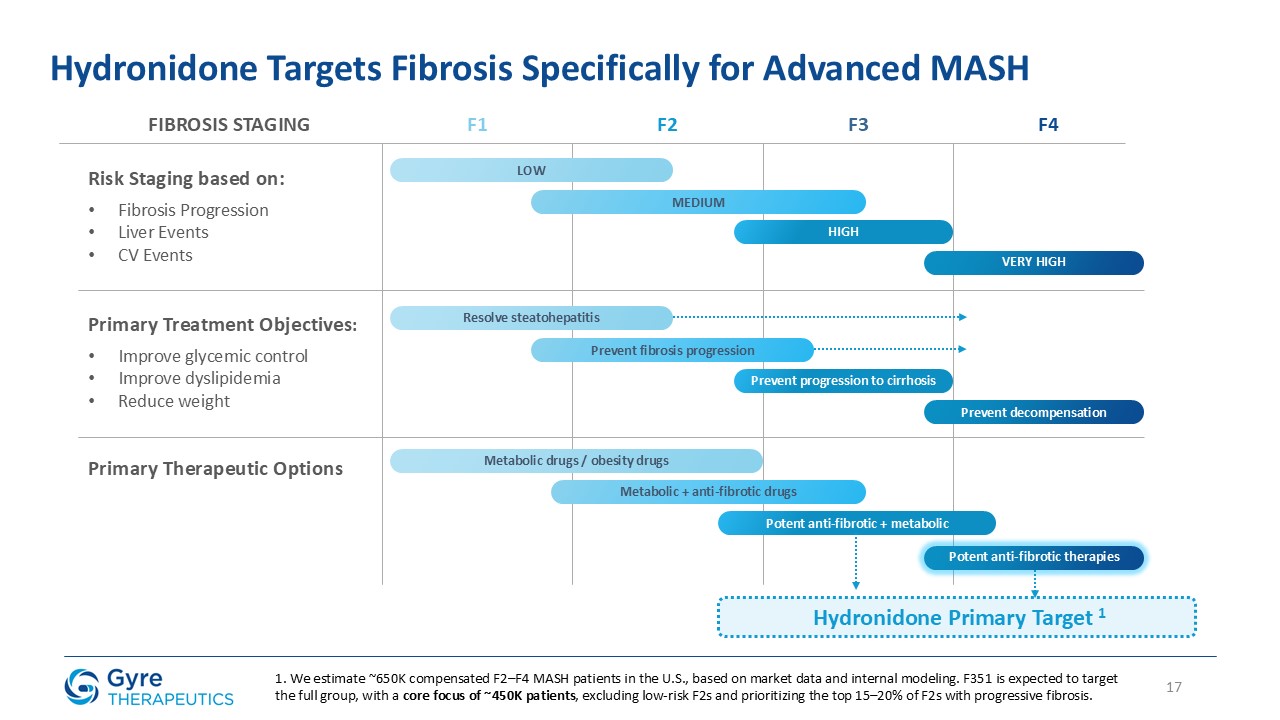
progressive fibrosis. Based on internal modeling of epidemiologic and staging data, Gyre estimates that approximately 2.6 million patients in China have been diagnosed with compensated F2-F4 CHB fibrosis, representing the initial addressable market
for Hydronidone. CHB remains the leading cause of liver fibrosis in China, and currently no approved anti-fibrotic therapies exist for this population.
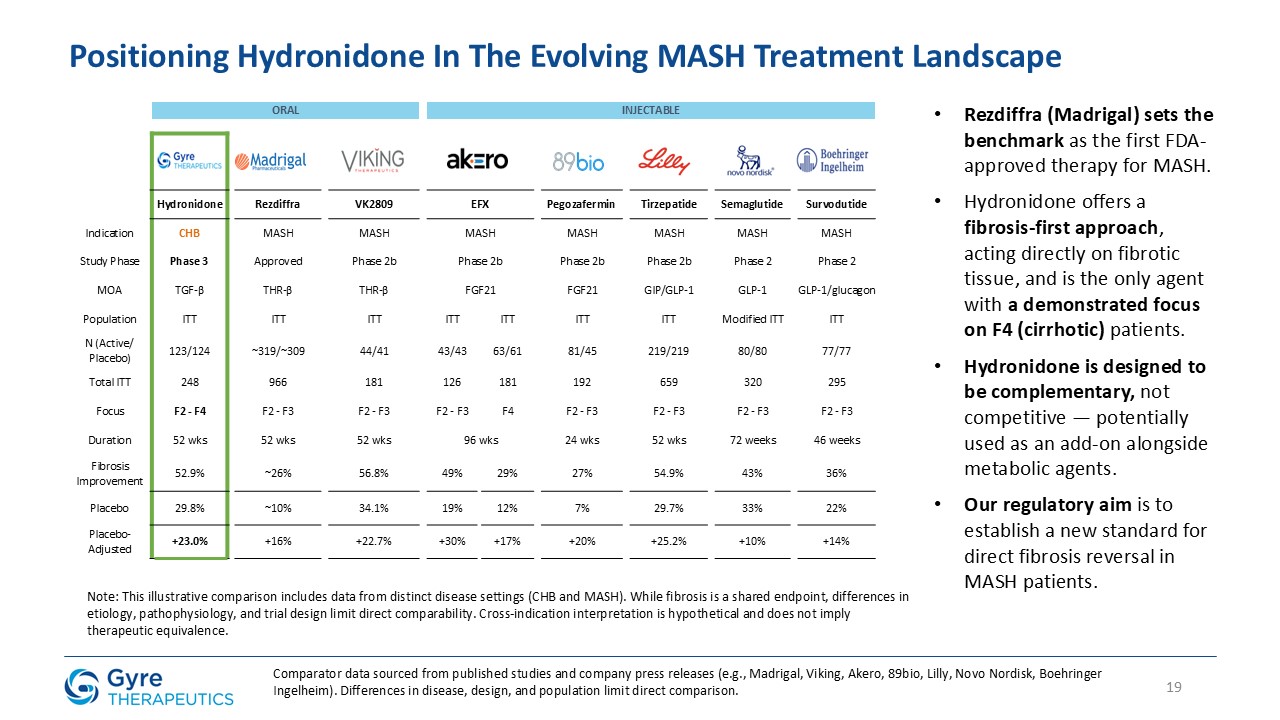
Hydronidone is uniquely positioned to address this significant and urgent unmet medical need. Gyre was granted Breakthrough Therapy Designation in 2021 by the NMPA.
About Hydronidone (F351)
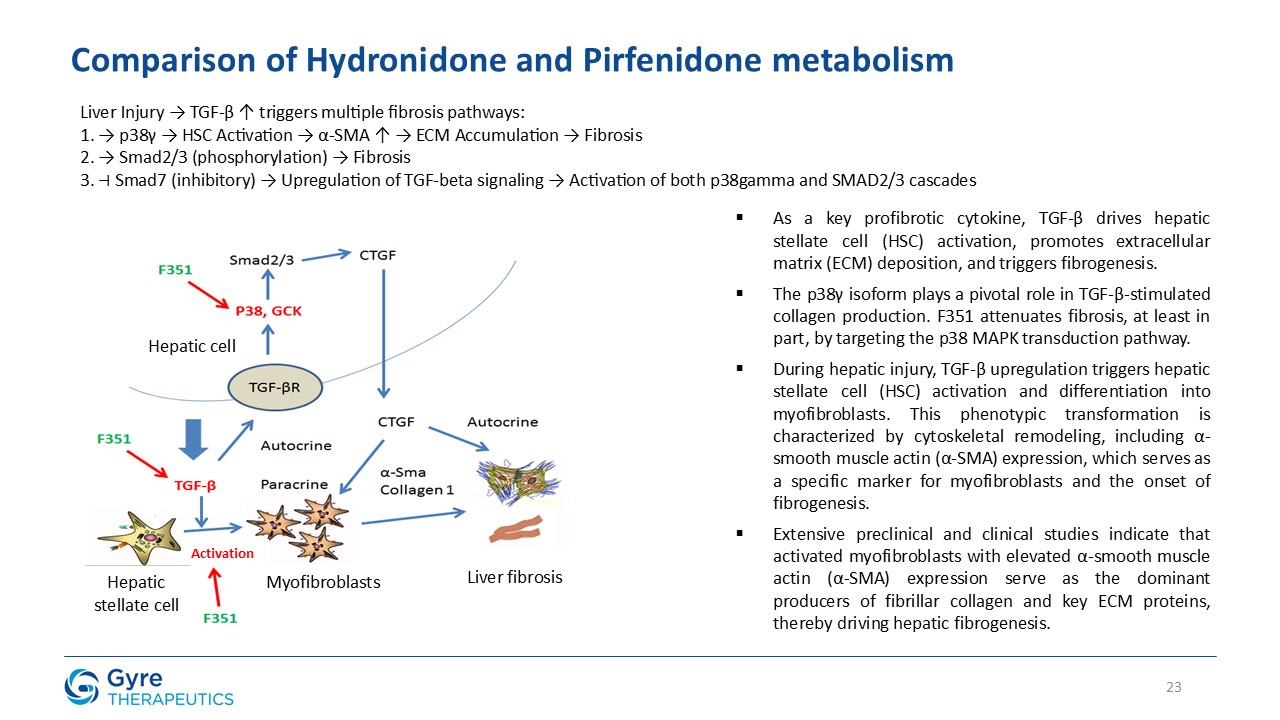
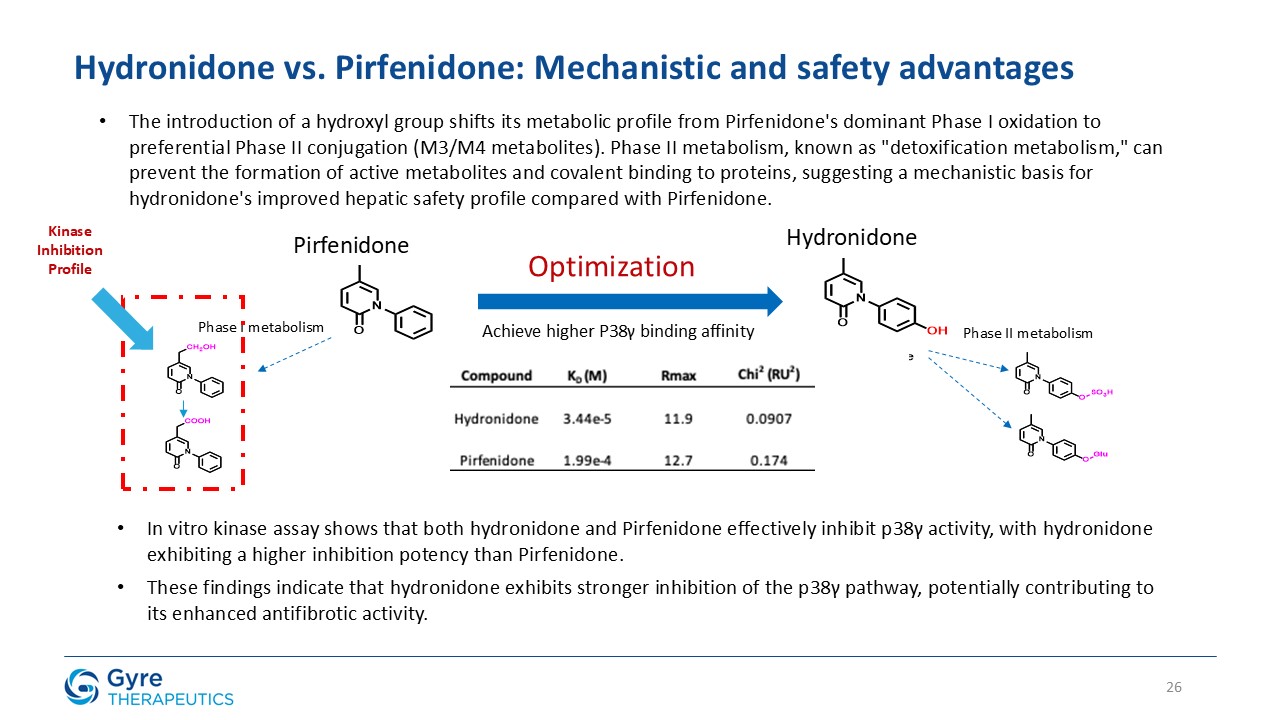
Hydronidone (F351) is a structural analogue of Pirfenidone, for which Gyre received first-in-class approval
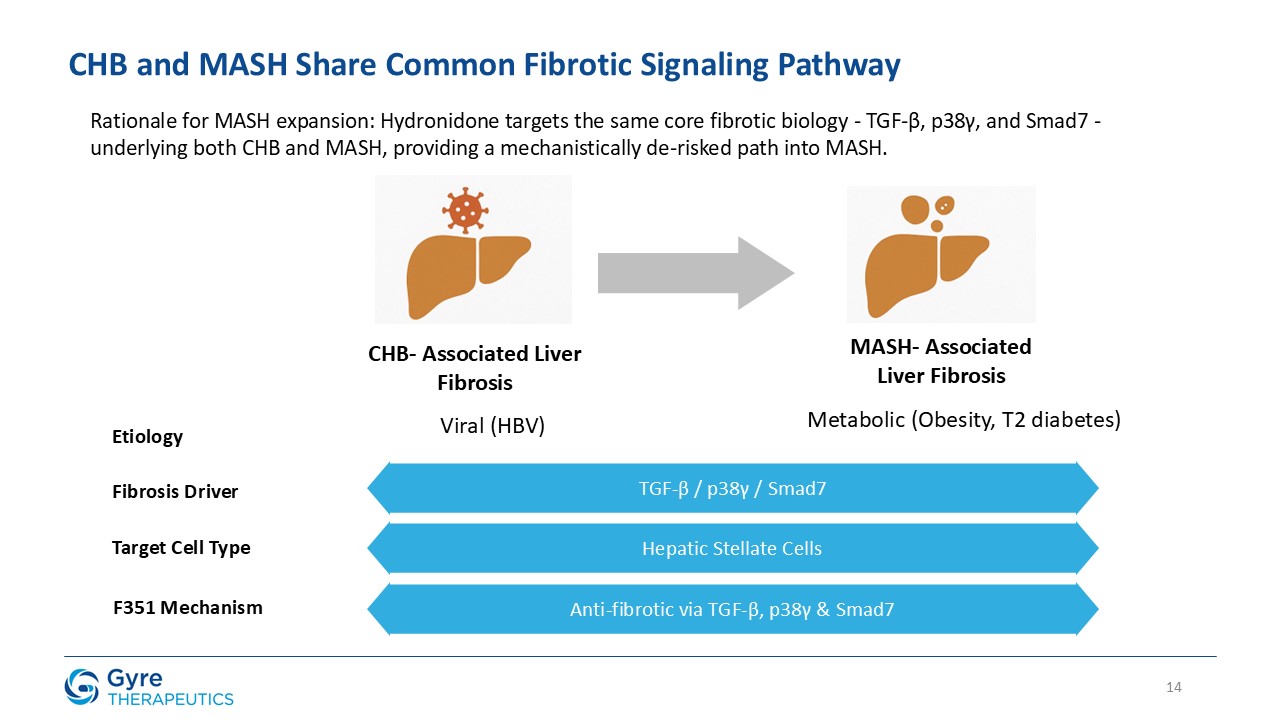
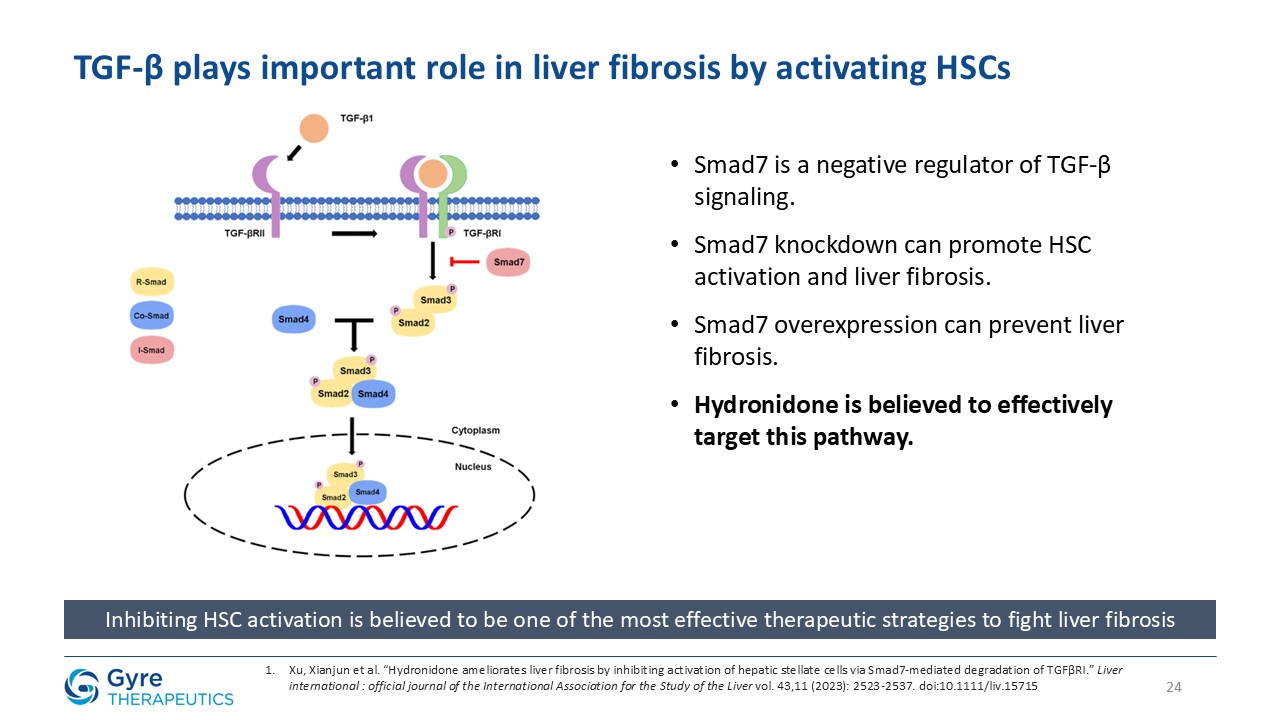
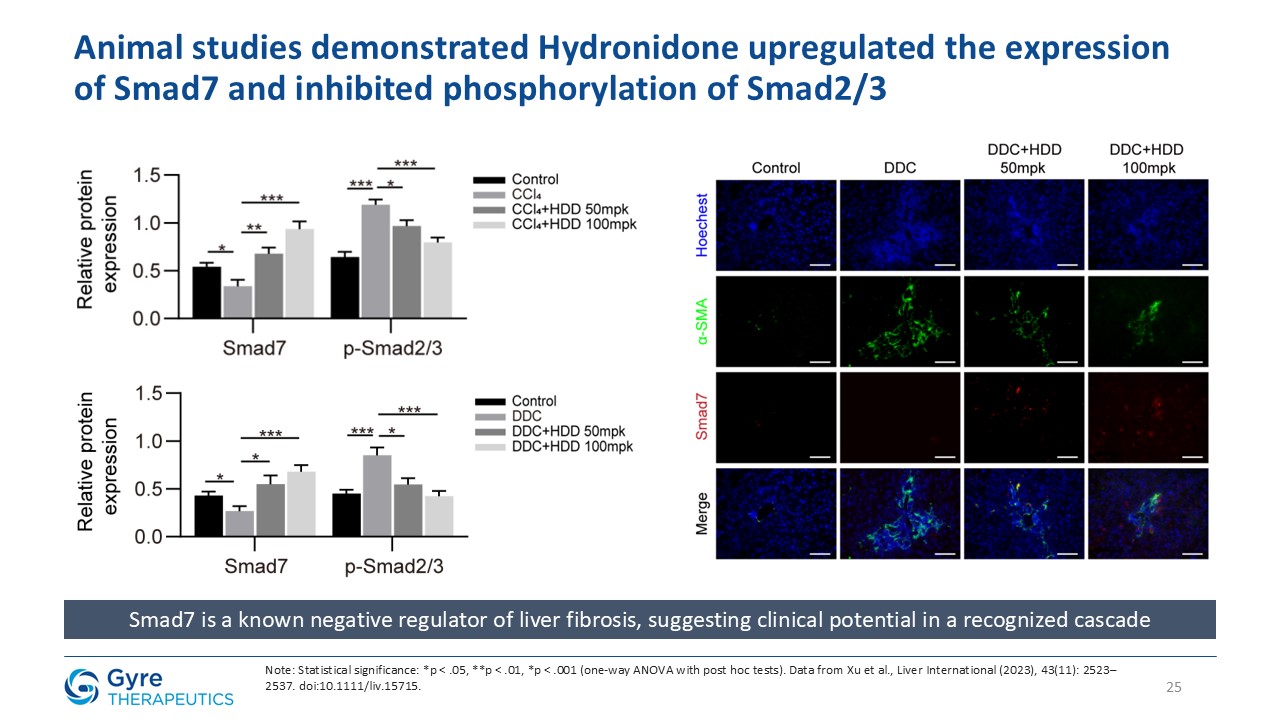
in China in 2011 for the treatment of idiopathic pulmonary fibrosis (IPF). Hydronidone exhibits enhanced potency in inhibiting p38γ kinase activity and TGF-β1-induced collagen synthesis in hepatic stellate cells (HSCs), key drivers of liver
fibrosis. It also demonstrates anti-proliferative activity in HSCs through upregulation of Smad7, which downregulates TGF-βRI, thereby suppressing both the p38γ and Smad2/3 fibrogenic pathways.
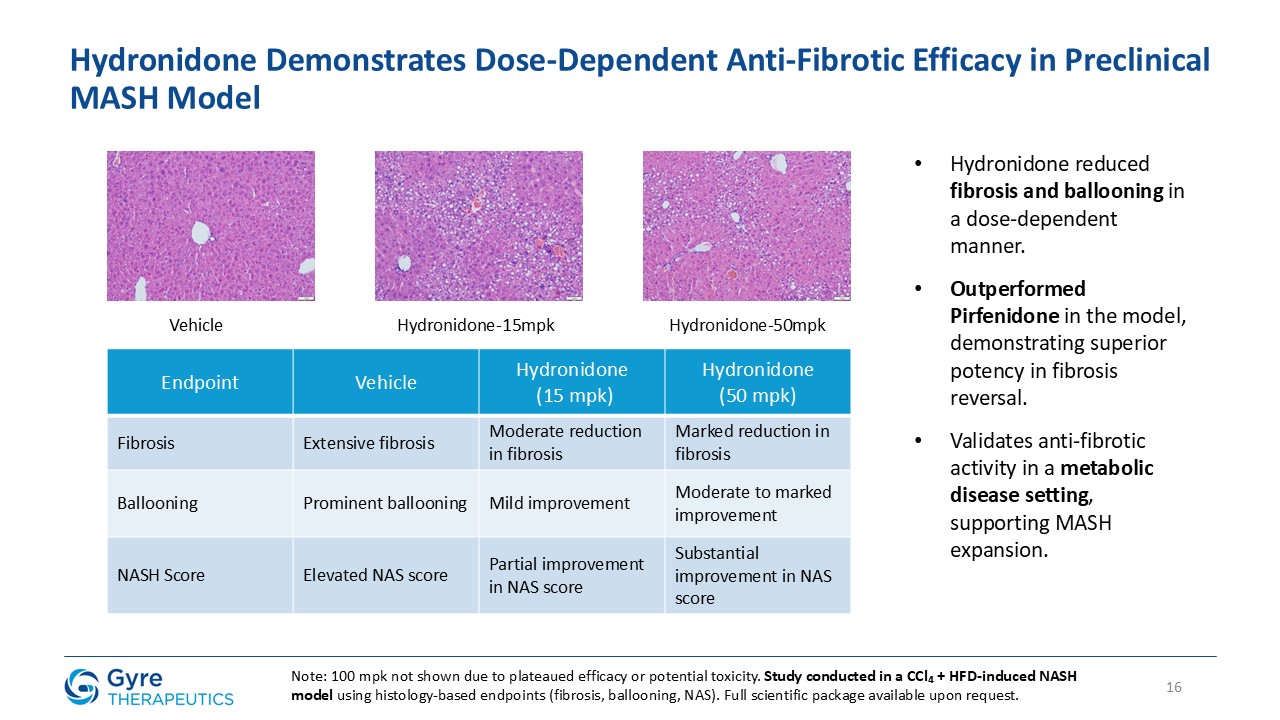
Hydronidone has shown robust anti-fibrotic activity in multiple preclinical models, including the CCl₄-induced mouse model, DMN- and HSA-induced rat models, and a MASH
model combining CCl₄ with a Western high-fat diet. In a randomized, double-blind, placebo-controlled Phase 3 trial, Hydronidone 270 mg/day significantly reduced liver fibrosis in CHB patients after 52 weeks of treatment. Compared to Pirfenidone,
Hydronidone’s unique Phase II conjugation metabolism may contribute to an improved hepatic safety profile.
About Gyre Therapeutics
Gyre Therapeutics is a biopharmaceutical company headquartered in San Diego, CA, primarily focused on the development and commercialization of Hydronidone for liver
fibrosis including MASH in the U.S. Gyre’s strategy builds on its experience in mechanistic studies using MASH rodent models and clinical studies in CHB-induced liver fibrosis. In the PRC, Gyre is advancing a broad pipeline through its indirect
controlling interest in Gyre Pharmaceuticals, including therapeutic expansions of ETUARY, and development programs for F573, F528, and F230.
Forward-Looking Statements
This press release contains “forward-looking statements” within the meaning of the “safe harbor” provisions of the Private Securities Litigation Reform Act of 1995, which
statements are subject to substantial risks and uncertainties and are based on estimates and assumptions. All statements, other than statements of historical facts included in this press release, are forward-looking statements, including statements
concerning: the expectations regarding Gyre’s research and development efforts and timing of expected clinical trials, including timing of a U.S. Phase 2 clinical trial initiation in the second half of 2025 and NDA submission to NMPA for F351. In
some cases, you can identify forward-looking statements by terms such as “may,” “might,” “will,” “objective,” “intend,” “should,” “could,” “can,” “would,” “expect,” “believe,” “design,” “estimate,” “predict,” “potential,” “plan” or the negative of
these terms, and similar expressions intended to identify forward-looking statements. These statements reflect our plans, estimates, and expectations, as of the date of this press release. These statements involve known and unknown risks,
uncertainties and other factors that could cause our actual results to differ materially from the forward-looking statements expressed or implied in this press release. Actual results and the timing of events could differ materially from those
anticipated in such forward-looking statements as a result of these risks and uncertainties, which include, without limitation: Gyre’s ability to execute on its clinical development strategies; positive results from a clinical trial may not
necessarily be predictive of the results of future or ongoing clinical trials; the timing or likelihood of regulatory filings and approvals; competition from competing products; the impact of general economic, health, industrial or political
conditions in the United States or internationally; the sufficiency of Gyre’s capital resources and its ability to raise additional capital. Additional risks and factors are identified under “Risk Factors” in Gyre’s Annual Report on Form 10-K for
the year ended December 31, 2024 filed on March 17, 2025 and in other filings the Company may make with the Securities and Exchange Commission.
Gyre expressly disclaims any obligation to update any forward-looking statements whether as a result of new information, future events or otherwise, except as required by
law.
David Zhang
david.zhang@gyretx.com