| ☒ |
Annual report pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934.
|
| ☐ |
Transition report pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934.
|
|
Delaware
|
11-3516358
|
|
(State or other jurisdiction of
incorporation or organization)
8 Davis Drive, Suite 220
Durham, NC
(Address of principal executive offices) |
(I.R.S. Employer
Identification No.)
(Zip Code)
|
|
Title of each class
|
Trading Symbol
|
Name of each exchange on which registered
|
|
Common Stock, $0.0001 par value per share
|
IRD
|
The Nasdaq Stock Market LLC
|
|
Large accelerated filer ☐
|
|
|
Accelerated filer ☐
|
|
|
|
|
|
||
|
Non-accelerated filer ☒
|
|
|
Smaller reporting company ☒
|
|
|
Emerging growth company ☐
|
||||
|
PART I
|
5 |
||
|
|
|||
|
ITEM 1.
|
5 | ||
|
ITEM 1A.
|
51 | ||
|
ITEM 1B.
|
95 | ||
|
ITEM 1C.
|
95 | ||
|
ITEM 2.
|
96 | ||
|
ITEM 3.
|
96 | ||
|
ITEM 4.
|
97 | ||
|
|
|
||
|
PART II
|
|
||
|
|
|||
|
ITEM 5.
|
97 | ||
|
ITEM 6.
|
98 | ||
|
ITEM 7.
|
98 | ||
|
ITEM 7A.
|
112 | ||
|
ITEM 8.
|
112 |
||
|
ITEM 9.
|
112 |
||
|
ITEM 9A.
|
112 |
||
|
ITEM 9B.
|
113 |
||
|
ITEM 9C.
|
113 |
||
|
|
|||
|
PART III
|
113 |
||
|
|
|||
|
ITEM 10.
|
113 |
||
|
ITEM 11.
|
114 |
||
|
ITEM 12.
|
114 |
||
|
ITEM 13.
|
114 |
||
|
ITEM 14.
|
114 |
||
|
|
|||
|
PART IV
|
114 |
||
|
|
|
||
|
ITEM 15.
|
114 |
||
|
ITEM 16.
|
115 |
||
|
|
|||
| 146 |
|||
|
|
• |
Failure to successfully integrate our businesses with Former Opus (as defined below) could have a material adverse effect on our business,
financial condition and results of operations.
|
|
|
• |
The Opus Acquisition (as defined below) significantly expanded our product pipeline and business operations and shifted our business
strategies, which may not improve the value of our common stock.
|
|
|
• |
Our gene therapy product candidates are based on a novel technology that is difficult to develop and manufacture, which may result in delays and difficulties in obtaining regulatory approval.
|
|
|
• |
Our planned clinical trials may face substantial delays, result in failure, or provide inconclusive or adverse results that may not satisfy FDA requirements to further develop our therapeutic products.
|
|
|
• |
Delays or difficulties associated with patient enrollment in clinical trials may affect our ability to conduct and complete those clinical trials and obtain necessary regulatory approvals.
|
|
|
• |
Changes in regulatory requirements could result in increased costs or delays in development timelines.
|
|
|
• |
We depend heavily on the success of our product pipeline; if we fail to find strategic partners or fail to adequately develop or commercialize our pipeline products, our business will be materially harmed.
|
|
|
• |
Others may discover, develop, or commercialize products similar to those in our pipeline before or more successfully than we do or develop generic variants of our products even while our product patents remain
active, thereby reducing our market share and potential revenue from product sales.
|
|
|
• |
We do not currently have any sales or marketing infrastructure in place and we have limited drug research and discovery capabilities.
|
|
|
• |
The future commercial success of our products could significantly depend upon several uncertain factors, including third-party reimbursement practices and the existence of competitors with similar products.
|
|
|
• |
Product liability lawsuits against us or our suppliers or manufacturers could cause us to incur substantial liabilities and could limit commercialization of any product candidate that we may develop.
|
|
|
• |
Failure to comply with health and safety laws and regulations could lead to material fines.
|
|
|
• |
We have not generated significant revenue from sales of any products and expect to incur losses for the foreseeable future.
|
|
|
• |
Our future viability is difficult to assess due to our short operating history and our future need for substantial additional capital, access to which could be limited by any adverse developments that
affect the financial markets.
|
|
|
• |
Raising additional capital may cause our stockholders to be diluted, among other adverse effects.
|
|
|
• |
We operate in a highly regulated industry and face many challenges adapting to sudden changes in legislative reform or the regulatory environment, which affects our pipeline stability and could impair our
ability to compete in international markets.
|
|
|
• |
We may not receive regulatory approval to market our developed product candidates within or outside of the U.S.
|
|
|
• |
With respect to any of our product candidates that receive marketing approval, we may be subject to substantial penalties if we fail to comply with applicable regulatory requirements.
|
|
|
• |
Our potential relationships with healthcare providers and third-party payors will be subject to certain healthcare laws and regulations, which could expose us to extensive potential liabilities.
|
|
|
• |
We rely on third parties for material aspects of our business, such as conducting our nonclinical and clinical trials and supplying and manufacturing bulk drug substances, which exposes us to certain risks.
|
|
|
• |
We may be unsuccessful in entering into or maintaining licensing arrangements or establishing strategic alliances on favorable terms, which could harm our business.
|
|
|
• |
Our current focus on the cash-pay utilization for future sales of RYZUMVI may limit our ability to increase sales or achieve profitability with this product.
|
|
|
• |
Inadequate patent protection for our product candidates may result in our competitors developing similar or identical products or technology, which would adversely affect our ability to successfully
commercialize.
|
|
|
• |
We may be unable to obtain full protection for our intellectual property rights under U.S. or foreign laws.
|
|
|
• |
We may become involved in lawsuits for a variety of reasons associated with our intellectual property rights, including alleged infringement suits initiated by third parties.
|
|
|
• |
We are dependent on our key personnel, and if we are not successful in attracting and retaining highly qualified personnel, we may not be able to successfully implement our business strategy.
|
|
|
• |
As we grow, we may not be able to operate internationally or adequately develop and expand our sales, marketing, distribution, and other corporate functions, which could disrupt our operations.
|
|
|
• |
The market price of our common stock is expected to be volatile and subject to certain dilutive risks associated with our Equity Line of Credit arrangement.
|
|
|
• |
Factors out of our control related to our securities, such as securities litigation or actions of activist stockholders, could adversely affect our business and stock price and cause us to incur significant
expenses.
|
|
|
• |
In April 2018, Ocuphire Pharma, Inc. merged with Ocularis Pharma, LLC, the original innovator of phentolamine mesylate ophthalmic solution.
|
|
|
• |
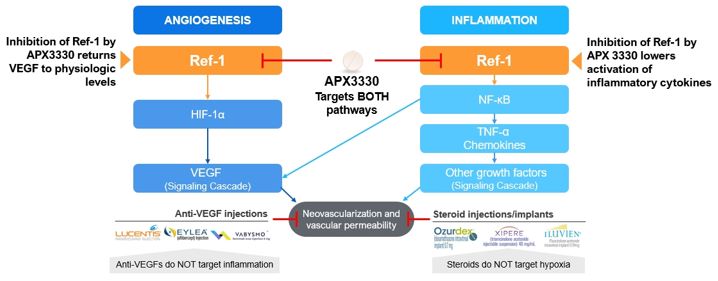
In January 2020, Ocuphire Pharma, Inc. obtained from Apexian Pharmaceuticals, Inc. certain rights to its Ref-1 inhibitor program, including APX3330.
|
|
|
• |
In November 2020, Ocuphire Pharma, Inc. completed a reverse merger into Rexahn Pharmaceuticals, Inc. (“Rexahn”), a publicly traded company that had ceased
its business of drug development activities, and simultaneously raised just over $21 million through an offering of common shares and warrants to purchase common shares. The combined company continued to operate under the name of
Ocuphire Pharma, Inc.
|
|
|
• |
On October 22, 2024, Ocuphire Pharma, Inc. acquired a private corporation then operating under the name of “Opus Genetics Inc.” (“Private Opus”) pursuant to the terms of an Agreement and Plan of Merger, dated as of October 22, 2024
(such agreement, the “Merger Agreement” and the transaction consummated via the Merger Agreement, the “Opus Acquisition”), by and among the Company, Opus, and certain merger subsidiaries party thereto.
|
|
•
|
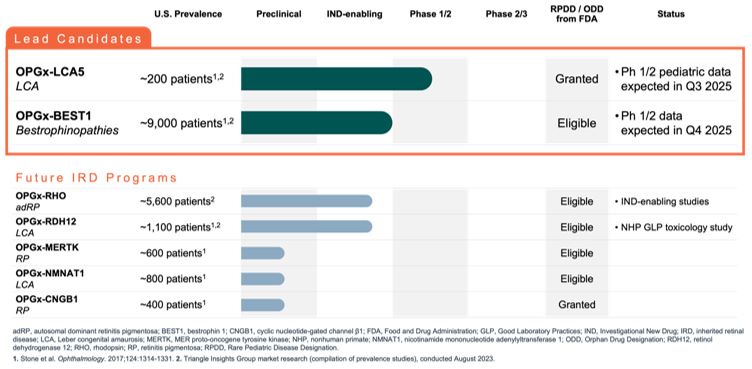
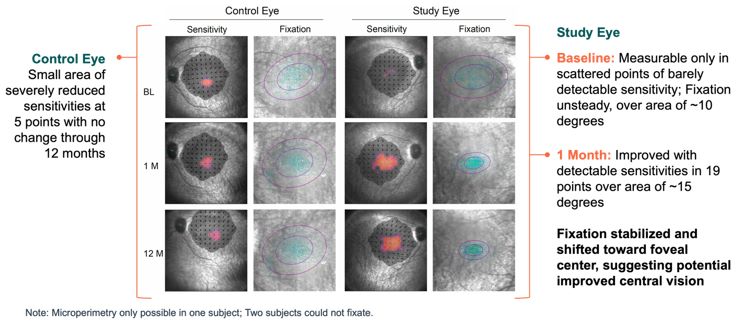
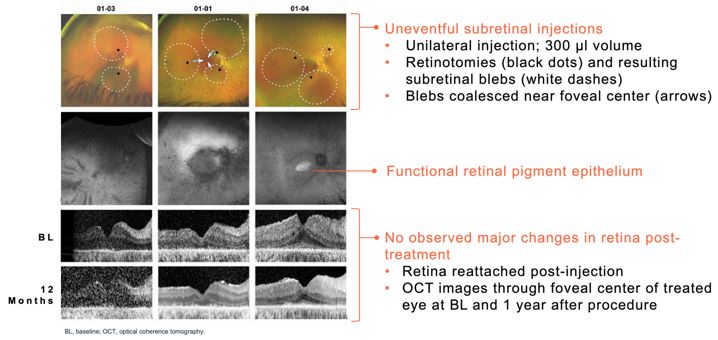
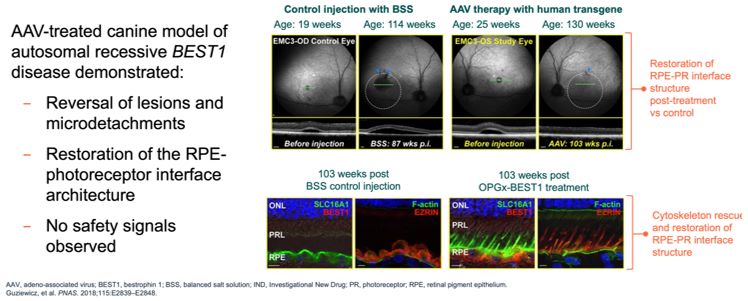
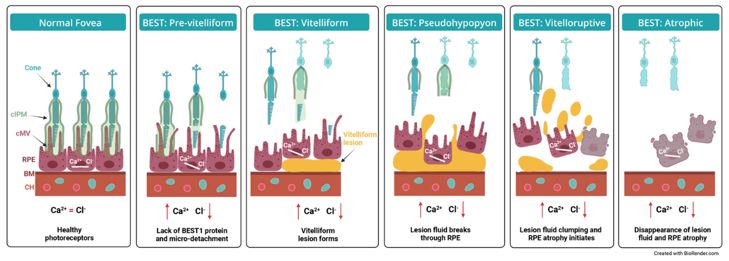
Advance the clinical development of our gene therapy products. OPGx-LCA5 is designed to address a form of LCA due to biallelic mutations in the LCA5, which
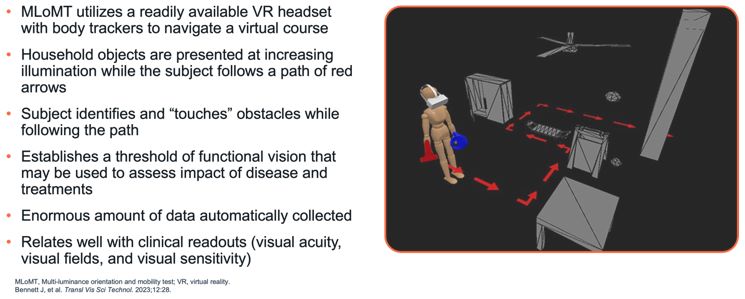
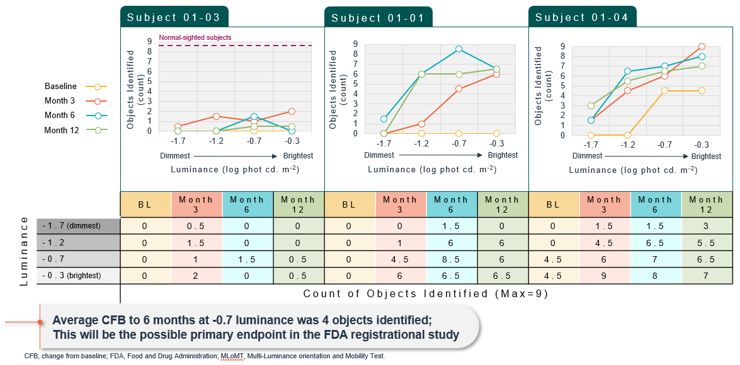
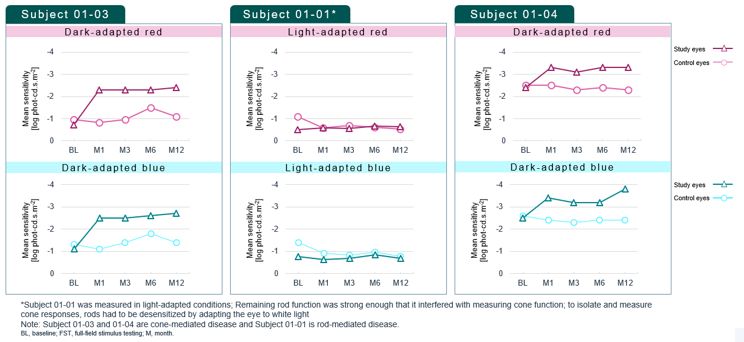
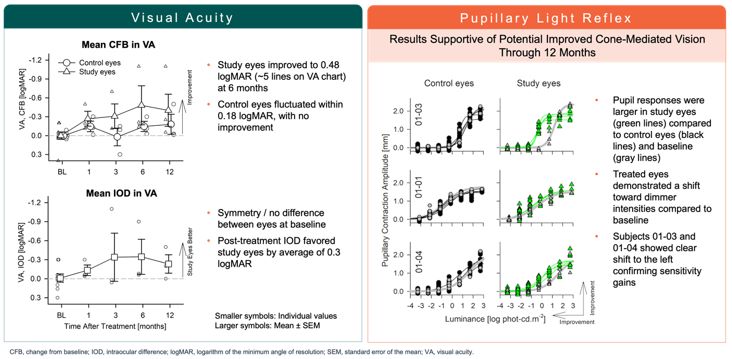
encodes the lebercilin protein. New six-month efficacy and safety data on OPGx-LCA5 were presented at a virtual KOL event on December 11, 2024 and showed improvement in visual function in the first three adult patient cohort treated,
each of whom has late-stage disease. Twelve-month data on OPGx-LCA5 in the adult patients cohort will be presented at ARVO in May 2025. A pediatric cohort to test the therapy in younger subjects is in progress
|
|
•
|
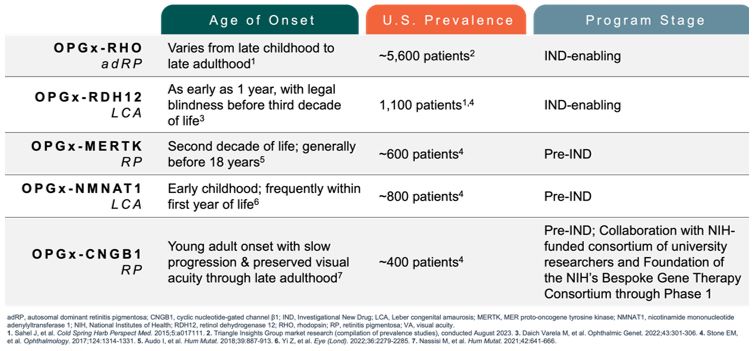
Future IRD programs. Beyond clinical development of OPGx-LCA5
and OPGx-BEST1, Opus has a preclinical portfolio of other AAV gene therapy candidates targeting different forms of vision threatening IRDs, including retinitis pigmentosa (e.g. adRP-RHO, CNGB1) and LCA (e.g., NMNAT1, RDH12). These
programs can be potentially further developed for clinical applications, subject to capital availability. We will continue IND-enabling work for these preclinical gene therapy programs, but these programs may require additional
funding to progress.
|
|
•
|
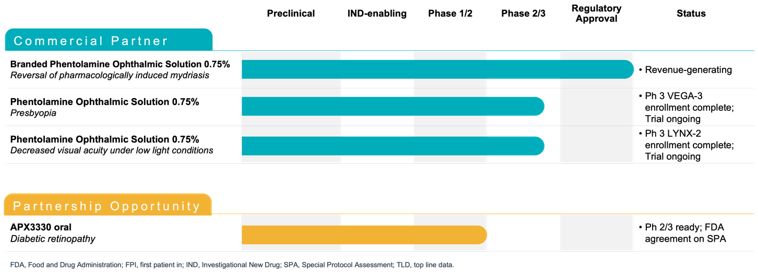
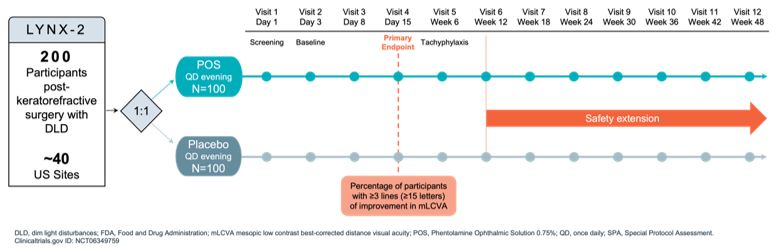
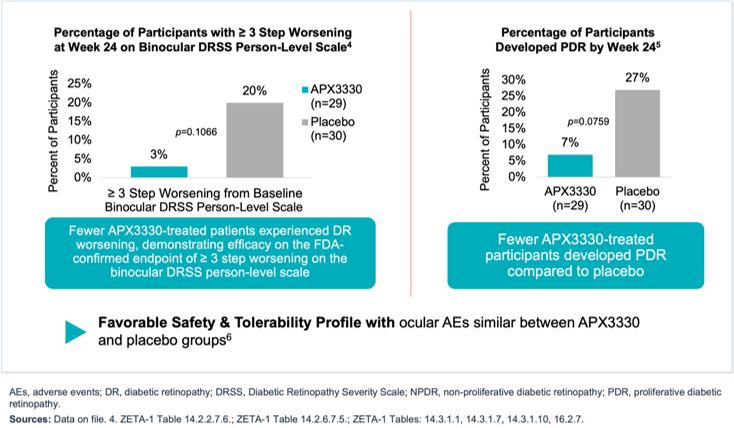
Maximize the value of APX3330 through partnership. In December 2024, we reached agreement with the FDA under SPA for a Phase 3 clinical trial evaluating oral
APX3330 for the treatment of moderate to severe NPDR. The SPA agreement reflects that the proposed Phase 3 trial design, and planned analyses adequately address the objectives necessary to support a New Drug Application (NDA)
submission for treatment of NPDR, subject to a successful outcome of the trial and review of all the data in the NDA, if submitted. The agreed primary endpoint for this clinical trial is a reduction in 3-step or greater worsening
on the binocular diabetic retinopathy severity scale (DRSS) score, compared to placebo. We are seeking a partner to advance the clinical development of APX3330, as we focus our resources on advancing our gene therapy candidates
for IRDs.
|
|
•
|
Complete late-stage development of PS programs. PS was approved by the FDA for the treatment for pharmacologically-induced mydriasis under the brand name
RYZUMVI in September 2023 and was launched commercially in April 2024.
|




|
•
|
Non-proliferative DR, or NPDR. NPDR is an earlier stage of DR and can progress into more severe forms of DR over time if left untreated and if
exposure to elevated blood sugar levels persist. Approximately 8 million patients in the U.S. have NPDR and are at risk of progressing to PDR (as defined below) if left untreated.
|
|
•
|
Proliferative DR, or PDR. PDR is a more advanced stage of DR than NPDR. It is characterized by retinal neovascularization and, if left
untreated, leads to permanent damage to the retina that results in loss of vision.
|
|
•
|
completion of preclinical laboratory tests, animal studies and formulation studies in compliance, as applicable, with the Animal Welfare Act and FDA’s good laboratory practice, or GLP, regulations;
|
|
•
|
submission to the FDA of an IND, which must take effect before human clinical trials may begin;
|
|
•
|
approval by an independent institutional review board, or IRB, representing each clinical site before each clinical trial may be initiated;
|
|
•
|
performance of human clinical trials, including adequate and well-controlled clinical trials, in accordance with good clinical practices, or GCP, and other applicable regulations to establish the safety
and efficacy of the proposed drug product, or the safety, purity, and potency of the proposed biologic, for each proposed indication;
|
|
•
|
manufacturing, packaging, labelling, and distribution of drug substances and drug products consistent with the FDA’s cGMP regulations, as well as GLP non-clinical and GCP clinical studies to investigate
the drug candidate;
|
|
•
|
development of product label, package inserts, and prescriber information that is intended to be used and included with the commercial product;
|
|
•
|
preparation and submission to the FDA of an NDA, BLA or supplements;
|
|
•
|
review of the product by an FDA advisory committee, where appropriate or if applicable;
|
|
•
|
satisfactory completion of one or more FDA inspections of the manufacturing facility or facilities at which the product, or components thereof, are produced to assess compliance with cGMP requirements
and to assure that the facilities, methods and controls are adequate to preserve the product’s identity, strength, quality and purity;
|
|
•
|
satisfactory completion of FDA audits of clinical trial site(s) to assure compliance with GCPs and the integrity of the clinical data;
|
|
•
|
FDA approval of application; and
|
|
•
|
compliance with any post-approval requirements, including Risk Evaluation and Mitigation Strategies, or REMS, and post-approval studies required by the FDA.
|
|
•
|
Phase 1. The drug or biological product is initially introduced into healthy human subjects or, in certain indications such as cancer, patients with the target
disease or condition and tested for safety, dosage tolerance, absorption, metabolism, distribution, excretion and, if possible, to gain an early indication of its effectiveness and to determine optimal dosage.
|
|
•
|
Phase 2. The drug or biological product is administered to a limited patient population to identify possible adverse effects and safety risks, to preliminarily
evaluate the efficacy of the product for specific targeted diseases and to determine dosage tolerance and optimal dosage.
|
|
•
|
Phase 3. The drug or biological product is administered to an expanded patient population, generally at geographically dispersed clinical trial sites, in
well-controlled clinical trials to generate enough data to statistically evaluate the efficacy and safety of the product for approval, to establish the overall risk-benefit profile of the product and to provide adequate information
for the labelling of the product.
|
|
•
|
restrictions on the marketing or manufacturing of the product, complete withdrawal of the product from the market or product recalls;
|
|
•
|
fines, warning letters or holds on post-approval clinical trials;
|
|
•
|
refusal of the FDA to approve pending NDAs or BLAs, or supplements to approved applications, or suspension or revocation of product license approvals;
|
|
•
|
product seizure or detention, or refusal to permit the import or export of products; or
|
|
•
|
injunctions or the imposition of civil or criminal penalties.
|
|
•
|
A special, non-deductible fee on any entity that manufactures or imports specified branded prescription drugs and biologic agents, apportioned among these entities according to their market share in
certain government healthcare programs, although this fee would not apply to sales of certain products approved exclusively for orphan indications;
|
|
•
|
Expansion of eligibility criteria for Medicaid programs by, among other things, allowing states to offer Medicaid coverage to certain individuals with income at or below 133% of the federal poverty
level, thereby potentially increasing a manufacturer’s Medicaid rebate liability;
|
|
•
|
Expansion of manufacturers’ rebate liability under the Medicaid Drug Rebate Program (“MDRP”) by (i) increasing the minimum rebate for both branded and generic drugs; (ii)
revising the definition of “average manufacturer price,” or AMP, which must be reported to the government for purposes of calculating Medicaid drug rebates on outpatient prescription drugs; and (iii) creating a new methodology by
which rebates owed by manufacturers under the MDRP are calculated for drugs that are inhaled, infused, instilled, implanted or injected;
|
|
•
|
Expansion of the types of entities eligible for the 340B drug discount program;
|
|
•
|
Provisions authorizing the creation of a new independent nonprofit organization called the Patient-Centered Outcomes Research Institute to oversee, identify
priorities in, and conduct comparative clinical effectiveness research, along with funding for such research; and
|
|
•
|
Establishment of the Center for Medicare and Medicaid Innovation within the Centers of Medicare and Medicaid Services (“CMS”) to test innovative payment and service
delivery models to lower Medicare and Medicaid spending, potentially including prescription drug spending.
|
|
•
|
The federal Anti-Kickback Statute (“AKS”), which is a criminal law that prohibits, among other things, persons and entities from knowingly and willfully soliciting, offering, receiving or providing
remuneration, directly or indirectly, in cash or in kind, to induce or reward either the referral of an individual for, or the purchase, order or recommendation of, any good or service, for which payment may be made, in whole or in
part, under a federal healthcare program such as Medicare or Medicaid. The AKS has been interpreted to apply to arrangements between pharmaceutical
manufacturers on the one hand and prescribers, pharmacies, purchasers, and formulary managers on the other, including, for example, consulting/speaking arrangements, discount and rebate offers, grants, charitable contributions, and
patient support offerings, among others. A person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation. Further, courts
have found that the AKS has been violated if any “one purpose” of an arrangement involving remuneration is to induce referrals of federal healthcare program business. Violations of the AKS can result in significant civil
monetary penalties and criminal fines, per each violation, additional civil penalties and treble damages under the federal Civil False Claims Act (“FCA”), as described in detail further below, as
well as imprisonment and mandatory exclusion from participation in government health care programs, meaning that federal healthcare programs would no longer reimburse (directly or indirectly) for
products or services furnished by the excluded entity or individuals. Although there are a number of statutory exceptions and regulatory safe harbors to the AKS that protect certain common industry activities from prosecution, these
exceptions and safe harbors are narrowly drawn. Arrangements that do not fully satisfy all elements of an available exception or safe harbor are evaluated based on the specific facts and circumstances and are typically subject to
increased scrutiny;
|
|
•
|
The FCA, which may be enforced through civil whistleblower or qui tam actions and imposes significant civil penalties, treble damages and potential exclusion from government health care programs against
individuals or entities for, among other things, knowingly presenting, or causing to be presented, to the federal government, claims for payment that are false or fraudulent or for making a false record or statement material to an
obligation to pay the federal government or for knowingly and improperly avoiding, decreasing or concealing an obligation to pay money to the federal government. Further, a violation of the AKS can serve as a basis for liability
under the FCA. Manufacturers can be held liable under the FCA even when they do not submit claims directly to government payors if they are deemed to “cause” the submission of false or fraudulent
claims. Pharmaceutical manufacturers have been investigated and/or subject to government enforcement actions asserting liability under the FCA for a variety of alleged activities, including alleged off-label promotion of drugs,
purportedly concealing price concessions in the pricing information submitted to the government for government price reporting purposes, and allegedly providing free product to customers with the expectation that the customers would
bill federal healthcare programs for the product. Violations of the FCA may result in significant civil fines and penalties for each false claim, currently ranging from $14,308 - $28,619 per false claim or statement for penalties
assessed after January 15, 2025, treble damages, and potential exclusion from participation in federal healthcare programs. There is also the federal Criminal False Claims Act, which is similar to the FCA and imposes criminal
liability on those that make or present a false, fictitious or fraudulent claim to the federal government;
|
|
•
|
The federal Civil Monetary Penalties Law, which authorizes the imposition of substantial civil monetary penalties against an entity that engages in activities including, among others (1) knowingly
presenting, or causing to be presented, a claim for services not provided as claimed or that is otherwise false or fraudulent in any way; (2) arranging for or contracting with an individual or entity that is excluded from
participation in federal health care programs to provide items or services reimbursable by a federal health care program; (3) violations of the AKS; and (4) failing to report and return a known overpayment;
|
|
•
|
The federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, which created additional federal criminal statutes that impose criminal liability for knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program, including
private third-party payors, or to obtain, by means of false or fraudulent pretenses, representations, or promises, any of the money or property owned by, or under the custody or control of, any healthcare benefit program; knowingly
and willfully embezzling or stealing from a healthcare benefit program; willfully preventing, obstructing, misleading, or delaying a criminal investigation of a healthcare offense; and knowingly and willfully falsifying, concealing
or covering up a material fact or making any materially false statements in connection with the delivery of or payment for healthcare benefits, items, or services. Similar to the federal Anti-Kickback Statute, a person or entity
need not have actual knowledge of the statute or specific intent to violate it in order to have committed a violation;
|
|
•
|
The federal Physician Payments Sunshine Act (“Sunshine Act”), implemented as the Open Payments Program, which requires certain manufacturers of drugs, devices, biologics and medical supplies for which
payment is available under Medicare, Medicaid, or the Children’s Health Insurance Program, among others, to track and report annually to CMS, within HHS, information related to payments and other “transfers of value” made by that
entity to US-licensed physicians (defined to include doctors, dentists, optometrists, podiatrists and chiropractors), physician assistants, nurse practitioners, clinical nurse specialists, anesthesiologist assistants, certified
registered nurse anesthetists, certified nurse midwives, and teaching hospitals. The Sunshine Act also requires certain manufacturers, among others, to track and report ownership and investment interests held by U.S.-licensed
physicians and their immediate family members. Failure to timely, accurately, and completely submit the required information for all payments, transfers of value and ownership or investment interests may result in civil monetary
penalties;
|
|
•
|
The federal Health Insurance Portability and Accountability Act of 1996 (“HIPAA”) which imposes criminal and civil liability for, among other things,
executing or attempting to execute a scheme to defraud any healthcare benefit program, including any third-party payors, knowingly and willfully embezzling or stealing from a healthcare benefit program, willfully obstructing a
criminal investigation of a healthcare offense, and knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statements or representations, or making
false statements relating to healthcare benefits, items or services. Similar to the AKS, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it to have committed a violation;
|
|
•
|
HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act of 2009, which mandates, among other things, the adoption of uniform standards for the electronic exchange of
information in common health care transactions as well as standards relating to the privacy and security of individually identifiable health information. These standards require the adoption of administrative, physical and technical
safeguards to protect such information. In addition, many states have enacted comparable laws addressing the privacy and security of health information, some of which are more stringent than HIPAA. Failure to comply with these laws
can result in the imposition of significant civil and criminal penalties;
|
|
•
|
Analogous state and foreign laws and regulations, such as state anti-kickback and false claims laws, may apply to sales or marketing arrangements and claims involving healthcare items or services
reimbursed by non-governmental third-party payors, including private insurers, and may be broader in scope than their federal equivalents; and
|
|
•
|
State laws that require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government;
require the reporting of certain pricing information, including information pertaining to and justifying price increases; prohibit prescription drug price gouging; or impose payment caps on certain pharmaceutical products deemed by
the state to be “high cost” in addition to requiring drug manufacturers to report information related to payments to physicians and other healthcare providers or marketing expenditures.
|
|
•
|
Additionally, we expect certain of our products, if and when approved, may be eligible for coverage under Medicare, the federal health care program that provides health care benefits to the aged and
disabled. Specifically, we expect our products would be primarily reimbursed under Medicare Part D, which provides an outpatient prescription drug benefit for Medicare beneficiaries. Medicare Part D is implemented through private
insurance plans under contractual arrangements between the plans and the federal government. Similar to pharmaceutical coverage through private health insurance, Part D plans develop formularies, impose utilization controls (such as
prior authorization, step therapy, and quantity limits), and negotiate discounts from drug manufacturers. Because of this, the list of prescription drugs covered by Part D plans varies by plan. However, with limited exceptions,
individual plans are required by statute to cover certain therapeutic categories and classes of drugs or biologics and to have at least two drugs in each unique therapeutic category or class. Our products may also be covered and
reimbursed under other government programs, including those discussed below:
|
|
•
|
We expect to be required to participate in the MDRP in order for federal payment to be available for our products under Medicaid. Medicaid is a government health insurance
program for eligible low-income adults, children, families, pregnant women, and people with certain disabilities and it is jointly funded by the federal and state governments. The MDRP requires pharmaceutical manufacturers
to enter into and have in effect a national rebate agreement with the Secretary of the HHS as a condition for states to receive federal matching funds for the manufacturer’s outpatient drugs furnished to Medicaid patients. Under the
MDRP, manufacturers must pay a rebate to each state Medicaid program for quantities of products utilized on an outpatient basis (with some exceptions) that are dispensed to Medicaid beneficiaries and paid for by a state Medicaid
program. MDRP rebates are calculated using a statutory formula, state-reported utilization data, and pricing data that are calculated and reported by manufacturers on a monthly and quarterly basis to CMS. These data include the AMP
and, in the case of single source and innovator multiple source products, the best price for each drug.
|
|
•
|
Under federal law, we further expect to be required to participate in the 340B drug pricing program, which 340B drug pricing program requires participating
manufacturers to agree to charge statutorily-defined covered entities no more than the 340B “ceiling price” for the manufacturer’s covered outpatient drugs. These 340B covered entities include health care organizations that have
certain federal designations or receive funding from specific federal programs, including Federally Qualified Health Centers, Ryan White HIV/AIDS Program grantees, and certain types of hospitals and specialized clinics, as well as
certain hospitals that serve a disproportionate share of low-income patients. The ACA expanded the 340B program to also include certain children’s hospitals, certain free-standing cancer hospitals, critical access hospitals, certain
rural referral centers and certain sole community hospitals, each as defined by ACA. The 340B ceiling price is calculated using a statutory formula, which is based on the AMP and rebate amount for the covered outpatient drug as
calculated under the MDRP, and in general, products subject to the MDRP are also subject to the 340B ceiling price calculation and discount requirement. Any changes to the definition of Medicaid AMP and the Medicaid rebate amount also
could affect our 340B ceiling price calculation for our products and could negatively impact our results of operations. In addition, after multiple delays, the final rule implementing civil monetary
penalties against manufacturers for instances of overcharging 340B covered entities became effective on January 1, 2019. Accordingly, if we have an approved product, we could be subject to such penalties if the government were to
find that we knowingly and intentionally overcharged a 340B covered entity.
|
|
•
|
Additionally, for a company to be eligible to have its products paid for with federal funds under the MDRP and Medicare Part B programs, as well as to be purchased by certain federal agencies and
grantees, it also must participate in the Department of Veterans Affairs (“VA”) Federal Supply Schedule (“FSS”) pricing program. To participate, manufacturers are required to enter into an FSS contract and other agreements with the VA
for any products which may qualify as “covered drugs.” Under these agreements, manufacturers must make such products available to the “Big Four” federal agencies-the VA, the Department of Defense
(“DoD”), the Public Health Service (including the Indian Health Service), and the Coast Guard-at pricing that is capped pursuant to a statutory federal ceiling price (“FCP”), formula set forth in Section 603 of the Veterans Health
Care Act of 1992 (“VHCA”). The FCP is based on a weighted average non-federal average manufacturer price (“Non-FAMP”), which manufacturers are required to report on a quarterly and annual basis to the VA.
|
|
•
|
Any failure to comply with price reporting and rebate payment obligations under federal healthcare programs could negatively impact our financial results. Civil monetary penalties can be applied if we
are found to have knowingly submitted any false price information to the government, if we are found to have made a misrepresentation in the reporting of our average sales price, or if we fail to submit the required price data on a
timely basis. Such conduct also could provide a basis for other potential liability under other federal laws such as the False Claims Act.
|
| ITEM 1A. |
RISK FACTORS
|
|
•
|
the inability to successfully combine our assets in a manner that permits us to expand our product pipeline or achieve the anticipated benefits from the Opus Acquisition, which would result in the
anticipated benefits of the Opus Acquisition not being realized partly or wholly in the time frame currently anticipated or at all;
|
|
•
|
the creation of uniform standards, controls, procedures, policies and information systems;
|
|
•
|
the addition of new personnel, including new management, which may be difficult to smoothly integrate; and
|
|
•
|
potential unknown liabilities and unforeseen increased expenses, delays or regulatory conditions associated with the Opus Acquisition.
|
|
•
|
regulatory authorities may suspend or withdraw approvals of such product candidate;
|
|
•
|
regulatory authorities may require additional warnings or limitations of use in product labeling;
|
|
•
|
we may be required to change the way a product candidate is administered or conduct additional clinical trials;
|
|
•
|
we could be sued and held liable for harm caused by our products to patients; and
|
|
•
|
our reputation may suffer.
|
|
•
|
delays in reaching a consensus with regulatory authorities on trial design;
|
|
•
|
delays in reaching agreement on acceptable terms with prospective CROs and clinical trial sites;
|
|
•
|
delays in opening clinical trial sites or obtaining required institutional review board or independent Ethics Committee approval at each clinical trial site;
|
|
•
|
delays in recruiting and enrolling suitable subjects to participate in our clinical trials, due to factors such as the size of the trial or subject population, process for identifying subjects, design
or expansion of protocols, eligibility and exclusive criteria, perceived risks and benefits of the relevant product candidate or gene therapy generally, availability of competing therapies and trials, severity of the disease under
investigation, need and length of time required to discontinue other potential therapies, availability of genetic testing, availability and proximity of trial sites for prospective subjects, ability to obtain subject consent and
referral practices of physicians;
|
|
•
|
imposition of a clinical hold by regulatory authorities, including as a result of a serious adverse event or after an inspection of our clinical trial operations or trial sites;
|
|
•
|
failure by us, any CROs we engage or any other third parties to adhere to clinical trial requirements;
|
|
•
|
failure to perform in accordance with GCP, or applicable regulatory guidelines in the European Union and other countries;
|
|
•
|
delays in the testing, validation, manufacturing and delivery of our product candidates to the clinical sites, including delays by third parties with whom we have contracted to perform;
|
|
•
|
delays in having subjects complete participation in a trial or return for post-treatment follow-up;
|
|
•
|
clinical trial sites or subjects dropping out of a trial;
|
|
•
|
selection of clinical endpoints that require prolonged periods of clinical observation or analysis of the resulting data;
|
|
•
|
occurrence of serious adverse events associated with the product candidate that are viewed to outweigh its potential benefits;
|
|
•
|
occurrence of serious adverse events in trials of the same class of agents conducted by other sponsors; or
|
|
•
|
changes in regulatory requirements and guidance that require amending or submitting new clinical protocols.
|
|
•
|
be delayed in obtaining marketing approval for our product candidates, if at all;
|
|
•
|
obtain approval for indications or patient populations that are not as broad as intended or desired;
|
|
•
|
obtain approval with labeling that includes significant use or distribution restrictions or safety warnings;
|
|
•
|
be subject to changes in the way the product is administered;
|
|
•
|
be required to perform additional clinical trials to support approval or be subject to additional post-marketing testing or other requirements;
|
|
•
|
have regulatory authorities withdraw, vary or suspend their approval of the product or impose restrictions on its distribution in the form of a modified risk evaluation and mitigation;
|
|
•
|
be subject to the addition of labeling statements, such as warnings or contraindications;
|
|
•
|
be sued; or
|
|
•
|
experience damage to our reputation.
|
|
•
|
perceived risks and benefits of gene therapy-based approaches or our product candidate under study;
|
|
•
|
availability of genetic testing for potential subjects;
|
|
•
|
availability and efficacy of medications already approved for the disease under investigation;
|
|
•
|
eligibility criteria and visit schedule for the trial in question;
|
|
•
|
competition for eligible patients with other companies conducting clinical trials for product candidates seeking to treat the same indication or patient population;
|
|
•
|
our payments for conducting clinical trials;
|
|
•
|
perceived risks and benefits of the product candidate under study;
|
|
•
|
efforts to facilitate timely enrollment in clinical trials;
|
|
•
|
patient referral practices of physicians;
|
|
•
|
the ability to monitor patients adequately during and after treatment; and
|
|
•
|
proximity and availability of clinical trial sites for prospective patients.
|
|
•
|
delays in, termination, or numerous unforeseen events during, or as a result of, manufacturing or clinical trials;
|
|
•
|
obtaining unfavorable results from nonclinical and clinical studies for our product candidates;
|
|
•
|
the cost of clinical trials being greater than anticipated;
|
|
•
|
the willingness of patients or medical investigators to follow our clinical trial protocols and the number of patients willing to participate;
|
|
•
|
delays in applying for and receiving marketing and NDA approvals from applicable regulatory authorities for our product candidates;
|
|
•
|
other government or regulatory delays and changes in regulatory requirements, policy and guidelines may require us to perform additional clinical trials or use substantial additional resources to obtain
regulatory approval;
|
|
•
|
issues with making arrangements with third-party manufacturers for commercial quantities of RYZUMVI and our product candidates and receiving regulatory approval of our manufacturing processes and our
third-party manufacturers’ facilities from applicable regulatory authorities;
|
|
•
|
establishing sales, marketing, and distribution capabilities and launching commercial sales of RYZUMVI and our product candidates, if and when approved, whether alone or in collaboration with others;
|
|
•
|
acceptance of RYZUMVI and our product candidates by patients, the medical community, and third-party payors;
|
|
•
|
effectively competing with other therapies, including the existing standard-of-care;
|
|
•
|
maintaining a continued acceptable safety profile of RYZUMVI and our product candidates following approval;
|
|
•
|
obtaining and maintaining coverage and adequate reimbursement from third-party payors;
|
|
•
|
obtaining and maintaining patent and trade secret protection and regulatory exclusivity;
|
|
•
|
protecting our rights in our intellectual property portfolio related to RYZUMVI and our product candidates; and
|
|
•
|
our ability to fulfill requests for additional data regarding our product candidates.
|
|
•
|
Viatris may not be able to manufacture our products in a timely or cost-effective manner;
|
|
•
|
Viatris may not timely perform its obligations under the Viatris License Agreement;
|
|
•
|
Viatris may fail to effectively commercialize our products;
|
|
•
|
Viatris may not be able to sublicense RYZUMVI or PS to one or more suitable parties outside the United States; or
|
|
•
|
contractual disputes or other disagreements between us and Viatris, including those regarding the development, manufacture, sub licensure and commercialization of our products, interpretation of the
License Agreement, and ownership of proprietary rights. Viatris may select a new development partner for RYZUMVI and PS in the U.S. upon 90 days’ notice to the Company.
|
|
•
|
we may discover that they are less effective, or identify undesirable side effects caused by our product candidates:
|
|
•
|
regulatory authorities may withdraw their approval of the product;
|
|
•
|
we may be required to recall the product, change the way this product is administered, conduct additional clinical trials, or change the labeling or distribution of the product (including REMS);
|
|
•
|
additional restrictions may be imposed on the marketing of, or the manufacturing processes for, the product;
|
|
•
|
we may be subject to fines, injunctions, or the imposition of civil or criminal penalties;
|
|
•
|
we could be sued and held liable for harm caused to patients;
|
|
•
|
the product may be rendered less competitive and sales may decrease; or
|
|
•
|
our reputation may suffer generally among both clinicians and patients.
|
|
•
|
the inability to recruit and retain adequate numbers of effective sales and marketing personnel or enter into distribution agreements with third parties;
|
|
•
|
the inability of sales personnel to obtain access to physicians or educate an adequate number of physicians as to the benefits of our products;
|
|
•
|
the lack of complementary products to be offered by sales personnel, which may put us at a competitive disadvantage relative to companies with more extensive product lines;
|
|
•
|
unforeseen costs and expenses associated with creating an independent sales and marketing organization; and
|
|
•
|
the inability to obtain sufficient coverage and reimbursement from third-party payors and governmental agencies.
|
|
•
|
efficacy and potential advantages compared to alternative treatments;
|
|
•
|
the ability to offer our product for sale at competitive prices;
|
|
•
|
the willingness of the target patient population to try new therapies and of physicians to prescribe these therapies;
|
|
•
|
any restrictions on the use of our product together with other medications;
|
|
•
|
interactions of our product with other medicines patients are taking;
|
|
•
|
inability of certain types of patients to take our product;
|
|
•
|
demonstrated ability to treat patients and, if required by any applicable regulatory authority in connection with the approval for target indications as compared with other available therapies;
|
|
•
|
the relative convenience and ease of administration as compared with other treatments available for approved indications;
|
|
•
|
the prevalence and severity of any adverse side effects;
|
|
•
|
limitations or warnings contained in the labeling approved by the FDA;
|
|
•
|
availability of alternative treatments already approved or expected to be commercially launched in the near future;
|
|
•
|
the effectiveness of our or our partners’ sales and marketing strategies;
|
|
•
|
our or our partners’ ability to increase awareness through marketing efforts;
|
|
•
|
guidelines and recommendations of organizations involved in research, treatment and prevention of various diseases that may advocate for alternative therapies;
|
|
•
|
our or our partners’ ability to obtain sufficient third-party coverage and adequate reimbursement;
|
|
•
|
the willingness of patients to pay out-of-pocket in the absence of third-party coverage; and
|
|
•
|
physicians or patients may be reluctant to switch from existing therapies even if potentially more effective, safe or convenient.
|
|
•
|
ability of physicians to identify patients with rare genetic diseases (IRDs).
|
|
•
|
limited genetic testing conducted on potential patients.
|
|
•
|
decreased demand for any product candidate that we are developing;
|
|
•
|
injury to our reputation and significant negative media attention;
|
|
•
|
withdrawal of clinical trial participants;
|
|
•
|
increased FDA warnings on product labels;
|
|
•
|
significant costs to defend the related litigation;
|
|
•
|
substantial monetary awards to trial participants or patients;
|
|
•
|
distraction of management’s attention from our primary business;
|
|
•
|
loss of revenue;
|
|
•
|
the inability to commercialize any product candidate that we may develop;
|
|
•
|
the initiation of investigations by regulators; and
|
|
•
|
the inability to take advantage of limitations on product liability lawsuits that apply to generic drug products, which could increase our exposure to liability for products deemed to be dangerous or
defective.
|
|
•
|
the successful launch and widespread commercialization of our gene therapy candidates and other product candidates;
|
|
•
|
obtain approvals on late-stage drugs in development and the receipt of associated financial payments from our partner;
|
|
•
|
obtain favorable results from and complete the nonclinical and clinical development of our product candidates for their planned indications, including successful completion of additional clinical trials
for these indications;
|
|
•
|
submit applications to regulatory authorities for both product candidates and receive timely marketing approvals in the United States and foreign countries;
|
|
•
|
establish and maintain commercially viable supply and manufacturing relationships with third parties that can provide adequate, in both amount and quality, products and services to support clinical
development and meet the market demand for our product candidates that we develop, if approved;
|
|
•
|
establish sales and marketing capabilities to effectively market and sell our product candidates in the United States or other markets, either alone or with a pharmaceutical partner;
|
|
•
|
address any competing products and technological and market developments;
|
|
•
|
obtain coverage and adequate reimbursement for customers and patients from government and third-party payors for our product candidates that we develop; and
|
|
•
|
achieve market acceptance of our product candidates.
|
|
•
|
Delayed access to deposits or other financial assets or the uninsured loss of deposits or other financial assets;
|
|
•
|
Loss of access to revolving existing credit facilities or other working capital sources and/or the inability to refund, roll over or extend the maturity of, or enter into new credit facilities or other
working capital resources;
|
|
•
|
Potential or actual breach of contractual obligations that require us to maintain letters or credit or other credit support arrangements; or
|
|
•
|
Termination of cash management arrangements and/or delays in accessing or actual loss of funds subject to cash management arrangements.
|
|
•
|
the scope, size, rate of progress, results, and costs of researching and developing our product candidates, and initiating and completing our nonclinical studies and clinical trials;
|
|
•
|
the cost, timing and outcome of our efforts to obtain further marketing approval for our product candidates in the United States and other countries, including to fund the preparation and filing of NDAs
with the FDA for our product candidates and to satisfy related FDA requirements and regulatory requirements in other countries;
|
|
•
|
the number and characteristics of any additional product candidates we develop or acquire, if any;
|
|
•
|
our ability to establish and maintain collaborations on favorable terms, if at all;
|
|
•
|
the amount of revenue, if any, from commercial sales, should our product candidates receive marketing approval;
|
|
•
|
the costs associated with commercializing our product candidates, if we receive marketing approval, including the cost and timing of developing sales and marketing capabilities or entering into
strategic collaborations to market and sell our product candidates;
|
|
•
|
the ability to secure grant funding from government and nongovernment foundations;
|
|
•
|
the cost of manufacturing our product candidates or products we successfully commercialize; and
|
|
•
|
the costs associated with general corporate activities, such as the cost of filing, prosecuting and enforcing patent claims and making regulatory filings.
|
|
•
|
litigation involving patients taking our drugs;
|
|
•
|
restrictions on such drugs, manufacturers, or manufacturing processes;
|
|
•
|
restrictions on the labeling or marketing of a drug;
|
|
•
|
restrictions on drug distribution or use;
|
|
•
|
requirements to conduct post-marketing studies or clinical trials;
|
|
•
|
warning letters or untitled letters;
|
|
•
|
withdrawal of the drugs from the market;
|
|
•
|
refusal to approve pending applications or supplements to approved applications that we submit;
|
|
•
|
product recall or public notification or medical product safety alerts to healthcare professionals;
|
|
•
|
fines, restitution, or disgorgement of profits or revenues;
|
|
•
|
suspension or withdrawal of marketing approvals;
|
|
•
|
damage to relationships with any potential collaborators;
|
|
•
|
unfavorable press coverage and damage to our reputation;
|
|
•
|
refusal to permit the import or export of drugs;
|
|
•
|
product seizure; or
|
|
•
|
injunctions or the imposition of civil or criminal penalties.
|
|
•
|
comply with the regulations of the FDA and applicable non-U.S. regulators;
|
|
•
|
provide accurate information to the FDA and applicable non-U.S. regulators;
|
|
•
|
comply with healthcare fraud and abuse laws and regulations in the United States and abroad;
|
|
•
|
report financial information or data accurately; or
|
|
•
|
disclose unauthorized activities to us.
|
|
•
|
fail to comply with contractual obligations;
|
|
•
|
experience regulatory compliance issues;
|
|
•
|
undergo changes in ownership or management;
|
|
•
|
undergo changes in priorities or become financially distressed; or
|
|
•
|
form relationships with other entities, some of which may be our competitors.
|
|
•
|
collaborators have significant discretion in determining the efforts and resources that they will apply to these collaborations;
|
|
•
|
collaborators may not perform their obligations as expected;
|
|
•
|
collaborators may not pursue development and commercialization or may elect not to continue or renew development or commercialization programs based on clinical trial results, changes in the
collaborator’s strategic focus or available funding, or external factors such as an acquisition that diverts resources or creates competing priorities;
|
|
•
|
collaborators may delay clinical trials, provide insufficient funding for a clinical trial program, stop a clinical trial or abandon a product candidate, repeat or conduct new clinical trials, or
require a new formulation of a product candidate for clinical testing;
|
|
•
|
collaborators could independently develop, or develop with third parties, products that compete directly or indirectly with our product candidate if the collaborators believe that competitive products
are more likely to be successfully developed or can be commercialized under terms that are more attractive than ours;
|
|
•
|
a collaborator with marketing and distribution rights to one or more product candidates may not commit sufficient resources to the marketing or distribution of any such product candidate;
|
|
•
|
collaborators may not properly maintain or defend our intellectual property rights or may use our proprietary information in such a way as to invite litigation that could jeopardize or invalidate our
proprietary information or expose us to litigation;
|
|
•
|
collaborators may infringe the intellectual property rights of third parties, which may expose us to litigation and potential liability;
|
|
•
|
disputes may arise between us and collaborators that result in the delay or termination of research, development, or commercialization of our product candidates, or in litigation or arbitration that
diverts management attention and resources;
|
|
•
|
we may lose certain valuable rights under circumstances identified in our collaborations, including if we undergo a change of control;
|
|
•
|
collaborations may be terminated and such terminations may create a need for additional capital to pursue further development or commercialization of the applicable product candidates;
|
|
•
|
collaborators may learn about our discoveries and use this knowledge to compete with us in the future;
|
|
•
|
the results of collaborators’ nonclinical or clinical studies could harm or impair other development programs;
|
|
•
|
there may be conflicts between different collaborators that could negatively affect those collaborations and potentially others;
|
|
•
|
the number and nature of our collaborations could adversely affect our attractiveness to potential future collaborators or acquirers;
|
|
•
|
collaboration agreements may not lead to development or commercialization of our product candidate in the most efficient manner or at all. If a present or future collaborator of us were to be involved
in a business combination, the continued pursuit and emphasis on our product development or commercialization program under such collaboration could be delayed, diminished, or terminated;
|
|
•
|
collaborators may be unable to obtain the necessary marketing approvals; and
|
|
•
|
collaborators may determine, as a part of product life-cycle management, that changes to a product are necessary or required, including regarding such
product’s formulation, container closure system, packaging, or other characteristics, which could affect the development or commercialization of the applicable product candidate.
|
|
•
|
increased operating expenses and cash requirements;
|
|
•
|
the assumption of indebtedness or contingent liabilities;
|
|
•
|
the issuance of our equity securities which would result in dilution to our stockholders;
|
|
•
|
assimilation of operations, intellectual property, products and product candidates of an acquired company, including difficulties associated with integrating new personnel;
|
|
•
|
the diversion of management’s attention from our existing product candidates and initiatives in pursuing such an acquisition or strategic partnership;
|
|
•
|
retention of key employees, the loss of key personnel, and uncertainties in our ability to maintain key business relationships;
|
|
•
|
risks and uncertainties associated with the other party to such a transaction, including the prospects of that party and their existing products or product candidates and regulatory approvals; and
|
|
•
|
our inability to generate revenue from acquired intellectual property, technology and/or products sufficient to meet our objectives or even to offset the associated transaction and maintenance costs.
|
|
•
|
any of our patents, or any of our pending patent applications, if issued, will include claims having a scope sufficient to protect our product candidates;
|
|
•
|
any of our pending patent applications will result in issued patents;
|
|
•
|
we will be able to successfully commercialize our product candidates, if approved, before our relevant patents expire;
|
|
•
|
we were the first to make the inventions covered by each of our patents and pending patent applications;
|
|
•
|
we were the first to file patent applications for these inventions;
|
|
•
|
others will not develop similar or alternative technologies that do not infringe our patents;
|
|
•
|
any of our patents will be valid and enforceable;
|
|
•
|
any patents issued to us will provide a basis for an exclusive market for our commercially viable products, will provide us with any competitive advantages or will not be challenged by third parties;
|
|
•
|
we will develop additional proprietary technologies or product candidates that are separately patentable; or
|
|
•
|
our commercial activities or products will not infringe upon the patents of others.
|
|
•
|
the scope of rights granted under the license agreement and other interpretation-related issues;
|
|
•
|
the extent to which our product candidates, technology and processes infringe on intellectual property of the licensor that is not subject to the licensing agreement;
|
|
•
|
the sublicensing of patent and other rights under our collaborative development relationships;
|
|
•
|
our diligence obligations under the license agreement and what activities satisfy those diligence obligations;
|
|
•
|
the inventorship and ownership of inventions and know-how resulting from the joint creation or use of intellectual property; and
|
|
•
|
the priority of invention of patented technology.
|
|
•
|
compliance with differing or unexpected regulatory requirements for our product candidates;
|
|
•
|
different medical practices and customs affecting acceptance of our product candidates, if approved, or any other approved product in the marketplace;
|
|
•
|
language barriers;
|
|
•
|
the interpretation of contractual provisions governed by foreign law in the event of a contract dispute;
|
|
•
|
difficulties in staffing and managing foreign operations, and an inability to control commercial or other activities where it is relying on third parties;
|
|
•
|
workforce uncertainty in countries where labor unrest is more common than in the United States;
|
|
•
|
potential liability under the Foreign Corrupt Practice Act of 1977 or comparable foreign regulations;
|
|
•
|
production shortages resulting from any events affecting raw material supply or manufacturing capability abroad;
|
|
•
|
foreign government taxes, regulations, and permit requirements;
|
|
•
|
U.S. and foreign government tariffs, trade restrictions, price and exchange controls, and other regulatory requirements, particularly changes that may occur as a result of the recent U.S. presidential
election;
|
|
•
|
economic weakness, including inflation, natural disasters, war, events of terrorism, or political instability in particular foreign countries;
|
|
•
|
fluctuations in currency exchange rates, which could result in increased operating expenses and reduced revenues;
|
|
•
|
compliance with tax, employment, immigration, and labor laws, regulations, and restrictions for employees living or traveling abroad;
|
|
•
|
changes in diplomatic and trade relationships; and
|
|
•
|
challenges in enforcing our contractual and intellectual property rights, especially in those foreign countries that do not respect and protect intellectual property rights to the same extent as the
United States.
|
|
•
|
the announcement of new products or product enhancements by us or our competitors;
|
|
•
|
changes in our relationships with our licensors or other strategic partners;
|
|
•
|
developments concerning intellectual property rights and regulatory approvals;
|
|
•
|
variations in ours and our competitors’ results of operations;
|
|
•
|
substantial sales of shares of our common stock due to the release of lock-up agreements;
|
|
•
|
the announcement of clinical trial results;
|
|
•
|
the announcement of potentially dilutive financings;
|
|
•
|
changes in earnings estimates or recommendations by securities analysts;
|
|
•
|
changes in the structure of healthcare payment systems;
|
|
•
|
developments and market conditions in the pharmaceutical and biotechnology industries;
|
|
•
|
any acquisitions or dispositions of products, product candidates or business; and
|
|
•
|
the results of clinical trials of our gene therapy products, PS, or any other product candidate that we may develop.
|
| ITEM 1B. |
UNRESOLVED STAFF COMMENTS
|
| ITEM 3. |
LEGAL PROCEEDINGS
|
| ITEM 4. |
MINE SAFETY DISCLOSURES
|
| ITEM 5. |
MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
|
| ITEM 6. |
[RESERVED]
|
| ITEM 7. |
MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
|
|
|
• |
continue clinical trials for LCA5, BEST1, PS and for any other product candidate in our future pipeline;
|
|
|
• |
continue nonclinical studies for our pipeline of gene therapies;
|
|
|
• |
develop additional product candidates that we identify, in-license or acquire;
|
|
|
• |
seek regulatory approvals for any product candidates that successfully complete clinical trials;
|
|
|
• |
contract to manufacture our product candidates;
|
|
|
• |
maintain, expand and protect our intellectual property portfolio;
|
|
|
• |
hire additional staff, including clinical, scientific, operational and financial personnel, to execute our business plan;
|
|
|
• |
add operational, financial and management information systems and personnel to support our product development and potential future commercialization efforts;
|
|
|
• |
continue to operate as a public company; and
|
|
|
• |
establish on our own or with partners, a sales, marketing and distribution infrastructure to commercialize any products for which we may obtain regulatory approval.
|
|
For the Year Ended
December 31,
|
||||||||||||
|
2024
|
2023
|
Change
|
||||||||||
|
License and collaborations revenue
|
$
|
10,992
|
$
|
19,049
|
$
|
(8,057
|
)
|
|||||
|
Operating expenses:
|
||||||||||||
|
General and administrative
|
18,215
|
11,959
|
6,256
|
|||||||||
|
Research and development
|
26,851
|
17,653
|
9,198
|
|||||||||
|
Acquired in-process research and development expenses
|
28,000
|
—
|
28,000
|
|||||||||
|
Total operating expenses
|
73,066
|
29,612
|
43,454
|
|||||||||
|
Loss from operations
|
(62,074
|
)
|
(10,563
|
)
|
(51,511
|
)
|
||||||
|
Financing costs
|
—
|
(1,328
|
)
|
1,328
|
||||||||
|
Fair value change in derivative liabilities
|
72
|
80
|
(8
|
)
|
||||||||
|
Other income, net
|
4,470
|
1,837
|
2.633
|
|||||||||
|
Loss before income taxes
|
(57,532
|
)
|
(9,974
|
)
|
(47,558
|
)
|
||||||
|
Provision for income taxes
|
—
|
(12
|
)
|
12
|
||||||||
|
Net loss
|
$
|
(57,532
|
)
|
$
|
(9,986
|
)
|
$
|
(47,546
|
)
|
|||
|
For the Year Ended
|
||||||||||||
|
December 31,
|
||||||||||||
|
2024
|
2023
|
Change
|
||||||||||
|
External costs:
|
||||||||||||
|
Phentolamine Ophthalmic Solution 0.75% (“PS”)
|
$
|
9,680
|
$
|
9,983
|
$
|
(303
|
)
|
|||||
|
APX 3330
|
11,466
|
4,818
|
6,648
|
|||||||||
|
IRD programs
|
902
|
—
|
902
|
|||||||||
|
Unallocated
|
414
|
678
|
(264
|
)
|
||||||||
|
Total external cost
|
22,462
|
15,479
|
6,983
|
|||||||||
|
Internal costs:
|
||||||||||||
|
Employee related expenses
|
4,216
|
2,148
|
2,068
|
|||||||||
|
Facilities, supplies and other
|
173
|
26
|
147
|
|||||||||
|
Total internal costs
|
4,389
|
2,174
|
2,215
|
|||||||||
|
Total research and development expenses
|
$
|
26,851
|
$
|
17,653
|
$
|
9,198
|
||||||
|
For the Year Ended
December 31,
|
||||||||
|
2024
|
2023
|
|||||||
|
Net cash used in by operating activities
|
$
|
(25,576
|
)
|
$
|
(1,112
|
)
|
||
|
Net cash provided by investing activities
|
1,210
|
—
|
||||||
|
Net cash provided by financing activities
|
4,186
|
8,979
|
||||||
|
Net (decrease) increase in cash and cash equivalents
|
$
|
(20,180
|
)
|
$
|
7,867
|
|||
| ITEM 7A. |
QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
|
| ITEM 8. |
FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
|
| ITEM 9. |
CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
|
| Item 9A |
CONTROLS AND PROCEDURES
|
| ITEM 9B. |
OTHER INFORMATION
|
| ITEM 9C. |
DISCLOSURE REGARDING FOREIGN JURISDICTIONS THAT PREVENT INSPECTIONS
|
| ITEM 10. |
DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
|
|
ITEM 12.
|
SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
|
|
|
(a) |
Financial Statements: The financial statements filed as part of this report are listed in Part II, Item 8.
|
|
|
(b) |
Financial Statement Schedules: The schedules are either not applicable or the required information is presented in the financial statements or notes thereto.
|
|
|
(c) |
Exhibits: The following exhibits are incorporated by reference or filed as part of this Annual Report on Form 10-K:
|
|
EXHIBIT
NUMBER
|
DESCRIPTION OF DOCUMENT
|
|
Agreement and Plan of Merger, dated as of October 22, 2024, by and among the Company, Former Opus, Orange Merger Sub I, Inc., and Orange Merger
Sub II, LLC (incorporated by reference to Exhibit 2.1 to Registrant’s Current Report on Form 8-K, filed on October 22, 2024).
|
|
|
Restated Certificate of Incorporation of Ocuphire Pharma, Inc., dated as of June 12, 2024 (incorporated by reference to Exhibit 3.1 to the
Registrant’s Quarterly Report on Form 10-Q, filed on August 13, 2024).
|
|
|
Certificate of Amendment to the Restated Certificate of Incorporation of the Company, effective as of October 23, 2024 (incorporated by
reference to Exhibit 3.2 to Registrant’s Current Report on Form 8-K, filed on October 22, 2024).
|
|
|
Certificate of Designation of Series A Non-Voting Convertible Preferred Stock, effective as of October 22, 2024 (incorporated by reference to
Exhibit 3.1 to Registrant’s Current Report on Form 8-K, filed on October 22, 2024).
|
|
|
Amended and Restated Bylaws, dated as of March 19, 2025 (incorporated by reference to Exhibit 3.1 to Registrant’s Current Report on Form 8-K,
filed on March 20, 2025).
|
|
|
Form of Common Stock Purchase Warrant (incorporated by reference to Exhibit 4.1 to the Registrant’s Current Report on Form 8-K, filed on October
13, 2017).
|
|
|
Form of Common Stock Purchase Warrant (incorporated by reference to Exhibit 4.1 to the Registrant’s Current Report on Form 8-K, filed on October
19, 2018).
|
|
|
Form of Common Stock Purchase Warrant (incorporated by reference to Exhibit 4.1 to the Registrant’s Current Report on Form 8-K, filed on January
25, 2019).
|
|
|
Form of Series A/B Warrants (incorporated by reference to Exhibit 4.1 to the Registrant’s Current Report on Form 8-K, filed on July 1, 2020).
|
|
|
Form of Warrant to purchase shares of common stock (incorporated by reference to Exhibit 4.1 to the Registrant’s Current Report on Form 8-K/A,
filed on June 7, 2021).
|
|
|
Form of Indenture (incorporated by reference to Exhibit 4.13 to the Registrant’s Registration Statement on Form S-3, filed on January 10, 2024).
|
|
|
Form of Common Stock Warrant Agreement and Warrant Certificate (incorporated by reference to Exhibit 4.15 to the Registrant’s Registration
Statement on Form S-3, filed on January 10, 2024).
|
|
|
Form of Preferred Stock Warrant Agreement and Warrant Certificate (incorporated by reference to Exhibit 4.16 to the Registrant’s Registration
Statement on Form S-3, filed on January 10, 2024).
|
|
|
Form of Debt Securities Warrant Agreement and Warrant Certificate (incorporated by reference to Exhibit 4.17 to the Registrant’s Registration
Statement on Form S-3, filed on January 10, 2024).
|
|
|
|
|
|
Form of Warrant (incorporated by reference to Exhibit 4.1 to the Registrant’s Current Report on Form 8-K, filed on March 24, 2025).
|
|
|
|
|
|
Form of Pre-Funded Warrant (incorporated by reference to Exhibit 4.2 to the Registrants’ Current Report on Form 8-K, filed on March 24, 2025).
|
|
|
Description of Securities.
|
|
|
Amended and Restated Employment Agreement by and among the Company and Bernhard Hoffmann, effective as of November 5, 2020 (incorporated by
reference to Exhibit 10.29 to the Registrant’s Registration Statement on Form S-4, filed on September 30, 2020).
|
|
First Amendment to the Amended and Restated Employment Agreement by and among the Company and Bernhard Hoffmann, effective as of March 26, 2023
(incorporated by reference to Exhibit 10.2.1 to the Registrant’s Annual Report on Form 10-K, filed on March 30, 2023).
|
|
|
Form of Indemnification Agreement (incorporated by reference to Exhibit 10.30 to the Registrant’s Registration Statement on Form S-4, filed on
September 30, 2020).
|
|
|
Sublicense Agreement, dated as of January 21, 2020, by and between Ocuphire Pharma, Inc. and Apexian Pharmaceuticals, Inc (incorporated by
reference to Exhibit 10.31 to the Registrant’s Registration Statement on Form S-4, filed on September 30, 2020).
|
|
|
First Amendment to Sublicense Agreement, dated as of June 4, 2020, by and between Apexian Pharmaceuticals, Inc. and Ocuphire Pharma, Inc
(incorporated by reference to Exhibit 10.32 to the Registrant’s Registration Statement on Form S-4, filed on September 30, 2020).
|
|
|
Lease Agreement, dated as of May 19, 2019, by and between Ocuphire Pharma, Inc. and Duke & Duke, LP (incorporated by reference to Exhibit
10.33 to the Registrant’s Registration Statement on Form S-4, filed on September 30, 2020).
|
|
|
First Amendment to Lease Agreement, dated as of October 29, 2019, by and between Ocuphire Pharma, Inc. and Duke & Duke, LP (incorporated by
reference to Exhibit 10.34 to the Registrant’s Registration Statement on Form S-4, filed on September 30, 2020).
|
|
|
Second Lease Amendment, dated as of November 17, 2020, by and between the Company and Duke & Duke (incorporated by reference to Exhibit
10.43 to the Registrant’s Annual Report on Form 10-K, filed on March 11, 2021).
|
|
|
Third Lease Amendment, dated as of September 9, 2021, by and between the Company and Duke & Duke (incorporated by reference to Exhibit 10.1
to the Registrant’s Quarterly Report on Form 10-Q, filed on November 12, 2021).
|
|
|
Fourth Lease Amendment, dated as of October 17, 2022, by and between the Company and Duke & Duke (incorporated by reference to Exhibit 10.1
to the Registrant’s Quarterly Report on Form 10-Q, filed on November 4, 2022).
|
|
|
Fifth Lease Amendment, dated as of November 29, 2023, by and between the Company and Duke & Duke (incorporated by reference to Exhibit
10.5.5 to the Registrant’s Annual Report on Form 10-K, filed on March 8, 2024).
|
|
Ocuphire Pharma, Inc. 2018 Equity Incentive Plan, dated as of April 9, 2018 (incorporated by reference to Exhibit 10.35 to the Registrant’s
Registration Statement on Form S-4, filed on July 6, 2020).
|
|
|
First Amendment to 2018 Equity Incentive Plan, dated as of December 23, 2019 (incorporated by reference to Exhibit 10.36 to the Registrant’s
Registration Statement on Form S-4 , filed on July 6, 2020).
|
|
|
Form of Option Agreement issuable under the Ocuphire Pharma, Inc. 2018 Equity Incentive Plan (incorporated by reference to Exhibit 10.37 to the
Registrant’s Registration Statement on Form S-4, filed on July 6, 2020).
|
|
|
Ocuphire Pharma, Inc. 2020 Equity Incentive Plan (incorporated by reference to Annex D to the Registrant’s Registration Statement on Form S-4,
filed on July 6, 2020).
|
|
|
Form of Restricted Stock Unit Grant Notice issued under the Ocuphire Pharma, Inc. 2020 Equity Incentive Plan (incorporated by reference to
Exhibit 10.7.1 to the Registrant’s Annual Report on Form 10-K, filed on March 30, 2023).
|
|
|
Form of Stock Option Grant Notice issued under the Ocuphire Pharma, Inc. 2020 Equity Incentive Plan (incorporated by reference to Exhibit 10.7.2
to the Registrant’s Annual Report on Form 10-K, filed on March 30, 2023).
|
|
|
Contingent Value Rights Agreement, dated as of November 5, 2020, by and among the Company, Shareholder Representative Services LLC and the Olde
Monmouth Stock Transfer Co., Inc. (incorporated by reference to Exhibit 10.4 to the Registrant’s Current Report on Form 8-K, filed on November 6, 2020).
|
|
|
Ocuphire Pharma, Inc. 2021 Inducement Plan (incorporated by reference to Exhibit 10.41 to the Registrant’s Annual Report on Form 10-K, filed on
March 11, 2021)
|
|
|
First Amendment to 2021 Inducement Plan (incorporated by reference to Exhibit 10.3 to the Registrant’s Current Report on Form 8-K, filed on
November 1, 2023).
|
|
|
Second Amendment to 2021 Inducement Plan (incorporated by reference to Exhibit 10.3 to the Registrant’s Quarterly Report on Form 10-Q, filed on
November 12, 2024).
|
|
|
Form of Stock Option Grant Notice issued under the Ocuphire Pharma, Inc. 2021 Inducement Plan (incorporated by reference to Exhibit 10.9.1 to
the Registrant’s Annual Report on Form 10-K, filed on March 30, 2023).
|
|
Employment Agreement dated November 11, 2020, by and between the Company and Amy Rabourn (incorporated by reference to Exhibit 10.42 to the
Registrant’s Annual Report on Form 10-K, filed on March 11, 2021).
|
|
|
First Amendment to the Employment Agreement by and among the Company and Amy Rabourn, effective as of March 26, 2023 (incorporated by reference
to Exhibit 10.10.1 to the Registrant’s Annual Report on Form 10-K, filed on March 30, 2023).
|
|
|
Capital on Demand™ Sales Agreement, dated March 11, 2021 between the Company and JonesTrading Institutional Services LLC (incorporated by
reference to Exhibit 1.1 to the Registrant’s Current Report on Form 8-K, filed on March 11, 2021).
|
|
|
Sales Agreement, dated January 13, 2025, between the Company and Leerink Partners LLC (incorporated by reference to Exhibit 1.1 to the
Registrant’s Current Report on Form 8-K, filed on January 14, 2025).
|
|
|
Form of Purchase Agreement, dated as of June 4, 2021, by and among Ocuphire Pharma, Inc. and the purchasers identified on the signature pages
thereto (incorporated by reference to Exhibit 10.1 to the Registrant’s Current Report on Form 8-K/A, filed on June 7, 2021).
|
|
|
Processa License Agreement dated June 16, 2021 (incorporated by reference to Exhibit 10.1 to the Registrant’s Current Report on Form 8-K, filed
on June 23, 2021).
|
|
|
Viatris (f/k/a Famy Life Sciences) License and Collaboration Agreement dated November 6, 2022 (incorporated by reference to Exhibit 10.1 to the
Registrant’s Current Report on Form 8-K, filed on November 7, 2022).
|
|
|
Consulting Agreement dated April 8, 2022, by and between the Company and Jay Pepose (incorporated by reference to Exhibit 10.1 to the
Registrant’s Quarterly Report on Form 10-Q, filed on May 13, 2022).
|
|
|
First Amendment to the Consulting Agreement dated September 19, 2022, by and between the Company and Jay Pepose (incorporated by reference to
Exhibit 10.15.1 to the Registrant’s Annual Report on Form 10-K, filed on March 30, 2023).
|
|
|
Amendment No. 2 to the Consulting Agreement dated December 1, 2022, by and between the Company and Jay Pepose (incorporated by reference to
Exhibit 10.15.2 to the Registrant’s Annual Report on Form 10-K, filed on March 30, 2023).
|
|
|
Third Amendment to the Consulting Agreement, dated January 1, 2024, by and between the Company and Jay Pepose, M.D (incorporated by reference to
Exhibit 10.1 to the Registrant’s Quarterly Report on Form 10-Q, filed on May 10, 2024).
|
|
Consulting Agreement, dated April 11, 2024, by and between the Company and Jay Pepose, M.D. (incorporated by reference to Exhibit 10.1 to the
Registrant’s Current Report on Form 8-K, filed on April 17, 2024).
|
|
|
Amended and Restated Non-Employee Director Compensation Policy dated July 1, 2022 (incorporated by reference to Exhibit 10.1 to the Registrant’s
Quarterly Report on Form 10-Q, filed on August 12, 2022).
|
|
|
Non-Employee Director Compensation Policy (incorporated by reference to Exhibit 10.2 to the Registrant’s Quarterly Report on Form 10-Q, filed on
May 15, 2023).
|
|
|
Second Amended and Restated Non-Employee Director Compensation Policy (incorporated by reference to Exhibit 10.2 to the Registrant’s Quarterly
Report on Form 10-Q, filed on May 15, 2023).
|
|
|
Third Amended and Restated Non-Employee Director Compensation Plan dated June 11, 2024 (incorporated by reference to Exhibit 10.1 to the
Registrant’s Quarterly Report on Form 10-Q, filed on August 13, 2024).
|
|
|
Purchase Agreement, dated as of August 10, 2023, by and between Ocuphire Pharma, Inc. and Lincoln Park Capital Fund, LLC (incorporated by
reference to Exhibit 10.1 to the Registrant’s Current Report on Form 8-K, filed on August 11, 2023).
|
|
|
Registration Rights Agreement, dated as of August 10, 2023, by and between Ocuphire Pharma, Inc. and Lincoln Park Capital Fund, LLC
(incorporated by reference to Exhibit 10.2 to the Registrant’s Current Report on Form 8-K, filed on August 11, 2023).
|
|
|
Employment Agreement entered into on October 31, 2023 by and between Ocuphire Pharma, Inc. and George Magrath (incorporated by reference to
Exhibit 10.1 to the Registrant’s Current Report on Form 8-K, filed on November 1, 2023).
|
|
|
|
|
| 10.24* |
Amended and Restated Employment Agreement, entered into on January 17, 2025, by and between the Company and George Magrath (incorporated by
reference to Exhibit 10.1 to the Registrant’s Current Report on Form 8-K, filed on January 24, 2025).
|
|
|
|
|
Form of Restricted Stock Unit Award and Form of Award Agreement (incorporated by reference to Exhibit 10.2 to the Registrant’s Current Report on
Form 8-K, filed on November 1, 2023).
|
|
Employment Agreement, effective as of February 12, 2024, by and between the Company and Nirav Jhaveri (incorporated by reference to Exhibit 10.1
to the Registrant’s Current Report on Form 8-K, filed on February 16, 2024).
|
|
|
First Amendment to Employment Agreement, effective as of February 12, 2024, by and between the Company and Nirav Jhaveri (incorporated by
reference to Exhibit 10.2 to the Registrant’s Current Report on Form 8-K, filed on February 16, 2024).
|
|
|
|
|
|
Second Amendment to Employment Agreement, effective as of January 17, 2025, by and between the Company and Nirav Jhaveri (incorporated by
reference to Exhibit 10.2 to the Registrant’s Current Report on Form 8-K, filed on January 24, 2025).
|
|
|
Offer Letter entered into on November 20, 2023 by and between Ocuphire Pharma, Inc. and Joseph Schachle (incorporated by reference to Exhibit
10.1 to the Registrant’s Current Report on Form 8-K, filed on November 27, 2023).
|
|
|
Employment Agreement, dated October 22, 2024, by and between the Company and Dr. Benjamin Yerxa (incorporated by reference to Exhibit 10.1 to
the Registrant’s Quarterly Report on Form 10-Q, filed on November 12, 2024).
|
|
|
Consulting Agreement, dated as of October 22, 2024, by and between the Company and Dr. Jean Bennett (incorporated by reference to Exhibit 10.2
to the Registrant’s Quarterly Report on Form 10-Q, filed on November 12, 2024).
|
|
|
Exclusive License Agreement with Know-How, dated as of April 10, 2019, by and among The Trustees of the University of Pennsylvania and The
University of Florida Research Foundation, Incorporated and the Company.
|
|
|
Amendment No. 1 to Exclusive License Agreement with Know-How, dated as of May 1, 2020, by and among The Trustees of the University of
Pennsylvania and The University of Florida Research Foundation, Incorporated and the Company.
|
|
|
Amendment No. 2 to Exclusive License Agreement with Know-How, dated as of July 1, 2022, by and among The Trustees of the University of
Pennsylvania and The University of Florida Research Foundation, Incorporated and the Company.
|
|
|
Amendment No. 3 to Exclusive License Agreement with Know-How, dated as of December 23, 2022, by and among The Trustees of the University of
Pennsylvania and The University of Florida Research Foundation, Incorporated and the Company.
|
|
Amendment No. 4 to Exclusive License Agreement with Know-How, dated as of April 15, 2024, by and among The Trustees of the University of
Pennsylvania and The University of Florida Research Foundation, Incorporated and the Company.
|
|
|
Amended and Restated License Agreement, dated as of June 15, 2022, by and between The Trustees of the University of Pennsylvania and the
Company.
|
|
|
Asset Purchase Agreement, dated as of December 23, 2022, by and between Iveric Bio Gene Therapy LLC and the Company.
|
|
|
Non-Exclusive License Agreement, dated as of March 2, 2023, by and between The Trustees of the University of Pennsylvania and the Company.
|
|
|
Insider Trading Compliance Policy.
|
|
|
|
|
|
Subsidiaries of the Registrant.
|
|
|
|
|
|
Consent of Ernst & Young, LLP.
|
|
|
|
|
|
Certification of Principal Executive Officer pursuant to Rules 13a-14(a) and 15d-14(a) promulgated under the Securities Exchange Act of 1934,
as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002.
|
|
|
|
|
|
Certification of Principal Financial Officer pursuant to Rules 13a-14(a) and 15d-14(a) promulgated under the Securities Exchange Act of 1934,
as adopted pursuant to section 302 of the Sarbanes-Oxley Act of 2002.
|
|
|
|
|
|
Certification of Principal Executive Officer and Principal Financial Officer pursuant to Rules 13a-14(b) and 15d-14(b) promulgated under the
Securities Exchange Act of 1934 and 18 U.S.C. Section 1350, as adopted pursuant to section 906 of The Sarbanes-Oxley Act of 2002.
|
|
Compensation Recovery Policy (incorporated by reference to Exhibit 97 to the Registrant’s Annual Report on Form 10-K, filed on March 8,
2024).
|
|
|
|
|
|
101.INS
|
Inline XBRL Instance Document (the instance document does not appear in the Interactive Data File because its XBRL tags are embedded within
the Inline XBRL document).
|
|
|
|
|
101.SCH
|
Inline XBRL Taxonomy Extension Schema Document.
|
|
|
|
|
101.CAL
|
Inline XBRL Taxonomy Extension Calculation Linkbase Document
|
|
|
|
|
101.DEF
|
Inline XBRL Taxonomy Extension Definition Linkbase Document
|
|
|
|
|
101.LAB
|
Inline XBRL Taxonomy Extension Label Linkbase Document
|
|
|
|
|
101.PRE
|
Inline XBRL Taxonomy Extension Presentation Linkbase Document
|
|
|
|
|
104
|
Cover Page Interactive Data File (formatted as inline XBRL and contained in Exhibit 101)
|
| * |
Indicates management contract or compensatory plan.
|
| ** |
Indicates exhibits that are being filed herewith.
|
| + |
Certain schedules and exhibits have been omitted pursuant to Item 601(b)(2) of Regulation S-K. A copy of any omitted schedule and/or exhibit will be furnished to the SEC upon request.
|
|
++
|
Portions of this exhibit have been omitted in compliance with Item 601 of Regulation S-K.
|
| ITEM 16. |
FORM 10-K SUMMARY
|
| ITEM 8. |
FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
|
|
Report of Independent Registered Public Accounting Firm (PCAOB ID: 42)
|
117 |
| 118 |
|
| 119 |
|
| 120 |
|
| 121 |
|
| 122 |
|
|
Acquisition of Opus Genetics
|
|
Description of the Matter
|
As described in Note 2 to the consolidated financial statements, on October 22, 2024, the Company completed the stock purchase of Private Opus for a purchase price of $25.8 million. The Company accounted for this transaction as
an asset acquisition.
Auditing the Company’s accounting for the stock purchase of Private Opus was complex due to the significant estimation used by management to determine the fair value of the acquired in-process
research and development (“IPR&D”) of $28 million. The significant estimation was primarily due to the complexity of the valuation model used by management to measure the fair value of the IPR&D and the sensitivity of the
respective fair value to the significant underlying assumptions. The Company used a multi-period excess earnings method to measure the IPR&D. The significant assumptions used to estimate the fair value of the IPR&D included
discount rate, projected EBITA margin, and revenue growth rates. These significant assumptions are forward-looking and could be affected by future economic and market conditions.
|
|
How We Addressed the Matter in Our Audit
|
To test the estimated fair value of the IPR&D, we performed audit procedures that included, among others, evaluating the Company’s selection of the valuation methodology, evaluating the methods
and significant assumptions used by the Company’s valuation specialist, and evaluating the completeness and accuracy of the underlying data supporting the significant assumptions and estimates. For example, we compared the revenue
growth rate significant assumption to industry data and external market research. We involved our specialist to assist with our evaluation of the methodologies used by the Company and significant assumptions included in the fair
value estimates. We also performed sensitivity analyses to evaluate the changes in the fair value of the IPR&D that would result from changes in the significant assumptions.
|
|
As of December 31,
|
||||||||
|
2024
|
2023
|
|||||||
|
Assets
|
||||||||
|
Current assets:
|
||||||||
|
Cash and cash equivalents
|
$
|
30,321
|
$
|
50,501
|
||||
|
Accounts receivable
|
3,563 | 926 | ||||||
|
Contract assets and unbilled receivables (Note
10)
|
2,209 | 1,407 | ||||||
|
Prepaids and other current assets
|
515
|
1,099
|
||||||
|
Short-term investments
|
2
|
15
|
||||||
|
Total current assets
|
36,610
|
53,948
|
||||||
|
Property and equipment, net
|
252
|
—
|
||||||
|
Total assets
|
$
|
36,862
|
$
|
53,948
|
||||
|
Liabilities and stockholders’ equity
|
||||||||
|
Current liabilities:
|
||||||||
|
Accounts payable
|
$
|
3,148
|
$
|
2,153
|
||||
|
Accrued expenses
|
8,145
|
1,815
|
||||||
| Derivative liability | 2 | 74 | ||||||
|
Total current liabilities
|
11,295
|
4,042
|
||||||
|
Total liabilities
|
11,295
|
4,042
|
||||||
|
Commitments and contingencies (Note 3 and Note 9)
|
||||||||
|
Series A preferred stock, par value $0.0001; 14,146 shares and no shares were designated as of December 31, 2024 and 2023, respectively; 14,145.374 and no shares issued and outstanding at December 31, 2024 and 2023, respectively.
|
18,843
|
—
|
||||||
|
Stockholders’ equity:
|
||||||||
|
Preferred stock, par value $0.0001; 9,985,854 and 10,000,000
shares authorized as of December 31, 2024 and 2023, respectively; no shares issued and
outstanding at December 31, 2024 and 2023.
|
— | — | ||||||
|
Common stock, par value $0.0001; 125,000,000 and 75,000,000 shares authorized as of December 31, 2024
and 2023, respectively; 31,574,657 and 23,977,491 shares issued and outstanding at December 31, 2024 and 2023, respectively.
|
3
|
2
|
||||||
|
Additional paid-in capital
|
145,719
|
131,370
|
||||||
|
Accumulated deficit
|
(138,998
|
)
|
(81,466
|
)
|
||||
|
Total stockholders’ equity
|
6,724
|
49,906
|
||||||
|
Total liabilities, series A preferred stock, and stockholders’ equity
|
$
|
36,862
|
$
|
53,948
|
||||
|
For the Year Ended
December 31,
|
||||||||
|
2024
|
2023
|
|||||||
| License and collaborations revenue |
$ | 10,992 | $ | 19,049 | ||||
| Operating expenses: | ||||||||
|
General and administrative
|
18,215
|
11,959
|
||||||
|
Research and development
|
26,851
|
17,653
|
||||||
|
Acquired in-process research and development
|
28,000 | — | ||||||
|
Total operating expenses
|
73,066
|
29,612
|
||||||
|
Loss from operations
|
(62,074
|
)
|
(10,563
|
)
|
||||
| Financing costs (Note 7) |
— | (1,328 | ) | |||||
|
Fair value change in derivative liabilities
|
72
|
80
|
||||||
|
Other income, net
|
4,470
|
1,837
|
||||||
|
Loss before income taxes
|
(57,532
|
)
|
(9,974
|
)
|
||||
|
Provision for income taxes
|
—
|
(12
|
)
|
|||||
|
Net loss
|
(57,532
|
)
|
(9,986
|
)
|
||||
|
Other comprehensive loss, net of tax
|
—
|
—
|
||||||
|
Comprehensive loss
|
$
|
(57,532
|
)
|
$
|
(9,986
|
)
|
||
|
Net loss per share (Note 11):
|
||||||||
|
Basic and diluted
|
$ | (2.15 | ) | $ | (0.46 | ) | ||
|
Number of shares used in per share calculations:
|
||||||||
|
Basic and diluted
|
26,715,526
|
21,589,821
|
||||||
|
Series A Preferred Stock
|
Common Stock
|
Additional
Paid–In
|
Accumulated
|
Total
Stockholders’
|
||||||||||||||||||||||||
| Shares |
Amount
|
Shares
|
Amount
|
Capital
|
Deficit
|
Equity
|
||||||||||||||||||||||
|
Balance at December 31, 2022
|
— | $ | — |
20,861,315
|
$
|
2
|
$
|
117,717
|
$
|
(71,480
|
)
|
$
|
46,239
|
|||||||||||||||
|
Issuance of common stock in connection with the at-the-market program and purchase agreement
|
— | — | 2,964,238 | — | 10,249 | — | 10,249 | |||||||||||||||||||||
| Issuance costs |
— | — | — | — | (136 | ) | — | (136 | ) | |||||||||||||||||||
|
Exercise of Series B warrants
|
— | — | 17,869 | — | — | — | — | |||||||||||||||||||||
|
Stock-based compensation
|
— | — | 106,600 | — | 3,510 | — | 3,510 | |||||||||||||||||||||
|
Exercise of stock options
|
— | — | 27,469 | — | 30 | — | 30 | |||||||||||||||||||||
|
Net and comprehensive loss
|
— | — | — | — | — | (9,986 | ) | (9,986 | ) | |||||||||||||||||||
|
Balance at December 31, 2023
|
— | — | 23,977,491 | 2 | 131,370 | (81,466 | ) | 49,906 | ||||||||||||||||||||
|
Issuance of common stock and Series A preferred stock to former private Opus Genetics Inc. stockholders and effect of asset acquisition.
|
14,145.374 | 18,843 | 5,237,063 | 1 | 6,964 | — | 6,965 | |||||||||||||||||||||
|
Issuance of common stock in connection with the at-the-market program and purchase agreement
|
— | — | 2,008,522 | — | 4,497 | — | 4,497 | |||||||||||||||||||||
|
Issuance costs
|
— | — | — | — | (395 | ) | — | (395 | ) | |||||||||||||||||||
|
Stock-based compensation
|
— | — | 395,396 | — | 3,362 | — | 3,362 | |||||||||||||||||||||
|
Share repurchases for the payment of employee taxes
|
— | — | (43,815 | ) | — | (79 | ) | — | (79 | ) | ||||||||||||||||||
|
Net and comprehensive loss
|
— | — | — | — | — | (57,532 | ) | (57,532 | ) | |||||||||||||||||||
|
Balance at December 31, 2024
|
14,145.374 | $ | 18,843 | 31,574,657 | $ | 3 | $ | 145,719 | $ | (138,998 | ) | $ | 6,724 | |||||||||||||||
|
For the Year Ended
December 31,
|
||||||||
|
2024
|
2023
|
|||||||
|
Operating activities
|
||||||||
|
Net loss
|
$
|
(57,532
|
)
|
$
|
(9,986
|
)
|
||
|
Adjustments to reconcile net loss to net cash used in operating activities:
|
||||||||
|
Stock-based compensation
|
3,362
|
3,510
|
||||||
|
Depreciation
|
10
|
6
|
||||||
|
Fair value change in derivative liabilities
|
(72
|
)
|
(80
|
)
|
||||
| Financing costs |
— | 1,328 | ||||||
|
Unrealized loss from short-term investments
|
13 | 34 | ||||||
|
Acquired in-process research and development
|
28,000 |
— |
||||||
|
Gain in connection with asset acquisition
|
(2,447 | ) | — |
|||||
|
Change in assets and liabilities:
|
||||||||
|
Accounts receivable
|
(2,637 | ) | 372 | |||||
| Contract assets and unbilled receivables |
(802 | ) | 2,145 | |||||
|
Prepaid expenses and other assets
|
634
|
354
|
||||||
|
Accounts payable
|
220
|
1,082
|
||||||
|
Accrued expenses
|
5,675
|
123
|
||||||
|
Net cash used in by operating activities
|
(25,576
|
)
|
(1,112
|
)
|
||||
|
Investing activities
|
||||||||
| Cash received in connection with asset acquisition |
1,210 |
— |
||||||
|
Net cash provided by investing activities
|
1,210
|
—
|
||||||
|
Financing activities
|
||||||||
| Proceeds from issuance of common stock |
4,497 | 9,227 | ||||||
|
Issuance costs attributed to common stock
|
(232
|
)
|
(278
|
)
|
||||
| Share repurchases for the payment of employee taxes |
(79 | ) | — | |||||
|
Exercise of stock options and Series B warrants
|
—
|
30
|
||||||
|
Net cash provided by financing activities
|
4,186
|
8,979
|
||||||
|
Net (decrease) increase in cash and cash equivalents
|
(20,180
|
)
|
7,867
|
|||||
|
Cash and cash equivalents at beginning of period
|
50,501
|
42,634
|
||||||
|
Cash and cash equivalents at end of period
|
$
|
30,321
|
$
|
50,501
|
||||
|
Supplemental disclosure of cash flow information:
|
||||||||
|
Cash paid for income taxes
|
$
|
—
|
$
|
344
|
||||
|
Cash paid for interest
|
$
|
—
|
$
|
—
|
||||
|
Supplemental non-cash financing transactions:
|
||||||||
|
Common stock and Series A preferred stock issued in connection with the asset acquisition
|
$ | 25,808 | $ | — | ||||
| Net liabilities assumed in connection with asset acquisition |
$ | 955 | $ | — | ||||
|
Non-cash issuance of common stock in connection with equity purchase agreement
|
$ | — | $ | 1,022 | ||||
|
Value of derivative established in connection with the equity purchase agreement
|
$ | — | $ | 154 | ||||
|
Unpaid issuance costs
|
$
|
163
|
$
|
10
|
||||
| 1. |
Company Description and Summary of Significant Accounting Policies
|
|
|
• |
Level 1 inputs: Unadjusted quoted prices for identical assets or liabilities in active markets;
|
|
|
• |
Level 2 inputs: Quoted prices for similar assets and liabilities in active markets, quoted prices in markets that are not active, or inputs which are observable, whether directly or indirectly, for substantially the full term of the
asset or liability; and
|
|
|
• |
Level 3 inputs: Unobservable inputs in which there is little or no market data available, which requires management to develop its own assumptions in pricing the asset or liability.
|
|
As of December 31, 2024
|
||||||||||||||||
|
Description
|
Total
|
Level 1
|
Level 2
|
Level 3
|
||||||||||||
|
Assets:
|
||||||||||||||||
|
Short-term investments
|
$
|
2
|
$
|
2
|
$
|
—
|
$
|
—
|
||||||||
|
Total assets at fair value
|
$
|
2
|
$
|
2
|
$
|
—
|
$
|
—
|
||||||||
|
Liabilities:
|
||||||||||||||||
|
Derivative liability
|
$ |
2 | $ |
— | $ |
— | $ | 2 | ||||||||
|
Total liabilities at fair value
|
$ |
2 | $ | — | $ |
— | $ | 2 | ||||||||
|
As of December 31, 2023
|
||||||||||||||||
|
Description
|
Total
|
Level 1
|
Level 2
|
Level 3
|
||||||||||||
|
Assets:
|
||||||||||||||||
|
Short-term investments
|
$
|
15
|
$
|
15
|
$
|
—
|
$
|
—
|
||||||||
|
Total assets at fair value
|
$
|
15
|
$
|
15
|
$
|
—
|
$
|
—
|
||||||||
| Liabilities: |
||||||||||||||||
| Derivative liability |
$ |
74 | $ |
— | $ |
— | $ |
74 |
||||||||
| Total liabilities at fair value |
$ |
74 | $ |
— | $ |
— | $ |
74 |
||||||||
|
|
2024
|
2023
|
||||||
|
Short-term investments
|
||||||||
|
Balance as of beginning of period
|
$
|
15
|
$
|
49
|
||||
|
Unrealized loss
|
(13
|
)
|
(34
|
)
|
||||
|
Balance as of end of period
|
$
|
2
|
$
|
15
|
||||
|
|
2024
|
2023
|
||||||
|
Derivative liability
|
||||||||
|
Balance as of beginning of period
|
$
|
74
|
$
|
—
|
||||
|
Purchase agreement execution
|
—
|
154
|
||||||
|
Unrealized gain
|
(72
|
)
|
(80
|
)
|
||||
|
Balance as of end of period
|
$
|
2
|
$
|
74
|
||||
| 2. |
Mergers
|
|
Number of common shares of the combined organization owned by the Company’s Pre-acquisition Private Opus stockholders
|
5,237,063
|
|||
|
Multiplied by the fair value per share of the Company’s common stock (1)
|
$
|
1.33
|
||
|
Fair value of common stock issued to affect the Opus Acquisition
|
$
|
6,965
|
||
|
Number of Series A preferred shares of the combined organization owned by the Company’s Pre-acquisition Private Opus stockholders
|
14,145.374
|
|||
|
Multiplied by the fair value per share of the Company’s Series A preferred stock (2)
|
$
|
1.3321
|
||
|
Fair value of preferred stock issued to affect the Opus Acquisition
|
$
|
18,843
|
||
|
Purchase price
|
$
|
25,808
|
|
|
(1) |
Based on the last reported sale price of the Company’s common stock on the Nasdaq Capital Market on October 22, 2024, the
closing date of the Opus Acquisition.
|
|
|
(2) |
Based on the fair market valuation of the Series A preferred stock that considered the reported sale price of the Company’s
common stock on the Nasdaq Capital Market on October 22, 2024 on an as converted basis (1,000 shares of common stock
for 1 share of preferred stock), the closing date of the Opus Acquisition, as well as the underlying dividend
provisions on a discounted cash flow basis.
|
|
Cash acquired
|
$
|
1,210
|
||
|
Net liabilities assumed
|
(955
|
)
|
||
|
IPR&D (3)
|
28,000
|
|||
|
Net assets and IPR&D acquired
|
$
|
28,255
|
|
|
(3) |
Represents the Private Opus Acquisition research and development projects which were in-process, but not yet completed, and which the Company may advance post the Opus Acquisition. This includes the
development of gene therapies for IRDs. Current accounting standards require that the fair value of IPR&D projects acquired in an asset acquisition with no alternative future use be charged to expense on the acquisition
date. The acquired IPR&D did not have outputs or employees. The fair value
of the IPR&D was recorded at fair value using Level 3 inputs. A Multi-Period
Excess Earnings Method (“MPEEM”) model was applied which incorporates assumptions such as future earnings and margins in connection with the further development and commercialization of IRD therapies, and a discount rate of
20%.
|
|
Purchase Price
|
$
|
25,808
|
||
|
Net assets and IPR&D acquired
|
28,255
|
|||
|
Gain recorded upon close of Opus Acquisition
|
$
|
2,447
|
|
|
• |
90% of all payments received by Rexahn or its affiliates during such
CVR Payment Period from or on behalf of BioSense Global LLC (“BioSense”) pursuant to that certain License and Assignment Agreement;
|
|
|
• |
90% of all payments received by Rexahn or its affiliates during such
CVR Payment Period from or on behalf of Zhejiang HaiChang Biotechnology Co., Ltd. (“HaiChang”) pursuant to that certain Exclusive License Agreement; and
|
|
|
• |
75% of the sum of (i) all cash consideration paid by a third party to
Rexahn or its affiliates during the applicable CVR Payment Period in connection with the grant, sale or transfer of rights to Rexahn’s pre-closing intellectual property (other than a grant, sale or transfer of rights involving a sale
or disposition of the post-Merger combined company) that is entered into during the 10-year period after the closing
(“Parent IP Deal”), plus (ii) with respect to any non-cash consideration received by Rexahn or its affiliates from a third party during the applicable CVR Payment Period in connection with any Parent IP Deal, all amounts received by
Rexahn or its affiliates for such non-cash consideration at the time such non-cash consideration is monetized by Rexahn or its affiliates, minus (iii) certain permitted deductions.
|
| 3. |
Commitments and Contingencies
|
| 4. |
Supplemental Balance Sheet Information
|
|
Accrued expenses consist of the following (in thousands):
|
December 31,
|
|||||||
|
2024
|
2023
|
|||||||
|
Payroll
|
$ |
1,481
|
$ |
753
|
||||
|
Professional services
|
1,608
|
591
|
||||||
|
R&D services and supplies
|
4,452
|
400
|
||||||
|
Other
|
604
|
71
|
||||||
|
Total
|
$
|
8,145
|
$
|
1,815
|
||||
| 5. |
Related Party Transactions
|
| 6. |
Series A Preferred Stock
|
| 7. |
Stockholders’ Equity
|
| 8. |
Stock-based Compensation
|
|
December 31,
|
||||||||
|
2024
|
2023
|
|||||||
|
General and administrative
|
$
|
2,382
|
$
|
2,435
|
||||
|
Research and development
|
980
|
1,075
|
||||||
|
Total stock-based compensation
|
$
|
3,362
|
$
|
3,510
|
||||
| |
Number of
Options
|
Weighted
Average
Exercise
Price
|
Weighted Average
Remaining
Contractual
Term (years)
|
Aggregate
Intrinsic
Value(1)
(in thousands)
|
||||||||||||
|
Outstanding at December 31, 2022
|
2,936,044
|
$
|
2.87
|
7.82
|
$
|
3,314
|
||||||||||
|
Granted
|
1,768,116
|
$
|
3.20
|
|
|
|||||||||||
|
Exercised
|
(27,469
|
)
|
$
|
1.09
|
|
|
||||||||||
|
Forfeited/Cancelled
|
(266,433
|
)
|
$
|
3.66
|
|
|
||||||||||
|
Outstanding at December 31, 2023
|
4,410,258
|
$
|
2.98
|
7.81
|
$
|
2,385
|
||||||||||
|
Granted
|
1,366,914
|
$
|
2.17
|
|
|
|||||||||||
|
Exercised
|
-
|
|
|
|
|
|
||||||||||
|
Forfeited/Cancelled
|
(703,436
|
)
|
$
|
3.58
|
|
|
||||||||||
|
Outstanding at December 31, 2024
|
5,073,736
|
$
|
2.68
|
7.37
|
|
124
|
||||||||||
| Vested and expected to vest at December 31, 2024 | 5,073,736 |
$ | 2.68 | 7.37 |
|
124 | ||||||||||
|
Vested and exercisable at December 31, 2024
|
2,887,816
|
$
|
2.80
|
6.03
|
|
124
|
||||||||||
| (1) |
The aggregate intrinsic value is
calculated as the difference between the exercise price of the underlying options and the fair value of our common stock as of December 31, 2024 and 2023 of $1.19 and $3.01 per share, respectively.
|
|
2024
|
2023
|
|||||||
|
Expected stock price volatility
|
98.1
|
%
|
96.0
|
%
|
||||
|
Expected life of options (years)
|
5.94
|
6.1
|
||||||
|
Expected dividend yield
|
0
|
%
|
0
|
%
|
||||
|
Risk free interest rate
|
4.2
|
%
|
4.2
|
%
|
||||
|
Number of
Shares
|
||||
|
Non-vested at December 31, 2022
|
—
|
|||
|
Granted
|
936,156
|
|||
| Forfeited |
(100,842 | ) | ||
|
Vested
|
(33,614
|
)
|
||
|
Non-vested at December 31, 2023
|
801,700
|
|||
| Granted | 1,025,022 | |||
| Forfeited | (119,330 | ) | ||
| Vested |
(314,162 | ) | ||
|
Non-vested at December 31, 2024
|
1,393,230 | |||
| 9. |
Apexian Sublicense Agreement
|
| 10. |
License and Collaboration Agreements
|
| 2024 | 2023
|
|||||||
| Contract Assets and Unbilled Receivables | ||||||||
|
Balance as of beginning of period
|
$
|
1,407
|
$ | 3,552 | ||||
|
Revenue recognized
|
10,992 | 19,049 | ||||||
|
Reclassification to accounts receivable related to costs billed
under the Viatris License Agreement
|
(10,190 | ) | (21,194 | ) | ||||
| Balance as of end of period | $ | 2,209 | $ | 1,407 | ||||
| 11. |
Net loss per share
|
|
2024
|
2023
|
|||||||
|
Series A and RDO warrants
|
7,204,299
|
7,204,299 | ||||||
|
Stock options
|
5,073,736
|
4,410,258
|
||||||
|
RSUs
|
1,393,230
|
801,700
|
||||||
|
Former Rexahn warrants
|
—
|
58,597 | ||||||
| 12. |
Income Taxes
|
|
2024
|
2023
|
|||||||
| Income tax (benefit) provision at federal statutory rate | (21.0 | )% | (21.0 | )% | ||||
|
Valuation allowance
|
26.5 |
23.8 | ||||||
|
State income tax, net of federal benefit
|
(4.9 | ) | (4.9 | ) | ||||
| Private Opus acquisition |
(11.2 | ) | — | |||||
|
Financing contracts
|
— |
3.2 |
||||||
|
Stock options
|
0.8 |
1.0 |
||||||
| Acquired IPR&D |
12.6 | — | ||||||
|
Research and development
|
(1.6 | ) | (3.9 | ) | ||||
|
Other
|
(1.2 | ) | 1.9 | |||||
|
Effective tax rate
|
— | % | 0.1 | % | ||||
| 2024 |
2023 |
|||||||
|
Loss before income taxes:
|
$ | (57,532 |
) | $ | (9,974 | ) | ||
|
Current:
|
||||||||
|
Federal
|
$ | — | $ | 2 | ||||
|
State
|
— | 10 | ||||||
|
Total current tax provision (benefit)
|
— | $ | 12 | |||||
|
Deferred:
|
||||||||
|
Federal
|
— | — | ||||||
|
State
|
— | — | ||||||
|
Total tax provision (benefit)
|
$ | — | $ | 12 | ||||
|
2024
|
2023
|
|||||||
|
Deferred tax assets:
|
||||||||
| Federal and state operating loss carryforwards | $ | 20,342 | $ | 12,780 | ||||
|
Acquired intangibles
|
547 |
547
|
||||||
| Deferral of research and development costs | 9,582 | 3,794 | ||||||
|
Organizational costs
|
5 |
6
|
||||||
| Other | 77 | 72 | ||||||
|
Stock-based compensation
|
2,035 |
1,835
|
||||||
|
Research and development credit carryforward
|
2,089 |
1,107
|
||||||
| Transaction costs in connection with Opus Acquisition | 753 | — | ||||||
|
Subtotal
|
35,430 |
20,141
|
||||||
|
Valuation allowance
|
(35,403
|
)
|
(20,141
|
)
|
||||
|
Total deferred tax assets, net of valuation allowance
|
27
|
—
|
||||||
|
Deferred tax liabilities:
|
||||||||
| Fixed assets | (27 | ) | — | |||||
|
Total deferred tax liabilities
|
(27
|
)
|
—
|
|||||
|
Net deferred tax assets
|
$ | — | $ | — | ||||
| 13. |
Deferred Compensation Plan
|
| 14. |
Subsequent Events
|
|
OPUS GENETICS, INC.
|
||
|
Dated: March 31, 2025
|
By:
|
/s/ George Magrath
|
|
George Magrath
|
||
|
Chief Executive Officer and Director
|
||
|
By
|
/s/ George Magrath
|
|
Date: March 31, 2025
|
|
|
George Magrath
|
|
|
|
|
Chief Executive Officer and Director (Principal Executive Officer)
|
|
|
|
|
|
|
|
|
By
|
/s/ Nirav Jhaveri
|
|
Date: March 31, 2025
|
|
|
Nirav Jhaveri
|
|
|
|
|
Chief Financial Officer (Principal Financial Officer and Principal Accounting Officer)
|
|
|
|
|
|
|
|
|
By
|
/s/ Sean Ainsworth
|
|
Date: March 31, 2025
|
|
|
Sean Ainsworth
|
|
|
|
|
Director
|
|
|
|
|
|
|
|
|
By
|
/s/ Dr. Jean Bennett
|
|
Date: March 31, 2025
|
|
|
Dr. Jean Bennett
|
|
|
|
|
Director
|
|
|
|
|
|
|
|
|
By
|
/s/ Susan K. Benton
|
|
Date: March 31, 2025
|
|
|
Susan K. Benton
|
|
|
|
|
Director |
|
|
|
|
|
|
|
|
By
|
/s/ Cam Gallagher
|
|
Date: March 31, 2025
|
|
|
Cam Gallagher
|
|
|
|
|
Director |
|
|
|
|
|
|
|
|
By
|
s/ Dr. Adrienne Graves
|
|
Date: March 31, 2025
|
|
|
Dr. Adrienne Graves
|
|
|
|
|
Director |
|
|
|
|
|
|
|
|
By
|
/s/ James S. Manuso
|
|
Date: March 31, 2025
|
|
|
James S. Manuso
|
|
|
|
|
Director |
|
|
|
|
|
||
|
By
|
/s/ Richard J. Rodgers
|
|
Date: March 31, 2025
|
|
|
Richard J. Rodgers
|
|
|
|
|
Director
|
|
|
|
|
|
|
|
|
By
|
/s/ Dr. Benjamin R. Yerxa
|
|
Date: March 31, 2025
|
|
|
Dr. Benjamin R. Yerxa
|
|
|
|
|
President and Director
|
|
|
|
|
• |
Directors may be removed with or without cause only by a stockholder vote of at least a majority of the voting power of the then-outstanding voting stock. Vacancies
on the Board may be filled by a majority of directors then in office, even if less than a quorum, unless the Board determines otherwise. The authorized number of directors may only be changed by a resolution of the Board.
|
|
|
• |
A special meeting of stockholders may be called only by a resolution adopted by a majority of our Board, by the Company’s Chief Executive Officer, by the Chair of
the Board (acting in his or her discretion), or by the Chair of the Board acting within 10 days of receipt of a written request on behalf of at least 20% or more of the stockholders of all of the then-outstanding voting stock.
|
|
|
• |
There is an advance notice procedure for stockholders to make nominations
of candidates for election as directors or to bring other business before an annual meeting of our stockholders. The notice must follow the form and content specified in the Bylaws and include, without limitation, the following information:
|
|
|
i. |
as to director nominations, all information relating to each director nominee that is required by the rules of the Securities and Exchange Commission to be
disclosed in solicitations of proxies, or is otherwise required by Regulation 14A of the Securities Exchange Act of 1934, as amended;
|
|
|
ii. |
as to any other business that the stockholder proposes to bring before the meeting, a brief description of the business to be proposed, the reasons for conducting
such business at the meeting and, if any, the stockholder’s material interest in the proposed business; and
|
|
|
iii. |
the name and address of the stockholder who intends to make the nomination and the class and number of our shares beneficially owned of record.
|
|
|
• |
The ability to authorize undesignated Preferred Stock makes it possible for our Board to issue Preferred Stock with voting or other rights or preferences that could
have the effect of delaying, deferring, preventing or otherwise impeding any attempt to change control of us.
|
|
Section 1
|
Definitions
|
2
|
|
Section 2
|
Grant
|
7 |
|
Section 3
|
Diligence Obligations
|
9 |
|
Section 4
|
Payments
|
11
|
|
Section 5
|
Representations and Disclaimers of Licensors and Licensee
|
16 |
|
Section 6
|
Record Keeping; Accounting
|
17 |
|
Section 7
|
Patent Prosecution
|
19 |
|
Section 8
|
Infringement and Invalidity
|
20 |
|
Section 9
|
Term and Termination
|
22 |
|
Section 10
|
No Discrimination
|
24 |
|
Section 11
|
Assignability
|
25 |
|
Section 12
|
Dispute Resolution
|
25 |
|
Section 13
|
Indemnification; Liability; Insurance
|
26
|
|
Section 14
|
Use of Names
|
28 |
|
Section 15
|
Miscellaneous
|
28 |
|
Section 16
|
Notices
|
30 |
|
Section 17
|
United States Government Interests; Foundation Fighting Blindness Rights
|
31 |
|
Section 18
|
Confidentiality
|
31 |
|
Section 19
|
University Rules and Regulations
|
33 |
|
Section 20
|
Contract Formation and Authority
|
33 |
|
Appendix A - Patent Rights and Know-How
|
35
|
|
Appendix B - Licensed Information
|
36
|
|
Appendix C - Initial Development Plan
|
37
|
|
Appendix D - Development Report
|
38
|
|
Appendix E - Royalty Report
|
39 |
|
Appendix F - Milestones
|
40 |
|
Appendix G - Subsequently Added Intellectual Property
|
41
|
|
Appendix H - [***]
|
42
|
| (i) |
Reasonable record keeping, audit and reporting obligations sufficient to enable Licensors and Licensee to reasonably verify the payments due to Licensee and to Licensors under such Sublicense and to reasonably monitor such Sublicensee’s
progress in developing and/or commercializing Licensed Products, provided that such obligations shall be no less stringent that those provided in this Agreement for Licensee.
|
|
|
(ii) |
Infringement and enforcement provisions that do not conflict with the restrictions and procedural requirements imposed on Licensee hereunder and do not provide greater rights to Sublicensee than as provided in Section 8.
|
|
|
(iii) |
Confidentiality provisions with respect to Confidential Information of Penn consistent with the restrictions on Licensee in Section 18 of this Agreement.
|
|
|
(iv) |
A requirement of indemnification of Licensors by Sublicensee that is equivalent to the indemnification of Licensors by Licensee under Section 13 of this Agreement.
|
|
|
(v) |
A requirement of obtaining and maintaining insurance by Sublicensee that is equivalent to the insurance requirements of Licensee under Section 13.3 of this Agreement, including coverage under such insurance of Penn and UFRF as provided in
Section 13.3.
|
|
|
(vi) |
Restriction on use of Licensors’ names etc. consistent with Section 14 of this Agreement.
|
|
Timing of Extension Request
|
Amount Spent by Licensee
under this Agreement in the
[***]
Months Prior to Extension
Request
|
|
[***]
|
[***]
|
|
[***]
|
[***]
|
|
[***]
|
[***]
|
|
Event
|
Milestone
Payment
|
|
One-Time Clinical or Regulatory Milestones
|
|
|
[***]
|
[***]
|
|
[***]
|
[***]
|
|
[***]
|
[***]
|
|
[***]
|
[***]
|
|
[***]
|
[***]
|
|
One-Time Worldwide Commercial Milestones on Net Sales
|
|
|
[***]
|
[***]
|
|
[***]
|
[***]
|
|
[***]
|
[***]
|
|
[***]
|
[***]
|
|
Event
|
Milestone
Payment
|
|
One-Time Clinical or Regulatory Milestones
|
|
|
[***]
|
[***]
|
|
[***]
|
[***]
|
|
[***]
|
[***]
|
|
[***]
|
[***]
|
|
[***]
|
[***]
|
|
One-Time Worldwide Commercial Milestones on Net Sales
|
|
|
[***]
|
[***]
|
|
[***]
|
[***]
|
|
[***]
|
[***]
|
|
[***]
|
[***]
|
|
Time period
|
Percentage (%) of
Licensor Allocated
Sublicense Income
payable to Penn, on
behalf of both
Licensors
|
||
|
[***]
|
[***]
|
||
|
[***]
|
[***]
|
||
|
[***]
|
[***]
|
||
|
[***]
|
[***]
|
|
[***]
|
[***]
|
|
If to UFRF:
|
If to Licensee:
|
|
President
University of Florida Research
Foundation, Incorporated
223 Grinter Hall University of Florida
P. O. Box 115500
Gainesville, FL 32611-5500
|
Ophthotech Corporation
One Penn Plaza, Suite 3520
New York, NY 10119
Attention: Legal Department
with a copy to:
WilmerHale LLP
60 State Street
Boston, MA 02109
Attention: [***]
|
|
with a copy to:
|
|
|
Office of Technology Licensing
University of Florida
Attn: Director (Rm. 112)
747 SW 2nd Avenue
Post Office Box 115575
Gainesville, Florida 32611-5575
|
|
|
If to Penn:
|
|
|
Penn Center for Innovation
University of Pennsylvania
3160 Chestnut Street, Suite 200
Philadelphia, PA 19104-6283
Attention: Managing Director
|
|
|
with a copy to:
|
|
|
University of Pennsylvania
Office of General Counsel
2929 Walnut Street, Suite 400
Philadelphia, PA 19104-5509
Attention: General Counsel
|
|
THE TRUSTEES OF THE UNIVERSITY OF PENNSYLVANIA
|
OPHTHOTECH CORPORATION
|
||
|
By:
|
/s/ Benjamin Dibling, Ph.D.
|
By:
|
/s/ Glenn Sblendorio
|
|
Name: Benjamin Dibling, Ph.D.
Title: . Executive Director of Licensing, Penn Center for Innovation
|
Name: Glenn Sblendorio
Title: CEO + President
|
||
|
Date: April 11, 2019
|
Date: April, 2019
|
||
|
UNIVERSITY OF FLORIDA RESEARCH FOUNDATION, INCORPORATED
|
|||
|
By:
|
/s/ Jim O’Connell
|
||
|
Name: Jim O’Connell
Title: Director of Technology Licensing
|
|||
|
Date: 4/11/2019
|
|||
|
|
1. |
The Patent Applications, which are described in the Appendix 1 of this Amendment, and all patent rights arising therefrom constitute Subsequently Added Intellectual Property and Appendix G to the License Agreement is hereby amended
by adding the information on Appendix 1 hereto to Appendix G.
|
|
|
2. |
The following definitions shall be added to Section 1 of the License Agreement in the appropriate alphabetical order:
|
|
|
3. |
The following subsections shall be added to Section 7 of the License Agreement (Patent Prosecution):
|
| (a) |
Penn or UFRF, as applicable, will solicit input from Licensee regarding [***]. Penn or UFRF, as applicable, will submit, or will cause to be submitted to Licensee (or its designated counsel) [***].
|
|
|
(b) |
Penn or UFRF, as applicable, shall maintain the Patent Rights related to Penn Subsequently Added Intellectual Property or UF Subsequently Added Intellectual Property, as applicable, in at least the following countries: [***]. Licensee
shall pay all costs and expenses incurred by Penn or UFRF, as applicable, related to the preparation, filing, prosecution (including interferences and oppositions), issuance, maintenance and reporting of the Patent Rights related to Penn
Subsequently Added Intellectual Property or UF Subsequently Added Intellectual Property, as applicable, in such countries and in any additional countries or jurisdictions in which Penn or UFRF, as applicable, and Licensee mutually agree to
pursue such preparation, filing, prosecution (including interferences and oppositions), issuance, maintenance and reporting, in each case that were not previously reimbursed, within [***] of receipt of an invoice from Penn or UFRF, as
applicable. Licensee shall keep Penn or UFRF, as applicable, fully apprised of the entity status of Licensee and all Sublicensees with respect to United States and applicable foreign patent laws. Licensee shall inform Penn or UFRF, as
applicable, of any changes in writing of the entity status from “small entity” to “large entity” or vice versa with respect to United States and applicable foreign patent laws within [***] of any change.
|
|
|
(c) |
Penn or UFRF, as applicable, shall, with respect to Penn Subsequently Added Intellectual Property or UF Subsequently Added Intellectual Property, as applicable, and any related Patent Rights, remain in compliance with The Patent and
Trademark Law Amendments Act of 1980 (Public Law 96-517; 35 U.S.C. §§ 200-212), including any amendments thereto and all regulations promulgated thereunder, and shall reasonably assist Licensee in complying with Licensee’s obligations under
such law, including, as applicable, that set forth in Section 17.1.
|
|
|
(d) |
Licensee shall have the sole right in electing which of the Patent Rights related to Subsequently Added Intellectual Property shall receive any patent term extension under 35 U.S.C. § 156 in the United States, supplemental protection
certificate in the European Union and similar rights in foreign jurisdictions. Penn or UFRF, as applicable, and Licensee shall cooperate in timely filing and obtaining the patent term extension, supplemental protection certificate and similar
rights for such Patent Right(s) elected by Licensee.
|
|
|
4. |
Reimbursement. Licensee shall pay Penn, within [***] after receipt of an invoice therefor, to reimburse expenses associated with preparation, filing, prosecution, issuance,
maintenance, and reporting of the Patent Rights relating to the Patent Applications incurred prior to the effective date of this Amendment.
|
|
|
5. |
Except as expressly set forth in this Amendment, the License Agreement remains in full force and effect in accordance with its terms.
|
|
|
6. |
Penn and UFRF each consents to Licensee filing a copy of this Amendment with the Securities and Exchange Commission, in accordance with its rules and regulations.
|
|
|
7. |
This Amendment may be executed in counterparts, each of which will be deemed an original, and all of which together will be deemed to be one and the same instrument. A portable document format (PDF) or electronic copy of this Amendment,
including the signature pages, will be deemed an original.
|
|
Licensors:
|
|
TRUSTEES OF THE UNIVERSITY OF PENNSYLVANIA
|
|
By: /s/ Benjamin Dibling, Ph.D.
|
|
Name: Benjamin Dibling, Ph.D.
|
|
Title: Executive Director of Licensing,
|
|
Penn Center for Innovation
|
|
UNIVERSITY OF FLORIDA RESEARCH FOUNDATION, INCORPORATED
|
|
By: /s/ Jim O’Connell
|
|
Name: Jim O’Connell
|
|
Title: Director, UF Innovate ǀ Tech Licensing
|
|
Licensee:
|
|
IVERIC BIO, INC.
|
|
By: /s/ Abraham Scaria, Ph.D.
|
|
Name: Abraham Scaria, Ph.D.
|
|
Title: Chief Scientific Officer
|
|
|
1. |
Payment. Licensee hereby agrees to pay to Penn a
non-creditable, non-refundable fee of [***] within [***] following the execution of this Amendment.
|
|
|
2. |
Section 3.2 First Commercial Sale — Milestones. The
first sentence of Section 3.2(a)(i) of the License Agreement is hereby deleted in its entirety and replaced with the following:
|
|
|
3. |
Appendix F — Milestones. Appendix F-1 is hereby
deleted in its entirety and replaced with the following:
|
|
Diligence Event
|
Achievement Date
|
||
|
[***]
|
[***]
|
||
|
[***]
|
[***]
|
||
|
[***]
|
[***]
|
|
|
4. |
Except as expressly set forth in this Amendment, the License Agreement remains in full force and effect in accordance with its terms.
|
|
|
5. |
Licensors consent to Licensee filing a copy of this Amendment with the U.S. Securities and Exchange Commission, in accordance with its rules and regulations.
|
|
|
6. |
This Amendment may be executed in counterparts, each of which will be deemed an original, and all of which together will be deemed to be one and the same instrument. A portable document
format (PDF) or electronic copy of this Amendment, including the signature pages, will be deemed an original.
|
|
Licensors:
|
|
TRUSTEES OF THE UNIVERSITY OF PENNSYLVANIA
|
|
By: /s/ Benjamin Dibling, Ph. D.
|
|
Name: Benjamin Dibling, Ph. D.
|
|
Title: Deputy Managing Director, Penn Center for Innovation
|
|
UNIVERSITY OF FLORIDA RESEARCH FOUNDATION, INCORPORATED
|
|
By: /s/ Jim O’Connell
|
|
Name: Jim O’Connell
|
|
Title: Director, UF Innovate ǀ Tech Licensing
|
|
License:
|
|
IVERIC BIO GENE THERAPY LLC
|
|
By: /s/ Kieth Westby
|
|
Name: Kieth Westby
|
|
Title: Chief Operating Officer
|
|
LICENSE CONTACT INFORMATION
|
|||
|
Company full legal name and notice address:
IVERIC bio Gene Therapy LLC
8 Sylvan Way
Parsippany, NJ 07054
Attention: [***]
Email: [***]
with a copy to:
Wilmer Cutler Pickering Hale and Dorr LLP
60 State Street
Boston, MA 02109
Attention: [***]
Email: [***]
|
Company primary phone number:
[***]
|
||
|
Company primary fax number:
[***]
|
|||
|
Company contact name:
[***]
|
Contact title:
[***]
|
Contact phone number:
[***]
|
|
|
ASSIGNEE CONTACT INFORMATION
|
|||
|
Company full legal name and notice address:
Opus Genetics Inc.
8 Davis Drive
Durham, NC 27709
Attention: [***]
Email: [***]
with a copy to:
Smith, Anderson, Blount, Dorsett, Mitchell & Jennigan, LLP
Wells Fargo, Capitol Center
150 Fayetteville Street, Suite 2300
Raleigh, NC 27601
Attention: [***]
Email: [***]
|
Company primary phone number:
|
||
|
Company primary fax number:
|
|||
|
Company contact name:
[***]
|
Contact title:
[***]
|
Contact phone number:
[***]
|
|
|
PENN CONTACT INFORMATION
|
||
|
Penn notice address:
University of Pennsylvania
Penn Center for Innovation
3600 Civic Center Blvd, 9th Flr
Philadelphia, PA 19104-6283
Attention: Managing Director
|
Penn primary phone number:
[***]
|
|
|
Company primary fax number:
[***]
|
||
|
Penn Investigator name:
[***]
|
Penn department:
[***]
|
|
|
Payments to Penn shall be made in accordance with Section 6.3 of the License Agreement:
[***]
|
||
|
UFRF CONTACT INFORMATION
|
||
|
UFRF notice address:
University of Florida Research Foundation. Incorporated
223 Grinter Hall, University of Florida
P.O. Box 115500
Gainesville, FL 32611-5500
Attn: President
With a copy to:
Office of Technology Licensing University of Florida
Attn: Director (Rm. 112) 747 SW 2nd Avenue Post Office Box 115575
Gainesville, Florida 32611-5575
|
UFRF primary phone number:
[***]
|
|
|
UFRF primary fax number:
[***]
|
||
|
UFRF Investigator name:
[***]
|
UFRF department:
[***]
|
|
LICENSE AGREEMENT
|
||
|
Patent Docket Numbers:
[***]
|
Effective Date of License:
April 10, 2019
|
|
|
Field of Use:
THERAPIES FOR THE PREVENTION, TREATMENT, CONTROL AND PALLIATION OF HUMAN DISEASES ASSOCIATED WITH THE BEST1 GENE.
|
Amendments/Effective Dates:
Amendment No. 1 May 1, 2020
Amendment No. 2. July 1, 2022
|
|
|
EFFECTIVE DATE OF ASSIGNMENT
|
||
|
Background: Opus is purchasing all or substantially all of the assets of Iveric related to products identified in the License Agreement (as defined below), pursuant to the Asset Purchase Agreement (defined below). The parties with to
provide for the assignment to Opus of all of Iveric’s rights under the License Agreement form and after the date hereof. The parties also desire to amend the License Agreement as provided in Section 5 hereof.
|
Effective Date of Assignment:
December 23, 2022
|
|
|
Assignment Consent Agreement
|
Page 2 of 5
|
|
|
SIGNATURES
|
|
|
This Assignment Consent Agreement and Third Amendment to the Exclusive License Agreement with Know-How includes this Signature Page and all of the attached Terms and Conditions. By signing below, Iveric, Opus, Penn and UFRF agree to all of
the provisions of this Agreement and intend to be bound hereby.
|
|
LICENSEE
IVERIC BIO GENE THERAPY LLC
By: /s/ Tony Gibney
(please sign)
Name: Tony Gibney
Title: Chief Business and Strategy Officer
|
ASSIGNEE
OPUS GENETICS INC.
By: /s/ Ben Yerxa
(please sign)
Name: Ben Yerxa
Title: President & Chief Executive Officer
|
||||||
|
Date:
|
12/23/2022 |
Date:
|
12/23/2022 | ||||
|
THE TRUSTEES OF THE UNIVERSITY OF PENNSYLVANIA
By: /s/ Benjamin Dibling
(please sign)
Name: Benjamin Dibling
Title: Deputy Managing Director
|
THE UNIVERSITY OF FLORIDA RESEARCH FOUNDATION INCORPORATED
By: /s/ Jim O’Connell
(please sign)
Name: Jim O’Connell
Title: Director, UF Innovate ǀ Licensing
|
||||||
|
Date:
|
12/23/2022 |
Date:
|
12/23/2022 | ||||
|
Assignment Consent Agreement
|
Page 3 of 5
|
|
|
1. Defined Terms. Capitalized terms used but not defined in this Assignment Agreement are defined in the Exclusive License
Agreement with Know-How, by and among Penn, UFRF and Iveric, dated as of April 10, 2019, as amended by Amendment No. 1, dated May 1, 2020, and Amendment No. 2, dated July 1, 2022 (the “License Agreement”).
|
(c) Anything in this Assignment Agreement to the contrary notwithstanding, in no event shall Iveric be released from any liabilities under, or failure to perform its
obligations pursuant to, the License Agreement prior to the Effective Date, and Iveric shall remain liable to Licensors for all such liabilities and obligations.
|
|||
|
2. Assignment and Consent. As of the Effective Date:
(a) Pursuant to the Asset Purchase Agreement between Iveric and Opus effective as of the Effective Date (the “Asset Purchase Agreement”),
Iveric has assigned to Opus all of Iveric’s right, title and interest in, to and under (i) the License Agreement, and Opus has assumed all of Opus’ obligations under the License Agreement from and after the Effective Date, (ii) the Penn SRA,
dated October 30, 2018, as amended by Amendment No. 1, dated October 1, 2019 and Amendment No. 2, dated October 31, 2022, the “Sponsored Research Agreement”, and collectively with the License Agreement,
the “Assigned Agreements”). The Licensors, on behalf of their respective institutions, hereby (x) consent to the assignment and assumptions of the License Agreement and (y) Penn acknowledges and agree
to the assignment of the Sponsored Research Agreements.
(b) In furtherance of the foregoing, effective as of the Effective Date:
(i) Opus hereby accepts, and agrees to assume, all of Iveric’s right, title and interest in, to and under
the Assigned Agreements;
(ii) Opus will become a party to the Assigned Agreements and will succeed to all of the rights and assume all
of the obligations of Licensee thereunder;
(iii) all references to “Licensee” in the License Agreement will refer to Opus; and
(iv) all references to “Sponsor” in the Sponsored Research Agreements will refer to Opus.
(b) The Patent Applications, which are described in Appendix 1 of this Assignment Agreement, and all patent rights arising therefrom constitute Subsequently Added Intellectual Property and
Appendix G to the License Agreement is hereby deleted in its entirety and replaced with Appendix 1..
All other terms and provisions of the License Agreement, except as expressly amended by this Section 5, remain in full force and effect.
6. Miscellaneous. Any notice must be in writing and sent to the address of the party listed on the Signature Page. The
parties do not intend that any agency or partnership relationship be created by this Assignment Agreement. This Assignment Agreement may only be modified by a written amendment that is executed by an authorized representative of each party
. Any waiver must be express and in writing. No waiver by a party of a breach by another party will constitute a waiver of any different or succeeding breach. This Assignment Agreement will be governed by and construed in accordance with
the laws of the Commonwealth of Pennsylvania without regard to conflicts of law principles of any jurisdiction.
|
3. Representations and Warranties. Each party to this Assignment Agreement represents and warrants to each other that the
person executing this Assignment Agreement on its behalf has all necessary power and authority to do so, and that upon such execution, this Assignment Agreement is a legal, valid and binding obligation enforceable against such party.
4. Consideration. In consideration for the Licensors’ consent to the assignment of the License Agreement from Iveric to
Opus, Opus agrees to pay Penn:
(a) [***], which payment will be made on the Effective Date;
(b) [***] of each milestone payment that it is required to pay to Iveric under Section 3.3 of the Asset Purchase Agreement, with
each such payment due to Penn within [***] of achieving the milestone and in addition to any amounts that may be owed to Penn under the License Agreement for the same milestone.
5. Amendments to License Agreement. The Licensors and Opus agree that the License Agreement shall be amended as follows, as of the Effective Date:
(a) Appendix F-1 of the License Agreement — Milestones — Diligence Events for Wildtype Only Products shall be amended by deleting the contents of such table and replacing them with the
following:
[***]
This Assignment Agreement, the Asset Purchase Agreement, the License Agreement (as amended hereby) and the Sponsored Research Agreements contain the entire agreement between the parties with
respect to subject matter of this Assignment Agreement and supersede all other oral or written representations, statements, or agreements with respect to such subject matter. This Assignment Agreement is binding upon the parties and their
respective heirs, successors, assigns, and personal representatives. No party may assign this Assignment Agreement without the prior written consent of the other parties. This Assignment Agreement may be signed in counterparts which, taken as
a whole, will constitute one agreement.
|
|
Assignment Consent Agreement
|
Page 4 of 5
|
|
|
Assignment Consent Agreement
|
Page 5 of 5
|
|
|
|
1. |
Appendix F — Milestones. Appendix F-1 is hereby
deleted in its entirety and replaced with the following:
|
|
Diligence Event
|
Achievement Date
|
|
[***]
|
[***]
|
|
[***]
|
[***]
|
|
[***]
|
[***]
|
|
|
2. |
Except as expressly set forth in this Amendment No. 4, the License Agreement remains in full force and effect in accordance with its terms.
|
|
|
3. |
This Amendment No. 4 may be executed in counterparts, each of which will be deemed an original, and all of which together will be deemed to be one and the same instrument. A portable
document format (PDF) or electronic copy of this Amendment No. 4, including the signature pages, will be deemed an original. The parties hereby agree that this Amendment No. 4 may be executed with electronic signatures and shall be valid and
binding on the parties to the extent electronically signed.
|
|
Licensors:
|
|
TRUSTEES OF THE UNIVERSITY OF PENNSYLVANIA
|
|
By: /s/ Benjamin Dibling, Ph.D.
|
|
Name: Benjamin Dibling, Ph.D.
|
|
Title: Associate Vice Provost for Research and Managing Director, Penn Center for Innovation
|
|
UNIVERSITY OF FLORIDA RESEARCH FOUNDATION, INCORPORATED
|
|
By: /s/ Jim O’Connell
|
|
Name: Jim O’Connell
|
|
Title: Director, UF Innovate | Tech Licensing
|
|
Licensee:
|
|
OPUS GENETICS INC.
|
|
By: /s/ Ben Yerxa
|
|
Name: Ben Yerxa
|
|
Title: Chief Executive Officer
|
|
Page
|
|
|
ARTICLE 1 DEFINITIONS
|
2
|
|
ARTICLE 2 LICENSES AND OTHER RIGHTS
|
6 |
|
ARTICLE 3 DILIGENCE
|
8 |
|
ARTICLE 4 FINANCIAL PROVISIONS
|
10 |
|
ARTICLE 5 INTELLECTUAL PROPERTY
|
15 |
|
ARTICLE 6 REPRESENTATIONS, WARRANTIES AND COVENANTS
|
18 |
|
ARTICLE 7 INDEMNIFICATION; INSURANCE AND LIMITATION OF LIABILITY
|
20 |
|
ARTICLE 8 TERM AND TERMINATION
|
23 |
|
ARTICLE 9 ADDITIONAL PROVISIONS
|
24 |
|
EXHIBIT A-1
|
PENN PATENT RIGHTS
|
|
EXHIBIT A-2
|
LICENSED INFORMATION
|
|
EXHIBIT B-1
|
DILIGENCE EVENTS
|
|
EXHIBIT B-2
|
MILESTONES
|
|
EXHIBIT B-3
|
ROYALTIES
|
|
EXHIBIT B-4
|
MINIMUM ANNUAL ROYALTIES
|
|
EXHIBIT B-5
|
PENN SUBLICENSE INCOME
|
|
APPENDIX I
|
EQUITY ISSUANCE AGREEMENT
|
|
APPENDIX II
|
FORM OF FINANCIAL REPORT
|
|
APPENDIX III
|
CLIENT & BILLING AGREEMENT
|
|
APPENDIX IV
|
DEVELOPMENT PLAN
|
|
APPENDIX V
|
SUBLICENSE DEVELOPMENT REPORT
|
| 1.1 |
“Achievement Date” means, with respect to a Diligence Event, the corresponding date such Diligence Event is to be achieved as provided in Exhibit B-1 attached hereto subject to modification pursuant to Section 3.3 below.
|
| 1.2 |
“Affiliate” means a Person that controls, is controlled by or is under common control with a Party, but only for so long as such control exists. For the
purposes of this Section 1.2, the word “control” (including, with correlative meaning, the terms “controlled by” or “under the common control with”) means the actual power, either directly or indirectly through one or more intermediaries, to
direct the management and policies of such Person or entity, whether by the ownership of more than fifty percent (50%) of the voting stock of such entity, or by contract or otherwise.
|
| 1.3 |
“Commercially Reasonable Efforts” means [***].
|
| 1.4 |
“Confidential Information” of a Party, means (i) information relating to the business, operations or products of a Party or any of its Affiliates, including
any know-how, that such Party has disclosed to the other Party under the Original Agreement or discloses to the other Party under this Agreement, or otherwise becomes known to the other Party by virtue of this Agreement, and (ii) the terms of
this Agreement; provided that Confidential Information shall not include information that:
|
|
|
(a) |
is or becomes generally available to the public other than as a result of disclosure by the recipient;
|
|
|
(b) |
is already known by or in the possession of the recipient at the time of disclosure by the disclosing Party;
|
|
|
(c) |
is independently developed by recipient without use of or reference to the disclosing Party’s Confidential Information; or
|
|
|
(d) |
is obtained by recipient from a Third Party that has not breached any obligations of confidentiality.
|
| 1.5 |
“Controlled” means, with respect to intellectual property rights, that a Party or one of its Affiliates owns or has a license or sublicense to such
intellectual property rights and has the ability to provide to, grant a license or sublicense to, or assign its right, title and interest in and to, such intellectual property rights as provided for in this Agreement without violating the
terms of any agreement or other arrangement with any Third Party.
|
| 1.6 |
“Development Plan” means the development plan provided by Licensee to Penn that provides the activities, and the associated timelines of when such activities
shall be conducted (including in detail the activities that shall be conducted in the calendar year following the submission of such Development Plan to Penn), in order to develop a Product for commercialization. The initial Development Plan
is attached hereto as Appendix IV.
|
| 1.7 |
“Diligence Event” means each of the events that Licensee is expected to accomplish in the development of a Product as provided in Exhibit B-1 attached hereto.
|
| 1.8 |
“Field of Use” means all fields of use.
|
| 1.9 |
“First Commercial Sale” means, on a country-by-country basis, the first commercial transfer or disposition for value of Product in such country to a Third
Party by Licensee, or any of its Affiliates or Sublicensees, in each case, after all Governmental Approvals have been obtained for such country.
|
| 1.10 |
“GAAP” means United States generally accepted accounting principles applied on a consistent basis.
|
| 1.11 |
“Governmental Approval” means, with respect to a Product in a country or region, the approval, clearance, license, registration or authorization (including but
not limited to emergency use authorization) by the relevant Governmental Body, if applicable, for the commercialization of such Product in such country.
|
| 1.12 |
“Governmental Body” means any: (a) nation, principality, state, commonwealth, province, territory, county, municipality, district or other jurisdiction of any
nature; (b) federal, provincial, state, local, municipal, foreign or other government; (c) governmental or quasi-governmental authority of any nature (including any governmental division, subdivision, department, agency, bureau, branch,
office, commission, council, board, instrumentality, officer, official, representative, organization, unit, body or entity and any court or other tribunal); (d) multi-national or supranational organization or body; or (e) individual, entity,
or body exercising, or entitled to exercise, any executive, legislative, judicial, administrative, regulatory, police, military or taxing authority or power of any nature.
|
| 1.13 |
“Intellectual Property” means the Penn Patent Rights.
|
| 1.14 |
“Law” or “Laws” means all applicable laws, statutes, rules, regulations,
ordinances and other pronouncements having the binding effect of law of any Governmental Body.
|
| 1.15 |
“Licensed Information” means [***].
|
| 1.16 |
“Limelight” means Limelight Bio, Inc.
|
| 1.17 |
“Net Sales” means [***].
|
| 1.18 |
“Patent Rights” means any of the following, whether existing now or in the future anywhere in the world: issued patent, including inventor’s certificates,
substitutions, extensions, confirmations, reissues, re-examination, renewal or any like governmental grant for protection of inventions, and any pending application for any of the foregoing.
|
| 1.19 |
“Penn Patent Rights” means (a) the Patent Rights listed in Exhibit A-1 Controlled by Penn as of the Effective
Date, (b) any continuations, provisionals, continued prosecution applications, substitutions, extensions and term restorations, registrations, confirmations, reexaminations, renewals or reissues thereof, including divisions, but excluding
continuations-in-part except to the extent of claims entirely supported in the specification and entitled to the priority date of the parent application, and (c) any corresponding foreign Patent Rights to the foregoing. Notwithstanding the
above, Penn Patent Rights does not include the Carve-Out Patent Rights.
|
| 1.20 |
“Person” means any natural person, corporation, firm, business trust, joint venture, association, organization, company, partnership or other business entity,
or any government or agency or political subdivision thereof.
|
| 1.21 |
“Product” means any (a) process, service or method covered by a Valid Claim or whose use or practice would, absent the License, constitute an infringement,
inducement of infringement or contributory infringement of any Valid Claim (“Method”), (b) article, composition, apparatus, substance, chemical or any other
material covered by a Valid Claim or whose manufacture, import, use offer for sale or sale would, absent the License, constitute an infringement, inducement of infringement or contributory infringement of any Valid Claim; or (c) service,
article, composition, apparatus, chemical, substance or any other material imported, made, used or sold by or utilizing or practicing a Method or (d) product, service, method, article, composition, apparatus, substance, chemical or any other
material which incorporates, consists of, makes use of or is made through use of or could not be made but for the use of Licensed Information.
|
| 1.22 |
“Product Category” means either (a) any Products directed towards treatment or correction of mutation of LCA5 (the “LCA5 Products”), or (b) any Products directed towards treatment or correction of mutation of RDH12 (the “RDH12 Products”). [***]
|
| 1.23 |
“Sale” means [***].
|
| 1.24 |
“Sublicensee” means a Person (including any Affiliate) to which a Sublicense is granted pursuant to the terms of Section 2.4. For further clarity, Sales by
Licensee, Affiliate or Sublicensee to a wholesaler or distributor other than for the account of the Licensee, Affiliate or Sublicensee or under a Sublicense are prohibited. In the event that Licensee wishes to amend this Agreement to permit
Sales by distributors other than for the account of Licensee, Licensee shall notify Penn in writing and Penn will promptly consider such request in good faith.
|
| 1.25 |
“Sublicense Documents” means any and all written agreements, amendments or written understandings entered into with a Sublicensee (including any of its
Affiliates) that are directly related to a Sublicense, Penn Patent Rights or Product. For clarity, a development agreement or distribution agreement for a Product is a Sublicense Document.
|
| 1.26 |
“Sublicense Income” means [***].
|
| 1.27 |
“Tax” means all taxes, duties, fees, premiums, assessments, imposts, levies, rates, withholdings, dues, government contributions and other charges of any kind
whatsoever, whether direct or indirect, together with all interest, penalties, fines, additions to tax or other additional amounts, imposed by any Governmental Body.
|
| 1.28 |
“Third Party” means any Person other than Penn, Licensee or any of their respective Affiliates.
|
| 1.29 |
“Third Party Royalties” means [***].
|
| 1.30 |
“United States” or “US” means the United States of America, its territories and
possessions.
|
| 1.31 |
“USD” or “$” means the lawful currency of the United States of America.
|
| 1.32 |
“Valid Claim” means a claim of (a) an issued and unexpired patent in Penn Patent Rights which claim has not been revoked or held unenforceable or invalid by a
decision of a court or governmental agency of competent jurisdiction from which no further appeal can be taken or has been taken within the time allowed for appeal, and has not been abandoned, disclaimed, denied or admitted to be invalid or
unenforceable through reissue or disclaimer; or (b) a pending patent application that is included in Penn Patent Rights which was filed and is being prosecuted in good faith, and has not been abandoned or finally disallowed without the
possibility of appeal or re-filing of the application (each a “Pending Patent Application”).
|
| 1.33 |
Other Terms. The definition of each of the following terms is set forth in the section
of the Agreement indicated below:
|
|
Defined Term
|
Section
|
|
Adjustment Equity
|
4.2.2(a)
|
|
Advance Payment
|
5.2.3
|
|
Agreement
|
Preamble
|
|
Bankruptcy Action
|
0
|
|
Carve-Out Patent Rights
|
3.3.2
|
|
Common Equity
|
4.2.1
|
|
Effective Date
|
Preamble
|
|
Financial Report
|
4.8
|
|
Historic Patent Costs
|
5.2.1
|
|
Infringement Notice
|
5.4.1
|
|
Inventor(s)
|
Recitals
|
|
License
|
2.1
|
|
Licensee
|
Preamble
|
|
Maintenance Fee
|
4.3
|
|
Method
|
1.21
|
|
Milestone
|
4.4.1
|
|
Milestone Payment
|
4.4.1
|
|
Minimum Annual Royalty
|
4.5.3
|
|
Ongoing Patent Costs
|
5.2.2
|
|
Parties
|
Preamble
|
|
Party
|
Preamble
|
|
Patent Costs
|
5.2.1
|
|
Patent Counsel
|
5.1.1
|
|
Patent Termination Notice
|
5.3
|
|
Penn
|
Preamble
|
|
Penn Indemnitees
|
7.1.1.1
|
|
Penn Sublicense Income
|
4.6
|
|
Progress Report
|
3.4.1
|
|
Prosecution Request
|
5.1.2
|
|
Royalty
|
4.5
|
|
Sublicense
|
2.4.1
|
|
Term
|
8.1
|
| 2.1 |
Grant of License. Subject to the terms and conditions of this Agreement, Penn hereby
grants to Licensee (i) an exclusive, royalty-bearing right and license (with the right to sublicense through multiple tiers as provided in, and subject to, the provisions of Section 2.4) under Penn Patent Rights, in all jurisdictions where
Penn Patent Rights exist (“Exclusive License”) and (ii) a non-exclusive, royalty-bearing right and license (with the limited right to sublicense as provided in and subject to, the provisions of Section 2.4) to Licensed Information (“Non-Exclusive License”), to make, have made, use, sell, offer for sale and import Product in the Field of Use during the Term (the Exclusive License and
Non-Exclusive License, collectively referred to hereafter as “License”).
|
| 2.2 |
Retained Rights. Notwithstanding the License, Penn retains the right under Penn Patent
Rights to: (a) conduct educational, research and clinical activities itself and (b) authorize non-commercial Third Parties to conduct educational, research and clinical activities. For clarity, Penn retains the right to use and authorize
Third Parties to use Licensed Information for any purpose.
|
| 2.3 |
U.S. Government Rights. The License is expressly subject to all applicable provisions of any license to the United States Government executed by Penn and is subject to any overriding obligations to the United States Federal
Government under 35 U.S.C. §§200-212, applicable governmental implementing regulations, and the U.S. Government sponsored research agreement or other guidelines, including that products that result from intellectual property funded by the
United States Federal Government that are sold in the United States be substantially manufactured in the United States. In the event that Licensee believes in good faith that substantial manufacture of such product is not commercially
feasible in the United States and makes a request to Penn in writing to assist in obtaining a waiver of such requirement from the United States Government, then Penn shall, at the expense of Licensee, use reasonable efforts to assist in
obtaining such waiver.
|
| 2.4 |
Grant of Sublicense by Licensee.
|
| 2.4.1 |
Penn grants to Licensee the right to grant sublicenses through multiple tiers, in whole or in part, under the License (each, a “Sublicense”) subject to the
terms and conditions of this Agreement and specifically this Section 2.4. For clarity, a sublicense to the Non-Exclusive License may only be granted in conjunction with a sublicense to the Exclusive License. The term Sublicense shall include
any grant of rights under the License by a Sublicensee to any downstream third party, such downstream third party shall also be considered a Sublicensee for purposes of this Agreement.
|
| 2.4.2 |
All Sublicenses will (i) be issued in writing, (ii) to the extent applicable, include all of the rights of Penn and require the performance of obligations due to Penn (and, if applicable, the U.S. Government under 35 U.S.C. §§200-212)
contained in this Agreement and (iii) include no less than the following terms and conditions:
|
|
|
(a) |
Reasonable record keeping, audit and reporting obligations sufficient to enable Licensee and Penn to reasonably verify the payments due to Licensee and Penn under such Sublicense and to reasonably monitor such Sublicensee’s progress in
developing and/or commercializing Product, provided that such obligations shall be no less stringent than those provided in this Agreement for Licensee.
|
|
|
(b) |
Infringement and enforcement provisions that do not conflict with the restrictions and procedural requirements imposed on Licensee and do not provide greater rights to Sublicensee than as provided in Section 5.4.
|
|
|
(c) |
Confidentiality provisions with respect to Confidential Information of Penn consistent with the restrictions on Licensee in Section 5.6 of this Agreement.
|
|
|
(d) |
Covenants by Sublicensee that are equivalent to those made by Licensee in Section 6.4.
|
|
|
(e) |
A requirement of indemnification of (x) Penn by Sublicensee that is equivalent to the indemnification of Penn by Licensee under Section 7.1.1 of this Agreement, and (y) Limelight by Sublicensee that is equivalent to the indemnification of
Limelight by Licensee under Section 7.1.2 of this Agreement.
|
|
|
(f) |
A requirement of obtaining and maintaining insurance by Sublicensee that is equivalent to the insurance requirements of Licensee under Section 7.2 of this Agreement, including coverage under such insurance of Penn as provided in Section
7.2.
|
|
|
(g) |
Restriction on use of Penn’s names etc. consistent with Section 9.4 of this Agreement.
|
|
|
(h) |
A requirement of antidiscrimination by Sublicensee no less stringent than that provided in Section 9.5 of this Agreement.
|
|
|
(i) |
A requirement that Penn is a third party beneficiary of such Sublicense; and that Limelight is a third party beneficiary of such Sublicense.
|
|
|
(j) |
[***]
|
| 2.4.3 |
Within [***] of the execution of a Sublicense Document, Licensee shall provide a complete and accurate copy of such Sublicense Document to Penn, in the English Language. Notwithstanding the foregoing, Licensee may redact any such
Sublicense Document only with respect to technology other than the Penn Patent Rights and to the extent necessary to preserve the confidentiality of confidential information of Sublicensee, provided that sufficient information remains
unredacted to allow Penn to assess whether Licensee and Sublicensee are in compliance with the terms and conditions of this Agreement and to verify amounts owed to Penn in connection with such Sublicense, provided that upon written request of
Penn, Licensee shall promptly provide a complete and accurate copy of such Sublicense Document to Penn, in the English Language. Penn’s receipt of a Sublicense Document, however, will constitute neither an approval nor disapproval of the
Sublicense Document nor a waiver of any right of Penn or obligation of Licensee under this Agreement.
|
| 2.4.4 |
Licensee shall provide an annual Sublicense Development Report on or before December 1 of each year during the Term (“SDR Report”) a form of which is attached
hereto as Appendix V.
|
| 2.5 |
No Implied License. Each Party acknowledges that the rights and licenses granted in
this Agreement are limited to the scope expressly granted. Accordingly, except for the rights expressly granted under this Agreement, no right, title, or interest of any nature whatsoever is granted whether by implication, estoppel,
reliance, or otherwise, by either Party to the other Party. All rights with respect to any know-how, patent or other intellectual property rights that are not specifically granted herein are reserved to the owner thereof.
|
| 3.1 |
Development Plan. No later than December 1 of each year during the Term, Licensee
shall submit an updated Development Plan, which shall include amendments and revisions to any long term development activities and detailed activities to be conducted in the following calendar year, provided that such updated Development
Plan for a given Product shall not be due after the First Commercial Sale of that Product in the United States. Notwithstanding the foregoing, Licensee shall provide to Penn such other information as is requested by a Governmental Body that
is required pursuant to Law, including 37 C.F.R. 401.
|
| 3.2 |
General Diligence. Licensee shall use Commercially Reasonable Efforts to develop and
commercialize a Product in the Field of Use. The efforts of an Affiliate or Sublicensee shall be considered the efforts of Licensee.
|
| 3.3 |
Diligence Events.
|
| 3.3.1 |
Licensee shall achieve each Diligence Event as set forth in Exhibit B-1 by the corresponding Achievement Date. Licensee may extend any Achievement Date for a Diligence Event by [***] at no
cost. After [***], Licensee may extend any Achievement Date for a Diligence Event by [***], but not more than [***] per Diligence Event, by making a [***] payment to Penn prior to the expiration of the Achievement Date for each such Diligence
Event. Beyond the [***] extensions, Licensee will be permitted [***] more increments of [***] per Diligence Event, by making a [***] payment to Penn prior to expiration of the Achievement Date for each such Diligence Event. Notwithstanding
the foregoing, the Achievement Dates for the Diligence Events relating to Licensee equity funding may not be extended by Licensee without the prior written consent of Penn.
|
| 3.3.2 |
Penn’s sole and exclusive remedy with respect to Licensee’s failure to achieve a Diligence Event by the corresponding Achievement Date and extensions per Section 3.3.1 shall be its right to terminate the License with respect to the Penn
Patent Rights corresponding to the Product Category for which the Licensee failed to achieve any such Diligence Event. Penn may terminate such Penn Patent Rights, upon written notice, with immediate effect. Such patent application and
patent(s) will thereafter not be part of the Penn Patent Rights (“Carve-Out Patent Rights”) and therefore not subject to this Agreement, including the License,
and Licensee will have no further rights to license them. Licensee shall cease using such Penn Patent Rights effective immediately. Penn may continue prosecution and maintenance of such Carve-Out Patent Rights in its sole discretion and use
and otherwise dispose of such rights without any further obligation to Licensee.
|
| 3.4 |
Progress Reports.
|
| 3.4.1 |
So long as Licensee continues to develop Products, Licensee on an annual basis, but in no event later than December 1st of each calendar year, shall submit to Penn a progress report (each, a “Progress
Report”) covering Licensee’s (and any Affiliates’ and Sublicensees’) activities related to the development of all Products and the obtaining of Governmental Approvals necessary for
commercialization of Products.
|
| 3.4.2 |
Each Progress Report must include all of the following for each annual period:
|
| 4.1 |
Issue Fee. [***]
|
| 4.2 |
Equity Issuance. As additional consideration for the License:
|
| 4.2.1 |
To the extent not previously issued under the Original Agreement, Licensee shall, within [***] of the Effective Date, and subject to Penn’s execution and delivery to Licensee of an Equity Issuance Agreement in substantially the form
attached hereto as Appendix I, issue to Penn shares of common stock of Licensee (“Common Equity”), which is
equal to [***] of stock of Licensee outstanding on a fully diluted basis as of the Original Agreement Effective Date, assuming the exercise, conversion and exchange of all outstanding securities of the Licensee for or into shares of Common
Equity of Licensee. To the extent not previously delivered under the Original Agreement, Licensee shall also within [***] of the Effective Date deliver a stock certificate in the name of Penn reflecting the Common Equity.
|
|
|
(a) |
Licensee shall also issue additional Common Equity to Penn (“Adjustment Equity”) until such time as [***] has been raised by Licensee in Net Proceeds from the
sales of equity securities of the Licensee or securities convertible into equity (which shall not include Licensee’s Series A Preferred Stock), so that after issuance of the Adjustment Equity, Penn still owns [***] of stock of Licensee
outstanding on a fully diluted basis. [***].
|
|
|
(b) |
Licensee shall also issue additional Adjustment Equity to Penn until such time as [***] has been raised by Licensee in Net Proceeds from the sales of equity securities of the Licensee, so that after issuance of the Adjustment Equity, Penn
still owns [***] of stock of Licensee outstanding on a fully diluted basis.
|
|
|
(c) |
Penn shall be issued Adjustment Equity within [***] after any issuance of stock or stock equivalent by Licensee. In all such adjustments, any increase in the number of shares of stock reserved for any option plan for employees,
consultants, directors and so forth authorized in connection with a financing shall be deemed to have been authorized prior to the sale of securities. For clarity, Adjustment Equity shall be issued at no additional consideration from Penn to
Licensee and Licensee shall within [***] of issuance of Adjustment Equity deliver a stock certificate in the name of Penn reflecting the Adjustment Equity.
|
| 4.3 |
License Maintenance Fee. As further consideration for the License, Licensee will pay
an annual maintenance fee (“Maintenance Fee”) of [***]. The Maintenance Fee will not be due and payable on any anniversary of the Original Agreement Effective
Date if on that date Licensee is commercially selling Product and paying an earned royalty to Penn on the Sales of that Product. For clarity, the Maintenance Fee is non-refundable, is not an advance against royalties due to Penn or any
other amounts due to Penn.
|
| 4.4 |
Milestone Payments.
|
| 4.4.1 |
As additional consideration for the License, Licensee will pay Penn the milestone payments (each, a “Milestone Payment”) provided in Exhibit B-2 attached hereto upon the first Product within each Product Category to achieve the corresponding milestone (each, a “Milestone”), whether
achieved by Licensee or an Affiliate or Sublicensee. Licensee shall promptly notify Penn in writing of the achievement of any such Milestone and Licensee shall pay Penn in full the corresponding Milestone Payment within [***] of such
achievement. For clarity, each Milestone Payment is non-refundable, is not an advance against royalties due to Penn or any other amounts due to Penn.
|
| 4.4.2 |
Each time a Milestone is achieved with respect to a particular Product Category, then any other Milestone Payments with respect to earlier Milestones for such Product Category that have not yet been paid will be due and payable together
with the Milestone Payment for the Milestone that is actually achieved.
|
| 4.4.3 |
For clarity, milestones are due and payable on Products and on products that, upon approval of the applicable Governmental Body, would become Products.
|
| 4.5 |
Royalties.
|
| 4.5.1 |
[***]
|
| 4.5.2 |
Licensee must pay Royalties owed to Penn on a calendar quarter basis on or before the following dates:
|
|
|
(a) |
[***] for any Sales that took place on or before the last day of the calendar quarter ending December 31, of the prior year;
|
|
|
(b) |
[***] for any Sales that took place on or before the last day of the calendar quarter ending March 31 of such calendar year;
|
|
|
(c) |
[***] for any Sales that took place on or before the last day of the calendar quarter ending June 30 of such calendar year; and
|
|
|
(d) |
[***] for any Sales that took place on or before the last day of the calendar quarter ending September 30 of such calendar year.
|
| 4.5.3 |
Licensee shall pay to Penn the minimum annual royalties (“Minimum Annual Royalty”) provided in Exhibit B-4
attached hereto during each of the following calendar years after the year in which the First Commercial Sale occurred in any country. Licensee will pay the Minimum Annual Royalty on January 15th of each calendar year it is due, provided that
the Minimum Annual Royalty paid for a calendar year shall be credited solely toward Royalties due in such calendar year.
|
| 4.5.4 |
If Licensee is obligated to pay Third Party Royalties, then Licensee may deduct [***] of such consideration paid to such Third Party, including royalties, for a license under such Patent Rights from any Royalties due under this Agreement,
provided that:
|
|
|
(a) |
[***]
|
|
|
(b) |
In no event shall all Royalties due to Penn in any reporting period be so reduced by more than [***] of the amount that would otherwise be due to Penn under this Agreement.
|
| 4.6 |
Penn Sublicense Income. Licensee will pay to Penn a percentage of Sublicense Income as
provided in Exhibit B-5 attached hereto (“Penn Sublicense Income”). Licensee will make such payment to Penn on
or before the following dates:
|
| 4.6.1 |
[***] for any Sublicense Income received by Licensee on or before the last day of the calendar quarter ending December 31, of the prior year;
|
| 4.6.2 |
[***] for any Sublicense Income received by Licensee on or before the last day of the calendar quarter ending March 31 of such calendar year;
|
| 4.6.3 |
[***] for any Sublicense Income received by Licensee on or before the last day of the calendar quarter ending June 30 of such calendar year; and
|
| 4.6.4 |
[***] for any Sublicense Income received by Licensee on or before the last day of the calendar quarter ending September 30 of such calendar year.
|
| 4.7 |
Mode of Payment and Currency. All payments to Penn hereunder shall be made by deposit
of USD in the requisite amount to the “The Trustees of the University of Pennsylvania” and will be made by delivery to any one of the following:
|
|
[***]
|
| 4.8 |
Royalty and Penn Sublicense Income Reports. Within [***] after the end of each calendar quarter [***], Licensee shall deliver
to Penn a report (“Financial Report”) setting out all details necessary to calculate the Royalty and Penn Sublicense Income due under this Article 4 for such
calendar quarter, including:
|
| 4.9 |
Late Payments. In addition to any other remedies available to Penn, including the
right to terminate this Agreement, any failure by Licensee to make a payment within [***] after the date when due shall obligate Licensee to pay computed interest, the interest period commencing on the due date and ending on the actual
payment date, to Penn at a rate per annum equal to [***], or the highest rate allowed by Law, whichever is lower.
|
| 4.10 |
Default Payment. In the event of default in payment of any payment owing to Penn under
the terms of this Agreement, and if it becomes necessary for Penn to undertake legal action to collect said payment, Licensee shall pay reasonable, documented out-of-pocket legal fees and costs incurred in connection therewith.
|
| 4.11 |
Accounting. Each Party shall calculate all amounts, and perform other accounting
procedures required, under this Agreement and applicable to it in accordance with GAAP.
|
| 4.12 |
Books and Records. Licensee will keep accurate books and records of all Products
developed, manufactured, used or sold and all Sublicenses, collaboration agreements and joint venture agreements entered into by Licensee that involved Penn Patent Rights. Licensee will preserve these books and records for at least [***]
from the date of the Financial Report to which they pertain. Upon reasonable notice, key personnel, books and records will be made reasonably available and will be open to examination by representatives or agents of Penn during regular
office hours to determine their accuracy and assess Licensee’s compliance with the terms of this Agreement, provided that Licensee shall not have an obligation to provide access more than [***] in any given [***] period.
|
| 4.13 |
Audits. In addition to the right of Penn to examine the books and records and
interview key personnel as provided in Section 4.12 above, Penn, at its own cost, through an independent auditor reasonably acceptable to Licensee (and who has executed an appropriate confidentiality agreement reasonably acceptable to
Licensee that requires the auditor to keep any information learned by it confidential except as needed to report its audit conclusions to Penn), may inspect and audit the relevant records of Licensee pertaining to the calculation of any
Milestones, Royalties and Penn Sublicense Income due to Penn under this Agreement. Licensee shall provide such auditors with access to the records during reasonable business hours. Such access need not be given to any such set of records
[***] after the date of any report to be audited. Penn shall provide Licensee with written notice of its election to inspect and audit the records related to the Milestones and Royalties due hereunder not less than [***] prior to the
proposed date of review of Licensee’s records by Penn’s auditors. Should the auditor find any underpayment of Milestones, Royalties or Penn Sublicense Income by Licensee, Licensee shall (a) promptly pay Penn the amount of such underpayment;
(b) shall reimburse Penn for the cost of the audit, if such underpayment equals or exceeds the higher of (i) [***] or (ii) [***]; and (c) provide such auditors with an audit right exercisable within [***] after Penn receives the audit
report. If the auditor finds overpayment by Licensee, then Licensee shall have the right to deduct the overpayment from any future milestones or royalties due to Penn by Licensee or, if no such future milestones or royalties are payable,
then Penn shall refund the overpayment to Licensee within [***] after Penn receives the audit report. Licensee may designate competitively sensitive information which such auditor may see and review but which it may not disclose to Penn;
provided, however, that such designation shall not restrict the auditor’s investigation or conclusions.
|
| 4.14 |
Taxes. All payments made by Licensee to Penn under the Agreement shall be made free
and clear of and without any deduction for or on account of any Taxes on or with respect to such payments.
|
| 5.1 |
Patent Filing Prosecution and Maintenance.
|
| 5.1.1 |
Penn Patent Rights will be held in the name of Penn and obtained with counsel selected by Penn and reasonably acceptable to Licensee (“Patent Counsel”). Penn
shall control all actions and decisions with respect to the filing, prosecution and maintenance of Penn Patent Rights and will consider any reasonable comments or suggestions by Licensee with respect to same. Penn will instruct Patent Counsel
to copy Licensee on all correspondence related to Penn Patent Rights (including copies of each patent application, office action, response to office action, request for terminal disclaimer, and request for reissue or reexamination of any
patent or patent application) and to interact with Licensee with respect to the preparation, filing, prosecution and maintenance of Penn Patent Rights. Penn has the right to take action to preserve rights and minimize cost whether or not
Licensee has commented, and will use reasonable efforts to not allow any Penn Patent Rights for which Licensee is licensed and is underwriting the costs to lapse or become abandoned without Licensee’s written authorization under this
Agreement, except for filing of continuations, divisionals, or the like that substitute for the lapsed application, provided that, Penn shall have no requirement to file, prosecute, or maintain Penn Patent Rights if Licensee is not current
with the Patent Cost obligations as set forth in this Agreement. For the purposes of this Agreement, “maintenance” of the Penn Patent Rights includes inter partes patent review proceedings before the USPTO or a similar patent administration
outside the US. For further clarity, validity challenges raised in infringement litigation will be handled per Section 5.4, Infringement.
|
| 5.1.2 |
Licensee has the right to request that a patent application be filed in a country or territory filing via a written request to Penn [***] prior to the deadline set by the patent office in the territory in which filing is to take place (“Prosecution Request”). [***]
|
| 5.1.3 |
If, and during such time that (i) Licensee is the only party to which Penn Patent Rights have been licensed by Penn, (ii) there are no unpaid Historic Patent Costs or Ongoing Patent Costs, and (iii) Licensee requests to manage the filing,
prosecution and maintenance of Penn Patent Rights, then Penn and Licensee will use reasonable efforts to enter into the Client and Billing Agreement with Patent Counsel in substantially the form attached hereto as Appendix III, which agreement upon execution shall, determine the management of Penn Patent Rights, in lieu of Section 5.1.1, provided that upon the termination of such agreement, the management of Penn Patent
Rights shall be in accordance with Section 5.1.1.
|
| 5.2 |
Patent Costs.
|
| 5.2.1 |
Within [***] of the Effective Date, Licensee will reimburse Penn for all documented out-of-pocket costs for the filing, prosecution and maintenance of Penn Patent Rights, including all accrued attorney fees, expenses, official and filing
fees (“Patent Costs”), incurred prior to the Effective Date (“Historic Patent Costs”) to
the extent not previously reimbursed by Licensee under the Original Agreement.
|
| 5.2.2 |
Licensee will bear all Patent Costs incurred during the Term (“Ongoing Patent Costs”).
|
| 5.2.3 |
At any time, at Penn’s request, Licensee shall pay in advance the Patent Counsel’s estimated costs for undertaking material patent actions before Penn authorizes the Patent Counsel to proceed (“Advance
Payment”). Notwithstanding whether Licensee makes an Advance Payment for any patent action, Licensee shall bear all Patent Costs incurred during the Term and shall pay such amounts within [***]
of receipt of invoice for such patent actions. For clarity, the term “Patent Costs” means and includes Historic Patent Costs and Ongoing Patent Costs. For further clarity, this Section 5.2.3 shall not apply during any period during the Term
where a Client and Billing Agreement is in effect.
|
| 5.3 |
Termination of Rights in, and Obligations with respect to, Certain Penn Patent Rights. Licensee may terminate its rights in, and obligations with respect to, any or all of Penn Patent Rights by providing written notice to Penn (“Patent Termination Notice”). Termination of Licensee’s rights in and obligation with respect to such Penn Patent Right will be effective [***] after receipt of such Patent Termination Notice by Penn. Penn will use reasonable efforts
to curtail Patent Costs chargeable to Licensee under this Agreement after the receipt of the Patent Termination Notice is received. Penn may continue prosecution and maintenance of such Patent Rights at its sole discretion and expense, and
such Patent Rights will then be Carve-Out Patent Rights and therefore not subject to this Agreement, including the License, and Licensee will have no further rights or license to them.
|
| 5.3.1 |
In the event that Penn terminates Licensee’s rights and license in certain Penn Patent Rights pursuant to Section 3.3.2 (Diligence Events), such that they become Carve -Out Patent Rights and therefore not subject to this Agreement,
Licensee will have no further patent reimbursement obligation with respect to Carve-Out Patent Rights except to the extent such patent reimbursement obligation accrued prior to the applicable Patent Termination Notice.
|
| 5.4 |
Infringement.
|
| 5.4.1 |
If either Party believes that an infringement by a Third Party with respect to any Penn Patent Right is occurring, the knowledgeable Party will provide the other Party with (a) written notice of such infringement or potential infringement
and (b) evidence of such infringement or potential infringement (the “Infringement Notice”). [***] Both Penn and Licensee will use their diligent efforts to
cooperate with each other to terminate such infringement without litigation.
|
| 5.4.2 |
If infringing activity of potential commercial significance has not been abated within [***] following the date the Infringement Notice for such activity was provided, then during the period in which, and in the jurisdiction where,
Licensee has exclusive rights under this Agreement, Licensee may institute suit for patent infringement against the infringer [***]. Penn may voluntarily join such suit at Licensee’s reasonable expense, but Penn may not thereafter commence
suit against the infringer for the acts of infringement that are the subject of Licensee’s suit or any judgment rendered in such suit. Licensee may not join Penn in a suit initiated by Licensee without Penn’s prior written consent, such
consent not to be unreasonably withheld. If in a suit initiated by Licensee, Penn is involuntarily joined, then Licensee will pay any costs incurred by Penn arising out of such suit, including any legal fees of counsel that Penn selects and
retains to represent it in the suit. Licensee shall be free to enter into a settlement, consent judgment or other voluntary disposition, provided that any settlement, consent judgment or other voluntary disposition that (i) limits the scope,
validity or enforcement of Penn Patent Rights or (ii) admits fault or wrongdoing on the part of Licensee or Penn must be approved in advance by Penn in writing. Licensee’s request for such approval shall include complete copies of final
settlement documents, a detailed summary of such settlement, and any other information material to such settlement. Penn shall provide Licensee notice of its approval or denial within [***] of any request for such approval by Licensee,
provided that (x) in the event Penn wishes to deny such approval, such notice shall include a detailed written description of Penn’s reasonable objections to the proposed settlement, consent judgment, or other voluntary disposition and (y)
Penn shall be deemed to have approved of such proposed settlement, consent judgment, or other voluntary disposition in the event it fails to provide such notice within such [***] period in accordance herewith.
|
| 5.4.3 |
If, within [***] following the date the Infringement Notice was provided, infringing activity of potential commercial significance has not been abated and if Licensee has not brought suit against the infringer, then Penn may institute suit
for patent infringement against the infringer. If Penn institutes such suit, then Licensee may not join such suit without the prior written consent of Penn and may not thereafter commence suit against the infringer for the acts of
infringement that are the subject of Penn’s suit or any judgment rendered in such suit.
|
| 5.4.4 |
Notwithstanding Sections 5.4.2 and 5.4.3, in the event that any Penn Patent Rights are infringed by a Third Party (a) prior to the First Commercial Sale of a Product in the United States or (b) if any of the infringed Penn Patent Rights
are also licensed by Penn to a Third Party prior to any enforcement action being taken by either Party regarding such infringement, the Parties shall discuss, and will mutually agree, in writing, as to how to handle such infringement by such
Third Party. Notwithstanding anything in this Section 5.4, Licensee shall not provide notice of infringement or potential infringement to a Third Party (including the infringer) or engage in any enforcement action or activities, or have any
rights under this Agreement related thereto, with regard to any Carve-Out Patent Rights.
|
| 5.4.5 |
Any recovery or settlement received in connection with any suit will first be shared by Penn and Licensee equally to cover any litigation costs each incurred and next shall be paid to Penn or Licensee to cover any litigation costs it
incurred in excess of the litigation costs of the other. Any remaining recoveries shall be allocated as follows:
|
| 5.4.6 |
[***] Each Party will reasonably cooperate and assist with the other in litigation proceedings instituted hereunder but at the expense of the Party who initiated the suit (unless such suit is being jointly prosecuted by the Parties). For
clarity, such requirement does not require a Party to join a suit unless otherwise specifically required under this Agreement. If Penn is subjected to third party discovery related to the Penn Patent Rights or Products licensed to Licensee
hereunder, Licensee will pay Penn’s documented out of pocket expenses with respect to same.
|
| 5.5 |
Patent Marking. Licensee shall place in a conspicuous location on any Product (or its
packaging where appropriate and practicable) made, imported and/or Sold under this Agreement a patent notice that is not in contravention of Laws concerning the marking of patented articles where such Product is made, used, imported and/or
sold, as applicable. Upon request from Penn, Licensee shall provide evidence of proper marking.
|
| 5.6 |
Confidentiality.
|
| 5.6.1 |
Each Party agrees that, for the Term and for [***] thereafter, such Party shall (a) use the same degree of care to maintain the secrecy of the Confidential Information of the other Party that it uses to maintain the secrecy of its
Confidential Information of like kind, (b) use the Confidential Information of the other Party only to accomplish the purpose of this Agreement or for audit or management purposes and (c) ensure that any employees, customers, and distributors
are bound to it by similar obligations of confidence and to make sure such disclosure occurs only as required to accomplish the purposes of this Agreement.
|
| 5.6.2 |
A Party may disclose the Confidential Information of the other Party to the extent required by Law or court order; provided, however, that the recipient promptly provides to the disclosing Party prior written notice of such disclosure and
provides reasonable assistance in obtaining an order or other remedy protecting the Confidential Information from public disclosure.
|
| 6.1 |
Mutual Representations and Warranties. Each Party represents and warrants to the other
Party that, as of the Effective Date:
|
| 6.1.1 |
such Party is duly organized and validly existing under the Laws of the jurisdiction of its incorporation or organization;
|
| 6.1.2 |
such Party has taken all action necessary to authorize the execution and delivery of this Agreement and the performance of its obligations under this Agreement;
|
| 6.1.3 |
this Agreement is a legal and valid obligation of such Party, binding upon such Party and enforceable against such Party in accordance with the terms of this Agreement, except as enforcement may be limited by applicable bankruptcy,
fraudulent conveyance, insolvency, reorganization, moratorium and other laws relating to or affecting creditors’ rights generally and by general equitable principles; and
|
| 6.1.4 |
such Party has all right, power and authority to enter into this Agreement, to perform its obligations under this Agreement.
|
| 6.2 |
[***]
|
| 6.3 |
Disclaimer of Representations and Warranties.
|
| 6.3.1 |
Other than the representations and warranties provided in Section 6.1 and Section 6.2 above, PENN MAKES NO REPRESENTATIONS AND WARRANTIES, WHETHER EXPRESS OR IMPLIED, AND EXPLICITLY DISCLAIMS ANY REPRESENTATION AND WARRANTY, INCLUDING WITH
RESPECT TO ANY ACCURACY, COMPLETENESS, MERCHANTABILITY, FITNESS FOR A PARTICULAR PURPOSE, COMMERCIAL UTILITY, NON-INFRINGEMENT OR TITLE FOR THE INTELLECTUAL PROPERTY, PATENT RIGHTS, LICENSED INFORMATION, LICENSE AND ANY PRODUCT.
|
| 6.3.2 |
Furthermore, nothing in this Agreement will be construed as:
|
|
|
(a) |
A representation or warranty by Penn as to the validity or scope of any Penn Patent Right;
|
|
|
(b) |
A representation or warranty that anything made, used, sold or otherwise disposed of under the License is or will be free from infringement of patents, copyrights, trademarks or any other forms of intellectual property rights or tangible
property rights of Third Parties;
|
|
|
(c) |
Obligating Penn to bring or prosecute actions or suits against Third Parties for patent, copyright or trademark infringement;
|
|
|
(d) |
Conferring by implication, estoppel or otherwise any license or rights under any Patent Rights of Penn other than Penn Patent Rights as defined herein, regardless of whether such Patent Rights are dominant or subordinate to Penn Patent
Rights; and,
|
|
|
(e) |
Obligating Penn to furnish any know-how.
|
| 6.4 |
Covenants of Licensee.
|
| 6.4.1 |
Licensee and its Affiliates will not, directly or indirectly (including where such is done by a Third Party on behalf of Licensee or its Affiliates, at the urging of Licensee or its Affiliates or with the assistance of the Licensee or its
Affiliates) challenge the validity, scope, or enforceability of or otherwise oppose any Penn Patent Right , provided that if any Penn Patent Right is asserted against Licensee or its Affiliate for activities authorized under this Agreement,
then Licensee or such Affiliate is entitled to all and any defenses available to it including challenging the validity or enforceability of such Patent Right.
|
| 6.4.2 |
Licensee will comply with all Laws that apply to its activities or obligations under this Agreement. For example, Licensee will comply with applicable United States export laws and regulations. The transfer of certain technical data and
commodities may require a license from the applicable agency of the United States government and/or written assurances by Licensee that Licensee will not export data or commodities to certain foreign countries without prior approval of the
agency.
|
| 6.4.3 |
Licensee will not grant a security interest in the License or this Agreement.
|
| 6.4.4 |
[***]
|
| 6.5 |
Participation Rights. If the Licensee proposes to sell any equity securities or
securities that are convertible into equity securities of the Licensee, then Penn and/or its Assignee (as defined below) will be offered the same terms and conditions as are offered to the other purchasers in each such financing, the right
to purchase up to such number of the securities in the offering as will cause Penn and its Assignee to own collectively stock representing at least Penn’s then current fully diluted percentage of the securities of Licensee. For example, if
Penn owned [***] of the securities of Licensee on a fully diluted basis before the offering of equity securities, then if Penn and/or Penn’s Assignee fully exercised its participation rights hereunder, Penn and its Assignee will
collectively own [***] of the securities of Licensee on a fully diluted basis after the offering of equity securities and Penn and/or its Assignee’s exercise of its participation rights in full. Penn shall exercise its rights within [***]
of receiving notice from Licensee of the applicable terms and conditions with respect to each financing round covered by this section, and failing that, such offer shall expire. The term “Assignee” means (a) any entity to which Penn’s
participation rights under this section have been assigned either by Penn or another entity, or (b) any entity that is an Affiliate of Penn. This section shall survive the termination of this Agreement.
|
| 7.1 |
Indemnification by Licensee.
|
| 7.1.1 |
Penn Indemnification.
|
| 7.1.1.1 |
Licensee shall defend, indemnify and hold Penn and its respective trustees, officers, faculty, students, employees, contractors and agents (the “Penn Indemnitees”) harmless
from and against any and all liability, damage, loss, cost or expense (including reasonable attorneys’ fees), including, without limitation, bodily injury, risk of bodily injury, death and property damage to the extent arising out of Third
Party claims or suits related to (a) the Original Agreement, this Agreement or any Sublicense, including (i) the development, testing, use, manufacture, promotion, sale or other disposition of any Product (including any product liability
claim), (ii) any enforcement action or suit brought by Licensee against a Third Party for infringement of Penn Patent Rights, (iii) any claim by a Third Party that the practice of Penn Patent Rights or use of Licensed Information or the
design, composition, manufacture, use, sale or other disposition of any Product infringes or violates any patent, copyright, trade secret, trademark or other intellectual property right of such Third Party, (iv) any breach of the Original
Agreement or this Agreement or Laws by Licensee, its Affiliates or Sublicensees and (b) Licensee’s gross negligence, omissions or willful misconduct, provided that Licensee’s obligations pursuant to this Section 7.1 shall not apply to the
extent such claims or suits result from the exercise by Penn of its retained rights under the Penn Patent Rights pursuant to Section 2.2 of this Agreement or the gross negligence or willful misconduct of any of the Penn Indemnitees, in each
case as determined by a court of law.
|
|
|
7.1.1.2 |
As a condition to a Penn Indemnitee’s right to receive indemnification under this Section 7.1, Penn shall: (a) promptly notify Licensee as soon as it becomes aware of a claim or suit for which indemnification may be sought pursuant hereto
(provided, that the failure to do so in a timely manner shall not affect the Licensee’s indemnification obligations hereunder); (b) reasonably cooperate, and cause the individual Penn Indemnitees to reasonably cooperate, with Licensee in the
defense, settlement or compromise of such claim or suit (at the expense of the Licensee); and (c) permit the Licensee to control the defense, settlement or compromise of such claim or suit, including the right to select defense counsel. In no
event, however, may Licensee compromise or settle any claim or suit in a manner which (a) admits fault or negligence on the part of Penn or any other Penn Indemnitee; (b) commits Penn or any other Penn Indemnitee to take, or forbear to take,
any action, without the prior written consent of Penn, or (c) grant any rights under the Penn Patent Rights except for Sublicenses permitted under Article 2. Penn shall reasonably cooperate with Licensee and its counsel in the course of the
defense of any such suit, claim or demand, such cooperation to include without limitation using reasonable efforts to provide or make available documents, information and witnesses.
|
|
|
7.1.1.3 |
Notwithstanding Section 7.1.1.2 above, in the event that a bona fide conflict exists between Licensee and Penn or any other Penn Indemnitee with respect to a claim or suit subject to indemnification hereunder, such that the same counsel
cannot represent the parties, then Penn or any other Penn Indemnitee shall have the right to defend against any such claim or suit itself, including by selecting its own counsel, with any documented attorney’s fees and litigation expenses
being paid for by [***]. [***].
|
| 7.1.2 |
[***]
|
| 7.2 |
Insurance.
|
| 7.2.1 |
Licensee, at its sole cost and expense, must insure its activities in connection with the exercise of its rights under this Agreement and obtain, and keep in force and maintain Commercial Form General Liability Insurance (contractual
liability included) with limits as follows:
|
|
|
7.2.1.1 Each occurrence |
$[***]; |
|
|
7.2.1.2 General aggregate | $[***] |
|
|
7.2.1.3 Clinical trials liability insurance
|
$[***]
|
|
|
7.2.1.4 Products liability insurance
|
$[***]
|
| 7.2.2 |
If the above insurance is written on a claims-made form, it shall continue for [***] following termination or expiration of this Agreement. The insurance shall have a retroactive date of placement prior to or coinciding with the Original
Agreement Effective Date, the date of commencement of clinical trials, or the date of First Commercial Sale, as applicable to the types of insurance required pursuant to Section 7.2.1.
|
| 7.2.3 |
Licensee expressly understands, however, that the coverages and limits in Section 7.2.1 do not in any way limit Licensee’s liability or indemnification obligations. Licensee’s insurance will:
|
| 7.2.3.1 |
Be issued by an insurance carrier with an A.M. Best rating of “A” or better;
|
|
|
7.2.3.2 |
Provide for [***] advance written notice to Penn of any reduction in such insurance;
|
|
|
7.2.3.3 |
State that Penn is endorsed as an additional insured with respect to the coverages in Section 7.2.1; and
|
|
|
7.2.3.4 |
Include a provision that the coverages will be primary and will not participate with nor will be excess over any valid and collective insurance or program of self insurance carried or maintained by Penn.
|
| 7.2.4 |
Licensee must furnish to Penn with (a) valid certificate of insurance evidencing compliance with all requirements of this Agreement and (b) additional insured endorsements for Licensee’s applicable policies naming “The Trustees of the
University of Pennsylvania” as an additional insured. Licensee must furnish both documents within [***] of the Effective Date (to the extent not previously provided under the Original Agreement), once per year thereafter and at any time there
is a modification in such insurance.
|
| 7.3 |
LIMITATION OF LIABILITY. IN NO EVENT SHALL EITHER PARTY OR ANY OF ITS AFFILIATES BE
LIABLE TO THE OTHER PARTY OR ANY OF ITS AFFILIATES FOR SPECIAL, INDIRECT, INCIDENTAL, CONSEQUENTIAL OR PUNITIVE DAMAGES, INCLUDING LOSS OF PROFITS, WHETHER IN CONTRACT, WARRANTY, TORT, NEGLIGENCE, STRICT LIABILITY OR OTHERWISE ARISING OUT
OF OR RELATING TO THIS AGREEMENT, THE TRANSACTIONS CONTEMPLATED HEREIN OR ANY BREACH HEREOF. NOTWITHSTANDING THE FOREGOING, NOTHING IN THIS AGREEMENT SHALL LIMIT LICENSEE’S INDEMNIFICATION OBLIGATIONS UNDER SECTION 7.1 OR SHALL LIMIT PENN’S
REMEDIES OR ABILITY TO RECOVER DAMAGES, INCLUDING INCREASED DAMAGES, FOR WILLFUL INFRINGEMENT IN THE EVENT PENN ASSERTS ITS INTELLECTUAL PROPERTY RIGHTS.
|
| 8.1 |
Term. The term of this Agreement (the “Term”) shall commence on the Effective Date and, unless terminated sooner as provided below, shall continue in full force and effect until the later of (i) expiration or abandonment of the last Penn Patent
Right or (ii) ten (10) years from the First Commercial Sale of a Product.
|
| 8.2 |
Termination of the Agreement by Licensee for Convenience. At any time during the Term, Licensee may, at its convenience,
terminate this Agreement upon providing at least [***] prior written notice to Penn of such intention to terminate, provided that Licensee ceases using the License or making, using, or selling Products following the effective date of such
termination.
|
| 8.3 |
Termination For Cause.
|
| 8.3.1 |
If Licensee fails to fulfill its obligations under Section 3.2 (i.e. use Commercially Reasonable Efforts to develop and commercialize a Product), Penn may provide written notice to Licensee of such failure. If Licensee fails to address
such failure to the reasonable satisfaction of Penn within [***] of receiving such written notice, Penn may terminate this Agreement upon written notice to Licensee.
|
| 8.3.2 |
In the event Licensee fails to achieve any Diligence Event by the corresponding Achievement Date in accordance to Section 3.3.1, Penn has the right and option to terminate this Agreement, upon written notice, with immediate effect.
|
| 8.3.3 |
If Licensee materially breaches any of its material obligations under this Agreement, Penn may give to Licensee a written notice specifying the nature of the default, requiring it to cure such breach, and stating its intention to terminate
this Agreement. If such breach is not cured within [***] of such notice, such termination shall become effective upon a notice of termination by Penn thereafter. For clarity, a breach of a material obligation includes:
|
| 8.3.4 |
[***] In addition to all other remedies available to it, Penn may terminate this Agreement, upon written notice, with immediate effect, upon a breach of [***], provided, however, that in the event that, in the sole discretion of Penn, such
breach is curable without adverse effect on Penn, Licensee will have [***] from receipt of such written notice to cure any breach under [***] and, if so cured, the Agreement shall not terminate.
|
| 8.3.5 |
Penn may terminate this Agreement, upon written notice, with immediate effect if, at any time, Licensee is unable to pay its debts, including any debts related to exclusive Sublicensees, when they come due, or files in any court or agency
pursuant to any statute or regulation of any state, country or jurisdiction, a petition in bankruptcy or insolvency or for reorganization or for an arrangement or for the appointment of a receiver or trustee of Licensee or of its assets, or
if Licensee proposes a written agreement of composition or extension of its debts, or if Licensee is served with an involuntary petition against it, filed in any insolvency proceeding, and such petition is not dismissed within [***] after the
filing thereof, or if Licensee proposes or is a party to any dissolution or liquidation, or if Licensee makes an assignment for the benefit of its creditors of all or substantially all its assets (in each case, “Bankruptcy Action”).
|
| 8.4 |
Effects of Termination.
|
| 8.4.1 |
Notwithstanding the termination of this Agreement, the following provisions shall survive: Sections 4.8-4.13, inclusive, 5.6, 6.3, and 8.4 and Articles 7 and 9.
|
| 8.4.2 |
Termination of this Agreement shall not relieve the Parties of any obligation or liability that, at the time of termination, has already accrued hereunder, or which is attributable to a period prior to the effective date of such
termination. Termination of this Agreement shall not preclude either Party from pursuing all rights and remedies it may have hereunder or at Law or in equity with respect to any breach of this Agreement nor prejudice either Party’s right to
obtain performance of any obligation.
|
| 8.4.3 |
If this Agreement is terminated for any reason, all outstanding Sublicenses (including all Sublicense Documents for each Sublicense) not in default will be assigned by Licensee to Penn, and such assignment will be accepted by Penn. Each
assigned Sublicense will remain in full force and effect with Penn as the licensor or sublicensor instead of Licensee, but the duties and obligations of Penn under the assigned Sublicenses will not be greater than the duties of Penn under
this Agreement, and the rights of Penn under the assigned Sublicenses will not be less than the rights of Penn under this Agreement, including all financial consideration and other rights of Penn. Penn may, at its sole discretion, amend such
outstanding Sublicenses to contain the terms and conditions found in this Agreement.
|
| 9.1 |
Relationship of the Parties. Nothing in this Agreement is intended or shall be deemed,
for financial, tax, legal or other purposes, to constitute a partnership, agency, joint venture or employer-employee relationship between the Parties. The Parties are independent contractors and at no time will either Party make commitments
or incur any charges or expenses for or on behalf of the other Party.
|
| 9.2 |
Expenses. Except as otherwise provided in this Agreement, each Party shall pay its own
expenses and costs incidental to the preparation of this Agreement and to the consummation of the transactions contemplated hereby
|
| 9.3 |
Third Party Beneficiary. The Parties agree that each Sublicensee is a third party
beneficiary of this Agreement with respect to Section 8.4.3. [***]
|
| 9.4 |
Use of Names. Licensee, its Affiliates and Sublicensees may not use the name, logo,
seal, trademark, or service mark (including any adaptation of them) of Penn or any Penn school, organization, employee, student or representative, without the prior written consent of Penn. Notwithstanding the foregoing, Licensee may use
the name of Penn in a non-misleading and factual manner solely in (a) executive summaries, business plans, offering memoranda and other similar documents used by Licensee for the purpose of raising financing for the operations of Licensee
as related to Product, or entering into commercial contracts with Third Parties, but in such case only to the extent necessary to inform a reader that the Penn Patent Rights has been licensed by Licensee from Penn, and to inform a reader of
the identity and published credentials of Inventors of the Intellectual Property, and (b) any securities reports required to be filed with the Securities and Exchange Commission.
|
| 9.5 |
No Discrimination. Neither Penn nor Licensee will discriminate against any employee or
applicant for employment because of race, color, sex, sexual or affectional preference, age, religion, national or ethnic origin, handicap, or veteran status.
|
| 9.6 |
Successors and Assignment.
|
| 9.6.1 |
The terms and provisions hereof shall inure to the benefit of, and be binding upon, the Parties and their respective successors and permitted assigns.
|
| 9.6.2 |
Licensee may not assign or transfer this Agreement or any of Licensee’s rights or obligations created hereunder, by operation of law or otherwise, without the prior written consent of Penn, provided that Penn shall not unreasonably
withhold, condition or delay its consent. Licensee may assign or transfer this Agreement in its entirety without the consent of Penn in connection with a merger, consolidation, or sale or transfer of all or substantially all of its assets
without any requirement to obtain Penn’s consent, to unrelated third party entity provided that: (i) [***]; (ii) there exists no breach by Licensee or its Affiliates of any term of this Agreement, including those caused by a Sublicensee, and
Licensee is not in breach of payment or diligence obligations hereunder that has not been cured as of the consummation of such transaction; (iii) the Licensee delivers to Penn [***] written notice of the proposed assignment when such notice
may be provided in accordance with applicable securities laws and non-disclosure agreements, (iv) the assignee agrees in writing to be legally bound by this Agreement and to deliver to Penn an updated Development Plan within [***] after the
closing of the proposed transaction and (v) the assignment is made as a part of and in connection with an asset sale, stock sale, merger or other combination, or any other transfer of Licensee’s entire business. Any permitted assignment will
not relieve Licensee of responsibility for performance of any obligation of Licensee that has accrued at the time of the assignment. For the avoidance of doubt, it is understood and agreed that this Section 9.6.2 shall not include the grant
of a sublicense.
|
| 9.6.3 |
Any assignment not in accordance with this Section 9.6 shall be void.
|
| 9.7 |
Further Actions. Each Party agrees to execute, acknowledge and deliver such further
instruments and to do all such other acts as may be necessary or appropriate in order to carry out the purposes and intent of this Agreement.
|
| 9.8 |
Entire Agreement of the Parties; Amendments. This Agreement, the Exhibits and Appendices or Schedules hereto, Equity Issuance
Agreement and, to the extent entered into, the Client & Billing Agreement constitute and contain the entire understanding and agreement of the Parties respecting the subject matter hereof and cancel and supersede any and all prior
negotiations, correspondence, understandings and agreements between the Parties, whether oral or written, regarding such subject matter. No waiver, modification or amendment of any provision of this Agreement shall be valid or effective
unless made in a writing referencing this Agreement and signed by a duly authorized officer of each Party.
|
| 9.9 |
Governing Law. This Agreement shall be governed by and interpreted in accordance with
the laws of the Commonwealth of Pennsylvania, excluding application of any conflict of laws principles that would require application of the law of a jurisdiction outside of the Commonwealth of Pennsylvania.
|
| 9.10 |
Dispute Resolution. If a dispute arises between the Parties concerning this Agreement,
then the Parties will confer, as soon as practicable, in an attempt to resolve the dispute. If such dispute remains unresolved, it will be escalated to Licensee’s Chief Executive Officer and Penn Center for Innovation’s Managing Director or
their respective designee(s), for discussion in good faith. If the Parties are not able to resolve the dispute within [***] of submission to such officers, then the Parties will submit to the exclusive jurisdiction of, and venue in, the
state and Federal courts located in the Eastern District of Pennsylvania.
|
| 9.11 |
Notices and Deliveries. Any notice, request, approval or consent required or permitted
to be given under this Agreement shall be in writing and directed to a Party at its address or facsimile number shown below or such other address or facsimile number as such Party shall have last given by notice to the other Party. A notice
will be deemed received: if delivered personally, on the date of delivery; if mailed, five (5) days after deposit in the United States mail; if sent via courier, one (1) business day after deposit with the courier service; or if sent via
facsimile, upon receipt of confirmation of transmission provided that a confirming copy of such notice is sent by certified mail, postage prepaid, return receipt requested.
|
|
For Penn:
Penn Center for Innovation
University of Pennsylvania
3600 Civic Center Blvd.,
9th Floor
Philadelphia, PA 19104-4310
Attention: Managing Director
|
with a copy to:
University of Pennsylvania
Office of General Counsel
2929 Walnut Street, Suite 400
Philadelphia, PA 19104-5509
Attention: General Counsel
|
|
For Licensee:
Opus Genetics, Inc.
223 S. West Street, Suite 900
Raleigh, NC 27603
Attention: [***]
|
with a copy to:
Smith, Anderson, Blount, Dorsett, Mitchell
& Jernigan, LLP
150 Fayetteville Street, Suite 2300
Raleigh, NC 27601
Attention: [***]
|
| 9.12 |
Waiver. A waiver by either Party of any of the terms and conditions of this Agreement in any instance shall not be deemed or construed to be a waiver of such term or condition for the future, or of any other term or
condition hereof. All rights, remedies, undertakings, obligations and agreements contained in this Agreement shall be cumulative and none of them shall be in limitation of any other remedy, right, undertaking, obligation or agreement of
either Party.
|
| 9.13 |
Severability. When possible, each provision of this Agreement will be interpreted in such manner as to be effective and valid
under law, but if any provision of this Agreement is held to be prohibited by or invalid under law, such provision will be ineffective only to the extent of such prohibition or invalidity, without invalidating the remainder of this
Agreement. The Parties shall make a good faith effort to replace the invalid or unenforceable provision with a valid one which in its economic effect is most consistent with the invalid or unenforceable provision.
|
| 9.14 |
Interpretation. The words “include,” “includes” and “including” shall be deemed to be
followed by the phrase “without limitation.” All references herein to Articles, Sections, Schedules, Appendices and Exhibits shall be deemed references to Articles and Sections of, Schedules, Appendices and Exhibits to, this Agreement
unless the context shall otherwise require. Except as otherwise expressly provided herein, all terms of an accounting or financial nature shall be construed in accordance with GAAP, as in effect from time to time. Unless the context
otherwise requires, countries shall include territories. References to any specific Law or article, section or other division thereof, shall be deemed to include the then-current amendments or any replacement Law thereto.
|
| 9.15 |
Counterparts. This Agreement may be executed in counterparts, each of which will be
deemed an original, and all of which together will be deemed to be one and the same instrument. A facsimile or a portable document format (PDF) copy of this Agreement, including the signature pages, will be deemed an original.
|
| 9.16 |
Timely Countersignature. The terms and conditions of this Agreement shall, at Penn's
sole option, be considered by Penn to be withdrawn from Licensee's consideration and the terms and conditions of this Agreement, and the Agreement itself to be null and void, unless this Agreement is executed by Licensee and a fully
executed original is received by Penn within thirty (30) days from the date of Penn's signature found below.
|
|
THE TRUSTEES OF THE UNIVERSITY OF PENNSYLVANIA
|
OPUS GENETICS INC.
|
||||
|
By:
|
/s/ Benjamin C. Dibling, Ph.D.
|
By:
|
/s/ Joe Schachle
|
||
|
Name:
|
Benjamin C. Dibling, Ph.D.
|
Name:
|
Joe Schachle
|
||
|
Title:
|
Deputy Managing Director, Penn Center for Innovation
|
Title:
|
Chief Operating Officer
|
||
|
Date:
|
6/17/2022
|
Date:
|
6/23/2022
|
||
|
Article I DEFINITIONS
|
1
|
||
|
1.1
|
Net Sales Definition
|
8
|
|
|
Article II PURCHASE AND SALE
|
12
|
||
|
2.1
|
Purchase and Sale of Assets
|
12
|
|
|
2.2
|
Assumption of Liabilities
|
12
|
|
|
2.3
|
Retained Liabilities
|
12
|
|
|
2.4
|
Purchase Price
|
12
|
|
|
2.5
|
Confidentiality Obligations
|
13
|
|
|
2.6
|
Freedom to Operate License
|
13
|
|
|
Article III PAYMENTS
|
13
|
||
|
3.1
|
Upfront Fee
|
13 | |
|
3.2
|
Equity
|
13 | |
|
3.3
|
Milestones
|
13 | |
|
3.4
|
Earn-out
|
14
|
|
|
3.5
|
Financial Records
|
15
|
|
|
3.6
|
Audits
|
15
|
|
|
3.7
|
Tax Matters
|
16
|
|
|
3.8
|
Foreign Derived Intangible Income Deduction
|
17 | |
|
3.9
|
Currency Exchange
|
17
|
|
|
3.10
|
Late Payments
|
17 | |
|
3.11
|
Blocked Payments
|
17 | |
|
3.12
|
Prohibitions on Payments
|
17 | |
|
Article IV IVERIC COVENANTS
|
17 | ||
|
4.1
|
Technology Transfer; Transition Assistance
|
18
|
|
|
4.2
|
Patent Matters
|
18
|
|
|
4.3
|
Regulatory Documentation
|
18
|
|
|
4.4
|
Manufacturing
|
18
|
|
|
4.5
|
Noncompetition Covenant
|
19
|
|
|
4.6
|
Deliverables
|
19
|
|
|
4.7
|
Debarment
|
19
|
|
|
Article V OPUS COVENANTS
|
19
|
||
|
5.1
|
Diligence
|
19
|
|
|
5.2
|
Discontinuation of Manufacturing Patent
|
20
|
|
|
5.3
|
Right of First Refusal
|
21
|
|
|
Article VI REPRESENTATIONS AND WARRANTIES; COVENANTS
|
21
|
||
|
6.1
|
The Parties’ Representations and Warranties
|
21
|
|
|
6.2
|
Iveric’s Representations and Warranties
|
23
|
|
|
Article VII INDEMNIFICATION
|
24 | ||
|
7.1
|
Iveric Indemnity
|
24
|
|
|
7.2
|
Opus Indemnity
|
25
|
|
|
7.3
|
Indemnification Procedure
|
25
|
|
|
7.4
|
Limitation of Liability; Exclusion of Damages; Disclaimer
|
26
|
|
|
Article VIII DISPUTE RESOLUTION
|
28 | ||
|
8.1
|
Resolution by Executives
|
28 | |
|
8.2
|
Arbitration
|
28 | |
|
Article IX GENERAL PROVISIONS
|
29 | ||
|
9.1
|
Governing Law
|
29 | |
|
9.2
|
Assignment
|
29 | |
|
9.3
|
Headings; Rules of Construction
|
29 | |
|
9.4
|
No Implied Waiver
|
30 | |
|
9.5
|
Notices
|
30 | |
|
9.6
|
Severability
|
30
|
|
|
9.7
|
Entire Agreement
|
31
|
|
|
9.8
|
Amendment; Waiver
|
31
|
|
|
9.9
|
Counterparts
|
31
|
|
|
9.10
|
Agency
|
31
|
|
|
9.11
|
Further Actions
|
31
|
|
|
9.12
|
Compliance with Laws
|
31
|
|
|
9.13
|
Press Release
|
32
|
|
|
Milestone Event
|
Milestone Payment
IC-100 Product
|
Milestone Payment
IC-200 Product
|
|
|
[***]
|
[***]
|
[***]
|
|
|
[***]
|
[***]
|
[***]
|
|
|
[***]
|
[***]
|
[***]
|
|
|
[***]
|
[***]
|
[***]
|
|
|
[***]
|
[***]
|
[***]
|
|
|
[***]
|
[***]
|
[***]
|
|
|
[***]
|
[***]
|
[***]
|
|
|
Annual Net Sales
|
Milestone Payment
IC-100 Product
|
Milestone Payment
IC-200 Product
|
|
|
[***]
|
[***]
|
[***]
|
|
|
[***]
|
[***]
|
[***]
|
|
|
[***]
|
[***]
|
[***]
|
|
|
[***]
|
[***]
|
[***]
|
|
|
[***]
|
[***]
|
[***]
|
|
|
If to Iveric:
|
IVERIC bio, Inc.
|
|
Attention: [***]
|
|
|
Email: [***]
|
|
|
with a copy to:
|
Wilmer Cutler Pickering Hale and Dorr LLP
|
|
60 State Street
|
|
|
Boston, MA 02109
|
|
|
Attention: [***]
|
|
|
Email: [***]
|
|
|
If to Opus:
|
Opus Genetics Inc.
|
|
8 Davis Drive
|
|
|
Durham, NC 27709
|
|
|
Attention: [***]
|
|
|
Email: [***]
|
|
|
with a copy to:
|
Smith, Anderson, Blount, Dorsett, Mitchell & Jernigan, LLP
|
|
Wells Fargo Capitol Center
|
|
|
150 Fayetteville Street, Suite 2300 Raleigh, NC 27601
|
|
|
Attention: [***]
|
|
|
Email: [***] and [***]
|
|
IVERIC bio Gene Therapy LLC
|
||
|
By:
|
/s/ Tony Gibney
|
|
|
Name:
|
Tony Gibney
|
|
|
Title:
|
Chief Business and Strategy Officer
|
|
Opus Genetics Inc.
|
||
|
By:
|
/s/ Ben Yerxa
|
|
|
Name:
|
Ben Yerxa
|
|
|
Title:
|
CEO
|
|
Page
|
|
|
ARTICLE 1 DEFINITIONS
|
2 |
|
ARTICLE 2 LICENSES AND OTHER RIGHTS
|
8
|
|
ARTICLE 3 DILIGENCE
|
8 |
|
ARTICLE 4 FINANCIAL PROVISIONS
|
8
|
|
ARTICLE 5 INTELLECTUAL PROPERTY
|
11
|
|
ARTICLE 6 REPRESENTATIONS, WARRANTIES AND COVENANTS
|
13
|
|
ARTICLE 7 INDEMNIFICATION; INSURANCE AND LIMITATION OF LIABILITY
|
14
|
|
ARTICLE 8 TERM AND TERMINATION
|
17
|
|
ARTICLE 9 ADDITIONAL PROVISIONS
|
18
|
|
EXHIBIT A
|
PENN PATENT RIGHTS
|
|
EXHIBIT B
|
COPY RIGHTS
|
|
EXHIBIT C
|
FORM OF JOINDER AGREEMENT
|
| 1.1 |
“Administration” means a visit, of a particular enrolled subject of a Clinical Trial, during which the Product collects Results from such enrolled subject as
part of such Clinical Trial. As used in this definition, “visit” is intended to align with the use of such term in the applicable Clinical Trial Protocol.
|
| 1.2 |
“Affiliate” means a Person that controls, is controlled by or is under common control with a Party, but only for so long as such control exists and, with
respect to Licensee, that signs a Joinder Agreement consistent with the form set forth in Exhibit C. For the purposes of this Section 1.2, the word “control” (including, with correlative
meaning, the terms “controlled by” or “under the common control with”) means the actual power, either directly or indirectly through one or more intermediaries, to direct the management and policies of such Person or entity, whether by the
ownership of more than fifty percent (50%) of the voting stock of such entity, or by contract or otherwise.
|
| 1.3 |
“Clinical Trial” means a clinical trial (whether denominated phase I, phase II, phase III or a combination of the foregoing) for the evaluation of treatments
for retinal disorders caused by a mutation or mutations in the lebercilin (LCA5) gene resulting in Leber congenital amaurosis 5 (such treatments, “LCA5 Therapy”), conducted pursuant to an applicable
Clinical Trial Protocol.
|
| 1.4 |
“Clinical Trial Protocol” means the project protocol applicable to a particular clinical trial, which shall have been approved by an applicable Institutional
Review Board or equivalent ethics committee, and which applies with respect to such clinical trial.
|
| 1.5 |
“Commercially Reasonable Efforts” means [***].
|
| 1.6 |
“Confidential Information” of a Party, means (a) information relating to the business, operations or products of a Party or any of its Affiliates, including
any know-how, that such Party discloses to the other Party under this Agreement, or otherwise becomes known to the other Party by virtue of this Agreement, and (b) the terms of this Agreement; provided that Confidential Information shall
not include information that:
|
|
|
i. |
is or becomes generally available to the public other than as a result of disclosure by the recipient;
|
|
|
ii. |
is already known by or in the possession of the recipient at the time of disclosure by the disclosing Party;
|
|
|
iii. |
is independently developed by recipient without use of or reference to the disclosing Party’s Confidential Information; or
|
|
|
iv. |
is obtained by recipient from a Third Party that has not breached any obligations of confidentiality.
|
| 1.7 |
“Controlled” means, with respect to intellectual property rights, that a Party or one of its Affiliates owns or has a license or sublicense to such
intellectual property rights and has the ability to provide to, grant a license or sublicense to, or assign its right, title and interest in and to, such intellectual property rights as provided for in this Agreement without violating the
terms of any agreement or other arrangement with any Third Party.
|
| 1.8 |
“Copyright(s)” means any (a) copyright rights in the written materials, software, computer programs, documentation, algorithms, or other materials subject to
copyright protection listed in Exhibit B Controlled by Penn as of the Effective Date, and (b) copyright rights in the written materials, software, computer programs, documentation, algorithms,
or other materials subject to copyright protection Controlled by Penn after the Effective Date with respect to any Derivative Work(s).
|
| 1.9 |
“Derivative Works” means any modifications, adaptations, translations, abridgements of, or collective works of the
written materials, software, computer programs, documentation, algorithms, or other materials subject to copyright protection listed in Exhibit B as defined by US copyright law.
|
| 1.10 |
“Field of Use” means the Administration of the Product and/or evaluation of one or more subjects enrolled in a Clinical Trial, including, without limitation,
data collection therefrom, preparation and maintenance of regulatory filings relating thereto, and any analysis or follow-up arising from such Clinical Trial.
|
| 1.11 |
“GAAP” means United States generally accepted accounting principles applied on a consistent basis.
|
| 1.12 |
“Governmental Approval” means, with respect to a Product in a country or region, the approval, clearance, license, registration or authorization (including
but not limited to emergency use authorization) by the relevant Governmental Body, if applicable, for the commercialization of such Product in such country.
|
| 1.13 |
“Governmental Body” means any: (a) nation, principality, state, commonwealth, province, territory, county, municipality, district or other jurisdiction of
any nature; (b) federal, provincial, state, local, municipal, foreign or other government; (c) governmental or quasi-governmental authority of any nature (including any governmental division, subdivision, department, agency, bureau, branch,
office, commission, council, board, instrumentality, officer, official, representative, organization, unit, body or entity and any court or other tribunal); (d) multi-national or supranational organization or body; or (e) individual,
entity, or body exercising, or entitled to exercise, any executive, legislative, judicial, administrative, regulatory, police, military or taxing authority or power of any nature.
|
| 1.14 |
“Intellectual Property” means the Penn Patent Rights and the Copyrights.
|
| 1.15 |
“Law” or “Laws” means all applicable laws, statutes, rules, regulations,
ordinances and other pronouncements having the binding effect of law of any Governmental Body.
|
| 1.16 |
“Patent Rights” means any of the following, whether existing now or in the future anywhere in the world: issued patent, including inventor’s certificates,
substitutions, extensions, confirmations, reissues, re-examination, renewal or any like governmental grant for protection of inventions, and any pending application for any of the foregoing.
|
| 1.17 |
“Penn Patent Rights” means (a) the Patent Rights listed in Exhibit A Controlled by Penn as of the Effective
Date, (b) any continuations, provisionals, continued prosecution applications, substitutions, extensions and term restorations, registrations, confirmations, reexaminations, renewals or reissues thereof, including divisions, but excluding
continuations-in-part except to the extent of claims entirely supported in the specification and entitled to the priority date of the parent application, and (c) any corresponding foreign Patent Rights to the foregoing. Notwithstanding the
above, Penn Patent Rights does not include the Carve-Out Patent Rights.
|
| 1.18 |
“Person” means any natural person, corporation, firm, business trust, joint venture, association, organization, company, partnership or other business
entity, or any government or agency or political subdivision thereof.
|
| 1.19 |
“Product” means: (a) any application, program, software, device, apparatus, product, process, service or method covered by a Valid Claim or whose use or
practice would, absent the License, constitute an infringement, inducement of infringement or contributory infringement of any Valid Claim (“Method”) (b) any application, program, software, device, article, apparatus, product, process or any other material covered by a Valid Claim or whose manufacture, import, use offer for sale or sale would,
absent the License, constitute an infringement, inducement of infringement or contributory infringement of any Valid Claim; (c) any application, program, software, device, service, article, apparatus, product, process or any other material
imported, made, used or sold by or utilizing or practicing a Method; or (d) any application, program, software, device, apparatus, product, process, service or method, article, or any other material which incorporates, consists of, makes
use of or is made through the use of Copyrights or constitutes an infringement, inducement of infringement or contributory infringement of any Copyright (this subsection (d), the “Copyright Product”).
|
| 1.20 |
“Result” means any observations or results of any Clinical Trial, including data, and the records created by or on behalf of Licensee pertaining thereto.
|
| 1.21 |
“Service Provider” means any Third Party providing services to Licensee in connection with or in preparation for a Clinical Trial, including but not limited
to a contract research organization.
|
| 1.22 |
“Tax” means all taxes, duties, fees, premiums, assessments, imposts, levies, rates, withholdings, dues, government contributions and other charges of any
kind whatsoever, whether direct or indirect, together with all interest, penalties, fines, additions to tax or other additional amounts, imposed by any Governmental Body.
|
| 1.23 |
“Third Party” means any Person other than Penn, Licensee or any of their respective Affiliates.
|
| 1.24 |
“United States” or “US” means the United States of America, its territories
and possessions.
|
| 1.25 |
“USD” or “$” means the lawful currency of the United States of America.
|
| 1.26 |
“Use” means Administration of the Product in a Clinical Trial.
|
| 1.27 |
“Valid Claim” means a claim of (a) an issued and unexpired patent in Penn Patent Rights which claim has not been revoked or held unenforceable or invalid by
a decision of a court or governmental agency of competent jurisdiction from which no further appeal can be taken or has been taken within the time allowed for appeal, and has not been abandoned, disclaimed, denied or admitted to be invalid
or unenforceable through reissue or disclaimer; or (b) a pending patent application that is included in Penn Patent Rights which was filed and is being prosecuted in good faith, and has not been abandoned or finally disallowed without the
possibility of appeal or re-filing of the application.
|
| 1.28 |
Other Terms. The definition of each of the following terms is set forth in the section of the Agreement indicated below:
|
|
Defined Term
|
Section
|
||
|
Advance Payment
|
5.2(c)
|
||
|
Agreement
|
Preamble
|
||
|
Bankruptcy Action
|
8.3.3
|
||
|
Carve-Out Patent Rights
|
5.1(b)
|
||
|
Copyright Product
|
1.19
|
||
|
Effective Date
|
Preamble
|
||
|
Fees
|
4.2(a)
|
||
|
Financial Report
|
4.4
|
||
|
Historic Patent Cost
|
5.2(a)
|
||
|
Inventor(s)
|
Recitals
|
||
|
Issue Fee
|
4.1
|
||
|
LCA5 Therapy
|
1.3
|
||
|
License
|
2.1
|
||
|
Licensee
|
Preamble
|
||
|
Method
|
1.19
|
||
|
Ongoing Patent Costs
|
5.2(b)
|
||
|
Parties
|
Preamble
|
||
|
Party
|
Preamble
|
||
|
Patent Costs
|
5.2(a)
|
||
|
Patent Counsel
|
5.1(a)
|
||
|
Patent Termination Notice
|
5.3
|
||
|
PCI
|
6.2
|
||
|
Penn
|
Preamble
|
||
|
Penn Indemnitees
|
7.1.1
|
||
|
Progress Reports
|
3.2(a)
|
||
|
Pro Rata Share
|
5.2(b)
|
||
|
Prosecution Request
|
5.1(b)
|
||
|
Provider Agreement
|
2.3
|
||
|
Providers
|
2.3
|
||
|
Term
|
8
|
| 2.1 |
Grant of License. Subject to the terms and conditions of this Agreement, Penn hereby grants to Licensee (i) a non-exclusive, non-sublicensable (except as extended to Providers as set forth in Section 2.3) license under Penn Patent Rights, in all jurisdictions where Penn Patent Rights exist, to make, have made,
use, and import Product in the Field of Use during the Term and (ii) a non-exclusive, non-sublicensable (except as extended to Providers as set forth in Section 2.3) license under the Copyrights, in all jurisdictions where Copyrights
exist, to, copy, reproduce, (as expressly authorized herein), distribute, publicly display (including to subjects enrolled in Clinical Trials), and publicly perform the Copyright Product in the Field of Use during the Term (collectively,
the “License”). Except as set forth in Section 2.3, the License does not include the right to sublicense.
|
| 2.2 |
Except as expressly permitted in this Agreement, Licensee shall not create Derivative Works of the Copyright Products in any manner or form; provided that Licensee may request prior written
permission from Penn to make any material changes to the Copyright Products, including Derivative Works, which permission shall not be unreasonably withheld. Licensee shall provide a copy to Penn of any Derivative Works created by, on
behalf of, or for the benefit of Licensee for purposes of Penn ensuring compliance with this Agreement. For clarity any Derivative Works created by, on behalf of, or for the benefit of Licensee (including by any Provider) are owned by Penn.
In the event, any Derivative Works are created, Licensee shall assign, and ensure that any third party assigns all right, title and interest in and to such Derivative Works to Penn.
|
| 2.3 |
Notwithstanding anything to the contrary herein, Licensee is permitted to extend its rights under the License to and contract with its Affiliates and/or Service Providers (collectively, “Providers”) for research and development purposes only and may grant rights to such Providers solely to copy, display, perform, make, have made, or use Products,
but shall not grant any rights to distribute, make derivatives works of, sell or have sold Products, provided that, in each case, (i) any work performed by Providers shall only be on behalf of and solely for the benefit of the Licensee, and
(ii) Licensee enters into a written agreement with such Providers (“Provider Agreement”) requiring the Providers to comply with the terms and conditions of this Agreement, and (iii) Licensee shall
provide Penn a copy of such Provider Agreement within [***] of such agreement’s execution, which may be redacted to the extent the terms thereof are not necessary to determine compliance with this Agreement, and (iv) Licensee remains
primarily liable to Penn for any acts or omissions of Providers, and (v) any acts or omissions of Providers that would be a breach of this Agreement if performed or omitted by Licensee will be deemed a breach of this Agreement by Licensee,
and (vi) Providers are prohibited from granting any rights to, distributing or otherwise transferring, the Copyrights or Penn Patent Rights to any party. Notwithstanding the foregoing, Penn reserves the right in its sole discretion to
review redacted information in any Provider Agreement, which Licensee shall promptly provide following Penn’s written request.
|
| 2.4 |
Retained Rights. Penn retains the right under the Intellectual Property rights to: (a) conduct educational, research and
clinical activities itself and (b) authorize non-commercial Third Parties to conduct educational, research and clinical activities.
|
| 2.5 |
U.S. Government Rights. The License is expressly subject to all applicable provisions of any license to the United States
Government executed by Penn and is subject to any overriding obligations to the United States Federal Government under 35 U.S.C. §§200-212, applicable governmental implementing regulations, and the U.S. Government sponsored research
agreement or other guidelines, including that products that result from intellectual property funded by the United States Federal Government that are sold in the United States be substantially manufactured in the United States. In the
event that Licensee believes in good faith that substantial manufacture of such product is not commercially feasible in the United States and makes a request to Penn in writing to assist in obtaining a waiver of such requirement from the
United States Government, then Penn shall, at the expense of Licensee, use reasonable efforts to assist in obtaining such waiver.
|
| 2.6 |
No Implied License. Each Party acknowledges that the rights and licenses granted in this Agreement
are limited to the scope expressly granted. Accordingly, except for the rights expressly granted under this Agreement, no right, title, or interest of any nature whatsoever is granted whether by implication, estoppel, reliance, or
otherwise, by either Party to the other Party. All rights with respect to any know-how, patent or other intellectual property rights that are not specifically granted herein are reserved to the owner thereof.
|
| 2.7 |
Restrictions on Use. Licensee acknowledges that THE PRODUCT MAY NOT BE USED BY LICENSEE OR PROVIDERS OR ANY THIRD PARTY FOR
PATIENT CARE OR DIAGNOSIS OR TREATMENT OF PATIENTS. USE IS LIMITED TO INTERNAL NON-COMMERCIAL INVESTIGATIONAL RESEARCH PURPOSES ONLY. For clarity, the foregoing is not intended to restrict the Use of the Product with respect to any
Clinical Trial as authorized hereunder.
|
| 3.1 |
General Diligence. Licensee shall use Commercially Reasonable Efforts to Use the Product in the Field of Use during the
Term, in accordance with the terms and conditions of this Agreement.
|
| 3.2 |
Progress Reports.
|
|
|
(a) |
On an annual basis, but in no event later than December 1st of each calendar year, Licensee shall submit to Penn a progress report (each, a “Progress Report”) covering Licensee’s (and any Providers’) activities related to the Use of all Products and the obtaining of Governmental Approvals necessary for Use of Products.
|
|
|
(b) |
Each Progress Report must include all of the following for each annual period: [***].
|
| 4.1 |
Issue Fee. In partial consideration of the License, Licensee will pay to Penn on the Effective Date a license issue fee of [***]. The Issue Fee is non-refundable and non-creditable against any other amounts due by Licensee.
|
| 4.2 |
Fees
|
|
|
(a) |
As further consideration for the License, Licensee shall pay to Penn a non-refundable, non-creditable fee of [***] per Administration of the Product to an enrolled subject in a Clinical Trial. (“Fees”).
|
|
|
(b) |
Licensee must pay Fees owed to Penn on a calendar quarter basis on or before the following dates:
|
|
|
i. |
[***] for any Administration that took place on or before the last day of the calendar quarter ending December 31, of the prior year;
|
|
|
ii. |
[***] for any Administration that took place on or before the last day of the calendar quarter ending March 31 of such calendar year;
|
|
|
iii. |
[***] for any Administration that took place on or before the last day of the calendar quarter ending June 30 of such calendar year; and
|
|
|
iv. |
[***] for any Administration that took place on or before the last day of the calendar quarter ending September 30 of such calendar year.
|
| 4.3 |
Mode of Payment and Currency. All payments to Penn hereunder shall be made by deposit of USD in the requisite amount to the
“The Trustees of the University of Pennsylvania” and will be made by delivery to any one of the following: [***]
|
| 4.4 |
Fee Reports. Within [***] after the end of each calendar quarter [***], Licensee shall deliver to Penn a report (“Financial Report”) setting out all details necessary to calculate the Fee due under this Article 4 for such calendar quarter, including:
|
| 4.5 |
Late Payments. In addition to any other remedies available to Penn, including the right to terminate this Agreement, any failure by Licensee to make a payment within [***] after the date when due shall obligate Licensee to pay computed interest, the interest period commencing on the due date and
ending on the actual payment date, to Penn at a rate per annum equal to [***], or the highest rate allowed by Law, whichever is lower.
|
| 4.6 |
Default Payment. In the event of default in payment of any payment owing to Penn under the terms of this Agreement, and if
it becomes necessary for Penn to undertake legal action to collect said payment, Licensee shall pay reasonable, documented out-of-pocket legal fees and costs incurred in connection therewith.
|
| 4.7 |
Accounting. Each Party shall calculate all amounts, and perform other accounting procedures required, under this Agreement
and applicable to it in accordance with GAAP.
|
| 4.8 |
Books and Records. Licensee will keep, and, as applicable, cause its Affiliates to keep, accurate books and records of all
Use of Products and all Provider Agreements and any agreements that involved Penn Patent Rights and/ or Copyrights. Licensee will preserve, and, as applicable, cause its Affiliates to preserve, these books and records for at least [***]
from the date of the Financial Report to which they pertain. Upon reasonable notice and to the extent not prohibited by applicable privacy laws, key personnel, books and records will be made reasonably available and will be open to
examination by representatives or agents of Penn during regular office hours to determine their accuracy and assess Licensee’s and, as applicable, its Affiliates’ compliance with the terms of this Agreement, provided that Licensee and, as
applicable, its Affiliates shall not have an obligation to provide access to any given records more than once in any given [***] period.
|
| 4.9 |
Audits. In addition to the right of Penn to examine the books and records and interview key personnel as provided in Section 4.8 above, Penn, at its own cost, through an independent auditor reasonably acceptable to Licensee (and who has executed an appropriate confidentiality agreement reasonably acceptable to
Licensee that requires the auditor to keep any information learned by it confidential except as needed to report its audit conclusions to Penn), may inspect and audit the relevant records of Licensee and its Affiliates, pertaining to the
calculation of any Fees due to Penn under this Agreement. Licensee and, as applicable, its Affiliates shall provide such auditors with access to the records during reasonable business hours. Such access need not be given to any such set
of records more often than [***] or more than [***] after the date of any report to be audited. Penn shall provide Licensee and, as applicable, its Affiliates with written notice of its election to inspect and audit the records related to
the Fees due hereunder not less than [***] prior to the proposed date of review of Licensee’s and, as applicable, its Affiliates’ records by Penn’s auditors. Should the auditor find any underpayment of Fees by Licensee, Licensee shall (a)
promptly pay Penn the amount of such underpayment; (b) shall reimburse Penn for the cost of the audit, if such underpayment equals or exceeds the higher of (i) [***] or (ii) [***]; and (c) provide such auditors with an audit right
exercisable within [***] after Penn receives the audit report. If the auditor finds overpayment by Licensee, then Licensee shall have the right to deduct the overpayment from any future Fees due to Penn by Licensee. Licensee may designate
competitively sensitive information which such auditor may see and review but which it may not disclose to Penn; provided, however, that such designation shall not restrict the auditor’s investigation or conclusions.
|
| 4.10 |
Taxes. All payments made by Licensee or its Affiliates to Penn under the Agreement shall be made free and clear of and
without any deduction for or on account of any Taxes on or with respect to such payments.
|
| 5.1 |
Patent Filing Prosecution and Maintenance.
|
|
|
(a) |
Penn Patent Rights will be held in the name of Penn and obtained with counsel selected by Penn and reasonably acceptable to Licensee (“Patent Counsel”). Penn
shall control all actions and decisions with respect to the filing, prosecution and maintenance of Penn Patent Rights and will consider any reasonable comments or suggestions by Licensee with respect to same. Penn will instruct Patent
Counsel to copy Licensee on all correspondence related to Penn Patent Rights (including copies of each patent application, office action, response to office action, request for terminal disclaimer, and request for reissue or reexamination
of any patent or patent application) and to interact with Licensee with respect to the preparation, filing, prosecution and maintenance of Penn Patent Rights. Penn has the right to take action to preserve rights and minimize cost whether or
not Licensee has commented, and will use reasonable efforts to not allow any Penn Patent Rights for which Licensee is licensed and is underwriting the costs to lapse or become abandoned without Licensee’s written authorization under this
Agreement, except for filing of continuations, divisionals, or the like that substitute for the lapsed application, provided that, Penn shall have no requirement to file, prosecute, or maintain Penn Patent Rights if Licensee is not current
with the Patent Cost obligations as set forth in this Agreement. For the purposes of this Agreement, “maintenance” of the Penn Patent Rights includes inter partes patent review proceedings before the USPTO or a similar patent administration
outside the US.
|
|
|
(b) |
Licensee has the right to request that a patent application be filed in a country or territory via a written request to Penn [***] prior to the deadline set by the patent office in the territory in which filing is to take place (“Prosecution Request”). [***].
|
| 5.2 |
Patent Costs.
|
|
|
(a) |
Within [***] of the Effective Date, Licensee will reimburse Penn for all out-of pocket costs for the filing, prosecution and maintenance of Penn Patent Rights, including all accrued attorney fees, expenses, official and filing fees (“Patent Costs”), incurred prior to the Effective Date (“Historic Patent Costs”), which are estimated to be approximately [***] as of the Effective Date.
|
|
|
(b) |
Licensee will bear a Pro Rata Share of all Patent Costs incurred during the Term (“Ongoing Patent Costs”). The “Pro Rata Share” will be calculated by
[***].
|
|
|
(c) |
At any time, at Penn’s request, Licensee shall pay in advance the Patent Counsel’s estimated costs for undertaking material patent actions before Penn authorizes the Patent Counsel to proceed (“Advance
Payment”). Notwithstanding whether Licensee makes an Advance Payment for any patent action, Licensee shall bear all Patent Costs incurred during the Term and shall pay such amounts within [***] of receipt of invoice for such
patent actions. For clarity, the term “Patent Costs” means and includes Historic Patent Costs and Ongoing Patent Costs.
|
| 5.3 |
Termination of Rights in, and Obligations with respect to, Certain Penn Patent Rights. Licensee may terminate its rights in,
and obligations with respect to any or all of Penn Patent Rights by providing written notice to Penn (“Patent Termination Notice”). Termination of Licensee’s rights in and obligation with respect
to such Penn Patent Right will be effective [***] after receipt of such Patent Termination Notice by Penn. Penn will use reasonable efforts to curtail Patent Costs chargeable to Licensee under this Agreement after the receipt of the
Patent Termination Notice is received. Penn may continue prosecution and maintenance of such Patent Rights at its sole discretion and expense, and such Patent Rights will then be Carve-Out Patent Rights and therefore not subject to this
Agreement, including the License, and Licensee will have no further rights or license to them.
|
| 5.4 |
Infringement.
|
|
|
(a) |
Notice. Each party will notify the other promptly of any infringement of the Intellectual Property rights that may come to
its attention. Both Penn and Licensee will use their diligent efforts to cooperate with each other to terminate such infringement without litigation.
|
|
|
(b) |
Control. As between Licensee and Penn, Penn shall have the exclusive right to initiate litigation with respect to
infringement of the Intellectual Property rights.
|
|
|
(c) |
Cooperation. In any litigation under this Paragraph 5.4, each Party, at the request and sole expense of the other Party,
will provide reasonable cooperation to such other Party. This Paragraph 5.4 will not be construed to require either Party to undertake any activities, including legal discovery, at the request of any Third Party, except as may be required
by lawful process of a court of competent jurisdiction.
|
| 5.5 |
Marking.
|
|
|
(a) |
Patent. Licensee shall maintain any patent notice included by Penn on any Product (or its packaging where appropriate and
practicable) Used under this Agreement.
|
|
|
(b) |
Copyright. Licensee shall maintain markings included by Penn on any Product (or its packaging where appropriate and
practicable) Used under this Agreement, including the following:
|
| 5.6 |
Confidentiality.
|
|
|
(a) |
Each Party agrees that, for the Term and for [***] thereafter, such Party shall (i) use the same degree of care to maintain the secrecy of the Confidential Information of the other Party that it uses to maintain the secrecy of its
Confidential Information of like kind, (ii) use the Confidential Information of the other Party only to accomplish the purpose of this Agreement or for audit or management purposes and (iii) ensure that any employees, customers, and
distributors are bound to it by similar obligations of confidence and to make sure such disclosure occurs only as required to accomplish the purposes of this Agreement.
|
|
|
(b) |
A Party may disclose the Confidential Information of the other Party to the extent required by Law or court order; provided, however, that the recipient promptly provides to the disclosing Party prior written notice of such disclosure
and provides reasonable assistance in obtaining an order or other remedy protecting the Confidential Information from public disclosure.
|
| 6.1 |
Mutual Representations and Warranties. Each Party represents and warrants to the other Party that, as of the Effective
Date:
|
|
|
(a) |
such Party is duly organized and validly existing under the Laws of the jurisdiction of its incorporation or organization;
|
|
|
(b) |
such Party has taken all action necessary to authorize the execution and delivery of this Agreement and the performance of its obligations under this Agreement;
|
|
|
(c) |
this Agreement is a legal and valid obligation of such Party, binding upon such Party and enforceable against such Party in accordance with the terms of this Agreement, except as enforcement may be limited by applicable bankruptcy,
fraudulent conveyance, insolvency, reorganization, moratorium and other laws relating to or affecting creditors’ rights generally and by general equitable principles; and
|
|
|
(d) |
such Party has all right, power and authority to enter into this Agreement, to perform its obligations under this Agreement.
|
| 6.2 |
[***].
|
| 6.3 |
Disclaimer of Representations and Warranties.
|
|
|
(a) |
Other than the representations and warranties provided in Section 6.1 and representations in Section 6.2 above, PENN MAKES NO REPRESENTATIONS AND WARRANTIES, WHETHER EXPRESS OR IMPLIED, AND EXPLICITLY
DISCLAIMS ANY REPRESENTATION AND WARRANTY, INCLUDING WITH RESPECT TO ANY ACCURACY, COMPLETENESS, MERCHANTABILITY, FITNESS FOR A PARTICULAR PURPOSE, COMMERCIAL UTILITY, NON-INFRINGEMENT OR TITLE FOR THE INTELLECTUAL PROPERTY, PATENT
RIGHTS, COPYRIGHT RIGHTS, LICENSE AND ANY PRODUCT.
|
|
|
(b) |
Furthermore, nothing in this Agreement will be construed as:
|
|
|
i. |
A representation or warranty by Penn as to the validity or scope of any Intellectual Property rights;
|
|
|
ii. |
A representation or warranty that anything made, used, sold or otherwise disposed of under the License is or will be free from infringement of patents, copyrights, trademarks or any other forms of intellectual property rights or tangible
property rights of Third Parties;
|
|
|
iii. |
Obligating Penn to bring or prosecute actions or suits against Third Parties for patent, copyright or trademark infringement;
|
|
|
iv. |
Conferring by implication, estoppel or otherwise any license or rights under any Intellectual Property rights of Penn other than as set forth herein, regardless of whether such Intellectual Property rights are dominant or subordinate to
the Intellectual Property rights under the License; and
|
|
|
v. |
Obligating Penn to furnish any know-how.
|
| 6.4 |
Covenants .
|
| 6.4.1 |
Licensee and its Affiliates will not, directly or indirectly (including where such is done by a Third Party on behalf of Licensee or its Affiliates, at the urging of Licensee or its Affiliates or with the assistance of the Licensee or
its Affiliates) challenge the validity, scope, or enforceability of or otherwise oppose any Intellectual Property right, provided that if any Intellectual Property rights are asserted against Licensee or its Affiliate for activities
authorized under this Agreement, then such Licensee or its Affiliates is entitled to all and any defenses available to it including challenging the validity or enforceability of such Intellectual Property rights.
|
|
|
6.4.2 |
Licensee and its Affiliates will comply with all Laws that apply to its activities or obligations under this Agreement. For example, Licensee and its Affiliates will comply with applicable United States export laws and regulations. The
transfer of certain technical data and commodities may require a license from the applicable agency of the United States government and/or written assurances by Licensee and/or its Affiliates that Licensee and/or its Affiliates will not
export data or commodities to certain foreign countries without prior approval of the agency.
|
|
|
6.4.3 |
Licensee will not grant a security interest in the License or this Agreement.
|
| 7.1 |
Indemnification by Licensee.
|
|
|
7.1.1 |
Licensee shall defend, indemnify and hold Penn and its respective trustees, officers, faculty, students, employees, contractors and agents (the “Penn Indemnitees”) harmless
from and against any and all liability, damage, loss, cost or expense (including reasonable attorneys’ fees), including, without limitation, bodily injury, risk of bodily injury, death and property damage to the extent arising out of Third
Party claims or suits related to (a) this Agreement, Provider Agreements, or Joinder Agreements, including (i) the use or other disposition of any Product (including any product liability claim), (ii) any claim by a Third Party that the
practice of Intellectual Property rights or creation of any Derivative Works or the design, use, or other disposition of any Product infringes or violates any patent, copyright, trade secret, trademark or other intellectual property right
of such Third Party, or (iii) any breach of this Agreement or Laws by Licensee or its Affiliates or Service Providers and (b) Licensee’s or its Affiliates’ or its Service Providers’ negligence, omissions or willful misconduct, provided that
Licensee’s obligations pursuant to this Section 7.1 shall not apply to the extent such claims or suits result from (1) the gross negligence or willful misconduct of any of Penn Indemnitees as determined by a court of law or (2) any exercise
of Penn’s retained rights pursuant to Section 2.4.
|
|
|
7.1.2 |
As a condition to a Penn Indemnitee’s right to receive indemnification under this Section 7.1, Penn shall: (a) promptly notify Licensee as soon as it becomes aware of a claim or suit for which indemnification may be sought pursuant
hereto; (b) reasonably cooperate, and cause the individual Penn Indemnitees to reasonably cooperate, with Licensee in the defense, settlement or compromise of such claim or suit; and (c) permit the Licensee to control the defense,
settlement or compromise of such claim or suit, including the right to select defense counsel. In no event, however, may Licensee compromise or settle any claim or suit in a manner which (i) admits fault or negligence on the part of Penn or
any other Penn Indemnitee; (ii) commits Penn or any other Penn Indemnitee to take, or forbear to take, any action, without the prior written consent of Penn, or (iii) grant any rights under the Intellectual Property rights. Penn shall
reasonably cooperate with Licensee and its counsel in the course of the defense of any such suit, claim or demand, such cooperation to include without limitation using reasonable efforts to provide or make available documents, information
and witnesses.
|
|
|
7.1.3 |
Notwithstanding Section 7.1.2 above, in the event that a bona fide conflict exists between Licensee and Penn or any other Penn Indemnitee with respect to a claim or suit subject to indemnification hereunder, then Penn or any other Penn
Indemnitee shall have the right to defend against any such claim or suit itself, including by selecting its own counsel, with any documented and reasonable attorney’s fees and litigation expenses being paid for by [***]. [***].
|
| 7.2 |
Insurance.
|
|
|
7.2.1 |
Licensee, at its sole cost and expense, must insure Licensee’s and its Providers’ (if any) activities in connection with the exercise of rights and performance of obligations under this Agreement and obtain, and keep in force and
maintain Commercial Form General Liability Insurance (contractual liability included) with limits as follows:
|
|
|
i. |
Each occurrence |
$[***]; |
|
|
ii. |
General aggregate |
$[***]; |
|
|
iii. |
Clinical trials liability insurance |
$[***] |
|
|
7.2.2 |
If the above insurance is written on a claims-made form, it shall continue for [***] following termination or expiration of this Agreement. The insurance shall have a retroactive date of placement prior to or coinciding with the
Effective Date of this Agreement (or the date of commencement of Clinical Trials, as applicable to the insurance required under Section 7.2.1.iii).
|
|
|
7.2.3 |
Licensee expressly understands, however, that the coverages and limits in Section 7.2.1 do not in any way limit Licensee’s liability or indemnification obligations. The insurance will:
|
|
|
i. |
Be issued by an insurance carrier with an A.M. Best rating of “A” or better;
|
|
|
ii. |
Provide for [***] advance written notice to Penn of any cancellation or termination of such coverages;
|
|
|
iii. |
State that Penn is endorsed as an additional insured with respect to the coverages in Section 7.2.1; and
|
|
|
iv. |
Include a provision that the coverages will be primary and will not participate with nor will be excess over any valid and collective insurance or program of self-insurance carried or maintained by Penn.
|
|
|
7.2.4 |
Licensee must furnish to Penn with (i) valid certificate of insurance evidencing compliance with all requirements of this Agreement and (ii) additional insured endorsements for applicable policies naming “The Trustees of the University
of Pennsylvania” as an additional insured. Licensee must furnish all documents within [***] of the Effective Date, once per year thereafter and at any time there is a modification in such insurance.
|
| 7.3 |
LIMITATION OF LIABILITY. IN NO EVENT SHALL EITHER PARTY OR ANY OF ITS AFFILIATES BE LIABLE TO THE OTHER PARTY OR ANY OF ITS
AFFILIATES FOR SPECIAL, INDIRECT, INCIDENTAL, CONSEQUENTIAL OR PUNITIVE DAMAGES, INCLUDING LOSS OF PROFITS, WHETHER IN CONTRACT, WARRANTY, TORT, NEGLIGENCE, STRICT LIABILITY OR OTHERWISE ARISING OUT OF OR RELATING TO THIS AGREEMENT, THE
TRANSACTIONS CONTEMPLATED HEREIN OR ANY BREACH HEREOF. NOTWITHSTANDING THE FOREGOING, NOTHING IN THIS AGREEMENT SHALL LIMIT LICENSEE’S INDEMNIFICATION OBLIGATIONS UNDER SECTION 7.1 ABOVE OR SHALL LIMIT PENN’S REMEDIES OR ABILITY TO
RECOVER DAMAGES, INCLUDING INCREASED DAMAGES, FOR WILLFUL INFRINGEMENT IN THE EVENT PENN ASSERTS ITS INTELLECTUAL PROPERTY RIGHTS.
|
| 8.1 |
Term. The term of this Agreement (the “Term”) shall commence on the Effective Date and, unless terminated sooner as
provided below, shall continue in full force and effect until six (6) months after the conclusion of all Clinical Trials.
|
| 8.2 |
Termination of the Agreement for Convenience. At any time during the Term, Licensee may, at its convenience, terminate this
Agreement upon providing at least [***] prior written notice to Penn of such intention to terminate, provided that Licensee (i) ceases and causes its Affiliates to cease, using the License and any Product and (ii) terminates any Joinder
Agreements and Provider Agreements.
|
| 8.3 |
Termination For Cause.
|
|
|
8.3.1 |
If Licensee fails to fulfill its obligations under Section 3.1 (i.e. use Commercially Reasonable Efforts to Use the Product), Penn may provide written notice to Licensee of such failure. If Licensee fails to address such failure to the
reasonable satisfaction of Penn within [***] of receiving such written notice, Penn may terminate this Agreement upon written notice to Licensee.
|
|
|
8.3.2 |
If Licensee materially breaches any of its material obligations under this Agreement (other than Section 3.1), Penn may give to Licensee a written notice specifying the nature of the default, requiring it to cure such breach, and stating
its intention to terminate this Agreement. If such breach is not cured within [***] of such notice, such termination shall become effective upon a notice of termination by Penn thereafter. For clarity, a breach of a material obligation
includes: [***].
|
|
|
8.3.3 |
In addition to all other remedies available to it, Penn may terminate this Agreement, upon written notice, with immediate effect, upon a breach of [***], provided, however, that in the event that, in the sole discretion of Penn, such
breach is curable without adverse effect on Penn, Licensee will have [***] from receipt of such written notice to cure any breach under [***] and, if so cured, the Agreement shall not terminate.
|
|
|
8.3.4 |
Penn may terminate this Agreement, upon written notice, with immediate effect if, at any time, Licensee is unable to pay its debts, when they come due, or files in any court or agency pursuant to any statute or regulation of any state,
country or jurisdiction, a petition in bankruptcy or insolvency or for reorganization or for an arrangement or for the appointment of a receiver or trustee of Licensee or of its assets, or if Licensee proposes a written agreement of
composition or extension of its debts, or if Licensee is served with an involuntary petition against it, filed in any insolvency proceeding, and such petition is not dismissed within [***] after the filing thereof, or if Licensee proposes
or is a party to any dissolution or liquidation, or if Licensee makes an assignment for the benefit of its creditors of all or substantially all its assets (in each case, “Bankruptcy Action”).
|
| 8.4 |
Effects of Termination.
|
|
|
8.4.1 |
Notwithstanding the termination of this Agreement, the following provisions shall survive: Sections 4.8 - 4.10, inclusive, 5.6, 6.1, 6.2, and 8.4 and Articles 7 and 9.
|
|
|
8.4.2 |
Termination of this Agreement shall not relieve the Parties of any obligation or liability that, at the time of termination, has already accrued hereunder, or which is attributable to a period prior to the effective date of such
termination. Termination of this Agreement shall not preclude either Party from pursuing all rights and remedies it may have hereunder or at Law or in equity with respect to any breach of this Agreement nor prejudice either Party’s right to
obtain performance of any obligation.
|
| 9.1 |
Relationship of the Parties. Nothing in this Agreement is intended or shall be deemed, for financial, tax, legal or other
purposes, to constitute a partnership, agency, joint venture or employer-employee relationship between the Parties. The Parties are independent contractors and at no time will either Party make commitments or incur any charges or expenses
for or on behalf of the other Party.
|
| 9.2 |
Expenses. Except as otherwise provided in this Agreement, each Party shall pay its own expenses and costs incidental to the
preparation of this Agreement and to the consummation of the transactions contemplated hereby.
|
| 9.3 |
Use of Names. Licensee and its Providers may not use the name, logo, seal, trademark, or service mark (including any
adaptation of them) of Penn or any Penn school, organization, employee, student or representative, without the prior written consent of Penn (except as required by Section 5.5). Notwithstanding the foregoing, Licensee may use the name of
Penn in a non-misleading and factual manner solely in (a) executive summaries, business plans, offering memoranda and other similar documents used by Licensee for the purpose of raising financing for the operations of Licensee as related
to Product, or entering into commercial contracts with Third Parties, but in such case only to the extent necessary to inform a reader that the Intellectual Property rights has been licensed by Licensee from Penn, and to inform a reader
of the identity and published credentials of Inventors of the Intellectual Property, and (b) any securities reports required to be filed with the Securities and Exchange Commission.
|
| 9.4 |
No Discrimination. Neither Penn nor Licensee or its Affiliates will discriminate against any employee or applicant for
employment because of race, color, sex, sexual or affectional preference, age, religion, national or ethnic origin, handicap, or veteran status.
|
| 9.5 |
Successors and Assignment.
|
|
|
9.5.1 |
The terms and provisions hereof shall inure to the benefit of, and be binding upon, the Parties and their respective successors and permitted assigns.
|
|
|
9.5.2 |
Licensee may not assign or transfer this Agreement or any of Licensee’s rights or obligations created hereunder, by operation of law or otherwise, without the prior written consent of Penn, provided that Penn shall not unreasonably
withhold, condition or delay its consent. Licensee may assign or transfer this Agreement in its entirety without the consent of Penn in connection with a merger, consolidation, or sale or transfer of all or substantially all of its assets,
without any requirement to obtain Penn’s consent, to an unrelated third party entity provided that: (i) [***]; (ii) there exists no breach by Licensee or its Affiliates of any term of this Agreement, including those caused by a Provider,
and Licensee is not in breach of payment or diligence obligations hereunder that has not been cured as of the consummation of such transaction; (iii) the Licensee delivers to Penn [***] written notice of the proposed assignment when such
notice may be provided in accordance with applicable securities laws and non-disclosure agreements, (iv) the assignee agrees in writing to be legally bound by this Agreement and to deliver to Penn an updated Clinical Trial Protocol within
[***] after the closing of the proposed transaction and (v) the assignment is made as a part of and in connection with an asset sale, stock sale, merger or other combination, or any other transfer of Licensee’s entire business. Any
permitted assignment will not relieve Licensee of responsibility for performance of any obligation of Licensee that has accrued at the time of the assignment.
|
|
|
9.5.3 |
Any assignment not in accordance with this Section 9.5 shall be void.
|
| 9.6 |
Further Actions. Each Party agrees to execute, acknowledge and deliver such further instruments and to do all such other
acts as may be necessary or appropriate in order to carry out the purposes and intent of this Agreement.
|
| 9.7 |
Entire Agreement of the Parties; Amendments. This Agreement, the Exhibits and Appendices or Schedules hereto constitute and
contain the entire understanding and agreement of the Parties respecting the subject matter hereof and cancel and supersede any and all prior negotiations, correspondence, understandings and agreements between the Parties, whether oral or
written, regarding such subject matter. No waiver, modification or amendment of any provision of this Agreement shall be valid or effective unless made in a writing referencing this Agreement and signed by a duly authorized officer of
each Party.
|
| 9.8 |
Governing Law. This Agreement shall be governed by and interpreted in accordance with the laws of the Commonwealth of
Pennsylvania, excluding application of any conflict of laws principles that would require application of the law of a jurisdiction outside of the Commonwealth of Pennsylvania.
|
| 9.9 |
Dispute Resolution. If a dispute arises between the Parties concerning this Agreement, then the Parties will confer, as
soon as practicable, in an attempt to resolve the dispute. If such dispute remains unresolved, it will be escalated to Licensee’s Chief Executive Officer and Penn Center for Innovation’s Managing Director or their respective designee(s),
for discussion in good faith. If the Parties are unable to resolve such dispute amicably within [***] of submission to such officers, then the Parties will submit to the exclusive jurisdiction of, and venue in, the state and Federal
courts located in the Eastern District of Pennsylvania.
|
| 9.10 |
Notices and Deliveries. Any notice, request, approval or consent required or permitted to be given under this Agreement
shall be in writing and directed to a Party at its address or facsimile number shown below or such other address or facsimile number as such Party shall have last given by notice to the other Party. A notice will be deemed received: if
delivered personally, on the date of delivery; if mailed, five (5) days after deposit in the United States mail; if sent via courier, one (1) business day after deposit with the courier service; or if sent via facsimile, upon receipt of
confirmation of transmission provided that a confirming copy of such notice is sent by certified mail, postage prepaid, return receipt requested.
|
|
For Penn
Penn Center for Innovation
University of Pennsylvania
3600 Civic Center Blvd.,
9th Floor
Philadelphia, PA 19104-4310
Attention: Managing Director
|
with a copy to:
University of Pennsylvania
Office of General Counsel
2929 Walnut Street, Suite 400
Philadelphia, PA 19104-5509
Attention: General Counsel
|
|
For Licensee:
Opus Genetics, Inc
223 S. West Street, Suite 900
Raleigh, NC 27603
Attention: Chief Executive Officer
|
with a copy to:
Smith, Anderson, Blount, Dorsett, Mitchel
& Jernigan, LLP
150 Fayetteville Street, Suite 2300
Raleigh, NC 27601
Attention: [***]
|
| 9.11 |
Waiver. A waiver by either Party of any of the terms and conditions of this Agreement in any instance
shall not be deemed or construed to be a waiver of such term or condition for the future, or of any other term or condition hereof. All rights, remedies, undertakings, obligations and agreements contained in this Agreement shall be
cumulative and none of them shall be in limitation of any other remedy, right, undertaking, obligation or agreement of either Party.
|
| 9.12 |
Severability. When possible, each provision of this Agreement will be interpreted in such manner as to be effective and
valid under law, but if any provision of this Agreement is held to be prohibited by or invalid under law, such provision will be ineffective only to the extent of such prohibition or invalidity, without invalidating the remainder of this
Agreement. The Parties shall make a good faith effort to replace the invalid or unenforceable provision with a valid one which in its economic effect is most consistent with the invalid or unenforceable provision.
|
| 9.13 |
Interpretation. The words “include,” “includes” and “including” shall be deemed to be followed by the phrase “without
limitation.” All references herein to Articles, Sections, Schedules and Exhibits shall be deemed references to Articles and Sections of, Schedules and Exhibits to, this Agreement unless the context shall otherwise require. Except as
otherwise expressly provided herein, all terms of an accounting or financial nature shall be construed in accordance with GAAP, as in effect from time to time. Unless the context otherwise requires, countries shall include territories.
References to any specific Law or article, section or other division thereof, shall be deemed to include the then-current amendments or any replacement Law thereto.
|
| 9.14 |
Counterparts. This Agreement may be executed in counterparts, each of which will be deemed an original, and all of which
together will be deemed to be one and the same instrument. A facsimile or a portable document format (PDF) copy of this Agreement, including the signature pages, will be deemed an original.
|
| 9.15 |
Timely Countersignature. The terms and conditions of this Agreement shall, at Penn’s sole option, be considered by Penn to
be withdrawn from Licensee’s consideration and the terms and conditions of this Agreement, and the Agreement itself to be null and void, unless this Agreement is executed by Licensee and a fully executed original is received by Penn
within thirty (30) days from the date of Penn’s signature found below.
|
|
By:
|
/s/ Benjamin C. Dibling, Ph.D. |
|
By:
|
/s/ Ben Yerxa |
| Name: |
Benjamin C. Dibling, Ph.D | Name: |
Ben Yerxa | |
|
Title:
|
Deputy Managing Director, Penn Center for Innovation |
Title:
|
Chief Executive Officer | |
|
Date:
|
3/2/2023 | Date: | Mar 3, 2023 |
| I. |
SUMMARY.
|
| II. |
STATEMENT OF POLICIES PROHIBITING INSIDER TRADING.
|
|
|
1. |
This Policy does not apply to the exercise of stock options or the vesting of restricted stock units or restricted stock, in each case granted under Company’s equity compensation plans.
This Policy does apply, however, to any sale of Company stock as part of a broker-assisted cashless option exercise, or any other market sale of the Company Stock received upon exercise or vesting of any equity award, whether or not for the
purpose of generating the cash needed to pay the exercise price of a stock option or to pay taxes. In addition, for purposes of this Policy, the Company considers sell-to-cover transactions solely for the purpose of paying withholding taxes
upon the vesting and delivery of restricted stock units to be exempt from this Policy if such sale is required by the Company in accordance with the terms of the equity award and not upon the directive of the employee.
|
|
|
2. |
This Policy does not apply to the surrender of shares directly to the Company to satisfy
tax withholding obligations as a result of the issuance of shares upon vesting or exercise of restricted stock units, stock options or other equity awards granted under the Company’s equity compensation plans. Of course, any market sale of
the Company Stock received upon exercise or vesting of any such equity awards remains subject to all provisions of this
Policy, whether or not for the purpose of generating the cash needed to pay the exercise price or pay taxes.
|
|
|
3. |
This Policy does not apply to acquisition of Company Stock on periodic designated dates
in accordance with the Company’s Non-Employee Directors’ Compensation Policy (the “Directors’
Compensation Policy”). This Policy does apply, however, to a director’s election to receive Company Stock in lieu of cash compensation under the Directors’ Compensation Policy. Accordingly, such elections may not be effected during a Black-Out Period or
when a director is otherwise in possession of material, non- public information relating to the Company or any of its securities.
|
| III. |
EXPLANATION OF INSIDER TRADING.
|
|
|
• |
Transactions by insiders while in possession of material, non-public information;
|
|
|
• |
Transactions by persons other than insiders while in possession of material, non-public information where the information either was given in breach of an insider’s fiduciary duty to keep
it confidential or was misappropriated; or
|
|
|
• |
Communicating or tipping material, non-public information to others, including recommending transactions in a security while in possession of such information.
|
|
A.
|
Definition of “Material”
|
|
|
• |
significant clinical trial results;
|
|
|
• |
regulatory approvals and significant discussions with the U.S. Federal Drug Administration;
|
|
|
• |
significant safety incidents;
|
|
|
• |
internal financial information which departs in any way from what the market would expect;
|
|
|
• |
changes in sales, earnings or dividends;
|
|
|
• |
an important financing transaction;
|
|
|
• |
stock splits or other transactions relating to Company securities;
|
|
|
• |
mergers, tender offers or acquisitions of other companies, or major purchases or sales of assets;
|
|
|
• |
major management changes;
|
|
|
• |
sales or purchases by the Company of its own securities;
|
|
|
• |
major litigation or regulatory developments;
|
|
|
• |
significant process or product developments;
|
|
|
• |
gain or loss of a major supplier;
|
|
|
• |
major transactions with other companies or entities, such as joint ventures, collaboration agreements or licensing agreements;
|
|
|
• |
the extent to which external events, including but not limited to pandemics, have had or will have a material impact on the Company’s operating results; and
|
|
|
• |
a major cybersecurity incident.
|
|
B.
|
Definition of “Non-Public”
|
|
C.
|
Who is an Insider?
|
|
D.
|
Trading by Persons Other than Insiders (Tipping).
|
|
E.
|
Penalties for Engaging in Insider Trading.
|
|
|
• |
SEC administrative sanctions;
|
|
|
• |
Securities industry self-regulatory organization sanctions;
|
|
|
• |
Civil injunctions;
|
|
|
• |
Damage awards to private plaintiffs;
|
|
|
• |
Disgorgement of all profits;
|
|
|
• |
Civil fines for the violator of up to three (3) times the amount of profit gained or loss avoided;
|
|
|
• |
Civil fines for the employer or other controlling person of a violator (i.e., where the violator is an employee or other controlled person) of up to the greater of $1,000,000 or three (3)
times the amount of profit gained or loss avoided by the violator;
|
|
|
• |
Criminal fines for individual violators of up to $5,000,000 ($25,000,000 for an entity); and
|
|
|
• |
Jail sentences of up to twenty (20) years.
|
|
F.
|
Examples of Insider Trading.
|
|
G.
|
Short-Swing Profits, Rule 144 and Short Sales.
|
| IV. |
STATEMENT OF PROCEDURES PREVENTING INSIDER TRADING.
|
|
A.
|
Pre-Clearance of Trades by Officers, Directors, Employees and Subject
Contractors.
|
|
|
• |
the vesting of restricted stock units or restricted stock;
|
|
|
• |
sell-to-cover transactions solely for the purpose of paying withholding taxes upon the vesting and delivery of restricted stock units, if such sale is required by the Company in accordance
with the terms of the equity award and not upon the directive of the employee;
|
|
|
• |
purchases of Company Stock under on periodic designated dates in accordance with the Directors’ Compensation Policy; and
|
|
|
• |
transactions effected under an approved Rule 10b5-1 Trading Plan as set forth in Section V below.
|
|
B.
|
Avoidance of Certain Aggressive or Speculative Trading.
|
|
|
1. |
PROHIBITION OF SPECULATIVE TRADING/HEDGING. All
directors, officers, employees and Subject Contractors are prohibited from engaging in short sales; transactions in put or call options, hedging or monetization transactions, including the purchase or sale of puts or calls or the use of any
other derivative instruments; or other inherently speculative transactions with respect to the securities of the Company at any time.
|
|
|
2. |
PROHIBITION ON PLEDGING. All directors, officers,
employees and Subject Contractors are prohibited from holding any securities of the Company in a margin account or otherwise pledging any securities of the Company as collateral for any loan.
|
| V. |
RULE 10B5-1 TRADING PLANS.
|
|
A.
|
Overview.
|
|
|
• |
First, before becoming aware of the information, the individual enters into a binding contract to purchase or sell the securities, provides instructions to another person to sell the
securities or adopts a written plan for trading the securities in good faith (i.e., the Trading Plan).
|
|
|
• |
Second, the Trading Plan must either:
|
|
|
° |
specify the amount of securities to be purchased or sold, the price at which the securities are to be purchased or sold and the date on which the securities are to be purchased or sold;
|
|
|
° |
include a written formula or computer program for determining the amount, price and date of the transactions; or
|
|
|
° |
prohibit the individual from exercising any subsequent influence over the purchase or sale of the Company’s Stock under the Trading Plan in question.
|
|
|
• |
Third, the purchase or sale must occur pursuant to the Trading Plan and the individual must not enter into a corresponding hedging transaction or alter or deviate from the Trading Plan.
|
|
B.
|
Termination/Amendments to Trading Plans.
|
|
C.
|
Discretionary Plans.
|
|
D.
|
Trades Outside of a Trading Plan.
|
|
E.
|
Disclosures.
|
|
F.
|
Policy Takes Precedence.
|
| VI. |
EXECUTION AND RETURN OF CERTIFICATION OF COMPLIANCE.
|
|
|
Transaction Vehicle (check one)
|
|
Transaction Initiated By (check one)
|
||
|
|
☐ | Open Market Transaction |
|
☐ | Employee or immediate family member directly |
|
|
☐ | Equity Compensation Plan |
|
☐ | Court or government decree (e.g., divorce decree) |
|
|
☐ | Other (specify): |
|
☐ | Broker (provide name, firm, telephone and e-mail): |
|
|
|
|
|
|
|
|
|
Type of Transaction (check one)
|
||||
|
|
☐ | Purchase or acquire common stock | |||
|
|
☐ | Sell or dispose of common stock | |||
|
|
☐ | Move Company Securities from one account to another (e.g., in or out of a trust) | |||
|
|
☐ | Dispose of fractional shares | |||
|
|
☐ | Pledge Company Securities for margin account, or otherwise | |||
|
|
☐ | Exercise options without subsequent sale | |||
|
|
☐ | Exercise options with subsequent sale (including a “cashless exercise”) | |||
|
|
☐ | Other (describe): | |||
|
|
Number of securities:
|
|
|
|
|
Estimated share price:
|
|
|
|
Contemplated execution date:
|
|
|
|
Date of your last “opposite way” transaction**:
|
|
|
|
Signature:
|
|
|
|
Print Name:
|
|
|
|
Date:
|
|
|
TO:
|
Compliance Officer
|
| FROM: |
|
|
|
RE:
|
INSIDER TRADING COMPLIANCE POLICY OF OCUPHIRE PHARMA, INC.
|
|
SIGNATURE
|
DATE
|
|
|
TITLE
|
|
TO:
|
Compliance Officer
|
|
RE:
|
INSIDER TRADING COMPLIANCE POLICY OF OCUPHIRE PHARMA, INC.
|
|
SIGNATURE
|
DATE
|
|
|
TITLE
|
|
TO:
|
Compliance Officer
|
|
RE:
|
INSIDER TRADING COMPLIANCE POLICY OF OCUPHIRE PHARMA, INC.
|
|
|
||
|
SIGNATURE
|
|
DATE
|
|
|
||
|
|
||
|
NAME
|
|
|
|
|
||
|
|
||
|
TITLE
|
|
|
Subsidiaries
|
Jurisdiction of Incorporation
|
|
Orange Merger Sub II, LLC
|
Delaware
|
|
(1)
|
Registration Statement (Form S-3 No. 333-276462) pertaining to the registration of Company debt and equity securities;
|
|
(2)
|
Registration Statement (Form S-3 No. 333-252715) as it pertains to the registration of Company common stock issuable upon the exercise of Series A/B Warrants;
|
|
(3)
|
Registration Statement (Form S-8 No. 333-282988) pertaining to the Ocuphire Pharma, Inc. 2021 Inducement Plan;
|
|
(4)
|
Registration Statement (Form S-8 No. 333-276471) pertaining to the Ocuphire Pharma, Inc. 2020 Equity Incentive Plan;
|
|
(5)
|
Registration Statement (Form S-8 No. 333-275673) pertaining to the Ocuphire Pharma, Inc. 2021 Inducement Plan;
|
|
(6)
|
Registration Statement (Form S-8 No. 333-271150) pertaining to the Ocuphire Pharma, Inc. 2020 Equity Incentive Plan;
|
|
(7)
|
Registration Statement (Form S-8 No. 333-264139) pertaining to the Ocuphire Pharma, Inc. 2020 Equity Incentive Plan;
|
|
(8)
|
Registration Statement (Form S-8 No. 333-254923) pertaining to the Ocuphire Pharma, Inc. 2021 Inducement Plan and Ocuphire Pharma, Inc. 2020 Equity Incentive Plan;
and
|
|
(9)
|
Registration Statement (Form S-8 No. 333-249978) pertaining to the Ocuphire Pharma, Inc. 2020 Equity Incentive Plan and Ocuphire Pharma, Inc. 2018 Equity Incentive
Plan.
|
| 1. |
I have reviewed this Annual Report on Form 10-K of Opus Genetics, Inc. (the “Company”);
|
| 2. |
Based on my knowledge, this report does not contain any untrue statement of a material fact or omit to state a material fact necessary to make the statements made, in light of the circumstances under which such
statements were made, not misleading with respect to the period covered by this report;
|
| 3. |
Based on my knowledge, the consolidated financial statements, and other financial information included in this report, fairly present in all material respects the financial condition, results of operations and
cash flows of the registrant as of, and for, the periods presented in this report;
|
| 4. |
The registrant’s other certifying officer(s) and I are responsible for establishing and maintaining disclosure controls and procedures (as defined in Exchange Act Rules 13a-15(e) and 15d-15(e)) and internal
control over financial reporting (as defined in Exchange Act Rules 13a-15(f) and 15d-15(f) for the registrant and have:
|
|
|
(a) |
Designed such disclosure controls and procedures, or caused such disclosure controls and procedures to be designed under our supervision, to ensure that material information relating to the registrant is made
known to us by others within those entities, particularly during the period in which this report is being prepared;
|
|
|
(b) |
Designed such internal control over financial reporting, or caused such internal control over financial reporting to be designed under our supervision, to provide reasonable assurance regarding the reliability of
financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles;
|
|
|
(c) |
Evaluated the effectiveness of the registrant’s disclosure controls and procedures and presented in this report our conclusions about the effectiveness of the disclosure controls and procedures, as of the end of
the period covered by this report based on such evaluation; and
|
|
|
(d) |
Disclosed in this report any change in the registrant’s internal control over financial reporting that occurred during the registrant’s most recent fiscal quarter (the registrant’s fourth fiscal quarter in the
case of an annual report) that has materially affected, or is reasonably likely to materially affect, the registrant’s internal control over financial reporting; and
|
| 5. |
The registrant’s other certifying officer(s) and I have disclosed, based on our most recent evaluation of internal control over financial reporting, to the registrant’s auditors and the audit committee of the
registrant’s board of directors (or persons performing the equivalent functions):
|
|
|
(a) |
all significant deficiencies and material weaknesses in the design or operation of internal control over financial reporting which are reasonably likely to adversely affect the registrant’s ability to record,
process, summarize and report financial information; and
|
|
|
(b) |
any fraud, whether or not material, that involves management or other employees who have a significant role in the registrant’s internal control over financial reporting.
|
|
Date: March 31, 2025
|
/s/ George Magrath
|
|
|
|
Name:
|
George Magrath
|
|
|
Title:
|
Chief Executive Officer
|
|
|
|
(Principal Executive Officer) |
| 1. |
I have reviewed this Annual Report on Form 10-K of Opus Genetics, Inc. (the “Company”);
|
| 2. |
Based on my knowledge, this report does not contain any untrue statement of a material fact or omit to state a material fact necessary to make the statements made, in light of the circumstances under which such
statements were made, not misleading with respect to the period covered by this report;
|
| 3. |
Based on my knowledge, the consolidated financial statements, and other financial information included in this report, fairly present in all material respects the financial condition, results of operations and
cash flows of the registrant as of, and for, the periods presented in this report;
|
| 4. |
The registrant’s other certifying officer(s) and I are responsible for establishing and maintaining disclosure controls and procedures (as defined in Exchange Act Rules 13a-15(e) and 15d-15(e)) and internal
control over financial reporting (as defined in Exchange Act Rules 13a-15(f) and 15d-15(f) for the registrant and have:
|
|
|
(a) |
Designed such disclosure controls and procedures, or caused such disclosure controls and procedures to be designed under our supervision, to ensure that material information relating to the registrant is made
known to us by others within those entities, particularly during the period in which this report is being prepared;
|
|
|
(b) |
Designed such internal control over financial reporting, or caused such internal control over financial reporting to be designed under our supervision, to provide reasonable assurance regarding the reliability of
financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles;
|
|
|
(c) |
Evaluated the effectiveness of the registrant’s disclosure controls and procedures and presented in this report our conclusions about the effectiveness of the disclosure controls and procedures, as of the end of
the period covered by this report based on such evaluation; and
|
|
|
(d) |
Disclosed in this report any change in the registrant’s internal control over financial reporting that occurred during the registrant’s most recent fiscal quarter (the registrant’s fourth fiscal quarter in the
case of an annual report) that has materially affected, or is reasonably likely to materially affect, the registrant’s internal control over financial reporting; and
|
| 5. |
The registrant’s other certifying officer(s) and I have disclosed, based on our most recent evaluation of internal control over financial reporting, to the registrant’s auditors and the audit committee of the
registrant’s board of directors (or persons performing the equivalent functions):
|
|
|
(a) |
all significant deficiencies and material weaknesses in the design or operation of internal control over financial reporting which are reasonably likely to adversely affect the registrant’s ability to record,
process, summarize and report financial information; and
|
|
|
(b) |
any fraud, whether or not material, that involves management or other employees who have a significant role in the registrant’s internal control over financial reporting.
|
|
Date: March 31, 2025
|
/s/ Nirav Jhaveri
|
|
|
|
Name:
|
Nirav Jhaveri
|
|
|
Title:
|
Chief Financial Officer
|
|
|
(Principal Financial Officer and Principal Accounting Officer)
|
|
|
Date: March 31, 2025
|
|
|
|
|
|
/s/ George Magrath
|
|
|
George Magrath
|
|
|
Chief Executive Officer
|
|
|
(Principal Executive Officer)
|
|
|
|
|
|
/s/ Nirav Jhaveri
|
|
|
Nirav Jhaveri
|
|
|
Chief Financial Officer
|
|
|
(Principal Financial Officer and Principal
|
|
| Accounting Officer) |
|