|
|
|
|
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
(Mark One)
|
☒
|
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
|
|
☐
|
TRANSITION REPORT UNDER SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
|
Commission File Number: 001-36728
|
Delaware |
|
56-2590442 |
|
(State or Other Jurisdiction of Incorporation or Organization) |
|
(I.R.S. Employer Identification No.) |
|
465 State Route 17, Ramsey, New Jersey |
|
07446 |
|
(Address of Principal Executive Offices) |
|
(Zip Code) |
Registrant’s telephone number, including area code: (201) 478-5552
Securities registered pursuant to Section 12(b) of the Act:
|
Title of each class |
Trading Symbol |
Name of each exchange on which registered |
|
Common stock, par value $0.0001 per share |
ADMA |
Nasdaq Global Market |
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☒ No ☐
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Act. Yes ☐ No ☒
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ☒ No ☐
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ☒ No ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company” and “emerging growth company” in Rule 12b-2 of the Exchange Act. (Check one):
|
Large Accelerated Filer
|
☒
|
Accelerated Filer
|
☐
|
|
Non-accelerated Filer
|
☐
|
Smaller Reporting Company
|
☒
|
|
|
|
Emerging Growth Company
|
☐
|
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act ☐
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report. Yes ☒ No ☐
If securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant included in the filing reflect the correction of an error to previously issued financial statements. ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes ☐ No ☒
The aggregate market value of the registrant’s common stock held by non-affiliates was $793,963,197 as of June 30, 2023 (the last business day of the registrant’s most recently completed second fiscal quarter), based on a total of 215,166,178 shares of common stock held by non-affiliates and a closing price of $3.69 as reported on the Nasdaq Global Market on June 30, 2023.
As of February 23, 2024, there were 228,220,236 shares of the issuer’s common stock outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the ADMA Biologics, Inc. definitive proxy statement to be filed pursuant to Regulation 14A within 120 days after the end of the fiscal year are incorporated by reference into Part III of this Annual Report on Form 10-K and certain documents are incorporated by reference into Part IV.
|
PART I
|
||
|
Item 1.
|
4
|
|
|
Item 1A.
|
31
|
|
|
Item 1B.
|
60
|
|
|
Item 1C.
|
60
|
|
|
Item 2.
|
61
|
|
|
Item 3.
|
61
|
|
|
Item 4.
|
61
|
|
|
PART II
|
||
|
Item 5.
|
62
|
|
|
Item 6.
|
63
|
|
|
Item 7.
|
64
|
|
|
Item 7A.
|
80
|
|
|
Item 8.
|
80
|
|
|
Item 9.
|
80
|
|
|
Item 9A.
|
80
|
|
|
Item 9B.
|
81
|
|
|
Item 9C.
|
81
|
|
|
PART III
|
||
|
Item 10.
|
81
|
|
|
Item 11.
|
81
|
|
|
Item 12.
|
82
|
|
|
Item 13.
|
82
|
|
|
Item 14.
|
82
|
|
|
PART IV
|
||
|
Item 15.
|
82
|
|
|
Item 16.
|
86
|
|
|
• |
our ability to manufacture ASCENIV and BIVIGAM on a commercial scale and further commercialize these products as a result of their approval by the U.S. Food and Drug Administration (the “FDA”) in 2019;
|
|
|
• |
our plans to develop, manufacture, market, launch and expand our commercial infrastructure and commercialize our current and future products and the success of such efforts;
|
|
|
• |
the safety, efficacy and expected timing of and our ability to obtain and maintain regulatory approvals for our current products and product candidates, and the labeling or nature of any such approvals;
|
|
|
• |
the achievement of or expected timing, progress and results of clinical development, clinical trials and potential regulatory approvals for our product candidates;
|
|
|
• |
our dependence upon our third-party customers and vendors and their compliance with applicable regulatory requirements;
|
|
|
• |
our belief that we have addressed the delays experienced with final drug product current Good Manufacturing Practices (“cGMP”) release testing by our third-party vendors by adding additional release testing laboratories to our
FDA-approved consortium listed in our drug approval documents;
|
|
|
• |
our ability to obtain adequate quantities of FDA-approved plasma with proper specifications;
|
|
|
• |
our plans to increase our supplies of source plasma, our ability to obtain and maintain regulatory compliance and reliance on third-party supply agreements as well as any extensions to such agreements;
|
|
|
• |
the potential indications for our products and product candidates;
|
|
|
• |
potential investigational new product applications;
|
|
|
• |
the acceptability of any of our products, including ASCENIV, BIVIGAM and Nabi-HB, for any purpose, including FDA-approved indications, by physicians, patients or payers;
|
|
|
• |
our plans to evaluate the clinical and regulatory paths to grow the ASCENIV franchise through expanded FDA-approved uses;
|
|
|
• |
Federal, state and local regulatory and business review processes and timing by such governmental and regulatory agencies of our business and regulatory submissions;
|
|
|
• |
concurrence by the FDA with our conclusions concerning our products and product candidates;
|
|
|
• |
the comparability of results of our hyperimmune and immune globulin (“IG”) products to other comparably run hyperimmune and immune globulin clinical trials;
|
|
|
• |
the potential for ASCENIV and BIVIGAM to provide meaningful clinical improvement for patients living with Primary Humoral Immunodeficiency (“PI”), also known as Primary Immunodeficiency Disease (“PIDD”) or Inborn Errors of Immunity, or
other immune deficiencies or any other condition for which the products may be prescribed or evaluated;
|
|
|
• |
our ability to market and promote Nabi-HB in a highly competitive environment with increasing competition from other antiviral therapies and to generate meaningful revenues from this product;
|
|
|
• |
our intellectual property position and the defense thereof, including our expectations regarding the scope of patent protection with respect to ASCENIV or other future pipeline product candidates;
|
|
|
• |
our ability to develop, manufacture, receive regulatory approval and commercialize our potential pipeline of any new hyperimmune globulins;
|
|
|
• |
our manufacturing capabilities, third-party contractor capabilities and vertical integration strategy;
|
|
|
• |
our plans related to the expansion and efficiencies of our manufacturing capacity, yield improvements, supply-chain robustness, in-house fill-finish capabilities, distribution and other collaborative agreements and the success of such
endeavors;
|
|
|
• |
our estimates regarding revenues, expenses, capital requirements, timing to profitability and positive cash flows and the potential need for and availability of additional financing;
|
|
|
• |
possible or likely reimbursement levels for our currently marketed products;
|
|
|
• |
estimates regarding market size, projected growth and sales of our existing products as well as our expectations of market acceptance of ASCENIV and BIVIGAM;
|
|
|
• |
pandemics, or a resurgence of a pandemic, may adversely affect our business, financial condition, liquidity or results of operations; and
|
|
|
• |
future domestic and global economic conditions including, but not limited to, supply chain constraints, inflationary pressures or performance or geopolitical conditions, including the continuing conflict in Europe or the evolving
conflict in the Middle East and surrounding areas.
|
| Item 1. |
Business
|
|
|
• |
Continue to expand the commercial production of our IG products, as well as the commercial presence, penetration and sales of ASCENIV and BIVIGAM for the treatment of patients with PI. We continue
to enhance our recruiting initiatives and expand our existing specialty commercial sales force and commercial-facing organization to market ASCENIV and BIVIGAM to appropriate sites of care including home healthcare infusion facilities,
hospitals, physician offices/clinics and other specialty treatment and infusion center organizations. We also anticipate staffing our Company with additional personnel for patient support, medical affairs, quality assurance, quality
control, inventory management, regulatory affairs, manufacturing, scientific affairs and third-party reimbursement. We currently use and may continue to partner with a network of national distributors to fulfill orders for ASCENIV and
BIVIGAM.
|
|
|
• |
Increase marketing efforts around Nabi-HB.We plan to increase our marketing efforts and attend relevant virtual or in-person medical conferences during 2024, raising awareness of the risks
associated with Hepatitis B and the benefits and efficacy of Nabi-HB in its indicated populations. We have published and may continue to publish scientific data supporting the use of Nabi-HB in at-risk and appropriate patient populations.
|
|
|
• |
Expand ASCENIV’s FDA-approved uses. Having received approval by the FDA for ASCENIV as a treatment for PIDD, we may elect to evaluate the clinical and regulatory paths to grow the ASCENIV franchise
through expanded FDA-approved uses. We believe that there may be other patient populations beyond PIDD that could potentially derive clinical benefit from ASCENIV, some of which may potentially be eligible for orphan status. We plan to
leverage our previously conducted randomized, double-blind, placebo-controlled Phase II clinical trial evaluating RI-001, RI-002’s predecessor product candidate, in immune-compromised, RSV-infected patients to explore ASCENIV for the
treatment of RSV or other potential respiratory viral pathogens, as well as in other patient populations we may believe are appropriate.
|
|
|
• |
Improve the Boca Facility’s and ADMA BioCenters’ operating efficiencies, yields and gross margins. During 2024, we plan to advance our biologic production yield enhancement initiative to capture
additional IG production yields from our manufacturing process with the same quantities of starting raw material. These initiatives are subject to further evaluation, validation of commercial-scale production and requisite regulatory
review. During 2021, we received FDA approvals for our 4,400L expanded IVIG production scale, as well as our in-house fill-finish and related operations production line using our aseptic filling machine. In 2023, ADMA commenced
manufacturing ASCENIV at the 4,400 Liter production scale for the first time in the Company’s history. We expect that this expansion should meaningfully improve the product’s margin profile and increase plant production capacity as fewer
batches will be needed to support revenue goals. Our successful experience in ramping up BIVIGAM to this production scale over the last two years lends confidence to its ability to leverage these same processes for ASCENIV. We began to
realize these benefits in the fourth quarter of 2023.
|
|
|
• |
Broadly Implement Innovative AI Program, ADMAlytics, to improve efficiencies across our supply chain and production operations. In February 2024, we announced the successful initial use of our
Artificial Intelligence (AI) program, named ADMAlytics. ADMAlytics combines AI and machine learning to improve and predict outcomes for production and operational processes. Recently, we successfully produced our first batch of ASCENIV
utilizing this innovative ADMAlytics software to prospectively automate and realize efficiency improvements to plasma pooling during commercial manufacturing. We expect broad implementation of the new ADMAlytics capabilities will provide
for rapid realization of efficiencies across our supply chain and production operations.
|
|
|
• |
Label expansion. Upon regulatory approval, the ongoing post-marketing clinical study of ASCENIV, which has successfully completed enrollment, may provide a label expansion opportunity to include
pediatric-aged PI patients as well as additional publications supporting product safety.
|
|
|
• |
Expand and develop our pipeline with additional specialty plasma and/or hyperimmune immunoglobulin products. Our core competency is in the development, manufacturing, testing and commercialization
of plasma-derived therapeutics. We believe there are a number of under-addressed medical conditions for which plasma-derived therapeutics may be beneficial. Utilizing our intellectual property patents, which include our proprietary testing
assay and other standardization methods and technologies, we have identified potential new product candidates that we may advance into preclinical activities. As part of expanding our product pipeline, we are looking to develop an S.
pneumonia hyperimmune globulin. S. pneumonia is the predominant cause of community-acquired pneumonia in the U.S., ranking as the ninth leading cause of overall mortality. We believe the strategic importance and unmet need are evident in
both the prophylactic and therapeutic settings where documented anti-infective resistance is on the rise. Annually, approximately one million U.S. adults contract pneumococcal pneumonia, resulting in 400,000 hospitalizations and a 5-7%
mortality rate, of which approximately 7,000 deaths annually are attributable to anti-infective resistance. Despite vaccine availabilities, vaccine-naive and immune-compromised patient populations remain at risk and could potentially
benefit from the immediately available neutralizing antibodies conferred with a hyperimmune globulin in both the in-patient and out-patient treatment settings. We estimate that an S. pneumonia hyperimmune globulin, if approved, has the
potential to generate peak revenue of $300-500 million.
|
|
|
• |
Secure new supply contracts for potential contract manufacturing organization (“CMO”) opportunities. We are exploring new potential CMO, contract testing and business development opportunities,
which include fill-finish capabilities, with our multi-faceted revenue generation platform, while continuing to fulfill our long-term CMO supply agreement to produce and sell plasma-derived intermediate fractions.
|
|
|
• |
antibody deficiency and recurrent bacterial infections;
|
|
|
• |
T-lymphocyte deficiency and opportunistic infections;
|
|
|
• |
other lymphocyte defects causing opportunistic infections;
|
|
|
• |
neutrophil defects causing immunodeficiency; and
|
|
|
• |
complement deficiencies.
|
|
|
• |
Red blood cells – Used to carry oxygen from the lungs to the body;
|
|
|
• |
White blood cells – Used by the immune system to fight infection;
|
|
|
• |
Platelets – Used for blood clotting; and
|
|
|
• |
Plasma – Used to carry the aforementioned components throughout the body and provide support in clotting and immunity.
|
|
|
• |
completion of extensive preclinical laboratory tests, preclinical, nonclinical and formulation studies performed in accordance with the FDA’s Good Laboratory Practice (“GLP”) regulations and other applicable laws and regulations;
|
|
|
• |
submission to the FDA of an Investigational New Drug (“IND”) application which must become effective before clinical trials may begin;
|
|
|
• |
obtaining approval by an Institutional Review Board (“IRB”) at each clinical site before a clinical trial may be initiated at that site;
|
|
|
• |
performance of adequate and well-controlled clinical trials meeting FDA requirements, commonly referred to as Good Clinical Practices (“GCP”), and other additional requirements for the protection of human research subjects and to
establish the safety and efficacy of the product candidate for each proposed indication;
|
|
|
• |
manufacturing (through an FDA-approved facility) of product in accordance with the FDA’s current Good Manufacturing Practices (“cGMP”) to be used in the clinical trials and providing manufacturing information needed in regulatory
filings;
|
|
|
• |
submission of a BLA to the FDA for marketing approval that includes substantial evidence of safety, purity and potency from results of clinical trials; the results of preclinical testing; detailed information about the Chemistry,
Manufacturing, and Controls (“CMC”) and proposed labeling and packaging for the product candidate;
|
|
|
• |
satisfactory completion of an FDA pre-approval inspection of the manufacturing facilities at which the product candidate is produced, and potentially other involved facilities as well, to assess compliance with cGMP regulations and other
applicable regulations;
|
|
|
• |
satisfactory completion of potential FDA inspections of the preclinical study and clinical trial sites that generate the data in support of the BLA; and
|
|
|
• |
FDA review and approval of a BLA prior to any commercial marketing, sale or shipment of the product, including agreement on post-marketing commitments, and compliance with any post approval commitments, such as Risk Evaluation and
Mitigation Strategies (“REMS”) and post approval studies required by the FDA.
|
|
|
• |
Phase I clinical trials are initially conducted in a limited population to test the product candidate for safety, dose tolerance, absorption, metabolism, distribution and excretion in healthy humans or, on occasion, in patients, such as
cancer patients.
|
|
|
• |
Phase II clinical trials are generally conducted in a larger but limited patient population to identify possible adverse effects and safety risks, to determine the efficacy of the product candidate for specific targeted indications and
to determine tolerance and optimal dosage. Multiple Phase II clinical trials may be conducted by the sponsor to obtain information prior to beginning larger and more expensive Phase III clinical trials.
|
|
|
• |
Phase III trials are conducted to establish the overall risk/benefit profile of the product. Certain Phase III clinical trials are referred to as pivotal trials. Phase III clinical trials aim to provide substantial evidence of
reproducibility of clinical efficacy and safety results for approval and to further test for safety in an expanded and diverse patient population at multiple, geographically dispersed clinical trial sites.
|
|
|
• |
We have a history of losses and we may, in the future, need to raise additional capital to operate our business, which may not be available on favorable terms, if at all.
|
|
|
• |
Although we achieved net income on a non-GAAP basis and generated positive cash flows for the year ended December 31, 2023 for the first time, we may not be able to achieve profitability and generate positive cashflows in the future.
|
|
|
• |
We contract with third parties for the filling, packaging, testing and labeling of the drug substance we manufacture. This reliance on third parties carries the risk that the services upon which we rely may not be performed in a timely
manner or according to our specifications, which could delay the availability of our finished drug product and could adversely affect our commercialization efforts and our revenues.
|
|
|
• |
The estimates of market opportunity and forecasts of market and revenue growth included in our filings may prove to be inaccurate, and even if the markets in which we compete achieve the forecasted growth, our business could fail to grow
at similar rates, if at all.
|
|
|
• |
Both of our business segments and our facilities are subject to periodic inspections by the FDA, which, depending on the outcome of such inspections, could result in certain FDA actions, including the issuance of observations, notices,
citations, warning letters or other enforcement actions.
|
|
|
• |
Business interruptions could adversely affect our business.
|
|
|
• |
Although we have received approval from the FDA to market ASCENIV as a treatment for PIDD, our ability to market or seek approval for ASCENIV for alternative indications could be limited and FDA could require clinical trials beyond what
we may deem to be reasonable. Unless additional clinical trials are successfully conducted and the FDA approves a BLA or other required submission for review, we may not be authorized to market ASCENIV for any other indication.
|
|
|
• |
With the approval to market ASCENIV, BIVIGAM and Nabi-HB, there can be no assurance that we will be successful in further developing and expanding commercial operations or balancing our research and development activities with our
commercialization activities.
|
|
|
• |
We depend on third-party researchers, developers and vendors to develop, manufacture, supply materials for or test our products and product candidates, and such parties are outside of our control.
|
|
|
• |
We may be unable to successfully expand our manufacturing processes to fulfill demand for our products or increase our production capabilities through the addition of new equipment, including if we do not obtain requisite approval from
the FDA.
|
|
|
• |
Our products, and any additional products for which we may obtain marketing approval in the future, could be subject to post-marketing restrictions or withdrawal from the market and we could be subject to substantial penalties if we fail
to comply with regulatory requirements or if we experience unanticipated problems with our products following approval.
|
|
|
• |
Historically, a few customers have accounted for a significant amount of our total revenue and accounts receivable and the loss of any of these customers could have a material adverse effect on our business, results of operations and
financial condition.
|
|
|
• |
Issues with product quality and compliance could have a material adverse effect upon our business, subject us to regulatory actions and cause a loss of customer confidence in us or our products.
|
|
|
• |
If physicians, payers and patients do not accept and use our current products or our future product candidates, our ability to generate revenue from these products will be materially impaired.
|
|
|
• |
Our long-term success may depend on our ability to supplement our existing product portfolio through new product development or the in-license or acquisition of other new products, product candidates and label expansion of existing
products, and if our business development efforts are not successful, our ability to achieve profitability may be adversely impacted.
|
|
|
• |
Our ADMA BioCenters operations collect information from donors in the U.S. that subjects us to consumer and health privacy laws, which could create enforcement and litigation exposure if we fail to meet their requirements.
|
|
|
• |
Our senior secured credit facility with Ares Capital Corporation and certain of its affiliates (“Ares”) is subject to acceleration in specified circumstances, which may result in Ares taking possession and disposing of any collateral.
|
|
|
• |
If we are unable to protect our patents, trade secrets or other proprietary rights, if our patents are challenged or if our provisional patent applications do not get approved, our competitiveness and business prospects may be materially
damaged.
|
|
|
• |
Cyberattacks and other security breaches could compromise our proprietary and confidential information, which could harm our business and reputation.
|
|
|
• |
Our ability to continue to produce safe and effective products depends on the safety of our plasma supply, testing by third parties and the timing of receiving the testing results, and the manufacturing processes we have in place to
counter transmittable diseases.
|
|
|
• |
We could become supply-constrained and our financial performance would suffer if we cannot obtain adequate quantities of FDA-approved source plasma with proper specifications or other necessary raw materials.
|
|
|
• |
The market price of our common stock may be volatile and may fluctuate in a way that is disproportionate to our operating performance.
|
|
|
• |
expand commercialization and marketing efforts;
|
|
|
• |
implement additional internal systems, controls and infrastructure;
|
|
|
• |
hire additional personnel; and
|
|
|
• |
expand production capacity at the Boca Facility.
|
| • |
we may be unable to identify contractors on acceptable terms or at all because the number of potential service providers is limited and the FDA must inspect and qualify any contract manufacturers for current cGMP compliance as part of
our marketing application;
|
| • |
a new fill/finisher would have to be educated in, or develop substantially equivalent processes for, the production of our products and product candidates;
|
| • |
a pandemic, or the resurgence of a pandemic such as the COVID-19 pandemic, could adversely affect our contracted fill/finishers’ operations, supply chain or workforce;
|
| • |
our contracted fill/finishers’ resources and level of expertise with plasma-derived biologics may be limited, therefore they may require a significant amount of support from us in order to implement and maintain the infrastructure and
processes required to deliver our finished drug product;
|
| • |
our third-party contractors might be unable to timely provide finished drug product or raw material plasma in sufficient quantity to meet our commercial needs;
|
| • |
contractors may not be able to execute our inspection procedures and required tests appropriately;
|
| • |
contractors are subject to ongoing periodic unannounced inspection by the FDA and corresponding state agencies to ensure strict compliance with cGMP and other government regulations, and we do not have control over third-party providers’
compliance with these regulations;
|
| • |
contractors may fail to comply with applicable regulatory requirements, placing them and us at risk of regulatory enforcement actions, recalls and other adverse consequences, and which place our patients at risk, which may negatively
impact our business and their ability to supply products to meet our development, clinical and commercial needs;
|
| • |
our third parties could breach or terminate their agreements with us; and
|
| • |
our contract fill/finishers may have unacceptable or inconsistent drug product quality success rates and yields, and we have no direct control over our contract fill/finishers’ ability to maintain adequate quality control, quality
assurance and qualified personnel.
|
|
|
• |
unforeseen safety issues;
|
|
|
• |
determination of dosing issues;
|
|
|
• |
lack of safety or effectiveness, or other adverse study results during clinical trials;
|
|
|
• |
slower than expected rates of patient recruitment or noncompliance with clinical trial requirements;
|
|
|
• |
inability to monitor patients adequately during or after treatment; and
|
|
|
• |
inability or unwillingness of medical investigators to follow our clinical protocols.
|
|
|
• |
Delays in reaching, or failure to reach, agreement on acceptable clinical trial contracts or clinical trial protocols with prospective trial sites and our CROs;
|
|
|
• |
Regulators requiring us to perform additional or unanticipated clinical trials to obtain approval or becoming subject to additional post-marketing testing, surveillance, or REMS requirements to maintain regulatory approval;
|
|
|
• |
Failure by our third-party contractors to comply with regulatory requirements or the clinical trial protocol, or meet their contractual obligations to us in a timely manner, or at all, or our being required to engage in additional
clinical trial site monitoring;
|
|
|
• |
The cost of clinical trials of our product candidates being greater than we anticipate or our having insufficient funds for a clinical trial or to pay the substantial user fees required by FDA upon the filing of a marketing application;
|
|
|
• |
Insufficient supply or inadequate quality of our product candidates or other materials necessary to conduct clinical trials;
|
|
|
• |
Inability to achieve sufficient study enrollment, subjects dropping out or withdrawing from our studies, delays in adding new investigators or clinical trial sites or a withdrawal of clinical trial sites;
|
|
|
• |
Flaws in our clinical trial design that are not discoverable until the clinical trial has progressed;
|
|
|
• |
Disagreement by the FDA or comparable foreign regulatory authorities with our intended indications or study design, including endpoints, or our interpretation of data from preclinical studies and clinical trials, finding that a product
candidate’s benefits do not outweigh its safety risks or requiring that we conduct additional development or study work;
|
|
|
• |
The need to make changes to our product candidates that require additional testing or that cause our product candidates to perform differently than expected;
|
|
|
• |
Global trade policies that may impact our ability to obtain raw materials and/or finished product for commercialization;
|
|
|
• |
FDA or comparable regulatory authorities taking longer than we anticipate to make decisions on our products or product candidates; and
|
|
|
• |
Potential inability to demonstrate that a product or product candidate provides an advantage over current standards of care or current or future competitive therapies in development.
|
|
|
• |
delay commercialization of, and our ability to derive revenues from, our product candidates;
|
|
|
• |
impose costly procedures on us; and
|
|
|
• |
diminish any competitive advantages that we may otherwise enjoy.
|
|
|
• |
restrictions on such products or manufacturing processes;
|
|
|
• |
restrictions on the labeling or marketing of a product;
|
|
|
• |
restrictions on product distribution or use;
|
|
|
• |
clinical holds or termination of clinical trials;
|
|
|
• |
requirements to conduct further post-marketing studies or clinical trials, implement risk mitigation strategies, or to issue corrective information;
|
|
|
• |
warning letters or untitled letters;
|
|
|
• |
withdrawal of the products from the market;
|
|
|
• |
refusal to approve pending applications or supplements to approved applications that we submit;
|
|
|
• |
recall of products;
|
|
|
• |
restrictions on coverage by third-party payers;
|
|
|
• |
fines, restitution or disgorgement of profits or revenues;
|
|
|
• |
suspension or withdrawal of marketing approvals;
|
|
|
• |
refusal to permit the import or export of products;
|
|
|
• |
FDA debarment, suspension and debarment from government programs, refusal of orders under existing government contracts, exclusion from participation in federal healthcare programs, consent decrees, deferred or non-prosecution agreements
or corporate integrity agreements;
|
|
|
• |
product seizure or detention; or
|
|
|
• |
injunctions or the imposition of civil penalties or criminal fines.
|
|
|
• |
our ability to sell our products at competitive prices;
|
|
|
• |
our ability to maintain features and quality standards for our products sufficient to meet the expectations of our customers;
|
|
|
• |
our ability to produce and deliver a sufficient quantity of our products in a timely manner to meet our customers’ requirements;
|
|
|
• |
the impact of a pandemic, or the resurgence of a pandemic, and government responses thereto on our customers and their businesses, operations and financial condition; and
|
|
|
• |
widespread economic conditions or geopolitical conditions, including the exacerbated conflicts in Europe, the Middle East and the surrounding areas.
|
|
|
• |
perceptions by members of the healthcare community, including physicians, about the safety and effectiveness of our products;
|
|
|
• |
cost-effectiveness of our products relative to competing products;
|
|
|
• |
availability of reimbursement for our products from government or other healthcare payers; and
|
|
|
• |
the effectiveness of marketing and distribution efforts by us and our licensees and distributors, if any.
|
|
|
• |
sales or potential sales of substantial amounts of our common stock;
|
|
|
• |
delay or failure in initiating or completing preclinical or clinical trials or unsatisfactory results of these trials;
|
|
|
• |
delay in a decision by Federal, state or local business regulatory authority;
|
|
|
• |
the timing of acceptance, third-party reimbursement and sales of BIVIGAM and ASCENIV;
|
|
|
• |
announcements about us or about our competitors, including clinical trial results, regulatory approvals or new product introductions;
|
|
|
• |
developments concerning our licensors or third-party vendors;
|
|
|
• |
litigation and other developments relating to our patents or other proprietary rights or those of our competitors;
|
|
|
• |
conditions in the pharmaceutical or biotechnology industries;
|
|
|
• |
governmental regulation and legislation;
|
|
|
• |
overall market volatility;
|
|
|
• |
global and economic uncertainty;
|
|
|
• |
variations in our anticipated or actual operating results; and
|
|
|
• |
change in securities analysts’ estimates of our performance, or our failure to meet analysts’ expectations.
|
|
|
• |
the inability of stockholders to call special meetings;
|
|
|
• |
classification of our Board and limitation on filling of vacancies could make it more difficult for a third party to acquire, or discourage a third party from seeking to acquire, control of our Company; and
|
|
|
• |
authorization of the issuance of “blank check” preferred stock, with such designation rights and preferences as may be determined from time to time by the Board, without any need for action by stockholders.
|
| Item 1B. |
Unresolved Staff Comments
|
| Item 1C. |
Cybersecurity
|
| Item 2. |
Properties
|
|
Location
|
Principal Business Activity
|
Approximate
Square Feet
|
Owned or expiration
date of lease
|
|
Ramsey, NJ
|
Corporate Headquarters
|
4,200
|
December 31, 2026 *
|
|
Boca Raton, FL
|
Manufacturing and Administration
|
84,462
|
Owned
|
|
Boca Raton, FL
|
Laboratory and Administration
|
44,495
|
Owned
|
| Item 3. |
Legal Proceedings
|
| Item 4. |
Mine Safety Disclosures
|
| Item 5. |
Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities
|
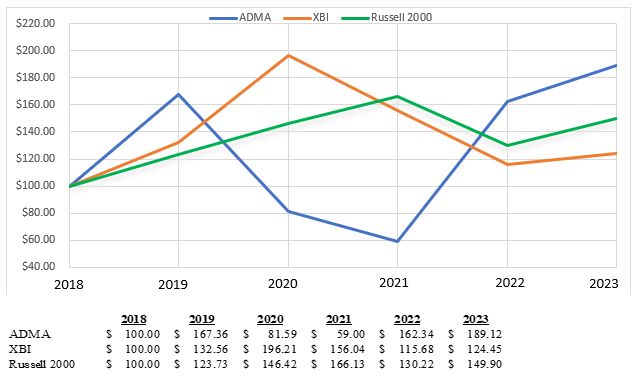
| Item 7. |
Management’s Discussion and Analysis of Financial Condition and Results of Operations
|
|
|
• |
A significant adverse change in legal factors or in the business climate that could affect the value of the asset.
|
|
|
• |
Significant and continued cash flow losses.
|
|
|
• |
A significant adverse change in the extent or manner in which an asset is used, such as a restriction imposed by the FDA or other regulatory authorities that could affect our ability to manufacture our products using a particular
asset.
|
|
|
• |
An expectation of losses or reduced profits associated with an asset. This could result, for example, from the introduction of a competitor’s product that impacts projected revenue growth, or a change in the acceptance of a product by
patients, physicians and payers that results in an inability to sustain projected product revenues.
|
|
|
|
Year Ended December 31,
|
|
|||||||||
|
(in thousands)
|
|
2023
|
|
|
2022
|
|
|
Increase (Decrease)
|
|
|||
|
Revenues
|
|
$
|
258,215
|
|
|
$
|
154,080
|
|
|
$
|
104,135
|
|
|
Cost of product revenue
|
|
|
169,273
|
|
|
|
118,815
|
|
|
|
50,458
|
|
|
Gross profit
|
|
|
88,942
|
|
|
|
35,265
|
|
|
|
53,677
|
|
|
Research and development expenses
|
|
|
3,300
|
|
|
|
3,614
|
|
|
|
(314
|
)
|
|
Plasma center operating expenses
|
|
|
4,266
|
|
|
|
17,843
|
|
|
|
(13,577
|
)
|
|
Amortization of intangibles
|
|
|
724
|
|
|
|
715
|
|
|
|
9
|
|
|
Selling, general and administrative expenses
|
|
|
59,020
|
|
|
|
52,458
|
|
|
|
6,562
|
|
|
Income (loss) from operations
|
|
|
21,632
|
|
|
|
(39,365
|
)
|
|
|
60,997
|
|
|
Interest expense
|
|
|
(25,027
|
)
|
|
|
(19,279
|
)
|
|
|
(5,748
|
)
|
|
Loss on extinguishment of debt
|
|
|
(26,174
|
)
|
|
|
(6,670
|
)
|
|
|
(19,504
|
)
|
|
Other income (expense), net
|
|
|
1,330
|
|
|
|
(590
|
)
|
|
|
1,920
|
|
|
Net loss
|
|
$
|
(28,239
|
)
|
|
$
|
(65,904
|
)
|
|
$
|
37,665
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Adjusted EBITDA *
|
|
$
|
40,251
|
|
|
$
|
(27,627
|
)
|
|
$
|
67,878
|
|
|
Adjusted net income (loss) *
|
|
$
|
705
|
|
|
$
|
(59,234
|
)
|
|
$
|
59,939
|
|
|
*
|
- See Non-GAAP Financial Measures appearing at the end of this discussion
|
|
|
|
Year Ended December 31,
|
|
|||||||||
|
(in thousands)
|
|
2022
|
|
|
2021
|
|
|
Increase (Decrease)
|
|
|||
|
Revenues
|
|
$
|
154,080
|
|
|
$
|
80,943
|
|
|
$
|
73,137
|
|
|
Cost of product revenue
|
|
|
118,815
|
|
|
|
79,770
|
|
|
|
39,045
|
|
|
Gross profit
|
|
|
35,265
|
|
|
|
1,173
|
|
|
|
34,092
|
|
|
Research and development expenses
|
|
|
3,614
|
|
|
|
3,646
|
|
|
|
(32
|
)
|
|
Plasma center operating expenses
|
|
|
17,843
|
|
|
|
12,289
|
|
|
|
5,554
|
|
|
Amortization of intangibles
|
|
|
715
|
|
|
|
715
|
|
|
|
-
|
|
|
Selling, general and administrative expenses
|
|
|
52,458
|
|
|
|
42,897
|
|
|
|
9,561
|
|
|
Loss from operations
|
|
|
(39,365
|
)
|
|
|
(58,374
|
)
|
|
|
19,009
|
|
|
Interest expense
|
|
|
(19,279
|
)
|
|
|
(13,057
|
)
|
|
|
(6,222
|
)
|
|
Loss on extinguishment of debt
|
|
|
(6,670
|
)
|
|
|
-
|
|
|
|
(6,670
|
)
|
|
Other expense, net
|
|
|
(590
|
)
|
|
|
(217
|
)
|
|
|
(373
|
)
|
|
Net loss
|
|
$
|
(65,904
|
)
|
|
$
|
(71,648
|
)
|
|
$
|
5,744
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Adjusted EBITDA *
|
|
$
|
(27,627
|
)
|
|
$
|
(49,608
|
)
|
|
$
|
21,981
|
|
|
Adjusted net loss *
|
|
$
|
(59,234
|
)
|
|
$
|
(71,648
|
)
|
|
$
|
12,414
|
|
|
*
|
- See Non-GAAP Financial Measures appearing at the end of this discussion
|
|
|
|
Years Ended December 31,
|
|
|||||||||
|
(in thousands)
|
|
2023
|
|
|
2022
|
|
|
2021
|
|
|||
|
Net loss
|
|
$
|
(28,239
|
)
|
|
$
|
(65,904
|
)
|
|
$
|
(71,648
|
)
|
|
Depreciation
|
|
|
7,608
|
|
|
|
6,398
|
|
|
|
4,780
|
|
|
Amortization
|
|
|
724
|
|
|
|
715
|
|
|
|
715
|
|
|
Interest expense
|
|
|
25,027
|
|
|
|
19,279
|
|
|
|
13,057
|
|
|
EBITDA
|
|
|
5,120
|
|
|
|
(39,512
|
)
|
|
|
(53,096
|
)
|
|
Stock-based compensation
|
|
|
6,187
|
|
|
|
5,215
|
|
|
|
3,488
|
|
|
IT systems disruption
|
|
|
2,770
|
|
|
|
-
|
|
|
|
-
|
|
|
Loss on extinguishment of debt
|
|
|
26,174
|
|
|
|
6,670
|
|
|
|
-
|
|
|
Adjusted EBITDA
|
|
$
|
40,251
|
|
|
$
|
(27,627
|
)
|
|
$
|
(49,608
|
)
|
|
|
|
Years Ended December 31,
|
|
|||||||||
|
(in thousands)
|
|
2023
|
|
|
2022
|
|
|
2021
|
|
|||
|
Net loss
|
|
$
|
(28,239
|
)
|
|
$
|
(65,904
|
)
|
|
$
|
(71,648
|
)
|
|
Loss on extinguishment of debt
|
|
|
26,174
|
|
|
|
6,670
|
|
|
|
-
|
|
|
IT systems disruption
|
|
|
2,770
|
|
|
|
-
|
|
|
|
-
|
|
|
Adjusted net income (loss)
|
|
$
|
705
|
|
|
$
|
(59,234
|
)
|
|
$
|
(71,648
|
)
|
|
|
• |
The collection and procurement of raw material source plasma, which includes plasma donor fees and plasma center supplies, and other raw materials necessary to maintain and scale up our manufacturing operations;
|
|
|
• |
Employee compensation and benefits;
|
|
|
• |
Capital expenditures for equipment upgrades and capacity expansion at the Boca Facility and to maintain our plasma collection facilities;
|
|
|
• |
Interest on our debt;
|
|
|
• |
Marketing programs, medical education and continued commercialization efforts;
|
|
|
• |
Boca Facility maintenance, repairs and supplies;
|
|
|
• |
Conducting required post-marketing clinical trials for our FDA-approved products; and
|
|
|
• |
Continuous improvements and updates to our IT infrastructure, laboratory equipment and assays, and facilities and engineering equipment.
|
|
|
|
Years Ended December 31,
|
|
|||||||||
|
(in thousands)
|
|
2023
|
|
|
2022
|
|
|
2021
|
|
|||
|
Net cash provided by (used in) operating activities
|
|
$
|
8,800
|
|
|
$
|
(59,508
|
)
|
|
$
|
(112,369
|
)
|
|
Net cash used in investing activities
|
|
|
(4,981
|
)
|
|
|
(13,911
|
)
|
|
|
(13,511
|
)
|
|
Net cash (used in) provided by financing activities
|
|
|
(38,989
|
)
|
|
|
108,852
|
|
|
|
121,048
|
|
|
Net change in cash and cash equivalents
|
|
|
(35,170
|
)
|
|
|
35,433
|
|
|
|
(4,832
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash and cash equivalents - beginning of year
|
|
|
86,522
|
|
|
|
51,089
|
|
|
|
55,921
|
|
|
Cash and cash equivalents - end of year
|
|
$
|
51,352
|
|
|
$
|
86,522
|
|
|
$
|
51,089
|
|
|
|
|
Years Ended December 31,
|
|
|||||||||
|
(in thousands)
|
|
2023
|
|
|
2022
|
|
|
2021
|
|
|||
|
Net loss
|
|
$
|
(28,239
|
)
|
|
$
|
(65,904
|
)
|
|
$
|
(71,648
|
)
|
|
Non-cash expenses, gains and losses
|
|
|
47,162
|
|
|
|
24,682
|
|
|
|
10,959
|
|
|
Changes in accounts receivable
|
|
|
(11,917
|
)
|
|
|
13,072
|
|
|
|
(15,340
|
)
|
|
Changes in inventories
|
|
|
(9,625
|
)
|
|
|
(38,556
|
)
|
|
|
(43,188
|
)
|
|
Changes in prepaid expenses and other current assets
|
|
|
(239
|
)
|
|
|
(756
|
)
|
|
|
(1,293
|
)
|
|
Changes in accounts payable and accrued expenses
|
|
|
11,369
|
|
|
|
8,334
|
|
|
|
9,697
|
|
|
Other
|
|
|
289
|
|
|
|
(380
|
)
|
|
|
(1,556
|
)
|
|
Cash provided by (used) in operations
|
|
$
|
8,800
|
|
|
$
|
(59,508
|
)
|
|
$
|
(112,369
|
)
|
| Item 7A. |
Quantitative and Qualitative Disclosures About Market Risk
|
| Item 8. |
Financial Statements and Supplementary Data
|
| Item 9. |
Changes in and Disagreements with Accountants on Accounting and Financial Disclosure
|
| Item 9A. |
Controls and Procedures
|
| Item 9C. |
Disclosure Regarding Foreign Jurisdictions that Prevent Inspections
|
| Item 10. |
Directors, Executive Officers and Corporate Governance
|
| Item 11. |
Executive Compensation
|
| Item 13. |
Certain Relationships and Related Transactions, and Director Independence
|
| Item 14. |
Principal Accounting Fees and Services
|
| Item 15. |
Exhibits, Financial Statement Schedules
|
|
|
Page
|
|
Management’s Annual Report on Internal Control Over Financial Reporting
|
F-2
|
|
Report of Independent Registered Public Accounting Firm (PCAOB ID 596)
|
F-3
|
|
Consolidated Balance Sheets as of December 31, 2023 and 2022
|
F-5
|
|
Consolidated Statements of Operations for the years ended December 31, 2023, 2022 and 2021
|
F-6
|
|
Consolidated Statements of Changes in Stockholders’ Equity for the years ended December 31, 2023, 2022 and 2021
|
F-7
|
|
Consolidated Statements of Cash Flows for the years ended December 31, 2023, 2022 and 2021
|
F-8
|
|
Notes to Consolidated Financial Statements
|
F-9
|
|
|
(2)
|
Financial Statement Schedules.
|
|
|
(3)
|
Index to Exhibits.
|
|
Exhibit
No.
|
|
Description
|
|
|
Second Amended and Restated Certificate of Incorporation of the Company (incorporated herein by reference to Exhibit 3.1 to the Company’s Current Report on Form 8-K, filed with the SEC on August 23, 2019).
|
|
|
|
Certificate of Amendment of the Second Amended and Restated Certificate of Incorporation of ADMA Biologics, Inc., dated as of May 27, 2021 (incorporated by reference to Exhibit 3.1 to the Company’s Current Report on Form 8-K filed on May
28, 2021).
|
|
|
|
Amended and Restated Bylaws (incorporated herein by reference to Exhibit 3.1 to the Company’s Current Report on Form 8-K, filed with the SEC on October 7, 2016).
|
|
|
|
Certificate of Designation of Series A Junior Participating Preferred Stock of ADMA Biologics, Inc. (incorporated by reference to Exhibit 3.1 to the Company’s Current Report on Form 8-K filed with the SEC on December 21, 2021).
|
|
|
|
Specimen Common Stock Certificate (incorporated herein by reference to Exhibit 4.1 to Amendment No. 1 to the Company’s Current Report on Form 8-K/A, filed with the SEC on March 29, 2012).
|
|
|
|
Form of Warrant Agreement, dated May 13, 2016, issued by the Company to Oxford Finance LLC (incorporated herein by reference to Exhibit 4.6 to the Company’s Quarterly Report on Form 10-Q, filed with the SEC on May 13, 2016).
|
|
|
|
Warrant to Purchase Stock, dated October 10, 2017, issued by the Company to Marathon Healthcare Finance Fund, L.P. (incorporated herein by reference to Exhibit 4.2 to the Company’s Current Report on Form 8-K, filed with the SEC on
October 11, 2017).
|
|
|
|
Warrant to Purchase Stock, dated February 11, 2019, issued by the Company to Perceptive Credit Holdings II, LP (incorporated herein by reference to Exhibit 4.2 to the Company’s Current Report on Form 8-K, filed with the SEC on February
12, 2019).
|
|
|
|
Warrant to Purchase Stock, dated May 3, 2019, issued by the Company to Perceptive Credit Holdings II, LP (incorporated by reference to Exhibit 4.2 to the Company’s Current Report on Form 8-K filed with the SEC on May 3, 2019).
|
|
|
|
Warrant to Purchase Stock, dated December 8, 2020, issued by the Company to Perceptive Credit Holdings II, LP (incorporated by reference to Exhibit 4.7 to the Company’s Annual Report on Form 10-K for the year ended December 31, 2020,
filed with the SEC on March 24, 2021).
|
|
|
|
Description of Securities Registered under Section 12 of the Securities Exchange Act of 1934 (incorporated by reference to Exhibit 4.11 to the Company’s Annual Report on Form 10-K, filed with the SEC on March 24, 2022).
|
|
|
|
Form of Warrant to Purchase Common Stock, in the form issued by the Company to various entities affiliated with Hayfin Services LLP, dated as of March 23, 2022 (incorporated by reference to Exhibit 4.13 to the Company’s Annual Report on
Form 10-K, filed with the SEC on March 24, 2022).
|
|
|
|
Form of Warrant to Purchase Common Stock, in the form issued by the Company to various entities affiliated with Hayfin Services LLP, dated as of May 1, 2023 (incorporated by reference to Exhibit 4.1 to the Company’s Current Report on
Form 8-K, filed with the SEC on May 2, 2023).
|
|
|
2007 Employee Stock Option Plan, as amended by Amendment No. 3 (incorporated herein by reference to Exhibit A to the Information Statement on Schedule 14C, filed with the SEC on October 29, 2012).
|
|
|
|
Amended and Restated ADMA Biologics, Inc. 2014 Omnibus Incentive Compensation Plan (incorporated herein by reference to Exhibit 10.1 to the Company’s Registration Statement on Form S-8, filed with the SEC on August 18, 2017).
|
|
|
|
Amended and Restated Employment Agreement, dated January 29, 2019, by and between ADMA Biologics, Inc. and Adam Grossman (incorporated herein by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K, filed with the SEC on
January 29, 2019).
|
|
|
|
Amendment to Employment Agreement, dated as of September 29, 2021, by and between ADMA Biologics, Inc. and Adam Grossman (incorporated by reference to Exhibit 10.2 to the Company’s Current Report on Form 8-K, filed with the SEC on
October 1, 2021).
|
|
|
|
Amended and Restated Employment Agreement, dated January 29, 2019, by and between ADMA Biologics, Inc. and Brian Lenz (incorporated herein by reference toExhibit 10.3 to the Company’s Current Report on Form 8-K, filed with the SEC on
January 29, 2019).
|
|
|
|
Amendment to Employment Agreement, dated as of September 29, 2021, by and between ADMA Biologics, Inc. and Brian Lenz (incorporated by reference to Exhibit 10.3 to the Company’s Current Report on Form 8-K, filed with the SEC on October
1, 2021).
|
|
|
|
Plasma Purchase Agreement, dated as of November 17, 2011, by and between Biotest Pharmaceuticals Corporation and ADMA Biologics, Inc., as amended by First Amendment to Plasma Purchase Agreement, dated as of December 1, 2011, by and
between Biotest Pharmaceuticals Corporation and ADMA Biologics, Inc. (incorporated herein by reference to Exhibit 10.9 to Amendment No. 3 to the Company’s Current Report on Form 8-K/A, filed with the SEC on June 22, 2012).
|
|
|
|
Second Amendment to Plasma Purchase Agreement, dated as of December 18, 2015, by and between Biotest Pharmaceuticals Corporation and ADMA Biologics, Inc. (incorporated herein by reference to Exhibit 10.3.1 to the Company’s Annual Report
on Form 10-K, filed with the SEC on March 23, 2016).
|
|
|
|
Third Amendment to Plasma Purchase Agreement, dated as of April 8, 2016, by and between Biotest Pharmaceuticals Corporation and ADMA Biologics, Inc. (incorporated herein by reference to Exhibit 10.3.2 to the Company’s Quarterly Report on
Form 10-Q, filed with the SEC on May 13, 2016).
|
|
|
|
Fourth Amendment to Plasma Purchase Agreement, dated as of June 6, 2017, by and between Biotest Pharmaceuticals Corporation and ADMA Biologics, Inc. (incorporated herein by reference to Exhibit 10.9 to the Company’s Quarterly Report on
Form 10-Q, filed with the SEC on August 11, 2017).
|
|
|
|
Fifth Amendment to Plasma Purchase Agreement, dated as of January 1, 2019, by and between Grifols Worldwide Operations Limited (as successor-in-interest to Biotest Pharmaceuticals Corporation) and ADMA Biologics, Inc. (incorporated
herein by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K, filed with the SEC on January 2, 2019).
|
|
|
|
Plasma Supply Agreement, dated as of June 6, 2017, by and between ADMA BioManufacturing, LLC and Biotest Pharmaceuticals Corporation (incorporated herein by reference to Exhibit 10.5 to the Company’s Quarterly Report on Form 10-Q, filed
with the SEC on August 11, 2017).
|
|
|
Amendment #1 to the Plasma Supply Agreement, dated as of July 19, 2018, by and between Biotest Pharmaceuticals Corporation and ADMA BioManufacturing, LLC (incorporated herein by reference to Exhibit 10.2 to the Company’s Quarterly Report
on Form 10-Q, filed with the SEC on August 10, 2018).
|
|
|
|
Amended and Restated Agreement for Services, effective as of January 1, 2016, as amended, by and between ADMA BioManufacturing, LLC and Areth LLC (incorporated herein by reference to Exhibit 10.18 to the Company’s Quarterly Report on
Form 10-Q, filed with the SEC on August 12, 2016).
|
|
|
|
Form of Indemnification Agreement (incorporated herein by reference to Exhibit 10.12 to the Company’s Current Report on Form 8-K, filed with the SEC on February 13, 2012).
|
|
|
|
License Agreement, effective as of December 31, 2012, by and between ADMA Biologics, Inc. and Biotest AG (incorporated herein by reference to Exhibit 10.21 to the Company’s Registration Statement on Form S-1, filed with the SEC on
February 11, 2013).
|
|
|
|
First Amendment to License Agreement, dated as of June 6, 2017, by and between the Company and Biotest AG (incorporated herein by reference to Exhibit 10.8 to the Company’s Quarterly Report on Form 10-Q, filed with the SEC on August 11,
2017).
|
|
|
|
Amendment 3 to the Amended and Restated Agreement for Services, effective as of November 7, 2019, by and between ADMA BioManufacturing, LLC and Areth LLC (incorporated herein by reference to the Exhibit 10.27
to the Company’s Annual Report on Form 10-K filed March 12, 2020).
|
|
|
|
Amendment 4 to the Amended and Restated Agreement For Services Between ADMA BioManufacturing, LLC and Areth LLC (incorporated by reference to Exhibit 10.1 to the Company’s Quarterly Report on Form 10-Q filed on November 9, 2022).
|
|
|
|
Distribution Agreement, dated September 3, 2021, by and between ADMA Biologics, Inc. and Raymond James & Associates, Inc. (incorporated by reference to Exhibit 1.1 to the Company’s Current Report on Form 8-K filed on September 3,
2021).
|
|
|
|
Form of Retention Bonus Agreement (incorporated by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on October 1, 2021).
|
|
|
|
ADMA Biologics, Inc. 2022 Equity Compensation Plan (incorporated herein by reference to Exhibit 10.1 to the Registrant’s Current Report on Form 8-K filed with the Commission on June 21, 2022)
|
|
|
|
Credit Agreement, dated as of December 18, 2023, by and among the Company, as Administrative Borrower, ADMA BioManufacturing, LLC, ADMA Plasma Biologics, Inc. and ADMA BioCenters Georgia Inc., each as Borrowers, certain subsidiaries of
the Company, as Guarantors, from time to time party thereto, the Lenders from time to time party thereto, Ares Capital Corporation, as Administrative Agent and as Collateral Agent, and ACF FINCO I LP, as Revolving Agent (incorporated herein
by reference to Exhibit 10.1 to the Registrant’s Current Report on Form 8-K filed with the Commission on December 18, 2023)
|
|
|
|
Security Agreement, dated as of December 18, 2023, by and among the Company, ADMA BioManufacturing, LLC, ADMA Plasma Biologics, Inc. and ADMA BioCenters Georgia, Inc., as Grantors, and Ares Capital Corporation, as Collateral Agent
(incorporated herein by reference to Exhibit 10.2 to the Registrant’s Current Report on Form 8-K filed with the Commission on December 18, 2023)
|
|
|
Subsidiaries of the Company.
|
|
|
|
Consent of CohnReznick LLP.
|
|
|
|
Certification of Principal Executive Officer pursuant to Rules 13a-14(a) and 15d-14(a), as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002.
|
|
|
|
Certification of Principal Financial Officer pursuant to Rules 13a-14(a) and 15d-14(a), as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002.
|
|
|
|
Certification of Principal Executive Officer pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002.
|
|
|
|
Certification of Principal Financial Officer pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002.
|
|
|
|
ADMA Biologics, Inc. Compensation Recoupment Policy
|
|
|
101*
|
|
The following materials from ADMA Biologics, Inc. Form 10-K for the year ended December 31, 2023, formatted in Extensible Business Reporting Language (XBRL): (i) Consolidated Balance Sheets at December 31, 2023 and December 31, 2022,
(ii) Consolidated Statements of Operations for the years ended December 31, 2023, 2022 and 2021, (iii) Consolidated Statements of Changes in Stockholders’ Equity for the years ended December 31, 2023, 2022 and 2021, (iv) Consolidated
Statements of Cash Flows for the years ended December 31, 2023, 2022 and 2021; and (v) Notes to Consolidated Financial Statements.
|
|
104
|
|
Cover Page Interactive Data File (formatted as Inline XBRL and contained in Exhibit 101).
|
| Item 16. |
Form 10-K Summary
|
|
|
ADMA Biologics, Inc.
|
|
|
|
|
|
|
Date: February 28, 2024
|
By:
|
/s/ Adam S. Grossman
|
|
|
Name:
|
Adam S. Grossman
|
|
|
Title:
|
President and Chief Executive Officer
|
|
Signature
|
|
Title
|
|
Date
|
|
|
|
|
|
|
|
/s/ Adam S. Grossman
|
|
|
|
|
|
Adam S. Grossman
|
|
President and Chief Executive Officer (Principal Executive Officer) and Director
|
|
February 28, 2024
|
|
|
|
|
|
|
|
/s/ Brian Lenz
|
|
|
|
|
|
Brian Lenz
|
|
Executive Vice President and Chief Financial Officer (Principal Financial Officer and Principal Accounting Officer)
|
|
February 28, 2024
|
|
|
|
|
|
|
|
/s/ Steven A. Elms
|
|
|
|
|
|
Steven A. Elms
|
|
Chairman of the Board of Directors
|
|
February 28, 2024
|
|
|
|
|
|
|
|
/s/ Dr. Jerrold B. Grossman
|
|
|
|
|
|
Dr. Jerrold B. Grossman
|
|
Vice Chairman of the Board of Directors
|
|
February 28, 2024
|
|
|
|
|
|
|
|
/s/ Alison Finger
|
|
|
|
|
|
Alison Finger
|
|
Director
|
|
February 28, 2024
|
|
|
|
|
|
|
|
/s/ Bryant E. Fong
|
|
|
|
|
|
Bryant E. Fong
|
|
Director
|
|
February 28, 2024
|
|
|
|
|
|
|
|
/s/ Lawrence P. Guiheen
|
|
|
||
|
Lawrence P. Guiheen
|
Director
|
February 28, 2024
|
||
|
|
||||
|
/s/ Young T. Kwon
|
||||
|
Young T. Kwon
|
Director
|
February 28, 2024
|
ADMA BIOLOGICS, INC. AND SUBSIDIARIES
CONSOLIDATED FINANCIAL STATEMENTS
TABLE OF CONTENTS
|
|
Page
|
|
F-2
|
|
|
F-3
|
|
|
F-5
|
|
|
F-6
|
|
|
F-7
|
|
|
F-8
|
|
|
F-9
|
|
/s/ Adam S. Grossman
|
/s/ Brian Lenz
|
|
Adam S. Grossman
|
Brian Lenz
|
|
President and Chief Executive Officer
|
Executive Vice President and Chief Financial Officer
|
|
February 28, 2024
|
February 28, 2024
|
|
/s/ CohnReznick LLP
|
|
We have served as the Company’s auditor since 2008.
|
|
Parsippany, New Jersey
|
|
February 28, 2024
|
ADMA BIOLOGICS, INC. AND SUBSIDIARIES
CONSOLIDATED BALANCE SHEETS
Years Ended December 31, 2023 and 2022
|
December 31, |
December 31, |
|||||||
|
|
2023 | 2022 | ||||||
| (In thousands, except share data) |
||||||||
|
ASSETS |
|
|
||||||
|
Current assets: |
||||||||
|
Cash and cash equivalents |
$ | 51,352 | $ | 86,522 | ||||
|
Accounts receivable, net |
27,421 | 15,505 | ||||||
|
Inventories |
172,906 | 163,280 | ||||||
|
Prepaid expenses and other current assets |
5,334 | 5,095 | ||||||
|
Total current assets |
257,013 | 270,402 | ||||||
|
Property and equipment, net |
53,835 | 58,261 | ||||||
|
Intangible assets, net |
499 | 1,013 | ||||||
|
Goodwill |
3,530 | 3,530 | ||||||
|
Right-to-use assets |
9,635 | 10,485 | ||||||
|
Deposits and other assets |
4,670 | 4,770 | ||||||
|
TOTAL ASSETS |
$ | 329,182 | $ | 348,461 | ||||
|
|
||||||||
|
LIABILITIES AND STOCKHOLDERS’ EQUITY
|
||||||||
|
Current liabilities: |
||||||||
|
Accounts payable |
$ | 15,660 | $ | 13,229 | ||||
|
Accrued expenses and other current liabilities |
32,919 | 24,990 | ||||||
|
Current portion of deferred revenue |
182 | 143 | ||||||
|
Current portion of lease obligations |
1,045 | 905 | ||||||
|
Total current liabilities |
49,806 | 39,267 | ||||||
|
Senior notes payable, net of discount |
130,594 | 142,833 | ||||||
|
Deferred revenue, net of current portion |
1,690 | 1,833 | ||||||
| End of term fee |
1,688 | 1,500 | ||||||
|
Lease obligations, net of current portion |
9,779 | 10,704 | ||||||
|
Other non-current liabilities |
419 | 350 | ||||||
|
TOTAL LIABILITIES |
193,976 | 196,487 | ||||||
|
|
||||||||
|
COMMITMENTS AND CONTINGENCIES |
||||||||
|
|
||||||||
|
STOCKHOLDERS’ EQUITY
|
||||||||
|
Preferred Stock, $0.0001 par value, 10,000,000 shares authorized, no shares issued and outstanding |
- | - | ||||||
|
Common Stock - voting, $0.0001 par value, 300,000,000 shares authorized, 226,063,032 and 221,816,930 shares issued and outstanding |
23 | 22 | ||||||
|
Additional paid-in capital |
641,439 | 629,969 | ||||||
|
Accumulated deficit |
(506,256 | ) | (478,017 | ) | ||||
|
TOTAL STOCKHOLDERS’ EQUITY |
135,206 | 151,974 | ||||||
|
TOTAL LIABILITIES AND STOCKHOLDERS’ EQUITY |
$ | 329,182 | $ | 348,461 | ||||
The accompanying notes are an integral part of these consolidated financial statements.
ADMA BIOLOGICS, INC. AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF OPERATIONS
|
|
Years ended December 31, |
|||||||||||
|
|
2023 |
2022 |
2021 |
|||||||||
|
|
(In thousands, except share and per share data) |
|||||||||||
|
REVENUES |
$ | 258,215 | $ | 154,080 | $ | 80,943 | ||||||
|
Cost of product revenue |
169,273 | 118,815 | 79,770 | |||||||||
|
Gross profit |
88,942 | 35,265 | 1,173 | |||||||||
|
|
||||||||||||
|
OPERATING EXPENSES: |
||||||||||||
|
Research and development |
3,300 | 3,614 | 3,646 | |||||||||
|
Plasma center operating expenses |
4,266 | 17,843 | 12,289 | |||||||||
|
Amortization of intangible assets |
724 | 715 | 715 | |||||||||
|
Selling, general and administrative |
59,020 | 52,458 | 42,897 | |||||||||
|
Total operating expenses |
67,310 | 74,630 | 59,547 | |||||||||
|
|
||||||||||||
|
INCOME (LOSS) FROM OPERATIONS |
21,632 | (39,365 | ) | (58,374 | ) | |||||||
|
|
||||||||||||
|
OTHER INCOME (EXPENSE): |
||||||||||||
|
Interest income |
1,617 | 45 | 35 | |||||||||
|
Interest expense |
(25,027 | ) | (19,279 | ) | (13,057 | ) | ||||||
|
Loss on extinguishment of debt |
(26,174 | ) | (6,670 | ) | - | |||||||
|
Other expense |
(287 | ) | (635 | ) | (252 | ) | ||||||
|
Other expense, net |
(49,871 | ) | (26,539 | ) | (13,274 | ) | ||||||
|
|
||||||||||||
|
NET LOSS |
$ | (28,239 | ) | $ | (65,904 | ) | $ |
(71,648 | ) | |||
|
|
||||||||||||
|
BASIC AND DILUTED LOSS PER COMMON SHARE |
$ | (0.13 | ) | $ | (0.33 | ) | $ |
(0.51 | ) | |||
|
|
||||||||||||
|
WEIGHTED AVERAGE COMMON SHARES OUTSTANDING: |
||||||||||||
|
Basic and Diluted |
223,977,315 | 197,874,895 | 139,578,538 |
|||||||||
The accompanying notes are an integral part of these consolidated financial statements.
ADMA BIOLOGICS, INC. AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF CHANGES IN STOCKHOLDERS’ EQUITY
|
|
Additional |
Total |
||||||||||||||||||
|
|
Common Stock |
Paid-in |
Accumulated |
Stockholders’ |
||||||||||||||||
|
|
Shares |
Amount |
Capital |
Deficit |
Equity |
|||||||||||||||
|
Balance at December 31, 2020 |
104,902,888 | $ | 10 | $ | 428,704 | $ | (340,465 | ) | $ | 88,249 | ||||||||||
|
Stock-based compensation |
- | - | 3,488 | - | 3,488 | |||||||||||||||
|
Issuance of common stock, net of offering expenses |
90,846,029 | 9 | 121,135 | - | 121,144 | |||||||||||||||
|
Vesting of Restricted Stock Units, net of shares withheld for taxes and retired
|
64,900 | 1 | (61 | ) | - | (60 | ) | |||||||||||||
|
Net loss |
- | - | - | (71,648 | ) | (71,648 | ) | |||||||||||||
|
Balance at December 31, 2021 |
195,813,817 | 20 | 553,266 | (412,113 | ) | 141,173 | ||||||||||||||
|
Stock-based compensation |
- | - | 5,215 | - | 5,215 | |||||||||||||||
|
Issuance of common stock, net of offering expenses
|
24,125,873 | 2 | 64,642 | - | 64,644 | |||||||||||||||
|
Vesting of Restricted Stock Units, net of shares withheld for taxes
|
1,808,561 | - | (2,899 | ) | - | (2,899 | ) | |||||||||||||
|
Warrants issued in connection with note payable
|
- | - | 9,570 | - | 9,570 | |||||||||||||||
|
Exercise of stock options |
68,679 | - | 175 | - | 175 | |||||||||||||||
|
Net loss |
- | - | - | (65,904 | ) | (65,904 | ) | |||||||||||||
|
Balance at December 31, 2022 |
221,816,930 |
|
22 |
|
629,969 |
|
(478,017 | ) |
|
151,974 | ||||||||||
|
Stock-based compensation
|
- | - | 6,187 | - | 6,187 | |||||||||||||||
|
Vesting of Restricted Stock Units, net of shares withheld for taxes
|
833,722 | - | (1,415 | ) | - | (1,415 | ) | |||||||||||||
|
Warrants issued in connection with notes payable
|
- | - | 5,595 | - | 5,595 | |||||||||||||||
| Exercise of stock options |
1,444,533 | 1 | 1,103 | - | 1,104 | |||||||||||||||
| Cashless exercise of warrants |
1,967,847 | - | - | - | - | |||||||||||||||
| Net loss |
- | - | - | (28,239 | ) | (28,239 | ) | |||||||||||||
| Balance at December 31, 2023 | 226,063,032 | $ |
23 | $ |
641,439 | $ |
(506,256 | ) | $ |
135,206 | ||||||||||
The accompanying notes are an integral part of these consolidated financial statements.
ADMA BIOLOGICS, INC. AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF CASH FLOWS
|
|
Year Ended December 31, |
|||||||||||
|
|
2023 |
2022 |
2021 | |||||||||
| (In thousands) |
||||||||||||
|
CASH FLOWS FROM OPERATING ACTIVITIES: |
||||||||||||
|
Net loss |
$ | (28,239 | ) | $ | (65,904 | ) | $ | (71,648 | ) | |||
|
Adjustments to reconcile net loss to net cash provided by (used in) operating activities: |
||||||||||||
|
Depreciation and amortization |
8,332 | 7,113 | 5,496 | |||||||||
|
Loss on disposal of fixed assets |
182 | 427 | 221 | |||||||||
|
Interest paid in kind
|
3,836 | 2,998 | - | |||||||||
|
Stock-based compensation |
6,187 | 5,215 | 3,488 | |||||||||
|
Amortization of debt discount |
2,594 | 2,402 | 1,897 | |||||||||
|
Loss on extinguishment of debt |
26,174 | 6,670 | - | |||||||||
|
Amortization of license revenue |
(143 | ) | (143 | ) | (143 | ) | ||||||
|
Changes in operating assets and liabilities: |
||||||||||||
|
Accounts receivable |
(11,916 | ) | 13,072 | (15,340 | ) | |||||||
|
Inventories |
(9,626 | ) | (38,556 | ) | (43,188 | ) | ||||||
|
Prepaid expenses and other current assets |
(239 | ) | (756 | ) | (1,293 | ) | ||||||
|
Deposits and other assets |
1,080 | 122 | (1,775 | ) | ||||||||
|
Accounts payable |
3,839 | 800 | 1,356 | |||||||||
|
Accrued expenses |
7,530 | 7,534 | 8,341 | |||||||||
|
Other current and non-current liabilities |
(791 | ) | (502 | ) | 219 | |||||||
|
Net cash provided by (used in) operating activities |
8,800 | (59,508 | ) | (112,369 | ) | |||||||
|
|
||||||||||||
|
CASH FLOWS FROM INVESTING ACTIVITIES: |
||||||||||||
|
Purchase of property and equipment |
(4,771 | ) | (13,911 | ) | (13,511 | ) | ||||||
|
Acquisition of intangible assets
|
(210 | ) | - | - | ||||||||
|
Net cash used in investing activities |
(4,981 | ) | (13,911 | ) | (13,511 | ) | ||||||
|
|
||||||||||||
|
CASH FLOWS FROM FINANCING ACTIVITIES: |
||||||||||||
|
Principal payments on notes payable |
(158,584 | ) | (100,000 | ) | - | |||||||
|
Proceeds from issuance of common stock, net of offering expenses |
- | 64,645 | 121,144 | |||||||||
|
Payment of debt refinancing fees |
(11,140 | ) | (2,000 | ) | - | |||||||
|
Proceeds from issuance of note payable |
135,000 | 151,750 | - | |||||||||
|
Taxes paid on vested Restricted Stock Units |
(1,415 | ) | (2,899 | ) | (62 | ) | ||||||
|
Payments on finance lease obligations |
(17 | ) | (36 | ) | (34 | ) | ||||||
|
Net proceeds from the exercise of stock options
|
1,104 | 175 | - | |||||||||
|
Payment of end of term fee
|
(1,586 | ) | - | - | ||||||||
|
Payment of deferred financing fees
|
(2,351 | ) | (2,783 | ) | - | |||||||
|
Net cash (used in) provided by financing activities |
(38,989 | ) | 108,852 | 121,048 | ||||||||
|
|
||||||||||||
|
Net (decrease) increase in cash and cash equivalents
|
(35,170 | ) | 35,433 | (4,832 | ) | |||||||
|
Cash and cash equivalents - beginning of year |
86,522 | 51,089 | 55,921 | |||||||||
|
Cash and cash equivalents - end of year |
$ | 51,352 | $ | 86,522 | $ | 51,089 | ||||||
The accompanying notes are an integral part of these consolidated financial statements.
|
1. |
ORGANIZATION AND BUSINESS |
ADMA Biologics, Inc. (“ADMA” or the “Company”) is an end-to-end commercial biopharmaceutical company dedicated to manufacturing, marketing and developing specialty biologics for the treatment of immunodeficient patients at risk for infection and others at risk for certain infectious diseases. The Company’s targeted patient populations include immune-compromised individuals who suffer from an underlying immune deficiency disorder or who may be immune-suppressed for medical reasons.
ADMA operates through its wholly-owned subsidiaries ADMA BioManufacturing, LLC (“ADMA BioManufacturing”) and ADMA BioCenters Georgia Inc. (“ADMA BioCenters”). ADMA BioManufacturing was formed in January 2017 to facilitate the acquisition of certain assets held by the Company’s former third-party contract manufacturer, which included the U.S. Food and Drug Administration (“FDA”)-licensed BIVIGAM and Nabi-HB immunoglobulin products, and an FDA-licensed plasma fractionation manufacturing facility located in Boca Raton, FL (the “Boca Facility”). ADMA BioCenters is the Company’s source plasma collection business with ten plasma collection facilities located throughout the U.S., all of which hold an approved license with the FDA.
The Company has three FDA-approved products, all of which are currently marketed and commercially available: (i) ASCENIV (Immune Globulin Intravenous, Human – slra 10% Liquid), an intravenous immune globulin (“IVIG”) product indicated for the treatment of Primary Humoral Immunodeficiency (“PI”), also known as Primary Immunodeficiency Disease (“PIDD”) or Inborn Errors of Immunity, for which the Company received FDA approval on April 1, 2019 and commenced first commercial sales in October 2019; (ii) BIVIGAM (Immune Globulin Intravenous, Human), an IVIG product indicated for the treatment of PI, and for which the Company received FDA approval on May 9, 2019 and commenced commercial sales in August 2019; and (iii) Nabi-HB (Hepatitis B Immune Globulin, Human), which is indicated for the treatment of acute exposure to blood containing Hepatitis B surface antigen (“HBsAg”) and other listed exposures to Hepatitis B. In addition to its commercially available immunoglobulin products, the Company generates revenues from the sale of intermediate by-products that result from the immunoglobulin production process and from time to time provides contract manufacturing and laboratory services for certain clients. The Company seeks to develop a pipeline of plasma-derived therapeutics, and its products and product candidates are intended to be used by physician specialists focused on caring for immune-compromised patients with or at risk for certain infectious diseases.
As of December 31, 2023, the Company had working capital of $207.2 million, including $51.4 million of cash and cash equivalents, accounts receivable of $27.4 million and $172.9 million of
inventories, partially offset by $49.8 million of current liabilities. Based upon the Company’s current projected revenue and
expenditures, including capital expenditures and continued implementation of the Company’s commercialization and expansion activities, the Company’s management currently believes that its cash, cash equivalents and accounts receivable, along
with its projected future operating cash flow, will be sufficient to fund ADMA’s operations, as currently conducted, through the end of the first quarter of 2025. However, the Company’s current outlook on cash flows and profitability may change
based upon several factors, including the success of the Company’s commercial sales of its products, whether or not the assumptions underlying the Company’s projected revenues and expenses are correct and the continued acceptability of ADMA’s
immune globulin products by physicians, patients or payers. The Company is subject to risks common to companies in the biotechnology and pharmaceutical manufacturing industries including, but not limited to, dependence on collaborative
arrangements, development by the Company or its competitors of new technological innovations, dependence on key personnel, inflationary pressures, supply chain constraints, protection of proprietary technology, and compliance with FDA and other
governmental regulations and approval requirements.
|
2. |
SIGNIFICANT ACCOUNTING POLICIES |
Principles of consolidation and basis of presentation
The accompanying consolidated financial statements include the accounts of ADMA and its wholly-owned subsidiaries, and have been prepared in conformity with accounting principles generally accepted in the United States of America (“U.S. GAAP”) and in accordance with Article 3 of Regulation S-X of the Securities and Exchange Commission (the “SEC”). All intercompany balances have been eliminated in consolidation. Any reference in these notes to applicable guidance is meant to refer to U.S. GAAP as found in the Accounting Standards Codification (“ASC”) and Accounting Standards Updates (“ASU”) of the Financial Accounting Standards Board (the “FASB”). During the years ended December 31, 2023, 2022 and 2021, comprehensive loss was equal to the net loss amounts presented for the respective periods in the accompanying consolidated statements of operations.
Use of estimates
The preparation of financial statements requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. Actual results could differ from those estimates. Significant estimates include rebates and chargebacks deducted from gross revenues, valuation of inventory, assumptions used in projecting future liquidity and capital requirements, assumptions used in the fair value of awards granted under the Company’s equity incentive plans and warrants issued in connection with the issuance of notes payable and the valuation allowance for the Company’s deferred tax assets.
Cash and cash equivalents
The Company considers all highly liquid instruments purchased with a maturity of three months or less to be cash equivalents.
The Company regularly maintains cash and cash equivalents at third-party financial institutions in excess of the Federal Deposit Insurance Corporation insurance limit. Although the Company monitors the daily cash balances in its operating accounts and adjusts the balances as appropriate, these balances could be impacted, and there could be a material adverse effect on the Company’s business, if one or more of the financial institutions with which the Company has deposits fails or is subject to other adverse conditions in the financial or credit markets. To date, the Company has not experienced a loss or lack of access to its deposited cash or cash equivalents; however, the Company cannot provide assurance that access to its cash and cash equivalents will not be impacted by adverse conditions in the financial and credit markets in the future.
Accounts receivable
Accounts receivable is reported at realizable value, net of allowances for contractual credits and doubtful accounts in the amount of $0.1 million at December 31, 2023 and 2022, which are recognized in the period the related revenue is recorded. The Company extends credit to its customers based upon an evaluation of each customer’s financial condition and credit history. Evaluations of the financial condition and associated credit risk of customers are performed on an ongoing basis. Based on these evaluations, the Company has concluded that its credit risk is minimal (see Note 16).
Inventories
Raw materials inventory consists of normal source plasma (“NSP”) and Respiratory Syncytial Virus (“RSV”) high titer plasma collected at the Company’s plasma collection facilities, along with various materials purchased from suppliers, used in the production of the Company’s products. Work-in-process and finished goods inventories (see Note 3) reflect the cost of raw materials as well as costs for direct and indirect labor, primarily salaries, wages and benefits for applicable employees, as well as an allocation of overhead costs related to the Boca Facility including utilities, property taxes, general repairs and maintenance, consumable supplies and depreciation. The allocation of Boca Facility overhead to inventory is generally based upon the estimated square footage of the Boca Facility that is used in the production of the Company’s products relative to the total square footage of the facility.
Property and equipment
Assets comprising property and equipment (see Note 4) are stated at cost less accumulated depreciation. Depreciation is calculated using the straight-line method over the asset’s estimated useful life. Land is not depreciated. The buildings have been assigned a useful life of 30 years. Property and equipment other than land and buildings have useful lives ranging from 3 to 15 years. Leasehold improvements are amortized over the lesser of the lease term or their estimated useful lives.
Goodwill
Goodwill represents the excess of purchase price over the fair value of net assets acquired by the Company. Goodwill at December 31, 2023 and 2022 was $3.5 million, all of which is attributable to the Company’s ADMA BioManufacturing business segment. There were no changes to the carrying amount of goodwill during the years ended December 31, 2023, 2022 and 2021.
Goodwill is not amortized but is assessed for impairment on an annual basis or more frequently if impairment indicators exist. The Company has the option to perform a qualitative assessment of goodwill to determine whether it is more likely than not that the fair value of its reporting unit is less than its carrying amount, including goodwill and other intangible assets. If the Company concludes that this is the case, then it must perform a goodwill impairment test by comparing the fair value of the reporting unit to its carrying value. An impairment charge is recorded to the extent the reporting unit’s carrying value exceeds its fair value, not to exceed the total amount of goodwill allocated to that reporting unit. The Company performs its annual goodwill impairment test as of October 1 of each year. The Company’s annual goodwill impairment tests as of October 1, 2023, 2022 and 2021 did not result in any impairment charges related to goodwill for the years ended December 31, 2023, 2022 and 2021.
Impairment of long-lived assets
The Company assesses the recoverability of its long-lived assets, which include property and equipment and finite-lived intangible assets, whenever significant events or changes in circumstances indicate impairment may have occurred. If indicators of impairment exist, projected future undiscounted cash flows associated with the asset are compared to its carrying amount to determine whether the asset’s carrying value is recoverable. Any resulting impairment is recorded as a reduction in the carrying value of the related asset in excess of fair value and a charge to operating results. For the years ended December 31, 2023, 2022 and 2021, the Company determined that there was no impairment of its long-lived assets.
Revenue recognition
Revenues for the years ended December 31, 2023, 2022 and 2021 are comprised of (i) revenues from the sale of the Company’s immunoglobulin products, ASCENIV, BIVIGAM and Nabi-HB, (ii) product revenues from the sale of human plasma collected by the Company’s Plasma Collection Centers business segment, (iii) contract manufacturing and laboratory services revenue, (iv) revenues from the sale of intermediate by-products and (v) license and other revenues primarily attributable to the out-licensing of ASCENIV to Biotest, AG (“Biotest”) in 2012 to market and sell this product in Europe and selected countries in North Africa and the Middle East. Biotest has provided the Company with certain services and financial payments in accordance with the related Biotest license agreement and is obligated to pay the Company certain amounts in the future if certain milestones are achieved. Deferred revenue is amortized into income over the term of the Biotest license, representing a period of approximately 22 years.
Product revenues from the sale of human plasma collected at the Company’s plasma collection centers are recognized at the time control of the product has been transferred to the customer, which generally occurs at the time of shipment. Product revenues are recognized at the time of delivery if the Company retains control of the product during shipment.
Cost of product revenue
Cost of product revenue includes costs associated with the manufacture of the Company’s FDA approved products and intermediates and for the collection of human source plasma, as well as expenses related to conformance batch production, process development and scientific and technical operations when these operations are attributable to marketed products. When the activities of these operations are attributable to new products in development, the expenses are classified as research and development expenses.
Research and development expenses
Research and development expenses consist of clinical research organization costs, costs related to clinical trials, post-marketing commitment studies for BIVIGAM and ASCENIV and salaries, benefits and stock-based compensation for employees directly related to research and development activities. All research and development costs are expensed as incurred.
Advertising and marketing expenses
Advertising and marketing expense includes cost for promotional materials and trade show expenses for the marketing of the Company’s products and expenses incurred for attracting donors to the Company’s plasma collection centers. All advertising and marketing expenses are expensed as incurred. Advertising and marketing expenses were $3.3 million, $2.2 million and $1.4 million for the years ended December 31, 2023, 2022 and 2021, respectively.
Stock-based compensation
The Company follows recognized accounting guidance which requires all equity-based payments, including grants of stock options and restricted stock unit awards (“RSUs”), to be recognized in the statement of operations as compensation expense based on their fair values at the date of grant. Compensation expense related to awards to employees and directors with service-based vesting conditions is recognized on a straight-line basis over the associated vesting period of the award based on the grant date fair value of the award. Stock options granted to employees under the Company’s equity incentive plans generally have a four-year vesting period and a term of 10 years. RSUs granted to employees also have a four-year vesting period. For milestone-based equity awards (see Note 8) the Company periodically assesses the probability of vesting for each milestone-based award and adjusts compensation expense based on its probability assessment. Pursuant to ASU No. 2016-09, Improvements to Employee Share-Based Payment Accounting (Topic 718), the Company has elected not to establish a forfeiture rate, as stock-based compensation expense related to forfeitures of unvested equity awards is fully reversed at the time of forfeiture.
Income taxes
The Company recognizes deferred tax assets and liabilities for the expected future tax consequences of events that have been included in the consolidated financial statements or its tax returns. Under this method, deferred tax assets and liabilities are recognized for the temporary differences between the tax bases of assets and liabilities and their respective financial reporting amounts at enacted tax rates in effect for the years in which the temporary differences are expected to reverse. The Company records a valuation allowance on its deferred tax assets if it is more likely than not that the Company will not generate sufficient taxable income to utilize its deferred tax assets (see Note 11). The Company is subject to income tax examinations by major taxing authorities for all tax years since 2019 and for previous periods as it relates to the Company’s net operating loss carryforwards.
Loss Per Share
Basic loss per share is computed by dividing net loss attributable to common stockholders by the weighted average number of shares of common stock outstanding during the period. Diluted loss per share is calculated by dividing net loss attributable to common stockholders as adjusted for the effect of dilutive securities, if any, by the weighted average number of shares of common stock and dilutive common stock outstanding during the period. Potentially dilutive common stock includes the shares of common stock issuable upon the exercise of outstanding stock options and warrants (using the treasury stock method). Potentially dilutive common stock in the diluted net loss per share computation is excluded to the extent that it would be anti-dilutive. No potentially dilutive securities are included in the computation of any diluted per share amounts as the Company reported a net loss for all periods presented. For the years ended December 31, 2023, 2022 and 2021, the following securities were excluded from the calculation of diluted loss per common share because of their anti-dilutive effects:
|
|
For the Years Ended December 31, |
|||||||||||
|
|
2023 |
2022 |
2021 | |||||||||
|
Stock Options |
5,906,184 | 8,256,211 | 7,862,722 | |||||||||
|
Restricted Stock Units |
4,657,297 | 2,866,987 | 4,485,133 | |||||||||
|
Warrants |
12,502,906 | 13,525,148 | 4,528,160 | |||||||||
|
|
23,066,387 | 24,648,346 | 16,876,015 | |||||||||
Fair value of financial instruments
The carrying amounts of certain of the Company’s financial instruments, including cash and cash equivalents, accounts receivable and accounts payable are shown at cost, which approximates fair value due to the short-term nature of these instruments. The debt outstanding under the Company’s senior notes payable (see Note 7) approximates fair value due to the variable interest rate on this debt.
Recent Accounting Pronouncements
|
3. |
INVENTORIES |
The following table provides the components of inventories:
|
|
December 31, |
December 31, |
||||||
| (In thousands) |
||||||||
|
Raw materials |
$ | 52,999 | $ | 48,644 | ||||
|
Work-in-process |
49,621 | 56,171 | ||||||
|
Finished goods |
70,286 | 58,465 | ||||||
|
Total inventories |
$ | 172,906 | $ | 163,280 | ||||
Raw materials includes plasma and other materials expected to be used in the production of ASCENIV, BIVIGAM and Nabi-HB. These materials will be consumed in the production of products expected to be available for sale or otherwise have alternative uses that provide a probable future benefit. All other activities and materials associated with the production of inventories used in research and development activities are expensed as incurred.
Work-in-process inventory primarily consists of the Company’s IVIG products that are manufactured to the bulk drug substance and unlabeled filled vials stage of production.
Finished goods inventory is comprised of the Company’s immunoglobulin products that have reached the filled, labeled and serialized vial stage of production and related intermediates that are available for commercial sale, as well as plasma collected at the Company’s plasma collection centers which is expected to be sold to third-party customers.
|
4. |
PROPERTY AND EQUIPMENT |
Property and equipment at December 31, 2023 and 2022 is summarized as follows:
|
|
December 31, 2023 |
December 31, 2022 |
||||||
| (In thousands) |
||||||||
|
Manufacturing and laboratory equipment |
$ | 21,093 | $ | 18,768 | ||||
|
Office equipment and computer software |
6,062 | 5,319 | ||||||
|
Furniture and fixtures |
5,776 | 5,110 | ||||||
|
Construction in process |
2,273 | 6,727 | ||||||
|
Leasehold improvements |
20,811 | 17,931 | ||||||
|
Land |
4,339 | 4,339 | ||||||
|
Buildings and building improvements |
20,218 | 19,544 | ||||||
|
|
80,572 | 77,738 | ||||||
|
Less: Accumulated depreciation |
(26,737 | ) | (19,477 | ) | ||||
|
Total property, plant and equipment, net |
$ | 53,835 | $ | 58,261 | ||||
The Company recorded depreciation expense on property and equipment of $7.6 million, $6.4 million and $4.8 million for the years ended December 31, 2023, 2022 and 2021, respectively.
|
5. |
INTANGIBLE ASSETS |
Intangible assets at December 31, 2023 and 2022 consist of the following:
|
|
December 31, 2023 |
December 31, 2022 |
||||||||||||||||||||||
| (In thousands) |
||||||||||||||||||||||||
|
|
Cost |
Accumulated Amortization |
Net |
Cost |
Accumulated Amortization |
Net |
||||||||||||||||||
|
Trademark and other intangible rights related to Nabi-HB |
$ | 4,100 | $ | 3,856 | $ | 244 | $ | 4,100 | $ | 3,270 | $ | 830 | ||||||||||||
| Internally developed software | 210 | 9 | 201 | - | - | - | ||||||||||||||||||
|
Rights to intermediates |
907 | 853 | 54 | 907 | 724 | 183 | ||||||||||||||||||
|
|
$ | 5,217 | $ | 4,718 | $ | 499 | $ | 5,007 | $ | 3,994 | $ | 1,013 | ||||||||||||
Under the previous contract manufacturing agreement between ADMA and Biotest, intermediate by-products derived from the manufacture of ASCENIV were property of Biotest. As a result of the acquisition of certain assets from Biotest on June 6, 2017, ADMA obtained the right to these intermediate products, which are being amortized over a period of seven years. The intangible rights to Nabi-HB are also being amortized over a period of seven years. During the year ended December 31, 2023, the Company implemented an internally developed data intelligence and analytics program at a cost of approximately $0.2 million which is being amortized over a period of four years.
Amortization expense related to the Company’s intangible assets for the years ended December 31, 2023, 2022 and 2021 was $0.7 million. Estimated aggregate future aggregate amortization expense is expected to be as follows (in thousands):
|
2024 |
$ |
351 | ||
|
2025 |
52 | |||
|
2026
|
52 | |||
| 2027 |
44 |
|
6. |
ACCRUED EXPENSES AND OTHER LIABILITIES |
Accrued expenses and other current liabilities at December 31, 2023 and 2022 are as follows:
|
|
December 31, 2023 |
December 31, 2022 | ||||||
| (In thousands) |
||||||||
|
Accrued rebates |
$ | 16,608 | $ | 11,437 | ||||
|
Accrued distribution fees |
5,954 | 3,167 | ||||||
|
Accrued incentives |
4,961 | 4,194 | ||||||
|
Accrued testing |
282 | 310 | ||||||
|
Accrued payroll and other compensation
|
2,203 | 4,086 | ||||||
|
Other |
2,911 | 1,796 | ||||||
|
Total accrued expenses and other current liabilities |
$ | 32,919 | $ | 24,990 | ||||
|
7. |
NOTES PAYABLE |
Senior Notes Payable
A summary of outstanding senior notes payable is as follows:
|
|
December 31, 2023 |
December 31, 2022 |
||||||
| (In thousands) |
||||||||
|
Term loan |
$ | 62,500 | $ | 154,748 | ||||
|
Revolving credit facility
|
72,500 | - | ||||||
|
Less: |
||||||||
|
Debt discount |
(4,406 | ) | (11,915 | ) | ||||
|
Senior notes payable |
$ | 130,594 | $ | 142,833 | ||||
On March 23, 2022 (the “Hayfin Closing Date”), the Company and all of its subsidiaries entered into the Hayfin Credit Agreement with Hayfin. The
Hayfin Credit Agreement provided for a senior secured term loan facility in a principal amount of up to $175.0 million (the “Hayfin Credit Facility”), composed of (i) a term loan made on the Hayfin Closing Date in the principal amount of
$ 150.0 million (the “Hayfin Closing Date Loan”), and (ii) a delayed draw term loan in the principal amount of $25.0 million (the “Hayfin Delayed Draw Loan” and, together with the Hayfin Closing
Date Loan, the “Hayfin Loans”). The Hayfin Delayed Draw Loan was not
drawn prior to the Ares Closing Date. The Hayfin Credit Facility had a maturity date of March 23, 2027 (the “Hayfin Maturity Date”),
subject to acceleration pursuant to the Hayfin Credit Agreement, including upon an Event of Default (as defined in the Hayfin Credit Agreement).
On the Ares Closing Date, the Company paid Hayfin the entire outstanding principal amount underlying the Hayfin Loans and all accrued and unpaid interest thereon, as well as the exit fee of 1.0% of the outstanding principal amount paid. This exit fee had been recorded separately as a non-current liability on the accompanying consolidated balance sheet as of December 31, 2022. In accordance with the terms of the Hayfin Credit Agreement, the Company also paid Hayfin the early prepayment fee in the amount equal 7.0% of the prepaid principal amount of $158.6 million, or $11.1 million.
All of the Company’s obligations under the Hayfin Credit Agreement were secured by a first-priority lien and security interest in substantially all of the Company’s tangible and intangible assets, including intellectual property, and all of the equity interests in the Company’s subsidiaries. The Hayfin Credit Agreement contained certain representations and warranties, affirmative covenants, negative covenants and conditions that are customarily required for similar debt financings. The negative covenants restricted or limited the ability of the Company and its subsidiaries to, among other things and subject to certain exceptions contained in the Hayfin Credit Agreement, incur new indebtedness; create liens on assets; engage in certain fundamental corporate changes, such as mergers or acquisitions, or changes to the Company’s or its subsidiaries’ business activities; make certain Investments or Restricted Payments (each as defined in the Hayfin Credit Agreement); change its fiscal year; pay dividends; repay certain other indebtedness; engage in certain affiliate transactions; or enter into, amend or terminate any other agreements that have the impact of restricting the Company’s ability to make loan repayments under the Hayfin Credit Agreement. In addition, the Company was required (i) at all times prior to the Hayfin Maturity Date to maintain a minimum cash balance of $6.0 million; and (ii) as of the last day of each fiscal quarter, report IVIG product and related revenues for the trailing 12-month period that exceed the amounts set forth in the Hayfin Credit Agreement, which ranged from $75.0 million for the fiscal quarter ended June 30, 2022 to $110.0 million for the fiscal quarter ended September 30, 2023. As of the Ares Closing Date and December 31, 2022, the Company was in compliance with all of the covenants contained in the Hayfin Credit Agreement.
As a result of the upfront fee and exit fee paid or payable to Hayfin, the expenses incurred by the Company in connection with this transaction and
the value of the Hayfin Warrants and Hayfin Second Amendment Warrants, the Company recognized a discount on the Hayfin Loans in the amount of $13.9
million for the year ended December 31, 2022 and an additional debt discount in the year ended December 31, 2023 in the amount of $5.7
million. The Company records debt discount as a reduction to the face amount of the debt, and the debt discount is amortized as interest expense over the life of the debt using the interest method. As of the Ares Closing Date, $15.0 million of the aggregate debt discount associated with the Hayfin Credit Facility was unamortized, and this amount is reflected in the loss on
extinguishment of debt for the year ended December 31, 2023. Based on the fair value of the Hayfin Warrants and the aggregate amount of fees and expenses associated with obtaining the Hayfin Credit Facility, the effective interest rate on the
Hayfin Loans as of the Hayfin Closing Date and as of December 31, 2022 was approximately 13.0% and 16.1%, respectively.
|
8. |
STOCKHOLDERS’ EQUITY |
Preferred Stock
The Company is currently authorized to issue up to 10 million shares of preferred stock, $0.0001 par value per share. There were no shares of preferred stock outstanding at December 31, 2023 and 2022.
Common Stock
|
|
Shares |
Weighted
Average
Exercise Price
|
||||||
|
Warrants outstanding at December 31, 2020
|
4,528,160
|
$
|
2.82
|
|||||
|
Expired
|
-
|
$
|
-
|
|||||
|
Granted
|
-
|
$
|
-
|
|||||
|
Exercised
|
-
|
$
|
-
|
|||||
|
Warrants outstanding at December 31, 2021
|
4,528,160
|
$
|
2.82
|
|||||
|
Expired
|
(106,059
|
)
|
$
|
8.23
|
||||
|
Granted
|
9,103,047
|
$
|
1.65
|
|||||
|
Exercised
|
-
|
$
|
-
|
|||||
|
Warrants outstanding at December 31, 2022
|
13,525,148
|
$
|
1.99
|
|||||
|
Expired
|
(24,800
|
)
|
$
|
6.37
|
||||
|
Granted
|
2,391,244
|
$
|
3.26
|
|||||
|
Exercised
|
(3,388,686
|
)
|
$
|
1.65
|
||||
|
Warrants outstanding at December 31, 2023
|
12,502,906
|
$
|
2.32
|
|||||
|
|
Years Ended |
|||||||||||
|
|
December 31, 2023 |
December 31, 2022 |
December 31,
2021
|
|||||||||
|
Expected term |
5.5-6.3 years |
5.5-6.3 years |
5.5-6.3 years
|
|||||||||
|
Volatility |
68 |
% |
68 | % | 68-70 | % | ||||||
|
Dividend yield |
0.0 |
0.0 |
0.0 | |||||||||
|
Risk-free interest rate |
4.20-4.62 |
% |
1.72-1.73 |
% |
0.80-1.27 | % | ||||||
The following table summarizes information about stock options outstanding as of December 31, 2023, 2022 and 2021:
|
|
Shares |
Weighted Average Exercise Price |
||||||
|
Options outstanding, vested and expected to vest at December 31, 2020
|
6,922,931 | $ | 4.40 | |||||
|
Forfeited
|
(529,202 | ) | $ | 2.89 | ||||
|
Expired
|
(426,557 | ) | $ | 4.91 | ||||
|
Granted
|
1,895,550 | $ | 2.14 | |||||
|
Exercised
|
- | $ | - | |||||
|
Options outstanding, vested and expected to vest at December 31, 2021 |
7,862,722 | $ | 3.93 | |||||
|
Forfeited |
(31,540 | ) | $ | 2.37 | ||||
|
Expired |
(700,324 | ) | $ | 6.86 | ||||
|
Granted |
1,194,032 | $ | 1.67 | |||||
|
Exercised |
(68,679 | ) | $ | 2.55 | ||||
|
Options outstanding, vested and expected to vest at December 31, 2022 |
8,256,211 | $ | 3.37 | |||||
|
Forfeited |
(99,345 | ) | $ | 2.73 | ||||
|
Expired |
(262,940 | ) | $ | 6.42 | ||||
|
Granted |
1,826,380 | $ | 3.36 | |||||
|
Exercised |
(3,814,122 | ) | $ | 3.15 | ||||
|
Options outstanding, vested and expected to vest at December 31, 2023 |
5,906,184 | $ | 3.38 | |||||
|
|
||||||||
|
Options exercisable |
3,410,131 |
$ | 3.79 | |||||
|
|
|
Stock Options Outstanding |
|
|
Stock Options Exercisable |
|
|||||||||||||||||||||||||||
|
Range of Exercise Prices |
|
Options Outstanding |
|
|
Weighted Average Remaining Contractual Life (Years) |
|
|
Weighted Average Exercise Price |
|
|
Aggregate Intrinsic Value ($000’s) |
|
|
Options Outstanding |
|
|
Weighted Average Remaining Contractual Life (Years) |
|
|
Weighted Average Exercise Price |
|
|
Aggregate Intrinsic Value ($000’s) |
|
|||||||||
|
$1.10 - $1.67 |
|
|
1,175,410 |
|
|
|
7.7 |
|
|
$ | 1.60 |
|
|
$ | 3,428 |
|
|
|
590,275 |
|
|
|
7.3 |
|
|
$ | 1.56 |
|
|
$ | 1,748 |
|
|
|
$1.73 - $2.60 |
|
|
1,028,392 |
|
|
|
6.6 |
|
|
$ | 2.33 |
|
|
|
2,253 |
|
|
|
712,704 |
|
|
|
6.4 |
|
|
$ | 2.34 |
|
|
|
1,551 |
|
|
|
$2.67 - $4.01 |
|
|
2,905,506 |
|
|
|
7.4 |
|
|
$ | 3.41 |
|
|
|
3,211 |
|
|
|
1,310,276 |
|
|
|
5.3 |
|
|
$ | 3.49 |
|
|
|
1,343 |
|
|
| $4.01 - $6.02 | 383,535 | 3.5 | $ |
5.09 | 22 | 383,535 | 3.5 | $ |
5.09 | 22 | |||||||||||||||||||||||
| $6.26 - $9.39 | 279,841 | 1.1 | $ |
8.48 | - | 279,841 | 1.1 | $ |
8.48 | - | |||||||||||||||||||||||
|
$10.80 - $16.20 |
|
|
133,500 |
|
|
|
1.1 |
|
|
$ | 10.80 |
|
|
|
- |
|
|
|
133,500 |
|
|
|
1.1 |
|
|
$ | 10.80 |
|
|
|
- |
|
|
|
|
|
|
5,906,184 |
|
|
|
6.6 |
|
|
$ | 3.38 |
|
|
$ | 8,914 |
|
|
|
3,410,131 |
|
|
|
5.2 |
|
|
$ | 3.79 |
|
|
$ | 4,664 |
|
|
|
|
Shares |
Weighted Average Grant Date Fair Value |
||||||
|
Balance at December 31, 2020
|
326,000 | $ | 2.81 | |||||
|
Granted
|
4,384,744 | $ | 1.30 | |||||
|
Vested
|
(92,750 | ) | $ | 2.82 | ||||
|
Forfeited
|
(132,861 | ) | $ | 2.51 | ||||
|
Balance at December 31, 2021 |
4,485,133 | $ | 1.34 | |||||
|
Granted |
1,174,266 | $ | 1.74 | |||||
|
Vested |
(2,727,412 | ) | $ | 1.25 | ||||
|
Forfeited |
(65,000 | ) | $ | 1.40 | ||||
| Balance at December 31, 2022 | 2,866,987 | $ | 1.59 | |||||
| Granted |
3,389,760 | $ | 3.42 | |||||
| Vested |
(1,199,445 | ) | $ | 1.63 | ||||
| Forfeited |
(400,005 | ) | $ | 2.71 | ||||
|
Balance at December 31, 2023 |
4,657,297 |
$ | 2.81 | |||||
Total stock-based compensation expense for all awards granted under the Company’s equity incentive plans for the years ended December 31, 2023, 2022 and 2021 was as follows (in thousands):
|
2023 |
2022 | 2021 | ||||||||||
|
Research and development |
$ | 40 | $ | 19 | $ | 154 | ||||||
|
Plasma center operating expenses |
146 | 82 | 60 | |||||||||
|
Selling, general and administrative |
5,331 | 4,717 | 2,958 | |||||||||
|
Cost of product revenue |
670 | 397 | 316 | |||||||||
|
Total stock-based compensation expense |
$ | 6,187 | $ | 5,215 | $ | 3,488 | ||||||
|
9. |
RELATED PARTY TRANSACTIONS |
The Company leases an office building and equipment from Areth, LLC (“Areth”) pursuant to an agreement for services effective as of January 1, 2016, as amended from time to time, and pays monthly rent on this facility in the amount of $10,000. On October 18, 2022, the Company amended the agreement to extend its term to December 31, 2026, with automatic successive one-year renewals thereafter. Either party may terminate the agreement by providing the other party with one year’s prior written notice. Rent expense for the years ended December 31, 2023, 2022 and 2021 amounted to $0.1 million. Areth is a company controlled by Dr. Jerrold B. Grossman, the Vice Chairman of the Board, and Adam S. Grossman, the Company’s President and Chief Executive Officer. The Company also reimburses Areth for office, warehousing and building related (common area) expenses, equipment and certain other operational expenses, which were not material to the consolidated financial statements for the years ended December 31, 2023, 2022 and 2021.
During the years ended December 31, 2023, 2022 and 2021, the Company purchased certain specialized medical equipment and services related to the Company’s plasma collection centers, as well as personal protective equipment, from GenesisBPS and its affiliates (“Genesis”) in the amount of $0.4 million, $0.2 million and $0.2 million, respectively. Genesis is owned by Dr. Grossman and Adam Grossman.
On August 15, 2023, two of the Company’s executive
officers exercised options to purchase 2,909,721 shares of the Company’s common stock on a cashless basis, and 688,657 shares of common stock were issued to these executive officers, net of 257,867 shares of common stock to cover a portion of their tax liabilities (see Note 8).
|
10. |
COMMITMENTS AND CONTINGENCIES |
General Legal Matters
From time to time the Company is or may become subject to certain legal proceedings and claims arising in connection with the normal course of its business. Management does not expect that the outcome of any such claims or actions will have a material effect on the Company’s liquidity, results of operations or financial condition.
IT Systems Disruption
Vendor Commitments
Pursuant to the terms of a plasma purchase agreement dated as of November 17, 2011 (the “2011 Plasma Purchase Agreement”), the Company agreed to purchase from its former contract manufacturer an annual minimum volume of source plasma containing antibodies to RSV to be used in the manufacture of ASCENIV. The Company must purchase a to-be-determined and agreed upon annual minimum volume from the counterparty, and under the original 2011 Plasma Purchase Agreement the Company was permitted to also collect high-titer RSV plasma from up to five wholly-owned ADMA plasma collection facilities. During 2015, the Company amended the 2011 Plasma Purchase Agreement to (i) allow the Company to collect its raw material RSV high-titer plasma from any number of wholly-owned ADMA plasma collection facilities and (ii) allow the Company to purchase its raw material RSV high-titer plasma from other third-party collection organizations, in each case, provided that the annual minimum volumes from the Company’s former contract manufacturer were met, thus allowing the Company to expand its reach for raw material supply as it executes its commercialization plans for ASCENIV. Unless terminated earlier, the 2011 Plasma Purchase Agreement expires in June 2027, after which it may be renewed for two additional five-year periods if agreed to by the parties. On December 10, 2018, the Company’s former contract manufacturer assigned its rights and obligations under the 2011 Plasma Purchase Agreement to Grifols Worldwide Operations Limited (“Grifols”) as its successor-in-interest, effective January 1, 2019.
On June 6, 2017, the Company entered into a Plasma Supply Agreement with its former contract manufacturer, pursuant to which the counterparty supplies, on an exclusive basis subject to certain exceptions, to ADMA BioManufacturing an annual minimum volume of hyperimmune plasma that contain antibodies to the Hepatitis B virus for the manufacture of Nabi-HB. The Plasma Supply Agreement has a 10-year term. On July 19, 2018, the Plasma Supply Agreement was amended to provide, among other things, that in the event the counterparty elects not to supply in excess of ADMA BioManufacturing’s specified amount of Hepatitis B plasma and ADMA BioManufacturing is unable to secure Hepatitis B plasma from a third party at a price that is within a low double- digit percentage of the price that ADMA BioManufacturing pays to the counterparty, then the counterparty shall reimburse ADMA BioManufacturing for the difference in price ADMA BioManufacturing incurs. On December 10, 2018, the Company’s former contract manufacturer assigned its rights and obligations under the Plasma Supply Agreement to Grifols, effective January 1, 2019.
Post-Marketing Commitments
In connection with the FDA approval of the BLA for BIVIGAM on December 19, 2012, Biotest committed to perform two additional post-marketing studies, a pediatric study to evaluate the efficacy and safety of BIVIGAM in children and adolescents, and a post-authorization safety study to further assess the potential risk of hypotension and hepatic and renal impairment in BIVIGAM-treated patients with primary humoral immunodeficiency. These studies were required to be completed by June 30, 2023. Both studies have been completed and the study reports have been submitted to the FDA. ADMA had assumed the remaining obligations, and the costs of the studies were expensed as incurred as research and development expenses. For the years ended December 31, 2023, 2022 and 2021, the Company incurred expenses related to these studies of $1.7 million, $2.2 million and $1.7 million, respectively.
In connection with the FDA approval of ASCENIV on April 1, 2019, the Company is required to perform a pediatric study to evaluate the safety and efficacy of ASCENIV in children and adolescents. For the years ended December 31, 2023, 2022 and 2021, the Company incurred expenses related to this study in the amount of $1.0 million, $0.5 million and $0.6 million, respectively. The Company expects to incur expenses of approximately $1.5 million to complete this study, which is required to be completed by June of 2026.
|
11. |
INCOME TAXES |
A reconciliation of income taxes at the U.S. federal statutory rate to the benefit for income taxes is as follows:
|
|
Years Ended December 31, |
|||||||||||
| (In thousands) |
||||||||||||
|
|
2023 |
2022 |
2021 |
|||||||||
|
Benefit at U.S. federal statutory rate |
$ | (5,930 | ) | $ | (13,840 | ) | $ | (15,046 | ) | |||
|
State taxes - deferred |
(763 | ) | (1,773 | ) | (252 | ) | ||||||
|
Increase in valuation allowance |
4,696 | 15,117 | 14,619 | |||||||||
|
Research and development credits |
- | (211 | ) | (240 | ) | |||||||
| Decrease in federal net operating loss |
- | - | 624 | |||||||||
| 162(m) disallowance |
1,183 | 862 | 64 | |||||||||
|
Other |
814 | (155 | ) | 231 | ||||||||
|
Benefit for income taxes |
$ | - | $ | - | $ | - | ||||||
A summary of the Company’s deferred tax assets is as follows:
|
|
Year Ended December 31, |
|||||||
|
|
2023 |
2022 |
||||||
|
Federal and state net operating loss carryforwards |
$ | 77,757 | $ | 81,526 | ||||
|
Federal and state research credits |
140 | 407 | ||||||
|
Interest expense limitation carryforwards |
21,165 | 12,194 | ||||||
|
Transaction costs |
778 | 882 | ||||||
|
Deferred revenue |
434 | 480 | ||||||
|
Accrued expenses and other |
1,148 | 1,236 | ||||||
|
Total gross deferred tax assets |
101,422 | 96,725 | ||||||
|
Less: valuation allowance for deferred tax assets |
(101,422 | ) | (96,725 | ) | ||||
|
Net deferred tax assets |
$ | - | $ | - | ||||
As of December 31, 2023, the Company had federal and state (post-apportioned basis) net operating losses (“NOLs”) of $315.6 million and $216.4 million, respectively, as well as federal research and development tax credit carryforwards of approximately $0.1 million. Approximately $35.6 million and $95.1 million of the foregoing Federal and state NOLs, respectively, will expire at various dates from 2028 through 2043, if not limited by triggering events prior to such time. Under the provisions of the Internal Revenue Code, changes in ownership of the Company, in certain circumstances, would limit the amount of federal NOLs that can be utilized annually in the future to offset taxable income. In particular, Section 382 of the Internal Revenue Code (“Section 382”) imposes limitations on an entity’s ability to use NOLs upon certain changes in ownership. If the Company is limited in its ability to use its NOLs in future years in which it has taxable income, then the Company will pay more taxes than if it were otherwise able to fully utilize its NOLs. The Company may experience ownership changes in the future as a result of subsequent shifts in ownership of the Company’s capital stock that the Company cannot predict or control that could result in further limitations being placed on the Company’s ability to utilize its Federal NOLs. As of December 31, 2023, the Company performed a substantive analysis of limitations imposed by Section 382 on historical information and concluded that no ownership changes occurred as of December 31, 2023 and 2022. As part of the substantive analysis, the Company did determine that an additional $14.2 million of state NOLs were not subject to limitation of prior ownership changes and are therefore available to offset future taxable income.
A valuation allowance, if needed, reduces deferred tax assets to the amount expected to be realized. When determining the amount of net deferred tax assets that are more likely than not to be realized, the Company assesses all available positive and negative evidence. This evidence includes, but is not limited to, prior earnings history, expected future earnings, carry-back and carry-forward periods and the feasibility of ongoing tax strategies that could potentially enhance the likelihood of the realization of a deferred tax asset. The weight given to the positive and negative evidence is commensurate with the extent the evidence may be objectively verified. As such, it is generally difficult for positive evidence regarding projected future taxable income, exclusive of reversing taxable temporary differences, to outweigh objective negative evidence of recent financial reporting losses. Based on these criteria and the relative weighting of both the positive and negative evidence available, management continues to maintain a full valuation allowance against its net deferred tax assets.
In accordance with U.S. GAAP, the Company is required to determine whether a tax position of the Company is more likely than not to be sustained upon examination by the applicable taxing authority, including resolution of any related appeals or litigation processes, based on the technical merits of the position. The tax benefit to be recognized is measured as the largest amount of benefit that is greater than 50% likely of being realized upon ultimate settlement. Derecognition of a tax benefit previously recognized could result in the Company recording a tax liability that would reduce net assets. The amount of the liability for which an exposure exists is measured as the largest amount of benefit determined on a cumulative probability basis that the Company believes is more likely than not to be realized upon ultimate settlement of the position. Components of the liability are classified as either a current or a long-term liability in the accompanying consolidated balance sheets based on when the Company expects each of the items to be settled. The Company does not have any unrecognized tax benefits as of December 31, 2023 and 2022 and does not anticipate a significant change in unrecognized tax benefits during the next 12 months.
|
12. |
LEASE OBLIGATIONS |
The Company leases certain properties and equipment for its ADMA BioCenters and ADMA BioManufacturing subsidiaries, which leases provide the right to use the underlying assets and require lease payments through the respective lease terms which expire at various dates through 2033. The Company’s lease agreements do not contain any material residual value guarantees or material restrictive covenants.
The Company determines if an arrangement is an operating lease at inception. Leases with an initial term of 12 months or less are not recorded on the balance sheet and lease expense for such leases are recognized on a straight-line basis over the lease term. All other leases are recorded on the balance sheet with assets representing the right to use the underlying asset for the lease term and lease liabilities representing the obligation to make lease payments arising from the lease. Right-to-use assets and lease liabilities are recognized at the lease commencement date based on the present value of lease payments over the lease term and include options to extend or terminate the lease when they are reasonably certain to be exercised. The present value of the lease payments is determined using the Company’s incremental borrowing rate as of the lease commencement date. For the lease liabilities recognized during the years ended December 31, 2023 and 2022, the Company used discount rates of 13% to 16% to determine the present value of its lease obligations. The Company’s operating lease expense is recognized on a straight-line basis over the lease term and is reflected in Plasma center operating expenses and Selling, general and administrative expenses in the accompanying consolidated statements of operations. Aggregate lease expense for the Company’s operating leases for the years ended December 31, 2023, 2022 and 2021 was $2.4 million, $2.1 million and $1.4 million, respectively. Aggregate cash paid on these leases for the years ended December 31, 2023, 2022 and 2021 was $2.4 million, $1.8 million and $1.4 million, respectively.
During the year ended December 31, 2023, the Company recognized one additional right-to-use asset and corresponding lease liability in the amount of $0.1 million for office equipment leased for the Boca Facility. During the year ended December 31, 2022, the Company recognized additional right-to-use assets and corresponding lease liabilities aggregating to approximately $4.0 million in connection with two new property leases where the Company has opened additional plasma collection facilities, a property lease for the storage of raw materials inventory and a property lease for the building that the Company utilizes as its corporate headquarters (see Note 9). Including a finance lease the Company entered into in June 2018, the Company has aggregate lease liabilities of $10.8 million and $11.6 million as of December 31, 2023 and 2022, respectively, which are comprised primarily of the leases for the Company’s plasma collection centers. The Company’s operating leases have a weighted average remaining term of 7.6 years. Scheduled payments under the Company’s lease obligations are as follows (in thousands):
|
Year ended December 31, 2024 |
$ | 2,397 | ||
|
2025 |
2,420 | |||
|
2026 |
2,157 | |||
|
2027 |
2,041 | |||
|
2028 |
2,088 | |||
|
Thereafter |
6,151 | |||
|
Total payments |
17,254 | |||
|
Less: imputed interest |
(6,430 | ) | ||
|
Current portion |
(1,045 | ) | ||
|
Balance at December 31, 2023 |
$ | 9,779 |
|
13. |
SEGMENTS |
The Company is dedicated to manufacturing, marketing and developing specialty plasma-derived biologics. The Company’s ADMA BioManufacturing segment reflects the Company’s immune globulin manufacturing and development operations in Florida, acquired on June 6, 2017. The Plasma Collection Centers segment consists of ten plasma collection facilities as of December 31, 2023, all of which were operational,collecting plasma, and hold an approved license with the FDA (and of which three facilities have received approvals from the Korean Ministry of Food and Drug Safety as well as FDA approval to implement a Hepatitis B immunization program). The Corporate segment includes general and administrative overhead expenses. The Company defines its segments as those business units whose operating results are regularly reviewed by the chief operating decision maker (“CODM”) to analyze performance and allocate resources. The Company’s CODM is its President and Chief Executive Officer. Summarized financial information concerning reportable segments is shown in the following tables:
|
Year Ended December 31, 2023 |
||||||||||||||||
|
(in thousands) |
ADMA BioManufacturing |
Plasma Collection Centers
|
Corporate |
Consolidated |
||||||||||||
|
|
||||||||||||||||
|
Revenues |
$ | 249,738 | $ | 8,334 | $ | 143 | $ | 258,215 | ||||||||
|
|
||||||||||||||||
|
Cost of product revenue |
161,157 |
8,116 |
- |
169,273 |
||||||||||||
|
|
||||||||||||||||
|
Income (loss) from operations |
47,525 | (4,048 | ) | (21,845 | ) | 21,632 | ||||||||||
|
|
||||||||||||||||
|
Interest and other expense, net |
(258 | ) | (1 | ) | (23,438 | ) | (23,697 | ) | ||||||||
|
|
||||||||||||||||
| Loss on extinguishment of debt |
- | - | (26,174 | ) | (26,174 | ) | ||||||||||
|
Net income (loss) |
47,267 | (4,049 | ) | (71,457 | ) | (28,239 | ) | |||||||||
|
|
||||||||||||||||
|
Capital expenditures |
2,952 | 1,819 | - | 4,771 | ||||||||||||
|
Depreciation and amortization expense |
5,156 | 3,176 | - | 8,332 | ||||||||||||
|
Total assets |
246,719 | 34,733 | 47,730 | 329,182 | ||||||||||||
|
Year Ended December 31, 2022 |
||||||||||||||||
|
(in thousands) |
ADMA BioManufacturing |
Plasma Collection
Centers
|
Corporate |
Consolidated |
||||||||||||
|
|
||||||||||||||||
|
Revenues |
$ | 144,070 | $ | 9,867 | $ | 143 | $ | 154,080 | ||||||||
|
|
||||||||||||||||
|
Cost of product revenue |
108,882 |
9,933 |
- |
118,815 |
||||||||||||
|
|
||||||||||||||||
|
Income (loss) from operations |
879 | (17,908 | ) | (22,336 | ) | (39,365 | ) | |||||||||
|
|
||||||||||||||||
|
Interest and other expense, net |
(505 | ) | (3 | ) | (19,361 | ) | (19,869 | ) | ||||||||
| Loss on extinguishment of debt | - | - | (6,670 | ) | (6,670 | ) | ||||||||||
|
|
||||||||||||||||
|
Net income (loss) |
374 | (17,911 | ) | (48,367 | ) | (65,904 | ) | |||||||||
|
|
||||||||||||||||
|
Capital expenditures |
5,247 | 8,664 | - | 13,911 | ||||||||||||
|
Depreciation and amortization expense |
4,709 | 2,404 | - | 7,113 | ||||||||||||
|
Total assets |
238,159 | 37,071 | 73,231 | 348,461 | ||||||||||||
|
Year Ended December 31, 2021 |
||||||||||||||||
|
(in thousands) |
ADMA BioManufacturing |
Plasma Collection Centers |
Corporate |
Consolidated |
||||||||||||
|
|
||||||||||||||||
|
Revenues |
$ | 74,936 | $ | 5,864 | $ | 143 | $ | 80,943 | ||||||||
|
|
||||||||||||||||
|
Cost of product revenue |
74,126 | 5,644 | - | 79,770 | ||||||||||||
|
|
||||||||||||||||
|
Loss from operations |
(29,294 | ) | (12,056 | ) | (17,024 | ) | (58,374 | ) | ||||||||
|
|
||||||||||||||||
|
Interest and other expense, net |
(218 | ) | (6 | ) | (13,050 | ) | (13,274 | ) | ||||||||
|
Net loss |
(29,512 | ) | (12,062 | ) | (30,074 | ) | (71,648 | ) | ||||||||
|
|
||||||||||||||||
|
Capital expenditures |
4,877 | 8,634 | - | 13,511 | ||||||||||||
|
Depreciation and amortization expense |
4,218 | 1,273 | 5 | 5,496 | ||||||||||||
|
Total assets |
208,391 | 24,682 | 43,180 | 276,253 | ||||||||||||
|
14. |
OTHER EMPLOYEE BENEFITS |
The Company sponsors a 401(k) savings plan. Under the plan, employees may make contributions which are eligible for a Company discretionary percentage contribution as defined in the plan and determined by the Board. The Company recognized $1.3 million, $1.3 million and $1.1 million of related compensation expense for the years ended December 31, 2023, 2022 and 2021, respectively.
|
15. |
SUPPLEMENTAL DISCLOSURE OF CASH FLOW INFORMATION |
Supplemental cash flow information for the years ended December 31, 2023, 2022 and 2021 is as follows:
|
|
2023 |
2022 |
2021 |
|||||||||
| (In thousands) |
||||||||||||
|
SUPPLEMENTAL CASH FLOW INFORMATION: |
||||||||||||
|
Cash paid for interest |
$ | 18,051 | $ | 13,880 | $ | 11,159 | ||||||
|
Noncash Financing and Investing Activities: |
||||||||||||
|
Equipment acquired reflected in accounts payable and accrued liabilities |
$ | 86 | $ | 1,495 | $ | 1,353 | ||||||
|
Right-to-use assets in exchange for lease obligations |
$ | 130 | $ | 4,048 | $ | 3,554 | ||||||
|
Warrants issued in connection with notes payable |
$ | 5,595 | $ | 9,570 | $ | - | ||||||
|
16. |
CONCENTRATIONS |
Financial instruments that potentially subject the Company to concentrations of credit risk consist of cash and cash equivalents and accounts receivable. At December 31, 2023, five customers accounted for approximately 98% of the Company’s consolidated accounts receivable. At December 31, 2022, two customers accounted for approximately 92% of the Company’s consolidated accounts receivable.
For the year ended December 31, 2023, two customers accounted for approximately 72% of the Company’s consolidated revenues. For the year ended December 31, 2022, two customers accounted for approximately 74% of the Company’s consolidated revenues. For the year ended December 31, 2021, four customers accounted for approximately 81% of the Company’s consolidated revenues.
During the years ended December 31, 2023 and 2022, plasma purchases from Grifols totaled approximately $9.5 million and $47.7 million, respectively, or approximately 21% and 65%, respectively, of the Company’s total inventory purchases.
|
Years Ended December 31,
|
||||||||||||
| (in thousands) |
2023
|
2022
|
2021 |
|||||||||
|
United States
|
$
|
244,881
|
$
|
146,427
|
$ |
70,626 | ||||||
|
International
|
13,334
|
7,653
|
10,317 | |||||||||
|
Total revenues
|
$
|
258,215
|
$
|
154,080
|
$ |
80,943 | ||||||
|
Additions
|
||||||||||||||||||||
| (in thousands) |
Balance at
beginning of year
|
Charged to costs
and expenses
|
Other
|
Deductions
|
Balance at
end of year
|
|||||||||||||||
|
Year ended December 31, 2023
|
||||||||||||||||||||
|
Accrued rebates
|
$
|
11,437
|
$
|
8,448
|
$
|
-
|
$
|
3,277
|
$
|
16,608
|
||||||||||
|
Inventory valuation allowance
|
$
|
5,400
|
$
|
6,963
|
$
|
6
|
$
|
9,377
|
$
|
2,992
|
||||||||||
|
Deferred tax asset valuation allowance
|
$
|
96,725
|
$
|
4,696
|
$
|
-
|
$
|
-
|
$
|
101,421
|
||||||||||
|
Year ended December 31, 2022
|
||||||||||||||||||||
|
Accrued rebates
|
$
|
5,040
|
$
|
8,227
|
$
|
1,830
|
$
|
11,437
|
||||||||||||
|
Inventory valuation allowance
|
$
|
8,577
|
$
|
2,744
|
$
|
-
|
$
|
5,921
|
$
|
5,400
|
||||||||||
|
Deferred tax asset valuation allowance
|
$
|
81,608
|
$
|
15,117
|
$
|
-
|
$
|
-
|
$
|
96,725
|
||||||||||
|
Year ended December 31, 2021
|
||||||||||||||||||||
|
Accrued rebates
|
$
|
2,604
|
$
|
2,815
|
$
|
-
|
$
|
379
|
$
|
5,040
|
||||||||||
|
Inventory valuation allowance
|
$
|
13,108
|
$
|
4,722
|
$
|
2
|
$
|
9,255
|
$
|
8,577
|
||||||||||
|
Deferred tax asset valuation allowance
|
$
|
66,990
|
$
|
14,618
|
$
|
-
|
$
|
-
|
$
|
81,608
|
||||||||||
|
Jurisdiction of incorporation:
|
Delaware
|
|
Name under which business conducted:
|
ADMA BioCenters Georgia Inc.
|
|
Jurisdiction of incorporation:
|
Delaware
|
|
Name under which business conducted:
|
ADMA Plasma Biologics, Inc.
|
|
Jurisdiction of incorporation:
|
Delaware
|
|
Name under which business conducted:
|
ADMA BioManufacturing, LLC
|
| 1. |
I have reviewed this Annual Report on Form 10-K of ADMA Biologics, Inc. for the year ended December 31, 2023;
|
|
2.
|
Based on my knowledge, this report does not contain any untrue statement of a material fact or omit to state a material fact necessary to make the
statements made, in light of the circumstances under which such statements were made, not misleading with respect to the period covered by this report;
|
|
3.
|
Based on my knowledge, the financial statements, and other financial information included in this report, fairly present in all material respects the
financial condition, results of operations and cash flows of the registrant as of, and for, the periods presented in this report;
|
|
4.
|
The registrant’s other certifying officer and I are responsible for establishing and maintaining disclosure controls and procedures (as defined in
Exchange Act Rules 13a-15(e) and 15d-15(e)) and internal control over financial reporting (as defined in Exchange Act Rules 13a-15(f) and 15d-15(f)) for the registrant and have:
|
|
(a)
|
Designed such disclosure controls and procedures, or caused such disclosure controls and procedures to be designed under our supervision, to ensure that
material information relating to the registrant, including its consolidated subsidiaries, is made known to us by others within those entities, particularly during the period in which this report is being prepared;
|
|
(b)
|
Designed such internal control over financial reporting, or caused such internal control over financial reporting to be designed under our supervision,
to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles;
|
|
(c)
|
Evaluated the effectiveness of the registrant’s disclosure controls and procedures and presented in this report our conclusions about the effectiveness
of the disclosure controls and procedures, as of the end of the period covered by this report based on such evaluation; and
|
|
(d)
|
Disclosed in this report any change in the registrant’s internal control over financial reporting that occurred during the registrant’s most recent
fiscal quarter (the registrant’s fourth fiscal quarter in the case of an annual report) that has materially affected, or is reasonably likely to materially affect, the registrant’s internal control over financial reporting; and
|
|
5.
|
The registrant’s other certifying officer and I have disclosed, based on our most recent evaluation of internal control over financial reporting, to the
registrant’s auditors and the audit committee of the registrant’s board of directors (or persons performing the equivalent functions):
|
|
(a)
|
All significant deficiencies and material weaknesses in the design or operation of internal control over financial reporting which are reasonably likely
to adversely affect the registrant’s ability to record, process, summarize and report financial information; and
|
|
(b)
|
Any fraud, whether or not material, that involves management or other employees who have a significant role in the registrant’s internal control over
financial reporting.
|
|
February 28, 2024
|
/s/ Adam S. Grossman
|
|
|
|
Name:
|
Adam S. Grossman
|
|
|
Title:
|
President and Chief Executive Officer
(Principal Executive Officer)
|
|
|
1. |
I have reviewed this Annual Report on Form 10-K of ADMA Biologics, Inc. for the year ended December 31, 2023;
|
|
|
2. |
Based on my knowledge, this report does not contain any untrue statement of a material fact or omit to state a material fact necessary to make the statements made, in light of the
circumstances under which such statements were made, not misleading with respect to the period covered by this report;
|
|
|
3. |
Based on my knowledge, the financial statements, and other financial information included in this report, fairly present in all material respects the financial condition, results
of operations and cash flows of the registrant as of, and for, the periods presented in this report;
|
|
|
4. |
The registrant’s other certifying officer and I are responsible for establishing and maintaining disclosure controls and procedures (as defined in Exchange Act Rules 13a-15(e) and
15d-15(e)) and internal control over financial reporting (as defined in Exchange Act Rules 13a-15(f) and 15d-15(f)) for the registrant and have:
|
|
|
(a) |
Designed such disclosure controls and procedures, or caused such disclosure controls and procedures to be designed under our supervision, to ensure that material information
relating to the registrant, including its consolidated subsidiaries, is made known to us by others within those entities, particularly during the period in which this report is being prepared;
|
|
|
(b) |
Designed such internal control over financial reporting, or caused such internal control over financial reporting to be designed under our supervision, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles; |
|
|
(c) |
Evaluated the effectiveness of the registrant’s disclosure controls and procedures and presented in this report our conclusions about the effectiveness of the disclosure controls
and procedures, as of the end of the period covered by this report based on such evaluation; and
|
|
|
(d) |
Disclosed in this report any change in the registrant’s internal control over financial reporting that occurred during the registrant’s most recent fiscal quarter (the registrant’s
fourth fiscal quarter in the case of an annual report) that has materially affected, or is reasonably likely to materially affect, the registrant’s internal control over financial reporting; and
|
|
|
5. |
The registrant’s other certifying officer and I have disclosed, based on our most recent evaluation of internal control over financial reporting, to the registrant’s auditors and
the audit committee of the registrant’s board of directors (or persons performing the equivalent functions):
|
|
|
(a) |
All significant deficiencies and material weaknesses in the design or operation of internal control over financial reporting which are reasonably likely to adversely affect the
registrant’s ability to record, process, summarize and report financial information; and
|
|
|
(b) |
Any fraud, whether or not material, that involves management or other employees who have a significant role in the registrant’s internal control over financial reporting.
|
|
February 28, 2024
|
/s/ Brian Lenz
|
|
|
Name:
|
Brian Lenz
|
|
|
Title:
|
Executive Vice President and Chief Financial Officer
(Principal Financial Officer)
|
|
|
|
(1) |
The Report fully complies with the requirements of Section 13(a) or 15(d) of the Securities Exchange Act of 1934, as amended; and
|
|
|
(2) |
The information contained in the Report fairly presents, in all material respects, the financial condition and results of operations of the Company.
|
|
February 28, 2024
|
/s/ Adam S. Grossman
|
|
|
Name:
|
Adam S. Grossman
|
|
|
Title:
|
President and Chief Executive Officer
(Principal Executive Officer)
|
|
|
|
(1) |
The Report fully complies with the requirements of Section 13(a) or 15(d) of the Securities Exchange Act of 1934, as amended; and
|
|
|
(2) |
The information contained in the Report fairly presents, in all material respects, the financial condition and results of operations of the Company.
|
|
February 28, 2024
|
/s/ Brian Lenz
|
|
|
Name:
|
Brian Lenz
|
|
|
Title:
|
Executive Vice President and Chief Financial Officer
(Principal Financial Officer)
|
|
| I. |
Defined Terms.
|
|
|
(a) |
“Applicable Rules” means Section 10D of the Exchange Act and Rule 10D-1 promulgated thereunder, Listing Rule 5608 of the Listing Rules of Nasdaq, and any other national stock exchange rules that the Company is or may become subject
to.
|
|
|
(b) |
“Clawback Event” means a required recoupment of Incentive-Based Compensation in the event of a Restatement under the Applicable Rules.
|
|
|
(c) |
“Exchange Act” means the Securities Exchange Act of 1934, as amended.
|
|
|
(d) |
“Executive Officer” means each officer of the Company who is the Company’s president, principal financial officer, principal accounting officer (or if there is no such accounting officer, the controller), any vice president of the
Company in charge of a principal business unit, division or function (such as sales, administration, or finance), any other officer who performs a policy-making function, or any other person who performs similar significant policy-making
functions for the Company, as determined under 17 CFR §229.401(b).
|
|
|
(e) |
“Financial Reporting Measures” means (i) measures that are determined and presented in accordance with the accounting principles used in preparing the Company’s financial statements, and any measures that are derived wholly or in
part from such measures, (ii) the Company’s stock price, and (iii) total shareholder return in respect of the Company. A “Financial Reporting Measure” need not be presented within the financial statements or included in a filing with the
SEC.
|
|
|
(f) |
“Incentive-Based Compensation” means any compensation that is granted, earned, or vested, based wholly or in part upon the attainment of a Financial Reporting Measure. Incentive-Based Compensation does not include, among other
forms of compensation, equity awards that vest exclusively upon completion of a specified employment period, without any performance condition, and bonus awards that are discretionary or based on subjective goals or goals unrelated to
Financial Reporting Measures.
|
|
|
(g) |
“Nasdaq” means the Nasdaq Stock Market LLC.
|
|
|
(h) |
“Received” – Incentive-Based Compensation is deemed “Received” for the purposes of this Policy in the Company’s fiscal period during which the Financial Reporting Measure applicable to the Incentive-Based Compensation award is
attained, even if the payment or grant of the Incentive-Based Compensation occurs after the end of that period.
|
|
|
(i) |
“Recovery Period” means the three completed fiscal years immediately preceding the date on which the Company is required to prepare a Restatement, which date is the earlier of (i) the date the Board, a committee of the Board, or the
officer(s) of the Company authorized to take such action if Board action is not required, concludes, or reasonably should have concluded, that the Company is required to prepare a Restatement or (ii) a date that a court, regulator, or other
legally authorized body directs the Company to prepare a Restatement.
|
|
|
(j) |
“Regulators” means, as applicable, the SEC and Nasdaq.
|
|
|
(k) |
“Restatement” means that the Company is required to prepare an accounting restatement due to a material noncompliance of the Company with any financial reporting requirement under applicable securities laws, including any required
accounting restatement to correct an error in previously issued financial statements (i) that is material to the previously issued financial statements, or (ii) that would result in a material misstatement if the error were corrected in the
current period or left uncorrected in the current period.
|
|
|
(l) |
“SEC” means the U.S. Securities and Exchange Commission.
|
| II. |
Purpose.
|
| III. |
Administration.
|
| IV. |
Recovery on a Restatement.
|
| V. |
Coverage and Application.
|
| VI. |
Exceptions to Policy.
|
|
|
(a) |
the direct expense paid to a third party to assist in enforcing this Policy would exceed the amount to be recovered; provided that prior to making a determination that it would be impracticable to
recover any Incentive-Based Compensation based on the expense of enforcement, the Company shall (i) have made a reasonable attempt to recover the Incentive-Based Compensation, (ii) have documented such reasonable attempts to recover, and
(iii) provide the documentation to the Regulators as required by the Applicable Rules;
|
|
|
(b) |
recovery would violate home country law where that law was adopted prior to November 28, 2022; or
|
|
|
(c) |
recovery would likely cause an otherwise tax-qualified retirement plan, under which benefits are broadly available to employees, to fail to meet the requirements of Section 401(a)(13) or Section 411(a) of the Internal Revenue Code of 1986,
as amended (the “Code”), and U.S. Treasury regulations promulgated thereunder.
|
| VII. |
Methods of Recovery.
|
|
|
(a) |
the reduction or cancellation of any Incentive-Based Compensation in the form of vested or unvested equity or equity-based awards that have not been distributed or otherwise settled prior to the date of determination;
|
|
|
(b) |
the recovery of any Incentive-Based Compensation that was previously paid to the Executive Officer;
|
|
|
(c) |
the recovery of any gain realized on the vesting, exercise, settlement, sale, transfer, or other disposition of any Incentive-Based Compensation in the form of equity or equity-based awards;
|
|
|
(d) |
the offset, withholding, or elimination of any amount that could be paid or awarded to the Executive Officer after the date of determination;
|
|
|
(e) |
the recoupment of any amount in respect of Incentive-Based Compensation contributed to a plan that takes into account Incentive-Based Compensation (excluding certain tax-qualified plans, but including long-term disability, life insurance,
supplemental executive retirement plans and deferred compensation plans, in each case to the extent permitted by applicable law, including Section 409A of the Code) and any earnings accrued to date on any such amount; and
|
|
|
(f) |
the taking of any other remedial and recovery action permitted by law, as determined by the Committee.
|
| VIII. |
Miscellaneous.
|
|
|
(a) |
Effective Date. This Policy shall be effective as of December 1, 2023 (“Effective Date”).
|
|
|
(b) |
Public Disclosure. The Company shall make all required disclosures and filings with the Regulators with respect to this Policy in accordance with the requirements of the Applicable Rules, and any
other requirements applicable to the Company, including any disclosures required in connection with SEC filings.
|
|
|
(c) |
Notice. Before the Company takes action to seek recovery of compensation pursuant to this Policy against an Executive Officer, the Company shall take commercially reasonable steps to provide such
individual with advance written notice of such clawback; provided that this notice requirement shall not in any way delay the reasonably prompt recovery of any erroneously awarded Incentive-Based Compensation.
|
|
|
(d) |
No Indemnification. The Company shall not indemnify any current or former Executive Officer against the loss of erroneously awarded compensation and shall
not pay or reimburse any Executive Officer for premiums incurred or paid for any insurance policy to fund such Executive Officer’s potential recovery obligations.
|
|
|
(e) |
No Substitution of Rights; Non-Exhaustive Rights. Any right of recoupment under this Policy is in addition to, and not in lieu of, any other remedies or rights of recoupment that may be available
to the Company pursuant to (i) any equity or equity-based incentive compensation plan or any successor plan thereto, any cash-based bonus plan or any other incentive plan of the Company or any of its subsidiaries or affiliates or (ii) the
terms of any similar policy or provision in any employment agreement, compensation agreement or arrangement, or similar agreement and any other legal remedies available to the Company. In addition to recovery of compensation as provided for
in this Policy, the Company may take any and all other actions as it deems necessary, appropriate and in the Company’s best interest in connection with a Clawback Event, including termination of an Executive Officer’s employment and
initiating legal action against an Executive Officer, and nothing in this Policy limits the Company’s rights to take any such or other appropriate actions.
|
|
|
(f) |
Governing Law. This Policy and all determinations made and actions taken pursuant hereto, to the extent not otherwise governed by mandatory provisions of the Applicable Rules, shall be governed by
and construed in accordance with the laws of the State of Delaware without regard to choice of law principles. If any provision of this Policy shall be held illegal or invalid for any reason, such illegality or invalidity shall not affect
the remaining parts of this Policy, but this Policy shall be construed and enforced as if the illegal or invalid provision had never been included in this Policy.
|
|
|
(g) |
Amendment; Termination; Sunset. The Board, based upon the recommendation of the Committee, may amend this Policy at any time for any reason, subject to any limitations under the Applicable Rules.
Unless otherwise required by applicable law, this Policy shall no longer be effective from and after the date that the Company no longer has a class of securities publicly listed on a U.S. national securities exchange or is otherwise not
subject to the Applicable Rules.
|