UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM 6-K
REPORT OF FOREIGN PRIVATE ISSUER
PURSUANT TO RULE 13a-16 OR 15b-16 OF
THE SECURITIES EXCHANGE ACT OF 1934
For the month of December 2025
Commission File Number: 001-41359
Belite Bio, Inc
(Exact name of registrant as specified in its charter)
Not Applicable
(Translation of Registrant’s name into English)
12750 High Bluff Drive Suite 475,
San Diego, CA 92130
(Address of principal executive office)
Indicate by check mark whether the registrant files or will file annual reports under cover Form 20-F or Form 40-F.
Form 20-F x Form 40-F ¨
On December 1, 2025, Belite Bio, Inc (the “Company”) issued a press release entitled “New Hope for People Living with a Disease Once Deemed Untreatable: Belite Bio Announces Positive Topline Results from the Pivotal Global, Phase 3 DRAGON Trial of Tinlarebant in Adolescents with Stargardt Disease.” A copy of this press release is attached hereto as Exhibit 99.1 and is incorporated herein by reference.
On December 1 2025, the Company updated its investor presentation. A copy of this presentation is attached hereto as Exhibit 99.2 and is incorporated herein by reference.
This Report on Form 6-K and the related exhibits are incorporated by reference into all effective registration statements filed by the registrant under the Securities Act of 1933 and shall be a part thereof from the date on which this report is filed, to the extent not superseded by documents or reports subsequently filed or furnished.
| Exhibit No. | Description of Exhibit |
| 99.1 | Press Release |
| 99.2 | Investor Presentation |
SIGNATURE
Pursuant to the requirements of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| Belite Bio, Inc | ||
| By: | /s/ Yu-Hsin Lin | |
| Name: | Yu-Hsin Lin | |
| Title: | Chief Executive Officer and Chairman | |
Date: December 1, 2025
Exhibit 99.1

New Hope for People Living with a Disease Once Deemed Untreatable: Belite Bio Announces Positive Topline Results from the Pivotal Global, Phase 3 DRAGON Trial of Tinlarebant in Adolescents with Stargardt Disease
| · | Tinlarebant is the first therapeutic candidate to demonstrate clinical efficacy in a global Phase 3 trial for Stargardt disease, achieving a statistically significant p-value of 0.0033 | |
| · | Tinlarebant met the primary efficacy endpoint, demonstrating clinical benefit by significantly reducing the lesion growth rate by 36% compared to placebo, as measured by retinal imaging | |
| · | Tinlarebant was well tolerated throughout the trial | |
| · | Stargardt disease impacts more than 50,000 patients in the U.S. | |
| · | Belite Bio plans to file an NDA with the US FDA in 1H 2026 | |
| · | Company will host a conference call and webcast today at 8:00 a.m. ET |
SAN DIEGO, December 1, 2025 (GLOBE NEWSWIRE) -- Belite Bio, Inc (NASDAQ: BLTE) (“Belite Bio” or the “Company”) today announced topline results from the global Phase 3 “DRAGON” trial of Tinlarebant, marking the first successful pivotal trial in patients with Stargardt disease type 1 (STGD1). STGD1 is an eye disease that leads to progressive vision loss, usually beginning in childhood or young adulthood, and currently has no approved treatment worldwide.
The Phase 3 DRAGON trial enrolled 104 patients with STGD1 and met its primary efficacy endpoint, demonstrating a statistically significant and clinically meaningful 36% reduction in the growth rate of retinal lesions, measured as definitely decreased autofluorescence (DDAF) by fundus autofluorescence imaging, compared with placebo. Statistical significance was reached when applying the pre-specified analysis (p-value = 0.0033). Considering the progressive nature typically seen in STGD1, a further post-hoc analysis providing a specific data correlation showed that the treatment effect remained consistent with a p-value < 0.0001.
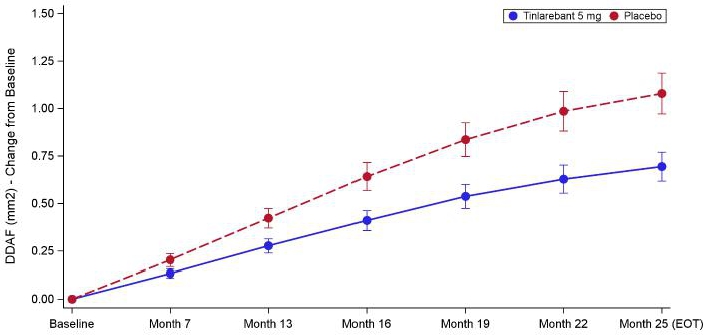
Figure: DDAF (mm2) - Least Squares Mean Change from Baseline by Visit - Study Eye
“The final results from the DRAGON trial mark a historic breakthrough in Stargardt disease, paving the way for the first potential treatment for this devastating condition and bringing new hope to patients and families who have long faced a disease once considered untreatable,” said Dr. Tom Lin, Chairman and CEO of Belite Bio. “Not only was Tinlarebant shown to be efficacious in slowing retinal degeneration, but this is also the first time that an oral treatment was able to demonstrate a clinically meaningful outcome in retinal degenerative disease. With this data, we are advancing our regulatory interactions globally and moving closer to delivering the first approved treatment for people living with Stargardt disease. We extend our sincere gratitude to the patients, families, and investigators whose dedication made this achievement possible.”
“The significant lesion growth reduction observed in the DRAGON study, along with the favorable safety profile, provide important validation of our therapeutic approach and the mechanism of Tinlarebant. These results underscore the team’s commitment to addressing the unmet need in Stargardt disease and the potential to meaningfully improve the quality of life for those affected,” said Dr. Nathan Mata, Chief Scientific Officer at Belite Bio.
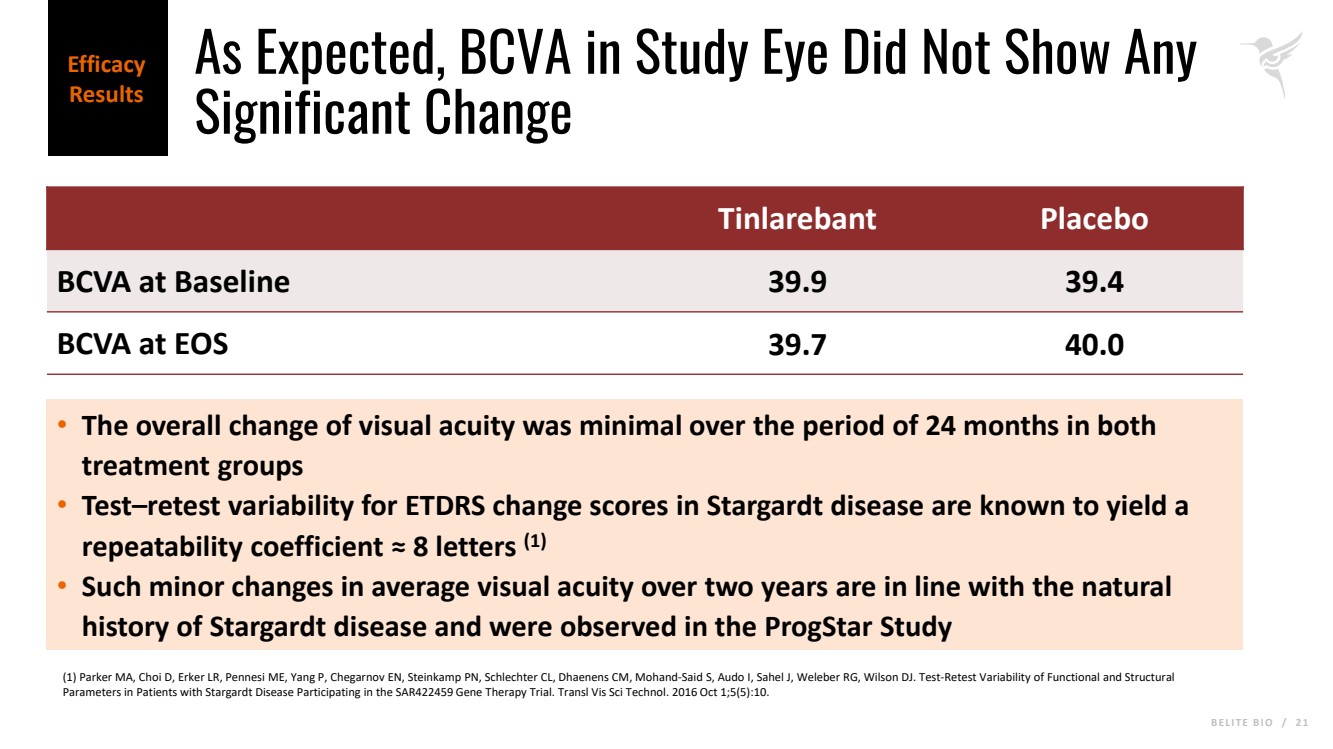
As expected, the overall change in visual acuity was minimal over the period of 24 months in both study groups, consistent with natural history data. The safety profile remains consistent with what the Company previously reported, and Tinlarebant was well tolerated with only four treatment-related discontinuations. After the full analysis is complete, the Company plans to share additional data at upcoming medical meetings.
“Seeing well-controlled Phase 3 data that shows a marked slowing of lesion growth in Stargardt disease is deeply encouraging,” said Professor Michel Michaelides, M.D., FRCOphth, Consultant Ophthalmologist at Moorfields Eye Hospital, the leading investigator in the UK and a top enroller in the DRAGON trial. “Given the strength and consistency of these findings, we believe an approved treatment option is on the horizon for people living with this devastating condition.”
“It is remarkable to recognize that with the robust results of the DRAGON trial, we may soon have Tinlarebant as the first treatment ever for Stargardt disease,” noted Quan Dong Nguyen, MD, MSc, FAAO, FARVO, FASRS, Professor of Ophthalmology at the Byers Eye Institute at Stanford, and Professor of Medicine and Pediatrics at Stanford University School of Medicine. “It is only a matter of time before the observed reduction in lesion growth translates into measurable benefits in visual function. Previous studies have demonstrated that if left untreated, progressive lesion enlargement caused by Stargardt disease is expected to compromise visual acuity and visual field. Tinlarebant has demonstrated that it can significantly reduce lesion growth.”
“The DRAGON trial delivers the most compelling evidence to date that an oral therapy can alter the course of Stargardt disease,” said Dr. Hendrik Scholl, Chief Medical Officer of Belite Bio. “These results validate the scientific approach behind Tinlarebant’s development, demonstrating that reducing the accumulation of toxic byproducts in the retina can meaningfully slow disease progression. Tinlarebant produced a clear and statistically significant treatment effect on DDAF lesion growth rate, not only in the study eye, but both eyes. Furthermore, the effect was supported by a statistically significant benefit in the key secondary endpoint, again in both eyes. Safety and tolerability of Tinlarebant was very favorable in the DRAGON trial. Collectively, these data reinforce Tinlarebant’s potential to change the treatment landscape for Stargardt disease and set a new benchmark for future research in inherited retinal disorders.”
Regulatory Highlights
The Company plans to engage regulatory authorities to discuss potential next steps and to submit New Drug Applications for Tinlarebant in the first half of 2026. Tinlarebant has been granted Breakthrough Therapy, Fast Track, and Rare Pediatric Disease Designations in the U.S.; Orphan Drug Designation in the U.S., Europe, and Japan; and Pioneer Drug Designation in Japan for STGD1.
DRAGON Data Highlights
The DRAGON trial was a 24-month, randomized (2:1, active: placebo), double-masked, placebo-controlled, global, multi-center, pivotal Phase 3 trial in adolescent STGD1 patients.
Patient Demographics
| · | 104 patients (n=69 in tinlarebant arm and n=35 in placebo arm), ranging in age from 12-20 years, were enrolled in the DRAGON trial. |
| · | All patients had been diagnosed with STGD1 with at least one mutation identified in the ABCA4 gene, an atrophic lesion size within three disc areas (7.62 mm2), and a best corrected visual acuity (BCVA) of 20/200 or better. |
Positive Efficacy Results
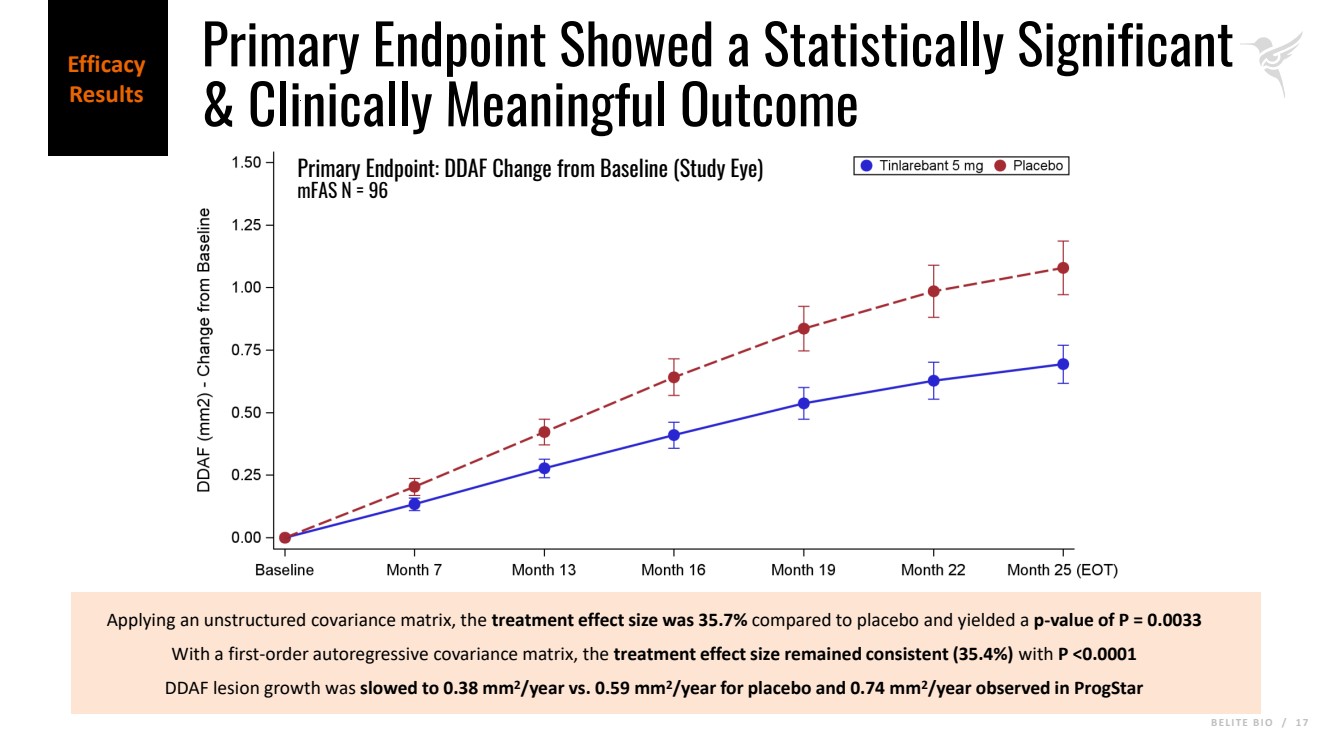
| · | Tinlarebant achieved the primary efficacy endpoint demonstrating a statistically significant reduction in lesion growth rate of 35.7% versus placebo (p-value of 0.0033) as measured by retinal imaging, when applying an unstructured covariance matrix under the Mixed Model for Repeated Measures (MMRM). To account for the longitudinal nature of the collected data while maintaining model stability given the sample size in the DRAGON trial, a post-hoc analysis using an autoregressive covariance matrix under MMRM yielded a treatment effect size of 35.4% with a p-value of <0.0001. |
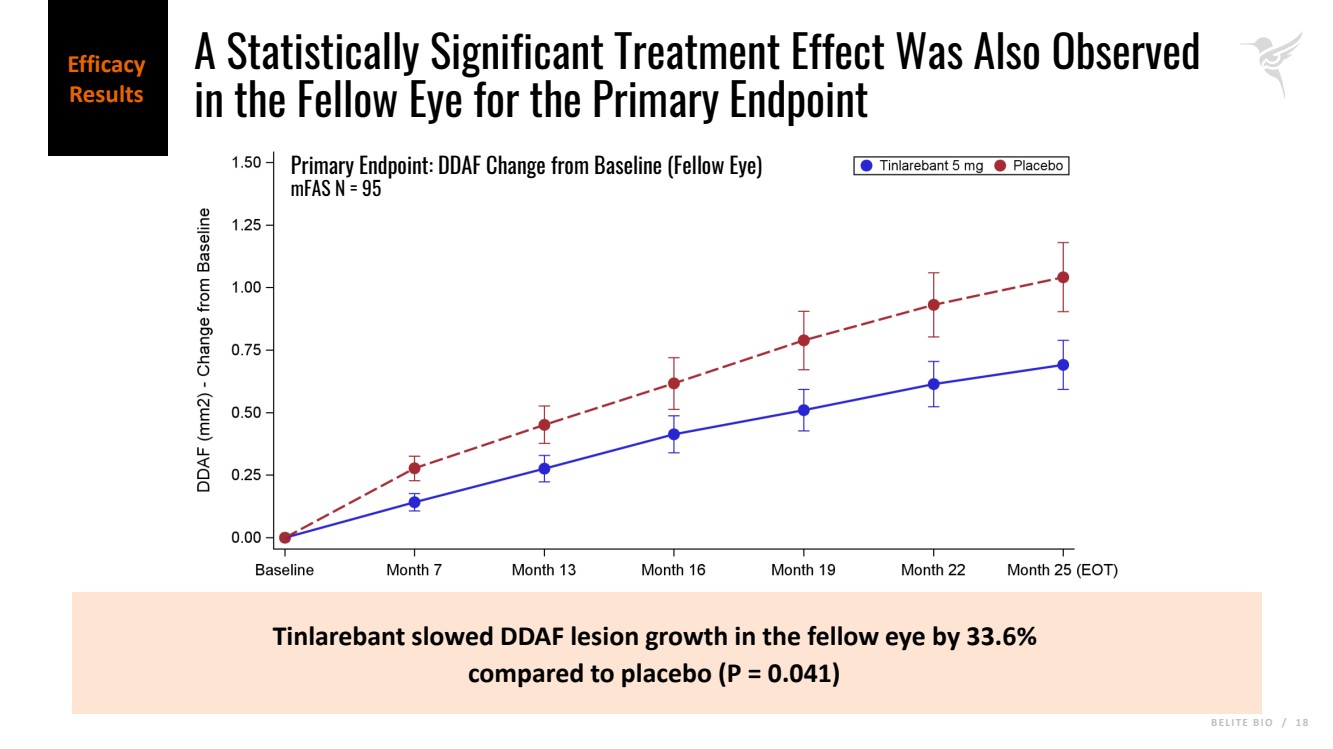
| · | A statistically significant treatment effect was also observed in the fellow eye for the primary endpoint with 33.6% lesion growth reduction (p = 0.041). |
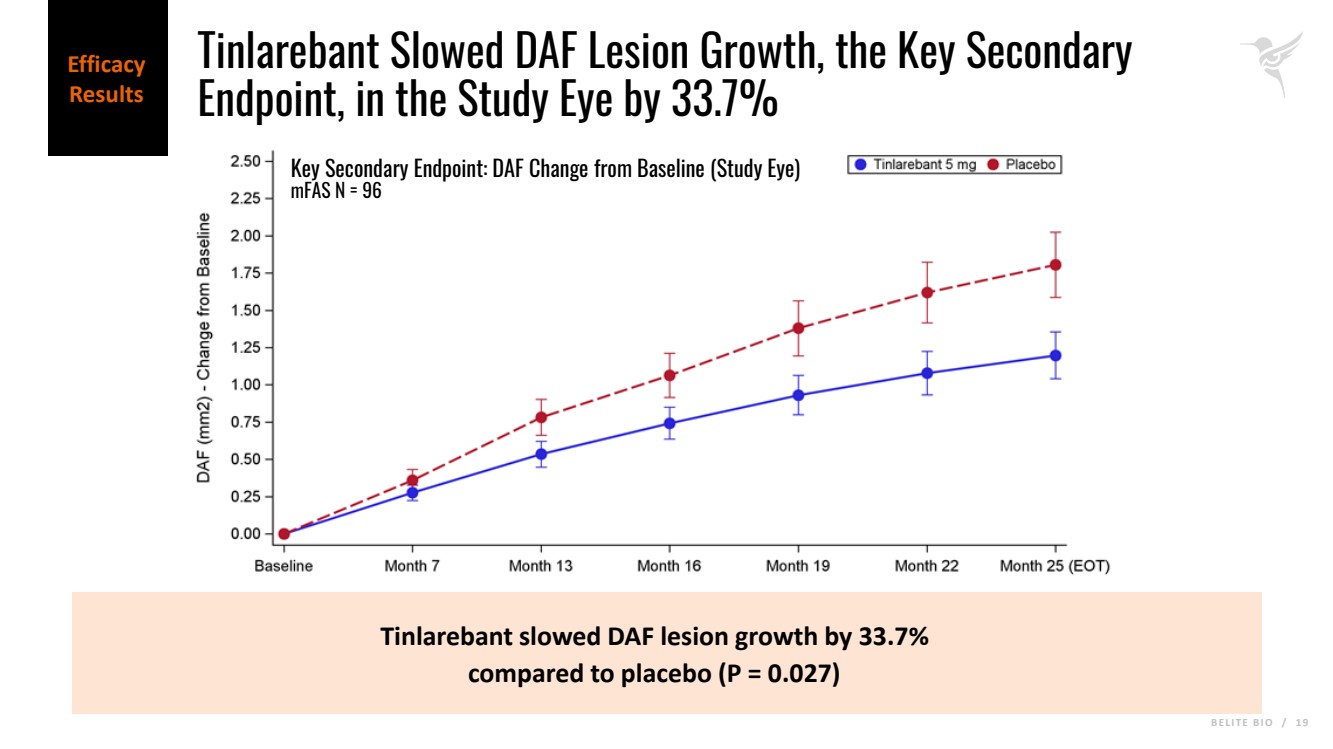
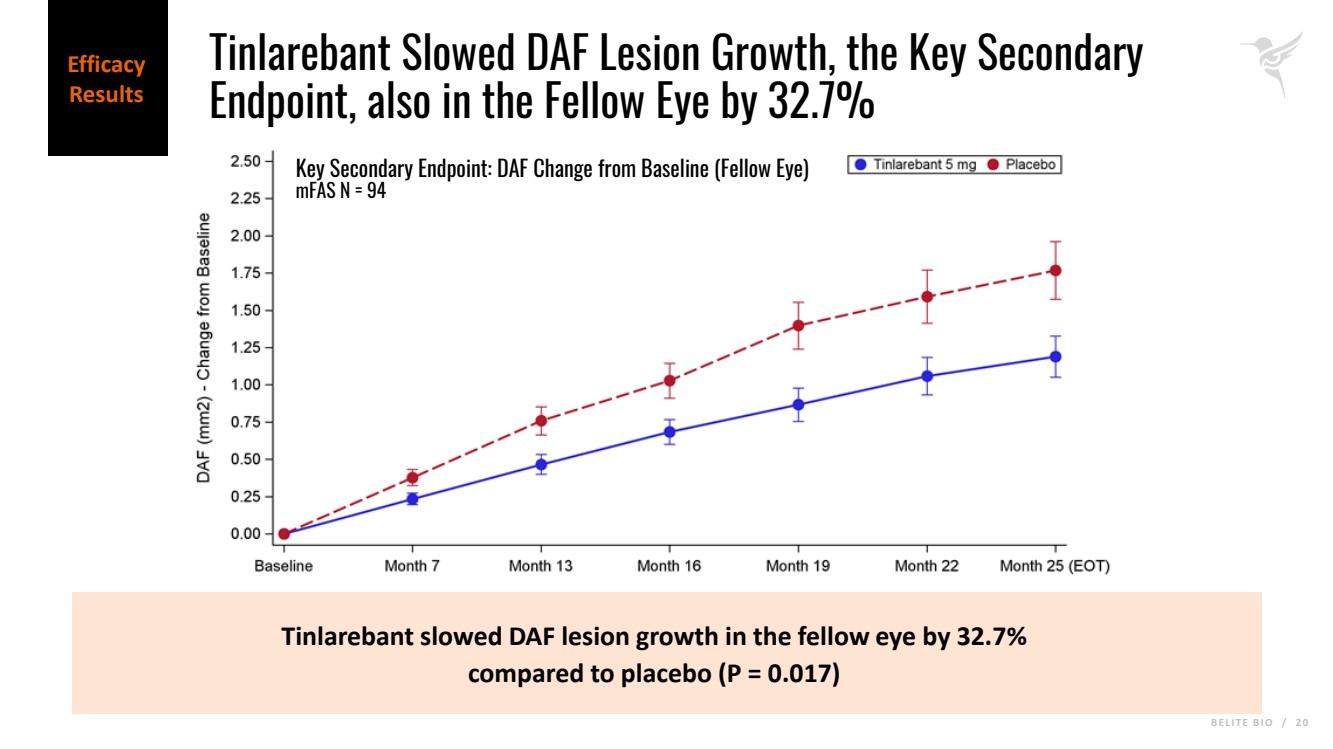
| · | In addition, Tinlarebant slowed decreased autofluorescence (DAF) lesion growth, the key secondary endpoint calculated as the sum of DDAF and questionably decreased autofluorescence (QDAF), in the study eye by 33.7% (p = 0.027) and in the fellow eye by 32.7% (p = 0.017). |
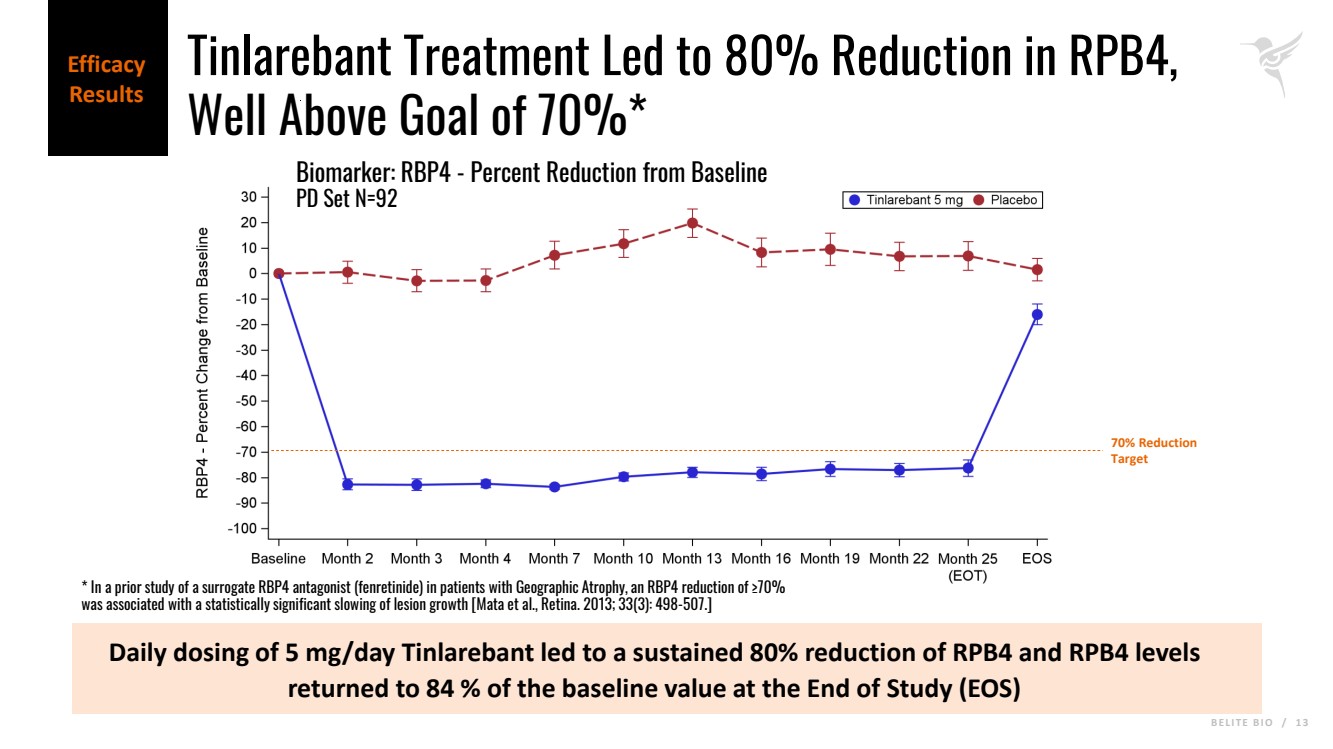
| · | The 5 mg daily dose achieved a reduction in RBP4 levels by a mean of approximately 80% relative to baseline. |
| · | Retinal binding protein 4 (RPB4) levels returned to 84% of the baseline value at End of Study (one- to three-months following drug cessation). Recovery of RBP4 concentration correlated well with the decreased Tinlarebant exposure. |
Strong Safety Profile Consistent with Past Trials
| · | Tinlarebant (5 mg orally, daily) was well tolerated in adolescent STGD1 patients. |
| · | There were no drug or trial discontinuations due to non-ocular adverse events (AE). There were 4 drug discontinuations that were related to the treatment. |
| · | Xanthopsia and delayed dark adaptation are the most common drug-related ocular AE. The majority of xanthopsia, delayed dark adaptation, and night vision impairment were mild, and most resolved during the trial. |
| · | Headaches were the most commonly reported treatment-related non-ocular AE. |
Webcast Information
Date: December
1, 2025
Time: 8:00 a.m. Eastern Time (5:00 a.m. Pacific Time)
Webcast Link: https://events.q4inc.com/attendee/851809284
Webcast Link Instructions
You can join the live webcast by visiting the link above or the “Presentations & Events” section of the Company’s Investor Relations website at https://investors.belitebio.com/presentations-events/events. A replay will be available following the event.
About Tinlarebant (a/k/a LBS-008)
Tinlarebant is a novel oral therapy that is intended to reduce the accumulation of vitamin A-based toxins (known as bisretinoids) that cause retinal disease in STGD1 and also contribute to disease progression in geographic atrophy (GA), or advanced dry age-related macular degeneration (AMD). Bisretinoids are by-products of the visual cycle, which is dependent on the supply of vitamin A (retinol) to the eye. Tinlarebant works by reducing and maintaining levels of serum retinol binding protein 4 (RBP4), the sole carrier protein for retinol transport from the liver to the eye. By modulating the amount of retinol entering the eye, Tinlarebant reduces the formation of bisretinoids. Tinlarebant has been granted Breakthrough Therapy Designation, Fast Track Designation and Rare Pediatric Disease Designation in the U.S., Orphan Drug Designation in the U.S. Europe, and Japan, and Sakigake Designation in Japan for the treatment of STGD1.
About Belite Bio
Belite Bio is a clinical-stage drug development company focused on advancing novel therapeutics targeting degenerative retinal diseases that have significant unmet medical need, such as Stargardt disease type 1 (STGD1) and Geographic Atrophy (GA) in advanced dry age-related macular degeneration (AMD), in addition to specific metabolic diseases. Belite’s lead candidate, Tinlarebant, an oral therapy intended to reduce the accumulation of toxins in the eye, has completed a Phase 3 trial (DRAGON) in adolescent STGD1 subjects and is currently being evaluated in a Phase 2/3 trial (DRAGON II) in adolescent STGD1 subjects and a Phase 3 trial (PHOENIX) in subjects with GA. For more information, follow us on X, Instagram, LinkedIn, and Facebook or visit us at www.belitebio.com.
Important Cautions Regarding Forward Looking Statements
This press release contains forward-looking statements about future expectations and plans, as well as other statements regarding matters that are not historical facts. These statements include but are not limited to statements regarding the potential implications of clinical data for patients, and Belite Bio’s advancement of, and anticipated preclinical activities, clinical development, regulatory milestones, timing of regulatory filings, and commercialization of its product candidates, the ability of Tinlarebant to treat STGD1 and GA, and any other statements containing the words “expect”, “hope” and similar expressions. Actual results may differ materially from those indicated in the forward-looking statements as a result of various important factors, including but not limited to Belite Bio’s ability to demonstrate the safety and efficacy of its drug candidates; the clinical results for its drug candidates, which may not support further development or regulatory approval; the timing to complete any ancillary clinical trials and/or to receive the interim/final data of such clinical trials; the timing to communicate with and submit trial data to regulatory authorities in various jurisdictions for drug approval; the content and timing of decisions made by the relevant regulatory authorities regarding regulatory approval of Belite Bio’s drug candidates; timing for Belite Bio to share additional data at upcoming medical meetings; the potential efficacy of Tinlarebant to set a new benchmark for future research in inherited retinal disorders, as well as those risks more fully discussed in the “Risk Factors” section in Belite Bio’s filings with the U.S. Securities and Exchange Commission. All forward-looking statements are based on information currently available to Belite Bio, and Belite Bio undertakes no obligation to publicly update or revise any forward-looking statements, whether as a result of new information, future events or otherwise, except as may be required by law.
Media and Investor Relations Contact:
Jennifer Wu /ir@belitebio.com
Sophie Hunt /belite@argotpartners.com
Exhibit 99.2

B E L I T E B I O / 1 December 1, 2025, 8:00 a.m. ET Nasdaq: BLTE Phase 3 DRAGON Trial in Stargardt Disease Topline Results |
|
B E L I T E B I O / 2 This presentation (including any oral briefing and any question-and-answer in connection with it) is not intended to, and does not constitute, represent or form part of any offer, invitation or solicitation of any offer to purchase, otherwise acquire, subscribe for, exchange, sell or otherwise dispose of, any securities of Belite Bio, Inc. (“Belite Bio”) from any investor or in any jurisdiction in which such an offer or solicitation is not authorized or would be unlawful. No shares or other securities of Belite Bio are being offered to the public by means of this presentation. No offering of securities shall be made in the United States except pursuant to registration under the U.S. Securities Act of 1933, as amended, or an exemption therefrom. This presentation is being given on the condition that it is for use by the recipient for information purposes and to evaluate Belite Bio and the proposed offering of securities of Belite Bio and for no other purpose. Any failure to comply with these restrictions may constitute a violation of applicable securities laws. Any statements in this presentation about Belite Bio’s future expectations, plans and prospects constitute forward-looking statements for purposes of the safe harbor provisions under the Private Securities Litigation Reform Act of 1995. Forward-looking statements include statements about the strategy, operations and future expectations and plans and prospects for Belite Bio, and any other statements containing the words “expect,” “intend”, “plan,” “predict,” “target,” “will,” “could,” “should,” “continue,” and similar expressions. This presentation contains forward-looking statements, including statements regarding the potential implications of clinical data for patients, and Belite Bio's advancement of, and anticipated preclinical activities, clinical development, regulatory milestones, and commercialization of its product candidates. Actual results may differ materially from those indicated in the forward-looking statements as a result of various important factors, including but not limited to Belite Bio's ability to demonstrate the safety and efficacy of its drug candidates; the clinical results for its drug candidates, which may not support further development or regulatory approval; the content and timing of decisions made by the relevant regulatory authorities regarding regulatory approval of Belite Bio's drug candidates; Belite Bio's ability to achieve commercial success for its drug candidates, if approved; Belite Bio's ability to obtain and maintain protection of intellectual property for its technology and drugs; Belite Bio's reliance on third parties to conduct drug development, manufacturing and other services; Belite Bio's limited operating history and Belite Bio's ability to obtain additional funding for operations and to complete the development and commercialization of its drug candidates; and Belite Bio's ability to enter into additional collaboration agreements beyond its existing strategic partnerships or collaborations, as well as those risks more fully discussed in the “Risk Factors” section in Belite Bio's filings with the U.S. Securities and Exchange Commission. All forward-looking statements are based on information currently available to Belite Bio, and Belite Bio undertakes no obligation to publicly update or revise any forward-looking statements, whether as a result of new information, future events or otherwise, except as may be required by law. Market data and industry information used throughout this presentation are based on the knowledge of the industry and the good faith estimates of Belite Bio’s management. Belite Bio also relied, to the extent available, upon management’s review of independent industry surveys and publications and other publicly available information prepared by a number of third-party sources. All of the market data and industry information used in this presentation involves a number of assumptions and limitations, and you are cautioned not to give undue weight to such estimates. Although the Belite Bio believes that these sources are reliable, it cannot guarantee the accuracy or completeness of, and has not independently conducted verification of the relevant market data and industry information used herein. While Belite Bio believes the estimated market position, market opportunity and market size information included in this presentation are generally reliable, such information, which is derived in part from the management’s estimates and beliefs, is inherently uncertain and imprecise. No representations or warranties are made by Belite Bio or any of its affiliates as to the accuracy of any such statements or projections. Projections, assumptions and estimates of our future performance and the future performance of the industry in which Belite Bio operates are necessarily subject to a high degree of uncertainty and risk due to a variety of factors, including those described above. These and other factors could cause results to differ materially from those expressed in our estimates and beliefs and in the estimates prepared by independent parties. Forward-Looking Statements and Legal Disclaimer |
|
B E L I T E B I O / 3 Today’s Speakers Tom Lin, MMED, PhD, MBA Chairman & Chief Executive Officer Nathan Mata, PhD Chief Strategy Officer Hendrik Scholl, MD, MA Chief Medical Officer Prof Leopold Schmetterer, PhD Prof Ruifang (Helen) Sui, MD, PhD • Consultant Ophthalmologist at Moorfields Eye Hospital in the Departments of Medical Retina, Inherited Eye Disease and Paediatric Ophthalmology • Professor of Ophthalmology at the UCL Institute of Ophthalmology Prof Michel Michaelides, BSc MB BS MD(Res) FRCOphth FACS • Head of Ocular Imaging and Scientific Director at Singapore Eye Research Institute • Professor at the Nanyang Technological University • DSMB Chair of the DRAGON study • Professor, Director of the Department of Ophthalmology at Peking Union Medical College Hospital • Vice President of the Chinese Medical Doctor Association Ophthalmology Committee & Counsellor of the Chinese Ophthalmology Genetics Alliance Belite Management Key Opinion Leaders |
|
B E L I T E B I O / 4 Agenda Stargardt Disease Overview Study Design Efficacy Results Safety Results Summary KOL Panel Discussion Q&A |
|
B E L I T E B I O / 5 Stargardt Disease Overview |
|
B E L I T E B I O / 6 Stargardt Disease Overview Stargardt disease (STGD1) and/or ABCA4-associated retinal dystrophy is a rare and progressive eye disease leading to legal blindness in almost all cases. There is no approved treatment for Stargardt disease • Autosomal recessive mutations in the ABCA4 gene, impairing retinoid transport and leading to toxic bisretinoid buildup in the retinal pigment epithelium (RPE) • Bisretinoid accumulation manifests as yellowish flecks underneath the retina and causes oxidative stress, toxicity, and eventually RPE cell death • RPE damage leads to secondary degeneration of macular photoreceptors • Progressive macular atrophy and retinal degeneration • Gradual central vision loss in both eyes, often starting in childhood or early adulthood • Blurry or distorted central vision • Central blind spots (scotomas) • Photophobia and delayed dark adaptation • Impaired color vision • Difficulty with detailed tasks like reading or recognizing faces • Peripheral vision preserved in the majority of cases • Challenges with reading, using screens, and recognizing faces, harming education and work • Typically, inability to drive, reducing mobility and independence • Restrictions in sports, leisure, and simple tasks like avoiding trips • Social struggles, including interactions, relationships, and events • Emotional effects like frustration, worry, anger, and depression • Career changes or job loss, affecting finances and productivity Pathophysiological Changes Key Symptoms Impact on Activities of Daily Living |
|
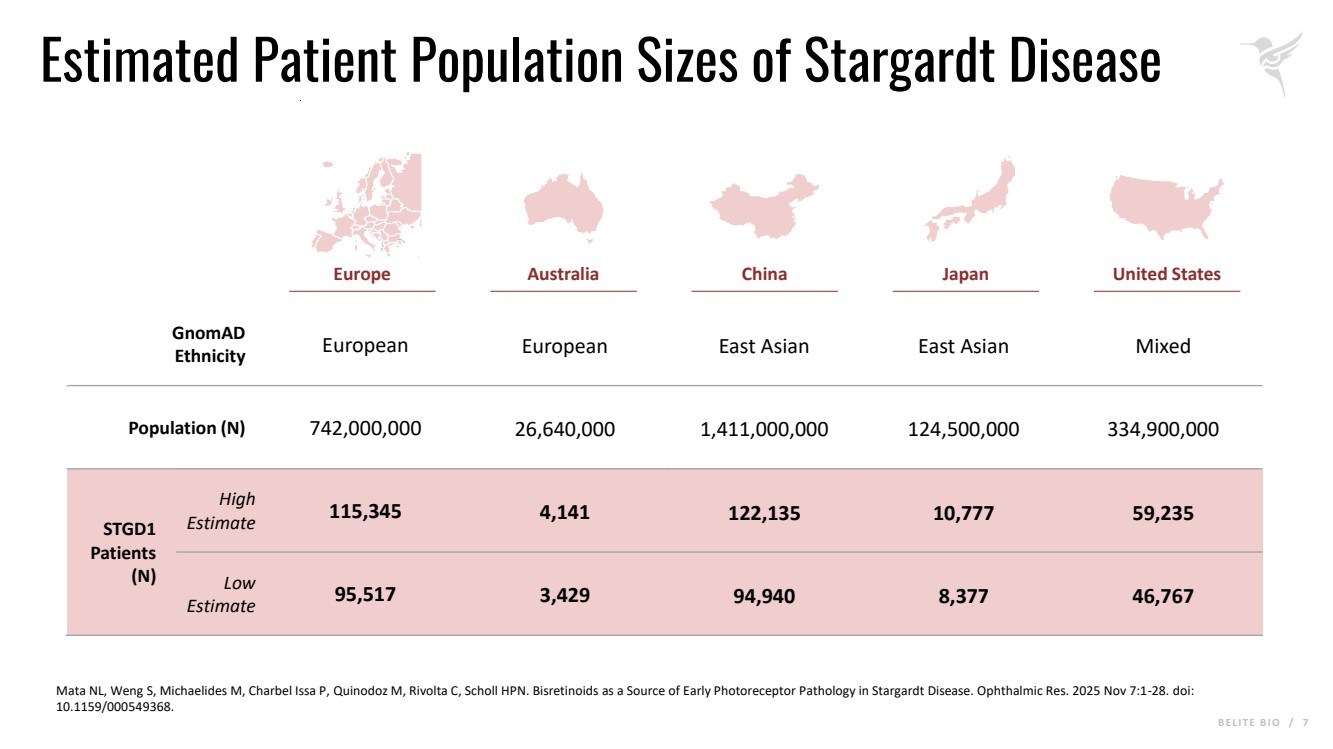
B E L I T E B I O / 7 Estimated Patient Population Sizes of Stargardt Disease Mata NL, Weng S, Michaelides M, Charbel Issa P, Quinodoz M, Rivolta C, Scholl HPN. Bisretinoids as a Source of Early Photoreceptor Pathology in Stargardt Disease. Ophthalmic Res. 2025 Nov 7:1-28. doi: 10.1159/000549368. Europe Australia China Japan United States GnomAD Ethnicity European European East Asian East Asian Mixed Population (N) 742,000,000 26,640,000 1,411,000,000 124,500,000 334,900,000 STGD1 Patients (N) High Estimate 115,345 4,141 122,135 10,777 59,235 Low Estimate 95,517 3,429 94,940 8,377 46,767 |
|
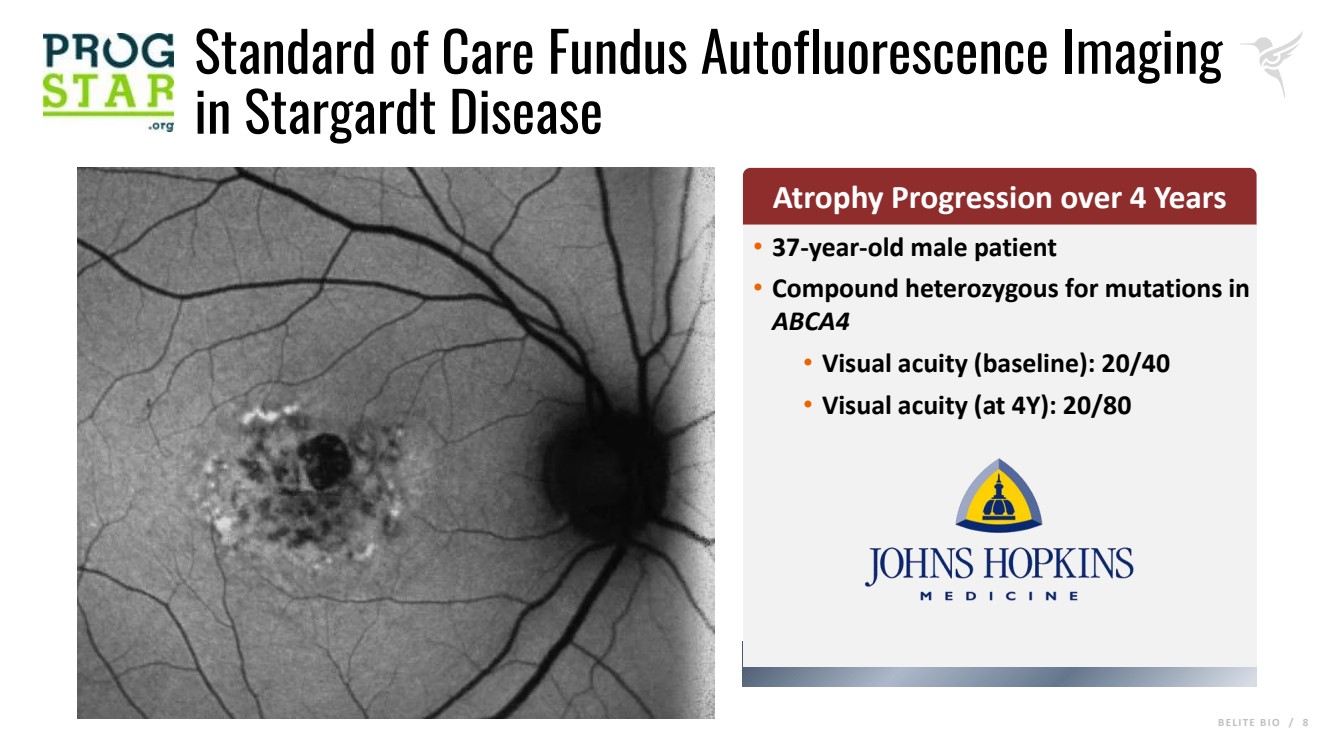
B E L I T E B I O / 8 Standard of Care Fundus Autofluorescence Imaging in Stargardt Disease • 37-year-old male patient • Compound heterozygous for mutations in ABCA4 • Visual acuity (baseline): 20/40 • Visual acuity (at 4Y): 20/80 Atrophy Progression over 4 Years |
|
B E L I T E B I O / 9 Study Design |
|
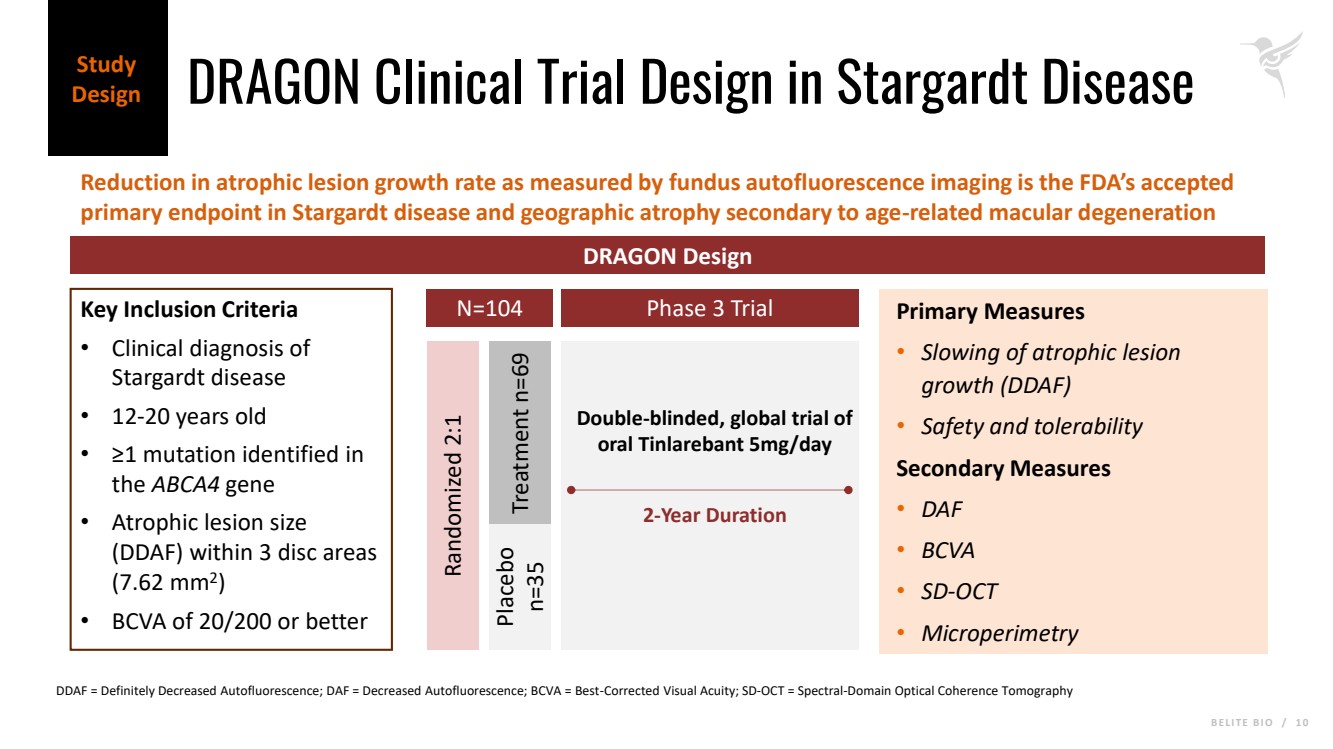
B E L I T E B I O / 1 0 DRAGON Clinical Trial Design in Stargardt Disease Reduction in atrophic lesion growth rate as measured by fundus autofluorescence imaging is the FDA’s accepted primary endpoint in Stargardt disease and geographic atrophy secondary to age-related macular degeneration Study Design DRAGON Design Key Inclusion Criteria • Clinical diagnosis of Stargardt disease • 12-20 years old • ≥1 mutation identified in the ABCA4 gene • Atrophic lesion size (DDAF) within 3 disc areas (7.62 mm2 ) • BCVA of 20/200 or better N=104 Randomized 2:1 Placebo n=35 Treatment n=69 Primary Measures • Slowing of atrophic lesion growth (DDAF) • Safety and tolerability Secondary Measures • DAF • BCVA • SD-OCT • Microperimetry Phase 3 Trial 2-Year Duration Double-blinded, global trial of oral Tinlarebant 5mg/day DDAF = Definitely Decreased Autofluorescence; DAF = Decreased Autofluorescence; BCVA = Best-Corrected Visual Acuity; SD-OCT = Spectral-Domain Optical Coherence Tomography |
|
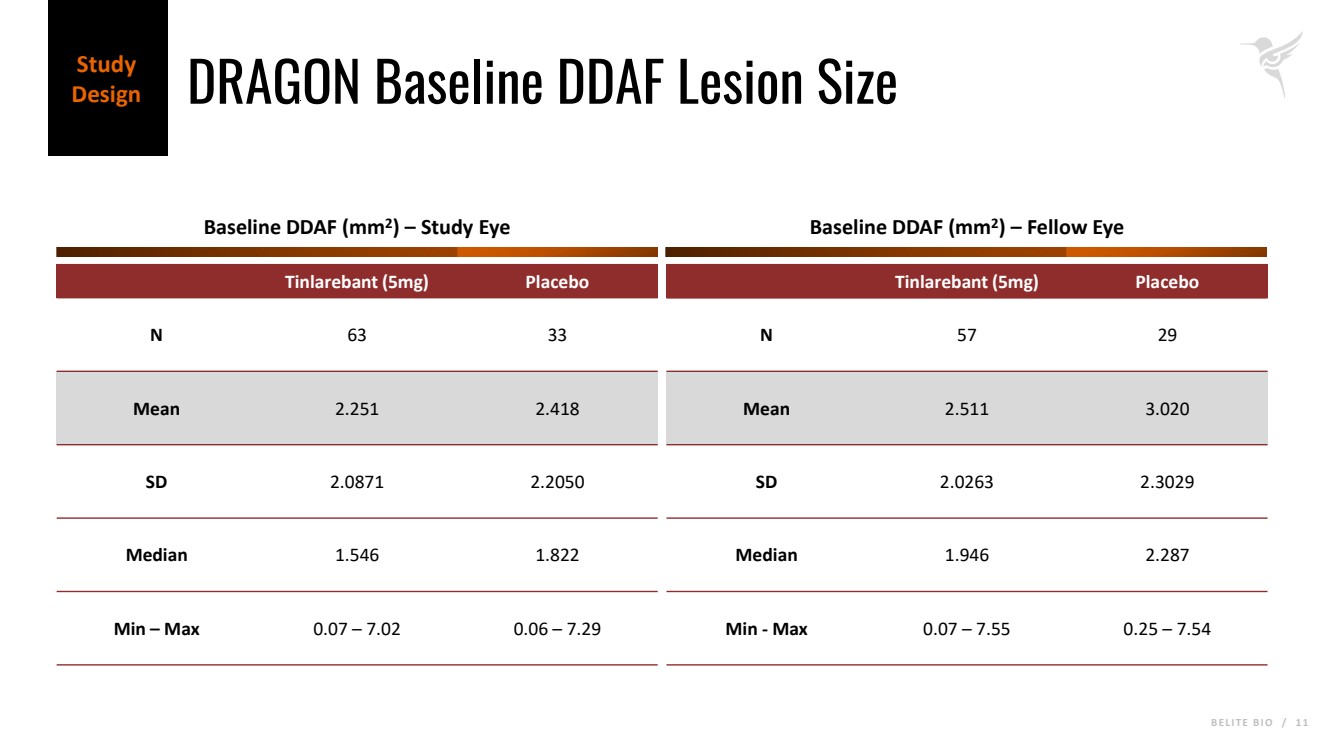
B E L I T E B I O / 1 1 DRAGON Baseline DDAF Lesion Size Study Design Tinlarebant (5mg) Placebo N 63 33 Mean 2.251 2.418 SD 2.0871 2.2050 Median 1.546 1.822 Min – Max 0.07 – 7.02 0.06 – 7.29 Baseline DDAF (mm2 ) – Study Eye Baseline DDAF (mm2 ) – Fellow Eye Tinlarebant (5mg) Placebo N 57 29 Mean 2.511 3.020 SD 2.0263 2.3029 Median 1.946 2.287 Min - Max 0.07 – 7.55 0.25 – 7.54 |
|
B E L I T E B I O / 1 2 Efficacy Results |
|
B E L I T E B I O / 1 3 Daily dosing of 5 mg/day Tinlarebant led to a sustained 80% reduction of RPB4 and RPB4 levels returned to 84 % of the baseline value at the End of Study (EOS) Biomarker: RBP4 - Percent Reduction from Baseline PD Set N=92 Efficacy Results 70% Reduction Target Tinlarebant Treatment Led to 80% Reduction in RPB4, Well Above Goal of 70%* * In a prior study of a surrogate RBP4 antagonist (fenretinide) in patients with Geographic Atrophy, an RBP4 reduction of ≥70% was associated with a statistically significant slowing of lesion growth [Mata et al., Retina. 2013; 33(3): 498-507.] |
|
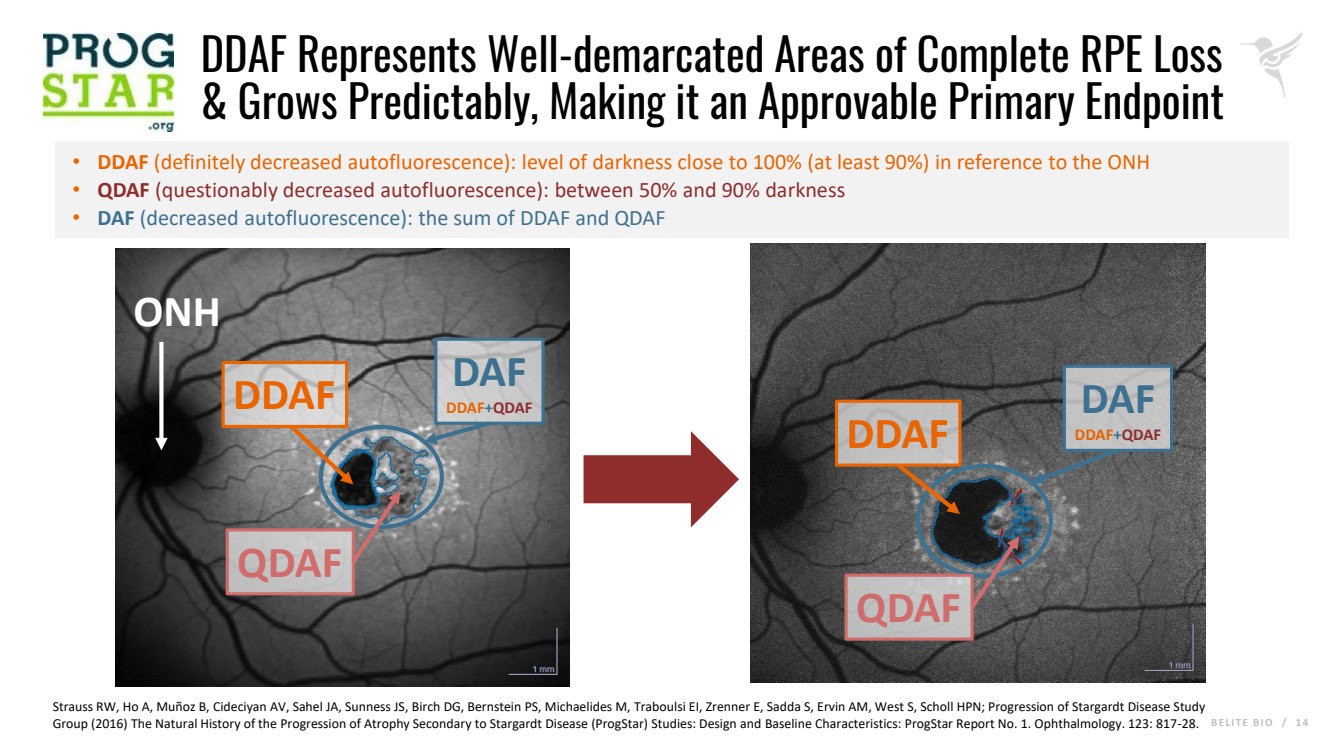
B E L I T E B I O / 1 4 DDAF Represents Well-demarcated Areas of Complete RPE Loss & Grows Predictably, Making it an Approvable Primary Endpoint • DDAF (definitely decreased autofluorescence): level of darkness close to 100% (at least 90%) in reference to the ONH • QDAF (questionably decreased autofluorescence): between 50% and 90% darkness • DAF (decreased autofluorescence): the sum of DDAF and QDAF DDAF QDAF DDAF QDAF ONH Strauss RW, Ho A, Muñoz B, Cideciyan AV, Sahel JA, Sunness JS, Birch DG, Bernstein PS, Michaelides M, Traboulsi EI, Zrenner E, Sadda S, Ervin AM, West S, Scholl HPN; Progression of Stargardt Disease Study Group (2016) The Natural History of the Progression of Atrophy Secondary to Stargardt Disease (ProgStar) Studies: Design and Baseline Characteristics: ProgStar Report No. 1. Ophthalmology. 123: 817-28. DAF DDAF+QDAF DAF DDAF+QDAF |
|
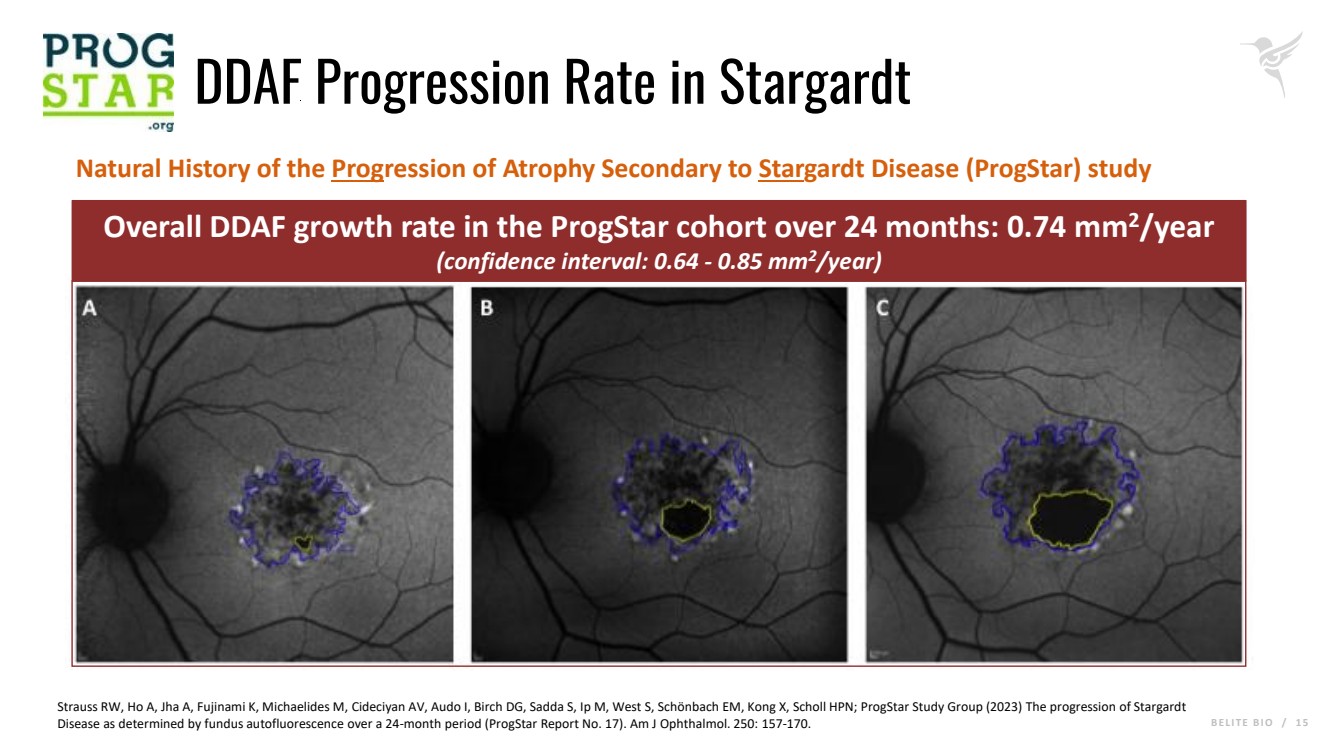
B E L I T E B I O / 1 5 DDAF Progression Rate in Stargardt Overall DDAF growth rate in the ProgStar cohort over 24 months: 0.74 mm2/year (confidence interval: 0.64 - 0.85 mm2/year) Natural History of the Progression of Atrophy Secondary to Stargardt Disease (ProgStar) study Strauss RW, Ho A, Jha A, Fujinami K, Michaelides M, Cideciyan AV, Audo I, Birch DG, Sadda S, Ip M, West S, Schönbach EM, Kong X, Scholl HPN; ProgStar Study Group (2023) The progression of Stargardt Disease as determined by fundus autofluorescence over a 24-month period (ProgStar Report No. 17). Am J Ophthalmol. 250: 157-170. |
|
B E L I T E B I O / 1 6 Primary Endpoint: DDAF in the Study Eye (Change from Baseline) • Annualized rate of lesion growth in the aggregate area of atrophy (DDAF) from baseline as assessed by fundus autofluorescence imaging at Month 25. • Data is shown for the modified full analysis set (mFAS) which consists of all subjects who were randomly assigned to receive study drug and have received at least one dose of study medication. In addition, the mFAS subjects must have a defined DDAF lesion meeting the eligibility criteria at baseline and have at least one post baseline assessment. • Data analysis used a Mixed Model for Repeated Measures (MMRM) measuring change from baseline in DDAF in the study eye and including terms for treatment, visit, treatment*visit interaction, baseline focality of lesions, and baseline DDAF lesion size. • The Statistical Analysis Plan (SAP) specified an unstructured covariance matrix for the MMRM. The CRO also performed a post-hoc analysis using a first-order autoregressive covariance matrix to account for the longitudinal nature of the data while maintaining model stability in a relatively small sample such as in the DRAGON trial. Efficacy Results |
|
B E L I T E B I O / 1 7 Applying an unstructured covariance matrix, the treatment effect size was 35.7% compared to placebo and yielded a p-value of P = 0.0033 With a first-order autoregressive covariance matrix, the treatment effect size remained consistent (35.4%) with P <0.0001 DDAF lesion growth was slowed to 0.38 mm2/year vs. 0.59 mm2/year for placebo and 0.74 mm2/year observed in ProgStar Primary Endpoint: DDAF Change from Baseline (Study Eye) mFAS N = 96 Efficacy Results Primary Endpoint Showed a Statistically Significant & Clinically Meaningful Outcome |
|
B E L I T E B I O / 1 8 Tinlarebant slowed DDAF lesion growth in the fellow eye by 33.6% compared to placebo (P = 0.041) Primary Endpoint: DDAF Change from Baseline (Fellow Eye) mFAS N = 95 Efficacy Results A Statistically Significant Treatment Effect Was Also Observed in the Fellow Eye for the Primary Endpoint |
|
B E L I T E B I O / 1 9 Tinlarebant slowed DAF lesion growth by 33.7% compared to placebo (P = 0.027) Key Secondary Endpoint: DAF Change from Baseline (Study Eye) mFAS N = 96 Efficacy Results Tinlarebant Slowed DAF Lesion Growth, the Key Secondary Endpoint, in the Study Eye by 33.7% |
|
B E L I T E B I O / 2 0 Tinlarebant slowed DAF lesion growth in the fellow eye by 32.7% compared to placebo (P = 0.017) Key Secondary Endpoint: DAF Change from Baseline (Fellow Eye) mFAS N = 94 Efficacy Results Tinlarebant Slowed DAF Lesion Growth, the Key Secondary Endpoint, also in the Fellow Eye by 32.7% |
|
B E L I T E B I O / 2 1 As Expected, BCVA in Study Eye Did Not Show Any Significant Change Tinlarebant Placebo BCVA at Baseline 39.9 39.4 BCVA at EOS 39.7 40.0 • The overall change of visual acuity was minimal over the period of 24 months in both treatment groups • Test–retest variability for ETDRS change scores in Stargardt disease are known to yield a repeatability coefficient ≈ 8 letters (1) • Such minor changes in average visual acuity over two years are in line with the natural history of Stargardt disease and were observed in the ProgStar Study (1) Parker MA, Choi D, Erker LR, Pennesi ME, Yang P, Chegarnov EN, Steinkamp PN, Schlechter CL, Dhaenens CM, Mohand-Said S, Audo I, Sahel J, Weleber RG, Wilson DJ. Test-Retest Variability of Functional and Structural Parameters in Patients with Stargardt Disease Participating in the SAR422459 Gene Therapy Trial. Transl Vis Sci Technol. 2016 Oct 1;5(5):10. Efficacy Results |
|
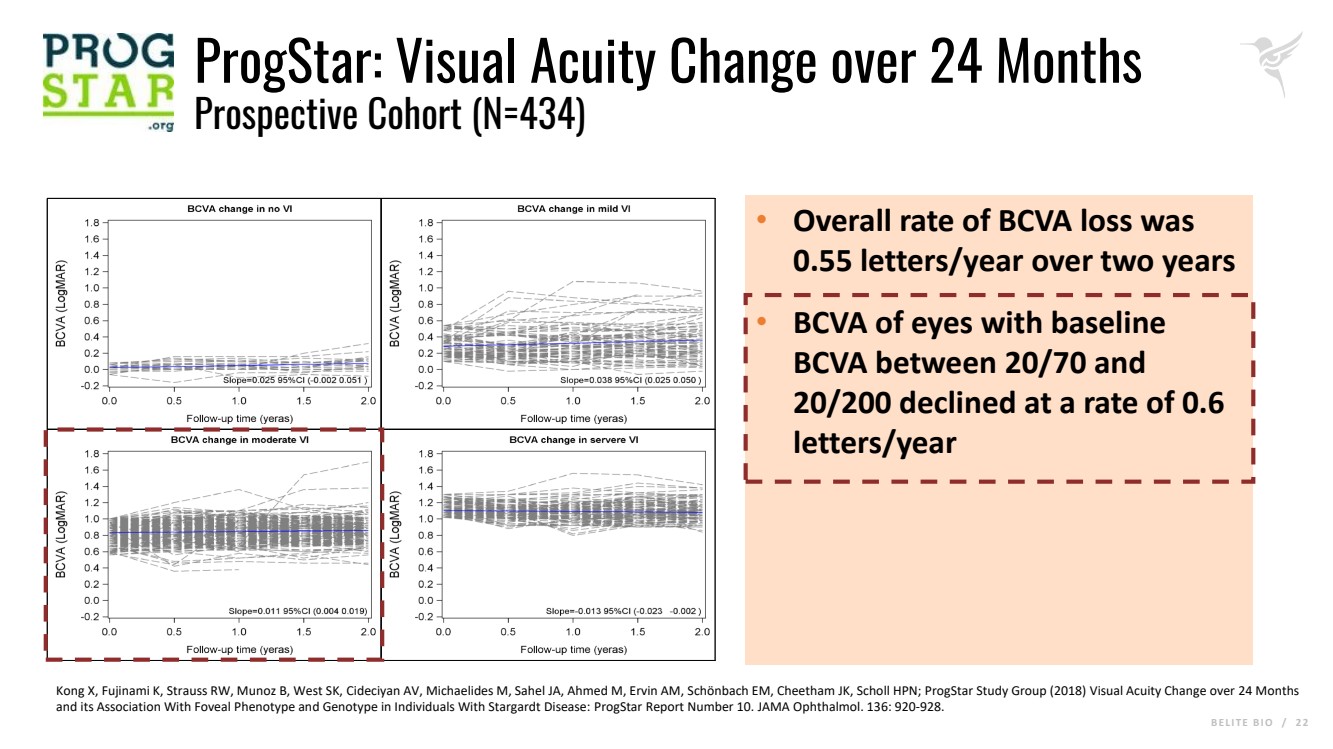
B E L I T E B I O / 2 2 ProgStar: Visual Acuity Change over 24 Months Prospective Cohort (N=434) • Overall rate of BCVA loss was 0.55 letters/year over two years • BCVA of eyes with baseline BCVA between 20/70 and 20/200 declined at a rate of 0.6 letters/year Kong X, Fujinami K, Strauss RW, Munoz B, West SK, Cideciyan AV, Michaelides M, Sahel JA, Ahmed M, Ervin AM, Schönbach EM, Cheetham JK, Scholl HPN; ProgStar Study Group (2018) Visual Acuity Change over 24 Months and its Association With Foveal Phenotype and Genotype in Individuals With Stargardt Disease: ProgStar Report Number 10. JAMA Ophthalmol. 136: 920-928. |
|
B E L I T E B I O / 2 3 Safety Results |
|
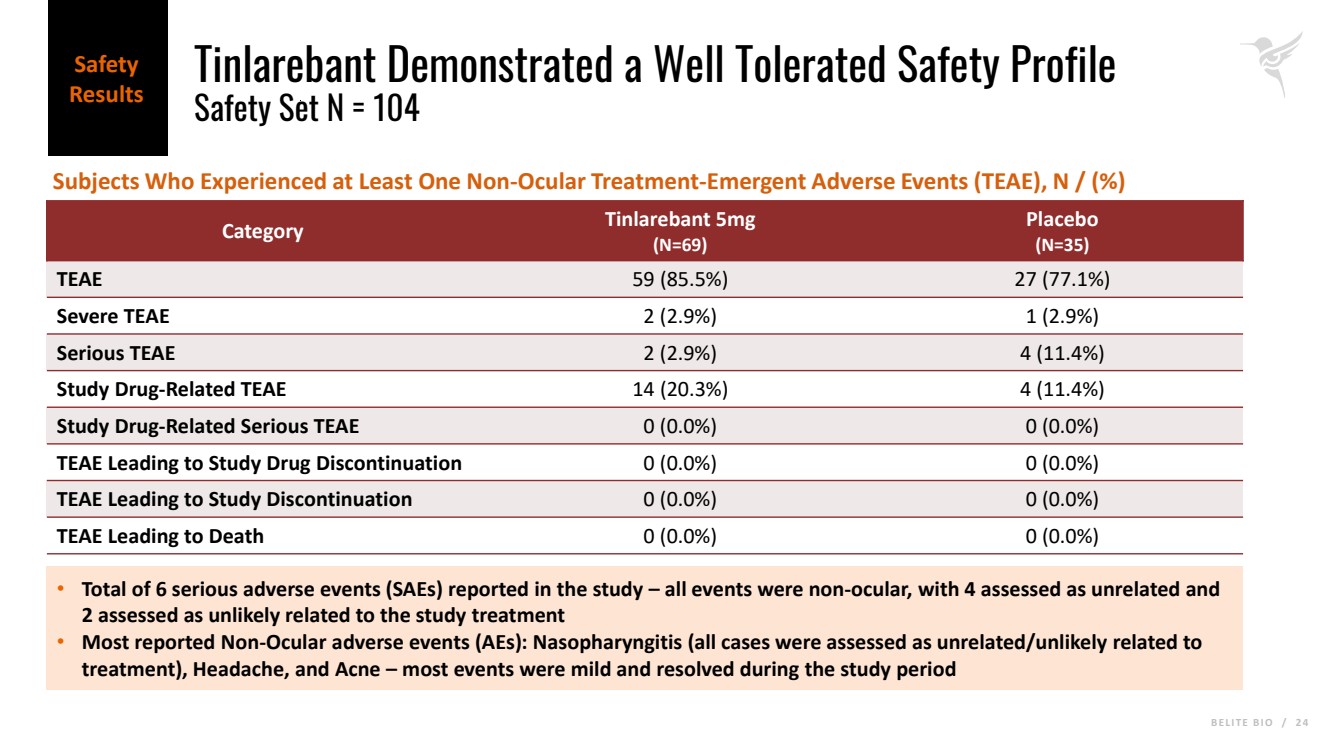
B E L I T E B I O / 2 4 Tinlarebant Demonstrated a Well Tolerated Safety Profile Safety Set N = 104 Category Tinlarebant 5mg (N=69) Placebo (N=35) TEAE 59 (85.5%) 27 (77.1%) Severe TEAE 2 (2.9%) 1 (2.9%) Serious TEAE 2 (2.9%) 4 (11.4%) Study Drug-Related TEAE 14 (20.3%) 4 (11.4%) Study Drug-Related Serious TEAE 0 (0.0%) 0 (0.0%) TEAE Leading to Study Drug Discontinuation 0 (0.0%) 0 (0.0%) TEAE Leading to Study Discontinuation 0 (0.0%) 0 (0.0%) TEAE Leading to Death 0 (0.0%) 0 (0.0%) • Total of 6 serious adverse events (SAEs) reported in the study – all events were non-ocular, with 4 assessed as unrelated and 2 assessed as unlikely related to the study treatment • Most reported Non-Ocular adverse events (AEs): Nasopharyngitis (all cases were assessed as unrelated/unlikely related to treatment), Headache, and Acne – most events were mild and resolved during the study period Subjects Who Experienced at Least One Non-Ocular Treatment-Emergent Adverse Events (TEAE), N / (%) Safety Results |
|
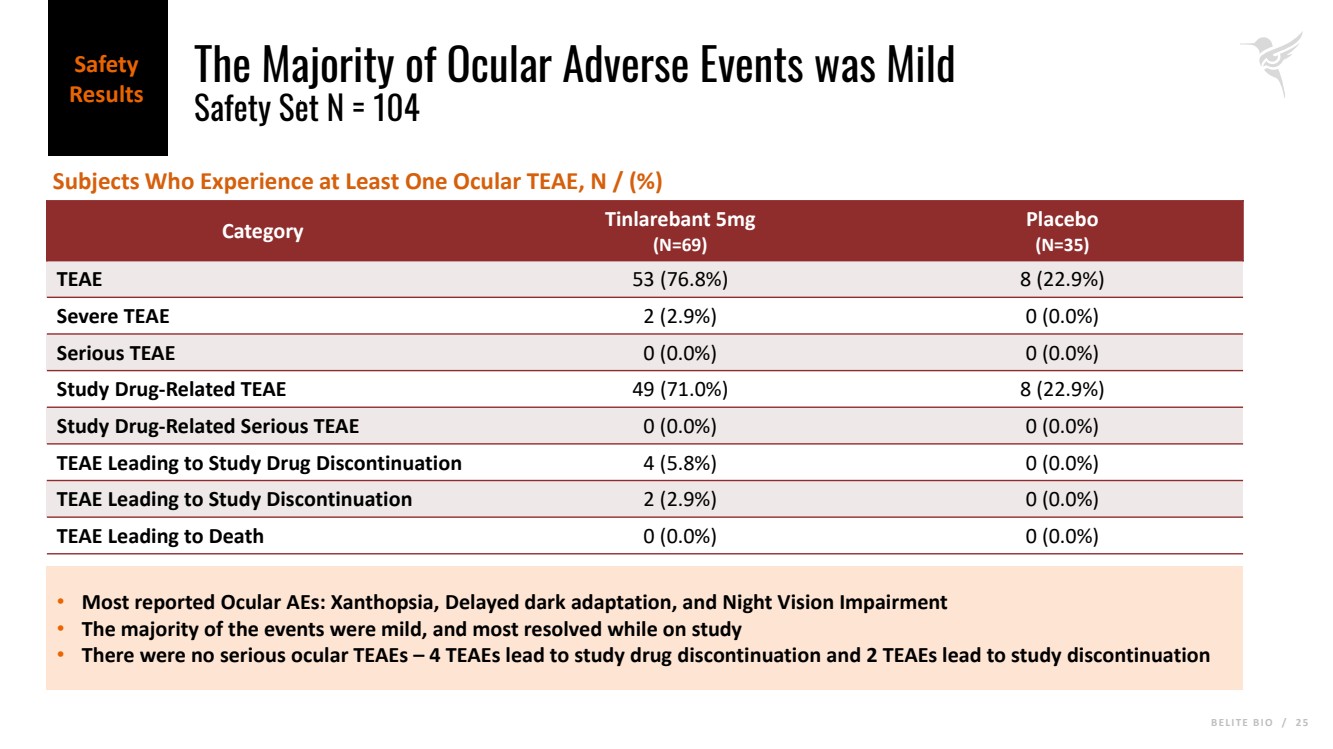
B E L I T E B I O / 2 5 The Majority of Ocular Adverse Events was Mild Safety Set N = 104 Category Tinlarebant 5mg (N=69) Placebo (N=35) TEAE 53 (76.8%) 8 (22.9%) Severe TEAE 2 (2.9%) 0 (0.0%) Serious TEAE 0 (0.0%) 0 (0.0%) Study Drug-Related TEAE 49 (71.0%) 8 (22.9%) Study Drug-Related Serious TEAE 0 (0.0%) 0 (0.0%) TEAE Leading to Study Drug Discontinuation 4 (5.8%) 0 (0.0%) TEAE Leading to Study Discontinuation 2 (2.9%) 0 (0.0%) TEAE Leading to Death 0 (0.0%) 0 (0.0%) • Most reported Ocular AEs: Xanthopsia, Delayed dark adaptation, and Night Vision Impairment • The majority of the events were mild, and most resolved while on study • There were no serious ocular TEAEs – 4 TEAEs lead to study drug discontinuation and 2 TEAEs lead to study discontinuation Subjects Who Experience at Least One Ocular TEAE, N / (%) Safety Results |
|
B E L I T E B I O / 2 6 Summary |
|
B E L I T E B I O / 2 7 Summary of Results of the DRAGON Trial • The DRAGON trial met its primary endpoint: A highly statistically significant slowing in DDAF lesion growth was observed in subjects treated with 5 mg/day oral Tinlarebant as compared to placebo • The treatment effect was 36% and must be considered clinically meaningful • The observed treatment effect was supported by the fellow eye data and the key secondary endpoint: a reduction of DAF area growth • The change in best-corrected visual acuity was minimal in both the treatment and the placebo group – and is in-line with natural history data • The biomarker of tinlarebant treatment, RBP4 reduction, showed a sustained 80% reduction with very little variability • Tinlarebant (5 mg p.o., daily) was safe and well tolerated in adolescent STGD1 patients Summary |
|
B E L I T E B I O / 2 8 Tinlarebant has the Potential to be the First-Ever Approved Treatment for Stargardt Disease • First-ever oral therapy in a retinal degenerative disease to demonstrate a clinically meaningful slowdown of neurodegeneration • 36% reduction in DDAF lesion growth rate, representing a robust and reproducible treatment effect in Stargardt disease • Excellent safety and tolerability profile across two years of treatment • Addresses the root pathogenic mechanism (bisretinoid accumulation), offering a rational, disease-modifying approach where no approved therapies currently exist • Broad applicability across disease stages, from early ABCA4-mediated changes to more advanced atrophy • Personal clinical impact: After >20 years caring for these patients, this represents a true game changer – a therapy I would confidently offer to all my Stargardt patients* Conclusions * Quoted from Dr. Hendrik Scholl, Chief Medical Officer of Belite Bio, based on his personal experience as an ophthalmologist treating STGD1 patients. |
|
KOL Panel Discussion For more info please visit: www.belitebio.com |
|
Q&A For more info please visit: www.belitebio.com |