UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM 8-K
CURRENT REPORT
Pursuant to Section 13 or 15(d) of the
Securities Exchange Act of 1934
Date of report (Date of earliest event reported): November 4, 2025
Verastem, Inc.
(Exact Name of Registrant as Specified in Charter)
| Delaware | 001-35403 | 27-3269467 | ||
| (State or Other Jurisdiction of Incorporation) |
(Commission File Number) |
(IRS Employer Identification No.) |
| 117 Kendrick Street, Suite 500, Needham, MA | 02494 | |
| (Address of Principal Executive Offices) | (Zip Code) |
Registrant’s telephone number, including area code: (781) 292-4200
(Former Name or Former Address, if Changed Since Last Report)
Check the appropriate box below if the Form 8-K filing is intended to simultaneously satisfy the filing obligation of the registrant under any of the following provisions:
| ¨ | Written communications pursuant to Rule 425 under the Securities Act (17 CFR 230.425) |
| ¨ | Soliciting material pursuant to Rule 14a-12 under the Exchange Act (17 CFR 240.14a-12) |
| ¨ | Pre-commencement communications pursuant to Rule 14d-2(b) under the Exchange Act (17 CFR 240.14d-2(b)) |
| ¨ | Pre-commencement communications pursuant to Rule 13e-4(c) under the Exchange Act (17 CFR 240.13e-4(c)) |
Securities registered pursuant to Section 12(b) of the Act:
| Title of each class | Trading Symbol(s) |
Name of each exchange on which registered | ||
| Common stock, $0.0001 par value per share | VSTM | The Nasdaq Capital Market |
Indicate by check mark whether the registrant is an emerging growth company as defined in Rule 405 of the Securities Act of 1933 (§230.405 of this chapter) or Rule 12b-2 of the Securities Exchange Act of 1934 (§240.12b-2 of this chapter).
Emerging growth company ¨
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ¨
Item 2.02. Results of Operations and Financial Condition.
On November 4, 2025, Verastem, Inc. (the “Company”) reported financial results for the quarter ended September 30, 2025, a copy of which is furnished as Exhibit 99.1 to this Current Report on Form 8-K.
Item 7.01 Regulation FD Disclosure
On November 4, 2025, the Company posted its updated corporate presentation on its website, a copy of which is furnished hereto as Exhibit 99.2 to this Current Report on Form 8-K.
Item 9.01 Financial Statements and Exhibits
| Exhibit No. | Description | |
| 99.1 | Earnings Release, dated November 4, 2025 | |
| 99.2 | Corporate Presentation, dated November 4, 2025 | |
| 104 | Cover Page Interactive Data File (embedded within the Inline XBRL document) |
SIGNATURES
Pursuant to the requirements of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned hereunto duly authorized.
| VERASTEM, INC. | ||
| Dated: November 4, 2025 | By: | /s/ Daniel W. Paterson |
| Daniel W. Paterson | ||
| Chief Executive Officer | ||
Exhibit 99.1
Verastem Oncology Reports Third Quarter 2025 Financial Results and Highlights Recent Business Updates
Achieved AVMAPKI™ FAKZYNJA™ CO-PACK net product revenue of $11.2 million
VS-7375 cleared first two monotherapy dose levels with no dose-limiting toxicities reported; no nausea, vomiting or diarrhea greater than Grade 1 were observed
Enrollment initiated for VS-7375 in combination with cetuximab in patients with advanced KRAS G12D mutant solid tumors, including colorectal cancer
Ended Q3 2025 with $137.7 million in cash and cash equivalents; with expected product revenue and exercise of cash warrants, Company cash runway would extend into the second half of 2026
Company to host a conference call and webcast today at 8:00 a.m. ET
BOSTON--(BUSINESS WIRE)—Nov. 4, 2025--Verastem Oncology (Nasdaq: VSTM), a biopharmaceutical company committed to advancing new medicines for patients with RAS/MAPK pathway-driven cancers, today announced business updates and reported financial results for the third quarter ended September 30, 2025.
"Our performance in Q3, which was the first full quarter since our accelerated approval and launch of AVMAPKI FAKZYNJA CO-PACK, exceeded expectations with net revenue of over $11 million and demonstrated the strength of our growing commercial business and consistent adoption by both academic and community oncologists for the first treatment approved by the FDA specifically for patients with KRAS- mutated recurrent LGSOC,” said Dan Paterson, president and chief executive officer of Verastem Oncology. “As we continue to build on this momentum and the fundamentals we have put into place to guide our commercial business, we’re simultaneously advancing our broader strategic priorities, and are very pleased with the progress of our clinical pipeline programs. Particularly for our KRAS G12D (ON/OFF) inhibitor, VS-7375, preliminary safety, tolerability, and anti-tumor activity are promising, and we believe in line as a potential best-in-class option for patients with pancreatic, lung, and other KRAS G12D-mutated solid tumor cancers. As we move ahead with opening the combination cohort with VS-7375 and cetuximab, we look forward to several important data readouts in the first half of 2026 that we believe will further demonstrate the breadth of our RAS/MAPK pathway-driven approach.”
Third Quarter 2025 and Recent Updates
AVMAPKI™ FAKZYNJA™ CO-PACK (avutometinib in combination with defactinib) U.S. Launch
| · | Achieved net product revenue of $11.2 million in the first full quarter of the launch. |
| · | Prescriptions for patients are being received from both academic and community centers, including both repeat prescriptions from physicians prescribing to multiple patients and refills for individual patients. |
| · | There has been broad payer coverage and reimbursement since launch. |
Avutometinib and Defactinib Combination in LGSOC
| · | In the ongoing Phase 3 RAMP 301 confirmatory trial, planned enrollment of the targeted 270 patients was completed a full quarter early. |
| · | A pre-planned Interim Analysis (IA) by an Independent Data Monitoring Committee (IDMC) was conducted for RAMP 301, and the IDMC recommended a modest one-time increase in enrollment. Based on the current total enrollment achieved to date, an additional 29 patients will be added across KRAS mutation status. The Company remains blinded to the IA results. |
| · | Preliminary safety and efficacy data from the Phase 2 RAMP 201J trial in Japan was accepted as an E-Poster (EP228/ #371) at the International Gynecologic Cancer Society (IGCS) 2025 Annual Meeting. In the published abstract, with a data extract date of April 11, 2025, no dose limiting toxicities were observed, and avutometinib and defactinib drug exposure levels were comparable to those observed in the global RAMP-201 study. Additional data, including efficacy (response rates) and updated safety will be available on November 5, 2025, when the embargo lifts. |
Key Milestone:
| · | Expect to complete patient enrollment of the IDMC recommended increase in Q1 2026. |
VS-7375, an Oral KRAS G12D (ON/OFF) Inhibitor, in Advanced Solid Tumors
| · | Announced a preliminary update on the Phase 1/2a monotherapy dose escalation trial of VS-7375 in patients with previously treated advanced KRAS G12D mutant solid tumors on Oct. 23, 2025. |
| o | In the study, VS-7375 cleared both the 400 mg daily (QD) and the 600 mg QD monotherapy doses with no dose-limiting toxicities (DLTs) observed. At the two dose levels evaluated in the U.S. cohort, no nausea, vomiting, or diarrhea greater than Grade 1 were reported. In addition, no new safety signals have been observed relative to earlier data presentations in both pancreatic ductal adenocarcinoma (PDAC) and non-small cell lung cancer (NSCLC) by our partner, GenFleet Therapeutics, in its ongoing Phase 1/ 2 clinical study in China evaluating VS-7375 (known as GFH375). The Company’s dose escalation study continues with evaluation of the monotherapy 900 mg QD dose level. |
| o | Of the five efficacy evaluable patients in the VS-7375-101 study with at least one scan, four out of five patients have had a tumor reduction and are still on treatment. The remaining patients receiving either the 400 mg QD or 600 mg QD doses have not yet reached their first response assessment. |
| · | The Company also announced it has initiated patient enrollment for the first dose escalation combination cohort evaluating VS-7375 with cetuximab in patients with advanced solid tumors, including colorectal cancer. |
| · | Announced updated data from partner GenFleet Therapeutics’ Phase 1 /2 study of GFH375 in China that was featured in a late-breaking oral presentation at the European Society for Medical Oncology (ESMO) Congress on October 19, 2025. |
| o | Among 59 heavily pre-treated patients with PDAC who received one or more prior lines of therapy, an overall response rate (ORR) of 41% was achieved at the monotherapy recommended Phase 2 dose (RP2D) of 600 mg QD. A disease control rate (DCR) of 96.7% (57/59) was also reported with the majority of patients (91.5%) experiencing a reduction in target lesions. |
| o | Overall survival (OS) observed at month four was 92.2%. The median OS was not reached as of the data cutoff, with a median follow-up time of 5.65 months. The median progression-free survival (mPFS) was 5.52 months with a median follow-up time of 5.65 months and a 4-month PFS rate of 78.2%. At evaluation, 31 (47%) patients were still on treatment with the longest duration of treatment eclipsing one year (367 days). The safety profile in PDAC patients was consistent with the previously reported data at recent medical congresses. |
| · | Announced updated data from GenFleet’s Phase 1 /2 study of GFH375 in China that was featured in a mini oral presentation at the IASLC 2025 World Conference on Lung Cancer (WCLC) on September 8, 2025. |
| o | At the RP2D of 600 mg QD, the ORR was 68.8% (11/16) (both confirmed and unconfirmed) and the DCR was 93.8% (15/16). Among the 26 evaluable patients with NSCLC treated across all dose levels, the ORR was 57.7% (15/26) (both confirmed and unconfirmed) and the DCR was 88.5% (23/26). |
| · | GenFleet shared the following additional analyses on Oct. 27, 2025, from previously presented data at recent medical congresses evaluating GFH375 in both advanced KRAS G12D mutant PDAC and NSCLC: |
| o | In a subgroup analysis, 12 patients with 2L PDAC at 600 mg QD achieved an ORR of 58.3% and a DCR of 100%. In the 3L+ setting, 47 PDAC patients receiving 600 mg QD achieved an ORR of 36.2% and a disease control rate (DCR) of 95.7%. In the 2L subgroup, the mPFS and mOS have not been reached. An additional analysis of gastrointestinal disorders, hematological toxicities, and liver enzyme abnormalities in 2L+ patients with PDAC (n=66) at 600 mg QD showed no adverse events Grade ≥3 occured at rates above 8.0%. |
| o | In an analysis of pre-treated patients with NSCLC at 600 mg QD, the four-month PFS rate was >75% and the mPFS has not been reached. The median follow-up time was 4.2 months. |
| · | GenFleet also shared that the first patient has been dosed in a Phase 1b/2 study of GFH375 combined with cetuximab or chemotherapy for advanced solid tumors on October 22, 2025. |
Key Milestones:
| · | Plan to initiate the dose escalation cohorts in combination with chemotherapy for PDAC and with chemotherapy plus anti-PD-1 for NSCLC in Q4 2025. |
| · | Plan to report an interim safety and efficacy update on the Phase 1/2a trial of VS-7375 in 1H 2026. |
| · | Expect to select the RP2D and plan to initiate monotherapy expansion cohorts in advanced PDAC, NSCLC, and other KRAS G12D-mutated solid tumors in 1H 2026. |
| · | Expect to select the RP2D and plan to initiate combination expansion cohorts in CRC, PDAC, and NSCLC in 1H 2026. |
| · | Plan to engage with the FDA to discuss our development path forward, including potential registration-directed clinical trials in PDAC and NSCLC in 1H 2026. |
RAMP 205: Avutometinib Plus Defactinib in Combination with Chemotherapy in First-Line Metastatic PDAC
| · | Completed enrollment in the RAMP 205 expansion cohort in Q3 2025. |
Key Milestone:
| · | Expect to report an update on the safety and efficacy of the RAMP 205 expansion cohort with 29 patients at the RP2D in 1H26. |
RAMP 203: Avutometinib Plus Defactinib in Combination with a KRAS G12C Inhibitor in NSCLC
| · | Patients continue to be evaluated in both the doublet and triplet combination cohorts of the study. |
Key Milestone:
| · | Report an interim update on the safety and efficacy results in RAMP 203 from both the doublet and triplet combinations in Q4 2025. |
Third Quarter 2025 Financial Results
Net product revenue for the three months ended September 30, 2025 (the “2025 Quarter”) was $11.2 million, compared to $0.0 million for the three months ended September 30, 2024 (the “2024 Quarter”). The Company began commercial sales of the AVMAPKI FAKZYNJA CO-PACK within the United States following receipt of FDA approval in May 2025.
Total operating expenses for the 2025 Quarter were $52.0 million, compared to $37.0 million for the 2024 Quarter. Cost of sales associated with product revenue was $1.7 million for the 2025 Quarter, compared to $0.0 for the 2024 Quarter.
Research & development expenses for the 2025 Quarter were $29.0 million, compared to $24.8 million for the 2024 Quarter. The increase of $4.2 million, or 16.9%, was primarily related to increased drug substance and drug product costs, increased contract research organization costs, and increased investigator trial costs.
Selling, general & administrative expenses for the 2025 Quarter were $21.0 million, compared to $12.3 million for the 2024 Quarter. The increase of $8.7 million, or 70.7%, was primarily related to commercialization costs required as part of the launch of AVMAPKI FAKZYNJA CO-PACK in KRAS-mutated recurrent LGSOC. This was comprised of increased consulting, personnel costs, and professional fees.
Net loss (GAAP basis) for the 2025 Quarter was $98.5 million, or $1.35 per share (basic and diluted), compared to $24.0 million, or $0.60 per share (basic and diluted) for the 2024 Quarter.
For the 2025 Quarter, non-GAAP adjusted net loss was $39.4 million, or $0.54 per share (diluted) compared to non-GAAP adjusted net loss of $35.3 million, or $0.88 per share (diluted), for the 2024 Quarter. Please refer to the GAAP to non-GAAP Reconciliation attached to this press release.
Verastem Oncology ended the third quarter of 2025 with cash, cash equivalents and investments of $137.7 million. With existing cash, product revenue, and exercise of cash warrants, Company has expected cash runway into the second half of 2026.
Conference Call and Webcast
Verastem will host a conference call and webcast today at 8:00 a.m. ET to review the third quarter 2025 financial results and recent business updates. To access the conference call, please dial ((888) 596-4144 (U.S.) or (646) 968-2525 (international) and enter the passcode 8194537 at least 10 minutes prior to the event start time. A live audio webcast of the call, along with accompanying slides, will be available under "Events & Presentations" in the Investor section of the Company's website, https://investor.verastem.com/events. A replay of the webcast will be archived and available following the event.
Use of Non-GAAP Financial Measures
To supplement Verastem Oncology’s condensed consolidated financial statements, which are prepared and presented in accordance with generally accepted accounting principles in the United States (GAAP), the Company uses the following non-GAAP financial measures in this press release: non-GAAP adjusted net loss and non-GAAP net loss per share. These non-GAAP financial measures exclude certain amounts or expenses from the corresponding financial measures determined in accordance with GAAP. Management believes this non-GAAP information is useful for investors, taken in conjunction with the Company’s GAAP financial statements, because it provides greater transparency and period-over- period comparability with respect to the Company’s operating performance and can enhance investors’ ability to identify operating trends in the Company’s business. Management uses these measures, among other factors, to assess and analyze operational results and trends and to make financial and operational decisions. Non-GAAP information is not prepared under a comprehensive set of accounting rules and should only be used to supplement an understanding of the Company’s operating results as reported under GAAP, not in isolation or as a substitute for, or superior to, financial information prepared and presented in accordance with GAAP. In addition, these non-GAAP financial measures are unlikely to be comparable with non-GAAP information provided by other companies. The determination of the amounts that are excluded from non-GAAP financial measures is a matter of management judgment and depends upon, among other factors, the nature of the underlying expense or income amounts. Reconciliations between these non-GAAP financial measures and the most comparable GAAP financial measures for the three and nine months ended September 30, 2025 and 2024 are included in the tables accompanying this press release after the unaudited condensed consolidated financial statements.
About AVMAPKI and FAKZYNJA Combination Therapy
AVMAPKI (avutometinib) inhibits MEK kinase activity while also blocking the compensatory reactivation of MEK by upstream RAF. RAF and MEK proteins are regulators of the RAS/RAF/MEK/ERK (MAPK) pathway. Blocking RAF and/or MEK activates FAK, a key mediator of drug resistance. FAKZYNJA (defactinib) is a FAK inhibitor and together, the avutometinib and defactinib combination was designed to provide a more complete blockade of the signaling that drives the growth and drug resistance of RAS/MAPK pathway-dependent tumors.
The U.S. Food and Drug Administration (FDA) approved AVMAPKI™ FAKZYNJA™ CO-PACK (avutometinib capsules; defactinib tablets) for the treatment of adult patients with KRAS-mutated recurrent LGSOC who have received prior systemic therapy on May 8, 2025. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial. Verastem is conducting RAMP 301 (GOG-3097/ENGOT-ov81/GTG-UK) (NCT06072781), an international Phase 3 confirmatory trial evaluating the combination of avutometinib and defactinib versus standard chemotherapy or hormonal therapy for the treatment of recurrent low-grade serous ovarian cancer (LGSOC) with and without a KRAS mutation. Verastem is also evaluating avutometinib in combination with defactinib and other agents as a potential treatment for patients with advanced pancreatic cancer (RAMP 205; NCT05669482) and advanced KRAS G12C mutant non-small cell lung cancer (RAMP 203; NCT05074810). Avutometinib and defactinib are not approved by the FDA or any other regulatory authority, either in combination or with other therapies, for any of these investigative uses. Neither avutometinib nor defactinib are approved by the FDA or any other regulatory authority on a stand-alone basis for any use.
AVMAPKI FAKZYNJA CO-PACK U.S. Indication
Indication
AVMAPKI FAKZYNJA CO-PACK is indicated for the treatment of adult patients with KRAS-mutated recurrent low-grade serous ovarian cancer (LGSOC) who have received prior systemic therapy.
This indication is approved under accelerated approval based on tumor response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial.
Important Safety Information
Warnings and Precautions
| · | Ocular Toxicities: Ocular toxicities, including visual impairment and vitreoretinal disorders, occurred. Perform comprehensive ophthalmic evaluation at baseline, prior to cycle 2, every three cycles thereafter, and as clinically indicated. Withhold AVMAPKI FAKZYNJA CO-PACK for ocular toxicities until improvement at the same or reduced dose. Permanently discontinue AVMAPKI FAKZYNJA CO-PACK for any grade 4 toxicity. |
| · | Serious Skin Toxicities: Skin toxicities, including photosensitivity and severe cutaneous adverse reactions (SCARSs) occurred. Adhere to concomitant medications. Monitor for skin toxicities and interrupt, reduce or permanently discontinue AVMAPKI FAKZYNJA CO-PACK based on severity, tolerability and duration. |
| · | Hepatotoxicity: Monitor liver function tests prior to each cycle, on day 15 of the first 4 cycles, and as clinically indicated. Withhold, reduce or discontinue AVMAPKI FAKZYNJA CO-PACK based on severity and persistence of abnormality. |
| · | Rhabdomyolysis: Monitor creatine phosphokinase prior to the start of each cycle, on day 15 of the first four cycles, and as clinically indicated. If increased CPK occurs, evaluate patients for rhabdomyolysis or other causes. Withhold, reduce or permanently discontinue AVMAPKI FAKZYNJA CO-PACK based on severity and duration of the adverse reaction. |
| · | Embryo-Fetal Toxicity: AVMAPKI FAKZYNJA CO-PACK can cause fetal harm. Advise patients of the potential risk to a fetus and to use effective contraception. |
Adverse Reactions
The most common (≥ 25%) adverse reactions, including laboratory abnormalities, were increased creatine phosphokinase, nausea, fatigue, increased aspartate aminotransferase, rash, diarrhea, musculoskeletal pain, edema, decreased hemoglobin, increased alanine aminotransferase, vomiting, increased blood bilirubin, increased triglycerides, decreased lymphocyte count, abdominal pain, dyspepsia, dermatitis acneiform, vitreoretinal disorders, increased alkaline phosphatase, stomatitis, pruritus, visual impairment, decreased platelet count, constipation, dry skin, dyspnea, cough, urinary tract infection, and decreased neutrophil count.
Drug Interactions
| · | Strong and moderate CYP3A4 inhibitors: Avoid concomitant use with AVMAPKI FAKZYNJA CO-PACK. |
| · | Strong and moderate CYP3A4 inducers: Avoid concomitant use with AVMAPKI FAKZYNJA CO-PACK. |
| · | Warfarin: Avoid concomitant use of AVMAPKI FAKZYNJA CO-PACK with warfarin and use an alternative to warfarin. |
| · | Gastric acid reducing agents: Avoid concomitant use of AVMAPKI FAKZYNJA CO-PACK with proton pump inhibitors (PPIs) or H2 receptor antagonists. If use of an acid-reducing agent cannot be avoided, administer FAKZYNJA 2 hours before or 2 hours after the administration of a locally acting antacid. |
Use in Specific Populations
| · | Lactation: Advise not to breastfeed. |
| · | Fertility: May impair fertility in males and females. |
Click here for full Prescribing Information.
About VS-7375, an Oral KRAS G12D (ON/OFF) Inhibitor
VS-7375 is a potential best-in-class, potent, and selective oral KRAS G12D dual ON/OFF inhibitor. VS-7375 is the lead program from the Verastem Oncology discovery and development collaboration with GenFleet Therapeutics. Verastem initiated VS-7375-101, a Phase 1/2a clinical trial, in June of 2025 in the U.S., with the potential to expand globally, that is evaluating the safety and efficacy of VS-7375 in patients with advanced KRAS G12D mutant solid tumors. Verastem announced in April 2025 that the U.S. Investigational New Drug (IND) application for VS-7375 was cleared.
About the GenFleet Therapeutics Collaboration
The collaboration with GenFleet Therapeutics aims to advance three oncology discovery programs related to RAS/MAPK pathway-driven cancers. The collaboration provides Verastem with an exclusive option to obtain a license for each of the three compounds in the collaboration after the successful completion of pre-determined milestones in a Phase 1 trial. Verastem selected VS-7375 (also known as GFH375), an oral KRAS G12D (ON/OFF) inhibitor, as its lead program in December 2023 and the license for VS-7375 that was exercised in January 2025 is the first one from this collaboration. The licenses would give Verastem development and commercialization rights outside the GenFleet markets of mainland China, Hong Kong, Macau, and Taiwan.
About Verastem Oncology
Verastem Oncology (Nasdaq: VSTM) is a biopharmaceutical company committed to developing and commercializing new medicines to improve the lives of patients diagnosed with RAS/MAPK pathway-driven cancers. Verastem markets AVMAPKI™ FAKZYNJA™ CO-PACK in the U.S. Our pipeline is focused on novel small molecule drugs that inhibit critical signaling pathways in cancer that promote cancer cell survival and tumor growth, including RAF/MEK inhibition, FAK inhibition, and KRAS G12D inhibition. For more information, please visit www.verastem.com and follow us on LinkedIn. This press release includes forward-looking statements.
Forward-Looking Statements Notice
These forward-looking statements generally can be identified by the use of words such as “anticipate,” “expect,” “plan,” “could,” “may,” “believe,” “estimate,” “forecast,” “goal,” “project,” and other words of similar meaning. Such forward-looking statements address various matters about, among other things, Verastem Oncology’s programs and product candidates, strategy, future plans and prospects, including statements related to the potential for and timing of commercialization of product candidates, the conduct of the Phase 1/2a study for VS-7375/GFH375, the expected outcome and benefits of the Company’s collaboration with GenFleet Therapeutics (Shanghai), Inc., the timing of commencing and completing trials and compiling data, the expected timing of the presentation of data by the Company and the potential clinical value of various of the Company’s clinical trials. Each forward-looking statement contained in this press release is subject to risks and uncertainties that could cause actual results to differ materially from those expressed or implied by such statements. Applicable risks and uncertainties include, among others: the uncertainties inherent in research and development, such as the possibility of negative or unexpected results of clinical trials; that we may not see a return on investment on the payments we have and may continue to make pursuant to the collaboration and option agreement with GenFleet, or that GenFleet may fail to fully perform under the agreement; that we may not be successful in our launch or commercialization of AVMAPKI FAKZYNJA CO-PACK; that the development and commercialization of our product candidates may take longer or cost more than planned, including as a result of conducting additional studies or our decisions regarding execution of such commercialization; that data may not be available when expected; risks associated with preliminary and interim data, which may not be representative of more mature data; risks associated with the recent changes in administration policy or actions that may create regulatory uncertainty that may adversely affect our business; risks associated with the current administration’s reductions to the FDA’s workforce and any subsequent reductions that may lead to disruptions and delays in the FDA’s review and oversight of our product candidates and impact the FDA’s ability to provide timely feedback on our development programs; that our product candidates may not receive regulatory approval, become commercially successful products, or result in new treatment options being offered to patients; and the risks identified under the heading "Risk Factors" as detailed in the Company’s Annual Report on Form 10-K for the year ended December 31, 2024, as filed with the Securities and Exchange Commission (SEC) on March 20, 2025, as well as the other information we file with the SEC, are possibly realized. We caution investors not to place considerable reliance on the forward-looking statements contained in this press release. You are encouraged to read our filings with the SEC, available at www.sec.gov, for a discussion of these and other risks and uncertainties. The forward-looking statements in this press release speak only as of the date of this press release, and we undertake no obligation to update or revise any of these statements. Our business is subject to substantial risks and uncertainties, including those referenced above. Investors, potential investors, and others should give careful consideration to these risks and uncertainties.
For Investor and Media Inquiries:
Julissa Viana
Vice President, Corporate Communications,
Investor Relations & Patient Advocacy (in thousands, except per share amounts)
investors@verastem.com or
media@verastem.com
Verastem Oncology
Condensed Consolidated Statements of Operations
(unaudited)
| Three months ended September 30, |
Nine months ended September 30, |
|||||||||||||||
| 2025 | 2024 | 2025 | 2024 | |||||||||||||
| Revenue: | ||||||||||||||||
| Product revenue, net | $ | 11,242 | $ | — | $ | 13,379 | $ | — | ||||||||
| Sale of COPIKTRA license and related assets | — | — | — | 10,000 | ||||||||||||
| Total revenue | 11,242 | — | 13,379 | 10,000 | ||||||||||||
| Operating expenses: | ||||||||||||||||
| Cost of sales - product | 1,670 | — | 1,988 | — | ||||||||||||
| Cost of sales - intangible amortization | 290 | — | 418 | — | ||||||||||||
| Research and development | 28,989 | 24,754 | 82,925 | 60,523 | ||||||||||||
| Selling, general and administrative | 21,008 | 12,276 | 56,702 | 32,843 | ||||||||||||
| Total operating expenses | 51,957 | 37,030 | 142,033 | 93,366 | ||||||||||||
| Loss from operations | (40,715 | ) | (37,030 | ) | (128,654 | ) | (83,366 | ) | ||||||||
| Other expense | (37 | ) | (77 | ) | (186 | ) | (131 | ) | ||||||||
| Interest income | 1,182 | 831 | 2,964 | 3,181 | ||||||||||||
| Interest expense | (319 | ) | (1,148 | ) | (723 | ) | (3,416 | ) | ||||||||
| Loss on debt extinguishment | — | — | (1,826 | ) | — | |||||||||||
| Change in fair value of preferred stock tranche liability | — | — | — | 4,189 | ||||||||||||
| Change in fair value of warrant liability | (55,881 | ) | 13,457 | (37,977 | ) | 13,457 | ||||||||||
| Change in fair value of Notes | (2,748 | ) | — | (10,153 | ) | — | ||||||||||
| Net loss | $ | (98,518 | ) | $ | (23,967 | ) | $ | (176,555 | ) | $ | (66,086 | ) | ||||
| Net loss per share—basic and diluted | $ | (1.35 | ) | $ | (0.60 | ) | $ | (2.73 | ) | $ | (2.11 | ) | ||||
| Weighted average common shares outstanding used in computing net loss per share—basic and diluted | 73,157 | 40,258 | 64,561 | 31,350 | ||||||||||||
Verastem Oncology
Condensed Consolidated Balance Sheets
(in thousands)
(unaudited)
| September 30, 2025 | December 31, 2024 | |||||||
| Cash & cash equivalents | $ | 137,706 | $ | 88,818 | ||||
| Accounts receivable, net | 6,716 | — | ||||||
| Inventory | 1,794 | — | ||||||
| Grant receivable | 200 | 200 | ||||||
| Prepaid expenses and other current assets | 7,640 | 5,943 | ||||||
| Property and equipment, net | 20 | 32 | ||||||
| Right-of-use asset, net | 730 | 1,405 | ||||||
| Intangible assets, net | 16,705 | — | ||||||
| Other assets | 5,341 | 5,140 | ||||||
| Total assets | $ | 176,852 | $ | 101,538 | ||||
| Current Liabilities | $ | 59,712 | $ | 30,973 | ||||
| Long term debt | 78,124 | 40,724 | ||||||
| Vendor financing arrangement, long-term | 6,250 | — | ||||||
| Lease liability, long-term | — | 535 | ||||||
| Accrued expenses, long-term | — | — | ||||||
| Warrant liability | 48,292 | 58,199 | ||||||
| Stockholders’ (deficit) | (15,526 | ) | (28,893 | ) | ||||
| Total liabilities, and stockholders’ (deficit) | $ | 176,852 | $ | 101,538 | ||||
Verastem, Inc.
Reconciliation of GAAP to Non-GAAP Financial Information
(in thousands, except per share amounts)
(unaudited)
| Three months ended September 30, |
Nine months ended September 30, |
|||||||||||||||
| 2025 | 2024 | 2025 | 2024 | |||||||||||||
| Net loss reconciliation | ||||||||||||||||
| Net loss (GAAP basis) | $ | (98,518 | ) | $ | (23,967 | ) | $ | (176,555 | ) | $ | (66,086 | ) | ||||
| Adjust: | ||||||||||||||||
| Stock-based compensation expense | 2,178 | 1,935 | 7,379 | 5,323 | ||||||||||||
| Non-cash interest, net | — | 201 | (62 | ) | (212 | ) | ||||||||||
| Change in fair value of preferred stock tranche liability | — | — | — | (4,189 | ) | |||||||||||
| Change in fair value of warrant liability | 55,881 | (13,457 | ) | 37,977 | (13,457 | ) | ||||||||||
| Non-cash change in fair value of Notes | 1,105 | — | 5,670 | — | ||||||||||||
| Loss on debt extinguishment | — | — | 1,826 | — | ||||||||||||
| Severance and Other | — | 10 | — | 619 | ||||||||||||
| Adjusted net loss (non-GAAP basis) | $ | (39,354 | ) | $ | (35,278 | ) | $ | (123,765 | ) | $ | (78,002 | ) | ||||
| Reconciliation of net loss per share | ||||||||||||||||
| Net loss per share – diluted (GAAP basis) | $ | (1.35 | ) | $ | (0.60 | ) | $ | (2.73 | ) | $ | (2.11 | ) | ||||
| Adjust per basic share | ||||||||||||||||
| Stock-based compensation expense | 0.03 | 0.05 | 0.11 | 0.17 | ||||||||||||
| Non-cash interest, net | — | — | — | (0.01 | ) | |||||||||||
| Change in fair value of preferred stock tranche liability | — | — | — | (0.13 | ) | |||||||||||
| Change in fair value of warrant liability | 0.76 | (0.33 | ) | 0.59 | (0.43 | ) | ||||||||||
| Non-cash change in fair value of Notes | 0.02 | — | 0.08 | — | ||||||||||||
| Loss on debt extinguishment | — | — | 0.03 | — | ||||||||||||
| Severance and Other | — | — | — | 0.02 | ||||||||||||
| Adjusted net loss per share – diluted (non-GAAP basis) | $ | (0.54 | ) | $ | (0.88 | ) | $ | (1.92 | ) | $ | (2.49 | ) | ||||
| Weighted average common shares outstanding used in computing net loss per share—diluted | 73,157 | 40,258 | 64,561 | 31,350 | ||||||||||||
Exhibit 99.2


Delivering Novel Therapies for RAS/MAPK Pathway Driven Cancers Corporate Presentation | November 2025 2 Forward - Looking Statements This presentation includes forward - looking statements about, among other things, Verastem Oncology’s (the “Company”) programs an d product candidates, strategy, future plans and prospects, including statements related to the approval and commercialization of AVMAPKIFAKZYNJACO - PACK ( avutometinib capsules; defactinib tablets) as a treatment for adult patients with Kirsten rat sarcoma viral oncogene homolog (KRAS) mutant - type (mt) recurrent Lo w - Grade Serous Ovarian Cancer(LGSOC),the expected outcome and benefits of collaborations, including with GenFleet Therapeutics (Shanghai), Inc. ( GenFleet ), including the conduct of a Phase 1/2a study with respect to VS - 7375, the status of enrollments for and potential of the resul ts of the RAMP 301 Phase 3 trial to confirm the results of the RAMP 201 study specific to KRAS mutant patients and to expand the indication rega rdl ess of KRAS mutation status, the structure and potential clinical value of our completed, planned and pending clinical trials , t he potential clinical value of various of the Company's clinical trials, including the RAMP 201, RAMP 201J, RAMP 203, RAMP 205, RAM P 301 and VS - 7375 trials, the timing of commencing and completing trials, including topline data reports, our interactions with regulators, the timeline and indications for clinical development, regulatory submissions and the potential for and timing of co mmercialization of our product candidates and potential for additional development programs involving the Company’s lead compound and the potential market opportunities thereof; the expected outcome and benefits of our collaboration with GenFleet Therapeutics (Shanghai), Inc. (“ GenFleet ”) and the estimated addressable markets for, and anticipated market opportunities of our drug candidates.. The words "anticipate," "believe," "estimate," "expect," "may," "plan," "target," "potential," "would," "c ould," "should," "continue," “can” and similar expressions are intended to identify forward - looking statements, although not all forward - looking statements contain these identifying words. Each forward - looking statement is subject to risks and uncertainties that co uld cause actual results to differ materially from those expressed or implied in such statement. Applicable risks and uncertainties include the risks and uncertainties, among other things, regarding: the success in the dev elo pment and potential commercialization of our product candidates, including avutometinib in combination with other compounds, including defactinib , LUMAKRAS, VS - 7375 and others; the uncertainties inherent in research and development, such as negative or unexpected results o f clinical trials; the occurrence or timing of applications for our product candidates that may be filed with regulatory authorities in any jurisdictions; whether and when regulatory authorities in any jurisdictions may approve any suc h a pplications that may be filed for our product candidates, and, if approved, whether our product candidates will be commercial ly successful in such jurisdictions; actions or advice of regulatory agencies to maintain regulatory approval of AVMAPKI FAKZYNJ A C O - PACK; our ability to obtain, maintain and enforce patent and other intellectual property protection for our product candidate s ; the scope, timing, and outcome of any legal proceedings; decisions by regulatory authorities regarding trial design, labeling an d other matters that could affect the timing, availability or commercial potential of our product candidates; whether preclin ica l testing of our product candidates and preliminary or interim data from clinical trials will be predictive of the results or s ucc ess of ongoing or later clinical trials; that the timing, scope and rate of reimbursement for our product candidates is uncer tai n; that the market opportunities of our drug candidates are based on internal and third - party estimates which may prove to be incorrect; tha t third - party payors (including government agencies) may not reimburse; that there may be competitive developments affecting our product candidates; that data may not be available when expected; that enrollment of clinical trials may take longer than ex pected; the risks that we will not satisfy our post - marketing requirements and commitments established and agreed to as part of the FDA's approval of AVMAPKI FAKZYNJA CO - PACK; the risks associated with preliminary and interim data, which may not be representat ive of more mature data, including with respect to interim duration of therapy data; that our marketed product candidates may cause adverse safety events and/or unexpected concerns may arise from additional data or analysis, or result in unmanagea ble safety profiles as compared to their levels of efficacy; that we may be unable to successfully validate, develop and obtain regulatory approval for companion diagnostic tests for our product candidates that require or would commercially benefit from su ch tests, or experience significant delays in doing so; that we may not be able to confirm the results from the RAMP 201 stud y o r expand the approved indication for AVMAPKI FAKZYNJA CO - PACK; that our product candidates may experience manufacturing or supply interruptions or failures; that any of our third - party contract research organizations, contract manufacturing organizations, clinical sites, or contractors, among others, who we rely on may fail to fully perform; that we face substantial competition, wh ich may result in others developing or commercializing products before or more successfully than we do which could result in red uced market share or market potential for our product candidates; that we may be unable to successfully initiate or complete the c lin ical development and eventual commercialization of our product candidates; that the development and commercialization of our product candidates may take longer or cost more than planned, including as a result of conducting additional studies or our d eci sions regarding execution of such commercialization; that we may not have sufficient cash to fund our contemplated operations , including certain of our product development programs; that we may not attract and retain high quality personnel; that we or Pfi zer, Inc. may fail to fully perform under the license agreement covering certain Pfizer FAK inhibitors, including defactinib ; that we or Chugai Pharmaceutical Co., Ltd. may fail to fully perform under the avutometinib license agreement; that our total addressable and target markets for our product candidates might be smaller than we are pres en tly estimating; that we or Secura Bio, Inc. may fail to fully perform under the asset purchase agreement with Secura Bio, Inc., including in relation to milestone payments; that we will not see a return on investment on the payments we have and may continue to make pursuant to the collaboration and option agreement with GenFleet , or that GenFleet may fail to fully perform under the agreement; that we may not be able to establish new or expand on existing collaborations or partnerships, including with respect to in - licensing of our product candidates, on favorable terms, or at all; that we may be unable to obtain adequate financing in the future through product licensing, co - promo tional arrangements, public or private equity, debt financing or otherwise; that we may not pursue or submit regulatory filin gs for our product candidates; that, due to the recent change in presidential administration and the significant reduction in the FD A's workforce and potential reductions to the FDA's budget, we may experience a materially impact to the FDA's ability to engage in a variety of activities that may affect our business, including routine regulatory and oversight activities; and that our produ ct candidates may not receive regulatory approval, become commercially successful products, or result in new treatment options b ein g offered to patients. Other risks and uncertainties include those identified under the heading “Risk Factors” in the Company’s Annual Report on For m 1 0 - K for the year ended December 31, 2024, as filed with the Securities and Exchange Commission (SEC) on March 20, 2025, and in any subsequent filings with the SEC, which are available at www.sec.govand www.verastem.com.The forward - looking statements in this presentation speak only as of the original date of this presentation, and we undertake no obligation to update or revise any of these statements whether as a result of new information, future events or otherwise, except as required by law. Our bu sin ess is subject to substantial risks and uncertainties, including those referenced above. Investors, potential investors, and oth ers should give careful consideration to these risks and uncertainties. Use of Non - GAAP Financial Measures This presentation contains references to our non - GAAP operating expense, a financial measure that is not calculated in accordanc e with generally accepted accounting principles in the US (GAAP). This non - GAAP financial measure excludes certain amounts or expenses from the corresponding financial measures determined in accordance with GAAP. Management believes this non - GAAP informa tion is useful for investors, taken in conjunction with the Company’s GAAP financial statements, because it provides greater transparency and period - over - period comparability with respect to the Company’s operating performance and can enhance in vestors’ ability to identify operating trends in the Company’s business. Management uses this measure, among other factors, to assess and analyze operational results and trends and to make financial and operational decisions. Non - GAAP information is no t prepared under a comprehensive set of accounting rules and should only be used to supplement an understanding of the Company’s operating results as reported under GAAP, not in isolation or as a substitute for, or superior to, financial inform ati on prepared and presented in accordance with GAAP. In addition, this non - GAAP financial measure is unlikely to be comparable wit h non - GAAP information provided by other companies. The determination of the amounts that are excluded from non - GAAP financial mea sures is a matter of management judgment and depends upon, among other factors, the nature of the underlying expense or income amounts. Reconciliations between this non - GAAP financial measure and the most comparable GAAP financial measure are in cluded in the footnotes to the slides in this presentation on which such non - GAAP number appears. Third - Party Sources Certain information contained in this presentation, including industry and market data and other statistical information, rel ate s to or is based on studies, publications, surveys and other data obtained from third - party sources and the Company’s own intern al estimates and research. While the Company believes these third - party sources to be reliable as of the date of this presentation, it has not independently verified, and makes no representation as to the adequacy, fairness, accuracy or completeness of, any information obtained from third - party sources.

In addition, all of the market data included in this presentation involves a numb er of assumptions and limitations, and there can be no guarantee as to the accuracy or reliability of such assumptions Disclaimers 3 3 Commercial Product and Pipeline Positioned to Deliver Long - Term Shareholder Value • Strong commercial launch execution and broad HCP adoption • AVMAPKI FAKZYNJA CO - PACK is the first - ever FDA approved therapy specifically for KRAS - mutated recurrent low - grade serous ovarian cancer (LGSOC), with a Category 2A listing in the NCCN guidelines • Consistent adoption by academic and community oncologists drove robust first full quarter of net product revenue • Advancing potential best - in - class oral KRAS G12D (ON/OFF) inhibitor for pancreatic, lung, colorectal, and other KRAS G12D mutant solid tumor cancers • VS - 7375 - 101: Encouraging preliminary data from the first two dose levels in U.S. trial with no DLTs reported, early signs of anti - tumor activity, and no nausea, vomiting or diarrhea greater than Grade 1 observed • Enrollment initiated for VS - 7375 in combination with cetuximab in advanced solid tumors, including colorectal cancer • Maximizing synergistic potential of avutometinib plus defactinib combination in other advanced solid tumors • RAMP 205: completed enrollment in DL1 expansion cohort of 29 patients in 1L metastatic pancreatic cancer; DL1 achieved confirmed ORR of 83% (10/12) • RAMP 203: advanced KRAS G12C mutant non - small cell lung cancer; no DLTs observed in the triplet combination cohort • Existing cash, investments, and anticipated future product sales give us a strong financial position and runway expected into the second half of 2026 KR AS: Kirsten Rat Sarcoma Virus; FDA: Food and Drug Administration; NCCN: National Comprehensive Cancer Network; DL: dose - level: DLT: dose - limiting toxicity; ORR: Overall Response Rate 4 Strong First Full Quarter Launch of AVMAPKI FAKZYNJA CO - PACK Please see the full Prescribing Information for more information PDUFA: Prescription Drug User Fee Action $11.2 million Of net product revenue in Q3 2025 FDA Approval Nearly Two Months Ahead of PDUFA Date Consistent Adoption Among Academic & Community Oncologists Patient Initiation & Retention Trending Positively Minimal Reimbursement Challenges


5 Anticipated Milestones FDA Approved Phase 3 Phase 2 Phase 1 Regimen Trial Name/ Therapeutic Area Asset RAF/MEK Clamp + FAKi RAMP 201, recurrent LGSOC Avutometinib + Defactinib Expect to complete enrollment of IDMC - recommended increase by Q1 2026 RAF/MEK Clamp + FAKi vs ICT RAMP 301, recurrent LGSOC Avutometinib + Defactinib Plan to report an interim safety and efficacy update in the 1H 2026 KRAS G12D (ON/OFF) inhibitor VS - 7375 - 101, Advanced solid tumors VS - 7375* Expect to report an update on the safety and efficacy of the expansion cohort in the 1H 2026 RAF/MEK Clamp + FAKi + gemcitabine, nab - paclitaxel PanCAN Collaboration RAMP 205, 1L mPDAC Avutometinib + Defactinib Report an interim safety and efficacy update in Q4 2025 RAF/MEK Clamp ± FAKi + KRAS G12Ci (sotorasib) Amgen Collaboration RAMP 203, advanced KRAS G12C NSCLC Avutometinib “ Defactinib RAF: Rapidly Accelerated Fibrosarcoma; MEK, Mitogen - Activated Extracellular Signal - regulated Kinase; RAS: Rat Sarcoma Virus; FAKi : focal adhesion kinase inhibitor; ICT: investigator choice of treatment; NSCLC: non - small cell lung cancer; mPDAC : metastatic Pancreatic Ductal Adenocarcinoma Our Pipeline: Addressing RAS/MAPK - Driven Cancers Not shown: *GenFleet Therapeutics has an ongoing Phase 1/2 clinical trial in China with VS - 7375, known as GFH375 in China. GenFleet retains greater China rights. Verastem has two undisclosed assets at discovery phase targeting the RAS/MAPK - pathway as part of the GenFleet collaboration.

6 Continued Progress Across All Key Milestones RAMP 301 Completed planned enrollment in the Phase 3 confirmatory study. Reported IDMC recommendation in Q4 2025. RAMP 201J Reported initial data from Phase 2 clinical trial being conducted in Japan in Q4 2025. RAMP 203 Report an interim data update on both doublet and triplet combinations in Q4 2025. RAMP 205 Completed enrollment in the expansion cohort in Q3 2025. PRODUCT LAUNCH • Effectively reach HCPs • Engage with patients • Ensure seamless access VS - 7375 - 101 Reported preliminary update on Phase 1/2a monotherapy dose escalation in Q4 2025. Initiated dose escalation combination cohort with cetuximab in Q4 2025.

IDMC: Independent Data Monitoring Committee; HCPs: health care professionals 7 A Look Ahead RAMP 301 Expect to complete modest IDMC recommended patient enrollment increase in Q1 2026 RAMP 205 Expect to report an update on the safety and efficacy of the expansion cohort in 1H 2026 Continued strong execution of product launch VS - 7375 - 101 Monotherapy Plan to report an interim update in 1H 2026 Expect to select the RP2D and initiate monotherapy expansion cohorts in both advanced PDAC, NSCLC and other solid tumors in 1H 2026 VS - 7375 - 101 Combinations VS - 7375 Regulatory Plan to engage with the FDA to discuss our development path forward , including potential registration - directed trials in PDAC and NSCLC in 1H 2026 Plan to initiate combo cohorts with chemotherapy in PDAC and chemo + anti - PD - 1 in NSCLC in Q4 2025 Expect to select the RP2D and plan to initiate combination cohorts in CRC, PDAC and NSCLC in 1H 2026 8 Commercially launched in the U.S. for KRAS - mutated recurrent LGSOC FDA Approval Date: May 8, 2025

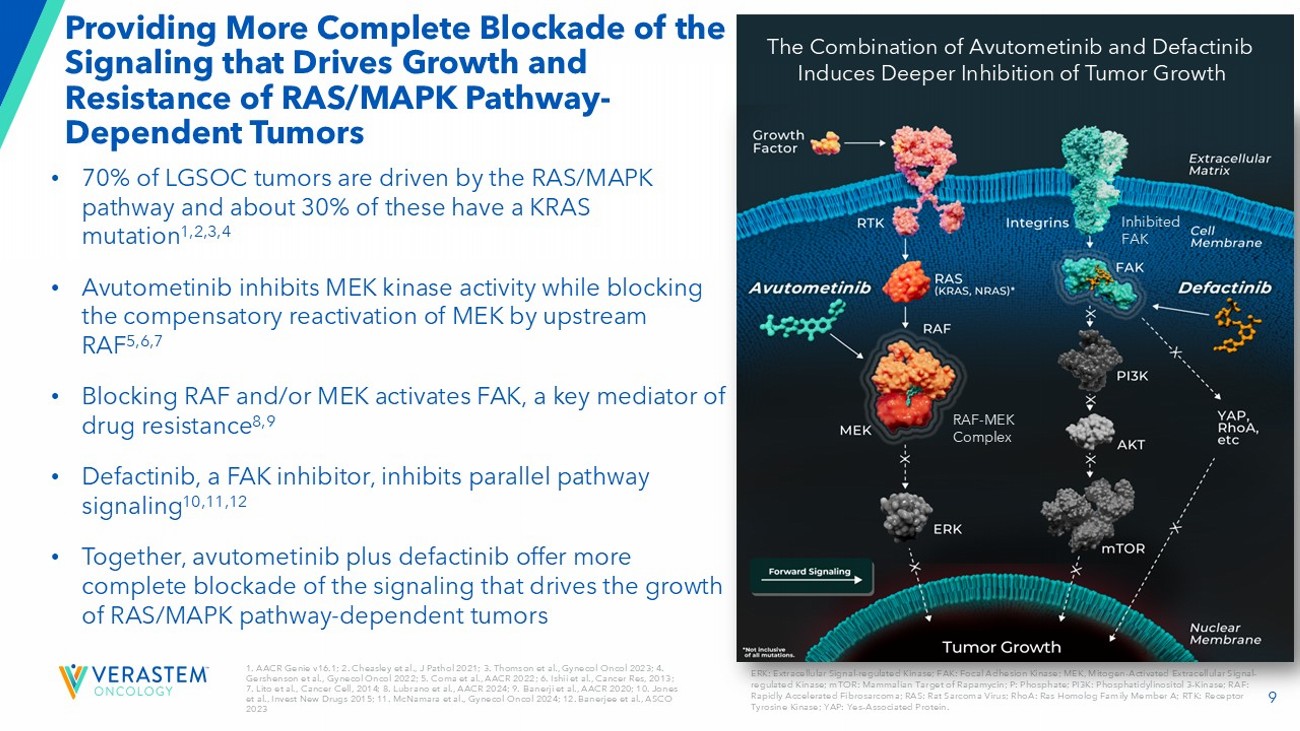
9 1. AACR Genie v16.1; 2. Cheasley et al., J Pathol 2021; 3. Thomson et al., Gynecol Oncol 2023; 4. Gershenson et al., Gynecol Oncol 2022; 5. Coma et al., AACR 2022 ; 6. Ishii et al., Cancer Res, 2013 ; 7. Lito et al., Cancer Cell, 2014 ; 8. Lubrano et al., AACR 2024 ; 9. Banerji et al., AACR 2020 ; 10. Jones et al., Invest New Drugs 2015; 11. McNamara et al., Gynecol Oncol 2024 ; 12. Banerjee et al., ASCO 2023 Providing More Complete Blockade of the Signaling that Drives Growth and Resistance of RAS/MAPK Pathway - Dependent Tumors ERK: Extracellular Signal - regulated Kinase; FAK: Focal Adhesion Kinase; MEK, Mitogen - Activated Extracellular Signal - regulated Kinase; mTOR: Mammalian Target of Rapamycin; P: Phosphate; PI3K: Phosphatidylinositol 3 - Kinase; RAF: Rapidly Accelerated Fibrosarcoma; RAS: Rat Sarcoma Virus; RhoA: Ras Homolog Family Member A; RTK: Receptor Tyrosine Kinase; YAP: Yes - Associated Protein.

• 70% of LGSOC tumors are driven by the RAS/MAPK pathway and about 30% of these have a KRAS mutation 1,2,3,4 • Avutometinib inhibits MEK kinase activity while blocking the compensatory reactivation of MEK by upstream RAF 5,6,7 • Blocking RAF and/or MEK activates FAK, a key mediator of drug resistance 8,9 • Defactinib, a FAK inhibitor, inhibits parallel pathway signaling 10,11,12 • Together, avutometinib plus defactinib offer more complete blockade of the signaling that drives the growth of RAS/MAPK pathway - dependent tumors RAF - MEK Complex Inhibited FAK The Combination of Avutometinib and Defactinib Induces Deeper Inhibition of Tumor Growth 10 High Unmet Need for an Effective and Tolerable Therapy in Recurrent LGSOC • U.S. incidence / prevalence : 1k - 2k 1 / 6k - 8k 2 • LGSOC affects younger women with bimodal peaks of diagnosis at ages between 20 - 30 and 50 - 60; disproportionately impacts health, fertility, and long - term quality of life 3,4 • 80 - 90% of patients will experience a recurrence 5 • Current standard of care offers low to moderate response rates (6 - 13%) 6,7,8 • Median overall survival (OS) of ~10 years from time of diagnosis 9: KRAS - mutated - ~12 years 10 and KRAS wild - type: ~7 years 10 1. Verastem DOF; 2. US Cancer Statistics. Accessed 2024 ; 3 . Slomovitz Gynecol Oncol 2020; 4. Manning - Geist B et al. Clin Cancer Res 2022;28(20):4456 - 4465; 5. Babaier 2022/p1/para1/ln6,7; 6. Gershenson Gynecol Oncol 2022; 7. Slomovitz Gynecol Oncol 2020; 8. Monk 2020/p3758/table2/footnote - b; 9 Banerjee SN). J Clin Oncol. 41. No 16_suppl (June 1, 2023) 5515 - 5515; 10. Manning - Geist B et al. Clin Cancer Res 2022;28(20):4456 - 4465; Calculated using figures in Gershenson Gynecol Oncol 2022. When you get told that you have a recurrence, the mental load is a lot . You’re thinking, okay, what did I have to do for treatment the first time? Now I have to repeat that .

And will there even be something available for me to take for a second, or a third recurrence? 11 Highly Targeted Approach Aimed to Drive a Successful Commercial Launch Effectively reach healthcare providers Top 100 commercial healthcare organizations contribute ~50% of patient claims 1 Engage and support patients Patients likely have progressed through other therapies, and many will be ready for a new treatment Ensure seamless access Support the patient to ensure any barriers to reimbursement are removed Surround Sound Support Programs HCPs PATIENTS ACCESS 1 VSTM DOF – Claims L G SOC Proxy 12 Continued Launch Momentum of AVMAPKI FAKZYNJA CO - PACK in Q325 Please see the full Prescribing Information for more information Source: VSTM DOF $11.2 million Of net product revenue in Q3 2025 133 Prescribers 65% Rx generated by top 100 organizations 60% Rx coming from GynOncs ; 40% from MedOncs 4 Specialty distributors now fully on board


13 Breadth and Reach Continue to Drive Impressive Results x Prescriptions coming from a mix of academic and community physicians x Seeing both repeat prescriptions and refills x Payer mix is a combination of commercial and Medicare Effectively reach healthcare providers Engage and support patients Ensure seamless access • High engagement among top 100 organizations and 100 offices, includes a mix of academic and community providers • 65% of Rx coming from tier 1 and 2 accounts • Repeat prescriptions from physicians prescribing to multiple patients and refills for patients • 800+ scientific exchanges and 100+ educational forums by medical team • Branded patient website is seeing high traffic with significant patient brochure downloads • Patients continue to opt - in to receive more details associated with product • Payer coverage has been broad and time to payment fast (~12 - 14 days) • Covered lives has now exceeded 80%; half commercial and half Medicare Source: VSTM DOF 14 NCCN Submission for Treatment Guideline Inclusion Under Review for the Entire Population Enrolled in RAMP 201 Study NCCN Category 3 NCCN Category 2b NCCN Category 2a NCCN Category 1 General % Commercial Payer Coverage Binimetinib • Study stopped early due to futility • 16% ORR byBICR • 31% discontinuation rate due to AEs • Supported by MILO study 3 Hormonal therapy (e.g., Anastrozole, Letrozole) & chemotherapy • 6 - 13% ORR 4 • 17 - 30% discontinuation rate due to AEs 4 Trametinib (2 - 4% US utilization rate 1 ) • 26% ORR by INV assessment, no BICR 5 • 36% discontinuation rate due to AEs 5 No category 1 recommendation Examples of Clinical Data in LGSOC and Current NCCN Guideline Category General source: NCCN; McGivney Global Advisory research and analysis; L.E.K.

research and analysis. NCCN categories of prefer enc e: Preferred intervention, Other recommended intervention, Useful in certain circumstances. High - level of evidence generally means large randomized controlled Phased 3 trials; Pie charts represent coverage by all major c ommercial players; 1. Data on File; 2. GOG 281 trial Gershenson et al., Lancet 2022; 3. MILO Study Monk et al., J Clin Oncol 2020; 4. Supported by GOG 281 and MILO studies 2,3 ; 5. Supported by GOG 281 2 Current Listing: Avutometinib + Defactinib Combination Therapy • KRAS mt recurrent LGSOC 15 Continuing Medical Progress in LGSOC


16 Across Patients With and Without KRAS Mutations in RAMP 201, 82% had a Reduction in Target Lesions while Receiving Avutometinib and Defactinib Source for all data: RAMP 201 data cut off as of June 30, 2024; Responses for 3 patients (KRAS wild type, n=1; KRAS mutant, n =2) were unknown Best Overall Response 17 *US FDA analysis plan will evaluate PFS independently in KRAS - mt and KRAS wt LGSOC.

BICR: blinded independent central review; BID: twice a day; BIW: twice a week; DCR: disease control rate; DoR : duration of response; INV: investigator; KRAS: kirsten rat sarcoma virus; MEKi : MEK inhibitor; mt: mutant; PO: per oral; pts, patients; ORR: objective response rate; OS: overall survival; PD: progressive di sease; PFS: progression - free survival; PROs: patient - reported outcomes; RECIST: response evaluation criteria in solid tumors; wt : wild type. RAMP 301: International Phase 3 Confirmatory Trial of Avutometinib + Defactinib in Recurrent LGSOC • Entry criteria similar to RAMP 201 patient population, KRAS mt and KRAS wt recurrent LGSOC; prior MEKi and bevacizumab use allowed and post at least one line of platinum chemotherapy • Study sites include the U.S., Canada, UK, Europe, Australia, New Zealand, Japan and South Korea Inclusion Criteria • Recurrent disease after prior platinum therapy • Documented KRAS mutation status • Measurable disease per RECIST v1.1 • Confirmed LGSOC diagnosis • Prior MEKi allowed • Prior bevacizumab allowed RAMP 301 (GOG - 3907/ENGOT - ov81/NCRI): Ongoing Randomized Controlled Trial (RCT) NCT06072781 Pegylated Liposomal Doxorubicin Paclitaxel Letrozole Anastrozole Investigator’s Choice n = 135 Avutometinib 3.2 mg PO BIW Defactinib 200 mg BID 3 weeks on, 1 week off Avutometinib + Defactinib n = 135 May cross over upon BICR - confirmed PD PFS (BICR by RECIST v1.1) Hierarchical Evaluation of PFS*: KRAS mutant LGSOC All recurrent LGSOC KRAS wt LGSOC Primary Endpoint: OS PFS by RECIST v1.1 per INV assessment ORR DoR DCR Safety Pharmacokinetics PROs Secondary Endpoints a a Unless otherwise specified, all tumor response - based endpoints will be analyzed using both BICR and INV assessments 1:1 Randomization n = 270 Stratification Factors: • KRAS mutation status ( wt vs. mt) • Geography (N. America/EU) vs. ROW • Number of prior therapies (1 - 3 vs. 4 or more)

18 Next Steps in LGSOC Clinical Program Continue to advance regulatory pathway in Japan and Europe Expect to complete modest IDMC recommended patient enrollment increase in Q1 2026 VS - 7375, oral KRAS G12D (ON/OFF) Inhibitor 19



20 Adapted from Hofmann et al., Cancer Discovery 2022 • The only approved KRAS inhibitors target KRAS G12C, which is largely restricted to NSCLC • KRAS G12D accounts for 26% of all KRAS mutations • KRAS G12D mutations are especially prevalent in pancreatic and colorectal cancers • Targeting KRAS G12D has historically been challenging due to the shallow pocket for drug interaction and lack of a cysteine for covalent binding KRAS G12D is the Most Frequent KRAS Mutation in Human Cancers 21 VS - 7375 is a Potent KRAS G12D Dual ON/OFF Inhibitor GEF GTP GDP GTP GDP VS - 7375 RAS/MAPK pathway signaling GEF: Guanine nucleotide exchange factor GAP: GTPase - activating protein GAP P KRAS G12D OFF (inactive) KRAS G12D ON (active) Assay VS - 7375 IC 50 ( nM ) KRAS G12D State RAF1 binding 2 1 GppNHp - bound (ON/active) Nucleotide exchange 6 1 GDP - bound (OFF/inactive) Assay VS - 7375 K D ( pM ) KRAS G12D State SPR affinity 18 GppNHp - bound (ON/active) SPR affinity 12 GDP - bound (OFF/inactive) Binding Assays* Functional Assays *Residence time for VS - 7375 is 18 - 24 hours compared to 1 hour for AZD0022 (G12Di) or AMG410 (pan - KRASi ) Pachter, Targeting RAS Salamanca 2025.

22 VS - 7375 Potently and Selectively Inhibits 3D Proliferation of KRAS G12D Mutant Cell Lines VS - 7375 IC50 (nM) KRAS status Cell Line 8 G12D SKLU1 2 G12D KP4 2 G12D GP2D 7 G12D HPAC 5 G12D HPAF - II 7 G12D AsPC1 8 G12D Panc08.13 7 G12D LS513 33 G12D LS180 63 G12D Panc04.03 133 G12C MiaPaca2 1471 G12C H358 3069 G12C H1373 >5000 G12C H2122 828 G12V H441 >5000 G12S A549 756 G13D HCT116 >5000 wild - type H1299 3287 wild - type GAK >5000 wild - type SKMEL2 >5000 wild - type PC9 >5000 wild - type H1975 >5000 wild - type A375 >5000 wild - type HT29 0.001 0.01 0.1 1 10 0 20 40 60 80 100 VS-7375 (µM) C e l l P r o l i f e r a t i o n ( % c o n t r o l ) LS180 (KRAS G12D) Panc04.03 (KRAS G12D) SKLU1 (KRAS G12D) AsPC1 (KRAS G12D) HPAF-II (KRAS G12D) KP4 (KRAS G12D) A549 (KRAS G12S) MiaPaca2 (KRAS G12C) H1975 (KRAS wt) A375 (KRAS wt) GAK (KRAS wt) H441 (KRAS G12V) VS-7375 HT29 (KRAS wt) H1373 (KRAS G12C) PC9 (KRAS wt) H358 (KRAS G12C) GP2D (KRAS G12D) Panc08.13 (KRAS G12D) H2122 (KRAS G12C) SKMEL2 (KRAS wt) KRAS G12D-mutant KRAS non-G12D-mutant KRAS wild-type HPAC (KRAS G12D) HCT116 (KRAS G13D) HT1299 (KRAS wt) LS513 (KRAS G12D) 0.001 0.01 0.1 1 10 0 20 40 60 80 100 VS-7375 (µM) C e l l P r o l i f e r a t i o n ( % c o n t r o l ) LS180 (KRAS G12D) Panc04.03 (KRAS G12D) SKLU1 (KRAS G12D) AsPC1 (KRAS G12D) HPAF-II (KRAS G12D) KP4 (KRAS G12D) A549 (KRAS G12S) MiaPaca2 (KRAS G12C) H1975 (KRAS wt) A375 (KRAS wt) GAK (KRAS wt) H441 (KRAS G12V) VS-7375 HT29 (KRAS wt) H1373 (KRAS G12C) PC9 (KRAS wt) H358 (KRAS G12C) GP2D (KRAS G12D) Panc08.13 (KRAS G12D) H2122 (KRAS G12C) SKMEL2 (KRAS wt) KRAS G12D-mutant KRAS non-G12D-mutant KRAS wild-type HPAC (KRAS G12D) HCT116 (KRAS G13D) HT1299 (KRAS wt) LS513 (KRAS G12D) Targeting RAS Salamanca 2025.

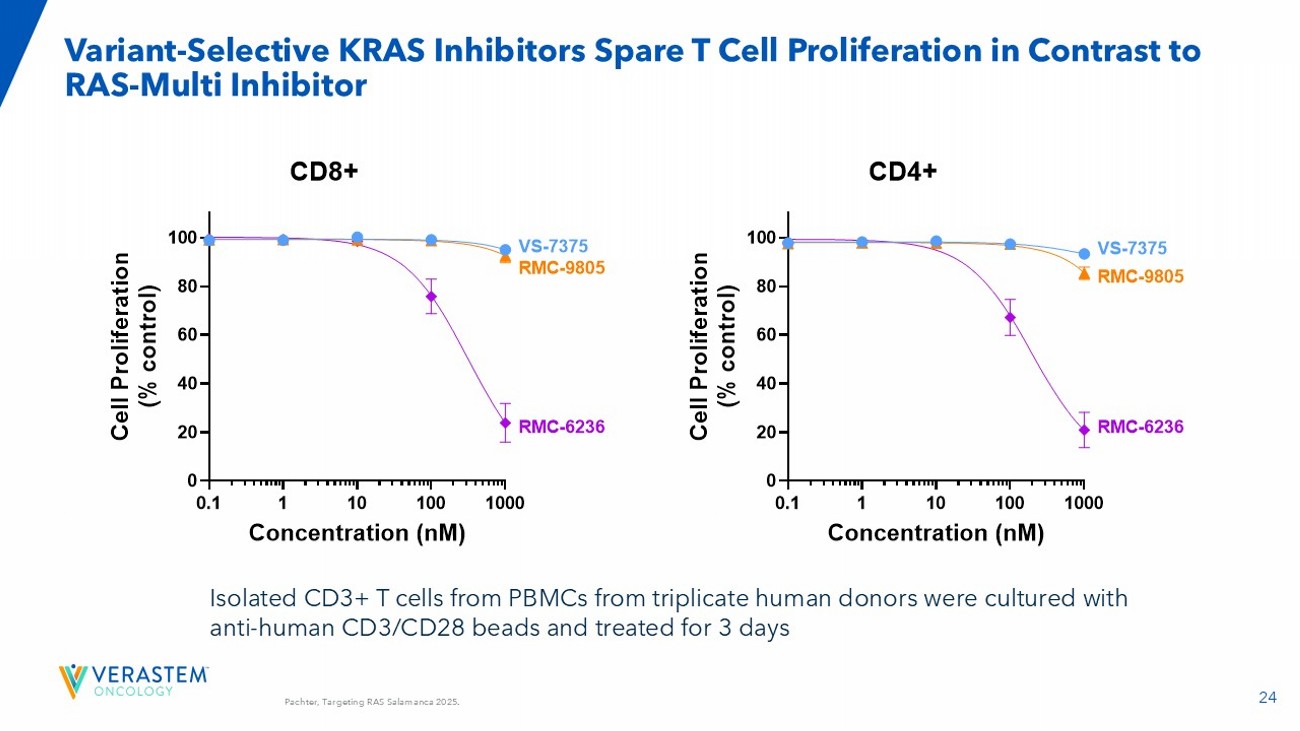
23 VS - 7375 (G12D ON/OFF inhibitor) is More Efficacious Than G12D ON and Pan - RAS ON Inhibitors in KRAS G12D Mutant Tumor Models KP4 Pancreatic Cancer Model LU0876 NSCLC PDX Model -100 0 100 200 500 1000 1500 2000 R e s p o n s e @ D a y 2 0 ( % c h a n g e f r o m b a s e l i n e ) v e h i c l e R M C - 9 8 0 5 1 0 0 m g / k g Q D V S - 7 3 7 5 5 0 m g / k g B I D 4 SDs R M C - 6 2 3 6 2 5 m g / k g Q D 8 PRs 4 SDs 0 10 20 30 0 500 1000 1500 2000 2500 Days after first dose T u m o r v o l u m e ( m m 3 + / - S E M ) Vehicle VS-7375 50 mg/kg BID RMC-9805 100 mg/kg QD RMC-6236 25 mg/kg QD vehicle RMC-9805 RMC-6236 VS-7375 -100 0 100 200 300 400 500 R e s p o n s e @ D a y 1 4 ( % c h a n g e f r o m b a s e l i n e ) v e h i c l e R M C - 9 8 0 5 1 0 0 m g / k g Q D V S - 7 3 7 5 3 0 m g / k g B I D 7 PRs 1 SD R M C - 6 2 3 6 2 5 m g / k g Q D 1 PR 2 SDs 2 PRs 4 SDs R M C - 9 8 0 5 6 0 m g / k g Q D V S - 7 3 7 5 1 0 0 m g / k g B I D 1 CR 7 PRs Equivalent daily dose Maximal dose 0 10 20 30 0 250 500 750 1000 1250 Days after first dose T u m o r v o l u m e ( m m 3 + / - S E M ) vehicle RMC-9805 RMC-6236 VS-7375 Vehicle VS-7375 30 mg/kg BID po RMC-9805 60 mg/kg QD po RMC-6236 25 mg/kg QD po LS513 Colorectal Cancer Model 24 Variant - Selective KRAS Inhibitors Spare T Cell Proliferation in Contrast to RAS - Multi Inhibitor 0.1 1 10 100 1000 0 20 40 60 80 100 Concentration (nM) C e l l P r o l i f e r a t i o n ( % c o n t r o l ) VS-7375 RMC-9805 RMC-6236 VS-7375 RMC-9805 RMC-6236 CD8+ 0.1 1 10 100 1000 0 20 40 60 80 100 Concentration (nM) C e l l P r o l i f e r a t i o n ( % c o n t r o l ) VS-7375 RMC-9805 RMC-6236 VS-7375 RMC-9805 RMC-6236 CD4+ Isolated CD3+ T cells from PBMCs from triplicate human donors were cultured with anti - human CD3/CD28 beads and treated for 3 days Pachter, Targeting RAS Salamanca 2025.

25 Source: GenFleet Therapeutics WCLC 2025 Mini Oral Presentation September 7. 2025; Efficacy and Safety of GFH375 in Advanced N on - Small Cell Lung Cancer Patients with KRAS G12D Mutation. WCLC 2025 Cut - off date: 01 August 2025. NSCLC at 600 mg QD (n=16) All NSCLC (n=28) 4 (25.0) 12 (42.9) Discontinued n(%) 1 (6.3) 1 (3.6) Adverse event 2 (12.5) 8 (28.6) Progress disease 1 (6.3) 2 (7.1) Patient’s decision 0 1 (3.6) Physician’s decision • 28 NSCLC patients with local tumor testing KRAS G12D+ were treated with GFH375 single agent, including 16 at 600 mg QD. • Majority (60.7%) were never smokers. • Among the 22 patients with baseline PD - L1 TPS data available, none had high expression (TPS ≥ 50%). • 17.9% had baseline brain metastases. • Majority (64.3%) had received at least 2 prior lines of therapies; all received prior platinum - based chemo therapies; 96.4% received prior ICIs. • Most patients were ongoing; the longest follow - up was 55.6 weeks. NSCLC at 600 mg (n=16) All NSCLC (n=28) 60.5 (36 - 74) 61 (36 - 74) Age, median (range), years 8 (50) 13 (46.4) Female, n(%) Smoking status, n(%) 8 (50) 11 (39.3) Current or former 8 (50) 17 (60.7) Never ECOG PS, n(%) 14 (87.5) 26 (92.9) 1 16 (100) 28 (100) Pathology adenocarcinoma, n(%) 16 (100) 28 (100) Baseline metastasis, n(%) 4 (25.0) 12 (42.9) Bone 1 (6.3) 5 (17.9) Brain 2 (12.5) 3 (10.7) Liver PD - L1 TPS, n(%) 11 (68.8) 22 (78.6) Known 6 (54.5) 2 13 (59.1) 1 < 1% 5 (45.5) 2 9 (40.9) 1 1 - 49% 0 0 ≥ 50% 1.5 (1 - 4) 2 (1 - 4) Prior lines of therapies, median (range) * 16 (100) 27 (96.4) Prior ICI, n(%) 16 (100) 28 (100) Prior platinum, n(%) 16 (100) 25 (89.3) Prior ICI +platinum concurrent, n(%) 1 Percentage denominator is 22. 2 Percentage denominator is 11. * A neoadjuvant and adjuvant therapy would be counted as a line of system therapy if patient relapsed or progressed during treatment or within 6 months of last dose. GFH375: NSCLC Baseline Characteristics & Patient Disposition 26 Source: GenFleet Therapeutics WCLC 2025 Mini Oral Presentation September 7.

2025; Efficacy and Safety of GFH375 in Advanced N on - Small Cell Lung Cancer Patients with KRAS G12D Mutation. WCLC 2025 600mg QD (N=16) All patients (N=26) 68.8% [41.3%, 89.0%] 57.7% [39.8%, 74.2%] ORR [90% CI] 93.8% [69.8%, 99.8%] 88.5% [72.8%, 96.8%] DCR [90% CI] • ORR is 68.8% in 16 patients at 600 mg QD and 57.7% in 26 evaluable patients * • Of the 11 patients at 600 mg QD with PR, 5 have confirmed and 5 have the potential to confirm the PR • 2 additional patients at 600 mg QD with 20 - 30% reduction are still on treatment with potential for response # # • Among the 5 patients with baseline brain metastases, 2 achieved PR • Median (range) time on treatment: 15.1 weeks (range: 4.6 - 55.6) • Median (range) time to response: 6.3 weeks (range: 6.0 - 48.1) C - PR confirmed → - On treatment BM - Brain metastasis at baseline BM BM BM BM BM Data cut - off date: 01 August 2025. All patients received first dose of GFH375 at least 10 weeks prior to data cut - off date. Median follow - up 21.8 weeks (range: 8.3 - 55.6). * One patient (200 mg QD) had no target lesion at baseline but remains stable; one patient (400 mg QD) dropped out early with out post - baseline tumor assessment due to patient’s decision and died 41 days after last dose due to disease progression. # Two patients achieved PR but discontinued before response confirmation. One (400 mg QD) had PD at the 2 nd assessment. The other dropped out due to G4 hepatic function abnormal (recovered after discontinuation). BOR, best overall response; CI, confidence interval; DCR, disease control rate; ORR, objective response rate; PD, progressive di sease; PR, partial response; SD, stable disease; PLoT : number of prior lines of therapies. GFH375: Efficacy in NSCLC 27 WCLC 2025 GFH375: Safety Data in All Patients from the GenFleet Phase 1/2 Study Source: GenFleet Therapeutics WCLC 2025 Mini Oral Presentation September 7.

2025; Efficacy and Safety of GFH375 in Advanced N on - Small Cell Lung Cancer Patients with KRAS G12D Mutation. • 142 patients, including 28 NSCLC, with advanced KRAS G12D mutation were treated with GFH375 single agent . » 93% had baseline ECOG PS = 1. » 75% had received at least ≥ 2 prior lines of therapies. • 97 patients, including 16 NSCLC, were treated at 600 mg QD. • No treatment related death. • 4.2% of patients discontinued treatment due to TRAEs. Dose intensity = 90%. • 27.5% of patients had grade 3/4 TRAEs. • Overall, GFH375 has manageable safety profile in previously treated advanced cancer patients. • Grade ≥3 TRAEs and SAEs are more frequent in NSCLC patients; however, discontinuation rate appears to be similar. 600 mg QD All dose levels Adverse events, n(%) NSCLC (N=16) All types (N=97) NSCLC 2 (N=28) All types 1 (N=142) TEAEs 16 (100) 96 (99.0) 28 (100) 141 (99.3) All grades 7 (43.8) 41 (42.3) 15 (53.6) 60 (42.3) Grade ≥ 3 3 (18.8) 19 (19.6) 6 (21.4) 27 (19.0) SAE 4 (25.0) 31 (32.0) 10 (35.7) 51 (35.9) Leading to interruption 3 (18.8) 7 ( 7.2) 5 (17.9) 12 ( 8.5) Leading to reduction 1 (6.3) 6 (6.2) 1 (3.6) 9 (6.3) Leading to discontinuation 0 3 (3.1) 0 4 (2.8) Leading to death TRAEs 16 (100) 96 (99.0) 28 (100) 139 (97.9) All grades 6 (37.5) 25 (25.8) 13 (46.4) 39 (27.5) Grade ≥ 3 2 (12.5) 8 (8.2) 4 (14.3) 11 (7.7) SAE 3 (18.8) 19 (19.6) 9 (32.1) 33 (23.2) Leading to interruption 3 (18.8) 7 (7.2) 5 (17.9) 11 (7.7) Leading to reduction 1 ( 6.3) 4 (4.1) 1 ( 3.6) 6 (4.2) Leading to discontinuation 0 0 0 0 Leading to death Data c ut - off date: 17 Jun 2025 The median exposure time to GFH375 in all patients was 9.6 (range: 0.9 - 49.1) week s by the cut - off date. 1 Including all patients treated at all dose levels: 100 mg QD (n=1), 200 mg QD (n=1), 400 mg QD (n=29), 600 mg QD (n=97), 750 m g QD (n=8) , 900 mg QD (n=3), and 300 mg BID (n=3). 2 Including NSCLC patients treated at all dose levels: 100 mg QD (n=1), 200 mg QD (n=1), 400 mg QD (n=7), 600 mg QD (n=16), 7 50 mg QD (n=1) , and 300 mg BID (n=2). SAE, serious adverse event; TEAE, treatment emergent adverse event; TRAE, treatment related adverse event.

28 WCLC 2025 Source: GenFleet Therapeutics WCLC 2025 Mini Oral Presentation September 7. 2025; Efficacy and Safety of GFH375 in Advanced N on - Small Cell Lung Cancer Patients with KRAS G12D Mutation. Abbreviations: ALT, alanine transaminase; AST, aspartate transferase; TRAE, treatment related adverse event; WBC, white blood cell. GFH375: Safety Data in All Patients from the GenFleet Phase 1/2 Study • No new safety signals were observed • The most frequent TRAEs were gastrointestinal AEs (diarrhea, vomiting, and nausea) and hematological AEs (anemia and neutropenia), mainly grade 1/2 and manageable with supportive medications • Most frequent G3/4 TRAEs were neutropenia (6.3%) and anemia (4.2%) • Multiple AEs had higher rates in NSCLC patients, potentially due to prior use of ICIs » 96.4% patients with NSCLC had received ICIs before GFH375 treatment » The median time of last dose of ICIs to first administration of GFH375 was 2.8 months (range: 1.0 - 40.0); the median time of ICI treatment was 4.3 months (range: 0.7 – 26.5) 0% 10% 20% 30% 40% 50% 60% 70% 80% 90% 100% Most frequent TRAEs (≥10% of all patients) Grade 1 Grade 2 Grade 3 Grade 4 Grade 5 Left bar: all patients (N=142) Right bar: NSCLC (N=28)

29 GFH375 600 mg QD (N=66) 60 (35, 74) Age, median (range), years 35 (53.0%) ≥ 60, n (%) 35 (53.0%) Male, n (%) 66 (100%) ECOG PS 1, n (%) Histological type, n (%) 64 (97.0%) Adenocarcinoma 2 (3.0%) Adenosquamous carcinoma 63 (95.5%) Stage IV at study entry, n (%) c Baseline metastasis, n (%) 52 (78.8%) Liver 19 (28.8%) Lung 19 (28.8%) Peritoneum 12 (18.2%) Bone (1, 5) Number of prior lines of anticancer therapy, (range) 21 (31.8%) 1, n (%) 45 (68.2%) ≥ 2, n (%) Prior anticancer therapy, n (%) 61 (92.4%) Gemcitabine - containing 50 (75.8%) Fluorouracil - containing 35 (53.0%) Irinotecan - containing 22 (33.3%) Immune checkpoint inhibitors • A total of 66 patients with advanced KRAS G12D mutant PDAC were treated at 600 mg QD of GFH375 monotherapy, all with ECOG PS 1. • The majority (95.5%) of patients were diagnosed with stage IV disease at study entry, with liver being the most common metastatic location (78.8%), followed by lung (28.8%), peritoneum (28.8%) and bone (18.2%). • 68.2% of patients had received at least 2 prior lines of anticancer therapies, mainly chemotherapies; 33.3% had previously received ICIs. • 31 (47.0%) patients were still on treatment; the longest duration of treatment was 367 days. 35 (53.0%) Patients discontinued from treatment: 21 (31.8%) • Disease progression 6 (9.1%) • Adverse event a 4 (6.1%) • Other b 2 (3.0%) • Investigator’s decision 1 (1.5%) • Initiation new anticancer therapy 1 (1.5%) • Withdrawal of consent Source: GenFleet Therapeutics. Data cut - off date: September 27, 2025. All patients received first dose of GFH375 at least 4 months prior to the cut - off date. a One due to treatment - related grade 3 hepatic function abnormalities; one due to treatment - related grade 3 renal failure; the oth ers due to AEs not related to GFH375. b All were by patient decision; c The rest of 3 were stage III with T4. Abbreviations: ICI, immune checkpoint inhibitor. ECOG, Eastern Cooperative Oncology Gro up. PS, performance status.

GFH375: Patient and Disease Characteristics - Pancreatic Ductal Adenocarcinoma (PDAC) ESMO 2025 30 GFH375: Best Overall Response (PDAC) Changes from baseline in target lesions 20 50 0 - 30 - 5 0 0 100 200 300 Time since study treatment initiation (days) 20 - 30 Best % changes from baseline in target lesions - 50 0 N=59 40.7% [30%, 52%] ORR [90%CI] Best overall response, n (%) 24 (40.7) Partial response 33 (55.9) a Stable disease 2 (3.4) b Progressive disease 96.7% [90%, 99%] DCR [90%CI] • ORR was 40.7% (24/59), 90%CI was [30%, 52%] in the 59 evaluable patients. • DCR was 96.7% (57/59), 90%CI was [90%, 99%]; Majority (91.5%) had reduction in target lesions. ESMO 2025 Partial response Stable disease Progressive disease Source: GenFleet Therapeutics. Data cut - off date: September 27, 2025. All patients received first dose of GFH375 at least 4 mont hs prior to the cut - off date. Seven patients had no post - treatment tumor assessments due to early dropout: 2 due to AEs not related to GFH375 (1 upper gastrointestinal hemorrhage and 1 respira tor y failure); 1 started new anticancer therapy; 4 early discontinued due to patient decision. Left figure: "C“ represents confirmed responders. Arrows indicate treatment ongoing. Abbreviations: CI, confidence in terval. DCR, disease control rate. ORR, objective response rate .

31 GFH375: Progression - Free Survival (PDAC) ESMO 2025 • Median PFS was 5.52 months ( 90%CI: 4.27, 7.20), with a median follow - up time 5.65 months ( 90%CI: 4.96, 6.08 ). • 4 - month PFS rate was 78.2% ( 90%CI: 69.8%, 87.5%). Source: GenFleet Therapeutics. Data cut - off date: September 27, 2025. All patients received first dose of GFH375 for at least 4 months prior to the cut - off date.
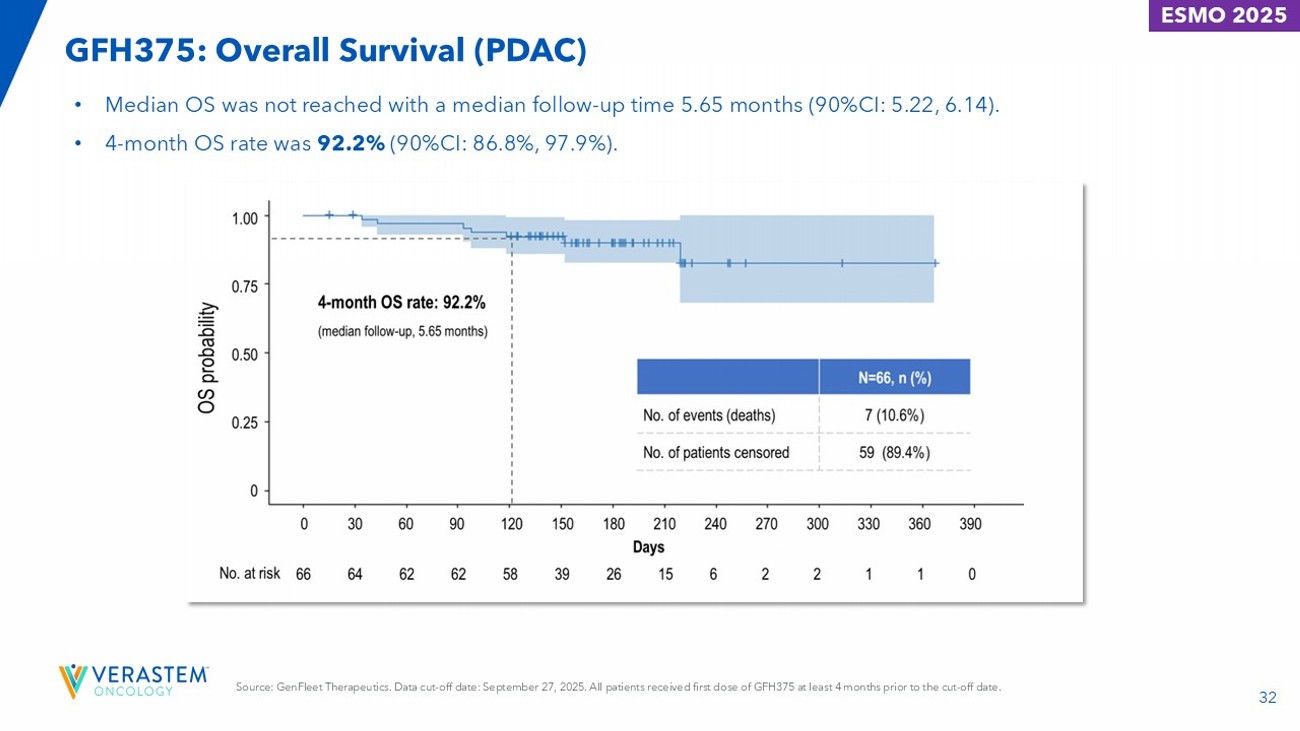
32 GFH375: Overall Survival (PDAC) ESMO 2025 • Median OS was not reached with a median follow - up time 5.65 months (90%CI: 5.22, 6.14). • 4 - month OS rate was 92.2% (90%CI: 86.8%, 97.9%). Source: GenFleet Therapeutics. Data cut - off date: September 27, 2025. All patients received first dose of GFH375 at least 4 months prior to the cut - off date.

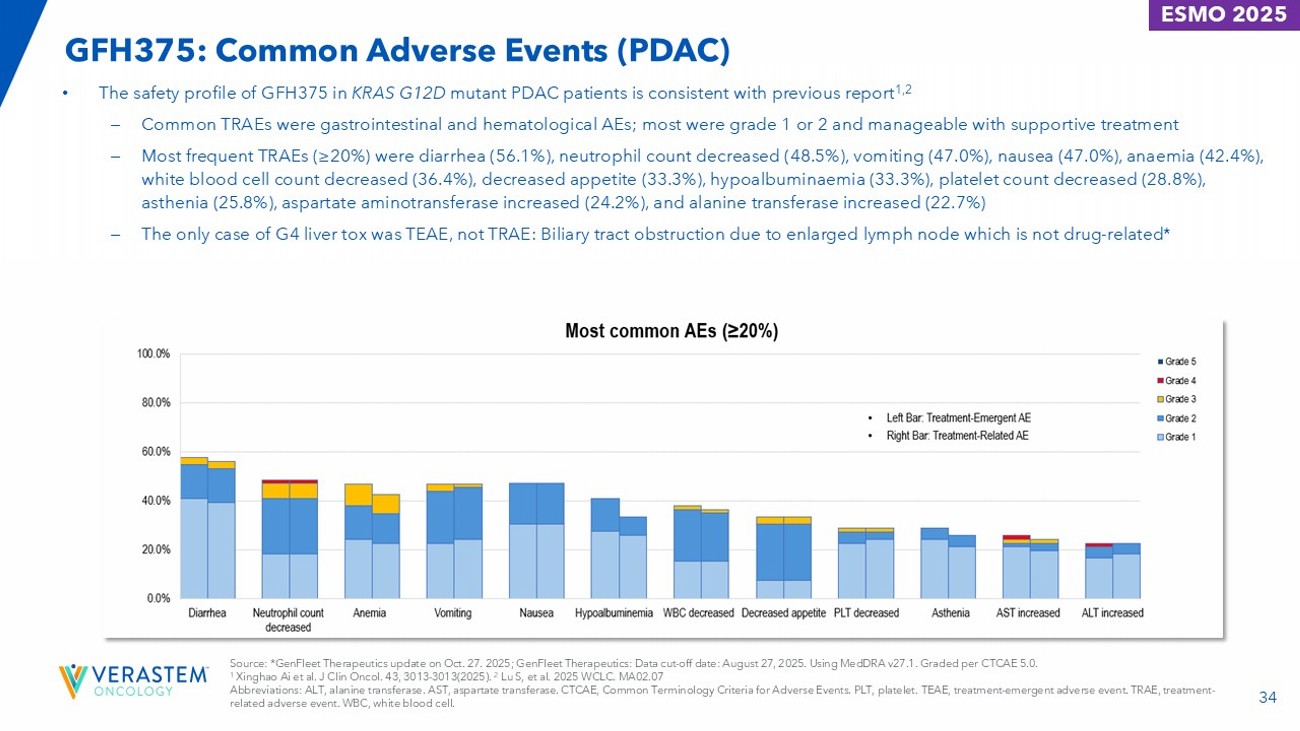
33 GFH375: Safety Overview (PDAC) • GFH375 presented a manageable safety profile at 600mg QD in previously heavily treated KRAS G12D mutant PDAC patients. • All patients experienced at least one TEAE and TRAE. • No fatal TRAE. Grade 3 TRAEs occurred in 20 patients (30.3%) and a grade 4 TRAE occurred in one patient (1.5%). • TRAE resulted in dose discontinuation in 2 patients (3.0%) and reduction in 4 patients (6.1%). • The mean relative dose intensity was 93%. GFH375 600mg QD (N=66) ≥ grade 3, n (%) a Any grade, n (%) 33 (50.0)/21 (31.8) a 66 (100)/66 (100) Patients with at least one TEAE/TRAE 14 (21.2)/7 (10.6) 17 (25.8)/9 (13.6) Patients with at least one serious TEAE/TRAE 20 (30.3)/12 (18.2) 28 (42.4)/21 (31.8) Patients with dose modifications due to TEAE/TRAE 5 (7.6)/2 (3.0) 5 (7.6)/2 (3.0) Discontinuation 1 (1.5)/1 (1.5) 4 (6.1)/4 (6.1) Dose reduction 18 (27.3)/11 (16.7) 25 (37.9)/18 (27.3) Dose interruption Source: GenFleet Therapeutics: Data cut - off date: August 27, 2025. Graded per CTCAE v5.0. a Only one patient experienced a grade 4 treatment - related neutropenia and recovered with treatment of G - CSF. Abbreviations: CTCAE, Common Terminology Criteria for Adverse Events. QD, once daily. TEAE, treatment - emergent adverse event. TRAE, treatment - related adverse event.
ESMO 2025 34 GFH375: Common Adverse Events (PDAC) ESMO 2025 • The safety profile of GFH375 in KRAS G12D mutant PDAC patients is consistent with previous report 1,2 – Common TRAEs were gastrointestinal and hematological AEs; most were grade 1 or 2 and manageable with supportive treatment – Most frequent TRAEs (≥20%) were diarrhea (56.1%), neutrophil count decreased (48.5%), vomiting (47.0%), nausea (47.0%), anaemia (42.4%), white blood cell count decreased (36.4%), decreased appetite (33.3%), hypoalbuminaemia (33.3%), platelet count decreased (28.8%), asthenia (25.8%), aspartate aminotransferase increased (24.2%), and alanine transferase increased (22.7%) – The only case of G4 liver tox was TEAE, not TRAE: Biliary tract obstruction due to enlarged lymph node which is not drug - related * Source: *GenFleet Therapeutics update on Oct. 27. 2025; GenFleet Therapeutics: Data cut - off date: August 27, 2025. Using MedDRA v27.1. Graded per CTCAE 5.0. 1 Xinghao Ai et al. J Clin Oncol. 43, 3013 - 3013(2025). 2 Lu S, et al. 2025 WCLC. MA02.07 Abbreviations: ALT, alanine transferase. AST, aspartate transferase. CTCAE, Common Terminology Criteria for Adverse Events. PLT, platelet. TEAE, treatment - emergent adverse event. TRAE, treatment - related adverse event. WBC, white blood cell.

35 Source: GenFleet Therapeutics Oct. 27, 2025. Data cut - off date: September 27, 2025. All participants received first dose of GFH3 75 for at least 4 months prior to the cut - off date. Seven participants had no post - treatment tumor assessments due to early dropout: 2 due to AEs not related to GFH375 (1 upper gastrointestinal hemorrhage and 1 respiratory failure); 1 started new anticancer therapy; 4 early discontinued due to participant decision. Figure: "C“ represents confirmed responders. Arrows indicate treatment ongoing. The bars for 3 participants are not shown in the waterfall plot: a 1 had best overall response (BOR) as SD, and the best percentage change from baseline in was 0%. b 2 had progressive disease as their BOR: one had a target lesion decrease by 0.4% from baseline with non - target lesion progression; the other had target lesions increase by 0.7% from baseline with non - target lesion progression. Abbreviations: CI, confidence interval. DC R, disease control rate. ORR, objective response rate. GFH375 PDAC Subgroup Analysis: ORR & PFS (Post ESMO Update) • 59 participants had measurable disease at baseline and at least one post - baseline tumor assessment • mPFS and mOS has not been reached for 2L PDAC patients. Median follow up time was 5.65 months. • mPFS for 3L+PDAC patients was 5.52 months and mOS has not been reached. 3L+ (N=47) 2L (N=12) All PDAC (N=59) 36.2% [24.52%, 49.18%] 58.3% [31.52%, 81.90%] 40.7% [29.87%, 52.22%] ORR [90%CI] 95.7% [87.20%, 99.24] 100% [77.91, 100%] 96.6% [89.71%, 99.39%] DCR [90%CI] PDAC: Subgroup Analysis – PFS 3L+ (N=54) 2L (N=12) All PDAC (N=66) 5.52 Not reached 5.52 Median PFS (months) 29 4 (maturity:33%) 33 No.

of Events 20 5 25 On Treatment without Event 36 GFH375 600mg QD 2L+ PDAC, N=66 Grade ≥ 3 Any grade Gastrointestinal disorders 3.0% 56.1% Diarrhea 1.5% 47.0% Vomiting 0 47.0% Nausea 3.0% 33.3% Decreased appetite Hematological toxicities 7.6% 48.5% Neutrophil count decreased 7.6% 42.4% Anemia 1.5% 36.4% WBC count decreased 1.5% 28.8% PLT decreased Liver enzyme abnormalities 1.5% 24.2% AST increased 0 22.7% ALT increased Skin toxicities 0 3.0% Rash GFH375 Breakdown of PDAC Safety Data (Post ESMO Update) Source: GenFleet Therapeutics.

37 • Efficacious starting dose: • 400 mg QD starting dose supported by monotherapy efficacy/safety in FIH study • Evaluating GI side effect mitigations: • Use prophylactic anti - emetics • Evaluate dosing with and without food • Evaluate additional formulation • Multiple, high - value indication strategy: • Monotherapy expansion cohorts in PDAC & NSCLC • Combination cohorts with cetuximab for CRC, chemotherapy for PDAC, chemo plus I - O for NSCLC • Expand to geographies outside of U.S. Accelerated Development Approach Builds on Preliminary First - in - Human Study Findings Multi - Arm Clinical Study Evaluating Monotherapy and Combinations FDA Engagement Plans • Granted Fast Track Designation for: • First - line in patients with KRAS G12D locally advanced or metastatic PDAC • Patients with KRAS G12D locally advanced or mPDAC who received at least one prior line of standard systemic therapy in July 2025 • Plan to pursue Breakthrough Therapy Designation • Accelerated clinical development with FDA input 38 VS - 7375 - 101 Study Schema, Efficiently Testing KRAS G12D+ Indications with Monotherapy and SOC Combinations Part A: Single Agent Dose Escalation – Any KRAS G12D Solid Tumor Cohort B1: 2L+ PDAC (N=20) Cohort D1: 2L+ CRC Cetuximab (N=20) VS - 7375 + Combinations Part B: Dose Expansion Part C: Combination Dose Escalations Part D: Combination Dose Expansion Dose Level 3: 900 mg QD Dose Level 2: 600 mg QD Dose Level 1: 400 mg QD Dose Level - 1 : 300 mg QD Cohort B2: 2L+ NSCLC (N=20) Cohort B3: 2L+ Solid Tumors (N=20) VS - 7375 Dose Escalation Anticipated dose levels range from 400 - 900 mg QD, with options for reduction if required.

+ Cohort C1: 2L+ Solid Tumors Cetuximab* Cohort D2: 1L NSCLC Carboplatin/Pemetrexed/ Pembrolizumab (N=20) Select RP2D Select RP2D Cohort C3: 2L+ PDAC Gemcitabine + Nab - Paclitaxel Cohort D3: 2L+ PDAC Gemcitabine/Nab - Paclitaxel (N=20) Select RP2D Select RP2D Cohort C4: 1L PDAC – 75+ yo Gemcitabine Cohort D4: 1L PDAC – 75+ yo Gemcitabine (N=20) Select RP2D Cohort C2: 1L NSCLC Carboplatin/Pemetrexed/ Pembrolizumab The study will evaluate dosing with meals and utilize prophylactic anti - emetics Dose - level cleared VS - 7375 Monotherapy 39 Source: Verastem press release Oct. 23, 2025; VSTM DOF VS - 7375 Clears Two Dose Levels, Demonstrates Promising Anti - Tumor Activity in Ongoing Phase 1/2a Trial Monotherapy Dose Escalation GI Adverse Events Combination Cohorts First two monotherapy dose levels (400 mg QD and 600 mg QD) cleared, with no dose - limiting toxicities reported No nausea, vomiting, or diarrhea greater than Grade 1 was observed in first two dose levels Enrollment initiated for VS - 7375 in combination with cetuximab in patients with advanced KRAS G12D mutant solid tumors, including colorectal cancer Anti - Tumor Activity Promising anti - tumor activity observed in patients with various solid tumors, including advanced pancreatic ductal adenocarcinoma


40 Next Steps in VS - 7375 Clinical Program Q4 2025 1H 2026 • Plan to report an interim safety and efficacy update on the Phase 1/2a trial of VS - 7375. ௗ • Expect to select the RP2D and plan to initiate monotherapy expansion cohorts in advanced PDAC, NSCLC, and other KRAS G12D - mutated solid tumors. • Expect to select the RP2D and plan to initiate combination expansion cohorts in CRC, PDAC, and NSCLC. • Continue enrollment in the combination cohort of VS - 7375 and cetuximab. • Plan to initiate the dose escalation cohorts in combination with chemotherapy in PDAC and chemotherapy with anti - PD - 1 in NSCLC. • Plan to engage with the FDA to discuss our development path forward , including potential registration - directed clinical trials in PDAC and NSCLC.

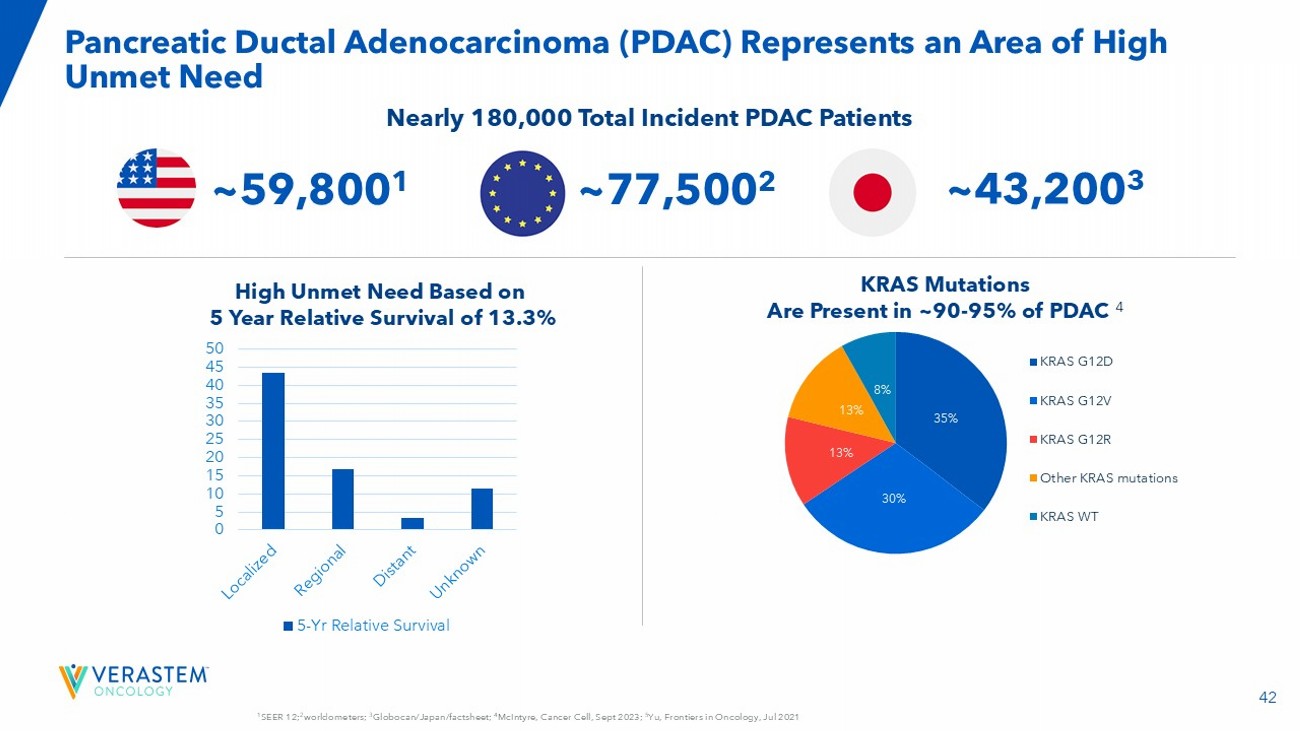

Topline Data from RAMP 205: Avutometinib + Defactinib + Standard of Care in First - Line Metastatic Pancreatic Cancer 42 1 SEER 12; 2 worldometers; 3 Globocan/Japan/factsheet; 4 McIntyre, Cancer Cell, Sept 2023; 5 Yu, Frontiers in Oncology, Jul 2021 Pancreatic Ductal Adenocarcinoma (PDAC) Represents an Area of High Unmet Need 0 5 10 15 20 25 30 35 40 45 50 5-Yr Relative Survival KRAS Mutations Are Present in ~90 - 95% of PDAC 4 High Unmet Need Based on 5 Year Relative Survival of 13.3% Nearly 180,000 Total Incident PDAC Patients 35% 30% 13% 13% 8% KRAS G12D KRAS G12V KRAS G12R Other KRAS mutations KRAS WT ~59,800 1 ~43,200 3 ~77,500 2 43 DLT: dose - limiting toxicity; RP2D: recommended phase 2 dose; CT: computed tomography; ECOG: European Cooperative Oncology Group; MRI: magnetic resonance imaging RAMP 205: Designed to Identify and Evaluate RP2D in Combination with Chemotherapy for Treatment of Newly Diagnosed mPDAC RAMP 205: Ongoing Phase 1/2 Evaluating Avutometinib + Defactinib with Gemcitabine and Nab - paclitaxel Inclusion Criteria • Histologic or cytologic confirmed mPDAC • Eligible for treatment in the first - line setting (no prior systemic therapy for advanced or metastatic disease) • Measurable by RECIST v1.1 by CT or MRI • ECOG Performance status of ≤1 • Part B only, adequate tissue sample to evaluate KRA mutational status Collaboration with PanCAN , NCT05669482 Part A: Dose Evaluation (3+3 DLT Assessment) RP2D Selection Part B: Dose Expansion (Primary Endpoint: ORR) Avutometinib + Defactinib + Gemcitabine + Nab - paclitaxel Dose Finding Cohorts n=3 - 6 Avutometinib + Defactinib + Gemcitabine + Nab - paclitaxel DLT - Cleared Dose Level Expansion • Patients with PDAC treated at RP2D • Stage 1: 17 patients if ≥7 responders, then • Simon’s 2 - stage design: expand to 29 patients Current status of trial 44 Updated 05/01/25 Enrolled N=60 Days Chemo Dosing Nab - Paclitaxel Gemcitabine Defactinib Avutometinib Dose Level 12 1 - 8 - 15 125 800 200 2.4 1 12 1 - 8 - 15 100 800 200 3.2 0 12 1 - 8 - 15 100 800 200 2.4 - 1 12 1 - 15 125 800 200 3.2 1a 12 1 - 15 125 1000 200 3.2 2a PART A Enrollment Summary Dose Levels & Administration Schedule (28 - day) Cycle RAMP 205: 12 Patients Enrolled in Each Dose Cohort • Enrolled: (n=60) • Treatment Ongoing: (n=19) • Ended Study: (n=30) • Survival Follow Up: (n=11) Dose level 1 selected as the RP2D
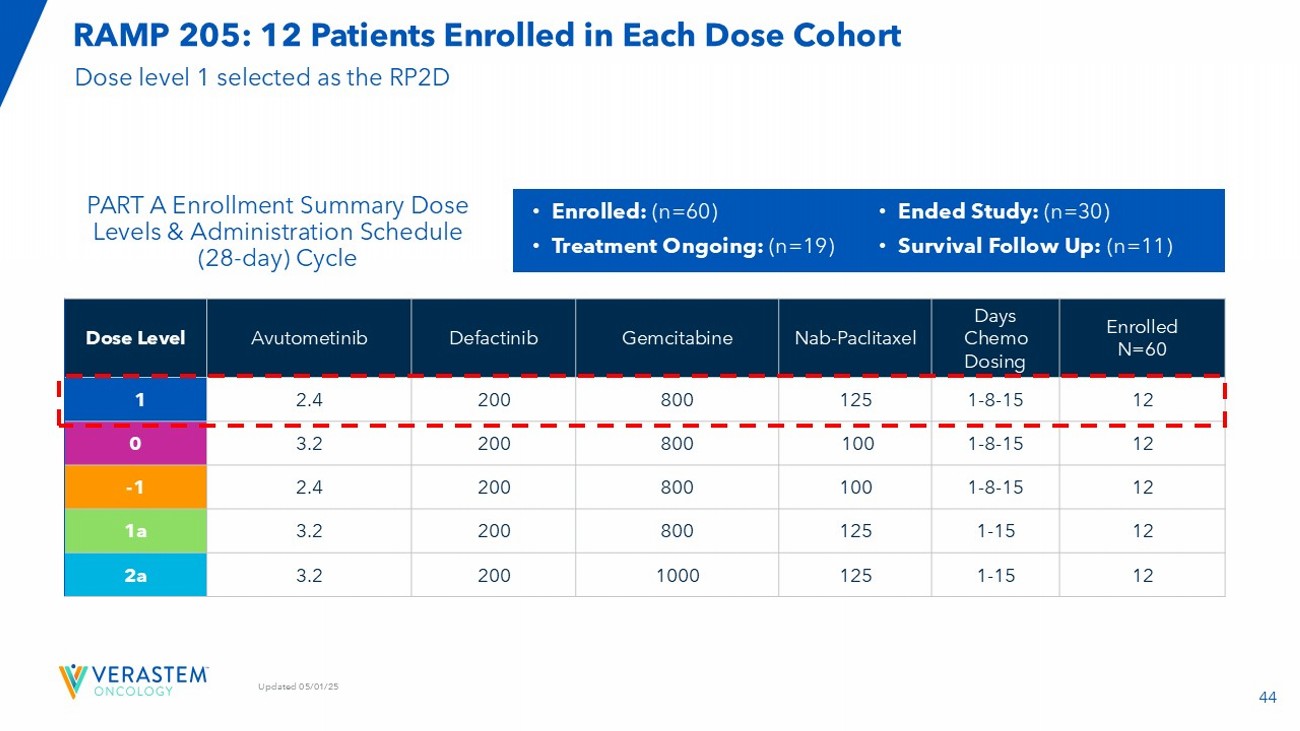
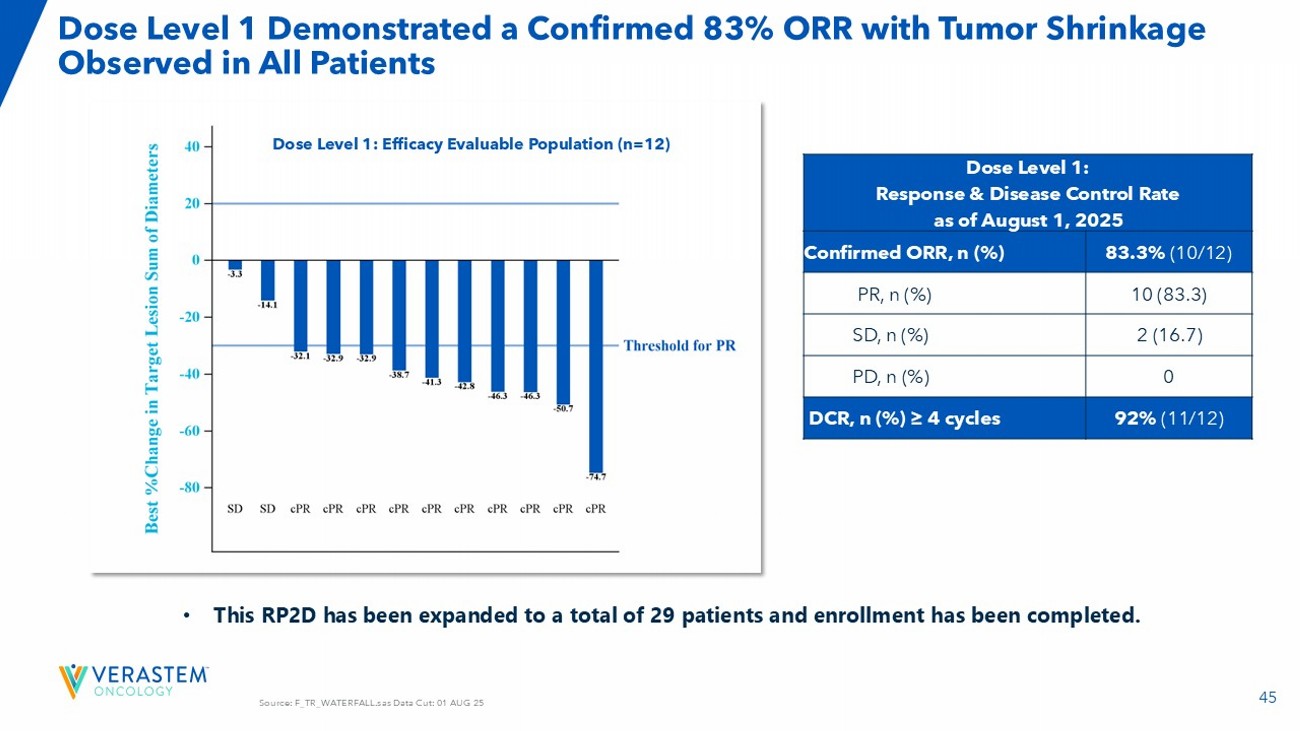
45 Source: F_TR_WATERFALL.sas Data Cut: 01 AUG 25 Dose Level 1 Demonstrated a Confirmed 83% ORR with Tumor Shrinkage Observed in All Patients Dose Level 1: Response & Disease Control Rate as of August 1, 2025 83.3% ( 10/12) Confirmed ORR, n (%) 10 (83.3) PR, n (%) 2 (16.7) SD, n (%) 0 PD, n (%) 92% (11/12 ) DCR, n (%) ≥ 4 cycles Dose Level 1: Efficacy Evaluable Population (n=12) • This RP2D has been expanded to a total of 29 patients and enrollment has been completed.

46 Source: Program:F_TX_RS_SWIMMER_DL1.sas Data Cut: 25 APR 25 Encouraging Duration of Treatment Observed to Date for Dose Level 1 Dose Level 1 Duration of Treatment Safety Population (n=12) Historical Gem/Nab PFS 95% CI mPFS Reference Study 4.5 - 5.9 5.5 MPACT N=421 Von Hoff 2013 5.3 - 5.8 5.6 NAPOLI 3 N=387 O’Reilly 2023 Historical Gem/Nab mPFS 47 Source: Dr. Lim, Siteman Cancer Center 7 Patient with mPDAC Showed Substantial Tumor Shrinkage in Dose Level 1 Before C1 After C2 After C4 After C6 metastasis primary tumor 63 year - old Female


48 AEs were Generally Manageable, Allowing Patients to Remain on Treatment DL1 Treatment Emergent AE All Grades ≥ 25% / Non - laboratory AEs NAPOLI 3* (N=379) Dose Level 1 (n=12) TEAEs Grade ≥3 n (%) All Grades n (%) Grade ≥3 n (%) All Grades n (%) Preferred term 10 (3) 162 (43) 0 10 (83) Nausea 17 (4) 139 (37) 0 9 (75) Diarrhoea 20 (5) 143 (38) 0 9 (75) Fatigue Not Listed** 119 (31) 0 8 (67) Alopecia 8 (2) 113 (30) 0 8 (67) Constipation Not Listed** 108 (29) 2 (17) 7 (58) Oedema peripheral Not Listed** Not Listed** 0 7 (58) Rash maculo - papular Not Listed** 45 (12) 1 (8) 7 (58) Stomatitis 8 (2) 100 (26) 1 (8) 7 (58) Vomiting Not Listed** 58 (15) 0 6 (50) Dysgeusia Not Listed** Not Listed** 0 6 (50) Hypotension Not Listed** 87 (23) 1 (8) 6 (50) Pyrexia 10 (3) 106 (28) 0 5 (42) Decreased appetite 8 (2) Not Listed** 1 (8) 5 (42) Hypertension 22 (6) 66 (17) 1 (8) 5 (42) Neuropathy peripheral Not Listed** Not Listed** 0 4 (33) Cough Not Listed** Not Listed** 0 4 (33) Dyspepsia 8 (2) 47 (12) 0 4 (33) Dyspnoea Not Listed** Not Listed** 1 (8) 4 (33) Retinopathy Not Listed** Not Listed** 1 (8) 4 (33) Vision blurred Not Listed** Not Listed** 0 3 (25) Abdominal Distension 14 (4) 77 (20) 0 3 (25) Abdominal Pain Not Listed** Not Listed** 0 3 (25) Depression Not Listed** 43 (11) 0 3 (25) Epistaxis 9 (2) Not Listed** 3 (25) 3 (25) Febrile Neutropenia Not Listed** Not Listed** 0 3 (25) Rash • No new or unexpected AEs observed • Most non - laboratory AEs were grade 1 or 2 • AEs were generally manageable, allowing patients to remain on treatment • The rates and severities of most AEs are consistent with the individual rates reported for Gem/Nab and Avutometinib/Defactinib • Nausea, diarrhea, constipation, febrile neutropenia, and anemia may be increased in comparison to those expected with Gem/Nab DL1 Treatment Emergent AE All Grades ≥ 25% / Laboratory - related AEs NAPOLI 3* (N=379) Dose Level (n=12) TEAEs Grade ≥3 n (%) All Grades n (%) Grade ≥3 n (%) All Grades n (%) Preferred term 66 (17) 153 (40) 5 (42) 8 (67) Anaemia 144 (38) 192 (51) 6 (50) 8 (67) Neutropenia*** 11 (3) Not Listed** 2 (17) 7 (58) Hyperbilirubinaemia*** 23 (6) 154 (41) 1 (8) 5 (42) Thrombocytopenia*** 8 (2) 40 (11) 1 (8) 3 (25) Aspartate aminotransferase increased 15 (4) 49 (13) 0 3 (25) Hypokalemia RAMP 205 data source:T_AE_GRADE_PT.sas : Data Cut: 11 APR 25 *Supplement Appendix: (NAPOLI 3). Wainberg et al. The Lancet, Volume 402, Issue 10409, 1272 – 1281 **If “Not Listed” then adverse event is either not reported or did not meet threshold of <10% all grade TEAE or <2% G3 or hig her TEAE. * **'Neutropenia' and 'Neutrophil count decreased' are grouped as 'Neutropenia'. 'Thrombocytopenia' and 'Platelet count decreas ed' are grouped as 'Thrombocytopenia'. ' Hyperbilirubinaemia ' and 'Blood bilirubin increased' are grouped as ' Hyperbilirubinaemia ’’. AE, adverse event; 49 Next Step for RAMP 205 Expect to report an update on the safety and efficacy of expansion cohort in 1H 2026


Avutometinib ± Defactinib with Sotorasib (G12Ci) in KRAS G12C mutant NSCLC 51 Collaboration Verastem & Mariano Barbacid , CNIO (Spain) Addition of FAK inhibitor Augments the Efficacy of Sotorasib + Avutometinib and Reverses Sotorasib Resistance in KRAS G12C NSCLC Preclinical Models AVUTOMETINIB ENHANCES SOTORASIB EFFICACY.

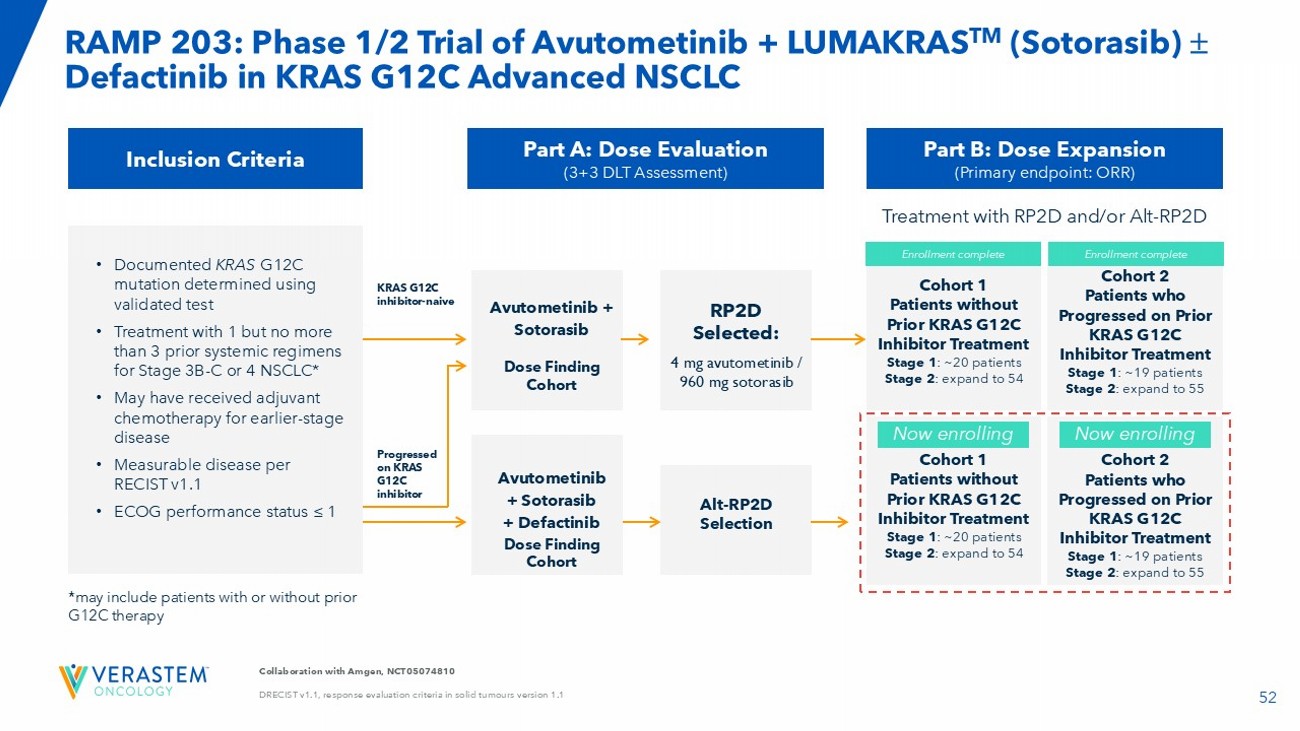
ADDITION OF FAK INHIBITOR INDUCES DEEP TUMOR REGRESSIONS IN ALL TREATED MICE ADDITION OF FAKi + AVUTOMETINIB REVERSES SOTORASIB RESISTANCE ADDITION OF AVUTOMETINIB IS INSUFFICIENT TO REVERSE SOTORASIB RESISTANCE -100 0 100 200 300 400 Response at Day 10 R e s p o n s e @ D a y 1 0 ( % c h a n g e f r o m b a s e l i n e ) v e h i c l e 10 PR a v u t o m e t i n i b F A K i s o t o r a s i b s o t o r a s i b + a v u t o m e t i n i b s o t o r a s i b + F A K i s o t o r a s i b + a v u t o m e t i n i b + F A K i 2 SD 1 SD1 SD 8 SD 2 PR 4 SD 3 PR Doses Tested Sotorasib : 30 mg/kg PO QD Avutometinib : 0.3 mg/kg PO QD FAKi : 50 mg/kg PO BID H2122 KRAS G12C NSCLC 0 50 100 150 0 5 10 15 20 25 Days after first sotorasib dose T u m o r v o l u m e ( m m 3 ) Sotorasib + Avutometinib + FAKiSotorasib 0 50 100 150 0 5 10 15 20 25 Days after first sotorasib dose T u m o r v o l u m e ( m m 3 ) Sotorasib + Avutometinib Sotorasib Mouse #1 Mouse #2 Mouse #3 Mouse #4 52 Collaboration with Amgen, NCT05074810 DRECIST v1.1, response evaluation criteria in solid tumours version 1.1 RAMP 203: Phase 1/2 Trial of Avutometinib + LUMAKRAS TM (Sotorasib) Defactinib in KRAS G12C Advanced NSCLC Inclusion Criteria • Documented KRAS G12C mutation determined using validated test • Treatment with 1 but no more than 3 prior systemic regimens for Stage 3B - C or 4 NSCLC* • May have received adjuvant chemotherapy for earlier - stage disease • Measurable disease per RECIST v1.1 • ECOG performance status ≤ 1 Part A: Dose Evaluation (3+3 DLT Assessment) Avutometinib + Sotorasib Dose Finding Cohort Part B: Dose Expansion (Primary endpoint: ORR) RP2D Selected: 4 mg a vutometinib / 960 mg sotorasib Avutometinib + Sotorasib + Defactinib Dose Finding Cohort Alt - RP2D Selection KRAS G12C inhibitor - naive Progressed on KRAS G12C inhibitor Cohort 1 Patients without Prior KRAS G12C Inhibitor Treatment Stage 1 : ~20 patients Stage 2 : expand to 54 Cohort 2 Patients who Progressed on Prior KRAS G12C Inhibitor Treatment Stage 1 : ~19 patients Stage 2 : expand to 55 Cohort 1 Patients without Prior KRAS G12C Inhibitor Treatment Stage 1 : ~20 patients Stage 2 : expand to 54 Cohort 2 Patients who Progressed on Prior KRAS G12C Inhibitor Treatment Stage 1 : ~19 patients Stage 2 : expand to 55 Enrollment complete *may include patients with or without prior G12C therapy Treatment with RP2D and/or Alt - RP2D Now enrolling Now enrolling Enrollment complete 53 RAMP 203: No Dose - Limiting Toxicities (DLTs) Were Observed in the First Triplet Combination Cohort in Patients Previously Treated with a G12C Inhibitor Triplet Combination Update: • As of a November 21, 2024, data cutoff, three patients whose cancer previously progressed on a G12C inhibitor have been treated with the triplet combination of sotorasib 960 mg administered daily on a continuous schedule and avutometinib 3.2 mg twice - weekly (BIW) plus defactinib 200 mg twice - daily (BID).

Avutometinib and defactinib are administered on a three out of four weeks schedule. • 2 of the 3 patients demonstrated initial tumor reductions of at least 20% at the first scan. As of the data cutoff, all three patients remain on treatment. • Completed enrollment of the planned dose level evaluation cohorts for the triplet combination in Q1 2025. Doublet Combination Update: • As previously reported, the doublet combination of avutometinib with sotorasib has completed enrollment (n=28) in the G12C inhibitor treatment - naïve Stage 1 Part B cohort. • Completed enrollment to the KRAS G 12 C inhibitor, prior - treated Stage 1 Part B doublet cohort in Q 1 2025 . • Patients enrolled in the doublet cohorts continue to be followed for safety and efficacy results (both the prior - treated and treatment - naïve cohorts) .

54 Next Steps for RAMP 203 Report an interim update on the efficacy and safety results from both the doublet and triplet combinations in Q4 2025 55 Anticipated Milestones & Financials 55


56 Execute Successful Commercial Launch in LGSOC, Followed by Meaningful Catalysts to Expand Into Larger, Underserved Patient Populations Anticipated Milestones & Activities Program x Complete planned enrollment in RAMP 301 by end of 2025. x Announce outcome of IDMC’s sample - size re - estimation recommendation for RAMP 301 in Q4 2025. x Report initial data from RAMP 201J Phase 2 clinical trial being conducted in Japan with JGOG in Q4 2025. □ Complete IDMC recommended patient enrollment increase in Q1 2026 . KRAS - mutated Recurrent LGSOC x Expect to report a preliminary update on the Phase 1 monotherapy dose escalation in Q4 2025. x Initiate the dose escalation cohorts in combination with cetuximab in advanced solid tumors, including CRC. □ Plan to initiate the dose escalation cohorts in combination with chemotherapy for PDAC and with chemotherapy for NSCLC in Q4 2025. □ Plan to report an interim safety and efficacy update in 1H 2026. □ Expect to select the RP2D and i nitiate monotherapy expansion cohorts in advanced PDAC, NSCLC and other KRAS G12D - mutated solid tumors in 1H 2026. □ Expect to select the RP2D and plan to initiate combination expansion cohorts in CRC, PDAC, and NSCLC in 1H 2026. □ Plan to engage with the FDA to discuss our development path forward , including potential registration - directed trials in PDAC and NSCLC in 1H 2026 . x Complete enrollment in the expansion cohort in Q3 2025. □ Expect to report an update on the safety and efficacy of the expansion cohort in 1H 2026. RAMP 205: Avutometinib + Defactinib + SOC in First - Line Metastatic Pancreatic Cancer □ Plan to share interim safety and efficacy results from both doublet and triplet combinations in Q 4 2025 . RAMP 203: Avutometinib ± Defactinib + Sotorasib: mKRAS G12C NSCLC VS - 7375, oral KRAS G12D (ON/OFF) Inhibitor 57 *Q3 2025 GAAP operating expenses of $51.96M less Q3 2025 stock - based compensation expense of $2.18M = $49.78M Q3 2025 non - GAAP operating expenses.

**Excludes unexercised warrants (9.0M shares upon exercise) and unexercised pre - funded warrants (9.8M shares upon exercise). Key Financial Statistics As of and for the quarter ended September 30, 2025 $137.7M Cash, cash equivalents & short - term investments $11.2M Net Product Revenue $52.0M GAAP Operating Expenses $49.8M * Non - GAAP Operating Expenses 66.7M ** Shares Outstanding Oberland Finance Credit Facility • Up to $150M available in a series of notes • $75M principal of notes outstanding • Remaining $75M available at Company’s option upon achievement of pre - defined milestones • $25M tranche upon FDA approval of avutometinib and defactinib for treatment of LGSOC • $50M tranche upon trailing six months revenue of at least $55M • Floating interest rate, subject to a floor and a cap • Interest only payments through January 2031 • No financial covenants 59 OB: Orange Book Listed Patent AVMAPKI FAKZYNJA CO - PACK Patent Portfolio OB: 7,897,792 Avutometinib COM (expiry 2/9/2027) OB: 11,400,090 – Avutometinib mono dosing - (expiry 10/29/2038) PTE OB: 7,928,109 Defactinib COM US (expiry 4/17/2028) PTA PTE (Aug, 2034) OB: 8,247,411 Defactinib – genus (expiry 4/17/2028) OB: 11,517,573 A+D combo dosing – (expiry 9/11/2040) OB: 11,873,296 Avuto polymorph – (expiry 12/29/2042) PTE (May 2039)

THANK YOU!
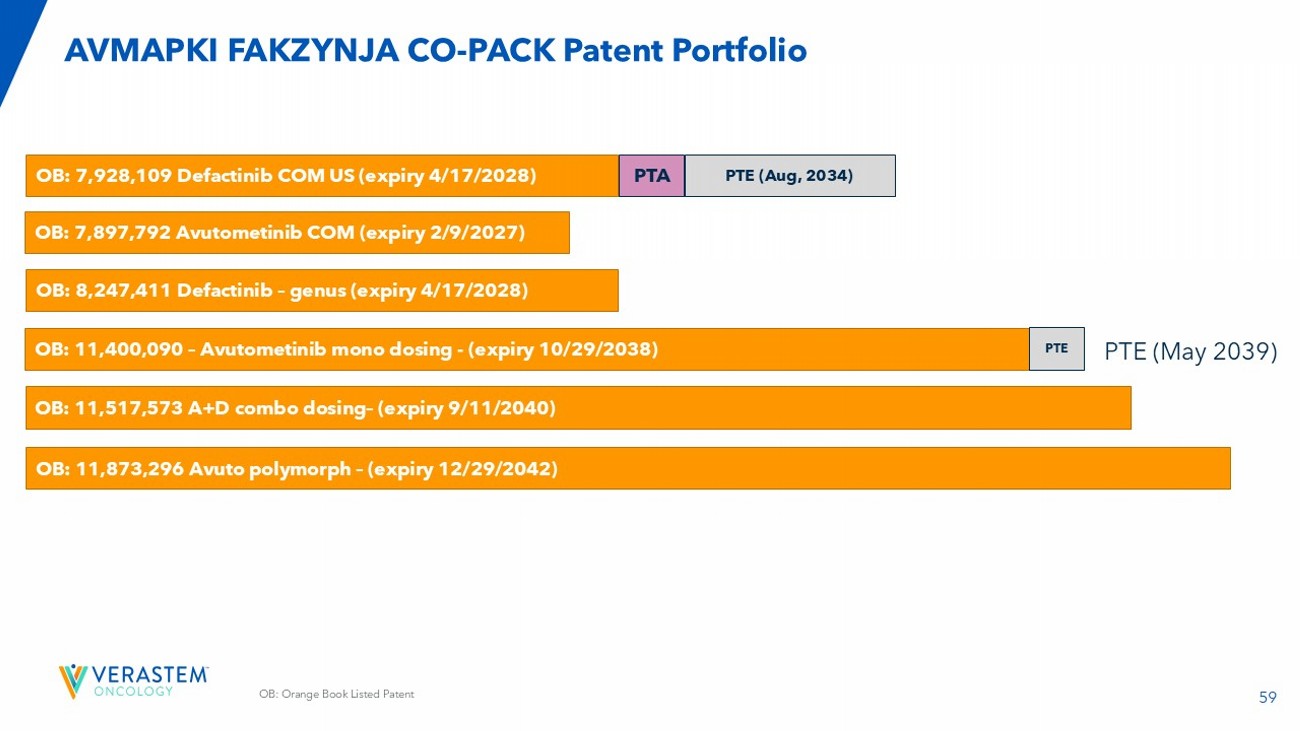

60 Experienced Senior Management Team Cathy Carew Chief Organizationa l Effectiveness Officer Colleen Mockbee Global Head of Regulatory Affairs and Development Nate Sanburn Chief Business Officer Dan Calkins Chief Financial Officer Matt Ros Chief Operating Officer Mike Crowther Chief Commercial & Strategy Officer Jonathan Pachter, Ph.D. Chief Scientific Officer John Hayslip, M.D. Chief Medical Officer Daniel Paterson President and CEO • Principal – HR Collaborative • Ironwood, ActiveBiotics , Dynogen , Tufts Health Plan • Chief Developmen t Officer & SVP of Regulatory, OncXerna • Head of Global Regulatory, Lilly Oncology • Associate VP, Head of Collaborations & Late Phase BD, Lilly Oncology • National Gene Vector Lab, Indiana Univer sity • Technical Accounting Consultant - CFGI • PwC LLP • CEO, FORE Biotherapeut ics • EVP & CSBO, Epizyme • COO, Sanofi - Genzyme • BMS • CBO, Minerva Biotechnolog ies • Interim US lead and VP of US Marketing, Kite Pharma • Celgene • Head of Cancer Biology – OSI (now Astellas) • Schering - Plough • CMO, I - MAB • Nektar Therapeutics , AbbVie • University of Kentucky’s Markey Cancer Center • CEO, The DNA Repair Co.

(now On - Q - ity ) • PharMetrics (now IMS) • Axion Previous experience 61 Oral Administration of VS - 7375 Inhibits Tumor Growth in a Dose - Dependent Manner in KRAS G12D models AsPC - 1 PDAC Panc 04.03 PDAC GP2D CRC LS513 CRC -100 -50 0 50 100 150 200 250 300 350 R e s p o n s e @ D a y 1 4 ( % c h a n g e f r o m b a s e l i n e ) v e h i c l e V S - 7 3 7 5 1 0 m g / k g V S - 7 3 7 5 3 0 m g / k g V S - 7 3 7 5 1 0 0 m g / k g 6 SDs 1 PR 5 SDs 8 PRs 8 PRs V S - 7 3 7 5 5 m g / k g -100 -50 0 50 100 150 200 250 300 350 R e s p o n s e @ D a y 1 4 ( % c h a n g e f r o m b a s e l i n e ) v e h i c l e V S - 7 3 7 5 1 0 m g / k g V S - 7 3 7 5 3 0 m g / k g V S - 7 3 7 5 1 0 0 m g / k g V S - 7 3 7 5 5 m g / k g 1 SD 4 SDs 5 PRs 3SDs 8 PRs -100 0 100 200 300 500 1000 1500 R e s p o n s e @ D a y 2 1 ( % c h a n g e f r o m b a s e l i n e ) v e h i c l e V S - 7 3 7 5 1 0 m g / k g V S - 7 3 7 5 3 0 m g / k g V S - 7 3 7 5 1 0 0 m g / k g 4 PRs 3 SDs 7 PRs 1 SD Presented at AACR 2024 • 10 mg/kg conferred strong tumor regressions in the most sensitive model (GP2D) • 30 mg/kg conferred strong tumor regressions across all models • 100 mg/kg conferred partial responses (>30% reduction) in >95% of all mice 62 Dose Levels of 400 and 600 mg QD Achieved Steady State Target Exposures in Almost all Patients Human equivalent to 100 mg/kg in mice (PR in all mice in all models) Human equivalent to 30 mg/kg in mice (Tumor regressions in all models) Human equivalent to 10 mg/kg in mice (Tumor regressions in most sensitive models)


63 Addition of Cetuximab with VS - 7375 Induces Complete Responses in All Mice in a KRAS G12D Colorectal Cancer Model LS513 Colorectal Cancer Model 0 10 20 30 40 0 500 1000 1500 Tumor growth Days after first dose T u m o r v o l u m e ( m m 3 + / - S E M ) Vehicle GFH375 Cetuximab GFH375 + cetuximab Last dose 0 500 1000 R e s p o n s e @ D a y 3 7 ( % c h a n g e f r o m b a s e l i n e ) v e h i c l e V S - 7 3 7 5 C e t u x i m a b V S - 7 3 7 5 + c e t u x i m a b 1 PR 1 SD 8 CRs -100 Presented at AACR 2025 64 NR: not reported; IP: intraperitoneal admiration; QD; once per day; QW; once per week; References: GenFleet /Verastem AACR 2024; Hallin et al (Mirati) 2022; RevMed AACR 2023; RevMed AACR 2024; Lilly ENA 2023; Lilly AACR 2024; AstraZeneca (AZ) AACR 2024; Incyte AACR 2024; Quanta AACR 2023; Tyligand AACR 2024; Betta AACR 2024; Zhou et al ( Hengrui ) 2024.
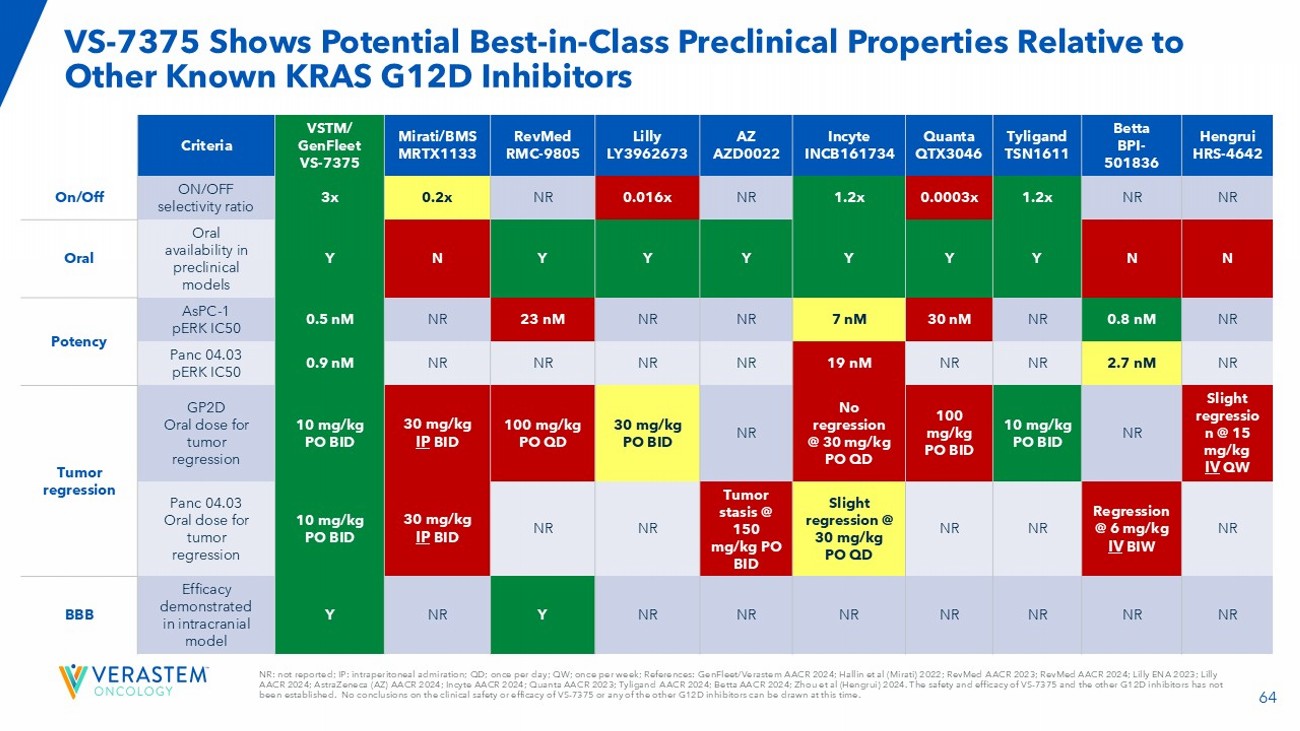
The safety and efficacy of VS - 7375 and the other G12D inhibitors has not been established. No conclusions on the clinical safety or efficacy of VS - 7375 or any of the other G12D inhibitors can be drawn at this time. VS - 7375 Shows Potential Best - in - Class Preclinical Properties Relative to Other Known KRAS G12D Inhibitors Hengrui HRS - 4642 Betta BPI - 501836 Tyligand TSN1611 Quanta QTX3046 Incyte INCB161734 AZ AZD0022 Lilly LY3962673 RevMed RMC - 9805 Mirati/BMS MRTX1133 VSTM/ GenFleet VS - 7375 Criteria NR NR 1.2x 0.0003x 1.2x NR 0.016x NR 0.2x 3x ON/OFF selectivity ratio On/Off N N Y Y Y Y Y Y N Y Oral availability in preclinical models Oral NR 0.8 nM NR 30 nM 7 nM NR NR 23 nM NR 0.5 nM AsPC - 1 pERK IC50 Potency NR 2.7 nM NR NR 19 nM NR NR NR NR 0.9 nM Panc 04.03 pERK IC50 Slight regressio n @ 15 mg/kg IV QW NR 10 mg/kg PO BID 100 mg/kg PO BID No regression @ 30 mg/kg PO QD NR 30 mg/kg PO BID 100 mg/kg PO QD 30 mg/kg IP BID 10 mg/kg PO BID GP2D Oral dose for tumor regression Tumor regression NR Regression @ 6 mg/kg IV BIW NR NR Slight regression @ 30 mg/kg PO QD Tumor stasis @ 150 mg/kg PO BID NR NR 30 mg/kg IP BID 10 mg/kg PO BID Panc 04.03 Oral dose for tumor regression NR NR NR NR NR NR NR Y NR Y Efficacy demonstrated in intracranial model BBB