UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
(Mark One)
☒ ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the fiscal year ended December 31, 2024
or
☐ TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the transition period from ______ to ______
Commission File Number 001-40133
ENVOY MEDICAL, INC.
(Exact name of registrant as specified in its charter)
| Delaware | 86-1369123 | |
| (State or other jurisdiction of incorporation or organization) | (I.R.S. Employer Identification No.) |
4875 White Bear Parkway, White Bear Lake, MN 55110
(Address of principal executive offices)
(877) 900-3277
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
| Title of each class | Trading Symbol(s) | Name of each exchange on which registered | ||
| Class A Common Stock, par value $0.0001 per share | COCH | The Nasdaq Stock Market LLC | ||
| Redeemable Warrants, each exercisable for one share of Class A Common Stock at an exercise price of $11.50 per share | COCHW | The Nasdaq Stock Market LLC |
Securities registered pursuant to Section 12(g) of the Act: None Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act.
Yes ☐ No ☒
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ☐ No ☒
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ☒ No ☐
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files). Yes ☒ No ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
| Large Accelerated Filer | ☐ | Accelerated Filer | ☐ |
| Non-accelerated Filer | ☒ | Smaller Reporting Company | ☒ |
| Emerging Growth Company | ☒ |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report. ☐
If securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant included in the filing reflect the correction of an error to previously issued financial statements. ☒
Indicate by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive-based compensation received by any of the registrant’s executive officers during the relevant recovery period pursuant to §240.10D-1(b). ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ☐ No ☒
The aggregate market value of the registrant’s Class A common stock, par value $0.0001 per share, held by non-affiliates of the registrant computed by reference to the last sales price of such stock, as of the last business day of the registrant’s most recently completed second fiscal quarter, which was June 30, 2024, was approximately $16.7 million. This calculation excludes shares of Class A common stock held by the registrant’s officers and directors and each person known by the registrant to beneficially own more than 5% of the registrant’s outstanding shares, as such persons may be deemed to be affiliates. This determination of affiliate status should not be deemed conclusive for any other purpose.
There were 21,326,619 shares of the registrant’s Class A common stock, par value $0.0001 per share, outstanding as of March 24, 2025.
DOCUMENTS INCORPORATED IN PART BY REFERENCE
Portions of the registrant’s definitive proxy statement relating to its 2025 Annual Meeting of Stockholders are incorporated by reference into Part III of this Annual Report on Form 10-K.
ENVOY MEDICAL, INC.
Annual Report on Form 10-K
For the Year Ended December 31, 2024
Table of Contents
CERTAIN TERMS
Unless otherwise stated in this Annual Report on Form 10-K (this “Report”), or the context otherwise requires, references to:
| ● | “Acclaim CI” means the Acclaim® fully implantable cochlear implant; |
| ● | “Anzu” means Anzu Special Acquisition Corp I, a Delaware corporation, which was renamed “Envoy Medical, Inc.” upon the closing of the Business Combination; |
| ● | “Anzu Class A Common Stock” means Anzu’s Class A common stock, par value $0.0001 per share, prior to the closing of the Business Combination; |
| ● | “Anzu Class B Common Stock” means Anzu’s Class B common stock, par value $0.0001 per share; |
| ● | “Board” means the board of directors of the Company; |
| ● | “Business Combination” means the merger and the other transactions contemplated by the Business Combination Agreement; |
| ● | “Business Combination Agreement” means the Business Combination Agreement, dated as of April 17, 2023, as amended by Amendment No. 1 to the Business Combination Agreement, dated May 12, 2023, and Amendment No. 2 to the Business Combination Agreement, dated August 31, 2023, by and among Anzu, Merger Sub and Legacy Envoy; |
| ● | “Bylaws” means the amended and restated bylaws of the Company; |
| ● | “Charter” means the second amended and restated certificate of incorporation of the Company; |
| ● | “Class A Common Stock” means the Company’s Class A common stock, par value $0.0001 per share; |
| ● | “Closing” means the closing of the Merger; |
| ● | “Esteem FI-AMEI” means the Esteem® fully implanted active middle ear implant (FI-AMEI); |
| ● | “Exchange Act” means the Securities Exchange Act of 1934, as amended; |
| ● | “Forward Purchase Agreement” means the Forward Purchase Agreement, dated April 17, 2023, as amended by Amendment No. 1 to the Forward Purchase Agreement, dated as of May 25, 2023, and Amendment No. 2 to the Forward Purchase Agreement, dated as of September 28, 2023, by and among Anzu, Legacy Envoy and the Meteora FPA Parties; |
| ● | “GAAP” means accounting principles generally accepted in the United States; |
| ● | “JOBS Act” means the Jumpstart Our Business Startups Act of 2012, as amended; |
| ● | “Legacy Envoy” means Envoy Medical Corporation, a Minnesota corporation, prior to the closing of the Business Combination; |
| ● | “Legacy Envoy Common Stock” means Legacy Envoy’s common stock, par value $0.01 per share; |
| ● | “Legacy Envoy Preferred Stock” means Legacy Envoy’s preferred stock, par value $0.01 per share; |
| ● | “Merger Sub” means Envoy Merger Sub, Inc., a Delaware corporation and a wholly owned subsidiary of Anzu; |
| ● | “Meteora FPA Parties” means Meteora Special Opportunity Fund I, LP, Meteora Capital Partners, LP, Meteora Select Trading Opportunities Master, LP and Meteora Strategic Capital, LLC; |
| ● | “Nasdaq” means The Nasdaq Capital Market; |
| ● | “Private Warrants” means warrants issued by Envoy Medical in 2024 in connection with debt financing transactions; |
| ● | “Public Warrants” means warrants issued by Anzu as part of its initial public offering; |
| ● | “Sarbanes-Oxley Act” means the Sarbanes-Oxley Act of 2002, as amended; |
| ● | “SEC” means the Securities and Exchange Commission; |
| ● | “Securities Act” means the Securities Act of 1933, as amended; |
| ● | “Series A Preferred Stock” means the Company’s Series A convertible preferred stock, par value $0.0001 per share; |
| ● | “Shortfall Warrants” means warrants issued to the Meteora FPA Parties for no additional consideration pursuant to the Forward Purchase Agreement; |
| ● | “Sponsor” means Anzu SPAC GP I LLC, a Delaware limited liability company and an affiliate of certain of Anzu’s officers and directors; |
| ● | “Subscription Agreement” means the subscription agreement, dated as of April 17, 2023, as amended by Amendment No. 1 to the Subscription Agreement, dated as of May 12, 2023, and Amendment No. 2 to the Subscription Agreement, dated as of August 23, 2023, by and between Anzu and the Sponsor; and |
| ● | “Warrants” means the Public Warrants, Shortfall Warrants, and Private Warrants. |
Additionally, references in this Report to the “Company,” the “registrant,” “Envoy Medical,” “we,” “us” and “our” in this Report refer to Envoy Medical, Inc. (formerly known as Anzu Special Acquisition Corp I), and references to our “management” or our “management team” refer to our officers and directors, other than certain historical information which refers to Legacy Envoy prior to the consummation of the Business Combination.
CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS
This Report contains certain “forward-looking statements” within the meaning of the United States Private Securities Litigation Reform Act of 1995, Section 27A of the Securities Act and Section 21E of the Exchange Act. All statements other than statements of historical fact contained in this Report, including statements as to future results of operations and financial position, revenue and other metrics, products, business strategy and plans, objectives of management for future operations of the Company, market size and growth, competitive position and technological and market trends, are forward-looking statements. The words “anticipate,” “believe,” “continue,” “could,” “estimate,” “expect,” “intends,” “may,” “might,” “plan,” “possible,” “potential,” “predict,” “project,” “should,” “will,” “would” and similar expressions may identify forward-looking statements, but the absence of these words does not mean that a statement is not forward-looking. All forward-looking statements are subject to risks, uncertainties, and other factors which could cause actual results to differ materially from those expressed or implied by such forward-looking statements. These risks and uncertainties include, but are not limited to:
| ● | Unpredictability in the medical device industry, the regulatory process to approve medical devices, and the clinical development process of the Company’s products; |
| ● | Potential need to make design changes to products to meet desired safety and efficacy endpoints; |
| ● | Changes in federal or state reimbursement policies that would adversely affect sales of the Company’s products; |
| ● | Introduction of other scientific advancements, including gene therapy or pharmaceuticals, that may impact the need for hearing devices such as cochlear implants or fully implanted active middle ear implants; |
| ● | Competition in the medical device industry, and the failure to introduce new products and services in a timely manner or at competitive prices to compete successfully against competitors; |
| ● | Disruptions in relationships with the Company’s suppliers, or disruptions in the Company’s own production capabilities for some of the key components and materials of its products; |
| ● | Changes in the need for capital and the availability of financing and capital to fund these needs; |
| ● | Changes in interest rates or rates of inflation; |
| ● | Legal, regulatory and other proceedings could be costly and time-consuming to defend; |
| ● | Changes in applicable laws or regulations, or the application thereof on the Company; |
| ● | A loss of any of the Company’s key intellectual property rights or failure to adequately protect intellectual property rights; |
| ● | The Company’s ability to maintain the listing of its securities on Nasdaq following the Business Combination; |
| ● | The effects of catastrophic events, including war, terrorism and other international conflicts; and |
| ● | Other risks and uncertainties indicated in this Report, including those set forth under the section entitled “Risk Factors.” |
Should one or more of these risks or uncertainties materialize, or should any of the underlying assumptions prove incorrect, actual results may vary in material respects from those expressed or implied by these forward-looking statements. Nothing in this Report should be regarded as a representation by any person that the forward-looking statements set forth herein will be achieved or that any of the contemplated results of such forward-looking statements will be achieved. You should not place undue reliance on these forward-looking statements. The Company does not give any assurance that it will achieve its expected results and does not undertake any duty to update these forward-looking statements, except as required by law.
Summary Risk Factors
Our Company is subject to numerous risks described in Item 1A. Risk Factors and elsewhere in this Report. You should carefully consider these risks before making an investment. Some of these risks relating to our business objectives, our organization and structure and our securities include:
| ● | We are an early-stage company with a history of losses. We have not been profitable historically and may not be able to achieve profitability in the future. |
| ● | We have generated limited revenue from product sales and may never be profitable. |
| ● | If the Acclaim CI contains design or manufacturing defects, our business and financial results could be harmed. |
| ● | We expect that we will need to raise substantial additional funding, which may not be available on acceptable terms, or at all. Failure to obtain funding on acceptable terms and on a timely basis may require us to curtail, delay or discontinue our product development efforts or other operations. |
| ● | Raising additional capital would cause dilution to our existing stockholders and may adversely affect the rights of existing stockholders. |
| ● | Failure of a key information technology system, process or site could have an adverse effect on our business. |
| ● | We have identified material weaknesses in our internal control over financial reporting. If we are unable to remediate these material weaknesses, or if we identify additional material weaknesses in the future or otherwise fail to maintain an effective system of internal control over financial reporting, we may not be able to accurately or timely report our financial condition or results of operations, which may adversely affect investor confidence in us and the value of our stock. |
| ● | Our financial statements contain an explanatory paragraph regarding substantial doubt about our ability to continue as a going concern, which could prevent us from obtaining new financing on reasonable terms or at all. |
| ● | Clinical failure can occur at any stage of clinical development. Our clinical experience to date does not necessarily predict future results and may not have revealed certain potential limitations of the technology or potential complications from the Acclaim CI and may require further clinical validation. Any product version we advance through clinical trials may not have favorable results in later clinical trials or receive regulatory approval. |
| ● | The successful commercialization of the Acclaim CI, if it receives FDA approval, will depend in part on the extent to which governmental authorities and health insurers establish coverage, adequate reimbursement levels and favorable pricing policies. Failure to obtain or maintain coverage and adequate reimbursement for our product candidates could limit our ability to market those products and decrease our ability to generate revenue. |
| ● | We operate in a very competitive business environment, and if we are unable to compete successfully against our existing or potential competitors, our business, financial condition and results of operations may be adversely affected. |
| ● | We expect to derive most of our revenues from sales of the Acclaim CI. Our inability to successfully commercialize this product, or any subsequent decline in demand for this product, could severely harm our ability to generate revenues. |
| ● | If healthcare professionals do not recommend the Acclaim CI to their patients, the Acclaim CI may not achieve market acceptance and we may not become profitable. |
| ● | We will be dependent upon contract manufacturing organizations and material suppliers, making us vulnerable to supply shortages and problems, increased costs and quality or compliance issues, any of which could harm our business. |
| ● | Our business plan relies on certain assumptions about the market for our product; however, the size and expected growth of our addressable market has not been established with precision and may be smaller than we estimate, and even if the addressable market is as large as we estimate, we may not be able to capture market share. |
| ● | We depend on third parties to manage our pre-clinical studies and clinical trials, perform related data collection and analysis, and to enroll patients for our clinical trials, and, as a result, we may face costs and delays that are beyond our control. |
| ● | We are highly dependent on key members of our executive management team. Our inability to retain these individuals could impede our business plan and growth strategies, which could have a negative impact on our business and the value of your investment. |
| ● | The market price of our Class A Common Stock and Public Warrants has been and may continue to be extremely volatile, which could cause purchasers of our securities to incur substantial losses. |
| ● | While we will pay dividends on shares of Series A Preferred Stock pursuant to the Certificate of Designation, we do not intend to pay dividends on shares of Class A Common Stock for the foreseeable future. |
| ● | We have been and in the future may become a defendant in one or more stockholder derivative, class-action and other litigation, and any such lawsuits may adversely affect our business, financial condition, results of operations and cash flows. |
PART I
ITEM 1. Business
Overview
We are a hearing health company focused on providing innovative medical technologies across the hearing loss spectrum. Our technologies are designed to shift the paradigm within the hearing industry and bring both providers and patients the hearing devices they desire. We are dedicated to pushing beyond the status quo to provide patients with improved access, usability, independence, and quality of life. We were founded in 1995 to create a fully implanted hearing device that leveraged the natural ear - not an artificial microphone - to pick up sound. The ear itself is an ideal way to capture sound from our environment.
To leverage the natural ear’s benefits, an implanted sensor was created to pick up incoming sound energy from the ossicular chain (i.e., the three tiny hearing bones that connect the eardrum to the cochlea). The sensor absorbs the mechanical energy from ossicular chain and turns it into a signal that can be processed, improved, and increased for a patient’s particular hearing needs.
Our first product, the Esteem Fully Implanted Active Middle Ear Implant (“Esteem FI-AMEI”), received FDA approval in 2010. The Esteem FI-AMEI remains the only FDA approved fully implanted active hearing device on the market. The Esteem FI-AMEI failed to gain commercial traction, primarily because the Centers for Medicaid and Medicare Services (“CMS”) classified it as a hearing aid and therefore not eligible for coverage. At an average total price (i.e., device and surgery) of over $25,000, very few individuals were willing or able to pay out-of-pocket for the Esteem FI-AMEI. We believe hearing aid classification is improper for the Esteem FI-AMEI and we continue to work towards having the Esteem FI-AMEI properly classified as a Fully Implanted Active Middle Ear Implant.
Despite the commercial challenges of the Esteem FI-AMEI, roughly 1,000 devices were implanted globally. Some devices were implanted in the early 2000s during clinical trials, providing us with nearly two decades of experience with its implantable sensor technology. Throughout our experience, our sensor technology proved a viable alternative to external or implanted microphones.
In late 2015, we made the decision to shift our focus from the Esteem FI-AMEI to a new product that would leverage our sensor technology and incorporate it into a cochlear implant. As a result, we have developed the investigational fully implanted Acclaim CI. We now believe we have the possibility to disrupt the cochlear implant market currently dominated by a small number of incumbents.
Business Combination
In September 2023, we completed the Business Combination pursuant to the Business Combination Agreement between Anzu and Legacy Envoy. As contemplated by the Business Combination Agreement: (a) each share of Legacy Envoy Preferred Stock issued and outstanding immediately prior to the Closing was converted into shares of Legacy Envoy Common Stock; (b) each share of Merger Sub Common Stock issued and outstanding immediately prior to the Closing was converted into and exchanged for one share of Legacy Envoy Common Stock; (c) each outstanding option to purchase shares of Legacy Envoy Common Stock outstanding as of immediately prior to the Closing was cancelled in exchange for nominal consideration; (d) each outstanding warrant to purchase shares of Legacy Envoy Common Stock outstanding as of immediately prior to the Closing automatically, depending on the applicable exercise price, was cancelled or exercised on a net exercise basis and converted into shares of Legacy Envoy Common Stock in accordance with its terms; (e) each outstanding Legacy Envoy convertible promissory note was automatically converted into shares of Legacy Envoy Common Stock in accordance with its terms; (f) each share of Legacy Envoy Common Stock issued and outstanding immediately prior to the Closing was cancelled and converted into the right to receive a number of shares of our Class A Common Stock equal to the Exchange Ratio; (g) the Sponsor forfeited 5,510,000 shares of Anzu Class B Common Stock and all 12,500,000 private warrants pursuant to the Sponsor Support Agreement; (h) the Sponsor exchanged 2,500,000 shares of Anzu Class B Common Stock for 2,500,000 shares of our Series A Preferred Stock; (i) an aggregate of 2,615,000 shares of Anzu Class B Common Stock held by the Sponsor and Anzu’s former independent directors automatically converted into our Class A Common Stock; (j) the Sponsor transferred an aggregate of 490,000 shares of our Class A Common Stock to the Legacy Forward Purchasers and the Extension Support Parties pursuant to the Side Letter Agreements and Extension Support Agreements, respectively; and (k) the Company issued an aggregate of 8,512 shares of Class A Common Stock to the Meteora FPA Parties pursuant to the Forward Purchase Agreement.
As of the open of trading on October 2, 2023, the Class A Common Stock and Public Warrants of the Company, formerly those of Anzu, began trading on Nasdaq as “COCH” and “COCHW,” respectively.
Our Product
Cochlear Implants - Fully Implanted vs. Partially Implanted
The cochlea converts vibrations from the ossicular chain into nerve signals that are transmitted through the auditory nerve for processing by the brain. Cochlear implants use electronic signals to stimulate the auditory nerve.
Partially implanted cochlear implants have two main components: a large external component that sits on or behind the patient’s ear and a surgically implanted internal component. The external component contains a microphone, sound processer, and batteries. A magnetic coil on the external component lines up with an internal magnetic coil in the internal component. The signal from the external component is transferred to the internal coil where it is delivered to the electrode array, which is implanted in the cochlea, to electrically stimulate the cochlea.
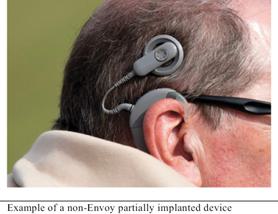 |
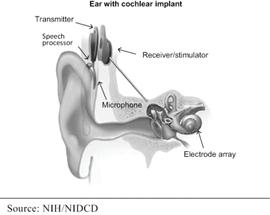 |
The Acclaim CI is fully implanted and does not have the need for any external component to be worn on the ear. Unlike partially implanted devices, the fully implanted Acclaim CI uses the ear to capture sound via a piezoelectric sensor that is implanted in the middle ear. The sound processor and power source are also implanted.

|
||
| CAUTION: Investigational Device – Limited by Federal (or United States) Law to Investigational Use. | ||
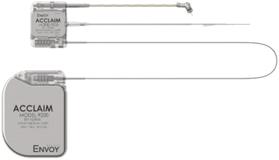 |
Acclaim CI - A Breakthrough Device
The fully implanted Acclaim CI received the Breakthrough Device Designation from the U.S. Food and Drug Administration (FDA) in 2019. However, the process of medical device development is inherently uncertain and there is no guarantee that this designation will accelerate the timeline for approval or make it more likely that the Acclaim CI will be approved.
Hearing loss is currently an irreversible and debilitating human condition. Significant hearing loss is correlated with increased anxiety, depression, social isolation, falls, and other costly health issues. An article published in the journal Acta Otorhinolaryngol Italica in June 2016 suggests that untreated or undertreated hearing loss correlates with earlier loss of cognitive function and poorer cardiovascular health.2 While some solutions for hearing loss already exist (e.g., hearing aids, traditional cochlear implants) these have inherent limitations in being fully or partially external, which may limit patients in initial time to adoption, hours of use during the day (inherent compliance restrictions), lifestyle, or quality of life.
We believe that the Acclaim CI will be able to offer hearing benefit over the patient’s baseline condition and may also offer other important advantages over alternative hearing loss treatments, such as:
| ● | Increased daily usage. We believe that the fully implanted nature of the Acclaim CI may facilitate an increase in daily usage over other types of cochlear implants because the device can be used 24-hours a day. |
| ● | Hearing at night. Unlike other types of available cochlear implants, the Acclaim CI can be used at night. This capability may support audibility of alarms, sirens, telephones, and other people for an added sense of security while they sleep. |
| ● | Hearing in and around water. Patients using the Acclaim CI will not need to worry about removing their device when showering, at the beach, or swimming laps. They will also not need to worry about damaging the device if caught in the rain. |
| ● | Hearing in active situations. A patient using the Acclaim CI will not need to worry about the external processor falling off during exercise or other physical activities. The patient will not need to preemptively remove the device prior to engaging in these types of activities, thus retaining audibility of the surrounding environment. |
| 2 | Source: Fortunato S, et al.; A Review of New Insights on the Association Between Hearing Loss and Cognitive Decline in Ageing; Acta Otorhinolaryngologica Italica (Jun 2016), finding that increasing evidence has linked age related hearing loss to more rapid progression of cognitive decline and incidental dementia and that many aspects of daily living of elderly people have been associated to hearing abilities, showing that hearing loss affects the quality of life, social relationships, motor skills, psychological aspects and function and morphology in specific brain areas. |
| ● | Lowered battery maintenance. Other cochlear implants require near-daily battery replacement or battery charging. In addition to the logistical hassle of worrying about keeping the batteries charged, this can be challenging for patients who have issues with dexterity or neuropathy, as the batteries and components are small and can be hard to handle. The Acclaim CI is designed with a battery contained within the implanted system components intended to be charged wirelessly through the skin. The Acclaim CI battery is expected to last for several days between charges and will not require the patient to use or handle small components like current cochlear implant systems do. |
| ● | No need for backup or secondary processors. Many patients who have partially implanted cochlear implants with external hardware desire or need a backup processor. The backup processor provides the patient with a sense of security because they know if their primary processor is lost or damaged, they will be left without hearing for a period of time while they wait for a replacement. In addition, lost or damaged components can be expensive to replace, with the cost of replacement often not covered by insurance. The Acclaim CI processor is implanted and therefore not susceptible to damage, discomfort or issues associated with moisture, germs, dirt, or other external causes of loss or physical damage due to having an externally worn processor. |
| ● | Use of equipment and accessories. The externally worn components of currently available cochlear implants can make wearing equipment or accessories difficult for existing cochlear implant patients. For example, wearing helmets, hats, headphones, stethoscopes, or other accessories can interfere with the placement of the external components and cause “coil offs” or prevent the patient from using the device altogether. |
| ● | Earlier adoption of cochlear implant technology from reduced stigma. For many potential users of hearing instruments like hearing aids and cochlear implants, the perception of stigma associated with those technologies can prevent or delay the adoption of the technology. We believe that the Acclaim CI, with no externally worn components, may help reduce or perhaps even eliminate such stigma. We believe we can increase penetration rates for adult cochlear implants in the U.S. |
| ● | Potential to significantly reduce overall costs while improving net healthcare outcomes. We believe a fully implanted cochlear implant could reduce cochlear implant costs over time by eliminating costly external components that are frequently replaced at the expense of the patient, the insurer, Medicare, or other third-party payor. There is also reason to believe that increasing compliance and use of cochlear implants, reducing time to adoption for candidates, and helping to support safety and security by providing the ability for true all-day hearing may improve the net healthcare outcome for society over time. |
The Acclaim CI is implanted by a surgeon through a procedure that we believe will average around two and a half to three hours under general anesthesia. We expect that patients may experience mild to moderate discomfort after the procedure. A four to eight week waiting period is required before the Acclaim CI can be activated to allow the middle ear to heal and fluid from surgery to dissipate. It is expected that the Acclaim CI battery pack will be replaced every 8-12 years via a less invasive surgical procedure that only replaces the Acclaim CI battery pack in the pectoral region (i.e., the whole system does not need to be replaced, just the Acclaim CI battery pack).
All of the competitive advantages referred to above require that the Acclaim CI obtain FDA approval in its current form and substantially on our planned timeline. If FDA approval is materially delayed for any reason, it is possible that competitors will offer products with similar features before we are able to market the Acclaim CI.
Market Overview
Overview of Hearing Loss
According to the National Center for Health Statistics, hearing loss impacts about 15% of the adult population in the United States.3 Among older adults, nearly 25% of people aged 65 to 74 have disabling hearing loss, and 50% of those aged 75 and older have disabling hearing loss, according to the National Institute on Deafness and Other Communications Disorders.4 Organizations such as the Centers for Disease Control and Prevention (CDC) and the World Health Organization (WHO) have recognized significant hearing loss as one of the most common disabilities impacting people around the world.5 The WHO estimates economic impact of untreated or undertreated hearing loss is approximately $750 billion each year.6
In common parlance, the terms “hearing loss,” “hard of hearing,” or “deafness” are often used to describe a variety of types, levels, and causes of hearing loss that are treated differently clinically. The hearing loss market can be classified based on causes and severity of hearing loss.
There are three main types of hearing loss: sensorineural, conductive, and mixed. Sensorineural hearing loss is due to problems of the inner ear and is often caused by damage to “hearing hair cells” in the cochlea. Common causes include normal aging, excessive noise exposure, viral infections, and exposure to drugs that are toxic to the hearing system. According to data published in the Journal of the American Medical Association, sensorineural hearing loss is the most common form of hearing loss, representing approximately 90% of all hearing loss.7
Conductive hearing loss is due to mechanical or structural problems with a part of the hearing system, generally a result of congenital issues with or damage to the ear canal, ear drum, or ossicular chain. Common causes include malformation of a particular part of the hearing system, middle ear infection, perforation of the eardrum, wax buildup, or dislocation of the ossicles. Conductive hearing loss represents approximately 10% of all hearing loss, according to data published in the Journal of the American Medical Association.8 Finally, mixed hearing loss has some combination of both sensorineural and conductive components.
In addition to the three main types of hearing loss, there are generally five levels of hearing loss severity: normal, mild, moderate, severe, and profound. Normal hearing is often defined as 0-20 decibels (dB) of hearing loss and even with a slight loss most people do not notice any impact. Mild hearing loss is often defined as 20-40 dB of hearing loss with some people reporting difficulty hearing soft spoken people. Most people with mild hearing loss do not address their hearing loss.
As hearing loss progresses, the impact on the individual becomes more noticeable. Moderate hearing loss is often defined as 40-70 dB of hearing loss and begins to show up with people reporting the ability to “hear but not understand” speech. More words are missed in conversations, and it is harder to hear in certain environments.
Severe hearing loss is often defined as 70-90 dB of hearing loss. People with severe hearing loss are unable to hear most speech and miss large portions of conversations without assistance. People with severe hearing loss may find that even with hearing aids they are not getting enough benefit to hear and understand most of the words in a conversation.
Profound hearing loss is often defined as 90 dB or more of hearing loss. People with profound hearing loss cannot hear speech or loud sounds such as sirens or horns. Most people who are considered clinically “deaf” would have severe to profound hearing loss.
| 3 | Source: National Health Interview Survey; Center For Disease Control And Prevention: National Center For Health Statistics (2022), finding that as of 2022 15.5% of US adults reported some level of difficulty hearing. |
| 4 | Source: Quick Statistics About Hearing; National Institute Of Health; National Institute On Deafness And Other Communications Disorders (https://www.nidcd.nih.gov/health/statistics/quick-statistics-hearing), summarizing statistics on hearing loss, including that 25% of people aged 65 to 74 have disabling hearing loss, and 50% of those aged 75 and older have disabling hearing loss. |
| 5 | Source: Preventing Noise-Induced Hearing Loss; Center For Disease Control And Prevention (2022); and Deafness and Hearing Loss, World Health Organization (2023), each providing an overview of the prevalence of hearing loss. |
| 6 | Source: Global Costs of Unaddressed Hearing Loss and Cost-Effectiveness of Intervention; World Health Organization (2017), providing an overview of the global costs of hearing loss, including components of cost and the monetary values attributable to such elements as costs typically incurred by health-care systems and patients, respectively, and reaching the conclusion that the cost of untreated or undertreated hearing loss is approximately $750 billion each year. |
| 7 | Source: Yueh B, et al.; Screening and Management of Adult Hearing Loss in Primary Care: Scientific Review; Journal Of The American Medical Association (2003), providing an epidemiology of types of hearing loss and identifying sensorineural hearing loss as the cause of 90% of hearing loss. |
| 8 | Source: Yueh B, et al.; Screening and Management of Adult Hearing Loss in Primary Care: Scientific Review; Journal Of The American Medical Association (2003), providing an epidemiology of hearing loss, including the allocation of hearing loss between sensorineural hearing loss and other types. |
Overview of Hearing Devices
There are several different types of hearing devices to address hearing loss. It is common for hearing loss to progress – continue to get worse – over the course of an individual’s life, so it is possible that a patient may have one or more hearing devices during the course of their lives.
Personal Sound Amplification Devices (PSAPs) are small electronic devices used to make sounds louder but with little sophistication. They are limited in ability and are only suitable for normal to mild hearing loss.
Hearing aids are the most common form of hearing device. These are small sound-amplifying devices that come in a variety of shapes and sizes. They are always external and pick up sound through a microphone and amplify the sound through a speaker in the ear canal. There are over-the-counter hearing aids (no prescription required) designed to treat mild to moderate hearing loss and prescription hearing aids designed to treat more significant hearing loss. Hearing aids can be used for all types of hearing loss and are typically the first device a person with hearing loss will try.
Active middle ear implants are implanted fully or partially in the middle ear (i.e., where the three ossicles or hearing bones are located). They are typically designed to treat moderate to severe sensorineural hearing loss, but some also can address a certain level of mixed hearing loss. Middle ear implants use mechanical energy to directly drive the cochlea with mechanical energy. Middle ear implants are not common due to the lack of reimbursement coverage throughout the world. The Esteem FI-AMEI is the only fully implanted active middle ear device currently with FDA approval and commercially available in the United States.
Cochlear implants are electrical hearing devices. They deliver electrical stimulation to the cochlea via an electrode array. The electrical stimulation is picked up by the hearing nerve and patients are able to perceive sound. Traditionally, all cochlear implants were partially implanted with an external component. We believe the fully implanted Acclaim CI will be the first-of-a-kind cochlear implant with no external component worn on the ear or required for daily hearing and that leverages the ear to pick up sound (i.e., versus a microphone).
Auditory osseointegrated implants (bone conduction implants) are used for conductive or certain types of mixed hearing loss. They are not used for sensorineural hearing loss. They address a patient’s conductive hearing loss by transferring sound information through the patient’s skull via vibration.
Acclaim CI’s Market Opportunity
The Acclaim CI is designed to address severe to profound sensorineural hearing loss that is not adequately addressed by hearing aids. We anticipate that the Acclaim CI will only be indicated for adults who have been deemed adequate candidates by a qualified physician.
We believe there is a significant population of adults in the United States who are cochlear implant candidates but choose not to get the therapy because of the external component required for daily hearing. We believe this is one of the main reasons why industry sources, such as a 2018 paper published in the journal Trends in Hearing, and our own market research estimate 5-8% penetration rate for cochlear implants in the adult population.9
| 9 | Sources: Holder JT, et al., Current Profile of Adults Presenting for Preoperative Cochlear Implant Evaluation; Trends In Hearing (2018), providing an analysis of implantation rates of cochlear implants among adults receiving preoperative screening, including a determination that “the market penetration for cochlear implantation was just 7.7% in the adult population of individuals with severe-to-profound sensory hearing loss.” We have also commissioned market research by S2N Health, which analyzed available literature and estimates from other market participants to reach the 5 - 8% penetration rate, based in part on an expansion of candidacy criteria since the publication of the Holder article. As an example of the effect of changing candidacy criteria, Nassiri AM, et al., determined penetration rates to be 12.1% based on the prior more restrictive criteria and 2.1% based on the current, broader criteria. Current Estimates of Cochlear Implant Utilization in the United States, Otol Neurotol (June 2022). |
Based on published literature and industry sources (prior to candidacy expansion for cochlear implant candidates), including the American Journal of Public Health, we believe there are approximately 6.6 million Americans age 12 or older with severe to profound hearing loss in at least one ear.10 Incorporating estimates for clinical indications (including limited benefit from hearing aids), we believe there are approximately 2.8 million adults in the United States who could qualify for a cochlear implant. Based on an assumed selling price in the United States for a traditional cochlear implant of $30,000 (a $5,000 premium over the average sale price of current partially-implanted devices), we believe the adult cochlear implant market in the United States alone represents a potential market opportunity of over $80 billion.
Based on the published literature and industry sources previously referenced, we believe there will be roughly 25,000 - 30,000 adults implanted with a cochlear implant in the United States every year by 2026. Based on an assumed selling price of $30,000, that is an annual market opportunity that exceeds $750 million for just the United States adult population.
In addition, many estimates from published literature and industry sources were made prior to changing candidacy within the cochlear implant market. Two major shifts in clinical candidacy have likely increased the market sizes: (a) the CMS has expanded coverage from 40% word recognition scores to 60% word recognition scores and (b) there is more acceptance of treating single sided deafness with a cochlear implant.
While these numbers represent the entire adult cochlear implant market in the United States, we believe that if we are able to establish distribution channels and strategic relationships with clinics and healthcare professionals the Acclaim CI will be in a unique position to capture existing market share quickly and to also capture a healthy portion of the unserved market - those who are not pursuing a cochlear implant because of the external components. Moreover, it is reasonable to believe that Acclaim CI will demand a higher average selling price than existing partially implanted cochlear implants.
We also believe there are substantial total market and annual market opportunities outside the United States. Currently, our analysis estimates that approximately 50% of the hearing device market is international. Given the greater number of hearing loss patients outside the United States, we also believe the international market is currently significantly underserved and offers significant opportunity for expansion if we are able to obtain the necessary regulatory approvals and expand our international distribution capabilities. However, we will be unable to expand into international markets if we are unable to obtain these regulatory approvals.
Market Competition
There are currently three major cochlear implant manufacturers - Cochlear Ltd., Advanced Bionics (Sonova), and Med-El. There are a few other minor regional players, such as Nurotron in China, which appears to be focused on developing countries.
Cochlear Ltd. (ASX: COH) is the leading cochlear implant device manufacturer with approximately 60% of global market share and a market capitalization of approximately $12 billion (US Dollars) as of December 31, 2024.
In comparison to Envoy Medical, the three current primary providers of cochlear implants have a greater penetration into the hearing loss treatment market, which has allowed them to develop relationships with audiologists, otolaryngologists (ENT physicians), hearing loss centers, and the other physicians on whom providers rely for referrals. The current providers also have existing relationships with patients who have used their devices. In addition, current providers also have substantially greater financial and operational resources, which may give them an advantage in capitalizing on new technology and responding to other changes to the marketplace.
| 10 | Source: Goman, AM and Frank RL, Prevalence of Hearing Loss by Severity in the United States, AMERICAN JOURNAL OF PUBLIC HEALTH (Oct 2016), estimating that 6.6 million (2.5%) of Americans aged 12 years or older have severe to profound hearing loss in at least one ear, with three quarters of these individuals being older than 60 years. We do not plan to market the Acclaim CI to patients under age 18. |
If we are able to obtain regulatory approval of the Acclaim CI, we believe physicians and patients may be receptive to it being a fully implanted cochlear implant. However, based on our lack of history in the market, we will need to make material investments in patient advertising, provider education and training, distribution capabilities, and physician strategic relationships to capitalize on such advantages and gain market share. We will be unable to begin investing in these areas until we obtain FDA approval.
Market Trends
The first documented cochlear implant was completed in 1961. The initial devices were crude single electrode cochlear implants with the intended purpose of giving some basic environmental and situational awareness to adults with profound hearing loss. A few years later, multi-channel devices were introduced. Over time, multi-channel devices evolved more quickly and allowed for more robust processing and mapping strategies. By the 1980s, cochlear implants were an accepted standard of care for adults with profound hearing loss with the multi-channel devices becoming the preferred design by most healthcare professionals.
The next two to three decades focused on the evolution of multi-channel electrodes and creating new sound processing and electrode mapping techniques to focus on speech understanding. As a result, most cochlear implant patients can understand speech quite well with the appropriate follow-up and speech therapy. Candidacy was expanded to include children and people with different levels or types of hearing loss.
Over the last few years, the trends of the cochlear implant industry have mirrored that of the hearing aid industry, with less emphasis on hardware design and more placed on appearance and usability. The physical form and function have not changed significantly, although new sound processing strategies have been implemented to improve patient outcomes. While product reliability has gradually improved, clinical efficacy seems to have plateaued.
To increase market share, manufacturers have focused on making cochlear implants more visibly appealing (e.g., slightly smaller external components, color “kits” for the external components), user friendly (e.g., connectivity), environmentally robust (e.g., water resistance), and more reliable (e.g., fewer recalls).
We believe that the trend over the next decade will be a continuation of the focus on usability, connectivity, lifestyle, and miniaturization. Artificial Intelligence and Machine Learning may also come into play as those technologies evolve. As cochlear implants become more accepted as a therapy for individuals with moderate to profound sensorineural hearing loss, manufacturers will pay attention to ways of making patients interested in their device over a similarly performing competing device.
Another major trend within the industry is a loosening of the clinical candidacy requirements. In addition to people with “better” hearing levels being considered for cochlear implants (e.g., people with moderate hearing in the lower frequencies) there has also been a movement to implant people with “single sided deafness” (“SSD”). Both Med El (in 2019) and Cochlear (in 2021) achieved FDA approval for treatment of those with SSD and asymmetric hearing loss. As a result, more patients are eligible for cochlear implants than ever before.
Finally, industry participants have made material investments to inform more adult candidates about cochlear implants to increase usage. Currently, industry sources, including a 2018 paper published in the journal Trends in Hearing,11 and our own market research estimate that less than 10% of adults who meet the indications for cochlear implant candidacy are implanted, leaving more than 90% of the current adult market as untapped potential for new technologies. However, we will require FDA approval for the Acclaim CI and significant investment in our training and distribution network before we can access such market.
Reimbursement Strategy
Cochlear implants enjoy a fully developed reimbursement pathway. Cochlear implants have been deemed a coverable benefit by CMS and enjoy an existing National Coverage Determination (“NCD”). In the United States, many private and public payors cover at least one cochlear implant per adult. There is existing coding, coverage, and payment for cochlear implants.
| 11 | Source: Holder JT, et al., Current Profile of Adults Presenting for Preoperative Cochlear Implant Evaluation; TRENDS IN HEARING (2018). |
Unlike the Esteem FI-AMEI, which was classified as a hearing aid by CMS and therefore statutorily excluded from being a coverable benefit under Medicare and Medicaid, the Acclaim CI is expected to be eligible for Medicare and Medicaid coverage as a cochlear implant.
As mentioned above, the Acclaim CI received Breakthrough Device Designation. There are potential reimbursement-related benefits to the designation (i.e., the ability to receive higher reimbursements than are received by incumbent devices); however, the implementation of these benefits has not been finalized by Congress and CMS and there is no guarantee that Breakthrough Device Designation will offer any benefit with respect to reimbursement.
Timeline to Commercialization of Acclaim CI
In the United States, before we can market a new Class III medical device, which the Acclaim CI is, we must first receive FDA approval via the premarket application (“PMA”) approval process. We currently anticipate obtaining FDA approval in late 2027 or early 2028, although the process of obtaining FDA approval is uncertain, and we may not obtain approval on that timeline or at all.
A large component of our PMA will be a successful pivotal clinical study. In order to begin a pivotal clinical study, you must have an Investigational Device Designation (“IDE”) approved by the FDA. We received approval for our IDE on October 31, 2024. However, FDA approved our IDE based on a staged clinical study that will require approval from the FDA to move from the first stage to the second stage.
The objective of this pivotal clinical study is to demonstrate the safety and efficacy of the fully implanted Acclaim cochlear implant for the treatment of severe to profound sensorineural hearing loss and is designed as a prospective, multicenter, non-randomized, open label clinical trial to evaluate the safety and efficacy of the Acclaim CI. The pivotal clinical study protocol currently requires 56 total patients enrolled and followed for 12 months. The first stage will have 10 patients enrolled. We will then provide a summary of effectiveness outcomes for these 10 patients to the FDA and request approval from the FDA to proceed to the second stage to implant the remaining 46 patients. There is no guarantee that the FDA will approve expansion to the second stage or that eventual PMA approval will be obtained.
The pivotal clinical study has a primary efficacy endpoint, a safety endpoint, many secondary endpoints and a couple of exploratory endpoints. The primary efficacy endpoint will compare speech perception (CNC words) from baseline to twelve-month follow-up and the safety endpoint will characterize incidence and frequency of adverse events. The total pivotal clinical study duration is estimated to be approximately two and a half years. There is no guarantee that we will meet any of the safety, efficacy, secondary, or exploratory endpoints or that the clinical study will proceed to the second stage or enroll all patients.
Once the pivotal study is completed, the data will be analyzed and sent to the FDA with the PMA submission. The FDA review may take 6-12 months depending on what comes up during the review and if the FDA review team recommends the device for a Panel Track review. There is no guarantee that PMA approval will be obtained.
If FDA approval is delayed, we will be unable to move forward with expansion of our corporate infrastructure, development of distribution capabilities, and implementation of product technical support and provider training, and the costs associated with delayed approval may limit the funds available for investment in these areas. Regulatory delays would also put us further behind our established competitors in the market and may allow additional competitors into the market with products that have competitive advantages over ours.
Moreover, if FDA approval is delayed beyond our current plan or if delay is based on safety or efficacy concerns that require product redesign, we will be required to raise significant additional capital to continue our operations. We may be unable to raise these additional funds on favorable terms or at all, especially if approval is delayed based on device performance or other issues with the Acclaim CI. Because the Acclaim CI is currently our only product candidate that we believe can be commercialized, we would be unable to continue operations if it were determined that we could not obtain FDA approval for the Acclaim CI.
Early Feasibility Study
The Acclaim CI has undergone extensive benchtop and laboratory testing throughout the design and development process. Animal testing was done to demonstrate the reliability of the Acclaim CI’s rechargeable battery and charging safety algorithm.
In the third quarter of 2022, we received an IDE to undergo a small Early Feasibility Study (“EFS”) at Mayo Clinic in Rochester, Minnesota. The principal investigator is Dr. Colin Driscoll, a respected veteran in the global cochlear implant industry. There were three patients enrolled, implanted, and activated in the fourth quarter of 2022.
The purpose of this early feasibility study was to demonstrate that the Acclaim CI is capable of operating as it was designed. In other words, there are no safety or efficacy endpoints. The study is essentially designed to elicit patient and professional feedback regarding their experience using the device and inform any necessary design changes prior to beginning the pivotal clinical study.
We believe that the initial results of the EFS were promising. A few device shortcomings have been identified and are in process of being addressed or appropriately mitigated. The primary concern is a signal to noise issue that subjects identify as a gurgling or sizzling background noise. Mitigation and resolution strategies have been implemented and further work is ongoing. We believe we have identified strategies to improve the signal to noise ratio. We have tested and implemented some of these strategies in EFS patients and they have shown improvement. We will not know the extent of signal to noise improvement of these strategies until they have been developed, tested, implemented and implanted into patients with a new or improved device.
The EFS patients have been implanted for over two years. They all have made it passed their 24 month follow-up appointments. They all use their devices daily. There have been reported adverse events, but no serious or unanticipated device effects.
Two of the three patients choose to wear a hearing aid on top of their Acclaim CI. This combination helps to mitigate the noise and provide patients with a signal to noise ratio that allows them to use and enjoy the performance of the device. It was an unanticipated discovery during the EFS that a hearing aid on top of the Acclaim CI could provide patients with additional improvement. We are intrigued by the possibility of offering a fully implanted cochlear implant that could also allow for the use of a hearing aid or other ear accessory (e.g., ear buds) because the Acclaim CI leverages the ear to pick up sound.
Go-To-Market Strategy
Assuming PMA approval is received, our commercialization strategy will be quality over quantity to facilitate the Acclaim CI gaining a meaningful foothold in the marketplace without unnecessary complications stemming from attempting to grow too quickly.
The surgical professionals believed to be best suited to implant the Acclaim CI are otologists and neurotologists (i.e., sub-specialties of otolaryngologists). This community is relatively small compared to other specialties, with only a few hundred active professionals in the United States. We anticipate carefully selecting roughly 30 sites to be trained and ready to implant upon commercialization. These 30 sites are expected to be spread throughout the country and focus on quality of surgical care and capacity to serve a sufficient number of qualified patients. Following the initial 30 sites, we intend to add additional sites every year until there are roughly 120-150 sites actively implanting the Acclaim CI. However, this strategy will require significant investments in the development of our management team, corporate infrastructure, and manufacturing capabilities, as well as expansion of our sales, distribution, and training network. We do not anticipate offering the Acclaim CI at every cochlear implant center.
The other key professional group is audiologists. Each surgical site will have its own audiology team familiar with cochlear implants. The audiology team is critical to the success of a surgical site’s performance. We will invest resources for in-person training, and technical and product support as well as virtual training, and technical and product support for audiologists servicing patients with our products.
Outside of surgical sites, there is a subset of audiologists who traditionally work with patients currently using hearing aids. These audiologists will be instrumental in identifying and referring potential Acclaim CI patients to surgical sites. One of the largest barriers to more cochlear implant candidates becoming cochlear implant recipients is the lack of awareness and understanding by the audiologists of the technology and associated benefits available for their patients. We believe strong relationships can be built with both surgical teams and audiologists to ensure both are able to understand the options and benefits of the technology and differentiate themselves from the marketplace by offering and working with the Acclaim CI. However, we will be unable to train, educate, and develop these relationships until we are able to obtain FDA approval for the Acclaim CI.
Commercial Activities Outside of the United States
We anticipate pursuing the Conformité Européenne mark (“CE Mark”) in the European Union shortly after FDA approval. The CE Mark will allow the Acclaim CI to be sold throughout the European Economic Area. We are currently focusing our resources on FDA approval and will address commercial activities outside of the United States when the FDA approval process is more advanced.
Eventually, we anticipate pursuing other markets based on the potential size of the markets and availability of reimbursement, such as Australia, Brazil, and parts of Asia, although no such approval is guaranteed, and approval may take longer and involve greater cost than we currently anticipate.
Product Evolution and Next Generation Products
The focus of research and development over the next several years will be to improve upon the existing product design of the Acclaim CI to aid the process of obtaining FDA approval. Quality and reliability will be a primary focus of the team in the initial years of market release. We will also focus on the growing need for robust software and user interfaces for both the patient and the professional.
It is possible that we will expand our portfolio to include a variety of cochlear electrode arrays similar to other cochlear implant companies. However, we do not anticipate expanding into as large of an electrode portfolio as some of our competitors as we are not convinced that a large electrode portfolio is efficient or effective.
Esteem FI-AMEI - a potentially viable product with reimbursement
The Esteem FI-AMEI is a unique technology that could serve a niche segment of the hearing market. FDA-approved since 2010, the Esteem FI-AMEI suffered from a lack of reimbursement due to categorization as a hearing aid. We believe that this categorization is inaccurate as, unlike a hearing aid which is essentially an externally worn microphone and speaker simply making sounds louder, the Esteem FI-AMEI is fully implanted and replaces the function of the middle ear. Although efforts to change that categorization have been unsuccessful to date, two bipartisan Congressional bills, both titled the Hearing Device Coverage Clarification Act were introduced in the House and the Senate in the 118th Congress. It is anticipated, although not guaranteed, that the bills will be reintroduced into the 119th Congress.
These bills seek to clarify that fully implanted active middle ear hearing devices (FI-AMEIs) are prosthetics and not subject to the current Medicare hearing aid coverage exclusion. If these bills are successful in clarifying that FI-AMEIs are eligible for coverage and a change does happen to reimbursement policy for fully implanted active middle ear implants, the Esteem FI-AMEI is an existing FDA approved product ready to capitalize on such a change.
Existing Esteem FI-AMEI patients and professionals who work with those patients will continue to be supported. It is not only important for the market to know our strategy for supporting patients for life, but it is the right thing to do for the patients.
New implantations of the Esteem FI-AMEI are not expected to be more than a few per year until, and if, the reimbursement policy changes. Absent a change in reimbursement policy, there only will be nominal revenue from replacement of the sound processor / battery assembly (the “Battery”) for existing patients who need a new Battery.
Intellectual Property
We rely on a combination of patent, copyright, trademark and trade secret laws and confidentiality and invention assignment agreements to protect our intellectual property rights. As of March 10, 2025, we had rights to 35 issued U.S. patents, which are estimated to expire between 2025 and 2043 assuming all required fees are paid, 13 pending U.S. patent applications, 33 issued foreign patents and 32 pending foreign and international patent applications. Our patents cover, among other things, aspects of our current Acclaim CI system and future product concepts. Some of the pending foreign and international patent applications preserve an opportunity to pursue patent rights in multiple countries.
Our pending patent applications may not result in issued patents, and we cannot assure you that any current or subsequently issued patents will protect our intellectual property rights or provide us with any competitive advantage. While there is no active litigation involving any of our patents or other intellectual property rights and we have not received any notices of patent infringement, we may be required to enforce or defend our intellectual property rights against third parties in the future. See Item 1A. Risk Factors - Risks Relating to our Intellectual Property for additional information regarding these and other risks related to our intellectual property portfolio and their potential effect on us.
Material Patents
As of March 10, 2025, our material patents, their jurisdiction, patent number, and expiration date are listed in the tables below:
| Jurisdiction | Patent No. | Expiration Date | Title | |||||
| U.S. | 7297101 | 01/17/2026 | Method and apparatus for minimally invasive placement of sensing and driver assemblies to improve hearing loss | |||||
| U.S. | 9782600 | 05/17/2033 | Self-regulating transcutaneous energy transfer | |||||
| U.S. | 7524278 | 08/15/2025 | Hearing aid system and transducer with hermetically sealed housing | |||||
| U.S. | 9497555 | 01/30/2035 | Implantable middle ear transducer having improved frequency response | |||||
| U.S. | 10129660 | 10/27/2028 | Implantable middle ear transducer having improved frequency response | |||||
| U.S. | 9036824 | 12/30/2033 | Transducer impedance measurement for hearing aid | |||||
| U.S. | 9521493 | 05/03/2032 | Transducer impedance measurement for hearing aid | |||||
| U.S. | 9682226 | 12/06/2033 | Electronic lead connection and related devices | |||||
| U.S. | 10549090 | 10/20/2037 | Communication system and methods for fully implantable modular cochlear implant system | |||||
| U.S. | 10646709 | 04/09/2038 | Fully implantable modular cochlear implant system | |||||
| U.S. | 10569079 | 09/04/2037 | Communication system and methods for fully implantable modular cochlear implant system | |||||
| U.S. | 10743812 | 03/25/2035 | Implantable middle ear diagnostic transducer | |||||
| U.S. | 11260220 | 02/28/2040 | Implantable cochlear system with integrated components and lead characterization | |||||
| U.S. | 11266831 | 06/13/2040 | Implantable cochlear system with integrated components and lead characterization | |||||
| U.S. | 9525949 | 03/16/2034 | Implantable middle ear transducer having diagnostic detection sensor | |||||
| Jurisdiction | Patent No. | Expiration Date | Title | |||||
| U.S. | 11051116 | 10/11/2032 | Implantable middle ear transducer having diagnostic detection sensor | |||||
| U.S. | 11471689 | 04/14/2041 | Cochlear implant stimulation calibration | |||||
| U.S. | 11564046 | 07/17/2041 | Programming of cochlear implant accessories | |||||
| U.S. | 9313590 | 03/13/2033 | Hearing aid amplifier having feed forward bias control based on signal amplitude and frequency for reduced power consumption | |||||
| U.S. | 9635478 | 03/09/2034 | Coulomb counter and battery management for hearing aid | |||||
| U.S. | 11672970 | 02/21/2040 | Implantable cochlear system with integrated components and lead characterization | |||||
| U.S. | 11697019 | 12/02/2040 | Combination hearing aid and cochlear implant system | |||||
| U.S. | 11711658 | 10/11/2032 | Implantable middle ear transducer having diagnostic detection sensor | |||||
| EP | 3500337 | 08/17/2037 | Implantable modular cochlear implant system with communication system and network | |||||
| DE | 3500337 | 08/17/2037 | Implantable modular cochlear implant system with communication system and network | |||||
| DK | 3500337 | 08/17/2037 | Implantable modular cochlear implant system with communication system and network | |||||
| AT | 3500337 | 08/17/2037 | Implantable modular cochlear implant system with communication system and network | |||||
| GB | 3500337 | 08/17/2037 | Implantable modular cochlear implant system with communication system and network | |||||
| BE | 3500337 | 08/17/2037 | Implantable modular cochlear implant system with communication system and network | |||||
| FR | 3500337 | 08/17/2037 | Implantable modular cochlear implant system with communication system and network | |||||
| IT | 3500337 | 08/17/2037 | Implantable modular cochlear implant system with communication system and network | |||||
| SE | 3500337 | 08/17/2037 | Implantable modular cochlear implant system with communication system and network | |||||
| MX | 421017 | 2/21/20240 | Implantable cochlear system with integrated components and lead characterization | |||||
| EP | 3927420 | 2/21/2040 | Implantable cochlear system with integrated components and lead characterization | |||||
| UP | 3927420 | 2/21/2040 | Implantable cochlear system with integrated components and lead characterization | |||||
| GB | 3927420 | 2/21/2040 | Implantable cochlear system with integrated components and lead characterization | |||||
| U.S. | 11633591 | 8/3/2041 | Combination implant system with removable earplug sensor and implanted battery | |||||
| U.S. | 11806531 | 4/11/2041 | Implantable cochlear system with inner ear sensor | |||||
| U.S. | 11839765 | 1/23/2042 | Cochlear implant system with integrated signal analysis functionality | |||||
| U.S. | 11865339 | 6/22/2042 | Cochlear implant system with electrode impedance diagnostics | |||||
| EP | 3858425 | 08/17/2037 | Implantable Modular Cochlear Implant System with Communication System and Network | |||||
| GB | 3858425 | 08/17/2037 | Implantable Modular Cochlear Implant System with Communication System and Network | |||||
| UP | 3858425 | 08/17/2037 | Implantable Modular Cochlear Implant System with Communication System and Network | |||||
| U.S. | 12090318 | 02/21/2040 | Implantable Cochlear System with Integrated Components and Lead Characterization | |||||
| U.S. | 12233256 | 10/09/2040 | Implantable Cochlear System with Integrated Components and Lead Characterization | |||||
| Jurisdiction | Patent No. | Expiration Date | Title | |||||
| HK | HK40066136 | 02/21/2040 | Implantable Cochlear System with Integrated Components and Lead Characterization | |||||
| JP | 7598401 | 02/21/2040 | Implantable Cochlear System with Integrated Components and Lead Characterization | |||||
| JP | 7597846 | 02/21/2040 | Implantable Cochlear System with Integrated Components and Lead Characterization | |||||
| JP | 7598327 | 02/21/2040 | Implantable Cochlear System with Integrated Components and Lead Characterization | |||||
| U.S. | 12081061 | 02/07/2043 | Recharge System For Implantable Battery | |||||
| U.S. | 12214195 | 12/02/2040 | Implantable Cochlear System with Inner Ear Sensor | |||||
| EP | 4204071 | 08/27/2041 | Programming Of Cochlear Implant Accessories | |||||
| GB | 4204071 | 08/27/2041 | Programming Of Cochlear Implant Accessories | |||||
| HK | HK40097814 | 08/27/2041 | Programming Of Cochlear Implant Accessories | |||||
| UP | 4204071 | 08/27/2041 | Programming Of Cochlear Implant Accessories | |||||
| U.S. | 12151102 | 12/02/2040 | Combination Hearing Aid and Cochlear Implant System | |||||
| EP | 4255554 | 11/24/2041 | Combination Hearing Aid and Cochlear Implant System | |||||
| GB | 4255554 | 11/24/2041 | Combination Hearing Aid and Cochlear Implant System | |||||
| UP | 4255554 | 11/24/2041 | Combination Hearing Aid and Cochlear Implant System | |||||
| EP | 4255555 | 11/24/2041 | Cochlear Implant Stimulation Calibration | |||||
| GB | 4255555 | 11/24/2041 | Cochlear Implant Stimulation Calibration | |||||
| UP | 4255555 | 11/24/2041 | Cochlear Implant Stimulation Calibration | |||||
| EP | 4319866 | 04/01/2042 | Cochlear Implant System with Electrode Impedance Diagnostics | |||||
| GB | 4319866 | 04/01/2042 | Cochlear Implant System with Electrode Impedance Diagnostics | |||||
| UP | 4319866 | 04/01/2042 | Cochlear Implant System with Electrode Impedance Diagnostics | |||||
Trademarks
As of February 28, 2025, we had trademark registrations, covering “Acclaim”, “Envoy”, “Envoy Medical”, “EnvoyCEM”, “Esteem”, “Invisible Hearing”, and “MEDCEM.” Our U.S. trademarks have registration dates between 2002 and 2021 and have upcoming renewal dates between 2027 and 2033. All of our trademarks are in current use, and we expect that they will remain in use for the foreseeable future. We also have pending trademark applications covering “Nature’s Microphone” and “Naturemic” with application dates in 2024 for use in 2025.
We also rely, in part, upon unpatented trade secrets, know-how and continuing technological innovation, and may in the future rely upon licensing opportunities, to develop and maintain our competitive position. We protect our proprietary rights through a variety of methods, including confidentiality and assignment agreements with suppliers, employees, consultants and others who may have access to our proprietary information.
Manufacturing and Supply
We currently do all final manufacturing at our facility in White Bear Lake, Minnesota. We rely on a limited number of technicians and have some critical equipment that would be difficult to replace in a timely manner. In order to scale quickly, we will need to expand our manufacturing capacity and add additional shifts.
We rely on third-party suppliers to manufacture some of our critical sub-assemblies. Outsourcing sub-assemblies manufacturing reduces our need for additional capital investment. We select our suppliers carefully and require they adhere to all applicable regulations. We monitor our suppliers and always inspect all components received. Our quality assurance process monitors and maintains supplier performance through qualification and periodic supplier reviews and audits.
Certain components used in our products are supplied by single-source suppliers, but we believe that we are able to plan supply in a manner that would minimize the effect of losing any of our existing suppliers. Our suppliers manufacture the components they produce for us and test our components and devices to our specifications. We intend to maintain sufficient levels of inventory to enable us to continue our operations while we qualify additional potential suppliers in the event that one or more of our single-source suppliers were to encounter a delay in supply or end supply. Due to our current limited production numbers, we order components and sub-assemblies on a purchase order basis and do not have supply agreements with any of our suppliers.
Government Regulation
The FDA’s policies may change and additional government laws and regulations may be enacted that could prevent, limit, or delay regulatory approval of our product candidates, that could limit the marketability of our product candidates, or that could impose additional regulatory obligations on us. For example, the current administration may implement new or revised laws, regulatory requirements, and associated compliance obligations, as well as postponed or frozen regulatory requirements. Changes in medical practice and standard of care may also impact the marketability of our product candidates. If we are slow or unable to adapt to changes in existing requirements, standards of care, or the adoption of new requirements or policies, or if we are not able to maintain regulatory compliance, we may lose any marketing approval that we may have obtained and be subject to regulatory enforcement action.
Our products and our operations are subject to extensive regulation by the FDA and other federal and state authorities in the U.S., as well as comparable authorities in the European Economic Area (“EEA”) and other countries in which we may sell our products. In the U.S., our products are subject to regulation as medical devices under the Federal Food, Drug, and Cosmetic Act (“FDCA”) as implemented and enforced by the FDA. The FDA regulates the development, design, non-clinical and clinical research, manufacturing, safety, efficacy, labeling, packaging, storage, installation, servicing, recordkeeping, premarket clearance or approval, import, export, adverse event reporting, advertising, promotion, marketing and distribution, and import and export of medical devices to ensure that medical devices distributed domestically are safe and effective for their intended uses and otherwise meet the requirements of the FDCA.
In addition to U.S. regulations, we are subject to a variety of regulations in the EEA governing clinical trials and the commercial sales and distribution of our products. Even if we obtain the required FDA clearance or approval for a product in the United States, we will be required to obtain authorization before commencing clinical studies and to obtain marketing authorization or approval of our products under the comparable regulatory authorities of countries outside of the U.S. before we can commence clinical studies or commercialize our products in those countries. The approval process varies from country to country and the time may be longer or shorter than that required for FDA clearance or approval.
FDA Premarket Clearance and Approval Requirements
Unless an exemption applies, each medical device commercially distributed in the U.S. requires either FDA clearance of a 510(k) premarket notification or PMA. Under the FDCA, medical devices are classified into one of three classes, Class I, Class II, or Class III, depending on the degree of risk associated with each medical device and the extent of manufacturer and regulatory control needed to ensure its safety and effectiveness. Class I includes devices with the lowest risk to the patient and are those for which safety and effectiveness can be assured by adherence to the FDA’s General Controls for medical devices, which include compliance with the applicable portions of the FDA’s Quality System Regulations (“QSR”), facility registration and product listing, reporting of adverse medical events, and truthful and non-misleading labeling, advertising, and promotional materials. Class II devices are subject to the FDA’s General Controls, and special controls as deemed necessary by the FDA to ensure the safety and effectiveness of the device. While most Class I devices are exempt from the 510(k) premarket notification requirement, manufacturers of most Class II devices are required to submit to the FDA a premarket notification under Section 510(k) of the FDCA requesting permission to commercially distribute the device. The FDA’s permission to commercially distribute a device subject to a 510(k) premarket notification is generally known as 510(k) clearance. Under the 510(k) process, the manufacturer must submit to the FDA a premarket notification demonstrating that the device is “substantially equivalent” to either a device that was legally marketed prior to May 28, 1976, the date upon which the Medical Device Amendments of 1976 were enacted, or another legally marketed device that was cleared through the 510(k) process.
Devices deemed by the FDA to pose the greatest risks, such as life-sustaining, life-supporting or some implantable devices, or devices that have a new intended use, or use advanced technology that is not substantially equivalent to that of a legally marketed device, are placed in Class III, requiring approval of a PMA.
Some pre-amendment devices are unclassified but are subject to the FDA’s premarket notification and clearance process in order to be commercially distributed.
The Acclaim CI will be regulated as a Class III device and will require approval of a PMA prior to commercialization.
PMA Approval Pathway
Class III devices require PMA approval before they can be marketed although some pre-amendment Class III devices for which the FDA has not yet required a PMA are cleared through the 510(k) process. The PMA process is more demanding than the 510(k) premarket notification process. In a PMA process, the manufacturer must demonstrate that the device is safe and effective, and the PMA must be supported by extensive data, including data from preclinical studies and human clinical trials. The PMA must also contain a full description of the device and its components, a full description of the methods, facilities and controls used for manufacturing, and proposed labeling. Following receipt of a PMA, the FDA determines whether the application is sufficiently complete to permit a substantive review. If the FDA accepts the application for review, it has 180 days under the FDCA to complete its review of a PMA, although in practice, the FDA’s review often takes significantly longer, and can take up to several years. An advisory panel of experts from outside the FDA may be convened to review and evaluate the application and provide recommendations to the FDA as to the approvability of the device. The FDA may or may not accept the panel’s recommendation. In addition, the FDA will generally conduct a preapproval inspection of the applicant or its third-party manufacturers’ or suppliers’ manufacturing facility or facilities to ensure compliance with the QSR.
The FDA will approve the new device for commercial distribution if it determines that the data and information in the PMA constitute valid scientific evidence and that there is reasonable assurance that the device is safe and effective for its intended use(s). The FDA may approve a PMA with post-approval conditions intended to ensure the safety and effectiveness of the device, including, among other things, restrictions on labeling, promotion, sale and distribution, and collection of long-term follow-up data from patients in the clinical study that supported the PMA or requirements to conduct additional clinical studies post-approval. The FDA may condition a PMA approval on some form of post-market surveillance when deemed necessary to protect the public health or to provide additional safety and efficacy data for the device in a larger population or for a longer period of use. In such cases, the manufacturer might be required to follow certain patient groups for a number of years and to make periodic reports to the FDA on the clinical status of those patients. Failure to comply with the conditions of approval can result in material adverse enforcement action, including withdrawal of the approval.
Certain changes to an approved device, such as changes in manufacturing facilities, methods, or quality control procedures, or changes in the design performance specifications, which affect the safety or effectiveness of the device, require submission of a PMA supplement. PMA supplements often require submission of the same type of information as a PMA, except that the supplement is limited to information needed to support any changes from the device covered by the original PMA and may not require as extensive clinical data or the convening of an advisory panel. Certain other changes to an approved device require the submission of a new PMA, such as when the design change causes a different intended use, mode of operation, and technical basis of operation, or when the design change is so significant that a new generation of the device will be developed, and the data that were submitted with the original PMA are not applicable for the change in demonstrating a reasonable assurance of safety and effectiveness.
Clinical Trials
Clinical studies are almost always required to support a PMA and are sometimes required to support a 510(k) submission. All clinical investigations of investigational devices to determine safety and effectiveness must be conducted in accordance with the FDA’s IDE regulations, which govern investigational device labeling, prohibit promotion of the investigational device, and specify an array of recordkeeping, reporting and monitoring responsibilities of study sponsors and study investigators. If the device presents a “significant risk” to human health, as defined by the FDA, the FDA requires the device sponsor to submit an IDE application to the FDA, which must become effective prior to commencing human clinical studies. A significant risk device is one that presents a potential for serious risk to the health, safety or welfare of a patient and either is implanted, used in supporting or sustaining human life, substantially important in diagnosing, curing, mitigating or treating disease or otherwise preventing impairment of human health, or otherwise presents a potential for serious risk to a subject. An IDE application must be supported by appropriate data, such as animal and laboratory test results, showing that it is safe to test the device in humans and that the testing protocol is scientifically sound. The IDE will automatically become effective 30 days after receipt by the FDA unless the FDA notifies the company that the investigation may not begin. If the FDA determines that there are deficiencies or other concerns with an IDE for which it requires modification, the FDA may permit a clinical study to proceed under a conditional approval. An IDE supplement must be submitted to, and approved by, the FDA before a sponsor or investigator may make a change to the investigational plan that may affect its scientific soundness, study plan or the rights, safety or welfare of human subjects.
In addition, the study must be approved by, and conducted under the oversight of, an Institutional Review Board (“IRB”) for each clinical site. The IRB is responsible for the initial and continuing review of the IDE, and may pose additional requirements for the conduct of the study. If an IDE application is approved by the FDA and one or more IRBs, human clinical studies may begin at a specific number of investigational sites with a specific number of patients, as approved by the FDA. If the device presents a non-significant risk to the patient, a sponsor may begin the clinical study after obtaining approval for the study by one or more IRBs without separate approval from the FDA, but must still follow abbreviated IDE requirements, such as monitoring the investigation, ensuring that the investigators obtain informed consent, and complying with labeling and record-keeping requirements.
During a study, the sponsor is required to comply with the applicable FDA requirements, including, for example, study monitoring, selecting clinical investigators and providing them with the investigational plan, ensuring IRB review, adverse event reporting, record keeping, and prohibitions on the promotion of investigational devices or on making safety or effectiveness claims for them. The clinical investigators in the clinical study are also subject to FDA regulations and must obtain patient informed consent, rigorously follow the investigational plan and study protocol, control the disposition of the investigational device, and comply with all reporting and recordkeeping requirements. Additionally, after a study begins, we, the FDA or the IRB could suspend or terminate a clinical study at any time for various reasons, including a belief that the risks to study subjects outweigh the anticipated benefits.
Expedited Development and Review Programs
Following passage of the 21st Century Cures Act, the FDA implemented the Breakthrough Devices Program, which is a voluntary program offered to manufacturers of certain medical devices and device-led combination products, including the Acclaim CI, that may provide for more effective treatment or diagnosis of life-threatening or irreversibly debilitating diseases or conditions. The goal of the program is to provide patients and health care providers with more timely access to qualifying devices by expediting their development, assessment and review, while preserving the statutory standards for FDA marketing authorization, although there is no guarantee that this designation will accelerate the timeline for approval or make it more likely that the Acclaim CI will be approved. The program is available to medical devices that meet certain eligibility criteria, including that the device provides more effective treatment or diagnosis of life-threatening or irreversibly debilitating diseases or conditions, and that the device meets one of the following criteria: (i) the device represents a breakthrough technology, (ii) no approved or cleared alternatives exist, (iii) the device offers significant advantages over existing approved or cleared alternatives, or (iv) the availability of the device is in the best interest of patients. Breakthrough Device designation provides certain benefits to device developers, including more interactive and timely communications with FDA staff, use of post-market data collection, when scientifically appropriate, to facilitate expedited and efficient development and review of the device, opportunities for efficient and flexible clinical study design, and prioritized review of premarket submissions. The Acclaim CI received Breakthrough Device designation in March 2019.
Post-market Regulation
After a device is cleared or approved for marketing, numerous and pervasive regulatory requirements continue to apply. These include:
| ● | establishment registration and device listing with the FDA; |
| ● | QSR requirements, which require manufacturers, including third-party manufacturers, to follow stringent design, testing, control, documentation and other quality assurance procedures during all aspects of the design and manufacturing process; |
| ● | labeling and marketing regulations, which require that promotion is truthful, not misleading, fairly balanced and provide adequate directions for use and that all claims are substantiated, and also prohibit the promotion of products for unapproved or “off-label” uses and impose other restrictions on labeling; FDA guidance on off-label dissemination of information and responding to unsolicited requests for information; |
| ● | the federal Physician Sunshine Act and various state and foreign laws on reporting remunerative relationships with health care customers; |
| ● | the federal Anti-Kickback Statute (and similar state laws) prohibiting, among other things, soliciting, receiving, offering or providing remuneration intended to induce the purchase or recommendation of an item or service reimbursable under a federal healthcare program, such as Medicare or Medicaid. A person or entity does not have to have actual knowledge of this statute or specific intent to violate it to have committed a violation; |
| ● | the federal False Claims Act (and similar state laws) prohibiting, among other things, knowingly presenting, or causing to be presented, claims for payment or approval to the federal government that are false or fraudulent, knowingly making a false statement material to an obligation to pay or transmit money or property to the federal government or knowingly concealing, or knowingly and improperly avoiding or decreasing, an obligation to pay or transmit money to the federal government. The government may assert that claim includes items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the false claims statute; |
| ● | clearance or approval of product modifications to 510(k)-cleared devices that could significantly affect safety or effectiveness or that would constitute a major change in intended use of one of our cleared devices, or approval of a supplement for certain modifications to PMA devices; |
| ● | medical device reporting regulations, which require that a manufacturer report to the FDA if a device it markets may have caused or contributed to a death or serious injury, or has malfunctioned and the device or a similar device that it markets would be likely to cause or contribute to a death or serious injury, if the malfunction were to recur; |
| ● | correction, removal and recall reporting regulations, which require that manufacturers report to the FDA field corrections and product recalls or removals if undertaken to reduce a risk to health posed by the device or to remedy a violation of the FDCA that may present a risk to health; |
| ● | complying with the new federal law and regulations requiring Unique Device Identifiers (“UDI”) on devices and also requiring the submission of certain information about each device to the FDA’s Global Unique Device Identification Database (“GUDID”); |
| ● | the FDA’s recall authority, whereby the agency can order device manufacturers to recall from the market a product that is in violation of governing laws and regulations; and |
| ● | post-market surveillance activities and regulations, which apply when deemed by the FDA to be necessary to protect the public health or to provide additional safety and effectiveness data for the device. |
We may be subject to similar foreign laws that may include applicable post-marketing requirements such as safety surveillance. Our manufacturing processes are required to comply with the applicable portions of the QSR, which cover the methods and the facilities and controls for the design, manufacture, testing, production, processes, controls, quality assurance, labeling, packaging, distribution, installation and servicing of finished devices intended for human use. The QSR also requires, among other things, maintenance of a device master file, device history file, and complaint files. As a manufacturer, our facilities, records and manufacturing processes are subject to periodic scheduled or unscheduled inspections by the FDA. Our failure to maintain compliance with the QSR or other applicable regulatory requirements could result in the shut-down of, or restrictions on, our manufacturing operations and the recall or seizure of our products.
The discovery of previously unknown problems with any of our products, including unanticipated adverse events or adverse events of increasing severity or frequency, whether resulting from the use of the device within the scope of its clearance or off-label by a physician in the practice of medicine, could result in restrictions on the device, including the removal of the product from the market or voluntary or mandatory device recalls.
The FDA has broad regulatory compliance and enforcement powers. If the FDA determines that we failed to comply with applicable regulatory requirements, it can take a variety of compliance or enforcement actions, which may result in any of the following sanctions:
| ● | warning letters, untitled letters, fines, injunctions, consent decrees and civil penalties; |
| ● | recalls, withdrawals, or administrative detention or seizure of our products; |
| ● | operating restrictions or partial suspension or total shutdown of production; |
| ● | refusing or delaying requests for 510(k) marketing clearance or PMA approvals of new products or modified products; |
| ● | withdrawing 510(k) clearances or PMA approvals that have already been granted; |
| ● | refusal to grant export or import approvals for our products; or |
| ● | criminal prosecution. |
Foreign Regulation
In order for us to market our products in countries outside the U.S., we must obtain regulatory approvals or certifications and comply with extensive product and quality system regulations in other countries. These regulations, including the requirements for approvals, clearance or certifications and the time required for regulatory review, vary from country to country. Some countries have regulatory review processes that are substantially longer than U.S. processes. Failure to obtain regulatory approval or certification in a timely manner and meet all of the local requirements including language and specific safety standards in any foreign country in which we plan to market our products could prevent us from marketing products in such countries or subject us to sanctions and fines.
Regulation of Medical Devices in the European Union
The European Union (“EU”) has adopted specific directives and regulations regulating the design, manufacture, clinical investigation, conformity assessment, labeling and adverse event reporting for medical devices.
Until May 25, 2021, medical devices were regulated by Council Directive 93/42/EEC (the “EU Medical Devices Directive”), and Directive 90/385/EEC (“AIMDD”) which have been repealed and replaced by Regulation (EU) No 2017/745 (the “EU Medical Devices Regulation”). Our current certificates have been granted under the EU Medical Devices Directive and the AIMDD whose regime is described below. However, as of May 26, 2021, some of the EU Medical Devices Regulation requirements apply in place of the corresponding requirements of the EU Medical Devices Directive and the AIMDD with regard to registration of economic operators and of devices, post-market surveillance and vigilance requirements. Pursuing marketing of medical devices in the EU will notably require that our devices be certified under the new regime set forth in the EU Medical Devices Regulation when our current certificates expire.
Medical Devices Directive
In the EU, there is currently no premarket government review of medical devices. However, all medical devices placed on the EU market must meet the essential requirements, including the requirement that a medical device must be designed and manufactured in such a way that it will not compromise the clinical condition or safety of patients, or the safety and health of users and others. In addition, the device must achieve the performance intended by the manufacturer and be designed, manufactured, and packaged in a suitable manner.
Compliance with the essential requirements is a prerequisite for the CE Mark without which medical devices cannot be marketed or sold in the EU. To demonstrate compliance with the essential requirements, medical device manufacturers must undergo a conformity assessment procedure, which varies according to the type of medical device and its (risk) classification. Except for low-risk medical devices (Class I non-sterile, non-measuring devices), where the manufacturer can self-assess the conformity of its products with the essential requirements (except for any parts which relate to sterility or metrology), a conformity assessment procedure requires the intervention of a notified body. Notified bodies are independent organizations designated by EU member states to assess the conformity of devices before being placed on the market. A notified body would typically audit and examine a product’s technical dossiers and the manufacturers’ quality system. If satisfied that the relevant product conforms to the relevant essential requirements, the notified body issues a certificate of conformity, which the manufacturer uses as a basis for its own declaration of conformity. The manufacturer may then apply the CE Mark to the device, which allows the device to be placed on the market throughout the EU.
Throughout the term of the certificate of conformity, the manufacturer will be subject to periodic surveillance audits to verify continued compliance with the applicable requirements. In particular, there will be a new audit by the notified body before it will renew the relevant certificate(s).
Medical Devices Regulation
On April 5, 2017, the EU Medical Devices Regulation was adopted with the aim of ensuring better protection of public health and patient safety. The EU Medical Devices Regulation establishes a uniform, transparent, predictable and sustainable regulatory framework across the EU for medical devices and ensures a high level of safety and health while supporting innovation. Unlike the EU Medical Devices Directive and the AIMDD, the EU Medical Devices Regulation is directly applicable in EU member states without the need for member states to implement into national law. This aims at increasing harmonization across the EU.
Devices lawfully placed on the market pursuant to the EU Medical Devices Directive or the AIMDD prior to May 26, 2021 may generally continue to be made available on the market or put into service until May 26, 2025, provided that the requirements of the transitional provisions are fulfilled. In particular, the certificate in question must still be valid. However, even in this case, manufacturers must comply with a number of new or reinforced requirements set forth in the EU Medical Devices Regulation, in particular the obligations described below.
The EU Medical Devices Regulation requires that before placing a device, other than a custom-made device, on the market, manufacturers (as well as other economic operators such as authorized representatives and importers) must register by submitting identification information to the electronic system (Eudamed), unless they have already registered. The information to be submitted by manufacturers (and authorized representatives) also includes the name, address and contact details of the person or persons responsible for regulatory compliance. The new Regulation also requires that before placing a device, other than a custom-made device, on the market, manufacturers must assign a unique identifier to the device and provide it along with other core data to the UDI database. These new requirements aim at ensuring better identification and traceability of the devices. Each device and, as applicable, each package will have a UDI composed of two parts: a device identifier (“UDI-DI”) specific to a device, and a production identifier (“UDI-PI”) to identify the unit producing the device. Manufacturers are also notably responsible for entering the necessary data on Eudamed, which includes the UDI database, and for keeping it up to date. The obligations for registration in Eudamed will become applicable at a later date (as Eudamed is not yet fully functional). Until Eudamed is fully functional, the corresponding provisions of the EU Medical Devices Directive and the AIMDD continue to apply for the purpose of meeting the obligations laid down in the provisions regarding exchange of information, including, and in particular, information regarding registration of devices and economic operators.
All manufacturers placing medical devices into the market in the EU must comply with the EU medical device vigilance system. Under this system, serious incidents and Field Safety Corrective Actions (“FSCAs”) must be reported to the relevant authorities of the EU member states. Manufacturers are required to take FSCAs defined as any corrective action for technical or medical reasons to prevent or reduce a risk of a serious incident associated with the use of a medical device that is made available on the market. An FSCA may include the recall, modification, exchange, destruction or retrofitting of the device.
The aforementioned EU rules are generally applicable in the EEA, which consists of the 27 EU member states plus Norway, Liechtenstein, and Iceland.
Brexit
Since January 1, 2021, the Medicines and Healthcare Products Regulatory Agency (“MHRA”) has become the sovereign regulatory authority responsible for Great Britain (i.e. England, Wales and Scotland) medical device market according to the requirements provided in the Medical Devices Regulations 2002 (SI 2002 No 618, as amended) that sought to give effect to EU Medical Devices Directive and AIMDD whereas Northern Ireland continues to be governed by EU rules according to the Northern Ireland Protocol. Following the end of the Brexit transitional period on January 1, 2021, new regulations require medical devices to be registered with the MHRA before being placed on Great Britain market. The MHRA only registers devices where the manufacturer or their United Kingdom (“UK”) Responsible Person has a registered place of business in the UK. Manufacturers based outside the UK need to appoint a UK Responsible Person that has a registered place of business in the UK to register devices with the MHRA.
On June 26, 2022, the MHRA published its response to a 10-week consultation on the post-Brexit regulatory framework for medical devices and diagnostics. MHRA seeks to amend the UK Medical Devices Regulations 2002 (which are based on EU legislation, primarily the EU Medical Devices Directive and the EU In Vitro Diagnostic Medical Devices Directive 98/79/EC), in particular to create new access pathways to support innovation, create an innovative framework for regulating software and artificial intelligence as medical devices, reform IVD regulation, and foster sustainability through the reuse and remanufacture of medical devices. Regulations implementing the new regime were originally scheduled to come into force in July 2023, but have recently been postponed to July 2025. Devices bearing CE Marks issued by EU notified bodies under the EU Medical Devices Regulation, the EU Medical Devices Directive or AIMDD are now subject to transitional arrangements. In its consultation response, the MHRA indicated that the future UK regulations will allow devices certified under the EU Medical Devices Regulation to be placed on the market in Great Britain under the CE Mark until either the certificate expires or for five years after the new regulations take effect, whichever is sooner. Devices certified under the EU Medical Devices Directive or AIMDD could continue to be placed on the market until either the certificate expires or for three years after the new regulations take effect, whichever is sooner. Following these transitional periods, it is expected that all medical devices will require a UK Conformity Assessed (“UKCA”) mark. Manufacturers may choose to use the UKCA mark on a voluntary basis until July 1, 2025. However, UKCA marking will not be recognized in the EU. The rules for placing medical devices on the market in Northern Ireland, which is part of the UK, differ from those in the rest of the UK. Compliance with this legislation is a prerequisite to be able to affix the UKCA mark to our products, without which they cannot be sold or marketed in Great Britain.
In addition, the Trade Deal between the UK and the EU generally provides for cooperation and exchange of information between the parties in the areas of product safety and compliance, including market surveillance, enforcement activities and measures, standardization-related activities, exchanges of officials, and coordinated product recalls. As such, processes for compliance and reporting should reflect requirements from regulatory authorities.
Similarly, we are subject to regulations and product registration requirements in many foreign countries in which we may sell our products, including in the areas of:
| ● | design, development, manufacturing, and testing; |
| ● | product standards; |
| ● | product safety; |
| ● | product safety reporting; |
| ● | marketing, sales, and distribution; |
| ● | packaging and storage requirements; |
| ● | labeling requirements; |
| ● | content and language of instructions for use; |
| ● | clinical studies; |
| ● | record keeping procedures; |
| ● | advertising and promotion; |
| ● | recalls and field corrective actions; |
| ● | post-market surveillance, including reporting of deaths or serious injuries and malfunctions that, if they were to recur, could lead to death or serious injury; |
| ● | import and export restrictions; |
| ● | tariff regulations, duties, and tax requirements; |
| ● | registration for reimbursement; and |
| ● | necessity of testing performed in country by distributors for licensees. |
The time required to obtain clearance or certification required by foreign countries may be longer or shorter than that required for FDA clearance, and requirements for licensing a product in a foreign country may differ significantly from FDA requirements.
Federal, State and Foreign Fraud and Abuse and Physician Payment Transparency Laws
In addition to FDA restrictions on marketing and promotion of drugs and devices, other federal, state, and foreign laws restrict our business practices. These laws include, without limitation, foreign, federal, and state anti-kickback and false claims laws, as well as transparency laws regarding payments or other items of value provided to healthcare providers.
The federal Anti-Kickback Statute prohibits, among other things, knowingly and willfully offering, paying, soliciting or receiving any remuneration (including any kickback, bribe or rebate), directly or indirectly, overtly or covertly, in cash or in kind to induce or in return for purchasing, leasing, ordering or arranging for or recommending the purchase, lease or order of any good, facility, item or service reimbursable, in whole or in part, under Medicare, Medicaid or other federal healthcare programs. The term “remuneration” has been broadly interpreted to include anything of value, including stock, stock options, and the compensation derived through ownership interests.
Although there are a number of statutory exceptions and regulatory safe harbors protecting some common activities from prosecution, the exceptions and safe harbors are drawn narrowly. Failure to meet all of the requirements of a particular applicable statutory exception or regulatory safe harbor does not make the conduct per se illegal under the federal Anti-Kickback Statute. Instead, the legality of the arrangement will be evaluated on a case-by-case basis based on a cumulative review of all its facts and circumstances. Conduct and business arrangements that do not fully satisfy one of these safe harbor provisions may result in increased scrutiny by government enforcement authorities. Several courts have interpreted the statute’s intent requirement to mean that if any one purpose of an arrangement involving remuneration is to induce referrals of federal healthcare covered business, the federal Anti-Kickback Statute has been violated. In addition, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation.
The majority of states also have anti-kickback laws which establish similar prohibitions and in some cases may apply more broadly to items or services covered by any third-party payor, including commercial insurers and self-pay patients.
The federal civil False Claims Act prohibits, among other things, any person or entity from knowingly presenting, or causing to be presented, a false or fraudulent claim for payment or approval to the federal government or knowingly making, using or causing to be made or used a false record or statement material to a false or fraudulent claim to the federal government. A claim includes “any request or demand” for money or property presented to the U.S. government. The federal civil False Claims Act also applies to false submissions that cause the government to be paid less than the amount to which it is entitled, such as a rebate. Intent to deceive is not required to establish liability under the civil federal civil False Claims Act. Moreover, a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the federal civil False Claims Act. In addition, private parties may initiate “qui tam” whistleblower lawsuits against any person or entity under the federal civil False Claims Act in the name of the government and share in the proceeds of the lawsuit. The government may further prosecute conduct constituting a false claim under the federal criminal False Claims Act. The criminal False Claims Act prohibits the making or presenting of a claim to the government knowing such claim to be false, fictitious or fraudulent and, unlike the federal civil False Claims Act, requires proof of intent to submit a false claim.
The Civil Monetary Penalties Law imposes penalties against any person or entity that, among other things, is determined to have presented or caused to be presented a claim to a federal healthcare program that the person knows or should know is for an item or service that was not provided as claimed or is false or fraudulent, or offering or transferring remuneration to a federal healthcare beneficiary that a person knows or should know is likely to influence the beneficiary’s decision to order or receive items or services reimbursable by the government from a particular provider or supplier.
The Health Insurance Portability and Accountability Act (“HIPAA”) also created additional federal criminal statutes that prohibit among other actions, knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program, including private third-party payors, knowingly and willfully embezzling or stealing from a healthcare benefit program, willfully obstructing a criminal investigation of a healthcare offense, and knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for healthcare benefits, items or services. Similar to the federal Anti-Kickback Statute, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation.
Many foreign countries have similar laws relating to healthcare fraud and abuse. Foreign laws and regulations may vary greatly from country to country. For example, the advertising and promotion of medical devices is subject to some general principles set forth in EU legislation. According to the EU Medical Devices Regulation, only devices that are CE marked may be marketed and advertised in the EU in accordance with their intended purpose. Directive 2006/114/EC concerning misleading and comparative advertising and Directive 2005/29/EC on unfair commercial practices, while not specific to the advertising of medical devices, also apply to the advertising thereof and contain general rules, for example, requiring that advertisements are evidenced, balanced and not misleading. Specific requirements are defined at a national level. EU member states’ laws related to the advertising and promotion of medical devices, which vary between jurisdictions, may limit or restrict the advertising and promotion of products to the general public and may impose limitations on promotional activities with healthcare professionals. These laws, which vary between jurisdictions (thus making compliance more complex), may limit or restrict the advertising and promotion of our products to the general public and may impose limitations on our promotional activities with healthcare professionals. Many EU member states have adopted specific anti-gift statutes that further limit commercial practices for our products, in particular vis-à-vis healthcare professionals and organizations. Additionally, there has been a recent trend of increased regulation of payments and transfers of value provided to healthcare professionals or entities and many EU member states have adopted national “Sunshine Acts” which impose reporting and transparency requirements (often on an annual basis), similar to the requirements in the United States, on medical device manufacturers. Certain countries also mandate implementation of commercial compliance programs. Also, many U.S. states have similar fraud and abuse statutes or regulations that may be broader in scope and may apply regardless of payor, in addition to items and services reimbursed under Medicaid and other state programs.
Additionally, there has been a recent trend of increased foreign, federal, and state regulation of payments and transfers of value provided to healthcare professionals or entities. In the U.S., the federal Physician Payments Sunshine Act imposes annual reporting requirements on certain drug, biologics, medical supplies and device manufacturers for which payment is available under Medicare, Medicaid or CHIP for payments and other transfers of value provided by them, directly or indirectly, to physicians, as defined by statute, certain other non-physician practitioners such as physician assistants and nurse practitioners, and teaching hospitals, as well as ownership and investment interests held by physicians and their immediate family members. Manufacturers must submit reports by the 90th day of each calendar year. Many EU member states have adopted national “Sunshine Acts” which impose similar reporting and transparency requirements (often on an annual basis) on certain drug, biologics and medical device manufacturers. Certain foreign countries and U.S. states also mandate implementation of commercial compliance programs, impose restrictions on device manufacturer marketing practices and require tracking and reporting of gifts, compensation, and other remuneration to healthcare professionals and entities.
Violation of any of the federal and state healthcare laws described above or any other governmental regulations that apply to device manufacturers may result in significant penalties, including the imposition of significant civil, criminal and administrative penalties, damages, disgorgement, monetary fines, imprisonment, possible exclusion from participation in Medicare, Medicaid and other federal healthcare programs, contractual damages, reputational harm, diminished profits and future earnings, additional reporting requirements and/or oversight if the entity becomes subject to a corporate integrity agreement or similar agreement to resolve allegations of non-compliance with these laws, and curtailment of operations.
Data Privacy and Security Laws
Numerous state, federal and foreign laws, regulations, and standards govern the collection, use, access to, confidentiality and security of health-related and other personal information and could apply now or in the future to our operations or the operations of our partners. In the United States, numerous federal and state laws and regulations, including data breach notification laws, health information privacy and security laws, including HIPAA, and consumer protection laws and regulations govern the collection, use, disclosure, and protection of health-related and other personal information. In addition, certain foreign laws govern the privacy and security of personal data, including health-related data. For example, the General Data Protection Regulation (the “GDPR”), imposes strict requirements for processing the personal data of individuals within the EEA. Privacy and security laws, regulations, and other obligations are constantly evolving, may conflict with each other to complicate compliance efforts, and can result in investigations, proceedings, or actions that lead to significant civil and/or criminal penalties and restrictions on data processing.
Healthcare Reform
The U.S. and some foreign jurisdictions are considering or have enacted a number of legislative and regulatory proposals to change the healthcare system in ways that could affect our ability to sell our products profitably. Among policy makers and payors in the U.S. and elsewhere, there is significant interest in promoting changes in healthcare systems with the stated goals of containing healthcare costs, improving quality or expanding access. Current and future legislative proposals to further reform healthcare or reduce healthcare costs may limit coverage of or lower reimbursement for the procedures associated with the use of our products. The cost containment measures that payors and providers are instituting and the effect of any healthcare reform initiative implemented in the future could impact our revenue from the sale of our products.
The implementation of the Affordable Care Act (“ACA”) in the U.S., for example, has changed healthcare financing and delivery by both governmental and private insurers substantially, and affected medical device manufacturers significantly. The ACA, among other things, provided incentives to programs that increase the federal government’s comparative effectiveness research, and implemented payment system reforms including a national pilot program on payment bundling to encourage hospitals, physicians, and other providers to improve the coordination, quality, and efficiency of certain healthcare services through bundled payment models. Additionally, the ACA expanded eligibility criteria for Medicaid programs and created a new Patient-Centered Outcomes Research Institute to oversee, identify priorities in, and conduct comparative clinical effectiveness research, along with funding for such research.
Since its enactment, there have been judicial, executive and Congressional challenges to certain aspects of the ACA. On June 17, 2021, the U.S. Supreme Court dismissed the most recent judicial challenge to the ACA without specifically ruling on the constitutionality of the ACA. Prior to the Supreme Court’s decision, President Biden issued an executive order to initiate a special enrollment period from February 15, 2021 through August 15, 2021 for purposes of obtaining health insurance coverage through the ACA marketplace. The executive order also instructed certain governmental agencies to review and reconsider their existing policies and rules that limit access to healthcare, including among others, reexamining Medicaid demonstration projects and waiver programs that include work requirements, and policies that create unnecessary barriers to obtaining access to health insurance coverage through Medicaid or the ACA.
In addition, other legislative changes have been proposed and adopted since the ACA was enacted. For example, the Budget Control Act of 2011, among other things, included reductions to Medicare payments to providers, which went into effect on April 1, 2013 and, due to subsequent legislative amendments to the statute, will remain in effect into 2032, with the exception of a temporary suspension from May 1, 2020 through March 31, 2022, unless additional Congressional action is taken. Additionally, the American Taxpayer Relief Act of 2012, among other things, reduced Medicare payments to several providers, including hospitals, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years. In addition, the Medicare Access and CHIP Reauthorization Act of 2015 enacted on April 16, 2015, repealed the formula by which Medicare made annual payment adjustments to physicians and replaced the former formula with fixed annual updates and a new system of incentive payments began in 2019 that are based on various performance measures and physicians’ participation in alternative payment models such as accountable care organizations.
We expect additional state, federal, and foreign healthcare reform measures to be adopted in the future, any of which could limit the amounts that federal, state, and foreign governments will pay for healthcare products and services, which could result in reduced demand for our products or additional pricing pressure.
Anti-Bribery and Corruption Laws
Our U.S. operations are subject to the Foreign Corrupt Practices Act (“FCPA”). We are required to comply with the FCPA, which generally prohibits covered entities and their intermediaries from engaging in bribery or making other prohibited payments to foreign officials for the purpose of obtaining or retaining business or other benefits. In addition, the FCPA imposes accounting standards and requirements on publicly traded U.S. corporations and their foreign affiliates, which are intended to prevent the diversion of corporate funds to the payment of bribes and other improper payments, and to prevent the establishment of “off books” slush funds from which such improper payments can be made. We also are subject to similar anticorruption legislation implemented in Europe under the Organization for Economic Co-operation and Development’s Convention on Combating Bribery of Foreign Public Officials in International Business Transactions.
Segment Information
We manage our business within one reportable segment. Segment information is consistent with how management reviews our business, makes investing and resource allocation decisions, and assesses our operating performance.
Facilities
Our principal office is located at 4875 White Bear Lake, Minnesota, where we lease approximately 11,540 square feet of office space. We lease this space under a lease that terminates on December 31, 2030. We believe that our existing facility is sufficient to meet our needs for the foreseeable future.
We also lease 1,100 square feet of office space in Ausbach, Germany pursuant to a lease that automatically renews each year for a successive one year period, unless the we notify the landlord six (6) months prior to the annual renewal. This lease renewed automatically on January 1, 2024 and again on January 1, 2025.
Employees and Human Capital
As of December 31, 2024, we had approximately 42 employees. A significant number of our employees have a technical background and hold advanced engineering or scientific degrees. We view our investment in human capital to be crucial to our success, and we are committed to ensuring a culture in which employees feel they are part of achieving a common goal.
Our work environment is highly collaborative and one that is based on trust and mutual respect. We believe that the relatively small size of our organization allows our employees to feel pride and ownership in their work and a sense of being part of fulfilling our mission more directly than with larger companies in our industry.
None of our employees is subject to a collective bargaining agreement or represented by a trade or labor union. We consider our relationship with our employees to be good.
Available Information
Our Annual Reports on Form 10-K, Quarterly Reports on Form 10-Q and Current Reports on Form 8-K, and all amendments to those reports, filed with or furnished to the SEC, are available free of charge through the investor relations sections of the Company’s website, https://www.envoymedical.com/investors, as soon as reasonably practicable after we have electronically filed such material with, or furnished it to, the SEC. In addition, the SEC maintains an Internet site that contains reports, proxy and information statements, and other information regarding issuers that file electronically with the SEC at www.sec.gov.
The information on our website is not, and shall not be deemed to be, part of this Report or incorporated into any other filings we make with the SEC, except as shall be expressly set forth by specific reference in any such filings.
ITEM 1A. Risk Factors
Risks Relating to Our Business and Operations
We are an early-stage company with a history of losses. We have not been profitable historically and may not be able to achieve profitability in the future.
We are a development-stage medical device company with a limited operating history. In recent years, we have focused almost exclusively on developing our lead product candidate, the Acclaim CI. We have funded our operations to date primarily through the issuance of our equity securities and convertible debt.
We have a limited operating history upon which you can evaluate our business and prospects. In addition, we have limited experience and have not yet demonstrated an ability to successfully overcome many of the risks and uncertainties frequently encountered by companies in new and rapidly evolving fields, particularly in the medical device industry. To date, we have not generated any revenue from the sale of the Acclaim CI. See Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations for additional information. We have incurred losses in each year since our inception, including net losses of approximately $20.8 million and $29.9 million for the years ended December 31, 2024 and 2023, respectively. As of December 31, 2024 and 2023, we had an accumulated deficit of approximately $284.7 million and $257.3 million, respectively. Substantially all of our operating losses in such years resulted from costs incurred in connection with the development of the Acclaim CI and from general and administrative costs associated with our operations.
We will incur significant expenses related to clinical trials to obtain approval of the FDA to market the Acclaim CI. If we obtain FDA marketing approval for the Acclaim CI we will likely incur significant sales, marketing, and outsourced manufacturing expenses, as well as continued research and development expenses. Furthermore, now that the Business Combination has been completed, we expect to incur additional costs associated with operating as a public company. As a result, we expect to continue to incur significant and increasing operating losses for the foreseeable future. Because of the numerous risks and uncertainties associated with developing a medical device, we are unable to predict the extent of any future losses or when we will become profitable, if at all.
We expect to continue to incur significant losses until we receive the necessary regulatory approvals to commercialize the Acclaim CI in the United States, which we may not be successful in achieving. We anticipate that our expenses will increase substantially if and as we:
| ● | continue the research and development of the Acclaim CI, including through clinical trials; |
| ● | seek additional regulatory and marketing approvals in jurisdictions outside the United States; |
| ● | establish a sales, marketing, and distribution infrastructure to commercialize our product candidate; |
| ● | rely on our third-party suppliers and manufacturers to obtain adequate supply of materials and components for our products; |
| ● | seek to identify, assess, acquire, license, and/or develop other product candidates and subsequent generations of our current product candidate; |
| ● | seek to maintain, protect, and expand our intellectual property portfolio; |
| ● | seek to identify, hire, and retain skilled personnel; |
| ● | create additional infrastructure to support our operations as a public company and our product candidate development and planned future commercialization efforts; and |
| ● | experience any delays or encounter issues with respect to any of the above, including, but not limited to, failed studies, complex results, safety issues or other regulatory challenges that require longer follow-up of existing studies or additional supportive studies in order to pursue marketing approval. |
The amount of any future operating losses will depend, in part, on the rate of our future expenditures and our ability to obtain funding through equity or debt financings, strategic collaborations, or grants. Even if we obtain regulatory approvals to market the Acclaim CI or any future product candidates, our future revenue will depend upon the size of any markets in which our products and product candidates receive approval and our ability to achieve sufficient market acceptance, pricing and reimbursement from third-party payors for our products and product candidates. Further, the operating losses that we incur may fluctuate significantly from quarter-to-quarter and year to year, such that a period-to-period comparison of our results of operations may not be a good indication of our future performance. Other unanticipated costs may also arise. If we continue to generate operating losses, there will be an adverse effect on our results of operations, financial condition, and the market price of our Class A Common Stock.
We have generated limited revenue from product sales and may never be profitable.
While we have historically obtained revenue from our legacy Esteem FI-AMEI product, such revenue has been limited, and we have not generated any revenue from sales of the Acclaim CI. Our ability to generate revenue and achieve profitability mainly depends on our ability to obtain FDA approval for the Acclaim CI and, if we obtain such approval, to successfully scale up production and market the device. We do not know when, or if, we will generate any such revenue. Our ability to generate future revenue from product sales will depend heavily on our success in many areas, including but not limited to:
| ● | completing research and development of the Acclaim CI in a timely and successful manner; |
| ● | completing our pivotal clinical study in the United States successfully; |
| ● | obtaining FDA approval for the Acclaim CI; |
| ● | maintaining and enhancing a commercially viable, sustainable, scalable, reproducible and transferable manufacturing process for the Acclaim CI that is compliant with current good manufacturing practices, (“cGMP”); |
| ● | establishing and maintaining supply and, if applicable, manufacturing relationships with third parties that can provide, in both amount and quality, adequate products to support development and the market demand for the Acclaim CI, if and when it is approved; |
| ● | identifying, assessing, acquiring and/or developing new product candidates; |
| ● | launching and commercializing any product candidates for which we obtain regulatory and marketing approval, either directly by establishing a sales force, marketing and distribution infrastructure, and/or with collaborators or distributors in the United States, Europe and other potential markets that we will target; |
| ● | accurately identifying demand for the Acclaim CI and any future product candidates; |
| ● | exposing and educating physicians and other medical professionals with respect to the use of our products; |
| ● | obtaining market acceptance of the Acclaim CI and any future product candidates from the medical community and third-party payors; |
| ● | ensuring our product candidates are approved for reimbursement from governmental agencies, health care providers and insurers in jurisdictions where they have been approved for marketing; |
| ● | addressing any competing technological and market developments that impact the Acclaim CI and any future product candidates or their prospective usage by medical professionals; |
| ● | negotiating favorable terms in any collaboration, licensing or other arrangements into which we may enter and performing our obligations under such arrangements; |
| ● | maintaining, protecting and expanding our portfolio of intellectual property rights, including patents, patent applications, trade secrets and know-how; |
| ● | avoiding and defending against third-party interference or infringement claims; and |
| ● | attracting, hiring and retaining qualified personnel. |
We anticipate incurring significant incremental costs associated with commercializing the Acclaim CI. Our expenses could increase beyond expectations if we are required by the FDA, or other domestic or foreign regulatory agencies, to change our product design or manufacturing processes or to perform studies in addition to those that we currently anticipate. Even if we are successful in obtaining regulatory approvals to market the Acclaim CI, our revenue earned from such product candidate will be dependent in part upon the size of the markets in the territories for which we gain regulatory approval for such product candidate, the accepted price for such product candidate, our ability to obtain reimbursement for such product candidate at any price, and the expenses associated with manufacturing and marketing such product candidate for such markets. Therefore, we may not generate significant revenue from the sale of the Acclaim CI, even if we obtain FDA approval. Further, if we are not able to generate significant revenue from the sale of our approved products, we may be forced to curtail or cease our operations, in which case our investors may lose the full amount of their investment in us. Due to the numerous risks and uncertainties involved in product development, it is difficult to predict the timing or amount of increased expenses, or when, or if, we will be able to achieve or maintain profitability.
If the Acclaim CI contains design or manufacturing defects, our business and financial results could be harmed.
To date, we have completed initial patient implants of the Acclaim CI as part of our early feasibility study, and we received approval from the FDA to begin our pivotal trial, which we began in the first quarter of 2025. As the Acclaim CI has no history of commercial operation, we have a limited frame of reference from which to evaluate its long-term performance. There can be no assurance that we will be able to detect and fix any defects in the Acclaim CI in time to maintain our FDA trial schedule. Once we have commenced with implantation in additional patients, we may discover latent defects in design, manufacture or construction that may cause our systems not to perform as expected or to cause side effects. The Acclaim CI also requires software to operate, which may need to be modified and updated over time.
There can be no assurance that we will be able to detect and fix any defects in the hardware or software of the Acclaim CI on the timescale necessary to maintain our clinical trial schedule, or at all. Further, such defects may not become apparent until our systems are implanted in patients and may cause adverse effects that cause harm to patients and require redesign of the Acclaim CI, which may result in great expense, harm to our reputation, and harm to our results of operations, financial condition, and the trading price of the Class A Common Stock.
We expect that we will need to raise substantial additional funding, which may not be available on acceptable terms, or at all. Failure to obtain funding on acceptable terms and on a timely basis may require us to curtail, delay or discontinue our product development efforts or other operations.
The expenses we were obligated to pay in relation to the Business Combination were substantial. As result, we will require substantial additional capital to commercialize the Acclaim CI. In addition, our operating plans may change as a result of many factors that may currently be unknown to us, and we may need to seek additional funds sooner than planned. Our future funding requirements will depend on many factors, including but not limited to:
| ● | the progress, results and costs of our planned studies and pivotal clinical trials; |
| ● | the cost, timing and outcomes of regulatory review of the Acclaim CI; |
| ● | the scope, progress, results and costs of product development, testing, manufacturing, preclinical development and, if applicable, clinical trials for any other product candidates that we may develop or otherwise obtain in the future; |
| ● | the costs of manufacturing the Acclaim CI, including costs related to engaging third-party manufacturers therefor; |
| ● | the cost of our future activities, including establishing sales, marketing and distribution capabilities for any product or product candidates in any particular geography where we receive marketing approval for such product candidates; |
| ● | the terms and timing of any collaborative, licensing and other arrangements that we may establish; |
| ● | the costs of preparing, filing and prosecuting patent applications, maintaining and enforcing our intellectual property rights and defending intellectual property-related claims; and |
| ● | the level of revenue, if any, received from commercial sales of any product candidates for which we receive marketing approval. |
Any additional fundraising efforts may divert our management from their day-to-day activities, which may adversely affect our ability to develop and commercialize the Acclaim CI. In addition, we cannot guarantee that future financing will be available in sufficient amounts or on terms acceptable to us, if at all. Moreover, the terms of any financing may adversely affect the holdings or the rights of holders of our securities and the issuance of additional securities, whether equity or debt, by us, or the possibility of such issuance, may cause the value of our securities to decline. The incurrence of indebtedness could result in increased fixed payment obligations, and we may be required to agree to certain restrictive covenants, such as limitations on our ability to incur additional debt, limitations on our ability to acquire, sell or license intellectual property rights and other operating restrictions that could adversely impact our ability to conduct our business.
If we are unable to obtain funding on a timely basis, we may be required to significantly curtail, delay or discontinue our research and development program or the development or commercialization, if any, of the Acclaim CI or be unable to expand our operations or otherwise capitalize on our business opportunities, as desired, which could materially and adversely affect our business, financial condition, results of operations and value of our securities.
Raising additional capital would cause dilution to our existing stockholders, which may adversely affect the rights of existing stockholders.
We may seek additional capital through a combination of private and public equity offerings, debt financings and collaborations, and strategic and licensing arrangements. To the extent that we raise additional capital through the issuance of equity or otherwise, including through additional preferred stock or convertible debt securities, your ownership interest will be diluted, and the terms may include liquidation or other preferences that adversely affect your rights as a stockholder. Future sales of our Class A Common Stock or of securities convertible into our Class A Common Stock, or the perception that such sales may occur, could cause immediate dilution and adversely affect the value of our Class A Common Stock.
Failure of a key information technology system, process or site could have an adverse effect on our business.
We rely extensively on information technology systems to conduct our business. These systems affect, among other things, ordering and managing materials from suppliers, summarizing and reporting results of operations, complying with regulatory, legal or tax requirements, data security, and other processes necessary to manage our business. Our information technology systems and those of our third-party service providers, vendors, strategic partners and other contractors or consultants are vulnerable to damage or interruption from computer viruses and malware (e.g., ransomware), natural disasters, terrorism, war, telecommunication and electrical failures, hacking, cyberattacks, phishing attacks and other social engineering schemes, malicious code, employee theft or misuse, human error, fraud, denial or degradation of service attacks, sophisticated nation-state and nation-state-supported actors or unauthorized access or use by persons inside our organization, or persons with access to systems inside our organization. The risk of a security breach or disruption, particularly through cyberattacks or cyber intrusion, including by computer hackers, foreign governments and cyber terrorists, has generally increased as the number, intensity and sophistication of attempted attacks and intrusions from around the world have increased and evolved. As a result of the COVID-19 pandemic, we and our third-party service providers and partners may also face increased cybersecurity risks due to our reliance on internet technology and the number of our employees who are working remotely, which may create additional opportunities for cybercriminals to exploit vulnerabilities. Although we have implemented cybersecurity protections to safeguard our data, including our patient and subject data, we can provide no assurances that these protections will prevent all cybersecurity breaches. We primarily use common off-the-shelf software systems, such as Microsoft 365, which receive frequent security updates from the software providers. We also utilize a third-party vendor to maintain our IT system networks, and as a result of limited internal IT resources, we are only able to perform limited due diligence on our third-party IT vendors. We receive periodic security monitoring from our cybersecurity insurance provider.
However, because the techniques used to obtain unauthorized access to, or to sabotage, systems change frequently and often are not recognized until launched against a target, we may be unable to anticipate these techniques or implement adequate preventative measures. We may experience security breaches that may remain undetected for an extended period. Even if identified, we may be unable to adequately investigate or remediate incidents or breaches due to attackers increasingly using tools and techniques that are designed to circumvent controls, to avoid detection, and to remove or obfuscate forensic evidence. Our third-party service providers and partners are also subject to these heightened risks. If our systems are damaged or cease to function properly due to any number of causes, ranging from catastrophic events to power outages to security breaches, and our business continuity plans do not effectively compensate on a timely basis, we may experience interruptions in our operations, which could have an adverse effect on our business.
We and certain of our service providers are from time to time subject to cyberattacks and security incidents. While we do not believe that we have experienced any significant system failure, accident or security breach to date, if such an event were to occur, it could lead to unauthorized access, disclosure and use of non-public information, including information from the patient information we create, receive, maintain or transmit, which are governed by HIPAA and other laws. Any such access, disclosure, or other loss of information could result in legal claims or proceedings, liability under laws that protect the privacy of personal information, and damage to our reputation, which would, in turn, materially and adversely affect our results of operations, financial condition, liquidity, and the value of our securities.
Unfavorable global economic conditions could adversely affect our business, financial condition or results of operations.
Our results of operations could be adversely affected by general conditions in the global economy and in the global financial markets. The global financial crisis caused extreme volatility and disruptions in the capital and credit markets. Factors such as geopolitical events (including the ongoing war in Ukraine and the military conflict in Israel and Gaza), inflationary pressures, impacts from the COVID-19 pandemic, and the U.S. election cycles have contributed to this volatility. Recently, among other effects, volatile economic conditions have caused high levels of inflation, increases in interest rates by central banks with the intent of slowing inflation, and a reduction of available capital following increased interest rates. These global economic conditions could result in a variety of risks to our business, including difficulty in raising funding from capital markets and increased interest rates on loans used to finance our business. Such impacts would materially and adversely affect our financial condition, liquidity and the value of our securities.
Our primary exposures to inflationary pressures to date have been through increases in the market cost of employee compensation, third-party vendor pricing, and component procurement. In particular, since 2022, we have had to increase employee salaries and benefits to aid employee retention and to compete for new employees. If labor costs in our market continue to rise, we expect we will need to continue to increase our compensation levels. We have also seen an increase in pricing from third-party vendors such as advisors, attorneys, and consultants. The per part pricing of components has also increased, and, in many instances, without advanced warning. If we increase production of the Acclaim CI for clinical trials and, if the Acclaim CI obtains FDA approval, eventual commercialization, we will also have greater exposure to rising costs of components if inflation rates remain high. These increases in expenses could materially and adversely affect our financial condition, liquidity and the trading price of our securities.
Recent increases in interest rates may also affect our ability to finance the continued development of the Acclaim CI, the cost of FDA trials, and additional costs of commercializing the Acclaim CI. In recent years, we have financed our operations through convertible loans from a related party, which we believe to have been favorable to us at below market interest rates. However, we expect that loans on such favorable terms will no longer be available to us now that the Business Combination has been consummated, and increased interest rates would make borrowing more expensive and may reduce the availability of equity financing. Our inability to raise additional funds on favorable terms, or at all, would materially and adversely affect our results of operations, financial condition, liquidity, the trading price of our securities, and our growth prospects.
As we have begun producing additional units of our Acclaim CI for the clinical trial process, we are exposed to the risk of supply chain disruptions from events such as tariffs and trade wars, the ongoing war in Ukraine and the military conflict in Israel and Gaza, and other global, national, regional, and local events that cannot yet be predicted. Our supply chain risk will be increased if we are able to obtain FDA approval for the Acclaim CI and begin commercial scale production. Supply constraints resulting from such events may also cause or exacerbate inflation. If such events prevent us from obtaining necessary components for production of Acclaim CI devices, or substantially raise the prices for such components, we may be delayed in the FDA trial process, or we may be unable to produce sufficient Acclaim CI devices to meet demand, which would materially and adversely affect our results of operations and financial condition.
We have identified material weaknesses in our internal control over financial reporting. If we are unable to remediate these material weaknesses, or if we identify additional material weaknesses in the future or otherwise fail to maintain an effective system of internal control over financial reporting, we may not be able to accurately or timely report our financial condition or results of operations, which may adversely affect investor confidence in us and the value of our common stock.
As a privately held company, we were not required to evaluate our internal control over financial reporting in a manner that meets the standards of publicly traded companies required by Section 404(a) of the Sarbanes-Oxley Act. As a public company, we are required to provide management’s attestation on internal control over financial reporting. If we are unable to establish or maintain appropriate internal control over financial reporting or implement these additional requirements in a timely manner or with adequate compliance, it could result in material misstatements in our consolidated financial statements, failure to meet our reporting obligations on a timely basis, increases in compliance costs, and subject us to adverse regulatory consequences, all of which may adversely affect investor confidence in us and the value of our Class A Common Stock.
In connection with the preparation and audit of our consolidated financial statements as of and for the years ended December 31, 2024, 2023 and 2022, material weaknesses were identified in our internal control over financial reporting. A material weakness is a deficiency, or combination of deficiencies, in internal control over financial reporting such that there is a reasonable possibility that a material misstatement of our financial statements will not be prevented or detected on a timely basis. The following material weaknesses were identified:
| ● | We do not maintain a sufficient complement of personnel with accounting knowledge, experience and training to appropriately analyze, record and disclose certain accounting matters to provide reasonable assurance of preventing material misstatements. |
| ● | Our management does not implement a formal risk assessment that addresses risks relevant to financial reporting objectives, including cybersecurity and fraud risks. |
| ● | We have not designed, documented and maintained formal accounting policies, procedures and controls over significant accounts and disclosures to achieve complete, accurate and timely financial accounting, reporting and disclosures, including segregation of duties and adequate controls related to the preparation, posting, modification and review of journal entries, and the accounting treatment of complex transactions, including fair value measurement under GAAP. |
| ● | We have not designed and maintained effective controls over certain information technology general controls for information systems that are relevant to the preparation of our consolidated financial statements, including ineffective controls around user access and segregation of duties. |
The material weaknesses related to the insufficient complement of personnel and formal accounting policies, and the lack of procedures and controls resulted in adjustments to several accounts and disclosures. The information technology deficiencies did not result in a material misstatement to the consolidated financial statements; however, the deficiencies, when aggregated, could result in potential misstatements that would not be prevented or detected. Each of these material weaknesses could result in a material misstatement to the annual or interim consolidated financial statements that would not be prevented or detected.
We have begun implementation of a plan to remediate these material weaknesses. These remediation measures are ongoing and include the following steps:
| ● | hiring additional accounting and financial reporting personnel with appropriate technical accounting knowledge and public company experience in financial reporting; |
| ● | designing and implementing effective processes and controls over significant accounts and disclosure; |
| ● | designing and implementing security management and change management controls over information technology systems, including adjusting user access levels and implementing external logging of activity and periodic review of such logs; and |
| ● | engaging an accounting advisory firm to assist with the documentation, evaluation, remediation and testing of our internal control over financial reporting based on the criteria established in “Internal Control — Integrated Framework’’ issued by the Committee of Sponsoring Organizations of the Treadway Commission. |
While we are designing and implementing measures to remediate our existing material weaknesses, we cannot predict the success of such measures or the outcome of its assessment of these measures at this time. Our current controls and any new controls that we develop may become inadequate because of changes in conditions in our business, personnel, information technology systems and applications, or other factors. If we fail to remediate our existing material weaknesses or identify new material weaknesses in our internal control over financial reporting, if we are unable to comply with the requirements of Section 404 of the Sarbanes-Oxley Act in a timely manner, or if we are unable to conclude that our internal control over financial reporting is effective, it is possible that a material misstatement of our financial statements would not be prevented or detected on a timely basis, investors may lose confidence in the accuracy and completeness of our financial reports, and the value of our securities could be materially and adversely affected.
Our financial statements contain an explanatory paragraph regarding substantial doubt about our ability to continue as a going concern, which could prevent us from obtaining new financing on reasonable terms or at all.
As described in our accompanying financial statements, our audited financial statements as of December 31, 2024 contain an explanatory paragraph regarding substantial doubt about our ability to continue as a going concern. This going concern opinion could materially limit our ability to raise additional funds through the issuance of equity or debt securities or otherwise. Future financial statements may include an explanatory paragraph with respect to our ability to continue as a going concern. Until we can generate significant recurring revenues, we expect to satisfy our future cash needs through debt or equity financing. We cannot be certain that additional funding will be available to us on acceptable terms, if at all. If funds are not available, we may be required to delay, reduce the scope of, or eliminate research or development plans for, or commercialization efforts with respect to our products. This continues to raise substantial doubt about our ability to continue as a going concern.
We are a development-stage company and are subject to all of the risks inherent in the establishment of a new product. We may not receive, or may be delayed in receiving, the necessary approval or clearance for the Acclaim CI.
Furthermore, even if our technology receives the necessary regulatory approvals and becomes commercially viable, our business models may not generate sufficient revenue necessary to support our business. If we are unable to address any issues mentioned above, or encounter other problems, expenses, difficulties, complications, and delays in connection with the establishment and expansion of our business, our entire business may fail, in which case you may lose part of, or your entire investment.
We have a history of net losses and negative cash flow from operations since our inception and we expect such losses and negative cash flows from operations to continue in the foreseeable future. We anticipate our losses will continue to increase from current levels because we expect to incur additional costs related to developing our business, including research and development costs, manufacturing costs, employee-related costs, costs of complying with government regulations, intellectual property development and prosecution costs, marketing and promotion costs, capital expenditures, general and administrative expenses, and costs associated with operating as a public company.
Our ability to generate revenue from our operations and, ultimately, achieve profitability will depend on, among other factors, whether we can complete the development and commercialization of our product candidate, whether we can manufacture the Acclaim CI on a commercial scale in such amounts and at such costs as we anticipate, and whether we can achieve market acceptance of our products, services and business models. We may never generate any revenue or operate on a profitable basis. Even if we achieve profitability, we may not be able to sustain it. If we are unable to achieve sustainable profitability, our financial condition and the price of our securities will be materially and adversely affected.
The FDA trial process is uncertain. Clinical failure can occur at any stage of clinical development. Our clinical experience to date does not necessarily predict future results and may not have revealed certain potential limitations of the technology or potential complications from the Acclaim CI and may require further clinical validation. Any product version we advance through clinical trials may not have favorable results in later clinical trials or receive regulatory approval. We cannot predict the timing of clinical trial results, availability of regulatory personnel, or delays, constraints or outcomes of any regulatory submissions or approvals.
Clinical failure can occur at any stage of clinical development. We have received approval from the FDA to begin our pivotal trial, which we began in the first quarter of 2025. As we have limited clinical experience, our ability to identify potential problems and/or inefficiencies concerning current and future versions of the Acclaim CI in advance of its use in general and expanded groups of patients may be limited, and we cannot assure you that actual clinical performances will be satisfactory to support proposed indications and regulatory approvals and clinical acceptance and adoption, or that its use will not result in unanticipated complications. If the results of our feasibility study are not satisfactory, our U.S. pivotal study could be delayed or may not occur. Furthermore, there can be no assurance that the implementation of our plan will be successful. In addition, the results of our clinical trials are subject to human analyses and interpretation of the data accumulated, which could be affected by various errors due to, among other factors, lack of sufficient clinical experience with the Acclaim CI, assumptions used in the statistical analysis of results, interpretation errors in the analysis of the clinical trials results, or uncertainty in the actual efficacy of the Acclaim CI in its current clinical stage. Therefore, the safety and efficacy of the Acclaim CI and the clinical results to date will require further independent professional validation and clinical study. If the Acclaim CI does not function as expected over time, we may not be able to develop the Acclaim CI at the rate or to the stage we desire, we could be subject to liability claims, our reputation may be harmed, the Acclaim CI may not achieve regulatory clearances, and the Acclaim CI may not be widely adopted by healthcare providers and patients. If the Acclaim CI is not widely adopted, our business, financial condition, and results of operations will be materially and adversely affected.
The FDA’s policies may change, and additional government laws, regulations, and policies may be enacted that could prevent, limit, or delay regulatory approval of our product candidates, limit the marketability of our product candidates, or impose additional regulatory obligations on us. The current U.S. presidential administration has proposed significant changes to the structure, operations, and staffing of the federal regulatory agencies, including the FDA. Although the proposals are for more efficient review and less regulation, it is possible that reductions and turnover in staffing, reductions in funding, changes to policy and procedure, and general uncertainty regarding the status of agencies, their staff, and their funding will cause delays in clinical trials for the Acclaim CI or result in the Acclaim CI not receiving FDA approval for commercialization. Any such delays will cause us significant expense by extending our time to commercialization if FDA approval is obtained, if we are able to obtain it at all.
The successful commercialization of the Acclaim CI, if it receives FDA approval, will depend in part on the extent to which governmental authorities and health insurers establish coverage, adequate reimbursement levels and favorable pricing policies. Failure to obtain or maintain coverage and adequate reimbursement for our product candidates could limit our ability to market those products and decrease our ability to generate revenue.
The availability of coverage and the adequacy of reimbursement by governmental healthcare programs such as Medicare and Medicaid, private health insurers and other third-party payors will be essential for most patients to be able to afford the Acclaim CI. Our ability to achieve coverage and acceptable levels of reimbursement for our products by third-party payors will affect our ability to successfully commercialize the Acclaim CI. Even if we obtain coverage for the Acclaim CI by a third-party payor, the resulting reimbursement payment rates may not be adequate. We can provide no assurance that coverage and reimbursement in the United States, the European Union, or elsewhere will be available for any product that we may develop, and any reimbursement that may become available may be decreased or eliminated in the future.
There is significant uncertainty related to third-party payor coverage and reimbursement of newly approved products. In the United States, third-party payors, including private and governmental payors, such as the Medicare and Medicaid programs, play an important role in determining the extent to which new products will be covered. Some third-party payors may require pre-approval of coverage for new or innovative devices before they will reimburse healthcare providers who use such therapies. Although we are confident that the Acclaim CI will be eligible for reimbursement, we cannot guarantee what third-party payors will decide with respect to the coverage and reimbursement for the Acclaim CI, if approved.
Obtaining and maintaining reimbursement status is time consuming, costly and uncertain. The Medicare and Medicaid programs increasingly are used as models for how private payors and other governmental payors develop their coverage and reimbursement policies for drugs and medical devices. However, no uniform policy for coverage and reimbursement for such products exists among third-party payors in the United States. Therefore, coverage and reimbursement for products can differ significantly from payor to payor. As a result, the coverage determination process is often a time consuming and costly process that may require us to provide scientific and clinical support for the use of our products to each payor separately, with no assurance that coverage and adequate reimbursement will be applied consistently or obtained in the first instance. Furthermore, rules and regulations regarding reimbursement change frequently, in some cases at short notice, and we believe that changes in these rules and regulations are likely.
Outside the United States, our international operations will generally be subject to extensive governmental price controls and other market regulations, and we believe the increasing emphasis on cost-containment initiatives in Europe and other countries has and will continue to put pressure on the pricing and usage of our products. In many countries, the prices of medical products are subject to varying price control mechanisms as part of national health systems. Other countries allow companies to fix their own prices for medical products but monitor and control company profits. Additional foreign price controls or other changes in pricing regulation could restrict the amount that we are able to charge for our product candidates, if approved. Accordingly, in markets outside the United States, the reimbursement for our product candidates may be reduced compared with the United States and may be insufficient to generate commercially reasonable revenue and profits.
Moreover, increasing efforts by governmental and third-party payors in the United States and abroad to cap or reduce healthcare costs may cause such organizations to limit both coverage and the level of reimbursement for newly approved products and, as a result, they may not cover or provide adequate payment for our products. We expect to experience pricing pressures in connection with the sale of any of our product candidates due to the trend toward managed healthcare, the increasing influence of health maintenance organizations and additional legislative changes. The downward pressure on healthcare costs in general, particularly prescription drugs and surgical procedures and other treatments, has become very intense. As a result, increasingly high barriers are being erected to the entry of new products.
If we are unable to obtain reimbursement coverage or adequate reimbursement levels, our results of operations, financial condition, the value of our securities, and our future prospects will be materially and adversely affected.
We operate in a very competitive business environment, and if we are unable to compete successfully against our existing or potential competitors, our business, financial condition and results of operations may be adversely affected.
The Acclaim CI will be subject to intense competition. The industry in which we operate is competitive, subject to change and sensitive to the introduction of new products, procedures or other market activities of industry participants. We will compete with large, diversified medical device companies, including Sonova, Demant, Cochlear, and others. We also compete with smaller companies similar to us.
At any time, these competitors and other potential market entrants may develop new products, procedures or treatment alternatives that could render our products obsolete or uncompetitive. In addition, one or more of such competitors may gain a market advantage by developing and patenting competitive products, procedures or treatment alternatives earlier than we can, obtaining regulatory clearances or approvals more rapidly than we can or selling competitive products at prices lower than ours. If medical research were to lead to the discovery of alternative therapies or technologies that better treat or cure hearing loss, our profitability could suffer through a reduction in sales or a loss in market share to a competitor. Many of our current and potential competitors have substantially greater sales and financial resources than we do. These competitors may also have more established distribution networks, a broader offering of products, entrenched relationships with physicians and distributors or greater experience in launching, marketing, distributing and selling products or treatment alternatives. Similarly, we cannot currently anticipate whether or how artificial intelligence may cause significant change in our industry, but our competitors will likely have greater resources than us to implement proprietary artificial intelligence solutions in their businesses, which may give them significant competitive advantages.
We also compete with our competitors to engage the services of independent sales agents, both those presently working with us and those with whom we hope to work as we expand. In addition, we compete with our competitors to acquire technologies and technology licenses complementary to our products or procedures or advantageous to our business. If we are unable to compete successfully against our existing or potential competitors, our business, financial condition and results of operations will be adversely affected, and we may not be able to grow at our expected rate, if at all.
We expect to derive most of our revenues from sales of the Acclaim CI. Our inability to successfully commercialize this product candidate or any subsequent decline in demand for this product candidate, could severely harm our ability to generate revenues.
We are currently dependent on the successful commercialization of the Acclaim CI to generate revenues. As a result, factors adversely affecting our ability to successfully commercialize, or the pricing of or demand for, this product could have a material adverse effect on our financial condition and results of operations. If we are unable to successfully commercialize or create market demand for the Acclaim CI, we will have limited ability to generate revenues.
Furthermore, we may be vulnerable to fluctuations in demand for the Acclaim CI, and a reduction in demand for the Acclaim CI would have a material adverse effect on our results of operations and financial condition. Such fluctuations in demand may be due to many factors, many of which are beyond our control, including, among others:
| ● | market acceptance of a new product, including healthcare professionals’ and patients’ preferences; |
| ● | market acceptance of the clinical safety and performance of the Acclaim CI; |
| ● | development of similarly cost-effective products by our competitors; |
| ● | development delays of the Acclaim CI; |
| ● | adverse medical side effects suffered by patients using the Acclaim CI, whether actually resulting from the use of the Acclaim CI or not; |
| ● | changes in regulatory policies toward hearing loss technologies; |
| ● | changes in regulatory approval, clearance requirements and licensure for our product; |
| ● | third-party claims of intellectual property infringement; |
| ● | budget constraints and the availability of reimbursement or insurance coverage from third-party payors for the Acclaim CI; |
| ● | any developments affecting the long-term implantation and use of the Acclaim CI; and |
| ● | responses from certain of our competitors to the offering of the Acclaim CI. |
If healthcare professionals do not recommend our product to their patients, the Acclaim CI may not achieve market acceptance and we may not become profitable.
If healthcare professionals, including physicians, do not recommend or prescribe our product to their patients, the Acclaim CI may not achieve market acceptance and we may not become profitable. In addition, physicians have historically been slow to change their medical diagnostic and treatment practices because of perceived liability risks arising from the use of new products. Delayed adoption of the Acclaim CI by healthcare professionals could lead to a delayed adoption by patients. Healthcare professionals may not recommend the Acclaim CI until certain conditions have been satisfied, including, among others:
| ● | there is sufficient long-term clinical and health-economic evidence to convince them to alter their existing hearing loss treatments and recommendations; |
| ● | there are recommendations from prominent physicians, educators and/or associations indicating that the Acclaim CI is safe and effective; |
| ● | we obtain favorable data from clinical and health-economic studies for the Acclaim CI; |
| ● | reimbursement or insurance coverage from government and private third-party payors is available; |
| ● | healthcare professionals obtain required approvals and licensures for the handling, storage, dispensing and disposal of the Acclaim CI; and |
| ● | healthcare professionals become familiar with the advantages of the Acclaim CI in comparison to other hearing loss solutions. |
We cannot predict when, if ever, healthcare professionals and patients will adopt the use of the Acclaim CI on a large scale. Even if favorable data is obtained from clinical studies for the regulatory approval of the Acclaim CI, there can be no assurance that prominent physicians would endorse it for use by their patients. If the Acclaim CI does not achieve an adequate level of acceptance by patients, healthcare professionals, and government and private third-party payors, we may not generate significant product revenues, we may not become profitable, in which case our results of operations, cash flows and the value of our securities will be materially and adversely affected.
We will be dependent upon contract manufacturing organizations and material suppliers, making us vulnerable to supply shortages and problems, increased costs and quality or compliance issues, any of which could harm our business.
Our production of Acclaim CI devices is currently limited to production of prototype devices and devices for our early feasibility study. As a result, our purchases of supplies and components are limited to date.
However, we expect that we will need to significantly increase our production rates to meet the supply of Acclaim CI devices needed for our clinical trials and, if the Acclaim CI obtains FDA approval, for eventual commercialization, which we are targeting to obtain in late 2027/early 2028. We also expect that some of the critical materials and components used in manufacturing the Acclaim CI may be sourced from single suppliers, which may expose us to greater risks as we increase production of Acclaim CI devices than if our supplier base were more diversified. For example, our suppliers may encounter problems during manufacturing for a variety of reasons, including, for example, failure to follow specific protocols and procedures, failure to comply with applicable legal and regulatory requirements, equipment malfunction and environmental factors, failure to properly conduct their own business affairs, and infringement of third-party intellectual property rights, any of which could delay or impede their ability to meet our increased requirements. An interruption in the supply of a key component could significantly delay our production of the Acclaim CI or increase our production costs.
When we increase production, our reliance on these third-party suppliers will also subject us to other risks that could harm our business, including:
| ● | we are not, and will not in the near future be, a major customer of many of our suppliers, and these suppliers may therefore give other customers’ needs higher priority than us; |
| ● | we may not be able to obtain an adequate supply of components in a timely manner, on commercially reasonable terms or at all; |
| ● | our suppliers, especially new suppliers, may make errors in manufacturing that could adversely affect the efficacy or safety of our products or cause delays in shipment; |
| ● | we may have difficulty locating and qualifying additional or alternative suppliers; |
| ● | switching components or suppliers may require product redesign and possibly resubmission to the FDA or other similar foreign regulatory agencies, which could impede or delay our commercial activities; |
| ● | one or more of our suppliers may be unwilling or unable to supply components for our products in a timely manner, on commercially reasonable terms or at all; |
| ● | the occurrence of a fire, natural disaster or other catastrophe impacting one or more of our suppliers may affect their ability to deliver products to us in a timely manner or at all; and |
| ● | our suppliers may encounter financial or other business hardships unrelated to our demand, which could inhibit their ability to fulfill our orders and meet our requirements. |
We may not be able to quickly establish additional or alternative suppliers if necessary, in part because we may need to undertake additional activities to establish such suppliers as required by the regulatory approval process. Any interruption or delay in obtaining products from our third-party suppliers, or our inability to obtain products from qualified alternate sources at acceptable prices in a timely manner, could materially impair our ability to meet the demand of our customers and cause them to switch to competing products. Given our reliance on a limited number of suppliers, we may be susceptible to supply shortages while looking for alternate suppliers, which could materially and adversely affect our business, financial condition, results of operations and the trading price of our securities.
Our business plan relies on certain assumptions about the market for our product; however, the size and expected growth of our addressable market has not been established with precision and may be smaller than we estimate, and even if the addressable market is as large as we have estimated, we may not be able to capture market share.
Our estimates of the addressable market for the Acclaim CI are based on a number of internal and third-party estimates and assumptions. While we believe our assumptions and the data underlying our estimates are reasonable, these assumptions and our estimates may not be correct. As a result, the projected demand for our products could materially differ from actual demand if our assumptions regarding these trends and acceptance of our products by the medical community prove to be incorrect or do not materialize, or if non-surgical treatments gain more widespread acceptance. In addition, even if the Acclaim CI gains acceptance, technological or medical advances could provide alternatives to address hearing loss that are less invasive or offer other benefits over Acclaim CI. As a result, our estimates of the addressable market for our current or future products and procedures may prove to be incorrect. If the addressable market is not as large as we believe, our business, financial condition and results of operations and business prospects would be materially and adversely affected.
We depend on third parties to manage our pre-clinical studies and clinical trials, perform related data collection and analysis, and to enroll patients for our clinical trials, and, as a result, we may face costs and delays that are beyond our control.
We rely upon third-party vendors, including Contract Research Organization (“CROs”), to monitor and manage data for our ongoing clinical trial. We also rely on CROs for execution of our clinical trial. Although we control only certain aspects of their activities, we are and will be responsible for ensuring that each of our studies is conducted in accordance with the applicable protocol, legal, regulatory and scientific standards, and our reliance on the vendors and CROs does not relieve us of our regulatory responsibilities. We and our CROs and other vendors are required to comply with good clinical practice (“GCP”), cGMP, the Helsinki Declaration, the International Conference on Harmonization Guideline for Good Clinical Practice, applicable European Commission Directives on Clinical Trials, laws and regulations applicable to clinical trials conducted in other territories, and good laboratory practices, which are regulations and guidelines enforced by the FDA, the Competent Authorities of the Member States of the EEA, and comparable foreign regulatory authorities for all of our product candidates in clinical development. Regulatory authorities enforce these regulations through periodic inspections of study sponsors, principal investigators, study sites and other contractors. If we or any of our CROs or vendors fail to comply with applicable regulations, including GCP and cGMP regulations, the clinical data generated in our clinical studies may be deemed unreliable and the FDA, European Medicines Agency (“EMA”), or comparable foreign regulatory authorities may require us to perform additional clinical trials before approving our marketing applications. Our failure to comply with these regulations may require us to repeat clinical trials, which would delay the regulatory approval process.
If any of our relationships with third-party CROs or vendors terminate, we may not be able to enter into arrangements with alternative CROs or vendors or do so on commercially reasonable terms. In addition, our CROs are not our employees, and, except for remedies available to us under our agreements with such CROs, we cannot control whether they devote sufficient time and resources to our ongoing clinical programs. If our CROs do not successfully carry out their contractual duties or obligations or meet expected deadlines, if they need to be replaced or if the quality or accuracy of the clinical data they obtain is compromised due to their failure to adhere to our clinical protocols, regulatory requirements or for other reasons, our clinical trials may be extended, delayed or terminated, and we may not be able to obtain regulatory approval for or successfully commercialize our product candidates. Our CROs may also generate higher costs than anticipated, which could adversely affect our results of operations and the commercial prospects for our product candidate, increase our costs and delay our ability to generate revenue.
Replacing or finding additional CROs involves additional cost and requires management time and focus. In addition, there is a natural transition period when a new CRO commences work. As a result, delays occur, which can materially impact our ability to meet our desired clinical development timelines. Though we carefully manage our relationships with our CROs, we may encounter similar challenges or delays in the future, which could have a material adverse effect on our business, financial condition and prospects.
We have been and in the future may become a defendant in one or more stockholder derivative, class-action, and other litigation, and any such lawsuits may adversely affect our business, financial condition, results of operations and cash flows.
We and certain of our officers and directors have been and may in the future become defendants in one or more stockholder derivative actions or other class-action lawsuits. For example:
| ● | A lawsuit was filed in November 2023 against Daniel Hirsch, Whitney Haring-Smith, the Sponsor and the Company, as successor to Anzu Special Acquisition Corp I alleging a claim for breach of Anzu’s Amended and Restated Certificate of Incorporation against the Company, a claim for breach of fiduciary duty against Mr. Hirsch, Dr. Haring-Smith and the Sponsor and claims for unjust enrichment, fraudulent misrepresentation and tortious interference with economic relations against the defendants. |
See Part I, Item 3. Legal Proceedings for more information on these lawsuits.
These lawsuits can divert our management’s attention and resources from our ordinary business operations, and we would likely incur significant expenses associated with their defense (including, without limitation, substantial attorneys’ fees and other fees of professional advisors and potential obligations to indemnify current and former officers and directors who are or may become parties to such actions). In connection with these lawsuits, we may be required to pay material damages, consent to injunctions on future conduct and/or suffer other penalties, remedies or sanctions, or issue additional shares upon the exercise of certain warrants, which may cause additional dilution. In addition, any such future lawsuits could adversely impact our reputation and/or ability to launch and commercialize our products, thereby harming our ability to generate revenue. Accordingly, the ultimate resolution of these matters and any future matters could have a material adverse effect on our business, financial condition, results of operation and cash flow and, consequently, could negatively impact the trading price of our Class A Common Stock.
We are highly dependent on key members of our executive management team. Our inability to retain these individuals could impede our business plan and growth strategies, which could have a negative impact on our business and the value of your investment.
Our ability to implement our business plan depends on the continued services of key members of our senior management. In particular, and to a critical extent, we are dependent on the continued efforts and services of the members of our management team. If we lose the services of such key members of our management team, we would likely be forced to expend significant time and money in the pursuit of replacement individuals, which may result in a delay in the implementation of our business plan and plan of operations. We may not be able to find satisfactory replacements on terms that would not be unduly expensive or burdensome to us. We do not currently carry a key-man life insurance policy that would assist us in recouping our costs in the event of the death or disability of our management team. The loss of members of our management team, or our inability to attract or retain other qualified individuals, could have a material adverse effect on our business, results of operations and financial condition.
Certain of our directors and/or officers may have interests that are different from holders of our Class A Common Stock.
Certain of our directors and officers may have different interests than other holders of Class A Common Stock.
As of March 24, 2025, Mr. Taylor, a member of the Board, holds approximately 48.2% of the currently outstanding shares of Class A Common Stock and approximately 24.2% of the outstanding shares of our Series A Preferred Stock. As a result of these holdings, Mr. Taylor has the ability to exert significant influence over matters submitted to a vote of our shareholders. Mr. Lucas, a member of the Board and the Chief Executive Officer, has interest in continued employment with the Company that is different from other holders of Class A Common Stock.
For additional information regarding related party transactions and potential conflicts of interest, see Item 13. Certain Relationships and Related Transactions, and Director Independence.
Our management team has limited experience managing a public company.
The members of our management team have limited experience managing a publicly traded company, interacting with public company investors, and complying with the increasingly complex laws pertaining to public companies in the United States. Our management team may not successfully or efficiently manage our transition to being a public company subject to significant regulatory oversight and reporting obligations under the U.S. federal securities laws and the continuous scrutiny of securities analysts and investors. These new obligations and constituents require significant attention from our senior management and could divert their attention away from the day-to-day management of our business, which could adversely affect our business, financial condition, results of operations and prospects.
Risks Relating to Our Intellectual Property
If we are unable to obtain significant patent protection for our products, or if our patents and other intellectual property rights do not adequately protect our products, we may be unable to gain significant market share and be unable to operate our business profitably.
We rely on patents, trade secrets, copyrights, know-how, trademarks, license agreements and contractual provisions to establish our intellectual property rights and protect our products. These legal means, however, afford only limited protection and may not completely protect our rights.
As of March 10, 2025, we had rights to 35 issued U.S. patents, which are estimated to expire between 2025 and 2043 assuming all required fees are paid, 13 pending U.S. patent applications, 33 issued foreign patents and 32 pending foreign and international patent applications. We cannot assure you that our intellectual property position will not be challenged or that all patents for which we have applied will be granted. The validity and breadth of claims in patents involve complex legal and factual questions and, therefore, may be highly uncertain. Uncertainties and risks that we face include the following:
| ● | our pending or future patent applications may not result in the issuance of patents; |
| ● | the scope of any existing or future patent protection may not exclude competitors or provide competitive advantages to us; |
| ● | our patents may not be held valid or enforceable if subsequently challenged; |
| ● | other parties may claim that our products and designs infringe the proprietary rights of others and even if we are successful in defending our patents and proprietary rights, the cost of such litigation may adversely affect our business; and |
| ● | other parties may develop similar products, duplicate our products, or design around our patents. |
The patent prosecution process is expensive and time-consuming, and we may not be able to file, prosecute, maintain, enforce or license all necessary or desirable patent applications at a reasonable cost or in a timely manner, or in all jurisdictions. We may choose not to seek patent protection for certain innovations and may choose not to pursue patent protection in certain jurisdictions, and under the laws of certain jurisdictions, patents or other intellectual property rights may be unavailable or limited in scope. It is also possible that we will fail to identify patentable aspects of our developments before it is too late to obtain patent protection.
In addition, the laws of foreign jurisdictions may not protect our rights to the same extent as the laws of the United States. For example, most countries outside of the United States do not allow patents for methods of treating the human body. This may preclude us from obtaining method patents outside of the United States having similar scope to those we have obtained or may obtain in the future in the United States. Changes in either the patent laws or interpretation of the patent laws in the United States and other countries may diminish the value of our patents or narrow the scope of our patent protection.
Moreover, we may be subject to a third-party pre-issuance submission of prior art to the U.S. Patent and Trademark Office (the “USPTO”) or patent offices in foreign jurisdictions, or become involved in opposition, derivation, reexamination, inter partes review, post-grant review or interference proceedings challenging our patent rights or the patent rights of others. An adverse determination in any such submission, proceeding or litigation could reduce the scope of, or invalidate, our patent rights, allow third parties to commercialize our technology and compete directly with us, without payment to us.
The issuance of a patent is not conclusive as to its inventorship, scope, validity or enforceability, and our patents may be challenged in the courts or patent offices in the United States and abroad. Such challenges may result in loss of exclusivity or freedom to operate or in patent claims being narrowed, invalidated or held unenforceable, in whole or in part, which could limit our ability to stop others from using or commercializing similar or identical products and techniques, or limit the duration of the patent protection of our technology.
While we are aware of several third-party patents of interest, we do not believe that any of our products infringe any valid claims of patents or other proprietary rights held by others. However, there can be no assurances that we do not infringe any patents or other proprietary rights held by third parties. If our products were found to infringe any proprietary right of another party, we could be required to pay significant damages or license fees to such party and/or cease production, marketing and distribution of those products.
We also rely on trade secrets and other unpatented proprietary technology. There can be no assurances that we can meaningfully protect our rights in our unpatented proprietary technology or that others will not independently develop substantially equivalent proprietary products or processes or otherwise gain access to our proprietary technology. We seek to protect our trade secrets and proprietary know-how, in part, with confidentiality agreements with employees and consultants that include customary intellectual property assignment obligations. Litigation may also be necessary to defend infringement claims of third parties or to enforce patent rights we hold or to protect trade secrets or techniques we own. There can be no assurances, however, that the agreements will not be breached, adequate remedies for any breach would be available or competitors will not discover our trade secrets or independently develop comparable intellectual property. If we are unable to successfully protect our intellectual property, our business, financial condition, and results of operations will be materially and adversely affected.
Obtaining and maintaining patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements.
The USPTO and various foreign governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other similar provisions during the patent application process. In addition, periodic maintenance fees, renewal fees, annuity fees and various other government fees on issued patents often must be paid to the USPTO and foreign patent agencies over the lifetime of the patent and/or applications and any patent rights we may obtain in the future. While an unintentional lapse of a patent or patent application can in many cases be cured by payment of a late fee or by other means in accordance with the applicable rules, there are situations in which noncompliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. Non-compliance events that could result in abandonment or lapse of a patent or patent application include, but are not limited to, failure to respond to official actions within prescribed time limits, non-payment of fees and failure to properly legalize and submit formal documents. If we fail to maintain the patents and patent applications covering our products, we may not be able to stop a competitor from marketing products that are the same as or similar to our products, which would have a material adverse effect on our business.
We may become a party to lawsuits or administrative proceedings involving patents or other intellectual property. If we were to lose any future intellectual property lawsuits, a court could require us to pay significant damages and/or prevent us from selling our products.
We may become a party to lawsuits or administrative proceedings involving patents or other intellectual property, including interference proceedings, post grant review and inter partes review before the USPTO or the equivalent foreign patent authority. A legal proceeding, regardless of the outcome, could drain our financial resources and divert the time and effort of our management. Protracted litigation to defend or prosecute our intellectual property rights could result in our customers or potential customers deferring or limiting their purchase or use of the affected products until resolution of the litigation.
If we are found to infringe a third party’s intellectual property rights, we could be required to obtain a license from such third party to continue selling, developing and marketing our products and techniques. However, we may not be able to obtain any required license on commercially reasonable terms or at all. Even if we were able to obtain a license, it could be non-exclusive, thereby giving our competitors access to the same technologies licensed to us. We could be forced, including by court order, to cease commercializing the infringing technology or product. In addition, we could be found liable for monetary damages, including treble damages and attorneys’ fees if we are found to have willfully infringed a patent. A finding of infringement could force us to cease some of our business operations, which could materially harm our business. Claims that we have misappropriated the confidential information or trade secrets of third parties could have a similar negative impact on our business. Intellectual property litigation may lead to unfavorable publicity that harms our reputation and causes the value of our securities to decline.
Because competition in our industry is intense, competitors may infringe or otherwise violate our issued patents, patents of our licensors or other intellectual property. To counter infringement or unauthorized use, we may be required to file infringement claims, which can be expensive and time consuming, and could distract our technical and management personnel from their normal responsibilities. Any claims we assert against perceived infringers could provoke these parties to assert counterclaims or file administrative actions against us alleging that we infringe their patents. In addition, in a patent infringement proceeding, a court may decide that a patent of ours is invalid or unenforceable, in whole or in part, construe the patent’s claims narrowly or refuse to stop the other party from using the technology at issue on the grounds that our patents do not cover the technology in question. An adverse result in any litigation proceeding or administrative action could put one or more of our patents at risk of being invalidated or interpreted narrowly. Our competitors may assert invalidity on various grounds, including lack of novelty, obviousness or that we were not the first applicant to file a patent application related to our product. We may elect to enter into license agreements in order to settle patent infringement claims or to resolve disputes before litigation, and any such license agreements may require us to pay royalties and other fees that could be significant. Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure.
Our competitors, many of which have made substantial investments in patent portfolios, trade secrets, trademarks and competing technologies, may have applied for or obtained, or may in the future apply for or obtain, patents or trademarks that may prevent, limit or otherwise interfere with our ability to make, use, sell and/or export our products or to use our technologies or product names. Moreover, individuals and groups that are non-practicing entities, commonly referred to as “patent trolls,” purchase patents and other intellectual property assets for the purpose of making claims of infringement in order to extract settlements. From time to time, we may receive threatening letters, notices or “invitations to license,” or may be the subject of claims that our products and business operations infringe or violate the intellectual property rights of others. The defense of these matters can be time consuming, costly to defend in litigation, divert management’s attention and resources, damage our reputation and brand and cause us to incur significant expenses or make substantial payments. Negative results in litigation regarding our intellectual property, or the requirement to make substantial expenditures in litigation (regardless of whether we ultimately prevail) would have material adverse effect on our liquidity, business, financial condition, results of operations, and the value of our securities.
If we fail to execute invention assignment agreements with our employees and contractors involved in the development of intellectual property or are unable to protect the confidentiality of our trade secrets, the value of our products and our business and competitive position could be harmed.
In addition to patent protection, we also rely on protection of copyright, trade secrets, know-how and confidential and proprietary information. We generally enter into confidentiality and invention assignment agreements with our employees, consultants and third parties upon their commencement of a relationship with us. However, we may not enter into such agreements with all employees, consultants and third parties who have been involved in the development of our intellectual property. In addition, these agreements may not provide meaningful protection against the unauthorized use or disclosure of our trade secrets or other confidential information, and adequate remedies may not exist if unauthorized use or disclosure were to occur. The exposure of our trade secrets and other proprietary information would impair our competitive advantages and could have a material adverse effect on our business, financial condition and results of operations. In particular, a failure to protect our proprietary rights may allow competitors to copy our products and procedures, which could adversely affect our pricing and market share. Further, other parties may independently develop substantially equivalent know-how and technology.
In addition to contractual measures, we try to protect the confidential nature of our proprietary information using commonly accepted physical and technological security measures. Such measures may not provide adequate protection for our proprietary information, such as in the case of misappropriation of a trade secret by an employee or third party with authorized access. Our security measures may not prevent an employee or consultant from misappropriating our trade secrets and providing them to a competitor, and recourse we take against such misconduct may not provide an adequate remedy to protect our interests fully. Unauthorized parties may also attempt to copy or reverse engineer certain aspects of our products that we consider proprietary. Enforcing a claim that a party illegally disclosed or misappropriated a trade secret can be difficult, expensive and time-consuming, and the outcome is unpredictable. Even though we use commonly accepted security measures, trade secret violations are often a matter of state law, and the criteria for protection of trade secrets can vary among different jurisdictions. In addition, trade secrets may be independently developed by others in a manner that could prevent legal recourse by us. While we have agreements with many of our employees, consultants and third parties that obligate them to assign their inventions to us, these agreements may not be self-executing, not all employees or consultants may enter into such agreements, or employees or consultants may breach or violate the terms of these agreements, and we may not have adequate remedies for any such breach or violation. If any of our intellectual property or confidential or proprietary information, such as our trade secrets, were to be disclosed or misappropriated, or if any such information was independently developed by a competitor, it could have a material adverse effect on our competitive position, business, financial condition, results of operations and prospects.
If our trademarks and trade names are not adequately protected, we may not be able to build name recognition in our markets of interest and our competitive position may be harmed.
We rely on our trademarks, trade names and brand names to distinguish our products from the products of our competitors and have registered or applied to register many of these trademarks. There can be no assurance that our trademark applications will be approved. Third parties may also oppose our trademark applications or otherwise challenge our use of the trademarks. In the event that our trademarks are successfully challenged, we could be forced to rebrand our products, which could result in loss of brand recognition, and could require us to devote resources to advertising and marketing new brands. Further, there can be no assurance that competitors will not infringe our trademarks or that we will have adequate resources to enforce our trademarks. We also license third parties to use our trademarks. In an effort to preserve our trademark rights, we enter into license agreements with these third parties, which govern the use of our trademarks and require our licensees to abide by quality control standards with respect to the goods and services that they provide under our trademarks. Although we make efforts to monitor the use of our trademarks by our licensees, there can be no assurance that these efforts will be sufficient to ensure that our licensees abide by the terms of their licenses. In the event that our licensees fail to do so, our trademark rights could be diluted. Any of the foregoing could have a material adverse effect on our competitive position, business, financial condition, results of operations and prospects.
Patent terms may not be sufficient to effectively protect our products and business for an adequate period of time.
Patents have a limited lifespan. In the United States, the natural expiration of a patent is generally 20 years after its first effective non-provisional filing date. Although various extensions may be available, the term of a patent, and the protection it affords, is limited. Even if patents covering our technologies and their uses are obtained, once the patent has expired, we may be open to competition. In addition, although upon issuance in the United States a patent’s term can be extended based on certain delays caused by the USPTO, this extension can be reduced or eliminated based on certain delays caused by the patent applicant during patent prosecution. Given the amount of time required for the development, testing and regulatory review of new products, patents protecting such products might expire before or shortly after such products are commercialized. If we do not have sufficient patent terms to protect our products, technologies and their uses, our business would be materially adversely affected.
We may be unable to enforce our intellectual property rights throughout the world.
The laws of some foreign countries do not protect intellectual property rights to the same extent as the laws of the United States. Many companies have encountered significant problems in protecting and defending their intellectual property rights in certain foreign jurisdictions. This could make it difficult for us to stop infringement of our foreign patents, if obtained, or the misappropriation of our other intellectual property rights. For example, some foreign countries have compulsory licensing laws under which a patent owner must grant licenses to third parties. In addition, some countries limit the enforceability of patents against certain third parties, including government agencies or government contractors. In these countries, patents may provide limited or no benefit. Patent protection must ultimately be sought on a country-by-country basis, which is an expensive and time-consuming process with uncertain outcomes. Accordingly, we may choose not to seek patent protection in certain countries, and we will not have the benefit of patent protection in such countries.
Proceedings to enforce our patent rights in foreign jurisdictions could result in substantial costs and divert our efforts and attention from other aspects of our business. Accordingly, our efforts to protect our intellectual property rights in such countries may be inadequate. In addition, changes in the law and legal decisions by courts in the United States and foreign countries may affect our ability to obtain adequate protection for our technology and the enforcement of our intellectual property. If we are unable to fully protect our intellectual property, our business will be materially and adversely affected.
We may be subject to claims that we or our employees have misappropriated the intellectual property of a third party, including trade secrets or know-how, or are in breach of non-competition or non-solicitation agreements with our competitors and third parties may claim an ownership interest in intellectual property we regard as our own.
Many of our employees and consultants were previously employed at or engaged by other medical device companies, including our competitors or potential competitors. Some of these employees, consultants and contractors may have executed proprietary rights, non-disclosure and non-competition agreements in connection with such previous employment. Although we try to ensure that our employees and consultants do not use the intellectual property, proprietary information, know-how or trade secrets of others in their work for us, we may be subject to claims that we or these individuals have, inadvertently or otherwise, misappropriated the intellectual property or disclosed the alleged trade secrets or other proprietary information of these former employers, competitors or other third parties. Additionally, we may be subject to claims from third parties challenging our ownership interest in or inventorship of intellectual property we regard as our own, for example, based on claims that our agreements with employees or consultants obligating them to assign intellectual property to us are ineffective or in conflict with prior or competing contractual obligations to assign inventions to another employer, to a former employer, or to another person or entity. Litigation may be necessary to defend against claims, and it may be necessary or we may desire to enter into a license to settle any such claim; however, there can be no assurance that we would be able to obtain a license on commercially reasonable terms, if at all. If our defense to those claims fails, in addition to paying monetary damages or a settlement payment, a court could prohibit us from using technologies, features or other intellectual property that are essential to our products, if such technologies or features are found to incorporate or be derived from the trade secrets or other proprietary information of the former employers, competitors or third parties. An inability to incorporate technologies, features or other intellectual property that are important or essential to our products could have a material adverse effect on our business and competitive position and may prevent us from selling our products. In addition, we may lose valuable intellectual property rights or personnel. Even if we are successful in defending against these claims, litigation could result in substantial costs and could be a distraction to management. Any litigation or the threat thereof may adversely affect our ability to hire employees or contract with independent sales representatives. A loss of key personnel or their work product could hamper or prevent our ability to commercialize our products, which could materially and adversely affect our business, financial condition, operating results, cash flows and prospects.
Risks Relating to Our Organization and Structure
Our Charter provides that the Court of Chancery of the State of Delaware will be the sole and exclusive forum for substantially all disputes between us and our stockholders, which could limit our stockholders’ ability to obtain a favorable judicial forum for disputes with us or our directors, officers, or employees.
Our Charter provides that, unless we consent in writing to the selection of an alternative forum, the (i) Court of Chancery of the State of Delaware (the “Court of Chancery”) shall, to the fullest extent permitted by law, be the sole and exclusive forum for: (a) any derivative action or proceeding brought on behalf of us, (b) any action asserting a claim of breach of a fiduciary duty owed by any of our directors, stockholders, officers or other employees to us or our stockholders, (c) any action asserting a claim against us, our directors, officers or employees arising pursuant to any provision of the Delaware General Corporation Law, our Bylaws or our Charter (as either may be amended from time to time), and (d) any action asserting a claim against us, our directors, officers or employees governed by the internal affairs doctrine; and (ii) subject to the foregoing, the federal district courts of the United States of America shall be the exclusive forum for the resolution of any complaint asserting a cause of action arising under the Securities Act. Notwithstanding the foregoing, such forum selection provisions shall not apply to suits brought to enforce any liability or duty created by the Exchange Act or any other claim for which the federal courts of the United States have exclusive jurisdiction. The choice of forum provision may limit a stockholder’s ability to bring a claim in a judicial forum that it finds favorable for disputes with us or our directors, officers, or other employees, and may potentially increase costs for investors to bring such a claim, both of which may discourage such lawsuits against us and our directors, officers, and other employees. Alternatively, if a court were to find the choice of forum provision contained in the Charter to be inapplicable or unenforceable in an action, we may incur additional costs associated with resolving such action in other jurisdictions, which could harm our business, results of operations, and financial condition.
Additionally, Section 22 of the Securities Act creates concurrent jurisdiction for federal and state courts over all suits brought to enforce any duty or liability created by the Securities Act or the rules and regulations thereunder. As noted above, the Charter provides that the federal district courts of the United States of America shall have jurisdiction over any action arising under the Securities Act. Accordingly, there is uncertainty as to whether a court would enforce such provision. Our stockholders will not be deemed to have waived our compliance with the federal securities laws and the rules and regulations thereunder.
As an “emerging growth company,” we cannot be certain if the reduced disclosure requirements applicable to “emerging growth companies” will make the Class A Common Stock less attractive to investors.
As an “emerging growth company,” we take advantage of certain exemptions from various reporting requirements that are applicable to other public companies that are not “emerging growth companies,” including not being required to obtain an assessment of the effectiveness of our internal control over financial reporting from our independent registered public accounting firm pursuant to Section 404 of the Sarbanes-Oxley Act, reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements, and exemptions from the requirements of holding a non-binding advisory vote on executive compensation and stockholder approval of any golden parachute payments not previously approved. In addition, the JOBS Act provides that an emerging growth company can take advantage of an extended transition period for complying with new or revised accounting standards, which we have elected to do.
We cannot predict if investors will find the Class A Common Stock less attractive because we rely on these exemptions. If some investors find the Class A Common Stock less attractive as a result, there may be a less active market for the Class A Common Stock, the share price of Class A Common Stock may be more volatile and the price at which our securities trade could be less than if we did not use these exemptions.
The requirements of being a public company may strain our resources and divert management’s attention.
As a public company, we are subject to the reporting requirements of the Exchange Act, the Sarbanes-Oxley Act, the Dodd-Frank Wall Street Reform and Consumer Protection Act, the listing requirements of Nasdaq and other applicable securities rules and regulations. Compliance with these rules and regulations increase our legal and financial compliance costs, make some activities more difficult, time-consuming or costly and increase demand on our systems and resources, particularly after we are no longer an “emerging growth company.” The Sarbanes-Oxley Act requires, among other things, that we maintain effective disclosure controls and procedures and internal control over financial reporting. In order to maintain and, if required, improve our disclosure controls and procedures and internal control over financial reporting to meet this standard, significant resources and management oversight may be required. As a result, management’s attention may be diverted from other business concerns, which could adversely affect our business and operating results. We may need to hire more employees in the future or engage outside consultants to comply with these requirements, which will increase our costs and expenses.
Risks Relating to Our Class A Common Stock and Warrants
We may not receive any proceeds from the exercise of Warrants, and if we do, we may be unable to invest the portion of the net proceeds from the exercise of Warrants on acceptable terms.
We will receive up to an aggregate of approximately $206.9 million from the exercise of our outstanding Warrants, assuming the exercise in full of all of the Warrants for cash. However, we will only receive proceeds to the extent holders of Warrants elect to exercise. We can provide no assurances as to the amount of proceeds we will receive from the exercise of Warrants or whether we will receive any proceeds. As of the date of March 10, 2025, nearly all of our Warrants are “out of the money,” which means that the trading price of the shares of Class A Common Stock underlying the Public Warrants, which was $1.33 on March 10, 2025 is below the $11.50 exercise price of the Public Warrants, the $2.00 exercise price of the lowest exercise price of the Shortfall Warrants, and the 1,500,000 Private Warrants with exercise prices of $3.04, $2.25, and $2.97. We have 500,000 outstanding Private Warrants with an exercise price of $1.24 per share, which are in the money. For so long as Warrants remain “out of the money,” we do not expect warrant holders to exercise their Warrants and, therefore, we do not expect to receive cash proceeds from any such exercise. We will have broad discretion in the use of any proceeds received from the exercise of Warrants. Delays in investing the net proceeds from the exercise of Warrants may impair our performance. We cannot assure you that we will be able to identify uses of proceeds that meet our investment objectives or that any investment that we make will produce a positive return. We may be unable to invest the net proceeds from the exercise of Warrants on acceptable terms within the time period that we anticipate or at all, which could harm our financial condition and operating results. Moreover, we will have significant flexibility in investing the net proceeds from the exercise of Warrants and may use the net proceeds from the exercise of Warrants in ways with which investors may not agree.
The sale of substantial amounts of our securities in the public market by our existing securityholders (including the shares of Class A Common Stock issuable upon exercise of the Warrants and conversion of the Series A Preferred Stock), or the perception that such sales may occur, may cause the market price of our securities to decline significantly.
We have registered the issuance of shares of Class A Common Stock representing approximately 102.9% of the total shares of Class A Common Stock outstanding as of the date of this Report (assuming that all Warrants are exercised and all outstanding shares of Series A Preferred Stock are converted into Class A Common Stock). In addition, we have registered the resale of Class A Common Stock representing 62.9% of the total shares of Class A Common Stock outstanding as of the date of this Report (assuming that no Public Warrants are exercised, all Shortfall Warrants are exercised and all outstanding shares of Series A Preferred Stock are converted into Class A Common Stock). Further, the shares of Class A Common Stock that we have registered for resale represent a significant percentage of our outstanding Class A Common Stock, including 11,159,614 shares of Class A Common Stock beneficially owned by Glen A. Taylor, which represent 52.3% of our outstanding Class A Common Stock (assuming that no Public Warrants, Shortfall Warrants, or Private Warrants are exercised and all shares of Series A Preferred Stock beneficially owned by Mr. Taylor are converted into Class A Common Stock.
The sale of all of these securities, including the shares of Class A Common Stock underlying the Warrants and Series A Preferred Stock, in the public market, or the perception that holders of a large number of securities intend to sell their securities, could significantly reduce the market price of our Class A Common Stock and Public Warrants and could impair our ability to raise capital through the sale of additional equity securities. Certain of our stockholders holding an aggregate of 12,905,049 shares of Class A Common Stock have agreed, subject to certain exceptions, not to sell their shares of Class A Common Stock during the period beginning on the Closing Date and ending on the first to occur of (a) March 29, 2024, (b) if the last sale price of our Class A Common Stock equals or exceeds $10.50 per share (as adjusted for stock splits, stock dividends, reorganizations, recapitalizations and the like) for any 20 trading days within any 30-trading day period or (c) such date on which the Company completes a liquidation, merger, stock exchange or other similar transaction that results in all of the Company’s stockholders having the right to exchange their shares of Common Stock for cash, securities or other property. Once such resale restrictions end, the market price of our Class A Common Stock could decline if such stockholders sell their shares or are perceived by the market as intending to sell them. Furthermore, despite such a decline in the public trading price, some of such stockholders may still experience a positive rate of return on the securities they purchased due to the price at which such stockholders initially purchased the securities.
The market prices of our Class A Common Stock and Public Warrants have been and may continue to be extremely volatile, which could cause purchasers of our securities to incur substantial losses.
The market prices and trading volume of our shares of Class A Common Stock have recently experienced, and may continue to experience, extreme volatility, which could cause purchasers of our Class A Common Stock and Public Warrants to incur substantial losses. Since the closing of the Business Combination, our Class A Common Stock has traded as low as $1.21 and as high as $9.60 as of March 10, 2025. In addition, the volume of trading of our Class A Common Stock has been inconsistent. For example, on October 21, 2024 our Class A Common Stock had trading volume of 2,670 shares and on November 1, 2024 our Class A Common Stock had trading volume of 11,770,940 shares. Our Public Warrants have not traded in tandem with our Class A Common Stock, and since the closing of the Business Combination, have traded within a range of $0.025 to $0.24 as of March 10, 2025.
We believe that the recent volatility and our current market prices reflect market and trading dynamics unrelated to our underlying business, or macro or industry fundamentals, and we do not know how long these dynamics will last. Under the circumstances, investors in our Class A Common Stock and Public Warrants are subject to the risk of losing all or a substantial portion of their investment.
The market volatility and trading patterns we have experienced create several risks for investors, including the following:
| ● | the market price of our Class A Common Stock has experienced and may continue to experience rapid and substantial increases or decreases unrelated to our operating performance or prospects, or macro or industry fundamentals, and substantial increases may be significantly inconsistent with the risks and uncertainties that we continue to face; |
| ● | factors in the public trading market for our Class A Common Stock may include the sentiment of retail investors, the direct access by retail investors to broadly available trading platforms, the amount and status of short interest in our securities, access to margin debt, trading in options and other derivatives on our Class A Common Stock and any related hedging and other trading factors; |
| ● | to the extent volatility in our Class A Common Stock is caused by a “short squeeze” in which coordinated trading activity causes a spike in the market price of our Class A Common Stock as traders with a short position make market purchases to avoid or to mitigate potential losses, investors purchase at inflated prices unrelated to our financial performance or prospects, and may thereafter suffer substantial losses as prices decline once the level of short-covering purchases has abated; and |
| ● | if the market price of our Class A Common Stock declines, you may be unable to resell your shares at or above the price at which you acquired them, and the Public Warrant you own may become out of the money. |
The trading prices of our Class A Common Stock and Public Warrants depend on many factors, including those described in this Item 1A. Risk Factors, many of which are beyond our control and may not be related to our operating performance. Any of the factors listed below could have a material adverse effect on investment in our Class A Common Stock and Public Warrants, and our Class A Common Stock and Public Warrants may trade at prices significantly below the price paid for them. In such circumstances, the trading prices of our Class A Common Stock and Public Warrants may not recover and may experience a further decline. Factors affecting the trading price of our Class A Common Stock and Public Warrants may include:
| ● | actual or anticipated fluctuations in our quarterly financial results or the quarterly financial results of companies perceived to be similar to us; |
| ● | changes in the market’s expectations about our operating results; |
| ● | the public’s reaction to our press releases, our other public announcements and our filings with the SEC; |
| ● | speculation in the press or investment community; |
| ● | actual or anticipated developments in our business or our competitors’ businesses or the competitive landscape generally; |
| ● | our operating results failing to meet the expectation of securities analysts or investors in a particular period; |
| ● | changes in financial estimates and recommendations by securities analysts concerning us or the market in general; |
| ● | operating and stock price performance of other companies that investors deem comparable to us; |
| ● | publications of research reports by securities analysts about us, our competitors, or the industry we operate in; |
| ● | changes in laws and regulations affecting our business; |
| ● | commencement of, or involvement in, litigation involving us; |
| ● | changes in our capital structure, such as future issuances of securities or the incurrence of additional debt; |
| ● | the volume of Class A Common Stock available for public sale; |
| ● | any major change in the Board or management; |
| ● | sales of substantial amounts of Class A Common Stock by directors, officers or significant stockholders or the perception that such sales could occur; |
| ● | general economic and political conditions such as recessions, interest rates, fuel prices, trade wars, pandemics (such as COVID-19), epidemics, currency fluctuations and acts of war (such as the conflict between Russia and Ukraine and the military conflict in Israel and Gaza) or terrorism; and |
| ● | other risk factors listed under this Item 1A. Risk Factors. |
There is no guarantee that the Public Warrants will be in the money, and they may expire worthless and the terms of our Public Warrants may be amended.
The exercise price for the Public Warrants is $11.50 per share of Class A Common Stock, which exceeds the market price of the shares of Class A Common Stock, which was $1.44 per share based on the closing price of the Class A Common Stock on March 24, 2025. There is no guarantee that the Public Warrants will be in the money at any given time prior to their expiration. Pursuant to the terms of Warrant Agreement, the Public Warrants will expire on September 29, 2028, at 5:00 p.m., New York City time, or earlier upon redemption or liquidation. If the trading price of Class A Common Stock declines, the Public Warrants may expire worthless. If all of the Public Warrants were exercised in full for cash, we would receive an aggregate of approximately $162.9 million. We do not expect the holders of the Public Warrants to exercise their Public Warrants and therefore, we do not expect to receive cash proceeds from any such exercise, for so long as the Public Warrants remain out of the money. We can provide no assurances that the trading price of our Class A Common Stock will remain at levels where it would be attractive to exercise our outstanding Public Warrants until the time that such Public Warrants become exercisable.
We may redeem unexpired Public Warrants prior to their exercise at a time that is disadvantageous to the holders of such Public Warrants, thereby making such Public Warrants worthless.
We have the ability to redeem outstanding Public Warrants at any time after they become exercisable and prior to their expiration, at a price of $0.01 per warrant, provided that the last reported sales price of our Class A Common Stock equals or exceeds $18.00 per share (as adjusted for stock splits, stock dividends, reorganizations, recapitalizations and the like) for any 20 trading days within a 30 trading day period ending on the third trading day prior to the date on which we give proper notice of such redemption and provided certain other conditions are met. Shares of our Class A Common Stock have never traded above $18.00 per share. If and when such Public Warrants become redeemable by us, we may not exercise our redemption rights if the issuance of shares of Class A Common Stock upon exercise of the Public Warrants is not exempt from registration or qualification under applicable state blue sky laws or we are unable to effect such registration or qualification. We will use our best efforts to register or qualify such shares of common stock under the blue sky laws of the state of residence in those states in which the Public Warrants were offered by Anzu. Redemption of the outstanding Public Warrants could force the holders of such Public Warrants (i) to exercise the Public Warrants and pay the exercise price therefor at a time when it may be disadvantageous for such holder to do so, (ii) to sell the Public Warrants at the then-current market price when you might otherwise wish to hold the Public Warrants or (iii) to accept the nominal redemption price which, at the time the outstanding Public Warrants are called for redemption, is likely to be substantially less than the market value of the Public Warrants.
We may amend the terms of the Public Warrants in a manner that may be adverse to holders of Public Warrants with the approval by the holders of at least 65% of the then outstanding Public Warrants.
The Public Warrants were issued in registered form under the Warrant Agreement. The Warrant Agreement provides that (a) the terms of the Public Warrants may be amended without the consent of any holder for the purpose of (i) curing any ambiguity or correcting any mistake or defective provision or (ii) adding or changing any provisions with respect to matters or questions arising under the Warrant Agreement as the parties to the Warrant Agreement may deem necessary or desirable and that the parties deem to not adversely affect the rights of the registered holders of the Public Warrants under the Warrant Agreement and (b) all other modifications or amendments require the vote or written consent of at least 65% of the then outstanding Public Warrants. Accordingly, we may amend the terms of the Public Warrants in a manner adverse to a holder if holders of at least 65% of the then outstanding Public Warrants approve of such amendment. Our ability to amend the terms of the Public Warrants with the consent of at least 65% of the then outstanding Public Warrants is broad. Examples of such amendments could be amendments to, among other things, increase the exercise price of the Public Warrants, shorten the exercise period or decrease the number of shares of Class A Common Stock purchasable upon exercise of a Public Warrant.
While we will pay dividends on shares of Series A Preferred Stock pursuant to the Certificate of Designation, we do not intend to pay dividends on shares of Class A Common Stock for the foreseeable future.
Except with respect to dividends on shares of Series A Preferred Stock pursuant to the terms of the Certificate of Designation, we currently intend to retain all available funds and any future earnings to fund the development and growth of our business. As a result, while we will pay dividends on shares of Series A Preferred Stock, we do not anticipate declaring or paying any cash dividends on shares of Class A Common Stock in the foreseeable future. Any decision to declare and pay dividends in the future will be made at the discretion of the Board and will depend on, among other things, the dividend rights of the Series A Preferred Stock pursuant to the Certificate of Designation, our business prospects, results of operations, financial condition, cash requirements and availability, certain restrictions related to our indebtedness, industry trends and other factors that the Board may deem relevant. Any such decision will also be subject to compliance with contractual restrictions and covenants in the agreements governing our current and future indebtedness. In addition, we may incur additional indebtedness, the terms of which may further restrict or prevent us from paying dividends on shares of Class A Common Stock. As a result, you may have to sell some or all of your shares of Class A Common Stock after price appreciation in order to generate cash flow from your investment, which you may not be able to do. Our inability or decision not to pay dividends, particularly when others in our industry have elected to do so, could also adversely affect the market price of shares of Class A Common Stock.
If analysts do not publish research about our business or if they publish inaccurate or unfavorable research, our stock price and trading volume could decline.
The trading market for our Class A Common Stock will depend in part on the research and reports that analysts publish about our business. We do not have any control over these analysts. If one or more of the analysts who cover us downgrade our Class A Common Stock or publish inaccurate or unfavorable research about our business, the price of our Class A Common Stock would likely decline. If few analysts cover us, demand for our Class A Common Stock could decrease and our Class A Common Stock price and trading volume may decline. Similar results may occur if one or more of these analysts stop covering us in the future or fail to publish reports on us regularly.
You may experience future dilution as a result of future equity offerings.
In order to raise additional capital, we may, in the future, offer additional shares of our Class A Common Stock or other securities convertible into or exchangeable for our Class A Common Stock at prices that may not be the same as the price per share paid by any investor. We may sell shares or other securities in any other offering at a price per share that is less than the price per share paid by any investor, and investors purchasing shares or other securities in the future could have rights superior to you. The price per share at which we sell additional shares of our Class A common Stock, or securities convertible or exchangeable into common stock, in future transactions may be higher or lower than the price per share paid by any investor.
We may be subject to securities litigation, which is expensive and could divert management attention.
The market price of Class A Common Stock may continue to be volatile and, in the past, companies that have experienced volatility in the market price of their stock have been subject to securities class action litigation. We may be the target of this type of litigation in the future. Securities litigation against us could result in substantial costs and divert management’s attention from other business concerns, which could seriously harm our business.
We are and may become involved in legal proceedings, and no assurance can be provided as to the outcome of these matters.
From time to time, we are involved in various legal proceedings, lawsuits, and other claims relating to matters incidental to our business. For example, we are currently a defendant in a lawsuit in the Court of Chancery of the State of Delaware involving a stockholder’s redemption request in connection with our special meeting of stockholders held on September 27, 2023. An unfavorable resolution of any litigation may have a material adverse effect on our business, results of operations and financial condition. Additionally, litigation may result in substantial costs and expenses and significantly divert the attention of management.
ITEM 1B. Unresolved Staff Comments
None.
ITEM 1C. Cybersecurity
Risk Management and Strategy
We have certain processes for the identification, assessment, and mitigation of cybersecurity risks which are incorporated into our overall risk management processes in coordination with our information technology function, which we rely on a third-party vendor who is associated with a Related Party to provide. Such processes include physical, procedural, and technical safeguards and routine review of our policies and procedures to identify risks and improve our practices. We use technology-based tools to mitigate cybersecurity risks and to bolster our employee-based cybersecurity programs. We consider the cybersecurity practices of our third-party service providers, including through a general security assessment and contractual requirements, as appropriate, before engaging them in order to help protect us from any related vulnerabilities.
We do not believe that there are currently any known risks from cybersecurity threats that are reasonably likely to materially affect us or our business strategy, results of operations or financial condition. For more information about the cybersecurity risks we face, see the risk factor entitled “Failure of a key information technology system, process or site could have an adverse effect on our business” in the section titled “Risk Factors” in Part 1, Item 1A of this Annual Report.
Governance
Our third-party vendor, alongside our senior management leads the operational oversight of the company-wide cybersecurity strategy, policy, standards and processes. As a smaller-reporting Company we do not have an employee who has significant and demonstrated professional IT management experience and possesses the requisite education, skills and experience expected to perform such a duty. The audit committee of the board of directors intends to provide oversight of our cybersecurity risk as part of its periodic review of enterprise risk management. Additionally, the board of directors intends to review our enterprise risk management processes and will be notified by management between management updates regarding significant new cybersecurity threats or incidents.
ITEM 2. Properties
Our principal office is located at 4875 White Bear Lake, Minnesota, where we lease approximately 11,540 square feet of office space. We lease this space under a lease that terminates on December 31, 2030. We believe that our existing facility is sufficient to meet our needs for the foreseeable future.
We also lease 1,100 square feet of office space in Ausbach, Germany pursuant to a lease that automatically renews each year for a successive one year period, unless the we notify the landlord six (6) months prior to the annual renewal. This lease renewed automatically on January 1, 2024 and again on January 1, 2025.
ITEM 3. Legal Proceedings
From time to time, we may be involved in various claims and legal actions in the ordinary course of business. Except as described below, we are not currently involved in any material legal proceedings outside the ordinary course of our business.
On November 14, 2023, the Company, Whitney Haring-Smith (the former chief executive officer and a former director of the Company), Daniel Hirsch (the former chief financial officer of the Company), and Anzu SPAC GP I LLC were named as defendants in a complaint filed by Atlas Merchant Capital SPAC Fund I LP (“Atlas”) in the Delaware Court of Chancery. Atlas alleges that it was not allowed to redeem its shares of the Company’s Common Stock and that Defendants acted to prevent Atlas’s attempt to redeem its shares. Defendants assert that Atlas did not comply with the requirements for redeeming shares set forth in the Company’s organizational documents. Atlas asserts damages in the amount of approximately $9.4 million, pre- and post-judgment interest, costs, and reasonable attorneys’ fees. The Company has standard indemnification obligations to Dr. Haring-Smith and Mr. Hirsch. The Company believes that the lawsuit is meritless and has been defending this matter vigorously. The Company is unable to predict the outcome of this legal proceeding.
ITEM 4. Mine Safety Disclosures
Not applicable.
PART II
ITEM 5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities Market Information
Our Class A Common Stock and Public Warrants are listed on Nasdaq under the symbols “COCH” and “COCHW,” respectively.
As of March 10, 2025, there were 397 holders of record of Class A Common Stock and one holder of record of Public Warrants. However, because many of the shares of Class A Common Stock and Public Warrants are held by brokers and other institutions on behalf of stockholders, we believe there are substantially more beneficial holders of Class A Common Stock and Public Warrants than record holders.
Dividends
Except with respect to dividends on shares of Series A Preferred Stock pursuant to the terms of the Certificate of Designation, we currently intend to retain all available funds and any future earnings to fund the development and growth of our business. As a result, while we will pay dividends on shares of Series A Preferred Stock, we do not anticipate declaring or paying any cash dividends on shares of Class A Common Stock in the foreseeable future. Any decision to declare and pay dividends in the future will be made at the discretion of the Board and will depend on, among other things, the dividend rights of the Series A Preferred Stock pursuant to the Certificate of Designation, our business prospects, results of operations, financial condition, cash requirements and availability, certain restrictions related to our indebtedness, industry trends and other factors that the Board may deem relevant. Any such decision will also be subject to compliance with contractual restrictions and covenants in the agreements governing our current and future indebtedness. In addition, we may incur additional indebtedness, the terms of which may further restrict or prevent us from paying dividends on shares of Class A Common Stock.
Securities Authorized for Issuance under Equity Compensation Plans
Information regarding the equity compensation plans of the Company is set forth in Item 11. Executive Compensation, which is incorporated by reference to our Definitive Proxy Statement.
Recent Sales of Unregistered Securities, Use of Proceeds from Registered Public Offering
During the year ended December 31, 2024, there were no unregistered sales of our securities that were not reported in a Current Report on Form 8-K or Quarterly Report on Form 10-Q.
Purchases of Equity Securities by the Issuer and Affiliated Purchasers
There were no repurchases of our equity securities during the three months ended December 31, 2024.
ITEM 6. [Reserved]
ITEM 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations
The following analysis of our financial condition and results of operations should be read in conjunction with the consolidated financial statements and the notes included elsewhere in this Report, and other filings with the SEC. Unless otherwise indicated or the context otherwise requires, references in this section to the “Company,” “Envoy Medical,” “we,” “us,” “our” and other similar terms refer (i) prior to the Closing Date, to Envoy Medical Corporation and (ii) after the Closing Date, to Envoy Medical, Inc. The following discussion contains forward-looking statements based upon Envoy Medical’s current expectations that involve risks, uncertainties, and assumptions. Our actual results may differ materially from those anticipated in these forward-looking statements as a result of various factors, including those set forth under the section of this Report titled “Risk Factors” and/or elsewhere in this Report. Our historical results are not necessarily indicative of the results that may be expected for any period in the future. All dollar amounts are expressed in thousands of United States dollars (“$”), unless otherwise indicated.
Overview
We are a hearing health company focused on providing innovative medical technologies across the hearing loss spectrum. Our technologies are designed to shift the paradigm within the hearing industry and bring both providers and patients the hearing devices they desire. Founded in 1995, our vision is to create fully implanted hearing devices that leverage the natural ear - not an artificial microphone - to pick up sound. In recent years, we have focused almost exclusively on developing the fully implanted Acclaim® cochlear implant (the “Acclaim CI”), our lead product candidate.
We believe that the Acclaim CI is a first-of-its-kind cochlear implant. Our fully implanted technology includes a sensor designed to leverage the natural anatomy of the ear instead of a microphone to capture sound. The Acclaim CI is designed to address severe to profound sensorineural hearing loss that is not adequately addressed by hearing aids. The Acclaim CI will only be indicated for adults who have been deemed adequate candidates by a qualified physician. The Acclaim CI received the Breakthrough Device Designation from the United States Food and Drug Administration (the “FDA”) in 2019.
Our first product, the Esteem ® Fully Implanted Active Middle Ear Implant (“Esteem FI-AMEI”), received FDA approval in 2010. The Esteem FI-AMEI is a fully implanted active middle ear hearing device and remains the only FDA approved fully implanted hearing device in the US market. Unfortunately, the Esteem FI-AMEI failed to gain commercial traction, primarily due to a lack of reimbursement or insurance coverage from third-party payors.
Despite the commercial challenges, approximately 1,000 Esteem FI-AMEI devices were implanted. Some devices were implanted in the early 2000s during clinical trials, providing Envoy Medical with over two decades of experience with our implantable sensor technology. Throughout our experience, our sensor technology proved a viable alternative and robust option to external or implanted microphones.
In late 2015, we made the decision to shift our focus from the Esteem FI-AMEI to a new product that would leverage our sensor technology and incorporate it into a cochlear implant. As a result, we now have the Acclaim CI, a fully implanted cochlear implant. We believe that Acclaim CI gives us the opportunity to disrupt the existing cochlear implant market. The cochlear implant market is one that already has established market acceptance and reimbursement pathways. In the United States, before we can market a new Class III medical device, like the Acclaim CI, we must first receive FDA approval via the premarket application approval process.
In October 2024, we received FDA approval of our application for an Investigational Device Exemption (“IDE”) for the Acclaim CI. The IDE application was approved for a staged clinical trial, which we began in the first quarter of 2025. The staged trial will allow 10 participants to be implanted before expanding the study to the full cohort. Institutional Site’s Investigational Review Board (“IRB”) approvals are needed before participants can be enrolled and implants can begin. IRB approvals can take several months. At the end of the study, a Premarket Approval (“PMA”) application will be submitted to the FDA. It is likely that a panel review will be requested by the FDA due to the novel nature of the Acclaim CI. As a result, we currently anticipate obtaining the FDA’s decision on our PMA in 2027. The FDA approval process is uncertain, and we cannot predict the effects that changes to federal regulatory staffing, funding, and policies and procedures will have on the timeline and ultimate FDA approval decision. As a result, we cannot guarantee that we will receive FDA approval on that timeline, or at all.
We had a net loss of $20.8 million and $29.9 million for the years ended December 31, 2024 and December 31, 2023, respectively, and had an accumulated deficit of $284.7 million and $257.3 million as of December 31, 2024 and December 31, 2023, respectively. We have funded our operations to date primarily through the issuance of equity securities, term debt and convertible debt and in September 2023, we received $11.7 million proceeds from the Business Combination (see Note 1, “Nature of the Business and Basis of Presentation” of the accompanying consolidated financial statements for the years ended December 31, 2024 and 2023 included elsewhere in this Report). We expect to continue to incur net losses for the foreseeable future, and expect our research and development expenses, sales and marketing expenses, general and administrative expenses, and capital expenditures will continue to increase. In particular, we expect our expenses to increase as we continue our development of the Acclaim CI and seek the necessary regulatory approvals for our product candidate, as well as hire additional personnel, pay fees to outside consultants, attorneys and accountants, and incur other increased costs associated with being a public company. In addition, if and when we seek and obtain regulatory approval to commercialize the Acclaim CI in the United States, we will also incur increased expenses in connection with commercialization and marketing of such product. Our net losses may fluctuate significantly from quarter-to-quarter and year-to-year, depending on the timing of our clinical trials, if any, and our expenditures on other research and development activities. We anticipate that our expenses will increase significantly in connection with our ongoing activities, if and as we:
| ● | continue our research and development efforts for the Acclaim CI product candidate, including through clinical trials; | |
| ● | seek additional regulatory and marketing approvals in jurisdictions outside the United States; | |
| ● | establish a sales, marketing and distribution infrastructure to commercialize our product candidate; | |
| ● | rely on our third-party suppliers and manufacturers to obtain adequate supply of materials and components for our products; | |
| ● | seek to identify, assess, acquire, license, and/or develop other product candidates and subsequent generations of our current product candidate; | |
| ● | seek to maintain, protect, and expand our intellectual property portfolio; | |
| ● | seek to identify, hire, and retain additional skilled personnel; | |
| ● | create additional infrastructure to support our operations as a public company and our product candidate development and planned future commercialization efforts; and | |
| ● | experience any delays or encounter issues with respect to any of the above, including, but not limited to, failed studies, complex results, safety issues or other regulatory challenges that require longer follow-up of existing studies or additional supportive studies in order to pursue marketing approval. |
We expect that our financial performance may fluctuate significantly from quarter-to-quarter and year-to-year due to the development status of our Acclaim CI product and our efforts to obtain regulatory approval and commercialize the Acclaim CI product.
The Acclaim CI has not yet been approved for sale. We do not expect to generate any product sales unless and until we successfully complete development and obtain regulatory approval for our product candidate. If we obtain regulatory approval for the Acclaim CI, we expect to incur significant commercialization expenses related to product sales, marketing, manufacturing and distribution. As a result, until such time, if ever, that we can generate substantial product revenue, we expect to finance our cash needs through equity offerings, debt financings or other capital sources, including collaborations, licenses or similar arrangements. However, we may be unable to raise additional funds or enter into such other arrangements when needed or on favorable terms, if at all. Any failure to raise capital as and when needed could have a negative impact on our financial condition and on our ability to pursue our business plans and strategies, including our research and development activities. If we are unable to raise capital, we will need to delay, reduce, or terminate planned activities to reduce costs.
Macroeconomic Conditions
Our business and financial performance are impacted by macroeconomic conditions. Global macroeconomic challenges, such as the effects of the ongoing war between Russia and Ukraine, the Middle East conflict, supply chain constraints, tariffs and trade wars, market uncertainty, volatility in exchange rates, inflationary trends, interest rates, and evolving dynamics in the global trade environment have impacted our business, financial performance, and our ability to raise capital.
Furthermore, a recession or market correction resulting from macroeconomic factors could materially affect our business and the value of our Class A common stock (“Common Stock”). The occurrence of any such events may lead to reduced disposable income which could adversely affect the number of Esteem FI-AMEI implants and replacement components sold as a result of customer and patient reluctance to seek treatment due to financial considerations.
Adverse macroeconomic conditions, including pandemics or international tensions, could also result in significant disruption of global economic conditions and consumer trends, as well as a significant disruption in financial markets, reducing our ability to access capital, which could in the future negatively affect our liquidity.
Key Components of Our Results of Operations
Revenue
Currently, we derive substantially all our revenue from the sale of the Esteem FI-AMEI implants and replacement components to Esteem FI-AMEI implants. We enter arrangements with patients to provide them with the Esteem FI-AMEI device, personal programmer devices, Battery replacements, and/or an optional Care Plan, each of which are outputs of our ordinary activities in exchange for consideration. Revenue from product sales is recognized upon transfer of control of the product to a customer, which occurs at a point in time, when we are notified the product has been implanted or used by the customer in a surgical procedure. New implantations of the Esteem FI-AMEI are not expected to be more than a few per year and may be as low as zero. Although we believe it to be unlikely, Esteem FI-AMEI implantations could potentially increase with favorable reimbursement policy and coverage changes. We will continue our efforts to pursue positive reimbursement changes for fully implanted active middle ear implants. There will be continued nominal revenue from replacement of sound processors for patients who need a new Battery.
Upon commercialization of our Acclaim CI product, we expect that Acclaim CI revenues will more than exceed our Esteem FI-AMEI revenue. We are targeting FDA approval for the Acclaim CI in 2027.
Cost of Goods Sold
Cost of goods sold includes direct and indirect costs related to the manufacturing and distribution of the Esteem FI-AMEI, including materials, labor costs for personnel involved in the manufacturing process, distribution-related services, indirect overhead costs, and charges for excess and obsolete inventory reserves and inventory write-offs.
We expect cost of goods sold to increase or decrease in absolute dollars primarily as, and to the extent, our revenue grows or declines, respectively.
Operating Expenses
Research and Development Expenses
Research and development (“R&D”) expenses consist of costs incurred for our research activities, primarily our discovery efforts and the development of the Acclaim CI product. We also incur R&D costs related to continuing to support, and improving upon where possible, our Esteem FI-AMEI product. We expense R&D costs as incurred, which include:
| ● | salaries, employee benefits, and other related costs for our personnel engaged in R&D functions; | |
| ● | service fees incurred under agreements with independent consultants, including their fees and related travel expenses engaged in R&D functions; | |
| ● | costs of laboratory testing including supplies and acquiring, developing, and manufacturing study materials; and | |
| ● | facility-related expenses, which include direct depreciation costs and allocated expenses for rent and maintenance of facilities and other operating costs. |
Costs for certain development activities are recognized based on an evaluation of the progress to completion of specific tasks using information and data provided to us by our vendors, service providers and our clinical sites.
Our R&D expenses are currently tracked on a program-by-program basis. The majority of our R&D expenses incurred during the years ended December 31, 2024 and 2023 were for the development of the Acclaim CI.
Our products require human clinical trials to obtain regulatory approval for commercial sales. We cannot determine with certainty the size, duration, or completion costs of future clinical trials, or if or when they may be completed. Furthermore, we do not know if the clinical trials will show positive or negative results, or what those results will mean for regulatory approval or commercialization efforts.
The duration, costs and timing of future clinical trials and development of our products will depend on a variety of factors, including:
| ● | the scope, rate of progress, and expense of our ongoing, as well as any additional, clinical trials and other R&D activities; | |
| ● | interest in or demand for both investigational site and subject enrollment; | |
| ● | future clinical trial results; | |
| ● | potential changes in government regulation; | |
| ● | potential changes in the reimbursement landscape; and | |
| ● | the timing and receipt of any regulatory approvals. |
A change in the outcome of any of these variables with respect to the development of our Acclaim CI product could mean a significant change in the costs and timing associated with the development of that implant. If the FDA or another regulatory authority were to require us to conduct clinical trials beyond those that we currently anticipate, or if we experience significant delays in the enrollment in any clinical trials, we could be required to expend significant additional financial resources and time on the completion of clinical development.
R&D activities are central to our business model. We expect that our R&D expenses will continue to increase for the foreseeable future as we initiate clinical trials for the Acclaim CI product and prepare the product for possible commercialization, should it gain regulatory approval(s). If the Acclaim CI product enters later stages of clinical trials and ongoing development, the product will generally incur higher R&D expenses than those in earlier stages of research and development, primarily due to simultaneously running clinical trials while also iterating the product for commercialization and preparing for the needs of commercialization. There are numerous factors associated with the successful commercialization of the Acclaim CI product or any products we may develop in the future, including future trial design and various regulatory requirements, many of which cannot be determined with accuracy at this time based on our stage of development. Additionally, future commercial and regulatory factors beyond our control will impact our clinical development program and plans.
Sales and Marketing Expenses
Sales and marketing expenses consist primarily of salaries, benefits, and other related costs for personnel in our sales and marketing functions. Sales and marketing expenses also include certain indirect costs associated with efforts to secure insurance reimbursement of our products. We expect our sales and marketing expenses to increase in the foreseeable future as we increase our sales and marketing personnel to support our continuing growth.
General and Administrative Expenses
General and administrative expenses consist primarily of salaries, benefits, and other related costs for personnel in our executive, operations, legal, human resources, finance, insurance premiums, and administrative functions. Administrative expenses also include professional fees for legal, patent, consulting, accounting, tax and audit services, travel expenses and facility-related expenses, which include direct depreciation costs and allocated expenses for rent and maintenance of facilities, technology, and other operating costs.
We expect our general and administrative expenses to continue to increase in the foreseeable future as we increase our administrative personnel to support our continuing growth, our costs of expanding our operations and operating as a public company. These increases will likely include the hiring of additional personnel and legal, regulatory, and other fees and services associated with maintaining compliance with Nasdaq and SEC requirements, director and officer insurance costs and investor relations costs associated with being a public company.
Change in Fair Value of Convertible Notes Payable (Related Party)
We previously elected the fair value option for convertible notes payable (related party), and accordingly, convertible notes payable (related party) were recorded at fair value at each reporting date on the consolidated balance sheets. Gain (loss) from changes in fair value of convertible notes payable consisted of changes in the fair value during each reporting period. Effective September 29, 2023, the convertible notes (related party) were converted upon completion of the Business Combination.
Change in Fair Value of Forward Purchase Agreement Put Option Liability
We recognized the forward purchase agreement put option liability at fair value at each reporting period. The liability is subject to re-measurement at each balance sheet date, and any change in fair value is recognized in our consolidated statements of operations and comprehensive loss during each reporting period. The forward purchase agreement put option liability has been derecognized as of December 31, 2024 due to the sale of the shares associated with the forward purchase agreement during the first quarter of 2024.
Change in Fair Value of Forward Purchase Agreement Warrant Liability
We recognize the forward purchase agreement warrant liability at fair value at each reporting period. The liability is subject to re-measurement at each balance sheet date, and any change in fair value is recognized in our consolidated statements of operations and comprehensive loss during each reporting period.
Change in Fair Value of Publicly Traded Warrant Liability
We recognize the publicly traded warrant liability at fair value at each reporting period. The liability is subject to re-measurement at each balance sheet date, and any change in fair value is recognized in our consolidated statements of operations and comprehensive loss during each reporting period.
Interest Expense, Related Party
Interest expense, related party consists of accrued interest for the 2024 Term Loans held by a related party, as well as amortization of the debt discount recorded as a result of the warrants issued with the 2024 Term Loans. Amortization of the debt discount is recorded over the respective terms of the 2024 Term Loans.
Other Income
Other income for the year ended December 31, 2024 consists of sales of internally created quality management documentation to a third party outside of our normal course of business. Other income for the year ended December 31, 2023 consists of changes in fair value of outstanding warrants prior to the closing of the Business Combination, interest earned on cash deposits, and other nonrecurring items.
Results of Operations
Comparison of the Years Ended December 31, 2024 and 2023
| Year ended December 31, | Change in | |||||||||||||||
| (In thousands, except percentages) | 2024 | 2023 | $ | % | ||||||||||||
| Net revenues | $ | 225 | $ | 316 | $ | (91 | ) | (28.8 | )% | |||||||
| Costs and operating expenses: | ||||||||||||||||
| Cost of goods sold | 742 | 789 | (47 | ) | (6.0 | )% | ||||||||||
| Research and development | 10,179 | 8,956 | 1,223 | 13.7 | % | |||||||||||
| Sales and marketing | 1,734 | 1,666 | 68 | 4.1 | % | |||||||||||
| General and administrative | 6,826 | 7,264 | (438 | ) | (6.0 | )% | ||||||||||
| Total costs and operating expenses | 19,481 | 18,675 | 806 | 4.3 | % | |||||||||||
| Operating loss | (19,256 | ) | (18,359 | ) | (897 | ) | 4.9 | % | ||||||||
| Other income (expense): | ||||||||||||||||
| Change in fair value of convertible notes payable (related party) | — | (13,332 | ) | 13,332 | (100.0 | )% | ||||||||||
| Change in fair value of forward purchase agreement put option liability | 103 | (69 | ) | 172 | (249.3 | )% | ||||||||||
| Change in fair value of forward purchase agreement warrant liability | 411 | 842 | (431 | ) | (51.2 | )% | ||||||||||
| Change in fair value of forward purchase agreement warrant liability due to modification | (881 | ) | — | (881 | ) | N/M | ||||||||||
| Change in fair value of publicly traded warrant liability | (330 | ) | 942 | (1,272 | ) | (135.0 | )% | |||||||||
| Interest expense, related party | (816 | ) | — | (816 | ) | N/M | ||||||||||
| Other income | (26 | ) | 54 | (80 | ) | (148.1 | )% | |||||||||
| Total other expense, net | (1,539 | ) | (11,563 | ) | 10,024 | (86.7 | )% | |||||||||
| Net loss | $ | (20,795 | ) | $ | (29,922 | ) | $ | 9,127 | (30.5 | )% | ||||||
N/M - not meaningful
Net Revenues
Net revenues decreased $91 thousand for the year ended December 31, 2024, compared to the year ended December 31, 2023, primarily due to the decrease in the number of Battery replacement sales due to supply chain limitations.
Cost of Goods Sold
Cost of goods sold decreased $47 thousand for the year ended December 31, 2024 compared to the year ended December 31, 2023. The decrease is aligned with the decrease in revenue resulting from the reduced number of Battery replacement sales.
Research and Development Expenses
The following table summarizes the components of our R&D expenses for the years ended December 31, 2024 and 2023:
| Years Ended December 31 | Change in | |||||||||||||||
| (In thousands, except percentages) | 2024 | 2023 | $ | % | ||||||||||||
| R&D product costs | $ | 5,843 | $ | 5,562 | $ | 281 | 5.1 | % | ||||||||
| R&D personnel costs | 3,558 | 2,909 | 649 | 22.3 | % | |||||||||||
| Other R&D costs | 778 | 485 | 293 | 60.4 | % | |||||||||||
| Total research and development costs | $ | 10,179 | $ | 8,956 | $ | 1,223 | 13.7 | % | ||||||||
R&D expenses increased $1.2 million for the year ended December 31, 2024 compared to the year ended December 31, 2023. The increase is primarily due to an increase in headcount and contractors in our engineering and clinical departments for the year ended December 31, 2024, as we increased headcount across our clinical and cochlear departments in preparation for our pivotal clinical study for the Acclaim CI. These increases in headcount included the addition of five new engineers, a clinical research associate, and a clinical research director. The increase in Other R&D costs for the year ended December 31, 2024 is attributable to additional purchases of computer equipment and supplies used in R&D and additional employee recruiting costs.
Sales and Marketing Expenses
Sales and marketing expenses increased $68 thousand for the year ended December 31, 2024 compared to the year ended December 31, 2023. The increase is primarily due to increased legal and professional fees to secure insurance reimbursement for the Esteem FI-AMEI product, partially offset by a reduction in headcount.
General and Administrative Expenses
General and administrative expenses decreased $438 thousand for the year ended December 31, 2024 compared to the year ended December 31, 2023. The decrease is primarily due to reduced professional service costs in 2024 compared to 2023 related to the Business Combination transaction occurring in September 2023.
Change in Fair Value of Convertible Notes Payable (Related Party)
There was a loss from changes in the fair value of convertible notes payable of $13.3 million for the year ended December 31, 2023. The notes payable were converted to Common Stock as a result of the completion of our Business Combination during September 2023.
Change in Fair Value of Forward Purchase Agreement Put Option Liability
The gain from changes in the fair value of the forward purchase agreement put option liability was $103 thousand for the year ended December 31, 2024 compared to a loss of $69 thousand for the year ended December 31, 2023. During the first quarter of 2024, the shares associated with the forward purchase agreement put option were sold.
Change in Fair Value of Forward Purchase Agreement Warrant Liability
The gain from changes in the fair value of the forward purchase agreement warrant liability was $411 thousand for the year ended December 31, 2024 compared to $842 thousand for the year ended December 31, 2023.
Change in Fair Value of Forward Purchase Agreement Warrant Liability Due to Modification
The loss from changes in the fair value of the forward purchase agreement warrant liability due to modification was $881 thousand for the year ended December 31, 2024 compared to $0 for the year ended December 31, 2023. The loss was due to a modification to the forward purchase agreement in December 2024 to extend the term of the warrants.
Change in Fair Value of Publicly Traded Warrant Liability
The loss from changes in the fair value of the publicly traded warrant liability was $330 thousand for the year ended December 31, 2024 compared to a gain of $942 thousand for the year ended December 31, 2023. This decrease is primarily due to an increase in the Company’s closing price for those warrants during the period.
Interest Expense, Related Party
Interest expense, related party was $816 thousand for the year ended December 31, 2024 due to interest incurred related to the issuance of the 2024 Term Loans.
Other Income
Other income decreased by $80 thousand for the year ended December 31, 2024 compared to the year ended December 31, 2023, primarily due to a reduction in interest earned on cash deposits since the completion of the Business Combination transaction in September 2023 and amounts recorded for the year ended December 31, 2023 related to changes in fair value of warrants and other items not recurring for the year ended December 31, 2024.
Liquidity and Capital Resources
Since inception, we have incurred significant operating losses. We expect to continue to incur significant expenses and operating losses for the foreseeable future as we advance the clinical development of our products and fund the process of clinical FDA trials. We have funded our operations to date primarily with proceeds from raising funds from issuing equity securities, term loans, convertible notes and proceeds from the Business Combination. As of December 31, 2024 and December 31, 2023, we had $5.5 million and $4.2 million of cash, respectively.
We proactively manage our access to capital to support liquidity and continued growth. Our sources of capital include issuances of our Common Stock, Series A preferred stock (“Preferred Stock”), warrants, convertible debt, term debt and other financing agreements such as the forward purchase agreement, and proceeds from the sales of the Esteem FI-AMEI implants and replacement components. See Note 1, “Nature of the Business and Basis of Presentation”, of the accompanying audited consolidated financial statements for the years ended December 31, 2024 and 2023 included elsewhere in this Report.
We may seek to raise any necessary additional capital through a combination of public or private equity offerings, debt financings, collaborations, strategic alliances, licensing arrangements and other marketing and distribution arrangements. There can be no assurance that we will be successful in acquiring additional funding at levels sufficient to fund our operations or on terms favorable to us. If we are unable to raise sufficient financing when needed or events or circumstances occur such that we do not meet our strategic plans, we may be required to reduce certain discretionary spending, be unable to develop new or enhanced production methods, or be unable to fund capital expenditures, which could have a material adverse effect on our financial position, results of operations, cash flows, and ability to achieve our intended business objectives. These matters raise substantial doubt about our ability to continue as a going concern. To the extent that we raise additional capital through additional collaborations, strategic alliances, or licensing arrangements with third parties, we may have to relinquish valuable rights to our Acclaim CI, future revenue streams, research programs or to grant licenses on terms that may not be favorable to us. If we do raise additional capital through public or private equity or convertible debt offerings, the ownership interest of our existing stockholders will be diluted, and the terms of these securities may include liquidation or other preferences that adversely affect our stockholders’ rights. If we raise additional capital through debt financing, we may be subject to covenants limiting or restricting our ability to take specific actions, such as incurring additional debt, making capital expenditures, or declaring dividends.
Our future capital requirements and the adequacy of available funds will depend on many factors, including those set forth in the section of this Report titled “Risk Factors - Risks Relating to Our Business and Operations.”
Cash Flows
The following table presents a summary of our cash flow for the periods indicated (in thousands):
| Years Ended December 31 | ||||||||
| 2024 | 2023 | |||||||
| Net cash (used in) provided by: | ||||||||
| Operating activities | $ | (17,949 | ) | $ | (17,091 | ) | ||
| Investing activities | (980 | ) | (153 | ) | ||||
| Financing activities | 20,198 | 21,282 | ||||||
| Effect of exchange rate on cash | (5 | ) | (3 | ) | ||||
| Net increase in cash | $ | 1,265 | $ | 4,035 | ||||
Cash Flows Used in Operating Activities
Net cash used in operating activities for the year ended December 31, 2024 was primarily used to fund a net loss of $20.8 million and $0.6 million of cash outflows from net changes in the levels of operating assets and liabilities, adjusted for non-cash expenses in an aggregate amount of $3.5 million.
The $0.6 million of cash outflows from net changes in the levels of operating assets and liabilities was primarily due to decreases of 1) $0.2 million in accrued expenses due to payment of final expenses related to the Business Combination, 2) $0.1 million in operating lease liability (related party) due to payments made for rent, and 3) $0.2 million in product warranty liability due to expected attrition of this liability over time, as well as increases of 4) $0.4 million in inventories related to the purchase of parts for the Esteem FI-AMEI product, 5) $0.6 million in other receivable due to the recognition of an incoming income tax refund and 6) $0.9 million in other liability due to the receipt of a tax refund and corresponding recognition of an uncertain tax benefit. We will continue to evaluate our capital requirements for both short-term and long-term liquidity needs, which could be affected by various risks and uncertainties, including, but not limited to, the effects of the current inflationary environment, rising interest rates, and other risks detailed in the section of this Report titled “Risk Factors.”
Net cash used in operating activities for the year ended December 31, 2023 was primarily used to fund a net loss of $29.9 million and $1.0 million of cash outflows from net changes in the level of operating assets and liabilities, adjusted for non-cash gains in an aggregate amount of $13.8 million.
The $1.0 million of cash outflows from net changes in the levels of operating assets and liabilities was primarily due to increases of 1) $0.2 million in other receivable related to an income tax receivable, 2) $0.9 million in prepaid expenses and other current assets related to an increase in prepaid insurance and 3) $0.6 million in accounts payable through normal business flows, offset by decreases of 4) $0.2 million in product warranty liability due to expected attrition of this liability over time.
Cash Flows Used in Investing Activities
Net cash used in investing activities for the year ended December 31, 2024 was $1.0 million and consisted of purchases of production equipment and lab equipment.
Net cash used in investing activities for the year ended December 31, 2023 was $0.2 million and consisted of purchases of computer equipment due to increased headcount and purchases of lab equipment.
Cash Flows Provided by Financing Activities
Net cash provided by financing activities for the year ended December 31, 2024 was $20.2 million and was primarily a result of proceeds from the issuance of the 2024 Term Loans in the amount of $20.0 million, the exercise of warrants in the amount of $1.8 million, and the sale of Common Stock in the amount of $1.7 million, partially offset by dividends paid to preferred stockholders in the amount of $2.4 million and payments made on insurance financing loans of $0.9 million.
Net cash provided by financing activities for the year ended December 31, 2023 was $21.3 million and was primarily a result of the $11.7 million net proceeds from the Business Combination and from $10.0 million of proceeds from the issuance of convertible notes payable to a related party, partially offset by payments made on insurance financing loans of $0.6 million.
Contractual Obligations and Commitments
Our principal commitments consist of our operating leases for office space, a litigation matter arising from the Company’s Business Combination, and term loans entered into during 2024 with GAT Funding, LLC in several installments totaling $20.0 million in outstanding principal as of December 31, 2024. Our obligations for leases are described in Note 7, “Operating Leases”, information on our open litigation matter is included in Note 16, “Commitments and Contingencies”, and details on the term loans are described in Note 9, “Debt (Related Party)” of the accompanying consolidated financial statements as of and for the years ended December 31, 2024 and 2023 included elsewhere in this Report.
Off-Balance Sheet Arrangements
During the periods presented, we did not have, nor do we currently have, any off-balance sheet arrangements as defined under the rules and regulations of the SEC.
Related Party Arrangements
Our related party arrangements consist of receiving term loan financings, leasing our headquarters office space, contracting for IT services from a stockholder, and receiving convertible loan financings from stockholders until September 29, 2023 at which point they were converted to Common Stock. For further information on the related party arrangements, refer to Note 7, “Operating Leases”, Note 9, “Debt (Related Party)” and Note 15, “Related Party Transactions”, of the accompanying consolidated financial statements as of and for the years ended December 31, 2024 and 2023 included elsewhere in this Report.
Critical Accounting Policies and Estimates
Our management’s discussion and analysis of our financial condition and results of our operations is based on our consolidated financial statements and accompanying notes, which have been prepared in accordance with accounting principles generally accepted in the United States. Certain amounts included in or affecting the consolidated financial statements presented in this Form 10-K and related disclosure must be estimated, requiring management to make assumptions with respect to values or conditions which cannot be known with certainty at the time the consolidated financial statements are prepared. Management believes that the accounting policies set forth below comprise the most important “critical accounting policies” for the Company. A “critical accounting policy” is one which is both important to the portrayal of our financial condition and results of operations and that involves difficult, subjective, or complex judgments, often as a result of the need to make estimates about the effect of matters that are inherently uncertain. Management evaluates such policies on an ongoing basis, based upon historical results and experience, consultation with experts and other methods that management considers reasonable in the particular circumstances under which the judgments and estimates are made, as well as management’s forecasts as to the manner in which such circumstances may change in the future.
Fair Value Measurements
We determine the fair value of financial assets and liabilities using the fair value hierarchy established in Accounting Standards Codification (“ASC”) Topic 820, Fair Value Measurement (“ASC 820”). ASC 820 identifies fair value as the exchange price, or exit price, representing the amount that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants. The hierarchy describes three levels of inputs that may be used to measure fair value, as follows:
| ● | Level 1 - Observable inputs, such as quoted prices in active markets for identical assets and liabilities. |
| ● | Level 2 - Observable inputs other than Level 1 that are observable, either directly or indirectly, such as quoted prices for similar assets or liabilities, quoted prices in markets that are not active, or other inputs that are observable or can be corroborated by observable market data for substantially the full term of the assets or liabilities. |
| ● | Level 3 - Unobservable inputs that are supported by little or no market activity and that are significant to the fair value of the assets or liabilities. |
Management uses valuation techniques in measuring the fair value of financial instruments, where active market quotes are not available.
The following table summarizes the activity for our Level 3 instruments measured at fair value on a recurring basis (in thousands):
| Forward Purchase Agreement Warrant Liability | Forward Purchase Agreement Put Option Liability | |||||||
| Balance as of December 31, 2023 | $ | 4 | $ | 103 | ||||
| Change in fair value | (411 | ) | (103 | ) | ||||
| Effect of amendments (see Note 10) | 975 | — | ||||||
| Extinguishment of excess warrant liability upon exercise of warrants associated with the forward purchase agreement | (96 | ) | — | |||||
| Balance as of December 31, 2024 | $ | 472 | $ | — | ||||
The fair values of the forward purchase agreement put option liability and the forward purchase agreement warrant liability, which are Level 3 fair value measurements, were estimated using Monte Carlo Simulation models. Key estimates and assumptions impacting the fair value measurement include (i) the Company’s stock price, (ii) the initial exercise price, (iii) volatility, (iv) the remaining term and (v) the risk-free rate.
Research and Development Expenses
We will incur substantial expenses associated with prototyping, improvements, testing and clinical trials. Accounting for clinical trials relating to activities performed by external vendors requires us to exercise significant estimates regarding the timing and accounting for these expenses. We estimate costs of R&D activities conducted by service providers, which include the conduct of sponsored research and contract manufacturing activities. The diverse nature of services being provided for our clinical trials and other arrangements, the different compensation arrangements that exist for each type of service and the lack of timely information related to certain clinical activities complicates the estimation of accruals for services rendered by third parties in connection with clinical trials. We record the estimated costs of R&D activities based upon the estimated amount of services provided but not yet invoiced and include these costs in accrued expenses or prepaid expenses on the consolidated balance sheets and within R&D expense on the consolidated statements of operations and comprehensive loss. In estimating the duration of a clinical study, we evaluate the start-up, treatment and wrap-up periods, compensation arrangements and services rendered attributable to each clinical trial and fluctuations are regularly tested against payment plans and trial completion assumptions.
We estimate these costs based on factors such as estimates of the work completed and budget provided and in accordance with agreements established with our collaboration partners and third-party service providers. We make significant judgments and estimates in determining the accrued liabilities and prepaid expense balances in each reporting period. As actual costs become known, we adjust our accrued liabilities or prepaid expenses. We have not experienced any material differences between accrued costs and actual costs incurred since our inception.
Our expenses related to clinical trials will be based on estimates of patient enrollment and related expenses at clinical investigator sites as well as estimates for the services received and efforts expended pursuant to contracts with multiple research institutions that may be used to conduct and manage clinical trials on our behalf. We will accrue expenses related to clinical trials based on contracted amounts applied to the level of patient enrollment and activity. If timelines or contracts are modified based upon changes in the clinical trial protocol or scope of work to be performed, we will modify our estimates of accrued expenses accordingly on a prospective basis.
Product Warranty
During 2013, we offered a lifetime warranty to clinical trial patients to cover Battery and surgery related costs. We estimate the costs that may be incurred under this lifetime warranty and record a liability in the amount of such costs at its present value. The assumptions utilized in developing the liability include an estimated cost per unit of $6 thousand, an average Battery life of five years, inflationary increases, discount rate, and an average patient life calculated on probabilities outlined in the PRI-2012 mortality tables, published from the Society of Actuaries.
Stock-based Compensation
Stock-based compensation is measured at the grant date, based on the fair value of the award, and is recognized as an expense over the requisite service period. The fair value of stock-based payment awards granted through June 30, 2024 is estimated using the Black-Scholes option model with a volatility figure derived from using a determined peer group of other companies’ stock prices since the trading history of our stock was too short to provide accurate data. The fair value of stock-based payment awards granted subsequent to June 30, 2024 is estimated using the Black-Scholes option model with a volatility figure derived from using the trading history of our Common Stock. We account for the expected term of options in accordance with the “simplified” method, which is used for “plain-vanilla” options, as defined in ASC Topic 718, Share-based Payment. The risk-free interest rate was determined from the implied yields of U.S. Treasury zero-coupon bonds with a remaining life consistent with the expected term of the options.
We adopted the guidance from Accounting Standards Update 2016-09, Compensation - Stock Compensation (Topic 718): Improvements to Employee Share-Based Compensation Accounting, and we determined not to apply a forfeiture rate and have made the accounting election that forfeitures will be recognized when the actual forfeiture takes place therefore no estimated forfeiture rate will be recorded.
Recently Issued/Adopted Accounting Pronouncements
A discussion of recently issued accounting pronouncements and recently adopted accounting pronouncements is included in Note 2, “Summary of Significant Accounting Policies” of the accompanying consolidated financial statements as of December 31, 2024 and 2023 and for the years then ended included elsewhere in this Report.
Emerging Growth Company
Section 102(b)(1) of the Jumpstart Our Business Startups Act (“JOBS Act”) exempts emerging growth companies from being required to comply with new or revised financial accounting standards until private companies (that is, those that have not had a Securities Act registration statement declared effective or no not have a class of securities registered under the Securities Exchange Act of 1934, as amended) are required to comply with the new or revised financial accounting standards. The JOBS Act provides that a company can elect to opt out of the extended transition period and comply with the requirements that apply to non-emerging growth companies but any such election to opt out is irrevocable. We have elected not to opt out of such extended transition period which means that when a standard is issued or revised and it has different application dates for public and private companies, we, as an emerging growth company, can adopt the new or revised standard at the time the private companies adopt the new or revised standard, until such time we are no longer considered to be an emerging growth company. At times, we may elect to early adopt a new or revised standard.
ITEM 7A. Quantitative and Qualitative Disclosures About Market Risk
We are exposed to a variety of market risks, including currency risk, credit and counterparty risk, and inflation risk, as set out below. We manage and monitor these exposures to ensure appropriate measures are implemented in a timely and effective manner.
Currency Risk
Foreign currency risk is the risk that the value of a financial instrument fluctuates because of the change in foreign exchange rates. We primarily operate in the United States and Germany with most of the transactions settled in the United States dollar. Our presentation and functional currency is the United States dollar. Certain bank balances, deposits and other payables are denominated in the Euro, which exposes us to foreign currency risk. However, any transactions that may be conducted in foreign currencies are not expected to have a material effect on our results of operations, financial position or cash flows.
Credit and Counterparty Risk
Financial instruments that potentially expose us to concentrations of credit risk consist primarily of cash and accounts receivable, net. Periodically, we maintain deposits in accredited financial institutions in excess of federally insured limits. We maintain cash with financial institutions that management believes to be of high credit quality. We have not experienced any losses on such accounts and do not believe we are exposed to any unusual credit risk beyond the normal credit risk associated with commercial banking relationships.
With respect to accounts receivable, we perform credit evaluations of our customers and do not require collateral. There have been no material losses on accounts receivable. There were no customers that accounted for 10% or more of sales for the years ended December 31, 2024 and 2023.
Inflation Risk
Inflationary factors, such as increases in our cost of goods sold and selling and operating expenses, may adversely affect our operating results. Although we do not believe that inflation has had a material impact on our financial position or results of operations to date, a high rate of inflation in the future may have an adverse effect on our ability to maintain and increase our gross margin and decrease our selling and marketing and operating expenses as a percentage of our revenue if the selling prices of our products do not increase as much as or more than these increased costs.
ITEM 8. Financial Statements and Supplementary Data
Index to Consolidated Financial Statements
Envoy Medical, Inc.
December 31, 2024 and 2023
F-
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
Board of Directors and Stockholders
Envoy Medical, Inc.
Opinion on the financial statements
We have audited the accompanying consolidated balance sheets of Envoy Medical, Inc. (a Delaware corporation) and subsidiaries (the “Company”) as of December 31, 2024 and 2023, the related consolidated statements of operations and comprehensive loss, stockholders’ deficit, and cash flows for each of the two years in the period ended December 31, 2024, and the related notes (collectively referred to as the “consolidated financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2024 and 2023, and the results of its operations and its cash flows for each of the two years in the period ended December 31, 2024, in conformity with accounting principles generally accepted in the United States of America.
Going concern
The accompanying consolidated financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 2 to the financial statements, the Company has incurred cumulative losses from operations, has an accumulated deficit of $284.7 million as of December 31, 2024, and relies on external sources of liquidity to sustain operations. These conditions, along with other matters set forth in Note 2, raise substantial doubt about the Company’s ability to continue as a going concern. Management’s plans regarding these matters are also described in Note 2 to the consolidated financial statements. The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Basis for opinion
These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s consolidated financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (“PCAOB”) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
/S/ GRANT THORNTON LLP
We have served as the Company’s auditor since 2023.
Fort Lauderdale, Florida
March 28, 2025
F-
ENVOY MEDICAL, INC.
CONSOLIDATED BALANCE SHEETS
(In thousands, except share and per share amounts)
| December 31, 2024 |
December 31, 2023 |
|||||||
| Assets | ||||||||
| Current assets: | ||||||||
| Cash | $ | 5,483 | $ | 4,218 | ||||
| Accounts receivable, net | 38 | 70 | ||||||
| Other receivable | 780 | 176 | ||||||
| Inventories | 1,708 | 1,404 | ||||||
| Prepaid expenses and other current assets | 1,375 | 1,588 | ||||||
| Total current assets | 9,384 | 7,456 | ||||||
| Property and equipment, net | 1,275 | 351 | ||||||
| Operating lease right-of-use asset (related party) | 879 | 464 | ||||||
| Total assets | $ | 11,538 | $ | 8,271 | ||||
| Liabilities and stockholders’ deficit | ||||||||
| Current liabilities: | ||||||||
| Accounts payable | $ | 1,652 | $ | 1,554 | ||||
| Accrued expenses | 4,416 | 4,613 | ||||||
| Other current liabilities | 573 | 645 | ||||||
| Forward purchase agreement warrant liability | 472 | 4 | ||||||
| Product warranty liability, current portion | 282 | 311 | ||||||
| Operating lease liability, current portion (related party) | 143 | 158 | ||||||
| Total current liabilities | 7,538 | 7,285 | ||||||
| Term loans payable (related party) | 18,716 | — | ||||||
| Product warranty liability, net of current portion | 1,771 | 1,923 | ||||||
| Operating lease liability, net of current portion (related party) | 802 | 404 | ||||||
| Publicly traded warrant liability | 662 | 332 | ||||||
| Forward purchase agreement put option liability | — | 103 | ||||||
| Other liability | 891 | — | ||||||
| Total liabilities | 30,380 | 10,047 | ||||||
| Commitments and contingencies (see Note 16) | ||||||||
| Stockholders’ deficit: | ||||||||
| Series A Preferred Stock, $0.0001 par value; 100,000,000 shares authorized and 10,000,000 shares designated as of December 31, 2024 and 2023; 4,126,667 and 4,500,000 shares issued and outstanding as of December 31, 2024 and 2023, respectively | — | — | ||||||
| Class A Common Stock, $0.0001 par value; 400,000,000 shares authorized as of December 31, 2024 and 2023; 21,326,609 and 18,599,982 shares issued and outstanding as of December 31, 2024 and 2023, respectively | 2 | 2 | ||||||
| Additional paid-in capital | 266,013 | 255,596 | ||||||
| Accumulated deficit | (284,734 | ) | (257,256 | ) | ||||
| Accumulated other comprehensive loss | (123 | ) | (118 | ) | ||||
| Total stockholders’ deficit | (18,842 | ) | (1,776 | ) | ||||
| Total liabilities and stockholders’ deficit | $ | 11,538 | $ | 8,271 | ||||
F-
ENVOY MEDICAL, INC.
CONSOLIDATED STATEMENTS OF OPERATIONS AND COMPREHENSIVE LOSS
(In thousands, except share and per share amounts)
| Year Ended December 31, |
||||||||
| 2024 | 2023 | |||||||
| Net revenues | $ | 225 | $ | 316 | ||||
| Costs and operating expenses: | ||||||||
| Cost of goods sold | 742 | 789 | ||||||
| Research and development | 10,179 | 8,956 | ||||||
| Sales and marketing | 1,734 | 1,666 | ||||||
| General and administrative | 6,826 | 7,264 | ||||||
| Total costs and operating expenses | 19,481 | 18,675 | ||||||
| Operating loss | (19,256 | ) | (18,359 | ) | ||||
| Other income (expense): | ||||||||
| Change in fair value of convertible notes payable (related party) | (13,332 | ) | ||||||
| Change in fair value of forward purchase agreement put option liability | 103 | (69 | ) | |||||
| Change in fair value of forward purchase agreement warrant liability | 411 | 842 | ||||||
| Change in fair value of forward purchase agreement warrant liability due to modification | (881 | ) | ||||||
| Change in fair value of publicly traded warrant liability | (330 | ) | 942 | |||||
| Interest expense, related party | (816 | ) | ||||||
| Other (expense) income | (26 | ) | 54 | |||||
| Total other expense, net | (1,539 | ) | (11,563 | ) | ||||
| Net loss | (20,795 | ) | (29,922 | ) | ||||
| Induced conversion of Series A Preferred Stock into Class A Common Stock | (1,162 | ) | ||||||
| Deemed dividend on waiver of restriction on Class A Common Stock | (495 | ) | ||||||
| Cumulative preferred dividends | (5,521 | ) | (1,349 | ) | ||||
| Net loss attributable to common stockholders, basic and diluted | $ | (27,973 | ) | $ | (31,271 | ) | ||
| Net loss per share attributable to common stockholders, basic and diluted | $ | (1.49 | ) | $ | (2.54 | ) | ||
| Weighted-average Class A Common Stock outstanding, basic and diluted | 18,790,448 | 12,295,391 | ||||||
| Other comprehensive loss: | ||||||||
| Foreign currency translation adjustment | (5 | ) | (3 | ) | ||||
| Other comprehensive loss | (5 | ) | (3 | ) | ||||
| Comprehensive loss | $ | (20,800 | ) | $ | (29,925 | ) | ||
F-
ENVOY MEDICAL, INC.
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ DEFICIT
(In thousands, except share amounts)
| Series A Preferred Stock |
Class A Common Stock |
Additional Paid-in |
Accumulated | Accumulated Other Comprehensive |
Total Stockholders’ |
|||||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | Capital | Deficit | Loss | Deficit | |||||||||||||||||||||||||
| Balance at December 31, 2022 | — | $ | — | 10,122,581 | $ | 1 | $ | 189,904 | $ | (225,985 | ) | $ | (115 | ) | $ | (36,195 | ) | |||||||||||||||
| Deemed capital contribution
from related party (Note 10) |
— | — | — | — | 18,702 | — | — | 18,702 | ||||||||||||||||||||||||
| Foreign currency translation adjustment | — | — | — | — | — | — | (3 | ) | (3 | ) | ||||||||||||||||||||||
| Conversion of Convertible Notes into Class A Common Stock in connection with Merger (Note 3) | — | — | 4,874,707 | 1 | 27,493 | — | — | 27,494 | ||||||||||||||||||||||||
| Conversion of Envoy Bridge Note into Series A Preferred Stock in connection with Merger (Note 3) | 1,000,000 | — | — | — | 10,982 | — | — | 10,982 | ||||||||||||||||||||||||
| Preferred stock subscriptions (Note 3) | — | — | — | — | 2,000 | — | — | 2,000 | ||||||||||||||||||||||||
| Net exercise of warrants (related party) (Note 10) | — | — | 2,702 | — | — | — | — | — | ||||||||||||||||||||||||
| Merger, net of redemptions
and transaction costs (Note 3) |
2,500,000 | — | 3,115,874 | — | (1,785 | ) | — | — | (1,785 | ) | ||||||||||||||||||||||
| Meteora forward purchase agreement shares (Note 3) | — | — | 434,118 | — | (1,384 | ) | — | — | (1,384 | ) | ||||||||||||||||||||||
| Issuance of Series A Preferred stock to PIPE Investors (Note 3) | 1,000,000 | — | — | — | 10,000 | — | — | 10,000 | ||||||||||||||||||||||||
| Issuance of Common Stock | — | — | 50,000 | — | 109 | — | — | 109 | ||||||||||||||||||||||||
| Dividends on the Series A Preferred Stock | — | — | — | — | — | (1,349 | ) | — | (1,349 | ) | ||||||||||||||||||||||
| Stock-based compensation | — | — | — | — | 1,575 | — | — | 1,575 | ||||||||||||||||||||||||
| Return of the subscription proceeds for the additional Series A Preferred Stock | — | — | — | — | (2,000 | ) | — | — | (2,000 | ) | ||||||||||||||||||||||
| Net loss | — | — | — | — | — | (29,922 | ) | — | (29,922 | ) | ||||||||||||||||||||||
| Balance at December 31, 2023 | 4,500,000 | $ | — | 18,599,982 | $ | 2 | $ | 255,596 | $ | (257,256 | ) | $ | (118 | ) | $ | (1,776 | ) | |||||||||||||||
| Dividends on the Series A Preferred stock | — | — | — | — | — | (5,521 | ) | — | (5,521 | ) | ||||||||||||||||||||||
| Sale of Class A Common Stock through forward purchase agreement | — | — | — | — | 1,683 | — | — | 1,683 | ||||||||||||||||||||||||
| Exercise of Shortfall Warrants | — | — | 664,883 | — | 1,815 | — | — | 1,815 | ||||||||||||||||||||||||
| Extinguishment of excess warrant liability upon exercise of warrants associated with the forward purchase agreement | — | — | — | — | 96 | — | — | 96 | ||||||||||||||||||||||||
| Modification of forward purchase agreement | — | — | — | — | (94 | ) | — | — | (94 | ) | ||||||||||||||||||||||
| Stock-based compensation | — | — | — | — | 562 | — | — | 562 | ||||||||||||||||||||||||
| Issuance of warrants associated with 2024 Term Loans | — | — | — | — | 1,397 | — | — | 1,397 | ||||||||||||||||||||||||
| Issuance of Class A Common Stock under employee stock purchase plan | — | — | 32,758 | — | 63 | — | — | 63 | ||||||||||||||||||||||||
| Waiver of accrued dividends associated with Sponsor Support Agreement | — | — | — | — | 3,733 | — | — | 3,733 | ||||||||||||||||||||||||
| Induced conversion of Series A Preferred Stock into Class A Common Stock | (373,333 | ) | — | 1,028,986 | — | 1,162 | (1,162 | ) | — | — | ||||||||||||||||||||||
| Waiver of restriction and vesting requirement of Common Stock | — | — | 1,000,000 | — | — | — | ||||||||||||||||||||||||||
| Foreign currency translation adjustment | — | — | — | — | — | — | (5 | ) | (5 | ) | ||||||||||||||||||||||
| Net loss | — | — | — | — | — | (20,795 | ) | — | (20,795 | ) | ||||||||||||||||||||||
| Balance at December 31, 2024 | 4,126,667 | $ | — | 21,326,609 | $ | 2 | $ | 266,013 |
$ | (284,734 |
) | $ | (123 | ) | $ | (18,842 |
) | |||||||||||||||
F-
ENVOY MEDICAL, INC.
CONSOLIDATED STATEMENTS OF CASH FLOWS
(In thousands)
| Year Ended December 31, |
||||||||
| 2024 | 2023 | |||||||
| Cash flows from operating activities | ||||||||
| Net loss | $ | (20,795 | ) | $ | (29,922 | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||
| Depreciation | 173 | 133 | ||||||
| Interest expense and amortization of debt discount on term loans payable (related party) | 816 | — | ||||||
| Amortization of prepaid insurance | 1,047 | 602 | ||||||
| Stock-based compensation | 562 | 1,575 | ||||||
| Change in fair value of convertible notes payable (related party) | — | 13,332 | ||||||
| Change in fair value of warrant liability (related party) | — | (127 | ) | |||||
| Change in fair value of publicly traded warrant liability | 330 | (942 | ) | |||||
| Change in fair value of forward purchase agreement warrant liability | (411 | ) | (842 | ) | ||||
| Change in fair value of forward purchase agreement put option liability | (103 | ) | 69 | |||||
| Change in fair value of forward purchase agreement warrant liability due to modification | 881 | — | ||||||
| Change in operating lease right-of-use asset (related party) | 113 | 113 | ||||||
| Change in inventory reserve | 76 | (99 | ) | |||||
| Changes in operating assets and liabilities: | ||||||||
| Accounts receivable, net | 32 | (29 | ) | |||||
| Other receivable | (604 | ) | (176 | ) | ||||
| Inventories | (380 | ) | (10 | ) | ||||
| Prepaid expenses and other current assets | 9 | (853 | ) | |||||
| Accounts payable | (19 | ) | 551 | |||||
| Operating lease liability (related party) | (145 | ) | (128 | ) | ||||
| Accrued expenses | (241 | ) | (94 | ) | ||||
| Product warranty liability | (181 | ) | (244 | ) | ||||
| Other liability | 891 | — | ||||||
| Net cash used in operating activities | (17,949 | ) | (17,091 | ) | ||||
| Cash flows from investing activities | ||||||||
| Purchases of property and equipment | (980 | ) | (153 | ) | ||||
| Net cash used in investing activities | (980 | ) | (153 | ) | ||||
| Cash flows from financing activities | ||||||||
| Proceeds from the issuance of convertible notes payable (related party) | — | 10,000 | ||||||
| Proceeds from the issuance of Common Stock | — | 109 | ||||||
| Proceeds from the PIPE Transaction, the forward purchase agreement, and the Business Combination, net of transaction costs | — | 11,736 | ||||||
| Payments on insurance financing loans | (916 | ) | (563 | ) | ||||
| Proceeds from the issuance of term loans (related party) | 20,000 | — | ||||||
| Dividends paid to stockholders of Series A Preferred Stock | (2,447 | ) | — | |||||
| Proceeds from issuance of Common Stock under employee stock purchase plan | 63 | — | ||||||
| Proceeds from exercise of forward purchase agreement warrants | 1,815 | — | ||||||
| Proceeds from the sale of Common Stock associated with forward purchase agreement, net of transaction costs | 1,683 | — | ||||||
| Net cash provided by financing activities | 20,198 | 21,282 | ||||||
| Effect of exchange rate changes on cash | (5 | ) | (3 | ) | ||||
| Net increase in cash | 1,265 | 4,035 | ||||||
| Cash, beginning of year | 4,218 | 183 | ||||||
| Cash, end of year | $ | 5,483 | $ | 4,218 | ||||
| Supplemental disclosures of cash flow information: | ||||||||
| Cash paid for interest | $ | 41 | $ | 26 | ||||
| Cash paid for income taxes | $ | — | $ | — | ||||
| Non-cash investing and financing activities: | ||||||||
| Property and equipment purchased on account | $ | 117 | $ | — | ||||
| Financing of prepaid insurance | $ | 843 | $ | 1,115 | ||||
| Deemed capital contribution from related party | $ | — | $ | 18,702 | ||||
| Accrued and unpaid dividends on Series A Preferred Stock | $ | 3,074 | $ | 1,349 | ||||
| SPAC excise tax liability recognized upon the Business Combination | $ | — | $ | 2,248 | ||||
| Warrants issued with term loans (related party) | $ | 1,397 | $ | — | ||||
| Convertible debt exchanged for equity | $ | — | $ | 27,493 | ||||
| Bridge note exchanged for equity | $ | — | $ | 10,982 | ||||
| Series A Preferred Stock issued to PIPE investor in connection with the Merger | $ | — | $ | 10,000 | ||||
| Prepaid forward purchase agreement | $ | — | $ | 1,384 | ||||
| Extinguishment of excess warrant liability upon exercise of warrants associated with the forward purchase agreement | $ | 96 | $ | — | ||||
| Modification of forward purchase agreement warrant liability | $ | 94 | $ | — | ||||
| Waiver of accrued dividends associated with Sponsor Support Agreement | $ | 3,733 | $ | — | ||||
| Deemed dividend on waiver of restriction on Class A Common Stock | $ | 495 | $ | — | ||||
| Induced conversion of Series A Preferred Stock to Common Stock | $ | 1,162 | $ | — | ||||
| Lease liability arising from obtaining right-of-use asset | $ | 528 | $ | — | ||||
F-
ENVOY MEDICAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
1. Nature of the Business and Basis of Presentation
Envoy Medical, Inc. (“Envoy Medical” or the “Company”) is a hearing health company focused on providing innovative medical technologies across the hearing loss spectrum. Envoy Medical’s technologies are designed to shift the paradigm within the hearing industry and bring both providers and patients the hearing devices they desire. The Company’s first commercial product, the Esteem® Fully Implanted Active Middle Ear Implant (“Esteem FI-AMEI”), is a fully implanted active middle ear hearing device. The Esteem FI-AMEI was approved for sale in 2010 by the United States Food and Drug Administration (“FDA”).
Envoy Medical believes the fully implanted Acclaim® Cochlear Implant (“Acclaim CI”) is a first-of-its-kind cochlear implant. Envoy Medical’s fully implanted technology includes a sensor designed to leverage the natural anatomy of the ear instead of a microphone to capture sound. The Acclaim CI is designed to address severe to profound sensorineural hearing loss that is not adequately addressed by hearing aids. The Acclaim CI will only be indicated for adults who have been deemed adequate candidates by a qualified physician. The Acclaim CI received the Breakthrough Device Designation from the FDA in 2019.
On September 29, 2023 (the “Closing Date”), a merger transaction between Envoy Medical Corporation, Anzu Special Acquisition Corp I (“Anzu”) and Envoy Merger Sub, Inc., a directly, wholly owned subsidiary of Anzu (“Merger Sub”) was completed (hereinafter, the “Merger” or “Business Combination”, see Note 3) pursuant to the business combination agreement, dated as of April 17, 2023 (as amended, the “Business Combination Agreement”). In connection with the closing of the Merger (the “Closing”), Merger Sub merged with Envoy Medical Corporation, with Envoy Medical Corporation surviving the merger as a wholly owned subsidiary of Anzu. In connection with the Closing, Anzu changed its name to Envoy Medical, Inc. The Company’s Class A common stock, par value $0.0001 per share (“Common Stock”), and the Company’s public warrants commenced trading on the Nasdaq Stock Market LLC (“Nasdaq”) on October 2, 2023 under the symbols “COCH” and “COCHW,” respectively.
On April 17, 2023, prior to entering into the Business Combination Agreement, Anzu and Envoy Medical Corporation entered into an agreement (as amended to date, the “Forward Purchase Agreement”) with Meteora Special Opportunity Fund I, LP (“MSOF”), Meteora Capital Partners, LP (“MCP”), Meteora Select Trading Opportunities Master, LP (“MSTO”) and Meteora Strategic Capital, LLC (“MSC” and, collectively with MSOF, MCP and MSTO, the “Sellers” or “Meteora parties”) for an over-the-counter equity prepaid forward transaction.
Pursuant to the terms of the Forward Purchase Agreement, on the Closing Date, the Sellers purchased 425,606 shares of the Company’s Common Stock (the “Recycled Shares”) directly from the redeeming stockholders of Anzu. Also, effective upon the Closing Date, the Company paid to the Sellers a prepayment amount of $4.5 million required under the Forward Purchase Agreement directly from the trust account and transferred to the Sellers 8,512 shares of the Company’s Common Stock (the “Share Consideration”). During the year ended December 31, 2024, the Sellers sold the full amount of the Recycled Shares, and, pursuant to the Forward Purchase Agreement, the Company received $4.00 per share sold, or $1.7 million.
In addition, pursuant to the subscription agreement dated April 17, 2023 (as amended to date, the “Subscription Agreement”), by and between Anzu and Anzu SPAC GP I LLC (the “Sponsor”), the Company issued, and certain affiliates of the Sponsor purchased, concurrently with the Closing, an aggregate of 1,000,000 shares of the Company’s Series A preferred stock, par value $0.0001 per share (“Series A Preferred Stock”) in a private placement (the “PIPE Transaction”) at a price of $10.00 per share for an aggregate purchase price of $10.0 million.
F-
ENVOY MEDICAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Pursuant to the convertible promissory note, dated April 17, 2023, between Envoy Medical Corporation and GAT Funding, LLC (“GAT”) (as amended to date, the “Envoy Bridge Note”), the Company issued 1,000,000 shares of the Company’s Series A Preferred Stock to GAT in exchange for the conversion of the Envoy Bridge Note in full, concurrently with the Closing.
The consolidated financial statements include the accounts of Envoy Medical, Inc. and its wholly-owned subsidiaries Envoy Medical Corporation and Envoy Medical GmbH (Ansbach) (GmbH), which operates a sales office in Germany. All intercompany accounts and transactions have been eliminated in consolidation.
Basis of Presentation
The accompanying consolidated financial statements are presented in U.S. dollars in conformity with accounting principles generally accepted in the United States of America (“U.S. GAAP”) for financial information and pursuant to the rules and regulations of the U.S. Securities and Exchange Commission.
2. Summary of Significant Accounting Policies
Going Concern
Since inception, the Company has incurred cumulative losses from operations and has an accumulated deficit of $284.7 million at December 31, 2024. In February 2024, May 2024, July 2024, August 2024, and December 2024, the Company received advances of $5.0 million, $2.5 million, $2.5 million, $5.0 million, and $5.0 million, respectively, from term loans provided by a related party (see Note 9). In February 2024, the Company received net proceeds of $1.7 million from the sale of 425,606 shares held by the Meteora parties (see Note 1). In September 2023, the Company received $11.7 million proceeds from the Business Combination, Forward Purchase Agreement, and the PIPE Transaction, net of transaction costs. The Company had cash of $5.5 million as of December 31, 2024.
Management believes that its existing cash balances combined with future capital raises and cash receipts from product sales will be sufficient to fund ongoing operations through at least one year from the date the consolidated financial statements are issued. However, there can be no assurance that the Company will be successful in achieving its strategic plans, that the Company’s cash balances and future capital raises will be sufficient to support its ongoing operations, or that any additional financing will be available in a timely manner or on acceptable terms, if at all. If the Company is unable to raise sufficient financing when needed or events or circumstances occur such that the Company does not meet its strategic plans, the Company may be required to reduce certain of its discretionary spending. The Company may be unable to develop new or enhanced production methods, or be unable to fund capital expenditures, which could have a material adverse effect on the Company’s financial position, results of operations, cash flows, and ability to achieve its intended business objectives. These matters raise substantial doubt about the Company’s ability to continue as a going concern. The consolidated financial statements have been prepared assuming the Company will continue as a going concern and do not include adjustments to reflect the possible effects on the recoverability and classification of assets or the amounts and classification of liabilities that may result from the outcome of this uncertainty.
Use of Estimates
The preparation of consolidated financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. Actual results could differ from those estimates. Significant estimates and assumptions reflected in these consolidated financial statements include but are not limited to the useful lives of property and equipment, the net realizable value of inventory, product warranty liability, stock-based compensation expense, the fair value of forward purchase agreement put option liability, the fair value of forward purchase agreement warrant liability, publicly traded warrant liability, and the outcome of litigation. Estimates and assumptions are reviewed periodically and the effect of changes, if any, are reflected in the consolidated statements of operations and comprehensive loss.
F-
ENVOY MEDICAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Revisions
Cumulative Preferred Dividends
The Company corrected the presentation of cumulative preferred dividends that were not included on the previously issued consolidated statement of operations and comprehensive loss for the year ended December 31, 2023. The Company now presents these cumulative preferred dividends as a component of net loss attributable to common stockholders. The Company determined that the correction was not material to the year ended December 31, 2023 and therefore, amendments of previously filed reports are not required.
Contingent Sponsor Shares
In the fourth quarter of 2024, the Company corrected the presentation of Class A Common Stock outstanding that previously included 1,000,000 contingent sponsor shares that were included in the previously issued consolidated statement of stockholders’ deficit for the year ended December 31, 2023. These contingent sponsor shares are now excluded from the number of shares of Class A Common Stock outstanding as of December 31, 2023. The Company determined that the correction was not material to any prior annual or interim periods and therefore, amendments of previously filed reports are not required.
The effect of the contingent sponsor shares revision on the Common Stock amounts on each of the impacted financial statement line items within the Company’s consolidated statement of stockholders’ deficit for the year ended December 31, 2023 was as follows:
| Year Ended December 31, 2023 | ||||||||||||
| As Previously Reported |
Adjustments | As Revised | ||||||||||
| Merger, net of redemptions and transaction costs (Note 3) | 4,115,874 | (1,000,000 | ) | 3,115,874 | ||||||||
| Balance at December 31, 2023 | 19,599,982 | (1,000,000 | ) | 18,599,982 | ||||||||
Financing of Prepaid Insurance
In the fourth quarter of 2024, the Company corrected the presentation of prepaid insurance expenses and insurance financing liabilities that were not included on the previously issued consolidated balance sheet as of December 31, 2023. The Company now presents these prepaid insurance expenses within prepaid expenses and other current assets and these insurance financing liabilities within other current liabilities on the consolidated balance sheet as of December 31, 2023. The Company determined that the correction was not material to any prior annual or interim periods and therefore, amendments of previously filed reports are not required.
F-
ENVOY MEDICAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
The effect of the financing of prepaid insurance revision on each of the impacted financial statement line items within the Company’s consolidated balance sheet as of December 31, 2023 was as follows:
| December 31, 2023 | ||||||||||||
| As Previously Reported |
Adjustments | As Revised | ||||||||||
| Prepaid expenses and other current assets | $ | 957 | $ | 631 | $ | 1,588 | ||||||
| Total current assets | 6,825 | 631 | 7,456 | |||||||||
| Total assets | $ | 7,640 | $ | 631 | $ | 8,271 | ||||||
| Other current liabilities | $ | $ | 645 | $ | 645 | |||||||
| Total current liabilities | 6,640 | 645 | 7,285 | |||||||||
| Total liabilities | 9,402 | 645 | 10,047 | |||||||||
| Accumulated deficit | (257,242 | ) | (14 | ) | (257,256 | ) | ||||||
| Total stockholders’ deficit | (1,762 | ) | (14 | ) | (1,776 | ) | ||||||
| Total liabilities and stockholders’ deficit | $ | 7,640 | $ | 631 | $ | 8,271 | ||||||
The effect of the cumulative preferred dividends, contingent sponsor shares, and financing of prepaid insurance revisions on each of the impacted financial statement line items within the Company’s consolidated statement of operations and comprehensive loss for the year ended December 31, 2023 was as follows:
| Year Ended December 31, 2023 | ||||||||||||
| As Previously Reported |
Adjustments | As Revised | ||||||||||
| General and administrative | 7,276 | (12 | ) | 7,264 | ||||||||
| Total costs and operating expenses | 18,687 | (12 | ) | 18,675 | ||||||||
| Operating loss | (18,371 | ) | 12 | (18,359 | ) | |||||||
| Other (expense) income | 80 | (26 | ) | 54 | ||||||||
| Total other expense, net | (11,537 | ) | (26 | ) | (11,563 | ) | ||||||
| Net loss | (29,908 | ) | (14 | ) | (29,922 | ) | ||||||
| Cumulative preferred dividends | (1,349 | ) | (1,349 | ) | ||||||||
| Net loss attributable to common stockholders, basic and diluted | $ | (29,908 | ) | $ | (1,363 | ) | $ | (31,271 | ) | |||
| Net loss per share attributable to common stockholders, basic and diluted | $ | (2.38 | ) | $ | (0.16 | ) | $ | (2.54 | ) | |||
| Weighted-average Class A Common Stock outstanding, basic and diluted | 12,552,925 | (257,534 | ) | 12,295,391 | ||||||||
F-
ENVOY MEDICAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
The effect of the financing of prepaid insurance revision on the accumulated deficit amounts on each of the impacted financial statement line items within the Company’s consolidated statement of stockholders’ deficit for the year ended December 31, 2023 was as follows:
| Year Ended December 31, 2023 | ||||||||||||
| As Previously Reported | Adjustments | As Revised | ||||||||||
| Net loss | $ | (29,908 | ) | $ | (14 | ) | $ | (29,922 | ) | |||
| Balance at December 31, 2023 | $ | (257,242 | ) | $ | (14 | ) | $ | (257,256 | ) | |||
The effect of the financing of prepaid insurance revision on each of the impacted financial statement line items within the Company’s consolidated statements of cash flows for the year ended December 31, 2023 was as follows:
| Year Ended December 31, 2023 | ||||||||||||
| As Previously Reported | Adjustments | As Revised | ||||||||||
| Amortization of prepaid insurance | $ | $ | 602 | $ | 602 | |||||||
| Prepaid expenses and other current assets | (828 | ) | (25 | ) | (853 | ) | ||||||
| Net cash used in operating activities | (17,668 | ) | 577 | (17,091 | ) | |||||||
| Payments on insurance financing loans | (563 | ) | (563 | ) | ||||||||
| Net cash provided by financing activities | 21,845 | (563 | ) | 21,282 | ||||||||
| Supplemental disclosures of cash flow information: | ||||||||||||
| Cash paid for interest | $ | $ | 26 | $ | 26 | |||||||
| Non-cash investing and financing activities: | ||||||||||||
| Financing of prepaid insurance | $ | $ | 1,115 | $ | 1,115 | |||||||
Concentration of Credit Risk and Significant Customers
Financial instruments that potentially expose the Company to concentrations of credit risk consist primarily of cash and accounts receivable, net. Periodically, the Company maintains deposits in accredited financial institutions in excess of federally insured limits. The Company maintains its cash with financial institutions that management believes to be of high credit quality. The Company has not experienced any losses on such accounts and does not believe it is exposed to any unusual credit risk beyond the normal credit risk associated with commercial banking relationships.
With respect to accounts receivable, the Company performs credit evaluations of its customers and does not require collateral. There have been no material losses on the Company’s accounts receivable. There were no customers that accounted for 10.0% or more of sales for the years ended December 31, 2024 and 2023. There were no customers that accounted for 10.0% or more of the accounts receivable balance as of December 31, 2024 and 2023.
Cash
The Company maintains cash balances in bank accounts which, at times, may exceed federally insured limits.
F-
ENVOY MEDICAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Accounts Receivable, Net
Accounts receivable are recorded at the invoiced amount and do not bear interest. The Company grants credit to customers in the normal course of business, but generally does not require collateral or other security to support amounts due. Accounts receivable are presented net of an allowance for credit losses. Management performs ongoing credit evaluations of its customers based on financial information provided by the customer. Accounts receivable outstanding longer than the contractual payment terms are considered past due. The Company estimates its allowance for credit losses by considering numerous factors, including delinquency trends along with ongoing customer credit evaluations. The Company writes off accounts receivable when they become uncollectible. Payments subsequently received on such receivables are credited to the allowance for credit losses. The Company had no material bad debt expense for the years ended December 31, 2024 and 2023. The allowance for credit losses was not material as of December 31, 2024 and 2023.
Inventories
Inventories are stated at the lower of cost or net realizable value. Cost is determined using the first-in, first-out method. The Company records write-downs of inventories that are obsolete, past the manufacturer’s recommended ‘use by’ date, or in excess of anticipated demand or net realizable value based on a consideration of marketability and product life cycle stage, historical net sales and demand forecasts which consider the assumptions about future demand and market conditions. Inventory on hand that is not expected to be sold or utilized is considered excess, and the Company recognizes the write-down in cost of goods sold at the time of such determination. The write-down is determined by the excess of cost over net realizable value. Net realizable value is the estimated selling price in the ordinary course of business, less reasonably predictable cost of completion, disposal and transportation. At the time of loss recognition, a new cost basis is established and subsequent changes in facts and circumstances would not result in an increase in the cost basis.
Property and Equipment, Net
Property and equipment are stated at cost, net of accumulated depreciation. Additions and improvements that extend the lives of the assets are capitalized, while expenditures for repairs and maintenance are expensed as incurred. When assets are retired or otherwise disposed of, their costs and related accumulated depreciation are removed from the accounts and resulting gains or losses are included in operating results. Depreciation is calculated using the straight-line method over the estimated useful life of the asset, which ranges from three to seven years for property and equipment.
Operating Leases
The Company determines if an agreement is a lease at inception. The Company elected not to recognize the right to use an underlying asset (“ROU asset”) and lease liabilities for short-term leases, which are those that have a lease term of twelve months or less, and includes renewal options in the measurement of lease liabilities only when the option to purchase or renew a lease for the underlying asset is reasonably certain to be exercised. The Company has elected as an accounting policy to account for lease components and associated non-lease components as a single component.
The Company leases its headquarters office space under an operating lease with a related party and also leases office space in Germany under an operating lease (see Note 7). The determination of whether an arrangement is, or contains, a lease is performed at the inception of the arrangement and as necessary at modification. An operating lease is recorded on the consolidated balance sheets with the operating lease asset representing the right to use the ROU asset for the lease term and the lease liability representing the obligation to make lease payments arising from the lease. The Company excludes variable lease payments when measuring the ROU asset and lease liability, except for those that depend on an index, a rate or are in-substance fixed payments.
F-
ENVOY MEDICAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
ROU assets and lease liabilities are recognized at the commencement date based on the present value of lease payments over the lease term. In addition, ROU assets include initial direct costs incurred by the lessee as well as any lease payments made at or before the commencement date and exclude lease incentives. The discount rate implicit within the Company’s leases is generally not determinable; therefore, the Company determines the discount rate using its incremental borrowing rate based on the information available at the commencement date in determining the present value of lease payments.
Impairment of Long-Lived Assets
Long-lived assets held and used by the Company, including property and equipment and ROU assets, are reviewed for impairment whenever events or changes in business circumstances indicate that the carrying amount of an asset may not be recoverable. An impairment loss is recognized when estimated future undiscounted cash flows related to the assets are less than its carrying value. The amount of the impairment loss to be recorded, if any, is calculated by the excess of the asset’s carrying value over its fair value. The Company did not incur any impairment charges during the years ended December 31, 2024 and 2023.
Fair Value of Financial Instruments
The Company calculates the fair value of its assets and liabilities that qualify as financial instruments and includes this additional information in the notes to the consolidated financial statements when the fair value is different than the carrying value of these financial instruments. The estimated fair value of cash, accounts receivable, other receivable, prepaid expenses and other current assets, accounts payable, and accrued expenses approximate their carrying amounts due to the relatively short maturity of these instruments. The carrying value of the operating lease liability also approximates fair value since the instrument bears market rates of interest. The carrying value of the term loans payable also approximates fair value based upon current borrowing rates with similar maturities. None of these instruments are held for trading purposes.
Fair Value Measurement
The Company determines the fair value of financial assets and liabilities using the fair value hierarchy established in Accounting Standards Codification (“ASC”) Topic 820, “Fair Value Measurement” (“ASC 820”). ASC 820 identifies fair value as the exchange price, or exit price, representing the amount that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants. The hierarchy describes three levels of inputs that may be used to measure fair value, as follows:
| ● | Level 1 - Observable inputs, such as quoted prices in active markets for identical assets and liabilities. |
| ● | Level 2 - Observable inputs other than Level 1 that are observable, either directly or indirectly, such as quoted prices for similar assets or liabilities, quoted prices in markets that are not active, or other inputs that are observable or can be corroborated by observable market data for substantially the full term of the assets or liabilities. |
| ● | Level 3 - Unobservable inputs that are supported by little or no market activity and that are significant to the fair value of the assets or liabilities. |
A financial instrument’s level within the fair value hierarchy is based on the lowest level of any input that is significant to the fair value measurement. Until the conversion at the Closing, the Company had elected the fair value option for the convertible notes payable (related party) under ASC Topic 825, Financial Instruments (“ASC 825”), with changes in fair value recorded in income (loss) each reporting period. The convertible notes payable (related party) were converted on the Closing Date and are no longer outstanding for any periods presented in the consolidated financial statements. The Company’s forward purchase agreement put option liability and forward purchase agreement warrant liability are considered to be Level 3 financial instruments measured at fair value and are described below (see Note 4).
F-
ENVOY MEDICAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Derivative Financial Instruments
The Company does not use derivative instruments to hedge exposures to cash flow, market, or foreign-currency risks. The Company evaluates its financial instruments to determine if such instruments are derivatives or contain features that qualify as embedded derivatives in accordance with ASC Topic 815, Derivatives and Hedging (“ASC 815”). For derivative financial instruments that are accounted for as liabilities, the derivative instrument is initially recorded at its fair value on the grant date and is then re-valued at each reporting date, with changes in the fair value reported in the other income (expense) section of the Company’s consolidated statements of operations and comprehensive loss. The classification of derivative instruments, including whether such instruments should be recorded as liabilities or as equity, is evaluated at the end of each reporting period. Derivative liabilities are classified in the consolidated balance sheets as current or non-current based on whether or not net-cash settlement or conversion of the instrument could be required within 12 months of the balance sheet date.
The Company accounts for its publicly traded warrant liability in accordance with ASC 815-40. Accordingly, the Company recognized the warrant instruments as a liability at fair value and adjusts the instruments to fair value at each reporting period. The publicly traded warrant liability is subject to re-measurement at each balance sheet date until exercised, and any change in fair value is recognized in the change in fair value of publicly traded warrant liability line item in the Company’s consolidated statements of operations and comprehensive loss.
The Company accounts for its Forward Purchase Agreement in accordance with ASC 815-40. Accordingly, the Company recognized the forward purchase agreement put option liability and the forward purchase agreement warrant liability at fair value at each reporting period. The forward purchase agreement put option liability and the forward purchase agreement warrant liability are subject to re-measurement at each balance sheet date, and any change in fair value is recognized in the change in fair value of forward purchase agreement put option liability and change in fair value of forward purchase agreement warrant liability line items, respectively, in the Company’s consolidated statements of operations and comprehensive loss.
SPAC Excise Tax Liability
The Company recognized an excise tax liability of $2.2 million upon completion of the Company’s Business Combination as an incremental cost to repurchase the Company’s treasury shares, with an offsetting tax liability recognized. The SPAC excise tax liability is recorded in accrued expenses in the Company’s consolidated balance sheets.
Revenue Recognition
The Company recognizes revenue in accordance with ASC Topic 606, Revenue from Contracts with Customers, which provides a five-step model for recognizing revenue from contracts with customers as follows:
| ● | identify the contract with a customer |
| ● | identify the performance obligations in the contract |
| ● | determine the transaction price |
| ● | allocate the transaction price to the performance obligations in the contract |
| ● | recognize revenue when or as performance obligations are satisfied. |
Revenue is recognized as performance obligations under the terms of a contract are satisfied, which generally occurs as control of the promised products or services is transferred to customers. Revenue is measured as the amount of consideration the Company expects to receive in exchange for transferring products or services to a customer (“transaction price”). To the extent the transaction price includes variable consideration, the Company estimates the amount of variable consideration that should be included in the transaction price using either the expected value or most likely amount method. Variable consideration is included in the transaction price if, in the Company’s judgment, it is probable that a significant future reversal of cumulative revenue under the contract will not occur. Estimates of variable consideration and determination of whether to include estimated amounts in the transaction price are based largely on an assessment of the Company’s anticipated performance and all information that is reasonably available.
F-
ENVOY MEDICAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
The Company primarily derives revenue from the sale of its hearing device products. Revenue from product sales is recognized upon transfer of control of the product to a customer, which occurs at a point in time, at the time the Company is notified the product has been implanted or used by the customer in a surgical procedure. The Company also sells prepaid Battery replacement options. Revenue from extended warranty plans is recognized ratably over time and was immaterial for each of the years ended December 31, 2024 and 2023. Amounts received from a customer prior to fulfillment of the performance obligation are included as accrued expenses on the consolidated balance sheets and are immaterial as of December 31, 2024 and 2023. The Company has elected to account for shipping and handling activities performed as activities to fulfill the promise to transfer the products; and therefore these activities are not assessed as a separate performance obligation to its customers.
Revenue is measured as the amount of consideration the Company expects to receive, which is based on the invoiced price. The majority of the Company’s contracts have a single performance obligation and are short term in nature. The Company’s contracts do not include variable consideration.
Payment terms differ by geography and customer, but payment is generally required within 30 days from the date of product utilization. The Company also offers extended payment plans on a limited basis. Amounts due to the Company under payment plans that extend beyond 12 months are immaterial as of December 31, 2024 and 2023, and therefore the Company did not adjust the promised amount of consideration for the effects of a significant financing component.
Cost of Goods Sold
Cost of goods sold is comprised of the costs of merchandise sold, as well as the related inbound freight costs and labor directly attributable to bringing certain goods to a salable condition. In categorizing costs, the Company captures applicable depreciation and costs to maintain and run revenue generating technology, equipment related costs and any personnel-related costs as cost of goods sold.
Product Warranty
The Company provides a limited warranty for its implantable components. At the time product revenue is recognized, the Company reserves for estimated future costs that may be incurred under its warranties based on historical experience. The limited warranty liability is recorded in accrued expenses in the consolidated balance sheets. As of December 31, 2024 and 2023, the amount of accrued limited warranty was immaterial and the Company’s warranty payments were immaterial.
During 2013, the Company offered a lifetime warranty to clinical trial patients to cover batteries and surgery related costs. The Company estimates the costs that may be incurred under this lifetime warranty and records a liability in the amount of such costs at its present value. The lifetime warranty is recorded in product warranty liability in the consolidated balance sheets. As of December 31, 2024 and 2023, the aggregate product warranty liability was $2.1 million and $2.2 million, respectively, of which $0.3 million and $0.3 million, respectively, was classified as a current liability in the consolidated balance sheets.
F-
ENVOY MEDICAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Patents
All patent-related costs incurred in connection with filing and prosecuting patent applications are expensed as incurred due to the uncertainty about the recovery of the expenditure. Amounts incurred are classified as general and administrative expenses.
Research and Development Costs
Expenditures for research and development activities are charged to operations as incurred. Research and development costs include salaries, employee benefits and laboratory testing expenses.
Income Taxes
Income taxes are accounted for under the asset and liability method. Deferred tax assets and liabilities are recognized for the future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax base and operating loss and tax credit carryforwards. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those items are expected to be recovered or settled. The Company has recorded a full valuation allowance against the net deferred tax asset due to the uncertainty of realizing the related benefits.
The Company recognizes the financial statement benefit of a tax position only to the extent the position is more likely than not to be sustained upon audit based on the technical merits of the position. For tax positions meeting the more-likely-than-not threshold, the amount recognized in the Company’s consolidated financial statements is the largest benefit that has a greater than 50% likelihood of being realized upon ultimate settlement with the relevant tax authority. The Company has elected to recognize interest and penalties related to uncertain tax positions in the provision for income taxes.
Foreign Currency Translation
The Euro is the functional currency for the Company’s foreign subsidiary in Germany. The assets and liabilities of the Company’s foreign operations are translated into U.S. dollars at the end-of-the-period exchange rates, and the revenues and expenses are translated at weighted-average rates for the respective reporting period. Unrealized translation gains and losses are recorded as a translation adjustment, which is included in the Company’s consolidated statements of stockholders’ deficit as well as a component of accumulated other comprehensive loss on the Company’s consolidated statements of operations and comprehensive loss.
Net Loss per Share
The Company’s Series A Preferred Stock certificate of designation entitles the holders to participate in dividends on an as converted basis when declared on Common Stock. As a result, the Series A Preferred Stock meets the definition of a participating security, which requires the Company to apply the two-class method to compute both basic and diluted net loss per share attributable to common stockholders. The two-class method is an earnings allocation formula that treats participating securities as having rights to earnings that would otherwise have been available to common stockholders. The two-class method requires income available to holders of the Company’s Common Stock for the period to be allocated between common and participating securities based upon their respective rights to share in the earnings as if all income for the period had been distributed. In periods where there is a net loss, no allocation of undistributed net loss to the Series A Preferred Stock is performed as the holders of the Series A Preferred Stock are not contractually obligated to participate in the Company’s losses. The Company reported net losses of $28.0 million and $31.3 million attributable to the stockholders of the Company’s Common Stock for the years ended December 31, 2024 and 2023, respectively.
Basic net loss per share of common stock is computed by dividing the net loss attributable to common stockholders by the weighted-average number of shares of Common Stock outstanding during the period. Diluted net loss per share is computed by dividing the net loss attributable to common stockholders by the weighted-average number of shares outstanding, plus the impact of potential common shares, if dilutive, resulting from the potential exercise of warrants or options, and the potential conversion of preferred stock or convertible notes, into Common Stock, under the if-converted method. In periods where a net loss is recorded, no effect is given to potentially dilutive securities, because the effect would be anti-dilutive.
F-
ENVOY MEDICAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Stock-based Compensation
Stock-based compensation is measured at the grant date, based on the fair value of the award, and is recognized as an expense over the requisite service period. The fair value of stock-based payment awards granted through June 30, 2024 is estimated using the Black-Scholes option model with a volatility figure derived from using a determined peer group of other companies’ stock prices since the trading history of the Company’s stock was too short to provide accurate data. The fair value of stock-based payment awards granted subsequent to June 30, 2024 is estimated using the Black-Scholes option model with a volatility figure derived from using the trading history of the Company’s Common Stock. Given limited historical exercise data, the Company accounts for the expected term of all options in all periods in accordance with the “simplified” method, which is used for “plain-vanilla” options, as defined in ASC Topic 718, Compensation - Stock Compensation. The simplified method calculates the expected term as the average of the time-to-vesting and the contractual life of the options. The risk-free interest rate was determined from the implied yields of U.S. Treasury zero-coupon bonds with a remaining life consistent with the expected term of the options.
The Company has adopted the guidance from Accounting Standards Update (“ASU”) 2016-09, Compensation – Stock Compensation (Topic 718): Improvements to Employee Share-Based Compensation Accounting, and has determined not to apply a forfeiture rate and has made the accounting election that forfeitures will be recognized when the actual forfeiture takes place and therefore no estimated forfeiture rate will be recorded.
Segments
Operating segments are identified as components of an enterprise about which discrete financial information is available for evaluation by the chief operating decision-maker (“CODM”) in deciding resource allocation and assessing performance. The Company has determined that its CODM is its Chief Executive Officer. The Company’s CODM reviews financial information presented on a consolidated basis for the purposes of making decisions, allocating resources and evaluating performance. Consequently, the Company has determined it operates in one operating and reportable segment.
Recently Adopted Accounting Pronouncements
In June 2016, the Financial Accounting Standards Board (“FASB”) issued ASU 2016-13, Measurement of Credit Losses on Financial Instruments (“ASC 326”). This guidance introduces a new model for recognizing credit losses on financial instruments based on an estimate of current expected credit losses. The Company adopted ASC 326 with an adoption date of January 1, 2023 using the modified retrospective approach. As a result, the Company changed its accounting policy for allowance for credit losses. The Company monitors accounts receivables and estimates the allowance for lifetime expected credit losses. Estimates of expected credit losses are based on historical collection experience and other factors, including those related to current market conditions and events. The adoption did not have a material effect on the Company’s accompanying consolidated financial statements.
In August 2020, the FASB issued ASU 2020-06, Debt—Debt with Conversion and Other Options (Subtopic 470-20) and Derivatives and Hedging—Contracts in Entity’s Own Equity (Subtopic 815-40) (“ASU 2020-06”) to simplify certain financial instruments. ASU 2020-06 eliminates the current models that require separation of beneficial conversion and cash conversion features from convertible instruments and simplifies the derivative scope exception guidance pertaining to equity classification of contracts in an entity’s own equity. The new standard also introduces additional disclosures for convertible debt and freestanding instruments that are indexed to and settled in an entity’s own equity. ASU 2020-06 amends the diluted earnings per share guidance, including the requirement to use the if-converted method for all convertible instruments. For smaller reporting companies, ASU 2020-06 is effective for fiscal years beginning after December 15, 2023 and should be applied on a full or modified retrospective basis. Early adoption is permitted, but no earlier than fiscal years beginning after December 15, 2020, including interim periods within those fiscal years. The Company adopted ASU 2020-06 effective January 1, 2024. The adoption of ASU 2020-06 did not have a material impact on the Company’s consolidated financial statements.
F-
ENVOY MEDICAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
In November 2023, the FASB issued ASU 2023-07, Segment Reporting (Topic 280): Improvements to Reportable Segment Disclosures (“ASU 2023-07”), which will add required disclosures of significant expenses for each reportable segment, as well as certain other disclosures to help investors understand how the CODM evaluates segment expenses and operating results. The new standard will also allow disclosure of multiple measures of segment profitability if those measures are used to allocate resources and assess performance. The Company adopted ASU 2023-07 effective for the year ended December 31, 2024. See Note 14 for related disclosures.
Accounting Pronouncements Not Yet Effective
In December 2023, the FASB issued ASU 2023-09, Income Taxes (Topic 740): Improvements to Income Tax Disclosures, which requires disaggregated information about a reporting entity’s effective tax rate reconciliation as well as information on income taxes paid. The standard is intended to benefit investors by providing more detailed income tax disclosures that would be useful in making capital allocation decisions. The standard will be effective for public companies for fiscal years beginning after December 15, 2024. Early adoption is permitted. The Company is currently evaluating the impact that this ASU will have on the Company’s disclosures within the consolidated financial statements.
In November 2024, the FASB issued ASU 2024-03, Income Statement - Reporting Comprehensive Income - Expense Disaggregation Disclosures (Subtopic 220-40): Disaggregation of Income Statement Expenses, that requires public companies to disclose, in interim and reporting periods, additional information about certain expenses in the financial statements. For public business entities, it is effective for annual periods beginning after December 15, 2026, and interim reporting periods beginning after December 15, 2027. Early adoption is permitted and is effective on either a prospective basis or retrospective basis. The Company is currently evaluating the impact that this ASU will have on the Company’s disclosures within the consolidated financial statements.
3. Merger
As discussed in Note 1, on September 29, 2023, the Company completed the Merger. Upon the Closing, the following occurred:
| ● | each share of Envoy Medical Corporation common stock (“Envoy Medical Corporation Common Stock”) immediately prior to the Business Combination was automatically cancelled and converted into the right to receive 0.063603 shares of Common Stock resulting in the issuance of 14,999,990 shares of Common Stock: |
| ○ | each share of outstanding Envoy Medical Corporation Common Stock, which totaled 139,153,144 shares, was cancelled and converted into 8,850,526 shares of Common Stock; |
| ○ | each outstanding warrant to purchase Envoy Medical Corporation Common Stock, depending on the applicable exercise price, was automatically cancelled or exercised on a net exercise basis and converted into 2,702 shares of Common Stock; |
| ○ | the outstanding Convertible Notes, as defined in Note 9, were automatically converted into 4,874,707 shares of Common Stock; |
| ○ | each share of Envoy Medical Corporation redeemable convertible preferred stock, par value $0.01 per share, issued and outstanding immediately prior to the Closing (“Envoy Medical Corporation Preferred Stock”), which totaled 4,000,000 shares, were converted into 20,000,000 shares of Envoy Medical Corporation Common Stock and subsequently exchanged for 1,272,055 shares of Common Stock; |
F-
ENVOY MEDICAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
| ● | each outstanding option to purchase shares of Envoy Medical Corporation Common Stock outstanding as of immediately prior to the Business Combination was cancelled in exchange for nominal consideration; |
| ● | each share of Merger Sub’s common stock, par value $0.0001 per share, issued and outstanding immediately prior to the Business Combination was converted into and exchanged for one share of Common Stock; |
| ● | the Sponsor forfeited 5,510,000 shares of Anzu’s Class B common stock, par value $0.0001 per share (“Anzu Class B Common Stock”), and all 12,500,000 private placement warrants pursuant to the sponsor support and forfeiture agreement dated April 17, 2023 by and between Anzu, Envoy Medical Corporation and the Sponsor, as amended or modified from time to time (the “Sponsor Support Agreement”); |
| ● | all of Anzu’s outstanding 14,166,666 public placement warrants were exchanged for warrants, each exercisable for one share of Common Stock at a price of $11.50 per share; |
| ● | the Sponsor exchanged 2,500,000 shares of Anzu Class B Common Stock for 2,500,000 shares of Series A Preferred Stock pursuant to the Sponsor Support Agreement; |
| ● | an aggregate of 2,615,000 shares of Anzu Class B Common Stock held by the Sponsor and Anzu’s former independent directors automatically converted into an equal number of shares of Common Stock; |
| ● | pursuant to the legacy forward purchase agreements and the extension support agreements of Anzu, the Sponsor transferred an aggregate of 490,000 shares of Common Stock to the parties to the legacy forward purchase agreements and the extension support agreements; |
| ● | the Company issued an aggregate of 8,512 shares of Common Stock as Share Consideration pursuant to the Forward Purchase Agreement; |
| ● | the Sellers in their sole discretion may request warrants of the Company exercisable for shares of Common Stock (the “Shortfall Warrants”) in an amount equal to 3,874,394 based on the terms of the Forward Purchase Agreement (as amended in July 2024 and December 2024); |
| ● | the Company issued, and certain affiliates of the Sponsor purchased, concurrently with the Closing, an aggregate of 1,000,000 shares of Series A Preferred Stock in the PIPE Transaction at a price of $10.00 per share for an aggregate purchase price of $10.0 million; and |
| ● | pursuant to the Envoy Bridge Note, the Company issued 1,000,000 shares of Series A Preferred Stock to GAT concurrently with the Closing. |
The proceeds received by the Company from the Merger, the PIPE Transaction, and the Forward Purchase Agreement, net of transaction costs, totaled $11.7 million.
The Merger was accounted for as a reverse recapitalization in accordance with U.S. GAAP. Under this method of accounting, Anzu was treated as the acquired company for financial reporting purposes. Accordingly, for accounting purposes, the Merger was treated as the equivalent of the Company issuing shares for the net assets of Anzu, accompanied by a recapitalization. The net assets of Anzu were stated at historical cost with no goodwill or other intangible assets recorded.
F-
ENVOY MEDICAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
The following tables present the total shares of Common Stock and Series A Preferred Stock outstanding immediately after the Closing:
| Class A Common Stock | Number of Shares |
|
| Exchange of Anzu Class A Common Stock subject to possible redemption that was not redeemed for Common Stock | 1,500,874 | |
| Conversion of Anzu Class B Common Stock held by the Sponsor and Anzu’s former independent director into Common Stock* | 1,615,000 | |
| Subtotal - Merger, net of redemptions | 3,115,874 | |
| Exchange of Envoy Medical Corporation Common Stock for Common Stock | 8,850,526 | |
| Exchange of Envoy Medical Corporation Preferred Stock for Common Stock | 1,272,055 | |
| Conversion of Convertible Notes as of September 29, 2023 into Common Stock | 4,874,707 | |
| Net exercise of Envoy Medical Corporation warrants outstanding | 2,702 | |
| Issuance of Share Consideration to Meteora parties | 8,512 | |
| Shares recycled by Meteora parties | 425,606 | |
| 18,549,982 |
| * | 1,000,000 shares of the Common Stock were unvested and subject to restrictions and forfeitures per the Sponsor Support Agreement and therefore excluded from the total shares of Common Stock outstanding immediately after the Closing. These shares would have vested upon the FDA approval of Acclaim CI or upon a change of control of the Company. On December 20, 2024, the Company and the Sponsor entered into an agreement to remove the vesting restriction (see Notes 10 and 11) and these shares are now considered outstanding as of that date. |
| Series A Preferred Stock | Number of Shares |
|||
| Exchange of Anzu Class B Common Stock for Series A Preferred Stock | 2,500,000 | |||
| Issuance of Series A Preferred Stock in connection with the PIPE Transaction | 1,000,000 | |||
| Issuance of Series A Preferred Stock in connection with the conversion of the Envoy Bridge Note | 1,000,000 | |||
| 4,500,000 | ||||
4. Fair Value Measurements
The following tables provide information related to the Company’s assets and liabilities measured at fair value on a recurring basis as of December 31, 2024 and 2023 (in thousands):
| December 31, 2024 | ||||||||||||||||
| Level 1 | Level 2 | Level 3 | Total | |||||||||||||
| Liabilities: | ||||||||||||||||
| Forward purchase agreement warrant liability | $ | $ | $ | 472 | $ | 472 | ||||||||||
| Publicly traded warrant liability | 662 | 662 | ||||||||||||||
| $ | 662 | $ | $ | 472 | $ | 1,134 | ||||||||||
F-
ENVOY MEDICAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
| December 31, 2023 | ||||||||||||||||
| Level 1 | Level 2 | Level 3 | Total | |||||||||||||
| Liabilities: | ||||||||||||||||
| Forward purchase agreement warrant liability | $ | $ | $ | 4 | $ | 4 | ||||||||||
| Forward purchase agreement put option liability | 103 | 103 | ||||||||||||||
| Publicly traded warrant liability | 332 | 332 | ||||||||||||||
| $ | 332 | $ | $ | 107 | $ | 439 | ||||||||||
The fair values of the forward purchase agreement warrant liability and forward purchase agreement put option liability, which are Level 3 fair value measurements, were estimated using Monte Carlo Simulation models. The use of significant unobservable inputs could result in those inputs being different at the reporting dates and which could result in a significantly higher or lower fair value measurement at the reporting dates. The following table presents the quantitative information regarding Level 3 fair value measurements of the forward purchase agreement warrant liability as of December 31, 2024 and the forward purchase agreement warrant liability and forward purchase agreement put option liability as of December 31, 2023:
| December 31, 2024 |
December 31, 2023 |
|||||||
| Stock price | $ | 1.43 | $ | 1.81 | ||||
| Initial exercise price | $ | 10.46 | $ | 10.46 | ||||
| Annual volatility | 130.0 | % | 46.9 | % | ||||
| Remaining term (in years) | 1.00 | 0.75 | ||||||
| Risk-free rate | 4.08 | % | 4.90 | % | ||||
The Company has classified the publicly traded warrant liability within Level 1 of the hierarchy as the warrant is separately listed and traded in an active market. The publicly traded warrant’s listed price in an active market was used as the fair value.
The following table summarizes the activity for the Company’s Level 3 instruments measured at fair value on a recurring basis (in thousands):
| Forward Purchase Agreement Warrant Liability |
Forward Purchase Agreement Put Option Liability |
|||||||
| Balance as of December 31, 2023 | $ | 4 | $ | 103 | ||||
| Change in fair value | (411 | ) | (103 | ) | ||||
| Effect of amendments (see Note 10) | 975 | |||||||
| Extinguishment of excess warrant liability upon exercise of warrants associated with the forward purchase agreement | (96 | ) | ||||||
| Balance as of December 31, 2024 | $ | 472 | $ | |||||
There were no transfers between Level 1 and Level 2, nor into and out of Level 3, during the periods presented.
F-
ENVOY MEDICAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Convertible Notes
Effective concurrently with the Merger, the outstanding balance of principal and accrued interest of the Convertible Notes was automatically converted into Common Stock and the outstanding balance of principal and accrued interest of the Envoy Bridge Note was converted into Series A Preferred Stock (see Note 3). As such, the Convertible Notes and Envoy Bridge Note were derecognized from the consolidated balance sheets. Immediately prior to the Merger, the fair value of the Convertible Notes was calculated by multiplying the amount of Common Stock the Convertible Notes converted into by the fair value of these shares. The fair value of the Common Stock was based on the listed prices for the shares, immediately prior to the Merger. Immediately prior to the Merger, the fair value of the Envoy Bridge Note was calculated by multiplying the amount of Series A Preferred Stock the Envoy Bridge Note converted into, by the fair value of these shares. The fair value of the Series A Preferred Stock was estimated using a Monte Carlo Simulation model, which is a Level 3 fair value measurement. The following table presents the quantitative information regarding Level 3 fair value measurements of the Series A Preferred Stock, which was valued at $10.98 per share.
| September 29, 2023 |
||||
| Underlying stock price | $ | 7.02 | ||
| Exercise price | $ | 11.50 | ||
| Expected term (in years) | 10.00 | |||
| Expected volatility | 48.9 | % | ||
5. Inventories
Inventories, consisted of the following (in thousands):
| December 31, 2024 |
December 31, 2023 |
|||||||
| Raw materials | $ | 1,386 | $ | 1,162 | ||||
| Work-in-progress | 203 | 158 | ||||||
| Finished goods | 119 | 84 | ||||||
| $ | 1,708 | $ | 1,404 | |||||
6. Property and Equipment, Net
Property and equipment consisted of the following (in thousands):
| December 31, 2024 |
December 31, 2023 |
|||||||
| Lab equipment | $ | 3,106 | $ | 2,750 | ||||
| Production equipment | 2,249 | 1,508 | ||||||
| Computer equipment | 648 | 648 | ||||||
| Office equipment | 102 | 102 | ||||||
| Total | 6,105 | 5,008 | ||||||
| Less: Accumulated depreciation | (4,830 | ) | (4,657 | ) | ||||
| Property and equipment, net | $ | 1,275 | $ | 351 | ||||
Depreciation expense was $0.2 million and $0.1 million for the years ended December 31, 2024 and 2023, respectively.
F-
ENVOY MEDICAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
7. Operating Leases
The Company leases its headquarters office space in Minnesota and leases office space in Germany. The headquarters office space lease is with a stockholder, which is considered a related party. During the year ended December 31, 2024, the Company and the landlord agreed to modify the lease to extend the lease term for three (3) additional years through December 31, 2030. Additionally, the Company requested and the landlord provided an additional 1,664 square feet of usable office space, for a total of 11,540 square feet of rentable space. Accordingly, base rent was increased to $6 thousand per month and increases each year by approximately four percent. Also, tenant improvements completed by the landlord totaling $0.15 million will be repaid in three $50 thousand annual payments beginning July 1, 2027. As a result of the modification, the Company recognized an increase to the ROU asset and operating lease liability of $0.5 million which is reflected in the consolidated balance sheets.
The lease of the office space in Germany is not with a related party and is immaterial.
The components of leases and lease costs were as follows (in thousands):
| December 31, 2024 |
December 31, 2023 |
|||||||
| Operating lease right-of-use asset (related party) | $ | 879 | $ | 464 | ||||
| Operating lease liability, current portion (related party) | $ | 143 | $ | 158 | ||||
| Operating lease liability, net of current portion (related party) | 802 | 404 | ||||||
| $ | 945 | $ | 562 | |||||
| Year Ended December 31, | ||||||||
| 2024 | 2023 | |||||||
| Operating lease cost | $ | 180 | $ | 127 | ||||
| $ | 180 | $ | 127 | |||||
Other supplemental information of lease amounts recognized in the consolidated financial statements is summarized as follows:
| Year Ended December 31, | ||||||||
| 2024 | 2023 | |||||||
| Cash paid for amounts included in the measurement of lease liability | $ | 201 | $ | 142 | ||||
| December 31, 2024 |
December 31, 2023 |
|||||||
| Weighted-average remaining lease term - in years | 6.0 | 3.9 | ||||||
| Weighted-average discount rate | 9.9 | % | 5.0 | % | ||||
F-
ENVOY MEDICAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Future minimum lease payments associated with these leases were as follows as of December 31, 2024 (in thousands):
| 2025 | $ | 229 | ||
| 2026 | 223 | |||
| 2027 | 230 | |||
| 2028 | 195 | |||
| 2029 | 198 | |||
| Thereafter | 151 | |||
| 1,226 | ||||
| Less: Imputed interest | (281 | ) | ||
| $ | 945 |
8. Product Warranty Liability
Changes in warranty liability were as follows (in thousands):
| Amount | ||||
| Balance as of December 31, 2023 | $ | 2,234 | ||
| Utilization | (181 | ) | ||
| Balance as of December 31, 2024 | $ | 2,053 | ||
The assumptions utilized in developing the liability as of December 31, 2024, include an estimated cost per unit of $6 thousand, an average Battery life of five years, inflationary increase of 3.8%, and an average patient life calculated based on probabilities outlined in the PRI-2012 mortality tables, published from the Society of Actuaries. Additionally, a discount rate of 5.2% was used in the calculation as of December 31, 2024.
9. Debt (Related Party)
Convertible Notes
In 2012, the Company issued a convertible note to a controlling stockholder and member of the board of directors (the “2012 Convertible Note”) and in 2013 the Company issued convertible notes to various stockholders (the “2013 Convertible Notes”) (collectively, the “Convertible Notes”). On April 17, 2023, the Convertible Notes were amended as part of the Business Combination Agreement, to provide for automatic conversion immediately prior to the Merger. The conversion formulae were not adjusted as part of these amendments. The loan amendments were accounted for as extinguishments with a related party and treated as deemed capital contributions.
Effective concurrently with the Merger, the outstanding balance of principal and any unpaid accrued interest on the Convertible Notes was automatically converted into Common Stock at a conversion price of $15.72 per share (see Note 3) and the fair value of the Convertible Notes was derecognized from the consolidated balance sheets.
The Company elected the fair value option for the Convertible Notes under ASC 825, with changes in fair value recorded in earnings each reporting period. The Company recorded an expense of $13.3 million related to the change in fair value prior to the conversion of the Convertible Notes during the year ended December 31, 2023.
Envoy Bridge Note
On April 17, 2023, the Company entered into the Envoy Bridge Note with a controlling stockholder and member of the board of directors for an aggregate borrowing capacity of $10.0 million, an interest rate of 4.5% per annum and maturity date of December 31, 2025. The Envoy Bridge Note was unsecured. According to this agreement, $4.0 million of the borrowing capacity was funded via the transfer of $4.0 million in principal from the 2012 Convertible Note. An additional $3.0 million was drawn upon during the second quarter of 2023 and $3.0 million was drawn upon during the third quarter of 2023. The transfer of $4.0 million in principal from the 2012 Convertible Note to the Envoy Bridge Note was accounted for as a debt modification.
F-
ENVOY MEDICAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
The difference between the proceeds received and the issuance-date fair value was recorded as a deemed capital contribution from related party in the consolidated statements of stockholders’ deficit.
The Company could have prepaid the Envoy Bridge Note in whole or in part without premium or penalty. Contingent upon, and effective concurrently with the Merger, the outstanding balance of principal and any unpaid accrued interest, automatically converted to Series A Preferred Stock at a conversion price of $10.00 per share.
If the Business Combination Agreement terminated pursuant to its terms, at the sole discretion of the noteholder, the outstanding principal amount plus accrued and unpaid interest could have been converted into shares of Envoy Medical Corporation Common Stock at a conversion price of $1.00 per share, subject to various adjustments as defined in the agreement.
If the Business Combination Agreement terminated pursuant to its terms and in the event that the Company obtained additional equity financing pursuant to which the Company sold shares of either common or preferred stock, at the sole discretion of the noteholder, the principal amount plus accrued and unpaid interest would have converted to the class of stock being offered in the financing at a price per share equal to 80% of the price per share paid by investors for the offered shares.
On August 23, 2023, the Envoy Bridge Note was amended pursuant to which the Company could have drawn an additional $5.0 million if the Company had less than $5.0 million in cash or net tangible assets immediately following the Merger. In addition, the Company could have drawn up to $2.0 million if the Merger did not occur by September 30, 2023.
Effective concurrently with the Merger, the outstanding balance of principal and any unpaid accrued interest, was automatically converted to Series A Preferred Stock at a conversion price of $10.00 per share and the fair value of the Envoy Bridge Note was derecognized from the consolidated balance sheets.
2024 Term Loans
In February 2024, the Company issued a promissory note (the “February 2024 Term Loan”) with a minimum principal amount of $5.0 million and up to $10.0 million to GAT, an entity controlled by Glen A. Taylor, a member of the Company’s board of directors and a controlling stockholder of the Company. At closing, the Company drew down $5.0 million from the February 2024 Term Loan. Provided that no event of default had occurred and the Company submitted a request for funding certifying that the Company had less than $7.5 million of net tangible assets, the Company had the ability to draw the remaining $5.0 million in $2.5 million tranches, as long as each request was made prior to February 1, 2025. In both May 2024 and July 2024, the Company requested and received additional advances of $2.5 million under the February 2024 Term Loan. As of December 31, 2024, the balance outstanding on the February 2024 Term Loan was $9.5 million, net of discount.
The February 2024 Term Loan has a five-year term and matures on February 27, 2029. The principal amount drawn bears interest at a rate of 8.0% per annum and is paid quarterly in arrears after the second anniversary of the February 2024 Term Loan. Interest will accrue and is not payable for the first two years of the term and will compound and be added to the principal balance of the February 2024 Term Loan both on the first and second anniversary of the February 2024 Term Loan. The Company may prepay the accrued interest and principal of the February 2024 Term Loan without penalty, with 10 days’ notice.
F-
ENVOY MEDICAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
In August 2024, the Company issued an additional promissory note (the “August 2024 Term Loan” and, collectively with the February 2024 Term Loan, the “2024 Term Loans”) with a principal amount of up to $10.0 million to GAT. At closing, the Company drew down $5.0 million from the August 2024 Term Loan. Provided that no event of default had occurred and the Company submitted a request for funding certifying that the Company had less than $7.5 million of net tangible assets, the Company had the ability to draw the remaining $5.0 million in $2.5 million tranches, as long as each request is made prior to August 1, 2025. In December 2024, the Company requested and received an additional advance of $5.0 million under the August 2024 Term Loan. As of December 31, 2024, the balance outstanding on the August 2024 Term Loan was $9.2 million, net of discount.
The August 2024 Term Loan has a five-year term and matures on August 27, 2029. The principal amount drawn bears interest at a rate of 8.0% per annum and is paid quarterly in arrears after the second anniversary of the August 2024 Term Loan. Interest will accrue and is not payable for the first two years of the term and will compound and be added to the principal balance of the August 2024 Term Loan both on the first and second anniversary of the August 2024 Term Loan. The Company may prepay the accrued interest and principal of the August 2024 Term Loan without penalty, with 10 days’ notice.
As a commitment fee, the Company is required to issue warrants to purchase 250,000 shares of its Common Stock for each $2.5 million of principal funded under the 2024 Term Loans. The warrants will have an exercise price equal to the closing price on the date of funding of the applicable tranche.
At closing of the initial funding of the February 2024 Term Loan, the Company issued warrants to purchase 500,000 shares of Common Stock at an exercise price of $1.24 per share. These warrants expire on February 27, 2026. Upon the second draw made in May 2024, the Company issued warrants to purchase 250,000 shares of Common Stock at an exercise price of $3.04 per share. These warrants expire on February 28, 2026. Upon the third draw made in July 2024, the Company issued warrants to purchase 250,000 shares of Common Stock at an exercise price of $2.25 per share. These warrants expire on July 23, 2026.
At closing of the initial funding of the August 2024 Term Loan, the Company issued warrants to purchase 500,000 shares of Common Stock at an exercise price of $2.97 per share. These warrants expire on August 27, 2026. Upon the second draw made in December 2024, the Company issued warrants to purchase 500,000 shares of Common Stock at an exercise price of $2.20 per share. These warrants expire on December 11, 2026.
The 2024 Term Loans were accounted for as a conventional debt instrument and are accounted for in accordance with ASU 2020-06.
As a result of the issuance of the warrants with the initial closing of the February 2024 Term Loan, which met the criteria for equity classification under applicable U.S. GAAP, the Company recorded the fair value of the warrants on the issuance date in the amount of $0.2 million as a debt discount and additional paid-in capital on the consolidated balance sheets. As a result of the additional draws made in May 2024 and July 2024, the Company recorded the fair value of the additional warrants in the amount of $0.2 million and $0.2 million, respectively, as debt discounts and additional paid-in capital. Subsequently, these debt discounts are being recorded to interest expense, related party over the term of the February 2024 Term Loan.
As a result of the issuance of the warrants with the initial closing of the August 2024 Term Loan, which met the criteria for equity classification under applicable U.S. GAAP, the Company recorded the fair value of the warrants on the issuance date in the amount of $0.5 million as a debt discount and additional paid-in capital on the consolidated balance sheets. As a result of the additional draw made in December 2024, the Company recorded the fair value of the additional warrant in the amount of $0.3 million as a debt discount and additional paid-in capital. Subsequently, this debt discount is being recorded to interest expense, related party over the term of the August 2024 Term Loan.
F-
ENVOY MEDICAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
The fair value of the warrants issued in connection with the 2024 Term Loans is estimated using the Black-Scholes option model using the following key inputs during the year ended December 31, 2024:
| Year Ended December 31, 2024 |
||||
| Risk-free interest rate | 3.8% - 4.9% | |||
| Expected term (years) | 1.77 - 2.00 | |||
| Volatility | 42.0% - 49.0% | |||
| Stock price | $1.24 - $3.05 |
During the year ended December 31, 2024, respectively, the Company recognized $0.7 million of interest expense in relation to the 2024 Term Loans, at an effective interest rate of 9.68% and 10.67% for the February 2024 Term Loan and August 2024 Term Loan, respectively. Additionally, during the year ended December 31, 2024, the Company recognized $0.1 million of amortization of the debt discount. As of December 31, 2024, accrued interest of $0.7 million is recorded in accrued expenses and unamortized debt discount of $1.3 million is recorded within term loans payable (related party) on the Company’s consolidated balance sheets.
10. Common Stock
As of December 31, 2024 and 2023, the Company was authorized to issue 400,000,000 shares of Common Stock. The voting, dividend and liquidation rights of the holders of the Company’s Common Stock are subject to and qualified by the rights, powers and preferences of the holders of the Series A Preferred Stock (see Note 11).
Contingent Sponsor Shares
Pursuant to the Sponsor Support Agreement, and as of the date of issuance, 1,000,000 shares of Common Stock held by the Sponsor shall be unvested and subject to the restrictions and forfeiture provisions set forth in the Sponsor Support Agreement (the “Contingent Sponsor Shares”). The Contingent Sponsor Shares shall vest upon the FDA’s approval of the Acclaim CI (the “FDA Approval”). If a change of control of the Company shall occur following the Closing, then the conditions for vesting of any Contingent Sponsor Shares that remain unvested as of immediately prior to the consummation of the change of control shall be deemed to have been achieved and such Contingent Sponsor Shares shall immediately vest as of immediately prior to the consummation of such change of control.
As of December 20, 2024 the Company and the Sponsor entered into an agreement to remove the vesting restriction on the Contingent Sponsor Shares, more fully described in Note 11 under “Sponsor Induced Conversion”.
The Contingent Sponsor Shares met the definition of a derivative, but met the criteria to be considered indexed to the Company’s stock and the equity-classification criteria. Accordingly, the Contingent Sponsor Shares were classified as permanent equity.
F-
ENVOY MEDICAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Outstanding Warrants
The following table summarizes the Company’s outstanding warrant activity for the years ended December 31, 2024 and 2023 (in number of warrant shares):
| Shortfall Warrants | Publicly Traded Warrants | 2024 Term Loan Warrants | Common Stock Warrants (Related Party) | |||||||||||||
| December 31, 2022 | 8,695,000 | |||||||||||||||
| Issued | 3,874,394 | 14,166,666 | ||||||||||||||
| Exercised | (70,000 | ) | ||||||||||||||
| Forfeited | (8,625,000 | ) | ||||||||||||||
| December 31, 2023 | 3,874,394 | 14,166,666 | ||||||||||||||
| Issued | 2,000,000 | |||||||||||||||
| Exercised | (664,883 | ) | ||||||||||||||
| Forfeited | ||||||||||||||||
| December 31, 2024 | 3,209,511 | 14,166,666 | 2,000,000 | |||||||||||||
Common Stock Warrants (Related Party)
Between November 2013 and July 2022, the Company issued warrants to purchase shares of Envoy Medical Corporation Common Stock to stockholders in connection with the issuance of the Convertible Notes and the issuance of Envoy Medical Corporation Preferred Stock.
On April 17, 2023, the Common Stock Warrants (Related Party) were amended to provide for automatic cashless exercise or cancellation of the warrants immediately prior to the Merger. On September 29, 2023, the Common Stock Warrants (Related Party) were canceled or converted on a net exercise basis into shares of Common Stock. There were no outstanding Common Stock Warrants (Related Party) as of December 31, 2024 and 2023.
2024 Term Loan Warrants
During the year ended December 31, 2024, the Company issued warrants to purchase 2,000,000 shares of its Common Stock to a related party in conjunction with the 2024 Term Loans (see Note 9) (the “2024 Term Loan Warrants”). The 2024 Term Loan Warrants are all outstanding as of December 31, 2024.
Forward Purchase Agreement Warrant Liability
Pursuant to the terms of the Forward Purchase Agreement, the Company issued to Meteora warrants to purchase 3,874,394 shares of Common Stock (the “Shortfall Warrants”). As issued, the Shortfall Warrants had an exercise price determined based on the volume weighted average price of the Common Stock, subject to a $4.00 price floor (the “Exercise Price Floor”), which Exercise Price Floor is adjustable for certain issuances of Common Stock at a price below the then-current Exercise Price Floor. The Shortfall Warrants had an expiration date of June 30, 2024 upon issuance. The fair value of the Shortfall Warrants are presented in the forward purchase agreement warrant liability line on the consolidated balance sheets. The change in fair value of the Shortfall Warrants each period is recorded within the change in fair value of forward purchase agreement warrant liability line on the consolidated statements of operations and comprehensive loss.
On June 24, 2024, the Company and Meteora entered into Amendment No. 1 to the Shortfall Warrants to extend the expiration date of the Shortfall Warrants to December 31, 2024. On December 19, 2024, the Company and Meteora entered into Amendment No. 2 to the Shortfall Warrants to extend the expiration date of the Shortfall Warrants to December 31, 2025. The Company concluded that a modification of the liability classified Shortfall Warrants had no impact on its balance sheet classification and is reflected in the change in fair value of forward purchase agreement warrant liability due to modification in the consolidated statements of operations and comprehensive loss and recognized in net loss for the year ended December 31, 2024.
F-
ENVOY MEDICAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
On various dates during the year ended December 31, 2024, Meteora exercised Shortfall Warrants resulting in the issuance of 664,883 shares of Common Stock, generating gross proceeds of $1.8 million. As of December 31, 2024, Shortfall Warrants to purchase 3,209,511 shares of Common Stock remained outstanding.
Sale of Common Stock Through Forward Purchase Agreement
Pursuant to the terms of the Forward Purchase Agreement, on the Closing Date of the Business Combination, the Meteora parties purchased 425,606 shares of the Company’s Common Stock directly from the redeeming stockholders of Anzu. During the year ended December 31, 2024, the Meteora parties sold the 425,606 shares of Common Stock and, pursuant to the Forward Purchase Agreement, the Company received $4.00 per share sold, or $1.7 million, net of transaction costs.
11. Series A Preferred Stock
As of December 31, 2024 and 2023, the Company’s certificate of incorporation, as amended and restated, authorized the Company to issue 100,000,000 shares of $0.0001 par value preferred stock, of which 10,000,000 shares have been designated as Series A Preferred Stock.
Pursuant to the Envoy Bridge Note, the Sponsor Support Agreement and the Subscription Agreement, the Company has outstanding an aggregate of 4,126,667 and 4,500,000 shares of Series A Preferred Stock (see Note 3) as of December 31, 2024 and 2023, respectively, originally issued to the following investors:
| ● | 1,000,000 shares of Series A Preferred Stock to GAT pursuant to the Envoy Bridge Note; |
| ● | 2,500,000 shares of Series A Preferred Stock to the Sponsor pursuant to the Sponsor Support Agreement; and |
| ● | 1,000,000 shares of Series A Preferred Stock to certain affiliates of the Sponsor in the PIPE transaction pursuant to the Subscription Agreement. |
The holders of the Series A Preferred Stock have the following rights and preferences:
Voting Rights
The holders of the Series A Preferred Stock are not entitled to vote or receive notice of any meeting of stockholders, except in the case that the Company creates any equity or debt instrument that ranks senior or pari passu to the rights of the Series A Preferred Stock or in the case of any adverse change to the powers, preferences or special rights of the Series A Preferred Stock.
Conversion Rights
Each share of Series A Preferred Stock shall be convertible, at the option of the holder, at any time after the date of issuance into such number of shares of Common Stock as determined by dividing the issuance price of the shares of Series A Preferred Stock of $10.00, by the conversion price, which was $11.50 per share as of December 31, 2024 and is adjustable for certain dilutive events.
At any time from and after 90 days following the Merger, if the closing price per share of Common Stock was greater than $15.00 for any twenty (20) trading days within a period of thirty (30) trading days, the Company could have elected, in its discretion, to convert all, but not less than all, of the then outstanding shares of Series A Preferred Stock into shares of Common Stock. In this case, each share of Series A Preferred Stock then outstanding would have been converted into the number of shares of Common Stock equal to the quotient of i) $10.00 divided by ii) $15.00.
F-
ENVOY MEDICAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Redemption
The holders of Series A Preferred Stock are not entitled to any redemption rights, other than those under their liquidation rights discussed below. The Company does not have the option to redeem the Series A Preferred Stock.
Dividend Rights
The holders of Series A Preferred Stock are entitled to a cumulative dividend which accrues at the rate of 12% of the original issuance price of $10.00 per share per annum (“Regular Dividend”). The Regular Dividend accrues on a daily basis from and including the issuance date of such shares, whether or not declared, and will be payable in cash on a quarterly basis. If the Company fails to pay the Regular Dividends on the dividend payment date, then an additional dividend on the amount of the unpaid portion shall automatically accrue at 12%.
The holders of Series A Preferred Stock are also entitled to dividends or distributions (“Participating Dividends”) senior to Common Stock of the Company when such dividends are declared. There were no Participating Dividends declared as of December 31, 2024 or 2023.
Specifically pursuant to the 2,500,000 shares of Series A Preferred Stock issued subject to the Sponsor Support Agreement, any dividends arising will accrue and not require timely payment at any time when the Company has less than $10.0 million of net tangible assets. During both the year ended December 31, 2024 and 2023, the Company did not meet the $10.0 million net tangible asset requirement and deferred payment on the dividends related to the $2.5 million Series A Preferred Stock shares held by the Sponsor. As of December 31, 2024 and 2023, the Company had accrued $0.1 million and $0.8 million, respectively, in unpaid dividends as a result of the Sponsor Support Agreement.
With respect to the holders of the Series A Preferred Stock other than the Series A Preferred Stock subject to the Sponsor Support Agreement held by the Sponsor, the Company had accrued $0.5 million and $0.6 million as of December 31, 2024 and 2023, respectively, in unpaid Regular Dividends.
Liquidation Preference
In the event of any liquidation, deemed liquidation, dissolution or winding up of the Company, whether voluntary or involuntary, the holders of the Series A Preferred Stock are entitled to receive, prior and in preference to any distribution of any of the assets or surplus funds of the Company to the holders of any security of the Company that ranks junior to the Series A Preferred Stock, including, but not limited to, the Common Stock, an amount per share of Series A Preferred Stock equal to the greater of i) $10.00 plus any unpaid cash dividends and ii) the amount the holder would have received, if such holder, immediately prior to such involuntary liquidation, dissolution or winding up of Company, had converted such shares of Series A Preferred Stock into Common Stock.
Sponsor Induced Conversion
On December 20, 2024 (the “Effective Date”), the Company entered into a Conversion and Waiver Agreement (the “Conversion Agreement”) with the Sponsor.
As of the Effective Date of the Conversion Agreement, Sponsor was the holder of 2,500,000 shares of the Company’s Series A Preferred Stock and the Contingent Sponsor Shares. The Contingent Sponsor Shares were unvested and subject to certain restrictions and risk of forfeiture under the Sponsor Support Agreement until certain milestones were achieved. Pursuant to the terms of the Sponsor Support Agreement, the Company may, under certain circumstances, accrue Regular Dividends and defer cash payment to the Sponsor until such circumstances are resolved.
F-
ENVOY MEDICAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Pursuant to the terms of the Conversion Agreement, the Sponsor and the Company agreed that, upon the Effective Date of the Conversion Agreement: (i) the Sponsor waives the Company’s obligation to pay the $3.7 million of dividends accrued on the Series A Preferred Stock as of the Effective Date; (ii) the Company waives the restriction and vesting requirement for the Contingent Sponsor Shares, making these shares unrestricted and freely tradable; (iii) the Company agreed to make a voluntary, temporary reduction in the conversion price, pursuant to the terms of the Certificate of Designation, of all of the outstanding shares of Series A Preferred Stock effective December 20, 2024 through January 20, 2025 from $11.50 per share of Common Stock issuable upon conversion of a share of Series A Preferred Stock to $3.63 (the “Conversion Price Reduction”), with the conversion ratio determined by dividing the $10.00 original issue price of the Series A Preferred Stock by such Conversion Price; and (iv) the Sponsor agreed to convert 373,333 shares of Series A Preferred Stock into 1,028,986 shares of Common Stock at the temporary Conversion Price.
As the Company was legally released from its obligation to pay certain accrued dividends to the Sponsor, the Company derecognized the accrued dividends in the amount of $3.7 million as a result of the Conversion Agreement. The Company determined that the release of the accrued dividends represents paid-in-kind consideration in the form of a stock dividend, as the Sponsor agreed to receive Common Stock at a reduced conversion price under the Conversion Agreement in exchange for waiving the Company’s obligation to pay the accrued dividends.
Additionally, the Company determined that the conversion of the Series A Preferred Stock into Common Stock was an induced conversion as the reduced conversion price was only offered for a limited time and included the issuance of all equity securities issuable pursuant to the conversion privileges included in the terms of the Series A Preferred Stock for each share of Series A Preferred Stock that was converted to Common Stock. As such, an inducement charge of $1.2 million related to the fair value of the additional shares issued compared to the original terms of the Series A Preferred Stock was recognized within accumulated deficit as well as a deduction from the net loss to arrive at net loss attributable to common stockholders in the Company’s consolidated statements of operations and comprehensive loss.
As a result of the the waiver of the restriction and vesting requirement for the Contingent Sponsor Shares, the Company recognizes the incremental fair value of the outstanding Contingent Sponsor Shares as a dividend to the Sponsor in the amount of $0.5 million. The deemed dividend is treated as an increase to net loss attributable to common stockholders on the Company’s consolidated statements of operations and comprehensive loss. The deemed dividend resulted in offsetting amounts recorded in additional paid-in capital on the Company's consolidated balance sheets.
12. Stock Options
The Company had a stock incentive plan (the “2003 Stock Option Plan”) that provided for the granting of stock options or other stock incentives to employees, officers, directors and consultants. In March 2013, the Company and its stockholders adopted a new plan (the “2013 Stock Option Plan”) on substantially the same terms and conditions of the 2003 Stock Option Plan. The Company and its stockholders reserved a total of 7,000,000 shares of Envoy Medical Corporation Common Stock for issuance under the 2013 Stock Option Plan and reduced the number of shares of Envoy Medical Corporation Common Stock available for issuance under the 2003 Stock Option Plan from 6,400,000 to 552,000. As of April 2013, the 2003 Stock Option Plan expired and no further stock options or shares may be granted under that plan. On April 17, 2023, the Company and the stock option holders agreed that the stock options granted under the 2003 Stock Option Plan will be cancelled and terminated for no consideration upon completion of the Merger.
On April 17, 2023, the Company’s board of directors adopted a new equity incentive plan, and the plan was approved by the stockholders on September 27, 2023 (the “2023 Equity Incentive Plan”). An aggregate of 4,000,000 shares of Common Stock are reserved and may be issued under the 2023 Equity Incentive Plan, provided that until such time as certain milestones are achieved the aggregate number of shares of Common Stock that may be issued pursuant to the 2023 Equity Incentive Plan will be 2,500,000 shares. As of December 31, 2024 there were options to acquire 2,214,769 shares of Common Stock outstanding under the 2023 Equity Incentive Plan. The Company initially values options at fair value on the grant date. For awards with periodic vesting, the Company recognizes the related expense on a straight-line basis over the requisite service period for the entire award, which generally vest based on continued service over four years and expire ten years from the date of grant, subject to periodic adjustments to ensure that the cumulative amount of expense recognized through the end of any reporting period is at least equal to the portion of the grant date value of the award that has vested through that date. Certain stock options granted in 2023 and 2024 under the 2023 Equity Incentive Plan have a certain percentage that are exercisable at any time following the date of grant and then vest based on continuous service over three years and expire ten years from the date of grant.
F-
ENVOY MEDICAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
The Company uses the Black-Scholes option model to estimate the fair value of stock options. In applying the Black-Scholes option model, the Company used the following assumptions in the valuation of options granted in 2024 and 2023:
| 2024 | 2023 | |||||||
| Expected volatility | 66.0% - 75.3 | % | 72.4% - 73.8 | % | ||||
| Expected dividend yield | ||||||||
| Expected life (years) | 6.08 - 6.25 | 5.77 - 6.25 | ||||||
| Risk-free rate | 3.9% - 4.4 | % | 4.3% - 4.7 | % |
The following table summarizes the Company’s stock option activity for the year ended December 31, 2024 and 2023:
| Options | Weighted- average Exercise Price per Option |
Weighted- average Remaining Contractual Term (Years) |
Aggregate Intrinsic Value |
|||||||||||||
| Outstanding at December 31, 2022 | 263,000 | $ | 1.25 | 1.0 | $ | — | ||||||||||
| Granted | 2,085,034 | $ | 2.39 | 9.8 | $ | — | ||||||||||
| Terminated | (380,300 | ) | $ | 1.60 | N/A | $ | — | |||||||||
| Outstanding at December 31, 2023 | 1,967,734 | $ | 2.38 | 9.8 | $ | — | ||||||||||
| Exercisable and vested at December 31, 2023 | 907,262 | $ | 2.40 | 9.8 | $ | — | ||||||||||
| Granted | 286,810 | $ | 2.23 | 9.6 | $ | 7,356 | ||||||||||
| Terminated | (39,775 | ) | $ | 1.61 | N/A | $ | — | |||||||||
| Outstanding at December 31, 2024 | 2,214,769 | $ | 2.38 | 8.9 | $ | 4,956 | ||||||||||
| Exercisable and vested at December 31, 2024 | 1,226,641 | $ | 2.39 | 8.8 | $ | — | ||||||||||
The aggregate intrinsic value of stock options vested during the years ended December 31, 2024 and 2023 is zero because the fair value of the underlying Common Stock was less than the exercise price for all options as of each date.
The stock-based compensation expense related to option grants was $0.6 million and $1.6 for the years ended December 31, 2024 and 2023, respectively.
F-
ENVOY MEDICAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
The weighted average grant date fair value of option activity during the years ended December 31, 2024 and 2023 is as follows:
| Shares | Weighted Average Grant Date Fair Value |
|||||||
| Unvested balance at December 31, 2022 | $ | |||||||
| Granted | 2,085,034 | $ | 1.61 | |||||
| Vested | (907,262 | ) | $ | 1.60 | ||||
| Forfeited | (117,300 | ) | $ | 1.66 | ||||
| Unvested balance at December 31, 2023 | 1,060,472 | $ | 1.62 | |||||
| Granted | 286,810 | $ | 2.41 | |||||
| Vested | (319,379 | ) | $ | 1.64 | ||||
| Forfeited | (39,775 | ) | $ | 3.03 | ||||
| Unvested balance at December 31, 2024 | 988,128 | $ | 1.81 | |||||
As of December 31, 2024, stock-based compensation related to unvested option awards of $1.8 million remains unamortized, which is expected to be recognized over a weighted-average period of 3.10 years.
Total stock-based compensation expense associated with stock options was classified as follows on the consolidated statements of operations and comprehensive loss for the years ended December 31, 2024 and 2023:
| 2024 | 2023 | |||||||
| Research and development expense | $ | 165 | $ | 156 | ||||
| Sales and marketing expense | 11 | 63 | ||||||
| General and administrative expense | 386 | 1,356 | ||||||
| Total stock-based compensation expense | $ | 562 | $ | 1,575 | ||||
Employee Stock Purchase Plan
In September 2023, the Company established an employee stock purchase plan under which eligible employees may direct the Company to withhold up to 15% of their gross pay to purchase shares of common stock at a price equal to 85% of the lower of the offering date stock price or exercise date stock price. During the year ended December 31, 2024, there were 32,758 shares of Common Stock purchased, representing $63 thousand in contributions made by employees, under the employee stock purchase plan.
13. Income Taxes
The components of the benefit from income taxes consist of the following (in thousands):
| Year Ended December 31, | ||||||||
| 2024 | 2023 | |||||||
| Deferred: | ||||||||
| Deferred benefit | $ | 2,585 | $ | 5,776 | ||||
| Deferred tax asset valuation allowance | (2,585 | ) | (5,776 | ) | ||||
| Total benefit from income taxes | $ | $ | ||||||
F-
ENVOY MEDICAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
The Company’s effective tax rate differs from the federal statutory rate primarily due to the tax expense impact of nondeductible equity compensation and other permanent differences, tax credits, state taxes, and the valuation allowance.
A reconciliation of the Company’s effective tax rate to the statutory federal income tax rate is as follows:
| Year Ended December 31, | ||||||||
| 2024 | 2023 | |||||||
| Tax benefit at statutory rate | 21.0 | % | 21.0 | % | ||||
| State income taxes, net of federal benefit | 0.2 | % | 1.5 | % | ||||
| Permanent items | (0.6 | )% | (11.8 | )% | ||||
| Federal business credits | (0.8 | )% | (0.4 | )% | ||||
| Valuation allowance | (14.9 | )% | (10.3 | )% | ||||
| Other | (4.9 | )% | % | |||||
| Effective income tax rate | % | % | ||||||
Deferred income taxes reflect the net tax effects of temporary differences between the carrying amount of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes, as well as net operating loss carryforwards and research and development credit carryforwards.
The components of deferred tax assets and liabilities consisted of the following (in thousands):
| Year Ended December 31, | ||||||||
| 2024 | 2023 | |||||||
| Deferred tax assets: | ||||||||
| Net operating loss carryforwards | $ | 43,283 | $ | 41,156 | ||||
| Startup/organization costs | 3,130 | 3,865 | ||||||
| Tax credits | 1,828 | 1,960 | ||||||
| Capitalized research and development | 3,720 | |||||||
| Other | 1,251 | 3,685 | ||||||
| Total deferred tax assets | 53,212 | 50,666 | ||||||
| Valuation allowance | (52,779 | ) | (50,228 | ) | ||||
| Net total deferred tax assets | 433 | 438 | ||||||
| Deferred tax liabilities: | ||||||||
| Derivative instruments | (194 | ) | (418 | ) | ||||
| Other | (239 | ) | (20 | ) | ||||
| Total deferred tax liabilities | (433 | ) | (438 | ) | ||||
| Net deferred tax assets | $ | $ | ||||||
F-
ENVOY MEDICAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
The reconciliation of tax contingencies is as follows (in thousands):
| Year Ended December 31, | ||||||||
| 2024 | 2023 | |||||||
| Gross tax contingencies - January 1 | $ | 516 | $ | 545 | ||||
| Gross decreases for current year | (29 | ) | ||||||
| Gross increases for current year | 964 | |||||||
| Gross tax contingencies - December 31 | $ | 1,480 | $ | 516 | ||||
The change in valuation allowance was $3.3 million and $5.8 million for the years ended December 31, 2024 and 2023, respectively.
As of December 31, 2024, the Company had federal tax net operating loss carryforwards of $193.0 million which will be available to offset earnings during the carryforward period. Additionally, as of December 31, 2024, the Company had state net operating loss carryforwards of $51.0 million. If not used, these carryforwards, including federal tax carryforwards generated prior to December 31, 2017, expire in 2025 continuing through 2035. As a result of the Tax Cuts and Jobs Act, the federal tax net operating loss carryforwards generated in the years ended December 31, 2018 through 2022 do not expire. In addition, significant changes in ownership of the Company as defined in Section 382 of the Internal Revenue Code could put limitations on the availability of the net operating loss carryforwards. Currently, no analysis has been performed to determine the applicability of the limitations if any that may have occurred to date.
As of December 31, 2024, the Company had federal research and development credits carryforwards of $1.6 million. Additionally, the Company had gross state research and development credits carryforwards of $0.8 million as of December 31, 2024. Both the federal and state research and development credits carryforwards will be available to offset earnings during the carryforward period. If not used, these credits expire in 2025 continuing through 2035.
The impact of an uncertain tax position taken or expected to be taken on an income tax return must be recognized in the consolidated financial statements at the largest amount that is more-likely-than-not to be sustained upon audit by the relevant taxing authority. An uncertain income tax position will not be recognized in the consolidated financial statements unless it is more likely than not of being sustained. As of December 31, 2024, the Company recorded an uncertain tax benefit within other liability on the consolidated balance sheet in the amount of $0.1 million resulting from a refund remitted to the Company during the year ended December 31, 2024 related to payments and carryforwards reported by Anzu on its final tax return prior to the Business Combination on September 29, 2023.
Subsequently, during the year ended December 31, 2024, the Company received a notice from the Internal Revenue Service (“IRS”) indicating an additional refund of $0.8 million is to be remitted to the Company. This refund is also considered to be the result of estimated tax payments made by Anzu as reported on its short period tax return filed for the period January 1, 2023 to September 29, 2023, prior to the Business Combination. The Company is uncertain whether the IRS treated the transaction as an IRC 368(a)(1)(F) reorganization (“F reorganization”) or as an IRC 368(a)(1)(A) reorganization (“A reorganization”). As of December 31, 2024, the Company recorded a receivable on its consolidated balance sheets in the amount of $0.8 for this refund. The Company has determined that it is appropriate to retain the funds, once received, under the premise that the transaction was treated as an F reorganization rather than an A reorganization, however, given the uncertainty of that outcome, the Company will establish an uncertain tax position liability as of December 31, 2024. The Company received the refund during the first quarter of 2025.
The Company has reduced its deferred tax asset for research and development credit by $0.5 million and $0.5 million for uncertain tax positions as of December 31, 2024 and 2023, respectively.
The Company currently files income tax returns in Arizona, Maryland, Michigan, Minnesota, Texas, and Vermont. Due to the previous losses incurred, the Company is subject to income tax examination by tax authorities since inception. The Company has not been, nor is it currently, under examination by any tax authorities.
F-
ENVOY MEDICAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
14. Segment Reporting
Operating segments are identified as components of an enterprise about which discrete financial information is available for evaluation by the CODM in deciding resource allocation and assessing performance. The Company has determined that its CODM is its Chief Executive Officer.
The Company has one reportable segment: hearing. The hearing segment derives revenue from the sale of the Esteem FI-AMEI implants and replacement components to Esteem FI-AMEI implants. The Company enters into arrangements with patients to provide them with the Esteem FI-AMEI device, personal programmer devices, sound processor replacements, and Battery replacements.
The Company derives revenue primarily in the United States and manages the business activities on a consolidated basis.
The accounting policies of the hearing segment are the same as those described in the summary of significant accounting policies. The CODM assesses performance for the hearing segment and decides how to allocate resources based on net loss that also is reported on the consolidated statements of operations and comprehensive loss. The measure of segment assets is reported on the consolidated balance sheets as total assets.
The CODM uses net loss to evaluate the results generated from segment assets (return on assets) in deciding whether to make investments into the hearing segment or into other parts of the entity, such as for entering into significant contracts, hiring of key management or executive personnel, making significant capital investment decisions, or changing Company-wide strategy.
Net loss is used to monitor budget versus actual results and assist the CODM in understanding the Company’s cash flows and liquidity position, which is critical as a development state entity. This allows the CODM to make the appropriate spending decisions for the Company.
F-
ENVOY MEDICAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
The following table summarizes the significant expense categories regularly reviewed by the CODM for the years ended December 31, 2024 and 2023.
| Year ended December 31, | ||||||||
| 2024 | 2023 | |||||||
| Net revenues | $ | 225 | $ | 316 | ||||
| Costs and operating expenses: | ||||||||
| Cost of goods sold | 742 | 789 | ||||||
| Research and development | 10,179 | 8,956 | ||||||
| Sales and marketing | 1,734 | 1,666 | ||||||
| General and administrative | 6,826 | 7,264 | ||||||
| Total costs and operating expenses | 19,481 | 18,675 | ||||||
| Operating loss | (19,256 | ) | (18,359 | ) | ||||
| Other segment items(1) | (697 | ) | (11,617 | ) | ||||
| Interest expense, related party | (816 | ) | ||||||
| Other income | (26 | ) | 54 | |||||
| Total other expense, net | (1,539 | ) | (11,563 | ) | ||||
| Net loss | $ | (20,795 | ) | $ | (29,922 | ) | ||
| (1) | Other segment items include the change in fair value of convertible notes payable (related party), change in fair value of forward purchase agreement put option liability, change in fair value of forward purchase agreement warrant liability, change in fair value of forward purchase agreement warrant liability due to modification, and change in fair value of publicly traded warrant liability. |
15. Related Party Transactions
The Company had various transactions with a member of the Company’s board of directors and a controlling stockholder of the Company, which is considered a related party.
| ● | The Company leases its headquarters office space in Minnesota from the stockholder. The lease is considered a common control leasing arrangement. The lease liability due to the stockholder was approximately $1.2 million and $0.6 million as of December 31, 2024 and 2023, respectively. See Note 7 for additional information related to this lease including the operating lease cost for the years ended December 31, 2024 and 2023. |
| ● | The Company received several loan financings from the stockholder or his affiliates between 2012 and 2024 (see Note 9), including the Convertible Notes, the Envoy Bridge Note, the 2024 Term Loans, and the March 2025 Term Loan (see Note 18). |
| ● | The Company has a shared services arrangement with a company that is indirectly owned by the stockholder, for certain support services used in the course of business. This arrangement originated on January 1, 2022 with a term of two years that automatically renews each year thereafter unless terminated by either party. In relation to the shared services arrangement, the Company expensed $0.1 million during both the years ended December 31, 2024 and 2023. |
F-
ENVOY MEDICAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
16. Commitment and Contingencies
The Company is party to various litigation matters arising from time to time in the ordinary course of business.
In January 2020, the Company’s controlling stockholder and convertible debt holder, along with current and former directors of the Company were named in a lawsuit brought by minority stockholders (the “Spearman Plaintiffs”). This lawsuit alleges our controlling stockholder of “self-dealing” in order to obtain control of the Company. In February 2020, there was a similar lawsuit referring to and citing the first lawsuit brought up by additional minority stockholders alleging our controlling stockholder and directors of similar wrong-doings. The February 2020 lawsuit was withdrawn in 2021. In June 2023, the Company received an additional complaint from additional stockholders affiliated or associated with the Spearman Plaintiffs, raising claims that were substantially the same as the claims raised in the existing litigation. On August 25, 2023, the Company entered into a binding agreement in principle to settle all claims and counterclaims in the lawsuit. On September 15, 2023, the parties entered into a binding settlement agreement. The settlement agreement included a transfer of all of the plaintiff’s stock holdings in Envoy Medical to an entity affiliated with the majority stockholder of the Company, which was completed on September 28, 2023. The settlement agreement did not require any payment to be made by the Company.
On November 14, 2023, the Company, Whitney Haring-Smith (the former chief executive officer and a former director of the Company), Daniel Hirsch (the former chief financial officer of the Company), and Anzu SPAC GP I LLC were named as defendants in a complaint filed by Atlas Merchant Capital SPAC Fund I LP (“Atlas”) in the Delaware Court of Chancery. Atlas alleges that it was not allowed to redeem its shares of the Company’s Common Stock and that Defendants acted to prevent Atlas’s attempt to redeem its shares. Defendants assert that Atlas did not comply with the requirements for redeeming shares set forth in the Company’s organizational documents. Atlas asserts damages in the amount of approximately $9.4 million, pre- and post-judgment interest, costs, and reasonable attorneys’ fees. The Company has standard indemnification obligations to Dr. Haring-Smith and Mr. Hirsch. The Company believes that the lawsuit is meritless and has been defending this matter vigorously. The Company is unable to predict the outcome of this legal proceeding.
The Company has business liability insurance to cover litigation costs exceeding $50 thousand. As of December 31, 2024 and 2023, the Company has not recorded accruals for potential losses related to any existing or pending litigation claims as the Company’s management determined that there are no matters where a potential loss is probable and reasonably estimable.
F-
ENVOY MEDICAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
17. Net Loss per Share
The following table sets forth the computation of basic and diluted loss per share (in thousands, except share and per share amounts):
| Year ended December 31, | ||||||||
| 2024 | 2023 | |||||||
| Numerator: | ||||||||
| Net loss | $ | (20,795 | ) | $ | (29,922 | ) | ||
| Less: Induced conversion of Series A Preferred Stock into Common Stock | (1,162 | ) | ||||||
| Less: Deemed dividend on waiver of restriction on Class A Common Stock | (495 | ) | ||||||
| Less: Cumulative preferred dividends | (5,521 | ) | (1,349 | ) | ||||
| Net loss attributable to common stockholders, basic and diluted | $ | (27,973 | ) | $ | (31,271 | ) | ||
| Denominator: | ||||||||
| Weighted-average Common Stock outstanding, basic and diluted | 18,790,448 | 12,295,391 | ||||||
| Net loss per share attributable to common stockholders, basic and diluted | $ | (1.49 | ) | $ | (2.54 | ) | ||
The Company’s potentially dilutive securities below, presented based on amounts outstanding at each period end, have been excluded from the computation of diluted net loss per share as the effect would be to reduce the net loss per share. Therefore, the weighted-average number of shares of Common Stock outstanding used to calculate both basic and diluted net loss per share attributable to stockholders of Common Stock for these periods is the same.
| Year ended December 31, | ||||||||
| 2024 | 2023 | |||||||
| Stock options | 2,214,769 | 1,967,734 | ||||||
| Series A Preferred Stock (as converted to common stock) | 3,588,406 | 3,913,043 | ||||||
| Publicly traded warrants | 14,166,666 | 14,166,666 | ||||||
| Shortfall Warrants | 3,209,511 | 3,874,394 | ||||||
| Contingent Sponsor Shares | — | 1,000,000 | ||||||
| 2024 Term Loan Warrants | 2,000,000 | |||||||
| 25,179,352 | 24,921,837 | |||||||
F-
ENVOY MEDICAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
18. Subsequent Events
The Company has evaluated all events occurring through the date on which these consolidated financial statements were issued, and during which time, nothing has occurred outside the normal course of business operations that would require disclosure, except for the following:
Loan from Related Party
Subsequent to the year ended December 31, 2024, the Company issued a promissory note (the “March 2025 Term Loan”) with a minimum principal amount of $5.0 million and up to $10.0 million to GAT. Upon meeting certain conditions, the Company may draw funds in $2.5 million tranches under the March 2025 Term Loan up to $10.0 million until the second anniversary of the March 2025 Term Loan. The March 2025 Term Loan has a five-year term and matures on March 6, 2030. The principal amount drawn bears interest at a rate of 8.0% per annum and is paid quarterly in arrears after the second anniversary of the March 2025 Term Loan. Interest will accrue and not be paid for the first two years of the term and will compound and be added to the principal balance of the March 2025 Term Loan on the first and second anniversary of the March 2025 Term Loan. The Company may prepay the accrued interest and principal of the March 2025 Term Loan without penalty, with 10 days’ notice. At closing, the Company requested and GAT agreed to fund an initial $5.0 million in principal under the March 2025 Term Loan. Additional draws will be made in $2.5 million tranches.
As a commitment fee, the Company will issue GAT warrants to purchase 375,000 shares of its Common Stock for each $2.5 million of principal funded under the March 2025 Term Loan. The warrants will have an exercise price equal to the closing price on the date of funding of the applicable tranche and a termination date as of the third anniversary of the initial closing for all warrants. At closing of the initial funding, the Company issued GAT warrants to purchase 750,000 shares of Common Stock at an exercise price of $1.35 per share.
F-
ITEM 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure
None.
ITEM 9A. Controls and Procedures
Evaluation of Disclosure Controls and Procedures
Disclosure controls and procedures are designed to ensure that information required to be disclosed by us in our Exchange Act reports is recorded, processed, summarized, and reported within the time periods specified in the SEC’s rules and forms, and that such information is accumulated and communicated to our management, including our principal executive officer and principal financial officer or persons performing similar functions, as appropriate to allow timely decisions regarding required disclosure.
Under the supervision and with the participation of our management, including our Chief Executive Officer and Chief Financial Officer, we evaluated the effectiveness of our disclosure controls and procedures as of December 31, 2024, as such term is defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act. Based on the evaluation of our disclosure controls and procedures as of December 31, 2024, our Chief Executive Officer and Chief Financial Officer concluded that, as of such date, our disclosure controls and procedures were not effective due to the existence of the material weaknesses in the Company’s internal control over financial reporting described below.
Notwithstanding the conclusion by our Chief Executive Officer and Chief Financial Officer that our disclosure controls and procedures as of December 31, 2024 were not effective, and notwithstanding the material weaknesses in our internal control over financial reporting described below, management believes that the consolidated financial statements and related financial information included in this Annual Report on Form 10-K fairly present in all material respects our financial condition, results of operations and cash flows as of the dates presented, and for the periods ended on such dates, in conformity with GAAP.
Management’s Report on Internal Controls Over Financial Reporting
As required by SEC rules and regulations implementing Section 404 of the Sarbanes-Oxley Act, our management is responsible for establishing and maintaining adequate internal control over financial reporting. Our internal control over financial reporting is designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of our financial statements for external reporting purposes in accordance with GAAP. Our internal control over financial reporting includes those policies and procedures that:
| (1) | pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the Company, |
| (2) | provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with GAAP, and that our receipts and expenditures are being made only in accordance with authorizations of our management and directors, and |
| (3) | provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of our assets that could have a material effect on the financial statements. |
Because of its inherent limitations, internal control over financial reporting may not prevent or detect errors or misstatements in our financial statements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree or compliance with the policies or procedures may deteriorate. Management assessed the effectiveness of our internal control over financial reporting as of December 31, 2024. In making these assessments, management used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) in Internal Control - Integrated Framework (2013). Based on our assessments and those criteria, management determined that our internal control over financial reporting was ineffective as of December 31, 2024 and that there were control deficiencies that constituted material weaknesses as described below. A material weakness is a deficiency, or combination of deficiencies, in internal control over financial reporting such that there is a reasonable possibility that a material misstatement of the Company’s annual or interim financial statements will not be prevented or detected on a timely basis.
This Report does not include an attestation report of our independent registered public accounting firm due to our status as an emerging growth company under the JOBS Act.
Material Weaknesses in Internal Control Over Financial Reporting
Management concluded the following material weaknesses existed as of December 31, 2024:
| ● | The Company does not maintain a sufficient complement of personnel with accounting knowledge, experience and training to appropriately analyze, record and disclose certain accounting matters to provide reasonable assurance of preventing material misstatements. |
| ● | The Company’s management does not implement a formal risk assessment that addresses risks relevant to financial reporting objectives, including cybersecurity and fraud risks. |
| ● | The Company has not designed, documented and maintained formal accounting policies, procedures and controls over significant accounts and disclosures to achieve complete, accurate and timely financial accounting, reporting and disclosures, including segregation of duties and adequate controls related to the preparation, posting, modification and review of journal entries, and the accounting treatment of complex transactions, including fair value measurement under GAAP. |
| ● | The Company has not designed and maintained effective controls over certain information technology general controls for information systems that are relevant to the preparation of its consolidated financial statements, including ineffective controls around user access and segregation of duties. |
Considering this, the Company performed additional procedures and analyses as deemed necessary to ensure that its financial statements were prepared in accordance with GAAP.
The Company has begun implementation of a plan to remediate these material weaknesses. These remediation measures are ongoing and include the following steps:
| ● | hiring additional accounting and financial reporting personnel with appropriate technical accounting knowledge and public company experience in financial reporting; |
| ● | designing and implementing effective processes and controls over significant accounts and disclosure; |
| ● | designing and implementing security management and change management controls over information technology systems, including adjusting user access levels and implementing external logging on activity and periodic review of such logs; and |
| ● | reviewing candidate accounting advisory firms to assist with the documentation, evaluation, remediation and testing of the Company’s internal control over financial reporting based on the criteria established in “Internal Control - Integrated Framework” issued by the Committee of Sponsoring Organizations of the Treadway Commission. |
Changes in Internal Control Over Financial Reporting
There was no change in the Company’s internal control over financial reporting that occurred during the fiscal quarter ended December 31, 2024 that has materially affected, or is reasonably likely to materially affect, its internal control over financial reporting.
ITEM 9B. Other Information
None of our directors or executive officers adopted or terminated a Rule 10b5-1 trading arrangement or adopted or terminated a non-Rule 10b5-1 trading arrangement (as defined in Item 408(c) of Regulation S-K) during the three months ended December 31, 2024.
ITEM 9C. Disclosure Regarding Foreign Jurisdictions that Prevent Inspections The following table sets forth information concerning our current directors and executive officers, including their ages as of March 24, 2025.
Not applicable.
PART III
ITEM 10. Directors, Executive Officers and Corporate Governance
Management
There are no family relationships among any of our directors or executive officers.
| Name | Age | Title | ||
| Brent T. Lucas | 43 | Chief Executive Officer and Director | ||
| David R. Wells | 62 | Chief Financial Officer | ||
| Charles R. Brynelsen | 68 | Chairman | ||
| Glen A. Taylor | 83 | Director and Chairman Emeritus | ||
| Michael Crowe | 61 | Director | ||
| Mona Patel | 57 | Director | ||
| Janis Smith-Gomez | 57 | Director | ||
| Susan J. Kantor | 69 | Director |
Executive Officers
Brent T. Lucas. Brent T. Lucas has served as our Chief Executive Officer and a member of the Board since the closing of the Business Combination in September 2023. At the time of the Business Combination Mr. Lucas was the Chief Executive Officer (since 2015) and Board member of Envoy Medical Corporation (since 2016). Mr. Lucas brings over 15 years of experience in active implantables in the hearing health industry. He has served in various roles with Envoy Medical Corporation and gained a tremendous amount of specialized experience. He has served in various roles with Envoy Medical Corporation and gained a tremendous amount of specialized experience, working his way up from an intern to CEO. Mr. Lucas received his bachelor’s degree from the University of St. Thomas and Juris Doctor degree from the Mitchell Hamline School of Law.
The Board believes Mr. Lucas is qualified to serve as a member of the Board given his specialized experience and knowledge in the hearing and medical device industries as Legacy Envoy’s Chief Executive Officer.
David R. Wells. David R. Wells has served as our Chief Financial Officer since the closing of the Business Combination. He possesses over 30 years of experience in finance, operations and administrative positions. While mainly focused on medical and technology companies, Mr. Wells has also worked in the water treatment, supply-chain management, manufacturing and professional services industries. In December 2022 Mr. Wells joined the Board of Directors of Heart Test Laboratories, Inc. (dba ‘HeartSciences’, NASDAQ: HSCS), which is developing a cardiac device which seeks to bridge today’s “diagnostic gap” in cardiac care by providing effective front-line solutions that assist in the detection of heart disease in at-risk patients. From June 2021 to September 2022, Mr. Wells served as Chief Financial Officer of GHS Investments, LLC, a privately held “super value” fund focused on investing in small-to-mid cap companies. From June 2014 to June 2021 he served as the Chief Financial Officer of ENDRA Life Sciences (Nasdaq: NDRA), a publicly traded clinical diagnostics technology company. Mr. Wells directed ENDRA’s initial public offering and subsequently helped raise an additional $55 million across multiple transactions. Also, in June 2021 David founded Atlas Bookkeeping, LLC, a technology-based financial services firm that provides bookkeeping and reporting services for emerging growth and small cap public and privately held companies. Mr. Wells holds an MBA from Pepperdine University and a BS in Finance and Entrepreneurship from Seattle Pacific University.
Non-Executive Directors
Charles R. Brynelsen. Charles R. Brynelsen has served as the Chairman of our Board since the closing of the Business Combination. Mr. Brynelsen has extensive experience in the medical device industry, including most recently serving as Senior Vice President and President of Abbott Vascular from 2017 to 2021. Since 2015 he has also been a Venture Partner of SpringRock Ventures, an investment firm that focuses on digital health, devices, services, oral health, SAAS, consumerization/ecommerce of healthcare, IT, wellness, HIPAA and other innovative companies improving general health. Mr. Brynelsen has also served on private companies’ boards of directors, including Alebra Technologies since 2010 and Neuspera Medical from 2022 to 2023. Mr. Brynelsen previously served as Senior Vice President and President of Medtronic Early Technologies from 2015 to 2016, as the Global President of Covidien Early Technologies from 2013 to 2015, and as the Chief Executive Officer of IntraPace from 2005 to 2012. Earlier in his career, Mr. Brynelsen held various commercial, corporate, international, and general management leadership roles across Medtronic from 1981 to 2005.
The Board believes Mr. Brynelsen is qualified to serve as a member of the Board due to his extensive experience in the management of a multinational public company in the medical device industry, including significant product development, clinical/regulatory, manufacturing, business development and strategic planning experience. He also provides valuable insights into operating in highly regulated global healthcare markets.
Glen A. Taylor. Glen A. Taylor has served as a member of our Board and Chairman Emeritus since the closing of the Business Combination. Mr. Taylor is the founder and chairman of Taylor Corporation, a global printing and communications company and one of the nation’s largest privately held companies. Among other investments, Mr. Taylor is owner of the Minnesota Star-Tribune, limited partner of Minnesota United FC and owner and chairman of Taylor Sports Group, Inc., the general partner of Minnesota Timberwolves Basketball Limited Partnership, which, in turn, owns the Timberwolves, Lynx, Iowa Wolves and T-Wolves Gaming. In addition, Mr. Taylor is a member (and former chair) of the Board of Governors of the National Basketball Association. Mr. Taylor served in the Minnesota State Senate from 1980 to 1990 and as Minority Leader from 1985 to 1988. Further, Mr. Taylor served as president of the YMCA, director of the Mankato Chamber of Commerce, director of the Greater Minnesota Corporation and the Minnesota Business Partnership, and served on the Minnesota State University Foundation Board of Directors. Additionally, Mr. Taylor attended Harvard Graduate School of Business and earned his Bachelor of Science at Minnesota State University in Mankato. He has an honorary doctorate from Minnesota State University, received the Distinguished Alumni Award from Mankato State University, and is a Laureate of the Minnesota Business Hall of Fame.
The Board believes Mr. Taylor is qualified to serve on the Board due to his extensive background and expertise in developing, operating and leading successful businesses across a variety of industries.
Michael Crowe. Michael Crowe has decades of experience in the medical device industry with a focus on operations. Since March 2023, Mr. Crowe has served as Senior Vice President Operations for Bioventus LLC. Previously, he served as Vice President Operations for Abbott Vascular from January 2015 to March 2023. Mr. Crowe earlier served in similar roles for Caris Life Sciences, Covidien Devices, Johnson & Johnson, Iomega Corporation, and SKF USA, Inc. Mr. Crowe earned a bachelor’s degree in engineering from the University of Louisville and an MBA from Duke University.
The Board believes Mr. Crowe is qualified to serve a member of the Board due to his wide-ranging experience in the management and operations of multinational public companies in the medical device industry, including significant experience overseeing distribution, supply chain and sourcing, facilities, engineering, customer service, reimbursement services, and product launch, which the Board believes will be vital to the Company’s next stages of development.
Mona Patel. Mona Patel has served as a member of our Board since the closing of the Business Combination. Ms. Patel has over 30 years of experience with medical devices in marketing, market development, clinical education and mergers and acquisitions. Currently, Ms. Patel works as a strategic advisor for med-tech start-ups, through which she helps companies raise funding, understand market opportunities, and develop go-to-market plans. Previously, she was the Vice President of Marketing and Clinical Education at Boston Scientific in their neuromodulation division, where she helped build the start-up into a market leader with approximately $1 billion in sales. While at Boston Scientific, she introduced the first rechargeable spinal cord stimulator into the market, helped convert the market from non-rechargeable to rechargeable stimulators, and launched the first rechargeable deep brain stimulator for Parkinson’s disease. Prior to joining Boston Scientific, Ms. Patel worked in various positions at Guidant in marketing and business development, through which she acquired and licensed a portfolio of technologies that became the Guidant Cardiac and Vascular surgery division, including the acquisition of two med-tech start-ups. As a marketing leader, she drove the adoption of a new procedure, endoscopic vessel harvesting, to become the gold standard for cardiac surgery. She began her career as an engineer for Abbott Labs. Ms. Patel earned a BSE in Mechanical Engineering from the University of Michigan and an M.B.A. from the University of Pennsylvania, Wharton School of Business.
The Board believes Ms. Patel is qualified to serve on the Board due to her extensive background in the medical devices field and expertise in marketing and business development.
Janis Smith-Gomez. Janis Smith-Gomez has served as a member of our Board since the closing of the Business Combination. Ms. Smith-Gomez has more than 30 years of experience in marketing and innovation, positioning global brands for growth and competitive advantage, contributing to her strong business acumen and stakeholder insights focus. From 2006 to 2022, Ms. Smith-Gomez held a variety of leadership positions at Johnson & Johnson across medical devices and consumer health, where she focused on building brands, launch excellence and innovative marketing strategies for revenue and market share growth. In her most recent role at Johnson & Johnson as the Vice President of Global Brand Experience, she led the brand identity efforts to evolve the $27 billion medical devices business into a leading patient-centered, customer-focused, digitally powered med-tech innovator. As the Vice President of U.S. Marketing for Ethicon LLC, a subsidiary of Johnson & Johnson, from 2014−2018, Ms. Smith-Gomez returned the business to growth and strengthened customer engagement. Prior to working at Johnson & Johnson, Ms. Smith-Gomez held the roles of Vice President of Marketing at Mars, Incorporated, Senior Director at Kraft Foods, and Director of Marketing at PepsiCo, Inc. Ms. Smith-Gomez started her career in consulting with Booz, Allen & Hamilton and completed a summer internship with Procter & Gamble. Ms. Smith-Gomez is currently a member of the board of trustees of several non-profit organizations, including the New York Academy of Medicine, Black Public Media, and the Vanderbilt University Parents and Family Association. She also previously served as a trustee for Kent Place School and Citymeals on Wheels. Ms. Smith-Gomez received her bachelor’s degree in Professional Option: Business and her M.B.A. from the University of Chicago.
The Board believes Ms. Smith-Gomez is qualified to serve on the board due to her extensive experience in the medical devices industry, her strategic planning expertise and her successful career as a senior executive, commercial leader and marketing strategist driving brand relevance and sustainable financial and operational performance.
Susan J. Kantor. Susan J. Kantor has served as a member of our Board since March 2021. Ms. Kantor has experience leading international finance, tax, treasury, risk, compliance and technology enablement for global services organizations. She was an Advisory Partner for PwC from 2011 to 2016, a Partner and CFO & Treasurer of PRTM Management Consultants from 1997 to 2011, and was previously a CFO/senior financial executive at corporate strategy and operations consulting firms Monitor Group and BCG, as well as Parexel International, a clinical research organization. She began her career in the audit practices at EY and PWC, where she provided audit services for over 12 years to privately held and publicly held companies across the industrial, life sciences and retail and consumer sectors. During her time at PRTM, she completed several successful M&A transactions in the U.S. and abroad, including the sale PRTM’s global business to PwC in 2011. Ms. Kantor is currently on the board of Teknor Apex Company, a privately-held material science company and on the board and as Audit Committee Chair of Guest Services Inc., a privately held hospitality company. She received her bachelor’s degree from Grove City College in Accounting and Business Administration and her CPA in Massachusetts.
The Board believes Ms. Kantor is qualified to serve on the Board due to her extensive background and expertise in global financial and tax matters. Ms. Kantor qualifies as a “financial expert” and has served as the Chair of the Audit Committee since March 2021.
Other Key Executives
Tom Hoegh. Tom Hoegh has served as our Director of Engineering since the closing of the Business Combination. Mr. Hoegh has over 25 years of experience in the medical device industry, primarily in the development and on-market support of active implantable devices such as neuromodulation systems for spinal, sacral, deep brain, and hypoglossal nerve stimulation. Mr. Hoegh’s previous experiences consist of leading engineering teams at Nuvectra, ICU/Smiths Medical, Medtronic, and Apnex Medical. Mr. Hoegh received a dual Bachelor of Science degree in Mechanical Engineering and Chemistry from Valparaiso University and a Master of Science degree in Technology Management from the University of St. Thomas.
Karin Simonson. Karin Simonson has served as our Vice President, General Counsel & Corporate Secretary since December, 2023. From April, 2023 to December, 2023, Ms. Simonson was General Counsel for Monarch Healthcare Management. She has over 20 years of diverse in-house counsel experience supporting clinical, regulatory, sales, marketing, compliance, data privacy, research and development, HR, IT, contracts and commercial operations with increasing responsibilities at both small and large companies including, Coloplast, Medtronic, American Medical Systems and Carlson Hotels Worldwide. She began her legal career over 25 years ago in commercial litigation, but over the last 15+ years has focused on the medical device industry. Ms. Simonson is currently and has been a board member of the Association of Corporate Counsel (ACC)- Minnesota Chapter for the last 9 years and has been an ACC member since 2007. Ms. Simonson has a BS, magna cum laude, from the University of Minnesota-Twin Cities and a JD, magna cum laude, from Mitchell Hamline School of Law. She is also a volunteer attorney with Children’s Law Center of Minnesota.
Involvement in Certain Legal Proceedings
To our knowledge, there have been no events under any bankruptcy act, no criminal proceedings and no federal or state judicial or administrative orders, judgments or decrees or findings, no violations of any federal or state securities law, and no violations of any federal commodities law material to the evaluation of the ability and integrity of any director (existing or proposed) or executive officer (existing or proposed) of the Company during the past 10 years.
Corporate Governance
Code of Ethics
The Board has adopted a Code of Ethics, which is applicable to our employees, directors and officers, including our principal executive officer, principal financial officer, principal accounting officer or controller and other persons performing similar functions. The Code of Ethics covers topics including conflicts of interest, confidentiality of information, full and fair disclosure, reporting of violations and compliance with laws and regulations. Our Code of Ethics is available, free of charge, on the investor relations page of our website, https://www.envoymedical.com/investors. This website address is intended to be an inactive, textual reference only; none of the material on this website is part of this Report.
If the Company amends or grants a waiver of one or more of the provisions of the Code of Ethics, it intends to satisfy the requirements under Item 5.05 of Item 8-K regarding the disclosure of amendments to or waivers from provisions of the Code of Ethics that apply to the Company’s principal executive officer, principal financial officer and principal accounting officer by posting the required information on the investor relations page of the Company’s website at https://www.envoymedical.com/investors.
ITEM 11. Executive Compensation
The information required by this Item 11 will be contained in the Definitive Proxy Statement and is incorporated herein by reference.
ITEM 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters
The information required by this Item 12 will be contained in the Definitive Proxy Statement and is incorporated herein by reference.
ITEM 13. Certain Relationships and Related Transactions, and Director Independence
The information required by this Item 13 will be contained in the Definitive Proxy Statement and is incorporated herein by reference.
ITEM 14. Principal Accounting Fees and Service
The information required by this Item 14 will be contained in the Definitive Proxy Statement and is incorporated herein by reference.
PART IV
ITEM 15. Exhibits and Financial Statement Schedules
The following is a list of documents filed as a part of this Report:
(1) Financial Statements
The consolidated financial statements of the Company, together with the independent registered public accounting firm’s report thereon, are included herein and are incorporated by reference. See Item 8. Financial Statements and Supplementary Data, filed herewith, for a list of financial statements.
(2) Financial Statement Schedules
All schedules for which provision is made in Regulation S-X are either not required to be included herein under the related instructions, are inapplicable or the related information is included in the footnotes to the applicable financial statement and, therefore, have been omitted.
(3) Exhibits
The exhibits required to be filed by Item 601 of Regulation S-K are listed in the accompanying Exhibit Index, which is incorporated by reference.
ITEM 16. Form 10-K Summary
None.
EXHIBIT INDEX
| (*) | Indicates a management contract or compensatory plan. |
| (#) | Filed herewith. |
| (+) | Certain schedules and exhibits to this Exhibit have been omitted pursuant to Item 601(a)(5) or Item 601(b)(10)(iv), as applicable, of Regulation S-K. The registrant agrees to furnish supplemental copies of all omitted exhibits and schedules to the Securities and Exchange Commission upon its request. |
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this Report to be signed on its behalf by the undersigned, thereunto duly authorized.
| March 28, 2025 | ENVOY MEDICAL, INC. | |
| /s/ Brent T. Lucas | ||
| Name: | Brent T. Lucas | |
| Title: | Chief Executive Officer | |
POWER OF ATTORNEY
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints Brent T. Lucas and David R. Wells and each or any one of them, his true and lawful attorney-in-fact and agent, with full power of substitution and resubstitution, for him and in his name, place and stead, in any and all capacities, to sign any and all amendments to this Report, and to file the same, with all exhibits thereto, and other documents in connection therewith, with the United States Securities and Exchange Commission, granting unto said attorneys-in-fact and agents, and each of them, full power and authority to do and perform each and every act and thing requisite and necessary to be done in connection therewith, as fully to all intents and purposes as he might or could do in person, hereby ratifying and confirming all that said attorneys-in-fact and agents, or any of them, or their or his substitutes or substitute, may lawfully do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this Report has been signed below by the following persons in the capacities and on the date indicated.
| Signature | Title | Date | ||
| /s/ Brent T. Lucas | Chief Executive Officer and Director | March 28, 2025 | ||
| Brent T. Lucas | (Principal Executive Officer) | |||
| /s/ David R. Wells | Chief Financial Officer | March 28, 2025 | ||
| David R. Wells | (Principal Financial and Accounting Officer) | |||
| /s/ Charles R. Brynelsen | Director | March 28, 2025 | ||
| Charles R. Brynelsen | ||||
| /s/ Glen A. Taylor | Director | March 28, 2025 | ||
| Glen A. Taylor | ||||
| /s/ Michael Crowe | Director | March 28, 2025 | ||
| Michael Crowe | ||||
| /s/ Mona Patel | Director | March 28, 2025 | ||
| Mona Patel | ||||
| /s/ Janis Smith-Gomez | Director | March 28, 2025 | ||
| Janis Smith-Gomez | ||||
| /s/ Susan J. Kantor | Director | March 28, 2025 | ||
| Susan J. Kantor |
Exhibit 4.4
Form of Private Warrant
NEITHER THIS SECURITY NOR THE SECURITIES INTO WHICH THIS SECURITY IS EXERCISABLE HAS BEEN REGISTERED WITH THE SECURITIES AND EXCHANGE COMMISSION OR THE SECURITIES COMMISSION OF ANY STATE IN RELIANCE UPON AN EXEMPTION FROM REGISTRATION UNDER THE SECURITIES ACT OF 1933, AS AMENDED (THE “SECURITIES ACT”), AND, ACCORDINGLY, MAY NOT BE OFFERED OR SOLD EXCEPT PURSUANT TO AN EFFECTIVE REGISTRATION STATEMENT UNDER THE SECURITIES ACT OR PURSUANT TO AN AVAILABLE EXEMPTION FROM, OR IN A TRANSACTION NOT SUBJECT TO, THE REGISTRATION REQUIREMENTS OF THE SECURITIES ACT AND IN ACCORDANCE WITH APPLICABLE STATE SECURITIES LAWS. THIS SECURITY AND THE SECURITIES ISSUABLE UPON EXERCISE OF THIS SECURITY MAY BE PLEDGED IN CONNECTION WITH A BONA FIDE MARGIN ACCOUNT WITH A REGISTERED BROKER-DEALER OR OTHER LOAN WITH A FINANCIAL INSTITUTION THAT IS AN “ACCREDITED INVESTOR” AS DEFINED IN RULE 501(a) UNDER THE SECURITIES ACT OR OTHER LOAN SECURED BY SUCH SECURITIES.
COMMON STOCK PURCHASE WARRANT
ENVOY MEDICAL, INC.
| Warrant Shares: [_____________] | Issue Date: [______], 202[_] |
THIS STOCK PURCHASE WARRANT (the “Warrant”) certifies that, for value received, GAT Funding, LLC or its permitted assigns (the “Holder”) is entitled, upon the terms and subject to the limitations on exercise and the conditions hereinafter set forth, at any time on or prior to 5:00 p.m. (New York City time) on [______] (the “Termination Date”) but not thereafter, to subscribe for and purchase from Envoy Medical, Inc., a Delaware corporation (the “Company”), up to [______] shares of Class A Common Stock, par value $0.0001 per share (as subject to adjustment hereunder, the “Common Shares,” and, the Common Shares issuable upon exercise of the Warrant, the “Warrant Shares”). The purchase price of one Warrant Share under this Warrant shall be equal to the Exercise Price, as defined in Section 2(b).
Section 1. Definitions. Capitalized terms used and not otherwise defined herein shall have the meanings set forth in that certain Promissory Note (the “Note”), dated [________], among the Company and the purchasers signatory thereto.
Section 2. Exercise.
a) Exercise of Warrant. Exercise of the purchase rights represented by this Warrant may be made, in whole or in part, at any time or times before the Termination Date by delivery to the Company of a duly executed facsimile copy or PDF copy submitted by e-mail (or e-mail attachment) of the Notice of Exercise in the form annexed hereto (the “Notice of Exercise”). In connection with delivery of the Notice of Exercise, the Holder shall deliver the aggregate Exercise Price for the shares specified in the applicable Notice of Exercise by wire transfer or cashier’s check drawn on a United States bank. Partial exercises of this Warrant resulting in purchases of a portion of the total number of Warrant Shares available hereunder shall have the effect of lowering the outstanding number of Warrant Shares purchasable hereunder in an amount equal to the applicable number of Warrant Shares purchased. The Holder and the Company shall maintain records showing the number of Warrant Shares purchased and the date of such purchases. The Holder and any assignee, by acceptance of this Warrant, acknowledge and agree that, by reason of the provisions of this paragraph, following the purchase of a portion of the Warrant Shares hereunder, the number of Warrant Shares available for purchase hereunder at any given time may be less than the amount stated on the face hereof.
b) Exercise Price. The exercise price per Warrant Share under this Warrant shall be $[________], subject to adjustment hereunder (the “Exercise Price”).
c) Mechanics of Exercise.
i. Delivery of Warrant Shares Upon Exercise. The Company shall cause the Warrant Shares purchased hereunder to be transmitted by the Transfer Agent to credit the account of the Holder’s, or its designee’s account, with the transfer agent.
ii. No Fractional Shares or Scrip. No fractional shares or scrip representing fractional shares shall be issued upon the exercise of this Warrant. As to any fraction of a share which the Holder would otherwise be entitled to purchase upon such exercise, the Company shall, at its election, either pay a cash adjustment in respect of such final fraction in an amount equal to such fraction multiplied by the Exercise Price or round up to the next whole share.
iii. Charges, Taxes and Expenses. Issuance of Warrant Shares shall be made without charge to the Holder for any issue or transfer tax or other incidental expense in respect of the issuance of such Warrant Shares, all of which taxes and expenses shall be paid by the Company, and such Warrant Shares shall be issued in the name of the Holder or in such name or names as may be directed by the Holder; provided, however, that, in the event that Warrant Shares are to be issued in a name other than the name of the Holder, this Warrant when surrendered for exercise shall be accompanied by the Assignment Form attached hereto duly executed by the Holder and the Company may require, as a condition thereto, the payment of a sum sufficient to reimburse it for any transfer tax incidental thereto. The Company shall pay all Transfer Agent fees required for same-day processing of any Notice of Exercise and all fees to the Depository Trust Company (or another established clearing corporation performing similar functions) required for same-day electronic delivery of the Warrant Shares.
Section 3. Certain Adjustments.
a) Stock Dividends and Splits. If the Company, at any time while this Warrant is outstanding: (i) pays a stock dividend or otherwise makes a distribution or distributions on shares of its Common Shares or any other equity or equity equivalent securities payable in Common Shares (which, for avoidance of doubt, shall not include any Common Shares issued by the Company upon exercise of this Warrant), (ii) subdivides outstanding Common Shares into a larger number of shares, (iii) combines (including by way of reverse stock split) outstanding Common Shares into a smaller number of shares, or (iv) issues by reclassification of shares of the Common Shares any shares of capital stock of the Company, then in each case the Exercise Price shall be multiplied by a fraction of which the numerator shall be the number of Common Shares (excluding treasury shares, if any) outstanding immediately before such event and of which the denominator shall be the number of Common Shares outstanding immediately after such event, and the number of shares issuable upon exercise of this Warrant shall be proportionately adjusted such that the aggregate Exercise Price of this Warrant shall remain unchanged. Any adjustment made pursuant to this Section 3(a) shall become effective immediately after the record date for the determination of stockholders entitled to receive such dividend or distribution and shall become effective immediately after the effective date in the case of a subdivision, combination or re-classification.
b) Subsequent Rights Offerings. In addition to any adjustments pursuant to Section 3(a) above, if at any time the Company grants, issues or sells any rights to purchase stock, warrants, securities or other property pro rata to the record holders of Common Shares (the “Purchase Rights”), then the Holder will be entitled to acquire, upon the terms applicable to such Purchase Rights, the aggregate Purchase Rights which the Holder could have acquired if the Holder had held the number of Warrant Shares acquirable upon complete exercise of this immediately before the date on which a record is taken for the grant, issuance or sale of such Purchase Rights, or, if no such record is taken, the date as of which the record holders of Common Shares are to be determined for the grant, issue or sale of such Purchase Rights.
c) Pro Rata Distributions. During such time as this Warrant is outstanding, if the Company shall declare or make any dividend or other distribution of its assets (or rights to acquire its assets) to holders of Common Shares, by way of return of capital or otherwise (including, without limitation, any distribution of cash, stock or other securities, property or options by way of a dividend, spin off, reclassification, corporate rearrangement, scheme of arrangement or other similar transaction) (a “Distribution”), at any time after the issuance of this Warrant, then, in each such case, the Holder shall be entitled to participate in such Distribution to the same extent that the Holder would have participated therein if the Holder had held the number of Warrant Shares acquirable upon complete exercise of this Warrant immediately before the date of which a record is taken for such Distribution, or, if no such record is taken, the date as of which the record holders of Common Shares are to be determined for the participation in such Distribution. To the extent that this Warrant has not been partially or completely exercised at the time of such Distribution, such portion of the Distribution shall be held in abeyance for the benefit of the Holder until the Holder has exercised this Warrant.
d) Calculations. All calculations under this Section 3 shall be made to the nearest cent or the nearest 1/100th of a share, as the case may be. For purposes of this Section 3, the number of Warrant Shares deemed to be issued and outstanding as of a given date shall be the sum of the number of Warrant Shares (excluding treasury shares, if any) issued and outstanding.
e) Notice to Holder. Whenever the Exercise Price is adjusted pursuant to any provision of this Section 3, the Company shall promptly deliver to the Holder by facsimile or email a notice setting forth the Exercise Price after such adjustment and any resulting adjustment to the number of Warrant Shares and setting forth a brief statement of the facts requiring such adjustment.
Section 4. Transfer of Warrant.
a) Transferability. With respect to any offer, sale or other disposition of this Warrant, Holder will give written notice to the Company prior thereto, describing briefly the manner thereof, together with a written opinion of Holder’s counsel, or other evidence if reasonably satisfactory to the Company, to the effect that such offer, sale or other distribution may be effected without registration or qualification (under any federal or state law then in effect). Upon receiving such written notice and reasonably satisfactory opinion, if so requested, or other evidence, the Company, as promptly as practicable, shall notify Holder that Holder may transfer this Warrant in accordance with the terms of the notice delivered to the Company. If a determination has been made pursuant to this Section 4(a) that the opinion of counsel for Holder, or other evidence, is not reasonably satisfactory to the Company, the Company shall so notify Holder promptly after such determination has been made. Upon an effective transfer, the Company shall execute and deliver a new Warrant or Warrants in the name of the assignee or assignees, as applicable, and in the denomination or denominations specified in such instrument of assignment, and shall issue to the assignor a new Warrant evidencing the portion of this Warrant not so assigned, and this Warrant shall promptly be cancelled.
b) New Warrants. This Warrant may be divided or combined with other Warrants upon presentation hereof at the aforesaid office of the Company, together with a written notice specifying the names and denominations in which new Warrants are to be issued, signed by the Holder or its agent or attorney. Subject to compliance with Section 4(a), as to any transfer which may be involved in such division or combination, the Company shall execute and deliver a new Warrant or Warrants in exchange for the Warrant or Warrants to be divided or combined in accordance with such notice. All Warrants issued on transfers or exchanges shall be dated the initial issuance date of this Warrant and shall be identical with this Warrant except as to the number of Warrant Shares issuable pursuant thereto.
Section 5. No Registration; Restricted Shares. Holder acknowledges that this Warrant and the Warrant Shares issuable upon exercise hereof have not been registered under the Securities Act in reliance upon a specific exemption therefrom, which exemption depends upon, among other things, the bona fide nature of the Holder’s investment intent as expressed herein. Holder understands that this Warrant and the Warrant Shares issued upon any exercise hereof must be held indefinitely unless subsequently registered under the Securities Act and qualified under applicable state securities laws, or unless exemption from such registration and qualification are otherwise available. Holder is aware of the provisions of Rule 144 promulgated under the Securities Act.
Section 6. Miscellaneous.
a) No Rights as Stockholder Until Exercise; No Settlement in Cash. This Warrant does not entitle the Holder to any voting rights, dividends or other rights as a stockholder of the Company prior to the exercise hereof as set forth in Section 2(d)(i), except as expressly set forth in Section 3. Without limiting any rights of a Holder to receive Warrant Shares to receive cash payments pursuant to Section 2(d)(i) and Section 2(d)(iv) herein, in no event shall the Company be required to net cash settle an exercise of this Warrant.
b) Loss, Theft, Destruction or Mutilation of Warrant. The Company covenants that upon receipt by the Company of evidence reasonably satisfactory to it of the loss, theft, destruction or mutilation of this Warrant or any stock certificate relating to the Warrant Shares, and in case of loss, theft or destruction, of indemnity or security reasonably satisfactory to it (which, in the case of the Warrant, shall not include the posting of any bond), and upon surrender and cancellation of such Warrant or stock certificate, if mutilated, the Company will make and deliver a new Warrant or stock certificate of like tenor and dated as of such cancellation, in lieu of such Warrant or stock certificate.
c) Saturdays, Sundays, Holidays, etc. If the last or appointed day for the taking of any action or the expiration of any right required or granted herein shall not be a business day, then such action may be taken or such right may be exercised on the next succeeding business day.
d) Authorized Shares.
The Company covenants that, during the period the Warrant is outstanding, it will reserve from its authorized and unissued Warrant Shares a sufficient number of shares to provide for the issuance of the Warrant Shares upon the exercise of any purchase rights under this Warrant. The Company further covenants that its issuance of this Warrant shall constitute full authority to its officers who are charged with the duty of issuing the necessary Warrant Shares upon the exercise of the purchase rights under this Warrant. The Company will take all such reasonable action as may be necessary to assure that such Warrant Shares may be issued as provided herein without violation of any applicable law or regulation, or of any requirements of the Trading Market upon which the Warrant Shares may be listed. The Company covenants that all Warrant Shares which may be issued upon the exercise of the purchase rights represented by this Warrant will, upon exercise of the purchase rights represented by this Warrant and payment for such Warrant Shares in accordance herewith, be duly authorized, validly issued, fully paid and nonassessable and free from all taxes, liens and charges created by the Company in respect of the issue thereof (other than taxes in respect of any transfer occurring contemporaneously with such issue).
Except and to the extent as waived or consented to by the Holder, the Company shall not by any action, including, without limitation, amending its certificate of incorporation or through any reorganization, transfer of assets, consolidation, merger, dissolution, issue or sale of securities or any other voluntary action, avoid or seek to avoid the observance or performance of any of the terms of this Warrant, but will at all times in good faith assist in the carrying out of all such terms and in the taking of all such actions as may be necessary or appropriate to protect the rights of Holder as set forth in this Warrant against impairment. Without limiting the generality of the foregoing, the Company will (i) not increase the par value of any Warrant Shares above the amount payable therefor upon such exercise immediately prior to such increase in par value, (ii) take all such action as may be necessary or appropriate in order that the Company may validly and legally issue fully paid and nonassessable Warrant Shares upon the exercise of this Warrant and (iii) use commercially reasonable efforts to obtain all such authorizations, exemptions or consents from any public regulatory body having jurisdiction thereof, as may be, necessary to enable the Company to perform its obligations under this Warrant.
Before taking any action which would result in an adjustment in the number of Warrant Shares for which this Warrant is exercisable or in the Exercise Price, the Company shall obtain all such authorizations or exemptions thereof, or consents thereto, as may be necessary from any public regulatory body or bodies having jurisdiction thereof.
e) Jurisdiction. All questions concerning the construction, validity, enforcement and interpretation of this Warrant shall be determined in accordance with the provisions of the Note.
f) Restrictions. The Holder acknowledges that the Warrant Shares acquired upon the exercise of this Warrant, if not registered, will have restrictions upon resale imposed by state and federal securities laws.
g) Nonwaiver and Expenses. No course of dealing or any delay or failure to exercise any right hereunder on the part of Holder shall operate as a waiver of such right or otherwise prejudice the Holder’s rights, powers or remedies. Without limiting any other provision of this Warrant or the Note, if the Company willfully and knowingly fails to comply with any provision of this Warrant, which results in any material damages to the Holder, the Company shall pay to the Holder such amounts as shall be sufficient to cover any costs and expenses including, but not limited to, reasonable attorneys’ fees, including those of appellate proceedings, incurred by the Holder in collecting any amounts due pursuant hereto or in otherwise enforcing any of its rights, powers or remedies hereunder.
h) Notices. Any notice, request or other document required or permitted to be given or delivered to the Holder by the Company shall be delivered in accordance with the notice provisions of the Note.
i) Limitation of Liability. No provision hereof, in the absence of any affirmative action by the Holder to exercise this Warrant to purchase Warrant Shares, and no enumeration herein of the rights or privileges of the Holder, shall give rise to any liability of the Holder for the purchase price of any Warrant Shares or as a stockholder of the Company, whether such liability is asserted by the Company or by creditors of the Company.
j) Remedies. The Holder, in addition to being entitled to exercise all rights granted by law, including recovery of damages, will be entitled to specific performance of its rights under this Warrant. The Company agrees that monetary damages would not be adequate compensation for any loss incurred by reason of a breach by it of the provisions of this Warrant and hereby agrees to waive and not to assert the defense in any action for specific performance that a remedy at law would be adequate.
k) Successors and Assigns. Subject to applicable securities laws, this Warrant and the rights and obligations evidenced hereby shall inure to the benefit of and be binding upon the successors and permitted assigns of the Company and the successors and permitted assigns of Holder. The provisions of this Warrant are intended to be for the benefit of any Holder from time to time of this Warrant and shall be enforceable by the Holder or holder of Warrant Shares.
l) Amendment. This Warrant may be modified or amended or the provisions hereof waived with the written consent of the Company and the Holder.
m) Severability. Wherever possible, each provision of this Warrant shall be interpreted in such manner as to be effective and valid under applicable law, but if any provision of this Warrant shall be prohibited by or invalid under applicable law, such provision shall be ineffective to the extent of such prohibition or invalidity, without invalidating the remainder of such provisions or the remaining provisions of this Warrant.
n) Headings. The headings used in this Warrant are for the convenience of reference only and shall not, for any purpose, be deemed a part of this Warrant.
********************
[Signature Page Follows]
IN WITNESS WHEREOF, the Company has caused this Warrant to be executed by its officer thereunto duly authorized as of the date first above indicated.
| ENVOY MEDICAL, INC. | ||
| By: | ||
| Name: | ||
| Title: | ||
EXHIBIT A
NOTICE OF EXERCISE
TO: ENVOY MEDICAL, INC.
(1) The undersigned hereby elects to purchase ________ Warrant Shares of the Company pursuant to the terms of the attached Warrant (only if exercised in full), and tenders herewith payment of the exercise price in full, together with all applicable transfer taxes, if any.
(2) Payment shall take the form of (check applicable box):
☐ in lawful money of the United States; or
☐ [if permitted the cancellation of such number of Warrant Shares as is necessary, in accordance with the formula set forth in subsection 2(c), to exercise this Warrant with respect to the maximum number of Warrant Shares purchasable pursuant to the cashless exercise procedure set forth in subsection 2(c).
(3) Please issue said Warrant Shares in the name of the undersigned or in such other name as is specified below:
_______________________________
The Warrant Shares shall be delivered to the following DWAC Account Number:
_______________________________
_______________________________
_______________________________
[SIGNATURE OF HOLDER]
Name of Investing Entity: __________________________________________________________________
Signature of Authorized Signatory of Investing Entity: ____________________________________________
Name of Authorized Signatory: _____________________________________________________________
Title of Authorized Signatory: ______________________________________________________________ FOR VALUE RECEIVED, the foregoing Warrant and all rights evidenced thereby are hereby assigned to
Date: ________________________________________________________________________________________
EXHIBIT B
ASSIGNMENT FORM
(To assign the foregoing Warrant, execute this form and supply required information. Do not use this form to purchase shares.)
| Name: | ______________________________________ |
| (Please Print) | |
| Address: | ______________________________________ |
| (Please Print) | |
| Phone Number: | ______________________________________ |
| Email Address: | ______________________________________ |
| Dated: _______________ __, ______ | |
| Holder’s Signature: | |
| Holder’s Address: |
Exhibit 10.23
Envoy Medical Corporation
Building Lease
THIS LEASE, Made and entered into this day of May 20, 2016, by and between Taylor Corporation, hereinafter called “Landlord”, and Envoy Medical Corporation, hereinafter called “Tenant.”
W I T N E S S E T H:
1. PREMISES. Landlord hereby lets to Tenant and Tenant hereby leases from Landlord approximately 8,290 square feet of space in the building located at 4875 White Bear Parkway, City of White Bear Lake, County of Ramsey, State of Minnesota, which the parties hereto acknowledge is non-homestead property, hereinafter called “leased premises” or “demised premises”. The amount of space leased may be adjusted by mutual consent between the Tenant and the Landlord during the term of the lease and the monthly rent in Section 3 adjusted accordingly along with proration of expenses.
Tenant acknowledges the receipt of the demised premises and the same to be in good and sanitary condition, and in good repair, the taking possession of the demised premises by the Tenant shall be conclusive evidence that the demised premises, and the equipment, fixtures, plumbing, etc., were at the time of so taking possession thereof in good, clean, sanitary and tenantable condition, and in all respects satisfactory and acceptable to the Tenant. Tenant hereby releases Landlord from any and all claims arising from any defect in the condition of said demised premises.
2. TERM. This lease shall be for the term of eleven (11) years, seven (7) months from and after the 1st day of June, 2016, to the last day of December 31, 2027, both dates inclusive, unless sooner terminated as hereinafter provided.
3. RENT. Tenant shall pay to Landlord at 1725 Roe Crest Drive, North Mankato, Minnesota, or at such place as Landlord may from time to time designate in writing the following base rent, repayment for build out cost for tenants space and building operating costs (common area maintenance, taxes, insurance and utilities which costs will be reviewed and calculated yearly), payable on or before the first of each month in accordance with the following schedule:
| Monthly | Repayment - Build-out Costs | Building operating | ||||||||||||||
| Period | Base Rent | monthly | balloon | expense | ||||||||||||
| 6/1/16 -12/31/16 | 3,496.00 | 2,072.50 | - | 2,349 | ||||||||||||
| 1/1/17 -12/31/17 | 3,496.00 | 2,072.50 | - | TBD | ||||||||||||
| 1/1/18 -12/31/18 | 3,631.00 | 2,072.50 | 10,000 | TED | ||||||||||||
| 1/1/19 -12/31/19 | 3,704.00 | 2,072.50 | 10,000 | TBD | ||||||||||||
| 1/1/20 -12/31/20 | 3,778.00 | 3,108.75 | 20,000 | TBD | ||||||||||||
| 1/1/21 -12/31/21 | 3,853.00 | 3,108.75 | 20,000 | TBD | ||||||||||||
| 1/1/22 -12/31/22 | 3,930.00 | 3,800.00 | 30, 000 | TBD | ||||||||||||
| 1/1/23 -12/31/23 | 4,009.00 | 3,800.00 | 30,000 | TED | ||||||||||||
| 1/1/24 -12/31/24 | 4,089.00 | 3,800.00 | 50,000 | TBD | ||||||||||||
| 1/1/25 -12/31/25 | 4,171.00 | 3,800.00 | 50,000 | TED | ||||||||||||
| 1/1/26 -12/31/26 | 4,254.00 | 3,800.00 | 50,000 | TBD | ||||||||||||
| 1/1/27 -12/31/27 | 4,339.00 | 3,146.00 | - | TBD | ||||||||||||
4. USE. It is agreed that the leased premises may be used and occupied by Tenant for its Research & Development and Manufacturing business or for any other lawful purpose. Said usage shall at all times be in compliance with all applicable laws, ordinances and governmental regulations, and requirements of insurers of the leased premises.
5. WARRANTIES OF TITLE AND QUIET POSSESSION. Landlord covenants that Landlord is seized of the demised premises in fee simple and has full right to make this lease and that Tenant shall have quiet and peaceable possession of the leased premises during the term hereof so long as it is not in default.
6. WASTE AND NUISANCE PROHIBITED. During the term of this lease, Tenant shall comply with all applicable laws affecting the demised premises, the breach of which might result in any penalty on Landlord or forfeiture of Landlord’s title to the leased premises. Tenant shall not commit, or suffer to be committed, any waste on the demised premises, or any nuisance.
7. ABANDONMENT OF PREMISES. Tenant shall not vacate or abandon the demised premises at any time during the term hereof; if Tenant shall abandon, vacate or surrender the demised premises, or be dispossessed by process of law, or otherwise, any personal property belonging to Tenant and left on the demised premises shall be deemed to be abandoned, at the option of Landlord, except such property as may be encumbered to Landlord.
8. LANDLORD’S RIGHT OF ENTRY. Tenant shall permit Landlord and the agents and employees of Landlord to enter into and upon the demised premises at all reasonable times for the purpose of inspecting the same, or for the purpose of posting notices of nonresponsibility for alterations, additions, or repairs, without any rebate of rent and without any liability to Tenant for any loss of occupation or quiet enjoyment of the premises thereby occasioned.
9. LEASE SUBORDINATE TO ENCUMBRANCES. This lease is subject and subordinate to any mortgages or trust deeds that may be heretofore or hereafter placed against the demised premises, and to all advances made or that may be made on account of the encumbrances, to the full extent of the principal sums secured thereby and interest thereon.
10. SUBLETTING AND ASSIGNMENT. Tenant may not sublet the leased premises in whole or in part without Landlord’s written consent. Tenant shall not assign or transfer this lease, or any interest herein without the prior written consent of Landlord, which consent will not be unreasonably withheld. Any such sublease or assignment without such consent shall be void, and shall at the option of Landlord, terminate this lease. Neither this lease nor the leasehold estate of Tenant or any interests of Tenant hereunder in the leased premises or any buildings or improvements thereon shall be subject to involuntary assignment, transfer, or sale, or to assignment, transfer or sale by operation of law in any manner whatsoever, and any such attempted involuntary assignment, transfer, or sale shall be void and of no effect and shall, at the option of Landlord, terminate this lease.
11. NOTICES. All notices, demands, or other writings in this lease provided to be given or made or sent, or which may be given or made or sent, by either party hereto to the other, shall be deemed to have been fully given or made or sent when made in writing and deposited in the United States mail, registered and postage prepaid, and addressed as follows:
| TO LANDLORD: | Taylor Corporation |
| 1725 Roe Crest Drive | |
| North Mankato, MN 56002-3728 | |
| TO TENANT: | Envoy Medical Corporation |
| 4875 White Bear Parkway | |
| White Bear Lake, MN 55110 |
12. REPAIRS AND MAINTENANCE. Tenant shall make all necessary incidental repairs to the interior of the demised premises, and maintain the interior in good condition. Tenant shall make no alterations or improvements to the demised premises without advance consent of Landlord. All such alterations or improvements shall remain with the demised premises and become the property of Landlord.
13. DAMAGE TO OR DESTRUCTION OF PREMISES. If during the term of this lease the leased premises or improvements thereon shall be injured or destroyed by fire or the elements, or through any other cause, so as to render the leased premises unfit for occupancy, or make it impossible to conduct the business of the Tenant thereof, or to such an extent that they cannot be repaired with reasonable diligence within ninety (90) days from the happening of such injury, then the Landlord may terminate this lease and term herein demised from the date of such damage or destruction, and the Tenant shall immediately surrender the demised premises and all interest therein to the Landlord, and the Tenant shall pay rent only to the time of such surrender. In case of any such destruction or injury, the Landlord may re-enter and repossess the leased premises discharged of this lease and may dispossess all parties then in possession thereof. If the leased premises can be restored within ninety (90) days from the happening of the injury thereto, and the Landlord within fifteen (15) days from the occurrence of such injury elects in writing to repair or restore said leased premises within ninety (90) days from the happening of the injury thereto, then this lease shall not end or terminate on account of such injury by fire or otherwise, but the rent shall not run or accrue after the injury and during the process of repairs, and up to the time when the repairs shall be completed, except only that Tenant shall during such time pay a pro rata portion of such rent apportioned to the portion of the demised premises which are in condition for occupancy or which may be actually occupied during such repairing period. If, however, the leased premises shall be so slightly injured by any cause aforesaid, as not to be rendered unfit for occupancy, then the Landlord shall repair the same with reasonable promptness, and in that case the rent shall not cease or be abated during such repairing period. All improvements or betterments placed by the Tenant on the leased premises shall, however, in any event be repaired and replaced by the Tenant at its own expense and not at the expense of the Landlord. Tenant is to pay its prorated share as determined by Landlord of these repairs and maintenance.
14. INSURANCE. (a) Insurance Companies. It is agreed that all policies of insurance to be maintained in force by the respective parties hereto shall be obtained from good and solvent insurance companies approved by Landlord.
(b) Tenant to Obtain Public Liability Insurance. Tenant shall, at its own expense, at all times during the term of this lease, maintain in force a policy or policies of insurance, written by one or more responsible insurance carriers designated by Landlord, which will insure Landlord against liability for injury to or death of persons or loss or damage to property occurring in or about the demised premises. The liability under such insurance shall not be less than Five Hundred Thousand Dollars ($500,000.00) for any person killed or injured, One Million Dollars ($1,000,000.00) for any one accident, and Two Hundred Thousand Dollars ($200,000.00) property damage.
(c) Tenant to Obtain Worker’s Compensation Insurance. Tenant shall maintain and keep in force all employees’ compensation insurance required under the laws of the State of Minnesota, and such other insurance as may be necessary to protect Landlord against any other liability to person or property arising hereunder by operation of law, whether such law be now in force or adopted subsequent to the execution hereof.
(d) Tenant’s Waiver of Casualty Insurance Proceeds. In the event the demised premises shall be damaged or destroyed by fire or other casualty so insured against, Tenant shall claim no interest in any insurance settlement arising out of any such loss and it will execute all documents required by Landlord or the insurance company or companies that may be necessary for use in connection with settlement of any such loss. Tenant shall also insure separately its own leasehold improvements.
(e) Tenant’s Failure to Insure. Should Tenant fail to keep in effect and pay for such insurance as it is in this section required to maintain, Landlord may do so, in which event the insurance premium paid by Landlord shall become due and payable forthwith and failure of Tenant to pay same on demand shall constitute a breach of this lease. All insurance policies applicable to the entire building shall have their cost prorated to the amount of space occupied as determined by the Landlord.
(f) Named Insureds. All policies of insurance required hereby shall be written in the names of Landlord, Tenant, and the mortgagee (if any), as their respective interests may appear, and a copy of each such policy shall be delivered to Landlord when issued.
15. LIENS. (a) Tenant’s Duty to Keep Premises Free of Liens. Tenant shall keep all of the demised premises and every part thereof and all building and other improvements at any time located thereon free and clear of any and all mechanic’s, materialmen’s, and other liens for or arising out of or in connection with work or labor done, services performed, or materials or appliances used or furnished for or in connection with any operations of Tenant, any alteration, improvement, or repairs or additions which Tenant may make or permit or cause to be made, or any work or construction, by, for, or permitted by Tenant on or about the leased premises, or any obligations of any kind incurred by Tenant, and at all times promptly and fully to pay and discharge any and all claims on which any such lien may or could be based, and to indemnify, defend and save harmless Landlord and all of the leased premises and all buildings and improvements thereon against all such liens and claims of liens and suits or other proceedings pertaining thereto. Tenant shall give Landlord written notice no less than sixty (60) days in advance of any construction, alteration, addition, improvement, or repair estimated to cost in excess of Twenty Thousand Dollars ($20,000.00), in order that Landlord may post appropriate notices of Landlord’s non-responsibility.
(b) Contesting Liens. If Tenant desires to contest any such lien, it shall notify Landlord of its intention to do so within thirty (30) days after filing of such lien. In such case, and provided that Tenant shall on demand protect Landlord by a good and sufficient surety bond against any such lien and any cost, liability, or damage arising out of such contest, Tenant shall not be in default hereunder until thirty (3 0) days after final determination of the validity thereof, within which time Tenant shall satisfy and discharge such lien to the extent held valid; but the satisfaction and discharge of any such lien shall not, in any case, be delayed until execution is had on any judgment rendered thereon, and such delay shall be a default of Tenant hereunder. In the event of such contest, Tenant shall protect and indemnify Landlord against all loss, expense, and damage resulting therefrom.
16. INDEMNIFICATION OF LANDLORD. Landlord shall not be liable for any loss, injury, death, or damage to persons or property which at any time may be suffered or sustained by Tenant or by any person whosoever may at any time be using or occupying or visiting the leased premises or be in, on, or about the same, whether such loss, injury, death, or damage shall be caused by or in any way result from or arise out of any act, omission, or negligence of Tenant or of any occupant, subtenant, visitor, or user of any portion of the leased premises, or shall result from or be caused by any other matter or thing whether of the same kind as or of a different kind than the matters or things above set forth, and Tenant shall indemnify Landlord against all claims, liability, loss, or damage whatsoever on account of any such loss, injury, death, or damage. Tenant hereby waives all claims against Landlord for damages to the building and improvements that are now on or hereafter placed or built on the premises and to the inventory, equipment or other property of Tenant in, on or about the leased premises, and for injuries to persons or property in or about the leased premises, from any cause arising at any time. The two preceding sentences shall not apply to loss, injury, death or damage arising by reason of the negligence or misconduct of Landlord. Nothing contained in this section shall, however, detract from Landlord’s rights to protection under the public liability insurance policy to be paid for by the Tenant as specified in Paragraph 15 hereof.
17. REDELIVERY OF PREMISES. Tenant shall pay the rent and all other sums required to be paid by Tenant hereunder in the amounts, at the times, and in the manner herein provided, and shall keep and perform all the terms and conditions hereof on its part to be kept and performed, and, at the expiration or sooner termination of the lease, peaceably and quietly quit and surrender to Landlord the leased premises in good order and condition, reasonable wear and tear excepted, subject to the other provisions of this lease. In the event of the nonperformance by Tenant of any of the covenants of Tenant undertaken herein, this lease may be terminated as herein provided.
18. REMEDIES CUMULATIVE. All remedies hereínbefore and hereafter conferred on Landlord shall be deemed cumulative and no one exclusive of the other, or of any other remedy conferred by law.
19. PROHIBITION OF INVOLUNTARY ASSIGNMENT; EFFECT OF BANKRUPTCY OR INSOLVENCY. (a) Prohibition of Involuntary Assignment. Neither this lease nor the leasehold estate of Tenant nor any interest of Tenant hereunder in the demised premises or in the building or improvements thereon shall be subject to involuntary assignment, transfer, or sale, or to assignment, transfer, or sale by operation of law in any manner whatsoever (except through statutory merger or consolidation) and any such attempt at involuntary assignment, transfer, or sale shall be void and of no effect.
(b) Effect of Bankruptcy. Without limiting the generality of the provisions of the preceding paragraph (a) of this section, Tenant agrees that in the event any proceedings under the Bankruptcy Act or any amendment thereto be commenced by or against Tenant, and, if against Tenant, such proceedings shall not be dismissed before either an adjudication in bankruptcy or the confirmation of a composition, arrangement, or plan of reorganization, or in the event Tenant is adjudged insolvent or makes an assignment for the benefit of its creditors, or if a receiver is appointed in any proceeding or action to which Tenant is a party, with authority to take possession or control of the demised premises or the business conducted thereon by Tenant, and such receiver is not discharged within a period of ninety (90) days after his appointment, any such event or any involuntary assignment prohibited by the provisions of the preceding paragraph (a) of this section shall be deemed to constitute a breach of this lease by Tenant and shall, at the election of Landlord, but not otherwise, without notice or entry or other action of Landlord terminate this lease and also all rights of Tenant under this lease and in and to the demised premises and also all rights of any and all persons claiming under Tenant.
20. DEFAULT. In the event of any breach of this lease by Tenant, Landlord, in addition to the other rights or remedies it may have, shall have the immediate right of re-entry and may remove all persons and property from the leased premises; such property may be removed and stored in a public warehouse or elsewhere at the cost of and for the account of Tenant. Should Landlord elect to re-enter, as herein provided, or should it take possession pursuant to legal proceedings or pursuant to any notice provided for by law, Landlord may either terminate this lease or it may from time to time, without terminating this lease, relet the demised premises or any part thereof for such term or terms (which may be for a term extending beyond the term of this lease) and at such rental or rentals and on such other terms and conditions as Landlord in the sole discretion of Landlord may deem advisable with the right to make alterations and repairs to the demised premises. On each such re-letting (a) Tenant shall be immediately liable to pay to Landlord, in addition to any indebtedness other than rent due hereunder, the expenses of such re-letting and of such alterations and repairs, incurred by Landlord, and the amount, if any, by which the rent reserved in this lease for the period of such re-letting (up to but not beyond the term of this lease) exceeds the amount agreed to be paid as rent for the demised premises for such period on such re-letting; or (b) at the option of Landlord, rents received by such Landlord from such re-letting shall be applied, first, to the payment of any indebtedness, other than rent due hereunder from Tenant to Landlord; second, to the payment of any expenses of such re-letting and of such alterations and repairs; third, to the payment of rent due and unpaid hereunder, and the residue, if any, shall be held by landlord and applied in payment of future rent as the same may become due and payable hereunder. If Tenant has been credited with any rent to be received by such re-letting under option (a) hereof, and such rent shall not be promptly paid to landlord by the new tenant, or if such rentals received from such re-letting under option (b) hereof during any month is less than that to be paid during the month by Tenant hereunder, Tenant shall pay any such deficiency to Landlord. Such deficiency shall be calculated and paid monthly. No such reentry or taking possession of the demised premises by Landlord shall be construed as an election on the part of landlord to terminate this lease unless a written notice of such intention is given to Tenant or unless the termination thereof is decreed by a court of competent jurisdiction. Notwithstanding any such reletting without termination, Landlord may at any time thereafter elect to terminate this lease for such previous breach. Should Landlord at any time terminate this lease for any breach, in addition to any other remedy it may have, Landlord may recover from Tenant all damages incurred by reason of such breach, including the cost of recovering the leased premises, and including the worth at the time of such termination of the excess, if any, of the amount of rent and charges equivalent to rent reserved in this lease for the remainder of the stated term over the then reasonable rental value of the leased premises for the remainder of the stated term, all of which amounts shall be immediately due and payable from Tenant to Landlord.
21. LANDLORD’S RIGHT TO PERFORM. In the event that Tenant by failing or neglecting to do or perform any act or thing herein provided by it to be done or performed, shall be in default hereunder and such failure shall continue for a period of thirty (30) days after written notice from Landlord specifying the nature of the act or thing to be done or performed, then Landlord may, but shall not be required to, do or perform or cause to be done or performed such act or thing (entering on the demised premises for such purposes, if Landlord shall so elect) and Landlord shall not be or be held liable or in any way responsible for any loss, inconvenience, annoyance, or damage resulting to Tenant on account thereof, and Tenant shall repay to Landlord on demand the entire expense thereof, including compensation to the agents and employees of Landlord. Any act or thing done by Landlord pursuant to the provisions of this section shall not be or be construed as a waiver of any such default by Tenant, or as a waiver of any covenants, term, or condition herein contained or the performance thereof, or of any other right or remedy of Landlord, hereunder or otherwise. All amounts payable by Tenant to Landlord under any of the provisions of this lease, if not paid when the same become due in this lease provided, shall bear interest from the date they became due until paid at the rate of ten percent (10%) per annum, compounded annually.
22. EFFECT OF EMINENT DOMAIN. (a) Effect of Total Condemnation. In the event the entire leased premises shall be appropriated or taken under the power of eminent domain by any public or quasi-public authority, this lease shall terminate and expire as of the date of such taking, and Tenant shall thereupon be released from any liability thereafter accruing hereunder.
(b) Effect of Partial Condemnation. In the event a substantial portion of the leased premises shall be so appropriated or taken and the remainder of the leased property shall not be suitable for the use then being made of the leased property by Tenant, or if the remainder of the leased property is not one undivided parcel of property, Tenant shall have the right to terminate this lease as of the date of such taking on giving to Landlord written notice of termination within sixty (60) days after Landlord has notified Tenant in writing that the leased property has been so appropriated or taken. In the event of such partial taking which does not so terminate this lease, then this lease shall continue in full force and effect as to the part not taken, and the rental to be paid by Tenant during the remainder of the term shall be adjusted by a majority of three (3) independent appraisers, one designated by Landlord, one designated by Tenant, and one designated by the first two appraisers named.
(c) Condemnation Award. In the event of the termination of this lease by reason of the total or partial taking of the leased premises by eminent domain, then in any such condemnation proceedings Landlord and Tenant each shall be free to make claim against the condemning or taking authority for the amount of any damage done to them, respective, as a result thereof.
23. SURRENDER OF LEASE. The voluntary or other surrender of this lease by Tenant, or a mutual cancellation thereof, shall not work a merger, and shall, at the option of Landlord, terminate all or any existing subleases or subtenancies, or may at the option of Landlord, operate as an assignment to it of any or all such subleases or subtenancies.
24. WAIVER. The waiver of Landlord, or the failure of Landlord to take action with respect to any breach of any term, covenant, or condition herein contained shall not be deemed to be a waiver of such term, covenant, or condition, or subsequent breach of the same, or any other term, covenant, or condition therein contained. The subsequent acceptance of rent hereunder by Landlord shall not be deemed to be a waiver of any preceding breach by Tenant of any term, covenant, or condition of this lease, other than the failure of Tenant to pay the particular rental so accepted, regardless of Landlord’s knowledge of such preceding breach at the time of acceptance of such rent.
25. EFFECT OF TENANT’S HOLDING OVER. Any holding over after the expiration of the term of this lease, with consent of Landlord, shall be construed to be a tenancy from month to month, at the same base monthly rental as required to be paid by Tenant for the period immediately prior to the expiration of the term hereof, and shall otherwise be on the terms and conditions herein specified so far as applicable.
26. PARTIES BOUND. The covenants and conditions herein contained shall, subject to the provisions as to assignment, transfer, and subletting, apply to and bind the heirs, successors, executors, administrators, personal representatives, and assigns of all of the parties hereto; and all of the parties hereto shall be jointly and severally liable hereunder.
27. TIME OF THE ESSENCE. Time is of the essence of this lease, and of each and every covenant, term, condition and provision hereof.
28. EFFECTIVE LAW. This lease is executed by a Minnesota Corporation as Tenant for the demise of real estate situated in the State of Minnesota and, accordingly, shall be enforced and interpreted under the laws of the State of Minnesota in effect from time to time.
29. TENANT WAIVER OF RIGHT TO TERMINATE LEASE. Notwithstanding any provision herein to the contrary, neither destruction nor condemnation of, nor damage to, all or any part of the property shall entitle or permit the Tenant to surrender or terminate this Lease or shall relieve the Tenant from its liability in full to pay rent and other sums and charges payable by the Tenant hereunder or from any of its other obligations under this Lease. The Tenant hereby waives any rights now or hereafter conferred upon it by statute or otherwise to surrender this Lease, to quit or surrender all or any part of the property, or to receive any suspension, diminution, abatement or reduction of the rent or other sums and charges payable by the Tenant hereunder on account of any such destruction, damage or condemnation.
30. SECTION CAPTIONS. The captions appearing under the section number designations of this lease are for convenience only and are not a part of this lease and do not in any way limit or amplify the terms and provisions of this lease.
IN WITNESS WHEREOF, the parties have executed this lease at North Mankato, Minnesota, on the day and year first above written.
| TENANT: | LANDLORD: | |||||
| Envoy Medical Corporation | Taylor Corporation | |||||
| By: | /s/ Brent Lucas | By: | /s/ Larry Taylor | |||
| Its: | Chief Executive Officer | Its: | Vice President | |||
AMENDMENT TO LEASE
The AMENDMENT TO LEASE, made and entered into this June 20, 2017, by and between Taylor Corporation hereinafter called the “LANDLORD” and Envoy Medical Corporation hereinafter called “TENANT”.
WITNESSETH
That the BUILDING LEASE AGREEMENT dated May 20, 2016 between the LANDLORD and the TENANT Covering a portion of a building located at 4875 White Bear Parkway, City of White Bear Lake, Minnesota, hereinafter called “Demised Premises”; and
Whereas LANDLORD and TENANT now desire to amend said lease in the following manner
The amount of space leased is increased from 8,290SF to 9,542SF.
TENANT agrees that the base monthly rent will be adjusted to the following:
| 7/1/17 - 12/31/17 | $ | 4,039.00 | ||
| 1/1/18 - 12/31/18 | $ | 4,104.00 | ||
| 1/1/19 - 12/31/19 | $ | 4,187.00 | ||
| 1/1/20 - 12/31/20 | $ | 4,270.00 | ||
| 1/1/21 - 12/31/21 | $ | 4,350.00 | ||
| 1/1/22 - 12/31/22 | $ | 4,443.00 | ||
| 1/1/23 - 12/31/23 | $ | 4,532.00 | ||
| 1/1/24 - 12/31/24 | $ | 4,622.00 | ||
| 1/1/25 - 12/31/25 | $ | 4,715.00 | ||
| 1/1/26 - 12/31/26 | $ | 4,809.00 | ||
| 1/1/27 - 12/31/27 | $ | 4,905.00 |
Except as modified herein all Terms and Conditions of the Building Lease Agreement dated May 20, 2016 shall remain in full force and effect.
In Witness whereof, the parties hereto have executed this AMENDMENT TO LEASE the day and year written above.
| TENANT: Envoy Medical Corporation | LANDLORD: Taylor Corporation | |||
| By: | /s/ Brent Lucas | By: | Larry Taylor | |
| Its: | Chief Executive Officer | Its: | Vice President | |
AMENDMENT #2
This Amendment #2 (“Second Amendment”) is entered into effective as of December 9. 2022 (“Amendment Date”) by and between Taylor Corporation (“Landlord”) and Envoy Medical Corporation (“Tenant”).
WHEREAS, Landlord and Tenant (each, a “Party” and collectively, “Parties”) intend to modify, amend, and supplement that certain Building Lease, dated May 20, 2106, by and between the Parties, as amended by that certain Amendment to Lease, dated June 20, 2017, between the Parties (“First Amendment”, and collectively, “Lease”) as set forth herein.
1. Amendment. The Parties hereby agree and affirm that they are parties to the Lease, which is hereby modified, amended, and/or supplemented as set forth in this Second Amendment. Any capitalized terms used but not specifically defined in this Second Amendment shall have the meanings ascribed to them in the Lease.
2. Leased Premises. The Leased Premises, as identified in Section 1 of the Lease and Section 1 of the First Amendment, is hereby amended to 9,876 square feet.
3. Base Monthly Rent. The monthly base rent for the remainder of the Term, as identified in Section 3 of the Lease and Section 2 of the First Amendment, is hereby amended as follows:
| Period | Monthly Base Rent | |||
| 1/112023-12/31/2023 | $ | 4,691.00 | ||
| 1/1/2024 - 12/31/2024 | $ | 4,784.00 | ||
| 1/1/2025 - 12/31/2025 | $ | 4,880.00 | ||
| 1/1/2026 - 12/31/2026 | $ | 4,977.00 | ||
| 1/1/2027 - 12/31/2027 | $ | 5,077.00 |
Tenant shall remain obligated to pay all other amounts due under the Lease in a timely manner, as specified therein thereby.
4. Construction: Except as expressly set forth herein, the terms and conditions of the Lease are hereby ratified, and all provisions of the Lease shall remain in full force and effect pursuant to their terms. No Party shall be bound by any condition, definition, warranty, or representations, other than as expressly set forth or provided for in the Lease or this Second Amendment, Wherever a conflict between this Second Amendment and the Lease exists, the provisions of this Second Amendment shall control.
5. Signature. This Second Amendment may be executed in any number of counterparts, which when taken together shall constitute an original document, and shall become effective when signed by each of the Parties. This Second Amendment may be accepted, executed, or agreed to through the use of an electronic signature or other reasonably reliable means and will be deemed an original and will be binding on the Parties in the same manner and shall have the same legal validity and enforceability as if it were physically or manually accepted, executed, delivered, or agreed (including, but not limited to, “wet-ink” signatures). The Parties hereby consent to the use of any third-party electronic signature capture service as may be reasonably selected by Landlord.
[Remainder of Page Intentionally Left Blank; Signature Page Follows]
IN WITNESS WHEREOF, the Parties have executed this Second Amendment as of the Amendment Date.
| LANDLORD: TAYLOR CORPORATION | |||
| Name: | Jason Greenfield | ||
| Its: | Director | ||
| Date: | December 9, 2022 | /s/ Jason Greenfield | |
| Signature | |||
| TENANT: ENVOY MEDICAL CORPORATION | |||
| Name: | Brent Lucas | ||
| Its: | CEO | ||
| Date: | December 9, 2022 | /s/ Brent Lucas | |
| Signature | |||
AMENDMENT #3
This Amendment #3 (“Third Amendment”) is entered into effective as of May 1, 2024 (“Amendment Date”) by and between Taylor Corporation (“Landlord”) and Envoy Medical Corporation (“Tenant”).
WHEREAS, Landlord and Tenant (each, a “Party” and collectively, “Parties”) intend to modify, amend, and supplement that certain Building Lease, dated May 20, 2016, by and between the Parties, as amended by that certain First Amendment to Lease dated June 20, 2017, and that Second Lease Amendment dated December 9, 2022, between the Parties (“collectively, “Lease”) as set forth herein.
1. Amendment. The Parties hereby agree and affirm that they are parties to the Lease, which is hereby modified, amended, and/or supplemented as set forth in this Third Amendment. Any capitalized terms used but not specifically defined in this Third Amendment shall have the meanings ascribed to them in the Lease.
2. Leased Premises. The Leased Premises (as identified in Exhibit A within the hash-marked areas of 107 sf, 714 sf, 615 sf, 9,042 sf and 1,062 sf), shall be increased from 9,876 square feet 11,540 square feet.
3. Term. The Term of Lease shall be extended for three (3) years and shall expire on December 31, 2030.
4. Rent. The monthly rent as identified in Section 3 of the Lease, is hereby amended as follows:
|
PERIOD |
MONTHLY BASE RENT | MONTHLY BUILD-OUT REPAYMENT | MONTHLY OPERATING EXPENSE | TOTAL MONTHLY RENT | BUILD-OUT BALLOON REPAYMENT |
||||||||||||||||
| 5/1/2024 - 12/31/2024 | $ | 5,587.28 | $ | 3,800.00 | $ | 5,244.69 | $ | 14,631.97 | $ | 50,000.00 - Due 7/1/2024 | |||||||||||
| 1/1/2025 - 12/31/2025 | $ | 5,702.68 | $ | 3,800.00 | $ | 5,454.48 | $ | 14,957.16 | $ | 50,000.00 - Due 7/1/2025 | |||||||||||
| 1/1/2026 - 12/31/2026 | $ | 5,818.08 | $ | 3,800.00 | $ | 5,672.66 | $ | 15,290.74 | $ | 50,000.00 - Due 7/1/2026 | |||||||||||
| 1/1/2027 - 12/31/2027 | $ | 5,933.48 | $ | 3,146.00 | $ | 5,899.56 | $ | 14,979.04 | $ | 50,000.00 - Due 7/1/2027 | |||||||||||
| 1/1/2028 - 12/31/2028 | $ | 6,173.90 | $ | 0.00 | $ | 6,135.55 | $ | 12,309.45 | $ | 50,000.00 - Due 7/1/2028 | |||||||||||
| 1/1/2029 - 12/31/2029 | $ | 6,423.93 | $ | 0.00 | $ | 6,380.97 | $ | 12,804.90 | $ | 50,000.00 - Due 7/1/2029 | |||||||||||
| 1/1/2030 - 12/31/2030 | $ | 6,673.97 | $ | 0.00 | $ | 6,626.21 | $ | 13,300.18 | $ | 0.00 | |||||||||||
Tenant shall remain obligated to pay all other amounts due under the Lease in a timely manner, as specified therein thereby.
5. Tenant Affiliate. Landlord and Tenant agree that Tenant’s parent company, Envoy Medical, Inc., (“Tenant Affiliate”) has the right to use the Leased Premises. Tenant shall be responsible for Tenant Affiliate’s compliance with the terms of the Lease and this Third Amendment.
6. Construction. Except as expressly set forth herein, the terms and conditions of the Lease are hereby ratified, and all provisions of the Lease shall remain in full force and effect pursuant to their terms. No Party shall be bound by any condition, definition, warranty, or representations, other than as expressly set forth or provided for in the Lease or this Third Amendment. Wherever a conflict between this Third Amendment and the Lease exists, the provisions of this Third Amendment shall control.
7. Signature. This Third Amendment may be executed in any number of counterparts, which when taken together shall constitute an original document, and shall become effective when signed by each of the Parties. This Third Amendment may be accepted, executed, or agreed to through the use of an electronic signature or other reasonably reliable means and will be deemed an original and will be binding on the Parties in the same manner and shall have the same legal validity and enforceability as if it were physically or manually accepted, executed, delivered, or agreed (including, but not limited to, “wet-ink” signatures). The Parties hereby consent to the use of any third-party electronic signature capture service as may be reasonably selected by Landlord.
[Signature Page Follows]
IN WITNESS WHEREOF, the Parties have executed this Third Amendment as of the Amendment Date.
| LANDLORD: TAYLOR CORPORATION | |||
| Name: | Jason Greenfield | ||
| Its: | Vice President | ||
| Date: | 6/6/2024 | /s/ Jason Greenfield | |
| Signature | |||
| TENANT: ENVOY MEDICAL CORPORATION | |||
| Name: | Brent Lucas | ||
| Its: | CEO | ||
| Date: | 6/6/2024 | /s/ Brent Lucas | |
| Signature | |||
EXHIBIT 10.24
SERVICES AGREEMENT
This Services Agreement (“Agreement”) is effective the 1st day of January, 2022 (“Effective Date”) between Envoy Medical Corporation and its subsidiaries (collectively “Company”) and Taylor Technology Services, Inc., a Minnesota corporation (“Provider”). Company and Provider hereby agree as follows:
| 1. | SCOPE OF AGREEMENT. Provider will supply information technology services set forth on Exhibit A, Exhibit B and include, without limitation any and all other information technology or security services provided by Provider to Company (collectively, the “Services”), pursuant to the terms and conditions of this Agreement. |
| 2. | TERM / TERMINATION. This Agreement shall commence on Effective Date and continue for a period of two (2) years. Thereinafter, it will automatically renew for one year unless terminated in writing by either party. Provider may terminate this Agreement for any or no reason on thirty (30) days written notice to Company. Company may terminate this Agreement for any or no reason on one hundred and eighty (180) days notice. |
| 3. | FEES. The fees for the Services are set forth in Exhibit A. Provider may change the fees set forth in Exhibit A by providing thirty (30) days notice to Company. If Company does not agree to Service Provider’s change in fees as set forth in the notice, Company may terminate this Agreement by providing thirty (30) days written notice to Service Provider. |
| 4. | CONFIDENTIAL INFORMATION. Any information that parties receive or otherwise have access to incidental to or in connection with this Agreement (collectively, the “Confidential Information”), shall be and remain the property of the disclosing party. Confidential Information shall not include information which: (i) was in the possession of the Receiving Party at the time it was first disclosed by the Disclosing Party; (ii) was in the public domain at the time it was disclosed to the Receiving Party; (iii) enters the public domain through sources independent of the Receiving Party and through no breach of this provision by the Receiving Party; (iv) is made available by the Disclosing Party to a Third Party on an unrestricted, non-confidential basis; (v) was lawfully obtained by the Receiving Party from a Third Party not known by the Receiving Party to be under an obligation of confidentiality to the Disclosing Party; or (vi) was at any time developed by the Receiving Party independently of any disclosure by the Disclosing Party. Confidential Information may be used to the extent necessary to perform this Agreement and the parties shall not disclose Confidential Information to any third party, except to its agents (who have executed confidentiality agreements containing terms substantially similar to the terms of this Agreement) as necessary to provide the Services hereunder. In no event shall Company acquire any right, title or interest in and to any product or process information, including related know how, either existing or developed during the course of the business relationship with Provider and Company and in no event shall Provider acquire and right, title, or interest in and to any materials or information provided to it by Company. |
| 5. | COMPANY WARRANTIES. Company represents, warrants and covenants that it: (a) has retained and will retain backup copies of all data, information, and/or items stored by Provider as part of Company’s use of the Services; (b) has implemented and regularly maintains its own disaster recovery plan; (c) has obtained all necessary liability insurance to protect Company from losses, damages and third party claims related to the Services and this Agreement; (d) is responsible for ensuring the security and confidentiality of all identification and passwords; (e) shall use industry standard efforts to prevent unauthorized access to, or use of, the Services; (f) will notify Provider promptly upon discovery of any unauthorized use of the Services or any breach, or attempted breach, of security that could impact Provider systems, networks, environment, equipment or the Services; (g) will not introduce any virus or other code, program, or sub-program that damages or interferes with the operation of the Provider’s network or environment that or halts, disables, or interferes with Provider’s network, systems, equipment or operations. |
| 6. | INDEMNIFICATION. Company (the “Indemnitor”) shall indemnify, defend and hold harmless Provider and its affiliates (collectively, “Indemnitees”) and their respective officers, directors, employees, agents and subsidiaries from and against any and all claims, damages, liabilities, and expenses (including attorney fees) arising from any third-party claim based on Indemnitor’s (or its agent’s) breach of any representation, warranty, covenant, agreement, or obligation under this Agreement, or Indemnitor’s (or its agent’s) negligent and/or willful acts in carrying out its obligations under this Agreement. In order to avail itself of this indemnity provision, Indemnitee shall provide notice to Indemnitor of any such claim, tender the defense of the claim to Indemnitor, and reasonably cooperate with Indemnitor in the defense of the claim. |
| 7. | WAIVER OF CLAIMS AGAINST PROVIDER’S INSURANCE CARRIERS; NO INDEMNIFICATION IN FAVOR OF COMPANY. COMPANY UNDERSTANDS, ACKNOWLEDGES AND AGREES THAT PROVIDER IS NOT PROVIDING: (A) ACCESS TO OR COVERAGE UNDER ANY PROVIDER INSURANCE POLICY IN PERFORMING THE SERVICES; OR (B) ANY INDEMNIFICATION AGAINST THIRD PARTY CLAIMS OR OTHERWISE IN FAVOR OF COMPANY. ACCORDINGLY, COMPANY AND ITS AFFILIATES IRREVOCABLY WAIVE AND FORFEIT ANY CLAIM: (A) FOR INDEMNIFICATION BY PROVIDER AND (B) AGAINST PROVIDER’S INSURANCE CARRIERS AND ASSOCIATED INSURANCE POLICIES FOR ANY PROVIDER SERVICES PERFORMED OR PROVIDER’S BREACH OR ALLEGED BREACH OF THIS AGREEMENT. |
| 8. | NO PROVIDER WARRANTIES. THE SERVICES ARE PROVIDED WITHOUT WARRANTY OF ANY KIND, EXPRESS OR IMPLIED. COMPANY ACKNOWLEDGES THAT THE SERVICES ARE PROVIDED “AS IS” AND FURTHER ACKNOWLEDGES THAT PROVIDER DOES NOT WARRANT: (A) THE SERVICES WILL BE UNINTERRUPTED, OR ERROR FREE; (B) THE SERVICES ARE NOT VULNERABLE TO FRAUD, BREACH OF CONFIDENTIALITY OR UNAUTHORIZED USE; (C) THE SERVICES WILL BE AVAILABLE AT ANY TIME IN THE FUTURE. COMPANY IS RESPONSIBLE AND PROVIDER SHALL HAVE NO RESPONSIBILITY FOR DETERMINING THAT THE USE OF THE SERVICES COMPLIES WITH APPLICABLE LAWS IN THE JURISDICTION(S) IN WHICH COMPANY MAY DEPLOY AND USE THE SERVICES. |
| 9. | PROVIDER LIMITATION OF LIABILITY. PROVIDER WILL NOT BE LIABLE TO COMPANY OR ANY THIRD PARTY FOR: (A) DAMAGES BASED ON USE OR ACCESS, INTERRUPTION, DELAY OR INABILITY TO USE THE SERVICES, LOST REVENUES OR PROFITS, LOSS OF SERVICES, BUSINESS OR GOODWILL, LOSS OR CORRUPTION OF DATA, LOSS OF PERSONAL INFORMATION OR LOSS OF OR UNAUTHORIZED ACCESS TO CONFIDENTIAL INFORMATION, LOSS RESULTING FROM SERVICES FAILURE, MALFUNCTION OR SHUTDOWN, OR BREACHES IN PROVIDER’S SECURITY; OR (B) ANY INDIRECT, SPECIAL, INCIDENTAL, PUNATIVE OR CONSEQUENTIAL DAMAGES. THESE LIMITATIONS SHALL APPLY WHETHER OR NOT PROVIDER HAS BEEN ADVISED OF THE POSSIBILITY OF SUCH DAMAGES AND NOTWITHSTANDING ANY FAILURE OF ESSENTIAL PURPOSE OF ANY REMEDY. IN NO EVENT WILL PROVIDER’S LIABLITY EXCEED THE AMOUNT OF FEES ACTUALLY PAID FOR THE SERVICES IN THE THREE (3) MONTHS IMMEDIATELY PRECEDING THE CLAIM. COMPANY ACKNOWLEDGES AND AGREES THAT THE LIMITATIONS OF LIABILITIES FOR PROVIDER’S BENEFIT SET FORTH IN THIS AGREEMENT ARE REASONABLE AND PROVIDER WOULD NOT HAVE ENTERED INTO THE AGREEMENT OR PROVIDE ANY SERVICES WITHOUT SUCH LIMITATIONS. |
| 10. | NOTICE. Any notice sent pursuant to this Agreement shall be sent by certified mail, return receipt requested, or by overnight mail to the addresses below or to such address as either party may in the future designate. A copy of any notice to Provider shall be also sent to Taylor Technology Services, Inc., attn: General Counsel, 1725 Roe Crest Drive, North Mankato, Minnesota 56003 together with a copy of this Agreement. Notices shall be effective upon receipt. |
| 11. | ASSIGNMENT. Except as otherwise provided, this Agreement shall be binding upon and inure to the benefit of the parties’ successors and lawful assigns. |
| 12. | STATUS. Company and Provider are separate entities. Nothing in this Agreement shall be construed as creating an employer-employee or joint venture relationship. |
| 13. | THIRD PARTY BENEFICIARIES. Company acknowledges and agrees that Provider’s parent company, Taylor Corporation, Taylor Corporation’s subsidiaries, and Provider’s current and future insurance carriers shall be third party beneficiaries and entitled to all the rights and benefits hereof as if they were direct parties to the Agreement. |
| 14. | GOVERNING LAW. This Agreement shall be governed by the laws of the State of Minnesota, without reference to conflicts of law principles. Any legal suit, action or proceeding arising out of or relating to this Agreement shall be commenced in a federal court in Minnesota or in state court in the County of Nicollet, Minnesota, and the appellate courts thereof, and each party hereto irrevocably submits to the exclusive jurisdiction and venue of any such court in any such suit, action or proceeding. With respect to any litigation arising out of this Agreement, the parties expressly waive any right they may have to a jury trial and agree that any such litigation shall be tried by a judge without a jury and the prevailing party shall be entitled to recover its expenses, including reasonable attorney’s fees, from the other party. |
| 15. | SURVIVAL. In the event any provision of this Agreement is held by a tribunal of competent jurisdiction to be contrary to the law, the remaining provisions of this Agreement will remain in full force and effect. All sections herein relating to payment, confidentiality, indemnification, representations and warranties, limitations of liability, insurance waiver, third party beneficiaries, waiver of jury trial and provisions which by their terms extend beyond the Term shall survive the termination of this Agreement. |
| 16. | NO PRESUMPTION AGAINST DRAFTSPERSON. The parties acknowledge that each party and its counsel have participated in the negotiation and preparation of this Agreement. This Agreement shall be construed without regard to any presumption or other rule requiring construction against the party causing the Agreement to be drafted |
| 17. | INCONSISTENT DOCUMENTS INEFFECTIVE. No proposal, purchase order, order confirmation, acceptance, or any other document provided by either Party to the other, nor any electronic click-wrap, terms of use or similar online consent or acceptance language accompanying or set forth as a prerequisite to any electronic interface or utility associated with any Services, shall be deemed to amend the terms hereof and any such contradictory or additional terms shall be ineffective. |
| 18. | ENTIRE AGREEMENT. This Agreement sets forth the entire agreement and understanding among the parties as to the subject matter hereof, and merges and supersedes all prior discussions, agreements, and understandings of every and any nature among them. This Agreement shall be effective only when signed by all of the parties on the signature pages. No party shall be bound by any condition, definition, warranty, or representations, other than as expressly set forth or provided for in this Agreement, or as may be, on or subsequent to the date hereof set forth in writing and signed by the party to be bound thereby. This Agreement may not be amended, supplemented, changed, or modified, except by agreement in writing signed by the parties to be bound thereby. |
Company and Provider acknowledge, agree and specifically intend this Agreement to be in full force and effect as of the Effective Date and govern all Services rendered as of the Effective Date, irrespective of the date fully executed.
| Company: Envoy Medical Corporation | Provider: Taylor Technology Services, Inc. | |||
| By: | /s/ Brad Anderson | By: | /s/ Chad Sorensen | |
| Print Name: | Brad Anderson | Print Name: | Chad Sorensen |
| Title: | Sr Accountant | Title: | Vice President | |
| Date: | Mar 4, 2022 | Date: | Mar 7, 2022 | |
EXHIBIT A
IT Services to Be Provided
| 1. | This Exhibit A is subject to the terms and conditions contained in the Services Agreement with an Effective Date of January 1, 2022, hereby incorporated by reference. |
| 2. | Description of the Services (attach additional information, as necessary): |
See Services Description set forth on Exhibit B, incorporated by reference. Services also include, without limitation any and all information technology or security services performed by Provider.
| 3. | Fees: |
| 3.1. Monthly Fee: | $4900 | |
| 3.2. Hourly Rate: | $70.00/hour or per quotation |
| 4. | Billing: Provider will bill monthly. All fees are due 30 days from Company’s receipt of Provider’s invoice. |
EXHIBIT B
Services Summary
This Exhibit B is subject to the terms and conditions contained in the Services Agreement, with an Effective Date of January 1, 2022, hereby incorporated by reference.
| Service | Description | Service | Price ($) | |
| Data Services | ● | Circuit Ordering: we will place the orders for all MPLS circuits | Included in Security Operations Allocation | Security Operations Allocation - |
| ● | Circuit Support: we initiate calls with AT&T for any outage on the MPLS network | $27.73/month/device | ||
| ● | Router Support: we provide support for all WAN routers, including setting up prioritization for your critical applications | |||
| ● | Solarwinds Orion: this tool is used to monitor routers and core switches; it’s also available to monitor your network equipment’s utilization and to look for errors | |||
| ● | WAN Engineering: we are responsible for the design of the WAN and how companies connect | |||
| Communication Services | ● | Account management: we provide account creations, deletions, and modifications for Active Directory and Exchange | Included in Internal Server Support Allocation | N/A |
| ● | Active Directory Support: we manage all Active Directory Domain Controllers | |||
| ● | Exchange Support: we manage all of the Exchange Servers in the organization | |||
| ● | SPAM services that process our inbound and outbound email and block junk email | |||
| ● | Central Fax Service: we provide incoming and outgoing fax capabilities | |||
| ● | Microsoft Office 365 for Email and Teams | |||
| Encryption Services | ● | Vormetric File and Database Server file encryption | Included in Security | Security Operations |
| ● | Implementation of role based rights to decrypt / encrypt files | Operations Allocation | Allocation - $27.73/month/device | |
| ● | Centralized reporting on file read and writes for Vormetric protected files | |||
| Internet Services | ● | Cisco Firewall support: we support the Cisco units that are used for Branch Office VPN connections | Included in Security Operations Allocation | Security Operations Allocation - |
| ● | IDS/IPS: we provide support for the IDS units that we deployed for Payment Card Industry compliancy | $27.73/month/device | ||
| ● | Internet Firewall: we support all Internet Firewalls | |||
| ● | Symantec Antivirus: we manage all Symantec update servers | |||
| ● | Window Software Update Services: we manage all Windows update servers | |||
| ● | Web content filtering | |||
| ● | Client VPN Server Management: we manage the device that provides VPN access through webconnect.taylorcorp.com and connect.taylorcorp.com | |||
| Server and Storage Management | ● | We provide support to build servers you need for applications and manage them so your environment is up-to-date and protected | Internal Server Support Allocation | $140/month/server | |
| ● | Server management locations | ||||
| ○ | Internal Data Center | ||||
| ○ | Your facility | ||||
| ● | Server build | ||||
| ○ | Software installation | ||||
| ○ | Disk storage configuration | ||||
| ○ | Networking configuration | ||||
| ○ | Licensing procurement and assignment | ||||
| ○ | Security scan | ||||
| ○ | Application set-up (working in conjunction with your development teams) | ||||
| ○ | Back-up configuration | ||||
| ○ | Server monitoring configuration | ||||
| ○ | Server documentation | ||||
| ● | Server management | ||||
| ○ | Monthly patching | ||||
| ○ | Monitoring | ||||
| ○ | 24/7 alerting and incident response | ||||
| ○ | Daily and monthly back-ups per our Back-up Policy | ||||
| ○ | Anti-virus definition updates | ||||
| Domain and SSL Certificate Registrations and Renewals | ● | We process all Domain Registrations and Renewals for Taylor companies. | Costs vary depending on type of renewal and length of term | ||
| ● | We process all SSL Certificate Purchases and Renewals for Taylor companies. | ||||
Changes and Review Process
One of our goals for continuous process improvement is to examine any changes to our processes as we find better ways and systems to update our service levels. At that time, we will update this Service Description appropriately.
Exhibit 21.1
LIST OF SUBSIDIARIES OF ENVOY MEDICAL, INC.
| Entity Name | Jurisdiction of Incorporation | |
| Envoy Medical Corporation | Minnesota | |
| Envoy Medical GmbH (Ansbach) (GmbH) | Germany |
Exhibit 23.1
CONSENT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
We have issued our report dated March 28, 2025, with respect to the consolidated financial statements included in the Annual Report of Envoy Medical, Inc. on Form 10-K for the year ended December 31, 2024. We consent to the incorporation by reference of said report in the Registration Statements of Envoy Medical, Inc. on Form S-1 (File No. 333-276590). Form S-3 (File No. 333-282474) and on Form S-8 (File No. 333-278662).
/s/ GRANT THORNTON LLP
Fort Lauderdale, Florida
March 28, 2025
Exhibit 31.1
CERTIFICATION PURSUANT TO
RULES 13a-14(a) AND 15d-14(a) UNDER THE SECURITIES EXCHANGE ACT OF 1934,
AS ADOPTED PURSUANT TO SECTION 302 OF THE SARBANES-OXLEY ACT OF 2002
I, Brent T. Lucas, certify that:
| 1. | I have reviewed this Annual Report on Form 10-K of Envoy Medical, Inc.; |
| 2. | Based on my knowledge, this report does not contain any untrue statement of a material fact or omit to state a material fact necessary to make the statements made, in light of the circumstances under which such statements were made, not misleading with respect to the period covered by this report; |
| 3. | Based on my knowledge, the financial statements, and other financial information included in this report, fairly present in all material respects the financial condition, results of operations and cash flows of the registrant as of, and for, the periods presented in this report; |
| 4. | The registrant’s other certifying officer(s) and I are responsible for establishing and maintaining disclosure controls and procedures (as defined in Exchange Act Rules 13a-15(e) and 15d-15(e)) and internal control over financial reporting (as defined in Exchange Act Rules 13a-15(f) and 15d-15(f)) for the registrant and have: |
| (a) | Designed such disclosure controls and procedures, or caused such disclosure controls and procedures to be designed under our supervision, to ensure that material information relating to the registrant, including its consolidated subsidiaries, is made known to us by others within those entities, particularly during the period in which this report is being prepared; |
| (b) | Designed such internal control over financial reporting, or caused such internal control over financial reporting to be designed under our supervision, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles; |
| (c) | Evaluated the effectiveness of the registrant’s disclosure controls and procedures and presented in this report our conclusions about the effectiveness of the disclosure controls and procedures, as of the end of the period covered by this report based on such evaluation; and |
| (d) | Disclosed in this report any change in the registrant’s internal control over financial reporting that occurred during the registrant’s most recent fiscal quarter (the registrant’s fourth fiscal quarter in the case of an annual report) that has materially affected, or is reasonably likely to materially affect, the registrant’s internal control over financial reporting; and |
| 5. | The registrant’s other certifying officer(s) and I have disclosed, based on our most recent evaluation of internal control over financial reporting, to the registrant’s auditors and the audit committee of the registrant’s board of directors (or persons performing the equivalent functions): |
| (a) | All significant deficiencies and material weaknesses in the design or operation of internal control over financial reporting which are reasonably likely to adversely affect the registrant’s ability to record, process, summarize and report financial information; and |
| (b) | Any fraud, whether or not material, that involves management or other employees who have a significant role in the registrant’s internal control over financial reporting. |
| Date: March 28, 2025 | By: | /s/ Brent T. Lucas |
| Brent T. Lucas | ||
| Chief Executive Officer |
Exhibit 31.2
CERTIFICATION PURSUANT TO
RULES 13a-14(a) AND 15d-14(a) UNDER THE SECURITIES EXCHANGE ACT OF 1934,
AS ADOPTED PURSUANT TO SECTION 302 OF THE SARBANES-OXLEY ACT OF 2002
I, David R. Wells, certify that:
| 1. | I have reviewed this Annual Report on Form 10-K of Envoy Medical, Inc.; |
| 2. | Based on my knowledge, this report does not contain any untrue statement of a material fact or omit to state a material fact necessary to make the statements made, in light of the circumstances under which such statements were made, not misleading with respect to the period covered by this report; |
| 3. | Based on my knowledge, the financial statements, and other financial information included in this report, fairly present in all material respects the financial condition, results of operations and cash flows of the registrant as of, and for, the periods presented in this report; |
| 4. | The registrant’s other certifying officer(s) and I are responsible for establishing and maintaining disclosure controls and procedures (as defined in Exchange Act Rules 13a-15(e) and 15d-15(e)) and internal control over financial reporting (as defined in Exchange Act Rules 13a-15(f) and 15d-15(f)) for the registrant and have: |
| (a) | Designed such disclosure controls and procedures, or caused such disclosure controls and procedures to be designed under our supervision, to ensure that material information relating to the registrant, including its consolidated subsidiaries, is made known to us by others within those entities, particularly during the period in which this report is being prepared; |
| (b) | Designed such internal control over financial reporting, or caused such internal control over financial reporting to be designed under our supervision, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles; |
| (c) | Evaluated the effectiveness of the registrant’s disclosure controls and procedures and presented in this report our conclusions about the effectiveness of the disclosure controls and procedures, as of the end of the period covered by this report based on such evaluation; and |
| (d) | Disclosed in this report any change in the registrant’s internal control over financial reporting that occurred during the registrant’s most recent fiscal quarter (the registrant’s fourth fiscal quarter in the case of an annual report) that has materially affected, or is reasonably likely to materially affect, the registrant’s internal control over financial reporting; and |
| 5. | The registrant’s other certifying officer(s) and I have disclosed, based on our most recent evaluation of internal control over financial reporting, to the registrant’s auditors and the audit committee of the registrant’s board of directors (or persons performing the equivalent functions): |
| (a) | All significant deficiencies and material weaknesses in the design or operation of internal control over financial reporting which are reasonably likely to adversely affect the registrant’s ability to record, process, summarize and report financial information; and |
| (b) | Any fraud, whether or not material, that involves management or other employees who have a significant role in the registrant’s internal control over financial reporting. |
| Date: March 28, 2025 | By: | /s/ David R. Wells |
| David R. Wells | ||
| Chief Financial Officer |
Exhibit 32.1
CERTIFICATION PURSUANT TO
18 U.S.C. SECTION 1350, AS ADOPTED PURSUANT TO
SECTION 906 OF THE SARBANES-OXLEY ACT OF 2002
In connection with the Annual Report of Envoy Medical, Inc. (the “Company”) on Form 10-K for the period ending December 31, 2024 as filed with the Securities and Exchange Commission on the date hereof (the “Report”), I certify, in the capacity and on the date indicated below, pursuant to 18 U.S.C. § 1350, as adopted pursuant to § 906 of the Sarbanes-Oxley Act of 2002, that:
| (1) | The Report fully complies with the requirements of Section 13(a) or 15(d) of the Securities Exchange Act of 1934; and |
| (2) | The information contained in the Report fairly presents, in all material respects, the financial condition and results of operations of the Company. |
| Date: March 28, 2025 | By: | /s/ Brent T. Lucas |
| Brent T. Lucas | ||
| Chief Executive Officer |
Exhibit 32.2
CERTIFICATION PURSUANT TO
18 U.S.C. SECTION 1350, AS ADOPTED PURSUANT TO
SECTION 906 OF THE SARBANES-OXLEY ACT OF 2002
In connection with the Annual Report of Envoy Medical, Inc. (the “Company”) on Form 10-K for the period ending December 31, 2024 as filed with the Securities and Exchange Commission on the date hereof (the “Report”), I certify, in the capacity and on the date indicated below, pursuant to 18 U.S.C. § 1350, as adopted pursuant to § 906 of the Sarbanes-Oxley Act of 2002, that:
| (1) | The Report fully complies with the requirements of Section 13(a) or 15(d) of the Securities Exchange Act of 1934; and |
| (2) | The information contained in the Report fairly presents, in all material respects, the financial condition and results of operations of the Company. |
| Date: March 28, 2025 | By: | /s/ David R. Wells |
| David R. Wells | ||
| Chief Financial Officer |