Document
CERTAIN INFORMATION IDENTIFIED WITH [***] HAS BEEN EXCLUDED FROM THIS EXHIBIT BECAUSE IT IS BOTH (I) NOT MATERIAL AND (II) IS OF THE TYPE THAT THE REGISTRANT TREATS AS PRIVATE OR CONFIDENTIAL.
COLLABORATION AND LICENSE AGREEMENT
BETWEEN
SANOFI PASTEUR INC.
AND
NOVAVAX, INC.
Effective Date: May 10, 2024
TABLE OF CONTENTS
SCHEDULES AND EXHIBITS
COLLABORATION AND LICENSE AGREEMENT
This Collaboration and License Agreement (this “Agreement”) is made effective as of May 10, 2024 (the “Effective Date”), by and between Sanofi Pasteur Inc., a corporation organized under the laws of the State of Delaware, having offices located at 1 Discovery Drive, Swiftwater, PA 18370 (“Sanofi”), and Novavax, Inc., a corporation organized under the laws of the State of Delaware, having offices located at 700 Quince Orchard, Gaithersburg, MD 20878 (“Novavax”). Sanofi and Novavax each may be referred to herein individually as a “Party” or collectively as the “Parties.”
RECITALS
WHEREAS, Novavax is a global biopharmaceutical company that has expertise in research, development and commercialization of products directed to pathogenic organisms including SARS-CoV- 2;
WHEREAS, Sanofi is a global biopharmaceutical company that has expertise in all aspects of the exploitation of pharmaceutical products, including a business unit dedicated to vaccines;
WHEREAS, the Parties wish to collaborate to further develop, commercialize, and otherwise exploit products referred to herein, in accordance with the terms and conditions set forth herein; and
WHEREAS, concurrently with the execution of this Agreement, the Parties have executed and delivered a Securities Subscription Agreement.
NOW, THEREFORE, in consideration of the respective covenants, representations, warranties, and agreements set forth herein, the Parties hereto agree as follows:
ARTICLE 1: DEFINITIONS
For purposes of this Agreement, the following capitalized terms will have the following meanings:
1.1“1974 Convention” has the meaning set forth in Section
17.10 (Governing Law).
1.2“2024/2025 Season” means the period of time during which (a) Research, Development, Manufacturing, Regulatory Activities, Commercial, or Medical Activities or (b) any decision to be taken hereunder, is performed or taken, in each case ((a) and (b)), with respect to a product Directed To Variant(s) of SARS-CoV-2 recommended by VBRPAC in its annual SARS-CoV-2/COVID-19 strain selection meeting held in 2024.
1.3“2025/2026 Season” means the period of time during which (a) Research, Development, Manufacturing, Regulatory Activities, Commercial, or Medical Activities to be performed hereunder or (b) any decision to be taken hereunder, is performed or taken, in each case ((a) and (b)), with respect to a product Directed To Variant(s) of SARS-CoV-2 recommended by VBRPAC in its annual SARS-CoV-2/COVID-19 strain selection meeting held in 2025.
1.4“Accounting Standards” means, with respect to a Party or its Affiliate or Sublicensee, GAAP or IFRS, as such Person uses for its financial reporting standards from time to time, in each case, as consistently applied.
1.5“Acquired Party” has the meaning set forth in Section
1.54 (Change of Control).
1.6“Acquirer” means, in the context of a Change of Control, a Third Party or its Affiliate that acquires a Party.
1.7“Actions” has the meaning set forth in Section
17.11 (Jurisdiction; Venue; Service of Process).
1.8“Adjuvant” means Matrix-M.
1.9“Adjuvant Development Milestone Event” has the meaning set forth in Section
10.4.1 (Development Milestone).
1.10“Adjuvant Development Milestone Payment” has the meaning set forth in Section
10.4.1 (Development Milestone).
1.11“Adjuvant Field” means each Field that is not Unavailable as assessed in accordance with Section
2.4.3 (Procedures), excluding all Excluded Adjuvant Fields, unless otherwise agreed in writing by Novavax.
1.12“Adjuvant Intermediate Know-How” means that Know-How Controlled by Novavax or its Affiliates as of the Effective Date or during the Term that is actually used for the Manufacture of the Adjuvant Intermediates.
1.13“Adjuvant Intermediate Technology” means (a) the Adjuvant Intermediate Know-How, (b) Materials Controlled by Novavax or its Affiliates as of the Effective Date or at any time during the Term, including raw materials and intermediates, used by or on behalf of Novavax or its Affiliate for the process steps set forth on Schedule
1.13 for the Manufacture of Adjuvant Intermediates, and
(c) [***].
1.14“Adjuvant Intermediate Escrow Trigger Event” means the failure of Novavax to deliver (in conformance with specifications and all other requirements for delivery under the Adjuvant Supply Agreement), at least (a) [***] of all amounts of Adjuvant Intermediates that Novavax is required to deliver pursuant to the terms of with the Adjuvant Supply Agreement in any [***], or (b) [***] of all amounts of Adjuvant Intermediates that Novavax is required to deliver pursuant to the terms of the Adjuvant Supply Agreement during any [***] period during the Term.
1.15“Adjuvant Intermediate Technology Escrow” means an escrow of Adjuvant Intermediate Technology held in confidence by a Third Party escrow agent that is designated by the Parties in accordance with this Agreement, which technology will be released to Sanofi upon the occurrence of an Adjuvant Intermediate Escrow Trigger Event in accordance with the terms of this Agreement.
1.16“Adjuvant Intermediates” means [***].
1.17“Adjuvant Launch Milestone Event” has the meaning set forth in Section
10.4.2 (Launch Milestone).
1.18“Adjuvant Launch Milestone Payment” has the meaning set forth in Section
10.4.2 (Launch Milestone).
1.19“Adjuvant License” has the meaning set forth in Section
2.1.4 (Licensed Adjuvanted Products).
1.20“Adjuvant Manufacturing Handover Date” has the meaning set forth in Section
5.3 (Adjuvant Technology Transfer).
1.21“Adjuvant Manufacturing Technology Transfer” has the meaning set forth in Section
5.3 (Adjuvant Technology Transfer).
1.22“Adjuvant Manufacturing Technology Transfer Plan” has the meaning set forth in Section
5.3 (Adjuvant Technology Transfer).
1.23“Adjuvant Product” means any Licensed Adjuvanted Product or Novavax Adjuvant Product.
1.24“Adjuvant Royalty Term” means, on a country-by-country and Licensed Adjuvanted Product-by- Licensed Adjuvanted Product basis, the period beginning upon the First Commercial Sale of a Licensed Adjuvanted Product in a country and terminating upon the later of the date (a) that is twenty (20) years following First Commercial Sale of such Licensed Adjuvanted Product in such country and of (b) expiration of the last Valid Claim of a Novavax Patent that Covers (i) the composition of matter or method of use of the Adjuvant in such Licensed Adjuvanted Product or
(ii) the making, having made, using, offering for sale, selling, or importing of the Adjuvant in such Licensed Adjuvanted Product.
1.25“Adjuvant Supply Agreement” has the meaning set forth in Section
6.3.2 (Supply Agreements).
1.26“Adjuvant Technology Escrow” means an escrow of (a) the Novavax Adjuvant Manufacturing Know-How and (b) Novavax Adjuvant Materials, held in confidence by a Third Party escrow agent that is designated by the Parties in accordance with this Agreement, which technology will be released to Sanofi upon the occurrence of an Other Escrow Trigger Event in accordance with the terms of this Agreement.
1.27“Adjuvant Third Party Patents” has the meaning set forth in Section
2.6 (Adjuvant In-Licenses).
1.28“Affiliate” means, as of any point in time and for so long as such relationship continues to exist with respect to any Person, any other Person that controls, is controlled by, or is under common control with such Person. A Person will be regarded as in control of another Person if it (a) owns or controls, directly or indirectly, more than fifty percent (50%) of the equity securities of the subject Person entitled to vote in the election of directors (or, in the case of a Person that is not a corporation, for the election of the corresponding managing authority); or (b) possesses, directly or indirectly, the power to direct or cause the direction of the management or policies of such Person (whether through ownership of securities or other ownership interests, by contract, or otherwise).
1.29“Agreement” has the meaning set forth in the Preamble.
1.30“Alliance Manager” has the meaning set forth in Section
9.20.1 (Appointment).
1.31“Allowable Overruns” means, with respect to any invoice and a budget agreed by the Parties for any activity to be performed under this Agreement, the amounts in such invoice that are less than or equal to [***] of the amounts set forth in the applicable budget for such activity.
1.32“Ancillary Agreement” means any agreement contemplated under this Agreement to be entered into between the Parties or any of their Affiliates, including [***].
1.33“Annual Net Sales” mean, with respect to a Licensed Product, the aggregate Net Sales of such Licensed Product invoiced by all Selling Entities in a given Calendar Year.
1.34“Antigen” means any protein, peptide, or other molecule that induces an adaptive immune response specific to such protein, peptide, or other molecule.
1.35“APA” means each advanced purchase agreement and related ancillary agreements between Novavax and any Third Party for the Manufacture or supply of any Licensed COVID-19 Mono Product for, to or on behalf of such Third Party. All APAs in effect as of the Effective Date are set forth on Schedule
1.35.
1.36“Applicable Law” means all applicable laws, statutes, ordinances, rules, regulations, and other pronouncements having the effect of law of any federal, national, multinational, state, provincial, county, city, or other political subdivision, agency, or other body, domestic or foreign, including any applicable rules, regulations, binding guidelines, or other requirements of the Regulatory Authorities that may be in effect from time to time, including the FD&C Act, GCP, GLP, and GMP, anti-bribery laws, such as the United States Anti-Kickback Statute, the United States Foreign Corrupt Practices Act, and the UK Bribery Act, as well as all applicable data protection and privacy laws, rules, and regulations, including the United States Department of Health and Human Services privacy rules under the Health Insurance Portability and Accountability Act, as amended, and the Health Information Technology for Economic and Clinical Health Act and the EU General Data Protection Regulation 2016/679 of the European Parliament and of the Council of 27 April 2016 on the protection of natural persons with regard to the processing of personal data and on the free movement of such data, and repealing Directive 95/46/EC, along with other country-level data protection laws, as may be applicable.
1.37“Audited Party” has the meaning set forth in Section
10.16 (Financial Records; Audits).
1.38“Auditing Party” has the meaning set forth in Section
10.16 (Financial Records; Audits).
1.39“Auditor” has the meaning set forth in Section
10.16 (Financial Records; Audits).
1.40“Bankrupt Party” has the meaning set forth in Section
16.4 (Bankruptcy).
1.41“Bankruptcy Code” has the meaning set forth in Section
16.4 (Bankruptcy).
1.42“Baseball Arbitration Procedure” has the meaning set forth in Section
9.18.5(d) (Budget Status Quo).
1.43“Biosimilar Product” means, with respect to a particular Licensed COVID-19 Product in a particular country, a pharmaceutical product that (a) is marketed by a Person other than Sanofi, Novavax, or their respective Affiliates or licensees or Sublicensees for the same Indication as such Licensed COVID-19 Product, and such Person did not purchase such product in a chain of distribution that included any of Sanofi or its Affiliates or Sublicensees, and that (b) (i) has been licensed as a biosimilar, bioequivalent or interchangeable product by the FDA under Section 351(k)
of the Public Health Service Act (42 U.S.C. § 201 et seq.), as amended, (ii) has been licensed as a similar biological medicinal product by the EMA as described in Directive 2001/83/EC, as may be amended (including by EU Directive 2004/27/EC), or any subsequent or superseding law, statute or regulation, or (iii) has received marketing authorization as a biosimilar, bioequivalent or interchangeable product by a Governmental Authority under any equivalent law, statute, or regulation in such jurisdiction; in each case ((b)(i)-(iii)), where the Licensed COVID-19 Product is the reference product for purposes of such licensure or marketing authorization.
1.44“BLA” means a biologics license application submitted to the FDA as defined in the Public Health Service Act and 21 C.F.R. 601.2 (including all additions, supplements, extensions, and modifications thereto).
1.45“BMF” means a biologics master file and all equivalents, and related proprietary dossiers, in any country or jurisdiction in the Territory for a biologic substance submitted (or to be submitted) by a Party to Regulatory Authorities.
1.46“Breaching Party” has the meaning set forth in Section
16.2.2(a) (Material Breach).
1.47“Budget Status Quo Matter” has the meaning set forth in Section
9.18.5(d)(ii) (Budget Status Quo).
1.48“Business Day” means a day on which the commercial banks in Gaithersburg, MD, U.S.A. or Swiftwater, Pennsylvania, U.S.A. are open for commercial banking business, other than a Saturday or a Sunday.
1.49“Calendar Quarter” means the respective periods of three (3) consecutive calendar months ending on March 31, June 30, September 30, or December 31, during the Term, or the applicable part thereof during the first or last calendar quarter of the Term.
1.50“Calendar Year” means any year commencing on January 1 and ending on December 31, or the applicable part thereof during the first or last year of the Term.
1.51“Cancellation Fees” means all monetary consideration received by any Selling Entity resulting from the cancellation of a purchase order or other binding purchase commitment for a Licensed Product by a customer of any Selling Entity.
1.52“CDA” means that Confidential Disclosure Agreement, by and between Sanofi and Novavax, dated [***].
1.53“CEPI Arrangements” means all contracts existing as of the Effective Date by and between Novavax and the Coalition for Epidemic Preparedness Innovations (CEPI), including the Outbreak Response Funding Agreements, COVAX Marketplace User Agreement and any amendments thereof, in each case relating to COVID-19.
1.54“Change of Control” means, [***].
1.55“CIC License” has the meaning set forth in Section
2.1.2 (COVID-19 and Influenza Combination Products).
1.56“CIC Product” means any Licensed CIC Product or Novavax CIC Product.
1.57“Clinical Trial” means any clinical investigation conducted on human subjects, as that term is defined in FDA regulations at 21 C.F.R. § 312.3, or a similar clinical investigation conducted on human subjects, as defined under Applicable Law outside the United States. Without limiting the foregoing, “Clinical Trial” includes any Phase 1 Clinical Trial, Phase 2 Clinical Trial, Phase 3 Clinical Trial, and Pivotal Clinical Trial.
1.58“CMC” means chemistry, manufacturing, and controls.
1.59“CMO” means a contract manufacturing organization.
1.60“Co-Exclusive” means, with respect to the grant of the COVID-19 Mono License hereunder by Novavax to Sanofi, that Sanofi, its Affiliates, and its Sublicensees shall be the only Persons who may practice the Licensed Novavax Assets to Exploit Licensed COVID-19 Mono Products other than: (a) Novavax and its Affiliates, in the Collaboration COVID-19 Territory and (b) Novavax, its Affiliates, and Third Parties under an Existing Strategic Partner Agreement in the Existing Partner Non-Exclusive Territory during the term of such Existing Strategic Partner Agreement, in each case ((a) and (b)), only as expressly permitted by this Agreement.
1.61“Collaboration COVID-19 Territory” means, with respect to any Licensed COVID-19 Mono Product, any country or region of the world other than Existing Partner Exclusive Territory.
1.62“Collaboration Term” means the period commencing on the Effective Date and ending upon the earlier of (a) termination of the COVID-19 Mono License under this Agreement, and (b) termination of this Agreement in its entirety.
1.63“Commercialize,” “Commercialized,” or “Commercializing” means, in respect of a drug product, to (a) (i) market, advertise, promote, detail, distribute, offer for sale, and sell, and (ii) import, or export, in each case, in connection with the activities described in clause (i) above; (b) conduct activities, other than Development, Medical Activities, and Manufacturing, in preparation for the foregoing activities, including obtaining Pricing Approval and interacting with Regulatory Authorities with respect to the foregoing; (c) conduct Post-Marketing Requirements; or (d) interact with recommending Governmental Authorities, such as local National Immunization Technical Advisory Groups, and supranational health organizations, such as World Health Organization, United Nations International Children’s Emergency Fund (UNICEF), Pan American Health Organization, Gavi, etc. When used as a noun, “Commercialization” means any activities involved in Commercializing. The term “Commercialize” does not include Medical Activities, Development activities, Manufacturing activities, or Regulatory Activities.
1.64“Commercially Reasonable Efforts” means, [***].
1.65“Committee” means the JSC and each Subcommittee.
1.66“Committee Dispute” has the meaning set forth in Section
9.18.3 (Decisions of the JSC).
1.67“Committee Matters” has the meaning set forth in Section
9.17 (Limitations of Committee Authority).
1.68“Confidential Information” means, with respect to a Disclosing Party, all non-public or proprietary information (including information communicated in oral, written, visual, graphic, or electronic form) that is disclosed or made available by or on behalf of such Party to the other Party pursuant to this Agreement, regardless of whether any of the foregoing are marked “confidential” or “proprietary”.
1.69“Control” or “Controlled” means with respect to a Party, other than pursuant to this Agreement,
(a) with respect to Materials, the legal authority or right to physical possession of such materials, with the right to provide such materials to the other Party on the terms set forth herein, and (b) with respect to Patents, Marketing Authorizations, Regulatory Filings, Know-How, or other intellectual property rights, the legal authority or right to grant a license, access or other right to use (as applicable) to the other Party under such Patents, Marketing Authorizations, Regulatory Filings, Know-How, or other intellectual property rights on the terms set forth herein, in each case (a) and (b), without breaching or otherwise violating the terms of any arrangement or agreement with a Third Party in existence as of the time such Party or its Affiliates would first be required hereunder to grant the other Party such access, right to use, licenses, or sublicense, provided that any Materials, Patents, Marketing Authorizations, Regulatory Filings, Know-How, or other intellectual property, as applicable, that comes into such Party’s or its Affiliates’ control as a result of a Change of Control of such Party, will not be deemed Controlled by such Party for purposes of this Agreement, except to the extent, and only to the extent, that such Materials, Patents, Marketing Authorizations, Regulatory Filings, Know-How, or other intellectual property, as applicable, are
(i) developed, acquired, or otherwise Controlled by the Acquirer pursuant to or in connection with a license or other agreement between the Acquirer or any of its Affiliates, on the one hand, and such Party, on the other hand, (ii) developed or acquired by the Acquirer following such Change of Control with the use of or access to, or otherwise incorporates, is derived from, or based upon, the subject matter used or made available by the Acquirer under this Agreement, (iii) actually used by such Party or its Affiliates, or the Acquirer, in the performance of activities under this Agreement or (iv) developed, acquired, or otherwise Controlled by the Acquirer in the course of or as a result of performing its Affiliate’s obligations or exercising its Affiliate’s rights under this Agreement.
1.70“Cover”, “Covered”, or “Coverage” means, in reference to a particular subject matter (such as a composition of matter, method, or process) and a Patent in a particular country or other jurisdiction, that such subject matter meets or practices all elements or limitations of at least one (1) claim (as
interpreted under principles of patent law in such country or jurisdiction or, in the case of applications filed under the Patent Cooperation Treaty, in the jurisdiction of the international searching authority) of such Patent.
1.71“COVID-19” means coronavirus disease, an infectious disease caused by the SARS-CoV-2 virus.
1.72“COVID-19 Development Milestone Event” has the meaning set forth in Section
10.3.1 (Development Milestone).
1.73“COVID-19 Development Milestone Payment” has the meaning set forth in Section
10.3.1 (Development Milestone).
1.74“COVID-19 Launch Milestone Event” has the meaning set forth in Section
10.3.2 (Launch Milestone).
1.75“COVID-19 Launch Milestone Payment” has the meaning set forth in Section
10.3.2 (Launch Milestone).
1.76“COVID-19 Manufacturing Handover Date” has the meaning set forth in Section
5.2.1 (COVID- 19 Manufacturing Handover).
1.77“COVID-19 Manufacturing Technology Transfer” has the meaning set forth in Section
5.2.1 (General).
1.78“COVID-19 Manufacturing Technology Transfer Plan” has the meaning set forth in Section
1.79“COVID-19 Mono License” has the meaning set forth in Section
2.1.1 (Licensed COVID-19 Mono Products).
1.80“COVID-19 Research and Development Budget” has the meaning set forth in Section
3.2.2 (COVID-19 Research and Development Budget).
1.81“COVID-19 Research and Development Budget Forecast” has the meaning set forth in Section
3.2.2 (COVID-19 Research and Development Budget).
1.82“COVID-19 Research and Development Plan” has the meaning set forth in Section
3.2.1 (COVID-19 Research and Development Plan).
1.83“COVID-19 Research and Development Plan Costs” means FTE Costs and Out-of-Pocket Costs incurred by or on behalf of a Party or its Affiliates after the Effective Date in the course of performing (a) Research or Development activities or (b) the specific Regulatory Activities or Medical Activities, in each case ((a) and (b)), included in the COVID-19 Research and Development Plan.
1.84“COVID-19 Royalty Term” means, on a country-by-country and Licensed COVID-19 Product- by-Licensed COVID-19 Product basis, the period beginning upon the First Commercial Sale of a Licensed COVID-19 Product in a country and terminating upon the latest of (a) expiration of the last Valid Claim of (i) a Novavax Patent or (ii) a Joint Arising Patent in such country that Covers, in whole or in part, the composition of matter or a method of use (of an Indication that has received Marketing Authorization) of such Licensed COVID-19 Product or the making, having made, using,
offering for sale, selling, or importing such Licensed COVID-19 Product; (b) expiration of all Regulatory Exclusivity applicable to such Licensed COVID-19 Product in such country; and (c) ten (10) years following First Commercial Sale of such Licensed COVID-19 Product in such country.
1.85“COVID-19 Supply Agreement” has the meaning set forth in Section
6.3.1 (Supply Agreements).
1.86“COVID-19 Technology Escrow” means an escrow of (a) the Novavax COVID-19 Manufacturing Know-How and (b) Novavax COVID-19 Materials, held in confidence by a Third Party escrow agent that is designated by the Parties in accordance with this Agreement, which technology will be released to Sanofi upon the occurrence of an Other Escrow Trigger Event in accordance with the terms of this Agreement.
1.87“Cure Period” has the meaning set forth in Section
16.2.2(a) (Material Breach).
1.88“Data” means all information and results, whether in raw or aggregate form, including preclinical data, in vitro and in vivo data, in silico data, clinical data, regulatory, biological, chemical, pharmacological, toxicological, pharmaceutical, physical, analytical, safety, and quality control data.
1.89[***].
1.90“Development” means, with respect to any drug product or drug substance contained therein or raw material or intermediate thereof, all development activities directed towards obtaining and maintaining Marketing Authorization of such drug product, including (a) the conduct of Clinical Trials, including all protocol and investigator brochure development; (b) test and assay method development and qualification; (c) process or formulation development; (d) delivery system development; (e) quality assurance and quality control development; and (f) statistical analysis in connection with any of the foregoing. “Development” does not include any Research activities, Commercialization activities, Medical Activities, Manufacturing activities, or Regulatory Activities. “Developing” means the act of performing Development and to “Develop” means to perform Development.
1.91“Directed To” and “Directed” means, with respect to an Antigen and a pathogen, organism, or disease, that such Antigen is designed to (a) in the case of a pathogen or organism, elicit a specific immune response to such pathogen or organism, and (b) in the case of a disease, elicit a prophylactic or therapeutic effect with respect to such disease.
1.92“Disclosing Party” has the meaning set forth in Section
11.1 (Confidentiality).
1.93“Dispute” has the meaning set forth in Section
17.12 (Dispute Resolution).
1.94“Distinct” means, with respect to a first Licensed Adjuvanted Product and a second Licensed Adjuvanted Product, that such first Licensed Adjuvanted Product is Directed To one (1) or more Antigen(s) from a different species of a pathogen or organism as compared to such second Licensed Adjuvanted Product (i.e., that the first Licensed Product and the second Licensed Product do not contain Antigens Directed To identical pathogens, organisms, or diseases). [***].
1.95“Distributor” means a Person who distributes, offers for sale, and sells Licensed Products in the Territory (with or without packaging rights), in circumstances where the Person purchases its requirements of Licensed Products from a Party or its Affiliates or Sublicensees pursuant to a written agreement but does not otherwise make any royalty or other payment to such Party or its Affiliates with respect to any proprietary rights in or to such Licensed Product, other than a license to use any trademarks in connection with the distribution, marketing, or sale of the Licensed Products. The term “packaging rights” in this Section
1.95 (Distributor) means the right for the Distributor to package and label Licensed Products supplied in unpackaged bulk form into individual ready-for-sale packs.
1.96“DMF” means a drug master file and all equivalents, and related proprietary dossiers, in any country or jurisdiction in the Territory for a drug substance submitted (or to be submitted) by a Party to Regulatory Authorities.
1.97“Dollars” means [***].
1.98“Effective Date” has the meaning set forth in the Preamble.
1.99“EMA” means the European Medicines Agency and any successor entity thereto.
1.100“Emergency Use Authorization” or “EUA” means a conditional Marketing Authorization granted pursuant to Section 564 of the FD&C Act to expedite access to medicines to address public health emergencies.
1.101“Enforcement Action” means, with respect to any Patent, Know-How, or trademark, any action, including bringing, maintaining, and terminating any suit, claim, or action against any Third Party engaged in any existing, alleged, or threated Infringement (with respect to Patents and trademarks), misappropriation, or other violation.
1.102“European Commission” means the European Commission or any successor entity thereto.
1.103“European Union” or “EU” means the countries of the European Economic Area, as it is constituted on the Effective date and as it may be modified from time to time after the Effective date.
1.104“Excluded Adjuvant Field” means (a) all Fields for (i) Influenza and (ii) SARS-CoV-2, and (b) any other Field that is Unavailable; provided that during the pendency of a Pandemic declared for a pathogen, organism, or disease that is not SARS-CoV-2, no Field for such pathogen, organism, or disease, regardless of whether Novavax has, as of the commencement of such Pandemic, granted exclusive rights to a Third Party (i.e., for any Field that is Unavailable due to operation of clause
(b) of the definition thereof), shall be deemed Excluded Adjuvant Fields. For clarity, nothing in this Section
1.104 (“Excluded Adjuvant Fields”) is intended to limit the COVID-19 Mono License, the CIC License, or the OC License.
1.105“Existing Novavax Patents” has the meaning set forth in Section
1.203 (Novavax Patent).
1.106“Existing Partner Exclusive Territory” means any country or region of the world where Novavax has, as of the Effective Date, granted a Third Party exclusive rights to Manufacture and Commercialize the Licensed COVID-19 Mono Product pursuant to an Existing Strategic Partner Agreement, provided that on the date of termination or expiration of all Existing Strategic Partner Agreements for a particular country in the Existing Partner Exclusive Territory, such country will cease to be included in the Existing Partner Exclusive Territory and such country will become included in the Collaboration COVID-19 Territory. Schedule
1.106 sets forth all countries and regions in the Existing Partner Exclusive Territory as of the Effective Date.
1.107“Existing Partner Non-Exclusive Territory” means any country or region of the world where Novavax has, as of the Effective Date, granted a Third Party non-exclusive rights to Manufacture and Commercialize the Licensed COVID-19 Mono Product pursuant to an Existing Strategic Partner Agreement, provided that on the effective date of termination or expiration of all Existing Strategic Partner Agreements for a particular country in the Existing Partner Non-Exclusive Territory, such country will cease to be included in the Existing Partner Non-Exclusive Territory. Schedule
1.107 sets forth all countries and regions in the Existing Partner Non-Exclusive Territory as of the Effective Date.
1.108“Existing Settlement Arrangements” has the meaning set forth in Section
1.270 (Settlement Arrangements).
1.109“Existing Strategic Partner Agreement” means any legally binding contract between Novavax and a Third Party (each such Third Party, a “Strategic Partner”) granting Manufacturing and Commercialization rights for Licensed COVID-19 Mono Products in a country or region to such Third Party, but excluding all APAs. Schedule
1.109 sets forth all Existing Strategic Partner Agreements in effect as of the Effective Date.
1.110“Existing Suppliers” means all suppliers of Materials for the Manufacturing of Licensed COVID- 19 Mono Products as of the Effective Date and any new suppliers engaged in accordance with Section
14.4.4 (New Arrangements).
1.111“Existing Supply Agreements” means the contracts between Novavax and Existing Suppliers that are material to the Manufacturing of Licensed COVID-19 Mono Products and for Materials that are not available on commercially available standard terms. The Existing Supply Agreements as of the Effective Date are set forth on Schedule
1.111 (Existing Supply Agreements).
1.112“Expert Budget Decision” has the meaning set forth in Section
9.18.5(d)(ii) (Budget Status Quo).
1.113“Exploit” means, with respect to a drug product or any drug substance contained therein or raw material or intermediate thereof, to use, have used, Research, have Researched, Develop, have Developed, Manufacture, have Manufactured, Commercialize, have Commercialized, import, export, transport, conduct Medical Activities with respect to, or otherwise make full use of such compound or product, and “Exploitation” means the act of exploiting such compound or product.
1.114“Extended Review Period” has the meaning set forth in Section
11.7.3(a) (Review Process).
1.115“Falsified Medicine” has the meaning set forth in Section
13.7 (Falsified Medicines).
1.116“FD&C Act” means the United States Federal Food, Drug, and Cosmetic Act, as amended, and the rules and regulations promulgated thereunder.
1.117“FDA” means the United States Food and Drug Administration and any successor entity thereto.
1.118“Field” means (a) with respect to a particular pathogen or organism, the treatment, diagnosis, or prevention of disease or medical condition in humans caused by or associated with infection with such pathogen or organism, and (b) in the case of targets other than a pathogen or organism, the treatment, diagnosis, or prevention of a particular Indication associated with such target.
1.119“Financial Records” has the meaning set forth in Section
10.16 (Financial Records; Audits).
1.120“First Commercial Sale” means with respect to a Licensed Product, the first sale of such Licensed Product by Sanofi, its Affiliate, or its Sublicensee to a Third Party, which sale is intended for end use or consumption, that results in Net Sales in a particular country and occurs after all required Marketing Authorizations, and if required for reimbursement in such country, Pricing Approvals, for the Licensed Product has been obtained in such country; provided that the following will not constitute a First Commercial Sale: (a) any sale or other disposition of a Licensed Product by Sanofi to any of its Affiliates or Sublicensees; (b) any sale or other disposition of a Licensed Product solely for use in Research or Development; (c) the disposal or transfer of a Licensed Product for a bona fide charitable purpose or disposition for no profit; or (d) sale or other disposition of a Licensed Product for compassionate use, “treatment IND sales”, “named patient sales”, “expanded access sales”, “right to try sales”, or similar, in each case, for no profit.
1.121“Force Majeure” means a condition, the occurrence and continuation of which is beyond the reasonable control of a Party, including an act of God, governmental acts, omissions or restrictions, embargoes, shortages, acts of war or terrorism, civil commotion, labor strike or lock-out, epidemic, pandemic, flood, failure or default of public utilities or common carriers, or destruction of production facilities or materials by fire, earthquake, explosion, storm, or like catastrophe.
1.122[***].
1.123[***].
1.124“FTE” means [***] hours of work per annum devoted to or in support of the Research or other Development activities, Technology Transfer activities, Manufacturing activities, Regulatory Activities, Medical Activities, or Commercialization activities, in each case, that are carried out by one or more qualified scientific or technical employees (excluding Service Providers) of either Party or any of its Affiliates, or its or their Sublicensees, as applicable. Notwithstanding the foregoing, the time of a single individual will not account for more than one (1) FTE for a given Calendar Year.
1.125“FTE Costs” means, for any period, the FTE Rate multiplied by the number of FTEs who perform a specified activity under this Agreement. FTEs will be calculated on a prorated basis, if necessary, based on the time spent performing the applicable activity.
1.126“FTE Rate” means [***] per annum, which includes (a) all wages and salaries, employee benefits, bonus, travel and entertainment, supplies, and other direct expenses; and (b) indirect allocations, including all general and administrative expenses, human resources, finance, occupancy, and depreciation, in each case ((a) and (b)), expended in connection with relevant activities. The FTE Rate will be increased on January 1 of each Calendar Year beginning in 2025 in accordance with the percentage year-over- year increase based on the then-current CORE CPI-US-U price index published by the U.S. Bureau of Labor Statistics.
1.127“Gatekeeper” has the meaning set forth in Section
2.4.2 (Appointment).
1.128“GAVI Arrangements” means the Termination and Settlement Agreement by and between Novavax and Gavi Alliance, dated February 16, 2024, and including any agreements with UNICEF and PAHO to effectuate the sales of Licensed COVID-19 Mono Products, Novavax CIC Products, or Novavax OC Products under such Termination and Settlement Agreement.
1.129“GCP” means good clinical practices, which are the then-current standards for Clinical Trials for pharmaceuticals, as set forth in the FD&C Act, ICH Guideline Q7A, or other Applicable Law, and such standards of good clinical practice as are required by the Regulatory Authorities of the European Union and other organizations and Governmental Authorities in countries for which the applicable Licensed Product is intended to be Developed, to the extent such standards are not less stringent than United States standards or ICH Guidelines.
1.130“GLP” means the then-current good laboratory practice standards promulgated or endorsed by the FDA as defined in 21 C.F.R. Part 58, or the successor thereto, or comparable regulatory standards in jurisdictions outside of the United States as they may be updated from time to time, to the extent such standards are not less stringent than United States standards.
1.131“GMP” means the then-current Good Manufacturing Practices as specified in Applicable Law, including the United States Code of Federal Regulations, ICH Guideline Q7A, or equivalent laws, rules or regulations of an applicable Regulatory Authority at the time of manufacture.
1.132“Governmental Authority” means any court, agency, department, authority, or other instrumentality of any national, federal, state, county, city, or other political subdivision, including any relevant Regulatory Authority.
1.133“HCP” means a health care practitioner, such as physicians, pharmacists, nurse practitioners, or other health professionals having prescribing or purchasing authority or who is able to influence prescribing or purchasing decisions. Whether a specific individual is considered as an HCP in any jurisdiction shall be determined in accordance with Applicable Law in such jurisdiction.
1.134“ICH” means the International Council on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use.
1.135“IND” means any Investigational New Drug application (including any amendment or supplement thereto) filed with the FDA pursuant to Part 312 of Title 21 of the U.S. Code of Federal Regulations, including any amendments thereto or, if applicable, a comparable application or submission filed with a Regulatory Authority outside the U.S. for the investigation of any product in any other country or group of countries (such as a Clinical Trial Application in the EU).
1.136“IND Clearance” means, with respect to a Novavax Adjuvant Product, the earlier of (a) in the U.S., the date that is thirty (30) days following the filing of the first IND for such Novavax Adjuvant Product, if Novavax has not received any notice of a clinical hold or any other administrative delay from the FDA during such thirty (30) day period, and if Novavax or its Affiliates or Sublicensee does receive a notice of a clinical hold or there is such other administrative delay, then the date of “IND Clearance” for the Novavax Adjuvant Product will be the date on which the FDA lifts such clinical hold or such other administrative delay is otherwise resolved and the FDA first allows the Novavax Adjuvant Product to be administered to a human as described in such IND and (b) in other regulatory jurisdictions outside the U.S., the date on which such Novavax Adjuvant Product is first permitted by the applicable Regulatory Authority of such jurisdiction to be administered to a human
pursuant to an IND filing in accordance with Applicable Laws (or, to the extent no IND filing is required under Applicable Law in such regulatory jurisdiction, the date of initiation of the first Clinical Trial in such jurisdiction using such Novavax Adjuvant Product).
1.137“Indemnified Party” has the meaning set forth in Section
15.1.3 (Procedure).
1.138“Indemnifying Party” has the meaning set forth in Section
15.1.3 (Procedure).
1.139“Indication” means, with respect to a product, a distinct disease, or medical condition in humans that such product is designed to address, such that a Regulatory Authority would approve such disease or medical condition as a discrete claim (as opposed to a variant or subdivision or subset of a claim) in the labeling of such product based on the results of a separate Clinical Trial(s) sufficient to support Marketing Authorization of such claim. The following will each be deemed to be part of the same Indication: (a) different diseases or medical condition with the same etiology, (b) different stages of disease (e.g., localized vs. systemic for the same disease or medical condition, etc.); (c) different biomarker status with respect to the same disease or medical condition; (d) variants or subdivisions or subclassifications of such disease or medical condition, including Variants of a pathogen or organism; or (e) treatment, modulation, and prophylaxis of a disease or medical condition in a different patient population or sub-population.
1.140“Influenza” means all species of influenza viruses in the Orthomyxoviridae family, including the A/H1N1 strains, A/H3N2 strains, and B-Victoria lineage and any other strains or Variants thereof.
1.141“Invalidity Action” means, with respect to a Patent, any action, claim, or demand brought by a Third Party, including a declaratory judgment action, opposition, inter partes review or other post- grant proceeding, alleging that such Patent is invalid or unenforceable.
1.142“JCC” has the meaning set forth in Section
9.5.1 (Formation).
1.143“JFC” has the meaning set forth in Section
9.7.1 (Formation).
1.144“JMAC” has the meaning set forth in Section
9.6.1 (Formation).
1.145“JMSC” has the meaning set forth in Section
9.4.1 (Formation).
1.146“Joint Arising Patent” has the meaning set forth in Section
12.1.3 (Joint Ownership).
1.147“JPC” has the meaning set forth in Section
9.8.1 (Formation).
1.148“JRDC” has the meaning set forth in Section
9.3.1 (Formation).
1.149“JSC” has the meaning set forth in Section
9.2.1 (Formation).
1.150“Know-How” means all non-public technical or scientific information, including Data, chemical structures, chemical sequences, formulas, methods, processes, procedures, practices, protocols, techniques, discoveries, inventions, specifications, designs, trade secrets, and supply chain sources, including any of the foregoing included or referenced in Regulatory Filings, and in each case, whether patentable or not, and in any tangible or intangible form whatsoever.
1.151“Knowledge Portal” has the meaning set forth in Section
5.1.1 (Transfer Method).
1.152“Liability” has the meaning set forth in Section
15.1.1 (Indemnification by Sanofi).
1.153“Licensed Adjuvanted Product” means a Proposed Adjuvanted Product in any dosage, strength, formulation, or presentation designed to treat any disease or condition that has been confirmed to be in the Adjuvant Field in accordance with Section
2.4 (Adjuvant Field). For clarity, Licensed Adjuvanted Product excludes Licensed COVID-19 Products and Novavax Products.
1.154“Licensed CIC Product” means any pharmaceutical product Exploited by Sanofi or its Affiliates or Sublicensees in any dosage, strength, formulation, or presentation that contains (a) a Licensed COVID-19 Component, (b) the Adjuvant, and (c) one (1) or more Antigens owned by, or licensed to, Sanofi or its Affiliate or Sublicensee and Directed To Influenza (excluding the Novavax Influenza Component), with or without any other Antigen Directed To any pathogen(s) other than SARS-CoV-2 and Influenza. For clarity, Licensed CIC Product excludes Licensed COVID-19 Mono Products, Licensed OC Products, Licensed Adjuvanted Products, and all Novavax Products.
1.155“Licensed COVID-19 Component” means (a) all Antigens Directed To SARS-CoV-2 formulated with any polysorbate to form a nanoparticle, and (b) all derivatives or modifications of the molecules of clause (a) generated by or on behalf of Novavax or Sanofi in the performance of Research and Development activities in accordance with this Agreement.
1.156“Licensed COVID-19 Mono Product” means any pharmaceutical product in any dosage, strength, formulation, or presentation that contains (a) a Licensed COVID-19 Component and (b) the Adjuvant, and does not include any Antigen Directed To any pathogen or organism other than SARS-CoV-2 and that is not for the prevention or prophylaxis of any disease other than COVID-
19. For clarity, Licensed COVID-19 Mono Product excludes Licensed CIC Products, Licensed OC Products and Licensed Adjuvanted Products, and all Novavax Products.
1.157“Licensed COVID-19 Mono Product Commercialization Framework” has the meaning set forth in Section
7.2 (Licensed COVID-19 Mono Product Commercialization Framework).
1.158“Licensed COVID-19 Product” means any Licensed COVID-19 Mono Product, Licensed CIC Product, or Licensed OC Product.
1.159“Licensed Novavax Assets” means collectively the Novavax Patents, Novavax Know-How, and Novavax Materials.
1.160“Licensed OC Product” means any pharmaceutical product Exploited by Sanofi or its Affiliates or Sublicensees in any dosage, strength, formulation, or presentation that contains (a) a Licensed COVID-19 Component, (b) the Adjuvant, and (c) at least one (1) Antigen Directed To any pathogen(s) other than SARS-CoV-2 or Influenza. For clarity, Licensed OC Product excludes Licensed COVID-19 Mono Products, Licensed CIC Products, Licensed Adjuvanted Products and all Novavax Products.
1.161“Licensed Product” means any (a) Licensed Adjuvanted Product or (b) Licensed COVID-19 Product.
1.162“Licensed Product Mark” means trademarks, designs, copyrights, domain names, trade dress, trade names, positioning, messages, logo, colors, and other visual branding elements a Party utilizes in connection with the Commercialization of Licensed COVID-19 Mono Product.
1.163“Major Markets” means [***].
1.164“Manufacture” or “Manufactured” or “Manufacturing” means with respect to a drug product or any drug substance contained therein or raw material or intermediate thereof, those activities directed to making, producing, manufacturing, processing, formulating, filling, finishing, packaging, labeling, conducting quality control testing and quality assurance release activities, supplying, shipping, or storing of such compound or product or any raw material or intermediate thereof. Manufacture excludes all Medical Activities, Commercialization activities, Research activities, Development activities, and Regulatory Activities.
1.165“Manufacturing Costs” means, with respect to any Licensed COVID-19 Mono Product, Licensed COVID-19 Component, Adjuvant, [***] and [***], or Adjuvant Intermediate, as applicable, including, to the extent applicable, in each case, works-in-progress or components or ingredients thereof, the fully-burdened cost incurred by a Party or any of its Affiliates for Manufacturing or purchasing from a Third Party, as applicable, such Licensed COVID-19 Mono Product, Licensed COVID-19 Component, or Adjuvant (including (a) the costs associated with inventory build-up in advance of launch of any thereof (including validation batches if deemed saleable), but only until technical feasibility is determined and such Licensed COVID-19 Mono Product, Licensed COVID-19 Component, or Adjuvant inventory can be capitalized, and thereafter such Licensed COVID-19 Mono Product, Licensed COVID-19 Component, or Adjuvant inventory will be charged as a cost only at the time such inventory is sold or destroyed; and (b) the cost to Manufacture samples of Licensed COVID-19 Mono Product, Licensed COVID-19 Component, or Adjuvant for distribution), which costs calculated as follows:
1.165.1[***];
1.165.2[***]:
(a)[***]; and
(b)[***].
[***].
1.166“Marketing Authorization” means, with respect to a Licensed Product in a particular country or group of countries, any approval, license, registrations, or authorization, including EUAs, granted by a Governmental Authority necessary for the commercial sale of a product in such country or other regulatory jurisdiction, excluding, in each case, Pricing Approval. For purposes of this Section
1.166 (Marketing Authorization), “Governmental Authority” will include the FDA in the U.S., the EC in the EU, or any governmental health authority in any country or other jurisdiction that is a counterpart to the foregoing agencies, in each case, that has jurisdiction in such country or group of countries to authorize the promotion, sale, and distribution of such Licensed Product for any Indication.
1.167“Marketing Authorization Application” or “MAA” means, with respect to a Licensed Product in a particular country or region, an application for Marketing Authorization, examples of which include, (a) with respect to the United States, a BLA; (b) with respect to the European Union, an MAA; and (c) with respect to countries and regions other than the United States and the European Union, equivalents to a BLA or MAA in such other country or region.
1.168“Materials” means all biological materials, chemical compounds, and other materials provided by or on behalf of a Party to the other Party, its Affiliate, or its Sublicensee, or their Service Providers for use in the conduct of activities pursuant to this Agreement or for incorporation into a Licensed Product, including drug products, drug substance, packaging, cell lines, Antigens, compounds, lipids, assays, viruses, and vectors.
1.169[***].
1.170[***].
1.171“Matrix-M” means the saponin-based adjuvant developed by Novavax or its Affiliates, (a) as described in Novavax’s DMF for Matrix-M filed with the FDA in the U.S. in existence as of the Effective Date, as such DMF may be updated with the FDA in the U.S. from time-to-time during the Term, including to reflect any changes to Matrix-M and (b) as described in Novavax’s or any Affiliate’s DMF for a saponin-based adjuvant that is a derivative, modification, or improvement of the saponin-based adjuvant in the DMF of the foregoing clause (a).
1.172“Medical Activities” means (a) activities directed to HCPs to provide them with objective, scientific information to educate and inform, including such information provided through congresses and other venues for peer-to-peer scientific discourse; (b) to convene and manage
advisory boards and relationships with key opinion leaders; and (c) evidence generation studies that do not include the administration of Licensed Product; in each case of (a)-(c), excluding promotional or other sales-related activities further described in
Article 7 (Commercialization) and excluding any Development activities, Manufacturing activities, or Regulatory Activities.
1.173

“Milestone Eligible Licensed COVID-19 Mono Product” means a Licensed COVID-19 Mono Product that has received Marketing Authorization pursuant to a BLA or MAA and that includes [***]. For purposes of this definition, Emergency Use Authorization by FDA or EC shall not be deemed Marketing Authorization.
1.174“Net Sales” means, with respect to a Licensed Product sold in a country during the Adjuvant Royalty Term or COVID-19 Royalty Term, as applicable, for such Licensed Product in such country, the gross amount invoiced by Sanofi or its Affiliates or Sublicensee (each such Person, a “Selling Entity”), in each case, to Distributors or other Third Party purchasers in an arms-length transaction, less the following customary deductions, [***]:
(a)[***];
(b)[***];
(c)[***];
(d)[***];
(e)[***];
(f)[***];
(g)[***];
(h)[***];
(i)[***];
(j)[***]; and
(k)[***];
in each case (a) through (k), on a country-by-country basis, to the extent consistent with applicable Accounting Standards and consistently and equitably applied across Sanofi’s pharmaceutical products.
[***]:
(i)[***].
(ii)[***].
(iii)[***].
Additionally, for the calculation of Net Sales of any Licensed CIC Product or Licensed OC Product, the following shall apply:
(A)[***].
(B)[***].
(C)[***].
1.175“NIH” means National Institutes of Health of the United States.
1.176[***].
1.177“Non-Bankrupt Party” has the meaning set forth in Section
16.4 (Bankruptcy).
1.178“Non-Breaching Party” has the meaning set forth in Section
16.2.2(a) (Material Breach).
1.179“Non-Party Guest” has the meaning set forth in Section
9.11 (Non-Party Guests).
1.180“Notifying Party” has the meaning set forth in Exhibit B (Data Safeguards).
1.181“Novavax” has the meaning set forth in the Preamble.
1.182“Novavax Adjuvant Know-How” means any Know-How Controlled by Novavax or any of its Affiliates, as of the Effective Date or at any time during the Term, that is [***] to Exploit the Adjuvant, excluding all Adjuvant Intermediate Technology.
1.183“Novavax Adjuvant Manufacturing Know-How” means any Novavax Adjuvant Know-How that [***] used to Manufacture the Adjuvant, excluding all Adjuvant Intermediate Technology.
1.184“Novavax Adjuvant Material” means any Materials Controlled by Novavax or any of its Affiliates, as of the Effective Date or at any time during the Term, that are [***] to Exploit the Adjuvant (excluding any Material related to the Manufacture of the Adjuvant Intermediate).
1.185“Novavax Adjuvant Patent” means any Patent Controlled by Novavax or any of its Affiliates, as of the Effective Date or at any time during the Term, that is [***] to Exploit the Adjuvant, excluding all Joint Arising Patents.
1.186“Novavax Adjuvant Product” means any pharmaceutical product Exploited by Novavax or its Affiliates or (sub)licensees in any dosage, strength, formulation, or presentation that contains the Adjuvant. For clarity, Novavax Adjuvant Product excludes Novavax CIC Products, Novavax OC Products, and Licensed Products.
1.187“Novavax CIC Product” means any pharmaceutical product Exploited by Novavax or its Affiliates or (sub)licensees in any dosage, strength, formulation, or presentation that contains (a) a Licensed COVID-19 Component and (b) a Novavax Influenza Component, with or without any Antigen Directed To any other pathogen(s) other than SARS-CoV-2 and Influenza. For clarity, Novavax CIC Product excludes Novavax OC Products, Novavax Adjuvant Products, and Licensed Products.
1.188“Novavax Commercialization Report” has the meaning set forth in Section
7.6.2 (Novavax Commercialization Reports).
1.189“Novavax COVID-19 Know-How” means any Know-How Controlled by Novavax or any of its Affiliates, as of the Effective Date or at any time during the Term, that is [***] to Exploit any Licensed COVID-19 Mono Product or Licensed COVID-19 Component, including all Know-How disclosed by Novavax to Sanofi pursuant to Section
3.1.1(a) (Strain Selection Activities), but excluding all Novavax Adjuvant Know-How and Adjuvant Intermediate Technology.
1.190“Novavax COVID-19 Manufacturing Know-How” means any Novavax COVID-19 Know-How that is [***] used to Manufacture any Licensed COVID-19 Mono Product or Licensed COVID-19 Component.
1.191“Novavax COVID-19 Material” means any Materials Controlled by Novavax or any of its Affiliates, as of the Effective Date or at any time during the Term, that are [***] to Exploit any Licensed COVID-19 Mono Product or Licensed COVID-19 Component, excluding any Novavax Adjuvant Material and any Material used for the Manufacture of the Adjuvant Intermediate.
1.192“Novavax COVID-19 Patent” means any Patent Controlled by Novavax or any of its Affiliates, as of the Effective Date or at any time during the Term, that is [***] to Exploit a Licensed COVID-19 Mono Product or Licensed COVID-19 Component (alone or in combination with a component Directed To another Antigen), excluding all Joint Arising Patents and Novavax Adjuvant Patents.
1.193“Novavax Domain Name” means the domain names set forth on Schedule
1.193.
1.194“Novavax General Combo Patent” means any Novavax Patent that Covers (a) Licensed COVID- 19 Component (in combination with a component Directed to an Antigen that is not an Influenza Antigen) and (b) a Licensed OC Product. The Novavax General Combo Patents that exist as of the Effective Date are set forth on Schedule
1.194.
1.195“Novavax Indemnified Party” has the meaning set forth in Section
15.1.1 (Indemnification by Sanofi).
1.196“Novavax Influenza Component” means an Antigen Directed To Influenza made using Novavax’s Baculovirus Platform.
1.197“Novavax Know-How” means Novavax Adjuvant Know-How, Novavax COVID-19 Know-How, Adjuvant Intermediate Know-How, any Unauthorized Inventions, and Novavax’s rights, title, and interests in Joint Arising Know-How.
1.198“Novavax License” has the meaning set forth in Section
2.1.5 (License to Novavax).
1.199“Novavax Manufacturing Know-How” means any Novavax Adjuvant Manufacturing Know- How and Novavax COVID-19 Manufacturing Know-How.
1.200“Novavax Material” means the Novavax COVID-19 Material and the Novavax Adjuvant Material.
1.201“Novavax Matter” has the meaning set forth in Section
9.18.5(a) (Novavax Matters).
1.202“Novavax OC Product” means any pharmaceutical product Exploited by Novavax or its Affiliates or (sub)licensees in any dosage, strength, formulation, or presentation that contains (a) a Licensed COVID-19 Component and (b) at least one (1) Antigen owned by, or licensed to, Novavax or its Affiliate or (sub)licensee and Directed To any other pathogen(s) other than SARS-CoV-2 or Influenza. For clarity, Novavax OC Product excludes Novavax CIC Products, Novavax Adjuvant Products, and Licensed Products.
1.203“Novavax Patent” means any Novavax Adjuvant Patent, Novavax COVID-19 Patent, Patent Covering an Unauthorized Invention, and Novavax’s rights, title, and interests in any Joint Arising Patent. The Novavax Patents that exist as of the Effective Date are set forth on Schedule
14.2.1 (such Patents, the “Existing Novavax Patents”), [***]. For the avoidance of doubt, any Patent that solely Covers a Novavax CIC Product or Novavax Influenza Component is not a Novavax Patent.
1.204“Novavax Product” means any Novavax Adjuvant Product, Novavax CIC Product, or Novavax OC Product.
1.205“Novavax Research and Development Report” has the meaning set forth in Section
3.5.1 (Novavax Research and Development Reports).
1.206“Novavax Trademark” means the trademarks set forth on Schedule
1.206.
1.207“Novavax’s Baculovirus Platform” means Novavax’s proprietary method for the expression of recombinant proteins using a baculovirus/Spodoptera frugiperda insect cell system. For clarity, as used in this Section
1.207, “proprietary” means that Novavax owns or is licensed to Patents, Know- How, or Materials necessary to express recombinant proteins using a baculovirus/Spodoptera frugiperda insect cell system.
1.208“Novavax’s Knowledge” means [***].
1.209“OC License” has the meaning set forth in Section
2.1.3 (Other Combination Products).
1.210“Other Escrow Trigger Event” means the occurrence of any of the following: (a) on or after [***], Sanofi’s written notice to Novavax, (b) Novavax makes an assignment for the benefit of creditors, appoints, or suffers appointment of a receiver or trustee over all or substantially all of its property, files a petition under any bankruptcy, or insolvency act, or has any such petition filed against it that is not discharged within [***] after the filing thereof, (c) the failure of Novavax to perform the technology transfer in all material respects as set forth in the Adjuvant Manufacturing Technology Transfer Plan or the COVID-19 Manufacturing Technology Transfer Plan or (d) the failure of Novavax to deliver (in conformance with specifications and all other requirements for delivery under the Adjuvant Supply Agreement or COVID-19 Supply Agreement), at least (i) [***] of all amounts of Adjuvant, Licensed COVID- 19 Mono Product or Licensed COVID-19 Component that Novavax is required to deliver pursuant to the terms of the Adjuvant Supply Agreement or COVID-19 Supply Agreement in any [***], or (ii) [***] of all amounts of Adjuvant, Licensed COVID-19 Mono Product or Licensed COVID-19 Component that Novavax is required to deliver pursuant to the terms of the Adjuvant Supply Agreement or COVID-19 Supply Agreement during any [***] period during the Term.
1.211“Out-of-Pocket Costs” means, with respect to a Party, those costs and expenses actually paid by such Party or its Affiliates to a Third Party, including contract personnel (or payable to Third Parties and accrued in accordance with such Party’s Accounting Standards) and incurred in the performance of activities under this Agreement. Out-of-Pocket Costs exclude each of the following:
(a) costs and expenses of a Party, or its Affiliates paid to its or their employees (excluding contractor personnel, which are included as Out-of-Pocket Costs); (b) capital expenditures and depreciation of assets purchased pursuant to such capital expenditures; and (c) costs incurred related to a Party’s or its Affiliates’ use of facilities, utilities, general office, or facility supplies. [***].
1.212“Pandemic” means with respect to a pathogen, organism, or disease, the period of time beginning when the World Health Organization declares a public health emergency of international concern, and ending when the World Health Organization declares that such pathogen, organism, or disease, is no longer a public health emergency of international concern. For example, with respect to COVID-19, the World Health Organization declared a pandemic on January 20, 2020, and declared that COVID-19 was no longer a public health emergency of international concern on May 5, 2023.
1.213“Parent Company” means, with respect to a Person, any other Person that controls (within the meaning of the second sentence of Section
1.26 (Affiliate)) such entity.
1.214“Party” or “Parties” has the meaning set forth in the Preamble.
1.215“Party Non-Member Guest” has the meaning set forth in Section
9.10 (Party Non-Member Guests).
1.216“Party Vote” has the meaning set forth in Section
9.13 (Committee Decisions).
1.217“Patent Authority” means any Governmental Authority responsible for the granting of Patents in any national, federal, regional, supranational, or international jurisdiction in the Territory, including the United States Patent and Trademark Office and the European Patent Office, including any adjudicatory bodies related to such Governmental Authorities such as the Patent Trial and Appeal Board (PTAB) and Unified Patent Court (UPC).
1.218“Patent Dispute” means any challenge to the validity or enforceability of a Patent by commencing any opposition proceeding, post-grant review, inter partes review, or declaratory action, or any foreign equivalent thereof, in any court, arbitration proceeding, or other tribunal, including the United States Patent and Trademark Office and any foreign counterpart thereof.
1.219“Patents” means (a) all national, regional, and international patents and patent applications, including provisional patent applications; (b) all patent applications filed from any of the foregoing provisional patent applications in clause (a); (c) all patent applications that claim priority to any patent or patent applications in clause (a) or clause (b), including divisionals, continuations, continuations-in-part, provisionals, converted provisionals, and continued prosecution applications; (d) any and all patents that have issued or in the future issue from any of foregoing patent applications in clause (a), clause (b), or clause (c), including utility models, petty patents, and design patents, and certificates of invention; and (e) any and all extensions or restorations by existing or future extension or restoration mechanisms, including revalidations, reissues, re- examinations, and extensions (including any supplementary protection certificates and the like) of any of the foregoing patents or patent applications in clause (a), clause (b), clause (c), or clause (d). The term “Patents” includes Unitary Patents.
1.220“Person” means an individual, sole proprietorship, partnership, limited partnership, limited liability partnership, corporation, limited liability company, business trust, joint stock company, trust, incorporated association, joint venture, or similar entity or organization, including a government or political subdivision or department or agency of a government.
1.221“Pharmacovigilance Agreement” has the meaning set forth in Section
4.9 (Pharmacovigilance).
1.222“Phase 1 Clinical Trial” means a Clinical Trial (or portion thereof in the case of a Phase 1/2 Clinical Trial) of a product that meets the definition of a Phase 1 study as described in 21 C.F.R.
§ 312.21(a), or its successor regulation, or the equivalent regulation in any other country where such Clinical Trial is conducted.
1.223“Phase 1 Full Clinical Trial” means a Phase 1 Clinical Trial that is (a) conducted under an IND and (b) intended by Sanofi to provide the basis for the conduct of a Phase 2 Clinical Trial or Phase 3 Clinical Trial without any additional Phase 1 Clinical Trials, excluding any Phase 1 Clinical Trials the main aim of which is to translate the results of Research into clinical practice in humans and includes the study of the biology of the disease to provide solid rationales for the development or improvement of new drugs, the evaluation of the biological effects of the drugs in animals to define how to best use those drugs in humans, and the study of the biological effects of those drugs in humans.
1.224“Phase 2 Clinical Trial” means a Clinical Trial (or portion thereof in the case of a Phase 1/2 Clinical Trial or Phase 2/3 Clinical Trial) of a product that meets the definition of a Phase 2 study as described in 21 C.F.R. § 312.21(b), or its successor regulation, or the equivalent regulation in any other country where such Clinical Trial is conducted.
1.225“Phase 3 Clinical Trial” means a Clinical Trial (or portion thereof in the case of a Phase 2/3 Clinical Trial) of a product that meets the definition of a Phase 3 study as described in 21 C.F.R.
§ 312.21(c), or its successor regulation, or the equivalent regulation in any other country where such Clinical Trial is conducted.
1.226“Pivotal Clinical Trial” means (a) any Phase 3 Clinical Trial or (b) any other Clinical Trial of a product the results of which, together with prior data and information concerning such product, are
intended to be sufficient, or are sufficient, without any additional Clinical Trial, to meet the evidentiary standard for demonstrating the safety, purity, and potency of such product established by a Regulatory Authority in any particular jurisdiction and that is intended to, or does, support the filing of a MAA in such jurisdiction.
1.227“Post-Marketing Requirements” means, with respect to a drug product, any activity that is required by a Regulatory Authority in a country or jurisdiction to be conducted after such Regulatory Authority grants Marketing Authorization for such drug product in such country or jurisdiction, the performance of which are a condition to obtaining or maintaining Marketing Authorization in such country or jurisdiction for such drug product; but expressly excluding those COVID-19 Research and Development activities conducted by or on behalf of a Party at its discretion and not as a condition to obtain or maintain Marketing Authorization in such country or jurisdiction.
1.228“Press Release” has the meaning set forth in Section
11.6 (Public Announcements).
1.229“Pricing Approval” means, in any country where a Governmental Authority authorizes reimbursement for, or approves or determines pricing for, pharmaceutical products, receipt (or, if required to make such authorization, approval or determination effective, publication) of any government approval, agreement determination, or decision establishing such reimbursement authorization or pricing approval or determination.
1.230“Proceeding” means any action, suit, litigation, arbitration, proceeding (including any civil, criminal, administrative, investigative, or appellate proceeding), prosecution, contest, hearing, inquiry, inquest, audit, examination, or investigation that is, has been, or may in the future be commenced, brought, conducted, or heard at law or in equity or before any Governmental Authority, excluding all administrative proceedings before any patent office.
1.231“Proposed Adjuvanted Product” has the meaning set forth in Section
2.4.2 (Gatekeeper).
1.232“Proposed Disclosure” has the meaning set forth in Section
11.7.3(a) (Review Process).
1.233“Proposed Field” has the meaning set forth in Section
2.4.3 (Procedures).
1.234“Prosecuting Party” means, as between the Parties and with respect to a particular Patent, the Party that has the right to take any action or make any final decision (other than a decision to enter into any settlement) to Prosecute and Maintain such Patent in accordance with the terms of this Agreement.
1.235“Prosecution and Maintenance,” “Prosecuted and Maintained,” or “Prosecute and Maintain” means, with regard to a Patent, the preparing, filing, prosecuting, and maintenance of such Patent, as well as handling re-examinations and reissues with respect to such Patent, together with the conduct of interferences, derivation proceedings, pre- and post-grant opposition proceedings, post- grant patent proceedings (such as inter partes review and post grant review). “Prosecution and Maintenance” or “Prosecute and Maintain” does not include any Enforcement Actions taken with respect to a Patent.
1.236“Publication Strategy” has the meaning set forth in Section
11.7.1 (Publication Strategy).
1.237“Publishing Party” has the meaning set forth in Section
11.7.3(a) (Review Process).
1.238“Recall” means, with respect to a product, (a) the notification, retrieval, and return of such returned product, (b) replacement of such returned product, and (c) distribution of any replacement of such returned product.
1.239“Receiving Party” has the meaning set forth in Section
11.1 (Confidentiality).
1.240“Region” means each of (a) the United States, (b) Europe (including the United Kingdom and Switzerland), and (c) all countries in the world other than the United States and countries in Europe.
1.241“Regulatory Activities” means activities directed to preparing and filing Regulatory Filings and other documentation for submission to Regulatory Authorities, communicating with Regulatory Authorities, and obtaining and fulfilling regulatory obligations as the holder of such Marketing Authorizations and Pricing Approvals. Regulatory Activities exclude any Medical Activities, Development activities, Manufacturing activities, and Commercialization activities.
1.242“Regulatory Authority” means, with respect to a country in the Territory, any national (e.g., the FDA), supra-national (e.g., the European Commission, the Council of the European Union, or the EMA), regional, state, or local regulatory agency, department, bureau, board, commission, council, or other Governmental Authority that holds responsibility for development, commercialization, or manufacturing of, and the granting of Marketing Authorization for, a pharmaceutical product in such country or region.
1.243“Regulatory Costs” means those FTE Costs and Out-of-Pocket Costs (including filing, user, maintenance, and other fees paid to Regulatory Authorities) incurred by or on behalf of Party or any of its Affiliates in the preparation or maintaining of Regulatory Filings in connection with Marketing Authorizations and Pricing Approvals for a product, including compliance with such Marketing Authorizations and Pricing Approvals.
1.244“Regulatory Exclusivity” means, with respect to a product in a country, any data exclusivity rights, market exclusivity rights, or other exclusive rights, other than a Patent, granted, conferred, or afforded by any Regulatory Authority in such country or otherwise under Applicable Law with respect to such product in such country, that (a) confers exclusive marketing rights with respect to a product that prevents such Regulatory Authority from accepting another Marketing Authorization Application (whether new or abbreviated) from another Person, other than a Party or any of its Affiliates or Sublicensees, for a biosimilar or generic version of such product; or (b) confers data protection for regulatory data submitted by the Parties (directly or through their Affiliates or Sublicensees) relating to such product against unfair commercial use or public release consistent with, or no less stringent than, TRIPS Article 39.3.
1.245“Regulatory Filings” means, collectively: (a) applications in connection with any Clinical Trials, including INDs (and all submissions made in connection therewith); (b) Marketing Authorization Applications; (c) establishment license applications; (d) DMFs or BMFs; (e) applications for designation (including as an “Orphan Product(s)” under the United States Orphan Drug Act, as amended, for “Fast Track” status under Section 506 of the FD&C Act (21 U.S.C. § 356) or for a Special Protocol Assessment under Section 505(b)(4)(B) and (C) of the FD&C Act (21 U.S.C.
§ 355(b)(4)(B)) and similar counterparts thereof in any country or region in the Territory); (f) applications for Pricing Approval; (g) any Patent-related filings made to Regulatory Authority; (h) all supplements and amendments to any of (a), (b), (c), (d), (e), (f), or (g); and (i) communications with any Regulatory Authority in connection with (a), (b), (c), (d), (e), (f), or (g), including confirmed meeting requests, meeting minutes, and correspondence.
1.246“Regulatory Responsible Party” means [***].
1.247“Regulatory Transfer Plan” has the meaning set forth in Section
4.1.2 (Regulatory Transfer Plan).
1.248“Representatives” means, with respect to a Party, such Party’s and each of its Affiliates’ directors, officers, employees, contractors, consultants, attorneys, auditors, agents, or other representatives.
1.249“Research” means, with respect to a compound or product or any raw material or intermediate thereof, those in vitro, in vivo (other than in humans), and in silico activities directed to discovering, designing, optimizing, synthesizing, and conducting initial characterization of such compound or product or any raw material or intermediate, including any in vivo (other than in humans) toxicology studies. When used as a verb, “Researching” means to engage in Research. Research excludes any Medical Activities, Commercialization activities, Manufacturing activities, or Regulatory Activities.
1.250“Research License” has the meaning set forth in Section
2.1.6 (Adjuvant Research License).
1.251“Reversion License” has the meaning set forth in Section
16.3.5 (Reversion License).
1.252“Review Period” has the meaning set forth in Section
11.7.3(a) (Review Process).
1.253“Reviewing Party” has the meaning set forth in Section
11.7.3(a) (Review Process).
1.254“Royalty Report” has the meaning set forth in Section
10.5.6 (Royalty Reports and Payments).
1.255“Sanofi” has the meaning set forth in the Preamble.
1.256“Sanofi Indemnified Party” has the meaning set forth in Section
15.1.2 (Indemnification by Novavax).
1.257“Sanofi Licensed COVID-19 Mono Assets” means all (a) Sanofi Arising Patents, (b) Sanofi Arising Know-How, and (c) other Know-How Controlled by Sanofi or its Affiliates, in each case
(b) or (c) that is actually disclosed by Sanofi, its Affiliates, or Sublicensees to Novavax hereunder, including pursuant to Section
3.1.1(a) (Strain Selection Activities); except any Know-How generated by or on behalf of Sanofi in the course of performing Manufacturing of any Licensed Product, other than Data generated through the performance of Strain Selection Activities (which Data, for clarity, will be included in the Sanofi Licensed COVID-19 Mono Assets).
1.258“Sanofi Licenses” means the Adjuvant License, the COVID-19 Mono License, the CIC License, the Research License, and the OC License.
1.259“Sanofi Matter” has the meaning set forth in Section
9.18.5(b) (Sanofi Matters).
1.260“Sanofi Research and Development Report” has the meaning set forth in Section
3.5.2 (Sanofi Research and Development Reports).
1.261“SARS-CoV-2” means severe acute respiratory syndrome coronavirus 2, as defined by the Coronaviridae Study Group of the International Committee on Taxonomy of Viruses (Nat. MicroBiol. 2020; 5(4): 536-544), including any Variants thereof.
1.262“SARS-CoV-2 Antigen” means an Antigen derived from SARS-CoV-2, including the spike protein of the SARS-CoV-2 capsid.
1.263“Scientific COVID-19 Mono Publications” has the meaning set forth in Section
11.7.1 (Publication Strategy).
1.264“Season” means, with respect to any product Directed To SARS-CoV-2, the period beginning on the date on which the VRBPAC announces in its annual SARS-CoV-2/COVID-19 strain selection meeting which Strain(s) of SARS-CoV-2 it believes are most immediate concern to public health as of such date and continuing for a period of approximately [***] thereafter.
1.265“Securities Subscription Agreement” means the Securities Subscription Agreement, dated as of the date of this Agreement, executed between the Parties.
1.266“Sell-Off Period” has the meaning set forth in Section
16.3.2 (Inventory Sell-Off Period).
1.267“Selling Entity” has the meaning set forth in Section
1.174 (Net Sales).
1.268“Senior Executives” means the senior executives or employees appointed by Novavax and Sanofi from time to time for the purpose of, and each of which possess the expertise and authority necessary for, resolving a matter pursuant to Section
9.18.4 (Escalation to Senior Executives for Expedited Decision) or Section
17.12.1 (Informal Dispute Resolution; Escalation to Senior Executives).
1.269“Service Provider” has the meaning set forth in Section
2.2.2(e) (Subcontracting).
1.270“Settlement Arrangements” means each settlement or similar agreement between Novavax and any Third Party to settle a dispute arising from (a) an APA or (b) other contract, that provides for supply by Novavax of Licensed COVID-19 Mono Product, including the GAVI Arrangements, (i) entered prior to the Effective Date (the “Existing Settlement Arrangements”) or (ii) approved by the JCC pursuant to Section
7.3 (Existing APAs and Settlement Arrangements). All Existing Settlement Arrangements are set forth on Schedule
1.270.
1.271[***].
1.272“Sole” means, with respect to the grant of the CIC License hereunder by Novavax to Sanofi, that Sanofi, its Affiliates, and its Sublicensees shall be the only Persons other than Novavax, its Affiliates and its licensees solely with respect to Novavax CIC Products, who may practice the Licensed Novavax Assets to Exploit a product that includes the Licensed COVID-19 Component and an Antigen Directed To Influenza, with or without any other Antigen Directed To any pathogen(s) other than SARS-CoV-2 and Influenza.
1.273“Sponsor” means a Person who initiates a clinical investigation and that otherwise meets the definition of a “sponsor” as described in 21 C.F.R. § 312.3, or its successor regulation.
1.274“Status Quo Matter” has the meaning set forth in Section 9.18.5(d)(i)(6) (In General).
1.275“Strain” has the meaning set forth in Section
1.307 (Variant).
1.276“Strain Selection Activities” means the activities listed in Part 1 of the COVID-19 Research and Development Plan, as amended from time to time in accordance with the terms of this Agreement.
1.277“Strain Selection Technology Transfer” has the meaning set forth in Section
5.2 (COVID-19 Manufacturing Technology Transfer).
1.278“Strategic Partner” has the meaning set forth in Section
1.108 (“Existing Strategic Partner Agreements”).
1.279“Subcommittee” means each committee, working group, or team formed from time to time by or at the direction of the JSC in accordance with Section
9.2.3 (Subcommittees). Each of the JRDC, JMSC, JCC, JFC, JMAC, and JPC shall be deemed a Subcommittee.
1.280“Subcommittee Dispute” has the meaning set forth in Section
9.18.2 (Escalation to JSC).
1.281“Subcontract” has the meaning set forth in Section
2.2.2(e) (Subcontracting).
1.282“Sublicense” means, directly or indirectly, to sublicense, grant any other right with respect to, or agree not to assert, the rights granted to a Party hereunder. When used as a noun, “Sublicense” means any agreement to Sublicense.
1.283“Sublicensee” means an Affiliate or Third Party that is not a Distributor to whom a Party (or its Affiliate or Sublicensee) grants a Sublicense under any intellectual property licensed to such Party pursuant to this Agreement in accordance with Section
2.2 (Sublicensing).
1.284“Successful Completion of Adjuvant Manufacturing Technology Transfer” means the date on which the Adjuvant Manufacturing Handover Date has occurred.
1.285“Successful Completion of COVID-19 Manufacturing Technology Transfer” means the date on which the COVID-19 Manufacturing Handover Date has occurred.
1.286“Technology Transfer” means, as applicable, the COVID-19 Manufacturing Technology Transfer, the Strain Selection Technology Transfer, or the Adjuvant Manufacturing Technology Transfer.
1.287“Technology Transfer Budget” means the budget included in any Technology Transfer Plan.
1.288“Technology Transfer Costs” means FTE Costs and Out-of-Pocket Costs incurred by or on behalf of Novavax or its Affiliates in the course of performing Technology Transfer activities in accordance with this Agreement.
1.289“Technology Transfer Costs Invoice” has the meaning set forth in Section
10.8.2 (Reporting Technology Transfer Costs).
1.290“Technology Transfer Plan” means, as applicable, the COVID-19 Manufacturing Technology Transfer Plan or Adjuvant Manufacturing Technology Transfer Plan.
1.291“Term” has the meaning set forth in Section
16.1 (Term; Expiration).
1.292“Terminated Product” means any Licensed Product with respect to which this Agreement has been terminated pursuant to Section
16.2 (Termination of the Agreement) after the effective date of termination of this Agreement in part as to such Licensed Product, and if this Agreement is terminated in its entirety, then all Licensed Products in the Territory will be Terminated Products.
1.293“Terminated Territory” means any Region with respect to which this Agreement has been terminated pursuant to Section
16.2 (Termination of the Agreement) after the effective date of termination of this Agreement in part as to such Region, and if this Agreement is terminated in its entirety, then all Regions in the Territory will be the Terminated Territory.
1.294“Territory” means any and all countries and regions of the world.
1.295“Third Party” means any Person other than Sanofi, Novavax, or their respective Affiliates.
1.296“Third Party License” has the same meaning set forth in Section
0 (Royalty Adjustments).
1.297“Third-Party Claim” has the same meaning set forth in Section
15.1.1 (Indemnification by Sanofi).
1.298“Transfer Date” means on a country-by-country, region-by-region, or jurisdiction-by-jurisdiction basis for Licensed COVID-19 Mono Products, the date set forth in the Regulatory Transfer Plan for such country, region, or jurisdiction.
1.299“Transferred Marks and Domains” has the meaning set forth in Section
16.3.4(c) (Licensed Product Marks and Licensed Product Domain Names).
1.300“Transferred Regulatory Filings” has the meaning set forth in Section
4.5 (Transfer of Regulatory Filings).
1.301\“Unavailable” means, with respect to a Field, that (a) Novavax has exchanged at least [***] with a Third Party to grant an exclusive license, option, or other right in relation to such Field, or pursuant to which Novavax would otherwise be subject to exclusivity obligations or covenants (such as an option to grant an exclusive license or other similar right) in relation to such Field for a Novavax Adjuvant Product that has received IND Clearance, in each case, that preclude the grant of rights to such Field to Sanofi pursuant to this Agreement, provided that Novavax and such Third Party execute a definitive agreement no later than [***] after the date such term sheet was first exchanged; (b) Novavax has granted [***] in relation to such Field for a Novavax Adjuvant Product that has received IND Clearance, or is otherwise subject to exclusivity obligations or covenants (such as an option to grant a license or other right) under a legally binding contract that preclude the grant of rights to such Field to Sanofi pursuant to this Agreement; or (c) (i) Novavax has received IND Clearance for a Novavax Adjuvant Product in such Field and (ii) Novavax, its Affiliates, or its or their licensees or sublicensees have not permanently abandoned or terminated Development of such Novavax Adjuvant Product with respect to such Field. Cognates of the word Unavailable shall have the correlative meaning.
1.302“Unitary Patent” means those European patents that are subject to EU Regulation No 1257/2012 (OJ EU L 361, 31.12.2012, p. 1). A “Unitary Patent” may also be referred to as a “European patent with unitary effect.”
1.303“United States” or “U.S.” means the United States of America and its territories, possessions, and military bases.
1.304“Upstream License Agreements” means [***].
1.305“Upstream Licensors” means the counterparties to Novavax under the Upstream License Agreements.
1.306“Valid Claim” means any claim in (a) any issued and unexpired Patent that has not been held unenforceable, unpatentable, or invalid by a decision of a court or other Governmental Authority of competent jurisdiction, and that has not been admitted to be invalid or unenforceable through abandonment, reissue, disclaimer, or otherwise; or (b) any Patent application that has not been cancelled, withdrawn, or abandoned, and has not been filed more than five (5) years from the earliest available priority date for said application in the applicable jurisdiction, provided that if such claim is later issued, then it will from the issuance date forward, if meeting the requirements of the foregoing clause (a), be deemed to be a Valid Claim.
1.307“Variant” means, with respect to a reference pathogen, another pathogen of the same recognized species that contains genetic mutations relative to the genome of such reference pathogen. For purposes of this Agreement, the term “Strain” as applied to a pathogen is used synonymously with Variant.
1.308“VBRPAC” means the Vaccines and Related Biological Products Advisory Committee of the FDA.
ARTICLE 2: LICENSE GRANTS; EXCLUSIVITY
1.1Licenses.
1.1.1Licensed COVID-19 Mono Products.
(a)Subject to the terms and conditions of this Agreement, Novavax hereby grants Sanofi a Co-Exclusive, sublicensable (subject to Section
2.2 (Sublicensing)), non- transferable (except in accordance with Section
17.1 (Assignment)), royalty- bearing license under the Licensed Novavax Assets to Exploit the Licensed COVID-19 Mono Products in the Collaboration COVID-19 Territory (the “COVID-19 Mono License”).
(b)Sanofi acknowledges that Novavax is a Party to certain Existing Strategic Partner Agreements under which Novavax has granted to certain Third Parties certain
rights to Exploit Licensed COVID-19 Mono Products in the Existing Partner Exclusive Territory and Existing Partner Non-Exclusive Territory and the COVID- 19 Mono License is subject to the non-exclusive rights granted in the Existing Partner Non-Exclusive Territory under such Existing Strategic Partner Agreements. Novavax covenants that it will not amend or modify any Existing Strategic Partner Agreement, in any manner that expands the scope, territory, exclusivity, product volume, or term, in each case, of any such agreement without Sanofi’s prior written consent, [***].
1.1.2COVID-19 and Influenza Combination Products. Subject to the terms and conditions of this Agreement, Novavax hereby grants Sanofi a Sole, sublicensable (subject to Section
1.2(Sublicensing)), non-transferable (except in accordance with Section
17.1 (Assignment)), royalty-bearing license under the Licensed Novavax Assets to Exploit the Licensed CIC Products in the Territory (the “CIC License”).
1.1.3Other Combination Products. Subject to the terms and conditions of this Agreement, Novavax hereby grants Sanofi a non-exclusive, sublicensable (subject to Section
2.2 (Sublicensing)), non-transferable (except as permitted under Section
17.1 (Assignment)), royalty-bearing license under the Licensed Novavax Assets to Exploit the Licensed OC Products in the Territory (the “OC License”).
1.1.4Licensed Adjuvanted Products. Subject to the terms and conditions of this Agreement, Novavax hereby grants Sanofi a non-exclusive, sublicensable (subject to Section
2.2 (Sublicensing)), non-transferable (except as permitted under Section
17.1 (Assignment)), royalty-bearing license under the Novavax Adjuvant Know-How, the Adjuvant Intermediate Know-How, the Novavax Adjuvant Patents, and the Novavax Adjuvant Materials to Exploit the Licensed Adjuvanted Products in the Adjuvant Field in the Territory (the “Adjuvant License”).
1.1.5License to Novavax. Subject to the terms and conditions of this Agreement, Sanofi hereby grants Novavax a non-exclusive, non-sublicensable (except as permitted under Section
2.2 (Sublicensing)), non-transferable (except as permitted under Section
17.1 (Assignment)), royalty-free, paid-up license under the Sanofi Licensed COVID-19 Mono Assets to Exploit the Licensed COVID-19 Mono Products, and Licensed COVID-19 Components in accordance with this Agreement in the Territory (the “Novavax License”).
1.1.6Research License for Selection of Adjuvant Fields. Subject to the terms and conditions of this Agreement, Novavax hereby grants Sanofi a non-exclusive, non-sublicensable, non- transferable (except as permitted under Section
17.1 (Assignment)), royalty-free license under the Novavax Adjuvant Know-How, Adjuvant Intermediate Technology, the Novavax Adjuvant Patents, and the Novavax Adjuvant Materials to conduct Research (but not any Development activities) for potential Proposed Adjuvanted Products and Proposed Adjuvanted Products in any Field (the “Research License”).
1.1.7Sanofi Covenants. Sanofi covenants that it will not, and it will not grant any rights to or authorize any Affiliate or Third Party under any of the Sanofi Licenses:
(a)to Exploit any product that contains (i) the Licensed COVID-19 Component without the Adjuvant, and (ii) except for Licensed Adjuvanted Products in the Adjuvant Field, the Adjuvant without the Licensed COVID-19 Component; or
(b)to Manufacture the Adjuvant Intermediates unless and until the occurrence of the Adjuvant Intermediate Escrow Trigger Event.
1.1.8Novavax Covenants. Novavax covenants that it will not, and it will not grant any rights, or authorize any Affiliate or Third Party to:
(a)practice under the Licensed Novavax Assets to Exploit the Licensed COVID-19 Mono Products except (i) in the Existing Partner Exclusive Territory or Existing Partner Non-Exclusive Territory, in each case, solely pursuant to the Existing Strategic Partner Agreements during the term thereof, and (ii) in the Collaboration COVID-19 Territory, solely in accordance with the COVID-19 Research and Development Plan, Regulatory Transfer Plan, Technology Transfer Plans, and Licensed COVID-19 Mono Product Commercialization Framework, or as otherwise expressly contemplated by this Agreement; or
(b)practice under the Licensed Novavax Assets to Exploit any product that contains the Licensed COVID-19 Component together with any Antigen Directed To Influenza other than the Novavax CIC Product; and
(c)grant an exclusive or co-exclusive (between Novavax and a Third Party licensee) license to any Third Party to Exploit a Novavax Adjuvant Product in a Field prior to IND Clearance of such Novavax Adjuvant Product.
1.2Sublicensing.
1.2.1To Affiliates.
(a)Sanofi may grant Sublicenses (through multiple tiers) under any of the Sanofi Licenses to its Affiliates without Novavax’s consent, but subject to the terms of Section
17.13 (Performance by Affiliates).
(b)Novavax may grant Sublicenses (through multiple tiers) under the Novavax License to its Affiliates without Sanofi’s consent, but subject to the terms of Section
17.13 (Performance by Affiliates).
1.2.2To Third Parties.
(a)Sanofi may grant Sublicenses (through multiple tiers) under the COVID-19 Mono License, CIC License, or OC License to one (1) or more Third Parties, including to any Service Provider, without Novavax’s consent.
(b)Sanofi may grant Sublicenses (through multiple tiers) under the Adjuvant License without Novavax’s prior written consent to any (i) Service Provider, or (ii) other Third Party who is granted a license under Patents or Know-How Controlled by Sanofi or its Affiliates to Exploit a Licensed Adjuvanted Product, except in the situation in which such Licensed Adjuvanted Product includes an Antigen that Sanofi or its Affiliate has in-licensed from a Third Party, in which case, unless Novavax otherwise agrees in writing, Sanofi may not grant a Sublicense under the Adjuvant License to such Third Party unless the applicable Licensed Adjuvanted Product also includes one or more Antigens or other pharmaceutically active
components that Sanofi or its Affiliate owns or has in-licensed from a different Third Party.
(c)Novavax may grant Sublicenses (through multiple tiers) under the Novavax License without Sanofi’s prior written consent to any (i) Service Provider and (ii) other Third Party who is concurrently granted a license to Exploit a Novavax Product.
(d)Notwithstanding the inclusion of “have used, have Research, have Developed, have Manufactured, or have Commercialized” in the definition of Exploit, neither Party may grant Sublicenses not expressly permitted under Sections
2.2.2(a) (To Third Parties) through
2.2.2(c) (To Third Parties) without the other Party’s prior written consent, such consent not to be unreasonably withheld, conditioned, or delayed.
(e)Each Sublicense granted to a Third Party must: (i) be in writing; (ii) be subject to, and consistent with, the relevant terms of this Agreement (including in accordance with the license grants set forth in Section
2.1 (Licenses), ownership of intellectual property set forth in
Article 12 (Patent and Know-How Matters), and obligations of confidentiality set forth in
Article 11 (Confidentiality and Non-Disclosure)); and (iii) require such Sublicensee to comply with all applicable terms of this Agreement.
1.2.3Subcontracting. Each Party (or its Affiliates or Sublicensees) may engage one or more Third Parties, including CMOs, to perform activities under this Agreement on behalf of such Party, its Affiliate, or its Sublicensee, as applicable (each such Third Party, a “Service Provider”) in accordance with this Section
2.2.3 (Subcontracting). Each Party subcontracting any of its activities hereunder to any Service Provider will: (a) reasonably oversee the performance by each such Service Provider of the subcontracted activities; and
(b) remain responsible for the performance of such activities in accordance with this Agreement. All subcontracted activities will be conducted pursuant to a written agreement between the subcontracting Party and the Service Provider (a “Subcontract”), which agreement will be consistent with the terms and conditions of this Agreement (including the terms of Section
2.2.2(e) (To Third Parties) if such Service Provider is also granted a sublicense of any rights granted hereunder to the Party entering into the Subcontract), and contain (i) confidentiality provisions no less restrictive than those set forth in
Article 11 (Confidentiality and Non-Disclosure), (ii) where applicable to the activities of such Service Provider, a certification that such Third Party and its officers, employees, and agents have not been debarred, and are not subject to debarment, pursuant to Section 306 of the FD&C Act, and are not the subject of a conviction described in such section, and (iii) a present assignment to such subcontracting Party of all of such Service Providers’ rights, title, and interests in and to any deliverables, work product, and any related property rights in such deliverables and work product, in each case arising out of such Subcontract.
1.2.4Retained Liability. Each Party will, notwithstanding any sublicensing of its rights or any subcontracting of its obligations permitted under this Section
2.2 (Sublicensing), remain liable to the other Party for the performance of its obligations hereunder, and notwithstanding the inclusion of “have used, have Research, have Developed, have Manufactured, or have Commercialized” in the definition of Exploit, neither Party may engage any Service Provider except as expressly provided in Section
2.2.3 (Subcontracting).
1.3Upstream License Agreements. Sanofi acknowledges that certain of the Licensed Novavax Assets are sublicensed to Sanofi from Upstream Licensors, and with respect to such sublicensed Licensed Novavax Assets, the Sanofi Licenses are no broader in scope than the rights granted by the applicable Upstream Licensor to Novavax under the applicable Upstream License Agreement. Sanofi will comply with all applicable provisions of the Upstream License Agreements, except that Novavax shall be solely responsible for all payments under such Upstream License Agreements, including all upfront, milestone, royalty, and other payments or consideration to the Upstream Licensors.
1.4Adjuvant Field.
1.4.1Gatekeeper. To ensure that Sanofi can Exploit the Adjuvant under the Adjuvant License while allowing Novavax to continue to Exploit the Adjuvant by itself or with Third Parties, with respect to any pharmaceutical product that contains (a) the Adjuvant and (b) one or more active pharmaceutical ingredients (a “Proposed Adjuvanted Product”) that Sanofi wishes to Develop or Commercialize, prior to conducting any Development activities with respect to any such Proposed Adjuvanted Product in a given Field that has not already been confirmed to be included in the Adjuvant Field, Sanofi must, in accordance with the procedures in Section
2.4.3 (Procedures), confirm that such Field is not Unavailable and therefore included in the Adjuvant Field.
1.4.2Appointment. The Parties shall agree upon an individual from a law firm, of whom neither Party is a client, to act as a gatekeeper (the “Gatekeeper”) solely for the purposes of verifying whether or not a particular Field is Unavailable. The Parties shall agree upon and enter into a customary three-way agreement with the Gatekeeper setting forth the procedures described in Section
2.4.3 (Procedures). The Parties shall agree on the identity of the Gatekeeper within [***] after the Effective Date. The Gatekeeper may be changed by written agreement of the Parties.
1.4.3Procedures.
(a)On an ongoing basis during the Term, promptly, but no later than [***] after a particular Field becomes Unavailable, Novavax shall provide the Gatekeeper with an updated list of the Excluded Adjuvant Fields at such time, together with reasonable documentary evidence to support the basis on which such Field is an Excluded Adjuvant Field. The Gatekeeper shall not disclose to Sanofi the list of Excluded Adjuvant Fields.
(b)At any time during the Term, prior to Sanofi’s [***] for a Proposed Adjuvanted Product, Sanofi shall submit to the Gatekeeper in writing the identity of all Fields intended for such Proposed Adjuvanted Product that has not been previously confirmed to be included within the Adjuvant Field (all such Fields for such Proposed Adjuvanted Product, to the extent that such Fields are not already confirmed to be included in the Adjuvant Field, the “Proposed Field”). The Gatekeeper will promptly notify Novavax that a request for an update of the Excluded Adjuvant Field has been received from Sanofi, but the Gatekeeper shall not disclose to Novavax the identity of the Proposed Field, and Novavax will, no later than [***] thereafter, provide in writing an updated Excluded Adjuvant Fields list to the Gatekeeper, together with reasonable documentary evidence of Unavailability thereof. The Gatekeeper will compare the Proposed Field to the Excluded Adjuvant Fields and (i) if such Proposed Field is
Unavailable, then the Gatekeeper will notify Sanofi that such Proposed Field is an Excluded Adjuvant Field and (ii) if such Proposed Field is not Unavailable, then the Gatekeeper will notify both Parties that such Proposed Field is included within the Adjuvant Field, in each case (i) and (ii), no later than [***] after the Gatekeeper’s receipt of Sanofi’s notice with respect to the applicable Proposed Field. If such Proposed Field is not Unavailable, then, as of the date that the Sanofi receives written notice from the Gatekeeper, (A) such Proposed Field will be deemed to be included within the Adjuvant Field and (B) such Proposed Adjuvanted Product will be deemed a Licensed Adjuvanted Product.
(c)[***].
(d)In addition, during the Term, no later than [***] after a Field that was previously Unavailable is no longer Unavailable, Novavax will notify the Gatekeeper of the availability of such Field and provide an updated list of Excluded Adjuvant Fields to the Gatekeeper to reflect such change. If such newly available Field is a Field that Sanofi previously proposed to the Gatekeeper to include in the Adjuvant Field, but was Unavailable at the time of Sanofi’s proposal thereof, then the Gatekeeper will notify Sanofi (but not Novavax) of the removal of such previously Unavailable Field from the Excluded Adjuvant Field. Upon receipt of such notice from the Gatekeeper, such previously Unavailable Field shall be confirmed as included within the Adjuvant Field.
(e)For avoidance of doubt, (i) once a Field is confirmed to be included within the Adjuvant Field in accordance with this Section
2.4.3 (Procedures), Sanofi need not repeat the procedures set forth in this Section
2.4.3 (Procedures) with respect to such Field, and any Proposed Adjuvanted Product in such Field will automatically be deemed a Licensed Adjuvanted Product and (ii) a Proposed Adjuvanted Product with respect to which all Fields intended for such Proposed Adjuvanted Product are already included in the Adjuvant Field may still be a Distinct Licensed Adjuvanted Product for purposes of Section
10.4 (Licensed Adjuvanted Product Milestone Payments).
(f)The decision of the Gatekeeper as to whether a Field is or is not Unavailable will be final and unappealable, but the Parties retain the right to resolve disputes as to each Party’s compliance with the terms of this Section
2.4 (Procedures) (including whether a Party properly designated a Field as Unavailable) in accordance with the procedures set forth in Section
17.12 (Dispute Resolution).
1.5No Adjuvant Improvements.
1.5.1Covenant. Sanofi will not, and will cause its Affiliates, Sublicensees, and Service Providers not to, modify, enhance, improve, or otherwise use the Adjuvant or any component thereof (including [***]) supplied by Novavax other than to conduct Research for selection of Adjuvant Fields or to Manufacture the Adjuvant as permitted hereunder and to incorporate and use the Adjuvant in Licensed Products in accordance to the terms and conditions of this Agreement.
1.5.2Assigned Inventions. If Sanofi becomes aware that it or its Affiliates, Sublicensees, or Service Providers is using the Adjuvant or any component thereof (including [***]) supplied by Novavax in any manner not consistent with, or otherwise in violation of, Section
2.5.1 (Covenant) during the Term or any time thereafter, then, in any such case, Sanofi will (a) promptly notify Novavax of such unauthorized use and will disclose to Novavax any inventions or other Know-How developed or invented in connection with such unauthorized use (each, an “Unauthorized Invention”), and (b) assign, and hereby does assign (and will use reasonable efforts to cause its Affiliates, Sublicensees, and Service Providers to assign), all of its and their respective rights, title, and interests in and to any and all such Unauthorized Inventions. For the avoidance of doubt, any use by Sanofi, its Affiliates, or Service Providers of the Adjuvant to develop, invent, or create a different form, formulation, or composition of Matrix-M from the specifications for Matrix-M included in the DMF will be deemed an Unauthorized Invention.
1.6Adjuvant In-Licenses. Each Party shall notify the other Party when such Party becomes aware of any Patents Controlled by a Third Party in a particular country or jurisdiction that may be [***] for the Development, Manufacture, Commercialization, or other Exploitation of the Adjuvant (“Adjuvant Third Party Patents”). Novavax shall have the first right, but not the obligation, to enter into an agreement for such Adjuvant Third Party Patents, and if Novavax exercises such right, then Novavax will use [***] efforts to obtain the right to sublicense such Adjuvant Third Party Patents to Sanofi and its Affiliates and its and their Sublicensees to Exploit Licensed Products. If Novavax does not enter into an agreement for Adjuvant Third Party Patents, or obtain the right under such agreement to sublicense such Adjuvant Third Party Patents to Sanofi and its Affiliates and its and their Sublicensees, in accordance with the terms of this Agreement, to Exploit Licensed Products, then to the extent such Adjuvant Third Party Patents are necessary (but not reasonably useful) for the Exploitation of Licensed Adjuvanted Products, Sanofi shall have the right, but not the obligation, to enter into an agreement for non-exclusive rights, or exclusive rights solely with respect to Licensed Adjuvanted Products, to such Adjuvant Third Party Patents for itself and its Affiliates and its and their Sublicensees to Exploit Licensed Product, and Sanofi will not enter any such agreement that would prevent Novavax from obtaining rights to the applicable Adjuvant Third Party Patents (other than for Licensed Adjuvanted Products).
1.7No Implied Licenses; Competing Products; Retained Rights.
1.7.1Except as expressly provided in this Agreement, no Party will be deemed by estoppel, implication, or otherwise to have granted the other Party any licenses or other rights with respect to any property owned or controlled by such Party. For clarity, no license or other right is granted hereunder by Sanofi or its Affiliates to Novavax or its Affiliates with respect to any property relating to any Sanofi adjuvant or any product that is proprietary to Sanofi ([***]) other than Sanofi Licensed COVID-19 Mono Assets. Neither Party will practice the Licensed Novavax Assets other than as expressly licensed and permitted under this Agreement.
1.7.2Except as expressly provided in this Agreement, each Party is free to engage in research, development, manufacturing, commercialization, and other activities relating to any products for which Marketing Authorization may be sought for any human pharmaceutical and diagnostic uses, including the prevention, prognosis, and diagnosis, and treatment of infection and other medical conditions and diseases caused by or mediated by SARS-CoV- 2, Influenza, or both SARS-CoV-2 and Influenza, whether alone or with other pathogens.
1.7.3Subject to the terms and conditions of this Agreement (including Section
2.1.8), Novavax retains all rights, title, and interests under the Licensed Novavax Assets not granted under the Sanofi Licenses
1.7.4Subject to the terms and conditions of this Agreement (including Section
2.1.7), Sanofi retains all rights, title, and interests under the Sanofi Licensed COVID-19 Mono Assets not granted under the Novavax License.
ARTICLE 3: RESEARCH AND DEVELOPMENT
1.1General.
1.1.1Licensed COVID-19 Component. Owing to the threat of new Variants of SARS-CoV-2 and their changing prevalence, it is the expectation of the Parties that continuous Research and Development of Licensed COVID-19 Components is necessary to ensure that the Licensed COVID-19 Components are Directed To the most applicable Variants of SARS- CoV-2. Accordingly, with respect to the Licensed COVID-19 Component, during the Collaboration Term, the Parties agree to collaborate on Research and Development for the Licensed COVID-19 Component in accordance with the COVID-19 Research and Development Plan and COVID-19 Research and Development Budget as further set forth in this
Article 3 (Research and Development).
(a)Strain Selection Activities. Unless the Parties agree otherwise, until completion of the Strain Selection Technology Transfer as provided for in
Article 5 (Technology Transfer), as between the Parties, Novavax will perform the Strain Selection Activities and the costs related thereto will be allocated in accordance with Section
10.6 (Research and Development Plan Costs). For so long as Novavax is performing the Strain Selection Activities hereunder, the Parties shall collaborate through a working group established by the JRDC to facilitate the exchange of information regarding SARS-CoV-2 Strains of interest, and generally to provide a forum for scientific exchange. Following completion of the Strain Selection Technology Transfer, as between the Parties, Sanofi will perform the Strain Selection Activities. The Party that performs the Strain Selection Activities under this Section
3.1 (General) shall, at least [***] provide a summary of the Data generated from such activities to the JRDC. The SARS-CoV-2 Variants that will be included in the Licensed COVID-19 Components after the Effective Date shall be determined in accordance with
Article 9 (Governance). After the completion of the Strain Selection Technology Transfer and while Novavax is still Commercializing the Licensed COVID-19 Mono Product in at least one country, Sanofi will transfer the master virus seed for such Strain to Novavax and disclose any Know-How that is [***] to Manufacture the selected Strain for use in Novavax Products and the Licensed COVID-19 Mono Product, as contemplated by this Agreement.
(b)Material Transfer. For each SARS-CoV-2 Strain that is selected as part of the Strain Selection Activities in accordance with Section
3.1.1(a) (Strain Selection Activities), Novavax will transfer to Sanofi all necessary quantities of the following Materials on a timely periodic basis, until the completion of the Strain Selection Technology Transfer: [***]
(c)
[***]. Such deliveries of Materials will be coordinated through the working group established by the JRDC per Section
3.1.1(a) (Strain Selection Activities).
(d)FDA working group on SARS-CoV-2 Variants. For so long as Novavax is performing the Strain Selection Activities hereunder, if permitted by the FDA, Sanofi shall have the right to attend meeting convened by the FDA of its strain selection technical working group on SARS-CoV-2 Variants together with Novavax. In the event that the FDA will not allow both Parties to participate in the FDA strain selection technical working group on SARS-CoV-2 Variants (or any successor working group thereto), Novavax’s personnel who participate in such working group shall coordinate with Sanofi with regard to such meetings, and Novavax shall keep Sanofi fully informed of the activities of the FDA strain selection technical working group on SARS-CoV-2 Variants (or any successor working group thereto). To the extent that Regulatory Authorities other than the FDA form working groups to monitor SARS-CoV-2 Variants, the provisions of this Section
3.1.1(c) shall apply to such other Regulatory Authorities, mutatis mutandis. Further, in the event that industry working groups are convened in respect of SARS-CoV-2 Variants (with or without the participation of Regulatory Authorities), both Parties shall have the right to attend such industry working groups, and shall, with respect to Licensed COVID-19 Mono Products, coordinate their communications for such industry working groups. The costs related to such activities will be allocated in accordance with Section
10.6 (Research and Development Plan Costs).
(e)Retired Strains. The Parties agree that, for the purposes of this Agreement, neither Party shall have any obligation to conduct any Research activities with respect to Licensed COVID-19 Components for Strains selected by VRBPAC prior to the annual SARS-CoV-2/COVID-19 strain selection meeting held in 2024.
(f)Activities Performed Outside of the COVID-19 Research and Development Plan for CIC Products and OC Products. Sanofi may, in its discretion, and at its cost and expense, conduct Research activities outside of the COVID-19 Research and Development Plan with respect to Licensed COVID-19 Components in support of the Development and Commercialization of Licensed CIC Products and Licensed OC Products throughout the Territory. Novavax may, in its discretion, and at its cost and expense, conduct Research activities outside of the COVID-19 Research and Development Plan with respect to Licensed COVID-19 Components in support of the Commercialization of Novavax CIC Products and Novavax OC Products throughout the Territory; provided that such Research by Novavax outside of the COIVD-19 Research and Development Plan shall not include any Strain Selection Activities after Sanofi assumes responsibility for such activities. To the extent that either Sanofi or Novavax conduct Research activities with respect to Licensed COVID-19 Components outside of the COVID-19 Research and Development Plan as permitted under this Section
3.1.1(e) (Activities Performed Outside of the COVID-19 Research and Development Plan), each Party shall, through the JRDC co-chairs, unless prohibited by Applicable Laws, inform the other Party in advance of the intended public disclosure of any material scientific Data with respect to any SARS-CoV-2 Antigen within a Licensed COVID-19 Component with respect to which such Party is independently
performing any Research or Development, if such Data could [***] have a material impact to the other Party’s Exploitation of Licensed COVID-19 Components hereunder.
1.1.2Licensed COVID-19 Mono Product. In addition to the Licensed COVID-19 Component as described in Section
3.1.1 (Licensed COVID-19 Component), during the Collaboration Term, the Parties agree to collaborate on COVID-19 Research and Development for the Licensed COVID-19 Mono Product in accordance with the COVID-19 Research and Development Plan and COVID-19 Research and Development Budget as further set forth in this
Article 3 (Research and Development), and neither Party shall independently outside of the COVID-19 Research and Development Plan and COVID-19 Research and Development Budget conduct any Research or Development of Licensed COVID-19 Mono Products. For clarity, Licensed COVID-19 Components that arise from the Research activities performed under Section
3.1.1 (Licensed COVID-19 Component) may be utilized by both Parties to Exploit Licensed COVID-19 Mono Products in accordance with
Article 2 (License Grants; Exclusivity). Allocation of costs related to such activities is set forth in Section
10.6 (Research and Development Plan Costs).
1.1.3CIC Products and OC Products. Sanofi shall have the right for Licensed CIC Products and Licensed OC Products (other than the Licensed COVID-19 Component incorporated therein), and Novavax shall have the right for Novavax CIC Products and Novavax OC Products (other than the Licensed COVID-19 Component incorporated therein), to conduct Research and Development for each such product that it is Exploiting in its sole discretion at it own cost and expense, but, in each case, within the scope of the CIC License and OC License, as applicable, and subject to other restrictions applicable to each Party as set forth in
Article 2 (License Grants; Exclusivity) and in this
Article 3 (Research and Development). For clarity, Sanofi may utilize in Licensed CIC Products and Licensed OC Products, and Novavax may utilize in Novavax CIC Products and Novavax OC Products, Licensed COVID-19 Components that arise from the Research and Development activities performed under Section
3.1.1 (Licensed COVID-19 Components) as permitted under
Article 2 (License Grants; Exclusivity).
1.1.4Licensed Adjuvanted Products. With respect to Licensed Adjuvanted Products, Sanofi shall have the right to conduct Research and Development in its sole discretion at its own cost and expense, within the scope of the Adjuvant License and subject to other restrictions applicable to Sanofi as set forth in
Article 2 (License Grants; Exclusivity) and in this
Article 3 (Research and Development). As between the Parties, with respect to Novavax Adjuvant Products, Novavax shall have the right to conduct Research and Development in its sole discretion at its own cost and expense.
1.2Collaborative Research and Development of Licensed COVID-19 Components and Licensed COVID-19 Mono Products.
1.2.1COVID-19 Research and Development Plan. As of the Effective Date, the Parties have agreed to an initial written plan for conducting (a) certain collaborative Research activities with respect to Licensed COVID-19 Components, and (b) Researching and Developing, and conducting certain Regulatory Activities and Medical Activities for, the Licensed COVID-19 Mono Products, which initial written plan is attached hereto as Schedule
3.2 (such plan, as it may be amended in accordance with the terms of this Agreement, the “COVID-19 Research and Development Plan”). The COVID-19 Research and Development Plan will include the allocation of Research and Development activities as
between the Parties and target timelines for achieving Research and Development milestone events. Each Party may propose amendments to the COVID-19 Research and Development Plan through the JRDC at any time to reflect any material adjustments to the applicable Research and Development activities described therein, and each proposed update to the COVID-19 Research and Development Plan will be subject to review and determine whether to approve by the JRDC in accordance with
Article 9 (Governance). At least [***], the JRDC will review, discuss, and determine if changes are required to the then-current COVID-19 Research and Development Plan (for example, to update the COVID-19 Research and Development Plan with respect to the Variants of SARS-CoV-2 that have been selected in accordance with
Article 9 (Governance) for inclusion in Licensed COVID-19 Mono Components or Licensed COVID-19 Mono Products for the current Season and any subsequent Season (i.e., the COVID-19 Research and Development Plan for the upcoming Seasons)) and the JRDC will prepare an update to the COVID-19 Research and Development Plan covering the period of the next [***]. Any amendments to the COVID-19 Research and Development Plan approved in accordance with
Article 9 (Governance) shall become effective and supersede the applicable portion(s) of previous COVID-19 Research and Development Plan as of the date such approval is given.
1.2.2COVID-19 Research and Development Budget. As of the Effective Date, the Parties have agreed to an initial rolling budget for the activities set forth in the COVID-19 Research and Development Plan for [***] and [***], which budget is attached hereto as Schedule
3.2.2 (COVID-19 Research and Development Budget) (such budget, as it may be updated in accordance with the terms of this Agreement, the “COVID-19 Research and Development Budget”). The Parties will collaboratively prepare the [***] update to the COVID-19 Research and Development Budget, and submit such initial draft of such update to the JRDC for the JRDC to review, discuss, and determine whether to approve in accordance with
Article 9 (Governance). The COVID-19 Research and Development Budget will be broken down by activity and costs, the break down of which, and any adjustments based on anticipated timing of events, will be determined by the JFC. For the initial COVID-19 Research and Development Budget attached hereto as Schedule
3.2.2 (COVID-19 Research and Development Budget), the period of such COVID-19 Research and Development Budget that includes [***] will be binding on the Parties in accordance with Section
10.6 (Research and Development Plan Costs) and the remainder of the COVID-19 Research and Development Budget will serve as non-binding guidance for the Parties. Each update to the COVID-19 Research and Development Budget will include the budgets of the FTE Costs and Out-of-Pocket Costs anticipated to be incurred in the performance of activities to be conducted by or on behalf of Novavax under the COVID-19 Research and Development Plan for [***]. The COVID-19 Research and Development Budget may also include a high-level non-binding estimate for the COVID-19 Research and Development Budget for [***] (the “COVID-19 Research and Development Budget Forecast”). [***].
1.2.3Budget Updates and Amendments. Each Party may propose amendments to the COVID- 19 Research and Development Budget through the JRDC to reflect proposed adjustments to the applicable COVID-19 Research and Development activities set forth in the COVID- 19 Research and Development Plan. No later than [***] of each Calendar Year, the JRDC will prepare (a) an updated COVID-19 Research and Development Budget covering the next [***] period (in accordance with Section
3.2.2 (Contents)) and
(b) an updated COVID-19 Research and Development Budget Forecast for Licensed COVID-19 Mono Products Directed To the then-current Variants of SARS-CoV-2. The Parties, through the JRDC, will use [***] to determine whether to grant their preliminary approval of such updates no later than [***] of each Calendar Year. [***] after the JRDC’s preliminary approval, such updates will be submitted to each Party for its internal budgeting process. After each Party performs its internal budgeting process, the JSC will review, discuss, and determine whether to grant its approval of such updates no later than [***] of each Calendar Year (or at a later time if agreed by the JSC) in accordance with, and subject to,
Article 9 (Governance), including Section
9.18 (Decision Making). Any amendments to the COVID-19 Research and Development Budget approved in accordance with
Article 9 (Governance) shall become effective and supersede the applicable amended portion(s) of the previous COVID-19 Research and Development Budget as of the date such approval is given.
1.3Day-to-Day Responsibility. Each Party will be responsible for routine, non-strategic, operational- level decisions relating to the Research and Development activities for which it is assigned responsibility under the COVID-19 Research and Development Plan, provided that such decisions will not conflict with the COVID-19 Research and Development Plan, or any decision of the JRDC or JSC with respect to such activity, or be a matter for which such Party does not have authority to take such decision per
Article 9 (Governance), or is otherwise inconsistent with Section
3.4 (Research and Development Diligence).
1.4Research and Development Diligence. Subject to the terms and conditions of this Agreement, each Party will use [***] to conduct all Research and Development activities allocated to it under the COVID-19 Research and Development Plan. Notwithstanding the generality of the foregoing, Sanofi will use [***] to Research and Develop at least (a) one (1) Licensed COVID-19 Mono Product in the U.S. and (b) one (1) Licensed CIC Product or Licensed OC Product in each Major Market country.
1.5Research and Development Reports.
1.5.1Novavax Research and Development Reports. To the extent Novavax is conducting Research or Development, certain Regulatory Activities, or certain Medical Activities, in each case, under the COVID-19 Research and Development Plan, Novavax will provide the JRDC with [***] reports, within [***] after the end of each [***], regarding its Research and Development activities (including associated Regulatory Activities), and such Medical Activities, with respect to each Licensed COVID- 19 Mono Product in the Territory, its progress compared to the COVID-19 Research and Development Plan, any anticipated or completed key Regulatory Filings, and a summary of all Novavax COVID-19 Know-How generated in the performance of such activities, in each case, during the applicable reporting period. Each [***] report of Research and Development activities delivered by Novavax in accordance with this Section
3.5.1 (Novavax Research and Development Reports) is a “Novavax Research and Development Report.” Novavax Research and Development Reports are the Confidential Information of Novavax.
1.5.2Sanofi Research and Development Reports. Sanofi will provide the JRDC with [***] reports, within [***] after the end of each [***], regarding (a) its Research and Development activities under the COVID-19 Research and Development Plan (to the extent Sanofi conducts Research and Development activities under the COVID-19 Research and Development Plan) and (b) a high-level summary of Development of Licensed CIC Products, Licensed OC Products, and Licensed Adjuvanted Products, in each case, during the applicable reporting period. Each [***] report of Research and Development activities delivered by Sanofi in accordance with this Section
3.5.2 (Sanofi Research and Development Reports) is a “Sanofi Research and Development Report.” Each Sanofi Research and Development Report will identify each Licensed Adjuvanted Product for which Sanofi filed an IND during the preceding [***]. Sanofi Research and Development Reports are the Confidential Information of Sanofi.
1.6Transfer of Materials. Each Party shall transfer to the other Party, in accordance with the COVID- 19 Research and Development Plan, Materials in its Control that are necessary to enable such other Party to carry out its activities under the COVID-19 Research and Development Plan.
ARTICLE 4: REGULATORY
1.1General.
1.1.1Licensed COVID-19 Mono Products. With respect to the Licensed COVID-19 Mono Products, during the Collaboration Term, the Parties agree to (a) conduct Regulatory Activities in accordance with the COVID-19 Research and Development Plan and this
Article 4 (Regulatory) and (b) transition to Sanofi the responsibility for all Regulatory Activities in accordance with the Regulatory Transfer Plan as further set forth in this
Article 4 (Regulatory). Neither Party shall conduct Regulatory Activities other than as set forth in the COVID-19 Research and Development Plan, the Regulatory Transfer Plan, or this
Article 4 (Regulatory).
1.1.2Regulatory Transfer Plan. As of the Effective Date, the Parties have agreed to an initial plan, on a country-by-country basis, for the transition to Sanofi of all Regulatory Activities for the Licensed COVID-19 Mono Products to enable Sanofi to become the Regulatory Responsible Party for such Licensed COVID-19 Mono Products, which initial plan includes (a) the Transfer Date, on a country-by-country basis, when Sanofi will become the Regulatory Responsible Party, and assume responsibility, for all Regulatory Activities in such country and (b) the process for transferring the Regulatory Filings and Marketing Authorizations for Licensed COVID-19 Mono Products to Sanofi, which initial plan is attached hereto as Schedule
4.1.2 (Regulatory Transfer Plan) (such plan, as it may be amended in accordance with this Agreement, the “Regulatory Transfer Plan”). Each Party may propose amendments to the Regulatory Transfer Plan through the JSC at any time to reflect any material adjustments to the applicable Regulatory Activities described therein, which amendments will be subject to review, discussion, and determination whether to approve in accordance with
Article 9 (Governance). Any amendments to the Regulatory Transfer Plan approved in accordance with
Article 9 (Governance) shall become effective and supersede the applicable amended portion(s) of the previous Regulatory Transfer Plan as of the date such approval is given.
1.1.3CIC Products and OC Products. With respect to (a) Licensed CIC Products and Licensed OC Products, Sanofi shall have the exclusive right, and (b) Novavax CIC Products and Novavax OC Products, Novavax shall have the exclusive right, in each case (a) and (b), to conduct Regulatory Activities, be the Regulatory Responsible Party for, and file for and obtain Marketing Authorization for each such product that it is Exploiting in its sole discretion at its own cost and expense, but in each case within the scope of the CIC License and OC License, as applicable, and subject to other restrictions applicable to each Party as set forth in
Article 2 (License Grants; Exclusivity) and in this
Article 4 (Regulatory).
1.1.4Adjuvant Products. With respect to Licensed Adjuvanted Products, Sanofi shall have the exclusive right to conduct Regulatory Activities, be the Regulatory Responsible Party for, and file for and obtain Marketing Authorization in its sole discretion at its own cost and expense, within the scope of the Adjuvant License and subject to other restrictions applicable to Sanofi as set forth in
Article 2 (License Grants; Exclusivity) and in this
Article 4 (Regulatory). Novavax shall have the exclusive right to conduct Regulatory Activities, be the Regulatory Responsible Party for, and file for and obtain Marketing Authorization in its sole discretion at its own cost and expense, for Novavax Adjuvant Products.
1.2Regulatory Responsible Party.
1.2.1The Regulatory Responsible Party for Licensed COVID-19 Mono Products shall, in each country, jurisdiction, or region with respect to a Licensed COVID-19 Mono Product, be, unless the Parties agree otherwise in a separate agreement, responsible for all Regulatory Activities for the Licensed COVID-19 Mono Product in such country, jurisdiction, or region, making all Regulatory Filings, obtaining and maintaining Marketing Authorization, and interacting with Regulatory Authorities. Without limiting the foregoing, the Regulatory Responsible Party shall: (i) be responsible to draft and file, in its own name or name of its designee, and shall own, the Regulatory Filings required to seek and maintain Marketing Authorization for such Licensed COVID-19 Mono Product in such country, jurisdiction, or region, and (ii) be responsible for, and the right to prepare and conduct, all communications with the applicable Regulatory Authorities in connection with (ii).
1.2.2On a country-by-country, jurisdiction-by-jurisdiction, and region-by-region basis, from the Effective Date until Sanofi becomes the Regulatory Responsible Party for Licensed COVID-19 Mono Products in a given country, jurisdiction, or region in the Collaboration COVID-19 Territory in accordance with the Regulatory Transfer Plan, Novavax shall be the Regulatory Responsible Party for all Licensed COVID-19 Mono Products in such country, jurisdiction, or region in the Collaboration COVID-19 Territory, and Sanofi shall have the right to participate (to the fullest extent permissible under Applicable Laws) in meetings between Novavax and Regulatory Authorities regarding the Licensed COVID- 19 Mono Products. The Parties shall undertake the transition process in accordance with Section
4.5 (Transfer of Regulatory Filings) and the Regulatory Transfer Plan to transfer the Regulatory Filings and Marketing Authorization (where granted) for the Licensed COVID-19 Mono Products in such country, jurisdiction, or region as of the applicable Transfer Date.
1.2.3With respect to a Licensed COVID-19 Mono Product in a country, jurisdiction, or region, a Party that is not the Regulatory Responsible Party shall cooperate with, and provide reasonable assistance to, the Regulatory Responsible Party in order for the Regulatory Responsible Party to seek and maintain Marketing Authorization for such Licensed
COVID-19 Mono Product in such country, jurisdiction, or region. The Regulatory Responsible Party for the Licensed COVID-19 Mono Product in a country, jurisdiction, or region will consult with the other Party regarding, and keep the other Party informed of, the status of the preparation of all Regulatory Filings it intends to submit, any feedback from any Regulatory Authority regarding any such Regulatory Filings once submitted, and all Marketing Authorizations that it obtains with respect to such Licensed COVID-19 Mono Product in such country, jurisdiction, or region. Without limiting the generality of the foregoing, Sanofi will share, and will cause its Affiliates and Sublicensees to share, copies of their Marketing Authorizations reasonably requested by Novavax to fulfill its obligations under any Existing Strategic Partner Agreement. The Regulatory Responsible Party in a given country, jurisdiction, or region will reimburse the other Party for all FTE Costs and Out-of-Pocket Costs incurred in providing reasonable assistance to such Regulatory Responsible Party under this Section
4.2.3 in accordance with Section
10.7 (Regulatory Costs).
1.2.4If a Party is the Regulatory Responsible Party for a Licensed COVID-19 Mono Product in a country, jurisdiction or region, and the other Party has been allocated Commercialization activities with respect to such Licensed COVID-19 Mono Products in accordance with this Agreement in such country, jurisdiction or region, then (a) the Regulatory Responsible Party must within [***] of receiving a written request from the other Party, deliver an executed all necessary letters of authorization or delegation of authority, in a form approved by the requesting Party, to enable the requesting Party to carry out its Commercialization activities in such country, jurisdiction or region and (b) the Parties will discuss in good faith expediting the timeline for Sanofi becoming the Regulatory Responsible Party in such country, jurisdiction, or region prior to Sanofi Commercializing in such country, jurisdiction, or region. [***].
1.3Manufacturing Changes. For so long as Novavax is the Regulatory Responsible Party for any Licensed COVID-19 Mono Product in any country, jurisdiction, or region, neither Party shall submit to any Regulatory Authority any proposed change to the process for Manufacturing such Licensed COVID-19 Mono Product (or any component thereof), without first discussing such proposed change with the other Party and considering, in good faith, any comments provided by the other Party (which comment shall be provided within a reasonable time based on the urgency of the proposed process change) prior to making the applicable submission to the applicable Regulatory Authority.
1.4Regulatory Diligence. Subject to the terms and conditions of this Agreement, each Party will use [***] to conduct the Regulatory activities for Licensed COVID-19 Mono Products for which such Party is the Regulatory Responsible Party, whether itself, or by or through or with any Affiliate, Sublicensee, or Service Provider. Each Party that is the Marketing Authorization holder for the Licensed COVID-19 Mono Product in a country, jurisdiction, or region will use [***] to obtain and maintain approval from the applicable Regulatory Authority in such country, jurisdiction, or region to sell Licensed COVID-19 Mono Products that are Manufactured by (or on behalf of) Sanofi (as opposed to Novavax) under the Marketing Authorization for the Licensed COVID-19 Mono Product in such country, jurisdiction, or region.
1.5Transfer of Regulatory Filings. Novavax will, in accordance with the Regulatory Transfer Plan, including the timelines set forth therein, initiate a process to transfer to Sanofi or its designated Affiliate, on a country-by-country, jurisdiction-by-jurisdiction, or region-by-region basis, no later than the applicable Transfer Date for such country, jurisdiction, or region, all Regulatory Filings for Licensed COVID-19 Mono Products held by Novavax or any of its Affiliates that are [***] for Sanofi to conduct the Regulatory Activities for which Sanofi is the Regulatory Responsible Party (such transferred Regulatory Filings, “Transferred Regulatory Filings”). The Parties will use [***] to complete such transfer in accordance with the timelines provided in the Regulatory Transfer Plan. Novavax and Sanofi shall cooperate to prepare and deliver any letters or other documents required by any Regulatory Authority to effectuate the transfer of such Transferred Regulatory Filings from Novavax to Sanofi. Notwithstanding any transfer of Regulatory Filings by Novavax or its Affiliate to Sanofi under this Section
4.5 (Transfer of Regulatory Filings), unless otherwise agreed by the Parties in a separate written agreement, for so long as Novavax continues to supply Sanofi with Licensed COVID-19 Mono Product or components thereof, (a) Novavax shall remain responsible for (i) preparing all CMC modules and other Manufacturing-related portions of Regulatory Filings for such Licensed COVID-19 Mono Product or components thereof and (ii) obtaining approvals required to conduct Manufacturing for such Licensed COVID-19 Mono Product or components thereof, and (b) Sanofi shall be responsible for all other portions of Regulatory Filings and Marketing Authorizations for such Licensed COVID-19 Mono Product or components thereof. The Parties shall cooperate to update the Regulatory Filings and approvals related to Manufacturing activities for any Licensed COVID-19 Mono Product or components thereof, as may be required to reflect Manufacturing improvements arising from activities conducted under this Agreement or an Ancillary Agreement with respect to any Licensed COVID-19 Mono Product or components thereof, or otherwise as required by Applicable Laws.
1.6Right of Reference; BMF; DMF.
1.6.1Novavax hereby grants to Sanofi and its Affiliates a right of reference to any INDs, Marketing Authorization Applications, BMFs, DMFs, and other Regulatory Filings and source documents Controlled by Novavax or any of its Affiliates for any Licensed COVID- 19 Mono Product, Licensed COVID-19 Component, or Adjuvant, for reference by Sanofi and its Affiliates, and its or their Sublicensees as [***] to support Sanofi’s Regulatory Activities with respect to the preparation, submission, and maintenance of any Regulatory Filings (including INDs, Marketing Authorization Applications, etc.) and in connection with obtaining and maintaining Marketing Authorization for any Licensed Products for which Sanofi and its Affiliates, and its or their Sublicensees is the Regulatory Responsible Party in accordance with this Agreement. Novavax will use [***] to deliver to the applicable Regulatory Authority, within [***] after a written request is received from Sanofi, a letter authorizing Sanofi and its Affiliates to exercise such right of reference. Novavax shall provide to Sanofi a draft of such letter prior to delivery to the applicable Regulatory Authority, and Sanofi may provide any comments thereto within [***] after receipt from Novavax. Novavax will consider any such timely comments provided by Sanofi in good faith prior to delivery of such letter to the applicable Regulatory Authority.
1.6.2Without limiting the foregoing, upon Sanofi’s request, to the extent any applicable Novavax Know-How and Regulatory Filings have not been disclosed to Sanofi (e.g., before the COVID-19 Manufacturing Handover Date or before release of the Adjuvant Intermediate Technology Escrow), Novavax shall prepare appropriate BMFs or DMFs, as applicable, for the Adjuvant, Licensed COVID-19 Component, or Licensed COVID-19
Mono Product, as applicable, and submit such BMFs or DMFs to the applicable Regulatory Authority in the Territory, and Novavax will be responsible for communicating with Regulatory Authorities in the Territory regarding such BMF or DMF, as applicable. Sanofi shall be entitled to reference such the BMF or DMF, as applicable for the Adjuvant, Licensed COVID-19 Component, or Licensed COVID-19 Mono Product, as applicable, in its Regulatory Filings for any Licensed Product in the Territory.
1.6.3Until the Successful Completion of COVID-19 Manufacturing Technology Transfer and Adjuvant Manufacturing Technology Transfer, Novavax shall also provide to Sanofi at no cost, (a) the open part of the BMF or DMF, as applicable, for the Adjuvant, Licensed COVID-19 Component, or Licensed COVID-19 Mono Product, as applicable, such as the sections containing general information, control of drug substance, container closure system, and stability, (b) an electronic dossier containing data and information related to the Manufacture of Licensed Products in accordance with this Agreement, and (c) upon Sanofi’s written request, information in the closed part of the BMF or DMF, as applicable, for the Licensed COVID-19 Components, Licensed COVID-19 Mono Products, or the Adjuvant, as applicable, that Sanofi reasonably believes is necessary for preparing Regulatory Filings for any Licensed Product in the Territory for which Sanofi is the Regulatory Responsible Party, in each case (a)-(c), excluding any Adjuvant Intermediate Know-How. At the reasonable request of Sanofi, Novavax shall send representative(s) of Novavax to attend meetings with Regulatory Authorities in the Territory relating to the Regulatory Activities regarding the BMFs or DMFs for the Adjuvant, Licensed COVID- 19 Component, or Licensed COVID-19 Mono Products, as applicable.
1.7Communications with Regulatory Authorities.
1.7.1Prompt Disclosures. Each Party will inform the other Party within [***], or such shorter time as is necessary to comply with the reporting requirements of any applicable Regulatory Authority or under Applicable Law, after notification of any action by, or notification or other information that it receives (directly or indirectly) from, any Regulatory Authority in the Territory to the extent such information: (a) raises any material concerns regarding the safety, efficacy, or quality of any Licensed COVID-19 Mono Product; (b) indicates or suggests a potential material liability of either Party to any Third Party in connection with any Licensed COVID-19 Mono Product; (c) is reasonably likely to lead to a clinical hold, recall, market withdrawal, or field alert with respect to any Licensed COVID-19 Mono Product; or (d) relates to expedited and periodic reports of adverse events with respect to any Licensed COVID-19 Mono Product, or Licensed COVID-19 Mono Product complaints, and may have an adverse impact on the receipt or maintenance of Marketing Authorization or the continued Commercialization of a Licensed COVID-19 Mono Product. The Parties will reasonably cooperate with and assist each other in complying with regulatory obligations and communications, including by providing to the Regulatory Responsible Party, promptly after a request and within [***] if such timing is required for the Regulatory Responsible Party for such product to comply with the reporting requirements of any applicable Regulatory Authority or under Applicable Law, such information and documentation that is in the other Party’s possession as may be [***] for the applicable Regulatory Responsible Party to prepare a response to an inquiry from a Regulatory Authority in the Regulatory Responsible Party’s purview with respect to a Licensed COVID-19 Mono Product. Each Party will also promptly provide the other Party with a copy of all correspondence received from a Regulatory Authority specifically regarding the matters
referred to in this Section
4.7.1 (Prompt Disclosures), including any matters regarding the safety, efficacy, or quality of any Licensed COVID-19 Mono Product.
1.7.2Other Material Communications. To the extent not provided pursuant to Section
4.7.1 (Prompt Disclosures), the Regulatory Responsible Party for the Licensed COVID-19 Mono Product will provide the other Party for its review and discussion a brief description of the principal issues raised in each material communication with Regulatory Authorities with respect to any Licensed COVID-19 Mono Product in the Territory [***] after receipt thereof, but in any event within [***] after receipt thereof. The Regulatory Responsible Party for the Licensed COVID-19 Mono Product will allow the other Party a reasonable opportunity to review and comment on such Regulatory Responsible Party’s proposed response to any material communications from any Regulatory Authority in the Territory with respect to any Licensed COVID-19 Mono Product in advance of the transmission of such response, and the Regulatory Responsible Party will reasonably consider all comments timely provided by the other Party in connection therewith.
1.7.3Other Disclosures. In addition to its other obligations under this Agreement, each Party will disclose to the other Party the following regulatory information:
(a)Regulatory Actions. All material information Controlled or otherwise received by such Party pertaining to actions taken by Regulatory Authorities related to any Licensed COVID-19 Mono Product in the Territory, including any notice, audit notice, notice of initiation by Regulatory Authorities of investigations, inspections, detentions, seizures, or injunctions concerning any Licensed COVID-19 Mono Product in the Territory, notice of violation letter (i.e., an untitled letter), warning letter, service of process, or other inquiry; provided that a Party will be entitled to redact those portions thereof to the extent not related to a Licensed COVID-19 Mono Product.
(b)Regulatory Non-Compliance. All information Controlled by such Party pertaining to notices from Regulatory Authorities in the Territory of non-compliance with Applicable Law in connection with any Licensed COVID-19 Mono Product, including receipt of a warning letter or other notice of alleged material non- compliance from any Regulatory Authority relating to any Licensed COVID-19 Mono Product; provided that a Party will be entitled to redact those portions thereof to the extent not related to a Licensed COVID-19 Mono Product.
1.8Compliance; Audit of Records. For any Licensed COVID-19 Mono Product for which (a) Sanofi is the Regulatory Responsible Party, and (b) Novavax, its Affiliates’, its Sublicensees’, and its and their Service Providers’ perform any Research, Development, Manufacturing, or Regulatory Activities hereunder, Sanofi shall have the right, but not the obligation, to conduct an audit of Novavax’s and its Affiliates’ records, maintained in connection with such activities on reasonable notice, during business hours, to confirm compliance with this Section
4.8 (Compliance; Audit of Records). Novavax will (i) use reasonable efforts to obtain a direct right for Sanofi to audit its Sublicensees and Service Providers, and (ii) if despite Novavax’s use of reasonable efforts, is unable to obtain a right for Sanofi to audit such Sublicensee or Service Provider, then Novavax will exercise its audit rights to the extent available under the applicable agreement with such Sublicensee or Service Provider to audit such Sublicensee or Service Provider in accordance with this Section
4.8 (Compliance; Audit of Records). Such audit shall not take place more than once every [***] and shall not include the same records more than once, unless otherwise
required by Applicable Law or at the request of a Regulatory Authority. Each Party will cause its Affiliates, its Sublicensees, and its and their Service Providers to cooperate with the inspecting Party in connection with any such audit.
1.9Pharmacovigilance.
1.9.1The Parties will cooperate with regard to the reporting and handling of safety information involving the Licensed Products in accordance with Applicable Law. Within such time to ensure that all regulatory requirements and timelines are met, the Parties shall negotiate in good faith and execute a pharmacovigilance agreement setting forth the pharmacovigilance procedures for the Parties with respect to Licensed COVID-19 Mono Products that shall include safety data exchange procedures governing the exchange of information affecting each applicable Licensed COVID-19 Mono Product (e.g., serious adverse events, emerging safety issues) to enable each Party to comply with all of its legal and regulatory obligations related to such Licensed COVID-19 Mono Products (the “Pharmacovigilance Agreement”). Each Party shall keep the other Party informed as to any matters related to patient safety and pharmacovigilance and any other material safety event (as defined in such Pharmacovigilance Agreement) in accordance with the timeline set forth therein.
1.9.2As of the Effective Date, as between the Parties, Novavax will initially own and maintain the global safety database for all Licensed COVID-19 Mono Products in the Territory. The Parties will transfer the contents of the global safety database from Novavax to Sanofi by no later than [***]. As and from such date (or such other date as the Parties may agree in writing), Sanofi will own and maintain the global safety database for all Licensed COVID-19 Mono Products that are Exploited by the Parties after the Effective Date at Sanofi’s sole cost and expense; provided that to the extent any information required to maintain such global safety database is in the possession or control of Novavax, its Affiliates, or its Strategic Partners, Novavax shall, within the timeline prescribed by Applicable Laws, provide such information to Sanofi. Each Party that is responsible under this Section
4.9.2 (Pharmacovigilance) to maintain the global safety database for Licensed COVID-19 Mono Products shall fully cooperate with the other Party (at such cooperating Party’s cost and expense) as reasonably requested by the maintaining Party. In the event that a Party is the Marketing Authorization holder for a Licensed COVID-19 Mono Product in a country, jurisdiction or region but is not the Party that is responsible to maintain the global safety database for the Licensed COVID-19 Mono Products in such country, jurisdiction or region, such Party shall, in accordance with Applicable Laws, remain responsible for local reporting obligations in such country, jurisdiction or region (and shall keep the other Party informed of such reporting). Each Party will bear and be responsible for costs and expenses incurred in the performance of its respective obligations under this Section
4.9.2 (Pharmacovigilance) in accordance with Section
10.7 (Regulatory Costs).
ARTICLE 5: TECHNOLOGY TRANSFER
1.1General Technology Transfer and Technical Assistance.
1.1.1Transfer Method. Within [***] after the Effective Date, Novavax will establish a secure data room platform, through which Novavax will transfer to Sanofi all Novavax COVID-19 Know-How and Novavax Adjuvant Know-How required to be transferred in accordance with this Agreement (such secure platform, the “Knowledge Portal”).
1.1.2Transfer of Novavax Know-How.
(a)Transfer of Know-How. Novavax will transfer to Sanofi, through the Knowledge Portal or such other means as the Parties may agree, all Novavax COVID-19 Know-How and Novavax Adjuvant Know-How (other than Novavax Manufacturing Know-How) that is [***] for Sanofi to Exploit the Licensed Products in accordance with the terms and conditions of this Agreement. All such Novavax COVID-19 Know-How and Novavax Adjuvant Know-How shall be in the format agreed to by the Parties through the applicable Committees and Novavax shall use [***] to provide such Know-How in accordance with the time frames set forth in the applicable plans, including the COVID-19 Research and Development Plan, the Regulatory Transfer Plan, and the Technology Transfer Plans, or as otherwise agreed to by the Parties through the applicable Committees.
(b)Confidentiality Obligations. Each Party will have the right to download and retain and make a copy(ies) of all such Know-How provided by the other Party, whether provided through the Knowledge Portal or otherwise (but, for clarity, such Know- How will remain subject to the confidentiality obligations set forth in
Article 11 (Confidentiality and Non-Disclosure)) and shall only be used by the Receiving Party in accordance with the rights, licenses, and terms set forth in this Agreement.
1.1.3Technical Assistance. During the Term, Novavax will make all of its relevant Representatives available (and, at the request of Sanofi, make the relevant Representatives of its or its Affiliates’ or Sublicensees’ and its and their Service Providers, as applicable) to Sanofi to answer Sanofi’s reasonable technical questions regarding the Licensed Novavax Assets, Licensed COVID-19 Mono Product, Licensed COVID-19 Component, and Adjuvant (including components therein but excluding any Adjuvant Intermediate Technology, unless the Adjuvant Intermediate Escrow Trigger Event occurs) to enable Sanofi to practice the full scope of its rights under the Sanofi Licenses, including to Develop, Manufacture, and obtain and maintain Marketing Authorization for Licensed Products. Novavax will provide answers [***] (i.e., within [***]) to any such reasonable requestions for Sanofi.
1.2COVID-19 Manufacturing Technology Transfer.
1.2.1General. Subject to this
Article 5 (Technology Transfer), for Licensed COVID-19 Components and Licensed COVID-19 Mono Products, Novavax and Sanofi shall, [***], conduct a full technology transfer of all Novavax Manufacturing Know-How and Novavax Materials and all related CMC information Controlled by Novavax or its Affiliates (but excluding any CMC information that is not Know-How relating to the Manufacture of Adjuvant except if the COVID-19 Manufacturing Technology Transfer and the Adjuvant Manufacturing Technology Transfer occur concurrently), that is [***] to fully enable Sanofi, its Affiliates, or its or their Service Providers to Manufacture the Licensed COVID-19 Components and Licensed COVID-19 Mono Products (“COVID-19 Manufacturing Technology Transfer”, and that portion of the COVID-19 Manufacturing Technology Transfer related to generating and selecting SARS-CoV-2 Strains for use in Licensed COVID-19 Components, the “Strain Selection Technology Transfer”); provided that for the purpose of the COVID-19 Manufacturing Technology Transfer, if the COVID-19 Manufacturing Technology Transfer occurs before the Adjuvant Manufacturing Technology Transfer, then the Adjuvant utilized to Manufacture Licensed COVID-19 Mono Products may be supplied by Novavax per Section
6.1.2 (Manufacture and Supply
to Sanofi), rather than Manufactured by Sanofi. The initial written plan for COVID-19 Manufacturing Technology Transfer to effectuate such transfer for each of the Licensed COVID-19 Components and Licensed COVID-19 Mono Product are attached hereto as Schedule
5.2.1 (such plans collectively, as may be amended or replaced from time to time in accordance herewith, the “COVID-19 Manufacturing Technology Transfer Plan”). The Parties shall, no later than [***] after the Effective Date, provide the JMSC with an initial draft of the Technology Transfer Budget for the COVID-19 Manufacturing Technology Transfer Plan, which budget the JMSC will review, discuss, and determine whether to approve in accordance with
Article 9 (Governance). Each Party may propose (a) that the JMSC prepare a new COVID-19 Manufacturing Technology Transfer Plan to replace the existing COVID-19 Manufacturing Technology Transfer Plan or (b) to amend the existing COVID-19 Manufacturing Technology Transfer Plan through the JMSC at any time to reflect any material adjustments to the applicable technology transfer activities described therein, and the new or amended COVID-19 Manufacturing Technology Transfer Plan will be subject to review, discussion, and approval by the JMSC in accordance with
Article 9 (Governance). Any new or amended COVID-19 Manufacturing Technology Transfer Plan approved in accordance with
Article 9 (Governance) shall become effective as of the date such approval is given.
1.2.2COVID-19 Manufacturing Handover. All activities under the applicable COVID-19 Manufacturing Technology Transfer Plan shall, unless otherwise agreed by the Parties through the JMSC, be deemed to be complete when Sanofi, its Affiliates, or its or their Service Providers have completed process performance qualification (PPQ) for the Manufacture of Licensed COVID-19 Mono Product in accordance with the COVID-19 Manufacturing Technology Transfer Plan, including Manufacturing and release of each of the following per its specifications: (a) Licensed COVID-19 Component and (b) Licensed COVID-19 Mono Products (such date, the “COVID-19 Manufacturing Handover Date”).
1.3Adjuvant Technology Transfer. Subject to this
Article 5 (Technology Transfer), for the Adjuvant, Novavax and Sanofi shall conduct a full technology transfer of all Novavax Adjuvant Manufacturing Know-How and Novavax Materials and all related CMC information Controlled by Novavax or its Affiliates (that is not Novavax Adjuvant Know-How and excluding any CMC information relating to the Manufacture of Adjuvant Intermediates), that is [***] to fully enable Sanofi, its Affiliates, or its or their Service Providers to Manufacture the Adjuvant, starting from Adjuvant Intermediates (“Adjuvant Manufacturing Technology Transfer”). The Parties will conduct the Adjuvant Manufacturing Technology Transfer in accordance with a written plan to be agreed by the Parties (such plan, as it may be amended or replaced from time to time in accordance with the terms of this Agreement, the “Adjuvant Manufacturing Technology Transfer Plan”). If the Parties are unable to agree to the terms of a proposed Adjuvant Manufacturing Technology Transfer Plan after good faith discussions, including at the JMSC, then the Parties shall appoint an independent technical expert acceptable to both Parties, with a minimum of [***] of experience in manufacturing operations and technology transfer in the pharmaceutical industry, to resolve any disagreements regarding the terms of a proposed Adjuvant Manufacturing Technology Transfer Plan. The decision of such technical expert shall be final and binding on both Parties and the Parties shall equally share the cost of engaging such an expert. The Parties shall provide the JMSC with draft of the Technology Transfer Budget for the Adjuvant Manufacturing Technology Transfer Plan, which budget the JMSC will review, discuss, and determine whether to approve in accordance with
Article 9 (Governance). Each Party may propose (a) that the JMSC prepare a new Adjuvant Manufacturing Technology Transfer Plan to replace the existing Adjuvant Manufacturing Technology Transfer
Plan or (b) to amend the existing Adjuvant Manufacturing Technology Transfer Plan through the JMSC at any time to reflect any material adjustments to the applicable technology transfer activities described therein, and the new or amended Adjuvant Manufacturing Technology Transfer Plan will be subject to review and discussion by the JMSC in accordance with
Article 9 (Governance) and the Parties will approve such plan (subject to Section
9.18 (Decision Making)). Any new or amended Adjuvant Manufacturing Technology Transfer Plan approved in accordance with
Article 9 (Governance) shall become effective as of the date such approval is given. All activities under the applicable Adjuvant Manufacturing Technology Transfer Plan shall, unless otherwise agreed by the Parties through the JMSC, be deemed to be complete when Sanofi, its Affiliates, or its or their Service Providers have completed process performance qualification (PPQ) for the Manufacture of the Adjuvant in accordance with the Adjuvant Manufacturing Technology Transfer Plan (such date, the “Adjuvant Manufacturing Handover Date”).
1.4Technology Transfer Diligence. Subject to the terms and conditions of this Agreement, each Party will use [***] to conduct the Technology Transfer activities allocated to it under each Technology Transfer Plan, including the timelines set forth in such Technology Transfer Plan(s) and otherwise pursuant to this Agreement, whether itself, or by or through or with any Affiliate or Sublicensee.
1.5Updates to Technology Transfer Plan for Manufacturing Slots. Sanofi may propose a reasonable acceleration to the timelines set forth in any Technology Transfer Plan, including for the purpose of meeting a Manufacturing slot deadline, which proposed amendment the JMSC will review, discuss, and determine whether to approve in accordance with
Article 9 (Governance) (subject to Section
9.18 (Decision Making)).
1.6Adjuvant Intermediates Technology Escrow.
1.6.1Establishment. Within [***] after the Effective Date, the Parties shall designate a neutral, independent Third Party agreed by the Parties to serve as an escrow agent to hold the Adjuvant Intermediate Technology Escrow. The Parties shall enter in a three (3)-way agreement with such escrow agent governing the confidentiality, maintenance, costs, release criteria, and technology transfer plan for escrow release, in each case, in accordance with this Agreement. Within [***] after entering into an escrow agreement with such escrow agent, Novavax shall deposit the Adjuvant Intermediate Technology into the Adjuvant Intermediate Technology Escrow.
1.6.2Maintenance. The Adjuvant Intermediate Technology Escrow will include (a) Adjuvant Intermediate Know-How that shall be stored electronically, with backups and disaster recovery protocols and other commercially reasonable safeguards, and (b) biological and chemical Material samples from the Manufacturing process for the Adjuvant Intermediate from raw material that shall be stored under appropriate conditions in compliance with GMP and Applicable Laws. Novavax shall be responsible for tracking and maintaining the viability and chemical purity of the stored Materials and shall replace any Materials that no longer meet the applicable specifications for such Materials for the intended applications.
1.6.3Updates. At least [***], Novavax shall deposit in the Adjuvant Intermediate Technology Escrow any new Adjuvant Intermediate Technology that is not already in the then-current version of the Adjuvant Intermediate Technology Escrow.
1.6.4Release from Escrow. Sanofi, pursuant to a written notice, may instruct the escrow agent to release all Adjuvant Intermediate Technology from the Adjuvant Intermediate Technology Escrow, in accordance with the terms of the applicable escrow agreement, only upon the occurrence of the Adjuvant Intermediate Escrow Trigger Event.
1.6.5Support Associated with Release from Escrow.
[***] after the occurrence of the Adjuvant Intermediate Escrow Trigger Event, Novavax and Sanofi shall conduct a full technology transfer with respect to all Know-How and Materials held in the Adjuvant Intermediate Technology Escrow in accordance with the plan agreed by the Parties and set forth in the Adjuvant Intermediate Technology Escrow agreement to enable Sanofi to Manufacture the Adjuvant Intermediate. The Adjuvant Intermediate Technology Escrow agreement will provide that Novavax shall, after the Adjuvant Intermediate Escrow Trigger Event, transfer to Sanofi any CMC information Controlled by Novavax or its Affiliates related to the Adjuvant Intermediates, to the extent not already included in the Adjuvant Intermediate Technology Escrow. Each Party will reasonably cooperate with the other Party to effectuate such transfers.
1.6.6[***].
(a)[***].
(b)[***].
1.7Other Escrows.
1.7.1Establishment. Within [***] after the Effective Date, the Parties shall designate a neutral, independent Third Party agreed by the Parties to serve as an escrow agent to hold the Adjuvant Technology Escrow and COVID-19 Technology Escrow. The Parties shall
enter in a three (3)-way agreement with such escrow agent governing the confidentiality, maintenance, costs, release criteria, and technology transfer plan for escrow release, in each case, in accordance with this Agreement. Within [***] after entering into an escrow agreement with such escrow agent, Novavax shall deposit (a) the Novavax Adjuvant Manufacturing Know-How and Novavax Adjuvant Materials that has not already been transferred to Sanofi as of such time into the Adjuvant Technology Escrow and (b) the Novavax COVID-19 Manufacturing Know-How and Novavax COVID-19 Materials that has not already been transferred to Sanofi as of such time into the COVID-19 Technology Escrow.
1.7.2Maintenance. All Know-How in the Adjuvant Technology Escrow and COVID-19 Technology Escrow shall be stored electronically, with backups and disaster recovery protocols and other commercially reasonable safeguards. All biological and chemical Material samples in the Adjuvant Technology Escrow and COVID-19 Technology Escrow shall be stored under appropriate conditions in compliance with GMP and Applicable Laws. [***].
1.7.3Updates. At least [***] prior to the Successful Completion of COVID-19 Manufacturing Technology Transfer or Adjuvant Manufacturing Technology Transfer, as applicable, Novavax shall deposit in each respective escrow any newly generated Novavax COVID-19 Manufacturing Know-How, Novavax Adjuvant Manufacturing Know-How, and Novavax Materials, in each case, that is not (a) already included in the Adjuvant Technology Escrow or COVID-19 Technology Escrow, as applicable and (b) has not been otherwise transferred to Sanofi pursuant to this
Article 5 (Technology Transfer).
1.7.4Release from Escrow. At anytime before Successful Completion of COVID-19 Manufacturing Technology Transfer or Adjuvant Manufacturing Technology Transfer, as applicable, Sanofi may instruct the escrow agent to release the Adjuvant Technology Escrow or COVID-19 Technology Escrow in accordance with the terms of the applicable escrow agreement only upon the occurrence of an applicable Other Escrow Trigger Event.
1.7.5Termination of Escrow. The Adjuvant Technology Escrow and COVID-19 Technology Escrow shall automatically terminate upon the Successful Completion of COVID-19 Manufacturing Technology Transfer or Adjuvant Manufacturing Technology Transfer, as applicable. Novavax shall be responsible for coordinating with the escrow agent for the return or destruction of any Know-How and Materials.
1.8Escrow Material License. Novavax hereby grants to Sanofi, and confirms the grant to Sanofi of (notwithstanding any release of any of the foregoing technology from the escrow agent and not directly from Novavax), the licenses set forth in Section
2.1 (Licenses) to use (a) the Adjuvant Intermediate Technology in the Adjuvant Intermediate Technology Escrow, (b) the Novavax Adjuvant Manufacturing Know-How and Novavax Adjuvant Materials in the Adjuvant Technology Escrow, and (c) the Novavax COVID-19 Manufacturing Know-How and Novavax COVID-19 Materials in the COVID-19 Technology Escrow, in each case ((a), (b), and (c)) solely to Exploit the Licensed Products as permitted under this Agreement. The foregoing license grant is effective as of the Effective Date, however, Sanofi covenants on behalf of itself and its Affiliates, successors, Service Providers, and Sublicensees, that it shall not exercise its rights under such
license until an Adjuvant Intermediate Escrow Trigger Event or an Other Escrow Trigger Event, as applicable, has occurred.
1.9Ancillary Agreements. To the extent necessary to effectuate the Parties’ obligations under this
Article 5 (Technology Transfer), the Parties will negotiate in good faith and execute any Ancillary Agreements that a Party may request be executed in order to comply with Applicable Law.
ARTICLE 6: MANUFACTURING
1.1Novavax Manufacturing.
1.1.1Supply for Novavax. Novavax shall be responsible for Manufacturing, whether itself or by or through any Affiliate or Service Provider, all supplies of the Licensed COVID-19 Mono Products and Licensed COVID-19 Component for Novavax’s and its Affiliates’ Exploitation of Novavax Products in the Territory as provided in this Agreement. After the Successful Completion of COVID-19 Manufacturing Technology Transfer, Novavax may provide written notice to Sanofi that Novavax is interested in receiving supply of Licensed COVID-19 Mono Products from Sanofi solely to comply with its obligations under one (1) or more APAs or Existing Strategic Partner Agreements. Upon receiving such written notice, (a) the Parties will negotiate in good faith the terms under which the Parties may enter a supply agreement under which Sanofi will supply the Licensed COVID-19 Mono Product to Novavax, and (b) until the Parties enter such agreement, Sanofi may, but is not obligated to, supply Licensed COVID-19 Mono Products to Novavax in accordance with the terms of the COVID-19 Supply Agreement, mutatis mutandis.
1.1.2Manufacture and Supply to Sanofi.
(a)Licensed COVID-19 Mono Product and Licensed COVID Component. As between the Parties, Novavax shall be responsible for the Manufacture of and supply to Sanofi, whether itself, or by any Affiliate, or through Service Providers, all non-GMP and GMP supplies of (i) Licensed COVID-19 Mono Product (in such forms or presentations as reasonably requested in writing by Sanofi) for any use (including use in Clinical Trials and for other Research or Development uses) or sale (or other Commercial use) of Licensed COVID-19 Mono Product in the Collaboration COVID-19 Territory and (ii) Licensed COVID-19 Component for use in Licensed CIC Product and Licensed OC Product in the Territory, in each case ((i) and (ii)), for the period starting from the Effective Date and ending on a country-by-country basis upon the later to occur of (A) the COVID-19 Manufacturing Handover Date, and (B) the date on which Sanofi sells a dose of Licensed COVID-19 Mono Products in such country that is Manufactured by or (or on behalf of) Sanofi (as opposed to Novavax).
(b)Adjuvant. Unless otherwise agreed in writing, including in the Adjuvant Supply Agreement, Novavax shall be responsible for the Manufacture of and supply to Sanofi, whether itself, or by any Affiliate, or through any Service Provider, for any use (including use in Clinical Trials and for other Research or Development uses) or sale (or other Commercial use) permitted under this Agreement, (i) [***] and [***] until the Adjuvant Manufacturing Handover Date, and (ii) Adjuvant Intermediates, from and after the Adjuvant Manufacturing Handover Date, and until both (A) the Adjuvant Intermediate Escrow Trigger Event has occurred and Novavax has transferred to Sanofi the Adjuvant Intermediate
Technology, and (B) Sanofi is able to obtain the Materials supplied by [***] to Novavax pursuant to the [***] that are used by Novavax to Manufacture the Adjuvant Intermediates.
(c)Capacity Allocation. For so long as Novavax is responsible for Manufacturing Licensed COVID-19 Mono Products, Licensed COVID-19 Components, ***] and [***], and [***] under this Section
6.1 (Novavax Manufacturing), Novavax and its Affiliates shall not allocate its and their Manufacturing capacity (including reactor space, labor and raw materials, and including capacity of its Service Providers available under Novavax’s agreements with such Service Providers) for any Licensed COVID-19 Mono Product, Licensed COVID-19 Component, [***] and [***], and Adjuvant Intermediates, as it exists as of the Effective Date, in a manner less favorable to Sanofi (including the timing of forecasting, submission of orders, and billing) than for (i) any other customer to whom Novavax supplies the same Licensed COVID- 19 Mono Product, Licensed COVID-19 Component, [***] and [***], or Adjuvant Intermediates, as applicable, or (ii) on and after the 2024/2025 Season, Novavax’s own use other than to fulfil its commitments under the APAs, Existing Strategic Partner Agreements, and GAVI Arrangements.
1.2Sanofi Manufacturing.
1.2.1Sanofi General Rights. Except for the Manufacturing and supply responsibilities that are allocated to Novavax under Section
6.1 (Novavax Manufacturing), after the COVID-19 Manufacturing Handover Date, Sanofi shall be responsible to Manufacture, whether itself or by or through any Affiliate, Sublicensee, or Service Provider, all supplies of the Licensed Products for Sanofi’s and its Affiliates’ and Sublicensees’ Exploitation of the Licensed Products in the Territory (or with respect to the Licensed COVID-19 Mono Product, the Collaboration COVID-19 Territory) as provided in this Agreement.
1.3Supply Agreements.
1.3.1Within [***] after the Effective Date, the Parties shall enter into one or more supply agreements governing the terms and conditions of Novavax’s supply to Sanofi of Licensed COVID-19 Mono Product (in such forms or presentations as reasonably requested in writing by Sanofi), including Licensed COVID-19 Component (each a “COVID-19 Supply Agreement”). Select key terms that will be included in the applicable COVID-19 Supply Agreement are set forth in the term sheet attached hereto as Schedule
6.3.1 (COVID-19 Supply Agreement Key Terms), which shall be binding on the Parties unless and until such time as the Parties have executed the applicable COVID-19 Supply Agreement. For clarity, within [***] after the Effective Date the Parties shall convene a meeting to discuss the contracting strategy (e.g., one (1) COVID-19 Supply Agreement for both Research and Development and Commercial supply or more than one
(1) separate agreement), and within [***] after such meeting, Novavax will deliver the initial draft(s) of the COVID-19 Supply Agreement(s) to Sanofi, and Sanofi shall have [***] after the receipt of such initial draft of the COVID- 19 Supply Agreement(s) to provide its comments, and the Parties shall continue with the same cadence until the COVID-19 Supply Agreement(s) is executed per the first sentence of this Section
6.3.1.
1.3.2Within [***] after the Effective Date, the Parties shall enter into a supply agreement governing the terms and conditions of Novavax’s supply to Sanofi of (a) [***] and [***] and (b) Adjuvant Intermediates, as applicable (the “Adjuvant Supply Agreement”). Select key terms that will be included the Adjuvant Supply Agreement are set forth in the term sheet attached hereto as Schedule
6.3.2 (Adjuvant Supply Agreement Key Terms), which shall be binding on the Parties unless and until such time as the Parties have executed the Adjuvant Supply Agreement. For clarity, within [***] after the Effective Date the Parties shall convene a meeting to discuss the contracting strategy (e.g., one Adjuvant Supply Agreement for both Research and Development and Commercial supply or more than one (1) separate agreement), and within [***] after such meeting, Novavax will deliver the initial drafts of the Adjuvant Supply Agreement(s) to Sanofi, and Sanofi shall have [***] after the receipt of such initial draft of the Adjuvant Supply Agreement(s) to provide its comments, and the Parties shall continue with the same cadence until the Adjuvant Supply Agreement(s) is executed per the first sentence of this Section
6.3.2.
1.4Quality Agreements. Concurrent with the execution of the COVID-19 Supply Agreement(s) and the Adjuvant Supply Agreement(s), as applicable, the Parties will negotiate in good faith and execute one or more quality agreements with respect to the supply of any Licensed COVID-19 Mono Product, Licensed COVID-19 Component, [***] and [***], Adjuvant Intermediates.
1.5Manufacturing Site Change. For so long as Novavax, by itself or by its Affiliates or Service Provider, Manufactures and supplies Licensed COVID-19 Mono Products (or any component thereof) to Sanofi hereunder (including under any Supply Agreement), Novavax will not change, and will not permit its Affiliate or Service Provider to change, the Manufacturing site from which Novavax supplies Sanofi the Licensed COVID-19 Products (or any component thereof), without the prior written consent of Sanofi.
ARTICLE 7: COMMERCIALIZATION
1.1General.
1.1.1Licensed COVID-19 Mono Products. With respect to Licensed COVID-19 Mono Products, during the Collaboration Term, the Parties agree to collaborate to conduct Commercialization activities for the Licensed COVID-19 Mono Products in the Collaboration COVID-19 Territory in accordance with the Licensed COVID-19 Mono Product Commercialization Framework as further described in Section
7.2 (Licensed COVID-19 Mono Product Commercialization Framework). Except as expressly set forth in this
Article 7 (Commercialization), neither Party shall conduct any Commercialization activities for any Licensed COVID-19 Mono Product in the Collaboration COVID-19 Territory.
1.1.2CIC Products and OC Products. Sanofi shall have the right for Licensed CIC Products and Licensed OC Products, and Novavax shall have the right for Novavax CIC Products and Novavax OC Products, in each case, to conduct Commercialization activities for each such product that such Party is Exploiting in its sole discretion at its own cost and expense, but, in each case, within the scope of the CIC License and OC License, as applicable, and subject to other restrictions applicable to each Party as set forth in
Article 2 (License Grants; Exclusivity) and in this
Article 7 (Commercialization).
1.1.3Adjuvant Products. Sanofi shall have the right for Licensed Adjuvanted Products, and Novavax shall have the right for Novavax Adjuvant Products, to conduct Commercialization activities for each such product that such Party is Exploiting in its sole discretion at its own cost and expense, but, in each case, within the scope of the Adjuvant License and subject to other restrictions as set forth in
Article 2 (License Grants; Exclusivity) and in this
Article 7 (Commercialization).
1.2Licensed COVID-19 Mono Product Commercialization Framework. The Parties have agreed to an initial framework for the Commercialization of Licensed COVID-19 Mono Products pursuant to which they have allocated between them responsibility for Commercialization of Licensed COVID-19 Mono Products throughout the Collaboration COVID-19 Territory (such initial framework, and any updated or amended version thereof, a “Licensed COVID-19 Mono Product Commercialization Framework”). The initial Licensed COVID-19 Mono Product Commercialization Framework is attached hereto as Schedule
7.2. Beginning [***] ([***]), and thereafter in each Calendar Year for the Season commencing the following Calendar Year, no later than [***], the JCC will review, discuss, and determine whether to approve updates to the Licensed COVID-19 Mono Product Commercialization Framework in accordance with
Article 9 (Governance), including which Party will be responsible for all Commercialization activities in the United States. Additionally, either Party may propose amendments to the Licensed COVID-19 Mono Product Commercialization Framework at any time, each of which updates the JCC will review, discuss, and determine whether to approve in accordance with
Article 9 (Governance). Each Licensed COVID-19 Mono Product Commercialization Framework will be developed in coordination with the Licensed COVID-19 Mono Product Medical Framework for the applicable Licensed COVID-19 Mono Product.
1.3Existing APAs and Settlement Arrangements. Sanofi acknowledges that Novavax has certain rights and obligations under the APAs and Existing Settlement Arrangements in certain countries in the Collaboration COVID-19 Territory for the Manufacture and supply of Licensed COVID-19 Mono Products to such countries. Novavax shall have the right, in accordance with the Licensed COVID-19 Mono Product Commercialization Framework, to Commercialize Licensed COVID-19 Mono Products pursuant to each APA and the Gavi Arrangement for the remainder of the term of each such APA and the Gavi Arrangement. If, for a particular country in the Collaboration COVID- 19 Territory for which Novavax has entered an APA or for which the Gavi Arrangement applies, Novavax has the right to Commercialize Licensed COVID-19 Mono Products in such country, then Sanofi shall have the right, but not the obligation, to Commercialize Licensed COVID-19 Mono Products in such country in accordance with the Licensed COVID-19 Mono Product Commercialization Framework. The Parties will collaborate in good faith to effectuate a smooth transition as Novavax winds-down performance under each APA and Sanofi assumes the sole right to Commercialize in the applicable country. [***]. Upon the JCC’s approval, such proposed Settlement Arrangement will be deemed a Settlement Arrangement for purposes of this Agreement.
1.4Commercialization Diligence. Each Party will, whether itself or by or through or with any Affiliate, Sublicensees, or its or their Service Providers, use [***] to perform the Commercialization activities allocated to it in the Licensed COVID-19 Mono Product Commercialization Framework. Notwithstanding the generality of the foregoing, Sanofi will use [***] to (a) obtain Marketing Authorization for and Commercialize at least one (1) Milestone Eligible Licensed COVID-19 Mono Product in the United States and each other Major Market country and (b) obtain Marketing Authorization and Commercialize one
(1) Licensed CIC Product or Licensed OC Product in each Major Market country, provided that on a Major Market-by-Major Market basis, once Sanofi is Commercializing a Licensed CIC Product or Licensed OC Product in a Major Market country, Sanofi’s obligations to Commercialize Licensed COVID-19 Mono Products in such Major Market under this Section
7.4 (Commercialization Diligence) will terminate.
1.5Commercialization Reports.
1.5.1Novavax Commercialization Reports. To the extent Novavax is conducting Commercialization activities in the Collaboration COVID-19 Territory during an applicable [***] for Licensed COVID-19 Mono Products, within [***] after the end of each Calendar Quarter, Novavax will provide the JCC with a summary report of Novavax’s Commercialization performance, including a summary of financial and market information related thereto, and any Regulatory activities in connection therewith. Each [***] Commercialization report delivered by Novavax in accordance with this Section
7.5.1 (Novavax Commercialization Reports) is a “Novavax Commercialization Report.” Novavax Commercialization Reports are the Confidential Information of Novavax.
1.5.2Sanofi Commercialization Reports. To the extent Sanofi is conducting Commercialization activities in the Collaboration COVID-19 Territory during an applicable [***] for Licensed Products, Sanofi will provide the JCC with a report summarizing Sanofi’s Commercialization activities for Licensed COVID-19 Mono Products, and with respect to other Licensed Products, Sanofi will provide information necessary for Novavax’s financial reporting requirements under Applicable Law. Each [***] Commercialization report delivered by Sanofi in accordance with this Section
7.5.2 (Sanofi Commercialization Reports) is a “Sanofi Commercialization Report.” Sanofi Commercialization Reports are the Confidential Information of Sanofi.
1.6Advertising and Promotional Materials.
1.6.1Promotional Materials. As between the Parties, Sanofi has the sole right with respect to the creation, preparation, production, reproduction, review (medical, legal, and regulatory), and filing with the applicable Regulatory Authorities of promotional materials relating to each Licensed Product that it is Commercializing. All rights, title, and interests, in and to any and all promotional materials for any Licensed Product created by or on behalf of Sanofi will be solely owned by Sanofi or its Affiliate. Sanofi will provide Novavax with the final draft of any major proposed promotional materials for the Licensed COVID-19 Mono Products and will consider in good faith comments provided by Novavax through the JCC that relate to such promotional materials for use in countries in which Novavax has obligations under an APA or has otherwise been allocated Commercialization activities in the Collaboration COVID-19 Territory under the Licensed COVID-19 Mono Product Commercialization Framework from time to time. Sanofi will supply Novavax an electronic copy of the promotional materials for the Licensed COVID-19 Mono Product under this Section
7.6.1 (Promotional Materials) for Novavax to service those countries in which Novavax has obligations under an APA or has otherwise been allocated Commercialization activities in the Collaboration COVID-19 Territory and Novavax may not, without the prior written consent of Sanofi, independently create or use promotional materials for Licensed COVID-19 Mono Products in any country in the Collaboration COVID-19 Territory.
1.6.2Labelling.
(a)Licensed COVID-19 Mono Products. For so long as Novavax is the Regulatory Responsible Party with respect to a Licensed COVID-19 Mono Product in a country, jurisdiction, or region within the Collaboration COVID-19 Territory, Novavax will develop and approve packaging and labeling for such Licensed COVID-19 Mono Product, including the core data sheet for such Licensed COVID-19 Mono Product in such country, jurisdiction, or region, and Novavax will afford Sanofi an opportunity to provide comments on initial and material updates to the packaging and labeling for Licensed COVID-19 Mono Products through the JRDC. Sanofi shall provide its comments on any such proposed packaging and labeling to Novavax within [***] after Sanofi receives a copy of such proposed packaging or labeling, and Novavax will consider in good faith any such timely comments provided by Sanofi. Where Sanofi is the Regulatory Responsible Party with respect to a Licensed COVID-19 Mono Products in a country, jurisdiction, or region within the Collaboration COVID-19 Territory, Sanofi will develop and approve packaging and labeling for such Licensed COVID-19 Mono Products, including the core data sheet for such Licensed COVID-19 Mono Product in such country, jurisdiction, or region, and Sanofi will afford Novavax an opportunity to provide comments on [***] updates to the packaging and labeling for Licensed COVID-19 Mono Products through the JRDC. Novavax shall provide its comments on any such proposed packaging and labeling to Sanofi within [***] after Novavax receives a copy of such proposed packaging or labeling, and Sanofi will consider in good faith any such timely comments provided by Novavax.
(b)CIC Products and Licensed OC Products. With respect to Licensed CIC Products and Licensed OC Products, as between the Parties, Sanofi shall have the sole right to develop and approve packaging and labeling for such products, including the core data sheet in the Territory. With respect to Novavax CIC Products and Novavax OC Products, as between the Parties, Novavax shall have the sole right to develop and approve packaging and labeling for such products, including the core data sheet in the Territory.
(c)Adjuvant Products. With respect to Licensed Adjuvanted Products, as between the Parties, Sanofi shall have the sole right to develop and approve packaging and labeling for such products, including the core data sheet in the Territory. With respect to Novavax Adjuvant Products, as between the Parties, Novavax shall have the sole right to develop and approve packaging and labeling for such products, including the core data sheet in the Territory.
1.7Sales and Distribution.
1.7.1Sales.
(a)Licensed COVID-19 Mono Products. With respect to the Licensed COVID-19 Mono Products, during the Collaboration Term, each Party shall book sales for Licensed COVID-19 Mono Products sold by or on behalf of such Party in such country in the Collaboration COVID-19 Territory in accordance with the then- current Licensed COVID-19 Mono Product Commercialization Framework for the applicable Seasons.
(b)CIC Products and Licensed OC Products. With respect to Licensed CIC Products and Licensed OC Products, as between the Parties, Sanofi shall have the sole right to book sales of such Licensed Products sold by or on behalf of Sanofi, its Affiliates, or its or their Sublicensees or licenses in the Territory. With respect to Novavax CIC Products and Novavax OC Products, as between the Parties, Novavax shall have the sole right to book sales of such Novavax Products sold by or on behalf of Novavax, its Affiliates, or its or their Sublicensees or licenses in the Territory.
(c)Adjuvant Products. With respect to Licensed Adjuvanted Products in the Adjuvant Field, as between the Parties, Sanofi shall have the sole right to book sales of such Licensed Adjuvanted Products sold by or on behalf of Sanofi, its Affiliates, or its or their Sublicensees or licenses in the Territory. With respect to Novavax Adjuvant Products, as between the Parties, Novavax shall have the sole right to book sales of such Novavax Adjuvant Products sold by or on behalf of Novavax, its Affiliates, or its or their Sublicensees or licenses in the Territory.
1.7.2Warehousing and Distribution. Each Party shall conduct all warehousing and distribution activities for Licensed COVID-19 Mono Products sold by or on behalf of such Party in such country in the Collaboration COVID-19 Territory in accordance with the Licensed COVID-19 Mono Product Commercialization Framework. For Licensed Products other than Licensed COVID-19 Mono Products, Sanofi shall, and for Novavax Products, Novavax shall, conduct all warehousing and distribution activities for such Licensed Products or Novavax Products, as applicable, sold by or on behalf of such Party, its Affiliates, its or their Sublicensees in the Territory.
1.8Training. Unless otherwise agreed by the Parties in writing or set forth in the Licensed COVID- 19 Mono Product Commercialization Framework, as between the Parties, Sanofi has the sole right to create the contents of all training materials for all sales forces for the Licensed COVID-19 Mono Products in the Collaboration COVID-19 Territory. Sanofi will supply Novavax with final versions of training materials for the Licensed COVID-19 Mono Product under this Section
7.8 (Training) to service those countries in which Novavax has obligations under an APA or has otherwise been allocated Commercialization activities in the Collaboration COVID-19 Territory.
1.9Recalls, Market Withdrawals, or Corrective Actions. In the event that any Regulatory Authority issues or requests a Recall with respect to a Licensed Product in the Territory, the Regulatory Responsible Party for such Licensed Product, on a country-by-country basis, has the sole right to decide whether to conduct a Recall and the manner in which any such Recall will be conducted.
1.10Market Access Strategy; Pricing Approvals.
1.10.1Pricing.
(a)[***] Season, except as specifically allocated to Sanofi in the Licensed COVID-19 Mono Product Commercialization Framework or as otherwise agreed to in writing between the Parties, Novavax shall have the sole right to determine the market access strategy, including all matters relating to setting prices (including all discounts, rebates, and other similar commercial pricing matters) for all Licensed COVID-19 Mono Products in the Collaboration COVID-19 Territory.
(b)[***] Season, except as specifically allocated to Novavax in the Licensed COVID-19 Mono Product Commercialization Framework or as otherwise agreed to in writing between the Parties, Sanofi shall have the sole right to determine the market access strategy, including all matters relating to setting prices (including all discounts, rebates, and other similar commercial pricing matters) for all Licensed COVID-19 Mono Products in the Collaboration COVID-19 Territory.
(c)[***] Season, the Party set forth in the Licensed COVID-19 Mono Product Commercialization Framework for the applicable Season as responsible for Commercialization in the United States shall have the sole right to determine the market access strategy, including all matters relating to setting prices (including all discounts, rebates, and other similar commercial pricing matters) for all Licensed COVID-19 Mono Products in the United States. For clarity, if Sanofi is the Party set forth in the Licensed COVID-19 Mono Product Commercialization Framework for the applicable Season, then Sanofi may offer to sell or sell Licensed COVID-19 Mono Products to customers in the United States together with other products (i.e., under the same contract); provided that in such case Sanofi will not apply discounts to the Licensed COVID-19 Mono Products disproportionately as compared to such other products.
(d)[***] Seasons, except as specifically allocated to Novavax in the Licensed COVID-19 Mono Product Commercialization Framework or as otherwise agreed to in writing between the Parties, Sanofi has the sole right to determine the market access strategy, including all matters relating to setting prices (including all discounts, rebates, and other similar commercial pricing matters) for all Licensed COVID-19 Mono Products in the Collaboration COVID-19 Territory other than in the United States (other than pricing for Licensed Products sold under existing APAs and Settlement Arrangements, provided that for Settlement Arrangements that are not Existing Settlement Arrangements, Novavax shall only have the right to submit confidential tenders, and any non-confidential tenders or other pricing by Novavax, in each case, will require Sanofi’s consent, not to be unreasonably withheld). For clarity, Sanofi may offer to sell or sell Licensed COVID-19 Mono Products to customers throughout the Collaboration COVID-19 Territory other than in the United States together with other products (i.e., under the same contract); provided that in such case Sanofi will not apply discounts to the Licensed COVID-19 Mono Products disproportionately as compared to such other products.
(e)Co-Commercialization. For clarity, in any Season in which both Parties have been allocated Commercialization activities under the Licensed COVID-19 Mono Product Commercialization Framework in the same country (other than pricing for Licensed Products sold under existing APAs and Settlement Arrangements, provided that for Settlement Arrangements that are not Existing Settlement Arrangements, Novavax shall only have the right to submit confidential tenders, and any non-confidential tenders or other pricing by Novavax, in each case, will require Sanofi’s consent, not to be unreasonably withheld), Sanofi shall have the sole right to determine the market access strategy, including all matters relating to setting prices (including all discounts, rebates, and other similar commercial
pricing matters) for all Licensed COVID-19 Mono Products in the Collaboration COVID-19 Territory.
(f)Cooperation; Review by JCC. Each Party that is the designated to have the right to determine prices of the Licensed COVID-19 Mono Products in a country for any specific Season under this Section
7.10.1 (Pricing) shall provide the other Party with an opportunity to review the [***] pricing policy created by such Party with respect to the applicable Licensed COVID-19 Mono Products in such country for such Season, [***] through the JCC, and the Party that creates such pricing policy shall consider in good faith the other Party’s reasonable comments with respect thereto that are delivered to such Party within [***] after such [***] pricing policy is distributed to the other Party for review by the JCC.
(g)Reporting. The Party that has been allocated responsibility to Commercialize Licensed COVID-19 Mono Products in a country for a given Season shall, unless the Parties agree otherwise in writing, be responsible for reporting its prices for such Licensed COVID-19 Mono Products to the applicable Governmental Authority in such country.
1.10.2Pricing Approvals.
(a)[***] Season, except as specifically allocated to Sanofi in the Licensed COVID-19 Mono Product Commercialization Framework or as otherwise agreed to in writing between the Parties, Novavax shall have the sole right to apply for, and update, Pricing Approvals for all Licensed COVID-19 Mono Products in the United States and all countries within the Collaboration COVID-19 Territory in which Novavax has an APA or Settlement Arrangement.
(b)[***] Season, except as specifically allocated to Novavax in the Licensed COVID-19 Mono Product Commercialization Framework or as otherwise agreed to in writing between the Parties, Sanofi shall have the sole right to apply for, and update, Pricing Approvals for such Licensed COVID-19 Mono Products in all countries within the Collaboration COVID-19 Territory.
(c)[***] Season, the Party set forth in the Licensed COVID-19 Mono Product Commercialization Framework for the applicable Season as responsible for Commercialization in the United States shall have the sole right to apply for, and update, Pricing Approvals for such Licensed COVID-19 Mono Products in the United States.
(d)[***] Season, except as specifically allocated to Novavax in the Licensed COVID-19 Mono Product Commercialization Framework or as otherwise agreed to in writing between the Parties, Sanofi shall have the sole right to apply for, and update, Pricing Approvals for such Licensed COVID-19 Mono Products in all countries within the Collaboration COVID-19 Territory other than the United States.
(e)Pricing Approval if not Regulatory Responsible Party. For clarity, in the event that a Party that is designated to have the right to apply for, and update, Pricing Approvals for such Licensed COVID-19 Mono Products in a country within the Collaboration COVID-19 Territory in accordance with paragraph (a)-(d) above is, at the relevant time, not the Regulatory Responsible Party, then the Regulatory Responsible Party shall act as the designated Party’s agent and shall fully cooperate and must apply for, and update, Pricing Approvals for such Licensed COVID-19 Mono Products in such country within the Collaboration COVID-19 Territory as directed by the other Party.
(f)CIC Products and Licensed OC Products. With respect to Licensed CIC Products and Licensed OC Products, as between the Parties, Sanofi shall have the sole right to apply for, and update, Pricing Approvals of such Licensed Products sold by or on behalf of Sanofi, its Affiliates, its or their Sublicensees or licenses in the Territory. With respect to Novavax CIC Products and Novavax OC Products, as between the Parties, Novavax shall have the sole right to apply for, and update, Pricing Approvals for such Novavax Products sold by or on behalf of Novavax, its Affiliates, or its or their Sublicensees or licenses in the Territory.
(g)Adjuvant Products. With respect to Licensed Adjuvanted Products in the Adjuvant Field, as between the Parties, Sanofi shall have the sole right to apply for, and update, Pricing Approvals for such Licensed Adjuvanted Products sold by or on behalf of Sanofi, its Affiliates, or its or their Sublicensees or licenses in the Territory. With respect to Novavax Adjuvant Products, as between the Parties, Novavax shall have the sole right to apply for, and update, Pricing Approvals for such Novavax Adjuvant Products sold by or on behalf of Novavax, its Affiliates, or its or their Sublicensees or licenses in the Territory.
1.11Housemarks.
1.11.1Licensed COVID-19 Products. The Parties, through the JCC, in consultation with regulatory experts, shall discuss whether it will be possible under Applicable Laws and regulatory guidance to include, in addition to Sanofi’s name, housemark, and logos, the name, housemark, and logos of Novavax on the packaging for the Licensed Products, and if so, how to implement such an arrangement on those particular packaging components. If it is permitted under Applicable Laws and regulatory guidance to include, in addition to Sanofi’s name, housemark, and logos, the name, housemark, and logos of Novavax on the packaging for a particular Licensed COVID-19 Product in a particular country, then Sanofi will, at Novavax’s request, include Novavax’s name, housemark and logo on the packaging for such Licensed COVID-19 Product in such country. If it is permitted under Applicable Laws and regulatory guidance to include, in addition to Sanofi’s name, housemark, and logos, the name, housemark, and logos of Novavax (e.g., Matrix-M) on the packaging for a Licensed Adjuvanted Product, then Sanofi will use [***] to include at Novavax’s request, such name, housemark, or logo of Novavax on such packaging.
1.11.2CIC Products and Licensed OC Products. With respect to Licensed CIC Products and Licensed OC Products, Sanofi will have the right to include its name, housemark, and logos, on the packaging for such products. With respect to Novavax CIC Products and Novavax OC Products, Novavax will have the right to include its name, housemark, and logos on the packaging for such products.
1.11.3Adjuvant Products. With respect to Licensed Adjuvanted Products in the Adjuvant Field, as between the Parties, Sanofi will have the right to include its name, housemark, and logos, shall appear on the packaging for such products. With respect to Novavax Adjuvant Products, Novavax will have the right to include its name, housemark, and logos on the packaging for such products.
ARTICLE 8: MEDICAL
1.1In General.
1.1.1Licensed COVID-19 Mono Products. With respect to Licensed COVID-19 Mono Products, during the Collaboration Term, the Parties agree to collaborate to conduct Medical Activities for the Licensed COVID-19 Mono Products in the Collaboration COVID-19 Territory in accordance with the Licensed COVID-19 Mono Product Medical Framework as further described in Section
8.2 (Licensed COVID-19 Mono Product Medical Framework). Except as expressly set forth in this
Article 8 (Medical), neither Party shall conduct any Medical Activities for any Licensed COVID-19 Mono Product in the Collaboration COVID-19 Territory.
1.1.2Licensed CIC Products and Licensed OC Products. Sanofi shall have the right for Licensed CIC Products and Licensed OC Products and Novavax shall have the right for Novavax CIC Products and Novavax OC Products, in each case, to conduct Medical Activities for each such product that such Party is Exploiting in its sole discretion at its own cost and expense, but, in each case, within the scope of the CIC License and OC License, as applicable, and subject to other restrictions applicable to each Party as set forth in
Article 2 (License Grants; Exclusivity) and in this
Article 8 (Medical).
1.1.3Licensed Adjuvanted Products. As between the Parties, Sanofi shall have the right for Licensed Adjuvanted Products and Novavax shall have the right for Novavax Adjuvant Products, to conduct Commercialization activities for each such product that such Party is Exploiting in its sole discretion at its own cost and expense, but, in each case, within the scope of the Adjuvant License and subject to other restrictions as set forth in
Article 2 (License Grants; Exclusivity) and in this
Article 8 (Medical).
1.2Licensed COVID-19 Mono Product Medical Framework. The Parties have agreed to an initial framework for Medical Activities to be conducted with respect to Licensed COVID-19 Mono Products and have allocated between them responsibility for performance of such Medical Activities for Licensed COVID-19 Mono Products throughout the Collaboration COVID-19 Territory (such initial framework, and any updated or amended version thereof, a “Licensed COVID-19 Mono Product Medical Framework”). The initial Licensed COVID-19 Mono Product Medical Framework is attached hereto as Schedule
8.2. The JMAC will review, discuss, and determine whether to approve any material amendments thereto in accordance with
Article 9 (Governance). Sanofi may make non-material amendments to the Licensed COVID-19 Mono Product Medical Framework from time to time in its reasonable discretion, provided that if Sanofi intends to make material amendments to Licensed COVID-19 Mono Product Medical Framework, then it will provide Novavax with a draft of such proposed amendments and Sanofi shall reasonably consider any comments from Novavax received within [***] of receipt thereof.
1.3Cooperation. Each Party will provide reasonable assistance, support, and information to the other Party as reasonably necessary in order to enable such other Party to conduct the Medical Activities related to Licensed COVID-19 Mono Products.
ARTICLE 9: GOVERNANCE
1.1General.
1.1.1The provisions of this
Article 9 (Governance) (other than those of Section
9.8 (Joint Patent Committee)) shall apply solely to (a) the Licensed COVID-19 Mono Product and (b) the Licensed COVID-19 Component, to the extent expressly provided in (i) the COVID-19 Research and Development Plan or (ii) a Technology Transfer Plan.
1.1.2Except as provided in the Section
9.1.1 (General), and notwithstanding any provision to the contrary set forth in this Agreement, the provisions of this
Article 9 (Governance) shall not apply to (a) any Licensed CIC Product, Licensed OC Product, or Licensed Adjuvanted Product or (b) any Novavax CIC Product, Novavax OC Product, or Novavax Adjuvant Product. For clarity, no Committee shall have any mandate hereunder with respect to any matter concerning any (a) Licensed CIC Product, Licensed OC Product, or Licensed Adjuvanted Product that is Exploited by Sanofi or (b) Novavax CIC Product, Novavax OC Product, or Novavax Adjuvant Product that is Exploited by Novavax.
1.1.3Except as provided in the Section
9.1.1 (General), all decisions with respect to (a) any Licensed CIC Product, Licensed OC Product, or Licensed Adjuvanted Product that is Exploited by Sanofi, its Affiliates, or its or their Sublicensees shall, as between the Parties, be made solely by, and in the discretion of, Sanofi, and (b) any Novavax CIC Product, Novavax OC Product, or Novavax Adjuvant Product that is Exploited by Novavax, its Affiliates, or its or their Sublicensees shall, as between the Parties, be made solely by, and in the discretion of, Novavax.
1.2Joint Steering Committee.
1.2.1Formation. Within [***] after the Effective Date, the Parties will establish a joint steering committee (the “JSC”) to act within the JSC’s mandate. The JSC will be comprised of [***] Representatives from each Party.
1.2.2JSC Responsibilities. Subject to the terms and conditions of this Agreement, including Section
9.1 (General), the JSC will coordinate and oversee the collaborative activities of the Parties hereunder with respect to the Licensed COVID-19 Component and Licensed COVID-19 Mono Products and will be responsible for those matters set forth on Schedule
9.2.2 (JSC Responsibilities). Any failure of the JSC to agree on matters within the purview of the JSC as set forth this Section
9.2.2 (JSC Responsibilities) will be resolved in accordance with the decision-making and escalation procedures set forth in Section
9.18 (Decision Making).
1.2.3Subcommittees. From time to time, the JSC may establish and delegate duties, including any responsibilities of the JSC set forth on Schedule
9.2.2 (JSC Responsibilities), to Subcommittees on an “as-needed” basis to oversee particular activities, which delegations will be reflected in the minutes of the meetings of the JSC. Such Subcommittees may be established on an ad hoc basis for purposes of a specific activity, or on such other basis as the JSC may determine and will be constituted and will operate as the JSC may determine; provided that each Subcommittee will have equal representation from each Party.
1.3Joint Research and Development Committee.
1.3.1Formation. Within [***] after the Effective Date, the Parties will establish a joint research and development committee (the “JRDC”) to coordinate and oversee the Parties’ respective Research and Development activities under the COVID-19 Research and Development Plan and COVID-19 Research and Development Budget, and technology transfer activities pursuant to the COVID-19 Technology Transfer Plan, until such time as the JRDC is disbanded as provided herein. The JRDC will be comprised of [***] Representatives from each Party.
1.3.2JRDC Responsibilities. Subject to the terms and conditions of this Agreement, including Section
9.1 (General), the JRDC will be responsible for those matters set forth on Schedule
9.3.2 (JRDC Responsibilities). Any failure of the JRDC to agree on matters within the purview of the JRDC as set forth this Section
9.3.2 (JRDC Responsibilities) will be resolved in accordance with the decision-making and escalation procedures set forth in Section
9.18 (Decision Making).
1.4Joint Manufacturing and Supply Committee.
1.4.1Formation. Within [***] after the Effective Date, the Parties will establish a joint manufacturing and supply committee (the “JMSC”) to act as a forum to coordinate and oversee the Parties’ Manufacturing activities, including technology transfer activities pursuant to the COVID-19 Manufacturing Technology Transfer Plan and the Adjuvant Manufacturing Technology Transfer Plan, and for the delivery, quality, and supply of Licensed COVID-19 Components, Licensed COVID-19 Mono Products, [***] and [***], and Adjuvant Intermediates that are Manufactured and supplied by Novavax to Sanofi pursuant to this Agreement (including under the COVID- 19 Supply Agreement and Adjuvant Supply Agreement entered into in furtherance of such Manufacturing activities) until such time as the JMSC is disbanded as provided herein. The JMSC will be comprised of [***] Representatives from each Party.
1.4.2JMSC Responsibilities. Subject to the terms and conditions of this Agreement, the JMSC will be responsible for those matters set forth on Schedule
9.4.2 (JMSC Responsibilities). Any failure of the JMSC to agree on matters within the purview of the JMSC as set forth in this Section
9.4.2 (JMSC Responsibilities) will be resolved in accordance with the decision-making and escalation procedures set forth in Section
9.18 (Decision Making).
1.5Joint Commercialization Committee.
1.5.1Formation. Within [***] after the Effective Date, the Parties will establish a joint commercialization committee (the “JCC”) to act as a forum to coordinate and oversee the Parties’ Commercialization activities for the Licensed COVID-19 Mono Product in the Collaboration COVID-19 Territory, until such time as the JCC is disbanded as provided herein. The JCC will be comprised of [***] Representatives from each Party.
1.5.2JCC Responsibilities. Subject to the terms and conditions of this Agreement, the JCC will be responsible for those matters set forth on Schedule
9.5.2 (JCC Responsibilities). Any failure of the JCC to agree on matters within the purview of the JCC as set forth this Section
9.5.2 (JCC Responsibilities) will be resolved in accordance with the decision-making and escalation procedures set forth in Section
9.18 (Decision Making).
1.6Joint Medical Affairs Committee.
1.6.1Formation. Within [***] after the Effective Date, the Parties will establish a joint medical affairs committee (the “JMAC”) to act as a forum to coordinate and oversee the Parties’ Medical Activities for the Licensed COVID-19 Mono Product until such time as the JMAC is disbanded as provided herein. The JMAC will be comprised of an equal number of Representative from each Party, with [***] Representatives from each Party. If Novavax is not performing any Medical Activities hereunder for a period of [***] and is not allocated any Medical Activities for Licensed COVID-19 Mono Products for the upcoming Calendar Year in the then-current Licensed COVID-19 Mono Product Medical Framework, then the JMAC shall be automatically disbanded and any responsibilities allocated to it under Section
9.6.2 (JMAC Responsibilities) shall automatically become responsibilities of the JSC.
1.6.2JMAC Responsibilities. Subject to the terms of this Agreement, the JMAC will be responsible for those matters set forth on Schedule
9.6.2 (JMAC Responsibilities). Any failure of the JMAC to agree on matters within the purview of the JMAC as set forth this Section
9.6.2 (JMAC Responsibilities) will be resolved in accordance with the decision- making and escalation procedures set forth in Section
9.18 (Decision Making).
1.7Joint Finance Committee.
1.7.1Formation. Within [***] after the Effective Date, the Parties will establish a joint finance committee (the “JFC”) to act as a forum to coordinate and oversee financial matters hereunder, until such time as the JFC is disbanded as provided herein. The JFC will be comprised of an equal number of Representative from each Party, with [***] Representatives from each Party. If Novavax has not, for a period of [***], performed any activities the costs of which are reimbursable by Sanofi hereunder, then the JFC shall be automatically disbanded and any responsibilities allocated to it under Section
9.7.2 (JFC Responsibilities) shall automatically become responsibilities of the JSC.
1.7.2JFC Responsibilities. Subject to the terms and conditions of this Agreement, the JFC will be responsible for those matters set forth on Schedule
9.7.2 (JFC Responsibilities). Any failure of the JFC to agree on matters within the purview of the JFC as set forth this Section
9.7.2 (JFC Responsibilities) will be resolved in accordance with the decision-making and escalation procedures set forth in Section
9.18 (Decision Making).
1.8Joint Patent Committee.
1.8.1Formation. Within [***] after the Effective Date, the Parties will establish a joint patent committee (the “JPC”) to coordinate between the Parties the oversight and handling of Patent matters (including Patent litigation) relating to Licensed COVID-19 Products until such time as the JPC is disbanded as provided herein. The JPC will be comprised of [***] from each Party that are each a patent counsel to such Party.
1.8.2External Counsel. Each Party may invite their respective external patent counsel to participate, as guests, in discussions and meetings of the JPC on an ad hoc basis. Each Party that intends to invite its external counsel to a meeting of the JPC for the first time shall notify the other Party through its JPC co-chair at least [***] in advance of the applicable JPC meeting.
1.8.3JPC Responsibilities. Subject to the terms of this Agreement, the JPC will be responsible for those matters set forth on Schedule
9.8.2 (JPC Responsibilities). Any failure of the Parties to agree on matters within the purview of the JPC as set forth in this Section
9.8.2 (JPC Responsibilities) will be resolved in accordance with the decision-making and escalation procedures set forth in Section
9.18 (Decision Making).
1.9Committee Members. Each Party’s Representatives on a Committee will have experience appropriate to the matters under the purview of such Committee. Each Party will appoint its initial representatives for a Committee at the time such Committee is established and may appoint the same Person to more than one Committee as long as such appointment is in accordance with this Section
9.9 (Committee Members). Each Party may replace any of its Representatives on a Committee at any time by providing notice in writing to the other Party. Each Party will appoint one (1) of its Representatives on such Committee to act as a co-chairperson of such Committee ([***]). The co-chairpersons of each Committee will be responsible for setting the agenda for meetings of such Committee with input from the other members of such Committee, and for conducting the meetings of such Committee. Each member of a Committee will conduct its responsibilities hereunder in good faith and with reasonable care and diligence. No Party will appoint a Representative to any Committee who: (a) is not a director, officer, or employee of such Party without the prior written consent of the other Party, which consent will not be unreasonably withheld, conditioned, or delayed; (b) has not executed a written confidentiality agreement or other agreement that imposes obligations of confidentiality on such Representative that are at least as stringent as those contained in
Article 11 (Confidentiality and Non-Disclosure); or (c) does not have the authority to make decisions on behalf of such Party on matters within the scope of such Committee’s authority.
1.10Party Non-Member Guests. Each Party may invite those of its (or its Affiliates’) employees having expertise in a particular subject matter that is being reviewed or discussed at a meeting of any Committee and who has executed a written confidentiality agreement or other agreement that imposes obligations of confidentiality on such employee that are at least as stringent as those contained in
Article 11 (Confidentiality and Non-Disclosure) (each, a “Party Non-Member Guest”) to attend such meeting and participate as guests in such meeting; provided that each Party shall notify the other Party through its co-chair (or designee) of the applicable Committee of the names of each of its Party Non-Member Guests who will attend a meeting at least [***] before the applicable meeting. If a Party desires to invite certain Party Non-Member Guests to multiple meetings of a given Committee, then such Party may request an advance waiver of such notice requirement with respect to the meetings that such Party expects that such Party Non-Member Guests will attend, and the co-chairs may agree in writing to so waive such notice obligation. Notwithstanding the foregoing, each Party’s Alliance Manager (or their designees) may attend any meeting of any Committee as a Party Non-Member Guest.
1.11Non-Party Guests. Except as set forth in Section
9.8.2 (External Counsel), no Third Party may attend a meeting of any Committee without the prior consent of both Parties (any such Third Party, a “Non-Party Guest”). Any Party that wishes to invite a Non-Party Guest to a Committee meeting must notify the other Party’s co-chair (or designee) of such Committee in writing at least [***] prior to any scheduled meeting. In the event a Party desires to invite certain Non- Party Guests to multiple meetings of a given Committee, then such Party may request an advance waiver of such notice requirement with respect to the meetings that such Party expects that Non- Party Guest will attend, and the co-chairs may agree in writing to so waive such notice obligation. No Non-Party Guest may attend any meeting of a Committee unless such Non-Party Guest has executed, prior to any meeting at which such Non-Party Guest would be present, a confidentiality agreement acceptable to both Parties. Any Non-Party Guest who attend any meeting of any
Committee pursuant to this Section
9.11 (Non-Party Guests) may not vote on any matter put to a vote by, or requiring the approval of, such Committee, and shall not be counted for such purposes.
1.12Meetings; Quorum. Except as otherwise provided herein, each Committee will meet [***] after its formation on such dates and at such times and by such means (i.e., whether in person or through teleconference or video conference or otherwise) as agreed by the members of such Committee. Except with respect to the JPC, at least [***] prior to each such [***] Committee meeting, the applicable co-chairs will circulate to each Representative of such Committee a proposed agenda for such Committee meeting. In the event that the co-chairs of any Committee agree that there are no particular items within the purview of such Committee to be reviewed or discussed in any particular [***], then such co- chairs may agree to not hold a meeting during such [***] upon reasonable advance notice to the members of such Committee. Upon [***] prior written notice, either Party may convene a special meeting of a Committee for the purpose of discussing any urgent matter within the purview of such Committee. Unless otherwise agreed by the Parties in writing, no meeting of any Committee formed hereunder may take place unless at least [***] Representative of each Party ([***]) appointed to such Committee is present for the entirety of the meeting and each Party will use reasonable efforts to cause its Representative(s) appointed to such Committee to participate in each Committee meeting of such Committee.
1.13Committee Decisions. Each Party’s Representatives on each Committee will, collectively, have one vote (the “Party Vote”) on all matters brought before such Committee for a decision by consensus. Each Committee will make decisions as to matters within its jurisdiction by unanimous Party Vote of both Parties, which Party Vote will be reflected in the minutes of the committee meeting (except that the JPC may elect to reflect its decisions in a form other than the minutes to protect legal privilege). No vote of any Committee will be effective for purposes of this Agreement unless each Party has at least [***] representatives in attendance at the time the vote is taken ([***]). Should a decision be made by a Committee outside of a meeting, the unanimous Party Vote of both Parties on such decision may be made either by a written resolution unanimously agreed to in writing by each Representative on such Committee or by authorized signatories of each Party. For clarity, the phrase “determine,” “designate,” “confirm,” “approve,” or “determine whether to approve” by a Committee and similar phrases used in this Agreement will mean approval in accordance with this Section
9.13 (Committee Decisions).
1.14Information Sharing. The Parties recognize that for the Committees to function optimally, each Party should provide the other Party with timely information that is relevant to the mandate of such Committee. For avoidance of doubt, neither Party has any obligation to share any competitively sensitive information with the other Party including with respect to Licensed CIC Products, Licensed OC Products, Licensed Adjuvanted Products, Novavax CIC Products, Novavax OC Products, or Novavax Adjuvant Products.
1.15Minutes. The responsibility for preparing the minutes for each Committee (other than the JPC) meeting will alternate between each Party on a meeting-by-meeting basis. The applicable Party’s designee responsible for the minutes will provide the other Party’s designee and the members of such Committee with draft written minutes for the Committee’s approval from each meeting within [***] days after each such meeting setting forth, among other things, a description, in reasonable detail, of the discussions at the meeting, each Party Vote, and a list of all decisions taken by such Committee, including any action assigned by such Committee. Such minutes will be effective only after being approved in writing by mutual consent of the co-chair of each Party for
such Committee. If the minutes of any meeting of the Committee are not approved by both co- chairs of the Committee within [***] days after the meeting, and the objecting Party, if any, has not appended a notice of objection with the specific details of the objection to the proposed minutes within such [***]-day period, then such minutes will be deemed to be approved by the Parties. Notwithstanding the foregoing, no Committee shall discuss any issue relating to any Patents (including with respect to any of their scope, patentability, validity, prosecution, or infringement), unless a JPC Representative for each Party is present. The Parties’ JPC Representatives shall be solely responsible for documenting, at their discretion, any issues discussed by the JPC or any other Committee relating to any Patents, which documents and the content of such discussions, shall each be held in strict confidence by the Parties to protect their common interests and preserve the privileged status of any attorney-client communication, advice, or legal opinion reflected therein.
1.16Expenses. Each Party will be responsible for its own expenses incurred to attend or participate in any Committee meeting, including the costs and expenses of such Party’s Third Party guests (including such Party’s external counsel with respect to the JPC).
1.17Limitations of Committee Authority. Each Committee shall only have authority to determine, approve, or resolve matters that such Committee is expressly authorized to determine, approve, or resolve under this Agreement (each a “Committee Matters”). No Committee has the authority to:
(a) modify or amend the terms and conditions of this Agreement; (b) waive or determine either Party’s compliance with the terms and conditions of this Agreement; (c) decide any issue in a manner that would conflict with the express terms and conditions of this Agreement (d) breach or violation an agreement with a Third Party or Applicable Law; (e) decide any issue for which this Agreement expressly requires the other Party’s approval or consent; or (f) resolve any Dispute (other than a Committee Dispute) under this Agreement, including a Dispute as to whether a Committee Matter is a Sanofi Matter, a Novavax Matter, or a Status Quo Matter.
1.18Decision Making.
1.18.1Discussion and General Decision-Making Process. The Parties acknowledge that each Party shall, in good faith, discuss each Committee Matter with a view of reaching consensus of the Parties with respect to such Committee Matter, and if such consensus is not reached, then the provisions of the remainder of this Section
9.18 (Decision Making) shall apply.
1.18.2Escalation to JSC. If a Subcommittee fails to reach consensus on a matter before it for decision within [***] from the date that the matter is first presented to such Subcommittee in writing (such matter, a “Subcommittee Dispute”), then the matter shall be referred in writing to the JSC. If a Subcommittee Dispute is escalated to the JSC by any Subcommittee, then each Party will provide an analysis to support its position with respect to the Subcommittee Dispute at the next meeting of the JSC (or a sooner ad hoc meeting of the JSC for any matter that is urgent, would be prejudiced by delay to the next scheduled JSC meeting, or that the Parties otherwise agree for which an ad hoc meeting should be held).
1.18.3Decisions of the JSC. The JSC has the authority (a) to make decisions as to matters specifically delegated to it or expressly specified in this Agreement, and (b) to resolve Subcommittee Disputes. The JSC has no other authority under this Agreement. The JSC will use good faith efforts, in compliance with this Section
9.18.3 (Decisions of the JSC), to promptly resolve any such matter for which it has authority, with a view of reaching
consensus of the Parties with respect to such matter. If, after the use of good faith efforts, the JSC fails to reach consensus regarding a Subcommittee Dispute or any other matter within the scope of the JSC’s authority as set forth in this Section
9.18.3 (Decisions of the JSC) (in either case, a “Committee Dispute”), then the matter shall be escalated in accordance with Section
9.18.4 (Escalation to Senior Executives for Expedited Decision), other than a Subcommittee Dispute as to Item 2 of Schedule
9.5.2, which Subcommittee Dispute shall be a Status Quo Matter in accordance with Section
9.18.5(d)(i)(1) (Status Quo Matters) without further escalation.
1.18.4Escalation to Senior Executives for Expedited Decision. If the JSC fails to reach consensus regarding a Committee Dispute within [***] after being presented with such matter for decision-making, then, at the election of either Party, the JSC shall refer such dispute to the Senior Executives of the Parties, and the Senior Executives of the Parties will confer in good faith on the resolution of the issue within [***] following first receipt of such written notice. If the Senior Executives of the Parties reach consensus regarding such dispute, then the decision of the Senior Executives shall be binding upon the Parties as a decision of the JSC. If the Senior Executives do not reach consensus regarding a dispute that is escalated to them in accordance with this Section
9.18.4 (Escalation to Senior Executives for Expedited Decision) within such [***] period, then the matter will be decided as set forth in Section
9.18.5 (Final Decisions).
1.18.5Final Decisions.
(a)Novavax Matters. As between the Parties, after compliance with the terms of Section
9.18.1 (Discussion and General Decision-Making Process) and Section
9.18.2 (Escalation to JSC) or Section
9.18.3 (Decisions of the JSC) (as applicable), Novavax will have the right to make the final decision with respect to:
(i)[***];
(ii)[***];
(iii)[***];
(iv)[***]; and
(v)[***];
(each such matter described in the foregoing clauses (i) through (v), a “Novavax Matter”).
(b)Sanofi Matters. As between the Parties, after compliance with the terms of Section
9.18.1 (Discussion and General Decision-Making Process) and Section
9.18.2 (Escalation to JSC) or Section
9.18.3 (Decisions of the JSC) (as applicable), Sanofi will have the right to make the final decision with respect to:
(i)[***];
(ii)[***];
(iii)[***];
(iv)[***];
(v)[***];
(vi)[***];
(vii)[***]; or
(viii)[***];
(each such matter described in the foregoing clauses (i) through (vii), a “Sanofi Matter”).
(c)Change of Control Matters. If, after compliance with the terms of Section
9.18.1 (Discussion and General Decision-Making Process), Section
9.18.3 (Decisions of the JSC), and Section
9.18.4 (Escalation to Senior Executives), the Parties are not able to reach consensus with respect to any matter related to reasonably determining whether an Acquirer of Novavax (i) is unable or unwilling to cooperate with Sanofi to implement Sanofi’s Commercial strategy for Licensed COVID-19 Products or (ii) is not able or willing to perform Novavax’s obligations under this Agreement with substantially the same degree of skill that Novavax was using prior to such Change of Control as required under Section
17.1.2(b) (Change of Control), then such matter will be resolved pursuant to Section
17.12 (Dispute Resolution).
(d)Status Quo Matters.
(i)In General. If, after compliance with the terms of Section
9.18.1 (Discussion and General Decision-Making Process), Section
9.18.2 (Escalation to JSC) or Section
9.18.3 (Decisions of the JSC) (as applicable), and Section
9.18.4 (Escalation to Senior Executives), the Parties are not able to reach consensus with respect to any matters related to:
(1)[***];
(2)[***];
(3)[***];
(4)[***];
(5)[***];
(6)[***]; or
(7)[***],
(any such matter described in (1) through (7), a “Status Quo Matter”), then neither Party will have unilateral final decision-making authority with respect to the final resolution of such Status Quo Matter, and unless and until consensus on such matter is reached, the status quo shall be maintained with respect to such Status Quo Matter and no further changes may be made or decisions may be taken unilaterally by either Party in relation to such Status Quo Matter, except with respect to the approval of any amendment or update to the COVID-19 Research and Development Budget or a Technology Transfer Budget (any such dispute, a “Budget Status Quo Matter”), which matter will be resolved in accordance Section
9.18.5(d)(ii) (Budget Status Quo).
(ii)Budget Status Quo. Notwithstanding Section
9.18.5(d)(i) (In General), if, after compliance with the terms of Section
9.18.1 (Discussion and General Decision-Making Process), Section
9.18.2 (Escalation to JSC), and Section
9.18.4 (Escalation to Senior Executives), the Parties are unable to reach consensus with respect to any Budget Status Quo Matter, then the already-existing budget (if any) will remain in effect until the earlier of the date on which (a) the Parties agree on such budget for the applicable period, or (b) either Party refers such dispute for resolution by an independent arbitrator in accordance with the procedure set forth in Schedule
(ii) (Baseball Arbitration Dispute Resolution) (such procedure, the “Baseball Arbitration Procedure”) (each such decision, an “Expert Budget Decision”) and such independent arbitrator renders an Expert Budget Decision; provided that until the Parties agree to a budget or the arbitrator renders an Expert Budget Decision, such already existing budget will be [***].
1.19Disbandment of Committees. The JSC may, upon consensus, disband any Subcommittee at any time and either allocate the responsibilities of such Subcommittee to another Subcommittee or assume such responsibilities itself. All Committees, including the JSC, will, if not already disbanded, automatically disband upon the expiration or termination of the Collaboration Term.
1.20Alliance Managers.
1.20.1Appointment. Within [***] after the Effective Date, each Party will appoint a representative of such Party to act as its alliance manager under this Agreement (each, an “Alliance Manager”). Each Party may replace its Alliance Manager at any time upon notice to the other Party.
1.20.2Specific Responsibilities. The Alliance Managers (or their designees) will, unless the co- chairs of the JSC designate an alternate Person, attend all meetings of the JSC and will take and disseminate minutes of meetings of the JSC, including any decisions made at such meeting. The co-chairs for each Subcommittee will designate a Person to take and disseminate minutes of meetings of such Subcommittee, including any decisions made at
such meeting. The Alliance Managers (or their designees) will serve as the primary contact point between the Parties for the purpose of providing each Party with information regarding the other Party’s activities pursuant to this Agreement and will have the following responsibilities: (a) schedule meetings of the JSC; (b) draft and circulate draft agendas for meetings of the JSC; (c) prepare the written minutes of any JSC meeting, and circulate drafts thereof, and ensure that approved minutes are archived and monitor and follow-up the execution of decisions and actions reflected in such minutes; (d) generally facilitate communication, coordination, and collaboration between the Parties; and (e) manage the dispute resolution process. Each Alliance Manager (and their designees) will conduct its responsibilities hereunder in good faith and with reasonable care and diligence.
ARTICLE 10: FINANCIAL
1.1Up-Front Fee. In partial consideration of the rights and licenses granted herein, Sanofi will pay Novavax a one-time, non-refundable upfront payment of Five Hundred Million Dollars ($500,000,000) within [***] days after the receipt of Novavax’s invoice, which invoice may be dated no earlier than the Effective Date.
1.2Equity Investment. In partial consideration for the grant of the rights and licenses herein, concurrent with the execution of this Agreement, the Parties or their designated Affiliates have executed the Securities Subscription Agreement.
1.3Licensed COVID-19 Product Milestone Payments
.
1.3.1Development Milestones. Novavax shall notify Sanofi of the achievement of the Development milestone event described in rows A and D of Table
10.3.1 and Sanofi shall notify Novavax of the achievement of each Development milestone event described in rows B, C, E, and F of Table
10.3.1 (the events of rows A-F “COVID-19 Development Milestone Event”) within [***] after the achievement thereof. Sanofi shall pay Novavax the COVID-19 Development milestone payment described in Table
10.3.1 (the “COVID-19 Development Milestone Payment”) within [***] after receipt of Novavax’s corresponding invoice dated after achievement of the COVID-19 Development Milestone Event:
|
|
|
|
|
|
|
|
|
Table 10.3.1 COVID-19 Development Milestones |
COVID-19 Development Milestone Event |
COVID-19 Development Milestone Payment |
A. |
Receipt of First Marketing Authorization by FDA of a Milestone Eligible Licensed COVID-19 Mono Product in the U.S. |
One-Hundred and Seventy Five Million Dollars ($175,000,000) |
| B. |
Completion of the transfer to Sanofi of Marketing Authorization granted to Novavax in the U.S. for
the first Milestone Eligible Licensed COVID-19 Mono Product
|
Twenty-Five Million Dollars ($25,000,000) |
| C. |
Completion of the transfer to Sanofi of Marketing Authorization granted to Novavax by the European Commission under the centralized procedure for the first Milestone Eligible Licensed COVID-19
Mono Product
|
Twenty-Five Million Dollars ($25,000,000) |
|
|
|
|
|
|
|
|
|
| D. |
Database lock of the 12-month interim analysis of cohort 1 (6-11 year olds) of the Phase 2/3 Clinical Trial with Novavax identifier 2019nCoV-503
(clinicaltrials.gov identifier NCT05468736)
|
Fifty Million Dollars ($50,000,000) |
| E. |
Successful Completion of COVID-19 Manufacturing Technology Transfer |
Seventy-Five Million Dollars ($75,000,000) |
| F. |
Dosing of the fifth (5th) patient in the first Pivotal Clinical Trial of the first Licensed CIC Product conducted by or on behalf of Sanofi, its Affiliates, or their respective Sublicensees in the United
States or any EU country.
|
One Hundred and Twenty-Five Million Dollars ($125,000,000) |
Each COVID-19 Development Milestone Payment in Table
10.3.1 is payable one (1)-time only, upon the first occurrence of each COVID-19 Development Milestone Event by the applicable Licensed COVID-19 Product. The maximum COVID-19 Development Milestone Payment that Sanofi will pay to Novavax under this Section
10.3.1 (Milestones for Licensed COVID-19 Products) is Four Hundred Seventy-Five Million Dollars ($475,000,000).
1.3.2Launch Milestones. Sanofi shall notify Novavax of the achievement of the launch milestone event described in Table
10.3.2 (each, a “COVID-19 Launch Milestone Event”) within [***] after the achievement thereof by Sanofi, its Affiliates, or their respective Sublicensees. Sanofi shall pay Novavax each COVID-19 launch milestone payment described in Table
10.3.2 (each, a “COVID-19 Launch Milestone Payment”) within [***] after receipt of Novavax’s corresponding invoice dated after achievement of the COVID-19 Launch Milestone Event:
|
|
|
|
|
|
Table 10.3.2 COVID-19 Launch Milestones |
| COVID-19 Launch Milestone Event |
COVID-19 Launch Milestone Payment |
| First Commercial Sale in the United States of the first Licensed CIC Product |
Two Hundred and Twenty-Five Million Dollars ($225,000,000) |
The COVID-19 Launch Milestone Payment set forth in Table
10.3.2 is payable one (1)- time only, upon the First Commercial Sale of the first Licensed CIC Product in the U.S.
1.4Licensed Adjuvanted Product Milestone Payments.
1.4.1Development Milestones. Sanofi shall notify Novavax of the achievement of each Development milestone event described in the table below (the “Adjuvant Development Milestone Event”) within [***] after the achievement thereof. Sanofi shall pay Novavax the Adjuvant Development Milestone payment described in the table below (the “Adjuvant Development Milestone Payment”) within [***] after receipt of Novavax’s corresponding invoice dated after achievement of the Adjuvant Development Milestone Event:
|
|
|
|
|
|
|
|
|
Table 10.4.1 Adjuvant Development Milestones |
| Adjuvant Development Milestone Event |
Adjuvant Development Milestone Payment |
| A. |
For a fifth (5th) and each subsequent Distinct Licensed Adjuvanted Product, the date on which either (i) the Gatekeeper provides written confirmation to Sanofi
that the Proposed Field for such Distinct Licensed
|
Three Million Dollars ($3,000,000) |
|
|
|
|
|
|
|
|
|
|
Adjuvanted Product is not Unavailable or (ii) Sanofi provides written notice to Novavax identifying such Distinct Licensed Adjuvanted Product in accordance
with Section 3.5.2 (Sanofi Research and Development Reports). |
|
B. |
Dosing of the fifth (5th) patient in the first Phase 1 Full Clinical Trial of the fifth (5th) and each subsequent Distinct Licensed Adjuvanted Product, conducted by or on behalf of Sanofi, its Affiliates, or their respective Sublicensees. |
Three Million Dollars ($3,000,000) |
C. |
Dosing of the fifth (5th) patient in the first Pivotal Clinical Trial of the first Distinct Licensed Adjuvanted Product conducted by or on behalf of Sanofi, its
Affiliates, or their respective Sublicensees.
|
Four Million Dollars ($4,000,000) |
Each Adjuvant Development Milestone Payment in Table
10.4.1 is payable one (1)- time only for each Licensed Adjuvanted Product that is Distinct, upon the achievement of the Adjuvant Development Milestone Event by such Distinct Licensed Adjuvanted Product. The maximum Adjuvant Development Milestone Payment that Sanofi will pay to Novavax under this Section
10.4.1 (Development Milestones) for each Distinct Licensed Adjuvanted Product is Ten Million Dollars ($10,000,000). For avoidance of doubt, Sanofi will provide written notice to Novavax for every Licensed Adjuvanted Product that achieves an Adjuvant Development Milestone Event, but (a) the first four (4) Distinct Licensed Adjuvanted Products Developed by or on behalf of Sanofi and (b) any non-Distinct Licensed Adjuvanted Product thereafter, in each case (a) and (b), do not trigger the Adjuvant Development Milestone Events set forth in Table
10.4.1.
1.4.2Launch Milestones. Sanofi shall notify Novavax of the achievement of each launch milestone event described in the table below (the “Adjuvant Launch Milestone Event”) within [***] after the achievement thereof. Sanofi shall pay Novavax the Adjuvant launch milestone payment described in the table below (the “Adjuvant Launch Milestone Payment”) within [***] after receipt of Novavax’s corresponding invoice dated after achievement of the Adjuvant Development Milestone Event:
|
|
|
|
|
|
Table 10.4.2 Adjuvant Launch Milestones |
Adjuvant Launch Milestone Event |
Adjuvant Launch Milestone Payment |
| First Commercial Sale in the United States of each Distinct Licensed Adjuvanted Product |
Fifty Million Dollars ($50,000,000) |
The Adjuvant Launch Milestone Payment for the Licensed Adjuvanted Product is payable upon the First Commercial Sale of each Distinct Licensed Adjuvanted Product.
1.4.3Sales Milestones. Sanofi will notify Novavax in writing of the achievement of each sales- based milestone event set forth in Table
10.4.3 (Adjuvant Sales Milestones) (each, a “Sales Milestone Event”) by Sanofi or its Affiliates or Sublicensees no later than [***] after the end of the Calendar Quarter in which such Sales Milestone Event is achieved. Sanofi will pay to Novavax the sales milestone payment corresponding to the applicable Sales Milestone Event set forth in Table
10.4.3 (Adjuvant Sales Milestones) (each, a “Sales Milestone Payment”) no later than [***] after receipt of the applicable invoice from Novavax.
|
|
|
|
|
|
Table 10.4.3 Adjuvant Sales Milestones |
| Sales Milestone Event |
Sales Milestone Payment |
| First occurrence of Annual Net Sales of each Licensed Adjuvanted Product greater than $500,000,000 |
Fifty Million Dollars ($50,000,000) |
|
First occurrence of Annual Net Sales of each Licensed
Adjuvanted Product greater than $1,000,000,000
|
One Hundred Million Dollars
($100,000,000)
|
For clarity, each Sales Milestone Payment will be paid only once for each Licensed Adjuvanted Product on the first achievement of the corresponding Sales Milestone Event by aggregate Net Sales of such Licensed Adjuvanted Product.
1.4.4Licensed Adjuvanted Products for pre-Pandemic. [***].
1.5Royalties. General. For purposes of calculating the royalties and royalty tiers in this Section
10.5 (Royalties) for a given Licensed Product, only the applicable portion of Annual Net Sales for such Licensed Product, adjusted on the basis set forth in Section
1.174 (“Net Sales”) shall be included for purposes of such calculation. Notwithstanding any provision to the contrary set forth in this Agreement, with respect to any Licensed CIC Product or Licensed OC Product in a given country, in no event will the reductions permitted under Section
10.5.4 (Royalty Adjustments) together with the Net Sales apportionment for Licensed CIC Products and Licensed OC Products as set forth in Section
1.174 (“Net Sales”) result in an effective royalty rate for a Licensed CIC Product or Licensed OC Product in such country below [***] of the Annual Net Sales of such Licensed CIC Product or Licensed OC Product in such country.
1.5.1Licensed COVID-19 Mono Product Royalty. Subject to Section
10.5.4 (Royalty Adjustments), during the COVID-19 Royalty Term, Sanofi will pay Novavax royalties on a tier-by-tier basis at the following royalty rates on Net Sales (including Cancellation Fees pursuant to Section
10.5.3 (Cancellation Fees)) of the applicable Licensed COVID-19 Product sold by Sanofi, its Affiliates, and their respective Sublicensees in the Territory as set forth in this Section
10.5.1 (Licensed COVID-19 Mono Product Royalty).
(a)Licensed COVID-19 Mono Product Royalty Rate. During the COVID-19 Royalty Term, Sanofi will pay Novavax royalties on Net Sales of Licensed COVID-19 Mono Products at the royalty rates set forth in Table
10.5.1(a) (Royalty Rate).
|
|
|
|
|
|
Table 10.5.1(a) Licensed COVID-19 Mono Product Royalty Rate |
| Portion of Aggregate Annual Net Sales for all Licensed COVID-19 Mono Products: |
Royalty Rate |
| [***] |
[***] |
| [***] |
[***] |
| [***] |
[***] |
(b)Licensed CIC Product and Licensed OC Product Royalty Rate. On a Licensed CIC Product-by-Licensed CIC Product or Licensed OC Product-by-Licensed OC Product basis, as applicable, during the COVID-19 Royalty Term, Sanofi will pay Novavax royalties on Net Sales (including Cancellation Fees pursuant to Section
10.5.3 (Cancellation Fees)) of Licensed CIC Products and Licensed OC Products at the royalty rates set forth in Table
10.5.1(b) (Royalty Rate).
|
|
|
|
|
|
Table 10.5.1(b) Licensed CIC Products and Licensed OC Products Royalty Rate |
| Portion of Aggregate Territory-wide Annual Net Sales for each Licensed CIC Product or each Licensed OC Product: |
Royalty Rate |
| [***] |
[***] |
| [***] |
[***] |
| [***] |
[***] |
1.5.2Licensed Adjuvanted Product Royalty Rates.
(a)Rate. Sanofi will pay Novavax royalties on Net Sales of all Licensed Adjuvanted Products in the Territory during the Adjuvant Royalty Term at a royalty rate of [***].
(b)Sole Reduction. For all Licensed Adjuvanted Products, the royalty rates set forth in Section
10.5.2(a) (Rate) may be reduced only in accordance with this Section
10.5.2(b) (Sole Reduction). On a Licensed Adjuvanted Product-by-Licensed Adjuvanted Product and country-by-country basis, Sanofi has the right to credit [***] of all consideration actually paid by Sanofi to a Third Party pursuant to an agreement between Sanofi and such Third Party entered into following the Effective Date to the extent in consideration for a license to Sanofi under Patents controlled by such Third Party that are necessary to Manufacture or Commercialize the Adjuvant (but not any Antigen or other component) in any Licensed Adjuvanted Product in such country against any royalties due in such Calendar Quarter to Novavax hereunder for the applicable Licensed Adjuvanted Product; provided that in no event may Sanofi credit any amounts under this Section
10.5.2(b) (Sole Reduction) that have already been, or that pursuant to the terms of Section
0 (Third Party Licenses) could be, credited against payments due to Novavax under Section
0 (Third Party Licenses).
(c)Cumulative Reductions Floor. In no event will the reductions set forth in Section
10.5.2(b) (Sole Reduction) cause the royalties otherwise due to Novavax for any Licensed Adjuvanted Product in a given Calendar Quarter during the applicable Adjuvant Royalty Term for such Licensed Adjuvanted Product to be reduced by more than [***] of the amount that would otherwise be due in such Calendar Quarter for such Licensed Adjuvanted Product but for such reductions, provided that any reductions not able to be made or taken in respect of royalties in a given Calendar Quarter as a result of this Section 10.5.3
(c) (Cumulative Reductions Floor) shall be carried forward for reduction in respect of royalties in future Calendar Quarters until fully exhausted.
1.5.3Cancellation Fees. On a Licensed Product-by-Licensed Product basis during the COVID- 19 Royalty Term or Adjuvant Product Royalty Term, as applicable, any Cancellation Fees
with respect to such Licensed Product shall be treated as Net Sales hereunder with respect to such Licensed Product.
1.5.4Royalty Adjustments. For all Licensed COVID-19 Products, the royalty rates set forth in Section
10.5.1 (Licensed COVID-19 Product Royalty Rates) may be reduced in accordance with this Section
10.5.4 (Royalty Adjustments).
(a)Lack of Valid Claim. Subject to Section
10.5.5 (Cumulative Reductions Floor) and the last sentence of the introductory paragraph of Section
10.5 (General), if, during the COVID-19 Royalty Term, on a country-by-country and Licensed COVID-19 Product-by-Licensed COVID-19 Product basis, for a given country and Licensed COVID-19 Product, (i) no Valid Claim of (A) a Novavax Patent or (B) a Joint Arising Patent Covers the composition of matter or a method of use (of an Indication that has received Marketing Authorization) of such Licensed COVID- 19 Product or the Manufacture or use thereof in such country and (ii) there is no Regulatory Exclusivity for such Licensed COVID-19 Product in such country, then the Net Sales in such country used for purposes of calculating worldwide Net Sales of such Licensed COVID-19 Product and royalty payments due to Novavax based on Net Sales of such Licensed COVID-19 Product in such country pursuant to Section
10.5.1 (Licensed COVID-19 Product Royalty Rates) will be reduced by [***].
(b)Biosimilar Product Competition. Subject to Section
10.5.5 (Cumulative Reductions Floor) and the last sentence of the introductory paragraph of Section
10.5 (General), if, during the COVID-19 Royalty Term, on a country-by-country Licensed COVID-19 Product-by-Licensed COVID-19 Product, and Calendar Quarter-by-Calendar Quarter basis, for a given country, Licensed COVID-19 Product and Calendar Quarter, (i) the MAA for a Biosimilar Product as to such Licensed COVID-19 Product has been approved and such Biosimilar Product is being sold in such country in such Calendar Quarter and (ii) the Net Sales of such Licensed COVID-19 Product in such country decline by the percentage described in a given row in the table below in a Calendar Quarter relative to the average Calendar Quarter Net Sales of such Licensed COVID-19 Product in such country achieved in the [***] consecutive Calendar Quarters immediately prior to the Calendar Quarter in which the initial sale of such Biosimilar Product after receipt of approval for the Marketing Authorization for such Biosimilar Product in such country occurred, then the Net Sales in such country used for purposes of calculating worldwide Net Sales of such Licensed COVID-19 Product will be reduced by the corresponding amount set forth in the table below on and after such Calendar Quarter:
|
|
|
|
|
|
Table 10.5.4(b) Biosimilar Reduction Percentage |
| Decline in Net Sales in a Country |
Net Sales Reduction in such Country |
| [***] |
[***] |
| [***] |
[***] |
| [***] |
[***] |
| [***] |
[***] |
(c)Third Party Licenses. Subject to Section
10.5.5 (Cumulative Reductions Floor) and the last sentence of the introductory paragraph of Section
10.5 (General), on a
Licensed COVID-19 Product-by-Licensed COVID-19 Product and country-by- country basis, Sanofi has the right to credit [***] of all consideration actually paid by Sanofi to a Third Party or any Acquirer pursuant to an agreement between Sanofi and such Third Party or Acquirer entered into following the Effective Date to the extent in consideration for a license to Sanofi under Patents controlled by such Third Party or Acquirer that (i) would be infringed by the sale of the Licensed COVID-19 Mono Product or (ii) Cover (A) the Licensed COVID-19 Component or (B) the Adjuvant, in each case, in such country against any royalties due to Novavax hereunder for the applicable Licensed Product in such country (“Third Party License”). Notwithstanding anything to the contrary in this Agreement, to the extent Sanofi is required to pay
(i) [***] or (ii) any royalty or other consideration under a Third Party License (including per any settlement of any Product Infringement Claim) that is attributable to Novavax’s breach of its representations and warranties hereunder, Sanofi shall have the right to deduct from the royalties owed to Novavax [***] of all such royalties and other consideration, and the cumulative reduction floor of Section
10.5.5 (Cumulative Reductions Floor) shall not apply to that portion of reduction attributable to such payments.
(d)Compulsory Licenses. During the COVID-19 Royalty Term, if for a given country and a given Licensed COVID-19 Product (i) Sanofi is compelled by Applicable Law to grant a compulsory license permitting a Third Party the right to sell a Biosimilar Product as to such Licensed COVID-19 Product in such country at a royalty rate that is lower than the applicable royalty rate set forth in Section
10.5.1 (Licensed COVID-19 Product Royalty Rates) for the corresponding Licensed COVID-19 Product and (ii) such Third Party sells a Biosimilar Product as to such Licensed COVID-19 Product in such country pursuant to such compulsory license, then Net Sales in such country used for purposes of calculating worldwide Net Sales of such Licensed COVID-19 Product and royalty payments due to Novavax based on Net Sales of such Licensed COVID-19 Product in such country pursuant to Section
10.5.1 (Licensed COVID-19 Product Royalty Rates) will be reduced by [***].
1.5.5Cumulative Reductions Floor. Subject to the last sentence of the introductory paragraph of Section
10.5 (General), in no event will the reductions set forth in Section
10.5.4(a) (Lack of Valid Claim),
10.5.4(b) (Biosimilar Product Competition), Section
0 (Third Party Licenses), and
10.5.4(d) (Compulsory Licenses) cause the royalties otherwise due to Novavax for any Licensed COVID-19 Product in a given Calendar Quarter in a given country during the applicable COVID-19 Royalty Term for such Licensed COVID-19 Product to be reduced by more than [***] of the amount that would otherwise be due in such Calendar Quarter for such Licensed COVID-19 Product in such country but for such reductions, except that if the Net Sales reduction under Section
10.5.4(b) (Biosimilar Product Competition) is [***], then the royalties due for such Licensed COVID-19 Product in such Calendar Quarter may be reduced by more than [***] of the amounts otherwise due for such Licensed COVID-19 Product in such Calendar Quarter but for such reductions. Any reductions not able to be made or taken in respect of royalties in a given Calendar Quarter as a result of this Section
10.5.5 (Cumulative Reductions Floor) shall be carried forward for reduction in respect of royalties in future Calendar Quarters until fully exhausted.
1.5.6Royalty Reports and Payments. Following the First Commercial Sale of a Licensed Product in the Territory and continuing until there are no Licensed Products being Commercialized in the Territory for which royalties are payable hereunder, Sanofi will deliver to Novavax within [***] a report summarizing on a Licensed Product-by-Licensed Product and country-by-country basis: (a) total Net Sales and Cancellation Fees, in each case (i) and (ii), in the relevant [***]; (b) to the extent such Net Sales and Cancellation Fees include sales or fees that are not denoted in Dollars, a summary of the amount of Net Sales and Cancellation Fees in local currency and any conversion per Section
10.14.2 (Exchange); and (c) royalties payable on such Net Sales and Cancellation Fees (each such report a “Royalty Report”). Within [***] following the receipt of each Royalty Report, Novavax will issue an invoice to Sanofi for the corresponding amount payable, and Sanofi shall pay such invoice (unless disputed) within [***] after receipt. In addition to the Royalty Reports, Sanofi shall provide Novavax with such other information requested by Novavax that is necessary for Novavax to comply with its royalty payment and financial reporting obligations under any Upstream License Agreement (including under Section 7.1 (Royalty Payments; Reports) of the Upstream License Agreement); provided however that Sanofi shall have no obligation to provide to Novavax or any Third Party with whom Novavax has entered into a Upstream License Agreement any competitively sensitive information.
1.6Research and Development Plan Costs.
1.6.1Reimbursement; In-Kind. As between the Parties, Novavax will bear the COVID-19 Research and Development Plan Costs it incurs in the performance of activities allocated to it under the COVID-19 Research and Development Plan from the Effective Date until December 31, 2024. Commencing on January 1, 2025, Sanofi will reimburse Novavax, on a Calendar Quarter-by-Calendar Quarter basis, (a) for all COVID-19 Research and Development Plan Costs incurred by or on behalf of Novavax in the applicable Calendar Quarter in accordance with the COVID-19 Research and Development Budget plus Allowable Overruns and (b) for all other amounts required to be reimbursed by Sanofi in accordance with this Agreement. In the event that Novavax incurs any COVID-19 Research and Development Plan Costs above the COVID-19 Research and Development Budget plus Allowable Overruns, then Novavax shall bear such COVID-19 Research and Development Plan Costs itself. Sanofi will bear the COVID-19 Research and Development Plan Costs it incurs in the performance of activities allocated to it under the COVID-19 Research and Development Plan.
1.6.2Reporting Research and Development Plan Costs. COVID-19 Research and Development Plan Costs will initially be borne by the Party incurring the cost or expense, subject to reimbursement as provided in Section
10.6.1 (Reimbursement; In-Kind). Within [***], Novavax will provide Sanofi with a detailed, itemized invoice of the COVID-19 Research and Development Plan Costs incurred by or on behalf of it and its Affiliates during such [***] in the form agreed by the Parties through the JFC, as may be updated by the JFC from time to time (each, a “Research and Development Plan Costs Invoice”). Within [***] following the receipt of each Research and Development Plan Costs Invoice, Sanofi will have the right to request reasonable additional information related to such COVID-19 Research and Development Plan Costs during such [***] in order to confirm that such reported COVID-19 Research and Development Plan Costs conform with the approved COVID-19 Research and Development Budget. Sanofi will pay the undisputed
amounts invoiced within [***] after receipt of such Research and Development Plan Costs Invoice.
1.6.3Sanofi will reimburse Novavax’s FTE Costs and Out-of-Pocket Costs that Novavax incurs in connection with transferring Novavax Materials pursuant to Section
3.6 (Transfer of Materials).
1.7Regulatory Costs.
1.7.1Reimbursement; In-Kind.
(a)Except as set forth in Section
10.6 (Research and Development Plan Costs), with respect to the specific Regulatory activities allocated to Novavax that are included in the COVID-19 Research and Development Plan and the corresponding COVID- 19 Research and Development Budget and this Section
10.7 (Regulatory Costs), Novavax will bear the FTE Costs and Out-of-Pocket Costs incurred by Novavax in the performance of Regulatory activities by Novavax with respect to Licensed COVID-19 Mono Products in the Collaboration COVID-19 Territory.
(b)Novavax will bear its own the Regulatory Costs incurred in the performance of the Regulatory activities allocated to it under the Regulatory Transfer Plan.
(c)In each country, jurisdiction, or region in which Sanofi is the Regulatory Responsible Party for a given Licensed COVID-19 Mono Product, Sanofi shall reimburse Novavax with respect to the FTE Costs and Out-of-Pocket Costs incurred by or on behalf of Novavax to provide assistance to Sanofi under Section
4.6.2 (Right of Reference) and Section
4.6.3 (Right of Reference).
(d)In each country, jurisdiction, or region in which Novavax (i) remains the Regulatory Responsible Party for a given Licensed COVID-19 Mono Product, and
(ii)is the sole Party that is Commercializing such Licensed COVID-19 Mono Product in such country, jurisdiction, or region, except for the specific Regulatory Costs referred to in paragraphs (a), (b), and (c) above, Novavax will bear all Regulatory Costs in connection with obtaining and maintaining Marketing Authorization for such Licensed COVID-19 Mono Product for such country, jurisdiction, or region.
(e)In each country, jurisdiction, or region in which Novavax (i) remains the Regulatory Responsible Party for a given Licensed COVID-19 Mono Product, and
(ii)both Parties are Commercializing such Licensed COVID-19 Mono Product, except for the specific Regulatory Costs referred to in paragraphs (a), (b), and (c) above, Sanofi will reimburse Novavax on a pro rata basis (based on doses of such Licensed COVID-19 Mono Product sold by each Party to final customers in such country, jurisdiction, or region) for all Regulatory Costs incurred by or on behalf of Novavax in connection with obtaining and maintaining Marketing Authorization for such Licensed COVID-19 Mono Product in such country, jurisdiction, or region.
(f)In each country, jurisdiction, or region in which Sanofi (i) is the Regulatory Responsible Party for a given Licensed COVID-19 Mono Product, and (ii) both Parties are Commercializing such Licensed COVID-19 Mono Product, except for
the specific Regulatory Costs referred to in paragraphs (a), (b), and (c) above, Novavax will reimburse Sanofi on a pro rata basis (based on doses of such Licensed COVID-19 Mono Product sold by each Party to final customers in such country, jurisdiction, or region) for all Regulatory Costs incurred by or on behalf of Sanofi in connection with obtaining and maintaining Marketing Authorization for such Licensed COVID-19 Mono Product in such country, jurisdiction, or region.
(g)In each country, jurisdiction, or region in which Sanofi is the Regulatory Responsible Party for a given Licensed COVID-19 Mono Product, and Sanofi the sole Party that is Commercializing such Licensed COVID-19 Mono Products in such country, jurisdiction, or region, except for the specific Regulatory Costs referred to in paragraphs (a), (b), and (c) above, Sanofi will bear all Regulatory Costs incurred in connection with obtaining and maintaining Marketing Authorization for such Licensed COVID-19 Mono Products in such country, jurisdiction, or region.
(h)Novavax will bear all FTE Costs and Out-of-Pocket Costs incurred by or on behalf of Novavax in maintaining the global safety database in Calendar Year 2024. Commencing in Calendar Year 2025 and continuing until the transfer of the global safety database to Sanofi in accordance with Section
4.9.2, Sanofi will reimburse Novavax for fifty percent (50%) of the FTE Costs and Out-of-Pocket Costs incurred by or on behalf of Novavax in maintaining the global safety database.
1.7.2Reporting Regulatory Costs. All Regulatory Costs will initially be borne by the Party incurring the cost or expense, subject to reimbursement as provided in Section
10.7.1 (Reimbursement; In-Kind). Within [***], Novavax will provide Sanofi with a detailed, itemized invoice of the costs incurred by or on behalf of it and its Affiliates during such [***] in the form agreed by the Parties through the JFC, as may be updated by the JFC from time to time (each, a “Regulatory Costs Invoice”). Sanofi will pay the undisputed amounts in each Regulatory Costs Invoice within [***] after receipt of such Regulatory Costs Invoice.
1.8Technology Transfer Costs.
1.8.1Reimbursement; In-Kind. Sanofi will reimburse Novavax, on a Calendar Quarter-by- Calendar Quarter basis for the Technology Transfer Costs incurred by or on behalf of Novavax in the applicable Calendar Quarter that are in accordance with Technology Transfer Budget plus Allowable Overruns. If Novavax incurs any Technology Transfer Costs under the applicable Technology Transfer Plan that are not in accordance with the applicable Technology Transfer Budget plus Allowable Overruns, then Novavax shall bear such Technology Transfer Costs itself. Sanofi will bear all FTE Costs and Out-of-Pocket Costs that it incurs in the performance of activities allocated to it under any Technology Transfer Plan.
5.1.3 (Technical Assistance). Each Party will bear its own FTE Costs and will each bear an equal (50%:50%) share of all Out-of-Pocket Costs incurred in connection with maintaining the Adjuvant Intermediate Technology Escrow pursuant to Section
5.6 (Adjuvant Intermediates Technology Escrow) and the Adjuvant Technology Escrow and COVID-19 Technology Escrow pursuant to Section
5.7 (Other Escrows).
1.8.2Technology Transfer Costs. Technology Transfer Costs will initially be borne by Novavax, subject to reimbursement as provided in Section 10.7.1 (Reimbursement; In-Kind). Within [***], Novavax will provide Sanofi with a detailed, itemized invoice of the Technology Transfer Costs reimbursable by Sanofi in accordance with Section
10.8.1 (Reimbursement; In-Kind) under the applicable Technology Transfer Budget that are incurred by or on behalf of it and its Affiliates during such [***] in the form agreed by the Parties through the JFC, as may be updated by the JFC from time to time (each, a “Technology Transfer Costs Invoice”). Within [***] following the receipt of each Technology Transfer Costs Invoice, Sanofi will have the right to request reasonable additional information related to such Technology Transfer Costs during such [***] in order to confirm that such reported Technology Transfer Costs are in accordance with the approved Technology Transfer Budget. Sanofi will pay the undisputed amounts invoiced within [***] after receipt of such Technology Transfer Costs Invoice.
1.9Manufacturing Costs. Novavax will bear its own Manufacturing Costs incurred in connection with the Manufacturing for (a) the Exploitation of any Novavax CIC Products, Novavax OC Products, or Novavax Adjuvant Products and (b) the Exploitation of Licensed COVID-19 Mono Products in any country for which Novavax is performing Commercialization activities pursuant to Article
7 (Commercialization) or the Licensed COVID-19 Mono Product Commercialization Framework. In addition, Novavax will reimburse Sanofi for all Licensed COVID-19 Mono Products that Sanofi may supply to Novavax in accordance with Section
6.1.1 (Supply for Novavax). Sanofi shall bear its own Manufacturing Costs incurred in connection with the Manufacturing for (a) the Exploitation of Licensed CIC Products, Licensed OC Products, and Licensed Adjuvanted Products, and (b) the Exploitation of Licensed COVID-19 Mono Products in any country for which Sanofi is performing Commercialization activities pursuant to Article
7 (Commercialization) or the Licensed COVID-19 Mono Product Commercialization Framework. In addition, Sanofi will reimburse Novavax for all Licensed COVID-19 Component, Licensed COVID-19 Mono Product, [***], [***] or Adjuvant Intermediate, as applicable, that Novavax supplies to Sanofi under the COVID-19 Supply Agreement or Adjuvant Supply Agreement, as applicable, in accordance with such agreement, and in the event that the Parties have not executed a COVID-19 Supply Agreement or Adjuvant Supply Agreement, as applicable, in accordance with this Agreement.
1.10Commercialization Costs. Except as set forth in Section
10.6 (Research and Development Plan Costs), with respect to the specific Medical Activities (e.g., post-Marketing Authorization clinical trials) allocated to Novavax that are included in the COVID-19 Research and Development Plan, Novavax will bear its own costs and expenses incurred in connection with the (a) Commercialization of any Novavax CIC Product, Novavax OC Product, or Novavax Adjuvant Product and (b) the Commercialization of Licensed COVID-19 Mono Products for which Novavax is performing Commercialization activities pursuant to Article
7 (Commercialization) or the Licensed COVID-19 Mono Product Commercialization Framework. Sanofi will bear its own costs and expenses incurred in connection with (a) the Commercialization of Licensed CIC Products, Licensed OC Products, and Licensed Adjuvanted Products, and (b) the Commercialization of Licensed COVID-19 Mono Products for which Sanofi is performing Commercialization activities pursuant to Article
7 (Commercialization) or the Licensed COVID-19 Mono Product Commercialization Framework. For clarity, each Party shall bear its own FTE Costs and Out-of- Pocket Costs incurred in connection with Section
7.10.1(g) (Pricing Approvals).
1.11Revenues and Expenses; Profits and Losses. Except as expressly provided pursuant to this
Article 10 (Financial), neither Party shall have any right to receive any payment from the other
Party hereunder, including that neither Party shall have any right to receive all or any portion of any revenues or profits of the other Party, and neither Party will have any obligation to bear any losses or to pay or reimburse for any costs or expenses incurred by the other Party.
1.12Medical Costs. With respect to the Licensed COVID-19 Mono Product, the costs of Medical Activities that are set forth in the COVID-19 Research and Development Plan will be deemed COVID-19 Research and Development Costs and will be accounted for in the COVID-19 Research and Development Budget. Novavax shall report to Sanofi the costs incurred by Novavax in connection with conducting such Medical Activities in accordance with Section
10.6.2 (Reporting Research and Development Plan Costs).
1.13Patent Costs. The Party Prosecuting and Maintaining a Patent will bear all FTE Costs and Out-of- Pocket Costs incurred by or on behalf of such Party in connection with such Prosecution and Maintenance.
1.14Other Payment Terms.
1.14.1Currency; Payment Method. All payments under this Agreement are expressed in Dollars and will be paid in Dollars, by wire transfer or automated clearing house payment to an account designated by the applicable Party, which account each Party may update from time to time in writing.
1.14.2Exchange. If any amounts that are relevant to the determination of amounts to be paid under this Agreement or any calculations to be performed under this Agreement are denoted in a currency other than Dollars, then such amounts will be converted to their Dollar equivalent using the paying Party’s (or, as applicable, its Sublicensee’s) then-current standard procedures and methodology, including its then-current standard exchange rate methodology for the translation of foreign currency expenses into Dollars consistently applied.
1.14.3Other Invoices. With respect to any payment owed by one Party to the other Party under this Agreement for which no other invoicing and payment procedure is specified elsewhere herein, the payee Party shall issue an invoice for such payment which invoice shall refer to this Agreement and the specific provision under which the payment is owed, and the payor Party shall pay such invoice (unless disputed) within [***] after receipt.
1.14.4Right to Reconcile Payments. If a court of competent jurisdiction has determined that Sanofi is entitled to any amount as damages for Novavax’s breach of this Agreement and Sanofi has not elected the alternative remedy set forth in Section
16.2.2(c)(ii) (Termination for Material Breach), then Sanofi will have the right to offset any such amount owed by it to Novavax against amounts payable to Novavax under this Agreement, provided that prior to exercising such offset right, Sanofi will notify Novavax of its intent to exercise such offset right. Such offsets will be in addition to any other rights or remedies available under this Agreement and Applicable Law.
1.14.5No Double Counting. To the extent any cost and expense of an activity conducted by a Party hereunder is reimbursable by the other Party under more than one (1) provision of this Agreement (e.g., the same activity is deemed an activity under the COVID-19 Research and Development Plan and a Technology Transfer Plan), such Party shall not seek reimbursement of such cost and expense under more than one (1) provision of this Agreement pursuant to which reimbursement may be sought, and the reimbursing Party
shall not be obligated to pay for any given reimbursable cost or expense for any activity more than once.
1.15Withholding Tax. If any sum due to be paid to or by a Party hereunder is subject to any withholding, then the payor Party will pay such withholding to the appropriate Governmental Authority and deduct the amount paid from the amount then due to the payee Party. The payor Party will timely transmit to the payee Party an official tax certificate or other evidence of such withholding sufficient to enable the payee Party to claim such payment of taxes. The Parties will cooperate with one another and use reasonable efforts to reduce or eliminate tax withholding in respect of royalties and other payments made by one Party to the other Party under this Agreement. The payee Party will provide the payor Party any tax forms that may be reasonably necessary in order for payor Party not to withhold tax or to withhold tax at a reduced rate under an applicable bilateral income tax treaty. Each Party will provide the other with reasonable assistance to enable the recovery, as permitted by Applicable Law, of withholding taxes, such recovery to be for the benefit of the Party bearing such withholding tax. In no event will the payor Party be obligated to make any adjustment or gross up payment to the payee Party as a result of any withholding or similar tax levied by any Governmental Authority in connection with any payments hereunder. If either Party is converted from a U.S. corporation to a legal entity that is not regarded for U.S. income tax purposes, and such conversion results in withholding tax that otherwise would not arise, then the converted Party will bear the cost of the resulting withholding tax. Novavax will provide Sanofi, from time to time, an executed US IRS W-9 form if requested by Sanofi to effectuate any payment hereunder, including with respect to Section
10.1 (Up-Front Fee) and subsequently upon request by Sanofi.
1.16Financial Records; Audits. Novavax will keep and maintain accurate and complete records regarding its COVID-19 Research and Development Plan Costs, Manufacturing Costs for Materials supplied to Sanofi, Technology Transfer Costs, and any other costs and expenses that are reimbursable by Sanofi under this Agreement, and Sanofi will keep and maintain accurate and complete records regarding Net Sales and Cancellation Fees of Licensed Products required to prepare the reports, including the Royalty Report, or calculate payments required by Section
0 (Milestone Payments) or owed under any Upstream License Agreement (collectively, the “Financial Records”). Each Party shall retain Financial Records for a period of at least [***]. Upon [***] prior written notice, a Party (the “Auditing Party”) will have the right to have an audit conducted of the other Party’s (the “Audited Party”) Financial Records; provided that such audit right may not be exercised more than [***] and shall be limited to the Financial Records of the Audited Party pertaining to any financial report or payment due in any Calendar Quarter ending not more than [***] prior to such Party’s request. The audit will be conducted by an independent certified public accounting firm of internationally recognized standing, selected by the Auditing Party and reasonably acceptable to the Audited Party (the “Auditor”). The Auditor shall examine the applicable Financial Records to determine the basis and accuracy of payments made or due under this Agreement during the period of time covered by the audit. The Auditor will be provided access to the Audited Party’s Financial Records at the Audited Party’s facility or facilities where such books and records are normally stored, and such examination will be conducted during the Audited Party’s normal business hours. The Audited Party has the right to require the Auditor to sign a customary non-disclosure agreement before providing the Auditor access to its Financial Records. Upon completion of the audit, the Auditor will provide both Parties a written report disclosing whether the Financial Records or the Royalty Reports, as applicable, covered by the audit, are correct or incorrect, and if incorrect, the specific details concerning any discrepancies. If the audit reveals an underpayment, then the Party that owes such payment will pay such underpayment to the other Party forthwith plus interest in accordance with Section
10.17 (Late Payment). If the audit reveals
an overpayment, then the Party that paid such overpayment may credit such amount against any future payments due to the other Party hereunder, and if none are expected within a Calendar Year, then such Party shall be paid by the other Party within [***] after receipt of an invoice for such amount that was overpaid. The costs and fees of any audit conducted by the Auditing Party under this Section
10.16 (Financial Records; Audits) will be borne by the Auditing Party, unless such audit reveals an underpayment of amounts owed to the Auditing Party exceeding the lesser of
(a) [***] and (b) [***] of the amount that was owed by the Audited Party with respect to the relevant period, in which case, the Audited Party will reimburse the Auditing Party for the reasonable expense incurred by the Auditing Party in connection with the audit.
1.17Late Payment. Any undisputed payment under this Agreement that is not paid on or before the date such undisputed payment is due will bear interest, to the extent permitted by Applicable Law, at [***].
ARTICLE 11: CONFIDENTIALITY AND NON-DISCLOSURE
1.1Confidential Information. Notwithstanding any provision to the contrary set forth herein:
1.1.1(a) the terms of this Agreement, Novavax COVID-19 Know-How, Novavax COVID-19 Materials, unpublished Novavax COVID-19 Patents, Joint Arising Patents, and Joint Arising Know-How; (b) information in any plan (whether for Research, Development, Manufacturing, Commercialization, Medical Activities, Technology Transfer activities, or otherwise, and whether in draft or final form) created by either Party if such plan is reviewed by any Committee; and (c) any budget (whether for Research, Development, Manufacturing, Commercialization, Medical Activities, Technology Transfer activities, or otherwise) created by either Party if such budget is reviewed by any Committee, in each case (a)-(c), will be treated as Confidential Information of both Parties with each Party treated as the Receiving Party with respect thereto; provided that, to the extent any Confidential Information of a Disclosing Party is included in any report, notes, analyses, plan, budget, or other documents prepared or delivered by or on behalf of either Party hereunder, whether with respect to any Research, Development, Manufacturing, Commercialization, or other activity carried out hereunder, the Confidential Information of the Disclosing Party contained in any such report notes, analyses, plan, budget, or other documents shall remain the Confidential Information of the Disclosing Party;
1.1.2the Novavax Adjuvant Know-How, Novavax Adjuvant Materials, Adjuvant Intermediate Technology, unpublished Novavax Adjuvant Patents and Novavax Commercialization Reports, will each be the Confidential Information of Novavax, with Sanofi deemed the Receiving Party with respect thereto; and
1.1.3the Sanofi Arising Know-How, Sanofi Arising Patents, Sanofi Commercialization Reports, and Sanofi Licensed COVID-19 Mono Assets will each be the Confidential Information of Sanofi, with Novavax deemed the Receiving Party with respect thereto.
1.2Confidentiality. Each Party (the “Receiving Party”) that receives or whose Affiliates receive any Confidential Information of the other Party (the “Disclosing Party”) hereunder will: (a) keep the
Disclosing Party’s Confidential Information confidential using not less than the efforts such Receiving Party uses to maintain in confidence its own confidential or proprietary information of similar kind and value (but in any event no less than reasonable efforts); (b) not publish, or allow to be published, and not otherwise disclose, or permit the disclosure of, the Disclosing Party’s Confidential Information; and (c) not use, or permit to be used, the Disclosing Party’s Confidential Information for any purpose, other than to exercise the Receiving Party’s rights or perform the Receiving Party’s obligations under this Agreement, in each case, strictly in accordance with this Agreement, except, in each case ((a) – (c)), to the extent permitted under this Agreement or otherwise agreed in writing by the Disclosing Party. Without limiting the generality of the foregoing, to the extent either Party provides the other Party any Confidential Information owned by any Third Party, the Receiving Party will handle such Confidential Information in accordance with the terms of this Article
11 (Confidentiality and Non-Disclosure) applicable to a Receiving Party. The Parties acknowledge and agree that any Confidential Information disclosed by the Disclosing Party to the Receiving Party prior to the Effective Date pursuant to the CDA will constitute the Confidential Information of the Disclosing Party hereunder.
1.3Exceptions. Confidential Information will not include any information to the extent the Receiving Party can demonstrate by competent written evidence that such information:
1.3.1was known to the Receiving Party or any of its Affiliates, without any obligation to the Disclosing Party or its Affiliates to keep it confidential, prior to disclosure to the Receiving Party or any of its Affiliates by the Disclosing Party;
1.3.2is subsequently disclosed to the Receiving Party or any of its Affiliates on a non- confidential basis by a Third Party that is not bound by a duty of confidentiality to the Disclosing Party or its Affiliate;
1.3.3is now, or hereafter becomes, through no act or failure to act on the part of the Receiving Party or any of its Affiliates in violation of this Agreement, generally known or available to the public, either before or after it is disclosed to the Receiving Party by the Disclosing Party; or
1.3.4is independently discovered or developed by or on behalf of the Receiving Party or any of its Affiliates, in each case, without the use of or reference to the Confidential Information of the Disclosing Party.
1.4Authorized Disclosure. Notwithstanding Section
11.1 (Confidentiality), the Receiving Party may disclose the Disclosing Party’s Confidential Information to the extent such disclosure is reasonably necessary to:
1.4.1prosecute or defend litigation, including responding to a subpoena in a Third Party litigation;
1.4.2Prosecute and Maintain Patents in accordance with the terms of this Agreement;
1.4.3generate Regulatory Filings and file for and obtain Marketing Authorizations or Pricing Approvals, in each case, in accordance with the terms of this Agreement;
1.4.4satisfy Novavax’s obligations under the Upstream License Agreements or any Existing Strategic Partner Agreements or APAs; provided that, to the extent practicable, Sanofi shall have the right to review any proposed disclosure to an Upstream Licensor, Strategic
Partner, or APA counterparty and request that Novavax redact, prior to any such disclosure, any commercially sensitive information that is not required to be shared with the Upstream Licensor, Strategic Partner, or APA counterparty in accordance with the Upstream License Agreement or any Existing Strategic Partner Agreements or APAs, as applicable, further provided that Sanofi provides such comments within [***] after Novavax provides such proposed disclosure to Sanofi;
1.4.5allow its Affiliates and actual or potential Sublicensees and actual or potential Service Providers, in each case, engaged in accordance with this Agreement to exercise the Receiving Party’s rights or perform the Receiving Party’s obligations under this Agreement strictly in accordance with this Agreement; provided that any such disclosure is covered by terms of confidentiality with such Affiliates, Sublicensees, and Service Providers that are consistent with the Receiving Party’s obligations under this
Article 11 (but of shorter duration if customary, but, in any event, not less than [***] after the expiration or termination of the agreement with such Third Party, provided that Confidential Information relating to manufacturing or CMC information, clinical data, or other information identified as, or customarily held in the pharmaceutical industry as, a trade secret, shall be held in confidence until such information is no longer deemed trade secret under Applicable Law). In any event, the Receiving Party will remain responsible for any violation of such confidentiality provisions by any Person who receives Confidential Information pursuant to this Section
11.4.5 (Authorized Disclosure);
1.4.6share with the Receiving Party’s advisors (including financial advisors, attorneys, and accountants), actual or potential acquisition or collaboration partners, financing sources, or investors, or underwriters on a need to know basis; provided that any such disclosure is covered by terms of confidentiality with such advisors, partners, sources, investors, and underwriters that are consistent with the Receiving Party’s obligations under this Article
11 (which may include professional ethical obligations) (but, in any event, not less than [***] after the expiration or termination of the agreement with such Third Party or as otherwise customary for the nature of the disclosure, provided that Confidential Information held as a trade secret, including relating to manufacturing or CMC information, clinical data, or other information identified as, or customarily held in the pharmaceutical industry as, a trade secret, shall be held in confidence until such information is no longer deemed trade secret under Applicable Law). In any event, the Receiving Party will remain responsible for any violation of such confidentiality provisions by any Person who receives Confidential Information pursuant to this Section
11.4.6 (Authorized Disclosure); or
1.4.7to the extent required to comply with Applicable Law or a court or administrative order (excluding the United States Securities and Exchange Commission or a similar regulatory agency in a country other than the United States (a “Stock Exchange”), which disclosures are subject to Section
11.5 (Required Disclosures)), in each case, to the extent applicable to such Party at such time; provided that the Party who is required to make such disclosure under this Section
11.4.7, if permitted under Applicable Law or order, (a) provides the other Party with reasonable prior written notice, (b) coordinates with the other Party with respect to the wording and timing of any such disclosure and affords the other Party an opportunity to oppose or limit, or secure confidential treatment for, such required disclosure, (c) if unsuccessful in its efforts pursuant to clause (b), then takes all reasonable and lawful actions to obtain confidential treatment for such disclosure, and (d) discloses the minimum amount and scope of the Confidential Information necessary to comply with Applicable Law.
If and whenever any Confidential Information is disclosed in accordance with this Section
11.4 (Authorized Disclosure), such disclosure will not cause any such information to cease to be Confidential Information for purposes of this Agreement, except to the extent that such disclosure results in a public disclosure of such information in accordance with Section
11.3.2.
With respect to Section
11.4.1 (Authorized Disclosure) or Section
11.4.7 (Authorized Disclosure), the Receiving Party will (a) except where impracticable or not legally permitted, notify the Disclosing Party of the Receiving Party’s intent to make any disclosure pursuant thereto sufficiently prior to making such disclosure so as to allow the Disclosing Party adequate time to take whatever action it may deem appropriate to protect the confidentiality of the information to be disclosed and (b) use not less than the same efforts to secure confidential treatment of such information as it would to protect its own Confidential Information from disclosure (but no less than reasonable efforts), including, if requested by the Disclosing Party, reasonably cooperating with, and at the expense of, the Disclosing Party to secure such confidential treatment.
1.5Required Disclosures. In addition to the disclosures permitted under Section
11.4 (Authorized Disclosure), either Party may disclose the terms of this Agreement and other information relating to this Agreement or the transactions contemplated by this Agreement to the extent required to comply with the rules and regulations promulgated by a Stock Exchange, subject to the remainder of this Section
11.5 (Required Disclosures). Before a Party discloses this Agreement or any of its terms or other any other Confidential Information in accordance with this Section
11.5 (Required Disclosures), to the extent reasonably practicable and to the extent not prohibited by Applicable Law or judicial or administrative process, such Receiving Party will (a) notify the Disclosing Party of such required disclosure, (b) take all steps reasonably necessary to seek the continued confidential treatment of, or redact, such Confidential Information, and (c) only disclose that portion of Confidential Information that is legally required to be disclosed. Promptly before disclosing this Agreement or any of the terms hereof pursuant to this Section
11.5 (Required Disclosures), the Parties shall, to the extent not prohibited by Applicable Law or judicial or administrative process and to the extent reasonably practicable, coordinate in advance with each other in connection with the redaction of certain provisions of this Agreement (together with all exhibits and schedules) with respect to filings required under Applicable Law, and the Parties shall use reasonable efforts to file redacted versions of this Agreement with the applicable governing bodies that are consistent with the coordinated redaction efforts between the Parties. In addition, the Parties hereby consent to the disclosure of a copy of this Agreement to any tax authority by the other Party (i) upon receipt of any legally enforceable information request by such tax authority,
(ii) in compliance with any legally enforceable filing requirement, or (iii) in connection with a submitted transfer pricing analysis. In the event of any such disclosure, the Party disclosing Confidential Information will make reasonable efforts to ensure that the information is maintained in confidence by the applicable tax authority, including marking any disclosed document as confidential.
1.6Public Announcements. On or about the Effective Date, each of Sanofi and Novavax will issue separate press releases or other media statements announcing that this Agreement has been signed in substantially the forms attached hereto as Exhibit A (Initial Press Releases). Except as set forth in the preceding sentence or as expressly required or permitted under Section
11.4 (Authorized Disclosure), Section
11.5 (Required Disclosures), this Section
11.6 (Public Announcements), or Section
11.7 (Scientific COVID-19 Mono Publications), (a) no Party shall issue any press release or other similar public communication relating to this Agreement, its subject matter, or the transactions covered by it, or the activities of the Parties under or in connection with this Agreement, without the prior written approval of the other Party, and (b) if a Party wishes to issue any press release or other similar public communication relating to this Agreement, its subject
matter, or the transactions covered by it, or the activities of the Parties under or in connection with this Agreement other than a press release or public announcement required to comply with the rules and regulations promulgated by a Stock Exchange, the issuance of which will be subject to the terms of Section
11.5 (Required Disclosures) (a “Press Release”) and such Party has obtained the other Party’s prior written approval to the extent required pursuant to the forgoing clause (a), then such Party shall provide the other Party reasonable opportunity to review and comment on any such press release or public communication at least [***] in advance thereof (to the extent permitted under Applicable Law), except that the period of review and comment shall, notwithstanding the foregoing, be [***] during the months of August and December (to the extent permitted under Applicable Law). The issuing Party shall act in good faith to incorporate any comments provided by the other Party on such press release or public communication. In addition, the disclosing Party may make subsequent public disclosures reiterating the text of any Press Release previously issued in accordance with this Agreement without having to obtain the other Party’s prior consent and approval so long as the information in such Press Release remains true, correct, and the most current information with respect to the subject matter set forth therein. Similarly, after a Proposed Disclosure has been made available to the public, each Party may post such Proposed Disclosure or a link to it on its corporate web site (or any website managed by such Party in connection with a Clinical Trial for a Licensed Product, as appropriate) without the prior written consent of the other Party, so long as the information in such Proposed Disclosure remains true, correct, and the most current information with respect to the subject matter set forth therein.
1.7Scientific COVID-19 Mono Publications.
1.7.1Publication Strategy. The publication and presentation of (a) any Data relating to any Licensed COVID-19 Mono Product and (b) any other scientific publications relating to or supportive of the Licensed COVID-19 Mono Product, whether or not peer-reviewed such as abstracts, manuscripts, commentaries, letters to the editor, review-articles, book- chapters, or pre-prints ((a) and (b), collectively, “Scientific COVID-19 Mono Publications”), in each case, shall be conducted in accordance with the terms of this Section
11.7 (Scientific COVID-19 Mono Publications). The JRDC will develop, discuss, and determine whether to approve a strategy for such publications for each Licensed COVID-19 Mono Product, including the appropriate timing (each, a “Publication Strategy”), which Publication Strategy will align with the advice and strategies of the JPC and will include the registration and reporting of results of pharmaceutical company sponsored Clinical Trials in accordance with each Party’s internal policies.
1.7.2Authorship. Authorship of any publication will be determined in accordance with the guidelines published periodically by the International Committee of Medical Journal Editors. The publishing Party will properly acknowledge the contribution of each Party to the Data being published.
1.7.3Prior Review.
(a)Review Process. Except for a disclosure of the Disclosing Party’s Confidential Information that is permitted under Section 11.4 (Authorized Disclosure), Section
11.5 (Required Disclosures), or Section 11.6 (Public Announcements), prior to publishing or presenting any (i) Confidential Information of the other Party, or (ii) Scientific COVID-19 Mono Publication (each of (i) and (ii), a “Proposed Disclosure”), a Party desiring to submit such Proposed Disclosure for publication (the “Publishing Party”) shall provide the other Party (the “Reviewing Party”)
with a copy of such Proposed Disclosure at least [***] (or at least [***] for any abstracts, posters, or presentations) prior to submission for publication (“Review Period”) in order to allow the Reviewing Party an opportunity to review and comment on the Proposed Disclosure, and such comments will be reasonably taken into account by the Publishing Party. The Reviewing Party shall notify the Publishing Party in writing if the Reviewing Party has an objection to the Proposed Disclosure, whether due to such Proposed Disclosure not aligning with the Publication Strategy, the inclusion of any of its Confidential Information, or to allow time for the Reviewing Party to file for patent protection on any potentially patentable inventions that may be disclosed by the Proposed Disclosure. If the Reviewing Party reasonably determines that such Proposed Disclosure does not align with the Publication Strategy, then the JSC will review, discuss, and determine if such Proposed Disclosure aligns with the Publication Strategy and the Publishing Party may only submit the Proposed Disclosure for publication if the JSC determines that the Proposed Disclosure aligns with the Publication Strategy. If the Reviewing Party reasonably determines that its Confidential Information or a potentially patentable invention would likely be disclosed by such Proposed Disclosure, then it shall so advise the Publishing Party within the Review Period, whereupon the Publishing Party shall postpone publication of such Proposed Disclosure for a period of [***] (the “Extended Review Period”) to afford the Reviewing Party the opportunity to further evaluate the patentability of any inventions that may be disclosed by the Proposed Disclosure; provided that, if such Confidential Information of the Reviewing Party may be valuable as a trade secret, then the Reviewing Party shall notify the Publishing Party of such determination during the Extended Review Period, and the Publishing Party will remove such Confidential Information of the Reviewing Party from the Proposed Disclosure prior to submission for publication. If the Reviewing Party gives notice to the Publishing Party during the Extended Review Period that it intends to prepare and file a Patent application that contains any of its Confidential Information contained in such Proposed Disclosure, then the Publishing Party will postpone submission of the Proposed Disclosure for publication by an additional [***] to afford the Reviewing Party the opportunity to prepare and file such a Patent application. Notwithstanding any provision to the contrary set forth in this Agreement, the Publishing Party will not submit for publication any Proposed Disclosure that contains Confidential Information of the Reviewing Party without the written consent of the Reviewing Party.
(b)Disclosure of Clinical Data. The Parties acknowledge that each Party, respectively, adheres to certain industry group positions and has adopted certain policies, in each case, regarding the registration and reporting of results of pharmaceutical company sponsored clinical studies. Accordingly, either Party may, in accordance with each Party’s internal compliance policies, Regulatory Authority requirements, and Applicable Law, disclose aggregate clinical results or summaries thereof generated with respect to Licensed COVID-19 Mono Products; provided that such Party complies with the review process set forth in Section
11.7.3(a) (Review Process).
ARTICLE 12: PATENT AND KNOW-HOW MATTERS
1.1Ownership.
1.1.1Novavax Ownership. As between the Parties, Novavax shall solely own all rights, title, and interests in and to any and all Know-How that is first generated, conceived, discovered, created, invented, or otherwise made during the Term by or on behalf of Novavax or any of its Affiliates in the course of performing activities in accordance with this Agreement (“Novavax Arising Know-How”). As between the Parties, Novavax shall solely own all rights, title, and interests in and to any and all Patents that Cover any invention included in the Novavax Arising Know-How (“Novavax Arising Patents”).
1.1.2Sanofi Ownership. As between the Parties, except for Unauthorized Inventions, Sanofi shall solely own all rights, title, and interests in and to any and all Know-How that is first generated, conceived, discovered, created, invented, or otherwise made during the Term by or on behalf of Sanofi or any of its Affiliates in the course of performing activities in accordance with this Agreement (“Sanofi Arising Know-How”). As between the Parties, Sanofi shall solely own all rights, title, and interests in and to any and all Patents that Cover any invention included in the Sanofi Arising Know-How (“Sanofi Arising Patents”, and together with the Novavax Arising Patents and the Joint Arising Patents, the “Arising Patents”).
1.1.3Joint Ownership. As between the Parties, except for Unauthorized Inventions, the Parties will jointly own all rights, title, and interests in and to all Know-How that is first generated, conceived, discovered, created, invented, or otherwise made during the Term jointly by or on behalf of Novavax or its Affiliates or its or their Sublicensees, on the one hand, and Sanofi or its Affiliates or its or their Sublicensees, on the other hand, in the course of performing activities in accordance with this Agreement (“Joint Arising Know-How”). As between the Parties, the Parties will jointly own all rights, title, and interests in and to any and all Patents that Cover any invention included in the Joint Arising Know-How (“Joint Arising Patents”). Except as expressly set forth in this Agreement, including
Article 2 (License Grants; Exclusivity), each Party will not practice the Joint Arising Patents and Joint Arising Know-How without the prior written consent of the other Party, such consent not to be unreasonably withheld, conditioned, or delayed.
1.2Prosecution and Maintenance of Patents.
1.2.1Arising Patents
(a)Sanofi shall have the sole right to Prosecute and Maintain the Sanofi Arising Patents.
(b)Except for any Novavax Arising Patent that is a Sanofi 1st Right COVID-19 Patent, in which case Section
12.2.2 (Sanofi 1st Right COVID-19 Patents) shall apply, Novavax shall have the sole right to Prosecute and Maintain Novavax Arising Patents.
1.2.2Sanofi 1st Right COVID-19 Patents.
(a)Sanofi’s First Right. As between the Parties, Sanofi shall have the first right to be the Prosecuting Party for all (i) Novavax COVID-19 Patents Covering the Licensed COVID-19 Mono Products or Licensed COVID-19 Component and (ii) Joint Arising Patents, but excluding any Novavax Adjuvant Patents and Novavax General Combo Patents (“Sanofi 1st Right COVID-19 Patents”). Sanofi shall,
through the JPC, keep Novavax reasonably informed of all Prosecution and Maintenance activities related thereto. Sanofi shall (i) provide to Novavax copies of all material communications from any Patent Authority regarding the Sanofi 1st Right COVID-19 Patents, and drafts of any material filings to be made to such Patent Authorities [***], or as soon as is practicable, before any final due date, (ii) afford Novavax a reasonable opportunity to review and comment on any drafts prior to their submission, and (iii) consider Novavax’s comments on such drafts in good faith.
(b)Backup Right. If Sanofi decides not to Prosecute and Maintain a Sanofi 1st Right COVID-19 Patent in a given jurisdiction, and such decision (i) is not made for a strategic business reason intended to benefit the Licensed COVID-19 Products (excluding where such reason is primarily for the purpose of (A) benefitting any product that is not a Licensed COVID-19 Product or (B) shortening or ending the COVID-19 Royalty Term for any Licensed COVID-19 Product) and (ii) will result in an irrevocable loss of rights in an invention disclosed or claimed in such Sanofi 1st Right COVID-19 Patent in such jurisdiction, then Sanofi shall provide reasonable prior written notice to Novavax of such decision (which notice shall, in any event, be given no later than [***] prior to the next deadline for any payment or filing that is necessary to avoid an irrevocable loss of rights). Novavax shall thereupon have the right, but not the obligation, to be the Prosecuting Party for such Sanofi 1st Right COVID-19 Patent at its expense in such country or other jurisdiction. Sanofi shall take all reasonable actions necessary to enable Novavax to become the Prosecuting Party in all respects, including executing all papers and instruments, or requiring its employees or contractors to execute such papers and instruments, so as to enable Novavax to Prosecute and Maintain the Sanofi 1st Right COVID-19 Patent in the applicable country or jurisdiction.
1.2.3Novavax General Combo Patents.
(a)Novavax’s First Right. As between the Parties, Novavax shall have the first right to be the Prosecuting Party for all Novavax General Combo Patents. Novavax shall, through the JPC, keep Sanofi reasonably informed of all Prosecution and Maintenance activities related thereto. Novavax shall (i) provide to Novavax copies of all material communications from any Patent Authority regarding the Novavax General Combo Patents, and drafts of any material filings to be made to such Patent Authorities [***], or as soon as is practicable, before any final due date, (ii) afford Sanofi a reasonable opportunity to review and comment on any drafts prior to their submission, and (iii) consider Sanofi’s comments on such drafts in good faith.
(b)Backup Right. If Novavax decides not to Prosecute and Maintain a Novavax General Combo Patent in a given jurisdiction, and such decision (i) is not made for a strategic business reason intended to benefit a Novavax OC Product (excluding abandoning any granted Patent where, in such jurisdiction, the presence or absence of a granted Patent is not reasonably anticipated to adversely impact pricing, approval, or Commercialization) and (ii) will result in an irrevocable loss of rights in an invention disclosed or claimed in such Novavax General Combo Patent in such jurisdiction, then Novavax shall provide reasonable prior written notice to Sanofi of such decision (which notice shall, in any event, be given no later than [***] prior to the next deadline for any payment or filing that is
necessary to avoid an irrevocable loss of rights). Sanofi shall thereupon have the right, but not the obligation, to be the Prosecuting Party for such Novavax General Combo Patents at its expense in such country or other jurisdiction. Novavax shall take all reasonable actions necessary to enable Sanofi to become the Prosecuting Party in all respects, including executing all papers and instruments, or requiring its employees or contractors to execute such papers and instruments, so as to enable Sanofi to Prosecute and Maintain the Novavax General Combo Patents in the applicable country or jurisdiction.
1.2.4Novavax Adjuvant Patents. Novavax will have the sole right (but not the obligation) to Prosecute and Maintain any Novavax Adjuvant Patent (including Joint Arising Patents that are Novavax Adjuvant Patents) at Novavax’s sole cost and expense.
1.2.5Cooperation Regarding Novavax CIC Product Patents. Upon Novavax’s written request, if any Novavax Patent being Prosecuted by Sanofi contains written description support for claims that would Cover a Novavax CIC Product or a Novavax Influenza Component but would not Cover a Licensed COVID-19 Product, then the Parties, through the JPC, will discuss (a) whether it is possible under Applicable Laws governing the Prosecution and Maintenance of Patent Rights, to file such claims in a separate Patent application, for example, by filing appropriate divisional(s), or continuations, that solely Cover the Novavax CIC Product or the Novavax Influenza Component and do not Cover a Licensed COVID-19 Product, (b) the potential for prejudice to the Prosecution and Maintenance, enforcement and defense of any claims of such Novavax Patent, and (c) a plan for coordination of the Prosecution and Maintenance of such separate Patent application and the Novavax Patent so as to minimize any such prejudice, which plan shall require at a minimum (i) that Novavax will diligently and timely provide Sanofi with all material communications with any Patent Authority in connection with such separate Patent application, including drafts of any proposed submissions to such Patent Authority, and shall consider in good faith Sanofi’s input on such communications, (ii) Novavax shall not take any action (including, for the avoidance of doubt, filing any terminal disclaimer) that would be reasonably likely to create a non-statutory double patenting issue between the separate Patent application and any other Novavax Patent Covering a Licensed COVID-19 Product, and (iii) Novavax will not make any remarks on the record that would disparage or call into question the scope, validity, enforceability, priority entitlement, or sufficiency of disclosure of a Novavax Patent Covering a Licensed COVID-19 Product. Following such JPC discussion, Sanofi shall consider in good faith whether to file such separate Patent application, provided that the choice of whether or not to file such separate Patent application will be at Sanofi’s sole discretion.
1.3Unitary Patent System.
1.3.1Definitions. Capitalized terms used in this Section
12.3 (Unitary Patent System) and not otherwise defined in this Agreement will have the meanings given in the Rules of Procedure of the Unified Patent Court as adopted by decision of the Administrative Committee on 8 July 2022 and established by the Agreement on a Unified Patent Court of 19 February 2013 (OJ C 175, 20.6.2013, p. 1) (including any subsequent amendments).
1.3.2Unified Patent Court: Opt-Out and Withdrawal of Opt-Out; Request for Unitary Effect. The Prosecuting Party of any Patent under Section
12.2 (Prosecution and Maintenance of Patents) shall have the sole right (but not the obligation) to (a) file an application to opt out any European Patent from the exclusive competence of the Unified Patent Court, (b) lodge
an application to withdraw an opt-out with respect to any European Patent from the exclusive competence of the Unified Patent Court, in accordance with Applicable Laws (including Article 83 of Unified Patent Court Agreement and Rule 5 of the Rules of Procedure of the Unified Patent Court), or (c) file a request for Unitary Effect for any European Patent. The Party that is not the Prosecuting Party of such Patent will cooperate with the Prosecuting Party in the preparation of any filing requested by the Prosecuting Party under this Section
12.3.2 (Unified Patent Court: Opt-Out and Withdrawal of Opt- Out; Request for Unitary Effect).
1.3.3Enforcement of Unitary Patents. If either Party receives notice in writing of any declaration of noninfringement in connection with the enforcement of any Unitary Patent under Section
12.6 (Patent Enforcement), then the Party receiving such notice shall notify the other Party as soon as is practicable and in any event within five (5) Business Days after the receipt of such notice. The Parties will cooperate to identify any available evidence of infringement or misappropriation of such Unitary Patent and to prepare and file a timely response to such declaration of noninfringement. For clarity, enforcement of any Unitary Patent shall be determined in accordance with Section
12.6 (Patent Enforcement).
1.4Patent Term Extension and Supplementary Protection Certificate. With respect to any Licensed COVID-19 Products, Sanofi shall have the sole right to make decisions regarding which Patent to file for patent term extensions, including supplementary protection certificates and any other extensions that are now or become available in the future, wherever applicable, in any country or other jurisdiction in the Territory. Sanofi shall have the right to extend any Sanofi 1st Right COVID-19 Patent, but shall not file for an extension of any Novavax Patent that is not a Sanofi 1st Right COVID-19 Patent without receiving Novavax’s prior written consent. Sanofi will, through the JPC, keep Novavax reasonably informed of its efforts to obtain such extension. Novavax shall provide prompt assistance, as requested by Sanofi, including by taking such action as Patent owner is required under any Applicable Law to obtain any such extension. Sanofi shall pay all expenses in regard to obtaining any such extension in any country or other jurisdiction in the Territory. For the avoidance of doubt, Novavax shall not have the right to extend the term of any Novavax Patent in connection with a Licensed Product of which the Marketing Authorization is in the name of Sanofi or its Affiliate or Sublicensees.
1.5Patent Listings. As between the Parties, Sanofi shall have the sole right to make all Patent listings and Patent-related submissions to Regulatory Authorities for Licensed Products in any country or other jurisdiction in the Territory, to the extent required or permitted by Applicable Law. Novavax shall provide prompt assistance to Sanofi to the extent necessary to effectuate the intent of this Section
12.5 by taking such action as Patent owner or as Regulatory Responsible Party as is required under any Applicable Law to enable such listing.
1.6Patent Enforcement.
1.6.1Notice. If a Party becomes aware of any infringement or other violation of a Sanofi 1st Right COVID-19 Patent by a Third Party in any country the Territory, or becomes aware of any Invalidity Action asserted by a Third Party against a Sanofi 1st Right COVID-19 Patent (each, an “Infringement”), then such Party shall promptly notify the other Party regarding such Infringement, which notice shall include a summary of all relevant evidence that such Party is aware of with respect thereto (each, an “Infringement Notice”).
1.6.2Enforcement Actions.
(a)

Sanofi 1st Right COVID-19 Patents. As between the Parties, Sanofi shall have the first right, but not the obligation, in its own name (or in the name of Novavax, to the extent required by Applicable Law), (i) to bring and control any Enforcement Action to enforce any Sanofi 1st Right COVID-19 Patent against any Infringement in the Territory (a “COVID-19 Product Competitive Infringement”), and (ii) to defend against any Invalidity Action brought against any Sanofi 1st Right COVID- 19 Patent in the Territory in response to a COVID-19 Product Competitive Infringement, in each case ((i) – (ii)), as it reasonably determines appropriate, using counsel of its own choice (any such Enforcement Action or defense against an Invalidity Action, a “COVID-19 Enforcement Action”). If Sanofi does not institute a COVID-19 Enforcement Action other than for a strategic business reason intended to benefit a Licensed COVID-19 Product (excluding where such reason is primarily for the purpose of benefitting any product that is not a Licensed COVID-19 Product) within [***] after a Party delivers an Infringement Notice to the other Party with respect to such Infringement or Invalidity Action or, if earlier, [***] prior to any final deadline for instituting a COVID-19 Enforcement Action before relinquishing any legal or equitable rights, defenses, or remedies with respect thereto that would otherwise be available, and Sanofi in its reasonable business judgment does not believe that a COVID-19 Enforcement Action would be detrimental to a Sanofi 1st Right COVID-19 Patent or the commercial success of the Licensed COVID-19 Products as a whole, then Sanofi will promptly notify Novavax, and Novavax may, upon written notice to Sanofi, which shall be delivered within [***] after receiving Sanofi’s notice of non-enforcement, institute a COVID-19 Enforcement Action in its own name using counsel of its choice.
(b)Novavax General Combo Patents. As between the Parties, Novavax shall have the first right, but not the obligation, in its own name, (i) to bring and control any Enforcement Action to enforce any Novavax General Combo Patent against any Infringement in the Territory (a “General Combo Product Competitive Infringement”), and (ii) to defend against any Invalidity Action brought against any Novavax General Combo Patent in the Territory in response to a General Combo Product Competitive Infringement, in each case ((i) – (ii)), as it reasonably determines appropriate, using counsel of its own choice (any such Enforcement Action or defense against an Invalidity Action, a “General Combo Product Enforcement Action”). If Novavax does not institute a General Combo Product Enforcement Action within [***] after a Party delivers an Infringement Notice to the other Party with respect to such Infringement or Invalidity Action or, if earlier, [***] prior to any final deadline for instituting a General Combo Product Enforcement Action before relinquishing any legal or equitable rights, defenses, or remedies with respect thereto that would otherwise be available, and Novavax in its reasonable business judgment does not believe that a General Combo Product Enforcement Action would be detrimental to a Novavax General Combo Product Patent or the commercial success of the Novavax OC Products as a whole, then Novavax will promptly notify Sanofi, and Sanofi may, upon written notice to Novavax, which shall be delivered within [***] after receiving Novavax’s notice of non-enforcement, institute a General Combo Product Enforcement Action in its own name using counsel of its choice.
(c)CIC and OC Product Enforcement Actions. Notwithstanding Section
12.6.2(a) (Sanofi 1st Right COVID-19 Patents) and Section
12.6.2(b) (Novavax General Combo Patents), for any Infringement by a product that includes both an Antigen Directed To SARS-CoV-2 and an Antigen Directed To one or more other pathogens or organisms, then:
(i)if Novavax has a Novavax Product being Commercialized, and Sanofi does not have a Licensed CIC Product or Licensed OC Product being Commercialized, in each case, directed to SARS-CoV-2 and to one of the same pathogens or organisms, then Novavax will have the first right, but not the obligation, in its own name, to bring and control any Enforcement Action;
(ii)if Novavax does not have a Novavax Product being Commercialized, and Sanofi does have a Licensed CIC Product or Licensed OC Product being Commercialized, in each case, directed to SARS-CoV-2 and to one of the same pathogens or organisms then Sanofi will have the first right, but not the obligation, in its own name, to bring and control any Enforcement Action; and
(iii)if Novavax has a Novavax Product being Commercialized, and Sanofi has a Licensed CIC Product or Licensed OC Product being Commercialized, in each case, directed to SARS-CoV-2 and to one of the same pathogens or organisms, then the Party that is not bringing a COVID-19 Enforcement Action or General Combo Product Enforcement Action pursuant to Section
12.6.2(a) (Sanofi 1st Right COVID-19 Patents) and Section
12.6.2(b) (Novavax General Combo Patents) shall have the right to join the applicable Enforcement Action, and the Party controlling such Enforcement Action will reasonably cooperate to allow such other Party to join the Enforcement Action.
The Parties will reasonably cooperate, including if necessary by executing common interest and other applicable agreements, to prosecute such Enforcement Action taking into account both Novavax Products and Licensed Products.
(d)Priority of Rights. A Party’s right to initiate and control an Enforcement Action under this Section
12.6.2 (Patent Enforcement) shall take precedence over a Party’s right as a Prosecuting Party under Section
12.2 (Prosecution and Maintenance of Patents) to defend any validity or enforceability or other challenges against a Novavax Patent in any forum, including in an interference, derivation proceeding, pre- and post-grant opposition proceeding, post-grant patent proceedings (such as inter partes review and post grant review). By way of example, if a Party has the right to control a COVID-19 Enforcement Action in a district court with respect to a Sanofi 1st Right COVID-19 Patent pursuant to Section
12.6.2(a) (Sanofi 1st Right COVID-19 Patents), then that same Party shall concurrently have the right to control the defense against an inter partes review against such Sanofi 1st Right COVID-19 Patent at the Patent Trial and Appeal Board, even if the other Party is the Prosecuting Party for such Sanofi 1st Right COVID-19 Patent.
(e)Novavax Sole Right. Novavax will have the sole right, but not the obligation, to bring an appropriate suit or other action to abate any existing, alleged, or threatened
infringement involving any (i) Sanofi 1st Right COVID-19 Patent against an infringement that is not a COVID-19 Product Competitive Infringement and (ii) Novavax Patent that is not a Sanofi 1st Right COVID-19 Patent or Novavax General Combo Product Patent against any infringement, excluding, in each case, General Combo Product Competitive Infringement.
1.7Misappropriation of Know-How.
1.7.1Notice. If a Party becomes aware that a Third Party has misappropriated any Novavax Know-How or Joint Arising Know-How (each, a “Misappropriation”), then each Party will promptly notify the other Party regarding such Misappropriation which notice shall include a summary of all evidence such Party is aware of with respect thereto (each, a “Misappropriation Notice”).
1.7.2Enforcement Actions.
(a)Novavax COVID-19 Know-How. As between the Parties, Sanofi shall have the first right, but not the obligation, in its own name (or in the name of Novavax to the extent required by Applicable Law), to bring and control any Enforcement Action against any Third Party in connection with any Misappropriation of any Novavax COVID-19 Know-How or Joint Arising Know-How that relates to the Licensed COVID-19 Mono Product or Licensed COVID-19 Component, as it reasonably determines appropriate, using counsel of its own choice. If Sanofi does not initiate an Enforcement Action to abate any such Misappropriation within [***] after a Party delivers a Misappropriation Notice to the other Party, and Sanofi in its reasonable business judgment does not believe that such Enforcement Action would be detrimental to the commercial success of a Licensed COVID-19 Products as a whole, then Sanofi will promptly notify Novavax, and Novavax may institute such Enforcement Action to abate such Misappropriation in its own name using counsel of its choice.
(b)Adjuvant Know-How. With respect to any Misappropriation of (i) any Novavax Adjuvant Know-How that is not otherwise Novavax COVID-19 Know-How or (ii) any Joint Arising Know-How that relates to the Adjuvant, Novavax shall have the sole right, but not the obligation, in its own name (or to the extent required by Applicable Law or with Sanofi’s written consent, in the name of Sanofi), to bring and control action against any Third Party in connection with such Misappropriation.
(c)Joint Arising Know-How. Except as set forth in Section 12.7
(a) (Novavax COVID- 19 Know-How) or Section 12.7
(b) (Adjuvant Know-How), for any other Joint Arising Know-How, unless the Parties agree otherwise, Sanofi shall have the first right to bring an action against such Misappropriation. If Sanofi does not initiate an Enforcement Action to abate any such Misappropriation within [***] after a Party delivers a Misappropriation Notice to the other Party, and Sanofi in its reasonable business judgment does not believe that such Enforcement Action would be detrimental to the commercial success of a Licensed Products, then Sanofi will promptly notify Novavax, and Novavax may institute such Enforcement Action to abate such Misappropriation in its own name using counsel of its choice.
(d)Novavax Sole Right. Except as set forth in Section 12.7
(a) (Novavax COVID-19 Know-How), Section 12.7
(b) (Adjuvant Know-How), or Section
12.7.2(c) (Joint Arising Know-How), Novavax will have the sole right, but not the obligation, to bring an appropriate suit or other action to abate any existing, alleged, or threatened Misappropriation involving any Novavax Know-How.
1.8Defense of Product Infringement Claims.
1.8.1Notice. If a Party receives any demand or claim (not in connection with an Enforcement Action, i.e., not a counterclaim or crossclaim) alleging that the Exploitation of any Licensed COVID-19 Product in the Territory infringes, misappropriates, or otherwise violates any Patents, Know-How, or other property rights of any Third Party (each, a “Product Infringement Claim”), then such Party will promptly notify the other Party and provide the other Party with a copy of such demand or claim, correspondence related thereto and summary of all evidence and other information that it is aware of relating to such demand or claim.
1.8.2Right to Defend.
(a)Licensed Products. Sanofi shall have the sole right, but not the obligation, using counsel of its choice, to develop the strategy for and otherwise institute a defense and settle any Product Infringement Claim with respect to any Licensed Product; provided that if such Product Infringement Claim escalates into a legal action and
(i) Novavax is a named co-defendant, (ii) Novavax joins as a co-defendant, or (iii) Novavax joins as a third party claimant, then Novavax shall reasonably cooperate with Sanofi in its defense using counsel that is reasonably acceptable to Sanofi, and Sanofi shall have the right to control the overall case strategy and lead both Parties’ defenses against such Product Infringement Claim; provided further, that Novavax shall not have the right to (A) join such legal action as a co-defendant or third-party claimant or (B) admit fault or infringement or settle any such dispute on its own, in each case (A) and (B), without Sanofi’s request or prior written consent. Novavax shall reasonably cooperate with Sanofi in the defense of all Product Infringement Claims, including by joining any action when requested by Sanofi and by making available to Sanofi all relevant Novavax personnel and records that Sanofi may request to serve as witnesses and otherwise satisfy its discovery burden.
(b)Novavax Products. Novavax shall have the sole right, but not the obligation, using counsel of its choice, to develop the strategy for and otherwise institute a defense and settle any Product Infringement Claim with respect to any (i) Novavax Product and (ii) on a country-by-country basis, any Licensed COVID-19 Mono Product Exploited, as between the Parties, solely by Novavax and not by Sanofi in a country.
(c)Further Cooperation. If a Third Party asserts a Product Infringement Claim against both Sanofi and Novavax alleging infringement by a Licensed COVID-19 Component common to Licensed COVID-19 Products Exploited by Sanofi and Novavax Products Exploited by Novavax, then the Parties shall confer in good faith to coordinate the defense and settlement of such claim and the allocation of expenses and costs or recoveries therefrom, in a manner that aligns with the rights
of each Party set forth in Sections
12.8.2(a) (Licensed Products) through
12.8.2(b) (Novavax Products).
1.9Information Sharing. The controlling Party in any action brought pursuant to Section
12.6 (Patent Enforcement), Section
12.7 (Misappropriation of Know-How), or Section
12.8 (Defense of Licensed Product Infringement Claims): (a) will discuss with the non-controlling Party the strategy for any such action and shall consider in good faith the non-controlling Party’s comments regarding such strategy; and (b) will keep the non-controlling Party reasonably informed of the status and progress of such action including by providing copies of any pleadings filed relating to such actions promptly upon their being filed or received and will reasonably consult with the non-controlling Party, and will consider in good faith the non-controlling Party’s comments with respect thereto; provided however, that the controlling Party shall have the final decision with regards to the institution, operation, status and progress of any such action, including whether to institute, settle, or dismiss any such actions, subject to Section
12.11 (Settlement). Notwithstanding any provision to the contrary set forth herein, Novavax shall notify and provide Sanofi with copies of any allegations of alleged patent invalidity, unenforceability, or non-infringement of any Novavax Patents, as soon as practicable following receipt thereof.
1.10Costs and Recoveries.
1.10.1Costs. For any action brought by or against a Party pursuant to Section
12.6 (Patent Enforcement), Section
12.7 (Misappropriation of Know-How), or Section
12.8 (Defense of Licensed Product Infringement Claims), the controlling Party will be responsible for all of its own costs and expenses incurred in relation to such action, including attorneys’ fees. The non-controlling Party will be responsible for all of its own costs if it participates in any such action, including attorneys’ fees.
1.10.2General Allocation of Recoveries. Any recoveries or amounts received as a result of any action brought by a Party pursuant to Section
12.6 (Patent Enforcement), Section
12.7 (Misappropriation of Know-How), or Section
12.8 (Defense Licensed Product Infringement Claims) shall be allocated as follows: (a) first, to reimburse the controlling Party’s costs and expenses incurred in connection with such action, including reasonable attorneys’ fees, (b) second, to reimburse the non-controlling Party’s costs and expenses incurred in connection with such action, if any, including reasonable attorneys’ fees, and
(c) third, all remaining recoveries or amounts to the controlling Party, provided that, (i) to the extent that any award or settlement amount (whether by judgment or otherwise) is attributable to loss of sales or profits with respect to a Licensed Product and retained by Sanofi, such amounts shall be included as Net Sales hereunder with respect to such Licensed Product and (ii) in the event both Parties are parties to an Enforcement Action in accordance with Section
12.6.2(c) (CIC and OC Product Enforcement Actions), Novavax will retain all award or settlement amount attributable to a Novavax Product.
1.11Settlement. A Party controlling any action or proceeding brought by or against a Party pursuant to Section
12.6 (Patent Enforcement), Section
12.7 (Misappropriation of Know-How), or Section
12.8 (Defense of Product Infringement Claims) may settle such action or proceeding in its sole discretion; provided that if such settlement is reasonably likely to materially adversely affect the other Party’s interests in the Licensed Novavax Assets in the Territory, including by way of admission of fault, infringement, misappropriation, invalidity, inequitable conduct or unenforceability that impacts the other Party, then such settlement may not be made without the prior written consent of the other Party, such consent not to be unreasonably withheld, conditioned or delayed.
1.12Common Interest Disclosures. With regard to any privileged or Confidential Information or opinions disclosed pursuant to this Agreement by a Party to the other Party regarding Patents, Know-How, Materials, or other property owned by the Disclosing Party or a Third Party, the Parties agree that they have a common legal interest in determining whether, and to what extent, such Patents, Know-How, Materials, or other property may affect any Licensed Product, and a further common legal interest in defending against any actual or prospective Third-Party Claims based on allegations of misuse or infringement of Patents, Know-How, Materials, or other property relating to any Licensed Product. Accordingly, the Parties agree that all such information and materials obtained by Sanofi and Novavax from each other in which they have such a common legal interest
(a) will be treated as protected by the attorney-client privilege, the work product privilege, and any other privilege or immunity that may otherwise be applicable; (b) by sharing such information, neither Party intends to waive or limit any privilege or immunity that may apply to the shared information and materials; and (c) neither Party shall have the authority to waive any privilege or immunity on behalf of the other Party without such other Party’s written consent, nor shall the waiver of privilege or immunity resulting from the conduct of one Party be deemed to apply against the other Party. The Parties may enter into a separate common interest agreement agreed by the Parties with respect to such information and materials, upon the request of either Party.
ARTICLE 13: TRADEMARK MATTERS
1.1Product Marks. As between the Parties and subject to Section
7.11 (Housemarks), each Party has the sole right, but not the obligation, to develop (and thereafter modify and update) and register (in its own name or the name of its Affiliates or Sublicensees or their designees) trademarks it determines appropriate in its sole discretion for the Licensed COVID-19 Mono Products for which it is the Party responsible for Commercialization; provided that on a Licensed COVID-19 Mono Product-by-Licensed COVID-19 Mono Product and country-by-country basis, if both Parties are Commercializing the same Licensed COVID-19 Mono Product in a country, then Sanofi shall have the sole right to develop and register trademarks for such Licensed COVID-19 Mono Product. As between the Parties, Sanofi has the sole right, but not the obligation, to develop (and thereafter modify and update), and register (in its own name or the name of its Affiliates or Sublicensees or their designees) trademarks it determines appropriate in its sole discretion for any Licensed CIC Product, Licensed OC Product, and Licensed Adjuvanted Product that it is Commercializing in the Territory. As between the Parties, Novavax has the sole right, but not the obligation, to develop (and thereafter modify and update) and register (in its own name or the name of its Affiliates or Sublicensees or their designees) trademarks it determines appropriate in its sole discretion for any Novavax Product.
1.2Review of Licensed COVID-19 Mono Product Marks. Each Party will provide final versions (and any material updates thereto) of the proposed trademarks for each Licensed COVID-19 Mono Product to the JCC for the JCC to review and discuss, provided that the Party responsible for Commercialization in each jurisdiction will have the sole right to choose the trademark for the Licensed COVID-19 Mono Product. Each Party will provide an update to the other Party through the JSC on an annual basis regarding the filing and prosecution of the trademarks used or intended for use with the Licensed COVID-19 Product.
1.3Domain Names; Websites. As between the Parties, each Party has the sole right, but not the obligation, to select, register (in its own name or the name of its Affiliates or Sublicensees or their designees), and maintain all domain names and websites used in connection with the Licensed COVID-19 Mono Products for which it is the Party responsible for Commercialization in accordance with the Licensed COVID-19 Mono Product Commercialization Framework; provided
that on a Licensed COVID-19 Mono Product-by-Licensed COVID-19 Mono Product and country- by-country basis, if both Parties are Commercializing the same Licensed COVID-19 Mono Product in a country, then Sanofi shall have the sole right to select and register domain names for such Licensed COVID-19 Mono Product. As between the Parties, Sanofi has the sole right, but not the obligation, to select, register (in its own name or the name of its Affiliates or Sublicensees or their designees) and maintain all domain names and websites used in connection with any Licensed CIC Product, Licensed OC Product, and Licensed Adjuvanted Product that it is Commercializing in the Territory. As between the Parties, Novavax has the sole right, but not the obligation, to select, register (in its own name or the name of its Affiliates or Sublicensees or their designees) and maintain all domain names and websites used in connection with any Novavax Product that it is Commercializing in the Territory. To effectuate the foregoing, promptly after Sanofi is responsible for Commercialization in accordance with the Licensed COVID-19 Mono Product Commercialization Framework, Novavax will transfer to Sanofi or its designee those domain names and websites that should be under Sanofi’s control in accordance with the foregoing, but are Controlled by Novavax as of the Effective Date.
1.4License to Sanofi. Subject to the terms and conditions of this Agreement, Novavax hereby grants to Sanofi a non-exclusive, sublicensable (through multiple tiers), non-transferable (except in accordance with Section
16.1 (Term; Expiration)) license to use Novavax Trademarks to Commercialize Licensed Products. Sanofi agrees that all trademark goodwill that accrues based on all uses of the Novavax Trademarks, by operation of law or otherwise, shall inure to the sole and exclusive benefit of Novavax.
1.5Trademark and Domain Name Infringement; Enforcement. As between the Parties, Novavax has the sole right, but not the obligation, to enforce all Novavax Trademarks. Each Party has the right, in any infringement action arising from the use of the Novavax Trademarks brought against such Party, or its Affiliates or Sublicensees, to defend by itself, at its own expense, and the other Party shall provide reasonable assistance if requested by the defending Party.
1.6Costs and Expenses. Novavax shall be solely responsible for costs associated with obtaining and maintaining the Novavax Trademarks. Novavax shall be solely responsible for costs incurred by or on its behalf in any action to enforce or defend its use of the Novavax Trademarks and shall have the right to retain recoveries associated therewith. Sanofi shall be solely responsible for costs incurred by or on its behalf in any action to defend its use of the Novavax Trademarks and shall have the right to retain recoveries associated therewith.
1.7Falsified Medicines. Each Party will promptly notify the other Party in writing if it becomes aware of any Third Party that is Manufacturing or Commercializing a medical product purporting to be a Licensed Product that deliberately or fraudulently misrepresents its identity, composition, or source (each, a “Falsified Medicine”). As between the Parties, Sanofi will have the first right, but not the obligation, to lead any detection program, investigation, or collaboration with any Governmental Authority and the first right, but not the obligation, to take action to enforce its rights against any Third Party that is Manufacturing or Commercializing such Falsified Medicines or contributing to any of these actions. If Sanofi declines to enforce its rights against such Third Party, then it will promptly notify Novavax, and Novavax will have the right, but not the obligation, to take such enforcement action. If requested by Sanofi, Novavax will reasonably cooperate with Sanofi with respect to any suspected Falsified Medicines to provide complementary information related to the applicable Licensed Product when necessary or requested by any Governmental Authority.
ARTICLE 14: REPRESENTATIONS AND WARRANTIES
1.1Representations and Warranties of Each Party. Each Party hereby represents and warrants to the other Party, as of the Effective Date, that:
1.1.1Corporate Existence and Power. Such Party is duly organized, validly existing, and in good standing under the Applicable Law of the jurisdiction of its formation and has full corporate power and authority and the legal right to own and operate its property and assets and to carry on its business as it is now being conducted and as contemplated in this Agreement, including the full right to grant the licenses and sublicenses granted by it hereunder.
1.1.2Authority. Such Party has the requisite organizational power and authority to enter into this Agreement and to consummate the transactions contemplated hereby. The execution and delivery of this Agreement by such Party and the consummation of the transactions contemplated hereby have been duly and validly authorized and no other organizational proceedings on the part of such Party or any of its Affiliates or any Parent Company are required therefor. Each Party has full shareholder and corporate power and authority to execute and deliver this Agreement and to perform its obligations hereunder.
1.1.3Binding Agreement. This Agreement has been duly executed and delivered on behalf of such Party and constitutes a legal, valid, and binding obligation, enforceable against it in accordance with its terms, except to the extent that enforcement of the rights and remedies created hereby is subject to: (a) bankruptcy, insolvency, reorganization, moratorium, or other similar laws of general application affecting the rights and remedies of creditors; or
(b) laws governing specific performance, injunctive relief, and other equitable remedies.
1.1.4No Conflicts. The execution, delivery, and performance of this Agreement by such Party does not breach, violate, or conflict with any agreement or any provision thereof (including any confidentiality or non-competition obligation, any exclusivity obligation, or any provisions with respect to the ownership, prosecution, maintenance, and enforcement of intellectual property rights), or any instrument or understanding, oral or written, to which such Party (or any of its Affiliates) is a party or by which such Party (or any of its Affiliates) is bound, nor violate any Applicable Law of any Governmental Authority having jurisdiction over such Party (or any of its Affiliates).
1.1.5Government Authorization. No other government authorization, consent, approval, license, exemption of, or filing or registration with, any court or governmental department, commission, board, bureau, agency, or instrumentality, under any Applicable Law currently in effect, is or will be necessary for, or in connection with, the transactions contemplated by this Agreement, or for the performance by it of its obligations under this Agreement, except as may be required to Exploit Licensed Products.
1.1.6Third Party Consent. It has obtained all necessary authorizations, consents, and approvals of any Third Party that are required to be obtained by it for, or in connection with, the execution and delivery of this Agreement and for the performance of its obligations hereunder (as contemplated as of the Effective Date), except as may be required to Exploit Licensed Products.
1.1.7No Debarment. (a) Neither it nor any of its Affiliates in the pharmaceutical industry, nor any of their officers, employees or agents, has been debarred or is subject to debarment pursuant to Section 306 of the FD&C Act or analogous provisions of Applicable Law
outside the United States or is listed on any excluded list; and (b) neither it nor any of its Affiliates has, to its knowledge, used in any capacity, in connection with the activities to be performed under this Agreement, any individual or entity that has been debarred pursuant to Section 306 of the FD&C Act or analogous provisions of Applicable Law outside the United States, or that is the subject of a conviction described in such Section or analogous provisions of Applicable Law outside the United States, or listed on any excluded list.
1.2Representations and Warranties of Novavax. Novavax hereby represents to Sanofi, as of the Effective Date that:
1.2.1Novavax Patents. All Novavax Patents existing as of the Effective Date are listed on Schedule
14.2.1.
1.2.2Ownership. Except as set forth on Schedule
14.2.2 (Ownership), Novavax solely and exclusively owns or otherwise exclusively Controls each Existing Novavax Patent, all Novavax Know-How, and Novavax Materials. As of the Effective Date, all exclusively owned or otherwise exclusively Controlled Existing Novavax Patents, Novavax Know- How, and Novavax Materials and, to Novavax’s Knowledge, all in-licensed Novavax Patents, Novavax Know-How, and Novavax Materials are free and clear of all financial liens, encumbrances, and security interests. Neither Novavax nor any of its Affiliates owns or holds rights to any Patents, Know-How, or Materials that would otherwise qualify as a Licensed Novavax Asset but for the fact that Novavax or such Affiliate owns or holds rights to, but does not Control, such Patent, Know-How or Materials.
1.2.3Validity and Enforceability. Novavax has complied in all material respects with Applicable Law with respect to the Prosecution and Maintenance of the Existing Novavax Patents. Neither Novavax nor its Affiliates have received any written notice of any claim or threat of a claim or litigation by any Third Party against Novavax or its Affiliates that alleges that any Existing Novavax Patent is invalid or unenforceable, and there is no pending adverse action, suit, or Proceeding against Novavax in relation to any Existing Novavax Patent. All Novavax Patents are: (a) subsisting, and, to the extent issued, to Novavax’s Knowledge, not invalid or unenforceable, in whole or in part; (b) in the case of pending applications, being diligently prosecuted in the respective patent offices in accordance with Applicable Law, and Novavax has presented all relevant references, documents and information of which it and the inventors are aware to the relevant patent examiner at the relevant patent office; and (c) being prosecuted and maintained in accordance with the policies and procedures of the applicable patent office, including that (i) every inventor of the claims thereof has been identified in accordance with the Applicable Laws of the relevant jurisdiction in which such Patent is issued or pending and (ii) all applicable fees required by such patent office have been paid by the due date for payment. To Novavax’s Knowledge, there is no reference or prior art that would preclude the issuance of any claim pending as of such date within any Novavax Patent.
1.2.4Litigation and Disputes. There are no claims, judgments, settlements, litigations, suits, actions, disputes, arbitration, or judicial, legal, administrative, or other Proceedings, or governmental investigations pending or, to Novavax’s Knowledge, threatened, against Novavax or its Affiliates that would reasonably be expected to adversely affect or restrict the ability of Novavax to consummate or perform the transactions and obligations contemplated under this Agreement, or that would adversely affect the Licensed Novavax
Assets, Novavax’s Control thereof, or the Licensed COVID-19 Mono Product, Licensed COVID-19 Component, and Adjuvant existing as of the Effective Date.
1.2.5No Misappropriation or Infringement. To Novavax’s Knowledge, Novavax has not misappropriated, infringed, or otherwise violated any intellectual property right of a Third Party in connection with developing and Exploiting the Adjuvant or the Licensed COVID- 19 Mono Product. Neither Novavax nor its Affiliates have received any written notice of any Third Party Claim that any intellectual property right controlled by a Third Party would be infringed, misappropriated, or otherwise violated by the performance of the activities contemplated hereunder or by the Exploitation of the Licensed COVID-19 Mono Product, Licensed COVID-19 Component, and Adjuvant as contemplated under this Agreement in accordance with this Agreement. To Novavax’s Knowledge, no Third Party is infringing, misappropriating, or otherwise violating the Licensed Novavax Asset existing as of the Effective Date.
1.2.6No Delinquency. To Novavax’s Knowledge, neither Novavax nor any of its Affiliates are in breach of any obligations to any Third Party, or engaged in any dispute with any Third Party, in connection with any Upstream License Agreement or any other agreement with a Third Party, in each case, that would materially limit Sanofi’s ability to Exploit the Licensed COVID-19 Mono Product, Licensed COVID-19 Component, and Adjuvant in the Territory.
1.2.7No Breach. To Novavax’s Knowledge, neither Novavax nor any of its Affiliates are, or have ever received written notice of being, in material breach of any term or condition of any Upstream License Agreement or any other agreement with a Third Party, in each case, that would have a material adverse impact on Sanofi’s rights under this Agreement.
1.2.8Assignments. To Novavax’s Knowledge, valid and enforceable written assignments (by assignment agreement or employment agreement) exist and are recorded from all individuals who are identified as inventors on any Novavax Patent existing as of the Effective Date. To Novavax’s Knowledge, each Person who claims to be an inventor of an invention claimed in an Existing Novavax Patent is identified as an inventor of such invention in the filed patent documents for such Novavax Patent existing as of the Effective Date. No dispute regarding inventorship, authorship, or ownership has been alleged or threatened with respect to any Novavax Patent existing as of the Effective Date.
1.2.9Protection of Know-How and Materials. Novavax and its Affiliates have taken commercially reasonable measures to protect the secrecy and confidentiality of all Novavax Know-How and the confidential and proprietary aspects of the Novavax Materials, in each case, existing as of the Effective Date, including by requiring relevant employees, consultants, and subcontractors to execute binding and enforceable agreements requiring all such employees, consultants, and subcontractors to maintain the confidentiality of all such Novavax Know-How and such confidential and proprietary aspects of such Novavax Materials. To Novavax’s Knowledge: (a) the Novavax Know- How and the confidential aspects of Novavax Materials have not been disclosed to, or used or discovered by, any Third Party except pursuant to such confidentiality agreements; and
(b) there has not been a breach by any party of such confidentiality agreements.
1.2.10Government Funding and Rights. To Novavax’s Knowledge, the inventions claimed in the Novavax Patents existing as of the Effective Date: (a) were not conceived, discovered, developed, or otherwise made as a result of any research activities funded, in whole or in
part, by the federal government of the United States (or any agency thereof) or the government of any other country; (b) are not a “subject invention” as that term is described in 35 U.S.C. §201(e); (c) are not otherwise subject to the provisions of the Patent and Trademark Law Amendments Act of 1980, codified at 35 U.S.C. §§200-212, or any regulations promulgated pursuant thereto, including in 37 C.F.R. Part 401; (d) in the case of clauses (b) or (c), are not subject to similar obligations or restrictions under the Applicable Law of any other country; and (e) are not the subject of any licenses, options or other rights of any Governmental Authority, within or outside the United States.
1.2.11Compliance with Applicable Law. Novavax and its Affiliates have conducted, and their respective Representatives have conducted, prior to the Effective Date, all Research, Development, and Manufacturing of the Licensed COVID-19 Mono Product, Licensed COVID-19 Component, and Adjuvant in accordance with all Applicable Laws in all material respects.
1.2.12Diligence Information. The information that Novavax has made available to Sanofi prior to the Effective Date in the electronic data room for Sanofi’s review prior to entering into this Agreement does not, to Novavax’s Knowledge, contain any untrue statement(s) of fact that Novavax would reasonably expect to be material to Sanofi’s decision to enter into this Agreement. To Novavax’s Knowledge, neither Novavax nor any of its Affiliates have knowingly withheld from Sanofi any correspondence received from any Regulatory Authority with respect to the Licensed COVID-19 Mono Product, Licensed COVID-19 Component, and Adjuvant that Novavax would reasonably expect to be material to Sanofi’s decision to enter into this Agreement.
1.2.13Upstream License Agreements. Novavax has provided Sanofi with a true, complete, and correct copy of each Upstream License Agreement. Each Upstream License Agreement is valid, binding, enforceable, and in full force and effect; no notice of breach, default, or termination has been received or given under any Upstream License Agreement, and, to Novavax’s Knowledge, there is no act or omission by Novavax or any of its Affiliates that would provide a right to terminate any such agreement; and to Novavax’s Knowledge, no circumstances or grounds exist that would reasonably be expected to give rise to a claim of material breach or a right of rescission, termination, revision, or amendment of any of the Upstream License Agreements, including the execution, delivery, and performance of this Agreement. Neither Novavax nor any of its Affiliates has waived any of their respective rights under any Upstream License Agreement in any manner that conflicts with or limits the scope of any of the rights or licenses granted to Sanofi hereunder, and, to Novavax’s Knowledge, no such rights have lapsed or otherwise expired or been terminated. Novavax has obtained all consents and provided all notices required to be provided under each Upstream License Agreement in connection with the execution and delivery of this Agreement and the performance by the Parties of their respective obligations hereunder, and the execution, delivery, and performance of this Agreement by the Parties does not and will not constitute a breach or default under any of the Upstream License Agreements.
1.2.14Novavax Trademarks and Domain Names.
(a)Existence. All Novavax Trademarks and Novavax Domain Names existing as of the Effective Date are listed on Schedule
14.2.14(a) (Existence).
(b)Ownership. Except as set forth on Schedule
14.2.14(b) (Ownership), Novavax solely and exclusively owns or otherwise exclusively Controls each Novavax
Trademarks and Novavax Domain Names. As of the Effective Date, all exclusively owned or otherwise exclusively Controlled Novavax Trademarks and Novavax Domain Names are free and clear of all financial liens, encumbrances, and security interests.
(c)Trademark Validity and Enforceability. Novavax has complied in all material respects with Applicable Law with respect to the Prosecution and Maintenance of the Novavax Trademarks. Neither Novavax nor its Affiliates have received any written notice of any claim or written threat of a claim or litigation by any Third Party against Novavax or its Affiliates that alleges that any Novavax Trademark is invalid or unenforceable, and there is no pending adverse action, suit, or Proceeding against Novavax in relation to any Novavax Trademark. All Novavax Trademark are, to Novavax’s Knowledge, subsisting, and, to the extent issued, not invalid or unenforceable in whole or in part. To Novavax’s Knowledge, there is no reference or prior art that would preclude the issuance of any claim pending as of such date within any Novavax Trademark.
(d)Litigation and Disputes. There are no claims, judgments, settlements, litigations, suits, actions, disputes, arbitration, or judicial, legal, administrative, or other Proceedings, or governmental investigations pending or, to Novavax’s Knowledge, threatened, against Novavax or its Affiliates that would reasonably be expected to adversely affect the Novavax Trademarks existing as of the Effective Date.
1.2.15Novavax Knowledge. With respect to representations of Novavax hereunder that are qualified by Novavax’s Knowledge, the Chief Executive Officer, Chief Scientific Officer, President of Research and Development, Executive Vice President Chief Operating Officer, Senior Vice President of Global Manufacturing, and Senior Director of Legal Affairs Intellectual Property & Transactions of Novavax have reasonably consulted with his or her direct reports as to such matters, and, with respect to Patent matters, have consulted with internal patent counsel(s) who attend to the Novavax Patent matters in the ordinary course.
1.3Covenants of Each Party. Each Party hereby covenants to the other Party that during the Term:
1.3.1Compliance with Applicable Law. Such Party and its Affiliates will perform its and their activities and obligations pursuant to this Agreement in compliance with all Applicable Law in all material respects, including, to the extent applicable, the United States Foreign Corrupt Practices Act, GCP, GLP, and GMP, and with applicable industry ethical codes. Further, each Party hereby covenants to the other Party that during the Term its and its Affiliates’ employees will not, and it will use reasonable efforts to cause its contractors to not, in connection with the performance of its obligations under this Agreement, directly or indirectly through Third Parties, pay, promise, or offer to pay, or authorize the payment of any money, or give, promise or offer to give, or authorize the giving of, anything of value to a government official, government employee, political party official, candidate for political office, or any other Person for the purpose of improperly obtaining or retaining business for or with, or directing business to, any Person, including either Party (it being understood that such Party and its Affiliates, and to its knowledge, its and its Affiliates’ Representatives, have not directly or indirectly promised, offered, or provided any bribe or any other corrupt payment, gratuity, emolument, kickback, gift or hospitality, or other
illegal or unethical benefit to a government official, government employee, political party official, candidate for political office or any other Person in connection with the performance of such Party’s obligations under this Agreement, and will not, directly or indirectly, engage in any of the foregoing).
1.3.2No Debarment. Such Party and its Affiliates will not, with respect to the performance of activities hereunder, knowingly employ, or otherwise use in any capacity, the services of any Person suspended, proposed for debarment or debarred under United States law, including under 21 U.S.C. § 335a, or any foreign equivalent thereof.
1.3.3No Conflicts. Such Party will not enter into any agreement, contract, commitment, or other arrangement, or otherwise take any action or fail to take any action, that would reasonably be expected to conflict with the rights granted to the other Party hereunder or otherwise prevent the other Party from exercising the rights granted to it hereunder.
1.3.4No Misappropriation. Such Party will not knowingly misappropriate any trade secret of a Third Party in connection with the performance of its activities hereunder.
1.3.5Government Authorization. Such Party will obtain and maintain all permits, licenses, registrations, and other forms of authorizations and approvals from any Governmental Authority that are necessary or required to be obtained or maintained by such Party in order for such Party to perform its obligations hereunder in a manner that complies with all Applicable Law in all material respects.
1.3.6Data Safeguards. Each Party will comply with the terms, conditions, and obligations set forth in Exhibit B (Data Safeguards).
1.4Covenants of Novavax. Novavax hereby covenants to Sanofi that during the Term:
1.4.1Upstream License Agreements. Novavax will not, without Sanofi’s prior written consent, which will be granted or withheld at Sanofi’s sole discretion, (a) amend, modify, or extend the Upstream License Agreements in any manner that would negatively affect Sanofi’s rights or obligations hereunder or (b) fail to comply with its obligations under the Upstream License Agreements in any manner that would negatively affect Sanofi’s rights or obligations hereunder.
1.4.2APAs, Existing Strategic Partner Agreements and Settlement Arrangements. Novavax will not, without Sanofi’s prior written consent, which will be granted or withheld at Sanofi’s sole discretion, amend, modify, extend, or otherwise make any change to any APA, Existing Strategic Partner Agreement, or any Settlement Arrangement, in each case, (i) to increase the scope of exclusivity, or (ii) in any other manner that would materially negatively affect Sanofi’s rights or obligations hereunder, including by increasing the quantity of products ordered or delivered, or increasing the price of products. Further, Novavax will not, in connection with any APA, Existing Strategic Partner Agreement, or Settlement Arrangement, order pre-filled syringes or unidose or single-dose vials of any Licensed COVID-19 Mono Product from a supplier that also supplies Licensed COVID- 19 Mono Products to Sanofi in any manner that would negatively affect Sanofi’s rights or obligations hereunder, without Sanofi’s prior written consent, such consent not to be unreasonably withheld.
1.4.3Existing Supply Agreements. Novavax will not, without Sanofi’s prior written consent, which will be granted or withheld at Sanofi’s sole discretion, amend, modify, extend, make any change to, or fail to comply with, any Existing Supply Agreements, in each case, (i) to increase the scope of exclusivity (but Novavax may extend the duration of any exclusivity in its sole discretion), or (ii) in any other manner that would materially negatively affect Sanofi’s rights or obligations hereunder. [***].
1.4.4New Arrangements. Except in connection with sales or tender offers for purposes of satisfying Novavax’s obligations under any Existing Settlement Arrangements, from and after the Effective Date, Novavax will not enter into any agreement or arrangement to Manufacture or Commercialize Licensed COVID-19 Mono Products, including any (i) APA without Sanofi’s prior written consent, which consent will be granted or withheld at Sanofi’s sole discretion or (ii) Settlement Arrangement, unless approved by the JCC pursuant to Section
7.3 (Existing APAs and Settlement Arrangements); except that Novavax may enter new agreements to Manufacture Licensed COVID-19 Mono Product that do not negatively affect Sanofi’s rights or obligations hereunder, and any such new agreement will be deemed an Existing Supply Agreement. [***].
1.4.5Assignment or Disposal of Licensed Novavax Assets. Except as permitted under Section
17.1 (Assignment), Novavax will not sell, assign, or otherwise dispose of any Licensed Novavax Assets to any Third Party unless such Third Party agrees in writing to be bound by all relevant terms of this Agreement, including Novavax’s covenants in respect of the Licensed Novavax Assets under
Article 2 (License Grants; Exclusivity),
Article 12 (Patent and Know-How Matters), and this Section
14.4 (Covenants of Novavax).
1.4.6Assignment Obligations. (a) All employees of each Party performing activities on behalf of such Party in connection with this Agreement are subject to a present obligation to assign to such Party all rights, title, and interests in and to any inventions developed by them in the conduct of such activities, whether or not patentable; and (b) all Service Providers of either Party performing activities on behalf of such Party in connection with this Agreement are subject to subcontracts that provide that such Party will obtain ownership or a sublicensable license of any Know-How and Patents that are discovered, developed, invented, or created by such subcontractor in their performance of such activities to the extent such Know-How and Patents would otherwise fall within the definition of Licensed Novavax Assets.
1.5Warranty Disclaimer. EXCEPT AS OTHERWISE EXPRESSLY PROVIDED IN THIS AGREEMENT, NEITHER PARTY MAKES ANY REPRESENTATION OR EXTENDS ANY WARRANTY OF ANY KIND, EITHER EXPRESS OR IMPLIED, TO THE OTHER PARTY WITH RESPECT TO ANY PATENTS, KNOW-HOW, MATERIALS, COMPOUND, PRODUCT, GOODS, SERVICES, RIGHTS, OR OTHER SUBJECT MATTER OF THIS AGREEMENT AND HEREBY DISCLAIMS ALL IMPLIED WARRANTIES OF
MERCHANTABILITY, FITNESS FOR A PARTICULAR PURPOSE, TITLE, OR NONINFRINGEMENT WITH RESPECT TO ANY AND ALL OF THE FOREGOING. THE PARTIES AGREE THAT THE MILESTONE EVENTS AND NET SALES LEVELS SET FORTH IN THIS AGREEMENT OR THAT HAVE OTHERWISE BEEN DISCUSSED BY THE PARTIES ARE MERELY INTENDED TO DEFINE THE MILESTONE PAYMENTS AND ROYALTY OBLIGATIONS IF SUCH MILESTONE EVENTS OR NET SALES LEVELS ARE ACHIEVED. NOVAVAX HEREBY ACKNOWLEDGES AND AGREES THAT NEITHER SANOFI NOR ITS AFFILIATES, OR ANY OF THEIR RESPECTIVE REPRESENTATIVES OR ANY OTHER PERSON HAS MADE OR IS MAKING, AND NOVANVAX HAS NOT RELIED UPON AND IS NOT RELYING UPON, ANY REPRESENATAIONS OR WARRANTIES, PROJECTION, FORECAST, OR STATEMENT, EITHER EXPRESS OR IMPLIED, THAT IT WILL BE ABLE TO SUCCESSFULLY ADVANCE ANY LICENSED PRODUCT OR DEVELOP, ACHIEVE MARKETING AUTHORIZATION FOR, MANUFACTURE, COMMERCIALIZE, OR OTHERWISE EXPLOIT ANY LICENSED PRODUCT OR, IF COMMERCIALIZED, THAT ANY PARTICULAR NET SALES LEVEL OF SUCH LICENSED PRODUCT WILL BE ACHIEVED.
ARTICLE 15: INDEMNIFICATION; INSURANCE; LIMITATIONS
1.1Indemnification.
Indemnification by Sanofi. Sanofi will indemnify, defend, and hold harmless Novavax, its Affiliates, and its and their respective employees, officers, directors, and agents and their respective successors, heirs, and assigns (each, a “Novavax Indemnified Party”) from and against any liability, loss, damage, or expense (including reasonable attorneys’ fees and expenses) (collectively, “Liability”) arising out of any Third Party suit, proceeding, investigation, claim, or demand (“Third Party Claim”) arising from or relating to:
(a)[***];
(b)[***];
(c)[***];
(d)[***]; or
(e)[***];
[***].
1.1.1Indemnification by Novavax. Novavax will indemnify, defend, and hold harmless Sanofi, its Affiliates, and its and their respective employees, officers, directors, and agents and their respective successors, heirs, and assigns (each, a “Sanofi Indemnified Party”) from and against any Liability arising out of any Third Party Claim arising from or relating to:
(a)[***]
(b)[***];
(c)[***];
(d)[***];
(e)[***];
(f)[***]; or
(g)[***];
[***].
1.1.2Procedure. Each Party will notify the other Party in writing if it becomes aware of a claim for which such Party may seek indemnification hereunder. If any Proceeding (including
any governmental investigation) is instituted against a Party with respect to which indemnity may be sought pursuant to Section
15.1.1 (Indemnification by Sanofi) or
15.1.2 (Indemnification by Novavax), as applicable, such Party (the “Indemnified Party”) will give prompt written notice of the claim to the other Party (the “Indemnifying Party”) and provide the Indemnifying Party with a copy of any complaint, summons, or other written notice that the Indemnified Party receives in connection with any such claim. An Indemnified Party’s failure to deliver such written notice will relieve the Indemnifying Party of liability to the Indemnified Party under Section
15.1.1 (Indemnification by Sanofi) or
15.1.2 (Indemnification by Novavax), as applicable, only to the extent such delay is prejudicial to the Indemnifying Party’s ability to defend such claim and allow the Indemnifying Party to assume the defense of the claim. Within thirty (30) days after delivery of such notice, the Indemnifying Party may, upon written notice thereof to the Indemnified Party, elect to assume control of the defense of such claim with counsel reasonably satisfactory to the Indemnified Party. If a Party believes that a claim presented to it for indemnification is one as to which the Party seeking indemnification is not entitled to indemnification under Section
15.1.1 (Indemnification by Sanofi) or
15.1.2 (Indemnification by Novavax), as applicable, then it shall so notify the Party seeking indemnification. Provided that the Indemnifying Party is not contesting the indemnity obligation, the Indemnified Party, upon notice of such election, will permit the Indemnifying Party to control any litigation relating to such claim and the disposition of such claim by negotiated settlement or otherwise (subject to this Section
15.1 (Indemnification)). Following such election, the Indemnified Party may participate in the defense of such claim at its own expense; provided that if the Indemnified Party reasonably concludes, based on advice from counsel, that the Indemnifying Party and the Indemnified Party have conflicting interests with respect to such claim, then the Indemnifying Party shall be responsible for the reasonable fees and expenses of counsel to the Indemnified Party (but no more than one law firm acting as such) solely in connection therewith. The Indemnifying Party will act reasonably and in good faith with respect to all matters relating to such claim, will keep the Indemnified Party informed of the status and progress of the defense and any settlement discussions concerning such claim, and will not settle such claim or consent to any judgment in respect thereof, or otherwise resolve such claim without the Indemnified Party’s prior written consent, which will not be unreasonably withheld, conditioned, or delayed; provided that such consent will not be required with respect to any settlement or consent to judgment that (a) imposes no obligation or liability other than the payment of monetary awards for which the Indemnifying Party will be fully responsible, (b) does not include an admission of liability on the part of the Indemnified Party, and (c) includes a complete and unconditional release of the applicable Novavax Indemnified Parties or the applicable Sanofi Indemnified Parties, as the case may be, from all liability with respect to such claim. The Indemnified Party will cooperate with the Indemnifying Party in the Indemnifying Party’s defense of any claim for which indemnity is sought under this Agreement, at the Indemnifying Party’s cost and expense. The Indemnified Party shall not agree to any settlement of such claim or consent to any judgment in respect thereof without the prior written consent of the Indemnifying Party, which shall not be unreasonably withheld, conditioned, or delayed.
1.2Insurance. Each Party will, during the Term and for a period of [***] thereafter, at its own cost and expense, obtain and maintain [***] insurance coverage for its activities under this Agreement from insurance carriers licensed to do business under the laws of the country, state, commonwealth, province, or territory in which such Party’s obligations are provided, with insurers that carry a rating of at least [***]. Each
Party will furnish to the other Party a certificate of insurance evidencing such insurance coverage upon request. Each Party will have the right to self-insure its activities under this Agreement.
1.3Limitation of Damages.
1.3.1[***].
1.3.2[***].
ARTICLE 16: TERM; TERMINATION
1.1Term; Expiration. The term of this Agreement shall commence on the Effective Date, and unless terminated earlier in accordance with Section
16.2 (Termination of this Agreement), will expire when neither Party is Exploiting or is planning to Exploit a Licensed Product or Novavax Product (such period, the “Term”).
1.2Termination of this Agreement.
1.2.1Sanofi’s Termination for Convenience. Sanofi has the right to terminate this Agreement on a Region-by-Region or Sanofi License-by-Sanofi License basis, at will and in its sole discretion on [***] prior written notice to Novavax, except such notice period shall be [***] with respect to termination of the COVID-19 Mono License.
1.2.2Termination for Material Breach.
(a)Material Breach. If either Party is in material breach of this Agreement (the “Breaching Party”), then the other Party (the “Non-Breaching Party”) may deliver notice of such material breach to the Breaching Party referencing this
Section
16.2.2(a) (Material Breach) with sufficient details regarding the specific actions or inactions of the Breaching Party giving rise to the material breach. Subject to Section
16.2.2(b) (Disagreement as to Material Breach), the Breaching Party shall have [***] from such notice to cure such material breach unless such breach is a payment breach, for which the Breaching Party shall have [***] to cure such breach (such applicable period to cure, the “Cure Period”). If the Breaching Party fails to cure such material breach within the Cure Period, then the Non-Breaching Party may terminate this Agreement in its entirety effective on written notice of termination to the Breaching Party, provided that if the material breach solely relates to (a) a Licensed Product, then the Non- Breaching Party may not terminate this Agreement in its entirety and instead shall only have the right to terminate the Sanofi License grant under which such Breaching Party was Exploiting such Licensed Product (and all rights and obligations and Licensed Products under such license will also terminate), and (b) a Region, then the Non-Breaching Party may not terminate this Agreement in its entirety and instead shall only have the right to terminate this Agreement solely with respect to such Region.
(b)Disagreement as to Material Breach. Notwithstanding Section
16.2.2(a) (Material Breach), if the Parties, reasonably and in good faith, disagree as to whether there has been a material breach of this Agreement, then: (i) the Party that disputes whether there has been a material breach may contest the allegation by referring such matter, prior to the expiration of the Cure Period, for resolution in accordance with Section
17.12 (Dispute Resolution); (ii) the relevant Cure Period with respect to such alleged material breach will be tolled in accordance with Section
17.12 (Dispute Resolution) from the date on which the Party that disputes whether there has been a material breach notifies the other Party of such Dispute and through the resolution of such Dispute in accordance with Section
17.12 (Dispute Resolution); and (iii) during the pendency of such Dispute, all of the terms and conditions of this Agreement will remain in effect and the Parties will continue to perform all of their respective obligations hereunder. If it is finally determined in accordance with Section
17.12 (Dispute Resolution) that such Breaching Party is in material breach, then from the date of such decision, the Cure Period will run and the Non- Breaching Party may terminate the Agreement if such breach is not cured in accordance with Section
16.2.2(a) (Material Breach).
(c)Sanofi’s Alternative Remedy for Novavax’s Material Breach. If Sanofi has the right to terminate this Agreement for Novavax’s material breach pursuant to pursuant to Section
16.2.2(a) (Material Breach), then Sanofi may, in lieu of terminating this Agreement elect (in its sole discretion) by written notice to Novavax, any or all of the following remedies solely with respect to the Licensed Products and Regions with respect to which Sanofi has the right to so terminate the Agreement pursuant to Section
16.2.2(a) (Material Breach):
(i)Where practicable, assume Novavax’s rights and obligations to conduct those activities for which Novavax’s failure to perform led to the material breach that gave rise to Sanofi’s termination right (such activities, the “Step-In Activities”). If Sanofi elects to perform the Step-In Activities, then the Parties will coordinate to transfer the Step-In Activities to Sanofi. Any amounts incurred by Sanofi or its Affiliates to assume such Step-In Activities will be at its sole cost and expenses; and
(ii)all royalties or milestone payments that become due to Novavax hereunder after Sanofi’s election of this alternative remedy shall be reduced by [***] of what would otherwise be due to Novavax hereunder (i.e., such that Novavax would receive [***] of what would otherwise be due Novavax).
The remedies set forth in this Section
16.2.2(c)Error! Reference source not found. (Sanofi’s Alternative Remedy for Novavax’s Material Breach) shall be without limitation to any other rights or remedies that may be available to Sanofi under this Agreement or at law or equity, except that if elected, then the remedy set forth in this Section
16.2.2(c)(ii) (Sanofi’s Alternative Remedy for Novavax’s Material Breach) shall be Sanofi’s sole and exclusive monetary remedy for such breach (including that Sanofi may not also exercise its right to receive a discount as a result of a supply delay under paragraph (a) of the Delivery Issues row of Schedule 6.3.1 / Schedule 6.3.2), if Sanofi exercises its rights under this Section
16.2.2(c)(ii) (Sanofi’s Alternative Remedy for Novavax’s Material Breach). In all cases, in the event that Sanofi has the right to terminate this Agreement pursuant to Section
16.2.2(a) (Material Breach), Sanofi will have the right to conduct the Step-In Activities pursuant to Section
16.2.2(c)(i) (Material Breach) whether or not Sanofi has elected the alternative remedy set forth in Section
16.2.2(c)(ii) (Sanofi’s Alternative Remedy for Novavax’s Material Breach) and whether or not Sanofi has exercised its offset right pursuant to Section
10.14.4 (Right to Reconcile Payments).
1.2.3Termination for Patent Challenge. Sanofi or any of its Affiliates files, assists a Third Party in filing or maintaining, or joins a Third Party in filing or maintaining, a Patent Dispute of any Novavax Patent, then Novavax may provide written notice of its intent to terminate; provided that a Patent Dispute under this Section 16.2.3 (Termination for Patent Challenge) shall not include: (a) any reissue, reexamination, post-grant proceeding or any other administrative proceeding filed or requested to be filed by Sanofi or its Affiliates or Sublicensees after prior consultation and agreement with Novavax with respect to any Novavax Patent in a good faith effort to (i) reinforce the patentability, validity or enforceability of such Novavax Patent or (ii) expand the claim scope of such Novavax Patent with respect to Licensed Products; (b) responses to compulsory discovery, subpoenas or other requests for information in a judicial or arbitration proceeding; (c) compliance with any Applicable Law or court order; (d) any Patent Dispute in which Sanofi submits all required documentation and otherwise uses best efforts to terminate or actually causes to be dismissed within [***] of receiving written notice from Novavax;
(e) any challenge to the validity or Valid Claim status of a claim of a Novavax Patent in defense of an action first brought by Novavax or its Affiliates or licensors (or any of their respective licensees) with respect to such Novavax Patent; (f) any Patent Dispute commenced by a Third Party that, after the Effective Date, becomes an Affiliate of Sanofi or its Sublicensee, provided that such Patent Dispute (i) was commenced prior to the closing of the transaction by which the Third Party becomes an Affiliate of Sanofi or its Sublicensee and (ii) Sanofi or such Sublicensee causes such Third Party to file a motion to dismiss such Patent Dispute within [***] after such closing; or (g) with respect to any Patent Dispute brought by a Sublicensee, if Sanofi acts to terminate the sublicense granted to such Sublicensee under the Novavax Patent within [***] of receiving written notice from Novavax. For clarity, a Patent Dispute hereunder shall not be deemed a material breach of Sanofi under this Agreement.
1.2.4Termination for Insolvency. If, at any time during the Term, either Party makes an assignment for the benefit of creditors, appoints, or suffers appointment of a receiver or trustee over all or substantially all of its property, files a petition under any bankruptcy, or insolvency act, or has any such petition filed against it that is not discharged within [***] after the filing thereof, then the other Party may terminate this Agreement in its entirety by providing written notice of its intent to terminate this Agreement to such first Party, in which case, this Agreement will terminate on the date on which such first Party receives such written notice.
1.3Consequences of Termination. Upon the termination of this Agreement in whole or in part by either Party pursuant to Section
16.2 (Termination of this Agreement), the following provisions shall apply on a Terminated Product-by-Terminated Product, and Terminated Territory-by- Terminated Territory basis:
1.3.1Termination of Rights. All rights and obligations of the Parties under this Agreement (including any licenses granted by a Party hereunder, except as necessary for the other Party to perform its surviving obligations hereunder) for the Terminated Products in the Terminated Territory shall cease except as otherwise set forth in this Section
0 (Consequences of Termination) or elsewhere in this Agreement.
1.3.2Inventory Sell-Off Period. Within [***] after the effective date of termination (or if this Agreement is terminated pursuant to Section
16.2.1 (Sanofi’s Termination for Convenience), then within [***] after the date of Sanofi’s termination notice), Sanofi will provide Novavax with a report of its currently on-hand and in-progress inventory with respect to the Terminated Products in the Territory. Sanofi may (with respect to the Terminated Products in the Terminated Territory) for a period of [***] after the date on which a notice of termination is effective (the “Sell-Off Period”) to (i) complete Manufacture with respect to any in-progress inventory of Terminated Products subject to a purchase order dated prior to the effective date of termination, and
(ii) continue to offer for sale and sell such on-hand and in-progress Terminated Products in such Terminated Territory (as applicable) to the extent related to such Terminated Product in Sanofi’s inventory as of such termination (or added to such inventory as a result of the completion described in clause (i)). Sanofi will be responsible for, and comply with, all payment obligations under this Agreement in connection with such inventory sell-off, including the payment of any royalties with respect to Net Sales of such Terminated Products in the Terminated Territory and any milestone payments associated with a milestone event achieved in the course of such inventory sell-off. Sanofi will not sell or otherwise Exploit any Terminated Product in a Terminated Territory except as expressly set forth in this Section
16.3 (Consequences of Termination).
1.3.3Clinical Trials.
(a)Solely for Terminated Products that are Licensed COVID-19 Mono Products, and not any other Licensed Products, if as of the effective date of termination, Sanofi or its Affiliates or Sublicensees are conducting any Clinical Trials of such a Terminated Product, then, upon Novavax’s request and at Novavax’s election, Sanofi and its Affiliates and Sublicensees shall (a) wind down any such ongoing Clinical Trials pursuant to Applicable Law or any requirements from the applicable Regulatory Authorities in compliance with Novavax’s instructions and Applicable Law, or (b) to the extent permitted by Applicable Law and any requirements from the applicable Regulatory Authorities, transfer such Clinical Trials to Novavax at
Novavax’s cost. If Novavax requests that any such Clinical Trials be transferred to Novavax as set forth in clause (b), then Novavax will assume any and all liabilities, including costs, for the conduct of such transferred Clinical Trials for Terminated Products that are Licensed COVID-19 Mono Products that arise after the effective date of such transfer.
(b)Sanofi and its Affiliates and Sublicensees have no obligation to, and will not, transfer to Novavax any Clinical Trials of a terminated Licensed CIC Product, Licensed OC Product, or Licensed Adjuvanted Product for any reason, and Sanofi shall wind down any ongoing Clinical Trials of such Licensed Products in accordance with Applicable Laws and reasonable medical practices.
1.3.4Assignment and Disclosure. Solely for Terminated Products that are Licensed COVID-19 Mono Products, and not any other Licensed Products, promptly following the effective date of termination:
(a)Regulatory Materials. Sanofi (i) hereby assigns and transfers to Novavax or its designee(s) all of Sanofi’s rights, title, and interests in and to all Regulatory Filings, Marketing Authorizations, and Pricing Approvals relating to the Terminated Product with respect to the Terminated Territory, and (ii) will disclose to Novavax or its designee(s) all documents, records, and Materials that are owned or controlled by Sanofi for the Terminated Product(s) in the Terminated Territory.
(b)Service Provider Agreement. If at the time of such termination Sanofi or its Affiliates are party to an agreement with a Service Provider solely related to any Terminated Product in the Terminated Territory, then Sanofi will, and will cause its Affiliates to, upon Novavax’s election, use good faith efforts to assign to Novavax or its designee Sanofi’s or such Affiliate’s rights and obligations under such agreement and that solely relate to such Terminated Product in the Terminated Territory.
(c)Licensed Product Marks and Licensed Product Domain Names. Sanofi hereby assigns and transfers to Novavax or its designee(s), as of the effective date of termination, all of its rights, title, and interests in and to (i) any trademarks (to the extent such Licensed Product Marks do not contain Sanofi’s name, logo or housemark) and domain names, in each case, solely used in connection with the Terminated Product, (ii) any copyrights solely related to or used in the branding of Terminated Product, and (iii) any registrations or applications for registration for the foregoing ((i) – (iii), together, the “Transferred Marks and Domains”). Subject to the terms and conditions of this Agreement, commencing on the effective date of termination and continuing for [***] thereafter, Sanofi hereby grants to Novavax a non-exclusive, sublicensable (through multiple tiers), non-transferable (except in accordance with Section
17.1 (Assignment)) license solely to use Sanofi’s name, logo, or housemark to the extent included in any Licensed Product Mark to Commercialize the applicable Terminated Products.
1.3.5Reversion License. Subject to the terms and conditions of this Agreement, upon Novavax’s reasonable request, Sanofi will grant to Novavax a non-exclusive, worldwide, sublicensable, transferable, royalty-bearing license under the Sanofi Licensed COVID-19 Mono Assets to Exploit the Licensed COVID-19 Mono Products and Licensed COVID-19 Component (the “Reversion License”). The Parties shall negotiate in good faith fair and
reasonable market royalty rate(s) and other market terms and conditions of the Reversion License. If the Parties are unable to reach agreement on the economics terms of the Reversion License within [***] of such license becoming effective, then the Parties shall resolve the dispute through the Baseball Arbitration Procedures set forth in Schedule
9.18.5(d)(ii) (Baseball Arbitration Dispute Resolution).
1.3.6Confidential Information. Following the expiration or termination of this Agreement, if requested by the Disclosing Party, the Receiving Party will use diligent efforts to destroy, all files, records, and other materials containing or comprising the Disclosing Party’s Confidential Information, except to the extent such Confidential Information is [***] to conduct surviving obligations or exercise surviving rights. Notwithstanding the foregoing, (a) the Receiving Party will be permitted to retain access to such files, records, and other materials solely for archival and legal compliance purposes, and (b) the Receiving Party will not be required to destroy or segregate any electronic back- up tapes or other electronic back-up files that have been created solely by the Receiving Party’s automatic or routine archiving and back-up procedures, to the extent created and retained in a manner consistent with its standard archiving and back-up procedures. Each Party’s obligations under Section
11.1 (Confidentiality) will survive for [***] after the effective date of termination or expiration of this Agreement, provided that such obligations will survive for Confidential Information held as a trade secret for so long as such Confidential Information remains trade secret.
1.4Bankruptcy. All rights and licenses granted under or pursuant to this Agreement by a Party to the other are and will otherwise be deemed to be, for purposes of Section 365(n) of the Bankruptcy Code, licenses of rights to “intellectual property” as defined under Section 101 of the Bankruptcy Code. The Parties agree that the Non-Bankrupt Party will retain and may fully exercise all of their rights and elections under the Bankruptcy Code and any foreign equivalent thereto. The Parties further agree that if (a) a bankruptcy proceeding by or against a Party (the “Bankrupt Party”) is commenced under the Bankruptcy Code; (b) this Agreement is rejected as provided in the Bankruptcy Code; and (c) the other Party (the “Non-Bankrupt Party”) elects to retain its rights hereunder as provided in Section 365(n) of the Bankruptcy Code, then, unless the Bankrupt Party continues to perform all of its obligations under this Agreement, the Non-Bankrupt Party will be entitled to a complete duplicate of, and complete access to (as the Non-Bankrupt Party deems appropriate), all such intellectual property and all embodiments thereof. Upon such occurrence, such intellectual property and all embodiments of such intellectual property will be promptly delivered to the Non-Bankrupt Party and the Non-Bankrupt Party will continue to pay all royalties and milestones hereunder to the fullest extent allowed under Section 365(n). The Bankrupt Party (in any capacity, including debtor-in-possession) and its successors and assigns (including any trustee) agrees not to interfere with the exercise by the Non-Bankrupt Party of its rights and licenses to such intellectual property and such embodiments of intellectual property in accordance with this Agreement, and agrees to assist the Non-Bankrupt Party and its Affiliates in obtaining such intellectual property and such embodiments of intellectual property in the possession or control of Third Parties. The foregoing provisions are without prejudice to any rights the Non-Bankrupt Party may have arising under the Bankruptcy Code or other Applicable Law. As used herein, “Bankruptcy Code” means Title 11, United States Code, as from time to time in effect.
1.5Surviving Provisions.
1.5.1Accrued Rights; Remedies. The expiration or termination of this Agreement for any reason will be without prejudice to any rights that will have accrued to the benefit of either Party prior to the date of such expiration or termination, and any and all damages or remedies
(whether at law or in equity) arising from any breach hereunder, each of which will survive expiration or termination of this Agreement, unless otherwise expressly set forth herein. Such expiration or termination will not relieve any Party from obligations that are expressly indicated to survive expiration or termination of this Agreement. Except as otherwise expressly set forth in this Agreement, the termination provisions of this
Article 16 (Term; Termination)are in addition to any other relief and remedies available to either Party under this Agreement, at law, or in equity.
1.5.2Survival. Without limiting the provisions of Section
16.5.1 (Accrued Rights; Remedies), the rights and obligations of the Parties set forth in the following Sections and Articles of this Agreement will survive the expiration or termination of this Agreement, in addition to those other terms and conditions that are expressly stated to survive termination or expiration of this Agreement: Article
1 (Definitions) (to the extent the definitions are used in other surviving provisions), Section
2.1.5 (License to Novavax), Section 2.1.7 (Sanofi Covenants) (solely for so long as Sanofi is continuing to sell Terminated Products during the Sell-Off Period), Section
2.5 (No Adjuvant Improvements), Section
2.7 (No Implied Licenses; Competing Products; Retained Rights), Article
10 (Financial) (solely with respect to any amounts that accrued prior to the effective date of expiration or termination of this Agreement or during the Sell-Off Period), Section
10.16 (Financial Records; Audits) (solely for thirty-six (36) months after the effective date of termination or expiration),
Article 11 (Confidentiality and Non-Disclosure), Section
12.1 (Ownership), Section
14.5 (Disclaimer), Article
15 (Indemnification; Insurance; Limitations), Section
16.3 (Consequences of Expiration or Termination), this Section
16.5 (Surviving Provisions), and Article
17 (Miscellaneous).
ARTICLE 17: MISCELLANEOUS
1.1Assignment.
1.1.1Assignment. This Agreement will not be assignable by either Party without the prior written consent of the non-assigning Party. Notwithstanding the foregoing, either Party may, subject to the terms of this Agreement, assign this Agreement in its entirety, without the written consent of the other Party, to an Affiliate that is a tax resident in the United States only or to a Third Party that acquires all or substantially all of the business or assets of such Party to which this Agreement relates (whether by merger, reorganization, acquisition, sale of equity or assets, or otherwise); provided that in the case of an assignment by Novavax to a Third Party, (a) Novavax only may so assign to a Third Party on or after the date that is [***] after the COVID-19 Manufacturing Handover Date, and (b) Novavax assigns to such Third Party its rights, title, and interests in the Licensed Novavax Assets. Notwithstanding any assignment of this Agreement by a Party to any Affiliate of such Party, such assigning Party shall remain fully liable for the performance of its obligations under this Agreement by such Affiliate. Novavax shall not, without the prior written consent of Sanofi (not to be unreasonably withheld), assign to any Third Party any of Novavax’s rights to receive payment from Sanofi under this Agreement, including the rights to receive royalty payments and milestones hereunder, except on or after the date that is [***] after the COVID-19 Manufacturing Handover Date to either (i) a single Third Party of Novavax’s entire interest in such royalty payments and milestones, and such Person may not further assign, in whole or in part, such royalty payments and milestones to any other Person or (ii) to more than one Third Party of Novavax’s entire interest in such royalty payments and milestones in a single transaction,
provided that Novavax does not grant, and each Third Party expressly waives all, rights to bring any claim or cause of action directly against Sanofi, and such Persons may not further assign, in whole or in part, such royalty payments and milestones to any other Persons. Upon the effectiveness of Novavax’s assignment of royalty payments and milestones to any Third Party, Sanofi may disband the JSC and all Subcommittees, and as from the date on which such Committees are disbanded, Sanofi will have all decision-making authority allocated to the Committees. Notwithstanding Section
10.15 (Withholding Tax), the tax consequences of any assignment of this Agreement will, as between the Parties, be borne by the assigning Party (who will make the non-assigning Party whole with respect to all such tax consequences to the extent arising as a result of such assignment). The assigning Party will promptly notify the non-assigning Party in writing of any permitted assignment or transfer under the provisions of this Section
17.1 (Assignment). This Agreement will be binding upon the successors and permitted assigns of the Parties. Any assignment not in accordance with this Section
17.1 (Assignment) will be null, void, and of no legal effect.
1.1.2Change of Control. Novavax will notify Sanofi within [***] following the date that there is a Change of Control of Novavax, in which case:
(a)Sanofi shall have the right, but not the obligation, to exercise those rights set forth in Section
17.1.2(b) (Change of Control), by providing written notice to Novavax and the applicable Acquirer, if, the JSC reasonably determines that the Acquirer
(i) is unable or unwilling to cooperate with Sanofi to implement Sanofi’s Commercial strategy for Licensed COVID-19 Products or (ii) is not able or willing to perform Novavax’s obligations under this Agreement with substantially the same degree of skill that Novavax was using prior to such Change of Control.
(b)If the JSC determines including, if applicable, through the dispute resolution process set forth in Section
9.18.5(c) (Change of Control Matters) and
17.12 (Dispute Resolution) that the criteria in clause (a) above are met then Sanofi may
(i) assume Novavax’s rights and obligations to perform any or all Novavax activities allocated to Novavax hereunder, excluding Novavax’s obligation to supply Licensed COVID-19 Products, Adjuvant, or Adjuvant Intermediates, as applicable, to Sanofi in accordance with
Article 6 (Manufacturing); and (ii) disband the JSC and all Subcommittees, and as from the date on which such Committees are disbanded, Sanofi shall have the sole right to make all decisions hereunder with respect to the Licensed COVID-19 Component and Licensed COVID-19 Products in the Collaboration COVID-19 Territory.
1.2Force Majeure. Each Party will not be deemed to have defaulted under or breached any of its obligations under this Agreement, except for the payment of any amounts owed under this Agreement, to the extent that such performance is prevented by Force Majeure and the nonperforming Party promptly provides written notice of the Force Majeure to the other Party. Such excuse will continue for so long as the condition constituting a Force Majeure continues, on the condition that the nonperforming Party continues to use [***] to remove or mitigate the Force Majeure and resume performance of its obligations under this Agreement; provided that after a period exceeding [***], the Parties will promptly meet to discuss in good faith mitigation strategies to proceed in a manner that maintains and abides by the Agreement.
1.3Representation by Legal Counsel. Each Party represents that it has been represented by legal counsel in connection with this Agreement and acknowledges that it has participated in the drafting
hereof. In interpreting and applying the terms and provisions of this Agreement, no presumption will exist or be implied against the Party that drafted such terms and provisions.
1.4Notices. All notices, requests, demands, waivers and other communications which are required or permitted hereunder will be in writing and sufficient if delivered personally or sent by nationally recognized overnight courier, addressed as follows:
If to Sanofi:
Sanofi Pasteur Inc. 1 Discovery Drive
Swiftwater, Pennsylvania 18370 USA Attn: [***]
[***]
with a copy (which does not constitute notice) to: Sanofi Global Alliance Management
Email: [***]
and
Latham & Watkins LLP
1271 Avenue of the Americas New York, NY 10020 Attention: [***]
Email: [***]
If to Novavax:
Novavax, Inc.
700 Quince Orchard
Gaithersburg, Maryland 20878 USA Attn: [***]
with copies (which will not constitute notice) to:
Novavax Legal Department
Email: [***]
and
Novavax’s Alliance Manager and
Ropes & Gray LLP
Boylston Street, Prudential Tower Boston, MA 02199
Attention: [***]
Email: [***]
or to such other address as the Party to whom written notice is to be given may have furnished to the other Party in writing in accordance herewith. In addition, each Party will deliver a courtesy copy to the other Party’s Alliance Manager concurrently with such notice. Any such notice will be deemed to have been given and received by the other Party: (a) when delivered if personally delivered; or (b) on receipt if sent by overnight courier.
1.5Amendment. No amendment, modification, or supplement of any provision of this Agreement will be valid or effective unless made in writing and signed by a duly authorized officer of each of Sanofi and Novavax. For clarity, references in this Agreement to a “written acknowledgement” will not be deemed to be an amendment, modification, or supplement of this Agreement.
1.6Waiver. No provision of this Agreement will be waived by any act, omission, or knowledge of a Party or its Representatives except by an instrument in writing expressly waiving such provision and signed by a duly authorized officer of the waiving Party. The waiver by either Party of any breach of any provision hereof by the other Party will not be construed to be a waiver of any succeeding breach of such provision or a waiver of the provision itself.
1.7Severability. If any clause or portion thereof in this Agreement is for any reason held to be invalid, illegal, or unenforceable, the same will not affect any other portion of this Agreement, as it is the intent of the Parties that this Agreement will be construed in such fashion as to maintain its existence, validity, and enforceability to the greatest extent possible. In any such event, this Agreement will be construed as if such clause or portion thereof had never been contained in this Agreement, and there will be deemed substituted therefor such provision as will most nearly carry out the intent of the Parties as expressed in this Agreement to the fullest extent permitted by Applicable Law.
1.8Descriptive Headings. The descriptive headings of this Agreement are for convenience only and will be of no force or effect in construing or interpreting any of the provisions of this Agreement.
1.9Export Control. This Agreement is made subject to any restrictions concerning the export of products or technical information from the United States or other countries that may be imposed upon or related to Novavax or Sanofi from time to time. Each Party agrees that it will not export, directly or indirectly, any technical information acquired from the other Party under this Agreement or any products using such technical information to a location or in a manner that at the time of export requires an export license or other governmental approval, without first obtaining the written consent to do so from the appropriate Governmental Authority.
1.10Governing Law. This Agreement, and all claims arising under or in connection therewith, will be governed by and interpreted in accordance with the substantive laws of the [***], excluding (a) the conflict of law principles thereof; (b) the United Nations Conventions on Contracts for the International Sale of Goods; (c) the 1974 Convention on the Limitation Period in the International Sale of Goods (the “1974 Convention”); and (d) the Protocol amending the 1974 Convention, done at Vienna April 11, 1980. Notwithstanding any provision to the contrary set forth in this Agreement, the interpretation and construction of any Patents shall be governed in accordance with the laws of the jurisdiction in which such Patents were filed or granted, as the case may be.
1.11Jurisdiction; Venue; Service of Process. Except as otherwise provided in Section
17.12.4 (Equitable Relief), (a) each Party irrevocably submits to the exclusive jurisdiction of (i) the courts
of the [***] located in [***], or (ii) the United States District Court for the [***], for the purposes of any actions, suits, and Proceedings (collectively, “Actions”) arising out of this Agreement (except for government agency actions to adjudicate registered intellectual property rights, e.g., post-grant proceedings at the United States Patent and Trademark Office or other foreign equivalent Proceedings); (b) each Party agrees to commence any such Action either in the United States District Court for the [***] or if such Action may not be brought in such court for jurisdictional reasons, in the courts of [***]; and (c) each Party irrevocably and unconditionally waives any objection to the laying of venue of any Action arising out of this Agreement in (i) the courts of [***], or (ii) the United States District Court for the [***], and hereby and thereby further irrevocably and unconditionally waives and agrees not to plead or claim in any such court that any such Action brought in any such court has been brought in an inconvenient forum. Each of the Parties agrees that process may be served upon it in the manner specified in Section
17.4 (Notices) and irrevocably waives and covenants not to assert or plead any objection that it might otherwise have to such jurisdiction, or to such manner of service of process.
1.12Dispute Resolution. If a dispute arises between the Parties in connection with or relating to this Agreement or any document or instrument delivered in connection herewith other than any dispute arising from a Novavax Matter, Sanofi Matter, or Status Quo Matter (a “Dispute”), then (a) any dispute arising from a Novavax Matter, Sanofi Matter, or Status Quo Matter, will be resolved pursuant to Section
9.18 (Decision Making), and (b) all other Disputes will be resolved pursuant to this Section
17.12 (Dispute Resolution).
1.12.1Informal Dispute Resolution; Escalation to Senior Executives. In the event any Dispute is not a Novavax Matter, a Sanofi Matter, or a Status Quo Matter, and is not otherwise finally resolved pursuant to Section
9.18 (Decision Making), then the Parties will first attempt in good faith to resolve such Dispute by negotiation and consultation between themselves. If, after [***] from receipt of the written notice of such Dispute, such Dispute has not been resolved on an informal basis, either Party may refer any such Dispute to the Senior Executives of the Parties by delivering written notice to the other Party, and the Senior Executives of the Parties will confer in good faith on the resolution of the issue for a [***] period following receipt of such written notice. If any such Dispute is not resolved within such [***] period by the Senior Executives, then each Party may, at its sole discretion, seek resolution of such Dispute in accordance with Section
1.12.2(Litigation).
1.1.2Litigation. If the Parties cannot resolve a Dispute in accordance with Section
17.12.1 (Informal Dispute Resolution; Escalation to Senior Executives), then either Party may commence litigation in accordance with Section
17.11 (Jurisdiction; Venue; Service of Process). The Parties agree to seek an early conference with the court and to advise the court of this Agreement.
1.1.3Jury Trial. EXCEPT AS LIMITED BY APPLICABLE LAW, EACH PARTY HEREBY IRREVOCABLY WAIVES ALL RIGHT TO TRIAL BY JURY IN ANY ACTION, PROCEEDING, OR COUNTERCLAIM (WHETHER BASED ON CONTRACT, TORT, OR OTHERWISE) ARISING OUT OF OR RELATING TO THIS AGREEMENT OR THE ACTIONS OF ANY PARTY HERETO IN THE NEGOTIATION, ADMINISTRATION, PERFORMANCE, AND ENFORCEMENT HEREOF.
1.1.4Equitable Relief. Notwithstanding the foregoing in this Section
17.12 (Dispute Resolution), nothing contained in this Agreement will in any way limit or preclude a Party from, at any time, seeking equitable relief hereunder, whether preliminary or permanent, including a temporary or permanent restraining order, preliminary or permanent injunction, specific performance, or any other form of equitable relief, from any United States court of competent jurisdiction if necessary to protect the interests of such Party. Each Party agrees that its unauthorized release of the other Party’s Confidential Information or its breach of Section
2.1 (Licenses), Section
2.5 (No Adjuvant Improvements),
Article 5 (Technology Transfer), Section
6.1.2 (Manufacture and Supply to Sanofi), Section
6.3 (Supply Agreements),
Article 11 (Confidentiality and Non-Disclosure), or Section
14.4 (Covenants of Novavax) of this Agreement may cause irreparable damage to the other Party for which recovery of damages may be inadequate, and that such other Party will be entitled to seek timely injunctive relief with respect to such breach and without the requirement of having to post bond or other security, as well as any further relief that may be granted by a court of competent jurisdiction.
1.1.5Tolling. The Parties agree that all applicable statutes of limitation and time-based defenses (such as estoppel and laches), as well as all time periods in which a Party must exercise rights or perform obligations hereunder, will be tolled once the dispute resolution procedures set forth in Section
9.18 (Decision Making) or this Section
17.12 (Dispute Resolution) have been initiated and for so long as they are pending, and the Parties will cooperate in taking all actions reasonably necessary to achieve such a result. Further, with respect to any time periods that have run during the pendency of the Dispute, the applicable Party will have a reasonable period of time, or any specific timeframe established by the applicable tribunal’s decision, to exercise any rights or perform any obligations affected by the running of such time periods.
1.1.6Certain Patent and other Disputes. Notwithstanding any provision to the contrary set forth in this Agreement, any Patent Dispute shall be subject to adjudication in accordance with Applicable Law of the country or jurisdiction in which the relevant Patent is pending or has been issued. The Parties agree that the venue of any such adjudication involving a Patent pending in or issued by the United States shall be a U.S. federal district court (or appellate body, as necessary) sitting in [***], and for a Patent pending in or issued by any other country, any competent court having jurisdiction over the subject of the Patent Dispute sitting in the capital of such country (or if there is not any such competent court in the capital, then in a location reasonably proximate to the capital), and each Party irrevocably submits to the jurisdiction of such court. Each Party agrees not to raise any objection at any time to the laying or maintaining of the venue of any action, suit, or proceeding for such purpose in any such court, irrevocably waives any claim that such action, suit, or other proceeding has been brought in an inconvenient forum, including any forum non conveniens argument, and further irrevocably waives the right to object, with respect to such action, suit or other proceeding, that such court does not have any jurisdiction over such Party.
1.13Performance by Affiliates. To the extent that this Agreement imposes obligations on Affiliates of a Party, such Party agrees to cause its Affiliates to perform such obligations. Each Party may use one (1) or more of its Affiliates to perform its obligations and duties under this Agreement, provided that such Party shall remain liable under this Agreement for the prompt payment and performance of all of its obligations under this Agreement and for the Affiliate’s compliance with the terms of this Agreement, including such Affiliate’s adherence to all waivers, disclaimers, and limitations in this Agreement in favor of the other Party hereunder.
1.14Entire Agreement. This Agreement (together with all schedules and exhibits attached hereto) constitutes and contains the complete, final, and exclusive understanding and agreement of the Parties and cancels and supersedes all prior negotiations (including as documented in any term sheet), correspondence, understandings, and agreements, whether oral or written, between the Parties respecting the subject matter hereof, including the CDA, which is hereby superseded and replaced in its entirety as of the Effective Date.
1.15Independent Contractors. Both Parties are independent contractors under this Agreement. Neither Party will have any express or implied power to enter into any contracts or commitments or to incur any liabilities in the name of, or on behalf of, the other Party, or to bind the other Party in any respect whatsoever.
1.16Interpretation. Except where the context expressly requires otherwise, (a) the use of any gender herein will be deemed to encompass references to either or both genders, and the use of the singular will be deemed to include the plural (and vice versa); (b) the words “include,” “includes,” and “including” will be deemed to be followed by the phrase “without limitation”; (c) the word “will” will be construed to have the same meaning and effect as the word “shall,” and vice versa; (d) any definition of or reference to any agreement, instrument, or other document herein will be construed as referring to such agreement, instrument, or other document as from time to time amended, supplemented, or otherwise modified (subject to any restrictions on such amendments, supplements, or modifications set forth herein); (e) any reference herein to any Person will be construed to include the Person’s successors and assigns; (f) the words “herein,” “hereof,” and “hereunder,” and words of similar import, will be construed to refer to this Agreement in its entirety and not to any particular provision hereof; (g) all references herein to Articles, Sections, or Schedules will be construed to refer to Articles, Sections, or Schedules of this Agreement, and references to this Agreement include all Schedules hereto; (h) except as otherwise expressly set forth herein, provisions that require that a Party, the Parties, or any Committee hereunder “agree,” “consent,” “approve,” or the like will require that such agreement, consent, or approval be specific and in writing, whether by written agreement, letter, approved minutes, or otherwise (but excluding e-mail and instant messaging); (i) references to any specific law, rule, or regulation, or article, section, or other division thereof, will be deemed to include the then-current amendments thereto or any replacement or successor law, rule, or regulation thereof; (j) any action or occurrence deemed to be effective as of a particular date will be deemed to be effective as of 11:59 PM ET on such date; and (k) the term “or” will be interpreted in the inclusive sense commonly associated with the term “or” (and/or). Unless otherwise specified, deadlines within which any payment is to be made or act is to be done within or following a specified time period after a date will be calculated by excluding the day, Business Day, month, or year of such date, as applicable, and including the day, Business Day, month, or year of the date on which the period ends. Whenever any payment to be made or action to be taken under this Agreement is required to be made or taken on a day other than a Business Day, such payment may be made, or such action may be taken on the next Business Day following such day. No prior draft of this Agreement may be used in the interpretation or construction of this Agreement.
1.17No Third Party Rights or Obligations. Except as expressly set forth in this Agreement (including in
Article 15 (Indemnification)), no provision of this Agreement will be deemed or construed in any way to result in the creation of any rights or obligations in any Person not a Party to this Agreement.
1.18Further Actions. Each Party agrees to execute, acknowledge, and deliver such further instruments, and to do all such other acts, as may be necessary or appropriate in order to carry out the purposes and intent of this Agreement.
1.19Counterparts. This Agreement may be executed in two (2) counterparts, each of which will be an original and both of which will constitute together the same document. Counterparts may be signed and delivered by digital transmission (e.g., .pdf), each of which will be binding when received by the applicable Party.
1.20Language; Translation.
1.20.1English. All meetings between the Parties shall be conducted in English. All documents exchanged by the Parties hereunder, including all reports, and all agendas and minutes of any Committee meetings, shall be written in English.
1.20.2Translation. In the event that a Party produces a document in a language other than English, such Party shall, upon written request of the other Party, provide a non-certified translation of such document to such Party within ten (10) Business Days. In the event that a Party wishes to receive a certified translation of any document provided by the other Party hereunder, such requesting Party shall retain a certified translator and the costs of such translation shall be borne by the Party requesting such certified transaction.
[Signature Page Follows]
IN WITNESS WHEREOF, the Parties have caused this Agreement to be executed by their representatives thereunto duly authorized as of the Effective Date.
|
|
|
|
|
|
Sanofi Pasteur Inc. |
Novavax, Inc. |
By: [***] |
By: /s/ John C. Jacobs |
|
Name: [***]
Title: [***]
|
Name: John C. Jacobs
Title: President and Chief Executive Officer
|
|
|
|
[Signature Page to Collaboration and License Agreement] |
Adjuvant Intermediate Technology
[Pursuant to Regulation S-K, Item 601(a)(5), this Schedule 1.13 setting forth the Adjuvant Intermediate Technology has not been filed. The Registrant agrees to furnish supplementally a copy of any omitted schedules to the Securities and Exchange Commission upon request; provided, however, that the Registrant may request confidential treatment of omitted items.]
[***]
Schedule 1.35
APAs
(as of the Effective Date)
[Pursuant to Regulation S-K, Item 601(a)(5), this Schedule 1.35 setting forth the APAs (as of the Effective Date) has not been filed. The Registrant agrees to furnish supplementally a copy of any omitted schedules to the Securities and Exchange Commission upon request; provided, however, that the Registrant may request confidential treatment of omitted items.]
[***]
Schedule 1.106
Existing Partner Exclusive Territory
[Pursuant to Regulation S-K, Item 601(a)(5), this Schedule 1.106 setting forth Existing Partner Exclusive Territory has not been filed. The Registrant agrees to furnish supplementally a copy of any omitted schedules to the Securities and Exchange Commission upon request; provided, however, that the Registrant may request confidential treatment of omitted items.]
[***]
Existing Partner Non-Exclusive Territory
[Pursuant to Regulation S-K, Item 601(a)(5), this Schedule 1.107 setting forth the Existing Partner Non-Exclusive Territory has not been filed. The Registrant agrees to furnish supplementally a copy of any omitted schedules to the Securities and Exchange Commission upon request; provided, however, that the Registrant may request confidential treatment of omitted items.]
[***]
Existing Strategic Partner Agreements
[Pursuant to Regulation S-K, Item 601(a)(5), this Schedule 1.109 setting forth the Existing Strategic Partner Agreements has not been filed. The Registrant agrees to furnish supplementally a copy of any omitted schedules to the Securities and Exchange Commission upon request; provided, however, that the Registrant may request confidential treatment of omitted items.]
[***]
Schedule 1.111
Existing Supply Agreements
[Pursuant to Regulation S-K, Item 601(a)(5), this Schedule 1.111 setting forth the Existing Supply Agreements has not been filed. The Registrant agrees to furnish supplementally a copy of any omitted schedules to the Securities and Exchange Commission upon request; provided, however, that the Registrant may request confidential treatment of omitted items.]
[***]
Schedule
1.193 Novavax Domain Names
[Pursuant to Regulation S-K, Item 601(a)(5), this Schedule 1.193 setting forth the Novavax Domain Names has not been filed. The Registrant agrees to furnish supplementally a copy of any omitted schedules to the Securities and Exchange Commission upon request; provided, however, that the Registrant may request confidential treatment of omitted items.]
[***]
Schedule
1.194 Novavax General Combo Patents
[Pursuant to Regulation S-K, Item 601(a)(5), this Schedule 1.194 setting forth the Novavax General Combo Patents has not been filed. The Registrant agrees to furnish supplementally a copy of any omitted schedules to the Securities and Exchange Commission upon request; provided, however, that the Registrant may request confidential treatment of omitted items.]
[***]
Schedule 1.206
Novavax Trademarks
[Pursuant to Regulation S-K, Item 601(a)(5), this Schedule 1.206 setting forth the Novavax Trademarks has not been filed. The Registrant agrees to furnish supplementally a copy of any omitted schedules to the Securities and Exchange Commission upon request; provided, however, that the Registrant may request confidential treatment of omitted items.]
[***]
Schedule
1.270 Settlement Arrangements
[Pursuant to Regulation S-K, Item 601(a)(5), this Schedule 1.270 setting forth the Settlement Agreements has not been filed. The Registrant agrees to furnish supplementally a copy of any omitted schedules to the Securities and Exchange Commission upon request; provided, however, that the Registrant may request confidential treatment of omitted items.]
[***]
COVID-19 Research and Development Plan
[Pursuant to Regulation S-K, Item 601(a)(5), this Schedule 3.2 setting forth the COVID-19 Research and Development Plan has not been filed. The Registrant agrees to furnish supplementally a copy of any omitted schedules to the Securities and Exchange Commission upon request; provided, however, that the Registrant may request confidential treatment of omitted items.]
[***]
COVID-19 Research and Development Budget
(as of the Effective Date)
[Pursuant to Regulation S-K, Item 601(a)(5), this Schedule 3.2.2 setting forth the COVID-19 Research and Development Budget has not been filed. The Registrant agrees to furnish supplementally a copy of any omitted schedules to the Securities and Exchange Commission upon request; provided, however, that the Registrant may request confidential treatment of omitted items.]
[***]
Schedule
4.1 Regulatory Transfer Plan (as of the Effective Date)
[Pursuant to Regulation S-K, Item 601(a)(5), this Schedule 4.1 setting forth the Existing Regulatory Transfer Plan has not been filed. The Registrant agrees to furnish supplementally a copy of any omitted schedules to the Securities and Exchange Commission upon request; provided, however, that the Registrant may request confidential treatment of omitted items.]
[***]
COVID-19 Manufacturing Technology Transfer Plan
(as of the Effective Date)
[Pursuant to Regulation S-K, Item 601(a)(5), this Schedule 5.2.1 setting forth the COVID-19 Manufacturing Technology Transfer Plan has not been filed. The Registrant agrees to furnish supplementally a copy of any omitted schedules to the Securities and Exchange Commission upon request; provided, however, that the Registrant may request confidential treatment of omitted items.]
[***]
Schedule 6.3.1 (COVID-19 Supply Agreement Key Terms)
and
Schedule 6.3.2 (Adjuvant Supply Agreement Key Terms)
[Pursuant to Regulation S-K, Item 601(a)(5), this Schedules 6.3.1 and 6.3.2 setting forth the COVID-19 Supply Agreement Key Terms and Adjuvant Supply Agreement Key Terms have not been filed. The Registrant agrees to furnish supplementally a copy of any omitted schedules to the Securities and Exchange Commission upon request; provided, however, that the Registrant may request confidential treatment of omitted items.]
[***]
Licensed COVID-19 Mono Product Commercialization Framework
(as of the Effective Date)
[Pursuant to Regulation S-K, Item 601(a)(5), this Schedule 7.2 setting forth the Licensed COVID-19 Mono Product Commercialization Framework has not been filed. The Registrant agrees to furnish supplementally a copy of any omitted schedules to the Securities and Exchange Commission upon request; provided, however, that the Registrant may request confidential treatment of omitted items.]
[***]
COVID-19 Product Medical Framework
(as of the Effective Date)
[Pursuant to Regulation S-K, Item 601(a)(5), this Schedule 8.2 setting forth the COVID-19 Product Medical Framework has not been filed. The Registrant agrees to furnish supplementally a copy of any omitted schedules to the Securities and Exchange Commission upon request; provided, however, that the Registrant may request confidential treatment of omitted items.]
[***]
Schedule
9.2.2 JSC Responsibilities
[Pursuant to Regulation S-K, Item 601(a)(5), this Schedule 9.2.2 setting forth the JSC Responsibilities has not been filed. The Registrant agrees to furnish supplementally a copy of any omitted schedules to the Securities and Exchange Commission upon request; provided, however, that the Registrant may request confidential treatment of omitted items.]
[***]
Schedule
9.3.2 JRDC Responsibilities
[Pursuant to Regulation S-K, Item 601(a)(5), this Schedule 9.3.2 setting forth the JRDC Responsibilities has not been filed. The Registrant agrees to furnish supplementally a copy of any omitted schedules to the Securities and Exchange Commission upon request; provided, however, that the Registrant may request confidential treatment of omitted items.]
[***]
Schedule 9.4.2
JMSC Responsibilities
[Pursuant to Regulation S-K, Item 601(a)(5), this Schedule 9.4.2 setting forth the JMSC Responsibilities has not been filed. The Registrant agrees to furnish supplementally a copy of any omitted schedules to the Securities and Exchange Commission upon request; provided, however, that the Registrant may request confidential treatment of omitted items.]
[***]
Schedule
9.7.2 JFC Responsibilities
[Pursuant to Regulation S-K, Item 601(a)(5), this Schedule 9.7.2 setting forth the JFC Responsibilities has not been filed. The Registrant agrees to furnish supplementally a copy of any omitted schedules to the Securities and Exchange Commission upon request; provided, however, that the Registrant may request confidential treatment of omitted items.]
[***]
Schedule
9.8.2 JPC Responsibilities
[Pursuant to Regulation S-K, Item 601(a)(5), this Schedule 9.8.2 setting forth the JPC Responsibilities has not been filed. The Registrant agrees to furnish supplementally a copy of any omitted schedules to the Securities and Exchange Commission upon request; provided, however, that the Registrant may request confidential treatment of omitted items.]
[***]
Baseball Arbitration Dispute Resolution
[Pursuant to Regulation S-K, Item 601(a)(5), this Schedule 9.18.5(d)(ii) setting forth the Baseball Arbitration Dispute Resolution has not been filed. The Registrant agrees to furnish supplementally a copy of any omitted schedules to the Securities and Exchange Commission upon request; provided, however, that the Registrant may request confidential treatment of omitted items.]
[***]
Schedule 14.2.1
Existing Novavax Patents
[Pursuant to Regulation S-K, Item 601(a)(5), this Schedule 14.2.1 setting forth the Existing Novavax Patents has not been filed. The Registrant agrees to furnish supplementally a copy of any omitted schedules to the Securities and Exchange Commission upon request; provided, however, that the Registrant may request confidential treatment of omitted items.]
[***]
Exhibit A Initial Press Releases
[Pursuant to Regulation S-K, Item 601(a)(5), this Exhibit A setting forth the Initial Press Releases has not been filed. The Registrant agrees to furnish supplementally a copy of any omitted schedules to the Securities and Exchange Commission upon request; provided, however, that the Registrant may request confidential treatment of omitted items.]
[***]
Exhibit B Data Safeguards
[Pursuant to Regulation S-K, Item 601(a)(5), this Exhibit B setting forth the Data Safeguards has not been filed. The Registrant agrees to furnish supplementally a copy of any omitted schedules to the Securities and Exchange Commission upon request; provided, however, that the Registrant may request confidential treatment of omitted items.]
[***]
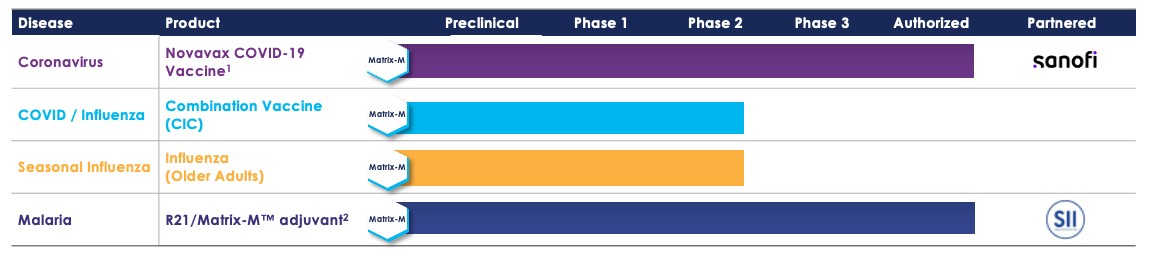




 Date: August 8, 2024 By: /s/ James P. Kelly James P. Kelly
Date: August 8, 2024 By: /s/ James P. Kelly James P. Kelly