UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM 20-F
(Mark One)
☐ REGISTRATION STATEMENT PURSUANT TO SECTION 12(b) OR 12(g) OF THE SECURITIES EXCHANGE ACT OF 1934
OR
☒ ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the fiscal year ended December 31, 2023
OR
☐ TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
OR
☐ SHELL COMPANY REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
Date of event requiring this shell company report
Commission File Number: 001-41636
OCULIS HOLDING AG
(Exact name of Registrant as specified in its charter)
|
|
|
Not applicable |
|
Switzerland |
(Translation of Registrant’s name into English) |
|
(Jurisdiction of incorporation or organization) |
Bahnhofstrasse 7
CH-6300
Zug, Switzerland
(Address of principal executive offices)
Riad Sherif, MD
EPFL Innovation Park, Bat D 3e Route J-D.
Colladon, CH-1015 Lausanne, Switzerland
+41-41-711-9325
(Name, Telephone, E-mail and/or Facsimile number and Address of Company Contact Person)
Securities registered or to be registered pursuant to Section 12(b) of the Act:
|
|
|
|
|
Title of each class |
|
Trading Symbol(s) |
|
Name of each exchange on which registered |
Ordinary Shares |
|
OCS |
|
The Nasdaq Stock Market LLC |
Warrants |
|
OCSAW |
|
The Nasdaq Stock Market LLC |
Securities registered or to be registered pursuant to Section 12(g) of the Act: None
Securities for which there is a reporting obligation pursuant to Section 15(d) of the Act: None
Indicate the number of outstanding shares of each of the issuer’s classes of capital or common stock as of the close of the period covered by the annual company report: 36,649,705 Ordinary Shares and 4,254,138 Warrants to purchase Ordinary Shares.
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☐ No ☒
If this report is an annual or transition report, indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934.
Yes ☐ No ☒ Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ☒ No ☐
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files). Yes ☒ No ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or an emerging growth company. See definition of “large accelerated filer,” “accelerated filer,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
|
|
|
|
|
|
|
Large accelerated filer |
|
☐ |
|
Accelerated filer |
|
☐ |
Non-accelerated filer |
|
☒ |
|
Emerging growth company |
|
☒ |
|
|
|
|
|
|
|
If an emerging growth company that prepares its financial statements in accordance with U.S. GAAP, indicate by check mark if the registrant has elected to use the extended transition period for complying with any new or revised financial accounting standards† provided pursuant to Section 13(a) of the Exchange Act. ☐
† |
The term “new or revised financial accounting standard” refers to any update issued by the Financial Accounting Standards Board to its Accounting Standards Codification after April 5, 2012. |
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report. ☐
If securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant included in the filing reflect the correction of an error to previously issued financial statements. ☐
Indicate by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive-based compensation received by any of the registrant’s executive officers during the relevant recovery period pursuant to §240.10D-1(b). ☐
Indicate by check mark which basis of accounting the registrant has used to prepare the financial statements included in this filing:
US GAAP ☐ |
|
International Financial Reporting Standards as issued |
|
|
|
|
|
Other ☐ |
|
|
by the International Accounting Standards Board ® |
|
☒ |
|
|
|
|
If “Other” has been checked in response to the previous question indicate by check mark which financial statement item the registrant has elected to follow. Item17 ☐ Item18 ☐
If this is an annual report, indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ☐ No ☒
TABLE OF CONTENTS
3 |
||||
7 |
||||
7 |
||||
9 |
||||
|
Item 1. Identity of Directors, Senior Management and Advisers. |
9 |
||
|
9 |
|||
|
9 |
|||
|
A. |
|
9 |
|
|
B. |
|
9 |
|
|
C. |
|
9 |
|
|
D. |
|
9 |
|
|
78 |
|||
|
A. |
|
78 |
|
|
B. |
|
79 |
|
|
C. |
|
121 |
|
|
D. |
|
121 |
|
|
122 |
|||
|
122 |
|||
|
A. |
|
131 |
|
|
B. |
|
133 |
|
|
C. |
|
138 |
|
|
D. |
|
138 |
|
|
E. |
|
138 |
|
|
140 |
|||
|
A. |
|
140 |
|
|
B. |
|
143 |
|
|
C. |
|
146 |
|
|
D. |
|
150 |
|
|
E. |
|
150 |
|
|
F. |
|
Disclosure of a Registrant’s Action to Recover Erroneously Awarded Compensation. |
150 |
|
150 |
|||
|
A. |
|
150 |
|
|
B. |
|
153 |
|
|
C. |
|
153 |
|
|
154 |
|||
|
A. |
|
154 |
|
|
B. |
|
154 |
|
|
154 |
|||
|
A. |
|
154 |
|
|
B. |
|
154 |
|
1
|
C. |
|
154 |
|
|
D. |
|
154 |
|
|
E. |
|
154 |
|
|
F. |
|
154 |
|
|
154 |
|||
|
A. |
|
154 |
|
|
B. |
|
155 |
|
|
C. |
|
155 |
|
|
D. |
|
156 |
|
|
E. |
|
156 |
|
|
F. |
|
164 |
|
|
G. |
|
164 |
|
|
H. |
|
165 |
|
|
I. |
|
165 |
|
|
J. |
|
165 |
|
|
Item 11. Quantitative and Qualitative Disclosures About Market Risk |
165 |
||
|
Item 12. Description of Securities Other than Equity Securities. |
165 |
||
|
A. |
|
165 |
|
|
B. |
|
165 |
|
|
C. |
|
165 |
|
|
D. |
|
166 |
|
166 |
||||
|
166 |
|||
|
Item 14. Material Modifications to the Rights of Security Holders and Use of Proceeds. |
166 |
||
|
166 |
|||
|
167 |
|||
|
167 |
|||
|
167 |
|||
|
167 |
|||
|
Item 16D. Exemptions from the Listing Standards for Audit Committees. |
168 |
||
|
Item 16E. Purchases of Equity Securities by the Issuer and Affiliated Purchasers. |
168 |
||
|
168 |
|||
|
168 |
|||
|
169 |
|||
|
Item 16I. Disclosure Regarding Foreign Jurisdictions that Prevent Inspections. |
169 |
||
|
169 |
|||
|
169 |
|||
171 |
||||
|
171 |
|||
|
171 |
|||
|
171 |
|||
2
DEFINEDTERMS
In this Annual Report:
“2023 Plan” means the Stock Option and Incentive Plan Regulation 2023 of the registrant.
“Acquisition Closing” means the closing of the First Merger, Second Merger and Oculis Share Contribution related to the Business Combination.
“Acquisition Closing Date” means March 2, 2023, the date upon which the Acquisition Closing occurred.
“Ancillary Agreements” means the Business Combination Agreement (together with the Oculis Disclosure Letter and the EBAC Disclosure Letter), the Subscription Agreements, the Convertible Loan Agreements, the Sponsor Support Agreement, the Non-Redemption Agreement, the Confidentiality Agreement, dated as of February 22, 2022, by and between Oculis and EBAC, the Oculis Shareholders Support Agreement and when entered into at the Acquisition Closing, the Registration Rights and Lock-Up Agreement and the Warrant Assignment and Assumption Agreement.
“Annual Report” means this annual report of Oculis on Form 20-F.
“Business Combination” means the transactions contemplated by the Business Combination Agreement, including the Mergers and the Oculis Share Contribution.
“Business Combination Agreement” means the Business Combination Agreement, dated as of October 17, 2022, as may be amended from time to time, by and among EBAC, Legacy Oculis, and Oculis.
“Closing” means the consummation of the Business Combination, which occurred on March 2, 2023.
“Closing Date” means March 2, 2023, the date upon which the Closing occurred.
“Code” means the Internal Revenue Code of 1986, as amended.
“Company” means the legal entity named Oculis Holding AG, individually or together with its consolidated subsidiaries.
“Company Share Capital” has the meaning ascribed to such term in the Business Combination Agreement.
“Continental” means Continental Stock Transfer & Trust Company, the transfer agent and warrant agent of EBAC and the Company.
“Convertible Loan Agreements” means the convertible loan agreements, dated as of October 17, 2022 and January 20, 2023 (as amended and restated on February 22, 2023), by and among Oculis and certain lenders party thereto.
“EBAC” means European Biotech Acquisition Corp., a Cayman Islands exempted company.
“EBAC Class A Common Stock” means Class A ordinary shares, par value $0.0001 per share, of EBAC.
“EBAC Class B Common Stock” or “Founder Shares” means Class B ordinary shares, par value $0.0001 per share, of EBAC.
“EBAC Common Stock” means EBAC Class A Common Stock and EBAC Class B Common Stock.
“EBAC Disclosure Letter” means that certain disclosure letter delivered to Oculis by EBAC on the date of the Business Combination Agreement.
“EBAC Private Placement Warrant” means a warrant to purchase one share of EBAC Class A Common Stock at an exercise price of $11.50 issued to the Sponsor.
3
“EBAC Public Warrant” means a warrant to purchase one share of EBAC Class A Common Stock at an exercise price of $11.50 that was included in the units sold as part of EBAC’s initial public offering.
“EBAC Shareholders” means the shareholders of EBAC as of any applicable determination time prior to the Acquisition Closing.
“EBAC Share Redemption” means the election of an eligible (as determined in accordance with EBAC’s amended and restated memorandum and articles of association) holder of shares of EBAC Class A Common Stock to redeem all or a portion of the shares of EBAC Class A Common Stock held by such holder in return for the right to receive a per-share price, payable in cash by Oculis, equal to a pro rata share of the aggregate amount on deposit in the Trust Account (including any interest earned on the funds held in the Trust Account) (as determined in accordance with EBAC’s amended and restated memorandum and articles of association) in connection with the Business Combination. The redeemed shares of EBAC Class A Common Stock shall be held in treasury for re-issuance to new investors.
“EBAC Share Redemption Amount” means the aggregate amount payable by Oculis with respect to all EBAC Share Redemptions.
“EBAC Warrants” means the EBAC Public Warrants and the EBAC Private Placement Warrants.
“Exchange Act” means the Securities Exchange Act of 1934, as amended.
“Exchange Agent” means Continental, which was selected by Oculis, Legacy Oculis and EBAC to act on behalf of EBAC, EBAC Shareholders, Oculis and Oculis Shareholders.
“Exchange Agent Contribution” means the contribution by the Exchange Agent of Surviving EBAC Shares to the Company.
“Exchange Agent Contribution Actions” means the distribution by the Exchange Agent of Ordinary Shares and Warrants to the holders of Surviving EBAC Shares and Surviving EBAC Warrants, respectively.
“Existing Warrant Agreement” means the Warrant Agreement, dated March 15, 2021, between EBAC and the Exchange Agent, as warrant agent.
“First Merger” means when Merger Sub 1 merged with and into EBAC, with EBAC as the surviving company.
“First Merger Effective Time” means the time at which the First Merger became effective pursuant to the filing and registration of the plan of merger with the Cayman Islands Registrar of Companies or at such later time as may be agreed by Oculis and Legacy Oculis in writing and specified in such plan of merger.
“IFRS” means IFRS Accounting Standards as adopted by the International Accounting Standards Board.
“Initial PIPE Financing” means the private placement pursuant to which the Initial PIPE Investors subscribed for EBAC Class A Common Stock, for a subscription price of $10.00 per share.
“Initial PIPE Investors” means the institutional investors that committed to subscribe for EBAC Class A Common Stock in the Initial PIPE Financing.
“Initial Subscription Agreements” means the subscription agreements, each dated as of October 17, 2022, by and among EBAC and the Initial PIPE Investors party thereto.
“Legacy Oculis” means Oculis SA, a stock corporation (Aktiengesellschaft), which was incorporated in Lausanne, Switzerland on December 11, 2017, individually or together with its consolidated subsidiaries, which together with its consolidated subsidiaries formed the Oculis group prior to the Closing.
“Lenders” means those certain Oculis Shareholders party to the Convertible Loan Agreements pursuant to which, among other things, such Oculis Shareholders agreed to grant Oculis a right to receive a convertible loan with certain conversion rights in an aggregate amount of $19,670,000.
4
“Mergers” means the two consecutive mergers between Merger Sub 1 and EBAC, and EBAC and Merger Sub 2, EBAC, after which Merger Sub 1 ceased to exist and Merger Sub 2 was the surviving company.
“Merger Sub 1” means Oculis Merger Sub I Company, a Cayman Islands exempted company that was a direct wholly owned subsidiary of Oculis prior to the Acquisition Closing.
“Merger Sub 2” means Oculis Merger Sub II Company, a Cayman Islands exempted company that is a direct wholly owned subsidiary of Oculis.
“Merger Sub 3” means Oculis Operations GmbH, a limited liability company (Gesellschaft mit beschränkter Haftung) incorporated and existing under the laws of Switzerland that is a direct wholly owned subsidiary of Oculis.
“Nasdaq” means the Nasdaq Stock Market LLC.
“New Parent Interests” means the Ordinary Shares and Warrants which were held by the Exchange Agent solely on behalf of holders of Surviving EBAC Shares and Surviving EBAC Warrants.
“Oculis” means as the context requires, (a) the registrant, a legal entity named Oculis Holding AG, a stock corporation (Aktiengesellschaft) incorporated and existing under the laws of Switzerland having its registered office at Bahnhofstrasse 7, CH-6300, Zug, Switzerland, individually or together with its consolidated subsidiaries; or (b) Legacy Oculis.
“Oculis Disclosure Letter” means that certain disclosure letter delivered to EBAC by Oculis on the date of the Business Combination Agreement.
“Oculis Shareholders” means, collectively, the holders of shares of Company Share Capital as of any applicable determination time prior to the Acquisition Closing.
“Oculis Shareholders Support Agreement” means that certain agreement entered into concurrently with the execution of the Business Combination Agreement, dated as of October 17, 2022, by and among Oculis, EBAC and the Oculis Shareholders party thereto.
“Oculis Share Contribution” means the contribution by the Oculis Shareholders of the full legal and beneficial ownership of the applicable Company Share Capital to Oculis.
“Ordinary Shares” means ordinary shares, nominal value CHF 0.01 per share of Oculis.
“PIPE Financing” means the Initial PIPE Financing and the Subsequent PIPE Financing, pursuant to which the PIPE Investors subscribed for EBAC Class A Common Stock, for a subscription price of $10.00 per share.
“PIPE Investors” means the Initial PIPE Investors and the Subsequent PIPE Investors.
“PIPE Shares” means the shares of EBAC Class A Common Stock purchased by the PIPE Investors and transferred to them by EBAC from treasury.
“Public Warrant” means a warrant to purchase an Ordinary Share at an exercise price of $11.50 originally issued as an EBAC Public Warrant.
“Private Placement Warrant” means a warrant to purchase an Ordinary Shares at an exercise price of $11.50 originally issued to Sponsor as an EBAC Private Placement Warrant.
“Registration Rights and Lock-Up Agreement” means the Amended and Restated Registration Rights and Lock-Up Agreement, dated as of the Acquisition Closing Date, by and among Oculis, Sponsor and certain Legacy Oculis Shareholders.
“SEC” means the U.S. Securities and Exchange Commission.
5
“Second Merger” means when EBAC merged with and into Merger Sub 2, with Merger Sub 2 as the surviving company.
“Second Merger Effective Time” means the time at which the Second Merger became effective pursuant to the filing and registration of the plan of merger with the Cayman Islands Registrar of Companies or at such later time as may be agreed by Oculis and Legacy Oculis in writing and specified in such plan of merger.
“Securities Act” means the Securities Act of 1933, as amended.
“Share Cancellation” means the cancellation of the Ordinary Shares held by EBAC concurrently with the Exchange Agent Contribution.
“Sponsor” means LSP Sponsor EBAC B.V. a Dutch limited liability company.
“Sponsor Support Agreement” means the Sponsor Support Agreement, dated October 17, 2022, by and among EBAC, Oculis and Sponsor.
“Subscription Agreements” means the Initial Subscription Agreements and the Subsequent Subscription Agreements.
“Subsequent PIPE Financing” means the private placement pursuant to which the Subsequent PIPE Investors subscribed for EBAC Class A Common Stock, for a subscription price of $10.00 per share.
“Subsequent PIPE Investors” means the institutional investors that committed to subscribe for EBAC Class A Common Stock in the Subsequent PIPE Financing.
“Subsequent Subscription Agreements” means the subscription agreements, entered into in January 2023, by and among EBAC and the Subsequent PIPE Investors party thereto.
“Surviving EBAC Shares” means EBAC Common Stock, including those held by the PIPE Investors, automatically converted into one class of common stock of EBAC, as the surviving company of the First Merger.
“Surviving EBAC Warrants” means EBAC Warrants outstanding immediately prior to the First Merger Effective Time automatically converted into warrants of EBAC, as the surviving company of the First Merger.
“Swiss Code of Obligations” means the Swiss Federal Act on the Amendment of the Swiss Civil Code of March 30, 1911.
“Third Merger” means when Legacy Oculis merged with and into Merger Sub 3, with Merger Sub 3 as the surviving company and wholly owned subsidiary of Oculis.
“Third Merger Effective Time” means the time at which the Third Merger became effective pursuant to the filing and the registration of the plan of merger in accordance with the provisions of the Swiss Code of Obligations or at such later time as may be agreed by Oculis and Legacy Oculis in writing and specified in such plan of merger.
“Transfer Agent” means Continental.
“Trust Account” means that certain trust account with Continental, as trustee, containing the cash proceeds of EBAC from its initial public offering and private placement of securities (and all accrued interest earned thereon), deposited therein for the benefit of EBAC and EBAC’s public shareholders.
“Warrant Agreement Assumption” means the assignment by EBAC of all its right, title and interest in the Existing Warrant Agreement to the Company and the acceptance by Company of such assignment.
“Warrant Assignment and Assumption Agreement” means the Warrant Assignment and Assumption Agreement entered into among EBAC, the Company and the Exchange Agent, which became effective immediately following the completion of the Exchange Agent Contribution and concurrent Share Cancellation.
6
“Warrant” means a right to acquire Ordinary Shares, on substantially the same terms as an EBAC Warrant.
GENERAL INFORMATION
Unless context otherwise requires, all references in this Annual Report on Form 20-F (“Annual Report”) to “Oculis,” the “Company,” “we,” “us” and “our” refer to Oculis and, where appropriate, its consolidated subsidiaries. Unless otherwise stated or unless the context otherwise requires, references to “Oculis” or the “Company” are to the registrant named “Oculis Holding AG” and its subsidiaries after the consummation of the Business Combination, whereas references to “Legacy Oculis” are to Oculis SA and its subsidiaries prior to the Closing.
This Annual Report includes trademarks, trade names and service marks, certain of which belong to us and others that are the property of other organizations. Solely for convenience, trademarks, trade names and service marks referred to in this Annual Report appear without the ®, ™ and SM symbols, but the absence of those symbols is not intended to indicate, in any way, that we will not assert our rights or that the applicable owner will not assert its rights to these trademarks, trade names and service marks to the fullest extent under applicable law. We do not intend our use or display of other parties’ trademarks, trade names or service marks to imply, and such use or display should not be construed to imply, a relationship with, or endorsement or sponsorship of us by, these other parties.
SPECIAL NOTE REGARDINGFORWARD-LOOKING STATEMENTS
This Annual Report on Form 20-F (the “Annual Report”) contains or may contain forward-looking statements as defined in Section 27A of the Securities Act of 1933, as amended (the “Securities Act”), and Section 21E of the Securities Exchange Act of 1934, as amended (the “Exchange Act”), that involve significant risks and uncertainties. All statements other than statements of historical facts are forward-looking statements. These forward-looking statements include information about our possible or assumed future results of operations or our performance. Words such as “may,” “might,” “will,” “could,” “would,” “should,” “expects,” “intends,” “plans,” “believes,” “anticipates,” “estimates,” “potential,” “continue,” “ongoing,” “targets”, “possible,” “project,” and “predict” and variations of such words and similar expressions are intended to identify the forward-looking statements. Forward-looking statements in this Annual Report may include, for example, statements about:
7
These forward-looking statements are based on information available as of the date of this Annual Report, and current expectations, forecasts and assumptions, and involve a number of judgments, risks and uncertainties. Accordingly, forward-looking statements should not be relied upon as representing our views as of any subsequent date, and we do not undertake any obligation to update forward-looking statements to reflect events or circumstances after the date they were made, whether as a result of new information, future events or otherwise, except as may be required under applicable securities laws. Accordingly, you should not place undue reliance on these forward-looking statements in deciding to invest in our securities. As a result of a number of known and unknown risks and uncertainties, our actual results or performance may be materially different from those expressed or implied by these forward-looking statements. You should refer to the section titled “Item 3.D Risk Factors” for a discussion of important factors that may cause our actual results to differ materially from those expressed or implied by our forward-looking statements. As a result of these factors, we cannot assure you that the forward-looking statements in this Annual Report will prove to be accurate. Furthermore, if our forward-looking statements prove to be inaccurate, the inaccuracy may be material. In light of the significant uncertainties in these forward-looking statements, you should not regard these statements as a representation or warranty by us or any other person that we will achieve our objectives and plans in any specified time frame, or at all. We undertake no obligation to publicly update any forward-looking statements, whether as a result of new information, future events or otherwise, except as required by law. You should, therefore, not rely on these forward-looking statements as representing our views as of any date subsequent to the date of this Annual Report.
8
PART I
Item 1. Identity of Directors, Senior Management and Advisers.
Not applicable.
Item 2. Offer Statistics and Expected Timetable.
Not applicable.
Item 3. Key Information.
Not applicable.
Not applicable.
An investment in our securities carries a significant degree of risk. In addition to the other information contained in this Annual Report on Form 20-F, including the matters addressed under the heading “Forward-Looking Statements,” you should carefully consider the following risk factors in deciding whether to invest in our securities. The occurrence of one or more of the events or circumstances described in these risk factors, alone or in combination with other events or circumstances, may have a material adverse effect relating to our business, financial condition, and results of operations and future prospects, in which event the market price of our securities could decline, and you could lose part or all of your investment. Additional risks and uncertainties of which we are not presently aware or that we currently deem immaterial could also affect our business operations and financial condition.
Summary Risk Factors
Our business is subject to a number of risks and uncertainties. If any of the following risks are realized, our business, financial condition and results of operations could be materially and adversely affected. You should carefully review and consider the full discussion of our risk factors in this section titled “Risk Factors” in Part I, Item 3.D. of this Annual Report. Set forth below is a summary list of the principal risk factors as of the date of the filing of this Annual Report:
9
10
Risk Factors
Risks related to our business, financial condition, capital requirements, or financial operations
We have a very limited operating history and no products approved for commercial sale, which may make it difficult to evaluate our current business and predict our future success and viability.
We are a clinical stage biopharmaceutical company specializing in novel therapeutics to treat ophthalmic diseases. We commenced operations in October 2003 and formed Legacy Oculis in December 2017, have no products approved for commercial sale and have not generated any revenue from product sales. Drug development is a highly uncertain undertaking and involves a substantial degree of risk. To date, we have not obtained marketing approval for any product candidates, manufactured a commercial scale product, or conducted sales and marketing activities necessary for successful product commercialization.
Our limited operating history as a company and early stage of drug development make any assessment of our future success and viability subject to significant uncertainty. We will encounter risks and difficulties frequently experienced by clinical-stage biopharmaceutical companies in rapidly evolving fields, and we have not yet demonstrated an ability to successfully overcome such risks and difficulties. If we do not address these risks and difficulties successfully, our business, financial condition, results of operations and growth prospects may be impaired.
We have incurred significant net losses in each period since our inception and anticipate that we will continue to incur significant and increasing net losses for the foreseeable future.
We have incurred net losses in each reporting period since our inception, including net losses of CHF 88.8 million and CHF 38.7 million for the fiscal years ended December 31, 2023 and 2022, respectively. As of December 31, 2023, we had accumulated losses of CHF 199.8 million.
We have invested significant financial resources in research and development activities, including for our product candidates. We do not expect to generate revenue from product sales in the foreseeable future, if at all. The amount of our future net losses will depend, in part, on the level of our future expenditures and our ability to generate revenue. Moreover, our net losses may fluctuate significantly from quarter to quarter and year to year, such that a period-to-period comparison of our results of operations may not be a good indication of our future performance quarter to quarter or year to year due to factors including the timing of clinical trials, any litigation that we may file or that may be filed against us, the execution of collaboration, licensing or other agreements and the timing of any payments we make or receive thereunder.
We expect to continue to incur significant and increasingly higher expenses and operating losses for the foreseeable future. We anticipate that our expenses will increase substantially if and as we:
11
Our prior losses and expected future losses have had and will continue to have an adverse effect on our shareholders’ deficit and working capital. In any particular quarter or quarters, our operating results could be below the expectations of securities analysts or investors, which could cause the share price of Ordinary Shares to decline.
As of December 31, 2023, we had cash, cash equivalents and short-term financial assets of CHF 91.7 million. We believe that these cash, cash equivalents and short-term financial assets will be sufficient to enable us to fund our current operations for at least the next twelve months.
Drug development is a highly uncertain undertaking and involves a substantial degree of risk. We have never generated any revenue from product sales, and we may never generate revenue or be profitable.
We have no products approved for commercial sale and have not generated any revenue from product sales. We do not anticipate generating any revenue from product sales until after we have successfully completed clinical development and received regulatory approval for the commercial sale of a product candidate, if ever.
Our ability to generate revenue, alone or with strategic collaboration, and achieve profitability depends significantly on many factors, including:
12
Because of the numerous risks and uncertainties associated with drug development, we are unable to predict the timing or amount of our expenses, or when we will be able to generate any meaningful revenue or achieve or maintain profitability, if ever. In addition, our expenses could increase beyond our current expectations if we are required by the FDA or non-U.S. regulatory agencies to perform studies in addition to those that we currently anticipate, or if there are any delays in any of our or our future collaborators’ clinical trials or the development of any of our product candidates. Even if one or more of our product candidates is approved for commercial sale, we anticipate incurring significant costs associated with commercializing any approved product candidate and ongoing compliance efforts.
Even if we are able to generate revenue from the sale of any approved products, we may not become profitable, and we will need to obtain additional funding through one or more debt or equity financings in order to continue operations. Revenue from the sale of any product candidate for which regulatory approval is obtained will be dependent, in part, upon the size of the markets in the territories for which we gain regulatory approval, the accepted price for the product, the ability to get reimbursement at any price and whether we own the commercial rights for that territory. If the number of addressable patients is not as significant as we anticipate, the indication approved by regulatory agencies is narrower than we expect, or the reasonably accepted population for treatment is narrowed by competition, physician choice or treatment guidelines, we may not generate significant revenue from sales of such products, even if approved. Even if we do achieve profitability, we may not be able to sustain or increase profitability on a quarterly or annual basis.
Our failure to become and remain profitable could decrease the value of our Company and could impair our ability to raise capital, expand our business, maintain our research and development efforts, diversify our pipeline of product candidates or continue our operations and cause a decline in the value of Ordinary Shares, all or any of which may adversely affect our viability.
Our operating and financial results are subject to concentration risk.
Our operational and financial results are subject to concentration risk. Our success will depend significantly on the development of OCS-01, OCS-02 (Licaminlimab) and OCS-05, their regulatory approval in a limited number of jurisdictions and their commercialization by a limited number of commercial partners. Even if we are successful in developing and commercializing all of these products, our revenue may be dependent on a limited number of products that would account for a significant majority of our revenues. This concentration risk would increase to the extent we are successful in developing and commercializing fewer products as we would be dependent on a lower number of products for the significant majority of our revenues. Unfavorable changes or the non-occurrence of certain anticipated events with respect to any of these limited number of products, jurisdictions or commercial partners may disproportionately affect our global results.
If we fail to obtain additional financing, we may be unable to complete the development and, if approved, commercialization of our product candidates.
13
Our operations have required substantial amounts of cash since inception. To date, we have financed our operations primarily through the sale of equity securities. Developing our product candidates is expensive, and we expect to substantially increase our spending as we advance our product candidates in clinical trials. Even if we are successful in developing our product candidates, obtaining regulatory approvals and launching and commercializing any product candidate will require substantial additional funding.
As of December 31, 2023, we had CHF 91.7 million in cash, cash equivalents and short-term financial assets. Although we believe that our existing cash, cash equivalents and short-term financial assets will be sufficient to fund our projected operations through at least the next 12 months, our estimate as to how long we expect our existing cash, cash equivalents and short-term financial assets to fund our operations is based on assumptions that may prove inaccurate, and we could use our available capital resources sooner than we currently expect. In addition, changing circumstances may cause us to increase our spending significantly faster than we currently anticipate, and we may need to spend more money than currently expected because of circumstances beyond our control. We may need to raise additional funds sooner than we anticipate if we choose to expand more rapidly than we presently foresee.
We will require additional capital for the further development and, if approved, commercialization of our product candidates. Additional capital may not be available when we need it, on terms acceptable to us or at all. We have no committed source of additional capital. If adequate capital is not available to us on a timely basis, we may be required to significantly delay, scale back or discontinue our research and development programs or the commercialization of any product candidates, if approved, or be unable to continue or expand our operations or otherwise capitalize on our business opportunities, as desired, which could materially affect our business, financial condition and results of operations and cause the price of Ordinary Shares to decline.
Our future success depends on our ability to retain key executives and to attract, retain and motivate qualified personnel.
We are highly dependent on the research and development, clinical and business development expertise of our chief executive officer as well as other principal members of our management, scientific and clinical team. Although we have entered into employment agreements with our executive officers, each of them may terminate their employment with us at any time.
Laws and regulations on executive compensation, including legislation in our home country, Switzerland, may restrict our ability to attract, motivate and retain the required level of qualified personnel. In Switzerland, legislation affecting public companies is in force that, among other things, (i) imposes an annual binding shareholders’ “say on pay” vote with respect to the compensation of our executive committee and board of directors, (ii) generally prohibits severance, advances, transaction premiums and similar payments to members of our executive committee and board of directors, and (iii) requires companies to specify certain compensation-related matters in their articles of association, thus requiring them to be approved by a shareholders’ vote.
Recruiting and retaining qualified scientific, clinical, manufacturing and sales and marketing personnel will also be critical to our success. The loss of the services of our executive officers or other key employees could impede the achievement of our research, development and commercialization objectives and seriously harm our ability to successfully implement our business strategy. Furthermore, replacing executive officers and key employees may be difficult and may take an extended period of time because of the limited number of individuals in our industry with the breadth of skills and experience required to successfully develop, gain regulatory approval of and commercialize products. Competition to hire from this limited pool is intense, and we may be unable to hire, train, retain or motivate these key personnel on acceptable terms given the competition among numerous pharmaceutical and biotechnology companies for similar personnel. We also experience competition for the hiring of scientific and clinical personnel from universities and research institutions. In addition, we rely on consultants and advisors, including scientific, medical, clinical and regulatory advisors, to assist us in formulating our research and development and commercialization strategy. Our consultants and advisors may be employed by employers other than us and may have commitments under consulting or advisory contracts with other entities that may limit their availability to us. If we are unable to continue to attract and retain high quality personnel, our ability to pursue our growth strategy will be limited.
14
We will continue to incur significant expenses and devote other significant resources and management time as a result of being a public company, which may negatively impact our financial performance and could cause our results of operations and financial condition to suffer.
We will continue to incur significant legal, accounting, insurance and other expenses as a result of being a public company. The rules implemented by the SEC, Nasdaq and Swiss corporate law require changes in corporate governance practices of public companies. We expect that compliance with these laws, rules and regulations will continue to substantially increase our expenses as a result of being a public company, including our legal, accounting and information technology costs and expenses, and make some activities more time consuming and costly. These obligations require attention from our executive officers and senior management and could divert their attention away from the day-to-day management of our business. These laws, rules and regulations and the move from a private to a public company has made, and we expect will continue to make, it more expensive for us to obtain director and officer liability insurance, and we may be required to accept reduced policy limits and coverage or incur substantially higher costs to obtain the same or similar coverage. Due to increased risks and exposure it may be more difficult for us to attract and retain qualified persons to serve on our board of directors or as officers. As a result of the foregoing, we expect to continue to experience a substantial increase in legal, accounting, insurance and certain other expenses in the future, which will negatively impact our financial performance and could cause our results of operations and financial condition to suffer. Furthermore, if we are unable to satisfy our obligations as a public company, we could be subject to delisting of Ordinary Shares, fines, sanctions and other regulatory action and potentially civil litigation, which could adversely impact our business, results of operation, financial condition and the price of Ordinary Shares.
We have been and will need to continue to expand our organization and we may experience difficulties in managing this growth, which could disrupt our operations.
As of December 31, 2023, we had 36 employees. Additionally, we may rely on a number of temporary workers and contractors from time-to-time as needed. As our development and commercialization plans and strategies develop, we expect to need additional managerial, operational, sales, marketing, financial, legal and other resources. Our management may need to divert a disproportionate amount of our attention away from our day-to-day activities and devote a substantial amount of time to managing these growth activities. We may not be able to effectively manage the expansion of our operations, which may result in weaknesses in our infrastructure, operational mistakes, loss of business opportunities, loss of employees and reduced productivity among remaining employees. In addition, our success depends on our ability to attract and retain a talented workforce with a specialized set of skills. Our expected growth could also require significant capital expenditures and may divert financial resources from other projects, such as the development of our current and potential future product candidates. If our management is unable to effectively manage our growth, our expenses may increase more than expected, our ability to generate and/or grow revenue could be reduced and we may not be able to implement our business strategy. Our future financial performance and our ability to commercialize product candidates and compete effectively will depend, in part, on our ability to effectively manage any future growth.
If we fail to maintain proper and effective internal controls, our ability to produce accurate financial statements on a timely basis could be impaired.
The Sarbanes-Oxley Act of 2022 requires, among other things, that we maintain effective disclosure controls and procedures and internal control over financial reporting. Our internal control over financial reporting will not prevent or detect all errors and all fraud. A control system, no matter how well designed and operated, can provide only reasonable, not absolute, assurance that the control system’s objectives will be met. Because of the inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that misstatements due to error or fraud will not occur or that all control issues and instances of fraud will be detected.
If we are not able to comply with the requirements of Section 404 of the Sarbanes-Oxley Act in a timely manner, or if we are unable to maintain proper and effective internal controls, we may not be able to produce timely and accurate financial statements. If that were to happen, the market price of our common stock could decline, and we could be subject to sanctions or investigations by Nasdaq, the SEC, or other regulatory authorities.
15
Economic, financial, geopolitical, epidemiological, or other conditions could result in business disruptions which could seriously harm our future revenue and financial condition and increase our costs and expenses.
Concerns over inflation, geopolitical issues, conflicts such as the Russia-Ukraine war and the Israel-Hamas conflict, the U.S. financial markets, foreign exchange rates, capital and exchange controls, unstable global credit markets and financial conditions, post-COVID-19 economic environment, supply chain disruptions and economic issues, have led to periods of significant economic instability, declines in consumer confidence and discretionary spending, diminished expectations for the global economy and expectations of slower global economic growth going forward, and increased unemployment rates. Our general business strategy may be adversely affected by any such economic downturns, volatile business environments and continued unstable or unpredictable economic and market conditions. If these conditions continue to deteriorate or do not improve, it may make any necessary debt or equity financing more difficult to complete, more costly and more dilutive. In addition, there is a risk that one or more of our current or future service providers, manufacturers, suppliers and other partners could be negatively affected by difficult economic times, which could adversely affect our ability to attain our operating goals on schedule and on budget or meet our business and financial objectives.
Our operations, and those of our contract research organizations (“CROs”), contract manufacturing organizations (“CMOs”), suppliers, and other third-party contractors and consultants upon which we rely, could be subject to wildfires, earthquakes, tsunamis, power shortages or outages, floods or monsoons, public health crises, such as pandemics and epidemics, political crises, such as terrorism, war (including trade wars), political instability or other conflicts, and other natural or man-made disasters or other events outside of our control that could disrupt our business.
In addition, our available cash, cash equivalents and investments (short-term financial assets) are held in accounts managed by third party financial institutions and consist of cash in our operating accounts and cash invested in money market funds. At any point in time, the funds in our U.S. operating accounts may exceed the Federal Deposit Insurance Corporation insurance limits. While we monitor the cash balances in our operating accounts and adjust the cash balances as appropriate, these cash balances could be impacted if the underlying financial institutions fail. Our active treasury strategy is to minimize risk through natural hedging of currencies, bank diversification and cash preservation. To date, we have experienced no loss or lack of access to cash in our operating accounts or our invested cash, cash equivalents or investments; however, we can provide no assurances that access to our operating cash or invested cash, cash equivalents or investments will not be impacted by adverse conditions in the financial markets.
The occurrence of any of these business disruptions could seriously harm our operations and financial condition and increase our costs and expenses. For example, we rely on third-party manufacturers to produce our product candidates. Our ability to obtain supplies of our product candidates, or other necessary supplies, could be disrupted if the operations of our suppliers are affected by a man-made or natural disaster or other business interruption. Damage or extended periods of interruption to our corporate, development or research facilities due to fire, natural disaster, power loss, communications failure, unauthorized entry or other events could cause us to cease or delay the marketing or development of some or all of our product candidates. Although we maintain property damage and business interruption insurance coverage, our business, financial condition, and results of operations may be seriously harmed should the losses we suffer as a result of such property damage and/or business interruption substantially exceed our insurance coverage and we are required to make up for this shortfall.
Our business, financial condition and results of operations would suffer in the event of computer system failures, security breaches or other disruptions to our information technology systems.
In the ordinary course of our business, we collect, store and transmit sensitive data, including protected health information (“PHI”), intellectual property, proprietary business information and other personal information. We rely on information technology systems, networks and services, some of which are managed, hosted or provided by third parties, to assist in conducting our business. While we have not previously experienced a security breach or computer failure resulting in destruction, theft, or other loss of this information, and we and our service providers have implemented a number of security measures designed to protect against security breaches, these measures could fail or may be insufficient, resulting in the unauthorized disclosure, modification, misuse, unavailability, destruction, or loss of confidential information or personal information we collect, store and transmit. Despite the implementation of security measures, our internal computer systems, and those of our contract research organizations ("CROs"), and other third parties on which we rely, are vulnerable to attack, damage or interruption from computer viruses, unauthorized access, cyberattacks, employee theft or misuse, human error, hacking, fraud, natural disasters, fire, terrorism, war and telecommunication and electrical failures.
16
Cyberattacks are increasing in their frequency, sophistication and intensity. Cyberattacks could include the deployment of harmful malware, “phishing attacks”, denial-of-service attacks, social engineering and other means to affect service reliability and threaten the confidentiality, integrity and availability of information. The use of cloud-based computing also creates opportunities for the unintentional dissemination or intentional destruction of confidential information stored in our or our third-party providers’ systems, portable media or storage devices. Furthermore, we may also face increased cybersecurity risks due to our reliance on internet technology and the number of our employees who are working remotely, which may create additional opportunities for cybercriminals to exploit vulnerabilities.
Significant disruptions of our information technology systems or security breaches could adversely affect our business operations and/or result in the loss, misappropriation, and/or unauthorized access, use or disclosure of, or the prevention of access to, confidential information (including trade secrets or other intellectual property, proprietary business information and personal information), and could result in financial, legal, business and reputational harm to us. If such disruptions were to occur and cause interruptions in our operations, it could result in a material disruption of our product development programs. For example, the loss of clinical trial data from completed, ongoing or planned clinical trials could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data. Despite our efforts to ensure the security, privacy, integrity, confidentiality, availability, and authenticity of our information technology networks and systems, processing and information, we may not be able to anticipate or to implement effective preventive and remedial measures against all data security and privacy threats. We cannot guarantee that the recovery systems, security protocols, network protection mechanisms and other security measures that we or our third-party providers have integrated into our or their systems, networks and physical facilities, which are designed to protect against, detect and minimize security breaches will be adequate to prevent or detect service interruption, system failures, data loss or theft, or other material adverse consequences. No security solution, strategy, or measures can address all possible security threats or block all methods of penetrating a network or otherwise perpetrating a security incident. The risk of unauthorized circumvention of our security measures has been heightened by advances in computer and software capabilities and the increasing sophistication of hackers who employ complex techniques, including without limitation, the theft or misuse of personal and financial information, counterfeiting, “phishing” or social engineering, ransomware, extortion, publicly announcing security breaches, account takeover attacks, denial or degradation of service attacks, malware, fraudulent payment and identity theft. Furthermore, because the techniques used to sabotage, disrupt or to obtain unauthorized access to our systems, networks, or physical facilities in which data is stored or through which data is transmitted change frequently and often are not recognized until launched against a target, we or our third-party providers may be unable to implement adequate preventative measures or stop security breaches while they are occurring. We or our third-party providers may also experience security breaches that may remain undetected for an extended period. Even if identified, we or our third-party providers may be unable to adequately investigate or remediate incidents or breaches due to attackers increasingly using tools and techniques that are designed to circumvent controls, to avoid detection, and to remove or obfuscate forensic evidence, or we or our third-party providers may be unable to repair our or their systems in an efficient and timely manner. In addition, laws, regulations, government guidance, and industry standards and practices are rapidly evolving to combat these threats, including new rules and regulations of the SEC. We expect to face increased compliance burdens regarding such requirements from regulators and incur additional costs for oversight and monitoring of security risks relating to our own supply chain.
If we or our third-party providers were to experience a significant cybersecurity breach of our or their information systems or data, the costs associated with the investigation, remediation and potential notification of the breach to counterparties and data subjects could be material. Unauthorized access to our systems, networks, or physical facilities could result in litigation with our customers or other relevant stakeholders, which may adversely affect our business. These proceedings could force us to spend money in defense or settlement, divert management’s time and attention, increase our costs of doing business, or adversely affect our reputation.
Further, we may not have adequate insurance coverage for security incidents or breaches, including fines, judgments, settlements, penalties, costs, attorney fees and other impacts that arise out of incidents or breaches. Depending on the facts and circumstances of such an incident, the damages, penalties and costs could be significant and may not be covered by insurance or could exceed our applicable insurance coverage limits.
17
If the impacts of a security incident or breach, or the successful assertion of one or more large claims against us, exceeds our available insurance coverage, or results in changes to our insurance policies (including premium increases or the imposition of large deductible or co-insurance requirements), it could have an adverse effect on our business. In addition, we cannot be sure that our existing insurance coverage and coverage for errors and omissions will continue to be available on acceptable terms, or at all, or that our insurers will not deny coverage as to all or part of any future claim or loss.
Further, the number of our employees and partners working remotely increases the risk of a data breach or issues with data and cybersecurity. To the extent that any disruption or security breach results in a loss of, or damage to, our data or applications, or inappropriate disclosure of confidential or proprietary information, we could incur liability and the further development of our future product candidates could be delayed.
We are subject to numerous laws, regulations, standards and other requirements related to personal information, privacy and data protection. Our actual or perceived failure to comply with such laws, regulations, standards and other requirements could negatively affect our business, financial condition or results of operations.
The global data protection landscape is rapidly evolving, and we are subject to numerous federal, state and foreign laws, regulations, standards and other requirements governing the collection, use, disclosure, retention and security of personal information, such as information that we may collect in connection with clinical trials in the United States and abroad. Implementation standards and enforcement practices are likely to remain uncertain for the foreseeable future, and we cannot yet determine the impact future laws, regulations, standards or requirements may have on our business. This evolution may create uncertainty in our business, affect our ability to operate in certain jurisdictions or to collect, store, transfer, use and share personal information, necessitate the acceptance of more onerous obligations in our contracts, result in liability or impose additional costs on us. Any failure or perceived failure by us to comply with federal, state or foreign laws or regulations, our internal or external policies and procedures or our contracts governing our processing of personal information could result in negative publicity, government investigations, enforcement actions, claims by third parties or damage to our reputation, any of which could have a material adverse effect on our business, results of operations and financial condition.
In the United States, numerous federal and state laws and regulations, including data breach notification laws, health information privacy laws, and consumer protection laws and regulations that govern the collection, processing, use, disclosure, and protection of health-related and other personal information could apply to our operations or the operations of our partners. For example, in the United States, the Health Insurance Portability and Accountability Act of 1996 (“HIPAA”) imposes among other things, certain standards relating to the privacy, security, transmission and breach reporting of individually identifiable health information. Entities that are found to be in violation of HIPAA, whether as the result of a breach of unsecured PHI, a complaint about privacy practices, or an audit by the U.S. Department of Health and Human Services ("HHS"), may be subject to significant civil, criminal, and administrative fines and penalties and/or additional reporting and oversight obligations if required to enter into a resolution agreement and corrective action plan with HHS to settle allegations of HIPAA non-compliance. Depending on the facts and circumstances, we could be subject to penalties if we violate HIPAA.
Even when HIPAA does not apply, according to the Federal Trade Commission (the “FTC”) failing to take appropriate steps to keep consumers’ personal information secure may constitute unfair acts or practices in or affecting commerce in violation of the Federal Trade Commission Act. The FTC expects a company’s data security measures to be reasonable and appropriate in light of the sensitivity and volume of consumer information it holds, the size and complexity of its business, and the cost of available tools to improve security and reduce vulnerabilities. Individually identifiable health information is considered sensitive data that merits stronger safeguards.
In addition, certain state laws govern the privacy and security of health-related and other personal information in certain circumstances, some of which may be more stringent, broader in scope or offer greater individual rights with respect to protected health information than HIPAA, many of which may differ from each other, thus, complicating compliance efforts. Such laws and regulations will be subject to interpretation by various courts and other governmental authorities, thus creating potentially complex compliance issues for us and our future customers and strategic partners. Failure to comply with these laws, where applicable, can result in the imposition of significant civil and/or criminal penalties and private litigation. For example, California enacted the California Consumer Privacy Act, (“CCPA”), which creates individual privacy rights for California consumers, including the right to opt out of certain disclosures of their information, and places increased privacy and security obligations on entities handling certain personal data of consumers or households and may apply to us in the future. The CCPA provides for civil penalties for violations and also creates a private right of action with statutory damages for certain data breaches, thereby potentially increasing risks associated with a data breach.
18
Further, the majority of the provisions of the California Privacy Rights Act ("CPRA"), went into effect on January 1, 2023 and additional compliance investment and potential business process changes may be required. The CRPA significantly amends the CCPA and imposes additional data protection obligations on covered businesses, including additional consumer rights processes, limitations on data uses, new audit requirements for higher risk data, and opt outs for certain uses of sensitive data. It also creates a new California data protection agency authorized to issue substantive regulations and could result in increased privacy and information security enforcement. The CCPA and CPRA could mark the beginning of a trend toward more stringent privacy legislation in the U.S., as other states or the federal government may follow California’s lead and increase protections for U.S. residents, which creates the potential for a patchwork of overlapping but different state laws and could increase our potential liability and adversely affect our business, financial condition and results of operations. The enactment of such laws could add layers of complexity to compliance in the U.S. market, increase our compliance costs and adversely affect our business, financial condition and results of operations.
Further, we are subject to international data protection laws and regulations, including the European Union General Data Protection Regulation and applicable national supplementing laws ("GDPR"), which may apply to health-related and other personal information obtained outside of the United States. The GDPR imposes strict requirements for collection, control, sharing, disclosure, transfer, use and other processing of the personal data of individuals located in the European Economic Area (“EEA”), including clinical trial data, as well as potential fines for noncompliant companies. The GDPR also imposes strict requirements relating to obtaining consent, providing information to individuals regarding data processing activities, implementing safeguards to protect the security and confidentiality of personal data, providing notification of data breaches, taking certain measures when engaging third-party processors. Compliance with the GDPR may increase our cost of doing business or require us to change our business practices, and despite those efforts, there is a risk that we may be subject to fines and penalties, litigation, and reputational harm in connection with our activities carried out in the context of our EEA operations.
Recent legal developments in Europe have created complexity and uncertainty regarding transfers of personal data from the EEA to the United States. On July 16, 2020, in a case known as Schrems II, the Court of Justice of the European Union ("CJEU"), invalidated the EU-U.S. Privacy Shield Framework under which personal data could be transferred from the EEA to U.S. entities who had self-certified under the Privacy Shield scheme. While the CJEU upheld the adequacy of the Standard Contractual Clauses (a standard form of contract approved by the European Commission as an adequate personal data transfer mechanism, and potential alternative to the Privacy Shield), it made clear that reliance on them alone may not necessarily be sufficient in all circumstances. Use of the standard contractual clauses must now be assessed on a case-by-case basis taking into account the legal regime applicable in the destination country, in particular applicable surveillance laws and rights of individuals and additional measures and/or contractual provisions may need to be put in place. Additionally, new Standard Contractual Clauses that repealed the Standard Contractual Clauses adopted under the Data Protection Directive have been adopted by the European Commission. As supervisory authorities issue further guidance on personal data export mechanisms, including on the new Standard Contractual Clauses, and/or start taking enforcement action, we could suffer additional costs, complaints and/or regulatory investigations or fines, and/or if we are otherwise unable to transfer personal data between and among countries and regions in which we conduct clinical trials, it could affect our business. U.S. President Joseph Biden and the President of the European Commission announced on March 25, 2022 that they had reached an agreement in principle for a Trans-Atlantic Data Privacy Framework, which would allow personal data to flow freely and safely between the EU and participating U.S. companies. To that end, U.S. President Joseph Biden signed the Executive Order on Enhancing Safeguard for United States Signals Intelligence Activities ("EO"), on October 7, 2022. The EO answers to certain shortcomings identified by the EU but it does not yet allow for the free transfers of personal data to the United States. Organizations must continue to implement a valid compliance mechanism for cross-border data transfers, such as the Standard Contractual Clauses, and conduct an assessment of the U.S. laws prior to transferring personal data to the United States. As the EO introduces safeguards for U.S. intelligence services’ access to European personal information, certain supplementary measures that have been implemented and are linked to these practices could be softened and the overall risk associated to the data transfer could be lowered. A new EU-U.S. data transfer framework is not yet available.
Relatedly, following the United Kingdom’s withdrawal from the EEA and the EU, we are required to comply with both the GDPR and, separately, the UK GDPR, which, together with the amended UK Data Protection Act 2018, retains the GDPR in UK national law. The UK GDPR sets out the UK-specific requirements related to data protection, including with respect to transfer of personal data outside of the UK, which increases our regulatory compliance burden.
19
Further, in July 2022, the UK government published a Data Reform Bill that will amend the UK GPDR. This creates uncertainty with regard to the data protection regulatory regime in the United Kingdom and could result in the introduction of data privacy laws that materially deviate from the EU GDPR. This would expose us to two parallel regimes. Further, the entry into force of the US-UK Data Access Agreement on 3 October 2022 may put at risk the European Commission’s adequacy decision granted to the UK. If such adequacy decision were to be withdrawn, personal data would not flow freely between the UK and the EU and additional safeguards would need to be adopted, which could result in additional costs for us.
Any failure or perceived failure by us to comply with our legal obligations concerning privacy, data protection or information security could result in claims by data subjects, governmental investigations and enforcement action against us, including fines, enforcement orders, imprisonment of company officials and public censure, (individual and collective) claims for damages by affected individuals and damage to our reputation, any of which could have a material adverse effect on our business, financial condition, and operating results. Companies that must comply with the GDPR and UK GDPR face increased compliance obligations and risk, including more robust regulatory enforcement of data protection requirements and potential fines for noncompliance of up to €20 million or 4.0% of the annual global revenues of the noncompliant company, whichever is greater, litigation (including private litigation related to processing of personal data brought by classes of data subjects or consumer protection organizations authorized at law to represent their interests), regulatory investigations, enforcement actions that require us to change the way we use personal data, and/or prohibitions on the use of personal data. Such penalties may be in addition to any civil litigation claims by data subjects. We may not be successful in avoiding potential liability or disruption of business resulting from the failure to comply with these laws and, even if we comply with laws, we may be subject to liability because of a security incident. Further, complying with the applicable notification requirements in the event of a security breach could result in significant costs. Furthermore, future interpretations of existing data protection laws or regulations could be inconsistent with our current interpretations, increase our compliance burden, make it more difficult to comply, and/or increase our risk of regulatory investigations and fines.
EU data protection laws also require opt-in consent to send marketing emails or use cookies and similar technologies for advertising, analytics and other purposes – activities on which our marketing strategies may rely. Enforcement of these requirements has increased and a new regulation that has been proposed in the EU, known as the Privacy Regulation, may make these requirements more stringent and increase the penalties for violating them. Such restrictions could increase our exposure to regulatory enforcement action, increase our compliance costs, and adversely affect our business. The relationship between the UK and the EU in relation to certain aspects of data protection law remains unclear, and it is unclear how UK data protection laws and regulations will develop in the medium to longer term, and how data transfers to and from the UK will be regulated in the long term. These changes will lead to additional costs and increase our overall risk exposure. On June 28, 2021, the European Commission adopted an adequacy decision permitting flows of personal data between the EU and the UK to continue without additional requirements. However, the UK adequacy decision will automatically expire in June 2025 unless the European Commission re-assesses and renews or extends that decision and remains under review by the European Commission during this period.
Additionally, we contract with, and are accountable for, third-party service providers we engage to process personal data on our behalf, including our CROs. We cannot assure you that our service providers with access to our or our customers’, suppliers’, trial patients’ and employees’ personal information, including health data and other sensitive or confidential information, will not breach contractual obligations imposed by us, or that they will not experience data security breaches or attempts thereof. If they were to breach their contractual obligations or experience a security incident, such event could have an adverse effect on our business, including putting us in breach of our obligations under privacy laws and regulations, which could in turn adversely affect our business, financial conditions and results of operations. We cannot assure you that our contractual measures and our own privacy and security-related safeguards will protect us from the risks associated with the third-party processing, storage and transmission of such information.
The Swiss Federal Act on Data Protection ("DPA") also applies to the collection and processing of personal data by companies located in Switzerland, or in certain circumstances, by companies located outside of Switzerland. The DPA, which was revised along with its ordinances, with effect per September 1, 2023 may lead to an increase in our costs of compliance, risk of noncompliance and penalties for noncompliance.
20
In addition to data privacy and security laws, we may be contractually subject to industry standards adopted by industry groups and may become subject to such obligations in the future. We may also be bound by other contractual obligations related to data privacy and security, and our efforts to comply with such obligations may not be successful.
We may publish privacy policies, marketing materials, and other statements, such as compliance with certain certifications or self-regulatory principles, regarding data privacy and security. If these policies, materials or statements are found to be deficient, lacking in transparency, deceptive, unfair, or misrepresentative of our practices, we may be subject to investigation, enforcement actions by regulators, or other adverse consequences.
Compliance with applicable United States and foreign data protection, privacy and security laws, regulations and standards could require us to take on more onerous obligations in our contracts, require us to engage in costly compliance exercises, restrict our ability to collect, use and disclose data, or in some cases, impact our ability, or our that of our partners or suppliers, to operate in certain jurisdictions. Each of these constantly evolving laws can also be subject to varying interpretations. Any failure or perceived failure to comply could result in government investigations and enforcement actions (which could include civil or criminal penalties), fines, private litigation, and/or adverse publicity, and could negatively affect our operating results and business. Moreover, patients about whom we or our partners obtain information, as well as the providers who share this information with us, may contractually limit our ability to use and disclose the information. Claims that we have violated individuals’ privacy rights, failed to comply with data protection laws, or breached our contractual obligations, even if we are not found liable, could be expensive and time-consuming to defend and could result in adverse publicity that could harm our business.
We may not realize the benefits of acquired assets or other strategic transactions.
We evaluate various strategic transactions on an ongoing basis. We may acquire other businesses, products or product candidates, intellectual property, or technologies as well as pursue joint ventures or investments in complementary businesses. The success of any potential future strategic transaction depends on various risks and uncertainties, including:
Foreign acquisitions and joint ventures are subject to additional risks, including those related to regulatory or compliance issues, integration of operations across different cultures and languages, currency risks, potentially adverse tax consequences of overseas operations, and the particular economic, political, and regulatory risks associated with specific countries.
Future acquisitions or dispositions could result in potentially dilutive issuances of our equity securities, the incurrence of debt, contingent liabilities, or amortization expenses or write-offs of goodwill, any of which could harm our financial condition. We could also incur losses resulting from undiscovered liabilities that are not covered by the indemnification we may obtain from the seller.
For existing in-licensed or future in-license product candidates or products or acquire businesses, we may not be able to realize the benefit of those transactions if we are unable to successfully integrate them with our existing operations and company culture.
21
We cannot be certain that, following a strategic transaction or license, we will achieve the results, revenue, or specific net income that justifies the transaction. Future acquisitions or dispositions could result in potentially dilutive issuances of our equity securities, the incurrence of debt, contingent liabilities, or amortization expenses or write-offs of goodwill, any of which could harm our financial condition.
We could be subject to securities class action litigation.
In the past, securities class action litigation has often been brought against a company following a decline in the market price of its securities. This risk is especially relevant for us because biopharmaceutical companies have experienced significant stock price volatility in recent years. If we face such litigation, it could result in substantial costs and a diversion of management’s attention and resources, which could harm our business.
Risks related to development and regulatory approval of our investigational therapies
The success of our product candidates, and our ability to generate revenue in the future, will depend upon a number of factors, many of which are beyond our control.
The success of our business, including our ability to finance and generate revenue in the future, primarily depends on the successful development, regulatory approval and commercialization of OCS-01, OCS-02 (Licaminlimab) and OCS-05. The clinical and commercial success of our product candidates depend on a number of factors, including the following:
22
The sizes of the market opportunities for our product candidates have not been established with precision and may be smaller than we estimate, possibly materially. If we overestimate the sizes of these markets, our sales growth may be adversely affected. We may also not be able to grow the markets for our product candidates as intended or at all.
Our assessment of the potential market opportunity the product candidates that we develop is based on industry and market data that we obtained from industry publications and research, surveys and studies conducted by third parties and our own internal epidemiology and market research studies. Industry publications and third-party research, surveys and studies generally indicate that their information has been obtained from sources believed to be reliable, although they do not guarantee the accuracy or completeness of such information. While we believe these industry publications and third-party research, surveys and studies are reliable, we have not independently verified such data. Similarly, although the studies we have conducted are based on information that we believe to be complete and reliable, we cannot guarantee that such information is accurate or complete. The potential market opportunities of our product candidates are difficult to precisely estimate. Therefore, our estimates of the potential market opportunities for our product candidates include several key assumptions based on our industry knowledge, industry publications, third-party research and our own epidemiology studies and market research, which may be based on a small sample size and fail to accurately reflect market opportunities. While we believe that our internal assumptions and the bases of the studies and research we have conducted are reasonable, no independent source has verified such assumptions or bases. If any of our assumptions or estimates, or these publications, research, surveys or studies prove to be inaccurate, then the actual market for our product candidates may be smaller than we expect, and as a result our product revenue may be limited and it may be more difficult for us to achieve or maintain profitability.
Our future growth may depend, in part, on our ability to penetrate foreign markets, where we would be subject to additional regulatory burdens and other risks and uncertainties.
Our future profitability may depend, in part, on our ability to commercialize our product candidates in foreign markets where we lack familiarity with local regulations, environment and procedures and for which we may rely on collaboration with third parties. We are evaluating the opportunities for the development and commercialization of our product candidates in other foreign markets. We are not permitted to market or promote any of our product candidates before we receive regulatory approval from the applicable regulatory authority in that foreign market, and we may never receive such regulatory approval for any of our product candidates. To obtain separate regulatory approvals in other countries we may be required to comply with numerous and varying regulatory requirements of such countries regarding the safety and efficacy of our product candidates and governing, among other things, clinical trials and commercial sales, pricing and distribution of our product candidates, and we cannot predict success in these jurisdictions. If we obtain approval of our product candidates and ultimately commercialize our product candidates in foreign markets, we would be subject to additional risks and uncertainties, including:
23
Our approach to the treatment of retinal disease with OCS-01 is unproven, and we do not know whether we will be able to successfully develop OCS-01.
OCS-01 is designed to deliver therapeutic drug levels to the retinal tissue by a topical route of administration as an eye drop formulation. There are currently no FDA-approved therapies that treat retinal diseases by a topical route of administration. Our future success partially depends on the successful development of OCS-01 which is based on this novel therapeutic approach. We have not yet obtained marketing approval of any product candidate. In Stage 2 of our DIAMOND Phase 3 clinical trial in Diabetic Macular Edema ("DME") and in OPTIMIZE-2 Phase 3 clinical trial in inflammation and pain following cataract surgery, OCS-01 may not demonstrate in patients any or all of the pharmacological benefits we believe it may possess. If we are unsuccessful in our development efforts, we may not be able to advance the development and commercialization of OCS-01.
Our potential approach to use OCS-02 (Licaminlimab) for the treatment of dry eye disease in patients identified with a biomarker is unproven, and we do not know whether we will be able to successfully confirm the role of the biomarker and successfully develop OCS-02.
OCS-02 (Licaminlimab) is in development for treating ophthalmic diseases including dry eye disease. One of our potential strategies for OCS-02 is also to develop it for patients identified with a biomarker to predict patients that may respond well to OCS-02 treatment. There are currently no FDA-approved therapies that treat dry eye disease in this “precision medicine” approach. If we choose to utilize this biomarker strategy, then our future success partially depends on the successful development of both OCS-02 and a companion diagnostic for the biomarker and our ability to demonstrate that patients with that biomarker are likely to respond well to OCS-02 treatment. We have not yet demonstrated efficacy and safety for OCS-02 or any other product candidates in patients with or without a biomarker in a pivotal trial or obtained marketing approval of any of our product candidates. OCS-02 may not demonstrate in patients with or without the biomarker any or all of the pharmacological benefits we believe it may possess. If we are unsuccessful in our development efforts, we may not be able to advance the development and commercialization of OCS-02.
24
Our approach to the treatment of ophthalmic disease with OCS-05 is unproven, and we do not know whether we will be able to successfully develop OCS-05.
OCS-05 is intended to prevent or reverse nerve damage (“neuroprotection”) in ophthalmic diseases in which patients lose vision due to nerve damage. There are currently no FDA-approved therapies that treat ophthalmic diseases in this “neuroprotective” way. Our future success partially depends on the successful development of OCS-05 which is based on this novel therapeutic approach. We have not yet demonstrated efficacy and safety for OCS-05 or any other product candidates in a pivotal trial or obtained marketing approval of any product candidate. OCS-05 may not demonstrate in patients any or all of the pharmacological benefits we believe it may possess. If we are unsuccessful in our development efforts, we may not be able to advance the development and commercialization of OCS-05.
We in-licensed OCS-05 from Accure in 2022. Accure was previously unable to establish a no-observed-adverse-effect-level (“NOAEL") for the product candidate. If our studies do not satisfy the FDA’s requirements, OCS-05 may not receive clearance from the FDA to proceed with human clinical trials, may never receive clearance from the FDA to proceed with human clinical trials and may never receive regulatory approval from the FDA, and we may be unable to market and commercialize OCS-05 in the United States.
We have not yet successfully completed any Phase 3 clinical trials, received any marketing approvals or commercialized any pharmaceutical products, which may make it difficult to evaluate our future prospects.
Our operations to date have been limited to financing and staffing our company, developing our technology and conducting preclinical research as well as Phase 1, Phase 2 and Phase 3 clinical trials for our product candidates. We have not yet demonstrated an ability to successfully complete Phase 3 clinical trials, obtain marketing approvals, manufacture a commercial-scale product or arrange for a third party to do so on our behalf, or conduct sales and marketing activities necessary for successful product commercialization. Accordingly, you should consider our prospects in light of the costs, uncertainties, delays and difficulties frequently encountered by clinical-stage biopharmaceutical companies such as ours. Any predictions made about our future success or viability may not be as accurate as they could be if we had a longer operating history or a history of successfully developing and commercializing pharmaceutical products.
We may encounter unforeseen expenses, difficulties, complications, delays and other known or unknown factors in achieving our business objectives. We will eventually need to transition from a company with a development focus to a company capable of supporting commercial activities. We may not be successful in such a transition.
We depend significantly on our product candidates, OCS-01, OCS-02 (Licaminlimab) and OCS-05, which are being developed for the treatment of multiple diseases. If we are unable to complete the clinical development of any of these product candidates, if we are unable to obtain marketing approvals for any of these product candidates, or if any of these product candidates are approved and we fail to successfully commercialize the product candidate or experience significant delays in doing so, our business will be materially harmed.
We depend significantly on the success of our lead product candidate, OCS-01, which we are developing for the treatment of patients with DME, for the treatment of patients with pain or inflammation following ocular surgery and for the treatment of patients with Cystoid Macular Edema ("CME"). In addition, we also depend on the success of OCS-02, which we are developing for the treatment of Dry Eye Disease ("DED") and non-infectious anterior uveitis and on the success of OCS-05, which we are initially developing for the treatment of Acute Optic Neuropathy ("AON").
We have invested a significant portion of our efforts and financial resources in the development of OCS-01 for the treatment of patients with DME as well as for the treatment of patients with pain or inflammation following ocular surgery. There remains a significant risk that we will fail to successfully develop OCS-01 in one or both of these indications. The results of our first Phase 3 clinical trials in each indication may not be predictive of the results of our Stage 2 Phase 3 clinical programs due, in part, to the fact that (i) we have no clinical data on OCS-01 therapy in DME in any clinical trial with treatment longer than 12 weeks, (ii) we have modified the methodology used to determine a patient’s eligibility under certain of the inclusion and exclusion criteria for our Phase 3 clinical trial as compared to our Phase 2 clinical trial, (iii) we have no clinical data from a trial of similar size to that anticipated for our Phase 3 clinical trial, and (iv) we plan to conduct our Phase 3 clinical trials at many clinical centers that were not included in our Phase 2 clinical trial. The results of our first Phase 3 clinical trial (“DIAMOND trial”) for DME may not be predictive of the results of the planned Stage 2 Phase 3 clinical trials (“DIAMOND-1 and DIAMOND-2”).
25
The results of our first Phase 3 clinical trial (“OPTIMIZE-1”) for the treatment of inflammation and pain following cataract surgery may not be predictive of the results of the second Phase 3 clinical trial (“OPTIMIZE-2”). Furthermore, despite consultation with regulatory agencies, no assurance can be provided that the FDA or non-U.S. regulatory agencies would consider the completed and planned Phase 3 DIAMOND clinical trials to be sufficient to serve as the basis for approval in DME, or that the completed and planned Phase 3 OPTIMIZE trials for inflammation and pain following cataract surgery will be adequate to support a New Drug Application (NDA) submission, with such a final determination only made by the FDA or non-U.S. regulatory agencies following review of the NDA.
We cannot accurately predict when or if any of our product candidates will prove effective or safe in humans or whether these product candidates will receive marketing approval. Our ability to generate product revenues sufficient to achieve profitability will depend heavily on our obtaining marketing approval for and commercializing OCS-01, OCS-02 (Licaminlimab) or OCS-05.
The success of OCS-01, OCS-02, OCS-05 and other product candidates will depend on many factors, including:
26
If we do not achieve one or more of these factors in a timely manner or at all, we could experience significant delays or an inability to successfully commercialize our product candidates, which would materially harm our business, financial condition, results of operations and growth prospects.
The results of previous clinical trials may not be predictive of future results, and the results of our current and planned clinical trials may not satisfy the requirements of the FDA or non-U.S. regulatory agencies.
The results from the prior preclinical studies and clinical trials for OCS-01, OCS-02 (Licaminlimab) and OCS-05 may not necessarily be predictive of the results of future preclinical studies or clinical trials. Even if we are able to complete our planned clinical trials of our product candidates according to our current development timelines, the results from our prior clinical trials of our product candidates may not be replicated in these future trials. Many companies in the pharmaceutical and biotechnology industries (including those with greater resources and experience than us) have suffered significant setbacks in late-stage clinical trials after achieving positive results in early-stage development, and we cannot be certain that we will not face similar setbacks. These setbacks have been caused by, among other things, preclinical findings made while clinical trials were underway or safety or efficacy observations made in clinical trials, including previously unreported adverse events. Moreover, preclinical and clinical data are often susceptible to varying interpretations and analyses, and many companies that believed their product candidates performed satisfactorily in preclinical studies and clinical trials nonetheless have failed to obtain FDA or non-U.S.-regulatory authority approval. If we fail to produce positive results in our clinical trials of any of our product candidates, the development timelines, regulatory approvals and commercialization prospects for our product candidates, as well as our business and financial prospects, would be adversely affected. For example, in May 2023, we announced topline data for Stage 1 of the DIAMOND Phase 3 clinical trial of OCS-01 in DME and in August 2023, we announced topline data from the OPTIMIZE Phase 3 clinical trial of OCS-01 in cataract surgery. Although OCS-01 met the primary and secondary endpoints in Stage 1 of the DME trial with robust statistical significance, there is no guarantee that these results will be replicated in Stage 2, which is the pivotal part of the trial. Similarly, although OCS-01 met both hierarchical primary endpoints of the OPTIMIZE trial, there is no guarantee that these results will be replicated in the second Phase 3 OPTIMIZE-2 trial that we are conducting.
Further, our product candidates may not be approved even if they achieve their respective primary endpoints in Phase 3 registration trials. The FDA or non-U.S. regulatory agencies may disagree with our trial designs or our interpretation of data from preclinical studies and clinical trials. In addition, any of these regulatory agencies may change requirements for the approval of a product candidate even after reviewing and providing comments or advice on a protocol for a pivotal clinical trial that has the potential to result in approval by the FDA or another regulatory agency. Furthermore, any of these regulatory agencies may also approve our product candidates for fewer or more limited indications than it requests or may grant approval contingent on the performance of costly post-marketing clinical trials.
Some of our clinical data results come from previous trials of less than 100 patients each, including a Phase 2a clinical trial of OCS-02 (Licaminlimab) for the treatment of DED, a Phase 2a clinical trial of OCS-02 for the treatment of non-infectious anterior uveitis, and a Phase 1 dose-ranging study of OCS-05 in healthy volunteers, making it difficult to predict whether the favorable results from such trials will be repeatable in larger, more advanced clinical trials. Moreover, preclinical and clinical data are often susceptible to varying interpretations and analyses, and many companies that have believed their product candidates performed satisfactorily in preclinical studies and clinical trials have nonetheless failed to obtain marketing approval of their products.
We cannot assure you that the FDA or non-U.S. regulatory agencies would consider our completed and planned clinical trials used for an NDA submission or comparable foreign submissions to be sufficient to serve as the basis for approval of our product candidates for any indication. Even if the results of future Phase 3 clinical trials are positive, the FDA and non-U.S. regulatory agencies retain broad discretion in evaluating the results of our clinical trials and in determining whether the results demonstrate that our product candidates are safe and effective. If we are required to conduct clinical trials of our product candidates in addition to those we have planned prior to approval, we will need substantial additional funds, and cannot assure you that the results of any such outcomes trial or other clinical trials will be sufficient for approval.
27
If we experience any or a number of possible unforeseen events in connection with our clinical trials, potential marketing approval or commercialization of our product candidates could be delayed or prevented.
We may experience numerous unforeseen events during, or as a result of, clinical trials that could delay or prevent our ability to receive marketing approval or commercialize any product candidate that we may develop, including:
If we are required to conduct additional clinical trials or other testing of our product candidates beyond those that we currently contemplate, if we are unable to successfully complete clinical trials or other testing of our product candidates, if the results of these trials or other tests are not favorable or are only modestly favorable or if there are safety concerns, we may:
Our product development costs will also increase if we experience delays in testing or marketing approvals. We do not know whether any of our preclinical studies or clinical trials will begin as planned, will need to be restructured or will be completed on schedule, or at all. Significant preclinical or clinical trial delays also could shorten any periods during which we may have the exclusive right to commercialize our product candidates or allow our competitors to bring products to market before we do and impair our ability to successfully commercialize our product candidates.
28
We may be required, or choose, to suspend, vary, repeat or terminate our clinical trials if they are not conducted in accordance with regulatory requirements, the results are negative or inconclusive, the trials are not well-designed, or research participants experience adverse safety outcomes.
Regulatory agencies, IRBs, ethics committees or data safety monitoring boards may at any time recommend the temporary or permanent discontinuation of our clinical trials or request that we vary clinical trials or cease using investigators in the clinical trials if they believe that the clinical trials are not being conducted in accordance with applicable regulatory requirements, or that they present an unacceptable safety risk to participants. Clinical trials must be conducted in accordance with Good Clinical Practices ("GCPs") and other applicable non-U.S. regulatory authority guidelines. Clinical trials are subject to oversight by the FDA, non-U.S. regulatory agencies, IRBs and ethics committees at the study sites where the clinical trials are conducted. In addition, clinical trials must be conducted with product candidates produced in accordance with applicable current good manufacturing practices. Clinical trials may be placed on a full or partial clinical hold by the FDA, non-U.S. regulatory agencies, or us for various reasons, including, but not limited to: deficiencies in the conduct of the clinical trials, including failure to conduct the clinical trial in accordance with regulatory requirements or clinical protocols; deficiencies in the clinical trial operations or trial sites; deficiencies in the trial designs necessary to demonstrate efficacy; fatalities or other adverse effects arising during a clinical trial due to medical problems that may or may not be related to clinical trial treatments; the product candidates may not appear to be more effective than current therapies; or the quality or stability of the product candidates may fall below acceptable standards.
If we elect or are forced to suspend, vary or terminate a clinical trial of any of our current or future product candidates, the commercial prospects for that product may be harmed and our ability to generate product revenue from that product may be delayed or eliminated. Furthermore, any of these events could prevent us or our partners from achieving or maintaining market acceptance of the affected product and could substantially increase the costs of commercializing our product candidates and impair our ability to generate revenue from the commercialization of these products either by us or by our collaboration partners.
Any additional serious adverse events ("SAEs") could result in the FDA or non-U.S. regulatory agencies delaying our clinical trials or denying or delaying clearance or approval of a product. Even though an adverse effect may not be the result of the failure of our drug candidate, the FDA, non-U.S. regulatory agency, IRB or ethics committee could delay or halt a clinical trial for an indefinite period of time while an adverse effect is reviewed, and likely would do so in the event of multiple such events. Any delay or termination of our current or future clinical trials as a result of the risks summarized above, including delays in obtaining or maintaining required approvals from IRBs, or positive opinions from ethics committees, delays in patient enrollment, the failure of patients to continue to participate in a clinical trial, and delays or termination of clinical trials as a result of protocol modifications or adverse effects during the trials, may cause an increase in costs and delays in the submission of any NDAs, to the FDA, or comparable foreign submissions to non-U.S. regulatory agencies, delay the approval and commercialization of our products or result in the failure of the clinical trial, which could adversely affect our business, financial condition, results of operations and growth prospects. Lengthy delays in the completion of clinical trials of our products would adversely affect our business and prospects and could cause us to cease operations.
If preliminary data demonstrate that any of our product candidates has an unfavorable safety profile and is unlikely to receive regulatory approval or be successfully commercialized, we may voluntarily suspend or terminate future development of such product candidate. Any one or a combination of these events could prevent us from obtaining approval and achieving or maintaining market acceptance of the affected product or could substantially increase the costs and expenses of commercializing the product candidate, which in turn could delay or prevent us from generating significant revenues from the sale of the product.
Our product candidates may cause undesirable side effects or have other unexpected properties that could delay or prevent their regulatory approval, limit the commercial profile of an approved label, or result in post-approval regulatory action. OCS-05 was placed on a clinical hold with the FDA in 2016. If we are unable to establish a NOAEL, or if our studies otherwise do not satisfy the FDA’s requirements, OCS-05 may not receive clearance from the FDA to proceed with human clinical trials, may never receive regulatory approval from the FDA, and we may not be able to market and commercialize OCS-05 in the United States, which could materially adversely affect our business, financial condition, results of operations and growth prospects.
29
Unforeseen side effects varying in severity (from minor reactions to death) and frequency (infrequent or prevalent) from OCS-01, OCS-02 (Licaminlimab), or OCS-05 could arise either during clinical development or, if approved, after marketing. Undesirable side effects could cause us, any partners with which we may collaborate, or regulatory agencies to interrupt, extend, modify, delay or halt clinical trials and could result in a more restrictive or narrower label or the delay or denial of regulatory approval by the FDA or comparable foreign agencies.
During the conduct of clinical trials, subjects report changes in their health, including illnesses, injuries, and discomforts, to their study doctor. Often, it is not possible to determine whether or not the product candidate being studied caused these conditions. It is possible that as we test our product candidates in larger, longer and more extensive clinical trials, or as use of these product candidates becomes more widespread if they receive regulatory approval, illnesses, injuries, discomforts and other adverse events that were not observed in earlier trials, as well as conditions that did not occur or went undetected in previous trials, will be reported by subjects. Many times, side effects are only detectable after investigational products are tested in large-scale, Phase 3 clinical trials or, in some cases, after they are made available to subjects on a commercial scale after approval.
If OCS-01, OCS-02 (Licaminlimab) or OCS-05 or any of our other product candidates are associated with SAEs or other undesirable side effects in clinical trials or have characteristics that are unexpected, we may need to abandon their development or limit development to more narrow uses or subpopulations in which the SAEs, undesirable side effects or other characteristics are less prevalent, less severe or more acceptable from a risk-benefit perspective.
In addition, OCS-05 was placed on a clinical hold by the FDA in 2016. We licensed OCS-05 from Accure in 2022. Accure had conducted a limited set of animal regulatory toxicology studies in 2016 and submitted them to the FDA in an IND requesting the initiation of human testing. Upon review, the FDA found the data insufficient and asked for more animal toxicology data to be generated prior to human studies, thereby placing OCS-05 on the regulatory status of “clinical hold” pending the availability of the requested data. In response, Accure chose to withdraw the IND in 2017 rather than invest in further toxicology studies to address the FDA’s request. Upon our license of OCS-05 from Accure in 2022, we reinstated the IND and are currently working on activities to enable a trial under the IND in the U.S., including conducting the necessary toxicology studies. Other regulatory agencies where clinical studies have been proposed, including the UK and France, have authorized us to commence clinical studies of selected doses and reinforced safety measures as in our European Phase 1 trial in AON. If our studies do not satisfy the FDA’s requirements, OCS-05 may not receive clearance from the FDA to proceed with human clinical trials, may never receive regulatory approval from the FDA, and we may be unable to market and commercialize OCS-05 in the United States, and our business, financial condition, results of operations and growth prospects could be materially adversely affected.
Results of clinical trials could reveal a high and unacceptable severity and prevalence of side effects. In such an event, trials could be suspended, varied or terminated, and the FDA or comparable non-U.S. regulatory agencies could order us to cease further development of or deny approval of a product candidate for any or all targeted indications. Such adverse event findings also could require us or our collaboration partners to perform additional studies or halt development or sale of these product candidates or expose us to product liability lawsuits which would harm our business, financial condition, results of operations and growth prospects. In such an event, we could be required by the FDA or other comparable regulatory agencies to conduct additional animal or human studies regarding the safety and efficacy of our product candidates which we have not planned or anticipated or our studies could be suspended, varied or terminated, and the FDA or comparable regulatory agencies could order us to cease further development of or deny, vary, or withdraw approval of our product candidates for any and all intended indications. The drug-related side effects could also affect patient recruitment or the ability of enrolled patients to complete the trial. There can be no assurance that we will resolve any issues related to any product-related adverse events to the satisfaction of the FDA or any comparable regulatory agency in a timely manner, if ever, and any of these occurrences may harm our business, financial condition, results of operations and prospects.
Additionally, if we or others identify undesirable side effects, or other previously unknown problems, caused by a product after obtaining U.S. or non-U.S. regulatory approval, a number of potentially negative consequences could result, including but not limited to, regulatory agencies suspending, withdrawing or varying approvals of such product, regulatory agencies requiring additional warnings on the label or otherwise requiring labeling to be updated or narrowed, us becoming liable for harm caused to patients and the diminution of our reputation, which could prevent us or our potential partners from achieving or maintaining market acceptance of the product candidate, if approved, and could substantially increase the costs of commercializing such product, which would have a material adverse effect on our business, results of operation, financial condition and prospects.
30
If any of our product candidates receives approval, regulatory agencies including the FDA and other non-U.S. regulatory agencies will require that we regularly report certain information, including information about adverse events that may have caused or contributed by those products. The timing of adverse event reporting obligations would be triggered by the date we become aware of the adverse event as well as the nature of the event. We may fail to report adverse events we become aware of within the prescribed timeframe especially if it is not reported to us as an adverse event or if it is an adverse event that is unexpected or removed in time from the use of our products. If we fail to comply with our reporting obligations, the FDA or other regulatory agencies could take action that may include criminal prosecution, the imposition of civil monetary penalties, seizure of our products, or suspension of market approval, and delay in approval or clearance of future products.
Interim, topline and preliminary data from our clinical trials may change as more patient data becomes available and are subject to audit and verification procedures that could result in material changes in the final data.
From time to time, we may publicly disclose preliminary, interim or topline data from our clinical trials. These interim updates are based on a preliminary analysis of then-available data, and the results and related findings and conclusions may be subject to change following a more comprehensive review of the data. We also may use assumptions and estimates as part of our preliminary analyses of the data, and we may not have received or had the opportunity to fully and carefully evaluate all data. Topline data also remain subject to audit and verification procedures before they can be finalized. In addition, the information we choose to publicly disclose regarding a particular study or clinical trial is typically selected from a more extensive amount of available information. For example, we may report interim analyses of only certain of the endpoints of the clinical trial, rather than all of the endpoints. Additional disclosure of interim data by us or by our competitors in the future could result in volatility in the price of Ordinary Shares. Further, investors may not agree with what we determine is the material or otherwise appropriate information to include in our public disclosures, and any information we determine not to disclose may ultimately be deemed significant by us or, if subsequently disclosed, by investors, with respect to future decisions, conclusions, views, activities or otherwise regarding a particular product candidate or our business. Further, others, including regulatory agencies and investors may not accept our conclusions regarding such preliminary or interim analyses, which could impact the value of a particular program or the approval or commercialization of the particular product candidate, or result in volatility in the price of Ordinary Shares.
The topline results that we report may differ significantly from the final results of the same studies, or different conclusions or considerations may qualify such results, once additional data have been received and fully evaluated. As a result, topline and interim data from clinical trials are subject to the risk that one or more of the reported clinical outcomes may materially change, and should be viewed with caution until the final data are available. For example, in May 2023, we announced topline data for Stage 1 of the DIAMOND Phase 3 clinical trial of OCS-01 in DME and in August 2023, we announced topline data from the OPTIMIZE Phase 3 clinical trial of OCS-01 in cataract surgery. These topline results may differ from the final results of the DIAMOND and OPTIMIZE trials. As a result, topline and interim data from clinical trials are subject to the risk that one or more of the reported clinical outcomes may materially change, and should be viewed with caution until the final data are available. If the preliminary or topline data that we report differ from the final results, or if others, including regulatory agencies, disagree with our conclusions, then our ability to obtain approval for, and to successfully commercialize our product candidates may be harmed, which could materially affect our business, financial condition, results of operations and growth prospects.
We may encounter substantial delays in our clinical trials, or may not be able to conduct or complete our clinical trials on the timelines we expect, if at all.
Clinical testing is expensive, time consuming, and subject to uncertainty. We cannot guarantee that any clinical trials will be conducted as planned or completed on schedule, if at all. We cannot be sure that submission of an investigational new drug application ("IND"), or a clinical trial application ("CTA"), will result in the FDA or comparable non-U.S. regulatory agencies, or any other regulatory authority as applicable, allowing clinical trials to begin in a timely manner, if at all. Moreover, even if these trials begin, issues may arise that could suspend or terminate such clinical trials. A failure of one or more clinical trials can occur at any stage of testing, and our future clinical trials may not be successful.
31
Any difficulties we experience relating to the initiation or completion of patient visits in clinical trials, could delay regulatory approval for our product candidates. Identifying and qualifying subjects to participate in clinical trials of our product candidates is critical to our success. The timing of clinical trials depends on our ability to recruit subjects to participate, as well as the completion of required follow-up periods. Patients may be unwilling to participate in clinical trials because of negative publicity from adverse events related to the biotechnology or pharmaceutical fields, competitive clinical trials for similar patient populations, the existence of current treatments or for other reasons. The timeline for recruiting patients, conducting studies and obtaining regulatory approval of our product candidates may be delayed, which could result in increased costs, delays in advancing our product candidates, delays in testing the effectiveness of our product candidates or termination of the clinical trials altogether. Patient enrollment for any of our future clinical trials may be affected by other factors, including:
32
Any inability to successfully initiate or complete clinical trials could result in additional costs to us or impair our ability to generate revenue. In addition, if we make manufacturing or formulation changes to our product candidates, we may be required to or we may elect to conduct additional studies to bridge our modified product candidates to earlier versions. Clinical trial delays could also shorten any periods during which our products have patent protection and may allow our competitors to bring products to market before we do, which could impair our ability to successfully commercialize our product candidates and may harm our business and results of operations.
We could also encounter delays if a clinical trial is suspended or terminated by us, by the data safety monitoring board for such trial or by the FDA, or comparable non-U.S. regulatory agencies, or any other regulatory authority, or if the IRBs or ethics committees of the institutions in which such trials are being conducted suspend or terminate the participation of their clinical investigators and sites subject to their review. Such agencies may suspend, vary or terminate a clinical trial due to a number of factors, including failure to conduct the clinical trial in accordance with regulatory requirements or our clinical protocols, inspection of the clinical trial operations or trial site by the FDA, or comparable non-U.S. regulatory agencies, or other regulatory agencies resulting in the imposition of a clinical hold, unforeseen safety issues or adverse side effects, failure to demonstrate a benefit from using a product candidate, changes in governmental regulations or administrative actions or lack of adequate funding to continue the clinical trial.
Delays in the commencement or completion of any clinical trial of our product candidates will increase our costs, slow down our product candidate development and approval process and delay or potentially jeopardize our ability to commence product sales and generate revenue. In addition, many of the factors that cause, or lead to, a delay in the commencement or completion of clinical trials may also ultimately lead to the denial of regulatory approval of our product candidates.
We do, and may in the future, conduct clinical trials for our product candidates outside the United States, and the FDA and applicable non-U.S. regulatory agencies may not accept data from such trials.
We and investigator sponsors have conducted clinical trials, are conducting clinical trials, and may in the future choose to conduct one or more clinical trials outside of the United States. Although the FDA or applicable non-U.S. regulatory agencies may accept data from clinical trials conducted outside the United States or the applicable jurisdiction, acceptance of such study data by the FDA or applicable non-U.S. regulatory agency may be subject to certain conditions or exclusions. Where data from foreign clinical trials are intended to serve as the basis for marketing approval in the United States, the FDA will not approve the application on the basis of foreign data alone unless such data are applicable to the U.S. population and U.S. medical practice; the studies were performed by clinical investigators of recognized competence; and the data are considered valid without the need for an on-site inspection by the FDA or, if the FDA considers such an inspection to be necessary, the FDA is able to validate the data through an on-site inspection or other appropriate means. Many non-U.S. regulatory agencies have similar requirements. In addition, such non-U.S. studies would be subject to the applicable local laws of the jurisdictions where the studies are conducted.
33
There can be no assurance the FDA or applicable non-U.S. regulatory agency will accept data from trials conducted outside of the United States or the applicable home country. If the FDA or applicable non-U.S. regulatory agency does not accept such data, it would likely result in the need for additional trials, which would be costly and time-consuming and delay aspects of our business plan.
We rely on and expect to continue to rely on third-party CROs and other third parties to conduct and oversee our clinical trials. If these third parties do not meet our requirements or otherwise conduct clinical trials as required, we may not be able to satisfy our contractual obligations or obtain regulatory approval for, or commercialize, our product candidates.
We rely on, and expect to continue to rely on, third-party CROs to conduct and oversee our clinical trials and other aspects of product development. We also expect to rely on various medical institutions, clinical investigators and contract laboratories to conduct our trials in accordance with our clinical protocols and applicable regulatory requirements, including the FDA’s regulations and good clinical practice, or GCP requirements, and equivalent non-U.S. and international standards, which are international standards meant to protect the rights and health of patients and to define the roles of clinical trial sponsors, administrators and monitors, and national, supranational, and state regulations governing the handling, storage, security and recordkeeping for drug and biologic products. These CROs and other third parties are expected to play a significant role in the conduct of these trials and the subsequent collection and analysis of data from the clinical trials. We expect to rely heavily on these parties for the execution of our clinical trials and preclinical studies and will control only certain aspects of their activities. We and our CROs and other third-party contractors will be required to comply with GCP and good laboratory practice ("GLP"), requirements, which are regulations and guidelines enforced by the FDA and comparable non-U.S. regulatory agencies. Regulatory agencies enforce these GCP and GLP requirements through periodic inspections of trial sponsors, principal investigators and trial sites. If we or any of these third parties fail to comply with applicable GCP and GLP requirements, or reveal noncompliance from an audit or inspection, the clinical data generated in our clinical trials may be deemed unreliable and the FDA or other comparable non-U.S. regulatory agencies may require us to perform additional clinical trials before approving our or our partners’ marketing applications. We cannot provide assurance that upon inspection by a given regulatory authority, such regulatory authority will determine whether or not any of our clinical or preclinical trials comply with applicable GCP and GLP requirements. In addition, our clinical trials generally must be conducted with product produced under cGMP regulations. Our failure to comply with these regulations and policies may require us to repeat clinical trials, which would delay the regulatory approval process, and adversely affect our operations.
If any of our CROs or clinical trial sites terminate their involvement in one of our clinical trials for any reason, we may not be able to enter into arrangements with alternative CROs or clinical trial sites or do so on commercially reasonable terms. In addition, if our relationship with clinical trial sites is terminated, we may experience the loss of follow-up information on patients enrolled in our ongoing clinical trials unless we are able to transfer the care of those patients to another qualified clinical trial site. In addition, principal investigators for our clinical trials may serve as scientific advisors or consultants to it from time to time and could receive cash or equity compensation in connection with such services. If these relationships and any related compensation result in perceived or actual conflicts of interest, the integrity of the data generated at the applicable clinical trial site may be questioned by the FDA and comparable non-U.S. regulatory agencies, which could delay the regulatory approval process and adversely affect our operations.
Even if we obtain regulatory approval for a product candidate, our products will remain subject to continuous subsequent regulatory obligations and scrutiny.
If our product candidates are approved, they will be subject to ongoing regulatory requirements for pharmacovigilance, manufacturing, labeling, packaging, storage, advertising, promotion, sampling, record-keeping, conduct of post-marketing studies (if any) and submission of other post-market information, including both federal and state requirements in the United States and equivalent requirements of comparable regulatory agencies.
Manufacturers and manufacturers’ facilities are required to comply with extensive FDA, and comparable regulatory authority requirements, including ensuring that quality control and manufacturing procedures conform to cGMP regulations. As such, we and our contract manufacturers will be subject to continual review and inspections to assess compliance with cGMP regulations and adherence to commitments made in any marketing authorization application ("MAA"). Accordingly, we and others with whom we work must continue to expend time, money and effort in all areas of regulatory compliance, including manufacturing, production and quality control.
34
Any regulatory approvals that we or our collaboration partners receive for our product candidates may be subject to limitations on the approved conditions of use for which the product may be marketed or to the conditions of approval or may contain requirements for potentially costly additional data generation, including clinical trials. We will be required to report certain adverse reactions and production problems, if any, to the FDA and comparable regulatory agencies, and to conduct surveillance to monitor the safety and efficacy of the product candidate. Any new legislation addressing drug safety or biologics issues could result in delays in product development or commercialization or increased costs to assure compliance.
We will have to comply with requirements concerning advertising and promotion for our product candidates, if approved. Promotional communications with respect to prescription drugs are subject to a variety of legal and regulatory restrictions that vary throughout the world and must be consistent with the information in the product’s approved label. As such, we may promote our products in ways that are not consistent with FDA-approved labeling, e.g., for indications or uses for which they do not have approval.
If a regulatory authority discovers previously unknown problems with one of our products such as adverse events of unanticipated severity or frequency, or if there are problems with the facility where the product is manufactured or the regulatory authority disagrees with the advertising, promotion, marketing or labeling of a product, such regulatory agency may impose restrictions on that product or us. If we fail to comply with applicable regulatory requirements, a regulatory authority such as FDA may, among other things:
Any government investigation of alleged violations of law or regulations could require us to expend significant time and resources in response and could generate negative publicity. Any failure to comply with ongoing regulatory requirements may significantly and adversely affect our ability to commercialize and generate revenue from our products. If regulatory sanctions are applied or if regulatory approval is suspended, varied or withdrawn, the value of us and our operating results will be adversely affected.
If we are not successful in discovering, developing, and commercializing additional product candidates beyond our current portfolio, our ability to expand our business and achieve our strategic objectives would be impaired.
A key element of our strategy is to discover, develop, and potentially commercialize additional product candidates beyond our current portfolio to treat various conditions in a variety of therapeutic areas. We intend to do so by investing in our own drug discovery efforts, exploring potential strategic alliances for the development of new products, and in-licensing technologies. Identifying new product candidates requires substantial technical, financial, and human resources. We may fail to identify promising product candidates and, even if we do identify such product candidates, we may fail to successfully develop and commercialize such product candidates for many reasons, including:
35
We have several early-stage programs in preclinical development as we seek to expand our pipeline. Preclinical development programs in the biotechnology industry carry high risk of failure. If any of these programs fails due to, among others, adverse formulation, pharmacokinetic, pharmacodynamics, or safety, we may need to terminate the program. If we are unsuccessful in identifying and developing additional product candidates and progressing those into clinical development, our potential for growth may be impaired.
We may expend our limited resources to pursue a particular product candidate or indication and fail to capitalize on product candidates or indications that may be more profitable or for which there is a greater likelihood of success.
Because we have limited financial and managerial resources, we focus on research programs and product candidates that we identify for specific indications. As a result, we may forego or delay pursuit of opportunities with other product candidates or for other indications that later prove to have greater commercial potential. Our resource allocation decisions may cause us to fail to capitalize on viable commercial products or profitable market opportunities. Our spending on current and future research and development programs and product candidates for specific indications may not yield any commercially viable products. If we do not accurately evaluate the commercial potential or target market for a particular product candidate, we may relinquish valuable rights to that product candidate through collaboration, licensing or other royalty arrangements in cases in which it would have been more advantageous for us to retain sole development and commercialization rights to such product candidate. As a result of the foregoing, our business, operations and prospects could be materially adversely affected.
We may choose to discontinue developing or commercializing any of our product candidates, or may choose not to commercialize product candidates in approved indications, at any time during development or after approval, which would reduce or eliminate our potential return on investment for those product candidates.
At any time, we may decide to discontinue the development of any of our product candidates for a variety of reasons, including the appearance of new technologies that make our product candidates obsolete, competition from a competing product, cost concerns, manufacturing challenges, analysis of preclinical and clinical trial results or changes in or failure to comply with applicable regulatory requirements. If we terminate a program in which we have invested significant resources, we will not receive any return on our investment and we will have missed the opportunity to have allocated those resources to potentially more productive uses. As a result, our business, financial condition, results of operations and growth prospects may be adversely affected.
Risks related to our manufacturing activities
We have no experience manufacturing any of our product candidates at a commercial scale. If we or any of our third-party manufacturers encounter difficulties in production, or fail to meet rigorously enforced regulatory standards, our ability to provide supply of our product candidates for clinical trials or our products for patients, if approved, could be delayed or stopped, or we may be unable to establish a commercially viable cost structure.
36
In order to conduct clinical trials of our product candidates, or supply commercial products, if approved, we need to manufacture them in small and large quantities. The manufacturing processes for OCS-02 (Licaminlimab) and OCS-05 have never been tested at commercial scale, and the process validation requirement (the requirement to consistently produce the active pharmaceutical ingredient used in these drug candidates in commercial quantities and of specified quality on a repeated basis and document our ability to do so) for each of OCS-01, OCS-02, and OCS-05 has not yet been satisfied. Our manufacturing partners may be unable to successfully increase the manufacturing capacity for any of our product candidates in a timely or cost-effective manner, or at all. In addition, quality issues may arise during scale-up activities. If our manufacturing partners are unable to successfully scale up the manufacture of our product candidates in sufficient quality and quantity, the development, testing and clinical trials of our product candidates may be delayed or become infeasible, and regulatory approval or commercial launch of any resulting product may be delayed or not obtained, which could significantly harm our business. The same risks would apply to any internal manufacturing facilities, should we in the future decide to build internal manufacturing capacity.
In addition, the manufacturing process for any products that we may develop is subject to FDA, competent agencies of EU member states, NMPA and other non-U.S. regulatory authority approval processes and continuous oversight. We will need to contract with manufacturers who can meet all applicable FDA, European Commission, EMA, NMPA and other non-U.S. regulatory authority requirements, including complying with cGMPs on an ongoing basis. If we or our third-party manufacturers are unable to reliably produce products to specifications acceptable to the FDA, EU, NMPA or other regulatory agencies, we may not obtain or maintain the approvals we need to commercialize such products. Even if we obtain regulatory approval for any of our product candidates, there is no assurance that either we or our CMOs will be able to manufacture the approved product to specifications acceptable to the FDA, European Commission, EMA, NMPA or other regulatory agencies, to produce it in sufficient quantities to meet the requirements for the potential launch of the product, or to meet potential future demand. Any of these challenges could delay completion of clinical trials, require bridging clinical trials or the repetition of one or more clinical trials, increase clinical trial costs, delay approval of our product candidate, impair commercialization efforts, increase our cost of goods, and have an adverse effect on our business, financial condition, results of operations and growth prospects.
In the event that we need to change our CMOs, our clinical trials or the commercialization of our product candidates could be delayed, adversely affected or terminated, or such a change may result in significantly higher costs.
Various steps in the manufacture of our product candidates may need to be sole-sourced. In accordance with cGMP, changing manufacturers may require the re-validation of manufacturing processes and procedures, and may require further clinical trials to show comparability between the materials produced by different manufacturers. Changing our current or future CMOs may be difficult for us and could be costly, which could result in our inability to manufacture our product candidates for an extended period of time and therefore a delay in the development of our product candidates. Further, in order to maintain our development time lines in the event of a change in our CMOs, we may incur significantly higher costs to manufacture our product candidates
The manufacture of OCS-02 (Licaminlimab), a biologic, is highly complex, costly and requires substantial lead time to produce.
Manufacturing OCS-02 (Licaminlimab), a biologic, involves complex processes, including developing cells or cell systems to produce the biologic, growing large quantities of such cells, and harvesting and purifying the biologic produced by them. These processes require specialized facilities, highly specific raw materials and other production constraints. As a result, the cost to manufacture a biologic is generally far higher than traditional small molecule chemical compounds, and the biologics manufacturing process is less reliable and is difficult to reproduce. Because of the complex nature of this product candidate, we need to oversee manufacture of multiple components that require a diverse knowledge base and specialized personnel.
Moreover, unlike chemical pharmaceuticals, the physical and chemical properties of a biologic such as OCS-02 generally cannot be adequately characterized prior to manufacturing the final product. As a result, an assay of the finished product is not sufficient to ensure that the product will perform in the intended manner. Accordingly, we expect to employ multiple steps to attempt to control our manufacturing process to assure that the process works and the product or product candidate is made strictly and consistently in compliance with the process.
37
Manufacturing biologics is highly susceptible to product loss due to contamination, equipment failure, improper installation or operation of equipment, vendor or operator error, improper storage or transfer, inconsistency in yields and variability in product characteristics. Even minor deviations from normal manufacturing, distribution or storage processes could result in reduced production yields, product defects and other supply disruptions. Some of the raw materials required in our manufacturing process are derived from biological sources. Such raw materials are difficult to procure and may also be subject to contamination or recall. A material shortage, contamination, recall or restriction on the use of biologically derived substances in the manufacture of our product candidates could adversely impact or disrupt commercialization. Production of additional drug substance and drug product for OCS-02 may require substantial lead time. In the event of significant product loss and materials shortages, we may be unable to produce adequate amounts of our product candidates or products for our operational needs, which would materially adversely affect our business, financial condition and results of operations.
Further, as product candidates are developed through preclinical studies to late-stage clinical trials towards approval and commercialization, it is common that various aspects of the development program, such as manufacturing methods, are altered along the way in an effort to optimize processes and results. Such changes carry the risk that they will not achieve these intended objectives, and any of these changes could cause our product candidates to perform differently and affect the results of planned clinical trials or other future clinical trials. We and our third-party manufacturing partner are engaged in efforts to reduce the expected costs for OCS-02. In the future, if the proposed manufacturing plans to reduce OCS-02 costs does not succeed when producing OCS-02 at commercial scale, we may not be able to proceed with OCS-02 commercialization, if approved.
Any of the foregoing could potentially materially adversely affect our business, financial condition, results of operations and growth prospects.
Risks related to our future commercialization activities
Even if a product candidate obtains regulatory approval, it may fail to achieve the broad degree of physician and patient adoption and use necessary for commercial success.
The commercial successes of OCS-01, OCS-02 (Licaminlimab) or OCS-05, if approved, will depend significantly on attaining broad adoption and use of the products by physicians and patients for approved indications, and any of these product candidates may not be commercially successful even if shown to be effective in clinical trials. The degree and rate of physician and patient adoption of a product, if approved, will depend on a number of factors, including but not limited to:
38
Even if we receive marketing approvals for OCS-01, OCS-02 (Licaminlimab), OCS-05 or any future product candidate, we may not be able to successfully commercialize our product candidates due to unfavorable pricing regulations or third-party coverage and reimbursement policies, which could make it difficult for us to sell our product candidates profitably.
Obtaining coverage and reimbursement approval for a product from a government or other third-party payor is a time consuming and costly process that could require us to provide supporting scientific, clinical and cost effectiveness data to the payor. There may be significant delays in obtaining such coverage and reimbursement for newly approved products, and coverage may be more limited than the purposes for which the product is approved by the FDA or comparable non-U.S. regulatory agencies. Moreover, eligibility for coverage and reimbursement does not imply that a product will be paid for in all cases or at a rate that covers costs, including research, development, intellectual property, manufacture, sale and distribution expenses. Interim reimbursement levels for new products, if applicable, may also not be sufficient to cover costs and may not be made permanent. Reimbursement rates may vary according to the use of the product and the clinical setting in which it is used, may be based on reimbursement levels already set for lower cost products and may be incorporated into existing payments for other services. Net prices for products may be reduced by mandatory discounts or rebates required by government healthcare programs or private payors, by any future laws limiting drug prices and by any future relaxation of laws that presently restrict imports of product from countries where they may be sold at lower prices than in the United States.
There is significant uncertainty related to the insurance coverage and reimbursement of newly approved products. Third-party payors in the United States often rely upon Medicare coverage policy and payment limitations in setting reimbursement policies, but also have their own methods and approval process apart from Medicare coverage and reimbursement determinations. Pricing and reimbursement outside of the United States vary widely and are constantly evolving, with requirements and limitations becoming increasingly strict.
Coverage and reimbursement by a third-party payor or competent foreign authority may depend upon a number of factors, including the third-party payor’s or competent foreign authority’s determination that use of a product is:
We cannot be sure that coverage and reimbursement will be available for any product that we commercialize and, if coverage and reimbursement are available, what the level of reimbursement will be. Our inability to promptly obtain coverage and adequate reimbursement rates from both government-funded and private payors for any approved products that we develop could have a material adverse effect on our operating results, our ability to raise capital needed to commercialize products and our overall financial condition.
Reimbursement may impact the demand for, and the price of, any product for which we obtain marketing approval. Assuming we obtain coverage for a given product by a third-party payor, the resulting reimbursement payment rates may not be adequate or may require co-payments that patients find unacceptably high.
39
Patients who are prescribed medications for the treatment of their conditions, and their prescribing physicians, generally rely on third-party payors or competent foreign authorities to reimburse all or part of the costs associated with those medications. Patients are unlikely to use our products unless coverage is provided and reimbursement is adequate to cover all or a significant portion of the cost of our products. Therefore, coverage and adequate reimbursement is critical to new product acceptance. Coverage decisions may depend upon clinical and economic standards that disfavor new products when more established or lower cost therapeutic alternatives are already available or subsequently become available. Coverage policies and third-party reimbursement rates may change at any time. Even if favorable coverage and reimbursement status is attained for one or more products for which we receive regulatory approval, less favorable coverage policies and reimbursement rates may be implemented in the future.
We expect to experience pricing pressures in connection with the sale of any of our product candidates due to the trend toward managed healthcare, the increasing influence of health maintenance organizations, and additional legislative changes. The downward pressure on healthcare costs in general, particularly prescription medicines, medical devices and surgical procedures and other treatments, has become very intense. As a result, increasingly high barriers are being erected to the successful commercialization of new products. Further, the adoption and implementation of any future governmental cost containment or other health reform initiative may result in additional downward pressure on the price that we may receive for any approved product.
Outside of the United States, many countries require approval of the sale price of a product before it can be marketed and the pricing review period only begins after marketing approval is granted. To obtain reimbursement or pricing approval in some of these countries, we may be required to conduct a clinical trial that compares the cost-effectiveness of our product candidate to other available therapies. In some foreign markets, prescription pharmaceutical pricing remains subject to continuing governmental control even after initial approval is granted. As a result, we might obtain marketing approval for a product candidate in a particular country, but then be subject to price regulations that delay our commercial launch of the product, possibly for lengthy time periods, and negatively impact the revenues, if any, we are able to generate from the sale of the product in that country. Adverse pricing limitations may hinder our ability to recoup our investment in one or more product candidates, even if such product candidates obtain marketing approval.
If, in the future, we are unable to establish sales and marketing capabilities or enter into agreements with third parties to sell and market any product candidates we may develop, we may not be successful in commercializing those product candidates if and when they are approved.
We do not have a sales or marketing infrastructure and have no experience in the sale, marketing or distribution of pharmaceutical products. To achieve commercial success for any approved product for which we retain sales and marketing responsibilities, we must either develop a sales and marketing organization or outsource these functions to third parties. In the future, we may choose to build a focused sales, marketing and commercial support infrastructure to sell, or participate in sales activities with our collaborators for, some of our product candidates if and when they are approved.
There are risks involved with both establishing our own commercial capabilities and entering into arrangements with third parties to perform these services. For example, recruiting and training a sales force or reimbursement specialists is expensive and time consuming and could delay any product launch. If the commercial launch of a product candidate for which we recruit a sales force and establish marketing and other commercialization capabilities is delayed or does not occur for any reason, we would have prematurely or unnecessarily incurred these commercialization expenses. This may be costly, and our investment would be lost if we cannot retain or reposition our commercialization personnel.
Factors that may inhibit our efforts to commercialize any approved product on our own include:
40
If we enter into arrangements with third parties to perform sales, marketing, commercial support and distribution services, our product revenue or the profitability of product revenue may be lower than if we were to market and sell any products we may develop ourselves. In addition, we may not be successful in entering into arrangements with third parties to commercialize our product candidates or may be unable to do so on terms that are favorable to us. We may have little control over such third parties, and any of them may fail to devote the necessary resources and attention to sell and market our products effectively. If we do not establish commercialization capabilities successfully, either on our own or in collaboration with third parties, we will not be successful in commercializing our product candidates if approved, which would materially adversely affect our business, results of operations, financial condition and growth prospects.
We face substantial competition, which may result in others discovering, developing or commercializing products before or more successfully than we do.
The development and commercialization of new drug products are highly competitive. We face competition with respect to our product candidates that we may seek to develop or commercialize, from major pharmaceutical companies, specialty pharmaceutical companies and biotechnology companies worldwide. Potential competitors may also include academic institutions, government agencies and other public and private research organizations that conduct research, seek patent protection and establish collaborative arrangements for research, development, manufacturing and commercialization.
The DME market is already served by multiple approved products, such as ranimizumab, aflibercept, brolucizumab, faricimab VEGF inhibitors as well as dexamethasone and fluocinolone acetonide intravitreal implants. These drugs are well established therapies and are widely accepted by physicians, patients and third-party payors, which may make it difficult to convince these parties to switch to OCS-01. Companies that we are aware are commercializing or are developing therapeutics for DME include large companies with significant financial resources, such as Roche (Genentech), Novartis, Bayer, Regeneron, Abbvie (Allergan) and Alimera Sciences, among others. In addition, OCS-01 will compete with the current status quo practice of treating DME, which is often observing and not treating milder patients before they often progress to invasive treatments.
The post-operative inflammation and pain market is already served by multiple approved steroid products, such as difluprednate ophthalmic emulsion, loteprednol etabonate ophthalmic gel and suspension, prednisolone acetate ophthalmic suspension, among others. These drugs are well established therapies with multiple generics in the market and are widely accepted by physicians, patients and third-party payors, which may make it difficult to convince these parties to switch to OCS-01. Companies that we are aware are commercializing or are developing therapeutics for post-operative inflammation and pain include large companies with significant financial resources, such as Bausch + Lomb, Kala Pharmaceuticals, Alcon Laboratories, Abbvie (Allergan), TEVA Pharmaceuticals, among others.
The DED market is already served by multiple approved products, such as cyclosporine ophthalmic emulsion and solution, lifitegrast ophthalmic solution, loteprednol etabonate ophthalmic suspension, varenicline solution and perfluorohexyloctane ophthalmic solution. These drugs are well established therapies and are widely accepted by physicians, patients and third-party payors. This, as well as the emerging development of generics, may make it difficult to convince these parties to switch to OCS-02 (Licaminlimab). Companies that we are aware are commercializing or are developing therapeutics for DED include large companies with significant financial resources, such as Abbvie (Allergan), Bausch + Lomb, Alcon, Sun Pharmaceuticals and Viatris, among others. In addition, over the counter products are currently available for the treatment of DED which may impact sales of our products.
The non-infectious anterior uveitis market is already served by multiple approved steroid products indicated to treat inflammation of the eyes, such as prednisolone acetate suspension, loteprednol etabonate ophthalmic formulations, dexamethasone sodium phosphate formulations, fluorometholone ophthalmic suspension, among others.
41
These drugs are well established therapies with multiple generics in the market and are widely accepted by physicians, patients and third-party payors, which may make it difficult to convince these parties to switch to OCS-02 (Licaminlimab). Companies that we are aware are commercializing or are developing therapeutics for non-infectious anterior uveitis include large companies with significant financial resources, such as Abbvie (Allergan) and Bausch + Lomb, among others.
The glaucoma market is already served by multiple approved drug classes to reduce elevated intraocular pressure ("IOP"), such as Alpha Agonists, Beta Blockers Carbonic Anhydrase Inhibitors, Cholinergic (Myotic), Prostaglandin Analogs, Rho Kinase Inhibitors and combination products, however no drug for neuroprotection has been approved so far. These drugs are well established therapies with multiple generics in the market and are widely accepted by physicians, patients and third-party payors. OCS-05 is not meant to replace IOP lowering but rather be an add-on to IOP lowering to tackle neuroprotection. Companies that we are aware are commercializing or are developing therapeutics for glaucoma include large companies with significant financial resources, such as Novartis, Abbvie (Allergan), Bausch + Lomb, Alcon, Akorn, Teva Pharmaceuticals, Pfizer, Merck and Sun Ophthalmics among others.
In addition to competition from other companies targeting the diseases which we target, any products we may develop may also face competition from other types of therapies, such as gene-editing therapies or drug delivery devices. Our commercial opportunity for any of our product candidates could also be reduced or eliminated if our competitors develop and commercialize new products that are safer, more effective, are more convenient, or are less expensive than our products. The competitors also may obtain FDA or other non-U.S. regulatory approval for their products more rapidly than we may obtain approval for our candidates, which could result in competitors establishing a strong market position before we are able to enter the market for a new product candidate. If our product candidates are not perceived as more effective, safe, cost-effective, or otherwise medically beneficial than current practices or products in their respective target market segments, then our commercial opportunities will be negatively impacted. If we are unable to demonstrate the value of our product candidates based on our clinical data, patient experience, or real-world evidence, future successful commercialization of such product candidates could be adversely affected.
In addition, our ability to compete may be affected in many cases by insurers or other third-party payors, including Medicare and equivalent foreign health insurance programs, seeking to encourage the use of generic products. For example, a generic version of Restasis® to treat DED received FDA approval and was launched in 2022. Generic products are generally offered at lower prices than branded products, and consequently, after the introduction of a generic competitor, a significant percentage of the sales of any branded product may be lost to the generic product. Accordingly, competition from generic products could have a material adverse impact on our ability to successfully commercialize OCS-02 (Licaminlimab) for DED or any other product candidate or indication, if approved, or negatively impact sales or pricing of our products or our ability to gain market acceptance or market share.
Many of our current and future competitors have significantly greater financial resources and expertise in research and development, manufacturing, preclinical testing, conducting clinical trials, obtaining regulatory approvals and marketing approved products than we do. Mergers and acquisitions in the pharmaceutical and biotechnology industries may result in even more resources being concentrated among a smaller number of our competitors. Smaller and other early-stage companies may also prove to be significant competitors, including through collaborative arrangements with large and established companies. These third parties compete with us in recruiting and retaining qualified scientific and management personnel, establishing clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies complementary to, or necessary for, our programs.
Product liability lawsuits against us could cause us to incur substantial liabilities and to limit commercialization of any products that we develop.
We face an inherent risk of product liability exposure related to the use of our product candidates that we develop in clinical trials. We face an even greater risk for any products we develop and sell commercially. Off-label use or misuse of our products if and when commercialized may harm our reputation in the marketplace, result in injuries that lead to costly product liability suits, or subject us to penalties if we fail to comply with regulatory requirements or experience unanticipated problems with any product. If we cannot successfully defend ourselves against claims that our product candidates or products caused injuries, we will incur substantial liabilities. Regardless of merit or eventual outcome, liability claims may result in:
42
We may need to purchase insurance coverage as we expand our clinical trials and should we eventually realize sales of any product candidate for which we obtain marketing approval. Insurance coverage is increasingly expensive, restrictive and narrow. We may not be able to maintain insurance coverage at a reasonable cost, upon adequate terms or in a sufficient amount necessary to protect us against losses due to product liability or other similar legal actions that may arise. A successful product liability claim or series of claims brought against us which substantially exceeds our insurance coverage will require us to make up the shortfall, which may in turn require us to drawdown on our cash reserve, and harm our business, financial condition, results of operations and growth prospects.
Risks related to our reliance on third parties
We may enter into collaborations with third parties for the development and commercialization of our product candidates. If our collaborations are not successful, we may not be able to capitalize on the market potential of these product candidates.
We may enter into a combination of exclusive and non-exclusive collaboration arrangements with third parties to develop or commercialize some or all of our product candidates. We also may enter into arrangements with third parties to perform these services in the United States and other jurisdictions if we do not establish our own sales, marketing and distribution capabilities in the United States and other jurisdictions for our product candidates or if we determine that such arrangements are otherwise beneficial. We also may seek collaborators for development and commercialization of other product candidates. Our likely collaborators for any sales, marketing, distribution, development, licensing or broader collaboration arrangements include large and mid-size pharmaceutical companies, regional and national pharmaceutical companies and biotechnology companies. While we are not currently party to any such arrangement, our ability to generate revenues from these arrangements will depend on our collaborators’ abilities and efforts to successfully perform the functions assigned to them in the future in these arrangements.
Collaborations that we enter into may pose a number of risks, including the following:
43
Collaboration agreements may not lead to development or commercialization of product candidates in the most efficient manner, or at all. If any collaborations that we enter into do not result in the successful development and commercialization of products or if one of our collaborators terminates its agreement with us, we may not receive any future research funding or milestone or royalty payments, or be able to recover any costs and expenses incurred by us under the collaboration arrangement. If we do not receive the funding we expect, or recover any costs and expenses incurred under these agreements, our development of our product candidates could be delayed and we may need additional resources to develop our product candidates. All of the risks relating to product development, regulatory approval and commercialization described herein also apply to the activities of our collaborators.
Additionally, subject to its contractual obligations to us, if a collaborator of ours were to be involved in a business combination, it might deemphasize or terminate the development or commercialization of any product candidate licensed to it by us. If one of our collaborators terminates its agreement with us, we may find it more difficult to attract new collaborators and our perception in the business and financial communities could be harmed.
We rely completely on third-party contractors to supply, manufacture and distribute clinical drug supplies for our product candidates, which may include sole-source suppliers and manufacturers; we intend to rely on third parties for commercial supply, manufacturing and distribution if any of our product candidates receives regulatory approval and for any future product candidates.
We do not currently have, nor do we plan to acquire, the infrastructure or capability to supply, store, manufacture or distribute preclinical, clinical or commercial quantities of drug substances or products. Additionally, we have not entered into a long-term commercial supply agreement to provide us with such drug substances or products.
44
As a result, our ability to develop our product candidates is dependent, and our ability to supply our products commercially will depend, in part, on our ability to obtain the active pharmaceutical ingredients "(APIs"), and other substances and materials used in our product candidates successfully from third parties and to have finished products manufactured by third parties in accordance with regulatory requirements and in sufficient quantities for preclinical and clinical testing and commercialization. If we fail to develop and maintain supply and other technical relationships with these third parties, and if we are unable to seek suitable replacements in a timely manner or at all, we may face delays or be unable to continue to develop or commercialize our products and product candidates.
We have limited control over whether or not our contract suppliers and manufacturers will maintain current pricing terms, be willing to continue supplying us with APIs and finished products or maintain adequate capacity and capabilities to serve our needs, including quality control, quality assurance and qualified personnel. We are dependent on our contract suppliers and manufacturers for day-to-day compliance with applicable laws and cGMP regulations for production of both APIs and finished products. If the safety or quality of any product or product candidate or component is compromised due to a failure to adhere to applicable laws or for other reasons, we may not be able to commercialize or obtain regulatory approval for the affected product or product candidate successfully, and we may be held liable for injuries sustained as a result.
We may be unable to establish any further agreements with third-party manufacturers or to do so on acceptable terms. Even if we are able to establish agreements with third-party manufacturers, reliance on third-party manufacturers entails additional risks, including:
Third-party manufacturers may not be able to comply with cGMP regulations or similar regulatory requirements outside the United States. We, or our contract manufacturers, or any future collaborators and their contract manufacturers could be subject to periodic unannounced inspections by the FDA, competent authorities of EU member states, or other comparable foreign regulatory agencies, to monitor and ensure compliance with cGMP. Despite our efforts to audit and verify regulatory compliance, one or more of our third-party manufacturing vendors may be found on regulatory inspection by the FDA, competent agencies of EU member states, or other comparable foreign regulatory agencies to be noncompliant with cGMP regulations. Our failure, or the failure of our third-party manufacturers, to comply with applicable regulations could result in clinical holds on our trials, sanctions being imposed on us, including shutdown of the third-party vendor or invalidation of drug product lots or processes, fines, injunctions, civil penalties, delays, suspension, variation or withdrawal of approvals, license revocations, seizures or recalls of product candidates or medicines, operating restrictions, and criminal prosecutions, any of which could significantly and adversely affect supplies of our products and harm our business, financial condition, results of operations, and prospects.
Any products that we may develop may compete with other product candidates and products for access to manufacturing facilities. There are a limited number of manufacturers that operate under cGMP regulations and that might be capable of manufacturing our products or product candidates.
Any performance failure on the part of our existing or future manufacturers could delay clinical development or marketing approval. We do not currently have arrangements in place for redundant supply for any of our product candidates. If any one of our current contract manufacturers cannot perform as agreed, we may be required to replace that manufacturer and may incur added costs and delays in identifying and qualifying any such replacement. Furthermore, securing and reserving production capacity with contract manufacturers may result in significant costs.
45
By relying on third-party manufacturers for outsourced, custom manufacturing, we may encounter difficulties in production, particularly with respect to formulation, process development or scaling up of manufacturing capabilities. If we, or our CMOs, encounter such difficulties, our ability to provide supply of our product candidates for preclinical studies, clinical trials or our products for patients, if approved, could be delayed or halted, or we may be unable to maintain a commercially viable cost structure, which would materially adversely affect our business, results of operations and financial condition.
If third-party suppliers on which we rely fail to successfully scale up their production of our product candidates, we may face delays and lost opportunities with our development or future commercialization efforts.
In order to conduct larger or late-stage clinical trials for a product candidate and supply sufficient commercial quantities of the resulting drug product and its components, if that product candidate is approved for sale, our contract manufacturers and suppliers will need to produce our drug substances and product candidates in larger quantities more cost-effectively and, in certain cases, at higher yields than they currently achieve. If our third-party contractors are unable to scale up the manufacture of any of our product candidates successfully in sufficient quality and quantity and at commercially reasonable prices, or are shut down or put on clinical hold by government regulators, and we are unable to find one or more replacement suppliers or manufacturers capable of production at a substantially equivalent cost in substantially equivalent volumes and quality, and we are unable to transfer the processes successfully on a timely basis, the development of that product candidate and regulatory approval or commercial launch for any resulting products may be delayed, or there may be a shortage in supply, either of which could significantly harm our business, financial condition, operating results and prospects.
We expect to continue to depend on third-party contract suppliers and manufacturers for the foreseeable future. Our supply and manufacturing agreements do not guarantee that a contract supplier or manufacturer will provide services adequate for our needs. Additionally, any damage to or destruction of our third-party manufacturers’ or suppliers’ facilities or equipment, may significantly impair our ability to have our products and product candidates manufactured on a timely basis. Our reliance on contract manufacturers and suppliers further exposes us to the possibility that they, or third parties with access to their facilities, will have access to and may misappropriate our trade secrets or other proprietary information. In addition, the manufacturing facilities of certain of our suppliers may be located outside of the United States. This may give rise to difficulties in importing our products or product candidates or their components into the United States or other countries.
We rely on third-party suppliers for key raw materials used in our manufacturing processes, and the loss of these third-party suppliers or their inability to supply us with adequate raw materials could harm our business.
We rely on third-party suppliers for the raw materials required for the production of our product candidates. Our reliance on these third-party suppliers and the challenges we may face in obtaining adequate supplies of raw materials involve several risks, including limited control over pricing, availability, quality and delivery schedules. As a small company, our negotiation leverage is limited and we are likely to get lower priority than our competitors who are larger than we are. We cannot be certain that our suppliers will continue to provide us with the quantities of these raw materials that we require or satisfy our anticipated specifications and quality requirements. Any supply interruption in limited or sole sourced raw materials could materially harm our ability to manufacture our product candidates until a new source of supply, if any, could be identified and qualified. We may be unable to find a sufficient alternative supply channel in a reasonable time or on commercially reasonable terms. Any performance failure on the part of our suppliers could delay the development and potential commercialization of our product candidates, including limiting supplies necessary for clinical trials and regulatory approvals, which would have a material adverse effect on our business.
Our rights to develop and commercialize our technology are subject, in part, to the terms and conditions of licenses granted to us by others. In particular, we depend on licenses for development and commercialization rights to OCS-02 (Licaminlimab) and OCS-05. If these rights are terminated or we fail to comply with our obligations under these agreements or any other license, collaboration or other agreement, we may be required to pay damages and we could lose intellectual property rights that are necessary for the development and protection of our product candidates.
We currently and may in the future license from third parties certain intellectual property relating to current and future product candidates. For example, we are party to various license agreements, including with Novartis and Accure, that we depend on for rights to OCS-02 and OCS-05, respectively. These agreements impose, and other potential agreements we may enter into with third parties may impose, diligence, development and commercialization timelines and milestone payment, royalty, insurance and other obligations on us.
46
Under the Novartis Agreement (as defined below) and Accure Agreement (as defined below), for example, we are obligated to make payments to the counterparty upon us achieving certain development or commercialization milestones and to make royalty payments to Novartis and Accure on net product sales of OCS-02 and OCS-05, respectively.
We also have diligence and development obligations under the Novartis Agreement and Accure Agreement. Generally, these diligence obligations require us to use commercially reasonable efforts to develop, manufacture, seek regulatory approval for and commercialize the licensed products. If we fail to comply with our obligations under current or future license agreements, use the licensed intellectual property in an unauthorized manner or otherwise breach a license agreement, our counterparties may have the right to terminate these agreements, in which event we might not have the rights or the financial resources to develop, manufacture or market any licensed product that is covered by these agreements. Future counterparties also may have the right to convert an exclusive license to non-exclusive in the territory in which we fail to satisfy our diligence obligations, which could materially adversely affect the value of the product candidate being developed under any such agreement. Termination of these agreements or reduction or elimination of our rights under these agreements may result in our having to negotiate new or reinstated agreements with less favorable terms, seek alternative sources of financing or cause us to lose our rights under these agreements, including our rights to OCS-02, OCS-05 or other important intellectual property or technology. Any of the foregoing could prevent us from commercializing OCS-02 or OCS-05 or cause a competitor to gain access to the licensed technology, which could have a material adverse effect on our operating results and overall financial condition.
Our license agreements are, and future license agreements are likely to be, complex, and certain provisions in such agreements may be susceptible to multiple interpretations. The resolution of any contract interpretation disagreement that may arise could narrow what we believe to be the scope of our rights to the relevant intellectual property or technology, or increase what we believe to be our financial or other obligations under the relevant agreement, either of which could have a material adverse effect on our business, financial condition, results of operations and prospects. Disputes may arise between us and our licensors or future licensors, including:
If disputes over intellectual property that we have licensed from third parties prevent or impair our ability to maintain our current licensing arrangements on acceptable terms, we may be unable to successfully develop and commercialize our product candidates.
The licensing or acquisition of third-party intellectual property rights is a competitive area, and several more established companies may pursue strategies to license or acquire third-party intellectual property rights that we may consider attractive or necessary. These established companies may have a competitive advantage over us due to their size, capital resources and greater clinical development and commercialization capabilities. In addition, companies that perceive us to be a competitor may be unwilling to assign or license rights to us. If we are unable to license such technology, or if we are forced to license such technology on unfavorable terms, our business could be harmed. If we are unable to obtain a necessary license, we may be unable to develop or commercialize the affected product candidates, which could harm our business, and the third parties owning such intellectual property rights could seek either an injunction prohibiting sales or an obligation on our part to pay royalties and/or other forms of compensation.
47
Even if we are able to obtain a license, it may be non-exclusive, thereby giving our competitors access to the same technologies licensed to us.
Additionally, our licensors may have relied on third-party consultants or collaborators or on funds from third parties such that our licensors are not the sole and exclusive owners of the patents we in-licensed. Some of our in-licensed patent rights are sublicensed to us pursuant to parent license agreements we are not a party to. If any such parent licenses terminate, whether due to our licensor’s breach of the parent license agreement or for other reasons outside of our control, we could lose our rights to such sublicensed patent rights. Furthermore, if other third parties have ownership rights to our in-licensed patents, the license granted to us in jurisdictions where the consent of a co-owner is necessary to grant such a license may not be valid, in any case, and such co-owners may be able to license such patents to our competitors, and our competitors could market competing products and technology. In addition, certain of our in-licensed patent rights are dependent, in part, on inter-institutional or other operating agreements between the joint owners of such in-licensed patent rights. If one or more of such joint owners breaches such inter-institutional or operating agreements, our rights to such in-licensed patent rights may be adversely affected. Any of these events could have a material adverse effect on our competitive position, business, financial conditions, results of operations and prospects.
Our current and future licenses may not provide us with exclusive rights to use the licensed intellectual property and technology, or may not provide us with exclusive rights to use such intellectual property and technology in all relevant fields of use and in all territories in which we may wish to develop or commercialize our technology. Patents licensed to us could be put at risk of being invalidated or interpreted narrowly in litigation filed by or against our licensors or another licensee or in administrative proceedings brought by or against our licensors or another licensee in response to such litigation or for other reasons. As a result, we may not be able to prevent competitors or other third parties from developing and commercializing competitive products, including in territories covered by our licenses. Some of our in-licensed patent rights are subject to pre-existing rights granted by the licensor to third parties and our acquired technologies and current or future licensed technology may also be subject to retained rights. Our predecessors or licensors may retain certain rights under their agreements with us, including the right to use the underlying technology for noncommercial academic and research use, to publish general scientific findings from research related to the technology, and to make customary scientific and scholarly disclosures of information relating to the technology. It is difficult to monitor whether our predecessors or future licensors limit their use of the technology to these uses, and we could incur substantial expenses to enforce our rights to our licensed technology in the event of misuse.
In addition, certain of our current or future agreements with third parties may limit or delay our ability to consummate certain transactions, may impact the value of those transactions, or may limit our ability to pursue certain activities. If we are limited in our ability to utilize acquired technologies or current or future licensed technologies, or if we lose our rights to critical acquired or in-licensed technology, we may be unable to successfully develop, out-license, market and sell our products, which could prevent or delay new product introductions. Our business strategy depends on the successful development of acquired technologies, and current or future licensed technology, into commercial products. Therefore, any limitations on our ability to utilize these technologies may impair our ability to develop, out-license or market and sell our product candidates.
For more information on our license agreements with third parties, please see the section entitled “Business Overview—Material Licenses, Partnerships and Collaborations.”
Risks related to our intellectual property
If we are unable to obtain, maintain, protect and enforce patent or other intellectual property protection for our current and future technology and products, or if the scope of the patent or other intellectual property protection obtained is not sufficiently broad, we may not be able to compete effectively in our markets.
We rely upon a combination of patents, trademarks, trade secrets and confidentiality agreements to protect the intellectual property related to our development programs and product candidates. These legal measures afford only limited protection, and competitors or others may gain access to our intellectual property and proprietary information. Our success depends in part on our ability to obtain, maintain, expand, enforce and defend the scope of our intellectual property protection in the United States and other countries with respect to our product candidates.
48
We have sought and will continue to seek to protect our proprietary position by filing patent applications in the United States and abroad related to our development programs and product candidates. However, the patent prosecution process is expensive and time-consuming, and we may not be able to file, prosecute, maintain, enforce or license all necessary or desirable patents or patent applications at a reasonable cost, in a timely manner, or in all jurisdictions where protection may be commercially advantageous, or we may not be able to protect our proprietary rights at all. Additionally, in some instances, we have submitted and expect to submit provisional patent applications. Corresponding non-provisional patent applications must be filed not later than 12 months after the provisional application filing date. While we intend to timely file non-provisional patent applications relating to our provisional patent applications, we cannot predict whether any such patent applications will result in the issuance of patents that provide us with competitive advantage. Any failure to obtain or maintain patent and other intellectual property protection with respect to our product candidates could harm our business, financial condition and results of operations. Additionally, although we seek to enter into non-disclosure and confidentiality agreements with parties who have access to patentable aspects of our research and development output, any of these parties may breach the agreements and disclose such output before a patent application is filed, thereby jeopardizing our ability to seek patent protection.
The patents and patent applications that we own or in-license may fail to result in issued patents with claims that protect our product candidates in the United States or in other foreign countries. There is no assurance that all of the potentially relevant prior art relating to our patents and patent applications has been found, which can prevent a patent from issuing from a pending patent application, or be used to invalidate a patent. Even if patents do successfully issue and even if such patents cover our product candidates, third parties may challenge their validity, enforceability or scope, which may result in such patents being narrowed, invalidated or held unenforceable. Any successful opposition to these patents or any other patents owned by or licensed to us could deprive us of rights necessary for the successful commercialization of any product candidates that we may develop. Further, if we encounter delays in regulatory approvals, the period of time during which we could market a product candidate under patent protection could be reduced.
The patent application process is subject to numerous risks and uncertainties, and there can be no assurance that we or any of our licensors will be successful in protecting our product candidates by obtaining, maintaining, enforcing and defending patents. These risks and uncertainties include the following:
We may also choose not to seek patent protection for certain innovations and may choose not to pursue patent protection in certain jurisdictions, and under the laws of certain jurisdictions, patents or other intellectual property rights may be unavailable or limited in scope. It is also possible that we will fail to identify patentable aspects of our research and development output before it is too late to obtain patent protection. If we fail to timely file for patent protection in any jurisdiction, we may be precluded from doing so at a later date.
49
Moreover, we are, and could become in the future, a licensee of a third party’s patents or patent applications and we may not have the right to control the preparation, filing or prosecution of such patent applications, or to maintain, enforce or protect the patents in-licensed from those third parties. We may also require the cooperation of our licensors in order to enforce the licensed patent rights, and such cooperation may not be provided. Therefore, any licensed patents or patent applications may not be prosecuted, maintained, enforced or protected in a manner consistent with the best interests of our business. We also cannot be certain that patent prosecution and maintenance activities by any of our licensors will be conducted in compliance with applicable laws and regulations, which may affect the validity and enforceability of such patents or any patents that may issue from such applications. If any of our licensors fail to do so, this could cause us to lose rights in any applicable intellectual property, and as a result our ability to develop and commercialize products or product candidates may be adversely affected and we may be unable to prevent competitors from making, using and selling competing products. In addition, even where we have the right to control the prosecution of patents and patent applications under a license from third parties, we may still be adversely affected or prejudiced by actions or inactions of our predecessors or licensors and their counsel that took place prior to us assuming control over patent prosecution. If our current or future licensors are not fully cooperative or disagree with us as to the prosecution, maintenance or enforcement of any patent rights, such patent rights could be compromised. If disputes over intellectual property that we license prevents or impairs our ability to maintain our licensing arrangements on acceptable terms, we may not be able to successfully develop and commercialize the affected product candidates. Any of these outcomes could impair our ability to prevent competition from third parties, which may have an adverse impact on our business.
If the patent applications we own, license, or may own or license in the future with respect to our development programs and product candidates fail to issue, if their breadth or strength of protection is threatened, or if they fail to provide meaningful exclusivity for our product candidates, it could dissuade other companies from collaborating with us to develop product candidates, and threaten our ability to commercialize our product candidates, if approved. Any such outcome could have a materially adverse effect on our business.
The patent position of biotechnology and pharmaceutical companies generally is highly uncertain, involves complex legal and factual questions, and has been and will continue to be the subject of litigation and new legislation. In addition, the laws of foreign countries may not protect our rights to the same extent as the laws of the United States. For example, many countries restrict the patentability of methods of treatment of the human body. Publications in scientific literature often lag behind the actual discoveries, and patent applications in the United States and other jurisdictions are typically not published until 18 months after filing, or in some cases not at all. Therefore, there is a risk that we cannot know with certainty whether we or our licensors were the first to make the inventions claimed in our owned or in-licensed patents or pending patent applications, or that we or our licensors were the first to file for patent protection of such inventions. As a result of these and other factors, the issuance, scope, validity, enforceability, and commercial value of our owned and in-licensed patent rights are highly uncertain. Our owned and in-licensed pending and future patent applications may not result in patents being issued which protect our technology or products, in whole or in part, or which effectively prevent others from commercializing competitive technologies and products. Changes in either the patent laws or interpretation of the patent laws in the United States and other countries may diminish the value of our owned and in-licensed patents or narrow the scope of patent protection for our product candidates.
Moreover, we or our licensors may be subject to a third-party pre-issuance submission of prior art to the USPTO or become involved in opposition, derivation, reexamination, inter partes review, post-grant review or interference proceedings challenging our owned or in-licensed patent rights or the patent rights of others. In particular, the costs of defending patents or enforcing our proprietary rights in post-issuance administrative proceedings and litigation can be substantial and the outcome can be uncertain. An adverse determination in any such submission, proceeding or litigation could reduce the scope of, or invalidate, our owned or in-licensed patent rights, allow third parties to commercialize our technology or products and compete directly with us, without payment to us, or result in our inability to manufacture or commercialize products without infringing third-party patent rights. In addition, if the breadth or strength of protection provided by our owned or in-licensed patents and patent applications is threatened, it could dissuade companies from collaborating to license, develop or commercialize current or future product candidates. We may not be aware of all third-party intellectual property rights potentially relating to our products, product candidates or their intended uses, and as a result the impact of such third-party intellectual property rights upon the patentability of our owned and in-licensed patents and patent applications, as well as the impact of such third-party intellectual property upon our freedom to operate, is highly uncertain. We cannot ensure that we do not infringe, misappropriate or otherwise violate any patents or other intellectual property or proprietary rights held by others or that we will not infringe, misappropriate or otherwise violate intellectual property or proprietary rights held by others in the future.
50
If our products were found to infringe, misappropriate or otherwise violate any proprietary intellectual property or right of another party, we could be required to pay significant damages or license fees to such party and/or cease production, marketing and distribution of those products. Litigation may also be necessary to defend infringement, misappropriation or other violation claims of third parties or to enforce patent or other intellectual property rights we hold or protect trade secrets or techniques or other intellectual property we own. Further, third parties may seek approval to market their own products similar to or otherwise competitive with our products. In these circumstances, we may need to defend and/or assert our patents or other intellectual property, including by filing lawsuits alleging patent infringement. In any of these types of proceedings, a court or agency with jurisdiction may find our owned or in-licensed patents invalid, unenforceable, or not infringed; competitors may then be able to market products and use manufacturing and analytical processes that are substantially similar. Even if we own or in-license valid and enforceable patents, these patents still may not provide protection against competing products or processes sufficient to achieve our business objectives.
The issuance of a patent is not conclusive as to its inventorship, scope, validity or enforceability, and patents in which we or our licensors have an interest may be challenged in the courts or patent offices in the United States and abroad. Such challenges may result in loss of exclusivity or in patent claims being narrowed, invalidated or held unenforceable, in whole or in part, which could limit our ability to stop others from using or commercializing similar or identical technology and products, or limit the duration of the patent protection of our technology and products. Generally, issued patents are granted a term of 20 years from the earliest claimed non-provisional filing date. In certain instances, patent terms can be adjusted to recapture a portion of delay incurred by the USPTO in examining the patent application (patent term adjustment). The scope of patent protection may also be limited. In addition, the laws of foreign jurisdictions may not protect our rights to the same extent as the laws of the U.S. For example, certain countries outside of the U.S. do not allow patents for methods of treating the human body. This may preclude us from obtaining method patents outside of the U.S. having similar scope to those we have obtained or may obtain in the future in the U.S.
It is possible that defects of form in the preparation or filing of our owned or in-licensed patents or patent applications may exist, or may arise in the future, for example with respect to proper priority claims, inventorship, claim scope, or requests for patent term adjustments. The acquisition or licensing of third-party intellectual property rights is a competitive area, and our competitors may pursue strategies to acquire or license third-party intellectual property rights that we may consider attractive or necessary, and our competitors could market competing products and technology. Our competitors may have a competitive advantage due to their size, capital resources and greater development and commercialization capabilities. In addition, companies may be unwilling to assign or license rights to us. We also may be unable to acquire or license third-party intellectual property rights on terms that would allow us to make an appropriate return on our investment or at all. If we are unable to successfully obtain rights to required third-party intellectual property rights or maintain the existing intellectual property rights we have, we may have to abandon development of the relevant product, and our customers may be forced to stop using the relevant products. If we or our current or future licensors fail to establish, maintain or protect such patents and other intellectual property rights, such rights may be reduced or eliminated. If there are material defects in the form, preparation, prosecution, or enforcement of our patents or patent applications, such patents may be invalid and/or unenforceable, and such applications may never result in valid, enforceable patents. Any of these outcomes could impair our ability to prevent competition from third parties, which may have an adverse impact on our business.
Without patent protection for our current or future product candidates, we may be open to competition from generic versions of such products. Given the amount of time required for the development, testing, and regulatory review of new product candidates, patents protecting such candidates might expire before or shortly after such candidates are commercialized. As a result, our patent portfolio may not provide us with sufficient rights to exclude others from commercializing products similar or identical to our own.
Depending upon the timing, duration and specifics of FDA marketing approval of future product candidates, one or more of our U.S. patents may be eligible for limited patent term restoration under the Drug Price Competition and Patent Term Restoration Act of 1984, referred to as the Hatch-Waxman Amendments. The Hatch-Waxman Amendments permit a patent restoration term of up to five years beyond the normal expiration of the patent as compensation for patent term lost during drug development and the FDA regulatory review process, which is limited to the approved indication (or any additional indications approved during the period of extension). A patent term extension cannot extend the remaining term of a patent beyond 14 years from the date of product approval. This extension is based on the first approved use of a product and is limited to only one patent that covers the approved product, the approved use of the product, or a method of manufacturing the product.
51
However, the applicable agencies, including the FDA and the USPTO in the U.S., and any equivalent regulatory authority in other countries, may not agree with our assessment of whether such extensions are available, and may refuse to grant extensions to our patents, or may grant more limited extensions than we request. We may not be granted an extension because of, for example, failing to apply within applicable deadlines, failing to apply prior to expiration of relevant patents or otherwise failing to satisfy applicable requirements. Moreover, the applicable time-period or the scope of patent protection afforded could be less than we request. If we are unable to extend the expiration date of our existing patents or obtain new patents with longer expiry dates, our competitors may be able to take advantage of our investment in development and clinical trials by referencing our clinical and preclinical data to obtain approval of competing products following our patent expiration and launch our product earlier than might otherwise be the case.
Obtaining and maintaining intellectual property, including patent protection, depends on compliance with various procedural, document submission, fee payment, and other requirements imposed by domestic and international governmental agencies, and our intellectual property, including patent protection, could be reduced or eliminated for noncompliance with these requirements.
The patent prosecution process is expensive, time-consuming and complex. Periodic maintenance, renewal, annuity and various other fees on any issued patent are due to be paid to the USPTO and other foreign governmental agencies in several stages over the lifetime of the intellectual property. The USPTO and various national or international agencies require compliance with a number of procedural, documentary, fee payment, and other similar provisions during the application process. While an inadvertent lapse can in many cases be cured by payment of a late fee or by other means in accordance with the applicable rules, there are situations in which noncompliance can result in abandonment or lapse of the intellectual property, resulting in partial or complete loss of rights in the relevant jurisdiction. Noncompliance events that could result in abandonment or lapse of rights include, but are not limited to, failure to timely file national and regional stage patent applications based on our international application, failure to respond to official actions within prescribed time limits, non-payment of fees, and failure to properly legalize and submit formal documents. If we or any of our licensors fail to maintain the intellectual property covering our product candidates, our competitors may be able to enter the market, which would have an adverse effect on our business, financial condition and results of operations.
We may not identify relevant third-party patents or may incorrectly interpret the relevance, scope or expiration of a third-party patent, which might adversely affect our ability to develop and market our products.
We cannot guarantee that any of our patent searches or analyses, including the identification of relevant patents, the scope of patent claims or the expiration of relevant patents, are complete or thorough, nor can we be certain that we have identified each and every third-party patent and pending application in the United States and abroad that is relevant to or necessary for the commercialization of our current and future product candidates in any jurisdiction. The scope of a patent claim is determined by an interpretation of the law, the written disclosure in a patent and the patent’s prosecution history. Our interpretation of the relevance or the scope of a patent or a pending application may be incorrect, which may negatively impact our ability to market our product candidates, if approved. We may incorrectly determine that our products are not covered by a third-party patent or may incorrectly predict whether a third-party’s pending application will issue with claims of relevant scope. Our determination of the expiration date of any patent in the United States or abroad that we consider relevant may be incorrect, and our failure to identify and correctly interpret relevant patents may negatively impact our ability to develop and market our products. Also, because the claims of published patent applications can change between publication and patent grant, there may be published patent applications that may ultimately issue with claims that we infringe. As the number of competitors in the market grows and the number of patents issued in this area increases, the possibility of patent infringement claims escalates. Moreover, in recent years, individuals and groups that are non-practicing entities, commonly referred to as “patent trolls,” have purchased patents and other intellectual property assets for the purpose of making claims of infringement in order to extract settlements. From time to time, we may receive threatening letters, notices or “invitations to license,” or may be the subject of claims that our products and business operations infringe, misappropriate or otherwise violate the intellectual property rights of others. The defense of these matters can be time consuming, costly to defend in litigation, divert management’s attention and resources, damage our reputation and brand and cause us to incur significant expenses or make substantial payments.
52
We may become subject to third-party claims or litigation alleging infringement, misappropriation or other violation of such third party’s patents or other intellectual property or proprietary rights, or seeking to invalidate our patents or other intellectual property or proprietary rights, which could be costly, time consuming, and, if successfully asserted against us, may delay or prevent the development and commercialization of any of our product candidates.
Our commercial success depends in part on us and our licensors avoiding infringement, misappropriation and other violations of the patents and other intellectual property or proprietary rights of third parties. There is a substantial amount of litigation, both within and outside the U.S., involving patent and other intellectual property rights in the biotechnology and pharmaceutical industries, including patent infringement lawsuits, interferences, derivation, and administrative law proceedings, inter partes review, and post-grant review before the USPTO, as well as oppositions and similar processes in foreign jurisdictions. Such challenges may result in loss of exclusivity or freedom to operate or in patent claims being narrowed, invalidated or held unenforceable, in whole or in part, which could limit our ability to stop others from using or commercializing similar or identical products and techniques without payment, or limit the duration of the patent protection of our technology. Numerous U.S. and foreign issued patents and pending patent applications, which are owned by third parties, exist in the fields in which we and our collaborators are developing product candidates. As the biotechnology and pharmaceutical industries expand and more patents are issued, and as we gain greater visibility and market exposure as a public company, the risk increases that our product candidates or other business activities may be subject to claims of infringement of the patent and other proprietary rights of third parties. Third parties may assert that we are infringing, misappropriating or otherwise violating their patents or other intellectual property or proprietary rights or employing their proprietary technology without authorization.
Also, there may be third-party patents or patent applications with claims to materials, formulations, methods of manufacture or methods of treatment related to the use or manufacture of our current and future product candidates. Because patent applications can take many years to issue, there may be currently pending patent applications which may later result in issued patents that our current or future product candidates may infringe.
In addition, third parties may obtain patent rights in the future and claim that use of our technologies infringes upon their rights. If any third-party patents were held by a court of competent jurisdiction to cover the manufacturing process of any of our product candidates, any molecules formed during the manufacturing process, methods of treating certain diseases or conditions that we are pursuing with our product candidates, our formulations including combination therapies, or any final product itself, the holders of any such patents may be able to block our ability to commercialize such product candidate unless we obtained a license under the applicable patents, or until such patents expire. Such a license may not be available on commercially reasonable terms or at all. In addition, we may be subject to claims that we are infringing, misappropriating or otherwise violating other intellectual property rights, such as trademarks or copyrights, or misappropriating the trade secrets of others, and to the extent that our employees, consultants or contractors use intellectual property or proprietary information owned by others in their work for us, disputes may arise as to the rights in related or resulting know-how and inventions.
Parties making claims against us may obtain injunctive or other equitable relief, which could effectively block our ability to further develop and commercialize one or more of our current and future product candidates. Defense of these claims, regardless of their merit, would involve substantial litigation expense and would be a substantial diversion of employee resources from our business. In the event of a successful infringement or other intellectual property claim against us, we may have to pay substantial damages, including treble damages and attorneys’ fees for willful infringement, obtain one or more licenses from third parties, pay royalties or redesign our affected products, which may be impossible or require substantial time and monetary expenditure. We cannot predict whether any such license would be available at all or whether it would be available on commercially reasonable terms. Furthermore, even in the absence of litigation, we may need to obtain licenses from third parties to advance our research or allow commercialization of our product candidates, and we have done so from time to time. We may fail to obtain any of these licenses at a reasonable cost or on reasonable terms, if at all. In that event, we would be unable to further develop and commercialize one or more of our product candidates, which could harm our business significantly. We cannot provide any assurances that third-party patents or other intellectual property or proprietary rights do not exist which might be enforced against our product candidates, resulting in either an injunction prohibiting sales, or, with respect to our sales, an obligation on our part to pay royalties or other forms of compensation to third parties.
During the course of any intellectual property litigation, there could be public announcements of the initiation of the litigation as well as results of hearings, rulings on motions, and other interim proceedings in the litigation. If securities analysts or investors regard these announcements as negative, the perceived value of our existing product candidates, programs or intellectual property could be diminished.
53
Accordingly, the market price of Ordinary Shares may decline. Such announcements could also harm our reputation or the market for future products, which could have a material adverse effect on our business.
Lawsuits or other proceedings to protect or enforce our patents, the patents of any licensors or our other intellectual property rights could be expensive, time consuming, and unsuccessful.
Competitors may infringe or otherwise violate our patents, the patents of our licensors or our other intellectual property rights. To counter infringement or unauthorized use or misappropriations, we may be required to file legal claims, which can be expensive and time-consuming. In addition, in an infringement proceeding, a court may decide that one or more patents of us or any of our current or future licensors is not valid or is unenforceable, or may refuse to stop the other party from using the technology at issue on the grounds that our patents do not cover the technology in question. An adverse result in any litigation or defense proceedings could put one or more of our patents at risk of being invalidated or interpreted narrowly and could put our patent applications at risk of not issuing. The initiation of a claim against a third party may also cause the third party to bring counterclaims against us, such as claims asserting that our patents are invalid or unenforceable. In patent litigation in the U.S., defendant counterclaims alleging invalidity or unenforceability are commonplace. Grounds for a validity challenge could be an alleged failure to meet any of several statutory requirements, including lack of novelty, obviousness, non-enablement or lack of statutory subject matter. Grounds for an unenforceability assertion could be an allegation that someone connected with prosecution of the patent withheld relevant material information from the USPTO, or made a materially misleading statement, during prosecution. Third parties may also raise similar validity claims before the USPTO in post-grant proceedings such as ex parte reexaminations, inter partes review, post-grant review or oppositions or similar proceedings outside the U.S., in parallel with litigation or even outside the context of litigation. The outcome following legal assertions of invalidity and unenforceability is unpredictable. We cannot be certain that there is no invalidating prior art, of which we and the patent examiner were unaware during prosecution. Additionally, for any patents and patent applications that we license from third parties, we may have limited or no right to participate in the defense of such licensed patents against challenge by a third-party. If a defendant were to prevail on a legal assertion of invalidity or unenforceability, we would lose at least part, and perhaps all, of the patent protection on our current or future product candidates. Such a loss of patent protection could harm our business.
Furthermore, even if our patents or other intellectual property or proprietary rights are found to be valid and infringed, a court may refuse to grant injunctive relief against the infringer and instead award us monetary damages or ongoing royalties. Such monetary compensation may be insufficient to adequately offset the damage to our business caused by the infringer’s competition in the market. Because of the expense and uncertainty of litigation, we may conclude that even if a third party is infringing our current or future owned or in-licensed patents, any patents that may be issued as a result of our current or future owned or in-licensed patent applications, or other intellectual property rights, the risk-adjusted cost of bringing and enforcing such a claim or action may be too high or not in the best interest of us or our shareholders. In such cases, we may decide that the more prudent course of action is to simply monitor the situation or initiate or seek some other non-litigious action or solution. Moreover, even if we are successful in any litigation, we may incur significant expense in connection with such proceedings, and the amount of any monetary damages may be inadequate to compensate us for damage as a result of the infringement and the proceedings.
In addition, third parties may assert infringement claims against our customers. These claims may require us to initiate or defend protracted and costly litigation on behalf of our customers or indemnify our customers for any costs associated with their own initiation or defense of infringement claims, regardless of the merits of these claims. If any of these claims succeed or settle, we may be forced to pay damages or settlement payments on behalf of our customers or may be required to obtain licenses for the products they use. If we cannot obtain all necessary licenses on commercially reasonable terms or at all, our customers may be forced to stop using our products.
We may not be able to prevent, alone or with our licensors, infringement, misappropriation or other violation of our intellectual property or other proprietary rights, particularly in countries where the laws may not protect those rights as fully as in the United States. Our business could be harmed if in litigation the prevailing party does not offer us a license on commercially reasonable terms or at all. Any litigation or other proceedings to enforce our intellectual property or proprietary rights may fail, and even if successful, may result in substantial costs and distract the management and other employees.
54
Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation. There could also be public announcements of the results of hearings, motions or other interim proceedings or developments. If securities analysts or investors perceive these results to be negative, it could have an adverse effect on the price of Ordinary Shares.
Changes in U.S. or foreign patent laws or their interpretations could diminish the value of patents in general, thereby impairing our ability to protect our products.
The United States government has enacted and implemented wide-ranging patent reform legislation. The U.S. Supreme Court has ruled on several patent cases in recent years, either narrowing the scope of patent protection available in certain circumstances or weakening the rights of patent owners in certain situations. In addition to increasing uncertainty with regard to our ability to obtain patents in the future, this combination of events has created uncertainty with respect to the value of patents, once obtained. Depending on actions by Congress, the federal courts, and the USPTO, the laws and regulations governing patents could change in unpredictable ways that would weaken our ability to obtain new patents or to enforce patents that we have licensed or that we might obtain in the future. Similarly, changes in patent law and regulations in other countries or jurisdictions or changes in the governmental bodies that enforce them or changes in how the relevant governmental authority enforces patent laws or regulations may weaken our ability to obtain new patents or to enforce patents that we have licensed or that we may obtain in the future.
In 2011, the Leahy-Smith America Invents Act (the “Leahy-Smith Act”) was signed into law. The Leahy-Smith Act includes a number of significant changes to U.S. patent law. These include provisions that affect the way patent applications are prosecuted and also affect patent litigation. These also include provisions that switched the U.S. from a “first-to-invent” system to a “first-to-file” system, allow third-party submission of prior art to the USPTO during patent prosecution and set forth additional procedures to attack the validity of a patent by the USPTO administered post grant proceedings. Under a first-to-file system, assuming the other requirements for patentability are met, the first inventor to file a patent application generally will be entitled to the patent on an invention regardless of whether another inventor had made the invention earlier. The USPTO recently developed new regulations and procedures to govern administration of the Leahy-Smith Act, and many of the substantive changes to patent law associated with the Leahy-Smith Act, and in particular, the first to file provisions, only became effective on March 16, 2013. A third-party that files a patent application in the USPTO after March 2013, but before the Company could therefore be awarded a patent covering an invention even if the Company had made the invention before it was made by such third-party. This will require the Company to be cognizant of the time from invention to filing of a patent application. Since patent applications in the U.S. and most other countries are confidential for a period of time after filing or until issuance, the Company cannot be certain that it was the first to file any patent application related to its products or invent any of the inventions claimed in its patents or patent applications.
The Leahy-Smith Act also includes a number of significant changes that affect the way patent applications will be prosecuted and also may affect patent litigation. These include allowing third-party submission of prior art to the USPTO during patent prosecution and additional procedures to attack the validity of a patent by USPTO administered post-grant proceedings, including post-grant review, inter partes review and derivation proceedings. Because of a lower evidentiary standard in USPTO proceedings compared to the evidentiary standard in U.S. federal courts necessary to invalidate a patent claim, a third-party could potentially provide evidence in a USPTO proceeding sufficient for the USPTO to hold a claim invalid even though the same evidence would be insufficient to invalidate the claim if first presented in a district court action. Accordingly, a third-party may attempt to use the USPTO procedures to invalidate the Company’s patent claims that would not have been invalidated if first challenged by the third-party as a defendant in a district court action. Therefore, the Leahy-Smith Act and its implementation could increase the uncertainties and costs surrounding the prosecution of the Company’s patent applications and the enforcement or defense of our issued patents. In addition, future actions by the U.S. Congress, the federal courts and the USPTO could cause the laws and regulations governing patents to change in unpredictable ways. The Leahy-Smith Act and its implementation could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents, all of which could have a material adverse effect on the Company’s business, financial condition, and results of operations.
We may not be able to protect our intellectual property rights throughout the world, which could impair our business.
55
Filing, prosecuting, and defending patents covering our product candidates throughout the world would be prohibitively expensive. Furthermore, the requirements for patentability and obtaining other intellectual property protection may differ in certain countries, particularly developing countries. In addition, the laws of many foreign countries will not protect our intellectual property or other proprietary rights to the same extent as the laws of the United States. Competitors may use our technologies in jurisdictions where we have not obtained patent or other intellectual property protection to develop their own products and, further, may export otherwise infringing products to territories where we may have or obtain patent or other intellectual property protection, but where patent or other intellectual property enforcement is not as strong as that in the United States. These unauthorized products may compete with our products in such jurisdictions and take away our market share where we do not have any issued or licensed patents or other intellectual property protection and any future patent claims or other intellectual property rights may not be effective or sufficient to prevent them from so competing. Our ability to protect and enforce our intellectual property rights may be adversely affected by unforeseen changes in foreign intellectual property laws.
Our reliance on third parties may require us to share our trade secrets, which increases the possibility that a competitor will discover them or that our trade secrets will be misappropriated or disclosed.
Because we rely on third parties for a wide variety of services, including the manufacture and continuing development of our product candidates, we must, at times, share trade secrets with them. We seek to protect our trade secrets in part by entering into agreements containing confidentiality and use restrictions and obligations prior to disclosing proprietary information. These agreements typically limit the rights of the third parties to use or disclose our confidential information, including our trade secrets. Despite the contractual provisions employed when working with third parties, the need to share trade secrets and other confidential information increases the risk that such trade secrets become known by our competitors, are inadvertently incorporated into the technology of others, or are disclosed or used in violation of these agreements. Given that our proprietary position is based, in part, on our know-how and trade secrets, a competitor’s discovery of our trade secrets or other unauthorized use or disclosure could impair our competitive position and may have an adverse effect on business and results of operations.
Despite our efforts to protect our trade secrets, our competitors may discover our trade secrets, either through breach of agreements with third parties, independent development or publication of information by any of the third-party collaborators. A competitor’s discovery of our trade secrets could impair our competitive position and have an adverse impact on our business.
If we fail to protect the confidentiality of our trade secrets and other proprietary information, the value of our product candidates and our business and competitive position may be harmed.
In addition to patent protection, we also rely on other proprietary rights, including protection of trade secrets, know-how or other proprietary information that is not patentable or that we elect not to patent. Trade secrets can be difficult to protect, and some courts are less willing or unwilling to protect trade secrets. To maintain the confidentiality of our trade secrets and proprietary information, we rely heavily on confidentiality provisions that we have in contracts with our employees, consultants, collaborators and others upon the commencement of their relationship with us. However, we cannot guarantee that we have entered into such agreements with each party that may have or has had access to our trade secrets or proprietary technology and processes and we may not enter into such agreements with all employees, consultants and third parties who have been involved in the development of our intellectual property rights. In addition, monitoring unauthorized use and disclosure of our intellectual property rights by employees, consultants and other third parties who have access to such intellectual property or other proprietary rights is difficult. Therefore, we may not be able to prevent the unauthorized disclosure or use of our technical knowledge or other trade secrets by such employees, consultants, advisors or third parties, despite the existence generally of these confidentiality restrictions. There can be no assurance that such employees, consultants, advisors or third parties will not breach their agreements with us, that we will have adequate remedies for any breach, or that our trade secrets will not otherwise become known or independently developed by third parties, including our competitors.
We may be subject to claims that our employees, consultants or independent contractors have infringed, misappropriated or otherwise violated the intellectual property of a third party, including trade secrets or know-how of their former employers or other third parties.
We may be subject to claims that our employees or consultants have wrongfully used for our benefit or disclosed to us confidential information of third parties. We employ individuals who were previously employed at other biotechnology or pharmaceutical companies, or at research institutions.
56
Some of these employees, consultants and contractors may have executed proprietary rights, non-disclosure and non-competition agreements in connection with such previous employment. Although we try to ensure that our employees and consultants do not use the intellectual property rights, proprietary information, know-how or trade secrets of others in their work for us and seek to protect our ownership of intellectual property rights by ensuring that our agreements with employees, collaborators, and other third parties with whom we do business include provisions requiring such parties to assign rights in inventions to us, we may be subject to claims that we or our employees, consultants or independent contractors have inadvertently or otherwise used or disclosed confidential information of our employees’ former employers or other third parties. To the extent that our employees, consultants or contractors use intellectual property rights or proprietary information owned by others in their work for us, disputes may arise as to the rights in any related or resulting know-how and inventions. We may also be subject to claims that former employers or other third parties have an ownership interest in our patents or other intellectual property or proprietary rights. Litigation may be necessary to defend against any of these claims. There is no guarantee of success in defending these claims, and if we fail in defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights, such as exclusive ownership of, or right to use, valuable intellectual property. In addition, we may lose personnel as a result of such claims and any such litigation or the threat thereof may adversely affect our ability to hire employees or contract with independent contractors. Even if we are successful, litigation could result in substantial cost and be a distraction to our management and other employees.
If we fail to validly execute invention assignment agreements with our employees and contractors involved in the development of intellectual property, the value of our products, business and competitive position may be harmed. Our patent rights and other intellectual property may also be subject to priority, ownership or inventorship disputes, interferences, and similar proceedings.
To maintain the confidentiality of our trade secrets, proprietary information and other intellectual property rights, we generally have confidentiality and invention assignment provisions in place with our employees, consultants, suppliers, contract manufacturers, collaborators, and others upon the commencement of a relationship. However, we may not enter into such agreements with each party that may have or have had access to our trade secrets or proprietary technology and processes or who conceives or develops intellectual property rights that we regard as our own. Moreover, even when we obtain agreements assigning intellectual property to us, the assignment of intellectual property rights may not be self-executing, and we may be forced to bring claims against third parties or defend claims that they may bring against us to determine the ownership of what we regard as our intellectual property. There can be no assurance that such agreements will be upheld in the face of a potential challenge or that third parties will not breach their agreements with us, or that we will have adequate remedies for any breach.
We may also be subject to claims that former employees, collaborators, or other third parties have an interest in our current or future patents and patent applications or other intellectual property rights, including as an inventor or co-inventor. If we are unable to obtain an exclusive license to any such third-party co-owners’ interest in such patents and patent applications, such co-owners rights may be subject, or in the future subject, to assignment or license to other third parties, including competitors. In addition, we may need the cooperation of any such co-owners to enforce any such patents and any patents issuing from such patent applications against third parties, and such cooperation may not be provided. Additionally, we may be subject to claims from third parties challenging our ownership interest in or inventorship of intellectual property we regard as our own, for example, based on claims that our agreements with employees or consultants obligating them to assign intellectual property rights to us are ineffective or in conflict with prior or competing contractual obligations to assign inventions to another employer, to a former employer, or to another person or entity, despite the inclusion of valid, present-tense intellectual property assignment obligations. Litigation may be necessary to defend against claims, and it may be necessary or we may desire to enter into a license to settle any such claim.
If we or our licensors are unsuccessful in any priority, validity (including any patent oppositions), ownership or inventorship disputes to which we or they are subject, we may lose valuable intellectual property rights through the loss of one or more of our patents, or such patent claims may be narrowed, invalidated, or held unenforceable, or through loss of exclusive ownership of or the exclusive right to use our owned or in-licensed patents. In the event of loss of patent rights as a result of any of these disputes, we may be required to obtain and maintain licenses from third parties, including parties involved in any such interference proceedings or other priority or inventorship disputes. Such licenses may not be available on commercially reasonable terms or at all, or may be non-exclusive. If we are unable to obtain and maintain such licenses, we may need to cease the development, manufacture, and commercialization of one or more of the product candidates we may develop.
57
An inability to incorporate technologies, features or other intellectual property that are important or essential to our products could have a material adverse effect on our business and competitive position. The loss of exclusivity or the narrowing of our patent claims could limit our ability to stop others from using or commercializing similar or identical technology and product candidates. Even if we are successful in priority, inventorship or ownership disputes, such disputes could result in substantial costs and be a distraction to management and other employees. Any litigation or the threat thereof may adversely affect our ability to hire employees or contract with independent sales representatives. Any of the foregoing could result in a material adverse effect on our business, financial condition, results of operations or prospects.
Intellectual property rights do not necessarily address all potential threats to our competitive advantage.
The degree of future protection afforded by our intellectual property rights is uncertain because intellectual property rights have limitations, and may not adequately protect our business, or permit us to maintain our competitive advantage. The following examples are illustrative:
Should any of these events occur, they could have a material adverse effect on our business, financial condition, results of operations and prospects.
If our current and future trademarks and trade names are not adequately protected, then we may not be able to build name recognition in our markets and markets of interest and our business may be adversely affected.
We intend to use registered or unregistered trademarks or trade names to brand and market our products. Our trademarks or trade names may be challenged, infringed, circumvented or declared generic or determined to be infringing on other marks. We may not be able to protect our rights to these trademarks and trade names, which we need to build name recognition among potential partners or customers in the markets of interest.
58
During trademark registration proceedings, we may receive rejections of our applications by the USPTO or in other foreign jurisdictions. Although we would be given an opportunity to respond to those rejections, we may be unable to overcome such rejections. In the event that our trademarks are successfully challenged, we could be forced to rebrand our products, which could result in loss of brand recognition, and could require us to devote resources to advertising and marketing new brands. In addition, in the USPTO and in comparable agencies in many foreign jurisdictions, third parties are given an opportunity to oppose pending trademark applications and to seek to cancel registered trademarks. Opposition or cancellation proceedings may be filed against our trademarks, and our trademarks may not survive such proceedings. At times, competitors may adopt trade names or trademarks similar to us, thereby impeding our ability to build brand identity and possibly leading to market confusion. In addition, there could be potential trade name or trademark infringement claims brought by owners of other trademarks or trademarks that incorporate variations of our registered or unregistered trademarks or trade names. Over the long term, if we are unable to establish name recognition based on our trademarks and trade names, then we may not be able to compete effectively and our business may be adversely affected. We may license our trademarks and trade names to third parties, such as distributors. Though these license agreements may provide guidelines for how trademarks and trade names may be used, a breach of these agreements or misuse of such trademarks and trade names by our licensees may jeopardize our rights in or diminish the goodwill associated with our trademarks and trade names. Our efforts to enforce or protect our proprietary rights related to trademarks, trade names, trade secrets, domain names, copyrights or other intellectual property may be ineffective and could result in substantial costs and diversion of resources and could adversely affect our business, growth prospects, operating results and financial condition.
Risks related to government regulations
The regulatory approval processes of the FDA and non-U.S. regulatory agencies are highly complex, lengthy, and inherently unpredictable. If we are unable to obtain regulatory approval for our product candidates, or to do so in a timely manner, we will be unable to generate product revenue and our business will be substantially harmed.
The processes that must be followed to obtain approval by the FDA and non-U.S. regulatory agencies to market a pharmaceutical product are highly complex and unpredictable, and typically take many years following the commencement of clinical trials. A company’s ability to obtain such an approval, and the time necessary to obtain it, depends upon numerous factors, including the type, complexity and novelty of the product candidates involved. In addition, approval policies, regulations or the type and amount of clinical data necessary to gain approval may change during the course of a product candidate’s clinical development and may vary among jurisdictions, which may cause delays in the approval or the decision not to approve an application. Regulatory agencies have substantial discretion in the approval process and may refuse to accept any application or may decide that our data is insufficient for approval and require additional preclinical, clinical or other data. Even if we eventually complete clinical testing and receive approval of any regulatory filing for our product candidates, the FDA and non-U.S. regulatory agencies may approve our product candidates for a more limited indication or a narrower patient population than we originally requested.
Further, development of a company’s product candidates and/or regulatory approval may be impacted or delayed by events beyond our control. For example, our competitors may file citizens’ petitions with the FDA in an attempt to persuade the FDA that our product candidates, or the clinical trials that support their approval, contain deficiencies. Such actions by our competitors could delay or even prevent the FDA from approving any of our NDAs or biologics license applications ("BLAs").
Applications for our product candidates could fail to receive regulatory approval for many reasons, including the following:
59
This complex and lengthy approval process, as well as the unpredictability of the results of clinical trials, may result in us failing to obtain regulatory approval to market any of our product candidates, or a failure to obtain such approval in a timely manner, which could materially adversely affect our business, financial condition, results of operations and growth prospects.
We may face difficulties in commercializing and achieving reimbursement of our products from changes to current regulations and future legislation.
In the United States, the European Union and other jurisdictions there have been a number of legislative and regulatory changes and proposed changes to the healthcare system that could affect our future results of operations. We cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative action, either in the United States or abroad. If we are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we are unable to maintain regulatory compliance, we may be unable to successfully commercialize our products, and may not achieve or sustain profitability.
For example, the Patient Protection and Affordable Care Act of 2010, as amended by the Health Care and Education Reconciliation Act of 2010 (or collectively, the “ACA”), substantially affects the way healthcare is financed by both the government and private insurers, and significantly impacts the U.S. pharmaceutical industry. The ACA contains provisions that can reduce the profitability of drug products through increased rebates for drugs reimbursed by Medicaid programs, extension of Medicaid rebates to Medicaid managed care plans, mandatory discounts for certain Medicare Part D beneficiaries and annual fees based on pharmaceutical companies’ share of sales to federal health care programs. There have been extensive judicial, Congressional and executive branch challenges to certain aspects of the ACA, as well as efforts and proposals to revise or repeal the law and its application, to control the prices at which pharmaceutical products are sold, and to implement other healthcare reform measures. For example, on August 16, 2022, President Biden signed the Inflation Reduction Act of 2022 (the “IRA”) into law, which among other things, extends enhanced subsidies for individuals purchasing health insurance coverage in ACA marketplaces through plan year 2025. The IRA also eliminates the “donut hole” under the Medicare Part D program beginning in 2025 by significantly lowering the beneficiary maximum out-of-pocket cost and through a newly established manufacturer discount program. Such efforts can be expected to continue in the future, but it is unclear what measures will be enacted or implemented, or how they might affect our business.
In addition, other legislative and administrative changes have been adopted in the United States in recent years, and others continue to be proposed. These changes include reductions to payments made under the Medicare program. In addition, during 2021, the Biden administration proposed additional potential legislative and administrative actions to, among other things, reform drug pricing. For example, in July 2021, the Biden administration released an executive order, “Promoting Competition in the American Economy,” with multiple provisions aimed at prescription drugs. In response to Biden’s executive order, on September 9, 2021, the U.S Department of Health and Human Services (“HHS”) released a Comprehensive Plan for Addressing High Drug Prices that outlines principles for drug pricing reform and sets out a variety of potential legislative policies that Congress could pursue as well as potential administrative actions HHS can take to advance these principles. In addition, the IRA, among other things, (i) directs HHS to negotiate the price of certain drugs and biologics covered under Medicare, and subjects drug manufacturers to civil monetary penalties and a potential excise tax by offering a price that is not equal to or less than the negotiated “maximum fair price” under the law, and (ii) imposes rebates under Medicare Part B and Medicare Part D to penalize price increases that outpace inflation.
60
The IRA permits HHS to implement many of these provisions through guidance, as opposed to regulation, for the initial years. HHS has and will continue to issue and update guidance as these programs are implemented. These provisions take effect progressively starting in fiscal year 2023. On August 29, 2023, HHS announced the list of the first ten drugs that will be subject to price negotiations, although the Medicare drug price negotiation program is currently subject to legal challenges. It is currently unclear how the IRA will be effectuated but is likely to have a significant impact on the pharmaceutical industry. In response to the Biden administration’s October 2022 executive order, on February 14, 2023, HHS released a report outlining three new models for testing by the CMS Innovation Center which will be evaluated on their ability to lower the cost of drugs, promote accessibility, and improve quality of care. It is unclear whether the models will be utilized in any health reform measures in the future. Further, on December 7, 2023, the Biden administration announced an initiative to control the price of prescription drugs through the use of march-in rights under the Bayh-Dole Act. On December 8, 2023, the National Institute of Standards and Technology published for comment a Draft Interagency Guidance Framework for Considering the Exercise of March-In Rights which for the first time includes the price of a product as one factor an agency can use when deciding to exercise march-in rights. While march-in rights have not previously been exercised, it is uncertain if that will continue under the new framework.
These recent laws, administrative decisions and proposals, and any new ones that follow, may result in additional reductions in Medicare payments and other healthcare funding, which could have a material adverse effect on customers for our products and product candidates, if approved, and accordingly, on our results of operations.
At the state level, legislatures have increasingly passed legislation and implemented regulations designed to control pharmaceutical product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing. For example, on January 5, 2024, the FDA approved Florida’s Section 804 Importation Program (SIP) proposal to import certain drugs from Canada for specific state healthcare programs. It is unclear how this program will be implemented, including which drugs will be chosen, and whether it will be subject to legal challenges in the United States or Canada. Other states have also submitted SIP proposals that are pending review by the FDA.
We expect that the ACA, the IRA, as well as other healthcare reform measures that have been adopted, or may be adopted in the future, could result in more rigorous healthcare insurance coverage criteria and in additional downward pressure on the price that we receive for any approved product. Any reduction in reimbursement from Medicare or other government programs may result in a similar reduction in payments from private payors. The implementation of cost containment measures or other healthcare reforms may prevent us from being able to generate revenue, attain profitability or commercialize our product candidates, if approved.
In the European Union and other countries, similar political, economic and regulatory developments may affect our ability to profitably commercialize our product candidates, if approved. We cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative action, either in the United States or abroad. As an example, the regulatory landscape related to clinical trials in the EU has evolved. The EU Clinical Trials Regulation, or CTR, which was adopted in April 2014 and repeals the EU Clinical Trials Directive, became applicable on January 31, 2022. The CTR permits trial sponsors to make a single submission to both the competent authority and an ethics committee in each EU member state, leading to a single decision for each EU member state. The assessment procedure for the authorization of clinical trials has been harmonized as well, including a joint assessment of some elements of the application by all EU member states in which the trial is to be conducted, and a separate assessment by each EU member state with respect to specific requirements related to its own territory, including ethics rules. Each EU member state’s decision is communicated to the sponsor through a centralized EU portal, the Clinical Trial Information System, or CTIS. The CTR provides a three-year transition period. The extent to which ongoing clinical trials will be governed by the CTR varies. For clinical trials in relation to which an application for approval was made on the basis of the Clinical Trials Directive before January 31, 2023, the CTD will continue to apply on a transitional basis until January 31, 2025. By that date, all ongoing trials will become subject to the provisions of the CTR. The CTR will apply to clinical trials from an earlier date if the related clinical trial application was made on the basis of the CTR or if the clinical trial has already transitioned to the CTR framework before January 31, 2025.
61
In addition to continuing pressure on prices and cost containment measures, legislative developments at the European Union or at the EU member state level may result in significant additional requirements or obstacles that may increase our operating costs. In many EU member states, healthcare budgetary constraints have resulted in restrictions on the pricing and reimbursement of medicines by relevant health service providers. This could prevent or delay marketing approval of our product candidates, restrict or regulate post-approval activities and affect our ability to commercialize our product candidates, if approved. Moreover, in the European Union, some EU member states may require the completion of additional studies that compare the cost-effectiveness of a particular medicinal product to currently available therapies. This Health Technology Assessment ("HTA"), which is currently governed by the national laws of the individual EU member states, is the procedure according to which the assessment of the public health impact, therapeutic impact and the economic and societal impact of use of a given medicinal product in the national healthcare systems of the individual country is conducted. The outcome of HTA regarding specific medicinal product will often influence the pricing and reimbursement status granted to these products by the competent agencies of individual EU member states. On December 15, 2021, the Health Technology Regulation ("HTA Regulation"), was adopted. The HTA Regulation is intended to boost cooperation among EU member states in assessing health technologies, including new medicinal products, and providing the basis for cooperation at EU level for joint clinical assessments in these areas. When it enters into application in 2025, the HTA Regulation will be intended to harmonize the clinical benefit assessment of HTA across the European Union.
In addition, on April 26, 2023, the European Commission adopted a proposal for a new Directive and Regulation to revise the existing pharmaceutical legislation. If adopted in the form proposed, the recent European Commission proposals to revise the existing EU laws governing authorization of medicinal products may result in a decrease in data and market exclusivity opportunities for our product candidates in the EU. We cannot be sure whether additional legislative changes will be enacted, or whether FDA, European Union, or other jurisdictions’ regulations, guidance or interpretations will be changed, or what the impact of such changes on the marketing approvals of our product candidates, if any, may be. In addition, increased scrutiny by, for example, United States Congress of the FDA approval process may significantly delay or prevent marketing approval, as well as subject us to more stringent product labeling and post-marketing testing and other requirements.
If the FDA does not conclude that OCS-01 satisfies the requirements for the Section 505(b)(2) regulatory approval pathway, or if the requirements under Section 505(b)(2) are not as we expect, the approval pathway for OCS-01 will likely take significantly longer, cost significantly more and entail significantly greater complications and risks than anticipated, and in either case may not be successful.
We plan to seek FDA approval through the Section 505(b)(2) regulatory pathway for OCS-01. The Hatch-Waxman Amendments added Section 505(b)(2) (“Section 505(b)(2)”) to the Federal Food, Drug and Cosmetic Act (the “FDCA”). Section 505(b)(2) permits the submission of an NDA where at least some of the information required for approval comes from studies that were not conducted by or for the applicant and for which the applicant has not obtained a right of reference. Section 505(b)(2), if applicable to us under the FDCA, would allow an NDA to rely in part on data in the public domain or the FDA’s prior conclusions regarding the safety and effectiveness of approved drug products, which could expedite the development program for OCS-01 by potentially decreasing the amount of preclinical or clinical data that we would need to generate in order to obtain FDA approval.
If we cannot pursue the Section 505(b)(2) regulatory pathway for OCS-01, we may need to conduct additional clinical trials, provide additional data and information, and meet additional standards for regulatory approval. If this were to occur, the time and financial resources required to obtain FDA approval for OCS-01, and complications and risks associated with OCS-01, would likely substantially increase. Moreover, our inability to pursue the Section 505(b)(2) regulatory pathway would likely result in new competitive products reaching the market more quickly than OCS-01, which would likely adversely impact our competitive position and prospects. Even if we can pursue the Section 505(b)(2) regulatory pathway, we cannot assure you that OCS-01 will receive the requisite approvals for commercialization.
In addition, notwithstanding the approval of products by the FDA under Section 505(b)(2), certain pharmaceutical companies and others have objected to the FDA’s interpretation of Section 505(b)(2). If the FDA’s interpretation of Section 505(b)(2) is successfully challenged, the FDA may change its 505(b)(2) policies and practices, which could delay or even prevent the FDA from approving any NDA that we submit under Section 505(b)(2). In addition, the pharmaceutical industry is highly competitive, and Section 505(b)(2) NDAs are subject to special requirements designed to protect the patent rights of sponsors of previously approved drugs that are referenced in a Section 505(b)(2)
62
NDA. These requirements may give rise to patent litigation and mandatory delays in approval of our NDAs for up to thirty (30) months or longer depending on the outcome of any litigation. It is not uncommon for the owner of the NDA of an approved product to file a citizen petition with the FDA seeking to delay approval of, or impose additional approval requirements for, pending competing products. If successful, such petitions could significantly delay, or even prevent, the approval of a new product. However, even if the FDA ultimately denies such a petition, the FDA may substantially delay approval while it considers and responds to the petition. In addition, even if we are able to utilize the Section 505(b)(2) regulatory pathway, there is no guarantee this would ultimately lead to earlier approval.
Moreover, even if OCS-01 is approved under Section 505(b)(2), the approval may be subject to limitations on the indicated uses for which the product may be marketed or to other conditions of approval, or may contain requirements for costly post-marketing testing and surveillance to monitor the safety or efficacy of the product.
The U.S. Government and non-U.S. regulatory agencies actively enforce laws and regulations regarding the promotion of pharmaceutical products, and if we are found to have violated any such laws or regulations, we may be subject to significant liability.
The FDA and other U.S. Government agencies and non-U.S. regulatory agencies strictly regulate the manner in which medicinal products may be marketed. In particular, a medicinal product may not be promoted for uses that are not approved by the FDA or such other regulatory agencies as reflected in the product’s approved labeling. In addition, sales, marketing and business arrangements in the health care industry are subject to extensive laws and regulations intended to prevent fraud, misconduct, kickbacks, self-dealing and other abusive practices. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, customer incentive programs and other business arrangements. Such laws, and the application of those laws, are complex and evolving.
If we are found to have improperly promoted the sale of any of our product candidates, if approved, such as through the promotion of the off-label use of those products, or through kickbacks or fraud, or through any other conduct or activity deemed to be unlawful, then we may become subject to significant liability. For example, if we receive marketing approval for a product as a treatment for a disease, physicians may nevertheless choose to prescribe the product for their patients in a manner that is inconsistent with the approved label. If we are found to have promoted such off-label uses, we may become subject to significant liability. The U.S. federal government has levied large civil and criminal fines against companies for alleged improper promotion of off-label use and has enjoined several companies from engaging in off-label promotion. The FDA has also requested that companies enter into consent decrees or permanent injunctions under which specified promotional conduct is changed or curtailed. If we cannot successfully manage the promotion of our product candidates, if approved, we could become subject to significant liability, which would materially adversely affect our business, growth prospects, operating results and financial condition.
In the EU, the advertising and promotion of medicinal products are subject to both EU and EU member states’ laws governing promotion of medicinal products, interactions with physicians and other healthcare professionals, misleading and comparative advertising and unfair commercial practices. Although general requirements for advertising and promotion of medicinal products are established under EU legislation, the details are governed by regulations in individual EU member states and can differ from one country to another. For example, applicable laws require that promotional materials and advertising in relation to medicinal products comply with the product’s Summary of Product Characteristics ("SmPC"), as approved by the competent agencies in connection with a marketing authorization. The SmPC is the document that provides information to physicians concerning the safe and effective use of the product. Promotional activity that does not comply with the SmPC is considered off-label and is prohibited in the EU. Direct-to-consumer advertising of prescription medicinal products is also prohibited in all EU member states. The competent regulatory agencies of the EU member states actively enforce the laws and regulations governing promotion of medicinal products. If we are found to have undertaken improper promotional activities we may be subject to significant civil, criminal and administrative penalties, as well as reputational harm, which could materially adversely affect our business, financial condition, results of operations and growth prospects.
Our employees, independent contractors, consultants, principal investigators, CROs, suppliers and vendors may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements.
63
We are exposed to the risk that our employees, independent contractors, consultants, principal investigators, CROs, suppliers, vendors and other third parties with which we do business may engage in misconduct or other improper activities. Misconduct by these parties could include failures to comply with federal and state health care fraud and abuse laws and regulations and equivalent foreign laws, FDA regulations and equivalent regulations of foreign agencies, requirements to provide accurate information to the FDA or equivalent foreign agencies, data privacy and security laws and requirements to accurately report financial information or data or to disclose unauthorized activities to us. Misconduct by these parties could also involve the improper use of information obtained in the course of clinical trials, which could result in regulatory sanctions and serious harm to our reputation. Although we have adopted a code of business conduct and ethics with respect to our employees, agents and contractors, it is not always possible to identify and deter misconduct by these parties, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to comply with these laws or regulations. Additionally, we are subject to the risk that a person or government could allege such fraud or other misconduct, even if none occurred. If any such actions are instituted against us, and we are not successful in defending or asserting our rights, those actions could have a significant impact on our business, including the imposition of significant penalties, including civil, criminal and administrative penalties, damages, fines, disgorgement, individual imprisonment, exclusion from participation in government funded healthcare programs, such as Medicare and Medicaid and equivalent foreign health insurance programs, integrity oversight and reporting obligations, contractual damages, reputational harm, diminished profits and future earnings and the curtailment or restructuring of our operations.
Obtaining and maintaining regulatory approval of our product candidates in one jurisdiction does not mean that we will be successful in obtaining regulatory approval of our product candidates in other jurisdictions. The FDA and non-U.S. regulatory agencies may not accept data from trials conducted in locations outside of their jurisdiction.
Obtaining and maintaining regulatory approval of our product candidates in one jurisdiction does not guarantee that we will be able to obtain or maintain regulatory approval in any other jurisdiction. For example, even if the FDA approves a drug candidate for an indication in the U.S., comparable regulatory agencies in foreign jurisdictions must also approve the manufacturing, marketing and promotion and reimbursement of the product in those countries. In addition, a failure or delay in obtaining regulatory approval in one jurisdiction may have a negative effect on the regulatory approval process in others. Approval procedures vary among jurisdictions and can involve requirements and administrative review periods different from those in the U.S., including additional preclinical studies or clinical trials, since clinical trials conducted in one jurisdiction may not be accepted by regulatory agencies in other jurisdictions. In many jurisdictions outside the U.S., a product candidate must be approved for reimbursement before it can be approved for sale in that jurisdiction. In some cases, the price that we intend to charge for our products is also subject to approval.
Obtaining non-U.S. regulatory approvals and establishing and maintaining compliance with non-U.S. regulatory requirements could result in significant difficulties and costs for us and could delay or prevent the introduction of our product candidates, if approved, in certain countries. If we or any future collaborator fail to comply with the regulatory requirements in international markets or fail to receive applicable marketing approvals, then our target market will be reduced and our ability to realize the full market potential of our product candidates, if approved, will be harmed.
Our business operations and current and future relationships with healthcare professionals, clinical investigators, consultants, patient organizations, commercial partners, customers, CROs and third-party payors in connection with our current and future business activities may be subject to federal, state and foreign healthcare fraud and abuse laws, false claims laws, transparency laws, government price reporting, and health information privacy and security laws, which could expose us to, among other things, criminal sanctions, civil penalties, contractual damages, exclusion from governmental healthcare programs, reputational harm, administrative burdens and diminished profits and future earnings.
Healthcare providers and third-party payors play a primary role in the recommendation and prescription of any product candidates for which we obtain marketing approval. Our current and future arrangements with healthcare professionals, including physicians, clinical investigators, CROs, third-party payors and customers may expose it to broadly applicable fraud and abuse and other healthcare laws and regulations that may constrain the business or financial arrangements and relationships through which we market, sell and distribute our products for which we obtain marketing approval. Restrictions under applicable healthcare laws and regulations include the following:
64
State and foreign laws require biotechnology companies to comply with the biotechnology industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the government and may require drug manufacturers to report information related to payments and other transfers of value to physicians and other healthcare providers or marketing expenditures. Some state and foreign laws require biotechnology companies to report information on the pricing of certain drug products.
65
Certain state and local jurisdictions require the registration of pharmaceutical sales representatives. State, federal and foreign laws also govern the privacy and security of health information in some circumstances, many of which differ from each other in significant ways and often are not preempted by HIPAA, thus complicating compliance efforts.
Efforts to ensure that our current and future business arrangements with third parties will comply with applicable healthcare laws and regulations will involve ongoing substantial costs. It is possible that governmental agencies will conclude that our business practices, including the provision of compensation for consulting services to physicians and other healthcare providers, some of whom may be in a position to recommend, purchase and/or prescribe our product candidates, if approved, may not comply with current or future statutes, regulations or case law involving applicable fraud and abuse or other healthcare laws and regulations. If our operations are found to be in violation of any of these laws or any other governmental regulations that may apply to it, we may be subject to significant penalties, including civil, criminal and administrative penalties, damages, fines, disgorgement, imprisonment, exclusion from participation in government funded healthcare programs, such as Medicare and Medicaid or similar programs in other countries or jurisdictions, integrity oversight and reporting obligations, contractual damages, reputational harm, diminished profits and future earnings and the curtailment or restructuring of our operations. Defending against any such actions can be costly, time-consuming and may require significant financial and personnel resources. Therefore, even if we are successful in defending against any such actions that may be brought against it, our business may be impaired. Further, if any of the physicians or other healthcare providers or entities with whom we expect to do business is found to be not in compliance with applicable laws, they may be subject to criminal, civil or administrative sanctions, including exclusions from government funded healthcare programs, which could have an adverse effect on our business and reputation.
Our business activities are subject to the FCPA and similar anti-bribery and anti-corruption laws of other countries in which we operate, as well as U.S. and certain foreign export controls, trade sanctions, and import laws and regulations. Compliance with these legal requirements could limit our ability to compete in foreign markets and subject us to liability if we violate them.
We may conduct clinical trials in countries other than the United States. In addition, we have entered into a license agreement with Accure, a biotechnology company headquartered in Barcelona, Spain. Our business activities are subject to the U.S. Foreign Corrupt Practices Act ("FCPA"), and similar anti-bribery or anti-corruption laws, regulations or rules of Switzerland and other countries in which we operate. Anti-corruption laws, including the FCPA, generally prohibit offering, promising, giving or authorizing others to give anything of value, either directly or indirectly, to a government official in order to influence official action or otherwise obtain or retain business. The FCPA also requires public companies to make and keep books and records that accurately and fairly reflect the transactions of the corporation and to devise and maintain an adequate system of internal accounting controls. Our business is heavily regulated and therefore involves significant interaction with public officials, potentially including officials of foreign governments. Additionally, although none of our product candidates is yet approved for sale in any country, in many countries other than the U.S., the healthcare providers who prescribe pharmaceuticals like our product candidates are employed by their government, and the purchasers of pharmaceuticals are government entities. Therefore, any future dealings by us with these prescribers and purchasers may be subject to regulation under the FCPA and other applicable anti-corruption laws.
The SEC and Department of Justice have increased their FCPA enforcement activities with respect to biotechnology and pharmaceutical companies. There is no certainty that all of our employees, agents or contractors, or those of our affiliates, will comply with all applicable anti-corruption laws and regulations, particularly given the high level of complexity of these laws. Violations of these laws and regulations could result in fines, criminal sanctions against us, our officers or our employees, the closing down of our facilities, cessation of business activities in certain countries, implementation of compliance programs and prohibitions on the conduct of our business. Any such violations could include prohibitions on our ability to offer our products, if approved, in one or more countries and could materially damage our reputation, our brand, international activities, our ability to attract and retain employees and our business, growth prospects, operating results and financial condition.
In addition, our products may be subject to U.S. and foreign export controls, trade sanctions and import laws and regulations. Governmental regulation of the import or export of our products, or our failure to obtain any required import or export authorization for our products, when applicable, could harm our international sales and adversely affect our revenue. Compliance with applicable regulatory requirements regarding the export of our products may create delays in the introduction of our products in international markets or, in some cases, prevent the export of our products to some countries altogether.
66
Furthermore, export control laws and economic sanctions may prohibit the shipment of certain products and services to specified countries, governments, and persons. If we fail to comply with export and import regulations and such economic sanctions, we may be fined or other penalties could be imposed, including a denial of certain export privileges. Moreover, any new export or import restrictions, new legislation or shifting approaches in the enforcement or scope of existing regulations, or in the countries, persons, or technologies targeted by such regulations, could result in decreased use of our products by, or in our decreased ability to export products to existing or potential customers with international operations. Any decreased use of our products or limitation on our ability to export or sell access to our products could adversely affect our business.
Disruptions at the FDA, the SEC and other government agencies and comparable non-U.S. regulatory agencies caused by funding shortages or global health concerns could hinder their ability to hire and retain key leadership and other personnel, prevent new products and services from being developed or commercialized in a timely manner, or otherwise prevent those agencies from performing normal business functions on which the operation of our business may rely, which could negatively impact our business.
The ability of the FDA and comparable non-U.S. regulatory agencies to review and approve new products can be affected by a variety of factors, including government budget and funding levels, our ability to hire and retain key personnel and accept the payment of user fees, statutory, regulatory, and policy changes, and other events that may otherwise affect the ability of the FDA and comparable non-U.S. regulatory agencies to perform routine functions. Average review times at the FDA and comparable non-U.S. regulatory agencies have fluctuated in recent years as a result. In addition, government funding of the SEC and other government agencies on which our operations may rely, including those that fund research and development activities, is subject to the political process, which is inherently fluid and unpredictable.
Disruptions at the FDA, other agencies, and comparable non-U.S. regulatory agencies may slow the time necessary for new drugs to be reviewed or approved, which could adversely affect our business. For example, in recent years, including in 2013, 2018 and 2019, the U.S. government shut down several times, and in 2020 and 2021 the FDA diverted significant resources to handle the SARS-CoV-2 virus public health emergency and pandemic. Certain regulatory agencies, such as the FDA and the SEC, have had to furlough critical FDA, SEC and other government employees for a time, and to stop critical activities in response to such events, and may be required to do so again in the future.
If such disruptions recur, or if a prolonged government shutdown occurs, it could significantly impact the ability of the FDA and comparable non-U.S. regulatory agencies to timely review and process our regulatory submissions, which could have a material adverse effect on our business. Further, in our operations as a public company, future government disruptions or shutdowns could impact our ability to access the public markets and obtain necessary capital in order to properly capitalize and continue our operations.
If we fail to comply with environmental, health and safety laws and regulations, we could become subject to fines or penalties or incur costs that could have a material adverse effect on our business.
We and any contract manufacturers and suppliers we engage are subject to numerous federal, state, and local environmental, health, and safety laws, regulations, and permitting requirements, including those governing laboratory procedures; the generation, handling, use, storage, treatment, and disposal of hazardous and regulated materials and waste; the emission and discharge of hazardous materials into the ground, air, and water; and employee health and safety. Our operations may involve the use of hazardous and flammable materials, including chemicals and biological and radioactive materials. Our operations also may produce hazardous waste. We generally contract with third parties for the disposal of these materials and wastes. We cannot eliminate the risk of contamination or injury from these materials. In the event of contamination or injury resulting from our use of hazardous materials, we could be held liable for any resulting damages, and any liability could exceed our resources. Under certain environmental laws, we could be held responsible for costs relating to any contamination at our current or past facilities and at third-party facilities. We also could incur significant costs associated with civil or criminal fines and penalties.
Compliance with applicable environmental laws and regulations may be expensive, and current or future environmental laws and regulations may impair our research, product development and manufacturing efforts. In addition, we cannot entirely eliminate the risk of accidental injury or contamination from these materials or waste.
67
Although we maintain workers’ compensation insurance to cover us for costs and expenses we may incur due to injuries to our employees resulting from the use of hazardous materials, this insurance may not provide adequate coverage against potential liabilities. We do not currently maintain insurance for environmental liability or toxic tort claims that may be asserted against us in connection with storage or disposal of hazardous and flammable materials, including chemicals and biological materials. Accordingly, in the event of contamination or injury, we could be held liable for damages or be penalized with fines in an amount exceeding our resources, and our clinical trials or regulatory approvals could be suspended, which could have a material adverse effect on business, financial condition, results of operations and growth prospects.
In addition, we may incur substantial costs in order to comply with current or future environmental, health and safety laws and regulations. These current or future laws and regulations may impair research, development or commercialization efforts. Failure to comply with these laws and regulations also may result in substantial fines, penalties or other sanctions that could have a material adverse effect on our business, reputation and growth prospects.
Risks related to domicile in Switzerland and being foreign private issuer
We are a Swiss stock corporation. The rights of its shareholders may be different from the rights of shareholders in companies governed by the laws of U.S. jurisdictions.
We are a Swiss stock corporation. Our corporate affairs are governed by our articles of association and by the laws governing companies, including listed companies, incorporated in Switzerland. The rights of our shareholders and the responsibilities of members of our board of directors may be different from the rights and obligations of shareholders and directors of companies governed by the laws of the United States. In the performance of its duties, our board of directors is required by Swiss law to consider the interests of the Company, and may also have regard to the interests of our shareholders, our employees and other stakeholders, in all cases with due observation of the principles of reasonableness and fairness. It is possible that some of these parties will have interests that are different from, or in addition to, your interests as a shareholder. Swiss corporate law limits the ability of our shareholders to challenge resolutions made or other actions taken by our board of directors in court.
Our shareholders generally are not permitted to file a suit to reverse a decision or an action taken by our board of directors, but are instead only permitted to seek damages for breaches of fiduciary duty. As a matter of Swiss law, shareholder claims against a member of our board of directors for breach of fiduciary duty would have to be brought to the competent courts at our registered office, currently in Zug, Switzerland. In addition, under Swiss law, any claims by shareholders against the Company must be brought exclusively to the competent courts at our registered office, currently in Zug, Switzerland. U.S.-style class actions and derivative actions are not available under Swiss law. There can be no assurance that Swiss law will not change in the future, which could adversely affect the rights of our shareholders, or that Swiss law will protect our shareholders in a similar fashion as under U.S. corporate law principles.
Our Ordinary Shares are not listed in Switzerland, our home jurisdiction. As a result, certain Swiss law provisions designed to protect shareholders in the event of a public takeover offer or change of control transaction will not apply.
The Swiss rules that require investors to disclose their interest in a company if they reach, exceed or fall below certain ownership thresholds only applies to issuers that have a listing (including a secondary listing) for their equity securities in Switzerland. Since the Ordinary Shares are listed exclusively on The Nasdaq Global Market, a U.S. market, the disclosure obligations regarding major shareholdings according to art. 120 of the Swiss Financial Markets Infrastructure Act and its implementing provisions do not apply to us. Likewise, the Swiss takeover regime does not apply to us. In particular, the duty to make a mandatory bid offer for all outstanding listed equity securities of a company by any person or group of persons that acquires more than one third of a company’s voting rights does not apply to us. In addition, the Swiss takeover regime imposes certain restrictions and obligations on bidders in a voluntary public takeover offer that are designed to protect shareholders. However, these protections are applicable only to issuers that list their equity securities in Switzerland and, because the Ordinary Shares are listed exclusively on The Nasdaq Global Market, are not applicable to us. Furthermore, since Swiss law restricts our ability to implement rights plans or U.S.-style “poison pills,” Our ability to resist an unsolicited takeover attempt or to protect minority shareholders in the event of a change of control transaction may be limited. Therefore, our shareholders may not be protected in the same degree in a public takeover offer or a change-of-control transaction as are shareholders in a Swiss company listed in Switzerland.
68
U.S. shareholders may not be able to obtain judgments or enforce civil liabilities against us or our executive officers or members of our board of directors.
We are a corporation organized and incorporated under the laws of Switzerland with registered office and domicile in Zug, Switzerland, and the majority of its assets are located within Switzerland. Moreover, a number of our directors and executive officers are not residents of the United States, and all or a substantial portion of the assets of such persons are or may be located outside the United States. As a result, investors may not be able to effect service of process within the United States upon us or upon such persons, or to enforce judgments obtained against us or such persons in U.S. courts, including judgments in actions predicated upon the civil liability provisions of the federal securities laws of the United States. There is doubt that a lawsuit based upon United States federal or state securities laws could be brought in an original action in Switzerland and that a judgment of a U.S. court based upon United States securities laws would be enforced in Switzerland.
The United States and Switzerland currently do not have a treaty providing for the reciprocal recognition and enforcement of judgments, other than arbitration awards, in civil and commercial matters. Consequently, a final judgment for payment given by a court in the United States, whether or not predicated solely upon U.S. securities laws, may not be enforceable in Switzerland, please see the section entitled “Enforcement of Civil Liabilities.”
Our status as a Swiss stock corporation means that our shareholders enjoy certain rights that may limit its flexibility to raise capital, issue dividends and otherwise manage ongoing capital needs.
Swiss law reserves for approval by shareholders certain corporate actions over which a board of directors would have authority in some other jurisdictions. For example, the payment of dividends and the cancellation of treasury shares must be approved by shareholders. Swiss law also requires that our shareholders themselves resolve to, or authorize its board of directors to, increase our share capital. While its shareholders may introduce a capital band pursuant to which share capital that can be issued by its board of directors without additional shareholder approval, Swiss law limits this capital band to 50.0% of the share capital registered in the commercial register at the time of the introduction of the capital band. The capital band, furthermore, has a limited duration of up to five years and must be renewed by the shareholders from time to time thereafter in order to be available for raising capital. Additionally, subject to specified exceptions, including exceptions explicitly described in our articles of association, Swiss law grants pre-emptive rights to existing shareholders to subscribe for new issuances of shares, which may be limited or withdrawn under certain conditions. Swiss law also does not provide as much flexibility in the various rights and regulations that can attach to different classes of shares as do the laws of some other jurisdictions. These Swiss law requirements relating to our capital management may limit our flexibility, and situations may arise where greater flexibility would have provided benefits to its shareholders.
Shareholders outside of the United States may not be able to exercise pre-emptive rights in future issuances of equity or other securities that are convertible into equity.
Under Swiss corporate law, shareholders may receive certain pre-emptive rights to subscribe on a pro-rata basis for issuances of equity securities or other securities that are convertible into equity securities. Due to the laws and regulations in certain jurisdictions, however, shareholders who are not residents of the United States may not be able to exercise such rights unless we take action to register or otherwise qualify the rights offering, including, for example, by complying with prospectus requirements under the laws of that jurisdiction. There can be no assurance that we will take any action to register or otherwise qualify an offering of subscription rights or shares under the laws of any jurisdiction other than the United States where the offering of such rights is restricted. If shareholders in such jurisdictions were unable to exercise their subscription rights, their ownership interest in the Company will be diluted.
Anti-takeover provisions in our articles of association could make an acquisition of the Company, which may be beneficial to its shareholders, more difficult.
Our articles of association contain provisions that may have the effect of discouraging, delaying or preventing a change in control of the Company that shareholders may consider favorable, including transactions in which its shareholders may receive a premium for their shares. Our articles of association include provisions that:
69
These and other provisions of our articles of association, alone or together, could delay or prevent takeovers and changes in control. Please see the sections entitled “Description of Securities” and “Comparison of Shareholder Rights.” Any provision of the articles of association that has the effect of delaying or preventing a change in control could limit the opportunity for shareholders to receive a premium for their shares of our share capital and could also affect the price that some investors are willing to pay for Ordinary Shares.
We are a foreign private issuer and, as a result, not subject to U.S. proxy rules and are subject to Exchange Act reporting obligations that, to some extent, are more lenient and less frequent than those of a U.S. domestic public company.
We report under the Exchange Act as a non-U.S. company with foreign private issuer status. Because we qualify as a foreign private issuer under the Exchange Act, we are exempt from certain provisions of the Exchange Act that are applicable to U.S. domestic public companies, including: (i) the sections of the Exchange Act regulating the solicitation of proxies, consents or authorizations in respect of a security registered under the Exchange Act; (ii) the sections of the Exchange Act requiring insiders to file public reports of their share ownership and trading activities and liability for insiders who profit from trades made in a short period of time; and (iii) the rules under the Exchange Act requiring the filing with the SEC of quarterly reports on Form 10-Q containing unaudited financial and other specified information, or current reports on Form 8-K, upon the occurrence of specified significant events. In addition, foreign private issuers are not required to file their annual report on Form 20-F until four months after the end of each financial year, while U.S. domestic issuers that are accelerated filers are required to file their annual report on Form 10-K within 75 days after the end of each fiscal year. Foreign private issuers are also exempt from the Regulation Fair Disclosure, aimed at preventing issuers from making selective disclosures of material information. As a result of the above, you may not have the same protections afforded to shareholders of companies that are not foreign private issuers.
As a foreign private issuer and as permitted by the listing requirements of Nasdaq, we have the option to follow certain home country governance practices rather than the corporate governance requirements of Nasdaq.
We are a foreign private issuer. As a result, in accordance with Nasdaq Listing Rule 5615(a)(3), we may choose, and have chosen, to comply with home country governance requirements and certain exemptions thereunder rather than complying with certain of the corporate governance requirements of the Nasdaq.
Swiss law does not require that a majority of our board of directors consist of independent directors. Its board of directors therefore may include fewer independent directors than would be required if we were subject to Nasdaq Listing Rule 5605(b)(1). In addition, we are not subject to Nasdaq Listing Rule 5605(b)(2), which requires that independent directors regularly have scheduled meetings at which only independent directors are present.
70
Although Swiss law also requires that we set up a remuneration committee, we may follow home country requirements with respect to such committee. Among other things, Swiss law does not require that all or a majority of the remuneration committee consist of independent directors.
We may also choose to take advantage of other exemptions including but not limited to the exemption from the requirement to obtain shareholder approval for certain issuances of securities, including shareholder approval of share option plans using conditional share capital approved by the shareholders.
Our articles of association provide for an independent proxy elected by its shareholders, who may represent its shareholders of record at a general meeting of shareholders, and it must provide shareholders of record with an agenda and other relevant documents for the general meeting of shareholders. However, Swiss law does not have a regulatory regime for the solicitation of proxies, thus our practice may vary from the requirement of Nasdaq Listing Rule 5620(b), which sets forth certain requirements regarding the solicitation of proxies. Furthermore, in accordance with Swiss law and generally accepted business practices, our articles of association do not provide quorum requirements generally applicable to general meetings of shareholders. Our practice thus varies from the requirement of Nasdaq Listing Rule 5620(c), which requires an issuer to provide in its bylaws for a generally applicable quorum, and that such quorum may not be less than one-third of the outstanding voting stock.
For an overview of our corporate governance principles, please see the section entitled “Corporate Governance.” As a result of the above, you may not have the same protections afforded to shareholders of companies that are not foreign private issuers.
We may lose our foreign private issuer status, which would then require us to comply with the domestic reporting requirements of the Exchange Act and cause us to incur significant legal, accounting and other expenses.
We are a foreign private issuer and therefore are not required to comply with all of the periodic disclosure and current reporting requirements of the Exchange Act applicable to U.S. domestic issuers. In order to maintain our status as a foreign private issuer, either (i) a majority of its ordinary shares must be either directly or indirectly owned of record by non-residents of the United States; or (ii) (a) a majority of its executive officers or directors may not be United States citizens or residents, (b) more than 50.0% of its assets cannot be located in the United States, and (c) its business must be administered principally outside the United States. If it lost this status, it would be required to comply with the Exchange Act reporting and other requirements applicable to U.S. domestic issuers, which are more detailed and extensive than the requirements for foreign private issuers. Among other things, we would be required under current SEC rules to prepare its financial statements in accordance with generally accepted accounting principles in the United States, rather than IFRS Accounting Standards, which would involve significant time and cost and could result in variations, which could be material, between historical financial results reported under IFRS Accounting Standards and as reported under U.S. GAAP. It may also be required to make changes in its corporate governance practices in accordance with various SEC and stock exchange rules. The regulatory and compliance costs to us under U.S. securities laws if we are required to comply with the reporting requirements applicable to a U.S. domestic issuer may be significantly higher than the cost it would incur as a foreign private issuer. As a result, we expect that a loss of foreign private issuer status would increase our legal and financial compliance costs and would make some activities highly time-consuming and costly. If it loses its foreign private issuer status and is unable to devote adequate funding and the resources needed to maintain compliance with U.S. securities laws, while continuing its operations, we could be forced to deregister with the SEC. A deregistration would substantially reduce or effectively terminate the trading of its securities in the United States. We also expect that if we were required to comply with the rules and regulations applicable to U.S. domestic issuers, it would make it more difficult and expensive for it to obtain director and officer liability insurance, and it may be required to accept reduced coverage or incur substantially higher costs to obtain coverage. These rules and regulations could also make it more difficult for us to attract and retain qualified members of our board of directors.
As a result of changes in tax laws, treaties, rulings, regulations or agreements, or their interpretation, of Switzerland or any other country in which we operate, the loss of a major tax dispute or a successful challenge to our operating structure, intercompany pricing policies or the taxable presence of our key subsidiaries in certain countries, or other factors, our effective income tax rates may increase in the future, which could adversely affect our net income and cash flows.
71
We operate in multiple jurisdictions and our profits are taxed pursuant to the tax laws of these jurisdictions. The tax laws applicable to our business activities, however, are subject to changes in interpretation. Our tax position could be adversely impacted by changes in tax rates, tax laws, tax practice, tax treaties or tax regulations or changes in the interpretation thereof by the tax authorities in jurisdictions in which we do business. Our effective income tax rate may be affected by changes in or interpretations of tax laws, treaties, rulings, regulations or agreements in any given jurisdiction, the resolution of issues arising from any future tax audits with various tax authorities, utilization of net operating loss and tax credit carryforwards, changes in geographical allocation of income and expense, and changes in management’s assessment of matters such as the realizability of deferred tax assets. In the past, we have experienced fluctuations in our effective income tax rate. Our actual tax rate may vary from our expectation and that variance may be material. Our effective income tax rate in a given fiscal year reflects a variety of factors that may not be present in the succeeding fiscal year or years. There is no assurance that our effective income tax rate will not change in future periods.
We file Swiss and non-Swiss tax returns. We are subject to tax audits, examinations and assessments in various jurisdictions. If any tax authority successfully challenges our operational structure, allocation of income by tax jurisdiction, or amounts paid between our affiliated companies pursuant to our intercompany arrangements or transfer pricing policies, if any tax authority successfully asserts that we are subject to income, withholding or other taxes in a jurisdiction by reason of our activities and operations or our other taxable presence in such jurisdiction, if the terms of certain income tax treaties are interpreted in a manner that is adverse to our structure, or if we lose a material tax dispute in any country, our effective income tax rate could increase. A tax authority may take the position that material income or other tax liabilities, interest and penalties are payable by us, in which case, we expect that we might contest such assessment. Contesting such an assessment may be lengthy and costly and if we were unsuccessful in disputing the assessment, the implications could increase our anticipated effective tax rate, which could adversely affect our profitability. If our effective income tax rate increases in future periods, our net income and cash flows could be adversely affected, including in future tax years.
Due to the Swiss corporate tax law reform that took effect on January 1, 2020, all Swiss cantons, including the Canton of Zug and the Canton of Vaud, have abolished the cantonal tax privileges. Therefore, since January 1, 2020, we are subject to standard cantonal taxation. The standard effective corporate tax rate in Zug, Canton of Zug and Lausanne, Canton of Vaud, can change from time to time. The standard combined (federal, cantonal, communal) effective corporate income tax rate, except for dividend income for which we could claim a participation exemption, for 2023 in Zug, Canton of Zug will be approximately 11.8% and in Lausanne (Ecublens), Vaud will be approximately 13.6%.
The Organisation for Economic Co-operation and Development (OECD) minimum tax rate will be implemented in Switzerland by means of an ordinance. The people and cantons approved the necessary constitutional amendment in a popular vote on June 18, 2023. During its meeting on December 22, 2023, the Federal Council decided to implement the minimum tax rate with the introduction of a supplementary tax (Qualified Domestic Minimum Top-up Tax) in Switzerland as from January 1, 2024. The introduction of the international supplementary taxes (Income Inclusion Rule and Under Tax Payment Rule) has been postponed to a later date. Within six years, the Federal Council must additionally submit to the Swiss Parliament a federal act that replaces the ordinance. Since we do not meet the threshold of a worldwide turnover of 750 million euro, we are not affected by the OECD minimum tax rate.
We urge our shareholders to consult with their legal and tax advisors with respect to the potential tax consequences of investing in or holding the Ordinary Shares.
Exchange rate fluctuations or abandonment of the euro currency may materially affect our results of operations and financial condition.
Due to the international scope of our operations, our assets, earnings and cash flows are influenced by movements in exchange rates of several currencies, particularly regarding U.S. dollars, euros and Swiss francs. Our functional currency is the Swiss franc and the majority of our operating expenses are paid in Swiss francs. Further, potential future revenue may be derived from abroad, particularly from the United States and the European Union. As a result, our business and share price may be affected by fluctuations in foreign exchange rates between the Swiss franc, the euro, the U.S. dollar and these other currencies, which may also have a significant impact on our reported results of operations and cash flows from period to period. Besides our natural hedging, currently, we do not have any exchange rate hedging arrangements in place.
72
In addition, the possible abandonment of the euro by one or more members of the European Union could materially affect our business in the future. Despite measures taken by the European Union to provide funding to certain European Union member states in financial difficulties and by a number of European countries to stabilize their economies and reduce their debt burdens, it is possible that the euro could be abandoned in the future as a currency by countries that have adopted its use. This could lead to the re-introduction of individual currencies in one or more European Union member states, or in more extreme circumstances, the abandonment of the euro or the dissolution of the European Union. The effects on our business of a potential dissolution of the European Union, the exit of one or more European Union member states from the European Union or the abandonment of the euro as a currency, are impossible to predict with certainty, and any such events could have a material adverse effect on our business, financial condition and results of operations.
Risks Related to Ownership of our Ordinary Shares and Warrants and our Status as a Public Company
We have and will incur increased costs as a result of operating as a public company, and our management will devote substantial time to new compliance initiatives.
We have and will incur significant legal, accounting and other expenses that we did not incur as a private company, and these expenses may increase even more if and when we are no longer an emerging growth company, as defined in Section 2(a) of the Securities Act. As a public company, we are subject to the reporting requirements of the Exchange Act, the Sarbanes-Oxley Act, the Dodd-Frank Wall Street Reform and Consumer Protection Act, as well as rules adopted, and to be adopted, by the SEC and Nasdaq. Our management and other personnel have and will need to devote a substantial amount of time to these compliance initiatives. Moreover, we expect that compliance with these rules and regulations will continue to substantially increase our legal and financial compliance costs and to make some activities more time consuming and costly. The increased costs have and will increase our net loss. For example, we expect these rules and regulations will continue to make it more difficult and more expensive for us to obtain director and officer liability insurance and we may be forced to accept reduced policy limits or incur substantially higher costs to maintain the same or similar coverage. We cannot predict or estimate the amount or timing of additional costs we may incur to respond to these requirements. The impact of these requirements could also make it more difficult for us to attract and retain qualified persons to serve on our board of directors, on our board advisors or as executive officers.
Our management has limited experience in operating a public company.
Our executive officers have limited experience in the management of a publicly traded company. Our management team may not successfully or effectively manage the transition to a public company that is and will be subject to significant regulatory oversight and reporting obligations under federal securities laws. Their limited experience in dealing with the increasingly complex laws pertaining to public companies could be a disadvantage in that it is likely that an increasing amount of their time may be devoted to these activities. This in turn may result in less time being devoted to our management and growth. We may not have adequate personnel with the appropriate level of knowledge, experience, and training in the accounting policies, practices or internal controls over financial reporting required of public companies in the United States. The development and implementation of the standards and controls necessary for us to achieve the level of accounting standards required of a public company in the United States may require costs greater than expected. It is possible that we will be required to expand our employee base and hire additional employees to support our operations as a public company, which will increase our operating costs in future periods.
The market price and trading volume of our Ordinary Shares and Warrants may be volatile and could decline significantly, and may make it more difficult for you to sell your Ordinary Shares.
The Nasdaq stock market, on which our Ordinary Shares and Warrants are listed under the symbols OCS and OCSAW, respectively, has from time to time experienced significant price and volume fluctuations. The market price of Ordinary Shares and Warrants may be volatile and could decline significantly. Generally, securities of biopharmaceutical companies tend to be volatile and experience significant price and volume fluctuations.
In addition, the trading volume in Ordinary Shares and Warrants may fluctuate and cause significant price variations to occur. The average trading volume of our Ordinary Shares traded per day from April 1, 2023 to December 31, 2023 was approximately 19,244 shares per day. The average trading volume of our Ordinary Shares can be very sporadic and may impair the ability of holders of our Ordinary Shares to sell their shares at the time they wish to sell them or at a price that they consider reasonable. A low trading volume may also reduce the fair market value of our Ordinary Shares.
73
Also, due to the low volume of shares traded on any trading day, persons buying or selling in relatively small quantities may easily influence prices of our Ordinary Shares. Accordingly, there can be no assurance that the price of our Ordinary Shares will reflect our actual value. There can be no assurance that the daily trading volume of our Ordinary Shares will increase or improve.
We cannot assure you that the market price of the Ordinary Shares and Warrants will not fluctuate widely or decline significantly in the future in response to a number of factors, including, among others, the following:
In the past, securities class-action litigation has often been instituted against companies following periods of volatility in the market price of their shares. This type of litigation could result in substantial costs and divert our management’s attention and resources, which could have a material adverse effect on us.
We expect to issue additional Ordinary Shares, including under our management incentive plan. Any such issuances would dilute the interest of our shareholders and likely present other risks.
We expect to issue a substantial number of Ordinary Shares, including under the Company's Stock Option and Incentive Plan Regulation 2023 (”2023 Plan”).
Ordinary Shares reserved for future issuance under the 2023 Plan will become eligible for sale in the public market once those shares are issued, subject to provisions relating to various vesting agreements, lock-up agreements and, in some cases, limitations on volume and manner of sale applicable to affiliates under Rule 144, as applicable. The aggregate number of Ordinary Shares initially reserved for issuance under the 2023 Plan is 7,835,544 Ordinary Shares. As of December 31, 2023, we had awards issued and outstanding covering 3,466,210 ordinary shares.
Any such issuances of additional Ordinary Shares or securities convertible into Ordinary Shares:
74
We do not currently intend to pay dividends on our securities and, consequently, your ability to achieve a return on your investment will depend on appreciation in the price of the ordinary shares. In addition, Swiss law may limit the amount of dividends we are able to distribute.
We have never declared or paid any cash dividends on our ordinary shares and do not currently intend to do so for the foreseeable future. We currently intend to invest our future earnings, if any, to fund our growth. Therefore, you are not likely to receive any dividends on your shares for the foreseeable future and the success of an investment in the shares will depend upon any future appreciation in its value. Consequently, investors may need to sell all or part of their holdings of the shares after price appreciation, which may never occur, as the only way to realize any future gains on their investment. There is no guarantee that the shares will appreciate in value or even maintain the price at which our shareholders have purchased them. Investors seeking cash dividends should not purchase the shares.
In addition, exchange rate fluctuations may affect the amount of euro that we are able to distribute, and the amount in U.S. dollars that our shareholders receive upon the payment of cash dividends or other distributions we declare and pay in Swiss Francs, if any. These factors could harm the value of the shares, and, in turn, the U.S. dollar proceeds that holders receive from the sale of the shares.
Our Warrants are exercisable for Ordinary Shares, the exercise of which would increase the number of shares eligible for future resale in the public market and result in dilution to our shareholders.
As a result of the Business Combination being consummated, outstanding warrants to purchase an aggregate of 4,403,294 Ordinary Shares became exercisable in accordance with the terms of the Warrant Agreement. These warrants became exercisable on April 2, 2023. The exercise price of these warrants is $11.50 per share, or approximately $50.6 million, assuming none of the warrants are exercised through “cashless” exercise. To the extent such warrants are exercised, additional ordinary shares will be issued, which will result in dilution to the holders of Ordinary Shares and increase the number of shares eligible for resale in the public market. We believe the likelihood that warrant holders will exercise their warrants, and therefore the amount of cash proceeds that we would receive, is dependent upon the trading price of our ordinary shares. If the trading price for our ordinary shares is less than $11.50 per share, we believe holders of our Public Warrants and Private Placement Warrants will be unlikely to exercise their warrants. On December 31, 2023, the last reported closing price of our ordinary shares was $11.23 per share and the last reported closing price of our Public Warrants was $1.50 per warrant. Sales of substantial numbers of such shares in the public market or the fact that such warrants may be exercised could adversely affect the market price of ordinary shares. However, there is no guarantee that the Public Warrants will ever be in the money prior to their expiration, and as such, the warrants may expire worthless.
The Warrants may never be in the money, and they may expire worthless and the terms of the Public Warrants may be amended in a manner adverse to a holder if holders of at least 50.0% of the then outstanding Public Warrants approve of such amendment.
The exercise price for our Warrants is $11.50 per Ordinary Share. We believe the likelihood that warrant holders will exercise their Public Warrants and Private Placement Warrants, and therefore the amount of cash proceeds that we would receive, is dependent upon the trading price of our Ordinary Shares. If the trading price for our Ordinary Shares is less than $11.50 per share, we believe warrant holders will be unlikely to exercise their Warrants. There is no guarantee that the Warrants will be in the money following the time they become exercisable and prior to their expiration, and as such, the Warrants may expire worthless.
The Warrants were issued in registered form under a warrant agreement between Continental Stock Transfer & Trust Company, as warrant agent, and EBAC, and were assumed by us at the time of the Closing, pursuant to a warrant assignment, assumption and amendment agreement by and between us, EBAC, and Continental Stock Transfer & Trust Company. Continental Stock Transfer & Trust Company is currently the warrant agent. The warrant agreement provides that the terms of the warrants may be amended without the consent of any holder to cure any ambiguity, correct any defective provision or correct any mistake, amend the definition of “Ordinary Cash Dividend” or add or change any provisions with respect to matters or questions arising under the warrant as the parties may deem necessary or desirable and that the parties deem shall not adversely affect the rights of the warrant holders, but requires the approval by the holders of at least 50.0% of the then-outstanding Public Warrants to make any change that adversely affects the interests of the registered holders of Public Warrants.
75
Accordingly, we may amend the terms of the Public Warrants in a manner adverse to a holder if holders of at least 50.0% of the then-outstanding Public Warrants approve of such amendment and, solely with respect to any amendment to the terms of the Private Placement Warrants or any provision of the warrant agreement with respect to the private placement warrants, 50.0% of the number of the then outstanding Private Placement Warrants. Although our ability to amend the terms of the Public Warrants with the consent of at least 50.0% of the then-outstanding Public Warrants is unlimited, examples of such amendments could be amendments to, among other things, increase the exercise price of the warrants, convert the warrants into cash, shorten the exercise period or decrease the number of Ordinary Shares purchasable upon exercise of a warrant.
We may redeem the Public Warrants prior to their exercise at a time that is disadvantageous to the holder, thereby making such warrants worthless.
We may redeem the Public Warrants prior to their exercise at a time that is disadvantageous to the holder, thereby making such warrants worthless. We will have the ability to redeem outstanding Public Warrants at any time after they become exercisable and prior to their expiration, at a price of $0.01 per warrant, provided that the closing price of the Ordinary Shares equals or exceeds $18.00 per share (as adjusted for share subdivisions, share capitalizations, reorganizations, recapitalizations and the like) for any 20 trading days within a 30 trading day period ending on the third trading day prior to the date on which a notice of redemption is sent to the warrant holders. We will not redeem the warrants as described above unless a registration statement under the Securities Act covering the Ordinary Shares issuable upon exercise of such warrants is effective and a current prospectus relating to those Ordinary Shares is available throughout the 30-day redemption period. If and when the Public Warrants become redeemable by us, we may exercise the redemption right even if we are unable to register or qualify the underlying securities for sale under all applicable state securities laws. Redemption of the outstanding Public Warrants could force holders (i) to exercise the Public Warrants and pay the exercise price therefor at a time when it may be disadvantageous to do so, (ii) to sell the Public Warrants at the then-current market price when holders might otherwise wish to hold the Public Warrants, or (iii) to accept the nominal redemption price which, at the time the outstanding Public Warrants are called for redemption, is likely to be substantially less than the market value of the Public Warrants.
In addition, we will have the ability to redeem the outstanding Public Warrants at any time after they become exercisable and prior to their expiration, at a price of $0.10 per warrant if, among other things, the closing price of the Ordinary Shares equals or exceeds $10.00 per share (as adjusted for share sub-divisions, share dividends, rights issuances, subdivisions, reorganizations, recapitalizations and the like) on the trading day prior to the date on which a notice of redemption is sent to the warrant holders. Recent trading prices for the Ordinary Shares have exceeded the $10.00 per share threshold at which the Public Warrants would become redeemable. In such a case, the holders will be able to exercise their Public Warrants prior to redemption for a number of Ordinary Shares determined based on the redemption date and the fair market value of the Ordinary Shares.
The value received upon exercise of the Public Warrants (1) may be less than the value the holders would have received if they had exercised their Public Warrants at a later time when the underlying share price is higher and (2) may not compensate the holders for the value of the Public Warrants.
Risks Related to Taxation
If we are treated as a “passive foreign investment company” for any taxable year, U.S. investors could be subject to adverse U.S. federal income tax consequences.
A non-U.S. corporation generally will be treated as a “passive foreign investment company” (“PFIC”) for U.S. federal income tax purposes if either (i) at least 75.0% of its gross income in a taxable year, including its pro rata share of the gross income of any corporation in which it is considered to own at least 25.0% of the shares by value, is passive income or (ii) at least 50.0% of its assets in a taxable year (ordinarily determined based on fair market value and averaged quarterly over the year), including its pro rata share of the assets of any corporation in which it is considered to own at least 25.0% of the shares by value, are held for the production of, or produce, passive income. Passive income generally includes dividends, interest, rents and royalties (other than rents or royalties derived from the active conduct of a trade or business), and gains from the disposition of passive assets.
Based on our analysis of our income, assets, activities, and market capitalization, we believe that we were not a PFIC for our taxable year ended December 31, 2023. The determination of whether a non-U.S. corporation is a PFIC is a fact-intensive determination made on an annual basis and the applicable law is subject to varying interpretation.
76
In particular, the characterization of our assets as active or passive may depend in part on our current and intended future business plans, which are subject to change. The amount of passive income and passive assets we take into account for PFIC testing purposes depends, in part, on the size of our cash balance (taking into account the timing and manner in which such cash is used) and the interest rates applicable thereto. In addition, the total value of our assets for PFIC testing purposes may be determined in part by reference to our market capitalization from time to time, which may fluctuate considerably. As a result, there can be no assurance with respect to our PFIC status for any taxable year, and our U.S. counsel expresses no opinion with respect to our PFIC status for any taxable year.
If we are treated as a PFIC, U.S. investors may be subject to certain adverse U.S. federal income tax consequences, including additional reporting requirements. See “Material U.S. Federal Income Tax Considerations—Passive Foreign Investment Company Rules” for a more detailed discussion of the PFIC rules. U.S. investors should consult their tax advisors regarding the application of the PFIC rules in their particular circumstances.
Changes to tax laws in any of the jurisdictions in which we operate, including new proposals on taxing digital companies and the ongoing work by the Organization for Economic Co-operation and Development (the "OECD"), could have a material adverse effect on our business, operating results, and financial condition.
Tax laws, including tax rates, in the jurisdictions in which we operate may change as a result of macroeconomic or other factors outside of our control. New income, sales, use or other tax laws, statutes, rules, regulations or ordinances could be enacted at any time, which could affect the tax treatment of our domestic and foreign earnings. For instance, the Inflation Reduction Act of 2022 (the “IRA”) imposes, among other rules, a 15.0% minimum tax on the book income of certain large corporations and a 1.0% excise tax on certain corporate stock repurchases.
Our tax treatment may also be impacted by tax policy initiatives and reforms such as the Base Erosion and Profit Shifting ("BEPS") Project (including "BEPS 2.0") of the OECD, and the European Commission’s state aid investigations and other initiatives. Such changes may include (but are not limited to) the taxation of operating income, investment income, dividends received or, in the specific context of withholding tax dividends paid. The OECD has published a package of measures for reform as a product of BEPS, which includes the reallocation of global profits of large multinational companies to market jurisdictions based on customer location as well as the introduction of a global minimum tax. Many of the package’s proposed measures require amendments to the domestic tax legislation of various jurisdictions.
Changes in tax laws, treaties, or regulations or their interpretation or enforcement are unpredictable. Any of these occurrences could have a material adverse effect on our business, operating results, and financial condition, including changing the amount and recognition of our deferred tax assets and liabilities.
If we or any of our subsidiaries is treated as a “controlled foreign corporation,” certain U.S. investors could be subject to adverse U.S. federal income tax consequences.
Generally, under the Code, if a U.S. investor owns or is treated as owning, directly, indirectly, or constructively, 10.0% or more of the total value or total combined voting power of our stock, the U.S. investor may be treated as a “Ten Percent United States shareholder” with respect to each controlled foreign corporation (“CFC”) in our corporate structure, if any. A non-U.S. corporation generally will be a CFC if Ten Percent United States shareholders own, directly, indirectly, or constructively, 50.0% or more of the total value or total combined voting power of the stock of such corporation. The determination of CFC status is complex and includes certain “downward attribution” rules, pursuant to which our non-U.S. subsidiaries may be treated as constructively owned by our U.S. subsidiaries. Because our corporate structure includes a U.S. corporate subsidiary, the downward attribution rules may cause any non-U.S. subsidiaries, including any that we form or acquire in the future, to be treated as controlled foreign corporations. Because our corporate structure includes a U.S. corporate subsidiary, our non-U.S. corporate subsidiaries, including any non-U.S. corporate subsidiaries that may be formed or acquired in the future, will be treated as CFCs, regardless of whether we are treated as a CFC. A Ten Percent United States shareholder of a CFC may be required to annually report and include in its U.S. taxable income its pro rata share of the CFC’s “Subpart F income”, “global intangible low-taxed income,” and investments of earnings in U.S. property, regardless of whether the CFC makes any distributions to its shareholders. Furthermore, an individual Ten Percent United States shareholder with respect to a CFC generally will not be allowed certain tax deductions and foreign tax credits that are allowed to a corporate Ten Percent United States shareholder.
77
Failure to comply with CFC reporting obligations may also subject a Ten Percent United States shareholder to significant penalties, preventing the statute of limitations with respect to such Ten Percent United States shareholder’s U.S. federal income tax return for the year for which reporting was due from starting. There can be no assurance that the Company will provide to any Ten Percent United States shareholder information that may be necessary for the Ten Percent United States shareholder to comply with its CFC reporting and tax paying obligations. U.S. investors should consult their tax advisors regarding the application of the CFC rules in their particular circumstances.
If securities or industry analysts do not publish or cease publishing research or reports about us, our business, or our market, or if they change their recommendations regarding ordinary shares adversely, then the price and trading volume of our ordinary shares could decline.
The trading market for ordinary shares is influenced by the research and reports that industry or securities analysts may publish about us, our business, our market, or our competitors. If any of the analysts who may cover our company change their recommendation regarding ordinary shares adversely, cease to provide coverage or provide more favorable relative recommendations about our competitors, the price of our ordinary shares would likely decline.
The JOBS Act permits “emerging growth companies” like Oculis to take advantage of certain exemptions from various reporting requirements applicable to other public companies that are not emerging growth companies, which may make our ordinary shares less attractive to investors.
Oculis currently qualifies as an “emerging growth company” as defined in Section 2(a)(19) of the Securities Act, as modified by the Jumpstart Its Business Startups Act of 2012, which is referred to as the “JOBS Act.” As such, we take advantage of certain exemptions from various reporting requirements applicable to other public companies that are not emerging growth companies for as long as we continue to be an emerging growth company, including the exemption from the auditor attestation requirements with respect to internal control over financial reporting under Section 404 of the Sarbanes-Oxley Act. As a result, our shareholders may not have access to certain information they deem important.
We cannot predict if investors will find our ordinary shares less attractive because we rely on these exemptions. If some investors find our ordinary shares less attractive as a result, there may be a less active trading market and the share price for our ordinary shares may be more volatile. We may incur increased legal, accounting and compliance costs associated with Section 404 of the Sarbanes-Oxley Act.
Item 4. Information on the Company
A. History and Development of the Company
We are a stock corporation (Aktiengesellschaft) that was incorporated under the laws of Switzerland on October 31, 2022. We are registered with the commercial register of the Canton of Zug under company registration number CHE-396.695.611. The mailing address of our principal executive office after the Acquisition Closing is Oculis Holding AG, Bahnhofstrasse 7, CH-6300, Zug, Switzerland. Neither our articles of association nor the operation of law limit our duration.
Certain additional information about the Company is included in Item 4.B “Business Overview” and is incorporated herein by reference. The material terms of the Business Combination are described in Item 10 of this Report. The Company is subject to certain of the informational filing requirements of the Exchange Act. Since the Company is a “foreign private issuer”, it is exempt from the rules and regulations under the Exchange Act prescribing the furnishing and content of proxy statements, and the officers, directors and principal shareholders of the Company are exempt from the reporting and “short-swing” profit recovery provisions contained in Section 16 of the Exchange Act with respect to their purchase and sale of Ordinary Shares. In addition, the Company is not required to file reports and financial statements with the SEC as frequently or as promptly as U.S. public companies whose securities are registered under the Exchange Act. However, the Company is required to file with the SEC an Annual Report on Form 20-F containing financial statements audited by an independent accounting firm. The SEC also maintains a website at http://www.sec.gov that contains reports and other information that the Company files with or furnishes electronically to the SEC.
78
Our telephone number is +41-41-711-9325 and our website is www.oculis.com.
B. Business Overview
Company Overview
We are a late clinical-stage biopharmaceutical company, based in Switzerland, with substantial expertise in therapeutics used to treat ocular diseases, engaged in the development of innovative drug candidates which embrace the potential to address large unmet medical needs for many eye-related conditions. Our focus is on advancing therapeutic candidates intended to treat significant and prevalent ophthalmic diseases which result in vision loss, blindness or reduced quality of life. Our mission is to improve the health and quality of life of patients around the world by developing medicines that save sight and improve eye care for patients. To realize this mission, we intend to become a global leader in ocular therapeutics.
Our pipeline currently includes three clinical-stage therapeutic candidates: OCS-01, OCS-02 (Licaminlimab) and OCS-05. Our lead product candidate, OCS-01, is currently being evaluated in two ongoing Phase 3 clinical programs: as a topical option for the treatment of DME, and as a once- daily steroid for the treatment of inflammation and pain following cataract surgery. Our second product candidate is OCS-02, currently being evaluated in a Phase 2b clinical trial to assess its potential as a topical anti-TNFα treatment for dry eye disease (“DED”) and potentially the use of a particular genotype to predict treatment response, which could be considered as a biomarker in a precision medicine approach. A second clinical trial for OCS-02, designed to evaluate its use as a potential treatment for non-infectious anterior uveitis, is expected to follow thereafter. Our third product candidate is OCS-05, a potential disease modifying neuroprotective agent against neurological damage with potential application in multiple indications, including glaucoma, dry age-related macular degeneration (“AMD”) and diabetic retinopathy (“DR”). We are conducting a Phase 2 Proof-of-Concept (“PoC”) trial evaluating OCS-05 as a potential treatment for acute optic neuritis (“AON”) for which there is currently no approved therapeutic treatment.
Summary of Our Clinical Product Candidates Portfolio
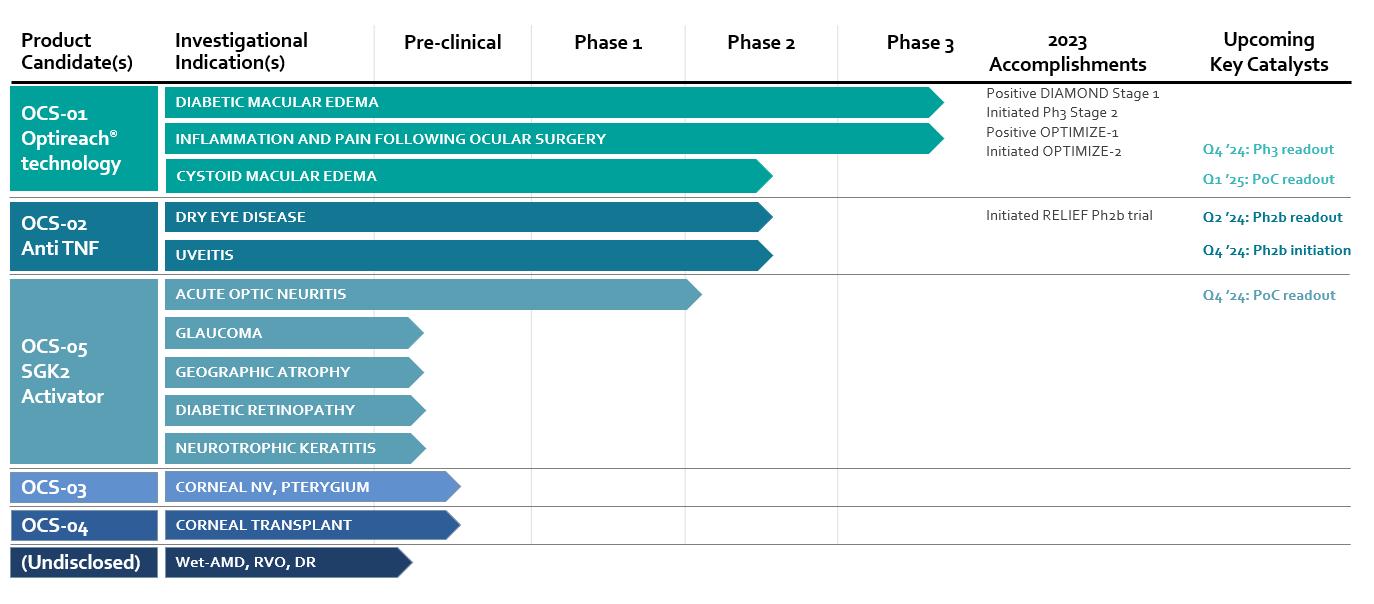
OCS-01 is based on the OPTIREACH technology, OCS-02 is a single chain antibody fragment (ScFv) against TNF alpha and OCS-05 is a SGK-2 activator peptidomimetic small molecule with novel MoA targeting the activation of the trophic factor pathways. The Company’s additional earlier stage development candidates are discussed in the section under the header “Our clinical development candidates” below.
Utilizing our internal core competency in formulation discovery and drug development capabilities, together with extensive licensing, collaboration and acquisition activities, we have assembled a pipeline of attractive development candidates that include both late-stage clinical candidates as well as earlier stage preclinical initiatives. Our clinical candidate portfolio includes:
79
OCS-01
Our lead candidate is OCS-01, a novel, high concentration, topical Optireach formulation (ophthalmic suspension) of dexamethasone, designed to enhance drug penetration into both the anterior and posterior segments of the eye with enhanced persistence following topical application. We are evaluating OCS-01 for use as a topical eye drop for the treatment of DME, as a once-daily steroid treatment for inflammation and pain following ocular surgery, and in treating two forms of cystoid macular edema (“CME”). LEOPARD, an investigator-initiated trial (“IIT”) for the treatment of Uveitic Macular Edema (“UME”) or Post-Surgical Macular Edema (“PSME”), both forms of CME, is ongoing and the related readout is expected in the first quarter of 2025. Using our proprietary Optireach® technology, OCS-01 was designed to enhance drug penetration into both the anterior and posterior segments of the eye. This therapeutic approach is in contrast to currently available therapies, which require the use of more invasive treatments such as ocular implants or intravitreal injections to deliver medication to the retina. Furthermore, current treatment of DME often involves multiple intravitreal injections per year. Given the burden of therapy, FDA-approved therapeutics are not widely used for early disease intervention. It has been reported that 60% of DME patients are not treated 12 months after the diagnostic (IRIS data base June 23), despite the deterioration in visual acuity in 19.0% of untreated patients within two years. In addition, approximately 40.0% of patients treated with anti-VEGF intravitreal injections have an inadequate response at 12 weeks.
OCS-01 is a topical dexamethasone Optireach formulation which is designed to deliver therapeutic levels of drug to the retina via an eye drop, a route of administration for DME treatment that may enable earlier intervention and thereby significantly increase the proportion of patients being treated as well as increase the prescribing physician base by providing a treatment option to general ophthalmologists. An eye drop treatment could also provide a new treatment option for patients with inadequate response to the current invasive standard of care. We are currently not aware of the existence of any other eye drop treatment for DME which is in a similar or more advanced stage of active clinical development; however, we cannot guarantee that OCS-01 will receive regulatory approval. We have two ongoing Phase 3 clinical trials: (i) DME and (ii) for the treatment of inflammation and pain following ocular surgery. For DME, the first stage of the DIAMOND Phase 3 clinical trial met its objective of validating the induction and maintenance dosing regimen designed to optimize OCS-01 efficacy potential with robust statistical significance. For the treatment of inflammation and pain following cataract surgery, the first Phase 3 clinical trial, OPTIMIZE-1, met both hierarchical primary efficacy endpoints with statistical significance, the absence of inflammation at day 15 and the absence of pain at day 4. Following the positive DIAMOND trial outcome, we are advancing the planned OCS-01 development program for DME into DIAMOND Stage 2, which includes two global pivotal Phase 3 clinical trials, DIAMOND-1 and DIAMOND-2. We announced first patient first visit in DIAMOND-1 and DIAMOND-2 in December 2023 and February 2024, respectively. Each trial is expected to enroll approximately 350 to 400 patients. Following the positive OPTIMZE-1 trial outcome, we are advancing the development program for inflammation and pain following cataract surgery into the second Phase 3 trial, OPTIMIZE-2. In December 2023, we also announced first patient first visit in OPTIMIZE-2. Data from the two OPTIMIZE trials are intended to support the our future NDA submission to the FDA.
In addition to the Phase 3 trials, we are conducting the LEOPARD trial, which is an IIT to investigate the safety and efficacy of OCS-01 in UME and PSME. LEOPARD is sponsored by Global Ophthalmic Research Center (GORC). This PoC trial’s data readout is expected in the first quarter of 2025.
The total U.S. prevalence of DME in 2023 is estimated at 3.0 million, with the diagnosed U.S. prevalence estimated at 1.8 million by the Decision Resources Group DME Landscape November 2020 report. The same report estimates that 0.9 million U.S. DME patients were treated with drugs in 2023, leaving 0.9 million U.S. patients diagnosed but untreated. These 0.9 million patients are a key addressable market segment for OCS-01. Additionally, OCS-01 is also intended to address the market segment of patients with inadequate response to anti-VEGF therapy. A study published in the American Journal of Ophthalmology in 2016 found that nearly 40.0% of patients treated with anti-VEGF therapy had inadequate responses at 12 weeks. By applying this figure to the number of treated U.S. patients, we estimate that inadequate response occurs in approximately 0.4 million patients. In total, we estimate that 1.3 million DME patients in the United States are addressable by OCS-01.
The Informa Meddevicetracker Ophthalmic Surgical Products Market 2017 report projected that ophthalmic surgeries are on the rise, mainly due to the aging population and lifestyle changes, and are expected to reach close to 10 million procedures per year in the U.S. alone by 2037.
80
Cataract surgeries are the most prevalent procedures of all medical specialties with an estimated 5 million procedures in 2021 in the U.S. Ophthalmic surgeries cause the release of inflammatory factors and can be associated with ocular pain. Cataract surgery, even with a very small incision, creates inflammation in the cornea, anterior chamber and iris. Given our observations in Stage 1 of the DIAMOND Phase 3 trial that OCS-01 treatment led to improvements in visual acuity and macular thickness in patients with DME, we believe OCS-01, if approved for inflammation and pain following ocular surgery, may be of benefit to patients at risk of retinal complications following ocular surgery. An exploratory investigator initiated trial is ongoing in the U.S. to assess its benefits in treating CME. A pilot study demonstrated favorable effects of OCS-01 on eyes with non-infectious uveitic macular oedema and vitritis (Schulman et al, Acta Ophthalmologica 2015).
OCS-02 (Licaminlimab)
We are also advancing the clinical development of licaminlimab, or OCS-02, a next-generation biologic treatment for ocular inflammation, specifically as a treatment for DED and non-infectious chronic anterior uveitis. Differentiating OCS-02 is its use of a single chain antibody fragment specifically formulated for topical delivery in ophthalmology, TNF inhibitors are directed against the cytokine human tumor necrosis factor alpha (“TNFα”). Furthermore, the small size of the fragment enables the topical delivery of an anti-TNFα construct with increased concentrations and enhanced ocular tissue penetration. The anti-inflammatory and anti-necrotic properties of therapeutics inhibiting TNFα activity are well established with anti-TNF pharmaceuticals already approved as systemic treatments for ocular disease. While OCS-02 is intended to be developed for all comers, we are advancing the development of OCS-02 in conjunction with the development of a potentially novel genetic biomarker intended to identify patients who may have a greater response to OCS-02 therapy. Two Phase 2 clinical trials in patients with symptoms of DED were conducted (the first with the predecessor of OCS-02, and the second with OCS-02), as well as one Phase 2 clinical trial in acute anterior uveitis. Topical ocular administration of OCS-02 was associated with improvements in the global ocular discomfort score versus vehicle in patients with DED, and with reaching a pre-specified responder rate in patients with non-infectious anterior uveitis, as well as being well tolerated in all three studies. In February 2024, we completed enrollment in the Phase 2b RELIEF trial evaluating OCS-02 for the treatment of DED, with topline results expected in the second quarter of 2024. We plan to commence a Phase 2b trial for OCS-02 as a treatment for chronic anterior uveitis thereafter.
We estimate the segment of DED patients in the United States addressable by OCS-02 (patients with moderate or severe DED) to be approximately 10 million patients. This comprises an estimated 7 million patients with moderate DED and 3 million patients with severe DED (based on the rates of approximately 35.0% moderate and 14.0% severe patients as reported by the Dry Eye Products Market Report published in Market Scope 2023 of approximately 20.0 million diagnosed prevalent cases of DED in the U.S. as estimated for 2024 by Decision Resources Group Dry Eye Disease Landscape and Forecast, December 2020).
We also estimate OCS-02 could help address a medical need in patients suffering from either chronic or recurring non-infectious anterior uveitis. This addressable patient population is estimated to be approximately 170 thousand in the United States based on a prevalence rate of non-infectious uveitis of 121 per 100,000, applied to the U.S. population and the fact that anterior uveitis is the most prevalent form representing 81.0% of all cases, as found in a study published in the Journal of the American Medical Association Ophthalmology in 2016, and based on a prevalence of recurrent and chronic disease being estimated at 51.0%, as found in a study published in the Journal of the American Medical Association Ophthalmology in 2013.
OCS-05
Our third clinical candidate is OCS-05, a novel serum/glucocorticoid-regulated protein kinase 2 (“SGK2”) activator peptidomimetic small molecule, in development as a potential disease modifying neuroprotective agent against neurological damage to the optic nerve. We are initially developing OCS-05 as a potential therapeutic to treat AON, a rare disease with high unmet medical need, as currently there is no treatment which is approved by the FDA or European Commission for AON. OCS-05 has been granted Orphan Drug Designation by both the FDA and the European Commission for this indication. OCS-05 has been studied in preclinical studies suggesting efficacious neuroprotective and remyelinating activity, as well as in a UK Phase 1 clinical trial under the Medicines and Healthcare products Regulatory Agency (“MHRA”) in healthy volunteers in which OCS-05 was observed to be well tolerated. We are currently conducting a First-in-Patient clinical trial of OCS-05 in AON in France to test the candidate’s safety and tolerability, and we are currently conducting IND-enabling activities for OCS-05 in the United States.
81
Should the clinical results of our AON trial prove sufficiently compelling, we intend to evaluate the promise of OCS-05 to treat other neuro-ophthalmic disorders such as geographic atrophy, glaucoma, diabetic retinopathy and neurotrophic keratitis.
Additional development initiatives
In addition to these six clinical development programs involving the three clinical candidates, we also are engaged in a number of earlier preclinical development initiatives, including:
Our Executive Management Team
We are led by an experienced management team, composed of individuals who have extensive backgrounds in drug discovery and development, clinical trial design and operations, regulatory affairs, business development and commercial and general management at both large pharmaceutical companies and emerging biopharmaceutical organizations. Collectively, our management team has a track record of advancing new drug candidates through regulatory approval and successful commercialization. The expertise of our management team is complemented by our board of directors, which includes many accomplished industry veterans with significant capabilities in guiding the success of emerging biopharmaceutical companies such as ours. Since our inception we have raised approximately CHF 239.2 million from leading North American, European and Asian life science venture capital investors including Brunnur Ventures (Brunnur vaxtarsjodur slhf.), BVCF Management (BEYEOTECH), EQT Life Sciences, Novartis Bioventures Ltd., LSP 7 Coöperatief, funds managed by Earlybird Capital, funds managed by Pivotal Partners, funds managed by Aberdeen (formerly Tekla Capital Management LLC), and VI Partners, among others. Please note that prospective investors should not rely on these named investors’ investment decisions, as each of such investor’s risk tolerance and investment strategy and goal may be different from those of other prospective investors.
Our Strategy
We intend to become a leader in developing therapeutics to address ocular diseases characterized by significant medical needs with large market opportunities. To accomplish this objective, we plan to focus on successful completion of our key strategic initiatives, which include:
Based on results achieved in the Stage 1 Phase 3 trial, we have progressed to the Stage 2 Phase 3 trials of OCS-01 in DME, DIAMOND-1 and DIAMOND-2, which are currently ongoing. We believe the use of OCS-01 formulated as a non-invasive, self-administered eye drop, could, if approved, promote a shift in the current treatment paradigm to allow earlier intervention and increase both the treated patient population and the prescribing physician base. In addition, OCS-01, if approved, could benefit patients who are diagnosed with DME and who have an inadequate response to anti-VEGF intravitreal injections.
Following positive results in the first Phase 3 trial, OPTIMIZE-1, we have initiated the second Phase 3 trial, OPTIMIZE-2, of OCS-01 in the treatment of inflammation and pain following ocular surgery, with first patient first visit achieved in December 2023. OCS-01 could be differentiated in the anterior segment by its potential ability to deliver therapeutic drug levels to the back of the eye. Topline results from OPTIMIZE-2 are expected in the fourth quarter of 2024. An investigator-initiated PoC trial is currently ongoing to explore further the potential of OCS-01 in treating edema in CME. We believe this potential benefit in CME, if supported by this study and validated by future studies, and if OCS-01 is approved, may enable us to achieve enhanced market access.
82
Based on results achieved in three Phase 2 clinical trials, we have advanced OCS-02 into a Phase 2b RELIEF clinical trial to assess its clinical benefit in treating DED. We intend to initiate a second Phase 2 trial for OCS-02 as a treatment for chronic anterior uveitis thereafter. OCS-02 is differentiated by its use of single-chain antibody fragment formulation technology, which enables the topical delivery of an anti-TNFα agent. We are advancing the development of OCS-02 in conjunction with further analysis of a potential novel genetic biomarker intended to identify patients who may demonstrate an enhanced response to OCS-02 therapy and believe this precision medicine approach may allow the candidate to deliver superior outcomes in this patient group, if approved.
The differentiated and novel mechanism of action of OCS-05, coupled with its potential disease modifying neuroprotective properties, suggests potential benefits across many of the more pervasive neurological pathologies of the eye including geographic atrophy, diabetic retinopathy, glaucoma and neurotrophic keratitis. We initially intend to assess the safety of OCS-05 as a treatment for AON and are currently evaluating OCS-05 in a First-in-Patient study called the ACUITY trial in France. There is currently no approved therapy for treatment of AON. OCS-05 has been granted Orphan Drug Designation by both the FDA and the European Commission. We believe that demonstration of therapeutic benefits in AON may provide compelling support for the exploration of OCS-05 in larger market opportunities.
We intend to complement our ongoing development programs by accessing additional innovative product candidates and technologies through in-licensing, strategic collaborations and acquisitions. We believe that the depth of our formulation discovery and drug development expertise specific to ocular therapeutics, coupled with the industry network of our executive management, board of directors and advisors, provide us with the differentiated set of capabilities necessary to identify and advance product candidates successfully in this therapeutic category.
We have retained rights globally to all of our indications, including our lead product candidate OCS-01, for the potential treatment of DME and inflammation and pain following ocular surgery; OCS-02 for the potential treatment of DED and non-infectious anterior uveitis; and OCS-05 as a neuroprotective agent. Given the potential to treat patients worldwide, we may opportunistically enter into strategic collaborations around certain product candidates, diseases or geographic regions.
Diseases and disorders of the eye
Numerous diseases and disorders, many of which represent significant medical needs, are associated with the human eye. Ocular diseases, which may result in visual impairment, blindness or reduced quality of life include retinal diseases such as DME, macular degeneration (including geographic atrophy), diabetic retinopathy, and retinal vein occlusion (“RVO”); disorders caused by swelling and inflammation such as DED, corneal keratitis and uveitis; and glaucoma, among other disease states. The global market for therapeutics used to treat eye disease is estimated to have exceeded $22 billion in 2020. We employ our substantial expertise in the development of therapeutics, in particular pharmaceuticals used to treat ocular diseases, to potentially address many eye-related conditions with high unmet medical needs. Our focus is on developing innovative drug candidates to address significant and growing ophthalmic diseases, which result in vision loss, blindness or reduced quality of life, for which there are currently limited treatment options.
Our clinical development candidates
83
Utilizing our internal formulation discovery and drug development capabilities, together with extensive licensing, collaboration and acquisition activities, we have assembled a pipeline of attractive development candidates that include both late-stage clinical candidates as well as earlier stage preclinical initiatives. Our clinical portfolio is made up of (i) OCS-01, currently in two ongoing Stage 2 Phase 3 clinical trials, one evaluating its use as a treatment for DME and the other assessing its utility to treat inflammation and pain following ocular surgery; (ii) OCS-02 (Licaminlimab), currently in one ongoing Phase 2b clinical trial evaluating its use as a potential treatment for DED and anticipated to enter a second Phase 2b trial evaluating its potential use as a therapy for the treatment of non-infectious anterior uveitis; and (iii) OCS-05, a novel neuroprotective agent with potential application in multiple indications, including glaucoma, GA and DR, which we are initially evaluating as a potential treatment for AON. A detailed assessment of each of these clinical candidates is contained in the descriptions provided below.
OCS-01
Key program highlights:
Our lead development candidate OCS-01 is a 1.5% suspension of the anti-inflammatory corticosteroid dexamethasone for use as a potential treatment for DME and for inflammation and pain following ocular surgery. In contrast to currently available formulations of dexamethasone, which require the use of more invasive treatments such as an implant or intravitreal injection to deliver the medication to the retina, differentiating OCS-01 is our use of the proprietary Optireach® technology, which enables the topical delivery, as an eye drop, of dexamethasone to the back of the eye for the treatment of diseases affecting the retina. OCS-01 is a topical dexamethasone formulation which we have observed in clinical trials to be capable of delivering therapeutic levels of drug to the retina via eye drop, a route of administration for DME treatment that may enable earlier treatment intervention and thereby significantly increase the proportion of patients being treated as well as increase the prescribing physician base by providing a treatment option to general ophthalmologists. We are currently not aware of the existence of any other eye drop treatment for DME which is in a similar or more advanced stage of active clinical development; however, we cannot guarantee that OCS-01 will receive regulatory approval.
During 2023, we reported data from two Phase 3 clinical trials in which we observed: in DME, a statistically significant improvement in BCVA and visual acuity; and in inflammation and pain following ocular surgery, a statistically significant increase in the proportion of subjects with absence of inflammation and pain under OCS-01 treatment versus vehicle. Following those results, we have initiated a Stage 2 OPTIMIZE Phase 3 trial for the treatment of inflammation and pain following ocular surgery and begun the first Stage 2 DIAMOND-1 Phase 3 trial for the treatment of DME.
84
Dexamethasone is a widely studied and well characterized pharmaceutical commonly used to treat a range of inflammatory conditions and is currently included on the World Health Organization’s List of Essential Medicines. It may be administered orally, by injection, or topically. Specific to ocular disorders, dexamethasone intravitreal implants have been approved by the FDA to treat DME, uveitis and macular edema caused by RVO. Dexamethasone is also used as an ophthalmic suspension for ocular inflammation though the required frequency of dosing in order to achieve a therapeutic effect often limits its utility.
We are developing OCS-01 as a g cyclodextrin-based formulation of dexamethasone, using the Optireach® delivery technology, in order to enhance its residence time at the anterior segment and its penetration into the posterior segment of the eye following topical application. The increased drug residence time produced by the delivery vehicle, combined with enhanced drug penetration allows for increases in drug concentration of more than 15-fold over conventional dexamethasone. We are currently not aware of the existence of any other topically administered formulation of dexamethasone or other active pharmaceutical ingredient in development intended to deliver sustained therapeutic levels of drug to diseased tissue at the back of the eye.
The Optireach® technology enables the topical delivery of therapeutics to the back of the eye.
OCS-01 for DME
We are advancing OCS-01 as a treatment for DME, which is a complication of diabetes and is caused by the progressive growth of new blood vessels under the retina that leak fluid and lipids, leading to swelling of the macula, which can result in significant blurring of vision and contribute to the risk of blindness from DR. DME is strongly associated with uncontrolled blood sugar levels, high blood pressure and high cholesterol. An estimated 5.5% of diabetics worldwide are affected by the disease. It is a leading cause of blindness among the U.S. adult population. In the G7 countries (the United States, France, Germany, Italy, Spain, UK and Japan), the market for the treatment of DME is estimated to have totaled approximately $4.4 billion in 2024.
DME is estimated to impact 3.0 million people in the United States alone. Of those three million, we estimate that 1.3 million patients in the United States are addressable by OCS-01.
We are currently conducting the Stage 2 DIAMOND-1 and DIAMOND-2 Phase 3 trials in study sites in the United States.
Limitations of current treatments for DME
The DME disease onset may initially go unnoticed and as a result an estimated 42.0% of patients with DME may go undiagnosed. A study by the American Academy of Ophthalmology indicates that, among diagnosed patients, fewer than half are treated, with therapeutic intervention used most commonly in the one-third of patients who have moderate to severe visual impairment.
85
Pharmacotherapy involves the invasive administration of a monoclonal antibody therapeutic targeting the vascular endothelial growth factor (“VEGF”) receptor to inhibit blood vessel growth. However, we estimate that approximately 40.0% of patients have an inadequate response to therapy after 12 weeks of anti-VEGF treatment, according to the results of a study published in the American Journal of Ophthalmology in 2016. Moreover, multiple intravitreal injections are required to maintain a therapeutic effect, which necessitates an increased treatment burden on patients, their caregivers and healthcare providers. Patients whose disease progresses while on anti-VEGF therapy may then receive a steroid implant, or laser photocoagulation of the retina.
Currently, physicians often do not treat patients who present with DME in its earlier stages of progression (patients with recent disease onset or mild visual impairment), a category that makes up approximately 67.0% of diagnosed patients with symptoms. We believe this decision to observe and not intervene is often driven by the significant burden current treatment options (laser photocoagulation, frequent intravitreal injections, intravitreal implants) place on the patient, as well as the expense and significant demands placed on healthcare resources. FDA approved therapeutics are not widely used for early disease intervention, despite the deterioration in visual acuity of approximately five letters, the equivalent of one line, or more in 19.0% of this observed/untreated patient population within two years.
OCS-01’s innovation and differentiation
OCS-01 is in development to be a topical treatment for DME, and we are currently not aware of the existence of any other eye drop treatment for DME which is in a similar or more advanced stage of active clinical development. In addition to this potential breakthrough advancement, we believe that an eye drop therapy would allow for an easy, accessible, low-burden, self-administered treatment for DME and would therefore significantly address the limitations of current, invasive therapies for DME. We expect that OCS-01, if approved, could address patients who are diagnosed with DME, with recent onset of disease or mild visual impairment and who are therefore currently observed and untreated, as well as patients who are diagnosed with DME and who have an inadequate response to anti-VEGF intravitreal injections. We estimate that both segments of patients combined totals 1.3 million in the United States alone.
OCS-01 has produced clinical trial results which support its continued development as a potential topical treatment for DME
In Stage 1 of our DIAMOND Phase 3 clinical trial which evaluated the use of OCS-01 as a treatment for DME, patients who received OCS-01 demonstrated a statistically significant improvement from baseline in key measurements of therapeutic efficacy. In this randomized, double masked trial of 148 DME patients with 2:1 randomization (OCS-01 vs. vehicle), 100 of the trial participants self-administered OCS-01 eye drops six times per day for a six-week induction phase then three times per day for a subsequent 6-week maintenance phase, with 48 participants administered vehicle only. As noted in the graphic presented below, OCS-01 demonstrated improvement in mean BCVA “Early Treatment Diabetic Retinopathy Study” chart (BCVA ETDRS) score from baseline to Week 6 versus (vs) vehicle (OCS-01: 7.2 letters vs vehicle: 3.1 letters, p=0.007) demonstrating strong visual gain in the treatment arm. The effect was sustained to Week 12 with statistical significance (OCS-01: 7.6 letters vs vehicle 3.7 letters, p= 0.016).
OCS-01 generated improvements in both CMT and BCVA measurements.
86
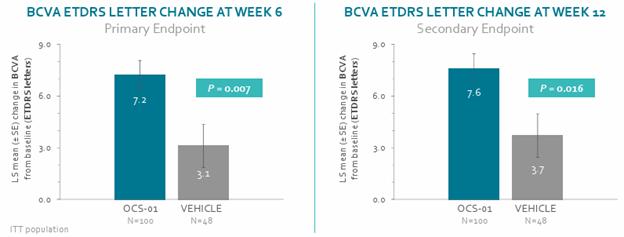
Furthermore, there was a higher percentage of patients in the OCS-01 group who achieved ≥15-letter improvement in BCVA from baseline vs vehicle at Week 6 (OCS-01: 25.3% vs vehicle: 9.8%, p=0.015), which was sustained to Week 12 (OCS-01: 27.4% vs vehicle 7.5%, p=0.009).
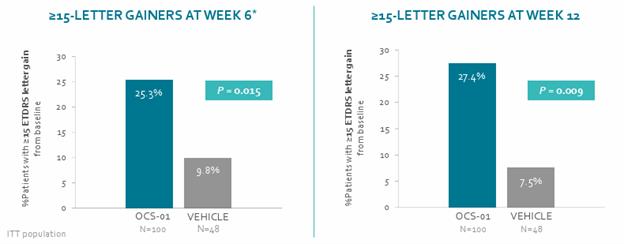
Improvements in both CMT and BCVA were greater among patients with lower baseline visual acuity.
A rapid reduction in retinal edema was observed in the OCS-01 treatment arm at week 2 of the study. The observed statistical significant treatment effect versus vehicle was preserved throughout the study.
87
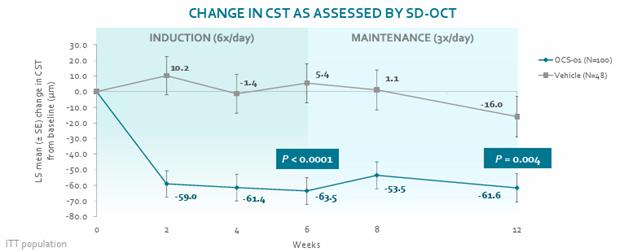
Treatment emergent adverse events (“TEAEs”) were noted in 70 of the 100 trial participants who received OCS-01, with the most prevalent AE being an increase in intraocular pressure (“IOP”), which was observed in 14 of the 100 patients in the active group. There was a small mean IOP increase, which was similar across induction and maintenance phase.
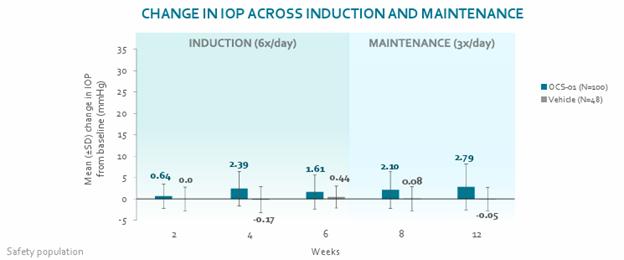
These findings of increased IOP were consistent with our expectations given glucocorticoids’ well-known ocular safety profiles, including the profile of an approved dexamethasone ocular implant. The findings were also consistent with current literature. Overall, the IOP effects observed in our trial were consistent with what is generally expected given established ophthalmic use of dexamethasone. Other AEs observed during clinical trials included diabetic and macular edema, which was noted more frequently in vehicle treated patients.
Except for increased IOP, AEs of a similar nature and number were noted among trial participants who received vehicle. The number of subjects with any ocular or non-ocular AEs leading to trial discontinuation was higher in the vehicle arm compared to the active arm. While OCS-01 may contribute to an accelerated onset of cataracts, no evidence of cataract formation was observed in the treatment arm up to 12 weeks.
The Stage 1 DIAMOND Phase 3 clinical trial results achieved with OCS-01 in treating DME follow outcomes achieved in the earlier Phase 2 study and two earlier small exploratory studies of DexNP (a previous formulation of OCS-01). In one of the studies, which was conducted in Japan, a 22-patient evaluation conducted in 2015 compared the use of a topically delivered g cyclodextrin-based formulation of dexamethasone to the posterior injection of 20 mg triamcinolone acetonide.
88
Used at the time of the trial as an off-label treatment for DME, the gcyclodextrin-based formulation generated significant improvements in visual acuity and decreased macular thickness, comparable to the results achieved using triamcinolone acetonide. The results of this 2015 study confirmed similar findings achieved in another 19-person exploratory Japanese study conducted in 2011.
Phase 3 trial design for OCS-01
Our DIAMOND program includes two stages: Stage 1 has been completed, and in Stage 2, we are conducting two, 52-week pivotal Phase 3 trials, DIAMOND-1 and DIAMOND-2. We anticipate that each of these global Phase 3 trials will enroll between an estimated 350 and 400 subjects. The primary endpoint of these studies is the mean change from baseline in BCVA at 52 weeks. Key secondary endpoints include the mean change in macular thickness (“CST”), as assessed by spectral domain optical coherence tomography and the percentage of participants that exhibit ETDRS improvement of 15 letters or more from baseline. Key inclusion criteria are similar to those used in Stage 1 of the program. The Phase 3 clinical trial protocol was reviewed by the FDA during an End-of-Phase 2 meeting.
OCS-01 has the potential to expand the number of treated patients and prescribing physicians
OCS-01 was designed to address two sizeable treatment gaps among the DME patient population in early on-set and in severe segments. Furthermore, the delivery of the drug to the back of the eye and non-invasive self-administration are unique differentiators to currently available treatments. Addressing the two existing treatment gaps may allow for increased early disease intervention with expanded treatment of retinal edema due to reduced treatment burden and improved access to care. Success in demonstrating therapeutic efficacy to treat the earlier-stages of DME disease progression may promote the use of OCS-01, if approved, among those DME patients whose treatment is currently restricted to observation. We believe that this potential expansion of the patient base to include earlier-stage DME patients may also increase the number of prescribing physicians, with general ophthalmologists, not just retina specialists, more likely to engage in disease management. If approved, OCS-01 may also be used as a non-invasive complement to currently approved therapeutic regimens, including anti-VEGF medications, potentially extending or enhancing the clinical benefit of those treatments particularly among those patients with more advanced diseases whose condition have not responded adequately to the current standard of care protocol.
OCS-01 for ocular surgery patients
There were approximately 6.8 million cataract, glaucoma, refractive, and vitrectomy surgical procedures performed in 2021 in the United States. Inflammation and pain remain an expected consequence of ocular surgery. While steroids have proven to be an effective treatment, compliance and potency are major issues with topical steroids dosed several times per day.
An estimated 30.0% of the patients who undergo cataract surgery are at an elevated risk for CME. Clinically significant CME occurs in up to 5.8% of cataract surgeries. Similar to DME, CME involves an accumulation of excess fluid in the macula which distorts central vision. CME is the most significant cause of postoperative vision loss among patients who undergo ocular surgery. Although the specific causes of CME are not well understood, comorbidities including diabetes and uveitis, among other factors, are believed to be significant contributors to disease emergence. In addition to developing OCS-01 to treat DME, we are also developing OCS-01 to treat inflammation and pain following from ocular surgery and conducting a PoC investigator initiated study to assess its potential in CME treatment. Prior to OCS-01’s commercial launch, if approved, we anticipate a PoC trial of OCS-01 (IIT) as a potential treatment for CME to be completed.
Limitations of current therapies for inflammation and pain post ocular surgery and OCS-01’s differentiation
Inexpensive steroids such as prednisone are currently widely prescribed after ocular surgery; however, since they are not formulated to reach the retina, their therapeutic benefit in treating or preventing complications related to CME has not been established. An investigator initiated PoC trial is currently ongoing to explore further the potential of OCS-01 in treating edema in CME. We believe this potential benefit in CME, if supported by this study and validated by future studies, and if OCS-01 is approved, may enable us to achieve enhanced market access.
OCS-01 has produced clinical trial results which support its continued development as a potential treatment for inflammation and pain post ocular surgery
89
During 2023, we conducted the Phase 3 OPTIMIZE-1 trial, which enrolled 241 subjects in a placebo-controlled, multi-center clinical trial in 25 sites across the United States, to assess the safety and efficacy of OCS-01, dosed once daily for 14 days, as a treatment for inflammation and pain following cataract surgery. After screening for an anterior chamber cell count of grade 2 or higher, an indication of intraocular inflammation, eligible trial participants were randomized into one of two cohorts, an active drug cohort administered OCS-01 once daily, and a cohort which received vehicle beginning one day after surgery for 15 consecutive days followed by a one-week observation period. The primary endpoints of the trial were the absence of anterior chamber cells at Day 15 and the absence of pain at Day 4. The key secondary endpoints were the absence of anterior chamber cells at Day 4 and 8, and the absence of pain at Days 2, 8, and 15.
The trial met both its hierarchical primary efficacy endpoints, the absence of inflammation at Day 15 and the absence of pain at Day 4, with robust statistical significance. A single daily application of OCS-01 was shown to reduce anterior chamber cells at Day 15 to zero in 57.2% of trial participants (p<0.0001), compared to 24.0% of subjects in the cohort that received vehicle alone. The elimination of pain at Day 4 was observed among 75.5% of subjects who received once-daily dosing of OCS-01 (p<0.0001), as compared to 52.0% in the vehicle only cohort.
OCS-01 was also well tolerated with a favorable safety profile. Overall, a higher number of ocular treatment emergent adverse events (TEAEs) were reported for the vehicle group (n=84) compared with the OCS-01 once-daily group (n=37). There was no meaningful difference in intraocular pressure (IOP) between treatment groups with a mean change from baseline to Day 15 of -0.90 mmHg in both the OCS-01 group and the vehicle group.
In December 2023, we announced first patient first visit in the second Phase 3 OPTIMIZE-2 trial. Data from both pivotal phase 3 trials, OPTIMIZE-1 (completed in 2023) and OPTIMIZE-2, are intended to support our future NDA submission to the FDA. OPTIMIZE-2 is identical in design compared with OPTIMIZE-1 (multi-center, randomized, double-masked, vehicle-controlled Phase 3 trial) and is evaluating OCS-01 for the treatment of inflammation and pain following cataract surgery. Similar to the OPTIMIZE-1 trial, patients in the OPTIMIZE-2 trial are being treated with once-daily OCS-01 post-cataract surgery versus vehicle for 2 weeks. Primary endpoints are the absence of anterior chamber cells (inflammation) on Day 15 and absence of pain on Day 4. Topline data readout is expected in the fourth quarter of 2024.
OCS-02 (Licaminlimab)
Key Program Highlights:
We are developing OCS-02 (Licaminlimab) as a next-generation biologic treatment for both DED, and as a treatment for non-infectious anterior uveitis. OCS-02 is differentiated by its use of a single chain antibody fragment formulation directed against the cytokine human TNFα to enable the topical delivery of an anti-TNFα construct at increased concentrations. The anti-inflammatory and anti-necrotic/anti-apoptotic properties of therapeutics inhibiting TNFα activity are well established with anti-TNF pharmaceuticals already approved as systemic treatments for ocular disease. While OCS-02 is intended to be developed for all comers with DED, we are advancing the development of OCS-02 in conjunction with the development of a potentially novel genetic biomarker intended to identify patients who may have a greater response to OCS-02 therapy and believe this precision medicine approach may allow the candidate to deliver superior outcomes in these patients if approved. Two Phase 2 clinical trials in patients with symptoms of DED were conducted (the first with the predecessor of OCS-02, and the second with OCS-02), as well one Phase 2 clinical trial in acute anterior uveitis. Topical ocular administration of OCS-02 demonstrated improvements in the global ocular discomfort score versus vehicle in patients with DED, and with reaching a pre-specified responder rate in patients with non-infectious anterior uveitis, as well as being well tolerated in all three studies.
90
In February 2024, we completed enrollment of the Phase 2b RELIEF study evaluating OCS-02 as a treatment for moderate-to-severe DED, with topline results anticipated in the second quarter of 2024. We plan to commence a second Phase 2 trial for OCS-02 as a treatment for chronic anterior uveitis thereafter.
TNFα performs important roles in the initiation and propagation of both normal and aberrant immune responses via mechanisms ranging from the stimulation of other cytokines to inflammatory cell recruitment to the alteration of vascular permeability. Inhibition of TNFα has demonstrated significant clinical benefit in the treatment of an array of diseases arising from dysfunctional immune system activity and anti TNFα therapeutics have become among the most widely prescribed biologics. Three anti-TNFα therapeutics (etanercept, sold under the brand name Enbrel®, infliximab, sold under the brand name Remicade®, and adalimumab, sold under the brand name Humira®), have each been studied for use in ocular disease. While the use of antagonists to TNFα have demonstrated favorable efficacy in the treatment of ocular inflammatory diseases, these drugs require intravenous infusion or subcutaneous injection and systemic anti-TNFα therapies are associated with a range of often serious adverse effects. Ocular diseases, such as DED and non-infectious anterior uveitis, involve a local TNFα driven inflammatory process which may not justify general, systemic TNFα-suppressive therapy. The novel design of OCS-02 embracing lower molecular weight single chain antibody fragment technology may enable it to be used in ocular disease as an eye drop for localized administration.
OCS-02 (Licaminlimab) for the treatment of DED
Keratoconjunctivitis sicca, also referred to as DED results from inflammation related to tear gland damage. DED is a multifactorial disease of the tears and ocular surface characterized by ocular surface inflammation and increased osmolarity of the tear film that results in ocular discomfort, visual disturbance and tear film instability. The etiology of DED can involve several deficiencies of the tear film, including the aqueous layer, the lipid layer, mucin layer or a combination of the three layers. The disease often presents as a complication of other diseases, prominently autoimmune disorders such as rheumatoid arthritis, diabetes and Sjogren’s syndrome, which may contribute to its manifestation. As such, DED may afflict individuals with differing severity of burning sensation, a feeling of dryness, and other symptoms of ocular discomfort. In severe cases, vision may be significantly impaired. Although the pathogenesis of DED includes a variety of causes, common consequences are a breakdown of corneal tear film with dehydration of the exposed outer corneal surfaces, ocular surface inflammation and subsequent damage to exposed tissues. Increased concentration of pro-inflammatory cytokines, such as TNFα, in patient tears or conjunctival tissue has been demonstrated to correlate with disease severity.
In 2024, the U.S. DED patient population is estimated to be approximately 39.3 million people and is expected to rise to 41.3 million patients by 2029. The market for prescription medications to treat DED is forecasted to increase to $7.3 billion in the G7 countries (the United States, France, Germany, Italy, Spain, UK and Japan) by 2029 from $3.9 billion in 2019. We estimate the segment of DED patients in the United States addressable by OCS-02 (patients with moderate or severe DED) to be approximately 10 million patients.
Limitations of current therapies and potential for OCS-02 (Licaminlimab) in DED
The DED patient population is significantly underpenetrated with only an estimated 13.0% of diagnosed U.S. patients expected to receive prescription treatments in 2024. The vast majority of patients who do receive treatment are treated with anti-inflammatory drugs, yet among treated patients only 13.0% feel that their chronic dry eye disease is well managed. Approved topical treatments for DED include Restasis®, Cequa® and Vevye®, which are formulations of cyclosporine. These drugs act only to increase tear production and are not indicated to reduce DED symptoms. Further limiting cyclosporine’s therapeutic utility is a delayed onset of action necessitating a two- to three-month steroid bridge, and a stinging sensation on application in some patients. Topical steroids, including Eysuvis®, are also often used to treat DED but are contraindicated for long-term use because of their side effects including glaucoma and cataracts. Furthermore, other treatments available for DED include Xiidra® and recently launched Tyrvaya® and Miebo®.
OCS-02’s differentiation as a potential treatment for DED
Given the central role of ocular inflammation in sustaining the pathology of DED and the utility of anti-TNFα as a highly effective anti-inflammatory agent, we believe the localized application of OCS-02 as an anti-TNFα therapeutic, if approved, may provide a differentiated DED treatment approach, which may effectively reduce ocular discomfort, avoid undesirable features of current therapies (such as stinging sensation, delayed onset of action, or steroid-related side effects), and provide benefit for many patients who do not receive lasting relief from current therapies.
91
We estimate the segment of DED patients in the United States addressable by OCS-02 (patients with moderate or severe DED) to be approximately 10 million patients.
OCS-02 has produced clinical trial results which support its continued development as a potential treatment for DED
Novartis, from whom we have obtained certain exclusive, worldwide rights to develop and commercialize OCS-02 through a December 19, 2018 licensing agreement (please see the section entitled “—Material Licenses, Partnerships and Collaborations” below), conducted a randomized, multi-center, double-masked, vehicle controlled Phase 2 clinical PoC trial designed to assess the safety and tolerability of OCS-02 and its efficacy in reducing DED symptoms. In the trial, patients were randomized on a 1:1 ratio into two cohorts. For a six-week period, the first trial cohort received a 60 mg/ml ophthalmic solution of OCS-02, while the second received vehicle. Participants in both cohorts self-administered one drop to each eye three times per day. The primary efficacy endpoint of the trial was improvement in the global ocular discomfort score as compared to vehicle. The global ocular discomfort score is a composite of discomfort frequency and severity as assessed by a visual analog scale using an electronic patient reported outcome. Improvement results in a reduction of the discomfort frequency or severity, or both, translating into a reduction of the resulting Global Ocular Discomfort Score as compared to baseline. A negative change from baseline indicates improvement. The secondary efficacy endpoint was an assessment of the number of patients that achieved more than 20 points improvement in the global ocular discomfort score. The data generated in this trial, consisting of 67 participants in the active group and 64 in the control group, are presented in the charts below.
OCS-02 generated statistically significant improvement in ocular discomfort as compared to vehicle.
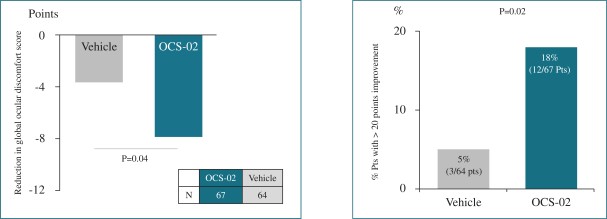
The trial met both primary and secondary endpoints. As is noted in the left chart above, administration of OCS-02 resulted in a statistically significant 7.9 mean point reduction in the global ocular discomfort score from baseline to treatment day 29 as compared to a 3.6 point mean reduction among patients that received vehicle only. In addition, as is noted in the right chart above, OCS-02 generated an improvement in the global ocular discomfort score of greater than 20 points in 12 of the 67 patients, or 18.0% of total trial participants. A similar level of response was achieved in only 5.0%, or three of the 64, patients included in the vehicle control group. The results of exploratory endpoints, which included physician graded conjunctival hyperemia, corneal staining, Meibomian gland assessment and tear film osmolarity, were similar across treatment groups. OCS-02 demonstrated a statistically significant improvement in the global ocular discomfort score compared to vehicle in patients with severe DED. It was well tolerated, with no increase in IOP and minimal systemic drug exposure.
Proprietary genetic biomarker may enable a precision medicine approach to DED
We conducted an exploratory pharmacogenetic analysis focused on the genes relevant to the TNF pathway and Sjogren’s syndrome among those 12 out of 86 patients who had the CC genotype gene variance or SNP. Among the gene variants analyzed, a correlation between one variant (rs1800693 CC genotype, “CC genotype”) in the TNFR1 gene, and a greater response (p<0.0001) to OCS-02 was observed at Day 29.
92
The below figure shows individual patient profiles by study days for change from baseline global ocular discomfort score for participants with the CC genotype.
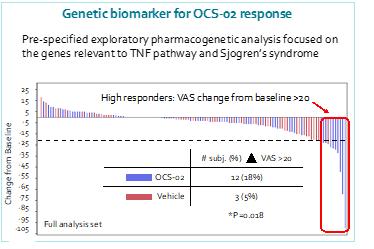
Patients with this variant displayed a significant reduction in inflammatory factors, including interleukin 1 beta (IL1B), interleukin 8 (IL8) and TNFα. This correlation is evidenced in the messenger RNA (“mRNA”) expression profiles of TNFα presented in the charts below which compared expression levels of patients with the various gene variants at Days 29 and 43 after dosing with either active drug candidate or vehicle. It was represented in 12 of 86 patients (14.0%) analyzed for the primary efficacy endpoint in this study, similar to the 13.0% of patients in the U.S. study.
A specific gene variant may enable biomarker based treatment
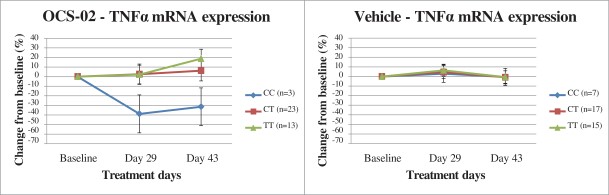
We believe that further validation of this genetic biomarker may enable us to identify a specific high response patient population which may allow us to enrich clinical trial enrollment and enhance our ability to evaluate the efficacy of OCS-02 in this indication and subpopulation. We intend to further evaluate the utility of this biomarker during our ongoing Phase 2b RELIEF trial of OCS-02.
Phase 2b trial design
In light of the results generated by OCS-02 in its Phase 2 PoC trials, we have advanced OCS-02 into an estimated 120 subject Phase 2b RELIEF clinical trial to evaluate the safety and efficacy of OCS-02 in treating the signs and symptoms of DED. This trial is randomized, multi-center, double masked and vehicle-controlled. Following initial screening, trial participants are randomized on a 1:1 basis into either the treatment cohort or the vehicle cohort and receive OCS-02 60mg/mL or vehicle three times daily for six weeks, followed by a two week follow up period. The efficacy measures and endpoints of the trial include a significant improvement in signs of DED, such as total corneal fluorescein staining, the percentage of patients with a 10 mm or greater increase in Schirmer’s test, as well as symptoms of DED such as global ocular discomfort compared to vehicle. Biomarker analyses (from impression cytology samples), as well as genotyping of subjects, are additional endpoints of the trial.
93
Topline results are anticipated in the second quarter of 2024.
OCS-02 (Licaminlimab) for the treatment of non-infectious anterior uveitis
In addition to its potential use as a therapeutic to treat DED, we are also evaluating OCS-02 (Licaminlimab) for use as a treatment option for patients with non-infectious anterior uveitis, including patients with chronic or recurrent non-infectious anterior uveitis who would benefit from a steroid-sparing option.
Uveitis is a condition characterized by the inflammation of the uveal tract but can also cause the inflammation of nearby tissues, such as the retina, the optic nerve, and the vitreous humor. Uveitis is caused by inflammatory responses inside the eye in response to an attack from the body’s own immune system, infection, or trauma and injury to the eye. Uveitis is closely associated with various systemic diseases, including autoimmune disorders, and infectious diseases. However, a significant proportion of uveitis is idiopathic, with no identifiable cause for the disease. It primarily affects people between 20 and 60 years of age but can present at any age. If left untreated, uveitis can cause complications including macular edema, retina scarring, glaucoma, cataracts, optic nerve damage, retinal detachment and permanent vision loss. Uveitis, which can affect one or both eyes, accounts for between 10.0% to 20.0% of all cases of blindness in the United States and Europe, and causes approximately 30,000 new cases of blindness each year in the United States alone.
Loss of vision is correlated with the severity, frequency and duration of inflammatory episodes. Accordingly, the objective of treatment is fast and complete suppression of inflammation. Uveitis is categorized as either anterior, intermediate or posterior uveitis depending on the location of inflammation, or as panuveitis if present in multiple locations. Anterior uveitis is the most prevalent form of the disease and is associated with visual impairment. We estimate that that approximately 51.0% of patients in the United States who are diagnosed with anterior uveitis experience chronic or recurrent disease.
We estimate OCS-02 to address a patient segment of 170,000 patients with chronic or recurrent, anterior, non-infectious uveitis in the United States for 2024.
Limitations with the standard of care to treat anterior uveitis
The standard of care for uveitis is corticosteroids, which are administered as topical, intravitreal, periocular or oral depending on the location and severity of the disease. Active non-infectious uveitis is treated with topical corticosteroids. While topical corticosteroids have demonstrated clinical efficacy, their use is associated with a number of adverse ocular and systemic events. Topical ocular corticosteroid use is estimated to cause an increase in IOP of more than 15 mmHg among 4.0% and 6.0% of the general population and an increase of between 6 and 15 mmHg in up to one-third of users after daily application for four to six weeks. Persistent elevation in IOP may result in glaucoma, characterized by visual field loss and optic nerve damage, or the formation of cataracts. Incidence of cataract worsening or formation is related to total topical dose and duration. Based on multi-year studies with ocular corticosteroid implants, we estimate that approximately 31.0 to 47.0% more patients developed or experienced worsening of cataracts compared to control arms (sham implants or standard of care).
OCS-02 differentiated as a steroid-sparing treatment for anterior uveitis
Given the limitations related to longer-term steroid use in patients with recurrent or chronic uveitis, we believe OCS-02 has potential as a steroid-sparing treatment alternative. In November 2019, we commissioned a market research report which involved interviews with 14 key opinion leaders, high volume practitioners of uveitis treatment (ophthalmologists and uveitis specialists) and payer experts. The results suggested that physicians are likely to be receptive to prescribing a topical, non-steroidal treatment after initial administration of a topical corticosteroid that may both shorten the duration of topical steroid use and obviate the potential need to advance patients to oral steroids. If approved, OCS-02 may also be appropriate for patients who demonstrate an inability to tolerate steroid treatment.
OCS-02 Phase 2 clinical trial results support its continued development as a potential treatment for non-infectious anterior uveitis
94
Novartis also conducted a Phase 2 clinical PoC trial to evaluate the use of OCS-02 as a potential treatment for acute anterior uveitis (“AAU”). This trial was a randomized, multi-center, double-masked, active controlled evaluation to assess the safety, tolerability and efficacy of OCS-02 administered for up to 21 days in resolving ocular inflammation in the anterior chamber associated with AAU. A 60 mg/ml ophthalmic solution of OCS-02 was administered to trial participants in the OCS-02 cohort and topical dexamethasone administered to patients in the active-control cohort. Trial participants received a maximum of eight drops daily per treated eye for the first two weeks with dosing tapered for the following two-week period. Response to treatment was defined as a reduction from baseline of 2 or more anterior chamber cell grades.
35 patients completed the trial, with 25 patients in the OCS-02 cohort and 10 patients in the active control cohort. OCS-02 achieved the primary endpoint established for the trial, which was a responder rate in excess of 30.0%. Among the 25 participants that completed the trial and were treated with OCS-02, 14 patients, or 56.0%, demonstrated a response to OCS-02 treatment at Day 22, specified as the PoC treatment period for the trial. In the trial, OCS-02 was observed to be well tolerated. No increase in IOP related to OCS-02 was observed, and no systemic adverse safety signals were observed.
Planned Phase 2b trial evaluating OCS-02 as a treatment for non-infectious anterior uveitis
Given the encouraging results generated by OCS-02 in the Phase 2 clinical PoC trial, we intend to advance this clinical candidate into a Phase 2b trial for evaluation as a therapeutic for non-infectious chronic anterior uveitis with potential as a steroid-sparing alternative to the currently used drugs. Trial parameters to be incorporated into this clinical evaluation are in development. The commencement of this trial will follow after the Phase 2b DED trial.
OCS-05
Key Program Highlights:
In addition to development candidates intended to modulate inflammatory conditions associated with ocular disease pathologies, we are also advancing OCS-05, a small molecule in development as a potential disease modifying neuroprotective agent designed to address neurological damage to the optic nerve. We are initially developing OCS-05 as a potential therapeutic to treat AON. OCS-05 has been granted Orphan Drug Designation by both the FDA and the European Commission for this indication. OCS-05 has been studied in preclinical studies suggesting neuroprotective and remyelinating activity, as well as in a UK Phase 1 clinical trial (with 48 healthy volunteers) in which OCS-05 was well tolerated and showed pharmacokinetics (“PK”) with good correlation with its pre-clinical animal studies. We are currently studying OCS-05 in a PoC trial in AON in France, for which we anticipate topline data readout in the fourth quarter of 2024. Should the clinical results of our AON trials prove sufficiently compelling, we intend to evaluate OCS-05 to treat other more pervasive neurological pathologies of the eye such as geographic atrophy, neurotrophic keratitis and glaucoma. We obtained an exclusive license, worldwide to develop OCS-05 through a licensing agreement we entered into with Accure Therapeutics SL (“Accure”), dated as of January 29, 2022 (Please see the section entitled “—Material Licenses, Partnerships and Collaborations” below).
OCS-05 is a small molecule peptidomimetic that has a differentiated mechanism of action through the activation of SGK2 which is hypothesized as part of the neurotrophic factor signaling pathways that supports neuronal cell development, survival and repair, including oligodendrocyte precursor differentiation and myelination. Enzymes in the SGK2 family are recognized to regulate a range of fundamental cellular processes such as cellular proliferation and survival.
95
SGK2 activation leads to an upregulation of signaling molecules forkhead box O3 (“FOXO3”), which reduces apoptosis, the downregulation of glycogen synthase kinase 3 beta (“GSK3B”), which improves anti-oxidation, and an upregulation of N-myc downstream-regulated gene 1 (“NDRG1”) involved in oligodendrocyte development and differentiation. The potential disease modulating activity of OCS-05 may distinguish it as a neuroprotective SGK2 activator.
OCS-05 was placed on a clinical hold by the FDA in 2016
Accure had conducted a limited set of animal regulatory toxicology studies in 2016 and submitted them to the FDA in an IND requesting the initiation of human testing. Upon review, the FDA found the data insufficient and asked for more animal toxicology data to be generated prior to human studies, thereby placing OCS-05 on the regulatory status of “clinical hold” pending the availability of the requested data. In response, Accure chose to withdraw the IND in 2017, rather than invest in further toxicology studies to address the FDA’s request, and pursue the development in the UK and France. Upon our license of OCS-05 from Accure in 2022, we reinstated the IND and are currently working on activities to enable a clinical trial under the IND in the U.S., including the necessary toxicology studies. Other health authorities where clinical studies have been proposed, including the UK and France, have authorized the initiation of clinical studies of selected doses and reinforced safety measures as in our European Phase 1 trial in AON.
OCS-05 for the treatment of acute optic neuritis
AON is an inflammation of the optic nerve that can cause the death of neurons, leading to vision impairment. A variety of infectious diseases, immune disorders, demyelinating disorders, non-inflammatory systemic disease or trauma can cause AON. AON is commonly associated with multiple sclerosis (“MS”) and shares similar physiopathology. AON is the presenting symptom of MS in 15.0-20.0% of patients and will impact over 50.0-65.0% of patients with MS at some time during their lifetime. However, the causes of AON are not always clear, as it can also arise in patients without MS.
The acute inflammation of the optic nerve causes the loss of myelin and oligodendrocytes, optic nerve conduction block and loss of vision. At the onset of AON, patients often suffer from ocular pain increasing with eye movement, associated with a variety of visual impairments. Deterioration of visual acuity, color vision or flashes of light are common. The loss of vision ranges considerably between patients from mild blurring to loss of perception of light. The condition tends to worsen over the first several days after the appearance of symptoms before starting to improve over the first two weeks. The recovery continues for as long as a year after onset. Even if high contrast visual acuity returns to near normal, patients often report that their vision has not completely recovered. There remains a persistent impairment of low contrast letter acuity and clinically meaningful reduction in vision-related quality of life.
When the inflammation recedes, remyelination often occurs but it is incomplete, the result of persistent demyelination and neuronal death. Without the myelin sheath which normally protects the axon, neurons located in demyelinated segments become fragile and prone to death. Thinning of the retinal neural fiber layer (“RNFL”), which is made up of unmyelinated axons originating from the retinal ganglion cell (“RGC”) bodies, indicates significant AON-induced axonal loss. RNFL thinning, most pronounced three to six months after an acute AON event, along with thinning of the ganglion cell bodies layer, correlates with diminished scores of visual acuity and visual field sensitivity.
No therapeutic is currently approved that preserves vision and ganglion/retinal nerve integrity after an acute episode of AON. Medication intended to treat the inflammation and related symptoms can be administered just after AON onset and patients often receive high doses of corticosteroids for a few days to alleviate disabling vision-related symptoms caused by the inflammation. Corticosteroids have become the current standard of care, as the therapy acts to shorten the attack and accelerate recovery of acute visual symptoms. However, vision loss persists in 10.0% to 20.0% of patients despite administration of IOP lowering therapy. We believe a neuroprotective therapeutic, such as OCS-05, if approved, could prevent long term axonal loss may promote enhanced clinical outcomes.
OCS-05 demonstrated compelling neuroprotective qualities in an animal model of AON
In a rat model of AON, animals were segregated into four groups. The first group of healthy animals represented a sham control. Three additional groups received lysolecithin via injection into the optic nerve of study animals to induce inflammation and demyelination. Rats in group two received no treatment and served as a pathological control group. Groups three and four were administered OCS-05 once daily over a five-day period. Animals in group three received a 35 mg/kg dose of OCS-05 while animals in the fourth group received a dose of 70 mg/kg.
96
The animals were sacrificed on the sixth day and assessed for a decline in RGC count.
As is noted in the results presented below, both groups of animals that received OCS-05 generated a statistically significant reduction in RGC loss when administered following the lysolecithin challenge, with rats administered the 35 mg/kg dose of OCS-05 demonstrating a 35.9% mean reduction of RGC loss. Animals in the higher dose treatment group who received a 70 mg/kg dose of OCS-05 displayed a more profound benefit from OCS-05 dosing, with RGC loss declining 64.0%.
RGC loss in animals treated with OCS-05 was significantly reduced in an animal model of AON.
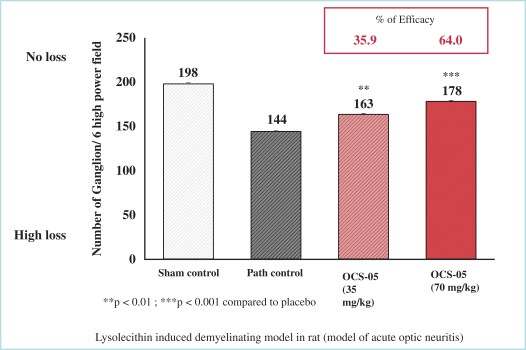
The reduction in RGC loss was also observed in a visual assessment of representative tissue samples collected from animals in three of the four study groups, the sham control group, the pathological control group and rats treated with the higher 70 mg/kg dose of OCS-05. As is depicted in the slides of the optic nerve presented below, normal ganglion cell density was observed in the evaluation of tissue taken from a healthy animal in the sham control group. In contrast, cell counts taken from samples of rats included the lysolecithin challenge group that made up the pathological control witnessed a prominent decrease. After completion of the five-day protocol, this decline was noted to have reversed, with rats who received the 70 mg/kg dose of OCS-05 observed to have retained a significantly higher number of ganglion cells. Similar results illustrating a reduction in axonal loss and demyelination, along with improvement in clinical function, have been achieved in animal models of AON.
97
OCS-05 was seen to bolster ganglion cell counts after lysolecithin challenge.
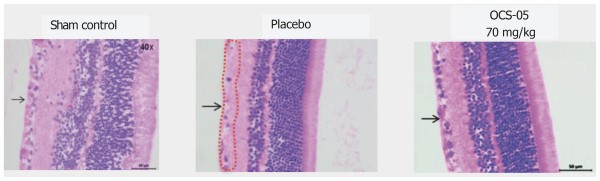
OCS-05 was well tolerated in a trial involving healthy volunteers
A randomized, double-masked, placebo controlled single-ascending dose and multiple-ascending dose trial was conducted in the United Kingdom to evaluate the safety, tolerability and PK and pharmacodynamics of OCS-05 dosing through the intravenous infusion of healthy volunteers with the drug candidate. This trial was designed to include four interlocking cohorts of eight adult subjects each to evaluate eight single ascending doses, with an additional two cohorts of eight adult subjects each included in the two multiple ascending dose trials. The single ascending dose cohorts were administered drug in doses ranging from .05 mg/kg to 3.2 mg/kg. The two cohorts in the multiple ascending dose trial received either a 2.4 mg/kg dose or a 3.0 mg/kg dose, once daily, for five consecutive days. In this trial, it was observed that OCS-05 was well tolerated with no serious AEs noted. Human PK data produced by this trial showed good correlation with data produced in animal studies of the compound. This trial was conducted under a clinical trial protocol approved by European regulatory authorities.
We are investigating OCS-05 as a treatment for AON in a First-in-Patient clinical trial
The results of prior clinical and preclinical trials of OCS-05 in promoting disease modifying effects, together with the safety and PK profile observed in this first-in-human clinical trial enabled us to advance the compound into a First-in-Patient clinical PoC trial. The Acute OptiC NeUrITis of DemYelinating Origin (“ACUITY”) trial, a randomized, double-masked, placebo controlled, multiple center trial, is a First-in-Patient trial enrolling patients diagnosed with AON within ten days of acute disease episode onset. The objective of this study is to assess the safety and tolerability of OCS-05 along with initial signs of efficacy. In addition to the trial’s primary safety endpoint, a key secondary endpoint will be the effect of OCS-05 on retinal layer thickness and other visual parameters in the affected eye. The study is currently being conducted in France under French regulatory guidance and we are anticipating topline data readout in the fourth quarter of 2024.
We believe that positive outcomes in this trial could support the compound’s possible development as a potential treatment in other ophthalmic conditions involving the posterior segment including glaucoma, geographic atrophy, DR as well as certain diseases of the anterior segment including corneal keratitis. The novel mechanism of action of OCS-05 may enable it to demonstrate benefit in treating these additional ocular conditions and may additionally allow its development in non-ocular neurological disorders involving neuronal inflammation such as MS.
In 2016, the OCS-05 development program was placed on clinical hold by the FDA related to the absence of no observed adverse effects levels (“NOAEL”), in prior preclinical studies conducted by the sponsor at that time. After we licensed the asset from Accure, our strategy has included plans to work with a CRO to complete the additional studies required to establish NOAEL, in order to enable our submission of an IND application with the FDA.
We are planning to investigate OCS-05’s potential as a treatment for Neurotrophic Keratitis and should the outcome of the AON trial be positive, we will also evaluate the potential as a treatment for neuro-ophthalmology diseases such as Glaucoma
Preclinical studies of OCS-05 in a model of glaucoma in Sprague rats showed results which support its potential to be developed as a treatment for glaucoma. In these two experiments, high intraocular pressure was induced in rats by injecting hypertonic saline solution into the episcleral vein of one eye of each rat, and then the rats were treated for six weeks.
98
In one experiment, rats in the active group were treated with OCS-05 as an eye drop twice daily for six weeks, rats in the positive control group received nerve growth factor (“NGF”), and rats in the control group received placebo of saline 5.0% dimethyl sulfoxide (“DMSO”). In the other experiment, rats in the active group were treated with OCS-05 as an intravitreal injection once every two weeks, for six weeks, and rats in the control group received placebo of saline 5.0% DMSO. Retinal ganglion cells (“RGCs”) count was measured via haematoxylin and eosin stain (“H & E”) histological quantification, and IOP was also measured.
Sprague rats displayed significant loss of RGCs one month after the induction of ocular hypertension. In animals treated with OCS-05, either as eye drops or through intravitreal injection, there were statistically significant increases in RGCs surviving compared with those that received the placebo. In the experiment which included a positive control of NGF, OCS-05 treatment showed a similar effect to that seen with NGF. In addition, IOP did not significantly decrease with administration of OCS-05. We believe this data suggests that OCS-05 may promote neuronal survival in this animal model of glaucoma via neuroprotection (and not by reversing the induced ocular hypertension).
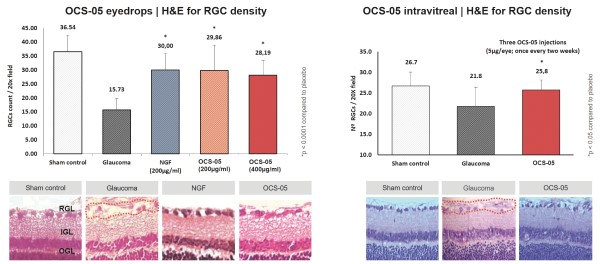
OCS-05 (eyedrops and intravitreal) prevents RGCs damage without reducing intraocular pressure
Given the results from these preclinical studies, we plan to further study OCS-05, and if results from our ACUITY trial in AON further support OCS-05’s potential as a neuroprotective compound, we may prepare for and initiate clinical development of OCS-05 in glaucoma. Glaucoma represents a large market, and we are not currently aware of the existence of any other compound in a similar or more advanced stage of development as a neuroprotective drug for glaucoma.
Additionally, we also plan to further study OCS-05 for its potential to enter clinical development as a treatment for neurotrophic keratitis (“NK”). NK is a rare eye disorder which results from damage or loss of function of nerves which innervate the cornea, which can lead to corneal perforation, corneal scarring, corneal melting, loss of vision, or loss of the eye. In 2018, the FDA approved the NGF drug cenergermin (“Oxervate”) to treat NK. However, Oxervate may be cost prohibitive for patients and payors, as ASCRS Eyeworld estimated in 2020 that Oxervate costs approximately $11,000 per week for an 8-week treatment course for NK.
Given that preclinical studies of OCS-05 have shown data suggesting that the OCS-05 could provide neuroprotective benefits, we believe it may have potential to treat the nerve impairment underlying NK. If results from our ACUITY trial in AON further support OCS-05’s potential as a neuroprotective compound, we may prepare for and initiate clinical development of OCS-05 in NK. We are currently not aware of the existence any other drugs except for Oxervate which are approved or in a similar or more advanced stage of development as a treatment for NK.
We are currently conducting formulation studies to develop a topical formulation of OCS-05 which can be used in further preclinical or in clinical development of OCS-05 in glaucoma or in NK.
Additional Development Initiatives
99
In addition to our six clinical development programs involving OCS-01, OCS-02 (Licaminlimab) and OCS-05, we also are engaged in a number of earlier preclinical development initiatives, including the evaluation of OCS-03 as a possible treatment for corneal neovascularization, a common disorder caused by the aberrant development of new blood vessels into the cornea and pterygium, a pink colored growth that originates in the conjunctiva. We are also assessing the preclinical candidate OCS-04, an innovative topical ophthalmic formulation project, preliminarily intended for corneal graft rejection prevention and possibly other inflammatory related conditions targeting the ocular surface.
Material Licenses, Partnerships and Collaborations
License Agreement with Novartis for OCS-02 (Licaminlimab)
Pursuant to a license agreement, dated as of December 19, 2018, as amended, by and between us and Novartis (the “Novartis Agreement”), we obtained an exclusive, royalty-bearing, sublicensable (subject to certain conditions), assignable (subject to certain conditions), worldwide license under certain patents, know-how and manufacturing platform technology to develop, manufacture and commercialize pharmaceutical, therapeutic or diagnostic products containing a specified single chain antibody fragment formulation as an active ingredient in the licensed field as defined in the Novartis Agreement. The license granted to us by Novartis includes sublicenses of rights granted to Novartis by certain third parties, and our license to such rights is expressly subject to the applicable terms and conditions of the agreements between Novartis and such third parties.
We are deemed the owner of any inventions that are (a) created solely by or on behalf of us pursuant to the Novartis Agreement and (b) severable from the licensed products, and grant Novartis a first right to negotiate a worldwide, royalty-bearing license under any patents directed at such inventions for purposes outside of the licensed field. We also grant Novartis a worldwide, non-exclusive, perpetual, irrevocable, royalty-free, fully paid-up license back under any patents owned by us that (i) cover inventions arising from the Novartis Agreement, the practice of which would infringe the patents licensed to us by Novartis, or (ii) otherwise incorporate Novartis’ proprietary information, in each case, for certain uses outside of the licensed field.
We made an upfront payment to Alcon of CHF 4.7 million ($4.7 million at the exchange rate at the time of payment) in cash and issued 401,709 ordinary shares (recast using the Exchange Ratio to reflect the impact of the BCA) for the residual between the fair value and the upfront payment. This was accounted for as a share-based payment transaction under IFRS 2. We originally entered into the Novartis Agreement with Alcon Research, Ltd. (“Alcon”), which subsequently assigned its rights and obligations under the Novartis Agreement to Novartis in connection with Alcon's spin-off from Novartis. As of December 31, 2023, we were obligated to pay Novartis additional up to CHF 81.6 million ($97.0 million at the December 31, 2023 exchange rate) in the aggregate upon the achievement of certain development, regulatory, sales and other milestones and tiered royalties ranging from a mid-single digit to a mid-teen percentage on net sales. In consideration for the exclusive sublicense from Novartis under certain third-party intellectual property rights, we are obligated to pay a low-single digit royalty on our net sales of the licensed product, however, such payments will be deducted from royalties payable to Novartis. Our royalty payment obligations are subject to certain reductions and expire with respect to any licensed product on a country-by-country basis upon the later of (a) the expiration of the last to expire valid claim of any licensed patent covering any such licensed product in such country; (b) the expiration of the period of data exclusivity in any country worldwide; or (c) twelve (12) years after first commercial sale of such licensed product in such country (“Royalty Term”).
Under the Novartis Agreement, we are obligated to use diligent efforts to develop, manufacture or have manufactured, and commercialize the licensed products in the licensed field worldwide. The Novartis Agreement will expire upon the last-to-expire Royalty Term. We may terminate the Novartis Agreement without cause at any time upon advance written notice to Novartis. Upon written notice to Novartis, we may terminate the Novartis Agreement for cause due to the following events: (a) an insolvency event occurs; (b) Novartis materially breaches its obligations under the Novartis Agreement and fails to cure such breach within a specified period of time; or (c) upon advance written notice for material scientific, technical or medical reasons or in case of a material adverse change that renders further continuation of the Novartis Agreement by us commercially unreasonable or otherwise not viable. Upon written notice to us, Novartis may terminate the Novartis Agreement for cause due to the following events: (i) we fail to pay any undisputed amount due under the Novartis Agreement and we fail to remedy such failure within a specified period of time; (ii) an insolvency event occurs; (iii) we materially breach our obligations under the Novartis Agreement and fail to cure such breach within a specified period of time; or (iv) following negative clinical trial results, we terminate development of the licensed product and do not pursue any further indications in the licensed field.
100
License Agreement with Accure for OCS-05
Pursuant to a license agreement, dated as of January 29, 2022, by and between us and Accure (the “Accure Agreement”), we obtained an exclusive, worldwide, sublicensable (subject to certain conditions) and transferable (subject to certain conditions) license under certain patents, know-how and inventory of Accure for any and all uses and purposes, including to perform research, development, manufacturing and commercialization activities in any manner and for any purpose. The licensed patents are co-owned by Accure with third parties who have reserved the right to use the licensed patents for education and research purposes pursuant to an inter-institutional agreement.
As of December 31, 2023, we have paid the full contractual non-refundable upfront fee of CHF 3.0 million and reimbursed costs in the amount of approximately CHF 0.5 million. As of December 31, 2023, we were obligated to pay Accure (a) up to CHF 94.3 million ($112.1 million at the December 31, 2023 exchange rate) in the aggregate upon the achievement of certain development, regulatory and sales milestones; (b) tiered royalties ranging from a mid-single digit to a low mid-teen percentage on net sales of licensed products; and (c) a percentage in the high teens on sublicensing revenues received any time after 36 months from the agreement effective date, and a higher percentage on sublicensing revenues received prior to such date, in all cases subject, in the case of this clause (c), to reduction for any amounts that were previously paid or are concurrently or later paid by us to Accure pursuant to our milestone payment obligations. Our royalty payment obligations are subject to certain reductions and expire on a licensed product-by-licensed product and country-by-country basis upon the later of (i) the expiration of the last valid claim of any licensed patent covering such licensed product in such country; (ii) the expiration of such licensed product’s Orphan Drug status, if any, in such country; or (iii) ten (10) years following the date of first commercial sale of such licensed product in such country (the “Payment Period”). Under the Accure Agreement, we are obligated to use commercially reasonable efforts to develop and seek regulatory approval for a licensed product in major countries of the territory as defined in the Accure Agreement.
The Accure Agreement will expire on a licensed product-by-licensed product and country-by-country basis upon the expiration of the applicable Payment Period with respect to such licensed product in such country. We may terminate the Accure Agreement in whole or in part at any time upon advance written notice (a) for documented reasonable scientific, regulatory, commercial reasons related to the licensed product without incurring any penalty or liability to Accure and (b) for no reason. Each party may terminate the Accure Agreement with immediate effect upon written notice to the other party (i) in the event such other party commits a material breach of its obligations under the Accure Agreement and fails to cure that breach within a specified period of time or (ii) with certain exceptions, upon such other party’s bankruptcy. Accure may terminate the Accure Agreement with immediate effect upon written notice to us if we file any action to invalidate any of the licensed patents or fail to maintain the licensed patents in major countries of the territory as defined in the Accure Agreement, or, subject to certain exceptions, if we fail to meet certain development obligations and are unable to agree upon modifications to the development plan with Accure.
Manufacturing Strategy
We oversee and manage third-party contract manufacturing organizations (“CMOs”), to support the development and manufacture of product candidates for our clinical trials, and, if any product candidates receive marketing approval, we expect to rely on such manufacturers to meet commercial demand. We expect this strategy will enable us to maintain a more efficient operating and cost infrastructure, avoiding dependence on our own manufacturing facility and equipment, while simultaneously enabling us to focus our expertise on the clinical development and future commercialization of our products, if approved. Currently, we rely on and have agreements with third-party contract manufacturers for developing and manufacturing API/drug substance/drug product for OCS-01, OCS-02 (Licaminlimab) and OCS-05, and we expect to enter into commercial supply agreements with such manufacturers prior to any potential approval. We continue to develop and improve the manufacturing processes for OCS-02 and OCS-05 and to address the requirements in these highly regulated markets. Improvement of manufacturing processes may involve transferring the development and manufacturing to another CMO, taking into account technical, quality and economic aspects.
Each of OCS-01, OCS-02 and OCS-05 is manufactured via conventional pharmaceutical processing procedures, employing commercially available excipients and packaging materials. The procedures and equipment employed for manufacture and analysis are consistent with standard pharmaceutical production, and are transferable to a range of manufacturing facilities, if needed.
101
Competition
We face substantial competition from multiple sources, including large and specialty pharmaceutical and biotechnology companies, academic research institutions and governmental agencies and public and private research institutions. Our competitors compete with us on the level of the technologies employed, or on the level of development of product candidates. In addition, many small biotechnology companies have formed collaborations with large, established companies to (i) obtain support for their research, development and commercialization of products or (ii) combine several treatment approaches to develop longer lasting or more efficacious treatments that may potentially directly compete with our current or future product candidates. We anticipate that we will continue to face increasing competition as new therapies and combinations thereof, technologies, and data emerge within the treatment of ocular conditions.
In addition to the current standard of care treatments for patients with ocular diseases, numerous commercial and academic preclinical studies and clinical trials are being undertaken by a large number of parties to assess novel technologies and product candidates.
Several large pharmaceutical and biopharmaceutical companies that have commercialized, or are developing treatments for ocular diseases, compete with us. Companies that compete with us directly on the level of the development of product candidates targeting DME include Abbvie, Alimera Sciences, Bayer, Novartis, Regeneron and Roche, among others. Companies that have commercialized or are developing drug candidates to treat inflammation and pain associated with ocular surgery include Abbvie, Alcon, Bausch + Lomb and Teva Pharmaceuticals, among others; companies that compete with us in the area of DED include Abbvie, Alcon, Bausch + Lomb, Viatris and Sun Pharmaceuticals, among others. Companies engaged in the commercialization or development of therapeutics to treat uveitis include Abbvie and Bausch + Lomb, among others. We are also aware of an eye drop product candidate in clinical development by OcuTerra Therapeutics for the treatment of diabetic retinopathy and DME, an indication related to that for which we are developing OCS-01.
Many of our competitors, either alone or in combination with their respective strategic partners, have significantly greater financial resources and expertise in research and development, manufacturing, regulatory approval process and marketing than we do. Mergers and acquisition activity in the pharmaceutical, biopharmaceutical and biotechnology sector is likely to result in greater resource concentration among a smaller number of our competitors. Smaller or early-stage companies may also prove to be significant competitors, particularly through sizeable collaborative arrangements with established companies. These competitors also compete with us in recruiting and retaining qualified scientific and management personnel, establishing clinical trial sites, patient registration for clinical trials and acquiring technologies complementary to, or necessary for, our programs.
Our commercial opportunities could be reduced or eliminated if one or more of our competitors develop and commercialize products that are safer, more effective, better tolerated, or of greater convenience or economic benefit than our proposed product offerings. Our competitors also may be in a position to obtain FDA or other regulatory approval for their products more rapidly, resulting in a stronger or dominant market position before we are able to enter the market. The key competitive factors affecting the success of all of our programs are likely to be product safety, efficacy, convenience and treatment cost.
Intellectual Property
Intellectual property is of vital importance in our field and in biotechnology generally. We seek to protect and enhance proprietary technology, inventions, and improvements that are commercially important to the development of our business by obtaining, maintaining, enforcing and defending intellectual property rights, including patent rights, whether owned or licensed from third parties. We will also seek to rely on regulatory protection afforded through inclusion in expedited development and review, data exclusivity, market exclusivity and patent term extensions where available.
We have sought patent protection in the United States and internationally related to our novel drug targets, composition of matter, formulations and other inventions and improvements that are central to our R&D efforts. For our product candidates, our strategy is to pursue patent protection covering compositions of matter, formulations and methods of use.
102
In addition, we seek to identify additional means of obtaining patent protection, including specific therapeutic indications and dosing regimen-related claims, which may enhance commercial success. We also rely on trade secrets that may be important to the development of our business. Trade secrets are difficult to protect and provide us with only limited protection.
As of December 31, 2023 and currently, we own and exclusively in-licensed a patent portfolio that included 11 issued U.S. patents, five issued European patents validated in multiple jurisdictions, and 56 issued patents in other foreign jurisdictions, as well as twelve pending non-provisional U.S. patent applications, and 65 foreign pending patent applications, including eight pending European patent applications, and four pending Patent Cooperation Treaty ("PCT") applications related to our different product candidates, namely, OCS-01, OCS-02 (Licaminlimab), OCS-03, OCS-04 and OCS-05.
OCS-01
Regarding our OCS-01 product candidate, as of December 31, 2023 and currently, we own a patent family that consisted of three issued U.S. patents and one granted European patent validated in 12 jurisdictions (Belgium, France, Germany, Great Britain, Iceland, Ireland, Italy, the Netherlands, Poland, Spain, Switzerland, Turkey) with claims covering the composition including dexamethasone. These patents will expire in 2026, without giving effect to any potential patent term extensions and patent term adjustments and assuming payment of all appropriate maintenance, renewal, annuity or other governmental fees.
As of December 31, 2023 and currently, we own a second patent family that consisted of two issued U.S. patents, two pending non-provisional U.S. patent applications, one granted European patent validated in 41 jurisdictions (Albania, Austria, Belgium, Bosnia and Herzegovina, Bulgaria, Croatia, Cyprus, Czech Republic, Denmark, Estonia, Germany, Great Britain, Greece, Finland, France, Hungary, Iceland, Ireland, Italy, Latvia, Lithuania, Luxembourg, Malta, Republic of Moldova, Monaco, Montenegro, Morocco, the Netherlands, North Macedonia, Norway, Poland, Portugal, Romania, San Marino, Serbia, Slovakia, Slovenia, Spain, Sweden, Switzerland, Turkey), sixteen issued patents in other foreign jurisdictions (Australia, China, Colombia, Eurasia, India, Japan, Mexico, South Africa (two patents),Taiwan (two patents), Ukraine, Hong Kong, Singapore, South Korea, Chili) and 14 pending foreign patent applications, including one pending European patent application, with claims covering the composition of matter of OCS-01. Patents (including any patents that issue from such patent applications) in this family will expire in 2037, without giving effect to any potential patent term extensions and patent term adjustments and assuming payment of all appropriate maintenance, renewal, annuity or other governmental fees.
As of December 31, 2023 and currently, we also own a patent family that consisted of six U.S. non-provisional patent applications and 21 additional foreign patent applications in other jurisdictions, including one European patent application, directed to specific formulations of OCS-01 and methods for stabilizing the composition for use as an eye drop. Patents, if issued from patent applications in this family, will expire in 2040, without giving effect to any potential patent term extensions and patent term adjustments and assuming payment of all appropriate maintenance, renewal, annuity or other governmental fees.
OCS-02 (Licaminlimab)
Regarding our OCS-02 (Licaminlimab) product candidate, as of December 31, 2023, we exclusively licensed from Novartis under the Novartis Agreement, in the licensed field as defined in the Novartis Agreement, one patent family that consisted of three issued U.S. patents and two granted European patents validated in 36 jurisdictions (Albania, Austria, Belgium, Bulgaria, Croatia, Cyprus, Czech Republic, Denmark, Estonia, Finland, France, Germany, Great Britain, Greece, Hungary, Iceland, Ireland, Italy, Latvia, Lithuania, Luxembourg, Malta, Monaco, the Netherlands, North Macedonia, Norway, Poland, Portugal, Romania, Serbia, Slovakia, Slovenia, Spain, Sweden, Switzerland, Turkey) and six jurisdictions (France, Germany, Great Britain, Italy, Spain, Switzerland), respectively, 22 issued patents in other foreign jurisdictions (Argentina, Australia, Brazil, Canada, Chile (two patents), China (two patents), India, Hong-Kong (two patents), GCC, Japan (two patents), Republic of Korea, Mexico (two patents), Philippines, Russia, South Africa, Taiwan, Ukraine) and two patent applications pending in other foreign jurisdictions, with claims covering composition of matter of OCS-02 or methods of use. Patents (including any patents that issue from such patent applications) will expire in 2031, without giving effect to any potential patent term extensions and patent term adjustments and assuming payment of all appropriate maintenance, renewal, annuity or other governmental fees.
103
In addition, as of December 31, 2023, we exclusively licensed from Novartis under the Novartis Agreement, in the licensed field as defined in the Novartis Agreement, one patent family directed on a biomarker for patient selection, that consists of one pending European and one U.S. patent application and four patent applications pending in Canada, China, Japan (two patent applications). Patents (including any patents that issue from such patent applications) will expire in 2037, without giving effect to any potential patent term extensions and patent term adjustments and assuming payment of all appropriate maintenance, renewal, annuity or other governmental fees.
In addition, as of December 31, 2023, we exclusively licensed from Novartis under the Novartis Agreement, in the licensed field as defined in the Novartis Agreement, six additional patent families covering composition of matter of OCS-02 or methods of use, which (including any patents that issue from patent applications in these families) will expire between 2023 and 2031, without giving effect to any potential patent term extensions and patent term adjustments and assuming payment of all appropriate maintenance, renewal, annuity or other governmental fees. Under the terms of the Novartis Agreement, Novartis is responsible for the prosecution and maintenance of these six patent families.
OCS-03
As of December 31, 2023 and currently, we own a patent family that consists of one pending U.S. non provisional application and one pending European application as well as one pending Taiwanese application, with claims covering composition of matter of OCS-03 and its use. Patents (including any patents that issue from patent application) will expire in 2041, without giving effect to any potential patent term extensions and patent term adjustments and assuming payment of all appropriate maintenance, renewal, annuity or other governmental fees.
OCS-04
As of December 31, 2023 and currently, we own one pending PCT application as well as pending applications in Argentina and Taiwan, with claims covering composition of matter of OCS-04 and manufacturing processes. In order for any future patent applications to claim the benefit of such PCT application, they must be filed not later than 30 or 31 months (depending on the jurisdiction) after the earliest priority date of such PCT application. Patents, if issued from the patent applications claiming the benefit of such priority application, if issued, will expire in 2043, without giving effect to any potential patent term extensions and patent term adjustments and assuming payment of all appropriate maintenance, renewal, annuity or other governmental fees.
OCS-05
Regarding our OCS-05 product candidate, as of December 31, 2023, we exclusively licensed from Accure under the Accure Agreement a patent family that consisted of three issued U.S. patents and one granted European patent validated in 24 jurisdictions (Austria, Belgium, Croatia, Czech Republic, Denmark, Finland, France, Germany, Great Britain, Greece, Hungary, Ireland, Italy, Luxembourg, Malta, the Netherlands, Norway, Poland, Portugal, Slovenia, Spain, Sweden, Switzerland, Turkey), as well as 10 issued patents (Australia, Brazil, Canada, China, India, Israel, Japan, Republic of Korea, Mexico, Russia) in other foreign jurisdictions, with claims covering composition of matter of OCS-05. These patents (including any patents that issue from such patent applications) will expire in 2031, without giving effect to any potential patent term extensions and patent term adjustments and assuming payment of all appropriate maintenance, renewal, annuity or other governmental fees.
As of December 31, 2023, we also exclusively licensed from Accure under the Accure Agreement a patent family that consisted of one pending non-provisional U.S. patent application and 15 pending foreign patent applications, including one pending European patent application, directed to the method of use of the composition of OCS-05 in combination with active compounds. Patents, if issued from such patent applications, will expire in 2040, without giving effect to any potential patent term extensions and patent term adjustments and assuming payment of all appropriate maintenance, renewal, annuity or other governmental fees.
As of December 31, 2023, we also exclusively licensed from Accure under the Accure Agreement a patent family consisting of one pending non-provisional U.S. patent application and six pending foreign patent applications, including one pending European patent application, with claims directed to specific dosage regimen for administering the active pharmaceutical ingredient of OCS-05. Patents, if issued from such patent applications, will expire in 2040, without giving effect to any potential patent term extensions and patent term adjustments and assuming payment of all appropriate maintenance, renewal, annuity or other governmental fees.
104
As of December 31, 2023 and currently, we also own a priority European patent application with claims covering a manufacturing process of OCS-05 and OCS-05’s intermediate synthesis products. In order for any future patent applications to claim the benefit of such priority application, such future patent application must be filed no later than 12 months after the filing date of such priority application. Patents, if issued from the patent applications claiming the benefit of such priority application, will expire in 2042 or 2043, assuming a filing within the 12-month priority period, without giving effect to any potential patent term extensions and patent term adjustments and assuming payment of all appropriate maintenance, renewal, annuity or other governmental fees.
Our commercial success will depend in part on obtaining, maintaining, protecting and enforcing patent protection and trade secret protection of our current and future product candidates and the methods used to develop and manufacture them, as well as successfully defending any such patents against third-party challenges, enforcing such patents against third-party infringers, and operating without infringing on, misappropriating or otherwise violating the intellectual property or proprietary rights of others. Our ability to stop third parties from making, using, selling, offering to sell or importing our product candidates will depend on the extent to which we have rights under valid and enforceable patents or trade secrets that cover these activities. We cannot be sure that patents will be issued with respect to any of our owned or in-licensed pending patent applications or with respect to any patent applications filed by us or our licensors in the future, nor can we be sure that any patents that may be granted to us or our licensors in the future will be commercially useful in protecting our product candidates, discovery programs and processes. For this and more comprehensive risks related to our intellectual property, please see the section entitled “Risk Factors—Risks Related to Our Intellectual Property.”
The terms of individual patents depend upon the legal term of the patents in the countries in which they are obtained. In most countries in which we file, including the United States, the patent term is 20 years from the earliest date of filing a non-provisional patent application. In the United States, a patent’s term may be lengthened by patent term adjustment, which compensates a patentee for administrative delays by the U.S. Patent and Trademark Office (“USPTO”), in examining and granting a patent, or may be shortened if a patent is terminally disclaimed over an earlier filed patent. In the United States, the term of a patent that covers an FDA-approved drug may also be eligible for extension, which permits patent term restoration as compensation for the patent term lost during the FDA regulatory review process. The Hatch-Waxman Act permits a patent term extension of up to five years beyond the expiration of the patent. The length of the patent term extension is related to the length of time the subject drug candidate is under regulatory review. U.S. patent term extension cannot extend the remaining term of a patent beyond a total of 14 years from the date of product approval, only one patent applicable to an approved drug may be extended and only those claims covering the approved drug, a method for using it, or a method for manufacturing it may be extended. Similar provisions to extend the term of a patent that covers an approved drug are available in Europe and other foreign jurisdictions. In the future, if and when our products receive FDA approval, we expect to apply for patent term extensions on patents covering those products. We plan to seek patent term extensions to any issued patents we may obtain in any jurisdiction where such patent term extensions are available, however there is no guarantee that the applicable authorities, including the FDA in the United States, will agree with our assessment that such extensions should be granted, and if granted, the length of such extensions. For more information regarding the risks related to our intellectual property, see section entitled “Risk Factors—Risks Related to Our Intellectual Property.”
We file U.S. non-provisional applications and PCT applications that claim the benefit of the priority date of earlier filed priority applications, when applicable. The PCT system allows a single application to be filed within 12 months of the original priority date of the patent application, and to designate all of the PCT member states in which national patent applications can later be pursued based on the international patent application filed under the PCT. The PCT searching authority performs a patentability search and issues a non-binding patentability opinion which can be used to evaluate the chances of success for the national applications in foreign countries prior to having to incur the filing fees. Although a PCT application is not issued as a patent, it allows the applicant to seek protection in any of the member states through national-phase applications. At the end of the period of two and a half years from the first priority date of the patent application, separate patent applications can be pursued in any of the PCT member states either by direct national filing or, in some cases by filing through a regional patent organization, such as the European Patent Office. The PCT system delays expenses, allows a limited evaluation of the chances of success for national/regional patent applications and enables substantial savings where applications are abandoned within the first two and a half years of filing.
105
For all patent applications, we determine claiming strategy on a case-by-case basis. Advice of counsel and our business model and needs are always considered. We seek to file patents containing claims for protection of all useful applications of our proprietary technologies and any product candidates, as well as all new applications and/or uses we discover for existing technologies and product candidates, assuming these are strategically valuable. We continuously reassess the number and type of patent applications in our portfolio, as well as the pending and issued patent claims to pursue maximum coverage and value for our processes and compositions, given existing patent office rules and regulations. Further, claims may be narrowed during patent prosecution, to the extent allowed, to meet our intellectual property and business needs.
We recognize that the ability to obtain patent protection and the degree of such protection depends on a number of factors, including the extent of the prior art, the novelty and non-obviousness of the invention, and the ability to satisfy the enablement requirement of the patent laws. In addition, the coverage claimed in a patent application can be significantly reduced before the patent is issued, and its scope can be reinterpreted or further altered even after patent issuance. Consequently, we or our licensors may not obtain or maintain adequate patent protection for any of our future product candidates or for our Optireach® technology platform. We cannot predict whether the owned or in-licensed patent applications we are currently pursuing will issue as patents in any particular jurisdiction or whether the claims of any issued patents we own or in-license will provide sufficient proprietary protection from competitors. Any patents that we own or in-license may be challenged, circumvented or invalidated by third parties.
The patent positions of biotechnology companies like ours are generally uncertain and involve complex legal, scientific and factual questions. Our commercial success will also depend in part on not infringing upon, misappropriating or otherwise violating the intellectual property or proprietary rights of third parties. Third-party patents could require us to alter our development or commercial strategies, or our product candidates or processes, obtain licenses or cease certain activities. Our breach of any license agreements or our failure to obtain a license to intellectual property or proprietary rights required to develop or commercialize our product candidates or future products may have a material adverse impact on us. If third parties prepare and file patent applications in the United States that also claim technology to which we have rights, we may have to participate in interference or derivation proceedings in the USPTO to determine priority of invention. For more information, please see the section entitled “Risk Factors—Risks Related to Intellectual Property.”
In addition to patent protection, we also rely on trademark registration, trade secrets, know how, other proprietary information and continuing technological innovation to develop and maintain our competitive position. As of December 31, 2023, we owned four registered U.S. trademarks (three of which being fractions of international registrations), four international trademark registrations (either granted or still under examination in several countries), 11 registered foreign trademarks as well as two pending foreign trademark applications. We seek to protect and maintain the confidentiality of proprietary information to protect aspects of our business that are not amenable to, or that we do not consider appropriate for, patent protection. Although we take steps to protect our proprietary information and trade secrets, including through contractual means with our employees and consultants, third parties may independently develop substantially equivalent proprietary information and techniques or otherwise gain access to our trade secrets or disclose our technology. Thus, we may not be able to meaningfully protect our trade secrets. It is our policy to require our employees, consultants, outside scientific collaborators, sponsored researchers and other advisors to execute confidentiality agreements upon the commencement of employment or consulting relationships with us. These agreements provide that all confidential information concerning our business or financial affairs developed or made known to the individual during the course of the individual’s relationship with us is to be kept confidential and not disclosed to third parties except in specific circumstances. Our agreements with employees also provide that all inventions conceived by the employee in the course of employment with us or from the employee’s use of our confidential information are our exclusive property. However, such confidentiality agreements and invention assignment agreements can be breached and we may not have adequate remedies for any such breach. In addition, our trade secrets may otherwise become known or be independently discovered by competitors. To the extent that our consultants, contractors or collaborators use intellectual property owned by others in their work for us, disputes may arise as to the rights in related or resulting trade secrets, know-how and inventions. For more information regarding the risks related to our intellectual property, please see the section entitled “Risk Factors—Risks Related to Intellectual Property.”
When available to expand market exclusivity, our strategy is to obtain, or license additional intellectual property or proprietary rights related to current or contemplated development platforms, core elements of technology and/or clinical candidates.
106
Government Regulation
Government authorities in the United States at the federal state and local level, and other countries extensively regulate, among other things, the research, development, nonclinical and clinical testing, manufacture, quality control, approval, labeling, packaging, storage, record-keeping, promotion, advertising, distribution, post-approval monitoring and reporting, marketing, and export and import of products such as those we are developing. Generally, before a new drug or biologic can be marketed, considerable data must be generated, which demonstrate the product’s quality, safety, and efficacy. Such data must then be organized into a format specific for each regulatory authority, submitted for review and approved by the regulatory authority.
U.S. Drug and Biologic Development Process
In the United States, the FDA regulates drugs and biologics under the federal Food, Drug, and Cosmetic Act (“FDCA”), and its implementing regulations. Biologics are additionally subject to regulations under the Public Health Service Act. The process of obtaining regulatory approvals and the subsequent compliance with appropriate federal, state, local and foreign statutes and regulations require the expenditure of substantial time and financial resources. Failure to comply with the applicable U.S. requirements at any time during the product development process, the approval process or after approval may subject an applicant to administrative or judicial sanctions. These sanctions could include the FDA’s refusal to approve pending applications, withdrawal of an approval, a clinical hold, warning letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, refusals of government contracts, restitution, disgorgement, or civil or criminal penalties. Any agency or judicial enforcement action could have a material adverse effect on us.
The process required by the FDA before a biopharmaceutical may be marketed in the United States generally involves the following:
Prior to beginning the first clinical trial with a product candidate in the United States, we must submit an IND to the FDA. An IND is a request for authorization from the FDA to administer an IND product to humans. The central focus of an IND submission is on the general investigational plan and the protocol(s) for clinical studies. The IND also includes results of animal and in vitro studies assessing the toxicology, PK, pharmacology, and pharmacodynamic characteristics of the product; chemistry, manufacturing, and controls information; and any available human data or literature to support the use of the investigational product.
107
An IND must become effective before human clinical trials may begin. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA, within the 30-day time period, raises safety concerns or questions about the proposed clinical trial. In such a case, the IND may be placed on clinical hold and the IND sponsor and the FDA must resolve any outstanding concerns or questions before the clinical trial can begin. Submission of an IND therefore may or may not result in FDA authorization to begin a clinical trial.
Clinical trials involve the administration of the investigational product to human subjects under the supervision of qualified investigators in accordance with GCPs, which include the requirement that all research subjects provide their informed consent for their participation in any clinical study. Clinical trials are conducted under protocols detailing, among other things, the objectives of the study, the parameters to be used in monitoring safety and the effectiveness criteria to be evaluated. A separate submission to the existing IND must be made for each successive clinical trial conducted during product development and for any subsequent protocol amendments. Furthermore, an independent IRB for each site proposing to conduct the clinical trial must review and approve the plan for any clinical trial and its informed consent form before the clinical trial begins at that site and must monitor the study until completed. Regulatory authorities, the IRB or the sponsor may suspend a clinical trial at any time on various grounds, including a finding that the subjects are being exposed to an unacceptable health risk or that the clinical trial is unlikely to meet its stated objectives. Some studies also include oversight by an independent group of qualified experts organized by the clinical study sponsor, known as a data safety monitoring board, which may review data and endpoints at designated check points, make recommendations and/or halt the clinical trial if it determines that there is an unacceptable safety risk for subjects or other grounds, such as no demonstration of efficacy. There are also requirements governing the reporting of ongoing clinical studies and clinical study results to public registries.
Human clinical trials are typically conducted in three sequential phases that may overlap or be combined:
Phase One: Phase 1 clinical trials are designed to test a new therapy in a small group of people for the first time to evaluate safety (e.g., to determine a safe dosage range and to identify adverse effects). It can include healthy participants or patients.
Phase Two: Phase 2 clinical trials are designed to study an investigational therapy in a larger group of people to determine efficacy and to further evaluate its safety. It is conducted in participants with the condition or disease under study and will determine common short-term adverse effects and risks.
Phase Three: Phase 3 clinical trials are designed to study the efficacy of the investigational therapy in large groups of patients by comparing the therapy to other standard or experimental therapies as well as to monitor adverse effects, and to collect information that will allow the therapy being studied to be used safely.
Post-approval clinical trials, sometimes referred to as Phase 4 studies, may be conducted after initial marketing approval. These clinical trials are used to gain additional experience from the treatment of patients in the intended therapeutic indication. In certain instances, the FDA may mandate the performance of Phase 4 clinical trials as a condition of approval of an NDA or BLA.
The FDA or the sponsor may suspend a clinical trial at any time on various grounds, including a finding that the research subjects or patients are being exposed to an unacceptable health risk. Similarly, an IRB can suspend or terminate approval of a clinical trial at its institution if the clinical trial is not being conducted in accordance with the IRB’s requirements or if the drug has been associated with unexpected serious harm to patients. In addition, some clinical trials are overseen by an independent group of qualified experts organized by the sponsor, known as a data safety monitoring board or committee. Depending on its charter, this group may determine whether a clinical trial may move forward at designated check points based on access to certain data from the clinical trial.
During the development of a new biopharmaceutical, sponsors are given opportunities to meet with the FDA at certain points. These points may be prior to submission of an IND, at the end of Phase 2, and before an NDA or BLA is submitted. Meetings at other times may be requested. These meetings can provide an opportunity for the sponsor to share information about the data gathered to date, for the FDA to provide advice, and for the sponsor and the FDA to reach agreement on the next phase of development. Sponsors typically use the meetings at the end of the Phase 2 clinical trial to discuss Phase 2 clinical results and present plans for the pivotal Phase 3 clinical trials that they believe will support approval of the new drug.
108
Concurrent with clinical trials, companies usually complete additional animal studies and must also develop additional information about the chemistry and physical characteristics of the drug and finalize a process for manufacturing the product in commercial quantities in accordance with cGMP regulations. The manufacturing process must be capable of consistently producing quality batches of the product candidate and, among other things, the manufacturer must develop methods for testing the identity, strength, quality, and purity of the final product. In addition, appropriate packaging must be selected and tested, and stability studies must be conducted to demonstrate that the product candidate does not undergo unacceptable deterioration over its shelf life.
While the IND is active and before approval, progress reports summarizing the results of the clinical trials and nonclinical studies performed since the last progress report must be submitted at least annually to the FDA, and written IND safety reports must be submitted to the FDA and investigators for serious and unexpected suspected AEs, findings from other studies suggesting a significant risk to humans exposed to the same or similar drugs, findings from animal or in vitro testing suggesting a significant risk to humans, and any clinically important increased incidence of a serious suspected adverse reaction compared to that listed in the protocol or investigator brochure.
NDA or BLA Review and Approval Process
Assuming successful completion of all required testing in accordance with all applicable regulatory requirements, the results of product development nonclinical and clinical trials, along with descriptions of the manufacturing process, analytical tests conducted on the chemistry of the drug, proposed labeling and other relevant information are submitted to the FDA as part of an NDA or BLA requesting approval to market the product. The submission of an NDA or BLA is subject to the payment of substantial user fees, although a waiver of such fees may be obtained under certain limited circumstances. Additionally, no user fees are assessed on NDAs or BLAs for products designated as orphan drugs, unless the product also includes a non-orphan indication.
The FDA reviews an NDA or BLA to determine, among other things, whether a product is safe and effective for its intended use and whether its manufacturing is cGMP-compliant to assure and preserve the product’s identity, strength, quality, and purity. Under the Prescription Drug User Fee Act (PDUFA) guidelines, the FDA has a goal of ten months from the date of “filing” of a standard NDA or BLA for a new molecular entity to review and act on the submission. This review typically takes 12 months from the date the NDA or BLA is submitted to FDA because the FDA has approximately two months to make a “filing” decision after the application is submitted. The FDA conducts a preliminary review of all NDAs or BLAs within the first 60 days after submission, before accepting them for filing, to determine whether they are sufficiently complete to permit substantive review The FDA may request additional information rather than accept an NDA or BLA for filing. In this event, the application must be resubmitted with the additional information. The resubmitted application is also subject to review before the FDA accepts it for filing.
The FDA may refer an application for a novel drug to an advisory committee. An advisory committee is a panel of independent experts, including clinicians and other scientific experts, that reviews, evaluates and provides a recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions.
Before approving an NDA or BLA, the FDA will typically inspect the facility or facilities where the product is manufactured. The FDA will not approve an application unless it determines that the manufacturing processes and facilities are in compliance with cGMP regulations and adequate to assure consistent production of the product within required specifications. Additionally, before approving an NDA or BLA, the FDA will typically inspect one or more clinical sites to assure compliance with GCPs. If the FDA determines that the application, manufacturing process, or manufacturing facilities are not acceptable, it will outline the deficiencies in the submission and often will request additional testing or information. Notwithstanding the submission of any requested additional information, the FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval.
After the FDA evaluates an NDA or BLA, it will issue an approval letter or a Complete Response Letter. An approval letter authorizes commercial marketing of the product with prescribing information for specific indications. A Complete Response Letter indicates that the review cycle of the application is complete, and the application will not be approved in its present form.
109
A Complete Response Letter usually describes the specific deficiencies in the application identified by the FDA and may require additional clinical data, such as an additional pivotal Phase 3 clinical trial or other significant and time-consuming requirements related to clinical trials, nonclinical studies, or manufacturing. If a Complete Response Letter is issued, the sponsor must resubmit the application, addressing all of the deficiencies identified in the letter, or withdraw the application. Even if such data and information are submitted, the FDA may decide that the application does not satisfy the criteria for approval.
If regulatory approval of a product is granted, such approval will be granted for particular indications and may entail limitations on the indicated uses for which such product may be marketed. For example, the FDA may approve the application with a Risk Evaluation and Mitigation Strategy ("REMS") to ensure the benefits of the product outweigh its risks. A REMS is a safety strategy to manage a known or potential serious risk associated with a medicine and to enable patients to have continued access to such medicines by managing their safe use. It could include medication guides, physician communication plans, or elements to assure safe use, such as restricted distribution methods, patient registries, and other risk minimization tools. The FDA also may offer conditional approval subject to, among other things, changes to proposed labeling or the development of adequate controls and specifications. Once approved, the FDA may withdraw the product approval if compliance with pre- and post-marketing requirements is not maintained or if problems occur after the product reaches the marketplace. The FDA may also require one or more Phase 4 post-market studies and surveillance to further assess and monitor the product’s safety and effectiveness after commercialization, and may limit further marketing of the product based on the results of these post-marketing studies. In addition, new government requirements, including those resulting from new legislation, may be established, or the FDA’s policies may change, which could impact the timeline for regulatory approval or otherwise impact ongoing development programs.
Post-Approval Requirements
Any products manufactured or distributed by us pursuant to FDA approvals are subject to pervasive and continuing regulation by the FDA, including, among other things, requirements relating to record-keeping, reporting of adverse experiences, periodic reporting, product sampling and distribution, and advertising and promotion of the product. After approval, most changes to the approved product, such as adding new indications or other labeling claims, are subject to prior FDA review and approval. There are continuing, annual program fees for any marketed products. Drug manufacturers and their subcontractors are required to register their establishments with the FDA and certain state agencies, and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with cGMP regulations, which impose certain procedural and documentation requirements upon us and our third-party manufacturers. Changes to the manufacturing process are strictly regulated, and, depending on the significance of the change, may require prior FDA approval before being implemented. FDA regulations also require investigation and correction of any deviations from cGMP regulations and impose reporting requirements upon us and any third-party manufacturers that we may decide to use. Accordingly, manufacturers must continue to expend time, money, and effort in the area of production and quality control to maintain compliance with cGMP regulations and other aspects of regulatory compliance.
The FDA may withdraw approval if compliance with regulatory requirements and standards is not maintained or if problems occur after the product reaches the market. Later discovery of previously unknown problems with a product, including AEs of unanticipated severity or frequency, or with manufacturing processes, or failure to comply with regulatory requirements, may result in revisions to the approved labeling to add new safety information; imposition of post-market studies or clinical studies to assess new safety risks; or imposition of distribution restrictions or other restrictions under a REMS program. Other potential consequences include, among other things:
110
The FDA closely regulates the marketing, labeling, advertising, and promotion of biopharmaceutical products. A company can make only those claims relating to safety and efficacy that are approved by the FDA and in accordance with the provisions of the approved label. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses. Failure to comply with these requirements can result in, among other things, adverse publicity, warning letters, corrective advertising, and potential civil and criminal penalties. Physicians may prescribe, in their independent professional medical judgment, legally available products for uses that are not described in the product’s labeling and that differ from those tested by us and approved by the FDA. Physicians may believe that such off-label uses are the best treatment for many patients in varied circumstances. The FDA does not regulate the behavior of physicians in their choice of treatments. The FDA does, however, restrict manufacturer’s communications on the subject of off-label use of their products. The federal government has levied large civil and criminal fines against companies for alleged improper promotion of off-label use and has enjoined companies from engaging in off-label promotion. The FDA and other regulatory agencies have also required that companies enter into consent decrees or permanent injunctions under which specified promotional conduct is changed or curtailed. However, companies may share truthful and not misleading information that is otherwise consistent with a product’s FDA-approved labelling.
Marketing Exclusivity
Market exclusivity provisions authorized under the FDCA can delay the submission and approval of certain marketing applications for products containing the same active ingredient. The FDCA permits patent term restoration of up to five years as compensation for a patent term lost during product development and FDA regulatory review process to the first applicant to obtain approval of an NDA for a new chemical entity in the United States. Patent-term restoration, however, cannot extend the remaining term of a patent beyond a total of 14 years from the product’s approval date. A drug is a new chemical entity if the FDA has not previously approved any other new drug containing the same active moiety, which is the molecule or ion responsible for the action of the drug substance. During the exclusivity period, the FDA may not approve or even accept for review an abbreviated new drug application (“ANDA”) or an NDA submitted under Section 505(b)(2) (“505(b)(2) NDA”), submitted by another company for another drug based on the same active moiety, regardless of whether the drug is intended for the same indication as the original innovative drug or for another indication, where the applicant does not own or have a legal right of reference to all the data required for approval. However, an application may be submitted after four years if it contains a certification of patent invalidity or non-infringement to one of the patents listed with the FDA by the innovator NDA holder.
The FDCA alternatively provides three years of marketing exclusivity for an NDA, or supplement to an existing NDA if new clinical investigations, other than bioavailability studies, that were conducted or sponsored by the applicant are deemed by the FDA to be essential to the approval of the application, for example new indications, dosages, or strengths of an existing drug. This three-year exclusivity covers only the modification for which the drug received approval on the basis of the new clinical investigations and does not prohibit the FDA from approving ANDAs or 505(b)(2) NDAs for drugs containing the active agent for the original indication or condition of use. Five-year and three-year exclusivity will not delay the submission or approval of a full NDA. However, an applicant submitting a full NDA would be required to conduct or obtain a right of reference to any nonclinical studies and adequate and well-controlled clinical trials necessary to demonstrate safety and effectiveness.
Pediatric exclusivity is another type of marketing exclusivity available in the United States. Pediatric exclusivity provides for an additional six months of marketing exclusivity attached to another period of exclusivity if a sponsor conducts clinical trials in children in response to a written request from the FDA. The issuance of a written request does not require the sponsor to undertake the described clinical trials. In addition, orphan drug exclusivity, as described above, may offer a seven-year period of marketing exclusivity, except in certain circumstances.
Section 505(b)(2) NDAs
111
A special type of NDA, commonly referred to as a Section 505(b)(2) NDA, enables the applicant in certain circumstances to rely, in part, on the FDA’s prior findings in approving a similar product or published literature in support of its application. A Section 505(b)(2) NDA may provide an alternate path to FDA approval for a new or improved formulation, a new route of administration, or a new use of a previously approved product. Section 505(b)(2) permits the submission of an NDA where at least some of the information required for approval comes from studies not conducted by, or for, the applicant and for which the applicant has not obtained a right of reference. If the Section 505(b)(2) applicant can establish that reliance on the FDA’s prior findings of safety and/or effectiveness is scientifically appropriate, it may eliminate the need to conduct certain preclinical or clinical studies of the new product. The FDA may also require companies to perform additional studies or measurements to support the change from the approved product. The FDA may then approve the new product candidate for all, or some, of the indications for which the referenced product has been approved, as well as for any new indication sought by the Section 505(b)(2) applicant. If we choose to rely on the 505(b)(2) process to seek approval for OCS-01, there can be no assurance that the FDA will agree with our use of that pathway.
To the extent that the Section 505(b)(2) applicant is relying on the FDA’s prior findings of safety or effectiveness for an already approved product, the applicant is required to certify to the FDA concerning any patents listed for the approved product in the Orange Book to the same extent that an ANDA applicant would. Thus, approval of a Section 505(b)(2) NDA can be stalled until all the listed patents claiming the referenced product have expired, until any non-patent exclusivity, such as exclusivity for obtaining approval of a new chemical entity, listed in the Orange Book for the referenced product has expired, and, in the case of a Paragraph IV certification and subsequent patent infringement suit, until the earlier of 30 months, settlement of the lawsuit or a decision in the infringement case that is favorable to the Section 505(b)(2) applicant.
FDA Approval and Regulation of Companion Diagnostics
A therapeutic product may rely upon an in vitro companion diagnostic for use in selecting the patients that will be more likely to respond to that therapy. If the FDA determines that a companion diagnostic device is essential to the safe and effective use of a novel therapeutic product or indication, the FDA generally will not approve the therapeutic product or new therapeutic product indication if the companion diagnostic device is not approved or cleared for that indication. Approval or clearance of the companion diagnostic device will ensure that the device has been adequately evaluated and has adequate performance characteristics in the intended population. The review of in vitro companion diagnostics in conjunction with the review of our therapeutic product candidate OCS-02 (Licaminlimab) will, therefore, likely involve coordination of review by the FDA’s Center for Biologics Evaluation and Research and the FDA’s Center for Devices and Radiological Health.
Under the FDCA, in vitro diagnostics, including companion diagnostics, are regulated as medical devices. In the United States, the FDCA and its implementing regulations, and other federal and state statutes and regulations govern, among other things, medical device design and development, preclinical and clinical testing, premarket clearance or approval, registration and listing, manufacturing, labeling, storage, advertising and promotion, sales and distribution, export and import, and post-market surveillance. Unless an exemption applies, diagnostic tests require marketing clearance or approval from the FDA prior to commercial distribution. The three primary types of FDA marketing authorization applicable to a medical device include premarket notification, also called 510(k) clearance, premarket approval (“PMA”), and de novo classification requests.
EU/Rest of World Regulation
Conduct of Clinical Trials in the EU
In addition to regulations in the United States, there are a variety of regulations in other jurisdictions governing, among other things, clinical trials, commercial sales and distribution of medicinal products. Even if FDA approval of a particular product is obtained, it must still obtain the requisite approvals from regulatory authorities in foreign countries prior to the commencement of clinical trials or marketing of the product in those countries. Certain countries outside of the United States have a similar process that requires the submission of a clinical trial application much like the IND prior to the commencement of human clinical trials.
In the EU, the Clinical Trials Regulation (EU) No 536/2014 entered into application on January 31, 2022. The Regulation is intended to harmonize and streamline clinical trial authorizations, simplify adverse-event reporting procedures, improve the supervision of clinical trials and increase their transparency.
112
Specifically, the new Regulation, which is directly applicable in all EU Member States, introduces a streamlined application procedure via a single entry point, the “EU portal”, the Clinical Trials Information System (“CTIS”); a single set of documents to be prepared and submitted for the application as well as simplified reporting procedures for clinical trial sponsors. A harmonized procedure for the assessment of applications for clinical trials has been introduced and is divided into two parts. Part I is assessed by the competent authorities of a reference member state selected by the trial sponsor largely of the type of clinical trial, risk-benefit analysis, and compliance with technical requirements. This assessment is then submitted to the competent authorities of all the concerned member states in which the trial is to be conducted for their review. Part II is assessed separately by the competent authorities and ECs in each EU member state concerned. Individual EU Member States shall retain the power to authorize the conduct of clinical trials on their territory. The extent to which on-going clinical trials will be governed by the Clinical Trials Regulation will depend on the duration of the individual clinical trial. If a clinical trial continues for more than three years from January 31, 2022, the Clinical Trials Regulation will at that time begin to apply to the clinical trial.
Pathways to Obtain a Marketing Authorization in the EU
In the European Economic Area (“EEA”), which consists of the 27 Member States of the European Union, as well as Norway, Iceland and Liechtenstein, medicinal products can only be commercialized after a related marketing authorization has been granted. A company may submit a marketing authorization application (“MAA”) on the basis of the centralized, decentralized or mutual recognition procedure. Under the centralized procedure, MAAs are submitted to the EMA for scientific review by the EMA’s Committee for Medicinal Products for Human Use (“CHMP”). The CHMP issues an opinion concerning whether the quality, safety and efficacy of the product has been demonstrated. The opinion is considered by the European Commission which is responsible for granting a centralized marketing authorization in the form of a binding European Commission decision. If the application is approved, the European Commission C grants a single marketing authorization that is valid throughout the EEA. The centralized procedure is mandatory for certain types of products, such as biotechnology medicinal products, orphan medicinal products, advanced-therapy medicines such as gene-therapy, somatic cell-therapy or tissue-engineered medicines and medicinal products containing a new active substance indicated for the treatment of HIV, AIDS, cancer, neurodegenerative disorders, diabetes, auto-immune and other immune dysfunctions and viral diseases. The centralized procedure is optional for products containing a new active substance not yet authorized in the EEA, or for products that constitute a significant therapeutic, scientific or technical innovation or which are in the interest of public health in the European Union.
National marketing authorizations, which are issued by the competent authorities of EEA countries and only cover their respective territory, are available for products not falling within the mandatory scope of the centralized procedure. Where a product has already been authorized for marketing in an EEA country, this national marketing authorization can be recognized in another EEA country through the mutual recognition procedure. The mutual recognition procedure provides for the EEA countries selected by the applicant to mutually recognize a national marketing authorization that has already been granted by the competent authority of another EEA country, referred to as the Reference Member State (“RMS”). The decentralized procedure is used when the product in question has yet to be granted a marketing authorization in any EEA country. Under this procedure the applicant can select the EEA country that will act as the RMS. In both the mutual recognition and decentralized procedures, the RMS reviews the application and submits its assessment of the application to the EEA countries for which marketing authorizations are being sought, referred to as Concerned Member States.
Within 90 days of receiving the application and assessment report, each Concerned Member State must decide whether to recognize the RMS assessment or reject it on the basis of potential serious risk to public health. If the disputed points cannot be resolved, the matter is first referred to the Heads of Medicines Agencies’ Coordination Group for Mutual Recognition and Decentralized Procedures for agreement. If the Heads of Medicines Agencies’ Coordination Group for Mutual Recognition and Decentralized Procedures cannot reach an agreement, a referral is made to the EMA. The CHMP will provide an opinion that will form the basis of a decision to be issued by the European Commission that is binding on all EEA countries. If the application is successful during the decentralized or mutual recognition procedure, national marketing authorizations will be granted by the competent authorities in each of the EEA countries chosen by the applicant.
In principle, a marketing authorization has an initial validity of five years. The marketing authorization may be renewed after five years on the basis of a re-evaluation of the risk-benefit balance by the EMA or by the competent authority of the EEA country in which the original marketing authorization was granted.
113
To support the application, the marketing authorization holder must provide the EMA or the competent authority with a consolidated version of the eCTD (Common Technical Document) providing up to date data concerning the quality, safety and efficacy of the product, including all variations introduced since the marketing authorization was granted, at least nine months before the marketing authorization ceases to be valid. The European Commission or the competent authorities of the EEA countries may decide, on justified grounds relating to pharmacovigilance, to proceed with one further five year renewal period for the marketing authorization. Once subsequently definitively renewed, the marketing authorization shall be valid for an unlimited period. Any authorization which is not followed by the actual placing of the medicinal product on the EU market (in case of centralized procedure) or on the market of the authorizing EEA country within three years after authorization ceases to be valid (the so-called sunset clause).
In the EU, conditional marketing authorizations may be granted in the centralized procedure for a limited number of medicinal products for human use in cases where the related clinical dataset is not yet complete. A conditional marketing authorization may be granted for a medicinal product, if (i) the risk-benefit balance of the product is positive, (ii) it is likely that the applicant will be in a position to provide the required comprehensive data after the authorization, (iii) the medicinal product fulfills unmet medical needs and (iv) the benefit to public health of the immediate availability on the market of the medicinal product outweighs the risk inherent in the fact that additional data are still required. The authorization is valid for one year and must be renewed annually until all related conditions have been fulfilled. Once any pending studies are provided, the conditional marketing authorization can be converted into a traditional marketing authorization. However, if the conditions are not fulfilled within the timeframe set by the EMA, the marketing authorization will cease to be renewed.
A marketing authorization may also be granted “under exceptional circumstances” where the applicant can show that it is unable to provide comprehensive data on the efficacy and safety under normal conditions of use even after the product has been authorized and subject to specific procedures being introduced. These circumstances may arise in particular when the intended indications are very rare and, in the state of scientific knowledge at that time, it is not possible to provide comprehensive information, or when generating data may be contrary to generally accepted ethical principles. Like a conditional marketing authorization, a marketing authorization granted in exceptional circumstances is reserved to medicinal products intended to be authorized for treatment of rare diseases or unmet medical needs for which the applicant does not hold a complete data set that is required for the grant of a standard marketing authorization. However, unlike the conditional marketing authorization, an applicant for authorization in exceptional circumstances is not subsequently required to provide the missing data. Although the marketing authorization “under exceptional circumstances” is granted definitively, the risk-benefit balance of the medicinal product is reviewed annually and the marketing authorization is withdrawn in case the risk-benefit ratio is no longer favorable.
Innovative products that target an unmet medical need and are expected to be of major public health interest may be eligible for a number of expedited development and review programs, such as the Priority Medicines (“PRIME”), scheme, which provides incentives similar to the breakthrough therapy designation in the U.S. PRIME is a voluntary scheme aimed at enhancing the EMA’s support for the development of medicinal products that target unmet medical needs. It permits increased interaction and early dialogue with companies developing promising medicinal products, to optimize their product development plans and speed up their evaluation to help the product reach patients earlier than normal. Product developers that benefit from PRIME designation are potentially eligible for accelerated assessment of their MAA although this is not guaranteed. Benefits accrue to sponsors of product candidates with PRIME designation, including but not limited to, early and proactive regulatory dialogue with the EMA, frequent discussions on clinical trial designs and other development program elements, and potentially accelerated MAA assessment once a dossier has been submitted.
In addition to an MAA, various other requirements apply to the manufacturing and placing on the EU market of medicinal products. Manufacture of medicinal products in the EU requires a manufacturing authorization, and import of medicinal products into the EU requires a manufacturing authorization allowing for import. The manufacturing authorization holder must comply with various requirements set out in the applicable EU laws, regulations and guidance. These requirements include compliance with EU GMP standards when manufacturing medicinal products and APIs, including the manufacture of APIs outside of the EU with the intention to import the APIs into the Union. Similarly, the distribution of medicinal products within the EU is subject to compliance with the applicable EU laws, regulations and guidelines, including the requirement to hold appropriate authorizations for distribution granted by the competent authorities of the EU member states. Marketing authorization holders and/or manufacturing and import authorization (MIA) holders and/or distribution authorization holders may be subject to civil, criminal or administrative sanctions, including suspension of manufacturing authorization, in case of non-compliance with the EU or EU member states’ requirements applicable to the manufacturing of medicinal products.
114
Data and Market Exclusivity
In the EU, innovative medicinal products that are subject to marketing authorization on the basis of a full dossier and do not fall within the scope of the concept of global marketing authorization qualify for eight years of data exclusivity upon marketing authorization and an additional two years of market exclusivity. The concept of global marketing authorization prevents the same marketing authorization holder or members of the same group, or companies that have concluded tacit or explicit agreements concerning the marketing of the same medicinal product, from obtaining separate data and market exclusivity periods for medicinal products that contain the same active substance. Data exclusivity, if granted, prevents regulatory authorities in the European Union from referencing the innovator’s data to assess a generic application or biosimilar application for eight years from the date of authorization of the innovative product, after which a generic or biosimilar marketing authorization application can be submitted, and the innovator’s data may be referenced. However, the generic product or biosimilar products cannot be marketed in the EU for a further two years thereafter. The overall ten-year period may be extended for a further year to a maximum of 11 years if, during the first eight years of those ten years, the marketing authorization holder obtains an authorization for one or more new therapeutic indications which, during the scientific evaluation prior to their authorization, are held to bring a significant clinical benefit in comparison with existing therapies.
Pediatric Development
In the EU, Regulation (EC) No 1901/2006 provides that all MAAs for new medicinal products must include the results of trials conducted in the pediatric population, in compliance with a pediatric investigation plan (“PIP”), agreed with the EMA’s Pediatric Committee (“PDCO”). The PIP sets out the timing and measures proposed to generate data to support a pediatric indication of the medicinal product for which marketing authorization is being sought. The PDCO may grant a deferral of the obligation to implement some or all of the measures provided in the PIP until there are sufficient data to demonstrate the efficacy and safety of the product in adults. Furthermore, the obligation to provide pediatric clinical trial data can be waived by the PDCO when these data are not needed or appropriate because the product is likely to be ineffective or unsafe in children, the disease or condition for which the product is intended occurs only in adult populations, or when the product does not represent a significant therapeutic benefit over existing treatments for pediatric patients. Once the marketing authorization is obtained in all EU Member States and study results are included in the product information, even when negative, the product is eligible for a six-month extension to the Supplementary Protection Certificate or SPC if any is in effect at the time of authorization or, in the case of orphan medicinal products, a two-year extension of orphan market exclusivity. For other countries outside of the European Union, such as certain countries in Eastern Europe, Latin America or Asia, the requirements governing the conduct of clinical trials, product approval, pricing and reimbursement vary from country to country. In all cases, the clinical trials are to be conducted in accordance with GCP and the applicable regulatory requirements and the ethical principles that have their origin in the Declaration of Helsinki.
Orphan Medicinal Products
Regulation (EC) No. 141/2000, as implemented by Regulation (EC) No. 847/2000 provides that a medicinal product can be designated as an orphan medicinal product by the European Commission if its sponsor can establish that: (i) the product is intended for the diagnosis, prevention or treatment of life-threatening or chronically debilitating conditions; (ii) either (a) such conditions affect not more than 5 in 10,000 persons in the EU when the application is made, or (b) the product without the benefits derived from orphan status, would not generate sufficient return in the EU to justify the necessary investment in developing the medicinal product; and (iii) there exists no satisfactory authorized method of diagnosis, prevention, or treatment of the condition that has been authorized in the EU, or even if such method exists, the product will be of significant benefit to those affected by that condition.
Orphan medicinal product designation entitles an applicant to incentives such fee reductions or fee waivers, protocol assistance, and access to the centralized marketing authorization procedure. Upon grant of a marketing authorization, orphan medicinal products are entitled to a ten-year period of market exclusivity for the approved therapeutic indication, which means that the EMA cannot accept another marketing authorization application, or grant a marketing authorization, or accept an application to extend a marketing authorization for a similar product for the same indication for a period of ten years.
115
The period of market exclusivity is extended by two years for orphan medicinal products that have also complied with an agreed PIP. No extension to any supplementary protection certificate can be granted on the basis of pediatric studies for orphan indications. Orphan medicinal product designation does not convey any advantage in, or shorten the duration of, the regulatory review and approval process.
The period of market exclusivity may, however, be reduced to six years if, at the end of the fifth year, it is established that the product no longer meets the criteria on the basis of which it received orphan medicinal product destination, including where it can be demonstrated on the basis of available evidence that the original orphan medicinal product is sufficiently profitable not to justify maintenance of market exclusivity or where the prevalence of the condition has increased above the threshold. Additionally, a marketing authorization may be granted to a similar medicinal product with the same orphan indication during the 10 year period if: (i) if the applicant consents to a second original orphan medicinal product application; (ii) if the manufacturer of the original orphan medicinal product is unable to supply sufficient quantities; or (iii) if the second applicant can establish that its product, although similar, is safer, more effective or otherwise clinically superior to the original orphan medicinal product. A company may voluntarily remove a product from the register of orphan products.
Post-Approval Requirements
Where a marketing authorization is granted in relation to a medicinal product in the EU, the holder of the marketing authorization is required to comply with a range of regulatory requirements applicable to the manufacturing, marketing, promotion and sale of medicinal products.
Similar to the United States, both marketing authorization holders and manufacturers of medicinal products are subject to comprehensive regulatory oversight by the EMA, the European Commission and/or the competent regulatory authorities of the individual EEA countries. The holder of a marketing authorization must establish and maintain a pharmacovigilance system and appoint an individual qualified person for pharmacovigilance who is responsible for oversight of that system. Key obligations include expedited reporting of suspected serious adverse reactions and submission of periodic safety update reports (“PSURs”).
All new marketing authorization applications must include a risk management plan (“RMP”), describing the risk management system that the company will put in place and documenting measures to prevent or minimize the risks associated with the product. The regulatory authorities may also impose specific obligations as a condition of the marketing authorization. Such risk-minimization measures or post-authorization obligations may include additional safety monitoring, more frequent submission of PSURs, or the conduct of additional clinical trials or post-authorization safety studies.
In the EU, the advertising and promotion of medicinal products are subject to both EU and EEA countries laws governing promotion of medicinal products, interactions with physicians and other healthcare professionals, misleading and comparative advertising and unfair commercial practices. Although general requirements for advertising and promotion of medicinal products are established under EU directives, the details are governed by regulations in each member state and can differ from one country to another. For example, applicable laws require that promotional materials and advertising in relation to medicinal products comply with the product’s Summary of Product Characteristics (“SmPC”), as approved by the competent authorities in connection with a marketing authorization. The SmPC is the document that provides information to physicians concerning the safe and effective use of the product. Promotional activity that does not comply with the SmPC is considered off-label and is prohibited in the EU. Direct-to-consumer advertising of prescription medicinal products is also prohibited in the EU.
Regulation of Companion Diagnostics in the EU
In the EU, despite the absence of a legal definition, companion diagnostics are deemed to be in vitro diagnostic medical devices and are governed by Directive 98/79/EC (“IVDD”). The IVDD currently regulates the placing on the market, the CE-marking, the essential requirements, the conformity assessment procedures, the registration obligations for manufacturers and devices as well as the vigilance procedure related to such products. In vitro diagnostic medical devices, including companion diagnostics, must comply with the requirements provided for in the IVDD, and with further requirements implemented at national level (as the case may be).
116
In vitro diagnostic medical devices (including companion diagnostics) are currently required to conform with the essential requirements of the IVDD. To demonstrate compliance with the essential requirements laid down in Annex I to the IVDD, the manufacturer must conduct a conformity assessment procedure.
For general in vitro diagnostic medical devices (i.e. all IVDs other than those covered by Annex II to the IVDD and IVDs for self-testing), the conformity assessment is performed through a self-assessment of the manufacturer without the intervention of a notified body which is an independent organization designated by the competent authorities of an EU member state to assess the conformity of devices before being placed on the market. The manufacturer must prepare an EC Declaration of Conformity confirming conformity of its products with the essential requirements laid down in the IVDD before placing the product on the EU market.
By contrast, the conformity assessment of in vitro diagnostic medical devices for self-testing or that are listed in Annex II (i.e. essentially moderate and high risk reagents and reagent products) to the IVDD requires the intervention of a notified body. Following successful completion of a conformity assessment procedure the notified body will issue a CE Certificate of Conformity. The device manufacturer may, after having completed remaining related procedures and obligations, affix the CE mark to its medical device after having prepared and signed a related EC Declaration of Conformity.
The regulation of companion diagnostics will be subject to further requirements once the in vitro diagnostic medical devices Regulation (No 2017/746), (“IVDR”), becomes applicable on May 26, 2022. The IVDR introduces a new classification system for companion diagnostics which are now specifically defined as diagnostic tests that support the safe and effective use of a specific medicinal product, by identifying patients that are suitable or unsuitable for treatment. Companion diagnostics will have to undergo a conformity assessment by a notified body. If the medicinal product has, or is in the process of, been authorized through the centralized procedure for the authorization of medicinal products, the notified body will, before it can issue a CE Certificate of Conformity, be required to seek a scientific opinion from the EMA on the suitability of the companion diagnostic for use in relation to the medicinal product concerned. For medicinal products that have or are in the process of authorization through any other route provided in EU legislation, the notified body must seek the opinion of the national competent authority of an EU Member State.
Other Healthcare Laws
Pharmaceutical manufacturers are subject to additional healthcare laws, regulation, and enforcement by the U.S. federal government and by authorities in the states and foreign jurisdictions in which they conduct their business. Such laws include, without limitation:
117
These laws and regulations are subject to change, which can increase the resources needed for compliance and delay drug approval or commercialization. Any action brought against us for violations of these laws or regulations, even successfully defended, could cause us to incur significant legal expenses and divert our management’s attention from the operation of our business. Also, we may be subject to private “qui tam” actions brought by individual whistleblowers on behalf of the federal or state governments. Actual or alleged violation of any such laws or regulations may lead to investigations and other claims and proceedings by regulatory authorities and in certain cases, private actors, and violation of any of such laws or any other governmental regulations that apply may result in penalties, including, without limitation, significant administrative, civil and criminal penalties, damages, fines, disgorgement, additional reporting obligations, and oversight if we become subject to a corporate integrity agreement or other agreement to resolve allegations of non-compliance with these laws, the curtailment or restructuring of operations, exclusion from participation in government healthcare programs and imprisonment.
118
The collection and use of personal health data in the EEA is governed by the General Data Protection Regulation ((EU) 2016/679), (“GDPR”), which became effective May 25, 2018. The GDPR applies to any company established in the EEA and to companies established outside the EEA that process personal data in connection with the offering of goods or services to data subjects in the EU or the monitoring of the behavior of data subjects in the EU. The GDPR enhances data protection obligations for controllers and processors of personal data, including stringent requirements relating to the consent of data subjects, expanded disclosures about how personal data is used, requirements to conduct privacy impact assessments for high-risk processing, limitations on retention of personal data and mandatory data breach notification and privacy by design requirements, and creates direct obligations on service providers acting as data processors. The GDPR also imposes strict rules on the transfer of personal data out of the EEA to countries that do not ensure the same level of protection, such as the United States. Failure to comply with the requirements of the GDPR and the related national data protection laws of the EEA countries may result in fines up to 20 million Euros or 4.0% of a company’s global annual revenues for the preceding financial year, whichever is higher. Moreover, the GDPR grants data subjects the right to claim compensation for damages resulting from infringement of the GDPR.
Coverage and Reimbursement
Sales of any pharmaceutical product depend, in part, on the extent to which such product will be covered by third-party payors, such as federal, state, and foreign government healthcare programs, commercial insurance, and managed healthcare organizations, and the level of reimbursement for such product by third-party payors. Significant uncertainty exists as to the coverage and reimbursement status of any newly approved product. Decisions regarding the extent of coverage and amount of reimbursement to be provided are made on a plan-by-plan basis. One third-party payor’s decision to cover a particular product does not ensure that other payors will also provide coverage for the product. As a result, the coverage determination process can require manufacturers to provide scientific details, information on cost-effectiveness, and clinical support for the use of a product to each payor separately. This can be a time-consuming process, with no assurance that coverage and adequate reimbursement will be applied consistently or obtained in the first instance. In addition, third-party payors are increasingly reducing reimbursements for pharmaceutical products and related services. The U.S. government and state legislatures have continued implementing cost-containment programs, including price controls, restrictions on coverage and reimbursement and requirements for substitution of generic products. Third-party payors are increasingly challenging the prices charged, examining the medical necessity and reviewing the cost-effectiveness of pharmaceutical products, in addition to questioning their safety and efficacy. Adoption of price controls and cost-containment measures, and adoption of more restrictive policies in jurisdictions with existing controls and measures, could further limit sales of any product. Decreases in third-party reimbursement for any product or a decision by a third-party payor not to cover a product could reduce physician usage and patient demand for the product.
In international markets, reimbursement and healthcare payment systems vary significantly by country, and many countries have instituted price ceilings on specific products and therapies. For example, the European Union provides options for its member states to restrict the range of medicinal products for which their national health insurance systems provide reimbursement and to control the prices of medicinal products for human use. A member state may approve a specific price for the medicinal product or it may instead adopt a system of direct or indirect controls on the profitability of the company placing the medicinal product on the market. Pharmaceutical products may face competition from lower-priced products in foreign countries that have placed price controls on pharmaceutical products and may also compete with imported foreign products. Furthermore, there is no assurance that a product will be considered medically reasonable and necessary for a specific indication, that it will be considered cost-effective by third-party payors, that an adequate level of reimbursement will be established even if coverage is available, or that the third-party payors’ reimbursement policies will not adversely affect the ability for manufacturers to sell products profitably.
Healthcare Reform
In the United States and certain foreign jurisdictions, there have been, and we expect there will continue to be, a number of legislative and regulatory changes to the healthcare system. In March 2010, the ACA was signed into law, which substantially changed the way healthcare is financed by both governmental and private insurers in the United States.
119
By way of example, the ACA increased the minimum level of Medicaid rebates payable by manufacturers of brand name drugs from 15.1% to 23.1%; it required collection of rebates for drugs paid by Medicaid managed care organizations; imposed a non-deductible annual fee on pharmaceutical manufacturers or importers who sell certain “branded prescription drugs” to specified federal government programs; it implemented a new methodology under which rebates owed by manufacturers under the Medicaid Drug Rebate Program are calculated for drugs that are inhaled, infused, instilled, implanted, or injected; it expanded the eligibility criteria for Medicaid programs; it created a new Patient-Centered Outcomes Research Institute to oversee, identify priorities in, and conduct comparative clinical effectiveness research, along with funding for such research; and it established a Center for Medicare Innovation at the Centers for Medicare & Medicaid Services ("CMS"), to test innovative payment and service delivery models to lower Medicare and Medicaid spending, potentially including prescription drug spending.
Since its enactment, there have been executive, judicial and Congressional challenges to certain aspects of the ACA, and we expect there will be additional challenges and amendments to the ACA in the future. Since January 2017, President Trump signed several Executive Orders and other directives designed to delay the implementation of certain provisions of the ACA or otherwise circumvent some of the requirements for health insurance mandated by the ACA. Concurrently, Congress has considered legislation that would repeal or repeal and replace all or part of the ACA. While Congress has not passed comprehensive repeal legislation, several bills affecting the implementation of certain taxes under the ACA have passed. For example, on June 17, 2021, the U.S. Supreme Court dismissed a challenge on procedural grounds that argued the ACA is unconstitutional in its entirety because the “individual mandate” was repealed by Congress. Thus, the ACA will remain in effect in its current form. Further, prior to the U.S. Supreme Court ruling, on January 28, 2021, President Biden issued an executive order that initiated a special enrollment period for purposes of obtaining health insurance coverage through the ACA marketplace. The executive order also instructed certain governmental agencies to review and reconsider their existing policies and rules that limit access to healthcare, including among others, reexamining Medicaid demonstration projects and waiver programs that include work requirements, and policies that create unnecessary barriers to obtaining access to health insurance coverage through Medicaid or the ACA. In addition, on August 16, 2022, President Biden signed the Inflation Reduction Act of 2022 (“IRA”), into law, which among other things, extends enhanced subsidies for individuals purchasing health insurance coverage in ACA marketplaces through plan year 2025 and eliminates the “donut hole” under the Medicare Part D program beginning in 2025, by significantly lowering the beneficiary maximum out-of-pocket cost through a newly established manufacturer discount program. It is possible that the ACA will be subject to judicial or Congressional challenges in the future. It is unclear how any such challenges or additional health reform measures of the Biden administration will impact the ACA.
Other legislative changes have been proposed and adopted since the ACA was enacted. These changes include aggregate reductions to Medicare payments to providers of up to 2.0% per fiscal year, effective April 1, 2013, which, due to subsequent legislative amendments, will stay in effect until 2031 with the exception of a temporary suspension implemented under various COVID-19 relief legislation from May 1, 2020 through March 31, 2021, unless additional congressional action is taken. Under current legislation, the actual reduction in Medicare payments will vary from 1.0% in 2022 to up to 4.0% in the final fiscal year of this sequester. On March 11, 2021, President Biden signed the American Rescue Plan Act of 2021 into law, which eliminates the statutory Medicaid drug rebate cap, currently set at 100% of a drug’s average manufacturer price, for single source and innovator multiple source drugs, beginning January 1, 2024. In January 2013, President Obama signed into law the American Taxpayer Relief Act of 2012, which, among other things, further reduced Medicare payments to several types of providers and increased the statute of limitations period for the government to recover overpayments to providers from three to five years.
Moreover, there has recently been heightened governmental scrutiny over the manner in which manufacturers set prices for their marketed products, which has resulted in several Congressional inquiries and proposed and enacted legislation designed, among other things, to bring more transparency to product pricing, to review the relationship between pricing and manufacturer patient programs, and to reform government program reimbursement methodologies for pharmaceutical products. For example, in July 2021, the Biden administration released an executive order, “Promoting Competition in the American Economy,” with multiple provisions aimed at prescription drugs. In response to Biden’s executive order, on September 9, 2021, the U.S. Department of Health and Human Services (“HHS”), released a Comprehensive Plan for Addressing High Drug Prices that outlines principles for drug pricing reform and sets out a variety of potential legislative policies that Congress could pursue to advance these principles. In addition, the IRA, among other things: (i) allows HHS to negotiate the price of certain single-source drugs and biologics covered under Medicare, and subjects drug manufacturers to civil monetary penalties and a potential excise tax by offering a price that is not equal to or less than the negotiated “maximum fair price” under the law, and (ii) imposes rebates under Medicare Part B and Medicare Part D to penalize price increases that outpace inflation. The IRA permits HHS to implement many of these provisions through guidance, as opposed to regulation, for the initial years.
120
These provisions will take effect progressively starting in 2023, although they may be subject to legal challenges.
In addition, individual states in the United States have also become increasingly active in implementing regulations designed to control pharmaceutical product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures and, in some cases, mechanisms to encourage importation from other countries and bulk purchasing. Furthermore, there has been increased interest by third-party payors and governmental authorities in reference to pricing systems and publication of discounts and list prices.
The Health Technology Assessment (“HTA”) process, which is currently governed by the national laws of the individual EU Member States, is the procedure according to which the assessment of the public health impact, therapeutic impact and the economic and societal impact of use of a given medicinal product in the national healthcare systems of the individual country is conducted. A new regulation adopted in December 2021 the HTA Regulation, is intended to boost cooperation among EU Member States in assessing health technologies, including new medicinal products, and to provide the basis for cooperation at EU level for joint clinical assessments in these areas. The Regulation will apply from 2025 followed by a phased roll-out ending in 2028.
C. Organizational Structure
Upon consummation of the Business Combination on March 2, 2023, Merger Sub 1 merged with and into EBAC, with EBAC as the surviving company of the First Merger, and EBAC merged with and into Merger Sub 2, with Merger Sub 2 as the surviving company of the Second Merger and wholly owned subsidiary of the Company. During the third quarter of 2023, the Company gave effect in its financial statements to the impending dissolution of Merger Sub 2, which will be completed in April 2024. On July 6, 2023, Legacy Oculis merged with and into Oculis Operations GmbH (“Oculis Operations”), and the separate corporate existence of Legacy Oculis ceased. Oculis Operations is the surviving company and remains a wholly-owned subsidiary of Oculis Holding AG. The following diagram illustrates our corporate structure as of December 31, 2023.
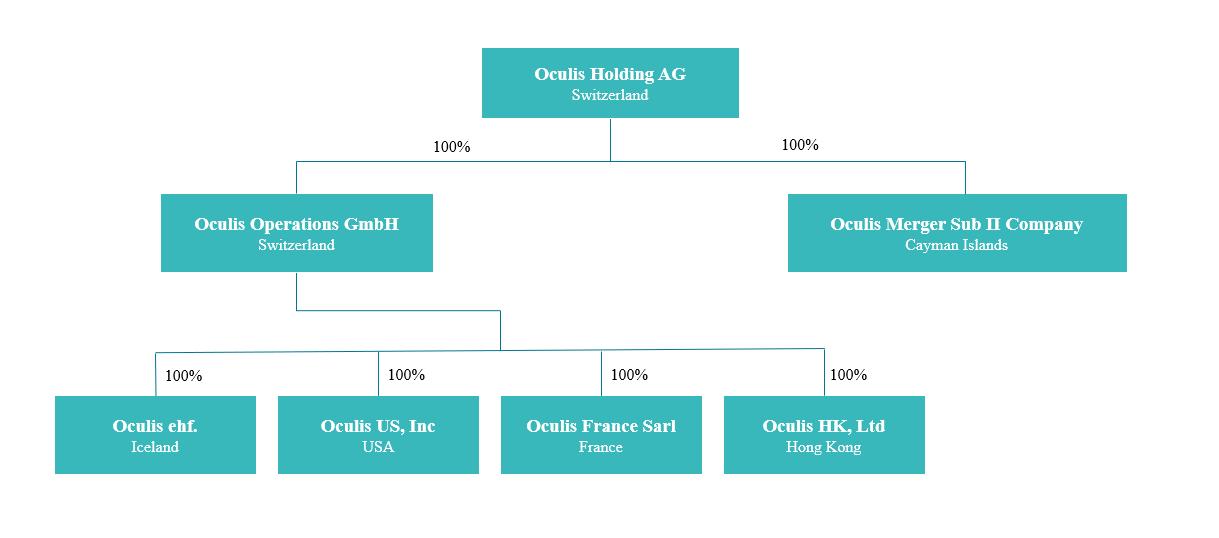
D. Property, Plants and Equipment
We currently lease approximately 8,800 square feet of facilities for our operations, including 4,300 square feet of laboratory and office space in Iceland, with main activities of research, business and clinical development, 2,740 square feet of office space in Switzerland, with main activities of business and clinical development and 1,725 square feet of office space in the United States, with main activities being general and administrative in nature.
121
We believe these facilities will be adequate for the foreseeable future and that suitable additional or substitute space will be available as and when needed. We believe that these facilities are adequate to meet our current needs, but we are constantly evaluating our needs for expanding and or adding to our existing facilities.
Item 4A. Unresolved Staff Comments
Not applicable.
Item 5. Operating and Financial Review and Prospects
You should read the following discussion and analysis of our audited financial condition and results of operations together with our consolidated financial statements appearing elsewhere in this Annual Report on Form 20-F. This Annual Report on Form 20-F contains forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended, or the Securities Act, and Section 21E of the Exchange Act, including, without limitation, statements regarding our expectations, beliefs, intentions or future strategies that are signified by the words “expect,” “anticipate,” “intend,” “believe,” or similar language. All forward-looking statements included in this Annual Report on Form 20-F are based on information available to us on the date hereof, and we assume no obligation to update any such forward-looking statements. In evaluating our business, you should carefully consider the information provided under “Item 3.D. Risk Factors.” Actual results could differ materially from those projected in the forward-looking statements. The terms “Company,” “Oculis,” “we,” “our” or “us” as used herein refer to Oculis and its consolidated subsidiaries unless otherwise stated or indicated by context.
Certain information called for by this Item 5, including a discussion of the year ended December 31, 2021, as well a comparison of the year ended December 31, 2022 against the year ended December 31, 2021, has been reported previously in our Annual Report on Form 20-F for the year ended December 31, 2022 filed on March 28, 2023 under Item 5. “Operating and Financial Review and Prospects”.
All amounts discussed are in Swiss francs, unless otherwise indicated.
Company Overview
We are a late clinical-stage biopharmaceutical company, based in Switzerland, with substantial expertise in therapeutics used to treat ocular diseases, engaged in the development of innovative drug candidates which embrace the potential to address large unmet medical needs for many eye-related conditions. Our focus is on advancing therapeutic candidates intended to treat significant and prevalent ophthalmic diseases which result in vision loss, blindness or reduced quality of life. Our mission is to improve the health and quality of life of patients around the world by developing medicines that save sight and improve eye care for patients. To realize this mission, we intend to become a global leader in ocular therapeutics.
Our pipeline currently includes three clinical-stage therapeutic candidates: OCS-01, OCS-02 (Licaminlimab) and OCS-05. Our lead product candidate, OCS-01, is currently being evaluated in two ongoing Phase 3 clinical programs: as a treatment for DME and as a once-daily topical for the treatment of inflammation and pain following cataract surgery. Our second product candidate is OCS-02, currently being evaluated in a Phase 2b clinical trial to assess its potential as a topical anti-TNFα treatment for DED and the potential use of a particular genotype to predict treatment response, which could be considered as a biomarker in a precision medicine approach. A second clinical trial for OCS-02, designed to evaluate its use as a potential treatment for non-infectious anterior uveitis, is expected to follow thereafter. Our third product candidate is OCS-05, a potential disease modifying neuroprotective agent against neurological damage with potential application in multiple indications, including glaucoma, dry AMD and DR. We are conducting a Phase 2 PoC trial evaluating OCS-05 as a potential treatment for AON for which there is currently no approved therapeutic treatment.
Numerous diseases and disorders, many of which represent significant medical needs, are associated with the human eye. The National Eye Institute, a part of the U.S. National Institutes of Health, estimates that in the United States, blindness or significant visual impairment impacts approximately seven million people, including those with vision loss resulting from retinal diseases such as DME, macular degeneration, DR, and retinal vein occlusion (“RVO”); disorders caused by swelling and inflammation such as DED, corneal keratitis and uveitis; and glaucoma, among other disease states.
122
It is estimated that the global spending for ophthalmology therapeutics will reach $33 billion in 2027, according to an industry source.
To date, we have primarily financed our operations through the proceeds from share issuances and grants. We have no products approved for commercialization and have never generated any revenues from product sales. Pharmaceutical and biopharmaceutical product development is a highly speculative undertaking and involves a substantial degree of risk. It may be several years, if ever, before we have a product candidate approved for commercialization, and we begin to generate revenue and royalties from product sales. We have also incurred significant operating losses. We incurred net losses of CHF 88.8 million for the year ended December 31, 2023, and an accumulated losses balance of CHF 199.8 million as of December 31, 2023.
Factors Affecting Our Performance
Business Environment
The biopharmaceutical industry is extremely competitive. We are subject to risks and uncertainties common to any clinical-stage biopharmaceutical company. These risks include, but are not limited to, the introduction of new products, therapies, standards of care or technological innovations, our ability to obtain, maintain, protect and enforce our licensed technology, data and other intellectual property and proprietary rights and compliance with extensive government regulation and oversight. Please see the section entitled “Risk Factors” for more information. We are also dependent upon the services of key personnel, including our Chief Executive Officer, executive team and other highly skilled employees. Demand for experienced personnel in the pharmaceutical and biotechnology industries is high and competition for talent is intense.
We face potential competition from many different sources, including pharmaceutical and biotechnology companies, academic institutions and governmental agencies as well as public and private research institutions. Many of our competitors are working to develop or have commercialized products similar to those we are developing and have considerable experience in undertaking clinical trials and in obtaining regulatory approval to market pharmaceutical products. Our competitors may also have significantly greater financial resources, established presence in the markets in which we hope to compete, expertise in research and development, manufacturing, preclinical and clinical testing, obtaining regulatory approvals and reimbursement and marketing approved products. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. These third parties also compete with us in recruiting and retaining qualified scientific and management personnel, establishing clinical trial sites and registering patients for clinical trials, entering into agreements with CMOs for the manufacture of our product candidates, as well as in acquiring technologies complementary to, or necessary for, our programs.
Business combination with European Biotech Acquisition Corp ("EBAC")
On March 2, 2023, we consummated a business combination with EBAC (the “Business Combination”) pursuant to the Business Combination Agreement ("BCA") between Legacy Oculis and EBAC dated as of October 17, 2022. We received gross proceeds of CHF 97.6 million or $103.7 million comprising CHF 12.0 million or $12.8 million of cash held in EBAC’s trust account and CHF 85.6 million or $90.9 million from private placement (“PIPE”) investments and conversion of notes issued under Convertible Loan Agreements (“CLA”) into our Ordinary Shares. In connection with the Business Combination, Oculis was listed on the Nasdaq Global Market with the ticker symbol "OCS" for its ordinary shares and “OCSAW” for its public warrants.
Under the terms of the BCA, EBAC formed four new legal entities (i) Oculis, (ii) Oculis Merger Sub I Company ("Merger Sub 1"), (iii) Merger Sub 2, and (iv) Oculis Operations. After two consecutive mergers between Merger Sub 1 and EBAC, and EBAC and Merger Sub 2, EBAC and Merger Sub 1 ceased to exist and Merger Sub 2 was the surviving company. During the third quarter of 2023, we gave effect in our financial statements to the impending dissolution of Merger Sub 2, which will be completed in April 2024. As a result, the cumulative translation adjustments related to Merger Sub 2 previously reported as equity and recognized in other comprehensive income, were reclassified from equity to the Consolidated Statement of Loss for the year ended December 31, 2023. The resulting foreign exchange impact of such reclassification amounted to CHF 5.0 million for the year ended December 31, 2023.
As a result of the BCA and as of the acquisition closing date on March 2, 2023:
123
PIPE and CLA Financing
In connection with the BCA, EBAC entered into subscription agreements with the PIPE investors for an aggregate of 7,118,891 shares of EBAC Class A ordinary shares at CHF 9.42 or $10.00 per share for aggregate gross proceeds of CHF 67.1 million or $71.2 million.
In connection with the BCA, Legacy Oculis and the investor parties thereto entered into CLAs pursuant to which the investor lenders granted Legacy Oculis a right to receive an interest free convertible loan with certain conversion rights with substantially the same terms as the PIPE investors. Following the mergers, we assumed the CLAs and the lenders exercised their conversion rights in exchange for 1,967,000 ordinary shares at CHF 9.42 or $10.00 per share for aggregate gross proceeds of CHF 18.5 million or $19.7 million.
Together, the PIPE and CLA financing resulted in aggregate gross cash proceeds of CHF 85.6 million or $90.9 million to Oculis in exchange for 9,085,891 ordinary shares.
Merger and listing expense
The Business Combination is accounted for as a capital re-organization. As EBAC does not meet the definition of a business in accordance with IFRS 3 Business Combinations, the BCA is accounted for within the scope of IFRS 2 Share-based Payment.
The Business Combination is treated as the equivalent of the Company issuing shares for the net assets of EBAC as of the acquisition closing date, accompanied by a recapitalization. The net assets of EBAC are stated at historical cost, with no goodwill or other intangible assets recorded. Any excess of the fair value of the Company’s shares issued considering a fair value of CHF 10.54 or $11.19 per share (price of EBAC ordinary share at the closing date) over the fair value of EBAC’s identifiable net assets acquired represents compensation for the service of a stock exchange listing for its shares.
124
This expense was incurred in the first quarter of 2023 and amounted to CHF 34.9 million, which was expensed to the statement of loss as operating expenses, “Merger and listing expense”. The expense is non-recurring in nature and represents a share-based payment made in exchange for a listing service and does not lead to any cash outflows.
|
|
Per share value |
|
|
Shares |
|
|
March 2, 2023 |
|
|||
Fair value of equity consideration issued by the Company |
|
|
|
|
|
|
|
|
|
|||
EBAC public shareholders |
|
|
10.54 |
|
|
|
12,754,784 |
|
|
|
134,435 |
|
EBAC sponsor class B |
|
|
10.54 |
|
|
|
3,188,696 |
|
|
|
33,609 |
|
EBAC sponsor class A |
|
|
10.54 |
|
|
|
455,096 |
|
|
|
4,797 |
|
Redemptions of EBAC public shareholders |
|
|
10.54 |
|
|
|
(11,431,606 |
) |
|
|
(120,489 |
) |
Sponsors shares forfeiture |
|
|
10.54 |
|
|
|
(1,596,490 |
) |
|
|
(16,827 |
) |
Total consideration transferred |
|
|
|
|
|
3,370,480 |
|
|
|
35,525 |
|
|
Less net assets of EBAC |
|
|
|
|
|
|
|
|
(662 |
) |
||
Merger and listing expense |
|
|
|
|
|
|
|
|
34,863 |
|
||
|
|
March 2, 2023 |
|
|
|
|
(in CHF thousands) |
|
|
Net assets of EBAC |
|
|
|
|
Cash and cash equivalents |
|
|
11,547 |
|
Public & private warrants |
|
|
(2,136 |
) |
Deferred underwriting fee |
|
|
(3,108 |
) |
Accrued transaction costs |
|
|
(4,400 |
) |
Others |
|
|
(1,241 |
) |
Net assets of EBAC |
|
|
662 |
|
Capitalization
The following summarizes the actual ordinary shares issued and outstanding and the ownership interests of Oculis immediately after the Business Combination:
|
|
Shares |
|
|
% |
|
||
Issuance of ordinary shares to Legacy Oculis shareholders in connection with BCA (1) |
|
|
20,277,002 |
|
|
|
61.9 |
% |
Issuance of ordinary shares in connection with closing of the PIPE financing |
|
|
7,118,891 |
|
|
|
21.7 |
% |
Issuance of ordinary shares under CLA |
|
|
1,967,000 |
|
|
|
6.0 |
% |
Ordinary shares owned by sponsors |
|
|
2,047,302 |
|
|
|
6.3 |
% |
Ordinary shares owned by EBAC public shareholders |
|
|
1,323,178 |
|
|
|
4.1 |
% |
Total (2) |
|
|
32,733,373 |
|
|
|
100.0 |
% |
(1) As a result of the BCA, Oculis issued 20,277,002 ordinary shares to Legacy Oculis shareholders in exchange for:
(2) In addition to the shares already issued, the following contingently issuable shares were granted: 3,793,995 earnout shares, 369,737 earnout options, 1,762,949 shares of outstanding conversion options, 4,251,595 public warrants and 151,699 private warrants. The earnout shares are contingently forfeitable if the price targets are not achieved during the earnout period.
125
|
|
Legacy Oculis shares |
|
|
Exchange ratios |
|
|
Oculis ordinary shares issued |
|
|||
Ordinary shares |
|
|
3,406,771 |
|
|
|
|
|
|
|
||
Treasury shares cancelled |
|
|
(100,000 |
) |
|
|
|
|
|
|
||
Ordinary shares after cancellation of treasury shares |
|
|
3,306,771 |
|
|
1.1432 |
|
|
|
3,780,399 |
|
|
Preferred shares: |
|
|
|
|
|
|
|
|
|
|||
Series A |
|
|
1,623,793 |
|
|
|
1.1432 |
|
|
|
1,856,370 |
|
Series B1 |
|
|
2,486,188 |
|
|
|
1.4154 |
|
|
|
3,518,922 |
|
Series B2 T1 |
|
|
1,675,474 |
|
|
|
1.3900 |
|
|
|
2,328,872 |
|
Series B2 T2 |
|
|
426,378 |
|
|
|
1.3310 |
|
|
|
567,508 |
|
Series B2 T3 |
|
|
603,472 |
|
|
|
1.3142 |
|
|
|
793,082 |
|
Series C T1 |
|
|
5,337,777 |
|
|
|
1.2658 |
|
|
|
6,756,580 |
|
Series C T2 |
|
|
362,036 |
|
|
|
1.2205 |
|
|
|
441,854 |
|
Series C T3 |
|
|
197,745 |
|
|
|
1.1804 |
|
|
|
233,415 |
|
Total preferred shares |
|
|
12,712,863 |
|
|
|
1.2976 |
|
|
|
16,496,603 |
|
|
|
|
|
|
|
|
|
|
|
|||
Total |
|
|
16,019,634 |
|
|
|
|
|
|
20,277,002 |
|
|
Earnout consideration
As a result of the BCA, Legacy Oculis preferred, ordinary and option holders (collectively “equity holders”) received consideration in the form of 3,793,995 earnout shares and 369,737 earnout options with an exercise price of CHF 0.01.
The earnout consideration is subject to forfeiture in the event of a failure to achieve the price targets during the earnout period defined as follows: (i) 1,500,000, (ii) 1,500,000 and (iii) 1,000,000 earned based on the achievement of post-acquisition closing share price targets of $15.00, $20.00 and $25.00, respectively, in each case, for any 20 trading days within any consecutive 30 trading day period commencing after the acquisition closing date and ending on or prior to March 2, 2028 (the “Earnout period”). A given share price target described above will also be deemed to be achieved if there is a change of control, as defined in the BCA, during the earnout period.
Public offering of ordinary shares
On May 31, 2023, we entered into an underwriting agreement with BofA Securities Inc. and SVB Securities, LLC, as representatives of several underwriters, and on June 5, 2023, closed the issuance and sale in a public offering of 3,500,000 ordinary shares at a public offering price of CHF 10.45 or $11.50 per share, for total gross proceeds of CHF 36.6 million or $40.3 million before deducting underwriting discounts, commissions and offering expenses.
In addition, we granted the underwriters an option to purchase additional ordinary shares which was partially exercised on June 13, 2023 for 154,234 ordinary shares and resulted in gross proceeds of CHF 1.6 million or $1.7 million before deducting underwriting discounts, commissions and offering expenses. After giving issuance to these additional shares, we sold a total of 3,654,234 ordinary shares in the offering for aggregate gross proceeds of CHF 38.2 million or $42.0 million, before deducting underwriting discounts, commissions and offering expenses. The unexercised portion of the underwriters’ options to purchase additional shares expired on June 30, 2023.
We intend to use the net proceeds from this offering, together with its existing resources, to advance our development programs, in particular Diabetic Macular Edema and for other ophthalmic indications, and for working capital and general corporate purposes.
126
Licensing agreement with Accure Therapeutics
Since our inception, we have entered into strategic collaboration agreements with a range of partners covering a number of our product candidates in our strategy to diversify our product portfolio and become a global ophthalmology company.
On January 29, 2022, Legacy Oculis entered into a License Agreement with Accure for the exclusive global licensing of its OCS-05 technology. Under this agreement, Oculis licensed a small molecule in development as a potential disease modifying neuroprotective agent designed to address neurological damage to the optic nerve.
As of December 31, 2023, we have paid the full contractual non-refundable upfront fee and reimbursed costs of CHF 3.5 million increasing our licenses intangible asset up to CHF 12.2 million as of December 31, 2023. We have not reached any milestones or royalties thresholds according to the agreement. If all predefined milestones will be reached, we will be obligated to pay up to CHF 94.3 million ($112.1 million ). In case of a commercialization, sublicense revenues will be subject to further royalty payments.
Components of Results of Operations
Revenue
We have not generated any revenue from the sale of products since our inception and do not expect to generate any revenue from the sale of products in the near future. If our development efforts for our product candidates are successful and result in regulatory approval or if we enter into collaboration or licensing agreements with third parties, we may generate revenue in the future from a combination of product sales or payments from such collaboration or licensing agreements. However, there can be no assurance as to when we will generate such revenue, if at all.
Grant Income
Grant income reflects reimbursement of research and development expenses and income from certain research projects managed by Icelandic governmental institutions. We maintain a subsidiary in Iceland that provides research and development for our product candidates. Certain expenses qualify for incentives from the Icelandic government in the form of tax credits or cash reimbursements. We do not anticipate generating significant grant income in the future.
Operating Expenses
Research and development expenses
Research and development expenses consist primarily of costs incurred in connection with the research and development of our product candidates and programs. We expense research and development costs and the cost of acquired intangible assets used in research and development activities as incurred. Research and development expenditures are capitalized only if they meet the recognition criteria of IAS 38 (“Intangible Assets”) and in such cases are amortized over the useful economic life on a straight-line basis. These expenses include:
127
We historically did not track our research and development costs by project category, primarily because we use our employee and infrastructure resources across multiple research and development programs that we are advancing in parallel, and therefore do not allocate salaries, stock-based compensation, employee benefit expenses or other indirect costs related to our research and development to specific product candidates.
Product candidates in later stages of clinical development generally have higher development costs than those in earlier stages of clinical development, primarily due to the increased size and duration of later-stage clinical trials. We expect that our research and development expenses will increase substantially in connection with our planned clinical development activities in the near term and in the future. At this time, we cannot accurately estimate or know the nature, timing and costs of the efforts that will be necessary to complete the clinical development of any current or future product candidates.
Prior to 2023, we did not track our total research and development costs by project category, primarily because our clinical development costs may vary significantly based on factors such as:
The successful development and commercialization of product candidates is highly uncertain. This is due to the numerous risks and uncertainties associated with product development and commercialization, including the following:
128
A change in the outcome of any of these variables with respect to the development of our product candidates could significantly change the costs and timing associated with the development of that product candidate. We may never succeed in obtaining regulatory approval for any of our product candidates or programs.
General and administrative expenses
General and administrative expenses consist primarily of salaries and related costs for personnel in executive management, finance, corporate and business development, and administrative functions. General and administrative expenses also include legal fees relating to patent and corporate matters; professional fees for accounting, auditing, tax, and administrative consulting services; insurance costs; marketing and communications expenses; and other operating costs.
Beginning in 2022 and continuing in 2023, we incurred increased accounting, audit, legal and other professional services costs associated with the Business Combination and the associated transition from a private company to a public company. We anticipate that our general and administrative expenses will continue to increase in the future in relation with costs associated with being a public company such as increased costs for fees to members of the board of directors, increased employee-related expenses, increased director and officer insurance premiums, audit and legal fees, investor relations fees and expenses for compliance with public company reporting requirements under the Exchange Act and Nasdaq rules.
129
Finance income and Finance expense
Finance income consists primarily of interest income earned from our short-term financial assets.
Prior to March 2023, Finance expense consisted primarily of accrued interest costs associated with the preferred dividend payment of 6.0% to the holders of Legacy Oculis preferred Series B and C shares. The preferred Series B and C shares are classified as liabilities under IAS 32 and the associated accrued dividend is recognized as interest expense. All preferred shares were converted into ordinary shares upon consummation of the Business Combination on March 2, 2023.
Fair value adjustment on warrant liabilities
Fair value adjustment on warrant liabilities reflects the changes in fair value of our warrant instruments. The fair value is dependent on the change in the underlying market price of the warrants and the number of outstanding warrants at the reporting date. The market price of the warrants is in general directly correlated with the market price of our ordinary shares. Assuming the number of outstanding warrants remains constant, we would expect a fair value loss due to an increase in the market price of the warrants, and a fair value gain due to a decrease in the market price of the warrants.
Foreign currency exchange gain (loss)
Foreign currency exchange gains and losses consisted of currency exchange differences that arise from transaction denominated in currencies other than Swiss Francs.
Income tax expense
We are subject to corporate Swiss federal, cantonal and communal taxation, respectively, in Switzerland, Canton of Zug, and Commune of Zug, as well as in the Canton of Vaud, and Commune of Ecublens, near Lausanne. We are also subject to taxation in other jurisdictions in which we operate, in particular the United States, France, China and Iceland where our wholly owned subsidiaries are incorporated.
We are entitled, under Swiss laws, to carry forward any losses incurred for a period of seven years and can offset our losses carried forward against future taxes owed. As of December 31, 2023, we had tax loss carry-forwards totaling CHF 170.4 million. There is no certainty that we will make sufficient profits to be able to utilize tax loss carry-forwards in full and no deferred tax assets have been recognized in the financial statements.
130
A. Operating Results
The following table summarizes our results of operations for the periods presented:
|
For the years ended December 31, |
|
|
|
|
|
|
|
|||||||
In CHF thousands |
2023 |
|
|
2022 |
|
|
Change |
|
|
% Change |
|
||||
Grant income |
|
883 |
|
|
|
912 |
|
|
|
(29 |
) |
|
|
(3.2 |
%) |
Operating income |
|
883 |
|
|
|
912 |
|
|
|
(29 |
) |
|
|
(3.2 |
%) |
Research and development expenses |
|
(29,247 |
) |
|
|
(22,224 |
) |
|
|
(7,023 |
) |
|
|
(31.6 |
%) |
General and administrative expenses |
|
(17,487 |
) |
|
|
(11,064 |
) |
|
|
(6,423 |
) |
|
|
(58.1 |
%) |
Merger and listing expense |
|
(34,863 |
) |
|
|
- |
|
|
|
(34,863 |
) |
|
|
(100.0 |
%) |
Operating expenses |
|
(81,597 |
) |
|
|
(33,288 |
) |
|
|
(48,309 |
) |
|
|
(145.1 |
%) |
Operating loss |
|
(80,714 |
) |
|
|
(32,376 |
) |
|
|
(48,338 |
) |
|
|
(149.3 |
%) |
Finance income |
|
1,429 |
|
|
|
126 |
|
|
|
1,303 |
|
|
|
1034.1 |
% |
Finance expense |
|
(1,315 |
) |
|
|
(6,442 |
) |
|
|
5,127 |
|
|
|
(79.6 |
%) |
Fair value adjustment on warrant liabilities |
|
(3,431 |
) |
|
|
- |
|
|
|
(3,431 |
) |
|
|
(100.0 |
%) |
Foreign currency exchange (loss) gain |
|
(4,664 |
) |
|
|
49 |
|
|
|
(4,713 |
) |
|
|
9618.4 |
% |
Finance result |
|
(7,981 |
) |
|
|
(6,267 |
) |
|
|
(1,714 |
) |
|
|
(27.3 |
%) |
Loss before tax for the period |
|
(88,695 |
) |
|
|
(38,643 |
) |
|
|
(50,052 |
) |
|
|
(129.5 |
%) |
Income tax expense |
|
(107 |
) |
|
|
(55 |
) |
|
|
(52 |
) |
|
|
(94.5 |
%) |
Loss for the period |
|
(88,802 |
) |
|
|
(38,698 |
) |
|
|
(50,104 |
) |
|
|
(129.5 |
%) |
Comparison of the Years Ended December 31, 2023 and 2022
Grant Income
Grant income for the years ended December 31, 2023 and 2022 was CHF 0.9 million for both years. The grant income is dependent upon the Icelandic government making such reimbursement available for research and development activities. While certain of our research and development expenses have historically qualified for reimbursement and we anticipate incurring a similar level of costs in the future, there is no assurance that the Icelandic government will continue with the tax reimbursement program.
Research and Development Expenses
|
For the years ended December 31, |
|
|
|
|
|
|
|
|||||||
In CHF thousands |
2023 |
|
|
2022 |
|
|
Change |
|
|
% Change |
|
||||
Personnel expenses |
|
6,509 |
|
|
|
4,608 |
|
|
|
1,901 |
|
|
|
41.3 |
% |
Payroll |
|
4,796 |
|
|
|
4,313 |
|
|
|
483 |
|
|
|
11.2 |
% |
Share-based compensation |
|
1,713 |
|
|
|
295 |
|
|
|
1,418 |
|
|
|
480.7 |
% |
Operating expenses |
|
22,738 |
|
|
|
17,616 |
|
|
|
5,122 |
|
|
|
29.1 |
% |
External service providers |
|
22,256 |
|
|
|
17,205 |
|
|
|
5,051 |
|
|
|
29.4 |
% |
Other operating expenses |
|
258 |
|
|
|
184 |
|
|
|
74 |
|
|
|
40.2 |
% |
Depreciation of property and equipment |
|
106 |
|
|
|
111 |
|
|
|
(5 |
) |
|
|
(4.5 |
%) |
Depreciation of right-of-use assets |
|
118 |
|
|
|
116 |
|
|
|
2 |
|
|
|
1.7 |
% |
Total research and development expense |
|
29,247 |
|
|
|
22,224 |
|
|
|
7,023 |
|
|
|
31.6 |
% |
Research and development expenses were CHF 29.2 million for the year ended December 31, 2023 compared to CHF 22.2 million for the year ended December 31, 2022. The net increase of CHF 7.0 million, or 31.6%, was primarily due to an increase in external CRO expenses as a result of the completion and subsequent startup activities and of multiple OCS-01 clinical trials and the commencement of the OCS-02 DED Phase 2b clinical trial, as well as an increase in research and development personnel costs. The increase in development expenses reflects the OCS-01 DIAMOND Phase 3 clinical trials, OCS-01 OPTIMIZE Phase 3 clinical trials, OCS-01 LEOPARD investigator-initiated trial ("IIT"), OCS-02 (Licaminlimab) drug development and OCS-05 ACUITY proof-of-concept ("PoC") clinical trial for AON. We anticipate that our research and development expenses will continue to increase as we advance our planned clinical development programs.
131
|
For the year ended |
|
|
|
December 31, 2023 |
|
|
OCS-01 |
|
15,135 |
|
OCS-02 |
|
8,793 |
|
OCS-05 |
|
3,354 |
|
Other development projects |
|
1,965 |
|
Total |
|
29,247 |
|
Prior to 2023, we did not track our total research and development costs by project category. For the year ended December 31, 2023, research and development expenses were primarily driven by our OCS-01 DME DIAMOND Phase 3 Stages 1 and 2 clinical trials, OCS-01 OPTIMIZE Phase 3 clinical trial for inflammation and pain following cataract surgery, the OCS-02 Phase 2b DED clinical and drug development, and OCS-05 ACUITY PoC clinical trial for AON.
General and Administrative Expenses (excluding Merger and Listing Expense)
|
For the years ended December 31, |
|
|
|
|
|
|
|
|||||||
In CHF thousands |
2023 |
|
|
2022 |
|
|
Change |
|
|
% Change |
|
||||
Personnel expenses |
|
7,029 |
|
|
|
4,449 |
|
|
|
2,580 |
|
|
|
58.0 |
% |
Payroll |
|
5,134 |
|
|
|
3,939 |
|
|
|
1,195 |
|
|
|
30.3 |
% |
Share-based compensation |
|
1,895 |
|
|
|
510 |
|
|
|
1,385 |
|
|
|
271.6 |
% |
Operating expenses |
|
10,458 |
|
|
|
6,615 |
|
|
|
3,843 |
|
|
|
58.1 |
% |
External service providers |
|
7,695 |
|
|
|
2,294 |
|
|
|
5,401 |
|
|
|
235.4 |
% |
Other operating expenses |
|
2,700 |
|
|
|
4,249 |
|
|
|
(1,549 |
) |
|
|
(36.5 |
%) |
Depreciation of property and equipment |
|
19 |
|
|
|
20 |
|
|
|
(1 |
) |
|
|
(5.0 |
%) |
Depreciation of right-of-use assets |
|
44 |
|
|
|
52 |
|
|
|
(8 |
) |
|
|
(15.4 |
%) |
Total |
|
17,487 |
|
|
|
11,064 |
|
|
|
6,423 |
|
|
|
58.1 |
% |
General and administrative expenses (excluding merger and listing expense) were CHF 17.5 million for the year ended December 31, 2023, compared to CHF 11.1 million for the year ended December 31, 2022. The increase of CHF 6.4 million, or 58.1%, was primarily due to non-capitalized financing transaction costs, public liability insurances, as well as personnel-related expenses. These expenses were largely attributable to the Business Combination, Nasdaq listing and operating as a public company.
Merger and listing Expense
|
For the years ended December 31, |
|
|
|
|
|
|
||||||
In CHF thousands |
2023 |
|
|
2022 |
|
|
Change |
|
|
% Change |
|||
Merger and listing expense |
|
(34,863 |
) |
|
|
- |
|
|
|
(34,863 |
) |
|
- |
We incurred a non-recurring merger and listing expense of CHF 34.9 million in the year ended December 31, 2023 in connection with the Business Combination. The Business Combination was accounted for as a share-based payment transaction involving the transfer of shares in Oculis for the net assets of EBAC. This expense represented one-time non-cash compensation for a stock exchange listing service equal to the excess of the fair value of the shares transferred compared to the fair value of the net assets.
Finance Income and Finance Expense
|
For the years ended December 31, |
|
|
|
|
|
|
|
|||||||
|
2023 |
|
|
2022 |
|
|
Change |
|
|
% Change |
|
||||
Finance income |
|
1,429 |
|
|
|
126 |
|
|
|
1,303 |
|
|
|
1034.1 |
% |
Finance expense |
|
(1,315 |
) |
|
|
(6,442 |
) |
|
|
5,127 |
|
|
|
(79.6 |
%) |
132
Finance income was CHF 1.4 million for the year ended December 31, 2023 compared to CHF 0.1 million for the year ended December 31, 2022. The increase of CHF 1.3 million was due to interest on short-term financial assets recorded during the year ended December 31, 2023. Finance expense was CHF 1.3 million for the year ended December 31, 2023, compared to CHF 6.4 million for the year ended December 31, 2022. The decrease of CHF 5.1 million was primarily due to two months of interest expense accrued during 2023 compared to twelve months of interest expense accrued for the comparative period in 2022, related to Legacy Oculis’ preferred Series B and C shares, which were converted into ordinary shares on March 2, 2023 under the BCA.
Fair Value Adjustment on Warrant Liabilities
|
For the years ended December 31, |
|
|
|
|
|
|
||||||
|
2023 |
|
|
2022 |
|
|
Change |
|
|
% Change |
|||
Fair value adjustment on warrant liabilities |
|
(3,431 |
) |
|
|
- |
|
|
|
(3,431 |
) |
|
- |
We incurred a fair value loss of CHF 3.4 million for the year ended December 31, 2023 primarily due to an increase in the market price of the warrants assumed by Oculis from March 2, 2023 to December 31, 2023.
Foreign Currency Exchange (Loss) Gain
|
For the years ended December 31, |
|
|
|
|
|
|
|
|||||||
|
2023 |
|
|
2022 |
|
|
Change |
|
|
% Change |
|
||||
Foreign currency exchange (loss) gain |
|
(4,664 |
) |
|
|
49 |
|
|
|
(4,713 |
) |
|
|
(9618.4 |
%) |
Foreign currency exchange loss was CHF 4.7 million for the year ended December 31, 2023, compared to a gain of CHF 49 thousand for the year ended December 31, 2022. For the year ended December 31, 2023, the unfavorable currency exchange was mainly due to the fluctuation of U.S. dollar against the Swiss Franc producing a foreign exchange loss over the year related to our U.S. dollar denominated cash balances, as well as a loss on the revaluation of the U.S dollar denominated Series C long-term financial debt (former preferred shares) from January to March 2023. The Series C long-term financial debt was fully converted to ordinary shares pursuant to the Business Combination in March 2023. For the year ended December 31, 2022, favorable currency exchange was mainly due to revaluation of U.S. dollar producing foreign exchange gains over the year related to our cash balances, offset by the full year 2022 revaluation of the Series C long-term debt.
Comparison of Years Ended December 31, 2022 and 2021
For a discussion of the financial results and condition for the fiscal year ended December 31, 2021, please refer to “Item 5. Operating and Financial Review and Prospects” for a comparison of years ended December 31, 2022 and 2021” of our Annual Report on Form 20-F for the year ended December 31, 2022 filed on March 28, 2023.
B. Liquidity and Capital Resources
Overview
Since our inception, we have incurred significant operating losses. We have not yet commercialized any products and we do not expect to generate revenue from sales of products for several years, if at all. As of December 31, 2023, we have funded our operations primarily with CHF 103.4 million of proceeds from the sale of our preferred stock, CHF 97.6 million of gross proceeds from the Business Combination, PIPE Financing and conversion of CLA and CHF 38.2 million of gross proceeds from the sale of our ordinary shares in the Public Offering. As of December 31, 2023 and 2022, we had cash, cash equivalents and short-term investments of CHF 91.7 million and CHF 19.8 million, respectively. We had accumulated losses of CHF 199.8 million and CHF 111.0 million as of December 31, 2023 and 2022, respectively.
133
We expect to incur additional operating losses in the near future and our operating expenses will increase as we continue to expand our organization through in-licensing, strategic collaboration and acquisition, and invest in the development of our product candidates through additional research and development activities and clinical trials. See “Risk Factors—Risks related to development and regulatory approval of our investigational therapies.” We will continue to incur additional costs associated with operating as a public company, including expenses related to legal, accounting, financial reporting and regulatory matters, maintaining compliance with exchange listing and SEC requirements, director and officer insurance premiums and investor relations.
Based on our current operating plan, we believe that our existing cash, cash equivalents and short-term financial assets will be sufficient to fund our operations and capital expenses through at least the next twelve months from the date that this Annual Report is filed with the SEC. We have based our estimate on assumptions that may prove to be wrong, and we may use our available capital resources sooner than we currently expect. We may require additional capital resources due to underestimation of the nature, timing and costs of the efforts that will be necessary to complete the development of our product candidates. We may also need to raise additional funds more quickly if we choose to expand our development activities, our portfolio or if we consider acquisitions or other strategic transactions, including licensing transactions. For more information regarding these risk and factors that could influence our future capital requirements and the timing thereof, please see the section entitled “Risk Factors.”
Future Funding Requirements
Product development is very expensive and involves a high degree of risk. Only a small number of research and development programs result in the commercialization of a product. We will not generate revenue from product sales unless and until we successfully complete clinical development and are able to obtain regulatory approval for and successfully commercialize the product candidates we are currently developing or that we may develop. We currently do not have any product candidates approved for commercial sale.
Our product candidates, currently under development or that we may develop, will require significant additional research and development efforts, including extensive clinical testing and regulatory approval prior to commercialization. These efforts require significant amounts of additional capital, adequate personnel infrastructure and extensive compliance and reporting capabilities. There can be no assurance that our research and development activities will be successfully completed, that adequate protection for our licensed or developed technology will be obtained and maintained, that products developed will obtain necessary regulatory approval or that any approved products will be commercially viable.
If we obtain regulatory approval for one or more of our product candidates, we expect to incur significant expenses related to developing our commercialization capabilities to support product sales, marketing and distribution activities, either alone or in collaboration with others. Further, as discussed further below, we expect to incur additional costs associated with operating as a public company. As a result, we will need substantial additional funding to support our continuing operations and pursue our growth strategy.
Until such time, if ever, we can generate substantial product revenue, we may finance our operations through a combination of private or public equity offerings, debt financings, collaborations, strategic alliances, marketing, distribution or licensing arrangements or through other sources of financing. Adequate capital may not be available to us when needed or on acceptable terms. To the extent that we raise additional capital through the sale of private or public equity or convertible debt securities, your ownership interest will be diluted, and the terms of these securities may include liquidation or other preferences that adversely affect your rights as a holder of Ordinary Shares. Debt financing and preferred equity financing, if available, may involve agreements that include covenants limiting or restricting our ability to take specific actions, such as incurring additional debt, making acquisitions or capital expenditures.
Debt financing would also result in fixed payment obligations. If we raise additional funds through collaborations, strategic alliances or marketing, distribution or licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies, future revenue streams, research programs or product candidates, or grant licenses on terms that may not be favorable to us. If we are unable to raise additional funds through equity or debt financings or other arrangements when needed, we may be required to delay, limit, reduce or terminate our research, product development or future commercialization efforts, grant rights to develop and market product candidates that we would otherwise prefer to develop and market ourselves, obtain funds through arrangement with collaborators on terms unfavorable to us or pursue merger or acquisition strategies, all of which could adversely affect the holdings or the rights of our shareholders.
134
Please see the section entitled “Risk Factors—Risks related to our business, financial condition, capital requirements, or financial operations” for additional risks associated with our substantial capital requirements.
We expect our expenses to increase substantially in connection with our ongoing activities, particularly as we advance the preclinical activities, manufacturing and clinical development of our product candidates. In addition, we have incurred additional costs associated with the Business Combination and will continue to incur additional costs associated with operating as a public company, including significant legal, accounting, investor relations and other expenses that we did not incur or incremental to operating a private company. Our expenses will also increase as we:
See the section of this Annual Report titled “Risk Factors” for additional risks associated with our substantial capital requirements.
Material Cash Requirements for Known Contractual Obligations and Commitments
We have certain payment obligations under various license and collaboration agreements. Under these agreements, we are required to pay non-refundable, upfront license fees, predefined development and commercial milestone payments and royalties on net sales of licensed products.
License Agreement with Novartis for OCS-02 (Licaminlimab)
Pursuant to a license agreement, dated as of December 19, 2018, as amended, by and between us and Novartis (the “Novartis Agreement”), we obtained an exclusive, royalty-bearing, sublicensable (subject to certain conditions), assignable (subject to certain conditions), worldwide license under certain patents, know-how and manufacturing platform technology to develop, manufacture and commercialize pharmaceutical, therapeutic or diagnostic products containing a specified single chain antibody fragment formulation as an active ingredient in the licensed field as defined in the Novartis Agreement.
135
The license granted to us by Novartis includes sublicenses of rights granted to Novartis by certain third parties, and our license to such rights is expressly subject to the applicable terms and conditions of the agreements between Novartis and such third parties.
We originally entered into the Novartis Agreement with Alcon Research, Ltd. (“Alcon”), which subsequently assigned its rights and obligations under the Novartis Agreement to Novartis in connection with its spin-off from Novartis.
We are deemed the owner of any inventions that are (a) created solely by or on behalf of us pursuant to the Novartis Agreement and (b) severable from the licensed products, and grant Novartis a first right to negotiate a worldwide, royalty-bearing license under any patents directed at such inventions for purposes outside of the licensed field. We also grant Novartis a worldwide, non-exclusive, perpetual, irrevocable, royalty-free, fully paid-up license back under any patents owned by us that (i) cover inventions arising from the Novartis Agreement, the practice of which would infringe the patents licensed to us by Novartis, or (ii) otherwise incorporate Novartis’ proprietary information, in each case, for certain uses outside of the licensed field.
We paid in full the contractual non-refundable upfront payment to Alcon of CHF 4.7 million ($4.7 million at the exchange rate at the time of payment) in cash and issued 401,709 ordinary shares (recast subsequent to the BCA) for the residual between the fair value and the upfront payment. This was accounted for as a share-based payment transaction under IFRS 2. As of December 31, 2023, we were obligated to pay Novartis additional up to CHF 81.6 million ($97.0 million at the December 31, 2023 exchange rate) in the aggregate upon the achievement of certain development, regulatory, sales and other milestones and tiered royalties ranging from a mid-single digit to a low mid-teen percentage on net sales. In consideration for the exclusive sublicense from Novartis under certain third-party intellectual property rights, we are obligated to pay a low-single digit royalty on our net sales of the licensed product, however, such payments will be deducted from royalties payable to Novartis. Our royalty payment obligations are subject to certain reductions and expire with respect to any licensed product on a country-by-country basis upon the later of (a) the expiration of the last to expire valid claim of any licensed patent covering any such licensed product in such country; (b) the expiration of the period of data exclusivity in any country worldwide; or (c) twelve (12) years after first commercial sale of such licensed product in such country (“Royalty Term”).
Under the Novartis Agreement, we are obligated to use diligent efforts to develop, manufacture or have manufactured, and commercialize the licensed products in the licensed field worldwide. The Novartis Agreement will expire upon the last-to-expire Royalty Term. We may terminate the Novartis Agreement without cause at any time upon advance written notice to Novartis. Upon written notice to Novartis, we may terminate the Novartis Agreement for cause due to the following events: (a) an insolvency event occurs; (b) Novartis materially breaches its obligations under the Novartis Agreement and fails to cure such breach within a specified period of time; or (c) upon advance written notice for material scientific, technical or medical reasons or in case of a material adverse change that renders further continuation of the Novartis Agreement by us commercially unreasonable or otherwise not viable. Upon written notice to us, Novartis may terminate the Novartis Agreement for cause due to the following events: (i) we fail to pay any undisputed amount due under the Novartis Agreement and we fail to remedy such failure within a specified period of time; (ii) an insolvency event occurs; or (iii) we materially breach our obligations under the Novartis Agreement and fail to cure such breach within a specified period of time; or (iv) following negative clinical trial results, we terminate development of the licensed product and do not pursue any further indications in the licensed field.
License Agreement with Accure for OCS-05
Pursuant to a license agreement, dated as of January 29, 2022, by and between us and Accure (the “Accure Agreement”), we obtained an exclusive, worldwide, sublicensable (subject to certain conditions) and transferable (subject to certain conditions) license under certain patents, know-how and inventory of Accure for any and all uses and purposes, including to perform research, development, manufacturing and commercialization activities in any manner and for any purpose. The licensed patents are co-owned by Accure with third parties who have reserved the right to use the licensed patents for education and research purposes pursuant to an inter-institutional agreement.
As of December 31, 2023, we had paid the full contractual non-refundable upfront fee of CHF 3.0 million and reimbursed costs in the amount of approximately CHF 0.5 million.
136
As of December 31, 2023, we were obligated to pay Accure (a) up to CHF 94.3 million ($112.1 million at the December 31, 2023 exchange rate) in the aggregate upon the achievement of certain development, regulatory and sales milestones; (b) tiered royalties ranging from a mid-single digit to a low mid-teen percentage on net sales of licensed products; and (c) high teens on sublicensing revenues received any time after 36 months from the agreement effective date, and a higher percentage on sublicensing revenues received prior to such date, in all cases subject to reduction for any amount that were previously paid or are concurrently or later paid by us to Accure pursuant to our milestone payment obligations and such amounts received from a sublicensee will be deduced from amounts owned to Accure. Our royalty payment obligations are subject to certain reductions and expire on a licensed product-by-licensed product and country-by-country basis upon the later of (i) the expiration of the last valid claim of any licensed patent covering such licensed product in such country; (ii) the expiration of such licensed product’s Orphan Drug status, if any, in such country; or (iii) ten (10) years following the date of first commercial sale of such licensed product in such country (the “Payment Period”).
Under the Accure Agreement, we are obligated to use commercially reasonable efforts to develop and seek regulatory approval for a licensed product in major countries of the territory as defined in the Accure Agreement.
The Accure Agreement will expire on a licensed product-by-licensed product and country-by-country basis upon the expiration of the applicable Payment Period with respect to such licensed product in such country. We may terminate the Accure Agreement in whole or in part at any time upon advance written notice (a) for documented reasonable scientific, regulatory, commercial reasons related to the licensed product without incurring any penalty or liability to Accure and (b) for no reason. Each party may terminate the Accure Agreement with immediate effect upon written notice to the other party (i) in the event such other party commits a material breach of its obligations under the Accure Agreement and fails to cure that breach within a specified period of time or (ii) with certain exceptions, upon such other party’s bankruptcy. Accure may terminate the Accure Agreement with immediate effect upon written notice to us if we file any action to invalidate any of the licensed patents or fail to maintain the licensed patents in major countries of the territory as defined in the Accure Agreement, or, subject to certain exceptions, if we fail to meet certain development obligations and are unable to agree upon modifications to the development plan with Accure.
Other Commitments
The majority of our near term cash needs relate to our clinical and Chemistry, Manufacturing and Controls ("CMC") projects. We have conducted research and development programs through collaborative programs that include, among others, arrangements with universities, CROs and clinical research sites. As of December 31, 2023, commitments for external research projects totaled CHF 50.5 million, with CHF 23.6 million due within one year and CHF 26.9 million due between one and five years.
In addition, we enter into agreements in the normal course of business with CROs for clinical trials and with vendors for preclinical studies, manufacturing services, and other services and products for operating purposes, which are generally cancelable upon written notice.
We have entered into three real estate lease agreements for lab and office facilities. At December 31, 2023, these lease agreements have aggregate lease liabilities of CHF 0.2 million due within one year and CHF 0.4 million due in more than one year.
Refer to Notes 10 and 19 to our audited consolidated financial statements included elsewhere in this Annual Report for further details on our obligations and timing of expected future payments.
Cash Flows
The following table summarizes our sources and uses of cash and cash equivalents for each of the periods presented:
|
For the years ended December 31, |
|
|
|
|
|
|
|
|||||||
|
2023 |
|
|
2022 |
|
|
Change |
|
|
% Change |
|
||||
Net cash outflow from operating activities |
|
(53,845 |
) |
|
|
(25,074 |
) |
|
|
(28,771 |
) |
|
|
114.7 |
% |
Net cash outflow from investing activities |
|
(54,211 |
) |
|
|
(3,548 |
) |
|
|
(50,663 |
) |
|
|
1427.9 |
% |
Net cash inflow from financing activities |
|
129,672 |
|
|
|
1,714 |
|
|
|
127,958 |
|
|
|
7465.5 |
% |
Increase/(Decrease) in cash and cash equivalents |
|
21,616 |
|
|
|
(26,909 |
) |
|
|
48,525 |
|
|
|
180.3 |
% |
137
Operating Activities
For the year ended December 31, 2023, operating activities used CHF 53.8 million of cash, primarily consisting of a loss before tax of CHF 88.7 million and a decrease in net working capital of CHF 13.0 million, partially offset by non-cash adjustments of CHF 46.8 million. Changes in net working capital were driven by an CHF 11.5 million increase in accrued expenses and other payables and a CHF 5.6 million increase in other current assets, partially offset by a CHF 3.7 million increase in trade payables. Non-cash charges primarily consisted of a non-recurring CHF 34.9 million of listing service expenses in connection with the Business Combination, CHF 3.5 million of foreign exchange transactions impacting net financial result, CHF 3.6 million of share-based compensation expense and CHF 3.4 million related to the fair value adjustment on warrant liabilities.
For the year ended December 31, 2022, operating activities used CHF 25.1 million of cash, primarily consisting of a net loss before tax of CHF 38.6 million partially offset by a decrease in net working capital of CHF 6.0 million and non-cash adjustments of CHF 7.6 million. Changes in net working capital were driven by a CHF 7.9 million increase in accrued expenses and other payables partly offset by CHF 1.8 million increase in other current assets. Non-cash charges primarily consisted of CHF 6.3 million from interest expense accrued on preferred Series B and C shares, CHF 0.8 million of share-based compensation expense and CHF 0.6 million from non-realized foreign exchange differences.
Investing Activities
For the years ended December 31, 2023 and 2022, investing activities used CHF 54.2 million and CHF 3.5 million, respectively. For the year ended December 31, 2023, CHF 54.2 million was used for the purchase of short term financial assets. For the year ended December 31, 2022, CHF 3.5 million was related to the license agreement with Accure for the exclusive global licensing of OCS-05 technology that was capitalized as an intangible asset.
Financing Activities
For the year ended December 31, 2023, net cash provided by financing activities was CHF 129.7 million, which relates primarily to the closing of the Business Combination, the PIPE Financing, the conversion of the CLAs and the Public Offering.
For the year ended December 31, 2022, net cash provided by financing activities was CHF 1.7 million, which primarily consisted of proceeds from the issuance of preferred Series C shares, classified as liabilities, net of transaction costs.
For a discussion of our cash flows for the year ended December 31,2021, see “Item 5. Operating and Financial Review and Prospects—B. Liquidity and Capital Resources” in our Annual Report on Form 20-F filed with the SEC on March 28, 2023.
C. Research and Development, Patents and Licenses, etc.
Full details of our research and development activities and expenditures are given in the “Item 4.B. Information on the Company—Business Overview” and “Item 5 Operating and Financial Review and Prospects” sections of this Annual Report.
D. Trend Information
Other than as described elsewhere in this Annual Report, we are not aware of any trends, uncertainties, demands, commitments or events that are reasonably likely to have a material adverse effect on our revenue, income from continuing operations, profitability, liquidity or capital resources, or that would cause our reported financial information not necessarily to be indicative of future operating results or financial condition.
E. Critical Accounting Estimates
We prepared our consolidated financial statements in accordance with IFRS Accounting Standards as issued by the IASB. Refer to Note 3 and 4 to our audited consolidated financial statements included elsewhere in this Annual Report for further details on the most significant accounting policies applied in the preparation of our consolidated financial statements and our critical accounting estimates and judgments.
138
139
Item 6. Directors, Senior Management and Employees
The following table sets forth the current executive officers and directors of Oculis as of the filing date. Unless otherwise noted, the business address of each of our directors and executive officers is EPFL Innovation Park, Bat D 3e Route J.A. Colladon, CH 1015, Lausanne, Switzerland.
Name |
|
Age |
|
Title |
Executive Officers |
|
|
|
|
Riad Sherif, M.D. |
|
56 |
|
Chief Executive Officer and Director |
Sylvia Cheung |
|
49 |
|
Chief Financial Officer |
Páll Ragnar Jóhannesson |
|
43 |
|
Chief Business Officer |
Non-Employee Directors |
|
|
|
|
Anthony Rosenberg |
|
71 |
|
Chairman of the Board of Directors |
Christina Ackermann |
|
59 |
|
Director |
Lionel Carnot |
|
56 |
|
Director |
Pravin Dugel, M.D. |
|
60 |
|
Director |
Martijn Kleijwegt |
|
69 |
|
Director |
Geraldine O'Keeffe |
|
58 |
|
Director |
Executive Officers
Riad Sherif, M.D., 56, has served as the Chief Executive Officer and Director of Oculis since December 2017. Previously, from June 2016 to September 2017, Dr. Sherif served as Entrepreneur in Residence at the Novartis Venture Fund. Before that, Dr. Sherif served as the President of Europe, Middle East and Africa of Alcon, Inc. from March 2014 to May 2016. Prior to that, from January 2002 to April 2014, Dr. Sherif held roles of increasing responsibility at Novartis AG, including as the Global Sales Head in the Transplant and Infectious Disease unit, as the Head for Latin America in transplant and infectious disease, as the President of the Novartis Vaccines and Diagnostics Division for Latin America and where he co-founded Synergium a leading biotech company, and most recently as the President of Novartis Pharmaceuticals, Canada. Prior to Novartis, Dr. Sherif worked for several pharmaceutical companies, holding positions of increasing seniority, mainly in marketing and general management with international scope. Dr. Sherif currently serves as a member of the board of directors of Revenio Group corporation. Dr. Sherif previously served as the Vice Chairman for the Innovative Medicine Canada Association, as the Chairman of In-Vivo Montreal, and as the Chairman of the Board Ophthalmic Surgery and Vision Care of Eucomed. Dr. Sherif is a Medical Doctor by training, and holds an MBA from IMD Business School and a Specialized Master’s Degree in Medical Management from ESCP.
Sylvia Cheung, 49, has served as the Chief Financial Officer of Oculis since September 2020. Prior to that, from October 2005 to August 2020, Ms. Cheung held executive positions at Anika Therapeutics, Inc., a publicly-traded joint preservation company. Most recently, from April 2013 to August 2020, Ms. Cheung served as the Chief Financial Officer of Anika Therapeutics, Inc. Previously, from 2000 to 2005, Ms. Cheung held a series of financial management positions of increasing responsibility at Transkaryotic Therapies, Inc., which was acquired by Shire Pharmaceuticals in 2005. Before that, from 1995 to 2000, Ms. Cheung served as a Senior Associate at PricewaterhouseCoopers. Ms. Cheung holds a Bachelor of Business Administration degree in Accounting from the University of Massachusetts in Amherst, an MBA from Boston University, and is a Certified Public Accountant in Massachusetts.
Páll Ragnar Jóhannesson, 43, has served as the Chief Business Officer of Oculis since January 2023. Previously, from September 2020 to January 2023, Mr. Jóhannesson served as the Chief Strategy Officer of Oculis. Previously, from January 2018 to September 2020, Mr. Jóhannesson served as the Chief Financial Officer of Oculis. Additionally, Mr. Jóhannesson has served as the Managing Director of Oculis Iceland ehf. since May 2015. Prior to that, from February 2012 to April 2015, Mr. Jóhannesson held a series of corporate finance positions of increasing responsibility at Straumur Investment Bank, and most recently, from September 2013 to April 2015, Mr. Jóhannesson served as the Managing Director, Corporate Finance. Before that, from January 2009 to November 2011, Mr. Jóhannesson served as a Director, Corporate Finance at Íslandsbanki and its predecessor Glitnir Bank. Mr. Jóhannesson holds a B.Sc. in Industrial Engineering from the University of Iceland, an M.Phil in Management Science from the University of Cambridge, and was certified as securities broker in Iceland.
140
Non-Employee Directors
Anthony Rosenberg, 71, has served as Chairman of the board of directors of Oculis since April 2018. Since April 2015, Mr. Rosenberg has served as the Chief Executive Officer of TR Advisory Services GmbH. Additionally, from April 2015 to April 2020, Mr. Rosenberg served as a Managing Director of MPM Capital. Prior to that, from 2005 to 2012, Mr. Rosenberg held a series of business development and licensing positions of increasing seniority at Novartis, and most recently, from 2012 to 2015, Mr. Rosenberg served as the Corporate Head of M&A and Licensing at Novartis International AG. Mr. Rosenberg currently serves on the boards of directors of Argenx BV and Cullinan Oncology. Mr. Rosenberg previously served on the boards of directors of SiO2 Materials Science, TriNetX and Radius Health, Inc. Mr. Rosenberg holds a B.Sc. (Hons) from the University of Leicester and a M.Sc. in Physiology from the University of London.
Christina Ackermann, 59, has served as a member of the board of directors of Oculis since March 2023. From January 2022 to May 2023, Ms. Ackermann served as Executive Vice President, General Counsel & President of Ophthalmic Pharmaceuticals at Bausch + Lomb. Ms. Ackermann joined Bausch Health as Executive Vice President, General Counsel, in August 2016. Prior to Bausch Health, Ms. Ackermann was part of the Novartis group of companies for 14 years, most recently serving as Senior Vice President, General Counsel for Alcon, where she was responsible for the legal, intellectual property and compliance functions, in addition to Trade Compliance Function, Enterprise Risk Management and Diversity & Inclusion. Previously, she served as Global Head, Legal and General Counsel at Sandoz, the generics division of Novartis, from 2007 to 2012. She joined Novartis Pharma in 2002 as Head, Legal Technical Operations and Ophthalmics, and assumed the role of Head Legal General Medicine in July 2005. Before Novartis, Ms. Ackermann served in Associate General Counsel roles with Bristol Myers Squibb and DuPont Pharmaceuticals, as well as in private practice, where she focused on securities, and mergers & acquisitions. From August 2021 to March 2023, Ms. Ackermann has served on the board of directors of Graybug Vision. Since September 2023, Ms. Ackermann also serves on the board of directors of Verona Pharma, where she is a member of the Audit Committee. Ms. Ackermann holds a LL.B in law from Queen’s University in Ontario, Canada and a post graduate degree in EU competition law from King’s College in London, England.
Lionel Carnot, 56, has served as a member of the board of directors of Oculis since December 2017. Since March 2012, Mr. Carnot has served as the Partner of Earlybird Venture Capital. Additionally, since 2005, Mr. Carnot has served as the Managing Director of Bay City Capital LLC. Prior to that, from 2000 to 2005, Mr. Carnot served as an Associate of The Pritzker Organization, LLC. Before that, from 1999 to 2000, Mr. Carnot served as a Principal of Oracle Partners. Prior to that, from 1997 to 1998, Mr. Carnot served as a Senior Associate of Booz Allen and Hamilton. Before that, from 1995 to 1997, Mr. Carnot served as a Product Manager of Eli Lilly & Co. Prior to that, from 1991 to 1994, Mr. Carnot served as a Senior Consultant of Accenture. Before that, from 1989 to 1991, Mr. Carnot served as a sales and marketing professional at Rhone-Poulenc. Mr. Carnot currently serves on the board of directors of iSTAR Medical, iQone Healthcare Group, and Priothera. Mr. Carnot previously served on the board of directors of Atlantic Therapeutics, Merus, Interleukin Genetics, Madrigal Pharmaceuticals Inc., Nabsys, Bioseek, Pathway Diagnostics, and Reliant Pharmaceuticals. Mr. Carnot holds an MBA with Distinction from INSEAD and a M.Sc. in Molecular Biology from the University of Geneva.
Pravin Dugel, M.D., 60, has served as a member of the board of directors of Oculis since March 2023. Mr. Dugel served as the President of Iveric Bio from May 2021 to October 2023. He joined as Executive Vice President in April 2020 and was promoted to President of the Company in May 2021. Dr. Dugel was previously Managing Partner, Retinal Consultants of Arizona and the Retinal Research Institute; Clinical Professor, USC Eye Institute, Keck School of Medicine, University of Southern California; and Founding Member, Spectra Eye Institute in Sun City, Arizona. Dr. Dugel has authored more than 200 papers, 35 book chapters and has been invited to lecture at several marquis medical meetings and to serve as a visiting professor at universities worldwide, including in Japan, India, China, Malaysia, Egypt, the United Kingdom, France, Germany, Austria, Italy, Poland, Denmark, Norway, Czechoslovakia, Canada and Australia. Dr. Dugel is internationally recognized as a major clinical researcher and has been a principal investigator in over 100 multicenter clinical trials. His research and educational contributions earned him the prestigious Senior Honor Award from the American Academy of Ophthalmology (AAO). He has been elected and previously served as the Retina Subspecialty Day Board Chairman for the American Academy of Ophthalmology Annual Meeting, as a member of the Board of Directors of the largest retina society in the United States, the American Society of Retina Specialists (ASRS), and the largest retina society in Europe, EURETINA.
141
Dr. Dugel graduated from Columbia University in New York City. He then attended UCLA School of Medicine where he obtained his M.D. He completed his residency in ophthalmology at the USC Eye Institute, Keck School of Medicine and completed his medical retina fellowship at the Bascom Palmer Eye Institute and his surgical retina fellowship at the USC Eye Institute, where he was elected to serve on the faculty as the Resident Director.
Martijn Kleijwegt, 69, has served as a member of the board of directors of Oculis since March 2023. Previously, he served as a member and the Chairman of the EBAC Board from EBAC’s inception in January 2021 to March 2023. Mr. Kleijwegt founded LSP in 1998 and is currently a partner at EQT Life Sciences (f/k/a Life Science Partners). Mr. Kleijwegt has over 30 years of hands-on finance and investment experience. Mr. Kleijwegt currently serves on the boards of Vico Therapeutics and A-M Pharma. Mr. Kleijwegt has a master’s degree in Economics from Amsterdam University.
Geraldine O’Keeffe, 58, has served as a member of the board of directors of Oculis since March 2023. Ms. O'Keeffe joined LSP in 2008. She became a Partner of the firm in 2010. Ms. O’Keeffe’s prime focus and responsibility within LSP is to invest in listed securities. Prior to joining LSP, she held the position of Senior Healthcare Analyst at Fortis Investment Banking. In that position, she researched a wide range of innovative life sciences companies, both in Europe and the US. Before joining the financial community, she worked within the life sciences industry for a number of years, gaining first-hand product development experience in a commercial setting. Prior to working in the industry, she lectured in Biomedical Sciences for several years at the Dublin Institute of Technology. Since July 2022, Ms. O’Keeffe serves on the board of directors of T-Knife Therapeutics, where she is a member of the Audit Committee. Ms. O’Keeffe has a Bachelor’s degree in Biochemistry and Microbiology from University College Cork and a Master’s degree in Biotechnology from University College Galway. She also conducted post-graduate research, inter alia at the prestigious Max Planck Institute for Biophysical Chemistry in Göttingen, Germany. In addition, Ms. O’Keeffe is also a graduate of The Dublin School of Business.
Diversity of the Board of Directors
The table below provides certain information regarding the diversity of our board of directors as of the filing date of this Annual Report. Our board diversity matrix for our prior fiscal year can be found in our Annual Report on Form 20-F for the year ended December 31, 2022, filed with the SEC on March 28, 2023.
Board Diversity Matrix |
||||
Country of Principal Executive Offices |
Switzerland |
|||
Foreign Private Issuer |
Yes |
|||
Disclosure Prohibited under Home Country Law |
No |
|||
Total Number of Directors |
7 |
|||
|
Female |
Male |
Non-Binary |
Did Not |
Part I: Gender Identity |
||||
Directors |
2 |
5 |
0 |
0 |
Part II: Demographic Background |
||||
Underrepresented Individual in Home Country Jurisdiction |
0 |
|||
LGBTQ+ |
0 |
|||
Did Not Disclose Demographic Background |
0 |
|||
142
Family Relationships
There are no family relationships among any of our executive officers or directors.
Corporate Governance
We structured our corporate governance in a manner we believe closely aligns our interests with those of our shareholders. Notable features of this corporate governance include:
Non-Classified Board of Directors
In accordance with our articles of association, our board of directors is not divided into classes of directors. The directors were appointed until the end of the general meeting of shareholders called to approve our annual accounts for the 2024 financial year.
Compensation of Executive Officers
Historically, our executive compensation program has reflected our innovative growth and development-oriented corporate culture. To date, the compensation of our Chief Executive Officer and our other executive officers has consisted of a combination of base salary, bonuses and long-term incentive compensation in the form of restricted common stock awards and/or stock options. Our executive officers who are full-time employees, like all other full-time employees, are participants in applicable retirement plans in the jurisdiction in which they reside. We evaluate our compensation values and philosophy and compensation plans and arrangements as circumstances merit. We review executive compensation periodically with input from a third-party compensation consultant. As part of this review process, the board of directors and the remuneration committee apply our values and philosophy, while considering the compensation levels needed to ensure our executive compensation program remains competitive with our peers. In connection with our executive compensation program, we also review whether we are meeting our retention objectives and the potential cost of replacing a key employee.
We use base salaries to recognize the experience, skills, knowledge and responsibilities required of all our executive officers. Base salaries are reviewed annually, typically in connection with our annual performance review process, and adjusted from time to time to align salaries with market levels after taking into account individual responsibilities, performance and experience. In addition, our executives are entitled to annual cash bonuses for their performance over the fiscal year, based on goals established by our board of directors. Furthermore, we have a formal process with respect to the grant of equity incentive awards to our employees, including our executive officers. We believe that equity incentive awards provide our employees with a strong link to our long-term performance, create an ownership culture and help to align the interests of our employees, including our executive officers, and our stockholders. In addition, we believe that equity incentive awards with time-based vesting features promote employee retention because this feature incentivizes our employees, including our executive officers, to remain in our employment during the vesting period.
Adoption of Clawback Policy
In October 2023, in accordance with Rule 10D-1 promulgated under the Exchange Act and Nasdaq Listing Rule 5608, we adopted an Incentive Compensation Recoupment Policy which is filed herewith as Exhibit 97.1.
143
Compensation of Directors
Our board of directors adopted a board of directors’ compensation policy that is designed to enable us to attract and retain, on a long-term basis, highly qualified non-employee directors. As of the filing date, we pay each eligible director who is not an employee of the Company annual cash retainers, as set forth below.
|
|
Annual |
||||
Board of Directors |
|
|
$ 45,200 |
|
|
|
Board of Directors Chair |
|
|
$ 84,750 |
|
|
|
Audit Committee Chair |
|
|
$ 22,600 |
|
|
|
Audit Committee Member |
|
|
$ 11,300 |
|
|
|
Remuneration Committee Chair |
|
|
$ 13,560 |
|
|
|
Remuneration Committee Member |
|
|
$ 6,780 |
|
|
|
Nomination and Governance Committee Chair |
|
|
$ 10,170 |
|
|
|
Nomination and Governance Committee Member |
|
|
$ 5,085 |
|
|
|
In addition, each eligible director elected or appointed to our board of directors is eligible to participate in the Stock Option and Incentive Plan Regulation 2023 of the Company (the “2023 Plan”), subject to its terms and conditions as approved and amended by our board of directors from time to time. Upon joining Oculis, we issue to eligible directors a one-time equity incentive award in the form of stock option or similar awards under the 2023 Plan or other equity incentive plans then in effect, at an estimated equity value of $240,000. The exact number of options to be granted and the vesting schedule shall be determined by the Board in the grant notice in its free discretion and only such grant notice shall have legal effect. We will also issue to eligible directors an annual equity incentive award in the form of stock option or similar awards under the 2023 Plan or other equity incentive plans then in effect, at an estimated equity value of $120,000, generally granted on the date of our annual general meeting.
The eligible directors are not eligible to any benefits other than those set out in the directors compensation policy, unless our board of directors decides otherwise. We reimburse all reasonable expenses in accordance with the terms and conditions of our travel and expense policy then in effect.
Compensation of Directors and Executive Officers
For the year ended December 31, 2023, the aggregate compensation earned by the members of our board of directors and our executive officers for services in all capacities was CHF 8.1 million.
For the year ended December 31, 2023, fees, salaries and other short-term employee benefits earned by the members of our board of directors and our executive officers was CHF 2.1 million.
The amount contributed by us to provide post-employment benefits to executive officers amounted to a total of CHF 0.2 million for the year ended December 31, 2023.
During the year ended December 31, 2023, 1,029,765 options, excluding earnout options, to purchase registered ordinary shares were granted to members of our board of directors and our executive officers for a total fair value of CHF 4.8 million.
144
See Note 13 to our audited consolidated financial statements included elsewhere in this Annual Report for further details regarding the share options, SARs and restricted stock, including the exercise price and the expiration date. The above compensation amounts may differ from those reported in our Swiss compensation report because the Swiss compensation report covers the period from March 2, 2023 through December 31, 2023, rather than the full fiscal year.
Risk Oversight
The board of directors is responsible for overseeing our risk management process. The board of directors focuses on our general risk management strategy, the most significant risks, and oversees the implementation of risk mitigation strategies by management. The audit committee is also responsible for discussing our policies with respect to risk assessment and risk management. The board of directors believes its administration of its risk oversight function has not negatively affected the board of directors’ leadership structure.
Code of Business Conduct and Ethics
Our board of directors adopted a Code of Business Conduct and Ethics applicable to the directors, executive officers and employees that complies with the rules and regulations of Nasdaq and the SEC. The Code of Business Conduct and Ethics is available on our website. In addition, we posted on the Corporate Governance section of our website all disclosures that are required by law or Nasdaq listing standards concerning any amendments to, or waivers from, any provision of the Code of Business Conduct and Ethics. The reference to our website address in this Annual Report does not include or incorporate by reference the information on our website into this Annual Report.
Stock Option and Incentive Plan Regulation 2023
The Stock Option and Incentive Plan Regulation 2023 (the “2023 Plan”) was approved by our board of directors on the Acquisition Closing Date and provides for the grant of options, restricted stock awards or units or stock appreciation rights to acquire the Ordinary Shares.
The purpose of the 2023 Plan is to attract and retain highly qualified personnel and to provide key employees with additional incentive to increase their efforts on behalf and in the best interest of us and our subsidiaries by giving them the opportunity to acquire a proprietary interest in us as an incentive for them to remain in the service of us. The terms of the 2023 Plan are described in more detail below.
The 2023 Plan shall be administered by a plan administrator (one or several persons) elected by our board of directors from time to time. The plan administrator acts within the guidelines set and approved by our board of directors or a committee thereof and is authorized to, among others, determine (i) which eligible persons are to receive awards under the 2023 Plan, (ii) the time or times when such options or rights grants are to be made, (iii) the nature of the shares and the number of awards covered by each such grant, (iv) the time or times at which each option or stock appreciation right is to become exercisable, (v) the vesting conditions applicable to the options or rights, (vi) the maximum term for which the options or rights are to remain outstanding, and (vii) any terms and conditions of any restricted stock award, in each case, subject to the guidelines set and approved by our board of directors or a committee thereof. Persons eligible to participate in our 2023 Plan are employees, members of the board of directors and consultants of Oculis or a subsidiary. The plan administrator determines within the guidelines set and approved by our board of directors or a committee which eligible persons are to receive rights to acquire options under the 2023 Plan.
The 2023 Plan provides for up to 7,835,544 registered shares corresponding to 16.0% of the Ordinary Shares on a fully diluted basis at the Acquisition Closing Date. In the event registered shares that otherwise would have been issuable under the 2023 Plan are withheld by us in payment of the exercise price or withholding obligations, such shares shall remain available for issuance under the 2023 Plan. In the event that an outstanding award expires or is cancelled, forfeited or terminated for any reason, the shares allocable to the unexercised or unsettled portion shall remain available for issuance under the 2023 Plan.
A participant may only exercise an option or stock appreciation right to the extent that the option or stock appreciation right has vested and has not lapsed under the 2023 Plan. Unless otherwise determined by our board of directors at the grant date or set forth in the grant notice, an option or an award in the form of a restricted stock unit or stock appreciation right granted under the 2023 Plan typically vests as to 25.0% of the award at the end of the first year following the vesting start date, with the remaining 75.0% of the award vesting monthly over the 3 years after the first year following the vesting start date.
145
Any restricted stock may not be transferred or pledged. Such restriction expires with the expiration of any repurchase right for the restricted stock. The 2023 Plan provides provisions that govern the exercise of any awards held by the participant at the time the legal relationship forming the basis of the service is coming to an end. Generally, any award not vested shall immediately lapse at the time a notice of termination has been received (regardless of which party gives notice) or at the end of the term in case of a board member. If indicated in the grant notice or otherwise resolved by the board of directors, upon the occurrence of a “Corporate Transaction” (as defined in the 2023 Plan ), all options and awards in the form of a restricted stock unit or stock appreciation rights (i) shall fully vest and (ii) in the case of options and stock appreciation rights must be immediately exercised, except if such options or awards in the form of a restricted stock unit or stock appreciation rights are repurchased by Oculis or a third party designated by Oculis for a cash consideration equivalent to the economic value applicable to such option or stock appreciation right under the 2023 Plan.
Our board of directors has complete and exclusive power and authority to amend or modify the 2023 Plan in any or all respects. Such amendment or modification shall be communicated in appropriate form as an amendment of the 2023 Plan. Unless such change is required to comply with applicable law, listing requirements, accounting rules or tax requirements, no such amendment or modification shall, without the consent of the concerned participant, adversely affect materially his/her rights and obligations under the 2023 Plan.
Composition of Our Board of Directors
Our board of directors is currently composed of seven members. In accordance with our articles of association, the board of directors is not divided into classes of directors. The directors were appointed until the end of the general meeting of shareholders called to approve our annual accounts for the 2024 financial year.
Six of seven directors are independent as defined in Nasdaq listing standards and applicable SEC rules and our board of directors has an independent audit committee, a nomination and governance committee, and a remuneration committee.
Committees of our Board of Directors
Our board of directors has three standing committees: an audit committee, a remuneration committee, and a nominating and nomination and governance committee. The board has adopted written charters that are available to shareholders on our website at https://investors.oculis.com/corporate-governance. The reference to our website address in this Annual Report on Form 20-F does not include or incorporate by reference the information on our website into this Annual Report on Form 20-F.
Audit Committee
The audit committee consists of Lionel Carnot, Geraldine O’Keeffe and Christina Ackermann. The audit committee assists the board of directors in overseeing our accounting and financial reporting processes and the audits of our financial statements. Mr. Carnot serves as chairperson of the audit committee. In addition, the audit committee is responsible for the appointment, compensation, retention and oversight of the work of our independent registered public accounting firm. Our board of directors has determined that Mr. Carnot, Ms. O’Keeffe and Ms. Ackermann satisfy the “independence” requirements set forth in Rule 10A-3 under the Exchange Act and Mr. Carnot qualifies as an “audit committee financial expert,” as such term is defined in the rules of the SEC.
146
Each of the members of our audit committee qualify as independent directors according to the rules and regulations of the SEC and Nasdaq with respect to audit committee membership. In addition, all of the audit committee members meet the requirements for financial literacy under applicable SEC and Nasdaq rules and at least one of the audit committee members qualifies as an “audit committee financial expert,” as such term is defined in Item 407(d) of Regulation S-K. The audit committee is governed by a charter that complies with applicable Nasdaq rules, which charter is posted on our website. We have adopted an audit committee charter, which details the principal functions of the audit committee, including:
147
Remuneration Committee
The remuneration committee consists of Christina Ackermann, Pravin Dugel and Lionel Carnot. The remuneration committee assists the board of directors in determining compensation for our executive officers and our directors. Ms. Ackermann serves as chairperson of the remuneration committee.
As of the first day of trading after the Business Combination, we were subject to the Swiss provisions regarding compensations for listed companies under the Swiss Code of Obligations, which require Swiss corporations listed on a stock exchange to establish a remuneration committee. In accordance with the Swiss Code of Obligations, the members of our remuneration committee must be elected by our general meeting of shareholders and the aggregate amount of compensation of each of our directors and our executive committee must also be approved by our general meeting of shareholders, in each case commencing with our first annual general meeting of shareholders as a public company. On March 2, 2023 the general meeting of shareholders approved the compensation packages for the Board and the executive committee until the general meeting of shareholders to be held in 2024. Our board of directors appointed Ms. Ackermann as the chair of the remuneration committee and will fill any vacancies on the remuneration committee until completion of the next annual general meeting of shareholders.
Each of the members of our remuneration committee qualifies as independent directors according to the rules and regulations of the SEC and Nasdaq with respect to remuneration committee membership, including the heightened independence standards for members of a remuneration committee. The remuneration committee is governed by a charter that is posted on our website. We have adopted a remuneration committee charter, which details the principal functions of the remuneration committee, including:
148
Nomination and Governance Committee
The nomination and governance committee consists of Dr. Pravin Dugel, Geraldine O’Keeffe and Martijn Kleijwegt. The nomination and governance committee assists our board of directors in identifying individuals qualified to become our directors consistent with criteria established by us and in developing our code of business conduct and ethics. Dr. Dugel serves as chairperson of the nomination and governance committee. The nomination and governance committee is governed by a charter that is posted on our website. We have adopted a nomination and governance committee charter, which details the principal functions of the nomination and governance committee, including:
149
As of December 31, 2023, we had 36 employees. Our headcount for R&D was 18, and our headcount for G&A was 18. Our employees include 16 executive leadership, administrative, and development personnel based in Switzerland; 8 executive leadership, administrative, and research personnel based in Iceland; 7 executives and administrators based in the United States; 5 management, research and administrative personnel based in France, UK and China. Pursuant to local laws, our employees in Iceland and France are represented by a labor union or covered under a collective bargaining agreement. We consider our relationship with our employees to be good.
Our human capital resources objectives include, as applicable, identifying, recruiting, retaining, incentivizing and integrating our existing and new employees, advisors and consultants. The principal purposes of our equity and cash incentive plans are to attract, retain and reward personnel through the granting of stock-based and cash-based compensation awards, in order to increase stockholder value and the success of our company by motivating such individuals to perform to the best of their abilities and achieve our objectives.
For information regarding the share ownership of our directors and executive officers, see “Item 7.A Major Shareholders” and “Item 6.B Compensation” for a discussion of the 2023 Plan.
Not applicable.
The following table sets forth information regarding the beneficial ownership of Ordinary Shares as of December 31, 2023:
Except as otherwise noted herein, the number and percentage of Ordinary Shares beneficially owned is determined in accordance with Rule 13d-3 of the Exchange Act, and the information is not necessarily indicative of beneficial ownership for any other purpose. Under such rule, beneficial ownership includes any Ordinary Shares as to which the holder has sole or shared voting power or investment power and also any Ordinary Shares which the holder has the right to acquire within 60 days of the Closing Date through the exercise of any option, warrant or any other right.
150
We have based percentage ownership on 36,649,705 Ordinary Shares outstanding as of December 31, 2023. The table below does not include earn-out shares which are issued and contingently forfeitable and are not deemed to be outstanding.
Name and Address of Beneficial Owners |
|
Number of Shares |
|
|
% Ownership |
|
||
Directors and Executive Officers (1) |
|
|
|
|
|
|
||
Riad Sherif(2) |
|
|
881,895 |
|
|
|
2.4 |
% |
Sylvia Cheung(3) |
|
|
201,067 |
|
|
* |
|
|
Páll Ragnar Jóhannesson(4) |
|
|
528,413 |
|
|
|
1.4 |
% |
Christina Ackermann |
|
|
11,718 |
|
|
* |
|
|
Lionel Carnot |
|
|
— |
|
|
|
— |
|
Pravin Dugel(5) |
|
|
23,819 |
|
|
* |
|
|
Martijn Kleijwegt(6) |
|
|
1,997,302 |
|
|
|
5.4 |
% |
Geraldine O'Keeffe |
|
|
— |
|
|
|
— |
|
Anthony Rosenberg(7) |
|
|
116,257 |
|
|
* |
|
|
All officers and directors as a group (9 individuals) |
|
|
|
|
|
|
||
Five Percent Holders of the Company |
|
|
|
|
|
|
||
LSP 7 Coöperatief U.A.(8) |
|
|
5,327,362 |
|
|
|
14.5 |
% |
Brunnur vaxtarsjóður slhf. (9) |
|
|
2,335,841 |
|
|
|
6.4 |
% |
BVCF Management (BEYEOTECH) (10) |
|
|
2,070,020 |
|
|
|
5.6 |
% |
Funds managed by Pivotal Partners(11) |
|
|
1,898,502 |
|
|
|
5.2 |
% |
* Indicates beneficial ownership of less than 1.0% of the total ordinary shares outstanding.
151
Significant Changes in Percentage Ownership
In March 2023, we experienced significant changes in the percentage ownership held by major shareholders as a result of the Business Combination.
Voting Rights
The voting rights of the principal shareholders do not differ from the voting rights of other shareholders.
Shareholders in the United States
As of February 15, 2023, to the best of our knowledge 40,503,780 of our outstanding ordinary shares, including earnout shares, were held by 104 shareholders of record in the United States. The actual number of holders is greater than these numbers of record holders, and includes beneficial owners whose ordinary shares are held in street name by brokers and other nominees.
152
This number of holders of record also does not include holders whose shares may be held in trust by other entities.
Policies and Procedures for Related Person Transactions
Our board of directors has adopted a written related person transaction policy that sets forth certain policies and procedures for the review and approval or ratification of transactions involving us in which a related person has or will have a direct or indirect material interest, as determined by the audit committee of the Board. A “related person” for purposes of the policy means: (i) enterprises that directly or indirectly through one or more intermediaries, control or are controlled by, or are under common control with, us; (ii) associates (defined as, unconsolidated enterprises in which we have a Significant Influence or which has Significant Influence over us); (iii) individuals owning, directly or indirectly, an interest in the voting power of us that gives them Significant Influence over us, and close members of any such individual’s family; (iv) key management personnel (i.e., having authority and responsibility for planning, directing and controlling our activities), including directors and close members of such individuals’ families; and (v) enterprises in which a substantial interest in the voting power is owned, directly or indirectly, by any person described in (iii) or (iv) above or over which such a person is able to exercise Significant Influence, including enterprises owned by our directors or major shareholders and enterprises that have a member of key management in common with us. “Significant Influence” for purposes of the policy means the power to participate in the financial and operating policy decisions of an enterprise but is less than control over those policies, provided that shareholders beneficially owning a 10.0% or more interest in the voting power of the enterprise concerned are presumed to have a significant influence on such enterprise.
Pursuant to the policy, each executive director, nominee for the position of executive director, and executive officer shall promptly notify the designated contact of any transaction involving us and a related person. The designated contact will present any new related person transactions, and proposed transactions involving related persons, to the audit committee of the board of directors at its next occurring regular meeting. If the audit committee determines that the related person involved has a direct or indirect material interest in the transaction, and therefore that the transaction is a related party transaction, the audit committee shall consider all relevant facts and circumstances, including the commercial reasonableness of the terms, the benefit and perceived benefit, or lack thereof, to Oculis, opportunity costs of alternate transactions, the materiality and character of the Related Person’s direct or indirect interest, and the actual or apparent conflict of interest of the Related Person. The audit committee will not approve or ratify a Related Person transaction unless it shall have determined that, upon consideration of all relevant information, the transaction is in, or not inconsistent with, our best interests . On an annual basis, the audit committee shall review previously approved related person transactions, under the standard described above, to determine whether such transactions should continue. If after the review described above, the audit committee determines not to approve or ratify a related person transaction (whether such transaction is being reviewed for the first time or has previously been approved and is being reviewed), the transaction will not be entered into or continued.
Agreements with our Executive Officers and Directors
Aside from standard employment agreements, there are no transactions between the Company and its directors and executive officers. The remuneration of the directors and executive officers (excluding non-executive directors), who are the key management personnel, is described in the section entitled “Compensation.”
Indemnification Agreements
The articles of association provide that we will indemnify our directors and officers to the fullest extent permitted by Swiss law, subject to certain exceptions contained in our articles of association.
In connection with the Business Combination, we also entered into indemnification agreements with each of our directors and executive officers. The indemnification agreements provide the indemnities with contractual rights to indemnification, and expense advancement and reimbursement, to the fullest extent permitted under Swiss law, subject to certain exceptions contained in those agreements.
153
Not applicable.
Item 8. Financial Information
A. Consolidated Statements and Other Financial Information
Our consolidated financial statements are appended at the end of this Annual Report, starting at page F-1.
Legal Proceedings
From time to time, we may be subject to legal proceedings. We are not currently a party to or aware of any proceedings that we believe will have, individually or in the aggregate, a material adverse effect on our business, financial condition or results of operations. Regardless of outcome, litigation can have an adverse impact on us because of defense and settlement costs, diversion of management resources, and other factors.
B. Significant Changes
Please see Note 23 Subsequent Events, included in the audited consolidated financial statements starting at page F-1 included elsewhere in this Form 20-F. Other than the events included in this note, no significant changes have occurred.
Item 9. The Offer and Listing.
A. Offer and Listing Details
Ordinary Shares and Warrants are listed on The Nasdaq Stock Market LLC under the symbols “OCS” and “OCSAW”, respectively. Prior to March 6, 2023, there was no public trading market for our Ordinary Shares or Warrants. Holders of Ordinary Shares and Warrants should obtain current market quotations for their securities.
B. Plan of Distribution
Not applicable.
C. Markets
Ordinary Shares and Warrants have been listed on The Nasdaq Stock Market LLC under the symbol “OCS” and “OCSAW”, respectively, since March 6, 2023. Prior to March 6, 2023, there was no public trading market for our Ordinary Shares or Warrants.
Not applicable.
E. Dilution
Not applicable.
F. Expenses of the Issue
Not applicable.
Item 10. Additional Information.
Not applicable.
154
B. Memorandum and Articles of Association
Please see the information set forth in Exhibit 2.5 “Description of Securities” and the copy of our Amended and Restated articles of association filed as Exhibit 1.1, which are each incorporated herein by reference.
C. Material Contracts
In addition to the contracts described elsewhere in this Annual Report, the following are summaries of each material contract to which we are a party for the two years preceding the date of this Annual Report. For additional information on our material contracts, please see “Item 4. Information on the Company,” “Item 6. Directors, Senior Management and Employees,” and “Item 7.B Related Party Transactions” of this Annual Report.
Material Contracts Relating to the Business Combination
Business Combination Agreement
On October 17, 2022, we entered into a business combination agreement (the “Business Combination Agreement”) with EBAC and Legacy Oculis. The Business Combination Agreement provided for, among other things, the following transactions on the closing date: (i) the PIPE Investors transferred $71,188,910 to EBAC in exchange for 7,118,891 PIPE Shares; (ii) EBAC underwent the First Merger; and as part of the First Merger, (1) each share of EBAC Common Stock (including those held by the PIPE Investors) was automatically converted into the Surviving EBAC Shares (2) each EBAC Warrant outstanding immediately prior to the First Merger Effective time was automatically converted into Surviving EBAC Warrants and (3) EBAC deposited, or cause to be deposited, with the Exchange Agent (held solely on behalf of the holders of EBAC Common Stock and EBAC Warrants) the Surviving EBAC Shares and Surviving EBAC Warrants on the terms and subject to the conditions set forth in the Business Combination Agreement and in the Ancillary Agreements; (iii) on the day before the Acquisition Closing Date and following the First Merger Effective Time but prior to the Second Merger Effective Time, the Exchange Agent, solely on behalf of the holders of Surviving EBAC Shares and Surviving EBAC Warrants, undertook the Exchange Agent Contribution Actions in exchange for receipt of the New Parent Interests Consideration; (iv) in connection with the Exchange Agent Contribution, on the day before the Acquisition Closing Date and prior to the Second Merger Effective Time, the Exchange Agent distributed (1) the Ordinary Shares as part of the New Parent Interests Consideration to the holders of Surviving EBAC Shares and (2) the Warrants as part of the New Parent Interests Consideration to the holders of Surviving EBAC Warrants; (v) on the day before the Acquisition Closing Date and following the completion of the Exchange Agent Contribution Actions, at the Second Merger Effective Time, EBAC underwent the Second Merger, pursuant to which, among other things, the separate corporate existence of EBAC ceased, and following the Acquisition Closing, Merger Sub 2 was liquidated and its assets distributed to Oculis; (vi) after the Second Merger Effective Time but before the Oculis Share Contribution, it was the intention of the parties to the Convertible Loan Agreements that Oculis assumed the Convertible Loan Agreements, pursuant to which the Lenders granted Oculis a right to receive a convertible loan with certain conversion rights in an aggregate amount of $19,670,000, and that immediately after such assumption but before the Oculis Share Contribution, the Lenders exercised their conversion rights in exchange for Ordinary Shares at $10.00 per share, on the same terms as the PIPE Investors; (vii) at approximately 10:00 a.m. Eastern Time on the Acquisition Closing Date, those Oculis Shareholders executing the Oculis Shareholders Support Agreements and the exchange notice contemplated by the Business Combination Agreement effected the contribution to Oculis of all Company Share Capital held by such Oculis Shareholders free and clear of all liens (other than general restrictions on transfer under applicable securities laws or the articles of association of Oculis) in exchange for Ordinary Shares on the terms and subject to the conditions set forth in the Business Combination Agreement and Oculis Shareholders Support Agreement; and (viii) on July 6, 2023, Legacy Oculis merged with and into Oculis Operations, and the separate corporate existence of Legacy Oculis ceased. Oculis Operations is the surviving company and remains a wholly-owned subsidiary of Oculis.
Subscription Agreements related to the PIPE Financing
In connection with the foregoing and concurrently with the execution of the Business Combination Agreement, EBAC entered into Initial Subscription Agreements with the Initial PIPE Investors pursuant to which the Initial PIPE Investors agreed to purchase from EBAC, severally and not jointly, and EBAC agreed to issue from treasury and sell to the Initial PIPE Investors, a number of EBAC Class A Common Stock equal to (i) the total subscription amount from the Initial PIPE Investors ($63,303,910) divided by (ii) $10.00.
155
Subsequent to the Initial PIPE Financing, in January 2023, EBAC entered into the Subsequent Subscription Agreements with the Subsequent PIPE Investors, pursuant to which the Subsequent PIPE Investors agreed to subscribe for, and EBAC agreed to issue to the Subsequent PIPE Investors, a number of EBAC Class A Common Stock equal to (i) the total subscription amount from the Subsequent PIPE Investors ($7,885,000) divided by (ii) $10.00. The aggregate amount of EBAC Class A Common Stock issued pursuant to the PIPE Financing was 7,118,891 for aggregate gross proceeds of $71,188,910. The shares of EBAC Class A Common Stock issued from treasury to the PIPE Investors pursuant to the Subscription Agreements was not registered under the Securities Act, in reliance upon the exemption provided in Section 4(a)(2) of the Securities Act (as defined below). In connection with the Acquisition Closing, we granted the PIPE Investors certain customary registration rights in connection with the PIPE Financing, including demand and piggyback rights as set forth in the Registration Rights and Lock-Up Agreement. The PIPE Financing was contingent upon, among other things, the Acquisition Closing.
The PIPE Financing closed on March 2, 2023.
The Convertible Loan Agreements
Concurrently with the execution of the Business Combination Agreement, Legacy Oculis and the Lenders party thereto entered into convertible loan agreements pursuant to which the Lenders granted Legacy Oculis a right to receive a convertible loan with certain conversion rights in an aggregate amount of $12.7 million. Subsequent to the execution of the Business Combination Agreement, on January 20, 2023 and as amended and restated on February 22, 2023, Legacy Oculis and an additional Lender entered into a convertible loan agreement in substantially the same form as the initial convertible loan agreement, pursuant to which, among other things, the Lender party thereto granted us a right to receive a convertible loan with certain conversion rights in an aggregate amount of $7.0 million. The aggregate amount raised under the Convertible Loan Agreements was $19.7 million. Following the Second Merger Effective Time on March 2, 2023, we assumed the Convertible Loan Agreements, and immediately after such assumption but before the Oculis Share Contribution, the Lenders exercised their conversion rights in exchange for Ordinary Shares at $10.00 per share, on substantially the same terms as the PIPE Investors. In accordance with the Convertible Loan Agreements, upon conversion, the Lenders were granted certain customary registration rights, substantially on the same terms as those offered pursuant to the Subscription Agreements.
D. Exchange Controls
There are no foreign exchange controls or foreign exchange regulations under the currently applicable laws of Switzerland.
E. Taxation
Material Swiss Tax Considerations
In the opinion of Vischer AG, the following are the material Swiss tax consequences of receiving, owning and disposing of Ordinary Shares and Warrants.
This summary is based upon Swiss tax laws, and the practices of the Swiss tax authorities, in effect on the date of this Annual Report. Such laws and administrative practice are subject to change at any time, possibly with retroactive effect. The summary does not constitute legal or tax advice and is intended only as a general guide. It is not exhaustive and shareholders should consult their own tax advisors about the Swiss tax consequences (and tax consequences under the laws of other relevant jurisdictions) of the acquisition, ownership and disposal of Ordinary Shares and Warrants and as to their tax position.
Please be aware that the residence concept used under the respective headings applies for Swiss tax assessment purposes only. Any reference in this section to a tax, duty, levy impost or other charge or withholding of a similar nature refers to Swiss tax law and/or concepts only.
Swiss Withholding Tax
Under present Swiss tax law, dividends and similar cash or in-kind distributions made by the Oculis to a holder of Ordinary Shares (including liquidation proceeds and bonus shares) are subject to Swiss federal withholding tax (the “Withholding Tax”), currently at a rate of 35.0% (applicable to the gross amount of taxable distribution), unless these payments are repayments of the par value of Ordinary Shares or, within the limitations accepted by the legislation in force and the respective administrative practice of the reserve from capital contribution (Reserve aus Kapitaleinlage).
156
Oculis is obliged to deduct the Withholding Tax from the gross amount of any taxable distribution and to pay the tax to the Swiss Federal Tax Administration within 30 days of the due date of such distribution, unless a notification procedure applies (the notification procedure does not apply to portfolio holdings).
Swiss resident individuals who hold their Ordinary Shares as private assets (“Resident Private Shareholders”) are in principle eligible for a full refund or credit against income tax of the Withholding Tax if they duly report the underlying income in their income tax return. In addition Domestic Commercial Shareholders who, among other things, are also the beneficial owners of the Ordinary Shares and the dividends or the other distributions made or paid by Oculis on the Ordinary Shares are in principle eligible for a full refund or credit against income tax of the Withholding Tax if they, inter alia, duly report the underlying income in their income statements or income tax return, as the case may be.
Shareholders who are not resident in Switzerland for tax purposes, and who, during the respective taxation year, have not engaged in a trade or business carried on through a permanent establishment with fixed place of business situated in Switzerland for tax purposes, and who are not subject to corporate or individual income taxation in Switzerland for any other reason (collectively, “Non-Resident Shareholders”) may be entitled to a total or partial refund of the Withholding Tax if the country in which such recipient resides for tax purposes maintains a bilateral treaty for the avoidance of double taxation with Switzerland and further conditions of such treaty are met. Non-Resident Shareholders should be aware that the procedures for claiming treaty benefits (and the time required for obtaining a refund) may differ from country to country. Non-Resident Shareholders should consult their own legal, financial or tax advisors regarding receipt, ownership, purchases, sale or other dispositions of Ordinary Shares and the procedures for claiming a refund of the Withholding Tax.
Swiss Federal Stamp Taxes
To the extent Oculis issues new shares, Oculis will bear the Swiss federal issue stamp tax (Emissionsabgabe) on the issuance of such Ordinary Shares of 1.0% of the offering price, net of certain deductions. The delivery of newly issued shares against payment of the offering price is generally not subject to Swiss federal securities turnover tax (Umsatzabgabe).
To the extent Oculis offers existing shares currently held by Oculis or certain existing shareholders of Oculis, the sale and delivery of any such existing shares will, subject to statutory exemptions, be subject to Swiss federal securities turnover tax (Umsatzabgabe) at an aggregate tax rate of up to 0.15% of the consideration paid on such sale and will be borne (or compensated) by the current holders of such existing Ordinary Shares.
Any subsequent transactions in Ordinary Shares in the secondary markets are subject to Swiss securities turnover tax at an aggregate rate of 0.15% of the consideration paid for such Ordinary Shares, however, only if a bank or other securities dealer in Switzerland or Liechtenstein, as defined in the Swiss Federal Stamp Tax Act (Stempelabgabengesetz), is a party or an intermediary to the transaction and no exemption applies.
Swiss Federal, Cantonal and Communal Individual Income Tax and Corporate Income Tax
Non-Resident Shareholders are not subject to any Swiss federal, cantonal or communal income tax on dividend payments and similar distributions because of the mere holding of Ordinary Shares. The same generally applies for capital gains on the sale of Ordinary Shares. For Withholding Tax consequences, please see the section entitled “—Material Swiss Tax Considerations—Swiss Withholding Tax.”
Resident Private Shareholders who receive dividends and similar cash or in-kind distributions (including liquidation proceeds as well as bonus shares or taxable repurchases of Ordinary Shares as described above), which are not repayments of the par value of Ordinary Shares or, within the limitations accepted by the legislation in force and the respective administrative practice, reserve from capital contribution (Kapitaleinlagereserven), are required to report such distributions in their individual income tax returns.
157
A gain or a loss by Resident Private Shareholders realized upon the sale or other disposition of ordinary shares to a third party will generally be a tax-free private capital gain or a not tax-deductible capital loss, as the case may be. Furthermore, the Swiss federal income tax on dividends is currently reduced to 70.0% of regular taxation (Teilbesteuerung), if the investment amounts to at least 10.0% of the total share capital of the issuer. On cantonal and communal level, the same provisions regarding partial taxation apply, with income reduced to between 50.0% and 80.0% depending on the canton of residency.
Domestic Commercial Shareholders who receive dividends and similar cash or in-kind distributions (including liquidation proceeds as well as bonus shares) are required to recognize such payments in their income statements for the relevant tax period and are subject to Swiss federal, cantonal and communal individual or corporate income tax, as the case may be, on any net taxable earnings accumulated (including the dividends) for such period. Domestic Commercial Shareholders who are corporate taxpayers may qualify for participation relief on dividend distributions (Beteiligungsabzug), if, inter alia, Ordinary Shares held have a market value of at least CHF 1 million. For cantonal and communal income tax purposes, the regulations on participation relief are broadly similar, depending on the canton of residency. For Domestic Commercial Shareholders who are individual taxpayers, the Swiss federal individual income tax on Dividends is reduced to 70.0% of regular taxation (Teilbesteuerung), if the investment is held in connection with the conduct of a trade or business or qualifies as an opted business asset (gewillkürtes Geschäftsvermögen) according to Swiss tax law and amounts to at least 10.0% of the total share capital of the Company. On cantonal and communal level the same provisions regarding partial taxation apply, with income reduced to between 50.0% and 80.0% depending on the canton of residency
Domestic Commercial Shareholders are required to recognize a gain or loss realized upon the disposal of ordinary shares in their income statement for the respective taxation period and are subject to Swiss federal, cantonal and communal individual or corporate income tax, as the case may be, on any net taxable earnings (including the gain or loss realized on the sale or other disposition of ordinary shares) for such taxation period.
Swiss Wealth Tax and Capital Tax
Non-Resident Shareholders holding Ordinary Shares are not subject to cantonal and communal wealth or annual capital tax because of the mere holding of Ordinary Shares.
Resident Private Shareholders are required to report the market value of their Ordinary Shares at the end of each tax period as part of their private wealth, which is subject to cantonal and communal wealth tax.
Domestic Commercial Shareholders are required to report their Ordinary Shares as part of their business wealth or taxable capital, as defined in the applicable cantonal and communal tax laws, which is subject to cantonal and communal wealth or annual capital tax.
Gift and Inheritance Taxes
The transfer of Ordinary Shares may be subject to cantonal and/or communal gift, estate or inheritance taxes if the donor is, or the deceased was, resident for tax purposes in a Swiss canton levying such taxes.
Automatic Exchange of Information in Tax Matters
On November 19, 2014, Switzerland signed the Multilateral Competent Authority Agreement. The Multilateral Competent Authority Agreement is intended to ensure the uniform implementation of Automatic Exchange of Information (the “AEOI”). The Swiss Federal Act on the International Automatic Exchange of Information in Tax Matters (the “AEOI Act”) entered into force on January 1, 2017. The AEOI Act is the legal basis for the implementation of the AEOI standard in Switzerland.
158
The AEOI is being introduced in Switzerland through bilateral agreements or multilateral agreements. The agreements have been, and will be, concluded on the basis of guaranteed reciprocity, compliance with the principle of specialty (i.e., the information exchanged may only be used to assess and levy taxes (and for criminal tax proceedings)) and adequate data protection.
Based on such multilateral and bilateral agreements and the implementing laws of Switzerland, Switzerland collects data in respect of financial assets, which may include Ordinary Shares, held in, and income derived thereon and credited to, accounts or deposits with a paying agent in Switzerland for the benefit of individuals resident in an EU member state or in a treaty state since 2017, and exchanges it since 2018. Switzerland has signed and is expected to sign AEOI agreements with other countries. A list of such agreements of Switzerland in effect or signed and becoming effective can be found on the website of the State Secretariat for International Finance.
Swiss Facilitation of the Implementation of the U.S. Foreign Account Tax Compliance Act
Switzerland has concluded an intergovernmental agreement with the United States to facilitate the implementation of U.S. Foreign Account Tax Compliance Act. The agreement ensures that the accounts held by U.S. persons with Swiss financial institutions are disclosed to the U.S. tax authorities either with the consent of the account holder or by means of group requests within the scope of administrative assistance. Information will not be transferred automatically in the absence of consent, but instead will be exchanged only within the scope of administrative assistance on the basis of the double taxation agreement between the United States and Switzerland. On September 20, 2019, the protocol of amendment to the double taxation treaty between Switzerland and the U.S. entered into force allowing the U.S. competent authority in accordance with the information reported in aggregated form to request all the information on U.S. accounts without a declaration of consent and on non-consenting non-participating financial institutions.
On October 8, 2014, the Swiss Federal Council approved a mandate for negotiations with the United States on changing the current direct-notification-based regime to a regime where the relevant information is sent to the Swiss Federal Tax Administration, which in turn provides the information to the U.S. tax authorities.
THE MATERIAL SWISS TAX DISCUSSION SET FORTH ABOVE IS INCLUDED FOR GENERAL INFORMATION ONLY AND MAY NOT BE APPLICABLE DEPENDING UPON A SWISS HOLDER'S PARTICULAR SITUATION. SWISS HOLDERS ARE URGED TO CONSULT THEIR TAX ADVISORS WITH RESPECT TO, THE OWNERSHIP AND DISPOSITION OF ORDINARY SHARES AND WARRANTS, INCLUDING THE TAX CONSEQUENCES UNDER NON-SWISS, AND OTHER TAX LAWS AND TAX TREATIES AND THE POSSIBLE EFFECTS OF CHANGES IN SWISS OR OTHER TAX LAWS.
Material U.S. Federal Income Tax Considerations for U.S. Holders
The following is a discussion of certain material U.S. federal income tax considerations generally applicable to the acquisition, ownership, and disposition of Ordinary Shares by a “U.S. Holder.” This discussion applies only to Ordinary Shares that are held by a U.S. Holder as “capital assets” within the meaning of Section 1221 of the Code (generally, property held for investment). This discussion does not describe all U.S. federal income tax considerations that may be relevant to a U.S. Holder in light of such U.S. Holder’s particular circumstances, nor does it address any state, local, or non-U.S. tax considerations, any non-income tax (such as gift or estate tax) considerations, the alternative minimum tax, the special tax accounting rules under Section 451(b) of the Code, the Medicare contribution tax on net investment income, or any tax consequences that may be relevant to U.S. Holders that are subject to special tax rules, including, without limitation:
159
If a partnership (including an entity or arrangement treated as a partnership for U.S. federal income tax purposes) holds Ordinary Shares, the tax treatment of a partner in such partnership generally will depend on the status of the partner and the activities of the partnership and the partner. Partnerships holding Ordinary Shares should consult their tax advisors regarding the tax consequences in their particular circumstances.
This discussion is based on the Code, the U.S. Treasury regulations promulgated thereunder, administrative rulings, and judicial decisions, all as currently in effect and all of which are subject to change or differing interpretation, possibly with retroactive effect. Any such change or differing interpretation could alter the tax consequences described herein. Furthermore, there can be no assurance that the Internal Revenue Service (the “IRS”) will not challenge the tax considerations described herein and that a court will not sustain such challenge.
For purposes of this discussion, a “U.S. Holder” is a beneficial owner of Ordinary Shares, that is, for U.S. federal income tax purposes:
160
THIS DISCUSSION IS FOR GENERAL INFORMATIONAL PURPOSES ONLY AND IS NOT TAX ADVICE. U.S. HOLDERS SHOULD CONSULT THEIR TAX ADVISORS REGARDING THE TAX CONSEQUENCES OF THE ACQUISTION, OWNERSHIP, AND DISPOSITION OF ORDINARY SHARES IN THEIR PARTICULAR CIRCUMSTANCES.
Distributions on Ordinary Shares
Subject to the PFIC rules discussed below under “—Passive Foreign Investment Company Rules,” distributions on Ordinary Shares generally will be taxable as a dividend for U.S. federal income tax purposes to the extent paid from our current or accumulated earnings and profits, as determined under U.S. federal income tax principles. Such distributions in excess of our current and accumulated earnings and profits will constitute a return of capital that will be applied against and reduce (but not below zero) the applicable U.S. Holder’s adjusted tax basis in its Ordinary Shares. Any remaining excess will be treated as gain realized on the sale or other taxable disposition of Ordinary Shares and will be treated as described below under “—Sale or Other Taxable Disposition of Ordinary Shares.” The amount of any such distributions will include any amounts required to be withheld by us (or another applicable withholding agent) in respect of any non-U.S. taxes. Any such amount treated as a dividend will be treated as foreign-source dividend income. Any such dividends received by a corporate U.S. Holder generally will not qualify for the dividends-received deduction generally allowed to U.S. corporations in respect of dividends received from other U.S. corporations. With respect to non-corporate U.S. Holders, any such dividends generally will be taxed at currently preferential long-term capital gains rates only if (i) Ordinary Shares are readily tradable on an established securities market in the United States or we are eligible for benefits under an applicable tax treaty with the United States, (ii) we are not treated as a PFIC with respect to the applicable U.S. Holder at the time the dividend was paid or in the preceding year, and (iii) certain holding period and other requirements are met. Any such dividends paid in a currency other than the U.S. dollar generally will be the U.S. dollar amount calculated by reference to the exchange rate in effect on the date of actual or constructive receipt, regardless of whether the payment is in fact converted into U.S. dollars at that time. A U.S. Holder may have foreign currency gain or loss if the dividend is converted into U.S. dollars after the date of actual or constructive receipt.
As noted above and subject to applicable limitations, taxing jurisdictions other than the United States may withhold taxes from distributions on Ordinary Shares, and a U.S. Holder may be eligible for a reduced rate of withholding to the extent there is an applicable tax treaty between the applicable taxing jurisdiction and the United States and/or may be eligible for a foreign tax credit against the U.S. Holder’s U.S. federal income tax liability. Recently issued U.S. Treasury regulations, which apply to foreign taxes paid or accrued in taxable years beginning on or after December 28, 2021, may in some circumstances prohibit a U.S. Holder from claiming a foreign tax credit with respect to certain foreign taxes that are not creditable under applicable tax treaties. In lieu of claiming a foreign tax credit, a U.S. Holder may, at such U.S. Holder’s election, deduct foreign taxes in computing such U.S. Holder’s taxable income, subject to generally applicable limitations under U.S. tax law. An election to deduct foreign taxes in lieu of claiming a foreign tax credit applies to all foreign taxes paid or accrued in the taxable year in which such election is made. The foreign tax credit rules are complex and U.S. Holders should consult their tax advisers regarding the application of such rules, including the creditability of foreign taxes, in their particular circumstances.
Sale or Other Taxable Disposition of Ordinary Shares
Subject to the PFIC rules discussed below under “—Passive Foreign Investment Company Rules,” upon any sale or other taxable disposition of Ordinary Shares, a U.S. Holder generally will recognize gain or loss in an amount equal to the difference, if any, between (i) the sum of (A) the amount of cash and (B) the fair market value of any other property received in such sale or disposition and (ii) the U.S. Holder’s adjusted tax basis in the Ordinary Shares. Any such gain or loss generally will be capital gain or loss and will be long-term capital gain or loss if the U.S. Holder’s holding period for such Ordinary Shares exceeds one year. Long-term capital gain recognized by non-corporate U.S. Holders generally will be taxed at currently preferential long-term capital gains rates. The deductibility of capital losses is subject to limitations. For foreign tax credit purposes, any such gain or loss generally will be treated as U.S. source gain or loss.
161
If the consideration received by a U.S. Holder upon a sale or other taxable disposition of Ordinary Shares is not paid in U.S. dollars, the amount realized will be the U.S. dollar value of such payment calculated by reference to the exchange rate in effect on the date of such sale or disposition. A U.S. Holder may have foreign currency gain or loss to the extent of the difference, if any, between (i) the U.S. dollar value of such payment on the date of such sale or disposition and (ii) the U.S. dollar value of such payment calculated by reference to the exchange rate in effect on the date of settlement.
U.S. Holders should consult their tax advisors regarding the tax consequences of a sale or other taxable disposition of Ordinary Shares, including the creditability of foreign taxes imposed on such sale or disposition by a taxing jurisdiction other than the United States, in their particular circumstances.
Passive Foreign Investment Company Rules
The U.S. federal income tax treatment of U.S. Holders could be materially different from that described above if we are treated as a PFIC for U.S. federal income tax purposes. A non-U.S. corporation generally will be treated as a PFIC for U.S. federal income tax purposes if either (i) at least 75.0% of its gross income in a taxable year, including its pro rata share of the gross income of any corporation in which it is considered to own at least 25.0% of the shares by value, is passive income or (ii) at least 50.0% of its assets in a taxable year (ordinarily determined based on fair market value and averaged quarterly over the year), including its pro rata share of the assets of any corporation in which it is considered to own at least 25.0% of the shares by value, are held for the production of, or produce, passive income. Passive income generally includes dividends, interest, rents and royalties (other than rents or royalties derived from the active conduct of a trade or business), and gains from the disposition of passive assets.
Based on our analysis of our income, assets, activities, and market capitalization, we believe that we were not a PFIC for our taxable year ended December 31, 2023. The determination of whether a non-U.S. corporation is a PFIC is a fact-intensive determination made on an annual basis and the applicable law is subject to varying interpretation. In particular, the characterization of our assets as active or passive may depend in part on our current and intended future business plans, which are subject to change. The amount of passive income and passive assets we take into account for PFIC testing purposes depends, in part, on the size of our cash balance (taking into account the timing and manner in which such cash is used) and the interest rates applicable thereto. In addition, the total value of our assets for PFIC testing purposes may be determined in part by reference to our market capitalization from time to time, which may fluctuate considerably. As a result, there can be no assurance with respect to our status as a PFIC for any taxable year, and our U.S. counsel expresses no opinion with respect to our PFIC status for any taxable year.
Although PFIC status is generally determined annually, if we are determined to be a PFIC for any taxable year (or portion thereof) that is included in the holding period of a U.S. Holder in its Ordinary Shares and the U.S. Holder did not make either a mark-to-market election or a qualified electing fund (“QEF”) election, which are referred to collectively as the “PFIC Elections” for purposes of this discussion, for the first taxable year in which we are treated as a PFIC, and in which the U.S. Holder held (or was deemed to hold) Ordinary Shares, or the U.S.
Holder does not otherwise make a purging election, as described below, the U.S. Holder generally will be subject to special and adverse rules with respect to (i) any gain recognized by the U.S. Holder on the sale or other taxable disposition of its Ordinary Shares and (ii) any “excess distribution” made to the U.S. Holder (generally, any distributions to the U.S. Holder during a taxable year of the U.S. Holder that are greater than 125.0% of the average annual distributions received by the U.S. Holder in respect of its Ordinary Shares during the three preceding taxable years of the U.S. Holder or, if shorter, the U.S. Holder’s holding period in its Ordinary Shares).
Under these rules:
162
PFIC Elections
If we are treated as a PFIC and Ordinary Shares constitute “marketable stock,” a U.S. Holder may avoid the adverse PFIC tax consequences discussed above if such U.S. Holder makes a mark-to-market election with respect to its Ordinary Shares for the first taxable year in which the U.S. Holder holds (or is deemed to hold) Ordinary Shares and each subsequent taxable year. Such U.S. Holder generally will include for each of its taxable years as ordinary income the excess, if any, of the fair market value of its Ordinary Shares at the end of such year over its adjusted tax basis in its Ordinary Shares. The U.S. Holder also will recognize an ordinary loss in respect of the excess, if any, of its adjusted tax basis in its Ordinary Shares over the fair market value of its Ordinary Shares at the end of its taxable year (but only to the extent of the net amount of previously included income as a result of the mark-to-market election). The U.S. Holder’s adjusted tax basis in its Ordinary Shares will be adjusted to reflect any such income or loss amounts, and any further gain recognized on a sale or other taxable disposition of its Ordinary Shares will be treated as ordinary income.
The mark-to-market election is available only for “marketable stock,” generally, stock that is regularly traded on a national securities exchange that is registered with the SEC, including the Nasdaq (on which Ordinary Shares are currently listed), or on a foreign exchange or market that the IRS determines has rules sufficient to ensure that the market price represents a legitimate and sound fair market value. As such, such election generally will not apply to any of our non-U.S. subsidiaries, unless the shares in such subsidiaries are themselves “marketable stock.” As such, U.S. Holders may continue to be subject to the adverse PFIC tax consequences discussed above with respect to any lower-tier PFICs, as discussed below, notwithstanding their mark-to-market election with respect to Ordinary Shares.
If made, a mark-to-market election would be effective for the taxable year for which the election was made and for all subsequent taxable years unless Ordinary Shares cease to qualify as “marketable stock” for purposes of the PFIC rules or the IRS consents to the revocation of the election. U.S. Holders should consult their tax advisors regarding the availability and tax consequences of a mark-to-market election with respect to Ordinary Shares in their particular circumstances.
If we are treated as a PFIC, a U.S. Holder may also avoid the adverse PFIC tax consequences discussed above with respect to Ordinary Shares if the U.S. Holder makes a valid QEF election for the first taxable year in which the U.S. Holder owns (or is treated as owning) Ordinary Shares.
If a U.S. Holder has made a QEF election with respect to Ordinary Shares, and the special tax and interest charge rules do not apply to such shares (because the QEF election was made in the U.S. Holder’s first taxable year in which the U.S. Holder owns (or is treated as owning) Ordinary Shares or a purging election was made, as described below), any gain recognized on the sale of Ordinary Shares will generally be taxable as capital gain and no interest charge will be imposed under the PFIC rules. U.S. Holders that make a QEF election with respect to Ordinary Shares are currently taxed on their pro rata shares of our earnings and profits, whether or not distributed. In such case, a subsequent distribution of such earnings and profits that were previously included in income should generally not be taxable as a dividend to such U.S. Holders. The tax basis of a U.S. Holder’s Ordinary Shares with respect to which a QEF election has been made will be increased by amounts that are included in taxable income, and decreased by amounts distributed but not taxed as dividends, under the above rules. Similar basis adjustments apply to property if by reason of holding such property the U.S. Holder is treated under the applicable attribution rules as owning Ordinary Shares with respect to which a QEF election has been made. A U.S. Holder generally can make a separate election to defer the payment of taxes on undistributed income inclusions under the QEF election rules, but if deferred, any such taxes will be subject to an interest charge.
In order to comply with the requirements of a QEF election with respect to Ordinary Shares, a U.S. Holder generally must receive a PFIC Annual Information Statement (as defined in Section 1.1295-1(g) of the Treasury Regulations) from us. If we are determined to be a PFIC for any taxable year, we will endeavor to make available to U.S. Holders a PFIC Annual Information Statement with respect to such taxable year. However, there is no assurance that we will have timely knowledge of our status as a PFIC in the future or that we will make available a PFIC Annual Information Statement.
163
U.S. Holders are urged to consult their tax advisors regarding the availability and tax consequences of a QEF election with respect to Ordinary Shares in their particular circumstances.
If we are treated as a PFIC and a U.S. Holder failed or was unable to timely make a PFIC Election for prior periods, the U.S. Holder might seek to make a purging election to rid its Ordinary Shares of the PFIC taint. Under the purging election, the U.S. Holder will be deemed to have sold its Ordinary Shares at their fair market value and any gain recognized on such deemed sale will be treated as an excess distribution, as described above. As a result of the purging election, the U.S. Holder will have a new adjusted tax basis and holding period in Ordinary Shares solely for purposes of the PFIC rules.
Related PFIC Rules
If we are treated as a PFIC and, at any time, have a non-U.S. subsidiary that is treated as a PFIC, a U.S. Holder generally would be deemed to own a proportionate amount of the shares of such lower-tier PFIC, and generally could incur liability for the deferred tax and interest charge described above if we receive a distribution from, or sell or otherwise dispose of all or part of our interest in, such lower-tier PFIC, or the U.S. Holder otherwise was deemed to have sold or otherwise disposed of an interest in such lower-tier PFIC. U.S. Holders should consult their tax advisors regarding the application of the lower-tier PFIC rules in their particular circumstances.
A U.S. Holder that owns (or is deemed to own) shares in a PFIC during any taxable year may have to file an IRS Form 8621 (whether or not a QEF election or a mark-to-market election is made) and to provide such other information as may be required by the U.S. Treasury Department. Failure to do so, if required, will extend the statute of limitations applicable to such U.S. Holder until such required information is furnished to the IRS and could result in penalties.
THE PFIC RULES ARE VERY COMPLEX AND U.S. HOLDERS SHOULD CONSULT THEIR TAX ADVISORS REGARDING THE APPLICATION OF SUCH RULES IN THEIR PARTICULAR CIRCUMSTANCES.
Information Reporting and Backup Withholding
Payments of dividends and sales proceeds that are made within the United States or through certain U.S.-related financial intermediaries are subject to information reporting, and may be subject to backup withholding, unless (i) the U.S. Holder is a corporation or other exempt recipient or (ii) in the case of backup withholding, the U.S. Holder provides a correct taxpayer identification number and certifies that it is not subject to backup withholding.
Backup withholding is not an additional tax. The amount of any backup withholding from a payment to a U.S. Holder will be allowed as a credit against the U.S. Holder’s U.S. federal income tax liability and may entitle the U.S. Holder to a refund, provided that the required information is timely furnished to the IRS.
U.S. Holders should consult their tax advisors regarding the information reporting requirements and the application of the backup withholding rules in their particular circumstances.
THIS DISCUSSION IS FOR GENERAL INFORMATIONAL PURPOSES ONLY AND IS NOT TAX ADVICE. U.S. HOLDERSSHOULD CONSULT THEIR TAX ADVISORS REGARDING THE U.S. FEDERAL, STATE, AND LOCAL AND NON-U.S. INCOME AND NON-INCOME TAX CONSEQUENCES OF THE ACQUISITION, OWNERSHIP, AND DISPOSITION OF ORDINARY SHARES, INCLUDING THE IMPACT OF ANY POTENTIAL CHANGE IN LAW, IN THEIR PARTICULAR CIRCUMSTANCES.
F. Dividends and Paying Agents
Not applicable.
G. Statement by Experts
Not applicable.
164
H. Documents on Display
We are subject to the information reporting requirements of the Exchange Act applicable to foreign private issuers and under those requirements will file reports with the SEC. Those reports may be inspected without charge at the locations described below. As a foreign private issuer, we are exempt from the rules under the Exchange Act related to the furnishing and content of proxy statements, and our officers, directors and principal shareholders are exempt from the reporting and short-swing profit recovery provisions contained in Section 16 of the Exchange Act.
In addition, we are not required under the Exchange Act to file periodic reports and financial statements with the SEC as frequently or as promptly as United States companies whose securities are registered under the Exchange Act. Nevertheless, we will file with the SEC an Annual Report on Form 20-F containing financial statements that have been examined and reported on, with and opinion expressed by an independent registered public accounting firm.
We maintain a corporate website at www.oculis.com. We intend to post our Annual Report on our website promptly following it being filed with the SEC. Information contained on, or that can be accessed through, our website does not constitute a part of this Annual Report. We have included our website address in this Annual Report solely as an inactive textual reference.
The SEC maintains a website (www.sec.gov) that contains reports, proxy and information statements and other information regarding registrants, such as us, that file electronically with the SEC.
With respect to references made in this Annual Report to any contract or other document of our company, such references are not necessarily complete, and you should refer to the exhibits attached or incorporated by reference to this Annual Report for copies of the actual contract or document.
I. Subsidiary Information
Not applicable.
J. Annual Report to Security Holders
We intend to submit an annual report provided to security holders in electronic format as an exhibit to a current report on Form 6-K.
Item 11. Quantitative and Qualitative Disclosures About Market Risk.
We are exposed to market risks that may result in changes of foreign currency exchange rates and interest rates, as well as the overall change in economic conditions in the countries where we conduct business.
The company takes a conservative approach to manage currency exchange risk by prioritizing long term stability and natural hedging of currencies with the underlying currency flow of operations. Other conservative measures include diversification of banks utilized by the Company and cash preservation in low risk short term investments.
For more information about financial risks we are exposed to, refer to Note 21 of our audited consolidated financial statements, included elsewhere in this Annual Report.
Item 12. Description of Securities Other than Equity Securities.
Not applicable.
Not applicable.
165
Not applicable.
Not applicable.
PART II
Item 13. Defaults, Dividend Arrearages and Delinquencies.
Not applicable.
Item 14. Material Modifications to the Rights of Security Holders and Use of Proceeds.
The net proceeds from our May 2023 offering have been used, and are expected to continue to be used, as described in our report on Form 6-K filed on May 30, 2023 and the final prospectus for such offering (File No. 333-272256), declared effective on May 31, 2023 and filed with the SEC on June 2, 2024.
Item 15. Controls and Procedures.
A. Disclosure Controls and Procedures
Our management evaluated, with the participation of the Chief Executive Officer and Chief Financial Officer, the effectiveness of the Company’s disclosure controls and procedures as of the end of the period covered by this report. The term "Disclosure Controls and Procedures," as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act means controls and other procedures of a company that are designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Exchange Act is recorded, processed, summarized and reported, within the time periods specified in the SEC's rules and forms. Disclosure controls and procedures include, without limitation, controls and procedures designed to provide reasonable assurance that the information is accumulated and communicated to management, including our principal executive and principal financial officers, as appropriate to allow timely decisions regarding our required disclosures. Based on the evaluation of our disclosure controls and procedures as of December 31, 2023, our Chief Executive Officer and Chief Financial Officer concluded that, as of such date, our disclosure controls and procedures were effective at a reasonable assurance level.
B. Management’s Annual Report on Internal Control Over Financial Reporting
Management is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Rule 13a-15(f) and 15d-15(f) under the Exchange Act.
Our management conducted an assessment of the effectiveness of our internal control over financial reporting based on the criteria set forth in “Internal Control-Integrated Framework (2013)” issued by the Committee of Sponsoring Organizations of the Treadway Commission ("COSO"). Based on this assessment, management concluded that, as of December 31, 2023, our internal control over financial reporting was effective based on criteria established in the COSO 2013 framework.
C. Attestation Report of the Registered Public Accounting Firm This Annual Report does not include an attestation report of the Company’s registered public accounting firm on management’s assessment of the Company’s internal control over financial reporting since we are an emerging growth company.
166
D. Changes in Internal Control Over Financial Reporting
Other than the remediation of the material weakness described below, there were no changes to internal control over financial reporting during the year ended December 31, 2023 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
Remediation of Previously Identified Material Weakness in Internal Control Over Financial Reporting
A material weakness is a deficiency, or a combination of deficiencies, in internal control over financial reporting such that there is a reasonable possibility that a material misstatement of a company’s annual or interim financial statements will not be prevented or detected on a timely basis by the Company’s internal controls. In connection with the preparation of our consolidated financial statements for the years ended December 31, 2022 and 2021, we identified material weaknesses in our internal control over financial reporting. One material weakness related to a lack of sufficient internal accounting personnel to support an efficient and structured financial statement close process and allow for the appropriate monitoring of financial reporting matters.
In connection with the preparation of our consolidated financial statements for the year ended December 31, 2023, management concluded that the previously-identified material weakness had been remediated. The remediation was accomplished through efforts undertaken by the Company to strengthen the organization, systems and processes of our accounting and finance function in 2022 and 2023. The remediation included the addition of internal accounting personnel and strengthening of external resources to support the Company’s financial statement preparation, implementation of various accounting systems and enhancement of reporting and control processes. Management has concluded that these efforts were sufficient to remediate the identified material weakness as of December 31, 2023 and that our controls over the components of the identified material weakness have been operating effectively for a sufficient period of time.
Item 16. [Reserved]
Item 16A. Audit Committee Financial Expert
Our Board has determined that Mr. Carnot (Chair) qualifies as an “audit committee financial expert” as defined by SEC rules and has the requisite financial sophistication under the applicable rules and regulations of the Nasdaq Stock Market. Mr. Carnot (Chair), Ms. Ackermann and Ms. O’Keeffe are independent as such term is defined in Rule 10A-3 under the Exchange Act and under the listing standards of the Nasdaq Stock Market.
Item 16B. Code of Ethics
Our board of directors adopted a Code of Business Conduct and Ethics applicable to the directors, executive officers and other team members, that complies with the rules and regulations of Nasdaq and the SEC. The Code of Business Conduct and Ethics is available on our website. In addition, we posted on the Corporate Governance section of our website all disclosures that are required by law or Nasdaq listing standards concerning any amendments to, or waivers from, any provision of the Code of Business Conduct and Ethics. The reference to our website address in this Annual Report on Form 20-F does not include or incorporate by reference the information on our website into this Annual Report on Form 20-F.
Item 16C. Principal Accountant Fees and Services
For the year ended December 31, 2023 and 2022, PricewaterhouseCoopers SA was our independent registered public accounting firm.
The following table shows the aggregate fees for services rendered by PwC to us and our subsidiaries, in the fiscal year ended December 31, 2023 and 2022.
167
|
|
For the years ended December 31, |
|
|||||
(in CHF thousands) |
|
2023 |
|
|
2022 |
|
||
Audit fees |
|
|
1,472 |
|
|
|
1,285 |
|
Audit-related fees |
|
|
12 |
|
|
|
5 |
|
Tax fees |
|
|
220 |
|
|
|
289 |
|
Other fees |
|
|
- |
|
|
|
- |
|
Total |
|
|
1,704 |
|
|
|
1,579 |
|
Auditor Name |
Auditor Location |
PricewaterhouseCoopers SA |
Pully, Switzerland |
Audit fees include fees billed for professional services rendered for audits of our annual consolidated financial statements, reviews of consolidated quarterly information, statutory audit of the Company and our subsidiaries, review of our securities offering documents in relation to the Business Combination, first registration statement in Nasdaq and IPO Follow-on transactions.
Audit-related fees include fees billed for assurance and related services in connection with capital increases and services that generally only the independent accountant can reasonably provide.
Tax fees include fees billed for professional services for tax compliance, tax advice and tax planning.
Audit Committee Pre-Approval Policies and Procedures
Our audit committee reviews and pre-approves the scope and the cost of audit services related to us and permissible non-audit services performed by the independent auditors. All of the services related to us provided by PricewaterhouseCoopers SA during the last fiscal year have been pre-approved by the audit committee.
Item 16D. Exemptions from the Listing Standards for Audit Committees.
Not applicable.
Item 16E. Purchases of Equity Securities by the Issuer and Affiliated Purchasers.
Not applicable.
Item 16F. Change in Registrant’s Certifying Accountant.
Not applicable.
Item 16G. Corporate Governance.
We are a “foreign private issuer,” as defined by the SEC. As a result, in accordance with Nasdaq rules, we will comply with home country governance requirements and certain exemptions thereunder rather than complying with Nasdaq corporate governance standards. While we expect to voluntarily follow most Nasdaq corporate governance rules, we may choose to take advantage of the following limited exemptions:
168
Furthermore, Nasdaq Rule 5615(a)(3) provides that a foreign private issuer, such as we, may rely on home country corporate governance practices in lieu of certain of the rules in the Nasdaq Rule 5600 Series and Rule 5250(d), provided that we nevertheless comply with Nasdaq’s Notification of Noncompliance requirement (Rule 5625), the Voting Rights requirement (Rule 5640) and that we have an audit committee that satisfies Rule 5605(c)(3), consisting of committee members that meet the independence requirements of Rule 5605(c)(2)(A)(ii). Although we are permitted to follow certain corporate governance rules that conform to Swiss requirements in lieu of many of the Nasdaq corporate governance rules, we intend to comply with the Nasdaq corporate governance rules applicable to foreign private issuers.
Accordingly, our shareholders will not have the same protections afforded to shareholders of companies that are subject to all of the corporate governance requirements of Nasdaq. We may utilize these exemptions for as long as we continue to qualify as a foreign private issuer. See the exhibit titled “Description of Securities” for additional information.
Item 16H. Mine Safety Disclosure.
Not applicable.
Item 16I. Disclosure Regarding Foreign Jurisdictions that Prevent Inspections.
Not applicable.
Item 16J. Insider Trading Policies.
Not applicable.
Item 16K. Cybersecurity.
Risk management and strategy
Cybersecurity and data privacy risks are evaluated through our annual risk management assessment. The Chief Business Officer oversees our cybersecurity risk management program, in partnership with a Cybersecurity Incident Management Team ("CSI Management Team"). The program has been developed to respond to the threat of security breaches and cyberattacks, and to protect and preserve the confidentiality, integrity, and continued availability of information owned by Oculis.
169
To address cybersecurity threats and prevent IT system interruptions, we have implemented a company-wide Cybersecurity Incident Response Policy that details the procedures to be followed in the event of a known or suspected incident. Depending on the environment, we implement and maintain various technical, physical, and organizational measures, processes, standards and policies designed to manage and mitigate material risks from cybersecurity threats to our information systems and data, including, for example access controls through multifactor authentication, regular back-ups of data and information, and cybersecurity awareness training of employees. We also have installed and regularly update antivirus software on all company-managed systems and computers to detect and prevent malicious code from impacting our systems. Where appropriate, any incidents would be escalated by the CSI Management Team to the audit committee of our board of directors, pursuant to our Cybersecurity Incident Response Policy. Oculis has not experienced any known material cybersecurity incidents during the years ended December 31, 2023, 2022 or 2021.
Our assessment and management of material risks from cybersecurity threats are integrated into the Company’s overall risk management processes. For example, all systems are evaluated by management to prioritize our risk management processes and mitigate cybersecurity threats that are more likely to lead to a material impact to our business. Furthermore, the Company is leveraging industry frameworks such as ISO 27001 and the SEC Cybersecurity Rules adopted in July 2023 to benchmark and work towards continues improvements of the Company’s cybersecurity practices.
We use third-party service providers to assist us from time to time to identify, assess, and manage material risks from cybersecurity threats, including for example by monitoring current information on system threats and vulnerabilities. We use third-party service providers to perform a variety of functions throughout our business, such as management of clinical studies, manufacturing and intellectual property management. Depending on the nature of the services provided, the sensitivity of the information systems and data at issue, and the identity of the provider, our vendor management process may involve different levels of assessment designed to help identify cybersecurity and data privacy risks associated with a provider and impose contractual obligations related to cybersecurity on the provider.
For a description of the risks from cybersecurity threats that may materially affect the Company and how they may do so, see our risk factors under Part 1. Item 3D. Risk Factors in this Annual Report on Form 20-F, including “Our business, financial condition and results of operations would suffer in the event of computer system failures, security breaches or other disruptions to our information technology systems.”
Governance
Our board of directors addresses our cybersecurity risk management as part of its general oversight function. The board of directors’ audit committee is responsible for overseeing our cybersecurity risk management processes, including oversight and mitigation of risks from cybersecurity threats.
Our cybersecurity risk assessment and management processes are implemented and maintained by certain Company management, including the Chief Business Officer and the Chief Financial Officer.
The Chief Business Officer, together with other company management, is responsible for hiring appropriate personnel, helping to integrate cybersecurity risk considerations into the Company’s overall risk management strategy, and communicating key priorities to relevant personnel. The Company’s board of directors and its audit committee is responsible for approving budgets, helping prepare for cybersecurity incidents, approving cybersecurity processes, and reviewing security assessments and other security-related reports.
Our Cybersecurity Incident Response Policy is designed to escalate certain cybersecurity incidents to members of management depending on the circumstances. The CSI Management Teams works on mitigating and remediating cybersecurity incidents of which they are notified. In addition, our Cybersecurity Incident Response Policy includes reporting to the audit committee of the board of directors for certain cybersecurity incidents.
The audit committee receives quarterly reports concerning any significant cybersecurity threats and risk and the processes we have implemented to address them. The audit committee also receives various reports, summaries or presentations related to cybersecurity threats, risk and mitigation.
170
PART III
Item 17. Financial Statements.
See pages F-1 through F-36 of this Annual Report.
Item 18. Financial Statements.
Not applicable.
Item 19. Exhibits
EXHIBIT INDEX
|
|
Incorporation By Reference |
|||
Exhibit |
Description |
Schedule/ Form |
File Number |
Exhibit |
File Date |
1.1* |
Amended and Restated Articles of Association of the Company. |
|
|
|
|
2.1 |
S-1 |
333-253220 |
4.3 |
03.04.2021 |
|
2.2 |
8-K |
001-40211 |
4.1 |
03.18.2021 |
|
2.3* |
|
|
|
|
|
4.1†+ |
Business Combination Agreement, dated as of October 17, 2022, by and among EBAC and Oculis |
8-K |
001-40211 |
2.1 |
10.17.2022 |
4.2†† |
License Agreement by and among Alcon Research, LTD., and Oculis, dated December 19, 2018 |
F-4 |
333-268201 |
10.8 |
12.12.2022 |
4.3†† |
F-4 |
333-268201 |
10.9 |
12.12.2022 |
|
4.4†† |
Letter Agreement by and among Novartis Technology LLC and Oculis, dated October 12, 2021 |
F-4 |
333-268201 |
10.11 |
12.12.2022 |
4.5†† |
License Agreement by and among Accure Therapeutics SL and Oculis, dated January 29, 2022 |
F-4 |
333-268201 |
10.12 |
12.12.2022 |
4.6 |
8-K |
001-40211 |
10.4 |
10.17.2022 |
|
4.7 |
Sponsor Support Agreement, dated as of October 17, 2022, by and among the Sponsor, EBAC and Oculis |
8-K |
001-40211 |
10.5 |
10.17.2022 |
171
4.8 |
Form of PIPE Subscription Agreement by and among EBAC and certain investors party thereto |
8-K |
001-40211 |
10.1 |
10.17.2022 |
4.9 |
Form of Convertible Loan Agreement by and among Oculis SA and certain shareholders party thereto |
8-K |
001-40211 |
10.2 |
10.17.2022 |
4.10 |
8-K |
001-40211 |
10.3 |
10.17.2022 |
|
4.11 |
20-F |
001-41636 |
4.11 |
03.07.2023 |
|
4.12# |
Stock Option and Incentive Plan Regulation 2023 of Oculis Holding AG |
F-4 |
333-268201 |
10.13 |
01.06.2023 |
4.13# |
Form of Indemnification Agreement with the officers and directors |
F-4 |
333-268201 |
10.10 |
01.06.2023 |
8.1* |
|
|
|
|
|
12.1* |
|
|
|
|
|
12.2* |
|
|
|
|
|
13.1** |
|
|
|
|
|
15.1* |
Consent of the Independent Registered Public Accounting Firm |
|
|
|
|
97.1* |
|
|
|
|
|
101.INS* |
Inline XBRL Instance Document (the instance document does not appear in the Interactive Data File because its XBRL tags are embedded within the Inline XBRL document) |
|
|
|
|
101.SCH* |
Inline XBRL Taxonomy Extension Schema Document |
|
|
|
|
172
104 |
Cover Page Interactive Data File (formatted as inline XBRL and contained in Exhibit 101) |
|
|
|
|
* Filed herewith.
** Furnished herewith.
† Certain schedules and exhibits to this Exhibit have been omitted pursuant to Company S-K Item 601(b)(2). The Registrant agrees to furnish supplementally a copy of any omitted schedule or exhibit to the SEC upon request.
†† Certain confidential portions (indicated by brackets and asterisks) have been omitted from this exhibit.
+ Certain schedules and exhibits to this Exhibit have been omitted pursuant to Regulation S-K Item 601(a)(5). The Company agrees to furnish supplementally a copy of any omitted schedule or exhibit to the SEC upon request.
# Indicates a management contract or any compensatory plan, contract or arrangement
SIGNATURES
The registrant hereby certifies that it meets all of the requirements for filing on Form 20-F and that it has duly caused and authorized the undersigned to sign this report on its behalf.
March 19, 2024
OCULIS HOLDING AG |
|
|
|
By: |
/s/ Riad Sherif |
|
|
Name: |
Riad Sherif |
|
|
Title: |
Chief Executive Officer |
173

Oculis Holding AG
Consolidated Financial Statements
Report of Independent Registered Public Accounting Firm
To the Board of Directors and Shareholders of Oculis Holding AG
Opinion on the Financial Statements
We have audited the accompanying consolidated statements of financial position of Oculis Holding AG and its subsidiaries (the “Company”) as of December 31, 2023 and 2022, and the related consolidated statements of loss, comprehensive loss, changes in equity and cash flows for each of the three years in the period ended December 31, 2023, including the related notes (collectively referred to as the “consolidated financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2023 and 2022, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2023 in conformity with IFRS Accounting Standards as issued by the International Accounting Standards Board.
Basis for Opinion
These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s consolidated financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits of these consolidated financial statements in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company's internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. We believe that our audits provide a reasonable basis for our opinion.
/s/ PricewaterhouseCoopers SA
Lausanne, Switzerland
March 19, 2024
We have served as the Company's auditor since 2019.
F-1
Oculis Holding AG, Zug
Consolidated Statements of Financial Position
(in CHF thousands)
|
|
|
|
As of December 31, |
|
|
As of December 31, |
|
||
|
|
Note |
|
2023 |
|
|
2022 |
|
||
ASSETS |
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
||
Non-current assets |
|
|
|
|
|
|
|
|
||
Property and equipment, net |
|
8 |
|
|
288 |
|
|
|
365 |
|
Intangible assets |
|
9 |
|
|
12,206 |
|
|
|
12,206 |
|
Right-of-use assets |
|
10 |
|
|
755 |
|
|
|
758 |
|
Other non-current assets |
|
|
|
|
89 |
|
|
|
74 |
|
Total non-current assets |
|
|
|
|
13,338 |
|
|
|
13,403 |
|
|
|
|
|
|
|
|
|
|
||
Current assets |
|
|
|
|
|
|
|
|
||
Other current assets |
|
11 |
|
|
8,488 |
|
|
|
2,959 |
|
Accrued income |
|
11 |
|
|
876 |
|
|
|
912 |
|
Short-term financial assets |
|
14 |
|
|
53,324 |
|
|
|
- |
|
Cash and cash equivalents |
|
14 |
|
|
38,327 |
|
|
|
19,786 |
|
Total current assets |
|
|
|
|
101,015 |
|
|
|
23,657 |
|
|
|
|
|
|
|
|
|
|
||
TOTAL ASSETS |
|
|
|
|
114,353 |
|
|
|
37,060 |
|
|
|
|
|
|
|
|
|
|
||
EQUITY AND LIABILITIES |
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
||
Shareholders' equity |
|
|
|
|
|
|
|
|
||
Share capital |
|
16 |
|
|
366 |
|
|
|
39 |
|
Share premium |
|
16 |
|
|
288,162 |
|
|
|
10,742 |
|
Reserve for share-based payment |
|
13 |
|
|
6,379 |
|
|
|
2,771 |
|
Actuarial loss on post-employment benefit obligations |
|
12 |
|
|
(1,072 |
) |
|
|
(264 |
) |
Treasury shares |
|
16 |
|
|
- |
|
|
|
(1 |
) |
Cumulative translation adjustments |
|
|
|
|
(327 |
) |
|
|
(300 |
) |
Accumulated losses |
|
|
|
|
(199,780 |
) |
|
|
(110,978 |
) |
Total equity |
|
|
|
|
93,728 |
|
|
|
(97,991 |
) |
|
|
|
|
|
|
|
|
|
||
Non-current liabilities |
|
|
|
|
|
|
|
|
||
Long-term lease liabilities |
|
10 |
|
|
431 |
|
|
|
491 |
|
Long-term financial debt |
|
15 |
|
|
- |
|
|
|
122,449 |
|
Long-term payables |
|
|
|
|
378 |
|
|
|
- |
|
Defined benefit pension liabilities |
|
12 |
|
|
728 |
|
|
|
91 |
|
Total non-current liabilities |
|
|
|
|
1,537 |
|
|
|
123,031 |
|
|
|
|
|
|
|
|
|
|
||
Current liabilities |
|
|
|
|
|
|
|
|
||
Trade payables |
|
|
|
|
7,596 |
|
|
|
3,867 |
|
Accrued expenses and other payables |
|
17 |
|
|
5,948 |
|
|
|
8,011 |
|
Short-term lease liabilities |
|
10 |
|
|
174 |
|
|
|
142 |
|
Warrant liabilities |
|
18 |
|
|
5,370 |
|
|
|
- |
|
Total current liabilities |
|
|
|
|
19,088 |
|
|
|
12,020 |
|
|
|
|
|
|
|
|
|
|
||
Total liabilities |
|
|
|
|
20,625 |
|
|
|
135,051 |
|
|
|
|
|
|
|
|
|
|
||
TOTAL EQUITY AND LIABILITIES |
|
|
|
|
114,353 |
|
|
|
37,060 |
|
The accompanying notes form an integral part of the consolidated financial statements.
F-2
Oculis Holding AG, Zug
Consolidated Statements of Loss
(in CHF thousands, except loss per share data)
|
|
|
|
For the years ended December 31, |
|
|||||||||
|
|
Note |
|
2023 |
|
|
2022 |
|
|
2021 |
|
|||
Grant income |
|
7. (A) / 11 |
|
|
883 |
|
|
|
912 |
|
|
|
960 |
|
Operating income |
|
|
|
|
883 |
|
|
|
912 |
|
|
|
960 |
|
Research and development expenses |
|
7. (B) |
|
|
(29,247 |
) |
|
|
(22,224 |
) |
|
|
(9,568 |
) |
General and administrative expenses |
|
7. (B) |
|
|
(17,487 |
) |
|
|
(11,064 |
) |
|
|
(4,624 |
) |
Merger and listing expense |
|
7. (B) |
|
|
(34,863 |
) |
|
|
- |
|
|
|
- |
|
Operating expenses |
|
|
|
|
(81,597 |
) |
|
|
(33,288 |
) |
|
|
(14,192 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|||
Operating loss |
|
|
|
|
(80,714 |
) |
|
|
(32,376 |
) |
|
|
(13,232 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|||
Finance income |
|
7. (C) |
|
|
1,429 |
|
|
|
126 |
|
|
|
21 |
|
Finance expense |
|
7. (C) |
|
|
(1,315 |
) |
|
|
(6,442 |
) |
|
|
(5,120 |
) |
Fair value adjustment on warrant liabilities |
|
7. (C) / 18 |
|
|
(3,431 |
) |
|
|
- |
|
|
|
- |
|
Foreign currency exchange (loss) gain |
|
7. (C) |
|
|
(4,664 |
) |
|
|
49 |
|
|
|
(193 |
) |
Finance result |
|
|
|
|
(7,981 |
) |
|
|
(6,267 |
) |
|
|
(5,292 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|||
Loss before tax for the period |
|
|
|
|
(88,695 |
) |
|
|
(38,643 |
) |
|
|
(18,524 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|||
Income tax expense |
|
7. (D) |
|
|
(107 |
) |
|
|
(55 |
) |
|
|
(27 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|||
Loss for the period |
|
|
|
|
(88,802 |
) |
|
|
(38,698 |
) |
|
|
(18,552 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|||
Loss per share: |
|
|
|
|
|
|
|
|
|
|
|
|||
Basic and diluted loss attributable to equity holders |
|
22 |
|
|
(2.97 |
) |
|
|
(11.32 |
) |
|
|
(5.84 |
) |
The accompanying notes form an integral part of the consolidated financial statements.
F-3
Oculis Holding AG, Zug
Consolidated Statements of Comprehensive Loss
(in CHF thousands)
|
|
|
|
For the years ended December 31, |
|
|||||||||
|
Note |
|
|
2023 |
|
|
2022 |
|
|
2021 |
|
|||
Loss for the period |
|
|
|
|
(88,802 |
) |
|
|
(38,698 |
) |
|
|
(18,552 |
) |
Other comprehensive loss |
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|||
Items that will not be reclassified to profit or loss: |
|
|
|
|
|
|
|
|
|
|
|
|||
Actuarial gains/(losses) of defined benefit plans |
12 |
|
|
|
(808 |
) |
|
|
744 |
|
|
|
88 |
|
Items that may be reclassified subsequently to profit or loss: |
|
|
|
|
|
|
|
|
|
|
|
|||
Foreign currency translation differences |
2. (D) |
|
|
|
(5,005 |
) |
|
|
3 |
|
|
|
(28 |
) |
Foreign currency translation differences recycling |
5 |
|
|
|
4,978 |
|
|
|
- |
|
|
|
- |
|
Other comprehensive profit/(loss) for the period |
|
|
|
|
(835 |
) |
|
|
747 |
|
|
|
60 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
Total comprehensive loss for the period |
|
|
|
|
(89,637 |
) |
|
|
(37,951 |
) |
|
|
(18,492 |
) |
The accompanying notes form an integral part of the consolidated financial statements.
F-4
Oculis Holding AG, Zug
Consolidated Statements of Changes in Equity
(in CHF thousands, except share numbers)
|
|
Legacy Oculis share capital |
|
Legacy Oculis treasury shares |
|
|
Oculis share capital |
|
|
|
|
|
|
|
|
|
|
|
|
||||||||
|
|
Note |
|
Shares |
|
Share capital |
|
Shares |
|
Treasury shares |
|
|
Shares |
|
Share capital |
|
Share premium |
|
Reserve for share-based payment |
|
Cumulative translation adjustment |
|
Actuarial loss on post-employment benefit obligations |
|
Accumulated losses |
|
Total |
Balance as of December 31, 2020 (as previously reported) |
|
|
|
2,967,155 |
|
297 |
|
(100,000) |
|
(100) |
|
|
- |
|
- |
|
9,609 |
|
1,640 |
|
(275) |
|
(1,096) |
|
(53,728) |
|
(43,654) |
Retroactive application of the recapitalization due to the business combination |
|
5 / 2 (B) / 16 |
|
424,985 |
|
(263) |
|
(14,323) |
|
99 |
|
|
- |
|
- |
|
164 |
|
- |
|
- |
|
- |
|
- |
|
- |
Balance as of January 1, (effect of the recapitalization) |
|
|
|
3,392,140 |
|
34 |
|
(114,323) |
|
(1) |
|
|
- |
|
- |
|
9,773 |
|
1,640 |
|
(275) |
|
(1,096) |
|
(53,728) |
|
(43,654) |
Loss for the period |
|
|
|
- |
|
- |
|
- |
|
- |
|
|
- |
|
- |
|
- |
|
- |
|
- |
|
- |
|
(18,552) |
|
(18,552) |
Other comprehensive profit/(loss): |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Actuarial gain on post-employment benefit obligations |
|
4. (C) / 12 |
|
- |
|
- |
|
- |
|
- |
|
|
- |
|
- |
|
- |
|
- |
|
- |
|
88 |
|
- |
|
88 |
Foreign currency translation differences |
|
2. (D) |
|
- |
|
- |
|
- |
|
- |
|
|
- |
|
- |
|
- |
|
- |
|
(28) |
|
- |
|
- |
|
(28) |
Total comprehensive loss for the period |
|
|
|
- |
|
- |
|
- |
|
- |
|
|
- |
|
- |
|
- |
|
- |
|
(28) |
|
88 |
|
(18,552) |
|
(18,492) |
Share-based compensation expense |
|
13 |
|
- |
|
- |
|
- |
|
- |
|
|
- |
|
- |
|
- |
|
328 |
|
- |
|
- |
|
- |
|
328 |
Restricted shares awards |
|
|
|
441,419 |
|
4 |
|
- |
|
- |
|
|
- |
|
- |
|
872 |
|
- |
|
- |
|
- |
|
- |
|
876 |
Transaction costs |
|
|
|
- |
|
- |
|
- |
|
- |
|
|
- |
|
- |
|
(12) |
|
- |
|
- |
|
- |
|
- |
|
(12) |
Balance as of December 31, 2021 (effect of the recapitalization) |
|
|
|
3,833,559 |
|
38 |
|
(114,323) |
|
(1) |
|
|
- |
|
- |
|
10,632 |
|
1,967 |
|
(303) |
|
(1,008) |
|
(72,280) |
|
(60,955) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance as of December 31, 2021 (as previously reported) |
|
|
|
3,353,271 |
|
335 |
|
(100,000) |
|
(100) |
|
|
- |
|
- |
|
10,434 |
|
1,967 |
|
(303) |
|
(1,008) |
|
(72,280) |
|
(60,955) |
Retroactive application of the recapitalization due to the business combination |
|
5 / 2 (B) / 16 |
|
480,288 |
|
(297) |
|
(14,323) |
|
99 |
|
|
- |
|
- |
|
198 |
|
- |
|
- |
|
- |
|
- |
|
- |
Balance as of January 1, 2022 (effect of the recapitalization) |
|
|
|
3,833,559 |
|
38 |
|
(114,323) |
|
(1) |
|
|
- |
|
- |
|
10,632 |
|
1,967 |
|
(303) |
|
(1,008) |
|
(72,280) |
|
(60,955) |
Loss for the period |
|
|
|
- |
|
- |
|
- |
|
- |
|
|
- |
|
- |
|
- |
|
- |
|
- |
|
- |
|
(38,698) |
|
(38,698) |
Other comprehensive profit/(loss): |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Actuarial gain on post-employment benefit obligations |
|
4. (C) / 12 |
|
- |
|
- |
|
- |
|
- |
|
|
- |
|
- |
|
- |
|
- |
|
- |
|
744 |
|
- |
|
744 |
Foreign currency translation differences |
|
2. (D) |
|
- |
|
- |
|
- |
|
- |
|
|
- |
|
- |
|
- |
|
- |
|
3 |
|
- |
|
- |
|
3 |
Total comprehensive loss for the period |
|
|
|
- |
|
- |
|
- |
|
- |
|
|
- |
|
- |
|
- |
|
- |
|
3 |
|
744 |
|
(38,698) |
|
(37,951) |
Share-based compensation expense |
|
13 |
|
- |
|
- |
|
- |
|
- |
|
|
- |
|
- |
|
- |
|
804 |
|
- |
|
- |
|
- |
|
804 |
Transaction costs |
|
|
|
- |
|
- |
|
- |
|
- |
|
|
- |
|
- |
|
(9) |
|
- |
|
- |
|
- |
|
- |
|
(9) |
Stock option exercised |
|
13 |
|
61,163 |
|
1 |
|
- |
|
- |
|
|
- |
|
- |
|
119 |
|
- |
|
- |
|
- |
|
- |
|
120 |
Balance as of December 31, 2022 (effect of the recapitalization) |
|
|
|
3,894,722 |
|
39 |
|
(114,323) |
|
(1) |
|
|
- |
|
- |
|
10,742 |
|
2,771 |
|
(300) |
|
(264) |
|
(110,978) |
|
(97,991) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance as of December 31, 2022 (as previously reported) |
|
|
|
3,406,771 |
|
340 |
|
(100,000) |
|
(100) |
|
|
- |
|
- |
|
10,540 |
|
2,771 |
|
(300) |
|
(264) |
|
(110,978) |
|
(97,991) |
Retroactive application of the recapitalization due to the business combination |
|
5 / 2 (B) / 16 |
|
487,951 |
|
(301) |
|
(14,323) |
|
99 |
|
|
- |
|
- |
|
202 |
|
- |
|
- |
|
- |
|
- |
|
- |
Balance as of January 1, 2023 (effect of the recapitalization) |
|
|
|
3,894,722 |
|
39 |
|
(114,323) |
|
(1) |
|
|
- |
|
- |
|
10,742 |
|
2,771 |
|
(300) |
|
(264) |
|
(110,978) |
|
(97,991) |
Loss for the period |
|
|
|
- |
|
- |
|
- |
|
- |
|
|
- |
|
- |
|
- |
|
- |
|
- |
|
- |
|
(88,802) |
|
(88,802) |
Other comprehensive profit/(loss): |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Actuarial loss on post-employment benefit obligations |
|
4. (C) / 12 |
|
- |
|
- |
|
- |
|
- |
|
|
- |
|
- |
|
- |
|
- |
|
- |
|
(808) |
|
- |
|
(808) |
Foreign currency translation differences |
|
2. (D) |
|
- |
|
- |
|
- |
|
- |
|
|
- |
|
- |
|
- |
|
- |
|
(5,005) |
|
- |
|
- |
|
(5,005) |
Foreign currency translation differences recycling |
|
5 |
|
- |
|
- |
|
- |
|
- |
|
|
- |
|
- |
|
- |
|
- |
|
4,978 |
|
- |
|
- |
|
4,978 |
Total comprehensive loss for the period |
|
|
|
- |
|
- |
|
- |
|
- |
|
|
- |
|
- |
|
- |
|
- |
|
(27) |
|
(808) |
|
(88,802) |
|
(89,637) |
Share-based compensation expense |
|
13 |
|
- |
|
- |
|
- |
|
- |
|
|
- |
|
- |
|
- |
|
3,608 |
|
- |
|
- |
|
- |
|
3,608 |
Conversion of Legacy Oculis ordinary shares and treasury shares into Oculis ordinary shares |
|
5 / 16 |
|
(3,894,722) |
|
(39) |
|
114,323 |
|
1 |
|
|
3,780,399 |
|
38 |
|
- |
|
- |
|
- |
|
- |
|
- |
|
- |
Conversion of Legacy Oculis long-term financial debt into Oculis ordinary shares |
|
5 / 15 / 16 |
|
- |
|
- |
|
- |
|
- |
|
|
16,496,603 |
|
165 |
|
124,637 |
|
- |
|
- |
|
- |
|
- |
|
124,802 |
Issuance of ordinary shares to PIPE investors |
|
5 / 16 |
|
- |
|
- |
|
- |
|
- |
|
|
7,118,891 |
|
71 |
|
66,983 |
|
- |
|
- |
|
- |
|
- |
|
67,054 |
Issuance of ordinary shares under CLA |
|
5 / 16 |
|
- |
|
- |
|
- |
|
- |
|
|
1,967,000 |
|
20 |
|
18,348 |
|
- |
|
- |
|
- |
|
- |
|
18,368 |
Issuance of ordinary shares to EBAC shareholders |
|
5 / 16 |
|
- |
|
- |
|
- |
|
- |
|
|
3,370,480 |
|
33 |
|
35,492 |
|
- |
|
- |
|
- |
|
- |
|
35,525 |
Transaction costs related to the business combination |
|
5 / 16 |
|
- |
|
- |
|
- |
|
- |
|
|
- |
|
- |
|
(4,821) |
|
- |
|
- |
|
- |
|
- |
|
(4,821) |
Proceeds from sale of shares in public offering |
|
5 / 16 |
|
- |
|
- |
|
- |
|
- |
|
|
3,654,234 |
|
36 |
|
38,143 |
|
- |
|
- |
|
- |
|
- |
|
38,179 |
Transaction costs related to the public offering |
|
5 / 16 |
|
- |
|
- |
|
- |
|
- |
|
|
- |
|
- |
|
(3,361) |
|
- |
|
- |
|
- |
|
- |
|
(3,361) |
Stock option exercised |
|
13 / 16 |
|
- |
|
- |
|
- |
|
- |
|
|
112,942 |
|
1 |
|
273 |
|
- |
|
- |
|
- |
|
- |
|
274 |
Issuance of shares in connection with warrant exercises |
|
16 / 18 |
|
- |
|
- |
|
- |
|
- |
|
|
149,156 |
|
2 |
|
1,726 |
|
- |
|
- |
|
- |
|
- |
|
1,728 |
Balance as of December 31, 2023 |
|
|
|
- |
|
- |
|
- |
|
- |
|
|
36,649,705 |
|
366 |
|
288,162 |
|
6,379 |
|
(327) |
|
(1,072) |
|
(199,780) |
|
93,728 |
The accompanying notes form an integral part of the consolidated financial statements.
F-5
Oculis Holding AG, Zug
Consolidated Statements of Cash Flows
(in CHF thousands)
|
|
|
|
For the years ended December 31, |
|
|||||||||
|
|
Note |
|
2023 |
|
|
2022 |
|
|
2021 |
|
|||
Operating activities |
|
|
|
|
|
|
|
|
|
|
|
|||
Loss before tax for the period |
|
|
|
|
(88,695 |
) |
|
|
(38,643 |
) |
|
|
(18,524 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|||
Non-cash adjustments: |
|
|
|
|
|
|
|
|
|
|
|
|||
- Financial result |
|
|
|
|
3,454 |
|
|
|
(500 |
) |
|
|
53 |
|
- Depreciation of property and equipment |
|
8 |
|
|
125 |
|
|
|
132 |
|
|
|
88 |
|
- Depreciation of right-of-use assets |
|
10 |
|
|
162 |
|
|
|
167 |
|
|
|
147 |
|
- Share-based compensation expense |
|
13 |
|
|
3,608 |
|
|
|
804 |
|
|
|
328 |
|
- Payroll expenses related to restricted stock |
|
13 / 16 |
|
|
- |
|
|
|
- |
|
|
|
876 |
|
- Interest expense on Series B and C preferred shares |
|
15 / 7.(C) |
|
|
1,266 |
|
|
|
6,343 |
|
|
|
4,996 |
|
- Interest on lease liabilities |
|
10 |
|
|
42 |
|
|
|
45 |
|
|
|
49 |
|
- Post-employment benefits |
|
12 |
|
|
(171 |
) |
|
|
(9 |
) |
|
|
(139 |
) |
- Non-realized foreign exchange differences |
|
15 / 7.(C) |
|
|
(30 |
) |
|
|
583 |
|
|
|
(792 |
) |
- Fair value adjustment on warrant liabilities |
|
18 |
|
|
3,431 |
|
|
|
- |
|
|
|
- |
|
- Merger and listing expense |
|
5 |
|
|
34,863 |
|
|
|
- |
|
|
|
- |
|
Working capital adjustments: |
|
|
|
|
|
|
|
|
|
|
|
|||
- De/(Increase) in other current assets |
|
11 |
|
|
(5,556 |
) |
|
|
(1,796 |
) |
|
|
(731 |
) |
- De/(Increase) in accrued income |
|
11 |
|
|
36 |
|
|
|
(152 |
) |
|
|
233 |
|
- Changes in receivables/payables from/to related parties |
|
|
|
|
- |
|
|
|
- |
|
|
|
29 |
|
- (De)/Increase in trade payables |
|
|
|
|
3,729 |
|
|
|
3,043 |
|
|
|
30 |
|
- (De)/Increase in accrued expenses and other payables |
|
17 |
|
|
(11,549 |
) |
|
|
4,903 |
|
|
|
(352 |
) |
- (De)/Increase in other operating assets/liabilities |
|
|
|
|
(29 |
) |
|
|
- |
|
|
|
- |
|
- (De)/Increase in long-term payables |
|
|
|
|
378 |
|
|
|
- |
|
|
|
- |
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
Interest received |
|
|
|
|
1,238 |
|
|
|
126 |
|
|
|
- |
|
Interest paid on lease liabilities |
|
|
|
|
(46 |
) |
|
|
(100 |
) |
|
|
(116 |
) |
Taxes paid |
|
|
|
|
(101 |
) |
|
|
(20 |
) |
|
|
- |
|
Net cash outflow from operating activities |
|
|
|
|
(53,845 |
) |
|
|
(25,074 |
) |
|
|
(13,825 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|||
Investing activities |
|
|
|
|
|
|
|
|
|
|
|
|||
Payment for purchase of property and equipment, net |
|
8 |
|
|
(48 |
) |
|
|
(65 |
) |
|
|
(28 |
) |
Payment for short-term financial assets, net |
|
14 |
|
|
(54,163 |
) |
|
|
- |
|
|
|
- |
|
Payment for purchase of intangible assets |
|
9 |
|
|
- |
|
|
|
(3,483 |
) |
|
|
- |
|
Net cash outflow from investing activities |
|
|
|
|
(54,211 |
) |
|
|
(3,548 |
) |
|
|
(28 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|||
Financing activities |
|
|
|
|
|
|
|
|
|
|
|
|||
Proceeds from the shares issued to PIPE investors |
|
5 |
|
|
67,054 |
|
|
|
- |
|
|
|
- |
|
Proceeds from the shares issued to CLA investors |
|
5 |
|
|
18,368 |
|
|
|
- |
|
|
|
- |
|
Proceeds from EBAC non-redeemed shareholders |
|
5 |
|
|
12,014 |
|
|
|
- |
|
|
|
- |
|
Transaction costs related to the business combination |
|
5 |
|
|
(4,607 |
) |
|
|
(214 |
) |
|
|
- |
|
Proceeds from sale of shares in public offering |
|
5 |
|
|
38,179 |
|
|
|
- |
|
|
|
- |
|
Transactions costs related to equity issuance in public offering |
|
5 |
|
|
(2,983 |
) |
|
|
- |
|
|
|
- |
|
Proceeds from exercises of warrants |
|
18 |
|
|
1,531 |
|
|
|
- |
|
|
|
- |
|
Proceeds from stock options exercised |
|
13 / 16 |
|
|
274 |
|
|
|
120 |
|
|
|
- |
|
Proceeds from issuance of preferred shares, classified as liabilities |
|
15 |
|
|
- |
|
|
|
2,030 |
|
|
|
56,096 |
|
Transaction costs for issuance of preferred shares, classified as liabilities/capital increase |
|
|
|
|
- |
|
|
|
(63 |
) |
|
|
(804 |
) |
Principal payment of lease obligations |
|
10 |
|
|
(158 |
) |
|
|
(159 |
) |
|
|
(98 |
) |
Net cash inflow from financing activities |
|
|
|
|
129,672 |
|
|
|
1,714 |
|
|
|
55,194 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
Increase/(Decrease) in cash and cash equivalents |
|
|
|
|
21,616 |
|
|
|
(26,909 |
) |
|
|
41,341 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
Cash and cash equivalents, beginning of period |
|
14 |
|
|
19,786 |
|
|
|
46,277 |
|
|
|
4,952 |
|
Effect of foreign exchange rate changes |
|
|
|
|
(3,075 |
) |
|
|
418 |
|
|
|
(16 |
) |
Cash and cash equivalents, end of period |
|
14 |
|
|
38,327 |
|
|
|
19,786 |
|
|
|
46,277 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
Net cash and cash equivalents variation |
|
|
|
|
21,616 |
|
|
|
(26,909 |
) |
|
|
41,341 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
Supplemental Non-Cash Financing Information |
|
|
|
|
|
|
|
|
|
|
|
|||
Transaction costs recorded in accrued expenses and other payables/trade payables |
|
|
|
|
378 |
|
|
|
356 |
|
|
|
- |
|
The accompanying notes form an integral part of the consolidated financial statements.
F-6
Oculis Holding AG, Zug
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
Oculis Holding AG ("Oculis" or "the Company") is a stock corporation ("Aktiengesellschaft") with its registered office at Bahnhofstrasse 7, CH-6300, Zug, Switzerland. It was incorporated under the laws of Switzerland on October 31, 2022.
The Company controls six wholly-owned subsidiaries: Oculis Operations GmbH ("Oculis Operations") with its registered office in Lausanne, Switzerland, which was incorporated in Zug, Switzerland on December 27, 2022, Oculis ehf ("Oculis Iceland"), which was incorporated in Reykjavik, Iceland on October 28, 2003, Oculis France Sàrl ("Oculis France") which was incorporated in Paris, France on March 27, 2020, Oculis US, Inc. ("Oculis US") with its registered office in Newton MA, USA, which was incorporated in Delaware, USA, on May 26, 2020, Oculis HK, Limited ("Oculis HK") which was incorporated in Hong Kong, China on June 1, 2021 and Oculis Merger Sub II Company ("Merger Sub 2") which was incorporated in the Cayman Islands on January 3, 2023 and is pending dissolution which will be completed in April 2024. The Company and its wholly-owned subsidiaries form the Oculis Group (the "Group"). Prior to the Business Combination (as defined in Note 5), Oculis SA ("Legacy Oculis"), which was incorporated in Lausanne, Switzerland on December 11, 2017, and its wholly-owned subsidiaries Oculis Iceland, Oculis France, Oculis U.S. and Oculis HK formed the Oculis group. On July 6, 2023, Legacy Oculis merged with and into Oculis Operations, and the separate corporate existence of Legacy Oculis ceased. Oculis Operations is the surviving company and remains a wholly-owned subsidiary of Oculis.
The purpose of the Company is the research, study, development, manufacture, promotion, sale and marketing of pharmaceutical products and substances as well as the purchase, sale and exploitation of intellectual property rights, such as patents and licenses, in the field of ophthalmology. As a global biopharmaceutical company, Oculis is developing treatments to save sight and improve eye care with breakthrough innovations. The Company’s differentiated pipeline includes candidates for topical retinal treatments, topical biologics and disease modifying treatments.
The consolidated financial statements of Oculis as of and for the year ended December 31, 2023, were approved and authorized for issue by the Company's Board of Directors on March 15, 2024.
The Group's accounts are prepared on a going concern basis. To date, the Group has financed its cash requirements primarily from share issuances, as well as government research and development grants. The recent business combination with European Biotech Acquisition Corp. (“EBAC”) and the listing in NASDAQ early in March 2023 raised additional funding to secure business continuity as explained under note 5. The Board of Directors believes that the Group has the ability to meet its financial obligations for at least the next 12 months.
The Company is a late clinical stage company and is exposed to all the risks inherent to establishing a business. Inherent to the Company’s business are various risks and uncertainties, including the substantial uncertainty as to whether current projects will succeed. The Company’s success may depend in part upon its ability to (i) establish and maintain a strong patent position and protection, (ii) enter into collaborations with partners in the biotech and pharmaceutical industry, (iii) successfully move its product candidates through clinical development, and (iv) attract and retain key personnel. The Company’s success is subject to its ability to be able to raise capital to support its operations. To date, the Company has financed its cash requirements primarily through the sale of its preferred stock, proceeds from the Business Combination, PIPE Financing and conversion of CLA and the sale of its common stock. Shareholders should note that the long-term viability of the Company is dependent on its ability to raise additional capital to finance its future operations. The Company will continue to evaluate additional funding through public or private financings, debt financing or collaboration agreements. The Company cannot be certain that additional funding will be available on acceptable terms, or at all. If the Company is unable to raise additional capital when required or on acceptable terms, it may have to (i) significantly delay, scale back or discontinue the development of one or more product candidates; (ii) seek collaborators for product candidates at an earlier stage than otherwise would be desirable and on terms that are less favorable than might otherwise be available; or (iii) relinquish or otherwise dispose of rights to product candidates that the Company would otherwise seek to develop itself, on unfavorable terms.
F-7
The consolidated financial statements of Oculis are prepared in accordance with IFRS Accounting Standards ("IFRS") as issued by the International Accounting Standards Board (“IASB”).
Prior to consummation of the Business Combination on March 2, 2023, the audited consolidated financial statements as of and for the year ended December 31, 2022 were issued for Legacy Oculis and its subsidiaries. Legacy Oculis became a wholly-owned subsidiary of the Company as a result of the Business Combination. In accordance with the BCA and described in Note 5, Oculis issued 3,780,399 ordinary shares to Legacy Oculis shareholders in exchange for 3,306,771 Legacy Oculis ordinary shares (after cancellation of 100,000 Legacy Oculis treasury shares) at the Exchange Ratio. The number of ordinary shares, and the number of ordinary shares within the loss per share held by the shareholders prior to the Business Combination have been adjusted by the Exchange Ratio to reflect the equivalent number of ordinary shares in the Company.
Reclassifications: given the immateriality of amounts recorded in financial assets and deferred income tax assets as of December 31, 2023 and 2022, these line items have been aggregated into Other non-current assets in the Consolidated Statements of Financial Position presented herein.
The policies set out below are consistently applied to all the years presented. The consolidated financial statements have been prepared under the historical cost convention, unless stated otherwise in the accounting policies in Note 3.
The totals are calculated with the original unit amounts, which could lead to rounding differences. These differences in thousands of units are not changed in order to keep the accuracy of the original data.
The consolidated financial statements of the Group are expressed in CHF, which is the Company's functional and presentation currency. The functional currency of the Company's subsidiaries is the local currency except for Oculis Iceland whose functional currency is CHF.
Assets and liabilities of foreign operations are translated into CHF at the rate of exchange prevailing at the reporting date and their statements of profit or loss are translated at yearly average exchange rates. The exchange differences arising on translation for consolidation are recognized in other comprehensive income.
The principal accounting policies adopted in the preparation of these financial statements are set out below. The policies set out below are consistently applied to all the years presented, unless otherwise stated.
The Company presents assets and liabilities in the balance sheet based on current/non-current classification. The Company classifies all amounts to be realized or settled within 12 months after the reporting period to be current and all other amounts to be non-current.
Foreign currency transactions are translated into the functional currency, Swiss Francs (CHF), using prevailing exchange rates at the dates of the transactions. Monetary assets and liabilities denominated in foreign currencies are translated into CHF at rates of exchange prevailing at reporting date. Any gains or losses from these translations are included in the statements of loss in the period in which they arise.
Oculis has six wholly owned subsidiaries, including Oculis Operations, Oculis Iceland, Oculis France, Oculis US, Oculis HK and Merger Sub 2. The Company's consolidated financial statements present the aggregate of the six Group entities, after elimination of intra-group transactions, balances, investments and capital.
F-8
The Company is managed and operated as one business. A single management team that reports to the Chief Executive Officer comprehensively manages the entire business and accordingly, has one reporting segment.
The Company has locations in five countries: Switzerland, Iceland, France, USA and Hong Kong. An analysis of non-current assets by geographic region is presented in Note 6.
All leases are accounted for by recognizing a right-of-use asset and a lease liability except for leases of low value assets and leases with a duration of 12 months or less.
Lease liabilities are measured at the present value of the expected contractual payments due to the lessor over the lease term, with the discount rate determined by reference to the rate inherent in the lease unless this is not readily determinable, in which case the Group’s incremental borrowing rate on commencement date of the lease is used. Variable lease payments are only included in the measurement of the lease liability if they depend on an index or rate and remain unchanged throughout the lease term. Other variable lease payments are expensed.
On initial recognition, the carrying value of the lease liability also includes amounts expected to be payable under any residual value guarantee, and the exercise price of any purchase option granted in favor of the group if it is reasonably certain to assess that option.
Right-of-use assets are initially measured at the amount of the lease liability, reduced for any lease incentives received, and increased for lease payments made at or before commencement of the lease and initial direct costs incurred.
Subsequent to the initial measurement, lease liabilities increase as a result of interest charged at a constant rate on the balance outstanding and are reduced for lease payments made. Right-of-use assets are depreciated on a straight-line basis over the remaining expected term of the lease or over the remaining economic life of the asset if this is judged to be shorter than the lease term.
When the Company revises its estimate of the term of any lease, it adjusts the carrying amount of the lease liability to reflect the expected payments over the revised term, which are discounted using a revised discount rate. The carrying value of lease liabilities is similarly revised if the variable future lease payments dependent on a rate or index is revised. In both cases, an equivalent adjustment is made to the carrying value of the right-of-use asset, with the revised carrying amount being amortized over the remaining lease term. If the carrying amount of the right-of-use asset is adjusted to zero, any further reduction is recognized in profit or loss.
Grant income is recognized where there is reasonable assurance that the grant will be received and all attaching conditions will be complied with, and in the year when the related expenses are incurred.
Taxes reported in the consolidated statements of loss include current and deferred taxes on profit. Taxes on income are accrued in the same periods as the revenues and expenses to which they relate.
Deferred tax is the tax attributable to the temporary differences that appear when taxation authorities recognize and measure assets and liabilities with rules that differ from those of the consolidated accounts. Deferred income tax is calculated using the liability method and determined using tax rates and laws that have been enacted or substantively enacted by the balance sheet date and are expected to apply when the related deferred income tax asset is realized, or the deferred income tax liability is settled. Any changes to the tax rates are recognized in the consolidated statements of loss unless related to items directly recognized in equity or other comprehensive loss.
Deferred income tax is recognized on temporary differences arising between the tax bases of assets and liabilities and their carrying amounts in the consolidated financial statements. Deferred income tax assets are recognized only to the extent that it is probable that future taxable profit will be available against which the temporary differences or the unused tax losses can be utilized. Deferred income tax assets from tax credit carry forwards are recognized to the extent that the national tax authority confirms the eligibility of such a claim and that the realization of the related tax benefit through future taxable profits is probable. Deferred income tax assets and liabilities are offset when there is a legally enforceable right to offset current tax assets against current tax liabilities and when the deferred income tax assets and liabilities relate to income taxes levied by the same taxation authority on either the same taxable entity or different taxable entities where there is an intention to settle the balances on a net basis.
F-9
The Company presents basic earnings / (loss) per share for each period in the financial statements. The earnings (loss) per share is calculated by dividing the earnings / (loss) of the period by the weighted average number of shares outstanding during the period. Diluted earnings per share, applicable in case of positive result, reflect the potential dilution that could occur if dilutive securities such as warrants or share options were exercised into common shares.
Judgment was required in determining the classification of the preferred shares issued by the Company as either equity or liabilities. The preferred shareholders hold certain preference rights that include preferential distribution of proceeds in the case of liquidity events as defined in the shareholder agreements. Under IAS 32 the Company classified the Preferred Shares as liabilities. This applied to Series A, B and C shares as per Note 15.
The Company considers all highly liquid investments with an original maturity of less than 3 months at the date of purchase to be cash equivalents. Cash and cash equivalents are recorded at cost, which approximates fair value.
Short-term financial assets consist of fixed term bank deposits with maturities between three and six months. Short-term financial assets are held in order to collect contractual cash flows made of payments of principal and interests.
Short-term financial assets are measured at amortized cost (approximates fair value) and are subsequently measured using the effective interest method. This method allocates interest income over the relevant period by applying the effective interest rate to the carrying amount of the asset. Gains and losses are recognized in the consolidated statements of loss when the asset is derecognized, modified or impaired.
The Company measures certain financial assets and liabilities at fair value on a recurring basis, including warrants. Fair value is the price the Company would receive to sell an asset or pay to transfer a liability in an orderly transaction with a market participant at the measurement date. The Company uses a three-level hierarchy that prioritizes fair value measurements based on the types of inputs used, as follows:
There was no change in the valuation techniques applied to financial instruments during all periods presented. There were no transfers between levels 1, 2 or 3 for recurring fair value measurements during the year. The Group recognizes transfers into and out of fair value hierarchy levels at the end of the reporting period.
All property and equipment are shown at cost, less subsequent depreciation and impairment. Cost includes expenditures that are directly attributable to the acquisition of the items. Subsequent costs are included in the asset’s carrying amount or recognized as a separate asset, as appropriate, only when it is probable that future economic benefits associated with the item will flow to the Group and the cost of the item can be measured reliably.
Depreciation is calculated on a straight-line basis over the useful life, according to the following schedule:
Category |
Useful life in years |
Laboratory equipment |
5 - 7 |
Laboratory fixtures and fittings |
10 |
Office - IT tools |
2 - 3 |
Office furniture and equipment |
5 |
The assets’ residual values and useful lives are reviewed, and adjusted if appropriate, at each balance sheet date. An asset’s carrying amount is impaired immediately to its recoverable amount if the asset’s carrying amount is greater than its estimated recoverable amount. Gains and losses on disposal or retirement of tangible fixed assets are determined by comparing the net proceeds received with the carrying amounts and are included in the consolidated statements of loss.
F-10
The Company recognizes the warrant instruments as liabilities at fair value and adjusts the instruments to fair value at each reporting period (refer to Note 18). Any change in fair value is recognized in the Company’s consolidated statements of loss. The fair value of the public warrants traded in active markets is based on the quoted market prices at the end of the reporting period for such warrants. Since the private placement warrants have identical terms to the public warrants, the Company determined that the fair value of each private placement warrant is equivalent to that of each public warrant. Public warrant instruments are included in Level 1 and private warrants in Level 2 in the fair value hierarchy. Warrants were classified as short-term liabilities given the Company cannot defer the settlement for at least 12 months.
Research expenditure is recognized in expense in the year in which it is incurred. Internal development expenditure is capitalized only if it meets the recognition criteria of IAS 38 “Intangible Assets”. Where regulatory and other uncertainties are such that the criteria are not met, which is almost invariably the case prior to approval of the drug by the relevant regulatory authority, the expenditure is recognized in the consolidated statements of loss. Where, however, recognition criteria are met, internal development expenditure is capitalized and amortized on a straight-line basis over its useful economic life. The amortization of the licenses will start when the market approval is obtained.
Licenses acquired are capitalized as intangible assets at historical cost and amortized over their useful lives, which are determined on a basis of the expected pattern of consumption of the expected future economic benefits embodied in the licenses and which therefore commence only once the necessary regulatory and marketing approval has been received. These licenses are tested for impairment in the last quarter of each financial period, or when there is any indication for impairment.
Amortization of capitalized licenses is charged to research and development expenses.
Impairment of capitalized licenses is charged to research and development expenses.
Assets that are subject to amortization are reviewed for impairment whenever events or changes in circumstances indicate that the carrying amount of the asset may not be recoverable.
An impairment loss is recognized for the amount by which the asset's carrying amount exceeds its recoverable amount. The recoverable amount is the higher of the asset's fair value less costs of disposal and value-in-use.
For the purpose of impairment testing, assets are grouped together into the smallest group of assets that generate cash inflows from continuing use that are largely independent of the cash flows of other assets ("cash-generating units"). Impairment losses are recognized in the consolidated statements of loss. Prior impairments of non-financial assets are reviewed for possible reversal of the impairment at each reporting date.
The principal financial instruments used by the Company are as follows:
F-11
Due to their short-term nature, the carrying value of cash and cash equivalents, short-term financial assets, other current assets, excluding prepaid expenses, accrued income, trade payables, accrued expenses and other payables approximates their fair value. For details of the fair value hierarchy, valuation techniques, and significant unobservable inputs related to determining the fair value of long-term financial debt, refer to Note 21.
The carrying amount of other receivables/current assets is reduced through the use of an allowance account, and the amount of the loss is recognized in the consolidated statements of loss. Subsequent recoveries of amounts previously written off are credited to the consolidated statements of loss.
Grant income reflects reimbursement of research and development expenses and income from certain research projects managed by Icelandic governmental institutions. Certain expenses qualify for incentives from the Icelandic government in the form of tax credits or cash reimbursements.
Short-term financial assets consist of fixed term bank deposits with maturities between three and six months. Short-term financial assets are held in order to collect contractual cash flows made of payments of principal and interests.
Cash and cash equivalents include cash on hand and highly liquid investments with original maturities of three months or less. These investments are readily convertible to known amounts of cash.
Trade payables are amounts due to third parties in the ordinary course of business. Trade payables are non-interest bearing and are normally settled on 45-day terms.
Accrued expenses and other payables are amounts provided for / due to third parties in the ordinary course of business. Accrued expenses and other payables are non-interest bearing.
Lease liabilities are measured at the present value of the expected contractual payments due to the lessor over the lease term, with the discount rate determined by reference to the rate inherent in the lease unless this is not readily determinable, in which case the Group’s incremental borrowing rate on commencement date of the lease is used.
Long-term financial debt exclusively results from the issuance of preferred shares that qualify as financial liabilities under IAS 32. Long-term financial debt is carried at amortized cost, plus the accrued interest/preferred dividend payments that are due by the Group under certain conditions. Refer to Note 15 for further information.
The Company operates a defined benefit pension plan for its Swiss-based employees, which is held in a multi-employer fund. The pension plan is funded by payments from employees and from the Company. The Company’s contributions to the defined contribution plans are charged to the consolidated statements of loss in the year to which they relate.
The liability recognized in the balance sheet in respect of defined benefit pension plans is the present value of the defined benefit obligation at the balance sheet date less the fair value of plan assets and the possible effect of the asset ceiling, together with adjustments for unrecognized past-service costs. The defined benefit obligation is calculated annually by independent actuaries using the projected unit credit method.
F-12
When the Company has a surplus in the defined benefit pension plans, it measures the net defined benefit asset at the lower of:
The Company does not expect any refunds or contribution reductions in case of a surplus in the defined benefit pension plan calculated per IAS 19, therefore no assets would be recognized in the Consolidated Statements of Financial Position.
The present value of the defined benefit obligation is determined by discounting the estimated future cash outflows using interest rates of high-quality corporate bonds that are denominated in the currency in which the benefits will be paid, and that have terms to maturity approximating to the terms of the related pension liability.
Actuarial gains and losses arising from experience adjustments and changes in actuarial assumptions are charged or credited to equity in other comprehensive income in the period in which they arise.
Past-service costs are recognized immediately in income, unless the changes to the pension plan are conditional on the employees remaining in service for a specified period of time (the vesting period). In this case, the past-service costs are amortized on a straight-line basis over the vesting period.
The Company operates an equity-settled, share-based compensation plan, under which the entity receives services from employees as consideration for equity instruments (e.g. options) of the Company. The fair value of the awards granted in exchange of the employee services received is recognized as an expense.
Non-market vesting conditions are included in assumptions about the number of options that are expected to vest. The total expense is recognized over the vesting period, which is the period over which all of the specified vesting conditions are to be satisfied. At the end of each reporting period, the entity revises its estimates of the number of options that are expected to vest based on the non-market vesting conditions. It recognizes the impact of the revision to original estimates, if any, in the consolidated statements of loss, with a corresponding adjustment to equity.
When the options are exercised, the Company issues new shares. The proceeds received net of any directly attributable transaction costs are credited to share capital (nominal value) and share premium when the options are exercised.
The Company recognizes the earnout consideration as a share-based contingent consideration within the scope of IFRS 2, and therefore equity classified as the earnout consideration ultimately settles in ordinary shares. The Company has determined that the fair value of the earnout shares should be accounted for as a component of the deemed cost of the listing services upon consummation of the Business Combination. The fair value of total consideration transferred included in the calculation of the IFRS 2 share listing service expense will not be subsequently adjusted regardless of whether the price target is achieved or not. The earnout options granted to employees were determined to be compensation for the dilution to their previously held Legacy Oculis equity instruments. No additional compensation charge is recognized under IFRS 2 because no additional fair value was granted as a result of the earnout options.
The Company capitalizes transaction costs within Other current assets in the Company’s consolidated balance sheet when costs are directly attributable to new equity financing instrument (including business combination related transactions) when it is highly probable that the financing transaction will take place in the future. If and when the Company completes the transaction, capitalized transaction costs will be offset against the proceeds and will be recorded as a reduction of share premium within the Company’s consolidated balance sheet. If the Company determines that it is not highly probable that the transaction will be completed, the Company will write-off capitalized transaction costs incurred during that respective quarter in the consolidated statement of loss.
There are no new IFRS standards, amendments or interpretations that are mandatory as of January 1, 2023 that are relevant to the Company. Additionally, the Company has not adopted any standard, interpretation or amendment that has been issued but is not yet effective.
F-13
Such standards are not currently expected to have a material impact on the entity in the current or future reporting periods and on foreseeable future transactions.
The Group’s principal accounting policies are set out in Note 3 of the Group’s consolidated financial statements and conform to IFRS Accounting Standards. Significant judgments and estimates are used in the preparation of the consolidated financial statements which, to the extent that actual outcomes and results may differ from these assumptions and estimates, could affect the accounting in the areas described in this section.
The Group assesses whether there are any indicators of impairment for all licenses at each reporting date, which refers exclusively to the licenses of two specific product candidates: OCS-02 (Licaminlimab) and OCS-05. Given the stage of Oculis’ development activities and the importance of both products in Oculis’ portfolio, the impairment test is performed first on the basis of a fair value model for the entire Company using a market approach, and second on the basis of the continued development feasibility of the relevant product candidate. Refer to Note 9.
Deferred income tax assets are recognized for all unused tax losses only to the extent that it is probable that taxable profits will be available against which the losses can be utilized. Judgment is required from management to determine the amount of tax asset that can be recognized, based on forecasts and tax planning strategies. Given the uncertainty in the realization of future taxable profits, no deferred tax asset on unused tax losses has been recognized as of December 31, 2023, 2022 and 2021. Refer to Note 7 (D).
The present value of the pension obligations depends on several factors that are determined on an actuarial basis using a number of assumptions. The assumptions used in determining the net cost (income) for pensions include the discount rate. Any changes in these assumptions will impact the carrying amount of pension obligations. The independent actuary of the Group uses statistical based assumptions covering future withdrawals of participants from the plan and estimates on life expectancy. The actuarial assumptions used may differ materially from actual results due to changes in market and economic conditions, higher or lower withdrawal rates or longer or shorter life spans of participants. These differences could have a significant impact on the amount of pension income or expenses recognized in future periods.
The Group determines the appropriate discount rate at the end of each year. This is the interest rate used to determine the present value of estimated future cash outflows expected to be required to settle the pension obligations. In determining the appropriate discount rate, the Group considers the interest rates of high-quality corporate bonds that are denominated in the currency in which the benefits will be paid, and that have terms to maturity approximating the terms of the related pension liability. Other key assumptions for pension obligations are based in part on current market conditions. Refer to Note 12.
(D) Share-based compensation
Stock options granted are valued using the Black-Scholes option-pricing model (see Note 13). This valuation model as well as parameters used such as expected volatility and expected term of the stock options are partially based on management’s estimates. The expected volatility is estimated using historical stock volatilities of comparable peer public companies within the Company's industry. The expected term represents the period that share-based awards are expected to be outstanding. The Company classifies its share-based payments as equity-classified awards as they are settled in ordinary shares. The Company measures equity-classified awards at their grant date fair value using a Black-Scholes option pricing model and does not subsequently remeasure them. Compensation costs related to equity-classified awards are equal to the fair value of the award at grant-date amortized over the vesting period of the award using the graded method. The Company reclassifies a portion of vested awards to reserve for share-based payment as the awards vest. The proceeds received net of any directly attributable transaction costs are credited to share capital (nominal value) and share premium when the options are exercised.
In relation to the Business Combination, the following critical estimates and judgments were made:
F-14
Despite EBAC being the legal acquirer, Legacy Oculis was determined to be the accounting acquirer for financial reporting purposes. This determination is primarily based on the fact that subsequent to the Business Combination, i) the shareholders of Legacy Oculis have a majority of the voting interest in the combined company; ii) Legacy Oculis’ operations comprise all of the ongoing operations of the combined company; and iii) Legacy Oculis’ management comprise all of the senior management of the combined company.
EBAC was a Special Purpose Acquisition Company and therefore does not meet the definition of a business under IFRS 3 as it has no operations and the related BCA cannot be treated as a business combination. The Business Combination was accounted for as a continuation of Legacy Oculis financial statements with a deemed issuance of shares by the Company accompanied by a recapitalization of the Company’s equity. The excess of fair value of the shares deemed issued by the Company over EBAC’s identifiable net assets has been recorded as share-based payment expense in accordance with IFRS 2 and represents a public listing service received by the Company.
Legacy Oculis and EBAC incurred costs such as legal, accounting, auditing, printer fees and other professional fees directly related to the Business Combination (“Transaction costs”). Transaction costs directly associated with equity issuance qualify for capitalization and are accounted for as a deduction of share premium. To capture costs associated with the new equity, the Company allocated capitalizable transaction costs to the various transaction components (equity issuance and listing) at the percentages of 38.0% and 62.0% for new shares and old shares, respectively.
Business combination with European Biotech Acquisition Corp (“EBAC”)
On March 2, 2023, the Company consummated a business combination with EBAC (the “Business Combination”) pursuant to the Business Combination Agreement ("BCA") between Legacy Oculis and EBAC dated as of October 17, 2022. The Company received gross proceeds of CHF 97.6 million or $103.7 million comprising CHF 12.0 million or $12.8 million of cash held in EBAC’s trust account and CHF 85.6 million or $90.9 million from private placement ("PIPE") investments and conversion of notes issued under Convertible Loan Agreements (“CLA”) into Oculis' ordinary shares. In connection with the Business Combination, Oculis was listed on the Nasdaq Global Market with the ticker symbol "OCS" for its ordinary shares and “OCSAW” for its public warrants.
Under the terms of the BCA, EBAC formed four new legal entities (i) Oculis, (ii) Oculis Merger Sub I Company ("Merger Sub 1"), (iii) Merger Sub 2, and (iv) Oculis Operations. After two consecutive mergers between Merger Sub 1 and EBAC, and EBAC and Merger Sub 2, EBAC and Merger Sub 1 ceased to exist and Merger Sub 2 was the surviving company. During the third quarter of 2023, the Company gave effect in its financial statements to the impending dissolution of Merger Sub 2, which is expected to be completed in the second quarter of 2024. As a result, the cumulative translation adjustments related to Merger Sub 2 previously reported as equity and recognized in other comprehensive income, were reclassified from equity to the Consolidated Statements of Loss for the year ended December 31, 2023. The resulting foreign exchange impact of such reclassification amounted to CHF 5.0 million for the year ended December 31, 2023.
As a result of the BCA and as of the acquisition closing date on March 2, 2023:
F-15
PIPE and CLA financing
In connection with the BCA, EBAC entered into subscription agreements with the PIPE investors for an aggregate of 7,118,891 shares of EBAC Class A ordinary shares at CHF 9.42 or $10.00 per share for aggregate gross proceeds of CHF 67.1 million or $71.2 million.
In connection with the BCA, Legacy Oculis and the investor parties thereto entered into CLAs pursuant to which the investor lenders granted Legacy Oculis a right to receive an interest free convertible loan with certain conversion rights with substantially the same terms as the PIPE investors. Following the mergers, Oculis assumed the CLAs and the lenders exercised their conversion rights in exchange for 1,967,000 ordinary shares at CHF 9.42 or $10.00 per share for aggregate gross proceeds of CHF 18.5 million or $19.7 million.
Together, the PIPE and CLA financing resulted in aggregate gross cash proceeds of CHF 85.6 million or $90.9 million to Oculis in exchange for 9,085,891 ordinary shares.
Merger and listing expense
The Business Combination is accounted for as a capital re-organization. As EBAC does not meet the definition of a business in accordance with IFRS 3 Business Combinations, the BCA is accounted for within the scope of IFRS 2 Share-based Payment.
The Business Combination is treated as the equivalent of the Company issuing shares for the net assets of EBAC as of the acquisition closing date, accompanied by a recapitalization. The net assets of EBAC are stated at historical cost, with no goodwill or other intangible assets recorded. Any excess of the fair value of the Company’s shares issued considering a fair value of CHF 10.54 or $11.19 per share (price of EBAC ordinary share at the closing date) over the value of EBAC’s identifiable net assets acquired represents compensation for the service of a stock exchange listing for its shares.
This expense was incurred in the first quarter of 2023 and amounted to CHF 34.9 million, which was expensed to the statement of loss as operating expenses, “Merger and listing expense”. The expense is non-recurring in nature and represents a share-based payment made in exchange for a listing service and does not lead to any cash outflows.
|
|
Per share value |
|
|
Shares |
|
|
March 2, 2023 |
|
|||
Fair value of equity consideration issued by the Company |
|
|
|
|
|
|
|
|
|
|||
EBAC public shareholders |
|
|
10.54 |
|
|
|
12,754,784 |
|
|
|
134,435 |
|
EBAC sponsor class B |
|
|
10.54 |
|
|
|
3,188,696 |
|
|
|
33,609 |
|
EBAC sponsor class A |
|
|
10.54 |
|
|
|
455,096 |
|
|
|
4,797 |
|
Redemptions of EBAC public shareholders |
|
|
10.54 |
|
|
|
(11,431,606 |
) |
|
|
(120,489 |
) |
Sponsors shares forfeiture |
|
|
10.54 |
|
|
|
(1,596,490 |
) |
|
|
(16,827 |
) |
Total consideration transferred |
|
|
|
|
|
3,370,480 |
|
|
|
35,525 |
|
|
Less net assets of EBAC |
|
|
|
|
|
|
|
|
(662 |
) |
||
Merger and listing expense |
|
|
|
|
|
|
|
|
34,863 |
|
||
F-16
|
|
March 2, 2023 |
|
|
|
|
(in CHF thousands) |
|
|
Net assets of EBAC |
|
|
|
|
Cash and cash equivalents |
|
|
11,547 |
|
Public & private warrants |
|
|
(2,136 |
) |
Deferred underwriting fee |
|
|
(3,108 |
) |
Accrued transaction costs |
|
|
(4,400 |
) |
Others |
|
|
(1,241 |
) |
Net assets of EBAC |
|
|
662 |
|
Capitalization
The following summarizes the actual ordinary shares issued and outstanding and the ownership interests of Oculis immediately after the Business Combination:
|
|
Shares |
|
|
% |
|
||
Issuance of ordinary shares to Legacy Oculis shareholders in connection with BCA (1) |
|
|
20,277,002 |
|
|
|
61.9 |
% |
Issuance of ordinary shares in connection with closing of the PIPE financing |
|
|
7,118,891 |
|
|
|
21.7 |
% |
Issuance of ordinary shares under CLA |
|
|
1,967,000 |
|
|
|
6.0 |
% |
Ordinary shares owned by sponsors |
|
|
2,047,302 |
|
|
|
6.3 |
% |
Ordinary shares owned by EBAC public shareholders |
|
|
1,323,178 |
|
|
|
4.1 |
% |
Total (2) |
|
|
32,733,373 |
|
|
|
100.0 |
% |
|
|
Legacy Oculis shares |
|
|
Exchange ratios |
|
|
Oculis ordinary shares issued |
|
|||
Ordinary shares |
|
|
3,406,771 |
|
|
|
|
|
|
|
||
Treasury shares cancelled |
|
|
(100,000 |
) |
|
|
|
|
|
|
||
Ordinary shares after cancellation of treasury shares |
|
|
3,306,771 |
|
|
1.1432 |
|
|
|
3,780,399 |
|
|
Preferred shares: |
|
|
|
|
|
|
|
|
|
|||
Series A |
|
|
1,623,793 |
|
|
|
1.1432 |
|
|
|
1,856,370 |
|
Series B1 |
|
|
2,486,188 |
|
|
|
1.4154 |
|
|
|
3,518,922 |
|
Series B2 T1 |
|
|
1,675,474 |
|
|
|
1.3900 |
|
|
|
2,328,872 |
|
Series B2 T2 |
|
|
426,378 |
|
|
|
1.3310 |
|
|
|
567,508 |
|
Series B2 T3 |
|
|
603,472 |
|
|
|
1.3142 |
|
|
|
793,082 |
|
Series C T1 |
|
|
5,337,777 |
|
|
|
1.2658 |
|
|
|
6,756,580 |
|
Series C T2 |
|
|
362,036 |
|
|
|
1.2205 |
|
|
|
441,854 |
|
Series C T3 |
|
|
197,745 |
|
|
|
1.1804 |
|
|
|
233,415 |
|
Total preferred shares |
|
|
12,712,863 |
|
|
|
1.2976 |
|
|
|
16,496,603 |
|
|
|
|
|
|
|
|
|
|
|
|||
Total |
|
|
16,019,634 |
|
|
|
|
|
|
20,277,002 |
|
|
F-17
Earnout consideration
As a result of the BCA, Legacy Oculis preferred, ordinary and option holders (collectively “equity holders”) received consideration in the form of 3,793,995 earnout shares and 369,737 earnout options with an exercise price of CHF 0.01.
The earnout consideration is subject to forfeiture in the event of a failure to achieve the price targets during the earnout period defined as follows: (i) 1,500,000, (ii) 1,500,000 and (iii) 1,000,000 earned based on the achievement of post-acquisition closing share price targets of Oculis of $15.00, $20.00 and $25.00, respectively, in each case, for any 20 trading days within any consecutive 30 trading day period commencing after the acquisition closing date and ending on or prior to March 2, 2028 (the “Earnout period”). A given share price target described above will also be deemed to be achieved if there is a change of control, as defined in the BCA, transaction of Oculis during the earnout period.
Public offering of ordinary shares
On May 31, 2023, the Company entered into an underwriting agreement with BofA Securities Inc. and SVB Securities, LLC, as representatives of several underwriters, and on June 5, 2023, closed the issuance and sale in a public offering of 3,500,000 ordinary shares at a public offering price of CHF 10.45 or $11.50 per share, for total gross proceeds of CHF 36.6 million or $40.3 million before deducting underwriting discounts, commissions and offering expenses.
In addition, the Company granted the underwriters an option to purchase additional ordinary shares which was partially exercised on June 13, 2023, leading to an additional purchase of 154,234 ordinary shares and gross proceeds of CHF 1.6 million or $1.7 million before deducting underwriting discounts, commissions and offering expenses. After giving issuance to these additional shares, Oculis sold a total of 3,654,234 ordinary shares in the offering for aggregate gross proceeds of CHF 38.2 million or $42.0 million, before deducting underwriting discounts, commissions and offering expenses. All of the underwriters' unexercised options to purchase additional shares expired on June 30, 2023.
The Company intends to use the net proceeds from this offering, together with its existing resources, to advance its development programs in particular Diabetic Macular Edema and for other ophthalmic indications, and for working capital and general corporate purposes.
The Company is managed and operated as one business. A single management team that reports to the Chief Executive Officer comprehensively manages the entire business and accordingly, the Company has one reportable segment.
The table below provides the carrying amount of certain non-current assets, by geographic area:
in CHF thousands |
|
Switzerland |
|
Iceland |
|
Others |
|
Total |
||||||||
|
|
As of December 31, 2023 |
|
As of December 31, 2022 |
|
As of December 31, 2023 |
|
As of December 31, 2022 |
|
As of December 31, 2023 |
|
As of December 31, 2022 |
|
As of December 31, 2023 |
|
As of December 31, 2022 |
Intangible assets |
|
12,206 |
|
12,206 |
|
- |
|
- |
|
- |
|
- |
|
12,206 |
|
12,206 |
Property and equipment, net |
|
17 |
|
24 |
|
253 |
|
338 |
|
18 |
|
3 |
|
288 |
|
365 |
Right-of-use assets |
|
- |
|
- |
|
687 |
|
758 |
|
68 |
|
- |
|
755 |
|
758 |
Total |
|
12,223 |
|
12,230 |
|
940 |
|
1,096 |
|
86 |
|
3 |
|
13,249 |
|
13,329 |
Grant income reflects reimbursement of research and development expenses and income from certain research projects managed by Icelandic governmental institutions. Certain expenses qualify for incentives from the Icelandic government in the form of tax credits or cash reimbursements. Icelandic government grant income for the year ended December 31, 2023, is CHF 0.9 million compared to CHF 0.9 million and CHF 1.0 million for the same periods in 2022 and 2021, respectively. Refer to Note 11.
The tables below show the breakdown of the Total operating expenses by category:
F-18
in CHF thousands |
|
For the years ended December 31, |
||||||||||||||||
|
|
Research and development expenses |
|
General and administrative expenses |
|
Total operating expenses |
||||||||||||
|
|
2023 |
|
2022 |
|
2021 |
|
2023 |
|
2022 |
|
2021 |
|
2023 |
|
2022 |
|
2021 |
Personnel expense |
|
6,509 |
|
4,608 |
|
4,407 |
|
7,029 |
|
4,449 |
|
2,416 |
|
13,538 |
|
9,056 |
|
6,823 |
Payroll |
|
4,796 |
|
4,313 |
|
4,189 |
|
5,134 |
|
3,939 |
|
2,306 |
|
9,930 |
|
8,252 |
|
6,495 |
Share-based compensation expense |
|
1,713 |
|
295 |
|
218 |
|
1,895 |
|
510 |
|
110 |
|
3,608 |
|
804 |
|
328 |
Operating expenses |
|
22,738 |
|
17,616 |
|
5,161 |
|
10,458 |
|
6,615 |
|
2,208 |
|
68,059 |
|
24,231 |
|
7,369 |
External service providers |
|
22,256 |
|
17,205 |
|
4,786 |
|
7,695 |
|
2,294 |
|
1,681 |
|
29,951 |
|
19,499 |
|
6,467 |
Other operating expenses |
|
258 |
|
184 |
|
189 |
|
2,700 |
|
4,249 |
|
478 |
|
2,958 |
|
4,433 |
|
667 |
Depreciation of property and equipment |
|
106 |
|
111 |
|
78 |
|
19 |
|
20 |
|
10 |
|
125 |
|
132 |
|
88 |
Depreciation of right-of-use assets |
|
118 |
|
116 |
|
108 |
|
44 |
|
52 |
|
39 |
|
162 |
|
167 |
|
147 |
Merger and listing expense(1) |
|
- |
|
- |
|
- |
|
- |
|
- |
|
- |
|
34,863 |
|
- |
|
- |
Total |
|
29,247 |
|
22,224 |
|
9,568 |
|
17,487 |
|
11,064 |
|
4,624 |
|
81,597 |
|
33,288 |
|
14,192 |
(1) Merger and listing expense is presented separately from research and development or general and administrative expenses on the consolidated statements of loss. The item relates to the BCA and is non-recurring in nature, representing a share-based payment made in exchange for a listing service.
in CHF thousands |
For the years ended December 31, |
|
|||||||||
|
2023 |
|
|
2022 |
|
|
2021 |
|
|||
Finance income |
|
1,429 |
|
|
|
126 |
|
|
|
21 |
|
Finance expense |
|
(1,315 |
) |
|
|
(6,442 |
) |
|
|
(5,120 |
) |
Fair value adjustment on warrant liabilities |
|
(3,431 |
) |
|
|
- |
|
|
|
- |
|
Foreign currency exchange gain (loss) |
|
(4,664 |
) |
|
|
49 |
|
|
|
(193 |
) |
Finance result |
|
(7,981 |
) |
|
|
(6,267 |
) |
|
|
(5,292 |
) |
Finance result in 2022 and 2021 represented mainly interest expense related to the preferred dividend owed to the holders of Legacy Oculis preferred Series B and C shares (refer to Note 15). Preferred Series B and C shares qualified as liabilities under IAS 32 and the related accrued dividends as interest expense. The preferred Series B and C shares were fully converted to ordinary shares at the closing of the Business Combination on March 2, 2023 (refer to Note 5).
Finance income consists primarily of interest income earned from the Company's short-term financial assets.
Refer to Note 18 for further discussions of the fair value gain/(loss) on warrant liabilities. The foreign currency translation differences recycling is related to the Merger Sub 2 entity and its impending dissolution, discussed further in Note 5.
Financial result as presented in the statements of cash flows is comprised of interest and the foreign exchange effect on cash and financial assets, net.
|
For the years ended December 31, |
|
||||||||
in CHF thousands |
2023 |
|
|
2022 |
|
2021 |
|
|||
Current income tax expense |
|
(127 |
) |
|
|
(90 |
) |
|
(22 |
) |
Deferred tax income (expense) |
|
20 |
|
|
|
35 |
|
|
(5 |
) |
Total tax expense |
|
(107 |
) |
|
|
(55 |
) |
|
(27 |
) |
The Group's expected tax expense for each year is based on the applicable tax rate in each individual jurisdiction, which ranged between approximately 8.3% and 25.0% for 2023, 2022 and 2021 in the tax jurisdictions in which the Group operates. The weighted average tax rates applicable to the profits of the consolidated entities were 12.7%, 13.9% and 13.6% for the years 2023, 2022 and 2021, respectively. This decrease is due to changes in the mix of the taxable results and the changes in tax rates of the individual group companies. The tax on the Group's profit / (loss) before tax differs from the statutory amount that would arise using the weighted average applicable tax rate as follows:
F-19
|
For the years ended December 31, |
|
||||||||
in CHF thousands |
2023 |
|
|
2022 |
|
2021 |
|
|||
Groups average expected tax rate |
|
12.7 |
% |
|
|
13.9 |
% |
|
13.6 |
% |
Accounting loss before income tax |
|
(88,695 |
) |
|
|
(38,643 |
) |
|
(18,524 |
) |
Taxes at weighted average income tax |
|
11,294 |
|
|
|
5,380 |
|
|
2,521 |
|
Effect of unrecorded tax losses |
|
(10,520 |
) |
|
|
(4,468 |
) |
|
(1,869 |
) |
Effect of non-deductible expenses |
|
(6,041 |
) |
|
|
(968 |
) |
|
(679 |
) |
Effect of non-taxable income |
|
5,103 |
|
|
|
- |
|
|
- |
|
Effect of other items |
|
57 |
|
|
|
- |
|
|
- |
|
Total tax expense |
|
(107 |
) |
|
|
(55 |
) |
|
(27 |
) |
As of December 31, 2023, 2022 and 2021, the Group has tax losses which arose mainly in Switzerland that are available for offset against future taxable profits of the company until expiration. Deferred tax assets have not been recognized in respect of these losses in Switzerland as it is not probable that future taxable profit will be available against which the unused tax losses can be utilized. Given the amount of tax losses has not been yet validated by the Tax Authorities, there could be potentially still be subject to material adjustments.
This does not affect the management assumption on the going concern hypothesis of the Group. Below is the maturity of the Group reportable losses:
|
As of December 31, |
|
|||||
in CHF thousands |
2023 |
|
|
2022 |
|
||
2025 |
|
16,733 |
|
|
|
16,733 |
|
2026 |
|
13,113 |
|
|
|
13,113 |
|
2027 |
|
12,437 |
|
|
|
12,437 |
|
2028 |
|
14,865 |
|
|
|
14,865 |
|
2029 |
|
31,786 |
|
|
|
31,790 |
|
2030 |
|
81,509 |
|
|
|
- |
|
Total |
|
170,443 |
|
|
|
88,938 |
|
The Group did not recognize the following temporary differences:
|
As of December 31, |
|
|||||
in CHF thousands |
2023 |
|
|
2022 |
|
||
Pension |
|
728 |
|
|
|
91 |
|
Tax losses in Switzerland |
|
170,443 |
|
|
|
88,938 |
|
Leasing |
|
(150 |
) |
|
|
(125 |
) |
Intangible asset |
|
(4,025 |
) |
|
|
(4,025 |
) |
Total |
|
166,996 |
|
|
|
84,879 |
|
As of December 31, 2023 and 2022 the Company had recognized deferred tax assets of CHF 44 thousand and CHF 24 thousand, respectively, and no deferred tax liabilities.
The following tables present the movements in the book values of property and equipment, net:
in CHF thousands |
Lab - equipment |
|
|
Lab - fixtures and fittings |
|
|
Office equipment & hardware |
|
|
Total |
|
||||
Acquisition cost: |
|
|
|
|
|
|
|
|
|
|
|
||||
Balance as of December 31, 2021 |
|
555 |
|
|
|
195 |
|
|
|
101 |
|
|
|
851 |
|
Acquisitions |
|
45 |
|
|
|
- |
|
|
|
20 |
|
|
|
65 |
|
Balance as of December 31, 2022 |
|
600 |
|
|
|
195 |
|
|
|
121 |
|
|
|
916 |
|
Acquisitions |
|
18 |
|
|
|
- |
|
|
|
30 |
|
|
|
48 |
|
Balance as of December 31, 2023 |
|
618 |
|
|
|
195 |
|
|
|
151 |
|
|
|
964 |
|
F-20
|
Lab - equipment |
|
|
Lab - fixtures and fittings |
|
|
Office equipment & IT tools |
|
|
Total |
|
||||
Accumulated depreciation: |
|
|
|
|
|
|
|
|
|
|
|
||||
Balance as of December 31, 2021 |
|
(305 |
) |
|
|
(59 |
) |
|
|
(55 |
) |
|
|
(420 |
) |
Depreciation expense |
|
(70 |
) |
|
|
(28 |
) |
|
|
(34 |
) |
|
|
(132 |
) |
Balance as of December 31, 2022 |
|
(375 |
) |
|
|
(87 |
) |
|
|
(89 |
) |
|
|
(551 |
) |
Depreciation expense |
|
(89 |
) |
|
|
(19 |
) |
|
|
(17 |
) |
|
|
(125 |
) |
Balance as of December 31, 2023 |
|
(464 |
) |
|
|
(106 |
) |
|
|
(106 |
) |
|
|
(676 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
||||
Carrying amount: |
|
|
|
|
|
|
|
|
|
|
|
||||
Balance as of December 31, 2022 |
|
225 |
|
|
|
108 |
|
|
|
32 |
|
|
|
365 |
|
Balance as of December 31, 2023 |
|
154 |
|
|
|
89 |
|
|
|
45 |
|
|
|
288 |
|
The following tables summarize the movement of intangibles assets:
in CHF thousands |
Licenses |
|
|
Total |
|
||
Acquisition cost: |
|
|
|
|
|
||
Balance as of December 31, 2021 |
|
8,724 |
|
|
|
8,724 |
|
Additions |
|
3,482 |
|
|
|
3,482 |
|
Balance as of December 31, 2022 |
|
12,206 |
|
|
|
12,206 |
|
Additions |
|
- |
|
|
|
- |
|
Balance as of December 31, 2023 |
|
12,206 |
|
|
|
12,206 |
|
|
|
|
|
|
|
||
Carrying amount: |
|
|
|
|
|
||
As of December 31, 2022 |
|
12,206 |
|
|
|
12,206 |
|
As of December 31, 2023 |
|
12,206 |
|
|
|
12,206 |
|
Intangible assets as of December 31, 2023 and 2022 were CHF 12.2 million and represent licenses purchased under license agreements with Novartis and Accure. Intangible assets as of December 31, 2021 were CHF 8.7 million and represented licenses purchased under a license agreement with Novartis. The Novartis license agreement was dated as of December 19, 2018 between Legacy Oculis and Novartis and relates to a novel topical anti-TNFα antibody, renamed OCS-02 (Licaminlimab), for ophthalmic indications. The license agreement between Legacy Oculis and Accure, dated as of January 29, 2022, relates to the exclusive global licensing of its OCS-05 (formerly ACT-01) technology. This license agreement contained an upfront payment of CHF 3.0 million and reimbursement of development related costs of CHF 0.5 million. The Company intends to advance the development of OCS-05 with a focus on multiple ophthalmology neuroprotective applications.
The products candidates related to the capitalized intangible assets are not yet available for use. The amortization of the licenses will start when the market approval is obtained.
Oculis performs an assessment of its licenses in the context of its annual impairment test. Given the stage of Oculis’ development activities and the importance of the relevant product candidates, OCS-02 (Licaminlimab) and OCS-05, in Oculis’ portfolio, the impairment test is performed first on the basis of a fair value model for the entire Company using a market approach and second on the basis of the continued development feasibility of both candidates.
Oculis performs its annual impairment tests on its entire portfolio of research and development assets, by deriving the fair value from an observable valuation for the entire Company determined via its stock market price quoted in Nasdaq as per the reporting date. The fair value of the asset portfolio is derived by deducting the carrying value of tangible assets and the remaining assets, which consist primarily of short-term financial assets and cash and cash equivalents, from the Company valuation.
OCS-02 and OCS-05, are additionally tested for impairment by assessing their probability of success. Assessments include reviews of the following indicators, and if the candidate fails any of these indicators the entire balance is written off:
F-21
In 2023, 2022 and 2021, reviews of all these indicators for OCS-02 and OCS-05 (in 2023 and 2022) was positive. No impairment losses were recognized in 2023, 2022 and 2021.
The following table presents the right-of-use assets, which are related to our Iceland and U.S. facilities:
in CHF thousands |
Right-of-use assets |
|
||||
|
2023 |
|
2022 |
|
||
Balance as of January 1, |
|
758 |
|
|
855 |
|
Indexation for the period |
|
47 |
|
|
70 |
|
New lease |
|
118 |
|
|
- |
|
FX revaluation |
|
(6 |
) |
|
- |
|
Depreciation charge for the period |
|
(162 |
) |
|
(167 |
) |
Balance as of December 31, |
|
755 |
|
|
758 |
|
There are no variable lease payments which are not included in the measurement of lease obligations. Expected extension options have been included in the measurement of lease liabilities.
The following table presents the lease obligations:
in CHF thousands |
Lease liabilities |
|
||||
|
2023 |
|
2022 |
|
||
Balance as of January 1, |
|
(633 |
) |
|
(770 |
) |
New lease |
|
(118 |
) |
|
- |
|
FX revaluation |
|
35 |
|
|
48 |
|
Indexation for the period |
|
(47 |
) |
|
(70 |
) |
Interest expense for the period |
|
(42 |
) |
|
(45 |
) |
Lease payments for the period |
|
200 |
|
|
204 |
|
Balance as of December 31, |
|
(605 |
) |
|
(633 |
) |
in CHF thousands |
As of December 31, 2023 |
|
As of December 31, 2022 |
|
||
Current |
|
(174 |
) |
|
(142 |
) |
Non-current |
|
(431 |
) |
|
(491 |
) |
Total |
|
(605 |
) |
|
(633 |
) |
The table below shows the breakdown of the Other current assets by category:
in CHF thousands |
|
As of December 31, 2023 |
|
|
As of December 31, 2022 |
|
||
Prepaid clinical and technical development expenses |
|
|
6,748 |
|
|
|
1,586 |
|
Prepaid general and administrative expenses |
|
|
1,412 |
|
|
|
1,208 |
|
VAT receivable |
|
|
328 |
|
|
|
165 |
|
Total |
|
|
8,488 |
|
|
|
2,959 |
|
The increase in prepaid clinical and technical development expenses as of December 31, 2023 compared to prior year relates to the commencement of significant clinical trials in the fourth quarter of 2023.
The table below shows the movement of the accrued income for the years ended December 31, 2023 and 2022:
F-22
in CHF thousands |
|
2023 |
|
|
2022 |
|
||
Balance as of January 1, |
|
|
912 |
|
|
|
760 |
|
Accrued income recognized during the period |
|
|
883 |
|
|
|
912 |
|
Payments received during the period |
|
|
(915 |
) |
|
|
(726 |
) |
Foreign exchange revaluation |
|
|
(4 |
) |
|
|
(34 |
) |
Balance as of December 31, |
|
|
876 |
|
|
|
912 |
|
Accrued income is generated by incentives for research and development offered by the Icelandic government in the form of tax credits for innovation companies. The aid in Iceland is granted as a reimbursement of paid income tax or paid out in cash when the tax credit is higher than the calculated income tax. The tax credit is subject to companies having a research project approved as eligible for tax credit by the Icelandic Centre for Research (Rannís).
The Company’s Swiss pension plan is classified as a defined benefit plan under IFRS Accounting Standards. Employees of the Icelandic, French, Hong Kong and American subsidiaries are covered by local post-retirement defined contribution plans.
Pension costs are charged to the consolidated statements of loss when incurred. Iceland pension expenses of CHF 0.1 million were recorded in 2023, 2022 and 2021.
Pension costs are charged to the consolidated statements of loss when incurred. In 2023, pension costs amounted to CHF 41 thousand, CHF 42 thousand in 2022 and CHF 47 thousand in 2021.
The U.S. entity adopted a 401(k) defined contribution plan effective December 1, 2020. Accrued employer contribution was CHF 48 thousand for 2023. There were no employer contributions made in 2022 and 2021.
Pension costs are charged to the consolidated statements of loss when incurred. CHF 3 thousand and CHF 4 thousand were recorded related to Hong Kong pension expenses in 2023 and 2022. The subsidiary in Hong Kong did not employ any personnel in 2021. Consequently, there was no pension expense in 2021.
The Company’s Swiss entity is affiliated to a collective foundation administrating the pension plans of various unrelated employers that qualifies as defined benefit plan under IAS 19. For employees in Switzerland, the pension fund provides post-employment, death-in-service and disability benefits in accordance with the Swiss Federal Law on Occupational Retirement, Survivor’s and Disability Pension Plans which specifies the minimum benefits that are to be provided.
The pension plan of the Company’s Swiss entity is fully segregated from the ones of other participating employers. The collective foundation has reinsured all risks with an insurance company. The most senior governing body of the collective foundation is the Board of Trustees. All governing and administration bodies have an obligation to act in the interests of the plan beneficiaries.
The retirement benefits are based on the accumulated retirement capital, which is made of the yearly contributions towards the old age risk by both employer and employee and the interest thereon until retirement. The employee contributions are determined based on the insured salary, depending on the age, staff level and saving amount of the beneficiary. The interest rate is determined annually by the governing body of the collective plan in accordance with the legal framework, which defines the minimum interest rates.
If an employee leaves the pension plan before reaching retirement age, the law provides for the transfer of the vested benefits to a new pension plan. These vested benefits comprise the employee and the employer contributions plus interest, the money originally brought into the pension plan by the beneficiary and an additional legally stipulated amount. On reaching retirement age, the plan beneficiary may decide whether to withdraw the benefits in the form of an annuity or (entirely or partly) as a lump-sum payment. The annuity is calculated by multiplying the balance of the retirement capital with the applicable conversion rate.
F-23
All actuarial risks of the plan, e.g. old age, invalidity and death-in-service or investment, are fully covered by insurance. However, the collective foundation is able to withdraw from the contract with the Company at any time, in which case the Company would be required to join another pension plan. In addition, the risk premiums may be adjusted by the insurance company periodically.
The Company's Swiss pension plan is fully reinsured with Swiss Life (“Swiss Life Business Protect”), therefore the plan assets are 100% covered by an insurance contract. The insurance company bearing the investment risk is also making these investments on behalf of the collective foundation. As a result, the assets of the plan consist of a receivable from the insurance police.
The assets are invested by the pension plan, to which many companies contribute, in a diversified portfolio that respects the requirements of the Swiss Law. The insurance policy has been treated as a qualifying insurance policy and therefore the pension assets are presented as one asset and are not desegregated and presented in classes that distinguish the nature and risks of those assets.
The following tables summarize the components of net benefit expense recognized in the consolidated statements of loss, amounts recognized in the balance sheet and gains/(losses) recognized in other comprehensive loss.
in CHF thousands |
|
|
|
|
|
|
|
For the years ended December 31, |
|
|||||
Actuarial gains / (losses) recognized in other comprehensive loss: |
|
|
|
|
|
|
|
|
2023 |
|
|
|
2022 |
|
On plan assets |
|
|
|
|
|
|
|
|
(70 |
) |
|
|
26 |
|
On obligation |
|
|
|
|
|
|
|
|
(738 |
) |
|
|
718 |
|
Total |
|
|
|
|
|
|
|
|
(808 |
) |
|
|
744 |
|
in CHF thousands |
|
|
|
|
|
|
|
For the years ended December 31, |
|
|||||
Net benefit expense (recognized in personnel costs): |
|
|
|
|
|
|
|
|
2023 |
|
|
|
2022 |
|
Current service cost |
|
|
|
|
|
|
|
|
(391 |
) |
|
|
(446 |
) |
Interest cost on benefit obligation |
|
|
|
|
|
|
|
|
(149 |
) |
|
|
(31 |
) |
Interest income |
|
|
|
|
|
|
|
|
147 |
|
|
|
26 |
|
Impact of plan changes |
|
|
|
|
|
|
|
|
- |
|
|
|
37 |
|
Administration cost |
|
|
|
|
|
|
|
|
(7 |
) |
|
|
(6 |
) |
Net benefit income / (expense) |
|
|
|
|
|
|
|
|
(400 |
) |
|
|
(420 |
) |
in CHF thousands |
|
|
|
|
|
|
|
As of December 31, |
|
|||||
Benefit asset / (liability) |
|
|
|
|
|
|
|
|
2023 |
|
|
|
2022 |
|
Defined benefit obligation |
|
|
|
|
|
|
|
|
(9,930 |
) |
|
|
(6,494 |
) |
Fair value of plan assets |
|
|
|
|
|
|
|
|
9,202 |
|
|
|
6,403 |
|
Net benefit asset / (liability) |
|
|
|
|
|
|
|
|
(728 |
) |
|
|
(91 |
) |
The impact of plan changes relates mainly to the changes of applicable rates for converting mandatory savings when employees retire (see also below).
Changes in the present value of the defined benefit obligation are as follows:
|
|
|
|
|
|
|
|
As of December 31, |
|
|||||
in CHF thousands |
|
|
|
|
|
|
|
|
2023 |
|
|
|
2022 |
|
Defined benefit obligation at January 1, |
|
|
|
|
|
|
|
|
(6,494 |
) |
|
|
(5,666 |
) |
Interest cost |
|
|
|
|
|
|
|
|
(149 |
) |
|
|
(31 |
) |
Current service cost |
|
|
|
|
|
|
|
|
(391 |
) |
|
|
(446 |
) |
Administrative expenses |
|
|
|
|
|
|
|
|
(7 |
) |
|
|
(6 |
) |
Contributions paid by participants |
|
|
|
|
|
|
|
|
(3,710 |
) |
|
|
(1,686 |
) |
Employees' contributions |
|
|
|
|
|
|
|
|
(247 |
) |
|
|
(185 |
) |
Benefits paid from plan assets |
|
|
|
|
|
|
|
|
1,806 |
|
|
|
770 |
|
Impact of plan changes |
|
|
|
|
|
|
|
|
- |
|
|
|
37 |
|
Actuarial gains / (losses) |
|
|
|
|
|
|
|
|
(738 |
) |
|
|
718 |
|
Defined benefit obligation at December 31, |
|
|
|
|
|
|
|
|
(9,930 |
) |
|
|
(6,494 |
) |
F-24
Changes in the fair value of plan assets are as follows:
|
|
|
|
|
|
|
|
As of December 31, |
|
|||||
in CHF thousands |
|
|
|
|
|
|
|
|
2023 |
|
|
|
2022 |
|
Fair value of plan assets at January 1, |
|
|
|
|
|
|
|
|
6,403 |
|
|
|
4,821 |
|
Expected return |
|
|
|
|
|
|
|
|
147 |
|
|
|
26 |
|
Contributions by employer |
|
|
|
|
|
|
|
|
571 |
|
|
|
429 |
|
Employees' contributions |
|
|
|
|
|
|
|
|
247 |
|
|
|
185 |
|
Benefits paid from plan assets |
|
|
|
|
|
|
|
|
(1,806 |
) |
|
|
(770 |
) |
Contributions paid by participants |
|
|
|
|
|
|
|
|
3,710 |
|
|
|
1,686 |
|
Actuarial gains / (losses) |
|
|
|
|
|
|
|
|
(70 |
) |
|
|
26 |
|
Fair value of plan assets at December 31, |
|
|
|
|
|
|
|
|
9,202 |
|
|
|
6,403 |
|
The Group expects to contribute CHF 0.6 million to its defined benefit pension plan in 2024. The average duration of the plan was 14.7 years and 14.0 years as of December 31, 2023 and 2022, respectively.
The principal assumptions used in determining pension benefit obligations for the Group's plan are shown below:
|
|
|
|
|
|
|
|
As of December 31, |
|
|||||
|
|
|
|
|
|
|
|
|
2023 |
|
|
|
2022 |
|
Discount rate |
|
|
|
|
|
|
|
|
1.45 |
% |
|
|
2.30 |
% |
Future salary increases |
|
|
|
|
|
|
|
|
1.20 |
% |
|
|
1.20 |
% |
Future pensions increases |
|
|
|
|
|
|
|
|
0.00 |
% |
|
|
0.00 |
% |
Retirement age |
|
|
|
|
|
|
|
M65/F64 |
|
|
M65/F64 |
|
||
Demographic assumptions |
|
|
|
|
|
|
|
BVG 2020 GT |
|
|
BVG 2020 GT |
|
||
In regard to the underlying estimates for the calculation of the defined benefit pension liabilities the Company updated, among other minor updates, the discount rate assumption to 1.45%, 2.30% and 0.35% as of December 31, 2023, 2022 and 2021, respectively. All the actuarial assumption changes resulted in an actuarial loss of defined benefit pension liabilities of CHF 0.8 million. The net result is an increase of defined benefit pension liabilities of CHF 0.1 million as of December 31, 2022 to CHF 0.7 million as of December 31, 2023. Other assumptions for defined benefit pension liabilities remain unchanged.
In 2023, the guaranteed interest to be credited to employees' savings was 1.00% (same as in 2022) for mandatory retirement savings, and 0.25% for supplementary retirement savings. Given current Swiss interest environment, the Company updated the estimated interest to be credited to employees’ savings up to 1.45%. The applicable rate for converting mandatory savings at age 65 for male and 64 for female employees retiring in 2023 was 6.20% and will be reduced to 5.90% for male and female for 2024 and 5.68% for women and 5.65% for men for 2025 and further reductions are expected in subsequent years. The rate for converting supplementary savings to an annuity remains stable at 4.49% for years 2023, 2024, 2025 and subsequent years for male employees and 4.54% in 2023, 2024 and 2025 and subsequent years for female employees.
Sensitivity analysis
A quantitative sensitivity analysis for significant assumptions as of December 31, 2023 and 2022 is shown below:
in CHF thousands |
Discount rate |
|
|
Future salary |
|
|
Mortality |
|
||||||||||||
Assumptions as of December 31, 2023 |
+0.25% |
|
-0.25% |
|
|
+0.50% |
|
-0.50% |
|
|
+1 year |
|
-1 year |
|
||||||
Potential defined benefit obligation |
|
(9,582 |
) |
|
(10,317 |
) |
|
|
(9,980 |
) |
|
(9,880 |
) |
|
|
(10,039 |
) |
|
(9,811 |
) |
Decrease/(increase) from actual defined benefit obligation |
|
348 |
|
|
(387 |
) |
|
|
(50 |
) |
|
50 |
|
|
|
(109 |
) |
|
119 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
Assumptions as of December 31, 2022 |
+0.25% |
|
-0.25% |
|
|
+0.50% |
|
-0.50% |
|
|
+1 year |
|
-1 year |
|
||||||
Potential defined benefit obligation |
|
(6,274 |
) |
|
(6,741 |
) |
|
|
(6,527 |
) |
|
(6,462 |
) |
|
|
(6,553 |
) |
|
(6,429 |
) |
Decrease/(increase) from actual defined benefit obligation |
|
221 |
|
|
(247 |
) |
|
|
(32 |
) |
|
32 |
|
|
|
(58 |
) |
|
65 |
|
The sensitivity analysis above is subject to limitations and has been determined based on a method that extrapolates the impact on net defined benefit obligation as a result of reasonable changes in key assumptions occurring at the end of the reporting period.
F-25
On March 2, 2023, the Company adopted a new Stock Option and Incentive Plan Regulation 2023 ("2023 Plan") which allows for the grant of equity incentives, including share-based options, stock appreciation rights ("SARs"), restricted shares and other awards. The 2023 Plan lays out the details for the equity incentives for talent acquisition and retention purposes.
Each grant of share-based options made under the 2023 Plan entitles the grantee to acquire ordinary shares from the Company with payment of the exercise price in cash. In the case of SARs, the intention of the Company is settling in equity. For each grant of share-based options or SARs, the Company issues a grant notice, which details the terms of the options or SARs, including number of shares, exercise price, vesting conditions and expiration date. Options granted under the 2023 Plan vest over periods ranging from one to four years and expire one day before the tenth anniversary of the grant date. The specific terms of each grant are set by the Board of Directors.
The 2023 Plan reflects the revised capital structure of the Company following completion of the Business Combination. As a result, all option holders holding options under the prior Stock Option and Incentive Plan Regulation 2018 (”2018 Plan”) prior to the close of the Business Combination exchanged their options held in Legacy Oculis for newly issued options to purchase ordinary shares of Oculis (“Converted Options”) and additional earnout options. The Converted Options continue to be subject to substantially the same terms and conditions except that the number of ordinary shares of Oculis issuable and related exercise prices were adjusted by the Exchange Ratio with all other terms remaining unchanged. The comparative fair value calculation of options using the Black-Scholes model before and after the merger concluded there was no significant change in value. The exchange of equity awards under the 2018 Plan for equity awards under the 2023 Plan was determined to be a modification in accordance with IFRS 2 – Share-based payment. The Group will continue to record the related expense per the original valuation and vesting period without incremental charges.
Option awards and SARs
Each share-based option or SAR granted under the 2023 Plan entitles the grantee to acquire common shares from the Company with cash payment of the exercise price. For each grant of share-based options or SARs, the Company provides a grant notice which details the terms of the option, including exercise price, vesting conditions and expiration date. The terms of each grant are set by the Board of Directors.
The fair value of option awards and SARs is determined using the Black-Scholes option-pricing model. The weighted average grant date fair value of options and SARs granted during the year ended December 31, 2023 was CHF 5.24 or $5.83 per share. The weighted average grant date fair value of options granted during the year ended December 31, 2022 was CHF 1.66 or $1.74 per share. The weighted average grant date fair value of options granted during the year ended December 31, 2021 was CHF 1.26 or $1.38 per share.
The Black-Scholes fair value of SARs was determined using assumptions that were not materially different from those used to value options. The following assumptions were used in the Black-Scholes option-pricing model for determining the fair value of options and SARs granted during the years indicated:
|
|
For the years ended December 31, |
||||
|
|
2023 |
|
2022 |
|
2021 |
Weighted average share price at the date of grant (1) |
|
USD 8.30 (CHF 7.46) |
|
USD 3.57 (CHF 3.41) |
|
USD 2.95 (CHF 2.70) |
Range of expected volatilities (%) (2) |
|
68.7-83.8 |
|
96.3 |
|
82.1 |
Expected term (years) (3) |
|
6.25 |
|
2.50 |
|
2.50 |
Range of risk-free interest rates (%) (1)(4) |
|
3.5-4.8 |
|
0.7 |
|
0.0 |
Dividend yield (%) |
|
0.0 |
|
0.0 |
|
0.0 |
(1) Following the NASDAQ listing in 2023, the equity award exercise price is now denominated in USD and the applicable risk-free interest rate has been adjusted accordingly.
(2) The expected volatility was derived from the historical stock volatilities of comparable peer public companies within the Company's industry.
(3) The expected term represents the period that share-based awards are expected to be outstanding.
(4) The risk-free interest rate in 2023 is based on the U.S. Treasury yield curve in effect at the measurement date with maturities approximately equal to the expected term. Prior to 2023, the risk-free interest rate was based on Switzerland Short-Term Government Bonds with maturities approximately equal to the expected term.
The following table summarizes the Company’s stock option and SAR activity under the 2023 Plan for the following periods:
F-26
|
|
Number of options (1) |
|
|
Weighted average exercise price (1) (CHF) |
|
|
Range of expiration dates |
||
Outstanding as of January 1, 2021 |
|
|
1,116,045 |
|
|
|
1.96 |
|
|
2026-2029 |
Granted |
|
|
341,794 |
|
|
|
2.36 |
|
|
2030 |
Forfeited |
|
|
(168,749 |
) |
|
|
2.09 |
|
|
2027-2028 |
Outstanding as of December 31, 2021 |
|
|
1,289,090 |
|
|
|
2.05 |
|
|
2026-2030 |
Exercisable at December 31, 2021 |
|
|
759,324 |
|
|
|
1.90 |
|
|
2026-2030 |
Outstanding as of January 1, 2022 |
|
|
1,289,090 |
|
|
|
2.05 |
|
|
2026-2030 |
Granted |
|
|
629,295 |
|
|
|
2.98 |
|
|
2031 |
Forfeited |
|
|
(94,273 |
) |
|
|
2.35 |
|
|
2023-2030 |
Exercised |
|
|
(61,163 |
) |
|
|
1.85 |
|
|
2026-2027 |
Outstanding as of December 31, 2022 |
|
|
1,762,949 |
|
|
|
2.39 |
|
|
2027-2031 |
Exercisable at December 31, 2022 |
|
|
819,603 |
|
|
|
1.97 |
|
|
2027-2031 |
Outstanding as of January 1, 2023 |
|
|
1,762,949 |
|
|
|
2.39 |
|
|
2027-2031 |
Options granted(2) |
|
|
1,614,000 |
|
|
|
7.49 |
|
|
2033 |
SARs granted |
|
|
134,765 |
|
|
|
7.27 |
|
|
2033 |
Earnout options granted |
|
|
369,737 |
|
|
|
0.01 |
|
|
2028 |
Forfeited(2)(3) |
|
|
(302,299 |
) |
|
|
2.62 |
|
|
2033 |
Exercised(3) |
|
|
(112,942 |
) |
|
|
2.43 |
|
|
2028-2032 |
Outstanding as of December 31, 2023 |
|
|
3,466,210 |
|
|
|
4.50 |
|
|
2027-2033 |
Exercisable at December 31, 2023 |
|
|
1,164,513 |
|
|
|
2.21 |
|
|
2028-2033 |
(1) Retroactive application of the recapitalization effect due to the BCA for activity prior to March 2, 2023, the Exchange Ratio was applied to the number of options and the weighted average exercise price was divided by the same exchange ratio.
(2) Pursuant to the BCA, all outstanding and unexercised options to purchase Legacy Oculis ordinary shares were assumed by Oculis and each option was replaced by an option to purchase ordinary shares of Oculis (the “Converted Options”). The exchange of Legacy Oculis 2018 Plan options for converted 2023 Plan options is not reflected in the table above. Refer to Note 5 - Business Combination and Financing Activities for further details.
(3) Forfeited amount includes earnout options forfeited during the year ended December 31, 2023. No SARs have been exercised or forfeited during the year ended December 31, 2023.
Excluding earnout options, which have an exercise price of CHF 0.01, options outstanding as of December 31, 2023 have exercise prices ranging from CHF 1.84 to CHF 11.66. The weighted average remaining contractual life of options and SARs outstanding as of December 31, 2023 was eight years. The weighted average contractual life of options outstanding as of December 31, 2022 was seven years.
Restricted Stock Awards
Each restricted stock award granted under the 2018 Plan was immediately exercised and the expense was recorded at grant date in full. The Company is holding call options to repurchase shares diminishing ratably on a monthly basis over three years from grant date. For each restricted stock award granted, the Company issues a grant notice, which details the terms of the grant, including the number of awards, repurchase right start date and expiration date. The terms of each grant are set by the Board of Directors. Restricted shares are granted and expensed at fair value. No restricted stock awards were granted under the 2023 Plan during the years ended December 31, 2023 and 2022.
The number and weighted average exercise prices of restricted stock awards outstanding under the 2023 Plan are as follows (recast after applying the Exchange Ratio to reflect the impact of the BCA):
|
|
Number of Restricted Stock Awards |
|
|
Weighted average exercise price (CHF) |
|
||
Issued and exercised as of January 1, 2021 |
|
|
745,512 |
|
|
|
1.58 |
|
Granted and exercised during 2021 |
|
|
441,419 |
|
|
|
1.99 |
|
Issued and exercised as of December 31, 2021 |
|
|
1,186,931 |
|
|
|
1.73 |
|
Not subject to repurchase at December 31, 2021 |
|
|
710,338 |
|
|
|
1.59 |
|
Issued and exercised as of January 1, 2022 |
|
|
1,186,931 |
|
|
|
1.73 |
|
Issued and exercised as of December 31, 2022 |
|
|
1,186,931 |
|
|
|
1.73 |
|
Not subject to repurchase at December 31, 2022 |
|
|
934,044 |
|
|
|
1.66 |
|
Issued and exercised as of January 1, 2023 |
|
|
1,186,931 |
|
|
|
1.73 |
|
Issued and exercised as of December 31, 2023 |
|
|
1,186,931 |
|
|
|
1.73 |
|
Not subject to repurchase at December 31, 2023 |
|
|
1,088,838 |
|
|
|
1.71 |
|
Share-based compensation expenses
The total expense recognized in the consolidated statements of loss for share options granted amounted to CHF 3.6 million for the year ended December 31, 2023, CHF 0.8 million for the year ended December 31, 2022, and CHF 0.3 million for the year ended December 31, 2021. No expense was recognized during the years ended December 31, 2023 or 2022 related to restricted stock awards. For the year ended December 31, 2021 the Company recognized CHF 1.0 million of expense related to restricted stock awards. The reserve for share-based payment increased from CHF
F-27
2.0 million as of December 31, 2021 to CHF 2.8 million as of December 31, 2022, and to CHF 6.4 million as of December 31, 2023.
Cash and cash equivalents consist primarily of cash balances held at banks and in the currencies:
in CHF thousands |
|
Cash and cash equivalents |
|
|
Short-term financial assets |
|
||||||||||
by currency |
|
As of December 31, 2023 |
|
|
As of |
|
|
As of December 31, 2023 |
|
|
As of |
|
||||
Swiss Franc |
|
|
19,144 |
|
|
|
7,216 |
|
|
|
33,532 |
|
|
|
- |
|
US Dollar |
|
|
16,610 |
|
|
|
9,741 |
|
|
|
15,148 |
|
|
|
- |
|
Euro |
|
|
2,020 |
|
|
|
2,350 |
|
|
|
4,644 |
|
|
|
- |
|
Iceland Krona |
|
|
542 |
|
|
|
383 |
|
|
|
- |
|
|
|
- |
|
Other |
|
|
11 |
|
|
|
96 |
|
|
|
- |
|
|
|
- |
|
Total |
|
|
38,327 |
|
|
|
19,786 |
|
|
|
53,324 |
|
|
|
- |
|
Short-term financial assets consist of fixed term bank deposits with maturities between three and six months.
As of December 31, 2022, Legacy Oculis had 12,712,863 preferred shares for a nominal amount of CHF 1.4 million. These shares were divided into 1,623,793 registered "A Series" shares of CHF 0.10 each, 5,191,512 registered "B Series" of CHF 0.10 each, 5,699,813 registered "C1a Series" shares (denominated in USD) of CHF 0.10 each and 197,745 registered "C1b Series" shares (denominated in USD) of CHF 0.50 each.
All preferred shares had a liquidation preference corresponding to their respective initial purchase price. Furthermore, the "B Series" and "C Series" shares included a preferred dividend payment of 6.0% (as a compounded interest) and the corresponding deemed interest expense of CHF 1.3 million was accrued for the period from January 1 to March 2, 2023. The cumulative interest expense accrued up to December 31, 2022 amounted to CHF 17.0 million. The nominal amounts (for "A, B and C Series") and the accrued preferred dividend resulted in a long-term debt of CHF 124.8 million as of March 2, 2023.
On March 2, 2023, at closing of the Business Combination, all preferred shares of Legacy Oculis were converted into ordinary shares of Oculis at the effective exchange ratios determined in accordance with the BCA and giving effect to the accumulated preferred dividends (refer to Note 5). The movement of the long-term financial debt is shown below:
in CHF thousands |
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
Series A shares |
|
|
Series B shares |
|
|
Series C shares |
|
|
Total |
|
||||
Balance as of December 31, 2021 |
|
|
8,179 |
|
|
|
48,569 |
|
|
|
56,754 |
|
|
|
113,502 |
|
Issuance of shares |
|
|
- |
|
|
|
- |
|
|
|
2,030 |
|
|
|
2,030 |
|
Transaction costs |
|
|
- |
|
|
|
- |
|
|
|
(54 |
) |
|
|
(54 |
) |
Interest |
|
|
- |
|
|
|
2,797 |
|
|
|
3,546 |
|
|
|
6,343 |
|
FX revaluation |
|
|
- |
|
|
|
- |
|
|
|
628 |
|
|
|
628 |
|
Balance as of December 31, 2022 |
|
|
8,179 |
|
|
|
51,366 |
|
|
|
62,904 |
|
|
|
122,449 |
|
Interest |
|
|
- |
|
|
|
519 |
|
|
|
747 |
|
|
|
1,266 |
|
FX revaluation |
|
|
- |
|
|
|
- |
|
|
|
1,087 |
|
|
|
1,087 |
|
Conversion of Legacy Oculis preferred shares into Oculis ordinary shares |
|
|
(8,179 |
) |
|
|
(51,885 |
) |
|
|
(64,738 |
) |
|
|
(124,802 |
) |
Balance as of December 31, 2023 |
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
|
- |
|
As a result of the Business Combination, the Company has retroactively restated the number of shares as of December 31, 2020, 2021 and 2022 to give effect to the Exchange Ratio under the BCA as explained in Note 5:
F-28
|
|
Number of shares |
|
|
In CHF thousands |
|
|||||||||||||||||||||||
|
|
Legacy Oculis ordinary shares |
|
|
Legacy Oculis restricted share awards |
|
|
Legacy Oculis treasury shares |
|
|
|
Oculis ordinary shares (1) |
|
|
Share capital (2) |
|
|
Treasury shares (2) |
|
|
Share premium |
|
|||||||
Balance as of December 31, 2020 (effect of the recapitalization) |
|
|
2,646,629 |
|
|
|
745,511 |
|
|
|
(114,323 |
) |
|
|
|
- |
|
|
|
34 |
|
|
|
(1 |
) |
|
|
9,773 |
|
Restricted shares awards |
|
|
- |
|
|
|
441,419 |
|
|
|
- |
|
|
|
|
- |
|
|
|
4 |
|
|
|
- |
|
|
|
872 |
|
Transaction costs |
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
|
(12 |
) |
Balance as of December 31, 2021 (effect of the recapitalization) |
|
|
2,646,629 |
|
|
|
1,186,930 |
|
|
|
(114,323 |
) |
|
|
|
- |
|
|
|
38 |
|
|
|
(1 |
) |
|
|
10,632 |
|
Stock option exercised |
|
|
61,163 |
|
|
|
- |
|
|
|
- |
|
|
|
|
- |
|
|
|
1 |
|
|
|
- |
|
|
|
119 |
|
Transaction costs |
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
|
(9 |
) |
Balance as of December 31, 2022 (effect of the recapitalization) |
|
|
2,707,792 |
|
|
|
1,186,930 |
|
|
|
(114,323 |
) |
|
|
|
- |
|
|
|
39 |
|
|
|
(1 |
) |
|
|
10,742 |
|
Reorganization following Business Combination |
|
|
(2,707,792 |
) |
|
|
(1,186,930 |
) |
|
|
114,323 |
|
|
|
|
- |
|
|
|
(39 |
) |
|
|
1 |
|
|
|
- |
|
Conversion of Legacy Oculis ordinary shares and treasury shares into Oculis ordinary shares |
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
|
|
3,780,399 |
|
|
|
38 |
|
|
|
- |
|
|
|
- |
|
Conversion of Legacy Oculis long-term financial debt into Oculis ordinary shares |
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
|
|
16,496,603 |
|
|
|
165 |
|
|
|
- |
|
|
|
124,637 |
|
Issuance of ordinary shares to PIPE investors |
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
|
|
7,118,891 |
|
|
|
71 |
|
|
|
- |
|
|
|
66,983 |
|
Issuance of ordinary shares under CLA |
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
|
|
1,967,000 |
|
|
|
20 |
|
|
|
- |
|
|
|
18,348 |
|
Issuance of ordinary shares to sponsor |
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
|
|
2,047,302 |
|
|
|
20 |
|
|
|
- |
|
|
|
34,843 |
|
Issuance of ordinary shares to non-redeemed shareholders |
|
|
|
|
|
|
|
|
|
|
|
|
1,323,178 |
|
|
|
13 |
|
|
|
- |
|
|
|
12,001 |
|
|||
Reorganization |
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
|
(11,352 |
) |
Transaction costs related to the business combination |
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
|
(4,821 |
) |
Proceeds from sale of shares in public offering |
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
|
|
3,654,234 |
|
|
|
36 |
|
|
|
- |
|
|
|
38,143 |
|
Transaction costs related to the public offering |
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
|
(3,361 |
) |
Stock option exercised |
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
|
|
112,942 |
|
|
|
1 |
|
|
|
- |
|
|
|
273 |
|
Issuance of shares in connection with warrant exercises |
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
|
|
149,156 |
|
|
|
2 |
|
|
|
- |
|
|
|
1,726 |
|
Balance as of December 31, 2023 |
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
|
|
36,649,705 |
|
|
|
366 |
|
|
|
- |
|
|
|
288,162 |
|
(1) Fully paid-in registered shares with a par value of CHF 0.01
(2) Recasted Legacy Oculis shares through the date of the Business Combination
The conditional capital at December 31, 2023 amounted to a maximum of CHF 176,089.41 split into 17,608,941 ordinary shares, in connection with the potential future issuances of:
CHF 50,000.00 through the issuance of a maximum of 5,000,000 fully paid-up registered shares, each with a par value of CHF 0.01 (ordinary shares), in connection with the exercise of convertible rights and/or option rights or warrants, new bonds and similar debt instruments.
F-29
CHF 78,355.44 through the issuance of a maximum of 7,835,544 fully paid-up registered shares, each with a par value of CHF 0.01 (ordinary shares), in connection with the exercise of option rights or other equity-linked instruments granted to any employee, consultant or member of the Board of Directors of Oculis.
As of December 31, 2023, 112,942 options have been exercised and associated ordinary shares have been issued using the conditional share capital for employee benefit plans (refer to Note 13). These shares were not registered yet in the commercial register as of December 31, 2023.
CHF 44,032.94 through the issuance of a maximum of 4,403,294 fully paid up registered shares, each with a par value of CHF 0.01 (ordinary shares), in connection with the exercise of warrants.
As of December 31, 2023, 149,156 warrants have been exercised and associated ordinary shares have been issued using the conditional share capital for EBAC public and private warrants (refer to Note 18). These shares were not registered yet in the commercial register as of balance sheet date.
CHF 3,701.03 through the issuance of a maximum of 370,103 fully paid up registered shares, each with a par value of CHF 0.01 (ordinary shares), in connection with the exercise of option rights or other equity-linked instruments granted to any employee, consultant or member of the Board of Directors of Oculis.
The Group cancelled 100,000 treasury shares effective March 2, 2023 as a result of the Business Combination. No treasury shares were outstanding as of December 31, 2023.
(D) Capital band
The Company has a capital band between CHF 365,273.68 (lower limit) and CHF 543,684.52 (upper limit). The Company may effect an increase of the Company’s share capital in a maximum amount of CHF 178,410.84 by issuing up to 17,841,084 ordinary shares with a par value of CHF 0.01 each out of the Company’s capital band. The Board of Directors is authorized to increase the share capital to the upper limit or decrease the share capital to the lower limit at any time and as often as required until March 2, 2028. In Q2 2023, 3,654,234 shares were issued from this capital band.
in CHF thousands |
As of December 31, 2023 |
|
As of December 31, 2022 |
|
||
Trade payables |
|
(7,596 |
) |
|
(3,867 |
) |
Total |
|
(7,596 |
) |
|
(3,867 |
) |
The increase in trade payables as of December 31, 2023 compared to prior year relates to the commencement of several clinical trials in the fourth quarter of 2023 requiring upfront invoicing by vendors.
The table below shows the breakdown of the Accrued expenses and other payables by category:
in CHF thousands |
|
As of December 31, 2023 |
|
|
As of December 31, 2022 |
|
||
Product development related expenses |
|
|
2,801 |
|
|
|
4,805 |
|
Personnel related expenses |
|
|
2,301 |
|
|
|
2,249 |
|
General and administration related expenses |
|
|
765 |
|
|
|
957 |
|
Other payables |
|
|
81 |
|
|
|
- |
|
Total |
|
|
5,948 |
|
|
|
8,011 |
|
Pursuant to the BCA and the Warrant Assignment and Assumption Agreement executed in connection with the BCA, the Company has assumed 4,251,595 EBAC public warrants and 151,699 EBAC private warrants from EBAC, and issued 4,403,294 warrants as of March 2, 2023 with substantially the same terms.
F-30
Each warrant entitles the registered holder to purchase one ordinary share at a price of CHF 9.68 or $11.50 per share, subject to certain adjustments, exercisable at any time commencing 30 days after the acquisition closing date, provided that the Company has an effective registration statement under the Securities Act covering the issuance of the ordinary shares issuable upon exercise of the warrants. This registration statement was filed with the SEC and declared effective on May 1, 2023. The warrants will expire on March 2, 2028.
As of March 2, 2023, the Company recognized the warrant liabilities at fair value of CHF 2.1 million. For the year ended December 31, 2023, the Company recognized a fair value loss in the Statement of Loss of CHF 3.4 million leading to an increase of the warrant liability up to CHF 5.4 million as of December 31, 2023. The exercise of 149,156 public warrants at a price of CHF 10.26 (average value of effective rate) or $11.50 per share for the year period ended December 31, 2023 resulted in a reduction of CHF 0.2 million to the liability, an additional CHF 1.5 million of cash and an increase of CHF 1.7 million in shareholder's equity (refer to Note 16).
The movement of the warrant liability is illustrated below:
in CHF thousands (except share number of warrants) |
|
Warrant liabilities |
|
|
Share number of outstanding public and private warrants |
|
||
Balance as of January 1, 2023 |
|
|
- |
|
|
|
- |
|
Issuance of assumed warrants from EBAC |
|
|
2,136 |
|
|
|
4,403,294 |
|
Fair value loss on warrant liability |
|
|
3,431 |
|
|
|
- |
|
Exercise of public and private warrants |
|
|
(197 |
) |
|
|
(149,198 |
) |
Balance as of December 31, 2023 |
|
|
5,370 |
|
|
|
4,254,096 |
|
The number of exercised warrants abovementioned of 149,198 warrants includes 149,156 EBAC warrants exercised in 2023 and an additional number of 42 EBAC warrants that are still formally part of the Company's conditional share capital, although they will not become exercisable because of the fractional conversion rate and rounding methodology applied when converting the initial warrants from EBAC into the Company's warrants.
Commitments related to Novartis license agreement
In December 2018, Oculis entered into an agreement with Novartis, under which Oculis licensed a novel topical anti-TNFα antibody, now named as Licaminlimab, or OCS-02, for ophthalmic indications. As consideration for the licenses, Oculis is obligated to pay non-refundable, upfront license fees, predefined development and commercial milestone payments and royalties on net sales of licensed products. Royalties range from high one digit to low teens, based on sales thresholds. As of December 31, 2019, Oculis has paid in full the contractual non-refundable upfront fee of CHF 4.7 million. Oculis has not reached any milestones or royalties thresholds according to the agreement. If all predefined milestones will be reached, Oculis will be obligated to pay additional CHF 81.6 million or $97.0 million. Oculis expects to reach the first milestone payment of CHF 4.2 million or $5.0 million in 2028. Royalties are based on net sales of licensed products, depending on the sales volumes reached.
Commitments related to Accure license agreement
On January 29, 2022, the Company entered into a License Agreement with Accure for the exclusive global licensing of its OCS-05 technology. Under this agreement, Oculis licensed a novel neuroprotective drug candidate, now renamed as OCS-05, for ophthalmic and other indications (refer to Note 9). As consideration for the licenses, Oculis is obligated to pay non-refundable, upfront license fees, predefined development and commercial milestone payments and royalties on net sales of licensed products. Royalties range from one digit to low teens, based on sales thresholds. As of December 31, 2023, Oculis has paid the full contractual non-refundable upfront fee of CHF 3.0 million and reimbursed costs in the amount of CHF 0.5 million. Oculis has not reached any milestones or royalties thresholds according to the agreement. If all predefined milestones will be reached, Oculis will be obligated to pay additional CHF 94.3 million or $112.1 million. In case of a commercialization, sublicense revenues will be subject to further royalty payments. The initial potential milestones under the agreement are IND approval by the U.S. FDA for the intravenous formulation of OCS-05 and completion of the first PoC clinical trial for AON for a combined amount of CHF 1.0 million or $1.2 million. These milestones are estimated to be reached toward the end of 2024 or beginning of 2025.
Commitments related to Rennes University Collaboration Research agreement
On January 31, 2022, the Company entered into a collaboration research agreement with the Rennes University and CNRS in France. This agreement is for the research of Antisense Oligonucleotide (ASO) to modulate gene expressions. As consideration for the research performed by Rennes University and CNRS, Oculis is obligated to pay a non-refundable cost contribution of CHF 0.2 million or EUR 0.2 million.
F-31
As of December 31, 2023, Oculis paid a contractual non-refundable cost contribution of CHF 0.1 million or EUR 0.1 million. Following completion of the research services, the parties shall sign a commercial agreement based on predefined development and commercial milestone payments and royalties on net sales of licensed products as defined in the collaboration research agreement. Oculis has not reached any milestones or royalties thresholds. If the commercial agreement was signed by the parties and development and commercial milestone payments were reached, Oculis would be obligated to pay additional CHF 6.5 million or EUR 7.0 million and royalties ranging from low to mid-single digit percentage on net sales. In case of sublicense revenues, Oculis shall be subject to further royalty payments.
Research and development commitments
The Group conducts product research and development programs through collaborative projects that include, among others, arrangements with universities, contract research organizations and clinical research sites. Oculis has contractual arrangements with these organizations. As of December 31, 2023, commitments for external research projects amounted to CHF 50.5 million, compared to CHF 13.1 million as of December 31, 2022, as detailed in the schedule below. The increase in commitments year over year is primarily due to the initiation of clinical trials in the last quarter of 2023.
in CHF thousands |
|
As of December 31, 2023 |
|
|
As of December 31, 2022 |
|
||
Within one year |
|
|
23,625 |
|
|
|
12,145 |
|
Between one and five years |
|
|
26,867 |
|
|
|
978 |
|
Total |
|
|
50,492 |
|
|
|
13,123 |
|
Key management, including the Board of Directors and the executive management team, compensation expenses were:
in CHF thousands |
|
For the years ended December 31, |
|
|||||||||
|
|
2023 |
|
|
2022 |
|
|
2021 |
|
|||
Salaries, cash compensation and other short-term benefits |
|
|
3,067 |
|
|
|
3,506 |
|
|
|
3,071 |
|
Payroll expenses related to restricted share |
|
|
- |
|
|
|
- |
|
|
|
951 |
|
Pension expense |
|
|
320 |
|
|
|
227 |
|
|
|
264 |
|
Share-based compensation expense |
|
|
2,543 |
|
|
|
535 |
|
|
|
251 |
|
Total |
|
|
5,930 |
|
|
|
4,268 |
|
|
|
4,537 |
|
Salaries, cash compensation and other short-term benefits include social security and board member fees.
The number of individuals reported as key management was reduced from 7 to 6 for the year ended December 31, 2023 as compared to the year ended December 31, 2022. The number of individuals reported for the Board of Directors increased from 1 to 3 for the year ended December 31, 2023 as compared to the year ended December 31, 2022.
Categories of financial instruments:
As indicated in Note 3, all financial assets and liabilities are shown at amortized cost, except for warrant liabilities that are held at fair value. The following table shows the carrying amounts of financial assets and liabilities:
in CHF thousands |
|
|
||||
Financial assets |
As of December 31, 2023 |
|
As of December 31, 2022 |
|
||
Financial assets - non-current |
|
45 |
|
|
50 |
|
Other current assets, excluding prepaids |
|
328 |
|
|
166 |
|
Accrued income |
|
876 |
|
|
912 |
|
Short-term financial assets |
|
53,324 |
|
|
- |
|
Cash and cash equivalents |
|
38,327 |
|
|
19,786 |
|
Total |
|
92,900 |
|
|
20,914 |
|
F-32
in CHF thousands |
|
|
||||
Financial liabilities |
As of December 31, 2023 |
|
As of December 31, 2022 |
|
||
Trade payables |
|
7,596 |
|
|
3,867 |
|
Accrued expenses and other payables |
|
5,948 |
|
|
8,011 |
|
Lease liabilities |
|
605 |
|
|
633 |
|
Warrant liabilities |
|
5,370 |
|
|
- |
|
Long-term financial debt related to preferred shares/accrued dividend |
|
- |
|
|
122,449 |
|
Total |
|
19,519 |
|
|
134,960 |
|
Below is the net debt table of liabilities from financing activities:
in CHF thousands |
Preferred shares |
|
Leasing |
|
Warrant liabilities |
|
Total |
|
||||
Net debt as of December 31, 2021 |
|
(113,502 |
) |
|
(770 |
) |
|
- |
|
|
(114,272 |
) |
Cashflows |
|
(2,030 |
) |
|
204 |
|
|
- |
|
|
(1,825 |
) |
Interest calculated on Series B & C shares |
|
(6,343 |
) |
|
- |
|
|
- |
|
|
(6,343 |
) |
Transaction costs related to 2021 |
|
54 |
|
|
- |
|
|
- |
|
|
54 |
|
Interest calculated on leases |
|
- |
|
|
(45 |
) |
|
- |
|
|
(45 |
) |
Indexation for the period |
|
- |
|
|
(70 |
) |
|
- |
|
|
(70 |
) |
FX revaluation |
|
(628 |
) |
|
48 |
|
|
- |
|
|
(580 |
) |
Net debt as of December 31, 2022 |
|
(122,449 |
) |
|
(633 |
) |
|
- |
|
|
(123,082 |
) |
Cashflows |
|
- |
|
|
200 |
|
|
- |
|
|
200 |
|
Interest calculated on Series B & C shares |
|
(1,266 |
) |
|
- |
|
|
- |
|
|
(1,266 |
) |
Issuance of warrants |
|
- |
|
|
- |
|
|
(2,136 |
) |
|
(2,136 |
) |
Fair value (gain)/loss on warrant liability |
|
- |
|
|
- |
|
|
(3,431 |
) |
|
(3,431 |
) |
Exercise of public and private warrants |
|
- |
|
|
- |
|
|
197 |
|
|
197 |
|
Addition of US lease |
|
- |
|
|
(118 |
) |
|
- |
|
|
(118 |
) |
Interest calculated on leases |
|
- |
|
|
(42 |
) |
|
- |
|
|
(42 |
) |
Indexation for the period |
|
- |
|
|
(47 |
) |
|
- |
|
|
(47 |
) |
FX revaluation |
|
(1,087 |
) |
|
35 |
|
|
- |
|
|
(1,052 |
) |
Conversion of Legacy Oculis preferred shares into Oculis ordinary shares |
|
124,802 |
|
|
- |
|
|
- |
|
|
124,802 |
|
Net debt as of December 31, 2023 |
|
- |
|
|
(605 |
) |
|
(5,370 |
) |
|
(5,975 |
) |
Fair values
Due to their short-term nature, the carrying value of cash and cash equivalents, short-term financial assets, other current assets, excluding prepaids, accrued income, trade payables and accrued expenses and other payables approximates their fair value.
The warrant liabilities are measured at fair value on a recurring basis, refer to Note 3.
Legacy Oculis preferred shares, presented as long-term financial debt as described in Note 15, had been converted into Oculis ordinary shares. The conversion occurred in March 2023. As of December 31, 2022, the fair value of preferred shares was determined from similar or identical instruments issued by the Company. This level 2 value resulted in a fair value of CHF 115.7 million compared to a book value of CHF 122.4 million.
Risk assessment
Since 2018 the Company implemented an Internal Control System (ICS), which includes a risk assessment. The ultimate responsibility of the risk management is of the Board of Directors and a yearly review takes place during one of the Board of Directors meetings.
Market risk
Market risk is the risk that changes in market prices, such as foreign exchange rates, interest rates and equity prices will affect the Company’s income or the value of its holdings of financial instruments. The objective of market risk management is to manage and control market risk exposures within acceptable parameters, while optimizing the return.
As of December 31, 2023, if the listed price of the warrants had moved by 5% with all other variables held constant, the net loss for the period would have been lower/higher by CHF 0.3 million. As of December 31, 2022, the Company did not hold any warrant liabilities.
F-33
Foreign currency risks
Since 2020, Oculis has established a presence in the U.S., France and Hong Kong with local currencies in U.S. Dollar (USD), Euro (EUR) and Hong Kong Dollar (HKD), respectively. In 2023, foreign currency risks primarily relate to cash and cash equivalents, short term financial assets, prepaid expenses, trade payables and accrued expenses denominated in U.S. Dollar and Euro, with immaterial amounts recorded in ISK and HKD.
The following table demonstrates the impact of a possible change in USD and EUR against CHF in regard to monetary assets and liabilities denominated in local functional currencies, as well as the impact of foreign currency risk on the Company's consolidated net loss:
|
As of December 31, / For the years ended |
|
|||||||||||
in CHF thousands |
2023 |
|
|
2022 |
|
||||||||
Change in rate |
Net exposure in CHF |
|
Impact on loss |
|
|
Net exposure in CHF |
|
Impact on loss |
|
||||
+5.0% USD |
|
21,667 |
|
|
1,083 |
|
|
|
9 577 |
|
|
479 |
|
-5.0% USD |
|
21,667 |
|
|
(1,083 |
) |
|
|
9 577 |
|
|
(479 |
) |
+5.0% EUR |
|
4,049 |
|
|
202 |
|
|
|
2,176 |
|
|
109 |
|
-5.0% EUR |
|
4,049 |
|
|
(202 |
) |
|
|
2,176 |
|
|
(109 |
) |
Interest rate risk
The Company’s long-term financial debt, which resulted from the issuance of preferred shares as indicated in Note 15, bore a deemed interest resulting from the preferred dividend, due under certain circumstances, at a fixed rate of 6.0% per year until their conversion on March 2, 2023 in connection with the Business Combination. The other financial instruments of the Group are not bearing interest and are therefore not subject to interest rate risk.
Hedging activities
There are no hedging activities within the Group.
Credit risk
As of December 31, 2023, the maximum exposure is the carrying amount of the Company’s cash, cash equivalents and short-term financial assets are mainly held with two financial institutions, each with a high credit rating of A+ assigned by international credit-rating agencies. Management focuses on diversification strategies and monitors counterparties’ ratings to minimize exposure.
Liquidity risk
Liquidity risk is the risk that the Group will encounter difficulty in meeting the obligations associated with its financial liabilities that are settled by delivering cash or another financial asset. Liquidity management is performed by Group finance based on cash flow forecasts which are prepared on a rolling basis and focuses mainly on ensuring that the Group has sufficient cash to meet its operational needs. The Group’s liquidity needs have been historically satisfied through the issuance of preferred shares, the Business Combination, PIPE and CLA financings, and the follow-on offering, discussed further in Note 5.
All of the Company’s financial instruments, except long-term financial debt and the long-term portion of the lease liabilities are due within one year.
in CHF thousands |
As of December 31, 2023 |
|
Less than one year |
|
Over |
|
|
As of December 31, 2022 |
|
Less than one year |
|
Over |
|
||||||
Trade payables |
|
7,596 |
|
|
7,596 |
|
|
- |
|
|
|
3,867 |
|
|
3,867 |
|
|
|
|
Accrued expenses and other payables |
|
5,948 |
|
|
5,948 |
|
|
- |
|
|
|
8,011 |
|
|
8,011 |
|
|
- |
|
Long-term financial debt |
|
- |
|
|
- |
|
|
- |
|
|
|
170,988 |
|
|
- |
|
|
170,988 |
|
Lease liability |
|
681 |
|
|
210 |
|
|
471 |
|
|
|
743 |
|
|
149 |
|
|
594 |
|
Total |
|
14,225 |
|
|
13,754 |
|
|
471 |
|
|
|
183,609 |
|
|
12,027 |
|
|
171,582 |
|
F-34
Long-term financial debt as of December 31, 2022 resulted from the issuance of preferred shares as indicated in Note 15.
Capital management
Since its incorporation, the Group has primarily funded its operations through capital increases, and at the current development stage, the Group frequently raises new funds to finance its projects. Refer to Notes 15 and 16 for further details.
As a result of the Business Combination, the Company has retroactively restated the weighted average number of outstanding shares prior to March 2, 2023 to give effect to the Exchange Ratio. The following table sets forth the loss per share calculations for the years ended December 31, 2023, 2022 and 2021.
|
For the years ended December 31, |
|
|||||||||
|
2023 |
|
|
2022 |
|
|
2021 |
|
|||
Net loss for the period attributable to Oculis shareholders - in CHF thousands |
|
(88,802 |
) |
|
|
(38,698 |
) |
|
|
(18,552 |
) |
|
|
|
|
|
|
|
|
|
|||
Weighted-average number of shares used to compute loss per share basic and diluted for the periods ended December 31, 2022 and December 31, 2021, Legacy Oculis ordinary shares |
|
- |
|
|
|
2,989,434 |
|
|
|
2,777,589 |
|
Exchange Ratio |
|
- |
|
|
|
1.1432 |
|
|
|
1.1432 |
|
Weighted-average number of shares used to compute basic and diluted loss per share for the periods ended December 31, 2022 and December 31, 2021, Legacy Oculis ordinary shares (as restated) |
|
- |
|
|
|
3,417,521 |
|
|
|
3,175,340 |
|
Weighted-average number of shares used to compute basic and diluted loss per share for the period ended December 31, 2023, Oculis ordinary shares |
|
29,899,651 |
|
|
|
- |
|
|
|
- |
|
|
|
|
|
|
|
|
|
|
|||
Basic and diluted net loss per share for the period, ordinary shares |
|
(2.97 |
) |
|
|
(11.32 |
) |
|
|
(5.84 |
) |
Since the Company has a loss for all periods presented, basic net loss per share is the same as diluted net loss per share. Potentially dilutive securities that were not included in the diluted loss per share calculations because they would be anti-dilutive were as follows:
The number of potentially dilutive securities prior to the Business Combination have been adjusted by the Exchange Ratio to reflect the equivalent number in the Company.
|
As of December 31, 2023 |
|
|
As of December 31, 2022 |
|
|
As of December 31, 2021 |
|
|||
Share options issued and outstanding |
|
3,096,473 |
|
|
|
1,762,949 |
|
|
|
1,289,090 |
|
Earnout options |
|
369,737 |
|
|
|
- |
|
|
|
- |
|
Share and earnout options issued and outstanding |
|
3,466,210 |
|
|
|
1,762,949 |
|
|
|
1,289,090 |
|
Restricted shares subject to repurchase |
|
98,094 |
|
|
|
252,880 |
|
|
|
476,581 |
|
Earnout shares |
|
3,793,995 |
|
|
|
- |
|
|
|
- |
|
Public warrants |
|
4,102,397 |
|
|
|
- |
|
|
|
- |
|
Private warrants |
|
151,699 |
|
|
|
- |
|
|
|
- |
|
Total |
|
11,612,395 |
|
|
|
2,015,829 |
|
|
|
1,765,671 |
|
There are no material subsequent events to report and no events out of the ordinary course of business.
F-35
Exhibit 1.1
|
ARTICLES OF ASSOCIATIONof Oculis Holding AG
with registered office in Zug (Translation; in case of controversy the German text shall prevail)
|
|
STATUTENder Oculis Holding AG
mit Sitz in Zug |
I. CORPORATE NAME, REGISTERED OFFICE, DURATION AND PURPOSE OF THE COMPANY |
|
I. FIRMA, SITZ, DAUER UND ZWECK DER GESELLSCHAFT |
|
Article 1 Corporate Name, Registered Office and Duration Under the name Oculis Holding AG a company limited by shares which is subject to the provisions of articles 620 et seq. of the Swiss Code of Obligations (CO) exists with registered office in Zug (Switzerland) (the "Company"). The duration of the Company is unlimited. |
|
Artikel 1 Firma, Sitz und Dauer Unter der Firma Oculis Holding AG besteht eine Aktiengesellschaft gemäss Artikeln 620 ff. OR mit Sitz in Zug (Schweiz) (die "Gesellschaft"). Die Dauer der Gesellschaft ist unbeschränkt. |
|
Article 2 Purpose The purpose of the Company is to acquire, hold, manage and sell interests in companies of all kinds in Switzerland and abroad, in particular in the areas of research and development in the field of pharmaceutical products, including biological and biotechnological products, |
|
Artikel 2 Zweck Die Gesellschaft bezweckt den Erwerb, das Halten, die Verwaltung und die Veräusserung von Beteiligungen an Gesellschaften aller Art in der Schweiz und im Ausland, insbesondere in den Bereichen Forschung und Entwicklung auf dem Gebiet von pharmazeutischen Produkten, einschliesslich bio-logischen und biotechnologischen Produkten, sowie |
2
as well as the production and commercialisation of such products. |
|
die Herstellung und Kommerzialisierung derartiger Produkte. |
The Company may purchase, hold and sell patents, copyrights, trademarks and other intellectual property rights as well as licenses of any kind. |
|
Die Gesellschaft kann Patente, Urheber-rechte, Marken und andere Immaterialgüterrechte sowie Lizenzen jeder Art erwerben, halten und veräussern. |
The Company may engage in and carry out any and all commercial, financial or other activity, which is directly or indirectly related to the purpose of the Company. The Company may purchase, hold and sell shares or interests in other companies in Switzerland or abroad. It may establish and maintain branches and subsidiaries in Switzerland and abroad. |
|
Die Gesellschaft kann alle kommerziellen, finanziellen und anderen Tätigkeiten ausüben, welche mit dem Zweck der Gesellschaft direkt oder indirekt im Zusammenhang stehen. Die Gesellschaft kann Beteiligungen an anderen Unternehmen im In- und Ausland erwerben, halten und veräussern. Sie kann Zweigniederlassungen und Tochtergesellschaften im In- und Ausland errichten. |
The Company may purchase, hold and sell real estate and carry out other investments. |
|
Die Gesellschaft kann Grundstücke erwerben, verwalten und veräussern sowie Vermögensanlagen anderer Art tätige. |
II. SHARE CAPITAL, SHARES AND SHARE REGISTER |
|
II. AKTIENKAPITAL, AKTIEN UND AKTIENBUCH |
|
Article 3 Share Capital and Shares The share capital of the Company is CHF 401'816.02 and is fully paid-in. It is divided into 40'181'602 registered shares with a nominal value of CHF 0.01 each. |
|
Artikel 3 Aktienkapital und Aktien Das Aktienkapital der Gesellschaft beträgt CHF 401'816.02 und ist voll liberiert. Es ist in 40'181'602 Namenaktien mit einem Nennwert von je CHF 0.01 eingeteilt. |
|
Article 3a Capital Band The Company has a capital band between CHF 365'273.68 (lower limit) and CHF 543'684.52 (upper limit). The Board of Directors is authorized to increase the share capital up to the upper limit at any time and as often as required until 2 March 2028. The increase must be effected by issuing a maximum of 17'841'084 registered shares with a par value of CHF 0.01, to be fully paid up. After a change in par value, the new par value shall also apply within the scope of the capital band. A capital |
|
Artikel 3a Kapitalband Die Gesellschaft hat ein Kapitalband zwischen CHF 365'273.68 (untere Grenze) und CHF 543'684.52 (obere Grenze). Der Verwaltungsrat ist ermächtigt, bis zum 2. März 2028 das Aktienkapital jederzeit und beliebig oft bis zur oberen Grenze zu erhöhen. Die Erhöhung hat durch Ausgabe von maximal 17'841'084 vollständig zu liberierenden Namenaktien im Nennwert von CHF 0.01 zu erfolgen. Nach einer Nennwertveränderung gilt der neue Nennwert auch im Rahmen des Kapitalbandes. Eine Kapitalherabsetzung wird ausgeschlossen. |
3
|
reduction is excluded. If the share capital increases as a result of an increase from conditional capital pursuant to Article 3b, 3c, 43d or 3e of these articles of association, the upper and lower limits of the capital range shall increase in an amount corresponding to such increase in the share capital. |
|
Erhöht sich das Aktienkapital aufgrund einer Erhöhung aus bedingtem Kapital gemäss Art 3b, 3c, 3d oder 3e der Statuten, so erhöhen sich die obere und die untere Grenze des Kapitalbands entsprechend dem Umfang der Erhöhung des Aktienkapitals. |
An increase of the share capital (i) by subscription of shares based on an offer signed by a financial institution, an association, another third party or third parties, followed by an offer to the then existing shareholders of the Company as well as (ii) in partial amounts is permitted. |
|
Eine Erhöhung des Aktienkapitals (i) durch die Zeichnung von Aktien aufgrund eines von einem Finanzinstitut, eines Verbandes, einer anderen Drittpartei oder Drittparteien unterzeichneten Angebots, gefolgt von einem Angebot gegenüber den zu diesem Zeitpunkt bestehenden Aktionären der Gesellschaft sowie (ii) in Teilbeträgen ist zulässig. |
The Board of Directors shall determine the time of the issuance, the issue price, the manner in which the new registered shares have to be paid up, the date from which the registered shares carry the right to dividends, the conditions for the exercise of the preemptive rights and the allotment of preemptive rights that have not been exercised. The Board of Directors may allow the preemptive rights that have not been exercised to expire, or it may place with third parties such rights or registered shares, the preemptive rights of which have not been exercised, at market conditions or use them otherwise in the interest of the Company. |
|
Der Verwaltungsrat soll den Ausgabezeitpunkt, den Bezugspreis, die Art und Weise der Liberierung, das Datum, ab welchem die Aktien zum Bezug einer Dividende berechtigen, die Bedingungen zur Ausübung der Bezugsrechte sowie die Zuteilung nicht ausgeübter Bezugsrechte festlegen. Der Verwaltungsrat kann bestimmen, dass nicht ausgeübte Bezugsrechte verfallen oder er kann Drittparteien solche Rechte oder Aktien, für welche die Bezugsrechte nicht ausgeübt wurden, zu Marktbedingungen zuteilen oder sie sonst im Interesse der Gesellschaft verwenden. |
|
The Board of Directors is authorized to withdraw or limit the preemptive rights of the shareholders and to allot them to third parties: a)
if the issue price of the new registered shares is determined by reference to the market price; or
b)
for the acquisition of an enterprise, part of an enterprise or participations, or for the financing or refinancing of any of such acquisition,
|
|
Der Verwaltungsrat ist ermächtigt, das Bezugsrecht der Aktionäre auszuschliessen oder Dritten zuzuteilen: a)
falls der Ausgabepreis der neuen Aktien anhand des Marktwertes festgelegt wird; oder
b)
für die Übernahme eines Unternehmens, den Teil eines Unternehmens oder Beteiligungen oder für die Finanzierung oder Refinanzierung solcher Erwerbe, oder im Falle einer Aktienplatzierung für die Finanzierung oder Refinanzierung solcher
|
4
|
or in the event of share placement for the financing or refinancing of such placement; or c)
for purposes of broadening the shareholder constituency of the Company in certain financial or investor markets, for purposes of the participation of strategic partners, or in connection with the listing or registration of new registered shares on domestic or foreign stock exchanges; or
d)
for purposes of granting an over-allotment option (Greenshoe) or an option to subscribe additional shares to the respective initial purchaser(s) or underwriter(s) in a placement or sale of registered shares; or
e)
for raising of capital (including private placements) in a fast and flexible way, which probably could not be reached without the exclusion of the statutory pre-emptive right of the existing shareholders;
f)
for other valid grounds in the sense of article 652b para. 2 CO; or
g)
following a shareholder or a group of shareholders acting in concert having accumulated shareholdings in excess of 15% of the share capital registered in the commercial register without having submitted to the other shareholders a takeover offer recommended by the Board of Directors, or for the defense of an actual, threatened or potential takeover bid, in relation to which the Board of Directors, upon consultation with an independent financial adviser retained by it, has not recommended to the shareholders acceptance on
|
|
Platzierungen; oder c)
zum Zweck der Erweiterung der Aktionärskreises der Gesellschaft in bestimmten finanziellen oder Investorenmärkten, für die Zwecke der Beteiligung von strategischen Partnern, oder im Zusammenhang mit der Auflistung oder Meldung neuer Namenaktien an inländischen oder ausländischen Börsen; oder
d)
zum Zweck der Gewährung einer Mehrzuteilungsoption (Greenshoe) oder eine Option zur Zeichnung von zusätzlichen Aktien an die betreffenden Erstkäufer oder Festübernehmer im Rahmen einer Aktienplatzierung oder eines Aktienverkaufs; oder
e)
um Kapital (inklusive durch private Vermittlung) in schneller und flexibler Weise zu beschaffen, welches wahrscheinlich ohne den Ausschluss der gesetzlichen Vorkaufsrechte der existierenden Aktionäre nicht erhoben werden könnte; oder
f)
aus anderen, gemäss Artikel 652 Abs. 2 OR zulässigen Gründen; oder
g)
einem Aktionär oder einer Gruppe von Aktionären folgend, die gemeinsam mehr als 15% des im Handelsregister eingetragenen Aktienkapitals halten und den übrigen Aktionären auf Empfehlung des Verwaltungsrats hin kein Übernahmeangebot unterbreitet haben, oder im Rahmen der Abwehr eines tatsächlichen, drohenden oder etwaigen Übernahmeversuchs, für den der Verwaltungsrat, nach Konsultation eines unabhängigen Finanzberaters, keine Zustimmungsempfehlung abgegeben hat, da das Übernahmeangebot vom Verwaltungsrat den Aktionären gegenüber als finanziell zu wenig angemessen betrachtet wird.
|
5
the basis that the Board of Directors has not found the takeover bid to be financially fair to the shareholders. |
|
|
The acquisition of registered shares and any transfers of registered shares shall be subject to the restrictions specified in article 4 of the articles of association. |
|
Der Erwerb von Namenaktien sowie jeder Transfer von Namenaktien unterliegen den Einschränkungen in Artikel 4 dieser Statuten. |
|
Article 3b Conditional Share Capital for Bonds and Similar Debt Instruments The share capital of the Company may be increased by the maximum amount of CHF 50'000.00 by issuing up to 5'000'000 fully paid-up registered shares with a nominal value of CHF 0.01 each, through the exercise of conversion and/or option rights or warrants or granted in connection with bonds or similar instruments, assumed, issued or to be issued by the Company or by its subsidiaries, including convertible debt instruments. The exercise of the conversion and/or option rights and the waiver of such right shall be made in writing on paper or in electronic form. |
|
Artikel 3b Bedingtes Aktienkapital für Anleihensobligationen oder ähnliche Instrumente Das Aktienkapital der Gesellschaft wird im Maximalbetrag von CHF 50'000.00 erhöht durch Ausgabe von höchstens 5'000'000 vollständig zu liberierenden Namenaktien mit einem Nennwert von je CHF 0.01 durch Ausübung von Wandlungs- und/oder Optionsrechten, welche im Zusammenhang mit von der Gesellschaft oder ihren Tochtergesellschaften übernommenen oder emittierten Anleihensobligationen oder ähnlichen Instrumenten eingeräumt wurden oder werden, einschliesslich Wandelanleihen. Die Form der Ausübung der Wandlungs- und/oder Optionsrechte und des Verzichts auf dieses Recht erfolgt auf schriftlichem Weg auf Papier oder in elektronischer Form. |
Shareholders' subscription rights for these shares are excluded. Shareholders' advance subscription rights with regard to the new bonds or similar instruments may be restricted or excluded by decision of the Board of Directors in order to finance or re-finance the acquisition of companies, parts of companies or holdings, or new investments planned by the Company, or in order to issue convertible bonds or similar instruments on the international capital markets or through private placement. If advance subscription rights are excluded, then (1) the instruments are to be placed at market conditions, (2) the exercise period is not to exceed ten years from the date of issue of option rights and twenty years |
|
Das Bezugsrecht der Aktionäre ist für diese Aktien ausgeschlossen. Das Vorwegzeichnungsrecht der Aktionäre in Bezug auf neue Anleihensobligationen oder ähnliche Instrumente kann durch Beschluss des Verwaltungsrates zu folgenden Zwecken eingeschränkt oder ausgeschlossen werden: Finanzierung und Refinanzierung des Erwerbs von Unternehmen, Unternehmensteilen, Beteiligungen, oder von der Gesellschaft geplanten neuen Investitionen, oder für die Ausgabe von Anleihensobligationen oder ähnlichen Instrumenten auf internationalen Kapitalmärkten oder mittels Privatplatzierungen. Falls Vorwegzeichnungsrechte ausgeschlossen werden, müssen (1) die Instrumente zu Marktkonditionen platziert werden, (2) der Ausübungszeitraum darf zehn Jahre seit dem Ausgabedatum der Optionsrechte und 20 Jahre seit dem Ausgabedatum der Wandlungsrechte nicht |
6
for conversion rights and (3) the conversion or exercise price for the new shares is to be set at least in line with the market conditions prevailing at the date on which the instruments are issued. |
|
überschreiten und (3) der Wandlungs- oder Ausübungspreis für die neuen Aktien muss mindestens gemäss den Marktbedingungen am Ausgabedatum der Instrumente festgelegt werden. |
The acquisition of registered shares through the exercise of conversion rights or warrants and any transfers of registered shares shall be subject to the restrictions specified in article 4 of the articles of Association. |
|
Der Erwerb von Namenaktien durch Ausübung von Wandelrechten oder Warrants sowie sämtliche weiteren Übertragungen von Namenaktien unterliegen den Übertragungsbeschränkungen gemäss Artikel 4 der Statuten. |
|
Article 3c Conditional Share Capital for Employee Benefit Plans The share capital of the Company shall be increased by an amount not exceeding CHF 78'355.44 through the issue of a maximum of 7'835'544 registered shares, payable in full, each with a nominal value of CHF 0.01, in connection with the exercise of option rights or other equity-linked instruments granted to any employee of the Company or a subsidiary, and any consultant, members of the Board of Directors, or other person providing services to the Company or a subsidiary. The exercise of the option rights and the waiver of such right shall be made in writing on paper or in electronic form. |
|
Artikel 3c Bedingtes Aktienkapital für Mitarbeiterbeteiligungspläne Das Aktienkapital kann durch die Ausgabe von höchstens 7'835'544 voll zu liberierenden Namenaktien im Nennwert von je CHF 0.01 um höchstens CHF 78'355.44 durch Ausübung von Optionsrechten oder anderen eigenkapitalbasierten Instrumenten erhöht werden, welche Mitarbeitenden der Gesellschaft oder ihrer Tochtergesellschaften, Personen in vergleichbaren Positionen, Beratern, Verwaltungsratsmitgliedern oder anderen Personen, welche Dienstleistungen zu Gunsten der Gesellschaft erbringen, gewährt wurden. Die Form der Ausübung der Optionsrechte und des Verzichts auf dieses Recht erfolgt auf schriftlichem Weg auf Papier oder in elektronischer Form. |
Shareholders' subscription rights shall be excluded with regard to these shares. These new registered shares may be issued at a price below the current market price. The Board of Directors shall specify the precise conditions of issue including the issue price of the shares. |
|
Das Bezugsrecht der Aktionäre ist für diese Aktien ausgeschlossen. Diese neuen Namenaktien können zu einem Preis unter dem aktuellen Marktpreis ausgegeben werden. Der Verwaltungsrat legt die genauen Bedingungen für die Ausgabe, einschliesslich des Ausgabepreises der Aktien fest. |
The acquisition of registered shares in connection with employee participation and any further transfers of registered shares shall be subject to the restrictions specified in article 4 of the articles of association. |
|
Der Erwerb von Namenaktien im Zusammenhang der Mitarbeiterbeteiligung sowie sämtliche weiteren Übertragungen von Namenaktien unterliegen den Übertragungsbeschränkungen gemäss Artikel 4 der Statuten. |
7
|
Article 3d Conditional Share Capital for EBAC-Warrants The share capital shall be increased by an amount not exceeding CHF 44'032.94 through the issue of a maximum of 4'403'294 registered shares, payable in full, each with a nominal value of CHF 0.01. The increase of the share capital shall occur in connection with the share capital increase of 1 March 2023 through the exercise of conversion and/or option rights, which were assumed from, and allocated by, European Biotech Acquisition Corp. with registered seat in George Town, Cayman Islands and business address at EPFL Innovation Park Building D, 1015 Lausanne, Switzerland (EBAC), at an exercise price of USD 11.50 on the basis of a Warrant Assumption Agreement between the Company and Continental Stock Transfer & Trust Company, with registered seat in New York (USA) as Warrant Agent. The exercise of conversion and/or option rights and the waiver of such right shall be made in writing on paper or in electronic form. |
|
Artikel 3d Bedingtes Aktienkapital für EBAC-Warrants Das Aktienkapital kann durch die Ausgabe von höchstens 4'403'294 voll zu liberierenden Namenaktien im Nennwert von je CHF 0.01 um höchstens CHF 44'032.94 erhöht werden. Die Erhöhung des Aktienkapitals erfolgt im Zusammenhang mit der Kapitalerhöhung vom 1. März 2023 durch die Ausübung von Wandlungs- und/oder Optionsrechten, welche von European Biotech Acquisition Corp. mit Sitz in George Town, Cayman Islands und Geschäftsadresse an der EPFL Innovation Park Building D, 1015 Lausanne, Schweiz (EBAC), mit einem Ausübungspreis von USD 11.50 auf der Grundlage eines Warrant Assumption Agreements zwischen der Gesellschaft und Continental Stock Transfer & Trust Company, mit Sitz in New York (USA) als Warrant Agent, übernommenen und eingeräumt wurden. Die Form der Ausübung der Wandlungs- und/oder Optionsrechte und des Verzichts auf dieses Recht erfolgt auf schriftlichem Weg auf Papier oder in elektronischer Form. |
Only the bearers of such conversion and/or option rights shall be entitled to obtain such new registered shares. The terms and conditions of the exercise and/or conversion rights, such as the exercise and/or conversion price and period, the time of entitlement to dividends and the type of contributions shall be defined by the Board of Directors |
|
Zum Bezug der neuen Namenaktien sind die Inhaber von Wandlungs- und/oder Optionsrechten berechtigt. Die Bezugsbedingungen, wie Ausübungs- und/oder Konvertierungspreis und -frist, Zeitpunkt der Dividendenberechtigung und Art der Einlagen, werden durch den Verwaltungsrat festgelegt. |
The shareholders' subscription rights are excluded for these shares. The shareholders’ advance subscription rights regarding these Warrants are excluded to abide by the obligations stemming from the Business Combination Agreement dated 17 October 2022 between EBAC and Oculis SA with registered seat in |
|
Das Bezugsrecht der Aktionäre ist für diese Aktien ausgeschlossen. Das Vorwegzeichnungsrecht der Aktionäre in Bezug auf diese Warrants ist ausgeschlossen, um die im Business Combination Agreement vom 17. Oktober 2022 zwischen EBAC und Oculis SA mit Sitz in Ecublens (VD), Schweiz, eingegangenen und von der Gesellschaft übernommenen Verpflichtungen zu erfüllen. |
8
Ecublens (VD), Switzerland, and assumed by the Company. |
|
|
The acquisition of registered shares through the exercise of conversion and/or option rights and the further transfer of registered shares shall be subject to the restrictions specified in article 4 of the articles of association. |
|
Der Erwerb von Namenaktien durch die Ausübung von Wandlungs- und/oder Optionsrechten sowie sämtliche weiteren Übertragungen von Namenaktien unterliegen den Übertragungsbeschränkungen gemäss Artikel 4 der Statuten. |
|
Article 3e Conditional Share Capital for Earn-Out Options The share capital of the Company shall be increased by an amount not exceeding CHF 3'701.03 through the issue of a maximum of 370'103 registered shares, payable in full, each with a nominal value of CHF 0.01, in connection with the exercise of option rights granted in connection with the Business Combination Agreement dated 17. October 2022 between European Biotech Acquisition Corp. with registered seat in George Town, Cayman Islands and business address at EPFL Innovation Park Building D, 1015 Lausanne, Switzerland (EBAC) and Oculis SA with registered seat in Ecublens (VD), Switzerland, to any employee of the Company or a subsidiary, and any consultant, members of the Board of Directors, or other person providing services to the Company or a subsidiary (earn-out options). The exercise of the option rights and the waiver of such right shall be made in writing on paper or in electronic form.
|
|
Artikel 3e Bedingtes Aktienkapital für Earn-Out Optionen Das Aktienkapital kann durch die Ausgabe von höchstens 370'103 voll zu liberierenden Namenaktien im Nennwert von je CHF 0.01 um höchstens CHF 3'701.03 durch Ausübung von Optionsrechten erhöht werden, welche Mitarbeitenden der Gesellschaft oder ihrer Tochtergesellschaften, Personen in vergleichbaren Positionen, Beratern, Verwaltungsratsmitgliedern oder anderen Personen, welche Dienstleistungen zu Gunsten der Gesellschaft erbringen, und im Zusammenhang mit dem Business Combination Agreement vom 17. Oktober 2022 zwischen European Biotech Acquisition Corp. mit Sitz in George Town, Cayman Islands und Geschäftsadresse an der EPFL Innovation Park Building D, 1015 Lausanne, Schweiz (EBAC) und Oculis SA mit Sitz in Ecublens (VD), Schweiz zusätzlich eingeräumt wurden (earn-out Optionen). Die Form der Ausübung der Optionsrechte und des Verzichts auf dieses Recht erfolgt auf schriftlichem Weg auf Papier oder in elektronischer Form. |
Shareholders' subscription rights shall be excluded with regard to these shares. These new registered shares may be issued at a price below the current market price. The Board of Directors shall specify the precise conditions of issue including the issue price of the shares. |
|
Das Bezugsrecht der Aktionäre ist für diese Aktien ausgeschlossen. Diese neuen Namenaktien können zu einem Preis unter dem aktuellen Marktpreis ausgegeben werden. Der Verwaltungsrat legt die genauen Bedingungen für die Ausgabe, einschliesslich des Ausgabepreises der Aktien. |
9
The acquisition of registered shares in connection with employee participation and any further transfers of registered shares shall be subject to the restrictions specified in article 4 of the articles of association. |
|
Der Erwerb von Namenaktien im Zusammenhang der Mitarbeiterbeteiligung sowie sämtliche weiteren Übertragungen von Namenaktien unterliegen den Übertragungsbeschränkungen gemäss Artikel 4 der Statuten. |
|
Article 4 Share Register The Company shall maintain a share register in which it shall register the name, first name and place of residence (in case of legal persons the place of incorporation) of the owners and usufructuaries of its registered shares. Natural and legal persons as well as legal representatives of minors etc. entitled by law to the voting rights of a share which they do not own will be noted in the share register upon request. |
|
Artikel 4 Aktienbuch Die Gesellschaft führt ein Aktienbuch, worin die Eigentümer und Nutzniesser von Namenaktien mit Namen, Vornamen und Wohnort (bei juristischen Personen Sitz) eingetragen werden. Natürliche und juristische Personen sowie gesetzliche Vertreter von Minderjährigen usw., welchen kraft Gesetzes Stimmrechte eines Anteils zukommen, den sie nicht besitzen, werden auf Anfrage im Aktienregister angemerkt. |
10
Upon request, acquirers of shares will be registered in the share register without limitation as shareholders if they expressly certify that they acquired the shares in their own name and for their own account. |
|
Erwerber von Aktien werden auf Gesuch hin ohne Begrenzung als Aktionäre mit Stimmrecht im Aktienregister eingetragen, falls sie ausdrücklich erklären, die Aktien im eigenen Namen und auf eigene Rechnung erworben zu haben. |
Persons who do not expressly declare in the registration application that they are holding the shares on their own account (thereafter: nominees) shall forthwith be entered on the share register as shareholders with voting rights up to a maximum of 3% of the share capital. Beyond that limit, registered shares of nominees shall only be entered as voting if the nominees in question confirm in writing that they are willing to disclose the names, addresses and shareholdings of the persons on whose account they hold 0.5% or more of the share capital. The Board of Directors concludes agreements with nominees that among other things govern the representation of shareholders and the voting rights. |
|
Personen, die im Eintragungsgesuch nicht ausdrücklich erklären, die Aktien für eigene Rechnung zu halten (nachstehend: Nominees) werden ohne weiteres bis maximal 3% des jeweils ausstehenden Aktienkapitals mit Stimmrecht im Aktienbuch eingetragen. Über diese Limite hinaus werden Namenaktien von Nominees nur dann mit Stimmrecht eingetragen, wenn sich der betreffende Nominee schriftlich bereit erklärt, gegebenenfalls die Namen, Adressen und Aktienbestände derjenigen Person offenzulegen, für deren Rechnung er 0.5% oder mehr des jeweils ausstehenden Aktienkapitals hält. Der Verwaltungsrat schliesst mit Nominees Vereinbarungen ab, die unter anderem die Vertretung der Aktionäre und der Stimmrechte regeln. |
After hearing the registered shareholder or nominee, the Board of Directors may remove entries in the share register with retroactive effect as per the date of entry, if such entry was based on false information. The party affected must be informed of such removal immediately. |
|
Nach Anhörung des eingetragenen Aktionärs oder Nominees, kann der Verwaltungsrat die Eintragungen im Aktienregister rückwirkend nach dem Datum der Eintragung entfernen, wenn ein solcher Eintrag aufgrund falscher Angaben erfolgte. Der Betroffene muss über eine solche Entfernung sofort informiert werden. |
No individual or legal entity may, directly or indirectly, formally, constructively or beneficially own (as defined in the next paragraph below) or otherwise control voting rights ("Controlled Shares") with respect to 15% or more of the registered share capital recorded in the Commercial Register except if such individual or legal entity has submitted prior to the acquisition of such Controlled Shares an orderly tender offer to all shareholders with a minimum price of the higher of (i) the volume weighted average price of the |
|
Weder eine Einzelperson, noch eine juristische Person kann, direkt oder indirekt, formell, konstruktiv oder vorteilhaft (wie im nächsten Abschnitt unten definiert) oder sonst wie das Stimmrecht ("Kontrollierte Aktien") hinsichtlich 15% oder mehr des im Handelsregister registrierten Aktienkapitals innehaben oder kontrollieren. Eine Ausnahme besteht dann, wenn diese Einzelperson oder juristische Person vor der Übernahme solcher Kontrollierter Aktien allen Aktionären eine ordentliche Offerte mit einem Minimalpreis stellt, wovon der höhere Preis, der entweder (i) dem gewichteten Durchschnittskurs der letzten 60 Handelstage vor der Veröffentlichung der |
11
last 60 trading days prior to the publication of the tender offer or (ii) the highest price paid by such individual or legal entity in the 12 months preceding to the publication of the tender offer. Those associated through capital, voting power, joint management or in any other way, or joining for the acquisition of shares, shall be regarded as one person. The registered shares exceeding the limit of 15% and not benefiting from the exemption regarding a tender offer shall be entered in the share register as shares without voting rights. |
|
Übernahmeofferte oder (ii) dem höchsten bezahlten Preis durch diese Einzelperson oder juristische Person während der 12 Monate vor der Veröffentlichung der Übernahmeofferte entspricht, der relevante Preis darstellt. Die durch Kapital, Stimmrecht, gemeinsame Führung oder in anderer Weise oder durch Beitritt zur Übernahme der Aktien verbundenen Personen, sind als eine Person zu betrachten. Die Namenaktien, welche die Limite von 15% übersteigen und nicht von der Ausnahme mit Bezug auf die Übernahmeofferte profitieren, sollen im Aktienbuch als Aktien ohne Stimmrecht verzeichnet werden. |
|
For the purposes of this article 4, "Controlled Shares" in reference to any individual or entity means: (a) all shares of the Company directly, indirectly or constructively owned by such individual or entity; provided that (i) shares owned, directly or indirectly, by or for a partnership, or trust or estate will be considered as being owned proportionately by its partners, or beneficiaries; and (ii) shares owned, directly or indirectly, by or for a corporation will be considered as being owned proportionately by any shareholder owning 50% or more of the outstanding voting shares of such corporation; and (iii) shares subject to options, warrants or other similar rights shall be deemed to be owned; and (b) all shares of the Company directly, indirectly or beneficially owned by |
|
Im Rahmen dieses Artikel 4 bedeuten "Kontrollierte Aktien" in Bezug auf jegliche Einzelperson oder juristische Person: (a) alle Aktien der Gesellschaft, die direkt, indirekt oder konstruktiv von einer solchen Einzelperson oder juristischen Person gehalten werden; vorausgesetzt dass (i) Aktien, die direkt oder indirekt durch oder für eine Personengesellschaft oder einen Trust oder eine Vermögensmasse gehalten werden, proportional auf die Partner oder Begünstigten aufgeteilt werden; und (ii) Aktien, die direkt oder indirekt durch oder für eine Gesellschaft gehalten werden, proportional auf jeden Aktionär, der 50% oder mehr der ausgegebenen Stimmrechtsaktien besitzt, aufgeteilt werden; und (iii) Aktien, die in Abhängigkeit zu Optionen, Bezugsrechten oder anderen ähnlichen Rechten stehen, als Eigentum gelten; und (b) alle Aktien der Gesellschaft, die direkt, indirekt oder vorteilhaft durch eine solche Einzelperson oder eine juristische Person gehalten werden, vorausgesetzt dass (i) ein begünstigter Eigentümer eines Wertpapiers jede Person umfasst, die direkt oder indirekt, durch jede Art von Vertrag, Vereinbarung, Einvernehmen, Bindung oder |
12
|
such individual or entity; provided that (i) a beneficial owner of a security includes any person who, directly or indirectly, through any contract, arrangement, understanding, relationship, or otherwise alone or together with other such persons has or shares: (1) voting power which includes the power to vote, or to direct the voting of, such security; and/or (2) investment power which includes the power to dispose, or to direct the disposition of, such security. (ii) Any person who, directly or indirectly, creates or uses a trust, proxy, power of attorney, pooling arrangement or any other contract, arrangement, or device with the purpose or effect of divesting such person of beneficial ownership of shares of the Company or preventing the vesting of such beneficial ownership as part of a plan or scheme to evade the provisions of these articles of association shall be deemed to be the beneficial owner of such shares. (iii) A person shall be deemed to be the beneficial owner of shares if that person has the right to acquire beneficial ownership of |
|
anderweitig allein oder mit anderen Personen gemeinsam hat oder teilt: (1) das Stimmrecht, welches das Recht zur Stimmabgabe, oder zur Leitung der Stimme eines solchen Wertpapiers umfasst; und/oder (2) das Investitionsrecht, welches die Verfügungsmacht oder ein Recht zur Bestimmung über die Verfügung eines solchen Wertpapiers umfasst. (ii) Jede Person, die, direkt oder indirekt, einen Trust, Stellvertretung, Vollmacht, Pooling-Vertrag oder jede andere Form von Vertrag, mit dem Zweck oder Ziel schafft oder benutzt, um eine Person von ihren wirtschaftlichen Begünstigungen aus dem Eigentum an den Aktien der Gesellschaft zu entheben oder zur Verhinderung der Ausübung eines solchen begünstigenden Eigentums als Teil eines Plans oder Vorhabens zur Umgehung der Regelungen in diesen Statuten, soll als begünstigter Eigentümer solcher Aktien gesehen werden. (iii) Eine Person soll als begünstigter Eigentümer von Aktien eingestuft werden, wenn diese Person das Recht hat, ein begünstigendes Eigentum an solchen Aktien innerhalb von 60 Tagen zu erwerben, inklusive, aber nicht beschränkt auf jegliches erworbenes Recht: (A) durch die Ausübung jeglicher Option, jedes Bezugsrechts oder sonstigen Rechts; (B) durch die Umwandlung eines Wertpapiers; (C) aufgrund der Befugnis, einen Trust, ein Vermögensverwaltungskonto oder ähnliche Verhältnisse zu widerrufen oder (D) in Zusammenhang mit der automatischen Auflösung eines Trusts, Vermögensverwaltungskontos oder eines ähnlichen Verhältnisses. |
13
such shares within 60 days, including but not limited to any right acquired: (A) through the exercise of any option, warrant or right; (B) through the conversion of a security; (C) pursuant to the power to revoke a trust, discretionary account, or similar arrangement; or (D) pursuant to the automatic termination of a trust, discretionary account or similar arrangement. |
|
|
The limit of 15% of the registered share capital also applies to the subscription for, or acquisition of, registered shares by exercising option or convertible rights arising from registered or bearer securities or any other securities issued by the Company or third parties, as well as by means of exercising purchased preemptive rights arising from either registered or bearer shares. The registered shares exceeding the limit of 15% shall be entered in the share register as shares without voting rights. |
|
Die Grenze von 15% des eingetragenen Aktienkapitals gilt auch für zur Zeichnung von, oder Akquisition von Namenaktien durch Ausübung einer Option oder umwandelbaren Rechte, welche aus Namen- oder Inhaberaktien hervor gehen oder jeder anderen von der Gesellschaft oder Dritten ausgegebenen Sicherheit, sowie durch die Ausübung von erworbenen Vorkaufsrechten, welche entweder aus Namen- oder Inhaberaktien hervorgehen. Die Namenaktien, welche die Grenze von 15% übersteigen, sind im Aktienbuch als Aktien ohne Stimmrecht einzutragen. |
The Board of Directors may in special cases approve exceptions to the above regulations. The Board of Directors is in addition authorized, after due consultation with the person concerned, to delete with retroactive effect entries in the share register which were effected on the basis of false information. |
|
Der Verwaltungsrat kann in besonderen Fällen Ausnahmen zu den oben genannten Regelungen genehmigen. Der Verwaltungsrat ist zusätzlich berechtigt, nach angemessener Anhörung der betreffenden Person, Einträge ins Aktienbuch, welche aufgrund falscher Informationen erfolgten, rückwirkend zu löschen. |
Article 5 Reporting Obligation of the Shareholder and Register of Beneficial Owners |
|
Artikel 5 Meldepflicht des Aktionärs und Verzeichnis der wirtschaftlich berechtigten Personen |
Any person who, alone or in concert with third parties, acquires shares in the Company and thereby reaches or exceeds the threshold of 25% of the share capital or voting rights must notify the Company within one month of the first name, last |
|
Wer allein oder in gemeinsamer Absprache mit Dritten Aktien der Gesellschaft erwirbt und dadurch den Grenzwert von 25% des Aktienkapitals oder der Stimmrechte erreicht oder überschreitet, muss der Gesellschaft innert Monatsfrist den Vor- und den Nach-namen und die Adresse der natürlichen Person |
14
name and address of the natural person on whose behalf he is ultimately acting (beneficial owner). |
|
melden, für die er letztendlich handelt (wirtschaftlich berechtigte Person). |
The shareholder must notify the company within three months of any change in the first or last name or address of the beneficial owner. |
|
Der Aktionär muss der Gesellschaft innert drei Monaten jede Änderung des Vor- oder des Nachnamens oder der Adresse der wirtschaftlich berechtigten Person melden. |
The Board of Directors shall keep a register of the beneficial owners reported to the Company. This register contains the first and last name as well as the address of the beneficial owners. The register must be kept in such a way that it can be accessed in Switzerland at any time. |
|
Der Verwaltungsrat führt ein Verzeichnis über die der Gesellschaft gemeldeten wirtschaftlich berechtigten Personen. Dieses Verzeichnis enthält den Vor- und den Nachnamen sowie die Adresse der wirtschaftlich berechtigten Personen. Das Verzeichnis muss so geführt werden, dass in der Schweiz jederzeit darauf zugegriffen werden kann. |
As long as the shareholder has not fulfilled his reporting obligations, the membership rights associated with the shares whose acquisition must be reported shall be suspended. The property rights attached to such shares may only be exercised by the shareholder once he has complied with his notification obligations. If the shareholder fails to comply with his reporting obligations within one month after the acquisition of the shares, the property rights shall be forfeited. If the shareholder makes the notification at a later date, he may assert the property rights accruing as of that date. The Board of Directors shall ensure that no shareholders exercise their rights in breach of the reporting obligations. |
|
Solange der Aktionär seinen Meldepflichten nicht nachgekommen ist, ruhen die Mitgliedschaftsrechte, die mit den Aktien verbunden sind, deren Erwerb gemeldet werden muss. Die Vermögensrechte, die mit solchen Aktien verbunden sind, kann der Aktionär erst geltend machen, wenn er seinen Meldepflichten nachgekommen ist. Kommt der Aktionär seinen Meldepflichten nicht innert eines Monats nach dem Erwerb der Aktien nach, so sind die Vermögensrechte verwirkt. Holt er die Meldung zu einem späteren Zeitpunkt nach, so kann er die ab diesem Zeitpunkt entstehenden Vermögensrechte geltend machen. Der Verwaltungsrat stellt sicher, dass keine Aktionäre unter Verletzung der Melde-pflichten ihre Rechte ausüben. |
15
|
Article 6 Share Certificates and Intermediated Securities The Company may issue its shares in any legally permissible form, namely in the form of individual certificates, global certificates, simple uncertificated securities pursuant to article 973c CO or registered uncertificated securities pursuant to article 973d CO and have them managed as intermediated securities. |
|
Artikel 6 Aktienzertifikate und Bucheffekten Die Gesellschaft kann ihre Aktien in jeder gesetzlich zulässigen Form, namentlich in Form von Einzelurkunden, Globalurkunden, einfachen Wertrechten nach Artikel 973c OR oder Registerwert-rechten nach Artikel 973d OR ausgeben und als Bucheffekten führen lassen. |
Within the legal framework, the Company is free to convert its shares issued in one of these forms into another form at any time and without the consent of the shareholders, and to withdraw shares held as intermediated securities from the custody system. It shall bear the costs thereof. |
|
Der Gesellschaft steht es im Rahmen der gesetzlichen Vorgaben frei, ihre in einer dieser Formen ausgegebenen Aktien jederzeit und ohne Zustimmung der Aktionäre in eine andere Form umzuwandeln sowie als Bucheffekten geführte Aktien aus dem Verwahrungssystem zurückzuziehen. Sie trägt dafür die Kosten. |
The shareholder shall not be entitled to the certification of membership rights in the form of physical securities or to the conversion of shares issued in a certain form into another form. However, the shareholder may at any time request the Company to issue a written confirmation of the shares held by him in accordance with the share register. |
|
Der Aktionär hat keinen Anspruch auf wertpapiermässige Verbriefung der Mitgliedschaftsrechte oder auf Umwandlung von in bestimmter Form ausgegebenen Aktien in eine andere Form. Der Aktionär kann jedoch von der Gesellschaft jederzeit die Ausstellung einer schriftlichen Bescheinigung über die von ihm gemäss Aktienbuch gehaltenen Aktien verlangen. |
The transfer of simple uncertificated securities pursuant to article 973c CO and registered uncertificated securities pursuant to article 973d CO as well as the provision of security for such uncertificated securities shall be governed by the provisions of the CO. |
|
Die Übertragung von einfachen Wertrechten nach Artikel 973c OR und Registerwertrechten nach Artikel 973d OR sowie die Bestellung von Sicherheiten an diesen Wert-rechten richten sich nach den Bestimmungen des OR. |
The transfer of intermediated securities and the provision of security for such intermediated securities shall be governed by the provisions of the Swiss Intermediated Securities Act. |
|
Die Übertragung von Bucheffekten und die Bestellung von Sicherheiten an diesen Bucheffekten richten sich nach den Bestimmungen des Bucheffektengesetzes. |
Article 7 Exercise of Shareholders Rights |
|
Artikel 7 Ausübung von Aktionärsrechten |
16
The shares are indivisible and the Company recognizes only one single representative per share. |
|
Die Aktien sind unteilbar und die Gesellschaft anerkennt nur einen einzigen Vertreter pro Aktie. |
The right to vote and the other rights pertaining to a registered share may only be exercised by a shareholder, an usufructuary or a nominee who is registered with the right to vote in the share register and by persons who are entitled by law to the voting rights of a share. |
|
Das Stimmrecht und die anderen zu einer Namenaktien gehörenden Rechte dürfen nur von einem Aktionär, einem Nutzniesser oder Nominee, dessen Stimmrecht im Aktienregister eingetragen ist und von Personen, welchen kraft Gesetzes die Stimmrechte einer Aktie zustehen, ausgeübt werden. |
III. CORPORATE STRUCTURE |
|
III. ORGANISATION DER GESELLSCHAFT |
|
Article 8 Corporate Bodies The corporate bodies are: A. the General Meeting; B. the Board of Directors; C. the Auditors. |
|
Artikel 8 Gliederung Die Gesellschaftsorgane sind: A. die Generalversammlung; B. der Verwaltungsrat; C. die Revisionsstelle. |
IV. GENERAL MEETING |
|
IV. GENERALVERSAMMLUNG |
|
Article 9 Powers The General Meeting is the supreme body of the Company. It has the following non delegable powers: a)
to adopt and amend the articles of association (articles 652g, 653g und 653i CO remain reserved);
b)
to elect and remove the members of the Board of Directors, the Chairman of the Board of Directors, the members of the Compensation Committee, the Auditors and the Independent Proxy;
c)
to approve the management report and the annual accounts and to determine the allocation of profits, in
|
|
Artikel 9 Befugnisse Oberstes Organ der Gesellschaft ist die Generalversammlung. Ihr stehen folgende unübertragbare Befugnisse zu: a)
Festsetzung und Änderung der Statuten (Artikel 652g, 653g und 653i OR bleiben vorbehalten);
b)
Wahl und Abberufung der Mitglieder des Verwaltungsrats, des Präsidenten des Verwaltungsrats, der Mitglieder des Vergütungsausschusses, der Revisionsstelle und des unabhängigen Stimmrechtsvertreters;
c)
Genehmigung des Lageberichts und der Jahresrechnung sowie Beschlussfassung über die Verwendung des Bilanzgewinnes,
|
17
|
particular with regard to dividends and bonus payments; d)
to determine the interim dividend and the approval of the required interim financial statements;
e)
to make a resolution on the repayment of the statutory capital reserve;
f)
to discharge the members of the Board of Directors and of the Executive Committee;
g)
delisting of the Company's equity securities;
h)
to approve the total compensation paid to the Board of Directors and the Executive Committee as per article 34 and article 35 below;
i)
to pass resolutions concerning all matters which are reserved to the authority of the General Meeting by law or by the articles of association.
|
|
insbesondere die Festsetzung der Dividende und der Tantieme; d)
Festsetzung der Zwischendividende und Genehmigung des dafür erforderlichen Zwischenabschlusses;
e)
Beschlussfassung über die Rückzahlung der gesetzlichen Kapitalreserve;
f)
Entlastung der Mitglieder des Verwaltungsrates und der Geschäftsleitung;
g)
Dekotierung der Beteiligungspapiere der Gesellschaft;
h)
Genehmigung der Gesamtvergütungen des Verwaltungsrats und der Geschäftsleitung nach Massgabe von Artikel 34 und Artikel 35 hiernach;
i)
Beschlussfassung über die Gegenstände, die der Generalversammlung durch das Gesetz oder die Statuten vorbehalten sind.
|
|
Article 10 Ordinary General Meeting The Ordinary General Meeting shall be held annually within six months after the end of the business year at such time and at such location, which may be within or outside Switzerland, as determined by the Board of Directors. |
|
Artikel 10 Ordentliche Generalversammlung Die ordentliche Generalversammlung findet jährlich innerhalb von sechs Monaten nach Abschluss des Geschäftsjahres statt, zum Zeitpunkt und an einem Ort, der innerhalb oder ausserhalb der Schweiz sein kann, gemäss Festlegung durch den Verwaltungsrat. |
18
|
Article 11 Extraordinary General Meeting Extraordinary General Meetings may be called by resolution of the General Meeting, the Auditors or the Board of Directors, or by shareholders with voting powers, provided they represent at least 5% of the share capital and who submit (a)(1) a request signed by such shareholder(s) that specifies the item(s) to be included on the agenda, (2) the respective proposals of the shareholders and (3) evidence of the required shareholdings recorded in the share register and (b) such other information as would be required to be included in a proxy statement pursuant to the rules of the country where the Company's shares are primarily listed. |
|
Artikel 11 Ausserordentliche Generalversammlung Ausserordentliche Generalversammlungen können einberufen werden durch Beschluss der ordentlichen Generalversammlung, durch die Revisionsstelle oder den Verwaltungsrat oder durch stimmberechtigte Aktionäre, sofern sie mindestens 5% des Aktienkapitals erreichen und die Folgendes einreichen: (a)(1) einen unterschriebenen Antrag dieser Aktionäre, welcher die Traktanden angibt, die auf die Traktandenliste gesetzt werden, (2) die entsprechenden Anträge der Aktionäre und (3) den Nachweis der erforderlichen Beteiligung dieser Aktionäre aufgrund des Aktienregisters und (b) alle anderen Informationen, die für eine Vollmacht nach den Regeln des Landes, in welchem die Aktien des Unternehmens hauptsächlich eingetragen sind, erforderlich wären. |
|
Article 12 Notice and Agenda of Shareholders' Meetings Notice of a General Meeting of Shareholders shall be given by the Board of Directors or, if necessary, by the Auditor, not later than 20 calendar days prior to the date of the General Meeting of Shareholders. Notice of the General Meeting of Shareholders shall be given by way of a one-time announcement in the official means of publication of the Company pursuant to article 48 of these articles of association. The notice period shall be deemed to have been observed if notice of the General Meeting of Shareholders is published in such official means of publication, it being understood that the date of publication shall not be computed in the notice period. Shareholders of record may in addition be informed of the General Meeting of Shareholders by ordinary mail or e-mail. |
|
Artikel 12 Mitteilung und Traktanden der Generalversammlung Die Mitteilung einer Generalversammlung erfolgt durch den Verwaltungsrat oder gegebenenfalls durch die Revisionsstelle, spätestens 20 Kalendertage vor dem Datum der Generalversammlung. Die Mitteilung der Generalversammlung erfolgt durch eine einmalige Bekanntmachung in den amtlichen Publikationsmitteln der Gesellschaft gemäss Artikel 48 dieser Statuten. Die Frist gilt als eingehalten, wenn Ankündigung der Generalversammlung im offiziellen Publikationsmittel veröffentlicht wurde, wobei das Datum der Veröffentlichung nicht in die Mitteilungsfrist eingerechnet werden darf. Eingetragene Aktionäre können zusätzlich per Post oder E-Mail über die Generalversammlung informiert werden. |
The convocation of an extraordinary general meeting may also be requested in |
|
Die Einberufung einer ausserordentlichen Generalversammlung kann auch von einem oder |
19
writing, indicating the agenda items and the proposals and, in case of elections, the names of the nominated candidates, by one or more shareholders together representing at least 5% of the share capital or the voting rights. |
|
mehreren Aktionären, die zusammen mindestens 5% des Aktienkapitals oder der Stimmen vertreten, schriftlich unter Angabe des Verhandlungsgegenstandes und des Antrages, bei Wahlen der Namen der vorgeschlagenen Kandidaten, verlangt werden. |
The Board of Directors shall state the matters on the agenda. |
|
Der Verwaltungsrat setzt die Verhandlungsgegenstände auf die Traktandenliste. |
The notice of a General Meeting of Shareholders shall specify the items on the agenda and the proposals of the Board of Directors and the shareholder(s) who requested that a General Meeting of Shareholders be held or an item be included on the agenda, and, in the event of elections, the name(s) of the candidate(s) that has or have been put on the ballot for election. |
|
Die Mitteilung der Genrealversammlung hat die Traktanden und die Anträge des Verwaltungsrates und der Aktionäre, welche beantragt haben, dass eine Generalversammlung abgehalten werden oder ein Traktandum auf die Traktandenliste gesetzt werden soll zu enthalten sowie, im Falle von Wahlen, die Namen der Kandidaten, welche auf den Wahlzettel gesetzt wurden. |
Shareholders, together representing more than 0.5% of the share capital or the voting rights, may demand that an item be placed on the agenda. Such request must be made in writing at least 70 days prior to the meeting by indicating the agenda items and the proposals. |
|
Aktionäre, die zusammen mindestens über 0.5% des Aktienkapitals oder der Stimmen vertreten, können die Traktandierung eines Verhandlungsgegenstandes verlangen. Dies hat mindestens 70 Tage vor der Versammlung schriftlich unter Angabe der Verhandlungsgegenstände und Anträge zu erfolgen. |
Each request for inclusion of an item on the agenda must include (i) a brief description of the business desired to be brought before the meeting and the reasons for conducting such business at the meeting; (ii) the name and address, as they appear on the Company's register of shareholders, of the shareholder proposing such business; (iii) the number of shares of the Company which are beneficially owned by such shareholder; (iv) the dates upon which the shareholder acquired such shares; (v) documentary support for any claim of beneficial ownership; (vi) any material interest of such shareholder in such business; and (vii) a statement in support of the matter and, for proposals sought to be included in the Company's proxy statement, any |
|
Jeder Antrag auf Aufnahme eines Traktandums hat zu enthalten: (i) eine kurze Zusammenfassung des Geschäfts, welches der Generalversammlung vorgelegt werden soll, sowie eine Begründung, weshalb an der Versammlung darüber entschieden werden soll; (ii) den Namen und die Adresse des Gesuchstellenden Aktionärs, wie sie im Aktienbuch der Gesellschaft eingetragen sind; (iii) die Anzahl Aktien der Gesellschaft, die in der wirtschaftlichen Berechtigung des Aktionärs stehen; (iv) die Daten, an denen der Aktionär seine Aktien erworben hat; (v) erforderliche Nachweise bei allfälligen Ansprüchen von wirtschaftlicher Berechtigung; (vi) jegliches materielle Interesse des Aktionärs im Zusammenhang mit diesem Geschäft; und (vii) eine Stellungnahme zum fraglichen Punkt und, für Anträge, welche der Aktionärsinformation durch die Gesellschaft beigefügt werden sollen, jede andere Information, welche die |
20
other information required by Securities and Exchange Commission Rule "14a-8". |
|
Securities and Exchange Commission Rule "14a-8" verlangt. |
In addition, if the shareholder intends to solicit proxies from the shareholders of the Company, such shareholder shall notify the Company of this intent in accordance with Securities and Exchange Commission Rule "14a-4" and/or Rule "14a-8". |
|
Für den Fall, dass ein Aktionär gedenkt, die Stimmrechtsvertretung von anderen Aktionären der Gesellschaft zu erlangen, hat dieser Aktionär die Gesellschaft über diese Absicht gemäss der Securities and Exchange Commission Rule "14a-4" und/oder Rule "14a-8" zu informieren. |
No resolution may be passed at a General Meeting of Shareholders concerning an item in relation to which due notice was not given. Proposals made during a General Meeting of Shareholders to (i) convene a extraordinary General Meeting or (ii) initiate a special investigation in accordance with article 697a of the Swiss Code of Obligations are not subject to the due notice requirement set forth herein. |
|
An der Generalversammlung darf kein Beschluss über ein Traktandum getroffen werden, über den nicht mit entsprechender Vorlaufzeit informiert worden ist. Anträge, die während der Generalversammlung gestellt werden, führen zu (i) einer ausserordentlichen Generalversammlung oder (ii) einer speziellen Untersuchung gemäss Artikel 697a OR und unterliegen nicht der hierin geforderten Voraussetzung der rechtzeitigen Information. |
No advance notice is required to propose motions on duly notified agenda items and to debate items without passing resolutions. |
|
Zur Stellung von Anträgen im Rahmen der Verhandlungsgegenstände und zu Ver-handlungen ohne Beschlussfassung bedarf es keiner vorherigen Ankündigung. |
|
Article 13 Documentation The annual business report, the compensation report and the Auditor's report must be submitted for examination by the shareholders at the registered office of the Company at least 20 days prior to the date of the Ordinary General Meeting. Each shareholder may request that a copy of this documentation be sent to him promptly. Such reference shall be included in the invitation to the General Meeting. |
|
Artikel 13 Unterlagen Spätestens zwanzig Tage vor der ordentlichen Generalversammlung sind der Geschäftsbericht, der Vergütungsbericht und der Revisionsbericht am Sitz der Gesellschaft zur Einsicht der Aktionäre aufzulegen. Jeder Aktionär kann verlangen, dass ihm unverzüglich eine Kopie dieser Unterlagen zugestellt wird. In der Einberufung zur Generalversammlung ist hierauf hinzuweisen. |
Article 14 Form of the General Meeting |
|
Artikel 14 Form der Generalversammlung |
The Board of Directors determines the location of the General Meeting. It may be abroad. |
|
Der Verwaltungsrat bestimmt den Ort der Generalversammlung. Er kann im Ausland liegen. |
21
The Board of Directors may provide that shareholders who are not present at the venue of the General Meeting may exercise their rights electronically (hybrid General Meeting). |
|
Der Verwaltungsrat kann vorsehen, dass Aktionäre, die nicht am Ort der Generalversammlung anwesend sind, ihre Rechte auf elektronischem Weg ausüben können (hybride Generalversammlung). |
A General Meeting may be held by electronic means without a meeting place (Virtual General Meeting). |
|
Eine Generalversammlung kann mit elektronischen Mitteln ohne Tagungsort durchgeführt werden (virtuelle Generalversammlung). |
22
|
The Board of Directors regulates the use of electronic means. It shall ensure that: a)
the identity of the participants is established; and
b)
the votes at the General Meeting are transmitted directly; and
c)
each participant can submit motions and take part in the discussion; and
d)
the voting results cannot be falsified.
|
|
Der Verwaltungsrat regelt die Verwendung elektronischer Mittel. Er stellt sicher, dass: a)
die Identität der Teilnehmer feststeht;
b)
die Voten in der Generalversammlung unmittelbar übertragen werden;
c)
jeder Teilnehmer Anträge stellen und sich an der Diskussion beteiligen kann; und
d)
das Abstimmungsergebnis nicht verfälscht werden kann.
|
If technical problems occur during a Virtual General Meeting so that the General Meeting cannot be held properly, it must be repeated. Resolutions passed by the General Meeting before the occurrence of the technical problems shall remain valid. |
|
Treten während einer Generalversammlung mit elektronischen Mitteln technische Probleme auf, sodass die Generalversammlung nicht ordnungsgemäss durchgeführt werden kann, so muss sie wiederholt werden. Beschlüsse, welche die Generalversammlung vor dem Auftreten der technischen Probleme gefasst hat, bleiben gültig. |
|
Article 15 Meeting of All Shareholders Shareholders or their proxies representing all shares issued may hold a General Meeting without observing the formalities required for calling a meeting, unless objection is raised. At such a meeting, discussions may be held and resolutions passed on all matters within the scope of the powers of a General Meeting for so long as the shareholders or proxies representing all shares issued are present. |
|
Artikel 15 Universalversammlung Die Eigentümer oder Vertreter sämtlicher Aktien können, falls kein Widerspruch erhoben wird, eine Generalversammlung ohne Einhaltung der für die Einberufung vorgeschriebenen Formvorschriften abhalten (Universalversammlung). Solange die Eigentümer oder Vertreter sämtlicher Aktien anwesend sind, kann in dieser Versammlung über alle in den Geschäftskreis der Generalversammlung fallenden Gegenstände verhandelt und gültig Beschluss gefasst werden. |
|
Article 16 Chairman, Secretary, Scrutineers The Chairman of the Board of Directors shall preside over the General Meeting. In his absence, a member of the Board of Directors or another Chairman of the Meeting designated by the General Meeting shall preside. |
|
Artikel 16 Vorsitz, Protokollführer, Stimmenzähler Den Vorsitz der Generalversammlung führt der Präsident, bei dessen Verhinderung ein anderes Mitglied des Verwaltungsrates oder ein anderer von der Generalversammlung gewählter Tagespräsident. |
The Chairman of the Meeting shall designate a Secretary and the scrutineers who need not be shareholders. |
|
Der Vorsitzende bezeichnet den Protokollführer und die Stimmenzähler, die nicht Aktionäre zu sein brauchen. |
23
The Chairman shall have all powers and authority necessary to ensure the orderly conduct of the General Meeting. |
|
Der Vorsitzende hat sämtliche Leitungsbefugnisse, die für die ordnungsgemässe Durchführung der Generalversammlung nötig sind. |
|
Article 17 Minutes The Board of Directors is responsible for the keeping of the minutes of the Meeting, which shall state the number, kind, nominal value of shares represented by the shareholders, by the corporate bodies and by the independent proxy and gives information on resolutions passed, elections, requests for information and information as well as declarations given by the shareholders. The minutes shall be signed by the Chairman and the Secretary. |
|
Artikel 17 Protokoll Der Verwaltungsrat sorgt für die Führung des Protokolls über die Generalversammlung, welches Anzahl, Art, Nennwert und Kategorie der von den Aktionären, von den Organen und von unabhängigen Stimmrechtsvertretern vertretene Aktien festhält und Aufschluss über Beschlüsse, Wahlergebnisse, Begehren um Auskunft und die darauf erteilten Auskünfte sowie die von den Aktionären zu Protokoll gegebenen Erklärungen gibt. Das Protokoll wird vom Vorsitzenden und vom Protokollführer unterzeichnet. |
The shareholders are entitled to inspect the minutes. |
|
Die Aktionäre sind berechtigt, das Protokoll einzusehen. |
|
Article 18 Right to Vote Each share entitles to one vote. Each shareholder may be represented at a General Meeting by any person who is so authorized by a written proxy. A proxy need not be a shareholder. |
|
Artikel 18 Stimmrecht Jede Aktie berechtigt zu einer Stimme. Jeder Aktionär kann sich in der Generalversammlung aufgrund einer schriftlichen Vollmacht durch eine andere handlungsfähige Person vertreten lassen, die nicht Aktionär zu sein braucht. |
Each shareholder may be represented by the Independent Proxy. The requirements regarding proxies and instructions are determined by the Board of Directors. |
|
Jeder Aktionär kann sich vom unabhängigen Stimmrechtsvertreter vertreten lassen. Die Anforderungen an Vollmachten und Weisungen werden vom Verwaltungsrat festgelegt. |
|
Article 19 Resolutions and Elections All voting and elections are held openly or electronically. A written voting or election shall be held if instructed so by the Chairman or if decided by the General Meeting. |
|
Artikel 19 Beschlussfassung und Wahlen Die Abstimmungen und Wahlen erfolgen offen oder elektronisch. Eine schriftliche Abstimmung oder Wahl wird durchgeführt, wenn dies vom Vorsitzenden angeordnet oder von der Generalversammlung beschlossen wird. |
The General Meeting shall pass its resolutions and carry out its elections with the simple majority of the votes cast regardless of abstentions and empty or invalid votes, unless law or articles of association state otherwise. In the event |
|
Die Generalversammlung fasst ihre Beschlüsse und vollzieht ihre Wahlen, soweit das Gesetz oder die Statuten es nicht anders bestimmen, mit der einfachen Mehrheit der abgegebenen Aktienstimmen ohne Berücksichtigung von Stimmenthaltungen oder leer eingelegten oder ungültigen Stimmen. Bei |
24
of tie votes, the request shall be refused. The Chairman shall not have a casting vote. |
|
Stimmengleichheit gilt ein Antrag als abgelehnt. Dem Vorsitzenden steht kein Stichentscheid zu. |
|
A resolution of the General Meeting passed by at least two thirds of the represented share votes and the absolute majority of the represented shares par value is required for: a)
The cases listed in article 704 para. 1 CO:
i.
the amendment of the purpose of the Company;
ii.
the consolidation of shares, insofar as this does not require the consent of all shareholders concerned;
iii.
the increase of the share capital against contributions in kind or by offsetting against a receivable and the granting of special benefits;
iv.
the limitation or withdrawal of subscription rights;
v.
the introduction of conditional capital, the creation of reserve capital pursuant to article 12 of the Swiss Banking Act or the introduction of a capital band;
vi.
the conversion of participation certificates into shares;
vii.
the restriction of the transferability of registered shares;
viii.
the creation of shares with privileged voting rights;
ix.
the change of currency of the share capital;
x.
the introduction of the casting vote of the chairman in the general assembly;
xi.
the introduction of a provision in
|
|
Ein Beschluss der Generalversammlung, durch mindestens zwei Drittel der vertretenen Aktienstimmen und die absolute Mehrheit der vertretenen Aktiennennwerte, ist erforderlich für: a)
die Fälle gemäss Artikel 704 Abs. 1 OR:
i.
die Änderung des Gesellschaftszweckes;
ii.
die Zusammenlegung von Aktien, soweit dafür nicht die Zustimmung aller betroffenen Aktionäre erforderlich ist;
iii.
die Kapitalerhöhung aus Eigenkapital, gegen Sacheinlagen oder durch Verrechnung mit einer Forderung und die Gewährung von besonderen Vorteilen;
iv.
die Einschränkung oder Aufhebung des Bezugsrechts;
v.
die Einführung eines bedingten Kapitals, die Schaffung von Vorratskapital gemäss Artikel 12 des Bankengesetzes oder die Einführung eines Kapitalbands;
vi.
die Umwandlung von Partizipationsscheinen in Aktien;
vii.
die Beschränkung der Übertragbarkeit von Namenaktien;
viii.
die Einführung von Stimmrechtsaktien;
ix.
der Wechsel der Währung des Aktienkapitals;
x.
die Einführung des Stichentscheids des Vorsitzenden in der Generalversammlung;
xi.
eine Statutenbestimmung zur Durchführung der Generalversammlung im Ausland;
xii.
die Verletzung des Sitzes der Gesellschaft;
xiii.
die Einführung einer statutarischen Schiedsklausel;
|
25
|
the articles of association to hold General Meetings abroad; xii.
the change of the registered office of the Company;
xiii.
the introduction of an arbitration clause in the articles of association;
xiv.
the delisting of the shares; or
xv.
the dissolution of the Company.
b)
the merger, de-merger or conversion of the Company (subject to mandatory law);
c)
the alleviating or withdrawal of restrictions upon the transfer of registered shares;
d)
the conversion of registered shares into bearer shares and vice versa; and
e)
the amendment or elimination of the provisions of articles 4 and 31 of the articles of association as well as those contained in this article 19.
|
|
xiv.
die Dekotierung der Beteiligungspapiere; oder
xv.
die Auflösung der Gesellschaft.
b)
die Fusion, Spaltung oder Umwandlung der Gesellschaft (vorbehalten zwingender gesetzlicher Bestimmungen);
c)
die Erleichterung oder den Entzug der Beschränkungen betreffend die Übertragung von Namenaktien;
d)
die Umwandlung von Namenaktien in Inhaberaktien und umgekehrt; und
e)
die Änderung oder Aufhebung der Bestimmungen der Artikel 4 und 31 der Statuten sowie dieses Artikels 19.
|
|
Article 20 Votes on Compensation Each year, the General Meeting approves in one or separate resolutions the total maximum amounts pursuant to articles 34 and 35 of the articles of association for: a)
the non-performance-related compensation of the Board of Directors for the next term of office;
b)
a possible additional compensation of the Board of Directors for the preceding business year;
c)
the non-performance-related compensation of the Executive Committee for the following business year;
d)
the variable compensation for the
|
|
Artikel 20 Abstimmung über Vergütungen Die Generalversammlung genehmigt jährlich in einem oder mehreren Beschlüssen die maximalen Vergütungen gemäss Artikel 34 und 35 der Statuten betreffend: a)
die nicht-erfolgsabhängige Vergütung des Verwaltungsrates für die Zeitperiode bis zur nächsten Generalversammlung;
b)
eine allfällige zusätzliche Vergütung für den Verwaltungsrat für das abgeschlossene Geschäftsjahr;
c)
die nicht-erfolgsabhängige Vergütung der Geschäftsleitung für das folgende Geschäftsjahr;
d)
die variable Vergütung der Geschäftsleitung für das folgende Geschäftsjahr; und
|
26
|
Executive Committee for the following business year; and e)
the grant of options, shares or other equity-linked instruments in the Company to the Board of Directors and the Executive Committee.
|
|
e) die Gewährung von Optionen, Aktien oder anderen eigenkapitalbasierten Instrumenten der Gesellschaft an den Verwaltungsrat oder die Geschäftsleitung.
|
The corresponding total compensation includes all social security, pension fund and other contributions payable by the receiving member of the Board of Directors or the Executive Board. |
|
Die entsprechenden Gesamtvergütungen umfassen alle vom empfangenden Mitglied des Verwaltungsrats oder der Geschäftsleitung zu bezahlenden Sozialversicherungs-, Pensionskassen- und andere Beiträge. |
If the General Meeting refuses to approve a respective motion by the Board of Directors, the Board of Directors may either submit a new motion at the same meeting or determine a maximum total remuneration or several maximum partial remunerations, subject to the relevant principles of the compensation, or submit a new motion to the next General Meeting for approval. The Company may pay remunerations within the framework of the maximum total or partial remuneration and subject to the approval by the General Meeting. |
|
Lehnt die Generalversammlung einen entsprechenden Antrag des Verwaltungsrats ab, kann der Verwaltungsrat entweder an der gleichen Versammlung einen neuen Antrag stellen, eine ausserordentliche Generalversammlung einberufen oder einen maximalen Gesamtbetrag oder mehrere maximale Teilbeträge unter Berücksichtigung der relevanten Grundsätze festsetzen und der nächsten Generalversammlung zur Genehmigung vorlegen. Die Gesellschaft kann im Rahmen des maximalen Gesamt- oder Teilbetrages und unter Vorbehalt der Genehmigung durch die Generalversammlung Vergütungen ausrichten. |
|
Article 21 Independent Proxy The Independent Proxy shall be elected by the Ordinary General Meeting for a term of one year until the end of the next Ordinary General Meeting. Re-election is permitted. The Independent Proxy informs the Company about number, type, par value and category of the represented shares. The Chairman of the Board discloses the information to the General Meeting. The other duties of the Independent Proxy are determined by the applicable statutory provisions. |
|
Artikel 21 Unabhängiger Stimmrechtsvertreter Der Unabhängige Stimmrechtsvertreter wird von der ordentlichen Generalversammlung für eine Amtsdauer von einem Jahr bis zum Ende der nächsten ordentlichen Generalversammlung gewählt. Wiederwahl ist möglich. Der unabhängige Stimmrechtsvertreter informiert die Gesellschaft über Anzahl, Art, Nennwert und Kategorie der vertretenen Aktien. Der Präsident des Verwaltungsrats gibt diese Informationen der Generalversammlung bekannt. Die Pflichten des Unabhängigen Stimmrechtsvertreters ergeben sich aus den anwendbaren gesetzlichen Bestimmungen. |
V. BOARD OF DIRECTORS |
|
V. VERWALTUNGSRAT |
27
|
Article 22 Number of Members, Term of Office The Board of Directors shall consist of at least 3 and not more than 9 members. The chairman and the members of the Board of Directors are individually elected by the General Meeting for a term of one year until the end of the next Ordinary General Meeting, provided that he/she does not resign or is not replaced during his term. |
|
Artikel 22 Anzahl der Mitglieder, Amtsdauer Der Verwaltungsrat besteht aus mindestens 3 und höchstens 9 Mitgliedern. Der Präsident sowie die Mitglieder des Verwaltungsrates werden jeweils für die Dauer von einem Jahr bis zum Ende der nächsten ordentlichen Generalversammlung einzeln gewählt. Vorbehalten bleiben vorheriger Rücktritt oder Abberufung. |
The members of the Board of Directors may be re-elected without limitation. The maximum age limit of members of the Board shall be 75 years. When a member of the Board of Directors reaches this age limit during his term of office, such term shall automatically extend to the next ordinary shareholders' meeting. The shareholders' meeting may resolve to grant an exception to the age limit. |
|
Die Mitglieder des Verwaltungsrates sind jederzeit wieder wählbar. Die oberste Altersgrenze von Mitgliedern des Verwaltungsrats beträgt 75 Jahre. Wenn ein Mitglied des Verwaltungsrats diese Altersgrenze während seiner Amtszeit erreicht, wird diese automatisch zur nächsten ordentlichen Generalversammlung verlängert. Die Generalversammlung kann eine Ausnahme von der Altersgrenze beschliessen. |
If the office of the Chairman becomes vacant, the board of directors shall appoint a new Chairman, from among its members for the remaining term of office. |
|
Wird das Amt des Präsidenten vakant, ernennt der Verwaltungsrat für die verbleibende Amtsdauer aus seiner Mitte einen neuen Präsidenten des Verwaltungsrates für die verbleibende Amtszeit. |
|
Article 23 Constitution Subject to the powers of the General Meeting, the Board of Directors determines its own organization. It appoints a Secretary who needs not be a member of the Board of Directors. |
|
Artikel 23 Konstituierung Der Verwaltungsrat konstituiert sich vorbehältlich der Befugnisse der Generalversammlung selbst. Er bezeichnet insbesondere einen Sekretär, der nicht Mitglied des Verwaltungsrates sein muss. |
|
Article 24 Function, Organization It is the Board of Director's duty to lead the Company and to supervise the management. The Board of Director represents the Company and may take decisions to all affairs which are not assigned to any other body of the Company by law, the articles of association or Regulations. |
|
Artikel 24 Funktion, Organisation Dem Verwaltungsrat obliegt die oberste Leitung der Gesellschaft und die Überwachung der Geschäftsführung. Er vertritt die Gesellschaft nach aussen und besorgt alle Angelegenheiten, die nicht nach Gesetz, Statuten oder Reglement einem anderen Organ der Gesellschaft übertragen sind. |
28
The Board of Directors shall adopt the organizational regulations and the corresponding contractual relationships. |
|
Der Verwaltungsrat erlässt das Organisationsreglement und ordnet die entsprechenden Vertragsverhältnisse. |
|
Article 25 Powers The Board of Directors has the following non-delegable and inalienable duties: a)
the overall management of the company and the issuing of all necessary directives;
b)
the determination of the company's organisation;
c)
the organisation of the accounting, financial control and financial planning systems as required for management of the company;
d)
the appointment and dismissal of the persons entrusted with the management and representation of the Company and grant of signatures;
e)
the overall supervision of the persons entrusted with managing the company, in particular with regard to compliance with the law, articles of association, operational regulations and directives;
f)
the compilation of the annual report, preparation for the general meeting and implementation of its resolutions;
g)
the preparation of the compensation report and to request approval by the General Meeting regarding compensation of the Board of Directors and the Executive Committee; and
h)
the notification of the court if liabilities exceed assets.
|
|
Artikel 25 Aufgaben Der Verwaltungsrat hat folgende unübertragbare und unentziehbare Aufgaben: a)
Oberleitung der Gesellschaft und Erteilung der nötigen Weisungen;
b)
Festlegung der Organisation der Gesellschaft;
c)
Organisation des Rechnungswesens, der Finanzkontrolle sowie der Finanzplanung zur Führung der Gesellschaft;
d)
Ernennung und Abberufung der mit der Geschäftsführung und der Vertretung betrauten Personen und Regelung der Zeichnungsberechtigung;
e)
Oberaufsicht über die mit der Geschäftsführung betrauten Personen, namentlich im Hinblick auf die Befolgung der Gesetze, Statuten, Reglemente und Weisungen;
f)
Erstellung des Geschäftsberichtes sowie Vorbereitung der Generalversammlung und Ausführung ihrer Beschlüsse;
g)
Erstellung des Vergütungsberichts sowie Antragsstellung betreffend die Genehmigung der Vergütungen des Verwaltungsrats und der Geschäftsleitung an die Generalversammlung;
h)
Benachrichtigung des Richters im Falle der Überschuldung.
|
The Board of Directors may assign responsibility for preparing and implementing its resolutions or monitoring transactions to committees or |
|
Der Verwaltungsrat kann die Vorbereitung und die Ausführung seiner Beschlüsse oder die Überwachung von Geschäften Ausschüssen oder einzelnen |
29
individual members. It must ensure appropriate reporting to its members. |
|
Mitgliedern zuweisen. Er hat für eine angemessene Berichterstattung an seine Mitglieder zu sorgen. |
|
Article 26 Representation of the Company The Board of Directors shall assign the persons with signatory power for the Company and the kind of signatory power. |
|
Artikel 26 Vertretung der Gesellschaft Der Verwaltungsrat bestimmt die für die Gesellschaft zeichnungsberechtigten Personen und die Art ihrer Zeichnung. |
|
Article 27 Delegation Moreover, the Board of Directors is authorized to delegate, in part or entirely, the management and the representation of the Company, within the limits of the law, to one or more individual directors (Delegates) or to third parties by pursuant to organizational regulations. |
|
Artikel 27 Delegation Der Verwaltungsrat kann die Geschäftsführung und alle Aufgaben und Befugnisse, die ihm nicht durch das Gesetz oder die Statuten zwingend zugewiesen sind, nach Massgabe des Organisationsreglements ganz oder zum Teil an einzelne oder mehrere Mitglieder oder Dritte übertragen. |
|
Article 28 Meetings, Resolutions and Minutes The organization of the meetings, the presence quorum and the passing of resolutions of the Board of Directors is determined by the organizational regulations. No presence quorum is required for the approval of a capital increase. |
|
Artikel 28 Sitzungen, Beschlussfassung und Protokoll Sitzungsordnung, Beschlussfähigkeit und Beschlussfassung des Verwaltungsrats richten sich nach dem Organisationsreglement. Für den Feststellungsbeschluss einer Kapitalerhöhung ist kein Präsenzquorum erforderlich. |
Resolutions may be passed via telephone or videoconference. Resolutions may also be passed by way of circulation, provided that no member requests oral deliberation. |
|
Beschlussfassung via Telefon- oder Videokonferenz ist zulässig. Beschlüsse können auch auf dem Zirkularweg gefasst werden, sofern nicht ein Mitglied die Durchführung einer Sitzung verlangt. |
Minutes are kept of the Board's discussions and resolutions and signed by the chairman and the minute-taker. |
|
Über Verhandlungen und Beschlüsse des Verwaltungsrats wird ein Protokoll erstellt, welches vom Vorsitzenden und vom Sekretär des Verwaltungsrates zu unterzeichnen ist. |
|
Article 29 Disclosure and Right of Inspection Any member of the Board of Directors may request information on any company business. |
|
Artikel 29 Recht auf Auskunft und Einsicht Jedes Mitglied des Verwaltungsrates kann Auskunft über alle Angelegenheiten der Gesellschaft verlangen. |
Outside meetings, any member may request information from the persons |
|
Ausserhalb der Sitzungen kann jedes Mitglied von den mit der Geschäftsführung betrauten Personen |
30
entrusted with managing the company's business concerning the Company's business performance and, with the Chairman's authorization, specific transactions. |
|
Auskunft über den Geschäftsgang und, mit Ermächtigung des Präsidenten, auch über einzelne Geschäfte verlangen. |
Where required for the performance of his duties, any member may request the Chairman to have books of account and documents made available to him for inspection. |
|
Soweit es für die Erfüllung einer Aufgabe erforderlich ist, kann jedes Mitglied dem Präsidenten beantragen, dass ihm Bücher und Akten vorgelegt werden. |
If the Chairman refuses a request for information, a request to be heard or an application to inspect documents, the Board of Directors rules on the matter. |
|
Weist der Präsident ein Gesuch auf Auskunft, Anhörung oder Einsicht ab, so entscheidet der Verwaltungsrat. |
31
|
Article 30 Compensation Committee The Compensation Committee shall comprise at least 2 members. The members of the Compensation Committee shall be individually elected by the Ordinary General Meeting from among the members of the Board of Directors for a term of one year until the next Ordinary General Meeting. Re-election is permitted. The Compensation Committee has the following duties: a)
to draw up principles for compensation of members of the Board of Directors and the Executive Committee and to submit them to the Board of Directors for approval;
b)
to propose to the Board of Directors the resolution to be submitted to the Ordinary General Meeting for the maximum total compensation of the Board of Directors and Executive Committee;
c)
subject to and within the bounds of the maximum compensation approved by the Ordinary General Meeting, to request approval by the Board of Directors of the individual remuneration packages to be paid to members of the Board of Directors and members of the Executive Committee;
d)
to request approval by the Board of Directors regarding the determination of the compensation-related targets for the Executive Committee;
e)
to request approval by the Board of Directors regarding the adjustments to the articles of association relating to remuneration; and
f)
to prepare the Compensation Report and submit it to the Board of
|
|
Artikel 30 Vergütungsausschuss Der Vergütungsausschuss umfasst mindestens 2 Mitglieder. Die Mitglieder des Vergütungsausschusses werden jährlich von der ordentlichen Generalversammlung aus den Mitgliedern des Verwaltungsrats für die Dauer von einem Jahr bis zur nächsten ordentlichen Generalversammlung einzeln gewählt. Wiederwahl ist zulässig. Der Vergütungsausschuss hat folgende Aufgaben: a)
Ausarbeiten der Grundsätze betreffend Vergütung an den Verwaltungsrat und an die Geschäftsleitung und Vorlegen derselben zur Genehmigung durch den Verwaltungsrat;
b)
Antragstellung an den Verwaltungsrat zur Unterbreitung an die Generalversammlung betreffend Gesamtvergütung des Verwaltungsrats und der Geschäftsleitung;
c)
Antragstellung an den Verwaltungsrat betreffend individuelle Vergütung der Verwaltungsratsmitglieder und der Mitglieder der Geschäftsleitung unter Vorbehalt und im Rahmen der Höhe der Gesamtvergütung;
d)
Antragstellung an den Verwaltungsrat hinsichtlich der für die Geschäftsleitung vergütungsrelevanten Ziele;
e)
Antragstellung an den Verwaltungsrat betreffend Anpassung der Statuten hinsichtlich des Vergütungssystems; und
f)
Entwurf des Vergütungsberichts und Unterbreitung des Vergütungsberichts an den Verwaltungsrat.
|
32
Directors. |
|
|
The Board of Directors shall set out any further duties and responsibilities vested on the Compensation Committee in the Company's organizational regulations. |
|
Der Verwaltungsrat kann weitere Aufgaben und Zuständigkeiten des Vergütungsausschusses im Organisationsreglement vorsehen. |
Article 31 Indemnification |
|
Artikel 31 Schadloshaltung. |
To the extent not included in insurance cover-age or paid by third parties, the Company shall indemnify and hold harmless, to the extent permitted by law, the existing and former members of the board of directors, the executive committee, and their heirs, executors and administrators, out of the assets of the Company from and against all threatened, pending or completed actions, suits or proceedings – whether civil, criminal, administrative or investigative – and all costs, charges, losses, damages, and expenses which they or any of them, their heirs, executors or administrators, shall or may incur or sustain by or by reason of any actual or alleged actions, consents or omissions in or about the execution of their du-ty, or alleged duty, or by reason of the fact that he/she is or was a member of the board of di-rectors or executive committee of the Company or the board of directors (or equivalent corporate body) or the management of one of its subsidiaries, or, while serving as a member of the board of directors or executive committee of the Company, is or was serving at the re-quest of the Company as a director, member of the executive committee, employee or agent of another corporation, partnership, joint venture, trust or other enterprise; provided, however, that this indemnity shall not extend to any matter in which any of said persons is found, in a final judgment or decree of a court or governmental or administrative authority of competent jurisdiction not subject to appeal, to have committed an intentional |
|
Soweit nicht durch Versicherungen gedeckt oder von Dritten bezahlt, hält die Gesellschaft soweit gesetzlich zulässig, die gegenwärtigen und bisherigen Mitglieder des Verwaltungsrates und der Geschäftsleitung sowie deren Erben, Testamentsvollstrecker und Verwalter aus dem Vermögen der Gesellschaft von allen angedrohten, hängigen, und abgeschlossenen Klagen, Prozessen oder Verfahren – ob zivil-, straf-, verwaltungs- oder untersuchungsrechtlich – schadlos, sowie von allen Kosten, Gebühren, Verlusten, Schäden und Ausgaben, die ihnen oder einem/einer von ihnen, ihren Erben, Testamentsvollstreckern oder Verwaltern durch oder aufgrund von tatsächlichen oder vermeintlichen Handlungen, Zustimmungen oder Unterlassungen im Zusammenhang mit der Ausübung ihrer Pflicht oder vermeintlichen Pflicht oder aufgrund der Tatsache, dass er/sie ein Mitglied des Verwaltungsrates oder der Geschäftsleitung der Gesellschaft oder des Verwaltungsrates (oder eines gleichwertigen Gesellschaftsorgans) oder der Geschäftsleitung einer ihrer Konzerngesellschaften ist oder war, oder dass er/sie während seiner/ihrer Tätigkeit als Mitglied des Verwaltungsrates oder der Geschäftsleitung der Gesellschaft, auf Ersuchen der Gesellschaft, als Mitglied des Verwaltungsrates oder der Geschäftsleitung, Angestellter oder Beauftragter einer anderen Kapitalgesellschaft, Personengesellschaft, eines Joint Ventures, eines Trusts oder eines anderen Unternehmens tätig ist oder war, entstanden sind, entstehen oder entstehen könnten, jedoch unter der Voraussetzung, dass sich diese Schadloshaltung nicht auf eine Angelegenheit erstreckt, in der eine der genannten Personen gemäss einem rechtskräftigen Urteil oder Beschluss eines Gerichts oder einer zuständigen Regierungs- oder Verwaltungsbehörde, gegen den kein Rechtsmittel eingelegt werden kann, eine vorsätzliche oder grobfahrlässige Verletzung ihrer gesetzlichen Pflichten |
33
or grossly negligent breach of his statutory duties as a member of the board of directors or executive committee. |
|
als Mitglied des Verwaltungsrates oder der Geschäftsleitung begangen hat. |
Without limiting the foregoing paragraph of this article 31, the Company shall advance costs and expenses indemnifiable thereunder to the existing and former members of the board of directors and executive committee to the ex-tent not included in insurance coverage or advanced by third parties. The Company may however recover such advanced costs if any of said persons is found, in a final judgment or decree of a court or governmental or administrative authority of competent jurisdiction not subject to appeal, to have committed an intentional or grossly negligent breach of his statutory duties as a member of the board of directors or executive committee. |
|
Ohne den vorstehenden Absatz dieses Artikels 31 einzuschränken, hat die Gesellschaft den gegenwärtigen und ehemaligen Mitgliedern des Verwaltungsrates und der Geschäftsleitung die Kosten und Auslagen zu erstatten, die nach diesem Artikel erstattungsfähig sind, soweit sie nicht durch Versicherungen gedeckt sind oder von Dritten vorab erstattet werden. Die Gesellschaft kann jedoch diese vorausbezahlten Konten zurückfordern, wenn eine der genannten Personen in einem rechtskräftigen Urteil oder Beschluss eines Gerichts oder einer zu-ständigen Regierungs- oder Verwaltungsbehörde, gegen das kein Rechtsmittel eingelegt werden kann, wegen vorsätzlicher oder grob-fahrlässiger Verletzung ihrer gesetzlichen Pflichten als Mitglied des Verwaltungsrates oder der Geschäftsleitung verurteilt wird. |
VI. AUDITORS |
|
VI. REVISIONSSTELLE |
|
Article 32 Election, Term The General Meeting shall elect one or more accountants as its Auditors in terms of articles 727 et seq. CO every year with the rights and duties determined by law. |
|
Artikel 32 Wahl, Amtsdauer Die Generalversammlung wählt jedes Jahr eine oder mehrere natürliche oder juristische Personen als Revisionsstelle im Sinne von Artikeln 727 ff. OR mit den im Gesetz festgehaltenen Rechten und Pflichten. |
The General Meeting may appoint Special Auditors for a term of up to three years who provide the attestations required for capital increases. |
|
Die Generalversammlung kann für die Dauer von bis zu drei Jahren Sonderrevisoren bestimmen, welche die bei Kapitalerhöhungen erforderlichen Bescheinigungen erbringen. |
|
Article 33 Duties The Auditors shall perform their duties to audit and report whether the accounting, the annual accounts and the proposal regarding allocation of profits is in accordance with law and the articles of association. |
|
Artikel 33 Aufgaben Die Revisionsstelle prüft, ob die Buchführung und die Jahresrechnung sowie der Antrag über die Verwendung des Bilanzgewinns Gesetz und Statuten entsprechen. |
34
VII. COMPENSATION AND RELATED PROVISIONS |
|
VII. VERGÜTUNGEN UND VERWANDTE BESTIMMUNGEN |
|
Article 34 Principles of the Compensation of the Board of Directors The compensation payable to the members of the Board of Directors comprises, subject to and within the bounds of the approval by the General Meeting of the total compensation, the following elements: a)
a fixed basic remuneration;
b)
a fixed committee fee for work in a committee of the Board of Directors;
c)
a lump sum compensation for expenses;
d)
a number of options, shares or other equity-linked instruments in the Company, as further outlined in article 43 of the articles of association.
|
|
Artikel 34 Grundsätze der Vergütung für die Mitglieder des Verwaltungsrats Die Vergütung für die Mitglieder des Verwaltungsrats umfasst, unter Vorbehalt der Genehmigung durch die Generalversammlung und im Rahmen der durch diese genehmigten Gesamtvergütung, folgende Elemente: a)
ein fixes Grundhonorar;
b)
eine fixe Entschädigung für Tätigkeiten als Mitglied eines Ausschusses des Verwaltungsrats;
c)
eine pauschale Spesenentschädigung;
d)
eine Anzahl von Optionen, Aktien oder anderen eigenkapitalbasierten Instrumenten der Gesellschaft, gemäss Artikel 43 der Statuten.
|
The compensation is paid in cash and in form of options, shares or other equity-linked instruments in the Company. The board of directors or, to the extent delegated to it, the Compensation Committee shall determine grant, exercise and forfeiture conditions. In particular, they may provide for continuation, acceleration or removal of vesting, exercise and forfeiture conditions, for payment or grant of compensation based upon assumed target achievement, or for forfeiture, in each case in the event of pre-determined events such as a change-of-control or termination of an employment or mandate agreement. The Company may procure the required shares through purchases in the market, from treasury |
|
Die Vergütung kann bar und in Form von Optionen, Aktien oder anderen eigenkapitalbasierten Instrumenten der Gesellschaft bezahlt werden. Der Verwaltungsrat oder, soweit an ihn delegiert, der Vergütungsausschuss legen Zuteilungs-, Ausübungs- und Verfallsbedingungen fest. Sie können insbesondere vorsehen, dass aufgrund des Eintritts im Voraus bestimmter Ereignisse, wie eines Kontrollwechsels oder der Beendigung des Arbeits- oder Mandatsverhältnisses, Vesting-, Ausübungs- und Verfallsbedingungen weitergelten, verkürzt oder aufgehoben werden, Vergütungen unter der Annahme der Erreichung von Zielwerten ausgerichtet werden oder Vergütungen verfallen. Die Gesellschaft kann die erforderlichen Aktien auf dem Markt erwerben, aus Beständen eigener Aktien entnehmen oder unter Verwendung von bedingtem oder genehmigtem Kapital bereitstellen. |
35
shares or by using contingent or authorized share capital. |
|
|
Subject to the approval by the General Meeting, the members of the Board of Directors may receive remuneration in cash at customary conditions for advisory services rendered outside their capacity as Board member for the benefit of the Company or companies under its control. The General Meeting may approve an additional bonus for the members of the Board of Directors in exceptional cases. |
|
Vorbehältlich der Genehmigung durch die Generalversammlung, kann den Mitgliedern des Verwaltungsrats eine Entschädigung in bar zu marktüblichen Konditionen für Beratungstätigkeiten, welche diese ausserhalb ihrer Funktion als Verwaltungsratsmitglied und zu Gunsten der Gesellschaft oder von ihr kontrollierter Gesellschaften erbringen, ausbezahlt werden. Die Generalversammlung kann in Ausnahmefällen einen zusätzlichen Bonus zu Gunsten der Verwaltungsratsmitglieder genehmigen. |
The compensation may also be paid for activities in companies that are directly or indirectly controlled by the Company and may be paid by the Company or by a company controlled by it. |
|
Die Vergütung kann auch ausgerichtet werden für Tätigkeiten in Unternehmen, die durch die Gesellschaft direkt oder indirekt kontrolliert werden und kann durch die Gesellschaft oder durch von ihr kontrollierte Unternehmen ausgerichtet werden. |
|
Article 35 Principles of the Compensation of the Executive Committee The compensation payable to the members of the Executive Committee is subject to the approval by the General Meeting and comprises the following elements: a)
a fixed remuneration payable in cash;
b)
a performance-related remuneration payable in cash (variable);
c)
a number of options, shares or equity-lined instruments in the Company (variable), as further outlined in article 43 of the articles of association.
|
|
Artikel 35 Grundsätze der Vergütung für die Mitglieder der Geschäftsleitung Die Vergütung für die Mitglieder der Geschäftsleitung ist von der Generalversammlung zu genehmigen und umfasst folgende Elemente: a)
eine fixe Vergütung in bar;
b)
eine erfolgsabhängige Vergütung in bar (variabel);
c)
eine Anzahl Optionen, Aktien oder anderen eigenkapitalbasierten Instrumenten der Gesellschaft (variabel), gemäss Artikel 43 der Statuten.
|
The performance-related remuneration depends on the Company's business success and the individual performance of the member of the Executive Committee based on the achievement of pre-determined targets during a business year. The Board of Directors determines annually at the beginning of each relevant |
|
Die erfolgsabhängige Vergütung richtet sich nach dem Geschäftserfolg und der individuellen Leistung gemessen nach dem Erreichen bestimmter vordefinierter Ziele über ein Geschäftsjahr. Der Verwaltungsrat definiert jährlich am Anfang jeder Leistungsperiode auf Antrag des Vergütungsausschusses hin die relevanten Ziele und deren Gewichtung. Die Höhe der erfolgsabhängigen |
36
business year the decisive targets and their weighting upon proposal by the Compensation Committee. The amount of the performance-related remuneration in cash for each member of the Compensation Committee is determined by the Board of Directors and may not exceed 100% of the respective individual fixed remuneration for the same year. |
|
Vergütung in bar für das jeweilige Geschäftsleitungsmitglied wird vom Verwaltungsrat festgelegt und darf 100% der im entsprechenden Geschäftsjahr relevanten individuellen, fixen Vergütung nicht überschreiten. |
The compensation may also be paid for activities in companies that are directly or indirectly controlled by the Company and may be paid by the Company or by a company controlled by it. |
|
Die Vergütung kann auch ausgerichtet werden für Tätigkeiten in Unternehmen, die durch die Gesellschaft direkt oder indirekt kontrolliert werden und kann durch die Gesellschaft oder durch von ihr kontrollierte Unternehmen ausgerichtet werden. |
|
Article 36 Compensation for new Members of the Executive Committee If new members of the Executive Committee are appointed and take up their position in the Company after the General Meeting has approved the maximum total compensation for members of the Executive Committee for the year in question, the new members may be paid an additional amount for the period until the next Ordinary Meeting of Shareholder. The additional amount payable to all new members of the Executive Committee may not exceed 50% of the respective total compensation already approved by the General Meeting. The additional compensation may only be paid if the total compensation amount that has been approved by the General Meeting for the compensation of the members of the Executive Committee is insufficient to compensate the newly appointed members. The General Meeting is not required to vote on this additional amount. |
|
Artikel 36 Vergütungen für neue Mitglieder der Geschäftsleitung Sofern neue Mitglieder der Geschäftsleitung ernannt werden und ihre Stelle antreten, nachdem die Generalversammlung die Gesamtvergütung für die Geschäftsleitungsmitglieder im entsprechenden Jahr genehmigt hat, darf diesen neuen Mitglieder ein zusätzlicher Betrag für die Dauer bis zur nächsten ordentlichen Generalversammlung vergütet werden. Dieser Zusatzbetrag an alle neuen Mitglieder der Geschäftsleitung darf 50% der von der Generalversammlung für das betreffende Jahr bereits genehmigten Gesamtvergütung nicht übersteigen. Der Zusatzbetrag darf nur ausgerichtet werden, sofern und soweit die von der Generalversammlung beschlossenen Vergütungsbeträge an die Geschäftsleitungsmitglieder bis zur nächsten ordentlichen Generalversammlung für die Vergütung der neuen Mitglieder nicht ausreicht. Über den verwendeten Zusatzbetrag stimmt die Generalversammlung nicht ab. |
This additional overall compensation is understood to include any settlements for any disadvantage suffered as a result of the change of job. |
|
Mit diesem Zusatzbetrag sind allfällige durch ein Geschäftsleitungsmitglied erlittene Nachteile aufgrund Stellenwechsel abgegolten. |
37
|
Article 37 Expenses Expenses which are not covered by the lump sum compensation pursuant to the Company's expense regulations shall be reimbursed following presentation of the supporting receipts. This additional remuneration is not subject to a separate vote by the General Meeting. |
|
Artikel 37 Spesen Spesen, welche nicht durch die pauschale Spesenentschädigung gemäss Spesenreglement abgedeckt sind, werden nach Vorlage der entsprechenden Belege rückvergütet. Diese Rückvergütung ist von der Generalversammlung nicht zu genehmigen. |
|
Article 38 Compensation Agreements Agreements on compensation with members of the Board of Directors may not exceed the term of maximal one year. |
|
Artikel 38 Verträge über die Vergütung Verträge, die den Vergütungen für die Mitglieder des Verwaltungsrats zugrunde liegen, sind auf maximal ein Jahr befristet. |
Employment agreements of the members of the Executive Committee are principally concluded for an indefinite period of time whereas a notice period may not exceed twelve months. If an employment agreement is concluded for a fixed term such term may not exceed one year. |
|
Die Arbeitsverträge der Geschäftsleitungsmitglieder sind grundsätzlich unbefristet, wobei die Kündigungsfrist maximal zwölf Monate betragen darf. Wird ein befristeter Vertrag abgeschlossen, so darf dieser die Dauer von ein Jahr nicht überschreiten. |
|
Article 39 Mandates of a Member of the Board of Directors outside the Company A member of the Board of Directors may cumulatively assume not more than the following number of mandates in the board of directors, the superior management or an administrative body of a legal entity which is obliged to be registered in the Swiss commercial register or an equivalent foreign register: a)
7 mandates for publicly traded companies pursuant to article 727 para. 1 number 1 CO; and
b)
8 mandates for companies pursuant to article 727 para. 1 number 2 CO; and
c)
5 mandates for companies which do not fulfil the criteria under a) and b) hereunder.
|
|
Artikel 39 Mandate eines Verwaltungsratsmitglieds ausserhalb der Gesellschaft Ein Mitglied des Verwaltungsrats darf kumulativ maximal folgende Mandate in einem obersten Leitungs- oder Verwaltungsorgan von Rechtseinheiten, die verpflichtet sind, sich ins Handelsregister oder in ein entsprechendes ausländisches Register eintragen zu lassen, übernehmen: a)
7 Mandate für Publikumsgesellschaften gemäss Artikel 727 Abs. 1 Ziff. 1 OR; und
b)
8 Mandate für Gesellschaften gemäss Artikel 727 Abs. 1 Ziff. 2 OR; und
c)
5 Mandate für Rechtseinheiten, welche die Kriterien gemäss lit. a) und b) hiervor nicht erfüllen.
|
38
Mandates held in several legal entities each operating under the same management or same beneficial owner (group) are deemed to be a single mandate. |
|
Mandate von verschiedenen Rechtseinheiten, welche aber derselben Führung oder derselben wirtschaftlichen Eigentümerin unterstehen (Konzern), gelten als ein Mandat, dürfen aber insgesamt vierzig nicht übersteigen. |
|
If a legal entity fulfills several of the above mentioned criteria, it can be freely counted towards any category. The following mandates are excepted from this restrictions: a)
mandates in legal entities which are controlled by the Company or which control the Company;
b)
honorary mandates in charitable legal entities.
|
|
Erfüllt eine Rechtseinheit mehrere der vorgenannten Kriterien, kann sie beliebig jeder auf sie zutreffenden Kategorie zugerechnet werden. Folgende Mandate sind von diesen Beschränkungen ausgenommen: a)
Mandate in Rechtseinheiten, welche von der Gesellschaft kontrolliert werden oder welche die Gesellschaft kontrollieren;
b)
Ehrenamtliche Mandate in gemeinnützigen Rechtseinheiten.
|
|
Article 40 Mandates of a Member of the Executive Committee outside the Company Each member of the Executive Committee may, with approval of the Board of Directors, cumulatively assume not more than the following number of mandates in the board of directors, the superior management or an administrative body of a legal entity which is obliged to be registered in the Swiss commercial register or an equivalent foreign register: a)
2 mandates for publicly traded companies pursuant to article 727 para. 1 number 1 CO; and
b)
3 mandates for companies pursuant to article 727 para. 1 number 2 CO; and
c)
5 mandates for companies which do not fulfil the criteria under litera a) and b) hereunder.
|
|
Artikel 40 Mandate eines Geschäftsleitungsmitglieds ausserhalb der Gesellschaft Jedes Mitglied der Geschäftsleitung darf mit Genehmigung des Verwaltungsrats kumulativ maximal folgende Mandate in einem obersten Leitungs- oder Verwaltungsorgan von Rechtseinheiten, die verpflichtet sind, sich ins Handelsregister oder in ein entsprechendes ausländisches Register eintragen zu lassen, übernehmen: a)
2 Mandate für Publikumsgesellschaften gemäss Artikel 727 Abs. 1 Ziff. 1 OR; und
b)
3 Mandate für Gesellschaften gemäss Artikel 727 Abs. 1 Ziff. 2 OR; und
c)
5 Mandate für Rechtseinheiten, welche die Kriterien gemäss lit. a) und b) hiervor nicht erfüllen.
|
Mandates held in several legal entities each operating under the same management or same beneficial owner |
|
Mandate von verschiedenen Rechtseinheiten, welche aber derselben Führung oder derselben |
39
(group) are deemed to be a single mandate. |
|
wirtschaftlichen Eigentümerin unterstehen (Konzern), gelten als ein Mandat. |
|
If a legal entity fulfills several of the above mentioned criteria, it can be freely counted towards any category. The following mandates are excepted from this restrictions: a)
mandates in legal entities which are controlled by the Company or which control the Company;
b)
honorary mandates in charitable legal entities.
|
|
Erfüllt eine Rechtseinheit mehrere der vorgenannten Kriterien, kann sie beliebig jeder auf sie zutreffenden Kategorie zugerechnet werden. Folgende Mandate sind von diesen Beschränkungen ausgenommen: a)
Mandate in Rechtseinheiten, welche von der Gesellschaft kontrolliert werden oder welche die Gesellschaft kontrollieren;
b)
Ehrenamtliche Mandate in gemeinnützigen Rechtseinheiten.
|
|
Article 41 Loans and Credits The members of the Board of Directors and the Executive Committee may not be granted any loans, credits or securities. Excepted from the above are advances in the maximum amount of CHF 500'000 per person for attorneys' fees, court and other similar costs required for the defence of third-party liability claims permitted by article 31. |
|
Artikel 41 Darlehen und Kredite Den Mitgliedern des Verwaltungsrats und der Geschäftsleitung dürfen keine Darlehen, Kredite oder Sicherheiten gewährt werden. Ausnahme davon bilden Vorschusszahlungen über einen Betrag von maximal CHF 500'000 pro Person für Anwalts-, Gerichts- und ähnliche Kosten zur Abwehr von Verantwortlichkeitsansprüchen, sofern zulässig nach Artikel 31. |
|
Article 42 Pension Funds The Company shall remunerate members of the Board of Directors only in respect of the employer's mandatory contributions to social insurance. Above and beyond this, the Company shall not make any contributions to pension funds or other such pension plans. In exceptional cases, contributions such as these may be made subject to a request by the Compensation Committee and the approval of the General Meeting. |
|
Artikel 42 Pensionskasse Die Gesellschaft leistet für die Mitglieder des Verwaltungsrats die gesetzlichen Arbeitgebersozialversicherungsbeiträge. Abgesehen davon richtet die Gesellschaft keine Beiträge an die Pensionskasse oder andere Vorsorgeeinrichtungen für die Mitglieder des Verwaltungsrats aus. Solche Beiträge können ausnahmsweise auf Antrag des Vergütungsausschusses und nach Genehmigung der Generalversammlung ausgerichtet werden. |
Members of the Executive Committee participate in the Company's pension plans (the Company's pension fund and the management pension plan). The pension plans conform to the legal requirements (BVG). For members of the Executive Committee, the insured income |
|
Die Mitglieder der Geschäftsleitung partizipieren am Pensionsplan der Gesellschaft (Pensionskasse sowie Management Pensionsplan). Der Pensionsplan hat den gesetzlichen Bestimmungen (BVG) zu entsprechen. Das versicherte Einkommen der Mitglieder der Geschäftsleitung entspricht jeweils dem Betrag der fixen Vergütung zuzüglich 50% der |
40
is defined as the fixed remuneration plus 50% of the target performance-related remuneration, up to the legal maximum. Equity-linked income components are not included. |
|
erfolgsabhängigen Vergütung bis zum gesetzlichen Maximum. Aktienbezogene Vergütungen werden nicht berücksichtigt. |
Within the overall compensation approved by the General Meeting, the Company may make additional payments into the Company's pension funds for the benefit of members of the Executive Committee in order to cover any disadvantage suffered as a result of the change of jobs or to purchase additional pension entitlements. In this context the Company may conclude life insurance policies on behalf of members of the Executive Committee and pay the insurance premiums either fully or in part. |
|
Die Gesellschaft kann zugunsten der Geschäftsleitungsmitglieder und im Rahmen der von der Generalversammlung genehmigten Gesamtvergütungen zusätzliche Einkäufe in die Pensionskasse tätigen, um Nachteile aufgrund von Stellenwechsel auszugleichen oder zugunsten zusätzlicher Rentenansprüche. In diesem Zusammenhang kann die Gesellschaft Lebensversicherungen zugunsten der Mitglieder der Geschäftsleitung abschliessen und die Versicherungsprämien vollumfänglich oder teilweise zahlen. |
Upon retirement, the Company may also grant members of the Executive Committee a bridging pension to cover the period between early retirement at 62 and the ordinary age of retirement, if such bridging pension does not exceed 100% of the total annual compensation of the respective member last paid. |
|
Die Gesellschaft kann ihren Geschäftsleitungsmitgliedern eine Überbrückungsrente zusichern, um die Zeitdauer zwischen einer Frühpensionierung ab dem 62. Altersjahr und dem ordentlichen Pensionsalter abzudecken, soweit eine solche Überbrückungsrente 100% der letztmalig an dieses Mitglied bezahlte Jahresvergütung nicht übersteigt. |
|
Article 43 Option and Share Plans Under the Company's Option Plan, the Board of Directors, upon proposal of the Compensation Committee, allocates the participating members of the Executive Committee and the Board of Directors a fixed number of options, shares or other equity-linked instruments with a vesting for a period to be determined by the Board of Directors (the vesting period). At the end of the vesting period, participants in the Option Plan are entitled to exercise the options granted against payment of the strike price. These options to acquire shares in the Company or allocated shares or other equity-linked instruments are subject to the basic principles set out in the following: |
|
Artikel 43 Options- und Aktienpläne Gemäss dem Optionsplan der Gesellschaft, teilt der Verwaltungsrat auf Antrag des Vergütungsausschusses den Mitgliedern der Geschäftsleitung und des Verwaltungsrats eine bestimmte Anzahl Optionen, Aktien oder anderen eigenkapitalbasierten Instrumenten zu, welche einer durch den Verwaltungsrat festzulegenden Sperrfrist unterliegen. Am Optionsplan partizipierende Mitglieder sind nach Ablauf der Sperrfrist berechtigt, die gewährten Optionen gegen Bezahlung des Ausübungspreises auszuüben. Die Optionen, welche zum Erwerb von Aktien an der Gesellschaft berechtigen, bzw. zugeteilten Aktien oder anderen eigenkapitalbasierten Instrumenten unterliegen den folgenden Grundsätzen: a)
Es liegt im freien Ermessen des Verwaltungsrats, ob und wem Optionen, Aktien oder anderen
|
41
|
a)
it is the sole discretion of the Board of Directors to decide whether to allocate options, shares and other equity-linked instruments and to whom;
b)
each year, the Board of Directors, upon proposal of the Compensation Committee, stipulates the number of options and shares to be allocated, the date of allocation and the strike price;
c)
each option incorporates a non-transferable, pre-emptive, and contingent right to acquire a certain number of Company's shares;
d)
in the case of a change of control (as defined in the Option Plan) or delisting of the Company's shares, the Board may decide that the vesting period shall end (accelerated vesting) and whether the participating member of the Executive Committee or the Board shall be entitled to exercise the options on a pro rata basis on the day the transaction that led to the change of control or delisting was executed. It is at the sole discretion of the Board of Directors to decide upon proposal of the Compensation Committee whether the objectives have been met;
e)
the individual members of the Executive Committee or the Board of Directors participating in the Option Plan are responsible for paying any taxes or social security contributions for which they are legally liable and for declaring income correctly to the authorities;
f)
it is at the sole discretion of the Board of Directors to decide whether to
|
|
eigenkapitalbasierten Instrumenten zugeteilt werden; b)
Der Verwaltungsrat bestimmt jährlich auf Antrag des Vergütungsausschusses Anzahl und Datum der Zuteilung sowie Ausübungspreis der Optionen und Aktien;
c)
Jede Option begründet ein unübertragbares, bedingtes Bezugsrecht eine bestimmte Anzahl Aktien der Gesellschaft zu erwerben;
d)
Im Falle eines Kontrollwechsels (gemäss Definition im Optionsplan) oder der Dekotierung der Aktien kann der Verwaltungsrat entscheiden, ob die Sperrfrist vorzeitig endet und das teilnehmende Geschäftsleitungsmitglied oder Mitglied des Verwaltungsrat entsprechend berechtigt wird, seine Optionen pro-rata basierend auf dem Stichtag der Transaktion, welche zum Kontrollwechsel geführt hat, oder der Dekotierung der Aktien auszuüben. Der Verwaltungsrat entscheidet nach freiem Ermessen und auf Antrag des Vergütungsausschusses, ob die Ziele in diesem Zusammenhang gegeben sind;
e)
Das jeweilige Mitglied der Geschäftsleitung oder des Verwaltungsrats, welches am Optionsplan teilnimmt, ist selber dafür verantwortlich, dass die vom Empfänger zu bezahlenden Steuern oder Sozialabgaben bezahlt und Einkommen der zuständigen Behörden korrekt gemeldet werden.
f)
Der Verwaltungsrat entscheidet nach freiem Ermessen über Ergänzungen des Optionsplans im Rahmen der obgenannten Grundsätze oder über dessen Beendigung.
|
42
supplement the Option Plan within the bounds of the principles set out above or to discontinue it. |
|
|
The Company may periodically offer shares in the Company to important and long-term employees for a price being at maximum 10% below the average volume-weighted price of the last 30 trading days at the stock exchange. Members of the Board of Directors and the Executive Committee may be included in this programme. The shares acquired thereby shall be blocked for a period of at least 3 years. |
|
Die Gesellschaft kann periodisch Aktien der Gesellschaft zu einem Preis, der maximal 10% unter dem über 30 Börsentage volumengewichteten durchschnittlichen Kurs an der Börse liegt, an wichtige und langjährige Mitarbeiter abgeben. Die Mitglieder des Verwaltungsrats und der Geschäftsleitung können in dieses Programm eingeschlossen werden. Die so erworbenen Aktien sind für mindestens 3 Jahre gesperrt. |
VIII. FISCAL YEAR, ACCOUNTING PRINCIPLES, ALLOCATION OF PROFITS |
|
VIII. GESCHÄFTSJAHR, RECHNUNGSLEGUNG, GEWINNVERTEILUNG |
|
Article 44 Fiscal Year The Board of Directors shall determine the start and the end of the Company's business year. |
|
Artikel 44 Geschäftsjahr Der Verwaltungsrat bestimmt, wann das Geschäftsjahr beginnt und wann es endet. |
|
Article 45 Accounting The annual accounts consist of the profit and loss statement, the balance sheet, the cash flow statement, the annex and the management report, and shall be drawn up pursuant to the provisions of the Swiss Code of Obligations, particularly of articles 958 et seq. CO, and the generally accepted commercial principles and customary rules in that business area. |
|
Artikel 45 Rechnungslegung Die Jahresrechnung besteht aus der Erfolgsrechnung, der Bilanz, der Geldflussrechnung, dem Anhang und dem Lagebericht und ist gemäss den Vorschriften des Schweizerischen Obligationenrechts, insbesondere Artikeln 958 ff. OR, sowie nach den allgemein anerkannten kaufmännischen und branchenüblichen Grundsätzen zu erstellen. |
If required by law, the consolidated financial statements shall be drawn in accordance with the provisions of article 962 CO. |
|
Die Konzernrechnung wird, sofern gesetzlich vorgeschrieben, gemäss den Bestimmungen von Artikeln 962 OR erstellt. |
|
Article 46 Allocation of Profits Subject to the legal provisions regarding distribution of profits, the profit as shown on the balance sheet shall be allocated by |
|
Artikel 46 Gewinnverteilung Die Generalversammlung beschliesst nach Entgegennahme der Anträge des Verwaltungsrates und des Berichtes der Revisionsstelle unter Vorbehalt |
43
the General Meeting at its discretion after receipt of the proposals of the Board of Directors and the Auditors. |
|
der gesetzlichen Bestimmungen über die Verwendung des Bilanzgewinnes und setzt die Dividende und den Zeitpunkt ihrer Auszahlung fest. |
In addition to the legal reserves, the General Meeting may create supplemental reserves. |
|
Zusätzlich zu den gesetzlichen Reserven kann die Generalversammlung zusätzliche Reserven bereitstellen. |
Dividends not claimed within five years after the due date shall remain with the Company and be allocated to the general reserves. |
|
Dividenden, die nicht innerhalb von fünf Jahren nach dem Fälligkeitstag beansprucht werden, verbleiben bei der Gesellschaft und werden den allgemeinen Rücklagen zugeführt. |
IX. DISSOLUTION AND LIQUIDATION |
|
IX. AUFLÖSUNG UND LIQUIDATION |
|
Article 47 Dissolution and Liquidation The dissolution and liquidation of the Company shall take place in accordance with the provisions of the Swiss Code of Obligations. |
|
Artikel 47 Auflösung und Liquidation Für die Auflösung und Liquidation der Gesellschaft gelten die Bestimmungen des Schweizerischen Obligationenrechts. |
X. NOTICES AND PUBLICATIONS |
|
X. MITTEILUNGEN UND BEKANNTMACHUNGEN |
|
Article 48 Notices and Publications The Swiss Official Gazette of Commerce (SOGC) is the official publication medium. |
|
Artikel 48 Mitteilungen und Bekanntmachungen Das Schweizerische Handelsamtsblatt (SHAB) ist das offizielle Publikationsmedium. |
Shareholder communications and notices the shareholders shall be made by publication in the Swiss Official Gazette of Commerce or sent by mail or e-mail to the addresses registered in the share register. |
|
Mitteilungen und Bekanntmachungen an die Aktionäre erfolgen durch Publikation im Schweizerischen Handelsamtsblatt oder durch Brief oder E-Mail an die im Aktienbuch verzeichneten Adressen. |
Unless the law provides otherwise, notices shall be given to creditors by publication in the Swiss Official Gazette of Commerce. The Board of Directors may assign further means of communication. |
|
Bekanntmachungen an die Gläubiger erfolgen in den vom Gesetz vorgeschriebenen Fällen durch Veröffentlichung im Schweizerischen Handelsamtsblatt, dem Publikationsorgan der Gesellschaft. Der Verwaltungsrat kann weitere Publikationsmittel bezeichnen. |
44
XI. QUALIFIED FACTS |
|
XI. QUALIFIZIERTE TATBESTÄNDE |
|
Artikel 49 Contribution in Kind In connection with the capital increase of 1 March 2023, and in accordance with the contribution in kind agreement as of 1 March 2023 (the Contribution in Kind Agreement), the Company acquires 10'489'371 ordinary shares in the nominal amount of UDS 0.001 each of European Biotech Acquisition Corp with registered seat in George Town, Cayman Islands (EBAC), from Continental Stock Exchange Corp (Contributor), acting in its own name but on behalf of the shareholders of EBAC. The shares of EBAC are acquired for a total value of USD 104'893'710.00. Based on the Contribution in Kind Agreement and as consideration, the Company issues to the Contributor, acting in its own name but for the account of the holders of ordinary shares of EBAC, a total of 10'489'371 fully paid registered shares with a with a par value of CHF 0.01 each. |
|
Artikel 49 Sacheinlage Die Gesellschaft übernimmt bei der Kapitalerhöhung vom 1. März 2023 gemäss Sacheinlagevertrag vom 1. März 2023 (Sacheinlagevertrag) 10'489'371 Aktien (ordinary shares) im Nennwert von USD 0.001 der European Biotech Acquisition Corp, mit Sitz in George Town, Cayman Islands (EBAC), von Continental Stock Exchange Corp (Einlegerin), handelnd im eigenen Namen aber auf Rechnung der Aktionäre der EBAC. Die Aktien der EBAC werden zu einem Übernahmewert von insgesamt USD 104'893'710.00 übernommen. Im Einklang mit dem Sacheinlagevertrag weist die Gesellschaft als Gegenleistung der Einlegerin, handelnd im eigenen Namen aber auf Rechnung der Aktionäre der EBAC, insgesamt 10'489'371 voll einbezahlte Namenaktien mit einem Nennwert von je CHF 0.01 der Gesellschaft zu. |
|
"In connection with the capital increase of 2 March 2023, and in accordance with the contribution in kind agreement as of 2 March 2023 (the Contribution in Kind Agreement), the Company acquires
3'306'771 registered shares (Common Shares) with a nominal value of CHF 0.10 each;
1'623'793 registered shares series A (Preferred Shares Series A) with a nominal value of CHF 0.10 each;
2'486'188 registered shares series B1 (Preferred Shares Series B1) with a nominal value of CHF 0.10 each;
1'675'474 registered shares series B2 (Preferred Shares Series B2 First Tranche) with a nominal value of CHF 0.10 each;
|
|
Die Gesellschaft übernimmt bei der Kapitalerhöhung vom 2. März 2023 gemäss Sacheinlagevertrag vom 2. März 2023 (Sacheinlagevertrag)
3'306'771 Namenaktien (Stammaktien) mit einem Nennwert von je CHF 0.10;
1'623'793 Namenaktien (Vorzugsaktien Serie A) mit einem Nennwert von je CHF 0.10;
2'486'188 Namenaktien (Vorzugsaktien Serie B1) mit einem Nennwert von je CHF 0.10;
1'675'474 Namenaktien (Vorzugsaktien Serie B2 erste Tranche) mit einem Nennwert von je CHF 0.10;
426'378 Namenaktien (Vorzugsaktien Serie B2 zweite Tranche) mit einem Nennwert von je CHF
|
45
|
426'378 registered shares series B2 (Preferred Shares Series B2 Second Tranche) with a nominal value of CHF 0.10 each;
603'472 registered shares series B2 (Preferred Shares Series B2 Third Tranche) with a nominal value of CHF 0.10 each
5'337'777 registered shares series C 1a (Preferred Shares Series C 1a First Tranche) with a nominal value of CHF 0.10 each
362'036 registered shares series C 1a (Preferred Shares Series C 1a Second Tranche) with a nominal value of CHF 0.10 each
197'745 registered shares series C 1b (Preferred Shares Series C 1b) with a nominal value of CHF 0.50 each
of Oculis SA with registered seat in Ecublens (VD), Switzerland, from Continental Stock Exchange Corp (Contributor), acting in its own name but on behalf of the shareholders Oculis SA. The shares of Oculis SA are acquired for a total value of USD 202'770.02 and CHF 37'939.95. Based on the Contribution in Kind Agreement and as consideration, the Company issues to the Contributor, acting in its own name but for the account of the shareholders of Oculis SA, a total of 24'070'997 fully paid registered shares with a with a par value of CHF 0.01. |
|
0.10;
603'472 Namenaktien (Vorzugsaktien Serie B2 dritte Tranche) mit einem Nennwert von je CHF 0.10;
5'337'777 Namenaktien (Vorzugsaktien Serie C 1a erste Tranche) mit einem Nennwert von je CHF 0.10;
362'036 Namenaktien (Vorzugsaktien Serie C 1a zweite Tranche) mit einem Nennwert von je CHF 0.10; sowie
197'745 Namenaktien (Vorzugsaktien Serie C 1b) mit einem Nennwert von je CHF 0.50
der Oculis SA mit Sitz in Ecublens (VD), Schweiz, von Continental Stock Exchange Corp (Einlegerin), handelnd im eigenen Namen aber auf Rechnung der Aktionäre der Oculis SA. Die Aktien der Oculis SA werden zu einem Übernahmewert von insgesamt USD 202'770.02 und CHF 37'939.95 übernommen. Im Einklang mit dem Sacheinlagevertrag weist die Gesellschaft als Gegenleistung der Einlegerin, handelnd im eigenen Namen aber auf Rechnung der Aktionäre der Oculis SA, insgesamt 24'070'997 voll einbezahlte Namenaktien mit einem Nennwert von je CHF 0.01 zu. |
Basel, 13 June 2023 Basel, 13. Juni 2023
Exhibit 2.3
DESCRIPTION OF SECURITIES
General
We were incorporated as a stock corporation (Aktiengesellschaft) organized under the laws of Switzerland in accordance with articles 620 et seqq. of the CO and registered with the Commercial Register of the Canton of Zug on October 31, 2022. Our corporate legal headquarters is located at Bahnhofstrasse 7, 6300 Zug, Switzerland. Neither the articles of association nor the operation of law limit the duration of Oculis Holding AG.
Capital Structure of Oculis Holding AG
Issued Share Capital
Immediately prior to the Business Combination, Oculis Holding AG’s share capital was CHF 356,821.68 divided into 35,682,168 fully paid-in registered shares with a nominal value of CHF 0.01 each.
In the context of the Business Combination, Oculis Holding AG increased its share capital in the Commercial Register of the Canton of Zug on the Acquisition Closing Date to CHF 365,273.68, divided into 36,527,368 Ordinary Shares, fully paid-up.
In the context of the public offering for the issuance and sale by Oculis Holding AG of ordinary shares based on that certain underwriting agreement entered into by Oculis Holding AG and BofA Securities Inc. and SVB Securities, LLC, as representatives of the several underwriters named therein, Oculis Holding AG increased its share capital in the Commercial Register of the Canton of Zug on 5 June 2023 to CHF 400,273.68, divided into 40,027,368 Ordinary Shares, fully paid-up.
As a result of the partial exercise by the underwriters to purchase additional ordinary shares as part of the abovementioned offering, Oculis Holding AG increased its share capital in the Commercial Register of the Canton of Zug on 13 June 2023 to CHF 401,816.02, divided into 40,181,602 Ordinary Shares, fully paid-up.
Share Classes
The Articles of Association provide for one class of Ordinary Shares with a nominal value of CHF 0.01 each. Each Ordinary Share will carry one vote in general meetings of shareholders, and the Ordinary Shares are listed on the Nasdaq Global Market.
Share Capital Increases (General)
Under Swiss law, we may increase our share capital and issue new shares through an ordinary capital increase, an increase by capital band (Kapitalband) or a conditional capital increase (Bedingte Kapitalerhöhung). In each case, the issue price for each share may not be less than the nominal value of the newly issued share. An ordinary capital increase is approved at a general meeting of shareholders. The required vote is generally the approval of simple majority of the votes cast at the general meeting of shareholders. At least two-thirds of the represented share votes and the absolute majority of the represented nominal value of the shares present in person or represented by proxy is required for capital increases against our equity, against contributions in kind, for the purposes of acquiring assets or the granting of special benefits, or for capital increases where the pre-emptive/subscription rights of shareholders are limited or excluded. The amount by which the capital can be increased in an ordinary capital increase is unlimited, provided that sufficient contributions are made to cover the capital increase. An ordinary capital increase that has been approved by the shareholders must be executed within six months of shareholder approval. In an ordinary capital increase, holders of Ordinary Shares have pre-emptive rights to obtain newly issued shares in an amount proportional to the nominal value of the shares they already hold, unless such rights are excluded in accordance with Swiss law. For further details on these circumstances, please see the section entitled “—Pre-emptive Rights and Advance Subscription Rights.”
Exhibit 2.3
Our shareholders can further authorize the Board of Directors by way of an amendment of the Articles of Association to increase or decrease the share capital within a capital band in an amount not to exceed 50% of the share capital registered in the commercial register for a period of five years without further shareholder approval. To create a capital band, a resolution of the general meeting of shareholders passed by a supermajority of at least two-thirds of the represented share votes and the absolute majority of the represented nominal value of the shares present in person or represented by proxy is required. Additional information regarding capital band is set forth below in the section entitled “—Capital band.”
Under Swiss law, conditional share capital is used to issue new shares in the context of employee benefit and incentive plans, debt instruments with conversion rights or warrants granted to creditors or options and warrants issued to third parties. To create conditional capital, a resolution of the general meeting of shareholders passed by a supermajority of at least two-thirds of the represented share votes and the absolute majority of the represented nominal value of the shares present in person or represented by proxy is required. The requirements for a conditional capital increase are set forth below in the section entitled “—Conditional Share Capital.”
Capital band
Under the Articles of Association, the Board of Directors is authorized to increase the share capital, at any time until March 2, 2028, at the latest, by a maximum amount of CHF 178,410.84 by issuing a maximum of 17,841,084 fully paid-up shares with a nominal value of CHF 0.01 each (Ordinary Shares). Such increase of the share capital (i) by means of an offering underwritten by a financial institution, a syndicate of financial institutions or another third party or third parties, followed by an offer to the then-existing shareholders of the Oculis Holding AG, and (ii) in partial amounts, are permissible.
The Board of Directors may determine the time of the issuance, the issue price, the manner in which the new shares have to be paid up, the date from which the shares carry the right to dividends, the conditions for the exercise of the pre-emptive rights and the allotment of pre-emptive rights that have not been exercised. The Board of Directors may allow the pre-emptive rights that have not been exercised to expire, or it may place such shares or the pre-emptive rights of which have not been exercised, at market conditions or use them otherwise in the interest of Oculis Holding AG.
The Board of Directors is authorized to withdraw or limit the pre-emptive rights of the shareholders with respect to the shares to be issued under the capital band and to allot them to individual shareholders or third parties:
|
1. |
if the issue price of the new registered shares is determined by reference to the market price; |
|
2. |
for the acquisition of an enterprise, part of an enterprise or participations, or for the financing or refinancing of any of such acquisition, or in the event of share placement for the financing or refinancing of such placement; |
|
3. |
for purposes of broadening the shareholders of our constituency in certain financial or investor markets, for purposes of the participation of strategic partners, or in connection with the listing or registration of new registered shares on domestic or foreign stock exchanges; |
|
4. |
for purposes of granting an over-allotment option (Greenshoe) or an option to subscribe additional shares to the respective initial purchaser(s) or underwriter(s) in a placement or sale of registered shares; |
|
5. |
for raising of capital (including private placements) in a fast and flexible way, which probably could not be achieved without the exclusion of the statutory pre-emptive right of the existing shareholders; |
|
6. |
for other valid grounds in the sense of article 652b para. 2 CO; or |
|
7. |
following a shareholder or a group of shareholders acting in concert having accumulated shareholdings in excess of 15% of the share capital registered in the commercial register without having submitted to the other shareholders a takeover offer recommended by the Board of Directors, or for the defense of an actual, threatened or potential takeover bid, in relation to which the Board of Directors, upon consultation with an independent financial adviser retained by it, has not recommended to the shareholders acceptance on the basis that the Board of Directors has not found the takeover bid to be financially fair to the shareholders. |
Exhibit 2.3
The authorization to withdraw or limit the pre-emptive rights is limited to the above listed items and exclusively linked to the particular available capital band (Kapitalband) set out in the articles of association. If the period to increase our share capital within the capital band lapses without having been used by the Board of Directors, the authorization to withdraw or to limit the pre-emptive rights lapses simultaneously with such capital.
Conditional Share Capital
Conditional Share Capital in Connection with Employee Benefit Plans
Under the articles of association, our share capital may be increased by an amount not exceeding CHF 78,355.44 through the issue of a maximum of 7,835,544 fully paid up registered shares, each with a nominal value of CHF 0.01 (Ordinary Shares), in connection with the exercise of option rights or other equity-linked instruments granted to any employee of Oculis Holding AG or a subsidiary, and any consultant, members of the Board of Directors, or other person providing services to us or a subsidiary.
Shareholders’ subscription rights are excluded with regard to these shares. These new registered shares may be issued at a price below the current market price. The Board of Directors shall determine the other conditions of issue including the issue price of the Ordinary Shares.
Conditional Share Capital for new Bonds and Similar Debt Instruments
Under the Articles of Association, our share capital may be increased by an amount not exceeding CHF 50,000 through the issuance from time to time of a maximum of 5,000,000 fully paid up registered shares, each with a par value of CHF 0.01 (Ordinary Shares), in connection with the exercise of convertible rights and/or option rights or warrants, which have been granted or will be granted in connection with new bonds and similar debt instruments, including convertible loans of Oculis SA which were issued prior to the date of the Business Combination in accordance with the Convertible Loan Agreements, that have been issued by us or our subsidiaries.
Shareholders’ advance subscription rights and subscription rights are excluded with regard to the new registered shares. These new registered shares may be issued at a price below the current market price. The Board shall determine the other conditions of issue including the issue price of the Ordinary Shares.
Conditional Share Capital for EBAC Warrants
Under the Articles of Association, our share capital may be increased by an amount not exceeding CHF 44,032.94 through the issuance, from time to time, of a maximum of 4,403,294 fully paid up registered shares, each with a par value of CHF 0.01 (Ordinary Shares), in connection with the exercise of warrants granted through the exercise of conversion and/or option rights, which were assumed from, and allocated by, EBAC, on the basis of the Warrant Assignment and Assumption Agreement.
Shareholders’ advance subscription rights and subscription rights are excluded with regard to the new registered shares. These new registered shares may be issued at a price below the current market price. The Board shall determine the other conditions of issue including the issue price of the Ordinary Shares.
Participation Certificates and Profit-sharing Certificates
As of the date of this proxy statement/prospectus, we have neither participation certificates (Partizipationsscheine) nor profit-sharing certificates (Genussscheine) outstanding.
Treasury Shares
As of the date of this proxy statement/prospectus, we may hold Ordinary Shares in treasury. Under Swiss law, a stock company may only hold 10% of its own shares in treasury and up to 20% under special circumstances.
Exhibit 2.3
Pre-emptive Rights and Advance Subscription Rights
Swiss law provides that any share issue, whether for cash or non-cash consideration, is subject to the prior approval at a general meeting of shareholders. Shareholders are granted certain pre-emptive rights (Bezugsrechte) to subscribe for new issues of shares and advance subscription rights (Vorwegzeichnungsrechte) to subscribe for warrants, convertible bonds or similar debt instruments with option rights in proportion to the nominal amount of shares held. Pursuant to the Articles of Association, a resolution adopted at a general meeting by a majority of two-thirds of the votes represented at the meeting is required to repeal, limit or suspend pre-emptive rights.
Warrants
Pursuant to the Business Combination Agreement and Warrant Assignment and Assumption Agreement, the Company has assumed and issued 4,403,294 Warrants. Each Warrant entitles the registered holder to purchase one Ordinary Share at a price of $11.50 per share, subject to adjustment as discussed below, exercisable at any time commencing 30 days after the completion of the Business Combination, provided that we have an effective registration statement under the Securities Act covering the issuance the Ordinary Shares issuable upon exercise of the Warrants. Pursuant to the Warrant Assignment and Assumption Agreement, a warrant holder may exercise its Warrants only for a whole number of Ordinary Shares. This means only a whole public warrant may be exercised at a given time by a Warrant holder. The Warrants will expire on March 2, 2028 (i.e. five years after the completion of the Business Combination), at 5:00 p.m. Eastern Time, or earlier upon redemption or liquidation.
We will not be obligated to deliver any Ordinary Shares pursuant to the exercise of a warrant and will have no obligation to settle such warrant exercise unless a registration statement under the Securities Act covering the issuance of the Ordinary Shares issuable upon exercise of the Warrants is then effective and a current prospectus relating thereto is current, subject to us satisfying our obligations described below with respect to registration, or a valid exemption from registration is available, including in connection with a cashless exercise permitted as a result of a notice of redemption described below under the section entitled “Redemption of warrants when the price per Ordinary Share equals or exceeds $10.00.” No Warrant will be exercisable for cash or on a cashless basis, and we will not be obligated to issue any shares to holders seeking to exercise their warrants, unless the issuance of the shares upon such exercise is registered or qualified under the securities laws of the state of the exercising holder, or an exemption is available. In the event that the conditions in the two immediately preceding sentences are not satisfied with respect to a Warrant, the holder of such warrant will not be entitled to exercise such warrant and such warrant may have no value and expire worthless.
We agreed to file with the SEC this registration statement covering the issuance, under the Securities Act, of the Ordinary Shares issuable upon exercise of the Warrants, and we will use our commercially reasonable efforts to cause this registration statement to become effective within 60 business days, and to maintain the effectiveness of such registration statement, and a current prospectus relating thereto, until the expiration of the Warrants in accordance with the provisions of the Warrant Assignment and Assumption Agreement. Notwithstanding the above, if the Ordinary Shares are, at the time of any exercise of a warrant, not listed on a national securities exchange such that they satisfy the definition of a “covered security” under Section 18(b)(1) of the Securities Act, we may, at our option, require holders of Warrants who exercise their warrants to do so on a “cashless basis” in accordance with Section 3(a)(9) of the Securities Act and, in the event we so elect, we will not be required to file or maintain in effect a registration statement, but will use commercially reasonable efforts to register or qualify the shares under applicable blue sky laws to the extent an exemption is not available. In such event, each holder would pay the exercise price by surrendering the Warrants for that number of Ordinary Shares equal to the lesser of (i) the quotient obtained by dividing (A) the product of the number of Ordinary Shares underlying the Warrants, multiplied by the excess of the “fair market value” (defined below) less the exercise price of the warrants by (B) the fair market value and (ii) 0.361. The “fair market value” as used in this proxy statement/prospectus shall mean the volume weighted average price of the Ordinary Shares for the 10 trading days ending on the trading day prior to the date on which the notice of exercise is received by the warrant agent.
We will not redeem the Warrants as described above unless a registration statement under the Securities Act covering the issuance of the Ordinary Shares issuable upon exercise of the warrants is then effective and a current prospectus relating to those Ordinary Shares is available throughout the 30-days redemption period. If and when the Warrants become redeemable by us, we may exercise our redemption right even if we are unable to register or qualify the underlying securities for sale under all applicable state securities laws.
Exhibit 2.3
We have established the last of the redemption criterion discussed above to prevent a redemption call unless there is, at the time of the call, a significant premium to the Warrant exercise price. If the foregoing conditions are satisfied and we issue a notice of redemption of the Warrants, each warrant holder will be entitled to exercise his, her or its warrants prior to the scheduled redemption date. However, the price of the Ordinary Shares may fall below the $18.00 redemption trigger price (as adjusted for adjustments to the number of shares issuable upon exercise or the exercise price of a Warrant as described under the heading “—Redeemable Warrants—Warrants—Anti-dilution Adjustments”) as well as the $11.50 (for whole shares) warrant exercise price after the redemption notice is issued.
Redemption of Warrants when the price per Ordinary Share equals or exceeds $10.00. Once the warrants become exercisable, we may redeem the outstanding Warrants:
|
• |
|
in whole and not in part; |
|
• |
|
at $0.10 per warrant upon a minimum of 30 days’ prior written notice of redemption provided that holders will be able to exercise their warrants on a cashless basis prior to redemption and receive that number of shares based on the redemption date and the “fair market value” of the Ordinary Shares, except as otherwise described below; |
|
• |
|
if, and only if, the Reference Value equals or exceeds $10.00 per share (as adjusted for adjustments to the number of shares issuable upon exercise or the exercise price of a warrant as described under the heading “—Redeemable Warrants—Warrants—Anti-dilution Adjustments”) for any 20 trading days within the 30-trading day period ending three trading days before we send the notice of redemption to the warrant holders; and |
|
• |
|
if the Reference Value is less than $18.00 per share (as adjusted for adjustments to the number of shares issuable upon exercise or the exercise price of a warrant, as described under the heading “—Redeemable Warrants—Warrants—Anti-dilution Adjustments”) the Private Placement Warrants must also be concurrently called for redemption on the same terms as the outstanding Warrants, as described above. |
During the period beginning on the date the notice of redemption is given, holders may elect to exercise their Warrants on a cashless basis. The numbers in the table below represent the number of Ordinary Shares that a warrant holder will receive upon such cashless exercise in connection with a redemption by us pursuant to this redemption feature based on the “fair market value” of the Ordinary Shares on the corresponding redemption date (assuming holders elect to exercise their warrants and such warrants are not redeemed for $0.10 per warrant), determined for these purposes based on volume weighted average price of the Ordinary Shares during the 10 trading days immediately following the date on which the notice of redemption is sent to the holders of warrants, and the number of months that the corresponding redemption is sent to the holders of warrants, each as set forth in the table below. We will provide its warrant holders with the final fair market value no later than one business day after the 10-trading day period described above ends.
Redemption of Warrants when the price per Ordinary Share equals or exceeds $18.00. Once the Warrants become exercisable, we may redeem the warrants (except as described herein with respect to the Private Placement Warrants):
|
• |
|
in whole and not in part; |
|
• |
|
at a price of $0.01 per warrant; |
|
• |
|
upon not less than 30 days’ prior written notice of redemption to each warrant holder; and |
|
• |
|
if, and only if, the last reported sale price of the Ordinary Shares for any 20 trading days within a 30-trading day period ending on the third trading day prior to the date on which we send the notice of redemption to the warrant holders (such price, the “Reference Value”) equals or exceeds $18.00 per share (as adjusted for adjustments to the number of shares issuable upon exercise or the exercise price of a warrant as described under the heading “—Redeemable Warrants—Public Shareholders’ Warrants—Anti-dilution Adjustments”). |
Exhibit 2.3
This redemption feature is structured to allow for all of the outstanding Warrants to be redeemed when the Ordinary Shares are trading at or above $10.00 per share, which may be at a time when the trading price of the Ordinary Shares is below the exercise price of the warrants. We have established this redemption feature to provide itself with the flexibility to redeem the Warrants without the Warrants having to reach the $18.00 per share threshold set forth above under the heading “—Redemption of Warrants when the price per Ordinary Share equals or exceeds $18.00.” Holders choosing to exercise their warrants in connection with a redemption pursuant to this feature will, in effect, receive a number of shares for their warrants based on an option pricing model with a fixed volatility input as of the date of this proxy statement/prospectus. This redemption right provides us with an additional mechanism by which to redeem all of the outstanding Warrants, and therefore have certainty as to our capital structure as the warrants would no longer be outstanding and would have been exercised or redeemed. We will be required to pay the applicable redemption price to warrant holders if we choose to exercise this redemption right and it will allow us to quickly proceed with a redemption of the Warrants if we determine it is in its best interest to do so. As such, we would redeem the Warrants in this manner when we believe it is in its best interest to update its capital structure to remove the Warrants and pay the redemption price to the warrant holders.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Redemption Date |
|
Fair Market Value of Ordinary Shares |
|
|||||||||||||||||||||||||||||||||
(period to expiration of warrants) |
|
≤$10.00 |
|
|
$11.00 |
|
|
$12.00 |
|
|
$13.00 |
|
|
$14.00 |
|
|
$15.00 |
|
|
$16.00 |
|
|
$17.00 |
|
|
≥$18.00 |
|
|||||||||
60 months |
|
|
0.261 |
|
|
|
0.281 |
|
|
|
0.297 |
|
|
|
0.311 |
|
|
|
0.324 |
|
|
|
0.337 |
|
|
|
0.348 |
|
|
|
0.358 |
|
|
|
0.361 |
|
57 months |
|
|
0.257 |
|
|
|
0.277 |
|
|
|
0.294 |
|
|
|
0.310 |
|
|
|
0.324 |
|
|
|
0.337 |
|
|
|
0.348 |
|
|
|
0.358 |
|
|
|
0.361 |
|
54 months |
|
|
0.252 |
|
|
|
0.272 |
|
|
|
0.291 |
|
|
|
0.307 |
|
|
|
0.322 |
|
|
|
0.335 |
|
|
|
0.347 |
|
|
|
0.357 |
|
|
|
0.361 |
|
51 months |
|
|
0.246 |
|
|
|
0.268 |
|
|
|
0.287 |
|
|
|
0.304 |
|
|
|
0.320 |
|
|
|
0.333 |
|
|
|
0.346 |
|
|
|
0.357 |
|
|
|
0.361 |
|
48 months |
|
|
0.241 |
|
|
|
0.263 |
|
|
|
0.283 |
|
|
|
0.301 |
|
|
|
0.317 |
|
|
|
0.332 |
|
|
|
0.344 |
|
|
|
0.356 |
|
|
|
0.361 |
|
45 months |
|
|
0.235 |
|
|
|
0.258 |
|
|
|
0.279 |
|
|
|
0.298 |
|
|
|
0.315 |
|
|
|
0.330 |
|
|
|
0.343 |
|
|
|
0.356 |
|
|
|
0.361 |
|
42 months |
|
|
0.228 |
|
|
|
0.252 |
|
|
|
0.274 |
|
|
|
0.294 |
|
|
|
0.312 |
|
|
|
0.328 |
|
|
|
0.342 |
|
|
|
0.355 |
|
|
|
0.361 |
|
39 months |
|
|
0.221 |
|
|
|
0.246 |
|
|
|
0.269 |
|
|
|
0.290 |
|
|
|
0.309 |
|
|
|
0.325 |
|
|
|
0.340 |
|
|
|
0.354 |
|
|
|
0.361 |
|
36 months |
|
|
0.213 |
|
|
|
0.239 |
|
|
|
0.263 |
|
|
|
0.285 |
|
|
|
0.305 |
|
|
|
0.323 |
|
|
|
0.339 |
|
|
|
0.353 |
|
|
|
0.361 |
|
33 months |
|
|
0.205 |
|
|
|
0.232 |
|
|
|
0.257 |
|
|
|
0.280 |
|
|
|
0.301 |
|
|
|
0.320 |
|
|
|
0.337 |
|
|
|
0.352 |
|
|
|
0.361 |
|
30 months |
|
|
0.196 |
|
|
|
0.224 |
|
|
|
0.250 |
|
|
|
0.274 |
|
|
|
0.297 |
|
|
|
0.316 |
|
|
|
0.335 |
|
|
|
0.351 |
|
|
|
0.361 |
|
27 months |
|
|
0.185 |
|
|
|
0.214 |
|
|
|
0.242 |
|
|
|
0.268 |
|
|
|
0.291 |
|
|
|
0.313 |
|
|
|
0.332 |
|
|
|
0.350 |
|
|
|
0.361 |
|
24 months |
|
|
0.173 |
|
|
|
0.204 |
|
|
|
0.233 |
|
|
|
0.260 |
|
|
|
0.285 |
|
|
|
0.308 |
|
|
|
0.329 |
|
|
|
0.348 |
|
|
|
0.361 |
|
21 months |
|
|
0.161 |
|
|
|
0.193 |
|
|
|
0.223 |
|
|
|
0.252 |
|
|
|
0.279 |
|
|
|
0.304 |
|
|
|
0.326 |
|
|
|
0.347 |
|
|
|
0.361 |
|
18 months |
|
|
0.146 |
|
|
|
0.179 |
|
|
|
0.211 |
|
|
|
0.242 |
|
|
|
0.271 |
|
|
|
0.298 |
|
|
|
0.322 |
|
|
|
0.345 |
|
|
|
0.361 |
|
15 months |
|
|
0.130 |
|
|
|
0.164 |
|
|
|
0.197 |
|
|
|
0.230 |
|
|
|
0.262 |
|
|
|
0.291 |
|
|
|
0.317 |
|
|
|
0.342 |
|
|
|
0.361 |
|
12 months |
|
|
0.111 |
|
|
|
0.146 |
|
|
|
0.181 |
|
|
|
0.216 |
|
|
|
0.250 |
|
|
|
0.282 |
|
|
|
0.312 |
|
|
|
0.339 |
|
|
|
0.361 |
|
9 months |
|
|
0.090 |
|
|
|
0.125 |
|
|
|
0.162 |
|
|
|
0.199 |
|
|
|
0.237 |
|
|
|
0.272 |
|
|
|
0.305 |
|
|
|
0.336 |
|
|
|
0.361 |
|
6 months |
|
|
0.065 |
|
|
|
0.099 |
|
|
|
0.137 |
|
|
|
0.178 |
|
|
|
0.219 |
|
|
|
0.259 |
|
|
|
0.296 |
|
|
|
0.331 |
|
|
|
0.361 |
|
3 months |
|
|
0.034 |
|
|
|
0.065 |
|
|
|
0.104 |
|
|
|
0.150 |
|
|
|
0.197 |
|
|
|
0.243 |
|
|
|
0.286 |
|
|
|
0.326 |
|
|
|
0.361 |
|
0 months |
|
|
— |
|
|
|
— |
|
|
|
0.042 |
|
|
|
0.115 |
|
|
|
0.179 |
|
|
|
0.233 |
|
|
|
0.281 |
|
|
|
0.323 |
|
|
|
0.361 |
|
As stated above, we can redeem the Warrants when the Ordinary Shares are trading at a price starting at $10.00, which is below the exercise price of $11.50, because it will provide certainty with respect to its capital structure and cash position while providing warrant holders with the opportunity to exercise their warrants on a cashless basis for the applicable number of shares. If we choose to redeem the Warrants when the Ordinary Shares are trading at a price below the exercise price of the warrants, this could result in the warrant holders receiving fewer Ordinary Shares than they would have received if they had chosen to exercise their warrants for Ordinary Shares if and when such Ordinary Shares were trading at a price higher than the exercise price of $11.50.
No fractional Ordinary Shares will be issued upon exercise. If, upon exercise, a holder would be entitled to receive a fractional interest in a share, we will round down to the nearest whole number of the number of Ordinary Shares to be issued to the holder. If, at the time of redemption, the Warrants are exercisable for a security other than the Ordinary Shares pursuant to the Warrant Assignment and Assumption Agreement (for instance, if we are not the surviving company after completion of a business combination), the warrants may be exercised for such security. At such time as the Warrants become exercisable for a security other than the Ordinary Shares, we (or the surviving company, as applicable) will use its commercially reasonable efforts to register under the Securities Act the security issuable upon the exercise of the warrants.
Exhibit 2.3
Redemption Procedures. A holder of a Warrant may notify us in writing in the event it elects to be subject to a requirement that such holder will not have the right to exercise such warrant, to the extent that after giving effect to such exercise, such person (together with such person’s affiliates), to the warrant agent’s actual knowledge, would beneficially own in excess of 9.8% (or such other amount as a holder may specify) of the Ordinary Shares issued and outstanding immediately after giving effect to such exercise.
Anti-dilution Adjustments. If the number of issued and outstanding Ordinary Shares is increased by a capitalization or share dividend payable in Ordinary Shares, or by a split-up of Ordinary Shares or other similar event, then, on the effective date of such capitalization or share dividend, split-up or similar event, the number of Ordinary Shares issuable on exercise of each Warrant will be increased in proportion to such increase in the issued and outstanding Ordinary Shares. A rights offering made to all or substantially all holders of Ordinary Shares entitling holders to purchase Ordinary Shares at a price less than the “historical fair market value” (as defined below) will be deemed a share dividend of a number of Ordinary Shares equal to the product of (i) the number of Ordinary Shares actually sold in such rights offering (or issuable under any other equity securities sold in such rights offering that are convertible into or exercisable for Ordinary Shares) and (ii) one minus the quotient of (a) the price per Ordinary Share paid in such rights offering and (b) the historical fair market value. For these purposes, (i) if the rights offering is for securities convertible into or exercisable for Ordinary Shares, in determining the price payable for Ordinary Shares, there will be taken into account any consideration received for such rights payable upon exercise or conversion and (ii) “historical fair market value” means the volume weighted average price of Ordinary Shares during the 10 trading day period ending on the trading day prior to the first date on which the Ordinary Shares trade on the applicable exchange or in the applicable market, regular way, without the right to receive such rights.
If the number of issued and outstanding Ordinary Shares is decreased by a consolidation, combination, reverse share sub-division or reclassification of Ordinary Shares or other similar event, then, on the effective date of such consolidation, combination, reverse share sub-division, reclassification or similar event, the number of Ordinary Shares issuable on exercise of each warrant will be decreased in proportion to such decrease in issued and outstanding Ordinary Shares. Whenever the number of Ordinary Shares purchasable upon the exercise of the Warrants is adjusted, as described above, the warrant exercise price will be adjusted by multiplying the warrant exercise price immediately prior to such adjustment by a fraction (i) the numerator of which will be the number of Ordinary Shares purchasable upon the exercise of the warrants immediately prior to such adjustment and (ii) the denominator of which will be the number of Ordinary Shares so purchasable immediately thereafter.
In addition, if (i) we issue additional Ordinary Shares or equity-linked securities for capital raising purposes in connection with the completion of the Business Combination at an issue price or effective issue price of less than $9.20 per ordinary share (with such issue price or effective issue price to be determined in good faith by the Board) (the “Newly Issued Price”), (ii) the aggregate gross proceeds from such issuances represent more than 60% of the total equity proceeds, and interest thereon, available for the funding of the Business Combination on the date of the completion of the Business Combination (net of redemptions), and (iii) the volume weighted average trading price of the Ordinary Shares during the 20 trading day period starting on the trading day prior to the day on which we consummate the Business Combination (such price, the “Market Value”) is below $9.20 per share, the exercise price of the Warrants will be adjusted (to the nearest cent) to be equal to 115% of the higher of the Market Value and the Newly Issued Price, the $18.00 per share redemption trigger prices described above under “—Redemption of Warrants when the price per Ordinary Share equals or exceeds $18.00” and “—Redemption of Warrants when the price per Ordinary Share equals or exceeds $10.00” will be adjusted (to the nearest cent) to be equal to 180% of the higher of the Market Value and the Newly Issued Price, and the $10.00 per share redemption trigger price described above under “—Redemption of Warrants when the price per Ordinary Share equals or exceeds $10.00” will be adjusted (to the nearest cent) to be equal to the higher of the Market Value and the Newly Issued Price.
Exhibit 2.3
In case of any reclassification or reorganization of the issued and outstanding Ordinary Shares (other than those described above or that solely affects the par value of such Ordinary Shares), or in the case of any merger or consolidation of Oculis Holding AG with or into another corporation (other than a merger or consolidation in which we are a continuing corporation and that does not result in any reclassification or reorganization of our issued and outstanding Ordinary Shares), or in the case of any sale or conveyance to another corporation or entity of the assets or other property of Oculis Holding AG as an entirety or substantially as an entirety in connection with which we are dissolved, the holders of the Warrants will thereafter have the right to purchase and receive, upon the basis and upon the terms and conditions specified in the warrants and in lieu of the Ordinary Shares immediately theretofore purchasable and receivable upon the exercise of the rights represented thereby, the kind and amount of shares, stock or other equity securities or property (including cash) receivable upon such reclassification, reorganization, merger or consolidation, or upon a dissolution following any such sale or transfer, that the holder of the Warrants would have received if such holder had exercised their warrants immediately prior to such event. However, if such holders are entitled to exercise a right of election as to the kind or amount of securities, cash or other assets receivable upon such merger or consolidation, then the kind and amount of securities, cash or other assets for which each warrant will become exercisable will be deemed to be the weighted average of the kind and amount received per share by such holders in such merger or consolidation that affirmatively make such election, and if a tender, exchange or redemption offer has been made to and accepted by such holders under circumstances in which, upon completion of such tender or exchange offer, the maker thereof, together with members of any group (within the meaning of Rule 13d-5(b)(1) under the Exchange Act) of which such maker is a part, and together with any affiliate or associate of such maker (within the meaning of Rule 12b-2 under the Exchange Act) and any members of any such group of which any such affiliate or associate is a part, own beneficially (within the meaning of Rule 13d-3 under the Exchange Act) more than 50% of the issued and outstanding Ordinary Shares, the holder of a warrant will be entitled to receive the highest amount of cash, securities or other property to which such holder would actually have been entitled as a shareholder if such warrant holder had exercised the warrant prior to the expiration of such tender or exchange offer, accepted such offer and all of the Ordinary Shares held by such holder had been purchased pursuant to such tender or exchange offer, subject to adjustment (from and after the consummation of such tender or exchange offer) as nearly equivalent as possible to the adjustments provided for in the Warrant Assignment and Assumption Agreement. Additionally, if less than 70% of the consideration receivable by the holders of Ordinary Shares in such a transaction is payable in the form of ordinary shares in the successor entity that is listed for trading on a national securities exchange or is quoted in an established over-the-counter market, or is to be so listed for trading or quoted immediately following such event, and if the registered holder of the Warrant properly exercises the warrant within 30 days following public disclosure of such transaction, the warrant exercise price will be reduced as specified in the Warrant Assignment and Assumption Agreement based on the per share consideration minus the Black-Scholes Warrant Value (as defined in the Warrant Assignment and Assumption Agreement) of the Warrant.
The Warrants will be issued in registered form under the Warrant Assignment and Assumption Agreement. The Warrant Assignment and Assumption Agreement provides that the terms of the Warrants may be amended without the consent of any holder for the purpose of (i) curing any ambiguity or correcting any mistake, including to conform the provisions of the Warrant Assignment and Assumption Agreement to the description of the terms of the warrants and the Warrant Assignment and Assumption Agreement set forth in this proxy statement/prospectus or defective provision or (ii) adding or changing any provisions with respect to matters or questions arising under the Warrant Assignment and Assumption Agreement as the parties to the Warrant Assignment and Assumption Agreement may deem necessary or desirable and that the parties deem not to adversely affect the rights of the registered holders of the warrants.
The warrant holders do not have the rights or privileges of holders of Ordinary Shares and any voting rights until they exercise their Warrants and receive Ordinary Shares. After the issuance of Ordinary Shares upon exercise of the Warrants, each holder will be entitled to one vote for each share held of record on all matters to be voted on by shareholders.
We have agreed that, subject to applicable law, any action, proceeding or claim against it arising out of or relating in any way to the Warrant Assignment and Assumption Agreement will be brought and enforced in the courts of the State of New York or the United States District Court for the Southern District of New York, and us irrevocably submits to such jurisdiction, which jurisdiction will be the exclusive forum for any such action, proceeding or claim. This provision applies to claims under the Securities Act but does not apply to claims under the Exchange Act or any claim for which the federal district courts of the United States of America are the sole and exclusive forum.
Exhibit 2.3
Dividends
General
Dividends may be paid only if we have sufficient distributable profit from previous years or sufficient free reserves to allow the distribution of a dividend. Swiss law requires that we retain at least 5% of its annual net profit as general reserves for so long as these reserves amount to less than 20% of its paid-in nominal share capital.
Annual Profit Distribution
Under Swiss law, dividends are proposed by the Board and require the approval at a meeting of shareholders. Our auditors must also confirm that the dividend proposal conforms to law and the Articles of Association. Dividends that have not been collected by shareholders within five years after the due date accrue to us.
For a description of certain tax considerations, including withholding taxes, in relation to dividend payments, please see the section entitled “Material Tax Consideration—Material Swiss Tax Considerations.”
Payment
The Board determines the date on which the dividend entitlement starts. Dividends are usually due and payable shortly after the shareholders have passed the resolution approving the payment, but shareholders may also resolve at an annual general meeting to pay dividends in quarterly or other instalments.
Capital Reduction
Distributions out of issued share capital (i.e., the aggregate nominal value of our issued shares) are not allowed and may be made only by way of a share capital reduction. Such a capital reduction requires a resolution passed by an absolute majority of the shares represented at a general meeting of shareholders or the introduction of a capital band (Kapitalband) pursuant to which the Board is empowered to make such resolution. The resolution of the shareholders must be recorded in a public deed and a special audit report must confirm that claims of our creditors remain fully covered despite the reduction in our share capital recorded in the Commercial Register. Our share capital may be reduced below CHF 100,000 only if and to the extent that at the same time the statutory minimum share capital of CHF 100,000 is re-established by sufficient new, fully paid-up capital. Upon approval or before the general meeting of the capital reduction, the Board must give public notice of the capital reduction resolution in the Swiss Official Gazette of Commerce (“SOGC”) and notify creditors that they may request, within thirty (30) days of the third publication, satisfaction of or security for their claims. The reduction of our share capital may be implemented only after expiration of this time limit.
Repurchases of Shares
Swiss law limits our right to purchase and hold our own shares. We may purchase our own shares only if and to the extent that: (i) We have freely distributable reserves in the amount of the purchase price; and (ii) the aggregate nominal value of all Ordinary Shares held by us does not exceed 10% of our share capital (or up to 20% under certain specific circumstances). Furthermore, according to Swiss accounting rules, we need to reflect the amount of the purchase price of the acquired Ordinary Shares as a negative position through the creation of a special reserve on its balance sheet. We may face negative tax consequences, if we hold more than 10% of our Shares for more than six years.
Ordinary Shares held by us or our subsidiaries do not carry any voting rights at general meetings of shareholders, but are entitled to the economic benefits, including dividends, pre-emptive rights (Bezugsrechte) in the case of share capital increases and advance subscription rights (Vorwegzeichnungsrechte) and in the case of issuance of debt instruments with option rights applicable to the Ordinary Shares generally.
Form and Transfer of Shares
Form of the Shares
Exhibit 2.3
Ordinary Shares may be issued as ordinary uncertificated securities within the meaning of article 973c CO (Wertrechte) and/or global certificates. In accordance with article 973c CO, we maintain a register of uncertificated securities (Wertrechtebuch). We may create intermediated securities (Bucheffekten) for Ordinary Shares.
Upon its registration with the share register, a shareholder may at any time request that we issue a written confirmation of the Ordinary Shares held by such shareholder. However, the shareholder has no right to request the printing and delivery of share certificates nor the conversion of Ordinary Shares issued in one form into another form. We may, however, at any time print and deliver certificates for registered (single certificates or global certificates) and, with the consent of the shareholder, delete without replacement issued share certificates, which have been returned to it. We may convert Ordinary Shares from one form into another form at any time and without the approval of the shareholders. We shall bear the cost associated with any such conversion.
Transfer of Shares
Ordinary Shares in uncertificated form (Wertrechte) may only be transferred by way of assignment. Ordinary Shares or the beneficial interest in Ordinary Shares, as applicable, credited in a securities account may only be transferred when a credit of the relevant intermediated securities to the acquirer’s securities account is made in accordance with applicable rules. For certain registration and voting right restrictions on the Ordinary Shares, please see the section entitled “Registration and Voting Right Restrictions.”
Share Register
We maintain a share register (Aktienbuch) (the “Share Register”) in which the owners of the Ordinary Shares are registered with name, address and nationality (in case of legal entities the registered office). In relation to Oculis Holding AG, only those shareholders registered in the Share Register are recognized as shareholders.
Pursuant to article 4 of the Articles of Association, acquirers of Ordinary Shares are, upon request and presentation of evidence of the transfer, registered as shareholders with voting rights in the Share Register if they explicitly declare to hold Ordinary Shares in their own name and for their own account.
The Board shall implement the necessary directions for maintaining the Share Register and it may issue corresponding regulations or guidelines. The Board may delegate such tasks.
In the invitation to the general meeting, the Board shall announce the record date for registration in the Share Register that is relevant with respect to the right to attend and vote.
We have the right to delete entries in the Share Register retroactively as of the date of the entry if the registration has been made on the basis of false information. We may give the relevant shareholder or nominee, in advance, the opportunity to be heard. The relevant shareholder or nominee must be informed of the deletion without delay.
Registration and Voting Right Restrictions
The Articles of Association contain the following registration restrictions:
Exhibit 2.3
Those associated through capital, voting power, joint management, beneficial ownership or in any other way, or joining for the acquisition of shares shall be regarded as one acquirer for the purposes of article 4 of the Articles of Association. Acquirers who do not meet the legal or regulatory requirements according to article 4 of the Articles of Association shall be entered in the Share Register as shareholder without voting rights for Ordinary Shares exceeding the limit of 15%. In case of an already occurred registration, Ordinary Shares exceeding the limit of 3% will be cancelled from the Share Register as Ordinary Shares with voting rights and instead be registered as Ordinary Shares without voting rights. The Board may enact regulations governing the details of such registration restriction. Nominees do not constitute acquirers within the meaning of article 4 of the Articles of Association. After hearing the person concerned, we may cancel the registrations in the Share Register if those registrations were based on false information of the acquirer. In addition, according to article 4 of the Articles of Association, the Board may refuse the exercise of voting rights of a shareholder in excess of 15% of the total number of voting rights of Oculis Holding AG pursuant to the entry in the commercial register, if such shareholder does not meet the legal or regulatory requirements according to article 4 of the Articles of Association
Further, the voting right restrictions pursuant to article 4 of the Articles of Association as set out above also apply to Ordinary Shares, which are held by a nominee for the account of a person exceeding the threshold of 15% (regulatory voting right restrictions).
Apart from the registration and voting rights restrictions as described above, there are no restrictions on the transferability of the Ordinary Shares in the Articles of Association.
General Meetings of Shareholders
Convocation of Meetings
Under Swiss law and article 10 of the Articles of Association, an annual general meeting of shareholders must be held each year within six months after the end of the business year. Extraordinary meetings of shareholders may be convened when required.
General meetings of shareholders must be convened by the Board of Directors at least 20 days before the date of the meeting. The general meeting of shareholders is convened by way of a notice appearing in our official publication medium, currently the SOGC. Registered shareholders may also be informed by ordinary mail or e-mail. The notice of a general meeting of shareholders must state the items on the agenda, the proposals to be acted upon and, in case of elections, the names of the nominated candidates. Except in the limited circumstances listed below, a resolution may not be passed at a general meeting without proper notice. This limitation does not apply to proposals to convene an extraordinary general meeting of shareholders or to initiate a special investigation. No previous notification is required for proposals concerning items included in the agenda or for debates that do not result in a vote.
Exhibit 2.3
In addition, one or several shareholders that represent at least 5% of the share capital may also request to convene a general meeting. Shareholders representing at least 0.5% of the share capital may request items to be put on the agenda, provided the request is submitted to the Board at least 70 calendar days in advance of the relevant general meeting. Convocation requests and requests for inclusion of agenda items need to be submitted to the Board in written form, indicating the agenda items and proposals. Swiss law and the Articles of Association do not prescribe that a particular quorum of shareholders is required for general meetings of shareholders to be validly held.
No resolutions may be passed on motions concerning agenda items which have not been duly announced, except for motions to convene an extraordinary general meeting, to initiate a special audit or to elect auditors upon a shareholders’ request. No prior notice is required to submit motions relating to items already on the agenda and to discuss matters on which no resolution is to be taken.
The general meeting will be chaired by the chairman of the Board, or, in his or her absence, by another member of the Board as appointed by the Board. If no member of the Board is present, the general meeting shall appoint the chairperson of the meeting.
Representation of Shareholders
Each shareholder may have its shares represented in the general meeting by itself or by a third person who does not need to be a shareholder by means of written proxy or by the independent proxy. The general meeting annually elects an independent proxy. The independent proxy’s term of office begins at the day of election and ends at the end of the following annual general meeting. Re-election is possible. If we do not have an independent proxy, the Board shall appoint the independent proxy for the next general meeting of shareholders.
Quorum and Majority Requirements at General Meetings of Shareholders
Except where the law or the Articles of Association provide otherwise, the general meeting passes its resolutions and performs elections with the absolute majority of the votes cast, excluding any abstentions, blank or invalid votes. The chairperson of the general meeting determines the voting procedure.
According to article 19 of the Articles of Association, a resolution of the general meeting passed with at least two-thirds of the votes represented at the meeting and the absolute majority of the nominal values of the Ordinary Shares represented at the meeting is required for:
|
1. |
the amendment of the purpose of the Company; |
|
2. |
the consolidation of shares, insofar as this does not require the consent of all shareholders concerned; |
|
3. |
the increase of the share capital against contributions in kind or by offsetting against a receivable and the granting of special benefits; |
|
4. |
the limitation or withdrawal of subscription rights; |
|
5. |
the introduction of conditional capital, the creation of reserve capital pursuant to article 12 of the Swiss Banking Act or the introduction of a capital band; |
|
6. |
the conversion of participation certificates into shares; |
|
7. |
the restriction of the transferability of registered shares; |
|
8. |
the creation of shares with privileged voting rights; |
|
9. |
the change of currency of the share capital; |
|
10. |
the introduction of the casting vote of the Chairman in the General Assembly; |
|
11. |
the introduction of a provision in the Articles of Association to hold general meetings outside of Switzerland; |
|
12. |
the change of the registered office of the Company; |
Exhibit 2.3
|
13. |
the introduction of an arbitration clause in the Articles of Association; |
|
14. |
the delisting of the Ordinary Shares; |
|
15. |
the dissolution of the Company; |
|
16. |
the merger, de-merger or conversion of the Company(subject to mandatory law); |
|
17. |
the alleviating or withdrawal of restrictions upon the transfer of registered shares; |
|
18. |
the conversion of registered shares into bearer shares and vice versa; and |
|
19. |
the amendment or elimination of the provisions of articles 4, 19 and 31 of the Articles of Association. |
Provisions of the Articles of Association which require higher majorities for the passing of certain resolutions than provided by law can only be adopted and removed with that same proposed majority.
Voting Rights
In principle, each Ordinary Share entitles a holder to one vote in our general meeting, irrespective of nominal value of such share (please see the section entitled “Comparison of Shareholder Rights—Voting Rights” for details on certain exceptions under Swiss law).
The Ordinary Shares are not divisible. The right to vote and the other rights of share ownership may only be exercised by shareholders (including any nominees) who are entered in the Share Register prior to the applicable cut-off date to be determined by the Board. Those entitled to vote in the general meeting may be represented by the independent proxy holder (annually elected by the general meeting of shareholders), by its legal representative or by another person with written authorization to act as proxy. The chairman of the general meeting has the power to decide whether to recognize a power of attorney. Only shareholders registered in the Share Register with voting rights are entitled to vote in an Ordinary Shareholders’ meeting.
Inspection of Books and Records
The annual report and the auditors’ report shall be made available for inspection by the shareholders at the registered office of the Company at the latest 20 days prior to the annual general meeting. Provided that the annual report and the auditors' report have not been made available electronically before the annual general meeting, each shareholder may demand a timely delivery of these documents. The notice to the shareholders must refer to this right. Furthermore, each shareholder may within one year after the annual general meeting demand the delivery of the auditors' report and the annual report in the form approved by the annual general meeting, provided that they have not been made available electronically.
Under Swiss law, a shareholder may also, upon request submitted to the Company, inspect the minutes of general meetings.
At general meetings, shareholders may further request information from the Board regarding the business and operations of the Company and may request information from our auditors regarding the performance and results of their examination of our financial statements. We may refuse to provide certain requested information to a shareholder if, in our opinion, the disclosure of the requested information would reveal confidential business secrets or infringe other protected interests.
Shareholders representing at least 5% of the share capital or votes have the right to inspect the company’s books. The Board of Directors must grant the inspection insofar as it is necessary for the exercise of shareholders’ rights and the disclosure would not reveal confidential business secrets or infringe other protected interests. Upon inspection of the books, the shareholders may make notes.
Special Investigations
Exhibit 2.3
If the shareholders’ inspection and information rights as outlined above prove to be insufficient, any shareholder may propose to the general meeting that specific facts be examined by a special commissioner in a special investigation. If the general meeting approves the proposal, the Company or any shareholder may, within 30 calendar days after the general meeting, request the court at our registered office to appoint a special commissioner. If the general meeting rejects the request, one or more shareholders representing at least 5% of the share capital or voting rights may request, within three months after the general meeting, a court to appoint a special commissioner as described in the Articles of Association. Such court will issue such order if the petitioners can demonstrate that the Board, any member thereof or an officer of the Company infringed the law or the Articles of Association and thereby damaged the Company or the shareholders. If admitted, the costs of the investigation by such court would generally be allocated to the Company and only in exceptional cases to the petitioners.
Notices
Official publications of the Company shall be made in the SOGC. The Board may designate additional means of publication.
Notices to the shareholders shall be made by official publications of the Company. Notices to shareholders may also be made by mail or email to the addresses recorded in the Share Register.
Takeover Regulation and Mandatory Bids
Swiss law provides for certain rules and protections of shareholders of domestic listed companies. Because the Ordinary Shares are listed exclusively on the Nasdaq Global Market, however, several of these rules do not apply to us as if we were a company listed in Switzerland. In particular, the Swiss rules under the Swiss Financial Market Infrastructure Act on disclosure of shareholdings and the tender offer rules under the Swiss Financial Market Infrastructure Act, including mandatory tender offer requirements and regulations regarding voluntary tender offers, which are typically available in relation to Swiss-listed companies, do not apply to us because we will not be listed in Switzerland.
Compulsory Acquisitions; Appraisal Rights
Business combinations and other transactions that are governed by the Switzerland’s Federal Act on Mergers, Demergers, Transformations and the Transfer of Assets of October 3, 2003, as amended (the “Swiss Merger Act”) (i.e., mergers, demergers, transformations and certain asset transfers) are binding on all shareholders. A statutory merger or demerger requires approval of two-thirds of the shares represented at a General Meeting of shareholders and the absolute majority of the nominal value of the shares represented.
If a transaction under the Swiss Merger Act receives all of the necessary consents, all shareholders are compelled to participate in such a transaction.
Swiss stock corporations may be acquired by an acquirer through the direct acquisition of the shares of the Swiss stock corporation. The Swiss Merger Act provides for the possibility of a so-called “cash-out” or “squeeze-out” merger with the approval of holders of 90% of the issued shares. In these limited circumstances, minority shareholders of the corporation being acquired may be compensated in a form other than through shares of the acquiring corporation (for instance, through cash or securities of a parent corporation of the acquiring corporation or of another corporation). For business combinations effected in the form of a statutory merger or demerger and subject to Swiss law, the Swiss Merger Act provides that if equity rights have not been adequately preserved or compensation payments in the transaction are unreasonable, a shareholder may request a competent court to determine a reasonable amount of compensation.
In addition, under Swiss law, the sale of “all or substantially all” of our assets may require the approval of two-thirds of the voting rights represented at a general meeting of shareholders and the absolute majority of the nominal value of the shares represented. Whether a shareholder resolution is required depends on the particular transaction, including whether the following test is satisfied:
Exhibit 2.3
Duration and Liquidation
Under Swiss law, unless the duration of a company is limited by its articles of association, a company may be dissolved at any time by way of liquidation, or, in the case of a merger with the Swiss Merger Act (Fusionsgesetz), based on a resolution of a general meeting of shareholders, which must be passed by a majority as provided by Swiss law or the relevant company’s articles of association, as the case may be. The Articles of Association do not limit the duration of the Company and provide that the majority required for the general meeting to resolve on the liquidation of the Company is set at two-thirds of the votes represented at the general meeting and the absolute majority of the nominal values of the shares represented at the meeting.
Dissolution and liquidation by court order is also possible if, among other things, (i) the Company becomes bankrupt or (ii) shareholders holding at least 10% of the Company’s share capital so request for important reasons. Under Swiss law, any surplus arising out of a liquidation (after settlement of all the claims of the Company’s creditors) is distributed in proportion to the paid-up nominal value of shares held. This surplus is subject to Swiss federal withholding tax, except if paid out of reserves from qualifying capital contributions (Reserven aus Kapitaleinlagen).
Comparison of Swiss and Delaware Shareholder Rights The Swiss laws applicable to Swiss corporations and their shareholders differ from laws applicable to U.S. corporations and their shareholders. The following table summarizes significant differences in shareholder rights pursuant to the provisions of the Swiss CO, by which our Company is governed, and the Delaware General Corporation Law applicable to companies incorporated in Delaware and their shareholders. Please note that this is only a general summary of certain provisions applicable to companies in Delaware and Switzerland.
|
|
|
Delaware Corporate Law |
|
Swiss Corporate Law |
|
Mergers and similar arrangements |
||
|
|
|
Exhibit 2.3
Under the Delaware General Corporation Law, with certain exceptions, a merger, consolidation, sale, lease or transfer of all or substantially all of the assets of a corporation must be approved by the board of di-rectors and a majority of the outstanding shares entitled to vote thereon. A shareholder of a Delaware corporation participating in certain major corporate transactions may, under certain circumstances, be entitled to appraisal rights pursuant to which such shareholder may receive cash in the amount of the fair value of the shares held by such shareholder (as determined by a court) in lieu of the consideration such shareholder would otherwise receive in the transaction. The Delaware General Corporation Law also provides that a parent corporation, by resolution of its board of directors, may merge with any subsidiary, of which it owns at least 90.0% of each class of capital stock without a vote by the shareholders of such subsidiary. Upon any such merger, dissenting shareholders of the subsidiary would have appraisal rights. |
|
Under Swiss law, with certain exceptions, a merger or a demerger of the corporation or a sale of all or substantially all of the assets of a corporation must be approved by two-thirds of the voting rights re-presented at the respective general meeting of shareholders as well as the majority of the par value of shares represented at such general meeting of shareholders. A shareholder of a Swiss corporation participating in a statutory merger or demerger pursuant to the Swiss Merger Act (Fusionsgesetz) can file a lawsuit against the surviving company. If the consideration is deemed “inadequate,” such shareholder may, in addition to the consideration (be it in shares or in cash) receive an additional amount to ensure that such shareholder receives the fair value of the shares held by such shareholder. Swiss law also provides that if the merger agreement provides only for a compensation payment, at least 90% of all members in the transferring legal entity who are entitled to vote shall approve the merger agreement.
|
|
||
Shareholders' suits |
||
|
|
|
Class actions and derivative actions generally are available to shareholders of a Delaware corporation for, among other things, breach of fiduciary duty, corporate waste and actions not taken in accordance with applicable law. In such actions, the court has discretion to permit the winning party to recover attorneys’ fees incurred in connection with such action. |
|
Class actions and derivative actions as such are not available under Swiss law. Nevertheless, certain actions may have a similar effect. A shareholder is entitled to bring suit against directors, officers or liquidators for breach of their duties and claim the payment of the company’s losses or damages to the corporation and, in some cases, to the individual shareholder. Likewise, an appraisal lawsuit won by a shareholder may indirectly compensate all shareholders. In addition, to the extent that U.S. laws and regulations provide a basis for liability and U.S. courts have jurisdiction, a class action may be available.
Under Swiss law, the prevailing party is generally entitled to recover a limited amount of attorneys’ fees incurred in connection with such action. The court has discretion to permit the shareholder who lost the lawsuit to recover attorneys’ fees incurred to the extent that he or she acted in good faith. |
|
||
Shareholder vote on board and management compensation |
||
|
|
|
Under the Delaware General Corporation Law, the board of directors has the authority to fix the compensation of directors, unless otherwise restricted by the certificate of incorporation or bylaws. |
|
Pursuant to Swiss law, the general meeting of shareholders has the non-transferable right, amongst others, to vote separately and bindingly on the aggregate amount of compensation of the members of the board of directors, of the executive committee and of the advisory boards
|
|
||
Annual vote on board renewal |
||
Exhibit 2.3
|
|
|
|
Unless directors are elected by written consent in lieu of an annual meeting, directors are elected in an annual meeting of shareholders on a date and at a time designated by or in the manner provided in the bylaws. Re-election is possible.
Classified boards are permitted. |
|
The general meeting of shareholders elects the members of the board of directors, the chairperson of the board of directors and the members of the compensation committee individually and annually for a term of office until the end of the following general meeting of shareholders. Re-election is possible. . |
|
||
Indemnification of directors and executive officers and limitation of liability |
||
|
|
|
|
The Delaware General Corporation Law provides that a certificate of incorporation may contain a provision eliminating or limiting the personal liability of directors and officers of the corporation for monetary damages for breach of a fiduciary duty as a director or officer, except no provision in the certificate of incorporation may eliminate or limit: •
the liability of a director or officer for any breach of the duty of loyalty to the corporation or its shareholder
•
the liability of a director or officer for acts or omissions not in good faith or which involve intentional misconduct or a knowing violation of law;
•
a director’s statutory liability for unlawful payment of dividends or unlawful share purchase or redemption;
•
the liability of a director or officer for any transaction from which the director or officer derived an improper personal benefit; or
•
the liability of an officer in any action by or in the right of the corporation.
|
|
Under Swiss corporate law, an indemnification by the corporation of a director or member of the executive committee in relation to potential personal liability is not effective to the extent the director or member of the executive committee intentionally or negligently violated his or her corporate duties towards the corporation (certain scholars advocate that at least a grossly negligent violation is required to exclude the indemnification). Furthermore, the general meeting of shareholders may discharge (release) the directors and members of the executive committee from liability for their conduct to the extent the shareholders have knowledge of the relevant facts of a potential claim. Such discharge is effective only with respect to claims of the company and of those shareholders who approved the discharge or who have since acquired their shares in full knowledge of the discharge. Most violations of corporate law are regarded as violations of duties towards the corporation rather than towards the shareholders. In addition, indemnification of other controlling persons is not permitted under Swiss corporate law, including shareholders of the corporation.
|
|
A Delaware corporation may indemnify any person who was or is a party or is threatened to be made a party to any proceeding, other than an action by or on behalf of the corporation, because the person is or was a director or officer, against liability incurred in connection with the proceeding if the director or officer acted in good faith and in a manner reasonably believed to be in, or not opposed to, the best interests of the corporation; and the director or officer, with respect to any criminal action or proceeding, had no reasonable cause to believe his or her conduct was unlawful.
Unless ordered by a court, any foregoing indemnification is subject to a determination that the director or officer has met the applicable standard of conduct:
•
by a majority vote of the directors who are not parties to the proceeding, even though less than a quorum;
|
|
The articles of association of a Swiss corporation may also set forth that the corporation shall indemnify and hold harmless, to the extent permitted by the law, the directors and executive managers out of assets of the corporation against threatened, pending or completed actions. Also, a corporation may enter into and pay for directors’ and officers’ liability insurance, which may cover negligent acts as well |
Exhibit 2.3
|
•
by a committee of directors designated by a majority vote of the eligible directors, even though less than a quorum;
•
by independent legal counsel in a written opinion if there are no eligible directors or if the eligible directors so direct; or
•
by the shareholders
Moreover, a Delaware corporation may not indemnify a director or officer in connection with any proceeding in which the director or officer has been adjudged to be liable to the corporation unless and only to the extent that the court determines that, despite the adjudication of liability but in view of all the circumstances of the case, the director or officer is fairly and reasonably entitled to indemnity for those expenses which the court deems proper |
|
|
|
||
Directors' fiduciary duties |
||
|
|
|
|
A director of a Delaware corporation has a fiduciary duty to the corporation and its shareholders. This duty has two components:
•
the duty of care; and
•
the duty of loyalty.
The duty of care requires that a director act in good faith, with the care that an ordinarily prudent person would exercise under similar circumstances. Under this duty, a director must inform himself or herself of, and disclose to shareholders, all material information reasonably available regarding a significant transaction.
The duty of loyalty requires that a director act in a manner he or she reasonably believes to be in the best interests of the corporation. He or she must not use his or her corporate position for personal gain or advantage. This duty prohibits self-dealing by a director and mandates that the best interest of the corporation and its shareholders take precedence over any interest possessed by a director, officer or controlling shareholder and not shared by the shareholders generally. In general, actions of a director are presumed to have been made on an informed basis, in good faith and in the honest belief that the action taken was in the best interests of the corporation. However, this presumption may be rebutted by evidence of a breach of one of the fiduciary duties.
Should such evidence be presented concerning a transaction by a director, a director must prove the procedural fairness of the transaction and that the transaction was of fair value to the corporation. |
|
The board of directors of a Swiss corporation manages the business of the corporation, unless responsibility for such management has been duly delegated to the executive committee based on organizational rules. However, there are several non-transferable duties of the board of directors:
•
the overall management of the corporation and the issuing of all necessary directives;
•
determination of the corporation’s organization;
•
the organization of the accounting, financial control and financial planning systems as required for management of the corporation;
•
the appointment and dismissal of persons entrusted with management and the representation of the corporation;
•
overall supervision of the persons entrusted with managing the corporation, in particular with regard to compliance with the law, articles of association, bylaws and internal directives;
•
compilation of the annual report, preparation for the general meeting of the shareholders, the compensation report and implementation of its resolutions;
•
the filing an application for a debt restructuring moratorium and notification of the court in the event that the company is over-indebted; and
•
the filing of the compensation report.
The members of the board of directors must perform their duties with all due diligence and safeguard the interests of the corporation in good faith. They must |
Exhibit 2.3
|
|
afford the shareholders equal treatment in equal circumstances.
The duty of care requires that a director act in good faith, with the care that an ordinarily prudent director would exercise under like circumstances.
The duty of loyalty requires that a director safeguard the interests of the corporation and requires that directors act in the interest of the corporation and, if necessarily, put aside their personal interests. The members of the board of directors and the executive committee are required to immediately and fully inform the board of directors about their conflicts of interests. If there is a risk of a conflict of interest, the board of directors must take appropriate measures to ensure that the interests of the company are duly taken into account. The burden of proof for a violation of these duties is with the corporation or with the shareholder bringing a suit against the director.
The Swiss Federal Supreme Court has established a doctrine that restricts its review of a business decision if the decision has been made after proper preparation, on an informed basis, and without conflicts of interest. |
|
||
Shareholder action by written consent |
||
|
|
|
A Delaware corporation may, in its certificate of incorporation, eliminate the right of shareholders to act by written consent. |
|
Shareholders of a Swiss corporation may exercise their voting rights in a general meeting of shareholders.
Shareholders may also exercise their rights by instructing an independent proxy, who is elected by the general meeting or the board of directors. The instruction of such (independent) proxies may occur in writing or electronically. The articles of association of a Swiss corporation may also provide for the possibility for shareholders to attend a general meeting electronically (virtual or hybrid general meeting) and cast their vote electronically.
|
|
||
Shareholder proposals |
||
|
|
|
A shareholder of a Delaware corporation has the right to put any proposal before the annual meeting of shareholders, provided it complies with the notice provisions in the governing documents. A special meeting may be called by the board of directors or any other person authorized to do so in the governing documents, but shareholders may be precluded from calling special meetings. |
|
At any general meeting of shareholders any shareholder may put proposals to the meeting if the proposal is part of an agenda item. No resolution may be taken on proposals relating to the agenda items that were not duly notified.
Unless the articles of association provide for a lower threshold or for additional shareholders’ rights: •
shareholders jointly representing at least 5% of the share capital or voting rights may
|
Exhibit 2.3
|
|
demand that a general meeting of shareholders be called for specific agenda items and specific proposals; and •
shareholders jointly representing at least 0.5% of the share capital or voting rights of the share capital or the voting rights may demand that an agenda item including a specific proposal, or a proposal with respect to an existing agenda item, be put on the agenda for a scheduled general meeting of shareholders, provided such request is made with appropriate lead time.
Any shareholder can propose candidates for election as directors or make other proposals within the scope of an agenda item without prior written notice. In addition, any shareholder is entitled, at a general meeting of shareholders and without advance notice, to (i) request information from the board of directors on the affairs of the company (note, however, that the right to obtain such information is limited), (ii) request information from the auditors on the methods and results of their audit, (iii) request that the general meeting of shareholders resolve to convene an extraordinary general meeting, or (iv) request that the general meeting of shareholders resolve to appoint an examiner to carry out a special examination (“Sonderuntersuchung”).
|
|
||
Cumulative voting |
||
|
|
|
Under the Delaware General Corporation Law, cumulative voting for elections of directors is not permitted unless the corporation’s certificate of incorporation provides for it. |
|
Cumulative voting is not permitted under Swiss corporate law. Pursuant to Swiss law, shareholders can vote for each proposed candidate, but they are not allowed to cumulate their votes for single candidates.
An annual individual election of (i) all members of the board of directors, (ii) the chairperson of the board of directors, (iii) the members of the compensation committee, (iv) the election of the independent proxy for a term of office of one year (i.e. until the following annual general meeting of shareholders), as well as the vote on the aggregate amount of compensation of the members of the board of directors, of the executive committee and of the members of any advisory board, is mandatory for listed companies. Re-election is permitted. |
|
||
Removal of directors |
||
|
|
|
A Delaware corporation with a classified board may be removed only for cause with the approval of a majority of the outstanding shares entitled to vote, unless the certificate of incorporation provides otherwise. |
|
A Swiss corporation may remove, with or without cause, any director at any time with a resolution passed by a majority of the shares represented at a general meeting of shareholders. The articles of |
Exhibit 2.3
|
|
association may provide that a larger majority is required. |
|
||
Transactions with interested shareholders |
||
|
|
|
|
The Delaware General Corporation Law generally prohibits a Delaware corporation from engaging in certain business combinations with an “interested shareholder” for three years following the date that such person becomes an interested shareholder. An interested shareholder generally is a person or group who or which owns or owned 15.0% or more of the corporation’s outstanding voting shares within the past three years.
|
|
No such rule applies to a Swiss corporation. |
|
||
Dissolution; Winding-up |
||
|
|
|
Unless the board of directors of a Delaware corporation approves the proposal to dissolve, dissolution must be approved by shareholders holding 100.0% of the total voting power of the corporation. Only if the dissolution is initiated by the board of directors may it be approved by a simple majority of the corporation’s outstanding shares. Delaware law allows a Delaware corporation to include in its certificate of incorporation a supermajority voting requirement in connection with dissolutions initiated by the board. |
|
A dissolution of a Swiss corporation requires the approval by two-thirds of the voting rights represented at the respective general meeting of shareholders as well as the majority of the par value of shares represented at such general meeting of shareholders. The articles of association may provide that a larger majority is required. |
|
||
Variation of rights of shares |
||
|
|
|
A Delaware corporation may vary the rights of a class of shares with the approval of a majority of the outstanding shares of such class, unless the certificate of incorporation provides otherwise. |
|
The general meeting of shareholders of a Swiss corporation may resolve that preference shares be issued or that existing shares be converted into preference shares with a resolution passed by a majority of the shares represented at the general meeting of shareholders.
Where a company has issued preference shares, further preference shares conferring preferential rights over the existing preference shares may be issued only with the consent of both a special meeting of the adversely affected holders of the existing preference shares and of a general meeting of all shareholders, unless otherwise provided in the articles of association. Shares with preferential voting rights are not regarded as preference shares for voting on such items.
|
|
||
Amendment of governing documents |
||
|
|
|
A Delaware corporation’s governing documents may be amended with the approval of a majority of the outstanding shares entitled to vote, unless the certificate of incorporation provides otherwise. |
|
The articles of association of a Swiss corporation may be amended with a resolution passed by a majority of the shares represented at a general meeting of shareholders, unless otherwise provided by law or in the articles of association. |
Exhibit 2.3
|
|
There are a number of resolutions, such as an amendment of the stated purpose of the corporation, the introduction of a capital band and conditional capital and the introduction of shares with preferential voting rights that require the approval by two-thirds of the votes and a majority of the par value of the shares represented at such general meeting of shareholders. The articles of association may increase these voting thresholds. . |
|
||
Inspection of books and records |
||
|
|
|
Shareholders of a Delaware corporation, upon written demand under oath stating the purpose thereof, have the right during the usual hours for business to inspect for any proper purpose and to obtain copies of list(s) of shareholders and other books and records of the corporation and its subsidiaries, if any, to the extent the books and records of such subsidiaries are available to the corporation. |
|
Shareholders of a Swiss corporation holding in the aggregate at least 5% of the nominal share capital or voting rights have the right to inspect books and records, subject to the safeguarding of the company’s business secrets and other interests warranting protection. A shareholder is only entitled to receive information to the extent required to exercise his or her rights as a shareholder. The board of directors has to decide on an inspection request within four months after receipt of such request. Denial of the request will need to be justified in writing. If the board of directors denies an inspection request, shareholders may request the order of an inspection by the court within thirty days. A shareholder’s right to inspect the share register is limited to the right to inspect his or her own entry in the share register. |
|
||
Payment of dividends |
||
|
|
|
|
The board of directors may approve a dividend without shareholder approval. Subject to any restrictions contained in its certificate of incorporation, the board may declare and pay dividends upon the shares of its capital stock either:
•
out of its surplus; or
•
in case there is no such surplus, out of its net profits for the fiscal year in which the dividend is declared and/or the preceding fiscal year.
Shareholder approval is required to authorize capital stock in excess of that provided in the charter. Directors may issue authorized shares without shareholder approval. |
|
Dividend (including interim dividend) payments and repayment of capital contributions (but not the nominal share capital) are subject to the approval of the general meeting of shareholders. The board of directors may propose to shareholders that a dividend shall be paid but cannot itself authorize the distribution.
Payments out of a corporation’s share capital (in other words, the aggregate par value of the corporation’s shares) in the form of dividends are not allowed and may be made only by way of a formal share capital reduction or a capital reduction within the capital band. Dividends may be paid only from the profits of the previous business year or brought forward from previous or current business years or if the corporation has distributable reserves, each as evidenced by the corporation’s audited stand-alone statutory balance sheet prepared pursuant to Swiss law and after allocations to reserves required by Swiss law and the articles of association have been deducted. |
|
||
Exhibit 2.3
Creation and issuance of new shares |
||
|
|
|
|
The creation of shares requires the board of directors to adopt a resolution or resolutions, pursuant to authority expressly vested in the board of directors by the provisions of the company’s certificate of incorporation.
|
|
All creation of shares require a shareholders’ resolution. The creation of a capital band or conditional share capital requires at least two-thirds of the voting rights represented at the general meeting of shareholders and a majority of the par value of shares represented at such meeting.
The board of directors may create and issue or cancel shares out of the capital band during a period of up to five years by a maximum amount of 50% of the current share capital.
Shares may be created and issued by the board of directors out of conditional share capital through the exercise of options or of conversion rights that the board of directors may grant to shareholders, creditors of bonds or similar debt instruments, employees, directors of the company or another group company or third parties. |
Exhibit 8.1
SUBSIDIARIES OF THE REGISTRANT
|
|
|
Name |
|
Jurisdiction of Formation/Organization |
|
|
|
Oculis Holding AG |
|
Switzerland |
Oculis Operations GmbH |
|
Switzerland |
Oculis ehf. |
|
Iceland |
Oculis U.S., Inc. |
|
United States |
Oculis France Sarl |
|
France |
Oculis HK, Ltd |
|
Hong Kong |
Oculis Merger Sub II Company |
|
Cayman Islands |
Exhibit 12.1
Certification by the Principal Executive Officer pursuant to
Securities Exchange Act Rules 13a-14(a) and 15d-14(a)
as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002
I, Riad Sherif, certify that:
|
1. |
I have reviewed this annual report on Form 20-F of Oculis Holding AG (the “Company”); |
|
2. |
Based on my knowledge, this report does not contain any untrue statement of a material fact or omit to state a material fact necessary to make the statements made, in light of the circumstances under which such statements were made, not misleading with respect to the period covered by this report; |
|
3. |
Based on my knowledge, the financial statements, and other financial information included in this report, fairly present in all material respects the financial condition, results of operations and cash flows of the Company as of, and for, the periods presented in this report; |
|
4. |
The Company’s other certifying officer and I are responsible for establishing and maintaining disclosure controls and procedures (as defined in Exchange Act Rules 13a-15(e) and 15d-15(e)) and internal control over financial reporting (as defined in Exchange Act Rules 13a-15(f) and 15d-15(f)) for the Company and have: |
|
(a) |
Designed such disclosure controls and procedures, or caused such disclosure controls and procedures to be designed under our supervision, to ensure that material information relating to the Company, including its consolidated subsidiaries, is made known to us by others within those entities, particularly during the period in which this report is being prepared; |
|
(b) |
Designed such internal control over financial reporting, or caused such internal control over financial reporting to be designed under our supervision, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles; |
|
(c) |
Evaluated the effectiveness of the Company’s disclosure controls and procedures and presented in this report our conclusions about the effectiveness of the disclosure controls and procedures, as of the end of the period covered by this report based on such evaluation; and |
|
(d) |
Disclosed in this report any change in the Company’s internal control over financial reporting that occurred during the period covered by the annual report that has materially affected, or is reasonably likely to materially affect, the Company’s internal control over financial reporting; and |
|
5. |
The Company’s other certifying officer and I have disclosed, based on our most recent evaluation of internal control over financial reporting, to the Company’s auditors and the audit committee of the Company’s board of directors (or persons performing the equivalent functions): |
|
(a) |
All significant deficiencies and material weaknesses in the design or operation of internal control over financial reporting which are reasonably likely to adversely affect the Company’s ability to record, process, summarize and report financial information; and |
|
(b) |
Any fraud, whether or not material, that involves management or other employees who have a significant role in the Company’s internal control over financial reporting.
|
Date: March 19, 2024
By: |
/s/ Riad Sherif |
|
|
Riad Sherif |
|
|
Chief Executive Officer |
|
|
(Principal Executive Officer) |
|
Exhibit 12.2
Certification by the Principal Financial Officer pursuant to
Securities Exchange Act Rules 13a-14(a) and 15d-14(a)
as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002
I, Sylvia Cheung, certify that:
|
1. |
I have reviewed this annual report on Form 20-F of Oculis Holding AG (the “Company”); |
|
2. |
Based on my knowledge, this report does not contain any untrue statement of a material fact or omit to state a material fact necessary to make the statements made, in light of the circumstances under which such statements were made, not misleading with respect to the period covered by this report; |
|
3. |
Based on my knowledge, the financial statements, and other financial information included in this report, fairly present in all material respects the financial condition, results of operations and cash flows of the Company as of, and for, the periods presented in this report; |
|
4. |
The Company’s other certifying officer and I are responsible for establishing and maintaining disclosure controls and procedures (as defined in Exchange Act Rules 13a-15(e) and 15d-15(e)) and internal control over financial reporting (as defined in Exchange Act Rules 13a-15(f) and 15d-15(f)) for the Company and have: |
|
(a) |
Designed such disclosure controls and procedures, or caused such disclosure controls and procedures to be designed under our supervision, to ensure that material information relating to the Company, including its consolidated subsidiaries, is made known to us by others within those entities, particularly during the period in which this report is being prepared; |
|
(b) |
Designed such internal control over financial reporting, or caused such internal control over financial reporting to be designed under our supervision, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles; |
|
(c) |
Evaluated the effectiveness of the Company’s disclosure controls and procedures and presented in this report our conclusions about the effectiveness of the disclosure controls and procedures, as of the end of the period covered by this report based on such evaluation; and |
|
(d) |
Disclosed in this report any change in the Company’s internal control over financial reporting that occurred during the period covered by the annual report that has materially affected, or is reasonably likely to materially affect, the Company’s internal control over financial reporting; and |
|
5. |
The Company’s other certifying officer and I have disclosed, based on our most recent evaluation of internal control over financial reporting, to the Company’s auditors and the audit committee of the Company’s board of directors (or persons performing the equivalent functions): |
|
(a) |
All significant deficiencies and material weaknesses in the design or operation of internal control over financial reporting which are reasonably likely to adversely affect the Company’s ability to record, process, summarize and report financial information; and |
|
(b) |
Any fraud, whether or not material, that involves management or other employees who have a significant role in the Company’s internal control over financial reporting.
|
Date: March 19, 2024
By: |
/s/ Sylvia Cheung |
|
|
Sylvia Cheung |
|
|
Chief Financial Officer |
|
|
(Principal Financial Officer) |
|
Exhibit 13.1
Certification by the Principal Executive Officer and Principal Financial Officer pursuant to
18 U.S.C. Section 1350, as adopted pursuant to
Section 906 of the Sarbanes-Oxley Act of 2002
In connection with the Annual Report on Form 20-F of Oculis Holding AG (the “Company”) for the fiscal year ended December 31, 2023, as filed with the Securities and Exchange Commission on the date hereof (the “Report”), I, Riad Sherif, Chief Executive Officer of the Company and Sylvia Cheung, Chief Financial Officer of the Company, pursuant to 18 U.S.C. § 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002, each hereby certifies that, to the best of his knowledge:
|
(1) |
The Report fully complies with the requirements of Section 13(a) or 15(d) of the Securities Exchange Act of 1934, as amended; and |
|
(2) |
The information contained in the Report fairly presents, in all material respects, the financial condition and results of operations of the Company.
|
Date: March 19, 2024
|
/s/ Riad Sherif |
|
|
Chief Executive Officer |
|
|
(Principal Executive Officer) |
|
|
/s/ Sylvia Cheung |
|
|
Chief Financial Officer |
|
|
(Principal Financial Officer) |
|
This certification accompanies the Form 20-F to which it relates, is not deemed filed with the Securities and Exchange Commission and is not to be incorporated by reference into any filing of the Company under the Securities Act of 1933, as amended, or the Securities Exchange Act of 1934, as amended (whether made before or after the date of the Form 20-F), irrespective of any general incorporation language contained in such filing.
Exhibit 15.1
CONSENT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
We hereby consent to the incorporation by reference in the Registration Statement on Form S-8 (No. 333-271938) of Oculis Holding AG of our report dated March 19, 2024 relating to the financial statements, which appears in this Form 20-F.
/s/ PricewaterhouseCoopers SA
Lausanne, Switzerland
March 19, 2024
Exhibit 97.1
Oculis Holding AG
Incentive Compensation Recoupment Policy
The Remuneration Committee (the “Remuneration Committee”) of the Board of Directors (the “Board”) of Oculis Holding AG, a stock corporation organized under the laws of Switzerland (the “Company”), has determined that it is in the best interests of the Company and its shareholders to adopt this Incentive Compensation Recoupment Policy (this “Policy”) providing for the Company’s recoupment of Recoverable Incentive Compensation that is received by Covered Management Personnel of the Company under certain circumstances. Certain capitalized terms used in this Policy have the meanings given to such terms in Section 3 below.
This Policy is designed to comply with, and shall be interpreted to be consistent with, Section 10D of the Exchange Act, Rule 10D-1 promulgated thereunder (“Rule 10D-1”) and Nasdaq Listing Rule 5608 (the “Listing Standards”).
This Policy shall apply to all Incentive Compensation that is received by any Covered Management Personnel on or after October 2, 2023 (the “Effective Date”). Incentive Compensation is deemed “received” in the Company’s fiscal period in which the Financial Reporting Measure specified in the Incentive Compensation award is attained, even if the payment or grant of such Incentive Compensation occurs after the end of that period.
“Accounting Restatement” means an accounting restatement that the Company is required to prepare due to the material noncompliance of the Company with any financial reporting requirement under the U.S. securities laws, including any required accounting restatement to correct an error in previously issued financial statements that is material to the previously issued financial statements, or that would result in a material misstatement if the error were corrected in the current period or left uncorrected in the current period.
“Accounting Restatement Date” means the earlier to occur of (a) the date that the Board, a committee of the Board authorized to take such action, or the officer or officers of the Company authorized to take such action if Board action is not required, concludes, or reasonably should have concluded, that the Company is required to prepare an Accounting Restatement, or (b) the date that a court, regulator or other legally authorized body directs the Company to prepare an Accounting Restatement.
“Administrator” means the Remuneration Committee or, in the absence of such committee, the Board.
“Code” means the U.S. Internal Revenue Code of 1986, as amended, and the regulations promulgated thereunder.
“Covered Management Personnel” means each current and former Executive Officer and any Vice President in the Company’s finance function.
“Exchange” means the Nasdaq Stock Market.
“Exchange Act” means the U.S. Securities Exchange Act of 1934, as amended.
“Executive Officer” means the Company’s president, principal financial officer, principal accounting officer (or if there is no such accounting officer, the controller), any vice-president of the Company in charge of a principal business unit, division, or function (such as sales, administration, or finance), any other officer who performs a policy-making function, or any other person who performs similar policy-making functions for the Company. Executive officers of the Company’s parent(s) or subsidiaries are deemed executive officers of the Company if they perform such policy-making functions for the Company. Policy-making function is not intended to include policy-making functions that are not significant. Identification of an executive officer for purposes of this Policy would include at a minimum executive officers identified pursuant to Item 401(b) of Regulation S-K promulgated under the Exchange Act.
“Financial Reporting Measures” means measures that are determined and presented in accordance with the accounting principles used in preparing the Company’s financial statements, and any measures derived wholly or in part from such measures, including Company share price and total shareholder return (“TSR”). A measure need not be presented in the Company’s financial statements or included in a filing with the SEC in order to be a Financial Reporting Measure.
“Incentive Compensation” means any compensation that is granted, earned or vested based wholly or in part upon the attainment of a Financial Reporting Measure.
“Lookback Period” means the three completed fiscal years immediately preceding the Accounting Restatement Date, as well as any transition period (resulting from a change in the Company’s fiscal year) within or immediately following those three completed fiscal years (except that a transition period of at least nine months shall count as a completed fiscal year). Notwithstanding the foregoing, the Lookback Period shall not include fiscal years completed prior to the Effective Date.
“Recoverable Incentive Compensation” means Incentive Compensation received by any Covered Management Personnel during the Lookback Period that exceeds the amount of Incentive Compensation that would have been received had such amount been determined based on the Accounting Restatement, computed without regard to any taxes paid (i.e., on a gross basis without regard to tax or social security withholdings and other deductions). For any compensation plans or programs that take into account Incentive Compensation, the amount of Recoverable Incentive Compensation for purposes of this Policy shall include, without limitation, the amount contributed to any notional account based on Recoverable Incentive Compensation and any earnings to date on that notional amount. For any Incentive Compensation that is based on share price or TSR, where the Recoverable Incentive Compensation is not subject to mathematical recalculation directly from the information in an Accounting Restatement, the Administrator will determine the amount of Recoverable Incentive Compensation based on a reasonable estimate of the effect of the Accounting Restatement on the share price or TSR upon which the Incentive Compensation was received. The Company shall maintain documentation of the determination of that reasonable estimate and provide such documentation to the Exchange in accordance with the Listing Standards.
“SEC” means the U.S. Securities and Exchange Commission.
2
3
Except as specifically set forth herein, this Policy shall be administered by the Administrator. The Administrator shall have full and final authority to make any and all determinations required under this Policy. Any determination by the Administrator with respect to this Policy shall be final, conclusive and binding on all interested parties and need not be uniform with respect to each individual covered by this Policy. In carrying out the administration of this Policy, the Administrator is authorized and directed to consult with the full Board or such other committees of the Board as may be necessary or appropriate as to matters within the scope of such other committee’s responsibility and authority. Subject to applicable law, the Administrator may authorize and empower any officer or employee of the Company to take any and all actions that the Administrator, in its sole discretion, deems necessary or appropriate to carry out the purpose and intent of this Policy (other than with respect to any recovery under this Policy involving such officer or employee).
If any provision of this Policy or the application of any such provision to any Covered Management Personnel shall be adjudicated to be invalid, illegal or unenforceable in any respect, such invalidity, illegality or unenforceability shall not affect any other provisions of this Policy, and the invalid, illegal or unenforceable provisions shall be deemed amended to the minimum extent necessary to render any such provision or application enforceable.
Nothing contained in this Policy, and no recoupment or recovery as contemplated herein, shall limit any claims, damages or other legal remedies the Company or any of its affiliates may have against any Covered Management Personnel arising out of or resulting from any actions or omissions by the Covered Management Personnel. This Policy does not preclude the Company or any subsidiary thereof from taking any other action to enforce any Covered Management Personnel’s obligations to the Company, including, without limitation, termination of employment and/or institution of civil proceedings.
4
This Policy is in addition to the requirements of Section 304 of the Sarbanes-Oxley Act of 2002 (“SOX 304”) that are applicable to the Company’s Chief Executive Officer and Chief Financial Officer and to any other compensation recoupment policy and/or similar provisions in any employment, equity plan, equity award, or other individual agreement, to which the Company or any subsidiary thereof is a party or which the Company or any subsidiary thereof has adopted or may adopt and maintain from time to time; provided, however, that compensation recouped pursuant to this Policy shall not be duplicative of compensation recouped pursuant to SOX 304 or any such compensation recoupment policy and/or similar provisions in any such employment, equity plan, equity award, or other individual agreement except as may be required by law.
The Administrator may amend, terminate or replace this Policy or any portion of this Policy at any time and from time to time in its sole discretion. The Administrator shall amend this Policy as it deems necessary to comply with applicable law or any Listing Standard.
This Policy shall be binding and enforceable against all Covered Management Personnel and, to the extent required by Rule 10D-1 and/or the applicable Listing Standards, their beneficiaries, heirs, executors, administrators or other legal representatives.
10. Required Filings
The Company shall make any disclosures and filings with respect to this Policy that are required by law, including as required by the SEC.
* * * * *
5