| ☒ |
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
|
| ☐ |
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES
EXCHANGE ACT OF 1934
|
|
Delaware
|
33-0336973
|
|
|
(State or other jurisdiction of incorporation or organization)
|
(IRS Employer Identification No.)
|
|
2855 Gazelle Court, Carlsbad, CA
|
92010
|
|
|
(Address of Principal Executive Offices)
|
(Zip Code)
|
|
Title of each class
|
Trading symbol
|
Name of each exchange on which registered
|
||
|
Common Stock, $.001 Par Value
|
“IONS”
|
The Nasdaq Stock Market LLC
|
|
Large Accelerated Filer ☒
|
Accelerated Filer ☐
|
|
|
Non-accelerated Filer ☐
|
Smaller Reporting Company ☐
|
|
|
Emerging Growth Company ☐
|
| * |
Excludes 39,747,443 shares of common stock held by directors and officers and by stockholders whose beneficial ownership is known by the Registrant to exceed 10 percent
of the common stock outstanding at June 30, 2023. Exclusion of shares held by any person should not be construed to indicate that such
person possesses the power, direct or indirect, to direct or cause the direction of the management or policies of the Registrant, or that such person is controlled by or under common control with the Registrant.
|
| ● |
Our ability to generate substantial revenue from the sale of our medicines;
|
| ● |
The availability of adequate coverage and payment rates for our medicines;
|
| ● |
Our and our partners’ ability to compete effectively;
|
| ● |
Our ability to successfully manufacture our medicines;
|
| ● |
Our ability to successfully develop and obtain marketing approvals for our medicines;
|
| ● |
Our ability to secure and maintain effective corporate partnerships;
|
| ● |
Our ability to sustain cash flows and achieve consistent profitability;
|
| ● |
Our ability to protect our intellectual property;
|
| ● |
Our ability to maintain the effectiveness of our personnel; and
|
| ● |
The impacts of pandemics, climate change, wars and other global events.
|
|
Page
|
||
|
Item 1.
|
4
|
|
|
Item 1A.
|
49
|
|
|
Item 1B.
|
65
|
|
|
Item 1C.
|
65
|
|
|
Item 2.
|
67
|
|
|
Item 3.
|
67
|
|
|
Item 4.
|
67
|
|
|
Item 5.
|
68
|
|
|
Item 6.
|
69
|
|
|
Item 7.
|
69
|
|
|
Item 7A.
|
79
|
|
|
Item 8.
|
79
|
|
|
Item 9.
|
79
|
|
|
Item 9A.
|
80
|
|
|
Item 9B.
|
82
|
|
|
Item 9C.
|
82
|
|
|
Item 10.
|
82
|
|
|
Item 11.
|
82
|
|
|
Item 12.
|
83
|
|
|
Item 13.
|
83
|
|
|
Item 14.
|
83
|
|
|
Item 15.
|
83
|
|
|
91
|
|
•
|
DEVOTE: In the Phase 2/3 DEVOTE study, Biogen is evaluating the safety and potential to achieve increased efficacy with a higher dose of SPINRAZA
compared to the currently approved dose. In 2022, Biogen reported final data from Part A of the ongoing, three-part DEVOTE study. Results from Part A, an open-label safety evaluation period in children and teens with later-onset SMA, suggest
that the higher dosing regimen of SPINRAZA leads to higher levels of the drug in the cerebrospinal fluid, or CSF, supporting further development of a higher dose of SPINRAZA. Additionally, the results indicated that SPINRAZA was generally
well-tolerated.
|
|
•
|
RESPOND: In the Phase 4 RESPOND study, Biogen is evaluating the benefit of SPINRAZA in infants and children with a suboptimal clinical response to
the gene therapy, onasemnogene abeparvovec. In 2023, Biogen presented interim results from the RESPOND study that showed improved motor function in most participants treated with SPINRAZA following treatment with onasemnogene abeparvovec.
|
|
•
|
ASCEND: In the Phase 3b ASCEND study, Biogen is evaluating the clinical outcomes and assessing the safety of a higher dose of SPINRAZA in children,
teens and adults with later-onset SMA following treatment with risdiplam.
|
|
•
|
33% and 21% reduction in SOD1 protein, the intended target for QALSODY, respectively
|
|
•
|
51% and 41% reduction in plasma neurofilament, a marker of neuron injury, respectively
|
|
•
|
WAINUA achieved a LS mean reduction of 82% in serum TTR concentration from baseline, compared to an 11% reduction from baseline in the external
placebo group (p<0.0001).
|
|
•
|
WAINUA stopped disease progression as measured by mNIS+7 resulting in a 0.28 point LS mean increase compared to a 25.06 point increase for the
external placebo group from baseline (24.8 point LS mean improvement; p<0.0001).
|
|
•
|
WAINUA improved quality of life demonstrating a 5.5 point LS mean decrease (improvement) on the Norfolk QoL-DN, compared to a 14.2 point increase
(worsening) in the external placebo group (19.7 point LS mean improvement; p<0.0001).
|




|
Jurisdiction
|
Patent No.
|
Title
|
Expiration
|
Description of Claims
|
||||
|
United States
|
10,266,822
|
SPINAL MUSCULAR ATROPHY (SMA) TREATMENT VIA TARGETING OF SMN2 SPLICE SITE INHIBITORY SEQUENCES
|
2025
|
Methods of increasing exon-7 containing SMN2 mRNA in a cell using an oligonucleotide having the sequence of SPINRAZA
|
||||
|
United States
|
8,110,560
|
SPINAL MUSCULAR ATROPHY (SMA) TREATMENT VIA TARGETING OF SMN2 SPLICE SITE INHIBITORY SEQUENCES
|
2025
|
Methods of using antisense oligonucleotides having sequence of SPINRAZA to alter splicing of SMN2 and/or to treat SMA
|
||||
|
Europe
|
1910395
|
COMPOSITIONS AND METHODS FOR MODULATION OF SMN2 SPLICING
|
2026
|
Sequence and chemistry (full 2’-MOE) of SPINRAZA
|
||||
|
Europe
|
3308788
|
COMPOSITIONS AND METHODS FOR MODULATION OF SMN2 SPLICING
|
2026
|
Pharmaceutical compositions that include SPINRAZA
|
||||
|
United States
|
7,838,657
|
SPINAL MUSCULAR ATROPHY (SMA) TREATMENT VIA TARGETING OF SMN2 SPLICE SITE INHIBITORY SEQUENCES
|
2027
|
Oligonucleotides having sequence of SPINRAZA
|
||||
|
United States
|
8,361,977
|
COMPOSITIONS AND METHODS FOR MODULATION OF SMN2 SPLICING
|
2030
|
Sequence and chemistry (full 2’-MOE) of SPINRAZA
|
||||
|
United States
|
8,980,853
|
COMPOSITIONS AND METHODS FOR MODULATION OF SMN2 SPLICING IN A SUBJECT
|
2030
|
Methods of administering SPINRAZA
|
||||
|
United States
|
9,717,750
|
COMPOSITIONS AND METHODS FOR MODULATION OF SMN2 SPLICING IN A SUBJECT
|
2030
|
Methods of administering SPINRAZA to a patient
|
||||
|
Europe
|
3449926
|
COMPOSITIONS AND METHODS FOR MODULATION OF SMN2 SPLICING IN A SUBJECT
|
2030
|
Pharmaceutical compositions that include SPINRAZA for treating SMA
|
||||
|
Europe
|
3305302
|
COMPOSITIONS AND METHODS FOR MODULATION OF SMN2 SPLICING IN A SUBJECT
|
2030
|
Antisense compounds including SPINRAZA for treating SMA
|
||||
|
United States
|
9,926,559
|
COMPOSITIONS AND METHODS FOR MODULATION OF SMN2 SPLICING IN A SUBJECT
|
2034
|
SPINRAZA doses for treating SMA
|
||||
|
United States
|
10,436,802
|
METHODS FOR TREATING SPINAL MUSCULAR ATROPHY
|
2035
|
SPINRAZA dosing regimen for treating SMA
|
|
Jurisdiction
|
Registration No.
|
Mark
|
||||
|
United States
|
5156572
|
SPINRAZA
|
_
|
(word mark)
|
||
|
Europe
|
013388145
|
SPINRAZA
|
(word mark)
|
|||
|
Europe
|
014812291 and 015309941
|
|
(design mark)
|
|||
|
Jurisdiction
|
Patent No.
|
Title
|
Expiration
|
Description of Claims
|
||||
|
United States
|
10,385,341
|
COMPOSITIONS FOR MODULATING SOD-1 EXPRESSION
|
2035
|
Composition of QALSODY
|
||||
|
United States
|
10,669,546
|
COMPOSITIONS FOR MODULATING SOD-1 EXPRESSION
|
2035
|
Methods of treating a SOD-1 associated neurodegenerative disorder by administering QALSODY
|
||||
|
United States
|
10,968,453
|
COMPOSITIONS FOR MODULATING SOD-1 EXPRESSION
|
2035
|
Methods of treating a SOD-1 associated neurodegenerative disorder by administering a pharmaceutical composition of QALSODY
|
||||
|
Europe
|
3126499
|
COMPOSITIONS FOR MODULATING SOD-1 EXPRESSION
|
2035
|
Composition of QALSODY
|
|
Jurisdiction
|
Registration No.
|
Mark
|
||||
|
United States
|
7164425
|
QALSODY
|
_
|
(word mark)
|
||
|
United States
|
7116182
|
|
(design mark)
|
|||
|
Europe
|
1542485
|
QALSODY
|
(word mark)
|
|||
|
Europe
|
018517819
|
|
(design mark)
|
|||
|
Jurisdiction
|
Patent No.
|
Title
|
Expiration
|
Description of Claims
|
||||
|
United States
|
10,683,499
|
COMPOSITIONS AND METHODS FOR MODULATING TTR EXPRESSION
|
2034
|
Composition of eplontersen
|
||||
|
Europe
|
3524680
|
COMPOSITIONS AND METHODS FOR MODULATING TTR EXPRESSION
|
2034
|
Composition of eplontersen
|
|
Jurisdiction
|
Application No.
|
Mark
|
||||
|
United States
|
98054331
|
WAINUA
|
_
|
(word mark)
|
||
|
United States
|
98228658
|
|
(design mark)
|
|||
|
Jurisdiction
|
Patent No.
|
Title
|
Expiration
|
Description of Claims
|
||||
|
United States
|
8,101,743
|
MODULATION OF TRANSTHYRETIN EXPRESSION
|
2025
|
Antisense sequence and chemistry of TEGSEDI
|
||||
|
United States
|
8,697,860
|
DIAGNOSIS AND TREATMENT OF DISEASE
|
2031
|
Composition of TEGSEDI
|
||||
|
United States
|
9,061,044
|
MODULATION OF TRANSTHYRETIN EXPRESSION
|
2031
|
Sodium salt composition of TEGSEDI
|
||||
|
United States
|
9,399,774
|
MODULATION OF TRANSTHYRETIN EXPRESSION
|
2031
|
Methods of treating transthyretin amyloidosis by administering TEGSEDI
|
||||
|
Europe
|
2563920
|
MODULATION OF TRANSTHYRETIN EXPRESSION
|
2031
|
Composition of TEGSEDI
|
|
Jurisdiction
|
Registration No.
|
Mark
|
||||
|
United States
|
5740635
|
TEGSEDI
|
_
|
(word mark)
|
||
|
Europe
|
017224742
|
TEGSEDI
|
(word mark)
|
|||
|
Jurisdiction
|
Patent No.
|
Title
|
Expiration
|
Description of Claims
|
||||
|
Europe
|
1622597
|
MODULATION OF APOLIPOPROTEIN C-III EXPRESSION
|
2024
|
Antisense sequence and chemistry of WAYLIVRA
|
||||
|
Europe
|
2441449
|
MODULATION OF APOLIPOPROTEIN C-III EXPRESSION
|
2024
|
Antisense compounds that hybridize within the nucleotide region of apo-CIII targeted by WAYLIVRA
|
||||
|
Europe
|
3002007
|
MODULATION OF APOLIPOPROTEIN C-III EXPRESSION
|
2024
|
Antisense compounds complementary to an apo-CIII nucleic acid for use in therapy
|
||||
|
United States
|
9,157,082
|
MODULATION OF APOLIPOPROTEIN C-III (APOCIII) EXPRESSION
|
2032
|
Methods of using apo-CIII antisense compounds for reducing pancreatitis and chylomicronemia and increasing HDL
|
||||
|
United States
|
9,593,333
|
MODULATION OF APOLIPOPROTEIN C-III (APOCIII) EXPRESSION IN LIPOPROTEIN LIPASE DEFICIENT (LPLD) POPULATIONS
|
2034
|
Methods of treating lipoprotein lipase deficiency with an apo-CIII specific inhibitor wherein triglyceride levels are reduced
|
||||
|
Europe
|
2956176
|
MODULATION OF APOLIPOPROTEIN C-III (APOCIII) EXPRESSION IN LIPOPROTEIN LIPASE DEFICIENT (LPLD) POPULATIONS
|
2034
|
Apo-CIII specific inhibitors including WAYLIVRA for treating lipoprotein lipase deficiency or FCS
|
|
Jurisdiction
|
Registration No.
|
Mark
|
||||
|
Europe
|
016409609
|
WAYLIVRA
|
(word mark)
|
|||
|
Jurisdiction
|
Patent No.
|
Title
|
Expiration
|
Description of Claims
|
||||
|
United States
|
9,163,239
|
COMPOSITIONS AND METHODS FOR MODULATING APOLIPOPROTEIN C-III EXPRESSION
|
2034
|
Composition of olezarsen
|
||||
|
Europe
|
2991656
|
COMPOSITIONS AND METHODS FOR MODULATING APOLIPOPROTEIN C-III EXPRESSION
|
2034
|
Composition of olezarsen
|
|
Jurisdiction
|
Patent No.
|
Title
|
Expiration
|
Description of Claims
|
||||
|
United States
|
9,315,811
|
METHODS FOR MODULATING KALLIKREIN (KLKB1) EXPRESSION
|
2032
|
Methods of treating HAE
|
||||
|
Europe
|
2717923
|
METHODS FOR MODULATING KALLIKREIN (KLKB1) EXPRESSION
|
2032
|
Compounds for use in treating an inflammatory condition, including HAE
|
||||
|
United States
|
10,294,477
|
COMPOSITIONS AND METHODS FOR MODULATING PKK EXPRESSION
|
2035
|
Composition of donidalorsen
|
||||
|
Europe
|
3137091
|
COMPOSITIONS AND METHODS FOR MODULATING PKK EXPRESSION
|
2035
|
Composition of donidalorsen
|
|
Jurisdiction
|
Patent No.
(Patent Application No.)
|
Title
|
Expiration
|
Description of Claims
|
||||
|
United States
|
11,786,546
|
COMPOUNDS AND METHODS FOR MODULATING GFAP
|
2041
|
Composition of zilganersen
|
||||
|
Europe
|
(20846055.0)
|
COMPOUNDS AND METHODS FOR MODULATING GFAP
|
2041
|
Composition of zilganersen
|
|
Jurisdiction
|
Patent Application No.
|
Title
|
Expiration
|
Description of Claims
|
||||
|
United States
|
17/613,183
|
COMPOUNDS AND METHODS FOR REDUCING FUS EXPRESSION
|
2040
|
Composition of ulefnersen
|
||||
|
Europe
|
20815459.1
|
COMPOUNDS AND METHODS FOR REDUCING FUS EXPRESSION
|
2040
|
Composition of ulefnersen
|
|
Jurisdiction
|
Patent No.
|
Title
|
Expiration
|
Description of Claims
|
||||
|
United States
|
9,574,193
|
METHODS AND COMPOSITIONS FOR MODULATING APOLIPOPROTEIN (A) EXPRESSION
|
2033
|
Methods of lowering Apo(a) and/or Lp(a) levels with an oligonucleotide
complementary within the nucleotide region of Apo(a) targeted by pelacarsen
|
||||
|
United States
|
10,478,448
|
METHODS AND COMPOSITIONS FOR MODULATING APOLIPOPROTEIN (A) EXPRESSION
|
2033
|
Methods of treating hyperlipidemia with an oligonucleotide complementary
within the nucleotide region of Apo(a) targeted by pelacarsen
|
||||
|
United States
|
9,884,072
|
METHODS AND COMPOSITIONS FOR MODULATING APOLIPOPROTEIN (A) EXPRESSION
|
2033
|
Oligonucleotides complementary within the nucleotide region of Apo(a) targeted by pelacarsen
|
||||
|
Europe
|
2855500
|
METHODS AND COMPOSITIONS FOR MODULATING APOLIPOPROTEIN (A) EXPRESSION
|
2033
|
Oligonucleotides complementary within the nucleotide region of Apo(a) targeted by pelacarsen for decreasing Apo(a) expression
|
||||
|
United States
|
9,181,550
|
COMPOSITIONS AND METHODS FOR MODULATING APOLIPOPROTEIN (a) EXPRESSION
|
2034
|
Composition of pelacarsen
|
||||
|
Europe
|
2992009
|
COMPOSITIONS AND METHODS FOR MODULATING APOLIPOPROTEIN (a) EXPRESSION
|
2034
|
Composition of pelacarsen
|
|
Jurisdiction
|
Patent No.
|
Title
|
Expiration
|
Description of Claims
|
||||
|
United States
|
8,642,752
|
MODULATION OF HEPATITIS B VIRUS (HBV) EXPRESSION
|
2032
|
Composition of bepirovirsen
|
||||
|
Europe
|
3505528
|
MODULATION OF HEPATITIS B VIRUS (HBV) EXPRESSION
|
2032
|
Composition of bepirovirsen
|
|
Jurisdiction
|
Patent No.
|
Title
|
Expiration
|
Description of Claims
|
||||
|
Europe
|
3043827
|
MODULATORS OF COMPLEMENT FACTOR B
|
2034
|
Compound comprising the antisense oligonucleotide portion of IONIS-FB-LRx.
|
||||
|
United States
|
10,280,423
|
COMPOSITIONS AND METHODS FOR MODULATING COMPLEMENT FACTOR B EXPRESSION
|
2035
|
Composition of IONIS-FB-LRx.
|
||||
|
Europe
|
3137596
|
COMPOSITIONS AND METHODS FOR MODULATING COMPLEMENT FACTOR B EXPRESSION
|
2035
|
Composition of IONIS-FB-LRx.
|
|
Jurisdiction
|
Patent No.
|
Title
|
Expiration
|
Description of Claims
|
||||
|
United States
|
7,399,845
|
6-MODIFIED BICYCLIC NUCLEIC ACID ANALOGS
|
2027
|
cEt nucleosides and oligonucleotides containing these nucleoside analogs
|
||||
|
United States
|
7,741,457
|
6-MODIFIED BICYCLIC NUCLEIC ACID ANALOGS
|
2027
|
cEt nucleosides and oligonucleotides containing these nucleoside analogs
|
||||
|
United States
|
8,022,193
|
6-MODIFIED BICYCLIC NUCLEIC ACID ANALOGS
|
2027
|
Oligonucleotides containing cEt nucleoside analogs
|
||||
|
Europe
|
1984381
|
6-MODIFIED BICYCLIC NUCLEIC ACID ANALOGS
|
2027
|
cEt nucleosides and oligonucleotides containing these nucleoside analogs
|
||||
|
Europe
|
2314594
|
6-MODIFIED BICYCLIC NUCLEIC ACID ANALOGS
|
2027
|
Oligonucleotides containing cEt nucleoside analogs and methods of use
|
||||
|
United States
|
7,569,686
|
COMPOUNDS AND METHODS FOR SYNTHESIS OF BICYCLIC NUCLEIC ACID ANALOGS
|
2027
|
Methods of synthesizing cEt nucleosides
|
||||
|
Europe
|
2092065
|
ANTISENSE COMPOUNDS
|
2027
|
Gapmer oligonucleotides having 2’-modifed and LNA nucleosides
|
||||
|
Europe
|
2410053
|
ANTISENSE COMPOUNDS
|
2027
|
Gapmer oligonucleotides having wings comprised of 2’-MOE and bicyclic nucleosides
|
||||
|
Europe
|
2410054
|
ANTISENSE COMPOUNDS
|
2027
|
Gapmer oligonucleotides having a 2’-modifed nucleoside in the 5’-wing and a bicyclic nucleoside in the 3’-wing
|
||||
|
United States
|
9,550,988
|
ANTISENSE COMPOUNDS
|
2028
|
Gapmer oligonucleotides having BNA nucleosides and 2’-MOE nucleosides
|
||||
|
United States
|
10,493,092
|
ANTISENSE COMPOUNDS
|
2028
|
Gapmer oligonucleotides having BNA nucleosides and 2’-MOE nucleosides and/or 2’-OMe nucleosides
|
||||
|
Europe
|
3067421
|
OLIGOMERIC COMPOUNDS COMPRISING BICYCLIC NUCLEOTIDES AND USES THEREOF
|
2032
|
Gapmer oligonucleotides having at least one bicyclic, one 2’-modified nucleoside and one 2’-deoxynucleoside
|
||||
|
United States
|
11,629,348
|
LINKAGE MODIFIED OLIGONUCLEOTIDES AND USES THEREOF
|
2040
|
Gapmer oligonucleotides having 2-4 mesyl phosphoramidate internucleoside linkages at specified positions in the gap
|
|
Jurisdiction
|
Patent No.
|
Title
|
Expiration
|
Description of Claims
|
||||
|
United States
|
9,127,276
|
CONJUGATED ANTISENSE COMPOUNDS AND THEIR USE
|
2034
|
Preferred THA LICA conjugated to any group of nucleosides, including gapmers, double-stranded siRNA compounds, and fully modified oligonucleotides
|
||||
|
United States
|
9,181,549
|
CONJUGATED ANTISENSE COMPOUNDS AND THEIR USE
|
2034
|
Preferred THA conjugate having our preferred linker and cleavable moiety conjugated to any oligomeric compound or any nucleoside having a 2’-MOE
modification or a cEt modification
|
||||
|
Europe
|
2991661
|
CONJUGATED ANTISENSE COMPOUNDS AND THEIR USE
|
2034
|
Preferred THA LICA conjugated to any group of nucleosides, including gapmers, double-stranded siRNA compounds, and fully modified oligonucleotides
|
| ● |
The federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act,
which governs the conduct of certain electronic healthcare transactions and protects the security and privacy of protected health information;
|
| ● |
Foreign and state laws governing the privacy and security of health information, such as the General Data Protection Regulation, or GDPR, in the EU; and the California
Consumer Privacy Act, or CCPA, in California, some of which are more stringent than HIPAA and many of which differ from each other in significant ways and may not have the same effect; and
|
| ● |
The Physician Payments Sunshine Act, which requires manufacturers of medicines, devices, biologics, and medical supplies to report annually to the HHS information related
to payments and other transfers of value to physicians (defined to include doctors, dentists, optometrists, podiatrists, and chiropractors), other healthcare providers (such as physician assistants and nurse practitioners), and teaching
hospitals, and ownership and investment interests held by physicians and their immediate family members.
|
|
Medicine
|
Company
|
Medicine Description (1)
|
Phase (1)
|
Route of Administration (1)
|
||||
|
Zolgensma
(Onasemnogene abeparvovec)
|
Novartis
|
Gene therapy targeting the genetic root cause of SMA by replacing the missing or nonworking SMN1 gene
|
Approved for pediatric SMA patients less than 2 years of age
|
Intravenous infusion
|
||||
|
Evrysdi
(Risdiplam)
|
Roche
|
A small molecule medicine that modulates splicing of the SMN2 gene
|
Approved for SMA in pediatric and adult patients
|
Oral
|
||||
|
OAV101
(Onasemnogene abeparvovec)
|
Novartis
|
Gene therapy targeting the genetic root cause of SMA by replacing the missing or nonworking SMN1 gene
|
Phase 3
|
Intrathecal injection
|
| (1) |
Taken from public documents including respective company press releases, company presentations, and scientific presentations.
|
|
Medicine
|
Company
|
Medicine Description (1)
|
Phase (1)
|
Route of Administration (1)
|
||||
|
NI-005 / AP-101
|
Neurimmune (AL-S Pharma) / Lilly
|
A human derived antibody targeting misfolded SOD1
|
Phase 2
|
Intravenous Infusion
|
| (1) |
Taken from public documents including respective company press releases, company presentations, and scientific presentations.
|
|
Medicine
|
Company
|
Medicine Description (1)
|
Phase (1)
|
Route of Administration (1)
|
||||
|
Onpattro
(Patisiran)
|
Alnylam
|
An RNAi medicine formulated with lipid nanoparticles to inhibit TTR mRNA
|
Received CRL in the U.S. for ATTR-CM
Approved in US, EU, Japan and select other markets for ATTRv-PN
|
Intravenous infusion
|
||||
|
Vyndaqel/Vyndamax
(Tafamidis and tafamidis meglumine)
|
Pfizer
|
A small molecule medicine to stabilize TTR protein
|
Approved in EU, Japan and select other markets for ATTRv-PN, ATTR-CM; indications vary by region
|
Oral
|
||||
|
Amvuttra
(Vutrisiran)
|
Alnylam
|
An RNAi medicine conjugated with GalNAc to inhibit TTR mRNA
|
Approved for ATTRv-PN in the U.S., EU and Japan, Phase 3 for ATTR-CM
|
Subcutaneous Injection
|
||||
|
Acoramidis
|
BridgeBio
|
Small molecule that binds and stabilizes TTR in the blood
|
Submitted in U.S., EU and Japan
|
Oral
|
||||
|
NTLA-2001
|
Intellia/ Regeneron
|
CRISPR therapeutic candidate designed to reduce circulating TTR protein levels
|
Phase 3 ATTR-CM
|
Intravenous Infusion
|
||||
|
ALXN2220
|
AstraZeneca
|
A monoclonal IgG1 which acts by targeting and depleting TTR protein
|
Phase 3 ATTR-CM
|
Intravenous Infusion
|
||||
|
NNC6019-0001
|
Novo Nordisk
|
A monoclonal antibody to deplete amyloid via antibody-mediated phagocytosis
|
Phase 2 ATTR-CM
|
Intravenous Infusion
|
| (1) |
Taken from public documents including respective company press releases, company presentations, and scientific presentations.
|
|
Medicine
|
Company
|
Medicine Description (1)
|
Phase (1)
|
Route of Administration (1)
|
||||
|
ARO-APOC3
(Plozasiran)
|
Arrowhead
|
Targets APOCIII by utilizing Targeted RNAi Molecule Platform
|
Phase 3 FCS,
Phase 2 SHTG
|
Subcutaneous Injection
|
||||
|
Pegozafermin
|
89bio
|
FGF21 analog
|
Phase 3 SHTG
|
Subcutaneous Injection
|
| (1) |
Taken from public documents including respective company press releases, company presentations, and scientific presentations.
|
|
Medicine
|
Company
|
Medicine Description (1)
|
Phase (1)
|
Route of Administration (1)
|
||||
|
Takhzyro
(lanadelumab-flyo)
|
Takeda
|
A monoclonal antibody that inhibits plasma kallikrein activity
|
Approved for HAE patients two years and older
|
Subcutaneous Injection
|
||||
|
Cinryze
(C1 esterase inhibitor)
|
Takeda
|
A human plasma protein that mediates inflammation and coagulation
|
Approved for HAE patients six years and older
|
Intravenous Infusion
|
||||
|
Orladeyo
(berotralstat)
|
BioCryst
|
Oral plasma kallikrein inhibitor
|
Approved for HAE patients 12 years and older
|
Oral
|
||||
|
Haegarda
(C1 esterase inhibitor)
|
CSL Behring
|
C1 esterase inhibitor
|
Approved for HAE patients 6 years and older
|
Subcutaneous Injection
|
||||
|
Garadacimab
|
CSL Behring
|
An anti-factor XIIa monoclonal antibody
|
Under regulatory review in the U.S. and EU
|
Subcutaneous Injection
|
||||
|
Deucrictibant
|
Pharvaris
|
An oral B2-receptor antagonist
|
Phase 2
|
Oral
|
||||
|
STAR-0215
|
Astria
|
A monoclonal antibody inhibitor of plasma kallikrein
|
Phase 2
|
Subcutaneous Injection
|
||||
|
NTLA-2002
|
Intellia
|
CRISPR therapeutic candidate designed to inactivate the kallikrein B1 gene
|
Phase 1/2
|
Intravenous Infusion
|
| (1) |
Taken from public documents including respective company press releases, company presentations, and scientific presentations.
|
|
Medicine
|
Company
|
Medicine Description (1)
|
Phase (1)
|
Route of Administration (1)
|
||||
|
Olpasiran
|
Amgen/ Arrowhead
|
RNAi therapeutic designed to lower Lp(a)
|
Phase 3
|
Subcutaneous Injection
|
||||
|
Zerlasiran
|
Silence
|
RNAi therapeutic designed to lower Lp(a)
|
Phase 2
|
Subcutaneous Injection
|
||||
|
Lepodisiran
|
Lilly
|
RNAi therapeutic designed to lower Lp(a)
|
Phase 2
|
Subcutaneous Injection
|
||||
|
Muvalaplin
|
Lilly
|
Small molecule therapy to lower Lp(a)
|
Phase 2
|
Oral
|
| (1) |
Taken from public documents including respective company press releases, company presentations, and scientific presentations.
|
|
Medicine
|
Company
|
Medicine Description (1)
|
Phase (1)
|
Route of Administration (1)
|
||||
|
Elebsiran
(VIR-2218)
|
Vir Biotech / Alnylam
|
RNAi therapeutic to reduce HBV viral antigens
|
Phase 2
|
Subcutaneous Injection
|
||||
|
Imdusiran
(AB-729)
|
Arbutus Biopharma
|
RNAi therapeutic to reduce HBV viral antigens
|
Phase 2
|
Subcutaneous Injection
|
||||
|
Xalnesiran
|
Roche
|
RNAi therapeutic to reduce HBV viral antigens
|
Phase 2
|
Subcutaneous Injection
|
| (1) |
Taken from public documents including respective company press releases, company presentations, and scientific presentations.
|
|
Medicine
|
Company
|
Medicine Description (1)
|
Phase (1)
|
Route of Administration (1)
|
||||
|
Tarpeyo (budesonide)
|
Calliditas
|
A corticosteroid indicated to reduce proteinuria in adults with primary IgAN
|
Approved for IgAN
|
Oral
|
||||
|
Filspari
(Sparsentan) |
Travere
|
An endothelin & angiotensin II receptor antagonist to reduce proteinuria in adults with primary IgAN
|
Approved for IgAN
|
Oral
|
||||
|
Atrasentan
|
Novartis (Chinook)
|
An endothelin A receptor antagonist
|
Phase 3 (IgAN)
|
Oral
|
||||
|
Iptacopan
|
Novartis (Chinook)
|
A factor B inhibitor of the alternative complement pathway
|
Phase 3 (IgAN)
|
Oral
|
||||
|
Zigakibart
|
Novartis (Chinook)
|
An anti-APRIL monoclonal antibody
|
Phase 3 (IgAN)
|
Subcutaneous Injection
|
||||
|
Sibeprenlimab
|
Otsuka (Visterra)
|
A humanized IgG2 monoclonal antibody that inhibits APRIL
|
Phase 3 (IgAN)
|
Intravenous Infusion
|
||||
|
Atacicept
|
Vera
|
A recombinant fusion protein a dual inhibitor of BLyS and APRIL
|
Phase 3 (IgAN)
|
Subcutaneous Injection
|
||||
|
Ravulizumab
|
Alexion (AstraZeneca)
|
A humanized monoclonal antibody to complement factor 5
|
Phase 2 (IgAN)
|
Subcutaneous Injection
|
||||
|
Vemircopan
|
Alexion (AstraZeneca)
|
A complement factor D inhibitor
|
Phase 2 (IgAN)
|
Oral
|
||||
|
Felzartamab
|
Hi-Bio
|
A monoclonal antibody directed against CD38
|
Phase 2 (IgAN)
|
Intravenous Infusion
|
| (1) |
Taken from public documents including respective company press releases, company presentations, and scientific presentations.
|
|
Medicine
|
Company
|
Medicine Description (1)
|
Phase (1)
|
Route of Administration (1)
|
||||
|
Ivervay (avacincaptad pegol)
|
Iveric Bio
|
A complement C5 inhibitor approved for GA secondary to AMD
|
Approved (GA)
|
Intravitreal
|
||||
|
Syfovre (pegcetacoplan)
|
Apellis
|
A complement C5 inhibitor approved for GA secondary to AMD
|
Approved (GA)
|
Intravitreal
|
||||
|
Tinlarebant
|
Belite Bio
|
A small molecule RBP4 antagonist
|
Phase 3 (GA)
|
Oral
|
||||
|
Danicopan
|
Alexion
|
A factor D inhibitor
|
Phase 2 (GA)
|
Oral
|
||||
|
PPY988 (GT005)
|
Novartis
|
A gene therapy with encoding for human complement factor I
|
Phase 2 (GA)
|
Intraocular
|
||||
|
AVD-104
|
Aviceda
|
A glycomimetic nanoparticle
|
Phase 2 (GA)
|
Intravitreal
|
||||
|
ANX007
|
Annexon Bio
|
A fragment antigen-binding (fab) antibody
|
Phase 2 (GA)
|
Intravitreal
|
| (1) |
Taken from public documents including respective company press releases, company presentations, and scientific presentations.
|
|
Innovate to improve the lives
of people with serious diseases
|
Empowering our
employees and communities
|
Operating
responsibly and sustainably
|
|
We innovate across the business and work tirelessly to discover, develop and deliver important new medicines for
people with serious diseases.
|
We are committed to fostering an inclusive culture that drives excellence, embraces diversity, and supports our
communities.
|
We operate with integrity to help create a better, more sustainable future for all through environmental
stewardship and responsible business practices and stakeholder interactions.
|
|
• Innovation and R&D
• Access and Affordability
• Patient Advocacy and Engagement
|
• Workplace Culture, Talent Attraction and Development
• Diversity, Equity and Inclusion
• Social Impact and Community Engagement
|
• Environmental Sustainability
• Governance and Integrity
• Data Privacy and Cybersecurity
|
| ● |
Comprehensive medical, dental and vision insurance;
|
| ● |
401(k) matching;
|
| ● |
Stock options, RSUs and an Employee Stock Purchase Plan, or ESPP;
|
| ● |
Vacation, holiday, sick time and paid time off for volunteering;
|
| ● |
Wellness programs;
|
| ● |
Flexible spending accounts for health and dependent day care needs;
|
| ● |
Family care benefits;
|
| ● |
Life, AD&D insurance and long-term disability insurance coverage options; and
|
| ● |
Employee Assistance Program, or EAP.
|
|
Name
|
Age
|
Position
|
||
|
Brett P. Monia, Ph.D.
|
62
|
Chief Executive Officer
|
||
|
Joseph T. Baroldi
|
46
|
Executive Vice President, Chief Business Officer
|
||
|
Brian Birchler
|
58
|
Executive Vice President, Corporate and Development Operations
|
||
|
C. Frank Bennett, Ph.D.
|
67
|
Executive Vice President, Chief Scientific Officer
|
||
|
Onaiza Cadoret-Manier
|
59
|
Executive Vice President, Chief Global Product Strategy and Operations Officer
|
||
|
Richard S. Geary, Ph.D.
|
66
|
Executive Vice President, Chief Development Officer
|
||
|
Elizabeth L. Hougen
|
62
|
Executive Vice President, Finance and Chief Financial Officer
|
||
|
Patrick R. O’Neil, Esq.
|
50
|
Chief Legal Officer, General Counsel and Corporate Secretary
|
||
|
Eugene Schneider, M.D.
|
51
|
Executive Vice President, Chief Clinical Development and Operations Officer
|
||
|
Eric E. Swayze, Ph.D.
|
58
|
Executive Vice President, Research
|
| ● |
receipt and scope of marketing authorizations;
|
| ● |
establishment and demonstration in the medical and patient community of the efficacy and safety of our medicines and their potential advantages over competing products;
|
| ● |
cost and effectiveness of our medicines compared to other available therapies;
|
| ● |
patient convenience of the dosing regimen for our medicines; and
|
| ● |
reimbursement policies of government and third-party payers.
|
| ● |
priced lower than our medicines;
|
| ● |
reimbursed more favorably by government and other third-party payers than our medicines;
|
| ● |
safer than our medicines;
|
| ● |
more effective than our medicines; or
|
| ● |
more convenient to use than our medicines.
|
| ● |
Onasemnogene abeparvovec and risdiplam compete with SPINRAZA;
|
| ● |
Taldefgrobep alfa, Evrysdi + GYM329 and NMD670 could compete with SPINRAZA;
|
| ● |
Patisiran, tafamidis, tafamidis meglumine and vutrisiran compete with TEGSEDI and WAINUA;
|
| ● |
Acoramidis, NTLA-2001 and NNC6019-0001 could compete with TEGSEDI and WAINUA;
|
| ● |
ARO-APOC3 and pegozafermin could compete with WAYLIVRA and olezarsen;
|
| ● |
Lanadelumab-flyo, C1 esterase inhibitor, berotralstat, C1 esterase inhibitor subcutaneous,
garadacimab, deucrictibant, NTLA-2002 and STAR-0215 could compete with donidalorsen;
|
| ● |
Olpasiran, zerlasiran, lepodisiran and muvalaplin could compete with pelacarsen;
|
| ● |
NI-005/AP-101 could compete with QALSODY;
|
| ● |
VIR-2218 + PEG-IFN-α, VIR-3434 ± VIR-2218 ± PEG-IFN-α, VIR-2218 + BRII-179, NI-204VIR-2218 + GS-9688 + nivolumab, AB-729, imdusiran + Peg-IFNa-2α + NA, xalnesiran +
RG6084 + NA, xalnesiran + NA, xalnesiran + pegIFN + NA, xalnesiran + RO7049389 + NA, xalnesiran + ruzotolimod + NA, RO7049389 + ruzotolimod + NA could complete with bepirovirsen; and
|
| ● |
Budesonide, sparsentan, atrasentan, iptacopan, zigakibart, sibeprenlimab, atacicept, ravulizumab, vemircopan, felzartamab, povetacicept, avacincaptad pegol,
pegcetacoplan, tinlarebant, danicopan, GT005, AVD-104 and ANX007 could compete with IONIS-FB-LRx.
|
| ● |
in the U.S., TEGSEDI’s label contains a boxed warning for thrombocytopenia and glomerulonephritis;
|
| ● |
TEGSEDI requires periodic blood and urine monitoring; and
|
| ● |
in the U.S., TEGSEDI is available only through a REMS program.
|
| ● |
fund our development activities for SPINRAZA and QALSODY;
|
| ● |
seek and obtain regulatory approvals for SPINRAZA and QALSODY; and
|
| ● |
successfully commercialize SPINRAZA and QALSODY.
|
| ● |
such authorities may disagree with the design or implementation of our clinical studies;
|
| ● |
we or our partners may be unable to demonstrate to the satisfaction of the FDA or other regulatory authorities that a medicine is safe and effective for any indication;
|
| ● |
such authorities may not accept clinical data from studies conducted at clinical facilities that have deficient clinical practices or that are in countries where the
standard of care is potentially different from the U.S.;
|
| ● |
we or our partners may be unable to demonstrate that our medicine’s clinical and other benefits outweigh its safety risks to support approval;
|
| ● |
such authorities may disagree with the interpretation of data from preclinical or clinical studies;
|
| ● |
such authorities may find deficiencies in the manufacturing processes or facilities of third-party manufacturers who manufacture clinical and commercial supplies for our
medicines; and
|
| ● |
the approval policies or regulations of such authorities or their prior guidance to us or our partners during clinical development may significantly change in a manner
rendering our clinical data insufficient for approval.
|
| ● |
the clinical study may produce negative or inconclusive results;
|
| ● |
regulators may require that we hold, suspend or terminate clinical research for noncompliance with regulatory requirements;
|
| ● |
we, our partners, the FDA or foreign regulatory authorities could suspend or terminate a clinical study due to adverse side effects of a medicine on subjects or lack of
efficacy in the trial;
|
| ● |
we or our partners may decide, or regulators may require us, to conduct additional preclinical testing or clinical studies;
|
| ● |
enrollment in our clinical studies may be slower than we anticipate;
|
| ● |
we or our partners, including our independent clinical investigators, contract research organizations and other third-party service providers on which we rely, may not
identify, recruit or train suitable clinical investigators at a sufficient number of study sites or timely enroll a sufficient number of study subjects in the clinical study;
|
| ● |
the institutional review board for a prospective site might withhold or delay its approval for the study;
|
| ● |
people who enroll in the clinical study may later drop out due to adverse events, a perception they are not benefiting from participating in the study, fatigue with the
clinical study process or personal issues;
|
| ● |
a clinical study site may deviate from the protocol for the study;
|
| ● |
the cost of our clinical studies may be greater than we anticipate;
|
| ● |
our partners may decide not to exercise any existing options to license and conduct additional clinical studies for our medicines; and
|
| ● |
the supply or quality of our medicines or other materials necessary to conduct our clinical studies may be insufficient, inadequate or delayed.
|
| ● |
AstraZeneca for the joint development and funding of WAINUA;
|
| ● |
Novartis for development and funding of pelacarsen;
|
| ● |
GSK for development and funding of bepirovirsen; and
|
| ● |
Roche for development and funding of IONIS-FB-LRx.
|
| ● |
conduct clinical studies;
|
| ● |
seek and obtain marketing authorizations; and
|
| ● |
manufacture and commercialize our medicines.
|
| ● |
pursue alternative technologies or develop alternative products that may be competitive with the medicine that is part of the collaboration with us;
|
| ● |
pursue higher-priority programs or change the focus of its own development programs; or
|
| ● |
choose to devote fewer resources to our medicines than it does to its own medicines.
|
| ● |
successful commercialization of our commercial medicines;
|
| ● |
the profile and launch timing of our medicines in development;
|
| ● |
changes in existing collaborative relationships and our ability to establish and maintain additional collaborative arrangements;
|
| ● |
continued scientific progress in our research, drug discovery and development programs;
|
| ● |
the size of our programs and progress with preclinical and clinical studies;
|
| ● |
the time and costs involved in obtaining marketing authorizations;
|
| ● |
competing technological and market developments, including the introduction by others of new therapies that address our markets; and
|
| ● |
our manufacturing requirements and capacity to fulfill such requirements.
|
| ● |
compliance with differing or unexpected regulatory requirements for our medicines and foreign employees;
|
| ● |
complexities associated with managing multiple payer reimbursement regimes, government payers or patient self-pay systems;
|
| ● |
difficulties in staffing and managing foreign operations;
|
| ● |
in certain circumstances, increased dependence on the commercialization efforts and regulatory compliance of third-party distributors or strategic partners;
|
| ● |
foreign government taxes, regulations and permit requirements;
|
| ● |
U.S. and foreign government tariffs, trade and export restrictions, price and exchange controls and other regulatory requirements;
|
| ● |
anti-corruption laws, including the Foreign Corrupt Practices Act, or the FCPA, and its equivalent in foreign jurisdictions;
|
| ● |
economic weakness, including inflation, natural disasters, war, events of terrorism, political instability or public health issues or pandemics, in particular foreign
countries or globally;
|
| ● |
fluctuations in currency exchange rates, which could result in increased operating expenses and reduced revenue, and other obligations related to doing business in
another country;
|
| ● |
compliance with tax, employment, privacy, immigration and labor laws, regulations and restrictions for employees living or traveling abroad;
|
| ● |
workforce uncertainty in countries where labor unrest is more common than in the U.S.; and
|
| ● |
changes in diplomatic and trade relationships.
|
| ● |
interruption of our research, development and manufacturing efforts;
|
| ● |
injury to our employees and others;
|
| ● |
environmental damage resulting in costly clean up; and
|
| ● |
liabilities under federal, state and local laws and regulations governing health and human safety, as well as the use, storage, handling and disposal of these materials
and resultant waste products.
|
| ● |
analyzing reports of certain threats and actors;
|
| ● |
conducting scans of the threat environment;
|
| ● |
evaluating our and our industry’s risk profile;
|
| ● |
evaluating certain threats reported to us;
|
| ● |
conducting internal and external audits;
|
| ● |
conducting threat assessments for certain internal and external threats; and
|
| ● |
conducting vulnerability assessments to identify vulnerabilities.
|
| ● |
incident response plan;
|
| ● |
disaster recovery/business continuity plans;
|
| ● |
risk assessments;
|
| ● |
encryption of certain data;
|
| ● |
network security and access controls for certain systems;
|
| ● |
physical security;
|
| ● |
asset management, tracking and disposal;
|
| ● |
systems monitoring; and
|
| ● |
employee training.
|
|
Property Description
|
Location
|
Square
Footage
|
Owned
or Leased
|
Initial Lease
Term End Date
|
Lease
Extension Options
|
|||||
|
Laboratory and office space facility
|
Carlsbad, CA
|
176,300
|
Leased
|
2037
|
Two, five-year options to extend
|
|||||
|
Office and meeting space facility
|
Carlsbad, CA
|
74,000
|
Leased
|
2037
|
Two, five-year options to extend
|
|||||
|
Manufacturing facility
|
Carlsbad, CA
|
26,800
|
Owned
|
|||||||
|
Manufacturing support facility
|
Carlsbad, CA
|
25,800
|
Leased
|
2026
|
One, five-year option to extend
|
|||||
|
Office space facility
|
Boston, MA
|
14,300
|
Leased
|
2029
|
One, five-year option to extend
|
|||||
|
Office space facility
|
Carlsbad, CA
|
5,800
|
Leased
|
2027
|
None
|
|||||
|
Warehouse facility
|
Carlsbad, CA
|
4,200
|
Leased
|
2028
|
None
|
|||||
|
Office space facility
|
Dublin, Ireland
|
3,900
|
Leased
|
2025
|
None
|
|||||
|
331,100
|
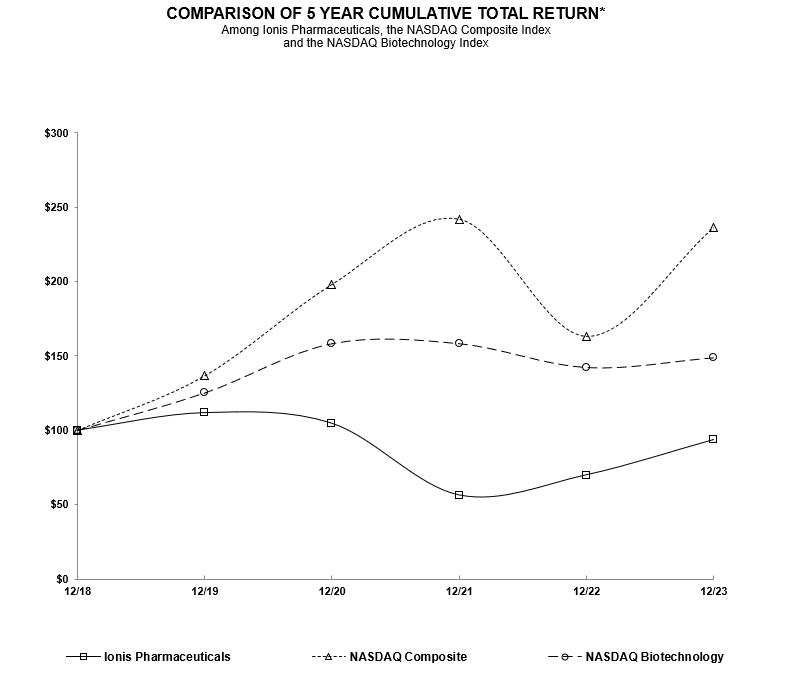
|
Dec-18
|
Dec-19
|
Dec-20
|
Dec-21
|
Dec-22
|
Dec-23
|
|||||||||||||||||||
|
Ionis Pharmaceuticals, Inc.
|
$
|
100.00
|
$
|
111.75
|
$
|
104.59
|
$
|
56.29
|
$
|
69.87
|
$
|
93.58
|
||||||||||||
|
Nasdaq Composite Index
|
$
|
100.00
|
$
|
136.69
|
$
|
198.10
|
$
|
242.03
|
$
|
163.28
|
$
|
236.17
|
||||||||||||
|
Nasdaq Biotechnology Index
|
$
|
100.00
|
$
|
125.11
|
$
|
158.17
|
$
|
158.20
|
$
|
142.19
|
$
|
148.72
|
||||||||||||
| (1) |
This section is not “soliciting material,” is not deemed “filed” with the SEC, is not subject to the liabilities of Section 18 of the Exchange Act and is not to be
incorporated by reference in any of our filings under the Securities Act or the Exchange Act, whether made before or after the date hereof and irrespective of any general incorporation language in any such filing.
|
|
Year Ended December 31,
|
||||||||
|
2023
|
2022
|
|||||||
|
Total revenue
|
$
|
787.6
|
$
|
587.4
|
||||
|
Total operating expenses
|
$
|
1,141.4
|
$
|
997.6
|
||||
|
Loss from operations
|
$
|
(353.7
|
)
|
$
|
(410.2
|
)
|
||
|
Net loss
|
$
|
(366.3
|
)
|
$
|
(269.7
|
)
|
||
|
Cash, cash equivalents and short-term investments
|
$
|
2,331.2
|
$
|
1,986.9
|
||||
|
Year Ended December 31,
|
||||||||
|
2023
|
2022
|
|||||||
|
Revenue:
|
||||||||
|
Commercial revenue:
|
||||||||
|
SPINRAZA royalties
|
$
|
240.4
|
$
|
242.3
|
||||
|
Other commercial revenue:
|
||||||||
|
TEGSEDI and WAYLIVRA revenue, net
|
34.9
|
30.1
|
||||||
|
Licensing and other royalty revenue
|
33.3
|
31.0
|
||||||
|
Total other commercial revenue
|
68.2
|
61.1
|
||||||
|
Total commercial revenue
|
308.6
|
303.4
|
||||||
|
R&D revenue:
|
||||||||
|
Amortization from upfront payments
|
125.3
|
68.6
|
||||||
|
Milestone payments
|
100.5
|
74.0
|
||||||
|
License fees
|
116.8
|
37.0
|
||||||
|
Other services
|
10.0
|
27.6
|
||||||
|
Collaborative agreement revenue
|
352.6
|
207.2
|
||||||
|
WAINUA joint development revenue
|
126.4
|
76.8
|
||||||
|
Total R&D revenue
|
479.0
|
284.0
|
||||||
|
Total revenue
|
$
|
787.6
|
$
|
587.4
|
||||
|
Year Ended December 31,
|
||||||||
|
2023
|
2022
|
|||||||
|
WAINUA joint development revenue
|
$
|
126.4
|
$
|
76.8
|
||||
|
Research and development expenses related to Phase 3 development expenses for WAINUA
|
150.8
|
147.1
|
||||||
|
Medical affairs expenses for WAINUA
|
4.1
|
2.0
|
||||||
|
Commercialization expenses for WAINUA
|
15.6
|
2.6
|
||||||
|
Year Ended December 31,
|
||||||||
|
2023
|
2022
|
|||||||
|
Operating expenses, excluding non-cash compensation expense related to equity awards
|
$
|
1,035.7
|
$
|
897.3
|
||||
|
Non-cash compensation expense related to equity awards
|
105.7
|
100.3
|
||||||
|
Total operating expenses
|
$
|
1,141.4
|
$
|
997.6
|
||||
|
Year Ended December 31,
|
||||||||
|
2023
|
2022
|
|||||||
|
Cost of sales, excluding non-cash compensation expense related to equity awards
|
$
|
8.7
|
$
|
13.4
|
||||
|
Non-cash compensation expense related to equity awards
|
0.4
|
0.7
|
||||||
|
Total cost of sales
|
$
|
9.1
|
$
|
14.1
|
||||
|
Year Ended December 31,
|
||||||||
|
2023
|
2022
|
|||||||
|
Research, development and patent expenses, excluding non-cash compensation expense related to equity awards
|
$
|
821.7
|
$
|
759.4
|
||||
|
Non-cash compensation expense related to equity awards
|
77.9
|
73.7
|
||||||
|
Total research, development and patent expenses
|
$
|
899.6
|
$
|
833.1
|
||||
|
Year Ended December 31,
|
||||||||
|
2023
|
2022
|
|||||||
|
Drug discovery expenses, excluding non-cash compensation expense related to equity awards
|
$
|
125.6
|
$
|
181.3
|
||||
|
Non-cash compensation expense related to equity awards
|
16.2
|
16.2
|
||||||
|
Total drug discovery expenses
|
$
|
141.8
|
$
|
197.5
|
||||
|
Year Ended December 31,
|
||||||||
|
2023
|
2022
|
|||||||
|
WAINUA
|
$
|
115.5
|
$
|
103.9
|
||||
|
TEGSEDI and WAYLIVRA
|
8.1
|
10.6
|
||||||
|
Olezarsen
|
138.3
|
68.1
|
||||||
|
Donidalorsen
|
24.9
|
14.1
|
||||||
|
Zilganersen
|
8.4
|
5.6
|
||||||
|
Ulefnersen
|
10.8
|
8.4
|
||||||
|
Other development projects
|
101.0
|
123.5
|
||||||
|
Development overhead expenses
|
123.3
|
92.0
|
||||||
|
Total drug development, excluding non-cash compensation expense related to equity awards
|
530.3
|
426.2
|
||||||
|
Non-cash compensation expense related to equity awards
|
34.5
|
31.5
|
||||||
|
Total drug development expenses
|
$
|
564.8
|
$
|
457.7
|
||||
|
Year Ended December 31,
|
||||||||
|
2023
|
2022
|
|||||||
|
Medical affairs expenses, excluding non-cash compensation expense related to equity awards
|
$
|
19.5
|
$
|
15.9
|
||||
|
Non-cash compensation expense related to equity awards
|
3.4
|
2.0
|
||||||
|
Total medical affairs expenses
|
$
|
22.9
|
$
|
17.9
|
||||
|
Year Ended December 31,
|
||||||||
|
2023
|
2022
|
|||||||
|
Manufacturing and development chemistry expenses, excluding non-cash compensation expense related to equity awards
|
$
|
65.3
|
$
|
76.2
|
||||
|
Non-cash compensation expense related to equity awards
|
8.8
|
9.9
|
||||||
|
Total manufacturing and development chemistry expenses
|
$
|
74.1
|
$
|
86.1
|
||||
|
Year Ended December 31,
|
||||||||
|
2023
|
2022
|
|||||||
|
Personnel costs
|
$
|
27.2
|
$
|
21.2
|
||||
|
Occupancy
|
28.7
|
19.2
|
||||||
|
Consulting
|
4.8
|
0.8
|
||||||
|
Patent expenses
|
4.3
|
4.7
|
||||||
|
Insurance
|
3.6
|
3.8
|
||||||
|
Computer software and licenses
|
2.7
|
1.9
|
||||||
|
Other
|
9.7
|
8.2
|
||||||
|
Total R&D support expenses, excluding non-cash compensation expense related to equity awards
|
81.0
|
59.8
|
||||||
|
Non-cash compensation expense related to equity awards
|
15.0
|
14.1
|
||||||
|
Total R&D support expenses
|
$
|
96.0
|
$
|
73.9
|
||||
|
Year Ended December 31,
|
||||||||
|
2023
|
2022
|
|||||||
|
Selling, general and administrative expenses, excluding non-cash compensation expense related to equity awards
|
$
|
205.1
|
$
|
124.4
|
||||
|
Non-cash compensation expense related to equity awards
|
27.5
|
25.9
|
||||||
|
Total selling, general and administrative expenses
|
$
|
232.6
|
$
|
150.3
|
||||
|
Year Ended December 31,
|
||||||||
|
2023
|
2022
|
|||||||
|
Convertible senior notes:
|
||||||||
|
Non-cash amortization of debt issuance costs
|
$
|
5.9
|
$
|
5.3
|
||||
|
Interest expense payable in cash
|
6.4
|
0.7
|
||||||
|
Interest on mortgage for primary R&D and manufacturing facilities
|
0.4
|
2.1
|
||||||
|
Total interest expense
|
$
|
12.7
|
$
|
8.1
|
||||
|
Contractual Obligations
|
Payments Due by Period
(in millions)
|
|||||||||||
|
(selected balances described below)
|
Total
|
Less than 1 year
|
More than 1 year
|
|||||||||
|
1.75% Notes (principal and interest payable)
|
$
|
620.3
|
$
|
10.1
|
$
|
610.2
|
||||||
|
0% Notes (principal payable)
|
632.5
|
—
|
632.5
|
|||||||||
|
0.125% Notes (principal and interest payable)
|
44.6
|
44.6
|
—
|
|||||||||
|
Building mortgage payments (principal and interest payable)
|
10.2
|
0.5
|
9.7
|
|||||||||
|
Operating leases
|
279.5
|
20.4
|
259.1
|
|||||||||
|
Other obligations (principal and interest payable)
|
0.8
|
0.1
|
0.7
|
|||||||||
|
Total
|
$
|
1,587.9
|
$
|
75.7
|
$
|
1,512.2
|
||||||
| ● |
Assessing the propriety of revenue recognition and associated deferred revenue;
|
| ● |
Determining the appropriate cost estimates for unbilled preclinical studies and clinical development activities; and
|
| ● |
Assessing the appropriate estimate of anticipated future royalty payments under our royalty purchase agreement
|
| ● |
Identifying the performance obligations contained in the agreement
|
| ● |
Determining the transaction price, including any variable consideration
|
| ● |
Allocating the transaction price to each of our performance obligations
|
| ● |
Whether a milestone payment is probable (discussed in detail above under “Determining the
transaction price, including any variable consideration”);
|
| ● |
Whether a milestone payment relates to services we are performing or if our partner is performing the services;
|
| ● |
If we are performing services, we recognize revenue over our estimated period of performance in a similar manner to the amortization of upfront payments (discussed above
under “R&D Services with Upfront Payments”); and
|
| ● |
Conversely, we recognize in full those milestone payments that we earn based on our partners’ activities when our partner achieves the milestone event and we do not have
a performance obligation.
|
| * |
Contract, instruction or written plan intended to satisfy the affirmative defense conditions of Rule 10b5-1(c) under the Exchange Act.
|
| ** |
“Non-Rule 10b5-1 trading arrangement” as defined in item 408(c) of Regulation S-K under the Exchange Act.
|
|
Action
|
Date
|
Trading Arrangement
|
Total Shares
to be Sold
|
Expiration Date
|
||||||||
|
Rule 10b5-1*
|
Non-Rule 10b5-1**
|
|||||||||||
|
Joseph Wender, Board Member
|
Adoption
|
November 30, 2023
|
X
|
104,079
|
February 28, 2025
|
|||||||
|
(1)
|
Any information that is included on or linked to our website is not part of this Form 10-K.
|
|
Plan Category
|
Number of Shares to
be Issued Upon Exercise
of Outstanding Options
|
Weighted Average
Exercise Price of
Outstanding Options
|
Number of Shares
Remaining Available
for Future Issuance
|
|||||
|
Equity compensation plans approved by stockholders (a)
|
14,090,732
|
$
|
48.43
|
9,976,286
|
(b)
|
|||
|
Total
|
14,090,732
|
$
|
48.43
|
9,976,286
|
||||
| (a) |
Consists of five Ionis plans: 1989 Stock Option Plan, Amended and Restated 2002 Non-Employee Directors’ Stock Option Plan, 2011 Equity Incentive Plan, 2020 Equity
Incentive Plan and Employee Stock Purchase Plan, or ESPP.
|
| (b) |
Of these shares, 386,792 were available for purchase under the ESPP as of December 31, 2023.
|
|
Exhibit Number
|
Description of Document
|
||
|
2.1
|
Agreement and Plan of Merger, dated as of August 30, 2020, among Akcea Therapeutics, Inc., Ionis Pharmaceuticals, Inc. and Avalanche Merger Sub, Inc., filed as an exhibit to the Registrant’s Current Report on Form 8-K filed August 31, 2020 and incorporated herein by reference.
|
||
|
3.1
|
Amended and Restated Certificate of Incorporation filed June 19, 1991,
filed as an exhibit to the Registrant’s Annual Report on Form 10-K for the year ended December 31, 2017 and incorporated herein by reference.
|
||
|
3.2
|
Certificate of Amendment to Restated Certificate of Incorporation, filed
as an exhibit to the Registrant’s Notice of Annual Meeting and Proxy Statement, for the 2014 Annual Meeting of Stockholders, filed on April 25, 2014 and incorporated herein by reference.
|
||
|
3.3
|
Certificate of Amendment to Restated Certificate of Incorporation, filed as an
exhibit to the Registrant’s Current Report on Form 8-K filed December 18, 2015 and incorporated herein by reference.
|
||
|
3.4
|
Amended and Restated Bylaws, filed as an exhibit to the
Registrant’s Current Report on Form 8-K filed March 29, 2021 and incorporated herein by reference.
|
||
|
4.1
|
Description of the Registrant’s Securities, filed as an exhibit to the
Registrant’s Annual Report on Form 10-K for the year ended December 31, 2021 and incorporated herein by reference.
|
||
|
4.2
|
Certificate of Designation of the Series C Junior Participating Preferred Stock, filed as an exhibit to Registrant’s Current Report on Form 8-K filed December 13, 2000 and incorporated herein by reference.
|
||
|
4.3
|
Specimen Common Stock Certificate, filed as an exhibit to the
Registrant’s Annual Report on Form 10-K for the year ended December 31, 2017 and incorporated herein by reference.
|
||
|
4.4
|
Indenture, dated as of December 19, 2019, by and between the Registrant and U.S. Bank National Association, as trustee, including Form of 0.125 percent
Convertible Senior Note due 2024, filed as an exhibit to the Registrant’s Current Report on Form 8-K filed December 23, 2019 and incorporated herein by
reference.
|
||
|
4.5
|
Form of Exchange and/or Subscription Agreement for Convertible Senior Notes due 2024, filed as an exhibit to the Registrant’s Current Report on Form 8-K filed December 12, 2019 and incorporated herein by reference.
|
||
|
4.6
|
Form of Convertible Note Hedge Transactions Confirmation for Convertible
Senior Notes due 2024, filed as an exhibit to the Registrant’s Current Report on Form 8-K filed December 12, 2019 and incorporated herein by reference.
|
||
|
4.7
|
Form of Warrant Transactions Confirmation for Convertible Senior Notes due
2024, filed as an exhibit to the Registrant’s Current Report on Form 8-K filed December 12, 2019 and incorporated herein by reference.
|
||
|
4.8
|
Indenture, dated as of April 12, 2021, by and between the Registrant and U.S. Bank National Association, as trustee, including Form of 0 percent
Convertible Senior Note due 2026, filed as an exhibit to the Registrant’s Current Report on Form 8-K filed April 13, 2021 and incorporated herein by reference.
|
||
|
4.9
|
Form of Warrant Confirmation for Convertible Senior Notes due 2026,
filed as an exhibit to the Registrant’s Current Report on Form 8-K filed April 13, 2021 and incorporated herein by reference.
|
||
|
4.10
|
Form of Convertible Note Hedge Confirmation for Convertible Senior Notes due 2026, filed as an exhibit to the Registrant’s Current Report on Form 8-K filed April 13, 2021 and incorporated herein by reference.
|
||
|
4.11
|
Indenture, dated as of June 12, 2023, by and between the Registrant and U.S. Bank Trust Company, a National Association, as trustee, including Form of
1.75 percent Global Note due in 2028, filed as an exhibit to the Registrant’s Current Report on Form 8-K filed June 12, 2023 and incorporated herein by
reference.
|
||
|
10.1*
|
Second Amended Non-Employee Director Compensation Policy, filed as an exhibit
to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended March 31, 2023 and incorporated herein by reference.
|
||
|
10.2*
|
Registrant’s Amended and Restated Severance Benefit Plan dated March 17, 2022, filed as an exhibit to the Registrant’s Quarterly Report on form 10-Q for the quarter ended March 31, 2022 and incorporated herein by reference.
|
||
|
Form of Indemnity Agreement entered into between the Registrant and its Directors and Executive Officers with related schedule
|
|||
|
10.4
|
Form of Employee Confidential Information and Inventions Agreement, filed as an exhibit to the Registrant’s Annual Report on Form 10-K for the year
ended December 31, 2017 and incorporated herein by reference.
|
||
|
10.5*
|
Registrant’s Amended and Restated 2000 Employee Stock Purchase Plan, filed as
an exhibit to Registrant’s Current Report on Form 8-K filed on March 26, 2019 and incorporated herein by reference.
|
||
|
10.6*
|
Registrant's Amended and Restated 2002 Non-Employee Directors’ Stock Option Plan, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended March 31, 2023 and incorporated herein by reference.
|
||
|
10.7*
|
Form of Option Agreement for Options granted under the Ionis Pharmaceuticals, Inc. Amended and Restated 2002 Non-Employee Directors’ Stock Option Plan, filed as an exhibit to the Registrant’s Annual Report on Form 10-K for the year ended December 31, 2022 and incorporated herein by reference.
|
||
|
10.8*
|
Forms of Restricted Stock Unit Grant Notice and Restricted Stock Unit Agreement for Restricted Stock Units granted under the Ionis Pharmaceuticals, Inc.
Amended and Restated 2002 Non-Employee Directors’ Stock Option Plan, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter
ended March 31, 2023 and incorporated herein by reference.
|
||
|
10.9*
|
Amended and Restated Ionis Pharmaceuticals, Inc. 2011 Equity Incentive Plan, filed as an exhibit to the Registrant’s Notice of 2021 Annual Meeting of Stockholders and Proxy Statement filed on April 23, 2021 and incorporated herein by reference.
|
||
|
10.10*
|
Form of Option Agreement under the 2011 Equity Incentive Plan, filed
as an exhibit to the Registrant’s Annual Report on Form 10-K for the year ended December 31, 2022 and incorporated herein by reference.
|
||
|
10.11*
|
Form of Time-Vested Restricted Stock Unit Agreement for Restricted Stock Units granted under the 2011 Equity Incentive Plan, filed as an exhibit to the Registrant’s Annual Report on Form 10-K for the year ended December 31, 2022 and incorporated herein by reference.
|
||
|
10.12*
|
Forms of Performance Based Restricted Stock Unit Grant Notice and Performance Based Restricted Stock Unit Agreement for Performance Based Restricted
Stock Units granted prior to January 1, 2023 under the 2011 Equity Incentive Plan, filed as an exhibit to the Registrant’s Annual Report on Form 10-K for
the year ended December 31, 2021 and incorporated herein by reference.
|
||
|
10.13*
|
Forms of Performance Based Restricted Stock Unit Grant Notice and Performance Based Restricted Stock Unit Agreement for Performance Based Restricted
Stock Units granted beginning January 1, 2023 under the 2011 Equity Incentive Plan, filed as an exhibit to the Registrant’s Annual Report on Form 10-K for the
year ended December 31, 2022 and incorporated herein by reference.
|
||
|
10.14*
|
Ionis Pharmaceuticals, Inc. 2020 Equity Incentive Plan, filed as
an exhibit to the Registrant’s Registration Statement on Form S-8 filed on December 31, 2020 and incorporated herein by reference.
|
||
|
10.15*
|
Form of Global Option Agreement for options granted under the Ionis Pharmaceuticals, Inc. 2020 Equity Incentive Plan, filed as an exhibit to the Registrant’s Registration Statement on Form S-8 filed on December 31, 2020 and incorporated herein by reference.
|
||
|
10.16*
|
Form of Global Restricted Stock Unit Agreement for restricted stock units granted under the Ionis Pharmaceuticals, Inc. 2020 Equity Incentive
Plan, filed as an exhibit to the Registrant’s Registration Statement on Form S-8 filed on December 31, 2020 and incorporated herein by reference.
|
||
|
10.17*
|
Forms of Restricted Stock Unit Grant Notice, Stock Option Grant Notice and Stock Option Exercise Notice for options granted under the Ionis
Pharmaceuticals, Inc. 2020 Equity Incentive Plan, filed as an exhibit to the Registrant’s Registration Statement on Form S-8 filed on December 31, 2020 and incorporated herein by reference.
|
||
|
10.18
|
Loan Agreement between Ionis Faraday, LLC and UBS AG dated July 18, 2017,
filed as an exhibit to the Registrant’s Current Report on Form 8-K filed July 21, 2017 and incorporated herein by reference.
|
||
|
10.19
|
Guaranty between the Registrant and UBS AG dated July 18, 2017, filed as an
exhibit to the Registrant’s Current Report on Form 8-K filed July 21, 2017 and incorporated herein by reference.
|
||
|
10.20
|
Purchase and Sale Agreement between Ionis Gazelle, LLC and 2850 2855 & 2859 Gazelle Owner (DE) LLC dated as of October 20, 2022, filed as an exhibit to the Registrant’s Annual Report on Form 10-K for the year ended December 31, 2022 and incorporated herein by reference. Portions of this exhibit
have been omitted because they are both (i) not material and (ii) is the type that the Registrant treats as private or confidential.
|
||
|
10.21
|
Purchase and Sale Agreement between the Registrant and Oxford I Asset Management USA Inc. dated as of October 20, 2022, filed as an exhibit to the Registrant’s Annual Report on Form 10-K for the year ended December 31, 2022 and incorporated herein by reference. Portions of this exhibit have been omitted because
they are both (i) not material and (ii) is the type that the Registrant treats as private or confidential.
|
||
|
10.22
|
First Amendment dated June 15, 2023 to the Purchase and Sale Agreement by and between the Registrant and Oxford I Asset Management USA Inc. dated as of
October 20, 2022, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2023 and incorporated herein by
reference.
|
||
|
10.23
|
Lease Agreement dated October 20, 2022 between the Registrant and 2850 2855 & 2859 Gazelle Owner (DE) LLC, filed as an exhibit to the Registrant’s Annual Report on Form 10-K for the year ended December 31, 2022 and incorporated herein by reference. Portions of this exhibit have been omitted because
they are both (i) not material and (ii) is the type that the Registrant treats as private or confidential.
|
||
|
10.24
|
Amended and Restated Lease Agreement between the Registrant and Lots 21 & 22 Owner (DE) LLC dated as of August 21, 2023, filed as an exhibit to the
Registrant’s Quarterly Report on Form 10-Q for the quarter ended September 30, 2023 and incorporated herein by reference. Portions of this exhibit have been omitted
because they are both (i) not material and (ii) is the type that the Registrant treats as private or confidential.
|
||
|
First Amendment dated as of November 6, 2023 to Amended and Restated Lease Agreement between the Registrant and Lots 21 & 22 Owner (DE) LLC dated
as of August 21, 2023.
|
||
|
10.26
|
Defeasance Pledge and Security Agreement dated as of October 20, 2022 by and among Ionis Gazelle, LLC, Wells Fargo Bank, National Association, as
Trustee for the Benefit of the Registered Holders of UBS Commercial Mortgage Trust 2017-C3, Commercial Mortgage Pass-Through Certificates, Series 2017-C3, and U.S. Bank Trust Company, National Association, filed as an exhibit to the Registrant’s Annual Report on Form 10-K for the year ended December 31, 2022 and incorporated herein by reference. Portions of this exhibit have been omitted because
they are both (i) not material and (ii) the type that the Registrant treats as private or confidential.
|
|
|
10.27
|
Defeasance Assignment, Assumption and Release Agreement dated as of October 20, 2022 by and among Ionis Gazelle, LLC, DHC UBSCM 17 C3 Successor
Borrower-R, LLC, Wells Fargo Bank, National Association, as Trustee for the Benefit of the Registered Holders of UBS Commercial Mortgage Trust 2017-C3, Commercial Mortgage Pass-Through Certificates, Series 2017-C3, Midland Loan Services, a
division of PNC Bank, National Association, and U.S. Bank Trust Company, National Association, filed as an exhibit to the Registrant’s Annual Report on Form 10-K
for the year ended December 31, 2022 and incorporated herein by reference.
|
|
|
10.28
|
Defeasance Account Agreement dated as of October 20, 2022 by and among Ionis Gazelle, LLC, U.S. Bank Trust Company, National Association, U.S. Bank
National Association, as Trustee for the Benefit of the Registered Holders of UBS Commercial Mortgage Trust 2017-C3, Commercial Mortgage Pass-Through Certificates, Series 2017-C3, and Midland Loan Services, a division of PNC Bank, National
Association, filed as an exhibit to the Registrant’s Annual Report on Form 10-K for the year ended December 31, 2022 and incorporated herein by reference.
Portions of this exhibit have been omitted because they are both (i) not material and (ii) the type that the Registrant treats as private or confidential.
|
|
|
10.29
|
Exclusive License Agreement between the Registrant and the University of Massachusetts dated January 14, 2010, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended September 30, 2014 and incorporated herein by reference. Portions of this exhibit have been omitted and separately
filed with the SEC with a request for confidential treatment.
|
|
|
10.30
|
Research, Development and License Agreement between the Registrant and Glaxo Group Limited dated March 30, 2010, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended March 31, 2010 and incorporated herein by reference. Portions of this exhibit have been omitted and
separately filed with the SEC with a request for confidential treatment.
|
|
|
10.31
|
Amendment #1 to the Research, Development and License Agreement dated May 11, 2011 by and between the Registrant and Glaxo Group Limited, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2011 and incorporated herein by reference. Portions of this exhibit
have been omitted and separately filed with the SEC with a request for confidential treatment.
|
|
|
10.32
|
Amendment #2 to the Research, Development and License Agreement by and between the Registrant and Glaxo Group Limited dated October 30, 2012, filed as an exhibit to the Registrant’s Annual Report on Form 10-K for the year ended December 31, 2012 and incorporated herein by reference. Portions of this exhibit
have been omitted and separately filed with the SEC with a request for confidential treatment.
|
|
|
10.33
|
Amendment No. 3 to the Research, Development and License Agreement by and between the Registrant and Glaxo Group Limited dated July 10, 2013, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2014 and incorporated herein by reference. Portions of this exhibit
have been omitted and separately filed with the SEC with a request for confidential treatment.
|
|
|
10.34
|
Amendment #4 to the Research, Development and License Agreement by and between the Registrant and Glaxo Group Limited dated April 10, 2014, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2014 and incorporated herein by reference. Portions of this exhibit
have been omitted and separately filed with the SEC with a request for confidential treatment.
|
|
|
10.35
|
Amendment #5 to the Research, Development and License Agreement by and between the Registrant, Glaxo Group Limited and GlaxoSmithKline Intellectual Property
Development Limited dated June 27, 2014, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2014 and
incorporated herein by reference. Portions of this exhibit have been omitted and separately filed with the SEC with a request for confidential treatment.
|
|
|
10.36
|
Amendment #6 to Research, Development and License Agreement by and between the Registrant, Glaxo Group Limited and GlaxoSmithKline Intellectual Property
Development Limited dated September 2, 2015, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended September 30, 2015 and
incorporated herein by reference. Portions of this exhibit have been omitted and separately filed with the SEC with a request for confidential treatment.
|
|
|
10.37
|
Amendment #7 to the Research, Development and License Agreement by and between the Registrant, Glaxo Group Limited and GlaxoSmithKline Intellectual Property
Development Limited dated March 4, 2016, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended March 31, 2016 and
incorporated herein by reference. Portions of this exhibit have been omitted and separately filed with the SEC with a request for confidential treatment.
|
|
10.38
|
Amendment #8 to the Research, Development and License Agreement by and between the Registrant, Glaxo Group Limited and Glaxosmithkline Intellectual Property
Development Limited, dated July 29, 2019, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended September 30, 2019 and
incorporated herein by reference. Portions of this exhibit have been omitted because they are both (i) not material and (ii) the type that the Registrant treats as private or confidential.
|
|||
|
10.39
|
Amended and Restated Collaboration and License Agreement between the Registrant and Cold Spring Harbor Laboratory dated October 26, 2011, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended September 30, 2014 and incorporated herein by reference. Portions of this
exhibit have been omitted and separately filed with the SEC with a request for confidential treatment.
|
|||
|
10.40
|
Amendment to Amended and Restated Collaboration and License Agreement between the Registrant and Cold Spring Harbor Laboratory dated March 14, 2014, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended September 30, 2014 and incorporated herein by reference. Portions of this
exhibit have been omitted and separately filed with the SEC with a request for confidential treatment.
|
|||
|
10.41
|
Development, Option and License Agreement between the Registrant and Biogen Idec International Holding Ltd. dated January 3, 2012, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended March 31, 2012 and incorporated herein by reference. Portions of this exhibit
have been omitted and separately filed with the SEC with a request for confidential treatment.
|
|||
|
10.42
|
Amendment #1 to the Development, Option and License Agreement between the Registrant and Biogen Idec International Holding Ltd. dated December 15, 2014, filed as an exhibit to the Registrant’s Annual Report on Form 10-K for the year ended December 31, 2014 and incorporated herein by reference. Portions of this exhibit
have been omitted and separately filed with the SEC with a request for confidential treatment.
|
|||
|
10.43
|
Collaboration, License and Development Agreement between the Registrant and AstraZeneca AB dated December 7, 2012, filed as an exhibit to the Registrant’s Annual Report on Form 10-K for the year ended December 31, 2012 and incorporated herein by reference. Portions of this exhibit have been omitted and
separately filed with the SEC with a request for confidential treatment.
|
|||
|
10.44
|
Amendment #1 to Collaboration, License and Development Agreement between the Registrant and AstraZeneca AB dated August 13, 2013, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended September 30, 2013 and incorporated herein by reference. Portions of this
exhibit have been omitted and separately filed with the SEC with a request for confidential treatment.
|
|||
|
10.45
|
Amendment No.2 to the Collaboration, License and Development Agreement between the Registrant and AstraZeneca AB dated October 15, 2014, filed as an exhibit to the Registrant’s Annual Report on Form 10-K for the year ended December 31, 2014 and incorporated herein by reference. Portions of this exhibit
have been omitted and separately filed with the SEC with a request for confidential treatment.
|
|||
|
10.46
|
Amendment No.3 to the Collaboration, License and Development Agreement between the Registrant and AstraZeneca AB dated January 18, 2016, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended March 31, 2016 and incorporated herein by reference. Portions of this exhibit
have been omitted and separately filed with the SEC with a request for confidential treatment.
|
|||
|
10.47
|
Amendment No. 4 to the Collaboration, License and Development Agreement by and between the Registrant and AstraZeneca AB, dated October 18, 2018, filed as an exhibit to the Registrant’s Annual Report on Form 10-K for the year ended December 31, 2018 and incorporated herein by reference. Portions of this exhibit
have been omitted and separately filed with the SEC with a request for confidential treatment.
|
|||
|
10.48
|
HTT Research, Development, Option and License Agreement among the Registrant, F. Hoffmann-La Roche Ltd and Hoffman-La Roche Inc. dated April 8,
2013, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended March 31, 2023 and incorporated herein by reference. Portions
of this exhibit have been omitted because they are both (i) not material and (ii) is the type that the Registrant treats as private or confidential.
|
|||
|
10.49
|
Amendment #1 to HTT Research, Development, Option and License Agreement between the Registrant, F. Hoffmann-La Roche Ltd and Hoffmann-La Roche Inc. dated
January 9, 2015, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended March 31, 2015 and incorporated herein by reference.
Portions of this exhibit have been omitted and separately filed with the SEC with a request for confidential treatment.
|
|||
|
10.50
|
Second Amended and Restated Strategic Collaboration and License Agreement between the Registrant and Alnylam Pharmaceuticals, Inc. dated January 8, 2015, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended March 31, 2015 and incorporated herein by reference. Portions of this exhibit
have been omitted and separately filed with the SEC with a request for confidential treatment.
|
|||
|
10.51
|
Amendment Number One to the Second Amended and Restated Strategic Collaboration and License Agreement between the Registrant and Alnylam Pharmaceuticals, Inc.
dated July 13, 2015, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended September 30, 2015 and incorporated herein by
reference. Portions of this exhibit have been omitted and separately filed with the SEC with a request for confidential treatment.
|
|
|
10.52
|
Strategic Collaboration Agreement between the Registrant and AstraZeneca AB dated July 31, 2015, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended September 30, 2015 and incorporated herein by reference. Portions of this exhibit have been omitted and separately filed with
the SEC with a request for confidential treatment.
|
|
|
10.53
|
Amendment No. 1 to the Strategic Collaboration Agreement by and between the Registrant and AstraZeneca AB, dated October 18, 2018, filed as an exhibit to the Registrant’s Annual Report on Form 10-K for the year ended December 31, 2018 and incorporated herein by reference. Portions of this exhibit
have been omitted and separately filed with the SEC with a request for confidential treatment.
|
|
|
10.54
|
Amendment No. 2 dated April 30, 2020 to the Strategic Collaboration Agreement by and between the Registrant and AstraZeneca AB dated July 31, 2015, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2020 and incorporated herein by reference. Portions of this exhibit have been omitted because they are both (i) not
material and (ii) is the type that the Registrant treats as private or confidential.
|
|
|
10.55
|
Amendment No. 3 dated December 17, 2020 to the Strategic Collaboration Agreement by and between the Registrant and AstraZeneca AB dated July 31, 2015, filed as an exhibit to the Registrant’s Annual Report on Form 10-K for the year ended December 31, 2020 and incorporated herein by reference. Portions of this exhibit have been omitted because they are both
(i) not material and (ii) the type that the Registrant treats as private or confidential.
|
|
|
10.56
|
Strategic Collaboration, Option and License Agreement by and among Akcea Therapeutics, Inc. and Novartis Pharma AG, dated January 5, 2017,
filed as an exhibit to Akcea Therapeutics, Inc.’s Form S-1 filed March 27, 2017 and incorporated herein by reference.
|
|
|
10.57
|
Amendment No. 1 to the Strategic Collaboration, Option and License Agreement between Akcea Therapeutics, Inc. and Novartis Pharma AG dated February
22, 2019, filed as an exhibit to Akcea Therapeutics, Inc.’s Quarterly Report on Form 10-Q for the quarter ended March 30, 2019 and incorporated herein by reference.
|
|
|
10.58
|
Stock Purchase Agreement among the Registrant, Akcea Therapeutics, Inc. and Novartis Pharma AG dated January 5, 2017, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended March 31, 2017 and incorporated herein by reference.
|
|
|
10.59
|
Research Collaboration, Option and License Agreement between the Registrant and Biogen MA Inc. dated December 19, 2017, filed as an exhibit to the Registrant’s Annual Report on Form 10-K for the year ended December 31, 2017 and incorporated herein by reference. Portions of this exhibit have been omitted and
separately filed with the SEC with a request for confidential treatment.
|
|
|
10.60
|
New Strategic Neurology Drug Discovery and Development Collaboration, Option and License Agreement by and between the Registrant and Biogen MA Inc., dated April 19, 2018, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2018 and incorporated herein by reference.
Portions of this exhibit have been omitted and separately filed with the SEC with a request for confidential treatment.
|
|
|
10.61
|
Amendment No. 1 to the New Strategic Neurology Drug Discovery and Development Collaboration, Option and License Agreement between the Registrant and Biogen MA
Inc., dated August 16, 2019, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended September 30, 2019 and incorporated
herein by reference. Portions of this exhibit have been omitted because they are both (i) not material and (ii) the type that the Registrant treats as private or confidential.
|
|
|
10.62
|
Side Letter dated December 31, 2020 to the New Strategic Neurology Drug Discovery and Development Collaboration, Option and License Agreement by and between the Registrant and Biogen MA Inc. dated April 19,
2018, filed as an exhibit to the Registrant’s Annual Report on Form 10-K for the year ended December 31, 2020 and incorporated herein by
reference. Portions of this exhibit have been omitted because they are both (i) not material and (ii) the type that the Registrant treats as private or confidential.
|
|
|
10.63
|
Factor B Development Collaboration, Option and License Agreement by and between the Registrant, F. Hoffmann-La Roche Ltd and Hoffmann-La Roche Inc.,
dated October 9, 2018, filed as an exhibit to the Registrant’s Annual Report on Form 10-K for the year ended December 31, 2018 and incorporated herein by
reference. Portions of this exhibit have been omitted and separately filed with the SEC with a request for confidential treatment.
|
|
10.64
|
First Amendment dated July 8, 2022 to Factor B Development, Collaboration, Option and License Agreement by and between the Registrant, F. Hoffmann-La Roche Ltd and Hoffmann-La Roche Inc., dated October 9, 2018, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2022. Portions of this exhibit have been
omitted because they are both (i) not material and (ii) the type that the Registrant treats as private or confidential.
|
||
|
10.65
|
Second Amended and Restated Strategic Neurology Drug Discovery and Development Collaboration, Option and License Agreement by and between the Registrant
and Biogen MA Inc., dated October 17, 2018, filed as an exhibit to the Registrant’s Annual Report on Form 10-K for the year ended December 31, 2018 and
incorporated herein by reference. Portions of this exhibit have been omitted and separately filed with the SEC with a request for confidential treatment.
|
||
|
10.66
|
Letter Agreement between the Registrant and Biogen MA Inc. dated October 28, 2016, filed as an exhibit to the Registrant’s Annual Report on Form 10-K for the year ended December 31, 2016 and incorporated herein by reference. Portions of this exhibit have been omitted and separately filed with the SEC with a
request for confidential treatment.
|
||
|
10.67
|
Amendment No. 1 to Second Amended and Restated Strategic Neurology Drug Discovery and Development Collaboration, Option and License Agreement by and between
the Registrant and Biogen MA Inc., dated May 2, 2019, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2019
and incorporated herein by reference.
|
||
|
10.68
|
Collaboration and License Agreement by and between the Registrant and Novartis Pharma AG dated as of August 2, 2023, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended September 30, 2023 and incorporated herein by reference. Portions of this exhibit have been omitted because they are both (i) not material and (ii) the type that the Registrant treats as private or confidential.
|
||
|
10.69
|
Research, Development, and License Agreement by and among the Registrant, F. Hoffmann-La Roche Ltd., and Hoffmann-La Roche Inc. dated as of September 26, 2023, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended September 30, 2023 and incorporated herein by reference. Portions of this exhibit have been omitted because they are both (i) not material and (ii) the type that the Registrant treats as private or confidential.
|
||
|
10.70
|
Side Letter dated June 11, 2020 to the Second Amended and Restated Strategic Neurology Drug Discovery and Development Collaboration, Option and License Agreement by and between the Registrant and Biogen MA Inc.
dated October 17, 2018, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2020 and incorporated
herein by reference. Portions of this exhibit have been omitted because they are both (i) not material and (ii) the type that the Registrant treats as private or confidential.
|
||
|
10.71
|
Collaboration and License Agreement by and between the Registrant and BicycleTX Limited dated July 9, 2021, filed as an exhibit to the Registrant’s
Quarterly Report on Form 10-Q for the quarter ended September 30, 2021 and incorporated herein by reference. Portions of this exhibit have been omitted because they are both (i) not material and (ii) the type that the Registrant treats as private or confidential.
|
||
|
10.72
|
Amendment No. 1 dated December 17, 2021 to the Collaboration and License Agreement by and between the Registrant and BicycleTX Limited dated July 9, 2021, filed as an exhibit to the Registrant’s Annual Report on Form 10-K for the year ended December 31, 2021 and incorporated herein by reference. Portions of this exhibit have been omitted because they are both (i) not material and (ii) the type that the Registrant treats as private or confidential.
|
||
|
10.73
|
Amendment No. 2 dated July 28, 2022 to the Collaboration and License Agreement by and between the Registrant and BicycleTx Limited dated July 9, 2021, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended September 30, 2022 and incorporated herein by reference. Portions of this exhibit have been omitted because they are both (i) not material and (ii) the type that the Registrant treats as private or confidential.
|
||
|
10.74
|
Amendment No. 3 dated April 27, 2023 to the Collaboration and License Agreement by and between the Registrant and BicycleTx Limited dated July 9, 2021, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2023 and incorporated herein by reference. Portions of this exhibit have been omitted because they are both (i) not material and (ii) the type that the Registrant treats as private or confidential.
|
||
|
10.75
|
Amended and Restated Neurology Drug Discovery and Development Collaboration, Option and License Agreement by and between the Registrant and Biogen MA Inc.
dated July 12, 2021, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended September 30, 2021 and incorporated herein by
reference. Portions of this exhibit have been omitted because they are both (i) not material and (ii) the type that the Registrant treats as private or confidential.
|
||
|
10.76
|
Collaboration and License Agreement by and between Akcea Therapeutics, Inc. and AstraZeneca AB dated December 6, 2021, filed as an exhibit to the Registrant’s Annual Report on Form 10-K for the year ended December 31, 2021 and incorporated herein by reference. Portions of this exhibit have been omitted because they are both (i) not material and (ii) the type that the Registrant treats as private or confidential.
|
||
|
10.77
|
Letter Agreement dated June 29, 2023 in reference to the Collaboration and License Agreement dated December 6, 2021 by and between Akcea Therapeutics, Inc.
and AstraZeneca AB, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2023 and incorporated herein by
reference. Portions of this exhibit have been omitted because they are both (i) not material and (ii) the type that the Registrant treats as private or confidential.
|
|
|
10.78
|
Collaboration and License Agreement between the Registrant and Metagenomi, Inc. dated November 10, 2022, filed as an exhibit to the Registrant’s Annual Report on Form 10-K for the year ended December 31, 2022 and incorporated herein by reference. Portions of this exhibit have been omitted because they are both
(i) not material and (ii) the type that the Registrant treats as private or confidential.
|
|
|
10.79
|
Royalty Purchase Agreement by and between the Registrant, Akcea Therapeutics, Inc. and Royalty Pharma Investments 2019 ICAV dated as of January 9, 2023, filed as an exhibit to the Registrant’s Annual Report on Form 10-K for the year ended December 31, 2022 and incorporated herein by reference. Portions of this exhibit
have been omitted because they are both (i) not material and (ii) the type that the Registrant treats as private or confidential.
|
|
|
License Agreement by and between the Registrant and Otsuka Pharmaceuticals Co., LTD. dated as of December 15, 2023. Portions of this exhibit have
been omitted because they are both (i) not material and (ii) the type that the Registrant treats as private or confidential.
|
||
|
List of Subsidiaries for the Registrant.
|
||
|
Consent of Independent Registered Public Accounting Firm.
|
||
|
Power of Attorney – Included on the signature page of this Annual Report on Form 10-K.
|
||
|
Certification by Chief Executive Officer Pursuant to 18 U.S.C. Section 1350 as Adopted Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002.
|
||
|
Certification by Chief Financial Officer Pursuant to 18 U.S.C. Section 1350 as Adopted Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002.
|
||
|
32.1+
|
Certification Pursuant to 18 U.S.C. Section 1350 as Adopted Pursuant to Section 906 of the Sarbanes-Oxley Act of 2002.
|
|
|
Registrant’s Amended and Restated Clawback Policy
|
||
|
101
|
The following financial statements from the Ionis Pharmaceuticals, Inc. Annual Report on Form 10-K for the year ended December 31, 2023, formatted in Extensive Business Reporting Language (XBRL): (i) consolidated balance sheets, (ii) consolidated statements of operations,
(iii) consolidated statements of comprehensive income (loss), (iv) consolidated statements of stockholders’ equity (v) consolidated statements of cash flows, and (vi) notes to consolidated financial statements (detail tagged).
|
|
|
104
|
Cover Page Interactive Data File (formatted in iXBRL and included in exhibit 101).
|
| * |
Indicates management compensatory plans and arrangements as required to be filed as exhibits to this Report pursuant to Item 14(c).
|
| + |
This certification is deemed not filed for purposes of Section 18 of the Securities Exchange Act of 1934, as amended, or otherwise subject to the liability of that
section, nor shall it be deemed incorporated by reference into any filing under the Securities Act of 133, as amended, or the Securities Exchange Act of 1934, as amended.
|
|
IONIS PHARMACEUTICALS, INC.
|
||
|
By:
|
/s/ BRETT P. MONIA
|
|
|
Brett P. Monia, Ph.D.
|
||
|
Chief Executive Officer (Principal executive officer)
|
||
|
Signatures
|
Title
|
Date
|
||
|
/s/ BRETT P. MONIA
|
Director and Chief Executive Officer
|
February 21, 2024
|
||
|
Brett P. Monia, Ph.D.
|
(Principal executive officer)
|
|||
|
/s/ ELIZABETH L. HOUGEN
|
Executive Vice President, Finance and Chief Financial Officer
|
February 21, 2024
|
||
|
Elizabeth L. Hougen
|
(Principal financial and accounting officer)
|
|||
|
/s/ JOSEPH LOSCALZO
|
Chairman of the Board
|
February 21, 2024
|
||
|
Joseph Loscalzo, M.D., Ph.D.
|
||||
|
/s/ SPENCER R. BERTHELSEN
|
Director
|
February 21, 2024
|
||
|
Spencer R. Berthelsen, M.D.
|
||||
|
/s/ ALLENE M. DIAZ
|
Director
|
February 21, 2024
|
||
|
Allene M. Diaz
|
||||
|
/s/ MICHAEL HAYDEN
|
Director
|
February 21, 2024
|
||
|
Michael Hayden, CM OBC MB ChB PhD FRCP(C) FRSC
|
||||
|
/s/ JOAN E. HERMAN
|
Director
|
February 21, 2024
|
||
|
Joan E. Herman
|
||||
|
/s/ JOSEPH KLEIN
|
Director
|
February 21, 2024
|
||
|
Joseph Klein, III
|
||||
|
/s/ B. LYNNE PARSHALL
|
Director
|
February 21, 2024
|
||
|
B. Lynne Parshall, J.D.
|
|
/s/ JOSEPH H. WENDER
|
Lead Independent Director
|
February 21, 2024
|
||
|
Joseph H. Wender
|
|
/s/ MICHAEL YANG
|
Director
|
February 21, 2024
|
||
|
Michael Yang
|
|
Page
|
|
|
Report of Independent Registered Public Accounting Firm (PCAOB ID 42)
|
F-2
|
|
Consolidated Balance Sheets at December 31, 2023 and 2022
|
F-4
|
|
Consolidated Statements of Operations for the years ended December 31, 2023, 2022 and 2021
|
F-5
|
|
Consolidated Statements of Comprehensive Loss for the years ended December 31, 2023, 2022 and 2021
|
F-6
|
|
Consolidated Statements of Stockholders’ Equity for the years ended December 31, 2023, 2022 and 2021
|
F-7
|
|
Consolidated Statements of Cash Flows for the years ended December 31, 2023, 2022 and 2021
|
F-8
|
|
F-9
|
|
Estimated Liability for Clinical Development Costs
|
||
|
Description of the Matter
|
As of December 31, 2023, the Company accrued $106 million for clinical development expenses. As discussed in Note 1 to the consolidated financial
statements, the Company records costs for clinical trial activities based upon estimates of costs incurred through the balance sheet date that have yet to be invoiced related to third-party clinical management costs, laboratory and analysis
costs, toxicology studies and investigator grants. The Company estimates its liability using assumptions about study and patient activities and the related expected expenses for those activities based on the contracted fees with service
providers.
Auditing the Company’s accruals for clinical and contract research organization costs is especially complex as the information necessary to estimate
the accruals is accumulated from multiple sources. In addition, in certain circumstances, the determination of the nature and level of services that have been received during the reporting period requires judgment because the timing and pattern
of vendor invoicing does not correspond to the level of services provided and there may be delays in invoicing from vendors.
|
|
How We Addressed the Matter in Our Audit
|
We obtained an understanding and evaluated the design and tested the operating effectiveness of controls over the accounting for accrued clinical
development expenses. This included controls over management's assessment of the assumptions and accuracy of data underlying the accrued clinical development expenses estimate.
To test the accuracy of the Company’s accrued clinical development expenses, we performed audit procedures that included, among other procedures,
obtaining supporting evidence of the research and development activities performed for significant clinical trials. We corroborated the status of significant clinical development expenses through meetings with accounting and clinical project
managers. We compared the costs for a sample of transactions against the related invoices and contracts, and examined a sample of subsequent payments to evaluate the accuracy of the accrued clinical development expenses and compared the results
to the current year accrual.
|
|
Accounting for the Royalty Pharma Sale of Future Royalties Transaction
|
||
|
Description of the Matter
|
As discussed in Note 7 to the consolidated financial statements, in January 2023, the Company entered into a royalty purchase agreement to monetize a
portion of future SPINRAZA and pelacarsen royalties that the Company is entitled to under existing agreements. As a result, the Company received an upfront payment of $500 million. The Company accounted for the sale of future royalties as a
liability. The Company determines the effective interest rate used to record interest expense based on the estimate of future royalty payments over the term of the agreement. The carrying value of the liability related to the sale of future
royalties at December 31, 2023 was $514 million.
Auditing the Company’s liability related to the sale of future royalties was complex due to the subjective judgments required to forecast the
expected royalty payments subject to the agreement and due to the nature and extent of audit effort required to address these matters. Specifically, as it related to pelecarsen, a product candidate that is not currently commercialized, these
estimates include significant assumptions such as market penetration, probability of success, and sales price, among others, that are affected by expectations about future market conditions.
|
|
How We Addressed the Matter in Our Audit
|
We obtained an understanding, evaluated the design, and tested the operating effectiveness of controls over the Company’s processes for estimating
the amount and timing of future royalty payments.
To test the liability balance and the amount of interest expense recognized, our audit procedures included, among others, evaluating the methodology
used and assessing the significant assumptions and the underlying data used by the Company in its effective interest model. We compared the significant assumptions in the estimate of future royalty payments to current industry and market
trends. We recalculated the current year interest expense based on the amortization schedules and estimates of royalties using the effective interest method, and performed sensitivity analyses to evaluate the changes in the effective interest
rate, and associated interest expense, that would result from changes in the assumptions.
|
|
December 31,
|
||||||||
|
2023
|
2022
|
|||||||
|
ASSETS
|
||||||||
|
Current assets:
|
||||||||
|
Cash and cash equivalents
|
$
|
399,266
|
$
|
276,472
|
||||
|
Short-term investments
|
1,931,935
|
1,710,397
|
||||||
|
Contracts receivable
|
97,778
|
25,538
|
||||||
|
Inventories
|
28,425
|
22,033
|
||||||
|
Other current assets
|
184,449
|
168,254
|
||||||
|
Total current assets
|
2,641,853
|
2,202,694
|
||||||
|
Property, plant and equipment, net
|
71,043
|
74,294
|
||||||
|
Right-of-use assets
|
171,896
|
181,544
|
||||||
|
Deposits and other assets
|
105,280
|
75,344
|
||||||
|
Total assets
|
$
|
2,990,072
|
$
|
2,533,876
|
||||
|
LIABILITIES AND
STOCKHOLDERS’ EQUITY
|
||||||||
|
Current liabilities:
|
||||||||
|
Accounts payable
|
$
|
26,027
|
$
|
17,921
|
||||
|
Accrued compensation
|
67,727
|
49,178
|
||||||
|
Accrued liabilities
|
147,894
|
140,101
|
||||||
|
Income taxes payable
|
2,151
|
6,249
|
||||||
|
0.125 percent
convertible senior notes, net
|
44,332
|
—
|
||||||
|
Current portion of deferred contract revenue
|
151,128
|
90,577
|
||||||
|
Other current liabilities
|
8,831
|
7,535
|
||||||
|
Total current liabilities
|
448,090
|
311,561
|
||||||
|
Long-term deferred contract revenue
|
241,184
|
287,768
|
||||||
|
1.75 percent
convertible senior notes, net
|
562,285
|
—
|
||||||
|
0 percent
convertible senior notes, net
|
625,380
|
622,242
|
||||||
|
0.125 percent
convertible senior notes, net
|
—
|
544,504
|
||||||
|
Liability related to sale of future royalties, net
|
513,736
|
—
|
||||||
|
Long-term lease liabilities
|
170,875
|
178,941
|
||||||
|
Long-term obligations
|
41,836
|
15,973
|
||||||
|
Total liabilities
|
2,603,386
|
1,960,989
|
||||||
|
Stockholders’ equity:
|
||||||||
|
Common stock, $0.001
par value; 300,000,000 shares authorized, 144,340,526 and 142,057,736 shares issued and outstanding at December 31,
2023 and December 31, 2022,
respectively
|
144
|
142
|
||||||
|
Additional paid-in capital
|
2,215,098
|
2,059,850
|
||||||
|
Accumulated other comprehensive loss
|
(32,645
|
)
|
(57,480
|
)
|
||||
|
Accumulated deficit
|
(1,795,911
|
)
|
(1,429,625
|
)
|
||||
|
Total stockholders’ equity
|
386,686
|
572,887
|
||||||
|
Total liabilities and stockholders’ equity
|
$
|
2,990,072
|
$
|
2,533,876
|
||||
|
Year Ended December 31,
|
||||||||||||
|
2023
|
2022
|
2021
|
||||||||||
|
Revenue:
|
||||||||||||
|
Commercial revenue:
|
||||||||||||
|
SPINRAZA royalties
|
$
|
240,379
|
$
|
242,314
|
$
|
267,776
|
||||||
|
Other commercial revenue
|
68,212
|
61,044
|
74,619
|
|||||||||
|
Total commercial revenue
|
308,591
|
303,358
|
342,395
|
|||||||||
|
Research and development revenue:
|
||||||||||||
|
Collaborative agreement revenue
|
352,657
|
207,222
|
468,061
|
|||||||||
|
WAINUA joint development revenue
|
126,399
|
76,787
|
—
|
|||||||||
|
Total research and development revenue
|
479,056
|
284,009
|
468,061
|
|||||||||
|
Total revenue
|
787,647
|
587,367
|
810,456
|
|||||||||
|
Expenses:
|
||||||||||||
|
Cost of sales
|
9,133
|
14,116
|
10,842
|
|||||||||
|
Research, development and patent
|
899,625
|
833,147
|
643,453
|
|||||||||
|
Selling, general and administrative
|
232,619
|
150,295
|
186,347
|
|||||||||
|
Total operating expenses
|
1,141,377
|
997,558
|
840,642
|
|||||||||
|
Loss from operations
|
(353,730
|
)
|
(410,191
|
)
|
(30,186
|
)
|
||||||
|
Other income (expense):
|
||||||||||||
|
Investment income
|
89,041
|
25,331
|
10,044
|
|||||||||
|
Interest expense
|
(12,660
|
)
|
(8,122
|
)
|
(9,349
|
)
|
||||||
|
Interest expense related to sale of future royalties
|
(68,797
|
)
|
—
|
—
|
||||||||
|
Gain (loss) on investments
|
(1,914
|
)
|
(7,333
|
)
|
10,103
|
|||||||
|
Gain (loss) on sale of real estate assets
|
(161
|
)
|
149,604
|
—
|
||||||||
|
Other income (expense)
|
14,256
|
(7,274
|
)
|
(9,760
|
)
|
|||||||
|
Loss before income tax benefit (expense)
|
(333,965
|
)
|
(257,985
|
)
|
(29,148
|
)
|
||||||
|
Income tax benefit (expense)
|
(32,321
|
)
|
(11,737
|
)
|
551
|
|||||||
|
Net loss
|
$
|
(366,286
|
)
|
$
|
(269,722
|
)
|
$
|
(28,597
|
)
|
|||
|
Basic and diluted net loss per share
|
$
|
(2.56
|
)
|
$
|
(1.90
|
)
|
$
|
(0.20
|
)
|
|||
|
Shares used in computing basic and diluted net loss per share
|
143,190
|
141,848
|
141,021
|
|||||||||
|
Year Ended December 31,
|
||||||||||||
|
2023
|
2022
|
2021
|
||||||||||
|
Net loss
|
$
|
(366,286
|
)
|
$
|
(269,722
|
)
|
$
|
(28,597
|
)
|
|||
|
Unrealized gains (losses) on investments, net of tax
|
24,484
|
(24,395
|
)
|
(11,486
|
)
|
|||||||
|
Currency translation adjustment
|
351
|
(417
|
)
|
(111
|
)
|
|||||||
|
Comprehensive loss
|
$
|
(341,451
|
)
|
$
|
(294,534
|
)
|
$
|
(40,194
|
)
|
|||
|
Common Stock
|
Additional
|
Accumulated Other
|
Accumulated
|
Total Ionis
Stockholders’
|
||||||||||||||||||||
|
Description
|
Shares
|
Amount
|
Paid in Capital
|
Comprehensive Loss
|
Deficit
|
Equity
|
||||||||||||||||||
|
Balance at December 31, 2020
|
140,366
|
$
|
140
|
$
|
1,895,519
|
$
|
(21,071
|
)
|
$
|
(1,131,306
|
)
|
$
|
743,282
|
|||||||||||
|
Net loss
|
—
|
—
|
—
|
—
|
(28,597
|
)
|
(28,597
|
)
|
||||||||||||||||
|
Change in unrealized losses, net of tax
|
—
|
—
|
—
|
(11,486
|
)
|
—
|
(11,486
|
)
|
||||||||||||||||
|
Foreign currency translation
|
—
|
—
|
—
|
(111
|
)
|
—
|
(111
|
)
|
||||||||||||||||
|
Issuance of common stock in connection with employee stock plans
|
1,132
|
1
|
11,563
|
—
|
—
|
11,564
|
||||||||||||||||||
|
Issuance of warrants
|
—
|
—
|
89,752
|
—
|
—
|
89,752
|
||||||||||||||||||
|
Purchase of note hedges
|
—
|
—
|
(136,620
|
)
|
—
|
—
|
(136,620
|
)
|
||||||||||||||||
|
Stock-based compensation expense
|
—
|
—
|
120,678
|
—
|
—
|
120,678
|
||||||||||||||||||
|
Payments of tax withholdings related to vesting of employee stock awards and exercise of employee stock options
|
(288
|
)
|
—
|
(16,725
|
)
|
—
|
—
|
(16,725
|
)
|
|||||||||||||||
|
Balance at December 31, 2021
|
141,210
|
$
|
141
|
$
|
1,964,167
|
$
|
(32,668
|
)
|
$
|
(1,159,903
|
)
|
$
|
771,737
|
|||||||||||
|
Net loss
|
—
|
—
|
—
|
—
|
(269,722
|
)
|
(269,722
|
)
|
||||||||||||||||
|
Change in unrealized losses, net of tax
|
—
|
—
|
—
|
(24,395
|
)
|
—
|
(24,395
|
)
|
||||||||||||||||
|
Foreign currency translation
|
—
|
—
|
—
|
(417
|
)
|
—
|
(417
|
)
|
||||||||||||||||
|
Issuance of common stock in connection with employee stock plans
|
1,194
|
1
|
6,372
|
—
|
—
|
6,373
|
||||||||||||||||||
|
Stock-based compensation expense
|
—
|
—
|
100,264
|
—
|
—
|
100,264
|
||||||||||||||||||
|
Payments of tax withholdings related to vesting of employee stock awards and exercise of employee stock options
|
(346
|
)
|
—
|
(10,953
|
)
|
—
|
—
|
(10,953
|
)
|
|||||||||||||||
|
Balance at December 31, 2022
|
142,058
|
$
|
142
|
$
|
2,059,850
|
$
|
(57,480
|
)
|
$
|
(1,429,625
|
)
|
$
|
572,887
|
|||||||||||
|
Net loss
|
—
|
—
|
—
|
—
|
(366,286
|
)
|
(366,286
|
)
|
||||||||||||||||
|
Change in unrealized gains, net of tax
|
—
|
—
|
—
|
24,484
|
—
|
24,484
|
||||||||||||||||||
|
Foreign currency translation
|
—
|
—
|
—
|
351
|
—
|
351
|
||||||||||||||||||
|
Issuance of common stock in connection with employee stock plans
|
2,283
|
2
|
49,439
|
—
|
—
|
49,441
|
||||||||||||||||||
|
Stock-based compensation expense
|
—
|
—
|
105,809
|
—
|
—
|
105,809
|
||||||||||||||||||
|
Balance at December 31, 2023
|
144,341
|
$
|
144
|
$
|
2,215,098
|
$
|
(32,645
|
)
|
$
|
(1,795,911
|
)
|
$
|
386,686
|
|||||||||||
|
Year Ended December 31,
|
||||||||||||
|
2023
|
2022
|
2021
|
||||||||||
|
Operating activities:
|
||||||||||||
|
Net loss
|
$
|
(366,286
|
)
|
$
|
(269,722
|
)
|
$
|
(28,597
|
)
|
|||
|
Adjustments to reconcile net loss to net cash provided by (used in) operating activities:
|
||||||||||||
|
Depreciation
|
10,292
|
14,328
|
15,487
|
|||||||||
|
Amortization of right-of-use operating lease assets
|
9,647
|
5,362
|
1,721
|
|||||||||
|
Amortization of other assets
|
2,559
|
2,415
|
2,352
|
|||||||||
|
Amortization of premium (discount) on investments, net
|
(28,885
|
)
|
7,389
|
17,776
|
||||||||
|
Amortization of debt issuance costs
|
6,330
|
5,373
|
4,958
|
|||||||||
|
Non-cash royalty revenue related to sale of royalties
|
(44,628
|
)
|
—
|
—
|
||||||||
|
Non-cash interest related to sale of future royalties
|
68,238
|
—
|
—
|
|||||||||
|
Stock-based compensation expense
|
105,809
|
100,264
|
120,678
|
|||||||||
|
Loss (gain) on early retirement of debt
|
(13,389
|
)
|
—
|
8,627
|
||||||||
|
Non-cash losses related to disposal of property, plant and equipment
|
16,649
|
531
|
—
|
|||||||||
|
Loss (gain) on sale of real estate assets
|
161
|
(150,135
|
)
|
—
|
||||||||
|
Loss (gain) on investments
|
1,589
|
224
|
(1,092
|
)
|
||||||||
|
Non-cash losses related to other assets
|
1,661
|
2,030
|
2,707
|
|||||||||
|
Changes in operating assets and liabilities:
|
||||||||||||
|
Contracts receivable
|
(72,059
|
)
|
36,358
|
14,308
|
||||||||
|
Inventories
|
(6,392
|
)
|
2,773
|
(2,841
|
)
|
|||||||
|
Other current and long-term assets
|
(29,840
|
)
|
(24,682
|
)
|
(877
|
)
|
||||||
|
Accounts payable
|
8,119
|
1,094
|
(6,000
|
)
|
||||||||
|
Income taxes
|
(4,098
|
)
|
6,213
|
(280
|
)
|
|||||||
|
Accrued compensation
|
18,549
|
10,368
|
(26,918
|
)
|
||||||||
|
Accrued liabilities and other current liabilities
|
(5,506
|
)
|
46,695
|
(8,381
|
)
|
|||||||
|
Deferred contract revenue
|
13,967
|
(71,248
|
)
|
(82,829
|
)
|
|||||||
|
Net cash provided by (used in) operating activities
|
(307,513
|
)
|
(274,370
|
)
|
30,799
|
|||||||
|
Investing activities:
|
||||||||||||
|
Purchases of short-term investments
|
(1,770,814
|
)
|
(1,485,772
|
)
|
(1,124,193
|
)
|
||||||
|
Proceeds from sale of short-term investments
|
1,584,676
|
989,152
|
1,344,185
|
|||||||||
|
Purchases of property, plant and equipment
|
(23,805
|
)
|
(15,721
|
)
|
(11,955
|
)
|
||||||
|
Proceeds from sale of real estate assets
|
22
|
254,083
|
—
|
|||||||||
|
Acquisition of licenses and other assets, net
|
(4,206
|
)
|
(4,378
|
)
|
(5,946
|
)
|
||||||
|
Purchases of strategic investments
|
—
|
—
|
(7,185
|
)
|
||||||||
|
Net cash provided by (used in) investing activities
|
(214,127
|
)
|
(262,636
|
)
|
194,906
|
|||||||
|
Financing activities:
|
||||||||||||
|
Proceeds from equity, net
|
49,442
|
6,373
|
11,565
|
|||||||||
|
Payments of tax withholdings related to vesting of employee stock awards and exercise of employee stock options
|
—
|
(10,953
|
)
|
(16,725
|
)
|
|||||||
|
Proceeds from issuance of 1.75 percent convertible senior notes
|
575,000
|
—
|
—
|
|||||||||
|
1.75 percent
convertible senior notes issuance costs
|
(14,175
|
)
|
—
|
—
|
||||||||
|
Repurchase of $504.4
million principal amount of 0.125 percent convertible senior notes
|
(487,943
|
)
|
—
|
—
|
||||||||
|
Proceeds from sale of future royalties
|
500,000
|
—
|
—
|
|||||||||
|
Payments of transaction costs related to sale of future royalties
|
(10,434
|
)
|
(29
|
)
|
—
|
|||||||
|
Proceeds from real estate transaction
|
32,352
|
—
|
—
|
|||||||||
|
Proceeds from the issuance of 0 percent convertible senior notes
|
—
|
—
|
632,500
|
|||||||||
|
0 percent
convertible senior notes issuance costs
|
—
|
—
|
(15,609
|
)
|
||||||||
|
Repurchase of $247.9
million principal amount of 1 percent convertible senior notes
|
—
|
—
|
(256,963
|
)
|
||||||||
|
Repayment of remaining principal amount of 1 percent convertible senior notes at maturity
|
—
|
—
|
(61,967
|
)
|
||||||||
|
Proceeds from issuance of warrants
|
—
|
—
|
89,752
|
|||||||||
|
Purchase of note hedges
|
—
|
—
|
(136,620
|
)
|
||||||||
|
Principal payments on debt
|
(160
|
)
|
(50,686
|
)
|
—
|
|||||||
|
Net cash provided by (used in) financing activities
|
644,082
|
(55,295
|
)
|
245,933
|
||||||||
|
Effects of exchange rates on cash
|
352
|
(418
|
)
|
(111
|
)
|
|||||||
|
Net increase (decrease) in cash and cash equivalents
|
122,794
|
(592,719
|
)
|
471,527
|
||||||||
|
Cash and cash equivalents at beginning of year
|
276,472
|
869,191
|
397,664
|
|||||||||
|
Cash and cash equivalents at end of year
|
$
|
399,266
|
$
|
276,472
|
$
|
869,191
|
||||||
|
Supplemental disclosures of cash flow information:
|
||||||||||||
|
Interest paid
|
$
|
6,512
|
$
|
2,898
|
$
|
4,778
|
||||||
|
Income taxes paid
|
$
|
48,334
|
$
|
5,010
|
$
|
38
|
||||||
|
Supplemental disclosures of non-cash investing and financing activities:
|
||||||||||||
|
Right-of-use assets obtained in exchange for lease liabilities
|
$
|
—
|
$
|
168,931
|
$
|
6,641
|
||||||
|
Amounts accrued for capital and patent expenditures
|
$
|
172
|
$
|
4,767
|
$
|
705
|
||||||
| 1. |
Identify the contract
|
| ● |
We and our partner approved the contract and we are both committed to perform our obligations;
|
| ● |
We have identified our rights, our partner’s rights and the payment terms;
|
| ● |
We have concluded that the contract has commercial substance, meaning that the risk, timing, or amount of our future cash flows is expected to change as a result of the
contract; and
|
| ● |
We believe collectability of the consideration is probable.
|
| 2. |
Identify the performance obligations
|
| 3. |
Determine the transaction price
|
| 4. |
Allocate the transaction price
|
| ● |
Estimated future product sales;
|
| ● |
Estimated royalties we may receive from future product sales;
|
| ● |
Estimated contractual milestone payments we may receive;
|
| ● |
Estimated expenses we may incur;
|
| ● |
Estimated income taxes; and
|
| ● |
A discount rate.
|
| ● |
The estimated number of internal hours we will spend performing these services;
|
| ● |
The estimated cost of work we will perform;
|
| ● |
The estimated cost of work that we will contract with third parties to perform; and
|
| ● |
The estimated cost of API we will use.
|
| 5. |
Recognize revenue
|
| 1) |
If the additional goods and/or services are distinct from the other performance obligations in the original agreement; and
|
| 2) |
If the goods and/or services are sold at a stand-alone selling price.
|
| ● |
Whether the agreements were negotiated together with a single objective;
|
| ● |
Whether the amount of consideration in one contract depends on the price or performance of the other agreement; or
|
| ● |
Whether the goods and/or services promised under the agreements are a single performance obligation.
|
| ● |
0 percent convertible senior notes, or 0% Notes;
|
| ● |
Note hedges related to the 0% Notes;
|
| ● |
0.125 percent convertible senior notes, or 0.125% Notes;
|
| ● |
Note hedges related to the 0.125% Notes;
|
| ● |
Dilutive stock options;
|
| ● |
Unvested restricted stock units, or RSUs;
|
| ● |
Unvested performance restricted stock units, or PRSUs; and
|
| ● |
Employee Stock Purchase Plan, or ESPP.
|
|
Estimated
|
|
|
Useful Lives
|
|
|
Computer software, laboratory, manufacturing and other equipment
|
3 to 10
|
|
Building, building improvements and building systems
|
15 to 40
|
|
Land improvements
|
20
|
|
Leasehold improvements
|
5 to 15
|
|
Furniture and fixtures
|
5 to 10
|
|
December 31,
|
||||||||
|
2023
|
2022
|
|||||||
|
Raw materials:
|
||||||||
|
Raw materials- clinical
|
$
|
20,985
|
$
|
17,061
|
||||
|
Raw materials- commercial
|
1,809
|
2,699
|
||||||
|
Total raw materials
|
22,794
|
19,760
|
||||||
|
Work in process
|
5,477
|
2,109
|
||||||
|
Finished goods
|
154
|
164
|
||||||
|
Total inventory
|
$
|
28,425
|
$
|
22,033
|
||||
|
December 31,
|
||||||||
|
2023
|
2022
|
|||||||
|
Computer software, laboratory, manufacturing and other equipment
|
$
|
79,885
|
$
|
74,351
|
||||
|
Building, building improvements and building systems
|
41,228
|
41,158
|
||||||
|
Leasehold improvements
|
28,276
|
28,357
|
||||||
|
Furniture and fixtures
|
9,844
|
9,575
|
||||||
|
159,233
|
153,441
|
|||||||
|
Less: Accumulated depreciation
|
(96,759
|
)
|
(87,716
|
)
|
||||
|
62,474
|
65,725
|
|||||||
|
Land
|
8,569
|
8,569
|
||||||
|
Total
|
$
|
71,043
|
$
|
74,294
|
||||
|
December 31,
|
||||||||
|
2023
|
2022
|
|||||||
|
Clinical expenses
|
$
|
105,967
|
$
|
116,460
|
||||
|
In-licensing expenses
|
7,454
|
7,945
|
||||||
|
Commercial expenses
|
4,875
|
3,498
|
||||||
|
Other miscellaneous expenses
|
29,598
|
12,198
|
||||||
|
Total accrued liabilities
|
$
|
147,894
|
$
|
140,101
|
||||
|
Year Ended December 31,
|
||||||||||||
|
2023
|
2022
|
2021
|
||||||||||
|
Revenue:
|
||||||||||||
|
Commercial revenue:
|
||||||||||||
|
SPINRAZA royalties
|
$
|
240,379
|
$
|
242,314
|
$
|
267,776
|
||||||
|
Other commercial revenue:
|
||||||||||||
|
TEGSEDI and WAYLIVRA revenue, net
|
34,913
|
30,051
|
55,500
|
|||||||||
|
Licensing and other royalty revenue
|
33,299
|
30,993
|
19,119
|
|||||||||
|
Total other commercial revenue
|
68,212
|
61,044
|
74,619
|
|||||||||
|
Total commercial revenue
|
308,591
|
303,358
|
342,395
|
|||||||||
|
Research and development revenue:
|
||||||||||||
|
Collaborative agreement revenue
|
352,657
|
207,222
|
468,061
|
|||||||||
|
WAINUA joint development revenue
|
126,399
|
76,787
|
—
|
|||||||||
|
Total research and development revenue
|
479,056
|
284,009
|
468,061
|
|||||||||
|
Total revenue
|
$
|
787,647
|
$
|
587,367
|
$
|
810,456
|
||||||
|
Year Ended December 31,
|
||||||||||||
|
2023
|
2022
|
2021
|
||||||||||
|
Revenue from our relationship with Biogen
|
$
|
350,146
|
$
|
366,696
|
$
|
428,784
|
||||||
|
Percentage of total revenue
|
44
|
%
|
62
|
%
|
53
|
%
|
||||||
|
Year Ended December 31,
|
||||||||||||
|
2023
|
2022
|
2021
|
||||||||||
|
Revenue from our relationship with AstraZeneca
|
$
|
202,236
|
$
|
79,160
|
$
|
254,591
|
||||||
|
Percentage of total revenue
|
26
|
%
|
13
|
%
|
31
|
%
|
||||||
|
Year Ended December 31,
|
||||||||||||
|
2023
|
2022
|
2021
|
||||||||||
|
Revenue from our relationship with GSK
|
$
|
15,000
|
$
|
—
|
$
|
—
|
||||||
|
Percentage of total revenue
|
2
|
%
|
0
|
%
|
0
|
%
|
||||||
| ● |
R&D services for pelacarsen;
|
| ● |
R&D services for olezarsen;
|
| ● |
API for pelacarsen; and
|
| ● |
API for olezarsen.
|
| ● |
$75 million from the upfront payment;
|
| ● |
$28.4 million for the premium paid by Novartis for its purchase
of our common stock at a premium in 2017; and
|
| ● |
$5.0 million for the potential premium Novartis would have paid
if they purchased our common stock in the future.
|
| ● |
$64.0 million for the R&D services for pelacarsen;
|
| ● |
$40.1 million for the R&D services for olezarsen;
|
| ● |
$1.5
million for the delivery of pelacarsen API; and
|
| ● |
$2.8
million for the delivery of olezarsen API.
|
|
Year Ended December 31,
|
||||||||||||
|
2023
|
2022
|
2021
|
||||||||||
|
Revenue from our relationship with Novartis
|
$
|
30,194
|
$
|
237
|
$
|
25,526
|
||||||
|
Percentage of total revenue
|
4
|
%
|
Less than 1%
|
3
|
%
|
|||||||
| ● |
$45 million for the R&D services for the investigational
medicine for AD; and
|
| ● |
$15 million for the R&D services for the investigational medicine for HD.
|
|
Year Ended December 31,
|
||||||||||||
|
2023
|
2022
|
2021
|
||||||||||
|
Revenue from our relationship with Roche
|
$
|
48,838
|
$
|
67,202
|
$
|
17,241
|
||||||
|
Percentage of total revenue
|
6
|
%
|
11
|
%
|
2
|
%
|
||||||
| ● |
$56 million for the license of donidalorsen; and
|
| ● |
$9 million for the R&D services for donidalorsen.
|
|
Year Ended December 31,
|
||||
|
2023
|
||||
|
Revenue from our relationship with Otsuka
|
$
|
56,480
|
||
|
Percentage of total revenue
|
7
|
%
|
||
|
Year Ended December 31,
|
||||||||||||
|
2023
|
2022
|
2021
|
||||||||||
|
TEGSEDI revenue from our distribution agreement with Sobi in North America
|
$
|
2,646
|
$
|
4,004
|
$
|
7,443
|
||||||
|
Percentage of total revenue
|
Less than 1%
|
1
|
%
|
1
|
%
|
|||||||
|
Year Ended December 31,
|
||||||||||||
|
2023
|
2022
|
2021
|
||||||||||
|
Revenue from our relationship with Alnylam
|
$
|
28,426
|
$
|
21,389
|
$
|
—
|
||||||
|
Percentage of total revenue
|
4
|
%
|
4
|
%
|
0
|
%
|
||||||
|
One year
or less
|
68
|
%
|
||
|
After one year
but within two years
|
24
|
%
|
||
|
After two years
but within three and a half years
|
8
|
%
|
||
|
Total
|
100
|
%
|
|
Amortized
|
Gross Unrealized
|
Estimated
|
||||||||||||||
|
December 31, 2023
|
Cost
|
Gains
|
Losses
|
Fair Value
|
||||||||||||
|
Available-for-sale securities:
|
||||||||||||||||
|
Corporate debt securities (1)
|
$
|
559,967
|
$
|
157
|
$
|
(2,625
|
)
|
$
|
557,499
|
|||||||
|
Debt securities issued by U.S. government agencies
|
224,711
|
64
|
(611
|
)
|
224,164
|
|||||||||||
|
Debt securities issued by the U.S. Treasury (1)
|
513,784
|
152
|
(1,889
|
)
|
512,047
|
|||||||||||
|
Debt securities issued by states of the U.S. and political subdivisions of the states
|
17,757
|
42
|
(113
|
)
|
17,686
|
|||||||||||
|
Total securities with a maturity of one year or less
|
1,316,219
|
415
|
(5,238
|
)
|
1,311,396
|
|||||||||||
|
Corporate debt securities
|
243,151
|
1,270
|
(692
|
)
|
243,729
|
|||||||||||
|
Debt securities issued by U.S. government agencies
|
110,138
|
547
|
(21
|
)
|
110,664
|
|||||||||||
|
Debt securities issued by the U.S. Treasury
|
294,873
|
1,239
|
(480
|
)
|
295,632
|
|||||||||||
|
Debt securities issued by states of the U.S. and political subdivisions of the states
|
3,466
|
7
|
(4
|
)
|
3,469
|
|||||||||||
|
Total securities with a maturity of more than one year
|
651,628
|
3,063
|
(1,197
|
)
|
653,494
|
|||||||||||
|
Total available-for-sale securities
|
$
|
1,967,847
|
$
|
3,478
|
$
|
(6,435
|
)
|
$
|
1,964,890
|
|||||||
|
Equity securities:
|
||||||||||||||||
|
Publicly traded equity securities included in other current assets (2)
|
$
|
11,897
|
$
|
236
|
$
|
(5,832
|
)
|
$
|
6,301
|
|||||||
|
Privately held securities included in deposits and other assets (3)
|
23,115
|
25,001
|
(5,125
|
)
|
42,991
|
|||||||||||
|
Total equity securities
|
$
|
35,012
|
$
|
25,237
|
$
|
(10,957
|
)
|
$
|
49,292
|
|||||||
|
Total available-for-sale and equity securities
|
$
|
2,002,859
|
$
|
28,715
|
$
|
(17,392
|
)
|
$
|
2,014,182
|
|||||||
|
Amortized
|
Gross Unrealized
|
Estimated
|
||||||||||||||
|
December 31, 2022
|
Cost
|
Gains
|
Losses
|
Fair Value
|
||||||||||||
|
Available-for-sale securities:
|
||||||||||||||||
|
Corporate debt securities (1)
|
$
|
513,790
|
$
|
23
|
$
|
(4,365
|
)
|
$
|
509,448
|
|||||||
|
Debt securities issued by U.S. government agencies
|
133,585
|
—
|
(1,829
|
)
|
131,756
|
|||||||||||
|
Debt securities issued by the U.S. Treasury (1)
|
512,655
|
23
|
(5,124
|
)
|
507,554
|
|||||||||||
|
Debt securities issued by states of the U.S. and political subdivisions of the states
|
57,484
|
18
|
(686
|
)
|
56,816
|
|||||||||||
|
Other municipal debt securities
|
6,008
|
—
|
(14
|
)
|
5,994
|
|||||||||||
|
Total securities with a maturity of one year or less
|
1,223,522
|
64
|
(12,018
|
)
|
1,211,568
|
|||||||||||
|
Corporate debt securities
|
227,631
|
14
|
(10,143
|
)
|
217,502
|
|||||||||||
|
Debt securities issued by U.S. government agencies
|
34,339
|
—
|
(1,040
|
)
|
33,299
|
|||||||||||
|
Debt securities issued by the U.S. Treasury
|
245,030
|
—
|
(4,109
|
)
|
240,921
|
|||||||||||
|
Debt securities issued by states of the U.S. and political subdivisions of the states
|
18,314
|
116
|
(329
|
)
|
18,101
|
|||||||||||
|
Total securities with a maturity of more than one year
|
525,314
|
130
|
(15,621
|
)
|
509,823
|
|||||||||||
|
Total available-for-sale securities
|
$
|
1,748,836
|
$
|
194
|
$
|
(27,639
|
)
|
$
|
1,721,391
|
|||||||
|
Equity securities:
|
||||||||||||||||
|
Publicly traded equity securities included in other current assets (2)
|
$
|
11,897
|
$
|
—
|
$
|
(1,358
|
)
|
$
|
10,539
|
|||||||
|
Privately held equity securities included in deposits and other assets (3)
|
23,115
|
17,257
|
—
|
40,372
|
||||||||||||
|
Total equity securities
|
$
|
35,012
|
$
|
17,257
|
$
|
(1,358
|
)
|
$
|
50,911
|
|||||||
|
Total available-for-sale and equity securities
|
$
|
1,783,848
|
$
|
17,451
|
$
|
(28,997
|
)
|
$
|
1,772,302
|
|||||||
| (1) |
Includes investments classified as cash equivalents in our consolidated
balance sheets.
|
| (2) |
Our publicly traded equity securities are included in other current
assets. We recognize publicly traded equity securities at fair value. In the year ended December 31, 2023, we recorded a $4.2 million net unrealized loss in our consolidated statements of
operations related to changes in the fair value of our investments in publicly traded companies.
|
| (3) |
Our privately held equity securities are included in deposits and other assets. We recognize our privately held equity securities at cost minus impairments, plus or minus changes resulting from observable price changes in
orderly transactions for the identical or similar investment of the same issuer, which are Level 3 inputs. In the year ended December 31, 2023, we recorded a $2.6 million net unrealized gain in our consolidated statements of operations related to changes in the fair value of our investments in privately held companies.
|
|
Less than 12 Months of
Temporary Impairment
|
More than 12 Months of
Temporary Impairment
|
Total Temporary
Impairment
|
||||||||||||||||||||||||||
|
Number of
Investments
|
Estimated
Fair Value
|
Unrealized
Losses
|
Estimated
Fair Value
|
Unrealized
Losses
|
Estimated
Fair Value
|
Unrealized
Losses
|
||||||||||||||||||||||
|
Corporate debt securities
|
297
|
$
|
323,708
|
$
|
(553
|
)
|
$
|
178,183
|
$
|
(2,764
|
)
|
$
|
501,891
|
$
|
(3,317
|
)
|
||||||||||||
|
Debt securities issued by U.S. government agencies
|
63
|
199,372
|
(246
|
)
|
14,777
|
(386
|
)
|
214,149
|
(632
|
)
|
||||||||||||||||||
|
Debt securities issued by the U.S. Treasury
|
34
|
325,966
|
(1,031
|
)
|
131,000
|
(1,338
|
)
|
456,966
|
(2,369
|
)
|
||||||||||||||||||
|
Debt securities issued by states of the U.S. and political subdivisions of the states
|
61
|
8,352
|
(17
|
)
|
7,888
|
(100
|
)
|
16,240
|
(117
|
)
|
||||||||||||||||||
|
Total temporarily impaired securities
|
455
|
$
|
857,398
|
$
|
(1,847
|
)
|
$
|
331,848
|
$
|
(4,588
|
)
|
$
|
1,189,246
|
$
|
(6,435
|
)
|
||||||||||||
|
At
December 31, 2023
|
Quoted Prices in
Active Markets
(Level 1)
|
Significant Other
Observable Inputs
(Level 2)
|
||||||||||
|
Cash equivalents (1)
|
$
|
185,424
|
$
|
185,424
|
$
|
—
|
||||||
|
Corporate debt securities (2)
|
801,228
|
—
|
801,228
|
|||||||||
|
Debt securities issued by U.S. government agencies (3)
|
334,828
|
—
|
334,828
|
|||||||||
|
Debt securities issued by the U.S. Treasury (3)
|
807,679
|
807,679
|
—
|
|||||||||
|
Debt securities issued by states of the U.S. and political subdivisions of the states (3)
|
21,155
|
—
|
21,155
|
|||||||||
|
Publicly traded equity securities included in other current assets (5)
|
6,301
|
6,301
|
—
|
|||||||||
|
Total
|
$
|
2,156,615
|
$
|
999,404
|
$
|
1,157,211
|
||||||
|
At
December 31, 2022
|
Quoted Prices in
Active Markets
(Level 1)
|
Significant Other
Observable Inputs
(Level 2)
|
||||||||||
|
Cash equivalents (1)
|
$
|
211,655
|
$
|
211,655
|
$
|
—
|
||||||
|
Corporate debt securities (4)
|
726,950
|
—
|
726,950
|
|||||||||
|
Debt securities issued by U.S. government agencies (3)
|
165,055
|
—
|
165,055
|
|||||||||
|
Debt securities issued by the U.S. Treasury (3)
|
748,475
|
748,475
|
—
|
|||||||||
|
Debt securities issued by states of the U.S. and political subdivisions of the states (3)
|
74,917
|
—
|
74,917
|
|||||||||
|
Other municipal debt securities (3)
|
5,994
|
—
|
5,994
|
|||||||||
|
Publicly traded equity securities included in other current assets (5)
|
10,539
|
10,539
|
—
|
|||||||||
|
Total
|
$
|
1,943,585
|
$
|
970,669
|
$
|
972,916
|
||||||
| (1) |
Included in cash and cash equivalents in our consolidated balance
sheets.
|
| (2) |
$33.0 million was included in cash and cash equivalents, with the difference included in short-term investments in our consolidated balance sheets.
|
| (3) |
Included in short-term investments in our consolidated balance sheets.
|
| (4) |
$11.0 million was included in cash and cash equivalents, with the difference included in short-term investments in our consolidated balance sheets.
|
| (5) |
Included in other current assets in our consolidated balance sheets.
|
|
December 31,
|
||||||||
|
2023
|
2022
|
|||||||
|
1.75%
convertible senior notes
|
$
|
562,285
|
$
|
—
|
||||
|
0%
convertible senior notes
|
625,380
|
622,242
|
||||||
|
0.125%
convertible senior notes
|
—
|
544,504
|
||||||
|
Liability related to sale of future royalties
|
513,736
|
—
|
||||||
|
Lease liabilities
|
178,969
|
186,156
|
||||||
|
Mortgage debt
|
8,859
|
8,998
|
||||||
|
Other obligations
|
33,714
|
7,295
|
||||||
|
Total
|
$
|
1,922,943
|
$
|
1,369,195
|
||||
|
Less: current portion
|
(8,831
|
)
|
(7,535
|
)
|
||||
|
Total Long-Term Obligations
|
$
|
1,914,112
|
$
|
1,361,660
|
||||
|
1.75% Notes
|
||||
|
Outstanding principal balance
|
$
|
575.0
|
||
|
Unamortized debt issuance costs
|
$
|
12.7
|
||
|
Maturity date
|
June 2028
|
|||
|
Interest rate
|
1.75
|
%
|
||
|
Effective interest rate
|
2.3
|
%
|
||
|
Conversion price per share
|
$
|
53.73
|
||
|
Total shares of common stock subject to conversion
|
10.7
|
|||
|
0% Notes
|
||||
|
Outstanding principal balance
|
$
|
632.5
|
||
|
Unamortized debt issuance costs
|
$
|
7.2
|
||
|
Maturity date
|
April 2026
|
|||
|
Interest rate
|
0
|
%
|
||
|
Effective interest rate
|
0.5
|
%
|
||
|
Conversion price per share
|
$
|
57.84
|
||
|
Effective conversion price per share with call spread
|
$
|
76.39
|
||
|
Total shares of common stock subject to conversion
|
10.9
|
|||
|
0.125% Notes
|
||||
|
Outstanding principal balance
|
$
|
44.5
|
||
|
Unamortized debt issuance costs
|
$
|
0.2
|
||
|
Maturity date
|
December 2024
|
|||
|
Interest rate
|
0.125
|
%
|
||
|
Effective interest rate
|
0.5
|
%
|
||
|
Conversion price per share
|
$
|
83.28
|
||
|
Effective conversion price per share with call spread
|
$
|
123.38
|
||
|
Total shares of common stock subject to conversion, excluding shares related to 0.125% Notes we have repurchased and are currently holding in treasury
|
0.5
|
|||
|
2024
|
$
|
55,298
|
||
|
2025
|
10,657
|
|||
|
2026
|
643,157
|
|||
|
2027
|
10,509
|
|||
|
2028
|
580,277
|
|||
|
Thereafter
|
8,462
|
|||
|
Total debt and mortgage maturities
|
$
|
1,308,360
|
||
|
Less: Current portion included in other current liabilities
|
(157
|
)
|
||
|
Less: Fixed and determinable interest
|
(47,138
|
)
|
||
|
Less: Debt issuance costs
|
(20,061
|
)
|
||
|
Total debt
|
$
|
1,241,004
|
|
At December 31, 2023
|
||||
|
Right-of-use operating lease assets
|
$
|
171.9
|
||
|
Operating lease liabilities
|
$
|
179.0
|
||
|
Weighted average remaining lease term
|
13.0 years
|
|||
|
Weighted average discount rate
|
6.9
|
%
|
||
|
Operating Leases
|
||||
|
Year ending December 31,
|
||||
|
2024
|
$
|
20,398
|
||
|
2025
|
20,645
|
|||
|
2026
|
20,781
|
|||
|
2027
|
20,800
|
|||
|
2028
|
20,774
|
|||
|
Thereafter
|
176,138
|
|||
|
Total minimum lease payments
|
279,536
|
|||
|
Less: Imputed interest
|
(100,567
|
)
|
||
|
Less: Current portion (included in other current liabilities)
|
(8,094
|
)
|
||
|
Total long-term lease liabilities
|
$
|
170,875
|
||
|
Proceeds from sale of future royalties
|
$
|
500,000
|
||
|
Royalty payments to Royalty Pharma
|
(44,628
|
)
|
||
|
Interest expense related to sale of future royalties
|
68,238
|
|||
|
Liability related to sale of future royalties as of December 31, 2023
|
523,610
|
|||
|
Issuance costs related to sale of future royalties
|
(10,434
|
)
|
||
|
Amortization of issuance costs related to sale of future royalties as of December 31, 2023
|
560
|
|||
|
Net liability related to sale of future royalties as of December 31, 2023
|
$
|
513,736
|
| ● |
arrange for assumption, continuation, or substitution of a stock award by a surviving or acquiring entity (or its parent company);
|
| ● |
arrange for the assignment of any reacquisition or repurchase rights applicable to any shares of our common stock issued pursuant to a stock award to the surviving or
acquiring corporation (or its parent company);
|
| ● |
accelerate the vesting and exercisability of a stock award followed by the termination of the stock award;
|
| ● |
arrange for the lapse of any reacquisition or repurchase rights applicable to any shares of our common stock issued pursuant to a stock award;
|
| ● |
cancel or arrange for the cancellation of a stock award, to the extent not vested or not exercised prior to the effective date of the corporate transaction, in exchange
for cash consideration, if any, as the Board, in its sole discretion, may consider appropriate; and
|
| ● |
arrange for the surrender of a stock award in exchange for a payment equal to the excess of (a) the value of the property the holder of the stock award would have
received upon the exercise of the stock award, over (b) any exercise price payable by such holder in connection with such exercise.
|
| ● |
An increase to the total number of shares reserved for issuance under the plan from 2.0 million to 2.8 million shares;
|
| ● |
A reduction to the amount of the automatic awards under the plan;
|
| ● |
A revision to the vesting schedule of new awards granted; and
|
| ● |
An extension of the term of the plan.
|
|
Number
of Shares
|
Weighted
Average Exercise
Price Per Share
|
Average
Remaining
Contractual Term
(Years)
|
Aggregate
Intrinsic
Value
|
|||||||||||||
|
Outstanding at December 31, 2022
|
14,970
|
$
|
50.57
|
|||||||||||||
|
Granted
|
2,407
|
$
|
38.80
|
|||||||||||||
|
Exercised
|
(1,444
|
)
|
$
|
45.06
|
||||||||||||
|
Cancelled/forfeited/expired
|
(1,842
|
)
|
$
|
55.90
|
||||||||||||
|
Outstanding at December 31, 2023
|
14,091
|
$
|
48.43
|
4.74
|
$
|
78,542
|
||||||||||
|
Exercisable at December 31, 2023
|
9,703
|
$
|
52.24
|
3.20
|
$
|
28,349
|
||||||||||
|
Number
of Shares
|
Weighted Average
Grant Date Fair
Value Per Share
|
|||||||
|
Non-vested at December 31, 2022
|
2,766
|
$
|
48.30
|
|||||
|
Granted
|
1,707
|
$
|
40.51
|
|||||
|
Vested
|
(1,055
|
)
|
$
|
51.64
|
||||
|
Cancelled/forfeited
|
(179
|
)
|
$
|
42.90
|
||||
|
Non-vested at December 31, 2023
|
3,239
|
$
|
43.40
|
|||||
|
Number
of Shares
|
Weighted Average
Grant Date Fair
Value Per Share
|
|||||||
|
Non-vested at December 31, 2022
|
143
|
$
|
52.59
|
|||||
|
Granted
|
158
|
$
|
57.43
|
|||||
|
Vested
|
(75
|
)
|
$
|
52.43
|
||||
|
Non-vested at December 31, 2023
|
226
|
$
|
56.04
|
|||||
|
Year Ended December 31,
|
||||||||||||
|
2023
|
2022
|
2021
|
||||||||||
|
Cost of sales
|
$
|
499
|
$
|
533
|
$
|
456
|
||||||
|
Research, development and patent
|
77,826
|
73,704
|
87,522
|
|||||||||
|
Selling, general and administrative
|
27,484
|
26,027
|
32,700
|
|||||||||
|
Total
|
$
|
105,809
|
$
|
100,264
|
$
|
120,678
|
||||||
|
Year Ended December 31,
|
||||||||||||
|
2023
|
2022
|
2021
|
||||||||||
|
Risk-free interest rate
|
3.8
|
%
|
2.1
|
%
|
0.6
|
%
|
||||||
|
Dividend yield
|
0.0
|
%
|
0.0
|
%
|
0.0
|
%
|
||||||
|
Volatility
|
46.8
|
%
|
54.5
|
%
|
54.0
|
%
|
||||||
|
Expected life
|
6.3 years
|
6.3 years
|
4.9 years
|
|||||||||
|
Year Ended December 31,
|
||||||||||||
|
2023
|
2022
|
2021
|
||||||||||
|
Risk-free interest rate
|
3.8
|
%
|
2.9
|
%
|
1.2
|
%
|
||||||
|
Dividend yield
|
0.0
|
%
|
0.0
|
%
|
0.0
|
%
|
||||||
|
Volatility
|
52.7
|
%
|
56.2
|
%
|
55.9
|
%
|
||||||
|
Expected life
|
7.7 years
|
7.4 years
|
7.3 years
|
|||||||||
|
Year Ended December 31,
|
||||||||||||
|
2023
|
2022
|
2021
|
||||||||||
|
Risk-free interest rate
|
5.3
|
%
|
1.2
|
%
|
0.1
|
%
|
||||||
|
Dividend yield
|
0.0
|
%
|
0.0
|
%
|
0.0
|
%
|
||||||
|
Volatility
|
36.0
|
%
|
50.1
|
%
|
42.4
|
%
|
||||||
|
Expected life
|
6 months
|
6 months
|
6 months
|
|||||||||
|
Year Ended December 31,
|
||||||||||||
|
2023
|
2022
|
2021
|
||||||||||
|
United States
|
$
|
(334,707
|
)
|
$
|
(258,493
|
)
|
$
|
(29,966
|
)
|
|||
|
Foreign
|
742
|
508
|
818
|
|||||||||
|
Loss before income taxes
|
$
|
(333,965
|
)
|
$
|
(257,985
|
)
|
$
|
(29,148
|
)
|
|||
|
Year Ended December 31,
|
||||||||||||
|
2023
|
2022
|
2021
|
||||||||||
|
Current:
|
||||||||||||
|
Federal
|
$
|
35,861
|
$
|
10,522
|
$
|
(200
|
)
|
|||||
|
State
|
(3,687
|
)
|
1,129
|
(690
|
)
|
|||||||
|
Foreign
|
147
|
86
|
339
|
|||||||||
|
Total current income tax expense (benefit)
|
32,321
|
11,737
|
(551
|
)
|
||||||||
|
Deferred:
|
||||||||||||
|
Federal
|
—
|
—
|
—
|
|||||||||
|
State
|
—
|
—
|
—
|
|||||||||
|
Total deferred income tax benefit
|
—
|
—
|
—
|
|||||||||
|
Total income tax expense (benefit)
|
$
|
32,321
|
$
|
11,737
|
$
|
(551
|
)
|
|||||
|
Year Ended December 31,
|
||||||||||||||||||||||||
|
2023
|
2022
|
2021
|
||||||||||||||||||||||
|
Pre-tax loss
|
$
|
(333,965
|
)
|
$
|
(257,985
|
)
|
$
|
(29,148
|
)
|
|||||||||||||||
|
Statutory rate
|
(70,133
|
)
|
21.0
|
%
|
(54,177
|
)
|
21.0
|
%
|
(6,121
|
)
|
21.0
|
%
|
||||||||||||
|
State income tax net of federal benefit
|
(22,597
|
)
|
6.8
|
%
|
(13,622
|
)
|
5.3
|
%
|
4,278
|
(14.7
|
)%
|
|||||||||||||
|
Foreign
|
(22
|
)
|
0.0
|
%
|
(49
|
)
|
0.0
|
%
|
143
|
(0.5
|
)%
|
|||||||||||||
|
Net change in valuation allowance
|
175,388
|
(52.5
|
)%
|
104,951
|
(40.7
|
)%
|
2,885
|
(9.9
|
)%
|
|||||||||||||||
|
Loss on debt transactions
|
—
|
—
|
—
|
—
|
262
|
(0.9
|
)%
|
|||||||||||||||||
|
Tax credits
|
(67,131
|
)
|
20.1
|
%
|
(39,729
|
)
|
15.4
|
%
|
(23,198
|
)
|
79.6
|
%
|
||||||||||||
|
Deferred tax true-up
|
4
|
0.0
|
%
|
(20
|
)
|
0.0
|
%
|
(24
|
)
|
0.1
|
%
|
|||||||||||||
|
Tax rate change
|
1,023
|
(0.3
|
)%
|
(3,091
|
)
|
1.2
|
%
|
12,838
|
(44.0
|
)%
|
||||||||||||||
|
Non-deductible compensation
|
3,814
|
(1.1
|
)%
|
3,023
|
(1.2
|
)%
|
5,085
|
(17.4
|
)%
|
|||||||||||||||
|
Other non-deductible items
|
327
|
(0.1
|
)%
|
57
|
0.0
|
%
|
84
|
(0.3
|
)%
|
|||||||||||||||
|
Foreign-derived intangible income benefit
|
(7,493
|
)
|
2.2
|
%
|
—
|
—
|
—
|
—
|
||||||||||||||||
|
Stock-based compensation
|
19,546
|
(5.9
|
)%
|
14,030
|
(5.4
|
)%
|
4,720
|
(16.2
|
)%
|
|||||||||||||||
|
Other
|
(405
|
)
|
0.1
|
%
|
364
|
(0.1
|
)%
|
(1,503
|
)
|
5.1
|
%
|
|||||||||||||
|
Effective rate
|
$
|
32,321
|
(9.7
|
)%
|
$
|
11,737
|
(4.5
|
)%
|
$
|
(551
|
)
|
1.9
|
%
|
|||||||||||
|
Year Ended December 31,
|
||||||||
|
2023
|
2022
|
|||||||
|
Deferred Tax Assets:
|
||||||||
|
Net operating loss carryovers
|
$
|
77,964
|
$
|
87,802
|
||||
|
Tax credits
|
239,962
|
277,436
|
||||||
|
Deferred revenue
|
71,683
|
85,700
|
||||||
|
Stock-based compensation
|
77,468
|
86,983
|
||||||
|
Intangible and capital assets
|
104,380
|
104,649
|
||||||
|
Convertible debt
|
16,849
|
34,384
|
||||||
|
Capitalized research and development expenses
|
238,738
|
119,635
|
||||||
|
Long-term lease liabilities
|
43,718
|
45,612
|
||||||
|
Sale of future royalties
|
144,608
|
—
|
||||||
|
Other
|
10,343
|
15,813
|
||||||
|
Total deferred tax assets
|
$
|
1,025,713
|
$
|
858,014
|
||||
|
Deferred Tax Liabilities:
|
||||||||
|
Fixed assets
|
(4,166
|
)
|
(4,475
|
)
|
||||
|
Right-of-use assets
|
(42,007
|
)
|
(44,504
|
)
|
||||
|
Other
|
(1,910
|
)
|
(313
|
)
|
||||
|
Net deferred tax asset
|
$
|
977,630
|
$
|
808,722
|
||||
|
Valuation allowance
|
(977,630
|
)
|
(808,722
|
)
|
||||
|
Total net deferred tax assets and liabilities
|
$
|
—
|
$
|
—
|
||||
|
Year Ended December 31,
|
||||||||||||
|
2023
|
2022
|
2021
|
||||||||||
|
Beginning balance of unrecognized tax benefits
|
$
|
56,567
|
$
|
55,085
|
$
|
54,163
|
||||||
|
Decrease for lapse of statute of limitations
|
(14,993
|
)
|
—
|
—
|
||||||||
|
Decrease for prior period tax positions
|
(737
|
)
|
(267
|
)
|
(695
|
)
|
||||||
|
Increase for prior period tax positions
|
429
|
259
|
263
|
|||||||||
|
Increase for current period tax positions
|
2,032
|
1,490
|
1,354
|
|||||||||
|
Ending balance of unrecognized tax benefits
|
$
|
43,298
|
$
|
56,567
|
$
|
55,085
|
||||||
|
Three Months Ended December 31,
|
2023
|
2022
|
||||||
|
Revenue (1)
|
$
|
324,505
|
$
|
151,890
|
||||
|
Operating expenses (2)
|
$
|
330,627
|
$
|
359,909
|
||||
|
Loss from operations
|
$
|
(6,122
|
)
|
$
|
(208,019
|
)
|
||
|
Net loss (3)
|
$
|
(9,263
|
)
|
$
|
(52,430
|
)
|
||
|
Basic net loss per share (4) (5)
|
$
|
(0.06
|
)
|
$
|
(0.37
|
)
|
||
|
Diluted net loss per share (4) (6)
|
$
|
(0.06
|
)
|
$
|
(0.37
|
)
|
||
| (1) |
Revenue was higher in the three months ended December 31, 2023
compared to the same period in 2022 primarily due to the $50 million milestone payment we earned from AstraZeneca when the FDA approved
WAINUA for ATTRv-PN in the U.S., $36 million payment we earned when AstraZeneca licensed ION826 and revenue we recognized in the fourth
quarter of 2023 from the upfront payments we received from our new collaborations with Otsuka, Roche and Novartis.
|
| (2) |
Operating expenses were lower in the three months ended December 31,
2023 compared to the same period in 2022 primarily due to the $80 million upfront payment we made for our collaboration with Metagenomi
in the fourth quarter of 2022.
|
| (3) |
Our net loss for the three months ended December 31, 2022 includes
the $150.1 million gain we recognized from the sale and leaseback transaction for our headquarters in Carlsbad, California.
|
| (4) |
We compute net loss per share independently for each quarter during the year.
|
| (5) |
As discussed in Note 1, Organization and Significant Accounting Policies, we compute basic net loss per share by dividing the total net loss by our weighted-average number of common shares outstanding
during the period.
|
| (6) |
We incurred a net loss for the fourth quarter of 2023 and 2022. As a
result, we did not include dilutive common equivalent shares in the computation of diluted net loss per share because the effect would have been anti-dilutive.
|
|
|
IONIS PHARMACEUTICALS, INC.
|
|
|
|
|
|
|
|
By:
|
|
|
|
Name:
|
Brett Monia
|
|
|
Title:
|
Chief Executive Officer
|
|
|
AGENT
|
|
|
|
By:
|
|
|
|
Name:
|
|
|
|
Address:
|
|
|
LANDLORD:
|
||
|
LOTS 21 & 22 OWNER (DE) LLC,
|
||
|
a Delaware limited liability company
|
||
|
By:
|
/s/ Tycho Suter
|
|
|
Name:
|
Tycho Suter
|
|
|
Title:
|
Vice President
|
|
|
By:
|
/s/ Kristen Binck
|
|
|
Name:
|
Kristen Binck
|
|
|
Title:
|
Vice President
|
|
|
TENANT:
|
||
|
IONIS PHARMACEUTICALS, INC.,
|
||
|
a Delaware corporation
|
||
|
By:
|
/s/ Beth Hougen
|
|
|
Name:
|
Beth Hougen
|
|
|
Title:
|
CFO
|
|
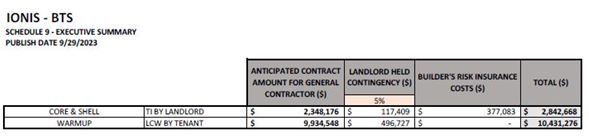
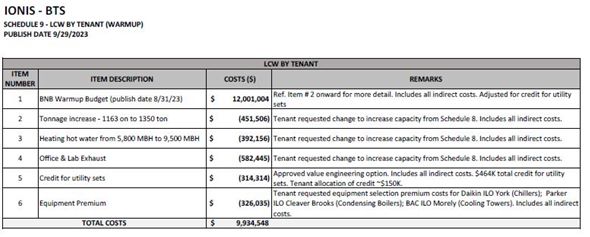
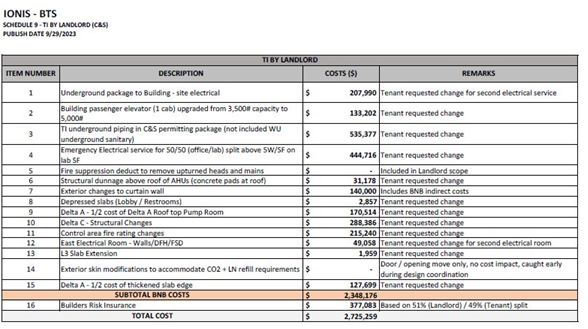
|
ARTICLE 1 OVERVIEW
|
1
|
||
|
1.1
|
Development and Commercialization
|
1
|
|
|
1.2
|
Governance
|
1
|
|
|
1.3
|
Purpose
|
1
|
|
|
ARTICLE 2 LICENSES
|
2
|
||
|
2.1
|
License Grants to Otsuka
|
2
|
|
|
2.2
|
License Grant to Ionis
|
2
|
|
|
2.3
|
Sublicensing and Subcontracting Terms
|
2
|
|
|
2.4
|
Collaboration Technology Enabling License
|
3
|
|
|
2.5
|
No Other Rights and Retained Rights; Negative Covenant
|
3 | |
|
2.6
|
Existing Third-Party IP Agreements
|
4
|
|
|
2.7
|
New Third-Party IP Agreements
|
5
|
|
|
2.8
|
Right of First Negotiation for Follow-On Products
|
6
|
|
|
ARTICLE 3 TECHNOLOGY TRANSFER
|
7
|
||
|
3.1
|
Initial Know-How Transfer
|
7
|
|
|
3.2
|
Technology Transfer Costs
|
7
|
|
|
ARTICLE 4 DEVELOPMENT
|
8
|
||
|
4.1
|
Development Diligence Obligations
|
8 | |
|
4.2
|
Development Plans
|
8
|
|
|
4.3
|
Otsuka Territory-Specific Development Plan
|
9
|
|
|
4.4
|
Development Costs
|
9
|
|
|
4.5
|
Development Reports
|
13
|
|
|
4.6
|
Development Records; Cooperation
|
13
|
|
|
4.7
|
Data Transfer
|
13
|
|
|
ARTICLE 5 REGULATORY AFFAIRS
|
13
|
||
|
5.1
|
Regulatory Responsible Party
|
13 | |
|
5.2
|
Regulatory Subcommittee
|
14
|
|
|
5.3
|
Correspondences with Regulatory Authorities
|
14
|
|
|
5.4
|
Regulatory Meetings
|
14
|
|
|
5.5
|
Regulatory Submissions
|
14
|
|
|
5.6
|
Cooperation
|
15
|
|
|
5.7
|
Cost of Regulatory Activities
|
15
|
|
|
5.8
|
No Harmful Actions
|
15
|
|
|
5.9
|
Right of Reference
|
15
|
|
|
5.10
|
Pharmacovigilance; Safety Information
|
16 | |
|
5.11
|
Pharmacovigilance Subcommittee
|
16
|
|
|
5.12
|
Ionis Internal Oligonucleotide Safety Database
|
16
|
|
|
5.13
|
Recall, Withdrawal, or Field Alerts
|
17
|
|
|
ARTICLE 6 COMMERCIALIZATION AND MEDICAL AFFAIRS
|
18
|
||
|
6.1
|
Commercialization Responsibilities for Licensed Product
|
18
|
|
|
6.2
|
Commercialization and Medical Affairs Reporting
|
18
|
|
|
6.3
|
Pricing
|
19 | |
|
6.4
|
Global Brand Strategic and Operating Plan
|
19 | |
|
6.5
|
Otsuka Territory Brand Strategic and Operating Plan
|
19 | |
|
6.6
|
Otsuka Commercialization Diligence Obligations
|
19 | |
|
6.7
|
Medical Affairs Plans.
|
19
|
|
|
6.8
|
Standards of Conduct; Compliance
|
20
|
|
|
6.9
|
Product Materials
|
20
|
|
|
6.10
|
Diversion
|
20
|
|
|
ARTICLE 7 MANUFACTURING
|
21
|
||
|
7.1
|
Responsibility
|
21
|
|
|
7.2
|
Supply and Quality Agreements; Manufacturing Costs
|
22
|
|
|
7.3
|
Audits and Inspections
|
23
|
|
|
7.4
|
Manufacturing Technology Transfer
|
23
|
|
|
ARTICLE 8 GOVERNANCE
|
24
|
||
|
8.1
|
Joint Steering Committee
|
24 | |
|
8.2
|
Additional Committees
|
26
|
|
|
8.3
|
Additional Participants
|
27
|
|
|
8.4
|
Decision-Making
|
27
|
|
|
8.5
|
Resolution of Committee Disputes
|
27
|
|
|
8.6
|
Day-to-Day Responsibilities
|
28
|
|
|
8.7
|
Alliance Managers
|
28
|
|
|
ARTICLE 9 PAYMENTS
|
29
|
||
|
9.1
|
Upfront Payment
|
29 | |
|
9.2
|
Milestone Payments
|
29
|
|
|
9.3
|
Royalties
|
30
|
|
|
9.4
|
Other Amounts Payable
|
32
|
|
|
9.5
|
Financial Records and Audits
|
33
|
|
|
9.6
|
No Refunds
|
33
|
|
|
9.7
|
Accounting Standards
|
33
|
|
|
9.8
|
Method of Payment; Exchange Rate
|
33
|
|
|
9.9
|
Blocked Payments
|
33
|
|
|
9.10
|
Taxes
|
34
|
|
|
9.11
|
Late Payments; Disputed Payments
|
34
|
|
|
ARTICLE 10 INTELLECTUAL PROPERTY
|
35
|
||
|
10.1
|
Inventions
|
35
|
|
|
10.2
|
Patent Prosecution
|
36
|
|
|
10.3
|
Enforcement Against Third Party Infringement or Misappropriation
|
38
|
|
|
10.4
|
Defense of Third-Party Patent Challenges
|
40
|
|
|
10.5
|
Third Party Infringement Claims
|
41
|
|
|
10.6
|
Patent Challenges of Third-Party Patent Rights
|
42
|
|
|
10.7
|
Patent Term Extensions
|
43
|
|
|
10.8
|
Unified Patent Court
|
43
|
|
|
10.9
|
Common Interest
|
43
|
|
|
10.10
|
Product Trademarks
|
43
|
|
|
ARTICLE 11 REPRESENTATIONS, WARRANTIES, AND COVENANTS
|
47
|
||
|
11.1
|
Mutual Representations and Warranties
|
47
|
|
|
11.2
|
Additional Ionis Representations and Warranties
|
48
|
|
|
11.3
|
Additional Otsuka Representations and Warranties
|
50
|
|
|
11.4
|
Additional Covenants
|
50
|
|
|
11.5
|
Disclaimer
|
51
|
|
|
11.6
|
Limitation of Liability
|
51
|
|
|
ARTICLE 12 CONFIDENTIALITY
|
52
|
||
|
12.1
|
Duty of Confidence
|
52
|
|
|
12.2
|
Confidential Information
|
52
|
|
|
12.3
|
Exemptions
|
53
|
|
|
12.4
|
Authorized Disclosures
|
53
|
|
|
12.5
|
Publications
|
54
|
|
|
12.6
|
Publicity; Use of Names
|
55
|
|
|
12.7
|
Acknowledgement
|
56 | |
|
ARTICLE 13 INDEMNIFICATION
|
57
|
||
|
13.1
|
Indemnification by Ionis
|
57
|
|
|
13.2
|
Indemnification by Otsuka
|
57 | |
|
13.3
|
Indemnification Procedure
|
57
|
|
|
13.4
|
Insurance
|
58 | |
|
ARTICLE 14 TERM AND TERMINATION
|
58 | ||
|
14.1
|
Term
|
58
|
|
|
14.2
|
Termination for Material Breach
|
58
|
|
|
14.3
|
Termination by Otsuka for Convenience
|
58
|
|
|
14.4
|
Discontinuation of Development and Commercialization
|
59
|
|
|
14.5
|
Termination For Patent Challenge
|
59
|
|
|
14.6
|
Termination for Insolvency
|
59
|
|
|
14.7
|
Rights in Bankruptcy
|
60
|
|
|
14.8
|
Full Force and Effect During Notice Period
|
60
|
|
|
14.9
|
Effects of Termination
|
60
|
|
|
14.10
|
Survival; Accrued Rights
|
65
|
|
|
ARTICLE 15 DISPUTE RESOLUTION; GOVERNING LAW
|
65
|
||
|
15.1
|
Executive Officers; Disputes
|
65
|
|
|
15.2
|
Arbitration
|
65
|
|
|
15.3
|
Intellectual Property Disputes
|
66
|
|
|
15.4
|
Equitable Remedies
|
67
|
|
|
15.5
|
Governing Law; English Language
|
67
|
|
|
ARTICLE 16 MISCELLANEOUS
|
67
|
||
|
16.1
|
Assignment
|
67
|
|
|
16.2
|
Entire Agreement; Amendment
|
67
|
|
|
16.3
|
No Strict Construction; Interpretation
|
68
|
|
|
16.4
|
Severability
|
69
|
|
|
16.5
|
Force Majeure
|
69
|
|
|
16.6
|
Notices
|
69
|
|
|
16.7
|
Further Assurances
|
70
|
|
|
16.8
|
Performance by Affiliates
|
70
|
|
|
16.9
|
Agency
|
70
|
|
|
16.10
|
Binding Effect; No Third-Party Beneficiaries or Obligors
|
70 | |
|
16.11
|
No Waiver
|
70
|
|
|
16.12
|
Cumulative Remedies
|
71
|
|
|
16.13
|
Counterparts
|
71
|
|
| 1.1 |
Development and Commercialization. As of the Effective Date, Ionis is Developing a Licensed Product in ongoing Clinical Trials in both the Otsuka Territory and the Ionis Territory (such studies, as
further described and defined in Section 4.2.1 (Cross-Territory Clinical Development Plan)). Under this Agreement, the Parties intend (a) that Ionis will continue to conduct the Ongoing Cross-Territory Studies in accordance with the
Cross-Territory Clinical Development Plan, at [***] and (b) to share the costs of all Future Cross-Territory Studies included in the Cross-Territory Clinical Development Plan as of the Effective Date and any additional Cross-Territory
Clinical Studies included in any updated version of the Cross-Territory Clinical Development Plan approved by [***], in each case, in accordance with the [***]. In addition, the Parties intend for Ionis to Commercialize the Licensed Products
in the Ionis Territory and Otsuka to Commercialize the Licensed Products in the Otsuka Territory, which Commercialization activities will be consistent with the global commercialization and global medical affairs strategy (as further
described in Article 6 (Commercialization and Medical Affairs)).
|
| 1.2 |
Governance. The Parties have agreed to form a joint steering committee to oversee and coordinate the Development, Manufacturing, and Commercialization activities with respect to the Licensed Products
under this Agreement.
|
| 1.3 |
Purpose. The purpose of this Article 1 (Overview) is to provide a high-level overview of the roles, responsibilities, rights, and obligations of each Party under this Agreement, and
therefore, this Article 1 (Overview) is qualified in its entirety by the more detailed provisions of this Agreement set forth below.
|
| 2.1 |
License Grants to Otsuka. Subject to the terms of this Agreement (including Ionis’ retained rights set forth in Section 2.5 (No Other Rights and Retained Rights; Negative Covenant)), Ionis
hereby grants to Otsuka:
|
|
|
2.1.1 |
an exclusive, royalty-bearing license, with the right to grant sublicenses solely in accordance with Section 2.3.1(a) (Rights of Otsuka to Grant Sublicenses), under the Ionis Technology and the Unitary Product Trademark, in each
case, to (a) Develop the Licensed Products in the Field in the Otsuka Territory in accordance with the Otsuka Territory-Specific Development Plan solely for Commercialization and for the conduct of Medical Affairs for such Licensed Products
in the Field in the Otsuka Territory and (b) Commercialize and conduct Medical Affairs for the Licensed Products in the Field in the Otsuka Territory. For clarity, the license grant under this Section 2.1.1 (License Grants to Otsuka)
does not include the right to Manufacture the Licensed Products.
|
|
|
2.1.2 |
a non-exclusive, royalty-bearing license, with the right to grant sublicenses solely in accordance with Section 2.3.1(b) (Rights of Otsuka to Grant Sublicenses), under the Ionis Technology and the Unitary Product Trademark solely
to (a) Package and Label the Licensed Products in the Field in the Territory and (b) Manufacture the Licensed Products in the Field in the Territory from and after the time Otsuka provides a Manufacturing Handover Notice, in each case ((a)
and (b)), solely for Commercialization and for the conduct of Medical Affairs for such Licensed Products in the Field in the Otsuka Territory.
|
| 2.2 |
License Grant to Ionis. Subject to the terms of this Agreement, including Otsuka’s retained rights set forth in Section 2.5 (No Other Rights and Retained Rights; Negative Covenants), Otsuka
hereby grants to Ionis an exclusive, royalty-free, fully-paid, perpetual license, with the right to grant sublicenses through multiple tiers, under the Otsuka Technology solely to (a) Develop the Licensed Products in the Ionis Territory and
the Otsuka Territory; provided that, unless this Agreement has been terminated, any such Development in the Otsuka Territory will be conducted solely in accordance with the Cross-Territory Clinical
Development Plan and the Non-Clinical HAE Development Plan, (b) Manufacture the Licensed Products worldwide in accordance with this Agreement, and (c) Commercialize the Licensed Products in the Ionis Territory in accordance with this
Agreement and, subject to Section 14.9.5 (Sublicenses), worldwide following any termination of this Agreement.
|
| 2.3 |
Sublicensing and Subcontracting Terms.
|
|
|
2.3.1 |
Rights of Otsuka to Grant Sublicenses.
|
|
|
(a) |
Subject to the terms of this Agreement, Otsuka will have the right to grant sublicenses of the rights granted under Section 2.1.1 (License Grants to Otsuka) (i) [***], and (ii) [***].
|
|
|
(b) |
Subject to the terms of this Agreement, Otsuka will have the right to grant sublicenses of the rights granted under Section 2.1.2 (License Grants to Otsuka) (i) [***], and (ii) [***].
|
|
|
2.3.2 |
Right to Subcontract. Each Party may engage one or more Third Party subcontractors to perform services in furtherance of the performance of its obligations or exercise of its rights under this
Agreement, including any Third Party contract manufacturer, contract research organization, contract sales organization, wholesaler or distributor (including a distributor that is engaged to conduct promotional activities with respect to the
Licensed Products on such Party’s behalf and under such Party’s control) (“Subcontractors”); provided that (a) neither Party will engage
any such Subcontractor that has been Debarred/Excluded; and (b) no engagement of any such Subcontractors will relieve the engaging Party of its obligations under this Agreement or any liability hereunder.
|
|
|
2.3.3 |
Sublicense and Subcontract Agreements. Each agreement pursuant to which a sublicense is granted to a Sublicensee by Otsuka pursuant to this Section 2.3 (Sublicensing and Subcontracting
Terms), each agreement pursuant to which a sublicense is granted to a Sublicensee by Ionis of the rights granted to it under Section 2.2 (License Grant to Ionis), and each agreement pursuant to which a Party engages any Subcontractor,
in each case after the Effective Date and during the Term, will (a) be subject and subordinate to this Agreement, (b) be consistent with the terms of this Agreement, (c) include obligations of confidentiality and non-use applicable to the
Confidential Information of the other Party that are at least as stringent as those set forth in Article 12 (Confidentiality), and (d) include terms that are consistent with the intellectual property provisions set forth in this
Agreement. As soon as reasonably practicable after execution of any sublicense agreement with a Sublicensee after the Effective Date, [***]. In addition, [***].
|
|
|
2.3.4 |
Responsibility for Sublicensees and Subcontractors. Notwithstanding any sublicense, the sublicensing or subcontracting Party will remain primarily liable to the other Party for the performance of all
of its obligations under, and such Party’s compliance with all provisions of, this Agreement. Each Party agrees that it will be fully responsible and liable for any breach of the terms of this Agreement by any of its Sublicensees or
Subcontractors to the same extent as if such Party itself has committed any such breach.
|
| 2.4 |
Collaboration Technology Enabling License. Subject to the terms and conditions of this Agreement (and without limiting the licenses granted to Otsuka under Section 2.1 (License Grants to
Otsuka)), Otsuka hereby grants Ionis a fully-paid, royalty-free, irrevocable, worldwide, non-exclusive, sublicensable (through multiple tiers) license under any Otsuka Collaboration Know-How and Otsuka Collaboration Patent Rights (excluding
any Product-Specific Patents) to Exploit products that include an oligonucleotide as an active pharmaceutical ingredient (other than a Licensed Product); provided that Ionis may only grant a
sublicense under the rights granted in this Section 2.4 (Collaboration Technology Enabling License) if such sublicense includes the grant of a license under Know-How or Patent Rights Controlled by Ionis or its Affiliates to Exploit
such products that include an oligonucleotide as an active pharmaceutical ingredient (other than a Licensed Product).
|
| 2.5 |
No Other Rights and Retained Rights; Negative Covenant.
|
|
|
2.5.1 |
No Other Rights and Retained Rights. Nothing in this Agreement will be interpreted to grant a Party any rights under any intellectual property rights owned or Controlled by the other Party, including
Ionis Technology or Otsuka Technology, in each case, that are not expressly granted herein, whether by implication, estoppel, or otherwise. Otsuka will not practice the Ionis Technology other than as expressly licensed and permitted under
this Agreement and Ionis will not practice the Otsuka Technology other than as expressly licensed and permitted under this Agreement. Any rights not expressly granted to a Party by the other Party under this Agreement are hereby retained by
such other Party. Without limiting the foregoing, (a) Ionis hereby expressly retains the right to perform (i) Development activities for the Licensed Products worldwide in accordance with the Cross-Territory Clinical Development Plan and the
Non-Clinical HAE Development Plan, (ii) Manufacturing activities worldwide, and (iii) Ionis’ other obligations under this Agreement, and (b) Otsuka hereby expressly retains the right to perform Development activities for the Licensed Products
in the Field in the Otsuka Territory in accordance with the Otsuka Territory-Specific Development Plan solely for Commercialization and for the conduct of Medical Affairs for such Licensed Products in the Field in the Otsuka Territory and to
Manufacture the Licensed Products in the Field in the Territory solely for Commercialization and for the conduct of Medical Affairs for such Licensed Products in the Field in the Otsuka Territory, in each case, in accordance with this
Agreement.
|
|
|
2.5.2 |
Negative Covenant. Ionis shall not, and shall cause its Affiliates to not, grant or convey any right to any Third Party (pursuant to a license grant, collaboration or services agreement, option
grant, or otherwise) that would be in conflict with, limit the scope of, or otherwise adversely affect the licenses granted to Otsuka pursuant to Section 2.1 (License Grants to Otsuka).
|
| 2.6 |
Existing Third-Party IP Agreements.
|
|
|
2.6.1 |
Compliance. Otsuka acknowledges and agrees that (a) the rights and licenses granted to Otsuka under this Agreement are subject to the applicable terms of all Existing Third-Party IP Agreements with
respect to the Ionis Technology that is being sublicensed thereunder to Otsuka, (b) Ionis’ ability to comply with its obligations, and grant rights and licenses to Otsuka, under this Agreement are limited by any and all requirements and
restrictions imposed on Ionis under the Existing Third-Party IP Agreements with respect to the Ionis Technology that is being sublicensed to Otsuka by Ionis under such Existing Third-Party IP Agreements, and (c) Ionis will not be required to
take any action or inaction pursuant to this Agreement that would cause Ionis to be in breach of any Existing Third-Party IP Agreement or to grant any rights to Otsuka hereunder that are in violation of, or inconsistent with, any Existing
Third-Party IP Agreement. Otsuka will abide by the applicable terms of the Existing Third-Party IP Agreements, and, subject to [***], the applicable terms of any amendments, in each case, to the extent such terms are disclosed in the copies
of the Existing Third-Party IP Agreements, and amendments thereto, that are provided or made available to Otsuka.
|
|
|
2.6.2 |
Existing Third-Party IP Amendments. During the Term, Ionis will promptly furnish Otsuka with copies of any amendment to any Existing Third-Party IP Agreement to the extent related to any of the
rights sublicensed to Otsuka hereunder, from which copies Ionis may redact confidential or commercially sensitive information or other information that is not relevant to the rights sublicensed to Otsuka pursuant to the applicable Existing
Third-Party IP Agreement. During the Term, Ionis shall: (a) [***]; (b) [***]; (c) [***]; and (d) [***]. Notwithstanding any provision to the contrary in this Agreement, as between the Parties, [***] shall be solely responsible for the payment
of all license fees, royalties, milestone payments, and other payment obligations under all Existing Third-Party IP Agreements.
|
| 2.7 |
New Third-Party IP Agreements.
|
|
|
2.7.1 |
Identification of New In-License Agreements. If either Party intends to obtain Control of any Patent Rights or Know-How from a Third Party (whether by acquisition or license) that such Party believes
are [***] to Exploit the Licensed Compound or a Licensed Product (other than in connection with a Change of Control of a Party or as a result of the acquisition by a Party of a Third Party by merger, acquisition, or similar transaction or
series of related transactions) (such Patent Rights and Know-How, “Identified Rights”), then such Party will notify the other Party of the Identified Rights.
|
|
|
2.7.2 |
Potential In-Licenses.
|
|
|
(a) |
Acquisition of Potential In-Licenses.
|
|
|
i. |
[***] that [***] in the Exploitation of a Licensed Product in [***] Territory, whether by license or acquisition, (each agreement to license or acquire such Identified Rights, a “Potential
In-License”) in accordance with this Section 2.7.2 (Potential In-Licenses). If [***] after the Effective Date, then [***] will [***]. If [***] pursuant to this Section 2.7.2(a) (Acquisition of Potential In-Licenses),
then [***] with respect to [***] to Exploit the Licensed Products. [***] such Potential In-License will [***]. [***]. If the Identified Rights to be licensed or acquired under a Potential In-License would constitute Ionis Core Technology or
Ionis Manufacturing and Analytical Technology if such Identified Rights were Controlled by Ionis (any such Identified Rights, “Core or Manufacturing Identified Rights”) then, [***]
such Potential In-License (a “Core or Manufacturing Potential In-License”) in accordance with Section 2.7.2(c)i (Non-Approved Potential In-Licenses).
|
|
|
ii. |
If either Party [***] and if the Parties [***], then [***]. At either Party’s request if [***] in accordance with this Section 2.7.2(a)ii (Acquisition of Potential In-Licenses), the Parties will [***]. If the [***], then the
Potential In-License [***] for all purposes of this Agreement. If [***], then Ionis will [***]. If Ionis [***] pursuant to this Section 2.7.2(a)ii (Acquisition of Potential In-Licenses) and Ionis [***] in accordance with this Section
2.7.2(a)ii (Acquisition of Potential In-Licenses), then [***]. If Ionis [***], then Otsuka will [***] in accordance with the terms of this Section 2.7.2(a)ii
(Acquisition of Potential In-Licenses) and Otsuka will [***].
|
|
|
(b) |
Collaboration In-Licenses. For any Potential In-License that [***] in accordance with Section 2.7.2(a) (Acquisition of Potential In-Licenses), and for [***] (i) such Potential In-License will
[***], (ii) the Party [***] will [***], to the extent set forth in Section 2.7.2(a)ii (Acquisition of Potential In-Licenses), (iii) the Patent Rights or Know-How in-licensed under such [***], and (iv) (A) each Party will [***], and
(B) the Parties will [***]. The Party that [***] will [***] pursuant to this Section 2.7.2(b) (Collaboration In-License), and such other Party will [***].
|
|
|
(c) |
[***] Potential In-Licenses.
|
|
|
i. |
If [***] a Potential In-License [***], then (A) such Potential In-License [***], (B) subject to Section 2.7.2(c)ii ([***] Potential In-Licenses), the Patent Rights and
Know-How in-licensed under such Potential In-License [***], (C) except as set forth in clause (D) of this Section 2.7.2(c)i ([***] Potential In-Licenses), Ionis will [***] and, subject to Section 2.7.2(c)ii ([***] Potential
In-Licenses), will [***]; provided that if such Potential In-License [***], then [***], and Ionis [***] in accordance with Section 2.7.2(c)ii ([***] Potential In-Licenses), then Otsuka [***],
and (D) if such Potential In-License (1) was [***] pursuant to Section 2.7.2(a) (Acquisition of Potential In-Licenses), (2) was not [***], and (3) is not [***], then [***] in accordance with this Agreement [***], and the terms of this
Section 2.7.2 (Potential In-Licenses) [***]. If Ionis [***] in accordance with Section 2.7.2(a) (Acquisition of Potential In-Licenses), then Otsuka [***]. If [***], then Otsuka [***] in accordance with this Section 2.7.2(c)i ([***] Potential In-Licenses) and Otsuka will [***].
|
|
|
ii. |
If Ionis [***], then Ionis [***]. If Ionis [***] in accordance with Section 2.7.2(c)i ([***] Potential In-Licenses), then Ionis will [***]. Ionis may [***]. Within [***], Otsuka will [***]. If Otsuka [***] in accordance with this Section
2.7.2(c)ii ([***] Potential In-Licenses), then such [***] and Section 2.7.2(b) (Collaboration In-Licenses) will apply, mutatis mutandis. If Otsuka [***] in accordance with this Section
2.7.2(c)ii ([***] Potential In-Licenses), then Otsuka [***].
|
| 2.8 |
Right of First Negotiation for Follow-On Products.
|
|
|
2.8.1 |
ROFN Exercise. If, during the period from the Effective Date until the [***], Ionis intends to grant rights to a Third Party that include the right to Commercialize [***] designed to bind to the RNA
encoding PKK for the treatment of HAE (any such compound, a “Follow-On Product”) in the Otsuka Territory, then Ionis will provide to Otsuka (a) notice of the proposed scope of
Commercialization rights that Ionis proposes to grant and (b) an information package containing, to the extent such information is in Ionis’ or its Affiliate’s Control: (i) summaries of [***] (ii) information about [***] (iii) a summary of
[***] and (iv) [***] related to the Follow-On Product to the extent necessary or reasonably useful for Otsuka to evaluate whether to obtain rights with respect to Follow-On Product (“ROFN
Notice and Package”). Promptly thereafter, Ionis will provide a high-level presentation to the JSC relating to the Follow-On Product and the rights Ionis proposes to grant. Otsuka will have an exclusive right, exercisable no later
than [***] after receipt of a ROFN Notice and Package from Ionis containing all information set forth in the foregoing clauses ((i) through (iv)) to the extent such information is in Ionis’ or its Affiliate’s Control, to notify Ionis in
writing as to whether Otsuka desires to negotiate for such rights to Commercialize such Follow-On Product in the Otsuka Territory (a “ROFN Exercise Notice”). During such [***], Ionis
will [***].
|
|
|
2.8.2 |
Negotiation. If Otsuka provides a ROFN Exercise Notice to Ionis within such [***], then the Parties will negotiate in good faith for [***] from the date of Ionis’ receipt of the ROFN Exercise Notice,
or such longer period as may be agreed upon in writing by the Parties (the “ROFN Negotiation Period”) the terms of a definitive agreement (or amendment to this Agreement) pursuant to
which Ionis would grant to Otsuka the rights to Commercialize (and, as agreed by the Parties, to otherwise Exploit) such Follow-On Product in the Otsuka Territory. Neither Party will have any obligation to enter into any agreement or
amendment to this Agreement granting rights to Otsuka to Commercialize or otherwise Exploit such Follow-On Product in the Otsuka Territory. If the ROFN Negotiation Period expires before the Parties have entered into an agreement or amendment
to this Agreement with respect to Otsuka’s Commercialization or other Exploitation of such Follow-On Product in the Otsuka Territory, and if such ROFN Negotiation Period [***], then Ionis will have the right to negotiate and enter into an
agreement with any Third Party with respect to a grant of rights to Exploit such Follow-On Product in the Otsuka Territory [***]. If Ionis does not grant rights to a Third Party that include the right to Commercialize such Follow-On Product
in the Otsuka Territory [***], then the terms of this Section 2.8 (Right of First Negotiation for Follow-On Products) will [***]. If the [***] expires before the Parties have entered into an agreement or amendment to this Agreement
with respect to Otsuka’s Commercialization or other Exploitation of such Follow-On Product in the Otsuka Territory, then Ionis will have no further obligation to negotiate with Otsuka with respect to any grant of such rights to Otsuka and
will be free to negotiate and enter into an agreement with any Third Party with respect to a grant of rights to Exploit such Follow-On Product in the Otsuka Territory.
|
|
|
2.8.3 |
Follow-On Product Activities. If Ionis enters into an agreement with a Third Party granting any rights to Exploit a Follow-On Product, then all Development, Commercialization, and Medical Affairs
activities related to such Follow-On Product (“Follow-On Product Activities”) will be subject to the following: (a) [***]
related to such Follow-On Product; and (b) Ionis and its Affiliates shall conduct the Follow-On Product Activities independently of the activities under this Agreement and [***].
|
| 3.1 |
Initial Know-How Transfer. At a time period to be agreed upon by the Parties after the Effective Date, Ionis will provide and transfer, and in any event will initiate such transfer within [***] after
the Effective Date, to Otsuka copies of the Ionis Know-How (other than Ionis Manufacturing and Analytical Know-How, the transfer of which will be conducted pursuant to Section 7.4 (Manufacturing Technology Transfer)) that (a) exists
on the Effective Date, (b) was not previously provided to Otsuka, and (c) is [***] to Develop, Commercialize or conduct Packaging and Labeling or Medical Affairs for a Licensed Product (such transfer, the “Initial Know-How Transfer”). Ionis may make such Ionis Know-How available in such reasonable form as maintained by Ionis. In addition to the Initial Know-How Transfer, upon Otsuka’s reasonable request during the
Term, Ionis will provide and transfer to Otsuka copies of or otherwise make available to Otsuka all Ionis Know-How (other than Ionis Manufacturing and Analytical Know-How) not previously provided to Otsuka hereunder to the extent such Ionis
Know-How is [***] to Develop, Commercialize or conduct Packaging and Labeling or Medical Affairs for a Licensed Product, including in accordance with Section 4.7 (Data Transfer), Section 5.6 (Cooperation), Section 6.1.2
(Commercialization in the Otsuka Territory), Section 6.2 (Commercialization and Medical Affairs Reporting), and Section 7.1.1 (Ionis Manufacturing) (the “Continuing Know-How
Transfer,” and together with the Initial Know-How Transfer, the “Technology Transfer”).
|
| 3.2 |
Technology Transfer Costs. Ionis will conduct the Technology Transfer, and will provide consultation and assistance with [***] to provide support set forth in Section 7.1.1 (Ionis
Manufacturing) (any such consultation, assistance, or support provided by [***]. Ionis will [***] in connection with the Initial Know-How Transfer, including, for clarity, [***]. In addition, Ionis will [***] in providing Requested Assistance
to Otsuka in connection with such Continuing Know-How Transfer, until [***] and, thereafter, [***]. After [***]. At all times, Otsuka will [***]. Ionis may [***] following receipt of such Regulatory Approval, to the extent [***]. Ionis shall
[***]. Notwithstanding the foregoing, [***]. For clarity, the terms of this Section 3.2 (Technology Transfer Costs) shall not apply with respect to [***] and, for clarity, the terms of this Section 3.2 (Technology Transfer
Costs) shall not apply to [***]. Notwithstanding any provision to the contrary, Ionis’ obligations to conduct the Technology Transfer and provide Requested Assistance will not require Ionis to conduct any additional Clinical Trials or
generate any additional data or information that is not expressly contemplated by the Cross-Territory Clinical Development Plan or Non-Clinical HAE Development Plan.
|
| 4.1 |
Development Diligence Obligations. Ionis will be responsible for conducting the activities under the Cross-Territory Clinical Development Plan and the Non-Clinical HAE Development Plan and will use
Commercially Reasonable Efforts to carry out such activities. For clarity, Ionis shall not conduct any [***]. Otsuka will be responsible for conducting the activities under the Otsuka Territory-Specific Development Plan [***]. Each Party will
conduct all Development activities for which it is responsible under this Agreement in a good scientific manner, in accordance with GLP and GCP, as applicable, and in compliance with Professional Requirements and Applicable Law.
|
| 4.2 |
Development Plans.
|
|
|
4.2.1 |
Cross-Territory Clinical Development Plan. The initial development plan for the clinical Development activities for both the Otsuka Territory and the Ionis Territory is set forth on Schedule 4.2.1 (such development plan as it may be modified in accordance with the terms and conditions of this Agreement, the “Cross-Territory
Clinical Development Plan”). The initial Cross-Territory Clinical Development Plan includes (and any updates to the Cross-Territory Clinical Development Plan will at all times include) all Clinical Trials that are intended to support
obtaining or maintaining Regulatory Approval for any Licensed Product in both the Otsuka Territory and the Ionis Territory (any such Clinical Trials, “Cross-Territory Clinical Studies”),
including (a) all Cross-Territory Clinical Studies that are ongoing as of the Effective Date (the “Ongoing Cross-Territory Studies”), (b) all Post-Approval Mandatory Studies that are
(i) required to support maintaining Regulatory Approval for any Licensed Product in both the Otsuka Territory and the Ionis Territory and (ii) designed to meet the requirements of the EMA for maintaining Regulatory Approval of the Licensed
Product in the Otsuka Territory (collectively ((i) and (ii)), “Post-Approval Cross-Territory Mandatory Studies”), and (c) all future Cross-Territory Clinical Studies for [***]. From
time to time during the Term, either Party may submit to the JSC any proposed update to the Cross-Territory Clinical Development Plan to include additional Cross-Territory Clinical Studies, including the study designs for such additional
Cross-Territory Clinical Studies. In addition, each Party shall submit to the JSC (or a designated Subcommittee) reasonably in advance proposed updates to the Cross-Territory Clinical Development Plan to take into account changed
circumstances, such as cessation of any Cross-Territory Clinical Study, or the need to amend any Cross-Territory Clinical Study, including amendments in response to Regulatory Authority requirements, for safety reasons or otherwise. The JSC
will review, discuss, and determine whether to approve each update to the Cross-Territory Clinical Development Plan. Once reviewed and approved by the JSC, each update to the Cross-Territory Clinical Development Plan will automatically become
effective and supersede the previous Cross-Territory Clinical Development Plan, as of the date of such approval by the JSC.
|
|
|
4.2.2 |
Non-Clinical HAE Development Plan. The initial development plan for all CMC Development and non-clinical Development, in each case, required to obtain and maintain Regulatory Approval for the
Licensed Product for the treatment of HAE in the Otsuka Territory is set forth on Schedule 4.2.2 (such development plan, as it may be modified in accordance with the terms and
conditions of this Agreement, the “Non-Clinical HAE Development Plan”). From time to time during the Term, either Party may submit to the JSC any proposed update to the Non-Clinical
HAE Development Plan to include additional CMC Development or non-clinical Development activities required to obtain or maintain Regulatory Approval for the Licensed Product for the treatment of HAE in the Otsuka Territory. In addition, each
Party shall submit to the JSC (or a designated Subcommittee) reasonably in advance proposed updates to take into account changed circumstances or the need to amend any CMC Development or non-clinical Development activities for the Licensed
Product for the treatment of HAE in the Otsuka Territory. The JSC will review, discuss, and determine whether to approve each update to the Non-Clinical HAE Development Plan. Once reviewed and approved by the JSC, each update to the
Non-Clinical HAE Development Plan will automatically become effective and supersede the previous Non-Clinical HAE Development Plan as of the date of such approval by the JSC.
|
| 4.3 |
Otsuka Territory-Specific Development Plan. Within [***] following the Effective Date, Otsuka will prepare and submit to the JSC a plan setting forth all Development activities that are intended to
support obtaining or maintaining Regulatory Approval for the Licensed Products solely in the Otsuka Territory other than the activities set forth in the Non-Clinical HAE Development Plan (the “Otsuka Territory-Specific Development Plan”). The JSC will review, discuss, and determine whether to approve the Otsuka Territory-Specific Development Plan. The Otsuka Territory-Specific Development Plan will, at all times
during the Term, be consistent with the then-current Cross-Territory Clinical Development Plan, except to the extent such inconsistency is (a) necessary to conform with any written requirement from any Regulatory Authority or with any
Applicable Law (including compliance requirements) in the Otsuka Territory or (b) approved by the JSC (by unanimous Party Vote). At least [***] during the Term (and more frequently as may be necessary), Otsuka will prepare an update to the
Otsuka Territory-Specific Development Plan to amend or include additional Development activities to be conducted during the [***] Calendar Year that are intended to support obtaining or maintaining Regulatory Approval for the Licensed
Products solely in the Otsuka Territory other than the activities set forth in the Non-Clinical HAE Development Plan (or otherwise update the Development activities under the Otsuka Territory-Specific Development Plan). The JSC will review,
discuss, and determine whether to approve each update to the Otsuka Territory-Specific Development Plan. Once approved by the JSC, the Otsuka Territory-Specific Development Plan and each update thereto will automatically become effective and,
in the case of an update, supersede the previous Otsuka Territory-Specific Development Plan as of the date of such approval. Notwithstanding the foregoing or anything to the contrary in this Agreement, the Otsuka Territory-Specific
Development Plan will at all times include Development activities that are (i) necessary to obtain and maintain Regulatory Approval for at least one Licensed Product in [***] consistent with Otsuka’s obligations under Section 4.1
(Development Diligence Obligations) and (ii) not included in the Cross-Territory Clinical Development Plan or the Non-Clinical HAE Development Plan.
|
| 4.4 |
Development Costs.
|
|
|
4.4.1 |
Overview.
|
|
|
(a) |
Ionis Costs. Ionis will be [***] responsible for all costs and expenses incurred in connection with the performance of the [***]. In addition, Ionis will be [***] responsible for all costs and
expenses incurred in connection with the performance of all activities under the [***].
|
|
|
(b) |
Otsuka Costs. Otsuka will be [***] responsible for all costs and expenses incurred in connection with the performance of all activities under the [***].
|
|
|
(c) |
Shared Development Costs. With respect to (i) all Post-Approval Cross-Territory Mandatory Studies for the treatment of HAE that are included in the
Cross-Territory Clinical Development Plan as of the Effective Date] and (ii) [any other Post-Approval Cross-Territory Mandatory Studies for the treatment of HAE or any Future Cross-Territory Studies, in each case, that are added to the
Cross-Territory Clinical Development Plan in accordance with Section 4.2.1 (Cross-Territory Clinical Development Plan), the Parties will share, [***], in each case, in accordance with the Cross-Territory Clinical Development Plan and
Shared Development Budget (such costs, “Shared Cross-Territory Development Costs”) in accordance with the terms of Section 4.4.3 (Shared Cross-Territory Development Costs).
|
|
|
4.4.2 |
Shared Development Budget; Cost Overruns.
|
|
|
(a) |
Shared Development Budget. The initial budget for the Shared Cross-Territory Development Costs (such budget as it may be modified in accordance with the terms and conditions of this Agreement, the “Shared Development Budget”) for the Post-Approval Cross-Territory Mandatory Studies that are included in the Cross-Territory Clinical Development Plan as of the Effective Date is set
forth in Schedule 4.4.2 (Shared Development Budget). With respect to each other Post-Approval Cross-Territory Mandatory Study for the treatment of HAE or Future Cross-Territory
Study that is subject to cost sharing by the Parties in accordance with Section 4.4.1(c) (Shared Development Costs), the JSC will develop, discuss, and determine whether to approve an update to the Shared Development Budget at the
time the JSC reviews, discusses, and determines whether to approve the update to the Cross-Territory Clinical Development Plan applicable to such additional Cross-Territory Clinical Study. Any update to the Shared Development Budget will at
all times include a detailed written budget for the performance of all Future Cross-Territory Studies and any additional Post-Approval Cross-Territory Mandatory Studies for the treatment of HAE, in each case, that are included in the
Cross-Territory Clinical Development Plan (as updated). From time to time during the Term, either Party may submit to the JSC any proposed update to the Shared Development Budget, including in connection with any update to the Cross-Territory
Clinical Development Plan or to address a potential Cost Overrun. The JSC will review, discuss, and determine whether to approve each update to the Shared Development Budget. Once reviewed and approved by the JSC, each update to the Shared
Development Budget will automatically become effective and supersede the previous Shared Development Budget, as of the date of such approval by the JSC.
|
|
|
(b) |
Cost Overruns. Ionis [***] for a given Calendar Year [***] for such Calendar Year. Ionis will notify the JSC (or any designated Subcommittee) without undue delay if it reasonably anticipates that the
Shared Cross-Territory Development Costs are reasonably likely to exceed the then-current Shared Development Budget by more than [***] (a “Cost Overrun”). Ionis will include [***]
and, to the extent reasonably possible, will [***]. Thereafter, the JSC (or designated Subcommittee) shall promptly hold an ad-hoc meeting to evaluate whether there are mitigation measures to prevent the Cost Overrun, and if not, the JSC (or
designated Subcommittee) will discuss what steps to take to address such Cost Overrun, including updating the Shared Development Budget or the Cross-Territory Clinical Development Plan, as applicable. If the JSC does not approve an update to
the Shared Development Budget to reflect the anticipated Cost Overrun, then [***] and, to the extent [***]. For clarity, [***].
|
|
|
4.4.3 |
Shared Cross-Territory Development Costs. The Parties will share, at the [***], all Shared Cross-Territory Development Costs incurred by or on behalf of Ionis or its Affiliates in accordance with the
Cross-Territory Clinical Development Plan and the amount budgeted therefor in the Shared Development Budget, plus [***] (“Eligible Cross-Territory
Development Costs”). No later than [***] after the end of each Calendar Quarter, Ionis will deliver to Otsuka a written report specifying in reasonable detail the Eligible Cross-Territory Development Costs incurred by or on behalf of
Ionis during such Calendar Quarter, together with reasonable supporting documentation (the “Development Cost Share Notice”) and an invoice (and, if there has been any change to a
Payment Form previously submitted, or if a previously submitted Payment Form has expired, an updated Payment Form) for Otsuka’s [***] share of such Eligible Cross-Territory Development Costs. No later than [***] after Otsuka’s receipt of the
Development Cost Share Notice and invoice (and updated Payment Form, if applicable) for such Calendar Quarter, Otsuka will make a balancing payment to Ionis equal to [***] of the total Eligible Cross-Territory Development Costs to effect the
[***] for such Eligible Cross-Territory Development Costs; provided that, if Otsuka disputes any invoiced amount, then Otsuka will pay the undisputed invoiced amount within such [***] and will pay any
disputed amounts within [***] following resolution of the dispute and determination that such amounts are owed.
|
|
|
4.4.4 |
Future Cross-Territory Studies for [***].
|
|
|
(a) |
Shared Costs. At any time during the Term, either Party may propose that the Parties clinically Develop a Licensed Product [***]. Such Party will submit a proposal to the JSC setting forth the
proposed clinical Development activities for such [***] and a timeline and budget for such activities (a “[***]”). The JSC will review, discuss, and determine whether to approve such [***], either as proposed or as may be revised by agreement
of the JSC, within [***] of the submission thereof. If the JSC approves such [***], as proposed or as revised per the agreement of the Parties, then (i) the JSC will approve an update to the Cross-Territory Clinical Development Plan in
accordance with Section 4.2.1 (Cross-Territory Clinical Development Plan) and to the Shared Development Budget in accordance with Section 4.4.2(a) (Shared Development Budget) to include any Future Cross-Territory Studies for
such [***] and (ii) the Parties will share the cost of any Eligible Cross-Territory Development Costs incurred in connection with the performance of such Future Cross-Territory Studies in accordance with Section 4.4.3 (Shared
Cross-Territory Development Costs).
|
|
|
(b) |
[***] by Ionis. If the JSC does not approve [***] for a given [***] or does not approve an updated Cross-Territory Clinical Development Plan in accordance with Section 4.2.1 (Cross-Territory
Clinical Development Plan) or Shared Development Budget in accordance with Section 4.4.2(a) (Shared Development Budget) to include the Future Cross-Territory Studies for such [***], then (i) Ionis will have the right, but not the
obligation, to proceed with the Development of such [***] in the Territory as contemplated by [***] with such modifications as Ionis deems appropriate [***] (“Ionis [***]”) and such
Development will be conducted outside the Cross-Territory Clinical Development Plan; and (ii) notwithstanding the licenses, rights of reference, and other rights granted to Otsuka under this Agreement, Otsuka will not have any license or
rights to use any Ionis [***] (including any right of reference to use such Ionis [***] contained in the related Regulatory Submissions by Ionis and notwithstanding the inclusion of any such data in the Ionis Technology) in support of any
Regulatory Submissions or Regulatory Approval for the Licensed Product in the Otsuka Territory or in the Commercialization of such Licensed Product in the Otsuka Territory, unless and until [***], provided
that Otsuka may use safety data in connection with such Ionis [***] solely to satisfy any safety-related reporting obligations to Regulatory Authorities related to Licensed Products in the Otsuka Territory without [***]. For clarity, Ionis
[***].
|
|
|
(c) |
Otsuka Opt-In. If Ionis conducts any Ionis [***] pursuant to Section 4.4.4(b) ([***] by Ionis), then Ionis shall provide Development reports to Otsuka related to such Ionis [***] in
accordance with Section 4.5 (Development Reports), and Otsuka shall have the right, [***] to opt-in with respect to such Ionis [***] in accordance with this Section 4.4.4(c) (Otsuka Opt-In). Upon Otsuka’s written request,
Ionis shall provide to Otsuka a written report of the Internal Costs and External Costs, in each case, incurred directly by or on behalf of Ionis or its Affiliates as of the date of such written request in the performance of such Ionis [***]
(the “Ionis [***] Costs”). Such written report shall include supporting documentation of the External Costs included
within the Ionis [***] Costs. Otsuka shall have the right, exercisable during the [***] period after receipt of such report, to provide notice to Ionis that Otsuka wishes to share the costs of such Ionis [***] (“Opt-In Notice”). Following the receipt of an Opt-In Notice, Ionis shall provide an invoice to Otsuka for [***] of such Ionis [***] (“Opt-In Fee”)
as follows: [***]. Otsuka shall pay the Opt-In Fee within [***] after receipt of such invoice and supporting documentation; provided that, if Otsuka disputes any invoiced amount, then Otsuka will pay
the undisputed invoiced amount within such [***] and will pay any disputed amounts within [***] following resolution of the dispute and determination that such amounts are owed. Following Otsuka’s payment of the Opt-In Fee, (A) the Ionis
[***] shall be deemed a Future Cross-Territory Study and will be added to the Cross-Territory Clinical Development Plan, (B) the Parties will share, [***], all Internal Costs reasonably incurred and External Costs incurred, in each case,
directly by or on behalf of Ionis or its Affiliates in the performance of the Ionis [***] from the date on which Ionis provides a written report of the Ionis [***] Costs, (C) the applicable Ionis [***] shall be included within Ionis Know-How
and subject to the licenses in Section 2.1 (License Grants to Otsuka), and (D) the Ionis [***] included in related Regulatory Submissions will be subject to Otsuka’s right of use and right of reference provided in Section 5.9
(Right of Reference).]
|
| 4.5 |
Development Reports. At each JSC meeting, Ionis and Otsuka will each provide the JSC with a written summary of the activities conducted by or on behalf of such Party under, as applicable, the
Cross-Territory Clinical Development Plan, the Non-Clinical HAE Development Plan, and the Otsuka Territory-Specific Development Plan, and with respect to Ionis, [***], in each case, since the last JSC meeting, including patient enrollment,
the ongoing status, and material results of all Clinical Trials for the Licensed Products conducted by or on behalf of such Party. Each Party will also promptly provide written notice to the other Party and keep the other Party reasonably
informed, through the JSC or Alliance Managers, of any significant Development events under the Cross-Territory Clinical Development Plan, the Non-Clinical HAE Development Plan, or the Otsuka Territory-Specific Development Plan, and with
respect to Ionis, any additional Development activities conducted by or on behalf of Ionis or any of its Affiliates for the Licensed Product that are not set forth in the Cross-Territory Clinical Development Plan or the Non-Clinical HAE
Development Plan, in each case, that [***] the Development activities of the other Party under this Agreement.
|
| 4.6 |
Development Records; Cooperation. Each Party and its Affiliates will maintain written or electronic records, in sufficient detail, in a good scientific manner, in accordance with Applicable Law
(including GLP, GCP, and GMP, as applicable), and appropriate for regulatory and patent purposes, and that are complete and accurate and reflect all Development work performed and results achieved, in each case, by or on behalf of such Party
and its Affiliates under, as applicable, the Cross-Territory Clinical Development Plan, the Non-Clinical HAE Development Plan, and the Otsuka Territory-Specific Development Plan, and with respect to Ionis, [***]. Each Party shall retain such
records for at least three years after the end of the Term or for such longer period as may be required by Applicable Law. The Parties will cooperate with each other to achieve the Development objectives contemplated herein in a timely,
accurate, and responsive manner. Without limiting the foregoing, [***].
|
| 4.7 |
Data Transfer. Upon Otsuka’s reasonable request, Ionis shall provide to Otsuka, [***] notwithstanding the terms of Section 3.2 (Technology Transfer Cost), true and complete copies of all
written, graphic or electronic embodiments of non-clinical data and clinical data generated by or on behalf of Ionis or any of its Affiliates in connection with the Development of Licensed Products, including all draft and final protocols and
final study reports and raw data, in each case, to the extent such data is (a) Controlled by Ionis or its Affiliates and (b) [***] to Exploit a Licensed Product. [***].
|
| 5.1 |
Regulatory Responsible Party. Ionis will be the Regulatory Responsible Party for the Licensed Products in the Ionis Territory. Otsuka will be the Regulatory Responsible Party for the Licensed
Products in the Otsuka Territory, provided that, Ionis shall have the right to conduct regulatory activities in the Otsuka Territory, including interacting with Regulatory Authorities, solely with
respect to (a) the Development activities for the Licensed Products in the Otsuka Territory for which Ionis is responsible under the Cross-Territory Clinical Development Plan and the Non-Clinical HAE Development Plan and (b) the Manufacturing
activities for the Licensed Products for which Ionis is responsible in accordance with Article 7 (Manufacturing), in each case subject to the remainder of this Article 5 (Regulatory Affairs) (“Ionis Regulatory Activities”). Subject to the obligations in this Article 5 (Regulatory Affairs), the Regulatory Responsible Party will be
responsible for, and [***] all Regulatory Submissions, communications, and other dealings with the Regulatory Authorities relating to the Licensed Products in the applicable Territory, and for seeking and maintaining all Regulatory Approvals
with respect to the Licensed Product in the applicable Territory. The Regulatory Responsible Party will not be required to delay any submission, correspondence, or communication with any Regulatory Authorities in a manner that affects such
Regulatory Responsible Party’s ability to comply with any Regulatory Authority requirement or deadline or Applicable Law in such jurisdiction. For clarity, Otsuka or its designee shall be the holder of all Regulatory Approvals for the
Licensed Product in the Otsuka Territory and will own all Regulatory Submissions in the Otsuka Territory, and Ionis or its designee shall be the holder of all Regulatory Approvals for the Licensed Product in the Ionis Territory and will own
all Regulatory Submissions in the Ionis Territory. Otsuka will only [***] and will not [***].
|
| 5.2 |
Regulatory Subcommittee. Within [***] after the Effective Date, the Parties will establish, through the JSC, a Subcommittee to (a) oversee the preparation and submission of any MAA for a Licensed
Product in the Otsuka Territory and (b) coordinate the regulatory responsibilities between the Parties in the Otsuka Territory, which allocation will be consistent with this Article 5 (Regulatory Affairs) (such Subcommittee, the “Regulatory Subcommittee”). The Regulatory Subcommittee will review and comment on any proposed MAA application for a Licensed Product sufficiently in advance of the filing or submission
thereof by Otsuka, and Otsuka will [***] any comments received from the Regulatory Subcommittee. The Regulatory Subcommittee will meet as often as necessary to carry out the activities described in this Section 5.2 (Regulatory
Subcommittee) and the terms of Section 8.2 (Additional Committees) will apply to the Regulatory Subcommittee.
|
| 5.3 |
Correspondences with Regulatory Authorities. Otsuka shall be solely responsible for communications with Regulatory Authorities in the Otsuka Territory regarding the Licensed Product and in no event
will [***] regarding the Licensed Product in the Otsuka Territory, except in connection with [***], and subject to the remainder of this Section 5.3 (Correspondences with Regulatory Authorities). Ionis will [***]. In addition, Ionis
will [***]. Furthermore, upon Otsuka’s reasonable request and at [***], Ionis will [***]. The Regulatory Responsible Party will provide the other Party with (a) copies of any material written correspondence submitted to or received from (i)
with respect to Otsuka, the EMA or any other Regulatory Authority in the Otsuka Territory, and (ii) with respect to Ionis, the FDA or any other Regulatory Authority in the U.S., and (b) summaries of any material oral communications with such
Regulatory Authority in clause (a), in each case ((a) and (b)), relating to Regulatory Submissions in support of Development of the Licensed Products in such jurisdiction or country, reasonably promptly after receipt or delivery by such
Regulatory Responsible Party of such correspondence or communication, as the case may be (but in any event, no later than [***] after receipt or delivery).
|
| 5.4 |
Regulatory Meetings. Ionis will [***] meetings pertaining to Regulatory Submissions for the Licensed Products relating to the Ionis Regulatory Activities [***] in the Otsuka Territory] to the extent
not prohibited by Applicable Law or the applicable Regulatory Authority. At Otsuka’s request, Ionis will [***]. With respect to all other meetings with any Regulatory Authority in the Otsuka Territory in support of Development of the Licensed
Products, (a) Ionis will have the right, but not the obligation, [***] to attend such meetings [***] and (b) at Otsuka’s reasonable request, Ionis will [***] and Otsuka will [***]. Further, Ionis will [***], unless Ionis reasonably believes
that [***], and will not [***] except as (a) required by Applicable Law, (b) permitted pursuant to Section 12.4.1(b) (Permitted Circumstances) or Section 12.4.1(c) (Permitted Circumstances), or (c) authorized by Otsuka in
writing. For clarity, the terms of this Section 5.4 (Regulatory Meetings) will apply solely with respect to any meetings with any Regulatory Authority in the Otsuka Territory in support of Development of the Licensed Products and do
not apply to any meetings with Regulatory Authorities in the Otsuka Territory pertaining to [***].
|
| 5.5 |
Regulatory Submissions. Each Party (the “Filing Party”) will provide the other Party with a copy of [***] (including, [***]) that the
Filing Party intends to file with or submit to any Regulatory Authority in support of Development in the Otsuka Territory for the other Party’s review and comment sufficiently in advance of the Filing Party’s filing or submission thereof. The
Filing Party will [***] any reasonable comments received from the other Party into such Regulatory Submissions. In addition and notwithstanding Section 3.2 (Technology Transfer Cost), Ionis will provide to Otsuka, [***].
|
| 5.6 |
Cooperation. The Parties will cooperate with each other to achieve the regulatory objectives contemplated herein in a timely, accurate, and responsive manner. Without limiting the foregoing or the
terms of Section 5.5 (Regulatory Submissions), at Otsuka’s reasonable request, Ionis will [***] of the Licensed Products in the Otsuka Territory or for obtaining and maintaining Regulatory Approval of the Licensed Products in the
Otsuka Territory. If Otsuka receives any inquiry from a Regulatory Authority in the Otsuka Territory pertaining to any activities for which Ionis is responsible hereunder (including Cross-Territory Clinical Studies, non-clinical or CMC
Development or Manufacturing (prior to the Manufacturing Handover Date)), then notwithstanding Section 3.2 (Technology Transfer Cost), upon Otsuka’s request, Ionis will, [***]. In addition, upon Otsuka’s reasonable request, Ionis
shall provide [***] in the Otsuka Territory (“Regulatory Support”). Ionis will provide all Regulatory Support [***] until [***]. Thereafter, [***], Ionis will [***], and [***], Ionis
will [***]. At all times, Otsuka will [***]. Ionis may [***] (i) [***], and (ii) [***] and, in each case ((i) and (ii)), [***] (and, with respect to [***]) therefor [***]. For clarity, if Otsuka requests Regulatory Support in connection
with [***], Ionis shall provide such Regulatory Support [***], and Otsuka [***]. Notwithstanding any provision to the contrary, this Section 5.6 (Cooperation) will not require Ionis to conduct any additional Clinical Trials or
generate any additional data or information that is not expressly contemplated by the Cross-Territory Clinical Development Plan or Non-Clinical HAE Development Plan.
|
| 5.7 |
Cost of Regulatory Activities. Except to the extent specified otherwise in this Article 5 (Regulatory Affairs), each Party will be responsible for all costs and expenses incurred in
connection with its activities under this Article 5 (Regulatory Affairs), including the preparation or maintenance of Regulatory Submissions and Regulatory Approvals with respect to the Licensed Products for which it is responsible,
including any filing fees and, with respect to Ionis, including all costs and expenses of Ionis Regulatory Activities and all costs and expenses related to the ASMF (if filed) and other regulatory affairs related to Manufacturing of Licensed
Products, which in each case will be borne solely by Ionis.
|
| 5.8 |
No Harmful Actions. If [***], then such first Party will have the right to bring such matter to the attention of the JSC and the Parties will discuss in good faith to resolve such concern. Without
limiting the foregoing and notwithstanding any provision to the contrary in this Agreement, Otsuka will not [***].
|
| 5.9 |
Right of Reference. Subject to the rules of the relevant Regulatory Authority and the terms of this Agreement, including Section 4.4.4(b) ([***] by Ionis), each Party hereby grants to the
other Party a “Right of Reference,” as that term is defined in 21 C.F.R. § 314.3(b) (or any successor rule or analogous Applicable Law recognized outside of the United States) to, and a right to copy, access, and otherwise use, all
information and data relating to the Licensed Products in any Regulatory Submission or Regulatory Approval Controlled by the grantor Party during the Term (including, with respect to the grant to Otsuka, a right of reference to Ionis’ Drug
Master File and ASMF, if filed), solely for the other Party’s or its Affiliates’ use in the Development or Commercialization of the Licensed Products in the other Party’s Territory in accordance with this Agreement. All such information and
data contained in any such Regulatory Submissions or Regulatory Approvals will be considered Confidential Information of the grantor Party and subject to the terms of Article 12 (Confidentiality). If requested by the grantee Party,
the grantor Party will provide a signed statement to this effect in accordance with 21 C.F.R. § 314.50(g)(3) (or any successor rule or analogous Applicable Law outside of the United States) to give effect to the intent of this Section 5.9
(Right of Reference).
|
| 5.10 |
Pharmacovigilance; Safety Information. Each Party will cooperate with the other Party, at no cost to the other Party (notwithstanding Section 3.2 (Technology Transfer Cost)), with regard to
the reporting and handling of safety information involving the Licensed Products in accordance with Applicable Law, regulatory requirements, and regulations on pharmacovigilance and clinical safety. Otsuka will be responsible for all
processing of information related to any adverse events for the Licensed Products in the Otsuka Territory and Ionis will be responsible for all processing of information related to any adverse events for the Licensed Products in the Ionis
Territory, in each case, including any information regarding such adverse events that is received from a Third Party. Each Party will provide to the other Party in a timely manner the relevant safety information it receives (either directly
or indirectly) related to the Licensed Products. At an appropriate time as agreed upon by the Parties following the Effective Date, but in any event prior to the [***], the Parties will negotiate in good faith and enter into a
Pharmacovigilance Agreement related to the Licensed Products, which will define the pharmacovigilance responsibilities of the Parties and include safety data exchange procedures governing the exchange of information affecting the class and
products (e.g., Serious Adverse Events, emerging safety issues) to enable each Party to comply with all of its legal and regulatory obligations related to such
Licensed Product. Prior to the execution of the Pharmacovigilance Agreement, each Party will have the right, upon reasonable notice to the other Party, to [***]. Ionis will own and maintain the global safety database for the Licensed Products
at [***], provided that at Otsuka’s reasonable request, Ionis will run queries of such global safety database and will provide copies of the data contained in such global safety database to the extent
necessary or reasonably useful to the Development and Commercialization of the Licensed Product in the Otsuka Territory. As part of the negotiation of the Pharmacovigilance Agreement, the Parties will [***], taking into account that Ionis
will own and maintain the global safety database, and the Parties’ determination of such matter will be set forth in the Pharmacovigilance Agreement. Subject to compliance with Applicable Law, each Party hereby agrees to comply with its
respective obligations under the Pharmacovigilance Agreement as the Parties may agree to modify it from time to time, and to cause its (sub)licensees to comply with such obligations. If there is a conflict between the terms and conditions of
this Agreement and any terms and conditions of the Pharmacovigilance Agreement, then the terms and conditions of the Pharmacovigilance Agreement will govern with respect to any pharmacovigilance matters and this Agreement will govern with
respect to any other matters.
|
| 5.11 |
Pharmacovigilance Subcommittee. The JSC shall establish a joint pharmacovigilance subcommittee (the “PV Subcommittee”) at an appropriate
time, but in any event prior to the [***]. In addition to any other matters that the JSC may delegate to the PV Subcommittee, the PV Subcommittee shall provide a forum for the Parties to discuss, share information, and escalate and attempt to
resolve safety issues regarding the Licensed Product, and any other pharmacovigilance matters, worldwide. The PV Subcommittee will meet as often as necessary to carry out such activities, and the terms of Section 8.2 (Additional
Committees) will apply to the PV Subcommittee.
|
| 5.12 |
Ionis Internal Oligonucleotide Safety Database.
|
|
|
5.12.1 |
Ionis maintains an internal database that includes information regarding the tolerability of its drug compounds, individually and as a class, including information discovered during non-clinical and clinical Development (the “Ionis Internal Oligonucleotide Safety Database”). To maximize understanding of the safety profile and pharmacokinetics of Ionis compounds, (a) Ionis will have the right to use any
safety-related information provided by Otsuka pursuant to the Pharmacovigilance Agreement or this Section 5.12 (Ionis Internal Oligonucleotide Safety Database) to maintain the Ionis Internal Oligonucleotide Safety Database and (b)
Otsuka will cooperate, at no cost to Ionis, with Ionis’ reasonable requests in connection with populating the Ionis Internal Oligonucleotide Safety Database, including by providing Ionis with reasonably requested safety-related supporting
data and answering any follow-up questions reasonably requested by Ionis or its Affiliates in connection with any information provided under the Pharmacovigilance Agreement, in each case to the extent such data and answers are reasonably
available to Otsuka. In addition, with respect to Clinical Trials of the Licensed Products conducted by or on behalf of Otsuka pursuant to the Otsuka Territory-Specific Development Plan (if any), Otsuka will provide Ionis with copies of
annual safety updates filed with each IND and the safety sections of any final Clinical Trial reports within [***] following the date such information is filed, as applicable. All such information disclosed by Otsuka to Ionis will be Otsuka
Confidential Information; provided, however, that so long as Ionis does not disclose the identity of a Licensed Product or Otsuka’s identity, Ionis may
disclose any such Otsuka Confidential Information to (i) Ionis’ other partners if such information is regarding class generic properties of oligonucleotides, (ii) any Third Party that contributes to the populating of the Ionis Internal
Oligonucleotide Safety Database, or (iii) any Regulatory Authority. Otsuka will also cause its Affiliates and Sublicensees to comply with this Section 5.12 (Ionis Internal Oligonucleotide Safety Database).
|
|
|
5.12.2 |
From time to time, Ionis utilizes the information in the Ionis Internal Oligonucleotide Safety Database to conduct analyses to keep Ionis and its partners informed regarding class generic properties of oligonucleotides, including with
respect to safety. As such, if and when Ionis identifies safety or other related issues that may be relevant to a Licensed Product (including any potential class-related toxicity), Ionis will promptly inform Otsuka of such issues and provide
the data supporting Ionis’ conclusions.
|
| 5.13 |
Recall, Withdrawal, or Field Alerts.
|
|
|
5.13.1 |
Notification and Determination. Each Party will notify the other Party immediately, and promptly confirm such notice in writing, if it obtains information indicating that any Licensed Product may be
subject to a recall (whether voluntary or mandated), corrective action, or similar regulatory action by any Governmental Authority or Regulatory Authority (a “Remedial Action”). The
Parties will assist each other in gathering and evaluating such information as is necessary to determine the necessity of conducting a Remedial Action with respect to the applicable Territory, and otherwise reasonably cooperate with each
other with respect to such Remedial Action or potential Remedial Action. Ionis will have sole discretion and final decision-making authority with respect to, and control over, any Remedial Action in the Ionis Territory, including any decision
to commence such Remedial Action in the Ionis Territory. Otsuka will have sole discretion and final decision-making authority with respect to, and control over, any Remedial Action in the Otsuka Territory, including any decision to commence
such Remedial Action in the Otsuka Territory; provided that if Ionis notifies Otsuka of [***] that Ionis reasonably believes could give rise to a Remedial Action, then Otsuka will initiate such
Remedial Action in accordance with Ionis’ request and at [***].
|
|
|
5.13.2 |
Cost Allocation. Except as otherwise set forth in Section 5.13.1 (Notification and Determination), all costs directly associated with implementing a Remedial Action with respect to a Licensed
Product will be allocated between Ionis and Otsuka as follows:
|
|
|
(a) |
If, and to the extent, that the Remedial Action arises as a result of [***], then [***] will bear all such costs and expenses; and
|
|
|
(b) |
in all other cases, Ionis will be responsible for such costs and expenses for such Licensed Product in the Ionis Territory and Otsuka will be responsible for such costs and expenses for such Licensed Product in the Otsuka Territory.
|
| 6.1 |
Commercialization Responsibilities for Licensed Product.
|
|
|
6.1.1 |
Commercialization in the Ionis Territory. Subject to the last sentence of Section 6.4 (Global Brand Strategic and Operating Plan) and the last sentence of Section 6.7.1 (Global
Medical Affairs Plan), and without limiting Ionis’ obligations under this Article 6 (Commercialization and Medical Affairs), Ionis and its Affiliates will have [***] with respect to the Commercialization of the Licensed Products in
the Ionis Territory, including, if applicable, [***].
|
|
|
6.1.2 |
Commercialization in the Otsuka Territory. Subject to the terms and conditions of this Agreement, and without limiting Otsuka’s obligations under this Article 6 (Commercialization and Medical
Affairs), Otsuka and its Affiliates will have [***] with respect to the Commercialization of the Licensed Products in the Otsuka Territory, including [***]. Upon Otsuka’s reasonable request, Ionis shall [***], including [***], and Ionis will
[***].
|
|
|
6.1.3 |
Coordination of Commercialization Activities. The Parties will coordinate global Commercialization activities with respect to Commercialization of the Licensed
Products in each Party’s Territory through the JSC, as further set forth in Section 8.1 (Joint Steering Committee) and this Article 6 (Commercialization and Medical Affairs).
|
| 6.2 |
Commercialization and Medical Affairs Reporting. At each JSC meeting following the first Regulatory Approval for a Licensed Product in the Otsuka Territory, Otsuka will provide to the JSC a
high-level summary (which may be in the form of a slide presentation) of the material Commercialization and Medical Affairs activities conducted by Otsuka or its Affiliates or Sublicensees for the Licensed Products in the Otsuka Territory
during the period since the last JSC meeting and the material Commercialization and Medical Affairs activities expected to be conducted by Otsuka or its Affiliates, or Sublicensees in the Otsuka Territory for the Licensed Products during the
period from the date of such update until the next JSC meeting, and shall answer any reasonable questions asked by Ionis to enable Ionis to assess Otsuka’s compliance with its Commercialization diligence obligations set forth in Section
6.6 (Otsuka Commercialization Diligence Obligations). In addition, no later than [***], Otsuka will provide to the JSC a report of the forecasted Net Sales anticipated to be generated by Otsuka or its Affiliates, licensees, or
Sublicensees in the Otsuka Territory during the upcoming Calendar Year, which forecast will be broken down on a country-by-country basis. At each JSC meeting following the first Regulatory Approval for a Licensed Product in the Ionis
Territory, Ionis will provide to the JSC a high-level summary (which may be in the form of a slide presentation) of the material Commercialization and Medical Affairs activities conducted by Ionis or its Affiliates for the Licensed Products
in the Ionis Territory during the period since the last JSC meeting and the material Commercialization and Medical Affairs activities expected to be conducted by Ionis or its Affiliates in the Ionis Territory for the Licensed Products during
the period from the date of such update until the next JSC meeting. Without limiting the foregoing, at Otsuka’s reasonable request, Ionis will provide to Otsuka, [***], all information relating to the [***].
|
| 6.3 |
Pricing. All decisions for the Licensed Products related to list price, targeted net pricing, sales-weighted average discounts and rebates, pricing strategy (including the approach to pricing with
different types of accounts and plans, including types of discounts and rebates), and modifications to any of the foregoing, will be made by (a) Ionis in the Ionis Territory and (b) Otsuka in the Otsuka Territory; provided that, [***].
|
| 6.4 |
Global Brand Strategic and Operating Plan. Ionis will develop, [***] a global brand strategic and operating plan with respect to the Commercialization of the Licensed Products throughout the
Territory (the “Global Brand Strategic and Operating Plan”). The Global Brand Strategic and Operating Plan shall at all times conform to applicable Professional Requirements and
Applicable Law (including compliance requirements). Ionis, through the JSC, will update the Global Brand Strategic and Operating Plan [***]. In addition, [***], either Party may propose material updates or modifications to the Global Brand
Strategic and Operating Plan to [***]. No update or modification to the Global Brand Strategic and Operating Plan will be effective unless and until [***]. Once [***], such updated version of the Global Brand Strategic and Operating Plan will
automatically become effective and replace the then-prior version of the Global Brand Strategic and Operating Plan. The Global Brand Strategic and Operating Plan will include, in reasonable detail, the Trademarks to be used by the Parties or
its Affiliates or its or their Sublicensees for the Commercialization of Licensed Product, trade dress, positioning, market access strategy, and marketing strategic imperatives, objectives and messaging with respect to the Licensed Products. At Otsuka’s reasonable request, Ionis will provide to Otsuka, [***]. Ionis will, [***], lead and conduct all Commercialization activities for the Licensed Products in the Ionis Territory [***].
|
| 6.5 |
Otsuka Territory Brand Strategic and Operating Plan. Within [***] after [***] approves the initial Global Brand Strategic and Operating Plan, Otsuka will prepare and submit to [***] a brand strategic
and operating plan with respect to the Commercialization of the Licensed Products in the Otsuka Territory (such plan, the “Otsuka Territory Brand Strategic and Operating Plan”).
[***] will review, discuss and determine whether to approve the initial Otsuka Territory Brand Strategic and Operating Plan. The Otsuka Territory Brand Strategic and Operating Plan will, at all times during the Term, be consistent with the
then-current Global Brand Strategic and Operating Plan, except to the extent such inconsistency is (a) necessary to (i) conform with any written requirement from any Regulatory Authority or with any Applicable Law or Professional Requirements
in the Otsuka Territory or (ii) avoid infringement of a Third Party Trademark in the Otsuka Territory, or (b) approved by the JSC (by unanimous Party Vote). On [[***] during the Term (and more frequently as may be necessary), Otsuka will
prepare an update to the Otsuka Territory Brand Strategic and Operating Plan. [***] will review, discuss and determine whether to approve each update to the Otsuka Territory Brand Strategic and Operating Plan. Once approved by [***], the
Otsuka Territory Brand Strategic and Operating Plan will automatically become effective and, in the case of an update, will supersede the previous Otsuka Territory Brand Strategic and Operating Plan as of the date of such approval by [***].
Otsuka will, at its cost and expense, lead and conduct all Commercialization activities in the Otsuka Territory [***].
|
| 6.6 |
Otsuka Commercialization Diligence Obligations. On a country-by-country basis in the Otsuka Territory, following [***], Otsuka will use Commercially Reasonable Efforts to obtain Reimbursement
Approval for and otherwise Commercialize such Licensed Product in such country.
|
| 6.7 |
Medical Affairs Plans.
|
|
|
6.7.1 |
Global Medical Affairs Plan. Ionis will develop, and will [***], a plan for the global Medical
Affairs activities for the Licensed Products throughout the Territory (the “Global Medical Affairs Plan”). The Global Medical Affairs Plan shall at all times conform to applicable
Professional Requirements and Applicable Law (including compliance requirements) with adjustments necessary to comply with local Applicable Law and Professional Requirements in the Otsuka Territory. Ionis, through the JSC, will update the
Global Medical Affairs Plan on [***] basis. In addition, between [***] updates, either Party may propose material updates or modifications to the Global Medical Affairs Plan [***]. No update or modification to the Global Medical Affairs Plan
will be effective unless and until approved by [***]. Once approved by [***], such updated version of the Global Medical Affairs Plan will become effective and replace the then-prior version of the Global Medical Affairs Plan. Ionis will, at
its cost and expense, lead and conduct all Medical Affairs activities for the Licensed Products in the Ionis Territory [***].
|
|
|
6.7.2 |
Otsuka Territory Medical Affairs Plan. Within [***] after [***] approves the initial Global
Medical Affairs Plan, Otsuka will prepare and submit to [***] a plan for the Medical Affairs activities for the Licensed Products in the Otsuka Territory (such plan, the “Otsuka Territory
Medical Affairs Plan”). [***] will review, discuss and determine whether to approve the initial Otsuka Territory Medical Affairs Plan. The Otsuka Territory Medical Affairs Plan will, at all times during the Term, be consistent with
the then-current Global Medical Affairs Plan, except to the extent such inconsistency is (a) necessary to conform with any written requirement from any Regulatory Authority or with any Applicable Law or Professional Requirements in the Otsuka
Territory or (b) approved by the JSC (by unanimous Party Vote). On [***] during the Term (and more frequently as may be necessary), Otsuka will prepare an update to the Otsuka Territory Medical Affairs Plan. [***] will review, discuss, and
determine whether to approve each update to the Otsuka Territory Medical Affairs Plan. Once approved by [***], the Otsuka Territory Medical Affairs Plan will automatically become effective and, in the case of an update, supersede the previous
Otsuka Territory Medical Affairs Plan as of the date of such approval by the JSC. Otsuka will, at its cost and expense, lead and conduct all Medical Affairs activities in the Otsuka Territory [***].
|
| 6.8 |
Standards of Conduct; Compliance. Each Party will perform, or will ensure that each of its Affiliates, Sublicensees, and Subcontractors perform, all Commercialization and Medical Affairs activities
in a professional and ethical business manner and in compliance with Applicable Law and applicable Professional Requirements.
|
| 6.9 |
Product Materials. Each Party will, at its cost and expense, be responsible for preparing, developing, producing, or otherwise obtaining, and utilizing promotional materials, training materials,
medical education materials, Packaging and Labeling, and all other literature or other information related to the Licensed Products (“Product Materials”) to support its
Commercialization and Medical Affairs activities in such Party’s Territory, which Product Materials will at all times [***]. From time to time, and in any event upon Otsuka’s request, Ionis will share with Otsuka samples of Product Materials
Controlled by Ionis and which are used by Ionis, its Affiliates, or licensees in connection with the Commercialization of or conduct of Medical Affairs activities for the Licensed Products. From time to time, and in any event upon Ionis’
request, Otsuka will share with Ionis samples of Product Materials Controlled by Otsuka and which are used by Otsuka, its Affiliates or sublicensees in connection with the Commercialization of or conduct of Medical Affairs activities the
Licensed Products in the Otsuka Territory.
|
| 6.10 |
Diversion. Neither Party nor its Affiliates will, and each Party will take reasonable measures to ensure that its Sublicensees, licensees, and Subcontractors do not, either directly or to such
Party’s knowledge, intentionally indirectly, promote, market, distribute, import, sell, or have sold any Licensed Product to any Third Party or to any address or Internet Protocol
address or the like outside of such Party’s Territory including via the Internet or mail order. Notwithstanding any provision to the contrary set forth in this Agreement, [***]. As applicable, (i) in the case of Otsuka, in any country or
jurisdiction outside of the Otsuka Territory, and (ii) in the case of Ionis, in any country or jurisdiction outside of the Ionis Territory:
|
|
|
6.10.1 |
such Party and its Affiliates will not engage, nor permit its Sublicensees, licensees, and Subcontractors to engage, in any advertising or promotional activities relating to any Licensed Product for use directed primarily to customers or
other buyers or users of the Licensed Products located in any such country or jurisdiction;
|
|
|
6.10.2 |
such Party and its Affiliates will not solicit orders of the Licensed Products from any prospective purchaser located in any such country or jurisdiction;
|
|
|
6.10.3 |
such Party and its Affiliates will not, and will take reasonable measures to cause its Sublicensees, licensees and Subcontractors to not, deliver or tender (or cause to be delivered or tendered)
any Licensed Product to Third Parties for use in such country or jurisdiction; and
|
|
|
6.10.4 |
if either Party or its Affiliates, Sublicensees, or licensees receive any order for any Licensed Product from a prospective purchaser located in any such country or jurisdiction, then such Party will immediately refer that order to the
other Party or its designee and will not accept any such orders.
|
| 7.1 |
Responsibility.
|
|
|
7.1.1 |
Ionis Manufacturing. Ionis will have sole control over and decision-making authority with respect to, at its cost and expense, the Manufacture of (a) all supplies of the Licensed Products required
for Ionis’ activities under the Cross-Territory Clinical Development Plan and the Non-Clinical HAE Development Plan, and for all Development activities in the Ionis Territory and (b) all supplies of the Licensed Products for Commercialization
purposes in the Ionis Territory. In addition, but subject to Section 7.1.2 (Otsuka Manufacturing), in accordance with the Supply Agreements and Quality Agreements, Ionis will Manufacture and supply Otsuka with all Licensed Product
that is necessary for Otsuka to (i) [***] and (ii) [***]. Upon Otsuka’s reasonable request prior to the Manufacturing Handover Date, Ionis will provide (or will use commercially reasonable efforts to cause its CMOs to provide) to Otsuka (A)
[***], (B) [***], and (C) [***]. Ionis will provide all such Requested Assistance to Otsuka in accordance with the terms of Section 3.2 (Technology Transfer Cost) and will provide such data and information to Otsuka [***]. For
clarity, and notwithstanding anything to the contrary herein, Ionis will provide to the Qualified Person for the Licensed Products in the Otsuka Territory, [***], Manufacturing audit or inspection reports as required under EU GMP, Annex 16,
section 2.2 for the purposes of batch certification in the Otsuka Territory.
|
|
|
7.1.2 |
Otsuka Manufacturing. Otsuka will have the right to assume responsibility for Manufacturing (a) all supplies of the Licensed Products required for Otsuka’s activities under the Otsuka
Territory-Specific Development Plan and (b) all supplies of the Licensed Products for Commercialization purposes in the Otsuka Territory, in each case, upon written notice to Ionis at any time after the earliest of (i) [***], (ii) [***] and
(iii) [***] (such notice, a “Manufacturing Handover Notice”). If Otsuka provides Ionis with a Manufacturing Handover Notice, then Ionis’ obligations to Manufacture and supply Otsuka
in accordance with Section 7.1.1 (Ionis Manufacturing) will terminate, following the completion of all activities under the Manufacturing Technology Transfer Agreement in accordance with Section 7.4 (Manufacturing Technology
Transfer), and the initiation of actual Manufacturing of the Licensed Products to be sold by or on behalf of Otsuka at Otsuka’s or its designee’s manufacturing facility (such date, the “Manufacturing
Handover Date”).
|
| 7.2 |
Supply and Quality Agreements; Manufacturing Costs.
|
|
|
7.2.1 |
Clinical Supply Agreement. Unless otherwise agreed by the Parties, within a timeframe following the Effective Date to be agreed by the Parties, the Parties will negotiate in good faith and enter into
a supply agreement on reasonable and customary terms for the supply of Licensed Products by Ionis to Otsuka for clinical use (the “Clinical Supply Agreement”), which agreement
(together with the related Quality Agreement) will govern the terms and conditions of the Manufacture and supply of the Licensed Products for Development purposes in the Otsuka Territory. Otsuka will pay a supply price to Ionis under the
Clinical Supply Agreement equal to [***].
|
|
|
7.2.2 |
Commercial Supply Agreement. Within a timeframe following the Effective Date to be agreed by the Parties, the Parties will negotiate in good faith and enter into a commercial supply agreement on
reasonable and customary terms for the commercial-grade supply of Licensed Products by Ionis to Otsuka (the “Commercial Supply Agreement” and together with the Clinical Supply
Agreement, the “Supply Agreements”), which agreement (together with the related Quality Agreement) will govern the terms and conditions of the Manufacture and supply of the Licensed
Products for Commercialization purposes in the Otsuka Territory. Otsuka will pay a supply price to Ionis under the Commercial Supply Agreement equal to [***]. The Commercial Supply Agreement shall allow Otsuka to order, and Ionis to supply, a
portion of a batch as minimum order quantity, for a duration of at least [***], as further detailed in such Commercial Supply Agreement.
|
|
|
7.2.3 |
Quality Agreements. The Parties will negotiate in good faith and enter into one or more quality technical agreements pertaining to clinical and commercial supply of Licensed Products to Otsuka (each,
a “Quality Agreement”) containing reasonable and customary terms and conditions regarding quality assurance, quality control, compliance with GMP, GDP and GCP (as applicable),
specifications, change control procedures, and provisions relating to audits and inspections.
|
|
|
7.2.4 |
Manufacturing Cost Increases. If the Manufacturing Costs, whether for clinical or commercial supplies of Licensed Product, are reasonably anticipated to increase, on a per unit basis, such that the
[***], then Ionis will provide prompt written notice to Otsuka of such increase. If such increase is anticipated to result in [***], on a per unit basis, then [***]. If such increase is anticipated to result in [***] on a per unit basis, then
[***].
|
|
|
7.2.5 |
Capital Expenditures. Ionis [***]. In addition, if any CMO requests or requires [***], then Ionis will notify Otsuka and the Parties will [***].
|
|
|
7.2.6 |
[***]. If the reasonable allocation of [***], then [[***].
|
| 7.3 |
Audits and Inspections.
|
|
|
7.3.1 |
By Otsuka. Prior to execution of the first Quality Agreement, Otsuka shall be entitled to conduct [***]. In addition, if Ionis elects to inspect or audit any facilities of its CMOs with respect to
the Manufacture of Licensed Products for the Otsuka Territory, Ionis shall notify Otsuka of such inspection or audit and, [***]. In addition, to the extent permitted under Ionis’ agreement with the applicable CMO and subject to any conditions
set forth in such agreement with respect to any inspection or audit (e.g., an obligation to enter into a confidentiality agreement with the applicable CMO), Ionis shall [***]. If Otsuka identifies the need to perform a “for cause” audit of
such facilities to address quality or compliance issues related to any Licensed Product Manufactured for the Otsuka Territory (including to address any notice from a Governmental Authority in the Otsuka Territory of noncompliance with
Applicable Laws), as well as in connection with the preparation of Regulatory Submissions for the Otsuka Territory and in response to Regulatory Authority requirements in the Otsuka Territory, then Otsuka shall notify Ionis and if Ionis
agrees with Otsuka’s determination that a “for cause” audit is needed, Ionis will schedule and conduct such audit and Otsuka will [***], in each case, to the extent permitted pursuant to the applicable agreement with the such CMO.
|
|
|
7.3.2 |
By Governmental Authority. If any Governmental Authority carries out or gives notice of its intention to carry out any inspection or audit of any of Ionis’ CMOs in relation to Manufacture of Licensed
Products for the Otsuka Territory and Ionis is aware of such upcoming inspection or audit, then Ionis shall promptly notify Otsuka thereof and Ionis shall, to the extent permitted by its agreement with the applicable CMO and the applicable
Governmental Authority, [***]. Following receipt by Ionis of the inspection results or audit observations of the Governmental Authority from such inspection or audit (a redacted copy of which Ionis will promptly provide to Otsuka to the
extent it relates to Licensed Products Manufactured for the Otsuka Territory), Ionis will (a) prepare any appropriate responses and (b) provide a copy of such responses to Otsuka [***] in advance of the date such responses are due, to the
extent such responses pertain to the Manufacture of Licensed Products for the Otsuka Territory, and Ionis shall [***], in each case ((a) and (b)), to the extent permitted under Ionis’ agreement with such CMOs and subject to any conditions set
forth in the applicable agreement with such CMOs with respect to any inspection or audit (e.g., an obligation to enter into a confidentiality agreement with the applicable CMO).
|
|
|
7.3.3 |
CMO Agreements. Ionis shall [***].
|
| 7.4 |
Manufacturing Technology Transfer. At Otsuka’s request any time after Otsuka provides a Manufacturing Handover Notice, Ionis will make available to Otsuka all Ionis Manufacturing and Analytical
Know-How and materials (the “Manufacturing Technology Transfer”). Otsuka will (a) use Ionis Manufacturing and Analytical Know-How and materials provided by Ionis in connection with
the Manufacturing Technology Transfer only in the fulfillment of obligations or exercise of rights under this Agreement, and (b) not transfer such Ionis Manufacturing and Analytical Know-How or materials or deliver the same to any Third
Party, without Ionis’ prior written consent. For purposes of the Manufacturing Technology Transfer, the Parties together with Ionis’ CMO (subject to the next sentence) will enter into a manufacturing technology transfer agreement, which will
also provide for reasonable technical assistance and support by Ionis and Ionis’ CMOs as reasonably requested by Otsuka to enable Otsuka or its Affiliates, or if agreed by Ionis, a Third Party manufacturer (other than Ionis’ CMOs), to
Manufacture the Licensed Products (“Manufacturing Technology Transfer Agreement”). Ionis will use reasonable efforts to [***]. If Ionis agrees to transfer Ionis Manufacturing and
Analytical Know-How to a Third Party manufacturer other than Ionis’ CMOs, such transfer shall be carried out pursuant to a direct license between Ionis and such Third Party manufacturer. If Otsuka [***]. Otsuka will [***]. Accordingly, Ionis
may [***]. Each Party will [***].
|
| 8.1 |
Joint Steering Committee.
|
|
|
8.1.1 |
Formation and Purpose of the JSC. Promptly, but not more than [***] after the Effective Date, Ionis and Otsuka will establish a Joint Steering Committee (“JSC”), which will have the responsibilities set forth in this Article 8 (Governance) and will oversee, review, monitor, coordinate, and, where specified in this Section 8.1 (Joint Steering Committee), approve
the Parties’ Development, Manufacturing, Medical Affairs, and Commercialization activities under this Agreement for the Licensed Products in the Territory in accordance with this Section 8.1 (Joint Steering Committee). The JSC will
dissolve upon the expiration of the Term.
|
|
|
8.1.2 |
Membership. The JSC will be composed of an equal number of representatives from each Party who have the appropriate and direct knowledge and expertise and requisite decision-making authority. Any
such representative who serves on the JSC or any committee under this Agreement may also serve on one or more other committees under this Agreement. Each Party may replace any of its representatives on the JSC and appoint a person to fill the
vacancy arising from each such replacement. A Party that replaces a representative will notify the other Party at least [***] prior to the next scheduled meeting of the JSC. Ionis will designate one of its JSC members as one of the
co-chairpersons of the JSC and Otsuka will designate one of its members as the other co-chairperson of the JSC (each, a “JSC Co-Chairperson”). The JSC Co-Chairpersons or their designees, in collaboration with the Alliance Managers, will be responsible for calling meetings, preparing and circulating an agenda
and related information in advance of each meeting, and preparing and issuing minutes of each meeting within [***] thereafter. Such minutes will not be finalized until the JSC Co-Chairpersons or their designees have had [***] to review and
confirm the accuracy of such minutes.
|
|
|
8.1.3 |
Meetings. The JSC will hold meetings at such times as it elects to do so, but will meet no less frequently than quarterly prior to [***] and thereafter no less frequently than [***], in each case,
unless otherwise agreed by the Parties. The JSC may meet in person or by means of teleconference, Internet conference, video conference, or other similar communication method. Each Party will be responsible for all of its own costs and
expenses of participating in any JSC meeting.
|
|
|
8.1.4 |
Meeting Agendas. Unless agreed otherwise by the Parties, the Parties will jointly prepare the agenda for each JSC meeting, facilitated by the Alliance Managers working closely with the JSC
Co-Chairpersons and, as appropriate, other JSC members and Subcommittee co-chairpersons, at least [***] in advance of each meeting of the JSC, and each Party will provide the other Party with all relevant materials to be presented at each JSC
meeting at least [***] in advance of each meeting of the JSC; provided that under exigent circumstances requiring JSC input, the agenda may be prepared or presentation materials may be provided within
a shorter period of time in advance of a meeting, with the approval of the JSC Co-Chairpersons. Either Party may propose that there not be a specific agenda for a particular meeting, so long as the other Party consents to the later addition
or modification of agenda items or the absence of a specific agenda for such JSC meeting.
|
|
|
8.1.5 |
Specific Responsibilities of the JSC. The responsibilities of the JSC will be to:
|
|
|
(a) |
manage the overall strategic alignment between the Parties under this Agreement and maintain the relationship between the Parties;
|
|
|
(b) |
review, discuss, and determine whether to [***];
|
|
|
(c) |
review, discuss, and determine whether [***];
|
|
|
(d) |
review, discuss, and determine whether [***];
|
|
|
(e) |
review, discuss, and determine whether [***];
|
|
|
(f) |
review, discuss and determine whether [***];
|
|
|
(g) |
review, discuss, and determine whether to approve any updates to the Shared Development Budget, as described in Section 4.4.2(a) (Shared Development Budget);
|
|
|
(h) |
review, discuss, and determine whether to approve [***] and the Shared Development Budget, as described in Section 4.4.4(a) (Shared Costs);
|
|
|
(i) |
share information related to, and review and discuss activities and progress of each Party in connection with the Development of Licensed Products in its Territory, including activities and progress under the Cross-Territory Clinical
Development Plan, Non-Clinical HAE Development Plan, and the Otsuka Territory-Specific Development Plan, including through updates from each Party of the status of Development for the Licensed Products in each Party’s Territory, as described
in Section 4.5 (Development Reports);
|
|
|
(j) |
review and discuss any matters related to the Development of the Licensed Products referred to the JSC by either Party’s representatives;
|
|
|
(k) |
discuss any concerns raised by either Party regarding any action that the other Party is taking or intends to take with respect to a Licensed Product that is [***], as described in Section 5.8 (No Harmful Actions);
|
|
|
(l) |
discuss [***];
|
|
|
(m) |
review, discuss, and determine whether to approve [***];
|
|
|
(n) |
review, discuss, and determine whether to approve [***];
|
|
|
(o) |
review, discuss, and determine whether to approve [***];
|
|
|
(p) |
review, discuss and determine whether to approve [***];
|
|
|
(q) |
review and discuss any matters related to the Commercialization of the Licensed Products referred to the JSC by either Party’s representatives;
|
|
|
(r) |
discuss the inclusion of Ionis’ logo, name, and housemark on the packaging for the Licensed Products in the Otsuka Territory, as described in Section 10.10.6 (Housemarks);
|
|
|
(s) |
establish and delegate specifically defined duties to any Subcommittees, as described in Section 8.2.1 (Formation; Authority);
|
|
|
(t) |
attempt to resolve any disputes or disagreements arising from matters within the jurisdiction of any Subcommittee; and
|
|
|
(u) |
perform such other functions as appropriate to further the purposes of this Agreement as determined by the Parties.
|
| 8.2 |
Additional Committees.
|
|
|
8.2.1 |
Formation; Authority. In addition to the Regulatory Subcommittee and the PV Subcommittee, the JSC may establish and delegate specifically-defined duties to operational committees or ad hoc subcommittees, on an “as needed” basis to oversee particular projects or activities (any such operational committees and subcommittees, including the Regulatory Subcommittee, a “Subcommittee”). Each such Subcommittee will be constituted and will operate as the JSC determines. Each Subcommittee and its activities will be subject to the oversight of, and will
report to, the JSC. The JSC or the JSC Co-Chairpersons, in each case as mutually agreed, may delegate to a Subcommittee any responsibilities of the JSC set forth in Section 8.1.5 (Specific Responsibilities of the JSC), and, in such
case, any agreement reached by unanimous Party Vote of the applicable Subcommittee with respect to such delegated responsibilities will be deemed to be approved by the JSC (to the extent such approval is required hereunder). The JSC or the
JSC Co-Chairpersons acting together may also reallocate any responsibility of a Subcommittee to any other Subcommittee. No Subcommittee’s authority may exceed that specified for the JSC in this Article 8 (Governance). Any disagreement
between the representatives of the Parties on a Subcommittee will be referred to the JSC for resolution in accordance with Section 8.4 (Decision-Making).
|
|
|
8.2.2 |
Subcommittee Leadership and Meetings. Ionis will designate a co-chairperson of each Subcommittee and Otsuka will designate a co-chairperson of each Subcommittee, each of whom will be a Party’s
representative who is a member of such Subcommittee (each, a “Subcommittee Co-Chairperson”).
The Subcommittee Co-Chairpersons or their designees, in collaboration with the Alliance Managers, will be responsible for calling meetings, preparing and circulating an agenda in advance of each meeting, and preparing and issuing minutes of
each meeting promptly thereafter. Such minutes will not be finalized until all Subcommittee members have had [***] to review and confirm the accuracy of such minutes. Each Party may replace its representatives and Subcommittee Co-Chairpersons
on each such Subcommittee at any time upon written notice to the other Party. The Alliance Manager of each Party (or his or her designee) will attend each meeting of each Subcommittee as a non-voting participant. Each Subcommittee will hold
meetings at such times as it elects to do so, and at such locations as the Parties may agree upon or by means of teleconference, Internet conference, video conference, or other similar communication method. Each Party will be responsible for
all of its own expenses of participating in any Subcommittee meeting.
|
| 8.3 |
Additional Participants. Employees of a Party or any of its Affiliates involved in the Exploitation of the Licensed Products may attend meetings of the JSC or any Subcommittee as non-voting
participants. In addition, with the prior consent of each Party, consultants, representatives, or advisors involved in the same activities and under written obligations of confidentiality and non-use applicable to the Confidential Information
of each Party that are at least as stringent as those set forth in Article 12 (Confidentiality) may attend meetings of the JSC or any Subcommittee as non-voting observers.
|
| 8.4 |
Decision-Making.
|
|
|
8.4.1 |
General Decision-Making Process. Each Party’s representatives on the JSC and each Subcommittee will, collectively, have one vote (the “Party Vote”)
on all matters brought before such committee for a decision by consensus. The JSC and each Subcommittee will make decisions as to matters within its jurisdiction by unanimous Party Vote, which may be reflected in the minutes of the committee
meeting or by an action by written consent signed by the JSC Co-Chairpersons or their designees identified in writing. Except as otherwise expressly set forth in this Agreement, the phrase “determine,” “designate,” “approve,” or “determine
whether to approve” by the JSC or any Subcommittee and similar phrases used in this Agreement will mean approval in accordance with this Section 8.4 (Decision‑Making), including the escalation and tie‑breaking provisions herein. For
the avoidance of doubt, matters that are specified in Section 8.1.5 (Specific Responsibilities of the JSC) to be reviewed and discussed (as opposed to reviewed, discussed, and approved) do not require any agreement or decision by
either Party and are not subject to the voting and decision-making procedures set forth in this Section 8.4 (Decision‑Making) or Section 8.5 (Resolution of Committee Disputes).
|
|
|
8.4.2 |
Decisions of the Subcommittees. If any Subcommittee cannot reach unanimous agreement using good faith efforts on any matter within their respective scope of authority within [***] of the meeting at
which such matter was discussed, then a Party may refer such matter to the JSC for resolution in accordance with Section 8.4.3 (Decisions of the JSC).
|
|
|
8.4.3 |
Decisions of the JSC. The JSC will use good faith efforts, in compliance with this Section 8.4.3 (Decisions of the JSC), to promptly resolve any such matter for which it has authority. If,
after the use of good faith efforts, including reasonable discussion and good faith consideration of each Party’s view on a particular matter, the JSC is unable to resolve any such matter referred to it by any Subcommittee or any matter with
respect to the matters within the scope of the JSC’s authority, in each case, within a period of [***], then either Party may refer such matter to the Party’s respective Executive Officer for resolution in accordance with Section 8.5.1
(Referral to Executive Officers).
|
| 8.5 |
Resolution of Committee Disputes.
|
|
|
8.5.1 |
Referral to Executive Officers. If a Party makes an election under Section 8.4.3 (Decisions of the JSC) to refer for resolution by the Executive Officers a matter as to which the JSC cannot
reach a consensus decision, then the JSC will submit in writing the respective positions of the Parties to their respective Executive Officers. The Executive Officers will use good faith efforts to resolve any such matter so referred to them
as soon as practicable but, in any event, within [***] after such matter is referred to them (or such longer period as the Executive Officers may agree upon), and any final decision that the Executive Officers agree to in writing will be
conclusive and binding on the Parties.
|
|
|
8.5.2 |
Final Decision-Making Authority. If the Executive Officers are unable to reach agreement on any such matter so referred within [***] after such matter is referred to them (or such longer period as
the Executive Officers may agree upon), then, subject to Section 8.5.3 (Limitations on Decision Making):
|
|
|
(a) |
No Change; Status Quo. Neither Party will have final decision-making authority with respect to the final resolution of any disagreement related to: (i) [***]; (ii) [***]; (iii) [***]; (iv) [***]; (v)
[***]; and (vi) [***].
|
|
|
(b) |
Ionis Final Decision-Making Authority. Ionis will have final decision-making authority over (i) [***], (ii) [***], (iii) [***], (iv) [***], and (vi) [***]. Notwithstanding the foregoing, [***].
|
|
|
(c) |
Otsuka Final Decision-Making Authority. Otsuka will have final decision making authority over (i) [***], (ii) [***], (iii) [***], and (iv)
[***].
|
|
|
8.5.3 |
Limitations on Decision Making. Notwithstanding anything to the contrary set forth in this Agreement, without the other Party’s prior written consent, no decision of the JSC, any Subcommittee, or a
Party’s Executive Officer (in the exercise of a Party’s decision‑making authority on any such matters), in each case may, without the other Party’s prior written consent, (a) be likely to [***], (b) impose any requirements that the other
Party take or decline to take any action that a Party reasonably believes would result in a violation of any Applicable Law, the requirements of any Regulatory Authority, or any agreement with any Third Party (including any Collaboration
In-License) or the infringement or misappropriation of intellectual property rights of any Third Party, or (c) conflict with, amend, interpret, modify, or waive compliance under this Agreement.
|
| 8.6 |
Day-to-Day Responsibilities. Each Party will: (a) be responsible for day-to-day implementation and conduct of the activities hereunder for which it has or is otherwise assigned responsibility under
this Agreement, provided that such implementation is consistent with the express terms of this Agreement or the decisions of the JSC within the scope of its authority as provided herein; and (b)
provide the other Party with information about material events related to the progress of such activities, as may be reasonably requested by the other Party from time to time.
|
| 8.7 |
Alliance Managers. Each of the Parties will appoint a representative of such Party to act as its alliance manager under this Agreement no later than [***] after the Effective Date (each, an “Alliance Manager”). The role of the Alliance Manager is to act as a single point of contact between the Parties to ensure a successful relationship under this Agreement. The Alliance
Managers will attend all JSC meetings and the Alliance Managers or their respective designees will attend all Subcommittee meetings and will support the JSC Co-Chairpersons and any Subcommittee Co-Chairpersons in the discharge of their
responsibilities. Alliance Managers will be non-voting participants in all JSC and Subcommittee meetings, but an Alliance Manager may bring any matter to the attention of the JSC or any Subcommittee if such Alliance Manager reasonably
believes that such matter warrants such attention. Each Party may change its designated Alliance Manager at any time upon written notice to the other Party. Any Alliance Manager may designate a substitute to temporarily perform the functions
of that Alliance Manager by written notice to the other Party. Each Alliance Manager will also: (a) be the point of first referral in all matters of conflict resolution; (b) provide a single point of communication for seeking consensus
between the Parties regarding key strategy and plan issues; (c) identify and bring disputes to the attention of the JSC in a timely manner; (d) plan and coordinate cooperative efforts and internal and external communications; and (e) take
responsibility for ensuring that governance activities, such as the conduct of required JSC and any Subcommittee meetings and production of meeting minutes, occur as set forth in this Agreement, and that the relevant action items resulting
from such meetings are appropriately carried out or otherwise addressed.
|
| 9.1 |
Upfront Payment. Following the Effective Date, within [***], Otsuka will pay to Ionis, by wire transfer of immediately available funds, a non-refundable, non-creditable upfront payment of $65,000,000
(the “Upfront Payment”).
|
| 9.2 |
Milestone Payments.
|
|
|
9.2.1 |
Regulatory Milestones. Subject to Section 9.2.1(a) (Regulatory Milestone Adjustment), after the first achievement of each regulatory milestone event set forth in Table 9.2.1 below by Otsuka
or its Affiliates or Sublicensees for the first Licensed Product, Otsuka will pay to Ionis the corresponding regulatory milestone payment set forth in Table 9.2.1 (the regulatory milestone events set forth in Table 9.2.1, the “Regulatory Milestone Events” and the regulatory milestone payments set forth in Table 9.2.1, the “Regulatory Milestone Payments”).
|
|
Table 9.2.1 –Regulatory Milestones
|
||
|
Regulatory Milestone Event
|
Regulatory Milestone
Payment (in U.S.
Dollars)
|
|
|
1. [***]
|
$[***]
|
|
|
2. [***]
|
$[***]
|
|
|
3. [***]
|
$[***]
|
|
|
|
(a) |
Regulatory Milestone Adjustment. If any [***], then the Regulatory Milestone Payments will be [***].
|
|
|
(b) |
Notice and Payment. Otsuka will notify Ionis in writing of the achievement of each Regulatory Milestone Event within [***] after achievement of such Regulatory Milestone Event by Otsuka or its
Affiliates or within [***] after Otsuka’s receipt of notification of such achievement by its Sublicensees. However, in no event will a failure or delay by Otsuka to deliver such
notice of achievement of a Regulatory Milestone Event relieve Otsuka of its obligation to pay Ionis the corresponding Regulatory Milestone Payment for achievement of such Regulatory Milestone Event. Following receipt of such notice, Ionis
will send Otsuka an invoice (and, if there has been any change to a Payment Form previously submitted, or if a previously submitted Payment Form has expired, then an updated Payment Form) for the applicable Regulatory Milestone Payment, and
Otsuka shall pay such Regulatory Milestone Payment within [***] after receipt of such invoice (and Payment Forms, if applicable). Each Regulatory Milestone Payment is payable only once, regardless of
the number of times the corresponding Regulatory Milestone Event is achieved. If Otsuka or its Affiliates or Sublicensees achieve all of the Regulatory Milestone Events, then the Regulatory Milestone Payments payable by Otsuka under this Section
9.2.1 (Regulatory Milestones) will not exceed $[***].
|
|
|
9.2.2 |
Sales Milestones. After each sales milestone event set forth in Table 9.2.2 below is achieved by Otsuka or its Affiliates or Sublicensees for the Licensed Products in the Otsuka Territory, Otsuka
will pay to Ionis the corresponding sales milestone payment, as set forth below (the sales milestone events set forth in Table 9.2.2, the “Sales Milestone Events” and the sales
milestone payments set forth in Table 9.2.2, the “Sales Milestone Payments”).
|
|
Table 9.2.2 – Sales Milestones
|
||
|
Sales Milestone Event
|
Sales Milestone
Payment
(in U.S. Dollars)
|
|
|
First Calendar Year in which the aggregate annual Net Sales of the Licensed Products in the Otsuka Territory equal or exceed [***]
|
$[***]
|
|
|
First Calendar Year in which the aggregate annual Net Sales of the Licensed Products in the Otsuka Territory equal or exceed [***]
|
$[***]
|
|
|
First Calendar Year in which the aggregate annual Net Sales of the Licensed Products in the Otsuka Territory equal or exceed [***]
|
$[***]
|
|
|
|
(a) |
Notice and Payment. Otsuka will notify Ionis in writing of the achievement of each Sales Milestone Event no later than (i) [***] or (ii) [***]. However, in no event will a failure or delay by Otsuka to deliver such notice of achievement of a Sales Milestone Event relieve Otsuka of its obligation to pay Ionis the corresponding Sales Milestone Payment for achievement of such Sale
Milestone Event. Following receipt of such notice, Ionis will send Otsuka an invoice (and, if there has been any change to a Payment Form previously submitted, or if a previously submitted Payment Form has expired, then an updated Payment
Form) for the applicable Sales Milestone Payment, and Otsuka shall pay such Sales Milestone Payment within [***] after receipt of such invoice (and Payment Forms, if applicable). If more than one of the Sales Milestone Events is achieved for
the first time in a given Calendar Quarter during the Term, then Otsuka will pay to Ionis a separate Sales Milestone Payment with respect to each such Sales Milestone Event. Each Sales Milestone Payment is
payable only once, regardless of the number of times the corresponding Sales Milestone Event is achieved. If Otsuka or its Affiliates or Sublicensees achieve all of the Sales Milestone Events, then the Sales Milestone Payments payable by
Otsuka under this Section 9.2.2 (Sales Milestones) will not exceed $[***].
|
| 9.3 |
Royalties.
|
|
|
9.3.1 |
Royalty Payments During the Initial Royalty Term. Subject to the provisions of Section 9.3.2 (Royalty Reductions), Otsuka will pay to Ionis royalties based on the Net Sales of a Licensed
Product by Otsuka and its Affiliates and Sublicensees in the Otsuka Territory at the rates set forth in Table 9.3.1 below (the “Initial Royalties”), on a Licensed Product-by-Licensed
Product and country-by-country basis, commencing on the first sale of such Licensed Product that results in Net Sales of such Licensed Product in such country and ending on the latest to occur of (a) the [***] anniversary of the First
Commercial Sale of such Licensed Product in such country, (b) the expiration of the last Valid Claim in the [***] that Cover such Licensed Product in such country [***], and (c) loss of Regulatory Exclusivity of such Licensed Product in such
country (the “Initial Royalty Term”).
|
|
Table 9.3.1– Royalty Rates for the Licensed Products
|
||
|
Calendar Year Net Sales of all Licensed Products in the Otsuka
Territory
|
Royalty
Rate
|
|
|
Portion of annual Net Sales of all Licensed Products in the Otsuka Territory that is [***]
|
[***]%
|
|
|
Portion of annual Net Sales of all Licensed Products in the Otsuka Territory that are [***]
|
[***]%
|
|
|
Portion of annual Net Sales of all Licensed Products in the Otsuka Territory that are [***]
|
[***]%
|
|
|
|
9.3.2 |
Royalty Reductions.
|
|
|
(a) |
Generic Approval. On a country-by-country and Licensed Product-by-Licensed Product basis, if at any time during the Initial Royalty Term a Generic Product receives Regulatory Approval in a country in
the Otsuka Territory, then, subject to Section 9.3.2(e) (Royalty Reductions Floor), the royalty rates set forth in Table 9.3.1 will be reduced by [***] for such Licensed Product in such country.
|
|
|
(b) |
Third Party Payments. Subject to Section 9.3.2(e) (Royalty Reductions Floor), Otsuka may credit [***] of [***] in a country in the Otsuka Territory in a Calendar Quarter during the
Royalty Term against the Royalties due and payable by Otsuka to Ionis on the Net Sales for such Licensed Product in such country in such Calendar Quarter; provided that the terms of this Section 9.3.2(b) (Third Party Payments) will not apply to any license agreement entered into without Ionis’ prior written consent in violation of the terms of Section 10.5.3
(Settlement). For clarity, Otsuka will not have the right to offset any Third Party Payments arising out of, or allocable to, the Manufacture of a Licensed Product.
|
|
|
(c) |
[***]. Subject to Section 9.3.2(e) (Royalty Reductions Floor), on a Licensed Product-by-Licensed Product and country-by-country basis, if during any Calendar Quarter during the Initial Royalty Term for such Licensed Product in such
country, (i) [***], and (ii) [***], then, commencing [***]; provided that, if [***], then [***].
|
|
|
(d) |
[***]. Subject to Section 9.3.2(e) (Royalty Reductions Floor), on a Licensed Product-by-Licensed Product and country-by-country basis, during the Initial Royalty Term for such Licensed Product in such country, if, [***].
|
|
|
(e) |
Royalty Reductions Floor. In no event will the Royalties due to Ionis for a Licensed Product in a country in the Otsuka Territory [***] set forth in this Section 9.3.2 (Royalty
Reductions). Notwithstanding the foregoing, [***].
|
|
|
9.3.3 |
Reduced Royalty Term. On a Licensed Product-by-Licensed Product and country-by-country basis in the Otsuka Territory, following expiration of the Initial Royalty Term for a Licensed Product in a
given country in the Otsuka Territory, Otsuka will pay Ionis a [***] royalty on the Net Sales of such Licensed Product by Otsuka and its Affiliates and Sublicensees in such country (the “Reduced
Royalties” and together with the Initial Royalties, the “Royalties”) until the later of (a) [***], and (b) [***] (the “Reduced
Royalty Term” and together with the Initial Royalty Term, the “Royalty Term”). For clarity, on a Licensed Product-by-Licensed Product and country-by-country basis, [***].
|
|
|
9.3.4 |
Royalty Payments and Reports.
|
|
|
(a) |
[***]. Commencing with the Calendar Quarter during which the first sale of a Licensed Product is made that results in Net Sales anywhere in the Otsuka Territory, [***].
|
|
|
(b) |
Royalty Report. Commencing with the Calendar Quarter during which the first sale of a Licensed Product is made that results in Net Sales anywhere in the Otsuka Territory, within [***] after the end
of each Calendar Quarter, Otsuka will provide to Ionis a written report (each, a “Royalty Report”) setting forth in reasonable detail: (i) the gross sales of the Licensed Products
sold by Otsuka or its Affiliate or Sublicensee in the Otsuka Territory in such Calendar Quarter; (ii) the aggregate Net Sales of the Licensed Products sold by Otsuka or its Affiliates or Sublicensees in the Otsuka Territory in such Calendar
Quarter; (iii) all deductions and reductions used to determine the Net Sales of the Licensed Products for such Calendar Quarter or the Royalties payable with respect to the Licensed Products for such Calendar Quarter, including any reduction
pursuant to Section 9.3.2 (Royalty Reductions) (if applicable); (iv) the exchange rates used to calculate the Royalties payable in U.S. Dollars; (v) any withholding taxes required to be made from such Royalties; and (vi) the quantity
and description of the Licensed Products sold by Otsuka or its Affiliate or Sublicensee in the Otsuka Territory during such Calendar Quarter comprising such Net Sales. The Parties will seek to resolve any questions or issues related to a
Royalty Report within [***] following receipt by Ionis of each Royalty Report.
|
|
|
(c) |
Royalty Payments. The information contained in each Royalty Report will be considered the Confidential Information of Otsuka. Following receipt of each Royalty Report, Ionis will [***] and, [***],
Otsuka will pay the Royalties due hereunder for the Calendar Quarter covered by the applicable Royalty Report.
|
| 9.4 |
Other Amounts Payable. With respect to any amounts owed under this Agreement by one Party to the other for which no other invoicing and payment procedure is specified hereunder, within [***] after
the end of each Calendar Quarter, each Party will provide an invoice, together with reasonable supporting documentation, to the other Party for such amounts owed in respect of such Calendar Quarter. The owing Party will pay any undisputed
invoiced amounts within [***] after the date of the invoice, and any disputed amounts owed by a Party will be paid within [***] following resolution of the dispute.
|
| 9.5 |
Financial Records and Audits. Each Party will, and will require its Sublicensees and Subcontractors to, maintain complete and accurate records in accordance with such Party’s Accounting Standards in
sufficient detail to permit the other Party to confirm the accuracy of any amounts payable under this Agreement for at least the preceding [***], including (as applicable) any Eligible Cross-Territory Development Costs, Milestone Payments,
Royalties, and sales of the Licensed Products (including all calculations of Net Sales). Upon reasonable prior notice, each Party agrees to permit such records to be open during regular business hours for examination by an independent
certified public accountant selected by the auditing Party and reasonably acceptable to the audited Party for the sole purpose of verifying the accuracy of the financial reports furnished by the audited Party pursuant to this Agreement or of
any payments made, or required to be made, by the audited Party pursuant to this Agreement; provided that such independent accounting firm is subject to
written obligations of confidentiality and non-use applicable to each Party’s Confidential Information that are at least as stringent as those set forth in Article 12 (Confidentiality). Such audit will not be (a) performed more
frequently than [***], or (b) repeated for any Calendar Year or with respect to the same set of records (in each case, except for cause). Such auditor will not disclose the audited Party’s Confidential Information to the auditing Party or to
any Third Party, except to the extent such disclosure is necessary to verify the accuracy of the financial reports furnished by the audited Party or the amount of payments by the audited Party under this Agreement. The audited Party will pay
any amounts shown to be owed to the auditing Party but unpaid within [***] after the accountant’s report, plus interest (as set forth in Section 9.11 (Late Payments; Disputed Payments)) from
the original due date solely if the audited Party is responsible for the discrepancy. If such examination of records reveals any overpayment by Ionis, then Otsuka will reimburse Ionis for the amount overpaid within [***] after the
accountant’s report, plus interest (as set forth in Section 9.11 (Late Payments; Disputed Payments)) from the original due date [***]. If such examination of records reveals any overpayment by
Otsuka, then [***]. The auditing Party will bear the full cost of such audit unless such audit reveals an underpayment by the audited Party of more than [***] of the amount actually due for the time period being audited, in which case the
audited Party will reimburse the auditing Party for the reasonable audit fees for such examination.
|
| 9.6 |
No Refunds. Except as expressly provided herein, all payments under this Agreement will be irrevocable, non-refundable, and non-creditable.
|
| 9.7 |
Accounting Standards. If a Party changes its general accounting principles from its then-current Accounting Standard (e.g., from GAAP to IFRS) at any time
during the Term, then at least [***] prior to adopting such change in principles, such Party will provide written notice to the other Party of such change. A Party may not change its general accounting principles to any accounting standard
other than GAAP or IFRS without the prior written approval of the other Party.
|
| 9.8 |
Method of Payment; Exchange Rate. All amounts to be paid pursuant to this Agreement will be made in U.S. Dollars and will be paid by wire transfer in immediately available funds to a bank account
designated by the receiving Party. The rate of exchange to be used in computing the amount of currency equivalent in U.S. Dollars owed to a Party under this Agreement will be the Selling Party’s then-current standard exchange rate methodology
employed for the translation of foreign currency sales into U.S. Dollars in accordance with its Accounting Standards and consistently applied during the period.
|
| 9.9 |
Blocked Payments. If by reason of Applicable Law in any country or jurisdiction, it becomes impossible or illegal for a Party to transfer, or have transferred on its behalf, payments owed the other
Party hereunder, then such Party will promptly notify the other Party of the conditions preventing such transfer and use reasonable efforts to deposit such payments in U.S. Dollars. If, after using reasonable efforts, such Party is not able
to deposit such payments in U.S. Dollars, then such payments will be deposited in local currency in the relevant country to the credit of the other Party in a recognized banking institution designated by the other Party or, if none is
designated by the other Party within [***], in a recognized banking institution selected by the transferring Party, as the case may be, and identified in a written notice given to the other Party.
|
| 9.10 |
Taxes.
|
|
|
9.10.1 |
Taxes on Income. Each Party will be solely responsible for the payment of any and all income Taxes levied on account of all payments it receives under this Agreement.
|
|
|
9.10.2 |
Withholding Tax. Any and all payments made pursuant to this Agreement will be paid without deduction or withholding for any Taxes, except as required by Applicable Law. To the extent a Party is
required by Applicable Law to deduct or withhold Taxes on any payment to the other Party (the “Withheld Amount”), such Party will remit such Withheld Amount to the proper
Governmental Authority in a timely manner and promptly transmit to the other Party an official Tax certificate or other evidence of any withholding sufficient to enable the other Party to claim available credits for such Withheld Amount. The
withholding Party will have the right to deduct such Withheld Amount from payment due to the other Party. For the avoidance of doubt, to the extent such Withheld Amount is so withheld and remitted in accordance with this Section 9.10.2
(Withholding Tax), such Withheld Amount will be treated for all purposes of this Agreement as having been paid to the other Party.
|
|
|
9.10.3 |
Tax Cooperation. The Parties agree to cooperate with one another in accordance with Applicable Law and use reasonable efforts to [***] in respect of payments made by each Party to the other Party
under this Agreement. Without limiting the generality of the foregoing, each Party will provide the other with any Tax forms and other information that may be reasonably necessary to [***] based on an applicable treaty or otherwise, including
a properly completed Internal Revenue Service (“IRS”) Form W-9 or appropriate IRS Form W-8, as applicable, before a payment is made. If any Tax form or other information a Party
previously delivered expires or becomes obsolete or inaccurate in any respect, then such Party will provide the other Party with an updated version of such form or certification or promptly notify the other Party in writing of its legal
inability to do so. Each Party will provide the other Party with reasonable assistance to enable the recovery, as permitted by Applicable Law, of withholding Taxes or similar obligations resulting from payments made under this Agreement, such
recovery to be for the benefit of the Party bearing such withholding Tax.
|
|
|
9.10.4 |
Changes in Domicile. Notwithstanding any provision to the contrary in this Agreement, including Section 9.10.2 (Withholding Tax), if as a result of a Party assigning, transferring, or
conveying rights under this Agreement to an Affiliate or changing its domicile, additional Taxes become due that would not otherwise have been due hereunder with respect to payments under this Agreement, then such Party will be responsible
for all such additional withholding Taxes.
|
| 9.11 |
Late Payments; Disputed Payments. Any undisputed payments or portions thereof due hereunder that are not paid on or before the date such payments are due under this Agreement will bear interest from
the due date until the date of payment at a per-annum rate equal to the lesser of: (a) [***] percentage points above the prime rate as published by The Wall Street Journal or any successor thereto;
or (b) the maximum rate permitted by Applicable Law. If a Party disputes an invoice or other payment obligation under this Agreement, then such Party will timely pay the undisputed amount of the invoice or other payment obligation, and the
Parties will resolve such dispute in accordance with Article 15 (Dispute Resolution; Governing Law).
|
| 10.1 |
Inventions.
|
|
|
10.1.1 |
Ownership of Background Intellectual Property. As between the Parties, and subject to the licenses granted under this Agreement, each Party retains all rights, title, and interests in and to all
Patent Rights and Know-How that such Party owns or Controls as of the Effective Date or that it develops or otherwise acquires after the Effective Date outside the performance of the activities under this Agreement.
|
|
|
10.1.2 |
Ownership of Arising Intellectual Property. As between the Parties, ownership of all Collaboration Know-How will be as follows:
|
|
|
(a) |
Ionis will be the sole owner of any (i) Collaboration Know-How that is developed or invented solely by Representatives of Ionis or its Affiliates or its or their licensees (other than Otsuka), Sublicensees, or Subcontractors, or any
Persons contractually required to assign or license such Collaboration Know-How to Ionis or any Affiliate of Ionis (“Ionis Collaboration Know-How”), and (ii) Patent Rights that Cover
the Ionis Collaboration Know-How (“Ionis Collaboration Patent Rights”), and will retain all of its rights thereto, subject to any rights or licenses expressly granted by Ionis to
Otsuka under this Agreement.
|
|
|
(b) |
Otsuka will be the sole owner of any (i) Collaboration Know-How that is developed or invented solely by Representatives of Otsuka or its Affiliates or its or their licensees (other than Ionis), Sublicensees, or Subcontractors, or any
Persons contractually required to assign or license such Collaboration Know-How to Otsuka or any Affiliate of Otsuka (“Otsuka Collaboration Know-How”), and (ii) Patent Rights that
Cover the Otsuka Collaboration Know-How (“Otsuka Collaboration Patent Rights”), and will retain all of its rights thereto, subject to any rights or licenses expressly granted by
Otsuka to Ionis under this Agreement.
|
|
|
(c) |
Each Party will own an equal, undivided share of all Joint Collaboration Technology.
|
|
|
10.1.3 |
Disclosure; Inventorship.
|
|
|
(a) |
Invention Disclosure. Each Party will promptly disclose to the other Party all Inventions within the Collaboration Know-How developed or invented during the Term by or on behalf of such Party, in
each case, as soon as practicable prior to an intended public disclosure of such Invention and prior to the filing of a patent application thereon. Each Party will also promptly respond to reasonable requests from the other Party for
additional information relating thereto.
|
|
|
(b) |
Inventions by a Party. Inventorship for Inventions and discoveries (including Know-How) first invented or developed during the course of the performance of activities under this Agreement will be
determined in accordance with United States Patent Laws for determining inventorship.
|
|
|
(c) |
Joint Research Agreement under the Leahy-Smith America Invents Act. If a Party intends to invoke its rights under 35 U.S.C. § 102(c) of the Leahy-Smith America Invents Act, then it will notify the
other Party and neither Party will make an election under such provision when exercising its rights under this Article 10 (Intellectual Property) without the prior written consent of the other Party (such consent not to be
unreasonably withheld, conditioned, or delayed), and the Parties will use reasonable efforts to cooperate and coordinate their activities with respect to any submissions, filings or other activities in support thereof. The Parties acknowledge
and agree that this Agreement is a “joint research agreement” as defined in 35 U.S.C. § 100(h).
|
|
|
10.1.4 |
Practice Under and Other Use of Joint Collaboration Technology. Subject to the rights granted under and the restrictions set forth in this Agreement (including the licenses granted under Article
2 (Licenses)), each Party will be entitled to the free use and enjoyment of all Joint Collaboration Technology and neither Party will have any obligation to account to the other Party for profits, or to obtain any approval of the other
Party to license, assign, or otherwise exploit any Joint Collaboration Technology by reason of joint ownership thereof. Each Party hereby waives any right it may have under the Applicable Law of any jurisdiction to require any such approval
or accounting. To the extent any further consent is required to enable a Party to so license or exploit its interest in the Joint Collaboration Technology, the other Party will grant consent promptly upon request. Without limitation, each
Party will cooperate with the other Party if the Parties determine to apply for U.S. or foreign patent protection for any Joint Collaboration Technology and will obtain the cooperation of the individual inventors of any such Joint
Collaboration Technology.
|
|
|
10.1.5 |
Representative Assignment. Each Party and its Affiliates will, and will cause its licensees, Sublicensees and Subcontractors to, enter into an agreement or employment policy with each of its
Representatives performing activities related to Development, Manufacture, or Commercialization of a Licensed Product that (a) compels prompt disclosure to such Party (or its Affiliate, licensee, Sublicensee or Subcontractor, as applicable)
of all Collaboration Know-How and Collaboration Patent Rights discovered, developed, invented, or filed by such Representative during any performance of such Development, Manufacture or Commercialization activities; and (b) automatically
assigns to such Party (or its Affiliate, licensee, Sublicensee or Subcontractor, as applicable) all rights, title, and interests in and to all Collaboration Know-How and Collaboration Patent Rights, and requires each Representative to execute
all documents and take such other actions as may be necessary to effectuate such assignment (or, if such assignment is not feasible, provides for such Party’s (or its Affiliate’s, licensee’s, Sublicensee’s or Subcontractor’s, as applicable)
joint ownership of, or an irrevocable, royalty-free license to such Party (or its Affiliate, licensee, Sublicensee or Subcontractor, as applicable) under, all Collaboration Know-How and Collaboration Patent Rights, with the right to
sublicense to the other Party as contemplated in this Agreement), provided that the foregoing will not apply with respect to improvements to background technology of a Subcontractor.
|
| 10.2 |
Patent Prosecution.
|
|
|
10.2.1 |
Ionis Patent Rights and Joint Collaboration Patent Rights.
|
|
|
(a) |
Right to Prosecute. As between the Parties, Ionis will have the (i) first right, in its sole discretion, to control the Patent Prosecution of all Ionis Product-Specific Patents in the Otsuka
Territory and all Joint Collaboration Patent Rights worldwide, and (ii) sole right, in its sole discretion, to control the Patent Prosecution of all (A) Ionis Product-Specific Patents in the Ionis Territory, and (B) Ionis Core Technology
Patents and Ionis Manufacturing and Analytical Patents, in each case, worldwide (collectively, ((i) and (ii)), the “Ionis Prosecuted Patent Rights”). Upon Ionis’ request, Otsuka will
obtain any necessary assignment documents for Ionis with respect to the Patent Prosecution of Ionis Prosecuted Patent Rights, will render all signatures that will be necessary for such patent filings, and will assist Ionis in all other
reasonable ways that are necessary for the issuance of Ionis Prosecuted Patent Rights as well as for the Patent Prosecution of Ionis Prosecuted Patent Rights, and Ionis will reimburse Otsuka’s reasonable External Costs incurred in connection
therewith. Ionis will be responsible for [***] of the costs and expenses incurred with respect to the Patent Prosecution of all Ionis Product-Specific Patents, Ionis Core Technology Patents and Ionis Manufacturing and Analytical Patents
throughout the world and for [***] of the costs and expenses incurred with respect to the Patent Prosecution of Joint Collaboration Patent Rights in the Ionis Territory. Otsuka will be responsible for [***] of the reasonable out-of-pocket
costs incurred by or on behalf of Ionis with respect to the Patent Prosecution of the Joint Collaboration Patent Rights in the Otsuka Territory (including any maintenance fees owed to local patent offices for the Joint Collaboration Patent
Rights in the Otsuka Territory), and Otsuka will reimburse Ionis for such costs within [***] after receiving an invoice with reasonable supporting documentation for such costs.
|
|
|
(b) |
Review and Consult. Ionis will consult with Otsuka and keep Otsuka reasonably informed regarding the Patent Prosecution of the Ionis Product-Specific Patents in the Otsuka Territory and the Patent
Prosecution of the Joint Collaboration Patent Rights worldwide and will provide Otsuka with all substantive correspondence received from any patent authority in connection therewith no later than [***] after receipt thereof. In addition,
Ionis will provide Otsuka with drafts of proposed substantive filings in the Otsuka Territory and correspondence to any patent authority in the Otsuka Territory in connection with the Patent Prosecution of the Ionis Product-Specific Patents
and with drafts of proposed substantive filings in the Territory and correspondence to any patent authority in the Territory in connection with the Patent Prosecution of Joint Collaboration Patent Rights, in each case for Otsuka’s review and
comment at least [***] prior to the submission of such proposed filings and correspondence, which comments (if any) Otsuka must provide no later than [***] after receipt of the applicable filing or correspondence. Ionis will consider in good
faith Otsuka’s reasonable comments on the Patent Prosecution of the Ionis Product-Specific Patents in the Otsuka Territory and the Joint Collaboration Patent Rights in the Territory, but Ionis will have final decision-making authority
regarding Patent Prosecution of such Patent Rights under this Section 10.2.1(b) (Review and Consult).
|
|
|
(c) |
Abandonment. If, at any time during the Term, Ionis decides to cease the Patent Prosecution of a particular Ionis Product-Specific Patent in the Otsuka Territory, or a particular Joint Collaboration
Patent Right in the Territory, then Ionis will provide written notice to Otsuka of such decision at least [***] prior to the date that such applicable Patent Right will become abandoned. Unless such written notice includes a reasonable
strategic reason for ceasing such Patent Prosecution (e.g., continuing such Patent Prosecution would adversely affect Ionis’ Patent Prosecution or litigation strategy), Otsuka may, upon written notice to Ionis, assume the Patent Prosecution
of any such Patent Right at Otsuka’s sole cost and expense. Without limiting the foregoing, with respect to any such Joint Collaboration Patent Right abandoned by Ionis, Ionis shall assign, and hereby does assign, to Otsuka all of its rights,
title and interests in and to such Joint Collaboration Patent Right, and upon such assignment, such Joint Collaboration Patent Right shall be deemed an Otsuka Patent Right for all purposes of this Agreement.
|
|
|
10.2.2 |
Otsuka Patent Rights.
|
|
|
(a) |
Right to Prosecute. As between the Parties, Otsuka will have the first right to control the Patent Prosecution of all Otsuka Patent Rights throughout the world. Otsuka will be responsible for [***]
of the costs and expenses incurred with respect to the Patent Prosecution of such Patent Rights throughout the world.
|
|
|
(b) |
Review and Consult. Otsuka will consult with Ionis and keep Ionis reasonably informed regarding the Patent Prosecution of the Otsuka Patent Rights and will provide Ionis with all substantive
correspondence received from any patent authority in connection therewith no later than [***] after receipt thereof. In addition, Otsuka will provide Ionis with drafts of all proposed substantive filings and correspondence to any patent
authority in connection with the Patent Prosecution of the Otsuka Patent Rights for Ionis’ review and comment at least [***] prior to the submission of such proposed filings and correspondence, which comments (if any) Ionis must provide no
later than [***] after receipt of the applicable filing or correspondence. Otsuka will consider in good faith Ionis’ reasonable comments on the Patent Prosecution of the Otsuka Patent Rights, but will have final decision-making authority
regarding Patent Prosecution of such Patent Rights under this Section 10.2.1(b) (Review and Consult).
|
|
|
(c) |
Abandonment. If, at any time during the Term, Otsuka ceases the Patent Prosecution of a particular Otsuka Patent Right, then Otsuka will provide written notice to Ionis of such decision at least
[***] prior to the date on which such Patent Right will become abandoned. Unless such written notice includes a reasonable strategic reason for ceasing such Patent Prosecution (e.g., continuing such Patent Prosecution would adversely affect
Otsuka’s Patent Prosecution or litigation strategy), Ionis may, upon written notice to Otsuka, assume the Patent Prosecution of any such Patent Right at Ionis’ sole cost and expense.
|
| 10.3 |
Enforcement Against Third Party Infringement or Misappropriation.
|
|
|
10.3.1 |
Notice of Infringement or Misappropriation. Each Party will promptly notify the other of any apparent, threatened, or actual Competitive Infringement of which it becomes aware.
|
|
|
10.3.2 |
Otsuka’s Enforcement Right. Otsuka will have the first right, but not the obligation, to enforce [***] against any Competitive Infringement in the Otsuka Territory and at its own cost and expense and
using counsel of its own choice; provided that, (a) [***] Ionis will be entitled to attend any substantive meetings, hearings, or other proceedings related to such infringement or misappropriation
suit (together with its own counsel, at its own expense) and to review and comment on all substantive documents related to such infringement or misappropriation suit prior to filing or submission of such documents, and (b) with respect [***]
Otsuka shall keep Ionis reasonably informed of the status of any substantive meetings, hearings, or other proceedings related to such infringement or misappropriation suit. If Otsuka fails to initiate a suit or take other action to abate any
such Competitive Infringement within the earlier of: (i) [***] and (ii) [***], then, in either case, Ionis will have the second right, but not the obligation, to attempt to resolve such Competitive Infringement, at its own expense, including
the filing of an infringement or misappropriation suit, as applicable, to enforce the applicable Patent Rights or Know-How using counsel of its own choice; provided that, if Otsuka notifies Ionis
during [***] that it is electing not to take steps to enforce the applicable Patent Rights against such Competitive Infringement [***].
|
|
|
10.3.3 |
Ionis’ Enforcement Right. Ionis will have the sole right, but not the obligation, to enforce [***] against any Competitive Infringement in the Territory, in each case ((a) and (b)), at its own cost
and expense and using counsel of its own choice; provided that Ionis shall keep Otsuka reasonably informed of the status of any substantive meetings, hearings, or other proceedings related to any
infringement or misappropriation suit to enforce [***] against any Competitive Infringement in the Otsuka Territory. Ionis will have the first right, but not the obligation, to enforce any [***] in the Ionis Territory, in each case, at its
own expense and using counsel of its own choice; provided that Otsuka will be entitled to attend any substantive meetings, hearings, or other proceedings related to such infringement or
misappropriation suit (together with its own counsel, at its own expense) and to review and comment on all substantive documents related to such infringement or misappropriation suit prior to filing or submission of such documents. If Ionis
fails to initiate a suit or take other action to abate any such Competitive Infringement with respect to [***] in the Ionis Territory within the earlier of: (a) [***] and (b) [***], then, in either case, Otsuka will have the second right, but
not the obligation, to attempt to resolve such Competitive Infringement, at its own expense, including the filing of an infringement or misappropriation suit, as applicable, to enforce the applicable Otsuka Technology or Joint Collaboration
Technology using counsel of its own choice; provided that, if Ionis notifies Otsuka during [***] that it is electing not to take steps to enforce the applicable Patent Rights against such Competitive
Infringement [***].
|
|
|
10.3.4 |
Allocation of Recoveries. Any recoveries resulting from an enforcement action relating to a claim of Competitive Infringement in the Territory will be first applied against payment of each Party’s
costs and expenses in connection therewith. Any such recoveries in excess of such costs and expenses will be [***]; provided that [***].
|
|
|
10.3.5 |
Cooperation; Procedures. At the request and expense of the Party bringing an infringement or misappropriation action under this Section 10.3 (Enforcement Against Third Party Infringement or
Misappropriation), the other Party will provide reasonable assistance and cooperation in any such action (including entering into a common interest agreement if reasonably deemed necessary by any Party) and agrees to be joined as a party to
the suit if necessary for the initiating Party to bring or continue an infringement or misappropriation action hereunder. In addition, the Party bringing an infringement or misappropriation action under this Section 10.3 (Enforcement
Against Third Party Infringement or Misappropriation) will provide the other Party with copies of all pleadings and other documents in advance of filing with the court and will consider reasonable input from the other Party during the course
of the action. For clarity, the Party bringing an infringement or misappropriation action under this Section 10.3 (Enforcement Against Third Party Infringement or Misappropriation) will control such infringement or misappropriation
action subject to the terms of this Section 10.3 (Enforcement Against Third Party Infringement or Misappropriation). Neither Party may settle any action or proceeding brought under this Section 10.3 (Enforcement Against Third
Party Infringement or Misappropriation) or knowingly take any other action in the course thereof that disclaims, limits the scope of, admits the invalidity or unenforceability of, or grants a license, covenant not to sue or similar immunity
under a Patent Right Controlled by the other Party without first obtaining the written consent of the Party that Controls the relevant Patent Right. Furthermore, Ionis may not [***]. The Parties will reasonably assist each other and cooperate
with each other, at their own expense, in any such investigation, pre-litigation preparation, or litigation to ensure that there is an aligned global litigation and enforcement strategy.
|
| 10.4 |
Defense of Third-Party Patent Challenges. Each Party will promptly notify the other Party in writing after becoming aware of an actual or threatened Patent Challenge by a Third Party of any Ionis
Patent Right, Otsuka Patent Right, and Joint Collaboration Patent Right (each, a “Third Party Patent Challenge”).
|
|
|
10.4.1 |
Otsuka’s Right to Defend. Subject to the terms of Section 10.4.3 (Cooperation; Procedures), and except as may be otherwise agreed by the Parties, Otsuka will have the first right, but not the
obligation, to control the defense of any Third Party Patent Challenge relating to an Otsuka Patent Right or Joint Collaboration Patent Right in the Otsuka Territory, and to compromise, litigate, settle, or otherwise dispose of any such
challenge, in each case at its own expense using counsel of its own choice; provided that (a) with respect to a Joint Collaboration Patent Right, Ionis will be entitled to attend any substantive
meetings, hearings, or other proceedings related to such Third Party Patent Challenge (together with its own counsel, at its own expense) and to review and comment on all substantive documents related to such Third Party Patent Challenge, and
(b) with respect to an Otsuka Patent Right, Otsuka shall keep Ionis reasonably informed of the status of any substantive meetings, hearings, or other proceedings related to such Third Party Patent
Challenge, and if Otsuka fails to initiate the defense of such Third Party Patent Challenge of a Patent Right in the Otsuka Territory within [***] after the notice provided under Section 10.4 (Defense of Third Party Patent
Challenges), or otherwise abandons or elects not to continue any such defense once initiated, then Ionis will have the second right, but not the obligation, to control the defense of such Third Party Patent Challenge at its own expense using
counsel of its own choice.
|
|
|
10.4.2 |
Ionis’ Right to Defend. Ionis will have the sole right, but not the obligation, to control the defense of any Third Party Patent Challenge relating to an (a) Ionis Product-Specific Patent in the
Ionis Territory or (b) Ionis Core Technology Patent or Ionis Manufacturing and Analytical Patent in the Territory, and to compromise, litigate, settle, or otherwise dispose of any such challenge, in each case, at its own expense using counsel
of its own choice. Subject to the terms of Section 10.4.3 (Cooperation; Procedures), Ionis will have the first right, but not the obligation, to control the defense of any Third Party Patent Challenge relating to an (i) Otsuka Patent
Right or Joint Collaboration Patent Right in the Ionis Territory or (ii) Ionis Product-Specific Patent in the Otsuka Territory and to compromise, litigate, settle, or otherwise dispose of any such challenge, at its own expense using counsel
of its own choice; provided that Otsuka will be entitled to attend any substantive meetings, hearings, or other proceedings related to such Third Party Patent Challenge (together with its own
counsel, at its own expense) and to review and comment on all substantive documents related to such Third Party Patent Challenge. If Ionis fails to initiate the defense of such Third Party Patent Challenge of an Otsuka Patent Right or Joint
Collaboration Patent Right in the Ionis Territory or an Ionis Product-Specific Patent in the Otsuka Territory, in each case, within [***] after the notice provided under Section 10.4 (Defense of Third Party Patent Challenges), or
otherwise abandons or elects not to continue any such defense once initiated, then Otsuka will have the second right, but not the obligation, to control the defense of such Third Party Patent Challenge at its own expense using counsel of its
own choice.
|
|
|
10.4.3 |
Cooperation; Procedures. At the request and expense of the Party controlling the defense of any Third Party Patent Challenge under this Section 10.4 (Defense of Third Party Patent
Challenges), the other Party will provide reasonable assistance and cooperation in any such action. In addition, the Party controlling the defense of any Third Party Patent Challenge under this Section 10.4 (Defense of Third Party
Patent Challenges) will provide the other Party with copies of all pleadings and other documents to be filed with the court and will consider reasonable input from the other Party during the course of the action. Otsuka may not settle any
action or proceeding brought or defended under this Section 10.4 (Defense of Third-Party Patent Challenges) or knowingly take any other action in the course thereof without Ionis’ prior written consent, unless such action or
proceeding solely concerns the Otsuka Patent Rights. Ionis may not settle any action or proceeding brought or defended under this Section 10.4 (Defense of Third-Party Patent Challenges) or knowingly take any other action in the course
thereof with respect to the Ionis Product-Specific Patents or Joint Collaboration Patent Rights in the Otsuka Territory, without Otsuka’s prior written consent not to be unreasonably withheld, conditioned or delayed. The Parties will
reasonably assist each other and cooperate with each other, at their own expense, in any such investigation, pre-litigation preparation, or litigation to ensure that there is an aligned global litigation strategy. Notwithstanding the above,
in the case of any invalidity or unenforceability claims arising in an enforcement action under Section 10.3 (Enforcement Against Third Party Infringement or Misappropriation), the Party controlling the enforcement action pursuant to
Section 10.3 (Enforcement Against Third Party Infringement or Misappropriation) shall control the response to such invalidity or unenforceability claims, provided such Party may not admit
invalidity or unenforceability of any Patent Right Controlled by the other Party without the prior written consent of the other Party.
|
| 10.5 |
Third Party Infringement Claims.
|
|
|
10.5.1 |
Infringement Claim; Patent Challenges of Third-Party IP. If a Third Party asserts that a Patent Right controlled by it is, or will be, infringed by the Exploitation of a Licensed Product in the
Territory in accordance with this Agreement, then the Party first obtaining knowledge of such claim will promptly provide the other Party with prompt written notice thereof and the related facts in reasonable detail.
|
|
|
10.5.2 |
Responsibility to Defend. During the Term of this Agreement, if a Third Party asserts that a Patent Right controlled by such Third Party is infringed, or will be infringed, by the Exploitation of a
Licensed Product, then the Parties will promptly discuss the matter and the appropriate course of action. If the Parties cannot agree on a course of action within [***] following the date on which the Parties receive notice of such Third
Party claim, then, subject to Article 13 (Indemnification): (a) Ionis will have the sole right, but not the obligation, to defend such claim in the Ionis Territory using counsel of its own choosing, and (b) Otsuka will have the first
right, but not the obligation, to defend such claim in the Otsuka Territory using counsel of its own choosing. If Otsuka does not take affirmative steps to defend such claim in the Otsuka Territory within [***] (or such shorter period of time
as is legally required to answer to such claim) and does not inform Ionis within such [***] period that it is electing not to defend such claim for strategic reasons intended to maintain the commercial value of the relevant Patent Rights or
any product or subject matter Covered thereby or relating thereto, then Ionis may defend such claim in the Otsuka Territory. The Party defending such claim in the Otsuka Territory will (i) keep the other Party reasonably informed regarding
any such assertion, including by providing the other Party with copies of all pleadings and other documents filed in any proceeding relating to such claim, (ii) consider reasonable input from the other Party during the course of the claim,
and (iii) provide the other Party with the opportunity to attend any substantive meetings, hearings, or other proceedings related to such claim (together with its own counsel, at its own expense) and to review and comment on all substantive
documents related to such claim prior to filing or submission of such documents. The Parties will reasonably assist each other and cooperate and share information with respect to any such claim. Each Party will bear its own costs and expenses
with respect to any such claim.
|
|
|
10.5.3 |
Settlement. Subject to Article 13 (Indemnification), neither Party will pursue or enter into any settlement or license agreement with any Third Party with respect to the Patent Rights that
are the subject of a claim brought by a Third Party that a Patent Right controlled by such Third Party is infringed by the Exploitation of a Licensed Product in the Otsuka Territory without the other Party’s prior written consent, not to be
unreasonably withheld, conditioned, or delayed. Subject to Article 13 (Indemnification), Otsuka will bear the costs of any amounts paid in settlement or to satisfy a judgment of a claim that the Exploitation of a Licensed Product
infringes any Third Party Patent Right in the Otsuka Territory, except to the extent such costs [***].
|
| 10.6 |
Patent Challenges of Third-Party Patent Rights.
|
|
|
10.6.1 |
Notice of Third-Party Patent Right. If either Party becomes aware of a Third Party Patent Right that might form the basis for a claim that the Exploitation of a Licensed Product anywhere in the world
infringes, or will infringe, such Patent Right, then the Party first obtaining knowledge of such Patent Right will promptly provide the other Party with written notice thereof and the related facts in reasonable detail, and the Parties will
promptly meet to discuss the matter.
|
|
|
10.6.2 |
Patent Challenges of Third-Party Patents. Ionis will have the sole right, but not the obligation, to initiate a Patent Challenge of any such Third Party Patent Right in the Ionis Territory using
counsel of its own choosing. Otsuka will have the first right, but not the obligation, to initiate a Patent Challenge of such Third Party Patent Right in the Otsuka Territory, and if Otsuka notifies Ionis that it does not intend to initiate
such a Patent Challenge, Ionis will have the second right, but not the obligation, to do so; provided that, [***]. The Party initiating such Patent Challenge will (a) keep the other Party reasonably
informed regarding any such Patent Challenge, including by providing the other Party with copies of all pleadings and other documents filed in any proceeding relating to such Patent Challenge, (b) consider reasonable input from the other
Party during the course of the Patent Challenge, and (c) provide the other Party with the opportunity to attend any substantive meetings, hearings, or other proceedings related to such Patent Challenge (together with its own counsel, at its
own expense) and to review and comment on all substantive documents related to such Patent Challenge prior to filing or submission of such documents. The Parties will reasonably assist each other and cooperate and share information with
respect to any such Patent Challenge. Each Party will bear its own costs and expenses with respect to any such Patent Challenge; provided, however, that the Parties will each bear [***] of the
reasonable out-of-pocket costs incurred with respect to any such Patent Challenge in the Otsuka Territory, and the non-controlling Party will reimburse the Party initiating such Patent Challenge in the Otsuka Territory for such costs within
[***] after receiving an invoice with reasonable supporting documentation for such costs.
|
|
|
10.6.3 |
Restrictions on Settlement. Neither Party nor its Affiliates will pursue or enter into any settlement or license agreement with any Third Party with respect to the Patent Rights that are the subject
of such Patent Challenge in the Otsuka Territory without the other Party’s prior written consent.
|
| 10.7 |
Patent Term Extensions. With respect to any system for extending the term of Patent Rights in the Otsuka Territory established by any applicable Regulatory Authority during the Term that is similar
to the patent term extension system in the U.S., [***] for making all decisions regarding patent term extensions of the Ionis Patent Rights or Joint Collaboration Patent Rights in the Otsuka Territory, including supplementary protection
certificates and any other extensions that are now or become available in the future, that are applicable to the Ionis Patent Rights or Joint Collaboration Patent Rights licensed hereunder and that become available directly as a result of the
Regulatory Approval of a Licensed Product in the Otsuka Territory; provided that Otsuka will consult with Ionis with respect to such decisions and consider in good faith the reasonable comments and
concerns of Ionis.
|
| 10.8 |
Unified Patent Court. Otsuka will be solely responsible for making all decisions regarding the opting-out or opting-in of existing Patent Rights into the jurisdiction of the Unified Patent Court or
the registration of Patent Rights with Unitary Effect; provided that Otsuka will consult with Ionis with respect to such decisions and will consider the comments and concerns of Ionis in good faith.
|
| 10.9 |
Common Interest. The Parties stipulate and agree that, with regard to such prosecution, maintenance, enforcement, and defense the interests of the Parties as collaborators and licensor and licensee
are to obtain the strongest patent protection possible, and as such, are aligned and are legal in nature. The Parties stipulate and agree that they have not waived, and nothing in this Agreement constitutes a waiver of, any legal privilege
concerning the Patent Rights under this Article 10 (Intellectual Property), including privilege under the common interest doctrine and similar or related doctrines. Notwithstanding any provision to the contrary set forth in this
Agreement, to the extent a Party has a good faith belief that any information required to be disclosed by such Party to the other Party under this Article 10 (Intellectual Property) is protected by attorney-client privilege or any
other applicable legal privilege or immunity, such Party will not be required to disclose such information and the Parties will in good faith cooperate to agree upon a procedure (including entering into a specific common interest agreement,
disclosing such information on a “for counsel eyes only” basis or similar procedure) under which such information may be disclosed without waiving or breaching such privilege or immunity.
|
| 10.10 |
Product Trademarks.
|
|
|
10.10.1 |
Ownership.
|
|
|
(a) |
Unitary Product Trademark. Ionis shall, at its sole cost and expense, develop, and shall use Commercially Reasonable Efforts to obtain and maintain, a unitary Trademark (and back-up Trademarks
thereof) to be used for the Licensed Products worldwide (the “Unitary Product Trademark”); provided however, [***]. Ionis will own all
right, title, and interest in and to the Unitary Product Trademark. Otsuka will use the Unitary Product Trademark in the Otsuka Territory to the extent required by and in accordance with the Otsuka Territory Brand Strategic and Operating
Plan, subject to Section 6.4 (Global Brand Strategic and Operating Plan) and Section 6.5 (Otsuka Territory Brand Strategic and Operating Plan).
|
|
|
(b) |
Ownership of Otsuka Product Trademarks. As between the Parties, Otsuka will have the sole right to determine and will own all right, title, and interest in and to any Trademarks, other than the
Unitary Product Trademark, to be created or used by Otsuka or its Affiliates or its or their Sublicensees for the Exploitation of Licensed Product in the Otsuka Territory excluding any trademarks, service marks, names, or logos that include
any corporate name or logo of the Parties or their Affiliates or its or their Sublicensees (“Otsuka Product Trademarks”); provided that
such Otsuka Product Trademarks are consistent with the Global Brand Strategic and Operating Plan. Ionis will not [***]. Ionis will not [***].
|
|
|
(c) |
Ownership of Ionis Product Trademarks. As between the Parties, Ionis will have the sole right to determine and will own all right, title, and interest in and to the Trademarks (other than the Unitary
Product Trademark) to be used by Ionis or its Affiliates or its or their Sublicensees or licensees for the Exploitation of Licensed Product in the Ionis Territory excluding any trademarks, service marks, names, or logos that include any
corporate name or logo of the Parties or their Affiliates or its or their Sublicensees or licensees (“Ionis Product Trademarks”); provided
that such Ionis Product Trademarks are consistent with the Global Brand Strategic and Operating Plan. Otsuka will not [***]. Otsuka will not [***].
|
|
|
10.10.2 |
Notice. Each Party will provide to the other Party prompt written notice of any actual or threatened infringement of the Otsuka Product Trademarks or Ionis Product Trademarks in the Territory and of
any actual or threatened claim that the use of the Otsuka Product Trademarks or Ionis Product Trademarks in the Territory violates the rights of any Third Party, in each case, of which such Party becomes aware.
|
|
|
10.10.3 |
Prosecution of Product Trademarks.
|
|
|
(a) |
Unitary Product Trademark. Ionis shall be responsible, at its sole discretion and cost and expense using counsel of its own choice, for the filing,
prosecution, registration and maintenance (including the defense of opposition proceedings and any equivalent proceedings and including any legal actions to prevent or exclude Third Party Trademark registrations that are confusingly similar
to the Unitary Product Trademark) of the Unitary Product Trademark in the Territory throughout the Term. Ionis shall keep Otsuka informed of material progress with regard to the prosecution, registration, and maintenance of the Unitary
Product Trademark in the Otsuka Territory, including the content and timing of the filing of the Unitary Product Trademark in the Otsuka Territory, [***], and Ionis shall [***] the Unitary Product Trademark in the Otsuka Territory.
|
|
|
(b) |
Otsuka Product Trademarks. Otsuka will have the sole right to register, prosecute and maintain the Otsuka Product Trademarks in the Territory using counsel of its own choice. All costs and expenses
of registering, prosecuting and maintaining the Otsuka Product Trademarks in the Territory will be borne solely by Otsuka.
|
|
|
(c) |
Ionis Product Trademarks. Ionis will have the sole right to register, prosecute and maintain the Ionis Product Trademarks in the Territory using counsel of its own choice. All costs and expenses of
registering, prosecuting and maintaining the Ionis Product Trademarks in the Territory will be borne solely by Ionis.
|
|
|
10.10.4 |
Enforcement of Product Trademarks.
|
|
|
(a) |
Unitary Product Trademark. During the Term, each Party will promptly notify the other Party in writing of any alleged, threatened or actual infringement,
dilution, misappropriation or other violation of or unfair trade practices or any other like offense by a Third Party relating to the Unitary Product Trademark in the Otsuka Territory (“Otsuka
Territory Trademark Infringement”). Otsuka will have the first right, but not the obligation, to take any reasonable measures it deems appropriate with respect to any Otsuka Territory Trademark Infringement, using counsel of its own
choice, and at its own cost and expense, including initiating or prosecuting an infringement, misappropriation or other appropriate suit or action to enforce the Unitary Product Trademark in the Otsuka Territory and, if requested by Otsuka,
Ionis shall (i) join as a party to such suit or action and execute and cause its Affiliates to execute all documents necessary for Otsuka to initiate and maintain such suit or action and (ii) provide reasonable assistance to Otsuka in
connection with such suit or action. Notwithstanding the foregoing, if Otsuka does not inform Ionis that it intends to initiate a suit or take other action against an Otsuka Territory Trademark Infringement within [***] after Otsuka becoming
aware of such Otsuka Territory Trademark Infringement and does not [***] within such [***], then Ionis will have the second right, but not the obligation, to initiate a suit or take other action against such Otsuka Territory Trademark
Infringement at its own cost and expense. Any recoveries resulting from such suit or other action will be first applied against payment of each Party’s costs and expenses in connection therewith. Any such recoveries in excess of such costs
and expenses will be [***].
|
|
|
(b) |
Otsuka Product Trademarks. Otsuka will have the sole right to take such action as Otsuka deems necessary against a Third Party based on any alleged, threatened or actual infringement, dilution,
misappropriation or other violation of or unfair trade practices or any other like offense relating to, the Otsuka Product Trademarks by a Third Party in the Territory, at its sole cost and expense and using counsel of its own choice. Otsuka
will retain any damages or other amounts collected in connection therewith.
|
|
|
(c) |
Ionis Product Trademarks. Ionis will have the sole right to take such action as Ionis deems necessary against a Third Party based on any alleged, threatened or actual infringement, dilution,
misappropriation or other violation of or unfair trade practices or any other like offense relating to, the Ionis Product Trademarks by a Third Party in the Territory, at its sole cost and expense and using counsel of its own choice. Ionis
will retain any damages or other amounts collected in connection therewith.
|
|
|
10.10.5 |
Third Party Claims.
|
|
|
(a) |
Unitary Product Trademark. If a Third Party brings suit alleging that Otsuka’s or its Affiliate’s or Sublicensee’s Exploitation of a Licensed Product in the Otsuka Territory infringes or will
infringe such Third Party’s Trademarks or that the use or registration of the Unitary Product Trademark in the Otsuka Territory infringes, dilutes, misappropriates or otherwise violates any Trademark or other right of such Third Party (“Trademark Infringement Suit”), then the Party against whom such suit is brought will promptly notify the other Party of such Trademark Infringement Suit and Otsuka will have the first
right, but not the obligation, to defend such Trademark Infringement Suit using counsel of its own choice. If Otsuka does not take affirmative steps to defend such Trademark Infringement Suit within [***] (or such shorter period of time as is
legally required to answer to such suit) and does not [***], then Ionis may defend such Trademark Infringement Suit. The Party defending such Trademark Infringement Suit will (i) keep the other Party reasonably informed regarding such suit,
including by providing the other Party with copies of all pleadings and other documents filed in any proceeding relating to such suit, (ii) consider reasonable input from the other Party during the course of the suit, and (iii) provide the
other Party with the opportunity to attend any substantive meetings, hearings, or other proceedings related to such suit (together with its own counsel, at its own expense) and to review and comment on all substantive documents related to
such suit prior to filing or submission of such documents. The Parties will reasonably assist each other and cooperate and share information with respect to any such suit. The Parties will [***] all of the costs incurred by either Party in
defending a Trademark Infringement Suit and any and all damages paid in settlement or to satisfy a judgment in a Trademark Infringement Suit. Neither Party will enter into any settlement of a Trademark Infringement Suit that is instituted or
threatened to be instituted against the other Party without the other Party’s prior written consent, not to be unreasonably withheld, conditioned or delayed; provided that such consent will not be
required if such settlement includes a release of all liability in favor of, and does not impose any obligation on, the other Party and contains no admission of liability by such settling Party. Further, neither Party shall settle or
compromise any Trademark Infringement Suit, or knowingly take any other action in the course thereof, in a manner that materially adversely affects the other Party’s rights or interests, without the other Party’s prior written consent.
|
|
|
(b) |
Otsuka Product Trademarks. Otsuka will have the sole right to defend against and settle any alleged, threatened or actual claim by a Third Party that the use or registration of the Otsuka Product
Trademarks in the Territory infringes, dilutes, misappropriates or otherwise violates any Trademark or other right of that Third Party or constitutes unfair trade practices or any other like offense or any other claims as may be brought by a
Third Party against a Party in connection with the use of the Otsuka Product Trademarks with respect to the Licensed Products in the Otsuka Territory, at its sole cost and expense and using counsel of its own choice. Otsuka will retain any
damages or other amounts collected in connection therewith.
|
|
|
(c) |
Ionis Product Trademarks. Ionis will have the sole right to defend against and settle any alleged, threatened or actual claim by a Third Party that the use or registration of the Ionis Product
Trademarks in the Territory infringes, dilutes, misappropriates or otherwise violates any Trademark or other right of that Third Party or constitutes unfair trade practices or any other like offense or any other claims as may be brought by a
Third Party against a Party in connection with the use of the Ionis Product Trademarks with respect to the Licensed Products in the Ionis Territory, at its sole cost and expense and using counsel of its own choice. Ionis will retain any
damages or other amounts collected in connection therewith.
|
|
|
10.10.6 |
Housemarks. The Parties, through the JSC, in consultation with regulatory experts, will [***].
|
|
|
10.10.7 |
Cooperation. Each Party will, and will cause its Affiliates to, promptly assist and cooperate with the other Party, as may be reasonably requested by a Party from time to time, in connection with its
activities set forth in this Section 10.10 (Product Trademarks), including where necessary, furnishing a power of attorney solely for such purpose or joining in, or being named as a necessary party to, such action, and providing
access to relevant documents and other evidence; provided that, except as provided otherwise in this Section 10.10 (Product Trademarks) with respect to [***], the requesting Party will
reimburse the other Party for its [***] incurred in connection therewith.
|
| 11.1 |
Mutual Representations and Warranties. Each of Otsuka and Ionis hereby represents and warrants to the other Party as of the Effective Date that:
|
|
|
11.1.1 |
It is a corporation or limited company duly organized, validly existing, and in good standing under the laws of the jurisdiction of its organization, and it has the full right, power, and authority to enter into this Agreement and to
perform its obligations hereunder.
|
|
|
11.1.2 |
All consents, approvals, and authorizations from all Governmental Authorities or other Third Parties required to be obtained by such Party in connection with this Agreement have been obtained.
|
|
|
11.1.3 |
The execution, delivery, and performance of this Agreement by it has been duly authorized by all requisite corporate action.
|
|
|
11.1.4 |
The execution and delivery of this Agreement and the performance of its obligations hereunder (a) do not conflict with or violate any requirement of Applicable Law or any provision of its articles of incorporation, bylaws, limited
partnership agreement, or any similar instrument, as applicable, in any material way, and (b) do not conflict with, violate, or breach or constitute a default or require any consent under, any Applicable Law or any contractual obligation or
court or administrative order by which it is bound.
|
|
|
11.1.5 |
It has not been debarred or suspended under 21 U.S.C. §335(a) or (b), is not the subject of a conviction described in Section 306 of the FD&C Act, has
not been and is not excluded from a federal or governmental health care program, debarred from federal contracting, convicted of or pled nolo contendere to any felony, or to any federal or state legal
violation (including misdemeanors) relating to prescription drug products or fraud, is not subject to OFAC sanctions or on the OFAC list of specially designated nationals, and is not subject to any similar
sanction of any Governmental Authority in the Territory (“Debarred/Excluded”), and no proceeding that could result in it being
Debarred/Excluded is pending, and neither it nor any of its Affiliates has used, in any capacity in the performance of obligations relating to the Licensed Products, any employee, subcontractor,
consultant, agent, representative, or other Person who has been Debarred/Excluded.
|
| 11.2 |
Additional Ionis Representations and Warranties. Ionis hereby represents and warrants as of the Effective Date to Otsuka that:
|
|
|
11.2.1 |
It has the right under the Ionis Technology to grant to Otsuka the licenses set forth in this Agreement, and it has not granted any license or other right under the Ionis Technology that is inconsistent with the licenses granted to Otsuka
hereunder.
|
|
|
11.2.2 |
Schedule 1.91 (Ionis Core Technology Patents), Schedule 1.96 (Ionis
Manufacturing and Analytical Patents), and Schedule 1.103 (Ionis Product-Specific Patents), collectively, list all Ionis Patent Rights existing as of the Effective Date. With respect to
any such Ionis Patent Right identified as being solely owned by Ionis, Ionis owns all rights, title, and interests in and to such Ionis Patent Rights.
|
|
|
11.2.3 |
As of the Effective Date, all issued Patent Rights within the Ionis Patent Rights are in full force and effect and, to Ionis’ Knowledge, are valid and enforceable. To Ionis’ Knowledge, all Ionis Patent Rights are being diligently
prosecuted in the respective patent offices in the Otsuka Territory in accordance with Applicable Law and have been filed, prosecuted and maintained properly and correctly, and all applicable fees have been paid on or before the due date for
payment.
|
|
|
11.2.4 |
There is no pending or, to Ionis’ Knowledge, threatened litigation, nor has Ionis received any written notice from any Third Party, asserting or alleging that the Exploitation of the Licensed Products prior to the Effective Date infringed
or misappropriated the Patent Rights, Know-How or other intellectual property rights of such Third Party or that the disclosing, copying, making, assigning, licensing or use of the Ionis Technology infringes or misappropriates any Patent
Right, Know-How or other intellectual property rights of such Third Party.
|
|
|
11.2.5 |
To Ionis’ Knowledge, the practice by Otsuka of the Ionis Technology and the Exploitation by Otsuka or its Affiliates or Sublicensee of the Licensed Products in the form existing as of the Effective Date for the treatment of HAE, in each
case, does not and will not infringe, misappropriate or otherwise violate any Patent Rights, Know-How or other intellectual property rights of any Third Party.
|
|
|
11.2.6 |
To Ionis’ Knowledge, there are no Third Party Know-How or Patent Rights that are necessary for the Exploitation of Licensed Products in the form existing as of the Effective Date for the treatment of HAE in the Otsuka Territory, other than
the Know-How and Patent Rights licensed to Ionis pursuant to the Existing Third-Party IP Agreements. To Ionis’ Knowledge, other than the Patent Rights and Know-How licensed to Ionis pursuant to the Existing Third-Party IP Agreements, there
are no Third Party Patent Rights that Cover the composition of matter of Licensed Compound or Licensed Products or any Third Party Patent Rights or Know-How that are used in the Manufacture of the Licensed Product in the form existing as of
the Effective Date.
|
|
|
11.2.7 |
There are no pending or, to Ionis’ Knowledge, threatened, adverse actions, suits, or proceedings against Ionis involving the Ionis Technology.
|
|
|
11.2.8 |
To Ionis’ Knowledge, no Person is infringing or threatening to infringe or misappropriating or threatening to misappropriate any Ionis Technology in the Otsuka Territory.
|
|
|
11.2.9 |
There are no legal claims, judgments, or settlements against or owed by Ionis or any of its Affiliates, or pending or, to Ionis’ Knowledge, threatened, legal claims or litigation, in each case, relating to antitrust, anti-competition, or
anti-corruption law violations.
|
|
|
11.2.10 |
Schedule 1.49 (Existing Third-Party IP Agreements) sets forth all Existing Third-Party IP Agreements in effect as of the Effective Date, redacted copies of which have been
provided to Otsuka prior to the date hereof. Other than the Existing Third-Party IP Agreements set forth in Schedule 1.49 (Existing Third-Party IP Agreements), as of the Effective
Date there are no agreements between Ionis and any Third Party pursuant to which Ionis Controls any Know-How or Patent Rights within the Ionis Technology.
|
|
|
11.2.11 |
Except for Existing Third-Party IP Agreements, Ionis is not obligated under any contract or other agreement with a Third Party as of the Effective Date to make any payments to any owner or licensor of, or other claimant to, any Patent
Right, Know-How or other intellectual property or proprietary right with respect to the Exploitation of the Licensed Product in the Otsuka Territory in the form existing as of the Effective Date for the treatment of HAE.
|
|
|
11.2.12 |
With respect to the Existing Third-Party IP Agreements, Ionis represents and warrants to Otsuka, as of the Effective Date, that: (a) it is in full force and effect; (b) neither Ionis nor any of its Affiliates is in material breach thereof;
(c) neither Ionis nor any of its Affiliates has received any notice from any counterparties thereto of any material breach or notice of threatened material breach thereof; (d) neither Ionis nor any of its Affiliates has received any notice
from any counterparties thereto of any intent to reduce the scope of the field thereunder or render any of the licenses thereunder non-exclusive or otherwise terminate such Existing Third-Party IP Agreements, and, to Ionis’s Knowledge no
event, act or omission has occurred which would reasonably give rise to the right of any counterparties thereto to reduce the scope of the field thereof or render any of the licenses thereunder non-exclusive or otherwise terminate such
agreement or any licenses thereunder (including with respect to any particular Patent Rights or other intellectual property); (e) neither Ionis nor any of its Affiliates have waived or relinquished any rights thereunder; (f) entering into
this Agreement and granting the rights and licenses granted (or purported to be granted) to Otsuka hereunder complies with and will not result in a breach of the terms and conditions of any Existing Third-Party IP Agreement; and (g) Ionis has
the right to grant sublicenses to Otsuka under the Existing Third-Party IP Agreements as contemplated herein, including to Develop, Manufacture, Commercialize and conduct Medical Affairs for the Licensed Products in the Field in the Otsuka
Territory.
|
|
|
11.2.13 |
Neither Ionis nor any counterparty to any Existing Third-Party IP Agreement has in writing alleged or threatened that the other party has breached an Existing Third-Party IP Agreement (which has not been cured) or, to Ionis’ Knowledge,
threatened in writing to terminate an Existing Third-Party IP Agreement.
|
|
|
11.2.14 |
Each [***].
|
|
|
11.2.15 |
to Ionis’ Knowledge: (a) [***]; and (b) [***].
|
|
|
11.2.16 |
All preclinical and clinical studies of the Licensed Products sponsored by Ionis or its Affiliates have been and as of the Effective Date are being conducted in material compliance with Applicable Law, including [***]. Neither Ionis nor
its Affiliates has received any written notice from the FDA, the EMA, or any other Regulatory Authority performing functions similar to those performed by those with respect to any ongoing clinical or pre-clinical studies or tests of the
Licensed Products requiring the termination, suspension, or material modification of such ongoing studies or tests, and no Governmental Authority has commenced any action to place a clinical hold order on, or otherwise terminate or suspend,
any ongoing Clinical Trial of the Licensed Products conducted by or on behalf of Ionis or its Affiliates as of the Effective Date.
|
|
|
11.2.17 |
As of the Effective Date, neither Ionis nor any of its Affiliates, and, to Ionis’ Knowledge, none of its or their respective officers, employees, or agents, has made an untrue statement of material fact or fraudulent statement to the FDA
or any other Regulatory Authority with respect to the Development of the Licensed Compounds or the Licensed Products, failed to disclose a material fact required to be disclosed to the FDA or any other Regulatory Authority with respect to the
Development of the Licensed Compounds or the Licensed Products, or committed an act, made a statement, or failed to make a statement with respect to the Development of the Licensed Compounds or the Licensed Products that could reasonably be
expected to provide a basis for the FDA to invoke its policy respecting “Fraud, Untrue Statements of Material Facts, Bribery, and Illegal Gratuities”, set forth in 56 Fed. Reg. 46191 (September 10, 1991) and any amendments thereto or any
analogous laws or policies in the Otsuka Territory.
|
|
|
11.2.18 |
As of the Effective Date, Ionis has no Knowledge of [***] in the Territory in the form existing as of the Effective Date for the treatment of HAE.
|
| 11.3 |
Additional Otsuka Representations and Warranties. Otsuka represents and warrants to Ionis as of the Effective Date that there are no Patent Rights Controlled by Otsuka or any of its Affiliates that
are necessary to Exploit a Licensed Product.
|
| 11.4 |
Additional Covenants. Each of Otsuka and Ionis hereby covenant to the other:
|
|
|
11.4.1 |
Assignment of Inventions. Each Party will require all of its and its Affiliates’ employees and consultants to assign all Inventions that are developed or invented by such employees according to the
ownership principles described in Section 10.1 (Inventions).
|
|
|
11.4.2 |
Compliance with Law. It will, and will ensure that its Affiliates, comply with all Applicable Law and, to the extent applicable, Professional Requirements, with respect to the performance of its
obligations under this Agreement, including, as applicable, the Approved Labeling, the European Data Protection Directive 95/46/EC, the European General Data Protection Regulation (Regulation (EU) 2016/679),
and any other applicable national data protection legislation.
|
|
|
11.4.3 |
No Bribery. It will not in the future offer, promise, pay, authorize, or give, money or anything of value, directly or indirectly, to any Government Official or Other Covered Party for the purpose,
pertaining to this Agreement, of: (a) influencing any act or decision of the Government Official or Other Covered Party; (b) inducing the Government Official or Other Covered Party to do or omit to do an act in violation of a lawful duty; (c)
securing any improper advantage; or (d) inducing the Government Official or Other Covered Party to influence the act or decision of a government or government instrumentality, in order to obtain or retain business, or direct business to, any
Person, in each case, in any way related to this Agreement.
|
|
|
11.4.4 |
Restricted Countries. Neither it nor its Affiliates will export, transfer, or sell any Licensed Product (a) to any country or territory that is subject to comprehensive economic sanctions
administered by OFAC, unless the sale of such Licensed Product would be permissible if Otsuka or its Affiliates or Sublicensees were subject to OFAC’s jurisdiction, (b) to any other country or territory in which such activity would violate
Applicable Law in the U.S., (c) to any Restricted Party unless the sale of such Licensed Product would be permissible if Otsuka or its Affiliates or Sublicensees was subject to OFAC’s jurisdiction, or (d) in such a manner that would violate
the Global Trade Control Laws.
|
|
|
11.4.5 |
FCPA Compliance. In performing under this Agreement, it and its Affiliates agree to comply with all applicable anti-corruption laws, including the Foreign Corrupt Practices Act of 1977 and the UK Bribery Act 2010, as amended from time-to-time; the anti-corruption laws of the Territory; and all laws enacted to implement the Organization for Economic Co-operation and Development
Convention on Combating Bribery of Foreign Officials in International Business Transactions.
|
|
|
11.4.6 |
Debarred/Excluded Persons. It will not engage, in any capacity in connection with this Agreement or any ancillary agreements, any officer, employee, contractor, consultant, agent, representative, or
other Person who has been Debarred/Excluded. Each Party will inform the other Party in writing promptly if it or any Person engaged by it or any of its Affiliates who is performing any obligations under this Agreement or any ancillary
agreements is Debarred/Excluded, or if any action, suit, claim, investigation, or legal or administrative proceeding is pending or, to each Party’s Knowledge, is threatened, pursuant to which a Party, any of its Affiliates or any such Person
performing obligations hereunder or thereunder may become Debarred/Excluded.
|
| 11.5 |
Disclaimer. EXCEPT AS EXPRESSLY SET FORTH HEREIN, THE INTELLECTUAL PROPERTY RIGHTS PROVIDED BY EACH PARTY ARE PROVIDED “AS IS” AND WITHOUT WARRANTY. EXCEPT AS EXPRESSLY SET FORTH HEREIN, EACH OF THE
PARTIES EXPRESSLY DISCLAIMS ANY AND ALL WARRANTIES OF ANY KIND, EXPRESS OR IMPLIED, INCLUDING THE WARRANTIES OF DESIGN, MERCHANTABILITY, FITNESS FOR A PARTICULAR PURPOSE, VALIDITY, OR ENFORCEABILITY OF THEIR RESPECTIVE INTELLECTUAL PROPERTY
RIGHTS, AND NONINFRINGEMENT OF THE INTELLECTUAL PROPERTY RIGHTS OF THIRD PARTIES, ARISING FROM A COURSE OF DEALING, USAGE, OR TRADE PRACTICES, IN ALL CASES WITH RESPECT THERETO.
|
| 11.6 |
Limitation of Liability. NEITHER OF THE PARTIES WILL BE ENTITLED TO RECOVER FROM THE OTHER PARTY ANY SPECIAL, INCIDENTAL, INDIRECT, CONSEQUENTIAL, OR PUNITIVE DAMAGES OR DAMAGES FOR LOSS OF PROFIT,
LOSS OF REVENUE, OR LOST OPPORTUNITY IN CONNECTION WITH THIS AGREEMENT, ITS PERFORMANCE OR LACK OF PERFORMANCE HEREUNDER, OR ANY LICENSE GRANTED HEREUNDER, EXCEPT TO THE EXTENT THE DAMAGES RESULT FROM A BREACH OF THE OBLIGATIONS OF A PARTY
UNDER ARTICLE 12 (CONFIDENTIALITY) OR BREACH OF SECTION 2.5.2 (NEGATIVE COVENANT) BY IONIS, MISAPPROPRIATION OR INFRINGEMENT OF INTELLECTUAL PROPERTY OWNED OR CONTROLLED BY THE OTHER PARTY, OR AMOUNTS REQUIRED TO BE PAID TO A
THIRD PARTY AS PART OF A CLAIM FOR WHICH A PARTY PROVIDES INDEMNIFICATION UNDER ARTICLE 13 (INDEMNIFICATION).
|
| 12.1 |
Duty of Confidence. Subject to the other provisions of this Article 12 (Confidentiality):
|
|
|
12.1.1 |
except to the extent expressly authorized by this Agreement, the Receiving Party shall maintain in confidence and otherwise safeguard, and not publish or otherwise disclosed, all Confidential Information of the Disclosing Party;
|
|
|
12.1.2 |
the Receiving Party will treat all Confidential Information provided by the Disclosing Party, at a minimum, with the same degree of care as the Receiving Party uses for its own similar information, but in no event less than a reasonable
degree of care;
|
|
|
12.1.3 |
the Receiving Party may only use any Confidential Information of the Disclosing Party for the purposes of performing its obligations or exercising its rights under this Agreement;
|
|
|
12.1.4 |
a Receiving Party may only disclose Confidential Information of the Disclosing Party to: (a) such Receiving Party’s Affiliates, licensees, and Sublicensees; and (b) employees, directors, officers, agents, contractors, attorneys,
accountants and consultants, of the Receiving Party and its Affiliates, licensees, and Sublicensees, in each case ((a) and (b)), to the extent reasonably necessary for the purposes of, and for those matters undertaken pursuant to, this
Agreement; provided that such Persons are bound by legally enforceable obligations of confidentiality and non-use with respect to the Disclosing Party’s Confidential Information, no less stringent
than the confidentiality and non-use obligations set forth in this Agreement, except that the term of such obligation will be customary for such recipient of Confidential Information. Each Party will remain responsible for any failure by its
Affiliates, licensees, and Sublicensees, and its and its Affiliates’, licensees’, and Sublicensees’ respective employees, directors, officers, agents, consultants, attorneys, accountants and contractors, in each case, to treat such
Confidential Information as required under this Section 12.1 (Duty of Confidence) (as if such Persons were Parties directly bound to the requirements of this Section 12.1 (Duty of Confidence)); and
|
|
|
12.1.5 |
each Party will promptly notify the other Party of any misuse or unauthorized disclosure of the other Party’s Confidential Information.
|
|
|
12.1.6 |
The confidentiality, non-use, and non-disclosure obligations set forth in this Section 12.1 (Duty of Confidence) will be in full force and effect from the Effective Date until [***] after expiration or termination of this
Agreement, provided that, with respect to any Know-How that is a trade secret and is identified as such by the Disclosing Party at the time of disclosure, the obligations of this Section 12.1
(Duty of Confidence) will continue for so long as such Know-How remains a trade secret.
|
| 12.2 |
Confidential Information. Notwithstanding anything to the contrary in the definition of “Confidential Information” set forth in Appendix 1 (Definitions), the Ionis Product-Specific Know-How,
any ROFN Exercise Notice, the Joint Collaboration Know-How and the terms of this Agreement will be the Confidential Information of both Parties, with each Party deemed to be the Receiving Party of such information; provided that Ionis Product-Specific Know-How will be deemed the Confidential Information of Ionis following any termination (but not expiration) of this Agreement. The Ionis Core Technology Know-How and the Ionis
Manufacturing and Analytical Know-How will be the Confidential Information of Ionis. The Otsuka Know-How will be the Confidential Information of Otsuka. Except as provided in Section 12.4 (Authorized Disclosures) and Section 12.6
(Publicity; Use of Names), neither Party nor its Affiliates may disclose the existence or the terms of this Agreement.
|
| 12.3 |
Exemptions. Information of a Disclosing Party will not be Confidential Information of such Disclosing Party to the extent that the Receiving Party can demonstrate through competent evidence that such
information:
|
|
|
12.3.1 |
was already known by the Receiving Party or any of its Affiliates without an obligation of confidentiality at the time of its receipt from the Disclosing Party, and not through a prior disclosure by or on behalf of the Disclosing Party, as
documented by the Receiving Party’s business records;
|
|
|
12.3.2 |
was generally available to the public or otherwise part of the public domain before its receipt from the Disclosing Party;
|
|
|
12.3.3 |
became generally available to the public or otherwise part of the public domain after its disclosure by the Disclosing Party other than through any act or omission of the Receiving Party or any of its Affiliates or disclosees in breach of
this Agreement;
|
|
|
12.3.4 |
is subsequently disclosed to the Receiving Party or any of its Affiliates without obligation of confidentiality by a Third Party who may rightfully do so and is not under a conflicting obligation of confidentiality to the Disclosing Party;
or
|
|
|
12.3.5 |
is developed by the Receiving Party or any of its Affiliates independently and without use of or reference to any Confidential Information received from the Disclosing Party, as documented by the Receiving Party’s business records.
|
| 12.4 |
Authorized Disclosures.
|
|
|
12.4.1 |
Permitted Circumstances. Notwithstanding the obligations set forth in Section 12.1 (Duty of Confidence), a Party may disclose the other Party’s Confidential Information (including this
Agreement and the terms herein) to the extent such disclosure is reasonably necessary in the following situations:
|
|
|
(a) |
the prosecution or enforcement of Ionis Patent Rights, Collaboration Patent Rights, or Otsuka Patent Rights, in each case, as contemplated by this Agreement;
|
|
|
(b) |
Regulatory Submissions and other filings or communications with Governmental Authorities (including Regulatory Authorities), as necessary for the Exploitation of the Licensed Products in connection with the exercise of the rights and the
performance of the obligations of the applicable Party under this Agreement;
|
|
|
(c) |
disclosure of this Agreement, its terms, and the status and results of Exploitation of the Licensed Products to actual or bona fide potential investors, acquirors, (sub)licensees (including any
counterparty to a Collaboration In-License), lenders, and other financial or commercial partners (including in connection with any royalty financing transaction), and their respective attorneys, accountants, banks, investors, and advisors,
solely for the purpose of evaluating or carrying out an actual or bona fide potential investment, acquisition, (sub)license, debt transaction, or collaboration transaction; provided that, in each such case, (i) such Persons are bound by obligations of confidentiality and non-use, or subject to professional ethical obligations of confidentiality, at
least as stringent as those set forth Article 12 (Confidentiality), except that the term of such obligation will be customary for such recipient of Confidential Information and such type of transaction and (ii) the scope of any such
disclosure is limited to the maximum extent practicable for the particular context in which it is being disclosed;
|
|
|
(d) |
such disclosure is required to comply with Applicable Law (whether generally or in pursuit of an application for listing of securities) including the United States Securities and Exchange Commission or equivalent foreign agency or
regulatory body, or otherwise required by judicial or administrative process, provided that in each such event, as promptly as reasonably practicable and to the extent not prohibited by Applicable
Law or judicial or administrative process, such Party will notify the other Party of such required disclosure and provide a draft of the disclosure to the other Party reasonably in advance of such filing or disclosure for the other Party’s
review and comment. The non-disclosing Party will provide any comments as soon as practicable, and the disclosing Party will consider in good faith any timely comments provided by the non-disclosing Party; provided
that the disclosing Party may or may not accept such comments in its reasonable discretion. Confidential Information that is disclosed in order to comply with Applicable Law or by judicial or administrative process pursuant to this Section
12.4.1(d) (Permitted Circumstances), in each case, will remain otherwise subject to the confidentiality and non-use provisions of this Article 12 (Confidentiality) with respect to the Party disclosing such Confidential
Information, and such Party will take all steps reasonably necessary, including seeking of confidential treatment or a protective order to the maximum extent permitted by Applicable Law or Governmental Authority, to ensure the continued
confidential treatment of such Confidential Information, and each Party will be responsible for its own legal and other external costs in connection with any such filing or disclosure pursuant to this Section 12.4.1(d) (Permitted
Circumstances); or
|
|
|
(e) |
disclosure pursuant to Section 12.6 (Publicity; Use of Names).
|
|
|
(f) |
If and whenever any Confidential Information is disclosed in accordance with this Section 12.4 (Authorized Disclosures), such disclosure will not cause any such information to cease to be Confidential Information except to the
extent that such disclosure results in a public disclosure of such information (other than by breach of this Agreement).
|
| 12.5 |
Publications.
|
|
|
12.5.1 |
Otsuka’s Right to Publish. Otsuka will have the right to publicly present or publish any Clinical Trial data, non-clinical or preclinical data, or any associated results or conclusions generated by
or on behalf of Ionis or Otsuka pursuant to this Agreement (each such proposed presentation or publication, an “Otsuka Publication”), [***]. If Otsuka desires to publicly present or
publish an Otsuka Publication in accordance with the foregoing sentence, then Otsuka will provide Ionis (including Ionis’ Alliance Manager and all Ionis members of the JSC) with a copy of such proposed Otsuka Publication at least [***] prior
to the earlier of its presentation or intended submission for publication (such applicable period, the “Review Period”). [***]. Notwithstanding any provision to contrary set forth in
this Agreement, Otsuka will [***]. Otsuka will provide Ionis a copy of any Otsuka Publication at the time of the submission or presentation thereof. Otsuka agrees to determine the authorship of all Otsuka Publications in accordance with all
applicable International Committee of Medical Journal Editors (ICMJE) guidelines. Otsuka will require its Affiliates and Sublicensees to comply with the obligations of this Section 12.5 (Publications) as if they were Otsuka, and
Otsuka will be liable for any non-compliance of such Persons.
|
|
|
12.5.2 |
Ionis’ Right to Publish. Ionis will have the right to publicly present or publish any Clinical Trial data, non-clinical or preclinical data, or any associated results or conclusions generated by or
on behalf of Ionis pursuant to this Agreement (each such proposed presentation or publication, a “Ionis Publication”) [***]. If Ionis desires to publicly present or publish a Ionis
Publication in accordance with the foregoing sentence, then Ionis will provide Otsuka (including Otsuka’s Alliance Manager and all Otsuka members of the JSC) with a copy of such proposed Ionis Publication for review during the applicable
Review Period. Ionis [***]. Notwithstanding any provision to contrary set forth in this Agreement, Ionis will [***]. Ionis will provide Otsuka a copy of any Ionis Publication at the time of the submission or presentation thereof. Ionis agrees
to determine the authorship of all Ionis Publications in accordance with all applicable International Committee of Medical Journal Editors (ICMJE) guidelines. Ionis will require its Affiliates to comply with the obligations of this Section
12.5 (Publications) as if they were Ionis, and Ionis will be liable for any non-compliance of such Persons.
|
|
|
12.5.3 |
Subsequent Publications. After any Otsuka Publication or Ionis Publication has been published or publicly presented in accordance with Section 12.5.1 (Otsuka’s Right to Publish) or Section
12.5.2 (Ionis’ Right to Publish), as applicable, either Party may make subsequent publications or presentations of the content of such previously published Otsuka Publication or Ionis Publication without further approval or review by
the other Party; provided, that such subsequent publication or presentation does not include any new data, information or conclusions, or present the content in a form or manner that materially alters
the conclusion or subject matter of the previous publication or public presentation.
|
| 12.6 |
Publicity; Use of Names.
|
|
|
12.6.1 |
Press Release. The Parties may issue a press release announcing this Agreement, on such date and time and in such form, in each case, as may be agreed by the Parties. Other than such press release and the public disclosures permitted by this Section 12.6.1 (Publicity) and Section 12.4 (Authorized Disclosures), the Parties agree that the portions of any other
news release or other public announcement relating to this Agreement or the performance hereunder that would disclose information other than that already in the public domain will require prior review and approval by both Parties (with such
approval not to be unreasonably withheld, conditioned, or delayed). However, the Parties agree that after (a) a disclosure pursuant to Section 12.6 (Publicity; Use of Names) or Section 12.4 (Authorized Disclosures) or (b) the
issuance of a press release (including the initial press release) or other public announcement pursuant to this Section 12.6.1 (Press Release) that has been reviewed and approved by the other Party, the disclosing Party may make
subsequent public disclosures reiterating such information without having to obtain the other Party’s prior consent and approval so long as the information in such press release or other public announcement remains true, correct, and such
disclosure is consistent with prior disclosures approved by the other Party pursuant to this Section 12.6 (Publicity; Use of Names) and which do not reveal non‑public information about the other Party. Similarly, after a Publication
has been made available to the public, each Party may post such Publication or a link to it on its corporate website or social media platforms (or any website managed by such Party in connection with a Clinical Trial for the Licensed
Products, as appropriate) without the prior written consent of the other Party, so long as the information in such Publication remains true, correct, and the most current information with respect to the subject matters set forth therein.
|
|
|
12.6.2 |
Disclosures by Ionis. Notwithstanding any provision to the contrary set forth in this Agreement, Ionis has the right to publicly disclose (in written, oral, or other form): (a) the achievement of any
Regulatory Milestone Event or Sales Milestone Event under this Agreement (including the timing of achievement of any such milestone event but without disclosing the amount of such milestone payment unless permitted pursuant to Section
12.4.1(d) (Permitted Circumstances)); (b) the commencement, completion, material data, or key results of any Clinical Trials for the Licensed Products conducted by or on behalf of Ionis; and (c) the achievement of Regulatory Approval
for any Licensed Product throughout the world; provided that, subject to Section 12.4.1(d) (Permitted Circumstances), [***].
|
|
|
12.6.3 |
Use of Names. Each Party will have the right to use the other Party’s name and logo in presentations, its website, collateral materials, and corporate overviews to describe the collaboration
relationship, as well as in taglines of press releases issued pursuant to this Section 12.6 (Publicity; Use of Names); provided that neither Party will [***], and each Party will [***].
Except as permitted under this Section 12.6 (Publicity; Use of Names) or with the prior express written permission of the other Party, neither Party will use the name, trademark, trade name, or logo of the other Party or its
Affiliates or their respective employees in any publicity, promotion, news release, or disclosure relating to this Agreement or its subject matter except as may be required by Applicable Law.
|
| 12.7 |
Acknowledgement.
|
|
|
12.7.1 |
To the extent permitted under Applicable Law in the Otsuka Territory, Otsuka will acknowledge in any press release, public presentation, or publication regarding a Licensed Product Ionis’ role in discovering and developing the Licensed
Products, that the Licensed Products are under license from Ionis, and [***].
|
|
|
12.7.2 |
Otsuka agrees that it will acknowledge Ionis’ role in the discovery of a Licensed Product in any scientific, medical, and other Licensed Product-related communications [***], by including the words “Discovered
by Ionis” or equivalent language (collectively, the “Ionis Attribution Language”) in any such communications; provided, however,
that [***].
|
| 13.1 |
Indemnification by Ionis. Ionis will indemnify, hold harmless, and defend Otsuka and its Affiliates and their respective directors, officers, employees, and agents (each, an “Otsuka Indemnitee”) from and against any and all Third Party suits, claims, actions, or demands (“Third Party Claims”) and all
liabilities, expenses, or losses (including reasonable attorneys’ fees, court costs, witness fees, damages, judgments, fines, and amounts paid in settlement) (“Losses”) arising
therefrom to the extent that the applicable Third Party Claims and such Losses arise out of (a) a breach of this Agreement by Ionis, (b) the Exploitation of the Licensed Products by or on behalf of Ionis or any of its Affiliates, licensees
(not including Otsuka or its Affiliates, Sublicensees, or its Subcontractors), Sublicensees, or Subcontractors, or (c) the negligence or willful misconduct of any Ionis Indemnitee. Notwithstanding the foregoing, Ionis will not have any
obligation to indemnify Otsuka Indemnitees to the extent that any Losses arise out of any Third Party Claim for which Otsuka is responsible for indemnifying Ionis pursuant to Section 13.2 (Indemnification by Otsuka).
|
| 13.2 |
Indemnification by Otsuka. Otsuka will indemnify, hold harmless, and defend Ionis and its Affiliates, and their respective directors, officers, employees, and agents (each, an “Ionis Indemnitee”) from and against any and all Third Party Claims and all Losses arising therefrom, to the extent that the applicable Third Party Claims and such Losses arise out of (a)
a breach of this Agreement by Otsuka, (b) the Exploitation of the Licensed Products by or on behalf of Otsuka or any of its Affiliates, Sublicensees, or Subcontractors, or (c) the negligence or willful misconduct of any Otsuka Indemnitee. Notwithstanding any provision to the contrary set forth in this Agreement, Otsuka will not have any obligation to indemnify the Ionis Indemnitees to the extent that
any Losses arise out of any Third Party Claim for which Ionis is responsible for indemnifying Otsuka pursuant to Section 13.1 (Indemnification by Ionis).
|
| 13.3 |
Indemnification Procedure. If either Party is seeking indemnification under Section 13.1 (Indemnification by Ionis) or Section 13.2 (Indemnification by Otsuka) (the “Indemnified Party”), then it will inform the other Party (the “Indemnifying Party”) of the Third Party Claim giving rise to
such indemnification obligations within [***] after receiving written notice of the Third Party Claim (it being understood and agreed, however, that the failure or delay by an Indemnified Party to give such notice of a Third Party Claim will
not affect the Indemnifying Party’s indemnification obligations hereunder except to the extent the Indemnifying Party will have been actually prejudiced as a result of such failure or delay to give notice). The Indemnifying Party will have
the right to assume the defense of any such Third Party Claim for which it is obligated to indemnify the Indemnified Party. The Indemnified Party will cooperate with the Indemnifying Party and the Indemnifying Party’s insurer as the
Indemnifying Party may reasonably request, and at the Indemnifying Party’s cost and expense. The Indemnified Party will have the right to participate, with counsel of its choice, in the defense of any Third Party that has been assumed by the
Indemnifying Party, which participation will be at the Indemnified Party’s expense unless (a) the Indemnifying Party has agreed to pay such fees and expenses, or (b) the Indemnified Party has been advised by counsel that there are actual or
potential conflicting interests between the Indemnifying Party and the Indemnified Party, including situations in which there are one or more legal defenses available to the Indemnified Party that are different from or additional to those
available to the Indemnifying Party. Neither Party will have the obligation to indemnify the other Party in connection with any settlement made without the Indemnifying Party’s written consent, which consent will not be unreasonably withheld,
conditioned, or delayed. The Indemnifying Party will not admit any fault or negligence on the part of the Indemnified Party, or impose any obligation on, or otherwise adversely affect, the Indemnified Party, without the Indemnified Party’s
prior written consent, which consent will not be unreasonably withheld, conditioned, or delayed. If the Parties cannot agree as to the application of Section 13.1 (Indemnification by Ionis) or Section 13.2 (Indemnification by
Otsuka) as to any Third Party Claim, then, pending resolution of the dispute pursuant to Article 15 (Dispute Resolution; Governing Law), then the Parties may conduct separate defenses of such Third Party Claims, with each Party
retaining the right to claim indemnification from the other Party in accordance with Section 13.1 (Indemnification by Ionis) or Section 13.2 (Indemnification by Otsuka), as applicable, upon resolution of the underlying Third
Party Claim.
|
| 13.4 |
Insurance. Each Party will, at its own expense, procure and maintain insurance, including product liability insurance, adequate to cover its obligations hereunder and that is consistent with normal
business practices of prudent companies similarly situated at all times during which any Licensed Product is being clinically tested in human subjects or commercially distributed or sold by such Party pursuant to this Agreement. It is
understood that such insurance will not be construed to create a limit of either Party’s liability with respect to its indemnification obligations under this Article 13 (Indemnification). Each Party will provide the other Party with
written evidence of such insurance upon request. Each Party will provide the other Party with [***].
|
| 14.1 |
Term. The term of this Agreement will begin on the Effective Date and, unless earlier terminated in accordance with this Article 14 (Term and Termination), will continue until Otsuka, its
Affiliates, and its Sublicensees are no longer Commercializing any Licensed Product in any country in the Otsuka Territory (the “Term”).
|
| 14.2 |
Termination for Material Breach.
|
|
|
14.2.1 |
Material Breach. If either Party believes in good faith that the other is in material breach of this Agreement, then the non-breaching Party may deliver notice of such breach to the other Party
stating the cause and proposed remedy (“Breach Notification”). For any breach alleged in any Breach Notification arising from a failure to make a payment set forth in this Agreement,
the allegedly breaching Party will have [***] from the receipt of the applicable Breach Notification to cure such breach. For all breaches other than a failure to make a payment as set forth in this Agreement, the allegedly breaching Party
will have [***] from the date of the Breach Notification to cure such breach. If the allegedly breaching Party fails to cure the applicable breach within the applicable period set forth above, then the Party originally delivering the Breach
Notification may terminate this Agreement effective on written notice of termination to such allegedly breaching Party.
|
|
|
14.2.2 |
Disagreement as to Material Breach. Notwithstanding Section 14.2.1 (Material Breach), if the Parties, reasonably and in good faith, disagree as to whether there has been a material breach of
this Agreement, then: (a) the Party that disputes whether there has been a material breach may contest the allegation by referring such matter, within the cure period applicable to such alleged material breach, for resolution in accordance
with Article 15 (Dispute Resolution; Governing Law); (b) the relevant cure period with respect to such alleged material breach will be tolled from the date on which the Party that disputes whether there has been a material breach
notifies the other Party of such dispute and through the resolution of such dispute in accordance with Article 15 (Dispute Resolution; Governing Law); and (c) during the pendency of such dispute, all of the terms and conditions of
this Agreement will remain in effect and the Parties will continue to perform all of their respective obligations hereunder.
|
| 14.3 |
Termination by Otsuka for Convenience. Otsuka will have the right to terminate this Agreement in its entirety at any time during the Term upon (a) [***] and (b) [***].
|
| 14.4 |
Discontinuation of Development and Commercialization. If Otsuka and its Affiliates and Sublicensees have not conducted any material Development or Commercialization activities with respect to the
Licensed Products in any country in the Otsuka Territory for a continuous period of [***], and such discontinuation of activity is not: (a) by written agreement of the Parties, (b) due to [***], or (c) due to a Force Majeure, then Ionis may, at its election, terminate this Agreement in its entirety upon [***] prior written notice to Otsuka. For purposes of this Section 14.4 (Discontinuation of
Development and Commercialization), the use of reasonable efforts, to the extent possible, by Otsuka or its Affiliates or Sublicensees (as applicable) to resolve a Force Majeure, clinical hold or other action or inaction of a Regulatory
Authority, or any scientific or technical issues, Manufacturing or supply interruption or other material adverse event outside of Otsuka’s control for the Licensed Product will, in each case, be considered material Development or
Commercialization activities.
|
| 14.5 |
Termination For Patent Challenge. Except to the extent unenforceable under Applicable Law, Ionis may terminate this Agreement in its entirety upon [***] prior written notice of termination to Otsuka
if Otsuka or its Affiliates or Sublicensees (individually or in association with any Person) commences or assists a Third Party in commencing or conducting a Patent Challenge with respect to any Ionis Patent Right, provided that, Ionis shall not have the right to terminate this Agreement on account of such Patent Challenge (a) if, within [***] after receipt by Otsuka of the written notice from Ionis as set forth above in this Section
14.5 (Termination for Patent Challenge), Otsuka or its Affiliate, as applicable, rescinds such Patent Challenge (or in the case of any ex-parte proceeding, multi-party proceeding, or other Patent Challenge that Otsuka or such Affiliate
does not have the power to unilaterally withdraw or cause to be withdrawn, Otsuka and its Affiliate, as applicable, knowingly ceases providing any direction to any Person with respect to such Patent Challenge and, to the extent Otsuka or any
of its Affiliates is a party to such Patent Challenge and to the extent permitted by the applicable tribunal, it withdraws from such Patent Challenge) and provided that neither Otsuka nor any of its
Affiliates thereafter continues such Patent Challenge or, knowingly provides any direction to any Person in respect of the same or (b) in the case of any Patent Challenge commenced by a Sublicensee of Otsuka or its Affiliate, if Otsuka or its
Affiliate, as applicable, terminates such Sublicensee’s sublicense under any Ionis Technology within [***] after receipt by Otsuka of the applicable written notice from Ionis as set forth above in this Section 14.5 (Termination for
Patent Challenge). If Ionis has the right to terminate this Agreement in accordance with this Section 14.5 (Termination for Patent Challenge) but such termination is prohibited under Applicable Law, then in lieu of such termination,
[***]. If Ionis [***], then [***]. Notwithstanding the foregoing, Ionis shall not have the right to terminate this Agreement pursuant to this Section 14.5 (Termination for Patent Challenge) if Otsuka or any of its Affiliates or its or
their Sublicensees commences a Patent Challenge (i) in a proceeding involving an Ionis Patent Right in which Otsuka or any of its Affiliates or its or their Sublicensees has been compelled to participate in the proceeding by a court, patent
office, or Third Party or (ii) that is necessary or reasonably required to assert a cross-claim or a counterclaim or to respond to a court request or order or administrative law request or order, including asserting any defense or
counterclaim in, or otherwise responding to, an action for infringement of intellectual property asserted, filed or threatened to be filed against Otsuka or any of its Affiliates or its or their Sublicensees by Ionis or any of its Affiliates
or its or their Sublicensees.
|
| 14.6 |
Termination for Insolvency. Each Party will have the right to terminate this Agreement upon delivery of written notice to the other Party if (a) such other Party files in any court or agency pursuant
to any statute or regulation of any jurisdiction a petition in bankruptcy or insolvency or for reorganization or similar arrangement for the benefit of creditors or for the appointment of a receiver or trustee of such other Party or its
assets, (b) such other Party is served with an involuntary petition against it in any insolvency proceeding and such involuntary petition has not been stayed or dismissed within [***] of its filing, or (c) such other Party makes an assignment
of substantially all of its assets for the benefit of its creditors.
|
| 14.7 |
Rights in Bankruptcy. The Parties intend to take advantage of the protections of Section 365(n) (or any successor provision) of the U.S. Bankruptcy Code or any analogous provisions in any other
country or jurisdiction to the maximum extent permitted by Applicable Law. All rights and licenses granted under or pursuant to this Agreement shall be deemed to be “intellectual property” for the purposes of Section 365(n) or any analogous
provisions in any other country or jurisdiction. The Parties shall retain and may fully exercise all of their rights and elections under the U.S. Bankruptcy Code or any analogous provisions in any other country or jurisdiction, including the
right to obtain the intellectual property from another entity. In the event of the commencement of a bankruptcy proceeding by or against either Party under the U.S. Bankruptcy Code or any analogous provisions in any other country or
jurisdiction, the Party that is not subject to such proceeding shall be entitled to a complete duplicate of (or complete access to, as appropriate) all such intellectual property (including all embodiments of such intellectual property),
which, if not already in the non-subject Party’s possession, shall be promptly delivered to it upon the non-subject Party’s written request (a) upon commencement of a bankruptcy proceeding, unless the Party subject to such proceeding
continues to perform all of its obligations under this Agreement, or (b) if not delivered pursuant to clause (a) above because the subject Party continues to perform, upon the rejection of this Agreement by or on behalf of the subject Party.
Unless and until the subject Party rejects this Agreement, the subject Party shall perform this Agreement or provide the intellectual property (including all embodiments of such intellectual property) to the non-subject Party, and shall not
interfere with the rights of the non-subject Party to such intellectual property, including the right to obtain the intellectual property from another entity.
|
| 14.8 |
Full Force and Effect During Notice Period. This Agreement will remain in full force and effect until the expiration of the applicable termination notice period. For clarity, if Otsuka or any of its
Affiliates or Sublicensees achieve any Milestone Events during the termination notice period, then the corresponding Milestone Payment is accrued and Otsuka will remain responsible for the payment of such Milestone Payment even if the due
date of such Milestone Payment occurs after the effective date of the termination.
|
| 14.9 |
Effects of Termination. If this Agreement is terminated by either Party pursuant to Section 14.2 (Termination for Material Breach) or Section 14.6 (Termination for Insolvency), by
Otsuka pursuant to Section 14.3 (Termination by Otsuka for Convenience), or by Ionis pursuant to Section 14.4 (Discontinuation of Development and Commercialization), or Section 14.5 (Termination for Patent Challenge),
then all rights in the Licensed Products will revert to Ionis, and the following will apply with respect to the Licensed Products:
|
|
|
14.9.1 |
Termination of Licenses. As of the effective date of termination of this Agreement, all rights licensed to Otsuka under Section 2.1 (License Grants to Otsuka) or otherwise under this
Agreement (except for the licenses granted under Section 2.4 (Collaboration Technology Enabling License)), in each case, will each terminate, but each Party will retain its joint ownership interests in the Joint Collaboration
Technology.
|
|
|
14.9.2 |
Reversion License.
|
|
|
(a) |
License Grant. Ionis will have, and Otsuka hereby grants to Ionis, effective upon such termination, a worldwide, [***] license under any Patent Rights and Know-How Controlled by Otsuka as of the
effective date of such termination, other than any Otsuka Technology, that, in each case, are used by Otsuka or its Affiliates in the Exploitation of any Licensed Product prior to or as of the effective date of such termination solely to
Exploit the Licensed Products in the Territory (the “Reversion License”). Except as otherwise provided in Section 14.9.8 (Sell-Off), Otsuka will not have the right to
Commercialize any Licensed Product in the Territory upon and following the effective date of termination of this Agreement.
|
|
|
(b) |
Reversion Royalty. Ionis will pay, on a Calendar Quarter basis during the applicable Royalty Term (defined mutatis mutandis with respect to the Reversion
License except that, for clarity, references to the Ionis Patent Rights in such definition will instead refer to any Patent Rights licensed by Otsuka to Ionis under the Reversion License) a [***] royalty on Ionis’ Net Sales (defined mutatis mutandis with respect to the Reversion License) of each Licensed Product in the Territory. The provisions of Section 9.3.4 (Royalty Payments and Reports) through Section 9.11 (Late
Payments; Disputed Payments) will apply to such payment obligation mutatis mutandis. Notwithstanding the foregoing, in no event will the total amount of the reversion royalty payments under this Section
14.9.2(b) (Reversion Royalty) exceed [***].
|
|
|
14.9.3 |
Transition Services.
|
|
|
(a) |
Scope. Ionis may request that Otsuka perform transition activities with respect to any Licensed Products in the Otsuka Territory that are necessary to transition the responsibilities under all
Regulatory Approvals and ongoing Clinical Trials for Licensed Products to Ionis or its designee. If Ionis requests that Otsuka perform any such transition activities, then the Parties will enter into a transition agreement containing a plan
for Otsuka to perform the transition services listed in Schedule 14.9.3 (Transition Services), to the extent applicable at the time of termination, and such other transition
services that the Parties mutually agree to (such plan, the “Transition Plan” and such activities, the “Transition Services”).
|
|
|
(b) |
Transition Plan. Ionis may elect to have Otsuka perform the Transition Services by providing written notice to Otsuka no later than [***] following the effective date of the termination. If Ionis
requests that Otsuka perform the Transition Services, then Ionis will propose a draft of the Transition Plan setting forth the Transition Services to be performed by Otsuka and the Parties will negotiate and enter into the Transition Plan,
which will be consistent with this Section 14.9.3 (Transition Services) and will include, to the extent applicable, the services listed on Schedule 14.9.3 (Transition
Services), within [***] after such request. In addition, the Parties will, within [***] after such request, establish a transition committee consisting of at least each Party’s Alliance Managers, a representative from each Party’s CMC group
who was responsible for the Licensed Product prior to the termination, and up to two additional representatives from each Party who are from other relevant functional groups to facilitate a smooth transition. While Otsuka is providing
Transition Services, Otsuka and Ionis will mutually agree on talking points and a communication plan to customers, specialty pharmacies, physicians, Regulatory Authorities, patient advocacy groups, and clinical study investigators, and Otsuka
will make all such communication to such entities in accordance with the mutually agreed talking points.
|
|
|
(c) |
Costs. Ionis will pay Otsuka for [***]. In addition, Ionis will reimburse Otsuka for [***]. Otsuka will submit an invoice, together with supporting documentation of [***], to Ionis quarterly for the
foregoing costs incurred by or on behalf of Otsuka in such Calendar Quarter, and Ionis will pay the undisputed invoiced amounts within [***] after the date of such invoice (and will pay any disputed amounts within [***] following resolution
of the dispute and determination that such amounts are owed).
|
|
|
14.9.4 |
Return of Confidential Information. Each Party will return or destroy (at the other Party’s election) all Confidential Information of the other Party in its possession upon termination of this
Agreement and, if applicable, the Receiving Party will provide a written confirmation of such destruction within [***] of such request. Notwithstanding the foregoing or any provision to the contrary set forth in this Agreement: (a) the
foregoing terms of this Section 14.9.4 (Return of Confidential Information) will not apply to any Confidential Information that is necessary to allow the Receiving Party to perform its obligations or exercise any of its rights that
expressly survive the applicable termination of this Agreement, and the Receiving Party may retain one copy of such Confidential Information for its legal archives; and (b) the Receiving Party will not be required to destroy electronic files
containing such Confidential Information that are made in the ordinary course of its business information back-up procedures pursuant to its electronic record retention and destruction practices that apply to its own general electronic files
and information.
|
|
|
14.9.5 |
Sublicenses. If this Agreement is terminated prior to expiration, then Ionis shall grant to each Sublicensee of Otsuka, at each such Sublicensee’s written request to Ionis within [***] of the
effective date of termination, a direct license, provided that such Sublicensee (a) is not then in default of its sublicense agreement or this Agreement, (b) agrees in writing to comply with the terms
of this Agreement to the extent applicable to the rights originally sublicensed to such Sublicensee by Otsuka, and (c) agrees to pay directly to Ionis such Sublicensee’s payments under such sublicense agreement. The scope of such direct
license shall be no less than the scope of the license granted herein and sublicensed to such Sublicensee, and Ionis shall have no obligation to perform any task for such Sublicensee beyond the obligations owed to Otsuka hereunder. Each
Sublicensee will be an intended Third Party beneficiary of this Section 14.9.5 (Sublicenses) with the right to enforce the same against Ionis.
|
|
|
14.9.6 |
Assignment. To the extent requested by Ionis in writing following the date that a Party provides notice of termination of this Agreement, Otsuka will promptly (and in any event no later than [***]
after the effective date of termination unless agreed otherwise in the Transition Plan or expressly specified otherwise below):
|
|
|
(a) |
provide to Ionis for its review unredacted copies of all clinical trial agreements and distribution agreements (to the extent assignable, not cancelled, and solely related to the Licensed Products), in each case, that are necessary or
reasonably useful for the Exploitation of the Licensed Products, and, following such review, upon Ionis’ written request within [***] after entering into a Transition Plan or [***] after the effective date of termination if Ionis does not
elect to enter into a Transition Plan, assign and transfer to Ionis or its designee all of Otsuka’s rights, title, and interests in and to any such agreements;
|
|
|
(b) |
assign to Ionis any Potential In-Licenses entered into by Otsuka pursuant to Section 2.7.2 (Potential In-Licenses);
|
|
|
(c) |
assign any agreements or arrangements with Third Party vendors (including distributors) solely related to the Licensed Products or, to the extent any such Third Party agreement or arrangement is not assignable to Ionis, reasonably
cooperate with Ionis to arrange to continue to provide such services for a reasonable time after termination of this Agreement to facilitate the orderly transition of all Commercialization and other activities then being performed by or on
behalf of Otsuka or its Affiliates or Sublicensees for the Licensed Products to Ionis or its designee;
|
|
|
(d) |
assign and transfer to Ionis or its designee, as of the effective date of termination, all of Otsuka’s rights, title, and interests in and to the Otsuka Product Trademarks and any domain names associated with the Otsuka Product Trademarks
(to the extent that Otsuka or its Affiliates has any) and promptly provide to Ionis all information necessary to maintain such domain names;
|
|
|
(e) |
assign and transfer to Ionis or its designee, as of the effective date of termination, all of Otsuka’s rights, title, and interests in and to any Product Materials specifically related to the Licensed Products, and copyrights and any registrations for the foregoing (to the extent that Otsuka or its Affiliates has any); and
|
|
|
(f) |
within [***] after entering into a Transition Plan or [***] after the effective date of termination if Ionis does not elect to enter into a Transition Plan, disclose to Ionis or its designee all documents, records, and materials that
embody any of the foregoing and that are Controlled by Otsuka.
|
|
|
14.9.7 |
Regulatory Submissions and Regulatory Approvals. Otsuka will and hereby does, and will cause its Affiliates and Sublicensees to, (a) no later than [***] after the effective date of termination of
this Agreement, assign and transfer to Ionis or its designee all of Otsuka’s rights, title, and interests in and to all Regulatory Submissions, Regulatory Approvals, and Reimbursement Approvals solely for the Licensed Products then Controlled
by Otsuka or any of its Affiliates or Sublicensees (for any Sublicensees that do not become a direct licensee of Ionis pursuant to Section 14.9.5 (Sublicenses)), and (b) to the extent assignment pursuant to clause (a) is delayed or is
not permitted by the applicable Regulatory Authority, permit Ionis to cross-reference and rely upon any such Regulatory Submissions, Regulatory Approvals, and Reimbursement Approvals filed by Otsuka or any of its Affiliates or Sublicensees
(for any Sublicensees that do not become a direct licensee of Ionis pursuant to Section 14.9.5 (Sublicenses)). Otsuka will execute and deliver, or will cause to be executed and delivered, to Ionis or its designee such endorsements,
assignments, commitments, acknowledgements, and other documents as may be necessary to effect the foregoing assignment, including submitting to each applicable Regulatory Authority or other Governmental Authority a letter or other necessary
documentation (with copy to Ionis) notifying such Regulatory Authority or other Governmental Authority of, or otherwise giving effect to, the transfer of ownership to Ionis of all such assigned Regulatory Submissions, Regulatory Approvals,
and Reimbursement Approvals. In addition, upon Ionis’ written request, Otsuka will, [***] (other than in the event of termination of this Agreement by Otsuka pursuant to Section 14.2 (Termination for Material Breach) or Section
14.6 (Termination for Insolvency), in which case Ionis shall [***]), provide to Ionis copies of all material related documentation, including material non-clinical, preclinical, and clinical data related to the Licensed Products that
are held by or reasonably available to Otsuka or its Affiliates or Sublicensees (for any Sublicensees that do not become a direct licensee of Ionis pursuant to Section 14.9.5 (Sublicenses)).
|
|
|
14.9.8 |
Sell-Off. If Otsuka is Commercializing any Licensed Product in any country in the Otsuka Territory as of the applicable effective date of termination, then, [***], either (a) Otsuka will appoint
Ionis or its designee as its exclusive distributor of such Licensed Product in such country and grant Ionis or its designee the right to appoint sub-distributors, to the extent not prohibited by any written agreement between Otsuka or any of
its Affiliates and a Third Party or (b) Otsuka will have the continued right to sell the Licensed Products in the Otsuka Territory from its inventory; provided, however,
that Otsuka’s obligations under this Agreement with respect to the Licensed Products that Otsuka sells, including the obligation to pay Royalties to Ionis hereunder, will continue in full force and effect during such period. If Ionis elects
to be appointed as the exclusive distributor pursuant to the foregoing clause (a), then the Parties will enter into a distribution agreement with respect to such appointment and Ionis will use good faith efforts to distribute such Licensed
Product in such country, or otherwise distribute such Licensed Product in such country, in accordance with the terms of the distribution agreement.
|
|
|
14.9.9 |
Inventory. [***].
|
|
|
14.9.10 |
Wind Down and Transition. Otsuka will be responsible, [***] (other than in the event of termination of this Agreement by Otsuka pursuant to Section 14.2 (Termination for Material Breach) or Section
14.6 (Termination for Insolvency), in which case Ionis shall [***]), for the wind-down of Otsuka’s and its Affiliates’ and Sublicensees (for any Sublicensees that do not become a direct licensee of Ionis pursuant to Section 14.9.5
(Sublicenses)) activities with respect to the Licensed Products. Otsuka will, and will cause its Affiliates and such Sublicensees to, reasonably cooperate with Ionis to facilitate orderly transition to Ionis or its designee of all
Commercialization and other activities then being performed by or on behalf of Otsuka or its Affiliates for the Licensed Products in the Otsuka Territory.
|
|
|
14.9.11 |
Cost of Transition Activities. Notwithstanding any provision to the contrary in this Section 14.9 (Effects of Termination), but without limiting Section 14.9.3(c) (Costs), if Otsuka
terminates this Agreement pursuant to Section 14.2 (Termination for Material Breach) or Section 14.6 (Termination for Insolvency), Ionis will be responsible for, and will pay Otsuka, [***]. Otsuka will submit an invoice,
together with supporting documentation of [***], to Ionis quarterly for the foregoing costs incurred by or on behalf of Otsuka in such Calendar Quarter, and Ionis will pay the undisputed invoiced amounts within [***] after the date of any
such invoice (and will pay any disputed amounts within [***] following resolution of the dispute and determination that such amounts are owed).
|
|
|
14.9.12 |
Other Assistance; Further Assurances. Otsuka will provide any other assistance reasonably requested by Ionis for the purpose of allowing Ionis or its designee to proceed expeditiously with the
Exploitation of the Licensed Products for a period of [***] after the effective date of termination of this Agreement. Otsuka will execute all documents, and take all such further actions as may be reasonably requested by Ionis in order to
give effect to the requirements in this Section 14.9 (Effects of Termination).
|
| 14.10 |
Survival; Accrued Rights. Expiration or termination of this Agreement will not relieve the Parties of any liability that accrued hereunder prior to the effective date of such expiration or
termination nor preclude either Party from pursuing all rights and remedies it may have hereunder or at law or in equity with respect to any breach of this Agreement, nor prejudice either Party’s right to obtain performance of any obligation.
Without limiting the foregoing, the following provisions of this Agreement will survive the expiration or termination of this Agreement: Section 2.2 (License Grant to Ionis); Section 2.4 (Collaboration Technology Enabling
License); Section 4.6 (Development Records; Cooperation) (solely with respect to the obligation to maintain records for at least [***] after the end of the Term or for such longer period as may be required by Applicable Law); Article
9 (Payments) (solely with respect to amounts that accrued prior to the effective date of termination and, with respect to Section 9.5 (Financial Records and Audits), solely for [***] after the effective date of termination), Section
10.1 (Inventions); Section 11.5 (Disclaimer); Section 11.6 (Limitation of Liability); Article 12 (Confidentiality, excluding Section 12.5 (Publications)); Article 13 (Indemnification); Section
14.9 (Effects of Termination); this Section 14.10 (Survival; Accrued Rights); Article 15 (Dispute Resolution; Governing Law); Article 16 (Miscellaneous); and Appendix 1 (Definitions).
|
| 15.1 |
Executive Officers; Disputes. Each Party will ensure that an Executive Officer is designated for such Party at all times during the Term for dispute resolution purposes, and will promptly notify the
other Party of any change in its designated Executive Officer. In the event of a dispute, controversy or claim arising under, relating to, or in connection with this Agreement (except for disputes arising at the JSC, which will be resolved in
accordance with Section 8.4 (Decision-Making) and Section 8.5 (Resolution of Committee Disputes)) (a “Disputed Matter”), then the Parties will refer such
dispute to their respective Executive Officer, and such Executive Officers or designees will attempt in good faith to resolve such dispute. If the Parties are unable to resolve any such dispute within [***] after both Parties have referred
such dispute to their designated Executive Officers pursuant to this Section 15.1 (Executive Officers; Disputes), then either Party will have the right to pursue any and all remedies available at law or equity, as set forth in Section
15.2 (Arbitration) or Section 15.3 (Intellectual Property Disputes), as applicable.
|
| 15.2 |
Arbitration.
|
|
|
15.2.1 |
If the Parties are unable to resolve a Disputed Matter using the process described in Section 15.1 (Executive Officers; Disputes) and Section 15.3 (Intellectual Property Disputes) does not
apply, then a Party seeking further resolution of the Disputed Matter will submit the Disputed Matter to resolution by final and binding arbitration in accordance with this Section 15.2 (Arbitration).
|
|
|
15.2.2 |
The seat, or legal place, of arbitration will be New York, New York. The arbitration will be administered by the International Chamber of Commerce pursuant to its Rules of Arbitration in effect at the
time of the arbitration, (the “Rules”), except they may be modified as set forth herein, and applying the substantive
law specified in Section 15.5 (Governing Law; English Language). The language of the arbitration will be English.
|
|
|
15.2.3 |
Unless a Party elects for application of the ICC’s Expedited Procedure Rules pursuant to Section 15.2.4 (Arbitration) or the Expedited Procedure Rules otherwise apply because of the amount in
dispute, the arbitration will be conducted by a tribunal of three arbitrators. The claimant will nominate an arbitrator in its request for arbitration; the respondent will nominate an arbitrator within [***] of receipt of the request for arbitration; and the two-party nominated arbitrators will nominate the third, who will serve as chair of the tribunal, within [***] of the second
arbitrator’s appointment. If any of the three arbitrators are not nominated within the time prescribed above, then the ICC will appoint the arbitrator(s). Within [***] of the commencement of
arbitration, the Parties will attempt in good faith to reach agreement upon and thereafter follow procedures directed at assuring that the arbitration will be concluded and the award rendered within no more than [***] from the date the ICC Secretariat transmits the file to the arbitral tribunal. Failing such agreement, the arbitral tribunal will design and the Parties will follow procedures directed at meeting such a time
schedule. Each arbitrator must have at least [***] of business or legal experience in the pharmaceutical industry. An arbitrator will be deemed to meet these qualifications unless a Party objects
within [***] after the arbitrator is nominated.
|
|
|
15.2.4 |
Notwithstanding Section 15.2.3 (Arbitration), if the Disputed Matter involves the dispute of a Breach Notification for any default other than a determination of an alleged failure to use
Commercially Reasonable Efforts to Develop or Commercialize the Licensed Product, the non-breaching Party may elect on notice to the breaching Party to apply the ICC Expedited Procedure Rules to the arbitration and, if such election is
made, the number of arbitrators will be one and the period for the rendering of the final award will be [***] from the date of the case management conference.
|
|
|
15.2.5 |
The Parties agree that any dispute concerning the propriety of the commencement of the arbitration or the scope and applicability of the agreement to arbitrate will be determined by the arbitrator(s).
|
|
|
15.2.6 |
No tribunal of arbitrators will have the power to award damages excluded pursuant to Section 11.6 (Limitation of Liability).
|
|
|
15.2.7 |
Article 38 of the Rules will apply with respect to the costs of the arbitration.
|
|
|
15.2.8 |
Except as may be required by Applicable Law, neither a Party nor any arbitrator may disclose the existence, content or results of any arbitration hereunder without the prior written consent of both Parties,
unless to protect or pursue a legal right. The arbitral award will be final and binding on the Parties and the Parties will carry out the award without delay. Judgment on the award so rendered may be entered in any court of competent
jurisdiction. No award or procedural order made in the arbitration shall be published.
|
| 15.3 |
Intellectual Property Disputes. Notwithstanding any provision to the contrary set forth in this Agreement, if a dispute arises under this Agreement with respect to the validity, scope,
enforceability, or ownership of any Patent Right or other intellectual property rights, and such dispute is not resolved in accordance with Section 15.1 (Executive Officers; Disputes), then such dispute will be submitted to a court of
competent jurisdiction in the jurisdiction in which such Patent Right or other intellectual property right was granted or arose.
|
| 15.4 |
Equitable Remedies. Notwithstanding any provision to the contrary set forth in this Agreement, the Parties each stipulate and agree that (a) the other Party’s Confidential Information includes highly
sensitive trade secret information such that a breach of Article 12 (Confidentiality) by a Party will cause irrevocable harm for which monetary damages would not provide a sufficient remedy; and (b) in such case of such breach of Article
12 (Confidentiality), the non-breaching Party will be entitled to seek equitable relief, including specific performance, temporary or permanent restraining orders, preliminary injunction, permanent injunction, or other equitable relief
without the posting of any bond or other security. In addition, and notwithstanding any provision to the contrary set forth in this Agreement, in the event of any other actual or threatened breach hereunder, the aggrieved Party may seek
interim equitable relief (including temporary restraining orders, or other provisional equitable relief) from any court of competent jurisdiction without first submitting to the dispute resolution procedures set forth in Article 15
(Dispute Resolution; Governing Law) and shall retain that right after the appointment of the arbitrator(s).
|
| 15.5 |
Governing Law; English Language. This Agreement and all amendments, modifications, alterations, or supplements hereto, and the rights of the Parties, will be construed under and governed by the laws
of the State of New York, United States, exclusive of its conflicts of laws principles. This Agreement has been prepared in the English language and the English language will control its interpretation. All consents, notices, reports, and
other written documents to be delivered or provided by a Party under this Agreement will be in the English language, and in the event of any conflict between the provisions of any document and the English language translation thereof, the
terms of the English language translation will control.
|
| 16.1 |
Assignment. This Agreement may not be assigned or otherwise transferred, nor may any right or obligation hereunder be assigned or transferred, by either Party without the prior written consent of the
other Party. Notwithstanding the foregoing, (a) Ionis may assign its rights to receive payments under this Agreement to one or more Persons (including as part of a royalty monetization transaction) (a “Payment Assignment”) without consent of Otsuka; provided that Ionis shall give prompt written notice to Otsuka upon making a Payment Assignment, and any assignee of a
Payment Assignment shall not have any rights, including any audit rights, hereunder (other than the right to receive payments under this Agreement) unless Otsuka provides express prior written consent, which Otsuka may grant or withhold in
its discretion, and (b) either Party may, without consent of the other Party, assign this Agreement and its rights and obligations hereunder (i) in whole or in part to an Affiliate
of such Party (for so long as such Affiliate remains an Affiliate), or (ii) in whole to its successor-in-interest in connection with the sale of all or substantially all of its assets, whether in a merger, acquisition, or similar transaction
or series of related transactions. If there is an assignment pursuant to the foregoing clauses (b)(i) or (b)(ii), then such assignment will only be effective if the Person to whom this Agreement is assigned agrees in writing to assume all of
the assigning Party’s obligations under this Agreement and the assigning Party provides written notice of such assignment to the non-assigning Party within [***] after the effective date of such assignment. Any attempted assignment of this
Agreement in violation of this Section 16.1 (Assignment) will be null, void, and of no legal effect. Any permitted assignee will assume all assigned obligations of its assignor under this Agreement. This Agreement will be binding on
and will inure to the benefit of the permitted successors and assigns of the Parties.
|
| 16.2 |
Entire Agreement; Amendment. This Agreement and the Ancillary Agreements, together with all exhibits and schedules attached hereto, constitutes the entire agreement between the Parties with respect
to the subject matter hereof, and supersedes and merges all prior and contemporaneous negotiations, representations, and understandings regarding the same, (including that certain mutual confidential disclosure agreement between the Parties
dated [***] (“Confidential Disclosure Agreement”)). All information shared by the Parties pursuant to the Confidential Disclosure Agreement will be Confidential Information under
this Agreement from and after the Effective Date, and the use and disclosure thereof will be governed by Article 12 (Confidentiality). This Agreement may not be modified or amended, except by another agreement in writing executed by
duly authorized signatories of each Party.
|
| 16.3 |
No Strict Construction; Interpretation. This Agreement has been prepared jointly and will not be strictly construed against either Party. Ambiguities, if any, in this Agreement will not be construed
against any Party, irrespective of which Party may be deemed to have authored the ambiguous provision. Except where the context expressly requires otherwise, (a) whenever any provision of this Agreement uses the term “including” (or
“includes”), such term will be deemed to mean “including without limitation” and “including but not limited to” (or “includes without limitations” and “includes but is not limited to”) regardless of whether the words “without limitation” or
“but not limited to” actually follow the term “including” (or “includes”); (b) “herein,” “hereby,” “hereunder,” “hereof,” and other equivalent words will refer to this Agreement in its entirety and not solely to the particular portion of this
Agreement in which any such word is used; (c) all definitions set forth herein will be deemed applicable whether the words defined are used herein in the singular or the plural; (d) wherever used herein, any pronoun or pronouns will be deemed
to include both the singular and plural and to cover all genders; (e) the schedules and exhibits to this Agreement, and the terms and conditions incorporated in such schedules and exhibits will be deemed integral parts of this Agreement and
all references in this Agreement to this Agreement will encompass such schedules and exhibits and the terms and conditions incorporated in such schedules and exhibits; provided that if there is a
conflict between the terms and conditions of this Agreement and any terms and conditions set forth in the schedules, or exhibits, then the terms of this Agreement will control; (f) in the event of any conflict between the terms and conditions
of this Agreement and any terms and conditions that may be set forth on any order, invoice, or verbal agreement by the Parties pursuant to this Agreement, the terms and conditions of this Agreement will govern; (g) unless otherwise provided,
all references to Sections, Articles, and Schedules in this Agreement are to Sections, Articles, and Schedules of and to this Agreement; (h) any reference to any federal, national, state, local, or foreign statute or law will be deemed to
also refer to all rules and regulations promulgated thereunder, and any reference to any law, rule, or regulation will be deemed to include the then‑current amendments thereto or any replacement or successor law, rule, or regulation thereof;
(i) wherever used, the word “shall” and the word “will” are each understood to be imperative or mandatory in nature and are interchangeable with one another; (j) the term “or” will be interpreted in the inclusive sense commonly associated
with the term “and/or”; (k) references to a particular Person include such Person’s successors and assigns to the extent not prohibited by this Agreement; (l) the section headings and captions used herein are inserted for convenience of
reference only and will not be construed to create obligations, benefits, or limitations; (m) any definition of or reference to any agreement, instrument, or other document herein will be construed as referring to such agreement, instrument,
or other document as from time to time amended, supplemented, or otherwise modified (subject to any restrictions on such amendments, supplements, or modifications set forth herein); (n) the word “notice” means notice in writing (whether or
not specifically stated) and will include notices, consents, approvals, and other written communications contemplated under this Agreement; and (o) provisions that require that a Party, the Parties, or any committee hereunder “agree,”
“consent,” or “approve” or the like will require that such agreement, consent, or approval be specific and in writing, whether by written agreement, letter, approved minutes, or otherwise (but excluding email and instant messaging).
|
| 16.4 |
Severability. If any provision of this Agreement is declared invalid by a court of last resort or by any court or other governmental body the decision of which an appeal is not taken within the time
provided by law, then and in such event, this Agreement will be deemed to have been terminated only as to the portion thereof that relates to the provision invalidated by that decision and only in the relevant jurisdiction, but this Agreement
will remain in force, in all other respects and all other jurisdictions; provided, however, that if the provision so invalidated is essential to this Agreement as a whole, then the Parties will
negotiate in good faith to amend the terms hereof as nearly as practical to carry out the original intent of the Parties, and, failing such amendment, either Party may submit the matter for resolution pursuant to Article 15 (Dispute Resolution; Governing Law).
|
| 16.5 |
Force Majeure. Neither Party will be held liable or responsible to the other Party nor be deemed to be in default under, or in breach of any provision of, this Agreement for failure or delay in
fulfilling or performing any obligation of this Agreement when such failure or delay is due to Force Majeure. For purposes of this Agreement, “Force Majeure” is defined as any cause
beyond the control of the affected Party and without the fault or negligence of such Party, which may include acts of God; material changes in Applicable Law; war; civil commotion; destruction of production facilities or materials by fire,
flood, earthquake, explosion or storm; labor disturbances; epidemic; pandemic; quarantine; and failure of public utilities or common carriers. Notwithstanding the foregoing, a Party will not be excused from making payments owed hereunder due
to any such Force Majeure circumstances affecting such Party. The Party affected by Force Majeure will immediately notify the other Party of such inability and of the period for which such inability is expected to continue. The Party giving
such notice will thereupon be excused from such of its obligations under this Agreement as it is thereby disabled from performing for so long as it is so disabled for up to [***], after which time the Parties will promptly meet to discuss in
good faith how to best proceed in a manner that maintains and abides by this Agreement. To the extent possible, each Party will use reasonable efforts to minimize the duration of any Force Majeure.
|
| 16.6 |
Notices. All notices that are required or permitted hereunder will be in writing and sufficient if delivered by internationally-recognized overnight courier or sent by registered or certified mail,
postage prepaid, return receipt requested, and in each case, addressed as follows (with a courtesy copy sent by email, which will not constitute notice):
|
| 16.7 |
Further Assurances. The Parties agree to reasonably cooperate with each other in connection with any actions required to be taken as part of their respective obligations under this Agreement, and
will (a) furnish to each other such further information; (b) execute and deliver to each other such other documents; and (c) do such other acts and things (including working collaboratively to correct any clerical, typographical, or other
similar errors in this Agreement), all as the other Party may reasonably request for the purpose of carrying out the intent of this Agreement.
|
| 16.8 |
Performance by Affiliates. Notwithstanding any provision to the contrary set forth in this Agreement, either Party will have the right to perform any or all of its obligations and exercise any or all
of its rights under this Agreement through any Affiliate. Each Party hereby guarantees the performance by its Affiliates of such Party’s obligations under this Agreement and will cause its Affiliates to comply with the provisions of this
Agreement in connection with such performance.
|
| 16.9 |
Agency. Neither Party is, nor will be deemed to be an employee, agent, or representative of the other Party for any purpose. Each Party is an independent contractor, not an employee or partner of the
other Party. Neither Party will have the authority to speak for, represent, or obligate the other Party in any way without prior written authority from the other Party.
|
| 16.10 |
Binding Effect; No Third-Party Beneficiaries or Obligors. As of the Effective Date, this Agreement will be binding upon and inure to the benefit of the Parties and their respective permitted
successors and assigns. Except as set forth in Article 13 (Indemnification), no Person other than Ionis, Otsuka, and their respective permitted successors and assigns hereunder will be deemed an intended beneficiary hereunder, nor
have any right to enforce any obligation of any Party to this Agreement, nor will any Person other than Ionis and Otsuka and their respective permitted successors and assigns have any obligations to any Party under this Agreement.
|
| 16.11 |
No Waiver. Any omission or delay by either Party at any time to enforce any right or remedy reserved to it, or to require performance of any of the terms, covenants, or provisions hereof, by the
other Party, will not constitute a waiver of such Party’s rights to the future enforcement of its rights under this Agreement. Any waiver by a Party of a particular breach or default by the other Party will not operate or be construed as a
waiver of any subsequent breach or default by the other Party.
|
| 16.12 |
Cumulative Remedies. No remedy referred to in this Agreement, including termination of this Agreement, is intended to be exclusive, but each will be cumulative and in addition to any other remedy
referred to in this Agreement or otherwise available under law.
|
| 16.13 |
Counterparts. This Agreement may be executed in one or more counterparts, all of which taken together will be regarded as one and the same instrument. Each Party may execute this Agreement in Adobe™
Portable Document Format (PDF) sent by electronic mail. PDF signatures of authorized signatories of the Parties will be deemed to be original signatures, will be valid and binding upon the Parties, and, upon delivery, will constitute due
execution of this Agreement.
|
|
Ionis Pharmaceuticals, Inc.
|
Otsuka Pharmaceutical Co., Ltd.
|
||||
|
By:
|
/s/ Brett Monia, PhD |
|
By:
|
/s/ Makoto Inoue |
|
|
Name: Brett Monia, PhD
|
Name: Makoto Inoue
|
||||
|
Title: Chief Executive Officer
|
Title: President and Representative Director
|
||||
|
|
Otsuka Pharmaceutical Co., Ltd.
|
||
|
By:
|
/s/ Takeshi Watanabe |
|
|
|
Name: Takeshi Watanabe, PhD, MBA
|
|||
|
Title: VP, Global Head of Business Development
|
|||
| 1.1 |
“Accounting Standards” means, with respect to a Party or its Affiliate or Sublicensee, GAAP or IFRS, as such Person uses for its financial reporting standards from time to time,
in each case, as consistently applied.
|
| 1.2 |
“Affiliate” means, with respect to a Person, any corporation or other business entity controlled by, controlling, or under common control with such Person, with “control” meaning
(a) direct or indirect beneficial ownership of more than 50% of the voting stock or other ownership interest of, or more than 50% interest in the income of, the applicable entity, or (b) the possession, directly or indirectly, of the power to
direct the management or policies of the applicable entity, whether through the ownership of voting securities or other equity rights, by contract relating to voting rights or corporate governance, or otherwise. Notwithstanding the foregoing,
for purposes of this Agreement, “Affiliates” will not include, (a) with respect to an entity, bona fide venture capital investors in such entity or bona fide
institutional investors in such entity, in each case, that routinely make venture capital investments for the potential financial return on such investments and not with any view to acquisition or for other strategic purpose, or Affiliates of
such venture capital or institutional investors, or (b) with respect to Otsuka, any entities that are controlled by Otsuka Holdings Co., Ltd. but are not subsidiaries of Otsuka.
|
| 1.3 |
“Alliance Manager” has the meaning set forth in Section 8.7 (Alliance Managers).
|
| 1.4 |
“[***]” means any [***].
|
| 1.5 |
“Ancillary Agreements” means the Pharmacovigilance Agreement, each Supply Agreement, and each Quality Agreement.
|
| 1.6 |
“Applicable Law” means applicable (with respect to the particular activity, task, or obligation under this Agreement to which such term applies) laws, statutes, rules,
regulations, and other pronouncements having the effect of law of any Governmental Authority that may be in effect from time to time, including for clarity any applicable rules, regulations, guidelines, or other requirements of any Regulatory
Authority that may be in effect from time to time.
|
| 1.7 |
“Approved Labeling” means, with respect to a Licensed Product and a jurisdiction: (a) the applicable Regulatory Authority-approved full prescribing information for such Licensed
Product in such jurisdiction; and (b) the applicable Regulatory Authority-approved labels and any other written, printed, or graphic materials on any container, wrapper, or any package insert that is used with or for such Licensed Product in
such jurisdiction.
|
| 1.8 |
“ASMF” has the meaning set forth in Section 5.3 (Correspondences with Regulatory Authorities).
|
| 1.9 |
“Blocking Identified Rights” has the meaning set forth in Section 2.7.2(a)ii (Acquisition of Potential In-Licenses).
|
| 1.10 |
“Breach Notification” has the meaning set forth in Section 14.2.1 (Material Breach).
|
| 1.11 |
“Business Day” means a day other than (a) a Saturday, Sunday, (b) a day on which banking institutions in California, Tokyo, Japan, or London, England are required by Applicable
Law to remain closed or (c) the nine consecutive days beginning on December 24 and continuing through January 1, to the extent not already covered in clause (a) or clause (b).
|
| 1.12 |
“Calendar Quarter” means, with respect to the first Calendar Quarter during the Term, the period beginning on the Effective Date and ending on the last day of the Calendar Quarter
within which the Effective Date falls, and thereafter each successive period of three calendar months ending on (and including) each of March 31, June 30, September 30, and December 31; except that the last Calendar Quarter during the Term
will end upon the expiration of the Term.
|
| 1.13 |
“Calendar Year” means the period of 12 consecutive calendar months beginning on January 1 and ending on December 31; except that (a) the first Calendar Year during the Term will
begin on the Effective Date and end on December 31 of the Calendar Year within which the Effective Date falls, and (b) the last Calendar Year during the Term will end upon expiration of the Term.
|
| 1.14 |
“Change of Control” means, with respect to a Party, that: (a) any Third Party acquires directly or indirectly the beneficial ownership of any voting security of such Party, or if
the percentage ownership of such Third Party in the voting securities of such Party is increased through stock redemption, cancellation, or other recapitalization, and immediately after such acquisition or increase such Third Party is,
directly or indirectly, the beneficial owner of voting securities representing more than 50% of the total voting power of all of the then outstanding voting securities of such Party; (b) a merger, consolidation, recapitalization, or
reorganization of such Party is consummated that would result in shareholders or equity holders of such Party immediately prior to such transaction owning 50% or less of the outstanding voting securities of the surviving entity (or its parent
entity) immediately following such transaction; or (c) the sale or transfer to a Third Party, in one or more related transactions, of all or substantially all of such Party’s consolidated assets taken as a whole.
|
| 1.15 |
“Clinical Supply Agreement” has the meaning set forth in Section 7.2.1 (Clinical Supply Agreement).
|
| 1.16 |
“Clinical Trial” means any clinical trial in humans.
|
| 1.17 |
“CMC” means chemistry, manufacturing, and controls.
|
| 1.18 |
“CMO” has the meaning set forth in Section 7.3.1 (By Otsuka).
|
| 1.19 |
“Collaboration In-License” means (a) any Potential In-License that [***] in accordance with Section 2.7.2(b) (Collaboration In-Licenses) and (b) any Existing Third-Party
IP Agreement.
|
| 1.20 |
“Collaboration Know-How” means all Know-How developed or invented by a Party’s or its Affiliates’, licensees’, Sublicensees’, or Subcontractors’ employees, agents, or independent
contractors, or any Persons contractually required to assign or license such Know-How to such Party or any Affiliate of such Party, either alone or jointly with the other Party’s or its Affiliates’, licensees’, Sublicensees’, or
Subcontractors’ employees, agents, or independent contractors, or any Persons contractually required to assign or license such Know-How to such other Party or any Affiliate of such other Party, in each case, in the performance of activities
under this Agreement during the Term.
|
| 1.21 |
“Collaboration Patent Rights” means any Patent Right that (a) has a priority date after the Effective Date and (b) Covers any Invention included in the Collaboration Know-How.
|
| 1.22 |
“Combination Product” has the meaning set forth in Section 1.136 of this Appendix 1 (Definitions).
|
| 1.23 |
“Combination Product Net Sales” has the meaning set forth in Section 1.136 of this Appendix 1 (Definitions).
|
| 1.24 |
“Commercial Supply Agreement” has the meaning set forth in Section 7.2.2 (Commercial Supply Agreement).
|
| 1.25 |
“Commercialization” means any and all activities directed to the marketing, promotion, distribution, pricing, reimbursement, offering for sale, and sale of a pharmaceutical or
biologic product and interacting with Regulatory Authorities following receipt of Regulatory Approval in the applicable country or region for such pharmaceutical or biologic product regarding the foregoing, including seeking and maintaining
any required Reimbursement Approval, but excluding activities directed to Manufacturing or Development. “Commercialize,” “Commercializing,”
and “Commercialized” will be construed accordingly.
|
| 1.26 |
“Commercially Reasonable Efforts” means, with respect to the Exploitation of a Licensed Product by a Party, [***].
|
| 1.27 |
“Competitive Infringement” means any infringement, unauthorized use, misappropriation or threatened infringement or misappropriation by a Third Party with respect to any Ionis
Technology, Otsuka Technology, or Joint Collaboration Technology by reason of the making, using, offering to sell, selling or importing of a compound, product, method, or process that would be competitive with a Licensed Product then being
Developed or Commercialized in the Field.
|
| 1.28 |
“Confidential Disclosure Agreement” has the meaning set forth in Section 16.2 (Entire Agreement; Amendment).
|
| 1.29 |
“Confidential Information” means, subject to Section 12.3 (Exemptions), (a) Know-How and any technical, scientific, trade, research, Manufacturing, business, financial,
marketing, product, supplier, intellectual property, and other non-public or proprietary data or information (including unpublished patent applications) that may be disclosed by one Party (the “Disclosing Party”) or its Affiliates to the other Party (the “Receiving Party”) or its Affiliates pursuant to this Agreement (including information disclosed prior
to the Effective Date pursuant to the Confidential Disclosure Agreement), regardless of whether such information is specifically marked or designated as confidential and regardless of whether such information is in written, oral, electronic,
or other form, and (b) the terms of this Agreement.
|
| 1.30 |
“Continuing Know-How Transfer” has the meaning set forth in Section 3.1 (Initial Know-How Transfer).
|
| 1.31 |
“Control” or “Controlled” means the possession by a Party (whether by
ownership, license, or otherwise other than pursuant to this Agreement) of, (a) with respect to any materials or other tangible Know-How, the legal authority or right to physical possession of such materials or tangible Know-How, with the
right to provide such materials or tangible Know-How to the other Party on the terms set forth herein, (b) with respect to Patent Rights, Regulatory Approvals, Regulatory Submissions, intangible Know-How, or other intellectual property, the
legal authority or right to grant a license, sublicense, access, or right to use (as applicable) to the other Party under such Patent Rights, Regulatory Approvals, Regulatory Submissions, intangible Know-How, or other intellectual property on
the terms set forth herein, in each case ((a) and (b)), without breaching or otherwise violating the terms of any arrangement or agreement with a Third Party in existence as of the time such Party or its Affiliates would first be required
hereunder to grant the other Party such access, right to use, licenses, or sublicense or incurring any additional payment obligations to a Third Party that would not be incurred but for such access, right to use, licenses, or sublicense,
other than payment obligations incurred under a Collaboration In-License, and (c) with respect to any product, the possession by a Party of the ability (whether by sole or joint ownership, license, or otherwise, other than pursuant to the
licenses granted under this Agreement) to grant an exclusive license or sublicense of Patent Rights that Cover such product or proprietary Know-How that is used in connection with the Exploitation of such product. Notwithstanding the
foregoing, [***].
|
| 1.32 |
“Core or Manufacturing Identified Rights” has the meaning set forth in Section 2.7.2(a)i (Acquisition of Potential In-Licenses).
|
| 1.33 |
“Core or Manufacturing Potential In-License” has the meaning set forth in Section 2.7.2(a)i (Acquisition of Potential In-Licenses).
|
| 1.34 |
“Cost Overrun” has the meaning set forth in Section 4.4.2(b) (Cost Overruns).
|
| 1.35 |
“Cover” means, with respect to a particular subject matter at issue and a relevant Patent Right or individual claim in such Patent Right, as applicable, that the manufacture, use,
sale, offer for sale, or importation of such subject matter would fall within the scope of one or more claims in such Patent Right.
|
| 1.36 |
“Cross-Territory Clinical Development Plan” has the meaning set forth in Section 4.2.1 (Cross-Territory Clinical Development Plan).
|
| 1.37 |
“Cross-Territory Clinical Studies” has the meaning set forth in Section 4.2.1 (Cross-Territory Clinical Development Plan).
|
| 1.38 |
“Debarred/Excluded” has the meaning set forth in Section 11.1.5 (Mutual Representations and Warranties).
|
| 1.39 |
“Development” means all internal and external research, development and regulatory activities regarding pharmaceutical or biologic products, including (a) research, process
development, non-clinical testing, toxicology, non-clinical activities, IND-enabling studies, and Clinical Trials, and (b) preparation, submission, review, and development of data or information for the purpose of submission to a Regulatory
Authority to obtain authorization to conduct Clinical Trials and to obtain, support, or maintain Regulatory Approval of a pharmaceutical or biologic product, but excluding activities directed to Manufacturing or Commercialization. Development
will include development and regulatory activities for additional presentations or indications for a product after receipt of Regulatory Approval of such product, including Post-Approval Mandatory Studies. “Develop,” “Developing,” and “Developed” will be construed accordingly.
|
| 1.40 |
“Development Cost Share Notice” has the meaning set forth in Section 4.4.3 (Shared Cross-Territory Development Costs).
|
| 1.41 |
“Disclosing Party” has the meaning set forth in Section 1.29 (Confidential Information) of this Appendix 1 (Definitions).
|
| 1.42 |
“Disputed Matter” has the meaning set forth in Section 15.1 (Executive Officers: Disputes).
|
| 1.43 |
“Effective Date” has the meaning set forth in the Preamble.
|
| 1.44 |
“Eligible Cross-Territory Development Costs” has the meaning set forth in Section 4.4.3 (Shared Cross-Territory Development Costs).
|
| 1.45 |
“EMA” means the European Medicines Agency or any successor agency thereto.
|
| 1.46 |
“European Union” or “E.U.” means the economic, scientific, and political organization of member states of the European Union as it may be
constituted from time to time.
|
| 1.47 |
“[***]” has the meaning set forth in Section 7.2.6 ([***]).
|
| 1.48 |
“Executive Officer” means (a) with respect to Otsuka, its President and Representative Director or their designee and (b) with respect to Ionis, the Chief Executive Officer or
their designee.
|
| 1.49 |
“Existing Third-Party IP Agreement” means any agreement between Ionis (or any of its Affiliates) and any Third Party entered into prior to the Effective Date under which Ionis (or
any of its Affiliates) obtained a license or other right to any of such Third Party’s Know-How or Patent Rights that fall within the definition of any of the Ionis Technology.
|
| 1.50 |
“Exploit” means to make, have made, use, offer to sell, sell, Develop, Manufacture, Commercialize, or otherwise exploit. “Exploitation”
will be construed accordingly.
|
| 1.51 |
“External Costs” mean, with respect to a Party, the documented actual expenses paid to Third Parties (or payable to Third Parties and accrued in accordance with such Party’s
Accounting Standards) by such Party (or its Affiliate) in consideration of the performance of activities under this Agreement, without mark-up, and excluding any costs or expenses included under the FTE Rate.
|
| 1.52 |
“FD&C Act” means the United States Federal Food, Drug and Cosmetic Act, as amended from time-to-time, together with any rules, regulations, and requirements promulgated
thereunder (including all additions, supplements, extensions, and modifications thereto).
|
| 1.53 |
“FDA” means the U.S. Food and Drug Administration or any successor agency thereto.
|
| 1.54 |
“Field” means for the treatment or prevention of any diseases and conditions in humans.
|
| 1.55 |
“Filing Party” has the meaning set forth in Section 5.5 (Regulatory Submissions).
|
| 1.56 |
“First Commercial Sale” means, with respect to a Licensed Product in a country, the first sale for end use or consumption to a Third Party of such Licensed Product in such country
by a Party, or its Affiliates or Sublicensees after the receipt of Regulatory Approval and Reimbursement Approval in the Field for such Licensed Product by the relevant Regulatory Authority in such country. First Commercial Sale excludes any
sale or other distribution for use in a Clinical Trial or other Development activity or for compassionate use, named-patient use, or expanded access, indigent or other patient access programs when sold or distributed at or below the
applicable Selling Party’s costs.
|
| 1.57 |
“[***]” has the meaning set forth in Section 9.3.4(a) ([***]).
|
| 1.58 |
“Follow-On Product” has the meaning set forth in Section 2.8.1 (ROFN Exercise).
|
| 1.59 |
“Follow-On Product Activities” has the meaning set forth in Section 2.8.3 (Follow-On Product Activities).
|
| 1.60 |
“Force Majeure” has the meaning set forth in Section 16.5 (Force Majeure).
|
| 1.61 |
“FTE” means the equivalent of the work of one duly qualified employee of a Party full time for one year (consisting of a total of [***] hours per year) directly carrying out [***]
activities under this Agreement. Overtime, and work on weekends, holidays and the like will not be counted with any multiplier (e.g., time-and-a-half or double time) toward the number of hours that are
used to calculate the FTE contribution, and no individual may be charged at greater than one FTE, regardless of that individual’s hours worked during that year. The portion of an FTE billable by a Party for one employee during a given
accounting period will be determined by dividing the number of hours worked directly by such employee on the work to be conducted under this Agreement during such accounting period by the number of FTE hours applicable for such accounting
period based on [***] working hours per Calendar Year. For clarity, travel time spent by an employee, unless also spent working directly on activities under this Agreement, will not be included in the number of hours used to calculate the FTE
contribution.
|
| 1.62 |
“FTE Rate” means [***] per FTE per hour. For the avoidance of doubt, such FTE Rate will be [***].
|
| 1.63 |
“Future Cross-Territory Studies” has the meaning set forth in Section 4.2.1 (Cross-Territory Clinical Development Plan).
|
| 1.64 |
“GAAP” means the generally accepted accounting principles in the United States.
|
| 1.65 |
“Generic Product” means, with respect to a Licensed Product in a country, a pharmaceutical product (other than such Licensed Product) that (a) is expected to be sold by a Third
Party other than a Sublicensee under license from Otsuka in such country, (b) is authorized for use in such country in one or more of the indications for which such Licensed Product has Regulatory Approval in such country; and (c) contains
the same active pharmaceutical ingredient(s) as such Licensed Product. A product will not be considered to be a Generic Product if (i) Otsuka or any of its Affiliates or Sublicensees was involved in or authorized the Development or
Commercialization of such product, (ii) Otsuka or any of its Affiliates or Sublicensees has granted a license to such Third Party in respect of such product, or (iii) such product is Commercialized by any Person who obtained such product in a
chain of distribution that included Otsuka or any of its Affiliates or Sublicensees.
|
| 1.66 |
“Global Brand Strategic and Operating Plan” has the meaning set forth in Section 6.4 (Global Brand Strategic and Operating Plan).
|
| 1.67 |
“Global Medical Affairs Plan” has the meaning set forth in Section 6.7.1 (Global Medical Affairs Plan).
|
| 1.68 |
“Global Trade Control Laws” means the U.S. Export Administration Regulations, the U.S. International Traffic in Arms Regulations, the economic sanctions regulations administered
by the U.S. Treasury Department’s Office of Foreign Assets Control, E.U. Council Regulations on export controls, including Nos. 428/2009, 267/2012, other E.U. Council sanctions regulations, as implemented in the E.U. member states, United
Nations sanctions policies, and all relevant regulations made under any of the foregoing.
|
| 1.69 |
“Good Clinical Practices” or “GCP” means the then-current good clinical practice standards, practices, and procedures
promulgated or endorsed by the applicable Regulatory Authority as set forth in the guidelines imposed by such Regulatory Authority, as may be updated from time-to-time.
|
| 1.70 |
“Good Laboratory Practices” or “GLP” means the then-current good laboratory practice standards, practices, and
procedures promulgated or endorsed by the applicable Regulatory Authority as set forth in the guidelines imposed by such Regulatory Authority, as may be updated from time-to-time.
|
| 1.71 |
“Good Manufacturing Practices” or “GMP” means the then-current good manufacturing practice standards, practices, and
procedures promulgated or endorsed by the applicable Regulatory Authority as set forth in the guidelines imposed by such Regulatory Authority, as may be updated from time-to-time.
|
| 1.72 |
“Government Official” means any official, officer, employee, or representative of: (a) any federal, state, provincial, administrative division, county, or municipal government or
any department or agency thereof; (b) any public international organization or any department or agency thereof; or (c) any company or other entity owned or controlled by any government or Governmental Authority.
|
| 1.73 |
“Governmental Authority” means any court, agency, department, authority, tribunal, or other instrumentality of any supra-national, national, state, provincial, county, city, or
other political subdivision. For clarity, Governmental Authorities include all Regulatory Authorities.
|
| 1.74 |
“HAE” means hereditary angioedema.
|
| 1.75 |
“Identified Rights” has the meaning set forth in Section 2.7.1 (Identification of New In-License Agreements).
|
| 1.76 |
“IFRS” means International Financial Reporting Standards, consistently applied.
|
| 1.77 |
“IND” means an Investigational New Drug application required pursuant to 21 C.F.R. Part 312 or any comparable filings outside of the U.S. required to commence human clinical
trials in such country or region (such as an application for a Clinical Trial Authorization in the E.U.), and all supplements or amendments that may be filed with respect to the foregoing.
|
| 1.78 |
“Indemnified Party” has the meaning set forth in Section 13.3 (Indemnification Procedure).
|
| 1.79 |
“Indemnifying Party” has the meaning set forth in Section 13.3 (Indemnification Procedure).
|
| 1.80 |
“Initial Know-How Transfer” has the meaning set forth in Section 3.1 (Initial Know-How Transfer).
|
| 1.81 |
“Initial Royalties” has the meaning set forth in Section 9.3.1 (Royalty Payments During the Initial Royalty Term).
|
| 1.82 |
“Initial Royalty Term” has the meaning set forth in Section 9.3.1 (Royalty Payments During the Initial Royalty Term).
|
| 1.83 |
“Initiation” means dosing of the first patient in a Clinical Trial.
|
| 1.84 |
“Internal Costs” means, for any period of time, the product obtained by multiplying (a) the total FTEs (or portion thereof) devoted to the performance of activity under this
Agreement during such period, by (b) the applicable FTE Rate for such period; provided that [***].
|
| 1.85 |
“Invention” means any process, method, composition of matter, article of manufacture, discovery, or finding that is conceived or reduced to practice (whether or not patentable).
|
| 1.86 |
“Ionis Attribution Language” has the meaning set forth in Section 12.7.2 (Acknowledgement).
|
| 1.87 |
“Ionis Collaboration Know-How” has the meaning set forth in Section 10.1.2(a) (Ownership of Arising Intellectual Property).
|
| 1.88 |
“Ionis Collaboration Patent Rights” has the meaning set forth in Section 10.1.2(a) (Ownership of Arising Intellectual Property).
|
| 1.89 |
“Ionis Core Technology” means Ionis Core Technology Know-How and the Ionis Core Technology Patents.
|
| 1.90 |
“Ionis Core Technology Know-How” means, subject to Section 4.4.4(b) ([***] by Ionis), all Know-How, including Ionis Collaboration Know-How but excluding Ionis
Product-Specific Know-How, Ionis Manufacturing and Analytical Know-How and Ionis’ interest in any Joint Collaboration Know-How, that (a) is Controlled by Ionis or its Affiliates as of the Effective Date or at any time during the Term, (b) is
necessary or reasonably useful to Exploit a Licensed Product, and (c) relates generally to oligonucleotide.
|
| 1.91 |
“Ionis Core Technology Patents” means, subject to Section 4.4.4(b) ([***] by Ionis), any Patent Rights, including Ionis Collaboration Patent Rights but excluding Ionis
Product-Specific Patents, Ionis Manufacturing and Analytical Patents and Ionis’ interest in any Joint Collaboration Patent Rights, that (a) are Controlled by Ionis or its Affiliates as of the Effective Date or at any time during the Term, (b)
are necessary or reasonably useful (or, with respect to patent applications, would be necessary or reasonably useful if such patent applications were to issue as patents) to Exploit a Licensed Product and (c) Cover subject matter generally
applicable to oligonucleotides. A list of the Ionis Core Technology Patents as of the Effective Date is set forth on Schedule 1.91 (Ionis Core Technology Patents); provided that, any Patent Right existing
as of the Effective Date that otherwise would be included in the definition of Ionis Core Technology Patents but is not included on Schedule 1.91 (Ionis Core Technology Patents) will still be considered a Ionis Core Technology Patent.
|
| 1.92 |
“Ionis Indemnitee” has the meaning set forth in Section 13.2 (Indemnification by Otsuka).
|
| 1.93 |
“Ionis Internal Oligonucleotide Safety Database” has the meaning set forth in Section 5.12.1 (Ionis Internal Oligonucleotide Safety Database).
|
| 1.94 |
“Ionis Know-How” means the Ionis Core Technology Know-How, Ionis Manufacturing and Analytical Know-How, and Ionis Product-Specific Know-How.
|
| 1.95 |
“Ionis Manufacturing and Analytical Know-How” means, subject to Section 4.4.4(b) ([***] by Ionis), Know-How, including Ionis Collaboration Know-How but excluding Ionis’
interest in any Joint Collaboration Know-How, that (a) is Controlled by Ionis or its Affiliates as of the Effective Date or at any time during the Term, (b) is necessary or reasonably useful to Exploit a Licensed Product, and (c) relates to
any Manufacturing Technology.
|
| 1.96 |
“Ionis Manufacturing and Analytical Patents” means, subject to Section 4.4.4(b) ([***] by Ionis), Patent Rights, including Ionis Collaboration Patent Rights but excluding
Ionis’ interest in any Joint Collaboration Patent Rights, that (a) are Controlled by Ionis or its Affiliates on the Effective Date or at any time during the Term, (b) are necessary or reasonably useful (or, with respect to patent
applications, would be necessary or reasonably useful if such patent applications were to issue as patents) to Exploit a Licensed Product, and (c) Cover Manufacturing Technology. A list of Ionis Manufacturing and Analytical Patents as of the
Effective Date is set forth on Schedule 1.96 (Ionis Manufacturing and Analytical Patents); provided that, any Patent Right existing as of the
Effective Date that otherwise would be included in the definition of Ionis Manufacturing and Analytical Patent but is not included on Schedule 1.96 (Ionis Manufacturing and Analytical
Patents) will still be considered a Ionis Manufacturing and Analytical Patent.
|
| 1.97 |
“Ionis Manufacturing and Analytical Technology” means Ionis Manufacturing and Analytical Know-How and Ionis Manufacturing and Analytical Patents.
|
| 1.98 |
“Ionis [***] Costs” has the meaning set forth in Section 4.4.4(c) (Otsuka Opt-In).
|
| 1.99 |
“Ionis [***]” has the meaning set forth in Section 4.4.4(b) ([***] by Ionis).
|
| 1.100 |
“Ionis [***]” means [***] generated by or on behalf of Ionis in the [***] in accordance with Section 4.4.4(b) ([***] by Ionis).
|
| 1.101 |
“Ionis Patent Rights” means the Ionis Core Technology Patents, Ionis Manufacturing and Analytical Patents, and Ionis Product-Specific Patents.
|
| 1.102 |
“Ionis Product-Specific Know-How” means, subject to Section 4.4.4(b) ([***] by Ionis), all Know-How, including Ionis Collaboration Know-How but excluding Ionis’ interest
in any Joint Collaboration Know-How, that is (a) Controlled by Ionis or its Affiliates as of the Effective Date or at any time during the Term, (b) necessary or reasonably useful to Exploit a Licensed Product in the Field, and (c)
specifically relating to (i) the composition of matter of a Licensed Product or (ii) methods of using a Licensed Product for the Field; provided however, Know-How that (i) consists of subject matter
applicable to oligonucleotide compounds or products in general or (ii) relates to an oligonucleotide compound that does not specifically modulate expression of PKK via the binding, partially or wholly, of such compound to RNA that encodes
PKK, will not be considered Ionis Product-Specific Know-How, and in each case of (i) and (ii), such Know-How will be considered Ionis Core Technology Know-How.
|
| 1.103 |
“Ionis Product-Specific Patents” means, subject to Section 4.4.4(b) ([***] by Ionis), all Product-Specific Patents, excluding Ionis’ interest in any Joint Collaboration
Patent Rights, that are (a) Controlled by Ionis or its Affiliates on the Effective Date or at any time during the Term and (b) necessary or reasonably useful (or, with respect to patent applications, would be necessary or reasonably useful if
such patent applications were to issue as patents) to Exploit a Licensed Product; provided, however, that Patent Rights that include only claims that are
directed to (i) subject matter applicable to oligonucleotide compounds or products in general or (ii) an oligonucleotide compound that does not specifically modulate expression of PKK via the binding, partially or wholly, of such compound to
RNA that encodes PKK, will not be considered Ionis Product-Specific Patents, and in each case of (i) and (ii), such Patent Rights will be considered Ionis Core Technology Patents. A list of Ionis
Product-Specific Patents as of the Effective Date is set forth on Schedule 1.103 (Ionis
Product-Specific Patents); provided that, any Patent Right existing as of the Effective Date that otherwise would be included in the definition of Ionis Product-Specific Patent but is not included on
Schedule 1.103 (Ionis Product-Specific Patents) will still be considered a Ionis Product-Specific Patent.
|
| 1.104 |
“Ionis Product-Specific Technology” means Ionis Product-Specific Know-How and Ionis Product-Specific Patents.
|
| 1.105 |
“Ionis Product Trademarks” has the meaning set forth in Section 10.10.1(c) (Ownership of Ionis Product Trademarks).
|
| 1.106 |
“Ionis Prosecuted Patent Rights” has the meaning set forth in Section 10.2.1(a) (Right to Prosecute).
|
| 1.107 |
“Ionis Publication” has the meaning set forth in Section 12.5.2 (Ionis’ Right to Publish).
|
| 1.108 |
“Ionis Regulatory Activities” has the meaning set forth in Section 5.1 (Regulatory Responsible Party).
|
| 1.109 |
“Ionis Technology” means the Ionis Know-How, the Ionis Patent Rights, and Ionis’ interest in the Joint Collaboration Technology.
|
| 1.110 |
“Ionis Territory” means worldwide, except for the Otsuka Territory.
|
| 1.111 |
“IRS” has the meaning set forth in Section 9.10.3 (Tax Cooperation).
|
| 1.112 |
“Joint Collaboration Know-How” means all Collaboration Know-How that is developed or invented jointly by a Party’s or its Affiliates’, licensees’, Sublicensees’, or
Subcontractors’ employees, agents, or independent contractors, or any Persons contractually required to assign or license such Collaboration Know-How to such Party or any Affiliate of such Party, on the one hand, and the other Party’s or its
Affiliates’, licensees’, Sublicensees’, or Subcontractors’ employees, agents, or independent contractors, or any Persons contractually required to assign or license such Collaboration Know-How to such Party or any Affiliate of such Party, on
the other hand.
|
| 1.113 |
“Joint Collaboration Patent Rights” means all Collaboration Patent Rights that Cover Joint Collaboration Know-How.
|
| 1.114 |
“Joint Collaboration Technology” means the Joint Collaboration Know-How and the Joint Collaboration Patent Rights.
|
| 1.115 |
“JSC” has the meaning set forth in Section 8.1.1 (Formation and Purpose of the JSC).
|
| 1.116 |
“JSC Co-Chairperson” has the meaning set forth in Section 8.1.2 (Membership).
|
| 1.117 |
“Know-How” means proprietary Inventions, discoveries, trade secrets, materials, information, experience, data, formulas, procedures, technology, and results (whether or not
patentable), including practices, knowledge, know-how, experience and test data (including physical, chemical, biological, toxicological, pharmacological, clinical and veterinary
data), dosage regimens, assays, diagnostics, product specifications, manufacturing techniques and costs, analytical and quality control data and marketing, pricing and distribution costs, and sales practices, methods, data, and descriptions.
|
| 1.118 |
“Knowledge” means the actual knowledge, without any inquiry or investigation, of (a) with respect to Ionis, its [***]; and (b) with respect to Otsuka, its [***].
|
| 1.119 |
“Licensed Compound” means the GaINAc-conjugated antisense oligonucleotide compound known as donidalorsen.
|
| 1.120 |
“Licensed Product” means any pharmaceutical product that contains, comprises, or incorporates the Licensed Compound, in all current and future formulations and in any dosage
strengths, presentations, or package configuration, and for any mode of administration. For clarity, any combination product comprised of an autoinjector pre-filled with the Licensed Compound is considered a Licensed Product. All products
containing or comprising the same Licensed Compound, regardless of the formulation, indication, line extension or otherwise, will be considered the same Licensed Product for all purposes of this Agreement.
|
| 1.121 |
“Losses” has the meaning set forth in Section 13.1 (Indemnification by Ionis).
|
| 1.122 |
“MAA” or “Marketing Authorization Application” means any (a) New Drug Application as defined in the FD&C Act, (b) a marketing authorization application filed with (i) the EMA
under the centralized EMA filing procedure to gain approval to market a biopharmaceutical in the E.U., or (ii) a Regulatory Authority in any country in the E.U. if the centralized EMA filing procedure is not used to gain approval to market a
biopharmaceutical in the E.U., or (c) substantially similar application or submission to those set forth in clause (a) or clause (b) filed with a Regulatory Authority in a country or group of countries to obtain Regulatory Approval to
Commercialize a biopharmaceutical or diagnostic product in that country or in that group of countries, in each case ((a) through (c)), including any amendments thereto, and supplemental applications, but excluding Reimbursement Approval
applications.
|
| 1.123 |
“MAA Acceptance” means, with respect to a Marketing Authorization Application filed for a Licensed Product, the receipt of written notice of acceptance by the EMA of such
Marketing Authorization Application for filing under the centralized filing procedure.
|
| 1.124 |
“[***]” means, [***].
|
| 1.125 |
“Manufacture” means activities directed to manufacturing, processing, packaging, labeling, filling, finishing, assembly, quality assurance, quality control, testing, and release,
shipping, or storage of any pharmaceutical or biologic product (or any components or process steps involving any product or any companion diagnostic), placebo, or comparator agent, as the case may be, including qualification, validation, and
scale-up, pre-clinical, clinical, and commercial manufacture and analytic development, product characterization, and stability testing, but excluding activities directed to Development, or Commercialization. “Manufacturing” and “Manufactured” will be construed accordingly.
|
| 1.126 |
“Manufacturing Costs” means, with respect to a Licensed Product [***].
|
| 1.127 |
“Manufacturing Handover Date” has the meaning set forth in Section 7.1.2 (Otsuka Manufacturing).
|
| 1.128 |
“Manufacturing Handover Notice” has the meaning set forth in Section 7.1.2 (Otsuka Manufacturing).
|
| 1.129 |
“Manufacturing Technology” means any or all of (a) methods or materials used in the synthesis or analysis of an oligonucleotide or a Licensed Product regardless of sequence or
chemical modification, (b) methods of manufacturing components of an oligonucleotide, and (c) methods or materials used in Manufacturing a Licensed Product.
|
| 1.130 |
“Manufacturing Technology Transfer” has the meaning set forth in Section 7.4 (Manufacturing Technology Transfer).
|
| 1.131 |
“Manufacturing Technology Transfer Agreement” has the meaning set forth in Section 7.4 (Manufacturing Technology Transfer).
|
| 1.132 |
“Medical Affairs” means activities conducted by a Party’s medical affairs department (or, if a Party does not have a medical affairs department, the equivalent function thereof),
including real world evidence, communications with key opinion leaders, continuing medical education, symposia, advisory boards (to the extent related to medical affairs or clinical guidance), activities performed in connection with patient
registries, review and approval of materials consistent with a Party’s or its Affiliate’s internal SOPs and Applicable Law, interactions and engagements with patient advocacy groups and other key stakeholders, and other similar medical
programs and communications.
|
| 1.133 |
“Milestone Events” means the Regulatory Milestone Events and the Sales Milestone Events.
|
| 1.134 |
“Milestone Payments” means the Regulatory Milestone Payments and the Sales Milestone Payments.
|
| 1.135 |
“Necessary Global In-License” has the meaning set forth in Section 2.7.2(a) (Acquisition of Potential In-Licenses).
|
| 1.136 |
“Net Sales” means, with respect to any Licensed Product, the amount invoiced by Otsuka or its Affiliates or Sublicensees (each a “Selling Party”) for sales of such Licensed Product in arm’s length transactions to Third Parties in all countries in the Otsuka Territory, less deduction (if not already deducted in the amount invoiced) of the following
items with respect to sales of such Licensed Product:
|
|
|
(a) |
Normal and customary trade, quantity, or cash discounts to non-affiliated brokers, agents or customers to the extent actually allowed and taken, provided that such discounts are not applied
disproportionately to the Licensed Products when compared to the other products of the Selling Party, as applicable;
|
|
|
(b) |
Actual amounts repaid or credited by reason of rejections, returns, defects, price adjustments, billing errors, or trial prescriptions, including amounts repaid, discounted or credited by reason of risk sharing schemes with respect to the
Licensed Product with any Governmental Authority;
|
|
|
(c) |
To the extent separately stated on purchase orders, invoices, or other documents of sale, any taxes, tariffs, duties, excises or other governmental charges (including any value added tax, sales tax, consumption tax or similar tax, other
than any taxes based on income) imposed or levied on the production, sale, transportation, delivery, or use, exportation or importation of the Licensed Products;
|
|
|
(d) |
Rebates, reimbursements, fees, clawbacks, discounts or chargeback payments paid, granted or credited to managed health care organizations, pharmacy benefit
managers (or equivalent thereof), national, state/provincial, local, and other governments or Governmental Authorities, their agencies/purchasers/reimbursement providers (including those requested by any Governmental Authority any time after
the actual sale of a Licensed Product), or to any Third Party payor, administrator, contractee or purchaser, including trade customers, including any fees levied by any Governmental Authority as a result of healthcare reform policies, and
including those offered as a result of the clinical or real-world performance of the Licensed Product after it is marketed and sold;
|
|
|
(e) |
Outbound transportation costs prepaid or allowed and costs of insurance in transit, with the exclusion of storage and warehousing costs;
|
|
|
(f) |
Any invoiced amounts that are not collected, including bad debts; and
|
|
|
(g) |
Any other deductions that are consistent with the Selling Party’s Accounting Standards and are not duplicative of the above deductions;
|
| 1.137 |
“[***]” has the meaning set forth in Section 4.4.4(a) (Shared Costs).
|
| 1.138 |
“[***]” has the meaning set forth in Section 4.4.4(a) (Shared Costs).
|
| 1.139 |
“Non-Clinical HAE Development Plan” has the meaning set forth in Section 4.2.2 (Non-Clinical HAE Development Plan).
|
| 1.140 |
“OFAC” means the Office of Foreign Assets Control of the United States Department of the Treasury or any successor agency thereto.
|
| 1.141 |
“Ongoing Cross-Territory Studies” has the meaning set forth in Section 4.2.1 (Cross-Territory Clinical Development Plan).
|
| 1.142 |
“Opt-In Fee” has the meaning set forth in Section 4.4.4(c) (Otsuka Opt-In).
|
| 1.143 |
“Opt-In Notice” has the meaning set forth in Section 4.4.4(c) (Otsuka Opt-In).
|
| 1.144 |
“Other Covered Party” means any political party or party official, or any candidate for political office.
|
| 1.145 |
“Other Global In-License” has the meaning set forth in Section 2.7.2(c) (Non-Approved Potential In-Licenses).
|
| 1.146 |
“Other Product” has the meaning set forth in Section 1.136 of this Appendix 1 (Definitions).
|
| 1.147 |
“Otsuka Collaboration Know-How” has the meaning set forth in Section 10.1.2(b) (Ownership of Arising Intellectual Property).
|
| 1.148 |
“Otsuka Collaboration Patent Rights” has the meaning set forth in Section 10.1.2(b) (Ownership of Arising Intellectual Property).
|
| 1.149 |
“Otsuka Indemnitee” has the meaning set forth in Section 13.1 (Indemnification by Ionis).
|
| 1.150 |
“Otsuka Know-How” means all Collaboration Know-How (excluding Otsuka’s interest in Joint Collaboration Know-How) that is (a) Controlled by Otsuka or any of its Affiliates during
the Term and (b) necessary or reasonably useful to Exploit a Licensed Product.
|
| 1.151 |
“Otsuka Patent Rights” means all Collaboration Patent Rights (excluding Otsuka’s interest in Joint Collaboration Patent Rights) that are (a) Controlled by Otsuka or any of its
Affiliates during the Term and (b) necessary or reasonably useful (or, with respect to patent applications, would be necessary or reasonably useful if such patent applications were to issue as patents) to Exploit a Licensed Product.
|
| 1.152 |
“Otsuka Product Trademarks” has the meaning set forth in Section 10.10.1(b) (Ownership of Otsuka Product Trademarks).
|
| 1.153 |
“Otsuka Publication” has the meaning set forth in Section 12.5.1 (Otsuka’s Right to Publish).
|
| 1.154 |
“Otsuka Technology” means Otsuka Know-How, Otsuka Patent Rights, and Otsuka’s interest in the Joint Collaboration Technology.
|
| 1.155 |
“Otsuka Territory” means (a) all members of the European Union or the European Economic Area (EEA) as of the Effective Date, and (b) the following countries: Iceland,
Liechtenstein, Norway, Switzerland, and the United Kingdom.
|
| 1.156 |
“Otsuka Territory Brand Strategic and Operating Plan” has the meaning set forth in Section 6.5 (Otsuka Territory Brand Strategic and Operating Plan).
|
| 1.157 |
“Otsuka Territory Medical Affairs Plan” has the meaning set forth in Section 6.7.2 (Otsuka Territory Medical Affairs Plan).
|
| 1.158 |
“Otsuka Territory-Specific Development Plan” has the meaning set forth in Section 4.3 (Otsuka Territory-Specific Development Plan).
|
| 1.159 |
“Otsuka Territory Trademark Infringement” has the meaning set forth in Section 10.10.4(a) (Unitary Product Trademark).
|
| 1.160 |
“Packaging and Labeling” means primary, secondary, or tertiary packaging and labeling of a Licensed Product (in its commercial packaging presentation) for sale or use in a
country, including the Approved Labeling and insertion of materials such as patient inserts, patient medication guides, and professional inserts and any other written, printed, or graphic materials accompanying such Licensed Product and any
brand security or anti-counterfeiting measures included in the packaging elements for such Licensed Product considered to be part of the finished packaged Licensed Product, and all testing and release thereof.
|
| 1.161 |
“Party Vote” has the meaning set forth in Section 8.4.1 (General Decision-Making Process).
|
| 1.162 |
“Patent Challenge” means, with respect to a Person, that such Person contests or assists a Third Party in contesting the scope, validity, or enforceability of a Patent Right or
any foreign counterpart thereof anywhere in the world in any court, tribunal, arbitration proceeding, or other proceeding, including the U.S. Patent and Trademark Office and the U.S. International Trade Commission. A Patent Challenge
includes: (a) filing an action under 28 U.S.C. §§ 2201-2202 seeking a declaration of invalidity or unenforceability of any such Patent Right; (b) filing, or joining in, a petition under 35 U.S.C. § 311 to institute inter partes review of any such Patent Right; (c) filing, or joining in, a petition under 35 U.S.C. § 321 to institute post-grant review of any such Patent Right or any portion thereof; (d) filing or commencing any
opposition, nullity, or similar proceedings challenging the validity of any such Patent Right in the Territory; or (e) any foreign equivalent of clauses (a), (b), (c), or (d), including any proceeding in any country or patent office in any
country or region in the Otsuka Territory.
|
| 1.163 |
“Patent Prosecution” means activities directed to (a) preparing, filing, and prosecuting applications (of all types) for any Patent Right, (b) maintaining any Patent Right, and
(c) deciding whether to abandon or maintain any Patent Right.
|
| 1.164 |
“Patent Rights” means (a) all patents, patent applications, and utility models in any country or jurisdiction, including provisional applications, priority applications, and
international applications, (b) all patent applications filed either from such patents or patent applications or from an application claiming priority from any of these, including divisionals, continuations, and continuations-in-part, (c) any
and all patents that have issued or in the future issue from the foregoing patent applications, (d) any and all substitutions, renewals, registrations, confirmations, revalidations, reissues, and re-examinations of the foregoing patents or
patent applications, and (e) extensions, restorations, supplemental protection certificates, and the like based on any of the foregoing patents or patent applications.
|
| 1.165 |
“Payment Assignment” has the meaning set forth in Section 16.1 (Assignment).
|
| 1.166 |
“Payment Forms” means one copy of each of the following documents which, at the time Ionis provides such documents to Otsuka, must be currently effective (un-expired), completed
and signed: the United States Internal Revenue Service Form 6166 (United States Residency Certification) as received from the United States Internal Revenue Service; Form 3 (Application Form for Income Tax Convention); and Form 17 (Attachment
Form for Limitation on Benefits Article).
|
| 1.167 |
“Person” means any corporation, limited or general partnership, limited liability company, joint venture, trust, unincorporated association, governmental body, authority, bureau,
or agency, or any other entity or body, or an individual.
|
| 1.168 |
“Pharmacovigilance Agreement” means an agreement regarding receipt, investigation, and reporting of product complaints, adverse events, product recalls, and any other information
related to the safety of the Licensed Products in the Territory.
|
| 1.169 |
“Phase 3 Clinical Trial” means a Clinical Trial of a Licensed Product that satisfies the requirements for a Phase 3 study as defined in 21 CFR § 312.21(c) (or any amended or
successor regulations) or that satisfies the requirements of similar laws or regulations outside the United States.
|
| 1.170 |
“[***]” means the [***].
|
| 1.171 |
“Post-Approval Cross-Territory Mandatory Studies” has the meaning set forth in Section 4.2.1 (Cross-Territory Clinical Development Plan).
|
| 1.172 |
“Post-Approval Mandatory Study” means any Clinical Trial or other study of a pharmaceutical or biologic product initiated following receipt of Regulatory Approval or to be
conducted after receipt of Regulatory Approval, in each case, that was mandated by the applicable Regulatory Authority in any country in the Territory as a condition of receiving or maintaining a Regulatory Approval for a product with respect
to a particular indication in such country (such as post-marketing approval studies and observational studies, if required by any Regulatory Authority in any country in the Territory to support or maintain Regulatory Approval for a product
in such country) or that is required for a label extension for a product in such country. For clarity, a [***] is a Post-Approval Mandatory Study.
|
| 1.173 |
“Potential In-License” has the meaning set forth in Section 2.7.2(a) (Acquisition of Potential In-Licenses).
|
| 1.174 |
“Product Materials” has the meaning set forth in Section 6.9 (Product Materials).
|
| 1.175 |
“Product-Specific Patents” means Patent Rights Controlled by a Party or any of its Affiliates as of or after the Effective Date claiming: (a) the composition of matter of a
Licensed Product, or (b) methods of using a Licensed Product.
|
| 1.176 |
“Professional Requirements” means (a) the codes and standards of the European Accreditation Council for Continuing Medical Education (EACCME) and the European Federation of
Pharmaceutical Industries and Associations (EFPIA), (b) the codes of the Prescription Medicines Code of Practice Authority (PMCPA) and the Association of the British Pharmaceutical Industry (ABPI), (c) FDA’s regulations, guidance, and
enforcement letters concerning the advertising of prescription drug products, (d) the American Medical Association’s Guidelines on Gifts to Physicians from Industry, (e) the Accreditation Council for Continuing Medical Education (ACCME)
Standards for Commercial Support of Continuing Medical Education, (f) the Pharmaceutical Supply Chain Initiative (PSCI) and Pharmaceutical Industry Principles for Responsible Supply Chain Management, (g) the Code on Interactions with
Healthcare Professionals promulgated by the Pharmaceutical Research and Manufacturers of America (PhRMA Code), (h) the Department of Health and Human Services Office of Inspector General Compliance Program Guidance for Pharmaceutical
Manufacturers (OIG Compliance Guidance), and (i) all other accepted national and international pharmaceutical industry codes of practice in and for the relevant countries in the Territory, as any of the foregoing may be amended from
time-to-time.
|
| 1.177 |
“Publication” has the meaning set forth in Section 12.5 (Publications).
|
| 1.178 |
“PV Subcommittee” has the meaning set forth in Section 5.11 (Pharmacovigilance Subcommittee).
|
| 1.179 |
“[***]” means [***].
|
| 1.180 |
“Quality Agreement” has the meaning set forth in Section 7.2.3 (Quality Agreements).
|
| 1.181 |
“Receiving Party” has the meaning set forth in Section 1.29 (Confidential Information) of this Appendix 1 (Definitions).
|
| 1.182 |
“Reduced Royalties” has the meaning set forth in Section 9.3.3 (Reduced Royalty Term).
|
| 1.183 |
“Reduced Royalty Term” has the meaning set forth in Section 9.3.3 (Reduced Royalty Term).
|
| 1.184 |
“[***]” has the meaning set forth in Section 9.3.2(c) ([***]).
|
| 1.185 |
“Regulatory Approval” means, with respect to a particular country or other regulatory jurisdiction, any approval of an MAA or other approval, product, or establishment license,
registration, or authorization of any Regulatory Authority necessary for the commercial sale of a pharmaceutical, diagnostic, or biologic product in such country or other regulatory jurisdiction, excluding, in each case, Reimbursement
Approval.
|
| 1.186 |
“Regulatory Authority” means, in a particular country or jurisdiction, any applicable Governmental Authority involved in granting Regulatory Approval in such country or
jurisdiction, including (a) in the U.S., the FDA and any other applicable Governmental Authority in the U.S. having jurisdiction over any pharmaceutical, diagnostic, or biologic product, (b) in the E.U., the EMA and any other applicable
Governmental Authority in the E.U. having jurisdiction over any pharmaceutical, diagnostic, or biologic product, and (c) in other countries, other analogous Governmental Authorities having jurisdiction over any pharmaceutical, diagnostic, or
biologic product.
|
| 1.187 |
“Regulatory Exclusivity” means, with respect to a Licensed Product in a country in the Otsuka Territory, the period of time during which: (a) Otsuka or its Affiliate or
Sublicensee has been granted the exclusive legal right by a Regulatory Authority (or is otherwise entitled to the exclusive legal right by operation of Applicable Law) in such country to market and sell such Licensed Product, and such right
precludes a Third Party from making such Licensed Product available for purchase for any indication; or (b) the data and information submitted by Otsuka or its Affiliate or Sublicensee to the relevant Regulatory Authority in such country for
purposes of obtaining Regulatory Approval of such Licensed Product may not be referenced, or relied upon in any way by a Third Party or such Regulatory Authority to support the Regulatory Approval or marketing of any product by a Third Party
in such country, or if such data and information is referenced, or relied upon to support a Regulatory Approval granted to a Third Party in such country, the product may not be placed on the market for any indication.
|
| 1.188 |
“Regulatory Milestone Events” has the meaning set forth in Section 9.2.1 (Regulatory Milestones).
|
| 1.189 |
“Regulatory Milestone Payments” has the meaning set forth in Section 9.2.1 (Regulatory Milestones).
|
| 1.190 |
“Regulatory Responsible Party” means the Party designated under Section 5.1 (Regulatory Responsible Party).
|
| 1.191 |
“Regulatory Subcommittee” has the meaning set forth in Section 5.2 (Regulatory Subcommittee).
|
| 1.192 |
“Regulatory Submission” means any filing, application, or submission with any Regulatory Authority in support of the Development, Manufacture, Commercialization, or other
Exploitation of a pharmaceutical, diagnostic, or biologic product (including to obtain, support, or maintain Regulatory Approval from that Regulatory Authority), and all written or electronic correspondence or communication with or from the
relevant Regulatory Authority, as well as minutes of any material meetings, telephone conferences, or discussions with the relevant Regulatory Authority. Regulatory Submissions include all INDs, MAAs, and other applications for Regulatory
Approval and their equivalents.
|
| 1.193 |
“Regulatory Support” has the meaning set forth in Section 5.6 (Cooperation).
|
| 1.194 |
“Reimbursement Approval” means, as applicable, (a) the Governmental Authority approval, agreement, determination, or other decision establishing prices that can be charged for a
product in regulatory jurisdictions where the applicable Governmental Authority approves or determines the prices charged to end-users for pharmaceutical, diagnostic, or biologic products, or (b) the Governmental Authority approval,
agreement, determination or decision establishing the prices at which a product will be reimbursed in regulatory jurisdictions where the applicable Governmental Authority approves, determines or recommends the reimbursement or use of
pharmaceutical, diagnostic, or biologic products.
|
| 1.195 |
“Remedial Action” has the meaning set forth in Section 5.13.1 (Notification and Determination).
|
| 1.196 |
“Representatives” means, with respect to a Person, such Person’s employees, officers, directors, consultants, contractors, Subcontractors and agents, in each case, who are
authorized to act on behalf of such Person.
|
| 1.197 |
“Requested Assistance” has the meaning set forth in Section 3.2 (Technology Transfer Costs).
|
| 1.198 |
“Restricted Party” means any individual or entity on one or more of the Restricted Party Lists.
|
| 1.199 |
“Restricted Party List” means the list of sanctioned entities maintained by the United Nations; the Specially Designated Nationals and Blocked Persons List, the Foreign Sanctions
Evaders List and the Sectoral Sanctions Identifications List, all administered by OFAC; the U.S. Denied Persons List, the U.S. Entity List, and the U.S. Unverified List, all administered by the U.S. Department of Commerce; and the entities
subject to restrictive measures and the consolidated list of Persons, Groups, and Entities Subject to E.U. Financial Sanctions, as implemented by the E.U. Common Foreign & Security Policy.
|
| 1.200 |
“Reversion License” has the meaning set forth in Section 14.9.2(a) (License Grant).
|
| 1.201 |
“Review Period” has the meaning set forth in Section 12.5.1 (Otsuka’s Right to Publish).
|
| 1.202 |
“ROFN Exercise Notice” has the meaning set forth in Section 2.8.1 (ROFN Exercise).
|
| 1.203 |
“ROFN Negotiation Period” has the meaning set forth in Section 2.8.2 (Negotiation).
|
| 1.204 |
“ROFN Notice and Package” has the meaning set forth in Section 2.8.1 (ROFN Exercise).
|
| 1.205 |
“Royalties” has the meaning set forth in Section 9.3.3 (Reduced Royalty Term).
|
| 1.206 |
“Royalty Report” has the meaning set forth in Section 9.3.4(b) (Royalty Report).
|
| 1.207 |
“Royalty Term” has the meaning set forth in Section 9.3.3 (Reduced Royalty Term).
|
| 1.208 |
“[***]” has the meaning set forth in Section 9.3.1 (Royalty Payments During the Initial Royalty Term).
|
| 1.209 |
“Rules” has the meaning set forth in Section 15.2.2 (Arbitration).
|
| 1.210 |
“Sales Milestone Events” has the meaning set forth in Section 9.2.2 (Sales Milestones).
|
| 1.211 |
“Sales Milestone Payments” has the meaning set forth in Section 9.2.2 (Sales Milestones).
|
| 1.212 |
“Selling Party” has the meaning set forth in Section 1.136 (Net Sales) of this Appendix 1 (Definitions).
|
| 1.213 |
“[***]” has the meaning set forth in Section 4.4.1(c) (Shared Development Costs).
|
| 1.214 |
“Shared Cross-Territory Development Costs” has the meaning set forth in Section 4.4.1(c) (Shared Development Costs).
|
| 1.215 |
“Shared Development Budget” has the meaning set forth in Section 4.4.2(a) (Shared Development Budget).
|
| 1.216 |
“Subcommittee” has the meaning set forth in Section 8.2.1 (Formation; Authority).
|
| 1.217 |
“Subcommittee Co-Chairperson” has the meaning set forth in Section 8.2.2 (Subcommittee Leadership and Meetings).
|
| 1.218 |
“Subcontractors” has the meaning set forth in Section 2.3.2 (Right to Subcontract).
|
| 1.219 |
“Sublicensee” means, with respect to a Party, any Third Party to which such Party or its Affiliate grants a sublicense under any of the rights licensed to the applicable Party
under this Agreement other than a Subcontractor.
|
| 1.220 |
“Supply Agreements” has the meaning set forth in Section 7.2.2 (Commercial Supply Agreement).
|
| 1.221 |
“Tax” or “Taxes” means any present or future taxes, levies, imposts, duties, charges, assessments or fees of any nature (including any
interest thereon), including value add, sales, excise or similar taxes.
|
| 1.222 |
“Technology Transfer” has the meaning set forth in Section 3.1 (Initial Know-How Transfer).
|
| 1.223 |
“Term” has the meaning set forth in Section 14.1 (Term).
|
| 1.224 |
“Territory” means (a) the Otsuka Territory, with respect to Otsuka, (b) the Ionis Territory, with respect to Ionis, and (c) collectively, worldwide.
|
| 1.225 |
“Third Party” means any Person other than a Party or its Affiliates.
|
| 1.226 |
“Third Party Claims” has the meaning set forth in Section 13.1 (Indemnification by Ionis).
|
| 1.227 |
“[***]” has the meaning set forth in Section 2.8.2 (Negotiation).
|
| 1.228 |
“Third Party Patent Challenge” has the meaning set forth in Section 10.4 (Defense of Third Party Patent Challenges).
|
| 1.229 |
“Third Party Payments” means, with respect to a Licensed Product, any (a) payments (including upfront payments, milestone payments, license fees, royalties and monetary damages)
made by Otsuka or its Affiliate to a Third Party (i) pursuant to an agreement between Otsuka or its Affiliate and such Third Party entered into following the Effective Date in accordance with Section 2.7.2 (Potential In-Licenses) to
obtain rights to Patent Rights or Know-How from such Third Party that would be infringed or misappropriated by the Exploitation of a Licensed Product in the Otsuka Territory or (ii) pursuant to an agreement between Otsuka or its Affiliate and
such Third Party, or otherwise, as part of a settlement or to satisfy a judgment in accordance with Section 10.5.3 (Settlement), or (b) amounts for which Otsuka reimburses Ionis under a Collaboration In-License, in each case ((a) or
(b)), that are directly in consideration for or reasonably allocable to a license or sublicense (as applicable) to Otsuka or its Affiliate under, or are paid in settlement or to satisfy a judgment of a claim relating to, Patent Rights or
Know-How Controlled by such Third Party that would, but for a license thereunder, be infringed or misappropriated by the Exploitation of such Licensed Product.
|
| 1.230 |
“Trademark” means any word, name, symbol, color, shape, designation or any combination thereof, including any trademark, service mark, trade name, brand name, sub-brand name,
domain name, trade dress, product configuration, program name, delivery form name, certification mark, collective mark, logo, tagline, slogan, design or business symbol, that functions as an identifier of source or origin, whether or not
registered and all statutory and common law rights therein and all registrations and applications therefor, together with all goodwill associated with, or symbolized by, any of the foregoing.
|
| 1.231 |
“Trademark Infringement Suit” has the meaning set forth in Section 10.10.5(a) (Unitary Product Trademark).
|
| 1.232 |
“Transition Plan” has the meaning set forth in Section 14.9.3(a) (Scope).
|
| 1.233 |
“Transition Services” has the meaning set forth in Section 14.9.3(a) (Scope).
|
| 1.234 |
“Unitary Product Trademark” has the meaning set forth in Section 10.10.1(a) (Unitary Product Trademark).
|
| 1.235 |
“Upfront Payment” has the meaning set forth in Section 9.1 (Upfront Payment).
|
| 1.236 |
“U.S.” means the United States of America (including all possessions and territories thereof, including Puerto Rico).
|
| 1.237 |
“U.S. Dollars” or “$” means the legal tender of the U.S.
|
| 1.238 |
“Valid Claim” means a claim of an issued and unexpired patent (as may be adjusted through a patent term adjustment or extended through supplementary protection certificate or
patent term extension or the like) that has not been revoked, held invalid, or held unenforceable by a patent office or other Governmental Authority of competent jurisdiction in a final and non-appealable judgment (or judgment from which no
appeal was taken within the allowable time period), and [***].
|
| 1.239 |
“Withheld Amount” has the meaning set forth in Section 9.10.2 (Withholding Tax).
|
|
Ionis File No.
|
Country/Treaty
|
Application No./
Patent No.
|
Filing Date
|
Grant Date
|
Title
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
Ionis File No.
|
Country
|
Application No./
Patent No.
|
Filing Date
|
Grant Date
|
Title
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
Ionis File No.
|
Country/
Treaty
|
Status
|
Application No./
Patent No.
|
Filing Date
|
Grant Date
|
Title
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
| Ionis File No. |
Country/
Treaty
|
Status |
Application No./
Patent No.
|
Filing Date | Grant Date | Title |
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
| Ionis File No. |
Country/
Treaty
|
Status |
Application No./
Patent No.
|
Filing Date | Grant Date | Title |
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
| Ionis File No. |
Country/
Treaty
|
Status |
Application No./
Patent No.
|
Filing Date | Grant Date | Title |
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
[***]
|
/s/ ERNST & YOUNG LLP |
|
San Diego, California |
|
February 21, 2024 |
1. |
I have reviewed this Annual Report on Form 10-K of Ionis Pharmaceuticals, Inc.; |
2. |
Based on my knowledge, this annual report does not contain any untrue statement of a material fact or omit to state a material fact necessary to make the statements made, in light of the circumstances under which such statements were made, not misleading with respect to the period covered by this annual report; |
3. |
Based on my knowledge, the consolidated financial statements, and other financial information included in this annual report, fairly present in all material respects the financial condition, consolidated results of operations and consolidated cash flows of the registrant as of, and for, the periods presented in this annual report; |
4. |
The registrant’s other certifying officer and I are responsible for establishing and maintaining disclosure controls and procedures (as defined in Exchange Act Rules 13a-15(e) and 15d-15(e)) and internal control over financial reporting (as defined in Exchange Act Rules 13a-15(f) and 15d-15(f)) for the registrant and have: |
a) |
Designed such disclosure controls and procedures, or caused such disclosure controls and procedures to be designed under our supervision, to ensure that material information relating to the registrant, including its consolidated subsidiaries, is made known to us by others within those entities, particularly during the period in which this report is being prepared; |
b) |
Designed such internal control over financial reporting, or caused such internal control over financial reporting to be designed under our supervision, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles; |
c) |
Evaluated the effectiveness of the registrant’s disclosure controls and procedures and presented in this report our conclusions about the effectiveness of the disclosure controls and procedures, as of the end of the period covered by this report based on such evaluation; and |
d) |
Disclosed in this report any change in the registrant’s internal control over financial reporting that occurred during the registrant’s most recent fiscal quarter (the registrant’s fourth fiscal quarter in the case of an annual report) that has materially affected, or is reasonably likely to materially affect, the registrant’s internal control over financial reporting; and |
5. |
The registrant’s other certifying officer and I have disclosed, based on our most recent evaluation of internal control over financial reporting, to the registrant’s auditors and the audit committee of registrant’s board of directors (or persons performing the equivalent functions): |
a) |
All significant deficiencies and material weaknesses in the design or operation of internal control over financial reporting which are reasonably likely to adversely affect the registrant’s ability to record, process, summarize and report financial information; and |
b) |
Any fraud, whether or not material, that involves management or other employees who have a significant role in the registrant’s internal control over financial reporting. |
Dated: February 21, 2024 |
|
/s/ BRETT P. MONIA |
|
Brett P. Monia, Ph.D. |
|
Chief Executive Officer |
1. |
I have reviewed this Annual Report on Form 10-K of Ionis Pharmaceuticals, Inc.; |
2. |
Based on my knowledge, this annual report does not contain any untrue statement of a material fact or omit to state a material fact necessary to make the statements made, in light of the circumstances under which such statements were made, not misleading with respect to the period covered by this annual report; |
3. |
Based on my knowledge, the consolidated financial statements, and other financial information included in this annual report, fairly present in all material respects the financial condition, consolidated results of operations and consolidated cash flows of the registrant as of, and for, the periods presented in this annual report; |
4. |
The registrant’s other certifying officer and I are responsible for establishing and maintaining disclosure controls and procedures (as defined in Exchange Act Rules 13a-15(e) and 15d-15(e)) and internal control over financial reporting (as defined in Exchange Act Rules 13a-15(f) and 15d-15(f)) for the registrant and have: |
a) |
Designed such disclosure controls and procedures, or caused such disclosure controls and procedures to be designed under our supervision, to ensure that material information relating to the registrant, including its consolidated subsidiaries, is made known to us by others within those entities, particularly during the period in which this report is being prepared; |
b) |
Designed such internal control over financial reporting, or caused such internal control over financial reporting to be designed under our supervision, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles; |
c) |
Evaluated the effectiveness of the registrant’s disclosure controls and procedures and presented in this report our conclusions about the effectiveness of the disclosure controls and procedures, as of the end of the period covered by this report based on such evaluation; and |
d) |
Disclosed in this report any change in the registrant’s internal control over financial reporting that occurred during the registrant’s most recent fiscal quarter (the registrant’s fourth fiscal quarter in the case of an annual report) that has materially affected, or is reasonably likely to materially affect, the registrant’s internal control over financial reporting; and |
5. |
The registrant’s other certifying officer and I have disclosed, based on our most recent evaluation of internal control over financial reporting, to the registrant’s auditors and the audit committee of registrant’s board of directors (or persons performing the equivalent functions): |
a) |
All significant deficiencies and material weaknesses in the design or operation of internal control over financial reporting which are reasonably likely to adversely affect the registrant’s ability to record, process, summarize and report financial information; and |
b) |
Any fraud, whether or not material, that involves management or other employees who have a significant role in the registrant’s internal control over financial reporting. |
Dated: February 21, 2024 |
|
/s/ ELIZABETH L. HOUGEN |
|
Elizabeth L. Hougen |
|
Chief Financial Officer |
| 1. | The Company’s Annual Report on Form 10-K for the year ended December 31, 2023, to which this Certification is attached as Exhibit 32.1 (the “Annual Report”), fully complies with the requirements of Section 13(a) or Section 15(d) of the Securities Exchange Act of 1934, as amended; and |
| 2. | The information contained in the Annual Report fairly presents, in all material respects, the financial condition of the Company at the end of the period covered by the Annual Report and the results of operations of the Company for the period covered by the Annual Report. |
Dated: February 21, 2024 |
|||
/s/ BRETT P. MONIA |
/s/ ELIZABETH L. HOUGEN |
||
Brett P. Monia, Ph.D. |
Elizabeth L. Hougen |
||
Chief Executive Officer |
Chief Financial Officer |
| 1. |
Introduction
|
| 2. |
Effective Date
|
| 3. |
Definitions
|
| 4. |
RECOUPMENT
|
| (c) |
Impracticability of Recovery. Recoupment may be determined to be impracticable if, and only if:
|
| 5. |
Administration
|
| 6. |
Severability
|
| 7. |
No Impairment of Other Remedies
|
| 8. |
Amendment; Termination
|
| 9. |
Successors
|
| 10. |
Required Filings
|
|
Agreed and Acknowledged:
|
||
|
|
||
| Name: |
|
|
| Title: |
|
|
| Date: |
|
|